Результат интеллектуальной деятельности: ПИКОЛИНАМИДНЫЕ И ПИРИМИДИН-4-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИХ
Вид РИД
Изобретение
Область техники
[1] Настоящее изобретение относится к новым амидным соединениям, т.е. пиколинамидным и пиримидин-4-карбоксамидным соединениям, способу их получения, фармацевтической композиции, включающей их, и к медицинскому применению с использованием соединения в качестве агента для предотвращения, регулирования и лечения заболеваний, связанных с регулированием глюкокортикоидов путем использования селективной ингибирующей активности соединения в отношении 11β-HSD1 фермента.
Предшествующий уровень техники
[2] Глюкокортикоиды (кортизол человека) выполняет важную роль в поддержании гомеостаза глюкозы и метаболизме липидов и белка в организме. В частности, избыточные глюкокортикоиды в печени и адипозных тканях вызывают метаболические синдромы, такие как резистентность к инсулину, висцеральное ожирение, гипертензию и дислипидемию.
[3] Известно, что 11β-гидроксистероидная дегидрогеназа (11β-HSD) имеет два вида изозимов, типа 1 и типа 2. Первый, (11β-HSD1) является NADPH-зависимой редуктазой, и важным ферментом для превращения неактивного глюкокортикоида, кортизона, в активный глюкокортикоид, кортизол, в тканях печени, адипозной и головного мозга. 11β-HSD2 выполняет действие противоположное 11β-HSD1 NAD-зависимым образом, и экспрессируется главным образом в почках.
[4] Сообщалось, что 11β-HSD1-сверхэкспрессированные трансгенные мыши имеют в крови нормальный уровень кортизола, но имеют повышенный уровень кортизола в адипозной или жировой ткани, что вызывает инсулиновую устойчивость, висцеральное ожирение, гиперлипидемию и гипертензию, и что они показывают гораздо большее увеличение веса тела и гораздо большую скорость увеличения веса тела, чем не-трансгенная группа мышей [Masuzaki H. Science 2001, 294, 2166-2170; Masuzaki H. J. Clin. Invest. 2003, 112, 83-90]. Дополнительно сообщалось, что 11β-HSD1 нокаутированные мыши показывают улучшение переносимости глюкозы, ухудшение триглицеридов в крови и увеличение HDL-холестерина [Morton N. M. J. Biol. Chem. 2001, 276, 41293-41300].
[5] Карбеноксолон (CBX), который является неселективным ингибитором 11β-HSD1, улучшает восприимчивость к инсулину у здоровых представителей и пациентов с диабетом типа 2, но нетучных [Andrew, R. C. J. Clin. Endocrinol. Metab. 2003, 88, 285-291]. Однако, сообщалось, что СВХ вызывает гипокалиемию и гипертензию вследствие неселективного ингибирования 11β-HSD1 и 11β-HSD2, и таким образом развитие их ограничивается терапевтическими агентами [Kotelevtsev, Y. J, Clin. Invest. 1999, 103, 683-689].
[6] Следовательно, эффективные и селективные ингибиторы 11β-HSD1 ферментов ингибируют превращение глюкокортикоидов в активный тип, подавляя действие глюкокортикоидов в тканях, и в результате они могут использоваться в качестве терапевтических агентов в отношении метаболических синдромов, вызываемых глюкокортикоидами, таких как не-инсулин зависимый диабет типа 2, ожирение, гиперлипидемия, гипертензия, переносимость глюкозы и аналогичные.
[7] По этой причине настоящие изобретатели исследовали соединения, обладающие эффективной и селективной ингибиторной активностью в отношении 11β-HSD1 ферментов с целью регулирования или лечения метаболических синдромов, таких как ожирение, диабет и пр. В результате, они смогли синтезировать новые пиколинамидные и пиримидин-4-карбоксамидные соединения, и удостоверились, что новые соединения проявляют эффективную и селективную ингибиторную активность в отношении 11β-HSD1 ферментов.
Раскрытие изобретения
Техническая проблема
[8] Целью настоящего изобретения является создание нового амидного соединения, т.е. пиколинамидных и пиримидин-4-карбоксамидных соединений, или их фармацевтически активной соли, сольвата, гидрата, пролекарства, рацемата или стереоизомера.
[9] Еще одной целью настоящего изобретения является создание фармацевтической композиции для ингибирования 11β-HSD1 ферментов человеческого происхождения, включающей новое амидное соединение или его фармацевтически активную соль, сольват, гидрат, пролекарство, рацемат или стереоизомер в качестве активного ингредиента.
[10] Еще одной целью настоящего изобретения является создание агента для предотвращения, регулирования и лечения заболеваний, связанных с регулированием глюкокортикоидов, в которые вовлечены 11β-HSD1 ферменты человеческого происхождения, например, метаболических синдромов, таких как диабет типа 1 и типа 2, осложнения после диабета, латентный аутоиммунный диабет (LADA), синдромы инсулинорезистентности, ожирение, нарушеная толерантность к глюкозе (IGT), нарушение гликемии натощак (IFG), поврежденная толерантности к глюкозе, дислипидемия, атеросклероз, гипертензия и др.
Решение проблемы
[11] Согласно одному общему аспекту настоящее изобретение предоставляет новые амидные соединения, представленные ниже формулой 1, т.е. пиколинамидные и пиримидин-4-карбоксамидные соединения, или их фармацевтически активную соль, сольват, гидрат, пролекарство, рацемат или стереоизомер.
[12] [Формула 1]
[13]
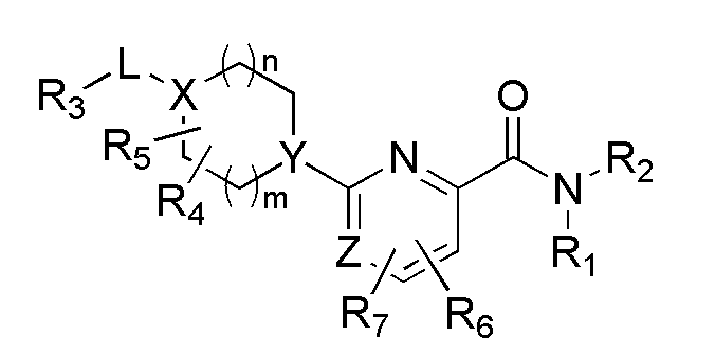
[14] [В формуле 1,
[15] Х представляет N или CR, и Y представляет N или CH, при условии, что X и Y не являются в одно и то же время углеродом;
[16] Z представляет N или CH;
[17] R1 и R2 представляют независимо водород, (С1-10)алкил, (С3-С10)циклоалкил, норборнил, адамантил, норадамантил, (С6-20)арил, (С6-С20)ар(С1-С10)алкил или (С3-С20)гетероарил, или R1 и R2 могут быть связаны друг с другом вместе с атомом азота, к которому они присоединены, образуя (С1-С10)насыщенный или ненасыщенный гетероцикл, бигетероцикл или сконденсированный гетероцикл, при условии, что R1 и R2 не являются в одно и то же время водородом;
[18] L представляет одинарную связь, -O-, -NR11-, -CO-, -SO2-, -(CR21R22)-(CH2)c- (с представляет целое число от 0 до 5), -CO(CR21R22)d- (d представляет целое число 1-6), (С3-С10)циклоалкилен, (С6-С20)арилен или (С3-С20)гетероарилен;
[19] R21 и R22 представляют независимо водород или (С1-С10)алкил, или R21 и R22 могут быть связаны через алкилен или алкенилен с образованием циклоалифатического кольца или ароматического кольца;
[20] R и R3 представляют независимо водород, (С1-С10)алкил, (С3-С10)циклоалкил, (С1-С10)алкокси, галоген, гидрокси, циано, -NR31R32, нитро, -CONH2, -CO2R33, -SO3H, -SO2NR34R35, -SO2R36, -O(CH2)aCO2H (а представляет целое число 1-3), O(CH2)bCONH2 (b представляет целое число 1-3), -NH(CO)R37, -NH(SO2)R38, 5- - 7-членный гетероцикл, (С6-С20)арил или (С3-С20)гетероарил;
[21] R4 и R5 независимо представляют водород, (С1-С10)алкил, (С3-С10)циклоалкил, (С1-С10)алкокси, галоген, гидрокси, циано, амино, нитро, -CONH2 или -CO2R12, и включают все изомеры и рацемические соединения их всех, или R4 и R5 могут быть замещены соседними атомами углерода, образуя (С1-С10) насыщенный или ненасыщенный карбоцикл, гетероцикл, бикарбоцикл, бигетероцикл, сконденсированный карбоцикл, или сконденсированный гетероцикл, или могут быть связаны с R3, образуя насыщенный или ненасыщенный карбоцикл;
[22] R6 и R7 представляют независимо водород, (С1-С10)алкил, (С3-С10)циклоалкил, (С1-С10)алкокси, галоген, гидрокси, циано, амино, нитро, -CONH2 или -CO2R12;
[23] циклоалкилен, арилен или гетероарилен группы L; алкил, циклоалкил, норборнил, адамантил, норадамантил, арил, аралкил или гетероарил групп R1 и R2; насыщенный или ненасыщенный гетероцикл, бигетероцикл или сконденсированный гетероцикл, образуемый связью R1 и R2; алкил, циклоалкил, алкокси, гетероцикл, арил или гетероарил групп R и R3; алкил, циклоалкил или алкокси групп R4 и R5; насыщенный или ненасыщенный карбоцикл, гетероцикл, бикарбоцикл, бигетероцикл, сконденсированный карбоцикл или сконденсированный гетероцикл, образуемый замещением R4 и R5 соседними атомами углерода; и алкил, циклоалкил или алкокси групп R6 и R7 могут быть дополнительно замещены одним или более заместителями, выбранными из группы, состоящей из (С1-С10)алкила, (С3-С10)циклоалкила, (С1-С10)алкокси, галоген(С1-С10)алкила, галоген(С1-С10)алкокси, галогена, гидрокси, циано, -NR41R42, нитро, -CO2R43, -CONH2, -SO3H, -SO2NR44R45, -SO2(CH2)cNR44R45 (c представляет целое число 1-3), -SO2R46,-O(CH2)cCO2H (с представляет целое число 1-3), -O(CH2)dCONH2 (d представляет целое число 1-3), -NH(CO)R47, -NH(SO2)R48, (С6-С20)арил и (С3-С20)гетероарил;
[24] R11, R12, R31, R32, R33, R34, R35, R36, R37, R38, R41, R42, R43, R44, R45, R46, R47 и R48 представляют независимо водород, (С1-С10)алкил, (С3-С10)циклоалкил или (С6-С20)арил; и
[25] m и n независимо представляют целое число 0-3, при условии, что m+n представляют целое число 2 или более.]
[26] В описании R1 и R2 независимо представляют водород, (С3-С10)циклоалкил, норборнил, адамантил, норадамантил или (С6-С20)ар(С1-С10)алкил, или R1 и R2 могут быть связаны друг с другом, образуя гетероцикл, выбранный из следующих:
[27]
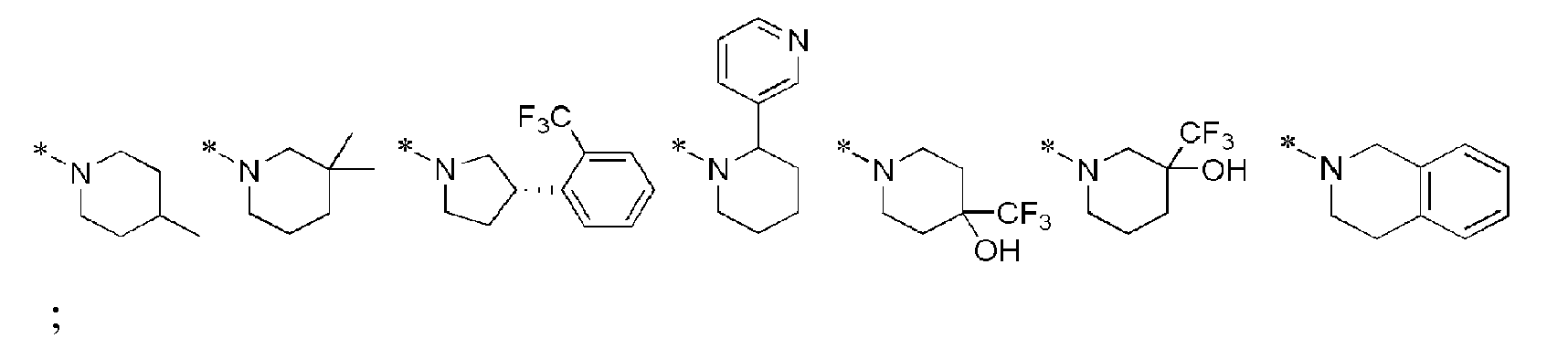 ;
;
[28] L представляет одинарную связь, -CO-, -SO2-, -(CR21R22)-(CH2)c- (c представляет целое число 0-5),
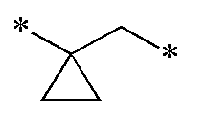 ,
,
-CO(CR21R22)d (d представляет целое число 1-6), (С3-С10)циклоалкилен, (С6-С20)арилен или (с3-С20)гетероарилен;
[29] R21 и R22 независимо представляют водород или (С1-С10)алкил;
[30] R3 представляет водород, (С1-С10)алкил, (С3-С10)циклоалкил, (С1-С10)алкокси, галоген, гидрокси, циано, -NR31R32, нитро, -CONH2, -CO2R33, -SO2NR34R35, -SO2R36, -O(CH2)aCO2H (a представляет целое число 1-3), -O(CH2)bCONH2 (b представляет целое число 1-3), -NH(CO)R37, -NH(SO2)R38, 5- - 7-членный гетероцикл, (С6-С20)арил или (С3-С20)гетероарил;
[31] R6 и R7 независимо представляют водород, (С1-С10)алкил или галоген;
[32] циклоалкилен, арилен или гетероарилен группы L; циклоалкил, норборнил, адамантил, норадамантил или аралкил групп R1 и R2; алкил, циклоалкил, алкокси, гетероцикл, арил или гетероарил группы R3; и алкил групп R6 и R7 могут быть дополнительно замещены одним или более заместителями, выбранными из группы, состоящей из (С1-С10)алкила, (С3-С10)циклоалкила, (С1-С10)алкокси, галоген(С1-С10)алкила, галоген(С1-С10)алкокси, галогена, гидрокси, циано, -NR41R42, нитро, -CO2R43, -CONH2, -SO3H, -SO2NR44R45, -SO2(CH2)cNR44R45 (c представляет целое число 1-3), -SO2R46, -O(CH2)cCO2H (с представляет целое число 1-3), -O(CH2)dCONH2 (d представляет целое число 1-3), -NH(CO)R47, -NH(SO2)R48, (С6-С20)арил и (С3-С20)гетероарил; и
[33] R11, R12, R31, R32, R33, R34, R35, R36, R37, R38, R41, R42, R43, R44, R45, R46, R47 и R48 представляют независимо водород, (С1-С10)алкил, (С3-С10)циклоалкил или (С6-С20)арил.
[34] Соединения настоящего изобретения могут быть получены с помощью использования известного метода органического синтеза, включая методы, описанные подробно в примерах.
[35] Пиколинамидное соединение настоящего изобретения можно получить согласно схеме 1 ниже
[36] [Схема 1]
[37]
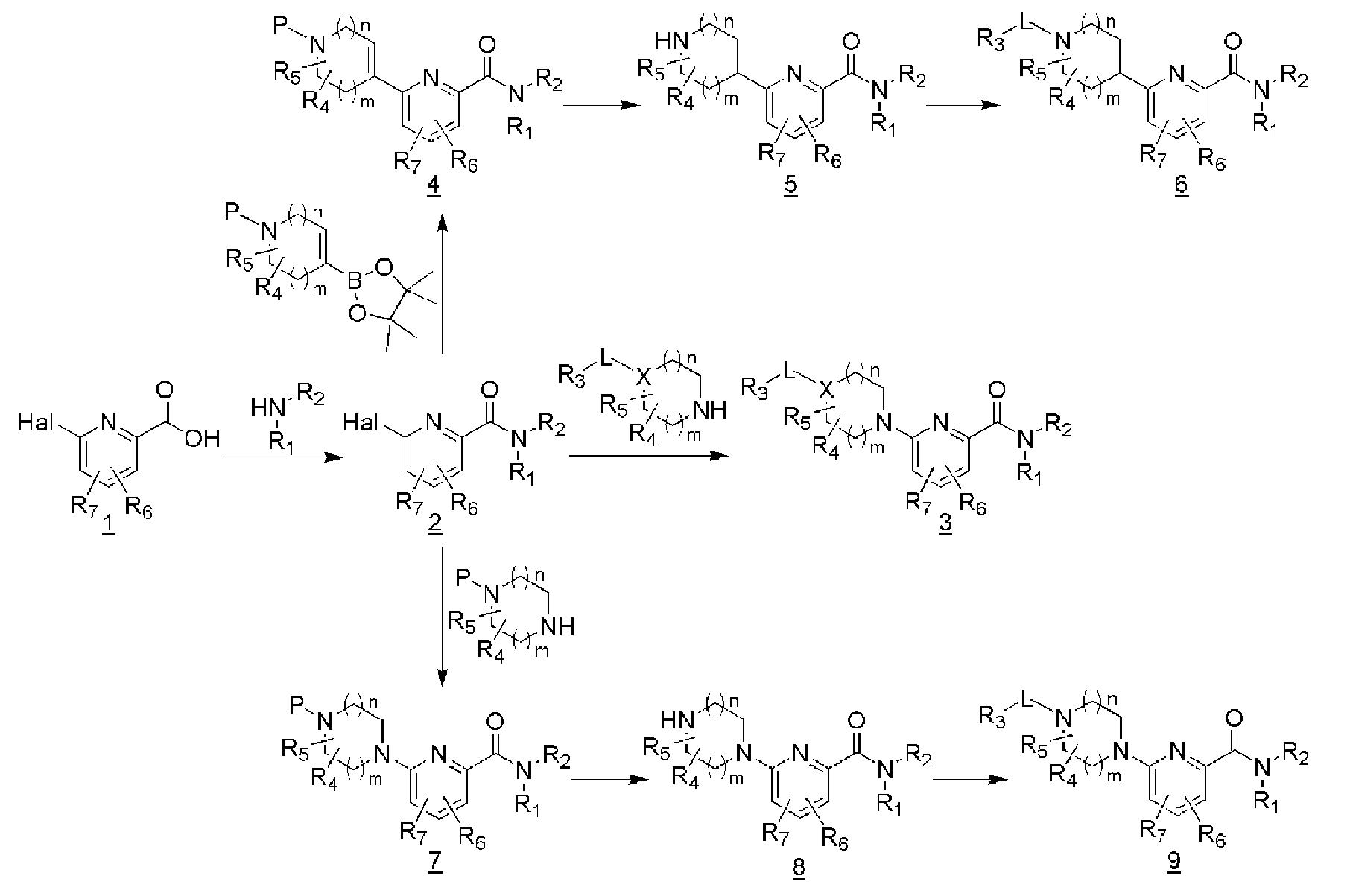
[38] (В схеме 1 Р представляет собой защитную группу.)
[39] Пиколиновую кислоту (1), замещенную в 6-положении галогеном или трифлатом, подвергали реакции с соответствующим амином и реагентами сочетания при комнатной температуре с получением промежуточного соединения 2. Далее вводили соответственно замещенный гетероцикл, содержащий атом азота, с последующей микроволновой реакцией, высокотемпературной реакцией или реакцией, катализируемой металлом, или аналогичным с получением конечного соединения 3.
[40] Дополнительно, промежуточное соединение 2 подвергали реакции с гетероциклом, включающим в себя один атом N, защищенный соответствующей защитной группой, и еще один атом N, с помощью микроволновой реакции, высокотемпературной реакции или катализируемой металлом реакции, с последующим снятием защиты с получением промежуточного соединения 8. Различные заместители (-L-R3) вводили в него с помощью метода алкилирования, карбонилирования, сульфонилирования, восстановительного аминирования, сочетания с использованием металлических катализаторов, или аналогичных, обеспечивая возможность эффективного синтеза различных производных.
[41] С другой стороны, чтобы получить соединение 6, в котором гетероцикл связан с 6-положением пиколинамида С-С связью, промежуточное соединение 2 подвергали реакции с гетероциклом, включающим 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан с N атомом, защищенным соответствующей защитной группой, с помощью катализируемой металлом реакции, с последующей реакцией гидрирования и снятия защиты, с получением промежуточного соединения 5. Различные заместители (-L-R3) могут вводиться туда с помощью метода алкилирования, карбонилирования, сульфонилирования, восстановительного аминирования, сочетания с использованием металлических катализаторов, или аналогичных.
[42] В случае, когда каждое из конечных соединений 3, 6 и 9 имеет функциональную группу из NO2, CN, CO2R, CO2H, NH2, OH или аналогичную, дальнейшее конечное соединение может быть получено с помощью реакции восстановления, гидролиза, аминирования, алкилирования, карбонилирования, сульфонилирования или аналогичных.
[43] Между тем, пиримидин-4-карбоксамидное соединение настоящего изобретения можно получить согласно схеме 2 ниже.
[44] [Схема 2]
[45]
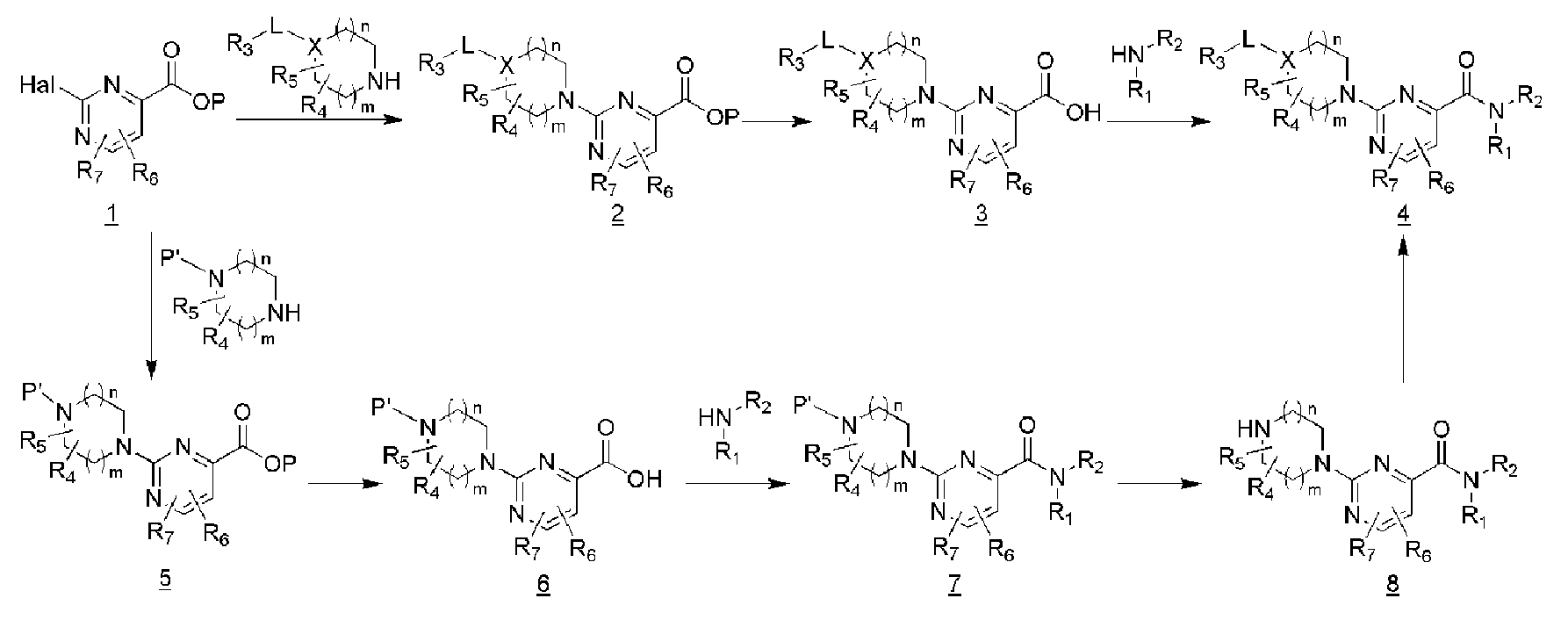
[46] (На схеме 2 Р и Р' являются защитными группами.)
[47] Пиримидин-4-карбоновую кислоту, замещенную в 2-положении галогеном или трифлатом защищали соответствующей защитной группой (1), а затем подвергали реакции с соответственно замещенным гетероциклом, включающим N атом с помощью микроволновой реакции или высокотемпературной реакции с последующим снятием защиты, обеспечивая возможность синтеза промежуточного соединения 3. Данное соединение вводили в реакцию с соответствующим амином и реагентами сочетания при комнатной температуре с получением конечного соединения 4.
[48] В дополнение, соединение 1 подвергали реакции с гетероциклом, включающим один N атом, защищенным соответствующей защитной группой, и другой N атом, с помощью микроволновой или высокотемпературной реакции с последующим снятием защиты защищяющей карбоновую кислоту группы с получением промежуточного соединения 6. Данное соединение подвергали реакции с соответствующим амином и реагентами сочетания при комнатной температуре с последующим снятием защиты защищающей амин группы, обеспечивая возможность синтеза промежуточного соединения 8. Различные заместители (-L-R3) вводили в него с помощью метода алкилирования, карбонилирования, сульфонилирования, восстановительного аминирования, сочетания с использованием металлических катализаторов, или аналогичных, обеспечивая возможность эффективного синтеза различных производных.
[49] В случае, когда конечные соединения 4 имеют функциональную группу, такую как NO2, CN, CO2R, CO2H, NH2, OH или аналогичную, дальнейшее конечное соединение может быть получено с помощью реакции восстановления, гидролиза, аминирования, алкилирования, карбонилирования, сульфонилирования или аналогичных.
[50] Согласно еще одному общему аспекту настоящее изобретение предоставляет фармацевтическую композицию, включающую амидное соединение формулы 1, или его фармацевтически приемлемую соль, сольват, гидрат, пролекарство, рацемат или стереоизомер и фармацевтически приемлемый носитель.
[51] Согласно одному общему аспекту настоящее изобретение предоставляет 11β-HSD1 ингибитор, включающий амидное соединение формулы 1, или его фармацевтически активную соль, сольват, гидрат, пролекарство, рацемат или стереоизомер. Более того, настоящее изобретение предоставляет фармацевтическую композицию для лечения и/или предотвращения заболеваний, вызываемых, опосредуемых и/или развивающихся под действием высокого уровня кортизола, фармацевтическую композицию, включающую амидное соединение формулы 1, или его фармацевтически приемлемую соль, сольват, гидрат, пролекарство, рацемат или стереоизомер и фармацевтически приемлемый носитель.
[52] Согласно еще одному общему аспекту настоящее изобретение предоставляет фармацевтическую композицию для лечения и/или предотвращения метаболических синдромов, диабета, особенно не-инсулин зависимого диабета, преддиабета, инсулиновой толерантности, низкой толерантности к глюкозе, гипергликемии, ожирения и связанных с весом расстройств, дислипидемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, липидных расстройств, таких как низкие уровни HDL или высокие уровни LDL, глаукомы, остеопороза, познавательных расстройств, глюкокортикоид-опосредуемых действий на функции нейронов, таких как беспокойство или депрессия, нейродегенеративных расстройств, иммунных расстройств, таких как туберкулез, лепра или псориаз, гипертензии, атеросклероза и их осложнений, рестеноза сосудов, сердечно-сосудистого заболевания, панкреатита, ретинита, нейропатии или нефропатии, фармацевтическую композицию, включающую амидное соединение формулы 1, или его фармацевтически приемлемую соль, сольват, гидрат, пролекарство, рацемат или стереоизомер и фармацевтически приемлемый носитель.
[53] Амидное соединение настоящего изобретения может обычно использоваться в виде свободной кислоты или свободного основания. В отличие от этого амидное соединение настоящего изобретения может использоваться в виде аддитивной соли кислоты или основания. Кислотно-аддитивная соль свободного аминового соединения настоящего изобретения может быть получена с помощью хорошо известного в технике метода, и образуется из органической кислоты и неорганической кислоты. Подходящие примеры органической кислоты включают малеиновую, фумаровую, бензойную, аскорбиновую, янтарную, метан-сульфоновую, уксусную, трифторуксусную, щавелевую, пропионовую, винную, салициловую, лимонную, глюконовую, молочную, миндальную, коричную, аспарагиновую, стеариновую, пальмитиновую, гликолевую, глютаминовую и бензолсульфоновую кислоту. Подходящие примеры неорганической кислоты включают соляную, бромистоводородную, серную, фосфорную и азотную кислоту. Примеры аддитивных солей основания включают соли, образуемые вместе с карбоксилатными анионами, и включают соли, образуемые с органическими и неорганическими катионами, например, катионами, выбранными из щелочных и щелочно-земельных металлов (например, лития, натрия, калия, магния, бария и кальция), и аммониевыми ионами и их замещенными производными (например, дибензиламмонием, бензиламмонием, 2-гидроксиэтиламмонием или аналогичными). Следовательно, имеется в виду, что термин “фармацевтически приемлемая соль” соединения формулы 1 включает любые приемлемые типы солей.
[54] Далее, в объем настоящего изобретения может быть включено пролекарство. Пролекарство, когда вводится пациенту, представляет собой ковалентно связанный носитель, в котором соединение формул 1 высвобождается in vivo. Пролекарство обычно получают модификацией функциональной группы, и данная модификация прерывается с помощью общепринятых операций или в организме, генерируя исходное соединение. Пролекарство, когда оно вводится пациенту, прерывается с образованием гидрокси, аминовой или сульфгидрильной группы, и таким образом пролекарство включает соединение настоящего изобретения, связанное с группой, образующей гидрокси, аминовую или сульфгидрильную группу. Следовательно, характерные представители примеров пролекарства включают, но не ограничиваются ими, ацетатные, формиатные и бензоатные производные спиртовых и аминовых функциональных групп соединений формулы 1. Кроме того, что касается карбоновокислотной группы (-СООН), могут использоваться группы сложных эфиров, таких как метилового эфира, этилового эфира или аналогичных.
[55] Что касается стереоизомера, соединение формулы 1 может иметь хиральный центр, и может существовать в виде рацемата, рацемической смеси и индивидуального энантиомера или диастереомера. Данные изомеры могут разделяться или расщепляться общепринятым методом, и любой конкретный изомер может быть получен с помощью общепринятого метода синтеза или стереоспецифического или асимметрического синтетического метода. В объем настоящего изобретения включены все данные изомерные типы и их смеси.
[56] Некоторые из кристаллических форм соединения формулы 1 могут существовать в полиморфной форме, и данная форма включена в настоящее изобретение. Кроме того, некоторые из соединений формулы 1 могут образовывать гидраты или сольваты вместе с водой или другими органическими растворителями. Данные гидраты или сольваты также включены в объем настоящего изобретения.
[57] Фармацевтическая композиция настоящего изобретения может включать в качестве активного ингредиента амидное соединение, представленное формулой 1, его фармацевтически приемлемую соль, сольват, гидрат, пролекарство, рацемат или стереоизомер, и к ним могут добавляться обычные нетоксичные фармацевтически приемлемые носители, адьюванты, наполнители или аналогичные для формулирования в области фармацевтики обычной рецептурной формы, такой как таблетки, капсулы, пастилки, жидкости, суспензии или аналогичные, или парентеральной рецептурной формы.
[58] Наполнители, используемые в фармацевтической композиции настоящего изобретения, могут включать подсластитель, связующее, растворитель, агент, способствующий растворению, смачивающий агент, эмульгатор, агент, придающий внешний вид, адсорбент, дезинтегрирующий агент, антиоксидант, консерваторы, агент скольжения, наполнитель, ароматизирующий агент и аналогичные. Например, в состав композиции могут включаться лактоза, декстроза, сахароза, манит, сорбит, целлюлоза, глицин, двуокись кремния, тальк, стеариновая кислота, стерин, стеарат магния, алюминийсиликат магния, крахмал, желатин, камедь трагаканта, альгиновая кислота, альгинат натрия, метилцеллюлоза, натриевая карбоксиметилцеллюлоза, агар-агар, вода, этанол, полиэтиленгликоль, поливинилпирролидон. Хлорид натрия, хлорид кальция, апельсиновая эссенция, земляничная эссенция, ванильный ароматизатор и аналогичные.
Преимущественные эффекты изобретения
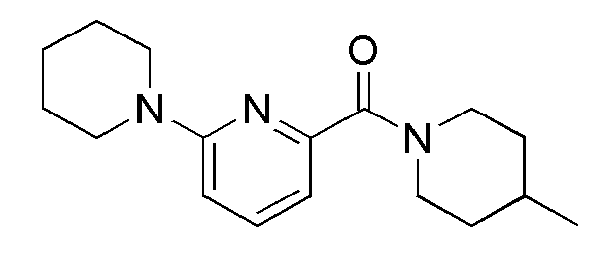
[59] Новые пиколинамидные и пиримидин-4-карбоксамидные соединения настоящего изобретения, их фармацевтически приемлемая соль, сольват, гидрат, пролекарство, рацемат или стереоизомер обладает селективной ингибиторной активностью в отношении 11β-HSD1 ферментов человеческого происхождения. Поэтому, соединения настоящего изобретения оказывают полезные действия как агенты для предотвращения, регулирования и лечения заболеваний, связанных с регулированием глюкокортикоидов, которые вызываются активностью 11β-HSD1 ферментов, например, метаболических синдромов, таких как диабет 1 и 2 типа, диабет после осложнений, латентный аутоиммунный диабет взрослых (LADA), синдромы инсулинорезистентности, ожирение, нарушенная толерантность к глюкозе (IGT), нарушенная гликемия натощак (IFG), поврежденная толерантность к глюкозе, дислипидемия, атеросклероз, гипертензия и проч.
Способ изобретения
[60] Описываемое выше настоящее изобретения будет описано подробно ниже на основе примеров, экспериментальных примеров и примеров получения. Однако, данные примеры, экспериментальные примеры и примеры получения являются только иллюстративными примерами и не предназначены для ограничения объема настоящего изобретения.
[61] [Примеры]
[62] Пример 1: Синтез N-циклогексил-6-(пиперидин-1-ил)пиколинамида
[63]

[64] Стадия 1: Синтез 6-бром-N-циклогексилпиколинамида (Промежуточное соединение 1)
[65] После того, как 6-бромпиколиновую кислоту (500 мг, 2,48 ммоль) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли циклогексиламин (0,34 мл, 2,97 ммоль), N,N-диизопропилэтиламин (0,65 мл, 3,72 ммоль) и HBTU (1,13 г, 2,97 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (EtOAc/гексаны), получая 624 мг бесцветного масла (94%).
[66] Стадия 2: Синтез N-циклогексил-6-(пиперидин-1-ил)пиколинамида
[67] 6-Бром-N-циклогексилпиколинамид (55 мг, 0,194 ммоль), пиперидин (18 мг, 0,213 ммоль), Pd2(dba)3 (3,5 мг, 0,00388 ммоль), ксантфос (6,7 мг, 0,0116 ммоль) и трет-бутоксид натрия (27,4 мг, 0,285 ммоль) суспендировали в толуоле (3 мл), и затем перемешивали при 100°С в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (30% EtOAc/гексаны), получая 51 мг бледно желтого масла (83%). MS (ESI): 288[M+H]+
[68]
[69] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 1 выше, с использованием промежуточного соединения 1 и соответствующего аминового исходного материала.
[70] Таблица 1
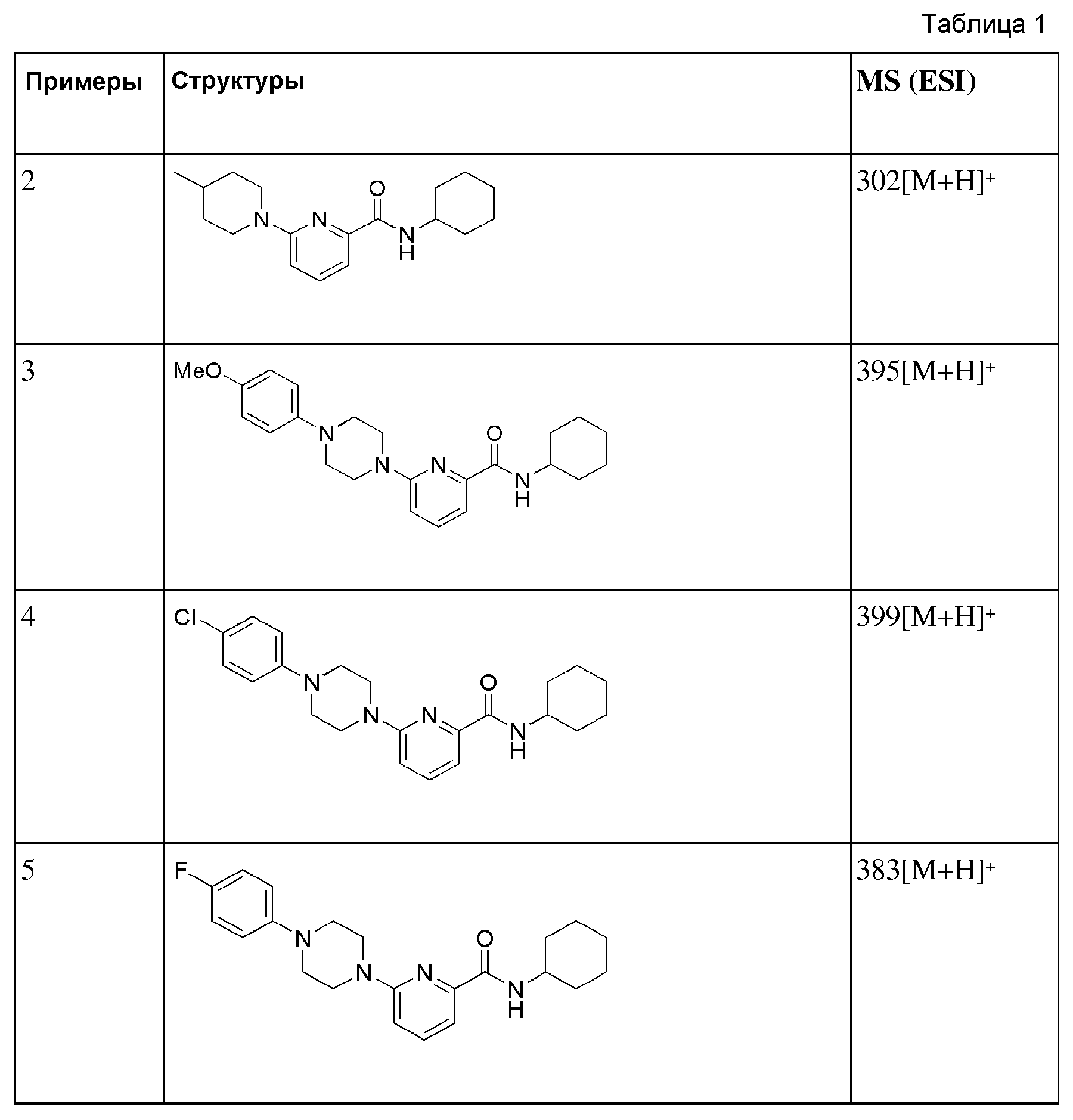
[71]
[72] Пример 6: Синтез (4-метилпиперидин-1-ил)(6-(пиперидин-1-ил)пиридин-2-ил)метанон
[73]
[74] Стадия 1: Синтез (6-бромпиридин-2-ил)(4-метилпиперидин-1-ил)метанона (промежуточное соединение 2)
[75] После того, как 6-бромпиколиновую кислоту (500 мг, 2,48 ммол) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли 4-метилпиперидинин (0,44 мл, 3,72 ммоль), N,N-диизопропилэтиламин (0,65 мл, 3,72 ммоль) и HBTU (1,13 г, 2,97 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 4 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (EtOAc/гексаны), получая 677 мг белого твердого вещества (97%).
[76] Стадия 2: Синтез (4-метилпиперидин-1-ил)(6-пиперидин-1-ил)пиридлин-2-ил)метанона
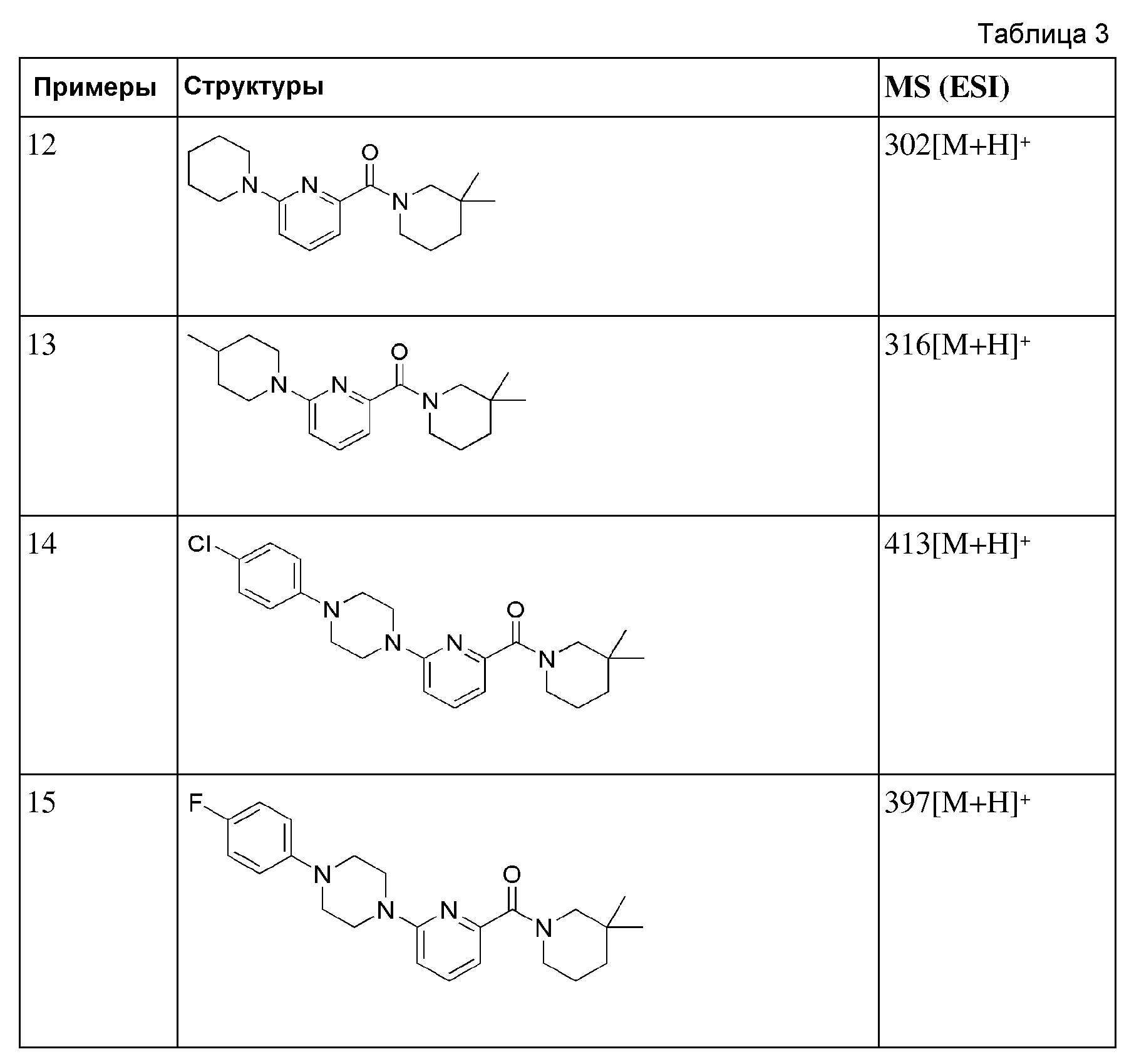
[77] (6-бромпиридин-2-ил)(4-метилпиперидин-1-ил)метанон (40 мг, 0,142 ммоль), пиперидин (13 мг, 0,156 ммоль), Pd2(dba)3 (2,6 мг, 0,00284 ммоль), ксантфос (5,0 мг, 0,00864 ммоль) и трет-бутоксид натрия (20,4 мг, 0,208 ммоль) суспендировали в толуоле (2 мл), и затем перемешивали при 100°С в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (30% EtOAc/гексаны), получая 31 мг бледно желтого масла (80%). MS (ESI): 288[M+H]+
[78]
[79] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 6 выше, с использованием промежуточного соединения 2 и соответствующего аминового исходного материала.
[80] Таблица 2
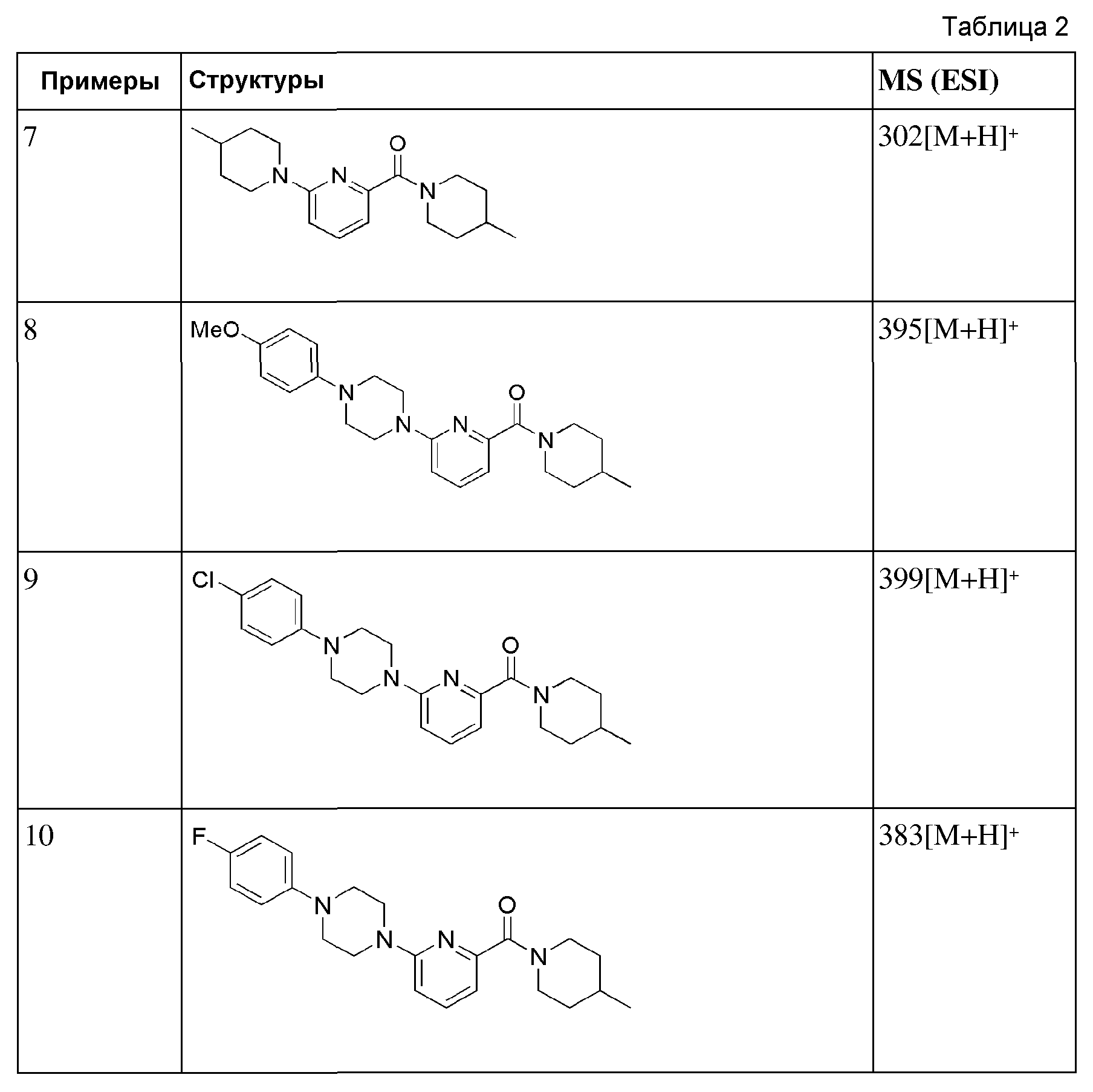
[81]
[82] Пример 11: Синтез (3,3-диметилпиперидин-1-ил)(6-(4-(4-метоксифенил)пиперазин-1-ил)пиридин-2-ил)метанона
[83]
[84] Стадия 1: Синтез (6-бромпиридин-2-ил)(3,3-диметилпиперидин-1-ил)метанона (промежуточное соединение 3)
[85] После того, как 6-бромпиколиновую кислоту (200 мг, 0,99 ммол) суспендировали в ацетонитриле (8 мл), к ней последовательно добавляли 3,3-диметилпиперидинин (0,17 мл, 1,19 ммоль), N,N-диизопропилэтиламин (0,26 мл, 1,49 ммоль) и HBTU (0,45 г, 1,19 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 3 часов.
[86] К получающейся в результате реакционной жидкости добавляли дистиллированную воду (15 мл) с последующей экстракцией МС (50 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (20% EtOAc/гексаны), получая 292 мг бесцветного масла (99%).
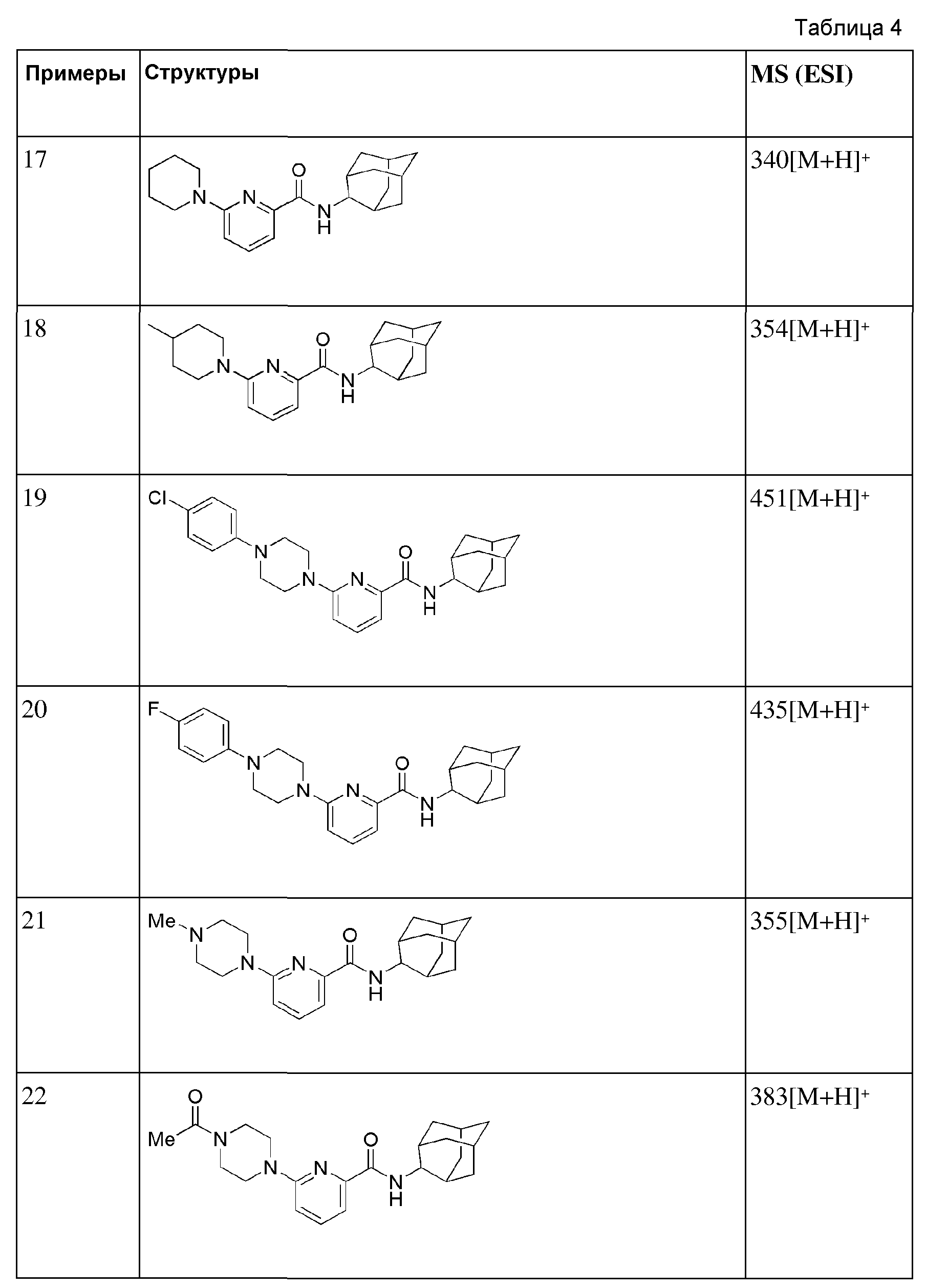
[87] Стадия 2: Синтез (3,3-диметилпиперидин-1-ил)(6-(4-(4-метоксифенил)ппперазин-1-ил)пиридин-2-ил)метанона
[88] (6-бромпиридин-2-ил)(3,3-диметилпиперидин-1-ил)метанон (40 мг, 0,135 ммоль), 1-(4-метоксифенил)пиперазин (29 мг, 0,149 ммоль), Pd2(dba)3 (2,5 мг, 0,00273 ммоль), ксантфос (4,7 мг, 0,00812 ммоль) и трет-бутоксид натрия (19,0 мг, 0,198 ммоль) суспендировали в толуоле (2 мл), и затем перемешивали при 100°С в токе азота в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 47 мг бледно желтого твердого вещества (85%). MS (ESI): 409[M+H]+
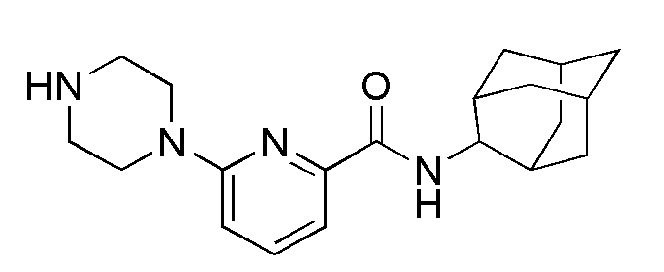
[90] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 11 выше, с использованием промежуточного соединения 3 и соответствующего аминового исходного материала.
[91] Таблица 3
[92]
[93] Пример 16: Синтез N(адамантан-2-ил)-6-(4-(4-метоксифенил)пиперазин-1-ил)пиколинамид
[94]
[95] Стадия 1: Синтез N(адамантан-2-ил)-6-бромопиколинамид (Промежуточное соединение 4)
[96] После того, как 6-бромпиколиновую кислоту (500 мг, 2,48 ммоль) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли 2-гидрохлорид адамантиламина (558 мг, 2,97 ммоль), N,N-диизопропилэтиламин (1,30 мл, 7,44 ммоль) и HBTU (1,13 г, 2,97 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (30%Teac/гексаны), получая 602 мг белого твердого вещества (73%).
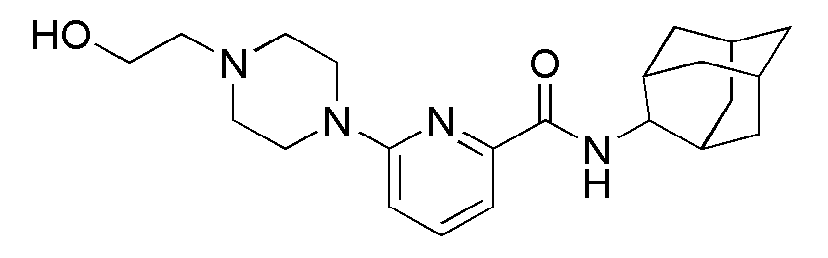
[97] Стадия 2: Синтез N(адамантан-2-ил)-6-(4-(4-метоксифенил)пиперазин-1-ил)пиколинамид
[98] N(адамантан-2-ил)-6-бромопиколинамид (55 мг, 0,194 ммоль), 1-(4-метоксифенил)пиперазин (41 мг, 0213 ммоль), PD2(dba)3 (3,5 мг 0,00388 ммоль), ксантфос (6,7 мг, 0,0116 ммоль) и трет бутоксид натрия (27,4 мг, 0,285 ммоль) суспендировали в толуоле (3 мл), и затем перемешивали при 100°С в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (30% EtOAc/гексаны), получая 64 мг бледно-желтого твердого вещества (84%). MS (ESI): 447[M+H]+
[99]
[100] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 16 выше, с использованием промежуточного соединения 4 и соответствующего аминового исходного материала.
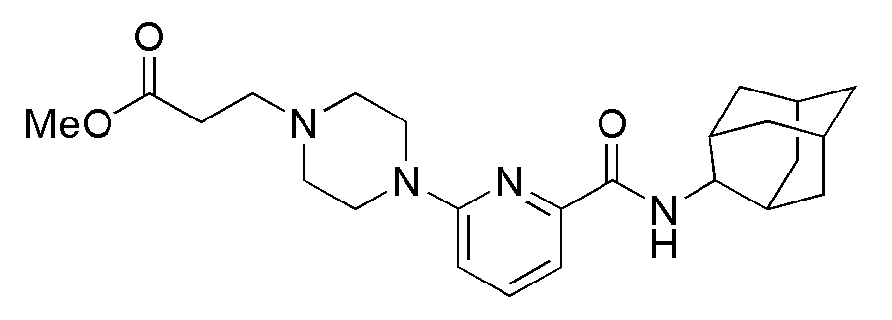
[101] Талица 4
[102]
[103] Пример 23: Синтез N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид
[104]
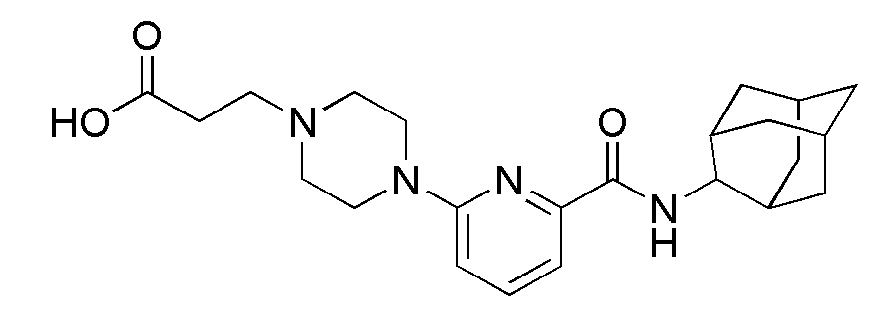
[105] Стадия 1: Синтез трет-бутил 4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-карбоксилат
[106] N(адамантан-2-ил)-6-бромопиколинамид (80 мг, 0,239 ммоль), трет-бутил пиперазин-1-карбоксилат (49 мг, 0,263 ммоль), PD2(dba)3 (4,4 мг 0,005 ммоль), ксантфос (8,3 мг, 0,014 ммоль) и трет бутоксид натрия (34 мг, 0,359 ммоль) суспендировали в толуоле (3 мл), и затем перемешивали при 100°С в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (40% EtOAc/гексаны), получая 86,8 мг бледно желтого масла (83%)
[107] Стадия 2: Синтез N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид
[108] После того как трет-бутил 4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-карбоксилат (86 мг, 0,195 ммоль) растворяли в МС (2 мл), с последующим последовательным добавлением трифторуксусной кислоты (2 мл), а затем получающуюся в результате жидкость перемешивали в потоке азота при комнатной температуре в течение 4 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением насыщенного водного раствора NaHCO3 (10 мл), а затем экстрагировали МС (15 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% MeOH/MC), получая 65 мг бесцветного масла (98%). MS (ESI): 341[M+H]+
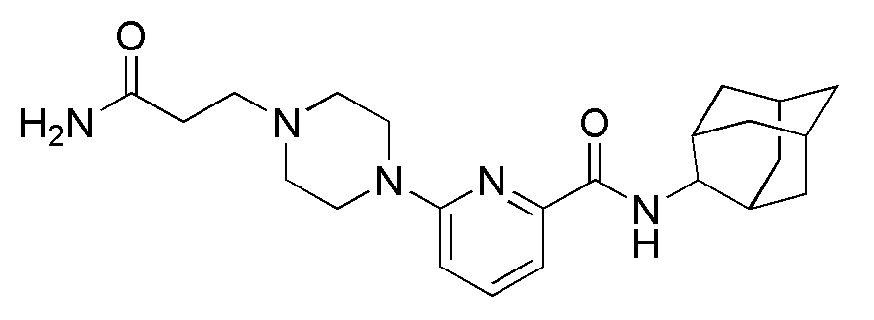
[109]
[110] Пример 24:Синтез N-(адамантан-2-ил)-6-(4-(2-гидроксиэтил)пиперазин-1-ил)пиколинамид
[111]
[112] После того как N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид (34 мг, 0,100 ммоль) растворяли в DMF (2 мг), с последующим последовательным добавлением 2-бромоэтанола (19 мг, 0,150 ммоль) и карбоната калия (41 мг, 0,300 ммоль), а затем получающуюся в результате смесь перемешивали при 100°С в токе азота в течение 18 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 37 мг бесцветного масла (96%). MS (ESI): 385[M+H]+.
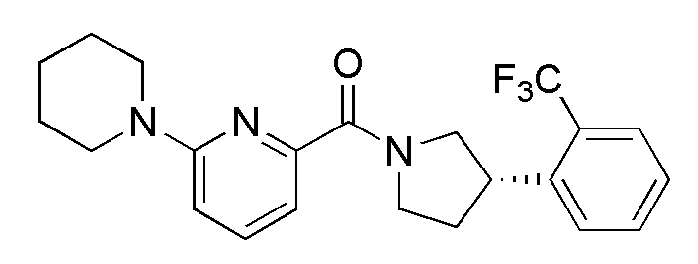
[113]
[114] Пример 25: Синтез метил 3-(4-(6(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропанат
[115]
[116] После того как N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид (34 мг, 0,100 ммоль) растворяли в ДМФ (2 мл), туда добавляли метиловый эфир 3-бромпропановой кислоты (25 мг, 0,150 ммоль)и карбонат калия (41 мг, 0,300 ммоль), а затем получающуюся в результате смесь перемешивали при 100°С в токе азота в течение 24 часов. Получающуюся в результате реакционную жидкость фильтровали и концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (60% EtOAc/гексаны), получая 41 мг белого твердого вещества (96%). MS (ESI): 427[M+H]+.
[117]
[118] Пример 26: Синтез (3-(4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропановая кислота
[119]
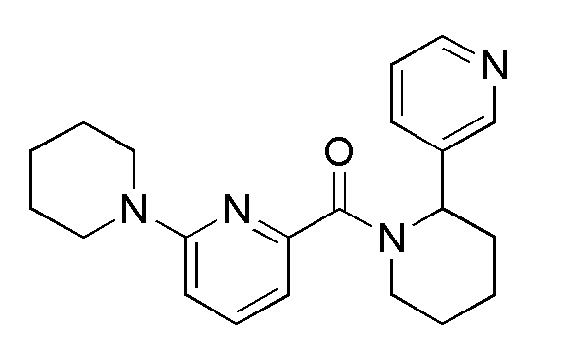
[120] Метил (3-(4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноат (43 мг, 0,101 ммоль) добавляли 4н водного раствора HCI (2 мл), а затем нагревали с обратным холодильником в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем нейтрализовали медленным добавлением насыщенного водного раствора NaHCO3, с последующей экстракцией раствором смеси (10 мл × 2) тетрагидрофуран:MC=4:1. Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% MeOH/MC), получая 27 мг бледно желтого масла (65%). MS (ESI):413[M+H]+
[121]
[122] Пример 27: Синтез N-(адамантан-2-ил)-6-4-(3-амино-3-оксопропил)пиперазин-1-ил)пиколинамид
[123]
[124] 7N аммония в метаноле (2мл) добавляли в метил 3-(4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноат (94 мг,0,22 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в течение 48 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении, а полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 54 мг бесцветного масла (60%). MS (ESI): 412[M+Н]+.
[125]
[126] Пример 28: Синтез (S)-(6-(пиперидин-1-ил)пиридин-2-ил)(3-(2-трифторметил)фенил)пирролидин-1-ил)метанона
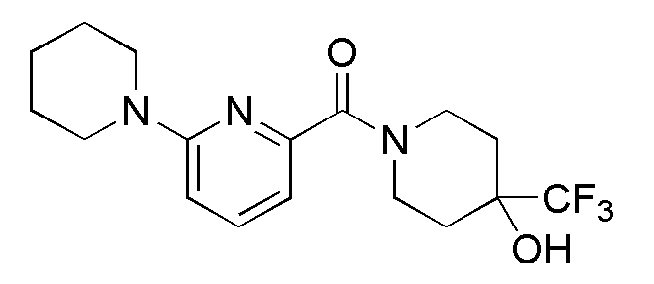
[127]
[128] Стадия 1: Синтез (S)-(6-бромпиридин-2-ил)(3-(2-трифторметил)фенил)пирролидин-1-ил)метанона (промежуточное соединение 5)
[129] После того, как 6-бромпиколиновую кислоту (430 мг, 2,13 ммоль) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли (S)-3-(2-трифторметил)фенил)пирролидин (503 мг, 2,34 ммоль), N,N-диизопропилэтиламин (0,56 мл, 3,20 ммоль) и HBTU (969 мг, 2,56 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (40% EtOAc/гексаны), получая 765 мг белого твердого вещества (90%).
[130] Стадия 2: Синтез (S)-(6-(пиперидин-1-ил)пиридин-2-ил)(3-(2-трифторметил)фенил)пирролидин-1-ил)метанона
[131] (S)-(6-бромпиридин-2-ил)(3-(2-трифторметил)фенил)пирролидин-1-ил)метанон (60 мг, 0,150 ммоль), пиперидин (15 мг, 0,180 ммоль), Pd2(dba)3 (3 мг, 0,003 ммоль), ксантфос (5 мг, 0,009 ммоль) и трет-бутоксид натрия (22 мг, 0,225 ммоль) суспендировали в толуоле (3 мл) и затем перемешивали при 100°С в токе азота в течение 3 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (15 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (450 EtOAc/гексаны), получая 39 мг бледно желтого масла (64%). MS (ESI): 404[M+Н]+.
[132]
[133] Пример 29: Синтез (6-(пиперидин-1-ил)пиридин-2-ил)(2-(пиридин-3-ил)пиперидин-1-ил)метанона
[134]
[135] Стадия 1: Синтез (6-бромпиридин-2-ил)(2-(пиридин-3-ил)пиперидин-1-ил)метанона (промежуточное соединение 6)
[136] После того, как 6-бромпиколиновую кислоту (565 мг, 2,80 ммоль) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли анабазин (499 мг, 3,07 ммоль), N,N-диизопропилэтиламин (0,73 мл, 4,20 ммоль) и HBTU (1,27 г, 3,36 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (40% EtOAc/гексаны), получая 255 мг белого твердого вещества (26%).
[137] Стадия 2: Синтез (6-(пиперидин-1-ил)пиридин-2-ил)(2-(пиридин-3-ил)пиперидин-1-ил)метанона
[138] (6-Бромпиридин-2-ил)(2-(пиридин-3-ил)пиперидин-1-ил)метанон (60 мг, 0,173 ммоль), пиперидин (18 мг, 0,208 ммоль), Pd2(dba)3 (3 мг, 0,003 ммоль), ксантфос (6 мг, 0,010 ммоль) и трет-бутоксид натрия (25 мг, 0,260 ммоль) суспендировали в толуоле (3 мл) и затем перемешивали при 100°С в токе азота в течение 3 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (15 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (90% EtOAc/гексаны), получая 39 мг бледно желтого масла (64%). MS (ESI): 351[M+Н]+.
[139]
[140] Пример 30: Синтез (4-гидрокси-4-(трифторметил)пиперидин-1-ил)(6-пиперидин-1-ил)пиридин-2-ил)метанона
[141]
[142] Стадия 1: Синтез 1-(6-бромпиколиноил)пиперидин-4-она
[143] После того, как 6-бромпиколиновую кислоту (600 мг, 2,97 ммоль) суспендировали в ацетонитриле (25 мл), к ней последовательно добавляли гидрохлорид моногидрата 4-пиперидона (500 мг, 3,27 ммоль), N,N-диизопропилэтиламин (1 мл, 7,43 ммоль) и HBTU (1,1 г, 3,56 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 13 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением МС (50 мл), а затем промывали дистиллированной водой. Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 752 мг желтого твердого вещества (89%).
[144] Стадия 2: Синтез (6-бромпиридин-2-ил)(4-гидрокси-4-(трифторметил)пиперидин-1-ил)метанона (промежуточное соединение 7)
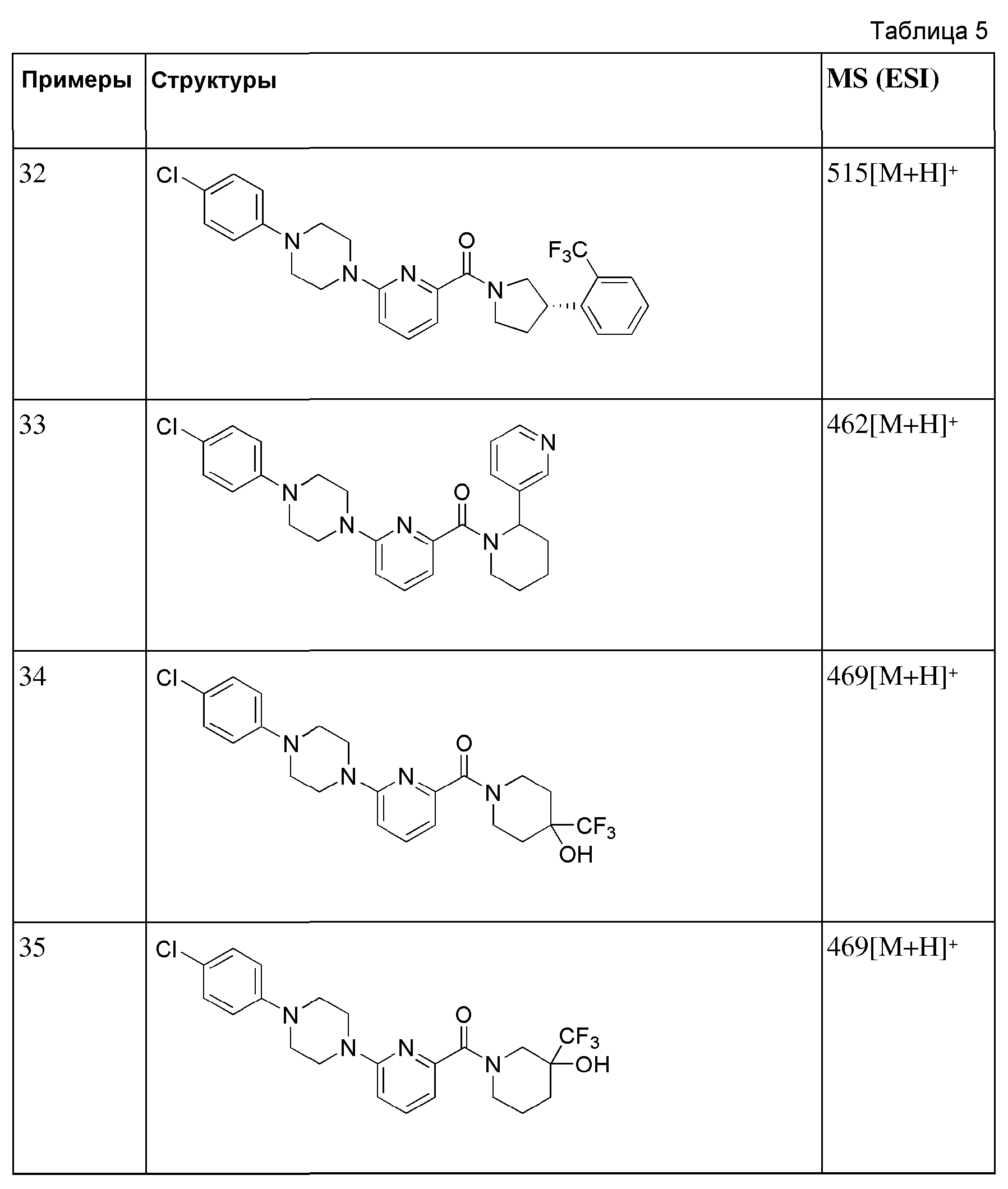
[145] После того, как 1-(6-бромпиколиноил)пиперидин-4-он (100 мг, 0,353 ммоль) растворяли в ТГФ (1,5 мл), к ней при 0°С последовательно добавляли триметил(трифторметил)силан (0,5М раствор в ТГФ, 1,4 мл, 0,706 ммоль) и фторид тетрабутиламмония (1,0М раствор в ТГФ, 0,74 мл, 0,741 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в течение 13 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (0,6 мл), а затем получающуюся смесь перемешивали в течение 10 минут с последующей экстракцией МС (10 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 120 мг белого твердого вещества (97%).
[146] Стадия 3: Синтез (4-гидрокси-4-(трифторметил)пиперидин-1-ил)(6-(пиперидин-1-ил)пиридин-2-ил)метанона
[147] (6-Бромпиридин-2-ил)(4-гидрокси-4-(трифторметил)пиперидин-1-ил)метанон (61 мг, 0,172 ммоль), пиперидин (0,02 мл, 0,189 ммоль), Pd2(dba)3 (3 мг, 0,003 ммоль), ксантфос (6 мг, 0,010 ммоль) и трет-бутоксид натрия (25 мг, 0,258 ммоль) суспендировали в толуоле (1,5 мл), а затем перемешивали при 100°С в токе азота в течение 2 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (5 мл) с последующей экстракцией смесью 5% МеОН/МС (10 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 37 мг желтого твердого вещества (61%). MS (ESI): 358[M+Н]+.
[148]
[149] Пример 31: Синтез (3-гидрокси-3-(трифторметил)пиперидин-1-ил)(6-(пиперидин-1-ил)пиридин-2-ил)метанона
[150]
[151] Стадия 1: Синтез трет-бутил 3-(трифторметил)-3-гидроксипиперидин-1-карбоксилата
[152] После того, как трет-бутил 3-оксопиперидин-1-карбоксилат (1 г, 5,02 ммоль) растворяли в ТГФ (20 мл), затем к нему при 0°С последовательно добавляли триметил(трифторметил)силан (0,5М раствор в ТГФ, 20 мл, 10,0 ммоль) и фторид тетрабутиламмония (1,0М раствор в ТГФ, 10,5 мл, 10,5 ммоль), с последующим перемешиванием при комнатной температуре в течение 2 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (5 мл) с последующим перемешиванием в течение 20 минут. Получающуюся реакционную жидкость концентрировали при пониженном давлении, а затем к полученному таким образом остатку добавляли МС (100 мл) с последующей промывкой дистиллированной водой (20 мл). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (20% EtOAc/гексаны), получая 709 мг белого твердого вещества (53%).
[153] Стадия 2: Синтез (6-бромпиридин-2-ил)(3-гидрокси-3-(трифторметил)пиперидин-1-ил)метанона (Промежуточное соединение 8)
[154] После того, как трет-бутил 3-(трифторметил)-3-гидроксипиперидин-1-карбоксилат (709 мг, 2,63 ммоль) растворяли в ТГФ (10 мл) с последующим добавлением HCl (2,0М раствор в диэтиловом эфире, 30 мл), а затем получающуюся в результате смесь перемешивали при комнатной температуре в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении и сушили в вакууме, а затем полученный таким образом остаток растворяли в ацетонитриле (30 мл), добавляли туда 6-бромпиколиновую кислоту (638 мг, 3,16 ммоль) и охлаждали до 0°С. Добавляли туда последовательно N,N-диизопропилэтиламин (1,1 мл, 6,58 ммоль) и HBTU (1,2 г, 3,16 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 20 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении с последующим добавлением МС (50 мл) и затем промывали дистиллированной водой. Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 800 мг белого твердого вещества (86%).
[155] Стадия 3: Синтез (3-гидрокси-3-(трифторметил)пиперидин-1-ил)(6-(пиперидин-1-ил)пиридин-2-ил)метанона
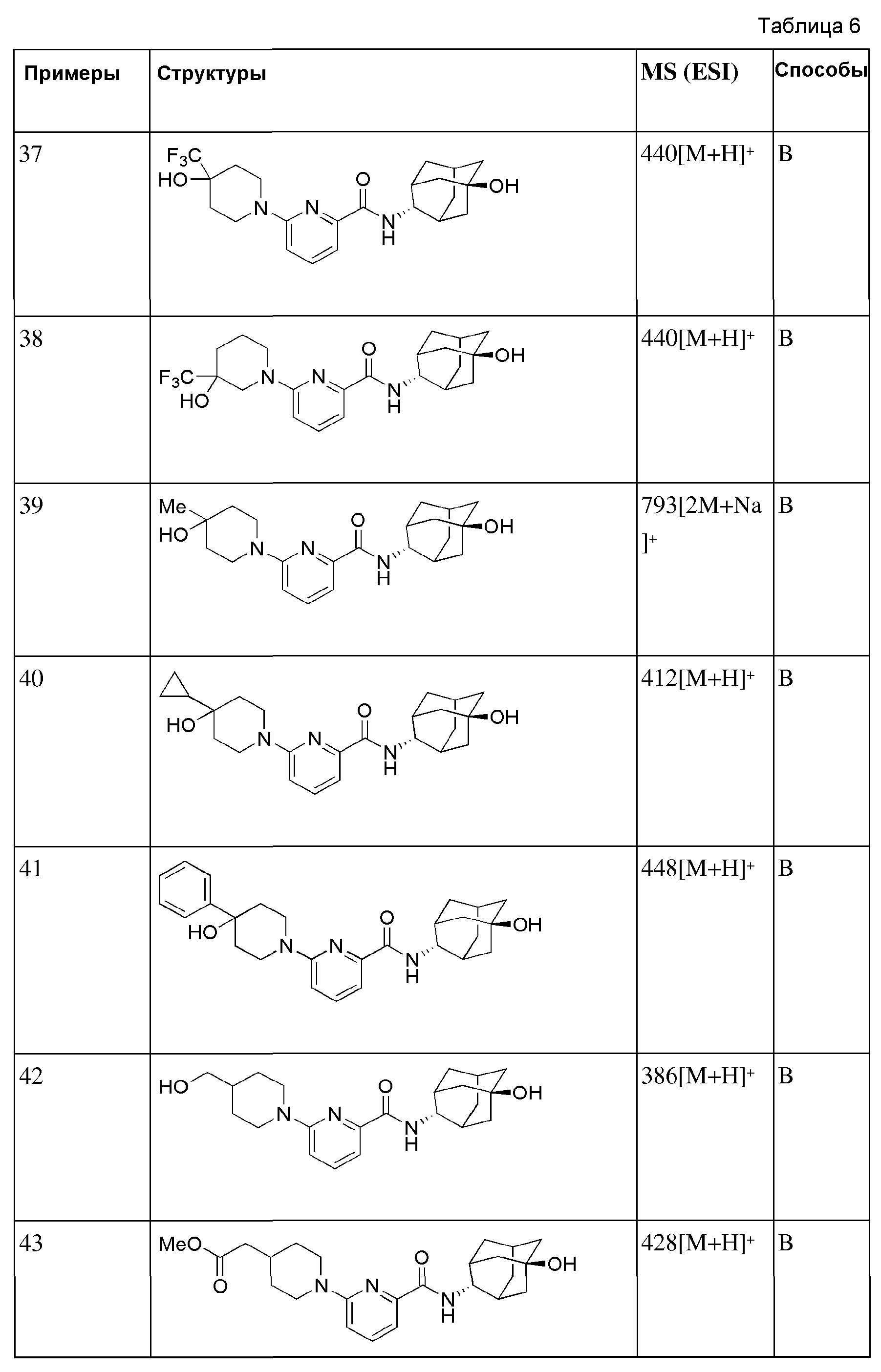
[156] После того, как (6-бромпиридин-2-ил)(3-гидрокси-3-(трифторметил)пиперидин-1-ил)метанон (60 мг, 0,170 ммоль) суспендировали в ацетонитриле (2 мл), к нему добавляли пиперидин (0,13 мл, 1,36 ммоль) и триэтиламин (0,05 мл, 0,340 ммоль), а затем получающуюся в результате смесь перемешивали при 100°С в течение 14 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 53 мг белого твердого вещества (87%). MS (ESI): 358[M+Н]+.
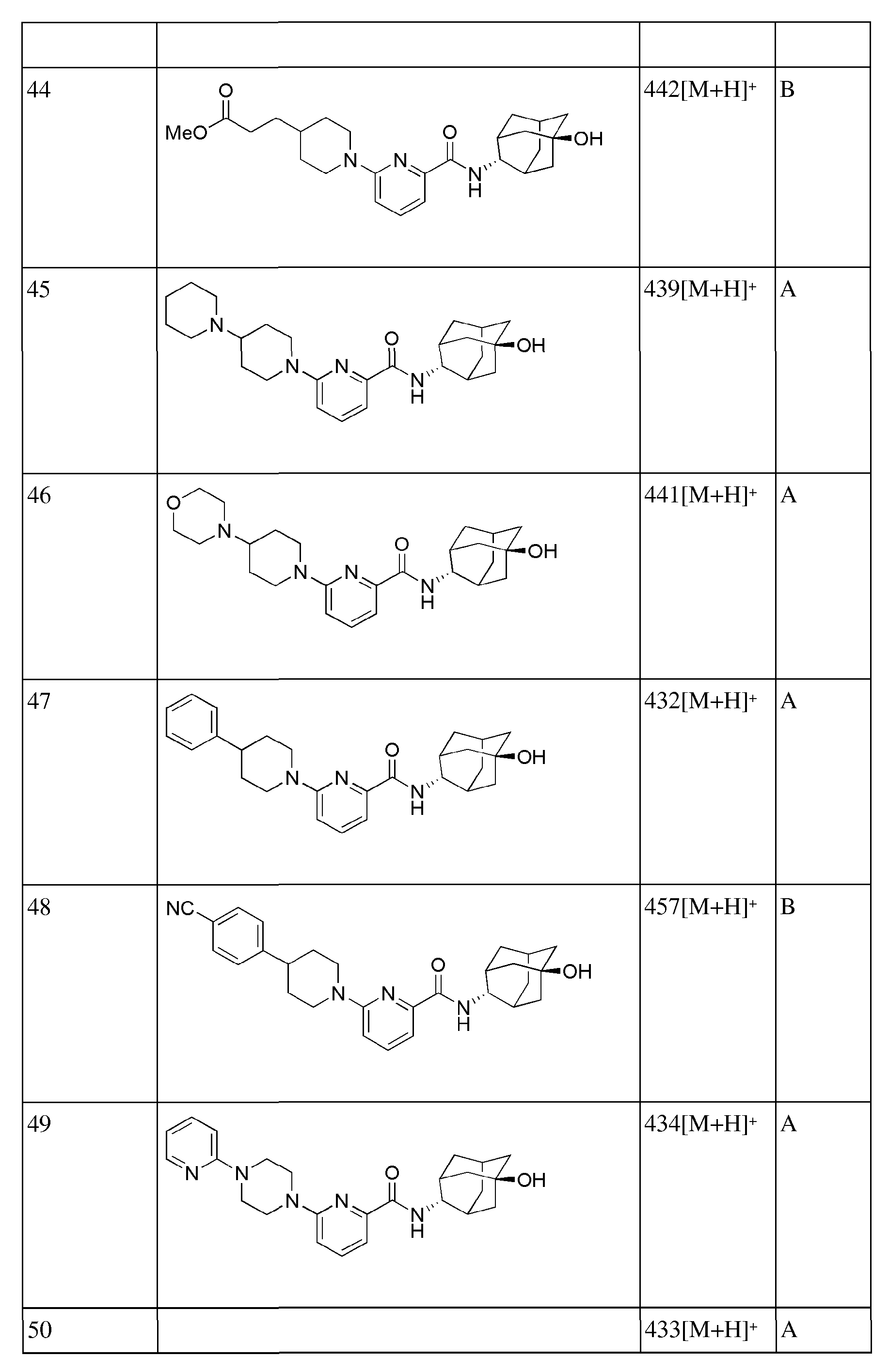
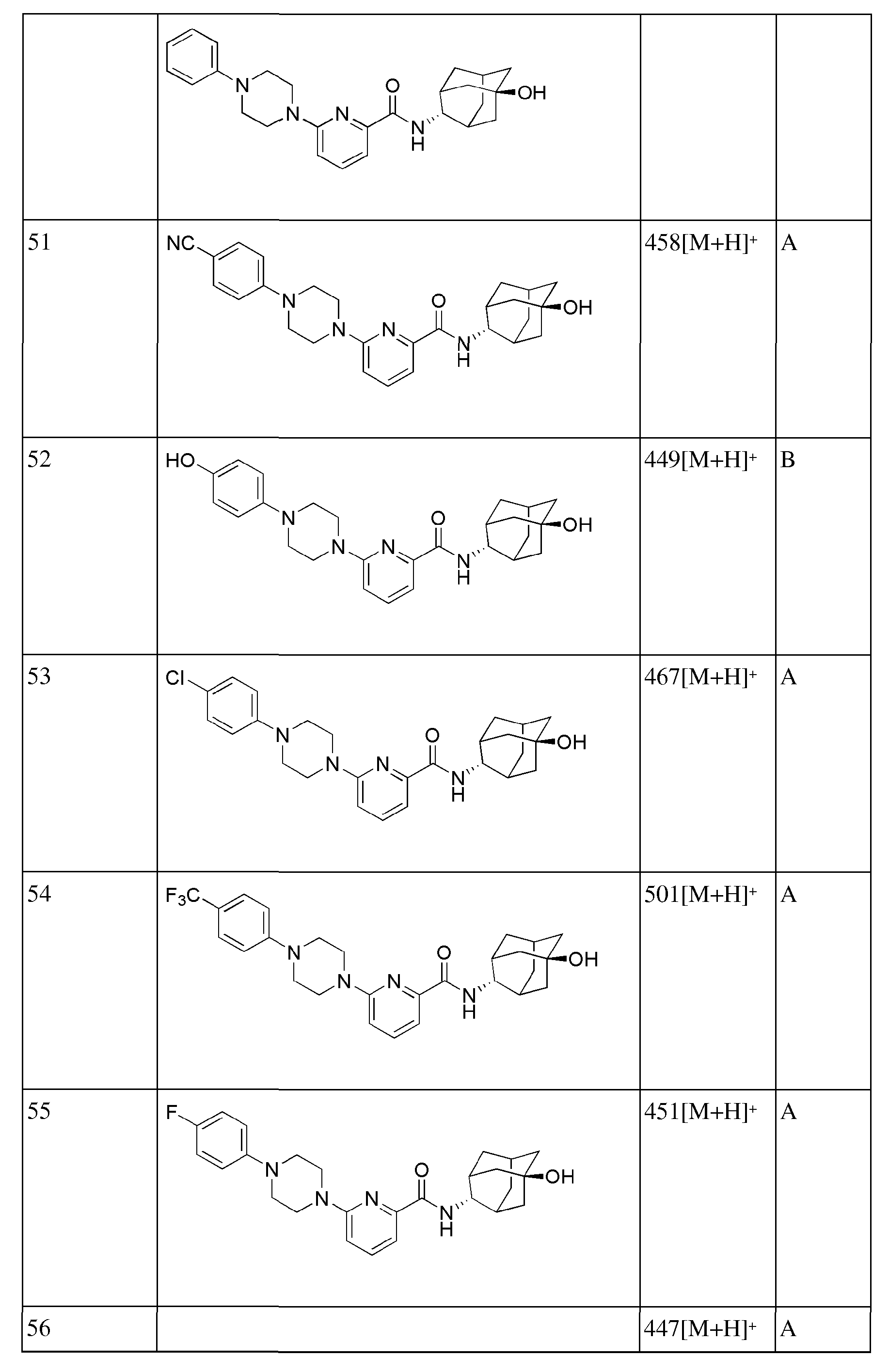
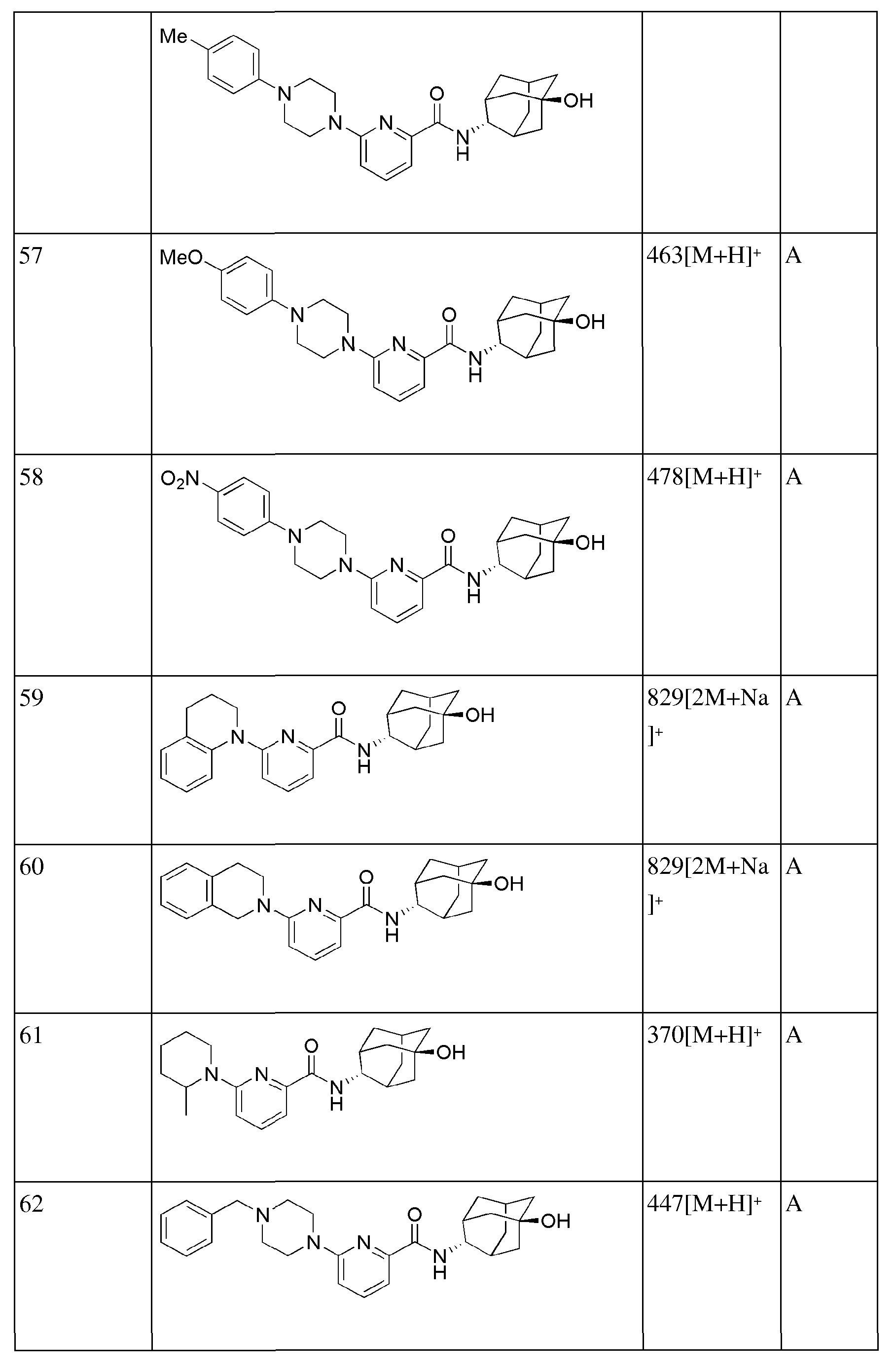
[157]
[158] Соединения следующих примеров синтезировали тем же самым способом, что и в примерах 28, 29, 30 или 31 выше, с использованием промежуточных соединений 5, 6, 7 или 8 и 1-(4-хлорфенил)пиперазина.
[159] Таблица 5
[160]
[161] Пример 36: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперидин-1-ил)пиколинамида)
[162]
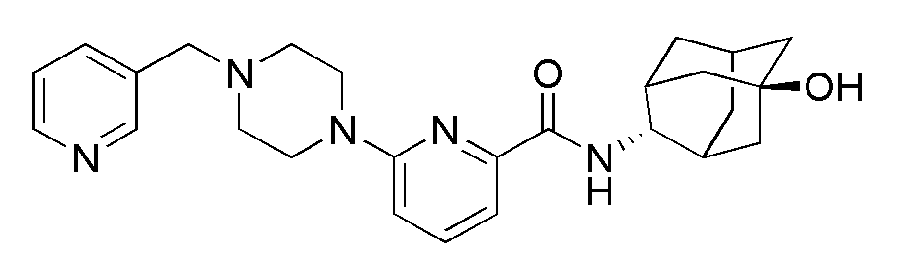
[163] Стадия 1: Синтез 6-бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида (Промежуточное соединение 9)
[164] После того, как 6-бромпиколиновую кислоту (17,5 г, 87 ммоль) суспендировали в ацетонитриле (500 мл), к ней последовательно добавляли 5-гидрокси-2-адамантанемин (2:1 E/Z смесь, 17,4 г, 104 ммоль), N,N-диизопропилэтиламин (18,1 мл, 104 ммоль) и HBTU (39,4 г, 104 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (200 мл) и экстрагировали 10% МеОН/МС (300 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (70% EtOAc/гексаны), получая 18,4 г белого твердого вещества (60%).
[165] Стадия 2: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперидин-1-ил)пиколинамида
[166] Способ А: 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (60 мг, 0,171 ммоль), пиперидин (17 мг, 0,205 ммоль), Pd2(dba)3 (3 мг, 0,003 ммоль), ксантфос (6 мг, 0,010 ммоль) и трет-бутоксид натрия (25 мг, 0,257 ммоль) суспендировали в толуоле (3 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 4 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (15 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 44 мг белого твердого вещества (73%).
[167] Способ В: 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (50 мг, 0,142 ммоль) растворяли в ацетонитриле (1 мл) с последующим добавлением пиперидина (48 мг, 0,568 ммоль) и триэтиламина (0,04 мл, 0,284 ммоль), а затем получающуюся жидкость подвергали микроволновому облучению при 150°С в течение 2 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении с последующим добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 32 мг белого твердого вещества (63%). MS (ESI): 356[M+H]+.
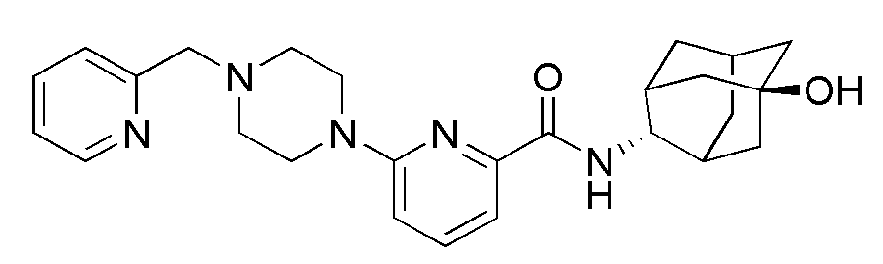
[168]
[169] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 36 выше, с использованием промежуточного соединения 9 и соответствующего аминового исходного материала.
[170] Таблица 6
[171]
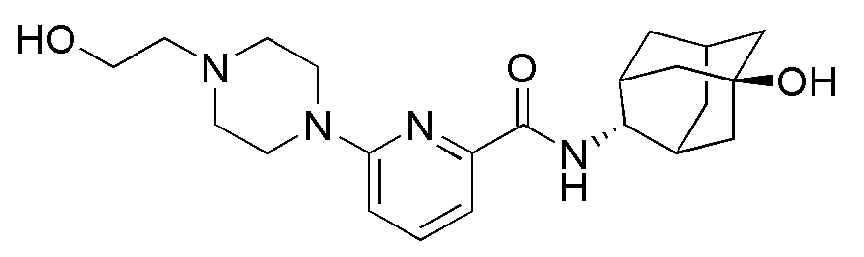
[172] Пример 63: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперазин-1-ил)пиколинамида)
[173]

[174] После того, как 6-(4-бензилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (150 мг, 0,336 ммоль) растворяли в метаноле с последующим добавлением Pd (10 вес.% на активированном угле, 50 мг), получающуюся смесь перемешивали при комнатной температуре в токе водорода в течение 15 часов. Получающуюся реакционную жидкость фильтровали, а затем концентрировали при пониженном давлении. Полученный таким образом остаток подвергали перекристаллизации (МС/Et2O), получая 114 мг белого твердого вещества (95%). MS (ESI): 357[M+H]+.
[175]
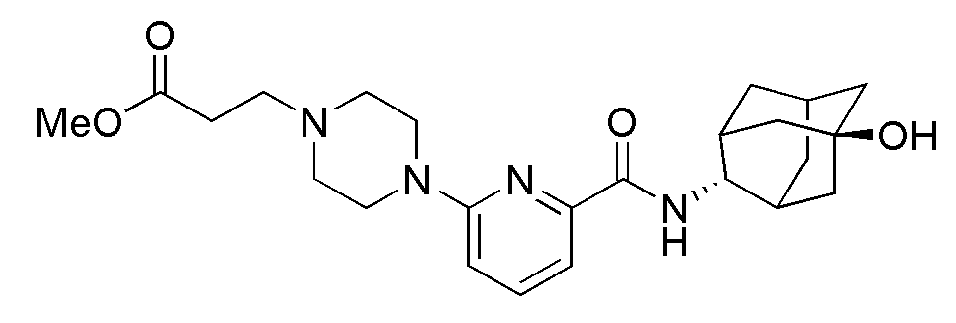
[176] Пример 64: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(пиридин-3-илметил)пиперазин-1-ил)пиколинамида
[177]
[178] После того, как N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид (40 мг, 0,112 ммоль) и гидробромид 3-(бромметил)пиридина (31 мг, 0,123 ммоль) суспендировали в 1,2-дихлорэтане (2 мл) с последующим добавлением N,N-диизопропилэтиламина (0,06 мл, 0,336 ммоль), а затем получающуюся в результате жидкость перемешивали при 70°С в токе азота в течение 2 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный хлорид аммония (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (6% МеОН/МС), получая 30,4 г бесцветного масла (60%). MS (ESI): 448[M+H]+.
[179]
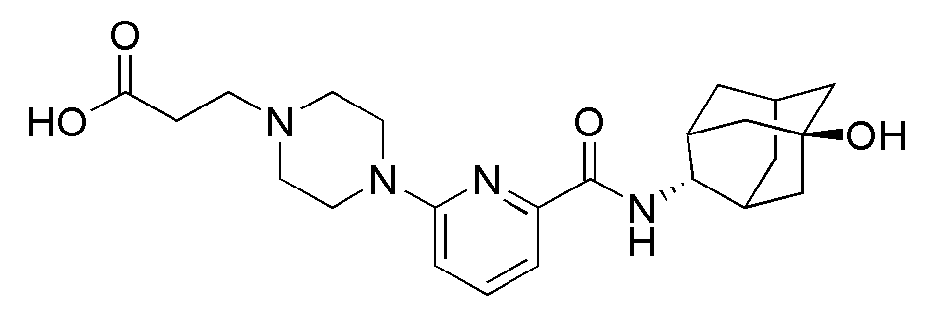
[180] Пример 65: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(пиридин-2-илметил)пиперазин-1-ил)пиколинамида
[181]
[182] Выполняли тот же способ, что в примере 64, за исключением того, что вместо 3-(бромметил)пиридингидробромида использовали гидробромид 2-(бромметил)пиридина, получая 36 мг бесцветного масла (72%). MS (ESI): 448[M+H]+.
[183]
[184] Пример 66: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(2-гидроксиэтил)пиперазин-1-ил)пиколинамида
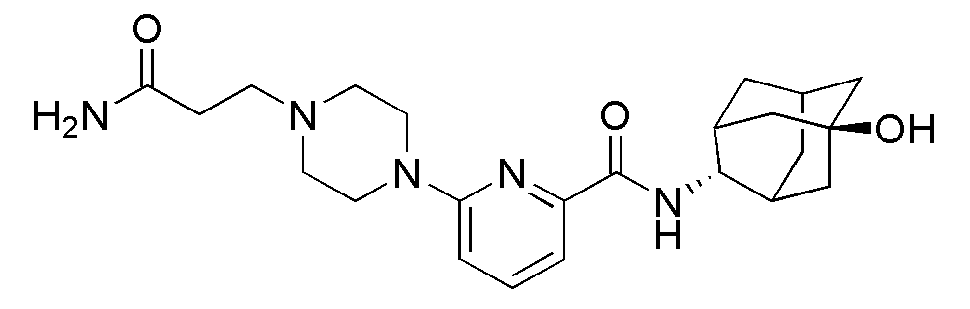
[185]
[186] Выполняли тот же способ, что в примере 24, за исключением того, что вместо N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамида использовали N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид (70 мг, 0,196 ммоль), получая 45 мг белого твердого вещества (57%). MS (ESI): 401[M+H]+.
[187]
[188] Пример 67: Синтез метил 3-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноата
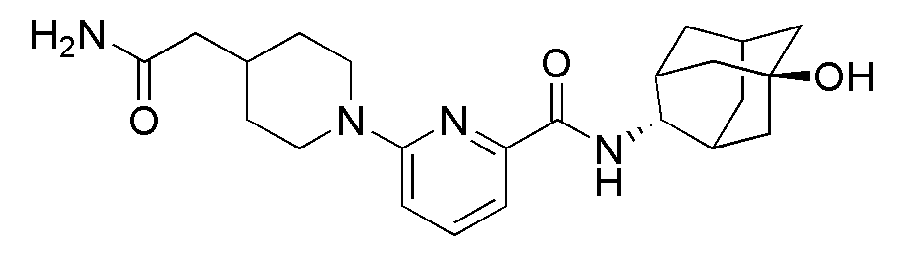
[189]
[190] Выполняли тот же способ, что в примере 25, за исключением того, что вместо N-(адамантан-2-ил)-6-(пиперазин-1-ил)пиколинамида использовали N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперазин-1-ил)пиколинамид (160 мг, 0,449 ммоль), получая 180 мг белого твердого вещества (81%). MS (ESI): 443[M+H]+.
[191]
[192] Пример 68: Синтез 3-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)пропановой кислоты
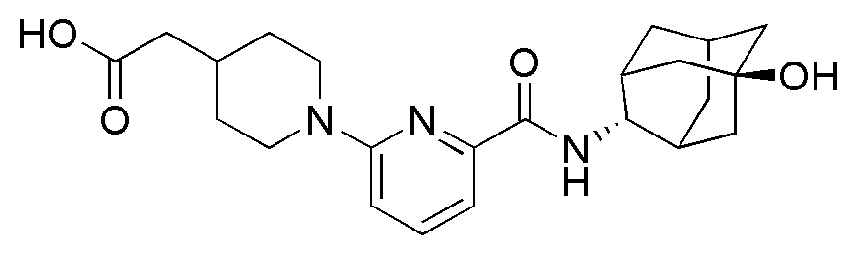
[193]
[194] Выполняли тот же способ, что в примере 26, за исключением того, что вместо метил 3-(4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноата использовали метил 3-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноат (70 мг, 0,158 ммоль), получая 41 мг бледно-желтого твердого вещества (60%). MS (ESI): 429[M+H]+.
[195]
[196] Пример 69: Синтез (6-(4-(3-амино-3-оксопропил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида)
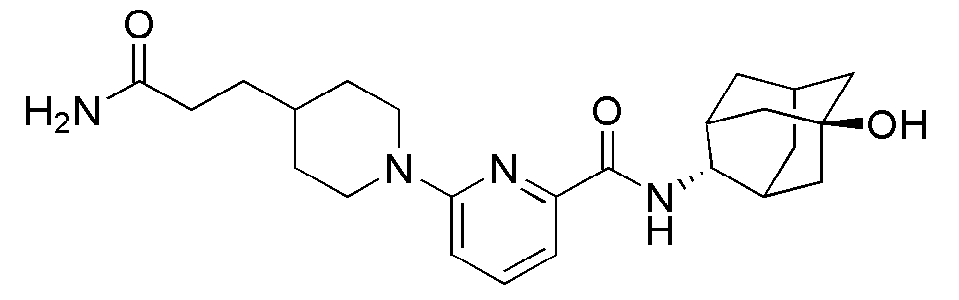
[197]
[198] Выполняли тот же способ, что в примере 27, за исключением того, что вместо метил 3-(4-(6-(адамантан-2-илкарбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноата использовали метил 3-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)пропаноат (100 мг, 0,225 ммоль), получая 10 мг белого твердого вещества (10%). MS (ESI): 428[M+H]+.
[199]
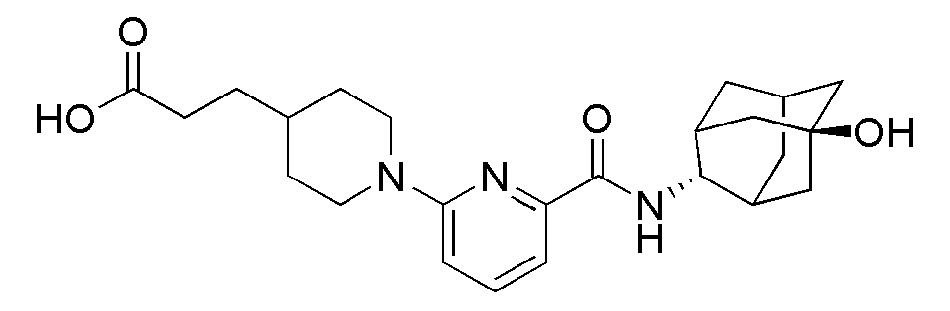
[200] Пример 70: Синтез 6-(4-(2-амино-2-оксоэтил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида)
[201]
[202] Метил 2-(1-(6-((((E)-5-гидроксиадамантан-2-ил)карбамоил))пиридин-2-ил)пиперазин-4-ил)ацетат (50 мг, 0,117 ммоль) растворяли в ДМФ (1 мл) с последующим добавлением формамида (0,02 мл, 0,526 ммоль). Добавляли туда по каплям NaOMe (25% раствор в МеОН, 0,03 мл, 0,129 ммоль) при перемешивании при 100°С в токе азота, а затем получающуюся в результате жидкость перемешивали в течение 2 часов. К получающейся реакционной жидкости добавляли дистиллированную воду (0,1 мл) с последующим концентрированием при пониженном давлении, а затем туда добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (7% МеОН/МС), получая 31 мг белого твердого вещества (64%). MS (ESI): 413[M+H]+.
[203]
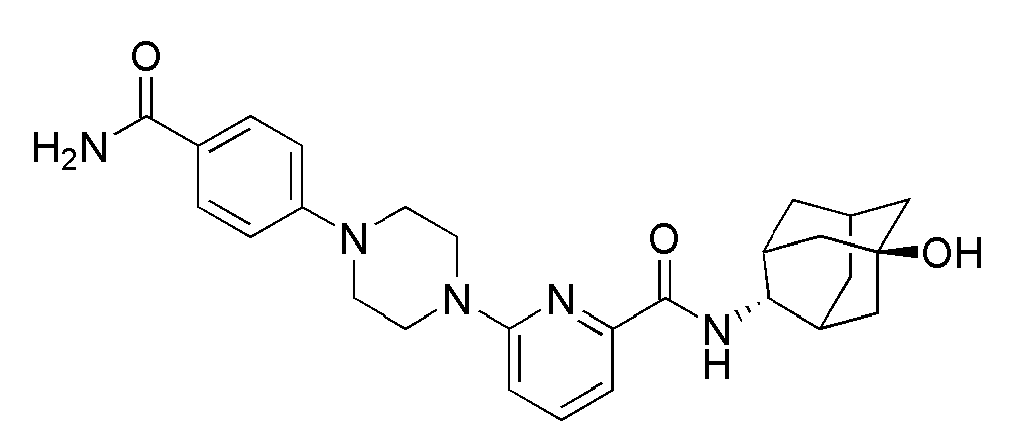
[204] Пример 71: Синтез 2-(1-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-4-ил)уксусной кислоты
[205]
[206] Метил 2-(1-(6-((((E)-5-гидроксиадамантан-2-ил)карбамоил))пиридин-2-ил)пиперидин-4-ил)ацетат (40 мг, 0,094 ммоль) растворяли в МеОН (2 мл) с последующим добавлением 10% водного раствора NaOH (0,17 мл, 0,468 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в течение 20 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении и растворяли добавлением дистиллированной воды (5 мл), а затем получающуюся жидкость нейтрализовали добавлением 1н водного раствора HCl при перемешивании при 0°С. Осажденную соль отфильтровали, с последующей вакуумной сушкой, получая 24 мг белого твердого вещества (62%). MS (ESI): 414[M+H]+.
[207]
[208] Пример 72: Синтез 6-(4-(3-амино-3-оксопропил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[209]
[210] Выполняли тот же способ, что в примере 70, за исключением того, что вместо метил 2-(1-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-4-ил)ацетата использовали метил 3-(1-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил(пиперазин-4-ил)пропаноат (50 мг, 0,113 ммоль), получая 38 мг белого твердого вещества (79%). MS (ESI): 427[M+H]+.
[211]
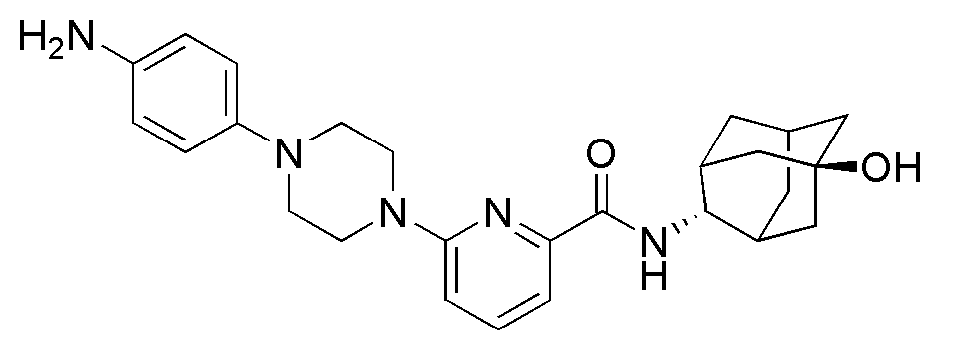
[212] Пример 73: Синтез 3-(1-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-4-ил)пропановой кислоты
[213]
[214] Выполняли тот же способ, что в примере 71, за исключением того, что вместо метил 2-(1-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-4-ил)ацетата использовали метил 3-(1-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-4-ил)пропаноат (40 мг, 0,091 ммоль), получая 30 мг белого твердого вещества (78%). MS (ESI): 428[M+H]+.
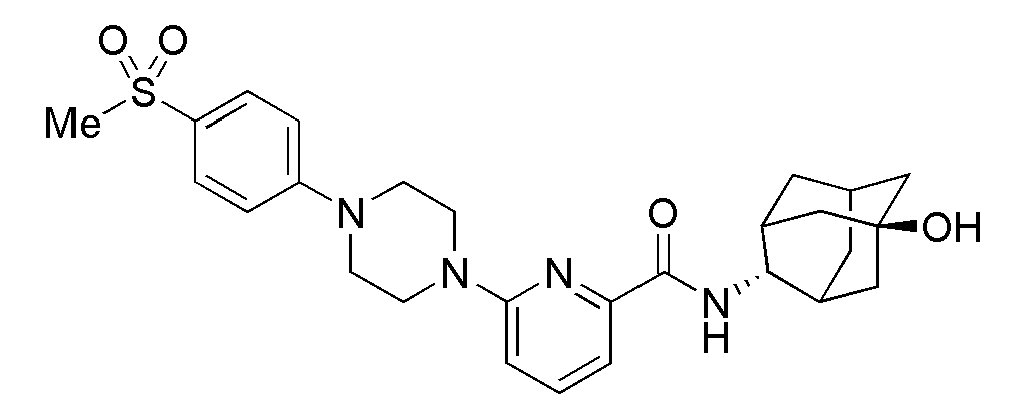
[215]
[216] Пример 74: Синтез 6-(4-(4-карбамоилфенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[217]
[218] 6-(4-(4-Цианофенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида) (53 мг, 0,116 ммоль) суспендировали в этаноле (0,53 мл) с последующим добавлением 1н водного раствора гидроксида натрия (0,46 мл, 0,463 ммоль) и перекиси водорода (30% раствор в воде, 0,024 мл, 0,232 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе водорода в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем нейтрализовали добавлением 1н водного раствор HCl с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 32 мг белого твердого вещества (58%). MS (ESI): 973[2M+Na]+
[219]
[220] Пример 75: Синтез 6-(4-(4-карбамоилфенил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[221]

[222] Выполняли тот же способ, что в примере 74, за исключением того, что вместо 6-(4-(4-цианофенил)пиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 6-(4-(4-цианофенил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (45 мг, 0,098 ммоль), получая 21 мг бесцветного масла (45%). MS (ESI): 475[M+H]+.
[223]
[224] Пример 76: Синтез 6-(4-(4-аминофенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
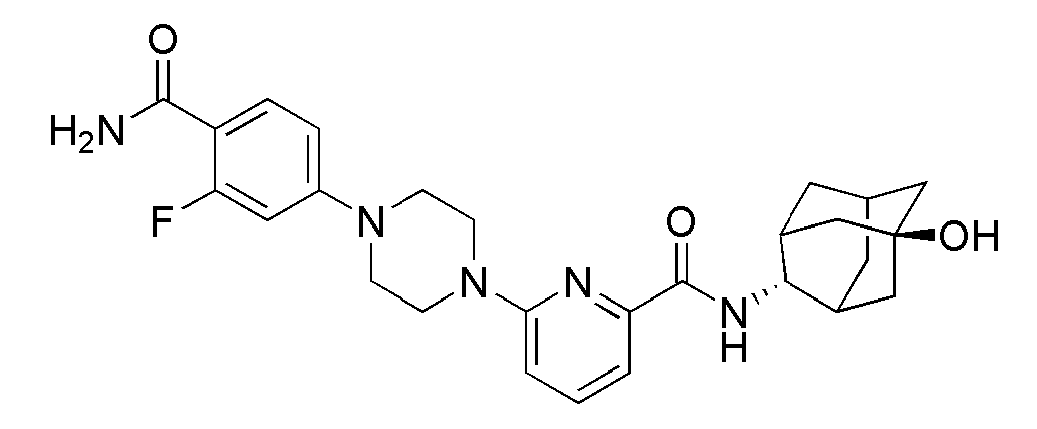
[225]
[226] N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(4-нитрофенил)пиперазин-1-ил)пиколинамид (40 мг, 0,084 ммоль) растворяли в 10% МеОН/МС с последующим добавлением палладия (10 вес.% на активированном угле, 2 мг), а затем получающуюся в результате жидкость перемешивали при комнатной температуре в токе водорода в течение 4 часов. Получающуюся в результате реакционную жидкость фильтровали, а затем концентрировали при пониженном давлении. Полученный таким образом остаток подвергали MPLC (100% EtOAc), получая 30 мг желтого твердого вещества (80%). MS (ESI): 448[M+Н]+.
[227]
[228] Пример 77: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(4-метилсульфонил)фенил)пиперазин-1-ил)пиколинамида
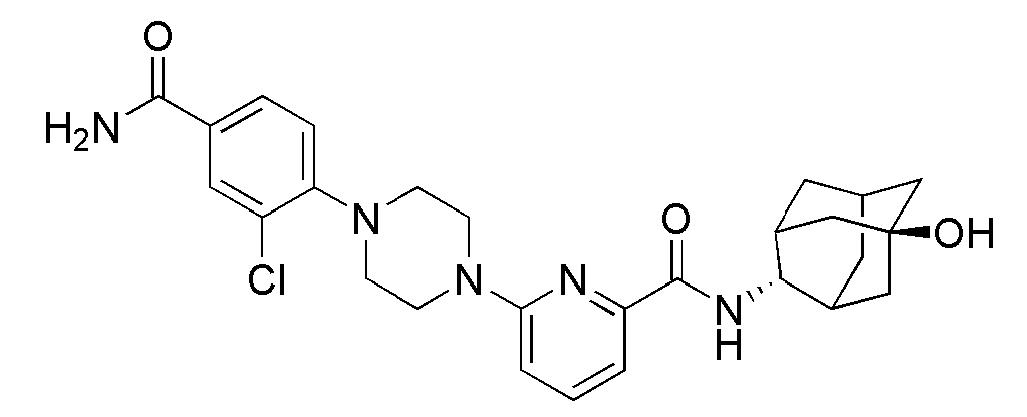
[229]
[230] Стадия 1: Синтез 1-(4-метилсульфонил)фенил)пиперазина
[231] 1-Бром-4-(метилсульфонил)бензол (275 мг, 0,169 ммоль), пиперазин (302 мг, 3,507 ммоль), Pd2(dba)3 (21 мг, 0,023 ммоль), BINAP (44 мг, 0,070 ммоль) и трет-бутоксид натрия (169 мг, 1,754 ммоль) суспендировали в толуоле (5 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 15 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (15 мл) с последующей экстракцией МС (20 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (15% МеОН/МС), получая 56 мг бледно желтого твердого вещества (20%).
[232] Стадия 2: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-(4-(4-метилсульфонил)фенил)пиперазин-1-ил)пиколинамида
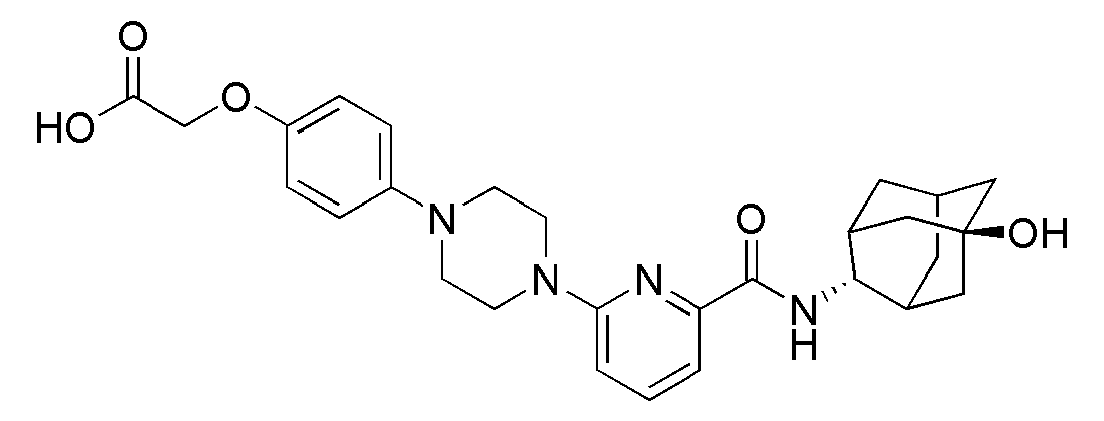
[233] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (60 мг, 0,171 ммоль), 1-(4-метилсульфонил)фенил)пиперазин (49 мг, 0,205 ммоль), Pd2(dba)3 (3,1 мг, 0,003 ммоль), ксантфос (5,9 мг, 0,010 ммоль) и трет-бутоксид натрия (25 мг, 0,257 ммоль) суспендировали в толуоле (3 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 4 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 34 мг бледно желтого твердого вещества (39%). MS (ESI): 511[M+Н]+.
[234]
[235] Соединения следующих примеров синтезировали тем же способом, что в примере 77 выше с использованием соответствующего бромбензольного исходного материала.
[236] Таблица 7
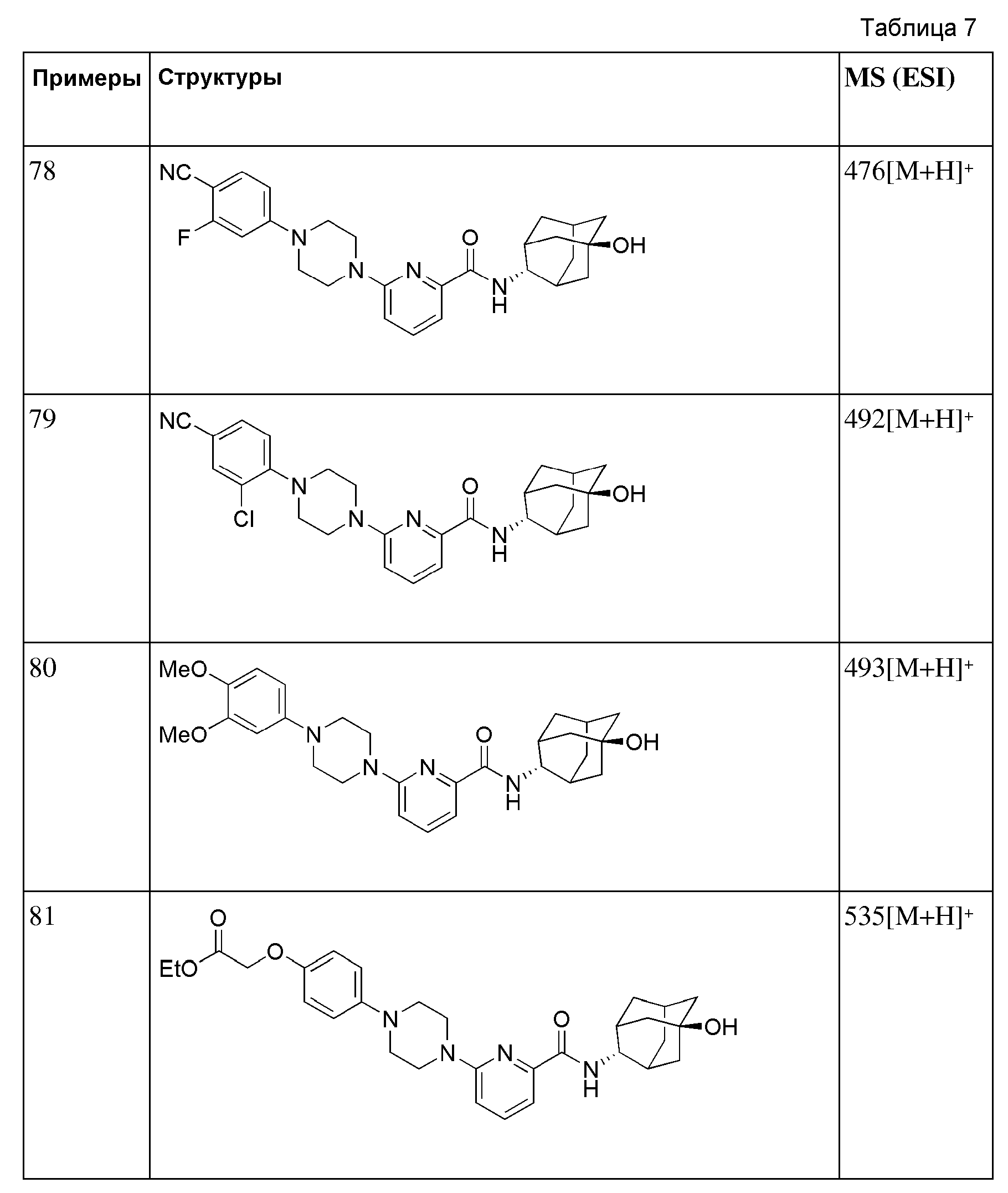
[237]
[238] Пример 82: Синтез 6-(4-(4-карбамоил-3-фторфенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[239]
[240] Выполняли тот же способ, что в примере 74, за исключением того, что вместо 6-(4-(4-цианофенил)пиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 6-(4-(4-циано-3-фторфенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (75 мг, 0,158 ммоль), получая 72 мг белого твердого вещества (92%). MS (ESI): 494[M+H]+.
[241]
[242] Пример 83: Синтез 6-(4-(4-карбамоил-2-хлорфенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[243]
[244] Выполняли тот же способ, что в примере 74, за исключением того, что вместо 6-(4-(4-цианофенил)пиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 6-(4-(4-циано-2-хлорфенил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (154 мг, 0,313 ммоль), получая 96 мг белого твердого вещества (60%). MS (ESI): 510[M+H]+.
[245]
[246] Пример 84: Синтез 2-(4-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)фенокси)уксусной кислоты
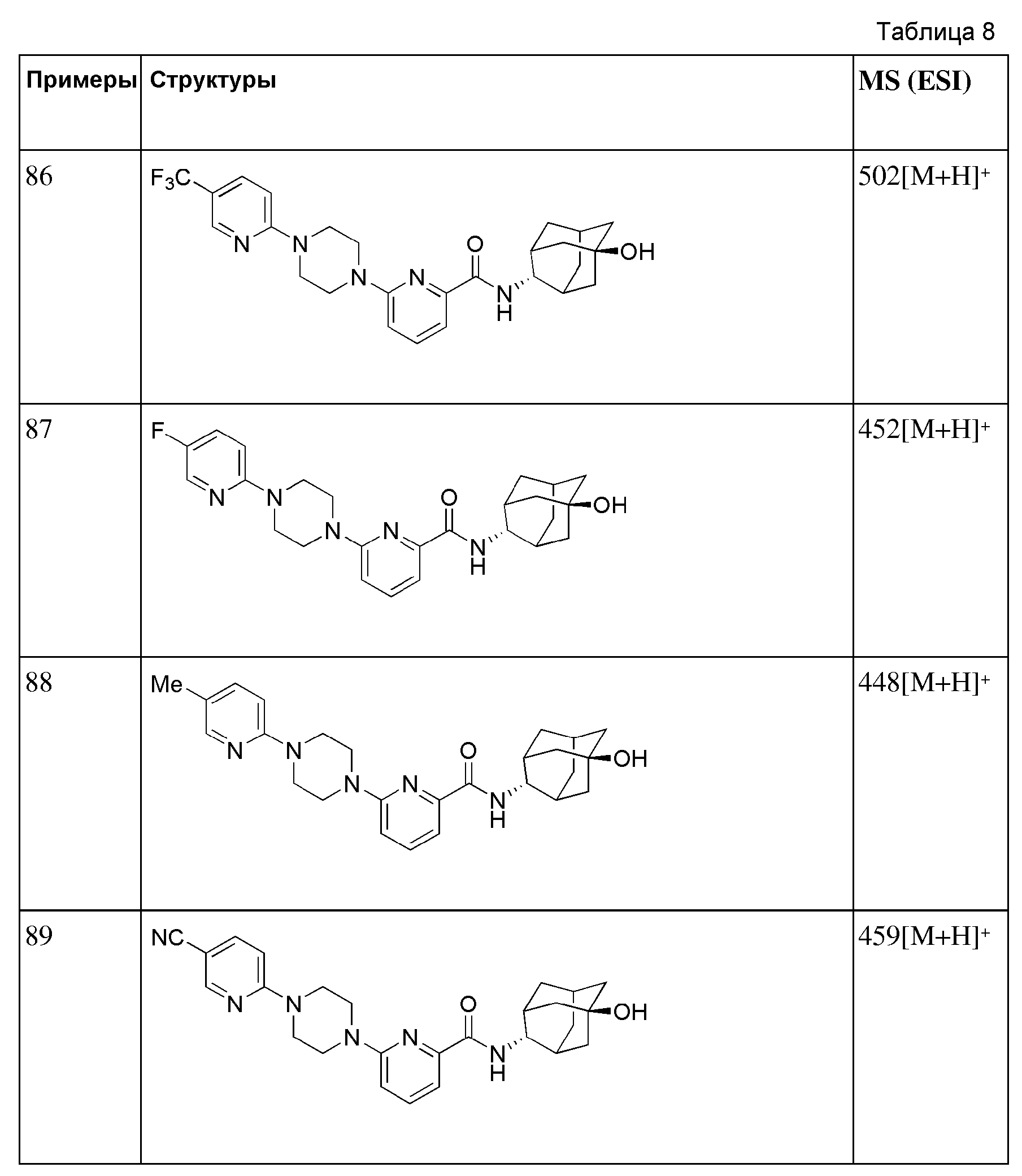
[247]
[248] Этил 2-(4-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)фенокси)ацетат (82 мг, 0,153 ммоль) растворяли в этаноле (3 мл) с последующим добавлением 1н водного раствора гидроксида натрия (0,46 мл, 0,460 ммоль), а затем получающуюся в результате жидкость перемешивали при 60°С в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении и растворяли добавлением дистиллированной воды (5 мл), а затем получающуюся жидкость нейтрализовали добавлением 1н водного раствора HCl при перемешивании при 0°С. Осажденное твердое вещество отфильтровывали с последующей вакуумной сушкой, получая 56 мг бледно желтого твердого вещества (72%). MS (ESI): 507[M+H]+.
[249]
[250] Пример 85: Синтез 6-(4-(5-хлорпиридин-2-ил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[251]
[252] Стадия 1: Синтез трет-бутил 4-(5-хлорпиридин-2-ил)пиперазин-1-карбоксилата
[253] Трет-бутил пиперазин-1-карбоксилат (300 мг, 1,611 ммоль) и 2-бром-5-хлорпиридин (465 мг, 2,416 ммоль) растворяли в ацетонитриле (3 мл) с последующим добавлением триэтиламина (0,45 мл, 3,222 ммоль), а затем получающуюся жидкость подвергали микроволновому облучению при 150°С в течение 2 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении с последующим добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (20% EtOAc/гексаны), получая 177 мг бесцветного масла (37%).
[254] Стадия 2: Синтез 1-(5-хлорпиридин-2-ил)пиперазина
[255] Трет-бутил 4-(5-хлорпиридин-3-ил)пиперазин-1-карбоксилат (170 мг, 0,571 ммоль) растворяли в МС (3 мл) с последующим добавлением трифторуксусной кислоты (3 мл), а затем получающуюся в результате смесь перемешивали при комнатной температуре в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением насыщенного водного раствора NaHCO3 (15 мл) и экстрагировали МС (15 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 112 мг белого твердого вещества (99%).
[256] Стадия 3: Синтез 6-(4-(5-хлорпиридин-2-ил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[257] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (50 мг, 0,142 ммоль), 1-(5-хлорпиридин-2-ил)пиперазин (34 мг, 0,171 ммоль), Pd2(dba)3 (2,6 мг, 0,003 ммоль), ксантфос (4,9 мг, 0,009 ммоль) и трет-бутоксид натрия (20 мг, 0,213 ммоль) суспендировали в толуоле (3 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 3 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (15 мл) с последующей экстракцией МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 24 мг желтого твердого вещества (36%). MS (ESI): 468[M+Н]+.
[258]
[259] Соединения следующих примеров синтезировали тем же способом, что в примере 85 выше с использованием соответствующего 2-бромпиридинового исходного материала.
[260] Таблица 8
[261]
[262] Пример 90: Синтез 6-(4-(5-карбамоилпиридин-2-ил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[263]
[264] Выполняли тот же способ, что в примере 74, за исключением того, что вместо 6-(4-(4-цианофенил)пиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 6-(4-(5-цианопиридин-2-ил)пиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (67 мг, 0,146 ммоль), получая 58 мг бледно желтого твердого вещества (83%). MS (ESI): 477[M+H]+.
[265]
[266] Пример 91: Синтез 6-(4-(4-фторфенил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[267]
[268] Стадия 1: Синтез трет-бутил 4-(((трифторметил)сульфонил)окси)-5,6-дигидропиридин-1(2Н)-карбоксилата
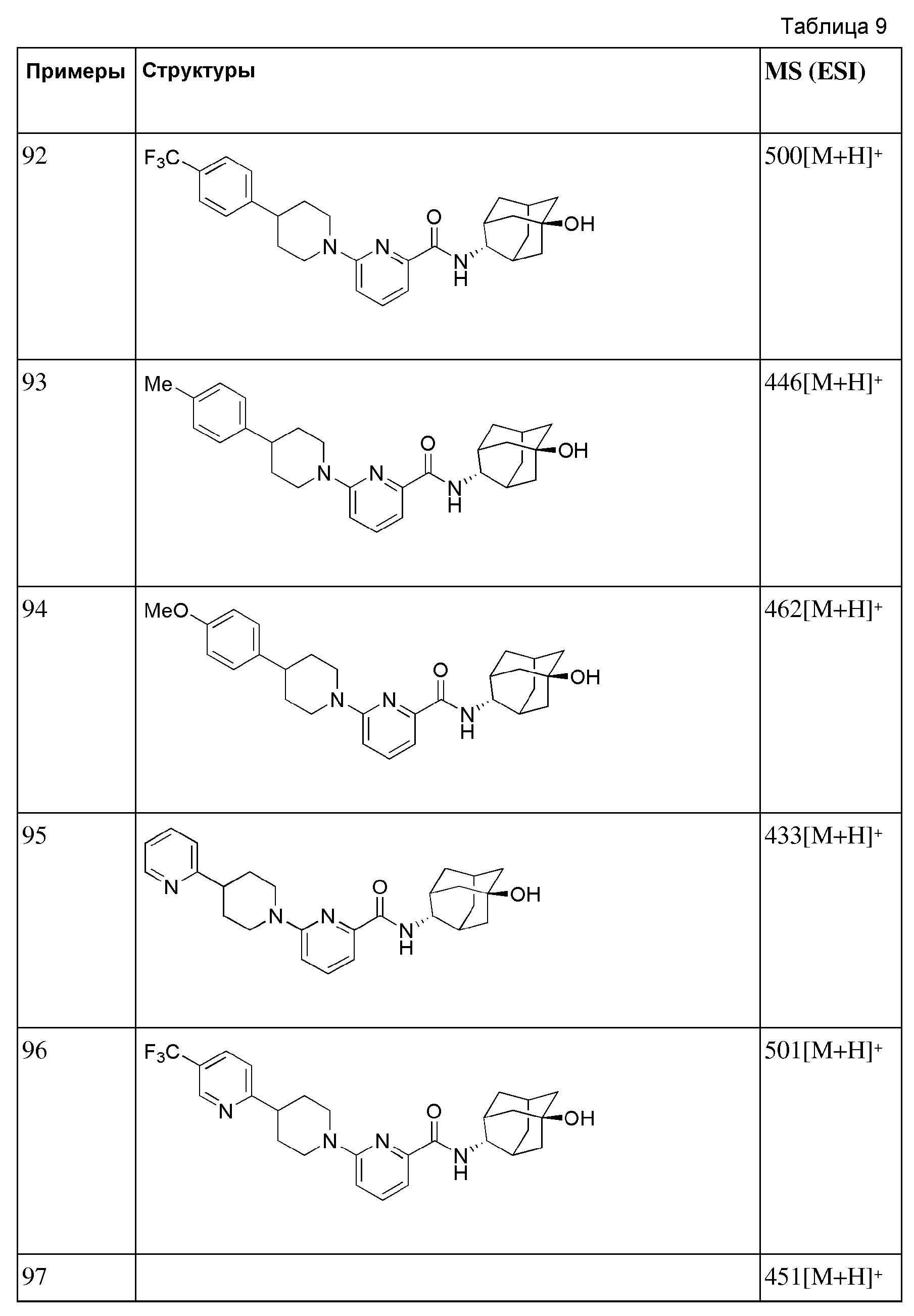
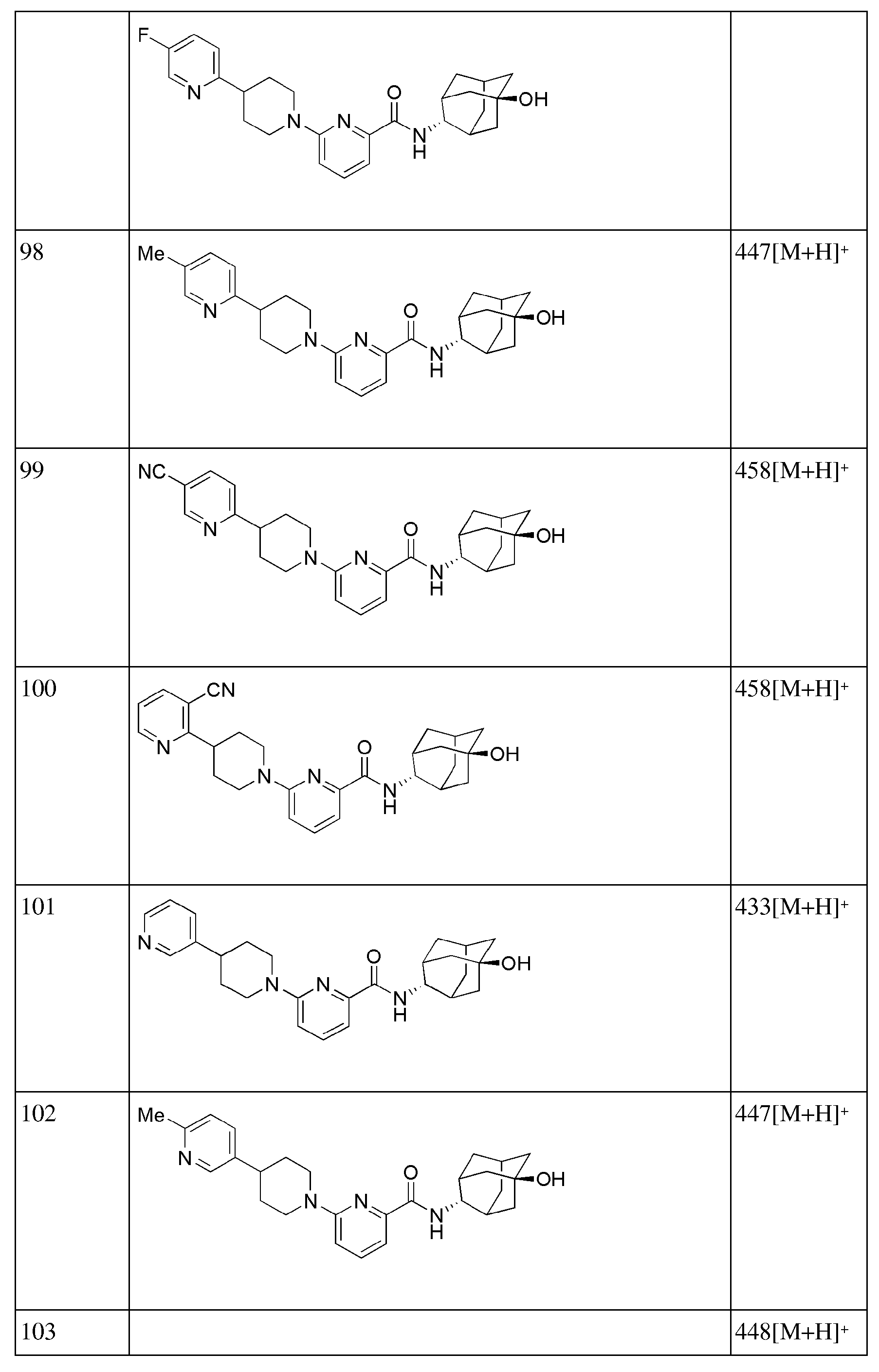
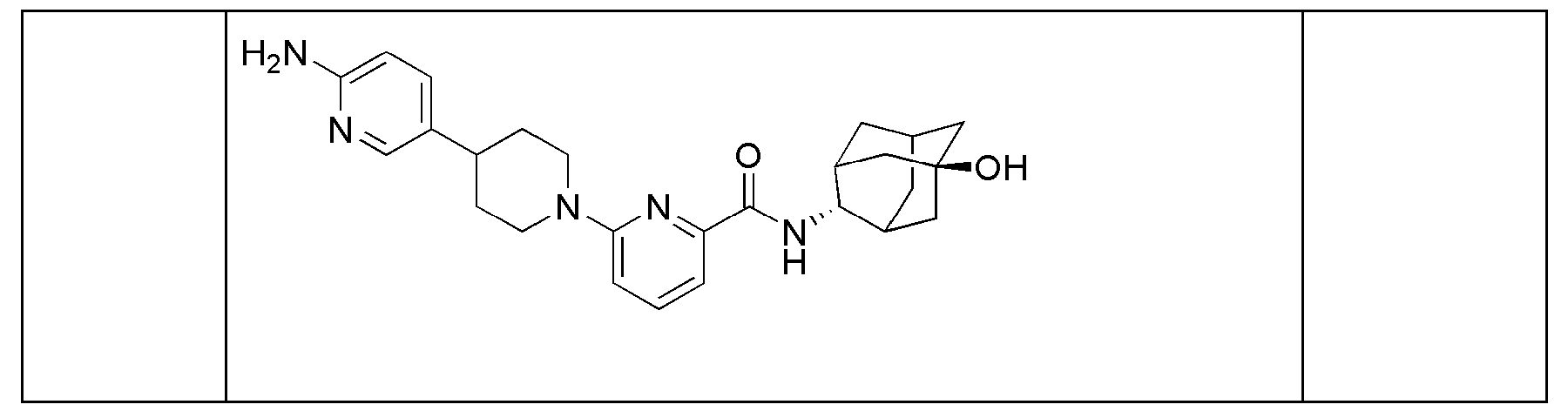
[269] Диизопропиламин (4,6 мл, 32,6 ммоль) растворяли в ТГФ (35 мл), а затем получающуюся в результате жидкость перемешивали при -78°С в токе азота с последующим медленным добавлением н-бутиллития (1,6М раствор в гексане, 20 мл, 32,6 ммоль). После перемешивания в течение 5 минут добавляли трет-бутоксикарбонил-4-пиперидон (5 г, 25,1 ммоль) и растворяли в ТГФ (25 мл) с последующим перемешиванием в течение 10 минут, а затем добавляли имид N-фенилтрифторметансульфона (9,8 г, 27,6 ммоль) и растворяли в ТГФ (25 мл) с последующим перемешиванием в течение 30 минут. Температуру реакции поднимали до комнатной температуры с последующим перемешиванием в течение двух с половиной часов, а затем реакцию прекращали добавлением к получающейся жидкости насыщенного водного раствора NaHCO3 (50 мл), туда добавляли 5% лимонную кислоту (50 мл) с последующей экстракцией диэтиловым эфиром (200 мл). Органический слой последовательно промывали 1н водным раствором гидроксида натрия (100 мл × 2), дистиллированной водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), а затем сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием. Полученный таким образом остаток подвергали MPLC (10% EtOAc/гексаны), получая 4,64 г желтого масла (56%).
[270] Стадия 2: Синтез трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата
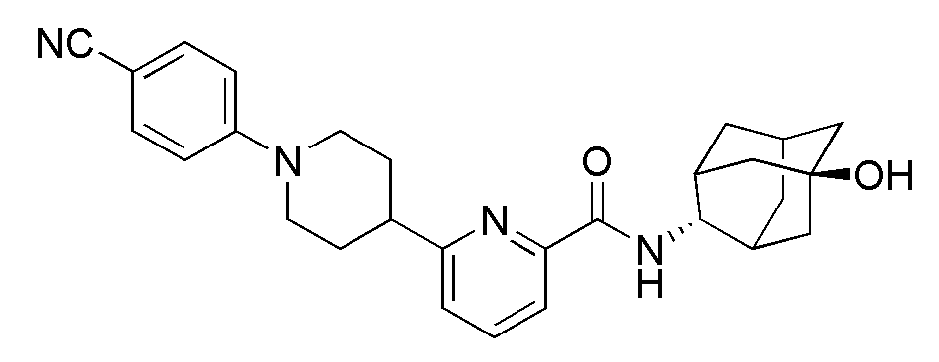
[271] Трет-бутил 4-(((трифторметил)сульфонил)окси)-5,6-дигидропиридин-1(2Н)-карбоксилат (4,64 г, 14 ммоль) растворяли в 1,4-диоксане (70 мл), а затем туда добавляли бис(пинахолато)диборон (3,91 г, 15,4 ммоль), КОАс (4,12 г, 42,0 ммоль), PdCl2dppf (343 мг, 0,42 ммоль) и dppf (233 мг, 0,42 ммоль). Получающуюся смесь перемешивали при 80°С в токе азота в течение 6 часов. К получающейся реакционной жидкости добавляли дистиллированную воду (50 мл) с последующей экстракцией МС (80 мл × 3). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% EtOAc/гексаны), получая 2,45 г желтого твердого вещества (57%).
[272] Стадия 3: Синтез трет-бутил 4-(4-фторфенил)-5,6-дигидропиридин-1(2Н)-карбоксилата
[273] Трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (120 мг, 0,388 ммоль) растворяли в смеси толуол/этанол (3 мл/0,15 мл), а затем туда последовательно добавляли 1-бром-4-фторбензол (0,05 г, 0,466 ммоль), Pd(PPh3)4 (22 мг, 0,02 ммоль) и К2СО3 (107 мг, 0,776 ммоль). Получающуюся смесь перемешивали при 100°С в токе азота в течение 2 часов. К получающейся реакционной жидкости добавляли дистиллированную воду (3 мл) с последующей экстракцией МС (10 мл × 3). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (6% EtOAc/гексаны), получая 48 мг бесцветного масла (45%).
[274] Стадия 4: Синтез трет-бутил 4-(4-фторфенил)пиперидин-1-карбоксилата
[275] Трет-бутил 4-(4-фторфенил)-5,6-дигидропиридин-1(2Н)-карбоксилат (48 мг, 0,173 ммоль) растворяли в этаноле (2 мл) с последующим добавлением палладия (10 вес.% на активированном угле, 5 мг), а затеи получающуюся жидкость перемешивали при комнатной температуре в токе водорода в течение 2 часов. Получающуюся в результате жидкость фильтровали, концентрировали при пониженном давлении и сушили в вакууме, получая 46 мг бесцветного масла (95%).
[276] Стадия 5: Синтез 4-(4-фторфенил)пиперидина
[277] Трет-бутил 4-(4-фторфенил)пиперидин-1-карбоксилат (46 мг, 0,165 ммоль) растворяли в МС (2 мл) с последующим добавлением трифторуксусной кислоты (1 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 4 часов. Получающуюся в результате реакционную жидкость нейтрализовали добавлением насыщенного водного раствора NaHCO3 при 0°С, а затем экстрагировали 5% МеОН/МС (10 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 22 мг бесцветного масла (74%).
[278] Стадия 6: Синтез 6-(4-(4-фторфенил)пиперидин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[279] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (40 мг, 0,114 ммоль), 4-(4-фторфенил)пиперидин (22 мг, 0,125 ммоль), Pd2(dba)3 (2 мг, 2 мол%), ксантфос (4 мг, 6 мол%) и трет-бутоксид натрия (16 мг, 0,171 ммоль) суспендировали в толуоле (1,5 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 2 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (3 мл) с последующей экстракцией МС (10 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 20 мг желтого твердого вещества (39%). MS (ESI): 450[M+Н]+.
[280]
[281] Соединения следующих примеров синтезировали тем же способом, что в примере 91 выше с использованием соответствующего бромбензольного или бромпиридинового исходного материала.
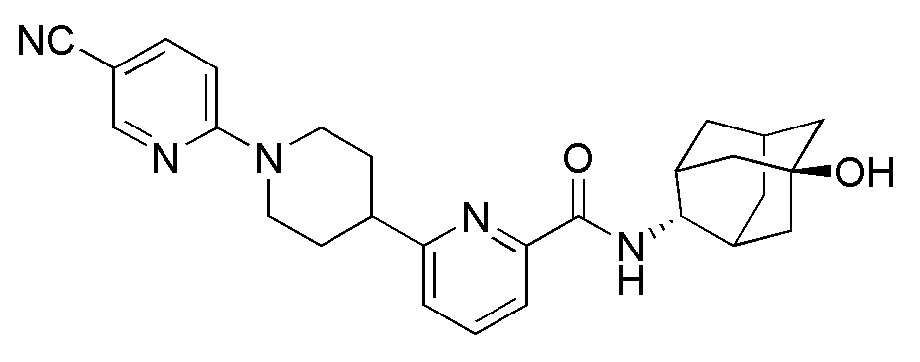
[282] Таблица 9
[283]
[284] Пример 104: Синтез 6-(1-(4-цианофенил)пиперидин-4-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[285]
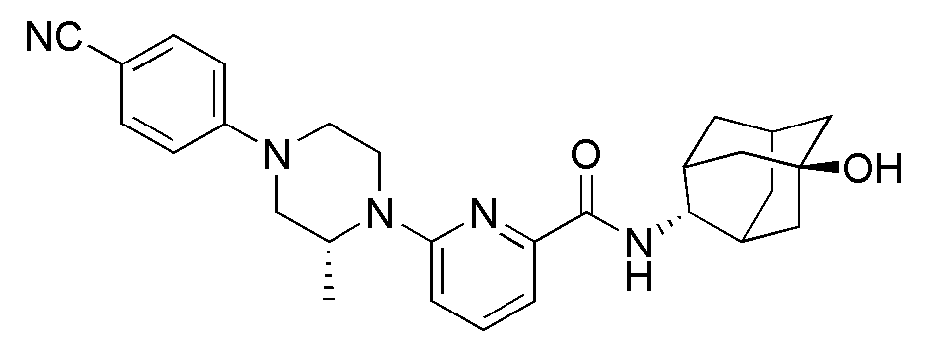
[286] Стадия 1: Синтез трет-бутил 6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)-5',6'-дигидро-[2,4'-бипиридин]-1'(2'H)-карбоксилата)
[287] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (300 мг, 0,854 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1-(2Н)-карбоксилат (317 мг, 1,025 ммоль) и карбонат калия (236 мг, 1,708 ммоль) суспендировали в толуоле (10 мл) с последующим добавлением EtOH (0,5 мл) и Pd(PPh3)4 (49 мг, 0,043 ммоль), а затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 4 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (20 мл) с последующей экстракцией МС (50 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 320 мг бледно желтого твердого вещества (83%).
[288] Стадия 2: Синтез трет-бутил 4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-1-карбоксилата
[289] Трет-бутил (6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)-5',6'-дигидро-[2,4'-бипиридин]-1'(2'H)-карбоксилат (320 мг, 0,706 ммоль) растворяли в этаноле (15 мл) с последующим добавлением палладия (10 вес.% на активированном угле, 200 мг), а затеи получающуюся жидкость перемешивали при комнатной температуре в токе водорода в течение 5 часов. Получающуюся в результате жидкость фильтровали, а затем концентрировали при пониженном давлении. Полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 285 мг белого твердого вещества (89%).
[290] Стадия 3: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-(пиперидин-4-ил)пиколинамида
[291] Трет-бутил 4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-карбоксилат (285 мг, 0,626 ммоль) растворяли в МС (4 мл) с последующим добавлением трифторуксусной кислоты (4 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 4 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (10 мл), а затем экстрагировали МС (15 мл). Водный слой нейтрализовали добавлением насыщенного водного раствора NaHCO3 c последующей экстракцией 5% МеОН/МС (25 мл × 4), а затем органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 186 мг белого твердого вещества (84%).
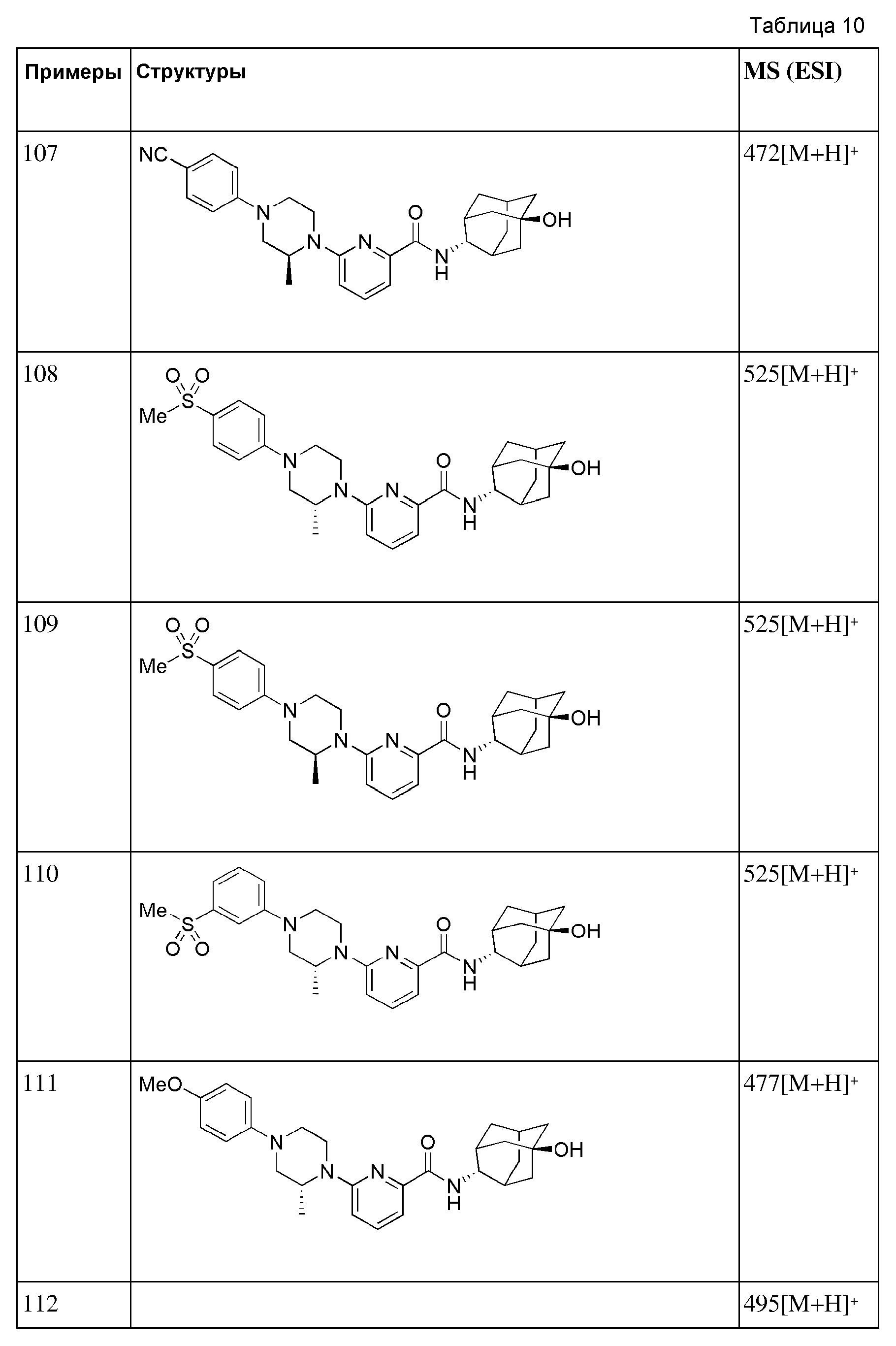
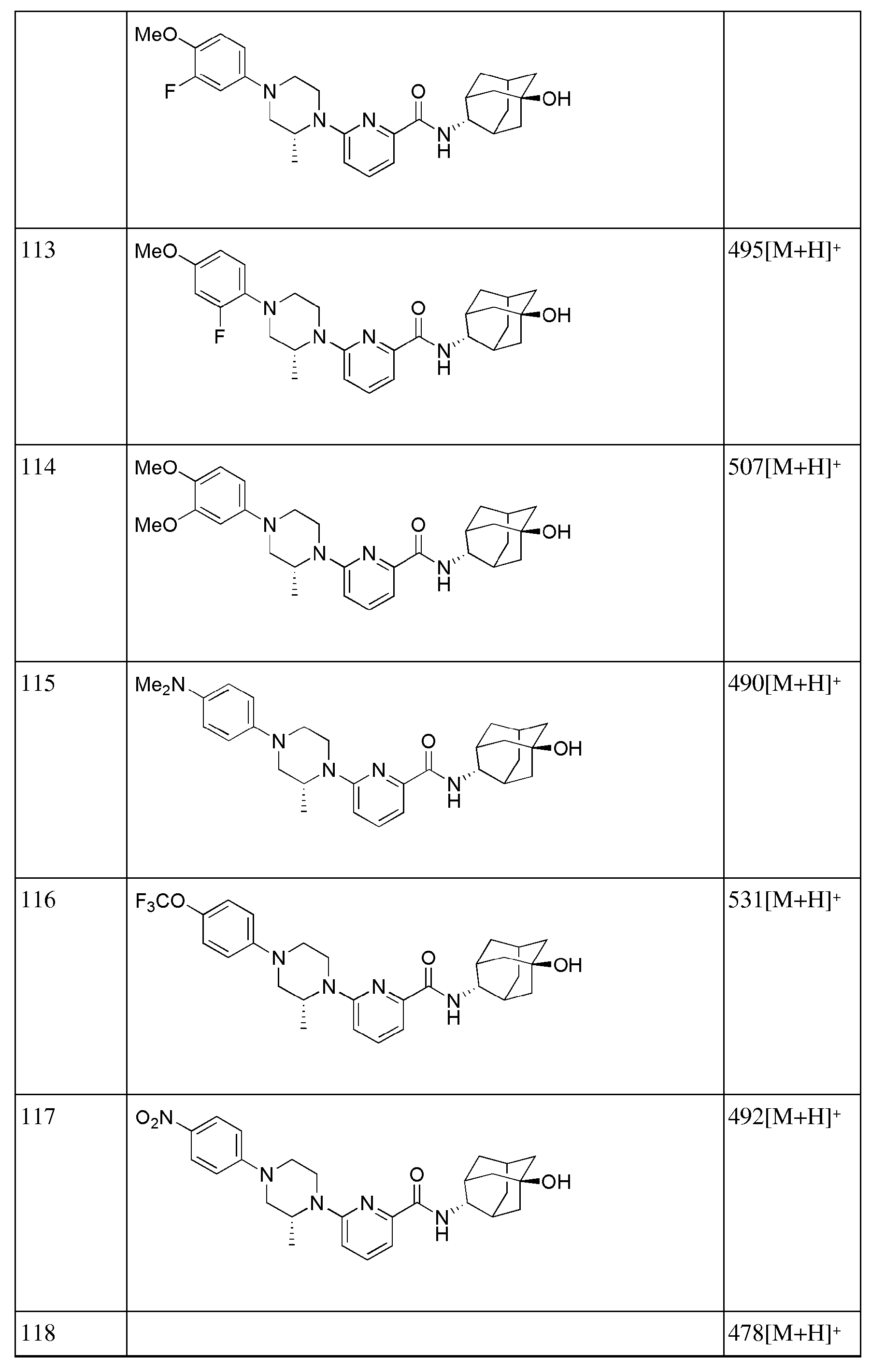
[292] Стадия 4: Синтез 6-(1-(4-цианофенил)пиперидин-4-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
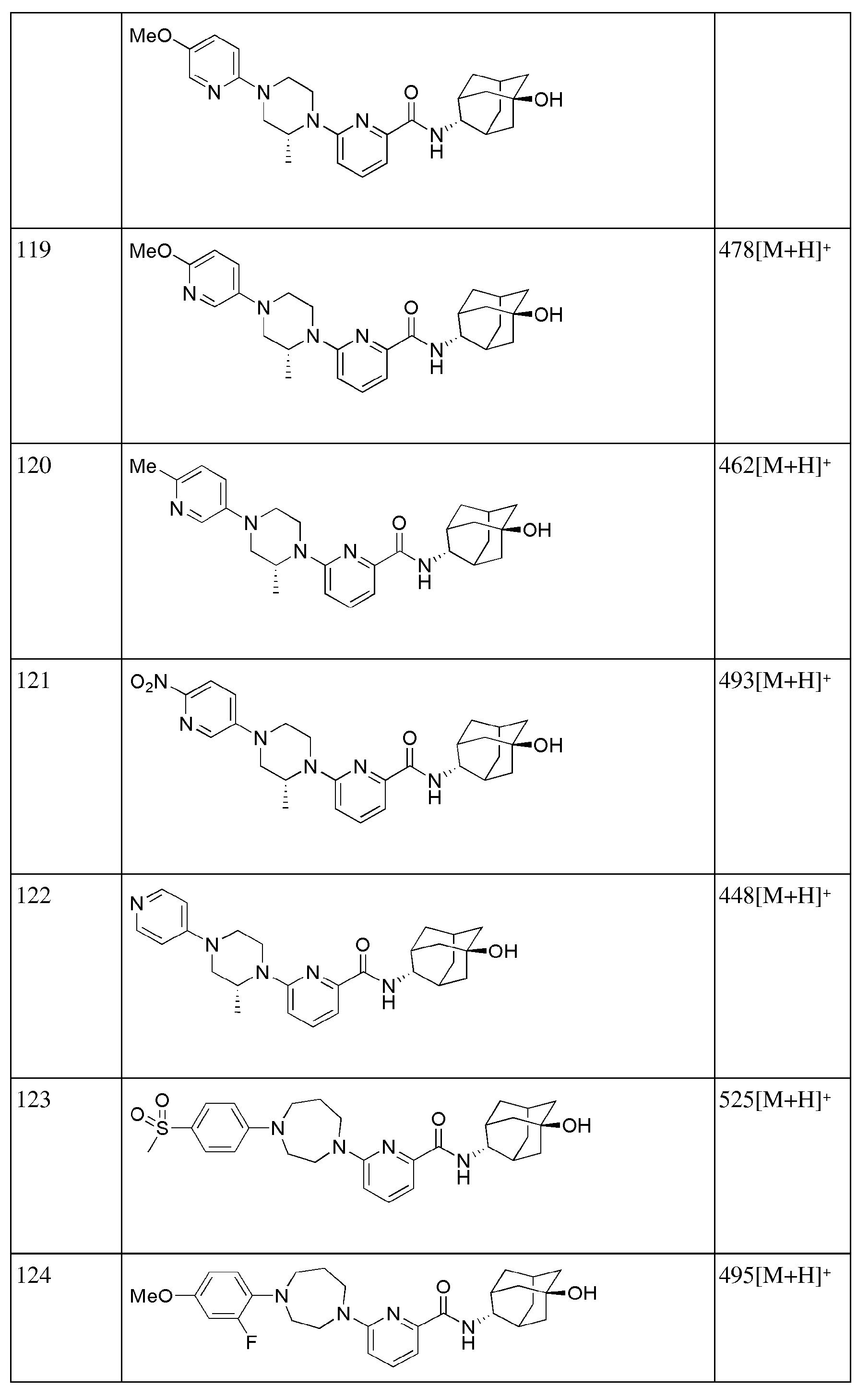
[293] N-((E)-5-гидроксиадамантан-2-ил)-6-(пиперидин-4-ил)пиколинамид (33 мг, 0,093 ммоль), 4-бромбензонитрил (19 мг, 0,102 ммоль), Pd2(dba)3 (2 мг, 0,002 ммоль), BINAP (3,5 мг, 0,006 ммоль) и трет-бутоксид натрия (13 мг, 0,140 ммоль) суспендировали в толуоле (2 мл) и затем получающуюся жидкость перемешивали при 80°С в токе азота в течение 4 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 31 мг белого твердого вещества (73%). MS (ESI): 457[M+Н]+.
[294]
[295] Пример 105: Синтез 6-(1-(5-цианопириддин-2-ил)пиперидин-4-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[296]
[297] N-((E)-5-Гидроксиадамантан-2-ил)-6-(пиперидин-4-ил)пиколинамид (40 мг, 0,113 ммоль) и 6-хлорникотинонитрил (19 мг, 0,135 ммоль) растворяли в ацетонитриле (1 мл) с последующим добавлением триэтиламина (0,03 мл, 0,226 ммоль) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 5 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 42 мг белого твердого вещества (82%). MS (ESI): 458[M+Н]+.
[298]
[299] Пример 106: Синтез 6-((R)-4-(4-цианофенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[300]
[301] Стадия 1: Синтез (R)-4-(3-метилпиперазин-1-ил)бензонитрила
[302] 4-Бромбензонитрил (200 мг, 1,099 ммоль), (R)-2-метилпиперазин (121 мг, 1,209 ммоль), Pd2(dba)3 (20 мг, 0,022 ммоль), BINAP (41 мг, 0,066 ммоль) и трет-бутоксид натрия (211 мг, 2,199 ммоль) суспендировали в толуоле (5 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 5 часов. К получающейся в результате реакционной жидкости добавляли 1н водный раствор HCl (20 мл) с последующей экстракцией МС (10 мл × 2). Водный слой нейтрализовали добавлением 5н водного раствора NaOH с последующей экстракцией 5% МеОН/МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 152 мг бледно желтого масла (69%).
[303] Стадия 2: Синтез 6-(R)-4-(4-цианофенил)-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[304] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (80 мг, 0,228 ммоль), (R)-4-(3-метилпиперазин-1-ил)бензонитрил (55 мг, 0,273 ммоль), Pd2(dba)3 (4,2 мг, 0,005 ммоль), ксантфос (7,9 мг, 0,014 ммоль) и трет-бутоксид натрия (33 мг, 0,342 ммоль) суспендировали в толуоле (5 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 15 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 20 мг бледно желтого твердого вещества (19%). MS (ESI): 472[M+Н]+.
[305] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 106 выше, с использованием соответствующего бромбензольного или бромпиридинового исходного материала и пиперазинового исходного материала.
[306] Таблица 10
[307]
[308] Пример 125: Синтез 6-((R)-4-(6-аминопиридин-3-ил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[309]
[310] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(6-нитропиридин-3-ил)пиперазин-1-ил)пиколинамид (85 мг, 0,173 ммоль) растворяли в 20% МеОН/EtOH (12 мл) с последующим добавлением Pd (10 вес.% на активированном угле, 40 мг) и затем получающуюся жидкость перемешивали при комнатной температуре в токе водорода в течение 3 часов. Получающуюся реакционную жидкость фильтровали, а затем концентрировали при пониженном давлении. Полученный таким образом остаток подвергали MPLC (7% МеОН/МС), получая 33 мг желтого твердого вещества (41%). MS (ESI): 463[M+Н]+.
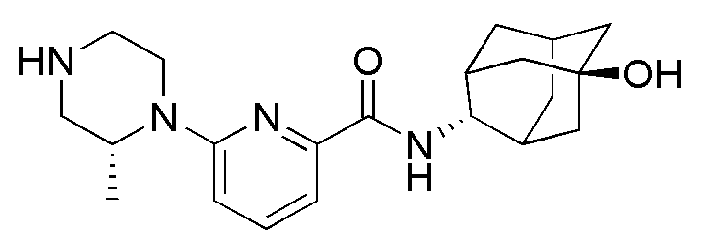
[311]
[312] Пример 126: Синтез 6-((R)-4-(4-аминофенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[313]
[314] Выполняли тот же способ, что в примере 125, за исключением того, что вместо N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(6-нитропиридин-3-ил)пиперазин-1-ил)пиколинамида использовали N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-нитрофенил)пиперазин-1-ил)пиколинамид (119 мг, 0,242 ммоль), получая 105 мг желтого твердого вещества (94%). MS (ESI): 462[M+H]+.
[315]
[316] Пример 127: Синтез 6-((R)-4-(4-ацетамидофенил)-2- метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[317]
[318] 6-((R)-4-(4-аминофенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (109 мг, 0,236 ммоль) растворяли в МС (5 мл) с последующим последовательным добавлением триэтиламина (0,049 мл, 0,354 ммоль) и уксусного ангидрида (0,022 мл, 0,236 ммоль) и затем получающуюся жидкость перемешивали при комнатной температуре в токе азота в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 94 мг белого твердого вещества (79%). MS (ESI): 504[M+Н]+.
[319]
[320] Пример 128: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-метилсульфонамидо)фенил)пиперазин-1-ил)пиколинамида
[321]

[322] 6-((R)-4-аминофенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (36 мг, 0,077 ммоль) растворяли в пиридине (1 мл) с последующим последовательным добавлением метансульфонилхлорида (0,010 мл, 0,129 ммоль) и затем получающуюся жидкость перемешивали при комнатной температуре в токе азота в течение 3 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 14 мг белого твердого вещества (34%). MS (ESI): 540[M+Н]+.
[323]
[324] Пример 129: Синтез N-((E)-5-гидроксиадамантан-2-ил) 6-((R)-2-метилпиперазин-1-ил)пиколинамида (промежуточное соединение 10)
[325]
[326] Стадия 1: Синтез (R)-трет-бутил 4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-карбоксилата
[327] 6-Бром-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (1,0 г, 2,847 ммоль), (R)-трет-бутил 3-метилпиперазин-1-карбоксилат (855 мг, 4,271 ммоль), Pd2(dba)3 (52 мг, 0,057 ммоль), ксантфос (99 мг, 0,171 ммоль) и трет-бутоксид натрия (410 мг, 4,271 ммоль) суспендировали в толуоле (20 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 3 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (20 мл) с последующей экстракцией МС (40 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (90% этилацетат/гексаны), получая 720 мг бледно желтого твердого вещества (54%).
[328] Стадия 2: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамида (промежуточное соединение 10)
[329] (R)-Трет-бутил 4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-карбоксилат (715 мг, 1,519 ммоль) растворяли в МС (10 мл) с последующим добавлением трифторуксусной кислоты (10 мл) и затем получающуюся жидкость перемешивали при комнатной температуре в течение 3 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (30 мл) с последующей экстракцией МС (15 мл × 2). Водный слой нейтрализовали добавлением 5н водного раствора NaOH с последующей экстракцией 5% МеОН/МС (40 мл × 3), а затем органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 516 мг белого твердого вещества (92%). MS (ESI): 371[M+Н]+.
[330]
[331] Пример 130: Синтез метил 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил0пиридин-2-ил)-3-метилпиперазин-1-ил)бензоата
[332]

[333] N-((E)-5-Гидроксиадамантан-2-ил)-6((R)-2-метилпиперазин-1-ил)пиколинамид (200 мг, 0,540 ммоль), метил 4-бромбензоат (174 мг, 0,810 ммоль), Pd(EAc)2 (5 мг, 0,022 ммоль), ксантфос (19 мг, 0,032 ммоль) и карбонат цезия (264 мг, 0,810 ммоль) суспендировали в толуоле (20 мл) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (100% этилацетат), получая 224 мг бледно желтого твердого вещества (82%). MS (ESI): 505[M+Н]+.
[334]
[335] Пример 131: Синтез 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензойной кислоты
[336]

[337] Метил 4-((R)-4-(6-(((E)-5-гидроадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензоат (100 мг, 0,198 ммоль) растворяли в МеОН (5 мл) с последующим добавлением 2н водного раствора NaOH (0,50 мл, 0,994 ммоль) и затем получающуюся смесь перемешивали при комнатной температуре в течение 20 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении и растворяли добавлением дистиллированной воды (10 мл), а затем получающуюся жидкость нейтрализовали добавлением 1н водного раствора HCl при перемешивании при 0°С. Осажденное твердое вещество отфильтровывали с последующей вакуумной сушкой, получая 72 мг бледно желтого твердого вещества (74%). MS (ESI): 491[M+Н]+.
[338]
[339] Пример 132: Синтез 6-((R)-4-(4-карбамоилфенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[340]
[341] 4-((R)-6-(((E)-5-Гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензойную кислоту (60 мг, 0,122 ммоль) растворяли в ацетонитриле (2 мл) с последующим последовательным добавлением аммиака (0,5М раствор в диоксане, 0,49 мл, 0,244 ммоль), N,N-диизопропилэтиламина (0,043 мл, 0,244 ммоль) и HBTU (56 мг, 0,146 ммоль) и затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией 10% МеОН/МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 47 мг бледно желтого твердого вещества (78%). MS (ESI): 512[M+Na]+.
[342]
[343] Пример 133: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-(4-(4-метилкарбамоил)фенил)пиперазин-1-ил)пиколинамида
[344]

[345] Выполняли тот же способ, что в примере 132, за исключением того, что вместо аммиака использовали метиламин (2М раствор в ТГФ), получая 17 мг бледно желтого твердого вещества (28%). MS (ESI): 504[M+H]+.
[346]
[347] Пример 134: Синтез 6-((R)-4-(4-циклопропилкарбамоил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[348]
[349] Выполняли тот же способ, что в примере 132, за исключением того, что вместо аммиака использовали циклопропиламин, получая 44 мг бледно желтого твердого вещества (70%). MS (ESI): 530[M+H]+.
[350]
[351] Пример 135: Синтез N-((E)-5-гидроксиадамантан-2-ил) 6-((R)-4-(4-(2-оксиэтил)карбамоил)фенил)-2-метилпиперазин-1-ил)пиколинамида
[352]
[353] Выполняли тот же способ, что в примере 132, за исключением того, что вместо аммиака использовали 2-аминоэтанол, получая 50 мг бледно желтого твердого вещества (77%). MS (ESI): 534[M+H]+.
[354]
[355] Соединения следующих примеров синтезировали тем же самым способом, что и в примерах 130, 131, 132 и 133 выше, с использованием промежуточного соединения 10 и соответствующего бромбензольного или бромпиридинового исходного материала.
[356] Таблица 11
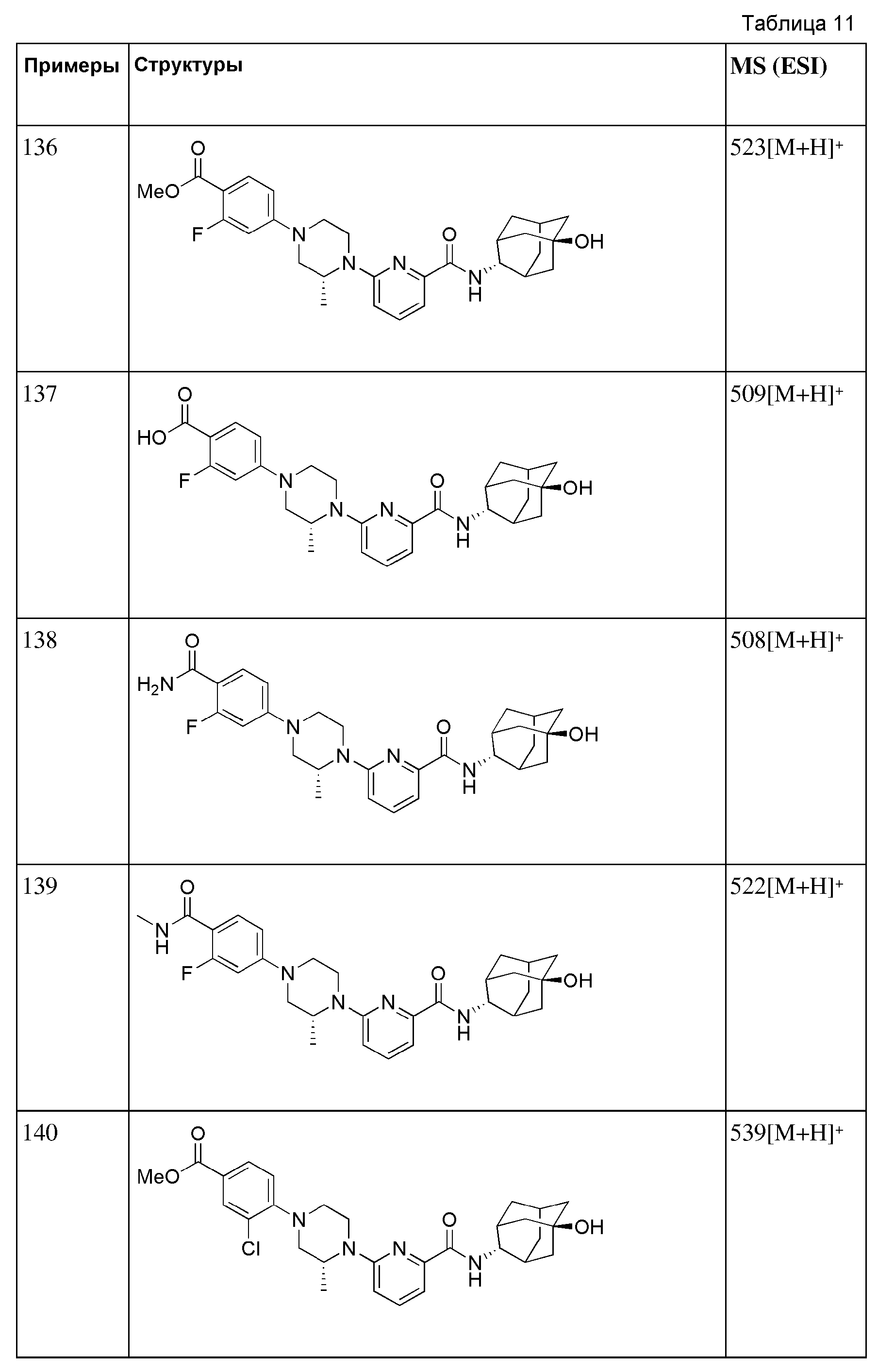
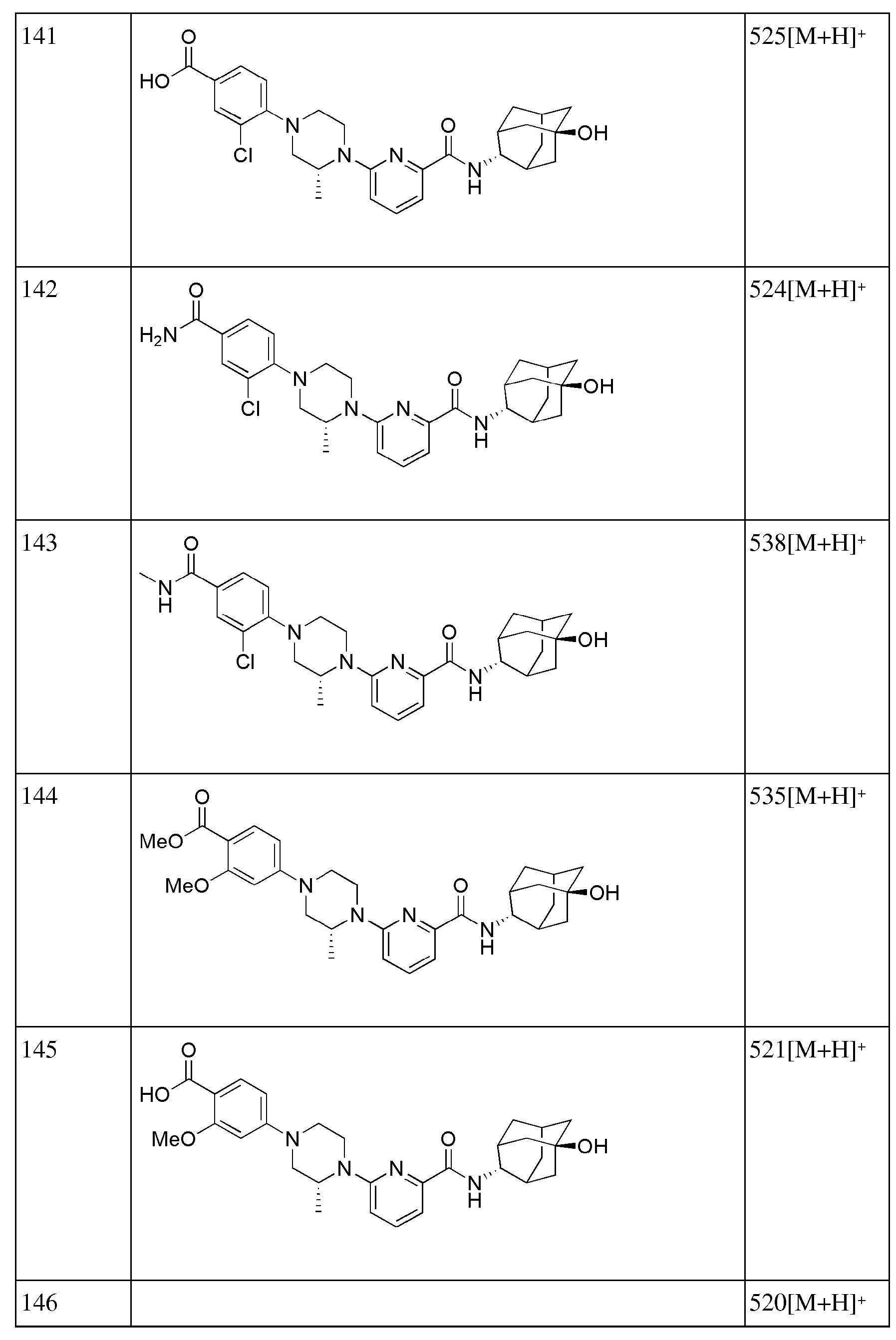
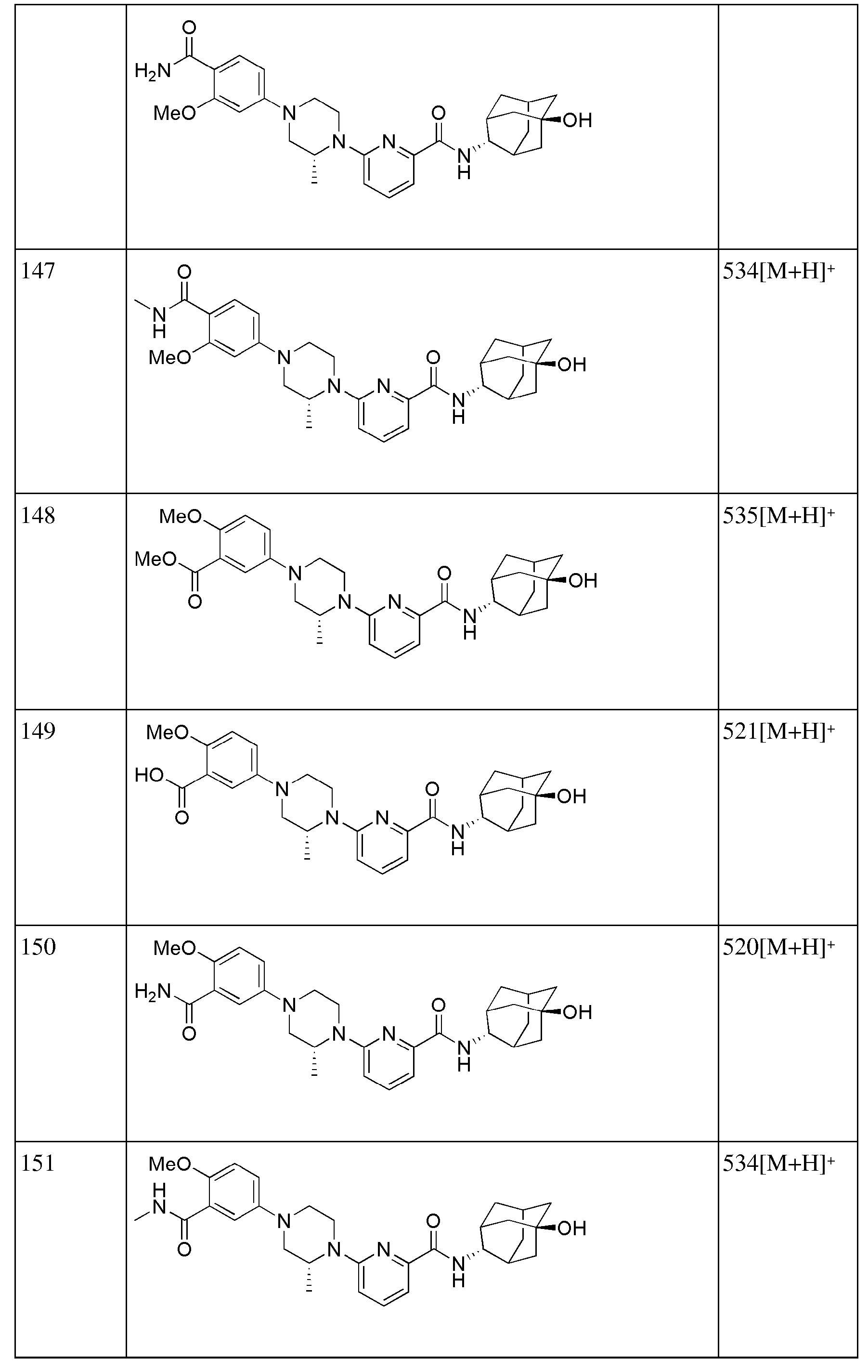

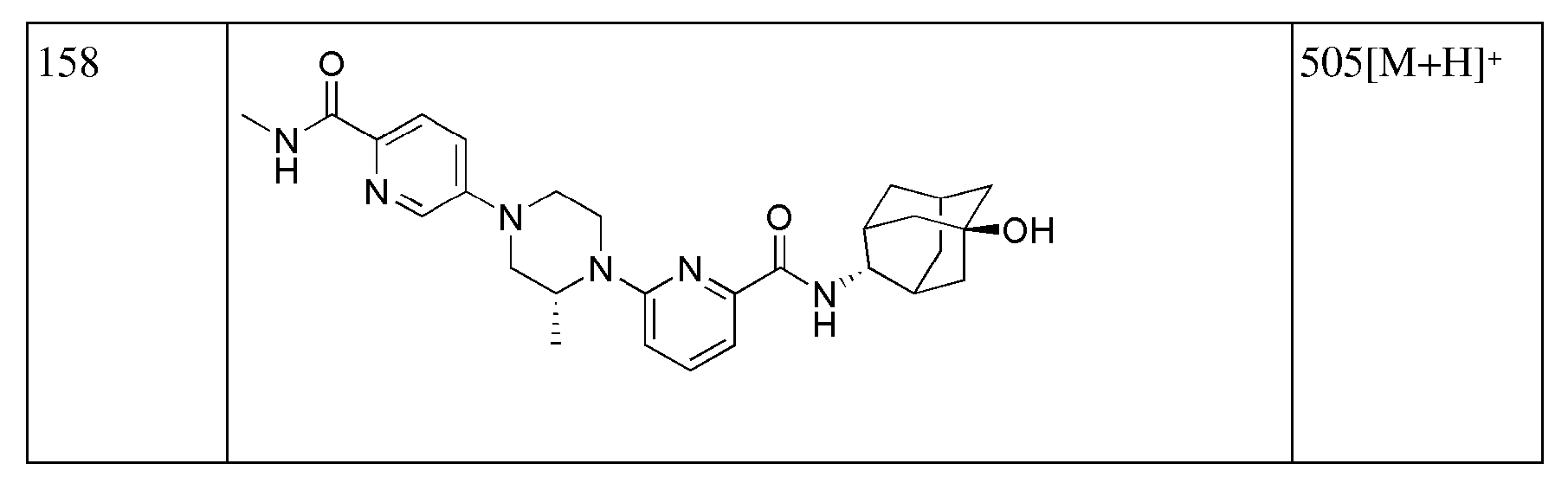
[357]
[358] Пример 159: Синтез метил 6-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)никотината
[359]
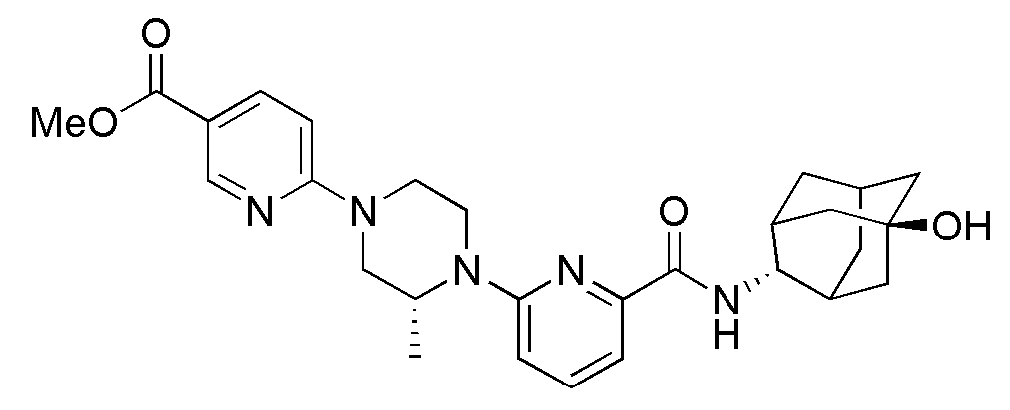
[360] N-(((E)-5-Гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (120 мг, 0,324 ммоль) и метил 6-бромникотинат (84 мг, 0,389 ммоль) суспендировали в ацетонитриле (4 мл) с последующим добавлением триэтиламина (0,09 мл, 0,648 ммоль) и затем получающуюся жидкость перемешивали при 95°С в токе азота в течение 24 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 160 мг бледно желтого масла (98%). MS (ESI): 506[M+Na]+.
[361]
[362] Пример 160: Синтез 6-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)никотиновой кислоты
[363]
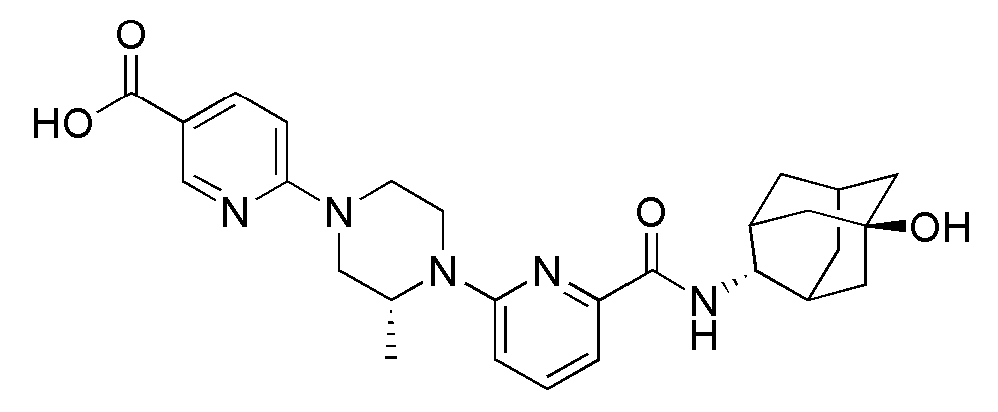
[364] Выполняли тот же способ, что в примере 131, за исключением того, что вместо 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензоата использовали метил 6-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)никотинат (155 мг, 0,307 ммоль), получая 135 мг бледно желтого твердого вещества (90%). MS (ESI): 492[M+H]+.
[365]
[366] Пример 161: Синтез 6-((R)-4-(5-карбамоилпиридин-2-ил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[367]
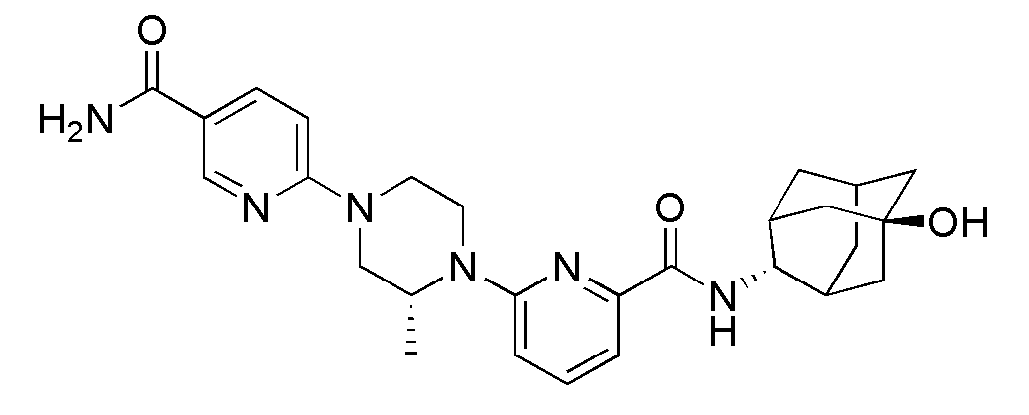
[368] Выполняли тот же способ, что в примере 132, за исключением того, что вместо 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензойной кислоты использовали 6-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)никотиновую кислоту (40 мг, 0,081 ммоль), получая 28 мг белого твердого вещества (70%). MS (ESI): 491[M+H]+.
[369]
[370] Пример 162: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(5-метилкарбамоил)пиридин-2-ил)пиперазин-1-ил)пиколинамида
[371]
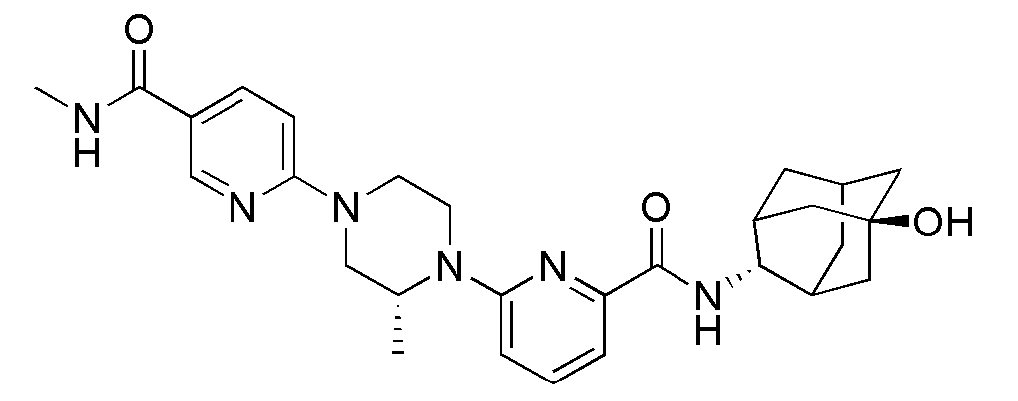
[372] Выполняли тот же способ, что в примере 133, за исключением того, что вместо 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензойной кислоты использовали 6-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)никотиновую кислоту (40 мг, 0,081 ммоль), получая 25 мг белого твердого вещества (61%). MS (ESI): 505[M+H]+.
[373]
[374] Соединения следующих примеров синтезировали тем же самым способом, что и в примерах 159, 160, 161 и 162 выше, с использованием промежуточного соединения 10 и этил 5,6-дихлорникотината.
[375] Таблица 12
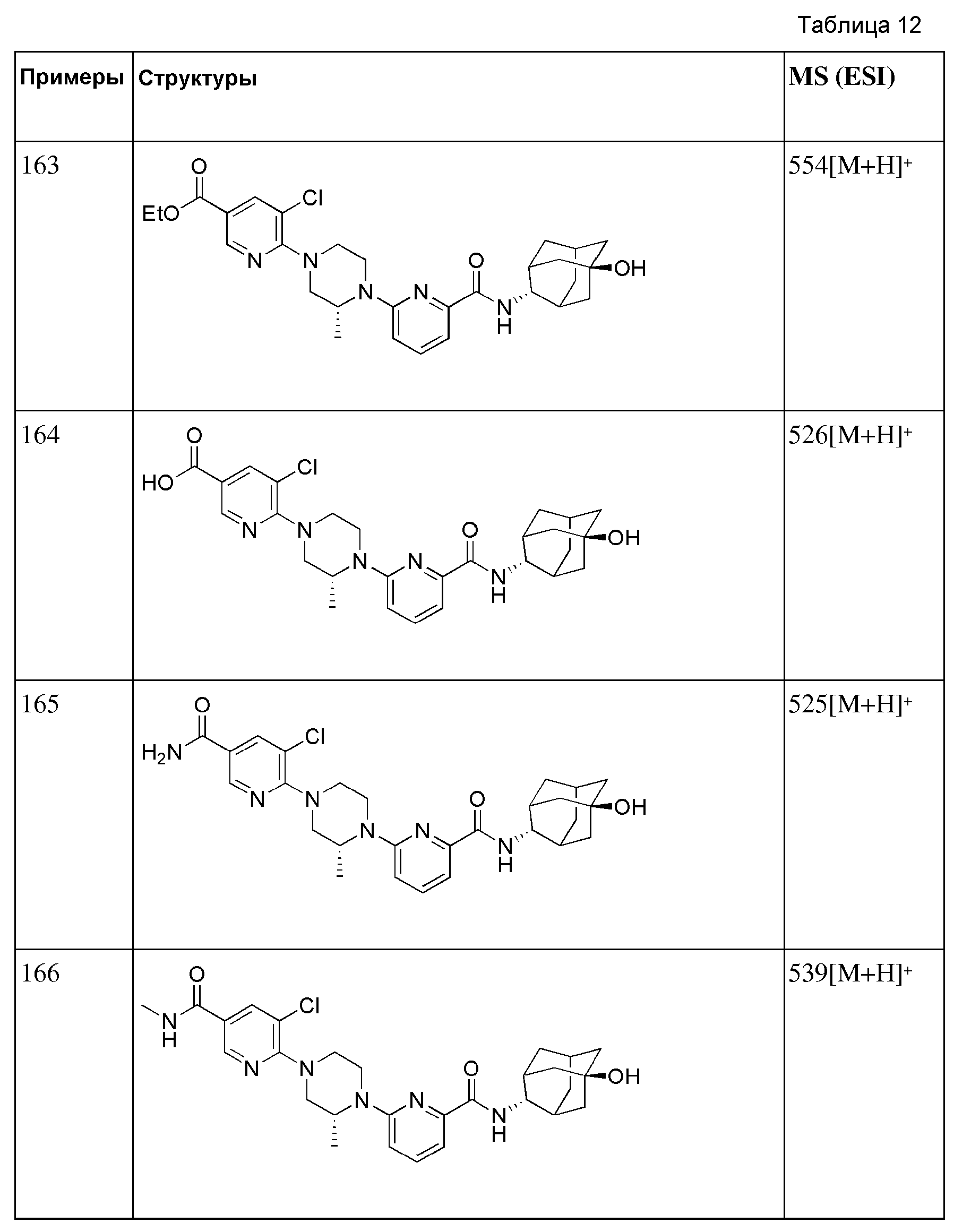
[376]
[377] Пример 167: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-сульфамоилфенил)пиперазин-1-ил)пиколинамида
[378]
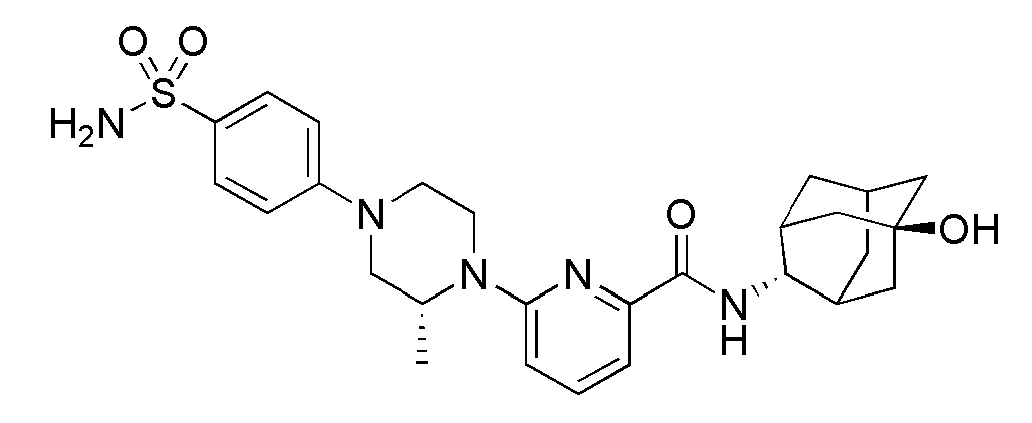
[379] Стадия 1: Синтез 4-бром-N-(трет-бутил)бензолсульфонамида
[380] 4-Бромбензолсульфонилхлорид (200 мг, 0,783 ммоль) растворяли в МС (10 мл) с последующим добавлением по каплям трет-бутиламина (0,41 мл, 3,91 ммоль) при 0°С, а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 1 часа. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (15 мл) с последующей экстракцией МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток сушили в вакууме, получая 228 мг белого твердого вещества (100%).
[381] Стадия 2: Синтез 6-((R)-4-(4-N-трет-бутил)сульфамоил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[382] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (30 мг, 0,0810 ммоль), 4-бром-N-(трет-бутил)бензолсульфониламид (28 мг, 0,0972 ммоль), Pd[P(o-толил)3]2Cl2 (1 мг, 0,000810 ммоль), BINAP (3 мг, 0,00486 ммоль) и карбонат цезия (26 мг, 0,0810 ммоль) суспендировали в толуоле (5 мл), а затем получающуюся жидкость перемешивали при 90°С в токе азота в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 45 мг бледно желтого твердого порошка (96%).
[383] Стадия 3: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-сульфамоилфенил)пиперазин-1-ил)пиколинамида
[384] 6-((R)-4-(4-N-(трет-бутил)сульфамоил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (69 мг, 0,119 ммоль) растворяли в МС (3 мл) с последующим добавлением трифторуксусной кислоты (3 мл) и затем получающуюся смесь перемешивали при комнатной температуре в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (10 мл), а затем экстрагировали МС (5 мл). Водный слой нейтрализовали добавлением насыщенного водного раствора NaHCO3, с последующей экстракцией 10% МеОН/МС (25 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 40 мг бледно желтого твердого вещества (64%). MS (ESI): 526[M+Na]+.
[385]
[386] Пример 168: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-N-метилсульфамоил)фенил)пиперазин-1-ил)пиколинамида
[387]
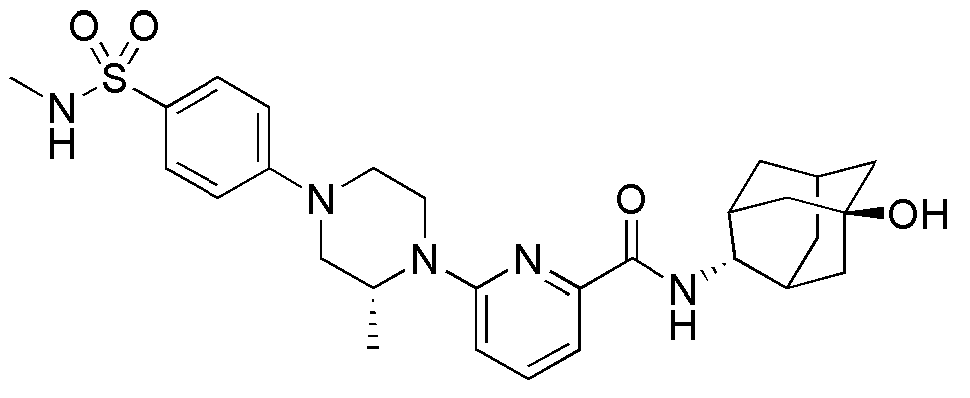
[388] Стадия 1: Синтез 4-бром-N-(трет-бутил)-N-метилбензолсульфонамида
[389] 4-Бром-N-(трет-бутил)бензолсульфониламид (100 мг, 0,342 ммоль) и карбонат калия (95 мг, 0,684 ммоль) растворяли в ДМФ (2 мл) с последующим добавлением иодометана (0,043 мл, 0,684 ммоль), а затем получающуюся жидкость перемешивали при комнатной температуре в течение 18 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (10 мл) с последующей экстракцией EtOAc (10 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (20% EtOAc/гексаны), получая 77 мг желтого масла (73%).
[390] Стадия 2: Синтез 6-((R)-4-(4-N-(трет-бутил)-N-метилсульфамоил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[391] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (70 мг, 0,189 ммоль), 4-бром-N-(трет-бутил)-N-метилбензолсульфонамид (69 мг, 0,227 ммоль), Pd[P(o-толил)3]2Cl2 (1,5 мг, 0,00189 ммоль), BINAP (7 мг, 0,0113 ммоль) и карбонат цезия (62 мг, 0,189 ммоль) суспендировали в толуоле (5 мл), а затем получающуюся жидкость перемешивали при 90°С в токе азота в течение 18 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 43 мг желтого твердого порошка (38%).
[392] Стадия 3: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-N-метилсульфамоил)фенил)пиперазин-1-ил)пиколинамида
[393] Трифторуксусную кислоту (3 мл) добавляли к 6-((R)-4-(4-(N-(трет-бутил)-(N-метилсульфамоил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамиду (43 мг, 0,0722 ммоль), а затем получающуюся смесь перемешивали при 90°С в течение 2 часов. Получающуюся в результате реакционную жидкость нейтрализовали медленным добавлением насыщенного водного раствора NaHCO3 c последующей экстракцией МС (30 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 14 мг бледно желтого твердого вещества (36%). MS (ESI): 562[M+Na]+.
[394]
[395] Соединения следующих примеров синтезировали тем же самым способом, что и в примерах 167 и 168 выше, с использованием соответствующего 4-бромбензолсульфонилхлорида.
[396] Таблица 13
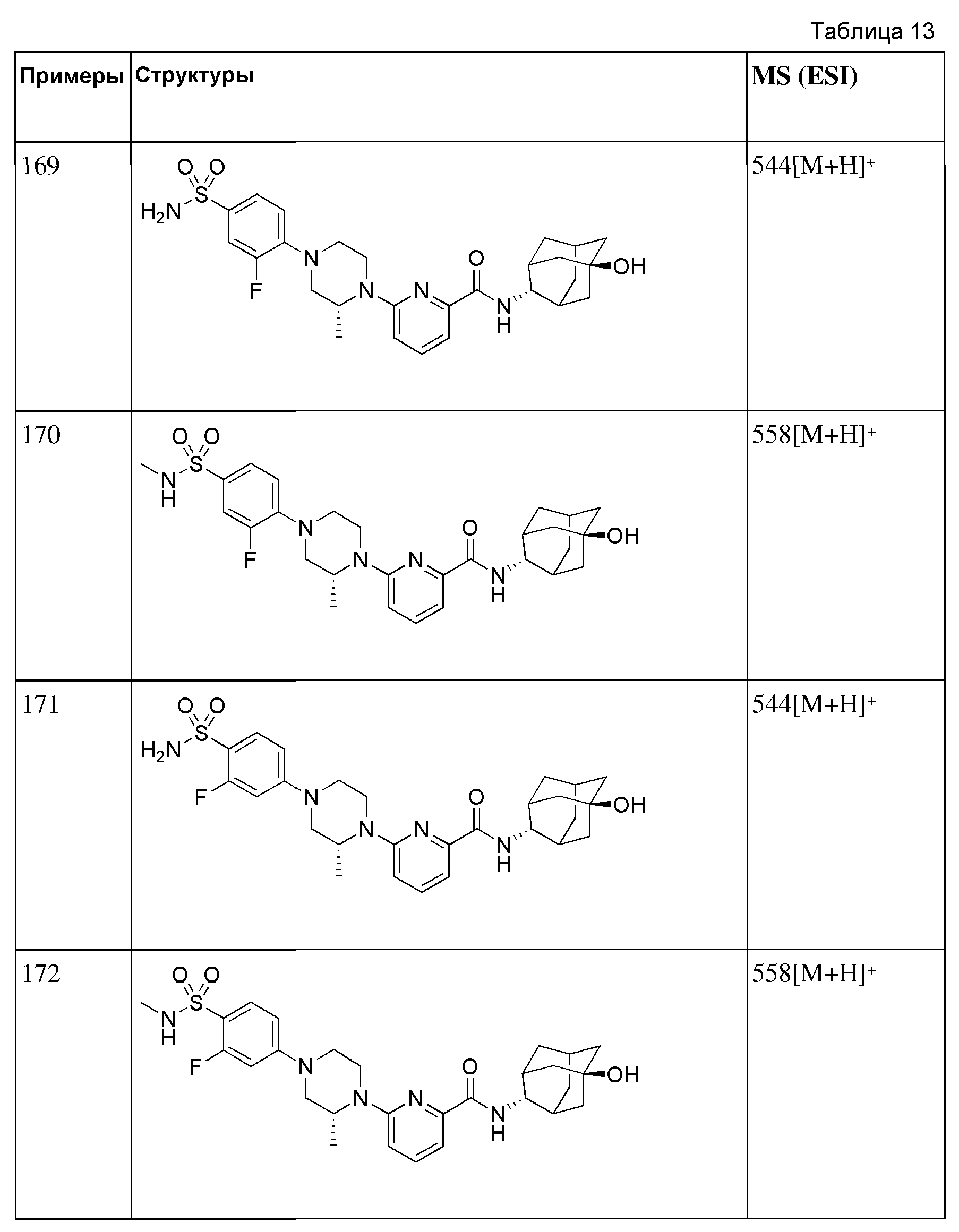
[397]
[398] Пример 173: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-4-(4-(2-гидроксипррпан-2-ил)фенил)-2-метилпиперазин-1-ил)пиколинамида
[399]
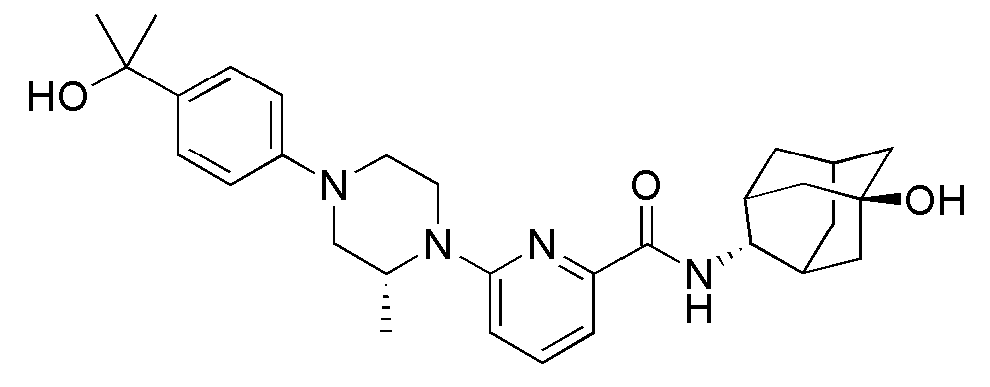
[400] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (30 мг, 0,081 ммоль), 2-(4-бромфенил)пропан-2-ол (21 мг, 0,097 ммоль), Pd(dba)3 (1,5 мг, 0,0016 ммоль), BINAP (3 мг, 0,0049 ммоль) и трет-бутоксид натрия (12 мг, 0,122 ммоль) суспендировали в толуоле (1 мл), а затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 15 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (10 мл) с последующей экстракцией МС (15 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 23 мг бледно желтого твердого порошка (56%). MS (ESI): 487[M-ОН]+.
[401]
[402] Пример 174: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-4-(6-(2-гидроксипропан-2-ил)пиридин-3-ил)-2-метилпиперазин-1-ил)пиколинамида
[403]
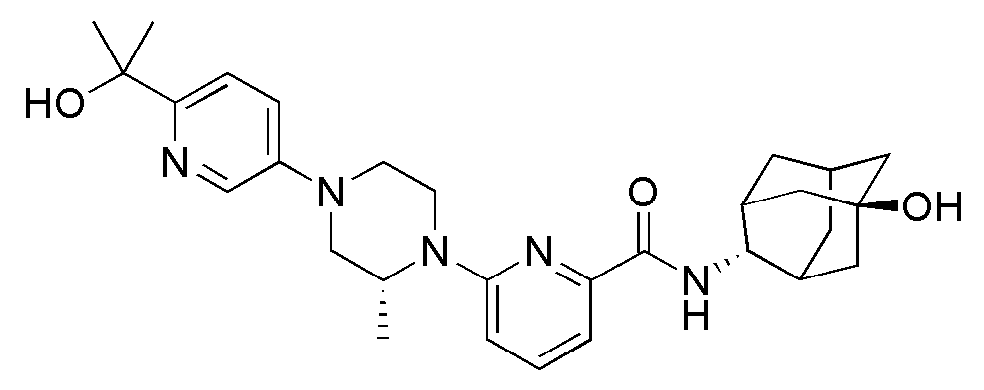
[404] Выполняли тот же способ, что в примере 173, за исключением того, что вместо 2-(4-бромфенил)пропан-2-ола использовали 2-(5-бромпиридин-2-ил)пропан-2-ол, получая 51 мг бледно желтого твердого вещества (29%). MS (ESI): 488[M-OH]+.
[405]
[406] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 173 выше, с использованием соответствующего бромбензола.
[407] Таблица 14
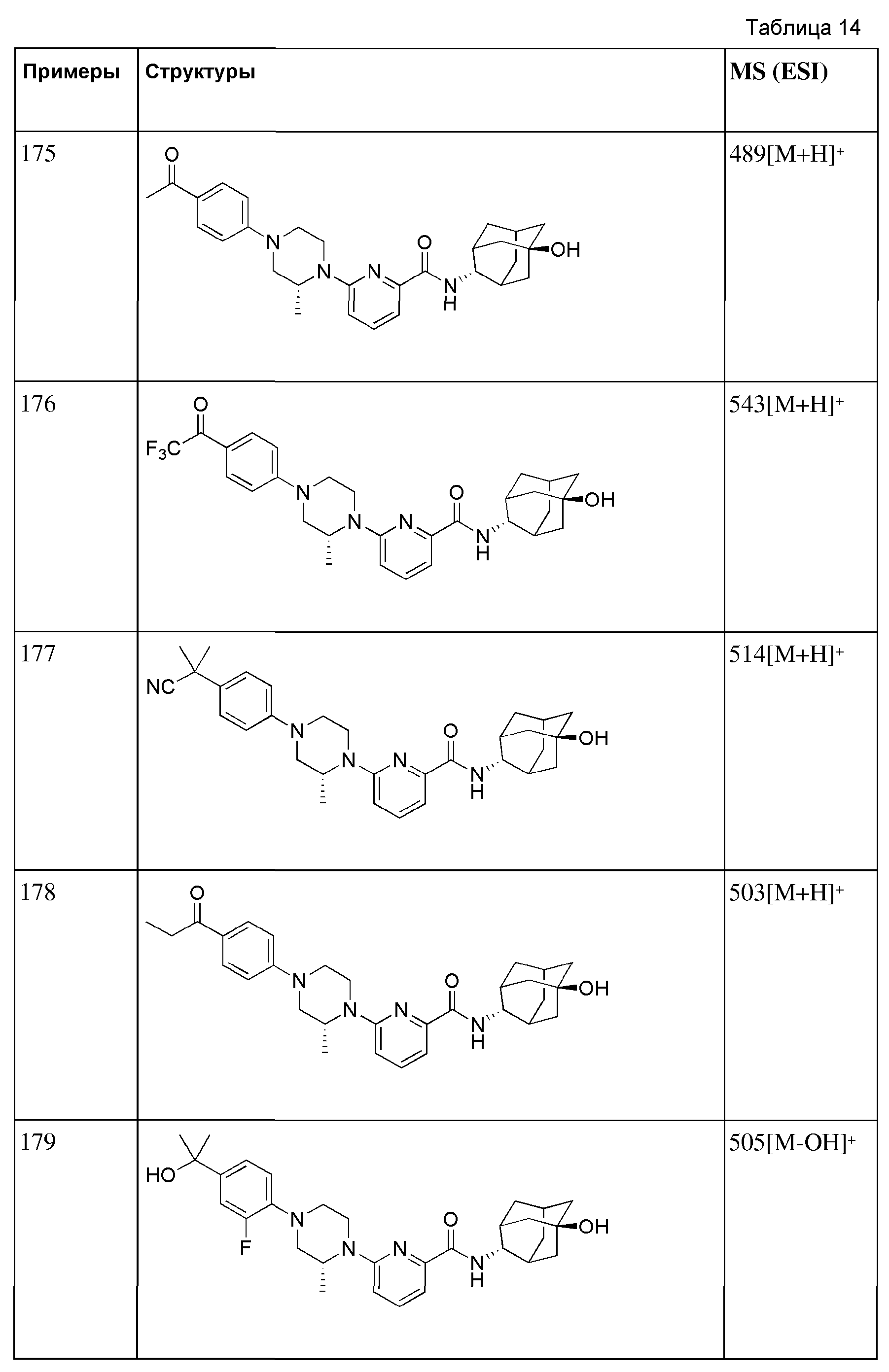

[408]
[409] Пример 184: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((2R)-4-(4-(1-гидроксиэтил)фенил)-2-метилпиперазин-1-ил)пиколинамида
[410]
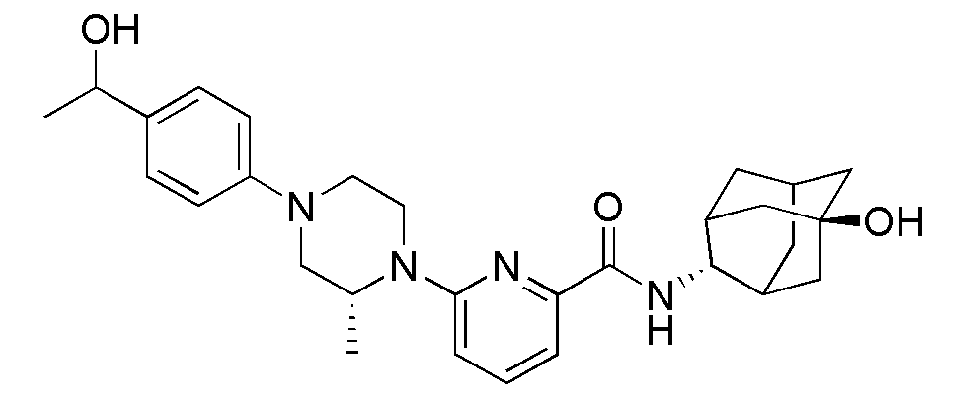
[411] 6-((R)-4-(4-ацетилфенил)-2-иметилпиперазин-1-ил)-N-(((E)-5-гидроксиадамантан-2-ил)пиколинамид (41 мг, 0,084 ммоль) растворяли в МеОН (2 мл), а затем к нему добавляли NaBH4 (4,8 мг, 0,126 ммоль) при комнатной температуре в токе азота. Выполняли повторно перемешивание получающейся жидкости в течение 30 минут, всего четыре раза. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (10 мл) с последующей экстракцией EtOAc (15 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 26 мг белого твердого вещества (63%). MS (ESI): 473[M-ОН]+.
[412]
[413] Пример 185: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((2R)-2-метил-4-(4-(2,2,2-трифтор-1-гидроксиэтил)фенил)пиперазин-1-ил)пиколинамида
[414]
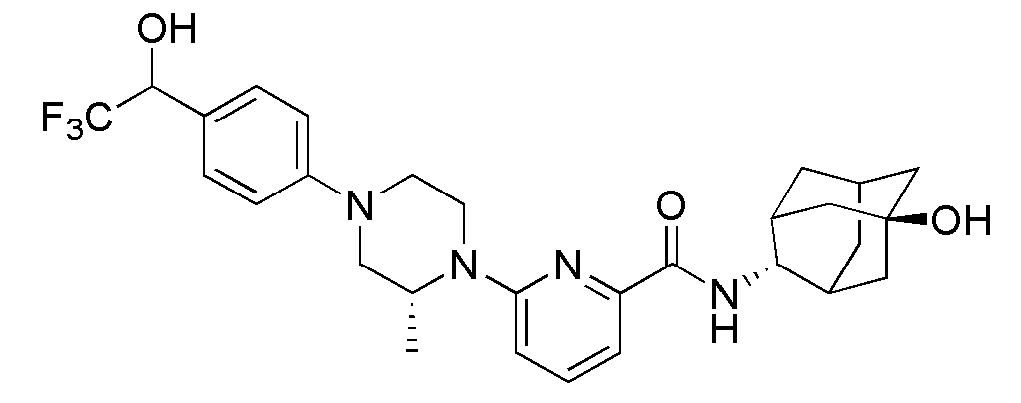
[415] Выполняли тот же способ, что в примере 184, за исключением того, что вместо 6-((R)-4-(4-ацетилфенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида использовали N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(4-(2,2,2-трифторацетил)фенил)пиперазин-1-ил)пиколинамид, получая 27 мг бледно желтого твердого вещества (20%). MS (ESI): 545[M+H]+.
[416]
[417] Пример 186: Синтез 6-((R)-4-(4-(1-амино-2-метил-1-оксопропан-2-ил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[418]
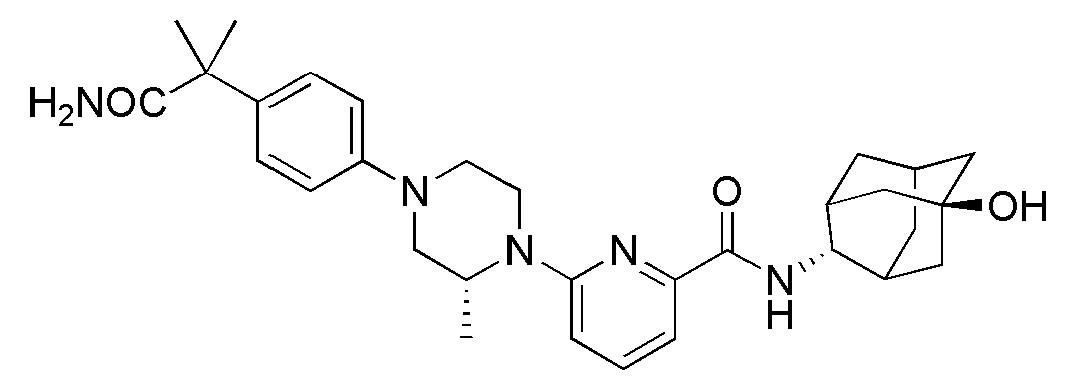
[419] 6-((R)-4-(4-(2-Цианопропан-2-ил)фенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (100 мг, 0,195 ммоль) растворяли в 2-метилпропан-2-оле (5 мл), с последующим добавлением КОН (257 мг, 3,894 ммоль), а затем получающуюся жидкость нагревали при температуре кипения с обратным холодильником в течение 4 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (20 мл) с последующей экстракцией 5% МеОН/МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 93 мг бледно желтого твердого вещества (90%). MS (ESI): 532[M+Н]+.
[420]
[421] Пример 187: Синтез 6-((R)-4-(3-хлор-4-гидроксифенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[422]
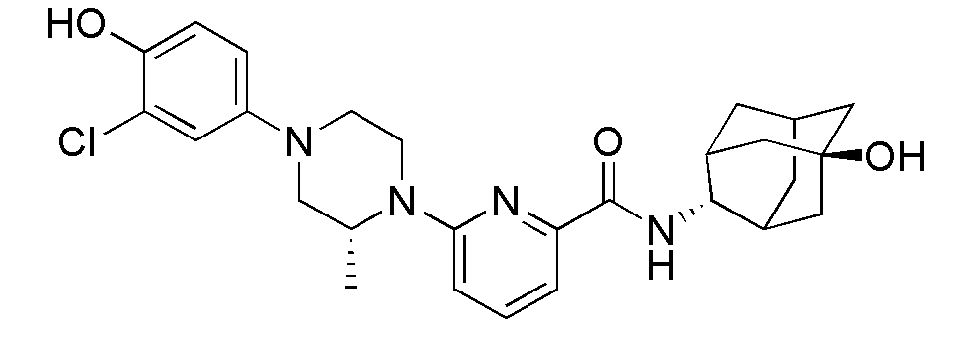
[423] Стадия 1: Синтез 4-бром-2-хлорфенокси)(трет-бутил)диметилсилана
[424] 4-Бром-2-хлорфенол (1,0 г, 4,82 ммоль) растворяли в МС (30 мл) с последующим добавлением трет-бутилдиметилсилилхлорида (1,09 г, 7,23 ммоль) и имидазола (492 мг, 7,23 ммоль) и затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 24 часов. Получающуюся в результате реакционную жидкость последовательно промывали дистиллированной водой (15 мл) и насыщенным водным раствором NaHCO3 (15 мл), а затем сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием. Полученный таким образом остаток подвергали MPLC (5% EtOAc/гексаны), получая 1,69 г бесцветного масла (99%).
[425] Стадия 2: Синтез 6-((R)-4-(4-трет-бутилдиметилсилил)окси)-3-хлорфенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[426] N-((E)-5-Гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (100 мг, 0,27 ммоль) и (4-бром-2-хлорфенокси)(трет-бутил)диметилсилан (130 мг, 0,405 ммоль) растворяли в толуоле (2 мл) с последующим добавлением Pd2(dba)3 (5 мг, 2 ммоль), BINAP (10 мг, 6 мол%) и трет-бутоксида натрия (39 мг, 0,405 ммоль) и затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 15 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (5 мл) с последующей экстракцией МС (20 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 42 мг желтого масла (25%).
[427] Стадия 3: Синтез 6-((R)-4-(3-хлор-14-гидроксифенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[428] 6-((R)-4-(4-((трет-бутилдиметилсилил)окси)-3-хлорфенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)карбамоил)пиколинамид (42 мг, 0,069 ммоль) растворяли в ТГФ (1 мл) с последующим добавлением тетрабутиламмонийфторида (1М раствор в ТГФ (0,137 мл, 0,137 ммоль), и затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 15 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (5 мл) с последующей экстракцией МС (20 мл × 2). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 16 мг желтого масла (47%). MS (ESI): 498[M+Н]+.
[429]
[430] Пример 188: Синтез этил 2-(2-хлор-4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)фенокси)ацетата
[431]
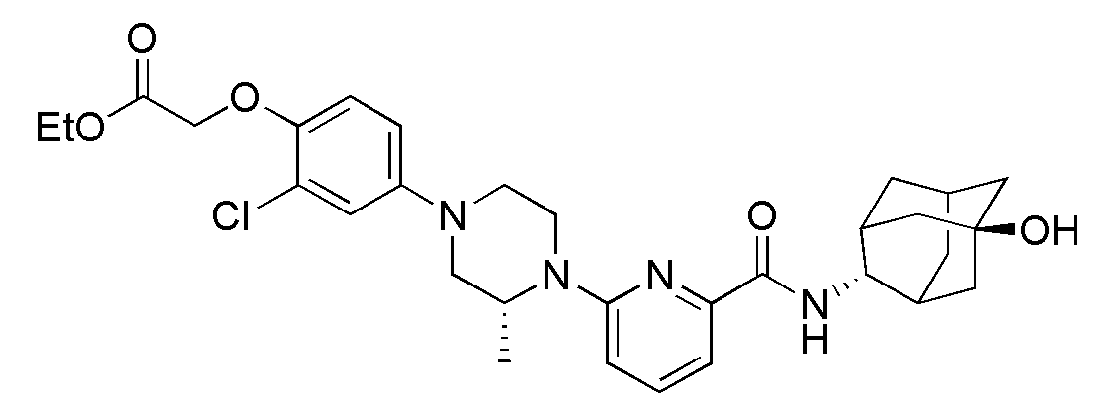
[432] 6-((R)-4-(3-Хлор-4-гидроксифенил)-2-метилпиперазин-1-мл)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (52 мг, 0,105 ммоль) и карбонат калия (20 мг, 0,126 ммоль) суспендировали в ДМФ (1 мл) с последующим добавлением этил-бромацетата (0,014 мл, 0,126 ммоль) и затем получающуюся смесь перемешивали при 100°С в токе азота в течение 2 часов. К получающейся в результате реакционной жидкости добавляли EtOAc (10 мл), а затем получающуюся жидкость последовательно промывали дистиллированной водой (5 мл × 2) и насыщенным водным раствором хлорида натрия (5 мл). Органический слой сушили над безводным сульфатом магния с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 61 мг бледно желтого масла (99%). MS (ESI): 583[M+Н]+.
[433]
[434] Пример 189: Синтез 2-(2-хлор-4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)феноксиуксусной кислоты
[435]
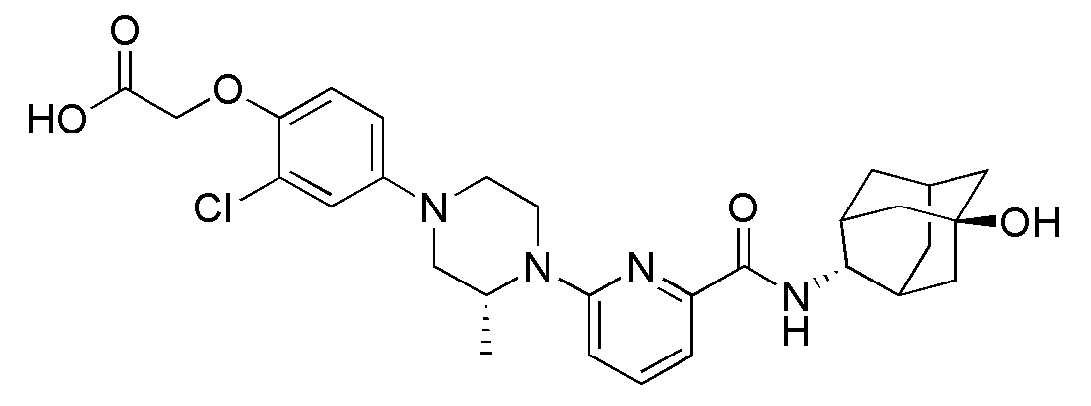
[436] Выполняли тот же способ, что в примере 84, за исключением того, что вместо этил 2-(4-(4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперазин-1-ил)фенокси)ацетата использовали метил этил 2-(2-хлор-4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)фенокси)ацетат (88 мг, 0,150 ммоль), получая 79 мг бледно желтого твердого вещества (94%). MS (ESI): 555[M+H]+.
[437]
[438] Пример 190: Синтез 6-((R)-4-(4-(2-амино-2-оксоэтокси)-3-хлорфенил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[439]
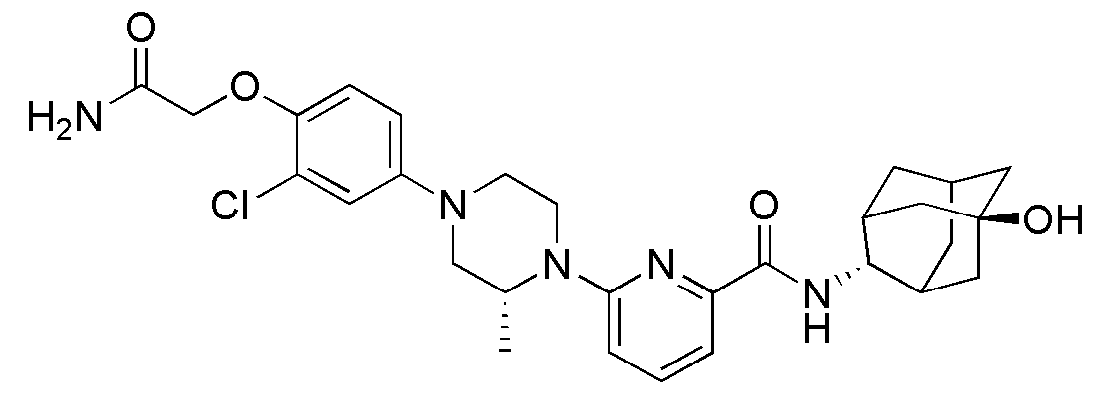
[440] Осуществляли тот же способ, что в примере 132, за исключением того, что 2-(2-хлор-4-((R)-4-(6-((((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)фенокси)уксусную кислоту (62 мг, 0,112 ммоль) использовали вместо 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)бензойной кислоты, получая 37 мг бледно желтого твердого вещества (60%). MS (ESI): 554[M+H]+.
[441]
[442] Пример 191: Синтез N-((E)-5-гидроксиадамантан-2-)-6-((R)-4-(2-гидроксиэтил)-2-метилпиперазин-1-ил)пиколинамида
[443]

[444] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (50 мг, 0,135 ммоль) и карбонат калия (37 мг, 0,270 ммоль) суспендировали в ацетонитриле (2 мл), с последующим добавлением 2-бромэтанола (0,014 мл, 0,202 ммоль), а затем получающуюся в результате смесь перемешивали при 90°С в токе азота в течение 10 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 44 мг белого твердого вещества (79%). MS (ESI): 415[M+H]+.
[445]
[446] Пример 192: Синтез 6-((R)-4-(2-гидрокси-2-метилпропил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[447]
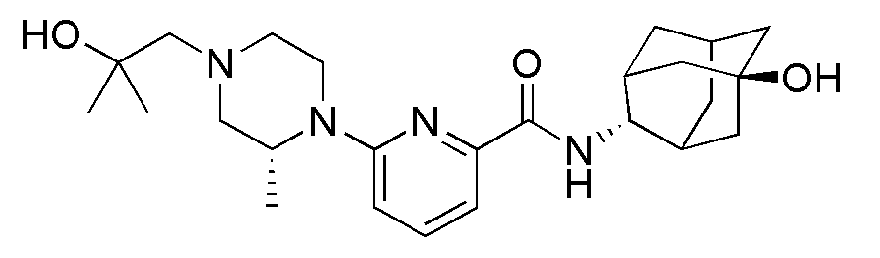
[448] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (60 мг, 0,162 ммоль), карбонат калия (45 мг, 0,324 ммоль) и иодид калия (27 мг, 0,162 ммоль) суспендировали в ацетонитриле (2 мл) с последующим добавлением 1-хлор-2-метилпропан-2-ола (0,10 мл, 0,192 ммоль), а затем получающуюся в результате смесь перемешивали при 100°С в токе азота в течение 72 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 10 мг белого твердого вещества (14%). MS (ESI): 443[M+H]+.
[449]
[450] Пример 193: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(2-(метилсульфонил)этил)пиперазин-1-ил)пиколинамида
[451]
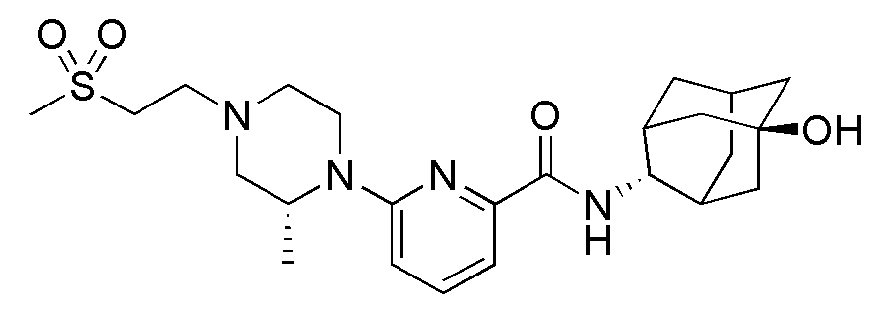
[452] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (60 мг, 0,162 ммоль) растворяли в ТГФ (1 мл), с последующим добавлением (метилсульфонил)этена (69 мг, 0,648 ммоль), а затем получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (3% МеОН/МС), получая 37 мг белого твердого вещества (48%). MS (ESI): 477[M+H]+.
[453]
[454] Пример 194: Синтез 6-((R)-4-((1-цианоциклопропил)метил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[455]
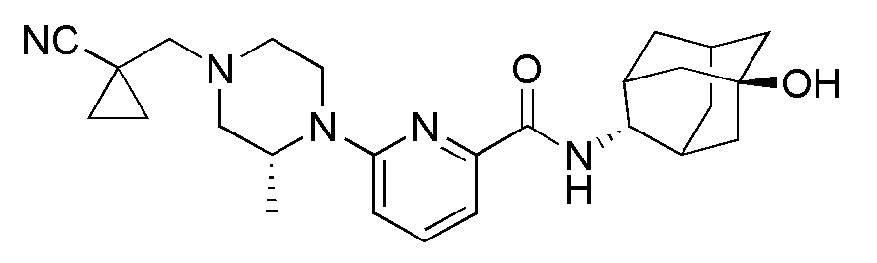
[456] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (150 мг, 0,405 ммоль) и (1-цианоциклопропил)метил-4-метилбензолсульфонат (122 мг, 0,486 ммоль) растворяли в ацетонитриле (3 мл) с последующим добавлением N,N-диизопропилэтиламина (0,14 мл, 0,810 ммоль), а затем получающуюся в результате жидкость перемешивали при 90°С в токе азота в течение 17 часов. К получающейся в результате реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл) с последующей экстракцией МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 157 мг бледно желтого твердого вещества (86%). MS (ESI): 450[M+H]+.
[457]
[458] Пример 195: Синтез 6-((R)-4-((1-карбамоилциклопропил)метил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[459]
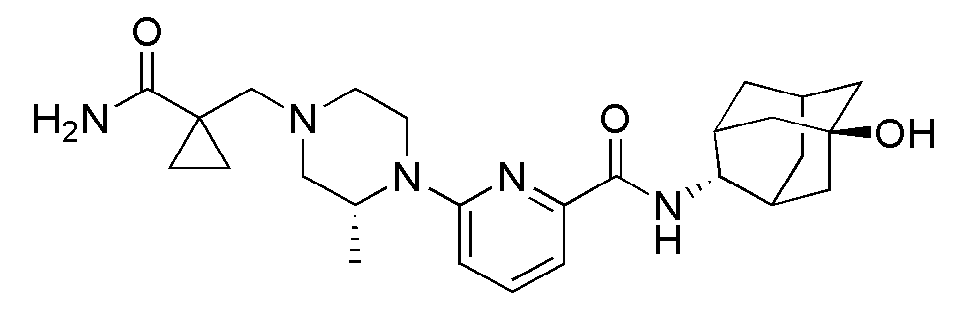
[460] 6-((R)-4-((1-цианоциклопропил)метил)-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамид (100 мг, 0,222 ммол) и KOH (294 мг, 4,448 ммоль) суспендировали в 2-метилпролпан-2-оле (5 мл), а затем получающуюся в результате реакционную жидкость перемешивали при 95°с в течение 4 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (15 мл) с последующей экстракцией 5% МеОН/МС (20 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (6% МеОН/МС), получая 81 мг белого твердого вещества (78%). MS (ESI): 468[M+H]+.
[461]
[462] Пример 196: Синтез 6-((R)-4-циклопропил-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиколинамида
[463]

[464] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (100 мг, 0,270 ммоль) растворяли в MeOH (2 мл), с последующим последовательным добавлением 1-этокси-1-триметилсилилоксициклопропана (0,324 мл, 1,62 ммоль), уксусной кислоты (0,155 мл, 2,70 ммоль) и цианоборгидрида натрия (76 мг, 1,21 ммоль), а затем получающуюся в результате жидкость перемешивали при 80°С в токе азота в течение 8 часов. К получающейся в результате реакционной жидкости добавляли EtOAc (30 мл), а затем получающуюся в результате жидкость последовательно промывали 1н водным раствором NaOH, насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (3% MeOH/MC), получая 87 мг белого твердого вещества (79%). MS (ESI): 411[M+H]+.
[465]
[466] Пример 197: Синтез N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(тетрагижро-2H-пиран-4-ил)пиперазин-1-ил)пиколинамида
[467]
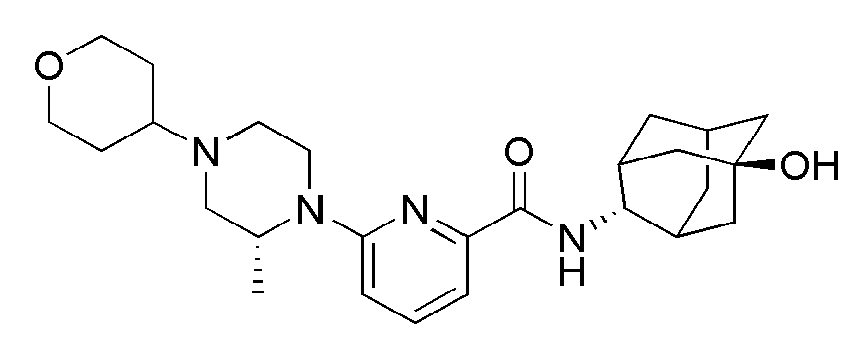
[468] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (100 мг, 0,270 ммоль) растворяли в 2:1 DCE/THF (1,5 мл), с последующим последовательным добавлением 2,3,5,6-тетрагидропиран-4-она (0,087 мл, 0,945 ммоль), уксусной кислоты (0,031 мл, 0,540 ммоль) и триацетоксиборгидрида натрия (286 мг, 1,35 ммоль), а затем получающуюся в результате жидкость перемешивали при 75°С в токе азота в течение 18 часов. К получающейся в результате реакционной жидкости добавляли EtOAc (30 мл), а затем получающуюся в результате жидкость последовательно промывали 1н водным раствором NaOH, насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5% MeOH/MC), получая 76 мг бледно желтого твердого вещества (62%). MS (ESI): 455[M+H]+.
[469]
[470] Пример 198: Синтез 6-((R)-4-(4-гидрокси-4-метилциклогексил)-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[471]
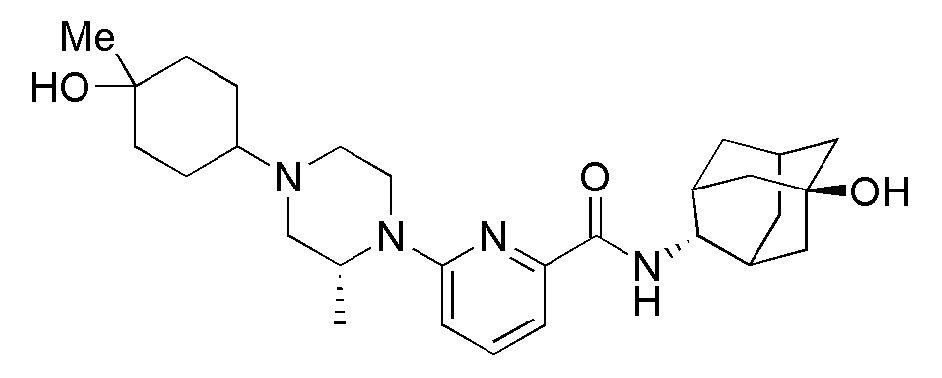
[472] Выполняли тот же способ, что и в примере 197, за исключением того, что вместо 2,3,5,6-тетрагидропиран-4-она использовали 4-гидрокси-4-метилциклогексанон, получая 117 мг белого твердого вещества (90%). MS (ESI): 483[M+H]+.
[473]
[474] Пример 199: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(пиперидин-4-ил)пиперазин-1-ил)пиколинамида
[475]
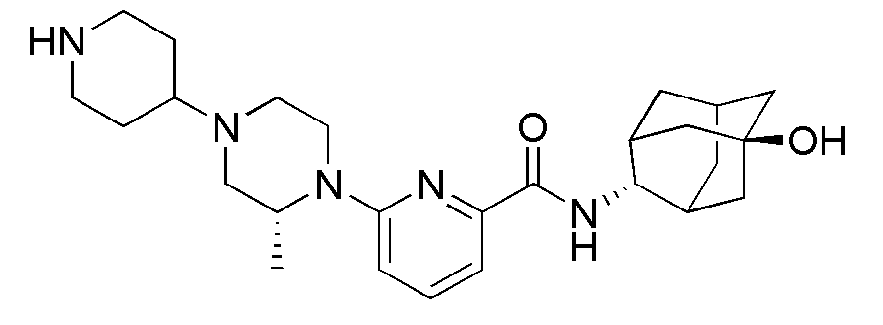
[476] Стадия 1: Синтез трет-бутил-4-N-((R)-4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)пиперидин-1-карбоксилата
[477] Выполняли тот же способ, что и в примере 197, за исключением того, что вместо 2,3,5,6-тетрагидропиран-4-она использовали 4-трет-бутоксикарбонилциклогексанон, получая 120 мг бесцветного масла (54%).
[478] Стадия 2: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(пиперидин-4-ил)пиперазин-1-ил)пиколинамида
[479] Трет-бутил 4-((R)-4-(6-(((E)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-мл)-3-метилпиперазин-1-ил)пиперидин-1-карбоксилат (120 мг, 0,217 ммоль) растворяли в МС (3 мл), с последующим последовательным добавлением трифторуксусной кислоты (3 мл), а затем получающуюся в результате жидкость перемешивали при комнатной температуре в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (10 мл), а затем экстрагировали МС (5 мл). Водный слой нейтрализовали 1н водным раствором гидроксида натрия с последующим экстрагированием 10% МеОН/МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% MeOH/MC), получая 75 мг белого твердого вещества (76%). MS (ESI): 454[M+H]+.
[480]
[481] Пример 200: Синтез 6-((R)-4-(1-ацетилпиперидин-4-ил)-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[482]
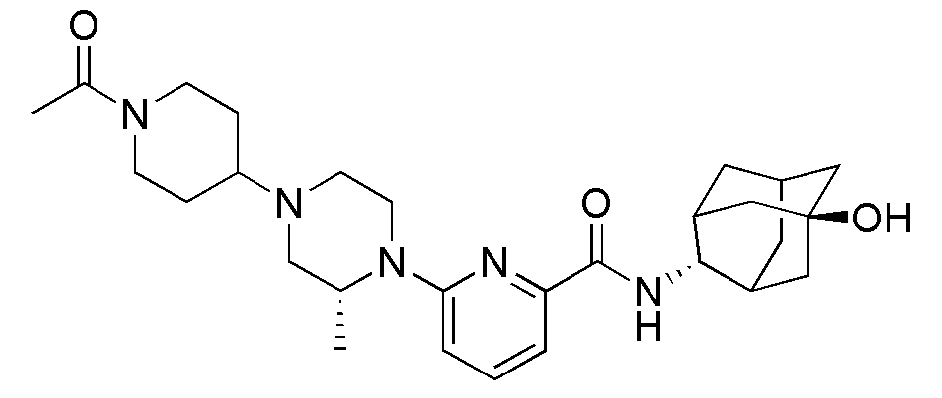
[483] N-((E)-5-Гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(пиперидин-4-ил)пиперазин-1-ил)пиколинамид (28 мг, 0,0617 ммоль) растворяли в МС (3 мл), с последующим последовательным добавлением триэтиламина (0,013 мл, 0,0926 ммоль) и уксусного ангидрида (0,006 мл, 0,0617 ммоль), а затем получающуюся в результате жидкость перемешивали при комнатной температуре в токе азота в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (10% MeOH/MC), получая 15 мг белого твердого вещества (49%). MS (ESI): 496[M+H]+.
[484]
[485] Пример 201: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(1-метилсульфонил)пиперидин-4-ил)пиперазин-1-ил)пиколинамида
[486]
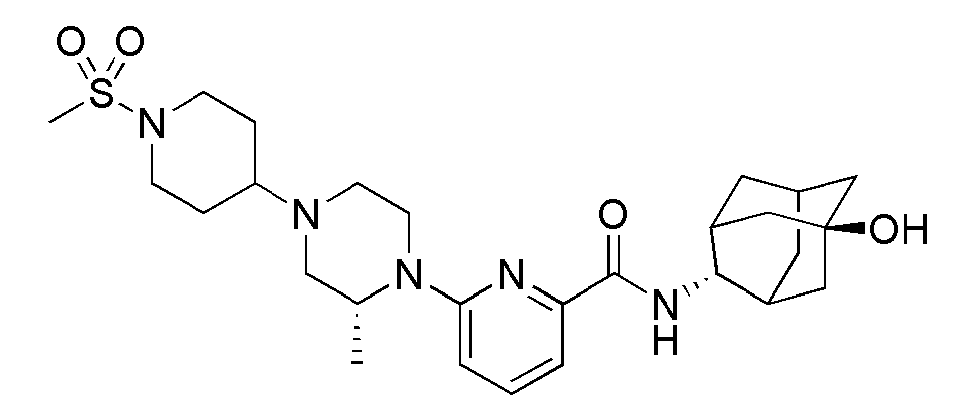
[487] N-((E)-5-Гидроксиадамантан-2-ил)-6-((R)-2-метил-4-(пиперидин-4-ил)пиперазин-1-ил)пиколинамид (29 мг, 0,0639 ммоль) растворяли в МС (3 мл), с последующим последовательным добавлением триэтиламина (0,009 мл, 0,0671 ммоль) и метансульфонилхлорида (0,005 мл, 0,0671 ммоль), а затем получающуюся в результате жидкость перемешивали при комнатной температуре в токе азота в течение 2 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% MeOH/MC), получая 14 мг белого твердого вещества (41%). MS (ESI): 532[M+H]+.
[488]
[489] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 200 или 201 выше, с использованием промежуточного соединения 10 и соответствующего хлорида кислоты или сульфонилхлорида.
[490] Таблица 15

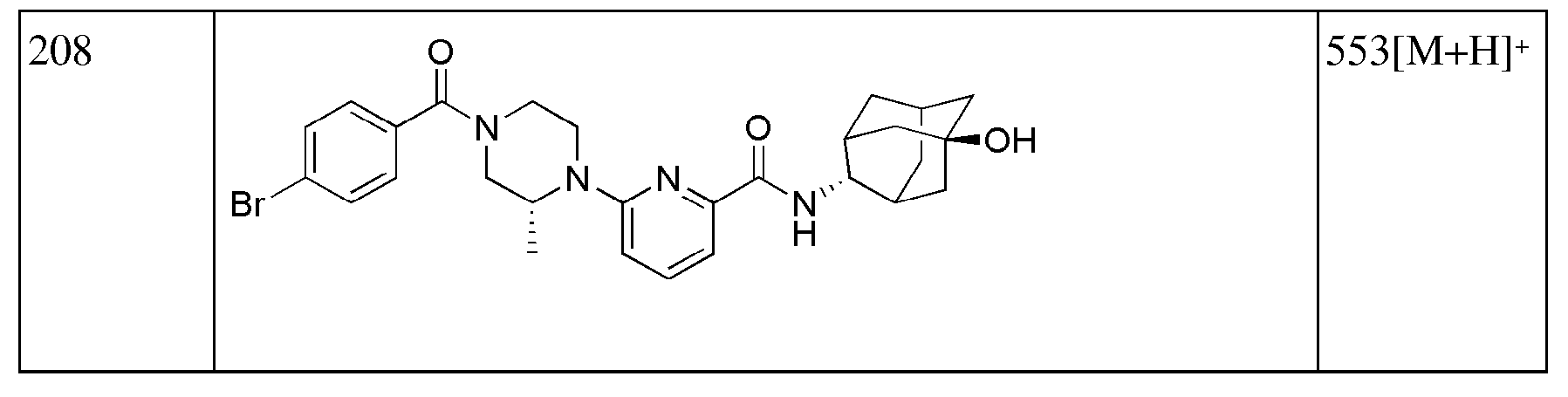
[491]
[492] Пример 209: Синтез 6-((R)-4-(4-гидроксиацетил)-2-метилпиперазин-1-ил)-N-ил((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[493]
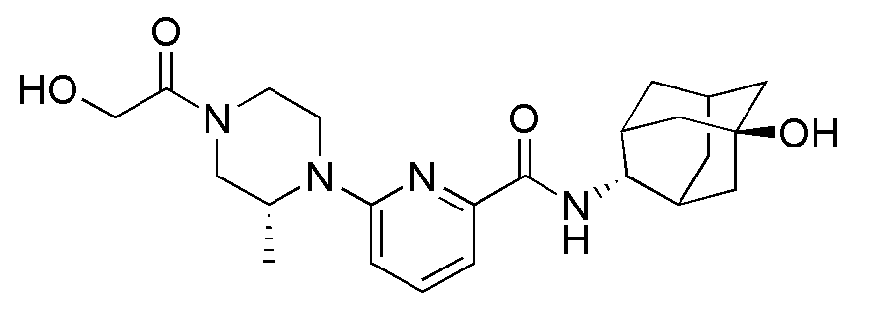
[494] N-((E)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамид (50 мг, 0,135 ммоль), 2-гидроксиуксусную кислоту (12 мг, 0,162 ммоль) и HBTU (61 мг, 0,162 ммоль) суспендировали в ацетонитриле (5 мл), с последующим добавлением N,N-диизопропилэтиламина (0,028 мл, 0,162 ммоль), а затем получающуюся в результате жидкость перемешивали при комнатной температуре в токе азота в течение 4 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 50 мг белого твердого вещества (89%). MS (ESI): 429[M+H]+.
[495]
[496] Соединения следующих примеров синтезировали тем же самым способом, что и в примере 209 выше, с использованием промежуточного соединения 10 и соответствующего кислотного исходного материала.
[497] Таблица 16
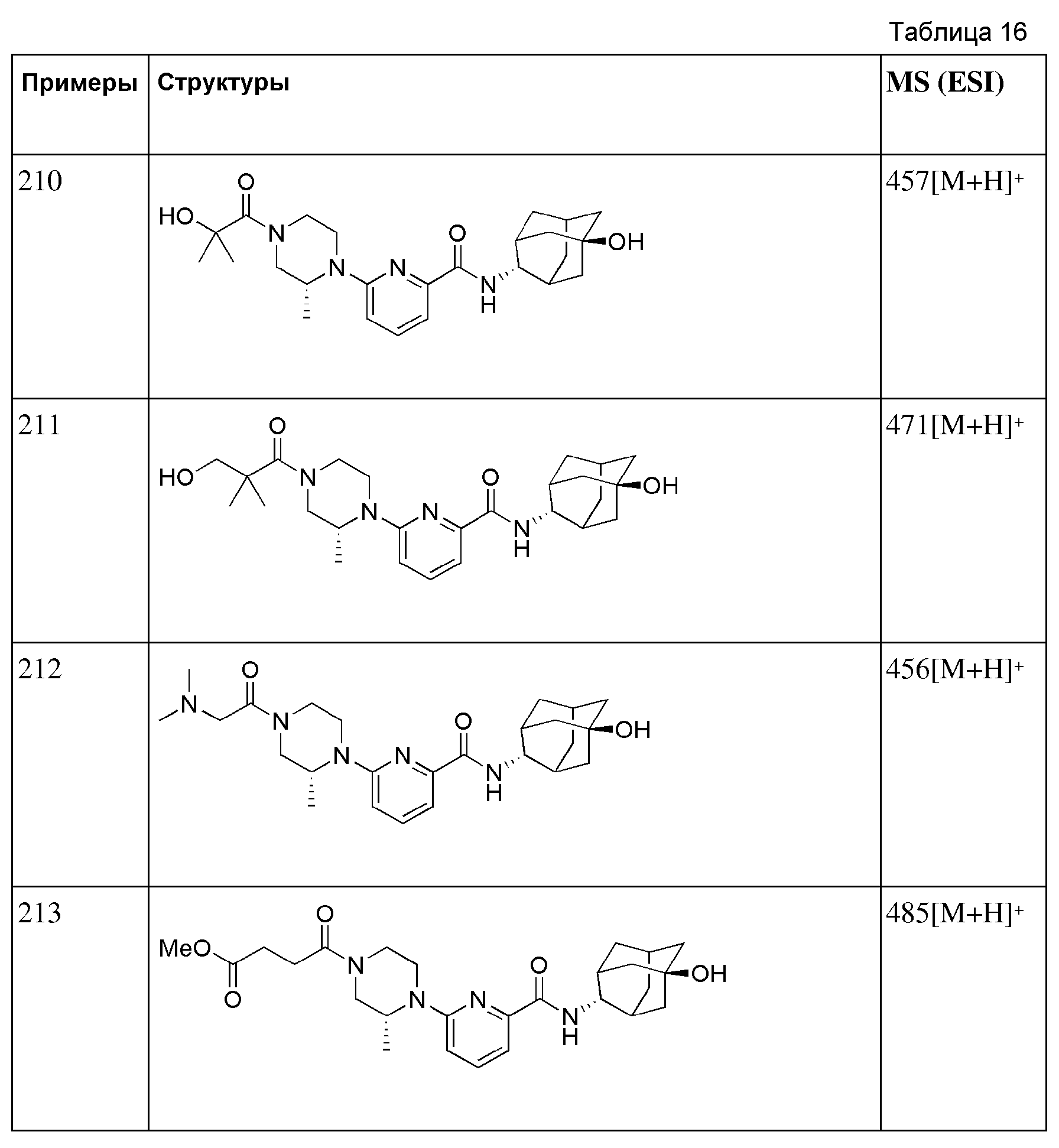
[498]
[499] Пример 214: Синтез 6-((R)-4-(4-амино-4-оксобутаноил)-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[500]
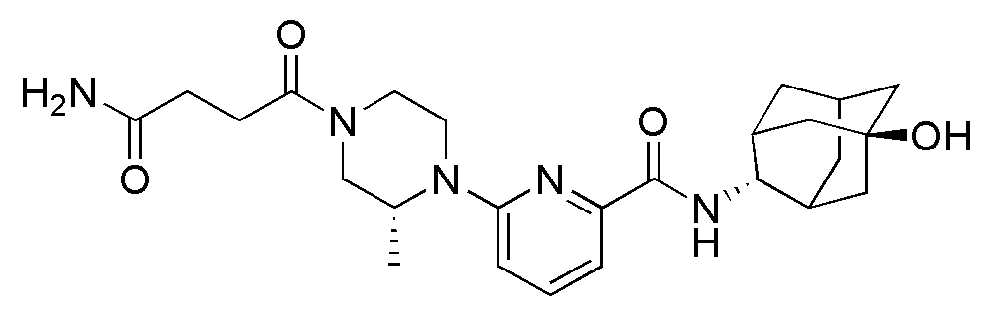
[501] Выполняли тот же способ, что в примере 70, за исключением того, что вместо метил 2-(1-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-4-ил)ацетата использовали метил 4-((R)-4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)-4-оксобутаноат (40 мг, 0,083 ммоль), получая 11 мг белового твердого вещества (28%). MS (ESI): 470[M+H]+.
[502]
[503] Пример 215: Синтез 4-((R)-4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)оксобутановой кислоты
[504]
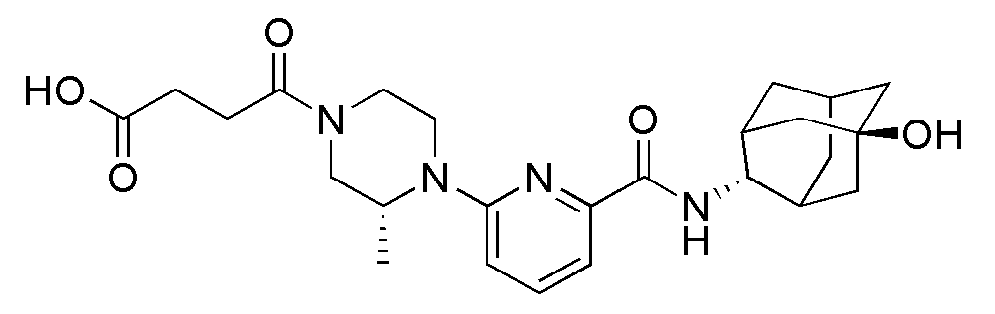
[505] Выполняли тот же способ, что в примере 71, за исключением того, что вместо метил 2-(1-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)пиперидин-4-ил)ацетата использовали метил 4-((R)-4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)пиридин-2-ил)-3-метилпиперазин-1-ил)-4-оксобутаноат (40 мг, 0,083 ммоль), получая 29 мг белого твердого вещества (75%). MS (ESI): 471[M+H]+.
[506]
[507] Пример 216: Синтез 5-фтор-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамида
[508]
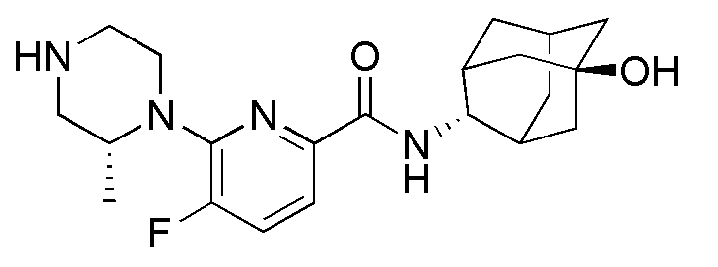
[509] Стадия 1: Синтез 1-оксида 5-фтор-2-(метоксикарбонил)пиридина
[510] Местил 5-фторпиколинат (40 мг, 2,836 ммоль) растворяли в CHCl3 (10 мл) с последующим добавлением mCPBA (954 мг, 4,254 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в течение 15 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор Na2S2O3 (15 мл) с последующим экстрагированием 5% МеОН/МС (30 мл × 2). Водный слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (2% МеОН/МС), получая 318 мг бесцветного масла (66%).
[511] Стадия 2: Синтез метил 6-хлор-5-фторпиколината
[512] 1-Оксид 5-фтор-2-(метоксикарбонил)пиридина (100 мг, 0,584 ммоль) растворяли в POCl3 (2 мл), а затем нагревали при температуре кипения с обратным холодильником в токе азота в течение 4 часов. Получающуюся реакционную жидкость медленно добавляли ко льду (15 г), а затем экстрагировали МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (20% EtOAc/гексаны), получая 88 мг белого твердого вещества (79%).
[513] Стадия 3: Синтез 6-хлор-5-фторпиколиновой кислоты
[514] метил 6-хлор-5-фторпиколинат (1,35 г, 7,095 ммоль) растворяли в ТГФ:H2O = 6:1 (42 мл) с последующим добавлением моногидрата гидроокиси лития (596 мг, 14,19 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в течение 3 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении, растворяли добавлением дистиллированной воды (20 мл), подкисляли медленным добавлением 1н водного раствора HCl, а затем экстрагировали 5% МеОН/МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 1,04 г белого твердого вещества (80%).
[515] Стадия 4: Синтез 6-хлор-5-фтор-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида
[516] Выполняли тот же способ, что в стадии 1 примера 36, за исключением того, что вместо 6-бромпиколиновой кислоты использовали 6-хлор-5-фторпиколиновую кислоту (1,04 г, 5,92 ммоль), получая 1,20 г белового твердого вещества (62%).
[517] Стадия 5: Синтез 5-фтор-N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиразин-1-ил)пиколинамида
[518] Выполняли тот же способ, что в примере 129, за исключением того, что вместо 6-бром-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 6-хлор-5-фтор-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамид (715 мг, 2,20 ммоль), получая 337 мг бледно желтого твердого вещества (39%). MS (ESI): 389[M+H]+.
[519]
[520] Соединения следующих примеров синтезировали тем же способом, что в примере 173 выше, с использованием 5-фтор-N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамида и соответствующего бромбензола или бромпиридина.
[521] Таблица 17
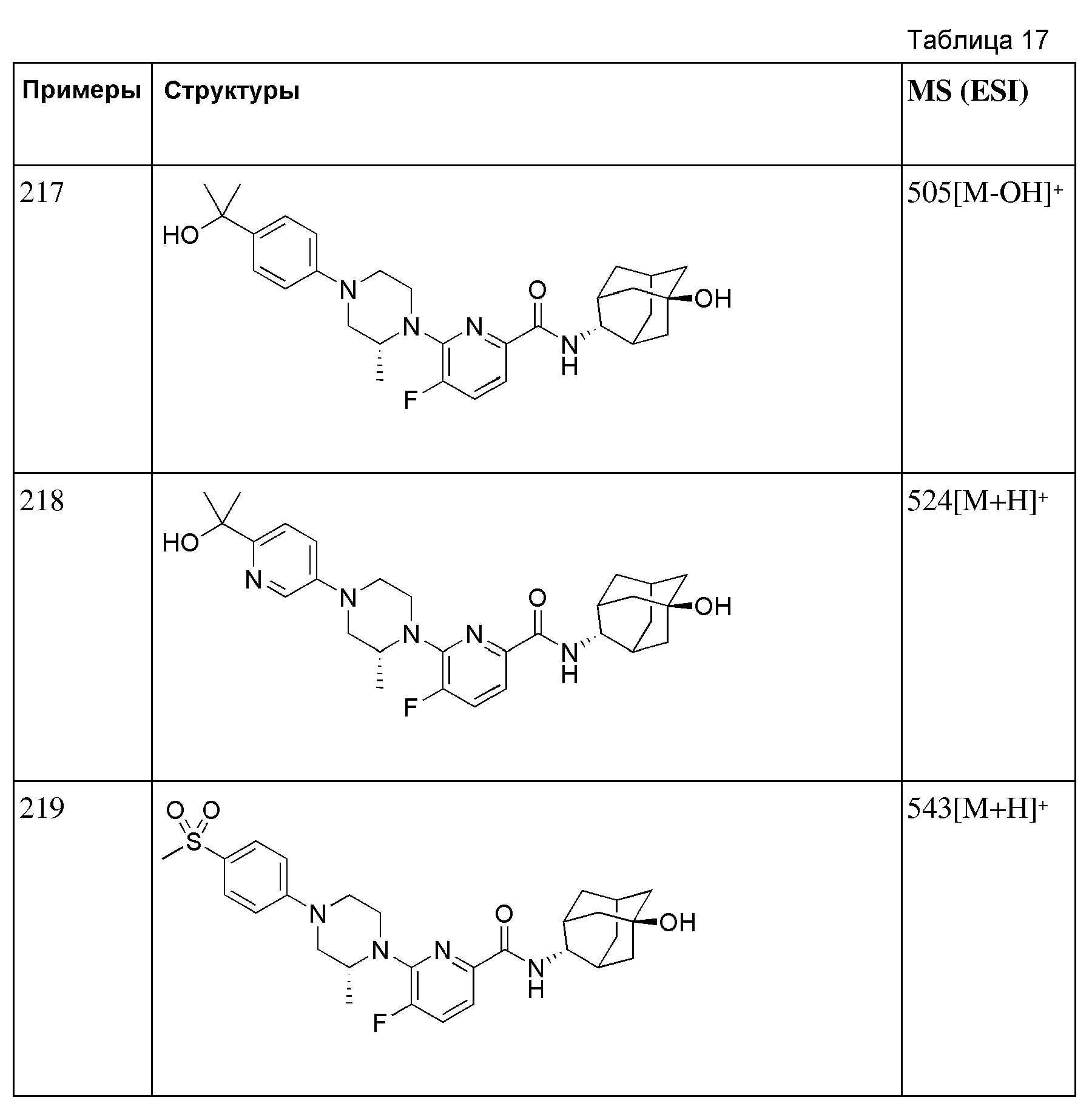
[522]
[523] Пример 220: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-5-метил-6-(пиперидин-1-ил)пиколинамида
[524]
[525] Стадия 1: Синтез 1-оксида 2-(метоксикарбонил)-5-метилпиридина
[526] Выполняли тот же способ, что на стадии 1 примера 216, за исключением того, что вместо метил 5-фторпиколината использовали метил-5-мтилпиколинат (761 мг, 5,03 ммоль), получая 484 мг белового твердого вещества (58%).
[527] Стадия 2: Синтез метил 6-хлор-5-метилпиколината
[528] Выполняли тот же способ, что на стадии 2 примера 216, за исключением того, что вместо 1-оксида 5-фтор-2-(метоксикарбонил)пиридина использовали 1-оксид 2-(метоксикарбонил)-5-метилпиридина (648 мг, 3,88 ммоль), получая 435 мг бледно желтого твердого вещества (60%).
[529] Стадия 3: Синтез 6-хлор-5-метилпиколиновой кислоты
[530] Выполняли тот же способ, что на стадии 3 примера 216, за исключением того, что вместо метил 6-хлор-5-фторпиколинатаата использовали метил 6-хлор-5-фторполинат (385 мг, 2,07 ммоль), получая 277 мг белового твердого вещества (78%).
[531] Стадия 4: Синтез 6-хлор-N-((Е)-5-гидроксиадамантан-2-ил)-5-мтилпиколинамида
[532] Выполняли тот же способ, что на стадии 1 примера 36, за исключением того, что вместо 6-бромпиколиновой кислоты использовали 6-хлор-5-метилпиколиновую кислоту (277 мг, 1,61 ммоль), получая 385 мг желтого твердого вещества (75%).
[533] Стадия 5: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-5-метил-6-(пиперидин-1-ил)пиколинамида
[534] 6-Хлор-N-((Е)-5-гидроксиадамантан-2-ил)-5-метилпиколинамид (79 мг, 0,25 ммоль), пиперидин (0,049 мл, 0,5 ммоль), Pd2(dba)3 (23 мг, 0,025 ммоль), триизобутилфосфатран (0,009 мл, 0,025 ммоль) и трет-бутоксид натрия (36 мг, 0,375 ммоль) суспендировали в толуоле (1,25 мл), а затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 24 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор NH4Cl (10 мл) с последующим экстрагированием МС (30 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% EtOAc/гексаны), получая 22 мг (24%). MS (ESI): 370[M+H]+.
[535]
[536] Пример 221: Синтез: N-((Е)-5-гидроксиадамантан-2-ил)-5-метил-6-(пиперидин-1-ил)пиколинамида
[537]
[538] Стадия 1: Синтез трет-бутил 4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил-3-метилпиридин-2-ил)пиперазин-1-карбоксилата
[539] 6-Хлор-N-((Е)-5-гидроксиадамантан-2-ил)-5-метилпиколинамид (385 мг, 1,20 ммоль), 1-ВОС-пиперазин (358 мг, 1,92 ммоль), Pd2(dba)3 (22 мг, 0,024 ммоль), триизобутилфосфатран (0,026 мл, 0,072 ммоль) и трет-бутоксид натрия (173 мг, 1,80 ммол) суспендировали в толуоле (6 мл), а затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 24 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор NH4Cl (15 мл) с последующим экстрагированием МС (50 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 410 мг бледно желтого твердого вещества (72%).
[540] Стадия 2: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-5-метил-6-пиперазин-1-мл)пиколинамида
[541] Трет-бутил 4-(6-(((Е)-5-гидроксиадамантан-2-ил)карбамоил)-3-метилпиридин-2-ил)пиперазин-1-карбоксилат (410 мг, 0,87 ммоль) растворяли в МС (1,5 мл) с последующим добавлением трифторуксусной кислоты (1,5 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 2 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (20 мл), а затем экстрагировали МС (10 мл). Жидкий слой нейтрализовали добавлением 15% водного раствора NaOH с последующим экстрагированием МС (50 мл × 2), а затем органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 277 мг (86%). MS (ESI): 371[M+H]+.
[542]
[543] Соединения следующих примеров синтезировали тем же способом, что в примере 173 выше, с использованием N-((Е)-5-гидроксиадамантан-2-ил)-5-метил-6-(пиперазин-1-ил)пиколинамида и соответствующего бромбензола или бромпиридина.
[544] Таблица 18
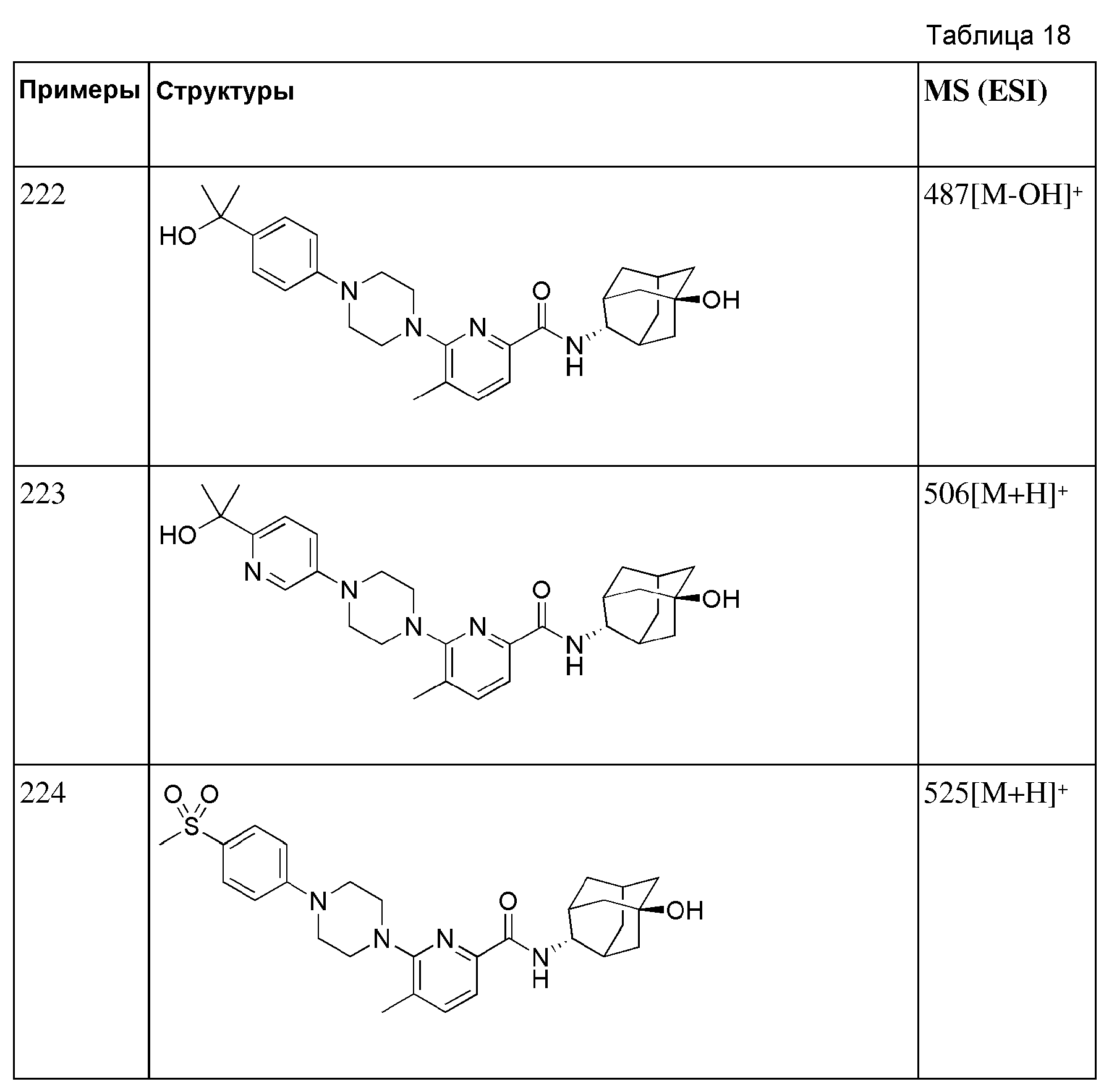
[545]
[546] Пример 225: Синтез N-((Е)-4-гидроксицтклогексил)-6-(пиперидин-1-ил)пиколинамида
[547]
[548] Стадия 1: Синтез 6-бром-N-((Е)-4-гидроксициклогексил)пиколинамида
[549] 6-Бромпиколиновую кислоту (600 мг, 2,97 ммоль) суспендировали в ацетонитриле (20 мл), с последующим последовательным добавлением гидрохлорида транс-4-аминоциклогексанола (500 мг, 2,48 ммоль), N,N-диизопропилэтиламина (1,0 мл, 6,19 ммоль) и HBTU (1,1 г, 2,97 ммоль), а затем получающуюся в результате смесь перемешивали при комнатной температуре в токе азота в течение 13 часов. К получающейся в результате реакционной жидкости добавляли дистиллированную воду (20 мл) с последующей экстракцией МС (40 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 538 мг белого твердого вещества (73%).
[550] Стадия 2: Синтез N-((Е)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамида
[551] 6-Бром-N-((Е)-4-гидроксициклогексил)пиколинамид (50 мг, 0,167 ммоль) растворяли в ацетонитриле (1 мл) с последующим добавлением пиперидина (0,13 мл, 1,336 ммоль), а затем получающуюся жидкость подвергали микроволновому облучению при 150°С в течение 2 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (5% МеОН/МС), получая 45 мг бледно желтого твердого вещества (89%). MS (ESI): 304[M+H]+.
[552]
[553] Пример 226: Синтез N-циклопропил-N-((Z)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамида и N-циклопропил-N-((E)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамида
[554]
[555] Стадия 1: Синтез 4-гидроксициклогексанона
[556] 1,4-циклогександион моно-этиленкеталь (1,0 г, 6,4 ммоль) растворяли в МеОН (30 мл) с последующим добавлением боргидрида натрия (750 мг, 19,2 ммоль) при 0°С, а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 2 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении с последующим добавлением насыщенного водного раствора хлорида натрия (30 мл) и экстрагировали этилацетатом (50 мл × 2). Органический слой сушили над безводным сульфатом натрия, а затем фильтровали, концентрировали и сушили в вакууме. Полученный таким образом остаток растворяли в ТГФ (30 мл) с последующим добавлением 1н водного раствора HCl (15 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 18 часов. Получающуюся реакционную жидкость нейтрализовали добавлением 10% водного раствора гидроксида натрия с последующим экстрагированием МС (30 мл) × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (60% EtOAc/гексаны), получая 450 мг бесцветного масла (62%).
[557] Стадия 2: Синтез 4-(циклопропиламино)циклогексанола
[558] 4-Гидроксициклогексанон (443 мг, 3,88 ммоль) растворяли в 1,2-дихлорэтане (20 мл) с последующим последовательным добавлением циклопропиламина (0,295 мл, 4,27 ммоль), NaBH(OAc)3 (1,3 г, 6,21 ммоль) и уксусной кислоты (0,2 мл, 3,88 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 13 часов. Получающуюся реакционную жидкость нейтрализовали добавлением 10% водного раствора гидроксида натрия и экстрагировали 1-% МеОН/МС (15 мл × 4). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 580 мг желтого твердого вещества (96%).
[559] Стадия 3: Синтез N-циклопропил-N-((Z)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамида и N-циклопропил-N-((E)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамида
[560] Выполняли тот же способ, что в примере 225, за исключением того, что вместо гидрохлорида транс-4-амииноциклогексанола использовали 4-(циклопропиламино)циклогексанол, получая N-циклопропил-N-((Z)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамид и N-циклопропил-N-((E)-4-гидроксициклогексил)-6-(пиперидин-1-ил)пиколинамид, соответственно. MS (ESI): 344[M+H]+, 344[M+H]+.
[561]
[562] Пример 227: Синтез N-циклопропил-N-((1s,4s)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамида и N-циклопропил-N-((1r,4r)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамида
[563]

[564] Стадия 1: Синтез 8-метил-1,4-диоксаспиро[4,5]декан-8-ола
[565] 1,4-Циклогександион-моно-этилен-кеталь (1,0 г, 6,4 ммоль) растворяли в ТГФ (30 мл) с последующим добавлением MeMgCl (3,0М раствор в ТГФ, 2,6 мл, 7,7 ммоль) при 0°С, а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 3 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием МС (50 мл × 4). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (5-% этилацетат/гексаны), получая 654 мг белого твердого вещества (59%).
[566] Стадия 2: Синтез 4-гидрокси-4-метилциклогексанона
[567] 8-Метил-1,4-диоксаспиро[4,5]декан-8-ол (650 мг, 3,77 ммоль) растворяли в ТГФ (10 мл), с последующим добавлением 1н водного раствора HCl (5 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 6 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем экстрагировали 10% МеОН/МС (20 мл × 5). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 443 мг желтого масла (92%).
[568] Стадия 3: Синтез 4-(циклопропиламино)-1-метилциклогексанола
[569] 4-Гидрокси-4-метилциклогексанон (440 мг, 3,43 ммоль) растворяли в 1,2-дихлорэтане (15 мл) с последующим последовательным добавлением циклопропиламина (0,26 мл, 3,78 ммоль), NaBH(OAc)3 (1,16 г, 5,40 ммоль) и уксусной кислоты (0,20 мл, 3,43 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 20 часов. Получающуюся в результате реакционную жидкость нейтрализовали добавлением 10% водного раствора NaOH и экстрагировали 5% МеОН/МС (15 мл × 4). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 417 мг желтого твердого вещества (72%).
[570] Стадия 4: Синтез N-циклопропил-N-((1s,4s)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамида и N-циклопропил-N-((1r,4r)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамида
[571] Выполняли тот же способ, что в примере 225, за исключением того, что вместо гидрохлорида транс-4-аминоциклогексанола использовали 4-(циклопропиламино)-1-метилциклогексанол, получая N-циклопропил-N-((1s,4s)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамид и N-циклопропил-N-((1r,4r)-4-гидрокси-4-метилциклогексил)-6-(пиперидин-1-ил)пиколинамид, соответственно. MS (ESI): 358[M+H]+, 358[M+H]+.
[572]
[573] Пример 228: Синтез N-циклопропил-N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида) и N-циклопропил-N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида
[574]
[575] Стадия 1: Синтез 8-(трифторметил)-1,4-диоксаспиро[4,5]декан-8-ола
[576] 1,4-Циклогександион-моно-этилен-кеталь (1,5 г, 9,6 ммоль) растворяли в ТГФ (35 мл) с последующим последовательным добавлением триметил(трифторметил)силана (2,8 мл, 19,2 ммоль) и фторида тетрабутиламмония (1,0М раствор в ТГФ, 20 мл, 20,0 ммоль) при 0°С, а затем получающуюся смесь перемешивали при комнатной температуре в течение 2 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (10 мл), а затем получающуюся смесь перемешивали в течение 10 минут с последующим концентрированием при пониженном давлении. К полученному таким образом остатку добавляли дистиллированную воду (10 мл) с последующей экстракцией МС (50 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (40% EtOAc/гексаны), получая 2,1 г желтого масла (97%).
[577] Стадия 2: Синтез 4-гидрокси-4-(трифторметил)циклогексанона
[578] 8-(Трифторметил)-1,4-диоксаспиро[4,5]декан-8-ол (2,0 г, 8,84 ммоль) растворяли в ТГФ (30 мл) с последующим добавлением 1н водного раствора HCl (15 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 24 часов. Получающуюся реакционную жидкость концентрировали при пониженном давлении, а затем экстрагировали 10% МеОН/МС (15 мл × 6). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (40% EtOAc/гексаны), получая 1,24 г белого твердого вещества (77%).
[579] Стадия 3: Синтез 4-(циклопропиламино)-1-(трифторметил)циклогексанола
[580] 4-Гидрокси-4-(трифторметил)циклогексанон (570 мг, 3,13 ммоль) растворяли в 1,2-дихлорэтане (20 мл) с последующим последовательным добавлением циклопропиламина (0,24 мл, 3,44 ммоль), NaBH(OAc)3 (1,06 г, 5,01 ммоль) и уксусной кислоты (0,18 мл, 3,13 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 13 часов. Получающуюся в результате реакционную жидкость нейтрализовали добавлением 10% водного раствора NaOH и экстрагировали 5% МеОН/МС (20 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием, концентрированием и вакуумной сушкой, получая 650 мг желтого твердого вещества (93%).
[581] Стадия 4: Синтез N-циклопропил-N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида и N-циклопропил-N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида
[582] Выполняли тот же способ, что в примере 225, за исключением того, что вместо гидрохлорида транс-4-аминоциклогексанола использовали 4-(циклопропиламино)-1-(трифторметил)циклогексанол, получая N-циклопропил-N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамид) и N-циклопропил-N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамид, соответственно. MS (ESI): 412[M+H]+, 412[M+H]+.
[583]
[584] Пример 229: Синтез N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида и N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида
[585]
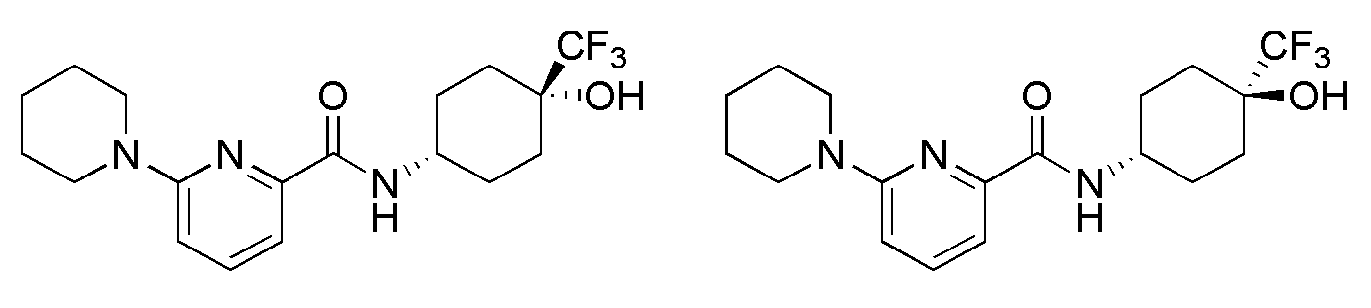
[586] Стадия 1: Синтез 4-амино-1-(трифторметил)циклогексанола
[587] 4-Гидрокси-4-(трифторметил)циклогексанон (570 мг, 3,13 ммоль) растворяли в 1,2-дихлорэтане (20 мл) с последующим последовательным добавлением бензиламина (0,38 мл, 3,44 ммоль), NaBH(OAc)3 (1,06 г, 5,01 ммоль) и уксусной кислоты (0,18 мл, 3,13 ммол), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 13 часов. Получающуюся в результате реакционную жидкость нейтрализовали добавлением 10% водного раствора NaOH и экстрагировали 5% МеОН/МС (20 мл × 3). Органический слой сушили над безводным сульфатом натрия, а затем фильтровали, концентрировали при пониженном давлении и сушили в вакууме. Полученный таким образом остаток растворяли в этаноле (20 мл) с последующим добавлением Pd (10 вес.% на активированном угле, 80 мг), а затем получающуюся смесь перемешивали при комнатной температуре в токе водорода в течение 15 часов. Получающуюся реакционную жидкость фильтровали, концентрировали при пониженном давлении и сушили в вакууме, получая 471 мг желтого твердого вещества (82%).
[588] Стадия 2: Синтез N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида и N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамида
[589] Выполняли тот же способ, что в примере 225, за исключением того, что вместо гидрохлорида транс-4-аминоциклогексанола использовали 4-амино-1-(трифторметил)циклогексанол, получая N-((1s,4s)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамид и N-((1r,4r)-4-гидрокси-4-(трифторметил)циклогексил)-6-(пиперидин-1-ил)пиколинамид, соответственно. MS (ESI): 372[M+H]+, 372[M+H]+.
[590]
[591] Пример 230: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-2-метил-4-(4-метилсульфонил)фенил)пиперазин-1-ил)пиримидин-4-карбоксамида
[592]
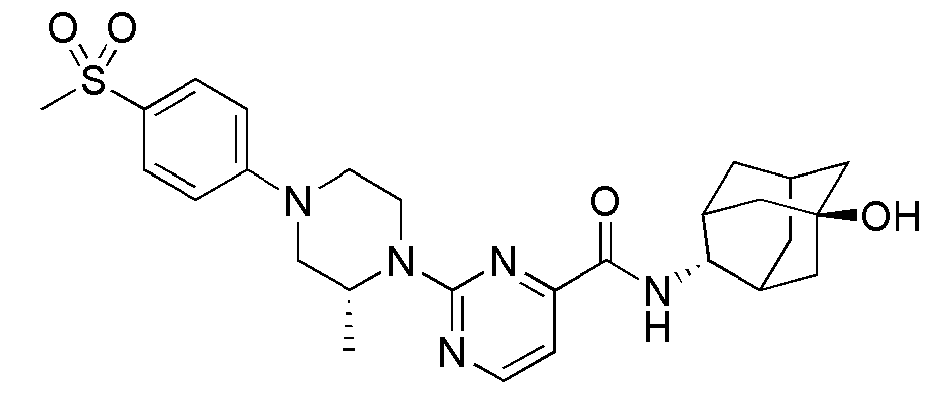
[593] Стадия 1: Синтез трет-бутил 2-хлорпиримидин-4-карбоксилата (промежуточное соединение 11)
[594] 2-Хлорпиримидин-4-карбоновую кислоту (500 мг, 3,154 ммоль) суспендировали в 2-метилпропан-2-оле (20 мл) с последующим добавлением пиридина (3 мл) и п-толуолсульфонилхлорида (1,2 г, 6,308 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 4 часов. Получающуюся в результате реакционную жидкость нейтрализовали медленным добавлением насыщенного водного раствора NaHCO3, а затем концентрировали при пониженном давлении с последующим добавлением дистиллированной воды (5 мл). Осажденное твердое вещество отфильтровывали вакуумной сушкой, получая 420 мг бледно желтого твердого вещества (62%).
[595] Стадия 2: Синтез (R)-трет-бутил 2-(2-метил-4-(4-иньтлсульфонил)фенил)пимперазин-1-ил)пиримидин-4-карбоксилата
[596] Трет-бутил 2-хлорпиримидин-4-карбоксилат (70 мг, 0,326 ммоль) и (R)-3-метил-1-(4-метилсульфонил)фенил)пиперазин (166 мг, 0,652 ммоль) растворяли в ацетонитриле (2 мл) с последующим добавлением N,N-диизопропилэтиламина (0,11 мл, 0,652 ммоль), а затем получающуюся жидкость перемешивали при 100°С в токе азота в течение 24 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении, а затем полученный таким образом остаток подвергали MPLC (50% EtOAc/гексаны), получая 120 мг бледно желтого твердого вещества (85%).
[597] Стадия 3: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-2-метил-4-(4-метилсульфонил)фенил)пиперазин-1-ил)пиримидин-4-карбоксамида
[598] (R)-трет-бутил 2-(2-метил-4-(4-метилсульфонил)фенил)пиперазин-1-ил)пиримидин-4-карбоксилат (120 мг, 0,277 ммоль) растворяли в МС (2 мл) с последующим добавлением трифторуксусной кислоты (2 мл), а затем получающуюся смесь перемешивали при комнатной температуре в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали при пониженном давлении и сушили в вакууме. Полученный таким образом остаток суспендировали в ацетонитриле (5 мл) с последующим последовательным добавлением 5-гидрокси-2-адамантанемина (2:1 E/Z смесь, 56 мг, 0,332 ммоль), N,N-диизопропилэтиламина (0,15 мл, 0,831 ммоль) и HBTU (126 мг, 0,332 ммоль), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 6 часов. К получающейся реакционной жидкости добавляли насыщенный водный раствор хлорида аммония (15 мл) с последующим экстрагированием МС (20 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (4% МеОН/МС), получая 90 мг бледно желтого твердого вещества (62%). MS (ESI): 526[M+H]+.
[599]
[600] Соединения следующих примеров синтезировали тем же способом, что в примере 230 выше, с использованием промежуточного соединения 11 и соответствующего пиперазинового исходного материала.
[601] Таблица 19

[602]
[603] Пример 234: Синтез 2-((R)-4-бензил-2-метилпиперазин-1-ил)-N-((E)-5-гидроксиадамантан-2-ил)пиримидин-4-карбоксамида
[604]
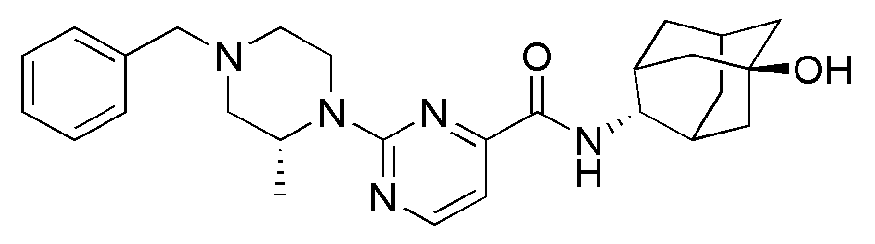
[605] Стадия 1: Синтез (R)-трет-бутил 2-(4-бензил-2-метилпиперазин-1-ил)пиримидин-4-карбоксилата
[606] Трет-бутил 2-хлорпиримидин-4-карбоксилат (4,12 г, 19,2 ммоль) и (R)-1-бензил-3-метилпиперазин (1,83 г, 9,6 ммоль) суспендировали в ацетонитриле (50 мл) с последующим добавлением N,N-диизопропилэтиламина (3,34 мл, 19,2 ммоль), а затем получающуюся жидкость нагревали при температуре кипения с обратным холодильником в токе азота в течение 15 часов. Получающуюся в результате реакционную жидкость концентрировали с последующим добавлением дистиллированной воды (50 мл), а затем экстрагировали МС (100 мл × 2). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (1% МеОН/МС), получая 3,34 г желтого твердого вещества (94%)
[607] Стадия 2: Синтез 2-((R)-4-бензил-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиримидин-4-карбоксамида
[608] Выполняли тот же способ, что на стадии 3 примера 230, за исключением того, что вместо (R)-трет-бутил-2-(2-метил-4-((4-метилсульфонил)фенил)пиперазин-1-ил)пиримидин-4-карбоксилата использовали (R)-трет-бутил-2-(4-бензил-2-метилпиперазин-1-ил)пиримидин-4-карбоксилат (3,34 г, 9,06 ммоль), получая 2,83 г белого твердого вещества (68%). MS (ESI): 462[M+H]+.
[609]
[610] Пример 235: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-4-(4-(2-гидроксипропан-2-ил)фенил)-2-метилпиперазин-1-ил)пиримидин-4-карбоксамида
[611]
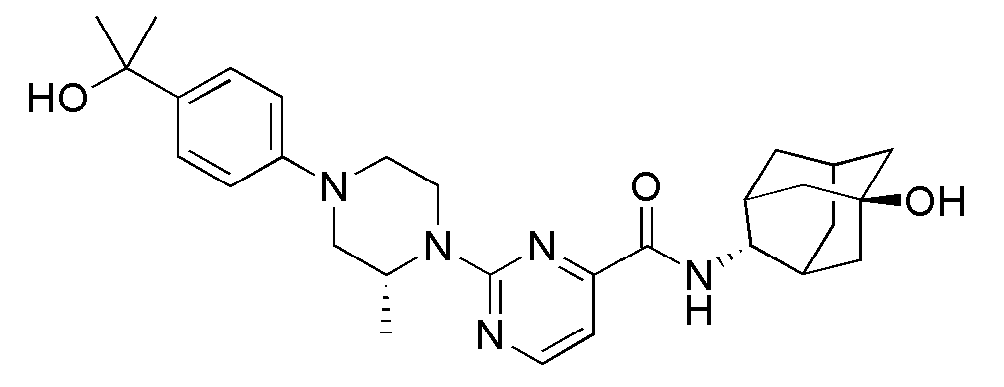
[612] Стадия 1: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-2-метилпиперазин-1-ил)пиримидин-4-карбоксамида (промежуточное соединение 12)
[613] Выполняли тот же способ, что в примере 63, за исключением того, что вместо 6-(4-бензилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиколинамида использовали 2-((R)-4-бензил-2-метилпиперазин-1-ил)-N-((Е)-5-гидроксиадамантан-2-ил)пиримидин-4-корбоксамид (2,83 г, 6,13 ммоль), получая 1,86 г белого твердого вещества (81%).
[614] Стадия 2: Синтез N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-4-(4-(2-гидроксипропан-2-ил)фенил)-2-метилпиперазин-1-ил)пиримидин-4-карбоксамида
[615] Выполняли тот же способ, что в примере 173, за исключением того, что вместо N-((Е)-5-гидроксиадамантан-2-ил)-6-((R)-2-метилпиперазин-1-ил)пиколинамида использовали N-((Е)-5-гидроксиадамантан-2-ил)-2-((R)-2-метилпиперазин-1-ил)пиримидин-4-карбоксамид (150 мг, 0,404 ммоль), получая 97 мг бледно желтого твердого вещества (47%). MS (ESI): 488[M+H]+.
[616]
[617] Соединения следующих примеров синтезировали тем же способом, что в примере 173 выше, с использованием промежуточного соединения 12 и соответствующего бромбензола или бромпиридина.
[618] Таблица 20
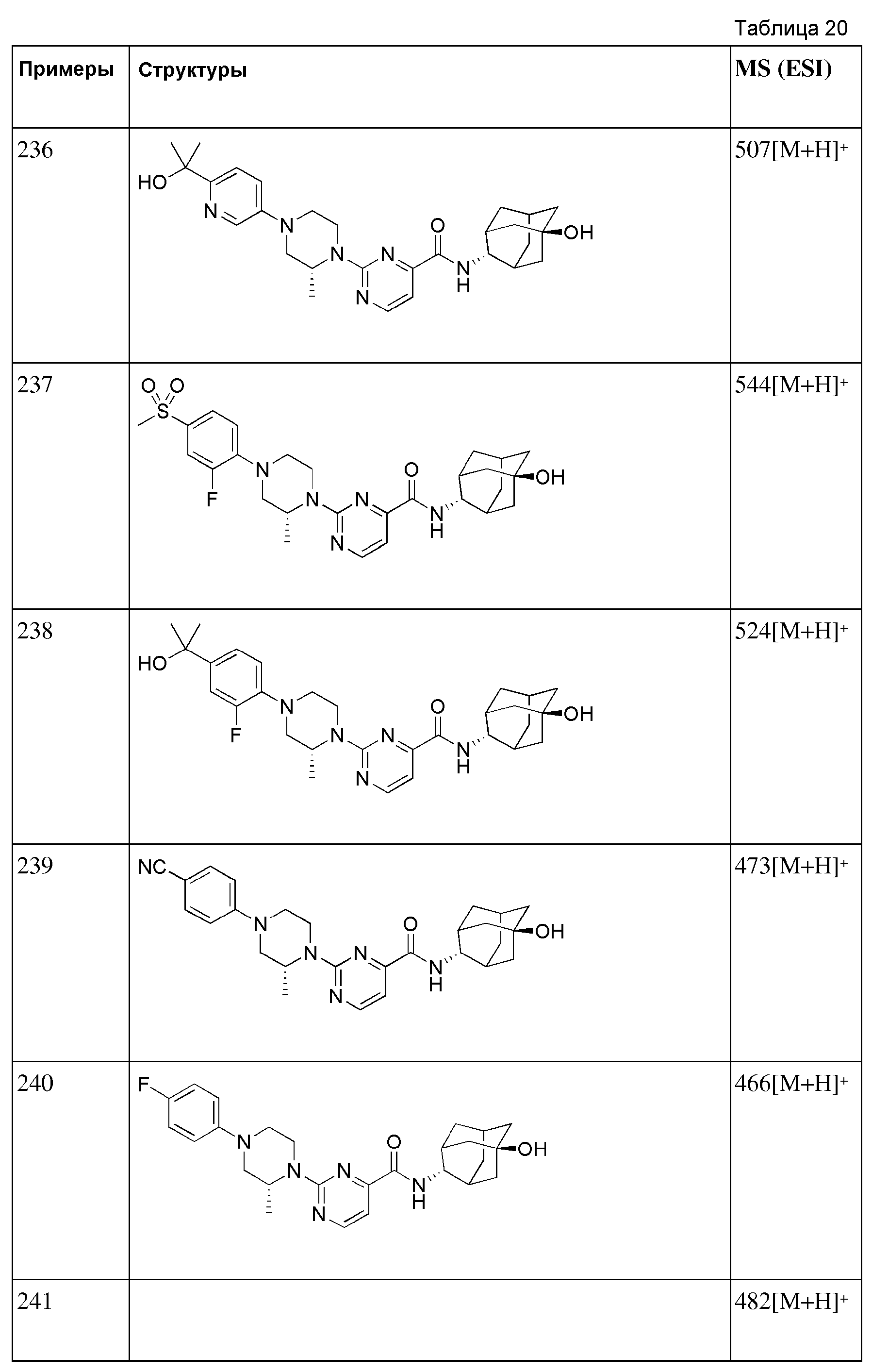
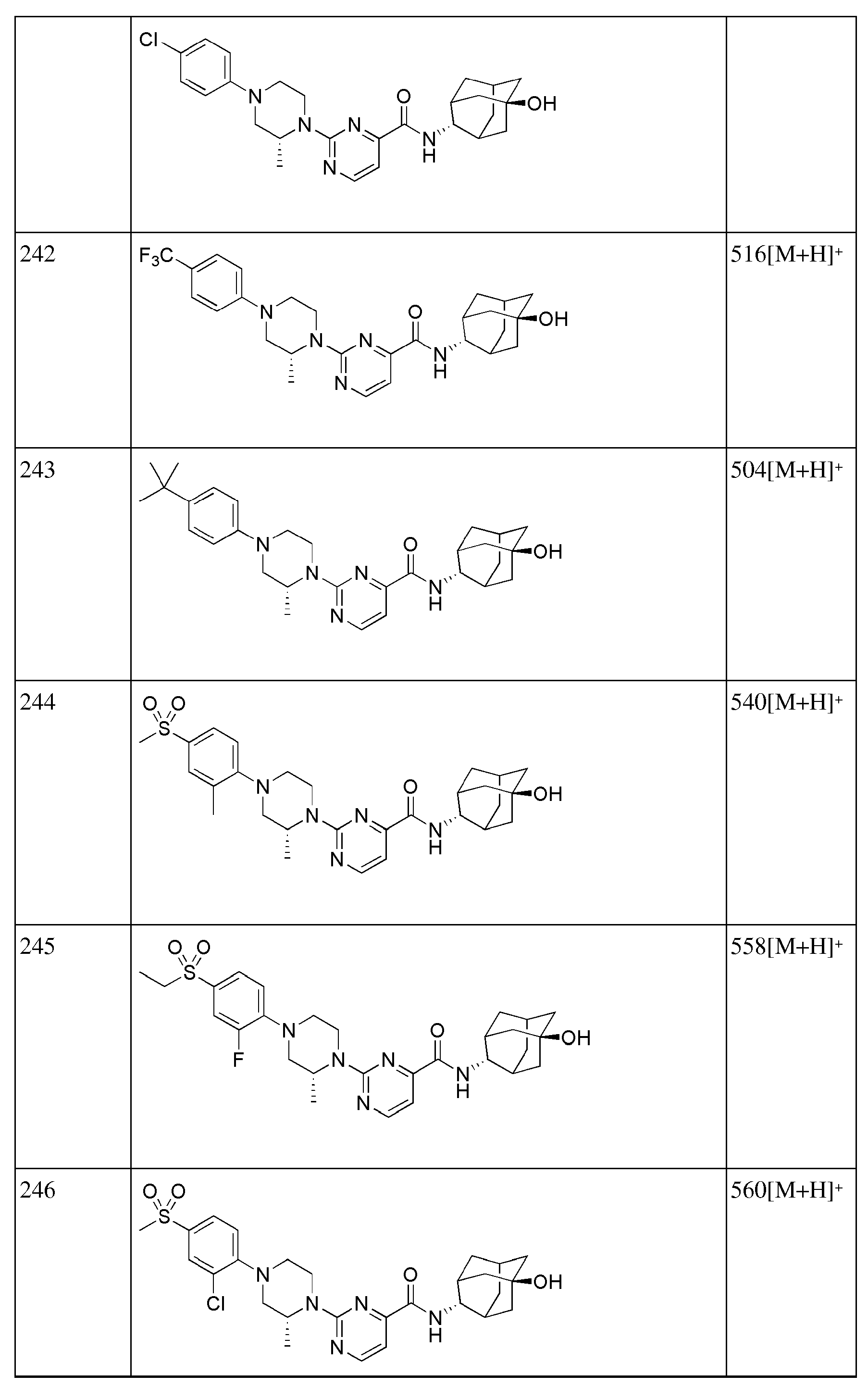
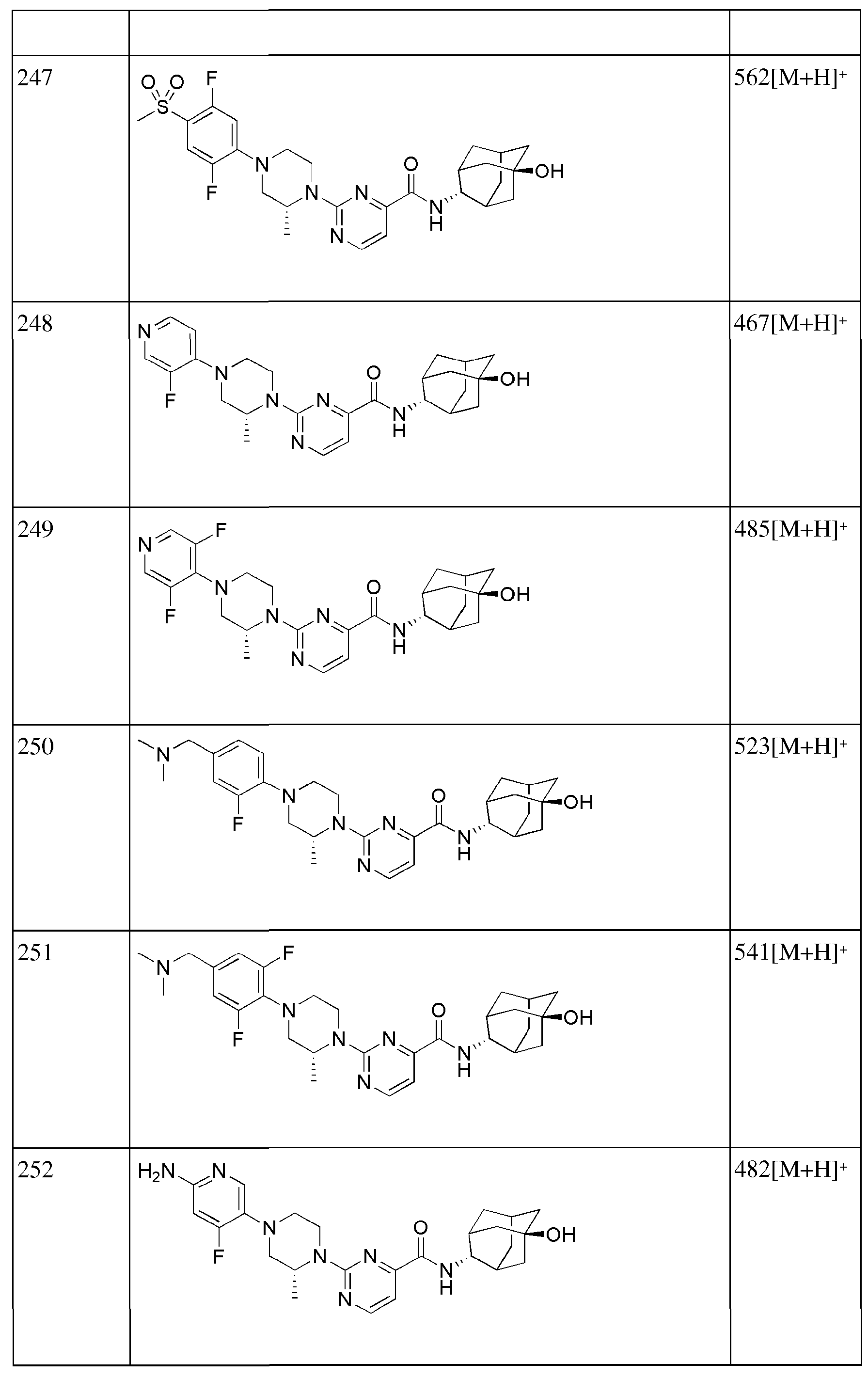
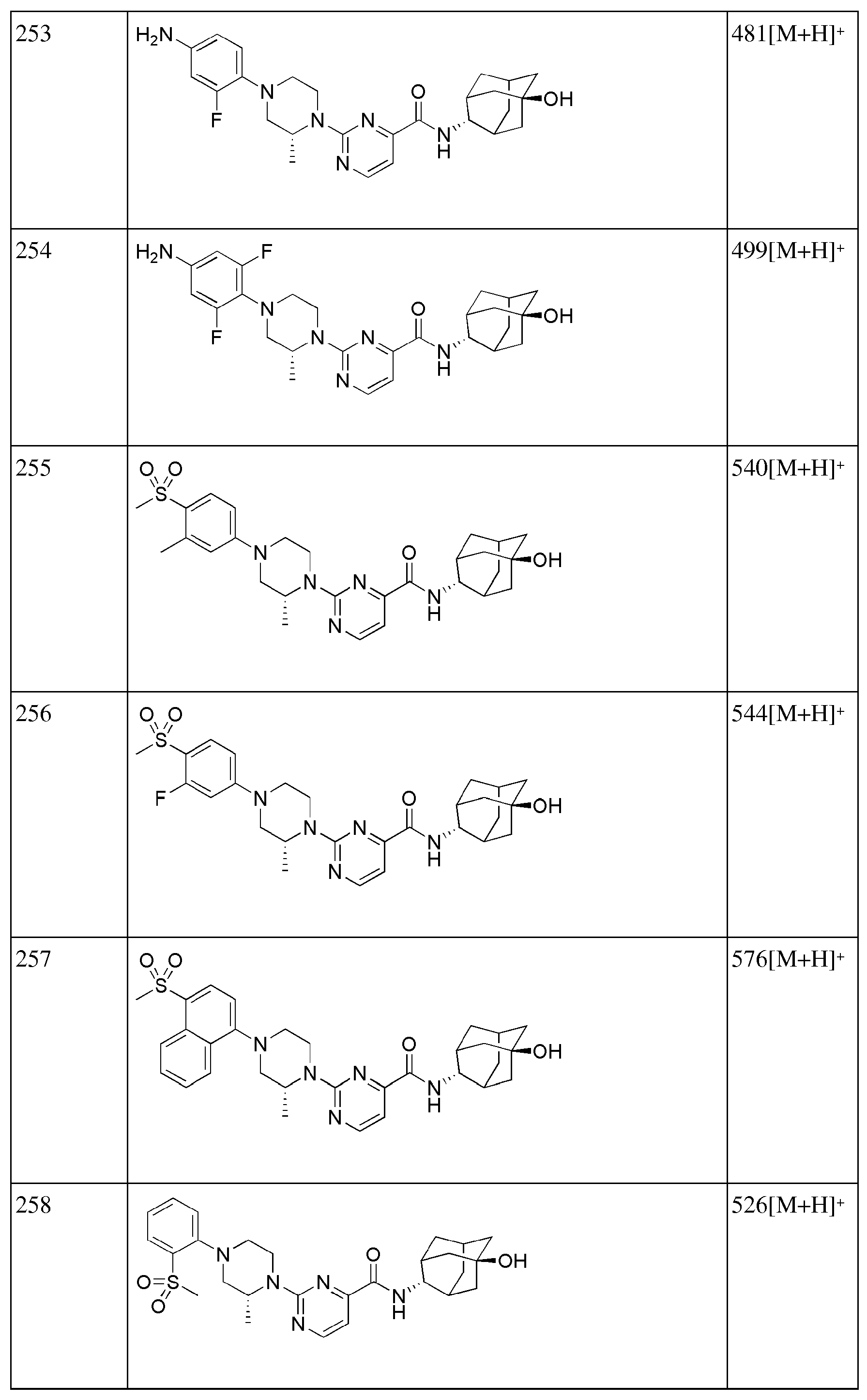
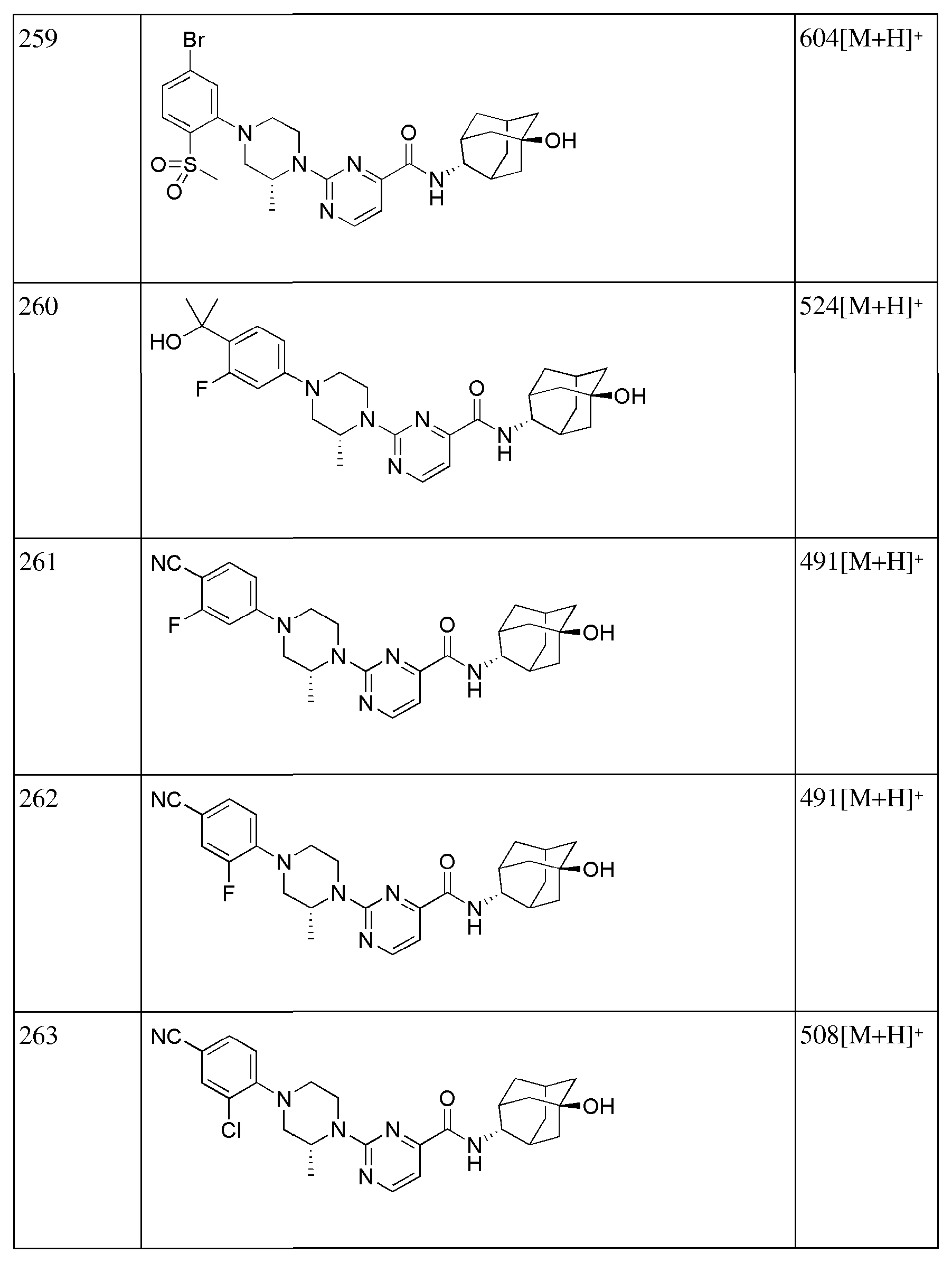
[619]
[620] Соединения следующих примеров синтезировали тем же способом, что в примере 230 выше, с использованием промежуточного соединения 11 и соответствующего аминового исходного материала
[621] Таблица 21
[Таблица 21]

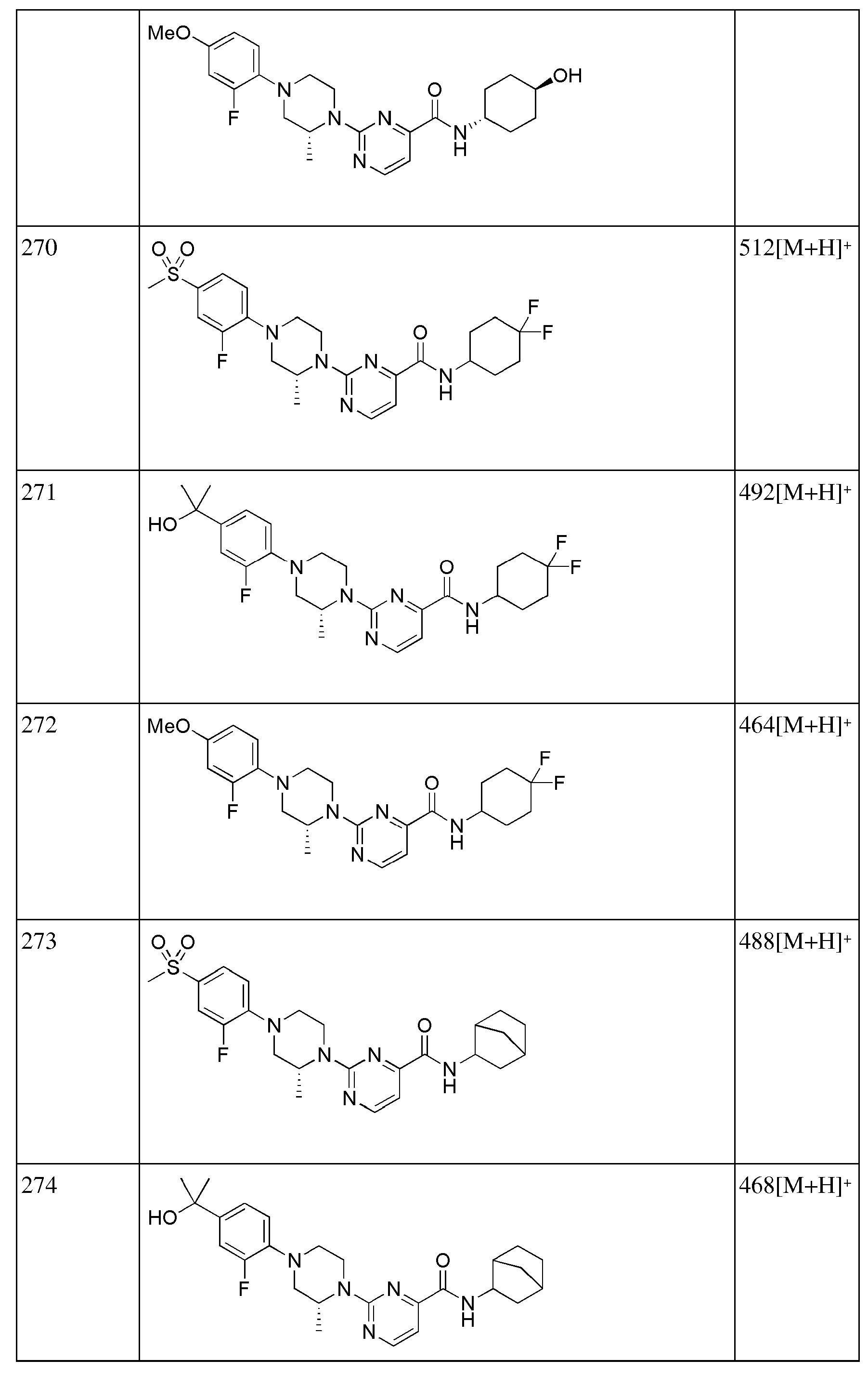

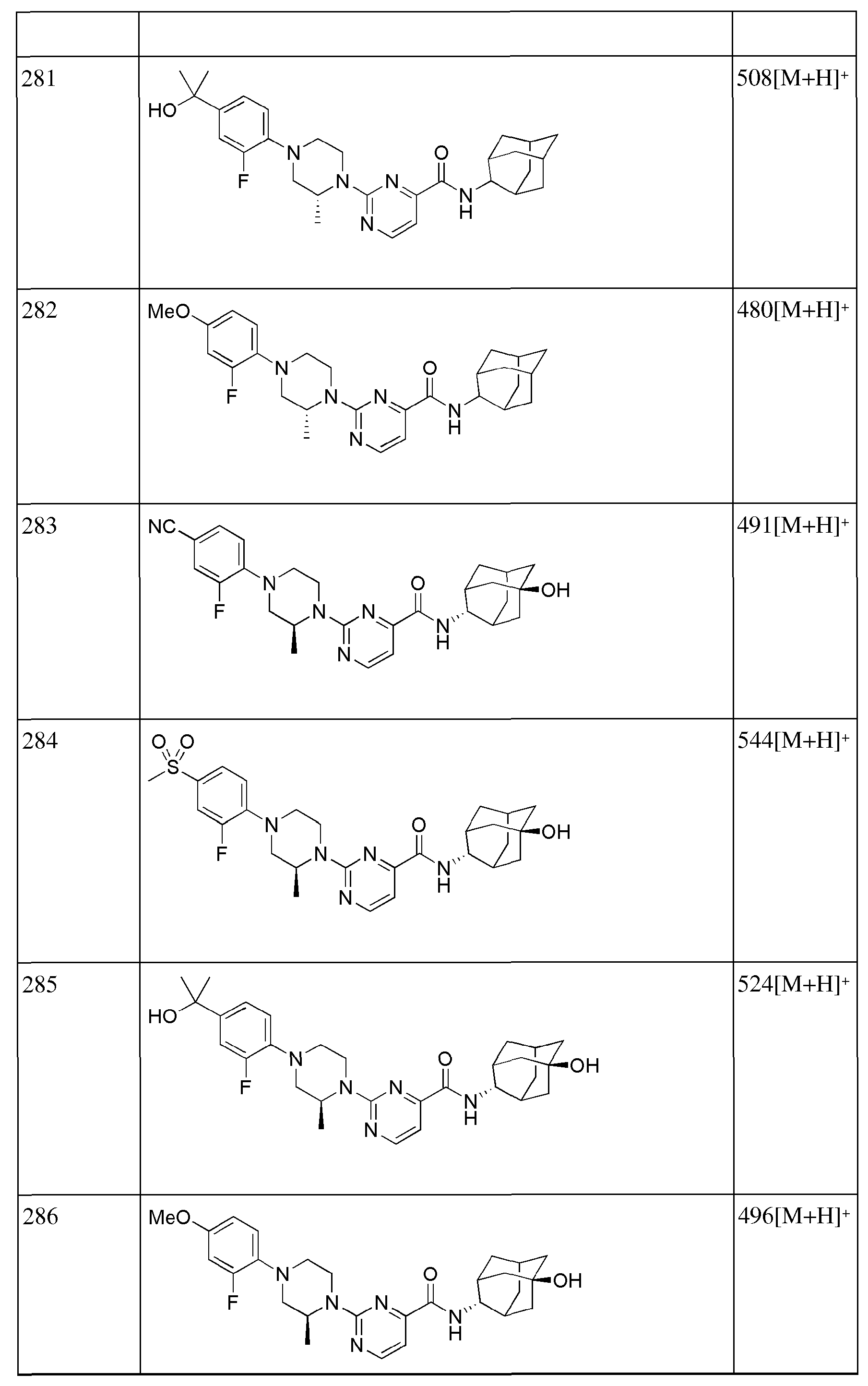
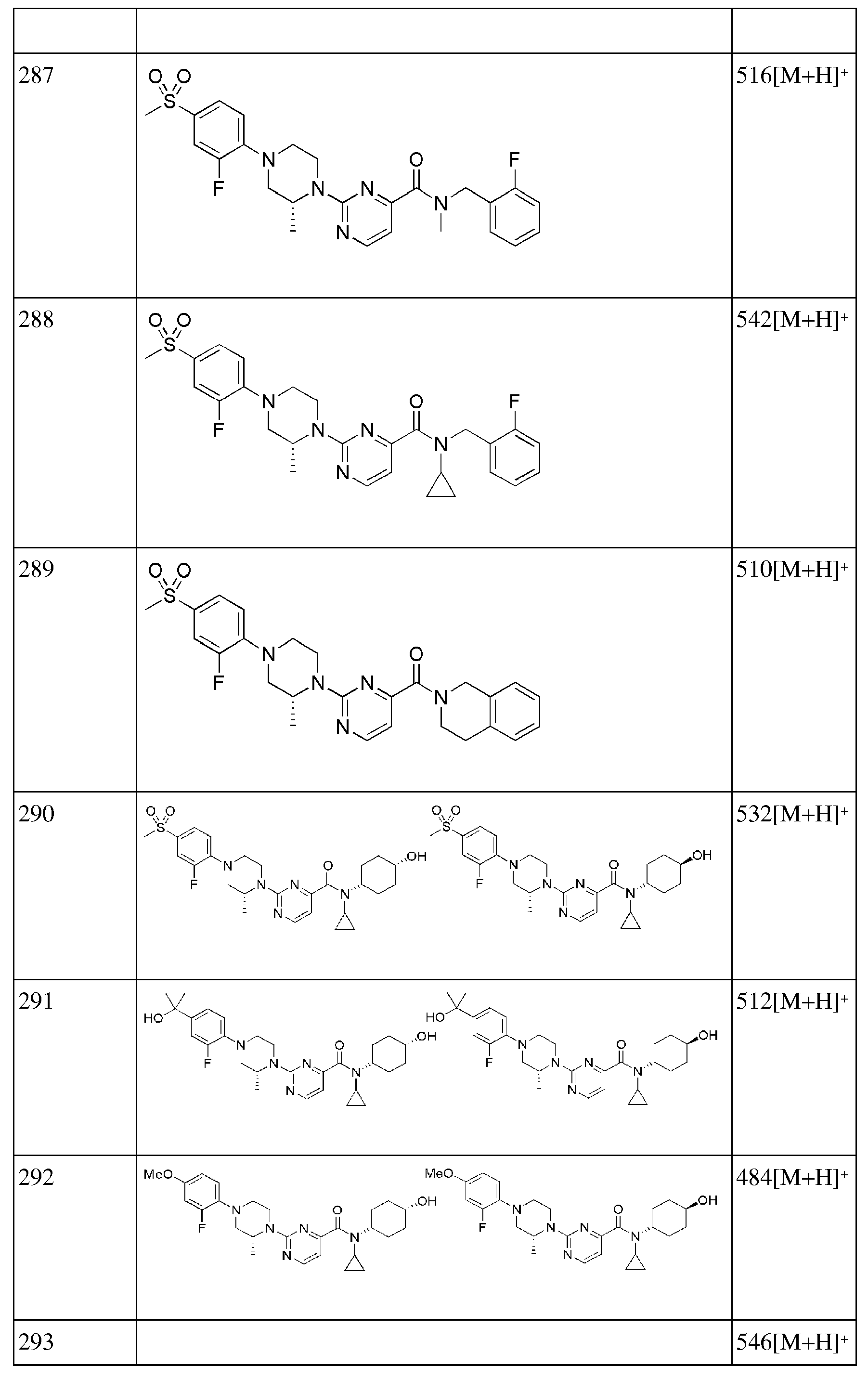
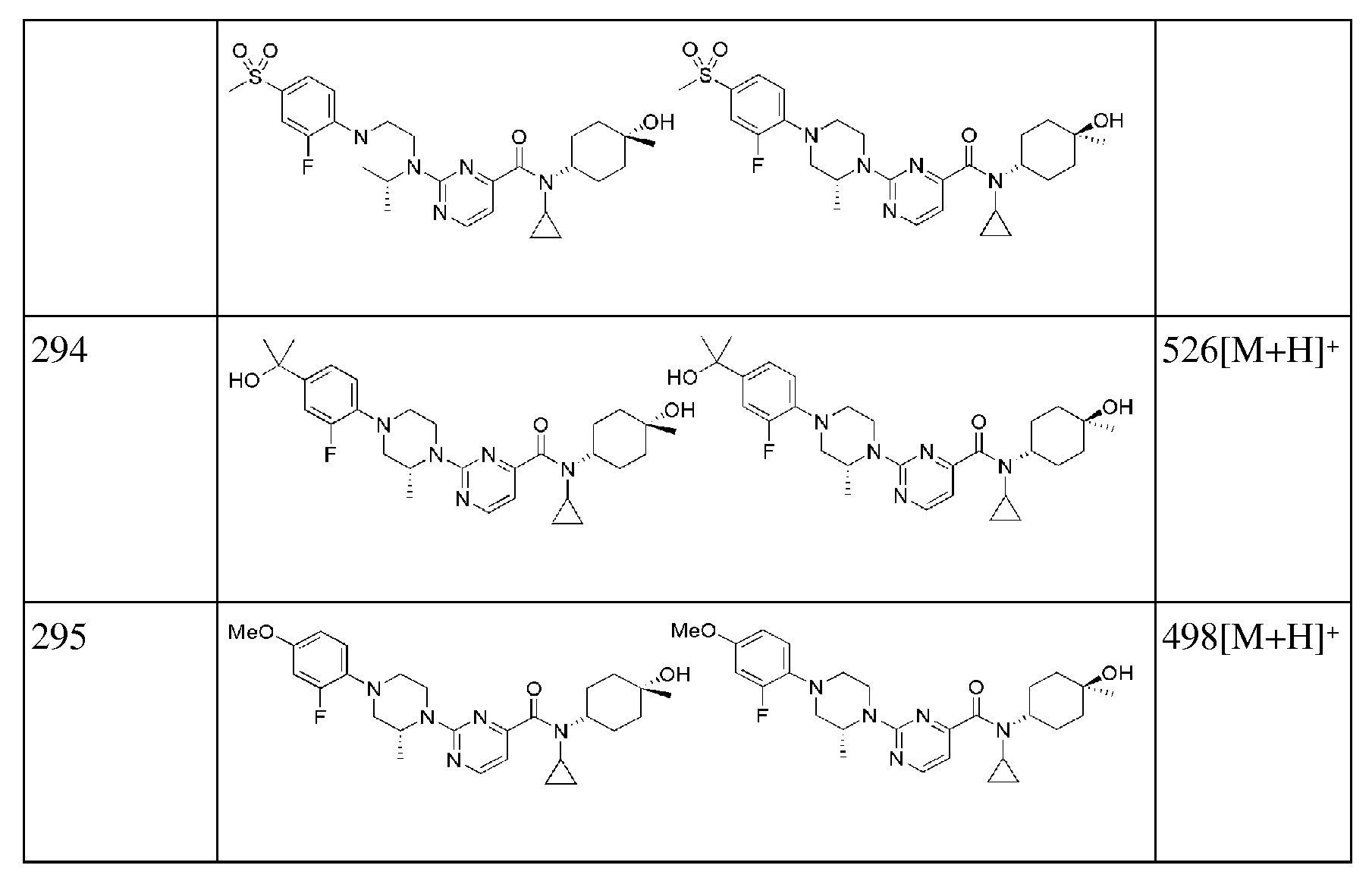
[622]
[623] Пример 296: Синтез 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1s,4S)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамида и 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1r,4R)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамида
[624]
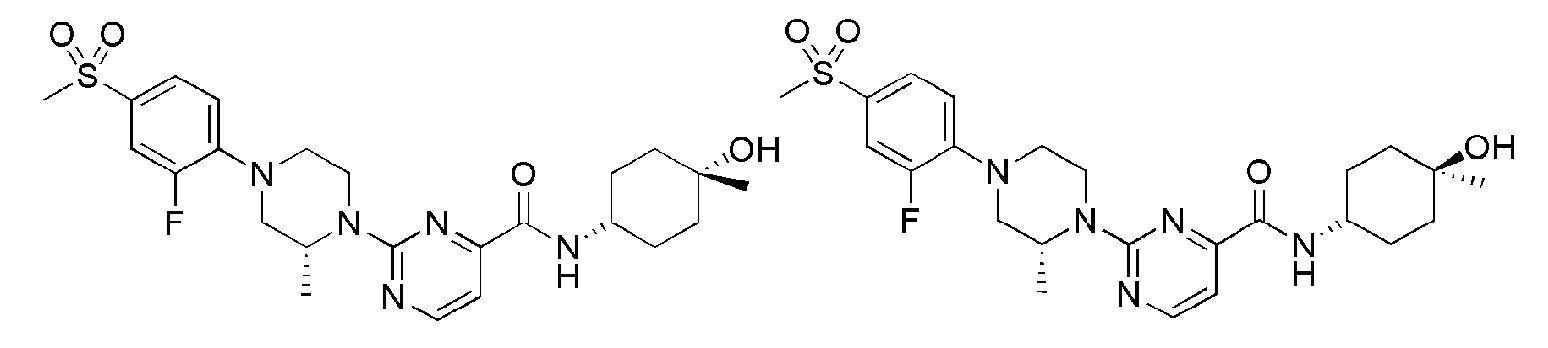
[625] Стадия 1: Синтез 4-(бензиламино)-1-метилциклогексанола
[626] 4-Гидрокси-4-метилциклогексанон (270 мг, 1,48 ммоль) растворяли в 1,2-дихлорэтане (8,3 мл) с последующим последовательным добавлением бензиламина (0,2 мл, 1,83 ммоль), NaBH(OAc)3 (560 мг, 2,66 ммоль) и уксусной кислоты (0,1 мл, 1,66 ммол), а затем получающуюся смесь перемешивали при комнатной температуре в токе азота в течение 72 часов. Получающуюся в результате реакционную жидкость нейтрализовали добавлением 10% водного раствора NaOH и экстрагировали 5% МеОН/МС (50 мл × 3). Органический слой сушили над безводным сульфатом натрия с последующим фильтрованием и концентрированием, а затем полученный таким образом остаток подвергали MPLC (10% МеОН/МС), получая 350 мг желтого твердого вещества (96%).
[627] Стадия 2: Синтез 4-амино-1-метилциклогексанола
[628] 4-(Бензиламино)-1-метилциклогексанол (335 мг, 1,53 ммоль) растворяли в этаноле (7,95 мл) с последующим добавлением Pd (10 вес.% на активированном угле, 35 мг), а затем получающуюся жидкость перемешивали при комнатной температуре в токе водорода в течение 31 часа. Получающуюся реакционную жидкость фильтровали, концентрировали при пониженном давлении и сушили в вакууме, получая 164 мг белого твердого вещества (83%).
[629] Стадия 3: Синтез 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1s,4S)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамида и 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1r,4R)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамида
[630] Выполняли тот же способ, что в примере 230 с использованием промежуточного соединения 11 и (R)-1-(2-фтор-4-(метилсульфонил(фенил)-3-метилпиперазин и 4-амино-1-метилциклогексанола, получая соответственно 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1s,4S)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамид и 2-((R)-4-(2-фтор-4-(метилсульфонил)фенил)-2-метилпиперазин-1-ил)-N-((1r,4R)-4-гидрокси-4-метилциклогексил)пиримидин-4-карбоксамид, соответственно MS (ESI): 506[M+H]+, 506[M+H]+.
[631]
[632] Соединения следующих примеров синтезировали с помощью того же способа, что выше в примере 296, с использованием промежуточного соединения 11 и соответствующего пиперазинового исходного материала.
[633] Таблица 22
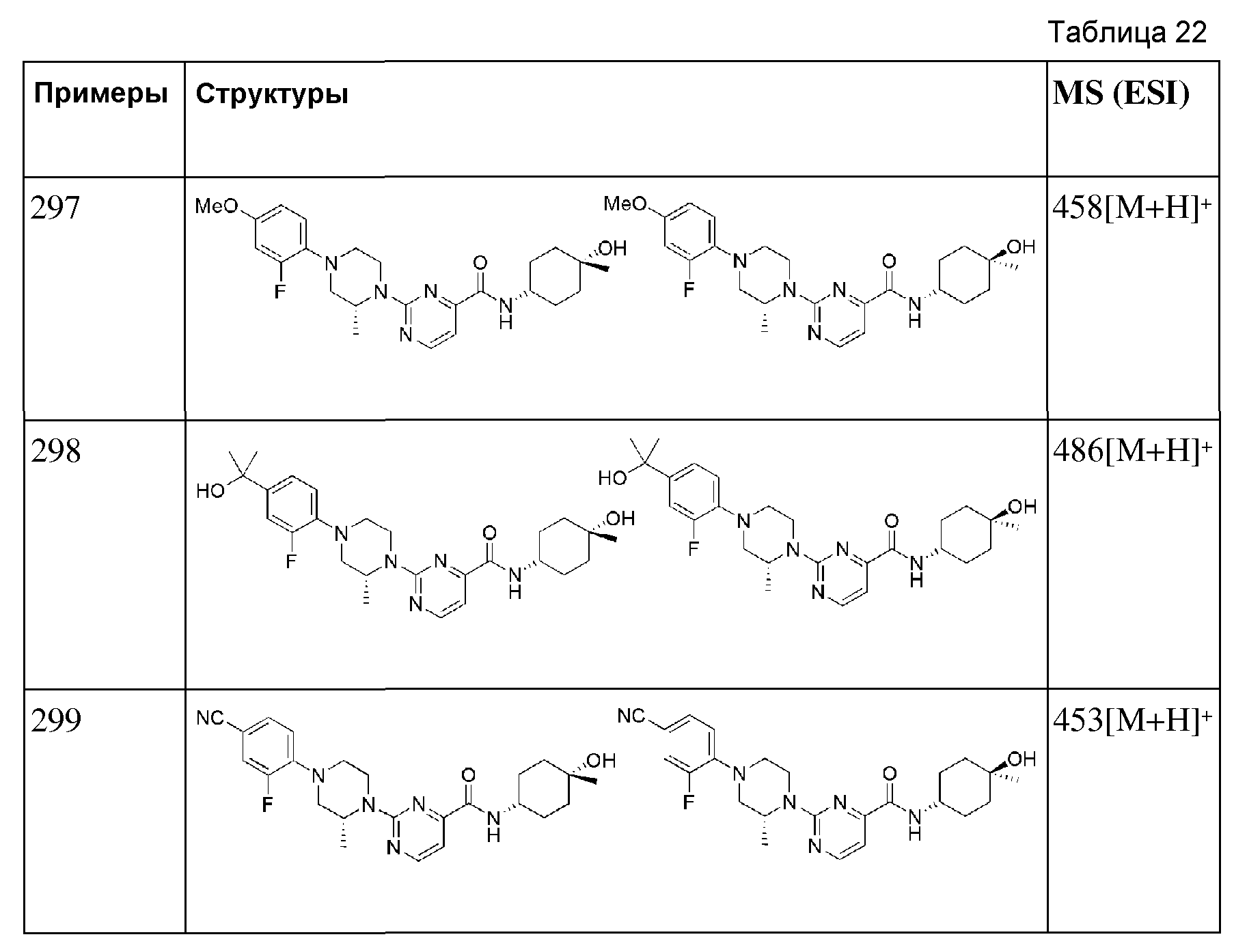
[634]
[635] Соединения следующих примеров синтезировали с помощью того же способа, что в примере 173 выше, с использованием промежуточного соединения 12 и соответствующего бромбензола, бромпиридина или бромпиразола.
[636] Таблица 23

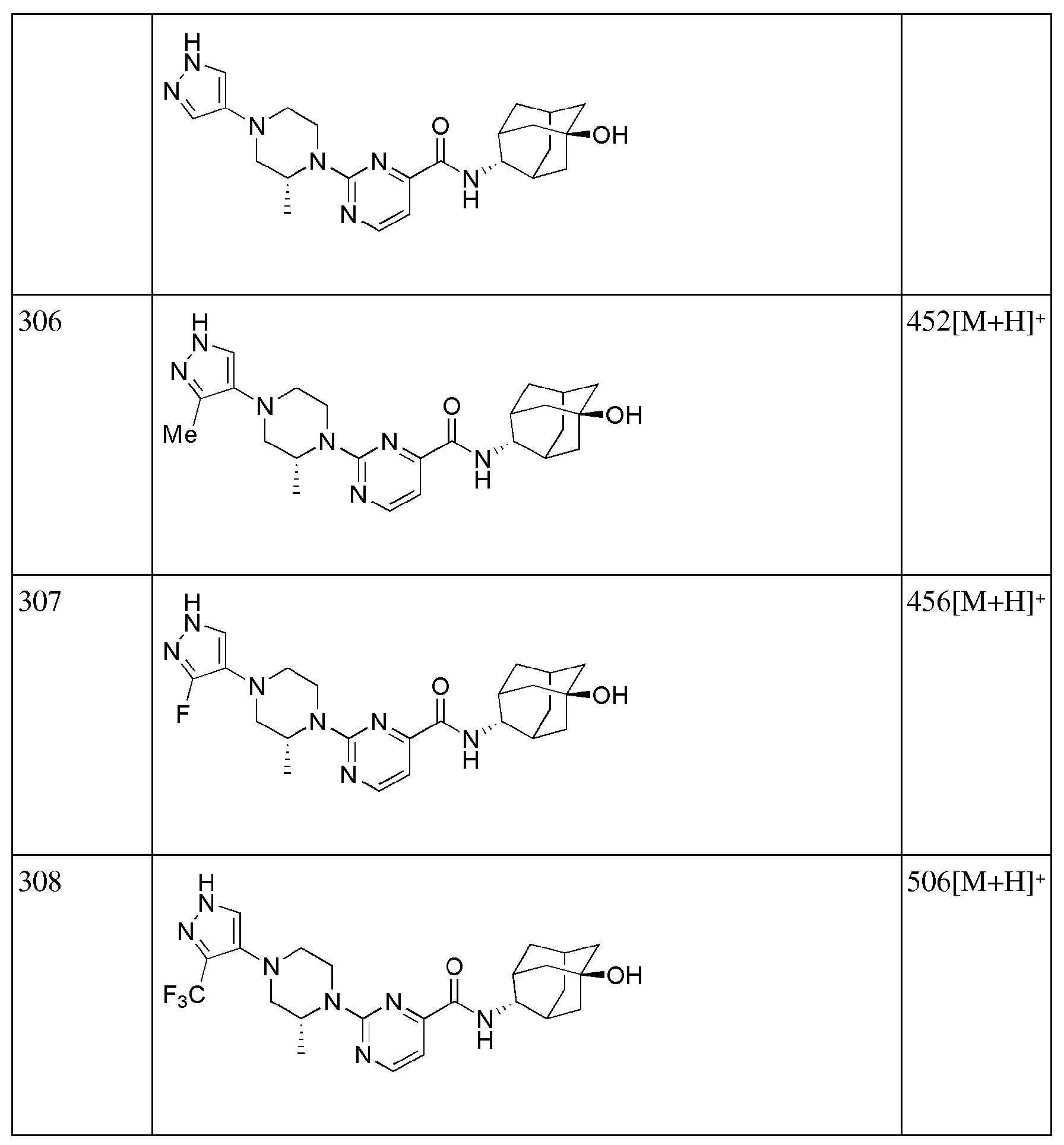
[637] [Экспериментальные примеры]
[638] Экспериментальный пример 1: Клеточный анализ на 11β-HSD1
[639] Для того, чтобы исследовать ингибирующие действия соединений настоящего изобретения на активность 11β-HSD1 фермента человеческого происхождения (h11β-HSD1), проводили следующий эксперимент.
[640] Для анализа активности 11β-HSD1 фермента, рекомбинантную ДНК, имеющую h11β-HSD1 ген, вводили в клетку животного для индуцирования сверхэкспрессии фермента [Arampatzirs, S. J. Mol Endocrinol. 2005, 35, 89-101]. Сначала HEK-293 клетки инкубировали с использованием инкубатора для клеток до тех пор, пока они не достигали 70-80% слияния на поверхности контейнера. Рекомбинантную ДНК, имеющую h11β-HSD1 ген, смешивали с Fugene 6, с последующей реакцией при комнатной температуре в течение 1 часа. Получающийся в результате материал использовали для обработки клеток, и таким образом в клетки инъецировали ДНК. На следующий день, клеточную надосадочную жидкость удаляли с последующим пополнением свежей средой, а затем клетки дополнительно инкубировали в течение 24 часов в клеточном инкубаторе.
[641] Когда клетки инкубировали при 70-80% слиянии, клетки разъединяли трипсином, и с использованием Гемацитометра измеряли число клеток. Инкубированную жидкость разбавляли до содержания 20000 клеток/мл, и 100 мкл разбавленной жидкости помещали в каждую лунку 96-луночной планшеты. С последующей инкубацией в течение 24 часов, соединение, растворенное в ДМСО, разбавляли до 1/100 инкубационной жидкостью. По 100 мкл получающейся в результате жидкости с разбавленным соединением распределяли в каждую лунку, а затем инкубировали в течение 30 минут. 10 мМ кортизона разводили до 21 мкМ инкубационной жидкостью и затем 5 мкл получающейся жидкости помещали в каждую лунку с последующей реакцией в течение 2 часов в клеточном инкубаторе [Jeffrey J. et al. J Med Chem, 2007, 50, 149-164].
[642] Количественный анализ генерируемого в данной реакции кортизола проводили с использованием системы, поставляемой фирмой Assay Designs Inc. Полученный в результате ферментный реагент добавляли на планшету, покрытую анти-мышиным IgG, а затем туда помещали вместе связывающимися с кортизолом специфические антитела и связанный с щелочной фосфатазой кортизол с последующей реакцией при комнатной температуре в течение 2 часов. Содержимое в лунках после реакции убирали и затем осуществляли три раза промывку буферным раствором, состоящим из поверхностно-активного вещества и трис-буферного солевого раствора. После этого раствор п-нитрофенил-фосфатазы, который является субстратом для щелочной фосфатазы, помещали туда для окрашивания реакционной смеси с последующей реакцией в течение 1 часа, а затем с использованием считывателя с планшеты измеряли световую абсорбцию при 405 нм. Результаты измерений отмечали в таблице 24.
[643] Таблица 24


[644]
[645] [Примеры рецептур]
[646] Пример рецептуры 1: Приготовление таблеток (прессованный тип)
[647] 5,0 мг соединения настоящего изобретения, представленного формулой 1, в качестве активного ингредиента просеивали и затем смешивали с 14,1 мг лактозы, 0,8 мг кросповидона USNF и 0,1 мг стеарата магния с последующим прессованием, формулируя таблетки.
[648]
[649] Пример рецептуры 2: Приготовление таблеток (мокрое гранулирование)
[650] 5,0 мг соединения настоящего изобретения, представленного формулой 1, в качестве активного ингредиента просеивали и затем смешивали с 16,0 мг лактозы и 4,0 мг крахмала. 0,3 мг полисольвата 80 растворяли в чистой воде, а затем к получающейся смеси добавляли соответствующее количество данного раствора с последующей микронизацией. Полученные таким образом микронизированные частицы просеивали, а затем смешивали с 2,7 мг коллоидной двуокиси кремния и 2,0 мг стеарата магния с последующим прессованием, формулируя таблетки.
[651]
[652] Пример рецептуры 3: Получение агентов в виде порошков и капсул
[653] 5,0 мг соединения настоящего изобретения, представленного формулой 1, в качестве активного ингредиента просеивали и затем смешивали с 14,8 мг лактозы, 10,0 мг поливинилпирролидона и 0,2 мг стеарата магния. Получающейся смесью заполняли твердые желатиновые капсулы №5 с использованием соответствующего устройства.
[654]
[655] Пример рецептуры 4: Приготовление инъецируемых препаратов
[656] Брали 100 мг соединения настоящего изобретения, представленного формулой 1, в качестве активного ингредиента, и помимо этого брали также 180 мг маннита, 26 мг Na2HPO4·12H2O и 2,974 мг дистиллированной воды для приготовления инъецируемого препарата.
[657]
[658] Хотя настоящее изобретение описано подробно со ссылкой на примеры, специалистам в данной области очевидно, что в технической сути настоящего изобретения могут производиться различные изменения и модификации, и таким образом очевидно, что данные изменения и модификации включены в объем прилагаемых пунктов формулы изобретения и их эквивалентов.
[659]
Промышленная применимость
[660] Новые пиколинамидные и пиримидин-4-карбоксамидные соединения настоящего изобретения, их фармацевтически приемлемые соли, сольваты, гидраты, пролекарства, рацематы или стереоизомеры обладают селективной ингибиторной активностью в отношении 11β-HSD1 ферментов человеческого происхождения. Следовательно, соединения настоящего изобретения оказывают полезные действия как агенты для предотвращения, регулирования и лечения заболеваний, связанных с регулированием глюкокортикоидов, которые вызываются в результате активности 11β-HSD1 ферментов, например, метаболических синдромов, диабета типа 1 и типа 2, диабета после осложнений, латентного аутоиммунного диабета взрослых (LADA), синдромов инсулинорезистентности, ожирения, нарушенной толерантности к глюкозе (IGT), нарушенной гликемии натощак (IFG), поврежденной толерантности к глюкозе, дислипидемии, атеросклероза, гипертензии и др.
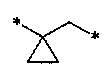
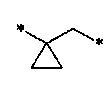
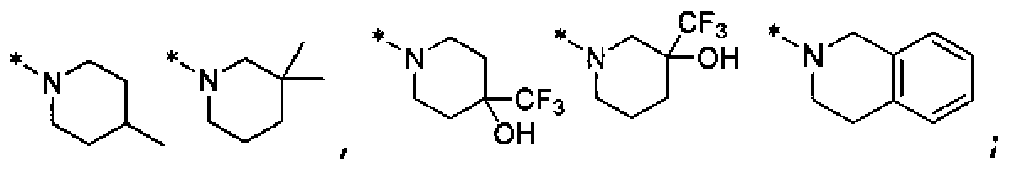
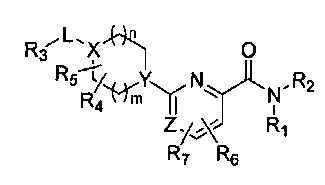
Способ управления туманом в сушильном устройстве для одежды
Слитый белок, обладающий активностью фактора vii
2-пиридилзамещенные имидазолы в качестве ингибиторов alk5 и/или alk4
Производные пирролпиримидинона, способ получения и их применение
Способ и устройство для получения алкильных эфиров жирных кислот
Способ управления туманом в сушильном устройстве для одежды
Слитый белок, обладающий активностью фактора vii
2-пиридилзамещенные имидазолы в качестве ингибиторов alk5 и/или alk4
Производные пирролпиримидинона, способ получения и их применение
Производное имидазопиримидина и имидазотриазина и фармацевтическая композиция, включающая это производное

