Результат интеллектуальной деятельности: СИЛИКОНОВЫЙ (МЕТ)АКРИЛАМИДНЫЙ МОНОМЕР, ПОЛИМЕР, ОФТАЛЬМОЛОГИЧЕСКАЯ ЛИНЗА И КОНТАКТНАЯ ЛИНЗА
Вид РИД
Изобретение
РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет по заявке на патент США № 13/048469, поданной 15 марта 2011 года, и заявке на патент Японии № JP-2010-061991, поданной 18 марта 2010 года.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к силиконовому (мет)акриламидному мономеру, молекулы которого содержат линейную силоксанильную группу и предпочтительно гидроксильную группу. Полимер, получаемый путем полимеризации данного мономера, подходит для изготовления различных изделий медицинского назначения, таких как офтальмологические линзы, эндоскопы, катетеры, трубки для переливания крови, трубки для подачи газа, стенты, защитные оболочки, манжеты, разъемы трубок, порты для забора проб, дренажные мешки, системы для переливания крови, материалы для закрытия ран, а также для изготовления различных видов носителей лекарственных препаратов, но в особенности подходит для изготовления контактных линз, офтальмологических линз и искусственных роговиц.
ОПИСАНИЕ УРОВНЯ ТЕХНИКИ
В последние годы в качестве материалов для изготовления контактных линз для непрерывного ношения приобрели известность силикон-гидрогели, получаемые путем сополимеризации силиконового мономера, гидрофильного мономера и перекрестносшивающего мономера. Силиконовые акриламидные мономеры, имеющие следующие формулы (x1) и (x2), рассматриваются в патентном документе 1 как допустимые для использования при получении силикон-гидрогелей.
[ФОРМУЛА 1]
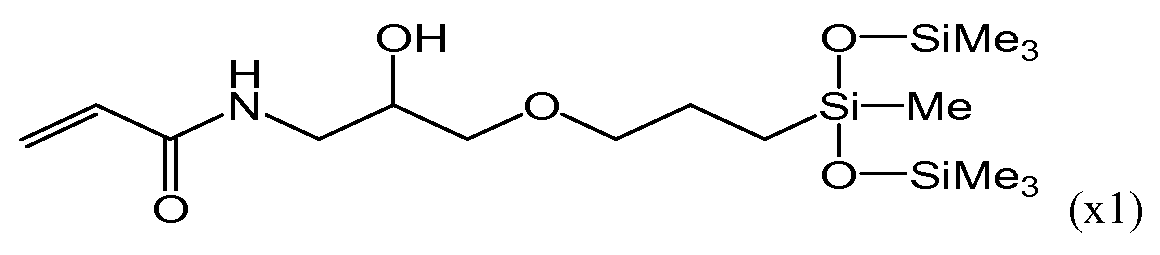
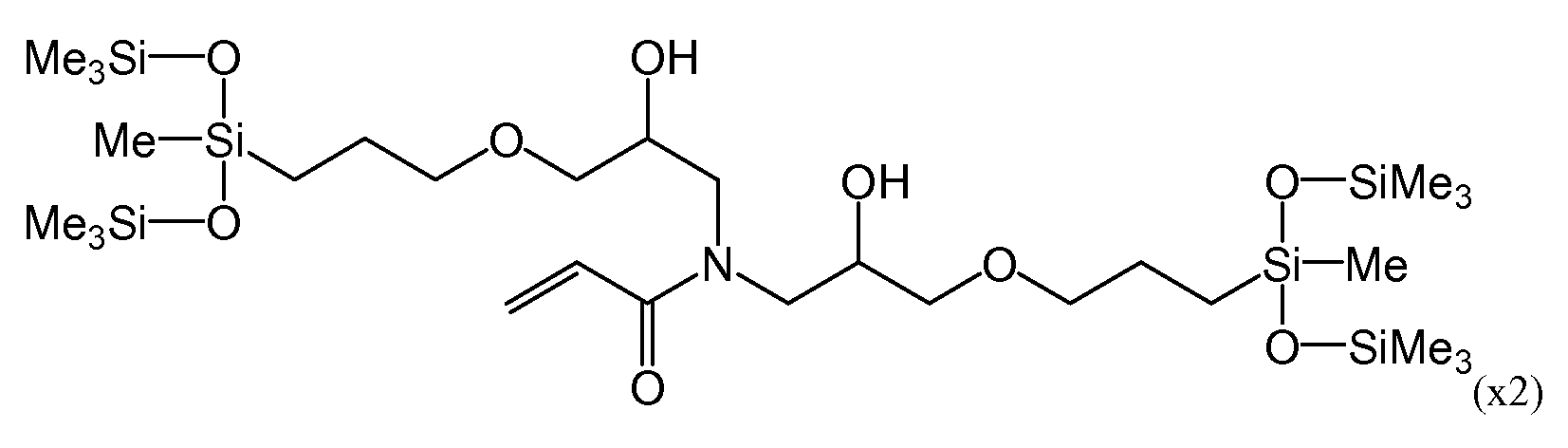
Однако силиконовый участок силиконового акриламидного мономера, описанного в патентном документе 1, представляет собой силоксанильную группу с разветвленной цепью, вследствие чего полимеры, получаемые в результате полимеризации данных мономеров, могут обладать низкими характеристиками восстановления формы. В настоящем документе термин «разветвленный» обозначает состояние, при котором связь Si-(O-Si)x (в которой x представляет собой целое число, равное 1 или более), начинающаяся у атома кремния, присоединенного к главной углеродной цепи молекулы, не является единой непрерывной цепью. Следовательно, при использовании линейной силоксанильной группы повышается гидрофобность и существует риск снижения совместимости с гидрофильными веществами.
Более того, композиция, описанная в патентном документе 1, содержит приблизительно от 30 до 40 весовых частей метакрилатного сложного эфира. В то время как акриламидный мономер во время гомополимеризации имеет более высокую константу скорости полимеризации, чем метакрилатный сложный эфир, скорость сополимеризации акриламида и метакрилата значительно ниже, в результате чего скорость полимеризации всей системы снижается.
С другой стороны, в патентном документе 2 рассматривается силиконовый акриламидный мономер, имеющий следующие формулы (y1) и (y2), и силикон-гидрогель, полученный из данного мономера и гидрофильного акриламидного мономера.
[ФОРМУЛА 2]
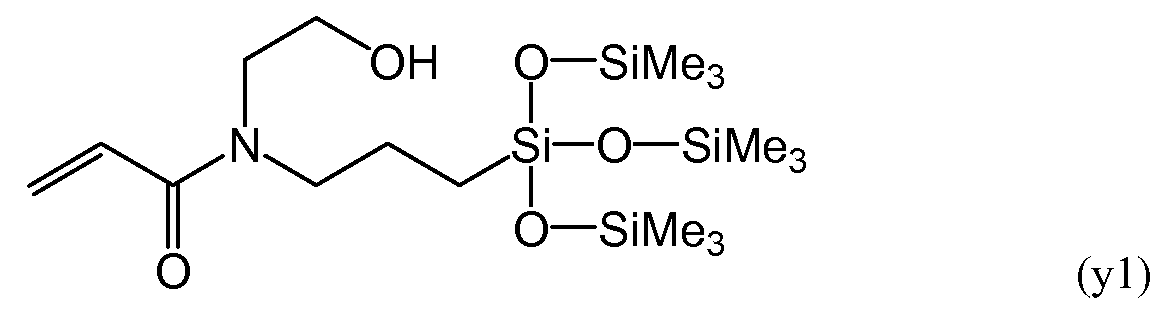
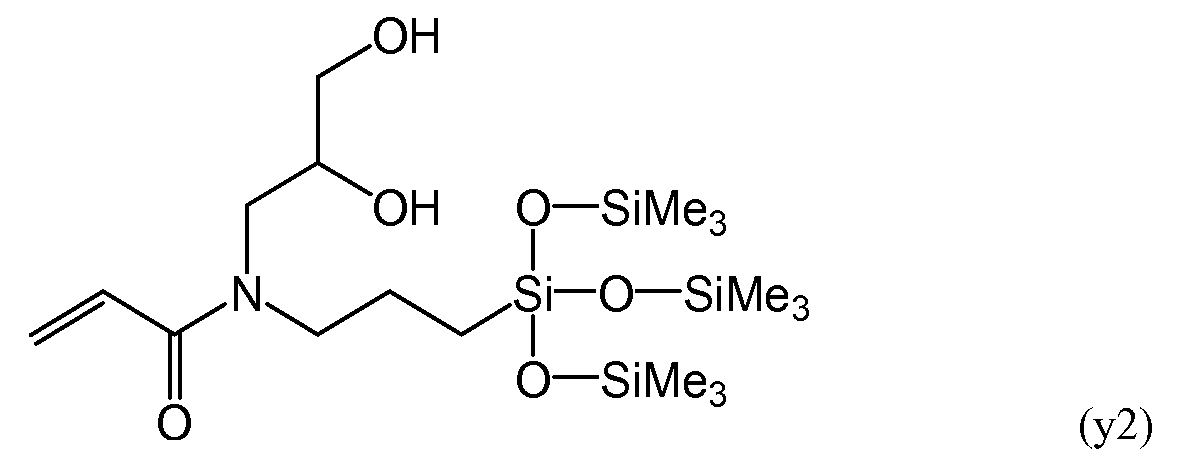
Большую часть композиции, используемой при получении силиконового акриламидного мономера, описанного в патентном документе 2, составляют акриламидные мономеры, и для всей системы предполагается более высокая скорость полимеризации. Тем не менее, силиконовый участок данных мономеров также имеет силоксанильную группу с разветвленной цепью, поэтому полимеры, получаемые в результате полимеризации данных мономеров, демонстрируют низкую способность к восстановлению формы. Более того, амидная связь акриламидной группы обладает высокой гидрофильностью, и, следовательно, получение прозрачной линзы может быть затруднено с точки зрения обеспечения одновременно достаточного количества силиконового компонента для обеспечения высокой кислородной проницаемости и достаточного содержания воды для обеспечения гибкости линзы. В частности, достижение прозрачности линзы особенно затрудняется при введении в материал линзы внутреннего смачивающего агента для повышения смачиваемости поверхности.
Ссылки смежной области техники
Патентная документация
Патентный документ 1, US 2005/0176911
Патентный документ 2, нерассмотренная патентная заявка Японии H10-212355
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к силиконовым (мет)акриламидным мономерам с высокой скоростью полимеризации и к получаемому из данных мономеров полимеру, обладающему благоприятной прозрачностью и способностью к восстановлению формы.
Настоящее изобретение дополнительно относится к следующим композициям, таким как:
(1) силиконовый (мет)акриламидный мономер, содержащий (мет)акриламидную группу и линейную силоксанильную группу, молекула которого имеет два или более повторяющихся звена -OSi;
(2) силиконовый (мет)акриламидный мономер по п. (1), который дополнительно содержит по меньшей мере одну гидроксильную группу;
(3) силиконовый (мет)акриламидный мономер по п. (2), имеющий следующую общую формулу (a).
[ФОРМУЛА 1]
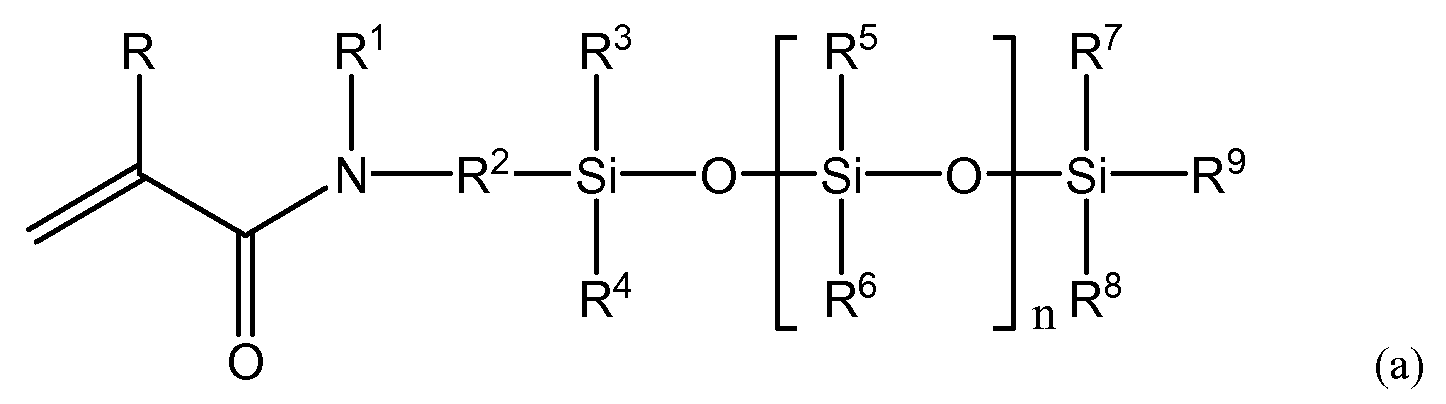
R представляет собой атом водорода или метильную группу; R1 представляет собой атом водорода или алкильную либо арильную группу, имеющую от 1 до 20 атомов углерода, которая может быть замещена гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями; R2 представляет собой C1-10-алкиленовую группу или ариленовую группу, которая может быть замещена гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями; причем по меньшей мере один из R1 или R2 имеет гидроксильную группу; R3-R9 независимо представляют собой C1-20-алкильную группу или арильную группу, имеющую от 1 до 20 атомов углерода, любая из которых может быть замещена фтором, гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями, а n представляет собой целое число в диапазоне от 1 до 10.
Настоящее изобретение дополнительно относится к полимерам, офтальмологическим линзам и контактным линзам, получаемым путем полимеризации смеси мономеров, содержащей по меньшей мере один силиконовый (мет)акриламидный мономер, как описано в настоящем документе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение содержит силиконовый (мет)акриламидный мономер, составляющий предмет настоящего изобретения, и, следовательно, может представлять собой силикон-гидрогель с высоким содержанием акриламидного мономера, обладающий прозрачностью, а также отличной способностью поддержания баланса между содержанием воды и смачиваемостью.
Полимер, получаемый путем полимеризации данного силикон-гидрогеля, подходит для изготовления различных видов изделий медицинского назначения, в частности, офтальмологических линз, таких как контактные линзы, интраокулярные линзы и искусственная роговица, и особенно для контактных линз.
Молекула силиконового (мет)акриламидного мономера, составляющего предмет настоящего изобретения, имеет (мет)акриламидную группу и линейную силоксанильную группу, а также предпочтительно имеет по меньшей мере одну гидроксильную группу. Термин «силоксанильная группа» обозначает группу, имеющую по меньшей мере одну связь Si-O-Si, термин «линейный» относится к структуре, в которой связь сформирована в виде одной линии, начинающейся с атома кремния, связанного с алкильной группой, имеющей (мет)акриламидную группу, или, другими словами, силиконовый (мет)акриламидный мономер, имеющий линейную силоксанильную группу, - это структура, имеющая следующую общую формулу (p).
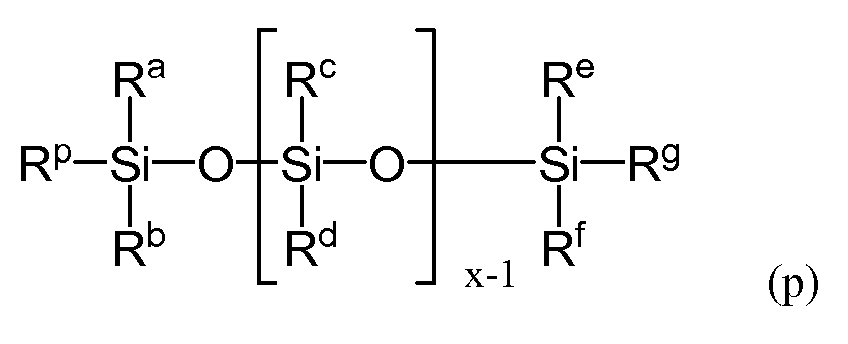
В формуле (p) Rp представляет собой алкильную группу с (мет)акриламидной группой. Ra-Rg представляют собой группы, не содержащие атома кремния. X представляет собой целое число, равное 2 или более. В отличие от силиконовых (мет)акриламидов, составляющих предмет настоящего изобретения, силиконовые (мет)акриламиды предшествующего уровня техники содержали по меньшей мере часть силикона в концевой цепи. В формуле (p) x представляет собой целое число, равное 2 или более, и, если значение x слишком низкое, невозможно достичь достаточной кислородной проницаемости, но если значение x слишком высокое, снизится совместимость с гидрофильным мономером, что затруднит достижение прозрачности линзы. Следовательно, в некоторых вариантах осуществления x имеет значение от 3 до 7, от 3 до 6 и от 3 до 5. При этом для повышения воспроизводимости физических характеристик получаемого полимера предпочтительно, чтобы по параметру x не было распределения. Для настоящего изобретения фраза «не было распределения» означает состояние, при котором при наличии нескольких пиков для различных значений x доля наибольшего пика составляет 80% или более, при измерении методом газовой хроматографии (ГХ) в случае мономера, который допускает измерение методом ГХ (при помощи ПИД - пламенно-ионизационного детектора), или при измерении методом жидкостной хроматографии (ЖХ) (при помощи РД - рефрактометрического детектора), в случае если мономер имеет настолько высокую точку кипения, что его невозможно измерить методом ГХ.
Более того, в данном описании термин «(мет)акрил» обозначает акрил, а термины «метакрил» и «(мет)акриламид» обозначают акриламид и метакриламид.
Силиконовый (мет)акриламидный мономер, составляющий предмет настоящего изобретения, без гидроксильной группы включает в себя мономер, имеющий следующие формулы (s1), (s5) и (s6):
[ФОРМУЛА]
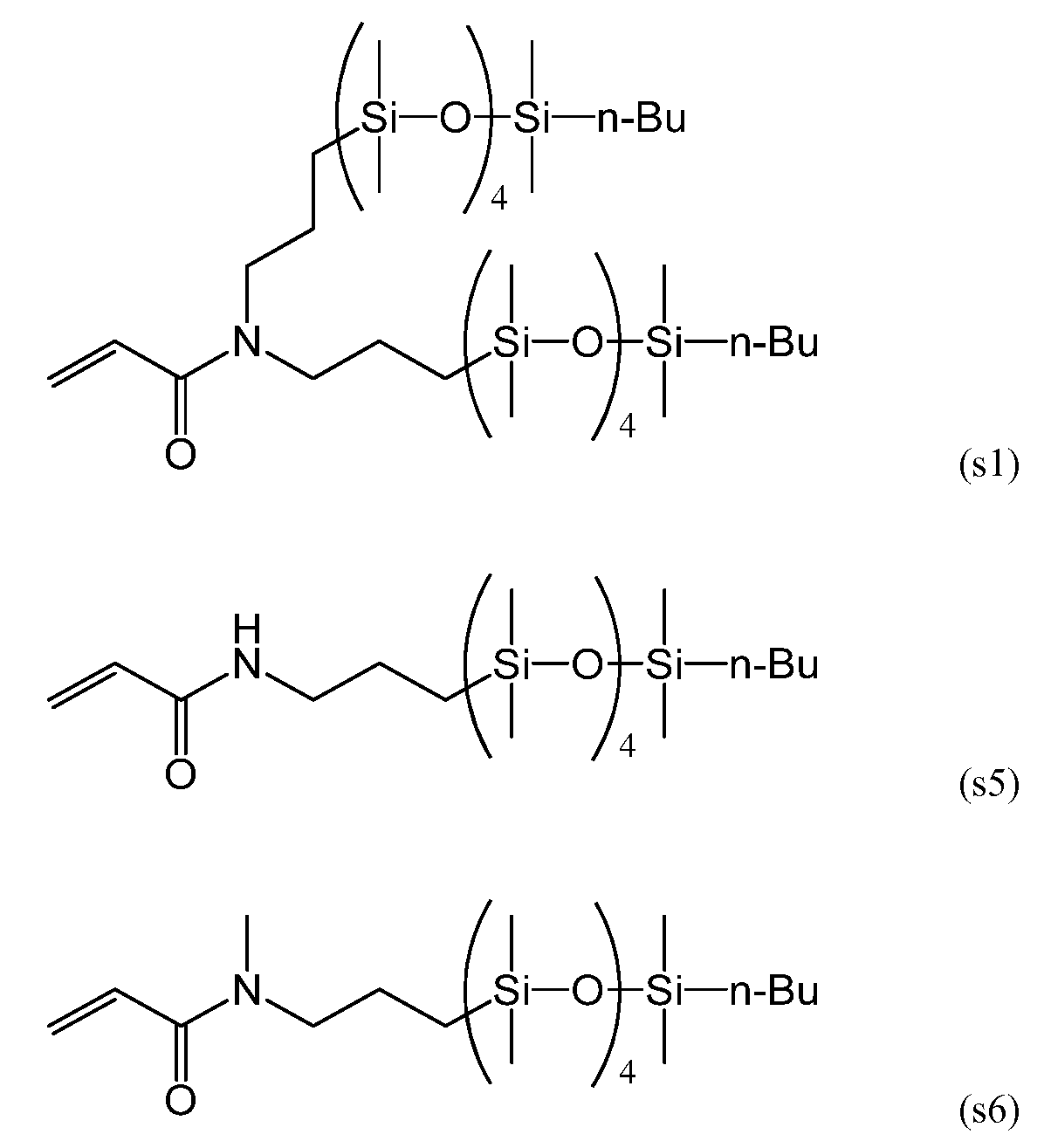
С точки зрения низкого модуля упругости получаемого силикон-гидрогеля, более предпочтительными из данных мономеров являются мономеры, имеющие формулы (s5) и (s6).
Что касается настоящего изобретения, существует риск, что, в случае если мономер содержит линейную силоксанильную группу, повысится гидрофобность и понизится совместимость с гидрофильными веществами, но гидрофильность можно повысить за счет присутствия амидной связи и гидроксильной группы. Однако если количество гидроксильных групп будет слишком большим, модуль упругости полимера будет нежелательно высоким, следовательно, количество гидроксильных групп на каждое мономерное звено составляет в одном варианте осуществления от 1 до 6, в другом - от 1 до 3, дополнительно - 1 или 2, а в случае, когда особенно важна смачиваемость (низкие значения углов смачивания) получаемого полимера, - 2.
Более того, если помимо линейной силоксанильной группы в молекуле присутствует силоксанильная группа с разветвленной цепью, гидрофобность может быть нежелательно высокой. Следовательно, в одном варианте осуществления силоксанильная группа с разветвленной цепью присутствует в количестве менее приблизительно 15% вес. от общей массы компонентов полимеризации, а в других вариантах осуществления силоксанильная группа с разветвленной цепью отсутствует.
Для повышения скорости полимеризации силиконовый (мет)акриламидный мономер, составляющий предмет настоящего изобретения, имеет в качестве полимерной группы (мет)акриламидную группу, благодаря чему можно ожидать повышения скорости полимеризации. Данный эффект особенно выражен при использовании акриламидного мономера в качестве гидрофильного вещества. В результате данного повышения скорости полимеризации сокращается время отверждения, необходимое для завершения полимеризации и получения конечного вещества. Из акриламидных и метакриламидных групп, акриамидные группы являются более предпочтительными с точки зрения дополнительного повышения скорости полимеризации. Как сказано выше, в присутствии амидной группы сложно добиться прозрачности, но падение уровня прозрачности в связи с присутствием амидной группы можно предотвратить при наличии гидроксильной группы. Подробные причины данного явления неясны, однако считается, что, помимо свойств компатибилизатора, гидроксильная группа также обладает способностью повышать смачиваемость.
Примером предпочтительной структуры силиконового (мет)акриламидного мономера, составляющего предмет настоящего изобретения, с по меньшей мере одной гидроксильной группой является мономер, имеющий следующую общую формулу (a).
[ФОРМУЛА 4]
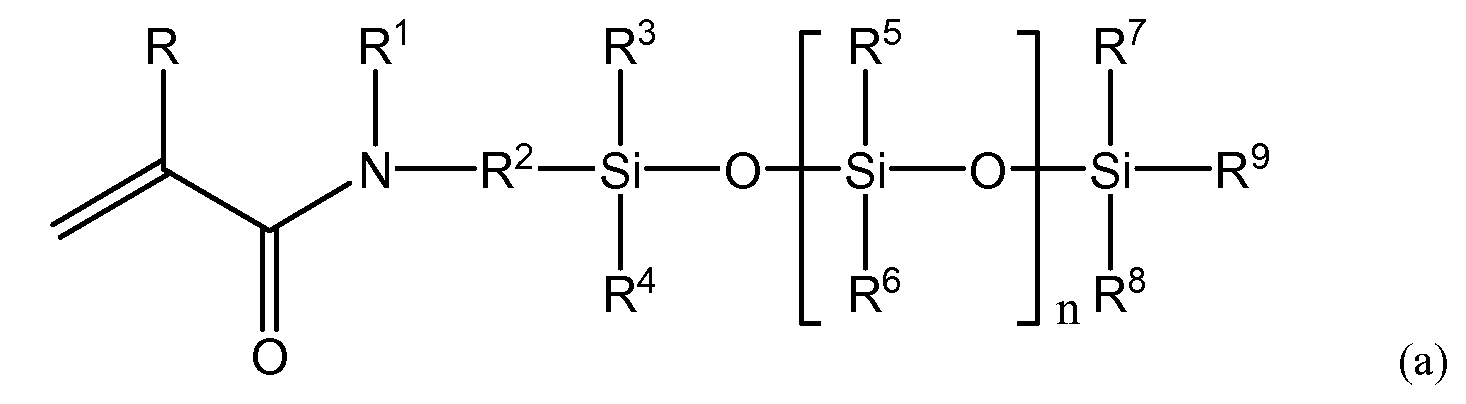
В формуле (a) R представляет собой атом водорода или метильную группу. В одном варианте осуществления R представляет собой атом водорода, а силиконовый (мет)акриламид демонстрирует более высокую скорость полимеризации. В формуле (a) R1 представляет собой атом водорода или алкильную либо арильную группу, имеющую от 1 до 20 атомов углерода, которая может быть замещена гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями.
В одном варианте осуществления R1 представляет собой атом водорода или алкил, имеющий от 1 до 5 атомов углерода, который может быть замещен гидроксильными, кислотными или сложноэфирными группами. В другом варианте осуществления R1 представляет собой алкильную группу, имеющую 1-5 атомов углерода, и может быть не замещен либо замещен по меньшей мере одной гидроксильной группой. Примеры R1 включают в себя атомы водорода, метильные группы, этильные группы, пропильные группы, н-пропильные группы, изопропильные группы, н-бутильные группы, втор-бутильные группы, трет-бутильные группы, н-пентильные группы, изопентильные группы, втор-пентильные группы, неопентильные группы, гексильные группы, гептильные группы, октильные группы, нонильные группы, децильные группы, додецильные группы, эйкозильные группы, фенильные группы, нафтильные группы, все из которых могут быть замещены в соответствии с вышеизложенным, и т.п. В одном варианте осуществления R1 выбирают из метильных групп, этильных групп, н-пропильных групп, изопропильных групп, н-бутильных групп, втор-бутильных групп или трет-бутильных групп, любая из которых может быть гидроксилзамещенной. В другом варианте осуществления R1 выбирают из атомов водорода, метильных групп и этильных групп. Атомы водорода, метильные группы и этильные группы обеспечивают повышение гидрофильности силиконового (мет)акриламидного мономера. Примеры гидроксилзамещенных групп R1 включают в себя 2-гидроксиэтильные группы, 2-гидроксипропильные группы, 3-гидроксипропильные группы, 2,3-дигидроксипропильные группы, 4-гидроксибутильные группы, 2-гидрокси-1,1-бис(гидроксиметил)этильные группы, 2-гидроксиметилфенильные группы, 3-гидроксиметилфенильные группы, 4-гидроксиметилфенильные группы и т.п. В одном варианте осуществления R1 выбирают из 2-гидроксиэтильных групп, 2-гидроксипропильных групп и 2,3-дигидроксипропильных групп, а в другом варианте осуществления R1 представляет собой 2,3-дигидроксипропильные группы. Силиконовый (мет)акриламид, содержащий 2,3-дигидроксипропильные группы, обладает повышенной гидрофильностью. Более того, если требуется дополнительно повысить прозрачность полимера, получаемого в ходе полимеризации силиконового (мет)акриламидного мономера, R1 предпочтительно не является атомом водорода.
R2 представляет собой бивалентную C1-10-алкиленовую или арильную группу, которая может быть замещена гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями. В другом варианте осуществления R2 выбирают из C1-5-алкиленовых групп, которые могут быть не замещены либо замещены гидроксилом, простыми эфирными группами и их комбинациями. В другом варианте осуществления R2 выбирают из C2-5-алкиленовых групп, которые могут быть не замещены либо замещены гидроксилом, простыми эфирными группами и их комбинациями, а еще в одном варианте осуществления R2 представляет собой C3-алкиленовые группы, которые могут быть не замещены либо замещены гидроксилом, простыми эфирными группами и их комбинациями. Еще в одном варианте осуществления R2 представляет собой группу формулы (d):
-CH2CH(OH)CH2- (d)
(d)
или группы, имеющие следующую формулу (b):
-CH2CH(OH)CH2OCH2CH2CH2-
(b)
Примеры допустимых групп R2 включают в себя метиленовые группы, этиленовые группы, пропиленовые группы, бутиленовые группы, пенталеновые группы, окталеновые группы, дециленовые группы и фениленовые группы, любая из которых может быть не замещена либо замещена по меньшей мере одним гидроксилом и группой, представленной следующей формулой (d):
-CH2CH(OH)CH2-
(d)
а также группами, имеющими следующую формулу (b):
-CH2CH(OH)CH2OCH2CH2CH2-
(b)
Данные алкиленовые и ариленовые группы могут иметь линейную или разветвленную цепь. В некоторых вариантах осуществления из данных групп предпочтительными являются этиленовые, пропиленовые, бутиленовые группы и группы, имеющие формулы (d) и (b), и силиконовые (мет)акриламиды, содержащие данные группы, полимеризуются с образованием полимера, обладающего низким модулем упругости. В другом варианте осуществления R2 выбирают из этиленовой группы, пропиленовой группы или бутиленовой группы, а еще в одном варианте осуществления R2 представляет собой н-пропиленовую группу, выбранную с точки зрения достижения баланса между гидрофобностью и сниженным модулем упругости полимера, получаемого в результате полимеризации силиконового (мет)акриламидного мономера.
В формуле (a) R3-R9 независимо представляют собой алкильную группу или арильную группу, имеющую от 1 до 20 атомов углерода, любая из которых может быть замещена фтором, гидроксилом, кислотой, сложным эфиром, простым эфиром, тиолом и их комбинациями. Примеры R3-R8 включают в себя атомы водорода, метильные группы, этильные группы, пропильные группы, н-пропильные группы, изопропильные группы, н-бутильные группы, втор-бутильные группы, трет-бутильные группы, н-пентильные группы, изопентильные группы, втор-пентильные группы, неопентильные группы, гексильные группы, гептильные группы, октильные группы, нонильные группы, децильные группы, додецильные группы, эйкозильные группы, фенильные группы, нафтильные группы, любые из которых могут быть замещены фтором, гидроксилом или их комбинациями, и т.п. В некоторых вариантах осуществления предпочтительна алкильная группа, имеющая от 1 до 4 атомов углерода, а наиболее предпочтительна, с точки зрения повышения кислородной проницаемости, метильная группа. R9 независимо представляет собой алкильную группу или арильную группу, имеющую от 1 до 20 атомов углерода. Примеры R9 включают в себя атомы водорода, метильные группы, этильные группы, пропильные группы, н-пропильные группы, изопропильные группы, н-бутильные группы, втор-бутильные группы, трет-бутильные группы, н-пентильные группы, изопентильные группы, втор-пентильные группы, неопентильные группы, гексильные группы, гептильные группы, октильные группы, нонильные группы, децильные группы, додецильные группы, эйкозильные группы, фенильные группы, нафтильные группы, любые из которых могут быть замещены фтором, гидроксилом или их комбинациями, и т.п. Что касается R9, то, исходя из баланса между кислородной проницаемостью и устойчивостью к гидролизу силоксанильной группы, предпочтительной является алкильная группа, имеющая от 1 до 4 атомов углерода, а в некоторых вариантах осуществления наиболее предпочтительна н-бутильная группа, втор-бутильная группа или трет-бутильная группа, которая может быть незамещенной или гидроксилзамещенной. В других вариантах осуществления R9 может быть метилом или этилом.
В формуле (a) n представляет собой целое число от 1 до 10, и, если значение n слишком низкое, невозможно достичь необходимой кислородной проницаемости, но если значение n слишком высокое, снизится совместимость с гидрофильным мономером, что затруднит достижение прозрачности линзы. Следовательно, в некоторых вариантах осуществления n имеет значение от 2 до 6, от 2 до 5 и от 2 до 4. Допускается объединение любых значений нижнего предела и любых предпочтительных значений верхнего предела. Более того, для повышения воспроизводимости физических характеристик получаемого полимера предпочтительно, чтобы по параметру n не было распределения. Для настоящего изобретения фраза «не было распределения» означает состояние, при котором при наличии нескольких пиков для различных значений n доля наибольшего пика составляет 80% или более, при измерении методом газовой хроматографии (ГХ) в случае мономера, который допускает измерение методом ГХ (при помощи ПИД - пламенно-ионизационного детектора), или при измерении методом жидкостной хроматографии (ЖХ) (при помощи РД - рефрактометрического детектора), в случае если мономер имеет настолько высокую точку кипения, что его невозможно измерить методом ГХ.
Силиконовые (мет)акриламидные мономеры, составляющие предмет настоящего изобретения, можно получать любым способом, известным специалистам в области синтетической органической химии. Пример способа получения силиконового (мет)акриламидного мономера, имеющего формулу (a)
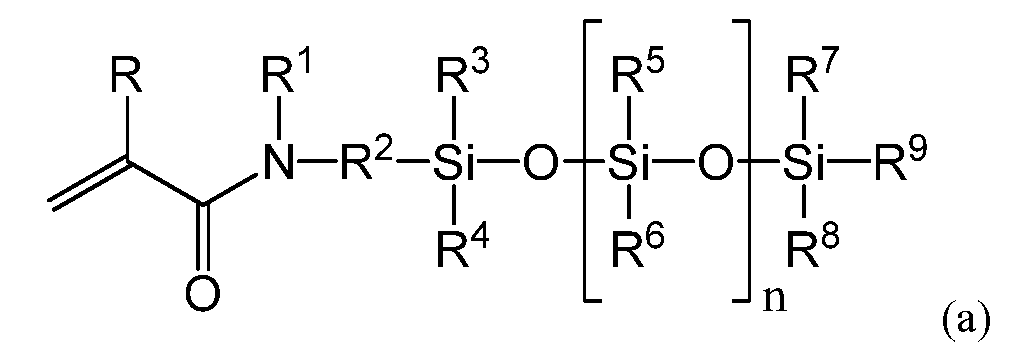
содержит гидросилилирование соединения, имеющего следующую формулу (a10)
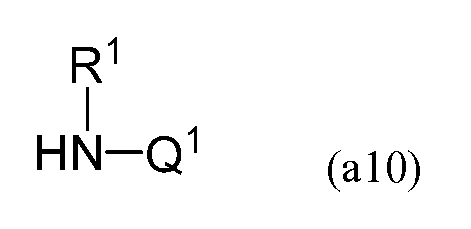
с линейным силоксаном, имеющим следующую формулу (a11)
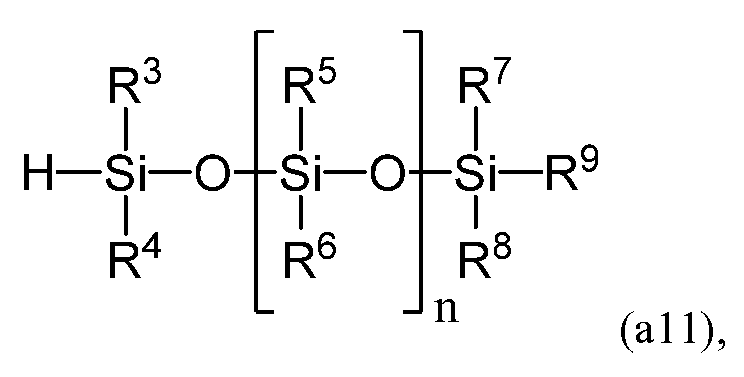
и содержит (мет)акрилирование соединения, имеющего следующую формулу (a12)
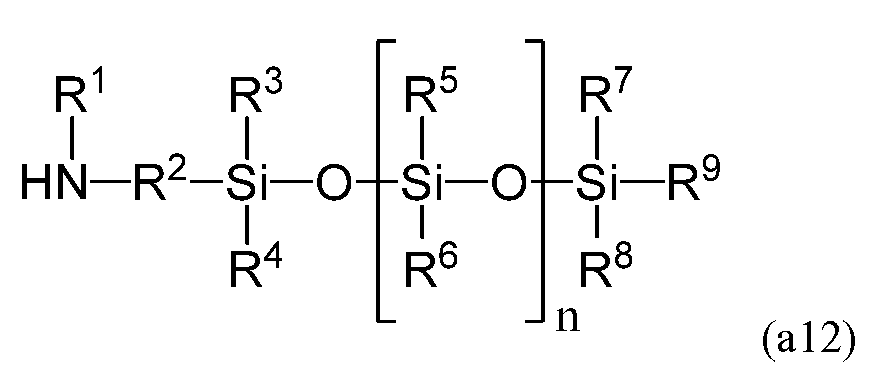
получаемого в результате гидросилилирования. В формуле (a10) Q1 содержит двойную углерод-углеродную связь и преобразуется в R2 формулы (a) в результате гидросилилирования.
Одним из более конкретных примеров способа получения силиконового (мет)акриламидного мономера, составляющего предмет настоящего изобретения, является способ с использованием мономеров, имеющих следующую формулу (a20).
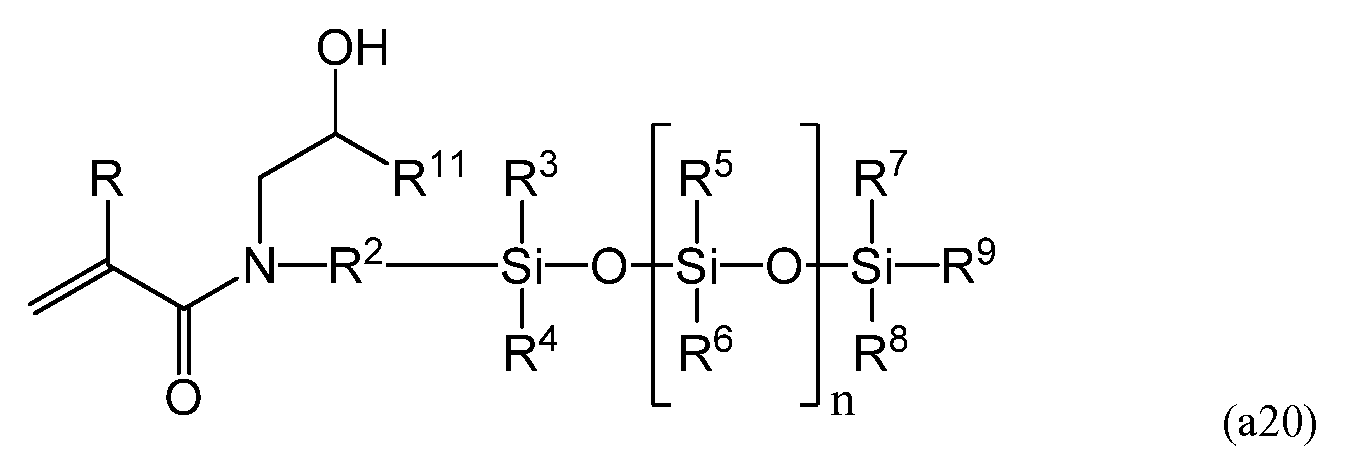
Способ получения содержит реакцию раскрытия цикла эпоксида при помощи амина, установку защиты на гидроксильную группу(ы), гидросилилирование, акрилирование и снятие защиты с гидроксильной группы (групп). В формуле (a20) R, R2, R3-R9 и n равносильны соответствующим элементам в представленной выше формуле (a), а -CH2CH(OH)R11 в формуле (a20) представляет собой R1 в формуле (a).
В процессе взаимодействия эпоксида, имеющего следующую формулу (a21), с амином, имеющим следующую формулу (a22), в результате реакции раскрытия цикла эпоксида при помощи амина на первой стадии можно получить соединение, имеющее формулу (a23).
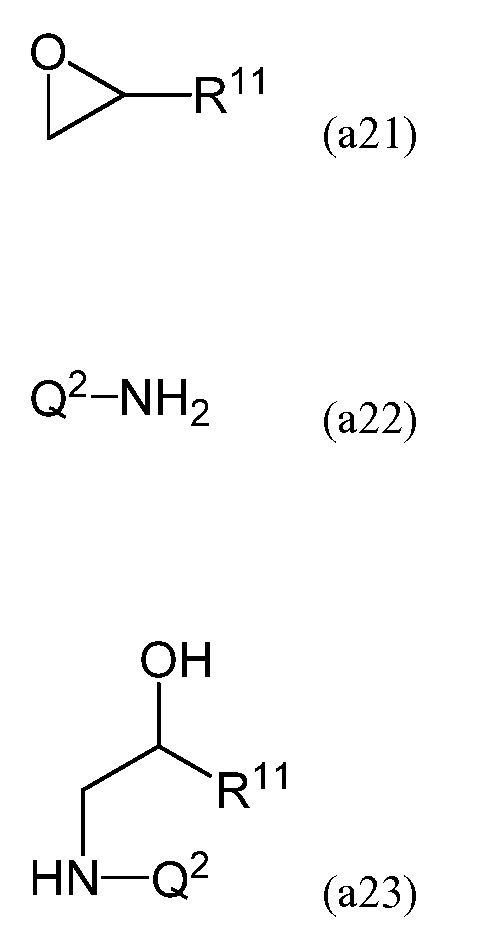
В формулах (a22) и (a23) Q2 содержит двойную углерод-углеродную связь и преобразуется в R2 в формуле (a20) в результате гидросилилирования двойной связи.
Для снижения количества побочных продуктов реакции - двузамещенного и трехзамещенного амина - соотношение компонентов реакции (a22)/(a21) предпочтительно составляет от приблизительно 1 до приблизительно 50, более предпочтительно - от приблизительно 1,5 до приблизительно 30, а наиболее предпочтительно - от приблизительно 2 до приблизительно 15. Во избежание испарения аминов температура реакции составляет предпочтительно от приблизительно -20°C до приблизительно 100°C, более предпочтительно - от приблизительно 0°C до приблизительно 50°C, а наиболее предпочтительно - от приблизительно 20°C до приблизительно 40°C. Время реакции составляет от приблизительно 1 часа до приблизительно 72 часов.
Во избежание побочной реакции гидросилилирования на второй стадии защищают гидроксильную группу(ы) полученного на первой стадии соединения формулы (a23). Одним допустимым примером защитной группы является триметилсилильная группа, и такую защиту можно установить и снять в неагрессивных условиях. Можно использовать любые известные реагенты для триметилсилилирования гидроксильной группы, такие как хлорид триметилсилила, трифторметансульфонат триметилсилила, N-метил-N-триметилсилилацетамид и гексаметилдисилазан. Среди данных соединений гексаметилдисилазан является предпочтительным, поскольку после реакции не остается соли. Температура реакции триметилсилилирования с гексаметилдисилазаном составляет от приблизительно 40°C до приблизительно 150°C, более предпочтительно - от приблизительно 50°C до приблизительно 130°C, а наиболее предпочтительно - от приблизительно 70°C до 120°C, а время проведения реакции составляет от приблизительно 0,5 часов до приблизительно 5 часов. Перед переходом к следующей стадии предпочтительно проводят очистку методом вакуумной перегонки.
На третьей стадии соединение, имеющее следующую формулу (a24)
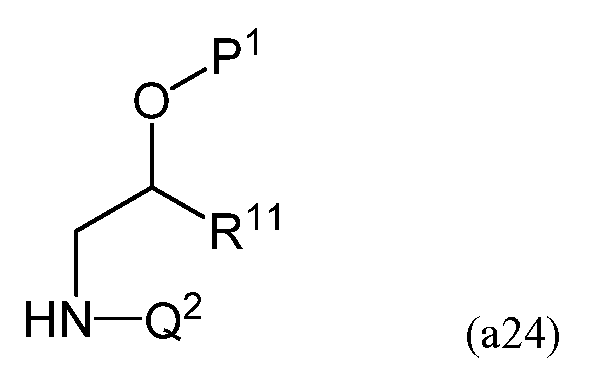
в которой P1 представляет собой защитную группу, и полученное на второй стадии, взаимодействует с линейным силоксаном, имеющим следующую формулу (a25)
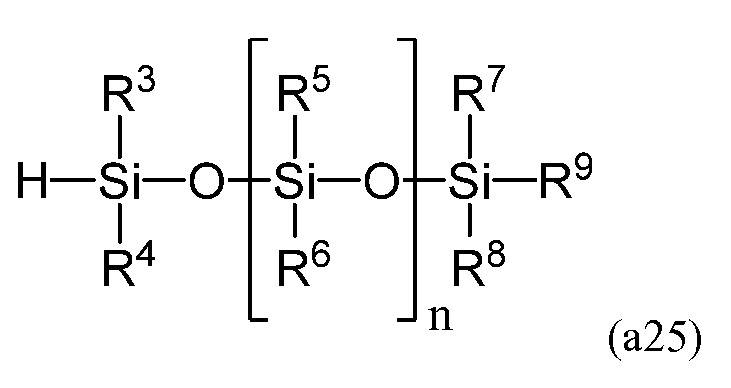
в которой R3-R9 и n равносильны соответствующим элементам формулы (a). Соотношение (a25)/(a24) составляет от приблизительно 0,5 до приблизительно 2,0, более предпочтительно - от приблизительно 0,8 до приблизительно 1,2, а наиболее предпочтительно - от 0,9 до 1,1. К допустимым катализаторам относятся 2,4,6,8-тетраметил-2,4,6,8-тетравинилциклотетрасилоксан платины (0), 1,3-диметил-1,3-дивинилдисилоксан платины (0), гексахлорплатинат водорода (IV), а более предпочтительно - 2,4,6,8-тетраметил-2,4,6,8-тетравинилциклотетрасилоксан платины (0), 1,3-диметил-1,3-дивинилдисилоксан платины (0). Катализатор используется в количестве от приблизительно 1 м.д. до приблизительно 10 000 м.д., более предпочтительно - от приблизительно 5 м.д. до приблизительно 5 000 м.д., а наиболее предпочтительно - от приблизительно 10 м.д. до приблизительно 1 000 м.д. Температура реакции предпочтительно составляет от приблизительно -20°C до приблизительно 180°C, более предпочтительно - от приблизительно -10°C до приблизительно 150°C, а наиболее предпочтительно - от приблизительно 0°C до приблизительно 130°C.
На четвертой стадии аминогруппа соединения, имеющего следующую формулу (a26)
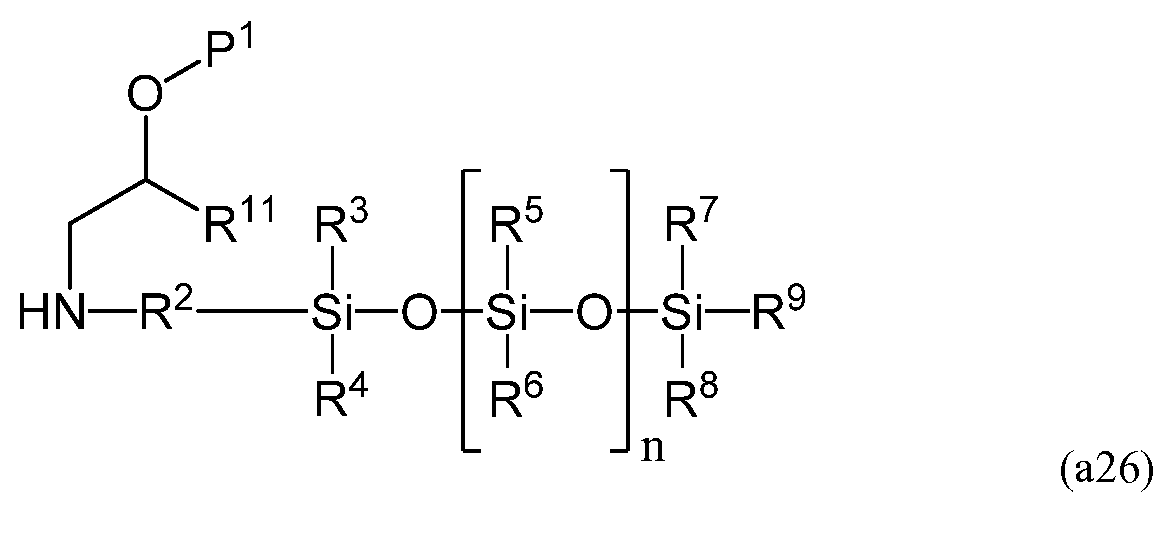
полученного на третьей стадии, может взаимодействовать с акрилирующим веществом, таким как (мет)акрилоилгалид и (мет)акриловый ангидрид. При использовании (мет)акрилоилгалида температура реакции предпочтительно составляет от приблизительно -80°C до приблизительно 40°C, более предпочтительно - от приблизительно -40°C до приблизительно 30°C, а наиболее предпочтительно - от приблизительно -20°C до приблизительно 10°C. Во избежание полимеризации во время синтеза мономеров можно добавлять ингибитор в количестве от 1 м.д. до 5 000 м.д., более предпочтительно - от 10 м.д. до 1 000 м.д., наиболее предпочтительно - от 50 м.д. до 500 м.д. К допустимым ингибиторам относятся монометиловый эфир гидрохинона, бутилированный гидрокситолуол, их смесь и т.п. В результате проведения данной стадии получают мономер, имеющий следующую формулу (a27).
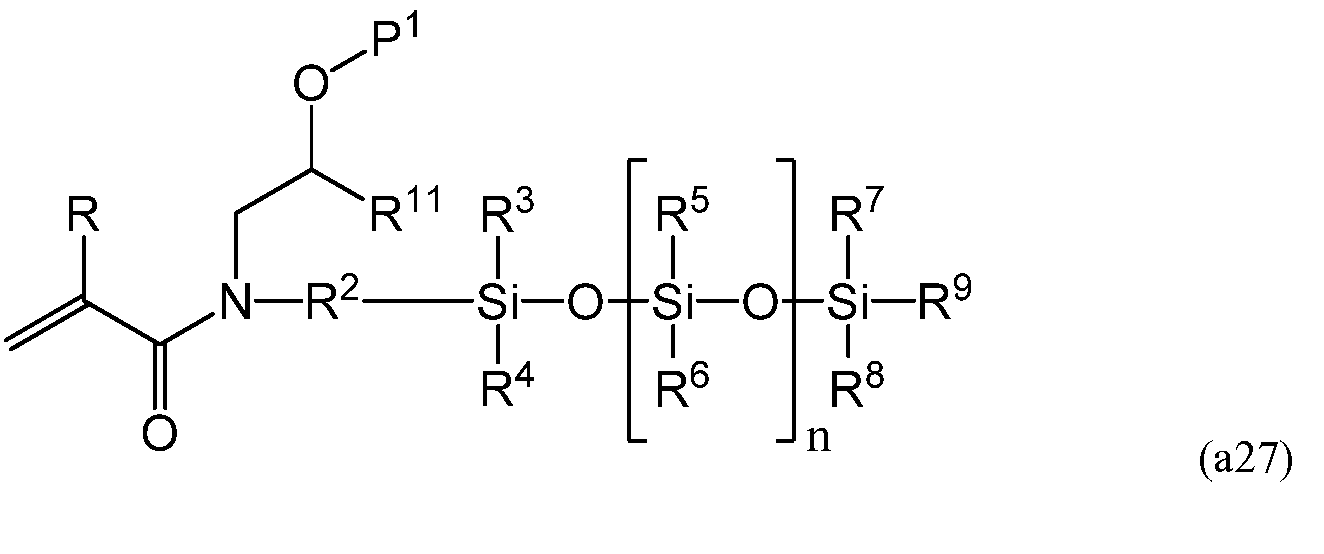
Мономер (a27) предпочтителен с точки зрения совместимости с широким диапазоном гидрофобных и гидрофильных сомономеров. После полимеризации с мономера (a27) можно снять защиту, при этом достигается необходимая совместимость в смеси мономеров, а также необходимая прозрачность полимера, обусловленная присутствием гидроксильных групп.
На пятой стадии снимают защиту с гидроксильной группы (групп) соединения, имеющего формулу (a27). Способ и условия снятия защиты зависят от P1. Если P1 представляет собой триметилсилильную группу, с точки зрения неагрессивных условий реакции предпочтительно снятие защиты при помощи уксусной кислоты в метаноле. Температура реакции предпочтительно составляет от приблизительно 20°C до приблизительно 40°C, а время проведения реакции предпочтительно составляет от приблизительно 0,5 часа до приблизительно 3 часов. После снятия защиты получаемый силиконовый (мет)акриламидный мономер можно очистить любым способом, включая колоночную хроматографию, вакуумную перегонку, молекулярную перегонку, обработку ионообменной смолой и их комбинацию. В результате проведения данной стадии получают силиконовый (мет)акриламидный мономер, имеющий формулу (a20).
Другим более конкретным примером способа получения силиконового (мет)акриламидного мономера, составляющего предмет настоящего изобретения, является способ с использованием мономеров, имеющих следующую формулу (a30)
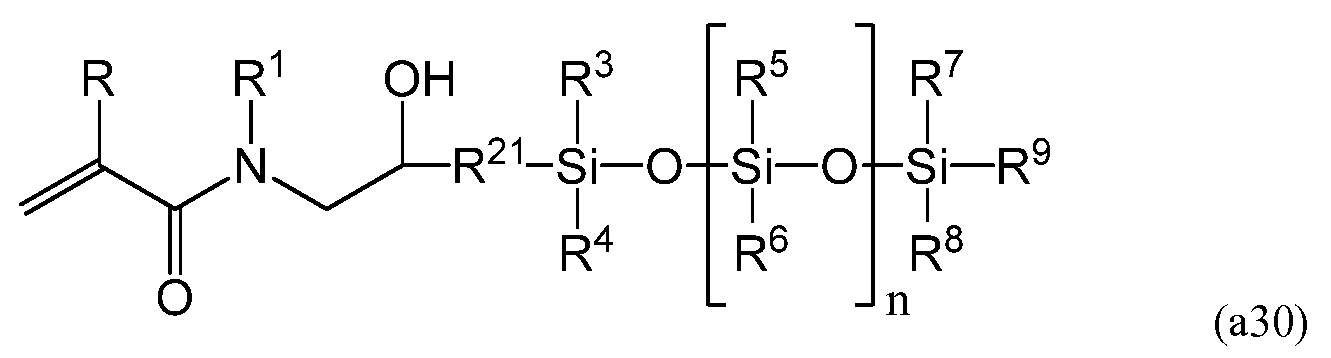
Способ получения содержит реакцию раскрытия цикла эпоксида при помощи амина, установку защиты на гидроксильную группу(ы), гидросилилирование, акрилирование и снятие защиты с гидроксильной группы (групп). В формуле (a30) R, R1, R3-R9 равносильны соответствующим элементам в представленной выше формуле (a), а -CH2CH(OH)R21- в формуле (a30) представляет собой R1 в формуле (a).
В процессе взаимодействия эпоксида, имеющего следующую формулу (a31), с амином, имеющим следующую формулу (a32), в результате реакции раскрытия цикла эпоксида при помощи амина на первой стадии можно получить соединение, имеющее формулу (a33).
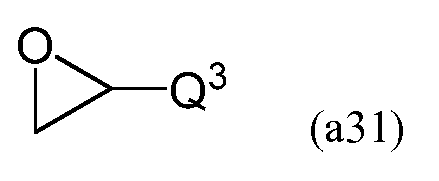
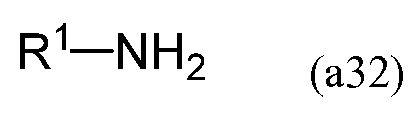
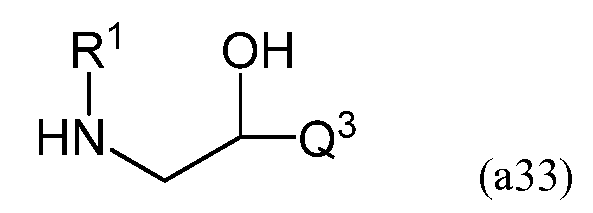
В формулах (a31) и (a33) Q3 содержит двойную углерод-углеродную связь и преобразуется в R2 в формуле (a30) в результате гидросилилирования двойной связи.
Для снижения количества побочных продуктов реакции - двузамещенного и трехзамещенного амина - соотношение компонентов реакции (a32)/(a31) предпочтительно составляет от приблизительно 1 до приблизительно 50, более предпочтительно - от приблизительно 1,5 до приблизительно 30, а наиболее предпочтительно - от приблизительно 2 до приблизительно 15. Во избежание испарения аминов температура реакции составляет предпочтительно от приблизительно -20°C до приблизительно 100°C, более предпочтительно - от приблизительно 0°C до приблизительно 50°C, а наиболее предпочтительно - от приблизительно 20°C до приблизительно 40°C. Время реакции составляет от приблизительно 1 часа до приблизительно 72 часов.
Во избежание побочной реакции гидросилилирования на второй стадии защищают гидроксильную группу(ы) полученного на первой стадии соединения формулы (a33). Одним допустимым примером защитной группы является триметилсилильная группа, и такую защиту можно установить и снять в неагрессивных условиях. Можно использовать любые известные реагенты для триметилсилилирования гидроксильной группы, такие как хлорид триметилсилила, трифторметансульфонат триметилсилила, N-метил-N-триметилсилилацетамид и гексаметилдисилазан. Среди данных соединений гексаметилдисилазан является предпочтительным, поскольку после реакции не остается соли. Температура реакции триметилсилилирования с гексаметилдисилазаном составляет от приблизительно 40°C до приблизительно 150°C, более предпочтительно - от приблизительно 50°C до приблизительно 130°C, а наиболее предпочтительно - от приблизительно 70°C до 120°C, а время проведения реакции составляет от приблизительно 0,5 часов до приблизительно 5 часов. Перед переходом к следующей стадии предпочтительно проводят очистку методом вакуумной перегонки.
На третьей стадии соединение, имеющее следующую формулу (a24)
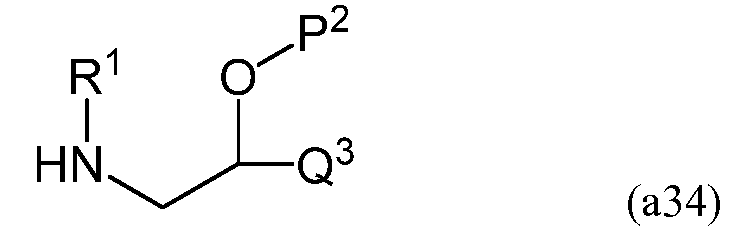
в которой P2 представляет собой защитную группу, и полученное на второй стадии, взаимодействует с линейным силоксаном, имеющим следующую формулу (a35)
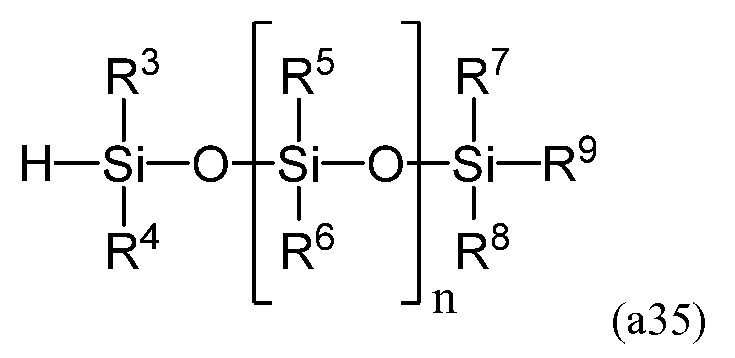
в которой R3-R9 и n равносильны соответствующим элементам формулы (a). Соотношение (a35)/(a34) составляет от приблизительно 0,5 до приблизительно 2,0, более предпочтительно - от приблизительно 0,8 до приблизительно 1,2, а наиболее предпочтительно - от 0,9 до 1,1. К допустимым катализаторам относятся 2,4,6,8-тетраметил-2,4,6,8-тетравинилциклотетрасилоксан платины (0), 1,3-диметил-1,3-дивинилдисилоксан платины (0), гексахлорплатинат водорода (IV), а более предпочтительно - 2,4,6,8-тетраметил-2,4,6,8-тетравинилциклотетрасилоксан платины (0), 1,3-диметил-1,3-дивинилдисилоксан платины (0). Катализатор используется в количестве от приблизительно 1 м.д. до приблизительно 10 000 м.д., более предпочтительно - от приблизительно 5 м.д. до приблизительно 5 000 м.д., а наиболее предпочтительно - от приблизительно 10 м.д. до приблизительно 1 000 м.д. Температура реакции предпочтительно составляет от приблизительно -20°C до приблизительно 180°C, более предпочтительно - от приблизительно -10°C до приблизительно 150°C, а наиболее предпочтительно - от приблизительно 0°C до приблизительно 130°C.
На четвертой стадии аминогруппа соединения, имеющего следующую формулу (a36)
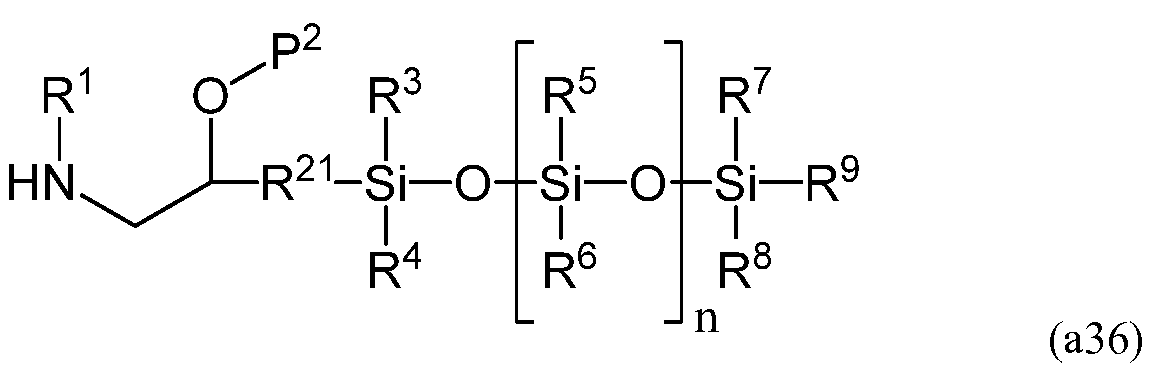
полученного на третьей стадии, может взаимодействовать с акрилирующим веществом, таким как (мет)акрилоилгалид и (мет)акриловый ангидрид. При использовании (мет)акрилоилгалида температура реакции предпочтительно составляет от приблизительно -80°C до приблизительно 40°C, более предпочтительно - от приблизительно -40°C до приблизительно 30°C, а наиболее предпочтительно - от приблизительно -20°C до приблизительно 10°C. Во избежание полимеризации во время синтеза мономеров можно добавлять ингибитор в количестве от 1 м.д. до 5 000 м.д., более предпочтительно - от 10 м.д. до 1 000 м.д., наиболее предпочтительно - от 50 м.д. до 500 м.д. К допустимым ингибиторам относятся монометиловый эфир гидрохинона, бутилированный гидрокситолуол, их смесь и т.п. В результате проведения данной стадии получают мономер, имеющий следующую формулу (a37)
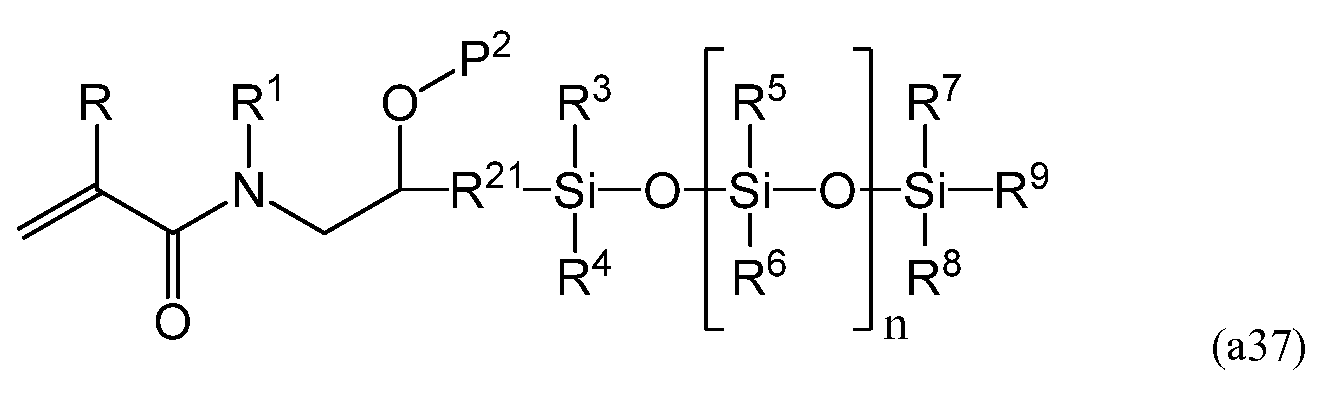
Мономер, имеющий формулу (a37), предпочтителен с точки зрения совместимости с широким диапазоном гидрофобных и гидрофильных сомономеров. После полимеризации с мономера (a37) можно снять защиту, при этом достигается необходимая совместимость в смеси мономеров, а также необходимая прозрачность полимера, обусловленная присутствием гидроксильных групп.
На пятой стадии снимают защиту с гидроксильной группы (групп) соединения, имеющего формулу (a37). Способ и условия снятия защиты зависят от P2. Если P2 представляет собой триметилсилильную группу, с точки зрения неагрессивных условий реакции предпочтительно снятие защиты при помощи уксусной кислоты в метаноле. Температура реакции предпочтительно составляет от приблизительно 20°C до приблизительно 40°C, а время проведения реакции предпочтительно составляет от приблизительно 0,5 часа до приблизительно 3 часов. После снятия защиты получаемый силиконовый (мет)акриламидный мономер можно очистить любым способом, включая колоночную хроматографию, вакуумную перегонку, молекулярную перегонку, обработку ионообменной смолой и их комбинацию. В результате проведения данной стадии получают силиконовый (мет)акриламидный мономер, имеющий формулу (a30).
Полимер, составляющий предмет настоящего изобретения, получают в ходе полимеризации базовой рецептуры, содержащей силиконовый (мет)акриламидный мономер. Если диапазон содержания силиконового (мет)акриламидного мономера включает в себя слишком низкие значения, кислородная проницаемость полимера может быть недостаточной, но если диапазон включает в себя слишком высокие значения, недостаточной может быть гидрофильность, поэтому соотношение мономерного и полимерного компонентов в мономерной смеси составляет от 30 до 98% вес., в некоторых вариантах осуществления - от приблизительно 40 до приблизительно 80% вес., в большинстве случаев в других вариантах осуществления - от приблизительно 50 до приблизительно 70% вес. Значения нижнего предела включают в себя приблизительно 30% вес., приблизительно 40% вес. и приблизительно 50% вес. Значения верхнего предела включают в себя приблизительно 98% вес., приблизительно 80% вес. и приблизительно 70% вес. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела.
Полимер, составляющий предмет настоящего изобретения, предпочтительно получают путем сополимеризации по меньшей мере одного гидрофильного мономера с по меньшей мере одним силиконовым (мет)акриламидным мономером. Примеры гидрофильного мономера включают в себя (мет)акрилатный мономер, такой как 2-гидроксиэтил(мет)акрилат, 2-(2-гидроксиэтокси)этил(мет)акрилат, глицерил(мет)акрилат и поли(этиленгликоль)моно(мет)акрилат, полимеризующийся мономер карбоновой кислоты, такой как (мет)акриловая кислота, итаконовая кислота, кротоновая кислота, винилбензойная кислота, N-виниламидный мономер, такой как N-винилпирролидон, N-винилформамид, N-винилацетамид и N-винил-N-метилацетамид, а также (мет)акриламидный мономер, такой как (мет)акриламид, N,N-диметил(мет)акриламид, N,N-диэтил(мет)акриламид, N-изопропил(мет)акриламид, (мет)акрилоилморфолин, N-метоксиметил(мет)акриламид, N-гидроксиметилакриламид и т.п. Среди данных гидрофильных мономеров в некоторых вариантах осуществления предпочтительно используется (мет)акриламидный мономер, поскольку он позволяет повысить скорость полимеризации всей системы и достичь благоприятных характеристик сополимеризации с силиконовым (мет)акриламидным мономером. С точки зрения скорости полимеризации и баланса между гидрофобностью и совместимостью с силиконовым мономером из (мет)акриламидных мономеров предпочтителен N,N-диметилакриламид.
Если диапазон количества используемого гидрофильного мономера включает в себя слишком высокие значения, кислородная проницаемость полимера может быть снижена, но, если диапазон включает в себя слишком низкие значения, силикон-гидрогель может быть слишком твердым, следовательно, содержание гидрофильного мономера составляет от приблизительно 1 до приблизительно 50% вес., в некоторых вариантах осуществления - от 10 до 40% вес., а в других вариантах осуществления - от приблизительно 15 до приблизительно 35% вес. от содержания мономерного и полимерного компонентов в мономерной смеси. Значения нижнего предела включают в себя приблизительно 1% вес., приблизительно 10% вес. и приблизительно 15% вес. Значения верхнего предела включают в себя приблизительно 50% вес., приблизительно 40% вес. и приблизительно 35% вес. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела.
В одном варианте осуществления в составе молекулы полимера, составляющего предмет настоящего изобретения, независимо от вышеупомянутого гидрофильного мономера или в дополнение к вышеупомянутому гидрофильному мономеру можно использовать гидрофильный (мет)акриламидный мономер с двумя или более гидроксильными группами. В частности, при использовании описанных ниже гидрофильных полимеров прозрачность полимера может беспрепятственно снижаться, поэтому предпочтительно использование гидрофильного (мет)акриламидного мономера. Количество гидрофильного (мет)акриламидного мономера при его использовании в составе молекулы составляет от приблизительно 1 до приблизительно 50% вес., более предпочтительно - от приблизительно 1 до приблизительно 30% вес., а наиболее предпочтительно - от приблизительно 1 до приблизительно 15% вес. от содержания мономерного и полимерного компонентов в мономерной смеси. Пример гидрофильного (мет)акриламидного мономера, содержащего две или более гидроксильные группы в составе молекулы, включает в себя мономеры, имеющие следующие общие формулы (c1)-(c3).
[ФОРМУЛА 5]
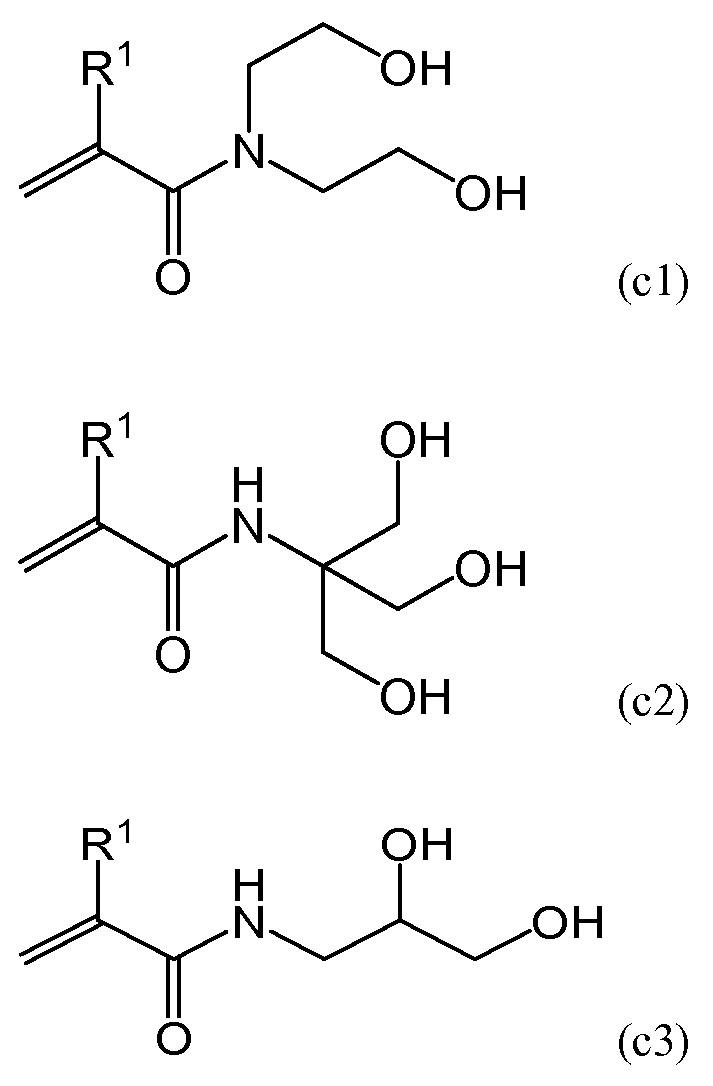
В формулах (c1)-(c3) R1 независимо представляет собой атом водорода или метильную группу. В одном варианте осуществления атомы водорода предпочтительны с точки зрения повышения скорости полимеризации. Более того, с точки зрения повышения прозрачности получаемого полимера, из данных мономеров предпочтительны мономеры, имеющие формулу (c1).
В другом варианте осуществления в составе полимера, составляющего предмет настоящего изобретения, независимо от вышеупомянутого гидрофильного мономера можно использовать гидрофильный N-(моногидроксилзамещенный C1-C20-алкил)метакриламидный или N-(моногидроксилзамещенный C6-C20-арил)метакриламидный мономер. Предпочтительно один из данных мономеров может использоваться в составе полимера в дополнение к вышеупомянутому гидрофильному мономеру. В одном варианте осуществления при использовании описанных ниже гидрофильных полимеров прозрачность полимера может беспрепятственно снижаться, поэтому используется гидрофильный метакриламидный мономер. Количество гидрофильного метакриламидного мономера составляет от приблизительно 1 до приблизительно 50% вес., в одном варианте осуществления - от приблизительно 1 до приблизительно 30% вес., а в другом варианте осуществления - от приблизительно 1 до приблизительно 15% вес. от содержания мономерного и полимерного компонентов в мономерной смеси. Пример гидрофильного метакриламидного мономера включает в себя N-гидроксиметилметакриламид, N-(2-гидроксиэтил)метакриламид, N-(2-гидроксипропил)метакриламид, N-(3-гидроксипропил)метакриламид, N-(2-гидроксибутил)метакриламид, N-(3-гидроксибутил)метакриламид, N-(4-гидроксибутил)метакриламид, N-(2-гидроксиметилфенил)метакриламид, N-(3-гидроксиметилфенил)метакриламид, N-(4-гидроксиметилфенил)метакриламид и т.п. Данные алкильные и арильные группы могут иметь линейную или разветвленную структуру. С точки зрения повышения прозрачности получаемого полимера, из данных мономеров предпочтителен N-(2-гидроксиэтил)метакриламидный мономер.
В одном варианте осуществления для повышения смачиваемости, устойчивости к адгезии белков, устойчивости к адгезии липидов и комбинаций данных характеристик мономерная смесь для получения полимера, составляющего предмет настоящего изобретения, дополнительно содержит в мономерном и полимерном компонентах мономерной смеси от приблизительно 1 до приблизительно 30% гидрофильного полимера с молекулярной массой приблизительно 1 000 или более.
Примеры гидрофильных полимеров, используемых в полимере, оставляющем предмет настоящего изобретения, включают в себя поли-N-винилпирролидон, поли-N-винил-2-пиперидон, поли-N-винил-2-капролактам, поли-N-винил-3-метил-2-капролактам, поли-N-винил-3-метил-2-пиперидон, поли-N-винил-4-метил-2-пиперидон, поли-N-винил-4-метил-2-капролактам, поли-N-винил-3-этил-2-пирролидон, поли-N-винил-4,5-диметил-2-пирролидон, поливинилимидазол, поли-N-винилформамид, поли-N-винилацетамид, поли-N-метил-N-винилацетамид, поли-N,N-диметилакриламид, поли-N,N-диэтилакриламид, поли-N-изопропилакриламид, поливиниловый спирт, полиакрилат, полиэтиленоксид, поли-2-этилоксазолин, гепарин, полисахариды, полиакрилоилморфолин, а также их смеси и сополимеры. Для повышения смачиваемости определенных силикон-гидрогелей особенно эффективными могут быть гидрофильные полимеры, выбранные из поливинилпирролидона, поли-N,N-диметилакриламида, полиакриловой кислоты, поливинилового спирта, а также их смесей и сополимеров. В определенных рецептурах поливинилпирролидон и поли-N,N-диметилакриламид обеспечивают баланс между смачиваемостью и совместимостью с полимеризационной смесью. Примеры допустимых смачивающих веществ описаны в публикациях US2006-0072069A1, US6367929 и US-2008-0045612A1.
Если количество гидрофильного полимера, используемого в составе силикон-гидрогеля, относящегося к настоящему изобретению, является слишком низким, необходимый уровень смачиваемости может быть не достигнут, но если его количество является слишком высоким, то растворение гидрофильного полимера в базовой жидкости для полимеризации может быть затруднено, следовательно, содержание гидрофильного полимера составляет от приблизительно 1 до приблизительно 30% вес., от приблизительно 2 до приблизительно 25% вес. и от приблизительно 3 до приблизительно 20% вес. от содержания мономерного и полимерного компонентов в полимеризационной смеси. Значения нижнего предела включают в себя приблизительно 1% вес., приблизительно 2% вес., предпочтительно - приблизительно 3% вес. и приблизительно 6% вес. Значения верхнего предела включают в себя приблизительно 30% вес., приблизительно 25% вес., приблизительно 20% вес., приблизительно 9% вес. Допускается объединение любых значений нижнего предела и любых значений верхнего предела.
Если диапазон значений молекулярной массы гидрофильного полимера, используемого в силикон-гидрогеле, составляющем предмет настоящего изобретения, включает слишком низкие значения, обеспечение необходимого уровня смачиваемости может быть невозможным, но если он будет включать слишком высокие значения, может понизиться растворимость в полимеризационной смеси и повыситься вязкость полимеризационной смеси. В одном варианте осуществления молекулярная масса составляет от приблизительно 1 000 дальтон до приблизительно 10 миллионов дальтон, в других вариантах осуществления - от приблизительно 100 000 дальтон до приблизительно 1 миллиона дальтон, а в других вариантах осуществления - от приблизительно 200 000 дальтон до приблизительно 800 000 дальтон. В вариантах осуществления, в которых гидрофильный полимер содержит по меньшей мере одну реакционноспособную группу, способную к образованию ковалентной связи с матрицей силикон-гидрогеля, молекулярная масса может составлять по меньшей мере приблизительно 2 000 дальтон, по меньшей мере приблизительно 5 000 дальтон; а в некоторых вариантах осуществления - от приблизительно 5 000 до приблизительно 180 000 дальтон или от приблизительно 5 000 дальтон до приблизительно 150 000 дальтон. Значения нижнего предела включают в себя приблизительно 1 000 дальтон, приблизительно 100 000 дальтон и приблизительно 200 000 дальтон. Значения верхнего предела включают в себя приблизительно 10 миллионов дальтон, приблизительно 1 миллион и приблизительно 800 000 дальтон. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела. Молекулярная масса гидрофильного полимера, составляющего предмет настоящего изобретения, выражается средневзвешенным значением молекулярной массы (Mw), измеренным при помощи гель-проникающей хроматографии (колонка: TSK gel GMPWXL производства компании Tosoh Corporation, подвижная фаза: вода/метанол = 50/50, с добавлением 0,1 Н нитрата лития, скорость потока: 0,5 мл/мин, детектор: дифференциальный рефрактометрический детектор, стандартный образец для определения молекулярной массы: полиэтиленгликоль).
Что касается содержания мономерных компонентов, используемых для полимеризации при получении полимера, составляющего предмет настоящего изобретения, если содержание акриламидного мономера слишком низкое, общая скорость полимеризации снижается, поэтому количество всех акриламидных мономеров в некоторых вариантах осуществления составляет приблизительно 90% вес. или более, приблизительно 95% вес. или более, а в некоторых вариантах осуществления - приблизительно 99% вес. или более.
Полимер, составляющий предмет настоящего изобретения, может включать в себя в качестве компонента сополимеризации мономер с двумя или более реакционноспособными группами. В данном случае полимер, составляющий предмет настоящего изобретения, приобретает устойчивость к воздействию растворителей. Предпочтительные примеры мономеров с двумя или более полимерными группами включают в себя бифункциональные и полифункциональные акрилаты, такие как этиленгликоль(мет)акрилат, диэтиленгликольди(мет)акрилат, триэтиленгликольди(мет)акрилат, неопентилгликольди(мет)акрилат, тетраэтиленгликольди(мет)акрилат, глицерилтри(мет)акрилат, пентаэритритолтетра(мет)акрилат и триметилолпропантри(мет)акрилат, а также бисакриламиды, такие как N,N'-метиленбисакриламид, N,N'-этиленбисакриламид, N,N'-пропиленбисакриламид и т.п. Из данных соединений с точки зрения повышенной скорости полимеризации предпочтительны бисакриламиды, а из бисакриламидов предпочтительны N,N'-метиленбисакриламид и N,N'-этиленбисакриламид. Диапазон количества используемого мономера, содержащего две или более полимерные группы, составляет от приблизительно 0,1 до приблизительно 10% вес., в некоторых вариантах осуществления - от приблизительно 0,5 до приблизительно 8% вес. и от приблизительно 0,8 до приблизительно 5% вес., поскольку это позволяет получить подходящую форму линзы. Значения нижнего предела включают в себя приблизительно 0,1% вес., приблизительно 0,5% вес. и приблизительно 0,8% вес. Значения верхнего предела включают в себя приблизительно 10% вес., приблизительно 8% вес. и приблизительно 5% вес. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела.
При получении полимера, составляющего предмет настоящего изобретения, путем полимеризации для ускорения процесса полимеризации можно также добавить в смесь инициатор полимеризации. Допустимые инициаторы включают в себя инициаторы термической полимеризации, такие как пероксидное соединение или азосоединение, или инициаторы фотополимеризации. В некоторых вариантах осуществления для ускорения процесса полимеризации добавляют фотоинициаторы. При использовании процесса термической полимеризации выбирают и используют инициатор термической полимеризации, обладающий оптимальными характеристиками распада при необходимой температуре реакции. Как правило, предпочтителен азоинициатор или пероксидный инициатор, характеризующийся 10-часовым периодом полураспада при температуре от приблизительно 40°C до приблизительно 120°C. Примеры инициаторов фотополимеризации включают в себя карбонильные соединения, пероксидные соединения, азосоединения, соединения серы, галогензамещенные соединения, соли металлов и т.п. Более конкретные примеры фотоинициаторов включают в себя ароматические альфа-гидроксикетоны, алкоксиоксибензоины, ацетофеноны, ацилфосфиноксиды, бисацилфосфиноксиды и третичный амин с дикетоном, их смеси и т.п. Показательными примерами фотоинициаторов являются 1-гидроксициклогексилфенилкетон, 2-гидрокси-2-метил-1-фенилпропан-1-он, бис(2,6-диметоксибензоил)-2,4-4-триметилпентилфосфиноксид (DMBAPO), бис(2,4,6-триметилбензоил)-фенилфосфиноксид (Irgacure 819), 2,4,6-триметилбензилдифенилфосфиноксид и 2,4,6-триметилбензоилдифенилфосфиноксид, бензоинметиловый эфир, а также комбинация камфорохинона и этил 4-(N,N-диметиламино)бензоата. К представленным в продаже инициаторам видимого света относятся Irgacure 819, Irgacure 1700, Irgacure 1800, Irgacure 819, Irgacure 1850 (все производства BASF) и инициатор Lucirin TPO (производства BASF). К представленным в продаже УФ-фотоинициаторам относятся Darocur 1173 и Darocur 2959 (BASF). Данные и другие фотоинициаторы, которые могут быть использованы в целях настоящего изобретения, описаны в справочнике Photoinitiators for Free Radical Cationic & Anionic Photopolymerization, том III, 2-е издание, J.V. Crivello и K. Dietliker; под редакцией G. Bradley; John Wiley and Sons; New York; 1998, содержание которого включено в настоящий документ путем ссылки. Данные инициаторы полимеризации можно использовать независимо или смешивать вместе в количестве приблизительно 1% вес. на 100% вес. мономерного компонента.
Другие компоненты, которые могут присутствовать в реакционной смеси, используемой для изготовления контактных линз, составляющих предмет настоящего изобретения, включают в себя соединения, поглощающие ультрафиолетовое излучение, лекарственные соединения, нутрицевтические соединения, противомикробные соединения, сополимеризуемые и неполимеризуемые красители, включая красители и соединения, обратимо изменяющие цвет или отражающие свет под воздействием света с различными длинами волны, антиадгезионные вещества, реакционноспособные оттеночные средства, пигменты, их комбинации и т.п.
При получении полимера, составляющего предмет настоящего изобретения, путем полимеризации можно использовать полимеризационный растворитель. В качестве растворителя можно использовать любой вид органического или неорганического растворителя. Растворители, подходящие для изготовления изделий данного изобретения, включают в себя простые эфиры, сложные эфиры, алканы, алкилгалиды, силаны и спирты. Примеры простых эфиров, подходящих для данного изобретения в качестве разбавителей, включают в себя тетрагидрофуран. Примеры сложных эфиров, подходящих для данного изобретения, включают в себя этилацетат. Примеры алкилгалидов, подходящих для данного изобретения в качестве разбавителей, включают в себя метиленхлорид. Примеры силанов, подходящих для данного изобретения в качестве разбавителей, включают в себя октаметилциклотетрасилоксан. Примеры спиртов, подходящих для данного изобретения в качестве разбавителей, включают в себя гексанол, гептанол, октанол, нонанол, деканол, трет-бутиловый спирт, 3-метил-3-пентанол, изопропанол и 3,7-диметил-3-октанол. Дополнительные разбавители, подходящие для данного изобретения, описаны в патенте США № 6020445, содержание которого включено в настоящий документ путем ссылки.
Примеры используемых разбавителей включают в себя воду, метанол, этанол, пропанол, 2-пропанол, бутанол, трет-бутанол, трет-амиловый спирт, 3,7-диметил-3-октанол, тетрагидролиналоол и другие спиртовые растворители; бензол, толуол, ксилол и другие виды ароматических углеводородных растворителей; гексан, гептан, октан, декан, петролейный эфир, керосин, лигроин, парафин и другие виды алифатических углеводородных растворителей; ацетон, метилэтилкетон, метилизобутилкетон и другие виды кетоновых растворителей; этилацетат, бутилацетат, метилбензоат, этиленгликольдиацетат и другие виды эфирных растворителей; диэтиловый эфир, тетрагидрофуран, диоксан, диалкиловый эфир этиленгликоля, диалкиловый эфир диэтиленгликоля, диалкиловый эфир триэтиленгликоля, диалкиловый эфир тетраэтиленгликоля, диалкиловый эфир полиэтиленгликоля, блок-сополимер полиэтиленгликоля и полипропиленгликоля, статистический сополимер полиэтиленгликоля и полипропиленгликоля и другие виды гликолевых эфирных растворителей. Растворители можно использовать по отдельности или в комбинациях. Из данных растворителей с точки зрения простоты удаления растворителей из получаемого полимера путем промывания водой предпочтительны спиртовые растворители и гликолевые эфирные растворители.
Полимер, составляющий предмет настоящего изобретения, можно использовать самостоятельно путем формования изделий необходимой формы, но также можно смешивать с другими веществами, а затем обрабатывать путем формования. Более того, на поверхность отформованных изделий предпочтительно нанести покрытие.
Области применения полимера, составляющего предмет настоящего изобретения, включают в себя изготовление офтальмологических линз, эндоскопов, катетеров, трубок для переливания крови, трубок для подачи газа, стентов, защитных оболочек, манжет, разъемов трубок, портов для забора проб, дренажных мешков, систем для переливания крови, материалов для закрытия ран, а также различных видов носителей лекарственных препаратов, но особенно подходящими являются контактные линзы, интраокулярные линзы, искусственная роговица, роговичные накладки (onlay) и вкладки (inlay), а наиболее подходящими являются контактные линзы.
Если полимер, составляющий предмет настоящего изобретения, обрабатывают формованием и используют для изготовления офтальмологической линзы, полимеризацию и формование осуществляют описанными ниже стандартными способами. Примеры включают в себя способ, при котором вначале в круглую заготовку или пластину отливают силикон-гидрогель, а затем осуществляют машинную обработку для придания материалу необходимой формы в процессе рассечения или т.п., способ полимеризации в форме, способ центробежного литья в формы и т.п.
В качестве одного примера ниже описывается способ изготовления офтальмологической линзы из полимера, составляющего предмет настоящего изобретения, методом полимеризации в форме.
Мономерную композицию впрыскивают в пространство между двумя половинками пресс-формы, имеющими форму линзы. Затем для формирования линзы проводят фотополимеризацию или термическую полимеризацию. Пресс-форму изготавливают из пластмассы, стекла, керамики, металла или т.п., но в случае фотополимеризации используют оптически прозрачное вещество и обычно используют пластмассу или стекло. При изготовлении полимера используют пространство, образованное двумя противолежащими половинками пресс-формы, в которое впрыскивают мономерную композицию. Затем пресс-форму, внутреннее пространство которой заполнено мономерной композицией, подвергают фотооблучению, например ультрафиолетом, видимым светом или их комбинацией, либо помещают в печь или баню и нагревают для полимеризации мономера. Также возможно использовать оба способа, проводя термическую полимеризацию после фотополимеризации или наоборот - проводя фотополимеризацию после термической полимеризации. В случае фотополимеризации, как правило, применяют кратковременное воздействие интенсивного излучения (обычно в течение 1 часа или меньше), испускаемого источником света, таким как ртутная лампа или люминесцентная лампа. Для сохранения оптической однородности и качества полимера, а также повышения воспроизводимости при проведении термической полимеризации предпочтительны условия, при которых температура постепенно повышается от близкой к комнатной до высоких значений от приблизительно 60°C до приблизительно 200°C в течение периода времени от нескольких часов до нескольких десятков часов.
Полимер, составляющий предмет настоящего изобретения, можно необязательно модифицировать различными способами. Если областью применения полимера является офтальмологическая линза и в его состав не входит гидрофильный полимер, модификацию можно осуществить для улучшения характеристик смачиваемости линзы.
К конкретным способам модификации относятся воздействие электромагнитным излучением (включая световое), плазменная обработка, парофазное осаждение, обработка химическим осаждением из паровой фазы, такая как напыление, термообработка, нанесение трансферного покрытия в форме, нанесение покрытий методом ассоциации зарядов, обработка основаниями, кислотная обработка, обработка другими допустимыми веществами для обработки поверхностей и их комбинации. Из данных способов модификации предпочтительны обработка основаниями и кислотная обработка благодаря своей простоте.
Примеры обработки основаниями или кислотной обработки включают в себя способ, при котором отформованное изделие вводят в контакт с основным или кислотным раствором, или способ, при котором отформованное изделие вводят в контакт с основным или кислотным газом. Более конкретные способы включают в себя, например, погружение отформованных изделий в основный или кислотный раствор, распыление основного или кислотного раствора либо основного или кислотного газа на отформованные изделия, нанесение основного или кислотного раствора на отформованное изделие при помощи лопатки, кисти или т.п., нанесение основного или кислотного раствора на отформованное изделие методом центрифугирования, нанесение покрытия погружением и т.п. Самым простым способом, обеспечивающим большой модификационный эффект, является способ погружения отформованного изделия в основный или кислотный раствор.
Температурный диапазон при погружении полимера в основный или кислотный раствор не ограничен особым образом, но обычно значения температуры находятся в диапазоне от приблизительно -50°C до приблизительно 300°C. С учетом простоты работы более предпочтителен температурный диапазон от приблизительно -10°C до приблизительно 150°C, а наиболее предпочтителен диапазон от приблизительно -5°C до приблизительно 60°C. Значение нижнего предела предпочтительно составляет приблизительно -50°C, более предпочтительно - приблизительно -10°C, а еще более предпочтительно - приблизительно 5°C. Значение верхнего предела предпочтительно составляет приблизительно 300°C, более предпочтительно - приблизительно 150°C, а еще более предпочтительно - приблизительно 60°C. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела.
Оптимальное время погружения полимера в основный или кислотный раствор варьируется в зависимости от температуры, но, как правило, предпочтительно составляет 100 часов или менее, более предпочтительно - 24 часа или менее, а наиболее предпочтительно - 12 часов или менее. Если время контакта слишком большое, это не только негативно отразится на удобстве работы и производительности, но также приведет к отрицательным последствиям, таким как снижение кислородной проницаемости и ухудшение механических характеристик линзы.
Примеры используемых оснований включают в себя гидроксиды щелочных металлов, гидроксиды щелочноземельных металлов, различные виды карбонатов, различные виды боратов, различные виды фосфатов, аммиак, различные аммонийные соли, различные амины, а также полимерные основания, такие как полиэтиленимин, поливиниламин и т.п. Из данных соединений наиболее предпочтительны гидроксиды щелочных металлов благодаря низкой стоимости и высоким результатам при обработке.
Примеры используемых кислот включают в себя различные виды неорганических кислот, такие как серная кислота, фосфорная кислота, соляная кислота и азотная кислота; различные виды органических кислот, такие как уксусная кислота, муравьиная кислота, бензойная кислота и фенол; а также различные виды полимерных кислот, такие как полиакриловая кислота, полистиролсульфоновая кислота и т.п. Из данных соединений наиболее предпочтительны полимерные кислоты благодаря высоким результатам при обработке и минимальному отрицательному воздействию на другие физические характеристики.
В качестве растворителя для приготовления основного или кислотного раствора можно использовать любой вид неорганических или органических растворителей. Примеры включают в себя воду, метанол, этанол, пропанол, 2-пропанол, бутанол, этиленгиколь, диэтиленгликоль, триэтиленгликоль, тетраэтиленгликоль, полиэтиленгликоль, глицерин и другие спирты, бензол, толуол, ксилол и другие ароматические углеводороды, гексан, гептан, октан, декан, петролейный эфир, керосин, лигроин, парафин и другие алифатические углеводороды, ацетон, метилэтилкетон, метилизобутилкетон и другие кетоны, этилацетат, бутилацетат, метилбензоат, диоктилфталат и другие сложные эфиры, диэтиловый эфир, тетрагидрофуран, диоксан, диалкиловый эфир этиленгликоля, диалкиловый эфир диэтиленгликоля, диалкиловый эфир триэтиленгликоля, диалкиловый эфир тетраэтиленгликоля, диалкиловый эфир полиэтиленгликоля и другие простые эфиры; диметилформамид, диметилацетамид, N-метил-2-пирролидон, диметилимидазолидинон, гексаметилфосфортриамид, диметилсульфоксид и другие полярные апротонные растворители, метиленхлорид, хлороформ, дихлорэтан, трихлорэтан, трихлорэтилен, другие галогенные растворители, фреоновые растворители и т.п. Из данных соединений наиболее предпочтительной с точки зрения экономии, простоты обращения, химической стабильности и т.п. является вода. В качестве растворителя также можно использовать смесь из двух или более видов растворителей.
Что касается настоящего изобретения, используемый основный или кислотный раствор может содержать другие компоненты помимо основания или кислоты и растворитель. Основание или кислоту можно удалять из полимера путем промывания после основной или кислотной обработки.
В качестве растворителя для промывания можно использовать любой вид неорганических или органических растворителей. Примеры включают в себя воду, метанол, этанол, пропанол, 2-пропанол, бутанол, этиленгиколь, диэтиленгликоль, триэтиленгликоль, тетраэтиленгликоль, полиэтиленгликоль, глицерин и другие спирты, бензол, толуол, ксилол и другие ароматические углеводороды, гексан, гептан, октан, декан, петролейный эфир, керосин, лигроин, парафин и другие алифатические углеводороды, ацетон, метилэтилкетон, метилизобутилкетон и другие кетоны, этилацетат, бутилацетат, метилбензоат, диоктилфталат и другие сложные эфиры, диэтиловый эфир, тетрагидрофуран, диоксан, диалкиловый эфир этиленгликоля, диалкиловый эфир диэтиленгликоля, диалкиловый эфир триэтиленгликоля, диалкиловый эфир тетраэтиленгликоля, диалкиловый эфир полиэтиленгликоля и другие простые эфиры; диметилформамид, диметилацетамид, N-метил-2-пирролидон, диметилимидазолидинон, гексаметилфосфортриамид, диметилсульфоксид и другие полярные апротонные растворители, метиленхлорид, хлороформ, дихлорэтан, трихлорэтан, трихлорэтилен, другие галогенные растворители и фреоновые растворители.
В качестве растворителя для промывания можно использовать смесь из двух или более видов растворителей. Растворитель для промывания может содержать другие компоненты помимо растворителя, такие как неорганические соли, поверхностно-активные вещества и моющие вещества.
Описанной выше модификационной обработке можно подвергать весь полимер или только часть полимера, например, только поверхность. При осуществлении модификаций только на поверхности возможно повысить только смачиваемость поверхности без значительного изменения физических характеристик всего полимера.
Если диапазон содержания воды в полимере, составляющем предмет настоящего изобретения, включает слишком низкие значения, силикон-гидрогель является твердым, но если содержание воды является слишком высоким, вода может испаряться с поверхности силикон-гидрогеля, вызывая у пользователя ощущение пересыхания линзы во время ее использования, поэтому содержание воды необходимо поддерживать в диапазоне от приблизительно 20 до приблизительно 50% вес., более предпочтительно - от приблизительно 25 до приблизительно 45% вес., а наиболее предпочтительно - от приблизительно 30 до приблизительно 40% вес. Значения нижнего предела включают в себя приблизительно 20% вес., приблизительно 25% вес. и приблизительно 30% вес. Значения верхнего предела включают в себя приблизительно 50% вес., приблизительно 45% вес. и приблизительно 40% вес. Допускается объединение любых предпочтительных значений нижнего предела и любых предпочтительных значений верхнего предела.
При использовании полимера, составляющего предмет настоящего изобретения, для производства офтальмологической линзы и, в частности, мягкой контактной линзы для достижения комфортного ощущения при ношении линз модуль упругости полимера составляет приблизительно 1 379 кПа (200 фунтов/кв. дюйм) или меньше, в некоторых вариантах осуществления - менее приблизительно 1 034 кПа (150 фунтов/кв. дюйм) или меньше, а в других вариантах осуществления - менее приблизительно 689 кПа (100 фунтов/кв. дюйм) или меньше.
Относительное удлинение полимера, составляющего предмет настоящего изобретения, составляет приблизительно 100% или более, в некоторых вариантах осуществления - приблизительно 150% или более, а в других вариантах осуществления - приблизительно 200% или более. Более высокие значения относительного удлинения означают, что силикон-гидрогель будет более устойчивым к разрыву. Значения модуля упругости и относительного удлинения полимера, составляющего предмет настоящего изобретения, определяют путем вырезания образца с шириной узкого участка 5 мм, а затем растягивания образца со скоростью 100 мм/мин при помощи динамометра.
Угол смачивания при натекании для полимера, составляющего предмет настоящего изобретения, составляет приблизительно 70 градусов или менее, приблизительно 60 градусов или менее, а в некоторых вариантах осуществления - приблизительно 50 градусов или менее, если областью применения полимера является офтальмологическая линза. Угол смачивания при натекании для полимера, составляющего предмет настоящего изобретения, определяют при помощи устройства для измерения динамического угла смачивания путем проведения измерений на образце в виде короткой полосы шириной 5 мм, вырезанной из образца в форме линзы, при скорости погружения 7 мм/мин.
Что касается кислородной проницаемости полимера, составляющего предмет настоящего изобретения, постоянная кислородной проницаемости предпочтительно равна 50×10-11 (см2/с) мл O2/(мл-гПа) или более, а в некоторых вариантах осуществления - 50×10-11 (см2/с) мл O2/(мл-гПа) или более. Значение постоянной кислородной проницаемости полимера, составляющего предмет настоящего изобретения, измеряют методом полярографии.
Что касается прозрачности полимера, составляющего предмет настоящего изобретения, значение полноспектрального светопропускания предпочтительно составляет приблизительно 85% или более, приблизительно 88% или более и приблизительно 91% или более, если областью применения полимера является офтальмологическая линза. Для получения значения полноспектрального светопропускания полимера, составляющего предмет настоящего изобретения, с образца в форме линзы толщиной 0,14-0,15 мм аккуратно вытирают воду, образец помещают в область воздействия луча света, испускаемого устройством для измерения светопропускания, а затем измеряют значение полноспектрального светопропускания.
Значение, описывающее характеристики восстановления формы полимера, составляющего предмет настоящего изобретения, - это время нулевой нагрузки, измеряемое описанным ниже способом. Меньшие значения времени нулевой нагрузки указывают на то, что характеристики восстановления формы силикон-гидрогеля являются благоприятными, и предпочтительным является значение, равное 1 секунде или менее, более предпочтительно - 0,95 секунды или менее, а наиболее предпочтительно - 0,9 секунды или менее. Измерение времени нулевой нагрузки для полимера, составляющего предмет настоящего изобретения, выполняют приведенным ниже способом. Из области возле центра линзы вырезали образец в форме полосы шириной 5 мм и длиной 1,5 см, после чего провели измерение при помощи устройства для измерения динамической вязкоупругости. Образец фиксировали зажимом при ширине 5 мм, и после растягивания на 5 мм со скоростью 100 мм/мин данный образец восстанавливал изначальную длину (5 мм) с такой же скоростью. Данный цикл повторяли 3 раза. Время нулевой нагрузки отсчитывали от момента, когда нагрузка становилась нулевой при частичном возврате образца к исходной длине во второй раз, и до момента применения нагрузки (нагрузка не равна нулю) после начала третьего цикла растяжения.
Полимер, получаемый путем полимеризации силиконового мономера, составляющего предмет настоящего изобретения, допустим для изготовления различных изделий медицинского назначения, таких как офтальмологические линзы, эндоскопы, катетеры, трубки для переливания крови, трубки для подачи газа, стенты, защитные оболочки, манжеты, разъемы трубок, порты для забора проб, дренажные мешки, системы для переливания крови, материалы для закрытия ран, а также различных видов носителей лекарственных препаратов, но в особенности допустим для изготовления контактных линз, интраокулярных линз и искусственных роговиц.
Ниже следует подробное описание настоящего изобретения на рабочих примерах, однако данное изобретение не ограничивается данными рабочими примерами.
Способ измерения
(1) Измерение методом ГХ
- Устройство
Shimadzu GC-18A (ПИД)
- Капиллярная колонка
Agilent HP-ULTRA2 (длина 25 м × внутренний диаметр 0,32 мм × толщина пленки 0,52 микрометра)
- Температурная программа
Температура инжектора: 300°C
Температура детектора: 320°C
Температура колонки: начальная температура 50°C (1 минута) → повышение температуры со скоростью 10°C/минуту → 300°C (поддерживается в течение 14 минут) (общее время 40 минут)
- Газ-носитель
Газообразный гелий (110 кПа)
- Приготовление образца
В качестве образца использовали 100 мкл реакционного раствора, разведенного 1 мл растворителя (толуола, 2-пропанола или этилацетата).
(2) Полноспектральное светопропускание
Полноспектральное светопропускание измеряли при помощи дифференциального колориметра SM-7-CH производство компании Suga Test Instruments Co. Ltd. С образца линзы аккуратно вытирали воду, а затем образец помещали в область воздействия луча света и проводили измерение. Толщину образца измеряли при помощи многооборотного индикатора ID-C112 производства компании Mitutoyo Corporation, при этом толщина образцов составляла 0,14-0,15 мм.
(3) Модуль упругости и относительное удлинение
Из образца линзы вырезали образец шириной 5 мм в самом узком участке, его толщину измеряли при помощи многооборотного индикатора ID-C112 производства компании Mitutoyo Corporation при скорости 100 мм/мин, а затем измеряли модуль упругости и относительное удлинение при помощи устройства Tensilon (модель RTM-100, производство компании Toyo Baldwin Co. Ltd., скорость движения ползуна 100 мм/мин).
(4) Содержание воды
Измеряли массу силикон-гидрогеля, содержащего воду (W1), и массу силикон-гидрогеля в сухом состоянии (W2), после чего вычисляли содержание воды при помощи приведенной ниже формулы.
Содержание воды (%)=(W1-W2)/W1×100
Тем не менее, что касается настоящего изобретения, под состоянием, когда силикон-гидрогель содержит воду, подразумевается состояние силикон-гидрогеля после погружения в физиологический раствор при 25°C на 6 часов или дольше. Более того, под сухим состоянием силикон-гидрогеля подразумевается состояние после его высушивания в вакуумной сушилке при 40°C в течение 16 часов или дольше.
(5) Угол смачивания при натекании
Из образца линзы вырезали образец в виде короткой полосы шириной 5 мм и измеряли динамический угол смачивания (при натекании (наступающий) и отступающий) (скорость погружения 7 мм/мин) при помощи устройства для измерения динамического угла смачивания WET-6000 производства компании Rhesca Corporation. После получения результатов измерения значение угла смачивания при натекании использовали в качестве показателя смачиваемости.
(6) Время нулевой нагрузки
Из области возле центра линзы вырезали образец в форме полосы шириной 5 мм и длиной 1,5 см и проводили измерения при помощи реометра CR-500DX производства компании Sun Scientific Co. Ltd. Образец фиксировали зажимом при ширине 5 мм, и после растягивания на 5 мм со скоростью 100 мм/мин данный образец восстанавливал изначальную длину (5 мм) с такой же скоростью. Данный цикл повторяли 3 раза. Время нулевой нагрузки отсчитывали от момента, когда нагрузка становилась нулевой при частичном возврате образца к исходной длине во второй раз, и до момента применения нагрузки (нагрузка не равна нулю) после начала третьего цикла растяжения.
(7) Фазовый переход, измеренный методом ДСК
Термический анализ отверждения реакционной мономерной смеси (RMM) проводили методом фотодифференциальной сканирующей калориметрии (фотоДСК). На предметном столике ДСК модели Q100 DSC производства компании TA Instruments в чашке взвешивали образец исследуемой RMM массой ~10 мг, а также определяли массу пустой эталонной чашки. Во время анализа камеру для образцов продували осушенным азотом (50 мл/мин). Образец нагревали до 70°C, для активации фотоинициатора приводили в действие источник света, светоизлучающий диод (~420 нм, 4,0 мВт/см2) и измеряли количество тепла, выделенного во время отверждения в изотермических условиях в течение 10 минут (энтальпия, Дж/г). Интегрированием площади под полученной на анализаторе ДСК зависимости энтальпии от времени получили полную энтальпию и рассчитали степень превращения в различные моменты времени.
Пример синтеза 1
В 4-горлую колбу объемом 500 мл, оснащенную механической мешалкой, капельной воронкой, обратным холодильником и стеклянной пробкой, помещали водный раствор аммиака концентрацией 28-30% вес. (320 мл) и метанол (48 мл). В колбу по каплям добавляли раствор аллилглицидилового эфира (59,6 г, 0,522 моль) в метаноле (48 мл) в течение приблизительно 9 часов, поддерживая температуру 4-горлой колбы на уровне приблизительно 25°C при помощи мешалки. Завершение реакции подтверждали методом ГХ.
После завершения реакции реакционный раствор концентрировали в испарителе. Полученную жидкость очищали путем перегонки при пониженном давлении (полный вакуум, 81°C). Выход составил 31,6 г (46,1%) при чистоте, измеренной методом ГХ, 99,0%.
Пример синтеза 2
В 4-горлую колбу объемом 200 мл помещали соединение, полученное в примере синтеза 1 (30,0 г, 0,229 моль), гексаметилдисилазан (22,2 г, 0,137 моль), бутилгидрокситолуол (0,09 г) и сахарин (0,09 г) и перемешивали в течение 2 часов при 100°C.
Реакцию завершали, убедившись в устранении пиков исходного вещества в результатах ГХ-анализа. После очистки перегонкой при пониженном давлении (полный вакуум, 72°C), выход составил 39,0 г (степень выхода 83,9%) при чистоте 99,4%, измеренной методом ГХ.
Пример синтеза 3
Соединение, полученное в примере синтеза 2 (38,0 г, 0,187 моль), 1-н-бутил-1,1,3,3,5,5,7,7,9,9-декаметилпентасилоксан, имеющее следующую формулу
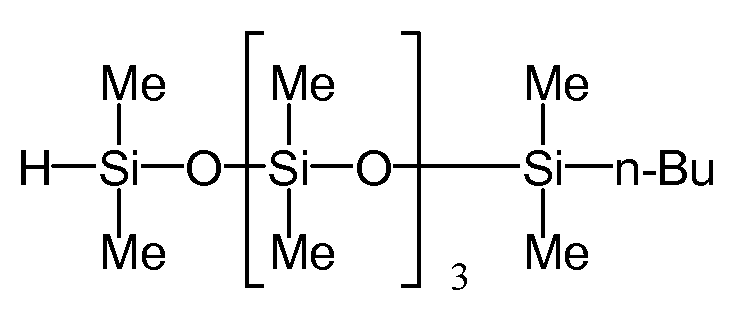
(далее обозначаемое «SiL5B») (77,2 г, 0,187 моль), раствор платины (0) 2,4,6,8-тетраметил-2,4,6,8-тетравинилциклотетрасилоксанового комплекса (производство компании Aldrich Corp., далее обозначаемое «раствор платинового комплекса») в метилвинилциклосилоксане концентрацией 0,104 M (610 мкл) и толуол (370 мкл) помещали в 4-горлую колбу объемом 200 мл, а затем нагревали до 120°C и перемешивали.
Через 2 часа добавляли дополнительно 610 мкл раствора платинового комплекса, поскольку по результатам ГХ-анализа выявили наличие исходного вещества, и реакцию продолжали в течение еще одного часа. Убедившись в устранении пиков исходного вещества в результатах ГХ-анализа, раствор охлаждали до комнатной температуры и концентрировали в испарителе. Полученную жидкость очищали перегонкой при пониженном давлении (8×10-2 Па, 156°C) при помощи масляного диффузионного насоса. Выход составил 65,5 г (56,9%) при чистоте, измеренной методом ГХ, 95,0%.
Пример синтеза 4
В 4-горлую колбу объемом 500 мл помещали соединение, полученное в примере синтеза 3 (64,0 г, 0,104 моль), триэтиламин (10,5 г, 0,104 моль) и гексан (170 мл), затем колбу помещали в ледяную баню (от -1 до 2°C) и в течение приблизительно 6 часов при помощи капельной воронки по каплям добавляли акрилоилхлорид (9,40 г, 0,104 моль) и гексан (130 мл). Через 40 минут после окончания добавления по каплям провели ГХ-анализ, подтвердивший почти полное расходование исходных веществ.
Через час после начала добавления по каплям реакционный раствор фильтровали и осадок отмывали гексаном, который был охлажден в холодильнике. Раствор для промывания объединяли с фильтратом, переносили в разделительную воронку и трижды промывали водой (300 мл), дважды - насыщенным водным раствором бикарбоната натрия (300 мл) и дважды - насыщенным водным раствором хлорида натрия (300 мл).
Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход полученного неочищенного вещества составил 58,7 г (84,3%).
Рабочий пример 1
В грушевидную колбу объемом 300 мл помещали неочищенный продукт, полученный в примере синтеза 4 (57,0 г, 0,085 моль), метанол (171 г) и уксусную кислоту (28,5 г) и перемешивали в течение приблизительно 1 часа при 40°C.
Методом тонкослойной хроматографии (ТСХ) подтверждали расходование исходных веществ, и после концентрации в испарителе добавляли 350 мл гексана и раствор переносили в разделительную воронку. Раствор по два раза промывали водой (250 мл), насыщенным водным раствором бикарбоната натрия (250 мл) и насыщенным водным раствором хлорида натрия (250 мл). Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход неочищенного вещества составил 50,83 г.
В колонке очищали 23,63 г неочищенного вещества. Для этого использовали силикагель массой приблизительно в 5 раз (120 г) больше массы неочищенного вещества. Элюирование выполняли методом ТСХ с использованием смеси гексана и этилацетата в соотношении 1/1 до исчезновения целевого пятна. Методом ГХ проанализировали несколько фракций, собранных перед началом выхода продукта и после окончания его выхода с колонки, и измерили и собрали те фракции, в которых все пики побочных продуктов составляли менее 1%. Добавляли 6,0 мг бутилгидрокситолуола и 2,0 мг монометилового эфира гидрохинона, раствор концентрировали, а затем перемешивали при 60°C с одновременным снижением давления при помощи вакуумного насоса в течение 1 дополнительного часа для удаления остатков растворителя. Было подтверждено, что в результатах ЯМР пик растворителя не наблюдался. Выход составил 20,14 г (85,2%) при чистоте, измеренной методом ГХ, 95,2%.
Пример синтеза 5
В 4-горлую колбу, оснащенную обратным холодильником, термометром, капельной воронкой и механической мешалкой, помещали аллиламин (112,4 мл, 85,54 г, 1,5 моль). В капельную воронку при перемешивании при 30°C по каплям добавляли глицидол (2,3-эпокси-1-пропанол, 37,8 г, 0,5 моль) в течение приблизительно 10 минут. Для контроля протекания реакции ГХ-анализ проводили каждый час, начиная с момента завершения добавления по каплям. Через 5 часов после завершения добавления по каплям было подтверждено, что пик глицидола составил 1% или менее, и реакцию прекращали. Раствор концентрировали в испарителе, а затем очищали при пониженном давлении (полный вакуум, точка кипения 52°C). Выход составил 27,43 г (41,8%) при чистоте, измеренной методом ГХ, 98,7%.
Пример синтеза 6
В 4-горлую колбу, оснащенную обратным холодильником, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 5 (23,2 г, 0,177 моль), гексаметилдисилазан (33,89 г, 0,210 моль), бутилгидрокситолуол (72 мг) и сахарин (70 мг). Реакцию осуществляли в течение 2 часов при 100°C и прекращали после того, как методом ГХ было подтверждено снижение концентрации или израсходование реагентов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (полный вакуум, точка кипения 77°C). Выход составил 36,31 г (70,2%) при чистоте, измеренной методом ГХ, 98,4%.
Пример синтеза 7
В 3-горлую колбу объемом 1 л, оснащенную обратным холодильником, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 6 (35,0 г, 0,127 моль), SiL5B (52,4 г, 0,127 моль), толуол (263 мл) и раствор платинового комплекса (350 мкл). Реакцию осуществляли при 120°C с проведением ГХ-анализа каждый час. Поскольку через 4 часа еще оставалось исходное вещество, добавляли дополнительно 350 мкл раствора платинового комплекса. Раствор нагревали и перемешивали в течение 4 дополнительных часов, а затем реакцию прекращали. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (8×10-2 Па, 176°C). Выход составил 58,22 г (66,6%) при чистоте, измеренной методом ГХ, 96,2%.
Пример синтеза 8
В 3-горлую колбу объемом 1 л, оснащенную капельной воронкой, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 7 (55 г, 0,080 моль), триэтиламин (8,08 г, 0,080 моль) и этилацетат (130 мл). В капельную воронку помещали акрилоилхлорид (7,23 г, 0,080 моль) и этилацетат (100 мл). 3-горлую колбу помещали в ледяную баню, содержащую соль, и после падения внутренней температуры до 0°C начинали добавление по каплям. Поддерживая внутреннюю температуру на уровне от -2 до 3°C, добавление по каплям осуществляли в течение приблизительно 3 часов и по завершении добавления по каплям реакцию продолжали в течение еще одного часа, а затем прекращали. Далее раствор фильтровали с одновременным промыванием небольшим количеством охлажденного этилацетата, который был охлажден в холодильнике. Фильтрат переносили в разделительную воронку и добавляли 400 мл гексана. Раствор дважды промывали водой (200 мл), дважды - насыщенным водным раствором бикарбоната натрия (200 мл) и дважды - насыщенным водным раствором хлорида натрия (200 мл). К органическому слою добавляли безводный сульфат натрия и оставляли высушиваться в течение ночи. После фильтрации данный раствор концентрировали в испарителе для получения неочищенного продукта. Выход неочищенного продукта составил 54,6 г при чистоте, измеренной методом ГХ, 88,5%.
Рабочий пример 2
На каждую часть неочищенного продукта, полученного в примере синтеза 8 (54,6 г, 0,074 моль), добавляли 3 части (по массе) метанола (163,8 г) и половину части (по массе) уксусной кислоты (27,3 г), а затем раствор перемешивали в течение 1 часа при 40°C. После взаимодействия раствор концентрировали в испарителе.
Полученную жидкость (70,5 г) переносили в разделительную воронку и на одну часть жидкости добавляли 4 части гексана (по объему) (280 г). Раствор по два раза промывали водой (210 мл), насыщенным водным раствором бикарбоната натрия (200 мл) и насыщенным водным раствором хлорида натрия (250 мл), а затем органический слой высушивали в течение ночи при помощи безводного сульфата натрия. Раствор концентрировали в испарителе для получения 35,03 г неочищенного вещества. В колонке очищали 35 г неочищенного вещества. Для этого использовали силикагель количеством приблизительно в пять раз больше количества неочищенного вещества (175 г). В качестве растворителя использовали смесь гексана и этилацетата в соотношении 5/1 и проводили очистку до удаления примесей (2,48 л), а после подтверждения удаления примесей целевого вещества элюировали при помощи смеси гексана и этилацетата в соотношении 1/3 (1,6 л). Диапазон фракции, содержащей целевое вещество, был подтвержден методом ТСХ, и несколько фракций на обоих концах данного диапазона были проанализированы методом ГХ, при этом отбирали только такие фракции, в которых содержание побочных продуктов составляло менее 1%. Добавляли 6,0 мг бутилгидрокситолуола и 2,0 мг монометилового эфира гидрохинона и раствор концентрировали в испарителе. Раствор перемешивали при 60°C с одновременным снижением давления при помощи вакуумного насоса в течение 1 часа для удаления остатков растворителя, и было подтверждено, что в результатах ЯМР пик остаточного растворителя не наблюдался. Выход составил 18,69 г (43%) при чистоте, измеренной методом ГХ, 95,2%.
Пример синтеза 9
В 4-горлую колбу объемом 500 мл, оснащенную механической мешалкой, капельной воронкой, обратным холодильником и стеклянной пробкой, помещали 40%-ный раствор (по массе) метиламина в воде (233 г, 3,0 моль) и метанол (35 мл). В колбу в течение приблизительно 5 часов по каплям добавляли раствор аллилглицидилового эфира (34,2 г, 0,3 моль) в метаноле (35 мл), поддерживая температуру 4-горлой колбы на уровне приблизительно 25°C при помощи мешалки. Завершение реакции подтверждали методом ГХ.
После завершения реакции реакционный раствор концентрировали в испарителе. Полученную жидкость очищали перегонкой при пониженном давлении (полный вакуум, 76°C). Выход составил 35,3 г (81%) при чистоте, измеренной методом ГХ, 99,2%.
Пример синтеза 10
В 3-горлую колбу объемом 100 мл помещали соединение, полученное в примере синтеза 9 (16,6 г, 0,114 моль), гексаметилдисилазан (11,2 г, 0,069 моль), бутилгидрокситолуол (0,05 г) и сахарин (0,05 г) и перемешивали в течение 3 часов при 100°C.
Реакцию завершали, убедившись в устранении пиков исходного вещества в результатах ГХ-анализа. После очистки перегонкой при пониженном давлении (полный вакуум, 68°C) выход составил 22,31 г (90%) при чистоте, измеренной методом ГХ, 99%.
Пример синтеза 11
В 3-горлую колбу объемом 300 мл, оснащенную обратным холодильником, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 10 (5,0 г, 0,023 моль), SiL5B (9,50 г, 0,023 моль), толуол (46 мл) и раствор платинового комплекса (80 мкл). Реакцию осуществляли при 50°C в течение 1,5 часов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (2,67 Па (0,02 мм рт. ст.), 135°C). Выход составил 8,3 г (57%) при чистоте, измеренной методом ГХ, 93%.
Пример синтеза 12
В 3-горлую колбу объемом 200 мл помещали соединение, полученное в примере синтеза 11 (20,0 г, 0,032 моль), триэтиламин (3,21 г, 0,032 моль) и гексан (54 мл), колбу помещали в ледяную баню (от -10 до -5°C) и при помощи капельной воронки в течение приблизительно 2 часов по каплям добавляли акрилоилхлорид (3,1 мл, 0,038 моль) и гексан (32 мл). Через 40 минут после окончания добавления по каплям провели ГХ-анализ, подтвердивший почти полное расходование исходных веществ.
Через час после начала добавления по каплям реакционный раствор фильтровали и осадок отмывали гексаном, который был охлажден в холодильнике. Раствор для промывания объединяли с фильтратом, переносили в разделительную воронку и трижды промывали водой (50 мл), дважды - насыщенным водным раствором бикарбоната натрия (50 мл) и дважды - насыщенным водным раствором хлорида натрия (50 мл).
Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход полученного неочищенного вещества составил 21,6 г (94%).
Рабочий пример 3
В грушевидную колбу объемом 100 мл помещали неочищенный продукт, полученный в примере синтеза 12 (12,0 г, 0,017 моль), метанол (36 г) и уксусную кислоту (5 мл) и перемешивали в течение приблизительно 1 часа при 40°C.
Методом ТСХ подтверждали расходование исходных веществ, и после концентрации в испарителе добавляли 50 мл гексана и раствор переносили в разделительную воронку. Раствор по два раза промывали водой (40 мл), насыщенным водным раствором бикарбоната натрия (40 мл) и насыщенным водным раствором хлорида натрия (40 мл). Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход неочищенного вещества составил 11,1 г.
В колонке очищали 10,00 г неочищенного вещества. Для этого использовали силикагель массой приблизительно в 5 раз (50 г) больше массы неочищенного вещества. Элюирование выполняли методом ТСХ с использованием смеси гексана и этилацетата в соотношении 1/3 до исчезновения целевого пятна. Методом ГХ проанализировали несколько фракций, собранных перед началом выхода продукта и после окончания его выхода с колонки, и измерили и собрали те фракции, в которых все пики побочных продуктов составляли менее 1%. Добавляли 3,0 мг бутилгидрокситолуола и 1,0 мг монометилового эфира гидрохинона, раствор концентрировали, а затем перемешивали при 60°C с одновременным снижением давления при помощи вакуумного насоса в течение 1 дополнительного часа для удаления остатков растворителя. Было подтверждено, что в результатах ЯМР пик растворителя не наблюдался. Выход составил 11,4 г (78%) при чистоте, измеренной методом ГХ, 98%.
Пример синтеза 13
В 3-горлую колбу объемом 200 мл, оснащенную обратным холодильником, термометром и механической мешалкой, помещали диаллиламин (2,49 мл, 0,02 моль), SiL5B (24,76 г, 0,06 моль), толуол (40 мл) и раствор платинового комплекса (0,07 мл). Реакцию осуществляли при 100°C в течение 3 часов. Реакционный раствор концентрировали в испарителе, а затем отгоняли низкокипящие примеси при пониженном давлении (2,67 Па (0,02 мм рт. ст.), 97°C). Выход составил 14,46 г (74%).
Рабочий пример 4
В 3-горлую колбу объемом 100 мл помещали соединение, полученное в примере синтеза 13 (9,2 г, 0,01 моль), триэтиламин (1,8 мл, 0,013 моль) и гексан (15 мл), после чего при помощи шприца по каплям добавляли акрилоилхлорид (1,05 мл, 0,013 моль). Через 40 минут после окончания добавления по каплям провели ГХ-анализ, подтвердивший почти полное расходование исходных веществ.
Через 2 часа после начала добавления по каплям реакционный раствор фильтровали и осадок отмывали гексаном, который был охлажден в холодильнике. Раствор для промывания объединяли с фильтратом, переносили в разделительную воронку и трижды промывали водой (50 мл), дважды - насыщенным водным раствором бикарбоната натрия (50 мл) и дважды - насыщенным водным раствором хлорида натрия (50 мл).
Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход полученного неочищенного вещества составил 9,99 г. Неочищенное вещество в количестве 3,1 г очищали методом препаративной гель-проникающей хроматографии (ГПХ) и получали 2,06 г очищенного силиконового акриламидного мономера, имеющего следующую формулу (s1).
[ФОРМУЛА]
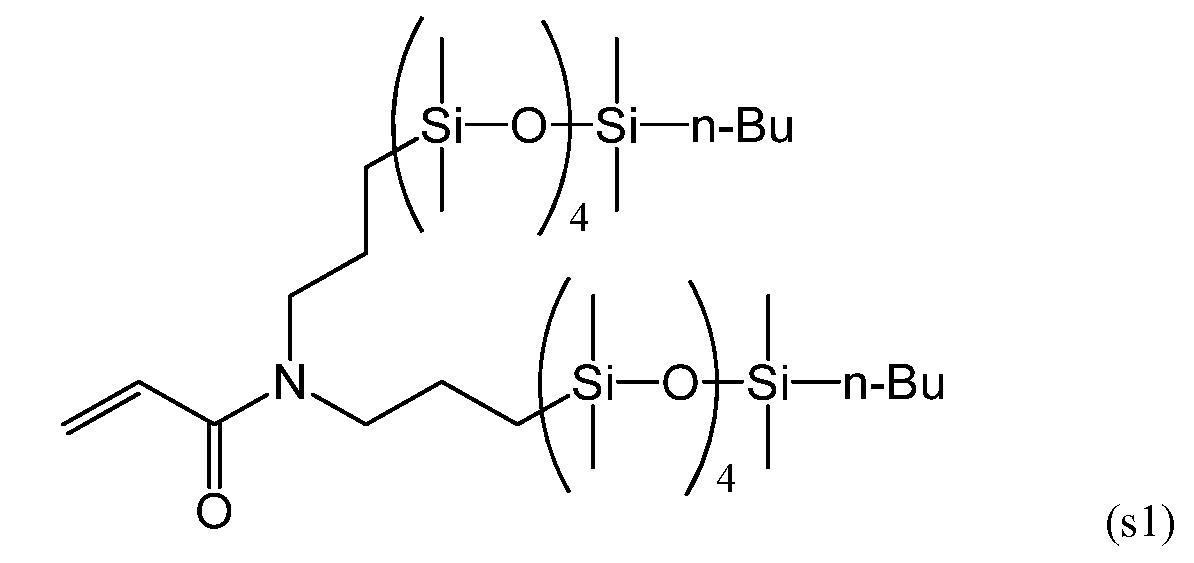
Пример синтеза 14
В 4-горлую колбу объемом 300 мл, оснащенную обратным холодильником, термометром и механической мешалкой, помещали аллиламин (5,0 г, 87 ммоль), SiL5B (39,50 г, 95,7 ммоль), толуол (100 мл) и раствор платинового комплекса (100 мкл). Реакцию осуществляли при 110°C в течение 3 часов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (22,7 Па (0,17 мм рт. ст.), 118°C). Выход составил 32,93 г (81%) при чистоте, измеренной методом ГХ, 96%.
Рабочий пример 5
В 3-горлую колбу объемом 100 мл помещали соединение, полученное в примере синтеза 14 (4,67 г, 0,01 моль), триэтиламин (1,8 мл, 0,013 моль) и гексан (15 мл), после чего колбу помещали в ледяную баню (от -10 до -5°C) и при помощи шприца по каплям добавляли акрилоилхлорид (1,05 мл, 0,013 моль) и гексан (32 мл). Через 40 минут после окончания добавления по каплям провели ГХ-анализ, подтвердивший почти полное расходование исходных веществ.
Через 2 часа после начала добавления по каплям реакционный раствор фильтровали и осадок отмывали гексаном, который был охлажден в холодильнике. Раствор для промывания объединяли с фильтратом, переносили в разделительную воронку и трижды промывали водой (50 мл), дважды - насыщенным водным раствором бикарбоната натрия (50 мл) и дважды - насыщенным водным раствором хлорида натрия (50 мл).
Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход полученного неочищенного вещества составил 5,31 г. Неочищенное вещество очищали в ходе колоночной хроматографии (элюэнт - смесь гексана и этилацетата в соотношении от 4/1 до 2/1, выход: 31,3%).
Пример синтеза 15
В 4-горлую колбу объемом 300 мл, оснащенную обратным холодильником, термометром и механической мешалкой, помещали N-метил-N-аллиламин (5,11 г, 71,8 ммоль), SiL5B (24,7 г, 60 ммоль), толуол (120 мл) и раствор платинового комплекса (100 мкл). Реакцию осуществляли при 110°C в течение 2 часов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (22,7 Па (0,17 мм рт. ст.), 115°C). Выход составил 20,34 г (70%) при чистоте, измеренной методом ГХ, 96%.
Рабочий пример 6
В 3-горлую колбу объемом 100 мл помещали соединение, полученное в примере синтеза 15 (4,84 г, 0,01 моль), триэтиламин (1,8 мл, 0,013 моль) и гексан (15 мл), после чего колбу помещали в ледяную баню (от -10 до -5°C) и при помощи шприца по каплям добавляли акрилоилхлорид (1,05 мл, 0,013 моль). Через 40 минут после окончания добавления по каплям провели ГХ-анализ, подтвердивший почти полное расходование исходных веществ.
Через 2 часа после начала добавления по каплям реакционный раствор фильтровали и осадок отмывали гексаном, который был охлажден в холодильнике. Раствор для промывания объединяли с фильтратом, переносили в разделительную воронку и трижды промывали водой (50 мл), дважды - насыщенным водным раствором бикарбоната натрия (50 мл) и дважды - насыщенным водным раствором хлорида натрия (50 мл).
Органический слой высушивали в течение ночи при помощи сульфата натрия, фильтровали, а затем концентрировали при помощи испарителя. Выход полученного неочищенного вещества составил 5,45 г. Неочищенное вещество очищали в ходе колоночной хроматографии (элюэнт - смесь гексана и этилацетата в соотношении от 5/1 до 3/1) с получением 1,06 г очищенного силиконового акриламидного мономера.
Рабочий пример 7
Силиконовый мономер, имеющий следующую формулу (s2)
[ФОРМУЛА 6]
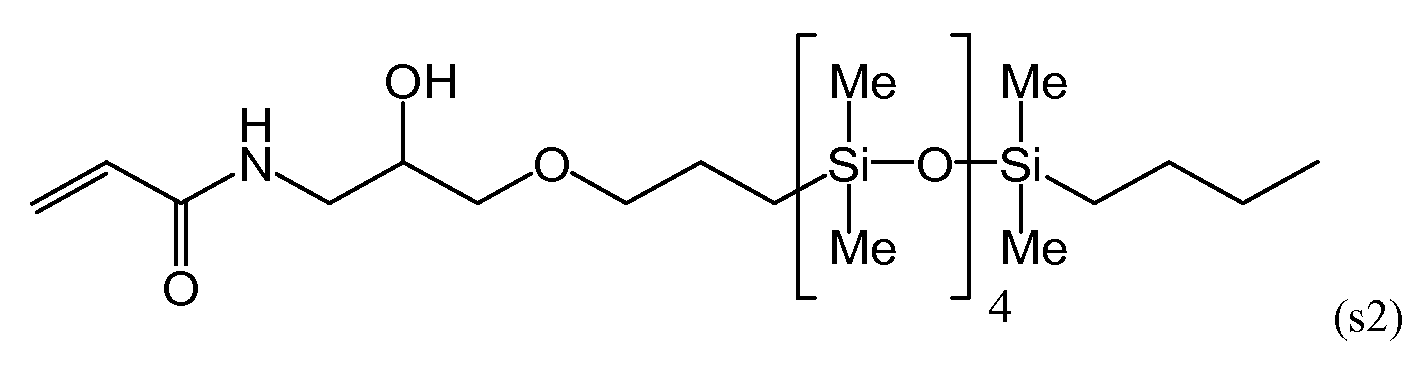
полученный в рабочем примере 1 (0,925 г, 56,06% вес.), N,N-диметилакриламид (0,510 г, 30,91% вес.) и гидрофильный акриламидный мономер, имеющий следующую формулу (h1)
[ФОРМУЛА 7]
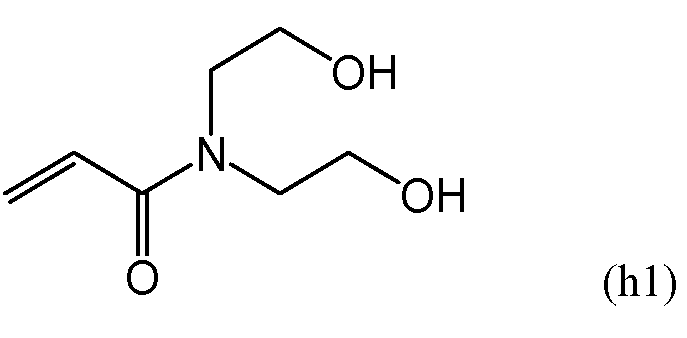
(0,0 17 г, 1% вес.), поливинилпирролидон (ПВП K90, 0,132 г, 8% вес.), N,N'-метиленбисакриламид (МБА, 0,018 г, 1,10% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,036 г, 2,20% вес.), 3-метил-3-пентанол (3М3П, 1,350 г) и фотоинициатор Irgacure 819 (0,004 г, 0,25% вес.) соединяли вместе и перемешивали. Полученную мономерную смесь дегазировали в аргоновой среде. Мономерную смесь впрыскивали в полость прозрачной пластмассовой пресс-формы (стенка формы со стороны кривизны передней поверхности: Zeonor, стенка со стороны базовой кривизны: полипропилен), имеющей форму линзы и помещенной в перчаточный бокс с азотной средой, и изготавливали линзу путем облучения светом (Philips TL03, 1,6 мВт/см2, 15 минут) для отверждения. Полученную линзу вынимали из пресс-формы и экстрагировали примеси, такие как остаточный мономер, путем погружения линзы в 70%-ный (по объему) водный раствор 2-пропанола (ИПС) на 70 минут при комнатной температуре. После погружения в воду на 10 минут образец помещали в буферный раствор борной кислоты (pH 7,1-7,3) во флаконе объемом 5 мл, флакон размещали в автоклаве и кипятили 30 минут при 120°C.
Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая прозрачностью и балансом благоприятных физических характеристик.
Рабочий пример 8
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер следующей формулы (s3)
[ФОРМУЛА 8]
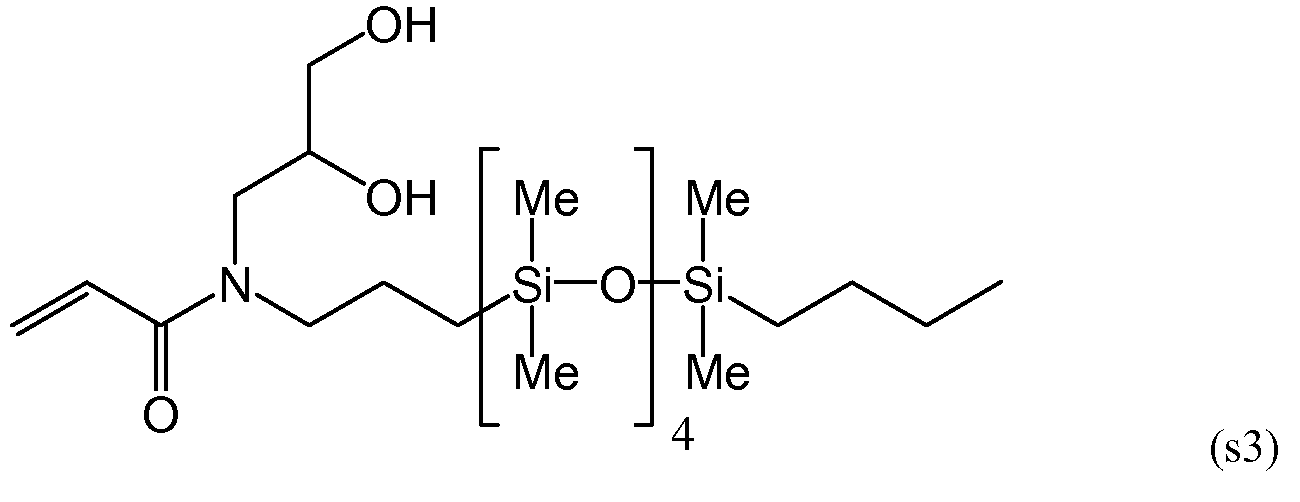
полученный в рабочем примере 2. Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая прозрачностью и балансом благоприятных физических характеристик.
Рабочий пример 9
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер следующей формулы (s4)
[ФОРМУЛА 9]
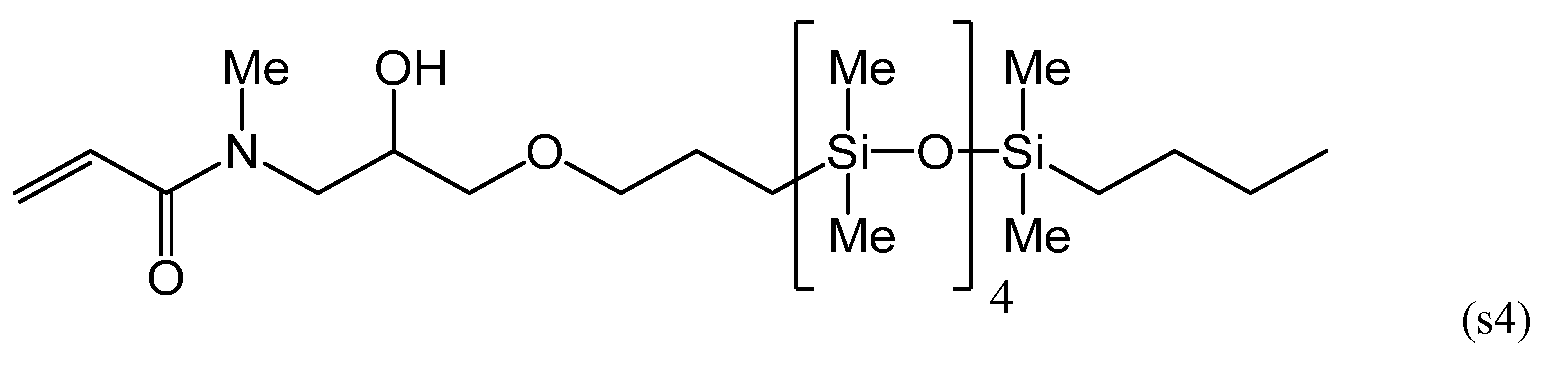
полученный в рабочем примере 3, и за исключением того, что мономерная смесь имела следующий состав: силиконовый акриламидный мономер (s4) (0,925 г, 56,06% вес.), N,N-диметилакриламид (0,552 г, 33,27% вес.), гидрофильный акриламидный мономер (h1) (0,116 г, 7% вес.), N,N'-метиленбисакриламид (МБА, 0,018 г, 1,10% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,036 г, 2,20% вес.), 3-метил-3-пентанол (3М3П, 1,350 г) и фотоинициатор Irgacure 819 (0,004 г, 0,25% вес.). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая прозрачностью и балансом благоприятных механических характеристик. Как показали результаты измерения угла смачивания, смачиваемость материала была выше обычно необходимых значений.
Рабочий пример 10
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что мономерная смесь имела следующий состав: силиконовый акриламидный мономер (s2) (1,21 г, 55% вес.), N,N-диметилакриламид (0,78 г, 35,53% вес.), поливинилпирролидон (ПВП K90, 0,088 г, 4% вес.), тетраэтиленгликольдиметакрилат (ТЭГМА, 0,066 г, 3% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,048 г, 2,20% вес.), 3-метил-3-пентанол (3М3П, 1,80 г) и фотоинициатор Irgacure 819 (0,006 г, 0,25% вес.). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза. Содержание воды и механические характеристики были благоприятными, но полноспектральное светопропускание образца линзы было низким, что указывает на нежелательную мутность линзы, а значения угла смачивания были выше обычно необходимых.
Рабочий пример 11
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 10, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер, имеющий формулу (s3). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза. Содержание воды и механические характеристики были благоприятными (включая необходимый низкий модуль упругости), и полноспектральное светопропускание линзы улучшилось в сравнении с рабочим примером 10. Значения угла смачивания были выше обычно необходимых.
Рабочий пример 12
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что мономерная смесь имела следующий состав: силиконовый акриламидный мономер (s2) (1,21 г, 55% вес.), N,N-диметилакриламид (0,43 г, 19,53% вес.), 2-гидроксиэтилметакрилат (0,176 г, 8% вес.), поливинилпирролидон (ПВП K90, 0,264 г, 12% вес.), тетраэтиленгликольдиметакрилат (ТЭГМА, 0,066 г, 3% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,048 г, 2,20% вес.), 3-метил-3-пентанол (3М3П, 1,80 г) и фотоинициатор Irgacure 819 (0,006 г, 0,25% вес.). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая прозрачностью и балансом благоприятных физических характеристик.
Рабочий пример 13
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 12, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер, имеющий формулу (s3). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая прозрачностью и балансом благоприятных физических характеристик.
Рабочий пример 14
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер следующей формулы (s5)
[ФОРМУЛА]
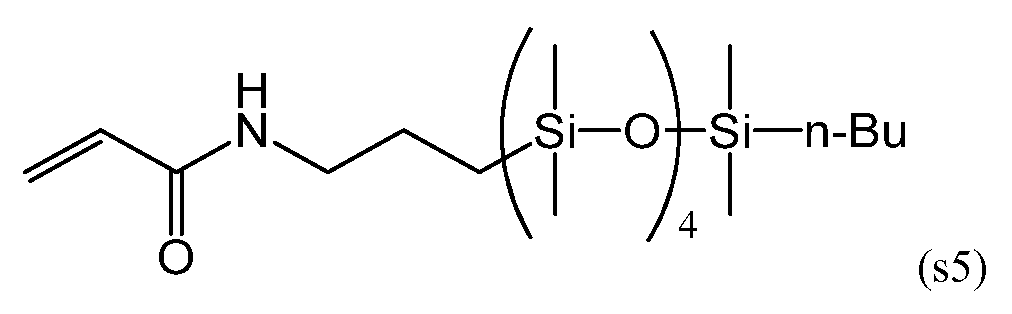
полученный в рабочем примере 18, и за исключением того, что мономерная смесь имела следующий состав: силиконовый акриламидный мономер (s5) (0,925 г, 56,06% вес.), N,N-диметилакриламид (0,419 г, 25,27% вес.), поливинилпирролидон (ПВП K90, 0,132 г, 8% вес.), гидрофильный акриламидный мономер (h1) (0,116 г, 7% вес.), N,N'-метиленбисакриламид (МБА, 0,018 г, 1,10% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,036 г, 2,20% вес.), трет-амиловый спирт (ТАС, 1,350 г) и фотоинициатор Irgacure 819 (0,004 г, 0,25% вес.). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая балансом благоприятных физических характеристик, включая необходимый модуль упругости. Значение полноспектрального светопропускания линзы было низким, что указывает на нежелательную мутность линзы.
Рабочий пример 15
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер следующей формулы (s6)
[ФОРМУЛА]
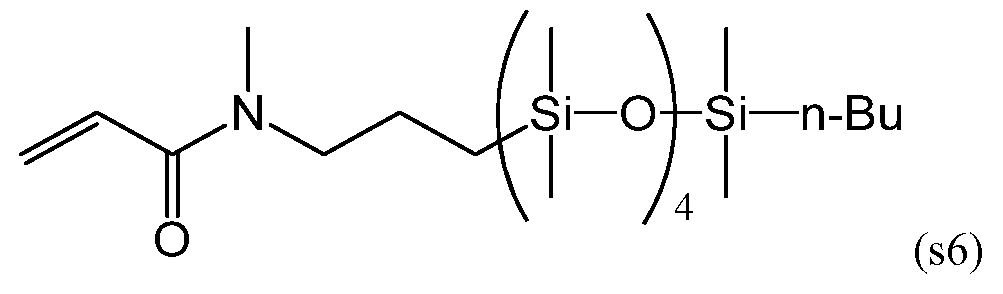
полученный в рабочем примере 18, и за исключением того, что мономерная смесь имела следующий состав: силиконовый акриламидный мономер (s6) (0,925 г, 56,06% вес.), N,N-диметилакриламид (0,419 г, 25,27% вес.), поливинилпирролидон (ПВП K90, 0,132 г, 8% вес.), гидрофильный акриламидный мономер (h1) (0,116 г, 7% вес.), N,N'-метиленбисакриламид (МБА, 0,018 г, 1,10% вес.), УФ-поглотитель 2-(2'-гидрокси-5'-метакрилоилоксиэтилфенил)-2H-бензотриазол (0,036 г, 2,20% вес.), трет-амиловый спирт (ТАС, 1,350 г) и фотоинициатор Irgacure 819 (0,004 г, 0,25% вес.). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца линзы показаны в таблице 1, и, таким образом, была получена линза, обладающая балансом благоприятных физических характеристик, включая необходимый модуль упругости. Значение полноспектрального светопропускания линзы было низким, что указывает на нежелательную мутность линзы.
Сравнительный пример 1
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что силиконовый акриламидный мономер следующей формулы (t1) (полученный способом, описанным в примерах 1 и 2 публикации WO 05/078482A1)
[ФОРМУЛА 10]
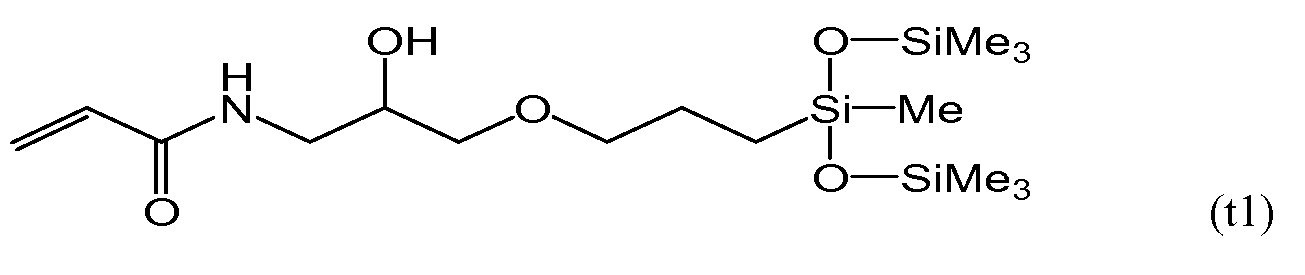
применяли в качестве силиконового акриламидного мономера вместо мономера, имеющего формулу (s2). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца в форме линзы показаны в таблице 1, и время нулевой нагрузки составляло более 1 секунды, что указывало на недостаточность характеристик восстановления формы.
Сравнительный пример 2
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что силиконовый акриламидный мономер следующей формулы (t2) (полученный способом, описанным в примерах 5 и 6 публикации WO05/078482A1)
[ФОРМУЛА 11]
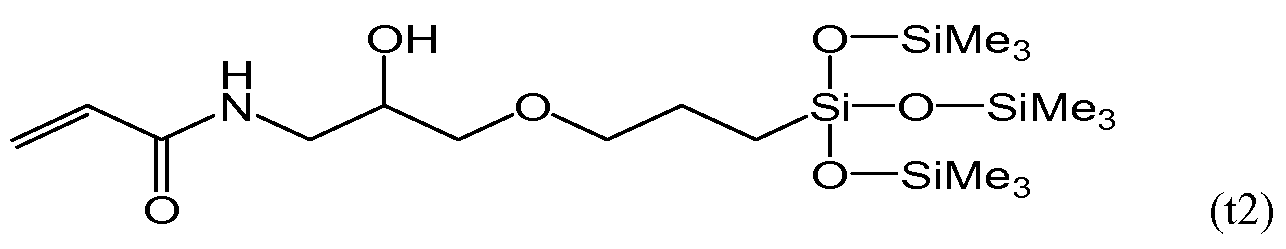
применяли в качестве силиконового акриламидного мономера вместо мономера, имеющего формулу (s2). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца в форме линзы показаны в таблице 1, и время нулевой нагрузки составляло более 1 секунды, что указывало на недостаточность характеристик восстановления формы.
Пример синтеза 16
В 3-горлую колбу объемом 500 мл, оснащенную обратным холодильником, термометром и механической мешалкой, помещали соединение, полученное таким же способом, что и в примере синтеза 6 (20,0 г, 0,072 моль), бис(триметилсилокси)метилсилан (16 г, 0,072 моль), толуол (150 мл) и раствор платинового комплекса (200 мкл). Реакцию осуществляли при 120°C в течение 2 часов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (полный вакуум, 160°C). Выход составил 21,13 г (58,7%) при чистоте, измеренной методом ГХ, 95,7%.
Пример синтеза 17
В 3-горлую колбу объемом 500 мл, оснащенную капельной воронкой, термометром и механической мешалкой, помещали соединение, полученное способом, описанным в примере синтеза 16 (20 г, 0,040 моль), триэтиламин (8,14 г, 0,040 моль) и этилацетат (130 мл). В капельную воронку помещали акрилоилхлорид (7,28 г, 0,040 моль) и этилацетат (100 мл). 3-горлую колбу помещали в ледяную баню, содержащую соль, и после падения внутренней температуры до 0°C начинали добавление по каплям. Поддерживая внутреннюю температуру на уровне от -2 до 3°C, добавление по каплям осуществляли в течение приблизительно 2 часов и по завершении добавления по каплям реакцию продолжали в течение еще одного часа, а затем прекращали. Далее раствор фильтровали с одновременным промыванием небольшим количеством охлажденного этилацетата, который был охлажден в холодильнике. Фильтрат переносили в разделительную воронку и добавляли 400 мл гексана. Раствор дважды промывали водой (180 мл), дважды - насыщенным водным раствором бикарбоната натрия (180 мл) и дважды - насыщенным водным раствором хлорида натрия (180 мл). К органическому слою добавляли безводный сульфат натрия и оставляли высушиваться в течение ночи. После фильтрации данный раствор концентрировали в испарителе для получения неочищенного продукта. Выход неочищенного продукта составил 27,5 г (125%).
Пример синтеза 18
На каждую часть неочищенного продукта, полученного в примере синтеза 17 (26,5 г, 0,048 моль), добавляли 3 части (по массе) метанола (79,5 г) и половину части (по массе) уксусной кислоты (13,3 г), а затем раствор перемешивали в течение 1 часа при 40°C. После взаимодействия раствор концентрировали в испарителе. Полученную жидкость (48,5 г) переносили в разделительную воронку и на одну часть жидкости добавляли 4 части гексана (по объему) (190 г). Раствор по два раза промывали водой (140 мл), насыщенным водным раствором бикарбоната натрия (140 мл) и насыщенным водным раствором хлорида натрия (170 мл), а затем органический слой высушивали в течение ночи при помощи безводного сульфата натрия. Раствор концентрировали в испарителе для получения 16,0 г неочищенного вещества. В колонке очищали 16,0 г неочищенного вещества. Для этого использовали силикагель количеством в пять раз больше количества неочищенного вещества (80 г). В качестве растворителя использовали смесь гексана и этилацетата в соотношении 2/1 и проводили очистку до удаления примесей (500 мл), а после подтверждения удаления примесей целевое вещество элюировали при помощи смеси гексана и этилацетата в соотношении 1/2 (750 мл). Диапазон фракции, содержащей целевое вещество, был подтвержден методом ТСХ, и несколько фракций на обоих концах данного диапазона были проанализированы методом ГХ, при этом отбирали только такие фракции, в которых содержание побочных продуктов составляло менее 1%. Добавляли 15 мг бутилгидрокситолуола и концентрировали раствор в испарителе. Раствор перемешивали при 60°C с одновременным снижением давления при помощи вакуумного насоса в течение 1 часа для удаления остатков растворителя, и было подтверждено, что в результатах ЯМР пик остаточного растворителя не наблюдался. Выход составил 7,5 г (38,3%) при чистоте, измеренной методом ГХ, 97,7%.
Пример синтеза 19
В 3-горлую колбу, оснащенную обратным холодильником, термометром и капельной воронкой, помещали 3-аминопропилтрис(триметилсилокси)силан (49 г, 0,14 моль). В капельную воронку при перемешивании при 30°C добавляли глицидол (2,3-эпокси-1-пропанол, 3,5 г, 0,05 моль) и по каплям добавляли его в течение приблизительно 40 минут. Для контроля протекания реакции ГХ-анализ проводили каждый час, начиная с момента завершения добавления по каплям. Через 20 часов после завершения добавления по каплям было подтверждено, что пик глицидола составляет 1% или менее, и реакцию прекращали. Раствор концентрировали в испарителе, а затем очищали при пониженном давлении (полный вакуум, точка кипения 190°C). Выход составил 13,65 г (67,8%) при чистоте, измеренной методом ГХ, 92,8%.
Пример синтеза 20
В 3-горлую колбу, оснащенную обратным холодильником, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 19 (12,5 г, 0,03 моль), гексаметилдисилазан (10,0 г, 0,06 моль) и сахарин (250 мг). Реакцию осуществляли в течение 2 часов при 100°C и прекращали после того, как методом ГХ было подтверждено снижение концентрации или израсходование реагентов. Реакционный раствор концентрировали в испарителе, а затем очищали перегонкой при пониженном давлении (полный вакуум, точка кипения 170°C). Выход составил 13,3 г (76%) при чистоте, измеренной методом ГХ, 89,5%.
Пример синтеза 21
В 3-горлую колбу объемом 1 л, оснащенную капельной воронкой, термометром и механической мешалкой, помещали соединение, полученное в примере синтеза 20 (13,0 г, 0,023 моль), триэтиламин (2,33 г, 0,023 моль) и этилацетат (50 мл). В капельную воронку помещали акрилоилхлорид (2,1 г, 0,023 моль) и этилацетат (10 мл). 3-горлую колбу помещали в ледяную баню, содержащую соль, и после падения внутренней температуры до 0°C начинали добавление по каплям. Поддерживая внутреннюю температуру на уровне от -2 до 3°C, добавление по каплям осуществляли в течение приблизительно 1 часа и по завершении добавления по каплям реакцию продолжали в течение еще одного часа, а затем прекращали. Далее раствор фильтровали с одновременным промыванием небольшим количеством охлажденного этилацетата, который был охлажден в холодильнике. Фильтрат переносили в разделительную воронку и добавляли 60 мл гексана. Раствор дважды промывали водой (50 мл), дважды - насыщенным водным раствором бикарбоната натрия (50 мл) и дважды - насыщенным водным раствором хлорида натрия (50 мл). К органическому слою добавляли безводный сульфат натрия и оставляли высушиваться в течение ночи. После фильтрации данный раствор концентрировали в испарителе для получения неочищенного продукта. Выход неочищенного продукта составил 14,2 г при чистоте, измеренной методом ГХ, 86,1%.
Пример синтеза 22
На каждую часть неочищенного продукта, полученного в примере синтеза 21 (14,0 г), добавляли 3 части (по массе) метанола (42,0 г) и половину части (по массе) уксусной кислоты (7,0 г), а затем раствор перемешивали в течение 4 часов при 40°C. После взаимодействия раствор концентрировали в испарителе. Полученную жидкость (16,95 г) переносили в разделительную воронку и на одну часть жидкости добавляли 4 части гексана (по объему) (68,0 г). Раствор по два раза промывали водой (50 мл), насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным водным раствором хлорида натрия (50 мл), а затем органический слой высушивали в течение ночи при помощи безводного сульфата натрия. Раствор концентрировали в испарителе для получения 9,95 г неочищенного вещества. В колонке очищали 9,5 г неочищенного вещества. Для этого использовали силикагель количеством в 5 раз больше количества неочищенного вещества (50 г). В качестве растворителя использовали смесь гексана и этилацетата в соотношении 2/1 и проводили очистку до удаления примесей (300 мл), а после подтверждения удаления примесей целевое вещество элюировали при помощи смеси гексана и этилацетата в соотношении 1/1 (500 мл). Диапазон фракции, содержащей целевое вещество, был подтвержден методом ТСХ, и несколько фракций на обоих концах данного диапазона были проанализированы методом ГХ, при этом отбирали только такие фракции, в которых содержание побочных продуктов составляло менее 1%. Добавляли 4,8 мг бутилгидрокситолуола и концентрировали раствор в испарителе. Раствор перемешивали при 60°C с одновременным снижением давления при помощи вакуумного насоса в течение 1 часа для удаления остатков растворителя, и было подтверждено, что в результатах ЯМР пик остаточного растворителя не наблюдался. Выход составил 3,14 г (28%) при чистоте, измеренной методом ГХ, 97,8%.
Сравнительный пример 3
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали силиконовый акриламидный мономер следующей формулы (t3)
[ФОРМУЛА 12]
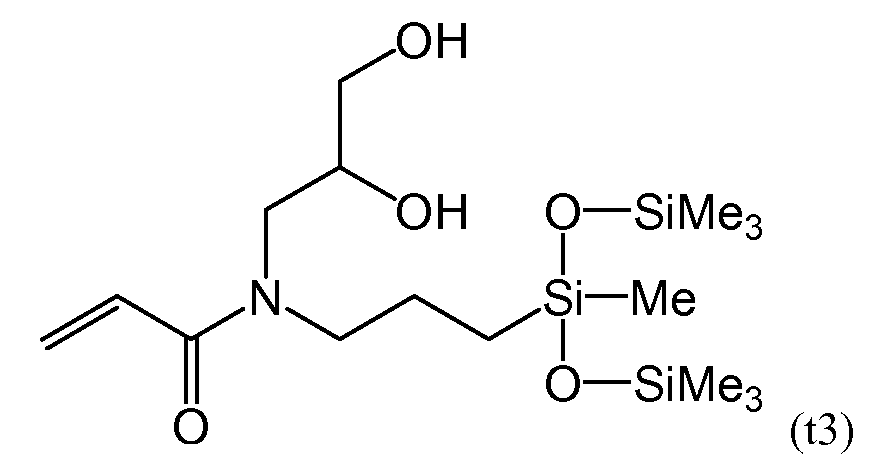
полученный в примере синтеза 16. Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца в форме линзы показаны в таблице 1, и время нулевой нагрузки составляло более 1 секунды, что указывало на недостаточность характеристик восстановления формы.
Сравнительный пример 4
Образец в форме линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что силиконовый акриламидный мономер следующей формулы (t4), полученный в примере синтеза 17
[ФОРМУЛА 13]
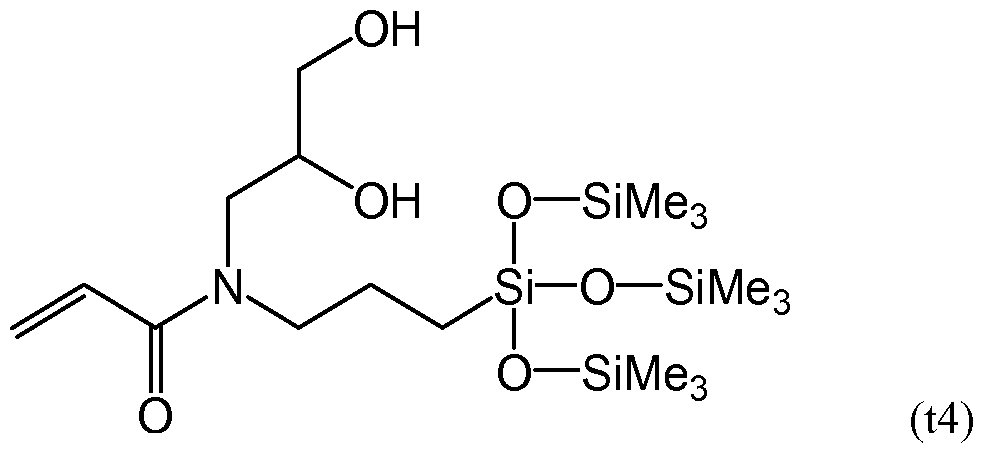
применяли в качестве силиконового акриламидного мономера вместо мономера, имеющего формулу (s2). Измеренные значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца в форме линзы показаны в таблице 1, и время нулевой нагрузки составляло более 1 секунды, что указывало на недостаточность характеристик восстановления формы.
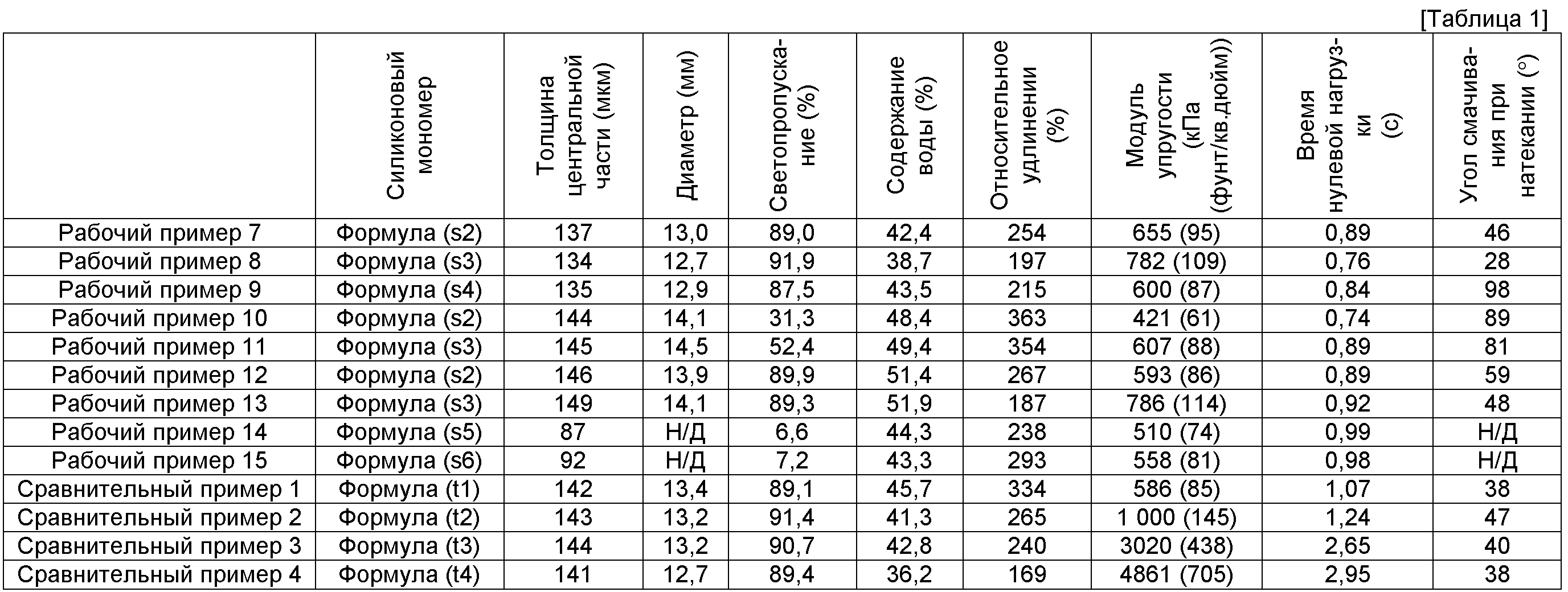
Рабочие примеры 16-19
Образец линзы изготавливали способом, аналогичным описанному в рабочем примере 7, за исключением того, что вместо мономера, имеющего формулу (h1), в качестве несиликонового акриламидного мономера использовали 2-гидроксиэтилметакриламид (ГЭМАА, приобретенный в компании Monomer Polymer Dajac Laboratories, штат Пенсильвания), имеющий формулу (h5)
[ФОРМУЛА 23]
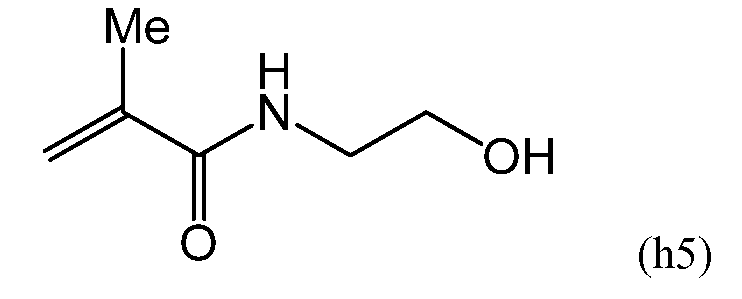
и за исключением того, что силиконовый мономер и композиция силиконового мономера, несиликоновый акриламид и N,N-диметилакриламид были изменены, как показано в таблице 2. Внешний вид, значения полноспектрального светопропускания, содержания воды, модуля упругости и относительного удлинения полученного образца показаны в таблице 2.
|
Рабочий пример 20
Приготавливали мономерную смесь со следующим молярным соотношением: мономер, имеющий формулу (s2) (32,4% мол.), N,N'-диметилакриламид (32,5% мол.), N-(2-гидроксиэтил)акриламид (32,4% вес.), N,N'-метиленбисакриламид (2,4% мол.) и фотоинициатор Irgacure 819 (0,2% мол.). В мономерную смесь (55% вес.) добавляли метиловый эфир трипропиленгликоля (ТПМЭ, 40% вес.) и поливинилпирролидон (ПВП K90, 5% вес.).
Полимеризацию мономерной смеси осуществляли при 70°C в атмосфере осушенного азота под воздействием света от светоизлучающего диода (~420 нм, 4,0 мВт/см2) в течение 10 минут в соответствии с описанной выше процедурой ДСК.
Скорость полимеризации мономерной смеси анализировали методом фотоДСК, результаты показаны на фиг. 1 под обозначением «SA1 RMM». Скорость полимеризации была намного выше скорости, полученной в сравнительном примере 5.
Рабочий пример 21
Приготавливали мономерную смесь способом, аналогичным описанному в рабочем примере 20, за исключением того, что вместо мономера, имеющего формулу (s2), в качестве силиконового акриламидного мономера использовали мономер, имеющий формулу (s3).
Скорость полимеризации мономерной смеси анализировали методом фотоДСК, результаты показаны на фиг. 1 под обозначением «SA2 RMM». Скорость полимеризации была намного выше скорости, полученной в сравнительном примере 5.
Сравнительный пример 5
Приготавливали мономерную смесь способом, аналогичным описанному в рабочем примере 20, за исключением того, что силиконовый метакрилатный мономер, имеющий следующую формулу (u1) (полученный способом, описанным в примере 29 публикации WO2008/005229)
использовали вместо силиконового акриламидного мономера, имеющего формулу (s2), за исключением того, что вместо 2-гидроксиэтилакриламида использовали 2-гидроксиэтилметакрилат, и за исключением того, что вместо N,N'-метиленбисакриламида использовали тетраэтиленгликольдиметакрилат. Данные мономеры использовались в таком же молярном соотношении, что и в рабочем примере 20.
Скорость полимеризации мономерной смеси анализировали методом фотоДСК, результаты показаны на фиг. 1 под обозначением «OH mPDMS RMM».
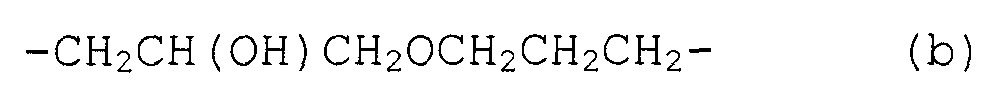
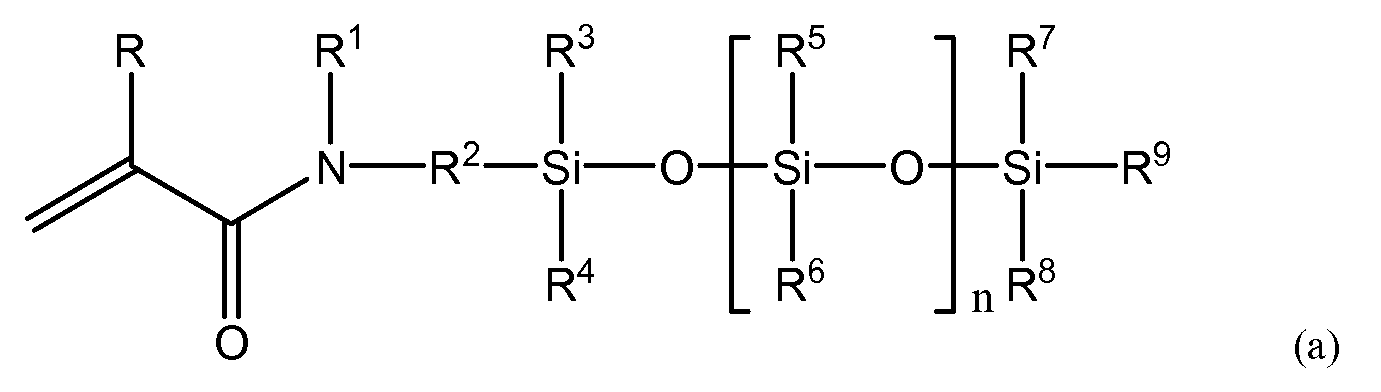
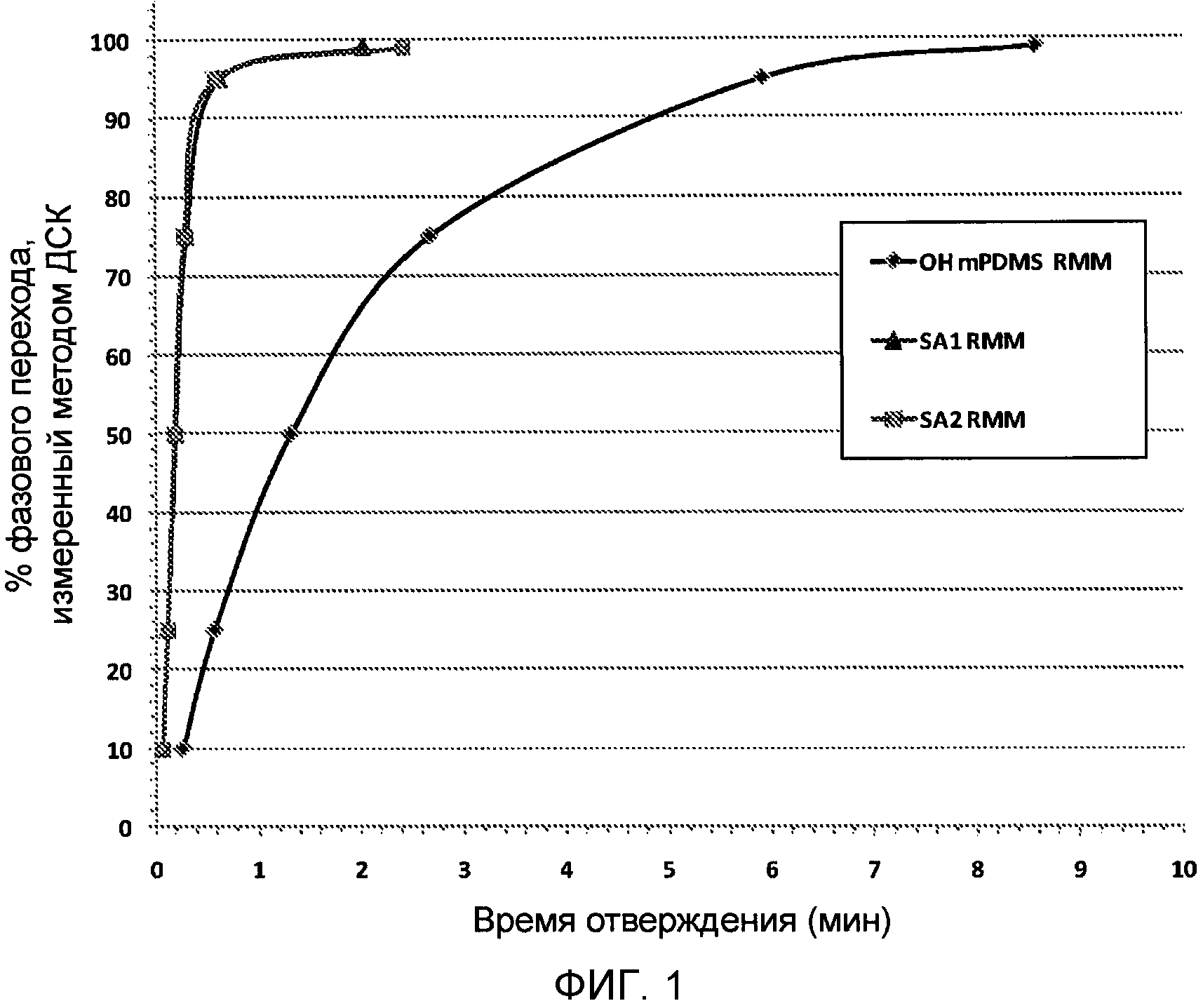
Способы получения антибактериальных контактных линз
Окрашенные контактные линзы, обладающие эффектом глубины
Антимикробные полимерные изделия, способы их получения и способы их применения
Контактные линзы со стабилизацией против смещения и способы их конструирования
Формы для применения при производстве контактных линз
Офтальмологические линзы для коррекции аберрации и способы изготовления таких линз
Тонированные офтальмологические устройства из силикона, способы их изготовления, процессы и используемые полимеры
Линзы для коррекции пресбиопии и способы конструирования линз
Способы формирования заготовки офтальмологической линзы и линзы
Прокалывающее откидное укупорочное средство
Способы получения антибактериальных контактных линз
Окрашенные контактные линзы, обладающие эффектом глубины
Антимикробные полимерные изделия, способы их получения и способы их применения
Контактные линзы со стабилизацией против смещения и способы их конструирования
Формы для применения при производстве контактных линз
Офтальмологические линзы для коррекции аберрации и способы изготовления таких линз
Тонированные офтальмологические устройства из силикона, способы их изготовления, процессы и используемые полимеры
Линзы для коррекции пресбиопии и способы конструирования линз
Способы формирования заготовки офтальмологической линзы и линзы
Прокалывающее откидное укупорочное средство

