АРИЛФТОРФОСФАТНЫЕ ИНГИБИТОРЫ КИШЕЧНОГО АПИКАЛЬНОГО МЕМБРАННОГО НАТРИЙ/ФОСФАТНОГО КОТРАНСПОРТА
Вид РИД
Изобретение
Перекрестная ссылка на родственные заявки
По данной заявке испрашивается приоритет по дате подачи предварительной заявки U.S. № 61/310902, поданной 5 марта 2010 г.
Область техники, к которой относится изобретение
Данное изобретение относится к гидрофильным арилфторфосфатам, которые действуют на ингибирование кишечного апикального мембранного Na-опосредованного фосфатного котранспорта, к эффективным путям лечения для снижения фосфата в крови, способам лечения гиперфосфатемии и способам изготовления ингибиторов.
Уровень техники
Вторичный гиперпаратиреоз представляет собой общее и тяжелое осложнение хронической почечной недостаточности (CRF), приводящее к почечной остеодистрофии, гипертензии, метаболическому ацидозу и возникновению заболевания сердца. Гиперфосфатемия из-за сниженной почечной экскреции фосфата, как полагают, способствует гиперпаратиреозу у пациентов с хронической почечной недостаточностью. Недавно было установлено, что снижение фосфатной нагрузки может уменьшать вторичный гиперпаратиреоз и возможно защищать функцию почек.
У млекопитающих кишечная абсорбция фосфата происходит в мембране щеточной каемки в проксимальном отделе кишечника (двенадцатиперстная кишка и тощая кишка). Абсорбция фосфата имеет активный компонент и пассивный компонент. Активное поглощение фосфата связано с поглощением Na+ со снижением градиента его электрохимического потенциала посредством Na+/фосфатного котранспортера. Активный компонент абсорбции фосфата регулируется фосфором в рационе питания и сывороточным 1,25 дигидроксивитамином D3. Сообщают, что изменения фосфора в рационе изменяют экспрессию NaPi II b в кишечнике. Na+-независимое поглощение фосфата происходит по неизвестному механизму со снижением градиента его электрохимического потенциала. Механизм фосфатного транспорта сквозь кишечную базолатеральную мембрану не был установлен.
Халконы представляют собой класс ароматических кетонов с важной биологической активностью, и их влияние на мембранный транспорт хорошо известно. Флоридзин, представитель халконов, является потенциальным ингибитором почечных и кишечных мембранных котранспортеров Na+/глюкоза щеточной каемки. Аглюкон флоридзина, флоретин, ингибирует множество мембранных транспортеров, включающих Band 3 (AE-1, 9, 10) и носитель для облегченной диффузии глюкозы (GLUT-4, 9, 14). Было показано, что фосфорилированное производное флоретина, 2'-PP, является потенциальным ингибитором кишечного Na+/фосфатного котранспортера, но не Na+/фосфатного котранспортера первичных почечных проксимальных канальцев. Главное ограничение 2'-РР для лечения гиперфосфатемии состоит в том, что он представляет собой сложный фосфатный эфир и поэтому он разлагается фосфатазами, включающими кишечную апикальную мембранную фосфатазу, щелочную фосфатазу.
Таким образом, новые или улучшенные средства, которые ингибируют кишечный апикальный мембранный Na-опосредованный фосфатный котранспорт, постоянно требуются для разработки новых и более эффективных фармацевтических препаратов, которые предназначены для лечения вторичного гиперпаратиреоза, вызванного CRF и приводящего к почечной остеодистрофии, гипертензии, метаболическому ацидозу и возникновению заболевания сердца. 2'-FPP, который в 30 раз более эффективен, чем 2'-PP, а также более устойчив к эстеразам, представляет собой главное достижение и улучшение по сравнению с 2'-PP, и, как хорошо известно, является эффективным в лечении CRF. Соединения, композиции и способы, описанные в настоящем описании, направлены на данные потребности и другие аспекты.
Сущность изобретения
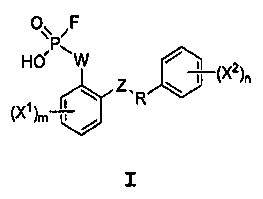
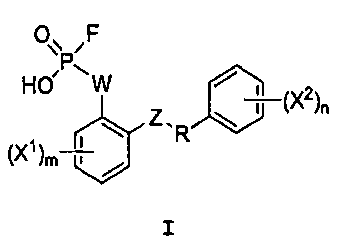
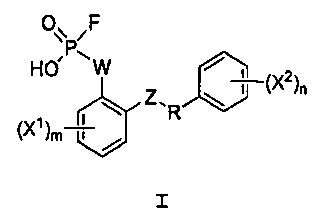
Настоящее изобретение относится, среди прочего, к соединениям, соответствующим формуле I:
I
или к их фармацевтически приемлемой соли, где составные элементы определены в настоящем описании. Соединения могут ингибировать кишечный апикальный мембранный натрий-опосредованный фосфатный котранспорт.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель. Данное соединение может присутствовать в количестве, которое обеспечивает клинически полезный результат у пациента, которому было введено соединение. Таким образом, композиции могут включать терапевтически эффективные количества соединений, описанных в настоящем изобретении.
Кроме того, настоящее изобретение относится к способам ингибирования кишечного апикального натрий/фосфатного котранспорта, включающим приведение в контакт кишечного эпителия с соединением формулы I или его фармацевтически приемлемой солью.
Кроме того, настоящее изобретение относится к способам лечения заболевания или расстройства, связанного с аномальными уровнями фосфата в крови у пациента, введением пациенту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к способам лечения заболеваний, таких как хроническая почечная недостаточность, почечная остеодистрофия, гипертензия, метаболический ацидоз и заболевание сердца, у пациента введением пациенту соединения формулы I или его фармацевтически приемлемой соли. Почечная недостаточность может представлять собой конечную стадию почечной недостаточности.
Кроме того, настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли для применения при получении лекарственного средства или при производстве лекарственного средства для применения в терапии, включающей терапию состояния, описанного в настоящем изобретении.
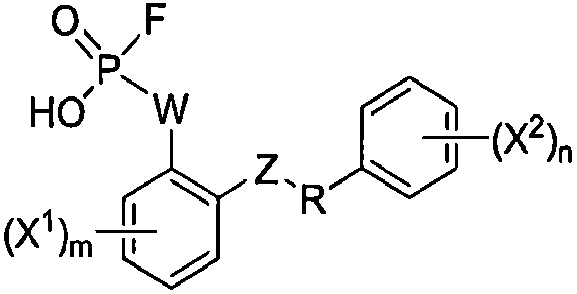
Настоящее изобретение относится к способам получения соединения формулы I:
I
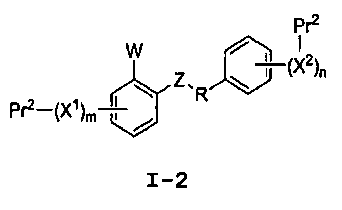
или его фармацевтически приемлемой соли, где R представляет собой С2-4алкил; W выбран из NRA, O и S; Z выбран из простой связи, -С(О)-, NRA, O и S; Х1 и Х2, каждый независимо, выбран из -ОН, -NHRA и -C(О)ОН; RA представляет собой Н или С1-3алкил; m равно 1, 2, 3 или 4; и n равно 1, 2, 3 или 4.
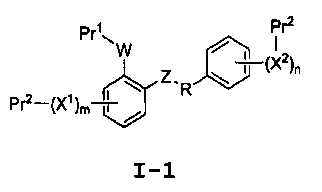
Настоящее изобретение относится к способам получения соединения формулы I-1:
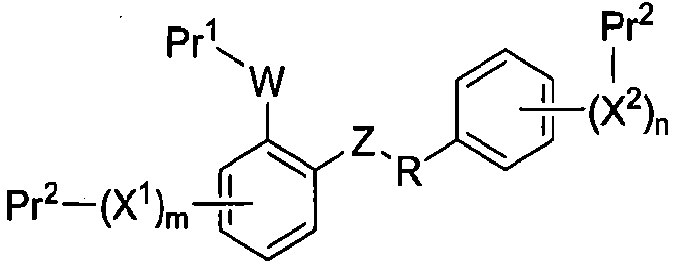
I-1
Настоящее изобретение относится к способам получения соединения формулы I-2:
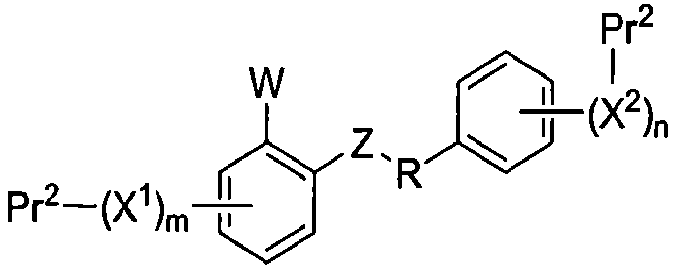
I-2
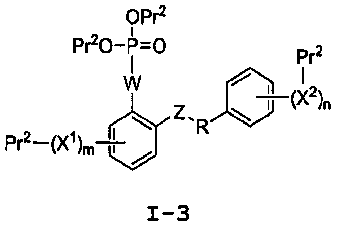
Настоящее изобретение относится к способам получения соединения формулы I-3:
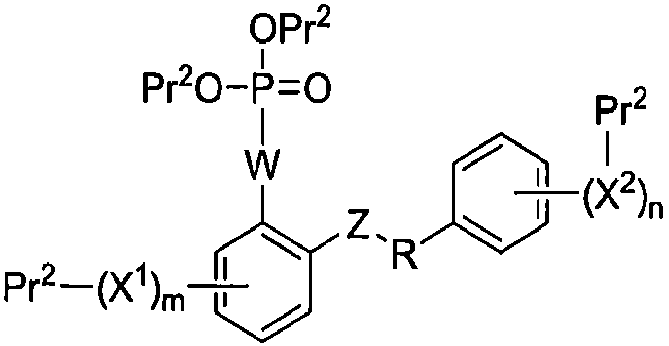
I-3
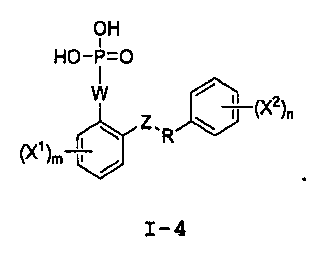
Настоящее изобретение относится к способам получения соединения формулы I-4:
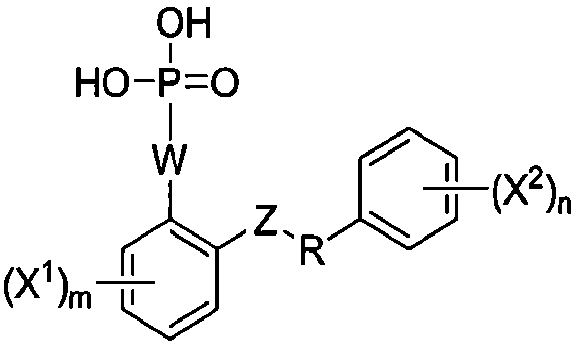
I-4
Подробное описание
Настоящее изобретение относится, среди прочего, к соединениям, которые ингибируют апикальный Na/фосфатный котранспорт и применимы, например, для снижения уровней фосфата в крови и в лечении гиперфосфатемии. Соединения изобретения включают соединения, имеющие формулу I:
I
или его фармацевтически приемлемую соль, где:
R представляет собой С2-4алкил;
W выбран из NRA, O и S;
Z выбран из простой связи, -С(О)-, NRA, O и S;
Х1 и Х2, каждый независимо, выбран из -ОН, -NHRA и -C(О)ОН;
RA представляет собой Н или С1-3алкил;
m равно 1, 2, 3 или 4; и
n равно 1, 2, 3 или 4.
В других вариантах осуществления Х1 и Х2, каждый независимо, выбраны из OR1 и OR2, где R1 и R2 независимо представляют собой Н или защитную группу. В других вариантах осуществления Х1 и Х2 независимо выбраны из COOR1 и COOR2, где R1 и R2 независимо представляют собой Н или защитную группу.
В некоторых вариантах осуществления R представляет собой -СН2-СН2- или -СН2-СН2-СН2-.
В некоторых вариантах осуществления W выбран из NRA и O.
В некоторых вариантах осуществления W представляет собой О.
В некоторых вариантах осуществления Z выбран из простой связи и -С(О)-.
В некоторых вариантах осуществления Z представляет собой -C(O)-.
В некоторых вариантах осуществления Х1 и Х2 независимо выбраны из OH и NH2.
В некоторых вариантах осуществления Х1 и Х2, оба, представляют собой ОН.
В некоторых вариантах осуществления n равно 1 или 2.
В некоторых вариантах осуществления m равно 1 или 2.
В некоторых вариантах осуществления m равно 2, и n равно 1.
В некоторых вариантах осуществления R представляет собой -СН2-СН2-, и Z представляет собой -С(О)-.
В некоторых вариантах осуществления W представляет собой O, и Х1 и Х2, оба, представляют собой ОН.
При использовании в настоящем описании термин “алкил” предназначен для обозначения насыщенной углеводородной группы, которая представляет собой прямую или разветвленную цепь. Примеры алкильных групп включают метил (Ме), этил (Et), пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, втор-бутил, трет-бутил), пентил (например, н-пентил, изопентил, втор-пентил, неопентил) и тому подобное. Алкильная группа может содержать от 1 до примерно 20, от 2 до примерно 20, от 1 до примерно 10, от 1 до примерно 8, от 1 до примерно 6, от 1 до примерно 4 или от 1 до примерно 3 атомов углерода.
При использовании в настоящем описании “арил” относится к моноциклическим или полициклическим (например, имеющим 2, 3 или 4 конденсированных кольца) ароматическим углеводородам, таким как, например, фенил, нафтил, антраценил, фенантренил, инданил, инденил и тому подобное. В некоторых вариантах осуществления арильные группы имеют от 6 до примерно 20 атомов углерода.
При использовании в настоящем описании “гетероарил” относится к ароматическому гетероциклу, имеющему по меньшей мере один кольцевой элемент в виде гетероатома, такой как сера, кислород или азот. Гетероарильные группы включают моноциклические и полициклические (например, имеющие 2, 3 или 4 конденсированных кольца) системы. Примеры гетероарильных групп включают, но без ограничения, пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, фурил, хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пиррил, оксазолил, бензофурил, бензотиенил, бензотиазолил, изоксазолил, пиразолил, триазолил, тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, бензотиенил, пуринил, карбазолил, бензимидазолил, индолинил и тому подобное. Примеры бициклических гетероарильных групп включают, но без ограничения, пуринил, индолил и тому подобное. В некоторых вариантах осуществления образующий кольцо N в гетероарильной группе может быть замещен оксогруппой. В некоторых вариантах осуществления гетероарильная группа имеет от 1 до примерно 20 атомов углерода, и в дополнительных вариантах осуществления примерно от 3 до примерно 20 атомов углерода. В некоторых вариантах осуществления гетероарильная группа содержит от 3 до примерно 14, от 4 до примерно 14, от 9 до примерно 10 или от 5 до 6 атомов, образующих кольцо. В некоторых вариантах осуществления гетероарильная группа имеет от 1 до примерно 4, от 1 до примерно 3 или от 1 до 2 гетероатомов.
Кроме того, следует понимать, что некоторые признаки изобретения, которые описаны, например, в контексте отдельных вариантов осуществления, могут быть также отнесены к комбинации в одном варианте осуществления. Напротив, различные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут быть также предусмотрены отдельно или в любой подходящей субкомбинации.
Соединения, описанные в настоящем описании, могут быть асимметрическими (например, имеющими один или несколько стереоцентров). Все стереоизомеры, такие как энантиомеры и диастереомеры, являются включенными, если не указано особо. Соединения настоящего изобретения, которые содержат асимметрически замещенные атомы углерода, могут быть выделены в оптически активной или рацемической формах. Способы получения оптически активных форм из оптически неактивных исходных продуктов известны в данной области, такие как разделение рацемических смесей или стереоселективный синтез. Многие геометрические изомеры олефинов с С=N двойными связями и тому подобные также могут присутствовать в соединениях, описанных в настоящем описании, и все такие стабильные изомеры предусмотрены в настоящем изобретении. Цис- и трансгеометрические изомеры соединений настоящего изобретения описаны и могут быть выделены в виде смеси изомеров или в виде отдельных изомерных форм.
Разделение рацемических смесей соединений может быть выполнено любым из многочисленных методов, известных в данной области. Пример метода включает фракционную перекристаллизацию при использовании хиральной разделяющей кислоты, которая является оптически активной, образующей соль органической кислотой. Подходящие разделяющие агенты для методов фракционной перекристаллизации представляют собой, например, оптически активные кислоты, такие как D и L формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты или различных оптически активных камфорсульфоновых кислот, таких как β-камфорсульфоновая кислота. Другие разделяющие агенты, подходящие для методов фракционной кристаллизации, включают стереоизомерно чистые формы α-метилбензиламина (например, S и R формы, или диастереомерно чистые формы), 2-фенилглицин, норэфедрин, эфедрин, N-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и тому подобное.
Разделение рацемических смесей может быть также выполнено элюированием на колонке, наполненной оптически активным разделяющим агентом (например, динитробензоилфенилглицином). Подходящая композиция растворителей для элюирования может быть определена специалистом в данной области.
Соединения данного изобретения также могут включать таутомерные формы. Таутомерные формы являются результатом замены простой связи соседней двойной связью вместе с сопутствующей миграцией протона. Таутомерные формы включают прототропные таутомеры, которые представляют собой изомерные состояния протонирования, имеющие такую же эмпирическую формулу и общий заряд. Примеры прототропных таутомеров включают пары кетон-энол, пары амид-имидокислота, пары лактам-лактим, пары енамин-имин и кольцевые формы, где протон может занимать одно или несколько положений гетероциклической системы, например, 1Н- и 3Н-имидазол, 1Н-, 2Н- и 4Н-1,2,4-триазол, 1Н- и 2Н-изоиндол и 1Н- и 2Н-пиразол. Таутомерные формы могут быть в равновесии или стерически заблокированы в одной форме соответствующим замещением.
Соединения данного изобретения могут также включать все изотопы атомов, находящихся в промежуточных продуктах или конечных соединениях. Изотопы включают атомы изотопов, имеющие тот же самый атомный номер, но разные номера масс. Например, изотопы водорода включают тритий и дейтерий.
Термин “соединение”, при использовании в настоящем описании, предназначен для включения всех стереоизомеров, геометрических изомеров, таутомеров и изотопов изображенных структур, включающих структуры в соответствии c общей формулой, представленной в настоящем описании.
Все соединения и их фармацевтически приемлемые соли могут быть обнаружены вместе с другими веществами, такими как вода и растворители (например, гидраты и сольваты), или могут быть выделены.
В некоторых вариантах осуществления соединения данного изобретения или их соли главным образом являются выделенными. Выражение “главным образом выделенное” означает, что соединение по меньшей мере частично или главным образом отделено от окружающей среды, в которой оно образовано или определено. Частичное отделение может включать, например, композицию, обогащенную соединениями по изобретению. Значительное отделение может включать композиции, содержащие по меньшей мере примерно 50%, по меньшей мере примерно 60%, по меньшей мере примерно 70%, по меньшей мере примерно 80%, по меньшей мере примерно 90%, по меньшей мере примерно 95%, по меньшей мере примерно 97% или по меньшей мере примерно 99% масс. соединений по изобретению или их соли. Способы выделения соединений и их солей являются общепринятой практикой в данной области.
Фраза “фармацевтически приемлемые” использована в настоящем описании для обозначения таких соединений, продуктов, композиций и/или лекарственных форм, которые подходят, в рамках качественной медицинской оценки, для применения в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримого с приемлемым соотношением польза/риск.
Выражения “температура окружающей среды” и “комнатная температура”, при их использовании в настоящем описании, понятны в данной области и обычно относятся к температуре, например температуре реакции, которая является примерно температурой помещения, в котором проводят реакцию, например температурой примерно от 20°С до примерно 30°С.
Настоящее изобретение также включает фармацевтически приемлемые соли соединений, описанных в настоящем описании. При использовании в настоящем описании выражение “фармацевтически приемлемые соли” относится к производным раскрытых соединений, где исходное соединение модифицировано преобразованием группы существующей кислоты или основания в его солевую форму. Примеры фармацевтически приемлемых солей включают, но без ограничения только ими, соли неорганических или органических кислот с остатками оснований, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и тому подобное. Фармацевтически приемлемые соли настоящего изобретения включают обычные нетоксичные соли исходного соединения, полученного, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли настоящего изобретения могут быть синтезированы из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Как правило, такие соли могут быть получены взаимодействием форм свободной кислоты или основания для данных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе, или в смеси двух растворителей; обычно предпочтительна безводная среда, а именно простой эфир, этилацетат, спирты (например, метанол, этанол, изопропанол или бутанол) или ацетонитрил (ACN). Перечень подходящих солей находится в публикациях Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 и Journal of Pharmaceutical Science, 66, 2 (1977), каждая из которых включена в настоящее описание посредством ссылки во всей своей полноте.
Синтез
Способы и промежуточные продукты настоящего изобретения пригодны для получения ингибиторов кишечного апикального натрий/фосфатного котранспорта, эффективных в снижении уровней фосфата в крови и гиперфосфатемии. Соединения изобретения, включая их соли, могут быть получены при использовании методов органического синтеза и могут быть синтезированы согласно любому из многочисленных возможных синтетических путей.
Реакции для получения соединений изобретения могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по существу нереакционноспособными по отношению к исходным продуктам (реагентам), промежуточным продуктам или продуктам реакций при температурах, при которых проводят реакции, например, температурах, которые могут изменяться от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси более чем из одного растворителя. В зависимости от конкретной стадии реакции, подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области.
Получение соединений данного изобретения может включать защиту или удаление защиты различных химических групп. Необходимость защиты или удаления защиты, выбор соответствующих защитных групп могут быть легко определены специалистом в данной области. Химию защитных групп можно найти, например, в публикации T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3th Ed., Wiley & Sons, Inc., New York (1999), которая включена в настоящее описание посредством ссылки во всей своей полноте.
Реакции могут быть отслежены любым подходящим методом, известным в данной области. Например, образование продукта может быть отслежено спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например, УФ-видимая), масс-спектрометрия, или хроматографическими методами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография(ТСХ).
Примеры синтетических способов получения соединений данного изобретения представлены на схемах ниже. Например, соединения данного изобретения могут быть получены по общей ретросинтетической схеме, показанной на схеме 1. Соединения формулы 1 могут быть получены взаимодействием соединения формулы 2 с фторирующим агентом. Соединения формулы 2 могут быть синтезированы удалением Pr2 из соединения формулы 3. Соединения формулы 3 могут быть легко получены при фосфорилировании соединений формулы 4. Наконец, соединения формулы 5 могут быть синтезированы через выявление точки присоединения (W) для селективного снятия защиты при фосфорилировании.
Схема 1
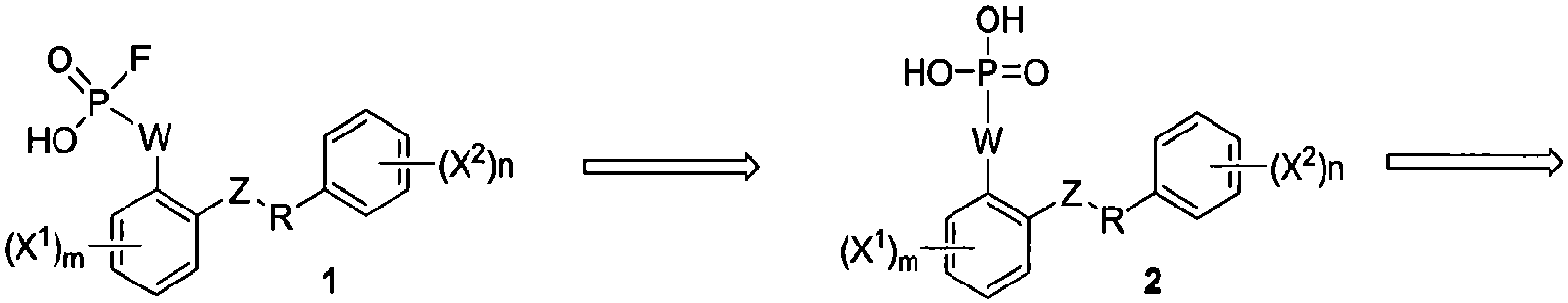
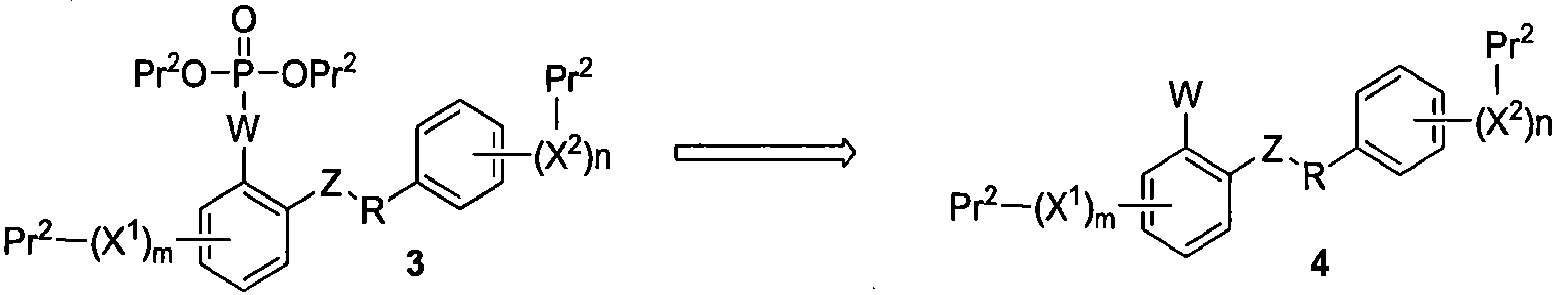
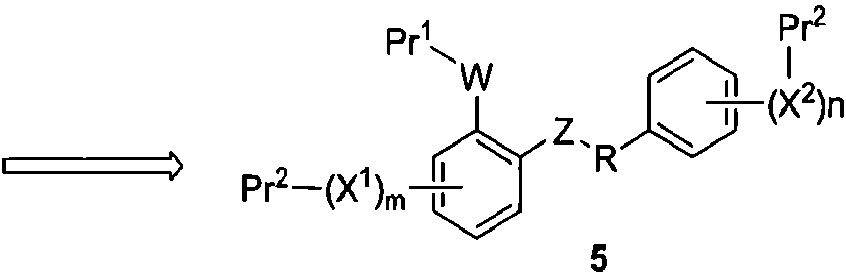
Таким образом, настоящее изобретение относится к способу получения соединения формулы I:
I
или его фармацевтически приемлемой соли, где R представляет собой С2-4алкил;
W выбран из NRA, O и S; Z выбран из простой связи, -С(О)-, NRA, O и S; Х1 и Х2, каждый независимо, выбран из -ОН, -NHRA и -C(О)ОН; RA представляет собой Н или С1-3алкил; m равно 1, 2, 3 или 4; и n равно 1, 2, 3 или 4;
включающему:
а) удаление Pr1 в присутствии Pr2 из соединения формулы I-1:
I-1
где Pr1 и Pr2 представляют собой защитные группы, для получения соединения формулы I-2:
I-2
b) взаимодействие соединения формулы I-2 с фосфорилирующим агентом, для получения соединения формулы I-3:
I-3
с) удаление Pr2 из соединения формулы I-3 восстановителем, для получения соединения формулы I-4:
I-4
d) взаимодействие соединения формулы I-4 c фторирующим агентом, для получения указанного соединения формулы I.
В некоторых вариантах осуществления Pr1 представляет собой:
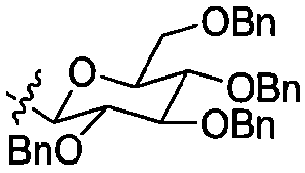
В некоторых вариантах осуществления Pr2 представляет собой бензил.
В некоторых вариантах осуществления фосфорилирующий агент представляет собой сложный эфир фосфита. В следующем варианте осуществления сложный эфир фосфита представляет собой дибензилфосфит. В другом варианте осуществления фосфорилирующий агент представляет собой фосфоновую кислоту. В следующем варианте осуществления фосфоновая кислота представляет собой фторфосфоновую кислоту.
В некоторых вариантах осуществления восстановитель составлен из газа водорода и переходного металла.
В некоторых вариантах осуществления фторирующий агент представляет собой динитрофторбензол. В следующем варианте осуществления фторирующий агент представляет собой фтористоводородную кислоту.
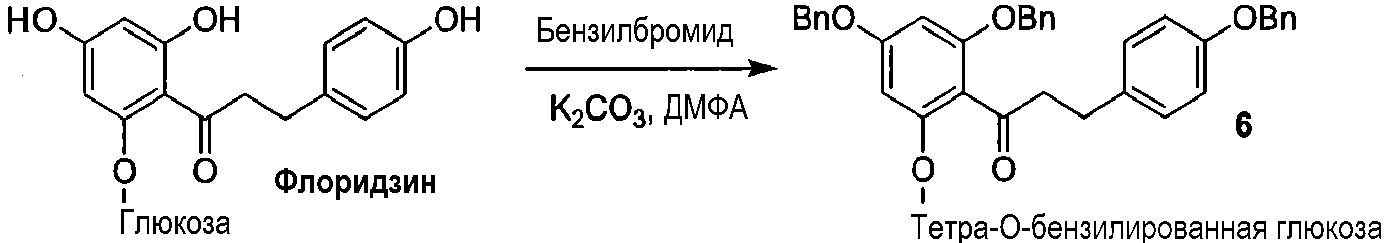
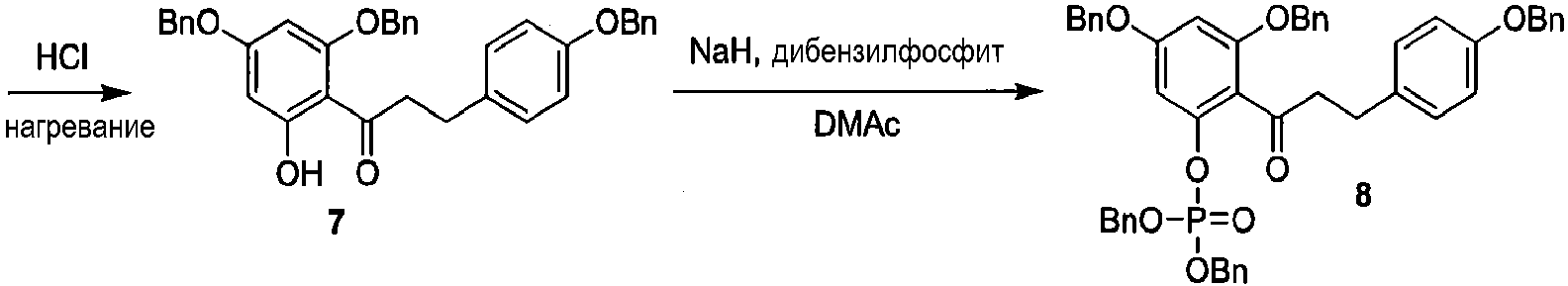
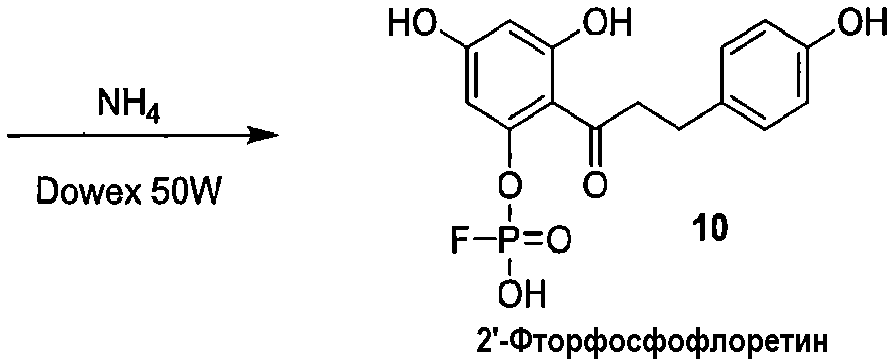
Как показано на схеме 2, известное соединение флоридзин бензилируют, с получением соединения 6, полностью защищенного бензильными группами. Обработка соединения 6 в кислых условиях давала фенол 7, который превращали в защищенное бензилом фосфатное соединение 8. Каталитическое гидрирование соединения 8 давало 2'-фосфофлоретин (9), который фторировали фторирующим агентом, таким как динитрофторбензол (DNFB) или фтористоводородная кислота (HF), для получения 2'-фторфосфофлоретина (10).
Схема 2
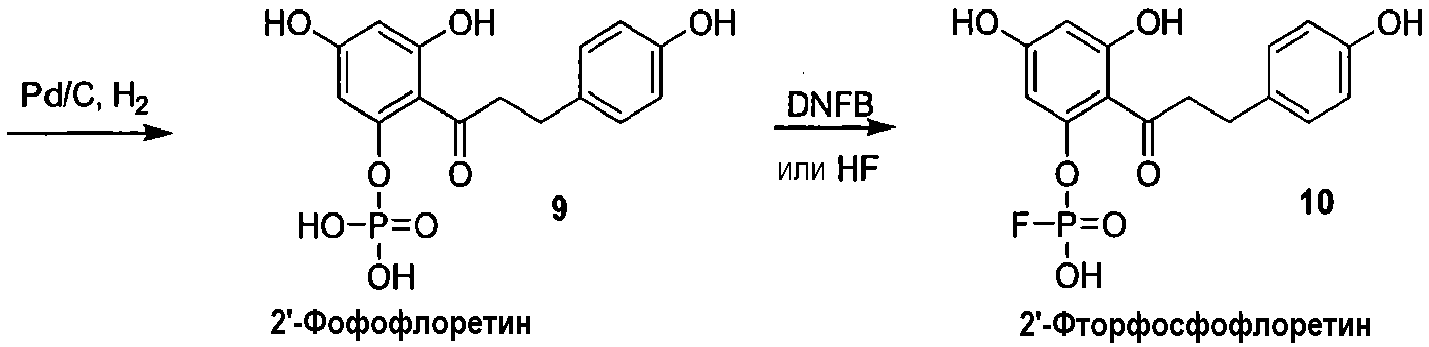
Альтернативно, 2'-фосфофлоретин (9) можно фторировать, используя безводную фтористоводородную кислоту, для получения 2'-фторфосфофлоретина (10) (схема 3).
Схема 3
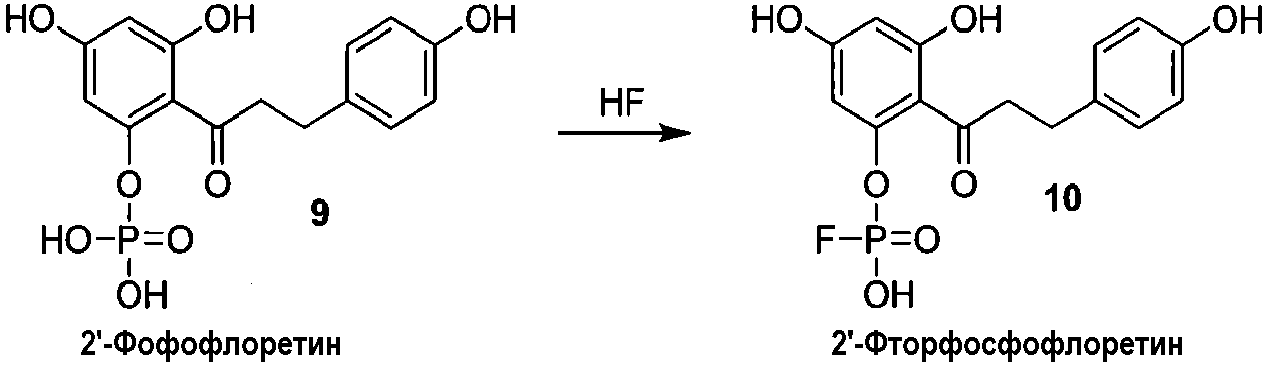
Альтернативный путь получения 2'-фторфосфофлоретина показан на схеме 4, исходя из известного соединения флоридзина. Бензилирование флоридзина давало соединение 6, с которого селективно удаляли защиту, с получением фенола 7. Обработка дихлорхроматом (DCC) и фторфосфоновой кислотой давала фторфосфоновую кислоту 8а. Удаление бензильных групп из соединения 8а выполняли, используя аммиак и Dowex 50W, c получением 2'-фторфосфофлоретина.
Схема 4
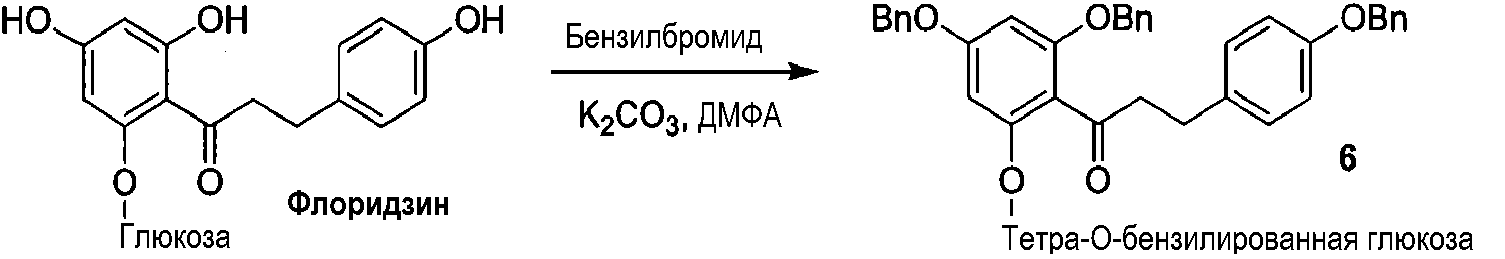
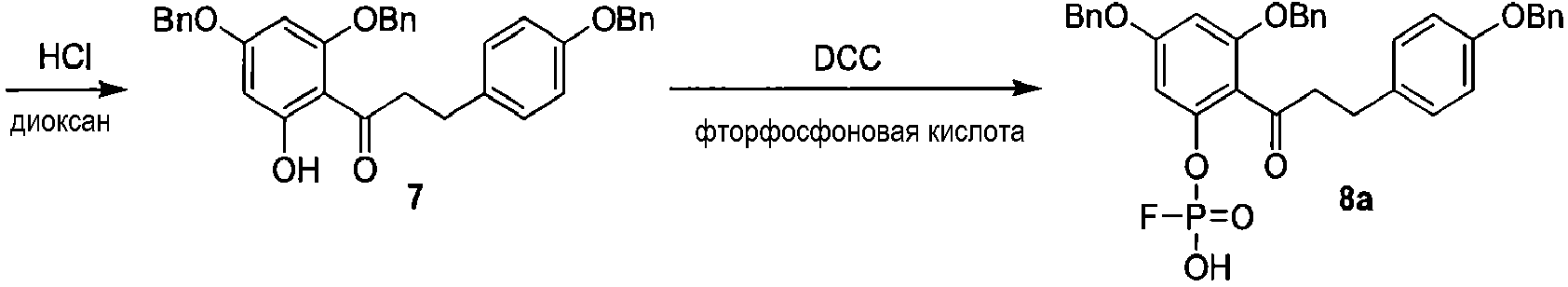
Способы применения
Соединения данного изобретения могут ингибировать кишечный апикальный натрий/фосфатный котранспорт. Соединения данного изобретения также могут быть эффективными при лечении для снижения уровней фосфата в крови и гиперфосфатемии.
Настоящее изобретение дополнительно относится к способам лечения заболевания, вызванного или связанного с повышенными уровнями фосфата в крови у млекопитающего субъекта, включающим идентификацию субъекта, в котором желательно снижение уровней фосфата в крови, и введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества или дозы соединения настоящего изобретения или его фармацевтической композиции. В некоторых вариантах осуществления субъект представляет собой млекопитающее. В некоторых вариантах осуществления субъект представляет собой человека. В некоторых вариантах осуществления соединение вводят перорально. В некоторых вариантах осуществления заболевание представляет собой хроническую почечную недостаточность. В некоторых вариантах осуществления хроническая почечная недостаточность связана с и/или сопровождается гиперфосфатемией, вторичным гиперпаратиреозом, почечной остеодистрофией, гипертензией, метаболическим ацидозом или заболеванием сердца. Хроническая почечная недостаточность может представлять собой почечную недостаточность на конечной стадии, и пациенты, страдающие почечной недостаточностью на конечной стадии, подлежат лечению, как описано в настоящем описании.
При использовании в настоящем описании термин “приведение в контакт” относится к совместному вводу указанных составляющих в in vitro систему или в in vivo систему. Например, “приведение в контакт” соединения изобретения с кишечным апикальным натрий/фосфатным котранспортом включает введение соединения настоящего изобретения индивидууму или пациенту, такому как человек, а также, например, введение соединения изобретения в образец, содержащий клеточный или очищенный препарат кишечного апикального натрий/фосфатного котранспорта.
При использовании в настоящем описании, термин “индивидуум” или “пациент”, используемый взаимозаменяемо, относится к любому животному, включая млекопитающих, предпочтительно мышей, крыс, других грызунов, кроликов, собак, кошек, свиней, крупный рогатый скот, овец, лошадей или приматов, и наиболее предпочтительно людей.
При использовании в настоящем описании фраза “терапевтически эффективное количество” относится к количеству активного соединения или фармацевтического средства, которое вызывает биологический или медицинский ответ, который фиксируется в ткани, системе, животном, индивидууме или человеке исследователем, ветеринаром, медицинским доктором или другим клиницистом.
При использовании в настоящем описании термин “лечение” или “терапия” относится к одному или нескольким выражениям, таким как: (1) предотвращение заболевания, например предотвращение заболевания, состояния или расстройства у индивидуума, который предрасположен к заболеванию, состоянию или расстройству, но еще не испытывает или не проявляет патологии или симптоматологии заболевания; (2) ингибирование заболевания, например ингибирование заболевания, состояния или расстройства у индивидуума, который испытывает или проявляет патологию или симптоматологию заболевания, состояния или расстройства; и (3) улучшение заболевания, например улучшение заболевания, состояния или расстройства у индивидуума, который испытывает или проявляет патологию или симптоматологию заболевания, состояния или расстройства (т.е. реверсию патологии и/или симптоматологии), а именно снижение тяжести заболевания.
Фармацевтические препараты и лекарственные формы
При использовании фармацевтических препаратов, соединения данного изобретения могут быть введены в форме фармацевтических композиций. Данные композиции могут быть получены способом, хорошо известным в фармацевтической области, и могут быть введены множеством путей, зависящих от того, желательно ли местное или системное лечение, и от области, предназначенной для лечения. Введение может быть пероральным.
Данное изобретение также включает фармацевтические композиции, которые содержат в качестве активного ингредиента соединение данного изобретения или его фармацевтически приемлемую соль, в комбинации с одним или несколькими фармацевтически приемлемыми носителями (эксципиентами). В некоторых вариантах осуществления композиция является подходящей для перорального введения. При изготовлении композиций данного изобретения активный ингредиент обычно смешивают с эксципиентом, разбавляют эксципиентом или помещают внутри такого носителя в форме, например капсулы, маленького пакетика, бумаги или другого контейнера. Когда эксципиент служит в качестве разбавителя, он может представлять собой твердое вещество, полутвердое вещество или жидкий продукт, который действует в качестве носителя, наполнителя или среды для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, лепешек, маленьких пакетиков, капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, мягких и твердых желатиновых капсул и стерильных упакованных порошков.
При приготовлении состава активное соединение может быть размолото для обеспечения соответствующего размера частиц перед объединением с другими ингредиентами. Если активное соединение, в основном, нерастворимо, то оно может быть размолото до размера частиц менее чем 200 меш. Если активное соединение, в основном, растворимо в воде, то размер частиц может быть отрегулирован размолом для обеспечения в основном однородного распределения в составе, например около 40 меш.
Соединения данного изобретения могут быть размолоты при использовании известных методик размола, таких как влажный размол, чтобы получить размер частиц, соответствующий образованию таблеток и для других типов составов. Тонко измельченные (из наночастиц) составы соединений данного изобретения могут быть приготовлены способами, известными в данной области, например, см. International App. № WO 2002/000196.
Некоторые примеры подходящих эксципиентов включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Составы могут дополнительно содержать: загустители, такие как тальк, стеарат магния и минеральное масло; смачиватели; эмульгирующие и суспендирующие средства; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители; и вкусовые добавки. Композиции изобретения могут быть приготовлены в виде состава, для обеспечения быстрого, пролонгированного или замедленного высвобождения активного ингредиента после введения пациенту с использованием методик, известных в данной области.
Композиции могут быть сформулированы в составы в единичной дозированной форме, причем каждая дозировка содержит примерно от 5 до примерно 1000 мг (1 г), чаще примерно от 100 до примерно 500 мг активного ингредиента. Термин “единичная дозированная форма” относится к физическим дискретным единицам, подходящим в качестве единичных дозировок для субъектов-людей и других млекопитающих, причем каждая единица содержит предварительно определенное количество активного продукта, рассчитанное для того, чтобы вызывать требуемый терапевтический эффект, в сочетании с подходящим фармацевтическим эксципиентом.
В некоторых вариантах осуществления соединения или композиции данного изобретения содержат примерно от 5 до примерно 50 мг активного ингредиента. Специалисту в данной области будет понятно, что это включает соединения или композиции, содержащие примерно 5 до примерно 10, примерно 10 до примерно 15, примерно 15 до примерно 20, примерно 20 до примерно 25, примерно 25 до примерно 30, примерно 30 до примерно 35, примерно 35 до примерно 40, примерно 40 до примерно 45, или примерно 45 до примерно 50 мг активного ингредиента.
В некоторых вариантах осуществления соединения или композиции данного изобретения содержат примерно от 50 до примерно 500 мг активного ингредиента. Специалисту в данной области будет понятно, что это включает соединения или композиции, содержащие примерно 50 до примерно 100, примерно 100 до примерно 150, примерно 150 до примерно 200, примерно 200 до примерно 250, примерно 250 до примерно 300, примерно 350 до примерно 400, или примерно 450 до примерно 500 мг активного ингредиента.
В некоторых вариантах осуществления соединения или композиции данного изобретения содержат примерно от 500 до примерно 1000 мг активного ингредиента. Специалисту в данной области будет понятно, что это включает соединения или композиции, содержащие примерно 500 до примерно 550, примерно 550 до примерно 600, примерно 600 до примерно 650, примерно 650 до примерно 700, примерно 700 до примерно 750, примерно 750 до примерно 800, примерно 800 до примерно 850, примерно 850 до примерно 900, примерно 900 до примерно 950, или примерно 950 до примерно 1000 мг активного ингредиента.
Активное соединение может быть эффективным на протяжении широкого интервала доз и обычно вводится в фармацевтически эффективном количестве. Однако следует понимать, что количество действительно вводимого соединения обычно будет определено врачом в соответствии с релевантными обстоятельствами, включающими состояние, предназначенное для лечения, выбранный путь введения, конкретное вводимое соединение, возраст, массу и реакцию отдельного пациента, тяжесть симптомов у пациента и тому подобное.
Для приготовления твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим эксципиентом для создания твердого предварительного состава композиции, содержащего гомогенную смесь соединения настоящего изобретения. Что касается данных гомогенных предварительных составов композиций, активный ингредиент обычно равномерно диспергируют по всей композиции, так что композиция может быть легко подразделена на равно эффективные единичные дозированные формы, такие как таблетки, пилюли и капсулы. Данный твердый предварительный состав затем подразделяют на единичные дозированные формы описанного выше типа, содержащие, например, примерно 0,1 до примерно 1000 мг активного ингредиента настоящего изобретения.
Таблетки или пилюли настоящего изобретения могут быть с покрытием или составлены иным образом для того, чтобы получить лекарственную форму, предоставляющую преимущественно пролонгированное действие. Например, таблетка или пилюля может содержать внутреннюю дозу и внешний дозированный компонент, причем последний находится в форме обертки, поверх первого. Два компонента могут отделяться энтеральным слоем, который служит для того, чтобы противостоять дезинтеграции в желудке и позволяет внутреннему компоненту проходить интактно в двенадцатиперстную кишку или быть замедленным по высвобождению. Множество продуктов может быть использовано для таких энтеральных слоев или покрытий, причем такие продукты включают ряд полимерных кислот и смесей полимерных кислот с такими продуктами как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которые соединения и композиции настоящего изобретения могут быть включены для введения перорально или инъекцией, включают водные растворы, в подходящем случае ароматизированные сиропы, водные или масляные суспензии, ароматизированные эмульсии со съедобными маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические наполнители.
В некоторых вариантах осуществления композиции вводят пероральным путем для местного эффекта. Композиции растворов, суспензий или порошков могут быть введены перорально с помощью устройств, которые доставляют состав соответствующим способом.
Количество соединения или композиции, вводимое пациенту, будет изменяться в зависимости от того, что вводится, цели введения, а именно профилактики или терапии, состояния пациента, способа введения и подобного. При терапевтических применениях, композиции могут быть введены пациенту, уже страдающему заболеванием, в количестве, достаточном для того, чтобы вылечить или по меньшей мере частично остановить симптомы заболевания и его осложнения. Эффективные дозы будут зависеть от состояния заболевания, которое подвергается лечению в настоящее время, а также заключения лечащего врача в зависимости от факторов, таких как тяжесть заболевания, возраст, масса и общее состояние пациента, и тому подобное.
Композиции, вводимые пациенту, могут быть в форме фармацевтических композиций, описанных выше. Данные композиции могут быть стерилизованы по обычным методикам стерилизации, или могут быть стерильно отфильтрованы. Водные растворы могут быть упакованы для применения как таковые, или лиофилизированы, причем лиофилизированный препарат объединяется со стерильным водным носителем перед введением. рН смешанных составов обычно будет составлять от 3 до 11, предпочтительнее от 5 до 9 и наиболее предпочтительно от 7 до 8. Следует понимать, что использование некоторых указанных выше эксципиентов, носителей или стабилизаторов будет приводить к образованию фармацевтических солей.
Терапевтическая дозировка соединения настоящего изобретения может меняться согласно, например, конкретному применению, для которого проводят лечение, способа введения соединения, здоровья и состояния пациента и заключения врача, прописывающего лекарственные средства. Пропорция или концентрация соединения данного изобретения в фармацевтической композиции может меняться в зависимости от ряда факторов, включающих дозировку, химические характеристики (например, гидрофобность) и способ введения. Например, соединения данного изобретения могут быть даны в водном физиологическом буферном растворе, содержащем примерно 0,1 до примерно 10% мас./об. соединения для парентерального введения. Некоторые конкретные интервалы доз представляют собой интервалы примерно от 1 мкг/кг до примерно 1 г/кг массы организма в сутки.
В исследованиях с 2'-фосфофлоретином авторы вводили животным 0,1 г/сутки и не наблюдали побочных эффектов на протяжении 3-месячного опыта на обычных крысах или на 5/6 крысах с нефрэктомией. В других исследованиях с 2'-фосфофторфлоретином авторы вводили животным 0,02 г/сутки. Авторы не наблюдали побочных эффектов на протяжении 2-месячного опыта на обычных крысах; на протяжении 1-месячного опыта на 5/6 крысах с нефрэктомией; и на протяжении 1-месячного опыта на крысах, линии Dahl, чувствительных к развитию гипертензии при употреблении солевой диеты. Соединения авторов благоприятно выдерживали сравнение с солями кальция (такими как TUMS®), LaCO3 (FOSRENOL®) и фосфор-связывающим полимером (RENAGEL®).
В некоторых вариантах осуществления интервалы доз представляют собой интервалы примерно от 0,01 мкг/кг до примерно 100 мг/кг массы организма в сутки. Дозировка вероятно зависит от таких переменных, как тип и степень прогрессирования заболевания или расстройства, общее состояние здоровья конкретного пациента, относительная биологическая эффективность отобранного соединения, состав эксципиента и путь его введения. Эффективные дозы могут быть экстраполированы из кривых доза-эффект, полученных in vitro или в тест-системах на моделях животных.
Примеры
Пример 1. Синтез 2'-фторфосфофлоретина (2'-FPP) при использовании динитрофторбензола (DNFB)
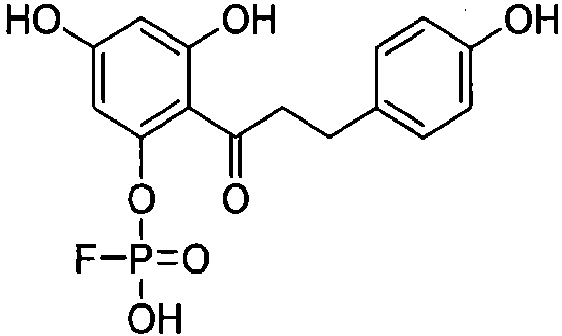
2'-Фторфосфофлоретин (2'-FPP) синтезировали по способу Peerce et al., Am. J. Physiol. Gastrointest. Liver Physiol. 283: G848-G855, 2002.
Стадия 1. 4,6,4-Три-О-бензилфлоретин
Флоридзин (2 г) растворяли в повторно перегнанном диметилформамиде (35 мл) и карбонате калия (3,1 г). К полученному раствору добавляли бензилбромид (2,7 мл) и реакционной смеси давали перемешиваться в течение 3 суток при 23°С. Полученный раствор перегоняли в вакууме, и остаток охлаждали до комнатной температуры. Остаток экстрагировали три раза смесью вода/этилацетат (2:1). Органические слои объединяли и концентрировали в вакууме. Остаток растворяли в 1,4-диоксане (200 мл), и к раствору добавляли по каплям HCl до конечной концентрации 0,4н. Смесь кипятили с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали, разбавляли 1М бикарбонатом натрия и экстрагировали этилацетатом натрия. Экстракцию этилацетатом повторяли три раза. Объединенные органические экстракты промывали водой с последующими двумя промывками NaCl (0,9%). Безводный Na2SO4 добавляли к органическому слою и перемешивали. Фильтрованием в вакууме удаляли Na2SO4. Свежий Na2SO4 добавляли к полученному раствору, который затем перемешивали в течение 12 часов при 23°С. Фильтрованием в вакууме удаляли Na2SO4, и фильтрат концентрировали, с получением 4,6,4-три-О-бензилфлоретина (2,1 г, выход 92%);
1H-ЯМР (CDCl3) δ 13,6 (с, 1H); 7,46-7,29 (м, 15H); 6,86 (д, J=8,8 Гц, 2H); 6,8 (д, J=8,8 Гц, 2H); 6,35 (д, J=2,3 Гц, 1H); 6,21 (д, J=2,3 Гц, 1H); 5,17 (с, 2H); 5,14 (с, 2H); 5,07 (с, 2H); 3,2 (n, J=7,1 Гц, 2H) м.д.
Стадия 2. Дибензилфосфотрибензилфлоретин
4,6,4-Три-О-бензилфлоретин (2,1 г) растворяли в диметилацетамиде (DMAc) и помещали на лед. Добавляли гидрид натрия (60%), и смесь перемешивали в течение 1 часа при 23°С. Раствор охлаждали, и гидрид натрия инактивировали тетрахлоридом углерода. Добавляли дибензилфосфит (1,31 мл) в N,N-диметилацетамиде, и раствор перемешивали в течение 30 мин. Раствор подкисляли и распределяли между водой и смесью гексан/этилацетат (1:1). Водный слой экстрагировали три раза смесью гексан/этилацетат. Объединенные органические экстракты промывали 0,9% NaCl и сушили над безводным сульфатом натрия. Сульфат натрия удаляли фильтрованием. Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/дихлорметан/гексаны (5:25:70). Получали дибензилфосфортрибензилфлоретин (750 мг, выход 43%);
1H-ЯМР (CDCl3) δ 7,42-7,29 (м, 25 H); 6,93 (д, J=8,8 Гц, 2H); 6,78 (д, J=8,8 Гц, 2H); 6,63 [двойной дублет (дд), J=1,2, 2 Гц, 1H]; 6,4 (дд, J=0,6, 2,1 Гц, 1H); 5,06 (с, 2H); 5,04 (с, 2H); 4,97 (д, J=4,8 Гц, 4H); 4,87 (с, 2H); 3,03 (т, J=8,4 Гц, 2H); 2,83 (т, J=8,2 Гц, 2H) м.д.
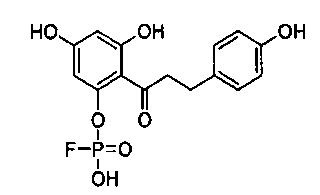
Стадия 3. 2'-Фторфосфофлоретин (2'-FPP)
Дибензилфосфотрибензилфлоретин растворяли в этилацетате. Добавляли палладий на углероде (200 мг, 10%-ный), и раствор перемешивали в атмосфере газообразного Н2 в течение 2 часов. Раствор фильтровали через целит (Sigma-Aldrich). Осадок на целитном фильтре дважды промывали этилацетатом, и объединенные промывки концентрировали при пониженном давлении с получением 2'-PP (400 мг, выход 29%) после сушки: температура плавления (т.пл.): 171-172°С;
1H-ЯМР (d6-ДМСО) δ 13,0 (с, 1H); 10,7 (ушир.с, 1H); 9,2 (ушир.с, 1H); 7,03 (д, J=8,6 Гц, 2H); 6,64 (д, J=8,4 Гц, 2H); 6,63 (дд, J=1,2, 2,1 Гц, 1H); 2,77 (д, J=7,6 Гц, 2H) м.д.
31P-ЯМР в D2O давал одиночный пик в 4 м.д., состоящий из 98% фосфорного сигнала. 31P-ЯМР в ДМСО давал одиночный пик в 4,3 импульсы/мин. Опыты с Н-несвязанными 13С-ЯМР давали 12 специфичных видов углерода, которые могли быть предназначены ингибитору.
Масс-спектрометрия с электрораспылением давала заряд масса/ион в 355 (масса+протон), согласующийся с вычисленной молекулярной массой 2'-РР, равной 354.
2'-РР пропускали через обменную колонку, уравновешенную 100 мМ бикарбонатом тетраметиламмония, концентрировали в вакууме и добавляли к 10 мМ DNFB. Реакционную смесь перемешивали при 23°С в течение 4 часов, и реагенты промывали через колонку с силикагелем при элюировании смесью хлороформ:метанол (70%:30%). Фракции сушили и осаждали 0,1М NaOH. Осадок собирали, буфер обменивали через колонку Sephadex G-10 и концентрировали в вакууме.
Пример 2. Синтез 2'-фторфосфофлоретина (2'-FPP) с использованием фтористоводородной кислоты
Альтернативный синтез 2'-FPP проводили, заменяя DNFB фтористоводородной кислотой.
Исходя из 20 мг 2'-РР, растворенного в 1 мл ДМФА, добавляли 0,1 мл 0,1М HF и раствору давали стоять при 23°С в течение 2 часов. Реакционную смесь охлаждали и нейтрализовали до рН 8 с использованием 12н. NaOH. Осажденные продукты пропускали через колонку Sephadex G-10, уравновешенную 100 мМ ТМАНСО3, и лиофилизировали.
Чистоту 2'-FPP оценивали тонкослойной хроматографией на силикагеле, используя смесь хлороформ:метанол (7:3) или хлороформ:3-пропанол (8:2). Анализ 2'-FPP после ТСХ проводили сравнением флоретина и 2'-PP. ТСХ-пластины исследовали на фосфат реакцией Фиске-Суббароу и на фторид смесью цирконий-ализариновый краситель HCl. Положительная реакция с ализариновым красителем требовала нагревания ТСХ-полоски в течение 1 часа при 60°С.
1H-ЯМР (d6-ДМСО) δ 10,7 (с, 1H); 9,3 (с, 1H); 7,17 (т, 2H J=8,6 Гц), 6,8 (т, 2H); 6,6 (д, 2H, J=8,4 Гц), 3,2 (т, 2H, J=2 Гц); 2,9 (т, 2H, J=2,1 Гц); 1,9 (слабый синглет) м.д.
Пример А: Воздействие 2'-FPP на Na-зависимое поглощение [32P]фосфата
Воздействие 2'-FPP на Na-зависимое поглощение [32P]фосфата в кишечные мембранные (апикальные) везикулы щеточной каемки оценивали, используя опыты с быстрой фильтрацией при быстром смешивании и подсчетом с помощью жидкостной сцинтилляции оставшихся на фильтре количеств в присутствии 100 мМ Na или 100 мМ К цис (внутри) направленных градиентов. 2'-PP и 2'-FPP оба ингибировали Na-зависимое поглощение фосфата в кишечные мембранные везикулы щеточной каемки. 2'-FPP был в 12 раз более эффективным, чем 2'-РР в ингибировании Na-зависимого поглощения фосфата. Кажущаяся IC50 для 2'-РР была равна 46 нМ±8 нМ (n=4) подобно предыдущим результатам (Peerce et al., Biochem. Biophys. Res. Comm. 301:8-12, 2003; Peerce and Clarke, Am. J. Physiol. 283:G848-G855, 2002). IC50 для 2'-FPP была равна 3,6 нМ±0,6 нМ (n=4).
Пример В: Действие фосфофлоретинов на гидролиз п-нитрофосфата посредством щелочной фосфатазы и кишечных фосфатаз BBMV крысы
Для результатов, показанных в примере А, использовали 3-секундное воздействие на везикулы и субстраты и 5-минутное воздействие на везикулы 2'-FPP. Предварительная экспозиция и деградация 2'-PP в противоположность 2'-FPP могла бы вносить вклад в повышенную эффективность 2'-FPP по сравнению с 2'-PP. Для оценки данной возможности исследовали эффект фосфофлоретинов на п-нитрофосфатный гидролиз посредством щелочной фосфатазы и кишечных фосфатаз BBMV крысы.
2'-PP является более хорошим ингибитором активности щелочной фосфатазы и активности кишечной фосфатазы BBMV, чем 2'-FPP. 2 мМ 2'-PP давал приблизительно 50% ингибирование активности кишечной фосфатазы BBMVB по сравнению с менее чем 10% ингибированием с использованием 2'-FPP. В соответствии с интерпретацией, что эффективность 2'-PP в качестве ингибитора Na-зависимого поглощения фосфата была ограничена опосредованным фосфатазой гидролизом 2'-PP, добавление ингибиторов щелочной фосфатазы (аскорбиновая кислота+цистеин) снижало IC50 для ингибирования посредством 2'-РР Na-зависимого поглощения фосфата до 34% (от 38 нМ до 25 нМ). Данные результаты указывают, что по сравнению с 2'-PP, 2 фактора являются ответственными за повышенную эффективность 2'-FPP в качестве ингибитора или Na-зависимого поглощения фосфата в кишечные мембранные везикулы щеточной каемки: 1) повышенная устойчивость 2'-FPP к катализируемой фосфатазой деградации сложного эфира фосфата, и 2) повышенная аффинность фторфосфатов для кишечного фосфатного сайта Na-фосфатного котранспортера по сравнению с фосфатом.
Пример С: Продолжительность действия разового воздействия на кишечник крысы 2'-FPP и 2'-PP
Для исследования продолжительности действия разовой дозы 2'-FPP на поглощение фосфата вырезали полоски кишечника (приблизительно 5 см квадратики) и инкубировали в 12 луночных планшетах для культуры клеток в присутствии 2'-FPP или 2'-PP в течение 1 часа, с последующей воздействием сырой DMEM (модифицированная по способу Дульбекко среда Игла), [32P]фосфатом и различными концентрациями 2'-PP 2'-FPP.
Разовое воздействие 2'-РР приводило к более чем 60% ингибированию поглощения фосфата, и такое ингибирование было устойчивым в течение 6 часов после лекарственного воздействия. Напротив, воздействие 2'-РР приводило к 40% ингибированию поглощения фосфата, которое уменьшалось до менее чем 35% ингибирования в течение 6 часов после лекарственного воздействия. Поглощение глюкозы не подвергалось воздействию как 2'-РР, так и 2'-FPP, согласно предыдущим сообщениям о том, что фосфофлоретины являются специфическими для Na-фосфатного котранспорта, и что полоски кишечника были жизнеспособными в течение проведения эксперимента.
Данные эксперименты были расширены для изучения действия концентрации 2'-FPP на поглощение фосфата.
Крысиные полоски двенадцатиперстной кишки/тощей кишки в 1 см квадратик инкубировали в DMEM при 37°С и 5% СО2 в течение одного часа. После инкубационного периода, DMEM заменяли включением 2'-FPP, и полоски инкубировали в течение дополнительного часа. После экспозиции в 1 час с 2'-FPP, DMEM заменяли свежей средой+5 мкКи [32P]фосфата. Полоски подвергали воздействию радиоактивного фосфата в течение 1 часа, осаждали 10% TCA и центрифугировали при 5000 g в течение 30 минут. Аликвоту супернатанта брали для подсчета сцинтилляции.
Na-зависимое поглощение фосфата в кишечнике составлено как из Na-зависимого, так и Na-независимого компонентов. Na-зависимое поглощение в двенадцатиперстную кишку и тощую кишку составляет приблизительно 70% Na-зависимого и 30% Na-независимого поглощения фосфата. Данные результаты согласуются с 2'-FPP, ингибирующим Na-зависимый компонент, который направляется кишечным мембранным Na/фосфатным котранспортером щеточной каемки, NaPi 2b. Максимальное ингибирование составляло 67%±5% (n=4) общей абсорбции фосфата. Кажущаяся IC50 для 2'-FPP составляла 160 нМ±20 нМ (n=4). Это сравнимо с максимальным ингибированием в идентичных условиях 43±6% (n=3) для 2'-PP, и IC50 в 15 мкМ±3 мкМ (n=3).
Пример D
Изучали действие 2'-FPP на фосфат сыворотки и кальций сыворотки на 4-5-месячных крысах с нормальной функцией почек. Крысам вводили через желудочный зонд ежедневно 3 мл 0,5 мкМ 2'-FPP в фосфатном буферном физиологическом растворе с рН 7. Приблизительно через 30 минут крыс подвергали кормлению в течение 3 часов. После периода кормления корм удаляли. Крысы имели по желанию доступ к воде. На третьи сутки опыта брали кровь из хвостовой вены, перед введением желудочного зонда. Животным затем, как и раньше, вводили желудочный зонд и подвергали кормлению. Данный способ повторяли на 5, 8, 11 и 14 сутки. На 14 сутки животных обезглавливали и заканчивали опыт. Определяли сывороточный фосфор, кальций и BUN, используя клинические комплекты.
Сывороточный фосфор снижался до 52%±6% (n=8) у взрослых крыс с нормальной функцией почек в пределах первой недели обработки 2'-FPP и остается постоянным при 3 мг/дл в течение остатка 2-недельного опыта. Сывороточный Са2+ не подвергался воздействию 2'-FPP, причем снижался менее чем на 2,5% в течение 2-недельного воздействия 2'-FPP. BUN составлял 10 мг/дл в начале опыта и не изменялся течение проведения опыта. Предыдущие опыты в идентичных условиях, выполненные с 25 мкМ 2'-PP, давали 30%±4% снижение сывороточного фосфора при отсутствии изменений в сывороточном Са2+.