Результат интеллектуальной деятельности: НАНЕСЕННЫЙ НА ДИОКСИД КРЕМНИЯ КАТАЛИЗАТОР
Вид РИД
Изобретение
Известный уровень техники
Область техники, к которой относится изобретение
Настоящее изобретение относится к нанесенному на диоксид кремния катализатору, используемому в производстве ненасыщенного нитрила.
Описание предшествующего уровня техники
Хорошо известен традиционный способ, в котором пропилен или изобутилен вступает в реакцию парофазного каталитического окисления или парофазного каталитического аммоксидирования, образуя соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил. В последние годы внимание привлекает способ, в котором в реакцию парофазного каталитического окисления вместо пропилена или изобутилена вступает пропан или изобутан или парофазного каталитического аммоксидирования, образуя соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил. Поэтому, в качестве катализатора для парофазного каталитического аммоксидирования пропана или изобутана предложены разнообразные оксидные катализаторы.
Патентный документ 1 описывает нанесенный на диоксид кремния катализатор, имеющий увеличенный объем пор в результате использования золя диоксида кремния и порошка диоксида кремния в качестве содержащих диоксид кремния исходных материалов.
Патентный документ 2 описывает композитный оксидный катализатор, используемый в производстве акролеина или акриловой кислоты, который имеет распределение пор, соответствующее определенному интервалу.
Патентный документ 3 описывает зернистый пористый катализатор аммоксидирования, в котором размеры пор ограничены определенным интервалом для повышения выхода целевого продукта.
Патентный документ 1: японская выложенная патентная заявка № 2002-219362
Патентный документ 2: японская выложенная патентная заявка № 2003-220334
Патентный документ 3: публикация международной патентной заявки WO 2004-078344
Проблемы, решаемые изобретением
Если золь диоксида кремния и порошок диоксида кремния смешивают, как описано в патентном документе 1, хотя объем пор может увеличиваться, средний размер пор не увеличивается. Следовательно, хотя наблюдается повышение текучести вследствие увеличения объема пор, это не приводит к повышению выхода целевого продукта. Кроме того, в патентном документе 1 не содержится описание сгорания содержащегося в исходном материале аммиака, что представляет собой одну из проблем при производстве нитрила посредством парофазного каталитического аммоксидирования алкана.
Патентный документ 2 описывает увеличение выхода путем регулирования размеров пор. Однако поскольку текучесть можно считать неудовлетворительной, принимая во внимание описание молоткового формования, можно понять, что данный катализатор представляет собой катализатор для реакции в неподвижном слое, и он не пригоден для реакции в псевдоожиженном слое.
Способ, описанный в патентном документе, 3 повышает выход целевого продукта путем обеспечения размеров пор в определенном интервале. На основании дополнительных экспериментов, проведенных в соответствии со способом, который описан в патентном документе 3, авторы настоящего изобретения обнаружили, что хотя распределение пор полученного катализатора удовлетворяет условию «суммарный объем пор, у которых размер составляет 80 Å или менее, составляет 20% или менее по отношению к суммарному объему всех пор катализатора, и суммарный объем пор, у которых размер составляет 1000 Å или более, составляет 20% или менее по отношению к суммарному объему всех пор катализатора», поры были распределены ближе к сравнительно малым размерам пор, в интервале от 80 до 1000 Å. Для катализатора аммоксидирования алканов, который является сильным окислителем, имеющие малые размеры поры не являются подходящими, потому что обычно происходит сгорание содержащегося в исходном материале аммиака и/или реакция разложения целевого продукта. Кроме того, поскольку размер кристаллитов оказывается неопределенным, можно полагать, что повышение выхода не является достаточным.
С учетом описанных выше обстоятельств, задача настоящего изобретения заключается в том, чтобы предложить катализатор, обеспечивающий низкую степень сгорания содержащегося в исходном материале аммиака и высокий выход целевого продукта.
Средства решения проблем
При таких обстоятельствах, в результате всесторонних исследований по решению описанных выше проблем предшествующего уровня техники авторы настоящей заявки обнаружили, что путем использования нанесенного на диоксид кремния катализатора, который содержит, по меньшей мере, Mo, V и Nb, и у которого значения определенных физических свойств, такие как средний размер пор, находятся в соответствующем интервале, выход целевого продукта значительно увеличивается, и, кроме того, что, поскольку можно подавлять сгорание содержащегося в исходном материале аммиака, ненасыщенный нитрил можно эффективно производить, и в результате этого они выполнили настоящее изобретение.
Таким образом, настоящее изобретение заключается в следующем.
[1] Нанесенный на диоксид кремния катализатор, используемый для производства соответствующего ненасыщенного нитрила в реакции парофазного каталитического аммоксидирования пропана или изобутана, причем катализатор содержит оксид металла, представленный следующей формулой (1):
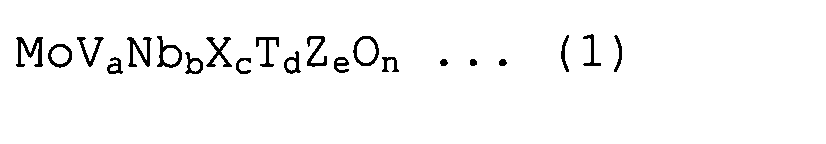 ,
,
в которой X представляет собой, по меньшей мере, один или несколько элементов, выбранных из Sb и Te; T представляет собой, по меньшей мере, один или несколько элементов, выбранных из Ti, W, Mn и Bi; Z представляет собой, по меньшей мере, один или несколько элементов, выбранных из La, Ce, Yb и Y; и a, b, c, d и e находятся в интервалах, составляющих 0,05≤a≤0,5, 0,01≤b≤0,5, 0,001≤c≤0,5, 0≤d≤1, и 0≤e≤1, соответственно, и n представляет собой значение, которое соответствует атомной валентности;
где у нанесенного на диоксид кремния катализатора средний размер пор составляет от 60 до 120 нм, суммарный объем пор составляет 0,15 см3/г или более, удельная поверхность составляет от до 25 м2/г, и размер кристаллитов составляет от 40 до 250 нм при определении методом рентгеноструктурного анализа по полуширине пика (001).
[2] Нанесенный на диоксид кремния катализатор согласно приведенному выше [1], в котором объем пор, у которых размер составляет менее чем 60 нм, по отношению к суммарному объему всех пор, составляет менее чем 30%, и объем пор, у которых размер пор превышает 120 нм, по отношению к суммарному объему всех пор составляет менее чем 30%.
[3] Нанесенный на диоксид кремния катализатор согласно приведенному выше пункту [1] или [2], в котором количество диоксида кремния в качестве носителя составляет от 20 до 70 мас.% по отношению к суммарной массе катализатора, изготовленного из оксида металла и диоксида кремния.
[4] Способ изготовления нанесенного на диоксид кремния катализатора, включающий следующие стадии:
(I) получение изготовленного из исходного материала раствора, содержащего Mo, V, Nb, X, T и Z, в котором атомное соотношение a количества атомов V и Mo представляет собой 0,05≤a≤0,5, атомное соотношение b количества атомов Nb и Mo представляет собой 0,01≤b≤0,5, атомное соотношение c количества атомов X и Mo представляет собой 0,001≤c≤0,5, атомное соотношение d количества атомов T и Mo представляет собой 0≤d≤1, и атомное соотношение e количества атомов Z и Mo представляет собой 0≤e≤1;
(II) высушивание изготовленного из исходного материала раствора для получения сухого порошка;
(III) предварительный обжиг сухого порошка при температуре от 200 до 400°C для получения предварительно обожженного материала; и
(IV) основной обжиг предварительно обожженного материала при температуре от 600 до 750°C для получения обожженного материала,
где изготовленный из исходного материала раствор содержит от 0 до 30 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (i) золя диоксида кремния, в котором средний размер первичных частиц составляет 3 нм или более и менее чем 20 нм, от 30 до 70 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (ii) золя диоксида кремния, в котором средний размер первичных частиц составляет 20 нм или более и 100 нм или менее, и от 30 до 70 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, порошка диоксида кремния, где средний размер первичных частиц составляет 50 нм или менее, и в котором суммарное количество золя диоксида кремния (i), золя диоксида кремния (ii) и порошка диоксида кремния составляет 100 мас.% по отношению к массе диоксида кремния.
[5] Способ производства соответствующего ненасыщенного нитрила путем осуществления реакции парофазного каталитического аммоксидирования пропана или изобутана с использованием нанесенного на диоксид кремния катализатора согласно любому из приведенных выше пунктов [1]-[3].
Преимущества изобретения
Настоящее изобретение может предложить катализатор, обеспечивающий низкую степень сгорания аммиака и высокий выход целевого продукта.
Подробное описание предпочтительного варианта осуществления
Далее будет подробно описан вариант осуществления настоящего изобретения (далее называется «настоящий вариант осуществления»). Следует отметить, что настоящее изобретение не ограничено следующим вариантом осуществления, и можно осуществлять множество вариантов в пределах объема настоящего изобретения.
Нанесенный на диоксид кремния катализатор согласно настоящему варианту осуществления представляет собой нанесенный на диоксид кремния катализатор, используемый для производства соответствующего ненасыщенного нитрила в реакции парофазного каталитического аммоксидирования пропана или изобутана, причем катализатор содержит оксид металла, представленный следующей формулой (1):
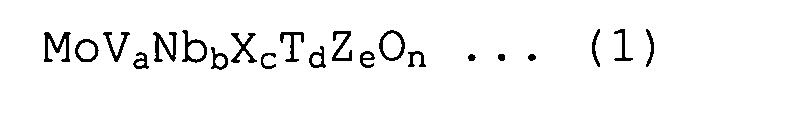 ,
,
в которой X представляет собой, по меньшей мере, один или несколько элементов, выбранных из Sb и Te; T представляет собой, по меньшей мере, один или несколько элементов, выбранных из Ti, W, Mn и Bi; Z представляет собой, по меньшей мере, один или несколько элементов, выбранных из La, Ce, Yb и Y; и a, b, c, d и e находятся в интервалах, составляющих 0,05≤a≤0,5, 0,01≤b≤0,5, 0,001≤c≤0,5, 0≤d≤1, и 0≤e≤1, соответственно, и n представляет собой значение, которое соответствует атомной валентности;
где у нанесенного на диоксид кремния катализатора средний размер пор составляет от 60 до 120 нм, суммарный объем пор составляет 0,15 см3/г или более, удельная поверхность составляет от до 25 м2/г, и размер кристаллитов составляет от 40 до 250 нм при определении методом рентгеноструктурного анализа по полуширине пика (001).
Поскольку нанесенный на диоксид кремния катализатор согласно настоящему варианту осуществления имеет оптимизированное соотношение металлов в составе оксида металла, содержащегося в катализаторе, он обладает хорошей каталитической активностью. Хотя способ изготовления нанесенного на диоксид кремния катализатора согласно настоящему варианту осуществления не ограничена определенным образом, оказывается предпочтительным изготовление нанесенного на диоксид кремния катализатора с использованием следующего способа, который включает стадии (I)-(IV).
Способ изготовления нанесенного на диоксид кремния катализатора, включающий следующие стадии:
(I) получение изготовленного из исходного материала раствора, содержащего Mo, V, Nb, X, T и Z, в котором атомное соотношение a количества атомов V и Mo представляет собой 0,05≤a≤0,5, атомное соотношение b количества атомов Nb и Mo представляет собой 0,01≤b≤0,5, атомное соотношение c количества атомов X и Mo представляет собой 0,001≤c≤0,5, атомное соотношение d чисел атомов T и Mo представляет собой 0≤d≤1, и атомное соотношение e количества атомов Z и Mo представляет собой 0≤e≤1;
(II) высушивание изготовленного из исходного материала раствора для получения сухого порошка;
(III) предварительный обжиг сухого порошка при температуре от 200 до 400°C для получения предварительно обожженного материала; и
(IV) основной обжиг предварительно обожженного материала при температуре от 600 до 750°C для получения обожженного материала,
где изготовленный из исходного материала раствор содержит от 0 до 30 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (i) золя диоксида кремния, в котором средний размер первичных частиц составляет 3 нм или более и менее чем 20 нм: от 30 до 70 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (ii) золя диоксида кремния, в котором средний размер первичных частиц составляет 20 нм или более и 100 нм или менее, и от 30 до 70 мас.% по отношению к суммарной массе содержащих диоксид кремния исходных материалов порошка диоксида кремния, в котором средний размер первичных частиц составляет 50 нм или менее, и где суммарное количество золя диоксида кремния (i), золя диоксида кремния (ii) и порошка диоксида кремния составляет 100 мас.% по отношению к массе диоксида кремния.
(Стадия (I) - стадия получения исходного материала)
Стадия (I) представляет собой стадию получения изготовленного из исходного материала раствора, содержащего Mo, V, Nb, X, T и Z, в котором атомное соотношение a количества атомов V и Mo представляет собой 0,05≤a≤0,5, атомное соотношение b количества атомов Nb и Mo представляет собой 0,01≤b≤0,5, атомное соотношение c количества атомов X и Mo представляет собой 0,001≤c≤0,5, атомное соотношение d количества атомов T и Mo представляет собой 0≤d≤1, и атомное соотношение e количества атомов Z и Mo представляет собой 0≤e≤1.
На стадии получения исходного материала изготовленный из исходного материала раствор получают путем растворения или диспергирования составляющих элементов нанесенного на диоксид кремния катализатора в растворителе и/или дисперсионной среде в определенном соотношении. Как правило, в качестве растворителя для изготовленного из исходного материала раствора можно использовать воду. Изготовленный из исходного материала раствор включает Mo, V, Nb, X, T и Z (причем X представляет собой, по меньшей мере, один или несколько элементов, выбранных из Sb и Te, T представляет собой, по меньшей мере, один или несколько элементов, выбранных из Ti, W, Mn и Bi, и Z представляет собой, по меньшей мере, один или несколько элементов, выбранных из La, Ce, Yb и Y). В качестве исходных материалов изготовленного из исходного материала раствора можно использовать соль или соединение, которые содержат составляющие элементы нанесенного на диоксид кремния катализатора.
В качестве исходного материала для Mo можно использовать такие соединения, как, например, гептамолибдат аммония [(NH4)6Mo7O24·4H2O], триоксид молибдена [MoO3], фосфорномолибденовая кислота [H3PMo12O40], кремнемолибденовая кислота [H4SiMo12O40], пентахлорид молибдена [MoCl5], и подобные соединения. Особенно предпочтительным является гептамолибдат аммония [(NH4)6Mo7O24·4H2O].
В качестве исходного материала для V можно использовать, например, метаванадат аммония [NH4VO3], пентаоксид ванадия [V2O5], хлориды ванадия [VCl4, VCl3] и подобные соединения. Особенно предпочтительным является метаванадат аммония [NH4VO3].
В качестве исходного материала для Nb можно использовать, например, ниобиевую кислоту, неорганический ниобат и органический ниобат. В частности, предпочтительной является ниобиевая кислота. Ниобиевая кислота представляет собой Nb2O5·nH2O, и ее также называют терминами «гидроксид ниобия» или «гидрат оксида ниобия». Кроме того, предпочтительно используют раствор исходного материала Nb, в котором молярное соотношение дикарбоновой кислоты и ниобия составляет от 1 до 4, а в качестве дикарбоновой кислоты предпочтительной является щавелевая кислота.
Исходные материалы для X (Sb и Te) не ограничены определенным образом, при том условии, что исходные материалы содержат данные элементы. Можно использовать соединение, содержащее данные элементы, и раствор, в котором металл из числа этих элементов солюбилизирован в соответствующем реагенте. В качестве соединения, содержащего эти элементы, могут быть обычно использованы аммониевая соль, нитрат, карбоксилат, аммониевая соль карбоновой кислоты, пероксокарбоксилат, аммониевая соль пероксокарбоновой кислоты, галогенированная аммониевая соль, галогенид, ацетилацетат и алкоксид этих элементов. Предпочтительно можно использовать водный исходный материал, такой как нитрат, а также карбоксилат.
Исходные материалы для T (Ti, W, Mn и Bi) не ограничены определенным образом, при том условии, что исходные материалы содержат данные элементы. Можно использовать соединение, содержащее данные элементы, и раствор, в котором металл из числа этих элементов солюбилизирован в соответствующем реагенте. В качестве соединения, содержащего эти элементы, могут быть обычно использованы аммониевая соль, нитрат, карбоксилат, аммониевая соль карбоновой кислоты, пероксокарбоксилат, аммониевая соль пероксокарбоновой кислоты, галогенированная аммониевая соль, галогенид, ацетилацетат и алкоксид этих элементов. Предпочтительно можно использовать водный исходный материал, такой как нитрат, а также карбоксилат.
Исходные материалы для Z (La, Ce, Yb и Y) не ограничены определенным образом, при том условии, что исходные материалы содержит данные элементы. Можно использовать соединение, содержащее данные элементы, и раствор, в котором металл из числа этих элементов солюбилизирован в соответствующем реагенте. В качестве соединения, содержащего эти элементы, могут быть обычно использованы аммониевая соль, нитрат, карбоксилат, аммониевая соль карбоновой кислоты, пероксокарбоксилат, аммониевая соль пероксокарбоновой кислоты, галогенированная аммониевая соль, галогенид, ацетилацетат и алкоксид этих элементов. Предпочтительно можно использовать водный исходный материал, такой как нитрат, а также карбоксилат.
При изготовлении исходного материала процедура растворения исходных материалов для образующих катализатор элементов, процедура перемешивания исходных материалов и процедура диспергирования исходных материалов не ограничены определенным образом. Исходные материалы можно растворять, перемешивать или диспергировать в одной и той же водной среде. В качестве альтернативы, исходные материалы можно растворять, перемешивать или диспергировать в раздельных водных средах, и эти водные среды можно смешивать. Когда это необходимо, можно осуществлять нагревание и/или перемешивание.
Для нанесенного на диоксид кремния катализатора одна из важных особенностей заключается в том, что компонент Z равномерно распределяется в частицах катализатора. Здесь равномерность означает, что распределение компонента Z в частицах катализатора не является неоднородным. Предпочтительно термин «равномерность» означает, что не менее чем 80% (массовое соотношение) оксидных частиц, содержащих компонент Z, существуют в частицах катализатора в форме тонкодисперсных частиц, причем размер этих частиц составляет не более чем 1 мм. Более подходящее определение термина «равномерность» заключается в том, что когда поперечное сечение частицы катализатора подвергают анализу состава, значение дисперсии (значение, полученное делением среднеквадратического отклонения на средний значение) соотношения интенсивностей сигналов компонентов Z и Si находится в интервале от 0 до 0,5. Здесь значение дисперсии представляет обозначение «Dx».
Для исследования состава можно использовать обычный способ анализа состава. Например, могут быть использованы сканирующая электронная микроскопия и энергодисперсионный рентгеноспектральный микроанализ (SEM-EDX), рентгеновская фотоэлектронная спектроскопия (XPS), масс-спектрометрия вторичных ионов (SIMS), электронно-зондовый микроанализ (EPMA) и т.п. Предпочтительно можно использовать EPMA. Здесь EPMA обычно обозначает электронно-зондовый рентгеновский микроанализатор (термин «рентгеновский» можно опустить при описании устройство). Аналитическое устройство представляет собой устройство, в котором наблюдают характеристическое рентгеновское излучение, полученное облучением вещества потоком ускоренных электронов, чтобы осуществлять анализ состава малой области (точки), облученной электронным пучком. Используя EPMA, обычно в поперечном сечении твердых частиц, таких как частицы катализатора и частицы носителя, можно получать информацию в отношении определенного элемента, такую как распределение концентрации и изменение состава.
Значение дисперсии (Dx) соотношения интенсивностей сигналов компонентов Z и Si в анализе EPMA представляет собой значение, полученное путем измерения поперечного сечения измеряемой частицы и осуществления вычисления согласно обычному способу для плоскостного анализа поперечного сечения частицы с помощью EPMA, который осуществляют в поле катализатора, следующим образом. А именно, сначала измеряют распределение интенсивности рентгеновского пика Si (число отсчетов ISi) в любом положении (x, y) поперечного сечения частицы катализатора таким образом, что покрывается полное поперечное сечение частицы катализатора. После этого аналогичным путем, распределение интенсивности рентгеновского пика (число отсчетов IX) компонента Z измеряют таким образом, что покрывается полное поперечное сечение частицы катализатора. На основании полученного ряда данных (x, y, ISi, IX) в отношении Si и компонента Z определяют соотношение интенсивностей пиков IR компонента Z и Si (IR=IX/ISi) в том же положении (x, y), и определяют среднее арифметическое значение (IR)av и среднеквадратическое отклонение S значения IR. Значение, полученное делением среднеквадратического отклонения S на среднее арифметическое значение (IR)av определяют как значение дисперсии (Dx). При этом среднее арифметическое значение и среднеквадратическое отклонение можно определять обычным способом. В настоящем описании термин «нанесенный на диоксид кремния катализатор» (иногда называется просто «катализатор») означает катализатор, в котором выступающие части, образованные на поверхности частиц, были удалены из обожженного материала после основного обжига. Поскольку измерение значения дисперсии основано на наблюдении поперечного сечения, и на него не влияет состояние поверхности, после основного обжига будет наблюдаться одинаковое значение, даже если измерение осуществляют перед стадией удаления выступающие части.
Предпочтительно во избежание неопределенности данных вследствие краевого эффекта поперечного сечения частицы при измерении, исключают область, которая составляет 10% площади поперечного сечения, в поперечном сечении частицы катализатора и соответствует внешней периферии частицы, а область, составляющую 90% в центре поперечного сечения частицы катализатора, используют в качестве эффективной области, и вычисляют данные для эффективной области. Разумеется, с самого начала плоскостной анализ способом EPMA можно осуществлять только для внутренней части поперечного сечения частицы катализатора, из которого исключена составляющая 10% область, соответствующая внешней периферии частицы, и из этих данных можно определять значение дисперсии Dx.
Поскольку катализатор согласно настоящему варианту осуществления представляет собой нанесенный на диоксид кремния катализатор, в котором оксид металла нанесен на диоксид кремния, изготовленный из исходного материала раствор изготавливают таким образом, что он включает содержащий диоксид кремния исходный материал. С точки зрения регулирования среднего размера пор катализатора с золем диоксида кремния, который составляет от 60 до 120 нм, оказывается предпочтительным, чтобы изготовленный из исходного материала раствор включал от 0 до 30 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (i) золя диоксида кремния, в котором средний размер первичных частиц составляет 3 нм или более и менее чем 20 нм, от 30 до 70 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, (ii) золя диоксида кремния, в котором средний размер первичных частиц составляет 20 нм или более и менее чем 100 нм, и от 30 до 70 мас.%, по отношению к суммарной массе содержащих диоксид кремния исходных материалов, порошка диоксида кремния, в котором средний размер первичных частиц составляет 50 нм или менее, где суммарное количество золя диоксида кремния (i), золя диоксида кремния (ii) и порошка диоксида кремния составляет 100 мас.% по отношению к массе диоксида кремния. Порядок, в котором эти содержащие диоксид кремния исходные материалы вводят в изготовленный из исходного материала раствор, не ограничен определенным образом, и эти содержащие диоксид кремния исходные материалы можно смешивать перед введением в изготовленный из исходного материала раствор. Хотя причина этого остается неясной, если используют определенное содержание золя диоксида кремния (i), золя диоксида кремния (ii) и порошка диоксида кремния, на основании экспериментов авторы настоящего изобретения обнаружили, что можно изготавливать катализатор, имеющий большой средний размер пор и высокое сопротивление истиранию.
Считается, что если используют золь диоксида кремния (i) и золь диоксида кремния (ii), имеющие малый диаметр частицы диоксида кремния поступают в пространство между имеющими большой диаметр частицами диоксида кремния, таким образом, что возникает эффект сокращения числа мелких пор в катализаторе. Кроме того, считается, что путем введения порошка диоксида кремния золь диоксида кремния защищается от агрегации, таким образом, что возникает эффект увеличения числа больших пор. Поскольку катализатор согласно настоящему варианту осуществления имеет больший средний размер пор, чем традиционный катализатор, предполагается, что температура реакционной смеси становится однородной вследствие увеличения скорости диффузии содержащегося в исходном материале аммиака и целевого продукта в частицах катализатора и/или увеличения диффузии тепла в частицах катализатора, и в результате этого можно подавлять сгорание содержащегося в исходном материале аммиака и разложение целевого продукта.
Металлы, присутствующие в качестве примесей в содержащих диоксид кремния исходных материалах, могут влиять на эффективность изготовленного нанесенного на диоксид кремния катализатора. Пример примеси в содержащем диоксид кремния исходном материале представляет собой натрий. Количество натрия составляет предпочтительно 0,02 атома или менее и предпочтительнее 0,01 атома или менее на 100 атомов кремния. Если присутствует более чем 0,02 атома на 100 атомов кремния, то когда полученный нанесенный на диоксид кремния катализатор используют в реакции аммоксидирования, исходные материалы и/или целевой продукт склонны вступать в реакцию разложения.
Когда производят ненасыщенный нитрил путем парофазного аммоксидирования алкана, вследствие низкой реакционной способности алканов, эту реакцию осуществляют в присутствии катализатора, обладающего высокой окислительной способностью, и/или при высокой температуре. Следовательно, обычно происходят реакции сгорания содержащегося в исходном материале аммиака и разложения целевого продукта. Если происходит сгорание содержащегося в исходном материале аммиака, то возникает недостаток аммиака для использования в производстве ненасыщенного нитрила, таким образом, что требуется введение большого количества аммиака по отношению к алкану. Следовательно, снижается производительность. В настоящем варианте осуществления путем подавления сгорания содержащегося в исходном материале аммиака можно эффективно производить ненасыщенный нитрил. Кроме того, очевидно, что при подавлении разложения ненасыщенного нитрила, который представляет собой целевой продукт, повышается выход.
Если средний размер пор катализатора составляет менее чем 60 нм, обычно происходят реакции сгорания содержащегося в исходном материале аммиака и разложения целевого продукта. С другой стороны, если средний размер пор катализатора составляет более чем 120 нм, уменьшается сопротивление истиранию, таким образом, что катализатор оказывается неподходящим для реакции в псевдоожиженном слое. С описанных выше точек зрения, средний размер пор катализатора согласно настоящему варианту осуществления находится в интервале от 60 до 120 нм и предпочтительно находится в интервале от 65 до 100 нм.
На основании дополнительных экспериментов, осуществленных авторами настоящего изобретения в отношении традиционных катализаторов, используемых в реакции аммоксидирования олефина, такого как пропилен или изопрен, объем пор, у которых размер составляет менее чем 60 нм, оказался составляющим 30% или более по отношению к суммарному объему пор. Для реакции аммоксидирования алканов, поскольку алканы имеют высокую окислительную способность, если объем пор, у которых размер составляет менее чем 60 нм, составляет 30% или более по отношению к суммарному объему пор, считается, что имется тенденция к протеканию реакций сгорания содержащегося в исходном материале аммиака и/или разложения целевого продукта. Таким образом, для реакции аммоксидирования алканов, оказывается предпочтительным, что катализатор имеет сравнительно большой размер, и что размеры пор являются однородными. С другой стороны, если установить объем пор, у которых размер составляет более чем 120 нм, составляющий менее чем 30% по отношению к суммарному объему пор, то становится большим сопротивление истиранию, таким образом, что катализатор обычно оказывается подходящим для использования в реакции в псевдоожиженном слое. С описанных выше точек зрения, оказывается предпочтительным, чтобы нанесенный на диоксид кремния катализатор согласно настоящему варианту осуществления имел объем пор, у которых размер составляет менее чем 60 нм, составляющий менее чем 30% по отношению к суммарному объему пор, и объем пор, у которых размер составляет более чем 120 нм, составляющий менее чем 30% по отношению к суммарному объему пор.
Для изготовления такого катализатора оказывается предпочтительным использование золей диоксида кремния, содержащих частицы различных размеров, и регулирование спекания диоксида кремния путем обжига при температуре от 600 до 750°C. Оказывается предпочтительным осуществление обжига при температуре, составляющей 600°C или более, потому что тогда спекание диоксида кремния происходит в достаточной степени, и увеличивается объем пор, у которых размер составляет 60 нм или более.
С точки зрения повышения активности катализатора, оказывается предпочтительным, чтобы количество диоксида кремния в качестве носителя, включенного в катализатор, составляло 20 мас.% или более по отношению к суммарной массе катализатора, изготовленного из оксида металла и диоксида кремния. С точки зрения придания достаточной активности, количество диоксида кремния в качестве носителя, включенного в катализатор, составляет предпочтительно 70 мас.% или менее и предпочтительнее от 40 до 65 мас.% по отношению к суммарной массе.
В качестве исходного материала для диоксида кремния в качестве носителя можно использовать только золь диоксида кремния. В качестве альтернативы, часть исходного материала можно заменить порошком диоксид кремния. Если используют порошок диоксида кремния как исходный материал для диоксида кремния в качестве носителя, предполагается эффект повышения активности катализатора и/или выхода целевого продукта. С другой стороны, если используют только порошок диоксида кремния без использования золя диоксида кремния для изготовления катализатора, сопротивление истиранию катализатора значительно уменьшается. В настоящем варианте осуществления термин «порошок диоксида кремния» означает тонкодисперсные частицы твердого SiO2. Если первичный размер частиц диоксида кремния является чрезмерно большим, получаемый катализатор, вероятно, окажется хрупким. Соответственно, порошок диоксида кремния с нанометровым размером частиц является предпочтительным. Порошок диоксида кремния предпочтительно изготавливают, используя высокотемпературный способ.
С точки зрения простоты введения и перемешивания с изготовленным из исходного материала раствором, порошок диоксида кремния предпочтительно диспергируют предварительно в воде. Способ диспергирования порошка диоксида кремния в воде не ограничен определенным образом, и порошок диоксида кремния можно диспергировать, используя обычный гомогенизатор, смеситель-гомогенизатор или ультразвуковой вибратор индивидуально или в сочетании нескольких устройств. При этом первичная форма частиц порошка диоксида кремния может быть сферической или несферической.
Средний размер первичных частиц золя диоксида кремния и порошка диоксида кремния, которые представляют собой исходные материалы носителя, можно определять, используя способ Брунауэра-Эммета-Теллера (BET), т.е. способ изотермы адсорбции. Диоксид кремния, который обычно имеется в продаже, можно считать имеющим интервал распределения частиц по размерам, где средний размер первичных частиц находится приблизительно в центре. Чтобы в достаточной степени проявлять эффект подавления сгорания аммиака, оказывается предпочтительным чтобы среднеквадратическое отклонение в распределении по размерам частиц диоксида кремния было минимально возможным. В частности, оказывается предпочтительным, чтобы среднеквадратическое отклонение составляло не более чем 30% среднего размера первичных частиц.
Для регулирования среднего размера пор катализатора оказывается эффективным изменение среднего размера первичных частиц золя диоксида кремния. Как правило, если средний размер первичных частиц золя диоксида кремния увеличивается, активность полученного катализатора имеет тенденцию к уменьшению. С другой стороны, поскольку высокая активность является желательной для промышленного катализатора в псевдоожиженном слое, традиционный золь диоксида кремния, в котором средний размер первичных частиц составляет порядка десятка нанометров, обычно используют в качестве содержащего диоксид кремния исходного материала. Когда катализатор изготавливают, используя такой золь диоксида кремния на основании традиционного способа, средний размер пор составляет приблизительно от 20 до 50 нм, что не соответствует интервалу от 60 до 120 нм для среднего размера пор, который определен в настоящем варианте осуществления. Кроме того, выход также оказывается недостаточным. Путем изменения условий обжига можно регулировать средний размер пор. Если температура обжига увеличивается, и/или увеличивается продолжительность обжига, то средний размер пор имеет тенденцию к увеличению. Однако если средний размер пор регулируют путем изменения условий обжига, удельная поверхность и/или размер кристаллитов также изменяется. Таким образом, оказывается затруднительным регулирование среднего размера пор, удельной поверхности и размера кристаллитов исключительно на основании условий обжига. Как описано выше, при использовании способов производства катализаторов, которые обсуждаются выше в разделе «Описание предшествующего уровня техники», практически невозможно получить катализатор, который удовлетворяет условиям среднего размера пор, удельной поверхности и/или размера кристаллитов. Оказывается предпочтительным регулирование среднего размера пор на основании способа с использованием золей диоксида кремния, имеющих различные средние размеры первичных частиц, поскольку удельную поверхность и/или размер кристаллитов можно регулировать на основании условий обжига. Удельную поверхность и размер кристаллитов можно регулировать путем регулирования условий обжига, потому что температурная область, в которой происходит спекание диоксида кремния, который производит большое воздействие на удельную поверхность, отличается от температурной области, в которой происходит рост кристаллов.
Средства регулирования среднего размера пор катализатора согласно настоящему варианту осуществления в соответствующем интервале не ограничены определенным образом. Можно использовать любые средства, при том условии, что средний размер пор можно регулировать в соответствующем интервале. Примеры средств для регулирования среднего размера пор катализатора включают способ, в котором средний размер первичных частиц описанных выше золей диоксида кремния, которые представляют собой содержащие диоксид кремния исходные материалы, способ, в котором порошок диоксида кремния используют в качестве части содержащих диоксид кремния исходных материалов, способ, в котором изменяется соотношение между содержащим диоксид кремния носителем и оксидом металла в катализаторе, и т.п.
Оказывается предпочтительным, чтобы изготовленный из исходного материала раствор содержал, в качестве части содержащих диоксид кремния исходных материалов, от 30 до 70 мас.% порошка диоксида кремния, в котором средний размер первичных частиц составляет 50 нм или менее по отношению к массе диоксида кремния. В настоящем варианте осуществления термин «по отношению к массе диоксида кремния» означает отношение к суммарной массе золя диоксида кремния и порошка диоксида кремния. Средний размер первичных частиц порошка диоксида кремния составляет предпочтительнее от 10 до 20 нм. Кроме того, количество порошка диоксида кремния составляет предпочтительнее от 30 до 50 мас.% по отношению к массе диоксида кремния. Если используют порошок диоксида кремния, удельная поверхность катализатора увеличивается. Для регулирования удельной поверхности и размера кристаллитов в соответствующем интервале, оказывается предпочтительным осуществление основного обжига при температуре обжига от 640 до 750°C, продолжительности обжига от 1 до 20 часов и средней скорости уменьшения температуры после окончания основного обжига от 0,05 до 20°C/мин.
Когда регулирование среднего размера пор осуществляют путем изменения соотношение между количеством диоксида кремния в качестве носителя и количеством оксида металла в катализаторе, оказывается предпочтительным установление количества диоксида кремния в качестве носителя, составляющего от 20 до 70 мас.% и предпочтительнее от 40 до 60 мас.% по отношению к суммарной массе катализатора, изготовленного из оксида металла и диоксида кремния. Как правило, если количество диоксида кремния в качестве носителя уменьшается, средний размер пор сдвигается в сторону большего размера пор, таким образом, что удельная поверхность уменьшается. Для регулирования удельной поверхности и размера кристаллитов в соответствующем интервале оказывается предпочтительным осуществление основного обжига при температуре обжига от 600 до 700°C, продолжительности обжига от 0,1 до 5 часов, и средней скорости уменьшения температуры после окончания основного обжига от 0,5 до 50°C/мин.
Примерами способов изменения распределения пор катализатора являются адсорбция газа, ртутная порометрия и т.п. Однако измеряемое значение зависит от способа измерения. Значение распределения пор катализатора согласно настоящему варианту осуществления определяют на основании способа ртутной порометрии, используя прибор Pore Master GT, изготовленный компанией Quantachrome Instruments. В данном способе ртутной порометрии измеряют распределение пор по размерам на основании соотношения между давлением и внедренным количеством ртути, которую вводят внутрь частиц катализатора. Используя полученные данные как первичные данные, получают интегральную кривую объема пор как функцию размера пор при вычислении на основании предположения о том, что поры имеют цилиндрическую форму. Значения, полученные путем вычисления первой производной этой интегральной кривой по отношению к объему пор от размера пор, наносят на кривую зависимости от соответствующего размера пор, и полученный график обычно называют термином «распределение пор». В частности, от 0,4 до 0,6 г образца (катализатора) помещают в дилатометр (прибор для измерения расширения), содержащийся в приборе газ откачивают до давления 6,67 Па или менее, используя вакуумный насос, и затем вводят ртуть. После этого дилатометр помещают в автоклав. Измеряют уменьшение уровня жидкой ртути при постепенном увеличении давления от атмосферного давления до 413 МПа, и распределение пор определяют на основании изменения давления и уровня жидкой ртути (количества ртути, введенной в поры катализатора).
В случае катализаторов, когда используют способ ртутной порометрии, расстояние между частицами катализатора, измеренное как поры, составляет от нескольких десятков тысяч до нескольких сотен тысяч ангстремов. Таким образом, поры, размеры которых составляют 200 нм или менее, учитываются в суммарном объеме. Кроме того, поскольку нижний предел измерения размеров пор составляет 6 нм, в суммарном объеме учитываются поры, размеры которых составляют 6 нм или более. Таким образом, в настоящем варианте осуществления суммарный объем пор рассматривают как суммарный объем пор, у которых размер составляет 6 нм или более и 200 нм или менее.
Суммарный объем пор катализатора согласно настоящему варианту осуществления составляет, с точки зрения текучести при реакции в псевдоожиженном слое, 0,15 см3/г или более. Если суммарный объем пор составляет менее чем 0,15 см3/г, текучесть уменьшается, что приводит к уменьшению выхода вследствие неоднородности температуры реакционной среды. Суммарный объем пор имеет тенденцию увеличиваться при увеличении среднего размера пор и/или при увеличении удельной поверхности. Примеры способов регулирования суммарного объема пор включают способ, в котором средний размер пор увеличивают, используя золи диоксида кремния, содержащие частицы различных размеров, и/или способ, в котором удельную поверхность увеличивают путем уменьшения температуры обжига и/или путем сокращения продолжительности обжига на стадии обжига.
Вычисление среднего размера пор катализатора осуществляют, используя формулу (i), на основании предположения, что поры являются цилиндрическими.
D=4V/S (i)
Здесь D представляет собой средний размер пор (м), V представляет собой суммарный объем пор (м3/г), и S представляет собой удельную поверхность (м2/г).
Далее стадия изготовления исходного материала будет описана с использованием примера, в котором растворитель и/или дисперсионная среда представляет собой воду, и получают изготовленный из исходного материала раствор для нанесенного на диоксид кремния катализатора, который содержит соединение Mo, соединение V, соединение Nb, соединение X, соединение T и соединение Z.
Соединение Mo, соединение V, соединение X и соединение компонента Z помещают в воду, и раствор нагревают, получая изготовленный из исходного материала раствор (A). Температуру нагревания и продолжительность нагревания в ходе получения изготовленного из исходного материала раствора (A) предпочтительно регулируют таким образом, что соединение исходного материала растворяется в достаточной степени. Температура нагревания составляет предпочтительно от 70°C до 100°C, и продолжительность нагревания составляет предпочтительно от 30 минут до 5 часов. Аналогичным образом, скорость вращения при перемешивании в течение нагревания устанавливают на уровне скорости вращения, при которой легко растворяется исходный материал. В том случае, когда исходный материал представляет собой соль, состояние перемешивания предпочтительно поддерживают с точки зрения достаточного растворения соли металла. При этом внутри контейнера может находиться воздушная атмосфера. С точки зрения регулирования степени окисления получаемого сложного оксидного катализатора, можно использовать атмосферу азота. Состояние, в котором завершается нагревание изготовленного из исходного материала раствора (A) называется термином «изготовленный из исходного материала раствор (A')». Изготовленный из исходного материала раствор (A') предпочтительно выдерживают при температуре, составляющей не менее чем 20°C и не более чем 80°C, и предпочтительнее не менее чем 40°C и не более чем 80°C. При температуре изготовленного из исходного материала раствора (A'), составляющей менее чем 20°C, можно осаждать соединение металла, растворенное в изготовленном из исходного материала растворе (A'). После завершения нагревания изготовленного из исходного материала раствора (A) вводят золь диоксида кремния в качестве исходного материала носителя. Когда вводят два или более видов золя диоксида кремния, имеющих различные средние размеры первичных частиц, порядок из введения не является ограниченным, и эти золи диоксида кремния можно смешивать перед введением в изготовленный из исходного материала раствор. Оказывается предпочтительным, чтобы температура изготовленного из исходного материала раствора (A') при введении золей диоксида кремния составляла не более чем 80°C. Если золи диоксида кремния вводят при температуре, превышающей 80°C, устойчивость золей диоксида кремния может уменьшаться таким образом, что изготовленный из исходного материала раствор может превращаться в гель. Хотя время введения золей диоксида кремния может наступать, когда начинается описанное ниже старение, в процессе старения или немедленно перед высушиванием изготовленного из исходного материала раствора, оказывается предпочтительным введение золя диоксида кремния в период пребывания в состоянии изготовленного из исходного материала раствора (A'). Кроме того, с точки зрения регулирования степени окисления полученного оксида металла, оказывается предпочтительным введение подходящего количества пероксида водорода в изготовленный из исходного материала раствор (A'), насколько это необходимо. Время введения пероксида водорода может наступать во время введения золя диоксида кремния в изготовленный из исходного материала раствор (A'), в ходе регулирования изготовленного из исходного материала раствора (A'), или до или после введения золя диоксида кремния. На этой стадии, с точки зрения регулирования степени окисления полученного оксидного катализатора в соответствующем интервале, добавляемое количество пероксида водорода составляет предпочтительно от 0,01 до 5, предпочтительнее от 0,5 до 3 и еще предпочтительнее 1 до 2,5 в расчете на молярное соотношение H2O2/Sb.
Температуру нагревания и продолжительность нагревания после введения пероксида в изготовленный из исходного материала раствор (A') предпочтительно регулируют таким образом, что жидкофазная реакция окисления раствором пероксида водорода может протекать в достаточной степени. Температура нагревания составляет предпочтительно от 30°C до 70°C, и продолжительность нагревания составляет предпочтительно от 5 минут до 4 часов. Аналогичным образом, скорость вращения при перемешивании в течение нагревания регулируют на уровне скорости вращения, при которой легко протекает жидкофазная реакция окисления раствором пероксида водорода. С точки зрения достаточного протекания жидкофазной реакции окисления раствором пероксида водорода, перемешивание предпочтительно продолжают в ходе нагревания. Изготовленный таким способом водный смешанный раствор называется термином «изготовленный из исходного материала раствор (A″)».
После этого соединение Nb и дикарбоновую кислоту нагревают и перемешивают в воде для получения смешанного раствора (B0). Примеры дикарбоновой кислоты включают щавелевую кислоту [(COOH)2]. Раствор пероксида водорода предпочтительно добавляют в смешанный раствор (B0) для получения изготовленного из исходного материала раствора (C). При этом молярное соотношение H2O2/Nb составляет предпочтительно от 0,5 до 20 и предпочтительнее от 1 до 10 с точки зрения образования комплекса с соединением Nb и стабилизации комплекса в растворенном состоянии, регулирования соответствующим образом состояния окисления и восстановления образующих катализатор элементов, а также оптимизации возможности получения катализатора.
В зависимости от целевого состава, изготовленный из исходного материала раствор (A″), изготовленный из исходного материала раствор (C), соединение T и порошок диоксида кремния соответствующим образом смешивают для получения изготовленного из исходного материала раствора (D). Полученный изготовленный из исходного материала раствор (D) подвергают старению для получения изготовленного из исходного материала раствора. Используемый здесь порошок диоксида кремния можно добавлять в неизменном виде. Предпочтительнее порошок диоксида кремния добавляют в форме водного раствора, в котором порошок диоксида кремния диспергирован в воде. Концентрация порошка диоксида кремния в воде при этом составляет предпочтительно от 1 до 30 мас.% и предпочтительнее от 3 до 20 мас.%. При концентрации порошка диоксида кремния, составляющей менее чем 1 мас.%, вязкость суспензии является чрезмерно низкой. По этой причине форма получаемых частиц может быть искаженной, и, вероятно, могут образовываться вмятины в частицах катализатора. С другой стороны, при концентрации порошка диоксида кремния, составляющей более чем 30 мас.%, вязкость изготовленного из исходного материала раствора является чрезмерно высокой, и изготовленный из исходного материала раствор может подвергаться гелеобразованию, образуя закупоривание внутри трубы. В результате может оказаться затруднительным получение сухого порошка, и активность катализатора может уменьшаться.
Старение изготовленного из исходного материала раствора (D) означает выдерживание в неподвижном состоянии или перемешивание изготовленного из исходного материала раствора (D) в течение заданного периода времени. Когда осуществляют промышленное производство нанесенного на диоксид кремния катализатора, распылительная сушилка имеет ограниченную скорость обработки, таким образом, что может потребоваться некоторое время для завершения распылительного высушивания всего смешанного раствора после того, как часть изготовленного из исходного материала раствора (D) была высушена распылением. В течение этого распылительного высушивания может продолжаться старение смешанного раствора, который подвергается распылительному высушиванию. В частности, продолжительность старения включает не только время старения перед распылительным высушиванием, но также и время от начала до конца распылительного высушивания.
Нанесенный на диоксид кремния катализатор является предпочтительным, например, с точки зрения достаточного растворения и/или диспергирования соединения, включающего составляющие катализатор элементы, с точки зрения соответствующего регулирования состояния окисления и восстановления составляющих катализатор элементов, с точки зрения приведения формы частиц и/или активности полученного катализатора в предпочтительное состояние, а также с точки зрения повышения каталитической эффективности полученного композитного оксида. Золь диоксида кремния можно добавлять соответствующим образом. Водную дисперсию порошка диоксида кремния можно использовать в качестве части золя диоксида кремния. Водную дисперсию порошка диоксида кремния можно также добавлять соответствующим образом.
Стадию получения состава из исходного материала можно осуществлять в периодическом режиме в зависимости от объема производства.
Стадия изготовления получения состава исходного материала в настоящем варианте осуществления предпочтительно включает следующие стадии (a)-(d):
(a) стадия получения изготовленного из исходного материала раствора, содержащего Mo, V, X и компонент Z;
(b) стадия введения золя диоксида кремния и раствора пероксида водорода в изготовленный из исходного материала раствор, полученный на стадии (a);
(c) стадия перемешивания водного раствора, содержащего Nb, дикарбоновую кислоту, раствор пероксида водорода и соединение T, с раствором, полученным на стадии (b); и
(d) стадия введения суспензии, содержащей порошок диоксида кремния, в раствор, полученный на стадии (c), и старения раствора.
(Стадия (II) - стадия высушивания)
Стадия (II) представляет собой стадию высушивания изготовленного из исходного материала раствора для получения сухого порошка.
Суспендированный изготовленный из исходного материала раствор, подвергнутый стадии получения состава из исходного материала, высушивают для получения сухого порошка. Высушивание можно осуществлять известным способом. Например, высушивание можно осуществлять путем распылительного высушивания или испарения досуха. В том случае, когда для реакции парофазного каталитического аммоксидирования используют способ реакции в псевдоожиженном слое, использование распылительного высушивания является предпочтительным, потому что оказывается предпочтительным получение микросферического сухого порошка с точки зрения предпочтительной текучести внутри реактора. Распыление в способе распылительного высушивания может осуществлять центробежная система, двухпоточная форсуночная система, или форсуночная система высокого давления. Воздух, нагреваемый паром, электрический нагреватель или подобное устройство можно использовать в качестве источника тепла для высушивания. Температура на впуске распылительной сушилки составляет предпочтительно от 150 до 300°C с точки зрения обеспечения предпочтительной формы и/или активности получаемых частиц катализатора, а также повышения эффективности получаемого комплексного оксидного катализатора. Температура на выпуске сушилки составляет предпочтительно от 100 до 160°C.
Предпочтительно, скорость распыления, скорость подачи изготовленного из исходного материала раствора и скорость вращения распылителя в случае центробежного типа регулируют таким образом, что получаемый сухой порошок имеет подходящий размер частиц. Средний размер частиц сухого порошка составляет предпочтительно от 5 мм до 200 мм и предпочтительнее от 10 до 150 мм.
Средний размер частиц сухого порошка можно определять следующим образом: согласно стандарту JIS R 1629-1997 «Способ измерения распределение частиц по размерам, использующий лазерное дифракционное рассеяние, для тонкодисперсного керамического исходного материала» измеряют распределение частиц по размерам и осуществляют усреднение по объему. Более конкретно, часть сухого порошка обжигают на воздухе при 400°C в течение одного часа, и полученные частицы измеряют, используя лазерное дифракционное рассеяние для определения распределения частиц по размерам с помощью устройства LS230, изготовленного компанией Beckman Coulter, Inc.
Средний размер частиц измеряют после того, как часть сухого порошка «обжигают на воздухе при 400°C в течение одного часа», потому что необходимо предотвращать растворение сухого порошка в воде. Таким образом, «обжиг на воздухе при 400°C в течение одного часа» предназначается, главным образом, для измерения и не имеет ничего общего со стадией обжига, описанной далее. Можно полагать, что размер частиц существенно не изменяется при сравнении состояния до и после обжига.
Более конкретно, средний размер частиц сухого порошка измеряют согласно руководству, прилагаемого к устройству, производимого под товарным наименованием LS230 компанией Beckman Coulter, Inc., для измерения распределение частиц по размерам методом лазерного дифракционного рассеяния следующим образом. Сначала после осуществления холостого измерения (скорость движения 60), 0,2 г частиц взвешивают и помещают в трубку с винтовой крышкой, которая имеет соответствующий размер, и добавляют 10 мл воды. Трубку с винтовой крышкой закрывают (плотно завинчивают) и встряхивают в достаточной степени, чтобы диспергировать частицы в воде. Устройство создает ультразвуковое воздействие мощностью 300 Вт, и затем трубку с винтовой крышкой снова встряхивают в достаточной степени. После этого в процессе ультразвукового воздействия частицы, диспергированные в воде, вводят в основной корпус устройства, используя пипетку, таким образом, чтобы получить соответствующую концентрацию (концентрация 10, PIDS 60). Когда показываемая концентрация стабилизируется, воздействие ультразвуковых волн прекращают. Трубку с винтовой крышкой выдерживают в данном состоянии в течение 10 секунд, и начинается измерение (продолжительность измерения составляет 90 секунд). Значение медианного размера по результатам измерения определяют как средний размер частиц.
((III) Стадия предварительного обжига и (IV) стадия основного обжига)
Стадия (III) представляет собой стадию предварительного обжига сухого порошка при температуре от 200 до 400°C для получения предварительно обожженного продукта.
Стадия (IV) представляет собой стадию основного обжига предварительно обожженного продукт при температуре от 600 до 750°C для получения обожженного продукта.
В настоящем документе в некоторых случаях стадия (III) и стадия (IV) вместе называются термином «стадия обжига».
На стадиях (III) и (IV) обжигают сухой порошок, полученный на стадии высушивания. Условия, такие как температура, продолжительность и атмосфера обжига, можно соответствующим образом определять с точки зрения удаления органических компонентов, содержащихся в сухом порошке, или роста кристаллов комплексного оксида, и они не ограничены определенным образом. В способе производства согласно настоящему варианту осуществления изменяются условия, такие как температура, и осуществляют многостадийный обжиг, такой как предварительный обжиг и основной обжиг, как описано далее.
В настоящем документе термин «выступающая часть» означает часть, которая выступает и/или прикрепляется на поверхности обожженного материала, полученного описанным ниже основным обжигом, и относится к части, которая выступает с поверхности обожженного материала или прикрепляется к ней. Здесь многие выступающие части представляют собой выступающие кристаллы оксидов и других примесей. В частности, в случае обожженного материала, содержащего множество металлов, оксиды, у которых состав отличается от состава кристаллов, образующих основную массу обожженного материала, могут образовываться в такой форме, что эти оксиды выступают из основной части обожженного материала. В данном случае выступающая часть часто образуется в форме множества выступающих частей (например, имеющих высоту от 0,1 мкм до 20 мкм) на поверхности сферической частицы обожженного материала (например, имеющей диаметр от 30 до 150 мкм). Удаление выступающих частей будет подробно описано ниже.
(Способ обжига сухого порошка)
В качестве обжигового устройства для обжига сухого порошка может быть использована, например, вращающаяся печь (вращающаяся обжиговая печь). Форма обжигового устройства не ограничена определенным образом. Трубчатая форма (обжиговая труба) является предпочтительной, и цилиндрическая форма является особенно предпочтительной с точки зрения обеспечения непрерывного обжига. В качестве способа нагревания, внешнее нагревание является предпочтительным с точки зрения простоты регулирования температуры обжига в предпочтительном режиме повышения температуры. Электрическую печь можно использовать соответствующим образом. Размер и материал обжиговой трубы можно выбирать соответствующим образом в зависимости от условий обжига и объема производства. Внутренний диаметр обжиговой трубы составляет предпочтительно от 70 до 2000 мм и предпочтительнее от 100 до 1200 мм с точки зрения обеспечения равномерного распределения температуры обжига в объеме слоя катализатора и регулирования продолжительности обжига и объема производства на соответствующем уровне. Длина обжиговой трубы составляет предпочтительно от 200 до 10000 мм и предпочтительнее 800 до 8000 мм с точки зрения уменьшения продолжительности выдерживания частиц сухого порошка и предшественника катализатора в объеме обжиговой трубы, а именно, распределения продолжительности обжига максимально возможным образом, предотвращения деформации обжиговой трубы и регулирования продолжительности обжига и объема производства на соответствующем уровне. Когда обжиговая труба подвергается ударной нагрузке, толщина обжиговой трубы составляет предпочтительно 2 мм или более и предпочтительнее 4 мм или более с точки зрения того, что обжиговая труба должна иметь достаточную толщину и не разрушаться от удара. Толщина обжиговой трубы составляет предпочтительно 100 мм или менее и предпочтительнее 50 мм или менее с точки зрения того, что удар в достаточной степени передается в обжиговую трубу. Материал обжигового устройства не ограничивается определенным образом при том условии, что обжиговое устройство предпочтительно имеет термическое сопротивление и прочность на таком уровне, чтобы не разрушаться от удара. Нержавеющую сталь (SUS) можно соответствующим образом использовать в качестве материала обжиговой трубы.
В настоящем документе термин «предшественник катализатора» означает соединение, изготовленное на промежуточном этапе стадии обжига.
На основной стадии обжига размер кристаллитов катализатора можно регулировать. Для регулирования размера кристаллитов в соответствующем интервале оказывается предпочтительным осуществление основного обжига при температуре от 600 до 750°C в течение от 0,1 до 20 часов и предпочтительнее от 650 до 720°C в течение 0,5 до 5 часов. На размер кристаллитов значительно влияет температура и/или продолжительность основного обжига. Чем выше температура обжига, и/или чем больше продолжительность обжига, тем больше размер кристаллитов. Нанесенный на диоксид кремния катализатор содержит столбчатые кристаллы, у которых соотношение площади боковых граней и суммарной площади кристаллических граней увеличивается, когда кристаллы растут в направлении (001). Известно, что реакция аммоксидирования происходит на верхних и нижних гранях, и что боковые грани представляют собой грани разложения. Размер кристаллитов, измеряемый в настоящем варианте осуществления, представляет собой длину в направлении (001). Если размер кристаллитов составляет более чем 250 нм, считается, что соотношение площади граней разложения и суммарной площади кристаллических граней увеличивается. Следовательно, упрощается протекание сгорания содержащегося в исходном материале аммиака и разложения целевого продукта. С другой стороны, чем ниже температура обжига, и/или чем короче продолжительность обжига, тем меньше размер кристаллитов. Если размер кристаллитов составляет менее чем 40 нм, образование активных центров оказывается недостаточным, и, таким образом, упрощается протекание сгорания содержащегося в исходном материале аммиака и разложения целевого продукта. Таким образом, размер кристаллитов катализатора составляет от 40 до 250 нм и предпочтительно от 40 до 180 нм. Поскольку катализатор, имеющий размер кристаллитов в соответствующем интервале, имеет высокую степень завершения кристаллов и малое соотношение площади граней разложения и суммарной площади кристаллических граней, сгорание содержащегося в исходном материале аммиака можно подавлять, и целевой продукт можно изготавливать с высоким выходом.
Размер кристаллитов катализатора можно определять на основании рентгеновской дифракции. Поскольку пики примесей, которые не принимают участия в реакции, перекрывают пик (001), который участвует в реакции, осуществляют предварительную обработку. Эту предварительную обработку осуществляют, помещая от 5 до 20 г катализатора, 200 мл воды и 2 мл азотной кислоты в устойчивый к давлению резервуар и выдерживают в течение 24 часов или более при температуре от 150 до 200°C в герметизированном состоянии для растворения примесей. После истечения периода, составляющего 24 часов или более, температуру устойчивого к давлению резервуара уменьшают до комнатной температуры, и осуществляют фильтрование, используя фильтровальную бумагу. Твердый продукт, полученный в результате фильтрования, высушивают в течение 24 часов или более в горячей ванне при температуре от 30 до 100°C. Высушенный порошок подвергают исследованию методом рентгеноструктурного анализа, и в результате этого можно получать пик (001) только для кристаллов, участвующих в реакции.
Способ измерения размер кристаллитов можно осуществлять, используя уравнение Шеррера (Scherrer) по полуширине пика, полученного путем рентгеноструктурного анализа дифракции после завершения предварительной обработки. Конкретные условия рентгеновского измерения можно описать следующим образом. Устройство: RIGAKU RINT 2500 HF/PC, источник излучения: CuKα, выходное напряжение: 40 кВ при токе -20 мА, интервал измерения (2θ): от 5 до 50°, скорость сканирования: 1°/мин, число повторов: 4. Для получения правильной полуширины перед измерением образца оказывается предпочтительным исправление разброса значений полуширины для конкретного устройства с использованием стандартного сравнительного вещества (LaB6).
Размер кристаллитов вычисляли, используя следующее уравнение Шеррера (ii) для полуширины пика (001) (межплоскостное расстояние d=4,02 Å), полученной на основании рентгеноструктурного анализа. Пик (001) (межплоскостное расстояние d=4,02 Å) представляет собой пик, обусловленный кристаллом, участвующим в реакции.
L=0,9λ/βcosθ (ii)
Здесь L представляет собой размер кристаллитов (Å), λ представляет собой длину волны (Å), β представляет собой ширину дифракционной линии (рад), и θ представляет собой дифракционный угол (рад).
Атмосфера обжига может представлять собой воздушную атмосферу, или обжиг можно осуществлять в потоке воздуха. Однако, по меньшей мере, часть обжига предпочтительно осуществлять в потоке инертного газа, в котором практически не содержится кислород, такого как азот, с точки зрения регулирования предпочтительной степени окисления/восстановления. В том случае, где обжиг осуществляют в периодическом режиме, количество поступающего инертного газа составляет не менее чем 50 Нл/ч на 1 кг сухого порошка с точки зрения регулирования предпочтительной степени окисления и восстановления. Поступающее количество инертного газа составляет предпочтительно от 50 до 5000 Нл/ч и предпочтительнее от 50 до 3000 Нл/ч. Здесь термин «Нл» означает объем в литрах, измеряемый при нормальных условиях температуры и давление, то есть при 0°C и 1 атм. (0,1 МПа).
В том случае, когда обжиг осуществляют в непрерывном режиме, количество поступающего инертного газа составляет не менее чем 50 Нл на 1 кг сухого порошка с точки зрения регулирования предпочтительной степени окисления и восстановления. Данное количество составляет предпочтительно от 50 до 5000 Нл и предпочтительнее от 50 до 3000 Нл. В данном случае потоки инертного газа и сухого порошка могут присутствовать в форме встречных потоков или сонаправленных потоков. Однако контакт встречных потоков является предпочтительным с учетом газообразных компонентов, образующихся из сухого порошка, и следового количества воздуха, поступающего вместе с сухим порошком.
Помимо влаги, сухой порошок обычно содержит радикалы аммония, органические кислоты, неорганические кислоты и т.п. В том случае, где обжиг осуществляют в потоке инертного газа, в котором практически не содержится кислород, составляющие катализатор элементы восстанавливаются, когда радикалы аммония, органические кислоты, неорганические кислоты и другие вещества испаряются или разлагаются. В том случае, где составляющий катализатор элемент в сухом порошке имеет близкую к наиболее высокой степень окисления, чтобы получить степень восстановления катализатора в желательном интервале, на стадии обжига осуществляют только восстановление, и это является простым в промышленных условиях.
С другой стороны, как описано ниже, окисляющий компонент или восстанавливающий компонент можно вводить в атмосферу обжига таким образом, чтобы степень восстановления предварительно обожженного материала находилась в желательном интервале. В способе производства согласно настоящему варианту осуществления обжиг предпочтительно осуществляют таким образом, что степень восстановления полученного предварительно обожженного материала составляет от 8 до 12%, и удельная поверхность катализатора составляет от 5 до 25 м2/г. Если сделать удельную поверхность катализатора, составляющую от 5 до 25 м2/г, обычно можно получать преимущественные эффекты повышенной активности и подавления сгорания содержащегося в исходном материале аммиака, а также значительно более высокий выход. Если удельная поверхность катализатора составляет более чем 25 м2/г, число центров разложения на поверхности диоксида кремния увеличивается, и, таким образом, обычно происходит сгорание содержащегося в исходном материале аммиака и разложение целевого продукта. Если удельная поверхность катализатора составляет менее чем 5 м2/г, достаточное число центров разложения не образуется, и, таким образом, выход обычно уменьшается. Кроме того, что касается эффекта введения соединения молибдена для сохранения выхода в реакции аммоксидирования, данный эффект проявляется в более значительной степени при отсутствии видимого внезапного ухудшения. Соответственно, это обычно позволяет уменьшать количество и частоту введения соединения молибдена. Хотя причина этого остается неясной, полагают, что это обусловлено тем, что при удельной поверхности катализатора, составляющей менее чем 5 м2/г, мала активная грань активной частицы, участвующей в реакции, и оказывается затруднительным проявление эффекта введения соединения молибдена. Кроме того, если удельная поверхность катализатора составляет более чем 25 м2/г, хотя активная грань активной частицы, участвующей в реакции и увеличивается, скорость выхода молибдена с активной грани также увеличивается. Таким образом, удельная поверхность катализатора составляет от 5 до 25 м2/г и предпочтительно от 8 до 18 м2/г. Удельную поверхность определяют на основании одноточечного способа BET, используя прибор Gemini 2360, который производит компания Micrometrics Instrument Corporation.
Степень восстановления предварительно обожженного продукта определяется следующим уравнением (2):
Степень восстановления (%)=((n0-n)/n0)·100... (2)
(в котором n представляет собой число атомов кислорода, которые соответствуют валентности других составляющих элементов, помимо кислорода, в предварительно обожженном продукте, и n0 представляет собой требуемое число атомов кислорода, когда соответствующие составляющие элементы, отличные от кислорода, в предварительно обожженном продукте имеют наиболее высокую степень окисления).
В частности, сухой порошок обжигают, используя следующие условия обжига: температура нагревания сухого порошка увеличивается от температуры, составляющей менее чем 400°C, в непрерывном или ступенчатом режим до температуры, находящейся в интервале от 600 до 750°C. При этом условия обжига регулируют таким образом, что степень восстановления предварительно обожженного продукта, который обжигают, когда температура нагревания достигает 400°C, составляет от 8 до 12%.
Хотя температура и продолжительность конечного обжига (нагревания) катализатора и содержание диоксида кремния влияют на удельную поверхность катализатора, степень восстановления, когда температура нагревания достигает 400°C, температура и/или продолжительность основного обжига, а также скорость уменьшения температуры после основного обжига производят особенно большой эффект. Если является низкой степень восстановления, когда температура нагревания достигает 400°C, удельная поверхность катализатора, как правило, уменьшается, в то время как если является высокой степень восстановления, когда температура нагревания достигает 400°C, удельная поверхность катализатора, как правило, увеличивается. Кроме того, основной обжиг осуществляют при температуре от 600 до 750°C в течение от 0,1 до 20 часов. Как правило, чем выше температура основного обжига, или чем больше его продолжительность, тем меньшей оказывается удельная поверхность катализатора. Хотя причина остается неясной, когда осуществление обжига происходит в две стадии, если температура основного обжига является постоянной, то чем выше максимальная температура предварительного обжига, тем большей становится удельная поверхность, в то время как чем ниже максимальная температура предварительного обжига, тем меньшей становится удельная поверхность. Кроме того, оказывается предпочтительным, чтобы скорость уменьшения температуры после основного обжига составляла от 0,05 до 50°C/мин и предпочтительнее от 0,05 до 20°C/мин. Как правило, чем меньше скорость уменьшения температуры после основного обжига, тем меньшей становится удельная поверхность.
Удельную поверхность и размер кристаллитов катализатора можно регулировать раздельно путем регулирования условий обжига. Поскольку температура, при которой происходит рост кристаллов, находится в области основного обжига, размер кристаллитов регулируют посредством регулирования температуры основного обжига и/или продолжительности основного обжига. Поскольку температурная область, в которой происходит спекание диоксида кремния, который производит большой эффект на удельную поверхность, является более широкой, чем температурная область, в которой происходит рост кристаллов, оказывается предпочтительным регулировать удельную поверхность путем регулирования скорости уменьшения температуры после завершения основного обжига. Кроме того, поскольку удельная поверхность оказывает большое влияние на степень окисления и восстановления, оказывается предпочтительным регулирование уровня восстановления на основании соответствующего показателя.
В том случае, где обжиг осуществляют, используя вращающуюся обжиговую печь, скорость введения сухого порошка можно регулировать в течение обжига для регулирования удельной поверхности катализатора. Если скорость введения является низкой, сухой порошок застаивается внутри системы в течение более продолжительного времени. По этой причине восстановление сухого порошка осуществляют, используя восстанавливающий газ, такой как аммиак, который получают путем нагревания сухого порошка в обжиговой трубе, и тогда повышается степень восстановления, и увеличивается удельная поверхность катализатора, получаемого после основного обжига. С другой стороны, когда является высокой скорость введения, снижается степень восстановления, и уменьшается удельная поверхность катализатора. В качестве альтернативы, удельную поверхность можно регулировать путем регулирования количества азота в течение предварительного обжига. Если количество азота увеличивается, газообразный компонент, который восстанавливает предварительно обожженный порошок в течение обжига, быстро выводится за пределы системы. Соответственно, считается, что восстановление предварительно обожженного продукта является затруднительным, и в результате этого получается небольшая удельная поверхность. С другой стороны, если количество азота уменьшается, то повышается степень восстановления, и увеличивается удельная поверхность катализатора.
Для получения удельной поверхности катализатора, составляющей от 5 до 25 м2/г, степень восстановления, когда температура нагревания достигает 400°C, предпочтительно находится в интервале от 8 до 12%, и температура конечного обжига составляет от 600°C до 750°C.
Стадия обжига включает предварительный обжиг и основной обжиг. Предпочтительно предварительный обжиг осуществляют при температуре в интервале от 200 до 400°C, и основной обжиг осуществляют при температуре в интервале от 600 до 750°C. Предварительный обжиг и основной обжиг можно осуществлять последовательно; или предварительный обжиг можно завершать, и основной обжиг можно осуществлять снова. В качестве альтернативы, каждый из предварительного обжига и основного обжига может включать несколько стадий.
В том случае, где измеряют степень восстановления предварительно обожженного продукта в ходе обжига, образец можно извлекать из обжигового устройства при высокой температуре. Однако образец может вступать в контакт с воздухом при высокой температуре и окисляться, и степень восстановления может изменяться. Предпочтительно после того, как обжиговое устройство охлаждается до комнатной температуры, предварительно обожженный продукт извлекают из обжигового устройства и используют в качестве представительного образца. Примеры способа регулирования в желательном интервале степени восстановления, когда температура нагревания достигает 400°C, включают, в частности, способ изменения температуры в предварительном обжиге, способ введения окисляющего компонента, такого как кислород, в атмосферу в течение обжига или способ введения восстанавливающего компонента в атмосферу в ходе обжига. Кроме того, эти способы можно использовать в сочетании.
Способ изменения температуры в предварительном обжиге представляет собой изменение температуры обжига в предварительном обжиге и способ изменения степени восстановления, когда температура нагревания достигает 400°C. Как правило, степень восстановления, вероятно, уменьшается при снижении температуры в предварительном обжиге и увеличивается при повышении температуры в предварительном обжиге. По этой причине температуру в предварительном обжиге можно изменять для регулирования степени восстановления. Степень восстановления можно также регулировать путем увеличения или уменьшения поступающего количества азота, путем увеличения или уменьшения поступающего количества сухого порошка и путем увеличения или уменьшения скорости вращения вращающейся обжиговой печи при обжиге с использованием вращающейся обжиговой печи. Считается, что если количество поступающего азота увеличивается, в окисленных компонентах, испаряющихся из сухого порошка при нагревании в печи, повышается пропорция окисленных компонентов, выходящих за пределы системы без окисления оксидом металла, который присутствует внутри обжиговой печи (оксид металла восстанавливается), и, таким образом, оказывается затруднительным восстановление обожженного продукта. Кроме того, можно полагать, что если восстанавливается поступающее количество сухого порошка, восстановление происходит во вращающейся обжиговая печь, потому что катализатор застаивается в течение более продолжительного времени во вращающейся обжиговой печи. Считается также, что в случае вращающейся обжиговой печи, если скорость ее вращения уменьшается, то уменьшается и скорость движения катализатора внутри вращающейся обжиговой печи; по этой причине происходит восстановление, потому что катализатор вступает в контакт с увеличенным количеством окисленных компонентов в течение более продолжительного времени.
Измерение степени восстановления предварительно обожженного продукта перед обжигом осуществляют следующим образом.
Приблизительно 200 мг предварительно обожженного продукта взвешивают и помещают в лабораторный стакан. Кроме того, добавляют избыточное количество водного раствора KMnO4, имеющего известную концентрацию. Кроме того, добавляют 150 мл чистой воды при 70°C и 2 мл раствора 1:1 серной кислоты (используют водный раствор серной кислоты, полученный путем смешивания концентрированной серной кислоты с водой в объемном соотношении 1/1), и лабораторный стакан накрывают часовым стеклом. Смешанный раствор перемешивают в горячей водяной бане при 70°C±2°C в течение одного часа для окисления образца. При этом присутствует избыток KMnO4, и в растворе остается непрореагировавший KMnO4. По этой причине проверяют, чтобы цвет раствора был фиолетовым. После завершения окисления раствор фильтруют, используя фильтровальную бумагу, чтобы получить полное количество фильтрата. Избыточное количество водного раствора оксалата натрия, имеющего известную концентрацию, добавляют к KMnO4, который присутствует в фильтрате, и нагревают при перемешивании таким образом, что температура раствора составляет 70°C. Проверяют, что раствор становится бесцветным и прозрачным, и добавляют 2 мл раствора 1:1 серной кислоты. Перемешивание продолжают, поддерживая при этом температуру раствора на уровне 70°C±2°C, и титруют водным раствором KMnO4, имеющим известную концентрацию. Когда цвет раствора сохраняется светло-розовым в течение приблизительно 30 секунд после добавления капли KMnO4, это считается конечной точкой титрования.
Зная суммарное количество KMnO4 и суммарное количество Na2C2O4, определяют количество KMnO4, израсходованное для окисления образца. По этому значению вычисляют (n0-n), и степень восстановления определяют на основании полученного значения.
Измерение степени восстановления обожженного продукта после завершения основного обжига можно осуществлять следующим образом.
Приблизительно 200 мг обожженного продукта, измельченного в агатовой ступке, взвешивают и помещают в лабораторный стакан. Добавляют 150 мл чистой воды при 95°C и 4 мл раствора 1:1 серной кислоты (используют водный раствор серной кислоты, полученный путем смешивания концентрированной серной кислоты с водой в объемном соотношении 1/1). Перемешивание продолжают, поддерживая при этом температуру раствора на уровне 95°C±2°C, и титруют водным раствором KMnO4, имеющим известную концентрацию. При этом, хотя цвет раствора временно становится фиолетовым при титровании KMnO4, добавление KMnO4 осуществляют медленно и малыми порциями, таким образом, что фиолетовый цвет не сохраняется в течение периода, составляющего более чем 30 секунд. Количество раствора уменьшают путем испарения воды. По этой причине чистую воду при 95°C добавляют таким образом, чтобы количество раствора оставалось постоянным. Когда цвет раствора сохраняется светло-розовым в течение приблизительно 30 секунд после добавления капли KMnO4, это считается конечной точкой титрования.
Таким образом, определяют количество KMnO4, израсходованное для окисления образца. По этому значению вычисляют (n0-n), и степень восстановления определяют на основании полученного значения.
Помимо данного способа измерения, измерение степени восстановления можно осуществлять для предварительно обожженного продукта перед осуществлением основного обжига и обожженного продукта после завершения основного обжига следующим образом.
При том условии, что составляющие элементы в образце не испаряются и не теряются, образец нагревают до температуры, превышающей температуру обжига, при которой обжигают предварительно обожженный продукт или обожженный продукт, и осуществляют полное окисление кислородом. Определяют увеличение массы (количество связанного кислорода). Отсюда определяют значение (n0-n). На основании этого определяют степень восстановления.
Обжиг осуществляют, используя инертный газ или предпочтительно окислительную/восстановительную атмосферу. Таким образом, предпочтительно используют обжиговое устройство, которое имеет соответствующую герметизирующую конструкцию, и которое способно в достаточной степени блокировать контакт с открытым воздухом.
Предварительный обжиг осуществляют предпочтительно в потоке инертного газа при температуре в предварительном обжиге в интервале, составляющем предпочтительно от 200°C до 400°C и предпочтительнее от 300°C до 400°C с точки зрения простоты поддержания получаемого катализатора в предпочтительном состоянии окисления и восстановления и повышения эффективности катализатора. Предпочтительно температуру в предварительном обжиге поддерживают на постоянном уровне в интервале от 200°C до 400°C. Температуру можно изменять в интервале от 200°C до 400°C или слегка повышать или снижать. Продолжительность удерживания температуры нагревания составляет предпочтительно не менее чем 30 минут и предпочтительнее от 3 до 12 часов с точки зрения простоты поддержания получаемого катализатора в предпочтительном состоянии окисления и восстановления и повышения эффективности катализатора. Что касается режима изменения температуры, которая достигает уровня температуры в предварительном обжиге, температуру можно увеличивать линейно, или ее можно изменять по восходящей или нисходящей дуге. Кроме того, температуру можно снижать на некоторое время в процессе повышения температуры, или температуру можно периодически повышать и снижать. Кроме того, эндотермическая реакция происходит в процессе повышения температуры за счет компонента, который содержит сухой порошок и/или предшественник катализатора, и температуру можно временно снижать.
Средняя скорость повышения температуры в процессе повышения температуры до уровня температуры в предварительном обжиге не ограничивается определенным образом. Средняя скорость повышения температуры обычно составляет приблизительно от 0,1 до 15°C/мин, предпочтительно от 0,5 до 5°C/мин и предпочтительнее от 1 до 2°C/мин с точки зрения простоты поддержания получаемого катализатора в предпочтительном состоянии окисления и восстановления и повышения эффективности катализатора.
Основной обжиг можно осуществлять предпочтительно в потоке инертного газа при температуре, составляющей предпочтительно от 600 до 750°C и предпочтительнее от 650 до 720°C с точки зрения простоты поддержания получаемого катализатора в предпочтительном состоянии окисления и восстановления, образования кристаллической структуры, имеющей достаточную активность для осуществления реакции, и повышения эффективности катализатора. Температуру основного обжига предпочтительно поддерживают на постоянном уровне в интервале от 650 до 720°C. Данную температуру можно изменять или слегка повышать или снижать в интервале от 650 до 720°C. Кроме того, температуру можно снижать на некоторое время в процессе повышения температуры, или температуру можно периодически повышать и снижать. Эндотермическая реакция происходит в процессе повышения температуры за счет компонента, который содержит сухой порошок, и температуру можно временно снижать в режиме, соответствующем развитию ситуации
Удельную поверхность катализатора можно регулировать посредством регулирования температуры обжига. Катализатор, имеющий заданную удельную поверхность, можно получать путем увеличения или уменьшения температуры предварительного обжига или удельной поверхности. Предпочтительно температуру обжига при основном обжиге, которая влияет на удельная поверхность, легко регулируют для получения целевого катализатора, имеющего заданную удельную поверхность.
Продолжительность основного обжига составляет предпочтительно от 0,1 до 20 часов и предпочтительнее от 0,5 до 5 часов. В том случае, где обжиговая труба разделена отбивной решеткой, предварительно обожженный продукт и/или обожженный продукт непрерывно проходит, по меньшей мере, через две зоны, предпочтительно от до 20 зон и предпочтительнее от 4 до 15 зон с точки зрения обеспечения продолжительности выдерживания сухого порошка или подобного материала в обжиговой трубе. Температуру можно регулировать, используя один или несколько регуляторов. Для получения желательного режима обжига нагреватель и регулятор предпочтительно устанавливают в каждой из зон, разделенных этими решетками, чтобы регулировать температуру. Например, в том случае, где семь отбивных решеток устанавливают таким образом, что часть обжиговой трубы, находящаяся внутри нагревательной печи, разделена на восемь зон равной длины, и используют обжиговую трубу, предпочтительно имеющую восемь разделенных зон, причем установленную температуру в каждой из восьми зон регулируют, используя нагреватель и регулятор, установленные в каждой из восьми зон, таким образом, что температуру частично обожженного порошка и/или обожженный порошок регулируют в соответствии с желательным температурным режимом обжига. Например, в том случае, где семь отбивных решеток установлены таким образом, что часть обжиговой трубы, находящаяся внутри нагревательной печи, разделена на восемь зон равной длины, и используют обжиговое устройство, имеющий восемь разделенных регулирование зон можно осуществлять следующим образом для получения желательного режима обжига. При предварительном обжиге температуру термопары, вставленной в центральную часть предварительно обожженного продукта, который выдерживают в каждой из зон обжигового устройства, предпочтительно регулируют таким образом, что температура составляет от 120 до 280°C в зоне 1, от 180 до 330°C в зоне 2, от 250 до 350°C в зоне 3, от 270 до 380°C в зоне 4, от 300 до 380°C в зоне 5, от 300 до 390°C в зона 6, от 320 до 390°C в зоне 7 и от 260 до 380°C в зоне 8, считая со стороны введения предварительно обожженного продукта. Аналогичным образом, при основном обжиге регулирование предпочтительно осуществляют таким образом, что температура составляет от 360 до 560°C в зоне 1, от 450 до 650°C в зоне 2, от 600 до 700°C в зоне 3, от 650 до 750°C в зоне 4, от 600 до 700°C в зоне 5, от 500 до 690°C в зона 6, от 480 до 630°C в зоне 7 и от 400 до 580°C в зоне 8.
Удельную поверхность предварительно обожженного продукта можно регулировать в некоторой степени согласно условиям предварительного обжига, но не в такой большой степени, как в случае обожженного продукта. Хотя причина остается неясной, степень восстановления пропорциональна удельной поверхности, и путем осуществления такого же регулирования, как описано выше, легко оптимизируется интервал значений удельной поверхности. Однако регулирование удельной поверхности катализатора в значительной степени зависит от способа обжига при основном обжиге.
Температура обжига, составляющая 650°C, значительно превышает температуры плавления оксидов составляющих металлов. По этой причине большое количество оксидов прикрепляется к поверхности стенки обжиговой трубы. Таким образом, продолжительность выдерживания предварительно обожженного продукта предпочтительно увеличивается при увеличении числа ударов, нанесенных по основной обжиговой трубе с использованием молота или аналогичного устройства, или при увеличении скорости вращения. Степень увеличения этих чисел можно произвольно устанавливать на основании массового баланса между количеством предварительно обожженного порошка, поступающим в основную обжиговую трубу, и количеством катализатора, выходящего из основной обжиговой трубы. Окисляющий компонент (например, кислород) или восстанавливающий компонент (например, аммиак) можно вводить в атмосферу обжига в потоке инертного газа, если это желательно.
Что касается режима повышения температуры до уровня температуры основного обжига, температуру можно увеличивать линейно, или ее можно изменять по восходящей или нисходящей дуге. Кроме того, температуру можно снижать на некоторое время в процессе повышения температуры, или температуру можно периодически повышать и снижать. Эндотермическая реакция происходит в течение повышения температуры за счет компонента, который содержится в предварительно обожженном продукте, и температуру можно снижать по режиму, определяемому в зависимости от развития ситуации.
Средняя скорость повышения температуры в процессе повышения температуры, при которой температура достигает уровня температуры основного обжига, не ограничивается определенным образом и предпочтительно составляет от 0,5 до 8°C/мин. Средняя скорость снижения температуры после завершения основного обжига составляет предпочтительно от 0,05 до 50°C/мин и предпочтительнее от 0,05 до 20°C/мин с точки зрения регулирования удельной поверхности, образования кристаллической структуры с достаточной активностью для реакции и повышения эффективности катализатора. Предпочтительно эту температуру выдерживают на уровне ниже температуры основного обжига, и отжиг осуществляют с точки зрения образования кристаллической структуры с достаточной активностью для реакции и повышения эффективности катализатора. Следует поддерживать температуру, которая на 5°C, предпочтительно на 10°C и предпочтительнее 50°C ниже, чем температура основного обжига. С такой же точка зрения, как указано выше, продолжительность выдерживания температуры составляет предпочтительно не менее чем 0,5 часа, предпочтительнее не менее чем 1 час, еще предпочтительнее не менее чем 3 часа и особенно предпочтительно не менее чем 10 часов.
Когда основной обжиг осуществляют снова после завершения предварительного обжига, низкотемпературную обработку можно осуществлять при основном обжиге. Время, требуемое для низкотемпературной обработки, то есть время, требуемое для уменьшения температуры предварительно обожженного продукта и/или обожженного продукта и повышения температуры до уровня температуры обжига, можно соответствующим образом регулировать посредством размера, толщины и материала обжигового устройства, количества производимого катализатора, последовательности периодов для непрерывного обжига предварительно обожженного продукта и/или обожженного продукта, а также путем установления скорости и установления количества и т.п. Время, требуемое для низкотемпературной обработки, составляет предпочтительно 30 суток или менее, предпочтительнее 15 суток или менее, еще предпочтительнее 3 суток или менее и особенно предпочтительно 2 суток или менее в течение последовательности непрерывного обжига обожженного продукта с точки зрения достаточного удаления предварительно обожженного порошка и/или обожженного продукта, прикрепленного к поверхности стенки обжиговой трубы, устойчивого поддержания температуры оксидного слоя и повышения эффективности получаемого катализатора. Температура оксидного слоя означает температуру, измеряемую с помощью термопары, вставленной в предварительно обожженный порошок и/или в основной обожженный порошок, находящийся внутри обжигового устройства. Кроме того, например, когда предварительно обожженный порошок поступает со скоростью 35 кг/ч, в то время как вращающаяся обжиговая печь, включающая обжиговую трубу, у которой внутренний диаметр составляет 500 мм, длина составляет 4500 мм, и толщина составляет 20 мм, которая изготовлена из нержавеющей стали и вращается со скоростью 6 об/мин, и температура основного обжига составляет 645°C, стадию снижения температуры до 400°C и повышения температуры до 645°C можно осуществлять в течение приблизительно одних суток. Когда обжиг непрерывно осуществляют в течение одного года, обжиг можно осуществлять путем проведения такой низкотемпературной обработки один раз в месяц при одновременном устойчивом поддержании температуры оксидного слоя.
Если ударное воздействие на обжиговое устройство происходит на стадии обжига, вероятно, должен усиливаться эффект растрескивания прикрепленных частей. В том случае, где осуществляют низкотемпературную обработку, ударное воздействие на обжиговое устройство является предпочтительным, потому что части с трещинами, вероятно, должны легко удаляться от обжигового устройства.
Выступающие части образуются на поверхности частиц обожженного материала, который был подвергнут основной стадии обжига. Поскольку обожженный материал согласно настоящему варианту осуществления имеет более соответствующий состав, чем состав традиционного обожженного материала, количество выступающих частей уменьшается, и эффект выступающих частей становится меньше, чем в случае традиционного катализатора. Однако если выступающие части присутствуют в реакторе в течение парофазной реакции аммоксидирования, потому что обычно происходят побочные реакции, и/или текучесть уменьшается вследствие отделения выступающих частей, оказывается предпочтительным удаление выступающих частей перед реакцией.
Оказывается предпочтительным снижать количество выступающих частей до 2 мас.% или менее по отношению к суммарной массе обожженного материала путем удаления выступающих частей. Можно рассматривать несколько способов в качестве способа удаления выступающих частей. Среди них предпочтительным является способ, в котором выступающие части удаляют, например, путем приведения частиц катализатора в контакт друг с другом в газовом потоке. Примеры такого способа включают способ, в котором газ протекает через бункер или подобное устройство, в котором хранится обожженный материал, и способ, в котором обожженный материал помещают реактор с псевдоожиженным слоем, и создают через него газовый поток. Хотя способ с использованием реактора с псевдоожиженным слоем является предпочтительным, поскольку в нем не требуется какое-либо специальное устройство для удаления выступающих частей, потому что такое устройство не было первоначально предназначено для приведения частицы катализатора в контакт друг с другом, если не предпринимаются особые меры, такие как введение небольшого количества катализатора и осуществление реакции в течение некоторого времени, в зависимости от условий, таких как поступающее количество катализатора и время для движения катализатора и количества газа, выступающие части не могут быть удалены в достаточной степени. Согласно исследованию авторов настоящего изобретения, воздушный поток при достаточной скорости потока может вступать в контакт с обожженным продуктом, имеющим выступающую часть, чтобы эффективно удалять выступающую часть. Если соответствующая скорость потока обеспечена в устройстве, имеющем структуру, в котором воздушный поток вступает в контакт с обожженным продуктом, выступающую часть можно эффективно удалять даже в большом масштабе.
Например, устройство способно эффективно удалять выступающие части в большом масштабе, причем данное устройство включает основной корпус, в котором находится обожженный продукт, извлекающий блок для извлечения обожженного продукта, находящегося в верхней части основного корпуса, и возвращающий блок для возвращения обожженного продукта, который присоединен к извлекающему блоку. Возвращающий блок сконструирован таким образом, что его нижний край находится в контакте с воздушным потоком. Часть обожженного продукта, которая вступает в контакт с воздушным потоком внутри основного корпуса, извлекают посредством извлекающего блока и возвращают в основной корпус посредством возвращающего блока.
Газ протекает через устройство, заполненное обожженным продуктом, такое как реактор с псевдоожиженным слоем. В результате этого обожженные продукты вступают в контакт друг с другом для удаления выступающих частей. Выступающая часть, удаленная с обожженного продукта, является значительно меньшей, чем обожженный продукт, и удаляется с потоком газа за пределы реактора с псевдоожиженным слоем. Предпочтительно обожженный продукт заполняет устройство таким образом, что плотность обожженного продукта при этом составляет от 300 до 1300 кг/м3. Площадь поперечного сечения корпуса используемого устройства составляет предпочтительно от 0,1 до 100 м2 и предпочтительнее от 0,2 до 85 м2.
Протекающий газ предпочтительно представляет собой инертный газ, такой как азот и воздух. Линейная скорость газа, протекающего через корпус устройства, заполненного обожженным продуктом, такого как бункер и реактор с псевдоожиженным слоем, составляет предпочтительно от 0,03 м/с до 5 м/с и предпочтительнее от 0,05 до 1 м/с. Продолжительность протекания газа составляет предпочтительно от 1 до 168 часов. В частности, устройство для удаления выступающих частей согласно настоящему варианту осуществления включает основной корпус, причем обожженный продукт, находящийся в основном корпусе, вступает в контакт с воздушным потоком, или частицы, переносимые воздушным потоком, вступают в контакт друг с другом для удаления выступающей части на поверхности обожженного продукта с обожженного продукта. Предпочтительно длина воздушного потока в направлении движения воздушного потока составляет не менее чем 10 мм, и средняя скорость потока для воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с, причем она представляет собой линейную скорость при температуре 15°C и давлении, составляющем одну атмосферу (0,1 МПа).
Если удаление выступающих частей, осуществляют, применяя граммовый масштаб, можно использовать следующее устройство. А именно, это устройство, которое содержит вертикальную трубу, оборудованную плитой, имеющей один или несколько отверстий, изготовленных в нижней части, и бумажный фильтр в верхней части. Обожженный материал поступает в вертикальную трубу, и воздушный поток движется из нижней части вертикальной трубы. Воздушный поток движется через соответствующие отверстия, что способствует контакту между частицами обожженного материала, и в результате этого удаляются выступающие части.
[Реакция парофазного каталитического аммоксидирования]
Реакция парофазного каталитического аммоксидирования согласно настоящему варианту осуществления представляет собой способ производства ненасыщенного нитрила, соответствующего пропану или изобутану, путем реакции парофазного каталитического аммоксидирования пропана или изобутана, в которой используют нанесенный на диоксид кремния катализатор.
Исходные материалы для пропана, изобутана и аммиака не всегда должны иметь высокую чистоту, и можно использовать газ технической чистоты. В качестве исходного материала для кислорода можно использовать воздух, чистый кислород или воздух, обогащенный чистым кислородом. Кроме того, в качестве разбавляющего газа можно вводить гелий, неон, аргон, диоксид углерода (углекислый газ), пар, азот и т.п.
Реакцию парофазного каталитического аммоксидирования пропана или изобутана можно осуществлять в следующих условиях.
Молярное соотношение поступающего для реакции кислорода и пропана или изобутана составляет от 0,1 до 6 и предпочтительно от 0,5 до 4. Молярное соотношение поступающего для реакции аммиака и пропана или изобутана составляет от 0,3 до 1,5 и предпочтительно от 0,7 до 1,2. Температура реакции составляет от 350 до 500°C и предпочтительно от 380 до 470°C. Давление реакции составляет от 5·104 до 5·105 Па и предпочтительно от 1·105 до 3·105 Па. Продолжительность контакта составляет от 0,1 до 10 (с·г/мл) и предпочтительно 0,5 до 5 (с·г/мл).
В настоящем варианте осуществления продолжительность контакта вычисляют, используя приведенное ниже уравнение:
Продолжительность контакта (с·г/мл)=(W/F)·273/(273+T)
Здесь W, F, и T определяют следующим образом:
W=количество используемого катализатора (г)
F=скорость потока исходный смешанного газообразного материала (Нмл/с) в стандартных условиях (0°C, 1,013·105 Па)
T=температура реакции (°C)
В качестве способа осуществления реакции парофазного каталитического аммоксидирования, можно использовать традиционный способ, такой как проведение реакции в неподвижном слое, в псевдоожиженном слое и в подвижном слое. Предпочтительным является реактор с псевдоожиженным слоем, в котором теплота реакция легко удаляется. Реакцию парофазного каталитического аммоксидирования можно осуществлять в однопроточной системе или в системе с рециркуляцией.
Примеры
Далее настоящий вариант осуществления будет подробно описан с представлением примеров и сравнительных примеров. Однако объем настоящего варианта осуществления не ограничен данными примерами.
В примерах и сравнительных примерах степень конверсии пропана, выход акрилонитрила и степень сгорания аммиака, соответственно, выражены следующими определениями.
Степень конверсии пропана (%)=(Число моль прореагировавшего пропана)/(Число моль исходного пропана)·100
Выход акрилонитрила (AN) (%)=(Число моль полученного акрилонитрила)/(Число моль исходного пропана)·100
Степень сгорания аммиака(%)=(Число моль полученного азота)·2/(Число моль исходного аммиака)
Число моль полученных акрилонитрила и азота измеряли методом газовой хроматографии.
(Пример 1)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали способом, который описан ниже. В 500 кг воды добавляли 76,33 кг ниобиевой кислоты, содержащей 80,2 мас.% ниобия в пересчете на Nb2O5, и 290,2 кг дигидрата щавелевой кислоты [H2C2O4·2H2O]. Молярное соотношение щавелевой кислоты и ниобия в качестве исходных материалов составляло 5,0, и концентрация исходного материала ниобий составляла 0,532 (моль Nb/кг раствора).
Полученный в результате раствор нагревали в течение одного часа при 95°C при перемешивании, и таким способом получали водный раствор, в котором было растворено соединение ниобия. Этот водный раствор выдерживали в неподвижном виде, охлаждали льдом, подвергали вакуумному фильтрованию для отделения твердого содержимого и в результате этого получали однородный водный раствор соединения ниобия. После того, как данную процедуру повторяли несколько раз, полученные в результате водные растворы соединения ниобия объединяли и получали раствор исходного материала ниобия. Молярное соотношение щавелевой кислоты и ниобия в данном растворе исходного материала ниобия составляло 2,40, как показал анализ, описанный ниже.
10 г этого раствора исходного материала ниобия точно взвешивали, помещали в тигель, высушенный в течение ночи при 95°C, подвергали термической обработке в течение одного часа при 600°C и в результате этого получали 0,8323 г Nb2O5. Согласно этому результату, концентрация ниобия составляла 0,626 (моль Nb/кг раствора).
3 г этого раствора исходного материала ниобия точно взвешивали и помещали в стеклянный лабораторный стакан, имеющий объем 300 мл, добавляли 200 мл горячей воды, у которой температура составляла приблизительно 80°C, и после этого добавляли 10 мл раствора 1:1 серной кислоты. Полученный в результате водный раствор титровали, используя ¼-нормальный раствор KMnO4 при перемешивании, поддерживая при этом температуру 70°C на горячей мешалке. Точку, в которой слабый светло-розовый цвет KMnO4 сохранялся в течение приблизительно 30 секунд или более, определяли как конечную точку титрования. Концентрацию щавелевой кислоты определяли на основании полученного в результате титра в соответствии со следующей формулой, и, таким образом, она составляла 1,50 (моль щавелевой кислоты/кг).
Полученный раствор исходного материала ниобия использовали в качестве раствора исходного материала ниобия в производство оксидного катализатора, которое будет описано ниже.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,9 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 34,2 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 3,60 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, устанавливали таким образом, что не происходило колебаний температуры выпуска распылительной сушилки, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 690°C со скоростью 2°C/мин, обжиг осуществляли при 690°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что он имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Удельную поверхность измеряли в соответствии с одноточечным методом BET, используя прибор Gemini 2360, изготовленный компанией Micrometrics Instrument Corporation.
Удельная поверхность составляла 10,8 м2/г.
(Удаление выступающих частей)
50 г оксидного катализатора пропускали в вертикальную трубу (внутренний диаметр: 41,6 мм, длина: 70 см), в котором перфорированный диск, имеющий три отверстия диаметром 1/64 дюйма (0,4 мм), сделанных в нижней части трубы, и бумажный фильтр устанавливали в ее верхней части. Длина воздушного потока в направлении его движения при этом составляла 52 мм, и средняя линейная скорость воздушного потока составляла 310 м/с. На основании исследования полученный оксидного катализатора через 24 часа с использованием сканирующего электронного микроскопа (SEM), не было обнаружено ни одной выступающей части, присутствующей на поверхности оксидного катализатора.
(Суммарный объем пор)
Суммарный объем пор определяли, используя ртутный поромер.
Суммарный объем пор составлял 0,297 см3/г.
(Распределение пор)
Распределение пор определяли, используя ртутный поромер.
Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 3,9%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 1,0%.
(Вычисление среднего размера пор)
Средний размер пор вычисляли, используя формулу (i) и предполагая, что поры имели цилиндрическую форму.
D=4·V/S (i)
Здесь D представляет собой средний размер пор (м), V представляет собой суммарный объем пор (м3/г), и S представляет собой удельную поверхность (м2/г).
Вычисленный средний размер пор составлял 110 нм.
(Измерение размера кристаллитов)
Способ измерения размера кристаллитов можно осуществлять, используя уравнение Шеррера, зная полуширину пика, полученную методом рентгеновской дифракции после завершения предварительной обработки. Конкретные условия рентгеновских измерений можно описать следующим образом. Устройство: RIGAKU RINT 2500 HF/PC, источник излучения: CuKα, выходное напряжение: 40 кВ при токе -20 мА, интервал измерения (2θ): от 5 до 50°, скорость сканирования: 1°/мин, число повторов: 4. Для получения правильной полуширины перед измерением образца оказывается предпочтительным исправление разброса значений полуширины для конкретного устройства с использованием стандартного сравнительного вещества (LaB6).
Размер кристаллитов вычисляли, используя следующее уравнение Шеррера (ii) для полуширины пика (001) (межплоскостное расстояние d=4,02 Å), полученной на основании рентгеновской дифракции. Пик (001) (межплоскостное расстояние d=4,02 Å) представляет собой пик, обусловленный кристаллом, участвующим в реакции.
L=0,9λ/βcosθ (ii)
Здесь L представляет собой размер кристаллитов (Å), λ представляет собой длину волны (Å), β представляет собой ширину дифракционной линии (рад), и θ представляет собой дифракционный угол (рад).
Вычисленный размер кристаллитов составлял 106 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор (Vycor) с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 89,8%, выход акрилонитрила составлял 54,8%, и степень сгорания аммиака составляла 18,8%.
(Пример 2)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 685°C со скоростью 2°C/мин, обжиг осуществляли при 685°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 12,8 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,288 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 6,8%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,6%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 90 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 98 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 90,1%, выход акрилонитрила составлял 54,9%, и степень сгорания аммиака составляла 18,6%.
(Пример 3)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 25,3 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 25 нм, и 12,5 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 10 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 13,6 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,221 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 18,7%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,2%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 65 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 102 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,5%, и выход акрилонитрила составлял 54,7%. Реакцию осуществляли в течение 3 месяцев. Выход акрилонитрила составлял 54,7%, и степень сгорания аммиака составляла 19,4%.
(Пример 4)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 1,96 кг триоксида теллура [TeO3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 45 нм, и 7,70 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 15 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 690°C со скоростью 2°C/мин, обжиг осуществляли при 690°C в течение трех часов, и температуру уменьшали со скоростью 0,5°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Te0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 10,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,235 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 7,1%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,7%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 92 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 185 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,8%, и степень сгорания аммиака составляла 19,0%.
(Пример 5)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II), 258 г водного раствора метавольфрамата аммония (чистота 50%), и 18,2 г диоксида титана (TiO2). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,005Ti0,002Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 12,8 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,288 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 6,6%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,5%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 90 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 98 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,6%, и степень сгорания аммиака составляла 19,5%.
(Пример 6)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II), 258 г водного раствора метавольфрамата аммония (чистота 50%) и 29,6 г диоксида марганца (MnO2). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,005Mn0,003Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 13,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Измерение суммарного объема пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,304 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 6,9%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,6%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 92 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 101 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,7%, и степень сгорания аммиака составляла 19,3%.
(Пример 7)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 155 г водного раствора метавольфрамата аммония (чистота 50%) и 220 г нитрата висмута [Bi(NO3)3·5H2O]. Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,003Bi0,004Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 13,3 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,313 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 7,3%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,8%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 94 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 103 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,6%, и степень сгорания аммиака составляла 19,2%.
(Пример 8)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 347 г нитрата церия [Ce(NO3)3·6H2O] и 147 г нитрата лантана [La(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 7,70 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,007La0,003. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 14,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,320 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 6,7%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,4%.
(Измерение распределения пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 90 нм.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 95 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,6%, и степень сгорания аммиака составляла 18,8%.
(Пример 9)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 397 г нитрата церия [Ce(NO3)3·6H2O] и 87 г нитрата иттрия [Y(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 7,70 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,008Y0,002. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 14,5 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,330 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 7,2%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,5%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 91 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 102 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,7%, и степень сгорания аммиака составляла 18,9%.
(Пример 10)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 297 г нитрата церия [Ce(NO3)3·6H2O] и 146 г нитрата иттербия [Yb(NO3)3·4H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 50 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 18 нм. После этого добавляли 3,80 г раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,006Yb0,003. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 15,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,334 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 8,2%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,4%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 88 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 98 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,8%, выход акрилонитрила составлял 54,6%, и степень сгорания аммиака составляла 19,0%.
(Пример 11)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 680°C со скоростью 2°C/мин, обжиг осуществляли при 680°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 14,6 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,350 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 5,8%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,9%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 96 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 61 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 88,9%, выход акрилонитрила составлял 54,4%, и степень сгорания аммиака составляла 19,1%.
(Пример 12)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 685°C со скоростью 2°C/мин, обжиг осуществляли при 685°C в течение двух с половиной часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 15,1 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,306 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 9,3%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,3%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 81 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 181 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 89,1%, выход акрилонитрила составлял 54,3%, и степень сгорания аммиака составляла 19,4%.
(Пример 13)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 34,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 55 нм, и 3,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 695°C со скоростью 2°C/мин, обжиг осуществляли при 695°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 8,0 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,168 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 8,8%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,4%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 84 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 156 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 89,2%, выход акрилонитрила составлял 54,0%, и степень сгорания аммиака составляла 19,2%.
(Пример 14)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 670°C со скоростью 2°C/мин, обжиг осуществляли при 670°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 16,7 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,342 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 9,0%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,3%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 82 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 52 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 89,1%, выход акрилонитрила составлял 54,0%, и степень сгорания аммиака составляла 19,5%.
(Пример 15)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 30,7 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 25 нм, и 7,1 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 12 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 695°C со скоростью 2°C/мин, обжиг осуществляли при 695°C в течение один час, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 9,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,170 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 10,6%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,3%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 74 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 55 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 89,1%, выход акрилонитрила составлял 54,1%, и степень сгорания аммиака составляла 19,3%.
(Пример 16)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 56,4 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,90 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Затем полученную в результате смесь выдерживали в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 685°C со скоростью 2°C/мин, обжиг осуществляли при 685°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 10,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,184 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 9,6%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,2%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 72 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 98 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 90,1%, выход акрилонитрила составлял 54,1%, и степень сгорания аммиака составляла 19,6%.
(Пример 17)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 550°C со скоростью 2°C/мин, обжиг осуществляли при 550°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 17,4 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,270 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 31,2%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,1%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 62 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 44 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 90,1%, выход акрилонитрила составлял 53,8%, и степень сгорания аммиака составляла 19,8%.
(Сравнительный пример 1)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
21,0 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,91 кг метаванадата аммония [NH4VO3], 3,46 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 524 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
134 г раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 937 г раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 26 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 16 нм. После этого добавляли 4,02 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 523 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,60Nb0,005Sb0,30W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 14,6 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,329 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 8,9%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0,4%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 90 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 120 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 87,5%, выход акрилонитрила составлял 51,5%, и степень сгорания аммиака составляла 21,1%.
(Сравнительный пример 2)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 108 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 16 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 14,0 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,543 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 1,3%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 3,2%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 155 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 105 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 86,9%, выход акрилонитрила составлял 52,6%, и степень сгорания аммиака составляла 19,3%.
(Сравнительный пример 3)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 12 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 5 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Классифицированные продукты обжигали таким же образом, как в примере 2.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 13,8 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,086 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 88,2%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 25 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 104 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 87,1%, выход акрилонитрила составлял 53,1%, и степень сгорания аммиака составляла 22,6%.
(Сравнительный пример 4)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 110 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 16 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 695°C со скоростью 2°C/мин, обжиг осуществляли при 695°C в течение четырех часов, и температуру уменьшали со скоростью 0,5°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 8,1 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,324 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 1,1%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 3,8%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 160 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 390 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 85,9%, выход акрилонитрила составлял 52,2%, и степень сгорания аммиака составляла 20,1%.
(Сравнительный пример 5)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 108 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 16 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 670°C со скоростью 2°C/мин, обжиг осуществляли при 670°C в течение один час, и температуру уменьшали со скоростью 2°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 16,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,559 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 2,3%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 3,2%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 138 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 20 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 86,5%, выход акрилонитрила составлял 52,1%, и степень сгорания аммиака составляла 19,3%.
(Сравнительный пример 6)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 12 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 8 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 695°C со скоростью 2°C/мин, обжиг осуществляли при 695°C в течение четырех часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 9,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,097 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 68,4%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 42 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 375 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 87,0%, выход акрилонитрила составлял 52,3%, и степень сгорания аммиака составляла 23,1%.
(Сравнительный пример 7)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 10 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 13 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 670°C со скоростью 2°C/мин, обжиг осуществляли при 670°C в течение один час, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 20,3 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,112 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 91,4%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 22 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 24 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 86,3,1%, выход акрилонитрила составлял 52,1%, и степень сгорания аммиака составляла 21,1%.
(Сравнительный пример 8)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 31,0 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 15 нм, и 6,80 кг золя диоксида кремния, содержащего 30,0 мас.% SiO2, у которого средний размер первичных частиц составлял 5 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 700°C со скоростью 2°C/мин, обжиг осуществляли при 700°C в течение двух часов, и температуру уменьшали со скоростью 0,2°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 4,2 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,055 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 54,6%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0%.
(Вычисление среднего размера пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 52 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 204 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 86,2%, выход акрилонитрила составлял 50,6%, и степень сгорания аммиака составляла 22,8%.
(Сравнительный пример 9)
(Изготовление раствора исходного материала ниобия)
Раствор исходного материала ниобия изготавливали таким же образом, как в примере 1.
(Составление изготовленного из исходного материала раствора в смесительном резервуаре)
19,9 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 2,75 кг метаванадата аммония [NH4VO3], 3,28 кг оксида сурьмы(III) [Sb2O3], а также водный раствор нитрата церия, в котором 495 г нитрата церия [Ce(NO3)3·6H2O] было растворено в 2 кг воды, добавляли в 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, в результате чего получали изготовленный из исходного материала раствор (I).
2,28 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, добавляли к 15,95 кг раствора исходного материала ниобия. Растворы смешивали и перемешивали, поддерживая температуру смешанного раствора на уровне приблизительно 20°C, в результате чего получали изготовленный из исходного материала раствор (II).
Полученный изготовленный из исходного материала раствор (I) охлаждали до 70°C, и добавляли 34,7 кг золя диоксида кремния, содержащего 30,2 мас.% SiO2, у которого средний размер первичных частиц составлял 23 нм. После этого добавляли 3,80 кг раствора пероксида водорода, содержащего 30 мас.% H2O2, и полученную смесь перемешивали в течение 30 минут при 55°C. После этого добавляли изготовленный из исходного материала раствор (II) и 516 г водного раствора метавольфрамата аммония (чистота 50%). Кроме того, 8,60 кг порошка диоксида кремния диспергировали в 77,4 кг воды, и полученную в результате смесь выдерживали в неизменном виде в течение одного часа при 50°C, в результате чего получали изготовленный из исходного материала раствор (III).
(Распылительное высушивание изготовленного из исходного материала раствора, полученного в смесительном резервуаре)
Воздух, нагретый до 210°C, и горячую воду при 50°C при скорости введения, установленной на уровне 80 кг/ч, направляли в центробежную распылительную сушилку до тех пор, пока не было завершено составление изготовленного из исходного материала раствора (III); причем у сушилки предварительно устанавливали температуру впуска на уровне 210°C и температуру выпуска на уровне 120°C.
Когда подаваемое количество изготовленного из исходного материала раствора, поступающего в распылительную сушилку, регулировали таким образом, что не происходило колебаний температуры материала, поступающего в распылительную сушилку, поступающее количество составляло 100 кг/ч. В течение этого периода температура выпуска составляла 120±5°C и не колебалась в значительной степени.
(Измерение ультрафиолетового и видимого спектра отражения)
Из полученного высушенного продукта ежедневно отбирали образцы. По 0,5 г из полученных 10 образцов исследовали в ультрафиолетовом и видимом диапазонах, используя спектрометр JASCO V-650, изготовленный компанией JASCO Corporation, в интервале от 200 до 800 нм, применяя метод рассеянного отражения. В качестве фонового эталонного материала использовали Spectralon, изготовленный компанией Labsphere. Максимальное значение коэффициента поглощения составляло 1,02. Коэффициент поглощения при 600 нм составлял от 0,31 до 0,36. Поскольку это был коэффициент поглощения, для которого можно было предполагать высокую эффективность на основании описания в японской выложенной патентной заявке № 2009-148749, весь полученный распылительным высушиванием продукт использовали в операции классификации без сортировки.
(Операция классификации)
Полученный высушенный продукт классифицировали, используя сито, у которого размер отверстий составлял 25 мкм, чтобы получить классифицированный продукт. Содержание частиц с размером 25 мкм или менее в полученном классифицированном продукте составляло 0,8 мас.%, и средний размер частиц составлял 55 мкм.
(Обжиг классифицированного продукта)
Полученный классифицированный продукт пропускали при скорости 20 кг/ч через цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали и имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина секции нагревательной печи была разделена на восемь равных отрезков. В потоке газообразного азота, составлявшем 600 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 370°C в течение приблизительно четырех часов и оставалась на уровне 370°C в течение трех часов, в то время как обжиговая труба вращалась при скорости 4 об/мин, чтобы подвергнуть классифицированный продукт предварительному обжигу, в результате чего получали предварительно обожженный продукт. После этого предварительно обожженный продукт пропускали при скорости 15 кг/ч через другую обжиговую трубу, изготовленную из нержавеющей стали, имеющую внутренний диаметр 500 мм, длину 3500 мм и толщину 20 мм, в которой было установлено семь отбивных решеток, имеющих высоту 150 мм, таким образом, что длина нагревательной секции была разделена на восемь равных отрезков, в то время как обжиговая труба вращалась при скорости 4 об/мин. При этом, в то время как часть предварительно обожженного продукта на стороне впуска порошка обжиговой трубы (часть, не закрытая нагревательной печью) обрабатывали молотом каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, используя молотовое устройство, оборудованное молотом, имеющим массу 14 кг, причем ударный наконечник молота был изготовлен из нержавеющей стали, и в потоке газообразного азота, составлявшем 500 Нл/мин, температуру нагревательной печи регулировали таким образом, чтобы получить режим изменения температуры, в котором температура увеличивалась до 685°C со скоростью 2°C/мин, обжиг осуществляли при 685°C в течение двух часов, и температуру уменьшали со скоростью 1°C/мин, осуществляли основной обжиг, и в результате этого получали оксидный катализатор.
(Состав оксидного катализатора)
Анализ оксидного катализатора показал, что оксид металла имел состав MoV0,21Nb0,09Sb0,20W0,01Ce0,01. Кроме того, количество диоксида кремния в качестве носителя составляло 47 мас.% по отношению к суммарному количеству катализатора, изготовленного из оксида металла и диоксида кремния.
(Измерение удельной поверхности)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, удельная поверхность составляла 15,1 м2/г.
(Удаление выступающих частей)
Выступающие части удаляли таким же образом, как в примере 1.
(Суммарный объем пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, суммарный объем пор составлял 0,174 см3/г.
(Распределение пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, Соотношение объема пор, у которых размер составляет менее чем 60 нм, составляло 63,8%, и соотношение объема пор, у которых размер пор превышает 120 нм, составляло 0%.
(Средний размер пор)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, средний размер пор составлял 46 нм.
(Измерение размера кристаллитов)
Согласно результатам измерения, осуществленного таким же образом, как в примере 1, размер кристаллитов составлял 98 нм.
(Реакция аммоксидирования пропана)
Пропан вводили в парофазную реакцию аммоксидирования следующим способом, используя оксидный катализатор, полученный, как описано выше. 35 г оксидного катализатора помещали в трубчатый реактор из стекла типа викор с псевдоожиженным слоем, имеющий внутренний диаметр 25 мм; и газовую смесь, содержащую пропан, аммиак, кислород и гелий в молярном соотношении 1:1:3:18, подавали в течение времени контакта 2,8 (с·г/мл), причем реакцию проводили при температуре 440°C и атмосферном давлении. Степень конверсии пропана после реакции составляла 90,1%, выход акрилонитрила составлял 53,1%, и степень сгорания аммиака составляла 22,3%.
Следующая таблица 1 иллюстрирует состав и физические свойства катализаторов согласно соответствующим примерам и сравнительным примерам, а также выход акрилонитрила и степень сгорания аммиака.
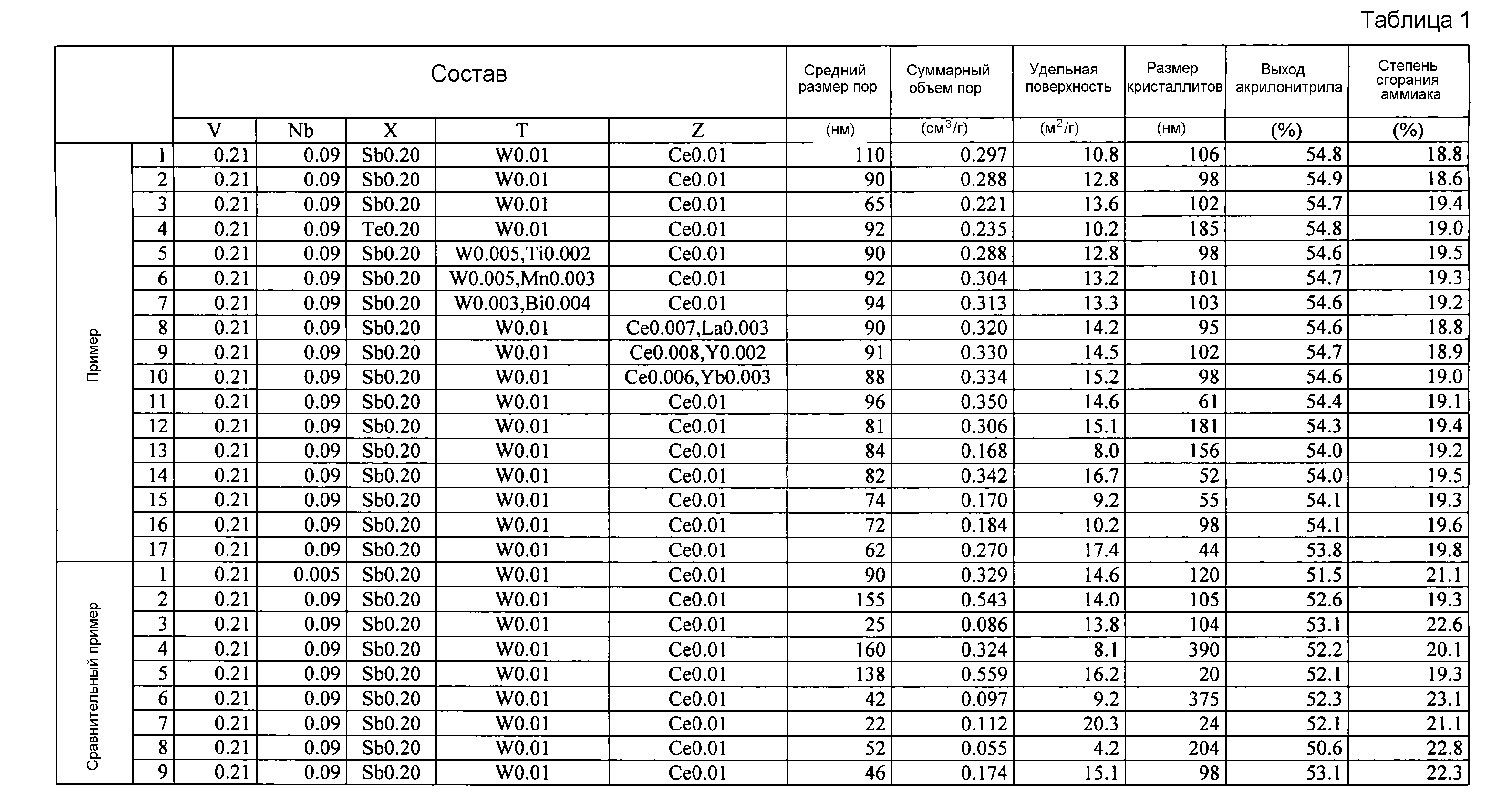
Настоящая заявка составлена на основании японской патентной заявки, поданной в японское патентное управление 21 апреля 2011 г. (японская выложенная патентная заявка № 2011-095422), содержание которой во всей своей полноте включается в настоящий документ посредством данной ссылки.
Промышленная применимость
Нанесенный на диоксид кремния катализатор согласно настоящему изобретению имеет промышленную применимость в качестве катализатора, используемого в производстве соответствующего ненасыщенного нитрила в реакции парофазного каталитического аммоксидирования пропана или изобутана.
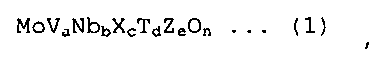
Половолоконный мембранный модуль с покрытой внешней периферией мембраны
Способ получения изоцианата
Метакриловая смола, литое изделие из нее и способ получения метакриловой смолы
Способ улавливания электроосаждаемой краски
Катионообменная мембрана, электролизер с ее использованием и способ изготовления катионообменной мембраны
Способ получения изоцианатов с использованием диарилкарбоната
Способ получения сложного эфира n-замещенной карбаминовой кислоты и способ получения изоцианата с использованием сложного эфира n-замещенной карбаминовой кислоты
Устройство для удаления поверхностных веществ катализатора
Устройство для получения смешанного раствора и способ приготовления смешанного раствора
Способ получения ненасыщенного нитрила
Половолоконный мембранный модуль с покрытой внешней периферией мембраны
Способ получения изоцианата
Метакриловая смола, литое изделие из нее и способ получения метакриловой смолы
Способ улавливания электроосаждаемой краски
Катионообменная мембрана, электролизер с ее использованием и способ изготовления катионообменной мембраны
Способ получения изоцианатов с использованием диарилкарбоната
Способ получения сложного эфира n-замещенной карбаминовой кислоты и способ получения изоцианата с использованием сложного эфира n-замещенной карбаминовой кислоты
Устройство для удаления поверхностных веществ катализатора
Устройство для получения смешанного раствора и способ приготовления смешанного раствора
Способ получения ненасыщенного нитрила

