Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ТЕТРАЗАМЕЩЕННЫХ 4-ДИАЗО-5,5-ДИАЛКИЛ-2,2-ДИАРИЛДИГИДРОФУРАН-3(2Н)-ОНОВ
Вид РИД
Изобретение
Изобретение относится к области органического синтеза, в частности, к способу получения 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов посредством пятистадийного синтеза из 1,1-диалкилпроп-2-ин-1-олов и паразамещенных бензофенонов. Эти диазокарбонильные соединения представляют значительный практический интерес, поскольку их можно использовать в реакциях сужения циклов и получать труднодоступные другими путями производные оксетана [1-4], входящие в состав природных соединений и многих фармакологически активных веществ.
Так, ключевой стадией синтеза оксетаноцина является перегруппировка Вольфа замещенного диазотетрагидрофуранона в производное оксетана [3]. Оксетаноцин - оптически активный четырехчленный нуклеозид-антибиотик, in vitro ингибирующий репликацию ВИЧ и обладающий очень сильным антивирусным действием. Его производные входят в состав фармацевтических препаратов против СПИДа, гепатита-B, герпеса и других вирусов [5-7].
Другим примером перспективного использования диазокетонов ряда ТГФ является получение на их основе 3(2H)-фуранонов - потенциальных высокоактивных нестероидных противовоспалительных препаратов [8-9]. 5-Арил-2,2-диалкил-4-фенил-3(2H)-фураноны с SO2NH2 и SO2Me группами в параположениях арильного кольца являются селективными ингибиторами фермента COX-2, что обуславливает их противовоспалительные свойства [10].
Таким образом, высокая биологическая активность соединений, получаемых из диазотетрагидрофуранонов, стимулирует разработку эффективных методов синтеза последних.
Известен способ получения диазосоединений окислением гидразонов надуксусной кислотой в двухфазной системе в присутствии катализаторов окисления и межфазного переноса - йода и четвертичных аммониевых солей [11]. Недостатком этого способа является пригодность метода для синтеза исключительно диазоалканов, а при его реализации используют токсичные реагенты. Так, йод в чистом виде может вызывать раздражение кожи [12], а надуксусная кислота и четвертичные аммониевые соли воздействуют на дыхательные пути [13], в отдельных случаях приводя к развитию астмы [14-15].
Другой способ получения диазосоединений, в частности диазокетонов, заключается во взаимодействии карбонилсодержащих третичных фосфинов с азидами и последующий термолиз или обработку основанием промежуточно образующихся ацилтриазенов в целевые диазокарбонильные соединения [16]. Недостатком этого аналога является сравнительно высокая токсичность третичных фосфинов [17] и потенциальная взрывоопасность азидов [18].
Наиболее близким аналогом заявляемого изобретения является способ получения диазокетонов, основанный на обработке α-дикетонов гидразин-гидратом и последующем окислении образующихся гидразонов окисью ртути(II) [19], который принят в качестве прототипа. При этом исходные дикетоны получают окислением α-оксикетонов ацетатом меди(II) в водно/спиртовом растворе.
К недостаткам прототипа следует отнести, во-первых, необходимость проведения процесса окисления гидразонов окисью ртути в течение весьма длительного времени (до 24 часов), а во-вторых, использование в качестве реагентов высокотоксичных оксида ртути(II) [20] и гидразин-гидрата [12], причисленного к вредным веществам первого класса опасности [21]. В-третьих, введение в реакцию оксида ртути(II) и ацетата меди(II) в качестве окислителей сопровождается образованием свободной ртути и оксида меди(II), которые загрязняют реакционную смесь и с трудом удаляются из продуктов реакции.
Техническая задача, решаемая заявляемым изобретением, направлена на устранение этих недостатков и заключается в использовании более эффективного, а также экологически более чистого и нетоксичного способа получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I
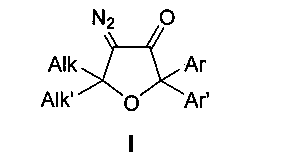
где Alk, Alk′ = Me, Et, cycloalkyl или другие алкильные группы, а Ar, Ar′ = Ph, p-MeO-C6H4, p-F-C6H4, p-Cl-C6H4 и другие замещенные или незамещенные арильные группы.
Технический результат, получаемый при реализации заявленного изобретения, заключается в более эффективном методе синтеза диазокетонов, пригодных в качестве прекурсоров при получении биологически активных и лекарственных препаратов, а также использовании нетоксичных реагентов и растворителей на каждой стадии процесса.
Указанный технический результат достигается тем, что:
во-первых, для получения 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов используется 5-стадийная схема синтеза из (3,3-диалкил-3-гидроксипроп-1-ин-1-ил)магний бромидов (II) и паразамещенных бензофенонов, взаимодействие которых по методу Йоцича (Стадия 1) приводит к образованию 1,1-диалкил-4,4-диарилбут-2-ин-1,4-диолов (III):
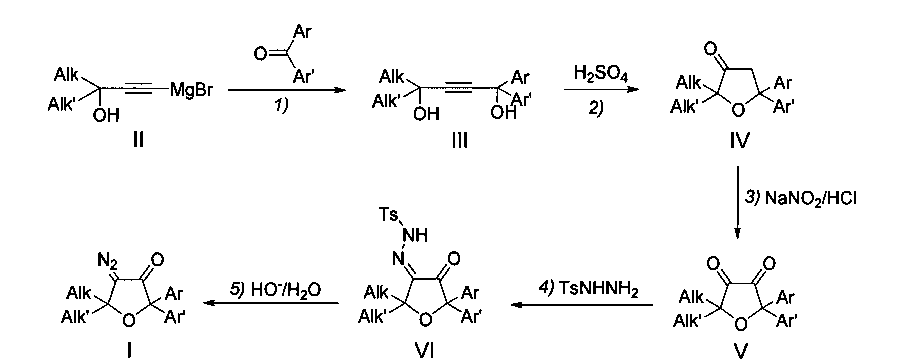
во-вторых, 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-оны (IV) получают дегидратацией с одновременной гидратацией ацетиленовых γ-гликолей (III) в присутствии катализатора - серной кислоты (Стадия 2);
в-третьих, при получении 2,2-диарил-5,5-диалкилтетрагидрофуран-3,4-дионов (V) используют нитрозирование 2,2-диарил-5,5-диалкил-тетрагидрофуран-3-онов (IV) нитритом натрия в водно-органическом растворе соляной кислоты и их последующий гидролиз в α-дикетоны (V) при 63°C (Стадия 3);
в-четвертых, для синтеза моногидразонов α-дикетонов (VI) вместо гидразин-гидрата применяют p-толуолсульфонилгидразид, который прибавляют непосредственно в реакционную смесь с α-дикетоном (V) в том же растворителе (ТГФ) и при той же температуре (63°C) (Стадия 4);
в-пятых, превращение тозилгидразонов (VI) в целевые 4-диазо-5,5-диарил-2,2-диалкилдигидрофуран-3(2H)-оны (I) на заключительной стадии синтеза (Стадия 5) проводят в водно-щелочном растворе при комнатной температуре вместо окисления гидразонов окисью ртути, используемого в известном прототипе.
Заявляемое изобретение имеет следующие отличительные признаки и преимущества:
1. значительное увеличение выходов диазокетонов (I) на последней стадии синтеза (до 97%);
2. многократное уменьшение длительности превращения гидразонов (VI) в диазокетоны (I) (с 8-24 часов до 1 часа);
3. заметное упрощение методики обработки реакционных смесей, выделение целевых продуктов (I) лишь путем перекристаллизации;
4. последовательное превращение монокетонов (IV) в тозилгидразоны (VI) без выделения промежуточных α-дикетонов (V) и отсутствие необходимости удаления твердых осадков из реакционной смеси, образующихся в известном способе;
5. отказ от использования токсичных и ядовитых реагентов на всех стадиях синтеза;
6. проведение реакций только в нетоксичных растворителях - диэтиловом эфире, этаноле и тетрагидрофуране.
Апробация заявленного изобретения была выполнена в Санкт-Петербургском государственном университете с использованием оборудования ресурсных центров СПбГУ «Магнитно-резонансные методы исследования» и «Методы анализа состава вещества». Результаты исследования представлены в виде конкретных примеров реализации.
Примеры выполнения способа
Спектры ЯМР 1H (400.13 МГц) и 13C (100.61 МГц) регистрировали на приборе "Bruker 400 МГц Avance" в растворе CDCl3, внутренний стандарт - (CH3)4Si. ИК-спектры получены на спектрофотометре "Perkin-Elmer BXII" в диапазоне 4000-400 см-1 в таблетках KBr. Для снятия масс-спектров использовали хромато-масс-спектрометры Brucker micrOTOF и «MaXis», Bruker Daltonik GmbH. Элементный анализ проводили на приборе Euro EA3028-HT. Температуру плавления измеряли на приборе Büchi B-540.
Реакции проводили в безводных растворителях, очищенных по стандартным методикам. Для препаративного разделения реакционных смесей использовали колоночную и флеш-хроматографию на силикагеле (Silicagel L, 40-100 µm; Chemapol и Alugram Sil G/UV254) в градиентном режиме. Аналитическую TCX проводили на пластинах Silica gel Merck 60 F254 (Германия) и Alugram Xtra Sil G/UV254 (Германия), элюенты - петролейный эфир, гексан и ацетон.
Соединения, приведенные без ссылок, приготовлены по стандартным лабораторным методикам либо являются коммерчески доступными веществами.
Пример 1.
Получение 4-диазо-5,5-диметил-2,2-ди(p-метоксифенил)тетрагидрофуран-3-она.
Стадия 1. К раствору этилмагний-бромида, полученному из 2.16 г (90 ммоль) магния и 9.81 г (90 ммоль) этилбромида в 35 мл диэтилового эфира, при 0°C прибавили по каплям раствор 2.95 г (35 ммоль) 2-метилбут-3-ин-2-ола в 35 мл эфира. Реакционную смесь кипятили в течение 3 часов, охладили и при 0°C прибавили в виде сухого порошка 6.05 г (25 ммоль) 4,4′-диметоксибензофенона. После этого смесь кипятили с обратным холодильником 4 часа (контроль по ТСХ), охладили, прибавили 40 мл соляной кислоты (3:1) при охлаждении, водный слой экстрагировали хлористым метиленом (3×35 мл), объединенные органические вытяжки сушили безводным K2CO3, растворитель отогнали в вакууме, остаток перекристаллизовали из смеси петролейный эфир/хлороформ. Выход 2-метил-5,5-ди(p-метоксифенил)пент-3-ин-2,5-диола 7.7 г (94%).
Бесцветное твердое вещество.
Т.пл. 112-113°C (петролейный эфир/хлороформ). Rf (петролейный эфир/ацетон, 5:3) 0.57.
FT-IR (KBr)  (cm-1): 3366 m, 3299 m, 2979 w, 2838 w, 1604 m, 1508 s, 1300 m, 1249 vs, 1172 m, 1080 w, 1030 m, 991 m, 956 m, 839 s, 682 m, 567 m.
(cm-1): 3366 m, 3299 m, 2979 w, 2838 w, 1604 m, 1508 s, 1300 m, 1249 vs, 1172 m, 1080 w, 1030 m, 991 m, 956 m, 839 s, 682 m, 567 m.
1H-NMR (400 MHz, CDCl3; 25°C): 1.54 (s, 6H, 2 Me); 2.52 (s, 1H, OH); 3.15 (s, 1H, OH); 3.76 (s, 6H, 2 OMe) 6.79-6.83 (m, 4 HAr), 7.42-7.46 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 31.3 (2 Me); 55.3 (2 OMe); 65.3, 73.5 (CMe2, CAr2); 85.2 (Me2C-C≡), 91.4 (≡C-CAr2,); 113.5, 127.3, 137.6, 158.9 (все CAr).
HRMS (ESI) для C20H22O4 (326.1518): вычисл. для [M+H]+ 327.1591, найдено 327.1591.
Аналит. вычисл. для C20H22O4 (326.1518): C 73.62, H 6.75; найдено: C 73.64, H 6.93.
Стадия 2. К раствору 6.52 г (20 ммоль) 2-метил-5,5-ди(p-метоксифенил)пент-3-ин-2,5-диола в 40 мл абсолютного этанола по каплям прибавили 4 мл серной кислоты (d 1.83 г/мл). Смесь перемешивали при 65-68°C в течение 7 часов, после чего охладили и поставили на ночь в холодильник. Образовавшиеся бесцветные кристаллы отфильтровали и промыли ледяной водой. К маточному раствору прибавили 50 мл воды, водный слой экстрагировали хлористым метиленом (3×25 мл), объединенные органические вытяжки промыли водой (2×30 мл), затем насыщенным раствором поваренной соли (30 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме, остаток перекристаллизовали из петролейного эфира. Общий выход 2,2-диметил-5,5-ди(p-метоксифенил)дигидрофуран-3(2H)-она 4.2 г (65%).
Бесцветное твердое вещество.
Т.пл. 76-77°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:1) 0.37.
FT-IR (KBr)
(cm-1): 3055 w, 3001 w, 2981 w, 2933 w, 2837 w, 1753 s, 1612 m, 1510 w, 1465 m, 1303 m, 1244 vs, 1180 s, 1172 m, 1126 m, 1029 m, 993 m, 833 m.
1H-NMR (400 MHz, CDCl3; 25°C): 1.20 (s, 6H, 2 Me); 3.25 (s, 2H, CH2); 3.77 (s, 3H, OMe); 6.81-6.85 (m, 4 HAr); 7.27-7.31 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 25.4 (2 CH3); 48.5 (CH2); 55.2 (OCH3); 81.2, 82.3 (CAr2, CMe2); 113.5, 127.4, 138.4, 158.8 (все CAr); 217.4 (C=O).
HRMS (ESI) для C20H22O4 (326.1518): вычисл. для [M+Na]+ 349.1410, найдено 349.1410.
Аналит. вычисл. для C20H22O4 (326.1518): C 73.60, H 6.79; найдено: C 73.62, H 6.75.
Стадии 3 и 4. К смеси 0.8 г (2.45 ммоль) 2,2-диметил-5,5-ди(p-метоксифенил)дигидрофуран-3(2H)-она и 0.5 г (7.35 ммоль) нитрита натрия в 15 мл ТГФ при перемешивании по каплям прибавили 3.7 мл (122 ммоль) концентрированной соляной кислоты. Нагревание до температуры 53-56°C привело к интенсивному выделению бурого газа и кипению реакционной смеси. После окончания выделения газа смесь перемешивали при 65°C в течение 1 часа (контроль по TCX), при этом она приобрела темно-малиновую окраску. Далее прибавили 0.46 г (2.45 ммоль) p-толуолсульфонилгидразида в 4 мл ТГФ, реакционная смесь изменила цвет на темно-желтый. Смесь кипятили еще 40 минут (контроль по TCX), охладили, прибавили 50 мл хлористого метилена. Реакционную смесь перенесли в делительную воронку, промыли водой (2×50 мл), затем насыщенным раствором поваренной соли (50 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме. Остаток хроматографировали на колонке с SiO2 (элюент : гексан/ацетон 10:1→4:1). Выход N′-(2,2-диметил-5,5-ди(p-метоксифенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида на двух стадиях составил 0.54 г (43%).
Желтое твердое вещество. Т.пл. 162-164° (гексан). Rf (гексан/ацетон, 3:1,2 раза) 0.24.
FT-IR (KBr)
(cm-1): 3234 w, 2983 w, 2933 w, 1710 m, 1604 m, 1595 m, 1508 vs, 1458 m, 1404 m, 1357 m, 1301 m, 1246 s, 1166 s, 1072 m, 1028 m, 871 m, 812 w, 779 m, 661 m, 541 m.
1H-NMR (400 MHz, CDCl3; 26°C): 1.40 (s, 6H, 2 Me); 2.42 (s, 3H, Me); 3.77 (s, 6H, 2 OMe); 6.82 (d, J=8.9 Hz, 4 CHAr); 7.28 (d, J=8.0 Hz, 4 CHAr); 7.31 (d, J=8.9 Hz, 2 CHTs); 7.80 (d, J=8.0 Hz, 2 CHTs); 12.22 (5, 1H, NH).
13C-NMR (100 MHz, CDCl3; 26°C): 21.6 (Me); 28.5 (2 Me); 55.3 (2 OMe); 78.7, 85.5 (CMe2, CAr2); 113.8, 127.8, 133.0, 159.4 (все CAr); 127.8, 129.8, 144.8, 146.8 (все CTs); 135.1 (C=N-NH2); 199.0 (C=O).
HRMS (ESI) для C27H28N2O6S (508.1668): вычисл. для [M+H]+ 509.1741, найдено 509.1741; вычисл. для [M+Na]+ 531.1560, найдено 531.1560.
Стадия 5. К раствору 508 мг (1 ммоль) N′-(2,2-диметил-5,5-ди(p-метоксифенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида в 15 мл диэтилового эфира прибавили раствор 60 мг NaOH в 10 мл H2O. Реакционную смесь интенсивно перемешивали в течение часа (контроль по TCX), перенесли в делительную воронку и прибавили еще 30 мл воды. Водный слой отделили от органического, экстрагировали Et2O (2×10 мл). Объединенные органические вытяжки промыли водой (2×20 мл), затем насыщенным раствором поваренной соли (20 мл), высушили безводным MgSO4. Растворитель отогнали в вакууме. Остаток перекристаллизовали из петролейного эфира. Выход 4-диазо-5,5-диметил-2,2-ди(p-метоксифенил)дигидрофуран-3(2H)-она 317 мг (90%).
Желтое твердое вещество. Т.пл. 131-132°C (петролейный эфир). Rf (циклогексан/ацетон, 10:1) 0.28.
FT-IR (KBr)
(cm-1): 2964 w, 2839 w, 2098 vs, 1678 s, 1608 m, 1510 s, 1462 m, 1352 s, 1298 m, 1249 s, 1174 m, 1031 m, 904 m, 833 m, 813 m, 567 m.
1H-NMR (400 MHz, CDCl3; 26°C): 1.61 (s, 6H, 2 Me); 3.78 (s, 6H, 2 OMe); 6.84 (d, J=8.7 Hz, 4 HAr), 7.37 (d, J=8.5 Hz, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 28.9 (2 Me); 55.2 (2 OMe); 65.1 (C=N2); 78.1, 90.2 (CMe2, CAr2); 113.5,128.0,134.0,159.2 (все CAr); 193.3 (C=O).
HRMS (ESI) для C20H20N2O4 (352.1423): вычисл. для [M+H]+ 353.1496, найдено 353.1496.
Аналит. вычисл. для C20H20N2O4 (352.1423): C 68.17, H 5.72, N 7.95; найдено: C 68.11, H 5.92, N 7.72.
Пример 2.
Получение 4-диазо-5,5-диметил-2,2-ди(p-фторфенил)дигидрофуран-3(2H)-она.
Стадия 1. К раствору этилмагний-бромида, полученному из 4.32 г (180 ммоль) магния и 19.62 г (180 ммоль) этилбромида в 75 мл диэтилового эфира, при 0°C прибавили по каплям раствор 5.88 г (70 ммоль) 2-метилбут-3-ин-2-ола в 70 мл эфира. Реакционную смесь кипятили в течение 3 часов, охладили и при 0°C прибавили в виде сухого порошка 10.9 г (50 ммоль) 4,4′-дифторбензофенона. Полученную смесь кипятили с обратным холодильником 4 часа (контроль по TCX), охладили, прибавили 55 мл соляной кислоты (3:1) при охлаждении, водный слой экстрагировали хлористым метиленом (3×70 мл). Объединенные органические вытяжки сушили безводным K2CO3, растворитель отогнали в вакууме, полученный остаток перекристаллизовали из петролейного эфира. Выход 2-метил-5,5-ди(p-фторфенил)пент-3-ин-2,5-диола 14.7 г (97%).
Бесцветное твердое вещество. Т.пл. 95-96°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:3) 0.66.
FT-IR (KBr)
(cm-1): 3383 s, 3264 m, 2987 w, 1601 m, 1507 s, 1227 s, 1158 m, 1077 w, 997 m, 955 w, 835 m, 683 w, 620 m.
1H-NMR (400 MHz, CDCl3; 25°C): 1.56 (s, 6H, 2 Me); 2.27 (s, 1H, OH); 3.11 (s, 1H, OH); 6.96-7.02 (m, 4 HAr), 7.48-7.53 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 31.3 (s, 2 Me); 65.4, 73.3 (CMe2, CAr2); 84.5 (Me2C-C≡), 92.2 (≡C-CAr2,); 115.1 (d, 2JC,F=21.6 Hz), 127.7 (d, 3JC,F=8.2 Hz); 140.7 (d, 4JC,F=2.7 Hz); 162.2 (d, 1JC,F=246.9 Hz).
HRMS (ESI) для C18H16F2O2 (302.1118): вычисл. для [M+H]+ 303.1191, найдено 303.1191.
Аналит. вычисл. для C18H16F2O2 (302.1118): C 71.52, H 5.29; найдено: C 71.26, H 5.38.
Стадия 2. К раствору 12.08 г (40 ммоль) 2-метил-5,5-ди(p-фторфенил)пент-3-ин-2,5-диола в 80 мл абсолютного этанола по каплям прибавили 7.5 мл серной кислоты (d 1.83 г/мл). Смесь перемешивали при 65-68°C в течение 6 часов, после чего охладили, поставили на ночь в холодильник. Образовавшиеся бесцветные кристаллы отфильтровали и промыли ледяной водой. К маточному раствору прибавили 100 мл воды, водный слой экстрагировали хлористым метиленом (3×30 мл), объединенные органические вытяжки промыли водой (2×60 мл), затем насыщенным раствором поваренной соли (60 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме, остаток перекристаллизовали из петролейного эфира. Общий выход 2,2-диметил-5,5-ди(p-фторфенил)дигидрофуран-3(2H)-она 10.8 г (89%).
Бесцветное твердое вещество. Т.пл. 57-58°C (петролейный эфир). Rf (петролейный эфир/ацетон, 5:1) 0.55.
FT-IR (KBr)
(cm-1): 2982 w, 2932 w, 1762 s, 1606 w, 1508 w, 1236 s, 1159 w, 1095 w, 995 w, 837 m, 736 w.
1H-NMR (400 MHz, CDCl3; 28°C): 1.20 (s, 6H, 2 Me); 3.27 (s, 2H, CH2); 6.97-7.03 (m, 4 HAr); 7.33-7.38 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 28°C): 25.3 (2 Me); 48.2 (CH2); 81.5, 81.9 (CMe2, CAr2); 115.2 (d, 2JC,F=21.5 Hz), 127.7 (d, 3JC,F=8.1 Hz); 141.8 (d, 4JC,F=3.2 Hz); 162.0 (d, 1JC,F=247.0 Hz); 216.3 (C=O).
HRMS (ESI) для C18H16F2O2 (302.1118): вычисл. для [M+H]+ 303.1191, найдено 303.1191; вычисл. для [M+Na]+ 325.1011, найдено 325.1011.
Аналит. вычисл. для C18H16F2O2 (302.1118): C 71.51, H 5.33; найдено: C 71.66, H 5.24.
Стадии 3 и 4. К смеси 0.6 г (2.0 ммоль) 2,2-диметил-5,5-ди(p-фторфенил)дигидрофуран-3(2H)-она и 0.41 г (6.0 ммоль) нитрита натрия в 15 мл ТГФ при перемешивании по каплям прибавили 3 мл (100 ммоль) концентрированной соляной кислоты. Нагревание до температуры 53-56°C привело к интенсивному выделению бурого газа и кипению реакционной смеси. После окончания выделения газа смесь перемешивали при 65°C в течение 1 часа (контроль по TCX), при этом она приобрела темно-малиновую окраску. Далее прибавили 0.37 г (2 ммоль) p-толуолсульфонилгидразида в 4 мл ТГФ, реакционная смесь изменила цвет на темно-желтый. Смесь кипятили еще 30 минут (контроль по TCX), охладили, прибавили 40 мл хлористого метилена. Реакционную смесь перенесли в делительную воронку, промыли водой (2×40 мл), затем насыщенным раствором поваренной соли (40 мл). Органический слой сушили безводным MgSO4, растворитель отогнали в вакууме. Остаток хроматографировали на колонке с SiO2 (элюент : гексан/ацетон 15:1→10:1). Выход Ν′-(2,2-диметил-5,5-ди(p-фторфенил)-4-оксодигидрофуран-3(2Н)-илиден)-4-метилбензолсульфоногидразида на двух стадиях составил 0.39 г (40%).
Желтое твердое вещество. Т.пл. 171-173° (гексан). Rf (гексан/ацетон, 3:1) 0.22.
FT-IR (KBr)
(cm-1): 3196 m, 2979 w, 1708 w, 1602 m, 1573 m, 1506 vs, 1388 m, 1363 s, 1230 s, 1170 vs, 1114 m, 1089 m, 1037 s, 869 s, 785 s, 678 m, 661 m, 590 s, 545 s.
1H-NMR (400 MHz, CDCl3; 26°C): 1.40 (s, 6H, 2 Me); 2.42 (s, 3H, Me); 6.98-7.02 (m, 4 HAr), 7.31 (d, J=8.0 Hz, 2 CHTs); 7.36-7.39 (m, 4 HAr); 7.81 (d, J=8.0 Hz, 2 CHTs); 12.18 (s, 1Η, ΝΗ).
13C-NMR (100 MHz, CDCl3; 27°C): 21.6 (Me); 28.5 (2 Me); 79.2, 84.8 (CMe2, CAr2); 115.5 (d, 2JC,F=21.6 Hz); 127.9, 129.8, 144.9, 145.9 (все CTs); 128.1 (d, 3JC,F=8.1 Hz); 134.9 (C=N); 136.5 (d, 4JC,F=2.9 Hz); 162.5 (d, 1JC,F=248.3 Hz); 197.9 (C=O).
HRMS (ESI) для C25H22F2N2O4S (484.1268): вычисл. для [M+H]+ 485.1341, найдено 485.1341; вычисл. для [M+Na]+ 507.1161, найдено 507.1161.
Стадия 5. К раствору 484 мг (1 ммоль) N′-(2,2-диметил-5,5-ди(p-фторфенил)-4-оксодигидрофуран-3(2H)-илиден)-4-метилбензолсульфоногидразида в 15 мл диэтилового эфира прибавили раствор 60 мг NaOH в 10 мл H2O. Реакционную смесь интенсивно перемешивали в течение 40 минут (контроль по TCX), перенесли в делительную воронку и прибавили еще 30 мл воды. Водный слой отделили от органического, экстрагировали Et2O (2×10 мл). Объединенные органические вытяжки промыли водой (2×20 мл), затем насыщенным раствором поваренной соли (20 мл), высушили безводным MgSO4. Растворитель отогнали в вакууме. Остаток перекристаллизовали из петролейного эфира. Выход 4-диазо-5,5-диметил-2,2-ди(p-фторфенил)дигидрофуран-3(2H)-она 308 мг (94%).
Желтое твердое вещество. Т.пл. 115-116°C (петролейный эфир). Rf (петролейный эфир/ацетон, 3:1) 0.49.
FT-IR (KBr)
(cm-1): 2091 s, 1694 w, 1550 s, 1508 m, 1350 m, 1251 m, 1007 m, 980 m.
1H-NMR (400 MHz, CDCl3; 25°C): 1.61 (s, 6H, 2 Me); 6.98-7.04 (m, 4 HAr); 7.43-7.48 (m, 4 HAr).
13C-NMR (100 MHz, CDCl3; 26°C): 28.9 (2 CH3); 65.5 (C=N2); 78.5, 89.3 (CAr2, CMe2); 115.1 (d, 2JC,F=21.5 Hz), 128.3 (d, 3JC,F=8.2 Hz); 137.4 (d, 4JC,F=3.1 Hz); 162.4 (d, 1JC,F=247.3 Hz); 196.5 (C=O).
HRMS (ESI) для C18H14F2N2O2 (328.1023): вычисл. для [M+H]+ 329.1096, найдено 329.1096.
Аналит. вычисл. для C18H14F2N2O2 (328.1023): C 65.85, H 4.30, N 8.53; найдено: C 65.63, H 4.31. N 8.18.
Таким образом, как показывают результаты апробации, заявленный способ получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I является более эффективным и исключает применение на всех стадиях процесса вредных для окружающей среды веществ, т.е. является экологически более чистым, чем известные аналоги, и может быть с уверенностью отнесен к Зеленой химии.
Использованные источники информации
1. W. Kirmse, 100 years of the Wolff rearrangement // Eur. J. Org. Chem., 2002, V. No. 14, pp. 2193-2256.
2. H.D. Stachel, H. Poschenrieder, J. Redlin, Thermolysis and photolysis of cyclic diazo compounds // Z. Naturforsch, Teil B, 1996, V. 51, No. 9, pp. 1325-1333.
3. D.W. Norbeck, J.B. Kramer, Synthesis of (-)-Oxetanocin // J. Am. Chem. Soc., 1988, V. 110, No. 21, pp. 7217-7218.
4. J. Beneke, R. Schobert, α-Carboxy-β-Lactones from Photoinduced Ring Contraction of 3-Diazodihydrofuran-2,4-diones // Synthesis, 2013, V. 45, No. 6, pp. 773-776.
5. Y. Nishiyama, N. Yamamoto, K. Takahash, N. Shimada, Selective inhibition of human cytomegalovirus replication by a novel nucleoside, oxetanocin G // Antimicrob. Agents Chemother., 1988, V. 32, No. 7, pp. 1053-1056.
6. S. Hayashi, D.W. Norbeck, W. Rosenbrook, R.L. Fine, M. Matsukura, J.J. Planner, S. Broder, H. Mitsuya, Cyclobut-A and cyclobut-G, carbocyclic oxetanocin analogs that inhibit the replication of human immunodeficiency virus in Τ cells and monocytes and macrophages in vitro // Antimicrob. Agents Chemother., 1990, V. 34, No. 2, pp. 287-294.
7. Y. Wang, G.W.J. Fleet, R. Storer, P.L. Myers, C.J. Wвсеis, O. Doherty, D.J. Watkin, K. Vogt, D.R. Witty, F.X. Wilson, J.M. Peach, Synthesis of the potent antiviral oxetane nucleoside epinoroxetanocin from D-lyxonolactone // Tetrahedron-Asymmetry, 1990, V. 1, No. 8, pp. 527-530.
8. L.L. Rodina, J.J. Medvedev, O.S. Galkina, V.A. Nikolaev, Thermolysis of 4-Diazotetrahydrofuran-3-ones: Total Change of Reaction Course Compared to Photolysis // Eur. J. Org. Chem., 2014, V. No. 14, pp. 2993-3000.
9. J.J. Medvedev, D.V. Semenok, L.L. Rodina, Effective approach to 4,5-Diaryl-3(2H)-furanones - a promising inhibitors for COX-2 // Journal of Medical & Biological Sciences, 2014, V. 1, Νο. pp. 84-88.
10. Патент WO 2000/061571. 4,5-Diaryl-3(2H)-furanone derivatives as cyclooxygenase-2 inhibitors. / Y.J. Byun, J.K. Choi, Y.H. Choi, S. Chung, J.Y. Ha, Y.S. Jeong, Y.H. Joo, S.H. Kang, J.Y. Kim, J.K. Kim. - Заявлено 12.04.2000. - Опубликовано 19.10.2000.
11. Патент US 4092306. Oxidation of hydrazones to the corresponding diazo compounds in the presence of a phase transfer and an oxidation catalyst which is iodine, an iodide or an iodonium salt. / D.T. Eastlick. - Заявлено 21.09.1976. - Опубликовано 30.05.1978.
12. Ν.Η. Proctor, J.P. Hughes, G.J. Hathaway, Proctor and Hughes′ Chemical hazards of the workplace. 5th ed. / [edited by] Gloria J. Hathaway and Nick H. Proctor, ed.; Wiley-Interscience: Hoboken; Chichester, 2004.
13. A. Bello, M.M. Quinn, M.J. Perry, D.K. Milton, Characterization of occupational exposures to cleaning products used for common cleaning tasks - a pilot study of hospital cleaners // Environ. Health, 2009, V. 8, No.
14. E. Cristofari-Marquand, M. Kacel, F. Milhe, A. Magnan, M. P. Lehucher-Michel, Asthma caused by peracetic acid-hydrogen peroxide mixture // J. Occup. Health, 2007, V. 49, No. 2, pp. 155-158.
15. A. Purohit, M.C. Kopferschmitt-Kubler, C. Moreau, E. Popin, M. Blaumeiser, G. Pauli, Quaternary ammonium compounds and occupational asthma // Int. Arch. Occup. Environ. Health, 2000, V. 73, No. 6, pp. 423-427.
16. Патент US 8350014. Preparation of diazo and diazonium compounds. / R.T. Raines, E.L. Myers. - Заявлено 17.11.2009. - Опубликовано 08.01.2013.
17. D.Ε.С. Corbridge, Phosphorus: chemistry, biochemistry and technology. 6th ed.; Taylor & Francis: Boca Raton, 2013.
18. S. Brase, C. Gil, K. Knepper, V. Zimmermann, Organic azides: An exploding diversity of a unique class of compounds // Angew. Chem., Int. Ed., 2005, V. 44, No. 33, pp. 5188-5240.
19 Патент US 2933494. Diazo pregnane compounds and method of preparing same. / B.G. Christensen, R.F. Hirschmann. - Заявлено 27.01.1958. - Опубликовано 19.04.1960 (Прототип).
20. A. Picot, N. Proust, Mercury and its compounds: from speciation to toxicity // Actual. Chim., 1998, V. No. 4, pp. 16-24.
21. ГОСТ 19503-88. Гидразин-гидрат технический. Технические условия. / - Введен в действие 01.07.1989.
Способ получения тетразамещенных 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-онов общей формулы I где Alk, Alk′ = Me, Et, циклоалкил или другие алкильные группы, а Ar, Ar′ = Ph, p-MeO-CH, p-F-CH, p-Cl-CH и другие замещенные или незамещенные арильные группы, мультистадийным процессом из 1,1-диалкилпроп-2-ин-1-олов и бензофенонов, отличающийся тем, что введение диазофункции в структуру молекулы осуществляют нитрозированием 2,2-диалкил-5,5-диарилдигидрофуран-3(2H)-онов нитритом натрия в водно-органическом растворе соляной кислоты, конденсацией образующихся 2,2-диалкил-5,5-диарилфуран-3,4(2H,5H)-дионов с p-толуолсульфонилгидразидом и превращением тозилгидразонов в целевые 4-диазо-5,5-диалкил-2,2-диарилдигидрофуран-3(2H)-оны в водно-щелочном растворе с выходами до 97% на заключительной стадии этого экологически чистого процесса.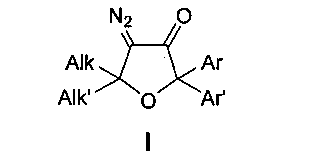
Способ получения безусадочного наномодифицированного конструкционного керамического материала
Способ радиографического контроля сварных соединений
Способ получения капсулированной каменной крошки и устройство для его реализации
Способ получения комплекса антимикробных пептидов насекомого
Способ получения 2,2-диалкил-4,5-диарилфуран-3(2н)-онов
Устройство для диагностики состояния микрогемолимфодинамики in vivo
Рекомбинантная плазмидная днк part27int6 и способ получения на ее основе инбредной линии растений табака, синтезирующего внутриклеточный гамма-интерферон быка
Способ подготовки сорбента для концентрирования паров полярных органических соединений
Способ получения нанокомпозитных материалов и устройство для его реализации
Способ разделения полидисперсных частиц в микронном и наноразмерном диапазоне и устройство для его реализации
Способ получения безусадочного наномодифицированного конструкционного керамического материала
Способ радиографического контроля сварных соединений
Способ получения капсулированной каменной крошки и устройство для его реализации
Способ получения комплекса антимикробных пептидов насекомого
Способ получения 2,2-диалкил-4,5-диарилфуран-3(2н)-онов
Устройство для диагностики состояния микрогемолимфодинамики in vivo
Рекомбинантная плазмидная днк part27int6 и способ получения на ее основе инбредной линии растений табака, синтезирующего внутриклеточный гамма-интерферон быка
Способ подготовки сорбента для концентрирования паров полярных органических соединений
Способ получения нанокомпозитных материалов и устройство для его реализации
Способ разделения полидисперсных частиц в микронном и наноразмерном диапазоне и устройство для его реализации

