Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 1-О-АЦИЛ-2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения 1-О-ацил-2-дезокси-2-фтор-4-тио-β-D-арабинофураноз и их промежуточных соединений.
Предпосылки создания изобретения
4'-Тионуклеозиды являются перспективными соединениями в отношении противовирусной и антинеопластической активности. Например, было показано, что 1-(2-дезокси-2-фтор-4-тио-β-D-арабинофуранозил)цитозин (4'-тио-FAC), обладает превосходной противоопухолевой активностью in vitro и in vivo [Y. Yoshimura et al; J. Org. Chem. 1997, 62, 3140-3152; S. Miura et al., Cancer Lett. 1998, 129, 103-110; S. Miura et al., Cancer Lett. 1999, 144, 177-182; Y. Yoshimura et al., Bioorg. Med. Chem. 2000, 8, 1545-1558; D.A. Zajchowski et al., Int. J. Cancer 2005, 114, 1002-1009].
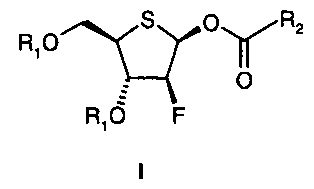
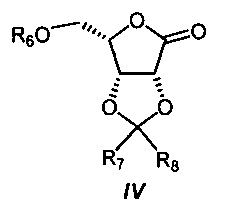
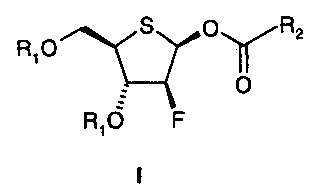
Изобретение относится в особенности к новому способу получения соединений формулы I
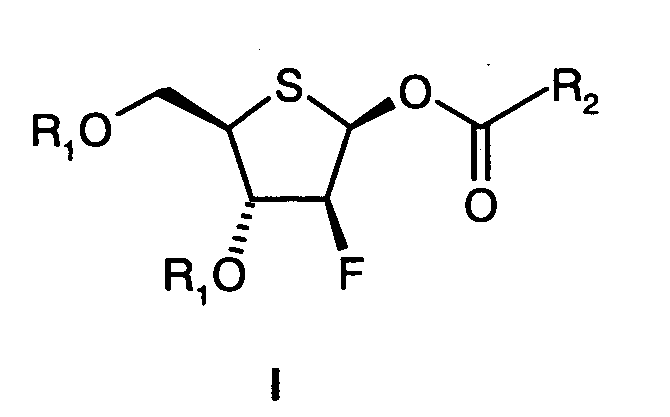
где
R1 представляет собой -С(О)-С1-С6-алкил или -С(О)-арил; и
R2 представляет собой С1-С6-алкил, С1-С4-перфторалкил или арил.
Соединения общей формулы I являются важнейшими промежуточными соединениями в получении 4'-тионуклеозидов.
1-(2-Дезокси-2-фтор-4-тио-β-D-арабинофуранозил)цитозин (4'-тио-FAC):
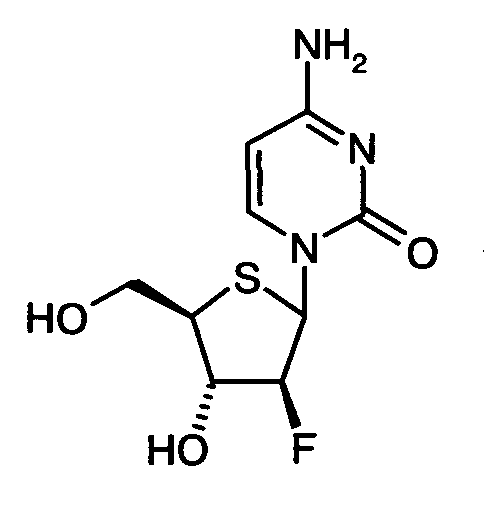
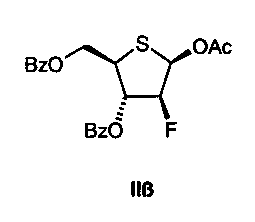
Существует особый интерес в получении 1-(2-дезокси-2-фтор-4-тио-β-D-арабинофуранозил)цитозина (4'-тио-FAC) в целях получения соединения II:
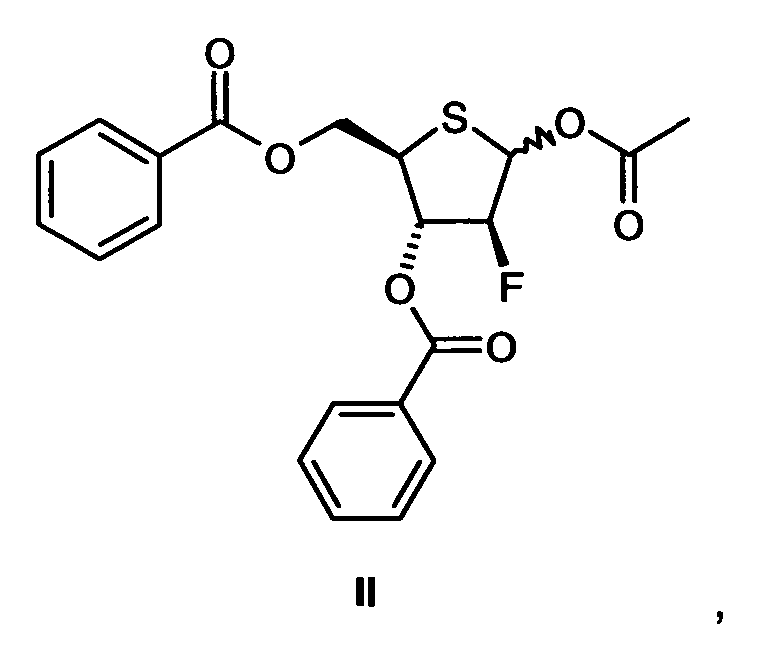
где в тексте, который следует ниже, α-диастереомер аномерного ацетата называют IIα; и IIβ используют для названия β-изомера.
Это соединение и его получение описано в первый раз в международных публикациях WO 97/73993, WO 97/038001 и в литературе, имеющей отношение к этому [Y. Yoshimura et al.; J. Org. Chem. 1999, 64, 7912-7920; Y. Yoshimura et al.; Nucleosides Nucleotides 1999, 18, 815-820; Y. Yoshimura et al.; Nucleic Acids Symposium Series 1998, 39, 11-12; Y. Yoshimura et al.; Tetrahedron Lett. 1999, 40, 1937-1940].
В этом контексте описан путь получения соединений формулы III
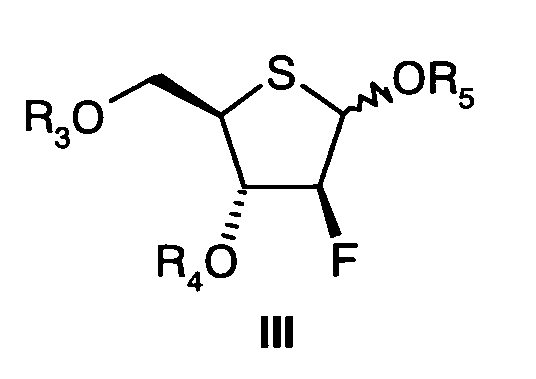
где
R3 и R4 представляют собой алкил, силил или ацил, и
R5 представляет собой ацил.
Схема 1
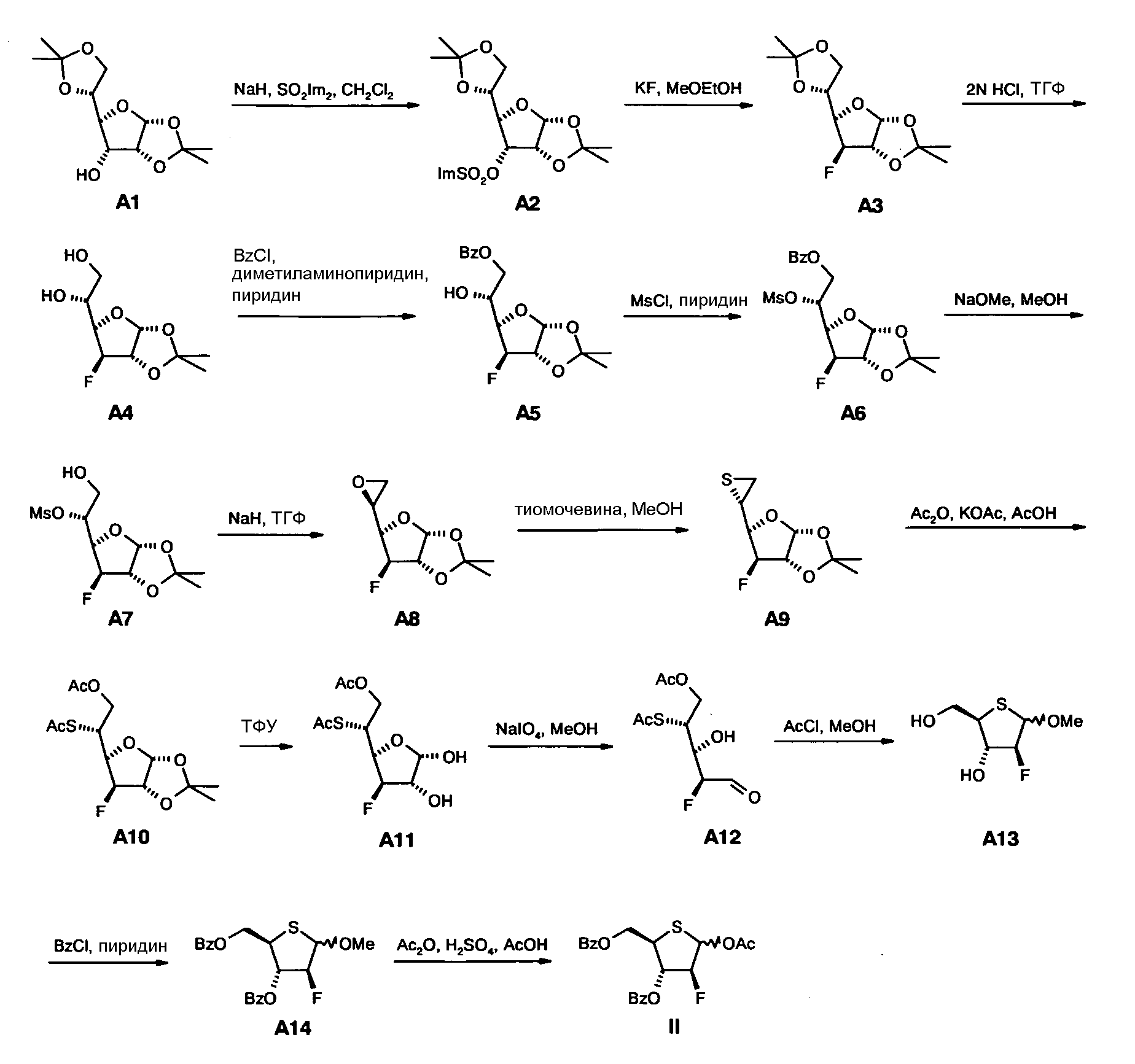
Исходным веществом в Схеме 1 является коммерчески доступная 1,2:5,6-ди-О-изопропилиден-α-D-аллофураноза (А1), которая в свою очередь может быть получена в четыре стадии из D-глюкозы [D.C. Baker et al., Carbohydr. Res. 1972, 24, 192-197]. Таким образом, соединение II может быть получено всего за 14 химических стадий, если исходить из 1,2:5,6-ди-О-изопропилиден-α-D-аллофуранозы, или за 18 химических стадий, если исходить из D-глюкозы. В данном изобретении соединение II получают в виде аномерной смеси, состоящей из IIα и IIβ. В литературе отсутствуют данные о соотношении IIα/IIβ [Y. Yoshimura et al., J. Org. Chem. 1999, 64, 7912-7920]. Лабораторные эксперименты, проведенные автором для получения II аналогично методике, приведенной в литературе, исходя из соединения А14, дали смеси IIα/IIβ 1:1-3:2.
Недостатком способа получения соединения II, известного из предшествующего уровня техники, является большое число требуемых химических стадий, что делает практическое применение способа в промышленном масштабе значительно более трудоемким. Кроме того, в особенности в том случае, когда синтез выполняют в промышленном масштабе, существуют следующие трудности и проблемы:
- Способ включает, по меньшей мере, препаративные хроматографические разделения (препаративная высокоэффективная жидкостная хроматография = prep-HPLC).
- Промежуточные соединения А6, А7, А9, А12 являются нестабильными.
- Работа с вязкими жидкостями на стадиях А2, А3, А4, А8, А9, А11, А12 является затруднительной.
- Соединение А6 растворяется в метаноле, только очень медленно. В присутствии метоксида натрия (NaOMe), нуклеофильное замещение мезилатной группы с помощью метоксигруппы в соединении А7 происходит как побочная реакция. Образование побочного продукта происходит в особенности в том случае, когда реакцию проводят в относительно большом масштабе.
- После отщепления изопропилиденовой группы, трифторуксусная кислота (TFA) должна быть отогнана при пониженном давлении, поскольку другие альтернативы для выделения продукта реакции приводят к образованию большого количества побочного продукта. В промышленном масштабе это сопровождается значительными трудностями.
В связи с длинной последовательностью синтеза и с тем фактом, что некоторые из ее стадий не могут быть выполнены в более крупном масштабе, способ, показанный на Схеме 1, не подходит для промышленного получения соединения II.
Альтернативный вариант получения соединения II описан в международной публикации WO 2007/068113 и в литературе, имеющей отношение к этому [J.K. Watts et al., J. Org. Chem. 2006, 71, 921-925], и кратко изложен здесь на Схеме 2.
Схема 2
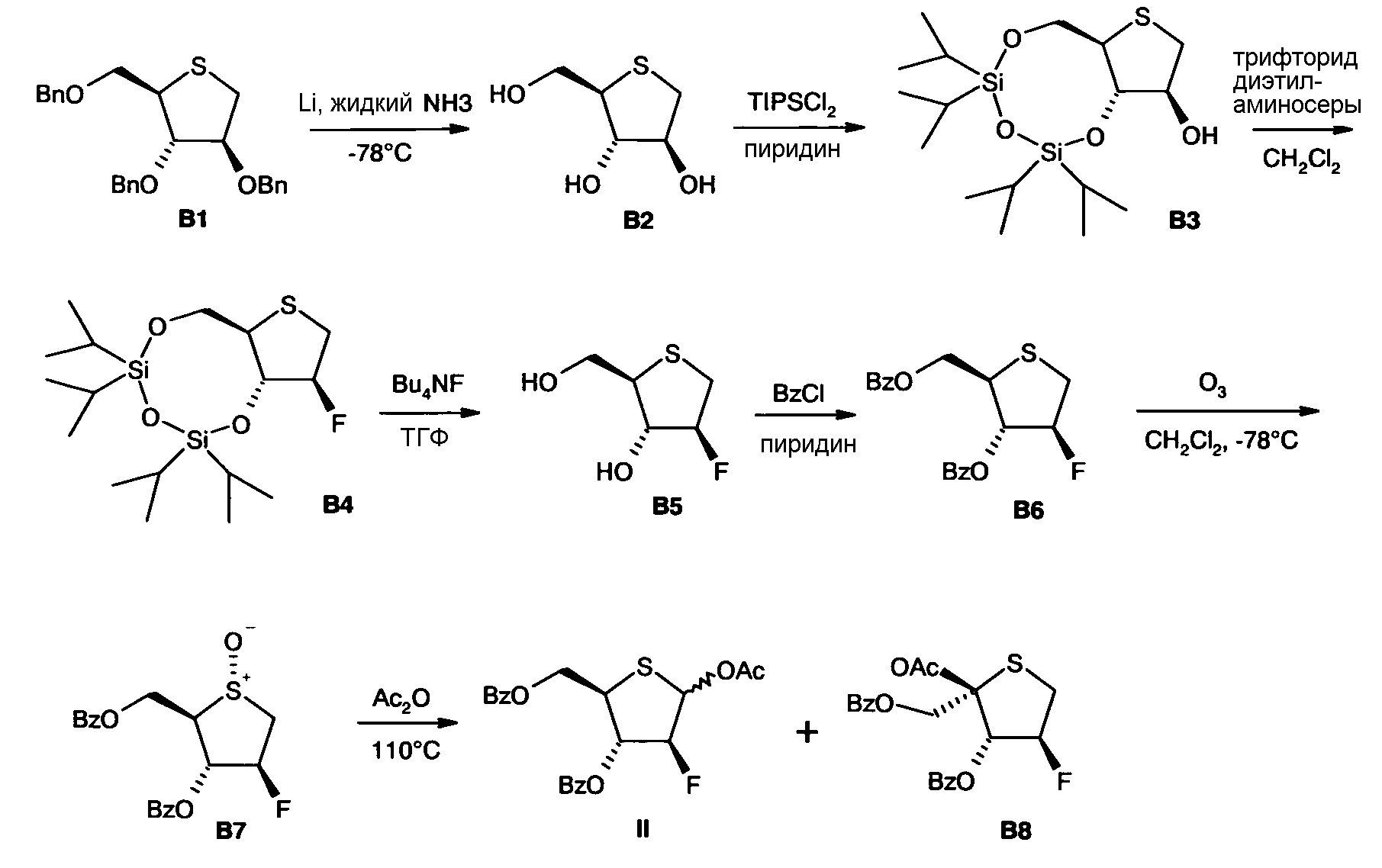
Здесь, на последней стадии синтеза, получают смесь аномерных ацетатов II в соотношении IIα/IIβ от 1:2 до 1:14 [см. также J.K. Watts et al., J. Org. Chem. 2006, 71, 921-925]. Побочный продукт В8 удаляют колоночной хроматографией.
Соединение В1 может быть получено в 6 стадий из L-ликсозы (которая не встречается в природе) [J.K. Watts et al., J. Org. Chem. 2006, 71, 921-925]. Таким образом, соединение II может быть получено в итоге за 13 химических стадий из L-ликсозы.
Конкретный недостаток такой альтернативы синтеза возникает вследствие того факта, что исходное вещество L-ликсоза является дорогим, и очень небольшое его количество является коммерчески доступным для синтеза в промышленном масштабе.
Кроме того, в особенности в том случае, когда синтез проводят в промышленном масштабе, имеют место следующие трудности и проблемы:
- На каждой стадии синтеза должны быть выполнены непростые превращения защитных групп и в каждом случае хроматографическая очистка.
- Применение жидкого аммиака и элементарного лития при очень низких температурах (стадия В2).
- Введение и удаление конкретной силильной защитной группы, которая имеет высокую молярную массу и промышленное получение которой является трудным (стадии В3 и В5).
- Применение DAST (трифторид диэтиламиносеры) в качестве фторирующего средства. В дополнение к тому факту, что получение DAST является трудным, в промышленном масштабе выполнения этой реакции играют важную роль вопросы безопасности (температура, при которой проводятся манипуляции, разложение DAST в экзотермической реакции с образованием газа) (стадия 4).
- Применение озона при очень низких температурах (В7).
- Высокие температуры (110ºС) во время перегруппировки Pummerer и образование приблизительно 20% побочного продукта В8 [J.K. Watts et al., J. Org. Chem. 2006, 71, 921-925].
Вследствие трудностей, описанных в этом документе, на отдельных стадиях синтеза, которые делают осуществление в промышленном масштабе затруднительным или невозможным, и вследствие ограниченной доступности исходного вещества, способ, показанный на Схеме 2, также не очень подходит для промышленного получения соединения II.
Краткое изложение сущности изобретения
На основании представленных предпосылок создания изобретения, целью настоящего изобретения было обеспечение альтернативного способа, позволяющего осуществить промышленное получение соединений формулы I.
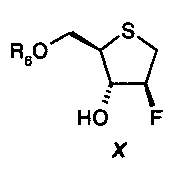
В соответствии с изобретением эта цель была достигнута с помощью способа, который предоставляет соединения формулы I с высокими выходами в 10 химических стадий, исходя из коммерчески легко доступных соединений формулы IV при использовании ключевых стадий «введение атома фтора посредством целевого раскрытия циклического сульфата» и «Pummerer-перегруппировка сульфоксида с использованием специального катализатора» (Схема 3).
Схема 3
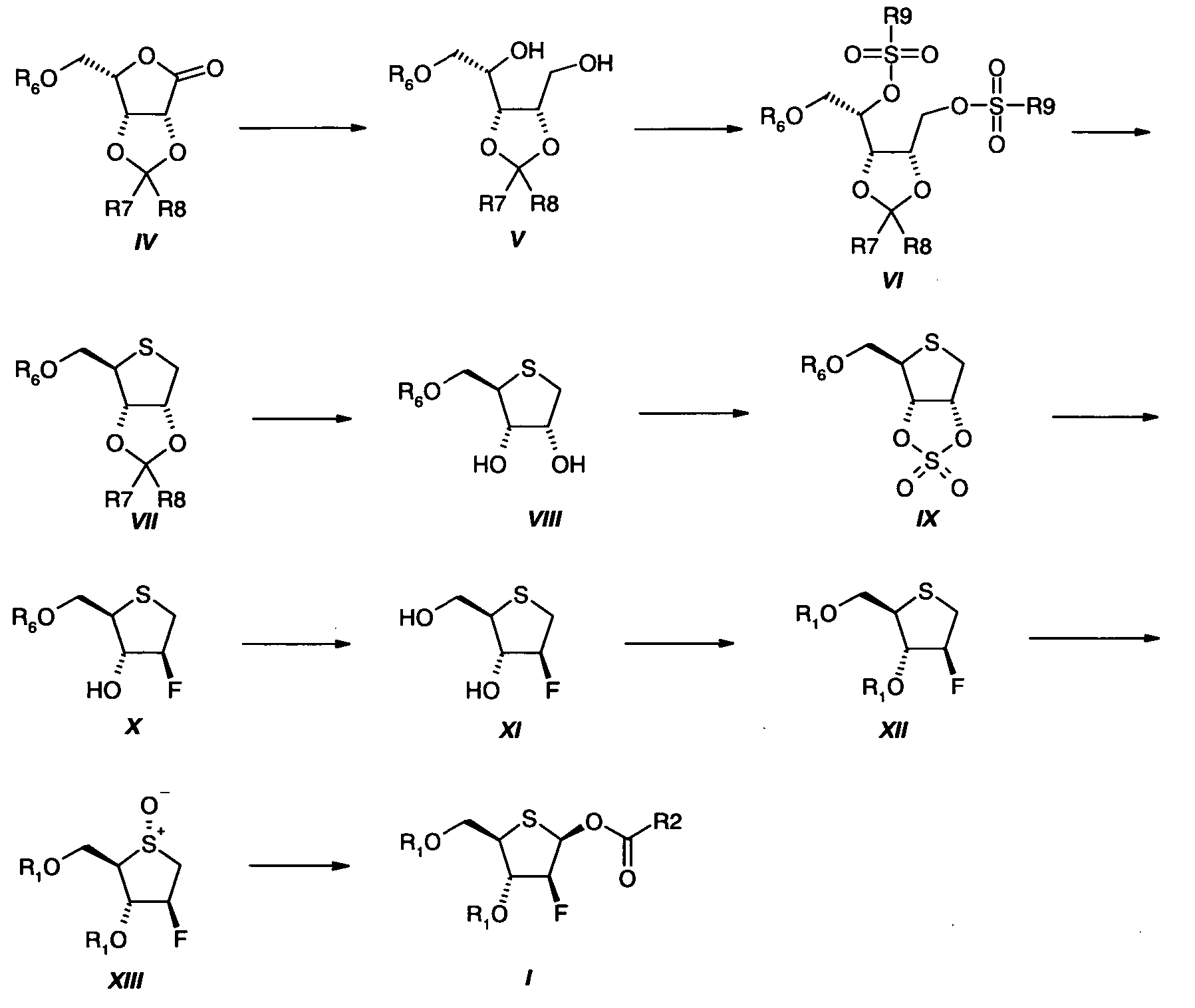
Соединения формулы IV, используемые в качестве исходных веществ для способа в соответствии с изобретением, могут быть синтезированы в 4 химические стадии из природной и, таким образом, легко доступной D-рибозы. Как D-рибоза, так и соединения типа IV (например, 5-О-бензил-2,3-О-изопропилиден-L-ликсоно-1,4-лактон) являются коммерчески доступными (Патент: US No. 6448415 B1).
Кроме того, в способе в соответствии с изобретением используются только реагенты, которые легко доступны даже в килограммовых количествах.
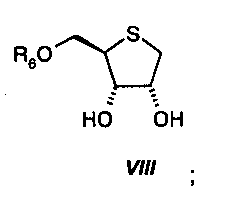
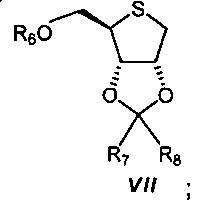
Промежуточные соединения V, VI и VII выделяют только в виде сырых продуктов и в каждом случае применяют непосредственно на следующей стадии. В итоге, соединение VIII кристаллизуется с высокой чистотой (>97%). Таким образом, можно обойтись без отнимающих много времени и ресурсоемких очисток (например, без препаративной хроматографии).
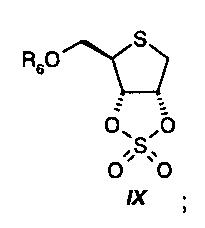
Соединения стадий IX, XI и XII также не выделялись и использовались непосредственно в виде сырых веществ на последующей стадии.
В соответствии с изобретением на протяжении всей последовательности синтеза, только 3 промежуточных соединения (VIII, X и XIII) должны быть выделены, и только в одном случае требуется препаративная хроматография (X). Промежуточные соединения VIII, XII и продукт I (II) выделяют с высокими выходами и высокими степенями чистоты (>93%) кристаллизацией.
Способ в соответствии с изобретением не требует никаких сложных превращений защитных групп.
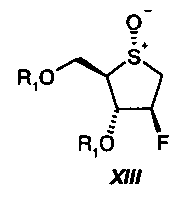
Окисление сульфида XII может быть проведено намеченным образом при комнатной температуре с использованием OXONE (тройной соли моноперсульфата калия, 2 KHSO5 * KHSO4 * K2SO4), переокисление может быть исключено без каких-либо проблем с помощью использования эквимолярных количеств (см. Схему 2, реакцию, которая дает В7).
В соответствии с изобретением перегруппировку Pummerer (XIII→I) проводят в присутствии каталитических количеств бисульфата калия. Посредством использования этого катализатора можно добиться высоких выходов (>80%) и в то же самое время образования очень небольшого количества побочных продуктов (<5%) при низких температурах реакции (<90ºС) [см. Схему 2, реакцию, которая дает II]. Полученный сырой продукт I содержит так мало примесей и также только очень небольшие доли α-аномера, что для очистки достаточно простой кристаллизации.
Способ в соответствии с изобретением действительно позволяет применять общие химические превращения, известные специалистам в данной области, для конструирования тиофуранозы через образование диола, его активацию с помощью биссульфоната и циклизацию с сульфидом натрия; для методов с использованием защитных групп и для окисления сульфида с помощью OXONE.
Однако конкретный аспект настоящего изобретения представляет собой целевое и высокоэффективное конструирование индивидуальных стереоцентров тиофураноз.
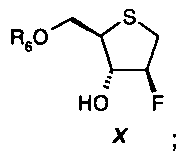
Дополнительным аспектом настоящего изобретения является стереоспецифическое введение атома фтора в положении атома С3 соединения X в результате стереоселективного раскрытия циклического сульфата IX.
Описание вариантов осуществления
На первой стадии способа, в соответствии с изобретением для получения соединения формулы I:
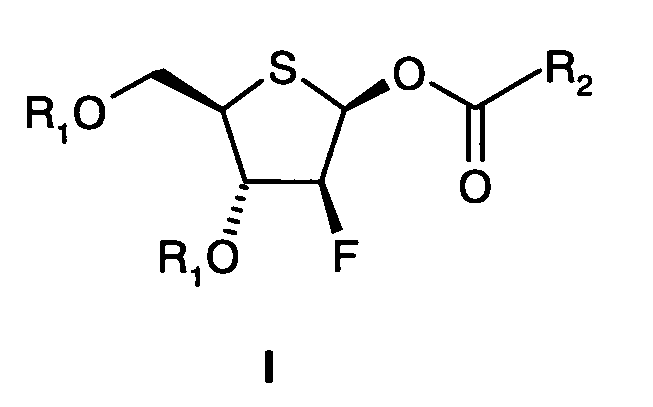
где
R1 представляет собой -С(О)-С1-С6-алкил или -С(О)-арил; и
R2 представляет собой С1-С6-алкил, С1-С4-перфторалкил или арил,
ликсонолактон формулы IV
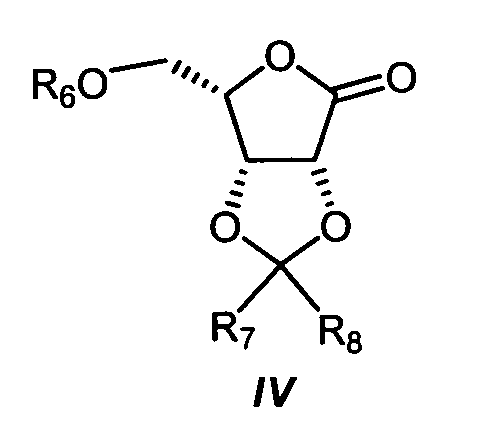
где
R6 представляет собой С1-С6-алкил или арилметилен; и
R7 и R8 независимо друг от друга представляют собой водород, С1-С6-алкил, С1-С4-перфторалкил или арил;
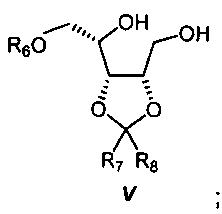
восстанавливают в присутствии 0,5-10 молярных эквивалентов гидридных доноров формулы А(AlH4) или А(ВН4), в которой А представляет собой щелочной металл, с получением диола формулы V:
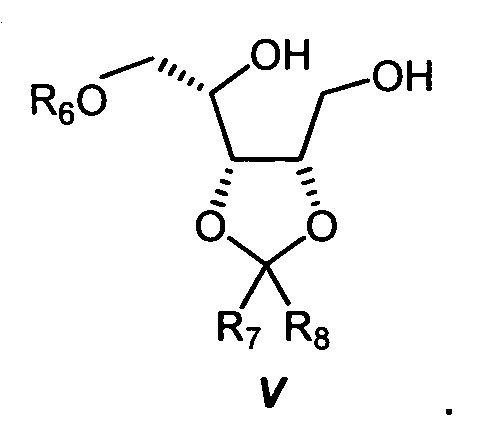
Здесь предпочтение отдают использованию 0,5-1,5 молярных эквивалентов алюмогидрида лития (LiAlH4). Реакционную стадию предпочтительно проводят при температуре от 0ºС до 30ºС.
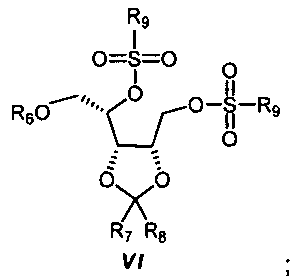
На второй стадии, диол V подвергают реакции с, по меньшей мере, 2 молярными эквивалентами сульфонилхлорида R9-SO2Cl или ангидрида сульфокислоты R9-SO2-O-SO2-R9, где R9 представляет собой С1-С6-алкил, С1-С4-перфторалкил или арил, предпочтительно с 2-5 молярными эквивалентами метансульфонилхлорида, в присутствии, по меньшей мере, 2 молярных эквивалентов третичного амина (например, триметиламина, триэтиламина, диизопропилэтиламина) или пиридина (например, пиридина, 4-N,N-диметиламинопиридина, коллидина, пиколинов, лутидинов), предпочтительно 2-5 молярных эквивалентов триэтиламина, с получением соединения формулы VI:
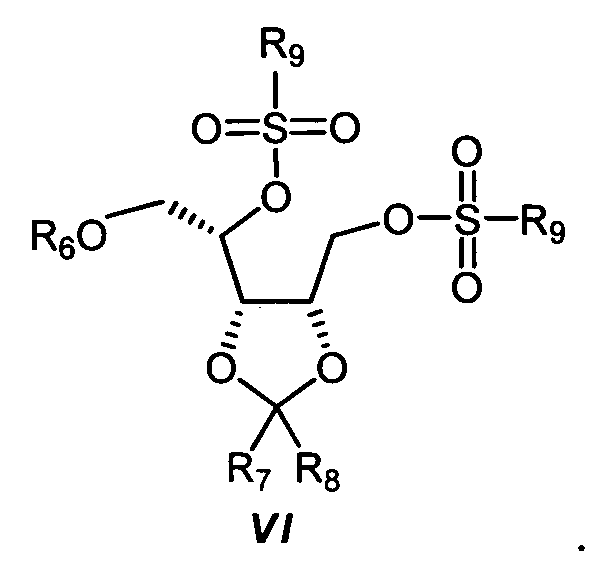
Реакционную стадию предпочтительно проводят при температуре от 0ºС до 30ºС.
На третьей стадии, биссульфонат VI подвергают реакции с, по меньшей мере, 1 молярным эквивалентом сульфида натрия (Na2S) в полярном апротонном растворителе, таком как, например, DMF (диметилформамид), NMP, DMA (диметиламин), DMSO (диметилсульфоксид), предпочтительно NMP (N-метилпирролидон), при температуре выше 50ºС, предпочтительно при температурах от 50 до 100ºС, с получением тиофуранозы формулы VII:
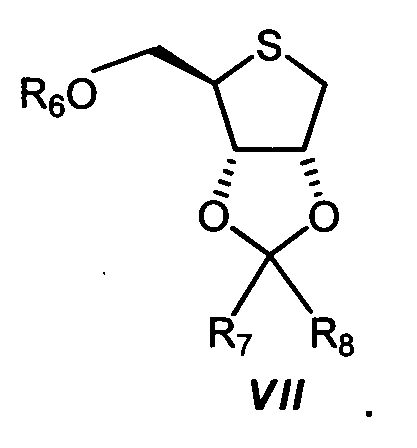
На стадии четыре, тиофуранозу формулы VII превращают в смесь растворителей, содержащей воду и органический растворитель, выбранный из группы простых эфиров (например, диэтилового эфира, THF, диоксана, MTBE (метил-трет-бутиловый эфир)), спиртов (например, метанола, этанола, изопропанола), карбоновых кислот (например, уксусной кислоты), ароматических углеводородов (например, бензола, толуола), предпочтительно в смеси тетрагидрофурана (THF) и воды, посредством 0,01-5 молярных эквивалентов кислоты, выбранной из группы минеральных кислот (например, HCl, H2SO4, H3PO4), алкансульфокислот (например, метансульфокислоты), арилсульфокислот (например, бензолсульфокислоты, толуолсульфокислоты), перфторсульфокислот (например, трифторметансульфокислоты, нонафторбутансульфокислоты) или перфторалканкарбоновых кислот (например, трифторуксусной кислоты), предпочтительно посредством 0,01-5 молярных эквивалентов H2SO4, в диол формулы VIII:
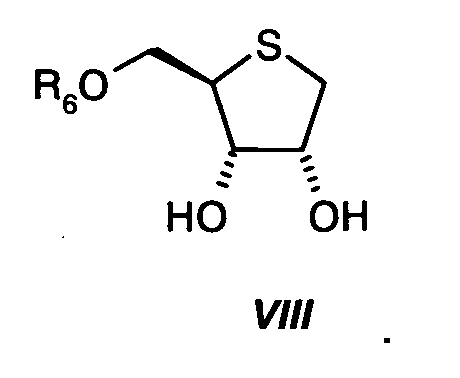
Реакционную стадию предпочтительно проводят при температуре от 20ºС до 100ºС.
Диол VIII выделяют кристаллизацией из смеси растворителей, состоящей из алкана или смеси алканов - предпочтительно гептана - и сложного эфира карбоновой кислоты - предпочтительно изопропилацетата или этилацетата.
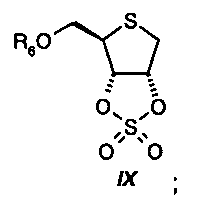
На стадии пять, диол общей формулы VIII подвергают реакции в присутствии, по меньшей мере, 0,2 молярных эквивалентов основания формулы AH, A2CO3 или A(OtBu), в которой А представляет собой щелочной металл; предпочтительно в присутствии 0,2-3 молярных эквивалентов гидрида натрия, с 1-2 молярными эквивалентами диол-активирующего реагента формулы Х1-SO2-X2, в которой Х1 и Х2 независимо друг от друга представляют собой Cl или имидазоил, предпочтительно с 1-2 молярными эквивалентами сульфонилдиимидазола, с получением циклического сложного эфира серной кислоты формулы IX:
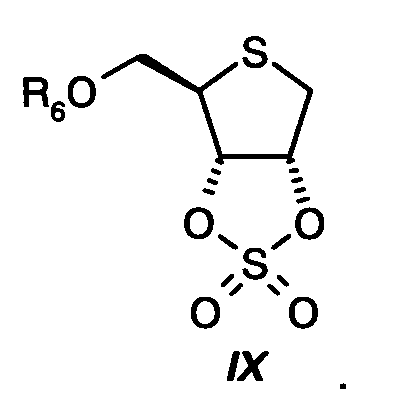
Реакционную стадию предпочтительно проводят при температуре от -5ºС до 20ºС.
На стадии шесть, циклический сложный эфир серной кислоты формулы IX сначала подвергают реакции с 1-3 молярными эквивалентами фторида аммония формулы N(R11)4F, где R11 представляет собой С1-С6-алкил, предпочтительно с 1-3 молярными эквивалентами фторида тетрабутиламмония, при температуре от 0ºС до 30ºС, и реакционную смесь, полученную таким образом, подвергают реакции с кислотой, выбранной из группы, состоящей из минеральных кислот (например, HCl, H2SO4), алкилсульфокислот (например, метансульфокислоты), арилсульфокислот (например, бензолсульфокислоты, толуолсульфокислоты), перфторсульфокислот (например, трифторметансульфокислоты, нонафторбутансульфокислоты) или перфторалканкарбоновых кислот (например, трифторуксусной кислоты), предпочтительно с серной кислотой (H2SO4),
с получением простого эфира формулы X:
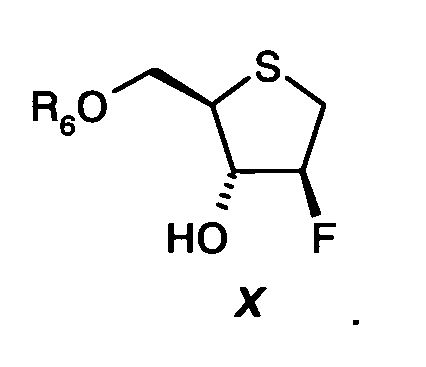
Вторую составляющую стадии реакции предпочтительно проводят при температуре от 20ºС до 70ºС.
На стадии семь, простой эфир формулы X сначала, на составляющей стадии а), подвергают реакции в присутствии, по меньшей мере, 1 молярного эквивалента галогенида бора BY3, где Y представляет собой F, Cl или Br, предпочтительно с использованием 1-4 молярных эквивалентов трихлорида бора, при температуре от 0ºС до -80ºС.
На составляющей стадии b), реакционную смесь, полученную на составляющей стадии а), подвергают реакции со смесью:
спиртового компонента, выбранного из группы, состоящей из С1-С6-алканола (например, метанола, этанола, н-пропанола, изопропанола, н-бутанола, трет-бутанола, изобутанола), и арилалканола (например, бензилового спирта) и фенолов (например, фенола); и
основания, выбранного из группы алифатических третичных аминов (например, триметиламина, триэтиламина, диизопропилэтиламина) или из группы пиридинов (например, пиридина, 4-N,N-диметиламинопиридина, коллидина, пиколинов, лутидинов); предпочтительно с использованием смеси метанола и пиридина, с получением диола формулы XI:
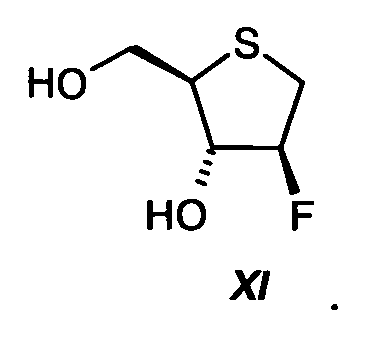
Процесс и разложение реакционной смеси предпочтительно проводят при температуре от 0ºС до -80ºС.
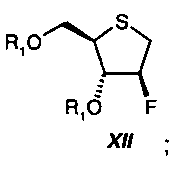
На стадии восемь, диол формулы XI подвергают реакции в присутствии, по меньшей мере, 2 молярных эквивалентов основания, выбранного из группы алифатических третичных аминов (например, триметиламина, триэтиламина, диизопропилэтиламина) или из группы пиридинов (например, пиридина, 4-N,N-диметиламинопиридина, коллидина, пиколинов, лутидинов); предпочтительно с использованием 2-10 молярных эквивалентов пиридина, с, по меньшей мере, 2 молярными эквивалентами хлорангидрида кислоты R1-Cl или ангидрида кислоты R1-O-R1, где R1 представляет собой -С(O)-C1-C6-алкил или -С(О)-арил; предпочтительно с использованием 2-5 молярных эквивалентов бензоилхлорида, с получением соединения формулы XII:
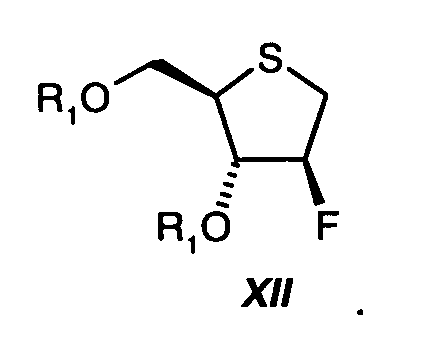
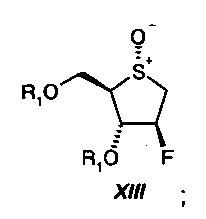
На стадии девять, сульфид формулы XII окисляют в смеси растворителей, состоящей из воды и кетона формулы R9-C(O)-R9 ', где R9 и R9 ' независимо друг от друга представляют собой С1-С6-алкил, С1-С4-перфторалкил или арил, предпочтительно ацетон, посредством 0,5-1 молярного эквивалента персульфата щелочного металла формулы AHSO5, где А* представляет собой щелочной металл, предпочтительно с использованием 0,5-1 молярного эквивалента OXONE (тройной соли моноперсульфата калия, 2 KHSO5*KHSO4 * K2SO4), при температуре от 0ºС до 50ºС, с получением сульфоксида формулы XIII:
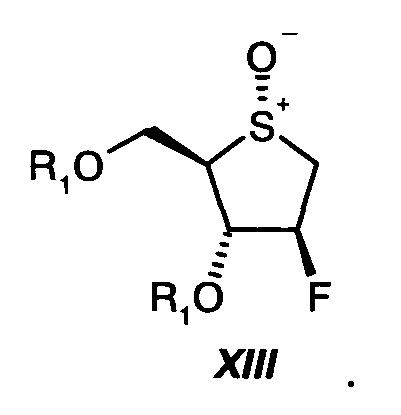
Продукт XIII выделяют кристаллизацией из подходящего растворителя, предпочтительно из метил-трет-бутилового эфира.
На стадии десять, сульфоксид формулы XIII подвергают реакции с, по меньшей мере, 1 молярным эквивалентом ангидрида кислоты R2-C(O)-O-C(O)-R2, где R2 представляет собой С1-С6-алкил, С1-С4-перфторалкил или арил; предпочтительно с использованием, по меньшей мере, 5 молярных эквивалентов ангидрида уксусной кислоты, в присутствии 0,01-2 молярных эквивалентов протонной кислоты, выбранной из группы, состоящей из минеральных кислот (например, HCl, HBr, H2SO4, H3PO4, бисульфатов щелочного металла, одноосновых фосфатов щелочного металла), алкансульфокислот (например, метансульфокислоты), арилсульфокислот (например, бензолсульфокислоты, толуолсульфокислоты), перфторсульфокислот (например, трифторметансульфокислоты, нонафторбутансульфокислоты) или перфторалканкарбоновых кислот (например, трифторуксусной кислоты), или в присутствии 0,01-2 молярных эквивалентов кислоты Льюиса (например, LiCl, MgBr2, Ti(OR13)4), где R13 представляет собой С1-С6-алкил, или арилметилена; предпочтительно в присутствии 0,01-2 молярных эквивалентов бисульфата калия; при температуре от 30ºС до 100ºС, с получением соединения формулы I:
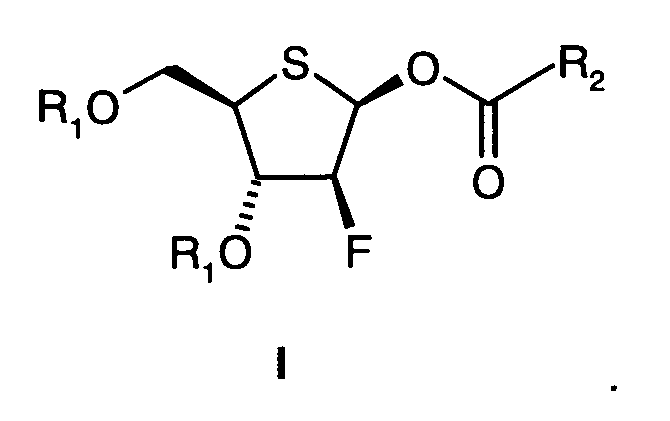
Продукт I выделяют кристаллизацией из подходящего растворителя, предпочтительно этанола.
До сих пор в литературе не предоставлено никаких примеров, где бисульфат калия катализирует Pummerer-перегруппировку сульфоксида в соответствующий тиоацеталь и предотвращает образование побочных продуктов.
С1-С6-алкильные группы радикальных групп R1, R2, R6, R7, R8, R9, R9', R11 и R13 могут представлять собой, например, метильные, этильные, н-пропильные, изопропильные, н-бутильные, изобутильные, втор-бутильные, трет-бутильные, н-пентильные или н-гексильные группы.
Арилметиленовые группы радикальных групп R6, R9 и R13 могут представлять собой, например, бензильные или 4-метоксибензильные группы.
Арильные группы радикальных групп R1, R2, R7, R8, R9 и R9' могут представлять собой, например, фенильные или замещенные фенильные группы.
С1-С4-перфторалкильные группы радикальных групп R2, R7, R8, R9 и R9' могут представлять собой, например, трифторметильные, пентафторэтильные или нонафторбутильные группы.
Щелочные металлы А гидридных доноров с формулами A(AlH4), А(ВН4); оснований AH, A2CO3, AOtBu; персульфатов щелочных металлов AHSO5 могут представлять собой, например, литий, натрий или калий.
-С(О)-С1-С6-алкильные группы радикальной группы R1 могут представлять собой, например, ацетильные, н-пропаноильные, изопропаноильные, н-бутаноильные, трет-бутаноильные, изобутаноильные, н-пентаноильные или н-гексаноильные группы.
-С(О)-арильные группы радикальной группы R1 могут представлять собой, например, бензоильные или замещенные бензоильные группы.
Предпочтительно, способ согласно изобретению используют для получения соединения I (в соответствии со Схемой 3), где
R1 представляет собой бензоил, и
R2 представляет собой метил,
то есть для получения соединения формулы IIβ.
Согласно настоящему изобретению особое предпочтение отдают способу для получения соединения формулы IIβ (в соответствии со Схемой 3), где
R7, R8 и R9 представляют собой метил, и
R6 представляет собой бензил.
Кроме того, настоящее изобретение также относится к промежуточным соединениям предпочтительного варианта осуществления способа в соответствии с изобретением (Схема 4), в частности, к
1-О-бензил-3,4-О-изопропилиден-L-арабинитолу (С2);
1-О-бензил-3,4-О-изопропилиден-2,5-ди-О-метансульфонил-L-арабинитолу (С3);
1,4-ангидро-5-О-бензил-2,3-О-изопропилиден-4-тио-D-рибитолу (С4);
1,4-ангидро-5-О-бензил-2,3-О-сульфонил-4-тио-D-рибитолу (С6);
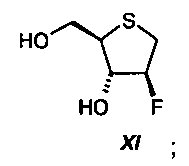
1,4-ангидро-5-О-бензил-2-дезокси-2-фтор-4-тио-D-арабинитолу (С7).
Схема 4
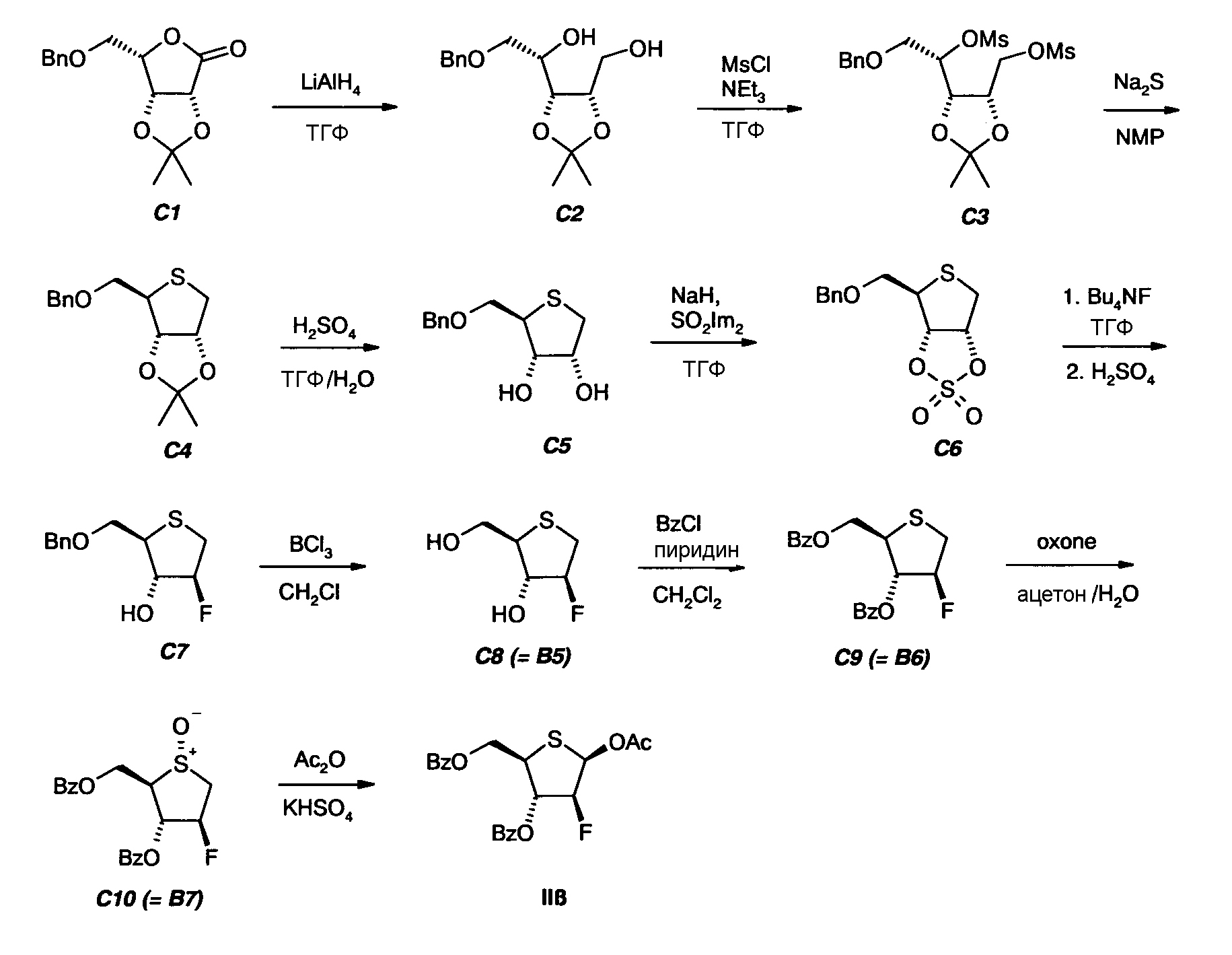
Соединение С1 (5-О-бензил-2,3-О-изопропилиден-L-ликсоно-1,4-лактон) является коммерчески доступным. Далее, описаны методики проведения реакций, показанных на Схеме 4.
Методики синтеза
1-О-Бензил-3,4-О-изопропилиден-L-арабинитол (С2):
При 0-10ºС, 44,68 г (116,78 ммоль) алюмогидрида лития отмеряют и подают в раствор, содержащий 50 г (179,66 ммоль) С1 в 450 мл THF, и смесь перемешивают до завершения реакции. При 20ºС затем добавляют воду и водный раствор гидроксида натрия, осажденное твердое вещество отфильтровывают и промывают до отсутствия в нем продукта с использованием тетрагидрофурана (THF). Раствор сырого продукта концентрируют и полностью освобождают от растворителя. Это дает 53,26 г С2 (105%) в виде сырого продукта, который используют в этой форме на последующей стадии С3.
1H-ЯМР (400 МГц, DMSO): 7,39-7,24 (м, 5H, H-7 до H-9), 4,81 (т, 1H, 5,46 Гц, 1-OH), 4,65 (д, 1H, 5,84 Гц, 4-OH), 4,5 (с, 2H, 2×H-6), 4,15-4,00 (м, 2H, H-2 и H-3), 3,76 (кв.д, 1H, 6 Гц, 2,5 Гц, H-4), 3,7-3,55 (м, 2H, 2×H-1), 3,49-3,35 (м, 2H, 2×H-5), 1,38 (с, 3H, 10-CH3), 1,25 (с, 3H, 10-CH3).
13C-ЯМР (75 МГц, CDCl3): 138,0, 128,8, 128,2, 128,1 (Ar), 108,9 (О-C-O), 77,7 (CH2), 76,9 (CH2), 73,8 (CH), 71,9 (CH), 68,9 (CH2), 61,0 (CH), 28,1 (CH3), 25,6 (CH3).
1-О-Бензил-3,4-О-изопропилиден-2,5-ди-О-метансульфонил-L-арабинитол (С3):
При 20ºС 54,54 г (538,97 ммоль) триэтиламина добавляют в раствор, содержащий 53,26 г (179,66 ммоль, при условии достижения выхода 100% на стадии С2) соединения С2 (сырой продукт) в 450 мл THF, и 39,39 г (431,17 ммоль) метансульфонилхлорида отмеряют и подают при 0-10ºС. Затем смесь перемешивают до завершения реакции, добавляют воду и фазы разделяют. Водную фазу экстрагируют посредством MTBE (метил-трибутиловый эфир) и объединенные органические фазы промывают насыщенным раствором хлорида натрия и разбавленным раствором бикарбоната натрия. Раствор сырого продукта концентрируют и полностью освобождают от растворителей. Это дает 86,66 г С3 (110%) в виде сырого продукта, который используют в этой форме на последующей стадии С4.
1H-ЯМР (400 МГц, CDCl3): 7,40-7,30 (м, 5H), 4,90 (дт, 1H, 11,5 Гц, 6,6 Гц), 4,56 (дд, 2H, 11,8 Гц), 4,42-4,39 (м, 2H), 4,38-4,35 (м, 2H), 3,83 (дд, 1H, 10,61 Гц, 6,06 Гц), 3,70 (дд, 1H, 10,61 Гц, 5,05 Гц), 3,10 (с, 3H), 3,02 (с, 3H), 1,50 (с, 3H), 1,37 (с, 3H).
13C-ЯМР (75 МГц, CDCl3): 136,8, 128,6, 128,2, 128,0 (Ar), 109,7 (О-C-O), 77,8, 75,6, 74,5, 73,7 (CH2), 69,6 (CH2), 67,7 (CH2), 39,0, 37,5, 27,2, 25,4.
1,4-Ангидро-5-О-бензил-2,3-О-изопропилиден-4-тио-D-рибитол (С4):
50,99 г (215,59 ммоль) сульфида натрия (33%) добавляют к раствору, содержащему 86,66 г (179,66 ммоль, при условии достижения выхода 100% на стадии С3) соединения С3 (сырой продукт) в 550 мл N-метил-2-пирролидона, и смесь нагревают до внутренней температуры 80ºС и перемешивают при этой температуре до завершения реакции. При внутренней температуре 20ºС затем добавляют воду и MTBE, фазы разделяют и водную фазу экстрагируют посредством MTBE. Объединенные органические фазы в конце промывают водой. Раствор сырого продукта концентрируют и полностью освобождают от растворителей. Это дает 56,41 г С4 (112%) в виде сырого продукта, который используют в этой форме на последующей стадии С5.
1H-ЯМР (400 МГц, DMSO): 7,40-7,25 (м, 5H, H-7 до H-9), 4,89 (т, 1H, 4,7 Гц, H-2), 4,73 (д, 1H, 5,65 Гц, H-3), 4,50 (с, 2H, 2×H-6), 3,55-3,42 (м, 2H, 2×H-5), 3,37 (т, 1H, 6,2 Гц, H-4), 3,10 (дд, 1H, 12,6 Гц, 4,7 Гц, H-1), 2,76 (д, 1H, 12,6 Гц, H-1').
13C-ЯМР (75 МГц, CDCl3): 137,9, 128,4, 127,9, 127,5 (Ar), 110,9 (О-C-O), 86,3 (CH), 83,8 (CH), 73,2 (CH2), 72,2 (CH2), 53,3 (CH), 38,3 (CH2), 27,2 (CH3), 25,3 (CH3).
1,4-Ангидро-5-О-бензил-4-тио-D-рибитол (С5):
Раствор, содержащий 50 мл воды и 9,22 г (0,094 ммоль) серной кислоты, добавляют к раствору, содержащему 56,41 г (179,66 ммоль при условии достижения выхода 100% на стадии С4) С4 (сырой продукт) в 450 мл THF, и смесь нагревают до 70ºС и перемешивают при этой температуре в течение нескольких часов. Для достижения полного превращения, отгоняют несколько миллилитров при 70ºС. Затем добавляют MTBE при 20ºС, фазы разделяют и водную фазу экстрагируют посредством MTBE. Объединенные органические фазы нейтрализуют насыщенным раствором карбоната калия. Твердое вещество отфильтровывают и промывают до отсутствия в нем продукта с использованием MTBE. Раствор сырого продукта дважды перегоняют в изопропилацетат и продукт кристаллизуют добавлением гептана. Это дает 33,67 г (78% за 4 стадии, если начинать с С1) соединения С5.
1H-ЯМР (400 МГц, CDCl3): 7,39-7,29 (м, 5H), 4,56 (с, 2H), 4,37 (дт, 1H, 7,33 Гц, 3,54 Гц), 4,04 (дт, 1H, 7,07 Гц, 6,82 Гц), 3,70 (дд, 1H, 9,09 Гц, 5,31 Гц), 3,62 (т, 1H, 9,09 Гц), 3,56-3,51 (м, 1H), 3,11 (д, 1H, 3,54 Гц), 3,05 (дд, 1H, 11,62 Гц, 4,55 Гц), 2,85 (дд, 1H, 11,62 Гц, 3,03 Гц), 2,65 (д, 1H, 3,54 Гц).
3C-ЯМР (75 МГц, CDCl3): 137,4, 128,6, 128,0, 127,8 (Ar), 80,4, 74,8, 73,6 (CH2), 73,0 (CH2), 47,0, 33,6 (CH2).
Температура плавления: 78-82ºС
1,4-Ангидро-5-О-бензил-2,3-О-сульфонил-4-тио-D-рибитол (С6):
При 0ºС раствор, содержащий 50 г (208,06 ммоль) С5 в 75 мл THF, добавляют к суспензии, содержащей 2,5 г гидрида натрия (в минеральном масле) в 100 мл THF, и смесь перемешивают при 0ºС в течение 1-2 часов. При 0ºС раствор, содержащий 45,36 г сульфонилдиимидазола в 450 мл THF, затем отмеряют и добавляют, смесь перемешивают при 20ºС до завершения реакции. В этой форме сырой продукт используют непосредственно на следующей стадии С7.
1H-ЯМР (400 МГц, CDCl3): 7,40-7,26 (м, 5H), 5,41 (дт, 1H, 6,06 Гц, 3,28 Гц), 5,33 (дд, 1H, 6,32 Гц, 2,78 Гц), 4,54 (д, 2H, 2,53 Гц), 3,81 (дд, 1H, 9,85 Гц, 4,29 Гц), 3,72 (м, 1H), 3,64 (дд, 1H, 9,85 Гц, 4,80 Гц), 3,47 (дд, 1H, 13,39 Гц, 5,81 Гц), 3,16 (дд, 1H, 13,39 Гц, 3,28 Гц).
13C-ЯМР (75 МГц, CDCl3): 137,0, 128,6, 128,2, 127,7 (Ar), 87,9, 86,1, 73,7 (CH2), 71,2 (CH2), 51,6, 36,8 (CH2).
1,4-Ангидро-5-О-бензил-2-дезокси-2-фтор-4-тио-D-арабинитол (С7):
При 20ºС раствор, содержащий 131,3 г тригидрата фторида тетрабутиламмония в 150 мл THF, отмеряют и подают в раствор сырого продукта стадии получения соединения С6. Смесь перемешивают при 30ºС до завершения реакции.
Реакционную смесь затем корректируют с доведением рН до 1 с помощью серной кислоты (концентрация 33%) и перемешивают при 50ºС до завершения реакции. При 20ºС рН затем доводят до 7-10 с использованием водного раствора гидроксида калия и образованный осадок отфильтровывают. Фазы разделяют и водную фазу экстрагируют дихлорметаном. Объединенные органические фазы концентрируют и сырой продукт, после полного удаления растворителя, очищают хроматографией. Это дает 22,68 г соединения С7 (45%).
1H-ЯМР (400 МГц, CDCl3): 7,4-7,27 (м, 5H, H-7 до H-9), 5,04 (кв.д, 1H, 51,7 Гц, 5,44 Гц, H-2), 4,55 (с, 2H, 2×H-6), 4,37 (тд, 1H, 11,7 Гц, 4,9 Гц, H-3), 3,68-3,55 (м, 2H, 2×H-5), 3,45-3,38 (м, 1H, H-4), 3,2-2,99 (м, 2H, 2×H-1).
13C-ЯМР (100 МГц, CDCl3): 137,65, 128,52, 127,91, 127,77 (Ar), 97,27 (C-2), 79,10 (C-3), 73,46 (C-6), 72,69 (C-5), 48,58 (C-4), 31,72 (C-1).
19F-ЯМР (376 МГц, CDCl3): -183,14 (м, 2-F).
Температура плавления: 83-85ºС
1,4-Ангидро-2-дезокси-2-фтор-4-тио-D-арабинитол (С8):
При температуре <-65ºС раствор, предварительно охлажденный до -10ºС, содержащий 50 г (206,35 ммоль) соединения С7 в 350 мл дихлорметана, добавляют к 546,82 г (412,69 ммоль) трихлорида бора (1 моль/л в дихлорметане). По истечении 30 минут при <-65ºС, реакционную смесь проверяют в отношении полноты превращения. Смесь, содержащую 150 мл метанола и 116 мл пиридина, затем добавляют при <-65ºС к реакционной смеси и через 15 минут эту смесь нагревают до 20ºС. Растворители удаляют полностью перегонкой при пониженном давлении. Дихлорметан затем добавляют к остатку и растворитель удаляют полностью. Это дает 133 г соединения С8 (423%) в виде сырого продукта (содержит остаточные количества пиридина и гидрохлорида пиридиния), который используют в этой форме на последующей стадии С9.
1H-ЯМР (400 МГц, DMSO): 5,49 (д, 1H, 4,77 Гц, 3-OH), 4,98 (кв.д, 1H, 51,55 Гц, 3,76 Гц, H-2), 4,94 (т, 1H, 5,2 Гц, 5-OH), 4,17 (ддд, 1H, 15,18 Гц, 4,14 Гц, 3,76 Гц H-3), 3,61-3,53 (м, 1H, H-5), 3,37-3,30 (м, 1H, H-5'), 3,19-3,02 (м, 2H, H-4 и H-1), 2,93 (ддд, 1H, 18,22 Гц, 12,1 Гц, 3,76 Гц, H-1').
13C-ЯМР (75 МГц, CDCl3): 97,1 (д, 185 Гц, C-2), 77,5 (д, 24 Гц, C-3), 63,3 (д, 3 Гц, C-5), 50,9 (д, 4 Гц, C-4), 31,1 (д, 22 Гц, C-1).
1,4-Ангидро-2-дезокси-2-фтор-3,5-ди-О-бензоил-4-тио-D-арабинитол (С9):
133 г (206,35 ммоль при условии достижения выхода 100% на стадии С8) соединения С8 (сырой продукт) растворяют в 400 мл дихлорметана и добавляют 97,92 г (1237,9 ммоль) пиридина. Затем при 10ºС добавляют по каплям 87,01 г (618,97 ммоль) бензоилхлорида и смесь перемешивают при 20ºС до завершения реакции. Затем добавляют метанол и смесь перемешивают в течение 1 часа. В конечном счете добавляют воду и фазы разделяют. Раствор сырого продукта концентрируют и полностью освобождают от растворителя. Это дает 114,53 г соединения С9 (154 %) в виде сырого продукта, который используют в этом виде на последующей стадии С10.
1H-ЯМР (400 МГц, CDCl3): 8,20-8,00 (м, 4H), 7,65-6,90 (м, 6H), 5,85 (дт, 1H, 9,98 Гц, 2,64 Гц), 5,40 (ддд, 1H, 48,79 Гц, 7,16 Гц, 3,01 Гц), 4,54 (с, 1H), 4,51 (с, 1H), 3,95-3,82 (м, 1H), 3,45-3,35 (м, 1H), 3,35-3,28 (м, 1H).
13C-ЯМР (75 МГц, CDCl3): 165,0 (C=0), 133,6, 133,1, 129,8, 129,7, 128,5, 128,4, 96,5 (д, 184 Гц, C-2), 79,0 (д, 29 Гц, C-3), 65,2 (д, 4 Гц, C-5), 48,5 (C-4), 34,9 (д, 23 Гц, C-1).
1,4-Ангидро-2-дезокси-2-фтор-3,5-ди-О-бензоил-4-сульфинил-D-арабинитол (С10):
114,53 г (206,35 ммоль при условии достижения выхода 100% на стадии С9) соединения С9 (сырой продукт) растворяют в 400 мл ацетона и добавляют 60 мл воды. При 20ºС затем добавляют небольшими порциями 69,77 г (113,49 ммоль) OXONE. Делают проверку реакции на полноту превращения и затем добавляют разбавленный раствор сульфита натрия. Реакционную смесь нейтрализуют с использованием насыщенного раствора бикарбоната натрия и затем ацетон удаляют полностью перегонкой при пониженном давлении. К суспензии добавляют дихлорметан, твердое вещество отфильтровывают и отфильтрованный осадок промывают до отсутствия в нем продукта с помощью дихлорметана. Раствор сырого продукта дважды перегоняют в MTBE и выделяют в этом растворителе. Это дает 56,4 г (72,6% за три стадии, если начитать с С7) соединения C10.
1H-ЯМР (400 МГц, CDCl3): 8,05 (м, 4H), 7,60 (м, 2H), 7,45 (м, 4H), 5,83 (м, 1H), 5,74 (м, 1H), 4,89 (ддд, 1H, 12,1 Гц, 5,1 Гц, 0,9 Гц), 4,75 (ддд, 1H, 12,4 Гц, 7,6 Гц, 0,9 Гц), 3,65 (м, 1H), 3,75 (м, 1H), 3,45 (м, 1H).
13C-ЯМР (75 МГц, CDCl3): 165,7, 165,2 (C=0), 134,0, 133,5, 130,0, 129,7, 128,6, 128,5 (Ar), 95,4 (д, 185 Гц, C-2), 77,2 (д, 33 Гц, C-3), 71,6 (C-4), 61,1 (д, 2 Гц, C-5), 55,8 (д, 19 Гц, C-1).
1-О-Ацетил-2-дезокси-2-фтор-3,5-ди-О-бензоил-4-тио-β-D-арабинофураноза (IIβ):
80 мл уксусного ангидрида и 361 мг (2,66 ммоль) бисульфата калия добавляют к 10 г (26,57 ммоль) соединения С10 и смесь перемешивают при 80ºС до завершения реакции. Затем реакционную смесь подвергают совместной перегонке сначала повторно с толуолом и затем с этанолом. В конце продукт кристаллизуют из этанола. Это дает 8,9 г (80 %) соединения IIβ.
1H-ЯМР (400 МГц, CDCl3): 8,10-7,90 (м, 4H, Ar), 7,63-7,29 (м, 6H, Ar), 6,18 (д, 1H, 4,4 Гц), 6,12-6,02 (м, 1H), 5,45 (дд, 1H, 9,04 Гц, 4,52 Гц), 5,28 (дд, 1H, 8,85 Гц, 4,52 Гц), 4,68 (дд, 1H, 11,49 Гц, 6,22 Гц), 4,49 (дд, 1H, 11,49 Гц, 6,41 Гц), 3,74 (дд, 1H, 13,56 Гц, 6,40 Гц), 2,12 (с, 3H).
13C-ЯМР (75 МГц, CDCl3): 169,6 (COCH3), 165,8, 165,4 (COPh), 133,6, 133,1 (Ar) 129,8, 129,7, 128,5, 128,2 (Ar), 92,5 (д, 207 Гц, C-2), 75,7 (д, 23 Гц, C-3), 74,0 (д, 17 Гц, C-1), 66,1 (C-5), 42,4 (д, 7 Гц, C-4), 21,0 (CH3).
Температура плавления: 130ºС
Промышленная применимость
Способ настоящего изобретения обеспечивает промышленно преимущественное и превосходное получение соединений формулы I.
Новые предшественники производных глутамата
Новые предшественники производных глутамата

