Результат интеллектуальной деятельности: СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к солям 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, к содержащим их фармацевтическим композициям, и к их использованию для лечения и/или профилактики заболеваний, связанных с сигма рецептором.
Известный уровень техники
Поиску новых терапевтических агентов в последние годы значительно способствовало лучшее понимание строения белков и других биомолекул, связанных с исследуемыми заболеваниями. Одним важным классом указанных белков является сигма (σ) рецептор, рецептор поверхности клеток центральной нервной системы (ЦНС), который может быть связан с дисфорическими, галлюциногенными и стимулирующими сердечную деятельность эффектами опиоидов. На основании изучения биологии и функций сигма рецепторов, были представлены доказательства того, что лиганды сигма рецепторов могут оказаться полезными при лечении психозов и двигательных нарушений, таких как дистония и старческая дискинезия, и двигательных расстройств, связанных с хореей Хантингтона или синдромом Тауретта, и при болезни Паркинсона (Walker, J.M. et al., Pharmacological Reviews, 1990, 42, 355). Сообщалось, что известный лиганд сигма рецептора римказол при клиническом применении демонстрирует эффекты при лечении психозов (Snyder, S.H., Largent, B.L. J. Neuropsychiatry 1989, 1, 7). Сайты связывания сигма рецепторов обладают предпочтительным сродством к правовращающим изомерам некоторых опиатных бензоморфанов, таких как (+)SKF 10047, (+)циклазоцин, и (+)пентазоцин и также к некоторым нарколептикам, таким как галоперидол.
Сигма рецепторы бывают по меньшей мере двух подтипов, которые можно дифференцировать, используя стереоселективные изомеры указанных фармакоактивных лекарственных средств. SKF 10047 обладает наномолярным сродством в отношении сигма 1 (σ-1) сайта, и обладает микромолярным сродством в отношении сигма 2 (σ-2) сайта. Галоперидол отличается аналогичными сродствами в отношении обоих подтипов. Эндогенные лиганды сигма рецепторов неизвестны, хотя было высказано предположение, что прогестерон является одним из них. Возможные эффекты лекарственных средств, опосредуемые сигма-сайтами, включают модулирование функций глутаматного рецептора, реакций нейротрансмиттера, нейропротекции, поведения и познавательных способностей (Quirion, R. et al. Trends Pharmacol. Sci., 1992, 13:85-86). Большинство исследований подразумевают, что сайты связывания сигма (рецепторы) представляют собой плазмалеммальные элементы каскада передачи сигналов. Лекарственные средства, о которых сообщалось, что они являются селективными лигандами сигма рецепторов, были оценены как антипсихотики (Hanner, M. et al. Proc. Natl. Acad. Sci., 1996, 93:8072-8077). Существование сигма рецепторов в ЦНС, иммунной и эндокринной системах предполагает вероятность того, что они могут служить связью между указанными тремя системами.
Учитывая потенциальные терапевтические применения агонистов или антагонистов указанных сигма рецепторов, было предпринято множество попыток, направленных на обнаружение селективных лигандов. Так, в известном уровне техники раскрыты различные лиганды сигма рецепторов. 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолин представляет собой один из таких многообещающих лигандов сигма рецептора. Указанное соединение и способ его получения раскрыты и заявлены в WO 2006/021462.
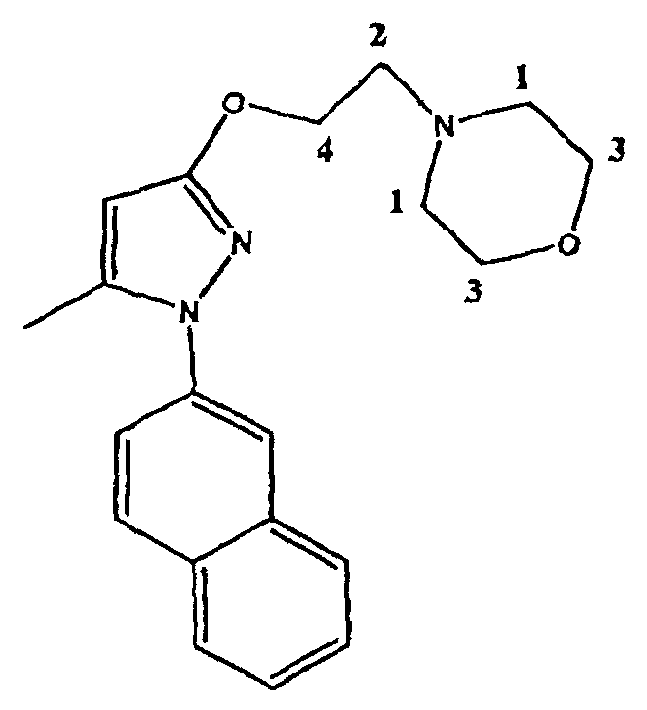
4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолин представляет собой высоко селективный антагонист сигма-1 (σ-1) рецептора. Он проявляет сильную анальгезирующую активность при лечении и профилактике хронической и острой боли, и особенно, невропатической боли. Молекулярный вес указанного соединения составляет 337,42 а.е.м.(uma). Структурная формула указанного соединения имеет вид:
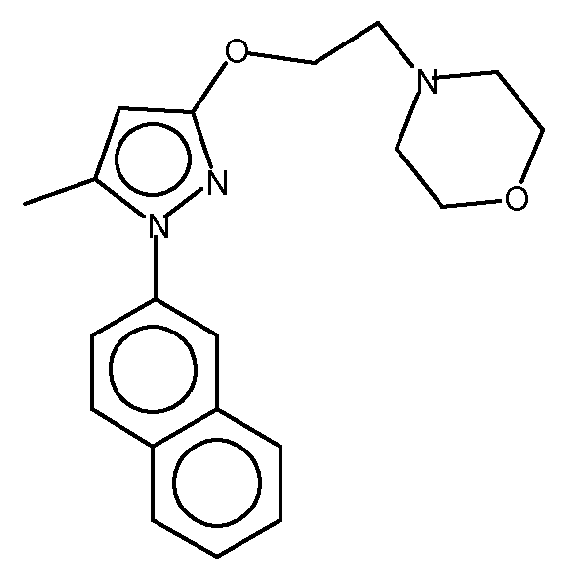
Для осуществления фармацевтической разработки и реализации потенциала указанного соединения существует необходимость в указанной области техники в дополнительных формах 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, которые облегчат получение лучшего качества композиций указанного активного фармацевтического ингредиента. Кроме того, новые формы указанного соединения могут также улучшить способы его производства, обработки и характеристики хранения, а также его терапевтические эффекты, такие как фармакологические характеристики. В этом плане альтернативные формы указанного соединения могут иметь весьма разнообразные свойства, такие как, например, повышенная термодинамическая стабильность, более высокая степень чистоты или улучшенная биодоступность (например, улучшенная абсорбция, улучшенные характеристики растворимости). Специфические формы соединения могут также облегчить получение (например, повысить сыпучесть), обработку и хранение (например, уменьшить гигроскопичность, увеличить срок хранения) композиций соединения или обеспечить использование более низких доз терапевтического агента, тем самым уменьшая возможные побочные эффекты. Таким образом важно создать такие формы, обладающие желательными характеристиками для фармацевтического использования.
Сущность изобретения
Авторы настоящего изобретения после обширных исследований различных форм 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина (именуемого здесь как "соединение 63"), с удивлением обнаружили и продемонстрировали, что некоторые из его солей, и особенно гидрохлоридная соль, обеспечивают благоприятные свойства получения, обработки, хранения и/или терапевтического применения.
Таким образом, в первом аспекте настоящее изобретение относится к соли 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, выбранной из группы, состоящей из этансульфоната, фумарата, гидрохлорида, малата, малеата, малоната и метансульфоната.
В предпочтительном варианте настоящее изобретение относится к гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина (именуемой здесь как "P027" или "соединение примера 1").
Соединение P027 имеет молекулярный вес 373,88 uma, pKa 6,73 и его температура плавления составляет 194,2°C. Указанное соединение очень хорошо растворяется в воде и хорошо растворяется в метаноле, 1н хлористоводородной кислоте и диметилсульфоксиде. Оно слабо растворяется в этаноле, слегка растворяется в ацетоне и практически не растворяется в этилацетате и 1н растворе гидроксида натрия. Указанный продукт демонстрирует лучший профиль растворимости и абсорбции in vivo нежели ответствующее ему основание.
В другом аспекте настоящее изобретение относится к способу получения гидрохлоридной соли 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, который включает:
a) смешивание 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина и раствора, содержащего хлористоводородную кислоту, и
b) выделение полученной гидрохлоридной соли.
Следующий аспект настоящего изобретения включает фармацевтические композиции, включающие гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина и фармацевтически приемлемый носитель, адъювант или средство доставки.
В следующем аспекте настоящее изобретение относится к гидрохлориду 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина для использования в качестве лекарственного средства, предпочтительно в качестве лиганда сигма рецептора, т.е. для использования для лечения и/или профилактики опосредованного рецептором сигма заболевания или состояния.
Другой аспект настоящего изобретения относится к способу лечения и/или профилактики заболеваний, опосредованных сигма рецептором, причем указанный способ включает введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения, как определено ранее, или его фармацевтической композиции.
Указанные аспекты и предпочтительные варианты настоящего изобретения дополнительно определены в формуле изобретения.
Краткое описание чертежей
Фиг.1: дифференциальная сканирующая калориметрия (DSC=ДСК) соединения примера 1.
Фиг.2: термогравиметрия (TGA=ТГМ) соединения примера 1.
Фиг.3: спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 1.
Фиг.4: спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения 63.
Фиг.5: спектр протонного ядерного магнитного резонанса (1H ЯМР) примера 2.
Фиг.6: дифференциальная сканирующая калориметрия (ДСК) примера 2.
Фиг.7: термогравиметрия (ТГМ) соединения примера 2.
Фиг.8: FTIR анализ соединения примера 2.
Фиг.9 спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 3.
Фиг.10: дифференциальная сканирующая калориметрия (ДСК) соединения примера 3.
Фиг.11: термогравиметрия (ТГМ) соединения примера 3.
Фиг.12: FTIR анализ соединения примера 3.
Фиг.13 спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 4.
Фиг.14: дифференциальная сканирующая калориметрия (ДСК) соединения примера 4.
Фиг.15: термогравиметрия (ТГМ) соединения примера 4.
Фиг.16: FTIR анализ соединения примера 4.
Фиг.17 спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 5.
Фиг.18: дифференциальная сканирующая калориметрия (ДСК) соединения примера 5.
Фиг.19: термогравиметрия (ТГМ) соединения примера 5.
Фиг.20: FTIR анализ соединения примера 5.
Фиг.21: спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 6.
Фиг.22: дифференциальная сканирующая калориметрия (ДСК) примера 6.
Фиг.23: термогравиметрия (ТГМ) соединения примера 6.
Фиг.24: FTIR анализ примера 6.
Фиг.25: спектр протонного ядерного магнитного резонанса (1H ЯМР) соединения примера 7.
Фиг.26: дифференциальная сканирующая калориметрия (ДСК) соединения примера 7.
Фиг.27: термогравиметрия (ТГМ) соединения примера 7.
Фиг.28: FTIR анализ соединения примера 7.
Фиг.29: Термодинамика растворимости для соединения примера 1. Калибровочная кривая.
Фиг.30: Концентрация соединения примера 1 в плазме крысы.
Подробное описание предпочтительного варианта настоящего изобретения
Авторы настоящего изобретения обнаружили, что соединение P027, которое представляет собой HCl соль 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, обладает преимуществами, связанными с тем фактом (наряду с другими), что оно представляет собой кристаллическое твердое вещество, что упрощает выделение, очистку и дальнейшую обработку.
Действительно, после обширного скринирования солей авторы обнаружили, что большое число кислот (например, серная кислота или L-винная кислота) не образуют твердого вещества при смешивании с 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолином, а образуют масло. Кроме того, среди кислот, которые можно использовать для получения солей в твердой форме, хлористоводородная кислота оказалась кислотой, которая обеспечивает лучшие результаты с точки зрения легкости получения, физической стабильности, увеличения масштаба производства, растворимости, и т.д. Таким образом, настоящее изобретение относится к соли 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина, выбранной из группы, состоящей из этансульфоната, фумарата, гидрохлорида, малата, малеата, малоната и метансульфоната. Указанные соли способны обеспечить получение кристаллического твердого вещества.
Предпочтительно, настоящее изобретение относится к гидрохлориду 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина (P027).
Гидрохлоридную соль 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина можно получить, добавляя раствор хлористоводородной кислоты к соответствующему ему основанию, растворенному в подходящем растворителе. В конкретном варианте соединение P027 удобно получать, растворяя указанное соединение в виде свободного основания в этаноле, насыщенном HCl.
Как было указано ранее, сообщалось, что 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолин представляет собой высоко селективный антагонист сигма-1 (σ-1) рецептора, демонстрируя высокую анальгезирующую активность при лечении и профилактике хронической и острой боли, и особенно, невропатической боли (см. WO 2006/021462). Теперь авторами было обнаружено, что гидрохлоридная соль 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина особенно подходит в качестве лекарственного средства. Поэтому в настоящем изобретении далее предложены лекарственные средства или фармацевтические композиции, включающие гидрохлорид 4-[2-[[5-метил-1-(2-нафталинил)-1H-пиразол-3-ил]окси]этил]морфолина вместе с фармацевтически приемлемым носителем, адъювантом или средством доставки для введения пациенту.
Более конкретно, соединение P027 можно использовать для лечения и/или профилактики заболевания или состояния опосредованного рецептором сигма.
В более предпочтительном варианте соединение P027 используют для получения лекарственного средства для лечения и/или профилактики заболевания, выбранного из группы, состоящей из диареи; липопротеиновых нарушений; мигрени; ожирения; артрита; гипертонии; аритмии; язвы; дефицитов обучаемости, памяти и внимания; нарушений познавательных способностей; нейродегенеративных расстройств; демиенилизирующих заболеваний; зависимости от лекарственных средств и химических веществ, включая кокаин, амфетамин, этанол и никотин; поздней дискинезии; ишемического удара; эпилепсии; удара; стресса; рака; психотических состояний, в частности депрессии, тревожных состояний или шизофрении; воспалений; или аутоиммунных заболеваний.
Вспомогательные материалы или добавки в фармацевтических композициях настоящего изобретения можно выбрать из носителей, эксципиентов, материалов основы, смазывающих агентов, наполнителей, растворителей, разбавителей, красителей, кондиционеров вкуса, таких как сахара, антиоксидантов, связующих, адгезивов, разрыхлителей, веществ, способствующих скольжению и/или агглютинирующих агентов. В случае суппозиториев могут быть использованы воски или сложные эфиры жирных кислот или консерванты, эмульгаторы и/или носители для парентеральных применений. Выбор указанных вспомогательных материалов и/или добавок и необходимые для использования количества будут зависеть от формы применения фармацевтической композиции.
Лекарственные средства или фармацевтические композиции в соответствии с настоящим изобретением могут быть в любой форме, пригодной для применения для людей и/или животных, предпочтительно людей, включая младенцев, детей и взрослых, и могут быть получены стандартными способами известными специалистам в данной области. Поэтому композиции в соответствии с настоящим изобретением могут быть адаптированы для местного или системного применения, в частности для кожного, трансдермального, подкожного, внутримышечного, внутрисуставного, внутрибрюшинного, внутривенного, внутриартериального, интравезикального, внутрикостного, интракавернозного, легочного, трансбуккального, сублингвального, внутриглазного, интраветриального, интраназального, чрескожного, ректального, вагинального, перорального, эпидурального, интратекального, интравентрикулярного, интрацеребрального, интрацеребровентрикулярного, интрацистернального, интраспинального, периспинального, интракраниального способов введения, или для доставки с помощью игл или катетеров с использованием или без использования помповых устройств.
Упомянутые композиции можно получить, используя стандартные способы, такие как те, что раскрыты или на которые имеются ссылки в фармакопеях Испании и США и в аналогичных ссылочных текстах.
В одном варианте настоящего изобретения предпочтительно, чтобы соединение P027 было использовано в терапевтически эффективных количествах. Врач определяет дозу присутствующего терапевтического агента, которая будет наиболее подходящей, и эта доза будет меняться в зависимости от формы введения и конкретного выбранного соединения, и кроме того, она будет меняться в зависимости от подлежащего лечению пациента, возраста пациента, подлежащего лечению типа заболевания или состояния. Если композицию вводят перорально, потребуются большие количества активного агента для достижения такого же эффекта, который достигается при введении меньших количеств, но парентерально. Указанное соединение можно использовать таким же образом, что и сравнимые терапевтические агенты, и уровни доз могут быть того же порядка величины, что и обычно используемые дозы для указанных других терапевтических агентов. Указанное активное соединение обычно вводят один или более раз в день, например, 1, 2, 3 или 4 раза ежедневно, причем обычная полная дневная доза находится в интервале от 0,1 до 1000 мг/кг/день.
Следующие примеры просто иллюстрируют некоторые варианты настоящего изобретения и их никоим образом не следует рассматривать как ограничивающие изобретение.
ПРИМЕРЫ
Аналитические методы
Для идентификации полученных различных солей соединения 63 в настоящем изобретении были использованы следующие методики:
- Дифференциальный сканирующий калориметрический анализ (ДСК).
ДСК анализы регистрируют с помощью дифференциального сканирующего калориметра Mettler Toledo DSC822e. Образцы массой 1-2 мг взвешивают в 40 мкл алюминиевые тигли с крышкой с маленькими отверстиями, и нагревают в атмосфере азота (50 мл/мин), от 30 до 300°C при скорости нагрева 10°C/мин. Сбор данных и их оценку осуществляют, используя программное обеспечение STARe.
- Термогравиметрический анализ (ТГМ).
Термогравиметрический анализ осуществляют, используя термогравиметрический анализатор Mettler Toledo SDTA851e. Образцы массой 3-4 мг взвешивают (используя микромасштабные весы MX5, Mettler) в открытые 40 мкл алюминиевые тигли, и нагревают со скоростью 10°C/мин от 30 до 300°C, в атмосфере азота (80 мл/мин). Сбор данных и их оценку осуществляют, используя программное обеспечение STARe.
- Протонный ядерный магнитный резонанс (1H-ЯМР).
Спектры протонного ядерного магнитного резонанса записывают в дейтерированном хлороформе или метаноле на спектрометре Bruker Avance 400 Ultrashield NMA, снабженном с z-градиентом 5 мм BBO (Broadband Observe) зондом с ATM и автоматическим BACS-120 автосэмплером. Спектры получают, растворяя 2-10 мг образца в 0,7 мл дейтерированного растворителя.
- Инфракрасная спектроскопия с преобразованием Фурье (FTIR).
FTIR спектры записывают, используя прибор Bruker Tensor 27, снабженный MKII с золотой решеткой однократного отражения (golden gate single reflection) ATR системой, источником излучения в среднем диапазоне инфракрасного излучения в качестве источника возбуждения и DTGS детектором. Спектры накапливают за 32 скана при разрешении 4 см-1. Для осуществления указанного анализа не требуется подготовки образцов.
Пример 1
Синтез 4-{2-[5-метил-1-(нафталин-2-ил)-1H-пиразол-3-илокси]этил}морфолина (соединение 63) и его гидрохлоридной соли (пример 1)
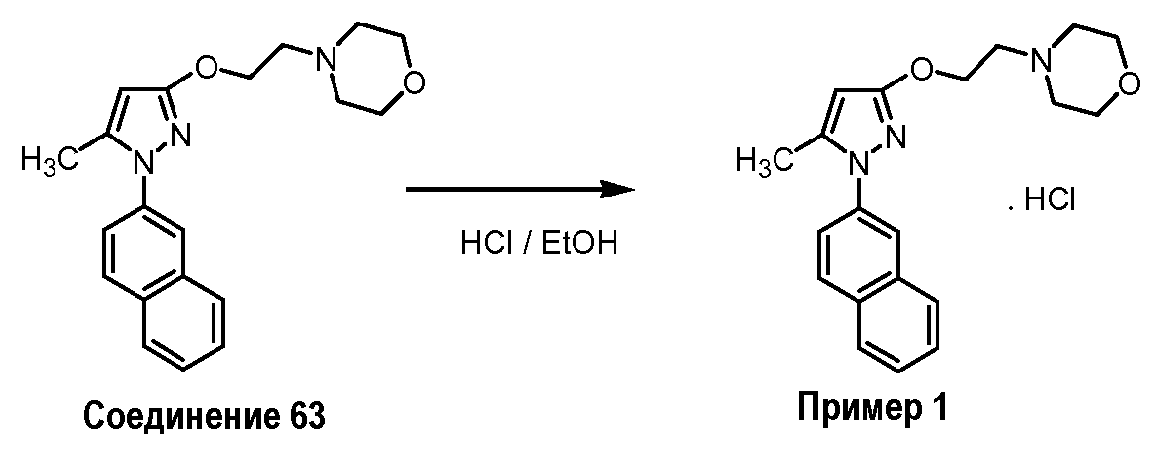
Соединение 63 можно получить способом, раскрытым в предыдущей заявке WO 2006/021462. Гидрохлорид соединения можно получить следующим способом:
Соединение 63 (6,39 г) растворяют в этаноле, насыщенном HCl, затем полученную смесь перемешивают в течение нескольких минут, и выпаривают досуха. Остаток кристаллизуют из изопропанола. Маточный раствор после первой кристаллизации обеспечивает возможность второй кристаллизации в результате концентрирования. В результате обеих кристаллизаций вместе получают 5,24 г (63%) соответствующей гидрохлоридной соли (Т.пл.=197-199°C).
1Н ЯМР (ДМСО-d6) δ м.д.: 10,85 (ушир.c, 1H), 7,95 (м, 4H), 7,7 (дд, J=2,2, 8,8 Гц, 1H), 7,55 (м, 2H), 5,9 (c, 1H), 4,55 (м, 2H), 3,95 (м, 2H), 3,75 (м, 2H), 3,55-3,4 (м, 4H), 3,2 (м, 2H), 2,35 (c, 3H).
Степень чистоты по данным ВЭЖХ составляет 99,8%.
Используя указанный способ, гидрохлоридную соль получают в виде кристаллического твердого вещества с очень хорошим выходом. Кроме того, его высокая температура плавления особенно удобна с фармацевтической точки зрения, так как это свойство обеспечивает хорошую физическую стабильность продукта.
Экстрагирование соединения 63 из его гидрохлоридной соли (пример 1)
Образцом, используемым в настоящем изобретении, является соединение примера 1. Основание (соединение 63) экстрагируют, используя CH2Cl2 из основного водного раствора (pH>10, используя 0,5 M водный раствор NaOH) соединения примера 1, получая масло оранжевого цвета.
Общий способ кристаллизации других солей соединения 63
Соли получают, вначале смешивая 1 мл 0,107 M раствора соединения 63, в виде полученного ранее масла оранжевого цвета (см. пример 1) в метаноле, с 1 мл 0,107 M раствора соответствующего противоиона в метаноле. Полученную смесь перемешивают в течение одного часа, и растворитель выпаривают в вакууме (Genevac, 8 мм рт.ст.), получая масло или твердое вещество белого цвета в зависимости от соли.
Продукт, полученный в начале процесса получения, растворяют в минимальном количестве кристаллизационного растворителя при его температуре кипения или максимально при температуре 75°C. Если после добавления 4 мл растворителя соль не растворяется полностью, суспензию перемешивают при высокой температуре в течение 30 минут, и остаток разделяют, используя горячее фильтрование или центрифугирование. Маточный раствор охлаждают до комнатной температуры и хранят в течение 24 часов.
Если образуется твердое вещество, его выделяют (используя фильтрование или центрифугирование). Если нет, раствор выдерживают в холодильнике (4°C) в течение нескольких дней. Если образуется твердое вещество, его выделяют из раствора. В том случае, если после всех указанных манипуляций твердое вещество не образуется, раствор оставляют выпариваться досуха.
Все полученные твердые вещества сушат в вакуумном термостате при 40°C (10 мм рт.ст.) в течение 4 часов, и, если получают достаточное количество, проводят анализы. Вначале осуществляют характеризацию, используя 1H-ЯМР для подтверждения получения соли. Использованные в настоящем изобретении растворители перечислены в таблице 1.
|
|
Кислоты, которые используют для исследования кристаллических солей соединения 63, выбирают в соответствии со следующими критериями (таблица 2):
- Кислоты, у которых pKa по меньшей мере на три единицы меньше, чем у соединения 63 (pKa 6,7).
- Кислоты, которые являются фармацевтически приемлемыми соединениями.
Хотя у некоторых из выбранных кислот возможны два или даже три (лимонная кислота) кислотных положения, в принципе, только серная кислота имеет второй кислотный протон достаточный для образования дисоли с соединением 63. Таким образом, всего существует одиннадцать различных солей, которые могут образоваться.
|
|
Общая стратегия, созданная для исследования кристаллических солей соединения 63, разделяется на три стадии:
Стадия 1: Скринирование по кристаллизации солей
Стадия 2: Оптимизация и характеризация солей
Стадия 3: Крупномасштабное получение выбранных солей
Вначале кристаллизационное скринирование осуществляют, используя выбранные противоионы, представленные в таблице 2, в поисках многообещающих кристаллических солей. Скринирование осуществляют в маленьком масштабе (40 мг соединения 63), используя широкий интервал кристаллизационных растворителей (таблица 1) и различные методики кристаллизации. При скринировании, условия кристаллизации строго не контролируются, и полученные твердые вещества характеризуют с помощью 1H-ЯМР. ЯМР спектроскопия является хорошим свидетельством образования солей, так как спектр 1H-ЯМР соли существенно отличается от спектра смеси кислоты и основания. Наблюдается четкий сдвиг сигналов, связанных с водородами, близкими к протонированному азоту. Кроме того, если кислотный противоион обладает характеристическим сигналом в спектре 1H-ЯМР, его можно идентифицировать, что позволяет определить стехиометрию соли и получить качественное представление о степени чистоты соли.
На второй стадии, все кристаллические соли получают в большем масштабе, в масштабе 100-500 мг в растворителях, которые показали наилучшие результаты во время процедуры скринирования. Кроме того, используют способ кристаллизации, который можно использовать для промышленного производства. Полученные соли полностью характеризуют, используя результаты 1H-ЯМР, ДСК, ТГМ и FTIR. Целью указанной стадии является, во-первых, создание процедуры с изменяющимся масштабом для получения выбранных солей с оптимизируемым выходом, и, во-вторых, полная характеризация полученных солей.
И, наконец, группу выбранных кристаллических солей с адекватными свойствами твердого состояния (кристалличность и термостабильность) получают в масштабе 2-3 г, начиная с соединения 63.
От кристаллизационного скринирования солей до получения солей в крупном масштабе (стадии 1-3)
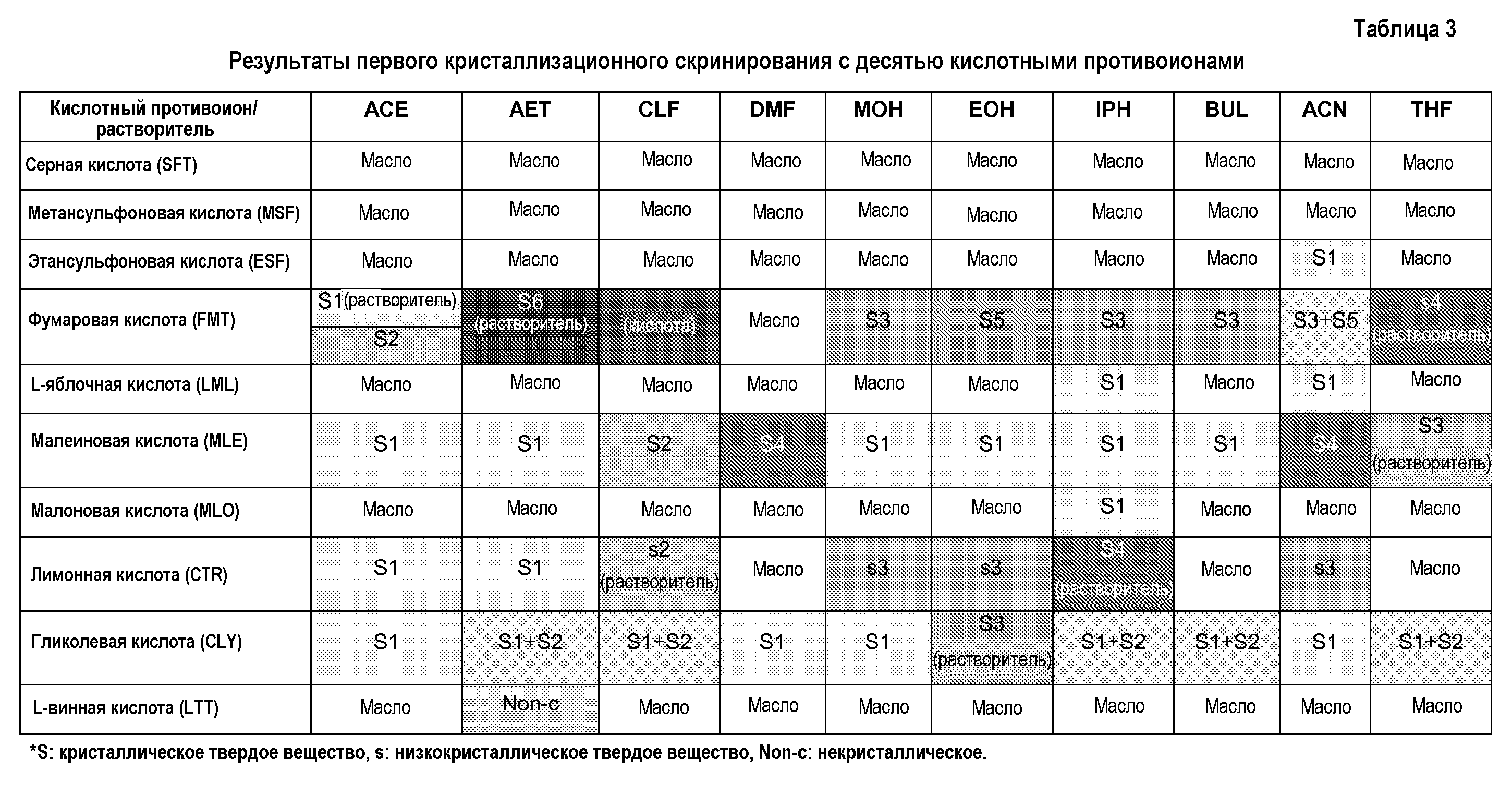
Вначале, кристаллизационное скринирование соединения 63 с десятью противоионами, представленными в таблице 2, осуществляют в масштабе 40 мг, в следующих десяти растворителях: в ацетоне, этилацетате, хлороформе, N,N-диметилформамиде, метаноле, этаноле, изопропаноле, 2-бутаноле, ацетонитриле и тетрагирофуране. Процедуру начинают с приготовления эквимолярных смесей, из известных концентраций метанольных растворов соединения 63 и различных противоинов кислот. Полученный сырой продукт после выпаривания метанола кристаллизуют из указанных ранее горячих растворителей. Используют различные стратегии кристаллизации в зависимости от растворимости смеси каждой кислоты и соединения 63, и поэтому твердые вещества получают, используя различные процедуры. Для некоторых кислот полученная смесь не растворяется в горячем кристаллизационном растворителе, образуя суспензию твердого вещества. В других случаях твердое вещество кристаллизуется при охлаждении раствора до комнатной температуры, или после нескольких дней хранения при 4°C или при -18°C. И, наконец, при некоторых попытках кристаллизации, твердое вещество образуется после медленного выпаривания растворителя при комнатной температуре. В некоторых случаях получают более одного твердого вещества за попытку кристаллизации.
Из указанного первого кристаллизационного скринирования (таблица 3), можно сделать следующие наблюдения:
- Кристаллические соли соединения 63 с фумаровой и малеиновой кислотами получают в большинстве анализированных растворителей. Для обоих кислотных противоионов получают несколько кристаллических твердых веществ, включая сольваты. Все твердые вещества соответствуют эквимолярным солям.
- Эквимолярная смесь соединения 63 и лимонной кислоты оказывается очень хорошо растворимой в подавляющем большинстве исследованных растворителей. Поэтому, большую часть твердых веществ получают после завершения выпаривания растворителя. Кроме того, полученные твердые вещества отличаются низкой кристалличностью или содержат значительные количества остаточных растворителей. Наиболее вероятно, что низкокристаллические твердые вещества образуются из десольватированных сольватов.
- Эквимолярная смесь соединения 63 и гликолевой кислоты оказалась очень хорошо растворимой в подавляющем большинстве исследованных растворителей. Поэтому, большинство твердых веществ получают после завершения выпаривания растворителя, и некоторые представляют собой смеси твердых веществ.
- Кристаллические соли соединения 63 с этансульфоновой, L-яблочной и малоновой кислотами получают только в одном или в двух из анализированных растворителей в условиях очень высоких концентраций. Большинство твердых веществ получают после завершения выпаривания растворителя.
- Не было получено кристаллических твердых веществ соединения 63 с серной, метансульфоновой и L-винной кислотами. Смеси основания и кислоты оказались хорошо растворимы во всех анализированных растворителях, и после завершения выпаривания растворителя получают или масла или некристаллические твердые вещества.
С учетом указанных результатов второе кристаллизационное скринирование осуществляют в девяти дополнительных растворителях. Менее полярные растворители (изобутилацетат, диметилкарбонат, хлорбензол, циклогексан, 3-пентанон, толуол, метил-трет-бутиловый эфир, диизопропиловый эфир) и воду выбирают для снижения растворимости солей (таблица 4).
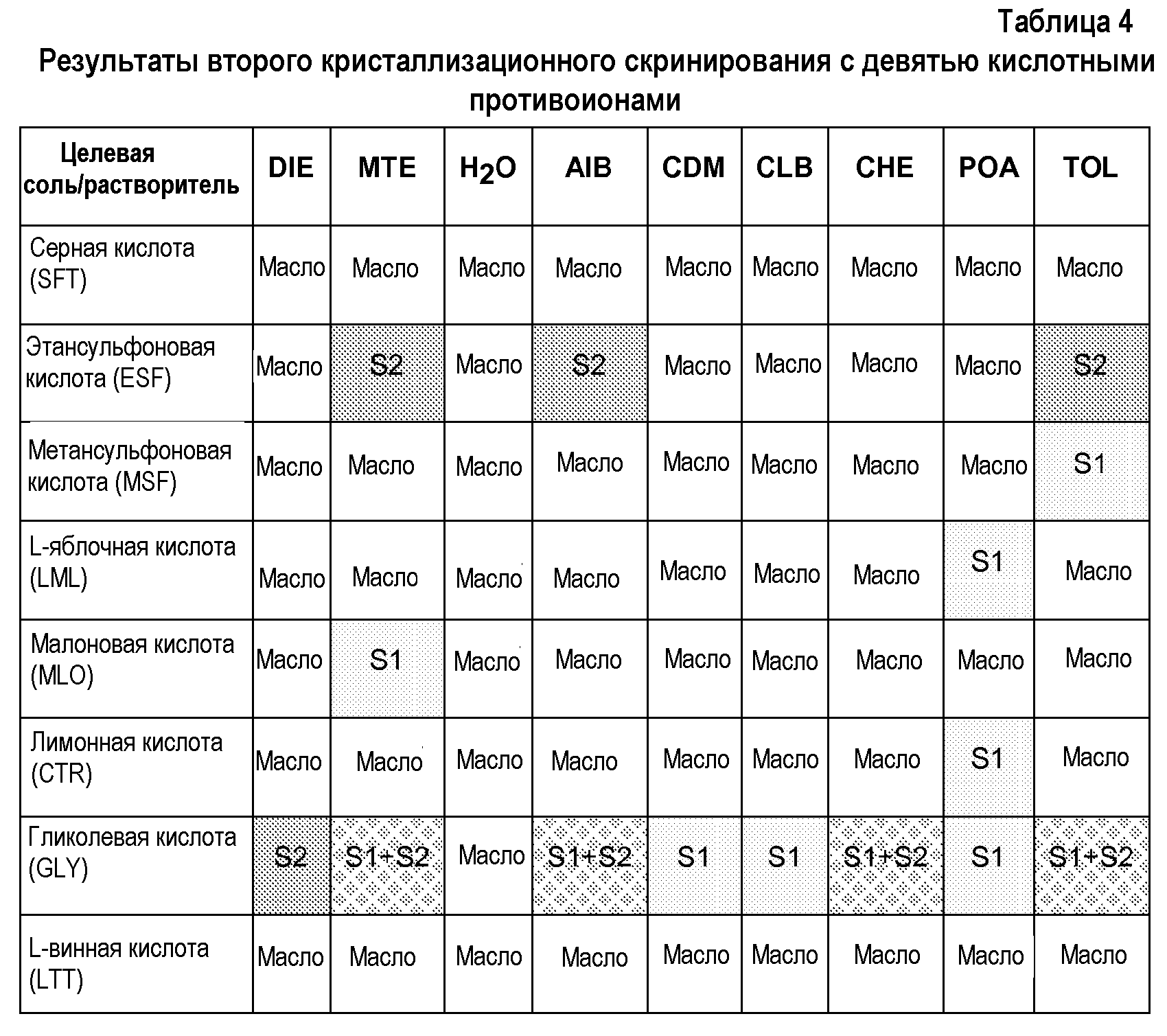
Из указанного второго кристаллизационного скринирования можно сделать следующие наблюдения:
- Хотя эквимолярная смесь соединения 63 и гликолевой кислоты оказалась менее растворимой в указанном втором наборе растворителей, ее поведение было весьма близко к результатам для первого блока кристаллизаций. Были получены несколько твердых веществ, соответствующих смесям твердых веществ. Твердое вещество 1 оказалось единственным, полученным после завершения выпаривания растворителя, и его не смогли полностью охарактеризовать.
- Кристаллические соли соединения 63 с L-яблочной, малоновой и лимонной кислотами получают только в одном растворителе, получая уже известное твердое вещество.
- Кристаллические соли соединения 63 с этансульфоновой кислотой получают в нескольких растворителях, получая, во всех случаях, новое твердое вещество, отличающееся от полученного в начальном кристаллизационном скринировании.
- Твердое вещество, соответствующее кристаллической соли соединения 63 с метансульфоновой кислотой, можно получить в толуоле.
- В указанном втором наборе растворителей не получают кристаллических твердых веществ соединения 63 с серной и L-винной кислотами.
Учитывая результаты двух описанных кристаллизационных скринирований, авторы оптимизировали получение наилучшим образом охарактеризованных не сольватированных солей соединения 63 с фумаровой, малеиновой, метансульфоновой, этансульфоновой, L-яблочной и малоновой кислотами. Оптимизацию экспериментов в увеличенных масштабах осуществляют, начиняя со 100 мг соединения 63. Процедуру в увеличенном масштабе также оптимизируют для солей фумаровой, малеиновой, метансульфоновой, этансульфоновой, L-яблочной и малоновой кислот.
Наконец, получение солей для шести выбранных противоионов осуществляют в масштабе 2-3 г, и они были полностью охарактеризованы. Общий процесс настоящего изобретения суммирован в следующей таблице.
|
Пример 2
Получение фумаратной соли соединения 63
Во время начального скринирования кристаллизации фумаратной соли были использованы в 10 различных растворителях. Кристаллические твердые вещества, соответствующие соли, получают во всех растворителях, за исключением ДМФ и хлороформа, используя различные кристаллизационные методики: суспендирование, охлаждение насыщенного раствора или после завершения выпаривания растворителя. В хлороформе исходную кислоту выделяют, тогда как в ДМФ соль выделяют в виде масла оранжевого цвета. Получают два несольватированных твердых вещества, первое вещество в метаноле, изопропаноле и бутаноле, и второе вещество только в этаноле. Наконец, сольваты получают в ацетоне, этилацетате и ТГФ, и смесь двух твердых веществ образуется в ацетонитриле.
Несольватируемое кристаллическое твердое вещество, в принципе любое из полученных при скринировании, выбирают для крупномасштабного производства. Вначале была предпринята попытка увеличения масштаба процесса в ацетонитриле, так как ацетонитрил оказался растворителем, который обеспечивает получение кристаллического продукта, в котором соль менее растворима. Хотя соль получают с очень хорошим выходом (83%), указанный процесс не является оптимальным для крупномасштабного процесса, так как кислота не растворяется в ацетонитриле, и конечная соль осаждается из смеси соединения 63 в виде масла и фумаровой кислоты в виде твердого вещества, причем оба суспендированы в растворителе. Затем предпринимают попытку кристаллизации в этаноле для получения чистого твердого вещества S5. К большому огорчению выяснилось, что при крупномасштабном процессе в этаноле, новое, малокристаллическое твердое вещество образуется с низким выходом. Наконец, кристаллизацию осуществляют в ацетонитриле, добавляя кислоту, растворенную в спирте (этаноле или изопропаноле). Несколько лучшие результаты получены в случае, когда фумаровую кислоту растворяют в этаноле и добавление осуществляют при комнатной температуре (таблица 6). С другой стороны, смесь фаз получают, если суспензию выдерживают при 4°C в течение двух дней (таблица 6, пункт 4).
|
Экспериментальная процедура, которую используют для получения фумаратной соли в масштабе 0,5 г (пункт 2 в таблице 6) является следующей:
Раствор фумаровой кислоты (153 мг, 1,32 ммоль) в 2 мл этанола медленно добавляют к раствору соединения 63 (456 мг, 1,35 ммоль) в 5 мл ацетонитрила при комнатной температуре. В полученный раствор желтого цвета вводят затравку и перемешивают при комнатной температуре в течение 15 минут. Быстро выпадает обильный твердый осадок белого цвета. Полученную суспензию перемешивают при комнатной температуре в течение 16 часов. Полученное твердое вещество отфильтровывают, промывают 1 мл ацетонитрила и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 6 часов, получая фумаратную соль в виде твердого вещества белого цвета (350 мг, 59%).
Образование солей можно легко охарактеризовать, используя результаты 1H-ЯМР спектров, которые существенно изменяются по сравнению со спектром свободного основания. В случае фумаратной соли сигналы атомов водорода близких к основному азоту (водороды 1 и 2 в приведенной далее формуле) отчетливо сдвинуты в слабое поле (таблица 7). Меньшие сдвиги можно также наблюдать для сигналов атомов водорода, расположенных дальше от азота (водороды 3 и 4 в приведенной далее формуле). Кроме того, сигнал для фумарата появляется с ожидаемым химическим сдвигом (δ: 6,72 м.д.). Объединение сигналов, соответствующих аниону и катиону однозначно подтверждает, что образуется эквимолярная соль, а не дисоль (фиг.5).
Молекулярная формула соединения 63 с указанием водородов, сдвиги которых изменяются в спектрах 1H-ЯМР после образования соли.
ДСК анализ при скорости нагревания 10°C/мин, демонстрирует небольшой эндотермический пик, после которого следует небольшой экзотермический пик и интенсивный эндотермический сигнал (фиг.6). Интенсивный сигнал, возникающий при 142°C, соответствует температуре плавления твердого вещества S5. Небольшой пик, появляющийся при 131°C, соответствует плавлению кристаллического твердого вещества S3. Указанный пик очень слабый, наиболее вероятно из-за того, что S3 частично трансформируется в твердое вещество S5 в процессе нагревания при проведении ДСК анализа. Таким образом, указанный пик соответствует плавлению остального S3, оставшегося при температуре плавления, которое легко кристаллизуется в S5 (небольшой экзотермический пик). Пик, соответствующий плавлению практически чистых твердых S3 образцов, имеет различные интенсивности в зависимости от конкретного образца. Наиболее вероятно, что переход твердого вещества S3 в твердое вещество S5 происходит в различной степени в зависимости от формы кристаллов и размеров кристаллов. Поэтому, образцы чистого S3 кристаллического твердого вещества демонстрируют ДСК профили, форма которых представлена на фиг.6.
При проведении ТГ анализа наблюдается небольшая потеря веса в 0,3% при температурах между 120 и 150°C, и наблюдается резкая потеря веса, начинающаяся при 190°C за счет разложения.
Далее приводится характеризация фумаратной соли (фиг.5-8):
1Н-ЯМР (400 МГц, d4-метанол) δ: 2,35 (c, 3H), 2,92-3,00 (м, 4H), 3,17 (т, J=5 Гц, 2H), 3,80 (т, J=5 Гц, 4H), 4,44 (т, J=5 Гц, 2H), 5,83 (c, 1H), 6,72 (c, 2H), 7,52-7,62 (м, 3H), 7,89-7,96 (м, 3H), 8,00 (д, J=9 Гц, 1H).
Остаточные растворители по данным 1H-ЯМР: 0,2% вес/вес ацетонитрила.
FTIR (ATR) ν: 3435, 3148, 3037, 2943, 2855, 1876, 1731, 1664, 1650, 1559, 1509, 1488, 1446, 1394, 1372, 1314, 1236, 1186, 1166, 1133, 1098, 1081, 1047, 1014, 981, 932, 917, 859, 816, 787, 769 и 748 см-1.
ДСК (10°C/мин): Два эндотермических пика слияния, возникающие при 131 и 142°C.
ТГМ (10°C/мин): Потеря веса 0,3% при температуре между 120 и 150°C. Процесс разложения начинается при 190°C.
Пример 3
Получение малеатной соли соединения 63
Во время начального скринирования попытки кристаллизации малеатной соли были предприняты в 10 различных растворителях. Указанная соль очень хорошо растворяется во всех анализированных растворителях. Наблюдаются растворимости в интервале от 50 и 200 мг/мл, за исключением варианта этилацетата, растворимость соли в котором составляет 20 мг/мл. Кристаллические твердые вещества получают во всех растворителях после охлаждения раствора до комнатной температуры или, для хлороформа, метанола и ДМФ, после завершения выпаривания растворителя. Были детектированы четыре различных твердых вещества. Несольватированную кристаллическую фазу получают в большинстве вариантов кристаллизации. Кроме того, сольват образуется в ТГФ и два других не полностью охарактеризованных твердых вещества образовались в трех экспериментах.
Учитывая температуры кипения и необходимые количества растворителя для кристаллизации (66 мг/мл), в качестве растворителя для увеличения масштаба и синтеза кристаллической соли выбирают изопропанол. Начальная попытка охлаждения смеси малеиновой кислоты и соединения 63 в изопропаноле с 60°C до комнатной температуры приводит к получению соли в виде масла (таблица 7). Полученное масло кристаллизуется после нового перемешивания смеси при 60°C в течение нескольких часов. Аналогичная методика в более разбавленных условиях приводит к получению соли непосредственно в виде твердого вещества. Наконец, процесс оптимизируют, вызывая непосредственное осаждение соли после добавления изопропанольного раствора кислоты к изопропанольному раствору соединения 63 при комнатной температуре.
|
Экспериментальная процедура, которую используют для получения малеатной соли в масштабе 2,5 г была следующей:
Раствор малеиновой кислоты (772 мг, 6,65 ммоль) в 15 мл изопропанола медленно добавляют к раствору соединения 63 (2,26 г, 6,69 ммоль) в 15 мл изопропанола при комнатной температуре. Быстро осаждается большое количество белого твердого вещества. Полученную суспензию перемешивают при комнатной температуре в течение 2 дней и фильтруют. Полученное твердое вещество промывают изопропанолом и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 10 часов, при 55°C в течение 6 часов и при 70°C в течение 17 часов, получая малеатную соль в виде белого твердого вещества (2,82 г, 96%; содержащую 1,1% изопропанола по данным 1H-ЯМР).
Полученную малеатную соль можно легко охарактеризовать, используя спектр 1H-ЯМР (фиг.9), изменения в котором аналогичны изменениям, подробно раскрытым для фумаратной соли. Кроме того, сигнал, характеризующий малеат, появляется с ожидаемым химическим сдвигом 6,30 м.д. Интегрирование сигналов, соответствующих аниону и катиону, однозначно подтверждает тот факт, что образуется эквимолярная соль, а не дисоль.
ДСК анализ (фиг.10), при скорости нагревания 10°C/мин, демонстрирует эндотермический интенсивный пик, возникающий при 139°C (101 Дж/г), соответствующей температуре плавления. В ТГМ наблюдается потеря веса в 1% (фиг.11) вблизи температуры плавления, что, вероятно, связано с удалением остаточного изопропанола. Отчетливое разложение соли наблюдается при температурах выше 150°C.
Далее приводятся характеристики малеатной соли (фиг.9-12):
1Н-ЯМР (400 МГц, d-хлороформ) δ: 2,35 (c, 3H), 3,02-3,64 (м, 6H), 3,99 (т, J=5 Гц, 4H), 4,61-4,66 (м, 2H), 5,70 (c, 1H), 6,30 (c, 2H), 7,50-7,58 (м, 3H), 7,79-7,82 (м, 1H), 7,84-7,95 (м, 3H).
Остаточные растворители по данным 1H-ЯМР: 1,1% вес/вес изопропанола.
FTIR (ATR) ν: 3043, 2853, 1707, 1619, 1599, 1557, 1487, 1445, 1374, 1357, 1340, 1302, 1237, 1163, 1135, 1096, 1041, 1022, 930, 919, 861, 817, 762 и 750 см-1.
ДСК (10°C/мин): Эндотермический пик слияния возникает при 139°C.
ТГМ (10°C/мин): Потеря веса 1,0% при температуре между 110 и 150°C. Процесс разложения начинается при 150°C.
Пример 4
Получение метансульфонатной соли соединения 63
Во время начального скринирования с использованием первого набора из десяти растворителей, метансульфонатную соль не удалось закристаллизовать. Соль хорошо растворяется во всех анализированных растворителях (>200 мг/мл), образуя масло после завершения выпаривания растворителя. Когда осуществляли попытки закристаллизовать соль во втором наборе из девяти менее полярных растворителей, в большинстве экспериментов также выделялись масла, или после выпаривания растворителя, или потому, что маслянистая соль не растворялась. Тем не менее, из толуольного раствора, охлажденного до -18°C, было получено соответствующее соли кристаллическое твердое вещество после выделения избытка соли в виде масла. Поэтому, толуол был выбран для оптимизации крупномасштабного синтеза соли.
В первой попытке крупномасштабного синтеза, метансульфоновую кислоту добавляют непосредственно к толуольному раствору соединения 63, но соль быстро выделяется в виде масла. Полученное масло кристаллизуется после того, как его перемешивают вместе с растворителем в течение нескольких часов при комнатной температуре. Для инициирования непосредственной кристаллизации соли в виде твердого вещества, тот же самый процесс повторяют в присутствии затравочных кристаллов указанной соли. Кроме того, для улучшения цвета соли метансульфоновую кислоту перегоняют непосредственно перед использованием (180°C, 1 миллибар).
Далее приводится экспериментальная процедура, которую используют для получения метансульфонатной соли в масштабе 2,5 г:
Метансульфоновую кислоту (0,45 мл, 6,94 ммоль) медленно добавляют к раствору соединения 63 (2,36 г, 6,98 ммоль) в 25 мл толуола при комнатной температуре в присутствии затравки. Быстро осаждается большое количество твердого вещества белого цвета. Полученную суспензию перемешивают при 0°C в течение 8 часов и затем фильтруют. Полученное твердое вещество промывают толуолом и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 2 дней и при 55°C в течение 6 часов, получая метансульфонатную соль в виде твердого вещества белого цвета (2,85 г, 98%; по данным 1H-ЯМР содержит 0,6% толуола).
Метансульфонатную соль можно легко охарактеризовать с помощью 1H-ЯМР спектра (фиг.13), который изменяется таким же образом, который был подробно раскрыт для фумаратной соли. Кроме того, сигнал метансульфоната появляется в спектре со значением химического сдвига 2,84 м.д.
ДСК анализ (фиг.14), при скорости нагревания 10°C/мин, демонстрирует эндотермический интенсивный пик, который появляется при 145°C (84 Дж/г), что соответствует температуре плавления. В ТГМ наблюдается потеря веса в 0,5% (фиг.15) вблизи температуры плавления, что вероятно связано с удалением остаточного толуола. Отчетливое разложение соли наблюдается при температурах выше 250°C.
Далее приводятся следующие характеристики метансульфонатной соли (фиг.13-16):
1Н-ЯМР (400 МГц, d-хлороформ) δ: 2,36 (c, 3H), 2,84 (c, 3H), 3,03-3,15 (м, 2H), 3,54-3,61 (м, 2H), 3,63-3,71 (м, 2H), 3,97-4,05 (м, 2H), 4,10-4,20 (м, 2H), 4,71-4,76 (м, 2H), 5,75 (c, 1H), 7,50-7,59 (м, 3H), 7,79-7,82 (м, 1H), 7,84-7,95 (м, 3H).
Остаточный растворитель толуол по данным 1H-ЯМР: 0,58% вес/вес.
FTIR (ATR) ν: 3018, 2957, 2920, 2865, 2693, 2627, 1634, 1602, 1562, 1509, 1485, 1435, 1392, 1376, 1265, 1221, 1164, 1131, 1098, 1049, 1033, 1007, 934, 914, 862, 822, 772 и 759 см-1.
ДСК (10°C/мин): Эндотермический пик слияния возникает при 145°C.
ТГМ (10°C/мин): Потеря веса 0,5% при температуре между 120 и 160°C. Процесс разложения начинается при 260°C.
Пример 5
Получение этансульфонатной соли соединения 63
Во время начального скринирования с первым набором из десяти растворителей этансульфонатную соль удалось закристаллизовать только в ацетонитриле. Но так как указанная соль очень хорошо растворяется во всех анализированных растворителях (>200 мг/мл) указанное твердое вещество получают только после завершения выпаривания растворителя. В остальных экспериментах после завершения выпаривания растворителя образуется масло. При осуществлении попыток кристаллизации во втором наборе из девяти менее полярных растворителей, получают три твердых вещества в метил-трет-бутиловом эфире, изобутилацетате и толуоле в смеси с маслянистой солью. В указанных экспериментах маслянистая соль полностью не растворялась. Толуол выбирают для оптимизации и синтеза соли в более крупном масштабе.
В начале крупномасштабного получения этансульфоната, маслянистую соль суспендируют в горячем толуоле и оставляют охлаждаться. Указанная соль не кристаллизуется и остается в виде масла. При второй попытке, в которой этансульфоновую кислоту медленно добавляют к раствору соединения 63 в толуоле, при охлаждении выделяется твердое вещество коричневого цвета. При повторении той же процедуры при комнатной температуре быстро образующееся масло медленно кристаллизуется после того, как его перемешивают вместе с растворителем в течение нескольких дней. Для того чтобы вызвать непосредственную кристаллизацию соли, тот же самый процесс повторяют при комнатной температуре в присутствии затравочных кристаллов соли. Кроме того, для улучшения цвета соли этансульфоновую кислоту перегоняют непосредственно перед использованием (200°C, 1 миллибар).
Далее приводится экспериментальная процедура, которую используют для получения этансульфонатной соли в масштабе 2,5 г:
Этансульфоновую кислоту (0,58 мл, 6,79 ммоль) медленно добавляют к раствору соединения 63 (2,29 г, 6,79 ммоль) в 40 мл толуола при комнатной температуре в присутствии затравочных кристаллов. Твердое вещество белого цвета быстро в большом количестве выпадает в осадок. Полученную суспензию перемешивают при 0°C в течение 12 часов и затем фильтруют. Полученное твердое вещество промывают толуолом и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 8 часов и при 55°C в течение 6 часов, получая этансульфонатную соль в виде твердого вещества белого цвета (2,90 г, 99%).
Образование этансульфонатной соли можно легко доказать, используя спектр 1H-ЯМР (фиг.17) изменения в котором при сравнении со спектром исходного соединения 63, происходят таким же образом, который был раскрыт подробно для фумаратной соли. Кроме того, сигналы, соответствующие этансульфонату, появляются в спектре с химическим сдвигом 1,37 и 2,93 м.д.
ДСК анализ (фиг.18), при скорости нагревания 10°C/мин, демонстрирует эндотермический интенсивный пик, который возникает при 133°C (85 Дж/г), что соответствует температуре плавления. Потеря веса в 0,3% наблюдается в ТГМ (фиг.19) вблизи температуры плавления, что, вероятно, связано с удалением остаточного толуола. Четкое разложение соли наблюдается при температурах выше 280°C.
Характеристики этансульфонатной соли (фиг.17-20):
1Н-ЯМР (400 МГц, d-хлороформ) δ: 1,37 (т, J=7 Гц, 3H), 2,36 (c, 3H), 2,93 (кв, J=1 Гц, 2H), 3,03-3,15 (м, 2H), 3,55-3,62 (м, 2H), 3,64-3,72 (м, 2H), 3,96-4,04 (м, 2H), 4,11-4,21 (м, 2H), 4,71-4,77 (м, 2H), 5,75 (c, 1H), 7,50-7,59 (м, 3H), 7,79-7,83 (м, 1H), 7,84-7,95 (м, 3H).
Остаточный растворитель по данным 1H-ЯМР: 0,35% вес/вес.
FTIR (ATR) ν: 3021, 2958, 2924, 2863, 2625, 2488, 1633, 1603, 1565, 1508, 1485, 1470, 1437, 1391, 1376, 1353, 1334, 1265, 1242, 1210, 1160, 1149, 1131, 1098, 1027, 1008, 978, 934, 916, 856, 819, 776 и 739 см-1.
ДСК (10°C/мин): Эндотермический пик слияния возникает при 133°C.
ТГМ (10°C/мин): Потеря веса в 0,3% при температурах между 110 и 160°C. Процесс разложения начинается при 280°C.
Пример 6
Получение малатной соли соединения 63
Во время начального скринирования с использованием первого набора из десяти растворителей малатную соль можно было закристаллизовать в ацетонитриле и изопропаноле. Тем не менее, указанная соль очень легко растворялась в обоих растворителях (>200 мг/мл) и указанные два твердых вещества получают только после полного выпаривания растворителя. В оставшихся экспериментах после завершения выпаривания растворителя образуется масло. При осуществлении попыток кристаллизации во втором наборе из девяти менее полярных растворителей, хотя соль и была менее растворимой, кристаллическое твердое вещество удалось получить только в 3-пентаноне. В остальных экспериментах удалось получить только масло. Учитывая полученные результаты, 3-пентанон выбирают для оптимизации и синтеза указанной соли в более крупном масштабе.
Начальные попытки получения солей в более крупном масштабе осуществляют, добавляя раствор L-яблочной кислоты в 3-пентаноне к раствору соединения 63 также в 3-пентаноне при температурах между 50 и 70°C. При использовании указанной процедуры соль иногда выделяется в виде масла при охлаждении. Полученное масло легко кристаллизуется после того, как его перемешивают вместе с растворителем при 50°C в течение нескольких часов. Непосредственное получение кристаллической соли можно вызвать введением затравочных кристаллов, как это раскрыто для процедуры получения малатной соли в масштабе 2,5 г, следующим образом:
Раствор L-яблочной кислоты (933 мг, 6,95 ммоль) в 10 мл 3-пентанона медленно добавляют к раствору соединения 63 (2,35 г, 6,95 ммоль) в 10 мл 3-пентанона при 50°C, используя затравочные кристаллы. Быстро осаждается большое количество твердого вещества белого цвета, и полученную суспензию разбавляют другой порцией в 10 мл 3-пентанона, медленно охлаждают до комнатной температуры, перемешивают в течение 12 часов и фильтруют. Полученное твердое вещество промывают 3-пентаноном и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 15 часов и при 55°C в течение 6 часов, получая малатную соль в виде твердого вещества белого цвета (3,03 г, 95%).
Образование малатной соли можно легко доказать, используя спектр 1H-ЯМР (фиг.21), который значительно изменяется по сравнению со спектром исходного соединения 63, как было подробно раскрыто для фумаратной соли. Кроме того, сигналы малата появляются в спектре с химическими сдвигами 2,59, 2,79 и 4,31 м.д.
В ДСК анализе (фиг.22), при скорости нагревания 10°C/мин, эндотермический интенсивный пик наблюдается при 125°C (119 Дж/г), что соответствует температуре плавления.
Кроме того, ТГМ анализ (фиг.23) не демонстрирует никаких потерь веса при температурах ниже температуры плавления, что свидетельствует об отсутствии летучих компонент. Отсутствие остаточных растворителей также подтверждается данными спектра 1H-ЯМР.
Далее приводятся характеристики малатной соли (фиг.21-24):
1Н-ЯМР (400 МГц, d4-метанол) δ: 2,35 (c, 3H), 2,59 (дд, J 1 =16 Гц, J 2 =7 Гц, 1H), 2,79 (дд, J 1 =16 Гц, J 3 = 5 Гц, 1H), 2,89-2,97 (м, 4H), 3,13 (т, J= 5 Гц, 2H), 3,80 (т, J=5 Гц, 4H), 4,39 (дд, J 2 =7 Гц, J 3 =5 Гц, 1H), 4,43 (т, J=5 Гц, 2H), 5,83 (c, 1H), 7,52-7,61 (м, 3H), 7,89-7,96 (м, 3H), 8,00 (д, J=9 Гц, 1H).
FTIR (ATR)ν: 3171, 3003, 2874, 1718, 1597, 1556, 1487, 1468, 1440, 1360, 1268, 1142, 1126, 1097, 1050, 1022, 1010, 986, 950, 920, 902, 863, 822, 797, 770, 746 и 742 см-1.
ДСК (10°C/мин): Эндотермический пик слияния возникает при 125°C.
ТГМ (10°C/мин): Потеря веса начинается с температуры 150°C в связи с разложением.
Пример 7
Получение малонатной соли соединения 63
Во время начального скринирования с использованием первого набора из десяти растворителей малонатную соль удалось закристаллизовать только в изопропаноле. Тем не менее, указанная соль оказалась очень хорошо растворимой в указанном растворителе (>200 мг/мл), что предвещало проблемы при увеличении масштабов синтеза. По этой причине была предпринята попытка кристаллизации во втором наборе из девяти менее полярных растворителей. В этой второй серии экспериментов, кристаллическое твердое вещество удалось получить только из метил-трет-бутилового эфира при охлаждении насыщенного раствора до -18°C после выделения, при более высоких температурах большая часть соли представляет собой масло.
Учитывая полученные результаты, вначале предпринимают попытку получения малонатной соли в большем масштабе. К большому разочарованию сразу после смешивания кислоты и соединения 63 сразу выделяется масло. Полученное масло кристаллизуется с малым выходом после того, как его перемешивают вместе с растворителем в течение нескольких часов. Выход можно повысить, если добавлять метил-трет-бутиловый эфир во время процесса кристаллизации после “вымасливания” (oiling out). Чтобы избежать образования соли вначале в виде масла и чтобы повысить выход, процесс кристаллизации модифицируют. Раствор малоновой кислоты в изопропаноле добавляют к раствору соединения 63 в метил-трет-бутиловом эфире. Если использовать указанную процедуру, соль образуется непосредственно в виде твердого вещества, но при этом все еще наблюдается некоторое вымасливание. И, наконец, непосредственное и полное образование соли можно обеспечить, используя затравочные кристаллы, как это раскрыто в следующей процедуре:
Раствор малоновой кислоты (736 мг, 7,07 ммоль) в 10 мл изопропанола медленно добавляют к раствору соединения 63 (2,38 г, 7,06 ммоль) в 15 мл метил-трет-бутилового эфира с введенными затравочными кристаллами при 0°C. Быстро выпадает в осадок большое количество твердого вещества белого цвета. Полученную суспензию перемешивают вначале при комнатной температуре в течение 12 часов, затем при 0°C в течение 2 часов, и фильтруют. Полученное твердое вещество промывают метил-трет-бутиловым эфиром и сушат в вакууме (10 мм рт.ст.) при 45°C в течение 7 часов и при 55°C в течение 6 часов, получая малонатную соль в виде твердого вещества белого цвета (2,42 г, 80%). Образование малонатной соли можно легко доказать, используя спектр 1H-ЯМР (фиг.25), который изменяется по сравнению со спектром исходного соединения 63, таким же образом, как было подробно раскрыто для фумаратной соли. Кроме того, сигнал, связанный с малонатом, появляется в спектре с химическим сдвигом 3,23 м.д.
ДСК анализ (фиг.26), при скорости нагревания 10°C/мин, демонстрирует эндотермический интенсивный пик, который возникает при 90°C (85 Дж/г), что соответствует температуре плавления. Потери веса не наблюдают в ТГМ (фиг.27) при температурах ниже температуры плавления. Тем не менее, в спектре 1H-ЯМР определяются примеси (0,2% вес/вес изопропанола и 0,2% метил-трет-бутилового эфира).
Далее приводятся характеристики малонатной соли (фиг.25-28):
1Н-ЯМР (400 МГц, d-хлороформ) δ: 2,35 (c, 3H), 3,10-3,40 (м, 4H), 3,23 (c, 2H), 3,40-3,46 (м, 2H), 3,97 (т, J=5 Гц, 4H), 4,59-4,64 (м, 2H), 5,70 (c, 1H), 7,49-7,58 (м, 3H), 7,79-7,82 (м, 1H), 7,84-7,95 (м, 3H).
Остаточными растворителями по данным 1H-ЯМР являются: 0,2% вес/вес изопропанола и 0,2% метил-трет-бутилового эфира.
FTIR (ATR) ν: 3148, 3027, 2942, 2857, 1718, 1621, 1599, 1561, 1488, 1443, 1374, 1343, 1308, 1260, 1165, 1135, 1097, 1080, 1046, 1022, 1011, 932, 918, 863, 819 и 752 см-1.
ДСК (10°C/мин): Эндотермический пик слияния возникает при 90°C.
ТГМ (10°C/мин): Потери веса начинаются со 100°C, что обусловлено разложением.
Результаты скринирования кристаллизации солей
Попытки получения солей соединения 63 с серной кислотой и L-винной кислотой окончились неудачей, и получались только масла. Другие соли, хотя и в форме твердых веществ, были получены только в результате комплексных процессов синтеза при сравнении их с экспериментальной частью для синтеза гидрохлорида, или в уникальных экспериментальных условиях. Кроме того, часто получали некристаллическое твердое вещество вместо кристаллической формы, которую получали для гидрохлорида. Все указанные недостатки подразумевают, что увеличение масштаба синтеза для указанных синтетических процессов обещает быть весьма сложным.
В приводимой далее таблице 8 суммированы ключевые результаты, относящиеся к каждой твердой соли, полученной в крупном масштабе в рассматриваемом изобретении: степень кристалличности, кристаллизационный растворитель, выход и температура плавления.
|
Как видно из вышеуказанного, гидрохлоридная соль всегда образуется в виде кристаллического твердого вещества с очень хорошим выходом (включая кристаллизацию) и его температура плавления выше на 50°C по сравнению с температурами плавления других солей, что четко обеспечивает преимущество в плане физической стабильности. Кроме того, при сравнении ТГМ анализов гидрохлорид имеет четкий профиль и не наблюдается никаких потерь растворителя. Далее, некоторые дополнительные эксперименты (термодинамическая растворимость, фармакинетика) были проведены для примера 1 (P027), чтобы подтвердить применимость указанного соединения для фармацевтических целей.
Пример 8
Термодинамическая растворимость
Общий протокол для термодинамической растворимости при pH 7,4 и pH 2 раскрыт далее.
A) Термодинамическая растворимость при pH 7,4
pH буфера 7,4 (50 мМ).
Буферные фосфаты с pH 7,4 приготавливают следующим образом:
- Приготавливают раствор 25 мМ Na2HPO4·12H2O (для 1 л воды, вес 8,96 г).
- Приготавливают раствор 25 мМ KH2PO4 (для 1 л воды, вес 3,4 г).
- Смешивают 812 мл раствора динатрийфосфата + 182 мл раствора калийфосфата, и соответствующее значение pH составляет 7,4.
Равновесие образцов
Образцы уравновешивают, используя:
- Термомиксер Эппендорфа (Stirrer Thermomixer Control of Eppendorf) 25°C и 1250 об/мин.
- pH-метр с комбинированным электродом pH, полумикро.
Процедура
Исследуемое соединение
Отвешивают 2 мг в ампулу для ВЭЖХ (в двойном экземпляре) и добавляют 1 мл буфера. Температуру ампулы поддерживают при 25°C, в термомиксере Thermomixer Comfort с перемешиванием, в течение 24 часов. Центрифугирование при 4000 об/мин проводят в течение 15 минут.
Образовавшийся верхний слой собирают стеклянной пипеткой и переносят в ампулы для ВЭЖХ. Снова центрифугируют, и инжектор программируют на 2,7 мм выше.
Стандарты (в дубликате)
Раствор A: 2 мг в 5 мл метанола (400 мкг/мл).
Раствор B: 1 мл раствора A до 10 мл с метанолом (40 мкг/мл).
Раствор C: 5 мл раствора B до 50 мл с метанолом (4 мкг/мл).
Раствор D: 4 мл раствора C до 10 мл с метанолом (1,6 мкг/мл).
Раствор E: 5 мл раствора D до 25 мл с метанолом (0,32 мкг/мл).
10 мкл всех приготовленных растворов инъектируют, начиная с более разбавленного стандарта. Инъектируют также плацебо для проверки отсутствия загрязнений.
Получают стандартную калибровочную кривую (см. фиг.29). Принимают Y = площадь y X = мкг инъектированного стандарта
10 мкл раствора исследуемого соединения (инъектируют в дубликате, и среднюю площадь пика (если она измерима) интерполируют в стандартную кривую (см. таблицы 9, 10 и 11 и приводимые далее примеры).
Условия хроматографической обработки
- Колонка: XBridge C18 (или аналогичная) 2,5 мкм 4,6×50 мм
- Температура: 35°C.
- Подвижная фаза ACN/бикарбонат аммония 10 мМ.
- Градиент: 0-3,5 мин: от 15% ACN до 95% ACN.
3,5-5 мин: 95% ACN.
5-6 мин: 95 и 15% ACN.
6-8 мин: 15% ACN.
- Скорость потока: 1,5 мл/мин.
- Детектирование: около максимума УФ поглощения.
B) Термодинамическая растворимость при pH 2.
Предыдущую процедуру выполняют, используя HCl 0,01н.
Термодинамическая растворимость для соединения примера 1
В соответствии с раскрытым протоколом получают 227 мкг/мл (pH=7,4). См. соответствующий график на фиг.29.
|
|
|
Пример 9
Параметры фармакокинетики C макс и AUC
Тестируют фармакокинетические характеристики соединения примера 1 для крыс штамма Wistar Hannover после однократного перорального введения 25 мг/кг (выражено как соединение 63). Для этой цели образцы плазмы отбирают в различные моменты времени и анализируют, используя метод ВЭЖХ (высокоэффективной жидкостной хроматографии) с детектированием флуоресценции.
Получение образцов
В указанном тесте используют две группы крыс. Группе 1 вводят носитель, а группе 2 вводят соединение примера 1 в дозе 25 мг/кг при вводимом объеме 10 мг/кг.
Образцы крови отбирают из ретро-орбитальной зоны в следующие моменты времени: перед введением дозы, через 15 мин, 30 мин, 1 час, 1,5 часа, 2 часа, 3 часа, 4 часа, 5 часов, 6 часов, 8 часов и 24 часа после введения дозы. Затем образцы крови переносят в пластиковые ампулы, содержащие гепарин. Плазму получают путем центрифугирования со скоростью примерно 3000 об/мин в течение 10 мин при 4°C. Полученные образцы плазмы маркируют и замораживают при температуре приблизительно -65°C до анализа.
Анализ образцов
Образцы анализируют, используя утвержденный ранее аналитический метод. Короче, образцы плазмы крыс оттаивают при комнатной температуре и центрифугируют при 3000 об/мин в течение 10 мин при приблизительно 4°C. Образцы по 300 мкл плазмы помещают в ампулы и вводят 30 мкл рабочего раствора внутреннего стандарта. Ампулы закрывают и тщательно перемешивают.
Для экстрагирования соединения примера 1 используют следующий метод твердофазной экстракции.
1. Картридж активируют метанолом в течение 1 мин при скорости 1,5 мл/мин.
2. Картридж активируют водой в течение 2 мин при скорости 1,5 мл/мин.
3. Загружают образец (80 мкл) в картридж с водой на 1,5 мин при скорости 1,0 мл/мин.
4. Смачивают смесью вода/ACN (90/10, об/об) в течение 30 сек при скорости 1,5 мл/мин.
5. Элюируют образцы подвижной фазой в течение 1 мин при скорости 0,5 мл/мин.
6. Картридж и капилляр промывают водой и метанолом.
Затем образцы хроматографируют, используя в качестве подвижной фазы смесь 20 мМ одноосновного калийфосфата при pH 3, и ацетонитрила (70-73%) A и (30-27%) B (об/об) при комнатной температуре. Используют скорость потока 0,5 мл/мин, и время анализа составляет около 17 мин.
Пики, соответствующие соединению примера 1 и его внутреннему стандарту, рассчитывают, используя детектирование флуоресценции на длине волны возбуждения 260 нм и на длине волны эмиссии 360 нм. Остальные параметры: Время реакции: >0,2 мин (4 сек стандарт) и PMT усиление 8.
Параметры фармакокинетики
Параметры фармакокинетики получают, используя кривые среднего уровня плазмы путем некомпартментальных кинетик с помощью программного обеспечения WinNonlin Professional version 5.0.1.
Величины пиковой концентрации плазмы (Cмакс) и времена, за которые указанные концентрации достигаются, (tмакс) получают непосредственно из экспериментальных результатов. Константу элиминации (kэл) рассчитывают, используя линейную регрессию последней фазы кривой (log концентрации от времени). Время полу-жизни (t1/2) определяют из уравнения t1/2=0,693/kэл. Площадь под кривой уровней плазмы в зависимость от времени от нуля до последнего времени определения (AUC0-t) рассчитывают, используя метод трапеций. Площадь под кривой уровней содержания в плазме в зависимости от времени от нуля до бесконечности (AUC0-∞) рассчитывают, используя выражение: AUC0-~=AUCo-t+Cконц/kэл, где Cконц. представляет собой концентрацию плазмы в последний момент измерения.
Фармакокинетические параметры C макс и AUC для соединения примера 1
В соответствии с описанным протоколом получают Cмакс: 1152,8 нг/мл, AUC0-t: 1218,4 нг.час/мл и AUC0-∞: 1249,6 нг.час/мл. См. соответствующие графики на фиг.30.
Результаты, полученные в последних двух тестах, (растворимость и фармакокинетика) поддерживают кандидатуру гидрохлорида как наилучшую соль для соединения 63 для соответствующих композиций и клинических исследований.
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/8bacb13550faa95992860361a2c5d0c6.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/ac0f40eed077949e066adbd03741e2ae.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/263fb4a51ebefdd2a8cf6e9abd5ed2eb.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/38fb7e541a16e05696cb0945e0f09167.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/e73a5f7b1ffb21a03859c4615043c906.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/16845044a314a5d6c4b4dafc4ad5e312.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/4516e947601be9f057cfdcf65af43fd2.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/1ad151979e25eaaa15d110b76813b908.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/400faab28e48592a5667c2a83810cce3.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/e6212fc9c352669b3e71931706b7bad3.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/6420d2a3d04355dfb70fbdc4bac160de.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/29af2d006e30932633377425d6929c94.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/e5cd4d68c86d12d3be7fb631f29ae71c.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/dce6a6286066d5a40dc85dd97483487d.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/65ed78f68cc53cd7c87cb4ce56c99573.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/9e248cc0089205f787b1b4886fd6ec43.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/5543975ca99e1d6a88ad2175253bdb53.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/bbafe9da9d06d8b163b5e379ef5a1b4a.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/b0e037de99ff548b9403a5f774307943.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/ef03e035fb36a53672bce391182cb091.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/cdd0eb7493232f6bb3a70401466ca2be.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/9809deb21420a145f620fafe128967a2.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/d9c9ca033a1823ee02ded22f7696983e.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/aaf0b5b0fc3014d50a8e1ab1bd1b3028.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/ce69a66e3086d6cd29c271101f22f730.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/95181c6a2adec6dc2c7a2a6e05dd511a.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/8e7659f5dc0c5e9d04b73120e27e232f.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/cbab18a7fc6e2cb980499cae54dfc544.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/d46632054335ee661a3e59520b6c0ba2.png)
![СОЛИ 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА](https://fips.edrid.ru/images/rid/5c/0b/5b/94689ff1609ada077ff470b292727226.png)
Слой для защиты твердых дозированных форм от механического воздействия
Способ получения промежуточных соединений нафталин-2-ил-пиразол-3-она, используемых в синтезе ингибиторов сигма рецептора
1-арил-3-аминоалкоксипиразолы как сигма-лиганды, усиливающие обезболивающее действие опиоидов и ослабляющие зависимость от них
Применение соединений, связывающихся с лигандами сигма-рецептора, для лечения развития невропатической боли вследствие химиотерапии
Лиганды сигма-рецепторов для предупреждения или лечения боли, вызванной химиотерапией
Сокристаллы трамадола и коксибов
Фармацевтические композиции, содержащие лиганды сигма рецептора
Полиморфы и сольваты гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1н-пиразол-3-ил]окси]этил]морфолина
Сигма-лиганды для потенцирования анальгетического эффекта опиоидов и опиатов при послеоперационной боли и для ослабления зависимости от них
Фармацевтические композиции сокристаллов трамадола и коксибов
Слой для защиты твердых дозированных форм от механического воздействия
Способ получения промежуточных соединений нафталин-2-ил-пиразол-3-она, используемых в синтезе ингибиторов сигма рецептора
1-арил-3-аминоалкоксипиразолы как сигма-лиганды, усиливающие обезболивающее действие опиоидов и ослабляющие зависимость от них
Применение соединений, связывающихся с лигандами сигма-рецептора, для лечения развития невропатической боли вследствие химиотерапии
Лиганды сигма-рецепторов для предупреждения или лечения боли, вызванной химиотерапией
Сокристаллы трамадола и коксибов
Фармацевтические композиции, содержащие лиганды сигма рецептора
Полиморфы и сольваты гидрохлорида 4-[2-[[5-метил-1-(2-нафталинил)-1н-пиразол-3-ил]окси]этил]морфолина
Сигма-лиганды для потенцирования анальгетического эффекта опиоидов и опиатов при послеоперационной боли и для ослабления зависимости от них
Фармацевтические композиции сокристаллов трамадола и коксибов

