Результат интеллектуальной деятельности: СИНТЕЗ ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ ПИРАЗОЛИН КАРБОКСАМИДИНА
Вид РИД
Изобретение
Настоящее изобретение относится к органической химии, в частности к способам получения производных пиразолин карбоксамидина, известных в качестве мощных антагонистов 5-HT6. Кроме того, изобретение относится к новым промежуточным продуктам, образующимся при получении этих соединений.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Производные сульфонилпиразолин карбоксамидина были впервые раскрыты в качестве эффективных антагонистов 5-HT6 в WO 2008/034863. Родственные (гетеро)арилсульфонилпиразолин карбоксамидины с аналогичной фармакологической активностью были раскрыты в WO 2009/115515. Описанные в этих заявках пути синтеза обеспечивают приемлемые выходы, но они не очень хорошо подходят для синтеза в масштабе, необходимом для клинических исследований лекарственных средств, не говоря уже о масштабе, необходимом для выхода лекарства на рынок.
Задача настоящего изобретения заключалась в разработке новых путей синтеза производных сульфонилпиразолин карбоксамидина с улучшенной атомной эффективностью [Trost, B.M. Science 1991, 254, 1471; Sheldon, R.A. Pure Appl. Chem.2000, 72, 1233] и более высокими выходами по сравнению с известными путями синтеза, с применением удобных или легко доступных структурных блоков, в мягких условиях и с ограниченным применением и образованием вредных химических веществ.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
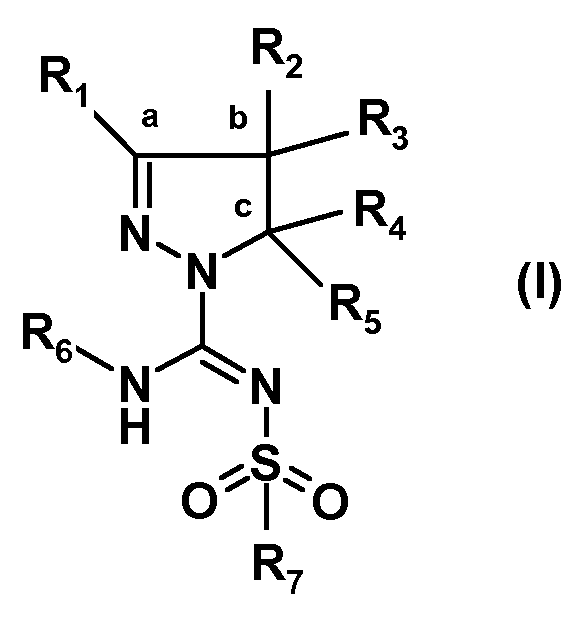
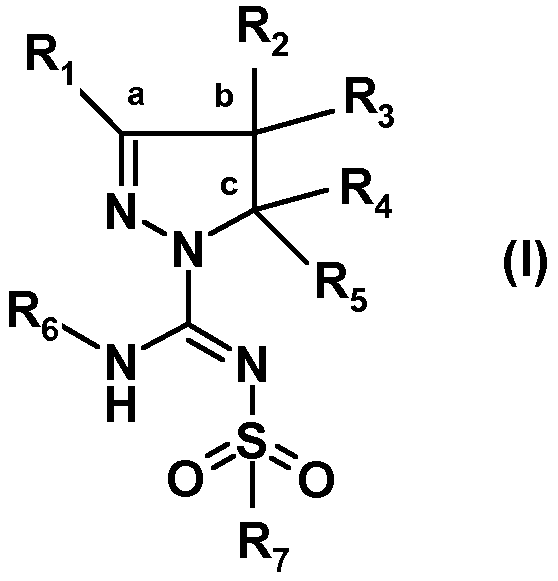
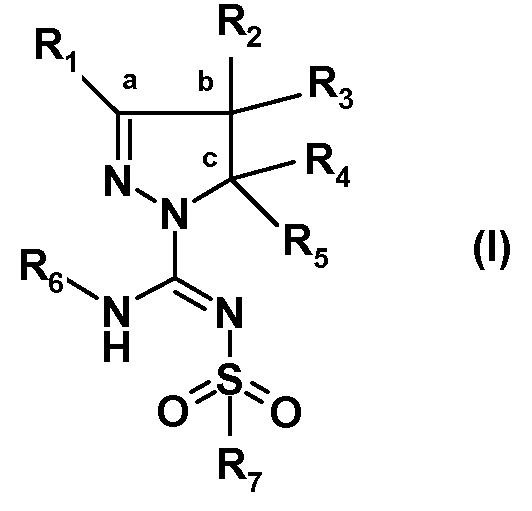
Было обнаружено, что новый путь синтеза производных (арил)сульфонилпиразолин карбоксамидина, характеризующийся более высокой атомной эффективностью и значительно более высокими выходами, чем у известных путей, осуществляемый в более мягких условиях, в большей степени поддается масштабированию. Настоящее изобретение относится к способу получения соединений формулы (I):

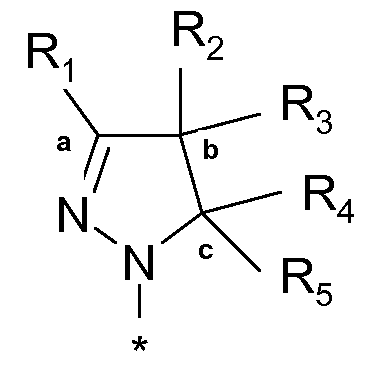
или таутомеров, стереоизомеров или фармацевтически приемлемых солей любого из этих соединений, где:
R1 выбран из атома водорода или (C1-4)алкильной группы, необязательно замещенной одним-тремя атомами фтора или гидроксигруппой,
R2 представляет собой атом водорода или (C1-4)алкильную группу, необязательно замещенную одним-тремя атомами фтора, гидроксигруппой, бензилоксиметильной группой, аминогруппой, монометиламиногруппой, диметиламиногруппой или Boc-, Fmoc- или Cbz-защищенной аминогруппой, где (C1-4)алкильная группа может включать кетогруппу, сульфонильную группу или атом N, O или S,
R3 представляет собой атом водорода или (C1-4)алкильную группу, необязательно замещенную одним-тремя атомами фтора, гидроксигруппой, бензилоксиметильной группой, аминогруппой, монометиламиногруппой, диметиламиногруппой или Boc-, Fmoc- или Cbz-защищенной аминогруппой, где (C1-4)алкильная группа может включать кетогруппу, сульфонильную группу или атом N, O или S, или
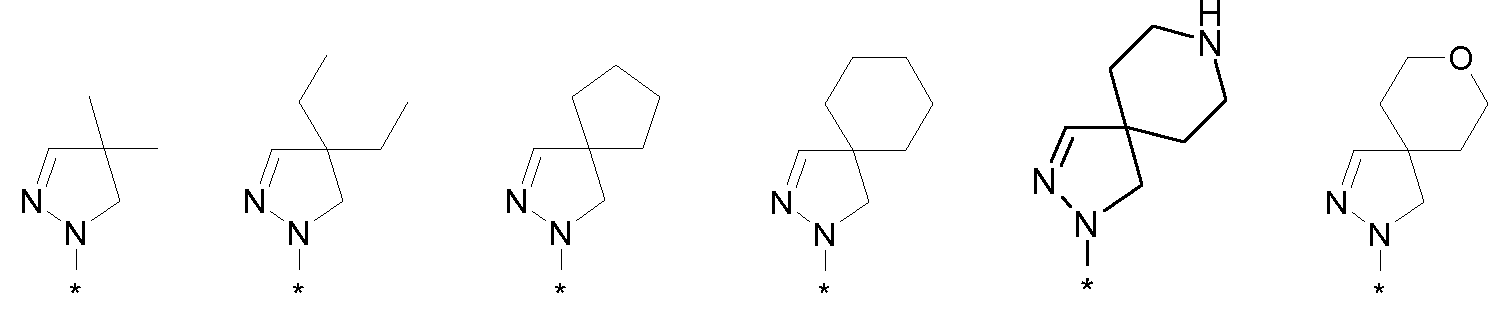
R1 и R2 совместно с атомами углерода, отмеченными символами 'a' и 'b', образуют C5-8 циклоалкил, необязательно замещенный одним-тремя атомами фтора, гидроксигруппой или (C1-4)алкильной группой, или
R2 и R3 совместно с атомом углерода, отмеченным символом 'b', образуют C3-8 циклоалкил, необязательно замещенный одним-четырьмя атомами фтора, одной или двумя метильными группами или гидроксигруппой, или
R2 и R3 совместно с атомом углерода, отмеченным символом 'b', образуют C5-8 гетероциклоалкил, необязательно замещенный одним-четырьмя атомами фтора, одной или двумя метильными группами, бензильной группой или гидроксигруппой,
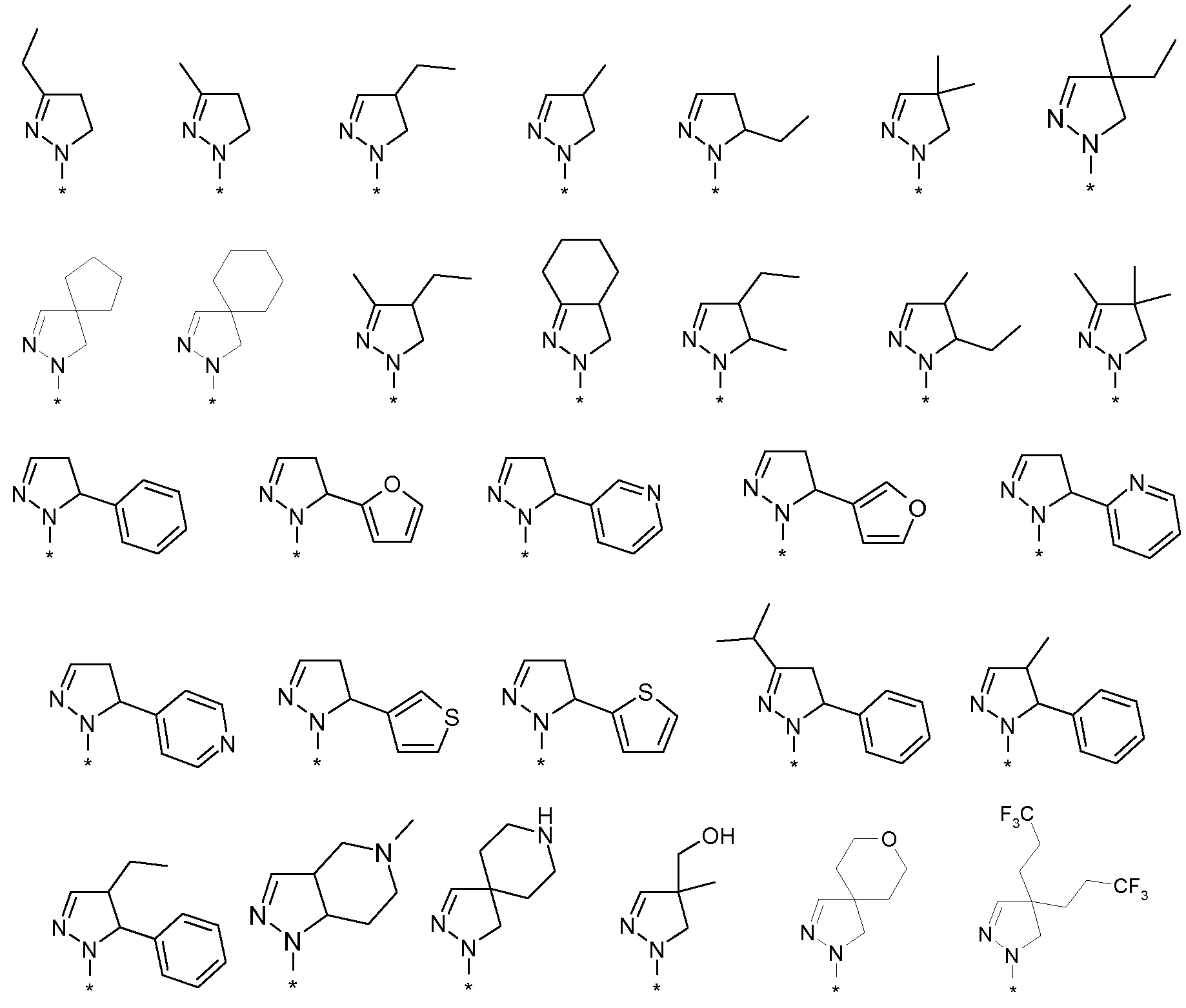
R4 представляет собой атом водорода или (C1-4)алкильную группу, необязательно замещенную одним-тремя атомами фтора или гидроксигруппой, или же R4 означает моноциклическую арильную или гетероарильную группу, необязательно замещенную одним-пятью заместителями Q, которые могут являться одинаковыми или различаться, выбранными из галогена, трифторметила, трифторметокси, циано, C1-3 алкила, C1-3 алкокси, гидрокси, амино, ацетила, ацетамидо, трифторацетамидо, -CONH2, -SO2NH2 или -CO2H, или
R3 и R4 совместно с атомами углерода, отмеченными символами 'b' и 'c', образуют C3-8 циклоалкил, необязательно замещенный одним-четырьмя атомами фтора, одной или двумя метильными группами или гидроксигруппой, или
R3 и R4 совместно с атомами углерода, отмеченными символами 'b' и 'c', образуют C5-8 гетероциклоалкил, необязательно замещенный одним-четырьмя атомами фтора, одной или двумя метильными группами, бензильной группой или гидроксигруппой,
R5 является атомом водорода или метилом,
R6 выбран из атома водорода или (C1-4)алкильной группы, необязательно замещенной одним-тремя атомами фтора или гидроксигруппой,
R7 представляет собой моноциклическую или конденсированную бициклическую ароматическую или гетероароматическую группу, где указанные группы являются незамещенными или замещены одним-пятью заместителями Q, которые определены выше, или
R7 представляет собой 2-арилэтенильную группу или 2-арилэтинильную группу, или
R7 представляет собой пиперидинильную группу, незамещенную или замещенную одним-четырьмя атомами фтора или группой CF3, или
R7 представляет собой 2,3-дигидроиндолильную группу или группу бензимидазол-2-она,
включающий стадии:
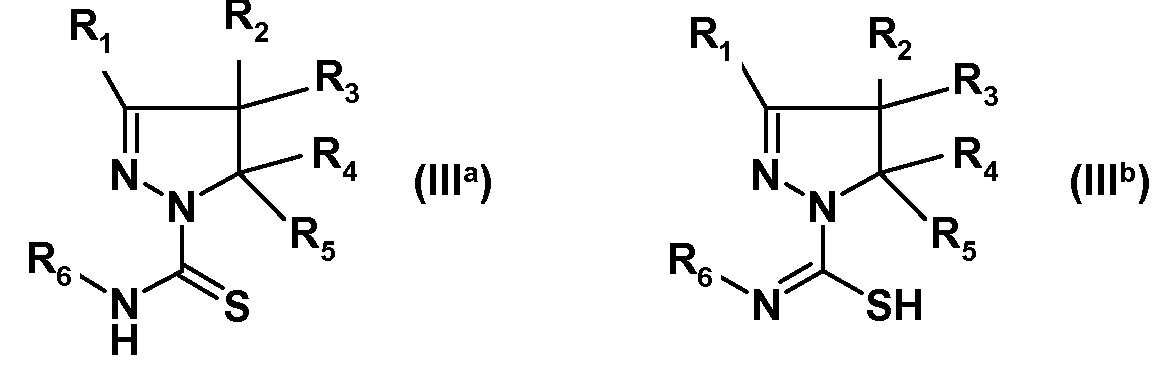
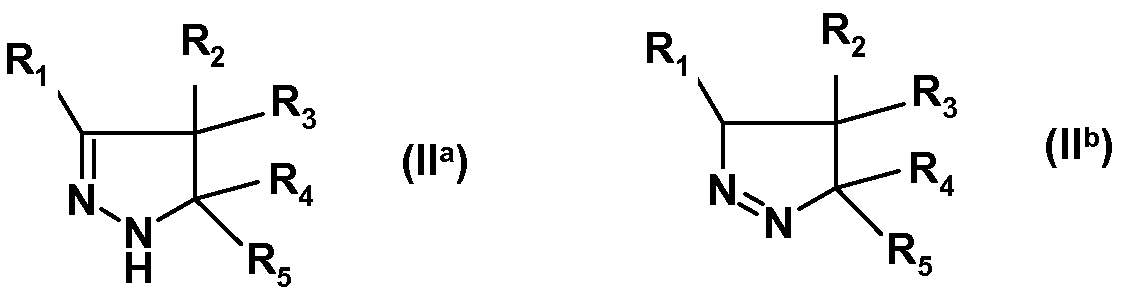
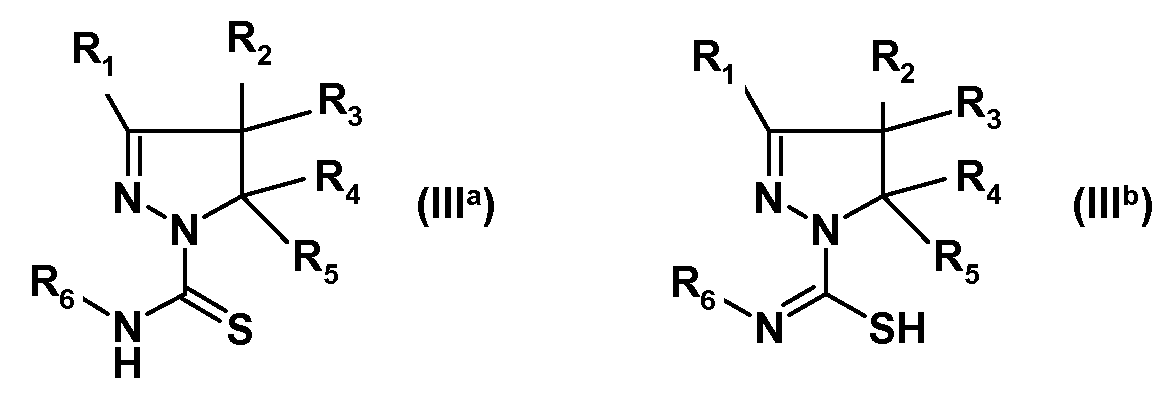
(i) взаимодействия замещенного 4,5-дигидро-(1H)-пиразола формулы (IIa) или изомерного замещенного 4,5-дигидро-3H-пиразола формулы (IIb):

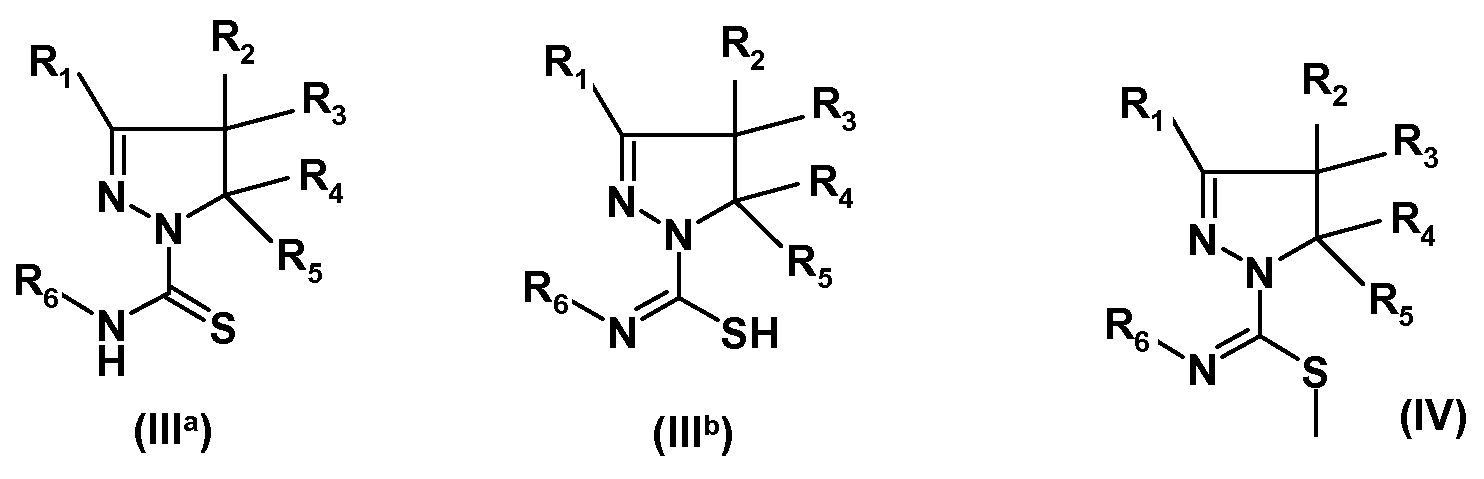
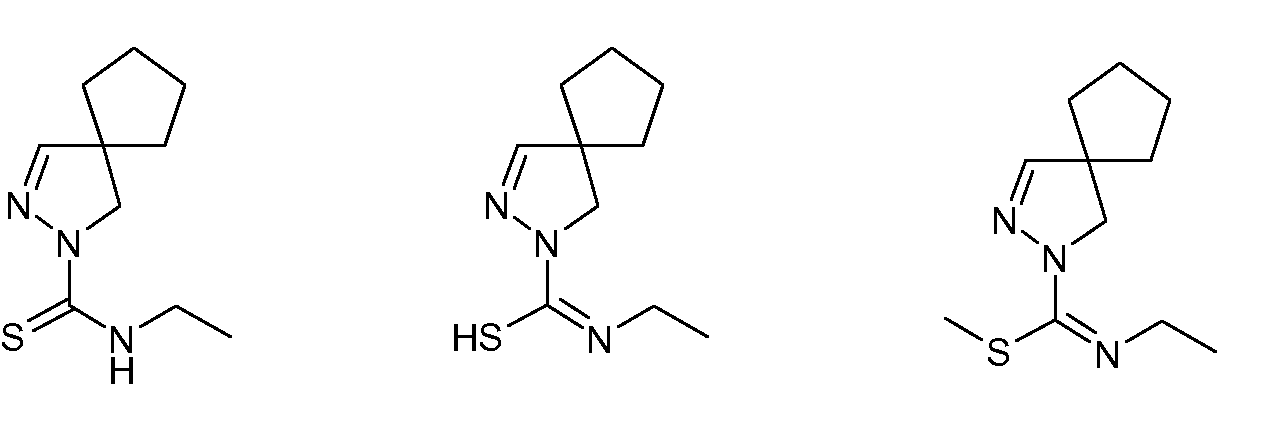
где R1, R2, R3, R4 и R5 имеют указанные выше значения, с изотиоцианатом формулы R6-N=C=S, где R6 имеет указанное выше значение, с получением амида замещенной 4,5-дигидро-(1H)пиразол-1-карботиокислоты формулы (IIIa) или таутомерной замещенной 4,5-дигидро-(1H)пиразол-1-карбоксимидотиокислоты формулы (IIIb):
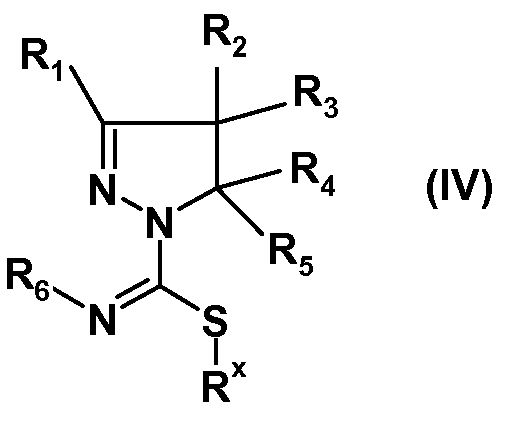
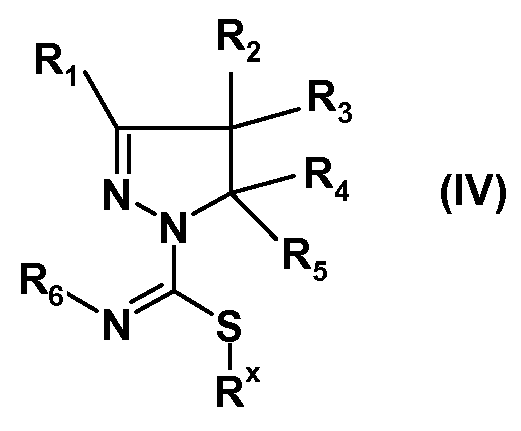
(ii) взаимодействия полученных соединений формулы (IIIa) или (IIIb) с алкилирующим реагентом общей формулы Rx-L, где Rx представляет собой линейную (C1-8)алкильную группу, и L означает уходящую группу, предпочтительно выбранную из Br, Cl или I, с получением соединения формулы (IV):
(iii) взаимодействия полученного соединения формулы (IV) с производным сульфонамида формулы R7SO2NH2, где R7 имеет указанное выше значение, с получением соединения формулы (I):

На стадии (i) реагенты в виде свободных оснований или их солей растворяют в подходящем растворителе, предпочтительно, полярном растворителе, более предпочтительно спирте (C1-8) или смеси таких спиртов, необязательно содержащей воду. Реакцию предпочтительно проводят при повышенной температуре, наиболее предпочтительно, в кипящем растворителе, в течение примерно 1-16 часов, предпочтительно от примерно 2,5 до примерно 5 часов.
Аналогично, на стадии (ii) реагенты в форме свободных оснований или их солей растворяют в подходящем растворителе, предпочтительно, полярном растворителе, например, ацетонитриле, метилэтилкетоне, спирте (C1-8), или смеси полярных растворителей, наиболее предпочтительно, метаноле или ацетонитриле. Реакцию предпочтительно проводят при повышенной температуре, но можно проводить реакцию и при комнатной температуре. Предпочтительной является температура от примерно 40°C до примерно 50°C. Наиболее предпочтительна температура реакционной смеси 50°C и время проведения реакции от примерно 1 до примерно 5 часов. Предпочтительными алкилирующими реагентами общей формулы RX-L, где RX означает линейную (C1-8)алкильную группу, и L означает «уходящую группу», предпочтительно выбранную из Br, Cl или I, являются метилгалогениды. Наиболее предпочтительными являются метилйодид.
На стадии (iii) реагенты в форме свободных оснований или их солей растворяют в подходящем растворителе, предпочтительно, полярном растворителе, наиболее предпочтительно, ацетонитриле. Эту реакцию предпочтительно проводят при повышенной температуре, предпочтительно при температуре кипения, в течение примерно 16-72 часов, предпочтительно в течение от примерно 10 до примерно 16 часов.
Настоящее изобретение относится к рацематам, смесям диастереомеров, а также к индивидуальным диастереомерам соединений формулы (I). Кроме того, настоящее изобретение относится к E-изомерам, Z-изомерам и смесям E/Z изомеров соединений формулы (I) и их солям. Помимо этого, изобретение относится к рацематам, смесям диастереомеров, а также к индивидуальным стереоизомерам соединений формул (IIIa), (IIIb) и (IV) и к солевым формам соединений формул (IIIa), (IIIb) и (IV).
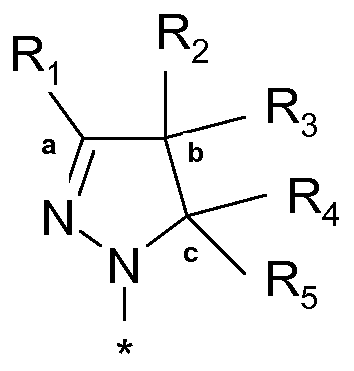
Далее, изобретение относится к способу получения соединения формулы (I), в котором:
R1 выбран из атома водорода или (C1-2)алкильной группы,
R2 представляет собой атом водорода или (C1-3)алкильную группу, необязательно замещенную одним-тремя атомами фтора или гидроксигруппой,
R3 представляет собой атом водорода или (C1-3)алкильную группу, необязательно замещенную одним-тремя атомами фтора или гидроксигруппой, или
R1 и R2 совместно с атомами углерода, отмеченными символами 'a' и 'b', образуют C5-8 циклоалкил, или R2 и R3 совместно с атомом углерода, отмеченным символом 'b', образуют C3-8 циклоалкил, необязательно замещенный одним-четырьмя атомами фтора или гидроксигруппой, или
R2 и R3 совместно с атомом углерода, отмеченным символом 'b', образуют C5-8 гетероциклоалкил, необязательно замещенный метильной или бензильной группой, или гидроксигруппой,
R4 представляет собой атом водорода или (C1-2)алкильную группу, или же R4 означает моноциклическую арильную или гетероарильную группу, необязательно замещенную одним-тремя заместителями Q, которые определены выше, или
R3 и R4 совместно с атомами углерода, отмеченными символами 'b' и 'c', образуют C5-8 циклоалкил, или
R3 и R4 совместно с атомами углерода, отмеченными символами 'b' и 'c', образуют C5-8 гетероциклоалкил, необязательно замещенный метильной или бензильной группой,
R5 является атомом водорода,
R6 выбран из атома водорода или (C1-3)алкильной группы, необязательно замещенной одним-тремя атомами фтора,
R7 представляет собой моноциклическую или конденсированную бициклическую ароматическую или гетероароматическую группу, где указанные группы являются незамещенными или замещены одним-пятью заместителями Q, которые определены выше, или
R7 представляет собой 2-арилэтенильную группу или 2-арилэтинильную группу, или
R7 представляет собой пиперидинильную группу, или
R7 представляет собой 2,3-дигидроиндолильную группу или группу бензимидазол-2-она.
Другой вариант осуществления изобретения относится к способу получения соединения формулы (I), в котором фрагмент :

выбран из:

R6 выбран из водорода или (C1-3)алкильной группы, необязательно замещенной одним-тремя атомами фтора,
R7 представляет собой моноциклическую или конденсированную бициклическую ароматическую или гетероароматическую группу, где указанные группы являются незамещенными или замещены одним-пятью заместителями Q, которые определены выше, или
R7 представляет собой 2-арилэтенильную группу или 2-арилэтинильную группу, или
R7 представляет собой пиперидинильную группу, или
R7 представляет собой 2,3-дигидроиндолильную группу или группу бензимидазол-2-она.
Другой вариант осуществления относится к способу получения соединения формулы (I), где фрагмент:

выбран из:

R6 выбран из водорода или (C1-2)алкильной группы, необязательно замещенной тремя атомами фтора,
R7 представляет собой моноциклическую или конденсированную бициклическую ароматическую или гетероароматическую группу, где указанные группы являются незамещенными или замещены одним или двумя заместителями, выбранными из метила, метокси, фтора, хлора, брома, циано, ацетамидо, трифторацетамидо, трифторметила, амино или гидрокси.
Конкретный вариант осуществления относится к способу получения соединения, имеющего формулу:

а также его таутомерных и солевых форм,
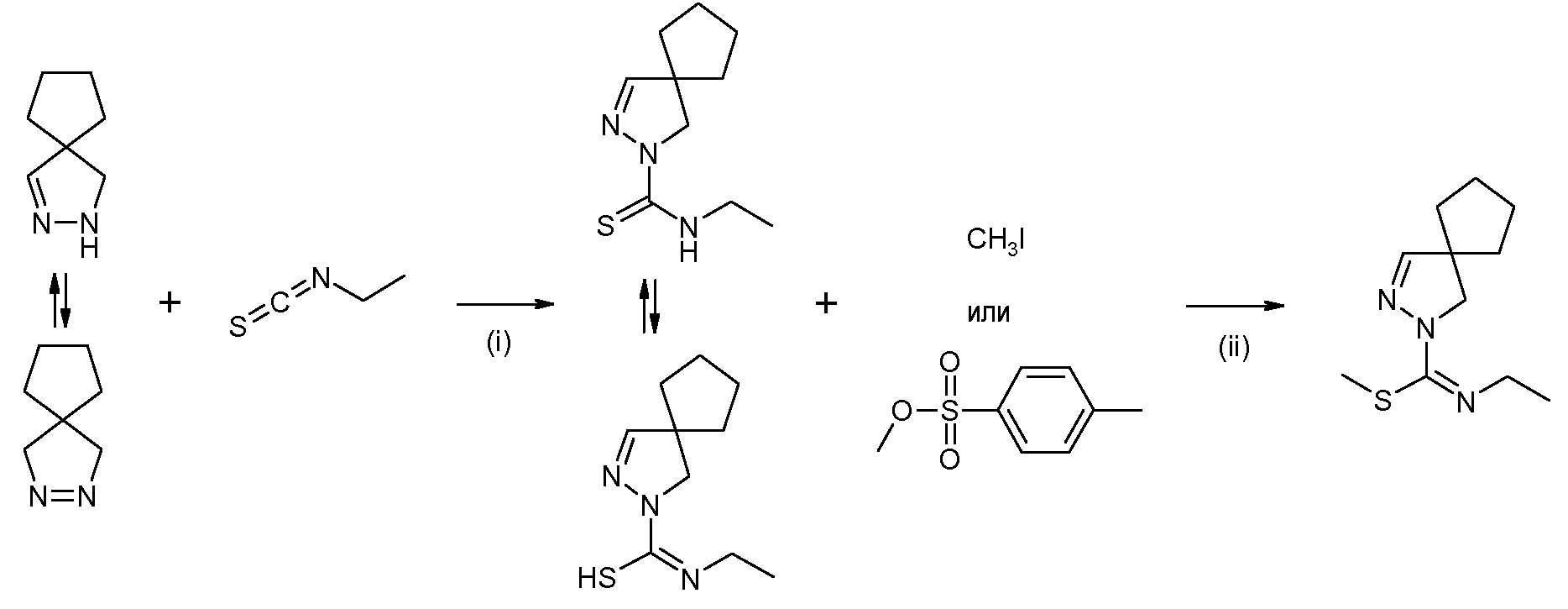
включающему стадии:
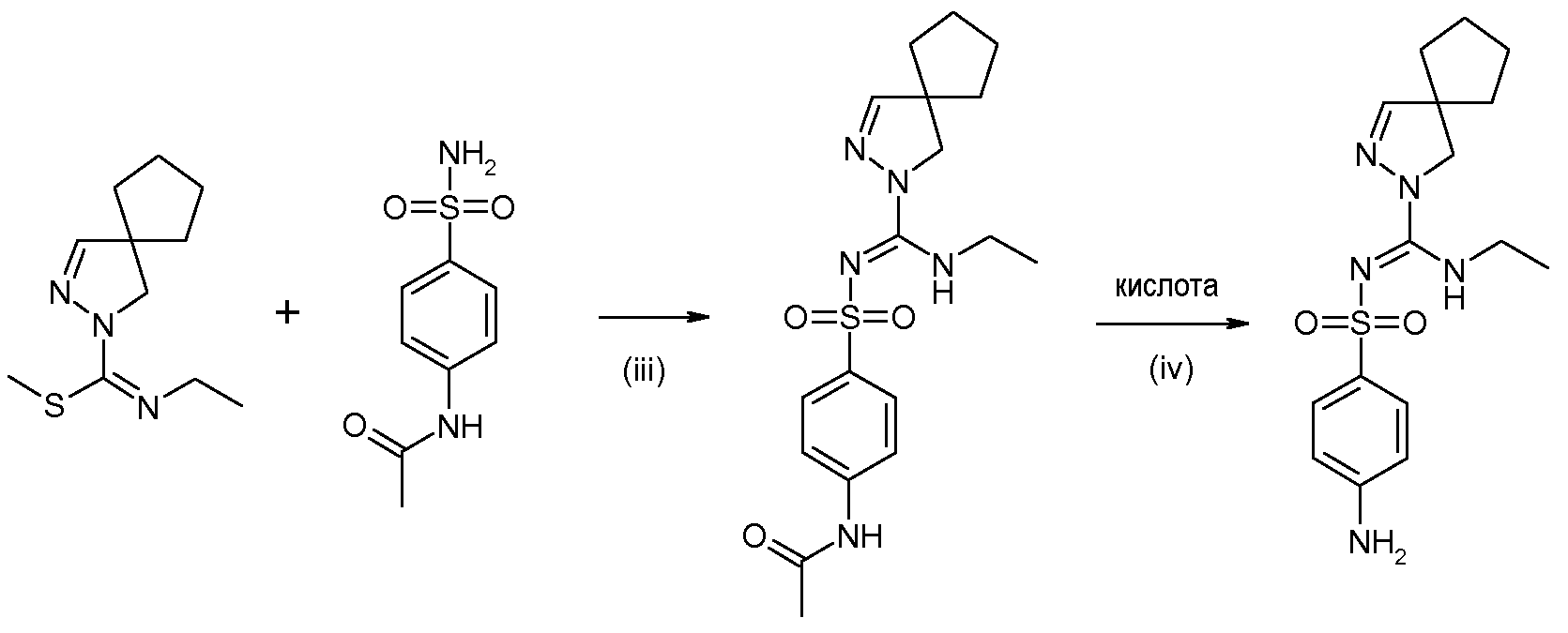
(i) взаимодействия 2,3-диазаспиро[4.4]нон-2-ена или 2,3-диазаспиро[4.4]нон-1-ена или их солей, синтезированных по способу, раскрытому в WO 2008/034863, с этилизотиоцианатом, с получением этиламида 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты или его таутомера
(ii) взаимодействия продукта стадии (i) с йодметаном или метил п-толуолсульфонатом, приводящее к получению метилового эфира N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты,

(iii) взаимодействия продукта стадии (ii) в форме свободного основания или соли, с 4-ацетамидобензолсульфонамидом (CAS 121-61-9, имеется в продаже) с получением N-(4-{[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамида
(iv) снятия защиты продукта стадии (iii) в кислой среде с получением 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамида

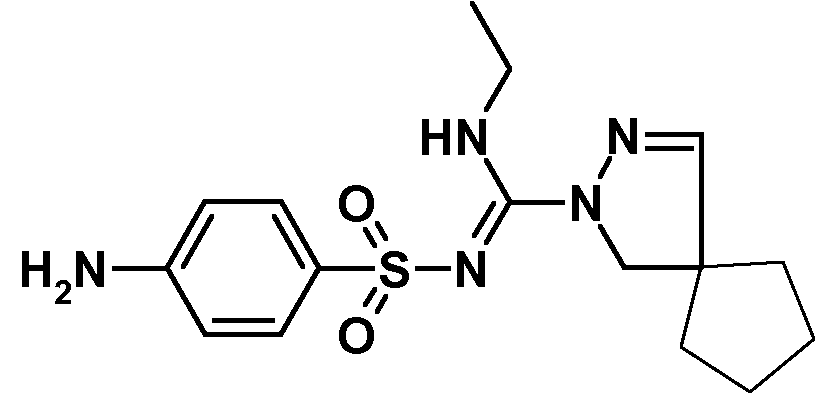
Другой конкретный вариант осуществления относится к описанному выше способу, в котором стадия (iii) заключается во взаимодействии метилового эфира N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты с сульфаниламидом (CAS 129-56-6, имеется в продаже) с получением 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамида:

Другой вариант осуществления изобретения относится к соединениям формул (IIIa), (IIIb) или (IV):

где R1, R2, R3, R4, R5 и R6 имеют указанные выше значения, а также к таутомерам, стереоизомерам и солям любых из указанных соединений, где перечисленные соединения находят применение в синтезе соединений формулы (I).
Описанные в настоящей заявке соединения и промежуточные продукты, если это желательно, могут быть выделены и очищены любым подходящим способом выделения или очистки, как, например, фильтрованием, экстракцией, кристаллизацией, колоночной хроматографией, тонкослойной хроматографией, толстослойной хроматографией, препаративной жидкостной хроматографией низкого или высокого давления или комбинацией указанных способов. В описании синтезов и в примерах показано как разделять и выделять соединения по настоящему изобретению, но кроме этого можно применять и другие эквивалентные методики.
Соединения по настоящему изобретению могут содержать один или несколько асимметрических центров и, вследствие этого, могут существовать в форме рацематов и рацемических смесей, чистых энантиомеров, диастереомерных смесей и индивидуальных диастереомеров.
В зависимости от природы имеющихся заместителей, в молекуле могут присутствовать дополнительные асимметрические центры. Каждый такой асимметрический центр будет независимо приводить к возникновению двух оптических изомеров. Все возможные оптические изомеры, энантиомеры и диастереомеры в виде смесей и чистых, а также частично очищенных соединений входят в объем настоящего изобретения. В настоящем изобретении рассматриваются все подобные изомерные формы соединений по настоящему изобретению. Формула (I) показывает структуру класса соединений без предпочтительной стереохимии. Независимый синтез указанных оптических изомеров или их хроматографическое разделение может быть осуществлено известными способами с соответствующими изменениями методик, раскрытых в настоящей заявке. Абсолютное пространственное строение можно установить с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, из которых, в случае необходимости, можно получать производные с реагентами, содержащими асимметрический центр известной абсолютной конфигурации. Рацемические смеси соединений можно разделять на индивидуальные энантиомеры хорошо известными способами, например, взаимодействием рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси, с последующим разделением индивидуальных диастереомеров стандартными способами, например фракционной кристаллизацией или хроматографией. Указанное взаимодействие часто заключается в образовании солей с применением энантиомерно чистой кислоты или основания, например, (-)-ди-п-толуоил-D-винной кислоты или (+)-ди-п-толуоил-L-винной кислоты. Диастереомерные производные затем можно превратить в чистые энантиомеры путем отщепления присоединенного хирального остатка. Рацемическую смесь соединений можно также разделить непосредственно, с помощью хорошо известных хроматографических методик с применением хиральных неподвижных фаз. В качестве альтернативы, любой энантиомер того или иного соединения можно получить с помощью стереоселективного синтеза, используя оптически чистые исходные материалы или реагенты известной конфигурации с применением методик, хорошо известных в технике.
Цис- и трансизомеры соединения формулы (I) или их фармацевтически приемлемые соли также входят в настоящее изобретение, и то же самое относится и к таутомерам соединений формулы (I).
Стратегия синтеза в новом способе по настоящему изобретению существенно отличается от известных путей синтеза введением заместителей R6 и R7 на других стадиях синтеза и/или с помощью других структурных блоков. Начиная с промежуточного продукта, имеющего таутомерные формы (IIa) или (IIb), который является общим структурным блоком как для нового пути синтеза, так и для ранее раскрытых путей заместитель R6 вводят с помощью изотиоцианатного, а не аминного (способы синтеза 1 и 3, раскрытые в WO 2009/115515) или тиомочевинного (способ 2, раскрытый в WO 2009/115515) блока. В результате этого, новые таутомерные промежуточные продукты (IIIa) или (IIIb) образуются в нейтральной среде с максимальной атомной эффективностью (100%), причем реакция легко поддается масштабированию. Из промежуточных продуктов (IIIa)/(IIIb) при алкилировании в мягких условиях легко получаются новые промежуточные продукты формулы (IV), в которых имеется легко замещаемая уходящая группа S-алкил. В противоположность этому, путь 3, раскрытый в WO 2009/115515, требует значительно более жестких условий для получения in situ соединения с галогеном в качестве уходящей группы, которую на завершающей стадии предполагается заместить блоком R6 амин. Заключительная стадия нового способа синтеза представляет собой замещение уходящей группы S-алкил в промежуточном продукте (IV), но в отличие от пути 1, раскрытого в WO 2009/115515, где блок R6 амин замещает фрагмент S-алкил, новый способ синтеза по настоящему изобретению завершается введением блока R7 сульфонамид, что примечательно, в нейтральной среде и незначительном нагревании. В путях синтеза 1 и 3, раскрытых в WO 2009/115515, заместитель R7 сульфонил вводится в более жестких условиях на более ранней стадии синтеза, в то время как в способе 2, раскрытом в WO 2009/115515, заместитель R7 сульфонил также вводится именно на заключительной стадии, но с помощью более реакционноспособного блока R7 сульфонилхлорид в основной среде (что ограничивает применение незащищенных нуклеофильных фрагментов в остатке R7). Поэтому новый способ синтеза включает усовершенствование с точки зрения более широкого выбора функциональных групп в заместителе R7, что показано в настоящей заявке в нескольких примерах, например, в синтезе соединения 4, где заместители R7, содержащие аминоарильные группы, вводились селективно без необходимости применения защиты.
Не говоря об очевидной разнице в стратегии синтеза и связанными с этой разницей мягкими условиями, в которых обычно могут проводиться стадии синтеза, новый путь синтеза по настоящему изобретению обеспечивает явное преимущество в ряде других аспектов, которые приобретают особую значимость при увеличении масштабов синтеза. В способе синтеза 3, раскрытом в WO 2009/115515, задействованы коррозионно-активные галогенирующие агенты, что ограничивает возможности его применения. В способе синтеза 1, раскрытом в WO 2009/115515 применяется токсичный компонент CS2 в сильно основной среде, причем другой недостаток этого способа заключается в том, что применяется два мольных эквивалента алкилирующего агента и в способ включены две стадии, на каждой из которых выделяется один мольный эквивалент алкантиола. В новом способе синтеза не применяются сильно основные или кислотные условия, не применяется CS2, применяется только один эквивалент алкилирующего агента и включена только одна стадия, на которой выделяется мольный эквивалент алкантиола. Хотя последние аргументы также верны для пути синтеза 2, раскрытого в WO 2009/115515, лимитирующим фактором в этом способе, не только при проведении реакций, но в отдельных случаях также и с точки зрения устойчивости тех или иных функциональных групп, может стать необходимость применения реакционноспособных сульфонилхлоридных блоков. Как показано для синтеза соединения 4, введение 4-аминофенилсульфонильных фрагментов в способе синтеза 2 заявки WO 2009/115515 требует защиты аминогруппы. Удаление N-ацетильной защитной группы (введенной с помощью N-ацетилсульфанилхлорида, CAS 121-60-8, имеется в продаже) подразумевает проведение дополнительной стадии синтеза в сильно кислой (коррозионно-активной) среде, что ведет к риску сопутствующего гидролиза сульфонамида, что, как показано, приводит лишь к умеренным выходам.
В эпоху, когда доступность сырья и забота об окружающей среде приобретают возрастающее значение, в особенности для крупномасштабных производств, атомная эффективность стала общепризнанным параметром для оценки способов синтеза. Атомная эффективность [Sheldon, R.A. Pure Appl.Chem. 2000, 72, 1233] (выраженная в процентах) может быть рассчитана путем вычисления соотношения молекулярной массы конечного продукта к суммарной молекулярной массе всех использованных строительных блоков, включающих атомы, из которых состоит продукт синтеза. При сравнении синтеза соединения 4 по настоящему изобретению, исходя из необходимых для получения конечных продуктов стадий синтеза, исключая синтез промежуточных продуктов формулы (IIa)/(IIb), общих для всех способов, становится очевидным, что новый способ синтеза по настоящему изобретению превосходит способы известного уровня техники, с точки зрения как атомной эффективности, так и суммарного выхода:
Способ 1, раскрытый в WO 2009/115515:

Атомная эффективность: [349,46/(172,21+76,14+(2×141,93)+124,19+45,08)]×100%=50%;
Выход: 40%×25%×67%=7%;
Способ 2, раскрытый в WO 2009/115515:
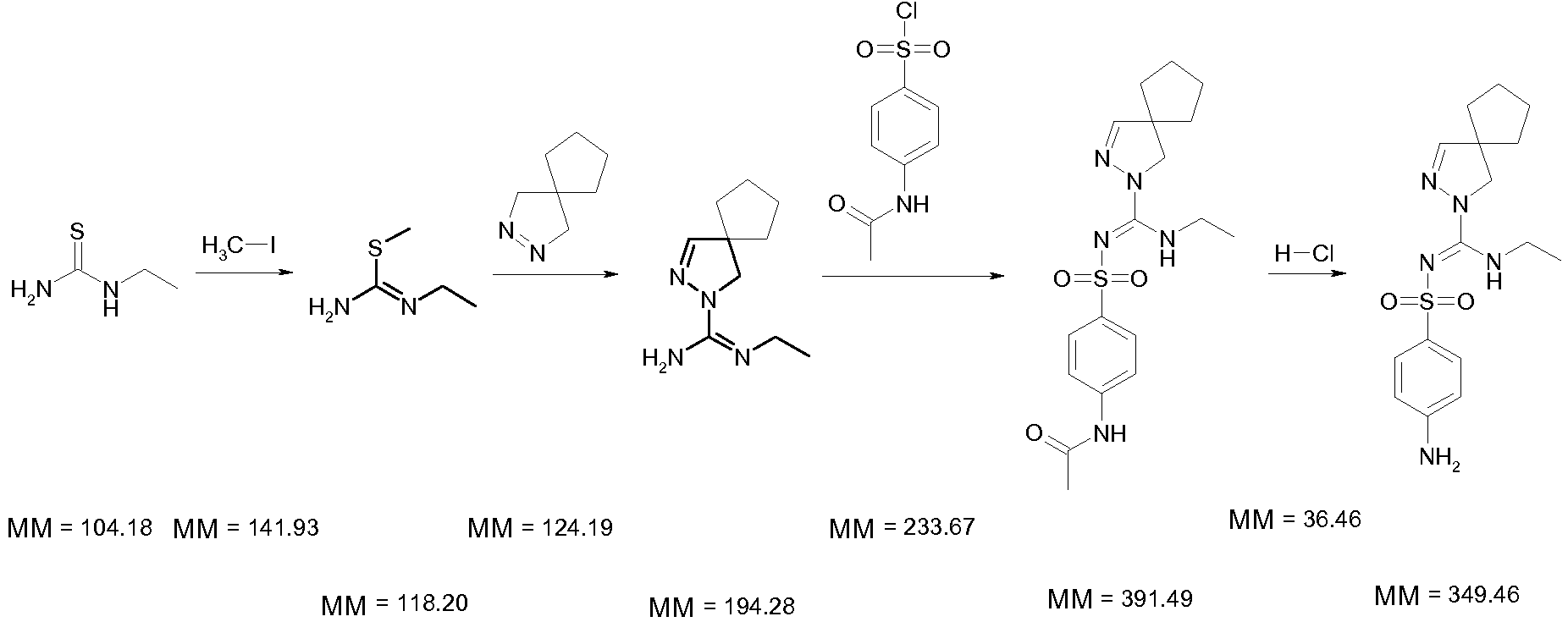
Атомная эффективность: [349,46/(104,18+141,93+124,19+233,67+36,46)]×100%=55%;
Выход: 100%×78%×77%×55%=33% (выходы 2 заключительных стадий в этом конкретном примере не приведены в WO 2009/115515, но определены в настоящей заявке).
Новый способ синтеза:

Атомная эффективность: [349,46/(124,19+87,15+141,93+172,21)]×100%=67%;
Выход: 83%×97%×67%=54% (Первая стадия: методика синтеза с большим количеством реагентов, с использованием в качестве исходного соединения соли пиразолин·HCl).
Определения
Основные термины, используемые в описании соединений, раскрытых в настоящей заявке, имеют свои обычные значения. Термин «алкил» означает одновалентный насыщенный, разветвленный или линейный углеводородный остаток. Если не указано иное, алкильные цепи могут содержать от 1 до 18 атомов углерода. Типовыми примерами таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, изогексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил и т.д. Если алкильная группа именуется «низшим алкилом», то она должна содержать от 1 до 6 атомов углерода. Аналогичное количество атомов углерода применимо к исходному термину «алкан» и к производным терминам, например «алкокси». Количество атомов углерода в различных углеродсодержащих фрагментах показано префиксом, указывающим на минимальное и максимальное количество атомов углерода во фрагменте, т.е. префикс Cx-y определяет число имеющихся атомов углерода от целого числа “x” до целого числа “y” включительно. Например, «(C1-3)алкил» включает метил, этил, н-пропил или изопропил, и «(C1-4)алкил» включает метил, этил, н-пропил, изопропил, изопропил, н-бутил, 2-бутил, изобутил или трет-бутил.
Термин «арил» охватывает моно- или полициклические ароматические группы, включая фенил, нафтил, 1,2,3,4-тетрагидронафтил, инденил, флуоренил, антраценил, фенантренил, нафтаценил и азуленил. Термин «гетероарил» охватывает моно- или полициклические гетероароматические группы, в том числе фурил, тиенил, пирролил, оксазолил, тиазолил, имидазолил, имидазо[2,1-b][1,3]тиазолил, пиразолил, изоксазолил, изотиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, 1,3,5-триазинил, индазолил, индолил, индолизинил, изоиндолил, бензо[b]фуранил, 1,2,3,4-тетрагидроизохинолинил, инданил, инденил, бензо[b]тиенил, 2,3-дигидро-1,4-бензодиоксин-5-ил, бензимидазолил, циннолинил, карбазолил, акридинил, феназинил, фенотиазинил, феноксазинил, бензотиазолил, бензо[1,2,5]тиадиазолил, пуринил, хинолинил, изохинолинил, хинолизинил, фталазинил, хиназолинил, хиноксалинил, 1,8-нафтиридинил и птеридинил.
Термин «галоген» относится к хлору, фтору, брому или йоду; термин «гетеро», например, в терминах «гетероалкил», «гетероароматический» и т.д. означает наличие одного или нескольких атомов N, O или S. Термин «гетероалкил» означает алкильную группу с гетероатомами в любом положении, включая, таким образом, алкильные группы, связанные через атомы N, O или S.
Термин «замещенный» означает, что определяемая им группа или фрагмент несет один или несколько заместителей. Если какая-либо группа может нести несколько заместителей и имеется возможность присутствия нескольких различных заместителей, эти заместители выбирают независимо и они необязательно должны быть одинаковыми. Термин «незамещенный» означает, что указанная группа не несет на себе заместителей. Термин «независимо», в отношении заместителей, означает, что если возможно присутствие более чем одного заместителя, эти заместители могут быть одинаковыми или отличаться друг от друга.
«C3-8 циклоалкил» включает циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил; термин «C5-8 гетероциклоалкил» относится к циклам, содержащим гетероатомы, включая пиперидинил, морфолинил, азепанил, пирролидинил, тиоморфолинил, пиперазинил, тетрагидрофурил, тетрагидропиранил;
Термины «окси», «тио» и «карбо», применяемые в качестве части наименования другой группы, относятся, соответственно, к атому кислорода, атому серы и карбонильной группе (C=O), которые играют роль линкеров между двумя группами, например, гидроксил, оксиалкил, тиоалкил, карбоксиалкил и т.д. Термин «амино», используемый сам по себе, или в качестве части наименования другой группы, относится к атому азота, который может быть либо концевым, либо линкером между двумя другими группами, где указанная группа может быть первичной, вторичной или третичной (соответственно, с атомом азота связаны два атома водорода, один атом водорода или ни одного атома водорода) аминогруппой. Термины «сульфинил» и «сульфонил» в качестве части наименования другой группы относятся к группами -SO- и -SO2-, соответственно.
С целью более краткого описания, термины «соединение» или «соединения» включают таутомеры, стереоизомеры, N-оксиды, изотопно-меченые аналоги или фармакологически приемлемые соли, даже в том случае, когда они не упомянуты в явном виде.
Термин «уходящая группа» (L) включает заряженный или незаряженный атом или группу, покидающую молекулу во время реакции замещения или вытеснения. Этот термин относится к группам, которые способны легко замещаться при действии нуклеофилов, например аминов, тиолов или спиртов. Подобные уходящие группы хорошо известны. Их примеры включают N-гидроксисукцинимид, N-гидроксибензотриазол, галогены (Br, Cl, I), трифлаты, мезилаты, тозилаты и т.д.
Для более лаконичного изложения, некоторые из количественных выражений, приведенных в настоящей заявке, не сопровождаются терминами «примерно» или «приблизительно». Имеется в виду, что независимо от того, используется ли любой из этих терминов в явном виде или нет, каждая приведенная в описании количественная величина может иметь приведенное значение, а также значения, приблизительно равные этой приведенной величине, о которых можно сделать разумное предположение, на основе стандартных знаний в данной области, включая приближения, связанные с экспериментальными условиями или условиями измерения данной конкретной величины. В тексте описания и формулы изобретения данной заявки не предполагается, что слово «включать» и его варианты, такие как «включающий» и «включает», исключают другие добавки, компоненты, целые числа или стадии.
Сокращения
|
|
Пример 1: аналитические методики
Спектры 1 H ЯМР регистрировали на приборе Varian UN400 (400 МГц) или приборе Bruker Avance DRX600 (600 МГц), используя в качестве растворителей ДМСО-d6, CD3CN или CDCl3 и тетраметилсилан в качестве внутреннего стандарта. Химические сдвиги приведены в м.д. (δ шкала) в слабое поле относительно тетраметилсилана. Константы спин-спинового взаимодействия (J) выражены в Гц. Флэш-хроматографию осуществляли с применением силикагеля 60 (0,040-0,063 мм, Merck). Колоночную хроматографию проводили с применением силикагеля 60 (0,063-0,200 мм, Merck) или оксида алюминия (act III). Хроматографическое разделение на системе Sepacore осуществляли с использованием оборудования Supelco, колонок VersaFLASHTM, картриджей с оксидом кремния VersaPakTM, УФ-монитора Büchi C-630, насосного модуля Büchi C-605, коллектора фракций Büchi C-660, системы управления насосом Büchi C-615. Температуры плавления регистрировали на приборе для измерения температур плавления Büchi B-545 или определяли с помощью методики DSC (дифференциальной сканирующей калориметрии).
Жидкостная хроматография - масс-спектрометрия (ЖХ-МС): Система ЖХ-МС включала два микронасоса Perkin Elmer series 200. Насосы соединялись друг с другом 50 мкл Т-образным миксером, соединенным с автоматическим дозатором Gilson 215. Применялась следующая методика:
|
A=100% вода с 0,025% HCOOH и 10 ммоль NH4COOH pH ±3;
B=100% ACN с 0,025% HCOOH.
Автодозатор был снабжен 2 мкл инъекционной петлей и соединен с колонкой Waters Atlantis C18 30*4,6 с частицами неподвижной фазы размером 3 мкм. Колонку термостатировали при 40°C в печи для колонок Perkin Elmer series 200. Колонка была соединена с УФ-детектором Perkin Elmer series 200, снабженным 2,7 мкл проточной ячейкой. Детектор работал на длине волны 254 нм. УФ-детектор был соединен с масс-спектрометром Sciex API 150EX. Масс-спектрометр функционировал при следующих параметрах: диапазон сканирования 150-900 ат.ед.массы; полярность: положительная; режим сканирования: profile; разрешение Q1: UNIT; величина шага: 0,10 ат.ед.массы.; время одного прохождения: 0,500 сек; NEB: 10; CUR: 10IS:5200; TEM: 325; DF: 30; FP: 225 и EP: 10. К масс-спектрометру Sciex API 150 был присоединен детектор светорассеяния. В качестве детектора светорассеяния применялся Sedere Sedex 55, работавший при 50°C и давлении N2, равном 3 бар. Вся система управлялась компьютером G3 powermac.
Пример 2: Общие аспекты синтеза
Замещенные 4,5-дигидро-(1H)-пиразолы формулы (IIa) или замещенные 4,5-дигидро-3H-пиразолы формулы (IIb) можно получать по методике, раскрытой в WO 2008/034863, и затем можно вводить в реакцию с изотиоцианатами формулы R6-N=C=S, где R6 имеет указанные выше значения, с получением замещенных амидов 4,5-дигидро-(1H)-пиразол-1-карботиокислот формулы (IIIa) или замещенных 4,5-дигидро-(1Р)-пиразол-1-карбоксимидотиокислот формулы (IIIb). Соединения формул (IIIa) или (IIIb) можно алкилировать по атому S, например, алкилгалогенидом, таким как метилйодид, с получением соединений формулы (IV). Последнее соединение можно ввести во взаимодействие с производным сульфонамида формулы R7SO2NH2, где R7 имеет указанное выше значение, с получением соединений формулы (I). Специалист в данной области заметит, что группа S-алкил в данной конкретной реакции играет роль уходящей группы. В упомянутой выше схеме, R1-R7 имеют указанные выше значения. Соединения (IIa) и (IIb), как и соединения (IIIa) и (IIIb) являются таутомерами, и поэтому все эти соединения входят в объем настоящего изобретения. Соединения формул (IIIa), (IIIb) и (IV) являются новыми.
На схеме 1 изображен синтез соединений формулы (I):

Фармацевтически приемлемые соли соединений можно получать с использованием хорошо известных стандартных методик, например, смешивая соединения по настоящему изобретению с подходящей кислотой, например неорганической кислотой, такой как хлористоводородная кислота, или органической кислотой, такой как фумаровая кислота.
Выбор конкретных способов синтеза зависит от факторов, известных специалистам в данной области. Например, от совместимости функциональных групп с применяемыми реагентами, возможности использования защитных групп, катализаторов, активирующих и сшивающих реагентов и итоговых структурных особенностей получаемого конечного соединения. Например, перед взаимодействием с R6-NCS можно защитить аминогруппы в R2, R3 или R4.
Пример 3: синтез соединений по настоящему изобретению
Этиламид 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты (соединение 1, синтез с небольшим количеством реагентов)

2,3-Диазаспиро[4.4]нон-2-ен (полученный по методике, описанной в WO 2008/034863) (1,05 г, 1 мольн. экв.) и этилизотиоцианат (0,95 мл, 1,3 мольн. экв.) добавляли к 10 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение 2,5 часов, перемешивая магнитной мешалкой. Добавляли силикагель и удаляли летучие компоненты в вакууме. Продукт очищали флэш-хроматографией на силикагеле (Et2O:PA=1:2) и после выпаривания летучих компонентов, перемешивали с диизопропиловым эфиром и собирали фильтрованием, получая 0,57 г (32%) этиламида 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,24 (т, J=7,2 Гц, 3H), 1,64-1,86 (м, 8H), 3,68 (д.кв, J=7,2, 5,5 Гц, 2H), 4,00 (с, 2H), 6,80 (с, 1H), 7,08-7,18 (ушир.с, 1H).
Этиламид 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты (соединение 1, синтез с большим количеством реагентов)

Гидрохлорид 2,3-диазаспиро[4.4]нон-1-ена (15,4 г, 95,9 ммоль; получен при реакции 2,3-диазаспиро[4.4]нон-1-ена, синтезированного по методике, описанной в WO 2008/034863, с HCl в смеси изопропанол/толуол) добавляли к смеси 70 мл метанола и 30 мл воды. К полученной смеси с помощью капельной воронки добавляли этилизотиоцианат (10,09 г, 115,1 ммоль) и промывали воронку 40 мл метанола. При 30°C по каплям в течение 10 минут добавляли диизопропилэтиламин (14,8 г, 114,5 ммоль) и промывали капельную воронку 7 мл воды. Реакционную смесь перемешивали при 30°C в течение 1 часа, затем смесь охлаждали до 10°C в течение 1 часа и затем перемешивали при этой температуре в течение еще 2 часов. Осадок отделяли фильтрованием, дважды промывали 20 мл холодной смеси метанола и воды 3:1 и высушивали при 50°C, получая 16,8 г (83%) этиламида 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты в виде твердого вещества цвета от не совсем белого до белого. Спектр 1H ЯМР идентичен спектру вещества, полученного в синтезе с малым количеством реагентов (см. выше).
Метиловый эфир N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты (соединение 2)

Этиламид 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты (0,55 г, 1 мольн. экв.) растворяли в 15 мл MeOH, добавляли йодметан (3,4 мл, 21 мольн. экв.) и перемешивали реакционную смесь магнитной мешалкой при нагревании до 45°C в течение 2 часов. Летучие компоненты удаляли в вакууме. Остаток смешивали с дихлорметаном (DCM) и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 0,57 г (97%) метилового эфира N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,16 (т, J=7,3 Гц, 3H), 1,64-1,80 (м, 8H), 2,46 (с, 3H), 3,54 (кв, J=7,3 Гц, 2H), 3,57 (с, 2H), 6,72 (с, 1H).
Метиловый эфир N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты (соединение 2)

К раствору этиламида 2,3-диазаспиро[4.4]нон-3-ен-2-карботиокислоты (1,0 г, 4,7 ммоль) в 10 мл метанола добавляли метил п-толуолсульфонат (1,1 г, 5,7 ммоль). Смесь кипятили с обратным холодильником в течение 48 часов и концентрировали при пониженном давлении. Остаток растирали с 30 мл диэтилового эфира и удаляли из выделенного маслянистого продукта все летучие компоненты при пониженном давлении. Оставшееся масло смешивали с 40 мл дихлорметана и 3 раза экстрагировали насыщенным водным раствором NaHCO3. Органический слой высушивали над MgSO4, фильтровали и упаривали досуха при пониженном давлении, получая 0,33 г (1,5 ммоль, 31%) метилового эфира N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты в виде светло-коричневого масла. Спектр 1H ЯМР идентичен спектру вещества, полученного в синтезе с использованием йодметана в качестве метилирующего агента (см. выше).
N-(4-{[(2,3-Диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамид (соединение 3, синтез новым способом)

Метиловый эфир N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты (157 мг, 1 мольн. экв.) и 4-ацетамидобензолсульфонамид (157 мг, 1,05 мольн. экв.) смешивали с 5 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи, перемешивая магнитной мешалкой, и удаляли летучие вещества в вакууме. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (этилацетат) позволяла получить 236 мг (87%) N-(4-{[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамида. 1H ЯМР (400 МГц, CDCl3) δ 1,14 (т, J=7,2 Гц, 3H), 1,62-1,83 (м, 8H), 2,20 (с, 3H), 3,43-3,51 (м, 2H), 3,80 (с, 2H), 6,80 (с, 1H), 6,87 (ушир.с, 1H), 7,56 (д, J=8,8 Гц, 2H), 7,77 (ушир.с, 1H), 7,83 (д, J=8,8 Гц, 2H).
N-(4-{[(2,3-Диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамид (соединение 3, синтез способом, раскрытым в WO 2009/115515)

Гидрохлорид N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксамидина (60 г, 260,08 ммоль; полученный реакцией N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксамидина, синтезированного способом, описанным в WO 2009/115515, с HCl в изопропаноле) растворяли в 1000 мл дихлорметана и добавляли 4-ацетиламинобензолсульфонилхлорид (60,7 г, 260,08 ммоль). В течение 20 минут при перемешивании механической мешалкой добавляли триэтиламин (131,6 г, 1300,4 ммоль) и затем перемешивали полученную смесь в течение ночи при комнатной температуре. Реакционную смесь экстрагировали водой (250 мл) и концентрировали органическую фазу при пониженном давлении (40°C, 600 мбар). Маслянистый остаток дважды упаривали совместно с 96% этанолом (250 мл) и смешивали с 500 мл дихлорметана. Органическую фазу экстрагировали 1 н. водным раствором HCl (75 мл), затем дважды водой (200 мл) и упаривали досуха при пониженном давлении, получая 78 г (199,2 ммоль, 77%) N-(4-{[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамида. Спектр 1H ЯМР идентичен спектру вещества, полученного новым способом синтеза (см. выше).
4-Амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамид (синтез соединения 4 из соединения 3)

N-(4-{[(2,3-Диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]сульфамоил}фенил)ацетамид (179 г) растворяли в 2685 мл EtOH и добавляли 1370 мл 1М HCl (3 мольн. экв.). Полученную смесь перемешивали при 55°C в течение 45 часов и концентрировали при пониженном давлении. Остаток смешивали с 2200 мл бутилацетата и добавляли 3800 мл 5% водного раствора NaHCO3 при перемешивании в течение 55 минут. Отделяли органическую фазу и водную фазу экстрагировали 200 мл бутилацетата. Объединенные органические слои промывали 1300 мл воды и упаривали досуха, получая 133 г неочищенного продукта. Остаток перекристаллизовывали из 800 мл EtOH и высушивали в вакууме при 50°C, получая 87,8 г (55%) 4-амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,14 (т, J=7,22 Гц, 3H), 1,47-1,89 (м, 8H), 3,35-3,57 (м, 2H), 3,79 (с, 2H), 4,02 (ушир.с, 2H), 6,65 (д, J=8,73 Гц, 2H), 6,78 (с, 1H), 6,91 (ушир.с,, 1H), 7,70 (д, J=8,73 Гц, 2H).
Этиламид 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты (соединение 5)

4,4-Диметил-4,5-дигидро-3H-пиразол (синтезированный по методике, описанной в WO 2008/034863) (10 г, 1 мольн. экв.) и этилизотиоцианат (11,6 мл, 1,3 мольн. экв.) добавляли к 100 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Добавляли силикагель и удаляли летучие вещества в вакууме. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:2) позволила получить 15,2 г (80%) этиламида 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,19-1,30 (м, 9H), 3,63-3,72 (м, 2H), 3,93 (с, 2H), 6,74 (с, 1H), 7,14 (ушир.с, 1H).
Метиловый эфир N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 6)

Этиламид 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты (15 г, 1 мольн. экв.) растворяли в 300 мл метанола, добавляли 50,4 мл (10 мольн. экв.) йодметана и нагревали реакционную смесь при 50°C в течение 3 часов. Летучие компоненты удаляли в вакууме. Остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 15,5 г (96%) метилового эфира N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,16 (т, J=7,3 Гц, 3H), 1,20 (с, 6H), 2,45 (с, 3H), 3,49 (с, 2H), 3,53 (кв, J=7,3 Гц, 2H), 6,66 (с, 1H).
3-Хлор-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамид (соединение 7)

Метиловый эфир N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,75 г, 1 мольн. экв.) и 3-хлорбензолсульфонамид (0,76 г, 1,05 мольн. экв.) добавляли к 10 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи и удаляли летучие компоненты в вакууме. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O) приводила к получению 1,26 г (98%) 3-хлор-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамида.
1H ЯМР (400 МГц, CDCl3) δ 1,17 (т, J=7,2 Гц, 3H), 1,23 (с, 6H), 3,43-3,52 (м, 2H), 3,79 (ушир.с, 2H), 6,77 (с, 1H), 6,60-6,90 (ушир.с, 1H), 7,37-7,42 (м, 1H), 7,43-7,47 (м, 1H), 7,81-7,85 (м, 1H), 7,94 (м, 1H).
3-Хлор-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]-4-метоксибензолсульфонамид (соединение 8)

Метиловый эфир N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,75 г, 1 мольн. экв.) и 3-хлор-3-метоксибензолсульфонамид (0,94 г, 1,05 мольн. экв.) добавляли к 10 мл ацетонитрила. Реакционную смесь кипятили в течение ночи с обратным холодильником и удаляли летучие компоненты в вакууме. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O) позволяла получить 1,43 г (97%) 3-хлор-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]-4-метоксибензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,17 (т, J=7,3 Гц, 3H), 1,22 (с, 6H), 3,43-3,52 (м, 2H), 3,77 (ушир.с, 2H), 3,95 (с, 3H), 6,75 (с, 1H), 6,96 (д, J=8,6 Гц, 1H), 6,70-6,90 (ушир.с, 1H) 7,82 (дд, J=8,6, 2,3 Гц, 1H), 7,95 (д, J=2,3 Гц, 1H).
Этиламид 3-этил-4,5-дигидропиразол-1-карботиокислоты (соединение 9)

3-Этил-4,5-дигидро-1H-пиразол (1,25 г, 1 мольн. экв.) (синтезированный по методике, описанной в WO 2008/034863) и этилизотиоцианат (1,45 мл, 1,3 мольн. экв.) добавляли к 10 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение 5 часов, добавляли силикагель и удаляли летучие компоненты в вакууме. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:1) приводила к получению 1,54 г (65%) этиламида 3-этил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,18 (т, J=7,5 Гц, 3H), 1,25 (т, J=7,2 Гц, 3H), 2,38 (кв, J=7,5 Гц, 2H), 2,83 (т, J=9,9 Гц, 2H), 3,63-3,72 (м, 2H), 4,19 (т, J=9,9 Гц, 2H), 7,06 (ушир.с, 1H).
Метиловый эфир 3,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 10)

Этиламид 3-этил-4,5-дигидропиразол-1-карботиокислоты (1,51 г, 1 мольн. экв.) растворяли в 30 мл метанола, добавляли йодметан (5,1 мл, 10 мольн. экв.) и нагревали реакционную смесь при 50°C в течение 1 часа. Летучие компоненты удаляли в вакууме. Остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 1,44 г (89%) метилового эфира 3,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,12-1,21 (м, 6H), 2,39 (кв, J=7,4 Гц, 2H), 2,48 (c, 3H), 2,70 (т, J=9,7 Гц, 2H), 3,52 (кв, J=7,2 Гц, 2H), 3,75 (т, J=9,7 Гц, 2H).
2-Хлор-N-[этиламино-(3-этил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамид (соединение 11)

Метиловый эфир 3,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты (1,42 г, 1 мольн. экв.) и 2-хлорбензолсульфонамид (1,43 г, 1,05 мольн. экв.) добавляли к 20 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи и удаляли в вакууме летучие компоненты. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Остаток, полученный после очистки флэш-хроматографией на силикагеле (Et2O), растирали с диизопропиловым эфиром, получая 2,08 г (81%) 2-хлор-N-[этиламино-(3-этил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,15 (т, J=7,3 Гц, 3H), 1,17 (т, J=7,3 Гц, 3H), 2,38 (кв, J=7,3 Гц, 2H), 2,80 (т, J=9,8 Гц, 2H), 3,44-3,53 (м, 2H), 4,11 (т, J=9,8 Гц, 2H), 6,73 (ушир.с, 1H), 7,33 (дт, J=7,6, 2,0 Гц, 1H), 7,38 (дт, J=7,6, 2,0 Гц, 1H), 7,46 (дд, J=7,6, 2,0 Гц, 1H), 8,17 (дд, J=7,6, 2,0 Гц, 1H).
N-(2-Бромфенил)-2,2,2-трифторацетамид (соединение 12)
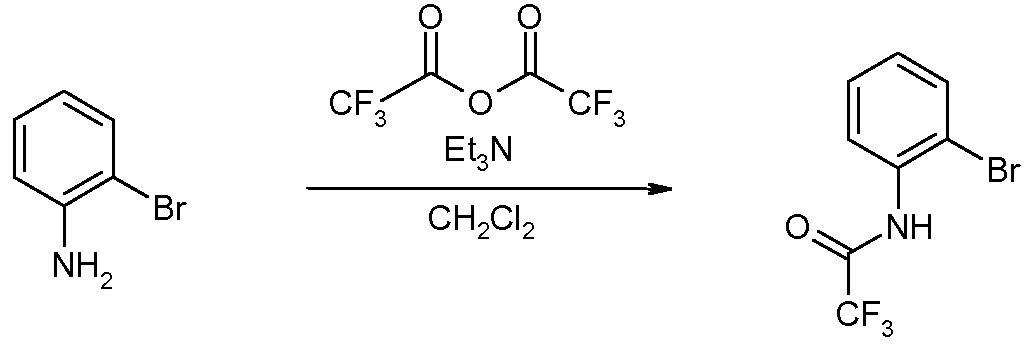
2-Броманилин (24,9 г, 1 мольн. экв.) растворяли в 200 мл дихлорметана, добавляли триэтиламин (28 мл, 1,4 мольн. экв.), охлаждали реакционную смесь до 0°C и по каплям добавляли трифторуксусный альдегид (24 мл, 1,2 мольн. экв.) (поддерживая температуру ниже 10°C). По окончании добавления смесь нагревали до комнатной температуры и перемешивали в течение еще 2 часов. Полученную смесь гасили водой, отделяли органический слой, высушивали над Na2SO4, фильтровали и упаривали при пониженном давлении. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:6) позволяла получить 34,6 г (89%) N-(2-бромфенил)-2,2,2-трифторацетамида. 1H ЯМР (400 МГц, CDCl3) δ 7,12 (дт, J=8,0, 1,3 Гц, 1H), 7,39 (дт, J=8,0, 1,3 Гц, 1H), 7,61 (дд, J=8,0, 1,3 Гц, 1H), 8,31 (дд, J=8,0, 1,3 Гц, 1H), 8,45 (ушир.с, 1H).
3-Бром-4-(2,2,2-трифторацетиламино)бензолсульфонилхлорид (соединение 13)

N-(2-Бромфенил)-2,2,2-трифторацетамид (15,0 г, 1,0 экв.) при охлаждении на ледяной бане четырьмя порциями добавляли к хлорсульфоновой кислоте (18,7 мл, 5 мольн. экв.). Убирали ледяную баню, нагревали смесь до комнатной температуры и затем до 80°C. После перемешивания в течение 1 часа смесь охлаждали до комнатной температуры и выливали на лед. Полученную смесь экстрагировали дихлорметаном, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 17,4 г, (85%) 3-бром-4-(2,2,2-трифторацетиламино)бензолсульфонилхлорида. 1H ЯМР (400 МГц, CDCl3) δ 8,09 (дд, J=9,0, 2,0 Гц, 1H), 8,30 (д, J=2,0 Гц, 1H), 8,69 (д, J=9,0 Гц, 1H), 8,71 (ушир.с,1H).
N-(2-Бром-4-сульфамоилфенил)-2,2,2-трифторацетамид (соединение 14)

3-Бром-4-(2,2,2-трифторацетиламино)бензолсульфонилхлорид (16,2 г, 1 мольн. экв.) растворяли в 150 мл ацетонитрила и охлаждали до 0°C. По каплям добавляли гидроксид аммония (20,8 мл, 3 мольн. экв.) и перемешивали реакционную смесь при комнатной температуре в течение 10 мин, причем за это время образовался белый осадок. Летучие компоненты удаляли при пониженном давлении, твердый остаток промывали водой и высушивали в вакууме, получая 14,3 г (94%) N-(2-бром-4-сульфамоилфенил)-2,2,2-трифторацетамида. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,59 (с, 2H), 7,69 (д, J=8,2 Гц, 1H), 7,88 (дд, J=8,2, 1,8 Гц, 1H), 8,14 (д, J=1,8 Гц, 1H), 11,55 (c, 1H).
N-(2-Бром-4-{[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]сульфамоил}фенил)-2,2,2-трифторацетамид (соединение 15)

Метиловый эфир N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (3,41 г, 1 мольн. экв.) и N-(2-бром-4-сульфамоилфенил)-2,2,2-трифторацетамид (6,24 г, 1,05 мольн. экв.) добавляли к 100 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи, после чего удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом, экстрагировали 2 н. NaOH, органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O) позволяла получить 7,1 г (83%) N-(2-бром-4-{[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]сульфамоил}фенил)-2,2,2-трифторацетамида. 1H ЯМР (400 МГц, CDCl3) δ 1,18 (т, J=7,3 Гц, 3H), 1,24 (с, 6H), 3,43-3,51 (м, 2H), 3,79 (ушир.с, 2H), 6,78 (с, 1H), 7,93 (дд, J=8,6, 2,0 Гц, 1H), 8,19 (д, J=2,0 Гц, 1H), 8,39 (д, J=8,6 Гц, 1H), 8,61 (ушир.с, 1H).
4-Амино-3-бром-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамид (соединение 16)

N-(2-Бром-4-{[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]сульфамоил}фенил)-2,2,2-трифторацетамид (7,0 г, 1 мольн. экв.) растворяли в 225 мл метанола, добавляли карбонат калия (10,3 г, 5 мольн. экв.) и воду (30 мл) и кипятили реакционную смесь с обратным холодильником в течение 2,5 часов. Выпаривали летучие компоненты при пониженном давлении, и остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и концентрировали на силикагеле. Очистка флэш-хроматографией на силикагеле (Et2O) позволяла получить 4,1 г (73%) 4-амино-3-бром-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,17 (т, J=7,3 Гц, 3H), 1,21 (с, 6H), 3,43-3,52 (м, 2H), 3,74 (ушир.с, 2H), 4,45 (ушир.с, 2H), 6,73 (с, 1H), 6,75 (д, J=8,4 Гц, 1H), 6,83-6,92 (ушир.с, 1H), 7,65 (дд, J=8,4, 2,0 Гц, 1H), 7,99 (д, J=2,0 Гц, 1H).
(4,4-Диметил-4,5-дигидропиразол-1-ил)этиламинометиленамид 2-трифторметил-1H-индол-5-сульфоновой кислоты (соединение 17)
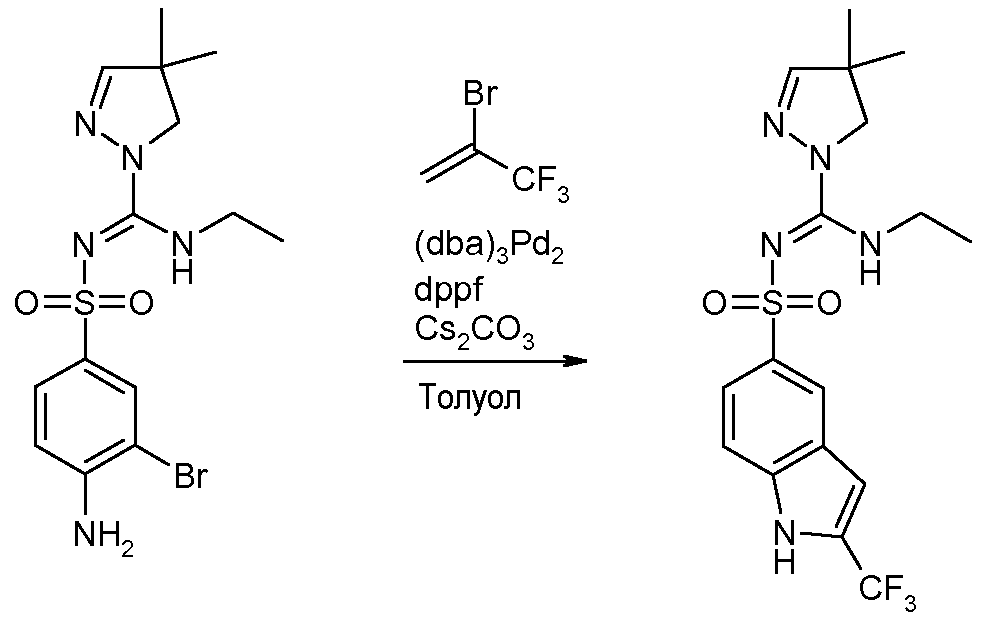
В сосуде из стекла Pyrex, продутом и заполненном азотом, растворяли 4-амино-3-бром-N-[(4,4-диметилпиразолидин-1-ил)этиламинометилен]бензолсульфонамид (2,23 г, 1 мольн. экв.) в 33 мл дегазированного толуола. После этого к реакционной смеси добавляли трис(дибензилиденацетон)дипалладий (0) (2,54 г, 0,5 мольн. экв.), 1,1'-бис(дифенилфосфино)ферроцен (4,61 г, 1,5 мольн. экв.), карбонат цезия (2,17 г, 1,2 мольн. экв.) и 2-бром-3,3,3-трифторпропен (1,94 г, 2 мольн. экв.). Реакционную смесь нагревали в течение ночи при 115°C, затем охлаждали, добавляли этилацетат и фильтровали смесь через hyflo. Очистка флэш-хроматографией на силикагеле (Et2O) с последующей очисткой препаративной ТСХ (Et2O) позволяли получить 254 мг (10%) (4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометиленамида 2-трифторметил-1H-индол-5-сульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,15 (т, J=7,3 Гц, 3H), 1,21 (c, 6H), 3,43-3,51 (м, 2H), 3,76 (ушир.с, 2H), 6,73 (с, 1H), 6,70-7,00 (ушир.с, 1H), 7,01 (с, 1H), 7,50 (д, J=8,7 Гц, 1H), 7,88 (дд, J=8,7, 1,5 Гц, 1H), 8,31 (ушир.с, 1H), 9,39 (ушир.с, 1H).
Этиламид 5-тиофен-3-ил-4,5-дигидропиразол-1-карботиокислоты (соединение 18)

5-Тиофен-3-ил-4,5-дигидро-1H-пиразол (1,82 г, 1 мольн. экв.) (синтезирован по методике, описанной в WO 2008/034863) и этилизотиоцианат (1,36 мл, 1,3 мольн. экв.) добавляли к 15 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение 5 часов и затем концентрировали на силикагеле при пониженном давлении. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:1) позволяла получить 0,70 г (26%) этиламида 5-тиофен-3-ил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,24 (т, J=7,2 Гц, 3H), 2,86 (ддд, J=18,5, 3,3, 1,7 Гц, 1H), 3,39 (ддд, J=18,5, 11,4, 1,7 Гц, 1H), 3,56-3,77 (м, 2H), 6,01 (дд, J=11,4, 3,3 Гц, 1H), 6,93 (дд, J=5,0, 1,0 Гц, 1H), 7,02 (т, J=1,7 Гц, 1H), 7,13 (м, 1H), 7,26 (м, 1H).
Метиловый эфир N-этил-5-тиофен-3-ил-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 19)

Этиламид 5-тиофен-3-ил-4,5-дигидропиразол-1-карботиокислоты (0,70 г, 1 мольн. экв.) растворяли в 14 мл метанола, добавляли йодметан (1,82 мл, 10 мольн. экв.) и реакционную смесь нагревали при 50°C в течение 1 часа. Летучие компоненты удаляли в вакууме, остаток смешивали с дихлорметаном и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (EtOAc:MeOH=9:1) позволяла получить 0,48 г (64%) метилового эфира N-этил-5-тиофен-3-ил-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,03 (т, J=7,3, 3H), 2,44 (с, 3H), 2,84 (ддд, J=18,1, 10,4, 1,5 Гц, 1H), 3,23-3,51 (м, 3H), 5,57 (т, J=10,4 Гц, 1H), 6,87 (ушир.с, 1H), 7,00 (д, J=4,8, 1H), 7,13 (д, J=3,0, 1H), 7,24 (дд, J=4,8, 3,0 Гц, 1H).
3-Хлор-N-[этиламино-(5-тиофен-3-ил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамид (соединение 20)
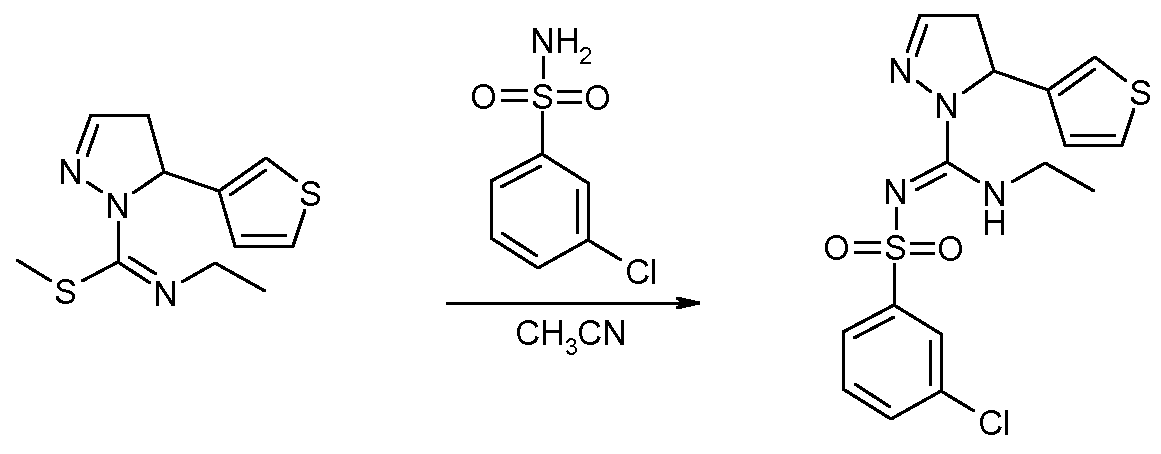
Метиловый эфир N-этил-5-тиофен-3-ил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,47 г, 1 мольн. экв.) и 3-хлорбензолсульфонамид (0,37 г, 1,05 мольн. экв. (0,37 г, 1,05 мольн. экв.) добавляли к 7 мл ацетонитрила. Реакционную смесь кипятили в течение ночи с обратным холодильником и удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O) позволяла получить 0,44 г (49%) 3-хлор-N-[этиламино-(5-тиофен-3-ил-4,5-дигидропиразол-1-ил)метилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,19 (т, J=7,2, 3H), 2,78 (ддд, J=18,6, 6,0, 1,4 Гц, 1H), 3,31 (ддд, J=18,6, 11,8, 1,4 Гц, 1H), 3,54-3,70 (м, 2H), 5,62 (дд, J=11,8, 6,0 Гц, 1H), 6,75 (д, J=4,3 Гц, 1H), 6,92 (д, J=2,0 Гц, 1H), 7,02 (ушир.с, 1H), 7,17-7,23 (м, 2H), 7,36 (м, 2H), 7,54 (ушир.с, 1H).
Амид 6-хлоримидазо[2,1-b]тиазол-5-сульфоновой кислоты (соединение 21)

6-Хлоримидазо[2,1-b]тиазол-5-сульфонилхлорид (2 г, 1 мольн. экв.) растворяли в 20 мл ацетонитрила и охлаждали до 0°C. По каплям добавляли гидроксид аммония (3,7 мл, 3 мольн. экв.) и перемешивали реакционную смесь при комнатной температуре в течение 10 мин, причем в течение этого времени образовывался белый осадок. Летучие вещества удаляли при пониженном давлении, твердый остаток промывали водой и высушивали в вакууме, получая 1,62 г (88%) амида 6-хлоримидазо[2,1-b]тиазол-5-сульфоновой кислоты. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,62 (д, J=4,6 1H), 7,98 (д, J=4,6 Гц, 1H), 8,00 (ушир.с, 2H).
Метиламид 8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карботиокислоты (соединение 22)

8-Окса-2,3-диазаспиро[4.5]дец-2-ен (0,8 г, 1 мольн. экв.) (синтезированный по методике, описанной в WO 2008/034863) и метилизотиоцианат (0,54 г, 1,3 мольн. экв.) добавляли к 10 мл этанола и кипятили реакционную смесь с обратным холодильником в течение 5 часов. Добавляли силикагель и удаляли летучие компоненты при пониженном давлении. Очистка флэш-хроматографией на силикагеле (Et2O) позволяла получить 0,52 г (35%) метиламида 8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,52-1,59 (м, 3H), 1,82-1,90 (м, 2H), 3,17 (д, J=5,0, 3H), 3,56-3,64 (м, 2H), 3,86-3,92 (м, 2H), 4,11 (с, 2H), 6,80 (с, 1H), 7,21 (ушир.с, 1H).
Метиловый эфир N-метил-8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карбоксимидотиокислоты (соединение 23)

Метиламид 8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карботиокислоты (0,50 г, 1 мольн. экв.) растворяли в 10 мл метанола, добавляли йодметан (1,2 мл, 10 мольн. экв.) и нагревали реакционную смесь при 50°C в течение 5 часов. Летучие компоненты удаляли при пониженном давлении, остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 0,43 г (99%) метилового эфира N-метил-8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,53-1,60 (м, 2H), 1,80-1,88 (м, 2H), 2,47 (с, 3H), 3,26 (с, 3H), 3,56-3,64 (м, 2H), 3,68 (с, 2H), 3,83-3,89 (м, 2H), 6,73 (с, 1H).
Метиламино-(8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-ил)метиленамид 6-хлоримидазо[2,1-b]тиазол-5-сульфоновой кислоты (соединение 24)

Метиловый эфир N-метил-8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-карбоксимидотиокислоты (0,42 г, 1 мольн. экв.) и амид 6-хлоримидазо[2,1-b]тиазол-5-сульфоновой кислоты (0,46 г, 1,05 мольн. экв.) добавляли к 7 мл ацетонитрила и кипятили реакционную смесь с обратным холодильником в течение ночи. Летучие компоненты удаляли при пониженном давлении и остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (EtOAc) позволяла получить 0,56 г (69%) метиламино-(8-окса-2,3-диазаспиро[4.5]дец-3-ен-2-ил)метиленамида 6-хлоримидазо[2,1-b]тиазол-5-сульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,51-1,57 (м, 2H), 1,80-1,89 (м, 2H), 3,10 (д, J=5,0 Гц, 3H), 3,51-3,59 (м, 2H), 3,83-3,90 (м, 4H), 3,89 (с, 2H), 6,89 (с, 1H), 6,99 (д, J=4,6 Гц, 1H), 7,12 (ушир.с, 1H), 8,01 (д, J=4,6 Гц, 1H).
Этиламид 4-этил-4,5-дигидропиразол-1-карботиокислоты (соединение 25)

4-Этил-4,5-дигидро-1H-пиразол (2,68 г, 1 мольн. экв.) (синтезированный по методике, описанной в WO 2008/034863) и этилизотиоцианат (3,11 мл, 1,3 мольн. экв.) добавляли к 20 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение ночи, добавляли силикагель и удаляли летучие компоненты при пониженном давлении. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:3) позволяла получить 1,80 г (36%) этиламида 4-этил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 0,99 (т, J=7,5 Гц, 3H), 1,25 (т, J=7,2 Гц, 3H), 1,47-1,71 (м, 2H), 3,08-3,18 (м, 1H), 3,63-3,72 (м, 2H), 3,86 (дд, J=11,5, 7,1 Гц, 1H), 4,25 (т, J=11,5 Гц, 1H), 6,90 (д, J=1,5 Гц, 1H), 7,12 (ушир.с, 1H).
Метиловый эфир 4,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 26)

Этиламид 4-этил-4,5-дигидропиразол-1-карботиокислоты (1,80 г, 1 мольн. экв.) растворяли в 36 мл метанола, добавляли йодметан (6,1 мл, 10 мольн. экв.) и нагревали реакционную смесь при 50°C в течение 4 часов. Летучие компоненты удаляли при пониженном давлении, остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 1,68 г (87%) метилового эфира 4,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 0,98 (т, J=7,5 Гц, 3H), 1,16 (т, J=7,3 Гц, 3H), 1,45-1,70 (м, 2H), 2,45 (с, 3H), 2,97-3,07 (м, 1H), 3,44 (дд, J=11,0, 8,3 Гц, 1H), 3,51-3,58 (м, 2H), 3,83 (т, J=11,0 Гц, 1H), 6,81 (с, 1H).
Этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамид пиперидин-1-сульфоновой кислоты (соединение 27)

Метиловый эфир 4,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,70 г, 1 мольн. экв.) и амид пиперидин-1-сульфоновой кислоты (0,61 г, 1,05 мольн. экв.) добавляли к 7 мл ацетонитрила и кипятили реакционную смесь с обратным холодильником в течение ночи. Летучие компоненты удаляли при пониженном давлении, остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:PA=2:1) позволяла получить 1,12 г (96%) этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида пиперидин-1-сульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 0,99 (т, J=7,5 Гц, 3H), 1,21 (т, J=7,2 Гц, 3H), 1,45-1,72 (м, 8H), 3,07-3,17 (м, 5H), 3,48-3,57 (м, 2H), 3,73 (дд, J=11,0, 7,7 Гц, 1H), 4,08-4,19 (м, 1H), 6,58 (ушир.с,1H), 6,87 (д, J=1,3 Гц, 1H).
Амид транс-2-фенилэтенсульфоновой кислоты (соединение 28)

Транс-2-фенилэтенсульфонилхлорид (3,3 г, 1 мольн. экв.) растворяли в 33 мл ацетонитрила и охлаждали до 0°C. По каплям добавляли гидроксид аммония (7,7 мл, 3 экв.) и перемешивали реакционную смесь при комнатной температуре в течение 10 мин. Летучие компоненты удаляли при пониженном давлении, твердый остаток промывали водой и высушивали в вакууме, получая 1,13 г (38%) амида транс-2-фенилэтенсульфоновой кислоты. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,11 (ушир.с, 2H), 7,23 (д, J=16,0 Гц, 1H), 7,31 (д, J=16,0 Гц, 1H), 7,41-7,45 (м, 3H), 7,64-7,71 (м, 2H).
Этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамид транс-2-фенилэтенсульфоновой кислоты (соединение 29)

Метиловый эфир 4,N-диэтил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,70 г, 1 мольн. экв.) и амид транс-2-фенилэтенсульфоновой кислоты (0,68 г, 1,05 мольн. экв.) добавляли к 7 мл ацетонитрила и кипятили реакционную смесь с обратным холодильником в течение ночи. Летучие компоненты удаляли при пониженном давлении и остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:PA=2:1) позволяла получить 1,00 г (81%) этиламино-(4-этил-4,5-дигидропиразол-1-ил)метиленамида транс-2-фенилэтенсульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 0,98 (т, J=7,5 Гц, 3H), 1,21 (т, J=7,2 Гц, 3H), 1,46-1,70 (м, 2H), 3,06-3,16 (м, 1H), 3,51-3,59 (м, 2H), 3,74 (дд, J=11,3, 7,5 Гц, 1H), 4,13 (т, J=11,3 Гц, 1H), 6,70-6,92 (м, 1H), 6,92 (д, J=1,3 Гц, 1H), 6,97 (д, J=15,4 Гц, 1H), 7,35-7,41 (м, 3H), 7,44 (д, J=15,4 Гц, 1H), 7,46-7,50 (м, 2H).
Амид 5-хлортиофен-2-сульфоновой кислоты (соединение 30)

5-Хлортиофен-2-сульфонилхлорид (3 г, 1 мольн. экв.) растворяли в 30 мл ацетонитрила и охлаждали до 0°C. По каплям добавляли гидроксид аммония (6,5 мл, 3 мольн. экв.) и перемешивали реакционную смесь при комнатной температуре в течение 10 мин. Летучие компоненты удаляли при пониженном давлении, твердый остаток промывали водой и высушивали в вакууме, получая 2,49 г (91%) амида 5-хлортиофен-2-сульфоновой кислоты. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,21 (д, J=4,0 Гц, 1H), 7,43 (д, J=4,0 Гц, 1H), 7,79 (ушир.с, 2H).
Амид 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты (соединение 31)

4,4-Диметил-4,5-дигидро-3H-пиразол (3,0 г, 1 мольн. экв.) (синтезирован по методике, описанной в WO 2008/034863) и триметилсилилизотиоцианат (5,6 мл, 1,3 мольн. экв.) добавляли к 30 мл этанола и кипятили реакционную смесь с обратным холодильником в течение 5 часов. Добавляли силикагель и удаляли летучие компоненты при пониженном давлении. Очистка флэш-хроматографией на силикагеле (Et2O:PA=2:1) позволяла получить 3,91 г (81%) амида 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,27 (с, 6H), 3,94 (с, 2H), 5,82-6,34 (ушир.с,1H), 6,50-7,00 (ушир.с,1H), 6,80 (с, 1H).
Метиловый эфир 4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 32)

Амид 4,4-диметил-4,5-дигидропиразол-1-карботиокислоты (1,50 г, 1 мольн. экв.) растворяли в 30 мл метанола, добавляли йодметан (5,9 мл, 10 мольн. экв.) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении, остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха, получая 1,53 г (94%) метилового эфира 4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,24 (с, 6H), 2,32 (с, 3H), 3,65 (с, 2H), 6,63 (с, 1H), 6,66-6,85 (ушир.с, 1H).
Амино-(4,4-диметил-4,5-дигидропиразол-1-ил)метиленамид 5-хлортиофен-2-сульфоновой кислоты (соединение 33)

Метиловый эфир 4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (1,0 г, 1 мольн. экв.) и амид 5-хлортиофен-2-сульфоновой кислоты (1,21 г, 1,05 мольн. экв.) добавляли к 10 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи и удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:PA=2:1) приводила к получению 1,58 г (80%) амино-(4,4-диметил-4,5-дигидропиразол-1-ил)метиленамида 5-хлортиофен-2-сульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,25 (с, 6H), 3,63 (с, 2H), 6,00-6,50 (ушир.с, 1H), 6,79 (с, 1H), 6,85 (д, J=4,0 Гц, 1H), 7,10-7,35 (ушир.с, 1H), 7,37 (д, J=4,0 Гц, 1H).
(2,2,2-Трифторэтил)амид 4-этил-4,5-дигидропиразол-1-карботиокислоты (соединение 34)

Раствор 2,2,2-трифторэтиламина (3,2 мл, 1 мольн. экв.) в 60 мл ацетонитрила добавляли при перемешивании и комнатной температуре к раствору 1,1'-тиокарбонилдиимидазола (7,4 г, 2,1 мольн. экв.) в 100 мл ацетонитрила. Реакционную смесь перемешивали в течение ночи и добавляли к ней 4-этил-4,5-дигидро-1H-пиразол (1,96 г, 1 мольн. экв.) (синтезирован по методике, описанной в WO 2008/034863). Через 1 час удаляли летучие компоненты при пониженном давлении и остаток очищали флэш-хроматографией на силикагеле (Et2O:PA=1:3), получая 2,85 г (60%) (2,2,2-трифторэтил)амида 4-этил-4,5-дигидропиразол-1-карботиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,5 Гц, 3H), 1,50-1,74 (м, 2H), 3,13-3,23 (м, 1H), 3,86 (дд, J=11,6, 7,1 Гц, 1H), 4,27 (т, J=11,6 Гц, 1H), 4,44 (м, 2H) 6,99 (д, J=1,5 Гц, 1H), 7,32-7,40 (ушир.с, 1H).
Метиловый эфир 4-этил-N-(2,2,2-трифторэтил)-4,5-дигидропиразол-1-карбоксимидотиокислоты (соединение 35)
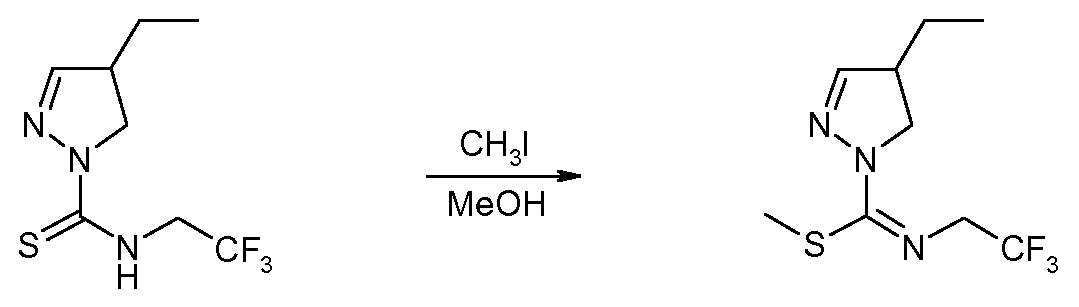
(2,2,2-Трифторэтил)амид 4-этил-4,5-дигидропиразол-1-карботиокислоты (2,80 г, 1 мольн. экв.) растворяли в 56 мл метанола, добавляли йодметан (7,3 мл, 10 мольн. экв.) и нагревали реакционную смесь при 50°C в течение 4 часов. Летучие компоненты удаляли при пониженном давлении, остаток смешивали с DCM и экстрагировали 5% водным раствором NaHCO3. Органический слой дважды промывали водой, высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:1) позволяла получить 0,57 г (19%) метилового эфира 4-этил-N-(2,2,2-трифторэтил)-4,5-дигидропиразол-1-карбоксимидотиокислоты. 1H ЯМР (400 МГц, CDCl3) δ 0,99 (т, J=7,5 Гц, 3H), 1,46-1,70 (м, 2H), 2,48 (с, 3H), 3,01-3,11 (м, 1H), 3,50 (дд, J=11,5, 7,8 Гц, 1H), 3,90 (т, J=11,5 Гц, 1H), 3,99-4,11 (м, 2H), 6,85 (д, J=1,5 Гц, 1H).
3-Хлор-N-[(4-этил-4,5-дигидропиразол-1-ил)-(2,2,2-трифторэтиламино)метилен]бензолсульфонамид (соединение 36)

Метиловый эфир 4-этил-N-(2,2,2-трифторэтил)-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,57 г, 1 мольн. экв.) и 3-хлор-бензолсульфонамид (3,0 г, 6,8 мольн. экв.) добавляли к 20 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение 72 часов и удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:PA=1:1) позволяла получить 0,36 г (38%) 3-хлор-N-[(4-этил-4,5-дигидропиразол-1-ил)-(2,2,2-трифторэтиламино)метилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,00 (т, J=7,5 Гц, 3H), 1,51-1,74 (м, 2H), 3,16-3,27 (м, 1H), 3,87 (дд, J=11,2, 7,5 Гц, 1H), 4,03-4,14 (м, 2H), 4,28 (т, J=11,2 Гц, 1H), 7,03 (д, J=1,5 Гц, 1H), 7,41 (т, J=7,8 Гц, 1H), 7,46-7,50 (м, 1H), 7,79-7,84 (м, 1H), 7,91-7,94 (м, 1H).
4-Амино-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамид (соединение 37)

Метиловый эфир N-этил-4,4-диметил-4,5-дигидропиразол-1-карбоксимидотиокислоты (0,75 г, 1 мольн. экв.) и сульфаниламид (0,65 г, 1,0 мольн. экв.) добавляли к 10 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение ночи и удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:EtOAc=1:1) позволяла получить 1,13 г (86%) 4-амино-N-[(4,4-диметил-4,5-дигидропиразол-1-ил)этиламинометилен]бензолсульфонамида. 1H ЯМР (400 МГц, CDCl3) δ 1,15 (т, J=7,2 Гц, 3H), 1,20 (с, 6H), 3,43-3,51 (м, 2H), 3,74 (ушир.с, 2H), 3,98 (ушир.с, 2H), 6,66 (д, J=8,6 Гц, 2H), 6,71 (с, 1H), 7,71 (д, J=8,6 Гц, 2H).
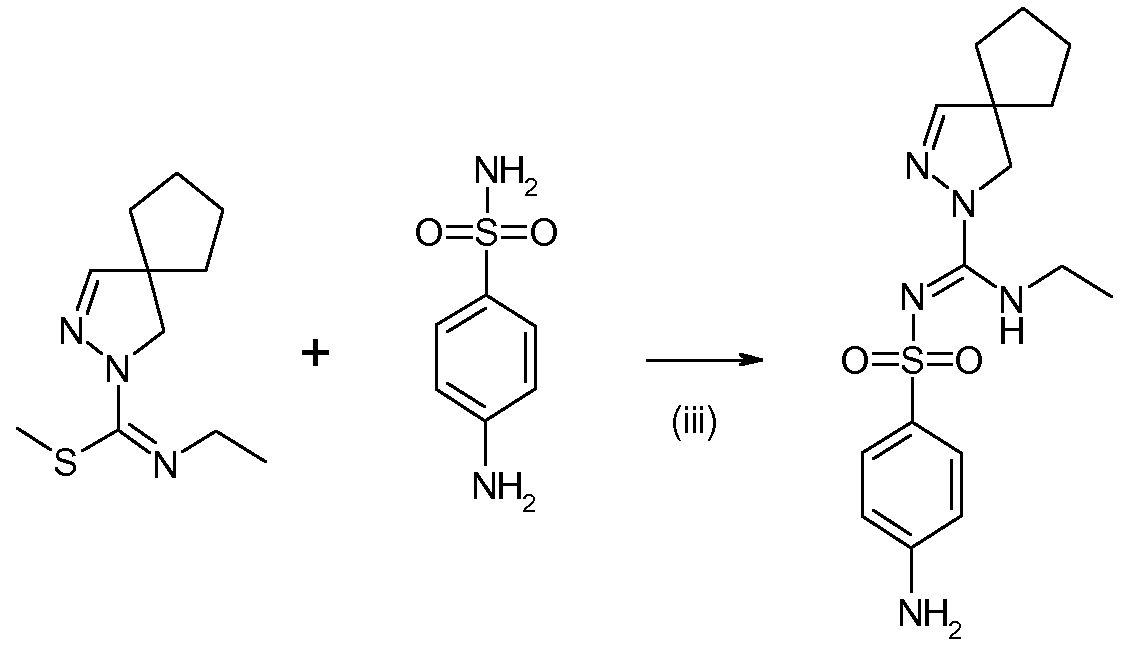
4-Амино-N-[(2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометилен]бензолсульфонамид (синтез соединения 4 из соединения 2)

В реакторе, снабженном газоочистителем, содержащем 50 мл 11% водного раствора NaOCl, 5 мл 50% водного раствора NaOH и 50 мл воды, смешивали метиловый эфир N-этил-2,3-диазаспиро[4,4]нон-3-ен-2-карбоксимидотиокислоты (4,00 г, 1 мольн. экв.) и сульфаниламид (3,06 г, 1 мольн. экв.) со 175 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение 18 ч и затем концентрировали примерно до половины объема отгонкой ацетонитрила при атмосферном давлении. После охлаждения до комнатной температуры добавляли 30 мл 2 н. NaOH и 100 мл DCM и перемешивали смесь в течение 5 минут. Разделяли слои и органическую фазу дважды промывали водой (твердый осадок, выделившийся во время второй промывки, объединяли с органической фазой). Органическую фазу концентрировали приблизительно до 1/3 исходного объема при пониженном давлении, твердые вещества отделяли фильтрованием, дважды промывали 5 мл DCM и высушивали в вакууме при 50°C, получая 3,14 г твердого вещества белого цвета. Еще 0,99 г твердого вещества выделяли из маточного раствора после оставления на ночь, что позволило поднять суммарный выход до 67%. 1H ЯМР (400 МГц, CD3CN) δ 1,04 (т, J=7,5 Гц, 3H), 1,58-1,83 (м, 8H), 3,36-3,44 (м, 2H), 3,68 (ушир.с, 2H), 4,63 (ушир.с, 2H), 6,64 (д, J=8,7 Гц, 2H), 6,95 (с, 1H), 3,96 (ушир.с, 1H), 7,54 (д, J=8,7 Гц, 2H). HR-MS [M+H]+ 350,1670; МС-МС [m/z] 257, 195, 178, 156 и 125 (идентично эталонному образцу соединения 4, полученного при снятии защиты соединения 3 действием кислоты).
(2,3-Диазаспиро[4.4]нон-3-ен-2-ил)этиламинометиленамид 1H-индол-5-сульфоновой кислоты (соединение 38)

Метиловый эфир N-этил-2,3-диазаспиро[4.4]нон-3-ен-2-карбоксимидотиокислоты (100 мг, 1 мольн. экв.) и амид 1H-индол-5-сульфоновой кислоты (92,5 мг, 1,05 мольн. экв.) добавляли к 3 мл ацетонитрила. Реакционную смесь кипятили в течение ночи с обратным холодильником и удаляли летучие компоненты при пониженном давлении. Остаток смешивали с этилацетатом и экстрагировали 2 н. NaOH. Органический слой высушивали над Na2SO4, фильтровали и упаривали досуха. Очистка флэш-хроматографией на силикагеле (Et2O:EtOAc=1:1) позволяла получить 152 мг (87%) (2,3-диазаспиро[4.4]нон-3-ен-2-ил)этиламинометиленамида 1H-индол-5-сульфоновой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,14 (т, J=7,2 Гц, 3H), 1,59-1,79 (м, 8H), 3,43-3,51 (м, 2H), 3,79 (ушир.с, 2H), 6,63-6,65 (м, 1H), 6,76 (с, 1H), 6,99 (ушир.с, 1H), 7,30 (т, J=2,8 Гц, 1H), 7,43 (д, J=8,6 Гц, 1H), 7,76 (дд, J=8,6, 1,8 Гц, 1H), 8,27 (ушир.с, 1H), 8,54 (ушир.с, 1H).
|


|

|

Индуцирующие апоптоз средства для лечения злокачественной опухоли и иммунных и аутоиммунных заболеваний
Апоптоз-индуцирующие средства для лечения рака и иммунных и аутоиммунных заболеваний
Производные (тио)морфолина в качестве модуляторов sip
Твердые дисперсии, содержащие ингибиторы киназ
Индуцирующие апоптоз средства для лечения злокачественной опухоли и иммунных и аутоиммунных заболеваний
Полученные экструзией расплава твердые дисперсии, содержащие индуцирующее апоптоз средство
Моногидрат производного азаадамантана
Полученные экструзией расплава твердые дисперсии, содержащие индуцирующее апоптоз средство
Интерлейкин-13-связывающие белки
Индуцирующие апоптоз средства для лечения злокачественной опухоли и иммунных и аутоиммунных заболеваний
Апоптоз-индуцирующие средства для лечения рака и иммунных и аутоиммунных заболеваний
Производные (тио)морфолина в качестве модуляторов sip
Твердые дисперсии, содержащие ингибиторы киназ
Индуцирующие апоптоз средства для лечения злокачественной опухоли и иммунных и аутоиммунных заболеваний
Полученные экструзией расплава твердые дисперсии, содержащие индуцирующее апоптоз средство
Моногидрат производного азаадамантана
Полученные экструзией расплава твердые дисперсии, содержащие индуцирующее апоптоз средство
Способ алкилирования при применении катализатора, содержащего цеолиты, имеющие в своем составе редкоземельные элементы, при высоком содержании церия, и гидрогенизирующий металл

