Результат интеллектуальной деятельности: СПОСОБЫ СИНТЕЗА СПИРО-ОКСИНДОЛЬНЫХ СОЕДИНЕНИЙ
Вид РИД
Изобретение
Перекрестная ссылка на родственную заявку
По данной формуле изобретения к заявке согласно § 119(e) 35 U.S.C. испрашивается приоритет по предварительной заявке на патент США № 61/251335, поданной 14 октября 2009 г. Раскрытие указанной заявки полностью включено сюда путем ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение касается улучшенных способов получения некоторых спиро-оксиндольных соединений, а также соответствующих различных промежуточных соединений. В частности, настоящее изобретение касается способов получения спиро-оксиндольных соединений, и соответствующих фармацевтически приемлемых солей, полезных при лечении опосредованных натриевыми каналами заболеваний или состояний, таких как боль, а также других заболеваний или состояний, связанных с посредничеством натриевых каналов.
Уровень техники изобретения
Натриевые каналы выполняют многочисленные функции в поддержании нормальных и патологических состояний, включая давно общепризнанную роль, которую потенциалзависимые натриевые каналы играют в генерации аномальной неврональной активности и невропатической или патологической боли. Повреждение периферических нервов после травмы или болезни может привести к изменениям активности натриевого канала и развитию аномальной афферентной активности, включая эктопические разряды от аксотомизированных афферентов и спонтанную активность сенсибилизированных интактных ноцицепторов. Такие изменения могут стать причиной длительной аномальной гиперчувствительности к обычно безвредному стимулу, или аллодинии. Примеры невропатической боли включают, но не в порядке ограничения, такие заболевания или состояния, как постгерпетическая невралгия, тригеминальная невралгия, диабетическая невропатия, хронический пояснично-крестцовый радикулит, фантомная боль в ампутированных конечностях и боль, возникающая при раке и химиотерапии, хроническая тазовая боль, комплексный региональный болевой синдром и сопутствующие невралгии.
Некоторые успехи в лечении симптомов невропатической боли были достигнуты при использовании лекарственных средств, таких как габапентин и, совсем недавно, прегабалин, в качестве краткосрочной терапии первой линии. Однако фармакотерапия невропатической боли обычно имеет ограниченный успех с незначительным откликом на обычно используемые болеутоляющие средства, такие как НПВС и опиаты. Следовательно, по-прежнему существует значительная потребность в изыскании новых методов терапии.
Продолжает существовать ограниченное число высокоэффективных блокаторов натриевых каналов с минимальными нежелательными клиническими действиями. Существует также нереализованная потребность медицины в эффективном и без нежелательных побочных действий лечении невропатической боли и других, связанных с натриевыми каналами патологических состояний.
Опубликованная патентная заявка PCT № WO 2006/110917, опубликованная патентная заявка PCT № WO 2010/45251 и патентная заявка PCT № PCT/US2010/040187 описывают некоторые спиро-оксиндольные соединения. Указанные соединения раскрыты там, как полезные для лечения заболеваний, опосредованных натриевыми каналами, преимущественно заболеваний, связанных с болью, состояний центральной нервной системы, таких как эпилепсия, тревожность, депрессия и биполярное расстройство; состояний сердечно-сосудистой системы, таких как аритмия, мерцательная аритмия и фибрилляция желудочков; состояний нервно-мышечной системы, таких как синдром усталых ног; для нейропротекции при ударе, травматическом повреждении нервов и рассеянном склерозе; и при каналопатиях, таких как эритромелалгия и наследственный синдром боли в прямой кишке.
Способы получения таких соединений и фармацевтические композиции, содержащие указанные соединения, также описаны в опубликованной патентной заявке PCT № WO 2006/110917, опубликованной патентной заявке PCT № WO 2010/45251 и патентной заявке PCT № PCT/US2010/040187.
Таким образом, существует потребность в улучшенных способах получения некоторых спиро-оксиндольных соединений.
Краткое описание изобретения
Настоящее изобретение касается способов получения некоторых спиро-оксиндольных соединений в виде отдельных стереоизомеров или отдельных энантиомеров, или их смесей, или в виде соответствующих фармацевтически приемлемых солей. Такие соединения полезны при лечении опосредованных натриевыми каналами заболеваний или состояний, таких как боль.
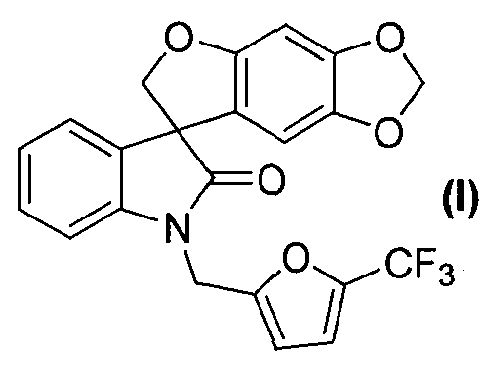
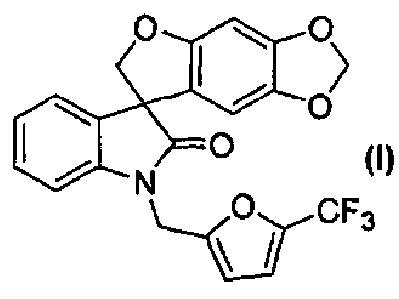
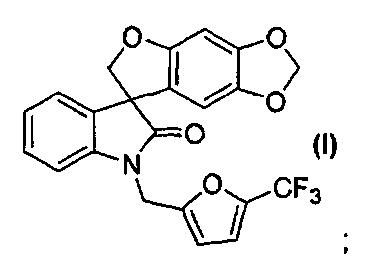
Таким образом, в одном из аспектов настоящее изобретение касается способа получения соединения формулы (I):

или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси;
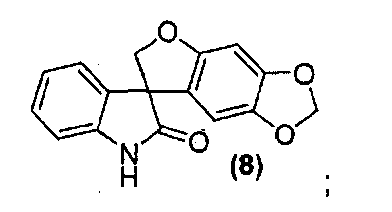
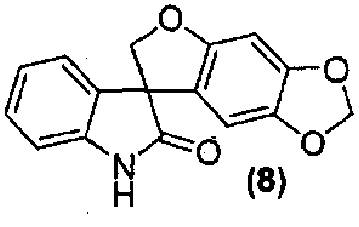
где способ включает обработку соединения формулы (8):
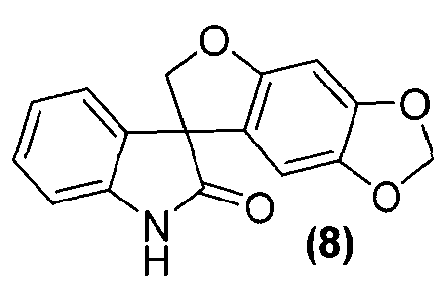
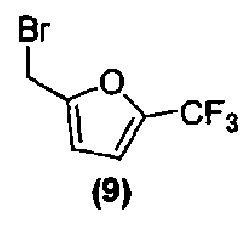
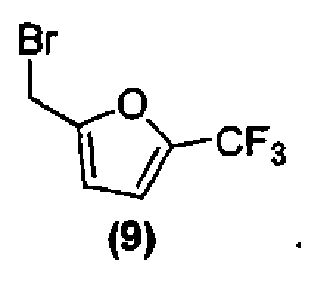
или его фармацевтически приемлемой соли соединением формулы (9):
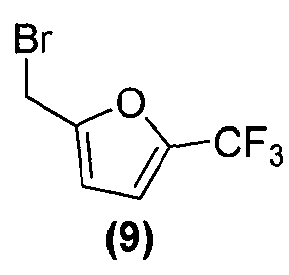
или его фармацевтически приемлемой солью, в подходящих условиях, с получением соединения формулы (I) или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси.
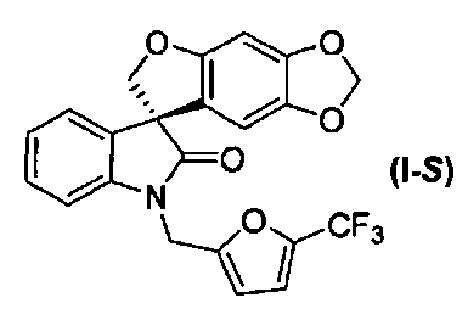
В другом аспекте настоящее изобретение касается способа получения соединения формулы (I-S):
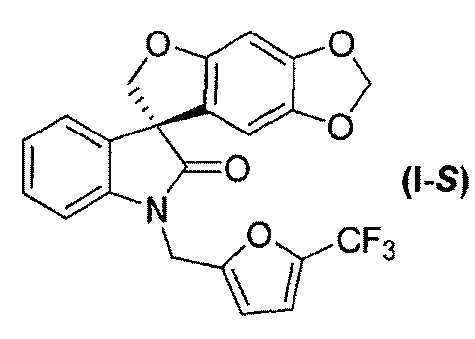
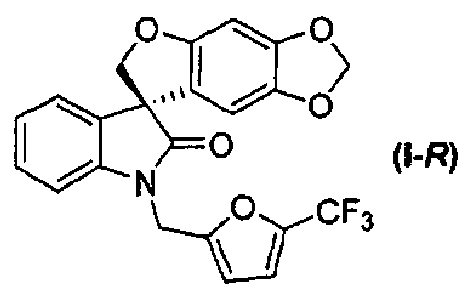
или его фармацевтически приемлемой соли и соединения формулы (I-R):
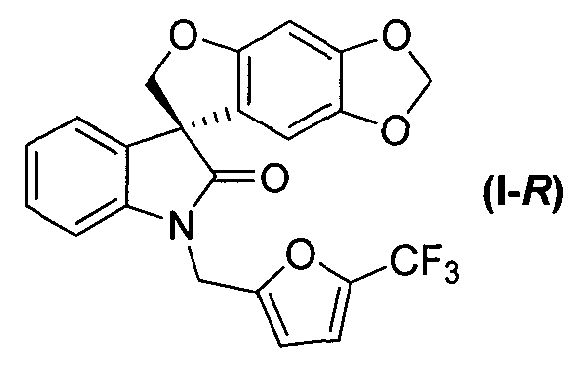
или его фармацевтически приемлемой соли, где способ включает разделение соединения формулы (I):
или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси в подходящих условиях с получением соединения формулы (I-S), или его фармацевтически приемлемой соли, и соединения формулы (I-R), или его фармацевтически приемлемой соли.
В другом аспекте настоящее изобретение касается способа получения соединения формулы (II):
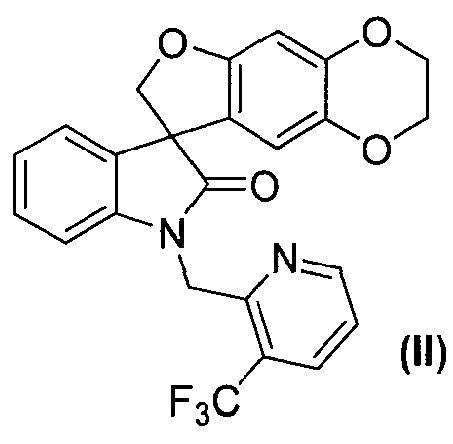
или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси;
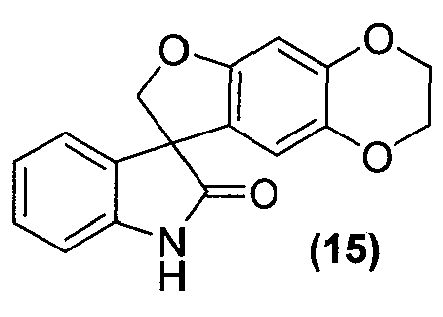
где способ включает обработку соединения формулы (15):
или его фармацевтически приемлемой соли соединением формулы (16):

или его фармацевтически приемлемой солью в подходящих условиях с получением соединения формулы (II) или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси.
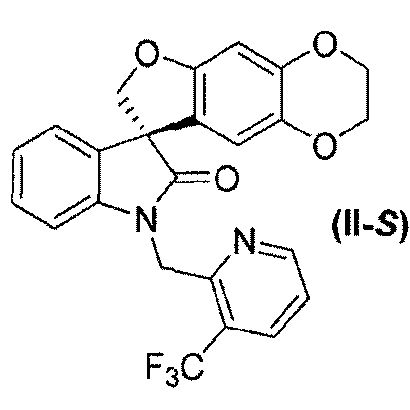
В другом аспекте настоящее изобретение касается способа получения соединения формулы (II-S):
или его фармацевтически приемлемой соли и соединения формулы (II-R):
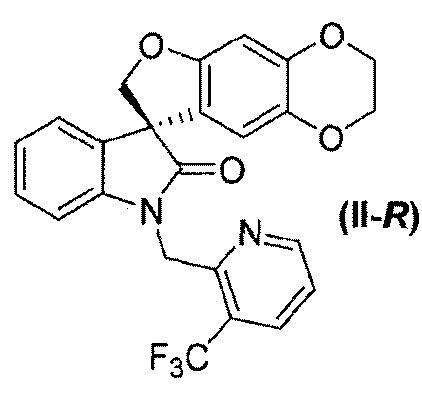
или его фармацевтически приемлемой соли, где способ включает разделение соединения формулы (II):
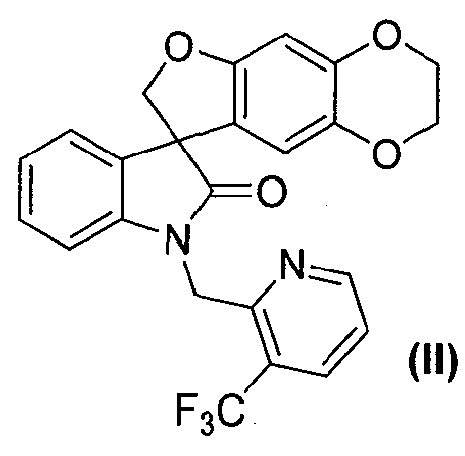
или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси в подходящих условиях с получением соединения формулы (II-S) или его фармацевтически приемлемой соли и соединения формулы (II-R) или его фармацевтически приемлемой соли.
Подробное описание изобретения
Определения
Как использовано в описании и приложенных пунктах, если не указано иное, следующие термины имеют приведенные значения:
"Амино" означает заместитель -NH2.
"Циано" означает заместитель -CN.
"Гидроксил" означает заместитель -OH.
"Имино" означает заместитель =NH.
"Нитро" означает заместитель -NO2.
"Оксо" означает заместитель =O.
"Трифторметил" означает заместитель -CF3.
"Аналгезия" означает отсутствие боли в ответ на стимул, который обычно должен причинять боль.
"Аллодиния" означает состояние, при котором обычно безвредное ощущение, такое как давление или легкое прикосновение, воспринимается как чрезвычайно болезненное.
Подразумевается, что "устойчивое соединение" и "устойчивая структура" означают соединение, являющееся достаточно прочным для того, чтобы выдерживать выделение до полезной степени чистоты из реакционной смеси и включение в состав эффективного терапевтического средства.
"Млекопитающее" включает людей и как домашних животных, таких как лабораторные животные и домашние животные (например, кошки, собаки, свинья, рогатый скот, овцы, козы, лошади и кролики), так и недомашних животных, таких как дикие животные и тому подобное.
"Фармацевтически приемлемая соль" включает как кислотно-, так и основно-аддитивные соли.
"Фармацевтически приемлемая кислотно-аддитивная соль" означает те соли, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются биологически или иным образом нежелательными и которые образованы неорганическими кислотами, такими как, но не в порядке ограничения, соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, и органическими кислотами, такими как, но не в порядке ограничения, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламовая кислота, додецилсерная кислота, этан-1,2-дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксоглутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, слизевая кислота, нафталин-1,5-дисульфоновая кислота, нафталин-2-сульфоновая кислота, 1-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и тому подобное.
"Фармацевтически приемлемая основно-аддитивная соль" означает те соли, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются биологически или иным образом нежелательными. Такие соли получают добавлением неорганического основания или органического основания к свободной кислоте. Соли, образованные неорганическими основаниями, включают, но не в порядке ограничения, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и тому подобное. Предпочтительными неорганическими солями являются соли аммония, натрия, калия, кальция и магния. Соли, образованные органическими основаниями, включают, но не в порядке ограничения, соли первичных, вторичных и третичных аминов, замещенных аминов, включая замещенные амины природного происхождения, циклических аминов и основных ионообменных смол, таких как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминовые смолы и тому подобное. В особенности предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
"Лечение" или "терапия", как использовано здесь, охватывает лечение рассматриваемого заболевания или состояния у млекопитающего, преимущественно человека, с соответствующим заболеванием или состоянием, и включает:
(i) предупреждение развития заболевания или состояния у млекопитающего, в частности, когда указанное млекопитающее предрасположено к состоянию, которое еще не диагностировано;
(ii) ингибирование заболевания или состояния, т.е. купирование развития;
(iii) облегчение заболевания или состояния, т.е. обеспечение регрессии заболевания или состояния, или
(iv) ослабление симптомов, обусловленных заболеванием или состоянием, т.е. обезболивание без устранения основного заболевания или состояния.
Как использовано здесь, термины "заболевание" и "состояние" могут быть использованы взаимозаменяемо или могут отличаться тем, что конкретное заболевание или состояние может не иметь известного возбудителя (следовательно, этиология еще не выяснена) и поэтому пока расценивается не как заболевание, а лишь как нежелательное состояние или синдром, для которого некоторое количество специфических симптомокомплексов установлено клиницистами.
Полученные согласно данному описанию соединения могут содержать один или несколько асимметрических центров и могут, таким образом, приводить к энантиомерам, которые могут быть определены, с точки зрения абсолютной стереохимии, как (R)-, или (S)-, или как (D)-, или (L)- для аминокислот. Подразумевается, что настоящее изобретение включает все возможные такие энантиомеры, равно как рацемические и оптически чистые формы указанных энантиомеров. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)-изомеры могут быть получены использованием хиральных синтонов или хиральных реагентов, либо разделением с использованием общепринятых методов, например хроматографии и фракционированной кристаллизации, либо описанными здесь методами. Обычные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего, оптически чистого, предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ).
"Стереоизомер" означает соединение, состоящее из одних и тех же атомов, соединенных теми же самыми связями, но имеющее различные трехмерные структуры, которые не являются взаимозаменяемыми. Настоящее изобретение рассматривает различные стереоизомеры и смеси таких стереоизомеров и включает "энантиомеры", что относится к двум стереоизомерам, молекулы которых являются несовместимыми при наложении зеркальными отображениями друг друга.
Используемые здесь химические названия в схемах синтеза и структурных формулах являются модифицированной формой номенклатурной системы ИЮПАК, использующей программу ACD/Name Version 9.07, в соответствии с которой соединения по изобретению названы как производные от структуры центрального ядра, т.е. структуры 2-оксиндола. В случае сложных химических названий, используемых здесь, группу заместителя называют перед группой, с которой связан заместитель. Например, циклопропилэтил включает основную этильную цепь с заместителем циклопропилом. В химических структурных формулах указаны все связи, за исключением некоторых углеродных атомов, которые, как подразумевается, связаны с достаточным количеством водородных атомов для заполнения валентности.
Так, например, соединение формулы (I):
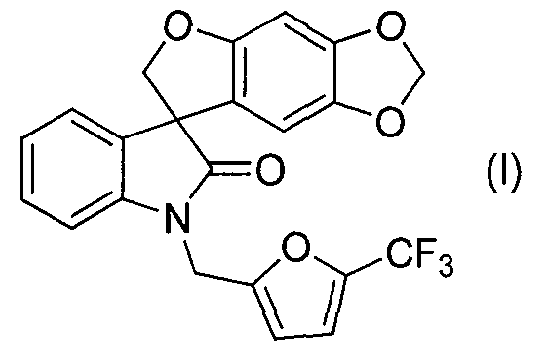
носит название 1'-{[5-(трифторметил)-2-фурил]метил}спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-он.
Варианты осуществления изобретения
Из числа различных аспектов изобретения, изложенных выше в кратком описании изобретения, некоторые варианты осуществления описанных здесь способов являются предпочтительными.
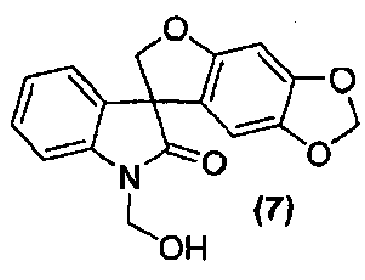
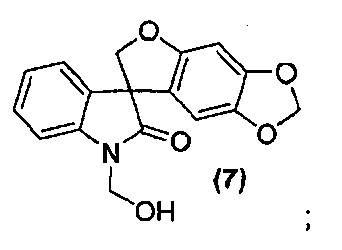
Для способа получения соединения формулы (I), изложенного выше в кратком описании изобретения, или его фармацевтически приемлемой соли в виде отдельного стереоизомера или энантиомера или их смеси один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (8) или его фармацевтически приемлемой соли, по которому соединение формулы (7):
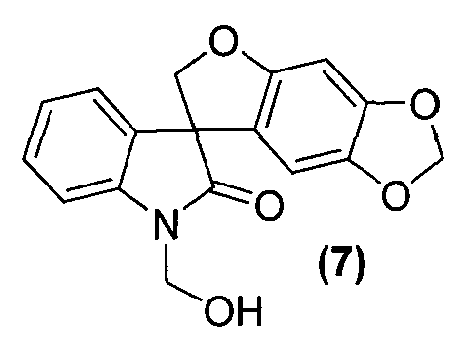
или его фармацевтически приемлемую соль обрабатывают основанием в подходящих условиях с образованием соединения формулы (8) или его фармацевтически приемлемой соли.
Для данного варианта осуществления дополнительный вариант осуществления представляет собой способ, включающий получение соединения формулы (7) или его фармацевтически приемлемой соли, по которому соединение формулы (6):
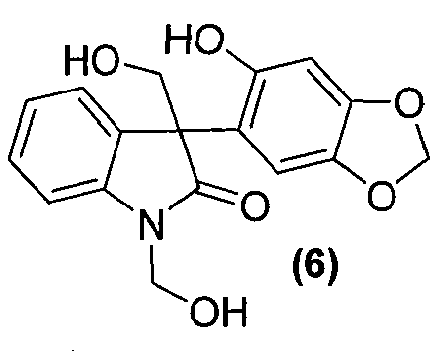
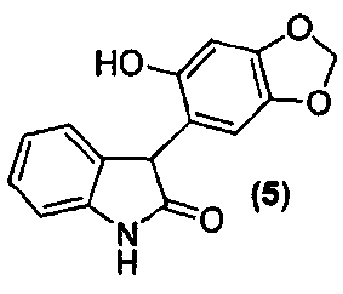
или его фармацевтически приемлемую соль обрабатывают в стандартных условиях реакции Мицунобу с образованием соединения формулы (5) или его фармацевтически приемлемой соли.
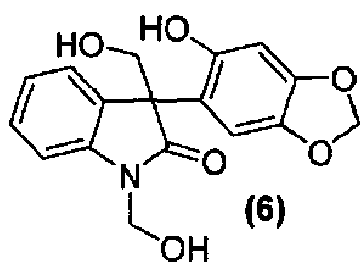
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (6) или его фармацевтически приемлемой соли, по которому соединение формулы (5):
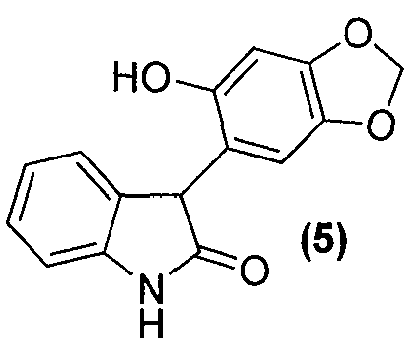
или его фармацевтически приемлемую соль обрабатывают альдегидом в подходящих условиях с образованием соединения формулы (6) или его фармацевтически приемлемой соли.
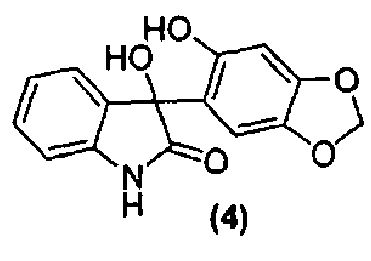
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (5) или его фармацевтически приемлемой соли, по которому соединение формулы (4):
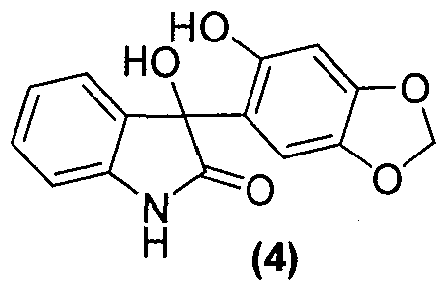
или его фармацевтически приемлемую соль обрабатывают в подходящих условиях с образованием соединения формулы (5) или его фармацевтически приемлемой соли.
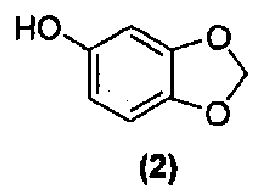
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (4) или его фармацевтически приемлемой соли, по которому соединение формулы (2):
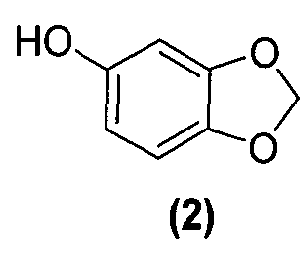
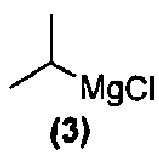
или его фармацевтически приемлемую соль обрабатывают реактивом Гриньяра формулы (3):
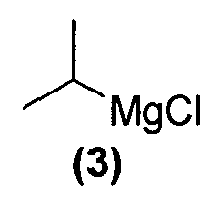
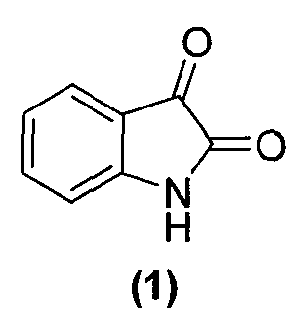
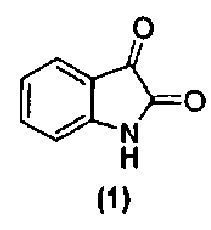
в подходящих условиях с образованием промежуточного продукта и промежуточный продукт затем подвергают взаимодействию с соединением формулы (1):
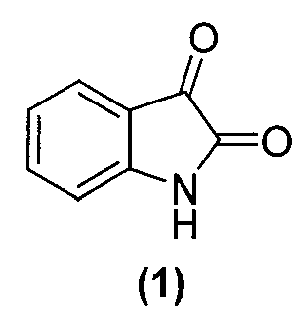
или его фармацевтически приемлемой солью в подходящих условиях с образованием соединения формулы (4) или его фармацевтически приемлемой соли.
Для способа получения соединения формулы (II), изложенного выше в кратком описании изобретения, или его фармацевтически приемлемой соли в виде отдельного стереоизомера, или энантиомера, или их смеси один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (15) или его фармацевтически приемлемой соли, по которому соединение формулы (14):
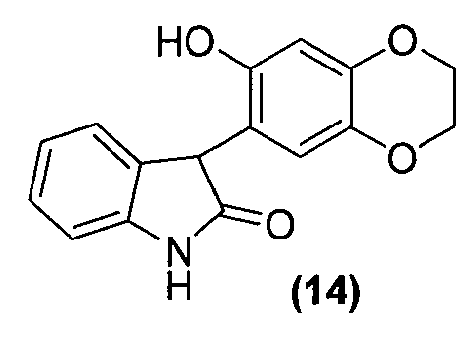
или его фармацевтически приемлемую соль обрабатывают алкилирующим агентом в подходящих условиях с образованием соединения формулы (15) или его фармацевтически приемлемой соли.
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (14) или его фармацевтически приемлемой соли, по которому соединение формулы (13):
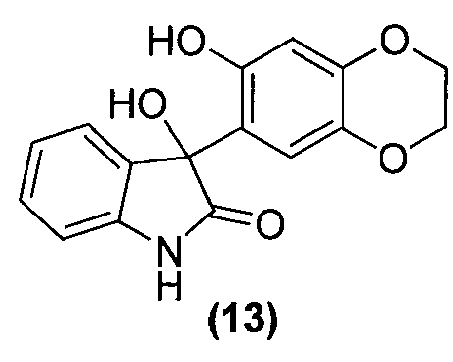
или его фармацевтически приемлемую соль обрабатывают в подходящих условиях с образованием соединения формулы (14) или его фармацевтически приемлемой соли.
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (13) или фармацевтически приемлемой соли, по которому соединение формулы (12):

обрабатывают реактивом Гриньяра формулы (3):
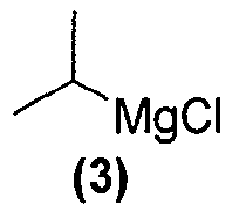
в подходящих условиях с образованием промежуточного продукта и затем промежуточный продукт подвергают взаимодействию с соединением формулы (1):
или его фармацевтически приемлемой солью в подходящих условиях с образованием соединения формулы (13) или его фармацевтически приемлемой соли.
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (12) или его фармацевтически приемлемой соли, по которому соединение формулы (11):
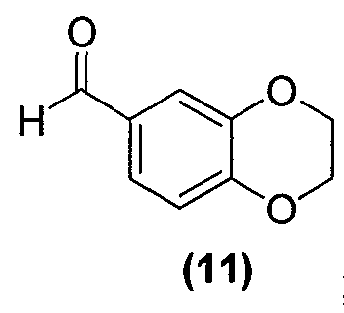
обрабатывают окисляющим агентом в подходящих условиях с образованием соединения формулы (12) или его фармацевтически приемлемой соли.
Для данного варианта осуществления еще один вариант осуществления представляет собой способ, дополнительно включающий получение соединения формулы (11), по которому соединение формулы (10):
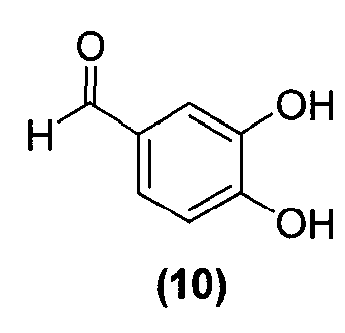
обрабатывают алкилирующим реагентом в подходящих условиях с образованием соединения формулы (11).
Конкретные варианты осуществления способов по изобретению, включающие подходящие условия для каждой из вышеуказанных стадий, описаны более подробно ниже в разделе способы по изобретению.
Способы по изобретению
Способы по изобретению касаются способов получения соединений формул (I) и (II) и соединений формул (I-S), (I-R), (II-S) и/или (II-R), как описано здесь, или их фармацевтически приемлемых солей.
В большинстве случаев исходные компоненты могут быть получены из таких источников, как Sigma Aldrich, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI и Fluorochem USA, и проч., или синтезированы согласно известным специалистам в данной области источникам (см., например, Smith, M.B. and J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)), или получены, как описано здесь, либо способами, описанными в опубликованной патентной заявке PCT № WO 2006/110917, описанными в опубликованной патентной заявке PCT № WO 2010/45251 и патентной заявке PCT № PCT/US2010/040187.
Получение соединений формул (I), (I-S) и (I-R)
Соединения формул (I), (I-S) и (I-R) получают, как представлено ниже реакционной схемой 1:
Реакционная схема 1
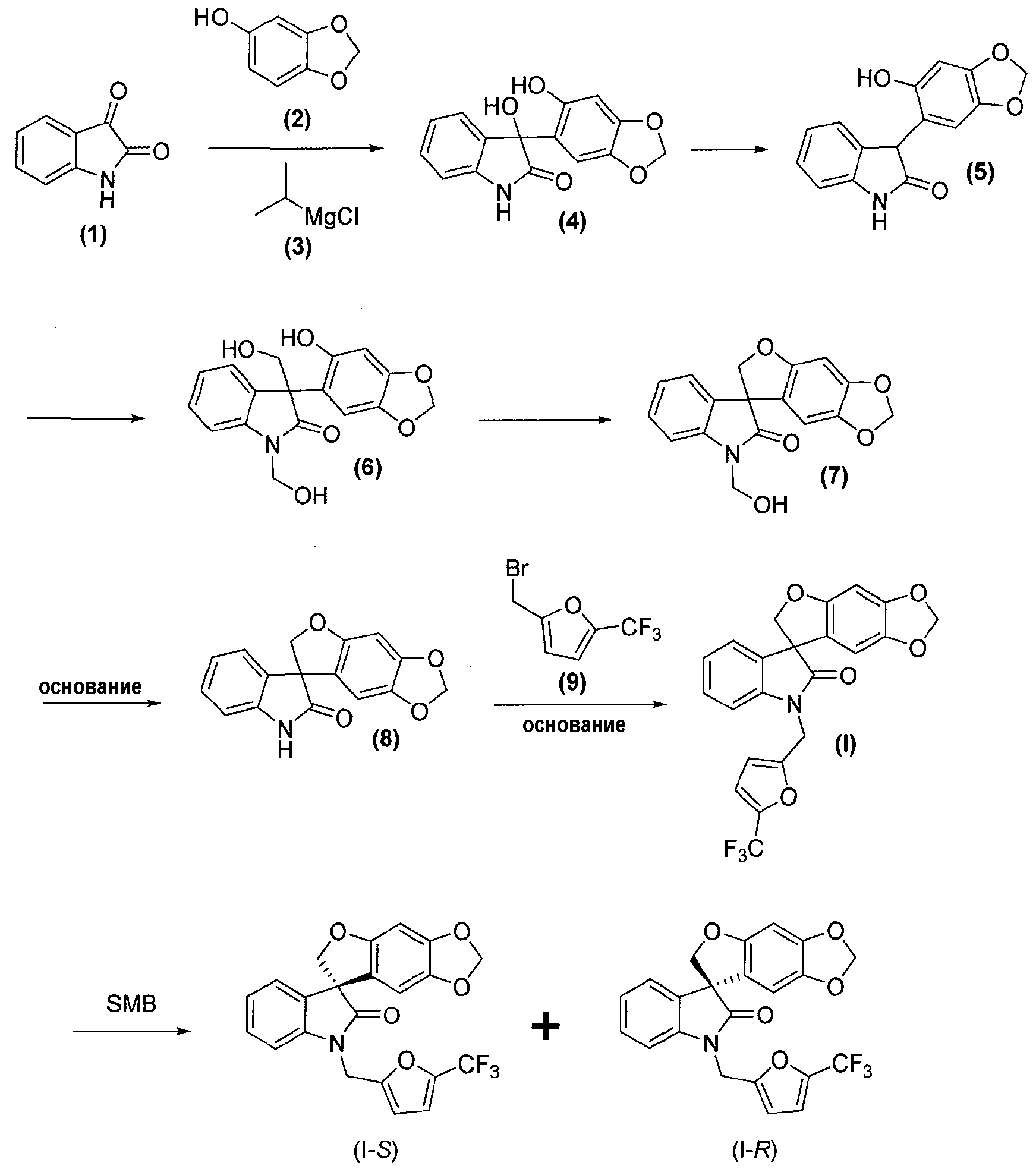
Соединения формулы (1), формулы (2), формулы (3) и формулы (9) являются коммерчески доступными, либо могут быть получены способами, известными специалистам в данной области.
Как правило, соединения формул (I), (I-S) и (I-R) получают способом, описанным выше реакционной схемой 1, первоначально взаимодействием соединения формулы (2) с подходящим реактивом Гриньяра (таким как реактив Гриньяра формулы (3)) в подходящих условиях, таких как при температуре в диапазоне примерно от -25°C до 25°C, предпочтительно примерно при 0°C, что обеспечивает образование магнийгалогенидного промежуточного продукта. Полученный промежуточный продукт подвергают реакции нуклеофильного присоединения с кетокарбонильной группой изатинового соединения формулы (1) в подходящих условиях, таких как в растворителе, предпочтительно, но не в порядке ограничения, тетрагидрофуране или дихлорметане, с получением оксиндольного соединения формулы (4).
Удаление гидроксильной группы в C-3-положении оксиндольного цикла соединения формулы (4) достигается обработкой соединения формулы (4) в подходящих условиях, таких как обработка силановым реагентом, таким как триэтилсилан, в присутствии кислоты, такой как, но не в порядке ограничения, трифторуксусная кислота, с получением соединения формулы (5). Удаление гидроксильной группы может также быть достигнуто обработкой соединения формулы (4) в подходящих условиях, таких как обработка SOCl2/NEt3, с последующим восстановлением полученного промежуточного соединения Zn-пылью, с получением соединения формулы (5). Альтернативно, удаление может быть достигнуто обработкой соединения формулы (4) йодистоводородной кислотой, что дает соединение формулы (5).
Затем соединение формулы (5) обрабатывают в подходящих условиях, таких как обработка основанием, предпочтительно, но не в порядке ограничения, таким как диизопропиламин, диизопропилэтиламин, диизопропиламид лития, гидроксид лития или гидроксид натрия, с последующим осуществлением взаимодействия с формальдегидом или параформальдегидом, получая гидроксиметильное промежуточное соединение формулы (6).
Внутримолекулярная циклизация соединения формулы (6), приводящая к соединению формулы (7), достигается обработкой соединения формулы (6) в стандартных условиях реакции Мицунобу, таких как использование фосфинового реагента, предпочтительно, но не в порядке ограничения, трифенилфосфина или трибутилфосфина, и азо-реагента, предпочтительно, но не в порядке ограничения, такого как диэтилазодикарбоксилат, диизопропилазодикарбоксилат, ди-трет-бутилазодикарбоксилат или тетраметилдиазендикарбоксамид, в растворителе, предпочтительно, но не в порядке ограничения, таком как тетрагидрофуран, дихлорметан или этилацетат. Полученное соединение формулы (7) может быть выделено из реакционной смеси стандартными методами выделения или использовано непосредственно на следующей стадии без выделения из реакционной смеси.
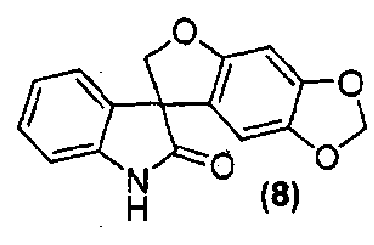
Альтернативно, внутримолекулярная циклизация достигается обработкой соединения формулы (6) подходящим бис-электрофилом, таким как, но не в порядке ограничения, хлорйодметан, в присутствии основания, такого как, но не в порядке ограничения, карбонат цезия, в подходящем растворителе, таком как, но не в порядке ограничения, N,N-диметилформамид или метилэтилкетон, с получением соединения формулы (8).
Удаление гидроксиметильной группы от азота соединения формулы (7) достигается обработкой соединения формулы (7) основанием, предпочтительно, но не в порядке ограничения, таким как гидроксид натрия, гидроксид лития или гидроксид аммония, в подходящих условиях с получением соединения формулы (8), которое может быть выделено из реакционной смеси стандартными методами выделения.
Затем соединение формулы (8) подвергают взаимодействию с электрофилом формулы (9) в подходящих условиях, таких как в присутствии основания, предпочтительно, но не в порядке ограничения, такого как гидрид натрия, карбонат цезия или гидроксид натрия, в растворителе, предпочтительно, но не в порядке ограничения, таком как N,N-диметилформамид, ацетонитрил, тетрагидрофуран или ацетон, получая соединение формулы (I), которое может быть выделено из реакционной смеси стандартными методами выделения.
Соединение формулы (I) может быть разделено на (S)-энантиомер (т.е. соединение формулы (I-S)) и соответствующий (R)-энантиомер (т.е. соединение формулы (I-R)) в подходящих условиях, например, хиральным хроматографическим разделением, таким как, но не в порядке ограничения, хроматография с псевдодвижущимся слоем или хиральная ВЭЖХ.
Получение соединений формул (II), (II-S) и (II-R)
Соединения формул (II), (II-S) и (II-R) получают, как представлено ниже реакционной схемой 2:
Реакционная схема 2
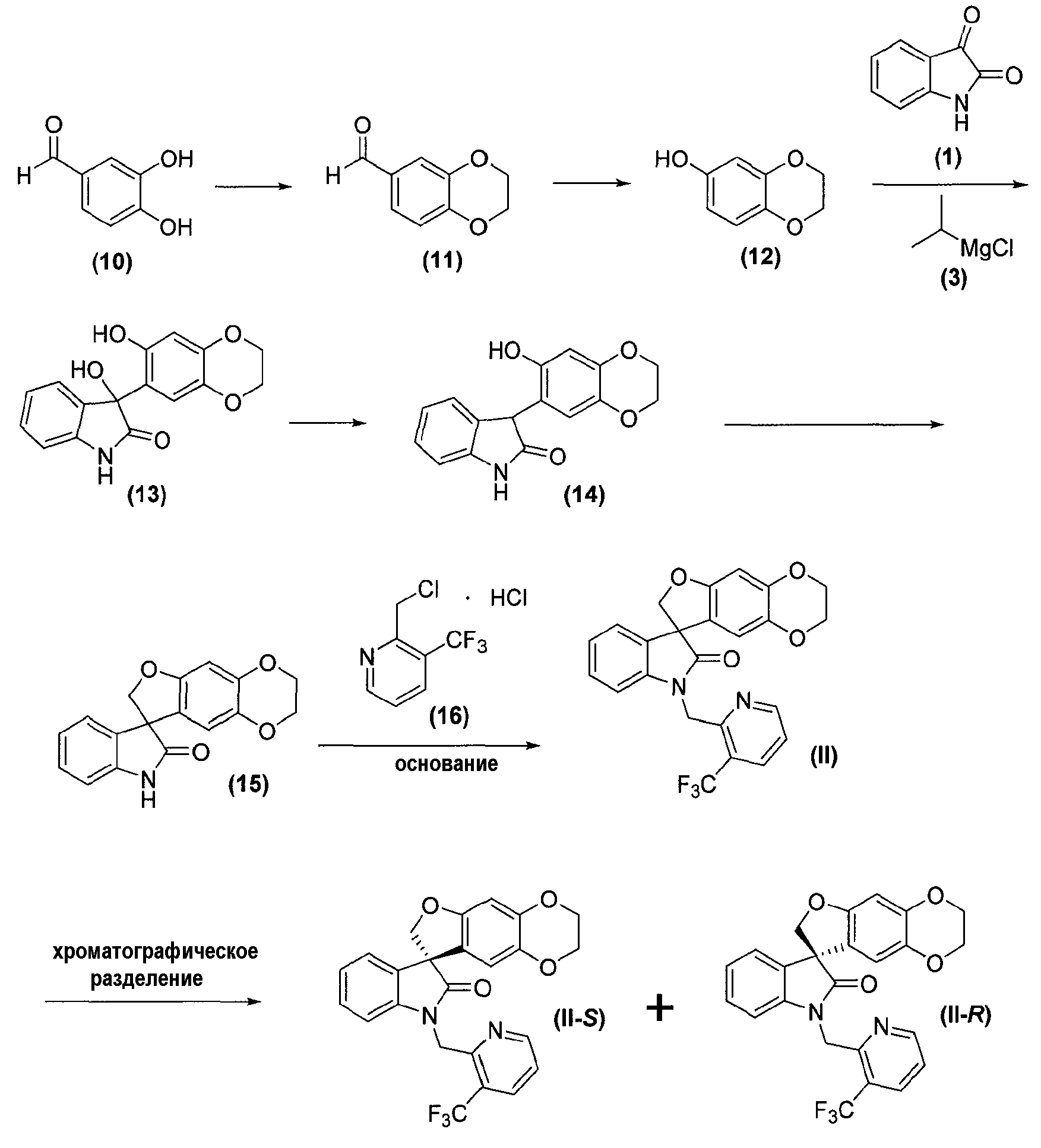
Соединения формулы (10), формулы (11), формулы (12), формулы (1), формулы (3) и формулы (16) являются коммерчески доступными либо могут быть получены способами, известными специалистам в данной области.
Как правило, соединения формул (II), (II-S) и (II-R) получают способом, описанным выше реакционной схемой 2, первоначально, нагреванием дигидроксиальдегида формулы (10) с подходящим бис-алкилирующим реагентом, таким как, но не в порядке ограничения, 1,2-дибромэтан, в присутствии основания, такого как, но не в порядке ограничения, карбонат калия, в растворителе, таком как, но не в порядке ограничения, ацетон, с получением соединения формулы (11).
Соединения формулы (12) получают обработкой соединения формулы (11) окисляющим агентом, таким как, но не в порядке ограничения, 3-хлорпероксибензойная кислота, в присутствии основания, такого как, но не в порядке ограничения, бикарбонат натрия, в подходящем растворителе, таком как, но не в порядке ограничения, дихлорметан, с последующей обработкой водой и основанием, таким как, но не в порядке ограничения, гидроксид натрия, с получением соединения формулы (12).
Соединение формулы (12) обрабатывают затем подходящим реактивом Гриньяра, таким как, но не в порядке ограничения, реактив Гриньяра формулы (3), в подходящих условиях, таких как при температуре в диапазоне примерно от -25°C до 25°C, предпочтительно, примерно при 0°C, с образованием фенилоксимагнийгалогенидного промежуточного продукта, который затем подвергают взаимодействию с соединением формулы (1) в подходящих условиях, таких как в полярном непротонном растворителе, предпочтительно, но не в порядке ограничения, дихлорметане или тетрагидрофуране, с получением оксиндольного соединения формулы (13).
Удаление гидроксильной группы в C-3-положении оксиндольного соединения формулы (13) достигается обработкой соединения формулы (13) в подходящих условиях, таких как обработка силановым реагентом, предпочтительно, но не в порядке ограничения, триэтилсиланом, в присутствии кислоты, предпочтительно, но не в порядке ограничения, такой как трифторуксусная кислота, с получением соединения формулы (14). Удаление гидроксильной группы в C-3-положении оксиндольного соединения формулы (13) может также быть достигнуто первоначальной обработкой соединения формулы (13) SOCl2/NEt3, затем восстановлением полученного промежуточного соединения Zn-пылью с получением соединения формулы (14). Альтернативно, удаление может быть достигнуто обработкой соединения формулы (13) йодистоводородной кислотой с получением соединения формулы (14).
Внутримолекулярная циклизация достигается обработкой соединения формулы (14) бис-алкилирующим агентом, предпочтительно, но не в порядке ограничения, хлорйодметаном, в подходящих условиях, таких как в присутствии основания, предпочтительно, но не в порядке ограничения, карбоната цезия, с получением соединения формулы (15), которое выделяют из реакционной смеси стандартными методами выделения.
Соединение формулы (15) затем подвергают взаимодействию с электрофилом формулы (16) в подходящих условиях, таких как в присутствии основания, предпочтительно, но не в порядке ограничения, такого как гидрид натрия, карбонат цезия или гидроксид натрия, в растворителе, предпочтительно, но не в порядке ограничения, таком как N,N-диметилформамид, ацетонитрил, тетрагидрофуран, 1,4-диоксан или ацетон, с получением соединения формулы (II), которое может быть выделено из реакционной смеси стандартными методами выделения.
Соединение формулы (II) может быть хроматографически разделено на (S)-энантиомер (т.е. соединение формулы (II-S)) и соответствующий (R)-энантиомер (т.е. соединение формулы (II-R)) хроматографией с псевдодвижущимся слоем, с применением подходящей хиральной неподвижной фазы, такой как, но не в порядке ограничения, ChiralPAK®-IC, и подходящей подвижной фазы, такой как, но не в порядке ограничения, смесь дихлорметан/ацетон.
Все вышеописанные соединения, которые на момент получения могут существовать в виде свободной основной или кислотной формы, могут быть превращены в соответствующие фармацевтически приемлемые соли путем обработки подходящим неорганическим или органическим основанием или подходящей неорганической или органической кислотой. Соли полученных выше соединений могут быть превращены в свободную основную или кислотную форму стандартными способами. Понятно, что все полиморфы, аморфные формы, ангидраты, гидраты, сольваты и соли соединений формул (I) и (II) рассматриваются, как охватываемые рамками объема изобретения. Кроме того, все соединения формул (I) и (II), которые содержат кислотную или сложноэфирную группу, могут быть превращены в соответствующий сложный эфир или кислоту соответственно способами, известными специалисту в данной области, или описанными здесь способами.
Следующие конкретные подготовительные синтезы (получение исходных веществ и промежуточных соединений) и синтетические примеры (получение соединения формулы (I) и формулы (II) способами по изобретению) представляют собой руководство, способствующее практическому воплощению изобретения, и не рассматриваются как ограничивающие рамки объема изобретения. Когда один или несколько спектров ЯМР приведены для конкретного соединения, каждый ЯМР-спектр может представлять отдельный стереоизомер, нерацемическую смесь стереоизомеров или рацемическую смесь стереоизомеров соединения.
Подготовительный синтез 1
Синтез 3-гидрокси-3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индола
Соединение формулы (4)
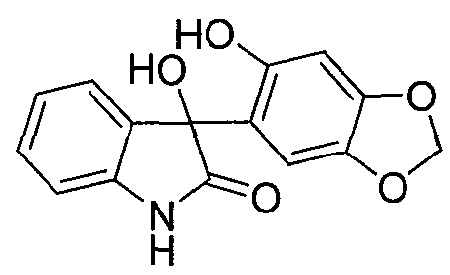
В реактор на 630 л загружают сезамол (42,6 кг, 299 моль). Добавляют тетрагидрофуран (400 кг) и полученный раствор охлаждают до 1°C за 42 минуты. Изопропилмагнийхлорид (2 М раствор в тетрагидрофуране, 173 кг, 337 моль) добавляют за 2 ч так, чтобы внутренняя температура поддерживалась в диапазоне от 0 до 4°C. После завершения добавления внутреннюю температуру снижают до -5°C и добавляют четырьмя порциями изатин (37,9 кг, 250 моль). Реакционную смесь перемешивают 2,75 ч при 1-3°C. В реактор на 1000 л загружают хлорид аммония (72 кг) с последующим добавлением деионизованной воды (356 кг). Смесь перемешивают при 15°C до полного растворения твердого вещества и полученный раствор охлаждают до 1°C за 1 ч. Содержимое реактора на 630 л переносят в реактор на 1000 л за 1 ч так, чтобы внутренняя температура оставалась в диапазоне 3-4°C. Реактор на 630 л промывают толуолом (133 кг) и промывочный раствор вносят в реактор на 1000 л. Содержимому реактора на 1000 л дают нагреться до 20-25°C за период 29 минут и перемешивают еще 15 минут. Перемешивание прекращают и содержимое реактора выдерживают при 25°C в течение 15 минут, давая фазам разделиться. Водную фазу удаляют и добавляют раствор хлорида натрия (42 кг) в деионизованной воде (218 кг) за 25 минут при внутренней температуре 22-24°C. Перемешивание прекращают и смесь выдерживают при 25°C в течение 1 ч, давая фазам разделиться. Органическую фазу дегазируют в течение 0,5 ч азотом и добавляют толуол (89 кг). В реакторе создают вакуум в 300 мбар и внутреннюю температуру реактора выводят на 50-60°C. Летучие компоненты смеси удаляют перегонкой за период 12 ч, собирая при этом 670 л дистиллята. Внешнюю температуру реактора устанавливают на 20-25°C. При охлаждении выпадает оранжевый осадок. Добавляют толуол (114 кг) и суспензию перемешивают 10 минут. Твердое вещество собирают фильтрованием, промывают трет-бутилметиловым эфиром (171 кг) и гептаном (85 кг) и сушат при 55-60°C, при пониженном давлении от 170 до 4 мбар, за период 10,5 ч, получая 3-гидрокси-3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индол-2-он (73,5 кг, количественный выход) в виде бледно-розового твердого вещества: чистота (ВЭЖХ-УФ при 300 нм) 99,3% a/a; 1Н ЯМР (300 МГц, ДМСО-d6) δ 10,18 (c, 1H), 9,08 (c, 1H), 7,21-7,07 (м, 2H), 6,88-6,74 (м, 3H), 6,38 (ушир.c, 1H), 6,23 (c, 1H), 5,92 (c, 2H); 13C ЯМР (75 МГц, ДМСО-d6) δ 178,4, 148,4, 146,6, 143,0, 139,4, 133,2, 128,6, 123,8, 121,1, 120,1, 109,0, 106,8, 100,8, 97,4, 75,1.
Подготовительный синтез 2
Синтез 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индол-2-она
Соединение формулы (5)
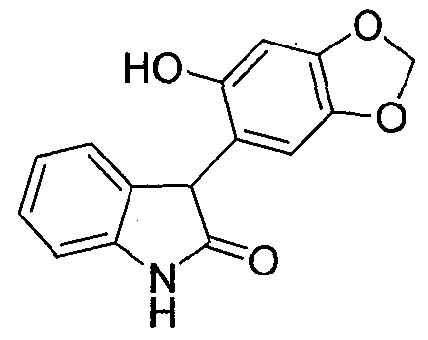
В реактор на 1600 л загружают 3-гидрокси-3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индол-2-он (113,1 кг, 396 моль), откачивают воздух и заполняют азотом. Добавляют двумя порциями трифторуксусную кислоту (679 кг) за 20 минут и внутреннюю температуру снижают до 10°C за 1 ч. Добавляют триэтилсилан (69,2 кг, 595 моль) за 2 ч 05 мин при 10-11°C и смесь перемешивают дополнительно 0,5 ч при 10-11°C. В реактор на 1000 л загружают гептан (524 кг) и трет-бутилметиловый эфир (63 кг). Содержимое реактора на 1000 л переносят в реактор на 1600 л за 13 минут при внутренней температуре 10-11°C. Полученной желто-оранжевой суспензии дают нагреться до 23°C за 1 ч. Твердое вещество собирают фильтрованием, промывают гептаном (464 кг) с последующей промывкой трет-бутилметиловым эфиром (57 кг) и сушат при 50°C при пониженном давлении от 58 до 7 мбар за период 25 ч, получая 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индол-2-он (82,8 кг, 75%) в виде не совсем белого твердого вещества: чистота (ВЭЖХ-УФ при 300 нм) 98,0% a/a; 1Н ЯМР (300 МГц, ДМСО-d6) δ 10,40 (c, 1H), 9,25 (c, 1H), 7,17-7,10 (м, 1H), 6,95-6,81 (м, 3H), 6,55 (c, 1H), 6,43 (c, 1H), 5,92-5,85 (м, 2H), 4,66 (c, 1H); 13C ЯМР (75 МГц, ДМСО-d6) δ 177,9, 150,1, 146,6, 142,7, 139,6, 130,9, 127,4, 123,8, 121,2, 115,9, 109,5, 109,0, 100,7, 97,8, 55,0.
Подготовительный синтез 3
Синтез 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-бис((гидроксиметил)-1,3-дигидро-2H-индол-2-она
Соединение формулы (6)

В реактор на 1000 л загружают 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-дигидро-2H-индол-2-он (56,3 кг, 209 моль) и затем параформальдегид (25,4 кг, 847 моль) и деионизованную воду (285 кг). Реакционную смесь охлаждают до внутренней температуры 5°C за 25 минут и добавляют 30 масс.% водный раствор гидроксида натрия (113 кг, 847 моль) при 5°C за 40 минут. Реакционную смесь перемешивают 1 ч при 5°C. Во второй реактор на 1000 л загружают деионизованную воду (260 кг) и 32% соляную кислоту (124 кг). Содержимое первого реактора добавляют к содержимому второго реактора при 1°C за 80 минут. Первый реактор промывают деионизованной водой (35 кг) и промывочный раствор переносят во второй реактор. Полученную суспензию перемешивают при 1°C в течение 1 ч и твердое вещество собирают фильтрованием, промывают смесью концентрированной соляной кислоты (11 кг) и воды (20 кг) и сушат при 55-60°C при пониженном давлении 50-6 мбар за период 24 ч, получая 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-бис(гидроксиметил)-1,3-дигидро-2H-индол-2-он (69,8 кг, 99%) в виде светло-коричневого твердого вещества: чистота (ВЭЖХ-УФ при 230 нм) 95,4% a/a; 1Н ЯМР (ДМСО-d6, 300 МГц) δ 9,06 (c, 1H), 7,17-6,84 (м, 5H), 6,19-6,10 (м, 2H), 5,86 (c, 2H), 5,12-4,92 (м, 3H), 4,11-4,06 (м, 1H), 3,79-3,73 (м, 1H); 13C ЯМР (ДМСО-d6, 75 МГц) δ 179,0, 150,5, 146,7, 143,9, 139,9, 132,6, 127,4, 124,0, 121,9, 118,2, 108,7, 108,6, 101,1, 98,0, 65,4, 63,2, 56,2.
Подготовительный синтез 4
Синтез спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-она
Соединение формулы (8)
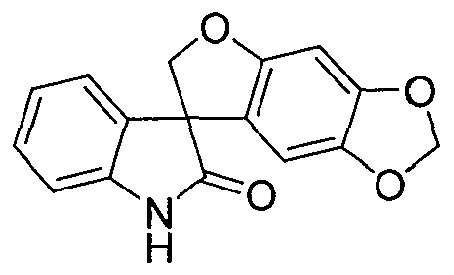
В реактор на 1000 л загружают 3-(6-гидрокси-1,3-бензодиоксол-5-ил)-1,3-бис(гидроксиметил)-1,3-дигидро-2H-индол-2-он (65,0 кг, 197 моль) и затем тетрагидрофуран (586 кг). Полученный раствор охлаждают до -4°C за 20 минут и добавляют три-н-бутилфосфин (40,0 кг, 197 моль) за 6 минут, с последующим добавлением раствора диизопропилазодикарбоксилата (44,8 кг, 197 моль) в тетрагидрофуране (75 кг) за 125 минут, таким образом, чтобы внутренняя температура оставалась ниже 0°C. Реакционную смесь перемешивают при -3°C еще 25 минут и содержимое реактора переносят в реактор на 2500 л. Реактор на 1000 л промывают тетрагидрофураном (16 кг) и промывочный раствор вносят в реактор на 2500 л. Добавляют 25 масс.% раствор аммиака в воде (118 кг) при температуре от -3 до -2°C за 30 минут. Реакционной смеси дают нагреться до 25°C за 1,25 ч и перемешивают еще 2 ч. Добавляют деионизованную воду (650 кг) и этилацетат (585 кг) и смесь нагревают до 40°C за 40 минут. После перемешивания в течение дополнительных 15 минут перемешивание прекращают и оставляют на 1 ч, давая разделиться фазам. Водную фазу удаляют и добавляют деионизованную воду (650 кг). Смесь перемешивают в течение 15 минут при 40°C. Перемешивание прекращают и оставляют на 1 ч, давая разделиться фазам. Водную фазу удаляют и добавляют деионизованную воду (325 кг). Смесь частично концентрируют перегонкой при пониженном давлении, при внутренней температуре 21-39°C и давлении 382-98 мбар, собирая 950 л дистиллята за период 4,5 ч. Добавляют метанол (1600 кг) и смесь нагревают до 60°C за 35 минут. Смесь частично концентрируют перегонкой при пониженном давлении, при внутренней температуре 32-58°C и давлении 530-170 мбар, собирая 1260 л дистиллята за период 9,33 ч. Полученную суспензию оставляют охлаждаться до 22°C на 2 ч и перемешивают дополнительно в течение 6 ч. Твердое вещество собирают фильтрованием, промывают смесью метанола (34 кг) и деионизованной воды (17 кг) и сушат при 55-60°C при пониженном давлении 50-3 мбар за период 31 ч, получая 35,8 кг коричневого твердого вещества, которое переносят в реактор на 400 л. Добавляют метанол (163 кг) и полученную суспензию перемешивают 0,5 ч. Смесь нагревают до температуры кипения с обратным холодильником за период 35 минут и нагревают при температуре кипения флегмы еще 15 минут. Добавляют деионизованную воду (33 кг) и смесь нагревают при температуре кипения флегмы 155 минут. Суспензию фильтруют еще горячей и плотный осадок на фильтре промывают смесью метанола (22 кг) и деионизованной воды (11 кг), и сушат при 55-60°C при пониженном давлении 50-4 мбар за период 8 ч, получая спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-он (30,44 кг, 49%) в виде светло-коричневого твердого вещества: чистота (ВЭЖХ-УФ при 230 нм) 89,4%; МС (ES+) m/z 282,3 (M+1); 1Н ЯМР (400 МГц, CDCl3) δ 10,56 (c, 1H), 7,27-6,92 (м, 4H), 6,67 (c, 1H), 6,26 (c, 1H), 5,93 (c, 2H), 4,79-4,64 (м, 2H).
Подготовительный синтез 5
Синтез 2,3-дигидро-1,4-бензодиоксин-6-карбальдегида
Соединение формулы (11)
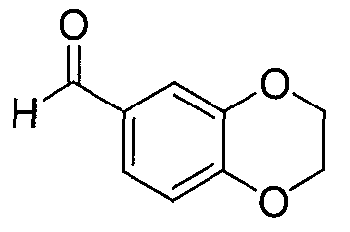
В реактор на 2000 л загружают ацетон (404,5 кг) и затем карбонат калия (256 кг, 1852 моль) и 1,2-дибромэтан (241,5 кг, 1298 моль). Смесь нагревают до температуры кипения с обратным холодильником. В реактор на 500 л загружают ацетон (606 кг) и 3,4-дигидроксибензальдегид (128 кг, 926 моль). Содержимое реактора на 500 л вносят в реактор на 2000 л со скоростью 150-180 кг/ч, поддерживая реакционную температуру при 50-60°C. Реакционную смесь перемешивают при 54-60°C в течение 12 ч, охлаждают до 20°C и фильтруют через нутч-фильтр на 500 л. Плотный осадок на фильтре промывают ацетоном (2×202 кг). Фильтратные и ацетоновые промывные воды объединяют в реакторе на 2000 л и полученный раствор концентрируют досуха при пониженном давлении при температуре <40°C. К остатку добавляют этилацетат (683 кг) и полученный раствор промывают 5 масс.% водным раствором карбоната калия (256 кг). Смесь перемешивают в течение 0,5 ч, оставляют осаждаться на 0,5 ч, и водную фазу удаляют. Такую процедуру промывки повторяют в общей сложности три раза. Органическую фазу оставляют на время в сборниках. В реактор на 2000 л загружают объединенные промывочные воды и затем этилацетат (113,9 кг). Смесь перемешивают 0,5 ч, дают отстояться в течение 0,5 ч и водную фазу удаляют. Органическую фазу из стерилизационных барабанов добавляют в реактор, с последующим добавлением 28 масс.% водного раствора хлорида натрия (192 кг). Смесь перемешивают 0,5 ч, дают отстояться в течение 0,5 ч и водную фазу удаляют. Органическую фазу концентрируют при пониженном давлении при температуре <45°C, пока содержание этилацетата в смеси не станет ниже 10% (устанавливается газовой хроматографией). К остатку добавляют петролейный эфир (268,8 кг) со скоростью 80-90 кг/ч, поддерживая температуру смеси при 35-45°C. Смесь охлаждают до 5°C за 3 ч и выдерживают при указанной температуре еще 1 ч, за указанное время выпадает осадок. Полученную суспензию фильтруют через центрифужный фильтр и сушат, получая 2,3-дигидро-1,4-бензодиоксин-6-карбальдегид (111,4 кг, 73%) в виде не совсем белого твердого вещества: чистота (ВЭЖХ-УФ при 230 нм) 99,3%.
Подготовительный синтез 6
Синтез 2,3-дигидро-1,4-бензодиоксин-6-ола
Соединение формулы (12)
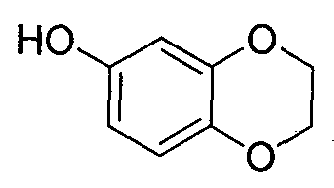
В реактор на 2000 л загружают дихлорметан (1303,4 кг) и затем 2,3-дигидро-1,4-бензодиоксин-6-карбальдегид (98,0 кг, 597 моль) и перемешивают до получения гомогенного раствора. Добавляют 3-хлорпероксибензойную кислоту (144,3 кг, 836 моль). Смесь нагревают до температуры кипения с обратным холодильником со скоростью 8-10°C/ч, нагревают при температуре кипения флегмы дополнительно 6 ч и дают охладиться до 20°C. Полученную суспензию фильтруют через нутч-фильтр на 500 л и плотный осадок на фильтре промывают дихлорметаном (391 кг). Фильтрат и промывной раствор переносят в реактор на 2000 л. Добавляют 7 масс.% водный раствор бикарбоната натрия (212,7 кг) и смесь перемешивают 0,5 ч. Перемешивание прекращают и оставляют на 0,5 ч, давая разделиться фазам. Водный слой удаляют. Процедуру промывки водным бикарбонатом натрия повторяют в общей сложности три раза. Органическую фазу концентрируют досуха при пониженном давлении при температуре <30°C. Добавляют метанол (116,1 кг) и полученную смесь охлаждают до 0°C. Добавляют 15,5 масс.% водный раствор гидроксида натрия (234,3 кг) со скоростью 30-40 кг/ч, поддерживая температуру смеси в диапазоне 0-10°C. Смесь перемешивают при указанной температуре еще 2,25 ч и pH смеси доводят до 6-7 добавлением 4н. соляной кислоты (266,5 кг), поддерживая температуру смеси в диапазоне 0-10°C. Смеси дают нагреться до температуры окружающей среды и экстрагируют в общей сложности три раза метил-трет-бутиловым эфиром (145 кг на каждую экстракцию), перемешивая 0,5 ч, прекращают перемешивание и оставляют на 0,5 ч, давая разделиться фазам. Объединенные органические экстракты промывают в общей сложности три раза 7% водным раствором бикарбоната натрия (212,7 кг на каждую промывку), перемешивая в течение 0,5 ч, перемешивание прекращают, оставляют на 0,5 ч, давая разделиться фазам, и удаляют водную фазу. Затем органическую фазу промывают 30 масс.% водным раствором хлорида натрия (212,7 кг), перемешивая в течение 0,5 ч, прекращают перемешивание, оставляют на 0,5 ч, давая разделиться фазам, и удаляют водную фазу. Органическую фазу концентрируют досуха при пониженном давлении при температуре <45°C. Добавляют тетрагидрофуран (170 кг) и полученный раствор концентрируют досуха при пониженном давлении при температуре <45°C. Добавляют дополнительное количество тетрагидрофурана (17,1 кг) и полученный раствор концентрируют досуха при пониженном давлении при температуре <45°C. Добавляют тетрагидрофуран (122,5 кг), получая коричнево-красный раствор 2,3-дигидро-1,4-бензодиоксин-6-ола (86,3 кг, 95%) в тетрагидрофуране, который используют в дальнейшем без дополнительной очистки: чистота (ВЭЖХ-УФ при 220 нм) 95,7%.
Подготовительный синтез 7
Синтез 3-гидрокси-3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-она
Соединение формулы (13)
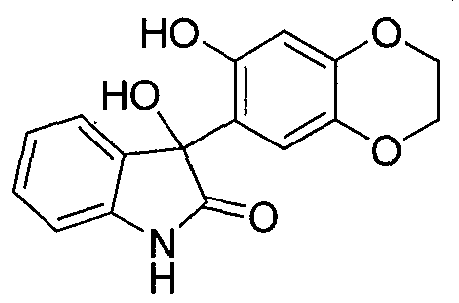
В реактор на 1000 л загружают тетрагидрофуран (296,8 кг). Тетрагидрофуран нагревают до температуры кипения с обратным холодильником в течение 1 ч и дают охладиться до температуры окружающей среды. Добавляют магний (15,0 кг, 625 моль), йод (19,5 г, каталитическое количество) и бромэтан (147,0 г, каталитическое количество) при температуре 15-30°C. Полученную смесь нагревают при 50-55°C в течение 0,5 ч и добавляют 2-хлорпропан (4,5 кг, 57 моль), с последующим добавлением 2 М раствора изопропилмагнийхлорида в тетрагидрофуране (7,6 кг, каталитическое количество). Добавляют 2-хлорпропан (39,2 кг, 500 моль) со скоростью 8-10 кг/ч, так, чтобы температура реакционной смеси поддерживалась в диапазоне 55-70°C. Реакционную смесь нагревают при 58-68°C в течение 3 ч, дают охладиться до температуры окружающей среды и перемешивают дополнительно 4 ч. В реактор на 2000 л загружают раствор 2,3-дигидро-1,4-бензодиоксин-6-ола (86,3 кг, 567 моль) в тетрагидрофуране (122,5 кг) и затем дополнительное количество тетрагидрофурана (804,1 кг). Полученный раствор охлаждают до 0°C и содержимое реактора на 1000 л добавляют в реактор на 2000 л со скоростью 30-50 кг/ч так, чтобы температура реакционной смеси поддерживалась в диапазоне 0-5°C. Реактор на 1000 л трижды промывают тетрагидрофураном (5 кг на каждую промывку) и промывочные растворы вносят в реактор на 2000 л. Реакционную смесь перемешивают при 7-13°C в течение 1 ч и охлаждают до -5°C. Добавляют тремя равными порциями изатин (69,5 кг, 472,5 моль) за 0,5 ч и смесь перемешивают 0,5 ч при -5-0°C. Реакционную смесь нагревают при 50-55°C в течение 7,5 ч и дают охладиться до температуры окружающей среды. В реактор на 5000 л загружают воду (576,9 кг) и хлорид аммония (118,2 кг). Полученный раствор охлаждают до 0-5°C. Содержимое реактора на 2000 л вносят в реактор на 5000 л со скоростью 300-500 кг/ч так, чтобы температура реакционной смеси поддерживалась в диапазоне 0-5°C. Смесь перемешивают при 15-25°C в течение 0,5 ч и перемешивание прекращают. Оставляют на 1 ч, давая разделиться фазам, и водную фазу удаляют. Добавляют 27 масс.% водный раствор хлорида натрия (69,6 кг) и смесь перемешивают 0,5 ч. Перемешивание прекращают, оставляют на 1 ч, давая разделиться фазам, и водный слой удаляют. Процедуру промывки водным хлоридом натрия повторяют в общей сложности три раза. Органическую фазу переносят в реактор на 2000 л и концентрируют при пониженном давлении при температуре 45-55°C. К остатку добавляют толуол (302,3 кг) со скоростью 90-130 кг/ч и при температуре 45-50°C. Полученную смесь охлаждают до 15°C со скоростью 8-10°C/ч и перемешивают при 10-15°C дополнительно 1 ч. Полученную суспензию фильтруют через центрифужный фильтр и плотный осадок на фильтре промывают водой (69,5 кг) и сушат при 45-50°C, получая 3-гидрокси-3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-он (130,8 кг, 93%) в виде не совсем белого твердого вещества: чистота (ВЭЖХ-УФ при 210 нм) 99,7%.
Подготовительный синтез 8
Синтез 3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-она
Соединение формулы (14)
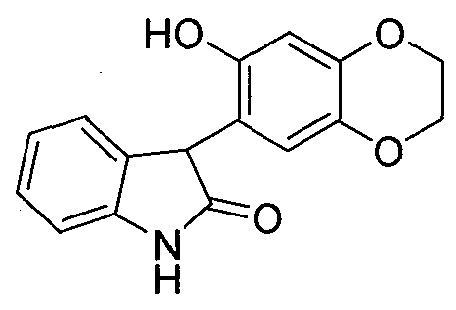
В реактор на 2000 л загружают дихлорметан (489,4 кг) с последующим добавлением 3-гидрокси-3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-она (92,0 кг, 307 моль) за четыре порции по 23 кг за 1 ч. Полученный раствор перемешивают при температуре окружающей среды в течение 1 ч и добавляют триэтилсилан (107,2 кг, 921 моль). Смесь охлаждают до -5°C и добавляют трифторуксусную кислоту (105,1 кг, 921 моль) со скоростью 25-30 кг/ч так, чтобы температура реакционной смеси поддерживалась ниже 0°C. Смесь перемешивают при -5-0°C в течение 2,5 ч, нагревают до 18-20°C, перемешивают дополнительно 6,5 ч и концентрируют досуха при пониженном давлении при температуре <30°C. К остатку добавляют метил-трет-бутиловый эфир (139,8 кг) при 15-20°C и смесь концентрируют почти досуха при пониженном давлении при температуре <35°C. Смесь фильтруют на центрифуге-фильтре и в реактор на 2000 л загружают плотный осадок с фильтра и затем метанол (72,7 кг). Смесь перемешивают при 10-15°C в течение 0,5 ч и фильтруют на центрифуге-фильтре. Плотный осадок на фильтре сушат при пониженном давлении при 40-50°C, получая 3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-он (68,0 кг, 78%) в виде бесцветного твердого вещества: чистота (ВЭЖХ-УФ при 254 нм) 99,3%.
Подготовительный синтез 9
Синтез 2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-она
Соединение формулы (15)
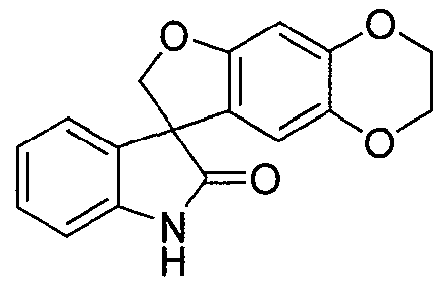
В кристаллизатор из нержавеющей стали на 2000 л загружают N,N-диметилформамид (113,7 кг) и тетрагидрофуран (1070,9 кг). Содержимое охлаждают до 0-5°C и добавляют 3-(7-гидрокси-2,3-дигидро-1,4-бензодиоксин-6-ил)-1,3-дигидро-2H-индол-2-он (12,0 кг, 42,4 моль), с последующим добавлением карбоната цезия (30,4 кг, 93,3 моль). Раствор хлорйодметана (9,4 кг, 53,7 моль) в N,N-диметилформамиде (16,9 кг) добавляют со скоростью 39,5 кг/ч так, чтобы температура реакционной смеси поддерживалась в диапазоне от 0 до 5°C. Реакционную смесь перемешивают при 0-5°C в течение 2 ч и нагревают при 20-25°C в течение 18,5 ч. Смесь фильтруют и плотный осадок с фильтра суспендируют в тетрагидрофуране (26,4 кг) и фильтруют вновь. Объединенные фильтраты собирают и концентрируют до объема 110 л при пониженном давлении при температуре <60°C. Смесь охлаждают до 20-25°C и добавляют очищенную воду (1200,8 кг) со скоростью 343,1 кг/ч. Смесь охлаждают до 0-5°C и фильтруют. Плотный осадок с фильтра суспендируют в воде (310,5 кг), фильтруют и сушат при температуре <60°C, пока содержание воды не достигнет 10,6 масс.% согласно титрованию по методу Карла Фишера. В реактор на 200 л загружают тетрагидрофуран (98,0 кг). Частично высушенный плотный осадок с фильтра (~11,0 кг) вносят в реактор на 200 л с помощью воронки для добавления твердых веществ. Смесь нагревают до температуры кипения с обратным холодильником в течение 4,5 ч, охлаждают до 10-15°C и перемешивают 3,5 ч при 10-15°C. Смесь фильтруют и плотный осадок на фильтре промывают охлажденным (0-5°C) тетрагидрофураном (2×0,7 кг) и сушат в центробежной сушилке при температуре <55°C, получая 2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-он (6,88 кг, 63%) в виде светло-желтого твердого вещества: чистота (ВЭЖХ-УФ при 210 нм) 98,3%; т.пл. >250°C; 1Н ЯМР (300 МГц, ДМСО-d6) δ 10,58 (ушир.c, 1H), 7,26-7,19 (м, 1H), 7,09 (д, J=7,2 Гц, 1H), 6,99-6,90 (м, 2H), 6,47 (c, 1H), 6,16 (c, 1H), 4,74, 4,60 (АВкв, JAB=9,2 Гц, 2H), 4,20-4,07 (м, 4H); 13C ЯМР (75 МГц, ДМСО-d6) δ 178,4, 154,7, 144,0, 141,8, 137,8, 132,6, 128,7, 123,8, 122,3, 121,5, 111,1, 109,8, 98,7, 79,5, 64,2, 63,6, 57,7; MC (ES+) m/z 295,9 (М+1).
Синтетический пример 1
Синтез 1'-{[5-(трифторметил)-2-фурил]метил}спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-она
Соединение формулы (I)
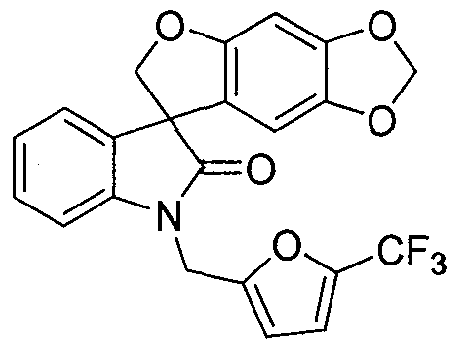
В реактор на 100 л загружают спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-он (6,03 кг, 19,5 моль) и затем карбонат цезия (16,02 кг, 48,7 моль). Добавляют ацетон (48,8 кг) и полученную суспензию нагревают до температуры кипения с обратным холодильником за 1 ч. Добавляют 2-бромметил-5-(трифторметил)фуран (4,92 кг, 21,2 моль) с помощью капельной воронки за период 2 ч, поддерживая реакционную смесь при температуре кипения флегмы. Реакционную смесь перемешивают при температуре кипения флегмы дополнительно 2 ч и ацетон удаляют перегонкой при атмосферном давлении, собирая 37 л дистиллята. Добавляют толуол (48,8 кг) и перегонку продолжают, сначала при атмосферном давлении, потом при пониженном давлении, пока не соберут 37 л дистиллята. Добавляют толуол (36,9 кг) и перегонку продолжают при 54-55°C и давлении 150-180 мбар, пока не соберут 37 л дистиллята. Содержимому реактора на 100 л дают охладиться до 25°C и добавляют толуол (40,9 кг). Содержимое реактора на 100 л переносят в реактор на 200 л и добавляют деионизованную воду (48,8 кг). Перемешиваемую смесь нагревают до 39°C, перемешивание прекращают и оставляют на 11 ч, давая разделиться фазам. Нижнюю фазу удаляют и оставшуюся толуольную фазу подвергают перегонке при 55-64°C при пониженном давлении 100 мбар, пока не соберут 18 л дистиллята. Полученный раствор разбавляют толуолом до общего объема 98 л. Содержимое реактора на 200 л пропускают через хроматографическую колонну, заполненную силикагелем (20 кг) и толуолом (40 кг). Колонну элюируют толуолом, так что собирают десять фракций по 30 кг. Колонну промывают ацетоном (100 кг). Фракции 2-10 последовательно переносят в реактор на 200 л, осуществляя перегонку при пониженном давлении. Содержимое реактора доводят с помощью толуола до объема 50 л и раствор нагревают до 79°C. Гептан (85 кг) добавляют за 15 минут и смесь охлаждают до 10°C за период 3 ч. Кристаллизация начинается при внутренней температуре 56°C. Твердое вещество собирают фильтрованием, промывают смесью гептана (10,2 кг) и толуола (5,1 кг) и сушат при 45-50°C при пониженном давлении 50 мбар за период 15 ч, получая 1'-{[5-(трифторметил)-2-фурил]метил}спиро[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-он (6,08 кг, 73%) в виде бесцветного твердого вещества: чистота (ВЭЖХ-УФ при 230 нм) 99,6%; т.пл. 139-141°C; 1Н ЯМР (300 МГц, CDCl3) δ 7,32-6,97 (м, 5H), 6,72 (д, J=3,3 Гц, 1H), 6,66 (c, 1H), 6,07 (c, 1H), 5,90-5,88 (м, 2H), 5,05, 4,86 (АВкв, JAB=16,1 Гц, 2H), 4,91 (д, J=9,0 Гц, 1H), 4,66 (д, J=9,0 Гц, 1H); 13C ЯМР (75 МГц, CDCl3) δ 176,9, 155,7, 153,5, 148,8, 142,2, 141,9, 140,8, 140,2, 139,7, 139,1, 132,1, 129,2, 124,7, 124,1, 123,7, 121,1, 120,1, 117,6, 114,5, 114,4, 110,3, 109,7, 103,0, 101,9, 93,8, 80,0, 57,8, 36,9; МС (ES+) m/z 430,2 (M+1), 452,2 (M+23); вычислено для C22H14F3NO5: C, 61,54%; H, 3,29%; N, 3,26%; найдено: C, 61,51%; H, 3,29%; N, 3,26%.
Синтетический пример 2
Разделение соединения формулы (I) хиральной ВЭЖХ
Соединение формулы (I) разделяют на соединение формулы (I-S) и соединение формулы (I-R) хиральной ВЭЖХ в следующих условиях:
Колонка: Chiralcel ® OJ-RH; 20 мм I.D. × 250 мм, 5 mic; Lot: OJRH CJ-EH001 (Daicel Chemical Industries, Ltd)
Элюент: смесь ацетонитрил/вода (60/40, об/об, изократический)
Скорость потока: 10 мл/мин
Время хроматографирования: 60 мин
Нагрузка: 100 мг соединения формулы (I) в 1 мл ацетонитрила
Температура: Окружающей среды
В вышеуказанных условиях хиральной ВЭЖХ соединение формулы (I-R), т.е. (R)-1'-{[5-(трифторметил)фуран-2-ил]метил}спиро[фуро[2,3-f][1,3]-бензодиоксол-7,3'-индол]-2'(1'H)-он выделяется в первой фракции как белое твердое вещество; ee (энантиомерный избыток) >99% (аналитическая OJ-RH, 55% ацетонитрил в воде); т.пл. 103-105°C; 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,32-6,99 (м, 5H), 6,71 (д, J=3,4 Гц, 1H), 6,67 (c, 1H), 6,05 (c, 1H), 5,89 (д, J=6,2 Гц, 2H), 5,13, 5,02 (АВкв, JAB=16,4 Гц, 2H), 4,82, 4,72 (АВкв, JАВ=9,4 Гц, 2H); 13C ЯМР (75 МГц, CDCl3) δ 177,2, 155,9, 152,0, 149,0, 142,4, 142,0, 141,3, 132,0, 129,1, 123,9, 120,6, 119,2, 117,0, 112,6, 109,3, 108,9, 103,0, 101,6, 93,5, 80,3, 58,2, 36,9; МС (ES+) m/z 430,2 (M+1), [α]D -17,46° (c 0,99, ДМСО). Соединение формулы (I-S), т.е. (S)-1'-{[5-(трифторметил)фуран-2-ил]метил}спиро-[фуро[2,3-f][1,3]бензодиоксол-7,3'-индол]-2'(1'H)-он, выделяется во второй фракции как белое твердое вещество; ee>99% (аналитическая OJ-RH, 55% ацетонитрил в воде); т.пл. 100-102°C; 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,32-6,99 (м, 5H), 6,71 (д, J=3,4 Гц, 1H), 6,67 (c, 1H), 6,05 (c, 1H), 5,89 (д, J=6,2 Гц, 2H), 5,13, 5,02 (АВкв, JAB=16,4 Гц, 2H), 4,82, 4,72 (ABq, JАВ=9,4 Гц, 2H); 13C ЯМР (75 МГц, CDCl3) δ 177,2, 155,9, 152,0, 149,0, 142,4, 142,0, 141,3, 132,0, 129,1, 123,9, 120,6, 119,2, 117,0, 112,6, 109,3, 108,9, 103,0, 101,6, 93,5, 80,3, 58,2, 36,9; МС (ES+) m/z 430,2 (M+1), [α]D +14,04° (c 0,99, ДМСО).
Синтетический пример 3
Разделение соединения формулы (I) ППС-хроматографией
Соединение формулы (I) разделяют на соединение формулы (I-S) и соединение формулы (I-R) ППС-хроматографией в следующих условиях:
Экстракт: 147,05 мл/мин
Рафинат: 86,13 мл/мин
Элюент: 183,18 мл/мин
Подача: 50 мл/мин
Рециркуляция: 407,88 мл/мин
Время хроматографирования: 0,57 мин
Температура: 25°C
Давление: 55 бар
Загрузочный раствор (25 г соединения формулы (I) в 1,0 л подвижной фазы (смесь ацетонитрил/метанол 25:75 (об:об)) непрерывно впрыскивают в систему ППС (Novasep Licosep Lab Unit), оборудованную восемью идентичными колонками в конфигурации 2-2-2-2, содержащими 110 г (на колонку, 9,6 см, 4,8 см I.D.) ChiralPAK-AD в качестве неподвижной фазы. Первый элюируемый энантиомер (соединение формулы (I-R)) содержится в потоке рафината и второй элюируемый энантиомер (соединение формулы (I-S)) содержится в потоке экстракта. Характеристики соединения формулы (I-R) и соединения формулы (I-S), полученных ППС-разделением, идентичны характеристикам, полученным выше с использованием хиральной ВЭЖХ.
Соединение формулы (I) разделяют на соединение формулы (I-R) и соединение формулы (I-S) на системе Waters preparative LCMS autopurification system. Элюируемый первым из хиральной колонки энантиомер является бромированным (по участку, значительно удаленному от стереогенного центра), что дает соответствующее 5'-бром-производное, которое впоследствии кристаллизуется, образуя монокристалл, пригодный для рентгеновской кристаллографии. Кристаллическая структура такого бромированного производного элюируемого первым энантиомера определена, и установлено, что абсолютная конфигурация является точно такой же, как и у соединения формулы (I-R). Следовательно, элюируемый вторым из хиральной колонки энантиомером является соединение формулы (I-S). Кроме того, вещество, полученное из потока экстракта ППС-разделения, имеет специфическое оптическое вращение того же знака (положительное, то есть правовращающее), что и вещество, полученное вышеупомянутым ЖХ-разделением.
Синтетический пример 4
Синтез 1'-{[3-(трифторметил)пиридин-2-ил]метил}-2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-она
Соединение формулы (II)
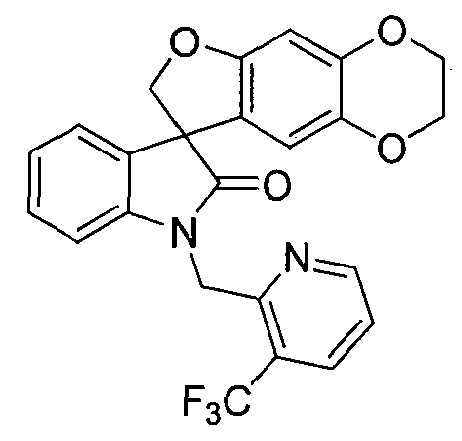
В реактор на 160 л загружают 1,4-диоксан (43 л) при температуре окружающей среды и затем 2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-он (6,80 кг, 23 моль). К полученной суспензии добавляют карбонат цезия (18,7 кг, 58 моль) и смесь нагревают до 82°C за 72 минуты. Контейнер промывают 1,4-диоксаном (7 л) и используют для введения порциями, за 35 минут, 2-(хлорметил)-3-(трифторметил)пиридингидрохлорида (5,88 кг, 25 моль). Температуру реакционной смеси повышают до 100°C за 43 минуты и смесь перемешивают при 100°C в течение 3 ч, охлаждают до 20°C за 90 минут и дополнительно перемешивают 16 ч. Добавляют деионизованную воду (40 л) и дихлорметан (40 л) и полученную смесь перемешивают при 22°C в течение 12 минут. Перемешивание прекращают и оставляют на 21 минуту, давая разделиться фазам. Водную и органическую фазы разделяют в сборниках. В реактор на 160 л загружают водную фазу и дихлорметан (41 л). Смесь перемешивают при 19°C в течение 10 минут, перемешивание прекращают и оставляют на 10 минут, давая разделиться фазам. Водную фазу удаляют, органическую фазу с предыдущей стадии переносят из сборника в реактор. Добавляют деионизованную воду (40 л) и смесь перемешивают при 22°C в течение 10 минут. Перемешивание прекращают и оставляют на 43 минуты, давая разделиться фазам. Водную фазу удаляют и органическую фазу концентрируют досуха при пониженном давлении 712-97 мбар при 19-38°C. К остатку добавляют метанол (56 л) за 19 минут. Полученную суспензию охлаждают до 3°C за 64 минуты и перемешивают 98 минут. Смесь фильтруют и плотный осадок на фильтре промывают охлажденным (0°C) метанолом (14 л) и сушат при пониженном давлении 90-9 мбар при 21-46°C в течение 10,5 ч, получая 1'-{[3-(трифторметил)пиридин-2-ил]метил}-2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-он (8,00 кг, 81%) в виде не совсем белого твердого вещества: чистота (ВЭЖХ-УФ) 99,8%; 1Н ЯМР (300 МГц, CDCl3) δ 8,67-8,63 (м, 1H), 8,01-7,96 (м, 1H), 7,35-7,28 (м, 1H), 7,22-7,13 (м, 2H), 7,05-6,98 (м, 1H), 6,63 (c, 1H), 6,62-6,58 (м, 1H), 6,49 (c, 1H), 5,42, 5,14 (АВкв, JAB=17,3 Гц, 2H), 5,00, 4,74 (АВкв, JAB=8,9 Гц, 2H), 4,22-4,10 (м, 4H); 13C ЯМР (75 МГц, CDCl3) δ 178,3, 155,3, 152,5, 144,6, 142,4, 138,4, 134,3 (кв, 3JC-F=5,2 Гц), 132,8, 128,7, 124,4, (кв, 2JС-F=32,6 Гц), 123,9 (кв, 1JC-F=273,3 Гц), 123,8, 123,4, 122,2, 121,7, 112,6, 108,6, 99,2, 80,2, 64,6, 64,0, 58,3, 42,3 (кв, 4JC-F=3,3 Гц); MC (ES+) m/z 454,8 (М+1).
Синтетический пример 5
Разделение соединения формулы (II) ППС-хроматографией
Соединение формулы (II) разделяют на соединение формулы (II-S) и соединение формулы (II-R) ППС-хроматографией в следующих условиях:
Экстракт: 182,67 мл/мин
Рафинат: 67,44 мл/мин
Элюент: 224,11 мл/мин
Подача: 26,0 мл/мин
Рециркуляция: 420 мл/мин
Время хроматографирования: 1,05 мин
Температура: 25°C
Давление: 50-55 бар
Загрузочный раствор (68,4 г соединения формулы (II)) в 1,0 л подвижной фазы (смесь дихлорметан/ацетон 97:3 (об:об)) непрерывно впрыскивают в систему ППС (Novasep Licosep Lab Unit), оборудованную восемью идентичными колонками в конфигурации 2-2-2-2, содержащими 110 г (на колонку, 10,0 см, 4,8 см I.D.) ChiralPAK®-AD в качестве неподвижной фазы. Соединение формулы (II-R) содержится в потоке рафината, и соединение формулы (II-S) содержится в потоке экстракта.
Все 10,62 кг соединения формулы (II) подвергают ППС-хроматографии, используя вышеуказанные условия. Все фракции экстракта хиральной чистоты (ВЭЖХ) >99,0 a/a объединяют, концентрируют до объема 26 л при пониженном давлении и переносят в реактор на 100 л. Раствор дополнительно концентрируют при пониженном давлении 700-590 мбар при 26-37°C, собирая 13 л дистиллята. Добавляют метанол (25 л) и смесь концентрируют при пониженном давлении 650-360 мбар при 30-38°C, собирая 15 л дистиллята. Смесь охлаждают до 20°C и добавляют метанол (15 л). Смесь концентрируют при пониженном давлении 650-320 мбар при 20-39°C, собирая 15 л дистиллята, и охлаждают до 1°C за 53 минуты, и перемешивают еще 70 минут. Суспензию фильтруют и плотный осадок на фильтре промывают охлажденным (0°C) метанолом (9 л), и сушат при температуре окружающей среды в токе газообразного азота 15,5 ч. Твердое вещество дополнительно сушат при пониженном давлении 40-1 мбар при 50°C 195 минут, что дает соединение формулы (II-S), т.е. (S)-1'-{[3-(трифторметил)пиридин-2-ил]метил}-2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-он (3,62 кг) в виде бесцветного твердого вещества: чистота (ВЭЖХ-УФ) 100%; 1Н ЯМР (300 МГц, CDCl3) δ 8,67-8,63 (м, 1H), 8,01-7,96 (м, 1H), 7,35-7,28 (м, 1H), 7,22-7,13 (м, 2H), 7,05-6,98 (м, 1H), 6,63 (c, 1H), 6,62-6,58 (м, 1H), 6,49 (c, 1H), 5,42, 5,14 (АВкв, JAB=17,3 Гц, 2H), 5,00, 4,74 (АВкв, JAB=8,9 Гц, 2H), 4,22-4,10 (м, 4H); 13C ЯМР (75 МГц, CDCl3) δ 178,3, 155,3, 152,5, 144,6, 142,4, 138,4, 134,3 (кв, 3JC-F=5,2 Гц), 132,8, 128,7, 124,4, (кв, 2JC-F=32,6 Гц), 123,9 (кв, 1JC-F=273,3 Гц), 123,8, 123,4, 122,2, 121,7, 112,6, 108,6, 99,2, 80,2, 64,6, 64,0, 58,3, 42,3 (кв, 4JC-F=3,3 Гц); MC (ES+) m/z 454,8 (M+1); [α]D +45,1° (c 2,02, ДМСО); ee (CHIRALPAK IC, смесь дихлорметан/ацетон 97/3 (об/об)) 100%.
Соединение формулы (II-R), т.е. (R)-1'-{[3-(трифторметил)пиридин-2-ил]метил}-2,3-дигидроспиро[фуро[2,3-g][1,4]бензодиоксин-8,3'-индол]-2'(1'H)-он выделяют из рафината стандартными способами.
Биологические испытания
Для более полного понимания описанного здесь изобретения предусмотрено следующее биологическое испытание, демонстрирующее полезность полученных здесь соединений. Следует понимать, что данный пример служит исключительно иллюстративным целям и никоим образом не рассматривается, как ограничивающий изобретение.
Биологический пример 1
Исследование инфлюкса с помощью гуанидина (испытание in vitro)
Данный пример описывает испытание in vitro для тестирования и анализа профиля испытуемых средств в отношении потенциалзависимых натриевых каналов человека или крысы, устойчиво экспрессируемых в клетках либо эндогенного, либо гетерологично экспрессируемого происхождения. Испытание также полезно для определения IC50 соединения, модулирующего (предпочтительно блокирующего) потенциалзависимые натриевые каналы. Испытание основано на исследовании инфлюкса с помощью гуанидина, описанном в Reddy, N.L., et al., J. Med. Chem. (1998), 41(17):3298-302.
Исследование инфлюкса с помощью гуанидина представляет собой исследование потока с помощью радиоактивной метки, используемое для определения активности ионного потока потенциалзависимых натриевых каналов в формате высокой производительности, основанном на микропланшетах. В исследовании используют 14C-гуанидингидрохлорид в комбинации с различными известными модуляторами потенциалзависимых натриевых каналов, которые создают постоянный приток, для изучения активности испытуемых средств. Активность определяют, рассчитывая IC50. Избирательность определяют, сравнивая активность соединения в отношении представляющего интерес потенциалзависимого натриевого канала с активностью соединения в отношении других потенциалзависимых натриевых каналов (также называемую 'селективность профилирования').
Каждое из испытуемых средств исследуют в отношении клеток, экспрессирующих представляющие интерес потенциалзависимые натриевые каналы. Потенциалзависимые натриевые каналы характеризуются как TTX-чувствительные или нечувствительные. Такое свойство полезно при оценке активностей представляющего интерес потенциалзависимого натриевого канала, когда указанный канал находится в смешанной популяции с другими потенциалзависимыми натриевыми каналами. В следующей таблице 1 сведены клеточные линии, полезные для скрининга активности отдельного потенциалзависимого натриевого канала в присутствии или отсутствии TTX.
|
Можно также использовать иммортализованные линии клеток, которые гетерологично экспрессируют потенциалзависимые натриевые каналы. Клонирование, стабильная трансфекция и размножение таких клеточных линий известны специалисту в данной области (см., например, Klugbauer, N, et al., EMBO J. (1995), 14(6):1084-90 и Lossin, C., et al., Neuron (2002), 34, pp.877-884).
Клетки, экспрессирующие представляющий интерес потенциалзависимый натриевый канал, выращивают согласно инструкциям поставщика или в случае рекомбинантной клетки в присутствии среды для избирательного выращивания клеток, такой как G418 (Gibco/Invitrogen). Клетки отделяют от чашек для культивирования с помощью ферментного раствора (1X) Трипсин/EDTA (Gibco/Invitrogen) и анализируют на плотность и жизнеспособность, используя счетную камеру (Neubauer). Отделенные клетки промывают и ресуспендируют в культуральных средах, затем высевают на чашку Петри в поли-D-лизин, покрытый Scintiplates (Perkin Elmer) (приблизительно 100000 клетки/ячейка), и инкубируют при 37°C/5% CO2 в течение 20-24 часов. После экстенсивной промывки HEPES-забуференным физиологическим раствором с низким содержанием натрия (LNHBSS) (холинхлорид 150 мМ, HEPES (Sigma) 20 нМ, хлорид кальция 1 мМ, хлорид калия 5 мМ, хлорид магния 1 мМ, глюкоза 10 мМ) испытуемые средства разбавляют LNHBSS и затем вносят в каждую ячейку при заданной концентрации. (Можно использовать изменяющиеся концентрации испытуемого средства). Активационная/меченная радиоактивным изотопом смесь содержит алкалоид, такой как вератридин или аконитин (Sigma), или пиретроид, такой как дельтаметрин, яд гигантского израильского скорпиона Leiurus quinquestriatus hebraeus (Sigma) и 14C-гуанидингидрохлорид (ARC), для измерения переноса через потенциалзависимые натриевые каналы.
После внесения в ячейки испытуемого средства и активационной/меченной радиоактивным изотопом смеси поли-D-лизин, покрытый Scintiplates, инкубируют при температуре окружающей среды. После инкубации поли-D-лизин, покрытый Scintiplates, экстенсивно промывают LNHBSS, пополненным гуанидином (Sigma). Поли-D-лизин, покрытый Scintiplates, сушат и затем считают на Wallac MicroBeta TriLux (Perkin-Elmer Life Sciences). Способность испытуемого средства блокировать активность потенциалзависимых натриевых каналов оценивают, сравнивая количество 14C-гуанидина, присутствующего в клетках, экспрессирующих различные потенциалзависимые натриевые каналы. На основании полученных данных ряд расчетов, как изложено в другом месте данного описания, может применяться для установления, является ли испытуемое средство селективным в отношении конкретного потенциалзависимого натриевого канала.
Значение IC50 испытуемого средства для конкретного потенциалзависимого натриевого канала может быть определено с использованием вышеуказанного общего метода. IC50 можно определить, используя 3-, 8-, 10-, 12- или 16-точечную кривую, в дубликате или трипликате, с исходной концентрацией 1, 5 или 10 мкМ, последовательно разбавляемой до конечной концентрации, достигающей субнаномолярного, наномолярного и низкого микромолекулярного диапазонов. Как правило, устанавливают срединную точку концентрации, равную 1 мкМ, и применяют последующие концентрации, равные большим или меньшим половинным разведениям (например, 0,5 мкМ; 5 мкМ и 0,25 мкМ; 10 мкМ и 0,125 мкМ; 20 мкМ и проч.). Кривую IC50 обсчитывают, используя 4-параметровую логистическую модель или сигмоидальную дозозависимую модельную формулу (подбор=(A+((B-A)/(1+((C/x)AD)))).
Кратную селективность, фактор селективности или коэффициент селективности рассчитывают делением значения IC50 для исследуемого потенциалзависимого натриевого канала на ссылочное значение для потенциалзависимого натриевого канала, например Nav1,5.
Соответственно соединения, полученные описанными здесь способами, демонстрирующие блокирующую активность потенциалзависимого натриевого канала против hNav1,7, приведены ниже в таблице 2.
|
Все патенты США, опубликованные патентные заявки США, патентные заявки США, иностранные патенты, иностранные патентные заявки и непатентные публикации, упомянутые в данном описании и/или перечисленные в перечне технических характеристик, полностью включены здесь в качестве ссылок.
Хотя вышеизложенное изобретение описано более детально, чтобы облегчить понимание, очевидно, что на практике могут быть осуществлены определенные изменения и модификации в рамках объема приложенных пунктов патентной формулы. Следовательно, описанные варианты осуществления рассматриваются как иллюстративные, а не ограничительные, и изобретение не ограничивается приведенными здесь подробностями, но может быть модифицировано в рамках объема и эквивалентов приложенных пунктов.
Органические соединения
Энантиомеры спиро-оксиндольных соединений и их применение в качестве терапевтических средств
Фармацевтические композиции спиро-оксиндольного соединения для местного введения и их применение в качестве терапевтических агентов
Соединения бензолсульфонамидов и их использование в качестве терапевтических средств
Соединения бензолсульфонамида и их применение в качестве терапевтических средств
Энантиомеры спиро-оксиндольных соединений и их применение в качестве терапевтических средств
Фармацевтические композиции спиро-оксиндольного соединения для местного введения и их применение в качестве терапевтических агентов

