Результат интеллектуальной деятельности: СПОСОБ НЕИНВАЗИВНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА МЕТОДОМ СЕКВЕНИРОВАНИЯ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Изобретение относится к области медицины, а именно неинвазивной пренатальной диагностике анеуплоидий плода по внеклеточной ДНК крови матери, и может быть использовано для определения генетических аномалий плода на первом триместре беременности безопасными как для ребенка, так и для матери неинвазивными методами.
Анеуплоидия является следствием изменений кариотипа, при котором число хромосом в клетках плода не кратно гаплоидному набору (в отличие от нормального состояния кариотипа, эуплоидии, при котором число хромосом равно двум гаплоидным наборам). Примерами анеуплоидий, которые могут быть выявлены с использованием заявленного способа, являются моносомия и трисомия, а также частичная трисомия или частичная моносомия (соответственно, приобретение дополнительных копий или делеция крупных участков хромосом, как правило, одного из хромосомных плеч). Частными примерами являются трисомия по 21-й хромосоме (синдром Дауна), трисомия по 13-й хромосоме (синдром Патау), трисомия по 18-й хромосоме (синдром Эдвардса), моносомия по Х-хромосоме (синдром Шерешевского-Тернера) или наличие более чем двух половых хромосом, например синдром Клайнфельтера (XXY), и т.д. Перечень связанных с анеуплоидией заболеваний, которые могут быть диагностированы заявленным способом, не ограничен каким-либо специальным образом.
Уровень техники
Из уровня техники известны широко применяемые инвазивные способы диагностики геномных аномалий плода (анеуплоидий), например, с помощью кариотипирования хорионной жидкости или образца плаценты.
Однако данные методы подразумевают физическое взаимодействие с плацентой или даже с плодом. Несмотря на общеприменимость и техническую зрелость этих методик, существует некоторая вероятность повреждения тканей плода, что в свою очередь может вести к различным нарушениям его развития.
Из уровня техники известны неинвазивные методы диагностики анеуплоидий - биохимические, ультразвуковые - которые дают нечеткие и/или недостоверные диагнозы. Данные методы обладают слишком низкой чувствительностью и/или селективностью по сравнению с генетическим подходом, основанным на исследовании внеклеточной ДНК крови матери, являющимся наиболее перспективным методом диагностики анеуплоидий.
Обнаружение плодного генного материала во внеклеточной ДНК в крови матери (Lo YM., et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350:485-7) позволило изучать геном будущего ребенка уже на ранних стадиях беременности, не нарушая при этом целостности плаценты. В первую очередь это вызвало интерес в области пренатальной диагностики анеуплоидий. С одной стороны, анеуплоидии - одни из самых тяжелых генетических заболеваний, с другой - одни из самых легко обнаруживаемых, будучи самыми отличающимися от нормы с точки зрения строения генома.
Развитию генетических методов диагностики различных заболеваний также способствовал прогресс в области секвенирования ДНК; с 2005 года появилось несколько принципиально схожих технологий массового параллельного секвенирования. Главной их особенностью явилась возможность, позволяющая не только читать сами последовательности ДНК, но и считать количества их фрагментов и сравнивать представленности разных участков генома.
В базовом полногеномном подходе диагностике анеуплоидий (например, Ehrich М., et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol 2011; 204:205.el-11) производится полногеномное секвенирование всей внеклеточной ДНК матери, после чего данные секвенирования картируются на референсный геном человека. Далее производится подсчет количеств чтений, относящихся к каждой хромосоме в каждом образце, эти количества нормализуются на общее количество полученных с образца чтений. Для определения статуса анеуплоидий по конкретной хромосоме необходимо собрать контрольную выборку секвенированных здоровых образцов и сравнить нормализованное количество чтений данной хромосомы между исследуемым образцом и распределением таких количеств в контрольной выборке (используется z-критерий).
Этот метод имеет следующие недостатки: во-первых, необходима готовая контрольная выборка, при этом подразумевается, что результаты работы секвенатора одинаковы по выходу данных, их качеству и структуре (чего не следует ожидать на практике); во-вторых, образец характеризуется единственным числом - нормализованным количеством чтений по исследуемой хромосоме, а статистическая поддержка исходит только от контрольной выборки, что не оптимально и снижает точность расчетов.
Наиболее близким к заявляемому способу является полногеномный подход с коррекцией GC-состава (патент US8296076). Данный метод отличается от предыдущего методикой расчета данных секвенирования. Во-первых, он учитывает неравномерность секвенирования, связанную с GC-составом читаемой ДНК; таковая зависимость обычно является нелинейной и варьируется не только между разными технологиями секвенирования, но также между разными приборами одной серии и версиями используемых реактивов. Во-вторых, вместо единой кумулятивной метрики по целой хромосоме используется разбиение генома на множество коротких участков (окон) и подсчет количества чтений, приходящихся на каждое такое окно, в результате чего определение анеуплоидии производится посредством сравнения двух выборок: окон с исследуемой хромосомы и окон со всех остальных хромосом. Это позволяет избавиться от необходимости в контрольной выборке, а также усиливает статистическую мощность анализа.
В настоящем изобретении предлагается ряд улучшений для последнего упомянутого подхода, заключающихся в оптимизации как экспериментальной части методики, так и интерпретации результата исследования.
Раскрытие изобретения
Ввиду тяжести заболеваний, связанных с анеуплоидией, постановка соответствующего диагноза может являться основанием для проведения аборта, в связи с чем имеет большое значение скорость и достоверность проведения такой диагностики (т.е. проведение исследований в более ранние сроки беременности безопасным как для ребенка, так и для матери неинвазивным методом, обеспечивающим получение достоверного результата). Генетические методы диагностики являются единственными методами из неинвазивных, которые дают достоверные результаты на ранних сроках беременности.
Задачей, решаемой авторами настоящего изобретения, является разработка нового способа неинвазивной диагностики анеуплоидии плода при исследовании образца, полученного у беременной женщины.
Технический результат настоящего изобретения заключается в повышении чувствительности и селективности определения анеуплоидии плода при использовании в качестве исследуемого материала образца, полученного у беременной женщины.
Поставленная задача решается тем, что способ определения анеуплоидии плода, включает следующие стадии:
а. выделение внеклеточной ДНК из образца, полученного у беременной женщины;
b. приготовление геномных библиотек с использованием выделенной внеклеточной ДНК, включающее достройку и затупление концов молекул ДНК, лигирование адаптеров и ПЦР-амплификацию;
с. секвенирование полученных геномных библиотек методом массового параллельного секвенирования;
d. картирование полученных чтений на референсный геном человека для определения их координат (номера хромосомы и позиции на ней, считая с 5'-конца хромосомы);
е. исключение чтений, координаты которых совпадают с координатами известных копийных полиморфизмов человека;
f. разбиение референсного генома на последовательно идущие, неперекрывающиеся «окна» (отрезки) равной длины, составляющей от 1 тыс. до 100 тыс. нуклеотидов; и сопоставление этим «окнам» чтений в зависимости от координат последних;
g. определение покрытия окон (числа сопоставленных окнам чтений) и отсев окон, не содержащих чтений;
h. определение GC-состава для нуклеотидной последовательности каждого оставшегося окна и отсев окон (оставшихся после стадии g), имеющих GC-состав менее 0,2 и более 0,8;
i. определение медианы покрытия чтениями (М) окон (оставшихся после стадии h) и стандартного отклонения этого покрытия (σ) с последующим отсевом окон, имеющих покрытие, большее чем М+2σ;
j. определение корректированного покрытия оставшихся по итогам стадии i окон по их GC-составу посредством деления их покрытия на коэффициент, характеризующий превышение среднего покрытия всех окон с таким же GC-составом над средним покрытием по всем окнам;
k. формирование из оставшихся окон выборки №1, в которую отбирают окна, принадлежащие исследуемой хромосоме, и выборки №2 - окна, принадлежащие к контрольным хромосомам;
l. анализ корректированного покрытия выборок №1 и №2 с использованием критерия, позволяющего выявить наличие статистически достоверных отличий между двумя выборками, при этом при выявлении статистически достоверных отличий делают вывод о наличии анеуплоидии у плода.
В качестве анеуплоидии определяют трисомию или моносомию по любой из 13, 18, 21, X или Y хромосом или по их комбинациям. В качестве образца исследуют кровь, плазму, сыворотку или мочу. Перед этапом выделения внеклеточной ДНК к образцу, полученному у беременной женщины, может быть добавлена молекулярная метка. В качестве молекулярной метки может быть использована молекула нуклеиновой кислоты, имеющая степень гомологии с любыми известными, природными либо технологическими, последовательностями ДНК не более 20%. При использовании в технологии молекулярной метки после стадии секвенирования осуществляют проверку наличия и идентификацию молекулярной метки в образце посредством сравнения чтений с последовательностью метки.
В одном из вариантов реализации изобретения протокол приготовления геномных библиотек включает дотирование адаптеров и ПЦР-амплификацию. Время лигирования адаптеров составляет не менее 5 часов. Выделение внеклеточной ДНК может быть реализовано с использованием наборов QIAamp Circulating Nucleic Acid Kit (Qiagen). Секвенирование может быть осуществлено с использованием технологии синтеза на молекулярных колониях. В процессе секвенирования получают чтения длиной не менее 25 нуклеотидов. Наилучший результат достигается в результате секвенирования при получении не менее 1 млн чтений на образец.
Перед стадией картирования может быть проведена проверка качества полученных в результате секвенирования чтений, при этом отсеваются чтения, средняя вероятность ошибки нуклеотидов которых составляет более 1%. Картирование чтений осуществляется на референсный геном человека hg19. В процессе картирования чтений допускается до 5% несоответствия между последовательностями чтений и последовательностями референсного генома человека. Могут быть исключены чтения, координаты которых совпадают с участками повторяющихся последовательностей генома человека.
GC-состав окна определяют как совокупный GC-состав всех сопоставленных данному окну чтений. При отборе окон на стадии i дополнительно отсевают окна с покрытием менее М-2σ.
В качестве контрольных хромосом анализируют одну или несколько хромосом, выбранных из группы, включающей 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 15, 16, 17, 19, 20 хромосомы. В качестве исследуемых хромосом анализируют одну хромосому из группы, включающей 13, 18, 21, X или Y хромосомы.
В качестве критерия, позволяющего выявить наличие статистически достоверных отличий между двумя выборками может быть использован t-критерий Стьюдента, U-критерий Уиттни-Манна или z-критерий Фишера, где в качестве генеральной совокупности используются окна контрольных хромосом; при этом вывод о наличии анеуплоидии делают при p-value, меньшем 0,05.
Заявляемое изобретение характеризуется новой совокупностью признаков, направленной на достижение указанного технического результата. В частности, повышение чувствительности и селективности обеспечивается напрямую за счет использования дополнительных стадий анализа, таких как исключение чтений (которое производят после картирования), координаты которых совпадают с координатами известных копийных полиморфизмов человека, и отсев окон, имеющих GC-состав менее 0,2 и более 0,8. Кроме того, эти параметры также улучшаются опосредованно ввиду применения измененной последовательности приготовления библиотек.
Таким образом, заявляемое изобретение включает ряд технических улучшений по сравнению с существующими способами.
- Модифицированные протоколы приготовления геномных библиотек позволяют избежать потерь или порчи исследуемого материала в процессе подготовки его к анализу, что в свою очередь снижает требования к количеству забираемого у пациента генетического материала. Поскольку для проведения секвенирования, как правило, требуется определенная концентрация геномных библиотек, поэтому их приготовление обязательно включает в себя процедуры разведения/концентрирования ДНК и прочих реагентов с целью доведения концентрации до необходимой. ПЦР-амплификация используется для экспоненциального увеличения количества молекул ДНК, однако несет в себе опасность внесения ошибок и их последующего накопления, также экспоненциального, в размножаемых таким способом последовательностях нуклеотидов. Меньшие потери исходного материала при приготовлении библиотек означают меньшую потребность в амплификации и, как следствие, меньшую вероятность порчи образца ДНК. Кроме того, ПЦР-амплификации присуща неравномерность, поскольку уже реплицированные молекулы имеют большую вероятность репликации в последующих циклах; поэтому уменьшение количества циклов амплификации также благоприятно отразится на последующем анализе из-за более равномерно распределенных по геному данных секвенирования.
- Использование молекулярных меток для маркировки исследуемых образцов является быстрым, экономичным и простым в реализации методом определения контаминации, смешения или подмены образцов. Хотя это улучшение и не является необходимым в случае анализа единственного образца, оно имеет большое значение на практике по причине использования пакетной обработки образцов и в связи с большим количеством экспериментальных стадий, необходимых для проведения диагностики.
- На этапе статистической обработки данных секвенирования используется несколько дополнительных стадий фильтрации данных, существенно повышающих точность анализа посредством исключения или снижения влияния особенностей, присущих используемым необходимым для анализа методикам (ПЦР-амплификация, неравномерное секвенирование, несовершенство референсной последовательности генома человека и алгоритмов картирования). В их числе: отсев чтений, совпадающих с известными копийными полиморфизмами; исключение окон со слишком высоким или отсутствующим покрытием или окон, имеющих критические значения GC-состава.
Подробное описание заявляемого способа.
Сущность способа определения анеуплоидий методом секвенирования заключается в цифровом количественном анализе внеклеточной ДНК посредством секвенирования. В основу способа легла методика массового параллельного полногеномного секвенирования, которая позволяет получать до миллиарда коротких чтений за счет случайной фрагментации и последующей амплификации геномной ДНК. Полученные короткие чтения последовательностей ДНК подвергаются статистическому компьютерному анализу. При этом количество чтений, приходящихся на каждую хромосому генома, определяет представленность данной хромосомы в геноме, и таким образом может быть определен статус эуплоидии или анеуплоидий. Заявляемый способ получения и анализа данных позволяет исключить необходимость разграничения ДНК по принадлежности к матери или плоду, а также способствует обнаружению минимальных отличий в представленности отдельных хромосом от среднего значения по геному. Наличие ряда стадий фильтрации и коррекции данных in silico позволяет нивелировать проблемы, связанные с особенностями биологических образцов и техническими погрешностями прибора.
Согласно настоящему изобретению в качестве исследуемого материала может быть использован образец, полученный от беременной женщины и содержащий геномную ДНК матери и плода. В частности, может быть исследован образец крови или плазмы.
Внеклеточная ДНК (вкДНК) плода выявляется в крови матери, начиная с первого месяца беременности и составляет в норме от 3 до 6% от общей вкДНК матери (Lo Y M, Corbetta N, Chamberlain P F, Rai V, Sargent I L, Redman C W, Wainscoat J S. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997 Aug 16;350(9076):485-7). Проведение диагностики посредством анализа вкДНК возможно, начиная с 8-й, предпочтительно с 10-й недели беременности, ввиду накопления достаточного количества плодной вкДНК. Плазма из крови должна быть получена в течение 4 часов после забора, при этом сам образец должен храниться при +4°С - + 6°С все время до получения плазмы. Получение плазмы двухстадийное: грубое отделение клеток крови центрифугированием при +4°С и 1600g в течение 10 минут; отбор плазмы и последующее центрифугирование полученной плазмы при 16000g в течение 10 минут при +4С с последующим отбором верхней надосадочной фракции.
Внеклеточная ДНК может быть выделена из плазмы любым хорошо известным специалисту в данной области техники методом выделения данного типа ДНК (Lo YM., et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350:485-7). В частности, вкДНК может быть выделена с использованием коммерчески доступных наборов, таких как QIAamp Circulating Nucleic Acid Kit (Qiagen).
Маркировка образцов молекулярными метками.
Согласно настоящему изобретению к образцу, полученному у беременной женщины, непосредственно перед стадией выделения вкДНК может быть добавлена молекулярная метка. Добавление молекулярной метки позволяет однозначно идентифицировать образец после проведения анализа и обеспечить контроль отсутствия смешения, контаминации или подмены образцов. Согласно частному варианту реализации настоящего изобретения молекулярная метка может представлять собой молекулу нуклеиновой кислоты, предпочтительно ДНК, имеющую степень гомологии с любыми известными, природными либо технологическими, последовательностями ДНК не более 10% (включая остальные используемые молекулярные метки). При этом предпочтительно, чтобы молекулярная метка была сходна по размеру с вкДНК и/или могла быть выделена способом, предназначенным для выделения вкДНК. В частности, предпочтительно, чтобы длина молекулярной метки составляла 30-400 нуклеотидов.
Модификации протокола приготовления геномных библиотек.
При работе с внеклеточной ДНК возникает ряд технологических особенностей, связанных с характеристикой данного материала. В первую очередь это размер молекул ДНК и их концентрация в растворе при выделении. В плазме крови вкДНК сильно фрагментирована и представлена в виде отрезков ДНК длиной от 150 до 170 пар нуклеотидов, поскольку появляется в кровотоке по причине апоптоза клеток тела, в том числе и плаценты в случае беременности. Концентрация вкДНК в плазме составляет около 50 пг/мл, при этом доля плодной ДНК в общем количестве вкДНК беременных составляет около 6%. ДНК в такой низкой концентрации подлежит лабораторному анализу только после амплификации, в противном случае концентрация ДНК окажется ниже порога чувствительности большинства методов анализа. Для преодоления этого затруднения для повышения концентрации исследуемой ДНК может быть модифицирован протокол приготовления геномных библиотек.
Геномная библиотека - приготовленный особым образом образец ДНК, доступный для чтения на секвенаторе.
Стандартная процедура приготовления геномных библиотек включает в себя следующие операции с молекулами ДНК: фрагментацию, достройку и затупление концов, лигирование адаптеров, отбор по длине и ПЦР-амплификацию. Согласно настоящему изобретению данная процедура может быть использована как в стандартном, предусмотренном производителем оборудования для проведения секвенирования виде, так и модифицирована.
В частном варианте воплощения настоящего изобретения в целях технологического упрощения способа, увеличения чувствительности метода и уменьшения количества крови пациента, необходимого для анализа, протокол приготовления геномных библиотек может быть модифицирован следующим образом.
1. Исключена стадия фрагментирования ДНК. ВкДНК имеет средний размер около 160 нуклеотидов, и дополнительное фрагментирование, будь то ферментативное или с помощью ультразвука, не оказывает какого-либо положительного влияния на процесс приготовления библиотек, наоборот, значительно уменьшает при этом количественный выход библиотек.
2. Увеличено время дотирования адаптеров. Стандартная процедура лигирования адаптера не приводит к достаточно полноценному лигированию. В нашей модификации время лигирования увеличено до 5 часов и более, что значительно увеличивает количество полученных библиотек с адаптерами.
3. Исключена стадия селективного отбора библиотек по размеру. ВкДНК имеет достаточно компактный диапазон длин. Кроме того, лигирование адаптеров увеличивает длину двухцепочечных отрезков вкДНК на фиксированную длину. По этой причине отпадает необходимость в дополнительном отборе участков необходимой длины, а используется весь объем библиотек с адаптерами. Применение техники селективного отбора приводит к дополнительным и неоправданным потерям материала.
Секвенирование.
Далее полученные геномные библиотеки подвергают секвенированию. Предпочтительно, чтобы в результате секвенирования было получено не менее 1 млн чтений на образец, наиболее предпочтительно - не менее 3 млн чтений. Для этого секвенирование должно быть осуществлено каким-либо из методов массового параллельного секвенирования, предполагающего массовое (от сотен до сотен миллионов) одновременное чтение коротких последовательностей ДНК, полученных путем случайного фрагментирования исходного исследуемого генетического материала.
Частными примерами технологий (приборов), которые могут быть использованы, являются: секвенирование синтезом на молекулярных колониях (Genome Analyzer, HiSeq, MiSeq (Illumina), патент US 8412467), лигазное секвенирование с использованием эмульсионного ПЦР (SOUD4, 5500-series (Applied Biosystems), патент US 8431691), полупроводниковое секвенирование (Ion Torrent, Ion Proton (Life Technologies), патент US 20130012399), пиросеквенирование (454 (Roche), патент СА 2513899) и т.д. При этом заявляемый способ не ограничивается перечисленными технологиями (приборами) секвенирования.
Предпочтительно, чтобы этап секвенирования включал предварительную амплификацию читаемых геномных библиотек, за исключением случаев, когда методика используемой технологии секвенирования рекомендует от этого воздерживаться.
Данные, полученные в результате секвенирования.
В результате секвенирования получают множество нуклеотидных последовательностей (чтений). Как было упомянуто выше, предпочтительно, чтобы в результате секвенирования было получено не менее 1 млн чтений на образец, наиболее предпочтительно - не менее 3 млн чтений для относительно равномерного покрытия ими всего генома (при общей длине генома в 3 млрд нуклеотидов это составило бы 1 чтение на каждую тысячу нуклеотидов).
Для более-менее однозначной локализации последовательности в геноме человека может быть достаточно около 25-30 нуклеотидов, причем чувствительность такого определения возрастает с длиной последовательности. По этой причине предпочтительно, чтобы длина чтений, полученных в результате массового параллельного секвенирования, составляла не менее 25 нуклеотидов, еще более предпочтительно не менее 50 нуклеотидов.
Проверка качества полученных данных.
Осуществляют проверку качества полученных чтений, согласно настоящему изобретению термин «проверка качества чтений» означает фильтрацию чтений с высокой средней вероятностью ошибки составляющих их нуклеотидов. Для анализа отбирают чтения, средняя вероятность ошибки нуклеотидов которых составляет не более 1%. Как правило, эта вероятность входит в состав данных, полученных при секвенировании наряду с самими чтениями, и представлена в виде метрики Phred (отрицательный десятичный логарифм вероятности ошибки) для каждого прочтенного нуклеотида. Расчет вероятностей ошибок производится непосредственно секвенатором и находится за рамками настоящего изобретения. При анализе с использованием метрики Phred отбирают чтения, среднее качество которых составляет не менее 20 по шкале Phred.
Процедура картирования данных.
Затем отобранные чтения картируют на референсный геном человека. Согласно настоящему изобретению термин «картирование» означает поиск места вхождения искомой последовательности (чтения) в референсную последовательность (ряд нуклеотидных последовательностей, составляющих гаплоидный набор хромосом человека), возможно с допущением небольшого несоответствия, вызванного генетическими отличиями между отдельными людьми или вероятными ошибками секвенирования, предпочтительно до 5%. Такие места вхождения, характеризуемые номером хромосомы и позицией относительно 5'-конца нуклеотидной последовательности этой хромосомы, далее называются «координатами». Существует ряд алгоритмических подходов к процедуре картирования и их программных реализаций (например, известна программа bowtie2 (Langmead В, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012, 9:357-359) или Burrows-Wheeler Aligner (Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25:1754-60)); различия между ними на практике обычно несущественны. При этом в качестве референсной последовательности могут быть использованы наборы данных GRCh37 (Genome Reference Consortium human genome assembly Build 37, RefSeq Assembly ID GCF_000001405.13), NCBI36 (RefSeq Assembly ID GCF_000001405.12) или аналогичные им.
Проверка соответствия молекулярных меток.
В случае использования молекулярных меток для маркировки отдельных исследуемых образцов настоящее изобретение предполагает их проверку. Для этого производится дополнительная процедура картирования полученных чтений, на этот раз в качестве референсных последовательностей должны быть взяты последовательности использованных молекулярных меток. Поскольку молекулы метки присутствовали в исследуемом образце ткани, они будут выделены вместе с вкДНК и секвенированы; таким образом, небольшая доля полученных чтений будет на самом деле происходить от меток, а не от вкДНК. В результате данного этапа для каждого образца станет известно количество чтений, совпавших с каждой из меток. В частной реализации настоящего изобретения показателем наличия определенной метки в образце считается превалирование количества чтений, совпавших с данной меткой, над суммой количеств чтений, совпавших с остальными использованными метками, не менее чем в 10 раз (предпочтительно - не менее чем в 100 раз). Заключение о подмене образцов или об их контаминации/смешении дается в случае несовпадения превалирующей в образце метки с ожидаемой или отсутствии превалирования какой-либо метки соответственно.
Исключение нежелательных участков генома.
Согласно настоящему изобретению из дальнейшего анализа могут быть исключены чтения, координаты которых совпадают с координатами известных копийных полиморфизмов человека. Термин «копийные полиморфизмы» означает, что разные индивидуумы могут иметь в своих геномах определенную последовательность нуклеотидов в разных количествах. Исключение таких участков позволяет избежать ситуаций, вызванных несовершенством построения референсного генома и/или алгоритмов выравнивания последовательностей, используемых в процедуре картирования; такие ситуации могут приводить к ошибочному определению координат чтений и/или к аномальным значениям покрытия отдельных регионов генома, что снижало бы точность проводимого анализа. Примерами копийных полиморфизмов могут служить таковые, описанные в базе данных Database of Genomic Variants (lafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004 Sep;36(9):949-51).
По аналогичным причинам предпочтителен также отсев чтений, координаты которых совпадают с координатами известных геномных повторов человека. Примеры таких повторов могут быть найдены в базе данных RepBase Update (Jurka J., et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 2005 110:462-467).
Группировка данных и отсев нежелательных групп.
Далее может быть проведено разбиение генома на последовательно идущие неперекрывающиеся участки (окна) равной длины. Длина (размер) окон может варьироваться от 1 до 100 тыс. нуклеотидов в зависимости от целей вычислений, требований точности или калибровки метода на выборке с известными диагнозами. Для последующего анализа отсеивают окна, потенциально способные повлиять на точность анализа. Для этого координаты чтений группируют по окнам и определяют покрытие окон (количество чтений, чьи координаты совпали с координатами окон). Отбор окон для анализа может включать в себя один или несколько этапов: 1) из анализа исключают окна, не содержащие чтений (поскольку разные хромосомы могут иметь разную встречаемость недоступных для процедур секвенирования и/или картирования участков); 2) для каждого окна определяют его GC-состав (отношение количества гуанинов и цитозинов в последовательности к общему количеству нуклеотидов в ней же), при этом из анализа исключают окна, имеющие GC-состав менее 0,2 и более 0,8 (пороговые значения GC-состава могут варьироваться); как правило, участки с экстремальными значениями GC-состава являются часто повторяющимися последовательностями и/или малодоступны для процедур секвенирования и/или картирования; 3) определяют медиану покрытия чтениями (М) окон и стандартное отклонение этого покрытия (σ) и исключают окна, имеющие покрытие, большее чем М+2σ, поскольку такие окна, как правило, содержат ранее неизвестные или неопределяемые предыдущими способами копийные участки или повторяющиеся последовательности. При этом очередность проведения и комбинация перечисленных выше этапов отсева окон могут быть различными.
Коррекция по GC-составу.
Для дальнейшего анализа проводят коррекцию покрытия отобранных окон с учетом их GC-состава. Для этого их покрытие делится на коэффициент превышения среднего покрытия всех окон с таким же GC-составом над средним покрытием по всем окнам. Коррекцию осуществляют следующим образом. Производят подсчет коэффициентов коррекции окон по GC-составу. Для всех окон каждого GC-состава в диапазоне от 0,2 до 0,8 с шагом 0,001 определяют среднее покрытие. Коэффициент коррекции для окон данного состава равен отношению этого среднего покрытия к среднему покрытию по всем окнам. Для дальнейших статистических расчетов используют корректированное покрытие окон, равное произведению покрытия окна на соответствующий его GC-составу коэффициент коррекции (Fan НС., Quake SR. Sensitivity of noninvasive prenatal detection of fetal aneuploidy from maternal plasma using shotgun sequencing is limited only by counting statistics. PLoS One. 2010 May 3; 5(5):e 10439).
Статистический анализ.
Далее окна группируют на следующие выборки: выборка №1 - окна, принадлежащие исследуемой хромосоме, выборка №2 - окна, принадлежащие к контрольным хромосомам, выборка №3 - окна, не принадлежащие к исследуемой и контрольным хромосомам. При этом в качестве исследуемой хромосомы может быть выбрана любая хромосома; в частности, исследуемой хромосомой может быть хромосома 13, 18, 21, X или Y. В качестве контрольных хромосом могут анализироваться одна или несколько аутосом (не половых хромосом); предпочтительно, чтобы в число контрольных хромосом не включались 13, 18, 21 хромосомы.
Корректированное покрытие окон из выборок №1 и №2 может быть проанализировано с использованием любого критерия, позволяющего выявить наличие статистически достоверных отличий между двумя выборками, при этом при выявлении статистически достоверных отличий делают вывод о наличии анеуплоидии у плода. В частном варианте воплощения настоящего изобретения в качестве указанного выше критерия может быть использован t-критерий Стьюдента, при этом вывод о наличии анеуплоидии делают при р-value, меньшем 0,05, предпочтительно р-value, меньшем 0,001.
В другом воплощении настоящего изобретения в качестве критерия может быть использован U-критерий Манна-Уитни (Mann, Н.В.; Whitney, D.R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Statistics 18, (1947). 50-60) с аналогичными порогами.
Примеры осуществление изобретения
Пример №1. Сбор материала
У женщины, проходящей пренатальную генетическую диагностику на 11-й неделе беременности, была собрана кровь в пробирки с ЭДТА, объемом 4 мл. Кровь хранили не более трех часов при +4°С. Не позднее чем через три часа после флеботомии пробирки с кровью центрифугировали в течение 10 мин при 2000g для получения плазмы, богатой тромбоцитами. Далее плазму повторно центрифугировали в течение 15 мин при 16000g для получения плазмы, свободной от целых клеток крови. Внеклеточную ДНК получали из очищенной плазмы крови с помощью набора реактивов QIAamp Circulating Nucleic Acid Kit (Qiagen), руководствуясь инструкцией к набору. Концентрацию полученной вкДНК определяли с помощью флюориметра Qubit 2.0 (Life Technologies).
Пример №2. Приготовление геномной библиотеки и секвенирование
Для приготовления библиотеки взяли 20 нг вкДНК, выделенной из плазмы крови. Приготовление библиотеки проводили с помощью наборов реактивов, совместимых с платформой Illumina: NEBNext DNA library prep reagent set for Illumina и NEBNext multiplex oligos for Illumina (North England Biolabs), руководствуясь инструкциями к набору. Процедура приготовления библиотеки включала в себя достройку и затупление концов вкДНК, лигирование адаптеров (в течение 10 часов) и ПЦР-амплификацию (15 циклов). Концентрацию полученной библиотеки проверяли с помощью флюориметра Qubit 2.0 (Life Technologies), она составила 13,5 нг/мкл. Определение размера и качества приготовления библиотеки проводили с помощью прибора Bioanalyzer 2100 (Agilent), длина составила 290±30 п.н. Полученную библиотеку подвергали полногеномному секвенированию на приборе HiSeq 1500 (Illumina) с использованием проточной ячейки HiSeq Rapid SR (Illumina).
Пример №3. Добавление молекулярной метки и ее определение в данных секвенирования
В качестве молекулярных меток использовали искусственно синтезированные нуклеотидные последовательности:
Метка А:
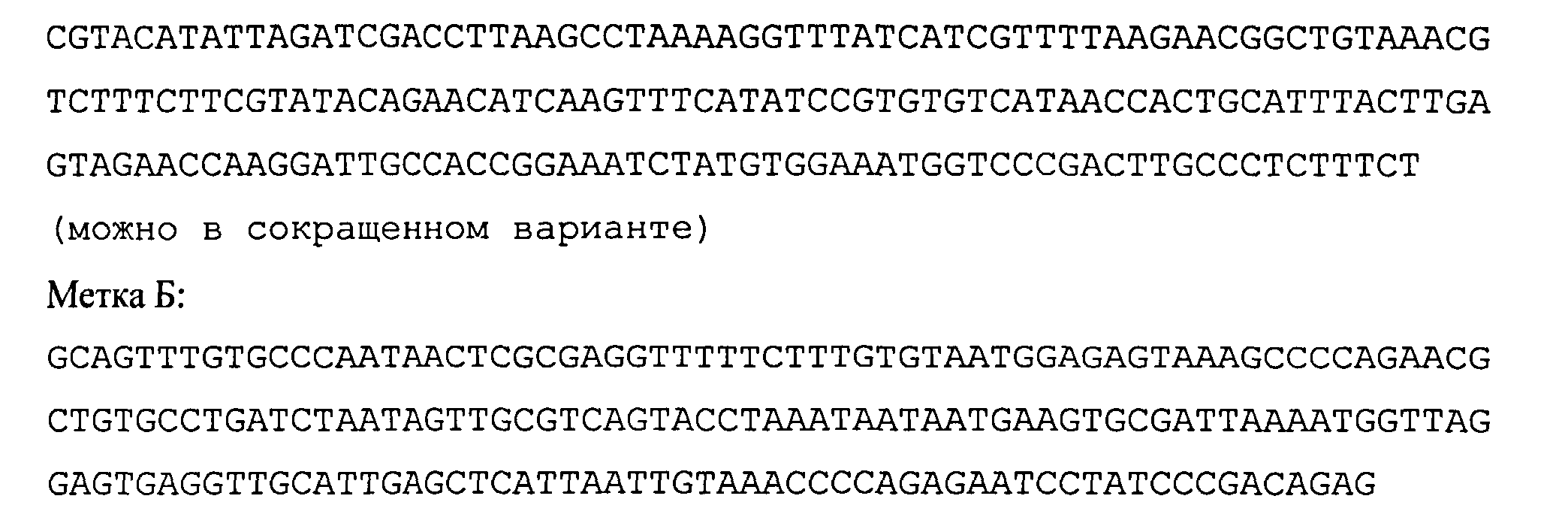
Два образца вкДНК, №1 и №2, были смешаны с молекулярными метками А и Б, соответственно, в пропорции 10 пг молекулярной метки на 20 нг вкДНК. Из обоих образцов вкДНК приготовили геномные библиотеки, которые поместили в две одинаковые неименованные пробирки и впоследствии секвенировали по отдельности. В результате проведения полногеномного секвенирования двух анонимных образцов было получено 5128319 и 7472622 чтений. Чтения каждого образца картировались с помощью программы bowtie2 на референсную последовательность, состоящую из нуклеотидных последовательностей меток А и Б. В результате этого было определено, что данные секвенирования одного анонимного образца содержали 0 чтений, совпадающих по всей длине с меткой А, и 1125 чтений, совпадающих с меткой Б. Для другого анонимного образца эти количества составили 837 и 0 соответственно. Это позволило сделать заключение, что в первом случае был секвенирован образец №2, а во втором - образец №1.
Пример №4. Анализ данных секвенирования и определение анеуплоидий
В результате проведения полногеномного секвенирования было получено 5128319 чтений. Для начала чтения проходили процедуру контроля качества, предполагающую отсев чтений со средней вероятностью ошибки нуклеотидов более 1%. Оставшиеся 4 854 348 чтений картировались с помощью программы bowtie2 на референсный геном человека hg19 для определения геномных координат. Чтения, для которых было невозможно определить геномные координаты, отбрасывались. Из успешно картированных 4252991 чтений исключались чтения, попадающие на регионы, известные как копийные полиморфизмы человека, описанные в базе данных Database of Genomic Variants, в результате чего было потеряно еще 96307 чтений. При дальнейшем анализе определялась степень покрытия референсного генома в окнах длиной 20 тыс. нуклеотидов (всего 157867 окон). В первую очередь производился подсчет GC-состава нуклеотидной последовательности для каждого окна. Исключались окна с GC-составом более 0,68 или менее 0,32, в результате чего осталось 154794 окон. Определялось покрытие оставшихся окон - количество картированных на каждое окно чтений, а также медиана М и стандартное отклонение от данного параметра по всем окнам; окна с покрытием вне пределов М±2σ исключались. Для оставшихся 102 504 окон производился подсчет коэффициентов коррекции по GC-составу их нуклеотидных последовательностей: для каждого GC-состава в диапазоне от 0,32 до 0,78 с шагом 0,001 определялось среднее наблюдаемое покрытие, которое нормировалось на общее покрытие по всем окнам. Для статистических расчетов использовалось корректированное покрытие, равное произведению покрытия окна на соответствующий его GC-составу коэффициент коррекции.
Для проверки анеуплоидии по какой-либо хромосоме все окна были разделены на две выборки: окна с целевой 21-й хромосомы (выборка №1, 1215 окон) и контрольные окна, в которые вошли окна хромосом 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 16, 17, 19, 20 (выборка №2, 88 239 окон). Использовался односторонний двухвыборочный t-критерий для независимых выборок. В качестве нулевой гипотезы принималось утверждение, что корректированные покрытия окон выборки №1 не выше, чем корректированные покрытия окон выборки №2. В конкретном примере было получено значение р-value, равное 0,00000008, что позволило сделать заключение о наличии трисомии по 21-й хромосоме у исследуемого плода.
Технология определения трисомии хромосом методом секвенирования
Технология определения анеуплоидии методом секвенирования
Способ неинвазивной пренатальной диагностики анеуплоидий плода
Способ селективного отбора целевых участков днк
Способ неинвазивной пренатальной диагностики анеуплоидий плода
Технология определения трисомии хромосом методом секвенирования
Технология определения анеуплоидии методом секвенирования
Способ неинвазивной пренатальной диагностики анеуплоидий плода
Способ селективного отбора целевых участков днк
Способ неинвазивной пренатальной диагностики анеуплоидий плода

