Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ
Вид РИД
Изобретение
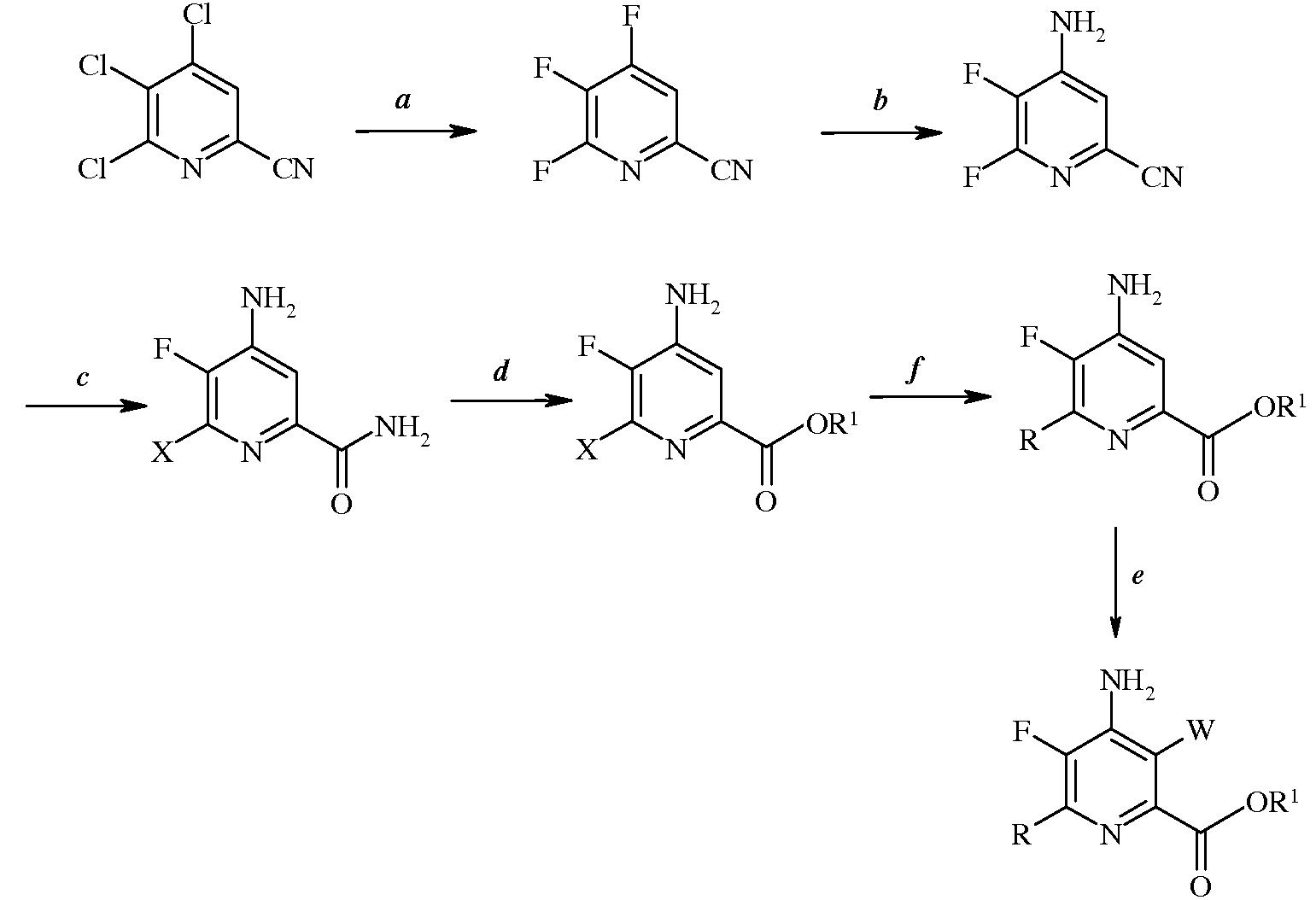
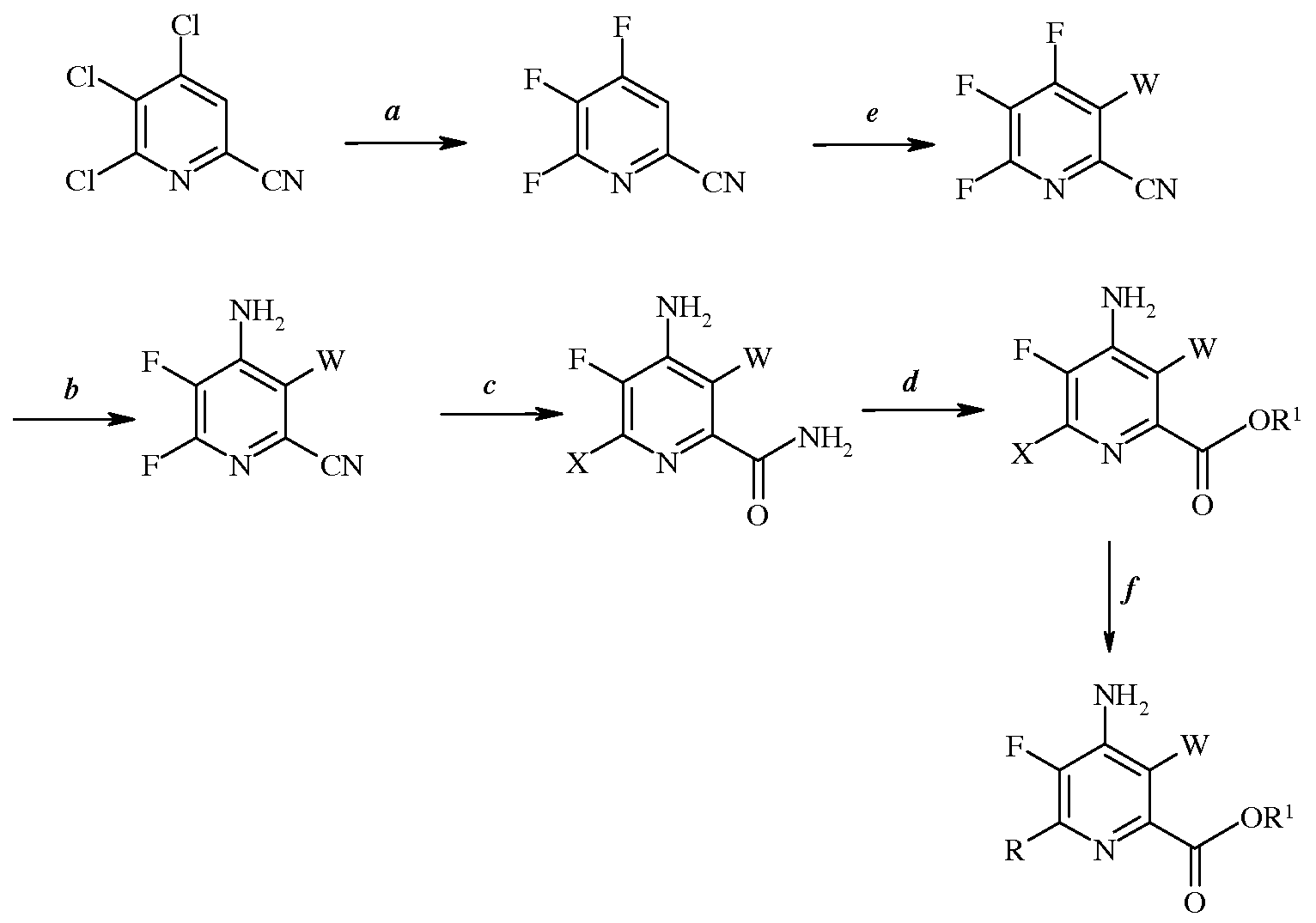
Настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов, в которых 5-фторзаместитель вводят путем обмена галогена на ранней стадии схемы способа.
В патенте США 6297197 В1, помимо прочего, описаны некоторые соединения 4-амино-3-хлор-5-фтор-6-(алкокси или арилокси)пиколинатов, и их применение в качестве гербицидов. В патентах США 6784137 В2 и 7314849 В2, помимо прочего, описаны некоторые соединения 4-амино-3-хлор-5-фтор-6-(арил)пиколинатов, и их применение в качестве гербицидов. В патенте США 7432227 В2, помимо прочего, описаны некоторые соединения 4-амино-3-хлор-5-фтор-6-(алкил)пиколинатов, и их применение в качестве гербицидов. В каждом из этих патентов описано получение исходных веществ на основе 4-амино-3-хлор-5-фторпиколината фторированием соответствующих 5-незамещенных пиридинов бис(тетрафторборатом) 1-(хлорметил)-4-фтор-1,4-диазониабицикло[2.2.2]октана. Было бы предпочтительно получать 4-амино-5-фтор-3-галоген-6-(замещенные) пиколинаты, не обращаясь к прямому фторированию 5-положения пиридинового цикла при помощи дорогостоящего фторирующего агента вроде бис(тетрафторбората) 1-(хлорметил)-4-фтор-1,4-диазониабицикло[2.2.2]октана.
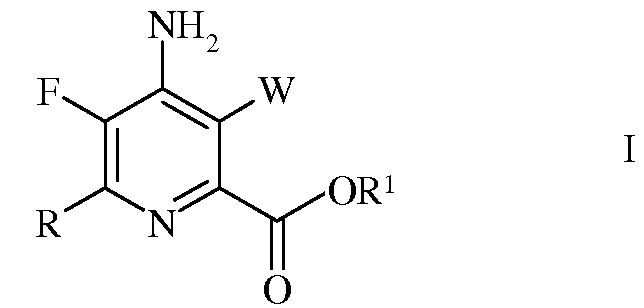
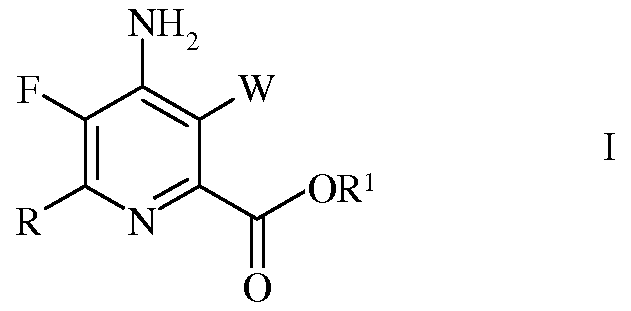
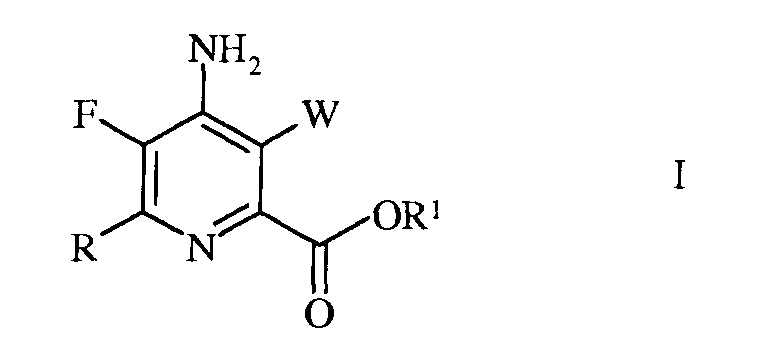
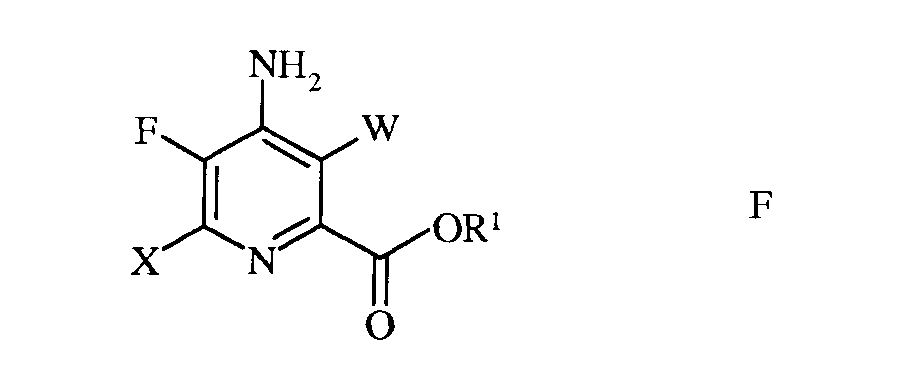
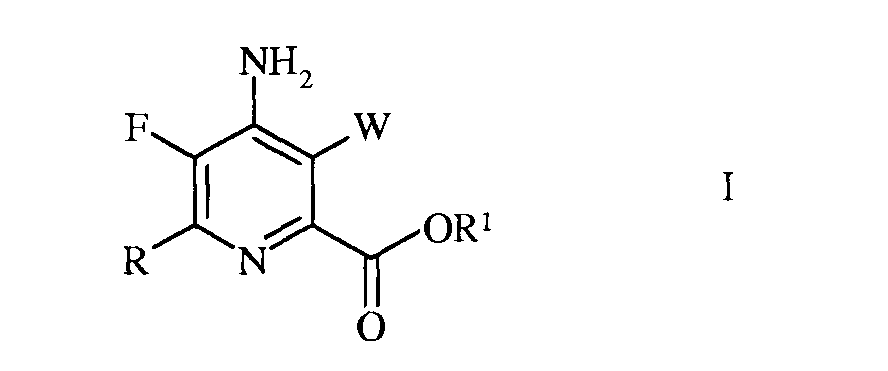
Настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенных)пиколинатов из 4,5,6-трихлорпиколинонитрила. Более конкретно, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
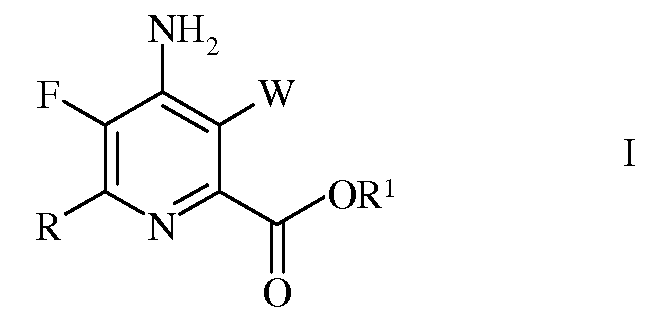
в которой
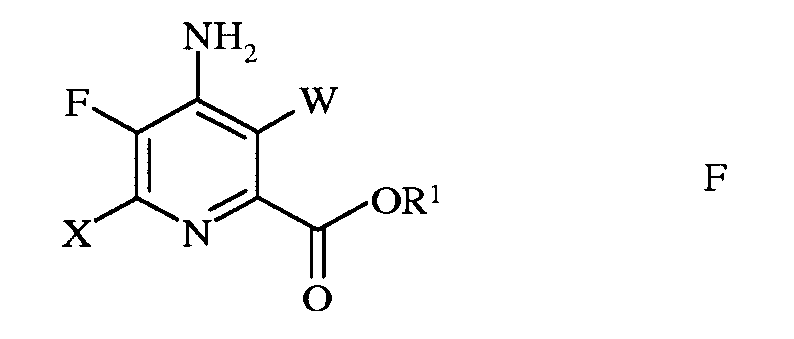
W представляет собой Cl, Br или I;
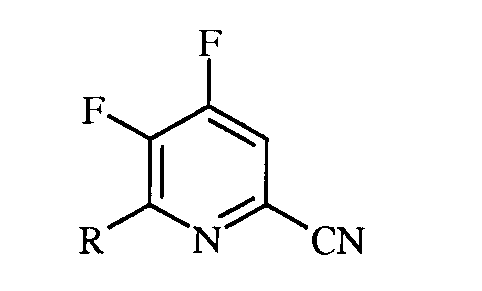
R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
который включает следующие стадии:
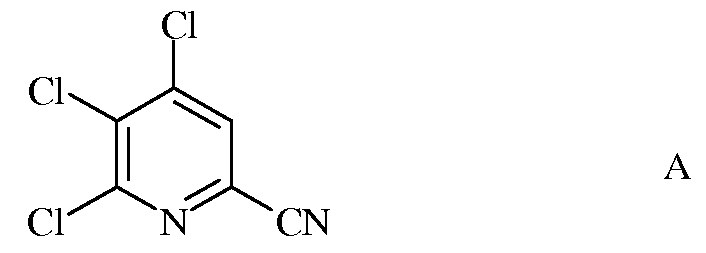
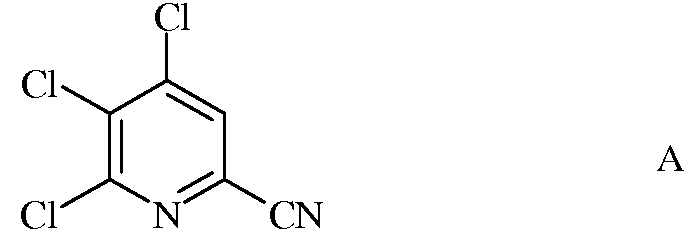
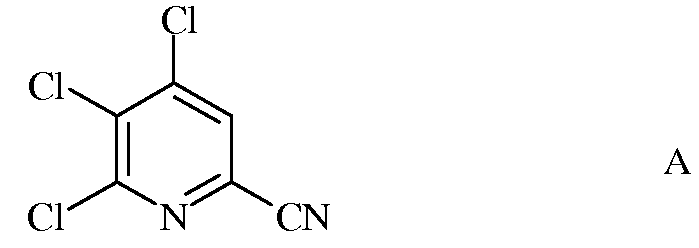
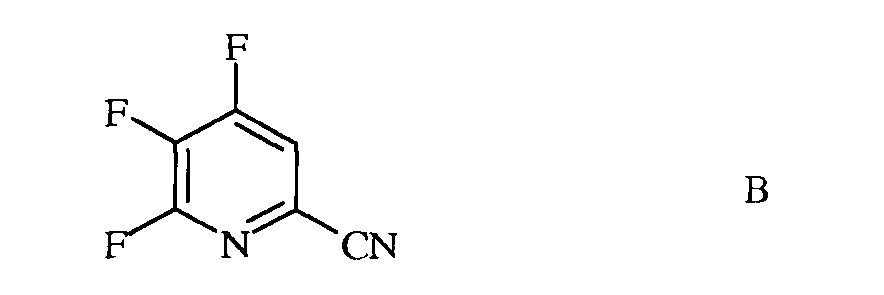
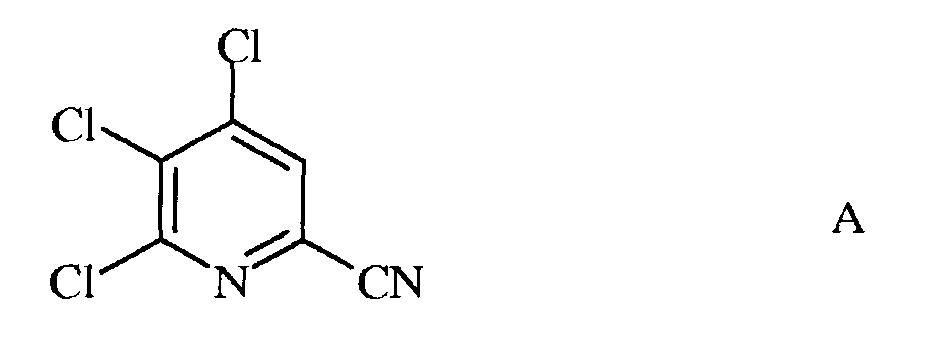
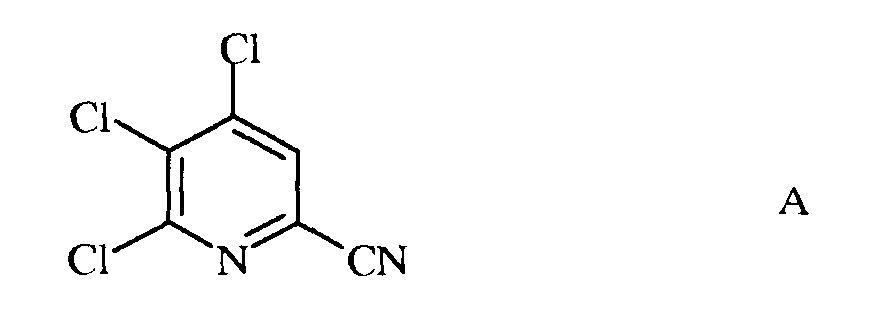
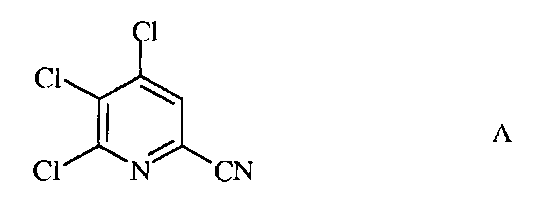
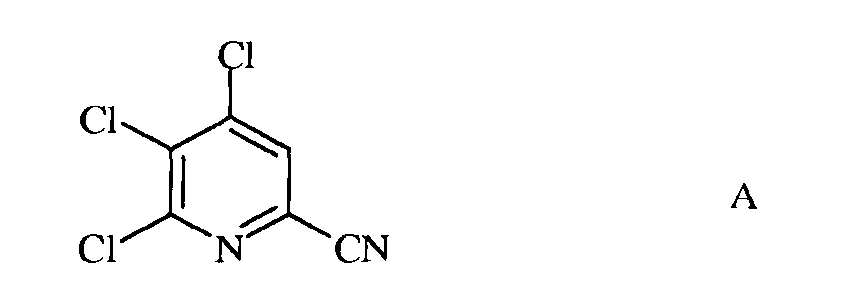
а) фторирование 4,5,6-трихлорпиколинонитрила (формула А)
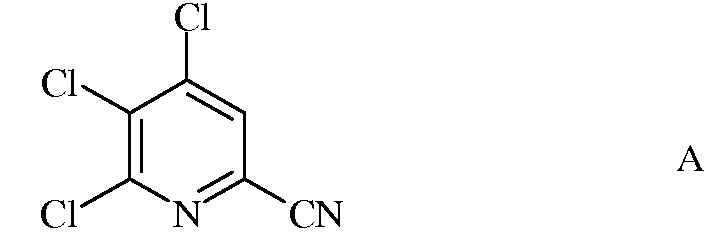
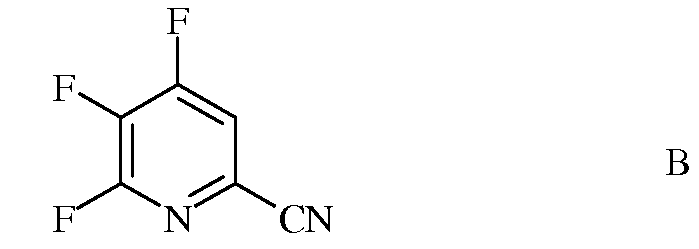
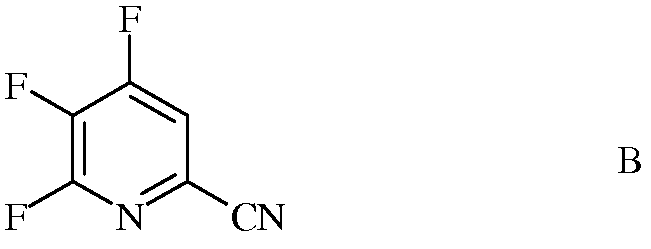
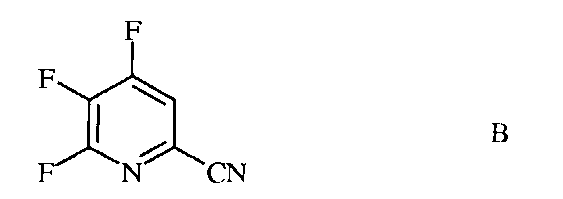
источником фторид-иона, с получением 4,5,6-трифторпиколинонитрила (формула В)
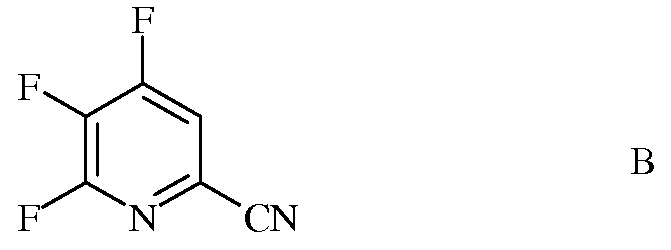
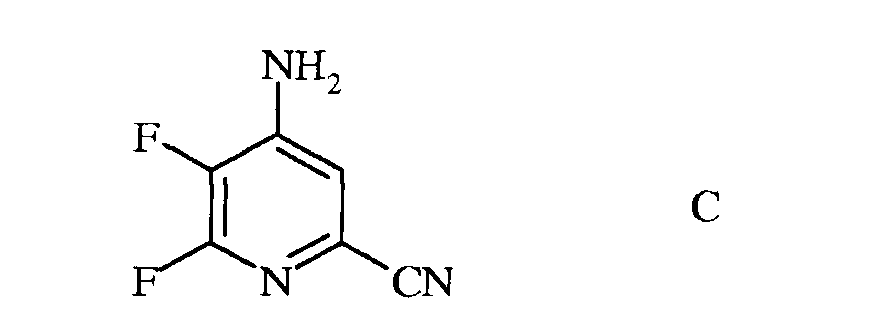
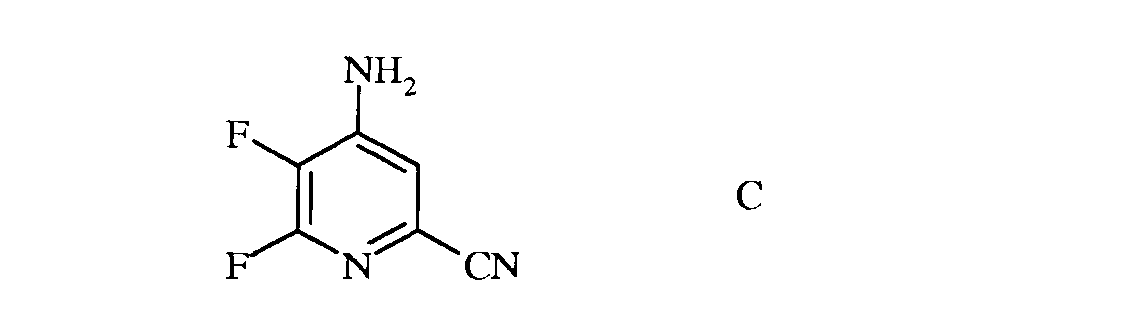
b) аминирование 4,5,6-трифторпиколинонитрила (формула В) аммиаком, с получением 4-амино-5,6-дифторпиколинонитрила (формула С)
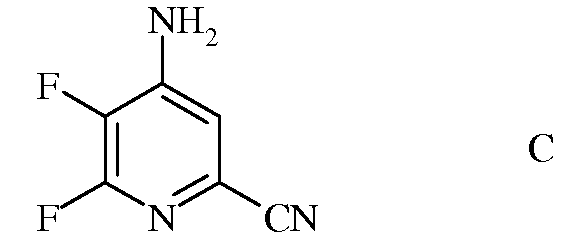
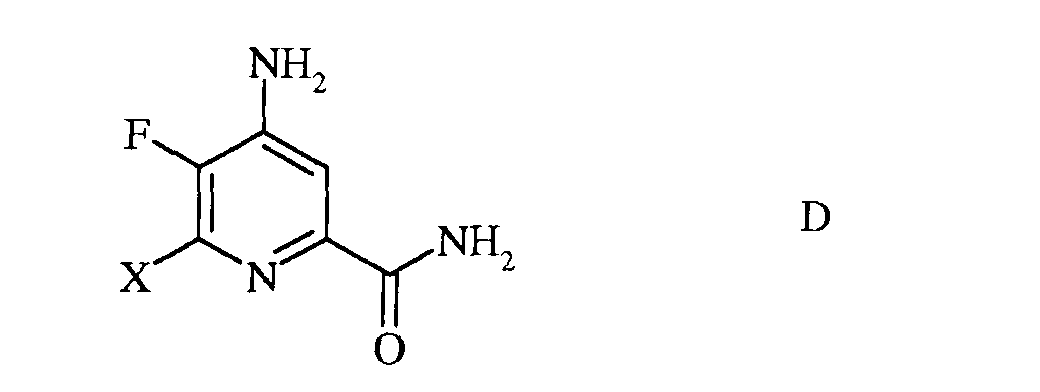
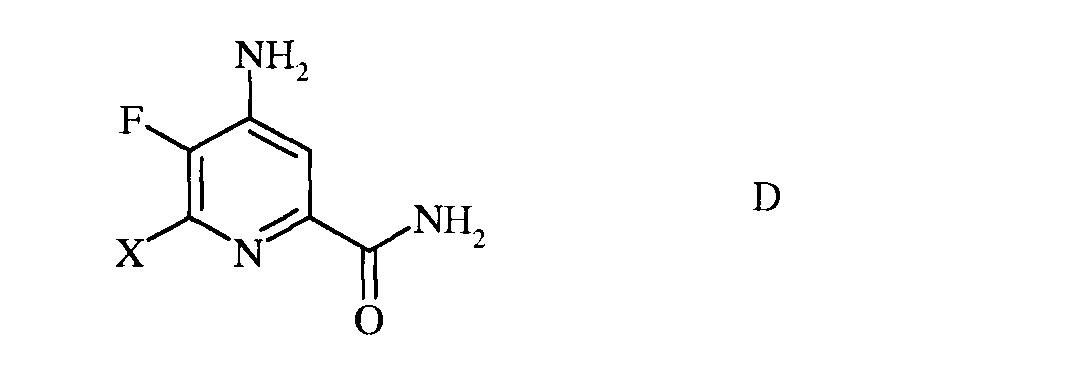
с) гидролиз нитрильного заместителя и замену фтор-заместителя в 6-положении 4-амино-5,6-дифторпиколинонитрила (формула С) на йод-, бром- или хлор-заместитель обработкой источником йода, брома или хлора, с получением 4-амино-5-фтор-6-галогенпиколинамида формулы D
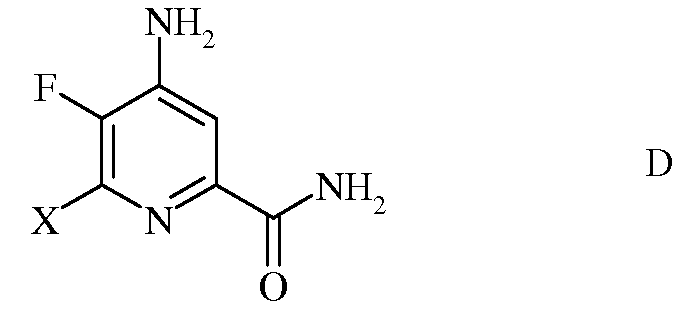
в которой Х представляет собой Cl, Br или I;
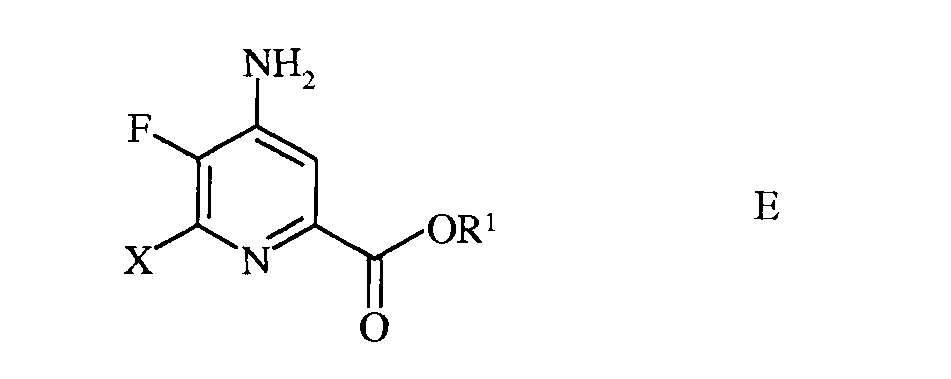
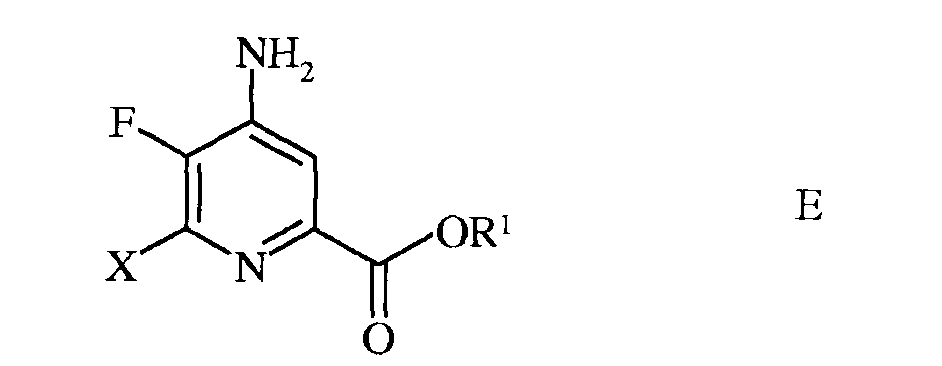
d) этерификацию 4-амино-5-фтор-6-галогенпиколинамида формулы D действием спирта (R1OH) и кислоты Бренстеда или Льюиса, с получением 4-амино-5-фтор-6-галогенпиколината формулы Е
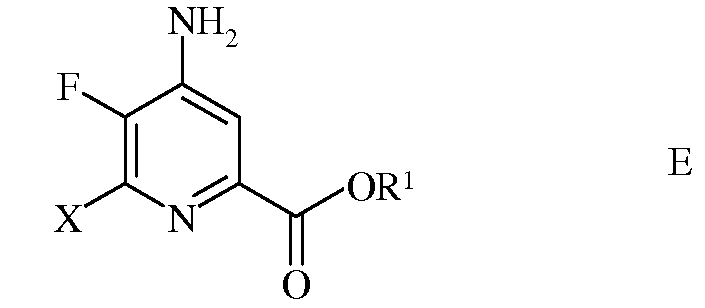
в которой R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
е) галогенирование 4-амино-5-фтор-6-галогенпиколината формулы Е источником галогена, с получением 4-амино-5-фтор-3,6-дигалогенпиколината формулы F
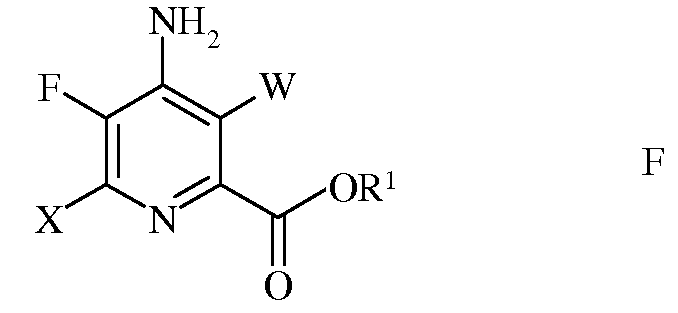
в которой W и Х независимо представляют собой Cl, Br или I;
а R1 такой, как определено ранее, и
f) сочетание 4-амино-5-фтор-3,6-дигалогенпиколината формулы F с арил-, алкил- или алкенилметаллоорганическим соединением формулы G

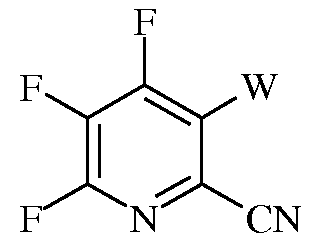
в которой R такой, как определено ранее, а Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь, или В(OR2)(OR3), где R2 и R3 независимы друг от друга, представляют собой водород, С1-С4алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла, с получением 4-амино-3-галоген-5-фтор-6-(замещенного)пиколината формулы I.
Стадии (а)-(f) можно осуществить в перечисленном порядке, как показано на схеме I.
Схема I
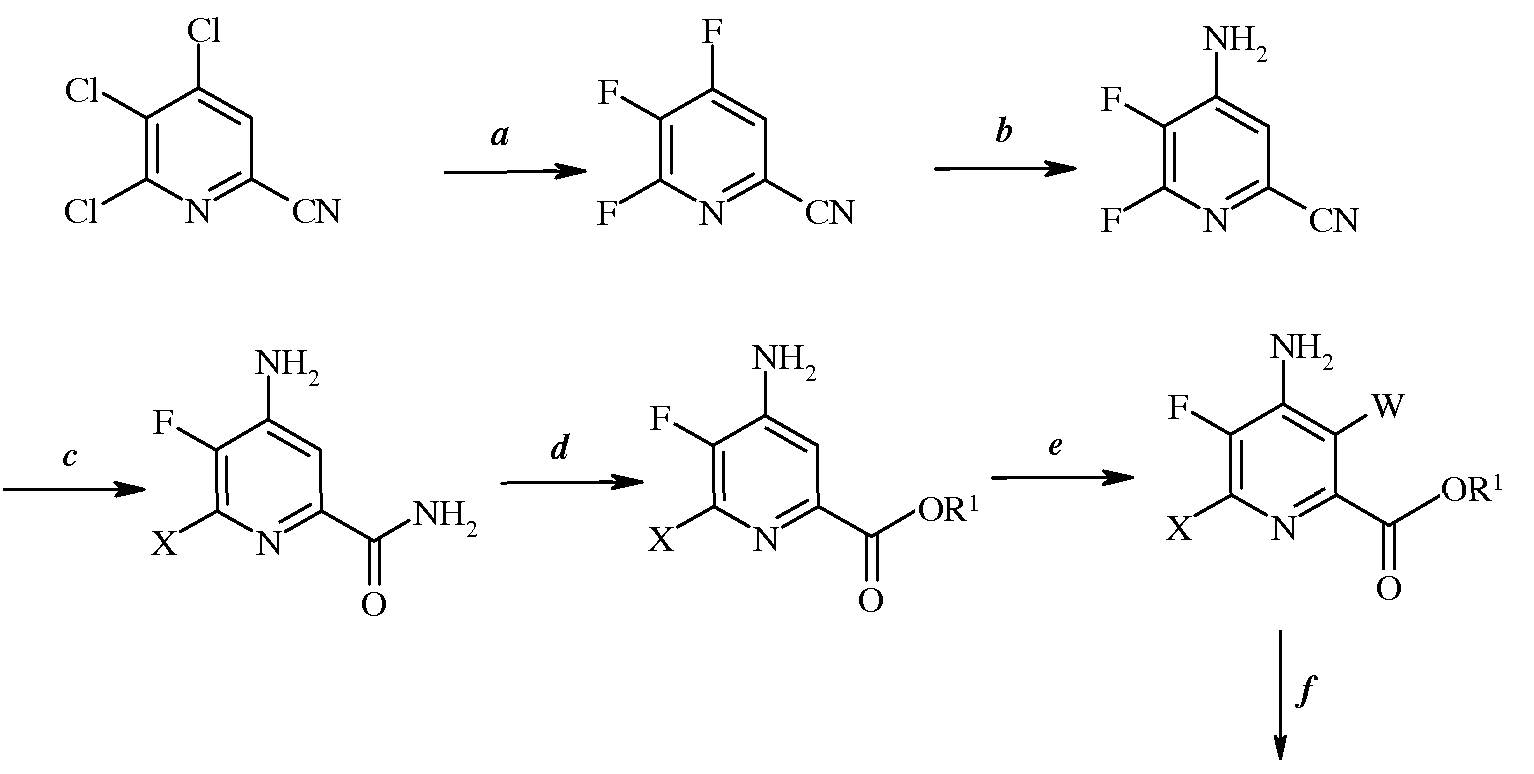

Альтернативным образом, порядок осуществления стадий можно изменить, как показано, например, на схемах II, III и IV.
Схема II
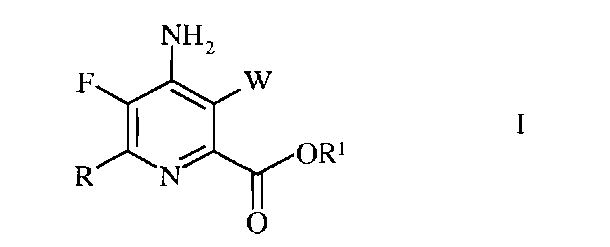
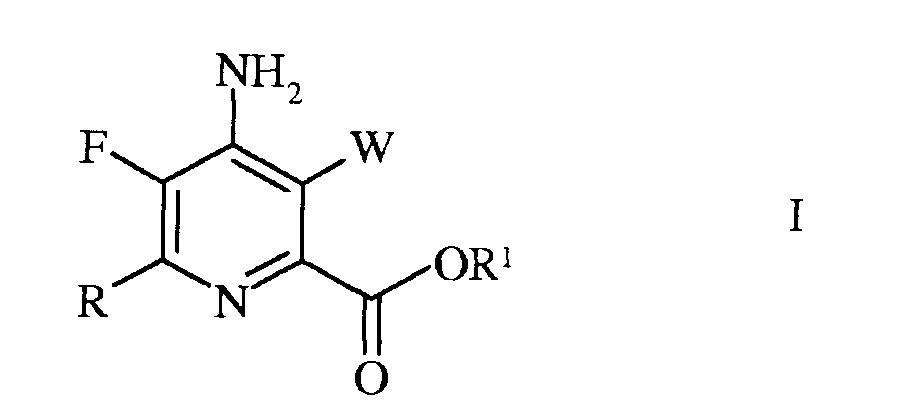
Согласно схеме II, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
в которой
W представляет собой Cl, Br или I;
R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
который включает следующие стадии:
а) фторирование 4,5,6-трихлорпиколинонитрила (формула А)
источником фторид-иона, с получением 4,5,6-трифторпиколинонитрила (формула В)
b) аминирование 4,5,6-трифторпиколинонитрила (формула В) аммиаком, с получением 4-амино-5,6-дифторпиколинонитрила (формула С)
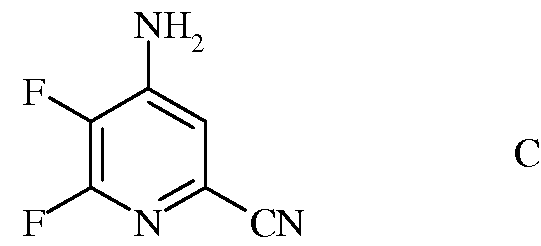
с) гидролиз нитрильного заместителя и замену фтор-заместителя в 6-положении 4-амино-5,6-дифторпиколинонитрила (формула С) на йод-, бром- или хлор-заместитель обработкой источником йода, брома или хлора, с получением 4-амино-5-фтор-6-галогенпиколинамида формулы D
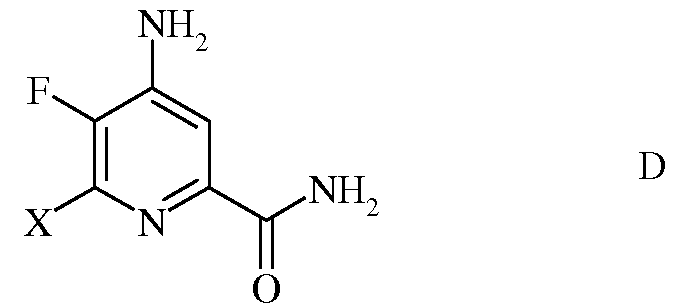
в которой Х представляет собой Cl, Br или I;
d) этерификацию 4-амино-5-фтор-6-галогенпиколинамида формулы D действием спирта (R1OH) и кислоты Бренстеда или Льюиса, с получением 4-амино-5-фтор-6-галогенпиколината формулы Е
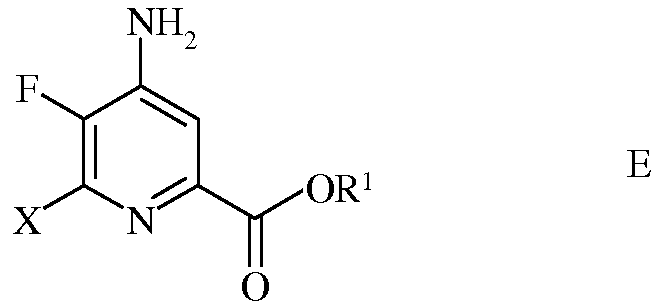
в которой Х представляет собой Cl, Br или I; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
е) сочетание 4-амино-5-фтор-6-галогенпиколината формулы F с арил-, алкил- или алкенилметаллоорганическим соединением формулы G

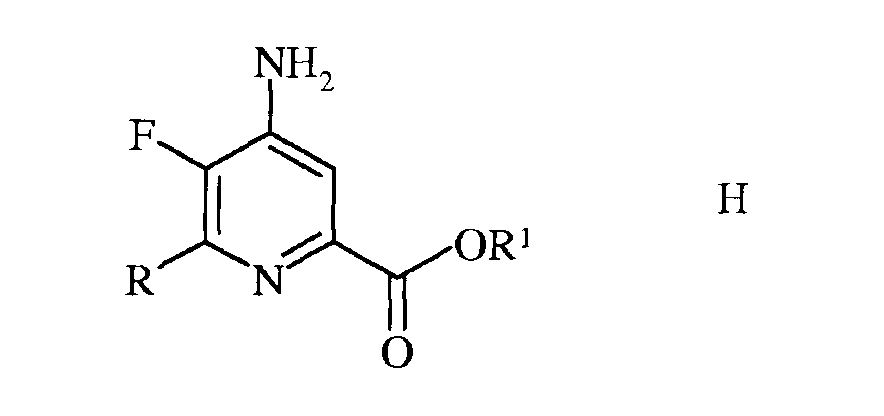
в которой R такой, как определено ранее, а Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь, или В(OR2)(OR3), где R2 и R3 независимы друг от друга, представляют собой водород, С1-С4алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла, с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы Н
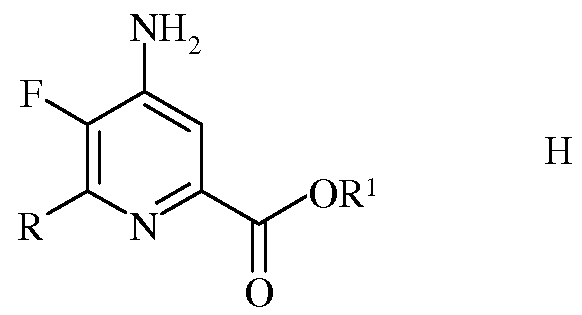
в которой R и R1 такие, как определено ранее; и
f) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы Н источником галогена, с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
Схема III
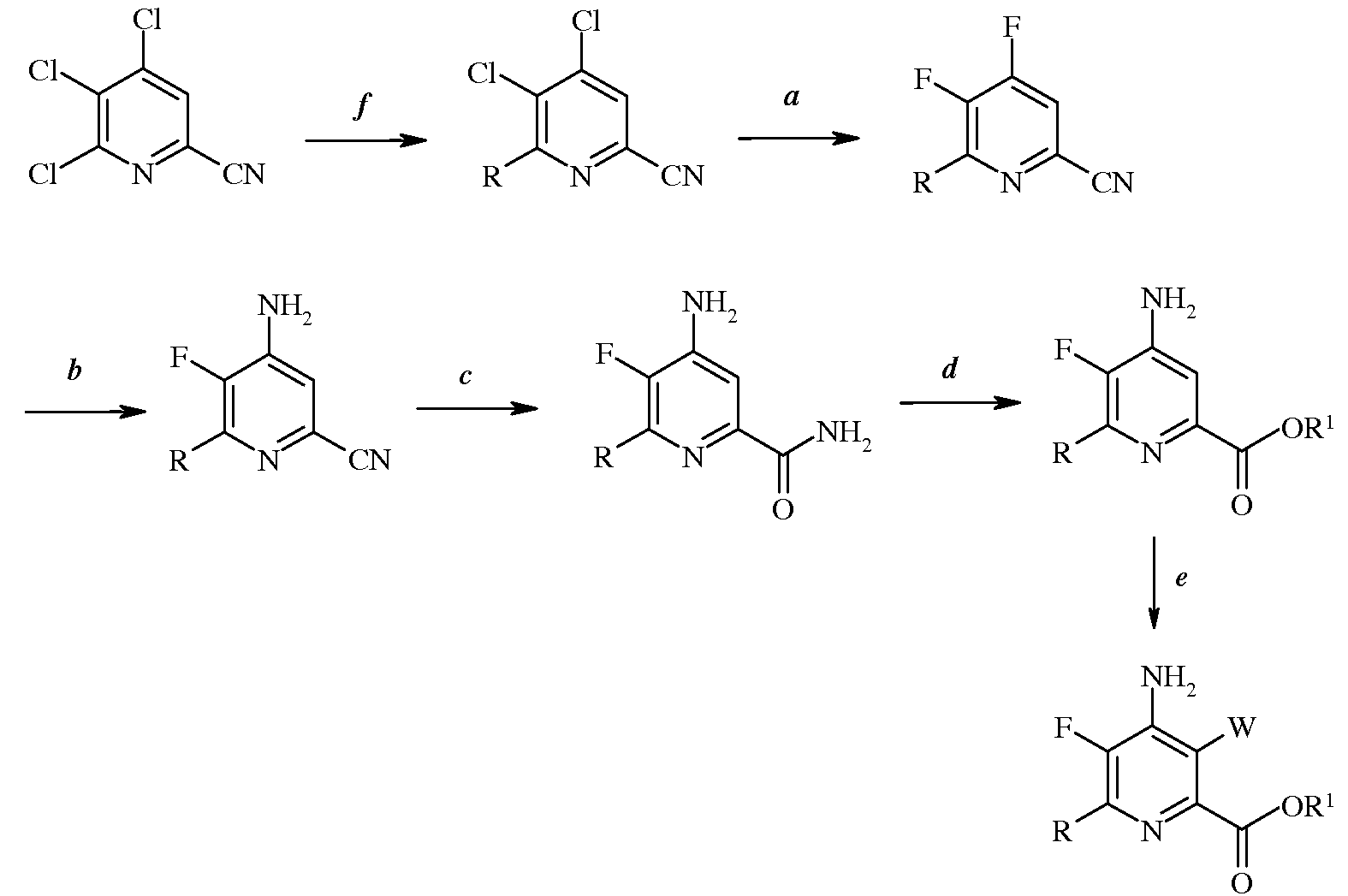
Согласно схеме III, часть стадии с), связанная с замещением атома йода, брома или хлора, не является необходимой. Таким образом, настоящее изобретение относится также к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
в которой
W представляет собой Cl, Br или I;
R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
который включает следующие стадии:
а) сочетание 4,5,6-трихлорпиколинонитрила формулы А
с арил-, алкил- или алкенилметаллоорганическим соединением формулы G

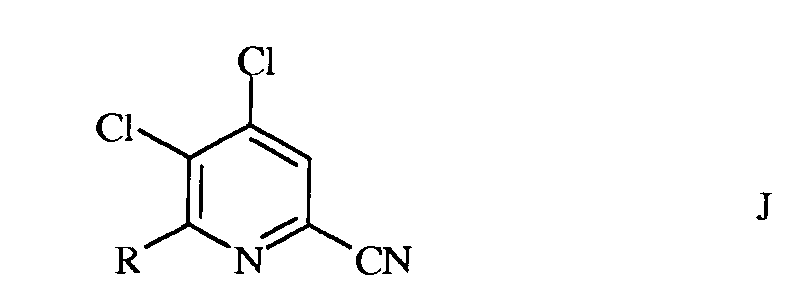
в которой R такой, как определено ранее, а Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь, или В(OR2)(OR3), где R2 и R3 независимы друг от друга, представляют собой водород, С1-С4алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла, с получением 4,5-дихлор-6-(замещенного)пиколинонитрила формулы J
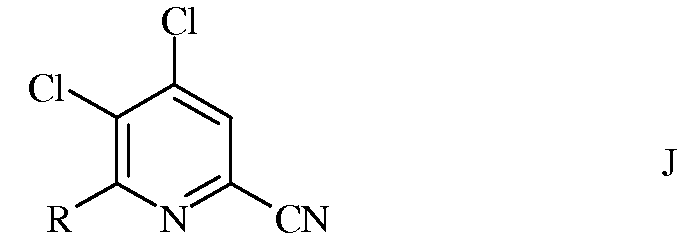
в которой R такой, как определено ранее;
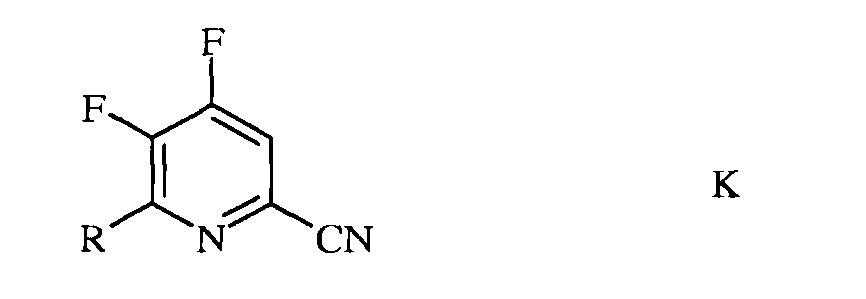
b) фторирование 4,5-дихлор-6-(замещенного)пиколинонитрила формулы J источником фторид-иона, с получением 4,5-дифтор-6-(замещенного)пиколинонитрила формулы К
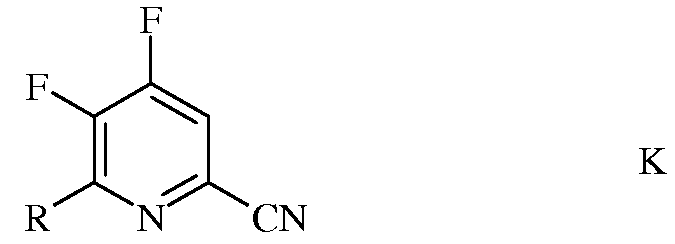
в которой R такой, как определено ранее;
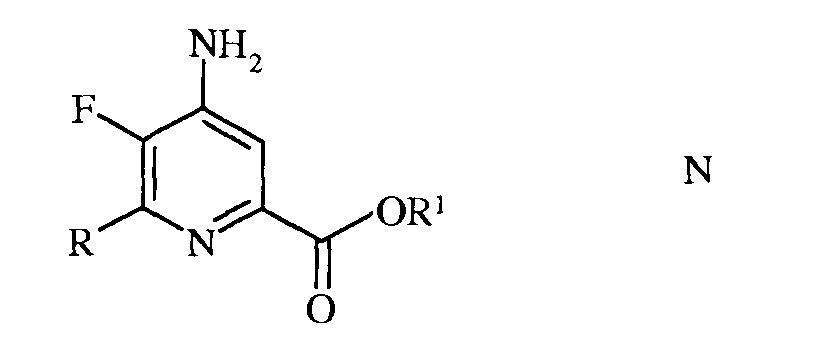
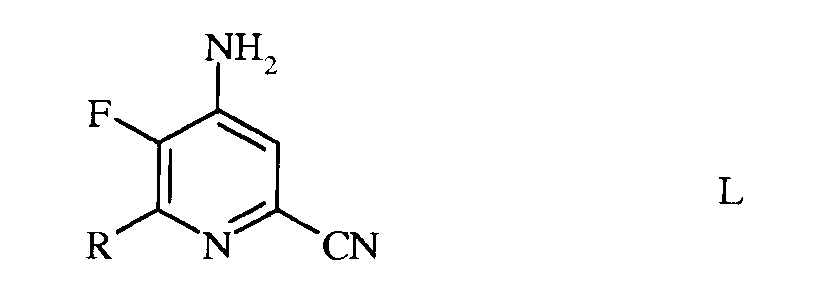
с) аминирование 4,5-дифтор-6-(замещенного)пиколинонитрила формулы К аммиаком, с получением 4-амино-5-фтор-6-(замещенного)пиколинонитрила формулы L
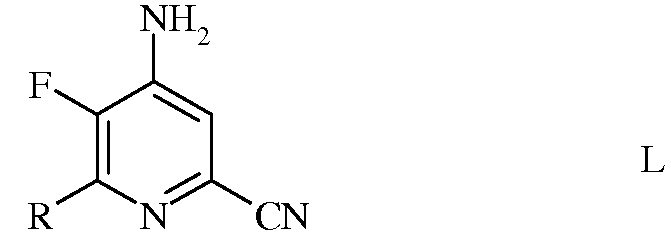
в которой R такой, как определено ранее;
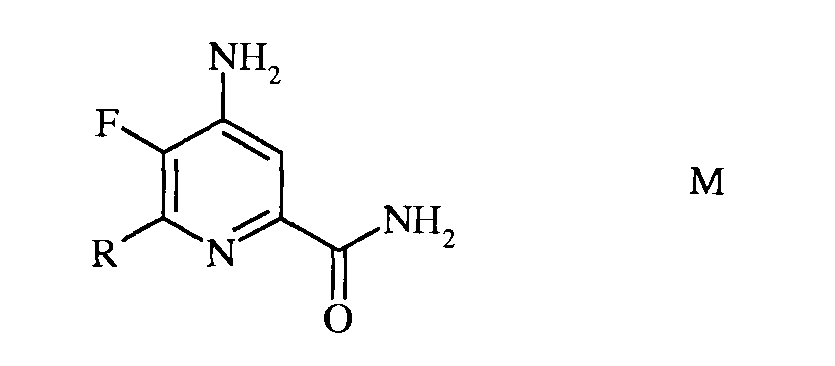
d) гидролиз 4-амино-5-фтор-6-(замещенного)пиколинонитрила формулы L под действием кислоты, с получением 4-амино-5-фтор-6-(замещенного)пиколинамида формулы М
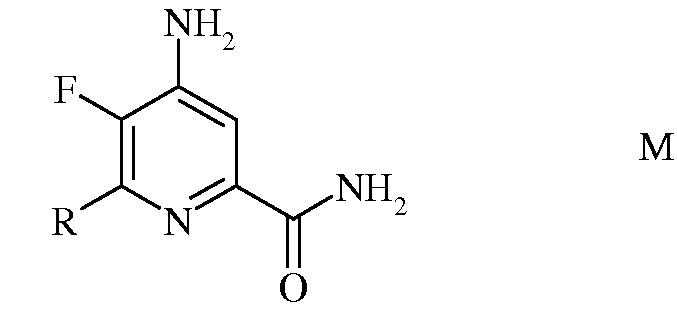
в которой R такой, как определено ранее;
е) этерификация 4-амино-5-фтор-6-(замещенного)пиколинамида формулы М действием спирта (R1OH) и кислоты Бренстеда или Льюиса, с получением 4-амино-5-фтор-6-(замещенного)пиколината формулы N
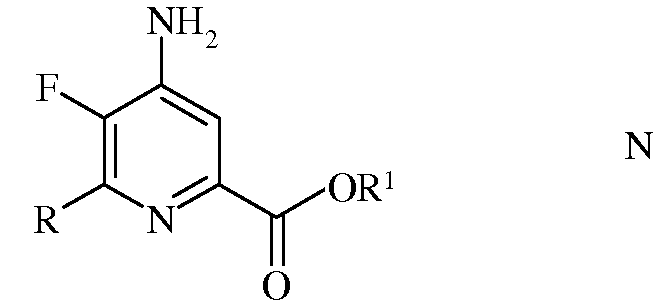
в которой R и R1 такие, как определено ранее; и
f) галогенирование 4-амино-5-фтор-6-(замещенного)пиколината формулы N источником галогена, с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
Схема IV
Согласно схеме IV, настоящее изобретение относится к способу получения 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I
в которой
W представляет собой Cl, Br или I;
R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
который включает следующие стадии:
а) фторирование 4,5,6-трихлорпиколинонитрила (формула А)
источником фторид-иона, с получением 4,5,6-трифторпиколинонитрила (формула В)
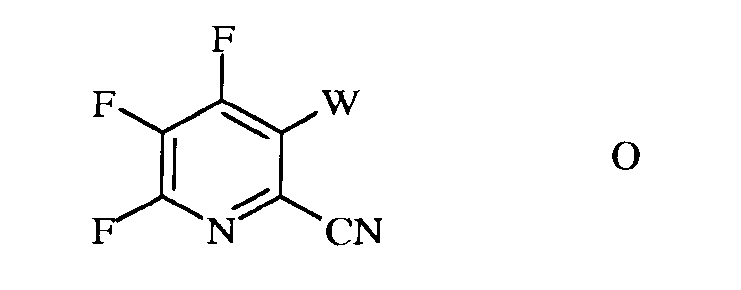
b) галогенирование 4,5,6-трифторпиколинонитрила (формула В) источником галогена, с получением 4,5,6-трифтор-3-галогенпиколинонитрила формулы О
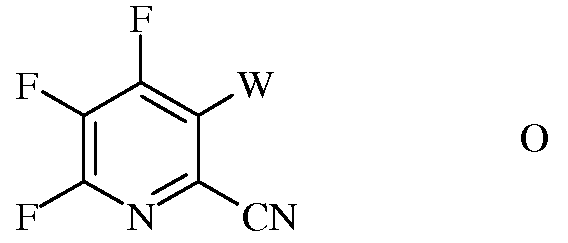
в которой
W представляет собой Cl, Br или I;
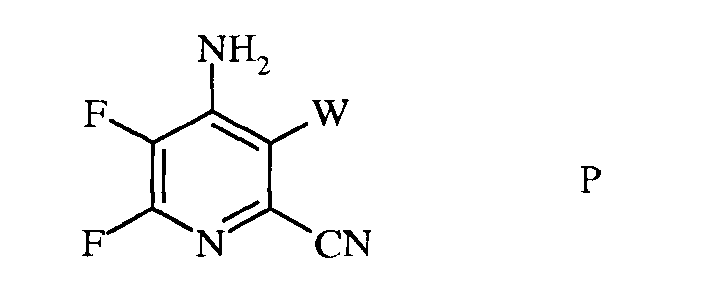
с) аминирование 4,5,6-трифтор-3-галогенпиколинонитрила формулы О аммиаком, с получением 4-амино-5,6-дифтор-3-галогенпиколинонитрила формулы Р
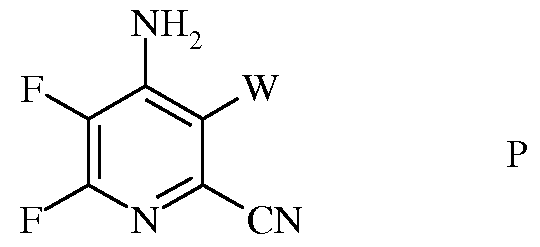
в которой
W представляет собой Cl, Br или I;
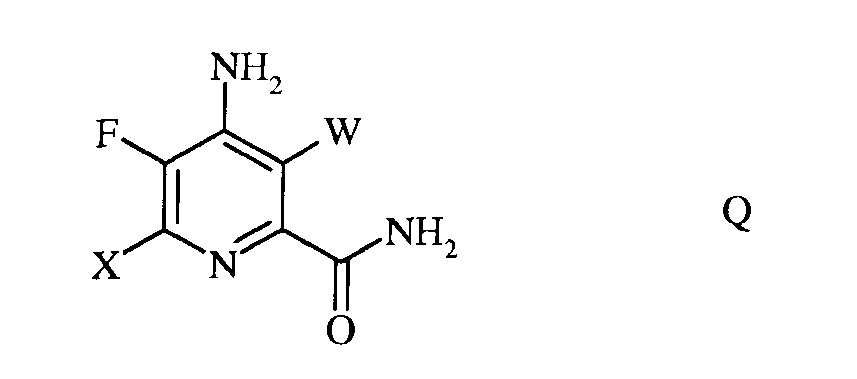
d) гидролиз нитрильного заместителя и замену фтор-заместителя в 6-положении 4-амино-5,6-дифтор-3-галогенпиколинонитрила формулы Р на йод-, бром- или хлор-заместитель путем обработки источником йода, брома или хлора, с получением 4-амино-5-фтор-3,6-дигалогенпиколинамида формулы Q
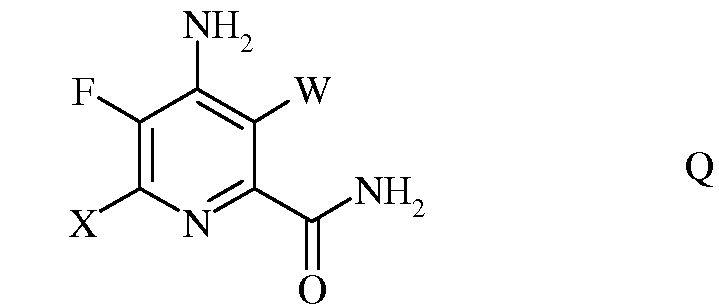
в которой W и Х независимо представляют собой Cl, Br или I;
е) этерификацию 4-амино-5-фтор-3,6-дигалогенпиколинамида формулы Q действием спирта (R1OH) и кислоты Бренстеда или Льюиса, с получением 4-амино-5-фтор-3,6-дигалогенпиколината формулы F
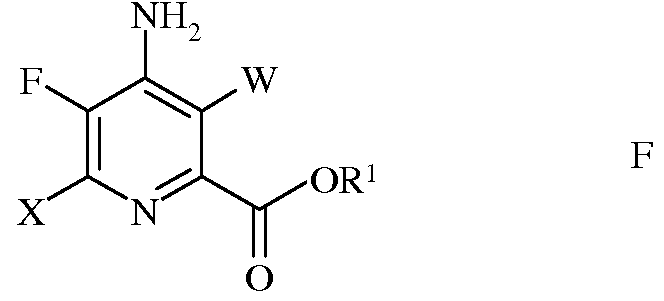
в которой W и Х независимо представляют собой Cl, Br или I; а
R1 представляет собой С1-С12алкил, или незамещенный или замещенный С7-С11арилалкил;
и
f) сочетание 4-амино-5-фтор-3,6-дигалогенпиколината формулы F с арил-, алкил- или алкенилметаллоорганическим соединением формулы G

в которой R такой, как определено ранее, а Met представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь, или В(OR2)(OR3), где R2 и R3 независимы друг от друга, представляют собой водород, С1-С4алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла, с получением 4-амино-5-фтор-3-галоген-6-(замещенного)пиколината формулы I.
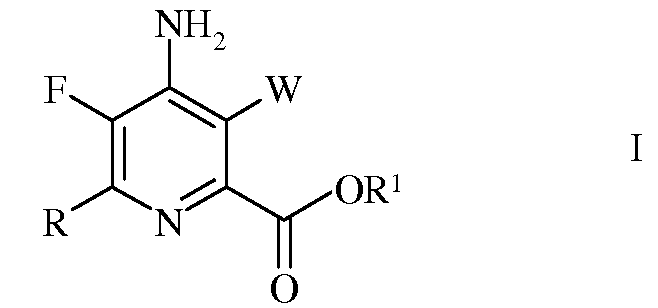
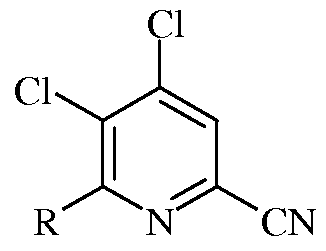
Следующий аспект настоящего изобретения составляют новые промежуточные соединения, полученные в процессе настоящего способа, а именно соединения, выбранные из группы, включающей:
а)
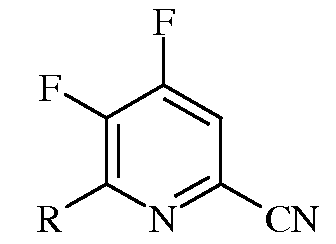
в которой R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси;
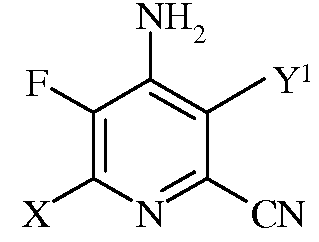
b)
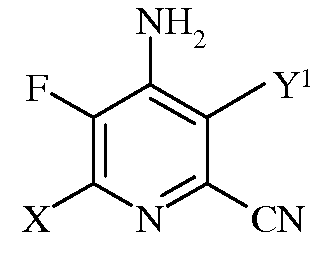
в которой Х представляет собой I, Br, Cl или F, а Y1 представляет собой Н, Cl, Br, или I;
с)
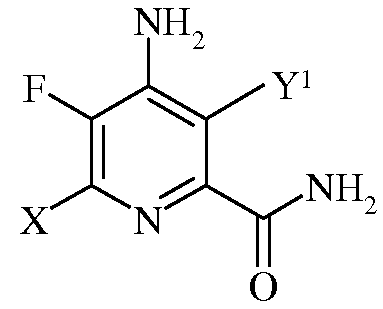
в которой Х представляет собой I, Br, Cl или F, а Y1 представляет собой Н, Cl, Br, или I;
d)
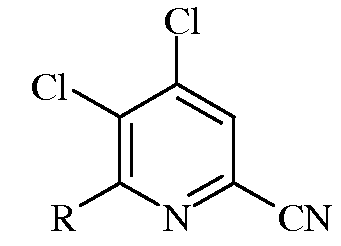
в которой R представляет собой С1-С4алкил, циклопропил, С2-С4алкенил, или фенил, содержащий от 1 до 4 заместителей, независимо выбранных из галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси;
е)
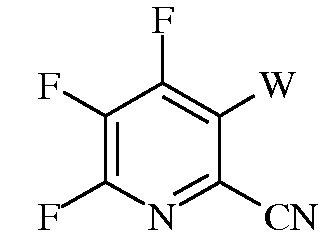
в которой W представляет собой Cl, Br, или I; и
f)
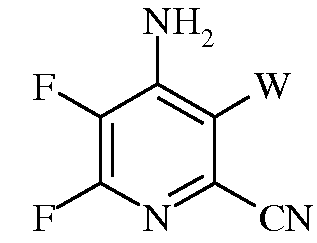
в которой W представляет собой Cl, Br, или I.
Подробное описание изобретения
Использованные в настоящем описании термины «алкил», «алкенил» и «алкинил», а также производные термины, такие как «алкокси», «ацил», «алкилтио» и «алкилсульфонил», включают в свои рамки группы с линейной цепью, разветвленной цепью, и циклические группы. Если специально не указано иначе, каждый из них может быть не замещен, или замещен одним или более заместителями, выбранными из числа, но не ограниченными, галогеном, гидрокси, алкокси, алкилтио, С1-С6ацилом, формилом, циано, арилокси или арилом, при условии, что данные заместители стерически совместимы и удовлетворяются правила химического связывания и энергии деформации. Подразумевается, что термины «алкенил» и «алкинил» включают в себя одну или более ненасыщенных связей.
Использованный в настоящем описании термин «арилалкил» относится к фенилу, замещенному алкильной группой, содержащему в целом от 7 до 11 атомов углерода, такому как бензил (-СН2С6Н5), 2-метилнафтил (-СН2С10Н7), и 1- или 2-фенетил (-СН2СН2С6Н5 или -СН(СН3)С6Н5). Сама фенильная группа может быть не замещена или замещена одним или более заместителями, независимо выбранными из числа галогена, нитро, циано, С1-С6алкила, С1-С6алкокси, галогензамещенного С1-С6алкила, галогензамещенного С1-С6алкокси, С1-С6алкилтио, С(О)ОС1-С6алкила, или случая, когда два соседних заместителя взяты вместе в виде -О(СН2)nO-, где n=1 или 2, при условии, что данные заместители стерически совместимы и удовлетворяются правила химического связывания и энергии деформации.
Если специально не указано иначе, термин «галоген», а также производные термины, такие как «галоид», относятся к фтору, хлору, брому и йоду.
Фенильные группы, содержащие от 1 до 4 заместителей, независимо выбранных из числа галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси, или С1-С4галогеналкокси, могут иметь любую ориентацию, но предпочтительными являются 4-замещенный фенильный, 2,4-дизамещенный фенильный, 2,3,4-тризамещенный фенильный, 2,4,5-тризамещенный фенильный, и 2,3,4,6-тетразамещенный фенильный изомеры.
4-Амино-5-фтор-3-галоген-6-(замещенные)пиколинаты получают из 4,5,6-трихлорпиколинонитрила при помощи ряда стадий, включающих замещение фтора, аминирование, замещение галогена, галогенирование, гидролиз нитрильной группы, этерификацию и сочетание под действием переходного металла. Отдельные стадии можно проводить в различной последовательности.
Исходное вещество 4,5,6-трихлорпиколинонитрил является известным соединением; смотри, например, пример 15 в патенте США 6784137 В2.
В реакции замещения атома фтора, фторированный пиколинонитрил получают, вводя во взаимодействие соответствующий хлорированный пиколинонитрил, по меньшей мере, с одним эквивалентом источника фторид-иона на каждый подлежащий обмену хлор-заместитель цикла.
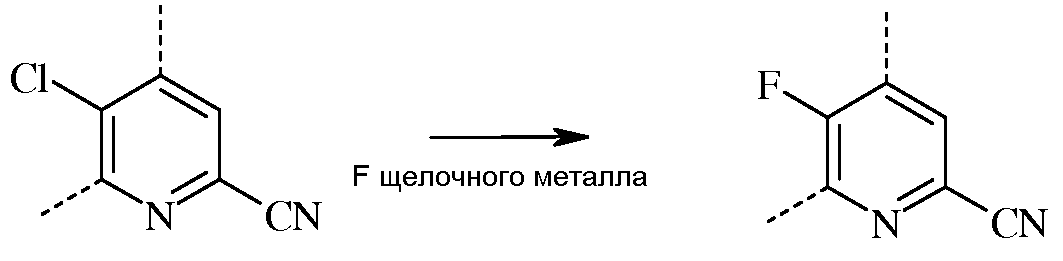
Стандартными источниками фторид-иона являются фториды щелочных металлов, которые включают в себя фторид натрия (NaF), фторид калия (KF) и фторид цезия (CsF), при этом предпочтительными являются KF и CsF. Кроме того, можно применять фторидные соли, такие как фторид тетрабутиламмония (н-Bu4NF). Предпочтительно, реакцию проводят в полярном апротонном растворителе или реакционной среде, такой как диметилсульфоксид (ДМСО), N-метилпирролидон (NMP), N,N-диметилформамид (ДМФА), гексаметилфосфорамид (ГМФТА), или сульфолан. Кроме того, можно применять добавки, такие как краун-эфиры или агенты межфазного переноса, которые, как известно, повышают скорость обмена фтора. Температура проведения реакции не является решающей, но обычно составляет от около 70°С до около 180°С, а предпочтительно, от около 80°С до около 120°С. Оптимальная температура будет изменяться в зависимости от того, какой растворитель используют в данной конкретной реакции. Вообще говоря, чем ниже температура, тем медленнее будет протекать реакция. Настоящую реакцию обычно проводят при энергичном перемешивании, достаточном для поддержания, по существу, равномерно распределенной смеси реагирующих веществ.
При проведении реакции фторирования ни скорость, ни порядок прибавления реагентов не является решающим. Как правило, растворитель и фторид щелочного металла смешивают перед прибавлением хлорированного пиколинонитрила к реакционной смеси. Стандартная реакция обычно занимает от около 4 до около 100 часов, и обычно проводится при атмосферном давлении окружающей среды.
Несмотря на то что точное количество реагентов не является решающим, предпочтительно использовать такое количество фторида щелочного металла, которое обеспечит, по меньшей мере, эквимолярное количество атомов фтора, исходя из количества атомов хлора, которые нужно заменить в исходном веществе, то есть, по меньшей мере, около одного эквивалента фторида щелочного металла. По завершении реакции, требуемый продукт выделяют, используя стандартные методики выделения и очистки.
В случае аминирования, 4-фторпиколинонитрил вводят во взаимодействие с аммиаком для замены атома фтора на аминогруппу.

Несмотря на то что необходимо лишь стехиометрическое количество аммиака, часто удобно применять большой избыток аммиака. Реакцию проводят в инертном растворителе, предпочтительно, полярном апротонном растворителе или реакционной среде, такой как ДМСО, NMP, ДМФА, ГМФТА или сульфолан. Альтернативным образом, можно применять водный гидроксид аммония (NH4OH), с использованием, или без использования органического растворителя. Температура проведения реакции не является решающей, но обычно составляет от около 0°С до около 45°С, а предпочтительно, от около 10°С до около 30°С.
При проведении реакции аминирования, 4-фторпиколинонитрил растворяют в растворителе и прибавляют к реакционной смеси аммиак при охлаждении. Обычно избыток газообразного аммиака барботируют в реакционную смесь. Как правило, стандартная реакция занимает от около 0,5 до около 5 часов, и обычно проводится при атмосферном давлении окружающей среды.
В случае реакции замещения галогена и гидролиза, 6-галогенпиколинамид получают, вводя во взаимодействие соответствующий 6-фторпиколинамид, по меньшей мере, с двумя эквивалентами галогеноводорода (гидройодида (HI), гидробромида (HBr), или гидрохлорида (HCl)).
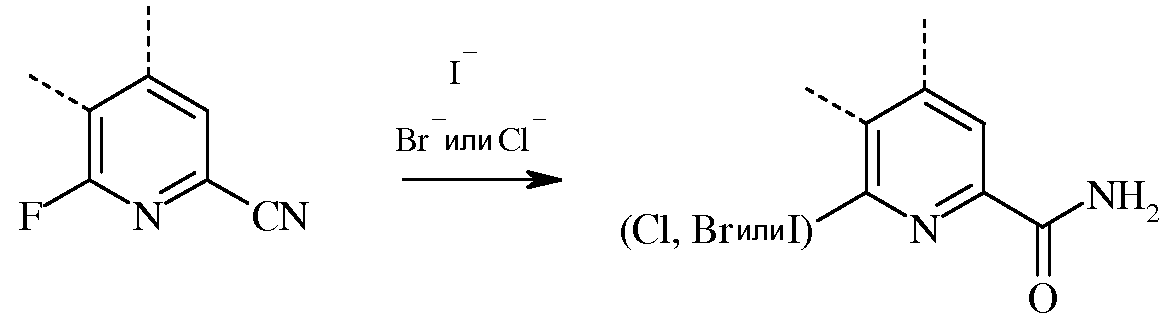
Несмотря на то что необходимо лишь два эквивалента галогеноводорода, часто удобно применять большой избыток галогеноводорода. Реакцию проводят в инертном органическом растворителе, при этом особенно предпочтительными являются С1-С4алкановые кислоты. Температура проведения реакции не является решающей, но обычно составляет от около 75°С до около 150°С, а предпочтительно, от около 100°С до около 130°С. Замещение галогена обычно проводят под давлением в герметизированном сосуде.
При проведении реакций галогенирования и гидролиза, 6-фторпиколинонитрил можно нагревать с галогеноводородом и растворителем на основе алкановой кислоты в герметизированном реакторе. Стандартная реакция обычно занимает от около 0,5 до около 24 часов. Требуемый продукт 6-галогенпиколинамид выделяют с использованием стандартных методик выделения и очистки.
В случае реакции этерификации, пиколинамид вводят во взаимодействие со спиртом в присутствии кислоты Бренстеда или Льюиса.
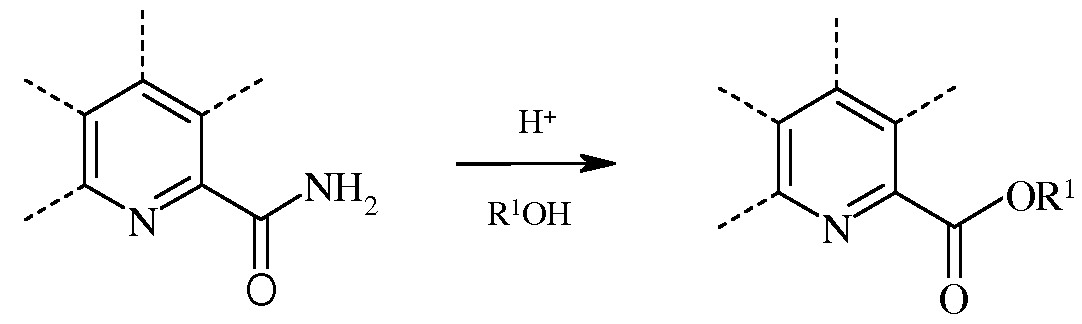
Сильные протонные кислоты Бренстеда вроде серной кислоты и фосфорной кислоты обычно применяют в стехиометрических количествах. Можно также использовать кислоты Льюиса вроде изопропилата титана(IV). Реакцию проводят при использовании С1-С12алкилового или незамещенного или замещенного С7-С11арилалкилового спирта для требуемого сложного эфира в качестве растворителя. Реакцию обычно проводят в герметизированном реакторе, как правило, при температуре, превышающей температуру кипения спиртового растворителя.
При проведении этерификации, пиколинамид или пиколинонитрил добавляют к смеси спирта и кислоты. Несмотря на то, что температура реакции не является решающей, часто осуществляют нагревание до температуры от около 80°С до около 140°С в течение от около 2 до около 24 часов, предпочтительно, от 100°С до 120°С в течение от 6 до 8 часов. Требуемый продукт выделяют с использованием стандартных методик разделения и очистки.
Иногда удобно проводить стадию этерификации в комбинации с обработкой на стадии замещения галогена.
В реакции галогенирования, атом хлора, брома или йода вводят в 3-положение либо пиколината, либо пиколинонитрила реакцией с источником галогена в инертном растворителе.
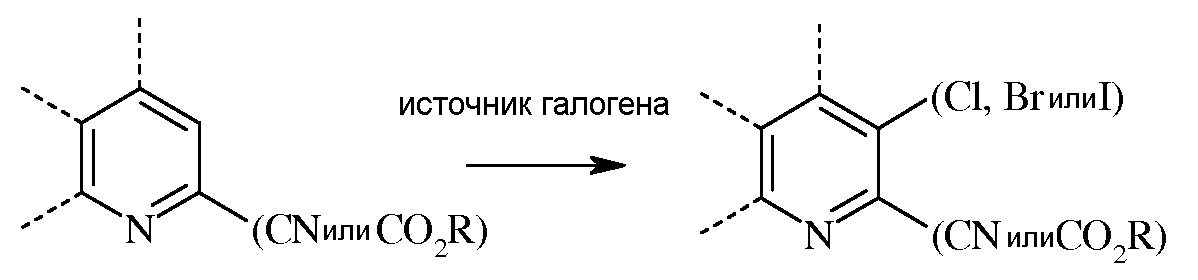
В случае, когда атомом галогена в 3-положении является Cl, источник хлора может представлять собой собственно хлор (Cl2), либо такие реагенты, как хлористый сульфурил, N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин. При использовании хлора или хлористого сульфурила применяют большой избыток хлорирующего агента. При использовании газообразного хлора реакцию проводят в инертном растворителе, предпочтительно, таком растворителе, как дихлорметан, смесь дихлорметан-вода, четыреххлористый углерод, или уксусная кислота. При использовании хлористого сульфурила реакцию можно проводить в инертном растворителе, таком как дихлорметан, или в чистом хлористом сульфуриле. Температура проведения реакции не является решающей, но обычно составляет от около 0°С до около 45°С, а предпочтительно, от около 10°С до около 30°С. Стандартная реакция, как правило, занимает от около 0,5 до около 5 часов. Реакцию хлорирования обычно проводят при атмосферном давлении окружающей среды. В некоторых случаях хлорирование не протекает в данных условиях. Вместо этого можно использовать хлорирование в газовой фазе в трубчатом реакторе с использованием газообразного хлора. Температура, при которой проводят данную реакцию, обычно составляет от около 350°С до около 600°С, а предпочтительно, от около 500°С до около 600°С. Реакцию хлорирования обычно проводят при атмосферном давлении окружающей среды.
В случае, когда применяемый хлорирующий агент представляет собой N-хлорсукцинимид или 1,3-дихлор-5,5-диметилгидантоин, реакцию можно проводить с использованием стехиометрического количества хлорирующего реагента. В случае реакций хлорирования с применением 1,3-дихлор-5,5-диметилгидантоина в качестве хлорирующего агента было найдено, что во взаимодействии принимают участие оба атома хлора гидантоина. Реакцию проводят в инертном полярном растворителе, таком как ДМФА или ацетонитрил. Температура проведения реакции не является решающей, но обычно составляет от около 20°С до около 85°С, а предпочтительно, от около 50°С до около 80°С. При использовании ацетонитрила в качестве растворителя, удобно проводить данную реакцию при температуре кипения. Стандартная реакция обычно занимает от около 0,5 до около 5 часов. Реакцию хлорирования обычно проводят при атмосферном давлении окружающей среды.
В случае, когда атомом галогена в 3-положении является Br, источник брома может представлять собой собственно бром (Br2), либо такие реагенты, как бромистый сульфурил, N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин. При использовании Br2 в качестве бромирующего агента можно применять большой избыток, а реакцию проводят в инертном растворителе, предпочтительно, таком растворителе, как дихлорметан, смесь дихлорметан-вода, или уксусная кислота. Температура проведения реакции не является решающей, но обычно составляет от около 0°С до около 45°С, а предпочтительно, от около 10°С до около 30°С. Стандартная реакция, как правило, занимает от около 0,5 до около 5 часов. Реакцию бромирования обычно проводят при атмосферном давлении окружающей среды.
В случае, когда применяемый бромирующий агент представляет собой N-бромсукцинимид или 1,3-дибром-5,5-диметилгидантоин, реакцию проводят с использованием стехиометрического количества бромирующего реагента. Реакцию проводят в инертном полярном растворителе, таком как ДМФА или ацетонитрил. Температура проведения реакции не является решающей, но обычно составляет от около 20°С до около 85°С, а предпочтительно, от около 50°С до около 80°С. При использовании ацетонитрила в качестве растворителя, удобно проводить данную реакцию при температуре кипения. Стандартная реакция обычно занимает от около 0,5 до около 5 часов. Реакцию бромирования обычно проводят при атмосферном давлении окружающей среды.
В случае, когда атомом галогена в 3-положении является I, источник иода может представлять собой собственно иод (I2), либо такие реагенты, как монохлорид йода или N-йодсукцинимид. Иодная кислота может быть использована совместно с I2. При использовании I2 в качестве йодирующего агента можно применять большой избыток, а реакцию проводят в инертном растворителе, предпочтительно, таком растворителе, как дихлорметан, смесь дихлорметан-вода, или уксусная кислота. Температура проведения реакции не является решающей, но обычно составляет от около 0°С до около 45°С, а предпочтительно, от около 10°С до около 30°С. Стандартная реакция, как правило, занимает от около 0,5 до около 5 часов. Реакцию йодирования обычно проводят при атмосферном давлении окружающей среды.
В реакции сочетания, 6-йод-, бром- или хлорпиколинату или пиколинонитрилу дают возможность взаимодействовать с арил-, алкил- или алкенилметаллоорганическим соединением, в котором металл представляет собой Zn-галогенид, Zn-R, три(С1-С4алкил)олово, медь, или В(OR2)(OR3), где R2 и R3 независимы друг от друга, водород, С1-С4алкил, или, взятые вместе, образуют этиленовую или пропиленовую группу, в присутствии катализатора на основе переходного металла.
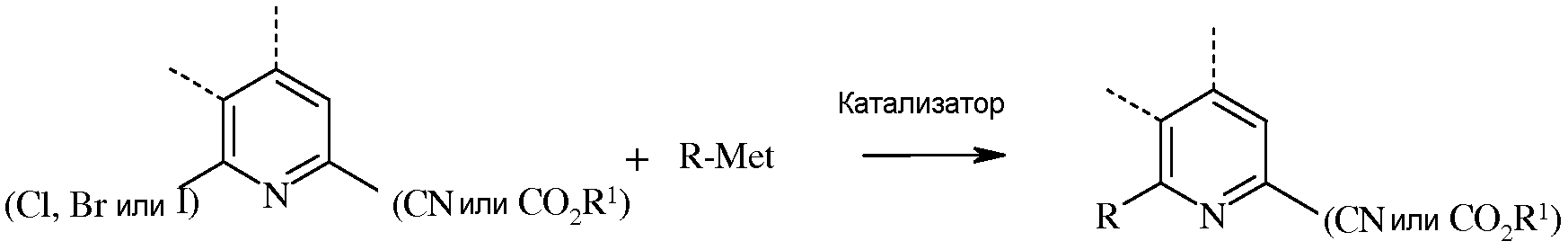
«Катализатор» представляет собой катализатор на основе переходного металла, в частности палладиевый катализатор, такой как ацетат палладия(II) или дихлорид бис(трифенилфосфин)палладия(II), или никелевый катализатор, такой как ацетилацетонат никеля(II) или дихлорид бис(трифенилфосфин)никеля(II). Кроме того, катализаторы можно получать in situ из солей металлов и лигандов, таких как ацетат палладия(II) и трифенилфосфин, или хлорид никеля(II) и трифенилфосфин. Данные полученные in situ катализаторы можно получать предварительной реакцией соли металла и лиганда, с последующим прибавлением к реакционной смеси, или путем отдельного прибавления соли металла и лиганда непосредственно к реакционной смеси.
Обычно реакцию сочетания можно осуществлять в отсутствие кислорода при использовании инертного газа, такого как азот или аргон. Методы, применяемые для удаления кислорода из реакционных смесей для проведения сочетания, такие как барботирование инертного газа, хорошо известны специалистам в данной области техники. Примеры таких методик описаны в The Manipulation of Air-Sensitive Compounds, 2nd ed., D.F. Shriver, M.A. Drezdzon, Eds.; Wiley-Interscience, 1986. Применяют субстехиометрические количества катализатора, обычно от около 0,0001 эквивалента до 0,1 эквивалента. Необязательно можно добавлять дополнительные количества лиганда для повышения стабильности и активности катализатора. Кроме того, в реакцию сочетания обычно вводят такие добавки, как карбонат натрия (Na2CO3), карбонат калия (K2CO3), KF, CsF и NaF. Для реакции сочетания обычно требуется от около 1 до около 5 эквивалентов подобной добавки, предпочтительно, от 1 до 2 эквивалентов. Необязательно, в реакцию сочетания можно добавить воду для повышения растворимости добавок. Для реакции сочетания обычно требуется от 1 до около 3 эквивалентов арил-, алкил- или алкенилметаллоорганического соединения, предпочтительно, от 1 до 1,5 эквивалента. Реакцию проводят в инертном растворителе, таком как толуол, тетрагидрофуран (ТГФ), диоксан или ацетонитрил. Температура, при которой проводят реакцию, не является решающей, но обычно составляет от около 25°С до около 150°С, а предпочтительно, от около 50°С до около 125°С. Стандартная реакция обычно занимает от около 0,5 до около 24 часов. Обычно не требуется определенного порядка прибавления реагентов. Часто бывает препаративно проще соединить все реагенты, за исключением катализатора, а затем дегазировать реакционный раствор. После удаления кислорода, для начала реакции сочетания можно добавить катализатор.
В случае, когда Met часть арил-, алкил- или алкенилметаллоорганического соединения представляет собой Zn-галогенид, Zn-R или медь, может потребоваться защита реакционноспособных функциональных групп. Например, при наличии амино-заместителя (-NHR или -NH2), может потребоваться защита данных реакционноспособных групп. Для защиты аминогрупп от взаимодействия с металлоорганическими реагентами в данной области известно множество групп. Примеры таких защитных групп описаны в Protective Groups in Organic Synthesis, 3nd ed.; Greene, T.W.; Wuts, P.G.M. Eds.; Wiley-Interscience, 1999. На выбор металла для применения в R-Met влияет ряд факторов, таких как стоимость, устойчивость, реакционноспособность и необходимость в защите реакционноспособных функциональных групп.
Продукты, полученные любым из данных способов, можно выделить обычными способами, такими как выпаривание или экстракция, и можно очистить стандартными методами, такими как перекристаллизация или хроматография.
Для иллюстрации данного изобретения предоставлены следующие примеры.
Примеры
Замещение атома фтора
Пример 1а 4,5,6-Трифторпиколинонитрил
В трехгорлую колбу на 100 миллилитров (мл), снабженную механической мешалкой, термопарой и насадкой для перегонки в вакууме, помещали безводный ДМСО (50 мл) и CsF (8,71 грамм (г), 57,4 миллимоль (ммоль)) в атмосфере азота. Прибор вакуумировали и нагревали до 60°С при перемешивании, чтобы было можно осушить систему путем отгонки ДМСО (15 мл) и следовых количеств воды. Прибавляли 4,5,6-трихлорпиколинонитрил (3,4 г, 16,3 ммоль). Реакционную смесь нагревали до 75°С (20,5 часов (ч)), а затем при 110°С (2,5 ч). Добавляли еще CsF (2,23 г) и продолжали нагревание при 110°С еще в течение часа. После охлаждения реакционную смесь выливали в холодный насыщенный (насыщ.) водный (водн.) раствор гидрокарбоната натрия (NaHCO3) при перемешивании и экстрагировали эфиром (Et2O). Объединенные органические экстракты промывали концентрированным раствором NaCl, сушили над сульфатом натрия (Na2SO4), фильтровали и концентрировали в вакууме, получая масло коричневого цвета (2,51 г): EIMS m/z 158.
Пример 1b 4,5,6-Трифторпиколинонитрил
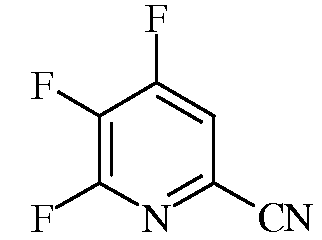
В автоклав на 45 мл из нержавеющей стали помещали 4,5,6-трихлорпиколинонитрил (1,0 г, 4,8 ммоль), сухой KF (1,3 г, 22,4 ммоль), 18-краун-6 (180 мг, 0,7 ммоль) и сухой ацетонитрил (10 мл). Автоклав герметизировали и нагревали при 135°С в течение 10 ч. После охлаждения из автоклава отбирали пробу, при этом по данным анализа методом газовой хроматографии (ГХ) в смеси содержалось 70% 4,5,6-трифторпиколинонитрила и 30% 5-хлор-4,6-дифторпиколинонитрила: EIMS (70 эВ) m/z 158 (М+, 100%), 131 (20%), 176 (М+, 30%), 174 (М+, 100%).
Пример 1с 6-(4-Хлорфенил)-4,5-дифторпиколинонитрил
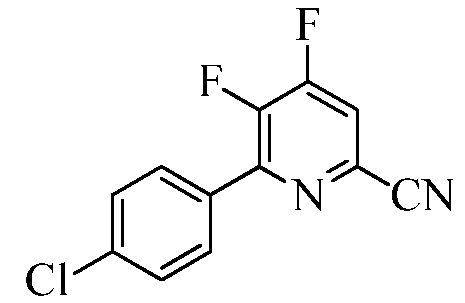
Трехгорлую колбу на 1000 мл снабжали насадкой для перегонки, вводом азота, механической мешалкой и термопарой. В колбу помещали CsF (93,6 г, 0,616 моль). Добавляли безводный ДМСО (500 мл) и суспензию вакуумировали/снова заполняли азотом. Суспензию нагревали при 80°С в течение 30 мин. ДМСО (100 мл) отгоняли в вакууме для удаления остаточной воды. Прибавляли 4,5-дихлор-6-(4-хлорфенил)пиколинонитрил (50 г, 0,1763 моль) и раствор вакуумировали/снова заполняли азотом. Реакционную смесь нагревали при 105°С в атмосфере азота. Спустя 4 ч при 105°С, по данным анализа методом ГХ, реакция завершалась. Реакционную смесь оставляли остывать до комнатной температуры. ДМСО удаляли перегонкой в вакууме. Остаток выливали в ледяную воду (500 г) и экстрагировали этилацетатом (EtOAc; 3×200 мл). Объединенные органические экстракты промывали водой (2×200 мл), а затем концентрированным раствором NaCl (100 мл). Экстракты сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, получая масло коричневого цвета, которое закристаллизовывалось при стоянии. В результате очистки колоночной хроматографией (силикагель 60-120 меш; элюирование при градиенте 0-20% EtOAc-гексан) получали твердое вещество белого цвета (17 г, 39%): т.пл. 89,0-90,8°С; 1H ЯМР (400 МГц, ДМСО-d6) δ 8,51-8,47 (м, 1H), 7,92 (д, JH-H=8,6 Гц, 2H), 7,64 (д, JH-H=8,6 Гц, 2H); 13C ЯМР (100 МГц, ДМСО-d6) δ 156,04 (дд, JF-C=262, 13 Гц), 148,16 (дд, JF-C=9, 2 Гц), 147,99 (дд, JF-C=267, 10 Гц), 135,72, 131,17 (дд, JF-C=15, 3 Гц), 130,4 (д, JF-C=6 Гц), 129,08 (дд, JF-C=11, 7 Гц), 128,91, 118,93 (д, JF-C=19 Гц), 115,96 (д, JF-C=3 Гц); 19F ЯМР (376,5 МГц, ДМСО-d6) δ -123,25 (д, JF-F=18,82 Гц),-141,07 (д, JF-F=18,82 Гц); ESIMS m/z 251 ([M]+). Анализ выч. для C12H5ClF2N2: C, 57,51; H, 2,01; N, 11,18. Найдено: C, 57,97; H, 2,15; N, 10,77.
Аминирование
Пример 2а 4-Амино-5,6-дифторпиколинонитрил
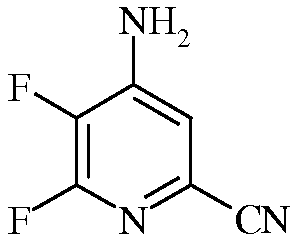
К сырому 4,5,6-трифторпиколинонитрилу (2,5 г, 15,8 ммоль) прибавляли гидроксид аммония (NH4OH) и перемешивали при комнатной температуре в течение 2 ч. Выпавший осадок коричневого цвета фильтровали, промывали водой и сушили, получая 0,72 г. По данным анализа методом хроматографии-масс-спектрометрии (ГХ-МС), имелось два изомерных продукта. Водный фильтрат экстрагировали EtOAc. Объединенные органические экстракты промывали концентрированным раствором NaCl, сушили над Na2SO4, и упаривали, получая дополнительное количество твердого вещества коричневого цвета. Две этих порции объединяли и очищали колоночной хроматографией, получая 4-амино-5,6-дифторпиколинонитрил (0,60 г, 24,4%): 1H ЯМР (400 МГц, CDCl3) δ 7,13 (д, J=5,3 Гц, 1H, ароматический), 6,62 (с, 2H, NH2); 13C ЯМР (101 МГц, CDCl3) δ 157,0 (дд, J=235, 12 Гц), 151,7 (дд, J=10, 7 Гц), 139,5 (дд, J=254, 29 Гц), 128,8 (д, J=19 Гц), 121,2 (с), 121,0 (с); 19F ЯМР (376 МГц, CDCl3) δ -85,29 (д, J=24,5 Гц), -156,98 (дд, J=24,5, 5,2 Гц).
Пример 2b 4-Амино-3-хлор-5,6-дифторпиколинонитрил
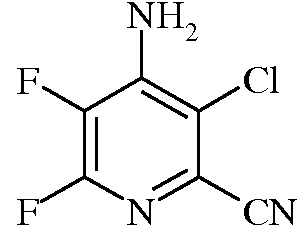
Раствор 3-хлор-4,5,6-трифторпиколинонитрила (200 г) в EtOAc (3 л) охлаждали до 10°С. К нему медленно прибавляли 14%-ный водн. NH4OH (1296 г), поддерживая температуру в интервале 18-23°С. Водный раствор отделяли от органического раствора. Органическую фазу последовательно промывали 50/50 раствором водного насыщенного NaCl и водой (500 мл) и насыщ. раствором NaCl (250 мл). Как только органическую фазу концентрировали в вакууме при 50°С до объема примерно 500 мл, продукт выкристаллизовывался. К данной суспензии добавляли гептан (1 л) и концентрировали смесь в вакууме. Осадок выделяли фильтрованием. Данный осадок промывали пентаном и сушили в вакууме, получая 4-амино-3-хлор-5,6-дифторпиколинонитрил (173,8 г, 90%, чистота 99,6%) в виде кристаллического вещества белого цвета: т.пл. 190-191,5°С; 13C ЯМР (101 МГц, ДМСО-d6) δ 150,03 (дд, J=232,4, 12,5 Гц, C6), 144,29 (дд, J=11,4, 6,9 Гц, C4), 133,72 (дд, J=257,9, 30,8 Гц, C5), 122,14 (дд, J=19,6, 4,9 Гц, C2), 119,31 (с, C3), 114,25 (с, CN); 19F ЯМР (376 МГц, ДМСО-d6) δ -91,24 (д, J=24,2 Гц),-154,97 (д, J=24,2 Гц); EIMS m/z 189 ([M]+). Анализ выч. для C6H2ClF2N3: C, 38,02; H, 1,06; N, 22,17. Найдено: C, 37,91; H, 1,00; N, 22,02.
Пример 2с 4-Амино-6-(4-хлорфенил)-5-фторпиколинонитрил
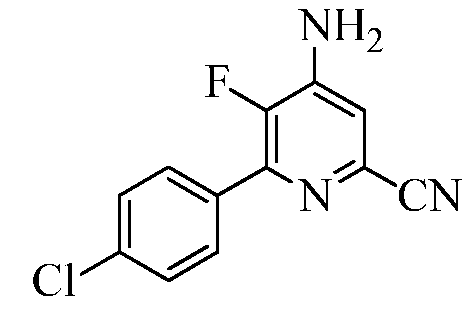
6-(4-Хлорфенил)-4,5-дифторпиколинонитрил (60 г, 0,24 моль) растворяли в ДМСО (1200 мл). Через раствор периодически барботировали аммиак в общей сложности в течение 24 ч за период времени 48 ч. Реакционную смесь выливали в ледяную воду (2000 г). Продукт экстрагировали EtOAc (3×500 мл). Объединенные органические слои промывали водой (5×500 мл), а затем концентрированным раствором NaCl (100 мл). Экстракты сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, получая твердое вещество белого цвета (50 г, 84%): т.пл. 185,3-187,8°С; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,85 (д, JH-H=8,5 Гц, 2H), 7,58 (д, JH-H=8,5 Гц, 2H), 7,21 (д, JH-H=6,0 Гц, 1H), 6,96 (ушир.с, 2H); 13C ЯМР (100 МГц, ДМСО-d6) δ 141,86 (д, JF-C=256 Гц), 144,81 (д, JF-C=14 Гц), 143,80 (д, JF-C=10 Гц), 134,34, 132,87 (д, JF-C=5 Гц), 130,3 (д, JF-C=6 Гц), 128,56, 128,38 (д, JF-C=5 Гц), 117,43, 115,08 (д, JF-C=5 Гц); 19F ЯМР (376,5 МГц, ДМСО-d6) δ -142,71; ESIMS m/z 248 ([M]+). Анализ выч. для C12H7ClFN3: C, 58,20; H, 2,85; N, 16,97. Найдено: C, 57,82; H, 3,022; N, 16,10.
Замещение галогена, гидролиз и этерификация
Пример 3а Метил 4-амино-6-бром-5-фторпиколинат
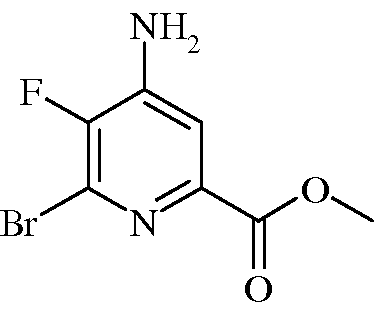
4-Амино-5,6-дифторпиколинонитрил (4,5 г, 6,45 ммоль) растворяли в 30%-ном растворе HBr в уксусной кислоте (40 мл). Раствор нагревали при 120°С в реакторе системы Parr в течение 3,0 ч. После охлаждения раствор концентрировали в вакууме. Остаток растворяли в метиловом спирте (СН3ОН; 40 мл) и переносили обратно в реактор системы Parr. Добавляли концентрированную серную кислоту (632 мг, 6,45 ммоль) и нагревали реактор при 110°С в течение 7 ч. После охлаждения растворитель выпаривали в вакууме. Остаток растворяли в EtOAc и нейтрализовали насыщ. водн. раствором NaHCO3. Органическую фазу отделяли, промывали концентрированным раствором NaCl, сушили (Na2SO4) и упаривали. Сырой продукт очищали колоночной хроматографией на силикагеле (10-100% EtOAc в гексане), получая твердое вещество желтого цвета (2,87 г, 40%): т.пл. 187-190°С; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,46 (д, J=6,2 Гц, 1H, ароматический), 6,88 (с, 2H, NH2), 3,83 (с, 3H, CH3); 13C ЯМР (101 МГц, ДМСО-d6) δ 163,8 (с), 144,7 (д, J=252 Гц), 144,3 (д, J=13 Гц), 143,1 (д, J=5 Гц), 127,7 (д, J=21 Гц), 113,1 (д, J=5 Гц), 52,4 (с); 19F ЯМР (376 МГц, ДМСО-d6) δ -133,77 (с).
Пример 3b 4-Амино-6-бром-5-фторпиколинамид и метил 4-амино-6-бром-3-хлор-5-фторпиколинат
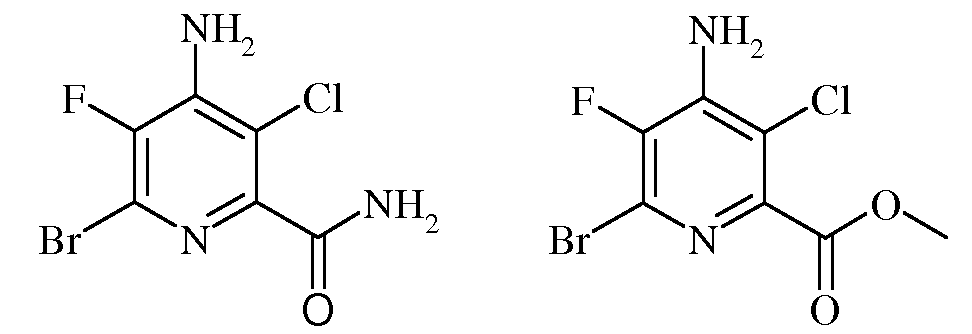
Смесь 4-амино-3-хлор-5,6-дифторпиколинонитрила (70 г, 0,37 моль) и 33%-ного HBr в уксусной кислоте (700 мл) нагревали до 120°С в герметизированном реакционном сосуде при перемешивании в течение 2 ч. После охлаждения до комнатной температуры надосадочную жидкость отделяли от большого количества твердого вещества желтовато-коричневого цвета, и концентрировали в вакууме, получая клейкий темный остаток. Данный остаток растворяли в СН3ОН (600 мл) и вновь добавляли к осадку желтовато-коричневого цвета, остававшемуся в автоклаве. К этой смеси медленно добавляли концентрированную серную кислоту (H2SO4; 40 г, 0,41 моль), и реактор вновь герметизировали и нагревали до 110°С в течение 6 ч. Охлажденную реакционную смесь медленно выливали в насыщ.водн. раствор карбоната натрия (Na2CO3; 2 л) и Et2O (1 л). Эфирный экстракт сушили над MgSO4, фильтровали и концентрировали до состояния твердого вещества желтовато-коричневого цвета. Данное твердое вещество очищали колоночной хроматографией, получая метил 4-амино-6-бром-3-хлор-5-фторпиколинат (78 г, 75%) в виде мелких кристаллов белого цвета: т.пл. 119-120°С; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,28 (с, 2H), 3,87 (с, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 163,54 (с, C=O), 144,63 (д, J=256,3 Гц, C5), 142,60 (д, J=4,9 Гц, C2), 140,55 (д, J=13,6 Гц, C4), 125,61 (д, J=21,0 Гц, C6), 116,65 (с, C3), 53,2 (с, OMe); 19F ЯМР (376 МГц, CDCl3) δ -128,86; EIMS m/z 284 ([M]+). Анализ выч. для C7H5BrClFN2O2: C, 29,66; H, 1,78; N, 9,88. Найдено: C, 30,03; H, 1,80; N, 9,91.
Кроме того, методом колоночной хроматографии выделяли 4-амино-6-бром-3-хлор-5-фторпиколинамид (200 мг) в виде твердого вещества светло-желтовато-коричневого цвета: т.пл. 215°С разл.; 13C ЯМР (101 МГц, ДМСО-d6) δ 165,64 (с, C=O), 148,02 (д, J=4,8 Гц, C2), 142,31 (д, J=233,2 Гц, C5), 141,86 (д, J=14,0 Гц, C4), 124,13 (д, J=19,9 Гц, C6), 112,55 (д, J=2,1 Гц, C3); 19F ЯМР (376 МГц, ДМСО-d6) δ -131,56; EIMS m/z 269 ([M]+). Анализ выч. для C6H4BrClFN3O: C, 26,84; H, 1,50; N, 15,65. Найдено: C, 26,95; H, 1,52; N, 15,16.
Гидролиз и этерификация
Пример 4а Метил 4-амино-6-(4-хлорфенил)-5-фторпиколинат
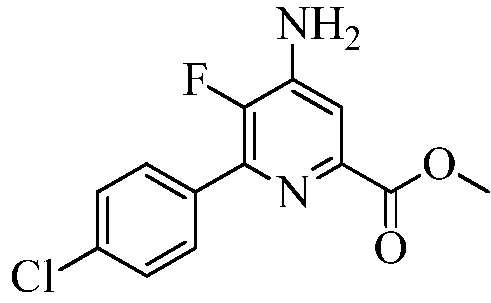
В запаянную трубку на 1000 мл помещали 4-амино-6-(4-хлорфенил)-5-фторпиколинонитрил (30 г, 0,1211 моль) и 90%-ную H2SO4 (30 мл) в СН3ОН (500 мл). Раствор нагревали при 110°С в течение 7 дней. При охлаждении до комнатной температуры выпадал осадок белого цвета. Реакционную смесь выливали в ледяную воду (300 г), нейтрализовывали насыщ. раствором NaHCO3, а затем экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты промывали водой (3×100 мл), а потом концентрированным раствором NaCl. Экстракты сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, получая твердое вещество белого цвета. В результате очистки колоночной хроматографией (силикагель 60-120 меш; элюирование при градиенте 0-20% EtOAc-гексан) получали твердое вещество белого цвета (25 г, 44%): т.пл. 176,8-178,9°С; 1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (д, JH-H=8,6 Гц, 2H), 7,56 (д, 8,6 Гц, 2H), 7,46 (д, JH-F=6,3 Гц, 1H), 6,64 (ушир.с, 2H), 3,38 (с, 3H); 13C ЯМР (100 МГц, ДМСО-d6) δ 165,44, 147,69 (д, JF-C=254 Гц), 144,82 (д, JF-C=13 Гц), 143,72 (д, JF-C=5 Гц), 142,56 (д, JF-C=11 Гц), 133,34 (д, JF-C=7 Гц), 130,75 (д, JF-C=6 Гц), 128,88, 112,44 (д, JF-C=5 Гц), 52,73; 19F ЯМР (376,5 МГц, ДМСО-d6) δ -145,01; ESIMS m/z 281 ([M]+). Анализ выч. для C13H10ClFN2O2: C, 55,63; H, 3,59; N, 9,98. Найдено: C, 55,59; H, 3,61; N, 9,98.
Сочетание
Пример 5а Метил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
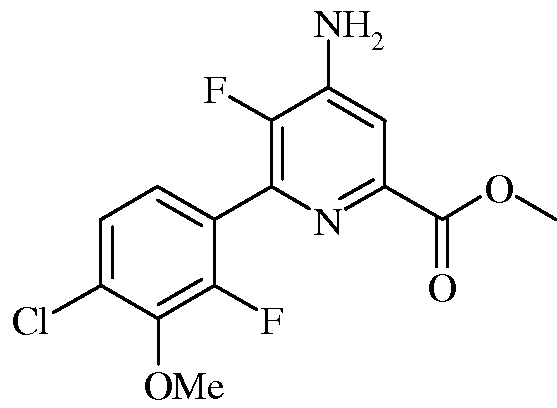
В круглодонную колбу на 50 мл, снабженную обратным холодильником, помещали твердый метил 4-амино-6-бром-5-фторпиколинат (1,0 г, 4,02 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (1,227 г, 5,02 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (Pd(PPh3)2Cl2: 0,141 г, 0,201 ммоль) и KF (0,467 г, 8,03 ммоль). Реакционную смесь продували азотом, а затем прибавляли растворитель (3:1 ацетонитрил-вода, 24 мл). Реакционную смесь нагревали при кипении в атмосфере азота в течение 2 ч. После охлаждения до комнатной температуры продукт фильтровали, промывали ацетонитрилом, после чего водой, и сушили в вакуумной печи в течение ночи, получая продукт (0,89 г) в виде твердого вещества почти белого цвета: т.пл. 204-206°С. Из фильтрата выделяли еще 0,29 г продукта с общим выходом 89%. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,54 (д, J=6,6 Гц, 1H), 7,46 (дд, J=8,5, 1,4 Гц, 1H), 7,30 (дд, J=8,4, 7,2 Гц, 1H), 6,71 (с, 2H), 3,93 (с, 3H), 3,83 (с, 3H), 3,31 (с, 4H); 13C ЯМР (101 МГц, ДМСО-d6) δ 164,82 (с), 153,17 (д, J=249,5 Гц), 146,87 (д, J=254,3 Гц), 143,83 (дд, J=13,4, 3,8 Гц), 143,69 (д, J=4,3 Гц), 139,05 (д, J=10,6 Гц), 128,20 (д, J=3,2 Гц), 125,96 (д, J=3,4 Гц), 125,42 (д, J=3,6 Гц), 123,83 (дд, J=14,3, 3,1 Гц), 112,54 (с), 61,56 (с), 52,25 (с); 19F ЯМР (376 МГц, ДМСО-d6) δ -129,37 (д, J=26,0 Гц), -142,56 (д, J=26,3 Гц).
Пример 5b Метил 4-амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
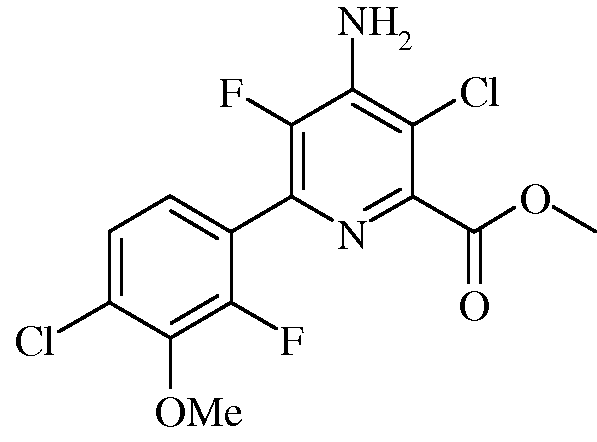
В трехгорлую колбу на 250 мл, снабженную обратным холодильником, вводом азота и термопарой, помещали метил 4-амино-3,6-дихлор-5-фторпиколинат (9,965 г, 41,7 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (12,74 г, 52,1 ммоль), и KF (4,84 г, 83 ммоль). Добавляли ацетонитрил (78 мл) и воду (26 мл). Реакционную смесь продували азотом. Прибавляли Pd(PPh3)2Cl2 (1,477 г, 2,10 ммоль, 5 мол.%), и нагревали раствор при 70°С в атмосфере азота в течение 2 ч. При охлаждении до комнатной температуры выпадал осадок, который фильтровали и промывали водой. Осадок растворяли в EtOAc (прибл. 500 мл) и промывали водой, а затем насыщенным раствором соли. Органический слой сушили (MgSO4) и удаляли растворитель на роторном испарителе, получая твердое вещество оранжевого цвета, которое сушили в вакуумной печи при 50°С (11,46 г, выход 76%): т.пл. 169-170,5°С. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,48 (д, J=8,4 Гц, 1H), 7,32 (т, J=7,7 Гц, 1H), 7,15 (с, 2H), 3,96 (с, 3H), 3,90 (с, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 164,85 (с), 153,11 (д, J=252,5 Гц), 146,29 (с), 144,52 (д, J=4,3 Гц), 143,74 (с), 142,75 (дд, J=227,1, 14,0 Гц), 136,38 (д, J=13,4 Гц), 128,58 (д, J=3,2 Гц), 125,87 (с), 125,54 (д, J=3,5 Гц), 122,89 (дд, J=13,8, 4,0 Гц), 113,01 (д, J=3,0 Гц), 61,61 (д, J=4,2 Гц), 52,70 (с); ESIMS m/z 364 ([M+Н]+). Анализ выч. для C14H10Cl2F2N2O3: C, 46,30; H, 2,78; N, 7,71. Найдено: C, 46,60; H, 2,68; N, 7,51.
Пример 5с 4-Амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
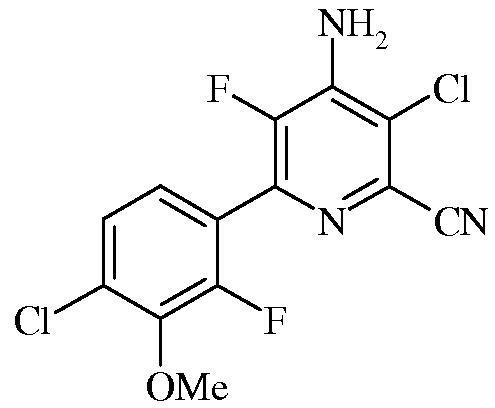
Смесь 4-амино-3,6-дихлор-5-фторпиколинонитрила (0,37 г, 1,80 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинана (0,549 г, 2,24 ммоль) и KF (0,209 г, 3,59 ммоль) растворяли в ацетонитриле (6,75 мл) и воде (2,25 мл). Смесь перемешивали и барботировали через нее азот. Прибавляли Pd(PPh3)2Cl2 (63 мг, 0,1 ммоль), и снова барботировали через смесь азота. После этого раствор нагревали при 75°С в атмосфере азота в течение 2 ч. При охлаждении выпадал осадок, который выделяли фильтрованием, промывали водой и сушили в вакууме, получая продукт (0,34 г) в виде твердого вещества почти белого цвета. Водную фазу экстрагировали EtOAc (3х), а объединенные органические экстракты промывали концентрированным раствором NaCl, сушили и концентрировали. В результате очистки колоночной хроматографией получали дополнительное количество продукта (0,12 г) в виде твердого вещества белого цвета. Общий выход 78%. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,50 (дд, J=8,5, 1,4 Гц, 1H), 7,45 (с, 2H), 7,33 (дд, J=8,5, 7,2 Гц, 1H), 3,94 (с, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 152,97 (д, J=253,2 Гц), 145,73 (д, J=260,8 Гц), 143,82 (д, J=13,7 Гц), 141,83 (д, J=14,7 Гц), 138,45 (д, J=14,8 Гц), 133,93-132,79 (м), 128,93 (д, J=3,3 Гц), 127,74 (с), 126,37-125,10 (м), 122,08 (дд, J=13,6, 3,9 Гц), 119,34 (д, J=4,5 Гц), 114,99 (с), 61,61 (с); 19F ЯМР (376 МГц, ДМСО-d6) δ -129,00 (дд, J=28,2, 7,0 Гц, 1F),-133,76 (д, J=28,2 Гц, 1F); ESIMS m/z 330,1 ([M+Н]+).
Пример 5d 4,5-Дихлор-6-(4-хлорфенил)пиколинонитрил
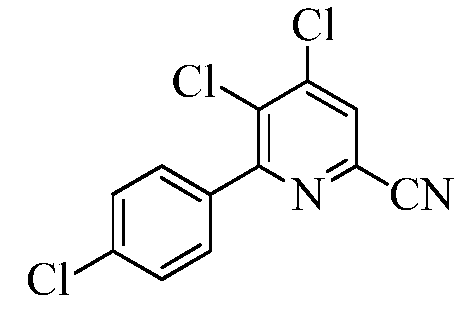
В круглодонную колбу на 3000 мл помещали 4,5,6-трифторпиколинонитрил (100 г, 0,482 моль), (4-хлорфенил)бороновую кислоту (106 г, 0,6797 моль), трифенилфосфин (11,4 г, 0,0433 моль) и гидрофосфат калия (К2НРО4; 252 г, 1,4462 моль). Добавляли ацетонитрил (900 мл) и воду (300 мл). Реакционную смесь вакуумировали/снова заполняли азотом. Прибавляли дихлорид бис(цианофенил)палладия(II) (Pd(PhCN)2Cl2; 9,2 г, 0,0241 моль). Раствор вакуумировали/снова заполняли азотом, а затем перемешивали при кипении в течение 5 ч. При охлаждении до комнатной температуры выпадал осадок белого цвета. Реакционную смесь выливали в ледяную воду (1000 г) и экстрагировали EtOAc (3×500 мл). Объединенные органические слои последовательно промывали водой (3×500 мл) и концентрированным раствором NaCl (100 мл). Экстракты сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, получая твердое вещество белого цвета (83 г, 61%): т.пл. 142,2-144,6°С. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,58 (с, 1H), 7,71 (д, J=8,56 Гц, 2H), 7,61 (д, J=8,56 Гц, 2H); 13C ЯМР (75 МГц, ДМСО-d6) δ 158,52, 144,49, 135,64, 135,26, 133,20, 131,64, 131,16, 129,84, 128,81, 116,5. Анализ выч. для C12H5Cl3N2: C, 50,83; H, 1,78; N, 9,88. Найдено: C, 51,54; H, 1,90; N, 9,19.
Галогенирование
Пример 6а Метил 4-Амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинат
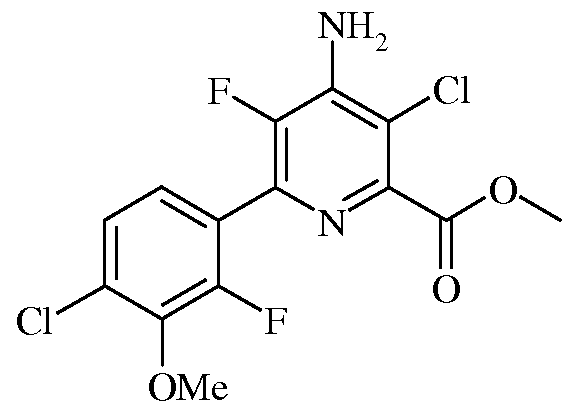
Смесь метил 4-амино-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколината (0,5 г, 1,52 ммоль) и 1,3-дихлор-5,5-диметилимидазолидин-2,4-диона (0,30 г, 1,52 ммоль) в ацетонитриле (10 мл) нагревали при кипении в течение 1,5 ч. После охлаждения до комнатной температуры реакционную смесь концентрировали в вакууме, а затем очищали колоночной хроматографией на силикагеле (градиент от 100% гексана до 100% EtOAc), получая твердое вещество светло-коричневого цвета (0,53 г, 100%): т.пл. 169-170,5°С. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,48 (д, J=8,4 Гц, 1H), 7,32 (т, J=7,7 Гц, 1H), 7,15 (с, 2H), 3,96 (с, 3H), 3,90 (с, 3H), 13C ЯМР (101 МГц, ДМСО-d6) δ 164,8 (с), 153,1 (д, J=253 Гц), 146,3 (с), 144,5 (д, J=4 Гц), 143,7 (с), 142,8 (дд, J=227, 14 Гц), 136,4 (д, J=13 Гц), 128,6 (д, J=3 Гц), 125,9 (с), 125,5 (д, J=4 Гц), 122,9 (дд, J=14, 4 Гц), 113,0 (д, J=3 Гц), 61,6 (д, J=4 Гц), 52,7 (с). Анализ выч. для C14H10Cl2F2N2O3: C, 46,30; H, 2,78; N, 7,71. Найдено: C, 46,60; H, 2,68; N, 7,51.
Пример 6b 3-Хлор-4,5,6-трифторпиколинонитрил
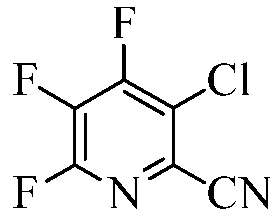
Использовали трубчатый реактор, состоящий из вертикальной трубки Hastelloy-C-276 с зоной смешения (длина = 48,26 сантиметров (см), ID=1,58 см, Т=290°С) и отдельно нагреваемой реакционной зоной (длина = 24,13 см, ID=1,58 см, Т=550°С). К выходному отверстию трубчатого реактора присоединяли нагреваемую зону испарения (180°С), состоящую из 4-футовой секции трубы из 1/8-дюймовой нержавеющей стали. Непосредственно в зону смешения отмеряли газообразный хлор (5 об.% Cl2 в N2) со скоростью 101 мл/мин. 4,5,6-Трифторпиколинонитрил (1,013 г, 79%, взвешенный в твердом состоянии в гексане), растворяли в четыреххлористом углероде (39,2 г). Данный раствор отмеряли при помощи насоса Гилсона со скоростью 0,1 мл/мин в зону испарения, чтобы гарантировать наличие реагентов в газовой фазе до их смешивания с газообразным хлором в зоне смешения. Конденсат из выходного потока реактора собирали в резервуар-ловушку, которую устанавливали примерно на 6 дюймов ниже реакционной зоны. Жидкую реакционную смесь выгружали из резервуара-ловушки после завершения реакции. Образец анализировали методами ГХ, ЯМР-спектроскопии и ГХ-МС. По данным ГХ-анализа (система Agilent 6890, колонка: 15 м × 0,32 мм J&W DB-5, 0,25 мкм; программа температуры: 80°С выдерживали 2 мин, линейное возрастание 20°/мин до 280°С, выдерживали 5 мин), соотношение площадей пиков исходного вещества (4,5,6-трифторпиколинонитрила) и продукта (3-хлор-4,5,6-трифторпиколинонитрила) составляло приблизительно 1:1,4. По результатам количественного ЯМР 13С, исходное вещество (4,5,6-трифторпиколинонитрил) и продукт (3-хлор-4,5,6-трифторпиколинонитрил) являлись основными компонентами смеси. Молярное соотношение исходного вещества и продукта составляло примерно 1,0:1,8. Данные ГХ-МС подтверждали идентичность продукта с 3-хлор-4,5,6-трифторпиколинонитрилом.
Выход продукта хлорирования (3-хлор-4,5,6-трифторпиколинонитрила) существенно возрастал, когда смесь из описанной выше реакции помещали в реакционную систему еще один раз. Применение хлорирования в два захода приводило к повышенной конверсии с соотношением продукта (3-хлор-4,5,6-трифторпиколинонитрила) и исходного вещества (4,5,6-трифторпиколинонитрила), составляющим 2,5:1,0.
Пример 6с Метил 4-амино-6-(4-хлорфенил)-5-фтор-3-йодпиколинат
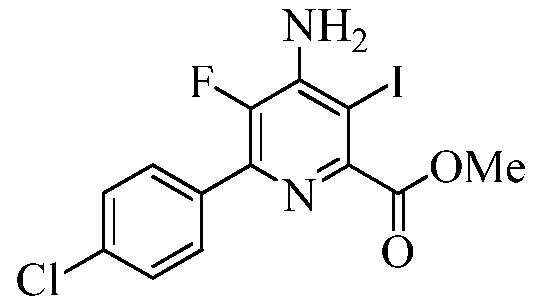
В круглодонную колбу на 250 мл помещали метил 4-амино-6-(4-хлорфенил)-5-фторпиколинат (25 г, 0,08906 моль), йод (18 г, 0,07125 моль) и йодную кислоту (H5IO6, 7,3 г, 0,03206 моль). Добавляли СН3ОН (100 мл). Реакционную смесь перемешивали при кипении в течение 16 ч. Реакционную смесь концентрировали в вакууме, а затем растворяли в Et2O (500 мл). Эфирный раствор промывали 10%-ным тиосульфатом натрия (3×100 мл), водой (3×100 мл), а затем насыщенным раствором соли. Органические экстракты сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, получая твердое вещество оранжевого цвета. В результате кристаллизации из смеси EtOAc-гексан (3:7) получали твердое вещество светло-оранжевого цвета (30,5 г, 83%): т.пл. 113,7-115,2°С. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,84 (д, J=8,4 Гц, 2H), 7,55 (д, J=8,4 Гц, 2H), 6,73 (ушир.с, 2H), 3,87 (с, 3H); 13C ЯМР (100 МГц, ДМСО-d6) δ 167,50, 151,48 (д, JF-C=5 Гц), 145,95 (д, JF-C=13 Гц), 144,05 (д, JF-C=257 Гц), 140,75 (д, JF-C=9 Гц), 134,56, 133,42 (д, JF-C=5 Гц), 130,63 (д, JF-C=6 Гц), 129,02, 112,44 (д, JF-C=5 Гц), 77,52, 53,05; 19F ЯМР (376,5 МГц, ДМСО-d6) δ -140,62; ESIMS m/z 407 ([M]+). Анализ выч. для C13H9ClFIN2O2: C, 38,40; H, 2,23; N, 6,89. Найдено: C, 38,40; H, 2,31; N, 6,85.




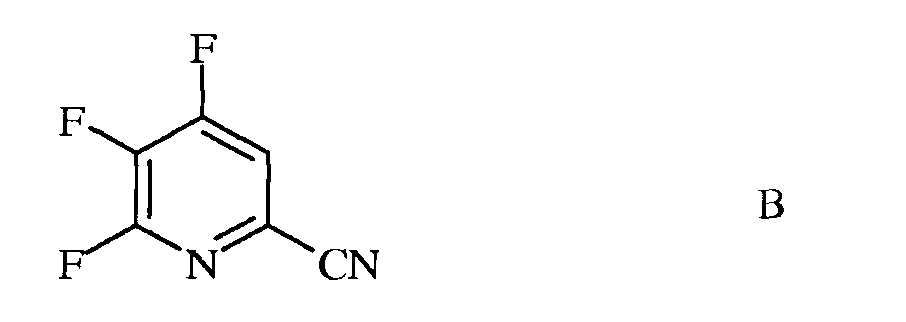
Высококонцентрированные препараты 1-метилгептилового эфира флуроксипира, стабильные при низких температурах
Пестициды, пестицидная композиция и способ контроля вредителей
Синергическая гербицидная композиция, содержащая хлорацетанилиды и пиколиновые кислоты
Подсолнечник с низким содержанием насыщенных жиров и соответствующие способы
Способы переноса молекулярных веществ в клетки растений
Стабилизированные эмульсии масло-в-воде, содержащие агрономически активные ингредиенты, и способы их применения
Пестицидные композиции
Защита от повреждения гербицидом 6-(трехзамещенный фенил)-4-амино-2-пиридинкарбоксилата посеянного семенами и рассадного риса-сырца
5-фторпиримидиновые производные в качестве фунгицидов
Пестицидные композиции
Высококонцентрированные препараты 1-метилгептилового эфира флуроксипира, стабильные при низких температурах
Пестициды, пестицидная композиция и способ контроля вредителей
Синергическая гербицидная композиция, содержащая хлорацетанилиды и пиколиновые кислоты
Подсолнечник с низким содержанием насыщенных жиров и соответствующие способы
Способы переноса молекулярных веществ в клетки растений
Стабилизированные эмульсии масло-в-воде, содержащие агрономически активные ингредиенты, и способы их применения
Пестицидные композиции
Защита от повреждения гербицидом 6-(трехзамещенный фенил)-4-амино-2-пиридинкарбоксилата посеянного семенами и рассадного риса-сырца
Стабильная пестицидная композиция на основе сульфоксимина и способ борьбы с насекомыми
5-фторпиримидиновые производные в качестве фунгицидов

