ВЫСОКОАКТИВНЫЙ И ВЫСОКОСЕЛЕКТИВНЫЙ КАТАЛИЗАТОР ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ ГЕКСЕНА ИЛИ ОКТЕНА С ПРИМЕНЕНИЕМ ДАННОГО КАТАЛИЗАТОРА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к высокоактивному и высокоселективному катализатору олигомеризации этилена для применения при олигомеризации этилена, такой как тримеризация или тетрамеризация, и к способу получения 1-гексена или 1-октена с применением данного катализатора и конкретнее к комплексному соединению хрома для селективной олигомеризации этилена, включающему соединение хрома и хиральный лиганд, имеющий особую стереоизомерную структуру, к композиции катализатора на основе комплекса хрома для селективной олигомеризации этилена, включающей комплексное соединение хрома для селективной олигомеризации этилена и сокатализатор, такой как метилалюминоксан (MAO), и к способу получения 1-гексена или 1-октена с высокой активностью и высокой селективностью с применением такой каталитической системы.
Уровень техники
1-гексен и 1-октен, которые являются мономерами или сомономерами для получения линейного полиэтилена низкой плотности, представляют собой важный промышленный материал, широко используемый в полимеризационном процессе, и их получают очисткой продуктов, получаемых олигомеризацией этилена. Однако традиционная олигомеризация этилена является неэффективной в терминах получения значительных количеств бутена, более высоких олигомеров и полиэтилена, а также 1-гексена и 1-октена. Такие традиционные технологии олигомеризации этилена типично дают разнообразные α-олефины в зависимости от распределения продуктов Шульца-Флори или Пуассона, причем, что нежелательно, желаемые продукты получаются с ограниченным выходом.
В настоящее время исследуется получение 1-гексена или 1-октена селективной тримеризацией или тетрамеризацией этилена с использованием катализа переходными металлами, где практически все известные катализаторы на основе переходных металлов представляют собой катализаторы на основе хрома. Международная патентная публикация WO 02/04119 раскрывает катализатор тримеризации этилена, а именно катализатор на основе хрома, использующий лиганд (R1)(R2)X-Y-X(R3)(R4), в котором X представляет собой фосфор, мышьяк или сурьму, Y представляет собой связующую группу, такую как -N(R5)-, и по меньшей мере один из R1, R2, R3 и R4 имеет полярный заместитель или электронодонорный заместитель.
Другая публикация раскрывает применение соединения (о-этилфенил)2PN(Me)P(о-этилфенил)2, в котором по меньший мере один из R1, R2, R3 и R4 не имеет полярного заместителя, в качестве лиганда, который не показывает каталитической активности по 1-гексену в каталитических условиях (Antea Carter et al., Chem. Commun., 2002, p. 858-859).
Кроме того, нерассмотренная публикация заявки на патент Кореи № 2006-0002741 раскрывает, что превосходная активность и селективность тримеризации этилена действительно возможна при использовании PNP-лиганда, содержащего неполярный заместитель в орто-положении фенильного кольца, присоединенного к фосфору, такого как (о-этилфенил)2PN(Me)P(о-этилфенил)2.
Также международная патентная публикация WO 04/056479 раскрывает увеличение селективности при получении 1-октена тетрамеризацией этилена с использованием катализатора на основе хрома, содержащего PNP-лиганд, не имеющий заместителя в фенильном кольце, присоединенном к фосфору. По существу, гетероатомный лиганд, использованный в катализаторе тетрамеризации этилена, иллюстрируется на примере (фенил)2PN(изопропил)P(фенил)2 или т.п.
Данная публикация традиционного уровня техники раскрывает, что катализатор на основе хрома, содержащий гетероатомный лиганд, имеющий гетероатомы азота и фосфора, допускает тетрамеризацию этилена даже в отсутствие полярного заместителя при гидрокарбильной или гетерогидрокарбильной группе, связанной с атомом фосфора, таким образом давая 1-октен с селективностью, превышающей 70% мас.
Однако традиционные технологии не предлагают конкретных примеров структуры лиганда, содержащего гетероатом, которая способна высокоселективно осуществлять тетрамеризацию или тримеризацию этилена, давая 1-октен или 1-гексен. Также, данные технологии предлагают лишь структуру с PNP-скелетом, такую как (R1)(R2)P-(R5)N-P(R3)(R4), в качестве лиганда, имеющего селективность по 1-октену примерно 70% мас. Более того, формы заместителей, которые являются замещаемыми в гетероатомных лигандах, представлены ограниченно. Конкретно, хотя селективность тетрамеризации сильно зависит от мостиковой структуры между атомом P и другим атомом P в структуре лигандного скелета, традиционные технологии раскрывают, что катализатор определяется как высокоселективный катализатор, если атомы P соответственно присоединены к обеим сторонам мостиковой структуры.
Кроме того, традиционные лиганды со скелетом PNP-типа, имеющие гетероатомы, проблематичны, поскольку при получении 1-октена или 1-гексена реакционная активность не остается одинаковой с течением времени и скорость реакции заметно снижается. Считается, что это может быть связано с тем, что N-атом структуры лигандного скелета может быть легко координирован переходным металлом благодаря присутствию его неподеленной электронной пары и таким образом может быть принят в качестве лиганда, но может быть побужден к легкой диссоциации от переходного металла посредством атома P, имеющего сравнительно плохую координационную способность. Публикация раскрывает, что превращение лигандного PNP-скелета из P-N-P в N=P-P облегчается в зависимости от условий синтеза, включая растворитель и полярность заместителей (Dalton Trans., 2003, 2772).
Кроме того, другая публикация раскрывает, что в случае лиганда со скелетом PNP-типа, содержащего гетероатомы, когда проводится олигомеризация этилена с использованием каталитического комплекса, уже синтезированного из лиганда и хромового предшественника, реакционная активность и селективность не сильно изменяются по сравнению с тем, когда лиганд и хромовый предшественник добавляют по отдельности (J. Am. Chem. Soc., 2004, 126, 14712).
Однако вышеупомянутые публикации раскрывают только весьма ограниченный катализатор для фактического получения 1-гексена и 1-октена с высокой активностью и высокой селективностью, в котором его активность низка, что имеет результатом ограниченную промышленную применимость. В частности, известные катализаторы проблематичны, поскольку используется дорогостоящий сокатализатор, примером которого является метилалюминоксан, что еще больше затрудняет достижение широкого применения в промышленности.
Раскрытие изобретения
Техническая проблема
Заявитель провел олигомеризацию этилена в условиях варьирования не только структуры между атомами P и P, но также заместителей R1, R2, R3, R4 атомов P, чтобы преодолеть проблемы со стабильностью традиционных катализаторов, раскрытые в заявке на патент Кореи № 2007-0005688, и таким образом обнаружил факт того, что каталитическая система на основе хрома, включающая лиганд с P-C-C-P-скелетом без азота, может быть использована для тримеризации или тетрамеризации этилена для получения таким образом 1-гексена или 1-октена с высокой селективностью, а также что активность катализатора может быть в значительной степени стабилизирована в течение времени реакции, так что скорость реакции постоянно поддерживается неизменной. Более того, заявитель обнаружил, что когда структуры, смежные с C-атомами между двумя P-атомами в лиганде с P-C-C-P-скелетом согласно настоящему изобретению, стерически изменены, активность и селективность тримеризации и тетрамеризации могут быть значительно увеличены.
Однако в традиционных технологиях, когда переходный металл и лиганд с P-C-C-P-скелетом добавляют по отдельности в среду олигомеризации этилена, на увеличение активности и селективности накладываются ограничения. Причина этого заключается в том, что атом углерода структуры скелета не имеет неподеленной электронной пары и направление координации переходного металла ограничено по причине хирального углерода структуры скелета вокруг P-атома, действующего как донор электронов для переходного металла, что таким образом затрудняет подход переходного металла в реакционной среде для образования координационной связи. Структура с P-C-C-P-скелетом оказывает стерические и электронные эффекты, при которых, когда предшественник переходного металла и лиганд с P-C-C-P-скелетом по отдельности добавляют в среду олигомеризации этилена в качестве катализатора подобно PNP-лиганду, число молекул предшественника переходного металла, которые должны быть превращены в каталитически активные участки, уменьшается в отличие от PNP-лиганда, что приводит к пониженным активности и селективности при тримеризации или тетрамеризации этилена.
Чтобы преодолеть ограничение увеличения активности катализатора, заявитель синтезировал, по существу, чистый комплекс переходного металла взаимодействием хирального лиганда, имеющего структуру с P-C-C-P-скелетом, с предшественником переходного металла и затем ввел синтезированный комплекс переходного металла в среду олигомеризации этилена и таким образом подтвердил, что активность и селективность катализатора значительно возрастают, тем самым приводя к настоящему изобретению.
Техническое решение
Следовательно, настоящее изобретение предназначено предоставить высокоактивный и высокоселективный катализатор олигомеризации этилена для применения в олигомеризации этилена, такой как тримеризация или тетрамеризация, а именно комплексное соединение хрома для проведения селективной олигомеризации этилена, включающее хиральный лиганд, имеющий структуру с P-C-C-P-скелетом и хром, связанные друг с другом, и предоставить каталитическую композицию на основе комплекса хрома для селективной олигомеризации этилена, включающую комплексное соединение хрома для селективной олигомеризации этилена и сокатализатор, такой как метилалюминоксан (MAO), а также предоставить способ получения 1-гексена или 1-октена с высокой активностью и высокой селективностью с применением такой каталитической системы.
Ниже будет дано подробное описание настоящего изобретения.
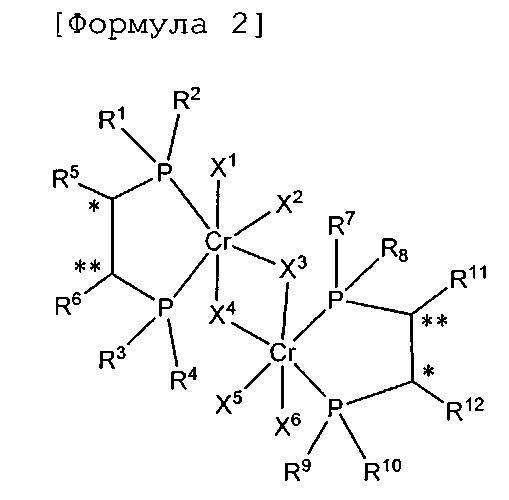
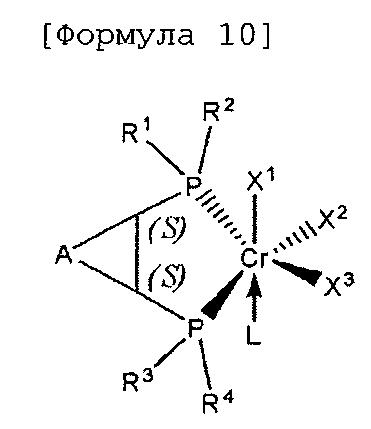
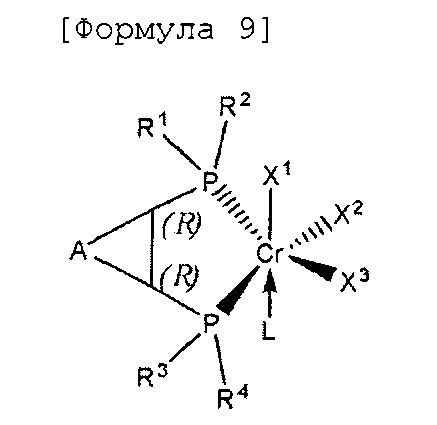
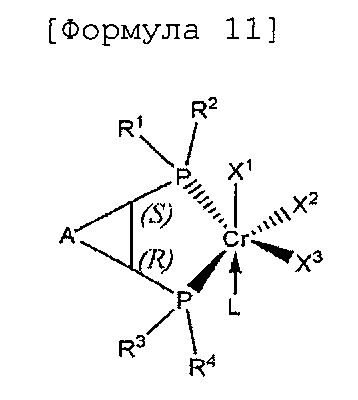
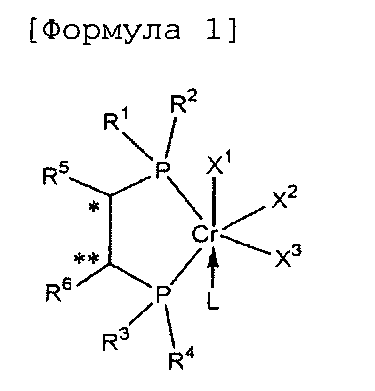
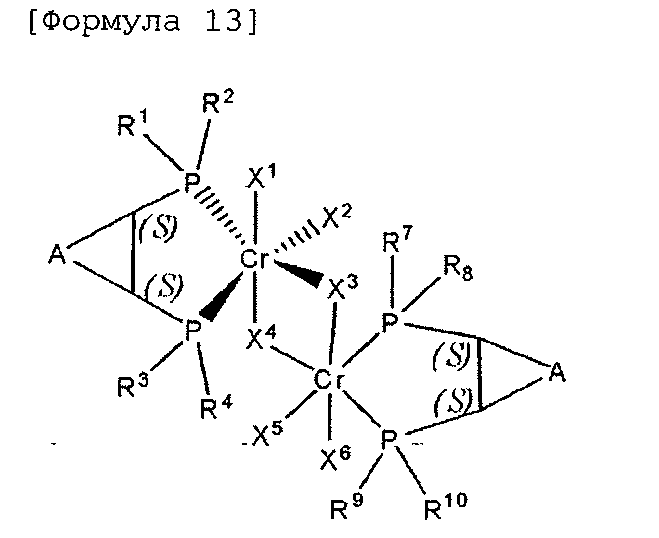
Настоящее изобретение предоставляет в качестве катализатора олигомеризации этилена комплексное соединение переходного металла, синтезированное координацией хирального лиганда, имеющего структуру с P-C-C-P-скелетом, с переходным металлом или с предшественником переходного металла. Конкретнее, настоящее изобретение предоставляет комплексное соединение хрома для селективной олигомеризации этилена, включающее хиральный лиганд, представленный нижеприведенной формулой 1 или 2.
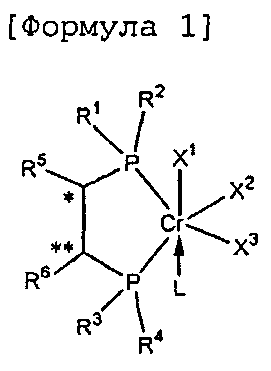
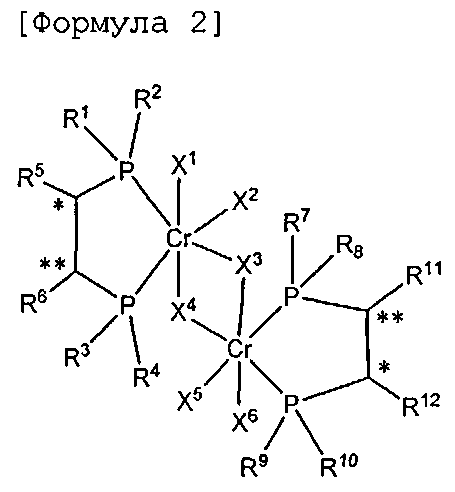
[В формуле 1 или 2 R1, R2, R3, R4, R7, R8, R9 и R10, каждый, независимо представляют собой гидрокарбил, замещенный гидрокарбил, гетерогидрокарбил или замещенный гетерогидрокарбил; R5, R6, R11 и R12, каждый, независимо представляют собой гидрокарбил или замещенный гидрокарбил, или R5 и R6 или R11 и R12 могут быть связаны с гидрокарбиленом, замещенным гидрокарбиленом, гетерогидрокарбиленом или замещенным гетерогидрокарбиленом; X1-X6, каждый, независимо представляют собой галоген, -OR21, -OCOR22 или -NR23R24, в которых R21, R22, R23 или R24 представляют собой водород, гидрокарбил или гетерогидрокарбил; L представляет собой углеводород или гетероуглеводород; и ∗, и ∗∗ обозначают положение хирального углерода и каждый независимо имеют (S)- или (R)-конфигурацию.].
Как таковой гидрокарбил или гетерогидрокарбил обозначает радикал, имеющий одно положение для связывания, произведенный от углеводорода или гетероуглеводорода, а гидрокарбилен обозначает радикал, имеющий два положения для связывания, произведенный от углеводорода, в котором гетеро- означает, что углерод замещен атомом O, S или N.
В хиральном лиганде по настоящему изобретению хиральные углероды в положениях * и ** имеют конфигурационные пары (R,R), (R,S), (S,R), (S,S).
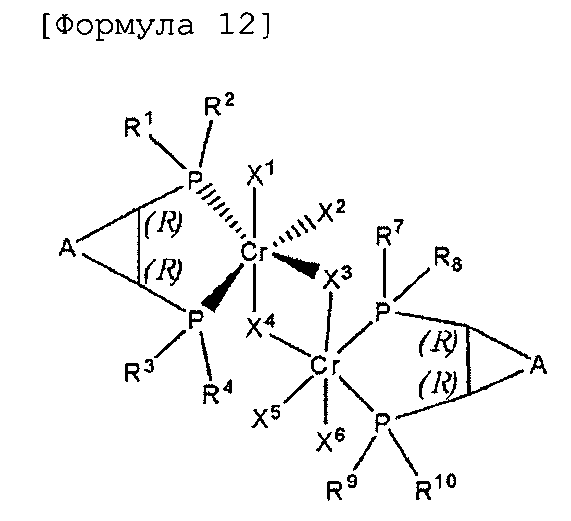
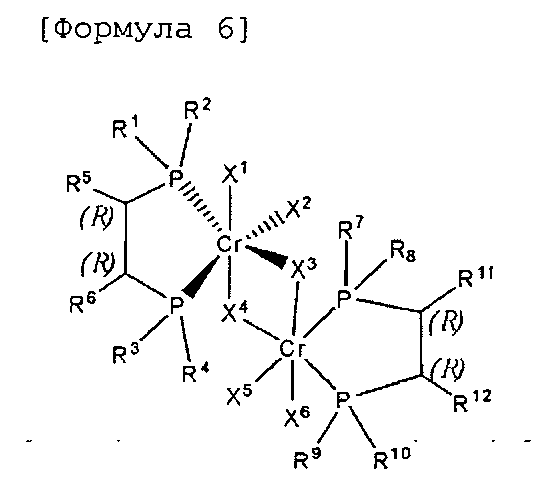
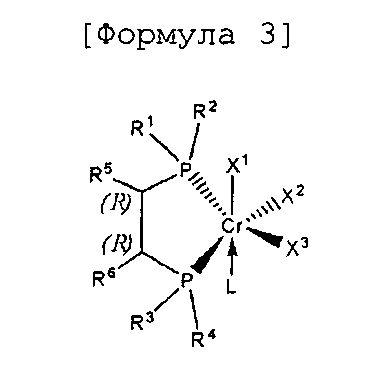
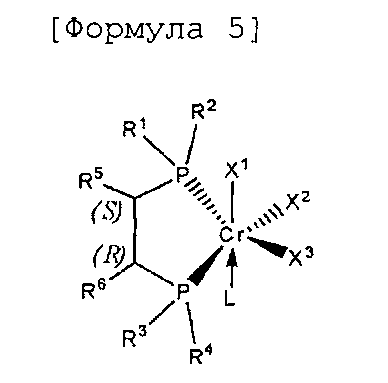
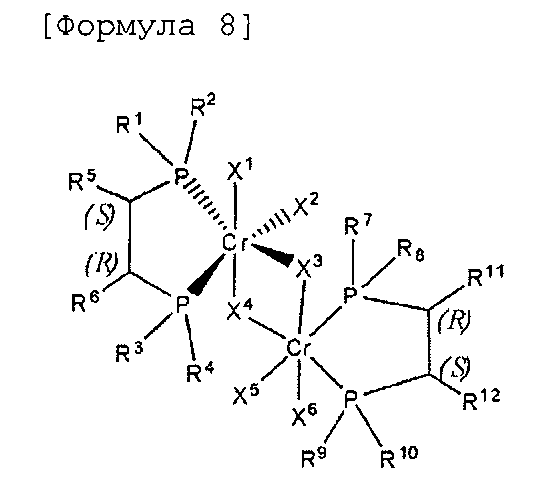
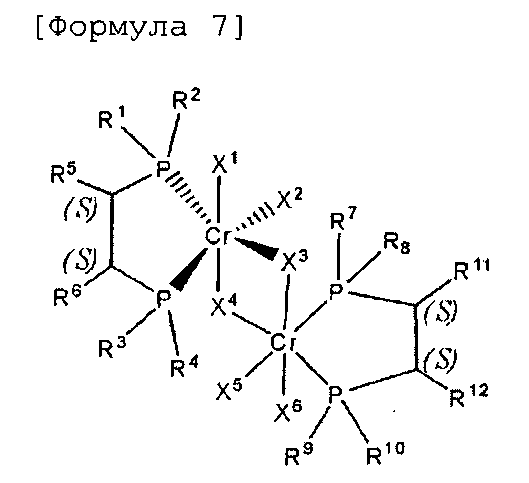
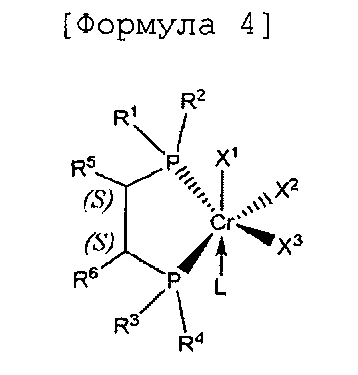
Согласно настоящему изобретению комплексное соединение хрома для селективной олигомеризации этилена формулы 1 или формулы 2 включает хиральный лиганд, представленный нижеприведенными формулами 3-8.
[Формула 3]
(R),(R)-Хирафос-моно, хромовый комплекс
[Формула 4]
(S),(S)-Хирафос-моно, хромовый комплекс
[Формула 5]
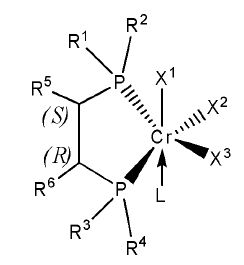
мезо-Хирафос-моно, хромовый комплекс
[Формула 6]
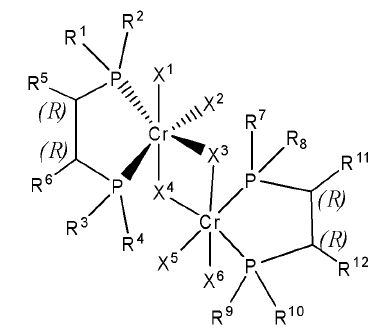
(R),(R)-Хирафос-бис, хромовый комплекс
[Формула 7]
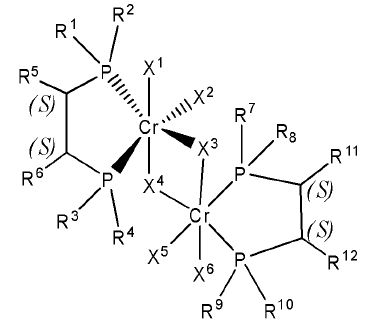
(S),(S)-Хирафос-бис, хромовый комплекс
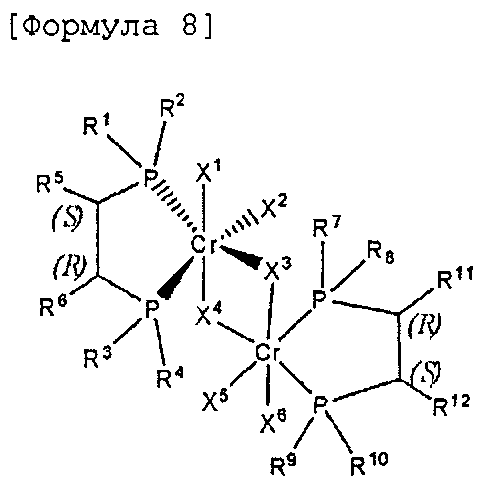
мезо-Хирафос-бис, хромовый комплекс
Как таковые, заместители формул 2-6 определены как в формулах 1 и 2.
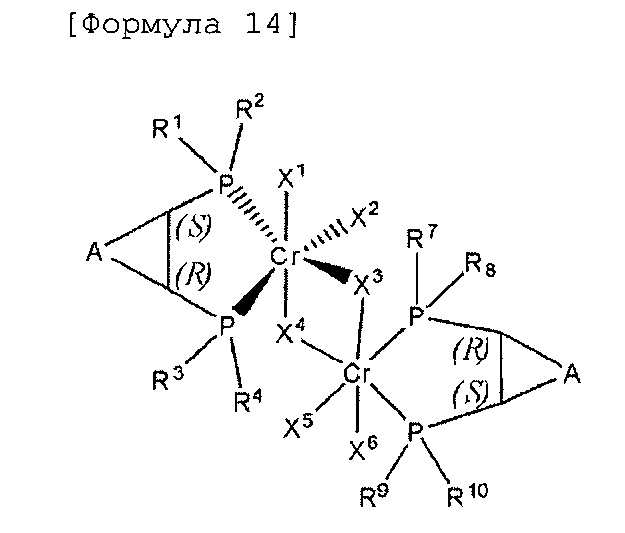
Комплексное соединение хрома для селективной олигомеризации этилена формулы 1 или 2 включает соединения нижеприведенных формул 9-14, когда R5 и R6 или R11 и R12 связаны с гидрокарбиленом, замещенным гидрокарбиленом, гетерогидрокарбиленом или замещенным гетерогидрокарбиленом.
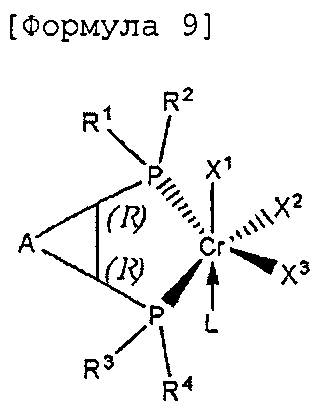
1,2-(R), (R)-транс-Циклофос-моно, хромовый комплекс
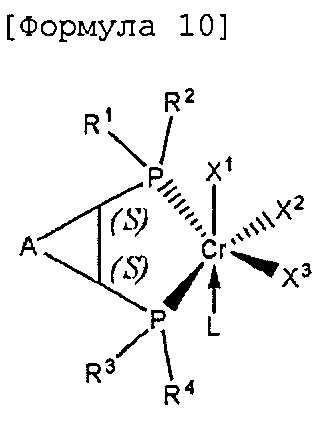
1,2-(S), (S)-транс-Циклофос-моно, хромовый комплекс
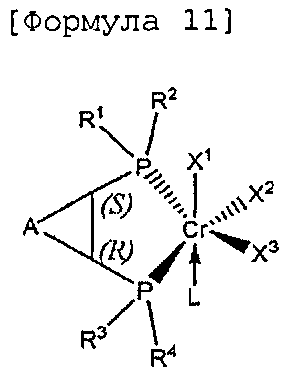
1,2-(R), (S)-цис-Циклофос-моно, хромовый комплекс

1,2-(R), (R)-транс-Циклофос-бис, хромовый комплекс
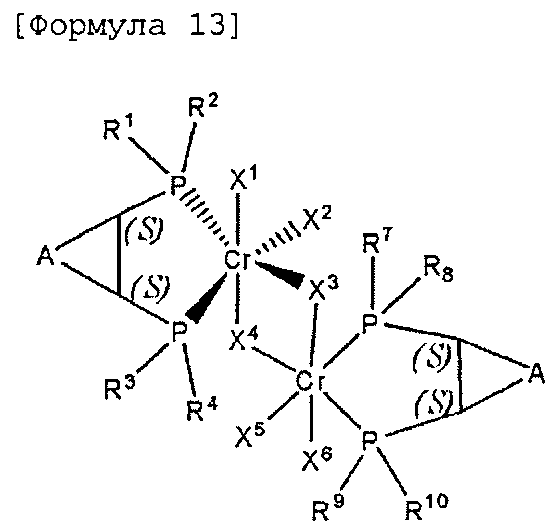
1,2-(S), (S)-транс-Циклофос-бис, хромовый комплекс
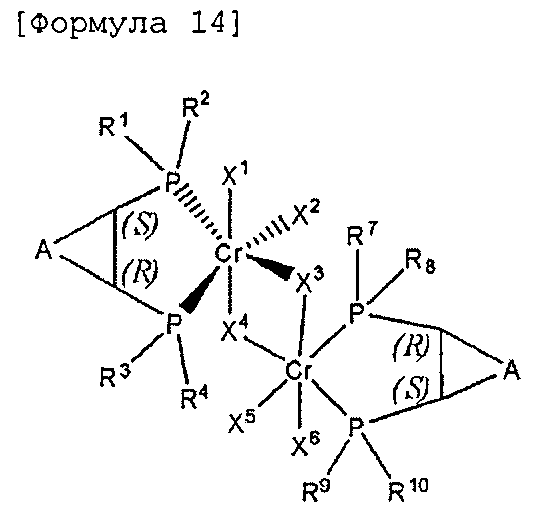
1,2-мезо-цис-Циклофос-бис, хромовый комплекс
В формулах 9-14 A выбран из (C1-C5) алкилена и (C1-C5) алкенилена, в которых включают такие алкилен и алкенилен в случае (C3-C5) алкилена и (C3-C5) алкенилена, которые образуют конденсированное кольцо со структурой, для которой смежным является (C5-C7) циклоалкил или (C6-C10) арил.
Заместители R1, R2, R3, R4, R7, R8, R9 и R10 хирального лиганда комплексного соединения хрома для селективной олигомеризации этилена, представленного формулами 1-14, каждый, независимо представляют собой (C6-C20) арил, (C6-C20)ар(C1-С10)алкил, (C1-C10)алкил, (C2-C10)алкенил, (C2-C10)алкинил, (C3-C7)циклоалкил, гетеро(C5-C20)арил, гетеро(C3-C7)циклоалкил или -NR23R24, в котором R23 или R24 представляет собой (C1-C10)алкил или (C6-C20)арил, и заместители R1, R2, R3, R4, R7, R8, R9 и R10 могут быть дополнительно замещены одним или более заместителями, выбранными из (C1-C10)алкила, (C1-C10)алкокси, (C6-C20)арилокси и галогена.
Конкретно, R1, R2, R3, R4, R7, R8, R9 и R10 хирального лиганда, каждый, независимо выбраны из группы, состоящей из фенила, нафтила, мезитила, метила, этила, этиленила, н-пропила, изопропила, пропенила, пропинила, н-бутила, трет-бутила, циклопропила, циклобутила, циклопентила, циклогексила, 4-метилциклогексила, 4-этилциклогексила, 4-изопропилциклогексила, бензила, толила, ксилила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила, 4-изопропоксифенила, кумила, метокси, этокси, фенокси, толилокси, диметиламино, тиометила, триметилсилила, диметилгидразила, 2-метилциклогексила, 2-этилциклогексила, 2-изопропилциклогексила, о-метилфенила, о-этилфенила, о-изопропилфенила, о-трет-бутилфенила, о-метоксифенила, о-изопропоксифенила, бифенила, нафтила и антраценила, и предпочтительно, каждый, независимо выбраны из группы, состоящей из фенила, бензила, нафтила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила, 4-изопропоксифенила, 2-метилциклогексила, 2-этилциклогексила, 2-изопропилциклогексила, о-метилфенила, о-этилфенила, о-изопропилфенила, о-трет-бутилфенила, о-метоксифенила и о-изопроксифенила.
В настоящем изобретении R5, R6, R11 и R12, которые связаны с хиральным углеродом хирального лиганда, содержащегося в комплексном соединении хрома для селективной олигомеризации этилена, каждый, независимо представляют собой (C6-C20)арил, (C6-C20)ар(C1-C10)алкил, (C1-C10)алкил, (C2-C10)алкенил, (C2-C10)алкинил, (C3-C7)циклоалкил, гетеро(C5-C20)арил, гетеро(C3-C7)циклоалкил, (C1-C10)алкокси, (C6-C20)арилокси, аминокарбонил, карбониламино, ди(C1-C10)алкиламино, (C1-C10)алкилсилил или (C6-C20)арилсилил, и заместители R5, R6, R11 и R12 могут быть замещены (C1-C10)алкилом, (C1-C10)алкокси, (C6-C20)арилокси и галогеном и предпочтительно выбраны из метила, этила, этиленила, н-пропила, изопропила, пропенила, пропинила, н-бутила, трет-бутила, изобутила, фенила, бензила, толила, ксилила, метокси, этокси, фенокси, метиламино и диметиламино.
В комплексном соединении хрома для селективной олигомеризации этилена X1, X2, X3, X4, X5 или X6, который координирован хромом, может быть выбран из Cl, Br, ацетоацетила и 2-этилгексаноила, но настоящее изобретение не ограничено ими.
В формуле 1 L, который координирован хромом, может быть выбран из тетрагидрофурана, диэтилового эфира, толуола, хлорбензола, дихлорбензола, ацетилацетона и 2-этилгексанона, и L является производным растворителей, которые могут являться средой для реакции между хиральным лигандом и солью хрома.
Примеры хирального лиганда комплексного соединения хрома для селективной олигомеризации этилена по настоящему изобретению включают следующие соединения, которые не ограничивают настоящее изобретение:
(S,S)-, (R,R)- или мезо-(фенил)2P-CH(метил)CH(метил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-метоксифенил)2P-CH(метил)CH(метил)-P(4-метоксифенил)2,
(S,S)-, (R,R)- или мезо-(4-метилфенил)2P-CH(метил)CH(метил)-P(4-метилфенил)2,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(метил)CH(метил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(этил)CH(метил)-P(4-этилфенил)2,
(S,S)-, (R,R)- или мезо-(4-метоксифенил)2P-CH(этил)CH(метил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(этил)CH(этил)-P(4-этилфенил)2,
(S,S)-, (R,R)- или мезо-(фенил)2P-CH(этил)CH(этил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(фенил)2P-CH(изопропил)CH(метил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-метоксифенил)2P-CH(изопропил)CH(метил)-P(4-метоксифенил)2,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(изопропил)CH(метил)-P(4-этилфенил)2,
(S,S)-, (R,R)- или мезо-(фенил)2P-CH(н-пропил)CH(метил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-метоксифенил)2P-CH(н-пропил)CH(метил)-P(4-метоксифенил)2,
(S,S)-, (R,R)- или мезо-4-(4-этилфенил)2P-CH(н-пропил)CH(метил)-P(4-этилфенил)2,
(S,S)-, (R,R)- или мезо-(фенил)2P-CH(изопропил)CH(этил)-P(фенил)2,
(S,S)-, (R,R)- или мезо-(4-метоксифенил)2P-CH(изопропил)CH(этил)-P(4-метоксифенил)2,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(изопропил)CH(этил)-P(4-этилфенил)2,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(фенил)2)циклогексан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(4-метоксифенил)2)циклогексан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(4-этилфенил)2)циклогексан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(фенил)2)циклопентан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(4-метоксифенил)2)циклопентан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(4-этилфенил)2)циклопентан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(фенил)2)пиррол,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(4-метоксифенил)2)пиррол,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(4-этилфенил)2)пиррол,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(4-этилфенил)2)имидазол,
(S,S)-, (R,R)- или мезо-(4-этилфенил)2P-CH(диметиламин)CH(диметиламин)-P(4-этилфенил)2,
(S,S)-, (R,R)- или мезо-(3-метоксифенил)2P-CH(метил)CH(метил)-P(3-метоксифенил)2,
(S,S)-, (R,R)- или мезо-(4-этоксифенил)2P-CH(метил)CH(метил)-P(о-этоксифенил)2,
(S,S)-, (R,R)- или мезо-(4-диметиламинофенил)2P-CH(метил)CH(метил)P(4-диметиламинофенил)2,
(S,S)-, (R,R)- или мезо-(4-этилциклогексил)2PCH(метил)CH(метил)P(4-этилциклогексил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(метил)CH(метил)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-изопропилфенил)2PCH(метил)CH(метил)P(2-изопропилфенил)2,
(S,S)-, (R,R)- или мезо-(2-метилфенил)2PCH(метил)CH(метил)P(2-метилфенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(метил)CH(метил)P(фенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(этил)CH(метил)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(этил)CH(этил)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(изопропил)CH(метил)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(н-пропил)CH(метил)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(изопропил)CH(этил)P(2-этилфенил)2,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(2-этилфенил)2)циклогексан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-1,2-ди-(P(2-этилфенил)2)циклопентан,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(2-этилфенил)2)пиррол,
(S,S)-транс-, (R,R)-транс- или мезо-цис-3,4-ди-(P(2-этилфенил)2)имидазол,
(S,S)-, (R,R)- или мезо-(2-этилфенил)2PCH(диметиламино)CH(диметиламино)P(2-этилфенил)2,
(S,S)-, (R,R)- или мезо-(2-метоксифенил)2PCH(метил)CH(метил)P(2-метоксифенил)2,
(S,S)-, (R,R)- или мезо-(2-этоксифенил)2PCH(метил)CH(метил)P(2-этоксифенил)2,
(S,S)-, (R,R)- или мезо-(2-диметиламинофенил)2PCH(метил)CH(метил)P(2-диметиламинофенил)2 и
(S,S)-, (R,R)- или мезо-(2-этилциклогексил)2PCH(метил)CH(метил)P(2-этилциклогексил)2.
Хиральные лиганды по настоящему изобретению могут быть получены с использованием различных способов, известных специалистам в данной области.
Стереоизомерная структура со скелетом P-C-C-P-типа лиганда по настоящему изобретению отличается от структуры известного традиционного гетеролиганда (R)nPN(R')P(R)m, и гетероатомом в структуре скелета лиганда является только фосфор (P). Конкретно, лиганд, используемый в каталитической системе по настоящему изобретению, представлен в форме структуры с углерод-углеродным скелетом без атома N между двумя атомами, P и таким образом пространственная структура должным образом приспосабливается в направлении расположения заместителей, присоединенных к атомам C, тем самым показывая превосходную каталитическую активность, достигающую высокой селективности 70% мас. или более по 1-гексену или 1-октену, и сохраняя стабильной реакционную активность.
Также, комплекс переходного металла, координированный хиральным лигандом, имеющим структуру с P-C-C-P-скелетом, может быть модифицирован с тем, чтобы быть прикрепленным к полимерной цепи, чтобы придать ему нерастворимость при комнатной или при более высокой температуре. Более того, данный комплекс может быть закреплен на основе, такой как диоксид кремния, силикагель, полисилоксан или оксид алюминия.
Кроме того, чтобы достичь более эффективной активности и более высокой селективности, настоящее изобретение предоставляет каталитическую композицию на основе комплекса хрома для селективной олигомеризации этилена, включающую вышеназванный катализатор на основе комплекса хрома для селективной олигомеризации этилена и известный сокатализатор.
Сокатализатор, использованный в каталитической композиции по настоящему изобретению, может представлять собой любое соединение, которое активирует комплекс переходного металла, координированный хиральным лигандом, имеющим структуру с P-C-C-P-скелетом. Активатор может быть использован также в смеси. Соединение, принятое для использования в качестве активатора, включает алюминийорганическое соединение, борорганическое соединение или органическую соль.
Примеры алюминийорганического соединения, подходящего для использования в качестве активатора в каталитической системе по настоящему изобретению, включают AlR3 (R представляет собой независимо (C1-C12) алкил, кислородсодержащий (C1-C12) алкил или галоген) или LiAlH4.
В каталитической композиции по настоящему изобретению сокатализатор включает триметилалюминий (ТМА), триэтилалюминий (TEA), триизобутилалюминий (TIBA), три-н-октилалюминий, дихлорид метилалюминия, дихлорид этилалюминия, хлорид диметилалюминия, хлорид диэтилалюминия, изопропоксид алюминия, сесквихлорид этилалюминия, сесквихлорид метилалюминия и алюмоксан.
Как обычно принято в данной области, алюмоксан широко известен в качестве олигомерного соединения, которое может быть получено надлежащим смешением воды и алкилалюминиевого соединения, например триметилалюминия. Полученное алюмоксановое олигомерное соединение может представлять собой линейное соединение, циклическое соединение, клеточное соединение или их сочетания.
Подходящие примеры борорганического соединения включают бороксин, NaBH4, триэтилборан, трифенилборан, аммонийные комплексы трифенилборана, трибутилборат, триизопропилборат, трис (пентафторфенил)боран, тритил(тетрапентафторфенил)борат, (тетрапентафторфенил)борат диметилфениламмония, (тетрапентафторфенил)борат диэтилфениламмония, (тетрапентафторфенил)борат метилдифениламмония или (тетрапентафторфенил)борат этилдифениламмония. Данное борорганическое соединение может быть использовано в смеси с вышеупомянутым алюминийорганическим соединением.
В сокатализаторе алюмоксан может быть выбран из алкилалюмоксана, например метилалюмоксана (МАО) и
этилалюмоксана (ЕАО), и модифицированного алкилалюмоксана, например модифицированного метилалюмоксана (ММАО).
Модифицированный метилалюмоксан (получаемый Akzo Nobel) содержит гибридную алкильную группу, такую как изобутильную или н-октильную группу, в дополнение к метильной группе.
Особенно подходящим в качестве сокатализатора является метилалюмоксан (МАО) или этилалюмоксан (ЕАО).
Комплекс хрома для селективной олигомеризации этилена и алюмоксан смешивают так, что молярное отношение алюминия к хрому находится в диапазоне от 1:1 до 10000:1, предпочтительно от примерно 1:1 до 1000:1.
Кроме того, настоящее изобретение дает способ получения олигомера, в частности 1-гексена или 1-октена с высокой активностью и высокой селективностью, который включает получение комплексного соединения хрома формулы 1 или 2 для селективной олигомеризации этилена, включающего предшественник переходного металла, координированный хиральным лигандом, имеющим структуру с P-C-C-P-скелетом, и добавление комплексного соединения хрома, полученного таким образом, к среде олигомеризации этилена, так что этилен олигомеризуется с высокой активностью и высокой селективностью, в котором лиганд с P-C-C-P-скелетом, принятый для использования с целью достижения высокой активности и высокой селективности, может являться (S,S)-, (R,R)- или мезо-изомерным линейным, транс- или цис-циклическим. Также подходящим в виде смеси некоторых изомеров может являться мультилиганд, в котором (S,S)-, (R,R)- или мезо-(R1)(R2)P-(R5)CHCH(R6)-P(R3)(R4)-лиганды связаны друг с другом.
Комплекс хрома для селективной олигомеризации этилена и сокатализатор, которые представляют собой индивидуальные компоненты каталитической системы, раскрытой в настоящем изобретении, могут быть смешаны одновременно или последовательно в любом порядке в присутствии или в отсутствие растворителя, с получением таким образом активного катализатора. Данные компоненты катализатора могут быть смешаны при температуре в диапазоне от -20 до 250°C. В ходе смешивания компонентов катализатора олефин может типично проявлять защитные эффекты, что имеет результатом улучшенные каталитические характеристики. Предпочтительно, смешивание проводят при температуре в диапазоне от 20 до 100°C.
Продукт реакции, раскрытый в настоящем изобретении, то есть олигомер этилена, в частности 1-гексен или 1-октен, может быть получен с использованием комплексного соединения хрома для селективной олигомеризации этилена по настоящему изобретению посредством типичного аппарата и каталитической технологии в присутствии или в отсутствие инертного растворителя гомогенной жидкостной реакцией, реакцией в суспензии, в которой каталитическая система частично или полностью растворена, двухфазной реакцией в среде жидкость/жидкость, реакцией в массе, в которой олефиновый продукт действует в качестве главной среды, или газовой реакцией.
Способ селективного получения олигомера по настоящему изобретению может быть осуществлен в присутствии инертного растворителя. Конкретно, может быть использован любой инертный растворитель, который не реагирует с каталитическим соединением и активатором, и инертный растворитель может включать любые насыщенные алифатические и ненасыщенные алифатические и ароматические углеводороды и галогениды углеводородов. Типичные примеры растворителя включают, но без ограничения, бензол, толуол, ксилол, хлорбензол, кумол, гептан, циклогексан, метилциклогексан, метилциклопентан, н-гексан, 1-гексен, 1-октен и так далее. В частности, когда использовано соединение формулы 1, то же соединение, что лиганд L, предпочтительно используют в качестве растворителя для реакции.
В способе получения по настоящему изобретению олигомеризация может быть проведена при температуре в диапазоне от -20 до 250°C, предпочтительно от 15 до 130°C и более предпочтительно от 30 до 70°C.
Более того, способ по настоящему изобретению осуществляют при давлении от атмосферного давления до 500 бар, предпочтительно от 10 до 70 бар и более предпочтительно от 30 до 50 бар.
В одном варианте осуществления настоящего изобретения координационный комплекс стереоизомерного лиганда с P-C-C-P-скелетом и условия реакции выбраны так, чтобы выход 1-гексена из этилена составлял 50% мас. или более и предпочтительно 70% мас. или более. В данном случае выход указывает количество граммов образовавшегося 1-гексена на 100 г всего продукта реакции.
В другом варианте осуществления настоящего изобретения координационный комплекс стереоизомерного лиганда с P-C-C-P-скелетом и условия реакции выбраны так, чтобы выход 1-октена из этилена составлял 30% мас. или более и предпочтительно 50% мас. или более. В данном случае выход указывает количество граммов образовавшегося 1-октена на 100 г всего продукта реакции.
В способе по настоящему изобретению в зависимости от лиганда с P-C-C-P-скелетом и условий реакции могут быть получены не только 1-гексен или 1-октен, но также различные количества 1-бутена, 1-гексена, метилциклопентана, метиленциклопентана, пропилциклопентана и ряда более высоких олигомеров и полиэтиленов.
Способ по настоящему изобретению может быть осуществлен с использованием установки, включающей любой тип реактора. Примеры реактора могут включать, но без ограничения, реактор периодического действия, реактор полупериодического действия и реактор непрерывного действия. Установка может включать сочетание реактора, впуска для ввода олефина и каталитической системы в реактор, линию для выпуска олигомеризованного продукта реакции из реактора и по меньшей мере один сепаратор для сепарации олигомеризованного продукта реакции, в которой каталитическая система может включать соединение переходного металла, активатор и координационный комплекс P-C-C-P-лиганда, раскрытый в настоящем описании.
В настоящем изобретении 1-гексен или 1-октен могут быть получены с высокой активностью и высокой селективностью олигомеризацией этилена с использованием каталитической системы олигомеризации этилена по настоящему изобретению.
Обеспечивающие преимущество эффекты
Когда лиганд и металлический хром или предшественник хрома по отдельности добавляют в реакционную среду олигомеризации, значительное количество лиганда не может приблизиться к атому хрома или не может нормально координироваться, что, и это нежелательно, делает невозможным получение активности и селективности, подходящих для промышленного применения. Однако по настоящему изобретению случай, когда этилен олигомеризуется с использованием комплексного соединения хрома для селективной олигомеризации этилена, имеющего хиральную структуру с P-C-C-P-скелетом, обеспечивает преимущество, поскольку используется катализатор, в котором хиральный лиганд нормально координирован хромом, реакционная активность повышена в 10 раз или более и повышена селективность, так что количество полимерного побочного продукта снижено до 1/10 или менее, и, следовательно, количество полимерного побочного продукта по завершении реакции может быть эффективно снижено до примерно 0,1% мас., что таким образом упрощает процесс очистки и тем самым позволяет выгодно осуществлять производственный процесс. Более того, количество дорогостоящего сокатализатора, такого как MAO, необходимого для активации катализатора, может быть снижено до 1/10 или более, что имеет результатом экономический производственный процесс.
Описание чертежей
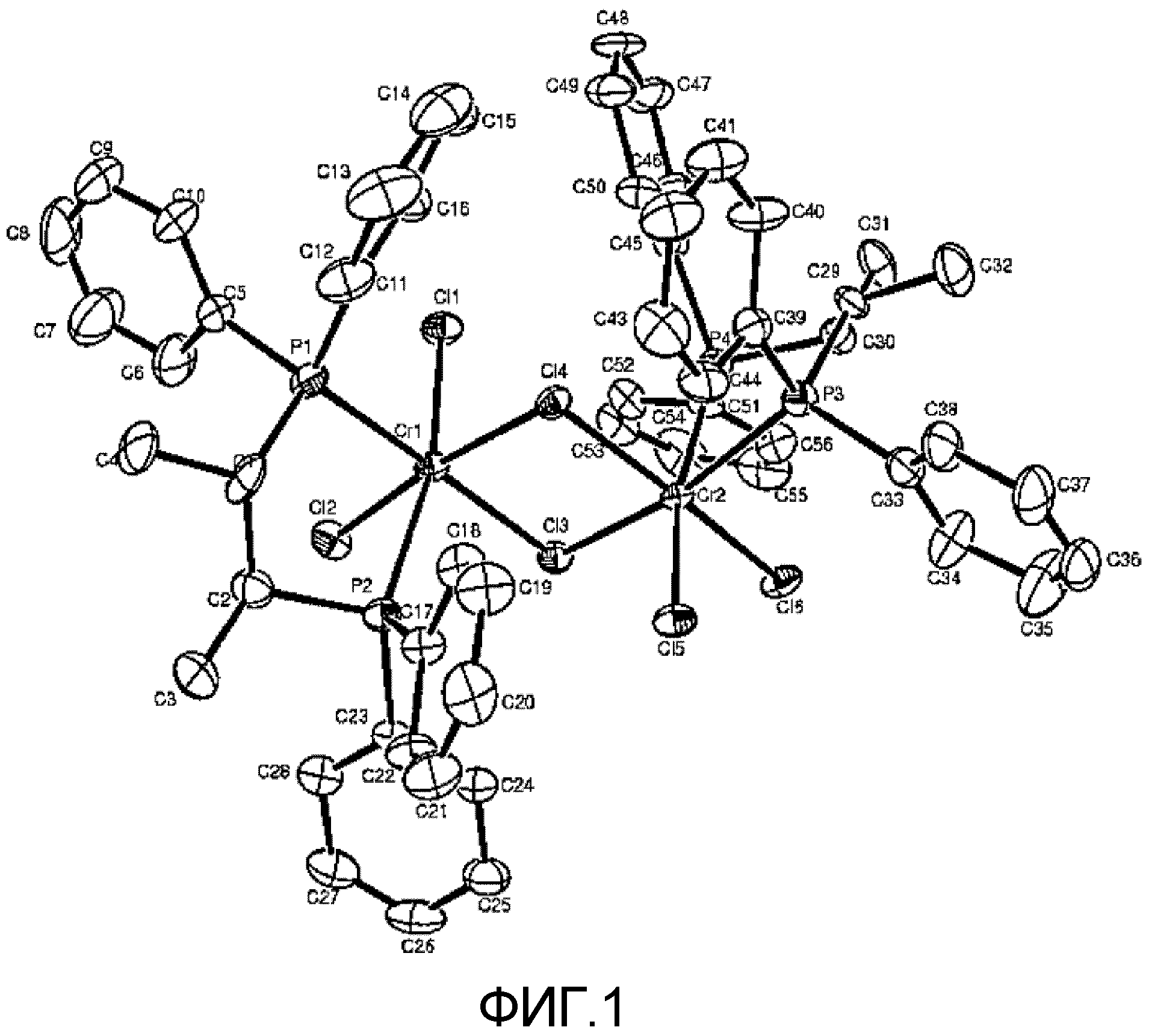
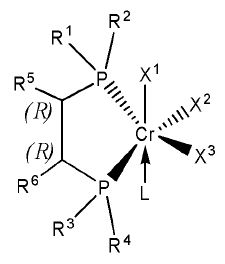
На Фиг.1 изображена структура, определенная рентгеновской дифракцией, соединения, полученного в примере 2 получения катализатора; и
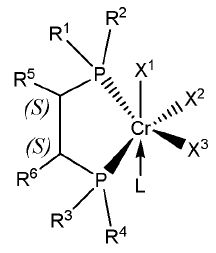
на Фиг.2 изображена структура, определенная рентгеновской дифракцией, соединения, полученного в примере 5 получения катализатора.
Варианты осуществления изобретения
Лучшее понимание настоящего изобретения может быть получено посредством следующих примеров, которые приведены для иллюстрации, но не истолковываются в качестве ограничивающих настоящее изобретение.
[Пример получения]
[Пример 1 получения катализатора] Получение (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - трихлорид хрома - тетрагидрофуран [CrCl3(THF){(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]
A. Получение лиганда (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 [(S,S)-Ph2PCH(Me)CH(Me)PPh2]
Хиральный лиганд [(S,S)-Ph2PCH(Me)CH(Me)PPh2] получали, как раскрыто в статье B. Bosnich et al., J. Am. Chem. Soc. 99(19) (1977) 6262. Ди-п-толуолсульфонат (2R,3R)-бутандиола получали из (2R,3R)-бутандиола. Данный способ осуществляли, как раскрыто в статье R.B. Mitra et al., J. Am. Chem. Soc. 84 (1962). В колбу на 1 л, охлаждаемую в бане со льдом, добавляли 100 мл (1,24 моль) сухого пиридина и затем смешивали со 100 г (0,525 моль) п-толуолсульфонилхлорида, после чего медленно по каплям добавляли 22 мл (0,245 моль) (2R,3R)-бутандиола. Смесь нагревали до комнатной температуры в течение 20 мин и затем оставляли стоять в состоянии полутвердой фазы при комнатной температуре в течение 12 часов. Избыток льда в кусках добавляли к смеси и колбу энергично встряхивали, чтобы смесь не превратилась в конгломерат. Контролировали, происходило ли медленное осаждение порошкообразных кристаллов, после чего смесь перемешивали в течение 2 часов вместе с кусками льда и затем к смеси добавляли куски дробленого льда и 70 мл раствора концентрированной соляной кислоты при энергичном перемешивании. Отделившуюся суспензию фильтровали, тщательно промывали водой и сушили, получая таким образом 85 г (86,3%) продукта ди-п-толуолсульфоната (2R,3R)-бутандиола (т.пл. 62-64°C).
С другой стороны, 95 г перекристаллизованного трифенилфосфора и 300 мл сухого тетрагидрофурана (THF) добавляли в трехгорлую круглодонную колбу на 1 л, оборудованную капельной воронкой на 250 мл, обратным холодильником и вводом азота. К раствору добавляли 5,0 г тонких кусков лития при 25°C в атмосфере азота при перемешивании, что таким образом приводило к образованию в растворе LiPPh2. Как таковой, раствор приобретал глубокий красновато-желтый цвет, причем выделялось большое количество теплоты. Температуру раствора медленно повышали до 55°C в течение 1 часа и раствор вновь охлаждали до 25°C в течение 2 часов при перемешивании. Образовавшийся фениллитий разлагали добавлением по каплям 33 г перегнанного и очищенного трет-бутилхлорида в течение 45 мин. Прозрачный красновато-желтый раствор кипятили в течение 5 мин и затем снова охлаждали до -4°C.
К охлажденному раствору по каплям в течение 1 часа добавляли 35 г полученного выше ди-п-толуолсульфоната (2R,3R)-бутандиола, растворенного в 100 мл сухого THF. Раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 мин. Затем добавляли к нему 300 мл продутой азотом воды и THF удаляли отгонкой при пониженном давлении, выделяя таким образом бесцветный маслообразный продукт. Продукт экстрагировали дважды 150 мл эфира и затем сушили Na2SO4. Эфирный экстракт фильтровали с раствором 15 г гексагидрата перхлората никеля в 50 мл этанола в атмосфере азота. Na2SO4, оставшийся на фильтре, тщательно промывали эфиром и затем эфирный раствор добавляли к никелевому раствору. Красновато-коричневый маслообразный продукт, который имел желтые кристаллы, представлял собой [Ni((S,S)-хирафос)2](ClO4)2. Смесь масла и кристаллов добавляли к раствору 15 г тиоцианата натрия (NaNCS) в 50 мл горячего этанола и раствор энергично перемешивали в течение нескольких часов до образования однородного желтовато-коричневого твердого вещества [Ni((S,S)-хирафос)2NCS]NCS. Данный твердый продукт полностью промывали этанолом и затем промывали эфиром.
15 г полученного таким образом никелевого комплекса суспендировали в 150 мл этанола под азотом и нагревали при перемешивании. 4 г цианида натрия (NaCN) быстро добавляли к 20 г воды. Никелевый комплекс медленно растворялся, давая прозрачный красный раствор иона [Ni((S,S)-хирафос)2CN3]‾, который затем превращался в мутный бежевого цвета раствор. Горячий раствор перемешивали до образования желтой суспензии. Раствор с суспензией охлаждали и твердое вещество непрерывно промывали дважды 25 мл воды и затем быстро охлаждали охлажденным льдом этанолом. Содержащее примеси, имеющее бежевый цвет твердое вещество сушили при 25°C, добавляли к 125 мл кипящего безводного этанола и затем фильтровали, используя фильтр Фрица. Фильтрование по Фрицу проводили при комнатной температуре в течение 12 часов, получая таким образом бесцветное блестящее твердое вещество. Затем твердое вещество перекристаллизовывали из 60 мл безводного этанола, получая тем самым 5,5 г совершенно бесцветного чистого лиганда (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2.
B. Получение (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - трихлорид хрома - тетрагидрофуран [CrCl3(THF){(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]
1,1 г (3,0 ммоль) трис-тетрагидрофурантрихлорида хрома (CrCl3(THF)3) растворяли в 100 мл тетрагидрофурана и затем медленно добавляли к этому раствору раствор 1,28 г (3,0 ммоль) полученного выше лигандного соединения (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 в 50 мл тетрагидрофурана и перемешивали образовавшийся реакционный раствор при комнатной температуре. Реакционный раствор дополнительно перемешивали в течение 1 часа и летучие соединения удаляли из него в вакууме, после чего к продукту реакции по каплям добавляли 100 мл петролейного эфира, получая таким образом осадок голубого твердого вещества. Проводили две промывки 100 мл петролейного эфира, что давало 1,77 г (выход 90%) продукта.
[Пример 2 получения катализатора] Получение бис-[(S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(µ-Cl){(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
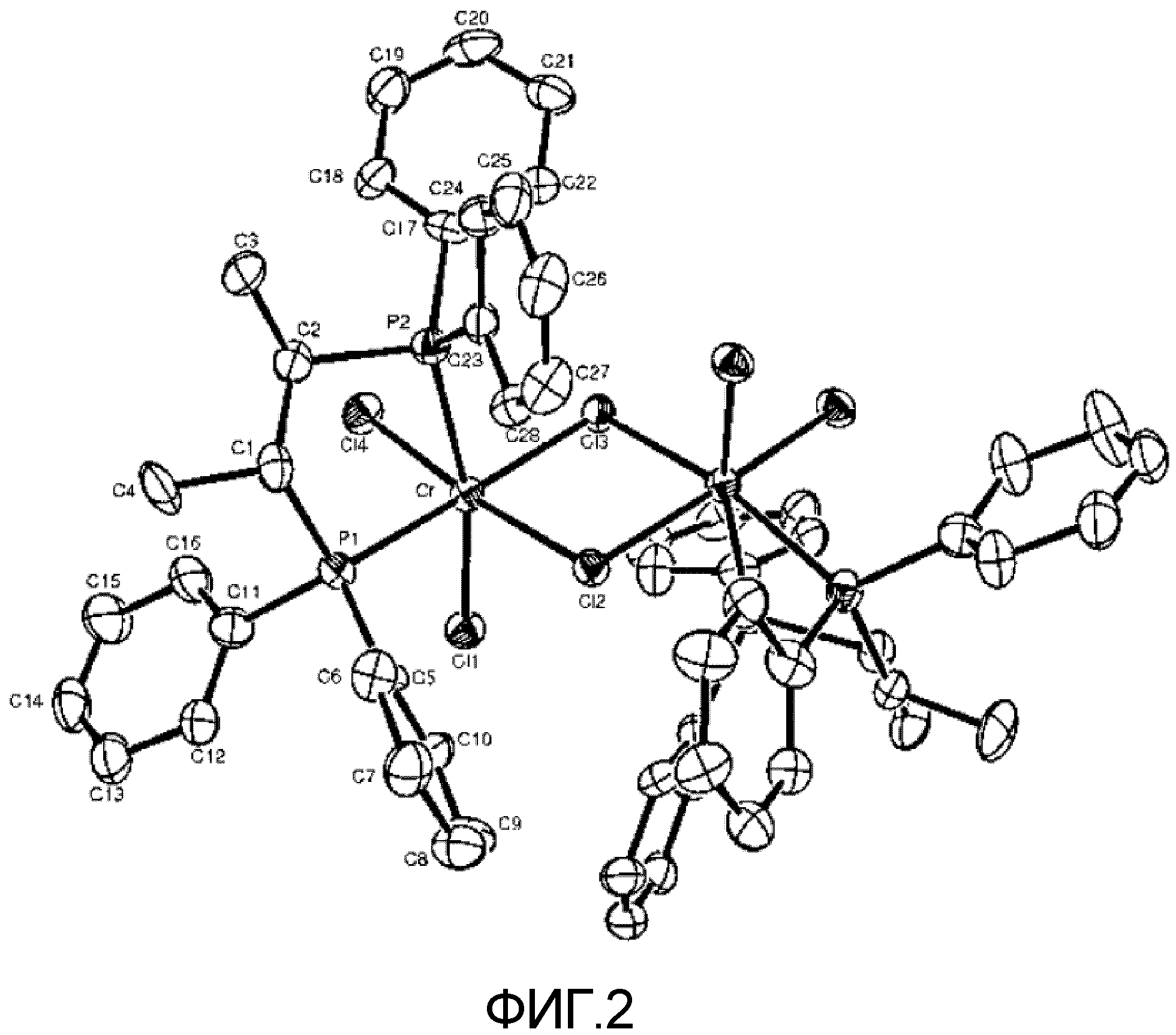
1,1 г (3,0 ммоль) трис-тетрагидрофурантрихлорида хрома (CrCl3(THF)3) растворяли в 100 мл дихлорметана и затем медленно добавляли к этому раствору раствор 1,28 г (3,0 ммоль) лигандного соединения (S, S)-(фенил)2PCH(метил)CH(метил)P(фенил)2, полученного в примере 1 получения катализатора, в 50 мл дихлорметана. Образовавшийся реакционный раствор дополнительно перемешивали в течение 1 часа и затем летучие соединения удаляли из него в вакууме. К продукту по каплям добавляли 100 мл петролейного эфира, получая таким образом голубой твердый осадок, который затем дважды промывали 100 мл петролейного эфира, что давало 1,58 г (выход 90%) названного соединения. Полученное таким образом комплексное соединение анализировали, используя рентгеновскую дифракцию на монокристалле. Его структура показана на Фиг.1.
[Пример 3 получения катализатора] Получение [(S,S)-(фенил)2РСН (метил)CH(метил)P(фенил)2 - диацетилацетонат хрома] [Cr{(O,O)-k2-(CH3COCH2COCH3)2}{(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]
1,62 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,1 г (3,0 ммоль) триацетилацетоната хрома (Cr(acac)3) использовали вместо трис-тетрагидрофурантрихлорида хрома (CrCl3(THF)3).
[Пример 4 получения катализатора] Получение [(S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - ди(2-этилгексаноат)хрома] [Cr{(OOCCH(C2H5)C4H9)2}{(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]
1,82 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,44 г (3,0 ммоль) три(2-этил)гексаноата хрома (Cr(OOCCH(C2H5)C4H9)3) использовали вместо трис-тетрагидрофурантрихлорида хрома (CrCl3(THF)3).
[Пример 5 получения катализатора] Получение бис-[(R,R)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(R,R)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
A. Получение лиганда (R,R)-(фенил)2PCH(метил)CH(метил)P(фенил)2 [(R,R)-Ph2PCH(Me)CH(Me)PPh2]
5,1 г названного соединения получали так же, как в примере 1 получения катализатора, за исключением того, что (2S,3S)-бутандиол использовали в качестве исходного материала вместо (2R,3R)-бутандиола.
B. Получение бис-[(R,R)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(R,R)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
1,58 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,28 г (3,0 ммоль) лигандного соединения (R,R)-(фенил)2PCH(метил)CH(метил)P(фенил)2 использовали вместо (S,S)-лигандного соединения. Полученное таким образом комплексное соединение анализировали, используя рентгеновскую дифракцию на монокристалле. Его структура показана на Фиг.2.
[Пример 6 получения катализатора] Получение бис-[мезо-(фенил)2PCH(метил)CH(метил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-мезо-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
A. Получение лиганда мезо-(фенил)2PCH(метил)CH(метил)P(фенил)2 [мезо-Ph2PCH(Me)CH(Me)PPh2]
5,7 г совершенно бесцветного чистого мезо-(фенил)2PCH(метил)CH(метил)P(фенил)2 получали так же, как в примере 1 получения катализатора, за исключением того, что мезо-бутандиол использовали в качестве исходного материала вместо (2R,3R)-бутандиола.
B. Получение бис-[мезо-(фенил)2PCH(метил)CH(метил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-мезо-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
1,43 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,28 г (3,0 ммоль) лигандного соединения мезо-(фенил)2PCH(метил)CH(метил)P(фенил)2 использовали вместо (S,S)-лигандного соединения.
[Пример 7 получения катализатора] Получение бис-[(R,R)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(R,R)-((4-MeOPh)2P(Me)CH-CH(Me)P(4-MeOPh)2)}]2
A. Получение лиганда (R,R)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2 [(R,R)-(4-MeOPh)2PCH(Me)CH(Me)P(4-MeOPh)2]
Названное соединение получали, как раскрыто в статье B. Bosnich et al., J. Am. Chem. Soc. 99(19) (1977).
Получение ди-п-толуолсульфоната (2S,3S)-бутандиола из (2S,3S)-бутандиола проводили так же, как в примере 1 получения катализатора.
Три(4-метоксифенил)фосфор получали следующим образом. Кусочки магния (91,1 г, 3,75 моль) добавляли порциями к 95 мл (0,75 моль) 4-броманизола в 2 л THF. По завершении интенсивной реакции реакционную смесь нагревали при кипении с обратным холодильником в течение 2 часов с получением реактива Гриньяра. Реактив Гриньяра добавляли по каплям к раствору 17,5 мл (0,2 моль) PCl3 в 2 л THF при -78°C в течение 2 часов при перемешивании. По завершении добавления по каплям удаляли баню с сухим льдом/ацетоном и реакционный раствор нагревали до комнатной температуры. Реакционный раствор перемешивали в течение ночи и в вакууме удаляли из него растворитель. Продукт фосфина полностью использовали на последующей стадии без извлечения.
В трехгорлую круглодонную колбу на 1 л, оборудованную капельной воронкой на 250 мл, обратным холодильником и вводом азота добавляли 70 г перекристаллизованного три(4-метоксифенил)фосфора и 300 мл сухого THF. К раствору добавляли 2,8 г тонких кусков лития при 25°C под азотом при перемешивании. В растворе немедленно образовывался Li(4-OMe-PPh)2, и раствор приобретал глубокий красновато-желтый цвет, причем выделялось большое количество теплоты. Температуру раствора медленно повышали до 55°C в течение 1 часа и раствор вновь охлаждали до 25°C в течение 2 часов при перемешивании. Образовавшийся 4-метоксифениллитий разлагали добавлением по каплям 18,5 г перегнанного и очищенного трет-бутилхлорида в течение 45 мин. Прозрачный красновато-желтый раствор кипятили в течение 5 мин и затем снова охлаждали до -4°C.
К охлажденному раствору по каплям в течение 1 часа добавляли 19,6 г полученного выше ди-п-толуолсульфоната (2S,3S)-бутандиола, растворенного в 100 мл сухого THF. Раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 мин. Затем добавляли к нему 300 мл продутой азотом воды, после чего удаляли THF отгонкой при пониженном давлении, выделяя таким образом бесцветный маслообразный продукт. Продукт экстрагировали дважды 150 мл эфира и затем сушили Na2SO4. Эфирный экстракт фильтровали с раствором 8,4 г гексагидрата перхлората никеля в 50 мл этанола в атмосфере азота. Na2SO4, оставшийся на фильтре, тщательно промывали эфиром и затем эфирный раствор добавляли к никелевому раствору. Красновато-коричневый маслообразный продукт представлял собой [Ni((2R,3R)-бис(ди-п-метоксифенил)фосфорбутан)2](ClO4)2. Смесь масла и кристаллов добавляли к 8,4 г тиоцианата натрия (NaNCS), растворенного в 50 мл горячего этанола, и раствор интенсивно перемешивали в течение нескольких часов до образования однородного желтовато-коричневого твердого вещества [Ni((2R,3R)-бис(ди-п-метоксифенил)фосфорбутан)2NCS]NCS. Твердый продукт полностью промывали этанолом и затем промывали эфиром.
17 г никелевого комплекса суспендировали в 150 мл этанола под азотом и нагревали при перемешивании. Раствор 4 г цианида натрия (NaCN) в 20 г воды быстро добавляли к суспензии никелевого комплекса. Никелевый комплекс медленно растворялся, давая прозрачный красный раствор иона [Ni((2R,3R)-бис(ди-п-метоксифенил)фосфорбутан)2CN3]‾, который затем превращался в мутный бежевого цвета раствор. Горячий раствор перемешивали до образования желтой суспензии. Раствор с суспензией охлаждали и твердое вещество непрерывно промывали дважды 25 мл воды и быстро охлаждали охлажденным льдом этанолом. Содержащее примеси твердое вещество бежевого цвета сушили при 25°C, добавляли к 125 мл кипящего безводного этанола и затем фильтровали, используя фильтр Фрица. Фильтрование по Фрицу проводили при комнатной температуре в течение 12 часов, посредством чего фильтрат полностью удалялся и оставалось лишь бесцветное блестящее твердое вещество. Твердое вещество перекристаллизовывали из 60 мл безводного этанола, что давало 6,2 г совершенно бесцветного чистого (R,R)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2.
B. Получение бис-[(R,R)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(R,R)-((4-MeOPh)2P(Me)CH-CH(Me)P(4-MeOPh)2)}]2
1,29 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,64 г (3,0 ммоль) (R,R)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2 использовали вместо (S,S)-(4-метоксифенил)2PCH(метил)CH(метил)P(4-метоксифенил)2.
[Пример 8 получения катализатора] Получение бис-[(S,S)-(4-метилфенил)2PCH(метил)CH(метил)P(4-метилфенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(S,S)-((4-MePh)2P(Me)CH-CH(Me)P(4-MePh)2)}]2
A. Получение лиганда (S,S)-(4-метилфенил)2PCH(метил)CH(метил)P(4-метилфенил)2 [(S,S)-(4-MePh)2PCH(Me)CH(Me)P(4-MePh)2]
3,9 г совершенно бесцветного чистого (S,S)-(4-метилфенил)2P-CH(метил)CH(метил)P(4-метилфенил)2 получали так же, как в примере 7 получения катализатора, за исключением того, что 4-толилбромид использовали для получения три(4-метилфенил)фосфора.
B. Получение бис-[(S,S)-(4-метилфенил)2PCH(метил)CH(метил)P(4-метилфенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(S,S)-((4-MePh)2P(Me)CH-CH(Me)P(4-MePh)2)}]2
1,31 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,45 г (3,0 ммоль) лигандного соединения (R,R)-(4-метилфенил)2PCH(метил)CH(метил)P(4-метилфенил)2 использовали вместо (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2.
[Пример 9 получения катализатора] Получение бис-[(S,S)-(фенил)2PCH(фенил)CH(фенил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(S,S)-(Ph2P(Ph)CH-CH(Ph)PPh2)}]2
A. Получение лиганда (S,S)-(фенил)2P-CH(фенил)CH(фенил)-P(фенил)2 [(S,S)-(Ph2P(Ph)CH-CH(Ph)PPh2)
3,3 г бесцветного названного соединения получали так же, как в примере 1 получения катализатора, за исключением того, что (1R,2R)-1,2-дифенилэтандиол использовали в качестве исходного материала.
B. Получение бис-[(S,S)-(фенил)2PCH(фенил)CH(фенил)P(фенил)2 - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(S,S)-(Ph2P(Ph)CH-CH(Ph)PPh2)}]2
0,9 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,35 г (3,0 ммоль) лигандного соединения (S,S)-(фенил)2P-CH(фенил)CH(фенил)-P(фенил)2 использовали вместо (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2.
[Пример 10 получения катализатора] Получение бис-[{(1S,2S)-транс-бис(дифенилфосфино)циклогексан} - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(1S,2S)-(Ph2P)2циклогексан}]2
A. Получение лиганда (1S,2S)-транс-бис(дифенилфосфино)циклогексан [(1S,2S)-(Ph2P)2циклогексан]2
3,6 г совершенно бесцветного чистого названного соединения получали так же, как в примере 1 получения катализатора, за исключением того, что (1R,2R)-транс-циклогександиол использовали в качестве исходного материала вместо (2R,3R)-бутандиола.
B. Получение бис-[{(1S,2S)-транс-бис(дифенилфосфино)циклогексан} - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(1S,2S)-(Ph2P)2циклогексан}]2
1,07 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,36 г (3,0 ммоль) лигандного соединения (1S,2S)-транс-бис(дифенилфосфино)циклогексан использовали вместо (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2.
[Пример 11 получения катализатора] Получение бис-[{(3S,4S)-транс-бис(дифенилфосфино)-1-бензилпирролидин} - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(3S,4S)-(Ph2P)2-1-бензилпирролидин}]2
A. Получение лиганда (3S,4S)-транс-бис(дифенилфосфино)-1-бензилпирролидин [(3S,4S)-(Ph2P)2-1-бензилпирролидин]
2,7 г бесцветного чистого (3S,4S)-транс-бис(дифенилфосфино)-1-бензилпирролидина получали так же, как в примере 1 получения катализатора, за исключением того, что (3R,4R)-транс-1-бензилпирролидиндиол использовали в качестве исходного материала вместо (2R,3R)-бутандиола.
B. Получение бис-[{(3S,4S)-транс-бис(дифенилфосфино)-1-бензилпирролидин} - дихлорид хрома - (μ-хлорид)] [CrCl2(μ-Cl){(P,P)-k2-(3S,4S)-(Ph2P)2-1-бензилпирролидин}]2
1,15 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,59 г (3,0 ммоль) лигандного соединения (3S,4S)-транс-бис(дифенилфосфино)-1-бензилпирролидин использовали вместо (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2.
[Пример 12 получения катализатора] Получение бис-[{(S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - хлорид хрома(II) - (μ-хлорид)} [Cr(II)Cl(μ-Cl){(P,P)-k2-(S,S)-((Ph)2P(Me)CH-CH(Me)P(Ph)2)}]2
1,51 г названного соединения получали так же, как в примере 2 получения катализатора, за исключением того, что 1,02 г (3,0 ммоль) Cr(II)Cl2 использовали вместо трис-тетрагидрофурантрихлорида хрома (CrCl3(THF)3).
[Пример 1] Олигомеризация этилена с применением полученного выше катализатора и MAO
Реактор из нержавеющей стали на 600 мл промывали азотом в вакууме и затем последовательно добавляли в него 200 мл циклогексана и MAO 1,5 ммоль-Al. Затем температуру повышали до 45°C. В колбе Шленка на 50 мл в перчаточном боксе смешивали 3,3 мг (0,005 ммоль) комплекса (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 - трихлорид хрома - тетрагидрофуран, описанного в примере 1 получения катализатора, с 10 мл циклогексана и полученную таким образом смесь добавляли в реактор. Реактор заряжали этиленом до 30 бар и смесь перемешивали при 300 об./мин. Спустя 120 мин подачу этилена в реактор останавливали, перемешивание останавливали, чтобы закончить реакцию, и реактор охлаждали до температуры ниже 10°C.
После выпуска избытка этилена из реактора этанол, содержащий 10% об. хлористоводородной кислоты, добавляли к жидкости в реакторе. Чтобы проанализировать жидкость методом GC-FID, в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали методом GC-FID. Оставшийся органический слой фильтровали, чтобы отделить твердый воск/полимерные продукты. Данные твердые продукты сушили в печи при 100°C в течение ночи и взвешивали, получая таким образом 1,2 г полиэтилена. GC-анализ показал, что суммарная масса реакционной смеси составляла 116,2 г. Распределение продуктов данного примера показано в нижеприведенной Таблице 1.
[Примеры 2-12] Олигомеризация этилена с применением полученного выше катализатора и MAO
Олигомеризацию этилена проводили так же, как в примере 1, за исключением того, что применяли соответствующие катализаторы примеров 2-12 получения в подходящих количествах и количество MAO и время реакции подбирали соответствующим образом. Условия реакции и результаты реакции показаны ниже в таблице 1.
[Сравнительный пример] Тетрамеризация этилена с применением CrCl3(тетрагидрофуран)3, (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 и MAO
Тетрамеризацию этилена проводили так же, как в примере 1, за исключением того, что использовали 2,2 мг (0,005 ммоль) лиганда (S,S)-(фенил)2PCH(метил)CH(метил)P(фенил)2 [(S,S)-Ph2PCH(Me)CH(Me)PPh2] примера 1 получения катализатора и 1,88 мг (0,005 ммоль) CrCl3(тетрагидрофуран)3. Условия реакции и результаты реакции показаны ниже в таблице 1.
|
Принимая во внимание вышеприведенные примеры и сравнительный пример, способ получения олигомера с использованием катализатора по настоящему изобретению увеличивал выход примерно в 15 раз и уменьшал количество полимерного побочного продукта с 5,3% до 0,5% или менее по сравнению с результатами сравнительного примера, в котором лиганд и предшественник катализатора добавляли по отдельности.
Результаты примеров показали, что структура и активность синтезированного каталитического комплекса может изменяться в зависимости от растворителя и условий, используемых при синтезе.