Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 4-ДЕМЕТОКСИДАУНОРУБИЦИНА
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к способам химического синтеза антрациклиновых соединений. В частности, настоящее изобретение относится к способу получения 4-деметоксидаунорубицина (идарубицина) из даунорубицина.
Уровень техники
Антрациклины представляют собой класс природных биологически активных соединений, продуцируемых бактериями рода Streptomyces. Было клинически доказано, что некоторые антрациклины являются эффективными противоопухолевыми агентами и могут быть использованы для лечения многих видов рака, включая, в частности, рак молочной железы, рак яичников, рак легких и гемобластозы, такие как лейкозы и лимфомы. Кроме того, было показано, что представители этого класса соединений могут подходить для применения в трансплантатах костного мозга и при трансплантации стволовых клеток. Примеры таких терапевтически значимых антрациклинов включают, помимо прочих, даунорубицин, идарубицин (т.е. 4-деметоксидаунорубицин), доксорубицин, эпирубицин, пирарубицин, зорубицин, акларубицин и карминомицин.
4-Деметоксидаунорубицин (идарубицин), имеющий химическую структуру согласно формуле (I) (см. ниже), является аналогом даунорубицина, который препятствует синтезу нуклеиновых кислот посредством интеркаляции в ДНК и взаимодействует с ферментом топоизомеразой II. За счет отсутствия метоксигруппы в положении 4 антрациклиновой структуры соединение обладает высокой липофильностью, в результате чего увеличивается скорость клеточного поглощения по сравнению с другими антрациклинами. В комбинации с цитозинарабинозидом 4-деметоксидаунорубицин в настоящее время является препаратом терапии первой линии при остром миелоидном лейкозе.
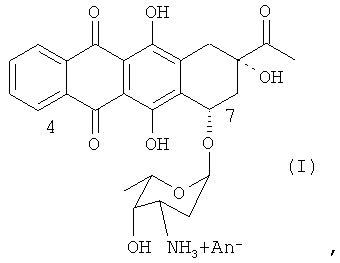
где An- представляет собой анион любой сильной кислоты, например, такой как хлороводородная кислота.
Доступные способы синтеза 4-деметоксидаунорубицина (идарубицина), как правило, основаны на реакции сочетания агликона данного соединения (т.е. неуглеводного компонента) и защищенного и активированного даунозамина (т.е. 3-амино-2,3,6-тридеокси-L-ликсогексозы, углеводного компонента) в присутствии трифлата серебра (AgOSO2CF3), триметилсилилтрифлата ((CH3)3SiOSO2CF3) или системы оксид ртути / бромид ртути (HgO/HgBr2). Агликон может быть синтезирован, например, с использованием или антрацентетрона, или изобензофурана в качестве исходного вещества. Тем не менее такие способы синтеза сложны вследствие образования оптически активных центров у атомов углерода С7 и С9.
В альтернативных способах синтеза 4-деметоксидаунорубицина применяют агликон даунорубицина, который получают кислотным гидролизом даунорубицина. В случае если даунорубицин подвергают кислотному гидролизу, можно отдельно получить аминосахар даунозамин, который далее, после химической модификации, применяют для гликозилирования модифицированного агликона.
Изначально способы, применяемые для замещения группы 4-СН3O (4-МеО) агликона на водород (и другие заместители, такие как NH2) включали деметилирование даунорубицинона, сульфирование полученного 4-деметилдаунорубицинона, замещение 4-ArSO2O группы на 4-ArCH2NH и последующее восстановление бензильной группы с получением 4-NH2 группы (см. патент США 4085548). Дальнейшее восстановительное дезаминирование приводит к получению агликона 4-деметоксидаунорубицина (см. Европейский патент 0328399 В1).
В патенте США 5587495 предложена реакция восстановительной конденсации 4-деметил-4-трифторметансульфонил-даунорубицинона (4-OTf-даунорубицинона) и фенилфосфина с применением комплексов палладия или никеля. Параллельно были получены 4-R замещенные даунорубициноны.
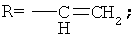
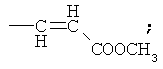
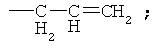
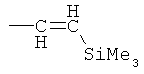
Аналогичным образом восстановительное карбонилирование 4-OTf-даунорубицинона с применением тех же самых комплексов позволяет получить 4-COCR-замещенные даунорубициноны (см. патент США 5218130). Если в качестве восстанавливающего агента применяют формиат, то происходит замещение 4-(OTf-остатка на водород с получением 4-деметоксидаунорубицинона (см. патент США 5103029).
Таким образом, все общепринятые способы синтеза 4-деметоксидаунорубицина включают фрагментацию молекулы даунорубицина на агликоновый компонент и компонент аминосахара, раздельную химическую модификацию этих двух компонентов с последующим сочетанием. Однако такая схема синтеза приводит к возникновению дополнительных сложностей, образованию оптически активного центра на атоме углерода С7. Как правило, подобные схемы синтеза включают 10-12 различных стадий, что снижает общий выход конечного продукта до 6-8%.
В патенте США 7053191 предложен альтернативный путь синтеза, в котором в качестве исходного соединения применяли производные 4-деметилдаунорубицина (т.е. карминомицина), в основном N-трифторацетил-4-деметилдаунорубицин. В этом случае 4-ОН-группу удаляют из целой молекулы антрациклина. На сегодняшний день, однако, N-трифторацетил-4-деметилдаунорубицин может быть получен в приемлемых количествах только путем сложного химического синтеза (см. патент США 4188377).
Предпочтительная модификация пути синтеза производных карминомицина может заключаться в применении в качестве исходного вещества даунорубицина с целью сокращения количества необходимых стадий синтеза. Тем не менее до сих пор не удавалось создать подобную схему синтеза в связи с отсутствием способов селективного деметилирования 4-МеО-группы антрациклинов без сопутствующего расщепления гликозидной связи при углеродном атоме С7.
Один из известных способов деметилирования алкилфениловых простых эфиров включает обработку алкилфениловых эфиров сильной кислотой Льюиса - AlCl3 - в инертных растворителях (в частности, в хлорированных углеводородах, таких как дихлорметан) при температуре кипения. Любая попытка реализовать подобный путь синтеза даунорубицина приводит к отщеплению даунозамина и последующей полной деструкции молекулы.
Таким образом, в настоящее время по-прежнему существует потребность в новых способах синтеза для получения клинически эффективных антрациклиновых соединений, таких как 4-деметоксидаунорубицин (идарубицин). В частности, по-прежнему существует потребность в менее сложных способах синтеза, включающих меньшее количество стадий реакции и, следовательно, обеспечивающих более высокий выход конечного продукта.
Соответственно, задачей настоящего изобретения является обеспечение таких способов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
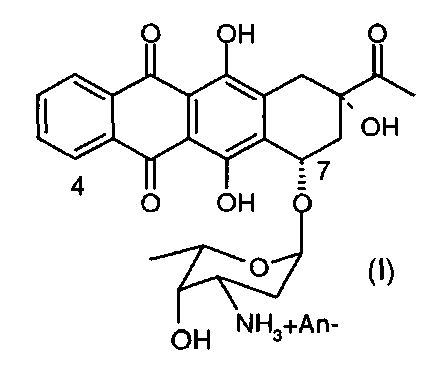
Настоящее изобретение относится к способу получения соли 4-деметоксидаунорубицина, имеющей химическую структуру согласно формуле (I):
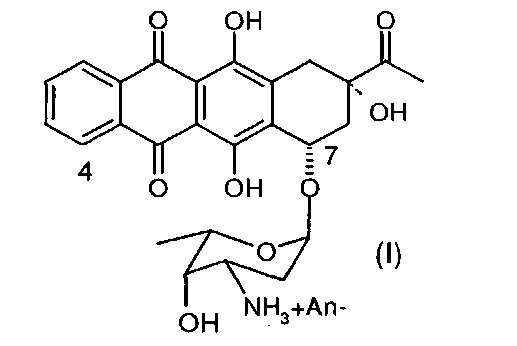
где An- представляет собой анион;
включающему:
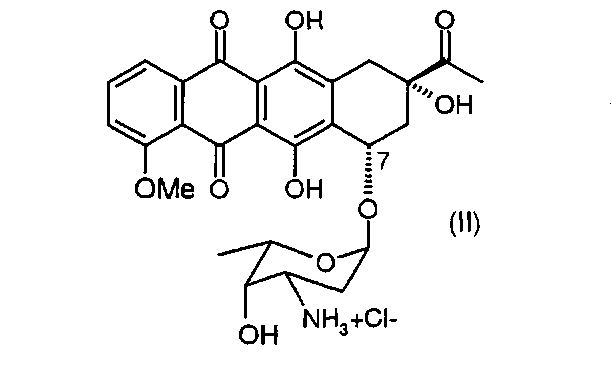
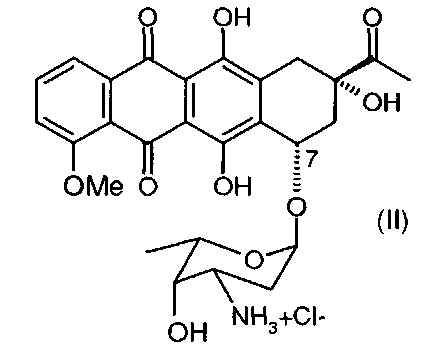
(а) превращение даунорубицина гидрохлорида, имеющего химическую структуру согласно формуле (II)
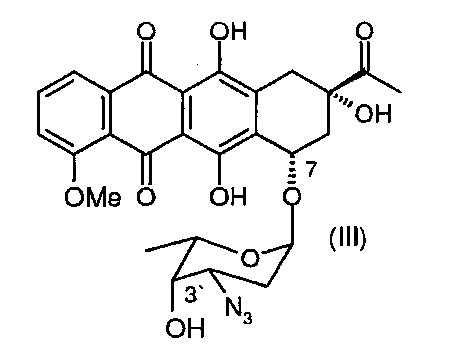
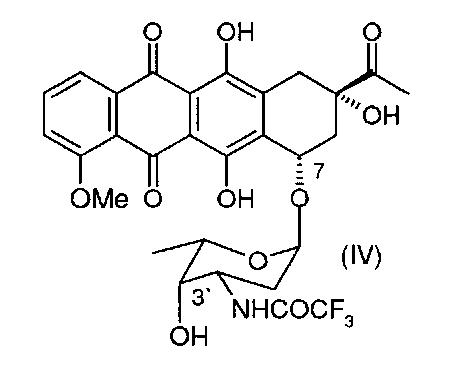
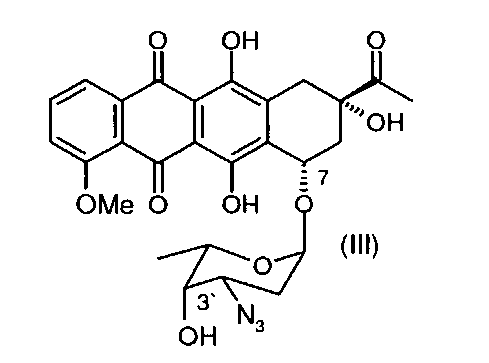
в 3′-защищенный даунорубицин (3′-защ.-даунорубицин), выбранный из группы, состоящей из соединений, имеющих химическую структуру согласно формулам (III) и (IV), причем превращение соли, образованной по 3′-аминогруппе, в 3′-азид путем приведения в контакт с азидобразующим реагентом приводит к получению соединения формулы (III), а превращение соли, образованной по 3′-аминогруппе, в 3′-трифторацетамид путем приведения в контакт с трифторацетилирующим реагентом приводит к получению соединения формулы (IV);
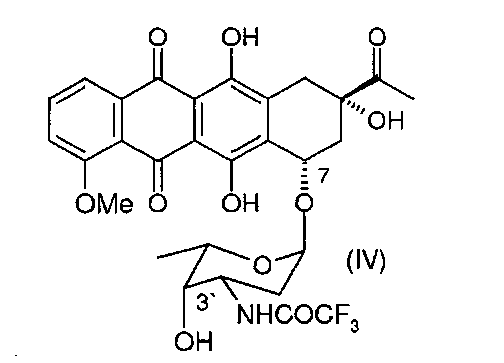
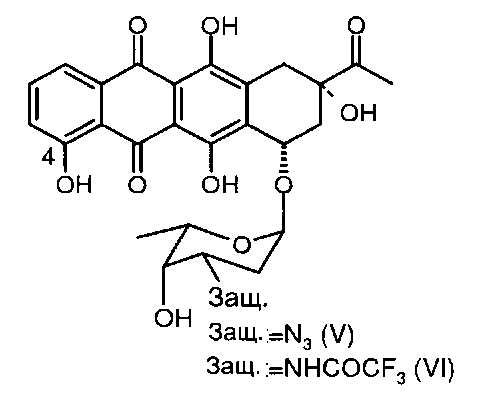
(b) деметилирование 3′-защ.-даунорубицина формулы (III) или (IV) путем приведения в контакт с «мягкой» кислотой Льюиса в безводном растворителе с получением 4-деметил-3′-защ.-даунорубицина, выбранного из группы, состоящей из соединений, имеющих химическую структуру согласно формулам (V) и (VI) соответственно, причем деметилирование соединения формулы (III) приводит к образованию соединения формулы (V), а деметилирование соединения формулы (IV) приводит к образованию соединения формулы (VI);
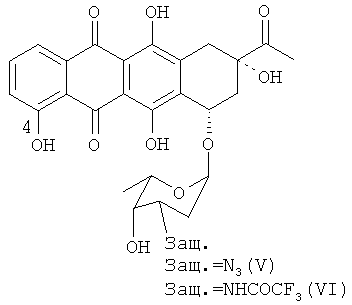
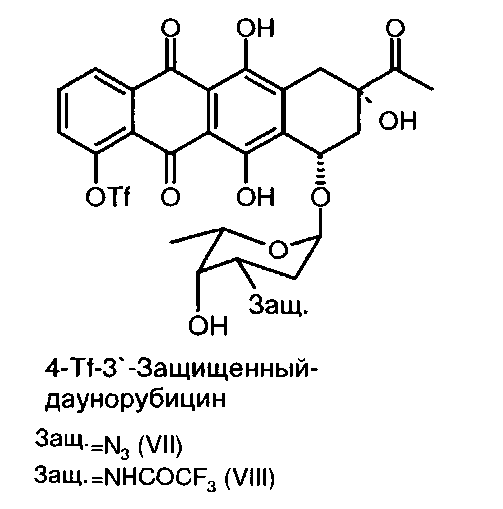
(с) трифторметансульфонилирование 4-деметил-3′-защ.-даунорубицина формулы (V) или (VI) путем приведения в контакт с трифторметансульфонилирующим реагентом с получением 4-О-трифторметансульфонил-3'-защ.-даунорубицина, выбранного из группы, состоящей из соединений, имеющих химическую структуру согласно формулам (VII) и (VIII) соответственно,
причем трифторметансульфонилирование соединения формулы (V) приводит к образованию соединения формулы (VII), а трифторметансульфонилирование (VI) приводит к образованию соединения формулы (VIII);
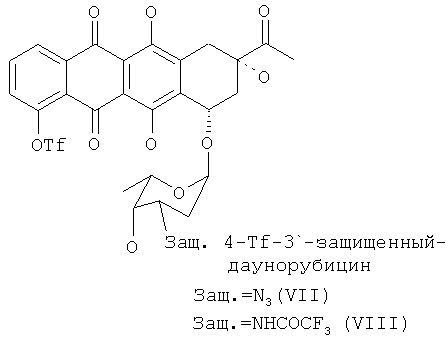
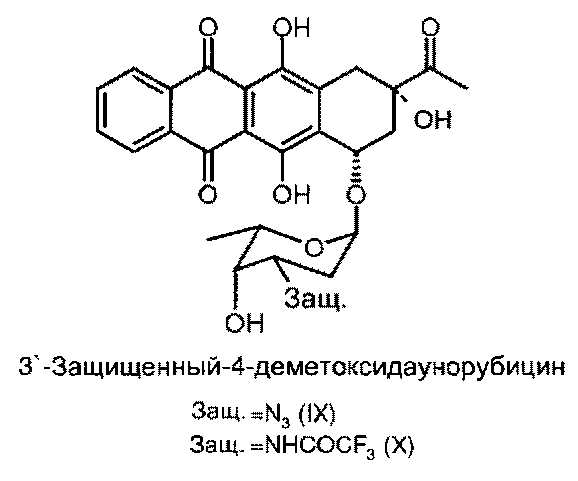
(d) восстановление 4-O-трифторметансульфонил-3′-защ.-даунорубицина формулы (VII) или (VIIII) путем приведения в контакт с восстанавливающим реагентом с получением 4-деметокси-3′-защ.-даунорубицина, выбранного из группы, состоящей из соединений, имеющих химическую структуру согласно формулам (IX) и (X) соответственно, причем восстановление соединения формулы (VII) приводит к получению соединения формулы (IX), а восстановление соединения формулы (VIII) приводит к получению соединения формулы (X); и
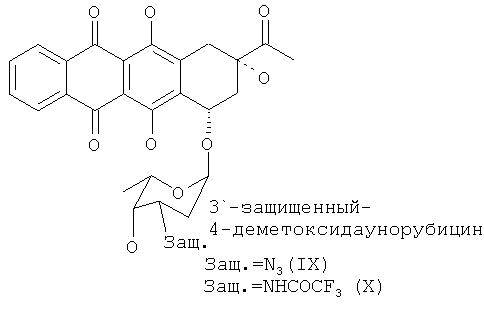
(е) удаление защитной группы 3′-защ. из 4-деметокси-3′-защ.-даунорубицина, выбранного из группы, состоящей из соединения формулы (IX) и соединения формулы (X),
при котором соединение формулы (IX) приводят в контакт с азид-восстанавливающим реагентом или соединение формулы (X) приводят в контакт с щелочным раствором с образованием, таким образом, 3′-аминогруппы и получением 4-деметоксидаунорубицина,
и
необязательно, приведение 4-деметоксидаунорубицина в контакт с кислотой формулы Н+Аn- с получением соли 4-деметоксидаунорубицина формулы (I).
Способ согласно настоящему изобретению не вызывает расщепления гликозидной связи при атоме углерода С7, что позволяет среднему специалисту в данной области техники получить продукт всего за пять стадий с общим выходом конечного продукта 30-45% в расчете на даунорубицина гидрохлорид.
На следующей схематичной иллюстрации (схема 1) представлен путь синтеза в соответствии со способом, заявленным в настоящем изобретении.
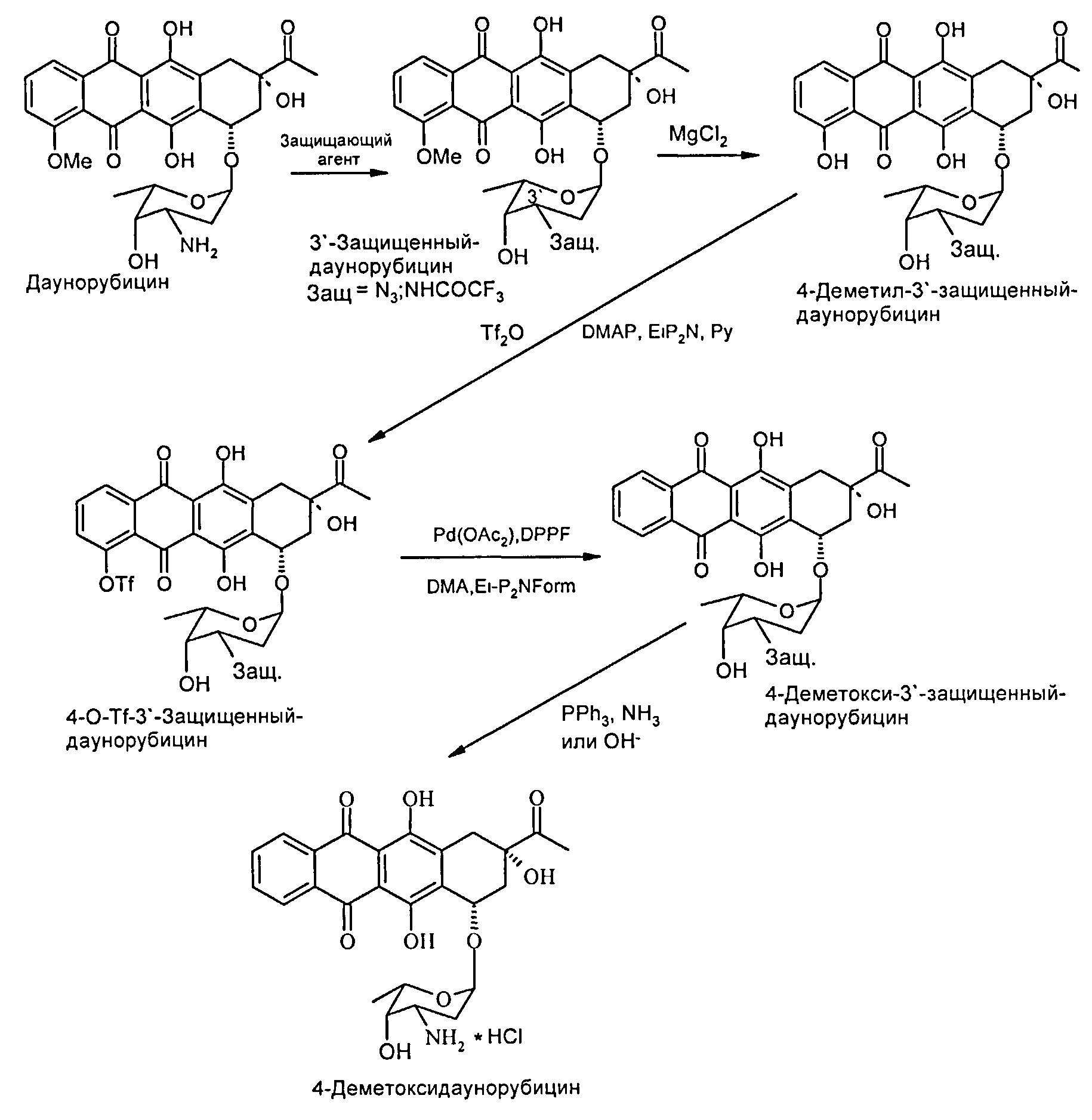
Соль даунорубицина, например даунорубицина гидрохлорид, имеющую химическую структуру формулы (II), защищают по 3′-аминогруппе (в остатке аминосахара), который на первой стадии превращают в азид (3′-N3) (см. соединение, имеющее химическую структуру формулы (III)) или трифторацетамид (3′-NHCOCF3) (см. соединение, имеющее химическую структуру формулы (IV)). Первое из альтернативных превращений может быть осуществлено путем взаимодействия соединения формулы (II) с азидобразующим реагентом. Примером является нитрит натрия/азид натрия. Предпочтительным примером является трифторметансульфонилазид (TfN3). Второе из альтернативных превращений может быть осуществлено путем взаимодействия соединения формулы (II) с трифторацетилирующим реагентом, таким как ангидрид трифторуксусной кислоты, или с активированным эфиром трифторуксусной кислоты, таким как N-гидроксисукцинимидным эфиром.
Затем 3′-защ.-даунорубицин формулы (III) или (IV), полученный таким образом, может быть О-деметилирован по 4-ому положению в присутствии «мягкой» кислоты Льюиса, предпочтительно MgCl2 (безводный). В частных вариантах реализации изобретения данную стадию реакции осуществляют в температурном диапазоне 10-80°С, предпочтительно в диапазоне 40-60°С. В других частных вариантах реализации изобретения реакцию проводят в присутствии KI в безводном растворителе, который может быть выбран из группы, состоящей из алканов, циклоалканов, галогеналканов, аренов, алкилоксидов, простых эфиров, С4-С6 спиртов и дисульфида углерода. Данная стадия позволяет осуществить деметилирование 4-ОМе-группы без расщепления гликозидной связи при углеродном атоме С7. При введении азидной защитной группы в 3′-положении достигают повышенных выходов.
4-Деметил-3′-защ.-даунорубицин формулы (V) или (VI), полученный таким образом, может быть далее трифторметансульфонилирован путем взаимодействия с трифторметансульфонилирующим реагентом с получением 4-O-трифторметансульфонил-3′-защ.-даунорубицина согласно формулам (VII) и (VIII) соответственно. Затем соединение формулы (VII) или формулы (VIII) может быть восстановлено с получением 4-деметокси-3′-защ.-даунорубицина согласно формулам (IX) и (X) соответственно. Реакция трифторметансульфонилирования может быть осуществлена посредством взаимодействия 4-деметил-3′-защ.-даунорубицина формулы (V) или (VI) с ангидридом трифторметансульфоновой кислоты, предпочтительно в пиридине в присутствии третичных аминов.
В частных вариантах реализации изобретения стадию восстановления соединения формулы (VII) или (VIII) с получением соединений формулы (IX) или (X) соответственно проводят с помощью восстанавливающего агента в присутствии каталитических количеств соединений, имеющих общую формулу PdLnL′m; где Pd представляет собой палладий, L и L′ независимо выбраны из группы, состоящей из фосфитов и фосфинов, а n и m могут независимо варьироваться от 0 до 4. Предпочтительно восстанавливающий реагент выбран из группы, состоящей из муравьиной кислоты и ее солей. В других предпочтительных вариантах реализации изобретения стадию восстановления осуществляют при температуре в диапазоне 30-100°С в полярном апротонном растворителе.
В некоторых вариантах реализации изобретения способ согласно настоящему изобретению дополнительно включает выделение 4-деметил-3′-защ.-даунорубицина, полученного путем обработки реакционной смеси сильными кислотами при рН 2,5±1,0 и последующей экстракции (нерастворимыми в воде органическими растворителями, включая, наряду с прочими, галогеналканы, циклоалканы, арены, С4-С6 спирты, а также их смеси). 4-Деметил-3′-защ.-даунорубицин выделяют путем испарения органической фазы в вакууме.
Окончательное удаление 3′-защитной группы из 4-деметокси-3'-защ.-даунорубицина осуществляют с помощью способов, описанных в уровне техники (см. ссылки, указанные выше), для модификации агликона 4-деметилдауномицинона. Восстановление 3′-N3 группы с образованием 3′-NH2 может быть осуществлено в присутствии трифенилфосфина (PPH3)-NH3, в то время как удаление защитной COCF3 группы осуществляют щелочным гидролизом амида.
В другом аспекте настоящее изобретение относится к способу получения и выделения ключевого промежуточного соединения - 4-деметил-3′-защищенного-даунорубицина формулы (V) или (VI) из даунорубицина гидрохлорида путем проведения реакционных стадий, описанных выше. Эти защищенные производные карминомицина могут быть превращены в карминомицин с применением методик снятия защиты, приведенных при описании снятия защиты с защищенных 4-деметоксидаунорубициновых (идарубициновых) соединений, представленных выше, с получением карминомицина или его соли.
Подробное описание изобретения
Настоящее изобретение относится к неожиданному обнаружению того факта, что деметилирование 3′-защищенного даунорубицина в присутствии «мягкой» кислоты Льюиса, предпочтительно MgCl2 (безводного), позволяет синтезировать 4-деметоксидаунорубицин из даунорубицина гидрохлорида без расщепления гликозидной связи при атоме углерода С7, что приводит, таким образом, к более быстрому циклу синтеза и увеличению выхода конечного продукта.
Настоящее изобретение далее будет описано в соответствии с частными вариантами реализации и со ссылкой на конкретные графические материалы, но следует понимать, что изобретение не ограничено ими, а ограничено только прилагаемой формулой изобретения. Описанные графические материалы являются сугубо схематичными и не должны рассматриваться как ограничивающие настоящее изобретение.
Термин «включающий», используемый в настоящем описании и формуле изобретения, не исключает других элементов или стадий. Для целей настоящего изобретения термин "состоящий из" считается предпочтительным вариантом термина "включающий". Если в дальнейшем группа определена как включающая по меньшей мере некоторое количество вариантов реализации, это следует понимать как раскрытие группы, которая предпочтительно состоит только из этих вариантов реализации.
Если существительное упоминается в единственном числе в определенной или неопределенной форме, это включает и множественное число данного существительного, если конкретно не указано иное.
В случае, если в контексте настоящего изобретения используются числовые значения, специалисту в данной области техники понятно, что технический результат, обеспечиваемый конкретным признаком, обеспечивается с учетом некоторого интервала погрешности, который обычно включает отклонения числовых значений в пределах ±10%, предпочтительно в пределах ±5%.
Кроме того, термины первый, второй, третий, (а), (b), (с) и т.п., используемые в настоящем описании и в формуле изобретения, применяются для различения сходных элементов и не обязательны для описания последовательности или хронологического порядка. Следует понимать, что используемые определения являются взаимозаменяемыми при соответствующих обстоятельствах и что варианты реализации изобретения, описанные здесь, могут быть реализованы в последовательности, отличной от описанной в настоящей заявке.
Дальнейшее определение терминов будет приведено ниже в контексте, в котором используются эти термины. Следующие термины и определения приведены исключительно для облегчения понимания изобретения. Не следует делать вывод, что указанные определения имеют более узкий смысл по сравнению со смыслом, очевидным среднему специалисту в данной области техники.
На первой стадии 3′-аминогруппу даунорубицина защищают путем переведения ее либо в азидогруппу, либо в трифторацетамидную группу.
В некоторых вариантах реализации изобретения превращение 3′-аминогруппы даунорубицина гидрохлорида в 3′-азидосоединение (3′-N3) может быть осуществлено при помощи азидобразующего реагента, такого как трифторметансульфонилазид (TfN3) в соответствии со следующей схемой 2:
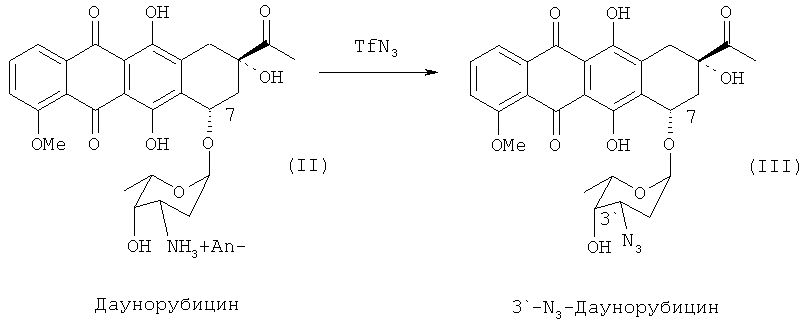
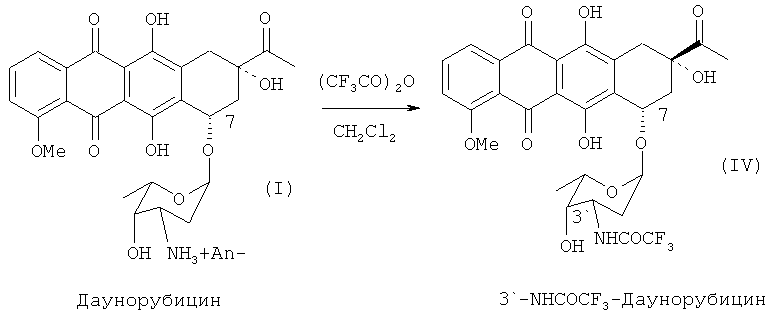
В альтернативных вариантах реализации изобретения защита 3′-аминогруппы даунорубицина гидрохлорида может быть осуществлена через соответствующий 3′-трифторацетамид (3′-NHCOCF3), который может быть получен путем взаимодействия 3′-аминосоединения (II) с ангидридом трифторуксусной кислоты в соответствии со следующей схемой 3:
Стадию деметилирования 4-ОМе-группы 3′-защ.-даунорубицина формул (III) и (IV) можно осуществить путем взаимодействия соединения формулы (III) или (IV) с кислотой Льюиса, в частности с «мягкой» кислотой Льюиса, такой как безводный MgCl2 с получением фенольных промежуточных соединений, производных карминомицина (V) или (VI) соответственно (см. схему 4 ниже).
Следующая схема 4 иллюстрирует реакцию 4-деметилирования:
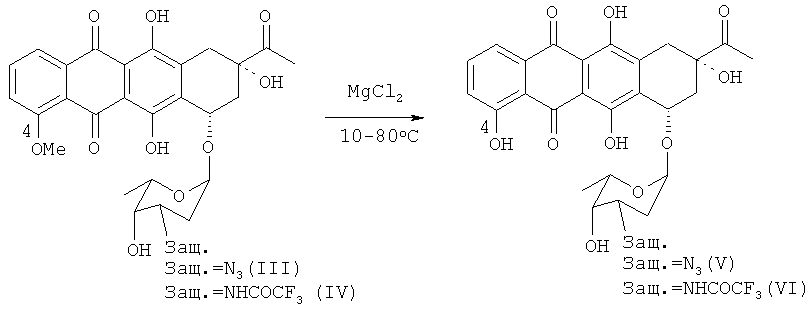
Термин «кислота Льюиса», используемый в настоящей заявке, обозначает молекулярную структуру (и соответствующее химическое соединение), которая является акцептором электронной пары и, следовательно, способна реагировать с основаниями Льюиса с образованием аддукта Льюиса путем принятия электронной пары, предоставляемой основанием Льюиса. Кислоты и основания Льюиса обычно подразделяют на «жесткие» и «мягкие». В данном контексте под «жесткой кислотой» следует понимать кислоту, содержащую небольшие или неполяризуемые атомы, а под «мягкой» - кислоту, содержащую атомы большего размера, которые более поляризуемы.
В таблице 1 ниже представлено взаимодействие 3′-защищенного даунорубицина с различными кислотами Льюиса.
|
Деметилирование 4-МеО-группы 3′-защищенного даунорубицина может быть осуществлено с применением «мягкой» кислоты Льюиса, MgCl2 (безводного). Сопутствующего расщепления гликозидной связи при углеродном атоме С7 при этом не наблюдалось. В противоположность этому, применение MgCl2∗6H2O, MgCl2∗4H2O и MgCl2∗2H2O не дало приемлемых результатов. Применение более «жестких» кислот Льюиса, таких как AlCl3, BF3 или TiCl4 приводило к нежелательному расщеплению гликозидной связи с образованием агликона.
В некоторых вариантах реализации изобретения реакцию 4-деметилирования осуществляют путем обработки 3′-защищенного даунорубицина, имеющего химическую структуру согласно формуле (III) или (IV), «мягкой» кислотой Льюиса - MgCl2 (безводным) при температуре реакции в диапазоне 10-80°С. Температура реакции зависит от активности применяемой кислоты Льюиса и выбранные температурные условия должны обеспечивать максимальную региоселективность реакции: деметилирование 4-ОМе-групп без сопутствующего расщепления гликозидной связи при атоме углерода С7.
Реакция 4-деметилирования может быть проведена в присутствии KI в безводном растворителе (устойчивом к кислотам Льюиса), который может быть выбран из группы, состоящей из алканов, циклоалканов, галогеналканов, аренов, алкилоксидов, простых эфиров, С4-С6 спиртов и дисульфида углерода. При этом растворители, выбранные из галогеналканов и простых эфиров, являются наиболее предпочтительными. Кислота Льюиса может присутствовать в 1-5 кратном молярном избытке по отношению к 3′-защ.-даунорубицину (последний, как правило, присутствует в количестве 1,5-3 молей).
В некоторых конкретных вариантах реализации изобретения продукт реакции 4-деметилирования, представляющий собой 4-деметил-3′-защ.-даунорубицин, имеющий структуру согласно формуле (V) или (VI), выделяют путем обработки реакционной смеси водным раствором сильных кислот (таких как, без ограничения, щавелевая кислота, трифторуксусная кислота, серная кислота и соляная кислота) при рН 2,5±1,0 и последующей экстракции нерастворимых в воде органических кислот (в случае применения водорастворимых эфиров). Далее соединения 4-деметил-3′-защ.-даунорубицина (V, VI) выделяют путем испарения органической фазы в вакууме.
В различных вариантах реализации изобретения соединение формулы (V) или (VI) могут быть преобразованы в карминомицин непосредственно со снятием защиты, таким как описано ниже для снятия защиты с производных 3′-защищенного 4-деметоксидаунорубицина. Соответственно, согласно настоящему изобретению предложен более «короткий» и эффективный способ получения карминомицина или его солей из даунорубицина или его солей.
При получении 4-деметоксидаунорубицина деметилированные 3′-защищенные-даунорубициновые соединения формулы (V) или (VI) сульфонилируют по 4-ОН группе с получением соединений формулы (VII) и (VIII) соответственно путем взаимодействия с трифторметансульфонилирующим агентом. Например, трифторметансульфонилирование может быть осуществлено путем проведения реакции соединения (V) или (VI) с ангидридом трифторметансульфоновой кислоты (Tf2O). Реакция может быть проведена в пиридине (Ру) в присутствии стерически затрудненных третичных аминов, таких как, но не ограничиваясь ими, N,N-диизопропилэтиламин, и каталитических количеств N,N-диметиламинопиридина. Гидроксильные группы при атомах углерода С6, С11 и С9 не реагируют в условиях приведенного здесь эксперимента (см. пример 4 ниже).
Продуктом реакции на данной стадии синтеза является 4-О-трифторметансульфонил-3′-защ.-даунорубицин, имеющий химическую структуру согласно формуле (VII) или (VIII).
Реакция показана на следующей схеме 5:
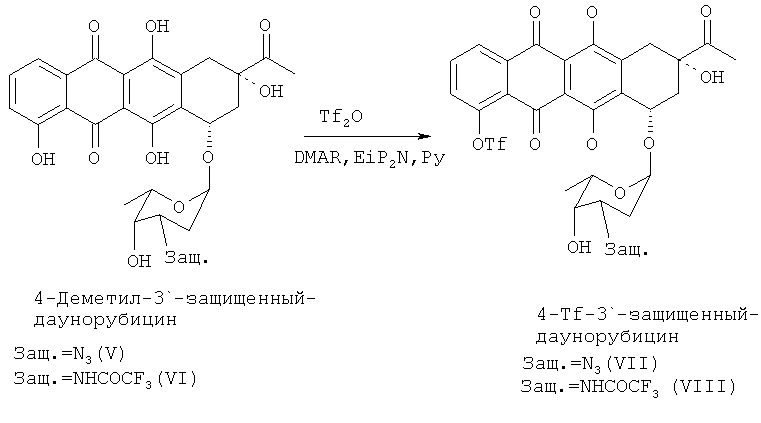
Далее 4-сульфопроизводное 4-деметилдаунорубицина (VII, VIII) подвергают реакции с восстанавливающим реагентом, выбранным из муравьиной кислоты и солей муравьиной кислоты в присутствии каталитических количеств (в молярном соотношении в диапазоне от 1:1 до 1:104, предпочтительно от 1:20 до 1:100) соединений, имеющих общую формулу PdLnL′m;
где Pd представляет собой палладий, L и L′ независимо выбраны из группы, состоящей из фосфитов и фосфинов, а n и m могут независимо варьировать от 0 до 4. Предпочтительными фосфинами для применения согласно настоящему изобретению являются, среди прочих, дифенилфосфинопропан и 1,1′-бис-(дифенилфосфино)ферроцен (DPPF). Предпочтительно, данную стадию осуществляют при температуре реакции в диапазоне 30-100°С в полярном апротонном растворителе, в частности в алкиламидах (диметилацетамид, DMA) в инертной атмосфере.
Продукт реакции на данной стадии синтеза показан на следующей схеме 6 и представляет собой 3′-защ.-4-деметоксидаунорубицин, имеющий химическую структуру согласно формуле (IX) или (X).
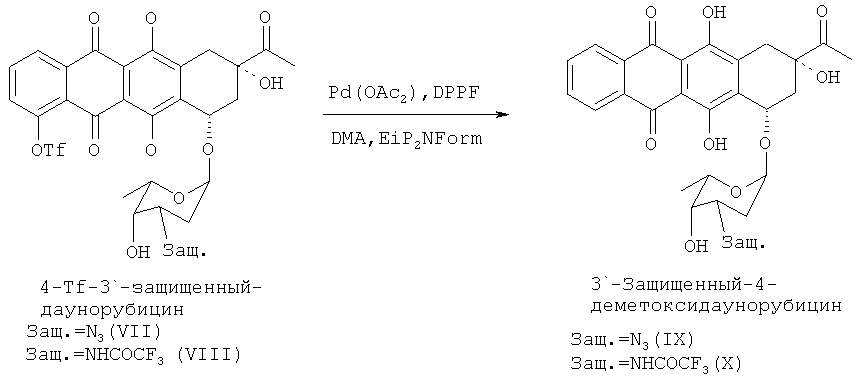
И окончательно, 3′-защитную группу из полученного 4-деметокси-3′-защ.-даунорубицина удаляют с образованием 3′-NH2 группы, применяя стандартные методики синтеза (см., например, примеры 6 и 7), с получением 4-деметоксидаунорубицина (I) в качестве конечного продукта.
Далее настоящее изобретение описано с помощью фигур и следующих примеров, которые служат исключительно для целей иллюстрации конкретных вариантов реализации данного изобретения и не должны толковаться как ограничивающие тем или иным образом объем изобретения.
Примеры
Пример 1
20 г гидрохлорида даунорубицина (II) растворяли в 125 мл МеОН. К 20 мл воды добавляли 7,5 г К2СO3, энергично перемешивали в течение 1 минуты и добавляли к раствору трифторметансульфонилазида (TfN3) в дихлорметане. Полученную смесь перемешивали на магнитной мешалке до полного растворения антрациклинового исходного соединения. Затем реакционную смесь добавляли к 300 мл воды. Органический слой удаляли, а водную фазу экстрагировали дихлорметаном. Оставшийся дихлорметан выпаривали на роторном испарителе.
Получили около 20 г 3′-N3-даунорубицина (III) с чистотой > 90%. Чистота полученного продукта являлась достаточной для дальнейшего синтеза.
Пример 2
20 г даунорубицина гидрохлорида (II) суспендировали в 400 мл дихлорметана (DCM), охлажденного до 0°С. К 45 мл DCM раствора при интенсивном перемешивании в течении 1 часа добавляли 28 мл ангидрида трифторуксусной кислоты. Смесь дополнительно выдерживали при температуре 0°С в течение 30 мин, затем добавляли к 750 мл дистиллированной воды и перемешивали. Органический слой удаляли.
К органическому слою добавляли 400 мл насыщенного раствора бикарбоната натрия и выдерживали при комнатной температуре при интенсивном перемешивании в течение 15-25 часов для гидролиза 4′O-3′N-ди-трифторацетилдауномицина. После завершения гидролиза (ВЭЖХ-контроль) органический слой отделяли и полностью упаривали растворитель при пониженном давлении.
После испарения растворителя получено 20 г N-трифторацетамидодаунорубицина (IV) с чистотой 93% (согласно ВЭЖХ). Чистота полученного продукта являлась достаточной для дальнейшего синтеза.
Пример 3
20 г 3′-защищенного-даунорубицина (III, IV) растворяли в 450 мл тетрагидрофурана. К раствору добавляли 25 г безводного хлорида магния и 20 г безводного йодида калия, исключая контакт с атмосферной влагой. Смесь выдерживали при 40°С в течение 1,5 часов, добавляли в ледяную воду и подкисляли до рН 2,5 трифторуксусной кислотой. Затем смесь экстрагировали 2×150 мл дихлорметана.
Органический слой отделяли и сушили над безводным MgSO4, а растворитель испаряли при пониженном давлении. Получили 15,8 г 4-деметил-3′-защ.-даунорубицина (V, VI) с чистотой > 90% (согласно ВЭЖХ).
Пример 4
Тщательно высушенный 4-деметил-3′-защ.-даунорубицин (V, VI) из примера 3 растворяли в 800 мл пиридина. Добавляли 28 мл диизопропилэтиламина и 3,5 г 4-диметиламинопиридина и смесь охлаждали до 0°С. Добавляли 7,5 мл свежеперегнанного ангидрида трифторметансульфоновой кислоты и выдерживали в течение 1 часа при комнатной температуре. Затем добавляли 650 мл концентрированной соляной кислоты, 0,8 кг льда и 800 мл дихлорметана. Органический слой промывали два раза 500 мл дистиллированной воды, проводили разделение и дихлорметан удаляли при пониженном давлении.
Получили 20 г 4-O-трифторметансульфонил-3′-защ.-даунорубицина (VII, VIII) с чистотой 75-90%, который использовали для последующих стадий синтеза без дополнительной очистки.
Пример 5
4-O-трифторметансульфонил-3′-защищенный-даунорубицин (VII, VIII), полученный в примере 4, растворяли в 500 мл диметилформамида. 12 г формиата триэтиламмония и 350 мг ацетата палладия добавляли при перемешивании в атмосфере аргона. Смесь нагревали до 50°С, к ней добавляли 1,2 г 1,1′-бис(дифенилфосфино)ферроцена и продолжали нагревание в течение 8 часов.
Реакционную смесь при интенсивном перемешивании добавляли в воду, прежде чем 4-деметокси-3′-защищенный-даунорубицин отделяли фильтрованием и очищали с помощью препаративной хроматографии. Получали 9,5-11,5 г 4-деметокси-3′-защ.-даунорубицина (IX, X) с чистотой 96-98%.
Пример 6
В случае когда 3′-защитная группа представляла собой N3 (IX), полученное промежуточное соединение растворяли в 200 мл тетрагидрофурана и добавляли 14 г трифенилфосфина. Раствор выдерживали при комнатной температуре для достижения полной конверсии 4-деметоксидаунорубицин азида. Затем добавляли 10 г раствора аммиака в метаноле и выдерживали до полной конверсии 3′-фосфинимина 4-демектоксидаунорубицина.
Реакционную смесь выпаривали и конечный продукт очищали с помощью препаративной хроматографии. После повторной хроматографической очистки и кристаллизации получили 8-8,5 г 4-деметоксидаунорубицина (I) с чистотой > 99,5% (т.е. общий выход составил 40-42,5% в расчете на исходное вещество).
Пример 7
В случае когда 3′-защитная группа представляла собой COCF3 (X), 9,5 г 4-деметокси-3′-трифторацетамидодаунорубицина суспендировали в 300 мл воды при температуре 30°С. Затем при перемешивании добавляли 30 мл 1,0 н. раствора NaOH. Смесь выдерживали в течение 30 мин, нейтрализовали до рН 7,0 раствором соляной кислоты.
Реакционную смесь выпаривали и конечный продукт реакции очищали с помощью препаративной хроматографии. Окончательно получали 8,2-8,8 г 4-деметоксидаунорубицина (I) с чистотой > 99,5% (т.е. общий выход составил 41-44% в расчете на исходное вещество).
Настоящее изобретение, иллюстративно описанное в данной заявке, может быть подходящим образом реализовано на практике в отсутствие любого элемента или элементов, ограничения или ограничений, конкретно не раскрытых в настоящей заявке. Так, например, термины «содержащий, «включающий» и т.п. следует понимать в широком смысле и без ограничений. Кроме того, применяемые в данной заявке термины и выражения были использованы в качестве терминов для описания, но не в качестве ограничения, и не следует применять данные термины и выражения, исключая любые эквиваленты представленных и описанных в данной заявке признаков или их частей, но при этом следует понимать, что в рамках заявленного объема настоящего изобретения возможны различные модификации. Таким образом, следует понимать, что, хотя настоящее изобретение и описано конкретно посредством вариантов реализации и необязательных признаков, модификации и вариации изобретения, предложенного в качестве вариантов реализации, могут быть осуществлены специалистом в данной области техники и такие модификации и вариации считаются находящимися в рамках настоящего изобретения.
В настоящей заявке изобретение описано широко и в общих чертах. Каждая из более узких интерпретаций и групп, входящих в общее описание изобретения, также является частью настоящего изобретения. Это включает общее описание изобретения с условием или отрицательным ограничением, исключающим любой объект из общего описания, независимо от того, упомянут ли данный исключенный материал конкретно в настоящей заявке.
Другие варианты реализации изобретения находятся в рамках следующей формулы изобретения. Кроме того, если признаки или аспекты настоящего изобретения описаны с применением групп Маркуша, специалистам в данной области будет понятно, что настоящее изобретение также описано для каждого индивидуального представителя или подгруппы из групп Маркуша.
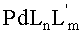
Термостойкий кристаллический гидрохлорид эпирубицина и способ его получения

