Результат интеллектуальной деятельности: ДИФОСФИНЫ, КАТАЛИЗАТОР СИНТЕЗА СЛОЖНЫХ ЭФИРОВ НА ИХ ОСНОВЕ И СПОСОБ СИНТЕЗА СЛОЖНЫХ ЭФИРОВ В ЕГО ПРИСУТСТВИИ
Вид РИД
Изобретение
Изобретение относится к получению сложных эфиров, которые широко используются в различных отраслях промышленности и быту.
Известно, что сложные эфиры, например различные эфиры циклогексанкарбоновой кислоты, находят приложение в качестве пластификаторов [К.Colle, J.E.Stanat, J.J.Reinoso, A.D.Godwin, WO Patent 2009/070398 to (2009) ExxonMobil], полупродуктов синтеза фармакологических препаратов [U.Hoffmann, M.Jansen, R.Reents, H.Stahr, US Patent 2009/0253927 (2009) to Hoffmann-La Roch Inc], жидких кристаллов [S.Sugimori, US Patent 4617142 to Chisso Corp.(1986)], душистых веществ [К.Jenni, О.Springer, US Patent 20100068160 to Evonik Goldschmidt GmbH (2010)] и др.
Одним из наиболее эффективных и современных методов синтеза сложных эфиров является каталитическое гидрокарбоалкоксилирование алкенов, которое протекает в одну стадию исходя из широкодоступных алкенов, спиртов и окиси углерода [G.Kiss, Chem. Rev., 2001, 101, pp.3435-3456]. Этот процесс уже нашел свое применение в промышленности. Так, например, несколько лет назад был коммерциализирован процесс гидрокарбометоксилирования этилена [Т.Fanjul, G.Eeastham, N.Fey, А.Hamilton, A.G.Orpen, P.G.Pringle, M. Waugh, Organometallics 2010, 29, pp.2292-2305].

Среди каталитических систем, используемых в гидрокарбоалкоксилировании алкенов, особый интерес представляют производные палладия, промотированные сильными протонными кислотами и различными фосфинами [G.Kiss, Chem. Rev., 2001, 101, pp.3435-3456]. В ряду последних, все большее значение приобретают дифосфины.
Известно использование трифенилфосфина (ТФФ) как промотора катализатора синтеза сложных эфиров, но эффективность полученного катализатора невелика.
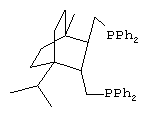
Наиболее близким аналогом настоящего изобретения являются транс-2,3-бис(дифенилфосфинометил)-норборнан (ТБДФФМН), отвечающий структурной формуле
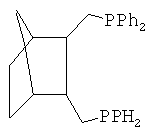
Катализатор синтеза сложных эфиров на основе производных палладия, модифицированный описанным дифосфином, проявляет активность в способе синтеза сложных эфиров, включающем смешение в среде растворителя алкена, СО и спирта с добавлением этого катализатора и кислоты и нагревание до температур 50÷150°C [I.E.Nifant′ev, N.T.Sevostyanova, V.A.Averyanov, S.A.Batashev, A.A.Vorobiev, S.A.Toloraya, V.V.Bagrov, A.N.Tavtorkin. Applied Catalysis, A: General. 2012, v.449, pp.145-152].
Однако для получения достаточно эффективного катализатора синтеза сложных эфиров на его основе требуется использовать существенные избытки дифосфина по отношению к взятому ацетату палладия. Это является важным недостатком из-за того, что стоимость дифосфинов, таких как ТБДФФМН, обычно является высокой.
Задачей настоящего изобретения является создание эффективной каталитической системы для производства сложных эфиров гидрокарбоалкоксилированием алкенов, на основе дифосфина, соли палладия и кислоты, проявляющей высокую активность при более низком содержании дорогостоящего дифосфина.
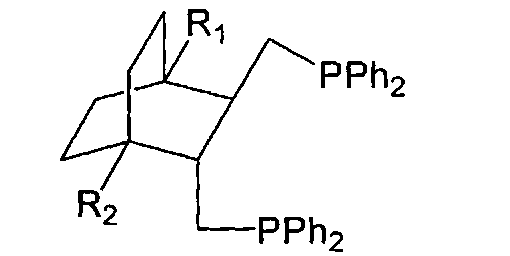
Поставленная задача решается тем, что впервые удалось синтезировать дифосфины общей структурной формулы:
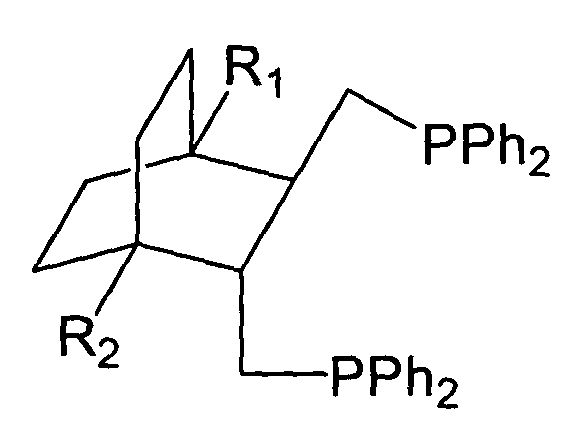
где R1,R2=H, Alk, Ar, ОМе, кроме R1=R2=H, Me - метил.
Структура полученных соединений установлена с помощью данных ЯМР (ядерно-магнитно-резонансной) спектроскопии и элементного анализа, приведенных в каждом конкретном примере получения соединений, а структурные формулы конкретных соединений представлены в табл.1.

Поставленная задача также решается тем, что предложен катализатор синтеза сложных эфиров на основе производных палладия, промотированных кислотами и фосфином, который в качестве фосфина содержит указанный дифосфин при соотношении дифосфин/Pd, равном 1-3.
Поставленная задача также решается тем, что предложен способ синтеза сложных эфиров путем гидрокарбоалкоксилирования алкенов в среде растворителя, СО и спирта в присутствии каталитических систем на основе производных палладия, промотированных кислотами и фосфином при повышенной температуре и давлении, в котором в качестве фосфина используют указанный дифосфин при соотношении дифосфин/Pd, равном 1-3.
В качестве алкенов используют циклические алкены или внутренние алкены, или терминальные алкены, или алкены, содержащие арильный заместитель.
В качестве производных палладия используют ацетат палладия.
В качестве кислот используют толуолсульфокислоту, а температуру гидрокарбоалкоксилирования поддерживают 80-115°C.
Техническим результатом изобретения является повышение степени конверсии реагентов при уменьшении расхода дорогостоящих дифосфинов на синтез сложных эфиров.
Данный результат может быть достигнут и в случае, если дифосфины содержат в структурной формуле OR вместо ОМе, где R - алкил.
Нижеследующие примеры иллюстрируют изобретение, но никоим образом не ограничивают его.
Полученные дифосфины синтезируют по представленным ниже методикам.
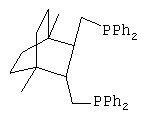
1. Синтез транс-({3-[(дифенилфосфино)метил]-1,4-диметил-бицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфина (Дифосфин 1).
Диметиловый эфир транс-1,4-диметилбицикло[2.2.2]окт-5-ен-2,3-дикарбоновой кислоты. К 12.4 г (0.12 моль) 1,4-диметил-1,3-циклогексадиена [Pearson, Anthony J.; Khetani, Vikram D. Journal of the American Chemical Society, 1989, vol. 111, #17, p.6778-6789] прибавляют по каплям 13 мл (0.12 моль) хлорангидрида фумаровой кислоты. Реакционную смесь перемешивают в течение часа, затем при охлаждении водой со льдом прибавляют по каплям 20 мл метанола. Через 16 часов избыток метанола отгоняют в вакууме, остаток растворяют в 100 мл бензола, промывают 100 мл насыщенного раствора Na2CO3, высушивают над MgSO4, отгоняют растворитель в вакууме. Остаток используют в дальнейшем без дополнительной очистки. Выход 30 г (94%).
Спектр ЯМР 31P (161 МГц, CDCl3, δ, м.д.): 6.03 (д, 1Н), 5.96 (д, 1Н), 3.69 (с, 3Н), 3.63 (с, 3Н), 2.78 (d, 1Н), 2.50 (дд, 1Н), 1.88 (м, 2Н), 1.60 (м, 2Н), 1.17 (с, 3Н), 1.18 (с, 3Н).
Диметиловый эфир транс-1,4-диметилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты. Смесь 30 г (0.12 моль) диметилового эфира 1,4-диметилбицикло[2.2.2]окт-5-ен-2,3-дикарбоновой кислоты и 0.70 г Pd/C в 150 мл метанола гидрируют водородом при комнатной температуре в течение 5 часов. По окончании реакции реакционную смесь отфильтровывают, растворитель отгоняют в вакууме, остаток высушивают. Выход 18 г (45%).
Спектр ЯМР 1Н (400МГц, CDCl3): δ=3.66 (с, 6Н), 1.95 (с, 2Н), 1.81 (м, 2Н), 1.54 (м, 2Н), 1.40 (м, 2Н), 1.20 (м, 2Н), 0.87 (с, 6Н).
Транс-2,3-бис(гидроксиметил)-1,4-диметилбицикло[2.2.2]октан. К суспензии 4 г (105 ммоль) LiAlH4 в 80 мл абсолютного эфира прикалывают раствор 18 г (70 ммоль) диметилового эфира 1,4-диметилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты в 40 мл абсолютного диэтилового эфира. Реакционную смесь кипятят в течение 4 часов. Затем при охлаждении обрабатывают 100 мл 10%-го раствора соляной кислоты, промывают 100 мл насыщенного раствора NaHCO3 и 100 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над Na2SO4, растворитель отгоняют в вакууме. Закристаллизовавшийся продукт в дальнейшем используют без дополнительной очистки. Выход 13.5 г (93%).
Спектр ЯМР 1Н (400МГц, CDCl3): δ=4.69 (с, 2Н), 3.85 (дд, 2Н), 3.33 (т, 2Н), 1.44 (м, 2Н), 1.35 (м, 4Н), 1.26 (м, 2Н), 0.84 (с, 6Н).
Дитозилат транс-2,3-бис(гидроксиметил)-1,4-диметилбицикло[2.2.2]октана. Смесь 13.5 г (68 ммоль) транс-2,3-бис(гидроксиметил)-1,4-диметилбицикло[2.2.2]октана, 39 г (204 ммоль) п-толуолсульфохлорида и 16.5 мл (204 ммоль) пиридина в 100 мл диэтилового эфира кипятят в течение 48 ч. Затем реакционную смесь отфильтровывают, обрабатывают 100 мл 10%-го раствора H2SO4, 150 мл насыщенного раствора NaHCO3, 200 мл воды и 100 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель отгоняют в вакууме, остаток перекристаллизовывают из этанола. Выход 33.5 г (94%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.80 (д, 4H), 7.38 (д, 4H), 4.07 (дц, 2H), 3.93 (дд, 2H), 2.47 (с, 6H), 1.45 (м, 2H), 1.30 (м, 4H), 1.22 (м, 4H), 0.73 (с, 6H).
Транс-({3-[(дифенилфосфино)метил]-4-метил-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин. К раствору 45 г (172 ммоль) трифенилфосфина в 230 мл абсолютного тетрагидрофурана добавляют 2.4 г (344 ммоль) мелкоизмельченного металлического лития и перемешивают реакционную смесь при комнатной температуре в течение 12 ч. Затем к реакционной смеси добавляют 33.5 г (66 ммоль) дитозилата 2,3-бис(гидроксиметил)-1,4-диметилбицикло[2.2.2]октана, растворенного в 100 мл абсолютного тетрагидрофурана и перемешивают при комнатной температуре в течение 2 часов. Затем обрабатывают 100 мл насыщенного водного раствора NH4Cl, 200 мл воды и 100 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель отгоняют в вакууме. Остаток перекристаллизовывают из этанола и бензола. Выход 12 г (34%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.56 (м, 4H), 7.39 (м, 10H), 7.33 (м, 6H), 2.42 (дд, 2H), 1.74 (м, 2H), 1.64 (м, 2H), 1.42 (м, 6H), 1.27 (м, 2H), 0.89 (с, 6H). Спектр ЯМР 31P (161МГц, CDCl3): β=-21.62.
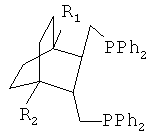
Получают дифосфин 1 - Транс-({3-[(дифенилфосфино)метил]-4-метил-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин, соответствующий структурной формуле
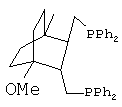
2. Синтез транс-({3-[(дифенилфосфино)метил]-4-метокси-1-метил-бицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфина (Дифосфин 2).
Диметил 1-метил-4-метоксибицикло[2.2.2]окт-5-ен-2,3-дикарбоксилат. К 1-метил-4-метокси-1,3-циклогексадиену [Birch, Journal of the Chemical Society, 1947, p.1642] (2.4 г, 22.6 ммоль) прибавляют по каплям 2.5 мл (23 ммоль) хлорангидрида фумаровой кислоты. Реакционную смесь перемешивают при нагревании (50°C) в течение часа, а затем при охлаждении водой со льдом прибавили по каплям 5 мл метанола. Через 2 часа избыток метанола удаляют в вакууме, а остаток растворяют в бензоле. Органический слой промывают 50 мл насыщенного раствора Na2CO3, затем 100 мл воды, а затем высушивают над MgSO4 и растворитель удаляют в вакууме. Остаток используют в дальнейшем без дополнительной очистки. Выход 6.3 г (96%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=6.40 (д, 1H), 5.90 (д, 1H), 3.70 (с, 3H), 3.62 (с, 3H), 3.42 (с, 3H), 2.98 (дд, 1H), 2.87 (д, 1H), 1.99 (м, 1H), 1.66 (м, 1H), 1.16 (с, 3H).
2,3-Бис(гидроксиметил)-1-метил-4 метоксибицикло[2.2.2]окт-5-ен. К 1.2 г (31 ммоль) суспензии LiAlH4 в 50 мл абсолютного эфира прикапывают раствор диметилового эфира 1-метил-4-метоксибицикло[2.2.2]окт-5-ен-2,3 дикарбоновой кислоты (6.3 г, 23.5 ммоль) в 40 мл абсолютного диэтилового эфира. Реакционную смесь кипятят в течение 3 часов. Затем при охлаждении обрабатывают 100 мл 10%-го раствора HCl, промывают 100 мл насыщенного раствора NaHCO3 и 100 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над Na2SO4, растворитель отгоняют в вакууме. Закристаллизовавшийся продукт используют без дополнительной очистки. Выход 4.7 г (94%).
Спектр ЯМР 1H (400МГц, CSCl3): δ=6.36 (д, 1H), 5.82 д, 1H), 3.90 (м, 1H), 3.73 (м, 2H), 3.61 (уш.с, 3H), 3.47 (м, 1H), 3.37 (с, 3H), 1.50 (м, 8H), 1.25 (м, 2H), 1.13 (с, 3H).
2,3-Бис(гидроксиметил)-1-метил-4-метоксибицикло[2.2.2]октан.
Смесь 4.7 г (22 ммоль) 2,3-Бис(гидроксиметил)-1-метил-4
метоксибицикло[2.2.2]окт-5-ена и 200 мг Pd/C в 30 мл метанола гидрируют водородом в течение 5 часов. По окончании реакции реакционную смесь отфильтровывают, растворитель отгоняют в вакууме. Выход продукта 4.4 г (94%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=4.07 (уш.с, 2H), 3.80 (м, 2H), 3.40 (м, 2H), 3.16 (с, 3H), 1.89 (м, 1H), 1.63 (м, 5H), 1.41 (м, 4H), 0.82 (с, 3H).
Дитозилат 2,3-бис(гидроксиметил)-1-метил-4-метоксибицикло[2.2.2]октана. Смесь 4.4 г (20 ммоль) 2,3-бис(гидроксиметил)-1-метил-4-метоксибицикло[2.2.2]октана, 15.2 г (80 ммоль) п-толуолсульфохлорида и 6.5 мл (80 ммоль) пиридина в 30 мл диэтилового эфира кипятят в течение 48 ч. Затем отфильтровывают выпавший осадок, фильтрат промывают 100 мл 5%-го раствора соляной кислоты, 100 мл насыщенного раствора NaHCO3, 200 мл воды и 50 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель отгоняют в вакууме. Остаток перекристаллизовают из этанола. Выход 4 г (38%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.73 (м, 4H), 7.31 (м, 4H), 4.03 (м, 2H), 3.93 (м, 2H), 2.91 (с, 3H), 2.39 (с, 6H), 1.76 (м, 1H), 1.55 (м, 4H), 1.35 (м, 5H), 0.69 (с, 3H).
({3-[(дифенилфосфино)метил]-4-метокси-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин. К раствору 4 г (15.3 ммоль) трифенилфосфина в 20 мл абсолютного тетрагидрофурана добавляют 0.2 г (31 ммоль) мелкоизмельченного металлического лития и перемешивают реакционную смесь при комнатной температуре в течение 12 ч. Затем прибавляют по каплям раствор 4 г (76.5 ммоль) дитозилата 2,3-бис(гидроксиметил)-1-метил-4-метоксибицикло[2.2.2]октана в 10 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Затем обрабатывают 100 мл водного насыщенного раствора NH4Cl, 200 мл воды и 50 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель отгоняют в вакууме. Продукт выделяют флэш-хроматографией на колонке с силикагелем элюированием системой бензол-петролейный эфир (1:5), а затем бензолом. Выход 1.5 г (36%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.60 (м, 4H), 7.36 (м, 16H), 2.83 (с, 3H), 2.53 (м, 2H), 2.10 (м, 1H), 1.95-1.42 (м., 11H), 0.81 (с, 3H). Спектр ЯМР 31P (161МГц, CDCl3, δ, м.д.): -20.21 (d, 1P), -23.15 (d, 1P).
Получают дифосфин 2 - ({3-[(дифенилфосфино)метил]-4-метокси-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин, соответствующий структурной формуле
3. Синтез транс-({3-[(дифенилфосфино)метил]-4-изопропил-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфина (Дифосфин 3).
Диметиловый эфир транс-1-изопропил-4-метилбицикло[2.2.2]окт-5-ен-2,3-дикарбоновой кислоты. К 32 г (0.23 моль) а-терпинена прибавляют по каплям 25 мл (0.23 моль) хлорангрида фумаровой кислоты. Реакционную смесь перемешивают в течение часа при нагревании (50°C). Затем к реакционной смеси прибавляют по каплям 50 мл метанола при охлаждении водой со льдом. Реакционную смесь кипятят 1 час, избыток метанола упаривают в вакууме, полученный продукт используют в дальнейшем без очистки. Выход 54 г (83%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=6.15 (дд, 1H), 6.03(дд, 1H), 4.08 (уш.с, 1H), 3.68 (с, 3H), 3.63(с, 1H), 2.71 (м, 2H), 1.81 (м, 2H), 1.58 (м, 3H), 1.18 (с, 3H), 1.04 (м, 3H), 0.95(м, 3H).
Диметиловый эфир транс-1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты. Смесь 54 г (0.19 моль) диметилового эфира 1-изопропил-4-метилбицикло[2.2.2]окт-5-ен-2,3-дикарбоновой кислоты и 1 г катализатора Pd/C в 250 мл метанола гидрируют водородом при комнатной температуре в течение 4 часов. По окончании реакции смесь отфильтровывают, растворитель упаривают в вакууме. Выход продукта 52.5 г (97%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=3.67 (с, 3H), 3.65 (с, 3H), 3.02 (дц, 1H), 2.65 (дд, 1H), 1.75 (м, 2H), 1.55 (м, 4H), 1.38 (м, 2H), 1.21 (м, 1H), 0.87 (д, 3H), 0.84 (с, 3H), 0.80 (д, 3H).
Транс-1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновая кислота. К раствору 24 г (0.085 моль) диметилового эфира 1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты в 200 мл тетрагидрофурана прибавляют небольшими порциями 11.2 г, (0.2 моль) тонкоизмельченного КОН и кипятят смесь в течение 7 часов. По окончании реакции смесь обрабатывают 500 мл воды, добавляют соляной кислоты до кислой реакции среды, выпавший осадок отфильтровывают, промывают гексаном и высушивают. Выход 14 г (64.7%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=3.11 (д, 1H), 2.76 (д, 1H), 1.37-1.18 (группа сигналов, 8H), 0.98 (с, 3H), 0.95 (д, 3H), 0.84 (д, 3H).
Диметиловый эфир 1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты. Смесь 30 г (0.19 моль) 1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты, 26 мл (0.35 моль) SOCl2, 100 мл бензола и нескольких капель ДМФА кипятят в течение часа. Затем растворитель и избыток SOCl2 отгоняют в вакууме, к остатку прибавляют по каплям 20 мл метанола и кипятят смесь в течение 2 часов. Затем бензол и избыток метанола упаривают в вакууме, полученный продукт используют без очистки. Выход 12.5 г (75%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=3.67 (с, 3H), 3.65 (с, 3H), 3.02 (дд, 1H), 2.65 (дд, 1H), 1.75 (м, 2H), 1.55 (м, 4H), 1.38 м, 2H), 1.21 (м, 1H), 0.87 (д, 3H), 0.84 (с, 3H), 0.80 (д, 3H).
Транс-2,3-бис(гидроксиметил)-1-изопропил-4 метилбицикло[2.2.2]октан. К суспензии 2.5 г (66 ммоль) LiAlH4 в 50 мл абсолютного диэтилового эфира добавляют по каплям раствор 12.5 г (44 ммоль) диметилового эфира 1-изопропил-4-метилбицикло[2.2.2]октан-2,3-дикарбоновой кислоты в 30 мл абсолютного диэтилового эфира при перемешивании. Через 12 часов реакционную смесь кипятят в течение 3 часов, охлаждают, обрабатывают 200 мл 20%-го раствора H2SO4, затем 100 мл воды и, наконец, 100 мл насыщенного водного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель упаривают в вакууме, полученный продукт используют без очистки. Выход 8.0 г (80%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=4.43 (с, 2H), 3.86(м, 2H), 3.27 (м, 2H), 1.73 (д, 1H), 1.56 (м, 2H), 1.41 (м, 5H), 1.21 (м, 3H), 0.85 (с, 6H), 0.78 (д, 3H).
Дитозилат./иранс-2,3-бис(гидроксиметил)-1-изопропил-4 метилбицикло[2.2.2]октана. Смесь 4 г (17.7 ммоль) транс-2,3-бис(гидроксиметил)-1-изопропил-4 метилбицикло[2.2.2]октана, 13.5 г (70.8 ммоль) п-толуолсульфохлорида и 5.7 мл (70.8 ммоль) пиридина в 30 мл диэтилового эфира кипятят в течение 48 часов. Затем отфильтровывают выпавший осадок, фильтрат промывают 50 мл 10%-го раствора соляной кислоты, 100 мл насыщенного раствора NaHCO3, 100 мл воды и 50 мл насыщенного раствора NaCl. Органический слой отделяют и высушивают над MfSO4, растворитель упаривают в вакууме. Остаток перекристаллизовывают из этанола. Выход 4.8 г (51%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.80 (дд, 4H), 7.37 (дд, 4H), 4.08 (м, 2H), 3.87 (м, 2H), 2.47 (д, 6H), 1.55 (м, 2H), 1.35 (м, 5H), 1.18 (м, 3H), 1.02 (м, 1H), 0.74 (с, 3H), 0.70 (д, 3H), 0.58 (с, 3H).
({3-[(дифенилфосфино)метил]-4-изопропил-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин. К раствору 11.8 г (45 ммоль) трифенилфосфина в 100 мл абсолютном тетрагидрофуране добавляют 0.63 г (90 ммоль) мелкоизмельченного металлического лития. Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Затем при охлаждении (-70°C) прибавляют раствор 14.5 г (27 ммоль) дитозилата транс-2,3-бис(гидроксиметил)-1 -изопропил-4 метилбицикло[2.2.2]октана в 50 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Затем обрабатывают 100 мл насыщенного водного раствора хлорида аммония, 30 мл 10%-го раствора H2SO4, 100 мл воды и 50 мл насыщенного водного раствора NaCl. Органический слой отделяют и высушивают над MgSO4, растворитель упаривают в вакууме. Остаток растворяют в петролейном эфире, профильтровывают через слой Al2O3, фильтрат упаривают в вакууме. Остаток промывают пентаном и высушивают. Выход 4.5 г (36%).
Спектр ЯМР 1H (400МГц, CDCl3): δ=7.63 (м, 4H), 7.34 (м, 16H), 2.52 (м, 2H), 1.87 (м, 1H), 1.71 (м, 3H), 1.60 (м, 2H), 1.40 (м, 5H), 1.19 (м, 2H), 0.81 (с, 3H), 0.73 (д, 3H), 0.40 (д, 3H). ЯМР 31Р (161МГц, CDCl3): δ=-23.37 (д, 1P), -25.23 (д, 1P).
Получают дифосфин 3- ({3-[(дифенилфосфино)метил]-4-изопропил-1-метилбицикло[2.2.2]окт-2-ил}метил)(дифенил)фосфин, соответствующий структурной формуле
Обнаружено, что синтезированные новые дифосфины обладают промотирующими свойствами по отношению к катализатору гидрокарбометоксилирования алкенов с синтезом сложных эфиров и исследованы в этой реакции.
Методика гидрокарбометоксилирования алкенов и анализа реакционной массы.
Синтез сложных эфиров гидрокарбоалкоксилированием алкенов проводят в периодическом реакторе, который представляет собой автоклав, снабженный рубашкой для обогрева и пробоотборником. В качестве спирта в настоящей работе используют метанол, а в качестве кислоты - толуолсульфокислоту (ТСК).
Гидрокарбоалкоксилирование алкенов (с синтезом сложных эфиров) проводят в периодическом реакторе, который представляет собой автоклав, снабженный рубашкой для обогрева и пробоотборником. Автоклав вместимостью 120 мл изготовлен из диамагнитной нержавеющей стали X18H10T, что обеспечивает возможность эффективного перемешивания с помощью магнитной мешалки. Температуру в автоклаве контролируют с помощью предварительно откалиброванной хромель-копелевой термопары. Поддержание заданной температуры в реакторе обеспечивают циркулированием через рубашку термостатируемого высокотемпературного органического теплоносителя (ВОТ). Давление в автоклаве измеряют образцовым манометром, закрепленным на его крышке.
Эксперимент включает следующую последовательность операций. В автоклав загружают раствор, содержащий все реагенты (см. Таблицу 2) и внутренний стандарт для хроматографии в растворителе (толуоле). Компоненты каталитической системы помещают в корзинку, которая располагается в верхней части автоклава над раствором; автоклав закрывают, дважды продувают инертным газом и дважды CO. Затем автоклав заполняют CO до достижения давления 0,5-1,0 МПа и нагревают реакционную массу до заданной температуры (см. Таблицу 2) при интенсивном ее перемешивании. При достижении заданной температуры давление доводят до установленного значения (2,1 МПа), в реакционную массу опускают шток с корзинкой, содержащей компоненты каталитической системы, обеспечивая ее ввод в реакционное пространство (момент ввода компонентов каталитической системы принимают за время начала реакции). По окончании опыта автоклав охлаждают, сбрасывают давление и извлекают реакционную массу.
Анализ реакционной массы гидрокарбометоксилирования проводят методом газо-жидкостной хроматографии на хроматографе «Цвет 162» с пламенно-ионизационным детектором.
Хроматографические расчеты проводят в программе МультиХром. Хроматографические пики реагентов и продуктов идентифицируют по временам удерживания, а содержание компонентов определяют методом внутреннего стандарта (в случае анализа реакционной массы гидрокарбометоксилирования циклогексена и октена-1) и внутренней нормализации (в случае анализа реакционной массы гидрокарбометоксилирования стирола). В качестве внутреннего стандарта используют о-ксилол. Точность определений концентраций реагентов и продуктов реакции составляет±3%.
Все опыты проводят в среде толуола при Рсо=2,1 МПа; концентрации, моль/л: [Pd(OAc)2]=0,001, [TsOH]=0,012, [мономер]=0,1, [СН3ОН]=0,45.
Примеры 1-4.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, при различных температурах - 80°C, 95°C, 105°C и 115°C соответственно в присутствии каталитической системы, промотированной дифосфином 1 при соотношении дифосфин/Pd, равном 1.
Результаты экспериментов приведены в Таблице 2.
Пример 5.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином 1 при соотношении дифосфин/Pd, равном 1 при 105°C, и времени гидрокарбоалкоксилирования, равном 90 мин.
Результаты экспериментов приведены в Таблице 2.
Пример 6.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием октена-1, как описано выше, в присутствии каталитической системы, промотированной дифосфином 1 при соотношении дифосфин/Pd, равном 1 при 80°C, и времени гидрокарбоалкоксилирования, равном 180 мин.
Результаты экспериментов приведены в Таблице 2.
Пример 7
Проводят синтез сложных эфиров гидрокарбоалкоксилированием стирола, как описано выше, в присутствии каталитической системы, промотированной дифосфином 1 при соотношении дифосфин/Pd, равном 1 при 80°C, и времени гидрокарбоалкоксилирования, равном 360 мин.
Результаты экспериментов приведены в Таблице 2.
Пример 8.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином 2 при соотношении дифосфин/Pd, равном 3 при 105°C, и времени гидрокарбоалкоксилирования, равном 180 мин.
Результаты экспериментов приведены в Таблице 2.
Пример 9.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином 2 при соотношении дифосфин/Pd, равном 1 при 105°C, и времени гидрокарбоалкоксилирования, равном 180 мин.
Результаты экспериментов приведены в Таблице 2.
Примеры 10-11.
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином 3 при соотношении дифосфин/Pd, равном 1 при 105°C, и времени гидрокарбоалкоксилирования, равном 90 и 180 мин соответственно.
Результаты экспериментов приведены в Таблице 2.
Пример 12 (сравнительный).
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином - транс-({3-[(дифенилфосфино)метил] бицикло[2.2.2]окт-2-ил}метил)-(дифенил)фосфина, соответсвующим структурной формуле
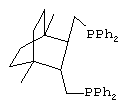 ,
,
и синтезированном по методике, описанной [Dang,T.P. et al. Journal of Organometallic Chemistry, 1975, vol. 91, p.105 - 115].
Этот дифосфин ближе по структуре к новым синтезированным дифосфинам, но хуже по достигаемому результату, чем дифосфин, выбранный за прототип.
Так, при оптимальных условиях гидрокарбоалкоксилирования циклогексена (при температуре 105°C и времени гидрокарбоалкоксилирования 180 мин) в присутствии каталитической системы, промотированной сравнительным дифосфином при соотношении дифосфин/Pd, равном 1, достигается конверсия 94.5% масс.
Результаты экспериментов приведены в Таблице 2.
Пример 13 (сравнительный по прототипу).
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной дифосфином - прототипом - транс-2,3-Бис-(дифенфосфинометил)норборнан, полученным по методике, и отвечающим структурной формуле, как указано ранее.
Так, при оптимальных условиях гидрокарбоалкоксилирования циклогексена (при температуре 105°C и времени гидрокарбоалкоксилирования 180 мин) в присутствии каталитической системы, промотированной дифосфином - прототипом при соотношении дифосфин/Pd, равном 3,5, достигается конверсия 100,0% масс. Однако при снижении соотношения дифосфин/Pd до 1 (пример 14), конверсия падает до 28.0% масс.
Результаты экспериментов приведены в Таблице 2.
Примеры 15-16 (сравнительные).
Проводят синтез сложных эфиров гидрокарбоалкоксилированием циклогексена, как описано выше, в присутствии каталитической системы, промотированной известным фосфином Ph3P при соотношении фосфин/Pd, равном 65/1 и 2/1, достигается конверсия 27,2 и 2,5% масс, соответственно.
Результаты экспериментов приведены в Таблице 2.
|
|
Таким образом, предлагаемое техническое решение позволяет проводить синтез сложных эфиров заданного состава эффективным способом с достижением высокой степени конверсии при отсутствии избытка дифосфина или же избытке не более 3 по отношению к палладию.
Способ получения синтетической нефти из природного или попутного нефтяного газа (варианты)
Способ измерения скорости циркуляции мелкодисперсного катализатора
Способ получения винилиденовых олефинов
Способ получения суспензии катализатора гидроконверсии тяжелого нефтяного сырья
Способ получения глюкозочувствительных полимерных гидрогелей
Способ получения композиционной мембраны и композиционная мембрана, полученная этим способом
Способ получения микро-мезопористого цеолита y и цеолит, полученный этим способом
Способ получения стирола из отходов полистирола
Способ удаления диоксида углерода из газовых смесей
Способ получения высокоплотного реактивного топлива для сверхзвуковой авиации
Способ получения винилиденовых олефинов
Металлоценовые соединения
Способ олигомеризации этилена в среде органического растворителя в присутствии хромового катализатора и алюминийорганического активатора
Способ получения противотурбулентных присадок для применения в условиях низких температур транспортируемой среды

