ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение относится к органическим соединениям, их фармацевтическим композициям и их применению для лечения и/или профилактики млекопитающих, в частности к гетероциклическим соединениям, которые модулируют сигнальный путь hedgehog (семейство белков, необходимых для передачи сигнала дифференцирования ткани).
Предпосылки создания изобретения
Автопротеолиз белка предшественника Shh человека в 45 кДа приводит к N-терминальному фрагменту в 20 кДа, который отвечает за нормальную передачу сигнала hedgehog и C-терминальный фрагмент в 25 кДа, вовлеченный в активность автопереработки, в которой N-терминальный фрагмент связывается с холестерином (Lee et al. Science 266: 1528-1537 (1994) и Bumcrot et al. Mol. Cell Biol. 15: 2294-2303 (1995)).
Обычно функционирующая передача сигнала Hedgehog (Hh) определяет зародышевую модель за счет направления клеточной дифференциации и пролиферации, о чем впервые сообщалось для Drosophilia melanogaster (Nusslein-Vollhard et al. Roux. Arch. Dev. Biol. 193: 267-282 (1984)). Клеточные ответные реакции на секретируемый полипептид Hh опосредуются двумя интегральными мембранными белками, Patched (Ptc) и Smothened (Smo). Hh связывается с двенадцатым трансмембранным белком Ptc и, следовательно, обращает Ptc-опосредованное подавление седьмого трансмембранного белка Smo. Такая активация Smo затем запускает ряд внутриклеточных событий, завершающийся стабилизацией фактора транскрипции Cubitus interruptus (Ci) и экспрессией Ci-зависимых генов. Данные события повторяются во время развития млекопитающего и генеза опухоли через множество гомологов белка, включая три различных члена семейства Hh [Sonic (Shh), Indian (Ihh) и Desert (Dhh)] и три Ci-подобных фактора транскрипции (Gli1, Gli2 и Gli3). Однако существует единственный гомолог Smo позвоночных, который вовлечен во все формы передачи сигнала Hh при генном анализе дрозофил, мышей и данио-рерио (zebrafish) (Chen et al. PNAS 99(22): 14071-14076 (2002)).
Smo инициирует сигнальный каскад, вызывая активацию факторов транскрипции Gli и их последующую транслокацию в ядре, приводящую к контролю транскрипции целевых генов. Через отрицательный контур обратной связи Gli оказывает влияние на транскрипцию Ptc и Hip1 (hedgehog-взаимодействующий белок 1 (Hip 1)), который ингибирует путь Hh. Утрата контроля за активацией пути Hh связана с увеличением ряда раковых заболеваний, включая те, которые поражают мозг, такие как медуллобластома (Romer and Curran, Cancer Res 65(12): 4975-4978 (2005)) и глиобластома (Bar et al. Stem Cells 25(10): 2524-33 (2007)); рак предстательной железы (Sanchez et al. PNAS 101(34): 12561-12566 (2004)); рак поджелудочной железы (Thayer et al. Nature 423: 851-856 (2003)); немелкоклеточная карцинома легких (Yuan et al. Oncogene 26: 1046-1055 (2007); мелкоклеточный рак легких (Watkins et al. Nature 422: 313-317 (2003)); рак молочной железы (Kubo et al. Cancer Res 64: 6071-6074 (2004)); различные опухоли пищеварительного тракта (Berman et al. Nature 425: 846-851 (2003)) и (Lees et al. Gastroenterology 129(5): 1696-1710 (2006)); базально-клеточная карцинома (Williams et al. PNAS 100(8): 4616-4621 (2003)); злокачественная меланома (Pons and Quintanilla Clin. Trans. Oncol. 8(7): 466-474 (2006)); плоскоклеточная карцинома (Xuan et al. Mod Pathol. 19(8): 1139-47 (2006)); злокачественные новообразования B-клеток, такие как множественная миелома и лимфомы (Dierks et al. Nat. Med. 13(8): 944-951 (2007); Peacock et al. PNAS 104(10): 4048-4053 (2007)); мезенхимальные виды рака, такие как хондросаркома (Tiet et al. Am. J. Pathol. 168(1): 321-330 (2006)), светлоклеточная карцинома почки (Cutcliffe et al. Clin Cancer Res. 11(22): 7986-94 (2005)) и рабдомиосаркома (Tostar et al. J. Pathol. 208(1): 17-25 (2006)); хроническая миелоидная лейкемия (Sengupta et al. Leukemia 21(5): 949-955 (2007)); эндометриальная карцинома (Feng et al. Clin. Cancer Res. 13(5): 1389-1398 (2007); гепатоклеточные карциномы (Huang et al. Carcinogenesis 27(7): 1334-1340 (2006)); опухоли яичников (Chen et al. Cancer Sci. 98(1): 68-76 (2007)).
Также было установлено, что Hh передача сигнала регулирует экспрессию транспортерных белков ABC, обладающего множественной устойчивостью к лекарственным средствам белка-1 (MDR1, ABCB1, P-гликопротеин) и (BCRP, ABCG2), и что направленный нокдаун экспрессии MDR1 и BCRP посредством небольшой перекрестно реагирующей РНК частично обращает вызванную Hh хеморезистентность. На этом основании можно было бы предположить, что путь Hh может являться мишенью для преодоления мультилекарственной резистентности и повышения химиотерапевтической ответной реакции (Sims-Mourtada et al. Oncogene 26(38): 5674-5679 (2007)). Было установлено, что блокирование sonic сигнального пути hedgehog усиливает антипролиферативное действие ингибиторов EGFR в раковых клетках поджелудочной железы (Hu et al. Acta Pharmacol Sin. 28(8): 1224-30 (2007)) и раковых клетках предстательной железы (Mimeault et al. Int. J. Cancer 118(4): 1022-31 (2006)).
Путь hedgehog также связан с повторным ростом опухоли после химио- и лучевой терапии и в качестве потенциальной мишени для улучшения ответной реакции на лучевую терапию (Sims-Mourtada et al. Clin. Cancer Res. 12(21): 6565-6572 (2006)), и циклопамин, антагонист пути hedgehog, повышает цитотоксическое действие паклитаксела и облучения в экспрессирующих Hh раковых клетках поджелудочной железы (Shafaee et al. Cancer Chemother. Pharmacol. 58(6): 765-70 (2006)).
Также сообщалось, что ингибирование сигнального пути Hedgehog может быть полезно для лечения ряда заболеваний, связанных с воспалением, гиперплазией эпителиальных клеток, фиброзом ткани, или иммунных нарушений (Lamb et al. EP1183040). Сообщалось, что ингибирование sonic передачи сигнала hedgehog снижает хроническое отторжение и продлевает долговечность аллотрансплантата на крысиной модели ортотопической трансплантации тонкой кишки. Хотя острое отторжение трансплантата можно контролировать с помощью иммуноподавляющих агентов, хроническое отторжение, которое характеризуется артериосклерозом в сосудах донорских органов, является основным препятствием для продолжительной долговечности аллотрансплантата. Долговечность трансплантата в крысиной модели ортотопической трансплантации тонкой кишки существенно продлевалась после обработки анти-Shh антителом по сравнению с иммуноглобулином G в качестве контроля. (116 против 77,5 дней). Осаждение коллагена и окклюзия сосудов в брыжейке существенно снижались у реципиентов анти-Shh антитела (Chen et al. Transplantation 83(10): 1351-1357 (2007); Lamb et al. EP1183040B1).
Также сообщалось, что sFRP-1 представляет собой расположенный по ходу транскрипции гена-мишени передачи сигнала Hh и что повышенная экспрессия секретируемого связанного с ожогом белка-1 (sFRP-1) после активации пути Hh обеспечивает молекулярную связь для ингибиторного действия на Wnt передачу сигнала (He et al. J. Biol. Chem. 281(47): 35598-35602 (2006)). Таким образом, модулирование Wnt-передачи сигнала за счет антагонистического действия на путь Hh через sFRP-1 могло бы обеспечить способ лечения ряда заболеваний, таких как остеопороз (Ai et al. Mol. Cell. Biol. 25(12): 4946-4955 (2005)), наряду с другими (Luo et al. Laboratory Investigation, 87, 97-103 (2007)).
Были исследованы различные ингибиторы пути Hh, включая природный продукт циклопамин, действие которого, как предполагается, заключается в связывании гептаспиралевидной области Smo. Кроме того, последние годы было описано несколько синтетических антагонистов с небольшими молекулами: обзор в этой области можно найти в Kiselyov, Anti-Cancer Agents in Medicinal Chemistry 6: 445-449 (2006).
Предшествующий уровень развития данной области
Lubisch и др. описывают ряд 2-фенилбензимидазолов в качестве ингибиторов PARP, которые можно использовать для лечения различных заболеваний, включая рак (WO2000026192), и в области косметики (WO2001082877). Повторяющейся характерной особенностью является присутствие карбамоильного фрагмента в 4-положении бензимидазольного кольца.
Arienti и др. (WO2003032984) и Ameriks и др. (WO2004093873 и US2004214857) описывают ряд производных 2-фенилбензимидазола в качестве ингибиторов контрольной точки киназы 2 для лечения рака, дополнительно характеризуемый тем, что 5-положение бензимидазольного кольца всегда является замещенным либо карбоксилатом, карбамоилом, либо сульфамоильной группой.
Ohemeng и др. (WO9911627 и US5942532) описывают ряд соединений 5-карбоксиимидамидо-2-фенилбензимидазолов в качестве антибактериальных агентов.
Mjalli и др. (WO2003075921) описывают фармацевтическое применение ряда производных 2-фенилбензимидазола.
Alekshun и др. (WO2004041209 и WO2006076009) описывают ряд производных 2-фенилбензимидазола, обладающих активностью в качестве антибиотиков.
Khaled и др. (Bulletin of the Faculty of Pharmacy (Cairo University), 40(1): 7-13, (2002)) описывают синтез и антигипертензивную активность производных 2-фенилбензимидазолов, тогда как ДНК-связывающие свойства некоторых других из них были описаны Kobuta и др. (Nucleic Acids Research Supplement, 2(Twenty-ninth Symposium on Nucleic Acids Chemistry), 193-194 (2002) и Nucleic Acids Symposium Series, 35(Twenty-third Symposium on Nucleic Acids Chemistry, 1996), 151-152 (1996)).
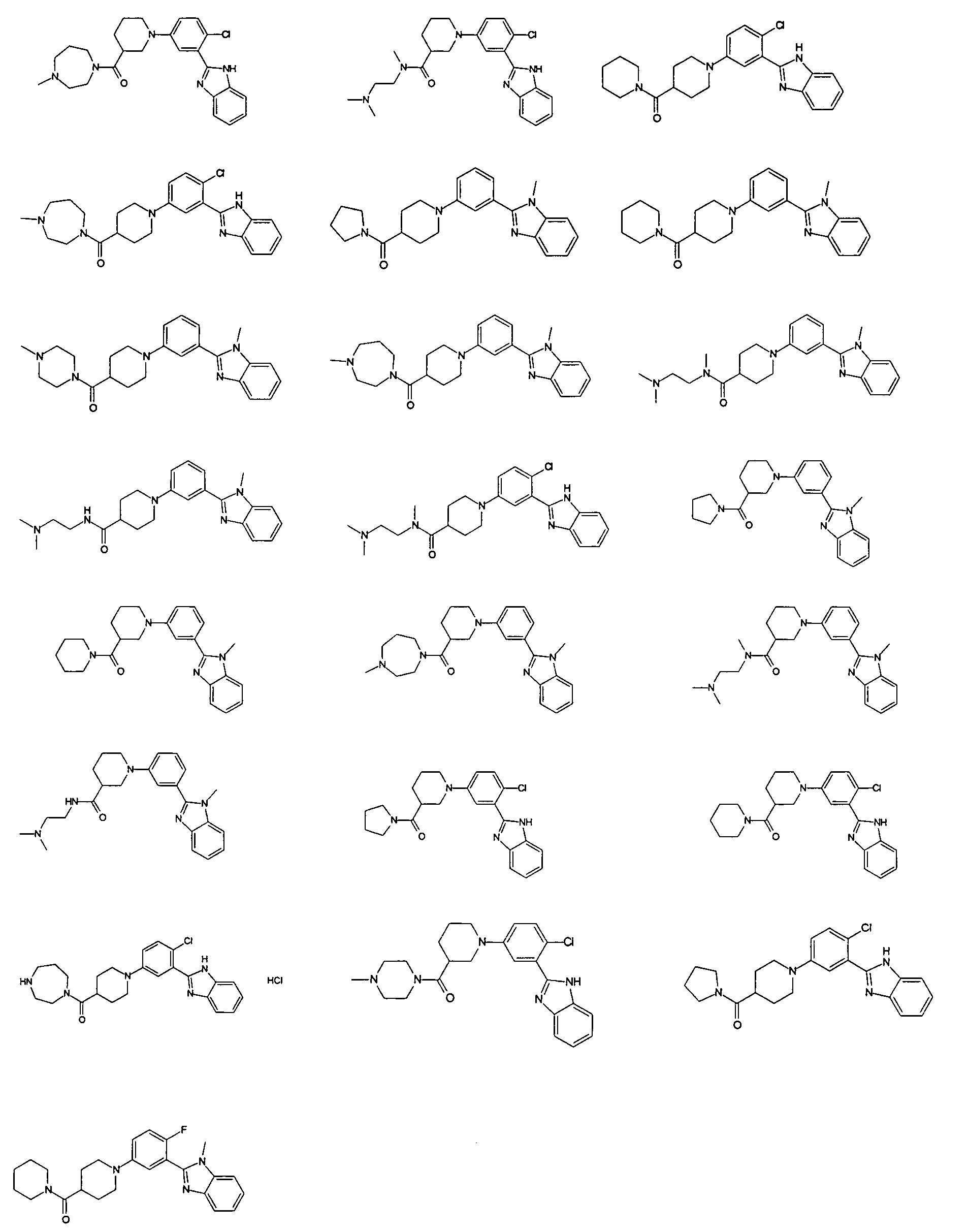
Guicherit и др. (WO2006050506), Beachy и др. (WO2003088970), Rubin и др. (WO2003011219), Yuach и др. (Nature, 455, 406 (2008) и Dakin и др. (WO2009027746) описывают арил- и алкиламидо/уреидопроизводные 2-фенилбензимидазола в качестве антагонистов сигнального пути Hedgehog для лечения различных форм рака. Guicherit и др. (WO2006050506) и Rubin и др. (WO2003011219) также описывают ариламидопроизводные 2-фенилимидазопиридина для той же цели. Следующие 22 соединения описаны в одновременно поданной заявке WO2009074300 от имени того же заявителя.
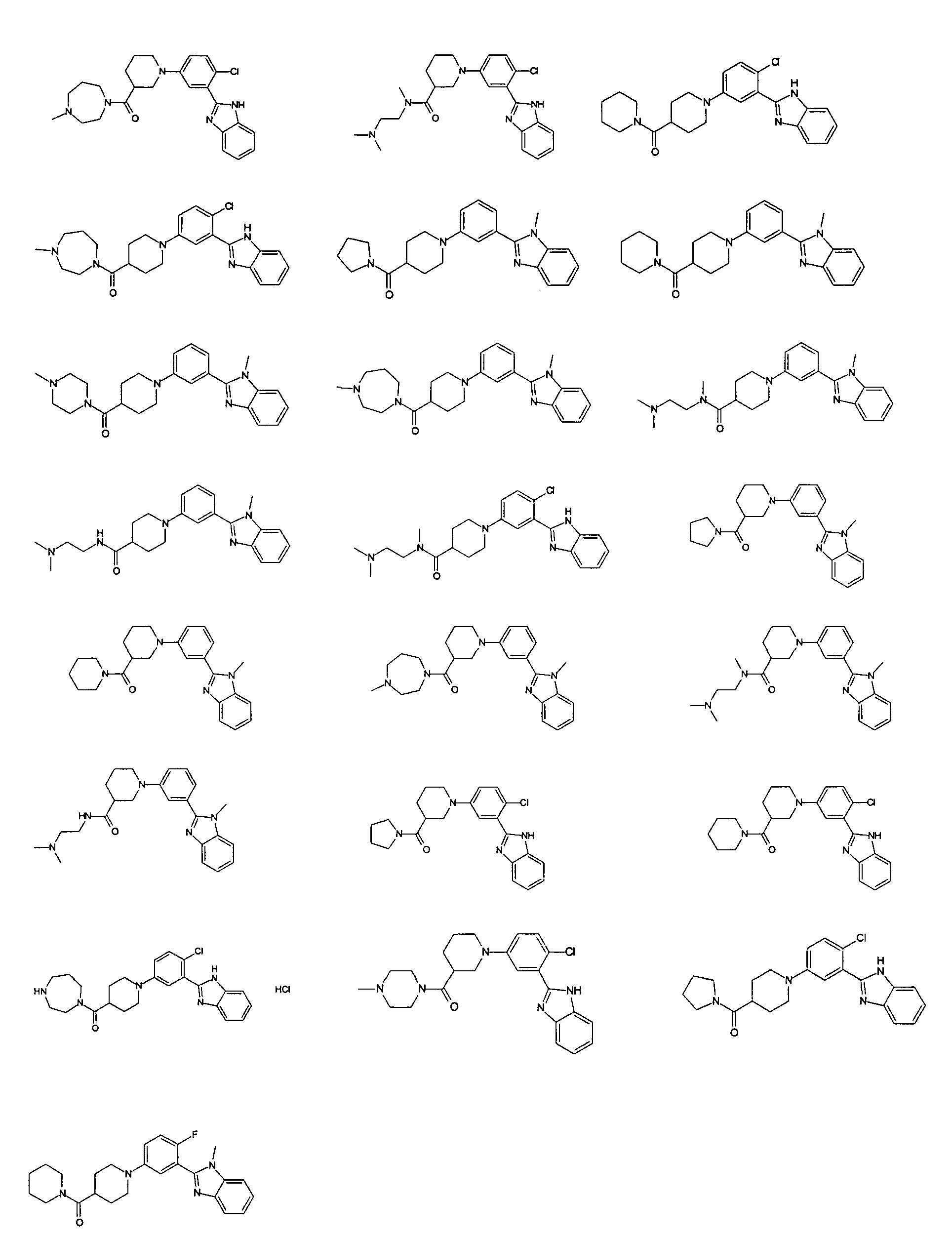
Подробное описание изобретения
Данное изобретение относится к соединениям формулы I
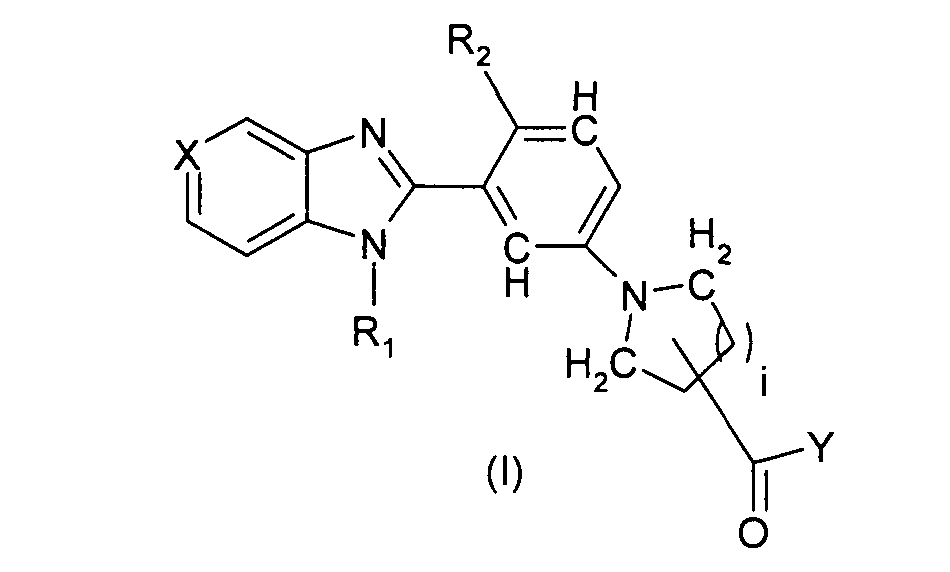
где, когда валентность и стабильность позволяют,
i может представлять собой 1 или 2,
R1 может представлять собой H; линейную, разветвленную или циклическую (C1-C4) алкильную группу,
R2 может представлять собой H, Cl или F,
X может представлять собой либо N, либо CR3,
R3 может представлять собой H; галоген; линейную, разветвленную или циклическую (C1-C4) алкильную или алкоксильную группу,
Y может представлять собой
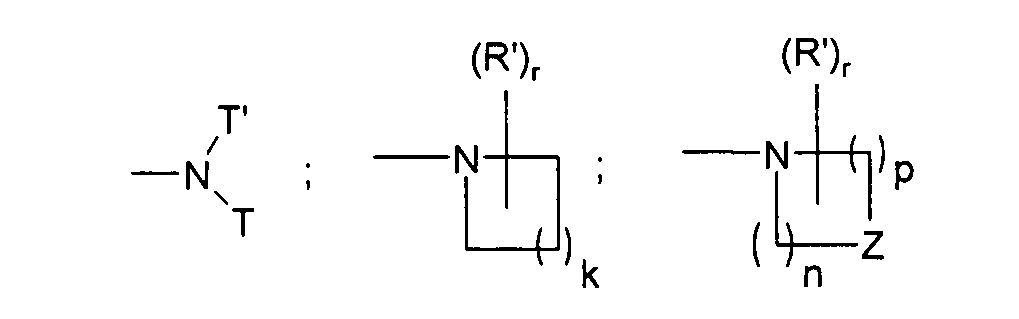
Z может представлять собой O или NRx,
Rx может представлять собой H или линейный, разветвленный или циклический (C1-C4) алкил,
k может представлять собой 1, 2, 3 или 4,
n и p независимо могут представлять собой 1, 2 или 3 и сумма n+p не может превышать 5,
T может представлять собой H или линейную или разветвленную (C1-C4) алкильную группу;
T' может представлять собой линейную или разветвленную C1-C3 алкильную цепь, замещенную либо (C1-C6)-диалкиламиногруппой, либо 4-6-членным насыщенным гетероциклом, содержащим один атом азота и необязательно содержащим второй гетероатом, выбранный из N и O, такое гетероциклическое кольцо необязательно является замещенным у атомов азота (C1-C4) алкильной цепью; или 4-6-членный насыщенный гетероцикл, содержащий один атом азота и необязательно содержащий второй гетероатом, выбранный из N и O, такое гетероциклическое кольцо необязательно является замещенным у атомов азота (C1-C4) алкильной цепью;
r может представлять собой ноль, 1, 2 или 3;
R' может представлять собой галоген; гидрокси; амино; циано; нитро; оксо; линейный или разветвленный (C1-C6) алкил, дигалогеналкил, азаалкил, оксаалкил, алкилкарбонил, оксаалкилкарбонил, алкоксикарбонил, алкиламинокарбонил, алкилкарбониламино, алкенил, оксаалкенил, азаалкенил, алкенилкарбонил, оксаалкенилкарбонил, алкенилоксикарбонил, алкениламинокарбонил, алкиламино, диалкиламино, меркаптоалкил, алкокси, алкилтиогруппу, необязательно замещенную одним или несколькими атомами фтора; где две группы R' могут образовывать 5-8-членное кольцо со спиро или конденсированным соединением.
И за исключением
В одном варианте осуществления i равно 2, -C(=О)-Y находится в 4-положении получающегося пиперидинового кольца и R1, R2, X, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено для формулы I выше. В другом варианте осуществления i равно 2, -C(=О)-Y находится в 3-положении получающегося пиперидинового кольца и R1, R2, X, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I; в одном варианте осуществления i равно 1 и R1, R2, X, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено для формулы I выше; R1 представляет собой H, R2 не является H, и i, X, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I; в другом варианте осуществления R2 представляет собой H, R1 не является H, и i, X, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I. В одном варианте осуществления X представляет собой N и i, R1, R2, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I; в другом варианте осуществления X представляет собой CR3, и i, R1, R2, R3, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I. В одном варианте осуществления R3 представляет собой H и i, R1, R2, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I. В другом варианте осуществления R3 представляет собой Cl, F, OMe и Me и i, R1, R2, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I. В другом варианте осуществления r равно нулю и i, R1, R2, Х, R3, Y, Z, Rx, k, n, p, T и T' являются такими, как определено выше для формулы I.
В предпочтительном варианте осуществления разработаны соединения формулы I выше, где Y представляет собой
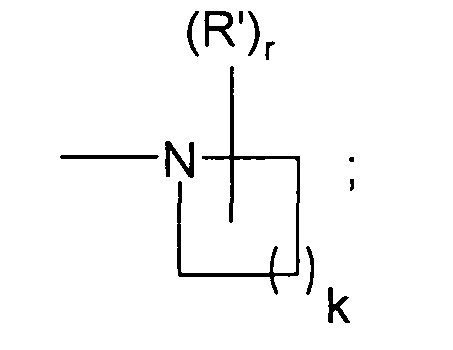
k равно 2, r равно 1, R' представляет собой диметиламино и i, R1, R2, X и R3 являются такими, как определено выше для формулы I.
Во втором предпочтительном варианте осуществления разработаны соединения формулы I выше, где Y представляет собой
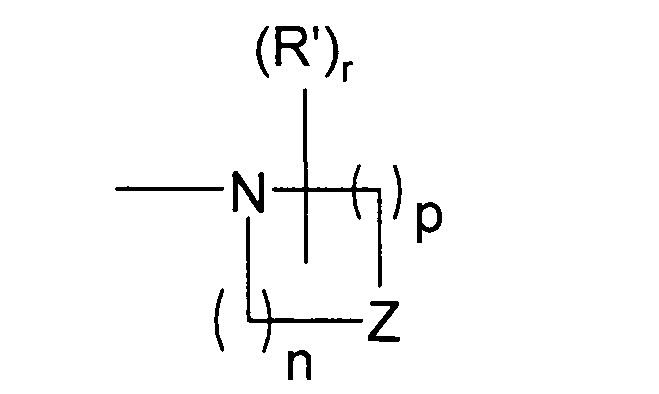
и где оба n и p равны 2, Z представляет собой O, r равно нулю и i, R1, R2, X и R3 являются такими, как определено выше для формулы I.
В третьем предпочтительном варианте осуществления разработаны соединения формулы I выше, где i равно 2 и -C(=О)-Y находится в 4-положении получающегося пиперидинового кольца, X представляет собой CR3, R3 представляет собой метил, R2 представляет собой F и где R1, Y, Z, Rx, k, n, p, T, T', r и R' являются такими, как определено выше для формулы I.
Особенно интересными являются следующие соединения:
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}пиперазин-1-илметанон;
азепан-1-ил-{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}метанон;
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}пирролидин-1-илметанон;
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}пиперидин-1-илметанон;
{(S)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}морфолин-4-илметанон;
{1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-4-ил}морфолин-4-илметанон;
(3-диметиламинопропил)метиламид 1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты;
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}-(4-метилпиперазин-1-ил)метанон;
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}-(4-пирролидин-1-илпиперидин-1-ил)метанон;
{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон;
{1-[4-хлор-3-(5-метокси-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон;
{(R)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}морфолин-4-илметанон;
{(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{(R)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{1-[4-фтор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон;
(3-диметиламинопирролидин-1-ил)-{(R)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон;
{(R)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{(S)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
(3-диметиламинопирролидин-1-ил)-{(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон;
(3-диметиламинопирролидин-1-ил)-{(R)-1-[3-(1-метил-1H-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон;
{(R)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{1-[4-хлор-3-(5-метокси-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-ил}морфолин-4-илметанон;
(3-диметиламинопирролидин-1-ил)-{1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-ил}метанон;
{(R)-1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{(R)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{(R)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{(S)-1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{(S)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{1-[4-хлор-3-(-1Н-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон;
{(R)-1-[4-хлор-3-(-1Н-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{(S)-1-[4-хлор-3-(-1Н-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон;
{1-[4-хлор-3-(-1Н-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{(S)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}-(3-диметиламинопирролидин-1-ил)метанон;
{1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон;
{1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}-(3-диметиламинопирролидин-1-ил)метанон
и {1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}морфолин-4-илметанон.
Фармакологическая активность иллюстративной группы соединений формулы I была продемонстрирована с использованием двух описанных ниже анализов in vitro. В соответствии со следующим аспектом настоящее изобретение относится к способу лечения рака или остеопороза, который включает введение субъекту, предпочтительно нуждающемуся в этом человеку, эффективного количества соединения формулы I. Типы рака, который можно лечить с использованием такого способа, не ограниченно включают немелкоклеточную карциному легких; мелкоклеточный рак легких; рак молочной железы; опухоли яичников; опухоли пищеварительного тракта; рак мозга, такой как медуллобластома и глиобластома; рак предстательной железы; рак поджелудочной железы; базально-клеточную карциному; злокачественную меланому; плоскоклеточную карциному; множественную миелому; лимфомы; мезенхимальные виды рака, такие как хондросаркома, светлоклеточная саркома почки и рабдомиосаркома; хроническую миелоидную лейкемию; эндометриальную карциному; гепатоклеточные карциномы.
Обычно соединения формулы I можно использовать для лечения любого заболевания, состояния или дисфункции, при которых может оказаться благоприятным ингибирование пути Hedgehog за счет связывания соединений с рецептором Smo, и включающих, но без ограничения, остеопороз и рак, выбранный из немелкоклеточной карциномы легких; мелкоклеточного рака легких; рака молочной железы; опухолей яичников; опухолей пищеварительного тракта; рака мозга, такого как медуллобластома и глиобластома; рака предстательной железы; базально-клеточной карциномы; злокачественной меланомы; плоскоклеточной карциномы; множественной миеломы; лимфом; мезенхимальных видов рака, таких как хондросаркома, светлоклеточная карцинома почки и рабдомиосаркома; хронической миелоидной лейкемии; эндометриальной карциномы; гепатоклеточных карцином.
Дозировка соединений для применения в терапии может изменяться в зависимости, например, от пути введения, природы и тяжести заболевания. Обычно приемлемый фармакологический эффект для людей может быть получен при дневной дозировке, колеблющейся от 0,01 до 200 мг/кг.
Еще в одном следующем аспекте изобретение относится к фармацевтической композиции, содержащей одно или несколько соединений формулы I в сочетании с фармацевтически приемлемыми носителями и эксципиентами. Фармацевтические композиции могут существовать в виде твердых, полутвердых или жидких препаратов, предпочтительно в виде растворов, суспензий, порошков, гранул, таблеток, капсул, сиропов, суппозиториев, аэрозолей или систем с контролируемой доставкой. Композиции можно вводить различными путями, включая пероральный, чрескожный, подкожный, внутривенный, внутримышечный, ректальный и интраназальный, и предпочтительно их получают в виде единичной препаративной лекарственной формы. Пероральные единичные препаративные лекарственные формы могут содержать от примерно 1 мг до примерно 1000 мг соединения по изобретению.
Для тех соединений, которые могут находиться в виде свободных оснований, данное изобретение также включает их кислотно-аддитивные соли, предпочтительно соли с фармацевтически приемлемыми кислотами. Изобретение также включает отдельные изомеры и диастереомеры соединений I или их смеси (например, рацемические смеси). Принципы и способы получения фармацевтических композиций описаны, например, в Remington's Pharmaceutical Science, Mack Publishing Company, Easton (PA).
Соединения формулы I, их оптические изомеры или диастереомеры могут быть очищены или разделены в соответствии с хорошо известными способами, включающими, но без ограничения, хроматографию с хиральной матрицей и дробную кристаллизацию.
Синтез соединений и экспериментальные способы
Соединения по настоящему изобретению могут быть получены с использованием различных синтетических путей, включая описанные ниже в качестве общих способов 1-11 и способов А-Т.
Материалы и методы
Все реагенты и растворители получены из коммерческих источников. Чувствительные к воздуху и влаге жидкие растворы переносили с помощью шприца. Ход реакций контролировали с помощью тонкослойной хроматографии (ТСХ) и/или жидкостной хроматографии с масс-спектрометрией (ЖХМС).
Все спектры ядерного магнитного резонанса регистрировали на спектрометре Varian Mercury Plus 400 МГц, оборудованном широкополосным зондом PFG ATB.
Методы с 10-минутным проходом осуществляли с использованием делительного модуля Waters 2795, обрудованного масс-спектрометром Micromass ZQ (ионизация методом электрораспыления (ES) и Waters PDA 2996, с использованием колонки Waters XTerra MS C18 3,5 мкм 2,1×50 мм.
Препаративную ВЭЖХ проводили с использованием системы Waters 2767 с бинарным насосом с градиентным модулем Waters 2525, соединенной с Waters Micromass ZQ (ES) или Waters 2487 DAD на колонке Supelco Discovery HS C18 5,0 м 10×21,2 мм.
Градиентное элюирование проводили с использованием либо способа а: 0,1% муравьиная кислота/вода и 0,1% муравьиная кислота/ацетонитрил с градиентом от 5/95 до 95/5 за указанное время прохода (поток: 1 мл/мин), либо способа b: 0,1% муравьиная кислота/вода и 0,1% муравьиная кислота/метанол с градиентом от 5/95 до 80/20 за указанное время прохода (поток: 0,8 мл/мин). Время прохода для конечных соединений составляло 10 минут.
Очистку проводили с использованием картриджей ISOLUTE Flash Si с силикагелем.
Все ТСХ анализы проводили на силикагеле (Merck 60 F254) и пятна проявляли с помощью УФ облучения при 254 нм и KMnO4 или нингидринового красителя.
ОБЩИЙ СПОСОБ 1
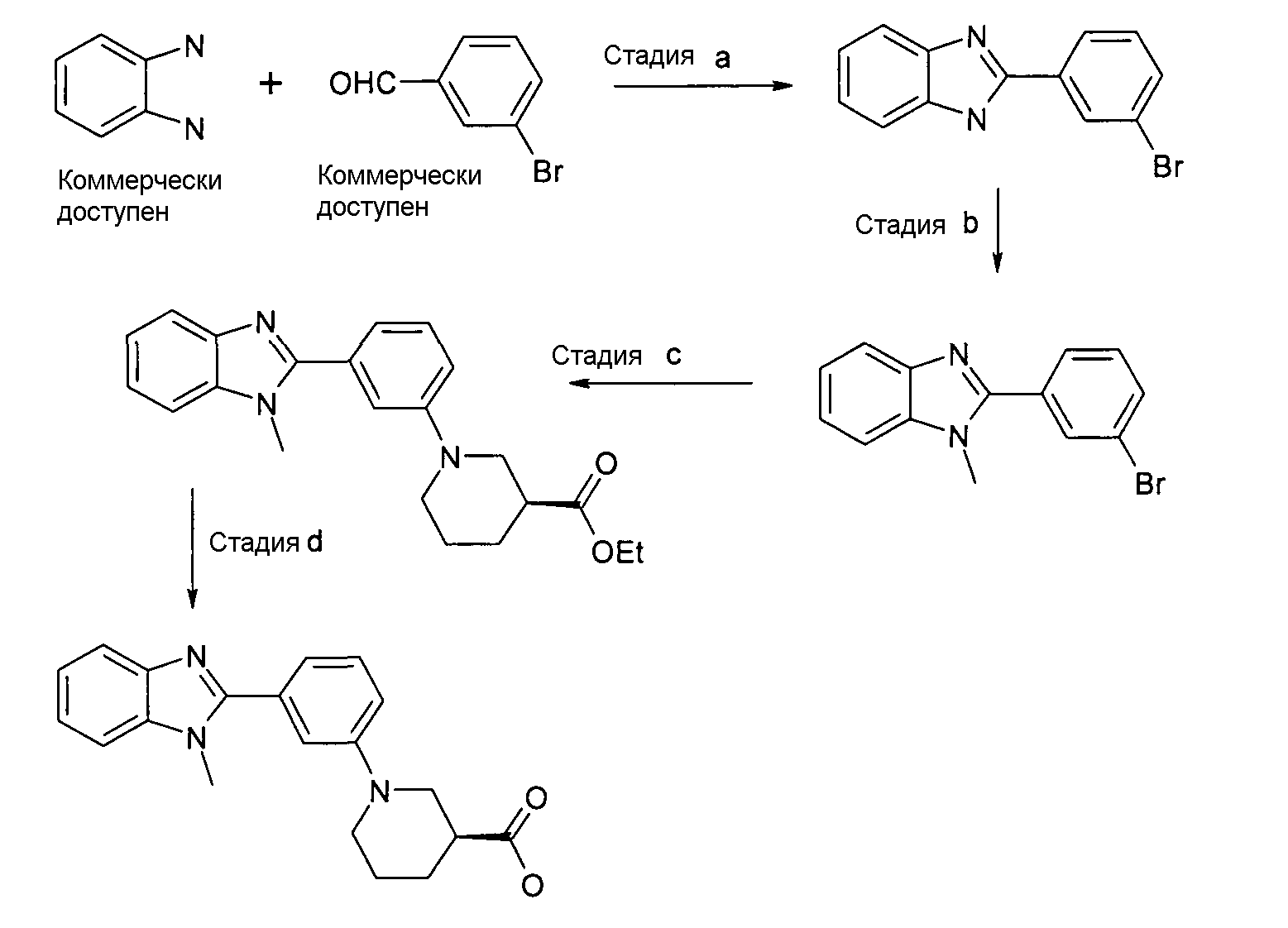
2-(3-бромфенил)-1Н-бензоимидазол
Способ 1 - Стадия a. О-фенилендиамин (81,8 г, 756,6 ммоль) и щавелевую кислоту (3,40 г, 37,8 ммоль) полностью растворяли в смеси EtOH-H2O/1:1 (2 л), предварительно нагретой до 80оC. Затем к раствору добавляли по каплям 3-бромбензальдегид (44,10 мл, 378,30 ммоль). Открытую для доступа воздуха реакционную смесь перемешивали в течение ночи при 70ºС. Через день твердое вещество отфильтровывали и растирали с MeOH (150 мл), получая продукт в виде бледно-желтого твердого вещества (27,50 г). 3,8 г выделяли из маточных растворов. Общий выход 31,30 г (30%).
1H-ЯМР (400 МГц, ДМСО): δ 7,24 (2H, м), 7,54 (2H, м), 7,70 (м, 2H), 8,19 (1Н, м), 8,37 (1Н, т), 13,2 (1Н, с); m/z 273 (М+H)+; время удерживания (способ а) = 8,60 (проход 10 минут).
2-(3-Бромфенил)-1-метил-1Н-бензоимидазол
Способ 1 - Стадия b. 2-(3-бромфенил)-1Н-бензоимидазол (7,8 г, 28,6 ммоль) полностью растворяли в сухом ТГФ (300 мл), затем к прозрачному желтому раствору добавляли порциями 60%-ный NaH в минеральном масле (1,49 г, 37,2 ммоль). Светло-коричневую суспензию перемешивали 1 час при комнатной температуре, затем добавляли по каплям CH3I (2,5 мл, 40,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили с использованием H2O (300 мл) и экстрагировали EtOAc (2×450 мл). Органические экстракты сушили над MgSO4, фильтровали и упаривали, получая соединение в виде коричнево-желтого твердого вещества (7,40 г, 70%).
1H-ЯМР (400 МГц, ДМСО): δ 3,90 (3H, с), 7,30 (2H, м), 7,55 (1Н, т), 7,64 (1Н, д), 7,70 (1Н, д), 7,77 (1Н, м), 7,88 (1Н, м), 8,05 (1Н, м); m/z= 287 [M+H]+, время удерживания (способ а) = 7,70 (проход 10 минут).
Этиловый эфир (S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3- карбоновой кислоты
Способ 1 - Стадия c. 2-(3-Бромфенил)-1-метил-1Н-бензоимидазол (0,85 г, 2,96 ммоль), этиловый эфир (S)-(+)-нипекотиновой кислоты (0,60 г, 3,85 ммоль) и карбонат цезия (4,82 г, 14,80 ммоль) помещали в сухую колбу Шленка в атмосфере азота. В то же время ацетат палладия (0,14 г, 0,60 ммоль) и рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP, БИНАП) (0,57 г, 0,90 ммоль) помещали в сухую колбу емкостью 7 мл в атмосфере азота. Затем добавляли безводный толуол (5 мл) и смесь перемешивали 20 минут в атмосфере азота, после чего добавляли смесь в первую колбу. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, отфильтровывали и нерастворимое вещество промывали EtOAC (3×10 мл). Органический раствор концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (элюент: циклогексан:AcOEt градиент от 100% циклогексана до смеси циклогексан 4:AcOEt 1), получая 0,75 г указанного в заголовке соединения (70%).
1H-ЯМР (400 МГц, CD3OD): δ 1,25 (3H, т), 1,66-1,88 (3H, м), 1,95-2,03 (1Н, м), 2,67-2,74 (1Н, м), 2,94-3,01 (1Н, м), 3,16-3,22 (1Н, м), 3,52-3,57 (1Н, м), 3,73-3,77 (1Н, м), 4,15 (2H, кв.), 7,16-7,19 (2H, м), 7,28-7,36 (3H, м), 7,41-7,45 (1Н, м), 7,53-7,56 (1Н, м), 7,66-7,68 (1Н, м).
Гидрохлорид (S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты
Способ 1 - Стадия d. Смесь этилового эфира (S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (0,76 г, 2,09 ммоль) в 6н. HCl (4,0 мл) нагревали в микроволновой печи при 120ºС в течение 20 минут; для завершения превращаения потребовалось 3 цикла. Затем растворитель удаляли и неочищенный продукт растирали со смесью ацетон/этилацетат (1:1), твердое вещество отфильтровывали и сушили в вакууме, получая 0,60 г указанного в заголовке соединения (86%).
1H-ЯМР (400 МГц, ДМСО): δ 1,52-1,84 (3H, м), 2,0 (1Н, м), 2,65 (1Н, м), 3,02 (1Н, т), 3,16 (1Н, т), 3,64 (1Н, д), 3,80 (1Н, д), 4,03 (3H, с), 7,35-7,51 (2H, м) 7,51-7,72 (4H, м), 7,83-7,90 (1Н, м), 8,01-8,09 (1Н, м); m/z 335 (м+H)+; время удерживания (способ a) = 1,27 (проход 5 минут).
ОБЩИЙ СПОСОБ 2
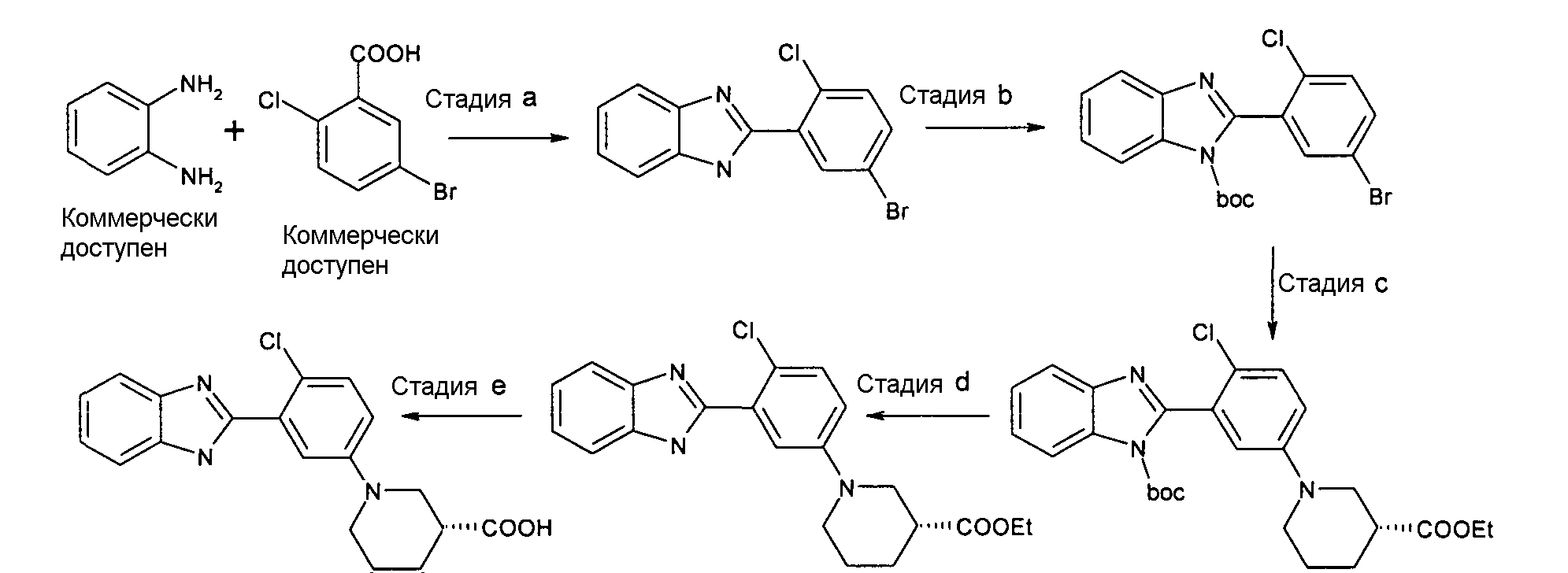
2-(5-бром-2-хлорфенил)-1Н-бензоимидазол
Способ 2 - Стадия a. В одногорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 5-бром-2-хлорбензойную кислоту (70,0 г, 297,3 ммоль), o-фенилендиамин (64,3 г, 594,6 ммоль) и метансульфоновую кислоту (140 мл) и нагревали до 170ºС для расплавления твердых веществ. Систему перемешивали 5 часов при данной температуре, затем оставляли охлаждаться до комнатной температуры. Голубое твердое вещество обрабатывали 35%-ным NaOH (200 мл), получая фиолетовую суспензию (рН 5), которую фильтровали и промывали 0,5 M NaOH (2 л) и H2O (2 л). Продукт сушили в вакууме (60ºC), получая 61,6 г чистого фиолетового твердого вещества. (67%).
m/z 307/309 (M+H)+; время удерживания (способ a) = 8,73 (проход 10 минут).
трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)бензоимидазол-1-карбоновой кислоты
Способ 2 - Стадия b. В трехгорлой круглодонной колбе, оборудованной магнитной мешалкой 2-(5-бром-2-хлорфенил)-1Н-бензимидазол (30,7 г, 99,8 ммоль) суспендировали в ТГФ (1 л). Затем добавляли 50% NaOH (72,0 г, 598 ммоль). Суспензию оставляли на 1 час при комнатной температуре при перемешивании. (Boc)2O (37,0 г, 169,7 ммоль) растворяли в ТГФ (200 мл) и добавляли к реакционной смеси. Реакционную смесь оставляли перемешиваться в течение ночи. Растворитель упаривали при пониженном давлении. Полученный остаток разбавляли водой (500 мл), фильтровали и сушили в вакууме (60оC), получая 39,8 г коричневого твердого вещества (98%).
m/z 407/409 (M+H)+; время удерживания (способ a) = 9,14 (проход 10 минут).
трет-Бутиловый эфир 2-[2-хлор-5-((R)-3-этоксикарбонилпиперидин-1-ил)фенил]бензоимидазол-1-карбоновой кислоты
Способ 2 - Стадия c. трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)бензоимидазол-1-карбоновой кислоты (1,50 г, 3,69 ммоль), этиловый эфир (R)-(-)-нипекотиновой кислоты (0,75 г, 4,79 ммоль) и карбонат цезия (6,00 г, 18,43 ммоль) помещали в сухую колбу Шленка в атмосфере азота. В то же время ацетат палладия (0,17 г, 0,74 ммоль) и БИНАП (0,71 г, 1,11 ммоль) помещали в сухую колбу емкостью 7 мл в атмосфере азота. Затем добавляли безводный толуол (5 мл) и смесь перемешивали 20 минут в атмосфере азота, после чего добавляли ее в первую колбу. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, отфильтровывали и нерастворимые вещества промывали EtOAC (3×10 мл). Органический раствор концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (элюент: циклогексан:AcOEt градиент от 100% циклогексана до циклогексан 4:AcOEt 1), получая 1,33 г указанного в заголовке соединения (74%).
1H-ЯМР (400 МГц, CD3OD): δ 1,23 (3H, т), 1,36 (9H, с), 1,64-1,84 (3H, м), 1,84-2,00 (1Н, м), 2,65-2,71 (1Н, м), 2,93-2,98 (1Н, м), 3,14-3,19 (1Н, м), 3,45-3,51 (1Н, м), 3,67-3,71 (1Н, м), 4,14 (2H, кв.), 7,11-7,16 (2H, м), 7,35-7,37 (1Н, м), 7,40-7,48 (2H, м), 7,71-7,73 (1Н, м), 8,13-8,15 (1Н, м).
Этиловый эфир (R)-1-[3-(1H-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-карбоновой кислоты
Способ 2 - Стадия d. К смеси трет-бутилового эфира 2-[2-хлор-5-((R)-3-этоксикарбонилпиперидин-1-ил)фенил]бензоимидазол-1-карбоновой кислоты (1,30 г, 2,68 ммоль) в дихлорметане (2 мл) добавляли 2M HCl в Et2O (10 мл) и полученную смесь перемешивали в течение ночи при комнатной температуре. Твердое вещество отфильтровывали, затем помещали в 10%-ный NaOH (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 0,81 г указанного в заголовке соединения без дополнительной очистки (85%).
1H-ЯМР (400 МГц, CD3OD): δ 1,25 (3H, т), 1,67-1,87 (3H, м), 1,98-2,03 (1Н, м), 2,66-2,72 (1Н, м), 2,94-3,01 (1Н, м), 3,17-3,22 (1Н, м), 3,50-3,55 (1Н, м), 3,70-3,75 (1Н, м), 4,14 (2H, кв.), 7,10-7,13 (1Н, м), 7,26-7,31 (2H, м), 7,40-7,42 (2H, м), 7,62 (2H, ушир.с); m/z=384 [M+H]+, время удерживания (способ a) = 1,82 (проход 5 минут).
Гидрохлорид (R)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-карбоновой кислоты
Способ 2 - Стадия e. Смесь этилового эфира (R)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-карбоновой кислоты (0,81 г, 2,11 ммоль) в 6н HCl (4,0 мл) нагревали в микроволновой печи при 120ºС в течение 20 минут; для полной конверсии потребовалось 2 цикла. Затем растворитель удаляли и неочищенный продукт растирали со смесью ацетон/этилацетат (1:1), твердое вещество отфильтровывали и сушили в вакууме, получая 0,60 г указанного в заголовке соединения (80%).
1H-ЯМР (400 МГц, ДМСО): δ 1,50-1,725 (2H, м), 1,72 (1Н, м), 2,54 (1Н, м), 2,93 (1Н, т), 3,06 (1Н, т), 3,63 (1Н, д), 3,80 (1Н, дд), 7,31 (1Н, дд), 7,54-7,58 (2H, м), 7,60-7,62 (2H, м), 7,86-7,90 (2H, м); m/z 355 (М+H)+, время удерживания (способ a) = 1,45 (проход 5 минут).
ОБЩИЙ СПОСОБ 3,4

N-(2-Амино-5-фторфенил)-5-бром-2-хлорбензамид
Способ 3, 4 - Стадия a. К смеси твердых 5-бром-2-хлорбензойной кислоты (7,00 г, 29,79 ммоль), 4-фторбензол-1,2-диамина (4,65 г, 36,94 ммоль) и гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) (11,89 г, 31,28 ммоль), добавляли триэтиламин (ТЭА) (4,60 мл, 32,77 ммоль), дихлорметан (120 мл) и диметилформамид (ДМФ) (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, добавляли воду (30 мл) и перемешивали до образования осадка. Осадок отфильтровывали, промывали дихлорметаном (3×10 мл) и сушили, получая 6,90 г указанного в заголовке соединения (69%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 5,27 (2H, с), 6,34 (1Н, тд), 6,50 (1Н, дд), 7,17 (1Н, дд), 7,50 (1Н, д), 7,67 (1Н, дд), 7,97 (1Н, д), 9,72 (1Н, с); m/z 345 (М+2)+.
2-(5-Бром-2-хлорфенил)-5-фтор-1Н-бензоимидазол
Способ 3, 4 - Стадия b. Раствор N-(2-амино-5-фторфенил)-5-бром-2-хлорбензамида (6,90 г, 20,12 ммоль) в уксусной кислоте (40 мл) перемешивали при 80ºС в течение ночи, затем растворитель удаляли при пониженном давлении и неочищенный продукт очищали осаждением из диэтилового эфира, получая 6,00 г указанного в заголовке соединения (92%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 7,10 (1Н, м), 7,35-7,50 (1Н, м), 7,56 и 7,70 (1Н, м), 7,61 (1Н, м), 7,73 (1Н, м), 8,08 (1Н, м), 12,92 (1Н, с). m/z 327 (М+2)+.
трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)-5-фторбензоимидазол-1-карбоновой кислоты
Способ 3 - Стадия c. В колбу, содержащую 2-(5-бром-2-хлорфенил)-5-фтор-1Н-бензоимидазол (6,00 г, 18,46 ммоль), добавляли 4-диметиламинопиридин (ДМАП) (0,23 г, 1,85 ммоль), ди-трет-бутилдикарбонат (Boc2O) (5,23 г, 24,00 ммоль) и дихлорметан (90 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, растворитель удаляли при пониженном давлении и неочищенный продукт осаждали из смеси циклогексан:AcOEt/10:1, получая 4,60 г указанного в заголовке соединения (59%).
1H-ЯМР (400 МГц, CDCl3): δ 1,40 (9H, с), 7,10-7,22 (1Н, м), 7,34 (1Н, дд), 7,47 и 7,83 (1Н, м), 7,55-7,59 (1Н, м), 7,71 (1Н, м), 7,74 и 8,06 (1Н, м); m/z 421 (М+2)+.
трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)-5-метоксибензоимидазол-1-карбоновой кислоты
Способ 4 - Стадия c. В колбу, содержащую 2-(5-бром-2-хлорфенил)-5-метокси-1Н-бензоимидазол (6,50 г, 19,29 ммоль), добавляли ДМАП (0,23 г, 1,93 ммоль), Boc2O (5,47 г, 25,07 ммоль) и дихлорметан (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (градиент элюента: от смеси циклогексан:AcOEt/5:1 до соотношения 1:2), получая 4,70 г указанного в заголовке соединения (56%).
1H-ЯМР (400 МГц, ДМСО): δ 1,32 (9H, с), 3,82 (3H, д), 7,04 (1Н, м), 7,29 и 7,65 (1Н, м), 7,56 (2H, м), 7,75 (1Н, м), 7,88 (1Н, м); m/z 438 (м+H)+, время удерживания (способ a) = 7,47 (проход 10 минут).
трет-Бутиловый эфир 2-[2-хлор-5-(4-этоксикарбонилпиперидин-1-ил)фенил]-5-фторбензоимидазол-1-карбоновой кислоты
Способ 3,4 - Стадия d. трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)-5-фторбензоимидазол-1-карбоновой кислоты (1,04 г, 2,46 ммоль), этиловый эфир пиперидин-4-карбоновой кислоты (0,49 мл, 3,19 ммоль) и карбонат цезия (3,99 г, 12,29 ммоль) помещали в сухую колбу Шленка в атмосфере азота. В то же время ацетат палладия (0,11 г, 0,49 ммоль) и БИНАП (0,46 г, 0,74 ммоль) помещали в сухую колбу емкостью 7 мл в атмосфере азота. Затем добавляли безводный толуол (4 мл) и смесь перемешивали 20 минут в атмосфере азота перед добавлением в первую колбу. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, соли отфильтровывали, органический раствор концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (элюент: циклогексан:AcOEt градиент от циклогексан:AcOEt/5:1 до 1:2), получая 0,84 г указанного в заголовке соединения (68%).
1H-ЯМР (400 МГц, CD3OD): δ 1,35 (3H, т), 1,45 (9H, с), 1,80 (2H, м), 1,99 (2H, м), 2,52 (1Н, м), 2,88 (2H, м), 3,72 (2H, м), 4,13 (2H, кв.), 7,10-7,28 (3H, м), 7,35 (1Н, д), 7,43 и 7,86 (1Н, м), 7,71 и 8,14 (1Н, дд); m/z 502 (М+H)+, время удерживания (способ a) = 3,10 (проход 5 минут).
Гидрохлорид 1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ 3, 4 - Стадия e. Смесь трет-бутилового эфира 2-[2-хлор-5-(4-этоксикарбонилпиперидин-1-ил)фенил]-5-фторбензоимидазол-1-карбоновой кислоты (0,42 г, 0,84 ммоль) в 6н. HCl (4 мл) перемешивали при комнатной температуре в течение нескольких минут и затем нагревали в микроволновой печи при 120ºС в течение 15 минут. Растворитель удаляли в вакууме, получая указанное в заголовке соединение с количественным выходом.
1H-ЯМР (400 МГц, CD3OD): δ 1,95 (2H, м), 2,13 (2H, м), 2,65 (1Н, м), 3,20 (2H, м), 3,83 (2H, м), 7,53 (2H, м), 7,69 (3H, м), 7,91 (1Н, м); m/z 374 (М+H)+, время удерживания (способ a) = 1,65 (проход 5 минут).
ОБЩИЙ СПОСОБ 5
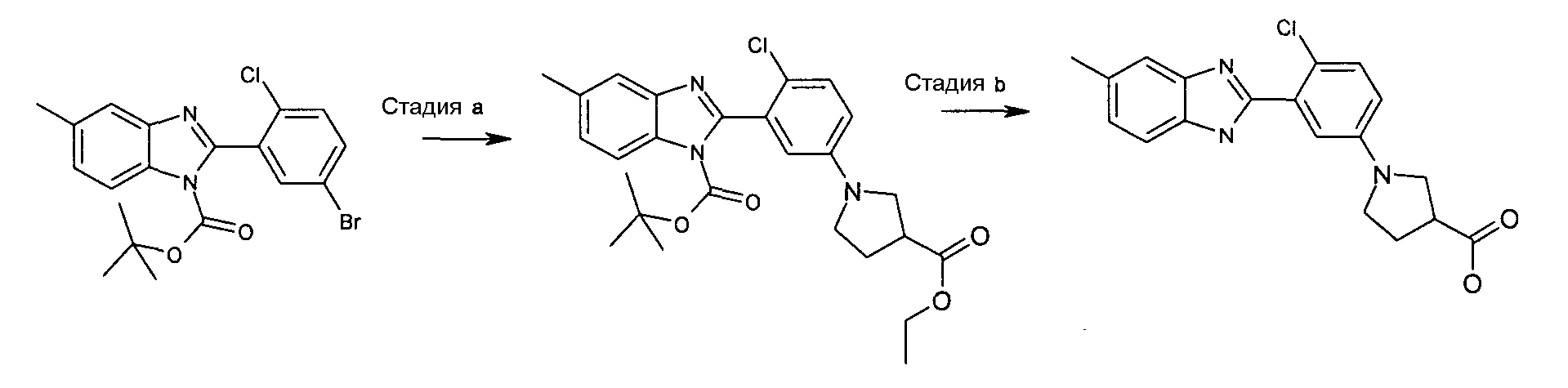
трет-Бутиловый эфир 2-[2-хлор-5-(3-этоксикарбонилпирролидин-1-ил)фенил]-5-метилбензоимидазол-1-карбоновой кислоты
Способ 5 - Стадия a. трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)-5-метилбензоимидазол-1-карбоновой кислоты (получен, как описано в общем способе 4, стадия c) (1,80 г, 4,28 ммоль), метиловый эфир пирролидин-3-карбоновой кислоты (0,92 г, 5,56 ммоль) и карбонат цезия (6,95 г, 21,38 ммоль) помещали в атмосфере азота в сухую колбу, содержащую ацетат палладия (0,19 г, 0,86 ммоль) и БИНАП (0,80 г, 1,28 ммоль) в безводном толуоле (11 мл), которые ранее перемешивали в течение 20 минут в атмосфере азота. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, разбавляли AcOEt (40 мл), соли отфильтровывали, органический слой промывали водой (1×30 мл) и насыщенным раствором соли (1×20 мл), сушили над Na2SO4 и затем концентрировали при пониженном давлении. К неочищенному продукту добавляли смесь растворителей циклогексан:AcOEt/4:1 (15 мл) и фильтровали через колонку с Na2SO4 для удаления всех солей, промывали смесью растворителей и органический слой очищали флэш-хроматографией (элюент: циклогексан:AcOEt/4:1), получая 1,71 г указанного в заголовке соединения (86%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 1,30 (9H, с), 1,38 (3H, с), 2,20 (2H, м), 3,24-3,51 (5H, м), 3,63 (3H, с), 6,68 (1Н, м), 6,74 (1Н, м), 7,23 (1Н, м), 7,30 (1Н, м), 7,54 и 7,82 (1Н, м), 7,62 и 7,86 (1Н, м); m/z 470 (М+H)+, время удерживания (способ a) = 5,12 (проход 10 минут).
1-[4-Хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-карбоновая кислота
Способ 5 - Стадия b. Как описано в общем способе 3,4, стадия e, исходя из трет-бутилового эфира 2-[2-хлор-5-(3-этоксикарбонилпирролидин-1-ил)фенил]-5-метилбензоимидазол-1-карбоновой кислоты получали указанное в заголовке соединение с количественным выходом.
ОБЩИЙ СПОСОБ 6
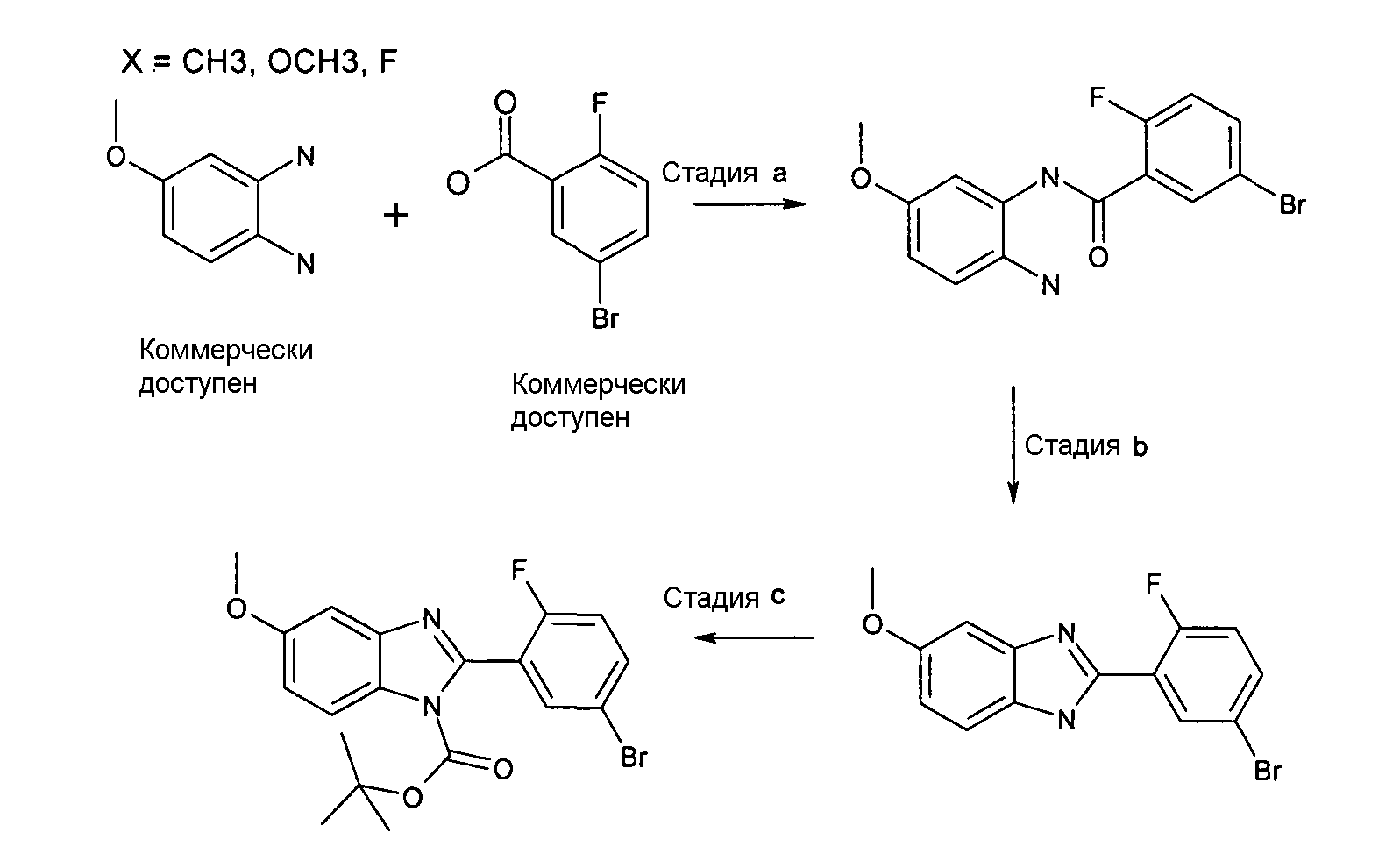
N-(2-Амино-5-метоксифенил)-5-бром-2-фторбензамид
Способ 6 - стадия a. Смесь 5-бром-2-фторбензойной кислоты (1,50 г, 6,85 ммоль), дигидрохлорида 4-метоксибензол-1,2-диамина (1,77 г, 8,49 ммоль), HATU (2,73 г, 7,19 ммоль) и ТЭА (2,88 мл, 20,76 ммоль) в дихлорметане (20 мл) и ДМФ (5 мл) перемешивали при комнатной температуре в течение ночи, затем добавляли воду (50 мл), смесь перемешивали в течение 2 часов и оставляли при комнатной температуре в течение ночи. Полученный осадок отфильтровывали и сушили, получая 1,45 г указанного в заголовке соединения (50%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 3,65 (3H, с), 4,96 (2H, ушир.с), 6,15 (1Н, дд), 6,31 (1Н, с), 7,05 (1Н, д), 7,31 (1Н, м), 7,71 (1Н, м), 7,91 (1Н, д), 9,50 (1Н, с).
2-(5-Бром-2-фторфенил)-5-метокси-1Н-бензоимидазол
Способ 6 - Стадия b. Смесь N-(2-амино-5-метоксифенил)-5-бром-2-фторбензамида (1,45 г, 4,28 ммоль) в уксусной кислоте (15 мл) нагревали при 80ºС в течение ночи. Растворитель удаляли при пониженном давлении и неочищенный продукт очищали осаждением из AcOEt (20 мл), сушили, заливали смесью дихлорметана (20 мл) и метанола (1 мл) и промывали насыщенным раствором NaHCO3 (3×5 мл), органический слой выделяли фильтрованием через фазовый сепаратор и растворитель удаляли при пониженном давлении, получая 0,86 г указанного в заголовке соединения (63%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 3,79 (3Н, с), 6,86 (1Н, д), 7,04 (1Н, ушир.с), 7,41 (1Н, дд), 7,55 (1Н, ушир.с), 7,69 (1Н, м), 8,30 (1Н, м), 11,93 (1Н, с).
трет-Бутиловый эфир 2-(5-бром-2-фторфенил)-5-метоксибензоимидазол-1-карбоновой кислоты
Способ 6 - Стадия c. К перемешиваемой смеси 2-(5-бром-2-фторфенил)-5-метокси-1Н-бензоимидазола (0,87 г, 2,70 ммоль) в ДХМ (10 мл), добавляли Boc2O (0,76 г, 3,50 ммоль) и ДМАП (0,03 г, 0,27 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение выходных. Затем добавляли дихлорметан (20 мл) и реакционную смесь промывали насыщенным раствором NaHCO3 (4 мл), лимонной кислоты (10% раствор), органический слой выделяли фильтрованием через фазовый сепаратор и растворитель удаляли при пониженном давлении. Неочищенный продукт затем очищали флэш-хроматографией (элюент циклогексан:этилацетат/10:1), получая 0,82 г указанного в заголовке соединения в виде смеси двух диастереоизомеров (72%).
1H-ЯМР (400 МГц, CDCl3): δ 1,45 (9H, с), 1,47 (9H, с), 3,88 (3H, с), 3,90 (3H, с), 6,98-7,06 (4H, м), 7,25-7,27 и 7,80-7,83 (3H, м), 7,54-7,61 (3H, м), 7,93 (1Н, м); m/z 423 (М+2H)+, время удерживания (способ a) = 3,02 (проход 5 минут).
ОБЩИЙ СПОСОБ 7
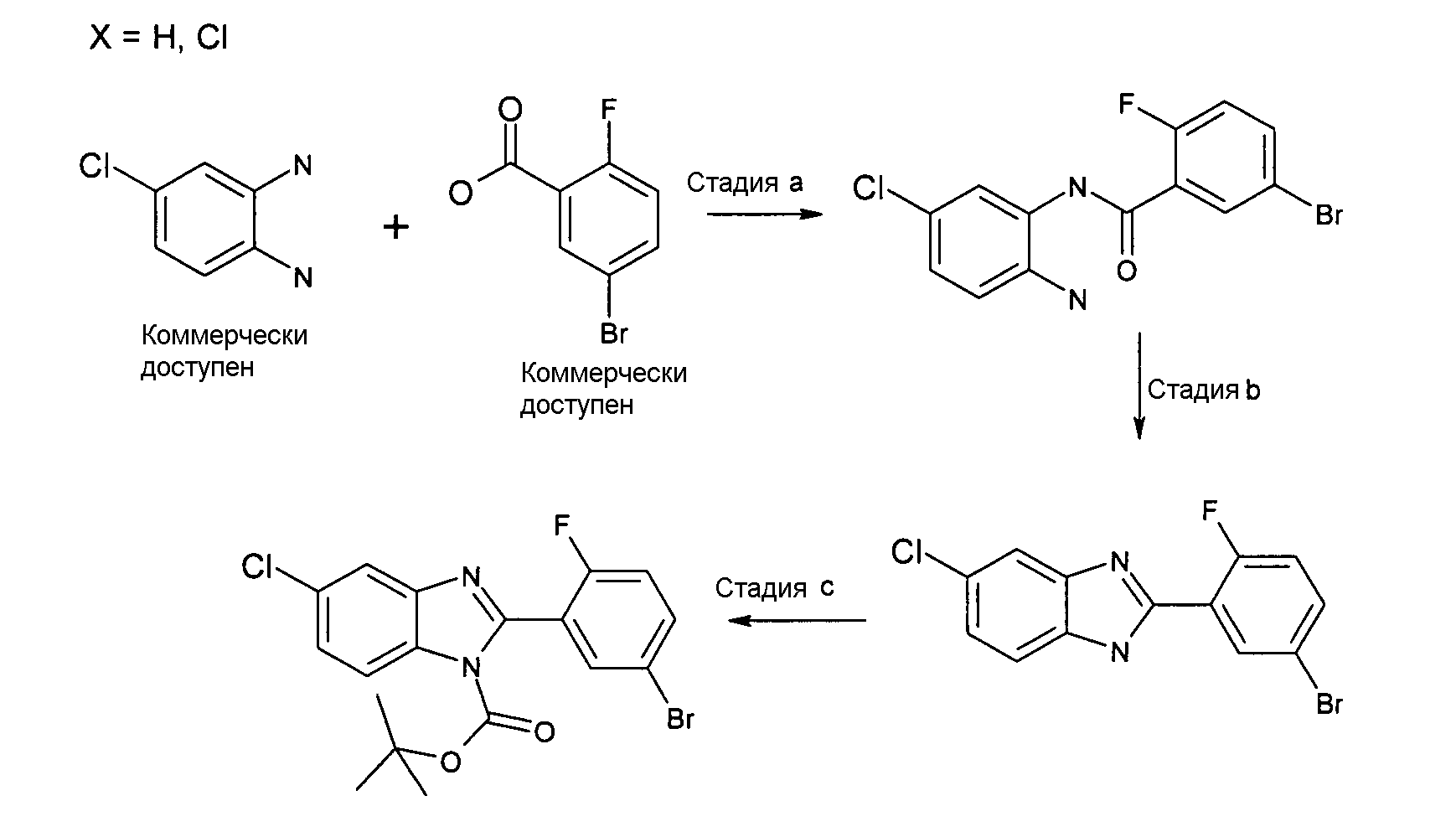
N-(2-Амино-5-хлорфенил)-5-бром-2-фторбензамид
Способ 7 - Стадия a. Смесь 5-бром-2-фторбензойной кислоты (3,00 г, 13,70 ммоль), 4-хлорбензол-1,2-диамина (2,42 г, 16,99 ммоль), HATU (5,47 г, 14,38 ммоль) и ТЭА (1,91 мл, 13,83 ммоль) в дихлорметане (70 мл) и ДМФ (16 мл) перемешивали при комнатной температуре в течение ночи, затем добавляли воду (80 мл) и оставляли при комнатной температуре на ночь. Органический слой отделяли и растворитель удаляли при пониженном давлении и неочищенный продукт в виде масла кристаллизовали из смеси растворителей дихлорметан:циклогексан/3:1 (30 мл), получая 2,19 г указанного в заголовке соединения (52%).
m/z 344 (M+H)+, время удерживания = 5,33 (проход 10 минут).а
2-(5-Бром-2-фторфенил)-5-хлор-1Н-бензоимидазол
Способ 7 - Стадия b. Смесь N-(2-амино-5-хлорфенил)-5-бром-2-фторбензамида (1,60 г, 4,67 ммоль) в уксусной кислоте (10 мл) нагревали при 85ºС в течение ночи. Растворитель удаляли при пониженном давлении и полученное твердое вещество промывали дихлорметаном и сушили, получая 1,40 г указанного в заголовке соединения (93%).
1H-ЯМР (400 МГц, CD3OD): δ 7,27-7,33 (2H, м), 7,60-7,64 (2H, м), 7,69 (1Н, м), 8,33 (1Н, дд).
трет-Бутиловый эфир 2-(5-бром-2-фторфенил)-5-хлорбензоимидазол-1-карбоновой кислоты
Способ 7 - Стадия c. К перемешиваемой смеси 2-(5-бром-2-фторфенил)-5-хлор-1Н-бензоимидазола (1,41 г, 4,33 ммоль) в ДХМ (28 мл) добавляли Boc2O (1,23 г, 5,63 ммоль) и ДМАП (0,05 г, 0,43 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь промывали насыщенным раствором NaH4Cl (2×5 мл) и неочищенный продукт затем очищали флэш-хроматографией (градиентное элюирование: циклогексан:EtOAc от 8:1 до 5:1), получая 1,31 г указанного в заголовке соединения (71%) в виде смеси двух региоизомеров.
1H-ЯМР (400 МГц, CDCl3): δ 1,45 (9H, 2с), 7,05 (1Н, м), 7,39 (1Н, м), 7,60 (1Н, м), 7,71 (0,5H, д), 7,78 (0,5H, д), 7,83 (1Н, м), 7,99 (0,5H, д), 8,10 (0,5H, д).
ОБЩИЙ СПОСОБ 8

трет-Бутиловый эфир 2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]-5-метилбензоимидазол-1-карбоновой кислоты
Способ 8 - Стадия a. трет-Бутиловый эфир 2-(5-бром-2-фторфенил)-5-метилбензоимидазол-1-карбоновой кислоты (получен, как описано в общем способе 6, стадия c) (0,94 г, 2,40 ммоль), этиловый эфир пиперидин-4-карбоновой кислоты (0,48 г, 3,12 ммоль) и карбонат цезия (3,90 г, 12,01 ммоль) помещали в сухую колбу Шленка и проводили 3 цикла вакуумирование/заполнение азотом, затем добавляли безводный толуол (4 мл). В то же время ацетат палладия(II) (0,82 г, 0,36 ммоль) и БИНАП (0,45 г, 0,72 ммоль) помещали в высушенную колбу Шленка в атмосфере азота и проводили 3 цикла вакуумирование/заполнение азотом. Затем при комнатной температуре в атмосфере азота добавляли безводный толуол (2 мл) и смесь добавляли в первую колбу. Реакционную смесь нагревали при 80ºС в течение ночи, добавляли воду (5 мл), органический слой фильтровали через Na2SO4 и затем очищали флэш-хроматографией (элюент: циклогексан:EtOAc 8:2), получая 1,15 г указанного в заголовке соединения с количественным выходом.
1H-ЯМР (400 МГц, CDCl3): δ 1,27 (3H, т), 1,43 (9H, с), 1,89 (2H, м), 2,02 (2H, м), 2,41 (1Н, м), 2,50 (3H, с), 2,80 (2H, м), 3,59 (2H, м), 4,16 (2H, кв.), 7,01 (2H, м), 7,21 (2H, м), 7,66 (1Н, д), 7,91 (1H, д).
Гидрохлорид 1-[4-(фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил-1-пиперидин-4-карбоновой кислоты
Способ 8 - Стадия b. Смесь трет-бутилового эфира 2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]-5-метилбензоимидазол-1-карбоновой кислоты (1,15 г, 2,39 ммоль) в 6н. HCl (10 мл) нагревали в микроволновой печи при 120ºС в течение 15 минут (необходимо 2 цикла). Растворитель удаляли в вакууме, получая указанное в заголовке соединение с количественным выходом.
1H-ЯМР (400 МГц, ДМСО-d6): δ 2,19 (2H, м), 2,34 (2H, м), 2,59 (3H, с), 2,80 (1Н, м), 3,55 (2H, м), 3,85 (2H, м), 7,53 (1Н, д), 7,63-7,70 (2H, м), 7,78 (1Н, д), 7,91-7,96 (1Н, м), 8,27-8,33 (1Н, м).
ОБЩИЙ СПОСОБ 9
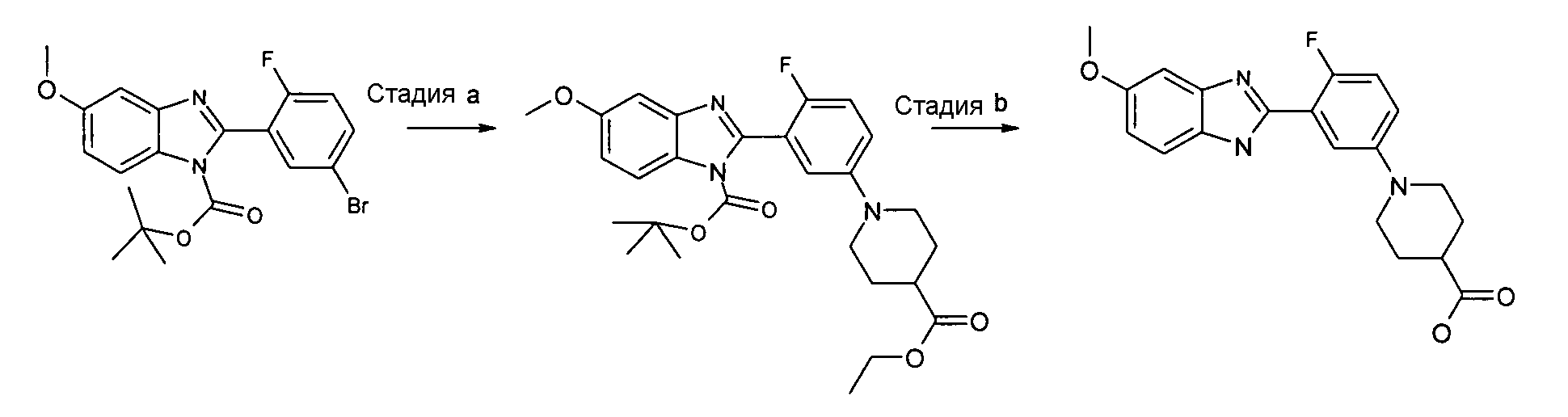
трет-Бутиловый эфир 2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]-5-метоксибензоимидазол-1-карбоновой кислоты
Способ 9 - Стадия a. трет-Бутиловый эфир 2-(5-бром-2-фторфенил)-5-метоксибензоимидазол-1-карбоновой кислоты (получен, как описано в общем способе 6, стадия c) (1,05 г, 2,50 ммоль), этиловый эфир пиперидин-4-карбоновой кислоты (0,51 г, 3,25 ммоль) и карбонат цезия (4,07 г, 12,50 ммоль) помещали в высушенную колбу Шленка и проводили 3 цикла вакуумирование/заполнение азотом, затем добавляли безводный толуол (4 мл). В то же время ацетат палладия(II) (0,11 г, 0,50 ммоль) и БИНАП (0,48 г, 0,75 ммоль) помещали в высушенную колбу Шленка в атмосфере азота и проводили 3 цикла вакуумирование/заполнение азотом. Затем при комнатной температуре в атмосфере азота добавляли безводный толуол (2 мл) и смесь добавляли в первую колбу Шленка. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, добавляли EtOAC (20 мл) и смесь фильтровали. Растворитель удаляли и неочищенный продукт очищали флэш-хроматографией (элюент градиент: циклогексан:EtOAc от 4:1 до 3:1), получая 0,82 г указанного в заголовке соединения (69%).
1H-ЯМР (400 МГц, CD3OD, два региоизомера): δ 1,25 (6H, т), 1,40 (18H, с), 1,76-1,88 (4H, м), 1,96-2,04 (4H, м), 2,42-2,51 (2H, м), 2,80 (4H, т), 3,58-3,65 (4H, м); 3,85 (3H, с); 3,87 (3H, с); 4,13 (4H, кв.); 6,99-7,06 (2H, м), 7,07-7,18 (4H, м), 7,19-7,23 (3H, м), 7,58 (1Н, д), 7,62 (1Н, д); 7,94 (1Н, д); m/z 498 (М+H)+.
Гидрохлорид 1-[4-(фтор-3-(5-метокси-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ 9 - Стадия b. Смесь трет-бутилового эфира 2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]-5-метоксибензоимидазол-1-карбоновой кислоты (0,92 г, 1,91 ммоль) в 6н. HCl (4 мл) перемешивали при комнатной температуре в течение 1 часа и затем нагревали в микроволновой печи при 120ºС в течение 15 минут. Растворитель удаляли в вакууме, затем помещали в смесь ацетон:диэтиловый эфир 1:1 (20 мл), твердое вещество отфильтровывали, промывали диэтиловым эфиром и сушили, получая 0,22 г указанного в заголовке соединения (29%).
1H-ЯМР (400 МГц, CD3OD): δ 1,98-2,10 (2H, м), 2,18-2,26 (2H, м), 2,63-2,71 (1Н, м), 3,24-3,32 (2H, м), 3,77-3,84 (2H, м); 3,95 (3H, с); 7,26-7,31 (2H, м), 7,54 (1Н, дд), 7,65-7,71 (1Н, м), 7,75 (1Н, дд), 7,92-7,98 (1Н, м); m/z 370 (М+H)+.
ОБЩИЙ СПОСОБ 10
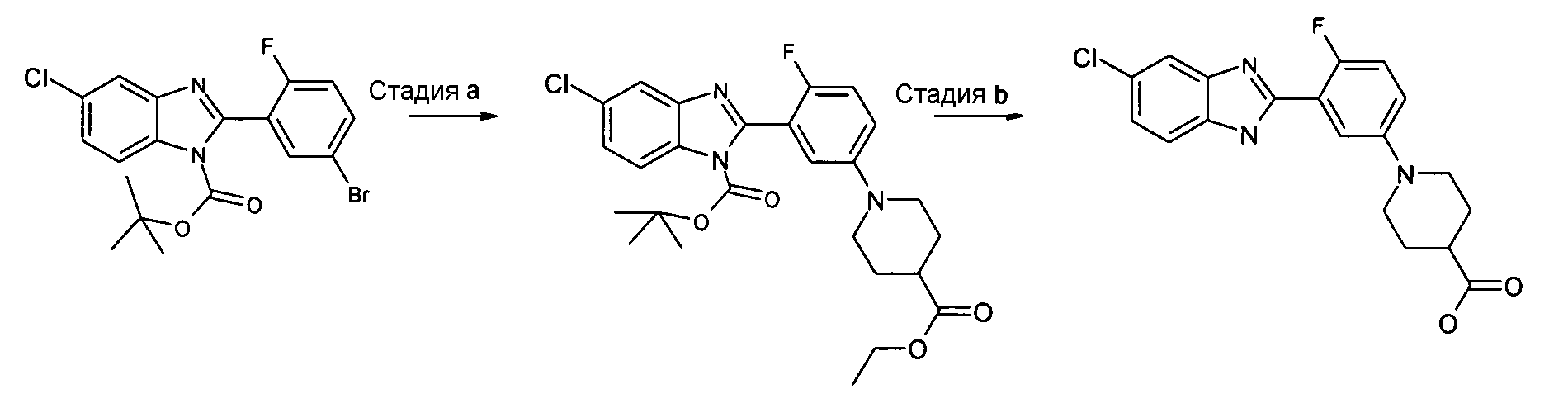
трет-Бутиловый эфир 5-хлор-2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]бензоимидазол-1-карбоновой кислоты
Способ 10 - Стадия a. трет-Бутиловый эфир 2-(5-бром-2-фторфенил)-5-хлорбензоимидазол-1-карбоновой кислоты (получен, как описано в общем способе 7, стадия c) (1,06 г, 2,50 ммоль), Pd2(dba)3 (0,36 г, 0,50 ммоль), 2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил (t-BuXphos) (0,32 г, 0,75 ммоль) и трет-бутоксид натрия (0,48 г, 5,00 ммоль) помещали в высушенную колбу и проводили несколько циклов вакуумирование/заполнение азотом. Затем добавляли безводный толуол (5 мл) и этиловый эфир пиперидин-4-карбоновой кислоты (0,50 мл, 3,25 ммоль), реакционную смесь нагревали при 85ºС в течение ночи и промывали насыщенным раствором Na2CO3 (3×5 мл) и водой (3×3 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, и неочищенный продукт очищали флэш-хроматографией (элюент: градиент от EtOA:циклогексан/1:5 до 1:2), получая 0,30 г указанного в заголовке соединения (24%).
1H-ЯМР (400 МГц, CDCl3): δ 1,26 (3H, т), 1,43 (9H, с), 1,60 (2H, м), 1,90 (2H, м), 2,42 (1Н, м), 2,81 (2H, м), 3,60 (2H, м), 4,16 (2H, кв.), 7,05 (2H, м), 7,36 (2H, м), 7,69 (1Н, д), 8,09 (1Н, с).
Гидрохлорид 1-[3-(5-хлор-1Н-бензоимидазол-2-ил)-4-фторфенил]пиперидин-4-карбоновой кислоты
Способ 10 - Стадия b. Смесь трет-бутилового эфира 5-хлор-2-[5-(4-этоксикарбонилпиперидин-1-ил)-2-фторфенил]бензоимидазол-1-карбоновой кислоты (0,30 г, 0,60 ммоль) в 6н HCl (10 мл) нагревали в микроволновой печи при 120ºС в течение 15 минут, потребовалось два цикла. Растворитель удаляли в вакууме, затем твердое вещество отфильтровывали, промывали диэтиловым эфиром и сушили, получая 0,17 г указанного в заголовке соединения (70%).
ОБЩИЙ СПОСОБ 11
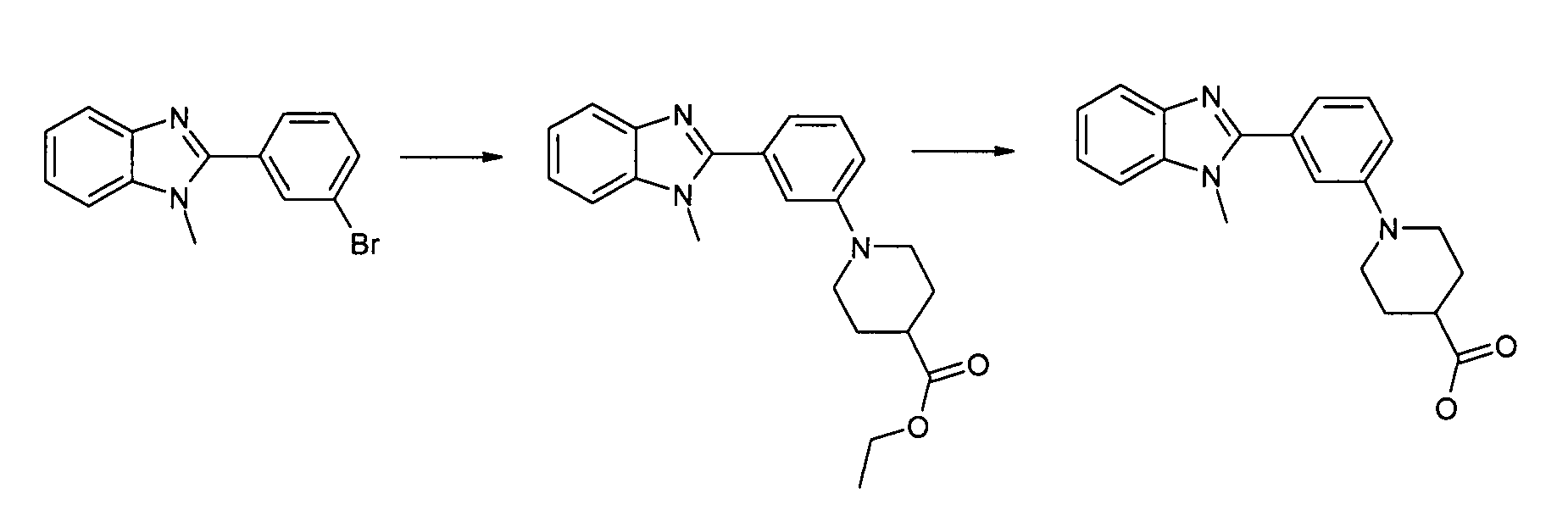
Этиловый эфир 1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ 11 - Стадия a. 2-(3-Бромфенил)-1-метил-1Н-бензоимидазол (получен, как описано в общем способе 1, стадия b) (5,00 г, 17,40 ммоль), этиловый эфир пиперидин-4-карбоновой кислоты (3,56 г, 22,62 ммоль) и карбонат цезия (28,34 г, 87 ммоль) помещали в круглодонную колбу в атмосфере азота. В то же время ацетат палладия (0,79 г, 3,48 ммоль) и БИНАП (3,33 г, 5,22 ммоль) помещали в колбу в атмосфере азота. Затем добавляли безводный толуол (18 мл) и смесь перемешивали 20 минут в атмосфере азота, после чего добавляли в первую колбу. Реакционную смесь нагревали при 80ºС в течение двух дней, затем разбавляли этилацетатом (100 мл), фильтровали через Na2SO4 и промывали водой (2×50 мл) и насыщенным раствором соли (1×50 мл). Органический раствор концентрировали при пониженном давлении и неочищенный продукт очищали флэш-хроматографией (элюент: циклогексан:AcOEt/4:1), получая 3,45 г указанного в заголовке соединения (55%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 1,19 (3H, т), 1,68 (2H, кв.д), 1,93 (2H, дд), 2,49-2,58 (1Н, м), 2,86 (2H, тд); 3,75 (2H, дт); 3,86 (3H, с); 4,09 (2H, кв.), 7,10-7,42 (6H, м), 7,59 (1Н, д), 7,67 (1Н, д).
Гидрохлорид 1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ 11 - Стадия b. Смесь этилового эфира 1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты (3,10 г, 8,54 ммоль) в 6н. HCl (25,0 мл) нагревали в микроволновой печи при 120ºС в течение 20 минут. Затем растворитель удаляли, получая указанное соединение с количественным выходом.
1H-ЯМР (400 МГц, MeOD): δ 1,65-1,85 (2H, м), 1,94 (2H, д), 2,48-2,57 (1Н, м), 3,04 (2H, т), 3,79 (2H, д), 4,05 (3H, с), 7,37-7,54 (2H, м), 7,55-7,71 (4H, м), 7,84-7,91 (1Н, м), 8,03-8,09 (1Н, м); m/z 333 (М+H)+, время удерживания (способ b) = 1,95 (проход 10 минут).
СПОСОБ A
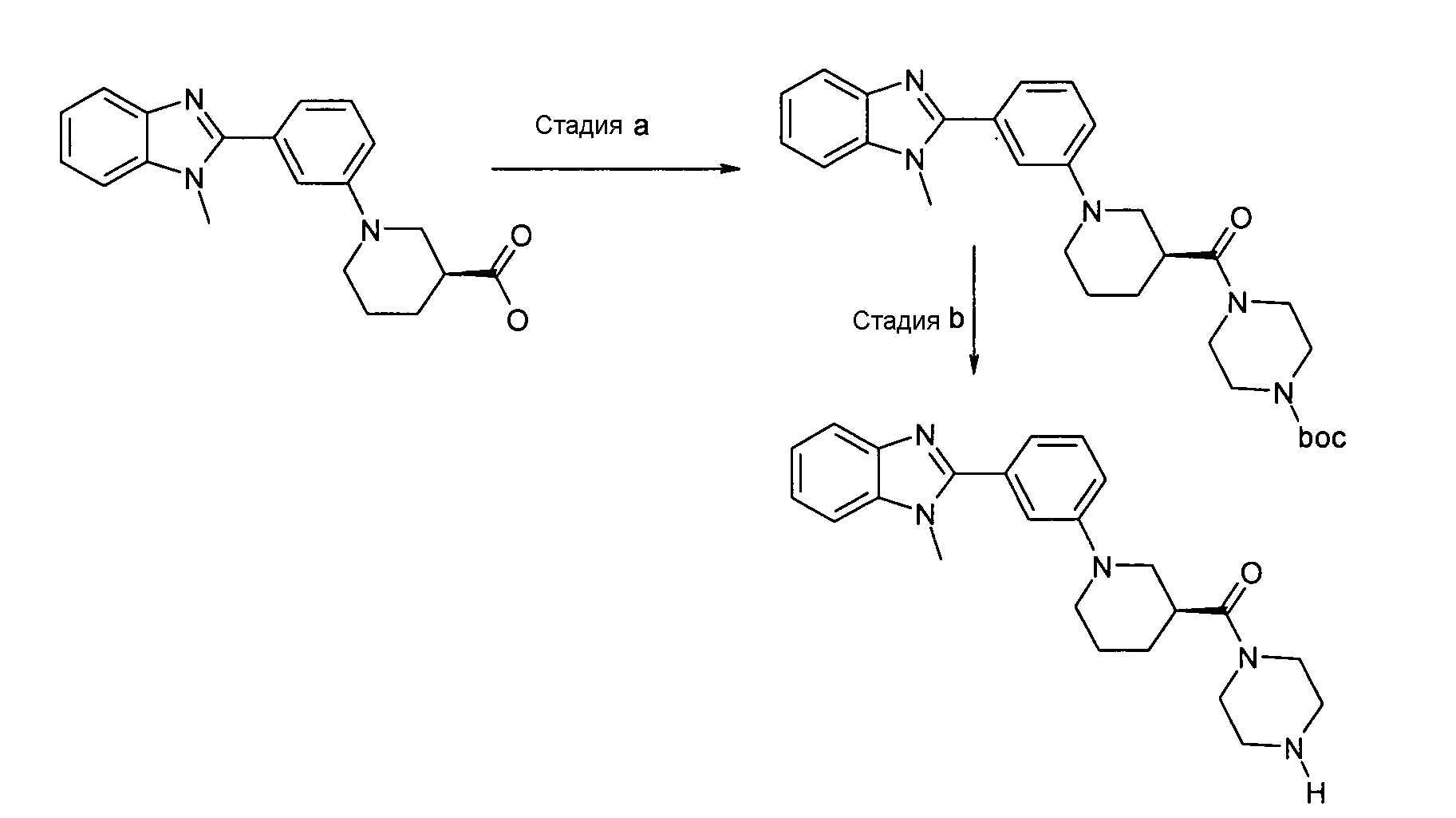
Пример 1: Гидрохлорид {(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}пиперазин-1-илметанон
трет-Бутиловый эфир 4-{(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбонил}пиперазин-1-карбоновой кислоты
Способ A - Стадия a. В колбу с (S)-1-[3-(1-метил-1H-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислотой (0,10 г, 0,30 ммоль) (получена, как описано в общем способе 1, стадия d), HATU (0,12 г, 0,31 ммоль), ТЭА (0,09 мл, 0,66 ммоль) и трет-бутил-1-пиперазинкарбоксилатом (0,07 г, 0,37 ммоль) добавляли дихлорметан (2 мл) и реакционную смесь нагревали при 35ºС в течение ночи. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли и неочищенный продукт очищали с помощью препаративной ВЭЖХ и фильтрования через NH2-колонку, получая 0,04 г указанного в заголовке соединения (27%).
m/z 503 (M+H)+, время удерживания (способ a) = 2,52 (проход 10 минут).
Гидрохлорид {(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}пиперазин-1-илметанон
Способ A - Стадия b. К смеси трет-бутилового эфира 4-{(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбонил}пиперазин-1-карбоновой кислоты (0,04 г, 0,08 ммоль) в дихлорметане (0,5 мл) добавляли 2M HCl в Et2O (2 мл) и полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли, получая 0,04 г указанного в заголовке соединения в виде соли хлористоводородной кислоты с количественным выходом.
1H-ЯМР (400 МГц, ДМСО): δ 1,74 (1Н, м), 1,92-2,17 (1Н, м), 3,03-3,51 (7H, м), 3,89 (6H, м), 4,14 (3H, с), 7,54-7,81 (4H, м), 7,83-7,91 (2H, м), 7,96-8,02 (2H, м); m/z 404 (М+H)+, время удерживания (способ а) = 0,20 (проход 10 минут).
СПОСОБ B
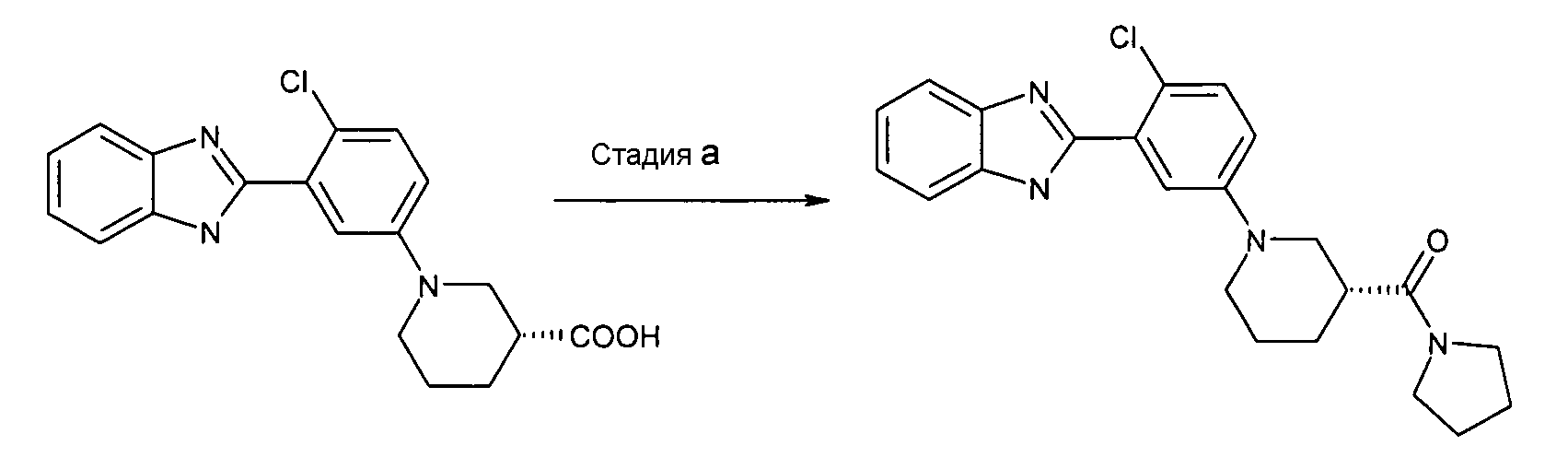
Пример 2: {(R)-1-[3-(1Н-Бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-ил}пирролидин-1-илметанон
Способ B - Стадия a. В колбу с гидрохлоридом (R)-1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 2, стадия e) (0,10 г, 0,28 ммоль) (получен, как описано в общем способе 2), HATU (0,11 г, 0,30 ммоль), триэтиламином (ТЭА) (0,09 мл, 0,66 ммоль) и трет-бутил-1-пиперазинкарбоксилатом (0,08 г, 0,62 ммоль) добавляли дихлорметан (2 мл) и реакционную смесь нагревали при 35ºС в течение ночи. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли и неочищенный продукт очищали с помощью препаративной ВЭЖХ и фильтрования через NH2-колонку, получая 0,06 г указанного в заголовке соединения (55%).
1H-ЯМР (400 МГц, ДМСО): δ 1,59-2,06 (8H, м), 2,75-2,89 (2H, м), 2,94 (2H, т), 3,35-3,48 (2H, м), 3,84 (2H, т), 7,12 (1Н, дд), 7,24-7,32 (2H, м), 7,37-7,44 (2H, м), 7,55-7,72 (1Н, ушир.с); m/z 409 (М+H)+, время удерживания (способ а) = 2,27 (проход 10 минут).
СПОСОБ C
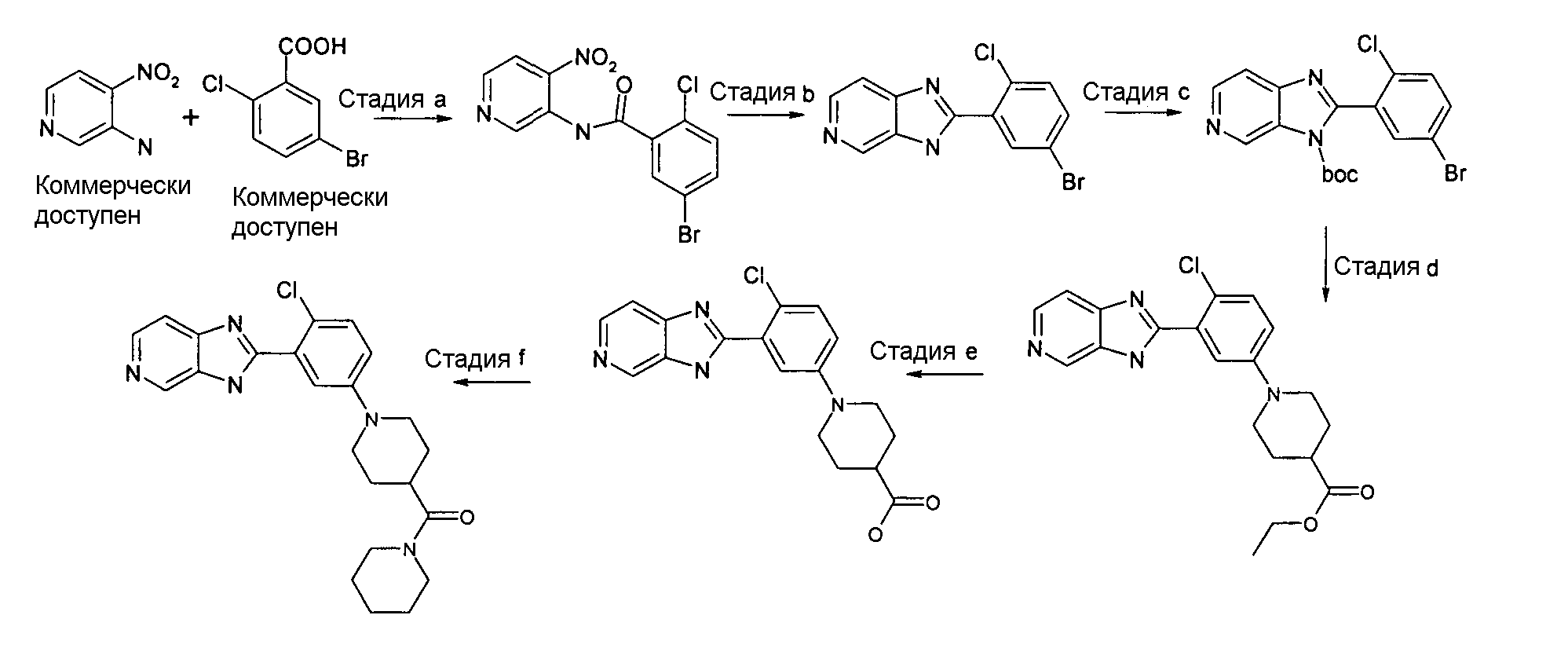
Пример 3: {1-[4-Хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-ил}пиперидин-1-илметанон
5-Бром-2-хлор-N-(4-нитропиридин-3-ил)бензамид
Способ C - Стадия a. К смеси 5-бром-2-хлорбензойной кислоты (5,00 г, 21,23 ммоль) в ДМФ (40 мл) добавляли HATU (8,48 г, 22,29 ммоль) и триэтиламин (2,97 мл, 21,44 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляли 4-нитропиридин-3-иламин (2,36 г, 16,99 ммоль), реакционную смесь перемешивали при 40ºС в течение ночи и растворитель удаляли. Неочищенный продукт затем разбавляли EtOAc (40 мл) и промывали первоначально насыщенным раствором Na2CO3 (6×30 мл), затем 1н. HCl (3×30 мл). Органический слой сушили над Na2SO4, фильтровали и оставляли. Образовавшийся осадок отфильтровывали, получая 4,75 г указанного в заголовке соединения (63%).
1H-ЯМР (400 МГц, ДМСО): δ 7,56 (1Н, д), 7,77-7,92 (3H, м), 8,82 (1Н, д), 9,14 (1Н, с), 11,35 (1Н, с); m/z 355 (М+H)+, время удерживания (способ а) = 2,32 (проход 5 минут).
2-(5-Бром-2-хлорфенил)-3H-имидазо[4,5-c]пиридин
Способ C - Стадия b. К смеси 5-бром-2-хлор-N-(4-нитропиридин-3-ил)фениламина (0,50 г, 1,47 ммоль) в уксусной кислоте (6 мл) добавляли железо (0,16 г, 2,94 ммоль) и реакционную смесь нагревали при 80ºС в течение 1,5 часов. Затем реакционную смесь охлаждали до комнатной температуры. Добавляли воду (30 мл) и проводили экстракцию с использованием ДХМ (20 мл) для удаления непрореагировавшего исходного вещества. Затем к водному слою добавляли насыщенный раствор соли Рошелле (50 мл) и насыщенный раствор Na2CO3 (30 мл) и затем проводили экстракцию с использованием ДХМ (20 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 0,28 г указанного в заголовке соединения (65%) без дополнительной очистки.
1H-ЯМР (400 МГц, MeOD): δ 7,45 (1Н, д), 7,59-7,68 (2H, м), 7,98 (1Н, д), 8,26 (1Н, д), 8,87 (1Н, с); m/z 309 (М+H)+, время удерживания (способ а) = 1,13 (проход 5 минут).
трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)имидазо[4,5-c]пиридин-3-карбоновой кислоты
Способ C - Стадия c. К смеси 2-(5-бром-2-хлорфенил)-3H-имидазо[4,5-c]пиридина в ДХМ (50 мл) добавляли Boc2O (0,36 г, 16,36 ммоль) и ДМАП (0,20 г, 1,63 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Растворитель затем удаляли при пониженном давлении и неочищенный продукт очищали путем фильтрования через Si-колонку (этилацетат в качестве элюента), получая 4,80 г указанного в заголовке соединения (80%).
1H-ЯМР (400 МГц, MeOD): δ 1,41 (18H, д), 7,52 (2H, дд), 7,73-7,87 (5H, м), 8,15 (1Н, дд), 8,56 (2H, т), 9,02 (1Н, с), 9,38 (1Н, с); m/z 409 (М+H)+, время удерживания (способ b) = 2,03 (проход 5 минут).
Этиловый эфир 1-[4-хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ C - Стадия d. трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)имидазо[4,5-c]пиридин-3-карбоновой кислоты (0,50 г, 1,23 ммоль), трис(дибензилиденацетон)дипалладий(0) (Pd2dba3) (0,13 г, 0,18 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (Xphos) (0,17 г, 0,37 ммоль) и карбонат цезия (1,20 г, 3,68 ммоль) помещали в высушенную колбу Шленка и проводили несколько циклов вакуумирование/заполнение азотом. Затем добавляли безводный диоксан (2,00 мл) и этиловый эфир пиперидин-4-карбоновой кислоты (0,38 мл, 2,45 ммоль), реакционную смесь нагревали при 80ºС в течение 4 часов, оставляли охлаждаться до комнатной температуры и фильтровали через сульфат натрия (Na2SO4). Неочищенный продукт очищали флэш-хроматографией (градиент элюента: от EtOAc 100% до EtOAc:MeOH/95:5), получая 0,60 г смеси указанного в заголовке соединения и исходного вещества с удаленной защитной группой (7:3). Смесь использовали для следующей стадии.
1H-ЯМР (400 МГц, MeOD): δ 1,26 (3H, т), 1,71-1,92 (2H, м), 1,92-1,94 (2H, м), 2,45-2,59 (1Н, м), 2,88 (2H, т), 3,71-3,80 (2H, м), 4,06-4,22 (2H, кв.), 7,11-7,18 (дд, 1Н), 7,42 (2H, кв.), 7,67 (1Н, д), 8,35 (1Н, д), 8,94 (1Н, с); m/z 384 (м+H)+, время удерживания (способ b) = 2,42 (проход 5 минут).
Гидрохлорид 1-[4-хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ C - Стадия e. Смесь этилового эфира 1-[4-хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-карбоновой кислоты (0,60 г, 1,56 ммоль) в 6н. HCl (15 мл) нагревали в микроволновой печи при 120ºС в течение 20 минут; для завершения конверсии потребовалось 2 цикла. Затем растворитель удаляли в вакууме, получая 0,61 г смеси указанного в заголовке соединения и 2-(5-бром-2-хлорфенил)-3H-имидазо[4,5-c]пиридина (из предшествующей стадии) в соотношении 7:3.
1H-ЯМР (400 МГц, CD3OD): δ 2,27-2,38 (4H, м), 2,90 (1Н, м), 3,80-3,87 (4H, м), 7,63 (1Н, дд), 7,91 (1Н, м), 8,25 (1Н, д), 8,37 (1Н, м), 8,64 (1Н, д), 9,43 (1Н, с).
{1-[4-Хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-ил}пиперидин-1-илметанон
Способ C - Стадия f. Смесь гидрохлорида 1-[4-хлор-3-(3H-имидазо[4,5-c]пиридин-2-ил)фенил]пиперидин-4-карбоновой кислоты (0,09 г, 0,25 ммоль), HATU (0,10 г, 0,26 ммоль), триэтиламина (ТЭА) (0,08 мл, 0,55 ммоль), пиперидина (0,03 г, 0,31 ммоль) и дихлорметана (2 мл) нагревали при 35ºC в течение ночи. Реакционную смесь охлаждали до комнатной температуры, промывали раствором хлорида аммония (3 мл) и неочищенный продукт очищали с помощью SCX-колонки (элюент: NH3 2н. в MeOH) и флэш-хроматографии (элюент: AcOEt:MeOH, 9:1), получая 0,04 г указанного в заголовке соединения (37%).
1H-ЯМР (400 МГц, CD3OD): δ 1,54-1,70 (6H, м), 1,79-1,91 (4H, м), 2,83-2,92 (3H, м), 3,53-3,59 (4H, м), 3,86 (2H, д), 7,16 (1Н, дд), 7,43 (2H, м), 7,69 (1Н, д), 8,36 (1Н, д), 8,94 (1H, с); m/z 424 (М+H)+, время удерживания (способ b) = 3,38 (проход 10 минут).
СПОСОБ D

Пример 4: (3-Диметиламинопирролидин-1-ил)-{(S)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон
Способ D - Стадия a. В колбу с гидрохлоридом (S)-1-[3-(1-метил-1H-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 1, стадия d) (0,10 г, 0,27 ммоль) (получен, как описано в способ A, стадия d) и HATU (0,11 г, 0,28 ммоль) добавляли ТЭА (0,08 мл, 0,59 ммоль) и дихлорметан (2 мл), затем добавляли диметилпирролидин-3-иламин (0,04 мл, 0,33 ммоль). Реакционную смесь нагревали при 35ºС в течение ночи, затем добавляли раствор хлорида аммония (2 мл) и двухфазный раствор перемешивали в течение нескольких минут. Органический слой выделяли и сырой продукт очищали с использованием SCX-колонки (элюент от ДХМ:MeOH 1:1 до 2N NH3 в MeOH) и препаративной ВЭЖХ, получая 0,07 г диастереоизомерной смеси указанного в заголовке соединения в виде формиатной соли (65%).
1H-ЯМР (400 МГц, CD3OD): δ 1,62-2,11 (м, 10H); 2,66-2,39 (м, 2H); 2,53 (д, J=2,3 Гц, 6H); 2,61 (д, J=2,3 Гц, 6H); 2,78-2,99 (м, 6H), 3,35 (м, 4H), 3,50 (м, 1Н); 3,62-3,73 (м, 2H); 3,82-3,90 (м, 12H); 4,02 (м, 1Н), 7,19 (м, 4H); 7,32 (м, 6H); 7,44 (м, 2H); 7,56 (м, 2H); 7,68 (м, 2H), 8,33 (с, 2H), m/z 432 (М+H)+; время удерживания (способ а) = 0,70 (проход 10 минут).
СПОСОБ E
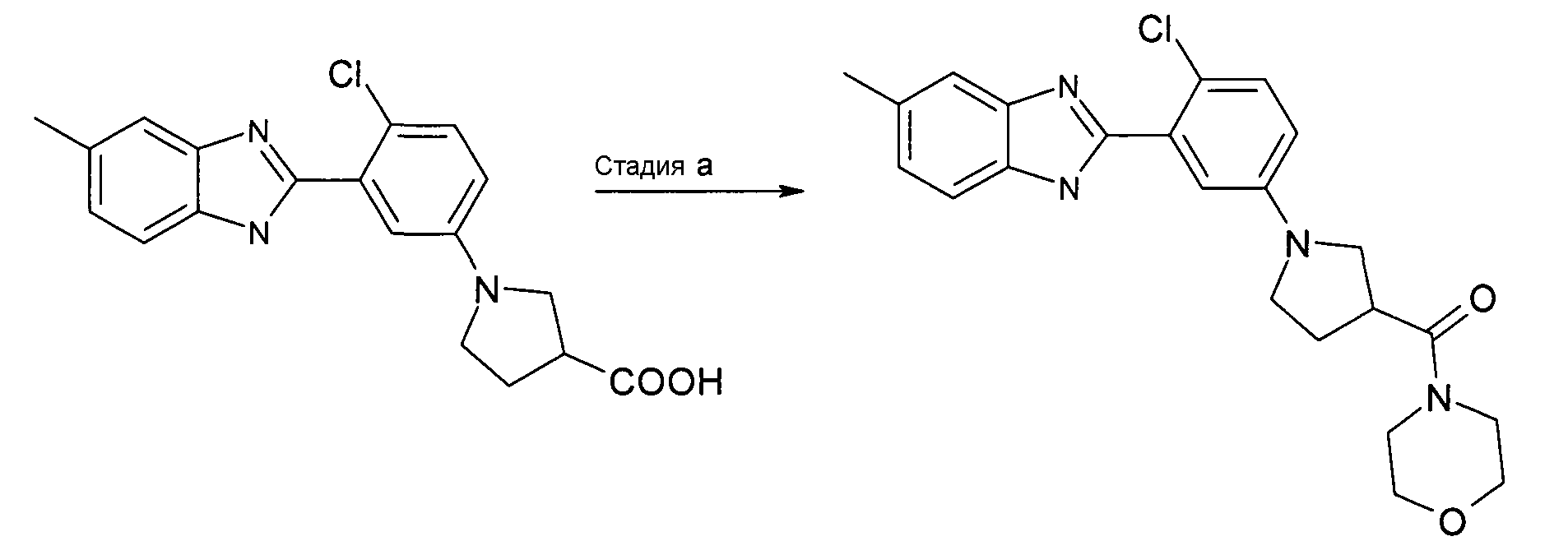
Пример 5: {1-[4-Хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-ил}морфолин-4-илметанон
Способ E - Стадия a. В колбу с 1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-карбоновой кислотой (получена, как описано в общем способе 5, стадия b) (0,09 г, 0,25 ммоль), HATU (0,10 г, 0,26 ммоль), ТЭА (0,07 мл, 0,53 ммоль) и морфолином (0,03 мл, 0,33 ммоль) добавляли дихлорметан (2 мл) и реакционную смесь нагревали при 35ºС в течение ночи. Реакционную смесь охлаждали до комнатной температуры, добавляли при перемешивании насыщенный раствор NaHCO3 (2 мл), органический слой выделяли фильтрованием через фазовый сепаратор и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали на NH2-колонке (элюент: дихлорметан:MeOH от 10:0 до 5:5) и SCX-колонке, получая 0,05 г указанного в заголовке соединения (46%).
1H-ЯМР (400 МГц, CD3OD): δ 2,20-2,32 (2H, м), 2,48 (3H, с), 3,34-3,52 (3H, м), 3,53-3,63 (4H, м), 3,64-3,72 (6H, м), 6,71 (1Н, дд), 7,01 (1Н, д), 7,12 (1Н, д), 7,34 (1Н, д), 7,41 (1Н, с), 7,50 (1Н, д); m/z 425 (М+H)+, время удерживания (способ а) = 2,13 (проход 10 минут).
СПОСОБ F
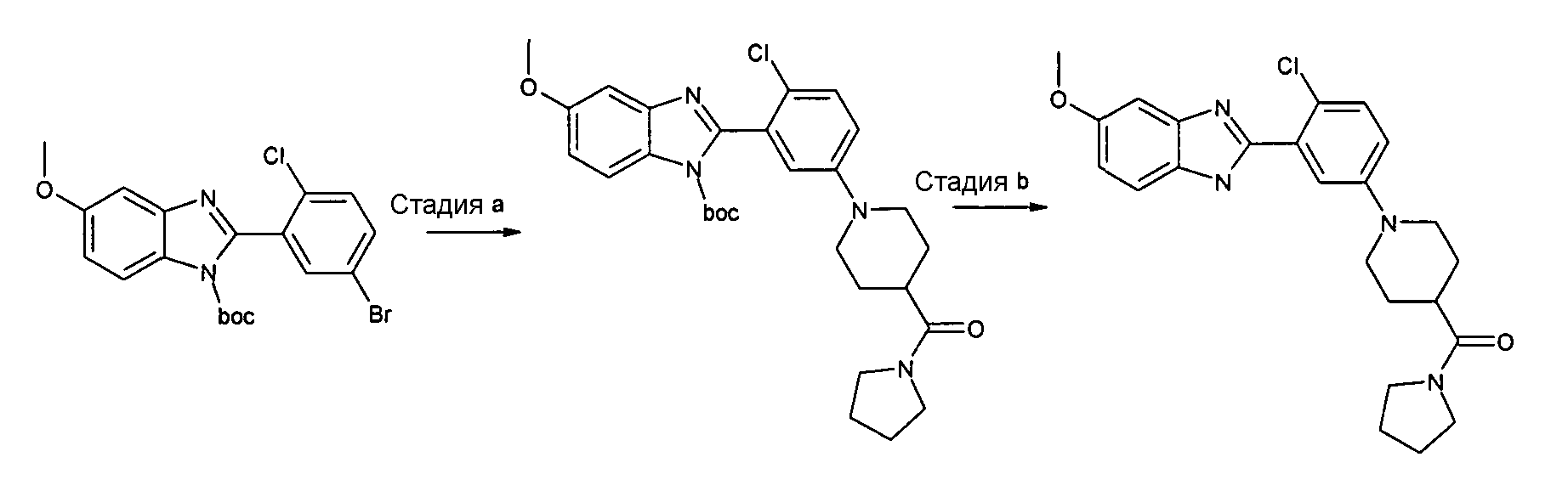
Пример 6: {1-[4-Хлор-3-(5-метокси-1Н-бензоимидазол-2-илфенил]пирролидин-4-ил}пирролидин-1-илметанонформиат
трет-Бутиловый эфир 2-{2-хлор-5-[4-(пирролидин-1-карбонил)пиперидин-1-ил]фенил}-5-метоксибензоимидазол-1-карбоновой кислоты
Способ F - Стадия a. трет-Бутиловый эфир 2-(5-бром-2-хлорфенил)-5-метоксибензоимидазол-1-карбоновой кислоты (получен, как описано в общем способе 4, стадия c) (0,10 г, 0,23 ммоль), пиперидин-4-илпирролидин-1-илметанон (0,05 г, 0,30 ммоль) и карбонат цезия (0,37 г, 1,14 ммоль) помещали в высушенную колбу и проводили 3 цикла вакуумирование/заполнение азотом, затем добавляли безводный толуол (0,20 мл). В то же время ацетат палладия (0,01 г, 0,05 ммоль) и БИНАП (0,04 г, 0,07 ммоль) помещали в высушенную колбу емкостью 4 мл в атмосфере азота и проводили 3 цикла вакуумирование/заполнение азотом. Затем при комнатной температуре в атмосфере азота добавляли безводный толуол (0,40 мл) и смесь добавляли в первую колбу. Реакционную смесь нагревали при 80ºС в течение ночи, охлаждали до комнатной температуры, добавляли EtOAC (3 мл) и смесь фильтровали. Растворитель удаляли и неочищенный продукт извлекали EtOAc (3,5 мл) и фильтровали через 2 г диоксида кремния на колонке (элюент EtOAc), получая 0,10 г указанного в заголовке соединения (82%) без дополнительной очистки.
(1-[4-Хлор-3-(5-метокси-1Н-бензоимидазол-2-ил)фенил]пирролидин-4-ил)пирролидин-1-илметанонформиат
Способ F - Стадия b. К смеси трет-бутилового эфира 2-{2-хлор-5-[4-(пирролидин-1-карбонил)пиперидин-1-ил]фенил}-5-метоксибензоимидазол-1-карбоновой кислоты (0,10 г, 0,19 ммоль) в 2M HCl в Et2O (2 мл), добавляли несколько капель дихлорметана и метанола для улучшения растворимости исходного вещества. Полученную смесь перемешивали в течение ночи при комнатной температуре, добавляли Et2O (5 мл), осадок отфильтровывали и затем очищали с помощью препаративной ВЭЖХ, получая 0,03 г указанного в заголовке соединения в виде гидрохлоридной соли с количественным выходом.
1H-ЯМР (400 МГц, CD3OD): δ 1,83-1,93 (6H, м), 1,95-2,04 (2H, м), 2,65-2,74 (1Н, м), 2,79-2,90 (2H, м), 3,41 (2H, т), 3,60 (2H, т), 3,81-3,88 (2H, м), 3,86 (3H, с), 6,92 (1Н, дд), 7,07-7,14 (2H, м), 7,37-7,41 (2H, м), 7,51 (1Н, д); m/z 439 (М+H)+, время удерживания (способ а) = 2,23 (проход 10 минут).
СПОСОБ G
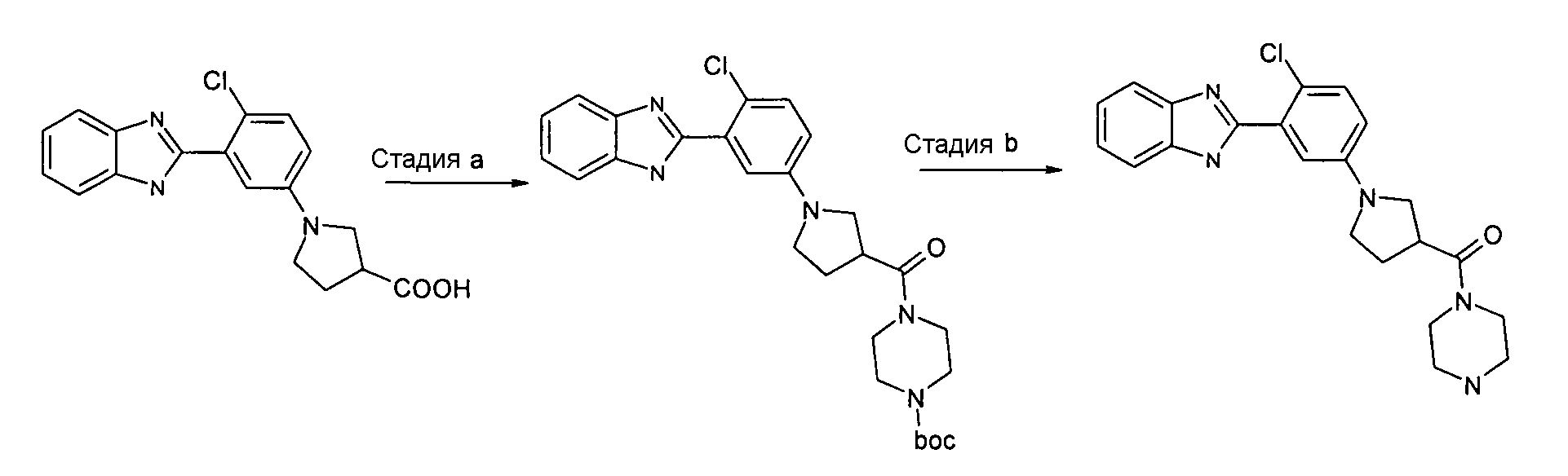
Пример 7: {1-[3-(1Н-Бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-ил}пиперазин-1-илметанон
трет-Бутиловый эфир 4-{1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-карбонил}пиперазин-1-карбоновой кислоты
Способ G - Стадия a. К смеси 1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-карбоновой кислоты (получена, как описано в общем способе 2, стадия e) (0,01 г, 0,26 ммоль) в ДХМ (4 мл), добавляли HATU (0,10 г, 0,29 ммоль), диизопропилэтиламин (DIPEA) (0,14 мл, 0,76 ммоль) и трет-бутил-1-пиперазинкарбоксилат (0,06 г, 0,32 ммоль). Реакционную смесь нагревали при 35ºС в течение ночи, охлаждали до комнатной температуры и промывали водой (2×5 мл) и насыщенным раствором Na2CO3 (2×5 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая неочищенное вещество, которое растирали с диэтиловым эфиром (3 мл), фильтровали и сушили. Осадок затем очищали флэш-хроматографией (элюент градиент: от EtOAc 100% до EtOAc:NH3 в MeOH (2M)/4:0,8), а затем проводили фильтрование через SCX картридж (элюент градиент: от ДХМ:MeOH/1:1 до NH3 в MeOH), получая 0,11 г указанного в заголовке соединения (67%).
(1-[3-(1Н-Бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-ил}пиперазин-1-илметанон
Способ G - Стадия b. К смеси трет-бутилового эфира 4-{1-[3-(1Н-бензоимидазол-2-ил)-4-хлорфенил]пирролидин-3-карбонил}пиперазин-1-карбоновой кислоты (0,11 г, 0,21 ммоль) в ДХМ (1 мл) добавляли 2M HCl в Et2O (4 мл). Смесь перемешивали при комнатной температуре в течение ночи, полученный осадок отфильтровывали и промывали Et2O. Осадок затем помещали в насыщенный раствор NaHCO3 (3 мл), экстрагировали ДХМ (2×3 мл), растворитель удаляли и неочищенный продукт фильтровали через SCX картридж, получая 0,05 г указанного в заголовке соединения (57%).
1H-ЯМР (400 МГц, CD3OD): δ 2,09-2,23 (2H, м), 2,69-2,78 (4H, м), 3,28-3,44 (3H, м), 3,47-3,56 (6H, м), 6,61-6,64 (1Н, м), 6,92-6,93 (1Н, м), 7,16-7,20 (2H, м), 7,24-7,27 (1Н, м), 7,53 (2H, ушир.с); m/z 410 (М+H)+, время удерживания (способ b) = 0,88 (проход 10 минут).
Способ Н
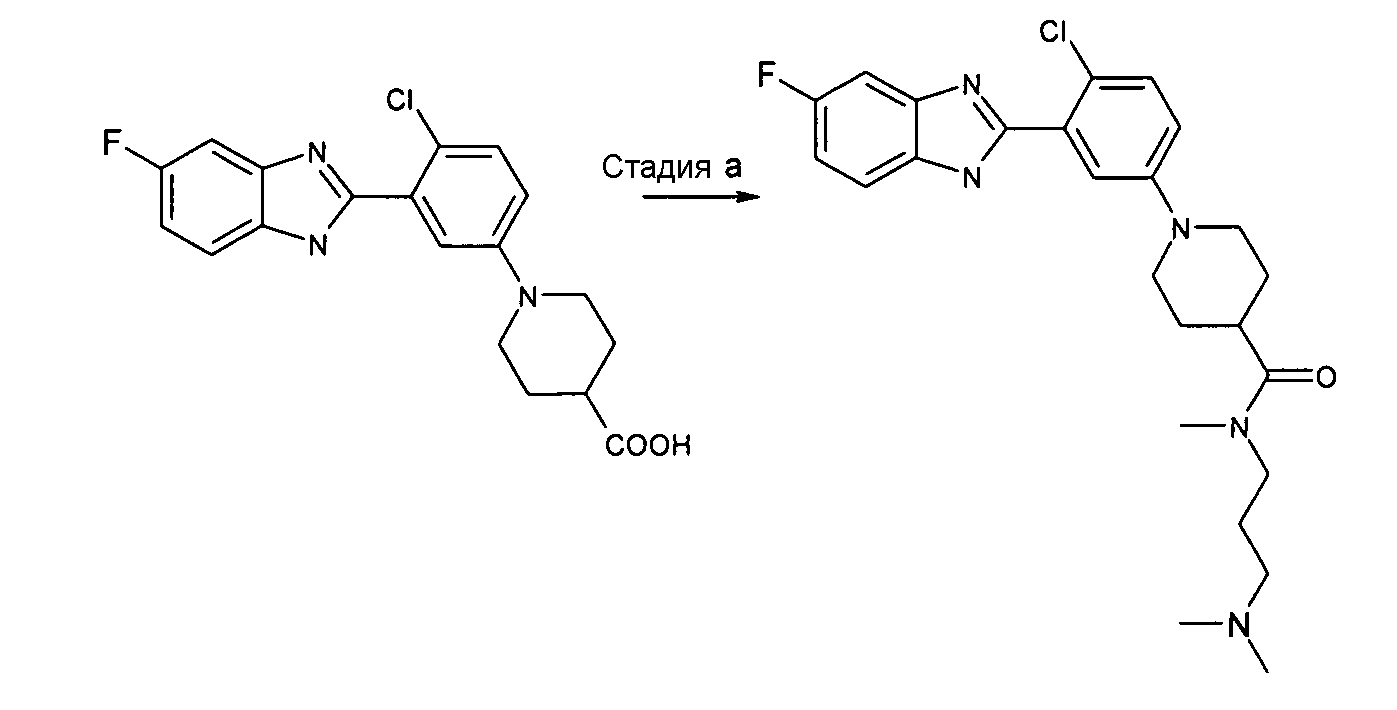
Пример 8: (3-Диметиламинопропил)метиламид 1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ H - Стадия a. В колбу с гидрохлоридом 1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты (получен, как описано в общем способе 3,4, стадия e) (0,10 г, 0,26 ммоль) и HATU (0,10 г, 0,27 ммоль) в дихлорметане (2 мл) добавляли ТЭА (0,07 мл, 0,54 ммоль) и N,N,N'-триметил-1,3-пропандиамин (0,32 ммоль, 0,05 мл). Реакционную смесь нагревали при 35ºС в течение ночи, растворитель удаляли и сырой продукт очищали с помощью препаративной ВЭЖХ и колонки SCX, получая 0,06 г указанного в заголовке соединения (49%).
1H-ЯМР (400 МГц, ДМСО): δ 1,50-1,72 (6H, м), 2,15 (8H, м), 2,80 (4H, м), 3,02 (2H, с), 3,30 (2H, м), 3,70 (2H, м), 7,06 (2H, м), 7,28-7,56 (3H, м), 7,69 (1Н, м), 12,74 (1Н, с); m/z 472 (М+H)+, время удерживания (способ a) = 1,68 (проход 10 минут).
СПОСОБ I
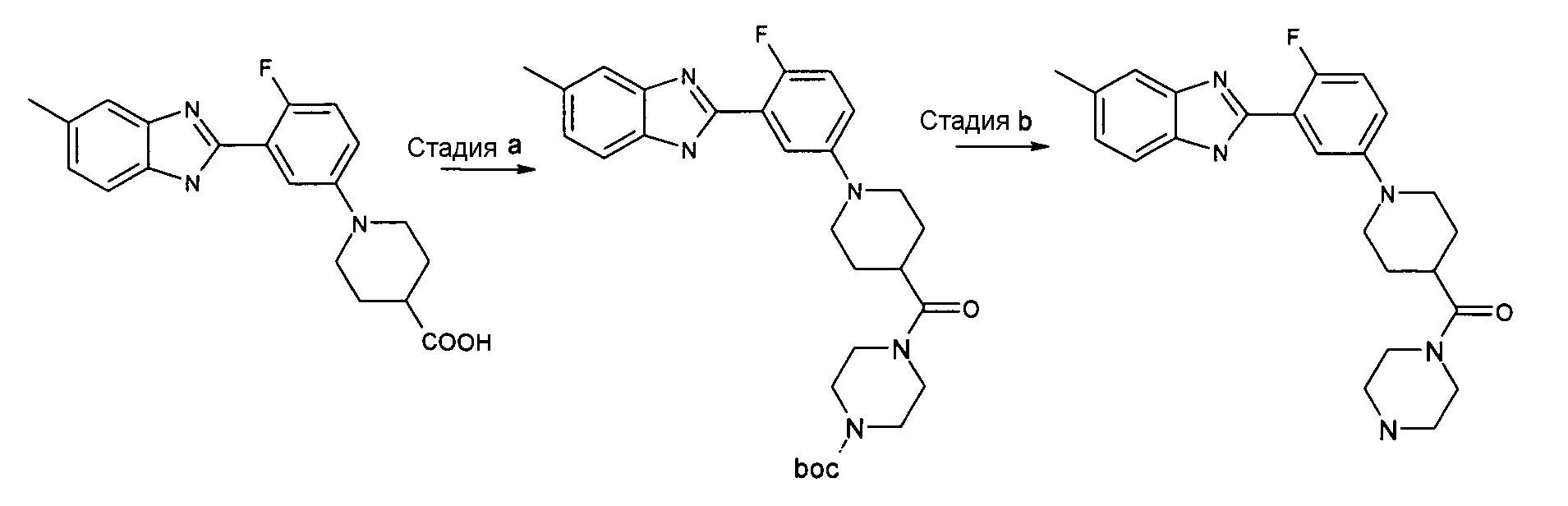
Пример 9: (1-[4-Фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}пиперазин-1-илметанон
трет-Бутиловый эфир 4-{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбонил}пиперазин-1-карбоновой кислоты
Способ I - Стадия a. В колбу с гидрохлоридом [1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты (получен, как описано в общем способе 8, стадия b) (0,10 г, 0,26 ммоль) и HATU (0,10 г, 0,27 ммоль) в дихлорметане (2 мл) добавляли ТЭА (0,08 мл, 0,56 ммоль) и трет-бутил-1-пиперазинкарбоксилат (0,32 ммоль, 0,06 г). Реакционную смесь нагревали при 35ºС в течение ночи, промывали водой (3×2 мл) и насыщенным раствором Na2CO3 (3×2 мл). Неочищенный продукт затем очищали флэш-хроматографией (элюент: EtOAc), а затем проводили фильтрование через NH2-картридж (элюент EtOAc), получая 0,02 г указанного в заголовке соединения (15%).
{1-[4-Фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-ил}пиперазин-1-илметанон
Способ I - Стадия b. Смесь трет-бутилового эфира 4-{1-[4-фтор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбонил}пиперазин-1-карбоновой кислоты (0,02 г, 0,04 ммоль) в 2M HCl в Et2O (3 мл) перемешивали в течение 2 дней при комнатной температуре, затем растворитель удаляли и неочищенный продукт фильтровали через NH2-картридж (элюент EtOAc), получая 0,02 г указанного в заголовке соединения с количественным выходом.
1H-ЯМР (400 МГц, CDCl3): δ 1,83 (2H, м), 1,96-2,07 (2H, м), 2,50 (3H, с), 2,56-2,63 (1Н, м), 2,77-2,93 (6H, м), 3,58 (4H, д), 3,79 (2H, д), 6,98 (1Н, м), 7,10 (2H, м), 7,29-7,39 (1Н, м), 7,62-7,72 (1Н, м), 8,00 (1Н, дд), 9,78 (1Н, ушир.с); m/z 421 (М+H)+, время удерживания (способ a) = 1,37 (проход 10 минут).
СПОСОБ J
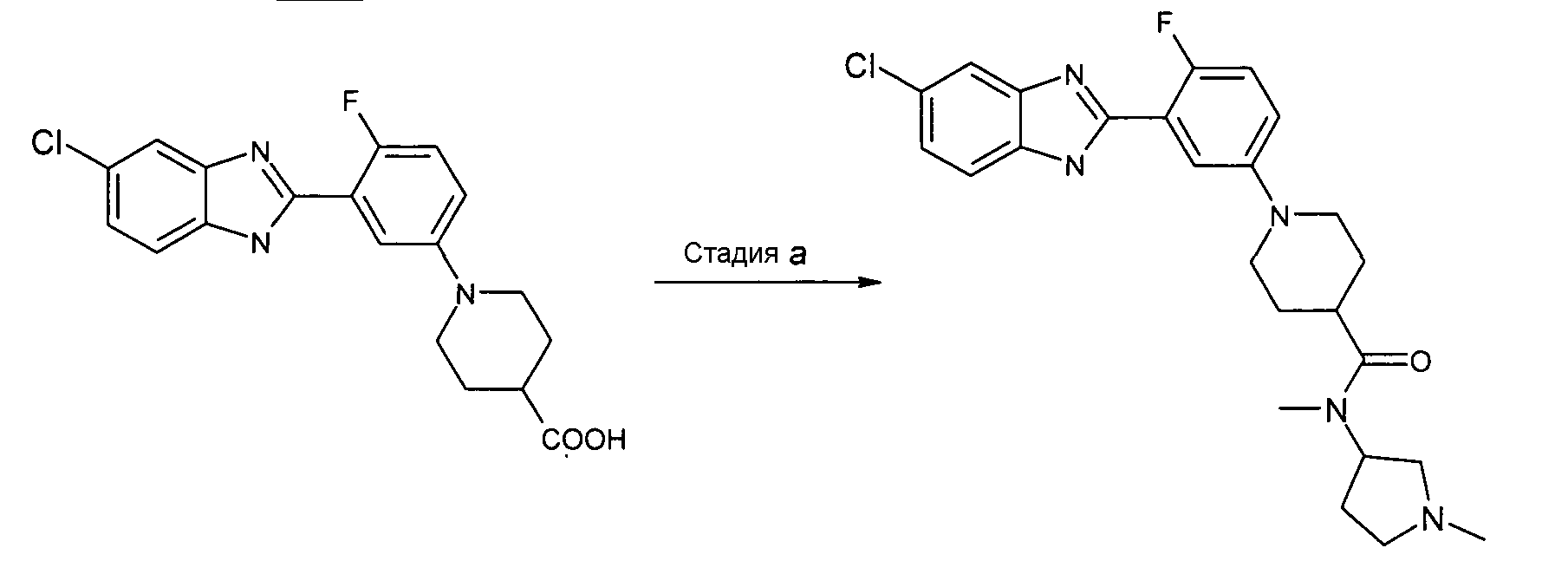
Пример 10: Метил-(1-метилпирролидин-3-ил)амид 1-[3-(5-хлор-1Н-бензоимидазол-2-ил)-4-фторфенил]пиперидин-4-карбоновой кислоты
Способ J - Стадия a. К смеси гидрохлорида 1-[3-(5-хлор-1Н-бензоимидазол-2-ил)-4-фторфенил]пиперидин-4-карбоновой кислоты (получен, как описано в общем способе 10, стадия b) (0,01 г, 0,25 ммоль) в ДХМ (2 мл), добавляли HATU (0,10 г, 0,26 ммоль), ТЭА (0,07 мл, 0,55 ммоль) и N,N'-диметил-3-аминопирролидин (0,04 г, 0,31 ммоль). Реакционную смесь нагревали при 35ºС в течение ночи, охлаждали до комнатной температуры и промывали раствором хлорида аммония (2 мл), насыщенным раствором Na2CO3 (2 мл) и водой (2 мл). Органический слой затем фильтровали через NH2-картридж и дополнительно очищали флэш-хроматографией (элюент: EtOAc:NH3 2н. в MeOH/9:1), получая 0,03 г указанного в заголовке соединения (30%).
1H-ЯМР (400 МГц, CD3OD): δ 1,79-1,94 (5H, м), 2,08-2,30 (2H, м), 2,37 (3H, с), 2,46-2,54 (2H, м), 2,62-2,69 (2H, м), 2,72-2,92 (2H, м), 3,10 (3H, с), 3,79 (2H, д), 5,16 (1Н, м), 7,14-7,20 (2H, м), 7,26 (1Н, дд), 7,58-7,63 (2H, м), 7,73 (1Н, дд); m/z 470 (М+H)+, время удерживания (способ b) = 1,80 (проход 10 минут).
СПОСОБ K
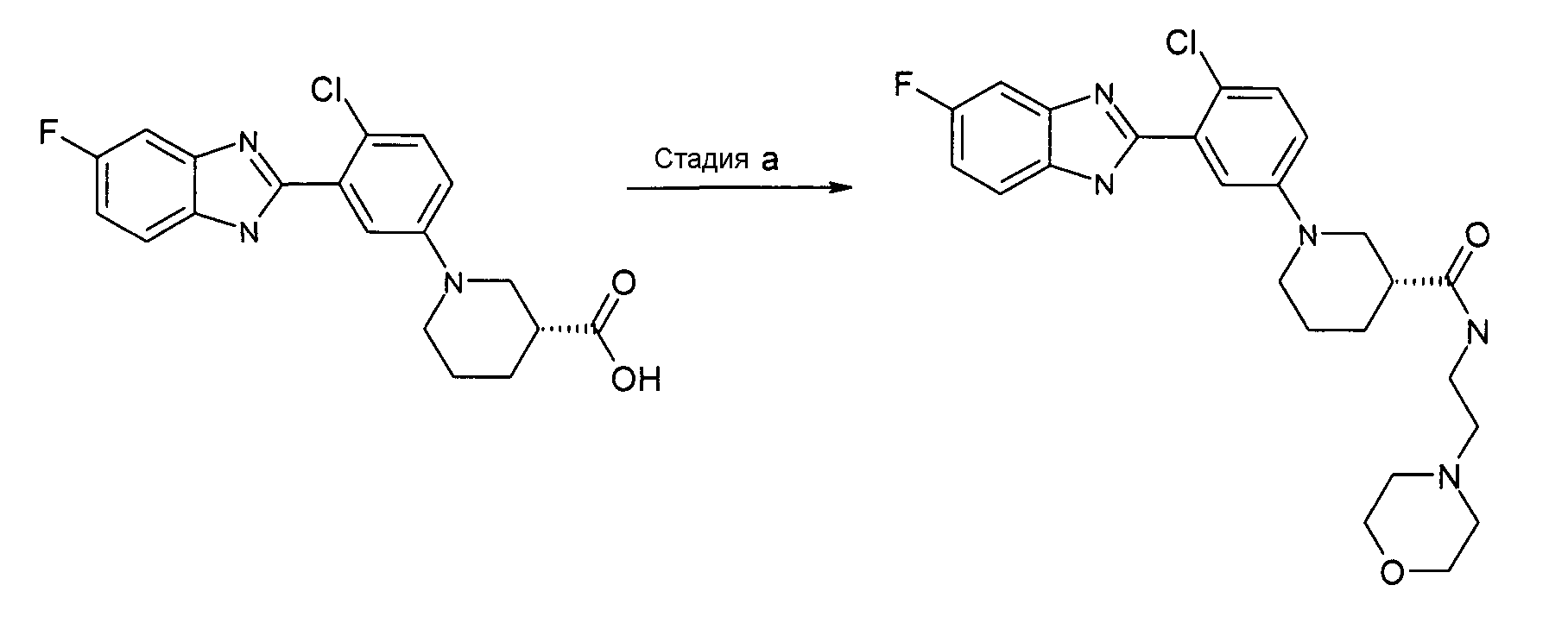
Пример 11: (2-Морфолин-4-илэтил)амид (R)-1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты
Способ K - Стадия a. К смеси гидрохлорида (R)-1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 3, 4, стадия e) (0,01 г, 0,27 ммоль) в ДХМ (2 мл) добавляли HATU (0,10 г, 0,28 ммоль), ТЭА (0,08 мл, 0,56 ммоль) и 2-морфолиноэтиламин (0,04 г, 0,33 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, промывали раствором хлорида аммония (2 мл), фильтровали через фазовый сепаратор и удаляли органический растворитель. Неочищенный продукт затем очищали на колонке SCX и флэш-хроматографией (элюент: градиент от EtOAc до EtOAc:NH3 в MeOH (2M)/10:1), получая 0,05 г указанного в заголовке соединения (40%).
1H-ЯМР (400 МГц, CD3OD): δ 1,66-1,98 (4H, м), 2,40-2,64 (7H, м), 2,94 (1Н, м), 3,06 (1Н, дд), 3,36 (2H, м), 3,57-3,80 (6H, м), 7,04-7,16 (2H, м), 7,33 (13H, ушир.с), 7,42 (2H, м), 7,62 (1Н, ушир.с); m/z 486 (М+H)+, время удерживания (способ b) = 2,97 (проход 10 минут).
СПОСОБ L
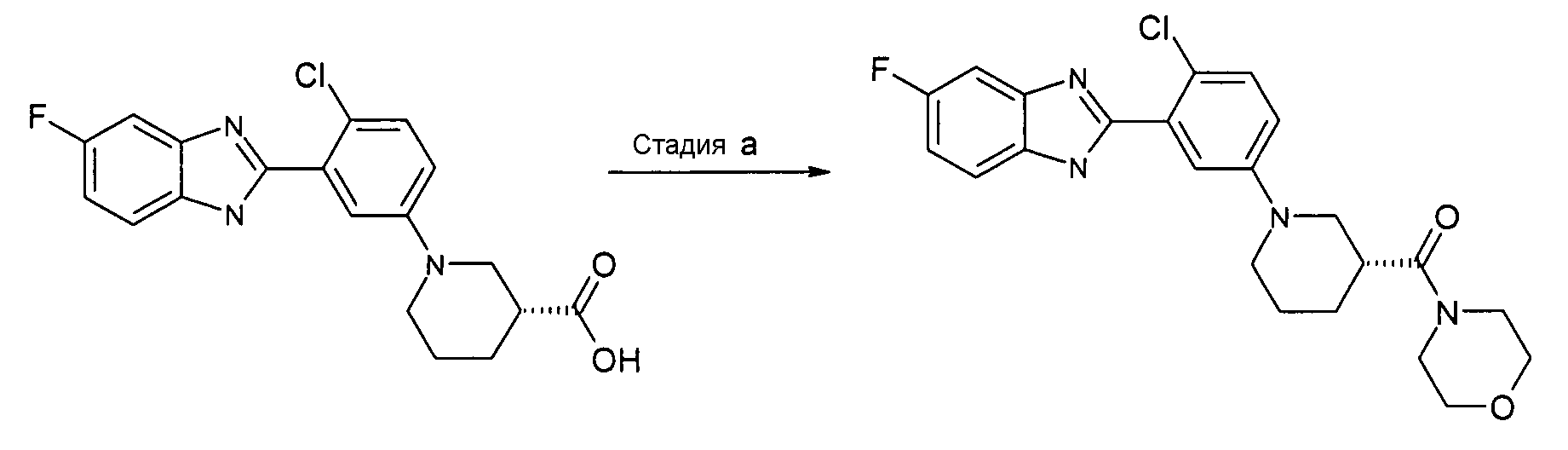
Пример 12: {(R)-1-[4-Хлор-3-(5-фтор-1Н-бензимидазол-2-ил)фенил]пиперидин-3-ил}морфолин-4-илметанон
Способ L - Стадия a. К смеси гидрохлорида (R)-1-[4-хлор-3-(5-фтор-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 3, 4, стадия e) (0,01 г, 0,27 ммоль) в ДХМ (2 мл) добавляли HATU (0,10 г, 0,28 ммоль), ТЭА (0,08 мл, 0,56 ммоль) и 2-морфолиноэтиламин (0,04 г, 0,33 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, промывали раствором хлорида аммония (2 мл), фильтровали через фазовый сепаратор и удаляли органический растворитель. Неочищенный продукт затем очищали на колонке SCX и флэш-хроматографией (элюент: градиент от EtOAc до EtOAc:NH3 в MeOH (2M)/10:1). Проводили дополнительную очистку с помощью препарартивной ВЭЖХ, получая 0,03 г указанного в заголовке соединения (29%).
1H-ЯМР (400 МГц, CD3OD): δ 1,57-1,88 (3H, м), 1,95 (1Н, м), 2,81 (1Н, тд), 2,98 (2H, м), 3,50-3,85 (10H, м), 7,10 (2H, м), 7,32 (1Н, м), 7,40 (2H, м), 7,60 (1Н, м); m/z 443 (М+H)+, время удерживания (способ b) = 4,98 (проход 10 минут).
СПОСОБ M

Пример 13: (4-Метоксипиперидин-1-ил)-{(R)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон
Способ M - Стадия a. К смеси гидрохлорида (R)-1-[3-(1-метил-1H-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 1, стадия d) (0,01 г, 0,30 ммоль) (получен, как описано в способ A, стадия d) в ДХМ (2,5 мл) добавляли HATU (0,12 г, 0,33 ммоль), ТЭА (0,09 мл, 0,63 ммоль) и 4-метоксипиперидин (0,04 г, 0,33 ммоль). Реакционную смесь нагревали при 35оC в течение ночи, охлаждали до комнатной температуры, промывали раствором хлорида аммония (3 мл), фильтровали через фазовый сепаратор и удаляли органический растворитель. Неочищенный продукт затем очищали на колонке SCX (элюент: первоначально ДХМ:MeOH/1:1, затем NH3 в MeOH (2н.)) и флэш-хроматографией (элюент: градиент от EtOAc: циклогексан/10:0 до 0:10), получая 0,05 г указанного в заголовке соединения (38%).
1H-ЯМР (400 МГц, CD3OD): δ 1,42-1,69 (3H, м), 1,73-1,96 (5H, м), 2,78-2,93 (1Н, м), 2,93-3,07 (2H, м), 3,25-3,50 (6H, м), 3,79-3,94 (7H, м), 7,16-7,19 (2H, м), 7,28-7,36 (3H, м), 7,42-7,46 (1Н, м), 7,54-7,56 (1Н, м), 7,66-7,68 (1Н, м); m/z 433 (М+H)+, время удерживания (способ b) = 3,63 (проход 10 минут).
СПОСОБ N
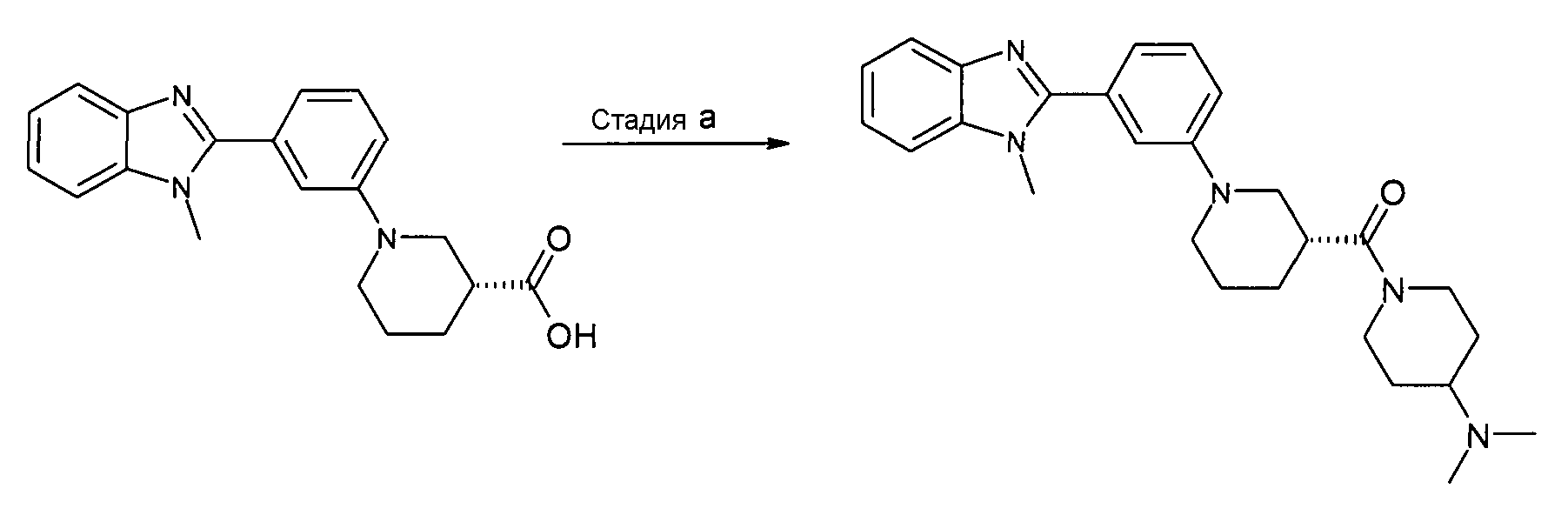
Пример 14: (4-Диметиламинопиперидин-1-ил)-{(R)-1-[3-(1-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-ил}метанон
Способ N - Стадия a. К смеси гидрохлорида (R)-1-[3-(1-метил-1H-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 1, стадия d) (0,01 г, 0,30 ммоль) (получен, как описано в способ A, стадия d) в ДХМ (2,5 мл) добавляли HATU (0,12 г, 0,33 ммоль), ТЭА (0,09 мл, 0,63 ммоль) и 4-(N,N-диметиламино)пиперидин (0,04 г, 0,35 ммоль). Реакционную смесь нагревали при 35оC в течение ночи, охлаждали до комнатной температуры, промывали раствором хлорида аммония (3 мл), фильтровали через фазовый сепаратор и удаляли органический растворитель. Неочищенный продукт затем очищали на колонке SCX (элюент: первоначально ДХМ:MeOH/1:1, затем NH3 в MeOH (2н.)) и флэш-хроматографией (элюент: градиент от EtOAc:NH3 в MeOH (2н.)/10:0 до 9:1), получая 0,05 г указанного в заголовке соединения (37%).
1H-ЯМР (400 МГц, CD3OD): δ 1,26-1,43 (2H, м), 1,59-2,00 (6H, м), 2,26-2,31 (6H, м), 2,43-2,49 (1Н, м), 2,57-2,64 (1Н, м), 2,79-3,18 (4H, м), 3,79-3,86 (2H, м), 3,89 (3H, с), 4,12-4,16 (1Н, м), 4,58-4,61 (1Н, м), 7,16-7,20 (2H, м), 7,28-7,37 (3H, м), 7,42-7,46 (1Н, м), 7,54-7,56 (1Н, м), 7,66-7,68 (1Н, м); m/z 446 (М+H)+, время удерживания (способ b) = 1,63 (проход 10 минут).
СПОСОБ O
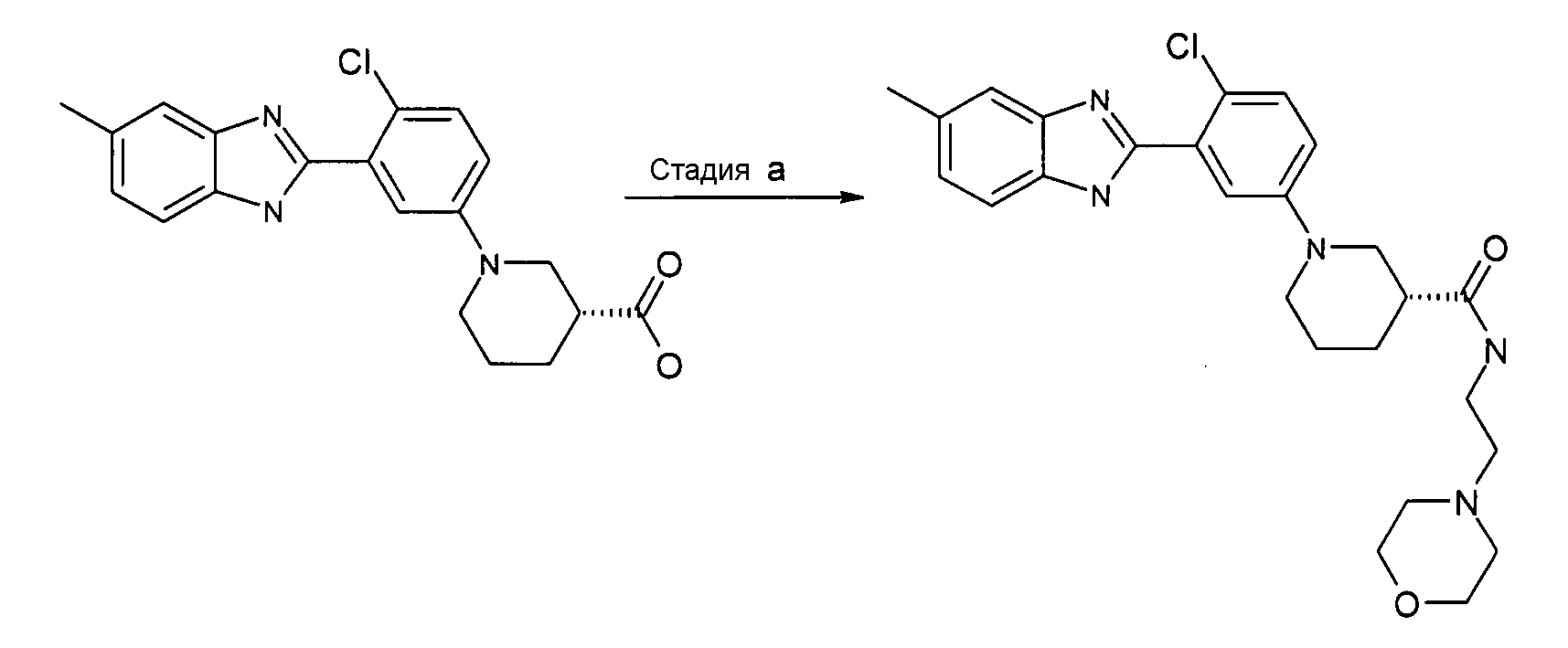
Пример 15: (2-Морфолин-4-илэтил)амид (R)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты
Способ O - Стадия a. В колбу с гидрохлоридом (R)-1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-3-карбоновой кислоты (получен, как описано в общем способе 3,4, стадия e) (0,10 г, 0,25 ммоль) добавляли HATU (0,10 г, 0,26 ммоль), ТЭА (0,07 мл, 0,53 ммоль) и 2-морфолин-4-илэтиламин (0,04 мл, 0,33 ммоль), дихлорметан (2 мл) и реакционную смесь нагревали при 35ºС в течение ночи. Реакционную смесь охлаждали до комнатной температуры, добавляли при перемешивании насыщенный раствор NaHCO3 (2 мл), органический слой выделяли путем фильтрования через фазовый сепаратор и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью SCX и флэш-хроматографии (элюент; градиент циклогексан:этилацетат от 100:0 до 3:1), получая 0,05 г указанного в заголовке соединения (42%).
1H-ЯМР (400 МГц, CD3OD): δ 1,66-1,97 (4H, м), 2,42-2,50 (6H, м), 2,48 (3H, с), 2,55-2,62 (1Н, м), 2,89-2,97 (1Н, м), 3,06 (1Н, дд), 3,28-3,41 (2H, м), 3,60 (4H, дд), 3,63-3,69 (1Н, м), 3,71-3,77 (1Н, м), 7,09-7,15 (2H, м), 7,38-7,54 (4H, м); m/z 482 (М+H)+, время удерживания (способ b) = 2,57 (проход 10 минут).
СПОСОБ P
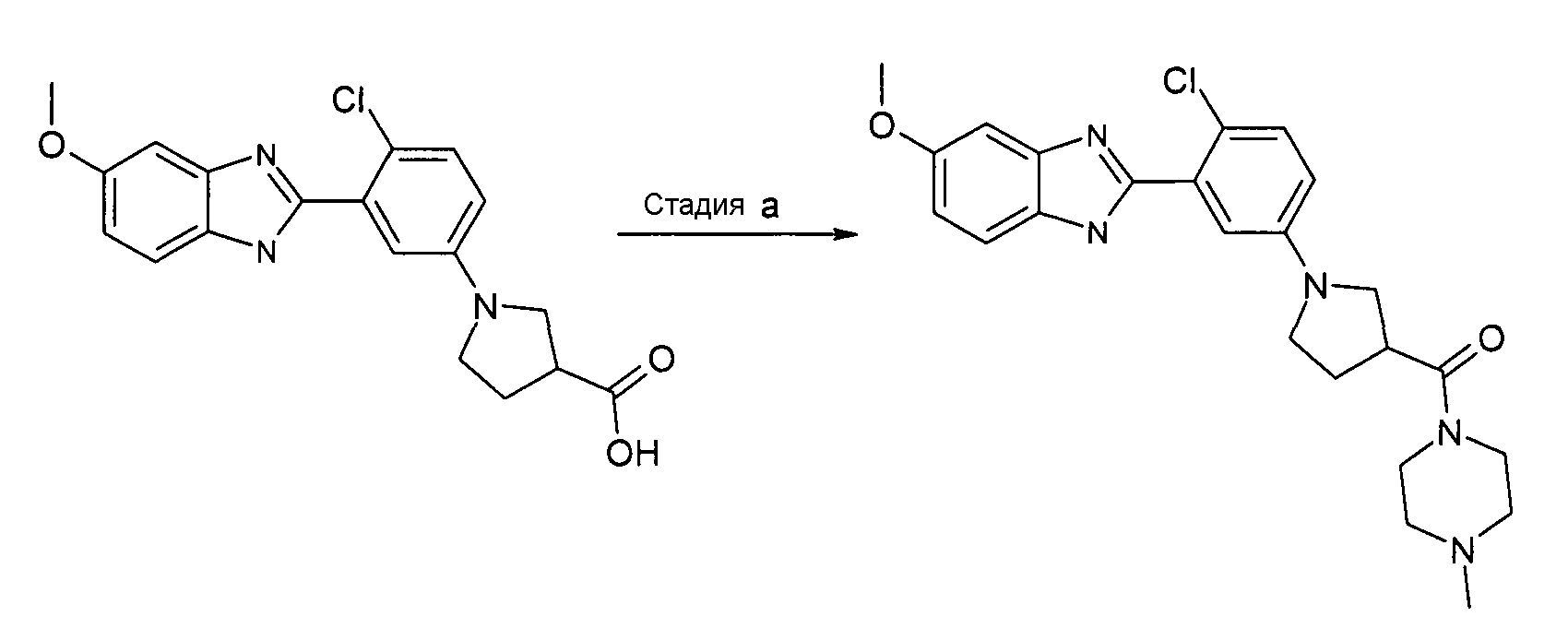
Пример 16: {1-[4-Хлор-3-(5-метокси-1Н-бензимидазол-2-ил)фенил]пирролидин-3-ил}-(4-метилпиперазин-1-ил)метанон
Способ P - Стадия a. В колбу с гидрохлоридом 1-[4-хлор-3-(5-метокси-1Н-бензоимидазол-2-ил)фенил]пирролидин-3-карбоновой кислоты (получен, как описано в общем способе 3,4, стадия e) (0,10 г, 0,25 ммоль) добавляли HATU (0,10 г, 0,26 ммоль), ТЭА (0,07 мл, 0,53 ммоль) и 1-метилпиперазин (0,04 мл, 0,33 ммоль), дихлорметан (2 мл) и реакционную смесь нагревали при 35ºС в течение ночи. Реакционную смесь охлаждали до комнатной температуры, добавляли при перемешивании насыщенный раствор NaHCO3 (2 мл), органический слой выделяли путем фильтрования через фазовый сепаратор и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на SCX колонке, растирания в диэтиловом эфире и, наконец, с помощью препаративной ВЭЖХ, получая 0,04 г указанного в заголовке соединения (34%).
1H-ЯМР (400 МГц, CD3OD): δ 2,20-2,33 (2H, м), 2,41 (3H, с), 2,51-2,66 (4H, м), 3,37-3,54 (3H, м), 3,56-3,76 (6H, м), 3,86 (3H, с), 6,71 (1Н, дд), 6,93 (1Н, ддд), 7,01 (1Н, д), 7,12 (1Н, д), 7,34 (1Н, дд), 7,52 (1Н, д); m/z 454 (М+H)+, время удерживания (способ b) = 2,13 (проход 10 минут).
СПОСОБ Q
Пример 17: Метил-(1-метилпирролидин-3-ил)амид 1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты
Способ Q - Стадия a. К смеси гидрохлорида 1-[4-хлор-3-(5-метил-1Н-бензоимидазол-2-ил)фенил]пиперидин-4-карбоновой кислоты (получен, как описано в общем способе 3,4, стадия e) (0,01 г, 0,25 ммоль) в ДХМ (2 мл) добавляли HATU (0,10 г, 0,26 ммоль), ТЭА (0,07 мл, 0,53 ммоль) и метил-(1-метилпирролидин-3-ил)амин (0,03 г, 0,31 ммоль). Реакционную смесь нагревали при 35ºС в течение ночи, охлаждали до комнатной температуры, промывали раствором хлорида аммония (3 мл), фильтровали через фазовый сепаратор и органическую фазу фильтровали через колонку SCX (элюент: первоначально ДХМ:MeOH/1:1, затем NH3 в MeOH (2н.)). Данную обработку проводили с использованием Zinsser Speedy (версия 6.1.3). Неочищенный продукт затем очищали флэш-хроматографией (элюент: градиент от EtOAc до EtOAc:NH3 в MeOH (2н.)/10:1), получая 0,04 г указанного в заголовке соединения (37%).
1H-ЯМР (400 МГц, CD3OD): δ 1,76-1,93 (5H, м), 2,08-2,23 (2H, м), 2,36-2,48 (6H, м), 2,51-3,09 (9H, м), 3,83-3,86 (2H, м), 5,13-5,20 (1Н, м), 7,09-7,13 (2H, м), 7,38-7,40 (3H, м), 7,51 (1Н, ушир.с); m/z 466 (М+H)+, время удерживания (способ b) = 2,30 (проход 10 минут).
В таблице показаны некоторые синтезированные соединения, которые были получены в соответствии со способом, указанным в третьей колонке таблицы, и приведенным выше обсуждением синтеза в примерах 1-17.
|
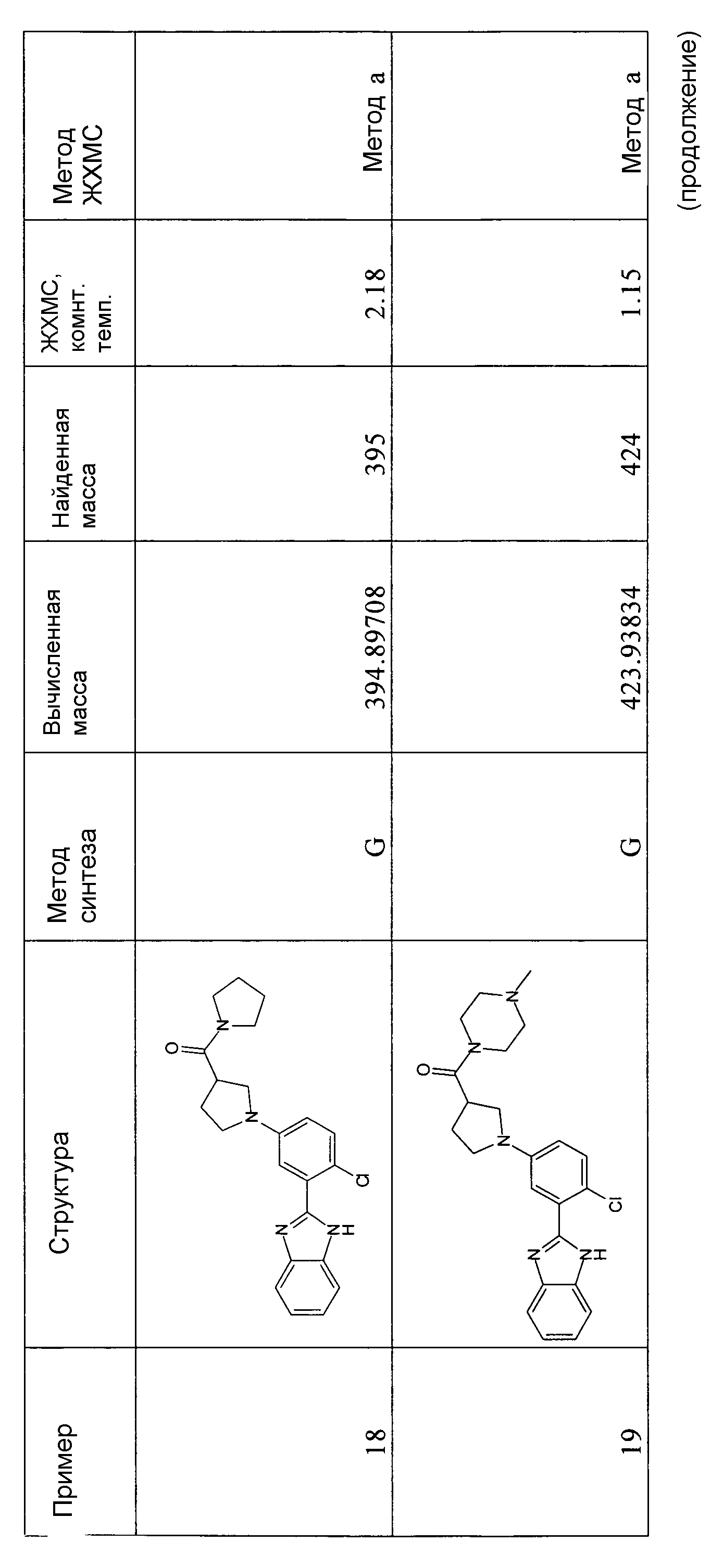
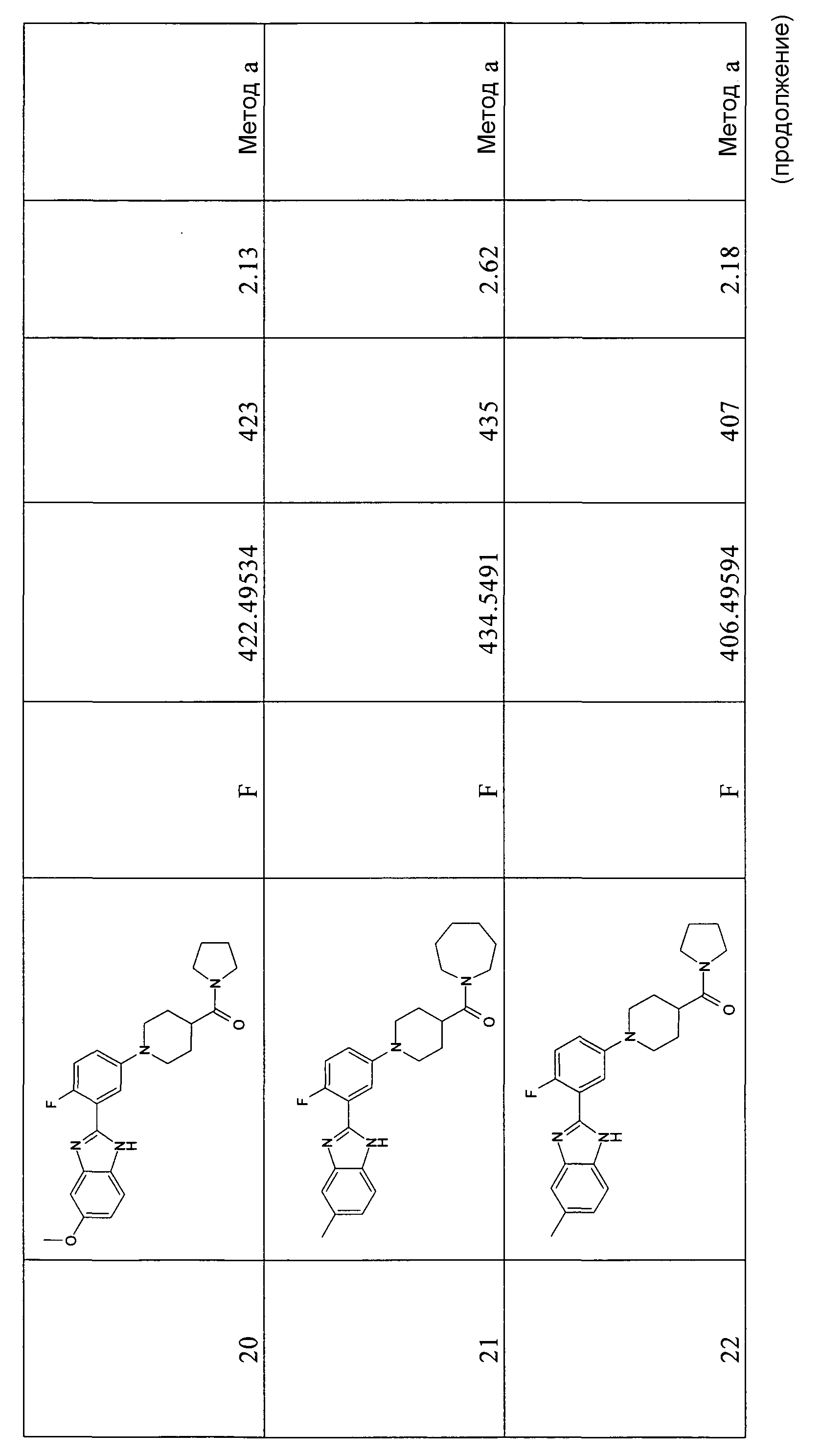
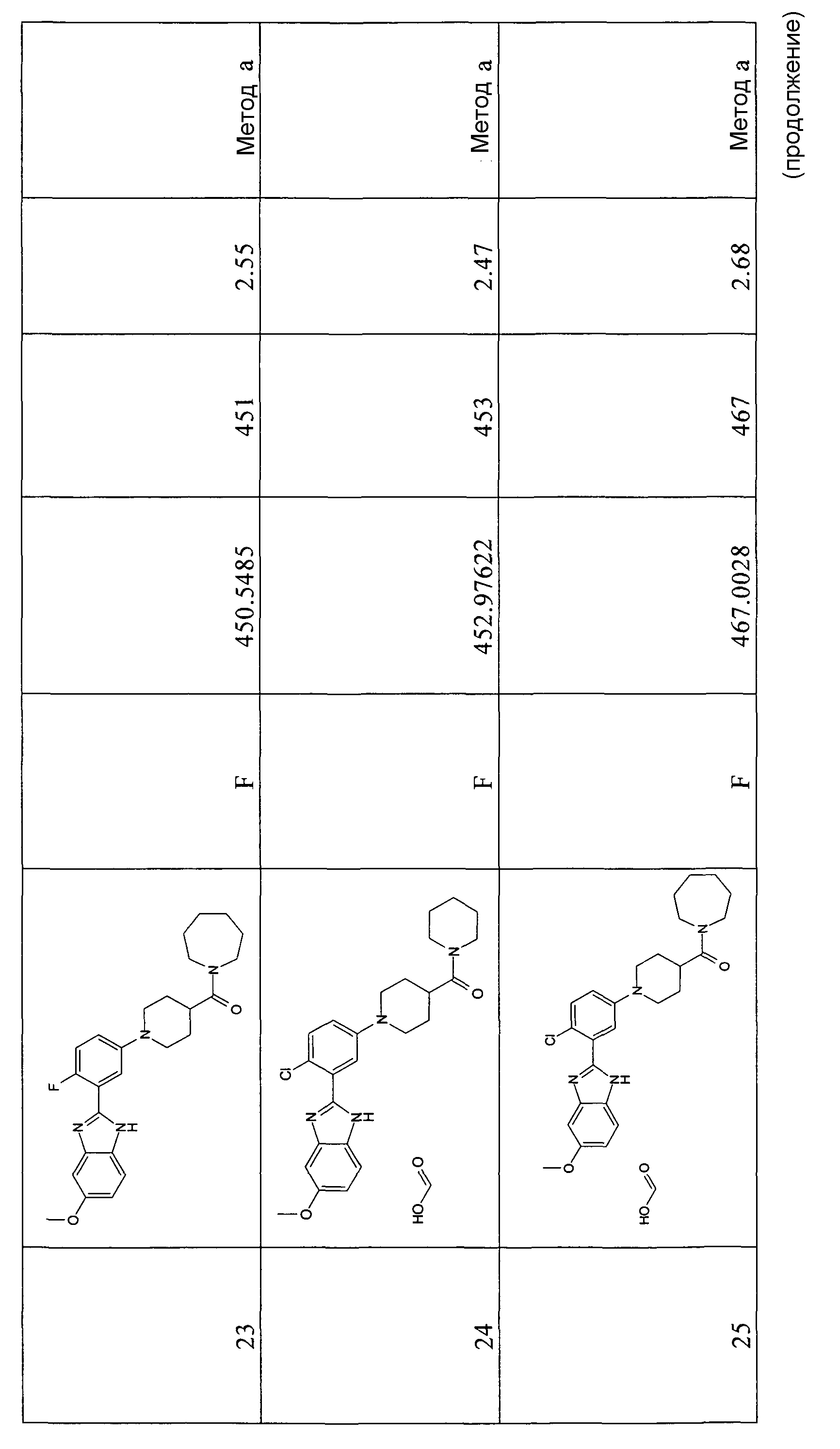
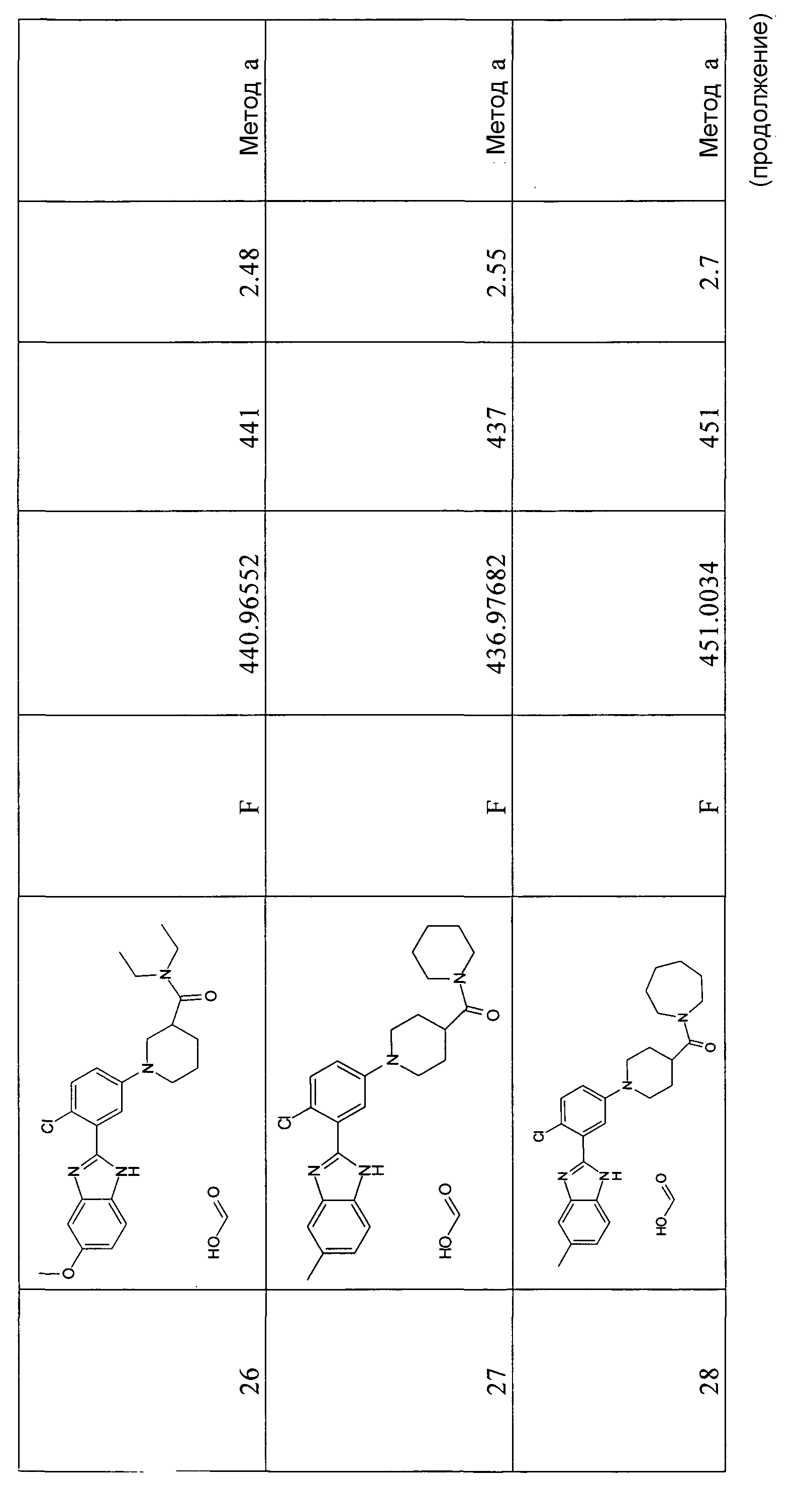
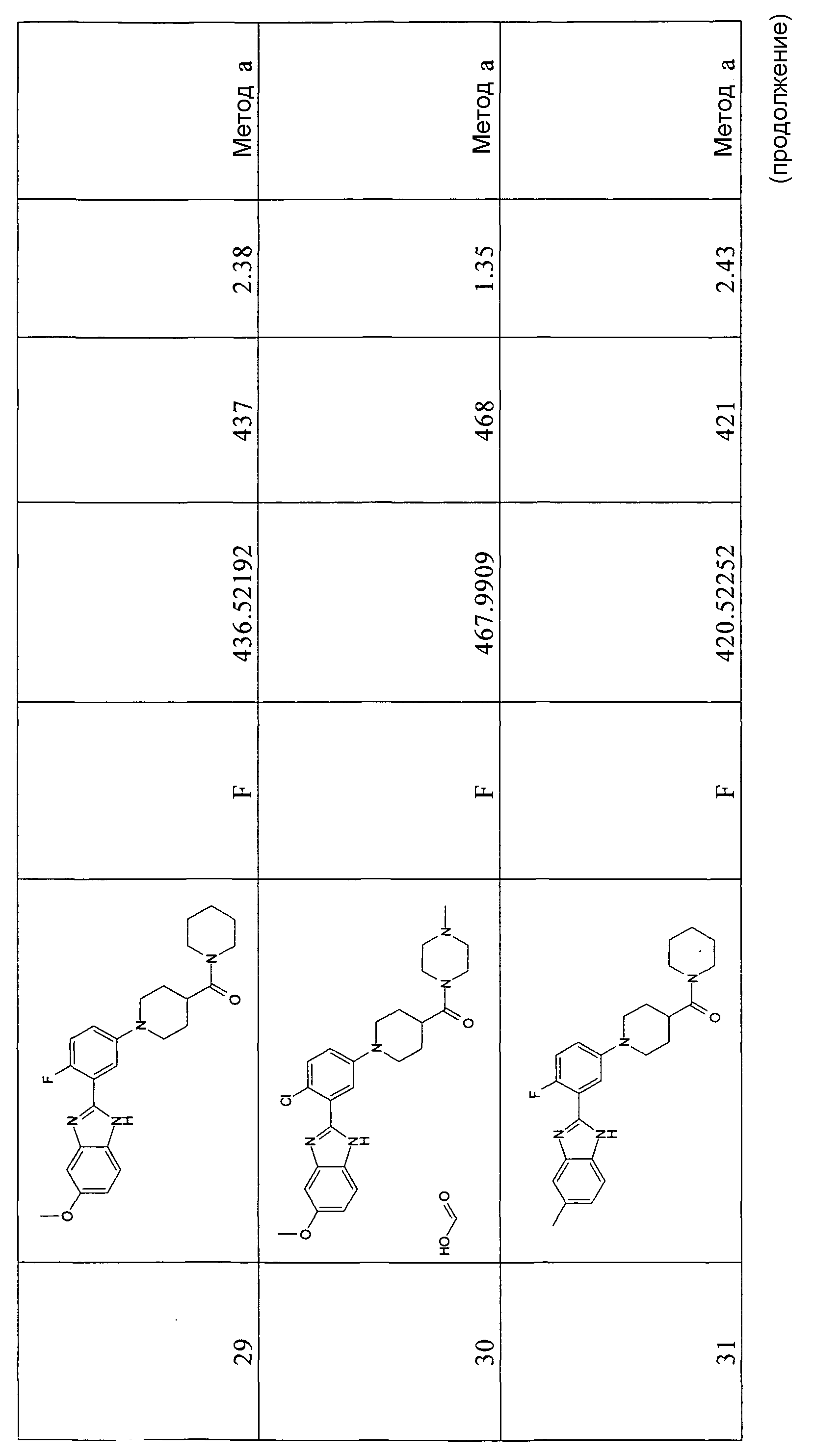
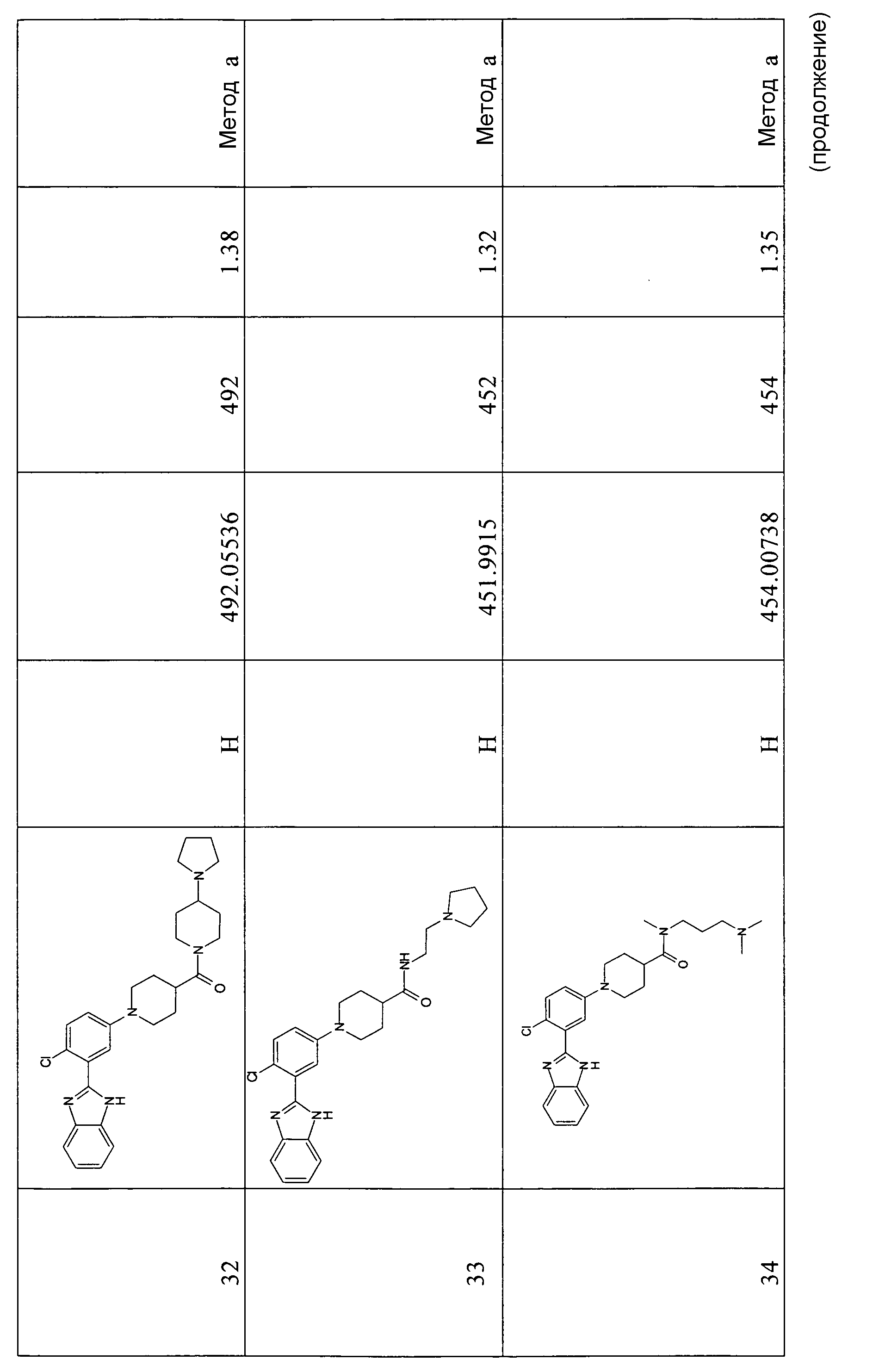
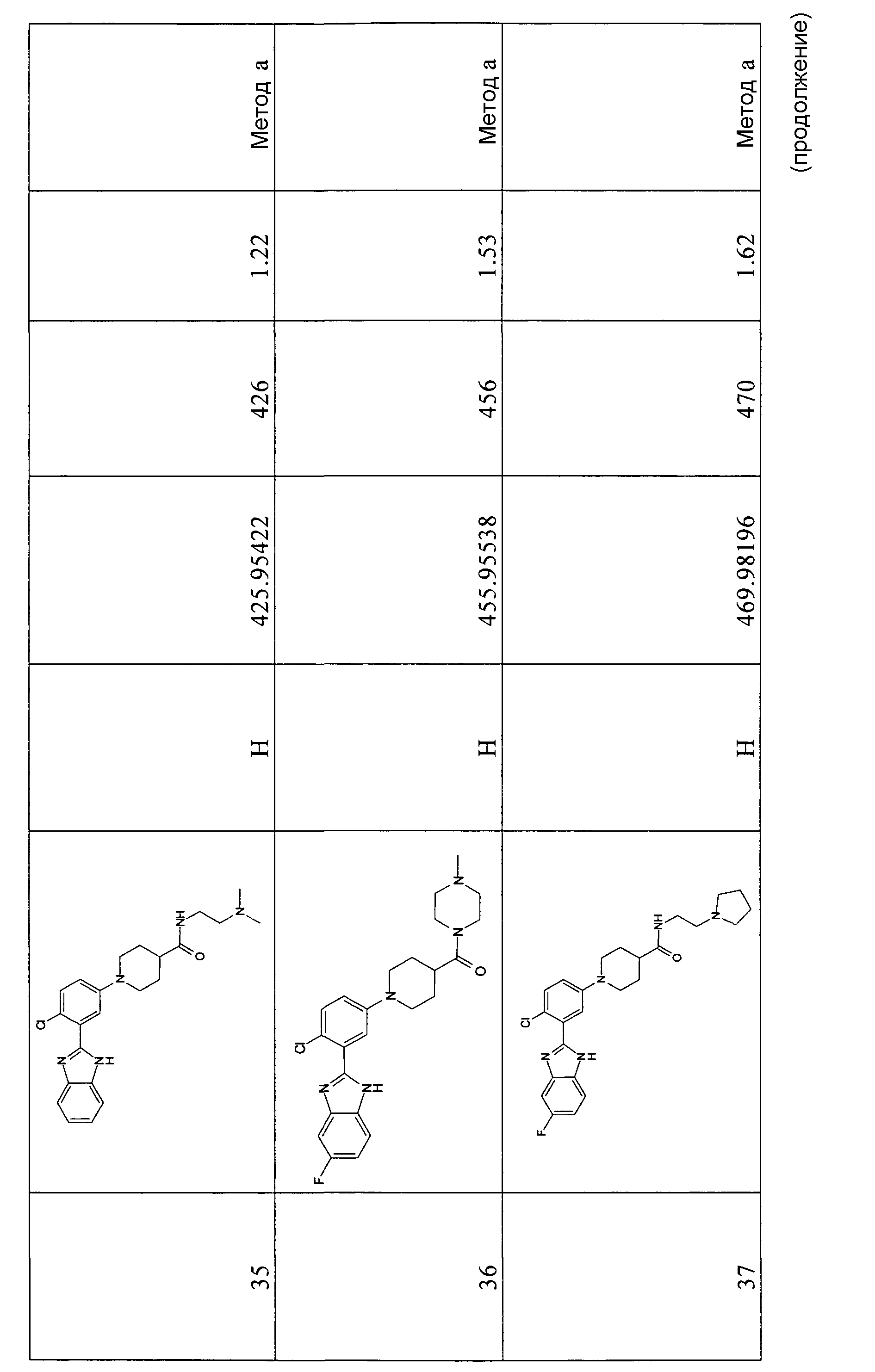
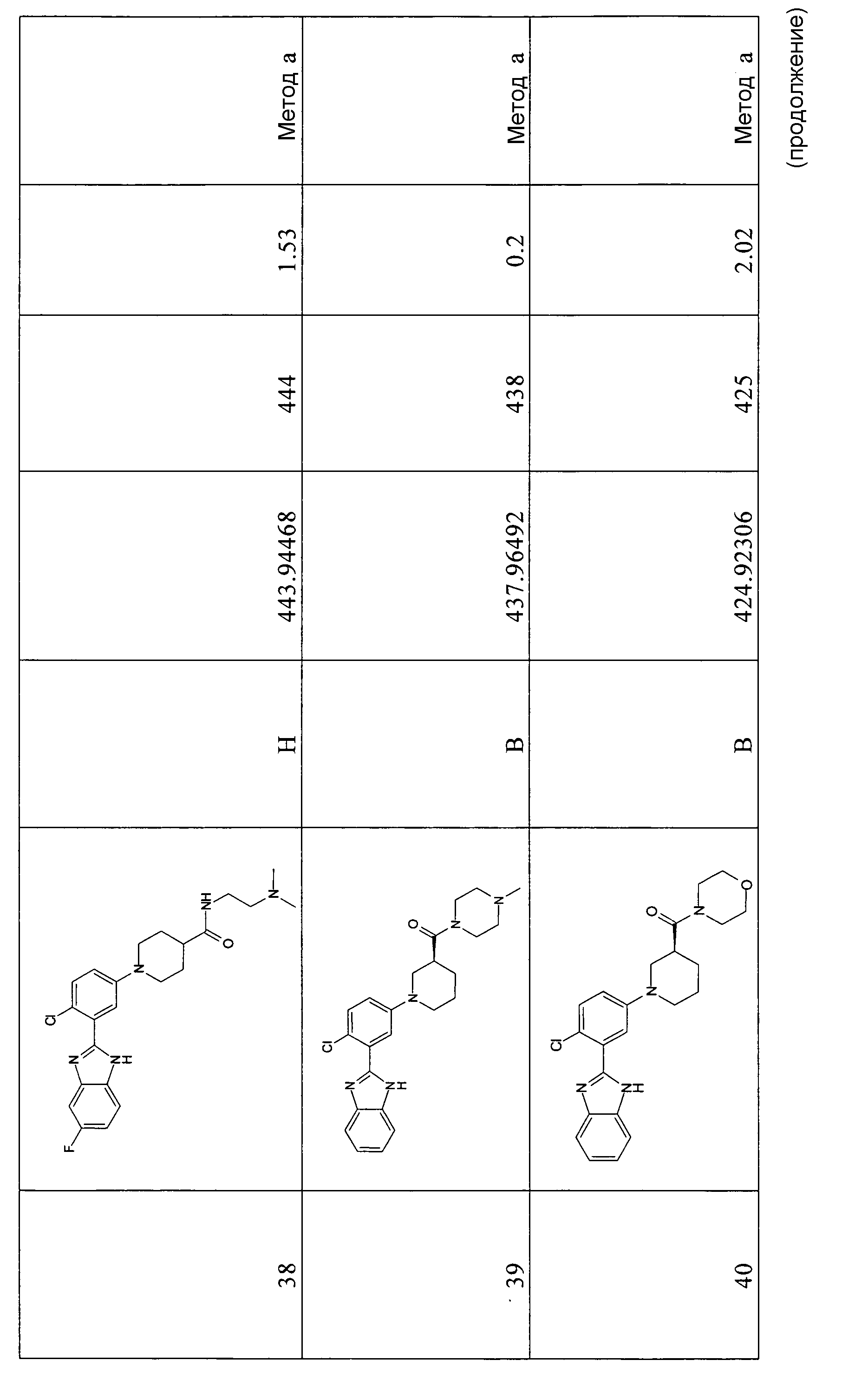
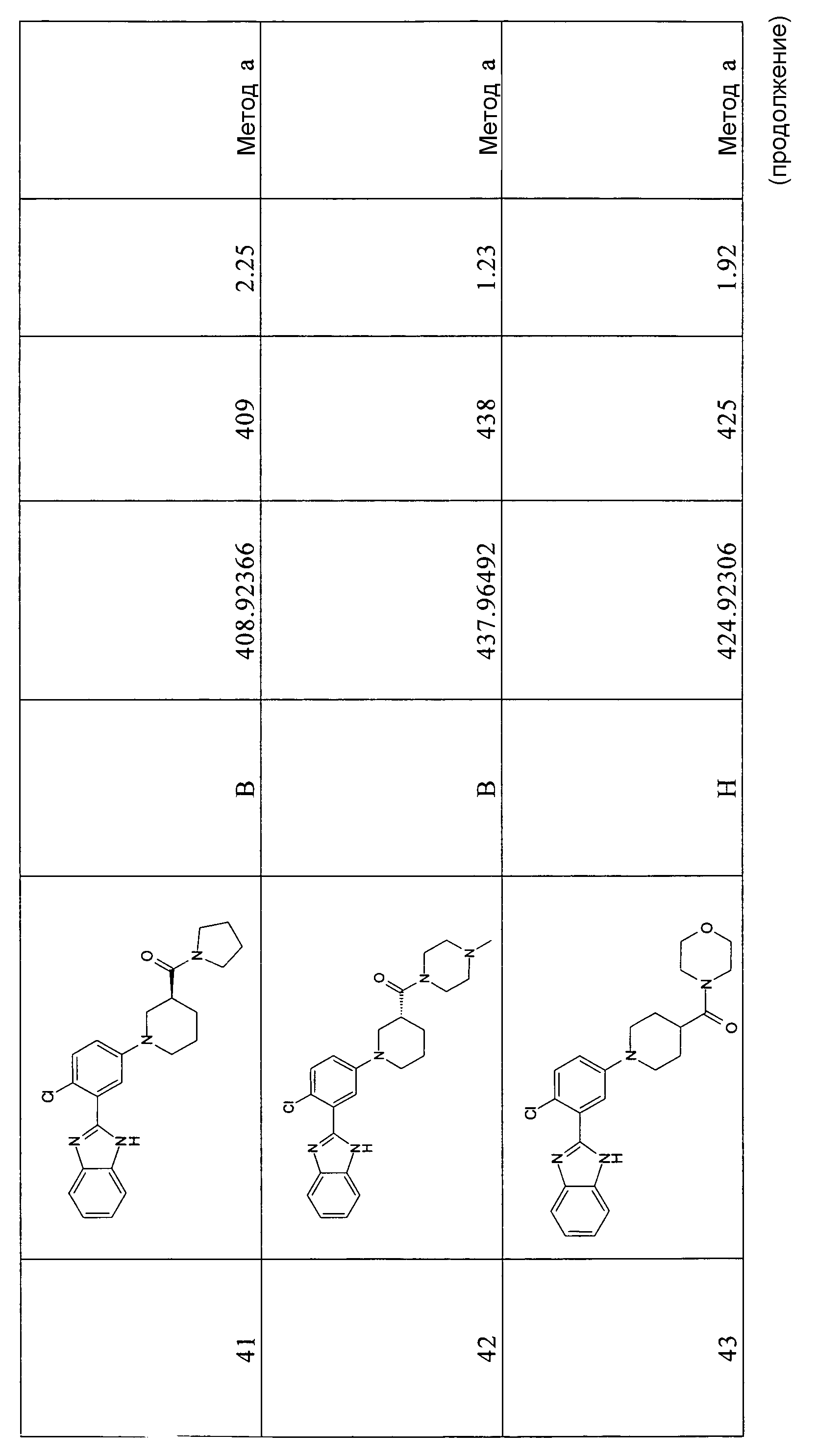
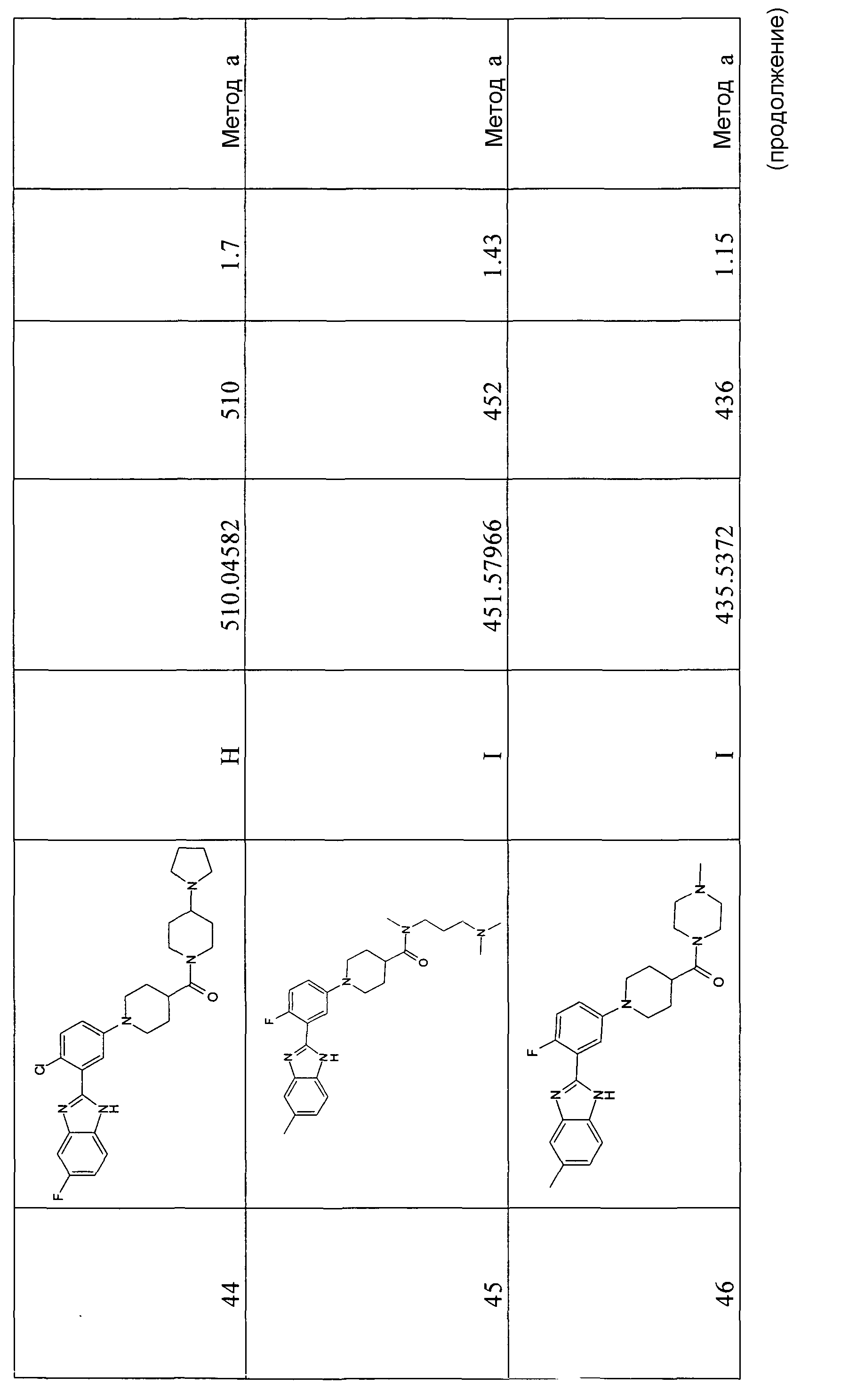
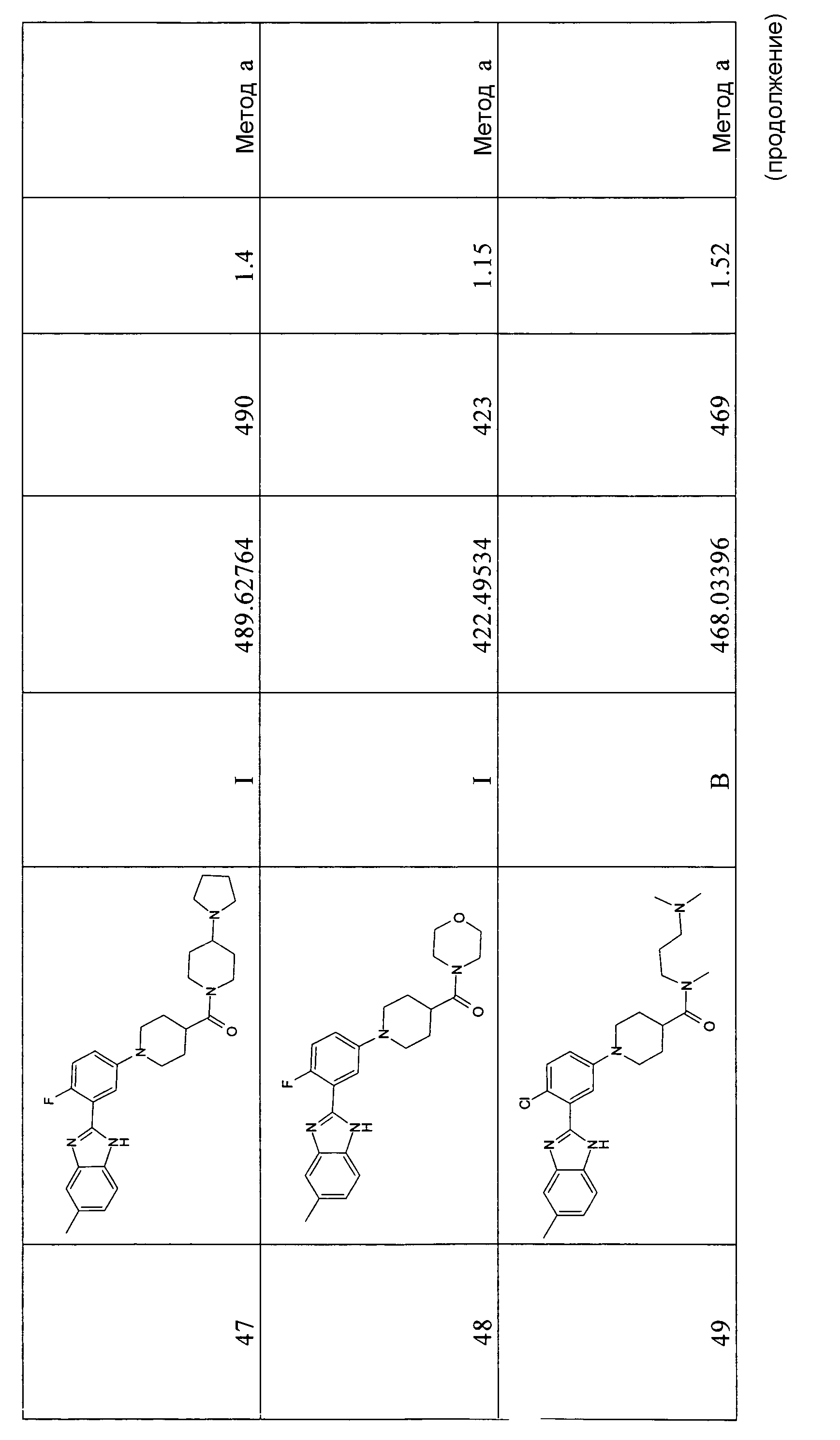
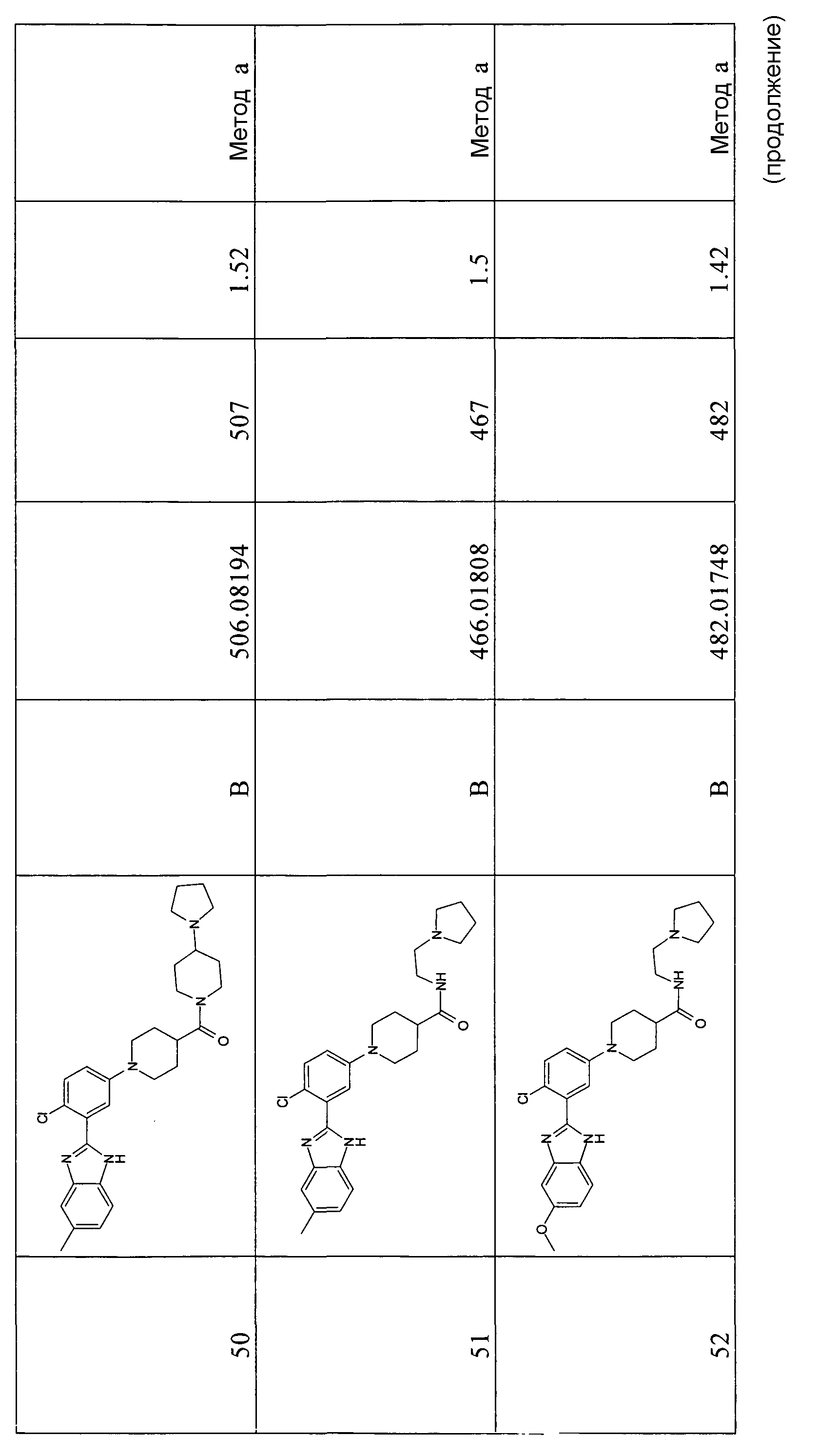
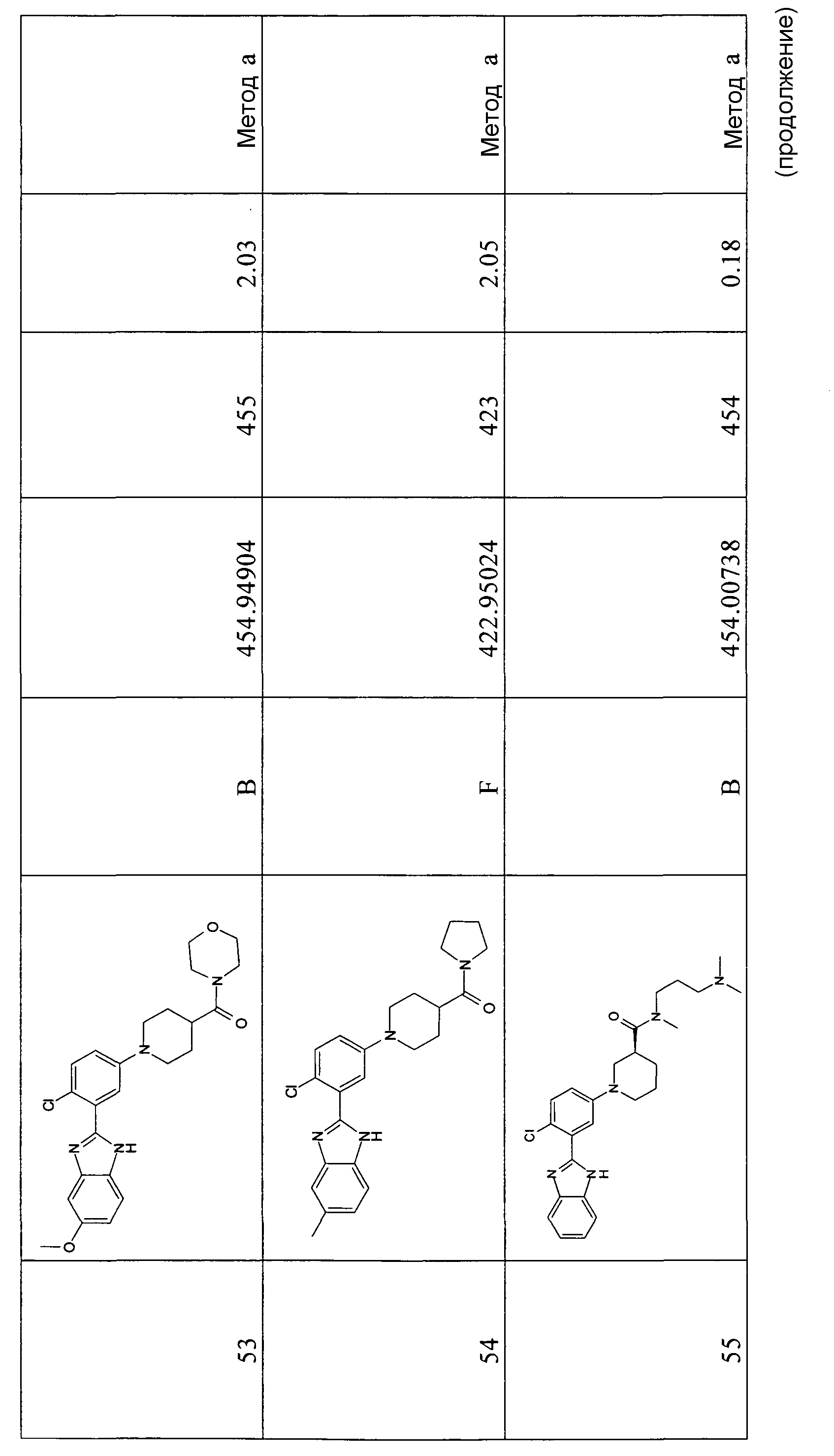
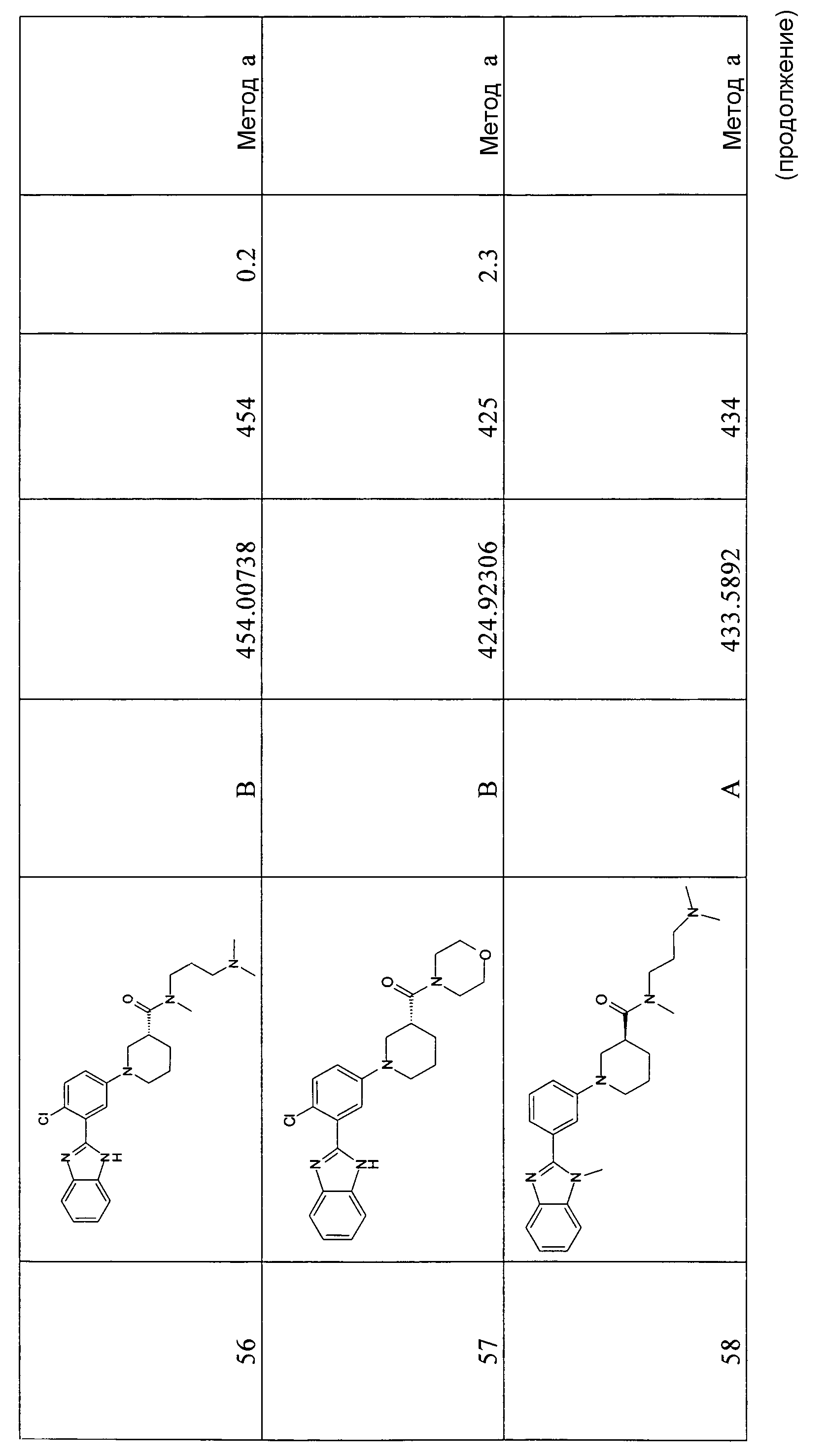

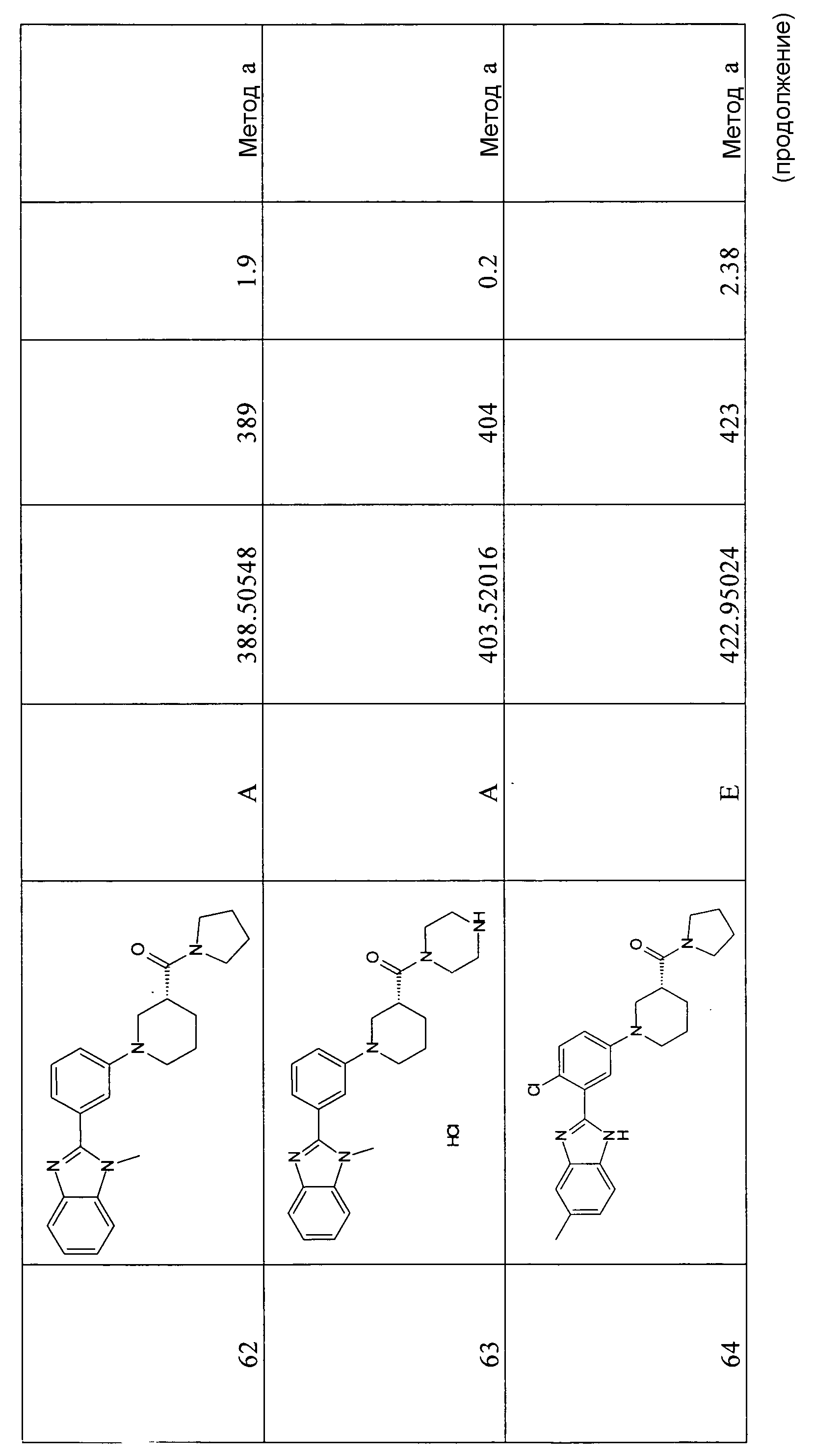

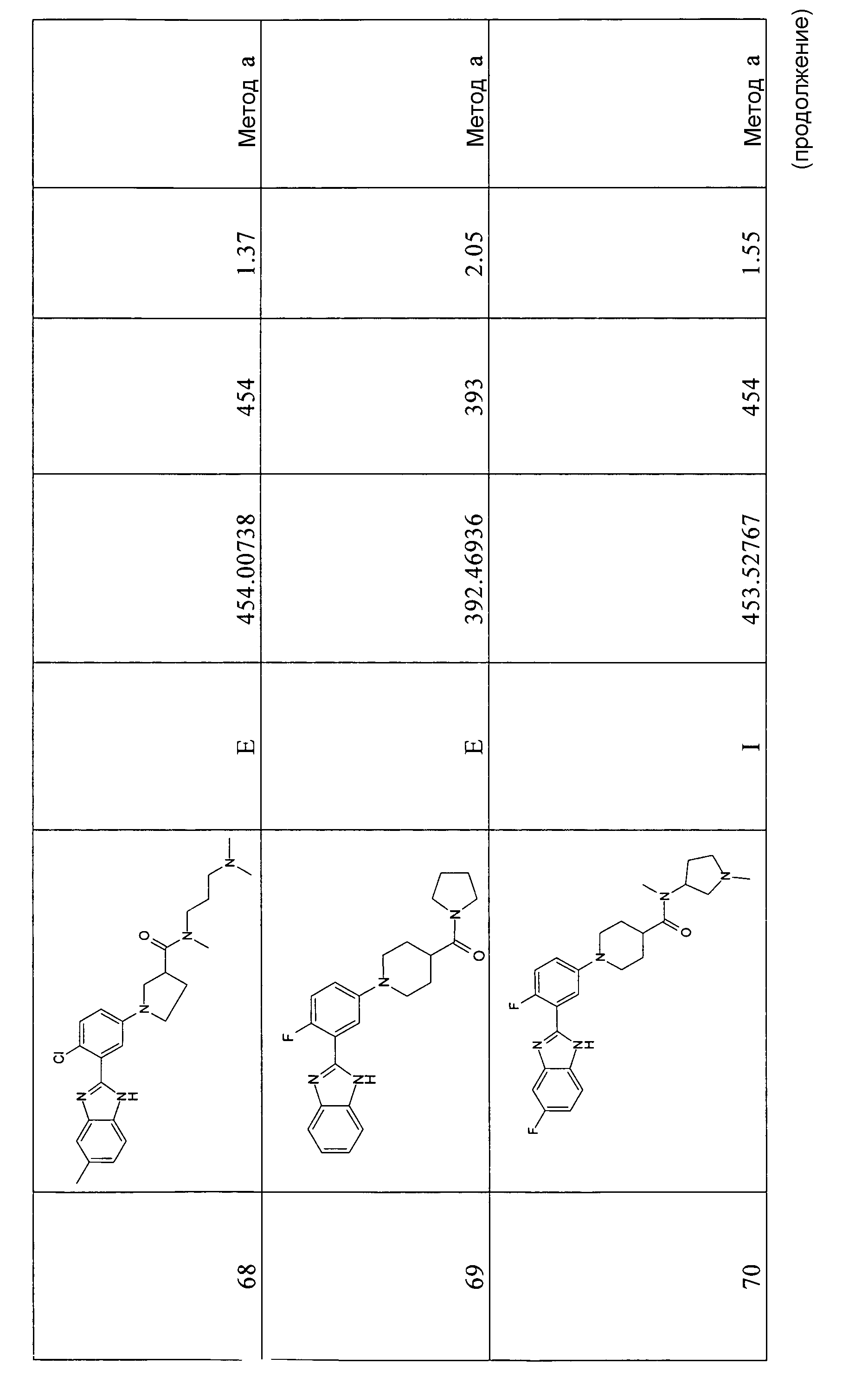
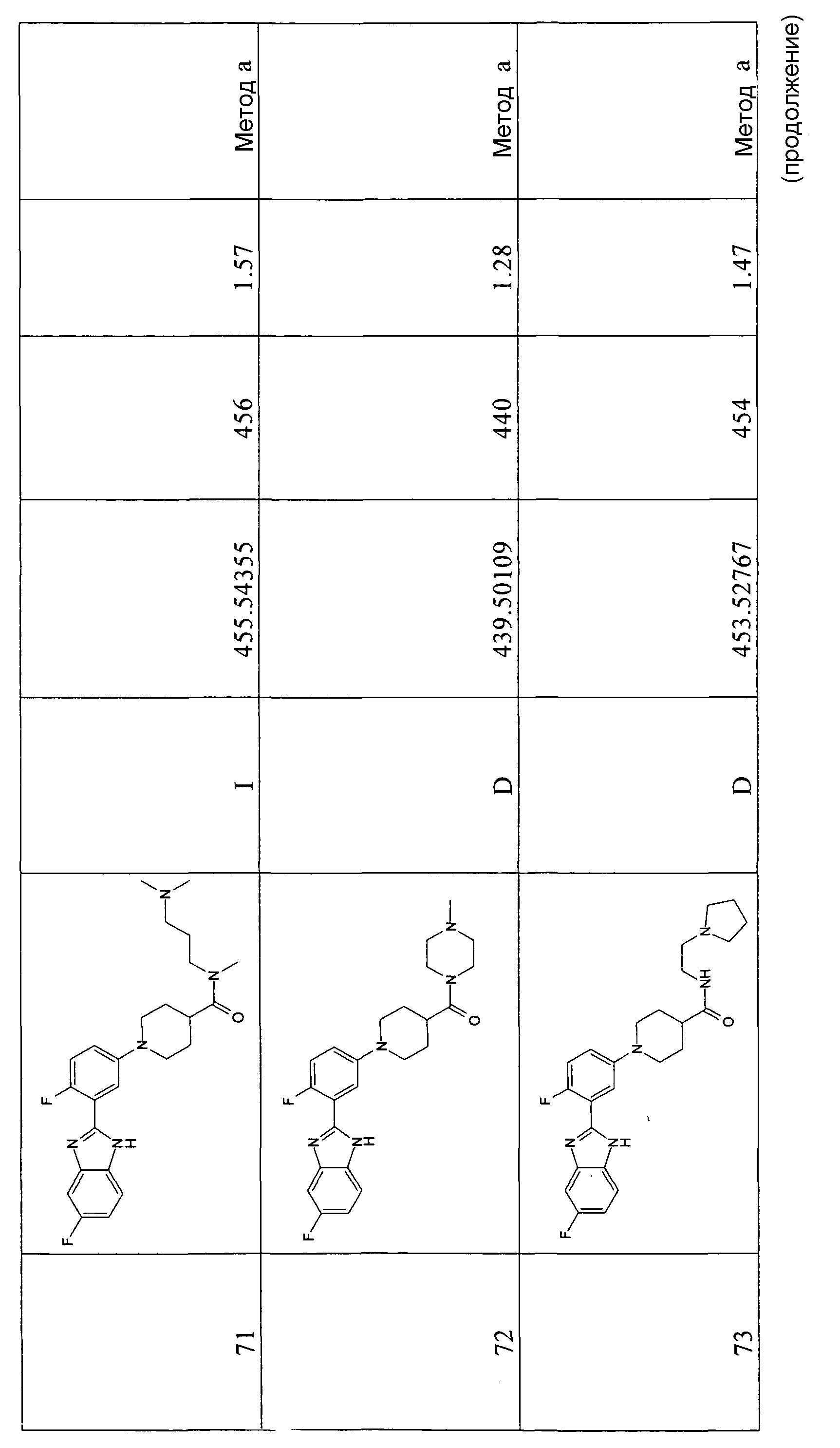
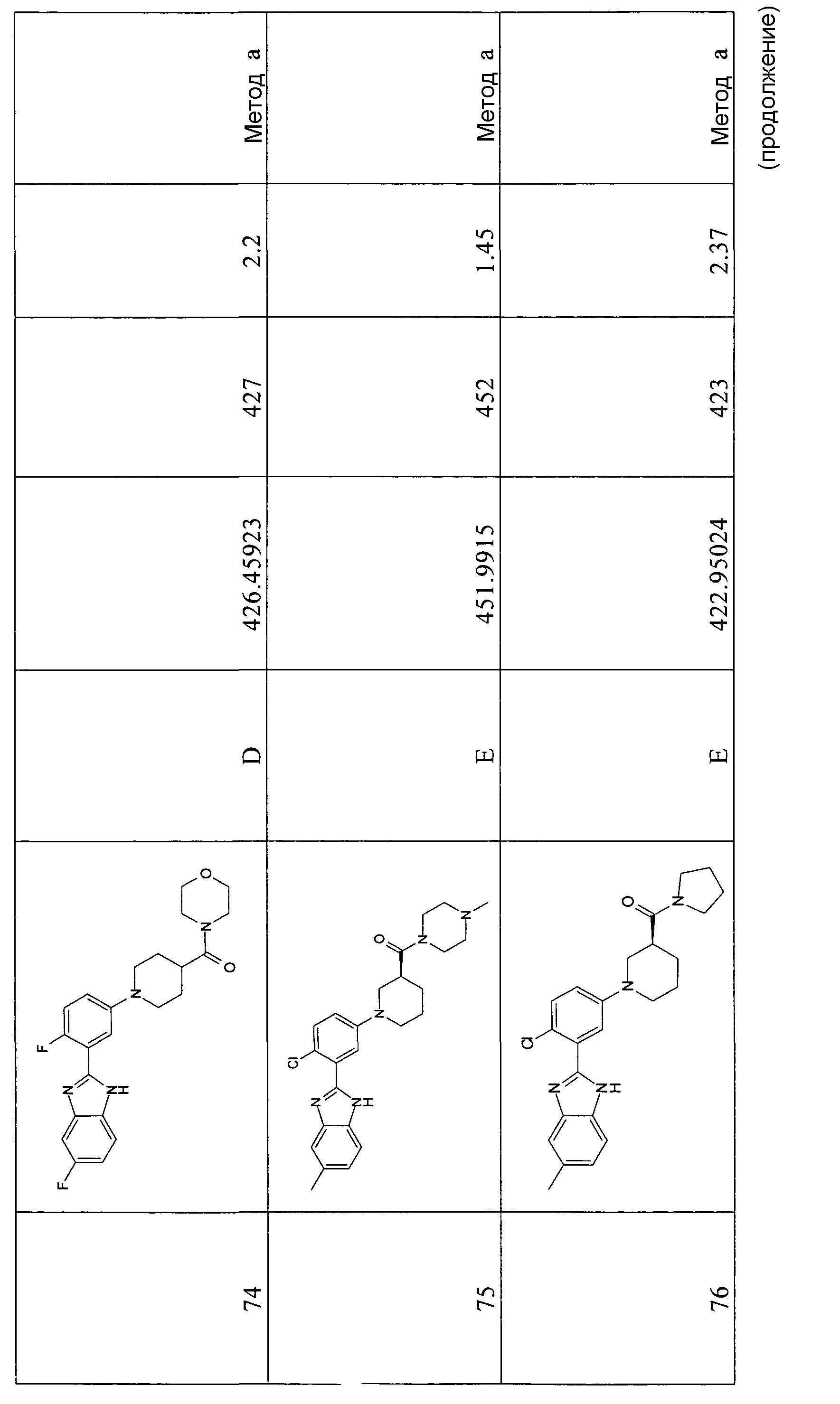
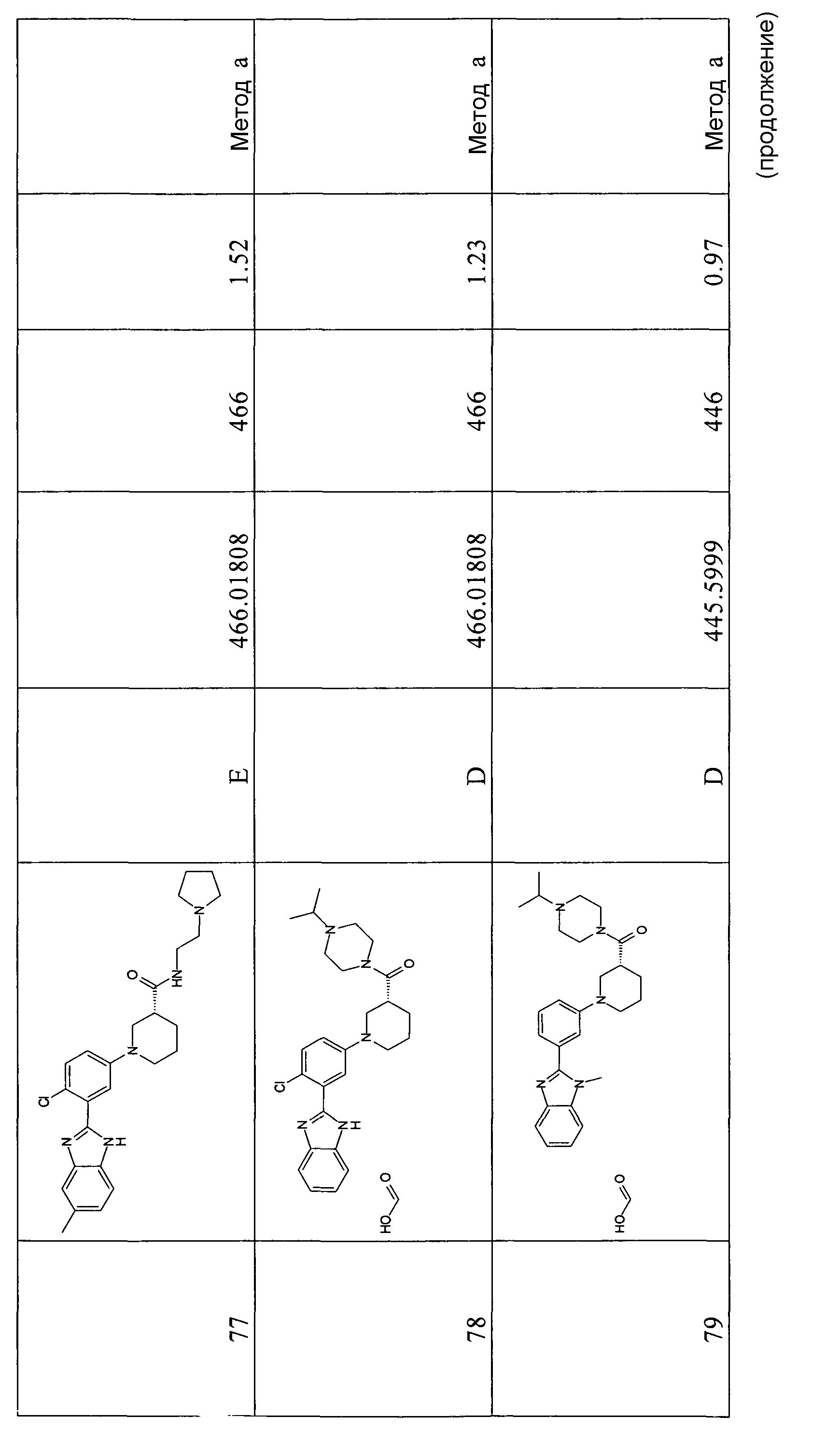
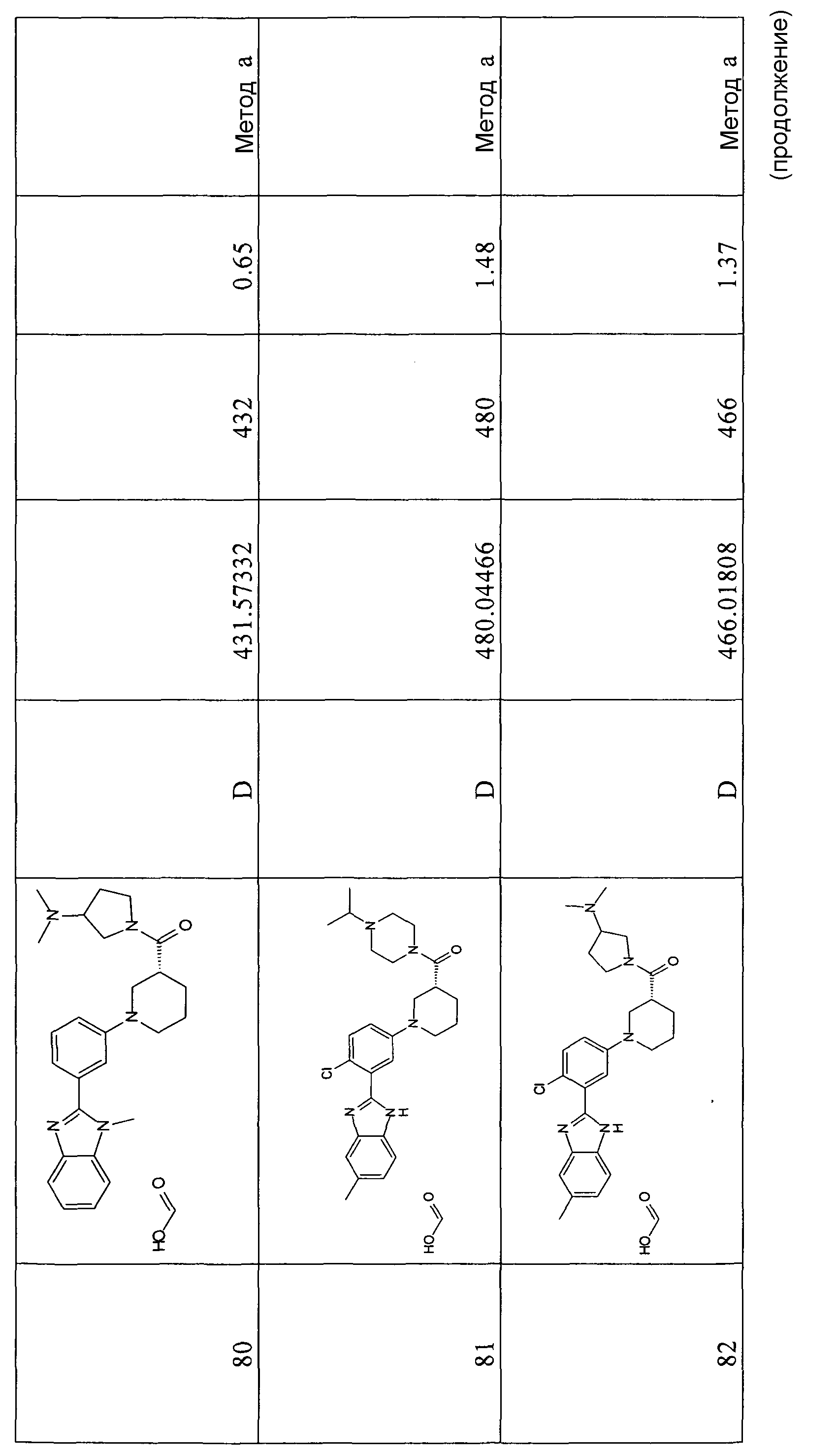
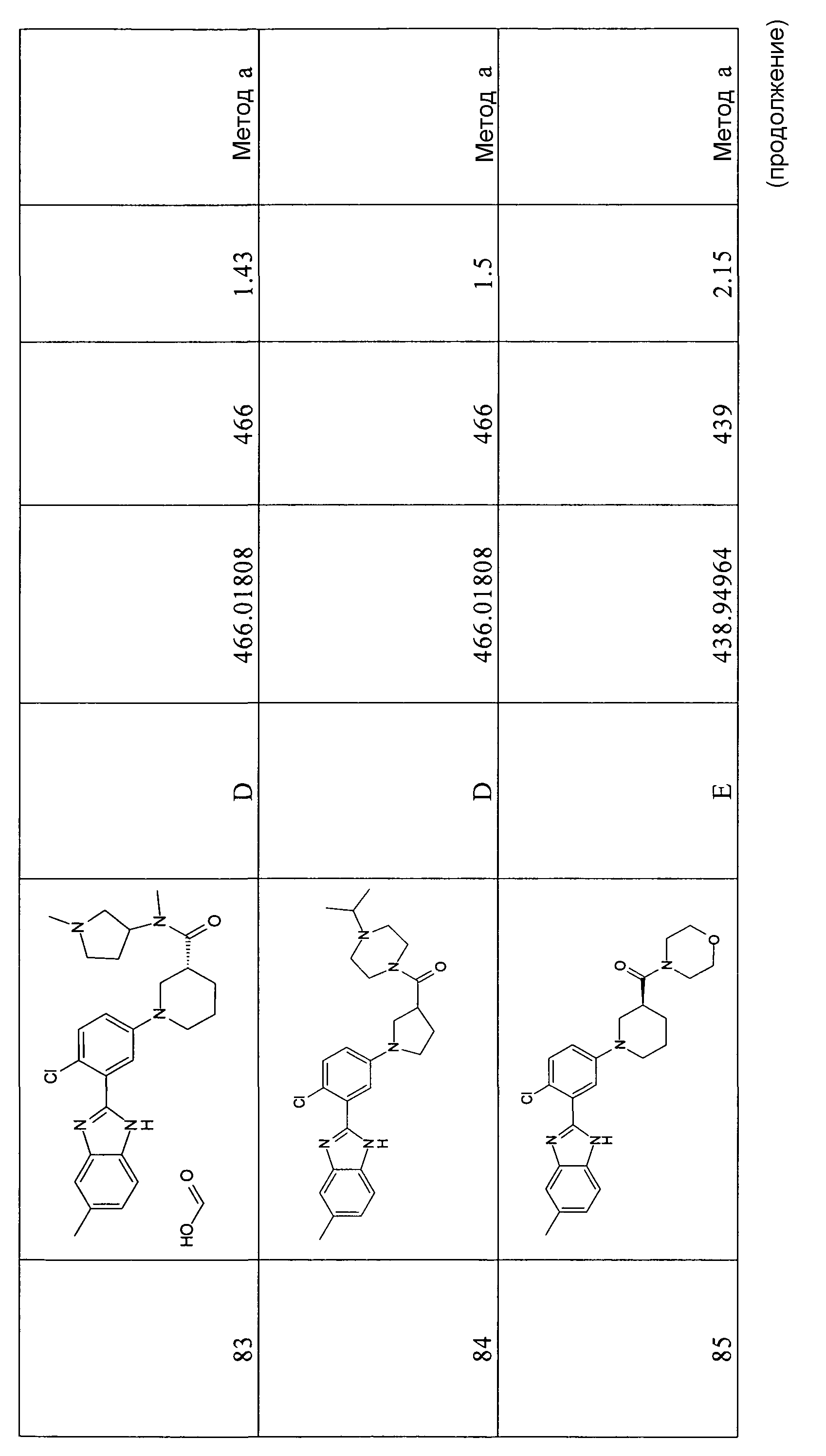
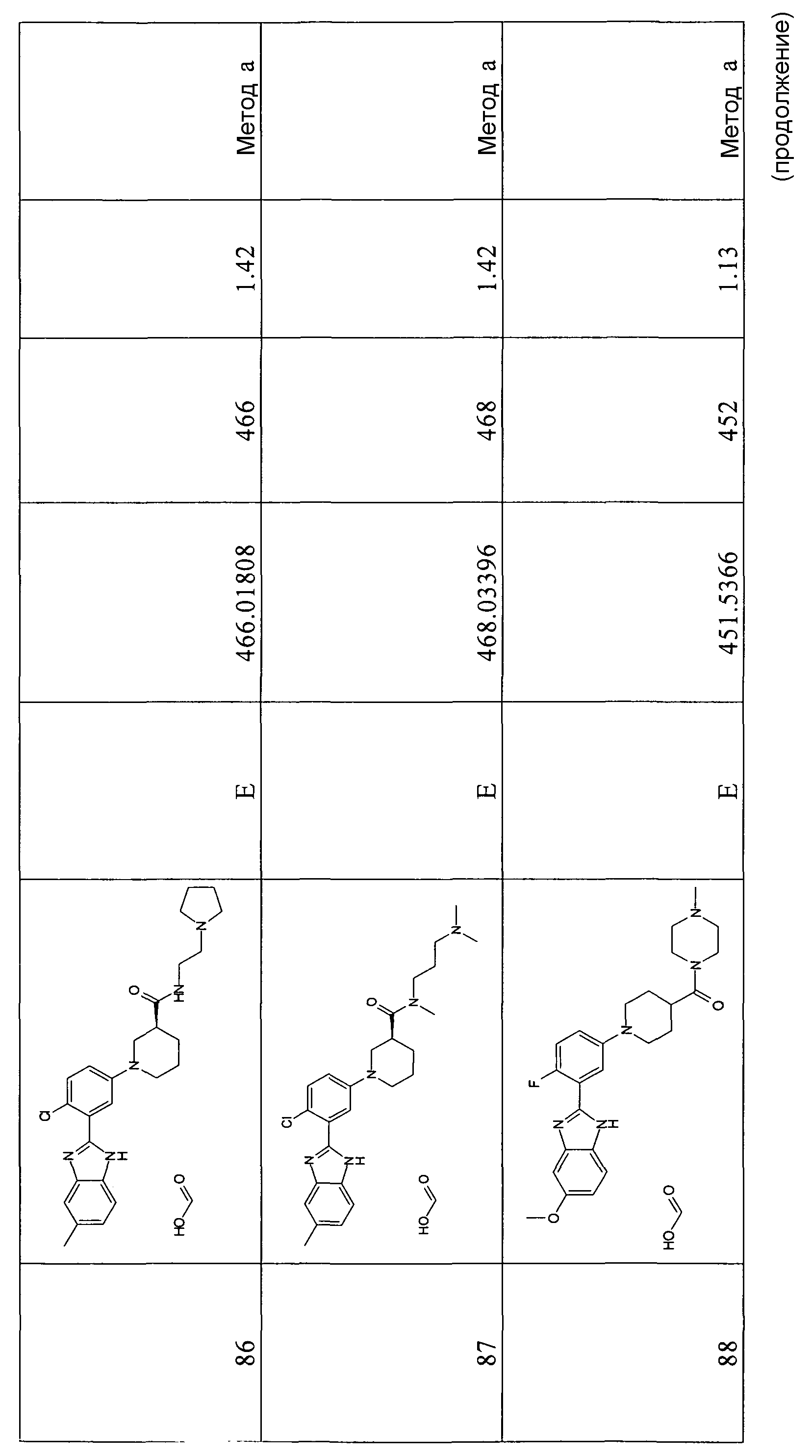
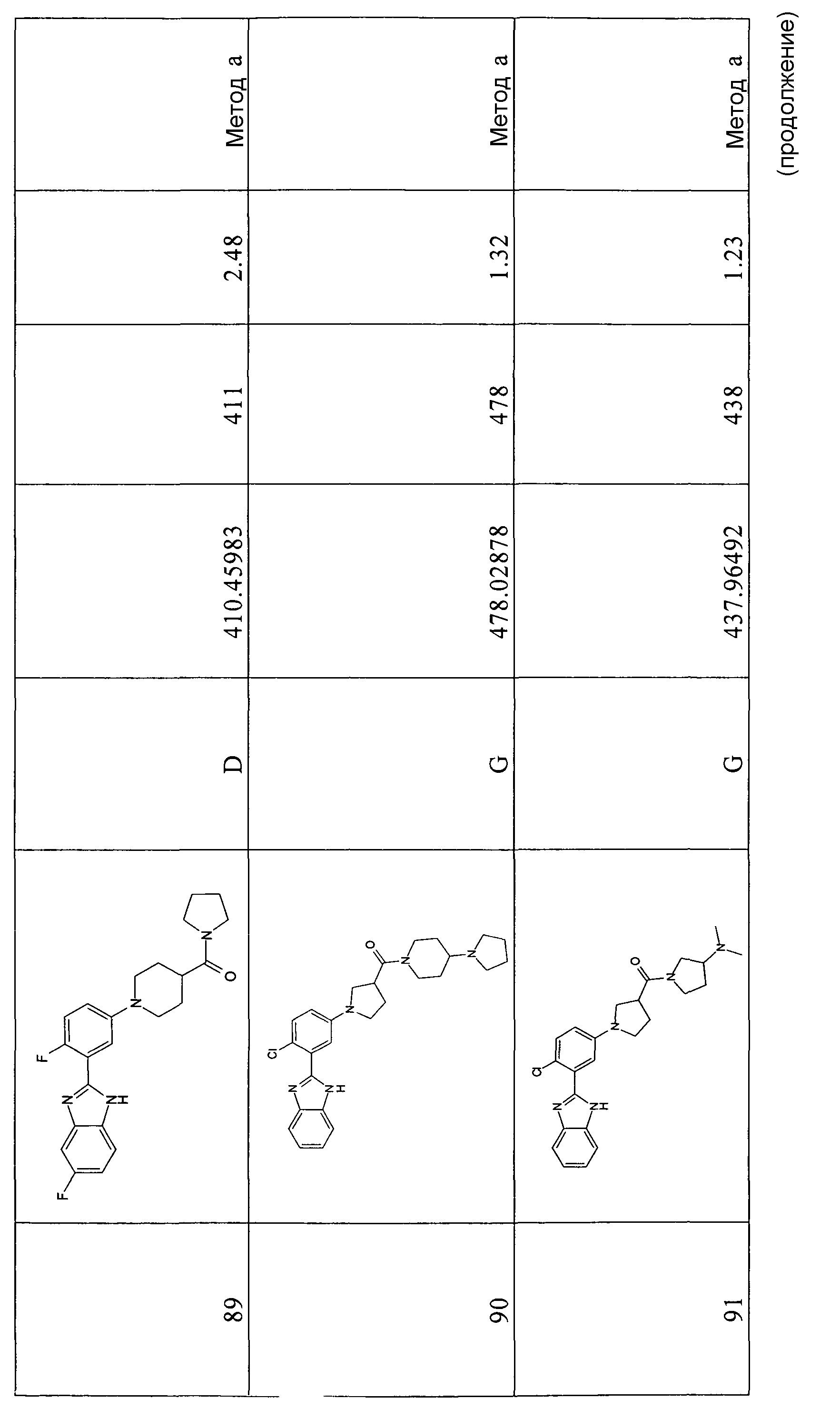
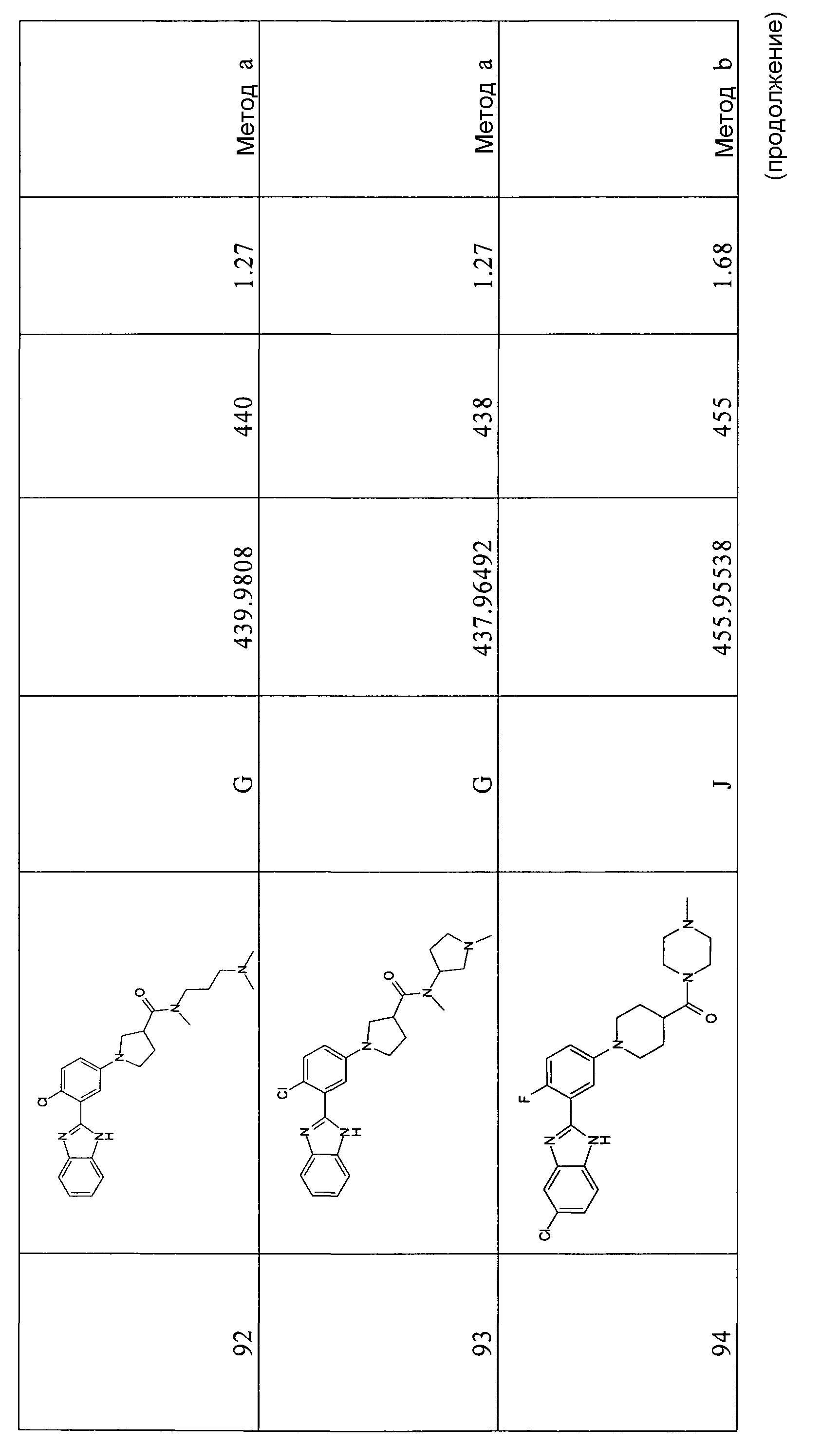
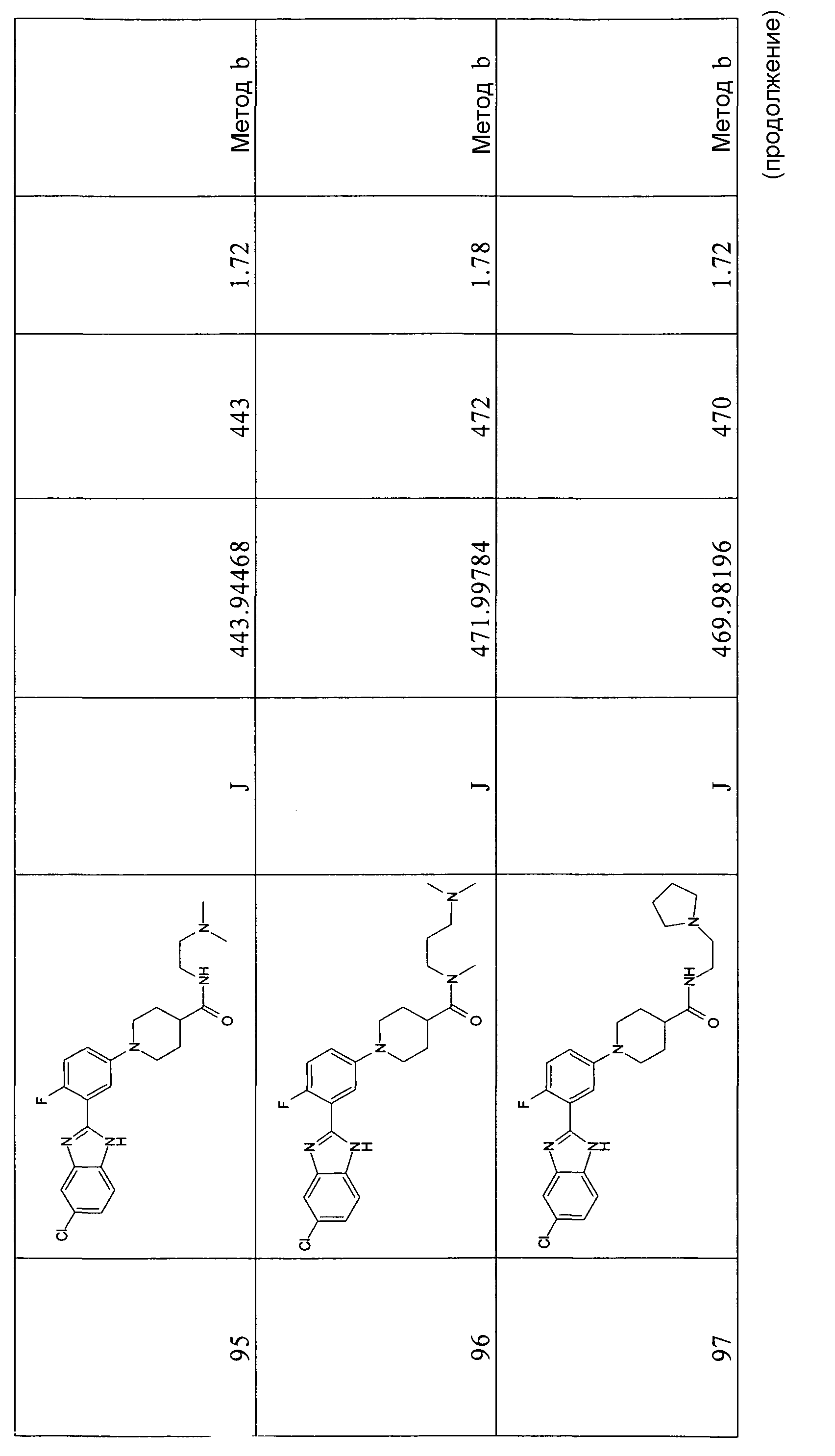
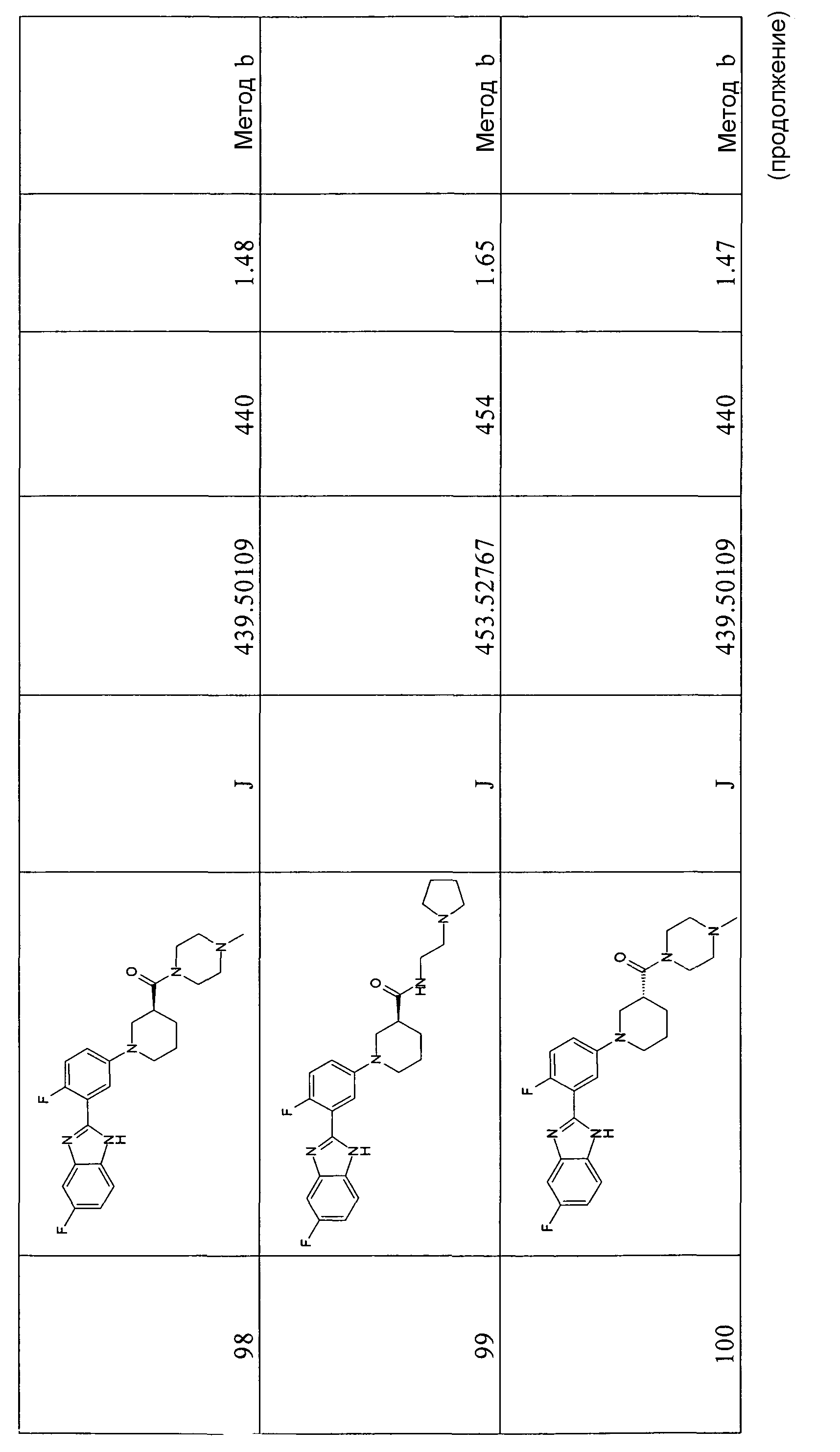

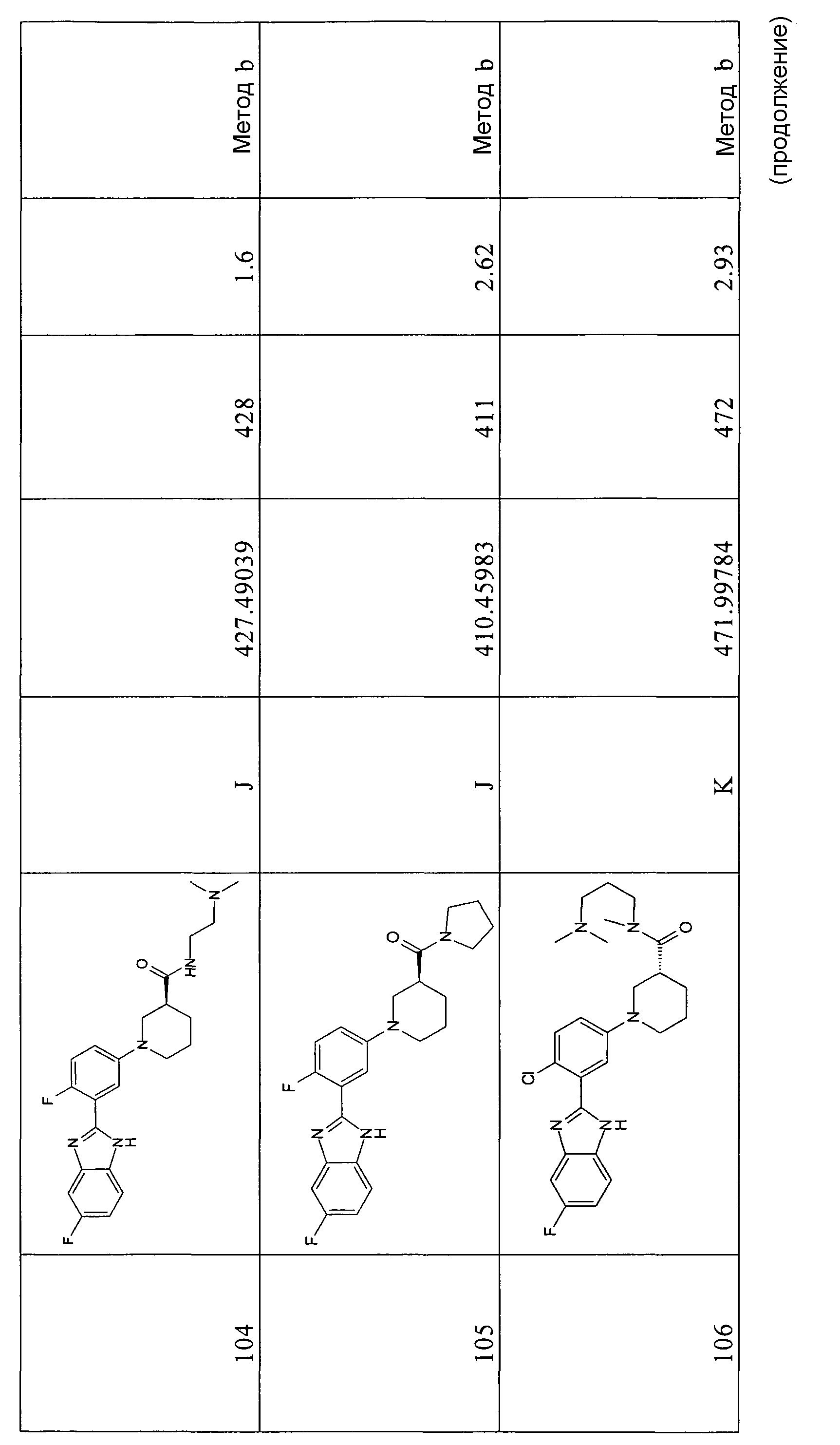
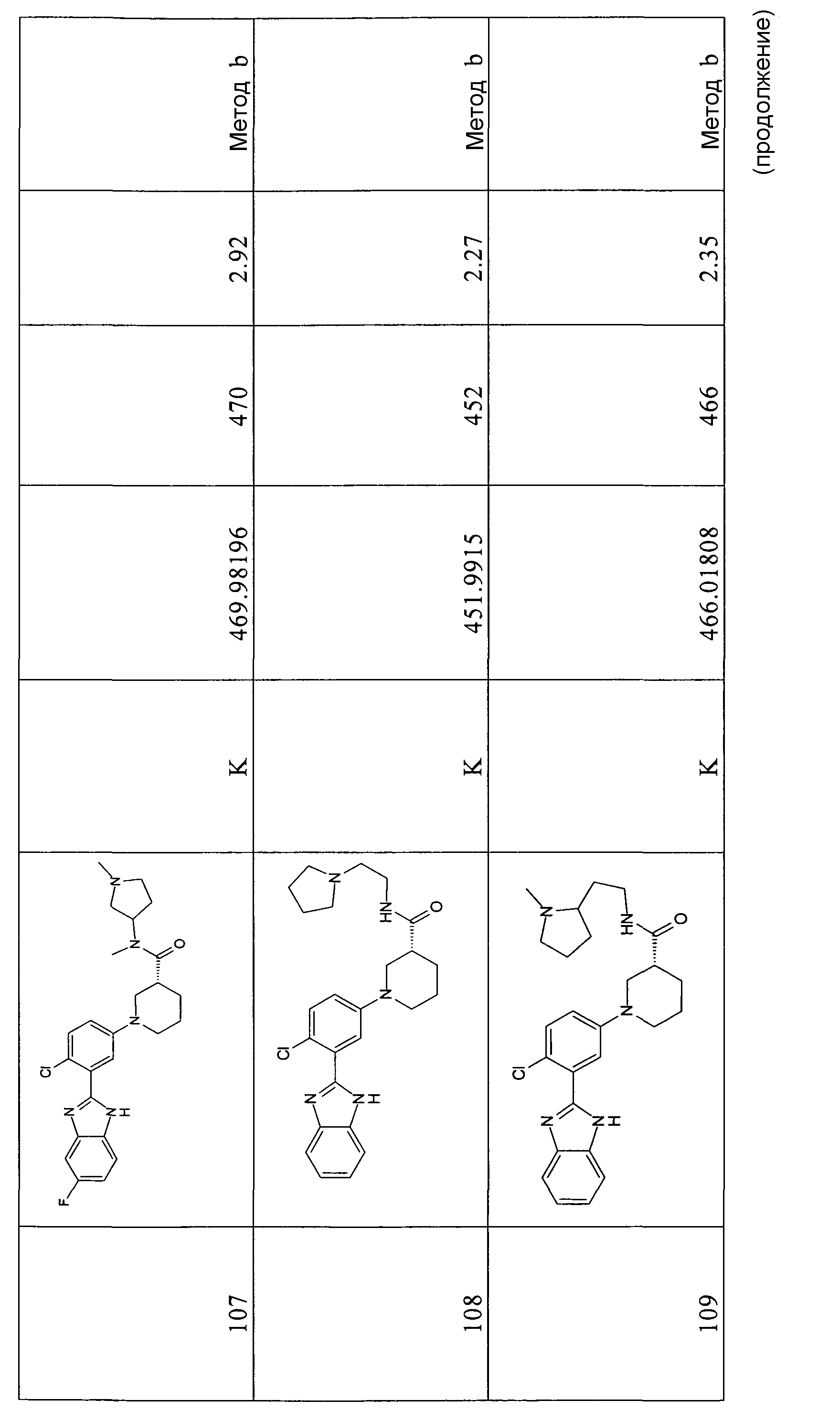
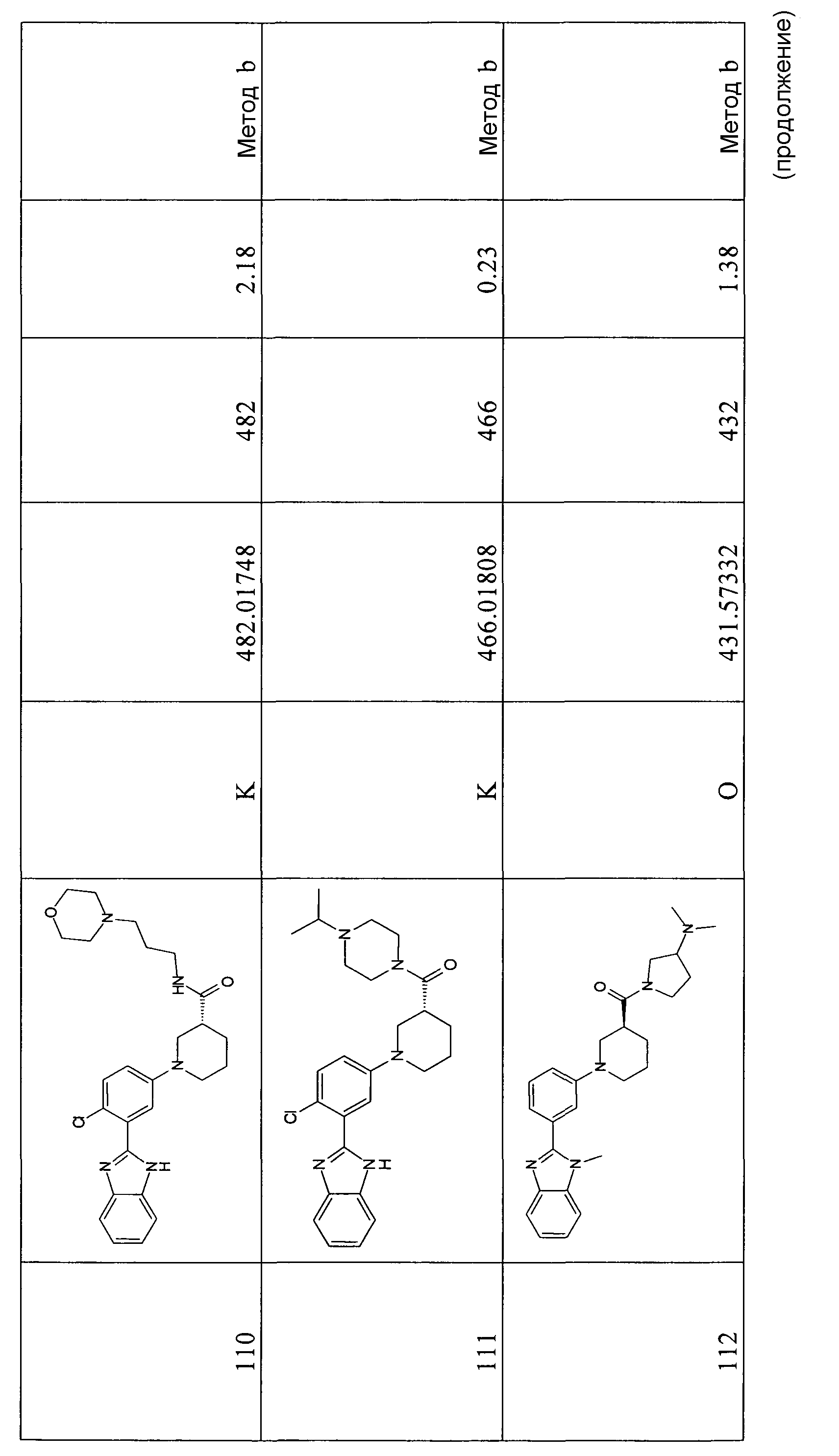

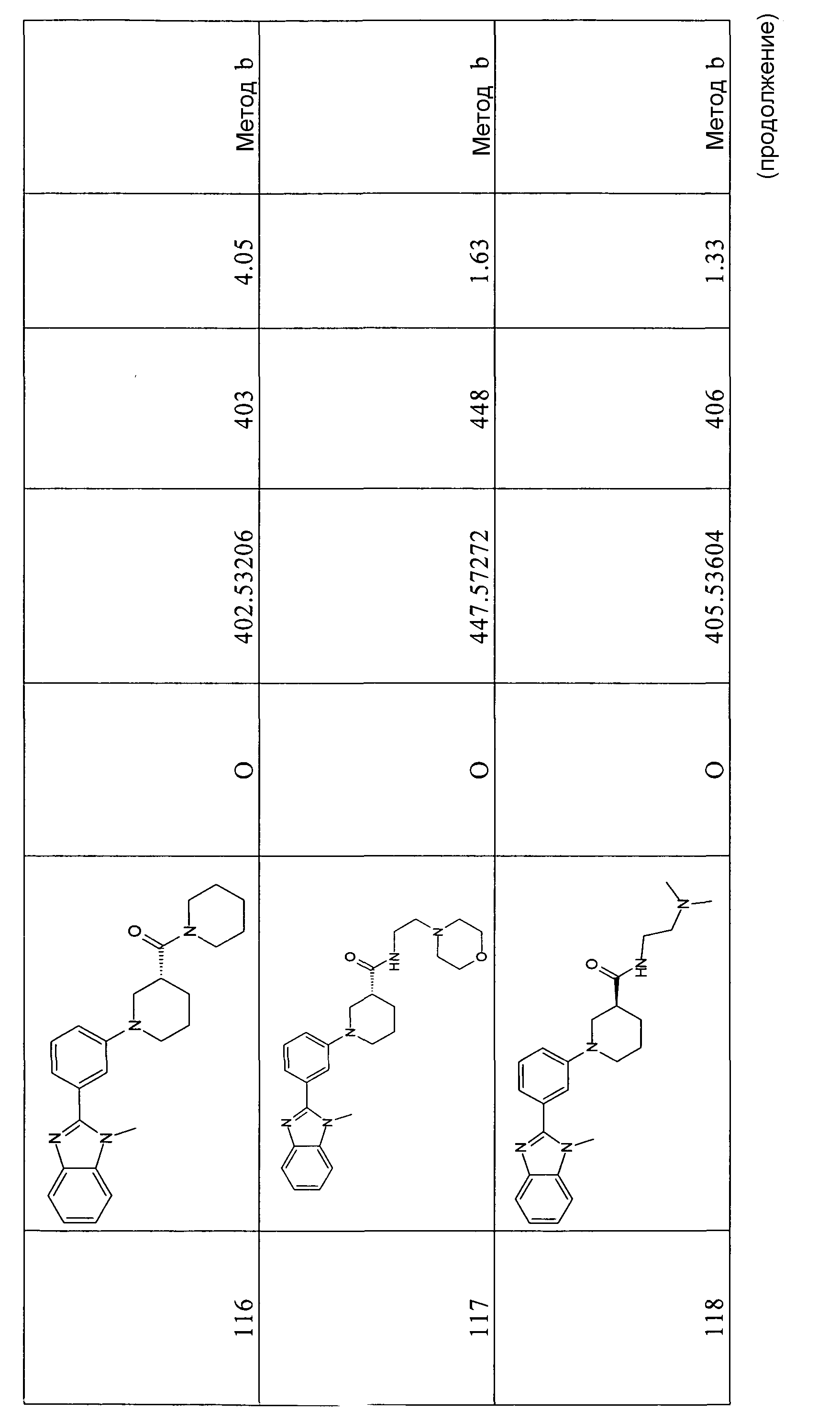
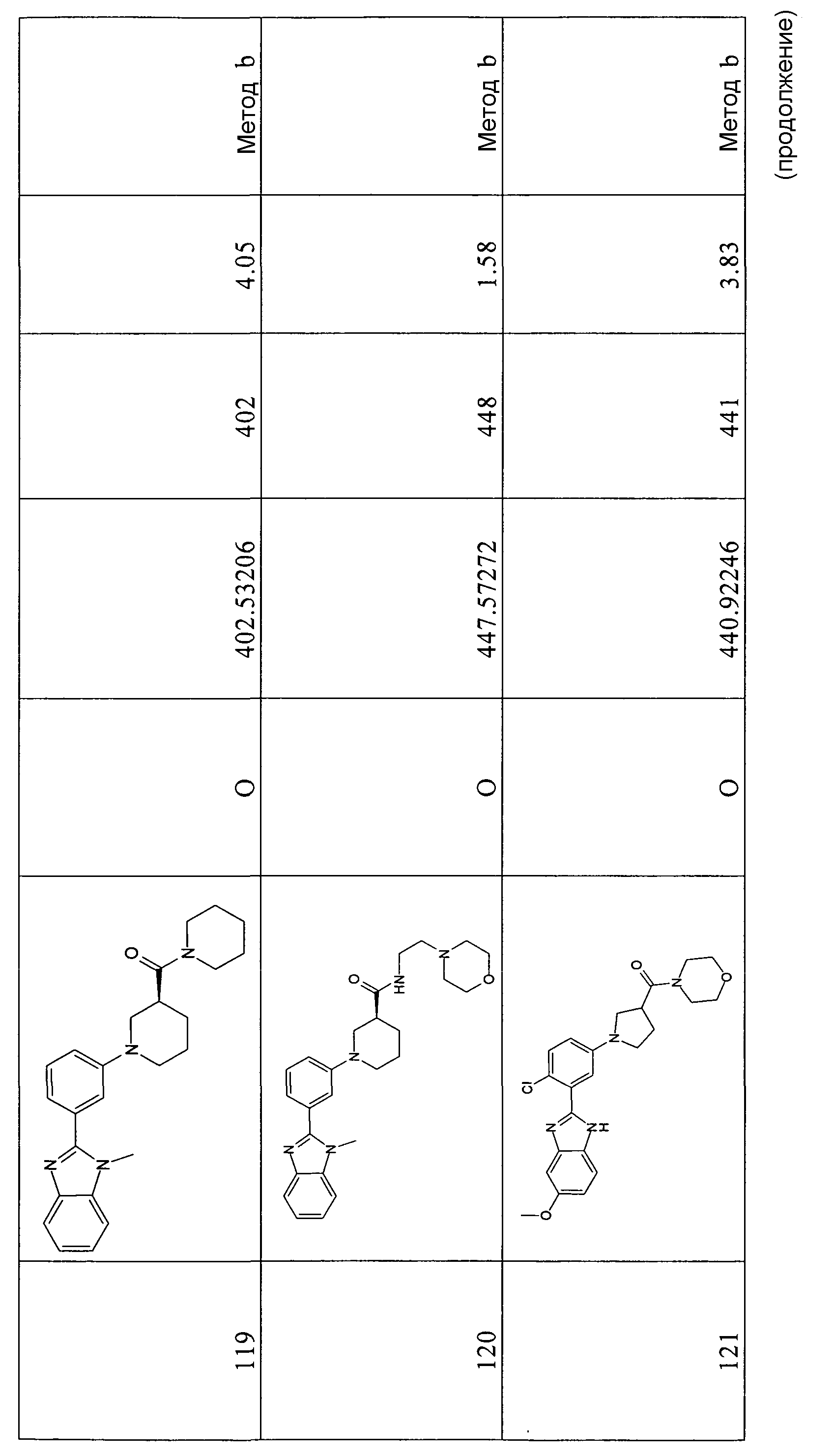
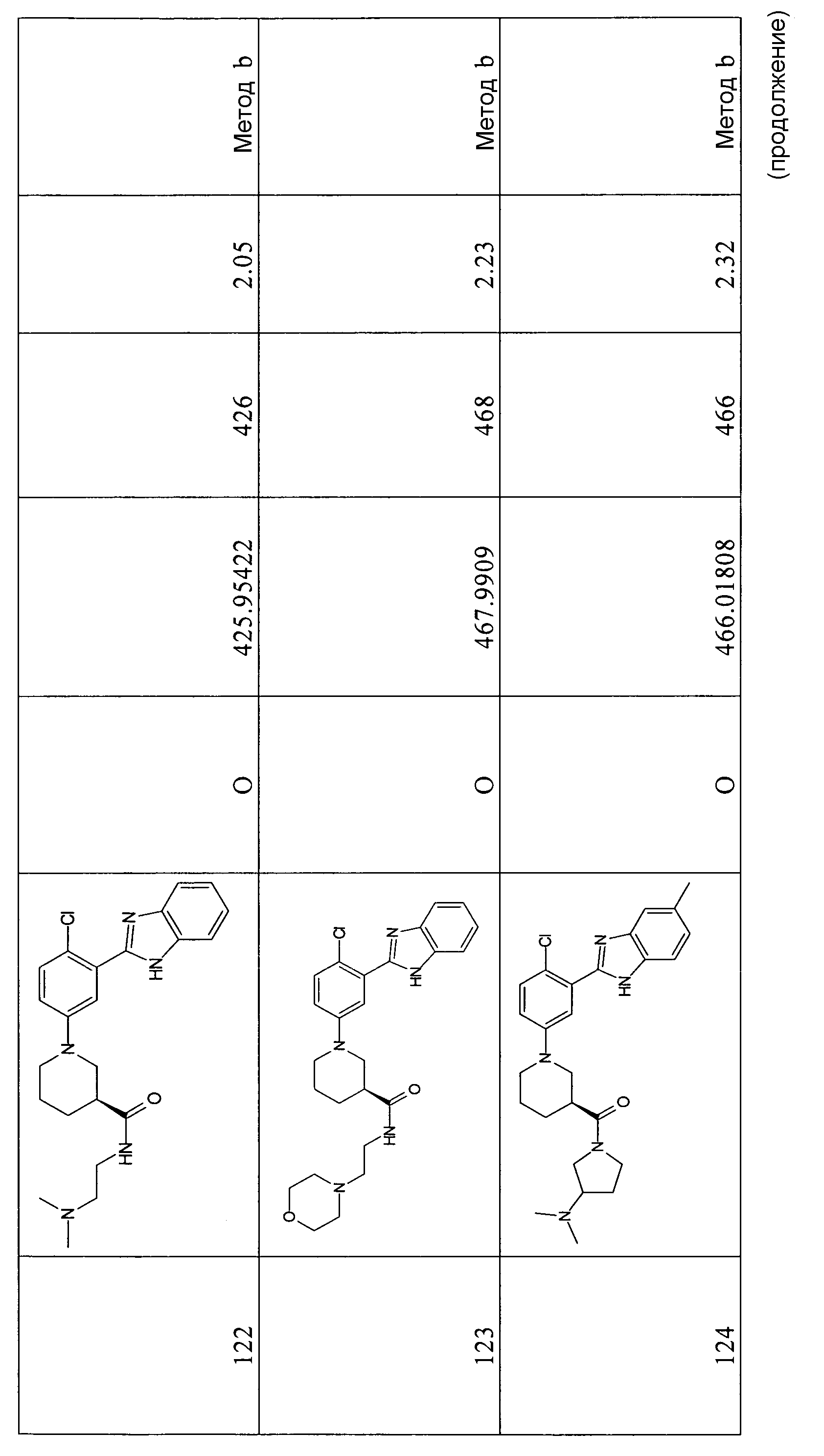

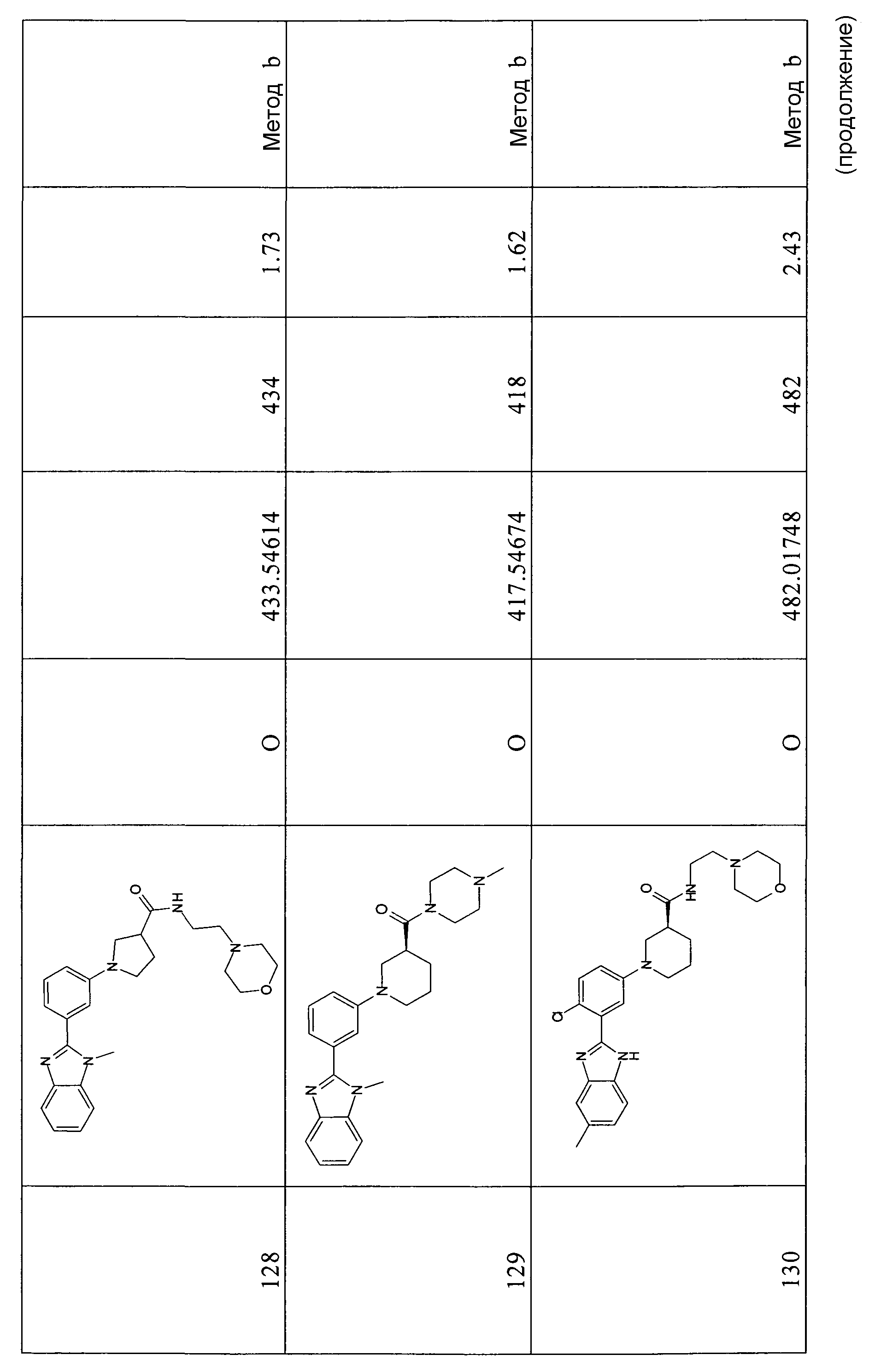
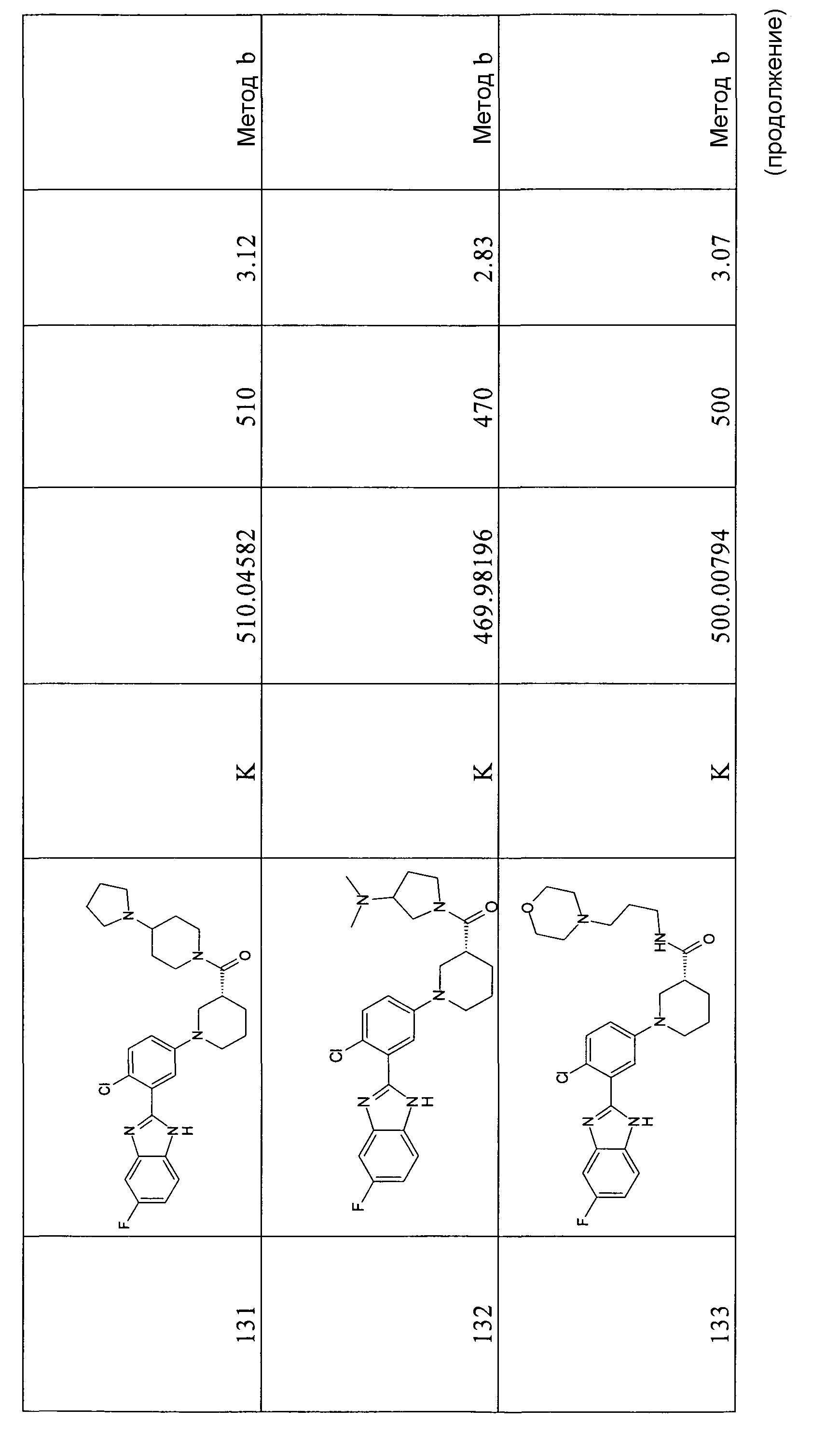

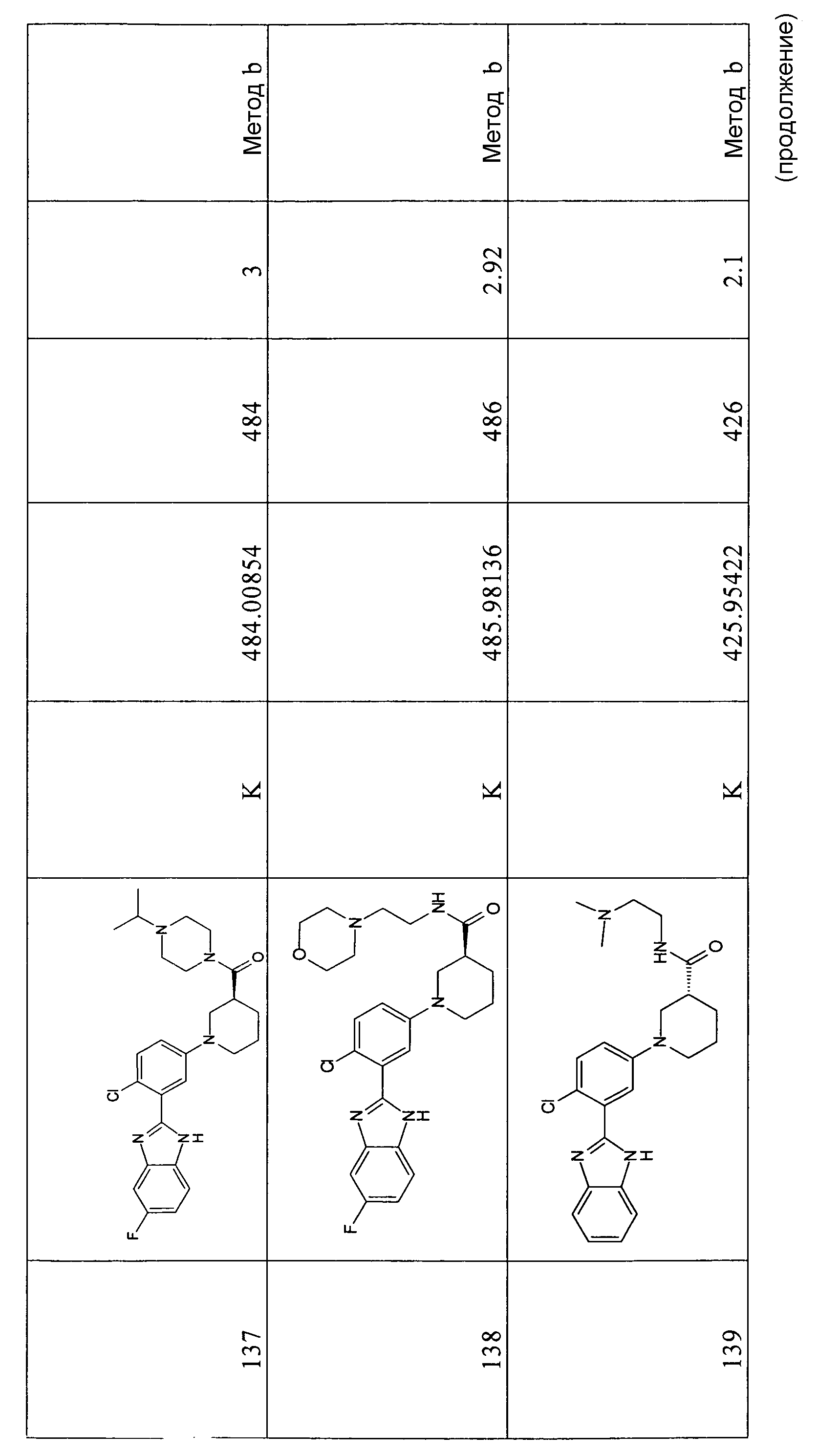
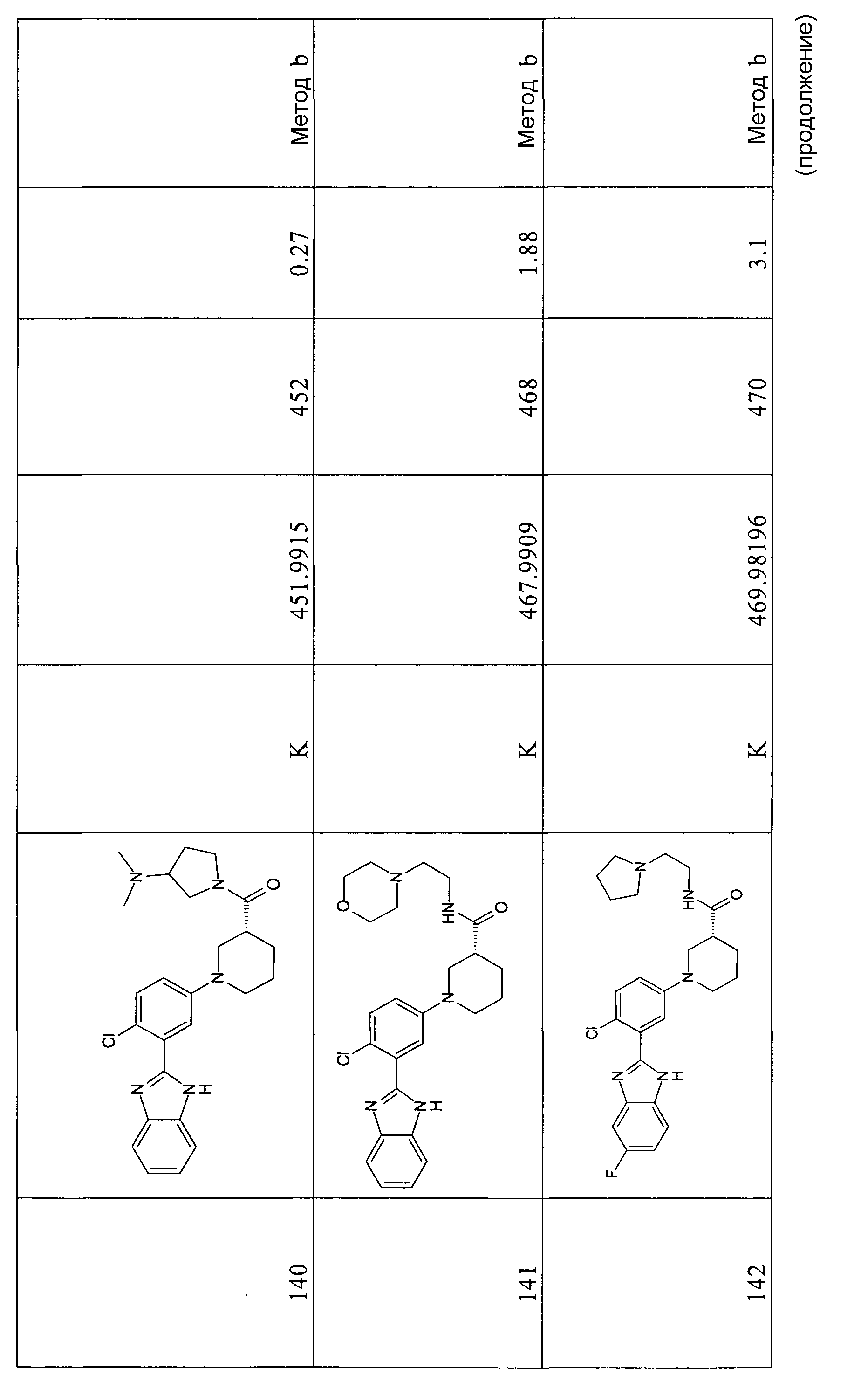
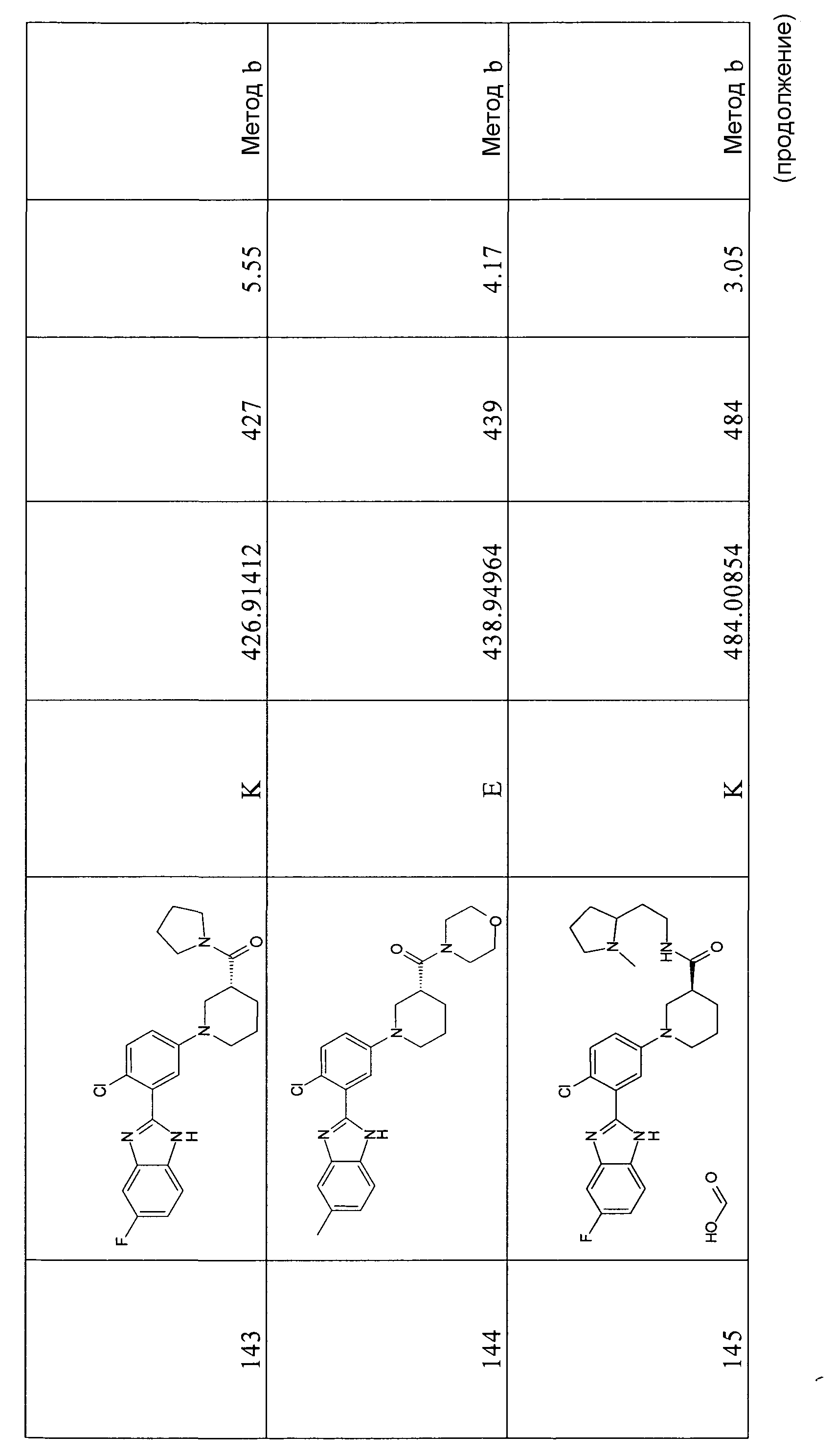
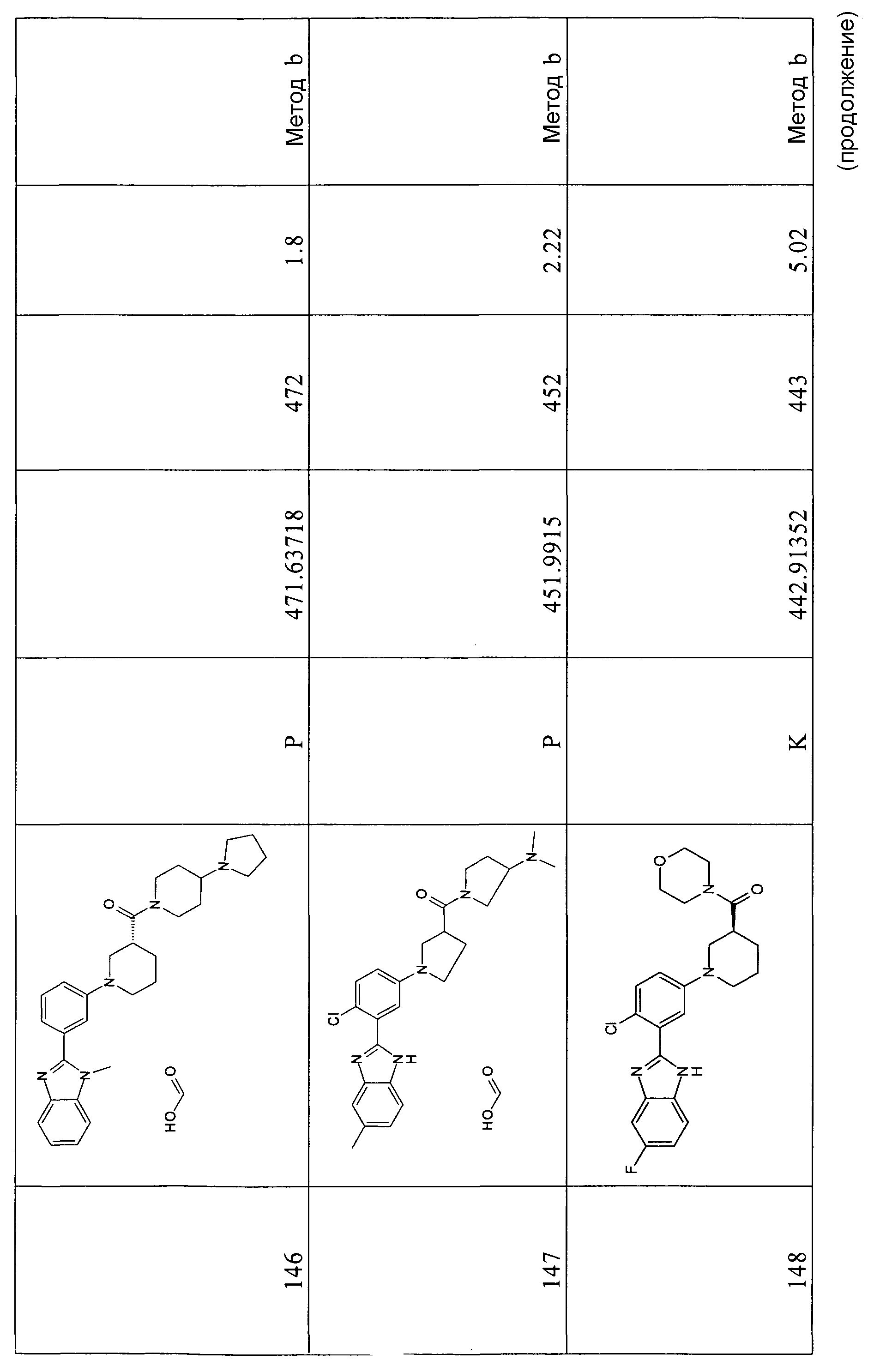
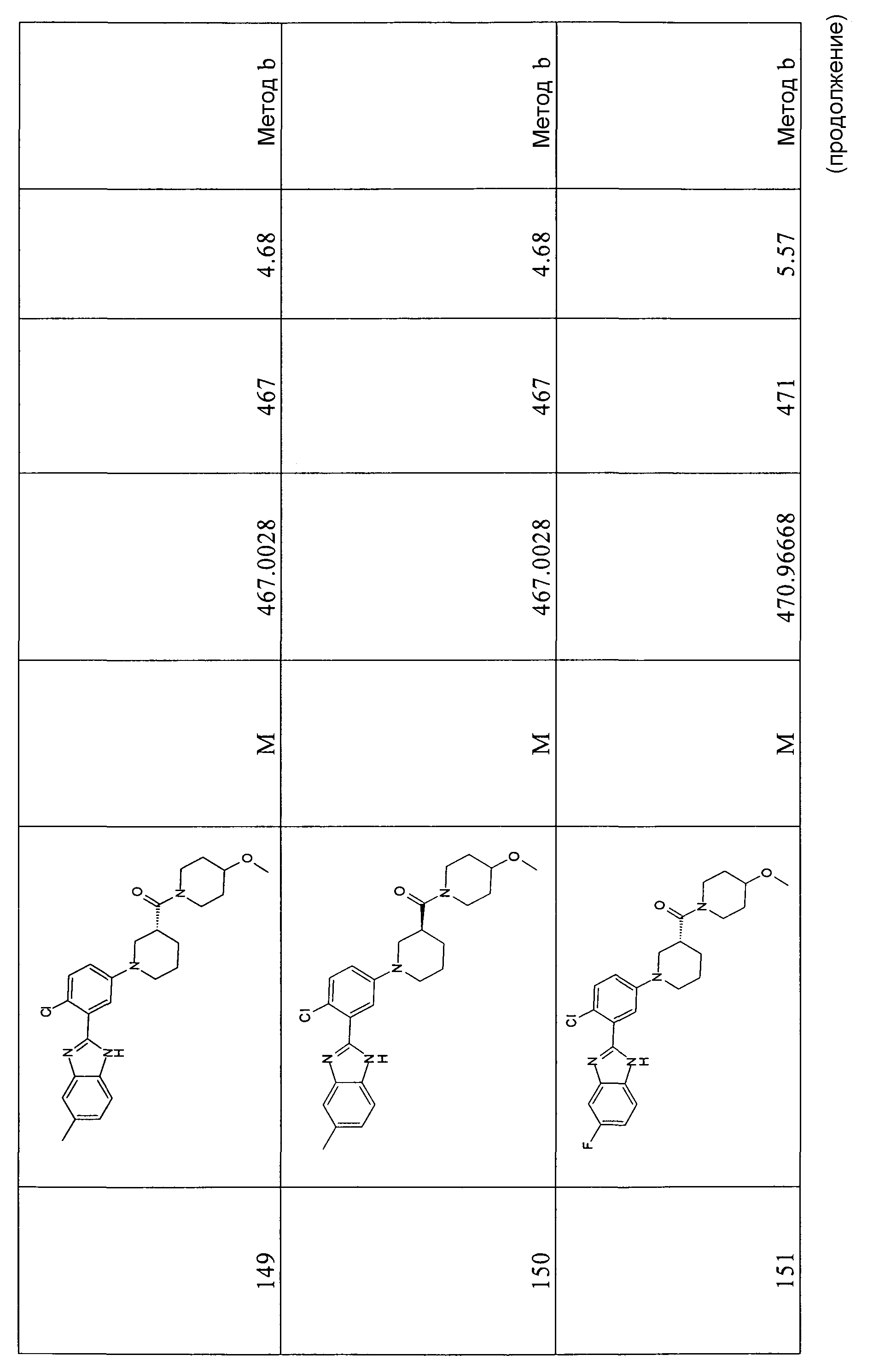
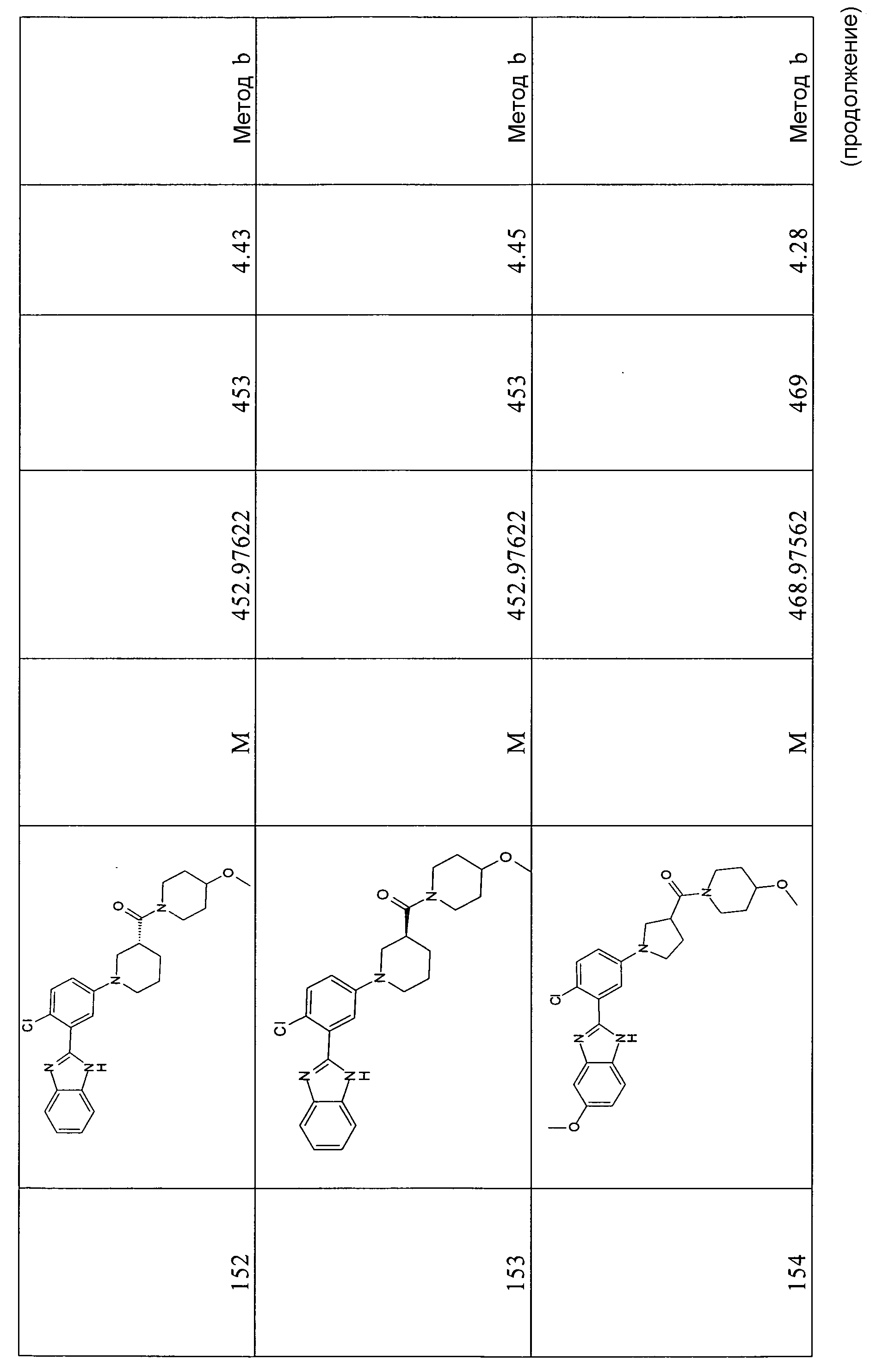
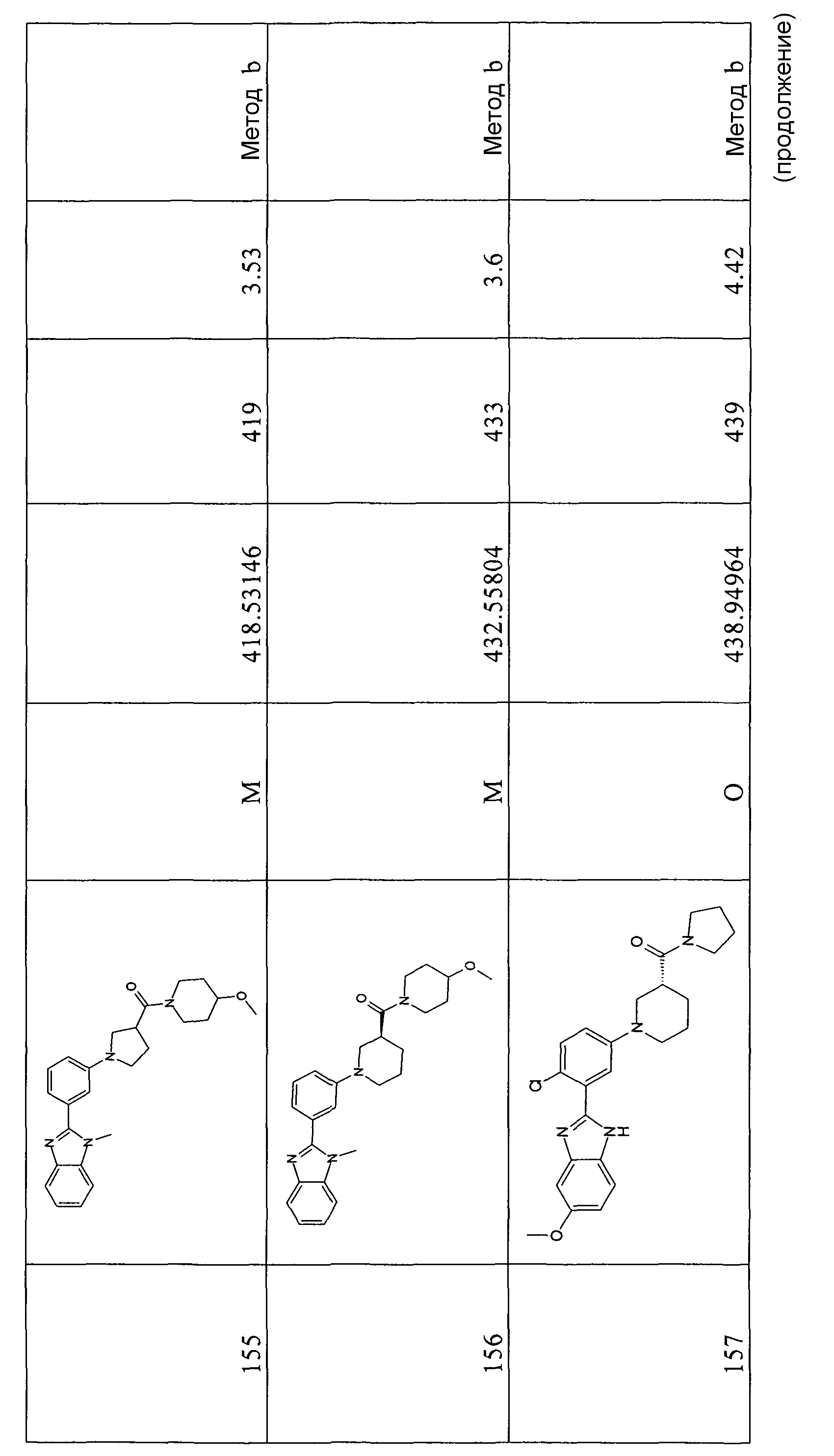
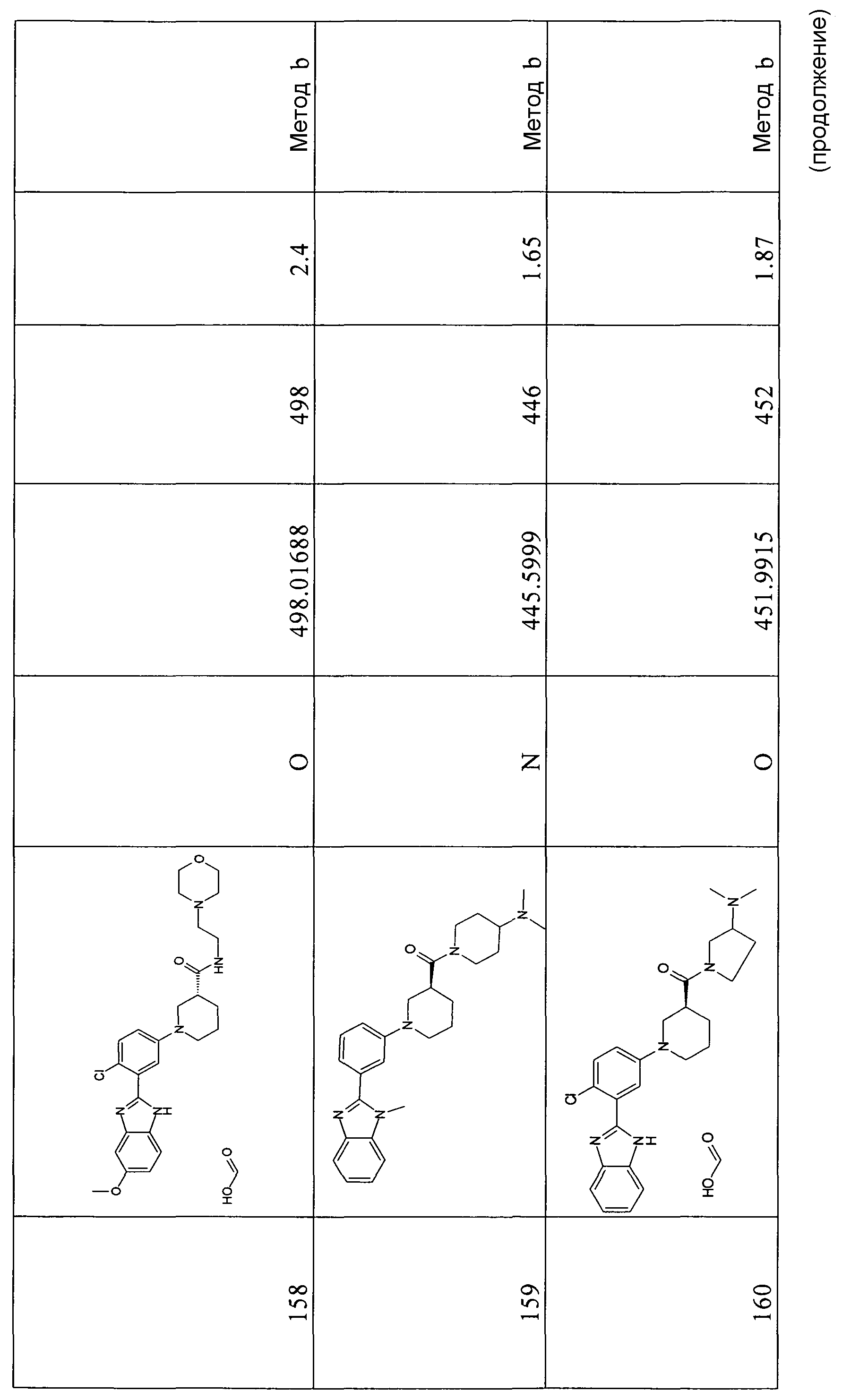
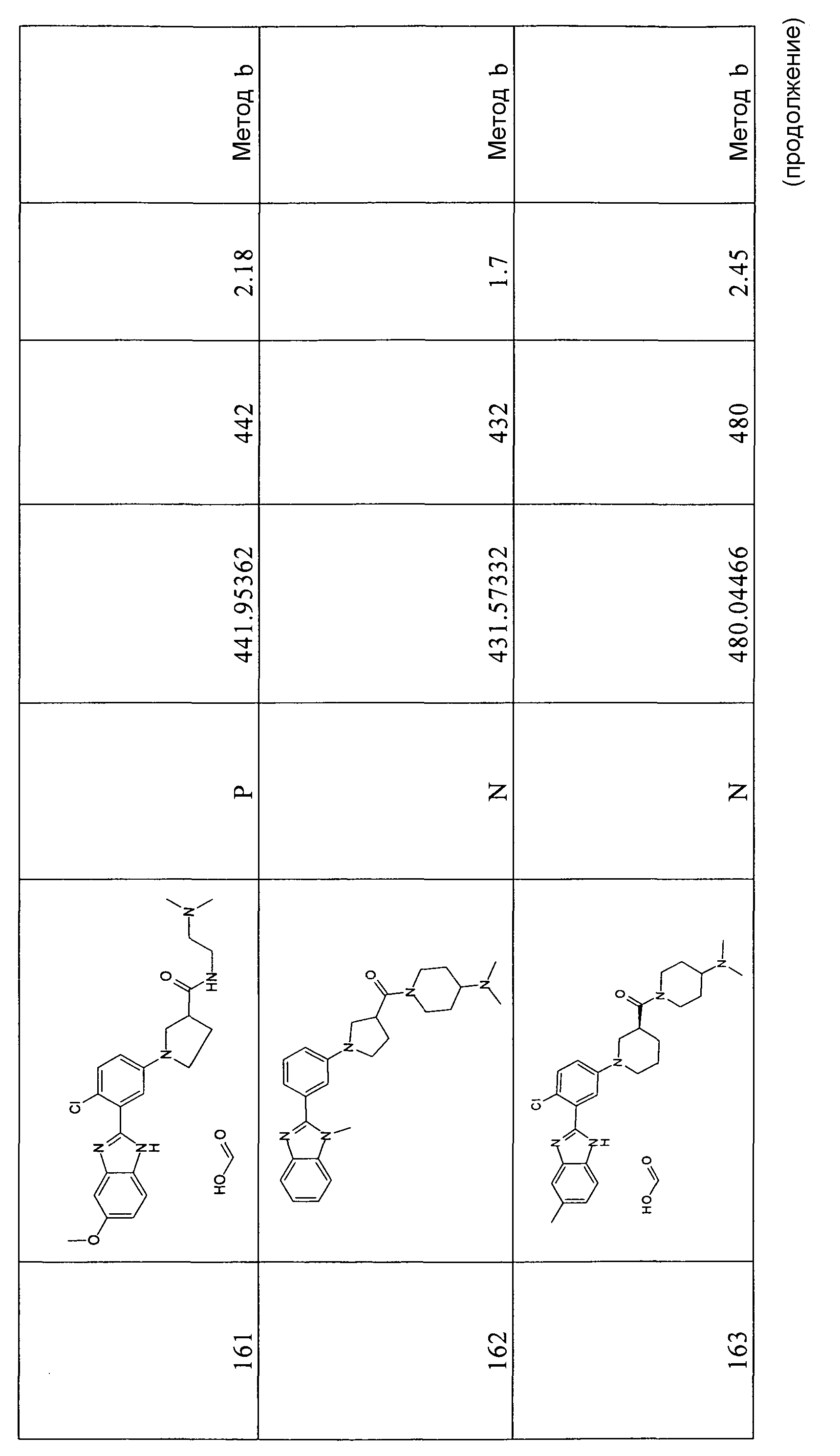

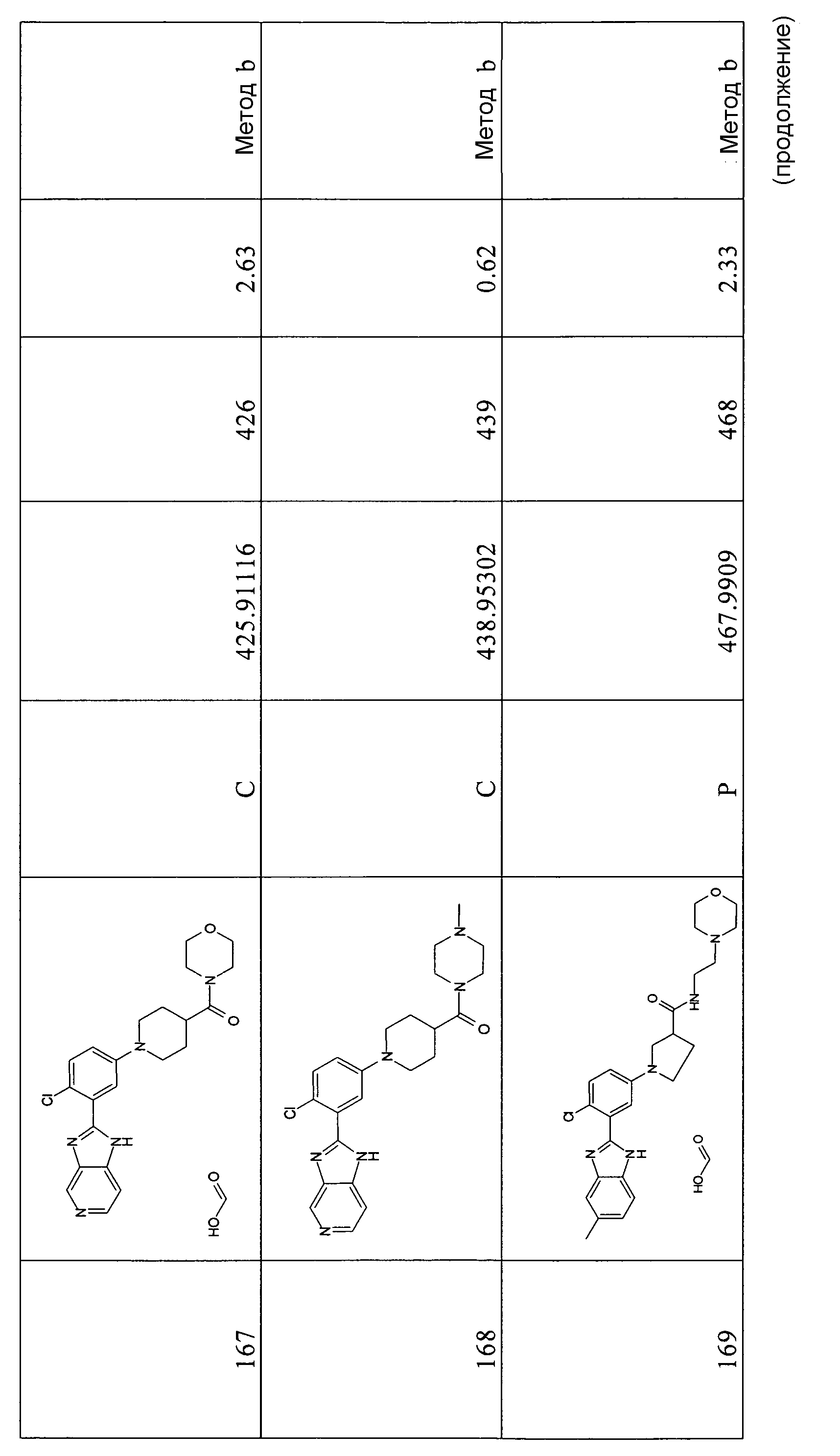
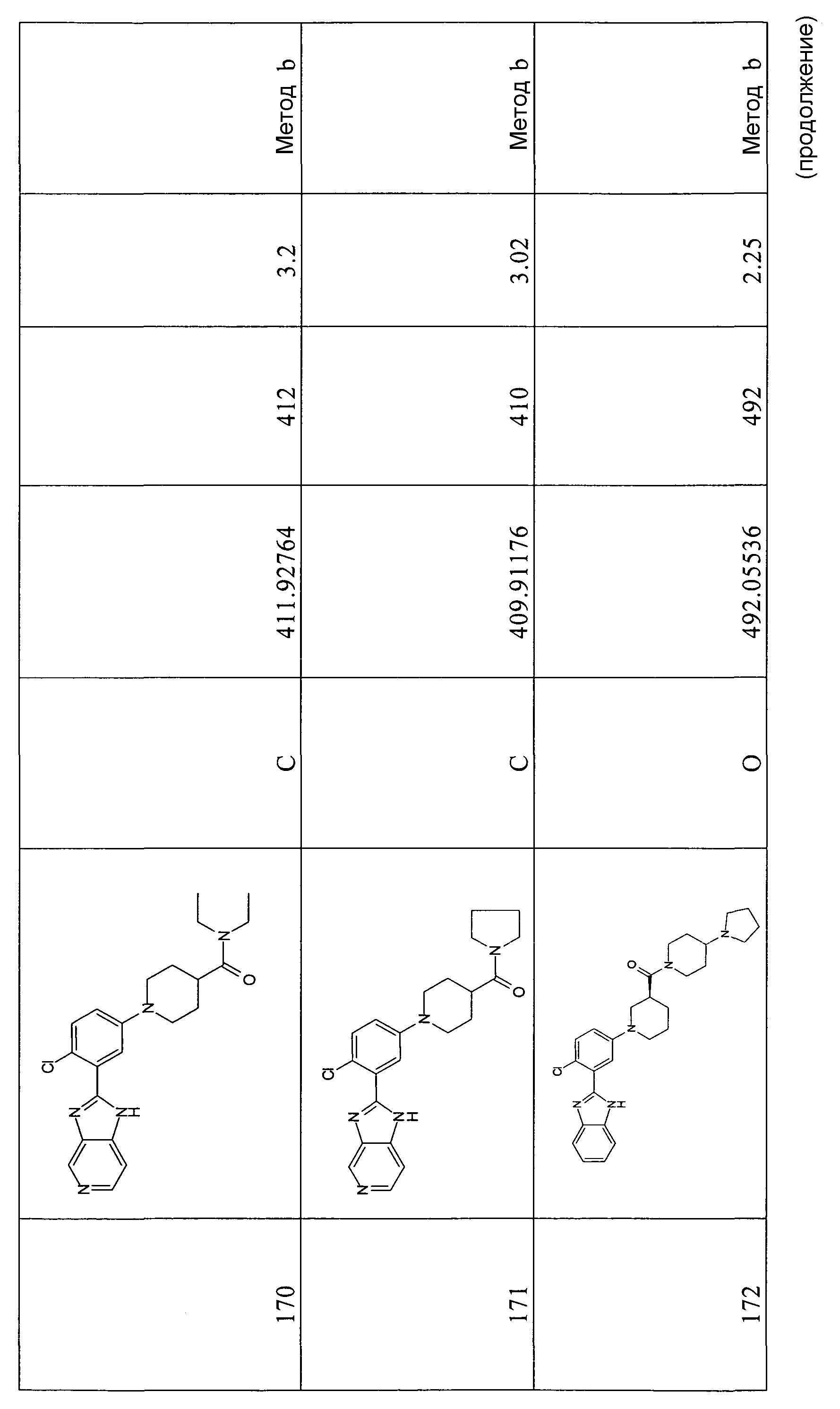
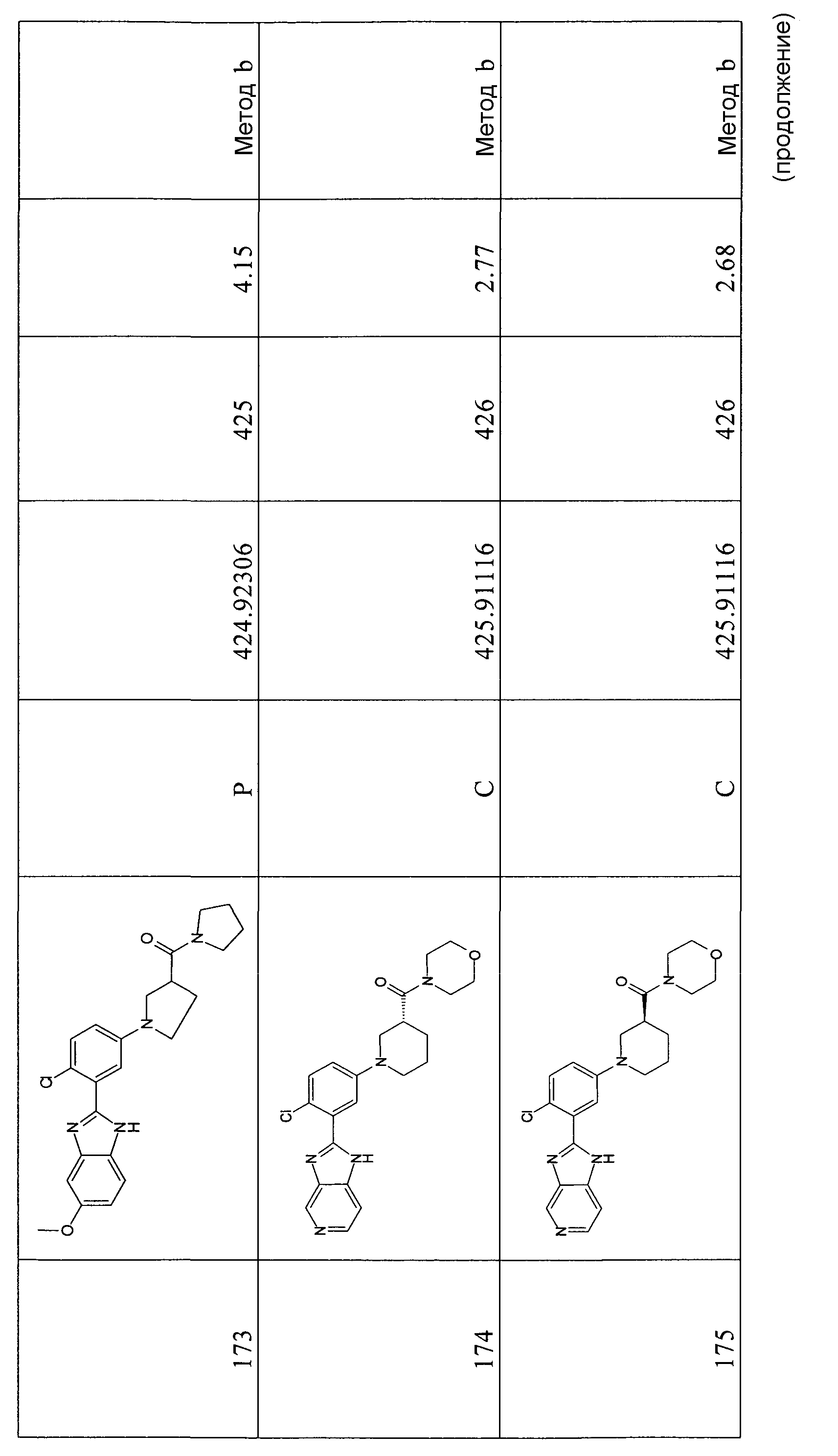
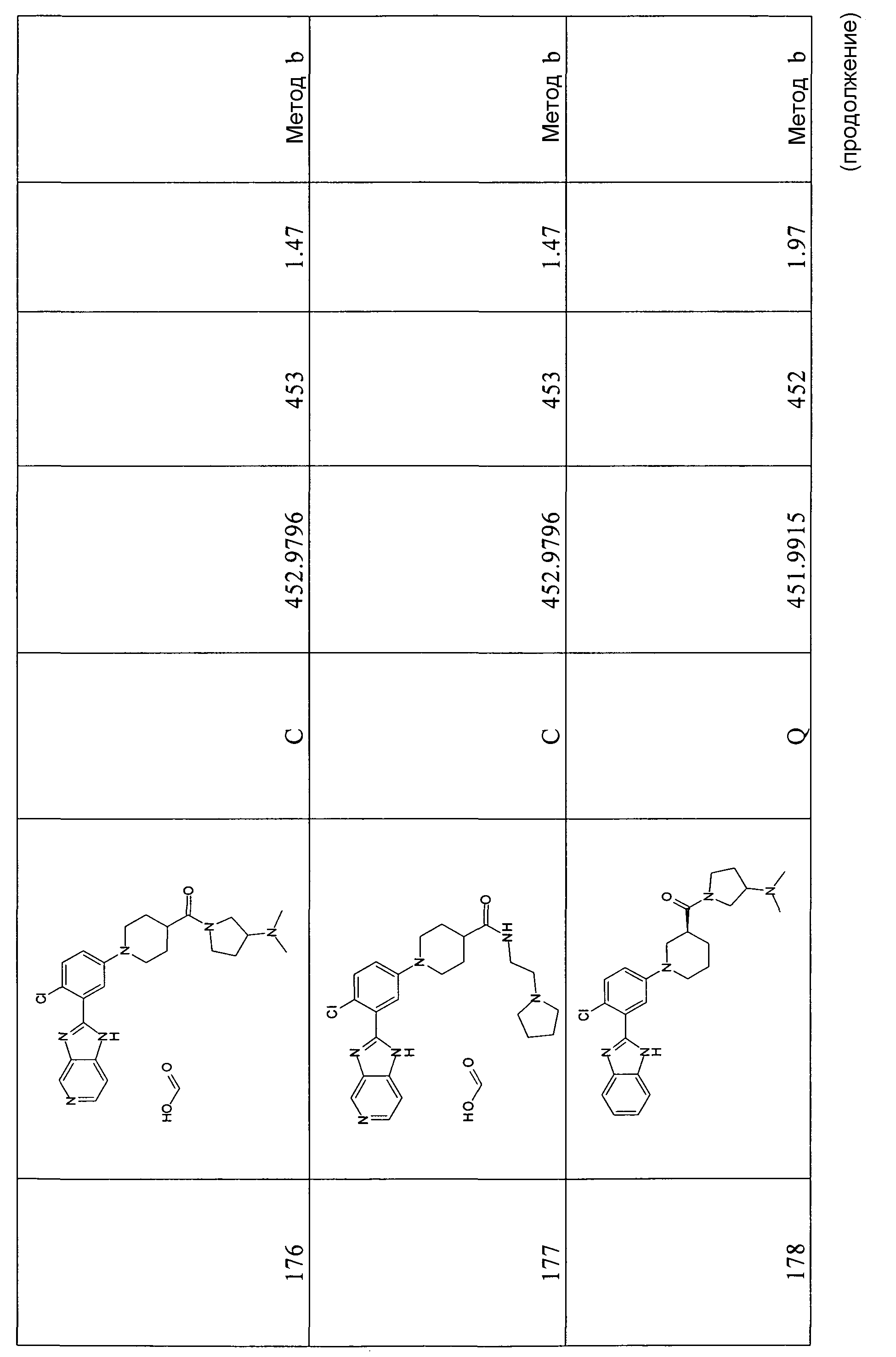
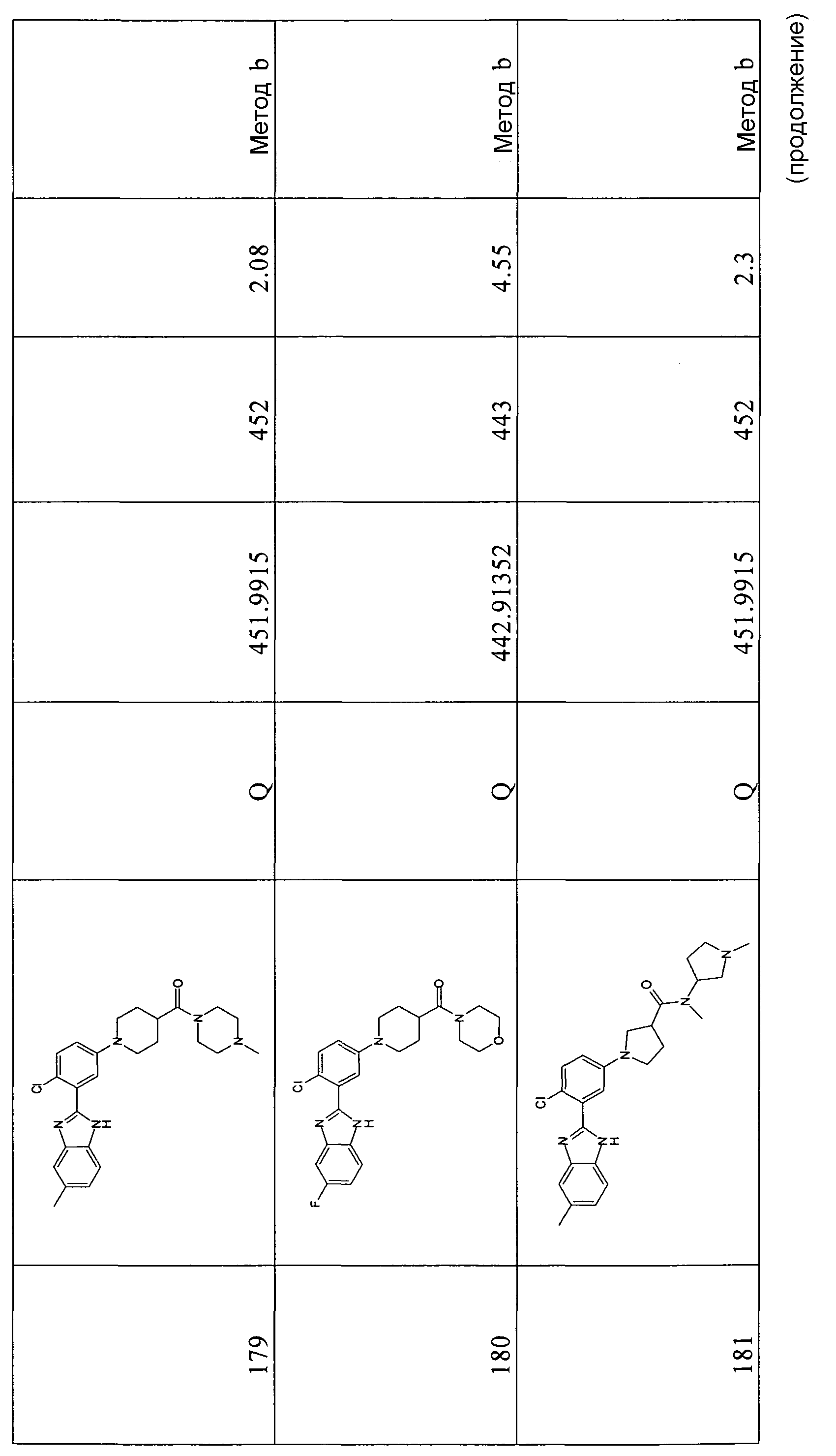
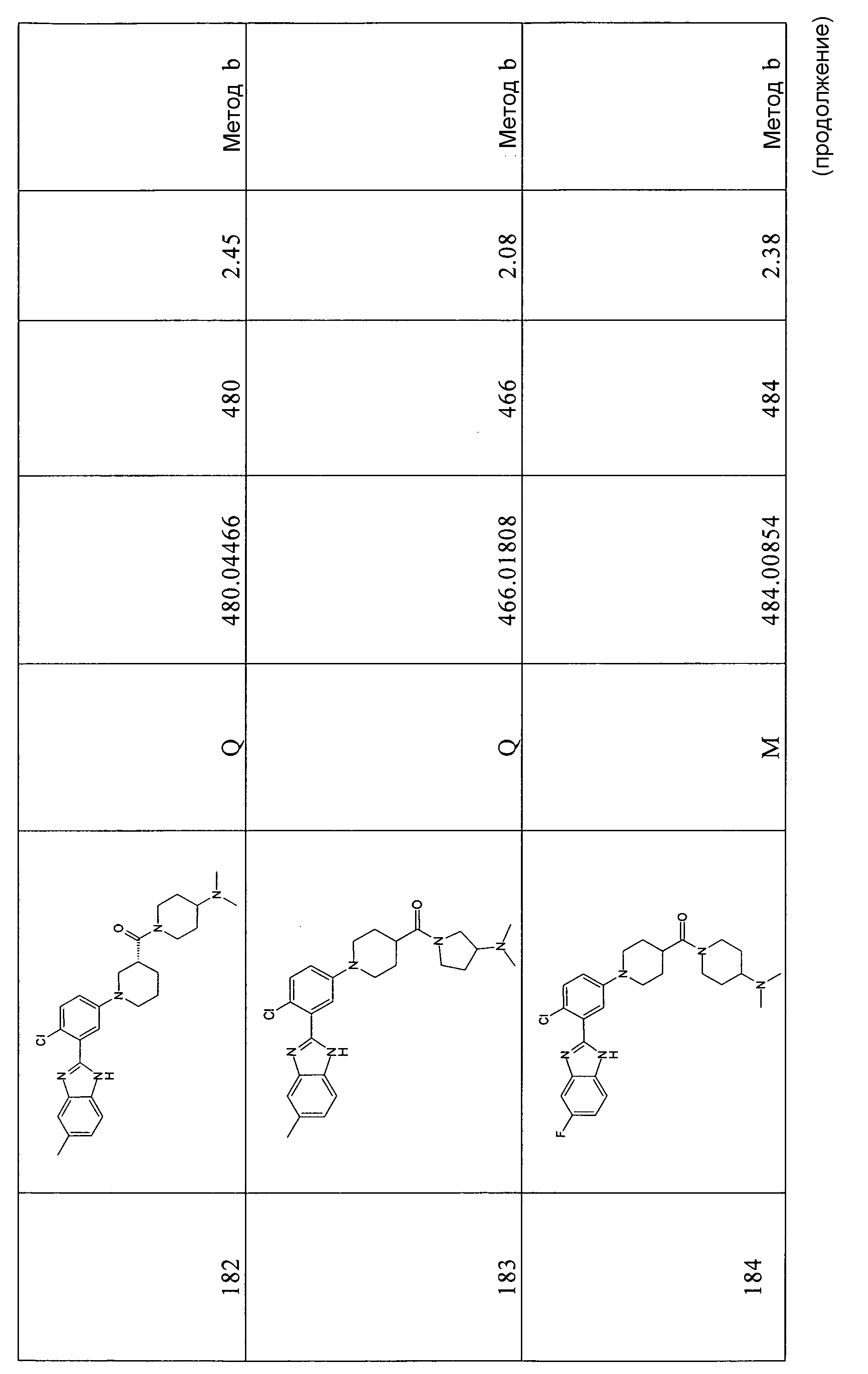
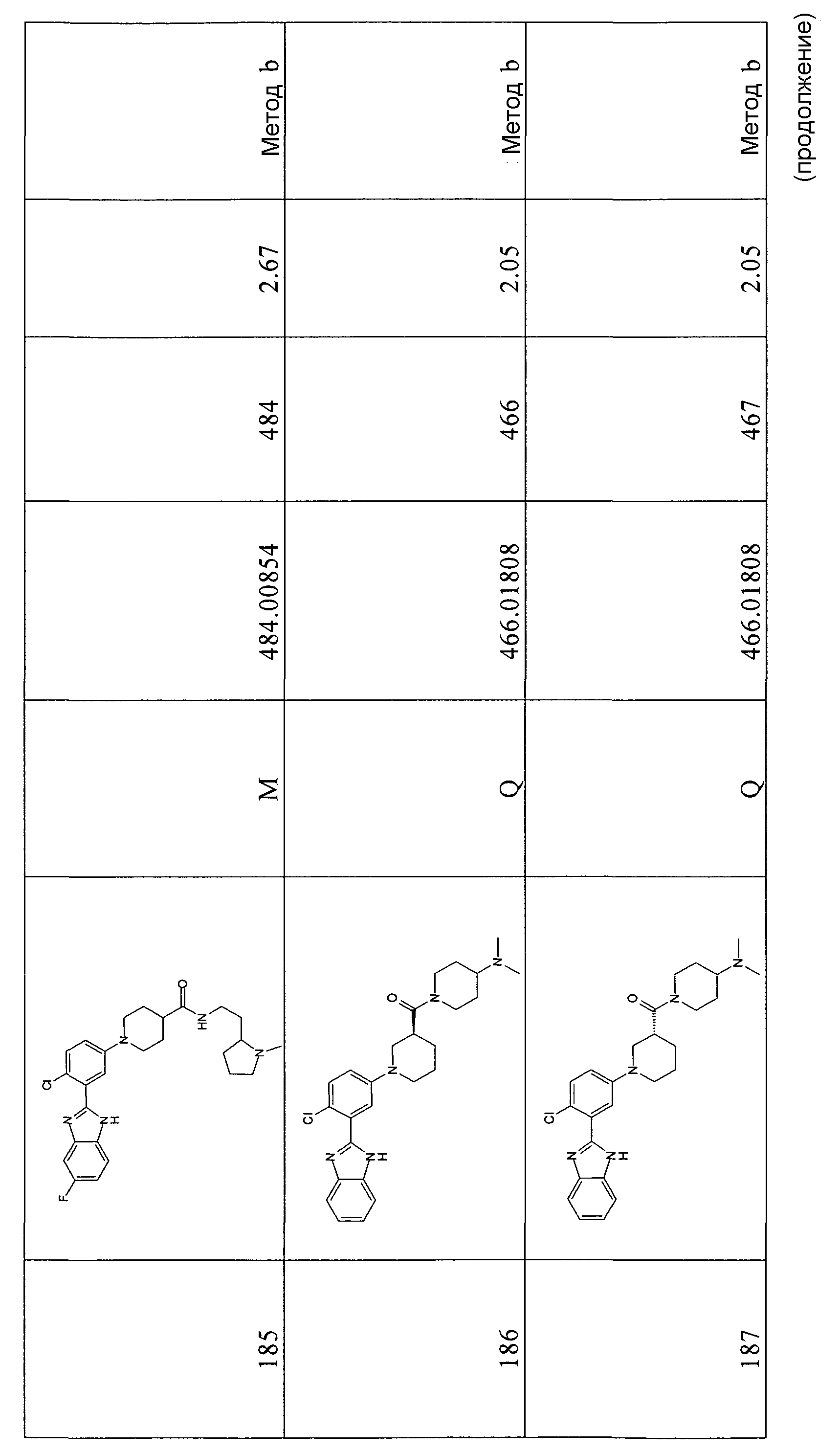
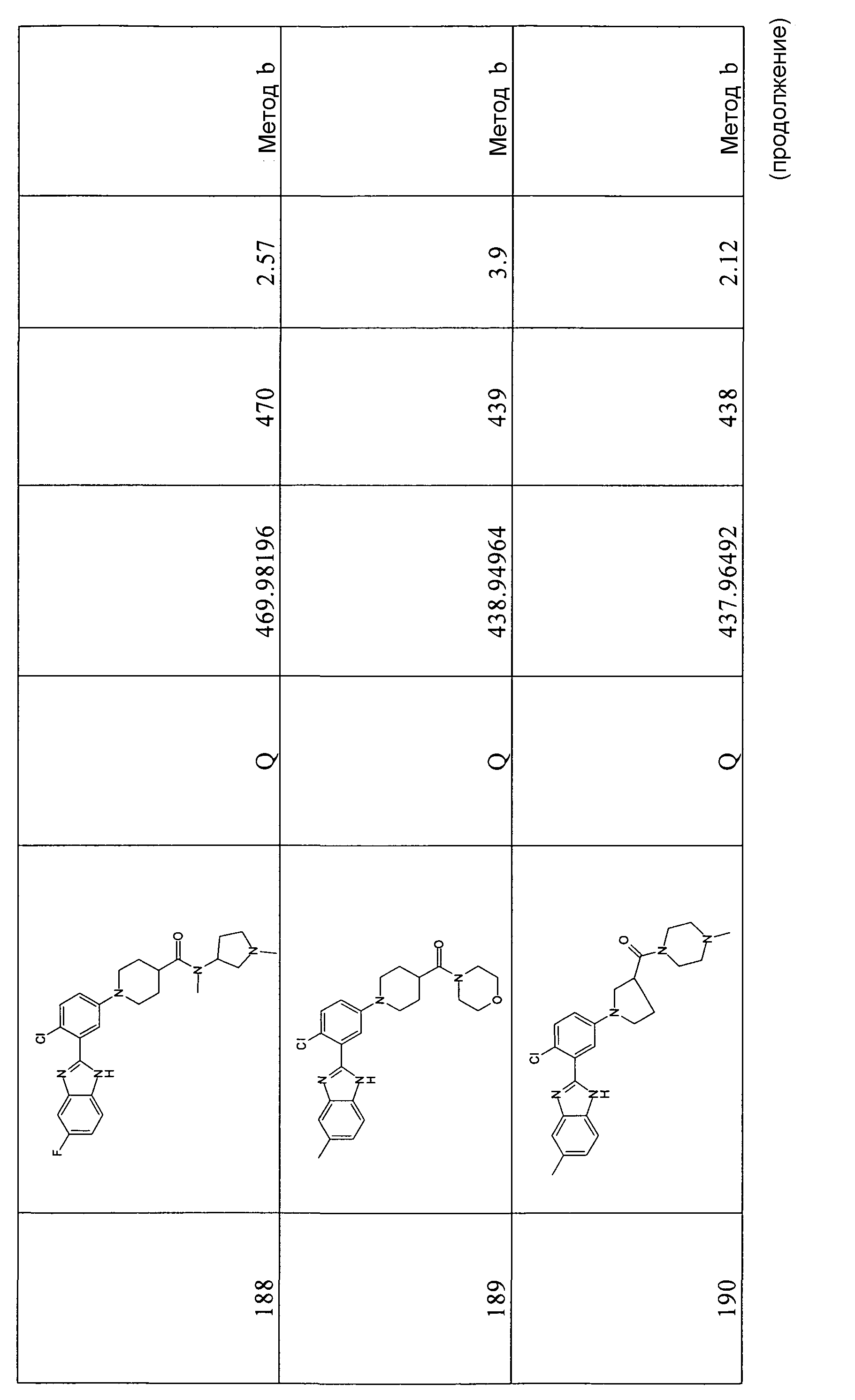
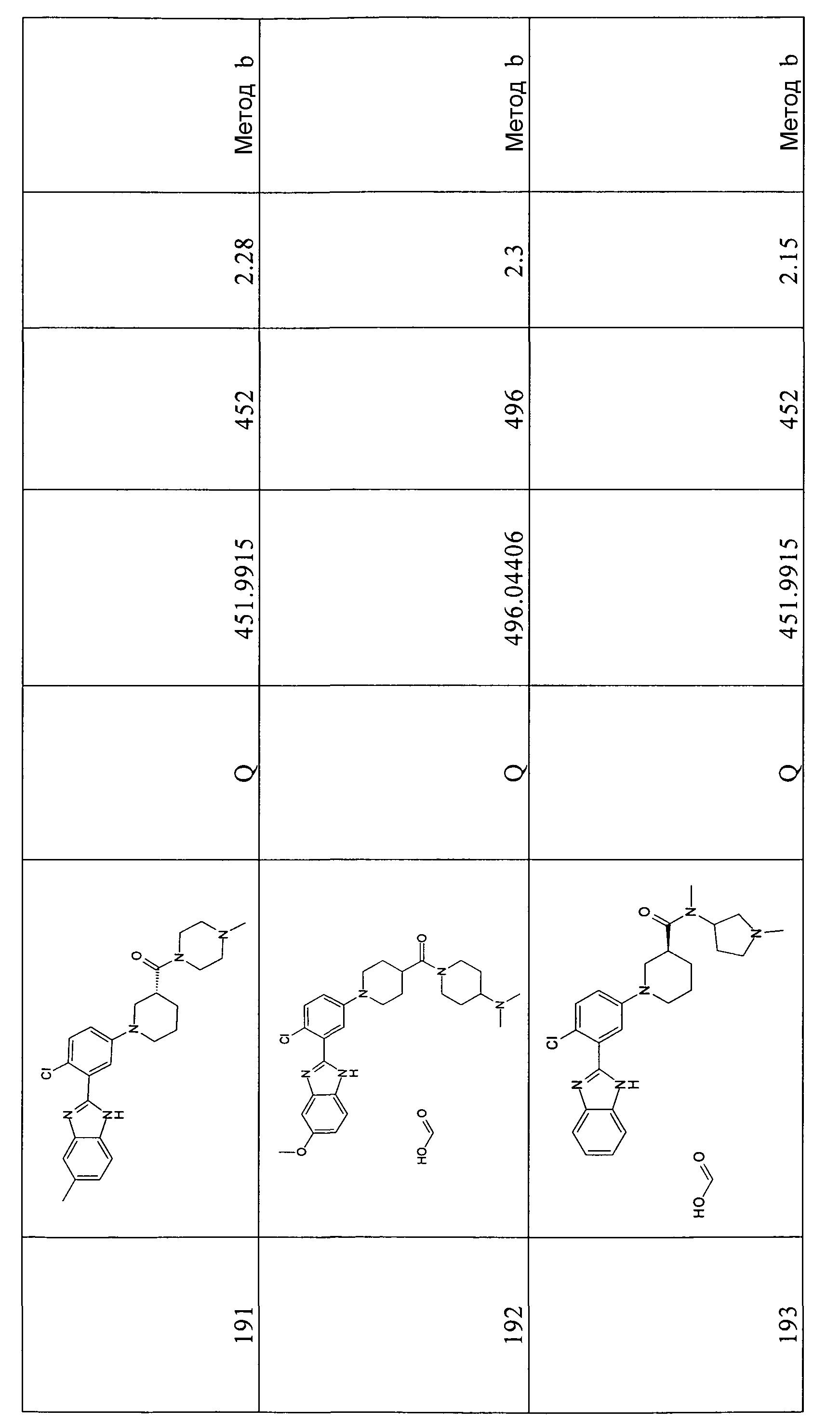
Клонирование Smo и генерирование стабильных рекомбинантных экспрессирующих Smo клеточных линий
Кодирующую последовательность Smo человека амплифицировали методом ПЦР в стандартных условиях. В качестве темплата использовали pcMV6-XL5-Smo от Origene (кат. TC 122724). Были сконструированы следующие праймеры:
прямой (5' GATCGGTACCGGGCTTTTGCTGAGTT 3') имел сайт рестрикции KpnI;
обратный (5' GATCGCGGCCGCCTACTTATCGTCGTCATCCTTGTAATCGAAGTCCGAGTCTGC 3') имел сайт рестрикции NotI, стоп-кодон и FLAG-кодирующую последовательность на 5'-конце.
Полученный ампликон был длиной 2424 пары оснований и содержал полную Smo-кодирующую последовательность, FLAG-метку и два сайта рестрикции на каждом из концов. Ампликон дважды подвергали обработке KpnI и NotI рестрикционными ферментами, а также плазмидой pcDNA5/FRT (Invitrogen), выбранной для клонирования. Лигирование и клонирование Smo-FLAG кодирующей последовательности в плазмиду pcDNA5/FRT давало плазмиду, которую назвали pcDNA5/FRT_Smo-FLAG и которая имела длину в 7432 пары оснований.
Метод FlpIN (Invitrogen) использовали для создания стабильно экспрессирующей Smo-FLAG клеточной линии с использованием клеточной линии FlpIN293 (Invitrogen, RT50-07). Эта линия получена из клеток HEK293 посредством стабильной трансфекции с использованием плазмиды pFRT/lacZeo для генерирования зеоцин-устойчивой FlpIN293 линии клетки-хозяина. Клетки FlpIN293 являются подходящими для создания стабильной клеточной линии млекопитающих, содержащей интегрированный Flp-рекомбинантный сайт-мишень (FRT) (Invitrogen).
Трансфекцию с использованием плазмиды pcDNA5/FRT_Smo-FLAG (или, в случае имитирующих трансфицированных клеток, трансфекция с использованием пустой плазмиды) проводили вместе с трансфекцией плазмиды pOG44, несущей Flp-рекомбиназу, которая катализирует гомологичную рекомбинацию между сайтом FRT в клетках-хозяевах и вектором экспрессии pcDNA5/FRT_Smo-FLAG или пустым вектором pcDNA5/FRT соответственно. Экспрессирующие Smo-FLAG клетки, а также имитирующие трансфицированные клетки обладают устойчивостью к гигромицину В и отрицательны к β-гал окрашиванию. Экспрессию Smo и FLAG антигенов контролировали также методом вестерн-блоттинга. Две генерированные клеточные линии названы 293FlpIN/клон E-3, указывая, что он является имитацией трансфицированной линии, и 293FlpIN/клон 3-5, указывая, что он является Smo-FLAG трансфицированной клеточной линией.
Условия культивирования клеток
Клетки поддерживали в среде DMEM, содержащей 10% фетальной бычьей сыворотки (оба от Invitrogen), с добавлением 0,25 мг/мл гигромицина B (Invitrogen). Клетки поддерживали при 37ºС в состоящей из 95% воздуха - 5% диоксида углерода полностью увлажненной среде и использовали до 20-25 циклов после оттаивания.
Разработка анализа связывания
Взаимодействие соединений с рецептором Smo тестировали путем замещающего анализа связывания с использованием флуоресцентного лиганда для рецептора Smo (бодипай-циклопамин, Bodipy-Cyclopamine), Toronto Research Chemical Inc., кат. № B674800) в качестве меченого лиганда, предназначенного для замещения.
Для определения Kd (концентрация лиганда, при которой достигается 50% максимального связывания) и Bmax (максимальное количество лиганда, которое может специфически связываться с рецептором в биологическом препарате) флуоресцентного лиганда, специфическое связывание (SB, СС) рассчитывали путем вычитания неспецифического связывания (NSB, НСС) из общего связывания (TB, ОС). ОС определяли путем добавления повышающейся концентрации бодипай-циклопамина к клеткам, тогда как НСС определяли путем добавления к клеткам смеси повышающейся концентрации бодипай-циклопамина с концентрацией насыщения хорошо описанного антагониста (в данном случае был выбран N-[3-(1Н-бензимидазол-2-ил)-4-хлорфенил]-3,5-диметоксибензамид (Rubin et al. WO2003011219) при концентрации 10 мкМ). Для каждой концентрации бодипай-циклопамина СС рассчитывали путем вычитания значения НСС из ОС. Из кривой СС рассчитывали Bmax и Kd. В данном случае было установлено, что стабильный имитирующий трансфицированную клеточную линию клон Е-3 имеет Kd, равную 115 нМ, тогда как клон 3-5 стабильной Smo-FLAG трансфицированной клеточной линии имеет Kd 44,3 нМ. Ki представляет собой концентрацию немеченого лиганда, которая ингибирует 50% специфического связывания (СС) меченого лиганда и корректирована для эффективной используемой концентрации меченого лиганда. Ki рассчитывали в соответствии с уравнением Ченга-Прусова (Cheng-Prusoff) как Ki=IC50/[1+[бодипай-циклопамин]/Kd)].
Тестирование соединений в анализе связывания
Клетки 293FlpIN/клон E-3 и 293FlpIN/клон 3-5 подсчитывали с использованием камеры Burker и переносили в два 96-луночных планшета из расчета 100000 клеток/100 мкл среды DMEM с 1% FBS (U-дно, Sigma Aldrich, кат. № M8185-100EA). Клетки 293FlpIN/клон E-3 использовали в качестве внутреннего контроля для контролирования изменения флуоресценции (FLU) с течением времени для сверхэкспрессирующих Smo клеток 293FlpIN/клон 3-5.
Контроли и соединения готовили в среде DMEM с 1% FBS и по 100 мкл добавляли к клеткам. Все контроли и соединения инкубировали при конечной концентрации бодипай-циклопамина в 5 нМ.
Соединения растворяли в ДМСО (10 мМ исходный раствор) и тестировали первоначально при концентрации 10 мкМ (анализ для одной концентрации); для каждого соединения тестирование проводили по меньшей мере дважды (на двух различных планшетах). Когда бодипай-циклопамин был замещен выше 30% порога, соединения повторно тестировали в анализе концентрация - ответная реакция при пропускной способности из расчета 8 соединений на планшет, и диапазон концентрации был 100, 10, 1, 0,5, 0,1, 0,01, 0,05, 0,001 и 0,0001 мкМ.
В качестве отрицательного контроля использовали клетки 293FlpIN/клон 3-5, к которым добавляли ДМСО, разведенный в соотношении 1:1000 для анализа одной концентрации и в соотношении 1:100 для анализа концентрационной ответной реакции.
В качестве положительного контроля для полного замещения связывания с бодипай-циклопамином использовали N-[3-(1Н-бензимидазол-2-ил)-4-хлорфенил]-3,5-диметоксибензамид (Rubin et al. WO2003011219) в концентрации 10 мкМ.
Два планшета инкубировали в течение 4 часов при комнатной температуре при защите от света на качающейся платформе. После инкубирования планшеты центрифугировали в течение 5 минут при скорости 1600 об/мин и промывали дважды PBS, содержащим 2% FBS. В конце клетки повторно суспендировали в 170 мкл буфера для промывки и сигналы флуоресценции регистрировали с использованием системы FACScalibur HTS (Becton Dickinson).
Параметры получения данных устанавливали в начале считывания каждого планшета с использованием необработанных немеченых клеток 293FlpIN/клон E-3. Программа получения данных HTS представляла собой BDTM Plate Manager (BD Bioscience), и анализ данных проводили с использованием программного обеспечения BD CellQuestTM Pro (BD Bioscience).
Количественную оценку проводили путем накладывания гистограмм FL1-H для положительного и отрицательного контролей и установления маркера в точке пересечения двух кривых. Количественную оценку проводили только в тех случаях, где флуоресценция была больше, чем установленный маркер. Затем значения нормализовали в соответствии с отрицательным контролем (0% замещения бодипай-циклопамина) и положительным контролем (100% замещения бодипай-циклопамина).
Все соединения из примеров 1-193, будучи протестированными в вышеуказанных условиях, показали значения Ki в диапазоне между 0,8 нМ и 21,6 мкМ.
Тестирование соединение в анализе со щелочной фосфатазой
Было продемонстрировано, что Shh in vitro индуцирует щелочную фосфатазу (AP), маркер дифференциации остеобластов, в мышиной мезенхимальной клеточной линии C3H10T1/2 (Katsuura et al., 1999; Kinto et al., 1997; Murone et al., 1999; Nakamura et al., 1997, Wu et al., 2004). Соответственно, для анализа влияния небольших молекул, способных к Hedgehog-Gli передаче сигнала, проводили функциональный анализ, основанный на активации АР в данной мышиной клеточной линии. Для обнаружения Ар в растворе использовали субстрат из набора AttoPhos® (кат. S1000, Promega). Кратко, использовали следующую методику.
Покрытые полилизином прозрачные плоскодонные 96-луночные планшеты (Corning, кат. 3667) наполняли из расчета 10000 клеток в 100 мкл клеточного культурального раствора на лунку. Клеточная культуральная среда состояла из DMEM (кат. 21969-035) с 1% глутамакса (кат. 35050-038), 1% пенициллин/стрептомицин (кат. 15140-122) и 1% Hepes (15630-056). Все реагенты получены от Invitrogen. Планшеты инкубировали в течение ночи при 37ºС с 5% диоксида углерода. Затем среду удаляли и добавляли в лунки 100 мкл свежей среды, содержащей либо соединение, либо ссылочный антагонист (N-[3-(1Н-бензимидазол-2-ил)-4-хлорфенил]-3,5-диметоксибензамид (Rubin et al. WO2003011219)). Все растворы соединений и ссылочные растворы содержали агонист парморфамин (Sinha et al. Nature Chem. Biol. 2, 29-30 (2005)) в концентрации 2 мкМ. Соединения тестировали в десяти концентрациях в трехкратной повторности в диапазоне между 100 пМ и 50 мкМ. Конечную концентрацию ДМСО в каждом образце доводили до 1% в культуральной среде. Клетки инкубировали с раствором соединения в течение 72 часов при 37ºС в присутствии 5% диоксида углерода. Клеточную культуральную среду удаляли из планшетов и 40 мкл разведенного 1:5 лизисного раствора (кат. E194A, Promega) добавляли в каждую лунку. Планшеты затем инкубировали в темноте в течение 20 минут на шейкере. Наконец, 40 мкл раствора восстановленного субстрата AttoPhos добавляли в лунки с последующим периодом инкубирования в течение 15 минут на шейкере. Влагосодержание субстрата AttoPhos восстанавливали в соответствии с инструкциями поставщика, но раствор субстрата всегда хранили при -80ºС. Планшетный ридер Safire 2 (Tecan) использовали для измерения изменений в интенсивности флуоресценции в образцах, при длине волны возбуждения 430 нм и длине волны испускания при 560 нм.
Все соединения из примеров 1-193, будучи протестированы в вышеуказанных условиях, показали значения IC50, колеблющиеся в диапазоне между 1,1 мкМ и 14,8 мкМ.
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/a719a29f645d3f9e84f30ad911218cc0.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/4067eaa9435cbf1b2bcd08285445560d.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/c87b8c387031a674673bab59bdd3a987.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/8b04c50d382b29bd55b5b5307019f043.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/5608c2d0c4d0eb1e87197d43c1aaa1d7.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/c5a0b133402645b2ca6f68c7ee72b1da.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/8e9b0c87ffef2081c0bd5cc99b7e9372.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/cd0045c7cf727de718b5a0f276a65c13.png)
![ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗО[4,5-с]ПИРИДИНА, В КАЧЕСТВЕ АНТАГОНИСТА ПУТИ HEDGEHOG](https://fips.edrid.ru/images/rid/0d/b7/a0/1ba007ae7193a8914738e7dabc041ec1.png)

