Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 11БЕТА, 17АЛЬФА, 21-ТРИГИДРОКСИ-16АЛЬФА-МЕТИЛ-9АЛЬФА-ФТОРПРЕГНА-1,4-ДИЕН-3,20-ДИОНА (ДЕКСАМЕТАЗОНА) ИЗ ФИТОСТЕРИНА
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области биотехнологии, конкретно касается получения стероидного соединения 11β,17α,21-тригидрокси-16α-метил-9α-фторпрегна-1,4-диен-3,20-диона (дексаметазона) из фитостерина и может быть использовано для производства субстанции дексаметазона и его производных.
УРОВЕНЬ ТЕХНИКИ
11β,17α,21-Тригидрокси-16α-метил-9α-фторпрегна-1,4-диен-3,20-дион (дексаметазон, 9α-фтор-16α-метилпреднизолон, CAS 50-02-2) является глюкокортикостероидным препаратом, обладающим сильным противовоспалительным и антиаллергическим действием [М.Д. Машковский. Лекарственные средства. В двух томах. Изд.13-е. Харьков: Торсинг, 1997 г., Т.2. с.35]. Кроме того, дексаметазон может быть использован в качестве исходного субстрата в синтезе его производных, например динатриевой соли 21-фосфата дексаметазона (динатрийфосфат дексаметазона, CAS 2392-39-4), применяемой для внутривенного и внутримышечного введения, а также в офтальмологии для лечения аллергических конъюнктивитов, кератитов, уменьшения воспалительных проявлений после глазных операций и травм.
К настоящему времени в литературных источниках нет опубликованных данных о схеме синтеза дексаметазона из фитостерина как таковой. Однако имеются сообщения о синтезе его различных интермедиатов. Поиск и изучение различных подходов в синтезе дексаметазона ведется ведущими фармацевтическими компаниями мира на протяжении последних 50 лет и обусловлен вариабельностью источников стероидного сырья и поиском возможных интермедиатов синтеза, общих для дексаметазона и ряда других стероидных препаратов.
Наиболее перспективным источником стероидного сырья являются стерины. Стерины являются структурными предшественниками в производстве стероидных гормональных препаратов (гестагенов, андрогенов, эстрогенов, кортикоидов и др.). Фитостерин - стерин растительного происхождения - может быть получен из отходов переработки древесины или соевых бобов. Слово «фитостерин» обозначает стерины растительного происхождения (β-ситостерин, кампестерин, стигмастерин и др.) или их смеси. В последние годы в мире разработаны эффективные методы окислительной деградации боковой цепи стеринов с образованием андрост-4-ен-3,17-диона (АД), которая может сопровождаться одновременной или последовательной его функционализацией с образованием 1,2-дегидро- или 9α-гидрокси-производных АД.
9α-Гидроксиандрост-4-ен-3,17-дион (9-ОН-АД) является идеальным интермедиатом в синтезе кортикостероидов из стеринов (например, таких, как гидрокортизон, преднизолон, дексаметазон, триамцинолон, 6α-метилпреднизолон и др.). Это особенно важно, так как позволяет исключить из технологического процесса стадию микробиологического 11-гидроксилирования.
Синтез дексаметазона из фитостерина через 9-ОН-АД представляет собой многостадийную последовательность превращений, включающую следующие этапы функционализации:
- дегидратация 9α-гидроксигруппы с образованием ∆9(11)-связи;
- введение 17α-гидрокси-17β-прегнановой цепи;
- введение гидроксильной группы в 21-положение 17α-гидрокси-С20-кетопрегнана с образованием кортикостероидной (диоксиацетоновой) цепи;
- дегидратация 17α-гидроксигруппы с образованием ∆16-3,20-дикетопрегнана;
- введение 1,2-двойной связи (химическим или микробиологическим способом);
- введение 16α-метил-17α-гидрокси- фрагмента по ∆16 связи;
- введение 9α-фтор-11β-гидрокси- фрагмента по ∆9(11)-связи.
Как следует из литературных данных, эти этапы могут быть проведены в любом порядке. Выбор очередности этапов и методов синтеза является сложной задачей, решение которой определяет успех в создании экономически эффективной технологии производства.
Методы окислительной деградации боковой цепи фитостерина с образованием Δ4-3,17-дикетоандростанов, в том числе с образованием 9-ОН-АД, известны. 9-ОН-АД получают из фитостерина микробиологической трансформацией методами и культурами микроорганизмов, описанными в литературных источниках [Прикл. биохим. микробиол., 2011, 47(3), 297-301; Biochim. Biophs. Acta, 1978, 539, 308; US4035236, 1977; US4175006, 1979; US4397946, 1983; DD298278, 1992; DD298279, 1992; RU2077590, 1997; Appl. Microbiol. Biotechnol., 2005, 67(5), 671-678; J. Chem. Technol. Biotechnol., 2005, 80, 55-60 и др.].
Дегидратация 9α-гидроксигруппы с образованием ∆9(11)-связи
В синтезе кортикостероидной цепи на основе 17-кетогруппы 9-ОН-АД дегидратацию 9α-гидроксигруппы обычно проводят на первом этапе, так как образующийся андроста-4,9(11)-диен-3,17-дион (Δ9(11)-АД) более стабилен в дальнейшем синтезе (например, в синтезе ацетата гидрокортизона из стерина [Tetrahedron Lett., 1990, 31(26), 3669-3672]). Это позволяет избежать на следующих стадиях синтеза возможных нежелательных реакций с участием третичной гидроксильной группы при С9. Кроме того, наличие 9,11-двойной связи - необходимое условие для дальнейшего введения галоида в положение 9α в синтезе 9-галоидированных кортикостероидов, в том числе и дексаметазона. Также целесообразно проводить дегидратацию в отсутствие гидроксильных групп в других положениях. Таким образом, Δ9(11)-АД является ключевым соединением в синтезе кортикостероидов из природных стеринов (холестерина или фитостерина).
Способы получения Δ9(11)-АД дегидратацией 9-ОН-АД известны. Особенностью дегидратации 9-ОН-АД является возможность образования нежелательного изомера андроста-4,8(9)-диен-3,17-диона (Δ8(9)-АД).
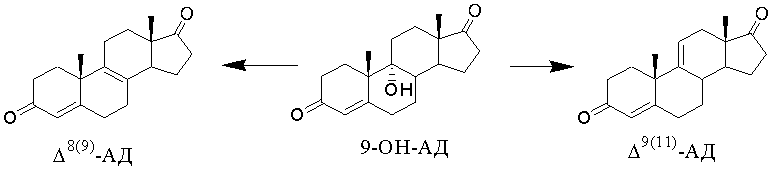
При этом изомерные ∆9(11)- и ∆8(9)- олефины имеют одинаковое значение Rf на ТСХ и не могут быть разделены кристаллизацией. Определение примеси ∆8(9)-изомера может быть проведено, например, спектроскопически (1Н ЯМР, 13С ЯМР) [EP0253415, 1988; EP0294911, 1988] или хроматографически (ВЭЖХ [EP0253415, 1988; EP0294911, 1988] , ГЖХ [US4102907, 1978]).
В качестве дегидратирующего агента используют минеральные и органические кислоты, ангидриды и хлорангидриды минеральных и органических кислот, а также другие реагенты.
Реакция дегидратации 9-ОН-АД с образованием ∆9(11)-АД может быть проведена или в среде дегидратирующего агента без использования растворителя, или в среде органического растворителя.
Сочетанием существенных признаков по заявляемому изобретению в части проведения дегидратации 9α-гидроксигруппы с образованием ∆9(11)-связи является проведение реакции дегидратации 9-ОН-АД с образованием ∆9(11)-АД в среде инертного апротонного органического растворителя, конкретно, в среде ароматического углеводорода (например, бензола, толуола) или хлорсодержащего углеводорода (например, дихлорметана, дихлорэтана, хлороформа), с применением в качестве дегидратирующего агента минеральной кислоты, в том числе содержащей воду, конкретно, ортофосфорной, серной или хлорной кислоты.
Известны способы проведения дегидратации 9-ОН-АД с образованием ∆9(11)-АД в среде органического растворителя. Известно, что в качестве среды могут быть использованы хлорсодержащие углеводороды, ароматические углеводороды, циклические эфиры, органические основания. Однако при этом в качестве дегидратирующего агента использованы N,N1-тионил-диимидазол, образующийся из тионилхлорида и имидазола (в среде тетрагидрофурана) [Journal für Praktische Chemie, 1988, 330(2), 309-312]; или п-толуолсульфокислота (п-ТСК, в среде бензола) [Chem.and Ind., 1961, 1530]; или адсорбат ароматической сульфокислоты на силикагеле (в среде ароматического углеводорода) [EP0253415, 1988]; или кислоты Льюиса - хлорид железа, трифторид бора или их комплексы, пентахлорид сурьмы, тетрахлорид титана - и смеси этих соединений с силикагелем (в среде инертного органического растворителя) [EP0294911, 1988]; или хлорсульфоновая кислота (в среде хлористого метилена) [US4127596, 1978, примеры 1-8].
Одним из существенных признаков по настоящему изобретению является использование в качестве дегидратирующего агента минеральной кислоты, в том числе содержащей воду, более конкретно, ортофосфорной, серной или хлорной кислоты.
Известен способ получения ∆9(11)-стероидов ряда андростана, в том числе и ∆9(11)-АД, реакцией 9α-гидроксистероида (в частности, 9-ОН-АД) с неароматической кислородсодержащей кислотой с pKa ≤1,0 [US4127596, 1978]. При этом кислота выбрана из группы: хлорсульфоновая, серная, фосфорная, метансульфоновая, перхлорная и трифторуксусная. Реакцию проводят в среде кислородсодержащей кислоты.
Также известен способ получения ∆9(11)-АД из 9-ОН-АД дегидратацией с полифосфорной кислотой [HUT36138, 1985].
Общим недостатком методов дегидратации в среде дегидратирующего агента, особенно в присутствии воды, является использование большого избытка дегидратирующего агента, большая продолжительность реакции, а также в большинстве случаев необходимость применения колоночной хроматографии для очистки продукта и, как следствие, или низкий выход, или неудовлетворительное качество.
Таким образом, анализ методов дегидратации 9-ОН-АД, известных из доступных литературных источников, показывает, что при проведении реакции в среде органического растворителя, в частности в среде ароматического углеводорода или хлорсодержащего углеводорода, использование минеральных кислот, в том числе содержащих воду, неизвестно. Описано проведение реакции органическими кислотами или их производными, ангидридами или хлорангидридами минеральных кислот, при этом наличие безводной среды является условием проведения реакции, обеспечивающим положительный результат. Как следует из уровня техники, использование минеральных кислот, в том числе содержащих воду, для получения ∆9(11)-АД возможно и известно в процессах дегидратации 9-ОН-АД без применения органического растворителя, однако эти методы имеют существенные недостатки и полученные результаты нельзя считать удовлетворительными.
Таким образом, в доступных литературных источниках отсутствует информация о проведении реакции дегидратации 9-ОН-АД с образованием ∆9(11)-АД с использованием в качестве дегидратирующего агента минеральных кислот, в том числе содержащих воду, в среде апротонного органического растворителя.
Кроме того, в доступных литературных источниках отсутствует информация об интенсификации процесса дегидратации удалением избыточной воды в течение реакции дегидратации, например, методом азеотропной отгонки.
Кроме того, в доступных литературных источниках отсутствует информация о возможности получения ∆9(11)-АД из фитостерина через образование 9-ОН-АД без выделения последнего из экстракта культуральной жидкости, полученной в результате микробиологической трансформации.
Введение 17α-гидрокси-17β-прегнановой цепи
Гидроцианирование
Одним из хорошо известных методов синтеза 17α-гидрокси-17β-ацетилстероидов является циангидринный метод, который заключается во взаимодействии 17-кетостероидов с цианистым водородом и последующем алкилировании полученного 17α-гидрокси-17β-нитрила. Химические синтезы прегнанов из 17-кетоандростанов с применением циангидринного синтеза известны и описаны в литературе [Pharmaceutical Chemistry Journal, 1987, 21, (4), 225-300]. Донором цианид-аниона могут служить как соли цианистоводородной кислоты [DD281394, 1990], так и ацетонциангидрин [J. Am. Chem. Soc., 1953, 75 (3), 650-653; RU2156255, 2000], а также их смеси [US4500461, 1985; US3043833, 1962; US4548748, 1985] и другие циансодержащие органические соединения, например, диэтилалюминия цианид [US3496169, 1970]. Реакцию с циансодержащими соединениями проводят в щелочной среде, в качестве щелочного агента, кроме гидроксидов щелочных металлов, может быть использован триэтиламин [GB1109626, 1968].
Циангидринный вариант построения боковой цепи кортикостероидов из АД, 9α-ОН-АД и других 17-кетостероидов имеет ряд преимуществ: избирательность реакции гидроцианирования по 17-кетогруппе [Organic Reactions, 1977, 25, р.255]. Однако гидроцианирование 17-кетостероидов протекает с образованием равновесной смеси эпимерных 17β- и 17α-карбонитрилов. В зависимости от условий проведения процесса гидроцианирования в смеси может преобладать тот или иной изомер.
Следует отметить, что наибольшее развитие и применение этот метод получил в синтезе 17-циангидринов АД. Известен способ введения прегнановой боковой цепи в АД с применением ацетонциангидрина в качестве цианирующего агента [DD147669, 1979, пример 2].
Использование одного ацетонциангидрина как агента для гидроцианирования ∆9(11)-АД, хотя и заявлено в патентах [RU2156255, 2000; US4500461, 1985], однако не подтверждено примерами. Циангидринный способ построения прегнановой цепи на основе АД описан японскими авторами [Bull. Chem. Soc. Jpn., 1985, 58 (3), 978-980]. Однако гидроцианирование ∆9(11)-АД с ацетонциангидрином в описанных авторами условиях протекает с образованием исключительно нежелательного 17α-циан-17β-гидрокси-эпимера, в то время как реакция в присутствии 9-гидроксигруппы в этих же условиях дает исключительно желаемый 17β-циан-17α-гидрокси-эпимер [Bull. Chem. Soc. Jpn., 1985, 58 (3), 978-980]. 17β-Циан-17α-гидрокси-∆9(11)-эпимер может быть получен последующей изомеризацией первично образованного 17α-циан-17β-гидрокси-изомера действием, например, цианида натрия или триэтиламмоний гидроцианида в среде низшего спирта [JPS5762298, 1982; JPS5762299, 1982].
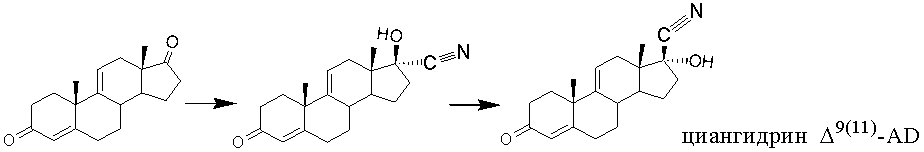
Таким образом, можно сделать вывод, что технический результат, полученный на примере АД в патентах [RU2156255, 2000 и RU2156256, 2000], не может быть очевидным при использовании ∆9(11)-АД в качестве исходного соединения в синтезе 17β-циан-17α-гидрокси-производного.
Авторам патента [DD281394, 1990] удалось получить 17β-циан-17α-гидрокси-эпимер с выходом 93%, исходя из ∆9(11)-АД, в среде водного спирта, содержащей ацетон, используя цианид натрия в качестве цианирующего агента и метод эпимеризации, сущность которого состояла в постепенном осаждении 17β-циан-эпимера изменением концентрации воды. Аналогичный метод получения циангидрина ∆9(11)-АД с применением цианида калия описан в патенте [US4548748, 1985], смеси ацетонциангидрина и цианида калия - в патенте [US4500461, 1985, пример 2].
Таким образом, о применении одного ацетонциангидрина для стереоселективного гидроцианирования ∆9(11)-АД информация нами не найдена.
Защита 3-кетогруппы
Для предотвращения возможного алкилирования 3-кетогруппы проводят ее защиту. Защита 3-кетогруппы может быть осуществлена образованием енолэфира (метилового или этилового) [J. Am. Chem. Soc., 1953, 75(3), 650-653; J. Org. Chem., 1961, 26(10), 3925-3928; US3516991, 1970; US4500461, 1985, EP263569, 1988; US4921638, 1990], циклического кеталя [US3516991, 1970; US4500461, 1985; http://www.springerlink.com/content/106464/?p=077e6ed5e8a848f38bd231d4c7dbdf27&pi=0, 1984, 20(3),302-304; JPS5762296, 1982; JPS5762298, 1982; JPS5762299, 1982; JPS5762300, 1982; Tetrahedron Letters, 1990, 31(26), 3669-3672] или енамина [US3629298, 1971]. Этиленкетальная защита нашла наибольшее применение и чаще других упоминается в литературных источниках. Так, в работах [US4500461, 1985; JPS5762296, 1982; , 1987, 21(4), 225- 300; EP263569, 1988] использована защита 3-кетогруппы образованием пятичленного 1,3-диоксолановаго цикла реакцией с этиленгликолем. Однако в синтезе 3.3-этилендиокси-17α-гидрокси-17β-цианоанодроста-5,9(11)-диена описано введение этиленкетальной защиты до стадии гидроцианирования [JPS5762296, 1982], а не после стадии гидроцианирования, как предлагается в заявляемом способе.
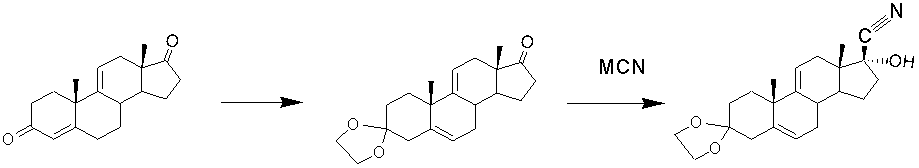
Способ гидроцианирования 17-кетона после защиты 3-кетогруппы ∆9(11)-АД образованием 3-метилового эфира в синтезе гидрокортизона из 9-ОН-АД описан в работе [Tetrahedron Lett., 1990, 31(26), 3669-3672].
Таким образом, описание примера защиты карбонила при атоме С3 образованием, например, этиленкеталя после получения 17α-гидрокси-17β-циан-производного из ∆9(11)-АД (циангидрина ∆9(11)-АД) в литературных источниках нами не найдено.
О применении 2,2-диметил-1,3-пропандиола (неопентилгликоля) для защиты 3-кетогруппы молекулы АД с образованием 1,3-диоксанового цикла впервые сообщено в патентах [RU2156255, 2000; RU2156256, 2000]. Однако, несмотря на то, что в общей формуле изобретения заявлена применимость этой защиты при алкилировании циангидрина ∆9(11)-АД, примеры получения и использования 1,3-диоксанового кеталя циангидрина ∆9(11)-АД отсутствуют.
Как в случае введения диоксановой, так и при образовании диоксолановой защиты применяют триалкилортоформиаты (триэтил- и триметилортофрмиат) [RU2156256, 2000] или диэтилацеталь диметилформамида. Это позволяет сдвинуть равновесие в реакции кетализации за счет химического связывания воды, образующейся в процессе реакции. В качестве катализатора используют п-ТСК или сульфосалициловую кислоту. Для кетализации 17α-гидрокси-17β-цианандрост-4-ен-3-она может быть применен классический способ кипячения с этиленгликолем в бензоле в присутствии каталитического количества п-ТСК с азеотропной отгонкой образующейся воды [Chemistry of Natural Compounds, 1984, 20(3), 302-304]. Однако он менее эффективен, чем описанный выше.
Защита 17α-гидроксигруппы
Известно, что в условиях алкилирования циангидринная группа может элиминироваться с регенерацией карбонильной группы с последующим образованием продуктов ее алкилирования. Содержание последних может достигать значительных количеств, что приводит к потере выхода и загрязнению целевого соединения. Для предотвращения этих нежелательных процессов прибегают к защите 17α-гидрокси-группы образованием эфира. В качестве этерифицирующего агента используют алкилвиниловые эфиры (этилвиниловый [US4500461. 1985; US4548748, 1985] или бутилвиниловый [JP57062296, 1982]). Также известны способы защиты образованием 17-тетрагидропиранилового эфира [J. Am. Chem. Soc., 1959, 81 (21), 5725-5727; EP263569, 1988] реакцией с 2,3-дигидропираном в условиях кислого катализа (п-ТСК или оксихлорид фосфора) или триметилсилилового эфира реакцией с триметилхлорсиланом [Tetrahedron Lett. 1971, 2005-2008]. Однако в процессе образования 17-тетрагидропиранилового эфира наблюдается частичная изомеризация 17β-циан-соединения с увеличением содержания примеси нежелательного 17α-циан-17β-тетрагидропиранилокси-изомера. Триметилсилильная же защитная группировка также нежелательна, так как является громоздкой и проявляет экранирующий эффект на последующей стадии алкилирования нитрильной группы. Это приводит к необходимости применения жестких условий алкилирования (длительное кипячение в ТГФ или применение высококипящих растворителей, например, анизола) и сопровождается побочной реакцией по 1,3-диоксолановому кольцу с образованием продукта его расщепления по С-О-связи 3ξ-метил-3ξ-(оксиэтокси)-прегнана с высоким выходом (в анизоле, например, с количественным) [Chemistry of Natural Compounds, 1984, 20(3), 302-304; J. Chem. Soc., Perkin Trans.I, 1993, (7), 783-793]. Поэтому наиболее приемлемой, по нашему мнению, является защита 17-гидроксигруппы 17β-карбонитрила образованием 17α-(1-алкокси)этилового эфира.
Введение 20-кето-прегнановой боковой цепи алкилированием карбонитрильной группы
Дальнейшее превращение 17β-циан-17α-гидрокси-соединения в 20-кетопрегнаны осуществляют взаимодействием соответствующих циангидринов с защищенными 3-кето- и 17α-гидрокси- группами с метиллитием [EP263569, 1988; Pharmaceutical Chemistry Journal, 1987, 21(4), 297-300; Bull. Chem. Soc. Jpn., 1985, 58(3), 978-980] или метилмагнийбромидом [Chemistry of Natural Compounds, 1984, 20(3), 302-304].
Наиболее близким по сущности к заявляемому способу в части построения прегнановой боковой цепи на основе 17-кетоандростанов с применением циангидринного метода, циклической защиты 3-кетогруппы и защиты 17α-гидроксильной группы образованием 17α-(1-алкокси)этилового эфира является способ, описанный в патенте [RU2156255, 2000].
Однако метод в формуле патента [RU2156255, 2000] заявлен в общем виде. Примерами подтверждено получение 17α-гидроксипрогестерона из АД. Применение в качестве исходного соединения ∆9(11)-АД не подтверждено примерами. ∆9(11)-АД не является гомологом АД. Как известно специалисту, работающему в данной области техники, и как упоминалось выше, 9,11-двойная связь является функциональной реакционн-оспособной группой, то есть дополнительным реакционным центром, поэтому технический результат, полученный на примере АД, не может быть очевидным при использовании ∆9(11)-АД в качестве исходного соединения.
Последовательность синтеза 17α-гидроксипрегна-4,9(11)-диен-3,20-диона (∆9(11)-ГПГ) из ∆9(11)-АД по настоящему изобретению в доступных источниках литературы не описана.
Введение гидроксильной группы в 21-положение 17α-гидрокси-С20-кетопрегнана с образованием кортикостероидной (диоксиацетоновой) цепи
Известно, что в синтезе кортикостероидов из 17α-гидроксипрегнанов предварительной стадией, как правило, является галоидирование 21-положения с образованием 21-галоид-17α-гидроксипрегнанового интермедиата, который далее подвергают реакции замещения на ацетоксигруппу действием ацетата щелочного металла.
Известен способ получения 21-ацетата вещества «S» из 17α-гидроксипрогестерона с предварительно защищенной 3-кетогруппой образованием N-морфолинил-производного, который заключается в том, что 20-кетопрегнан подвергают α-галоидированию по положению С21 путем введения атома брома в присутствии HCl, а затем полученную смесь 21-бром- и 21-хлор-производных обрабатывают ацетатом калия в присутствии небольшого количества иодистого натрия в среде ацетона [Хим. фарм.ж. 1990, 24(11), 55-57].
Синтез 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона (21-ацетокси-∆9(11)-ГПГ) также может быть осуществлен из 21-бромпроизводного ∆9(11)-ГПГ [US4041055, 1977] или 21-хлорпроизводного [US4342702,1982; US4357279, 1982] реакцией последовательного замещения атома брома или хлора сначала на атом иода реакцией с иодидом калия, а затем - на ацетоксигруппу взаимодействием 21-иод-∆9(11)-ГПГ с ацетатом калия в среде ацетона [US4041055, 1977] или смеси ацетона и дихлорметана [US4342702,1982].
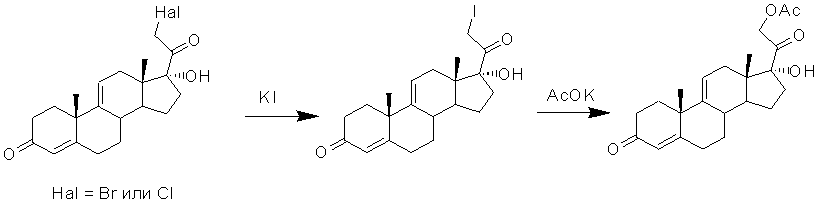
Однако при переходе от ∆9(11)-ГПГ к 21-бром- или 21-хлор-производному необходима защита ∆4-3-кетосистемы, для чего используют образование метилового или этилового 3,5-диенолэфиров, пирролидинового или морфолинового енаминов, или кетальной защиты. В противном случае наблюдается образование побочного продукта α-галоидирования (например, бромирования) по атому С2 и при большем избытке брома - продукта галоидирования и в положение С6.
 Последующая реакция замещения атома иода на ацетоксигруппу может быть проведена с применением как ацетата калия, так и ацетата натрия. Кроме того, может быть использован ацетат триэтиламина в среде ацетона [CN101125877, 2008] с общим выходом на стадиях иодирования и ацетоксилирования до 88% [Bull. Chem. Soc. Jpn., 1985, 58, 1081-1082].
Последующая реакция замещения атома иода на ацетоксигруппу может быть проведена с применением как ацетата калия, так и ацетата натрия. Кроме того, может быть использован ацетат триэтиламина в среде ацетона [CN101125877, 2008] с общим выходом на стадиях иодирования и ацетоксилирования до 88% [Bull. Chem. Soc. Jpn., 1985, 58, 1081-1082].
Способ 21-ацетоксилирования прегнанов с применением метода прямого иодирования без защиты ∆4-3-кетогруппировки также хорошо известен. Впервые этот метод был предложен Хоггом и соавторами в 1955 г. [J. Am. Chem. Soc., 1955, 77(16), 4436-4438] для получения 21-ацетата кортексолона и ацетата гидрокортизона из 17-гидроксипрогестерона, согласно которому С21-метильная группа стероидной молекулы реагирует с иодом в присутствии основания и образованное 21-моно-иодпроизводное в результате обработки ацилатом щелочного металла превращается в соответствующий 21-ацилоксистероид.
Наибольшее применение метод прямого иодирования получил в производстве ацетата кортексолона из 17α-гидроксипрогестерона [J. Am. Chem. Soc., 1958, 80(1), 250; http://www.springerlink.com/content/106490/?p=97444ae762a94bcfab26466d7f1502ed&pi=0, 1990, 24(11), 839-842; http://www.springerlink.com/content/0091-150x/" \o "Link to the Journal of this Article 1969, http://www.springerlink.com/content/0091-150x/3/12/" \o "Link to the Issue of this Article), 709-712; SU151331, 1966; RU2156256, 2000]. В патенте [GB882388, 1961] описано проведение этого процесса на 6-метилзамещенном 17α-гидроксипрогестерона, в патенте [US5565588, 1996] - на 9,17α-дигидроксипрогестероне.
Наиболее близким по сущности к заявляемому способу в части последовательности построения диоксиацетоновой боковой цепи на основе 17-кетоандростанов, а именно с применением циангидринного метода, циклической защиты 3-кетогруппы, защиты 17α-гидроксильной группы образованием 17α-(1-алкокси)этилового эфира, алкилирования нитрильной группы и 21-ацетоксилирования с применением метода прямого иодирования, является способ, описанный в патенте [RU2156256, 2000].
В патенте [RU2156256, 2000] заявлено применение метода прямого иодирования в синтезе кортикостероидов с образованием 21-моно-иодпроизводного и последующего ацетоксилирования в условиях Л.Г. Гаценко и В.Н. Петрова [Хим. фарм. ж., 1972, 10, 27]. Однако метод в формуле патента [RU2156256, 2000] заявлен в общем виде. Примерами подтверждено получение 21-ацетокси-17α-гидроксипрогестерона из АД. Примеры получения 21-иод-∆9(11)-ГПГ и далее 21-ацетокси-∆9(11)-ГПГ отсутствуют.
В литературных источниках отсутствует информация о получении 21-иод-∆9(11)-ГПГ методом прямого иодирования ∆9(11)-ГПГ.
Последовательность синтеза 21-ацетокси-∆9(11)-ГПГ из ∆9(11)-АД через образование 21-иод-∆9(11)-ГПГ по настоящему изобретению в доступных источниках литературы не описана.
Дегидратация 17α-гидроксигруппы с образованием ∆16-3,20-дикетопрегнана
Для перехода от 17α-гидрокси-20-кетопрегнана к соединению с ∆16-связью, необходимой для введения 17α-гидрокси-16α-метильного фрагмента, проводят дегидратацию 17α-гидроксигруппы.
Дегидратация 17α-гидроксигруппы в соответствующий 16,17-дегидропродукт может быть проведена, например, смесью оксихлорида фосфора и пиридина.
Известен способ введения ∆16-связи в молекулу 21-ацетокси-∆9(11)-ГПГ [US3493563, 1970]. Способ состоит в обработке 17-гидроксисоединения тионилхлоридом в пиридине и предусматривает предварительную защиту 3-кетогруппы кетализацией, а 20-кетогруппы - образованием метоксииминопроизводного.
Однако постадийные выходы интермедиатов невысокие.
В 1970 г. Salse, Hazen и Schoenewaldt сообщили, что нагревание 17α-ацилокси-производных 20-оксостероидов при температуре 100-105ºС в содержащем ацетат калия диметилформамиде в течение 7 ч приводит к элиминированию ацилокси-функции с образованием 16-дегидро-20-оксостероидов [J. Org. Chem., 1970, 35, p 1681; US 3631076, 1971]. Важным условием протекания этой реакции является наличие сложноэфирной группы при атоме С21, что было установлено в результате исследования влияния природы заместителя при С21 на элиминирование 17α-ацилоксигруппы у 17α-ацилокси-20-оксостероидов, проведенного Solo и Suto в 1980 г. [J. Org. Chem., 1980, 45, 2012].
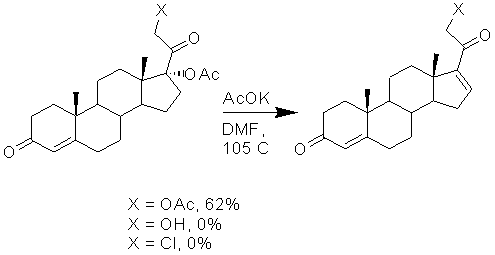
При этом выход продукта при наличии 21-ацетоксигруппы не превышал 62%. В присутствии 21-гидроксигруппы происходила ацильная миграция ацетоксигруппы из положения С17 в положение С21, а в случае 21-хлорзамещенного реакция элиминирования не шла вовсе.
Этерификацию пространственно затрудненной третичной 17α-гидроксигруппы проводят ацилированием преимущественно 21-ацетатов 20-оксопрегнанов смесью карбоновой кислоты и ангидрида карбоновой кислоты в присутствии кислотных катализаторов при температуре 18-35ºС. В качестве катализаторов могут быть использованы п-ТСК, 55-70% хлорная кислота, серная кислота и др. [SU668288, 1994; GB1375770, 1974]. Основной побочной реакцией 17-ацилирования ∆4-3-кетосоединений является енолацилирование по атому С3, которое протекает в тех же условиях кислотного катализа. С3-Ацилат может быть затем гидролизован с регенерацией ∆4-3-кетоструктуры. Однако в случае кортикоидов - соединений с диоксиацетоновой боковой цепью при С17 - при гидролизе 3-ацилата возникает новая проблема: гидролиз 21-ацетоксигруппы, ацильная миграция сложноэфирной группы из 17 положения к атому С21, и ее дальнейший гидролиз с образованием 17α,21-дигидроксисоединения.
Кроме того, реакция 17-ацетилирования может быть проведена в условиях основного катализа [US3678082, 1972] диметиламинопиридином в системе Ас2О/триэтиламин. Однако в патенте примеры использования этого метода для 17-ацетилирования кортикоидов отсутствуют.
Этерификация стерически затрудненных гидроксильных групп без затрагивания ∆4-3-кетогруппы возможна при проведении реакции в среде Ас2О в присутствии СаО при температуре кипения [JP53009755, 1978]. Кроме СаО, авторы использовали Al2O3, ZnO, а также ацетаты магния, кальция, марганца, цинка или бария. Однако получение 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона этим способом в формуле и описании отсутствует.
В патенте [DE2055221, 1972] описано проведение ацилирования 17α-гидроксипрегнанов с высоким выходом с использованием ангидрида или ацилхлорида в смеси с безводным SnCl4 в среде нитрометана. Могут быть также использованы галоидиды или ангидриды карбоновых кислот в присутствии фосфатного катализатора (например, H3PO4 или ее моно- или диалкиловых эфиров), например Ac2O/H3PO4. Однако описание получения 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона в этом патенте отсутствует.
Известен способ ацетилирования 17α-гидроксигруппы молекулы 21-ацетокси-∆9(11)-ГПГ [US4154748, 1979, пример 1], согласно которому образование 17α,21-ацетоксипрегна-4,9(11)-диен-3,20-диона проводят действием уксусного ангидрида в условиях катализа производными фосфорной кислоты. Отмечено, что при катализе 85% фосфорной кислотой (Ас2О/АсОН) продукт получен с выходом 97,5% и содержанием примесей исходного соединения и 3-енолацетата в следовых количествах.
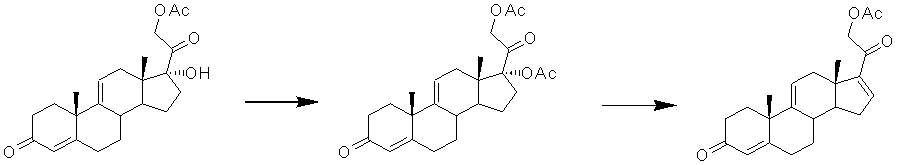
Наиболее близким по сущности метода ацетилирования, использованного в настоящем изобретении, является способ [GB1097164, 1967]. Ацилирование 17α-гидроксигруппы в присутствии 21-ацетоксигруппы проводят методом смешанных ангидридов, который заключается в обработке исходного стероида смесью трифторуксусного ангидрида и кислоты, получение сложного эфира которой является целью реакции, при температуре 80ºС в течение 1 ч [GB1097164, 1967]. Однако получение 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона этим способом в формуле и описании отсутствует. Более того, применение метода смешанных ангидридов для ацетилирования 17α-гидроксигруппы у 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона в доступных литературных источниках не найдено.
Известен способ [US3631076, 1971], согласно которому 17,21-диацетат может быть конвертирован в ∆16-производное реакцией с ацетатом калия при температуре 100°С в течение 7 ч в среде диметилформамида. Однако пример получения 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона в этом патенте не описан и выходы продуктов не указаны.
Более эффективный метод элиминирования 17α-ацетоксигруппы описан в патенте [SU 819119, 1981], который заключается в обработке исходного стероида ацетатом калия в среде диметилсульфоксида, и процесс проводят при температуре 80-85°С. Однако получение 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона этим способом в формуле и описании отсутствует.
Введение 1,2-двойной связи микробиологическим способом
Микробиологические способы проведения процесса 1,2-дегидрирования стероидов, включая стероиды прегнанового ряда, известны.
В качестве биокатализаторов процесса 1,2-дегидрирования используют культуры с 3-кетостероид-∆1-дегидрогеназой (3-КСД) (ЕС 1.3.99.4) родов: Arthrobacter, Alternaria, Alcaligenes, Calonectria, Ophiobolus, Corynebacterium, Bacillus, Nocardia, Streptomyces, Bacterium, Mycobacterium, Fusarium, Cylindrocarpon, Pseudomonas, Protaminobacter, Septomyxa, Didymella, Rhodococcus и др. [Charney W., Herzog H. Microbial transformation of Steroids. Academic Press. Inc. New York. 1967, p. 236-261; J. Basic. Microbiol. 1990, 30(6), 415-423; J. Biochem., 1999, 126, 662-667]. Наиболее часто для проведения процесса 1,2-дегидрирования стероидных соединений ряда прегнана и андростана используются штаммы вида Arthrobacter simplex.
Особенностью процесса 1,2-дегидрирования ацетилированных производных 21-гидрокси-∆4-3-кетостероидов микроорганизмами является сопровождение реакции 1,2-дегидрирования дезацетилированием (гидролизом сложноэфирной группы) [Steroids, 2003, 68(5), 415-421]. Это является существенным недостатком известных методов, так как появляется необходимость проведения реакции ацетилирования гидроксильной группы при С21.
Наиболее близким по сущности к предложенному способу в части введения 1,2-двойной связи является метод, описанный в патенте [US4524134, 1985, пример 4]. Авторы используют растущую культуру Arthrobacter simplex для 1,2-дегидрирования 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона с нагрузкой стероида 8 г/л в среде фосфатного буфера (рН 7,5) в присутствии менадиона, а стероидный субстрат вносят в среду для трансформации в растворе диметилформамида, концентрация которого в среде составляет 2 об.%. Однако 1,2-дегидропродукт получен лишь с выходом 18,1%, при этом содержание основного вещества составляет всего 59,1%. В продукте также содержится 40,9% нетрансформированного субстрата и значительное количество других нежелательных стероидных соединений. Авторы отмечают, что указанные недостатки связаны с использованием растущей культуры и их можно избежать, если использовать высушенные клетки в тех же условиях трансформации. Способ получения 1,2-дегидро-Δ4-3-кетостероидов, включающий обработку соответствующего 1-насыщенного Δ4-3-кетостероида высушенными клетками Arthrobacter simplex, содержащими от 1 до 10% влаги, причем клетки высушены в отсутствие какого-либо органического растворителя, описан в этом же примере [US 4524134, 1985, пример 4]. При этом 1,2-дегидро-Δ4-3-кетостероид или 21-моноэфир выбраны из группы, включающей и 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-дион (п.2 формулы). Согласно этому способу процесс 1,2-дегидрирования 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона проводят в фосфатном буфере (рН 7,5) высушенными клетками (10 г/л) с нагрузкой стероида 8 г/л в присутствии менадиона, а стероидный субстрат вносят в среду для трансформации в растворе диметилформамида, концентрация которого в среде составляет 2 об.%. Продолжительность инкубирования 24 ч при температуре 31°С. Однако, как отмечено в примере 3, в результате биоконверсии 21-ацетилированных стероидов получают смеси, состоящие из 21-ацетокси-Δ1,4-соединений и их 21-гидрокси-аналогов. Так, выход смеси продуктов - 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона (21-ацетата тетраена) и 21-гидроксипрегна-1,4,9(11),16-тетраен-3,20-диона (тетраена) - с использованием высушенных клеток составил 79,6%, содержание основного вещества, имеющего Δ1,4-структуру, 97,8%, содержание остаточного субстрата 2,1%.
Таким образом, основным недостатком указанного способа является необходимость проведения предварительного высушивания клеток до содержания влаги от 1 до 10% в отсутствие органического растворителя. Как описано в примере 2 патента [US 4524134, 1985] клетки культуры после инкубации подвергают центрифугированию, а затем высушиванию в вакууме при температуре 55°С в течение 24 ч. После чего высушенный клеточный материал помещают в герметичный контейнер и хранят при 50°С до использования в биоконверсии. Процесс высушивания клеток сложен и энергоемок. Кроме этого, несмотря на высокий выход продукта трансформации при использовании высушенных клеток (1,25 г клеток на 1 г стероидного субстрата), он содержит смесь 21-ацетата тетраена и его 21-дезацилированного аналога. Таким образом, анализ материалов, представленных в патенте, свидетельствует о том, что процесс 1,2-дегидрирования 21-ацетилированных производных ∆4-3-кето-стероидов ряда прегнана, как с применением высушенных клеток, так и растущей культурой, неселективен. 1,2-Дегидрирование сопровождается дезацетилированием. В проанализированном информационном материале отсутствуют данные о количественном выходе 1,2-дегидрированного ацетилированного целевого продукта. Не указана степень гидролиза и соотношение дегидрированного 21-ацетилированного и дегидрированного 21-дезацетилированного продуктов, что является существенными недостатками данного способа.
Наиболее близким к заявляемому способу в части введения 1,2-двойной связи по техническому результату является способ получения 1,2-дегидро-3-кетостероида, который заключается в том, что 1,2-насыщенный 3-кетостероид подвергают взаимодействию с A. simplex или Bacterium cyclooxydans в присутствии экзогенного носителя электронов и несмешивающегося с водой растворителя, включая ароматические углеводороды [GB2131811, 1984]. Согласно этому способу (пример 1) процесс трансформации 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона с нагрузкой 8 г/л проводят клетками A. simplex в присутствии 1% вес. менадиона в среде фосфатного буферного раствора (рН 7.5), содержащей 10 об.% толуола, в течение 25 ч при температуре 28±1°С. Толуольная фаза содержит 94,3% 21-ацетата тетраена, 4,2% тетраена и 1,5% исходного 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона. Однако в описании отсутствует информация о результате выделения продукта и, соответственно, о его выходе и качестве.
Также известен способ превращения 1,2-насыщенных 3-кетостероидов в 1,2-дегидро-3-кетостероиды, который заключается в том, что 1,2-насыщенные 3-кетостероиды подвергают взаимодействию со средством, имеющим стероид-1,2-дегидрогеназную активность A. simplex или B. cyclooxydans, в присутствии экзогенного носителя электронов и одного или более дополнительного поглотителя токсичного кислорода, выбранного из группы, содержащей каталазу, супероксиддисмутазу или платину [US4749649, 1988]. При этом 1,2-насыщенный 3-кетостероид является 1,2-насыщенным 3-кетопрегнаном (п.9 формулы), выбранным из группы, включающей и 21-ацетоксипрегна-4,9(11),16-триен-3,20-дион. Согласно этому способу процесс проводят в буферной водной системе, имеющей молярность от 0,01 М до 2,0 М и содержащей несмешиваемый с водой растворитель (толуол, ксилол или бензол) и препарат, обладающий стероид-1-дегидрогеназной активностью, каковым может быть культуральная жидкость, влажная паста клеток с содержанием влаги от 60% до 85% или сухая клеточная масса с содержанием влаги от 1 до 30%. Рассматриваемый способ [US4749649, 1988] предлагает также использование экзогенного фермента - каталазы. Однако способ проиллюстрирован единственным примером трансформации соединения ряда прегнана - гидрокортизона (пример 3) - в среде фосфатного буфера (50 mM) с применением сухих клеток и нагрузкой субстрата 0,5 г/л. При этом после 23 ч инкубации в присутствии каталазы конверсия гидрокортизона в преднизолон составляет 69%, а без каталазы - на 27% ниже. Удорожание технологии, обусловленное необходимостью добавления в среду поглотителя (фермента каталазы, или супероксиддисмутазы, или платины), устраняющего ингибирующее влияние синглетного кислорода на активность дегидрогеназы, определяет основной недостаток данного способа, что ограничивает применение данного способа в промышленном масштабе. Кроме того, несмотря на упоминание возможности 1,2-дегидрирования 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона известным способом, описание получения 21-ацетата тетраена отсутствует.
Таким образом, общими недостатками способов получения 21-ацетата тетраена микробиологическим 1,2-дегидрированием его Δ4-3-кетопредшественника в двухфазной среде с применением несмешивающихся с водой органических растворителей являются:
• низкая концентрация субстрата (до 8 г/л), большая продолжительность процесса трансформации (не менее 24 ч) и отсутствие информации о способе выделения, а также о выходе и качестве выделенного продукта;
• использование больших объемов легко воспламеняющихся органических растворителей на этапе трансформации, что требует специального ферментационного оборудования и соблюдения особых правил противопожарной безопасности; требование применения специального технологического оборудования фактически исключает практическую реализацию этих способов в условиях промышленного производства;
• сложность проведения технологического процесса в условиях необходимого регулирования состава газовой смеси, подаваемой в реакционную среду: необходимость создания и строгого поддержания минимально допустимой концентрации растворенного кислорода в среде, чтобы избежать вспышки паров газовоздушной смеси и в то же время обеспечить оптимальные условия для процесса окисления субстрата;
Еще одним близким по сущности к предложенному способу в части введения 1,2-двойной связи является метод, включающий трансформацию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона [J. Steroid Biochem. Mol. Biol., 2003, 87(4-5), 434-439]. Биоконверсию проводят без добавок метил-β-циклодекстрина (МЦД) в среде 0.01 М натрий-фосфатного буферного раствора (рН 7.4-8.1) при температуре 30°С. Исходный субстрат (нагрузка 1,0 г/л) добавляют в виде микронизированного порошка. При конверсии субстрата нативными клетками при содержании биомассы 330 мг/л наблюдается активное дезацетилирование, сопровождающееся последующим 1,2-дегидрированием. Через 20 ч трансформации происходит накопление 70% дезацетилированного 1,2-дегидрированного продукта тетраена, 30% составляет содержание дезацетилированного субстрата при полной конверсии загруженного субстрата.
Использование МЦД позволяет значительно повысить эффективность этого процесса [J. Steroid Biochem. Mol. Biol., 2003, 87(4-5), 434-439]. При конверсии субстрата (1,0 г/л) нативными клетками при содержании биомассы 330 мг/л и мольном соотношении стероид:МЦД, равном 1:2, в присутствии ФМС (0.4 мг/мл) максимальное накопление 21-ацетата тетраена наблюдается через 1 ч инкубации и составляет 75%. Последующее снижение его содержания до нулевого значения коррелирует с накоплением дезацетилированного 1,2-дегидрированного продукта - тетраена, - достигающего 90% за 5 ч. При этом содержание дезацетилированного субстрата - 21-гидроксипрегна-4,9(11),16-триен-3,20-диона - не превышает 10% (субстрат трансформируется полностью за 5 ч инкубации). Для снижения вклада дезацетилирования в начальные часы конверсии используют низкую биомассу, равную 33 мг/мл; при этом конверсия субстрата (1,0 г/л) при мольном соотношении стероид:МЦД, равном 1:2, в присутствии ФМС (0,4 мг/мл) завершается за 5,5 ч с содержанием 92% 21-ацетата тетраена и менее чем 5-7% его дезацетилированного аналога.
Основным недостатком этого процесса является низкая концентрация исходного субстрата (1 г/л).
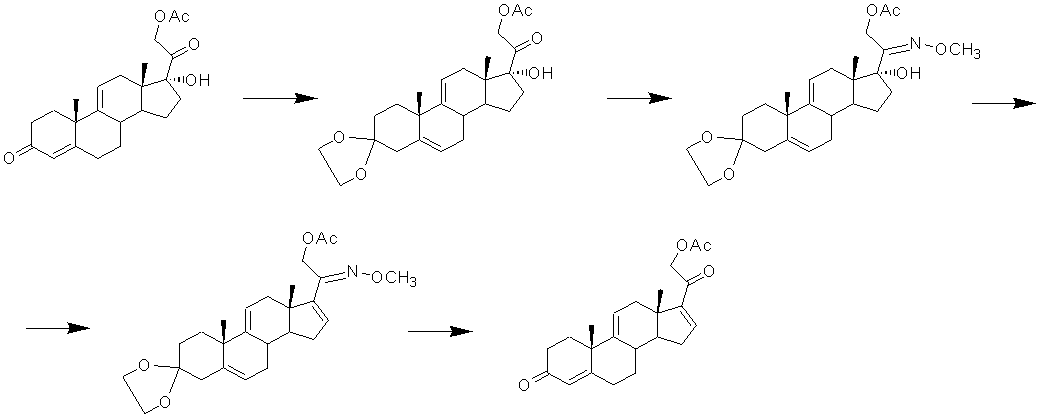
Введение 16α-метил-17α-гидрокси- фрагмента по ∆16 связи
К настоящему времени в литературных источниках имеется скудная информация о синтезе 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (1,4,9-триена) из 21-ацетата тетраена. Имеются литературные данные о функционализации положений С16 и С17 в 21-ацетокси-соединениях с кросс-сопряженной системой в кольце А. При этом введение Δ9(11)-связи обычно осуществляется в последнюю очередь.
Так, известен способ получения 1,4,9-триена [US3284477, 1966]. Однако синтез осуществляют из 21-ацетата 16α-метилпреднизолона дегидратацией 11β-гидроксильной группы действием метансульфонилхлорида в среде пиридина.
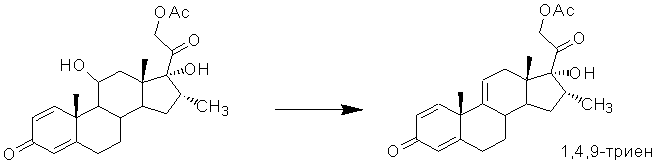
1,4,9-Триен является ключевым интермедиатом в синтезе кортикостероидов из 9-ОН-АД, имеющих метильную группу в α-конфигурации при атоме С16 и гидроксильную группу при атоме С17, например дексаметазона, икометазона (9-хлор-аналога дексаметазона), флуметазона (6α-фтор-дексаметазона), параметазона (6α-фтор-16α-метилпреднизолона), мометазона (11,17-дигидрокси-9,21-дихлор-16-метилпрегна-1,4-диен-3,20-диона) и др.
Введение 17α-гидрокси- и 16α-метил- заместителей в -∆16-20-кетопрегнан осуществляют, как правило, двухступенчатым процессом, первоначально проводя 16α-алкилирование. Введение 16α-метильной группы к Δ16-20-кето-фрагменту стероидной молекулы обычно осуществляют реакцией 1,4-присоединения метилмагнийгалогенида в присутствии соединений меди.
Известен процесс 16α-алкилирования 21-ацетата тетраена с образованием 21-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона, его последующее 21-ацетилирование [CA1029659, 1978].
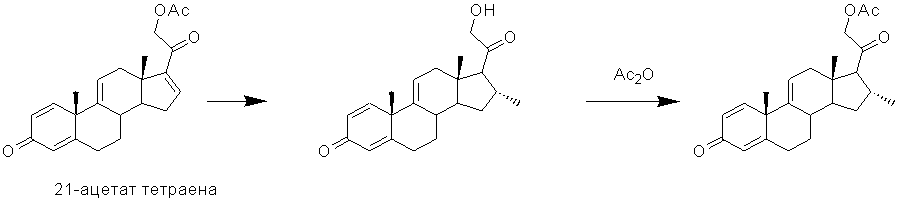
Однако из 100 г 21-ацетата тетраена авторы получают с использованием хроматографической очистки и перекристаллизации из этилацетата всего лишь 22,7 г 16α-метилпроизводного.
17α-Гидроксигруппа может быть введена в 16α-метил-Δ4-3,20-дикетосоединение микробиологической трансформацией с культурой Dactylium dendroides [GB891594, 1962]. Однако в литературных источниках отсутствует информация о возможности микробиологического 17-гидроксилирования 16α-метил-Δ1,4,9(11)-3,20-дикетосоединений.
Известно, что 17α-гидроксилирование 20-кетопрегнанов может быть проведено, например, методом прямого окисления по Бартону [GB963287, 1964] или методом Галлахера и Кричевского [US2562030, 1951]. В основе этих методов лежит идея окисления ∆17(20)-20-гидроксипроизводного, полученного предварительной енолизацией кетогруппы при атоме С20. В первом случае окисление проводят кислородом в присутствии трет-бутилата калия, во втором - действием надкислоты. Однако енолацетилирование 20-кетогруппы у 16α-метилстероидов затруднено. Реакция протекает медленно и с низким выходом.
Известны способы получения 1,4,9-триена из 21-ацетата тетраена с применением модифицированного метода Галлахера и Кричевского, который заключается в обработке 16α-метил-∆17(20)-20-енолята ацилирующим агентом [EP0165037, 1985, примеры 1 и 2; US4929395, 1990, примеры 1 и 2]. Однако выход 1,4,9-триена в обоих патентах не указан, и эффективность метода оценить невозможно.
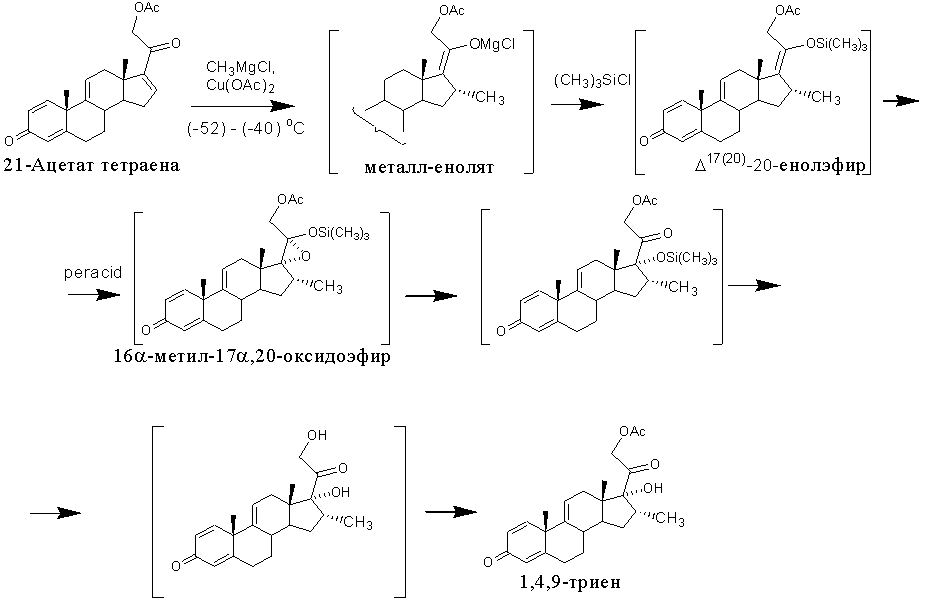
Известен метод получения 1,4,9-триена из 21-ацетата тетраена [US4530795, 1985, пример 2], включающий образование енолсульфината, способного подвергаться сульфенат-сульфоксидной перегруппировке. Последняя происходит самопроизвольно при комнатной температуре с образованием сульфенатного эфира, разложение которого небольшим избытком тиофила (метилксантат или тиоцианат натрия) приводит к 17α-гидрокси-16α-метилсоединению (в частности, к 1,4,9-триену).
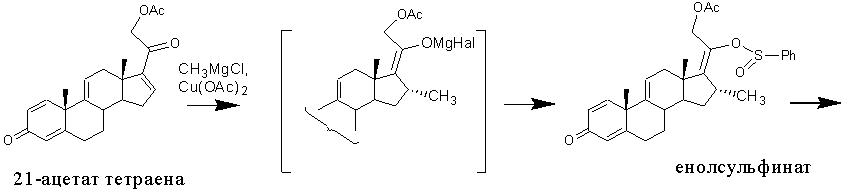
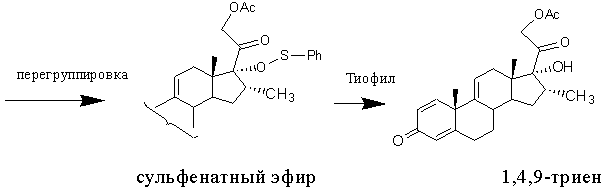
Этот способ позволяет устранить затруднения в енолацетилировании 16α-метил-20-кетопрегнанов, возникающие при 17α-гидроксилировании методом Галлахера и Кричевского, и, как следствие, приводит к повышению выхода 17α-гидрокси-16α-метилсоединений (из 3,66 г 21-ацетата тетраена получают 2.35 г 1,4,9-триена, что соответствует молярному выходу 59%). Однако этот способ имеет существенные недостатки: многостадийность, применение дорогостоящих и опасных реагентов, очистка продуктов реакции с применением колоночной хроматографии.
Известны способы введения 17α-гидрокси-16α-метилфрагмента в Δ16-20-кетостероиды восстановлением 17α-гидропероксипродуктов, полученных аутоокислением молекулярным кислородом промежуточных 16α-метил-∆17(20)-20-галоидмагниевых соединений [Lieb. Ann. Chem. 1971. B. 752(1). S. 78-85; DE1921396, 1970; GB2001990, 1979]. Известно, что промежуточные 16α-метил-∆17(20)-магниевые еноляты весьма реакционно-способны и легко окисляются кислородом воздуха с образованием 17α-гидроперокси-20-оксосоединений. Так, Керб и Вихерт [Lieb. Ann. Chem. 1971. B. 752(1). S. 78-85; DE1921396, 1970] показали, что 17α-гидропероксигруппа может быть легко введена одновременно с 16α-метильной группой в 18-метил-∆16-20-кетостероиды ∆5-ряда или ряда эстрана. При этом выход 17α-гидроперокси-16α-метил-соединения составлял от 40 до 65%.
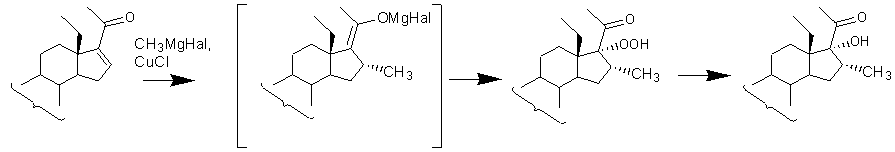 Восстановление 17α-гидропероксигруппы в 17α-гидроксильную может быть проведено известными методами, например, действием цинковой пыли в среде уксусной кислоты [DE1921396, 1970; DE2407967,1974; GB1377975, 1974; GB2001990, 1979] или каталитическим гидрированием над палладием на угле [DE1921396, 1970; US2562030, 1951; GB898093, 1962; GB2001990, 1979]. 17α-Гидропероксигруппа может быть восстановлена в 17α-гидроксильную триметилфосфитом [GB1196984, 1970] или триэтилфосфитом [Comptes Rendus, 1964, 258(23), 5669-5671; Tetrahedron, 1971, 27(10), 1909-1915]. В связи с неустойчивостью 17α-гидропероксисоединений обычно проводят их восстановление в 17α-гидроксианалоги без выделения из реакционной массы.
Восстановление 17α-гидропероксигруппы в 17α-гидроксильную может быть проведено известными методами, например, действием цинковой пыли в среде уксусной кислоты [DE1921396, 1970; DE2407967,1974; GB1377975, 1974; GB2001990, 1979] или каталитическим гидрированием над палладием на угле [DE1921396, 1970; US2562030, 1951; GB898093, 1962; GB2001990, 1979]. 17α-Гидропероксигруппа может быть восстановлена в 17α-гидроксильную триметилфосфитом [GB1196984, 1970] или триэтилфосфитом [Comptes Rendus, 1964, 258(23), 5669-5671; Tetrahedron, 1971, 27(10), 1909-1915]. В связи с неустойчивостью 17α-гидропероксисоединений обычно проводят их восстановление в 17α-гидроксианалоги без выделения из реакционной массы.
Известен способ получения производных 16α-метилпреднизолона, включая дексаметазон [GB2001990, 1979], с применением метода аутоокисления 16α-метил-∆17(20)-енолята и восстановления 17α-гидроперокси- в 17α-гидроксигруппу. Однако выход дексаметазона, полученного этими авторами, не превышает 26% вес., что косвенно свидетельствует о низком содержании 17α-гидропероксисоединения в техническом продукте. Кроме того, описание получения 1,4,9-триена из 21-ацетата тетраена известным способом отсутствует.
Наиболее близкими по сущности к заявляемому способу в части введения 16α-метил-17α-гидрокси- фрагмента по ∆16 связи являются способы получения 16α-метилированных стероидов [RU2125575, 1999] и [RU2127278, 1999], где функция кетона в положении 3 может быть защищена в виде кеталя, тиокеталя, гемитиокеталя или эфира енола. Получение 1,4,9-триена из 21-ацетата тетраена описано в примере 3 патента [RU2125575, 1999]. Этот способ включает метилирование 16-ненасышенного стероида в присутствии катализатора на основе меди, гидролиз полученного металл-енолята с образованием соответствующего 16α-метил-∆17(20)-енола, его окисление молекулярным кислородом в присутствии восстановителя и агента образования внутрикомплексного элементоорганического соединения меди и получение целевого соединения. В качестве катализатора используют моногидрат Cu(OAc)2 (2,73% вес. /5% мол.), реакцию 1,4-присоединения проводят в среде тетрагидрофурана действием 3 М раствора метилмагнийхлорида в эфире при температуре минус 20°С в течение 1 ч. По окончании алкилирования в реакционную массу при температуре 0°С добавляют 13,6% водный раствор гидрофосфата калия, а затем при температуре 15°С добавляют диметилсульфид в присутствии воздуха. В полученную смесь барботируют воздух в течение 18 часов, одновременно конденсируя перегоняемый диметилсульфид. 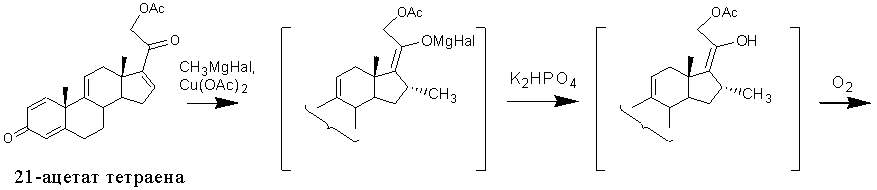
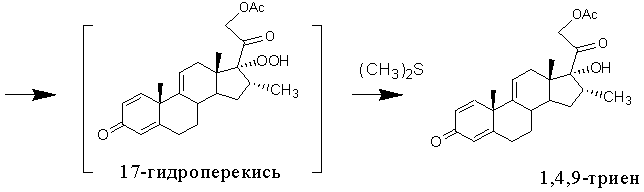
Реакционную массу экстрагируют этилацетатом. Продукт очищают хроматографией на силикагеле. Однако выход 1,4,9-триена крайне низок: из 0,733 г 21-ацетата тетраена авторы получили всего лишь 0,16 г 1,4,9-триена (что соответствует молярному выходу 20%).
Этой же компанией заявлен подобный способ получения 1,4,9-триена из 21-ацетата тетраена [RU2127278, 1999, пример 2], в котором гидролиз металл-енолята и аутоокисление проводят смесью фосфорной кислоты и едкого натра в присутствии перекиси водорода, а вместо диметилсульфида в качестве восстановителя используют суспензию надкислоты в ТГФ, приготовленную из фталевой кислоты и перекиси водорода. При этом из 0,733 г 21-ацетата тетраена авторы получают 0,4 г 1,4,9-триена (что соответствует молярному выходу 50%). Помимо низкого выхода целевого соединения существенным недостатком этого способа является применение опасных в промышленном производстве надкислот.
Таким образом, общим недостатком известных методов введения 16α-метил-17α-гидрокси- фрагмента по ∆16 связи является низкий выход целевого продукта.
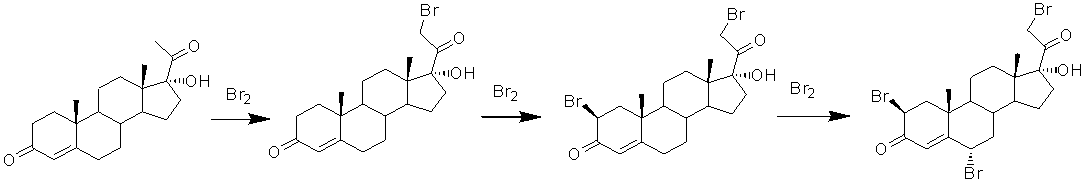
Введение 9α-фтор-11β-гидрокси-фрагмента по ∆9(11)-связи
Введение 9α-фтор- и 11β-гидрокси- фрагмента в Δ9(11)-стероиды осуществляют, как правило, трехступенчатым процессом, первоначально проводя гипогалоидирование (например, гипобромирование) Δ9(11)-связи, дегидрогалоидирование с образованием 9,11β-эпоксида, а затем раскрытие эпоксида фтористоводородной кислотой.
В реакции гипобромирования в качестве реагента используют дибромантин. Помимо дибромантина в качестве донора гипобромной кислоты могут быть использованы N-бромсукцинимид или N-бромацетамид [US3211758, 1965].
Замыкание в 9,11β-эпоксид может быть проведено обработкой 9α-бромпроизводного ацетатом калия в ацетоне [US3211758, 1965].
9,11β-Эпоксид реакцией с HF или с реагентом, содержащим HF, в безводных или водных условиях превращают в 9α-фтор-11β-гидроксисоединение. В качестве HF содержащего реагента может быть использован комплекс мочевины и фтористого водорода [US3211758, 1965], раствор безводного фтористого водорода в тетрагидрофуране [US3700702, 1972, US3780177, 1973]. Кроме того, синтез фторгидрина может быть осуществлен из Δ9(11)-соединения напрямую, минуя стадии гипобромирования и эпоксидирования. Однако в этом случае происходит образование значительного количества побочных соединений, что затрудняет очистку дексаметазона.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является упрощение и удешевление технологии получения дексаметазона из фитостерина.
Сущностью изобретения является оптимальное сочетание биотехнологических и химических стадий синтеза 11бета,17альфа,21-тригидрокси-16альфа-метил-9альфа-фторпрегна-1,4-диен-3,20-диона (дексаметазона) из фитостерина. Синтез дексаметазона из фитостерина представляет собой последовательность превращений, включающую следующие этапы функционализации:
- микробиологическое элиминирование боковой цепи фитостерина с образованием 9-ОН-АД культурой бактериальных клеток Mycobacterium sp. ВКМ Ac-1817Д;
- дегидратацию 9α-гидроксигруппы с образованием ∆9(11)-связи;
- введение 17α-гидрокси-17β-прегнановой цепи;
- введение гидроксильной группы в 21-положение 17α-гидрокси-С20-кетопрегнана с образованием кортикостероидной (диоксиацетоновой) цепи;
- ацетилирование 17α-гидроксигруппы;
- отщепление 17α-ацетоксигруппы с образованием ∆16-3,20-дикетопрегнана;
- микробиологическое 1,2-дегидрирование с применением клеток микроорганизма Nocardioides simplex ВКМ Ас-2033Д;
- введение 16α-метил-17α-гидрокси-фрагмента по ∆16 связи;
- введение 9α-фтор-11β-гидрокси-фрагмента по ∆9(11)-связи.
Технический результат настоящего изобретения заключается в повышении общего выхода целевого продукта, что обеспечивается заявляемой последовательностью проведения биотехнологических и химических реакций и совокупностью существенных признаков, каковыми являются:
Проведение дегидратации 9-ОН-АД, полученного из фитостерина микробиологической трансформацией, действием минеральной кислоты в среде апротонного органического растворителя предварительно выделенного в виде кристаллического порошка или в среде экстракта культуральной жидкости, из которой предварительно удалены бактериальные клетки штамма-трансформанта; при этом для экстракции 9-ОН-АД используют органические растворители, а именно ароматические углеводороды или хлорорганические углеводороды.
Использование в качестве дегидратирующего агента минеральных кислот, в том числе содержащих воду, более конкретно, использование ортофосфорной кислоты, или серной кислоты, или хлорной кислоты; удаление воды из реакционной массы в процессе реакции, например, азеотропной отгонкой.
Построение кортикостероидной (диоксиацетоновой) цепи проведением поэтапно реакций стереоселективного гидроцианирования, защиты кетона при атоме С3 образованием циклического кеталя, защиты 17α-гидроксигруппы образованием 17α-(1-алкокси)этилового эфира, алкилированием нитрильной группы, введением гидроксильной группы в 21-положение 17α-гидрокси-С20-кетопрегнана с использованием метода прямого С21-иодирования.
Ацетилирование 17α-гидроксигруппы методом смешанных ангидридов и элиминирование 17α-ацетоксигруппы с образованием ∆16-3,20-дикетопрегнана.
Введение 1,2-двойной связи микробиологическим способом в водной среде с применением клеток микроорганизма Nocardioides simplex ВКМ Ас-2033Д, с использованием полярного растворителя, смешивающегося с водой, в концентрации до 8 % об., в среде буферного раствора с рН от 5,0 до 5,5 при температуре 25°С в течение от 6 до 14 ч.
Введение 16α-метил-17α-гидрокси-фрагмента по ∆16 связи методом медь-катализируемого 1,4-присоединения реактива Гриньяра с последующим аутоокислением 16α-метил-Δ17(20)-енолята и восстановлением 17α-гидроперокси-группы в 17α-гидроксильную.
Введение 9α-фтор-11β-гидрокси-фрагмента по ∆9(11)-связи последовательностью, включающей гипобромирование, дегидробромирование и гидрофторирование образованного 9β,11-эпоксида.
Общий выход дексаметазона фармакопейного качества из фитостерина достигает 23%.
Предлагаемое техническое решение является новым (не известно из уровня техники), явным образом не следует из уровня техники, промышленно применимо.
Техническая задача получения дексаметазона из фитостерина решается способом получения 11β,17α,21-тригидрокси-16α-метил-9α-фторпрегна-1,4-диен-3,20-диона (дексаметазона) формулы (I)
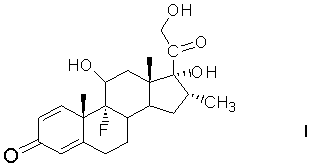
из фитостерина общей формулы (II)
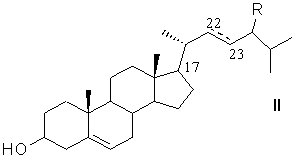
где R=C2H5 - β-ситостерин; R=CH3 - кампестерин; R=C2H5 и ∆22(23) - стигмастерин; R=CH3 и ∆22(23) - брассикастерин,
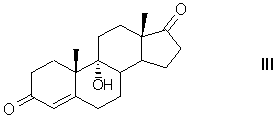
включающим микробиологическое окислительное элиминирование боковой цепи фитостерина при атоме С17 с образованием 9α-гидроксиандрост-4-ен-3,17-диона формулы (III)
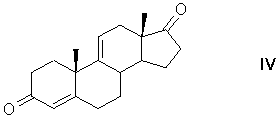
отделение биомассы, экстракцию 9α-гидроксиандрост-4-ен-3,17-диона (III) из осветленной культуральной жидкости, обработку 9α-гидроксиандрост-4-ен-3,17-диона дегидратирующим агентом в среде апротонного растворителя или в экстракте культуральной жидкости с образованием андроста-4,9(11)-диен-3,17-диона формулы (IV)
17-гидроцианирование андроста-4,9(11)-диен-3,17-диона взаимодействием с цианирующим агентом в среде протонного растворителя в условиях основного катализа c образованием 17α-гидрокси-17β-цианандроста-4,9(11)-диен-3-она формулы (V)
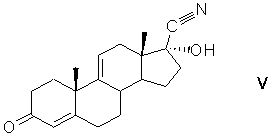
защиту 3-кетогруппы 17α-гидрокси-17β-цианандроста-4,9(11)-диен-3-она кетализацией с образованием циклического кеталя общей формулы (VI)
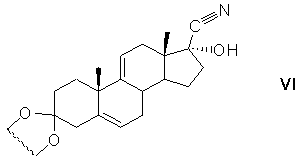
защиту 17α-гидроксигруппы действием алкоксиэтилена с образованием 17α-этерифицированного производного циклического кеталя общей формулы (VII)
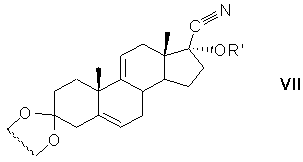
где R'=СН(СН3)OC2H5 или СН(СН3)OC4H9;
алкилирование карбонитрильной группы 17α-этерифицированного производного циклического кеталя общей формулы (VII) алкилирующим агентом в среде апротонного растворителя и удаление (снятие) защитных групп с образованием 17α-гидроксипрегна-4,9(11)-диен-3,20-диона формулы (VIII)
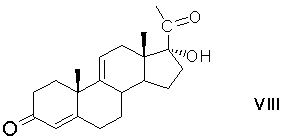
С21-иодирование 17α-гидроксипрегна-4,9(11)-диен-3,20-диона с образованием 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-диона формулы (IX)
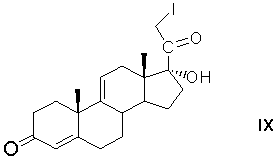
С21-ацетоксилирование 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-диона замещением атома иода на ацетоксигруппу действием ацетата щелочного металла с образованием 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона формулы (X)
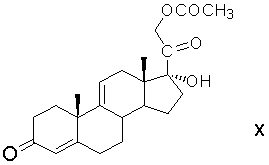
этерификацию 17α-гидроксигруппы 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона ацетилирующим агентом с образованием 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона формулы (XI)
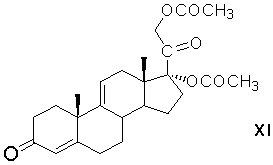
элиминирование 17α-ацетоксигруппы 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона с образованием 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона формулы (XII)
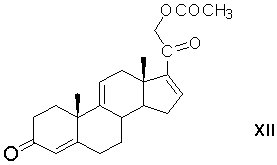
микробиологическое 1,2-дегидрирование 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона с образованием 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона формулы (XIII)
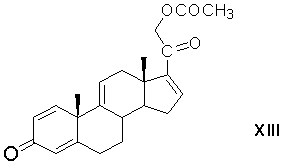
реакцию каталитического 1,4-присоединения метилмагнийгалогенида по Δ16-20-кетосистеме 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона в среде апротонного растворителя (или смеси растворителей) с образованием магнийгалоидпроизводного 21-ацетокси-20-гидрокси-16α-метилпрегна-1,4,9(11),17(20)-тетраен-3-она формулы (XIV)
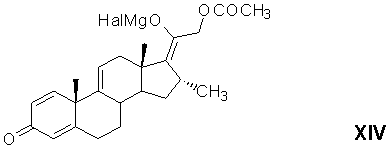
аутоокисление магнийгалоидпроизводного 21-ацетокси-20-гидрокси-16α-метилпрегна-1,4,9(11),17(20)-тетраен-3-она с образованием 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона формулы (XV)
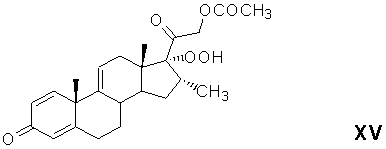
восстановление 17α-гидропероксигруппы 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона в 17α-гидроксильную группу восстанавливающим агентом с образованием 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона формулы (XVI)
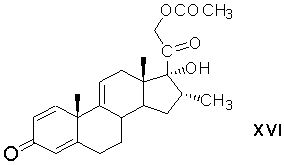
реакцию 9,11-бромгидроксилирования 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона действием гипобромной кислоты с образованием 21-ацетокси-9α-бром-11β,17-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона формулы (XVII)
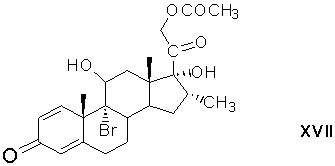
9β(11)-эпоксидирование 21-ацетокси-9α-бром-11β,17α-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона и одновременное удаление сложноэфирной защитной группировки при С21 с образованием 17α,21-дигидрокси-16α-метил-9β(11)-эпоксипрегна-1,4-диен-3,20-диона формулы (XVIII)
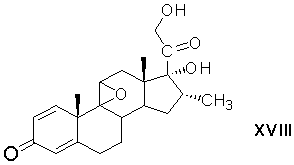
и реакцию раскрытия 9β(11)-эпокисного кольца 17α,21-дигидрокси-16α-метил-9β(11)-эпоксипрегна-1,4-диен-3,20-диона действием фтористоводородной кислоты.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Получение 11β,17α,21-Тригидрокси-16α-метил-9α-фторпрегна-1,4-диен-3,20-диона (дексаметазона) формулы (I) из фитостерина формулы (II) осуществляется по схеме, изображенной на рисунке 1.
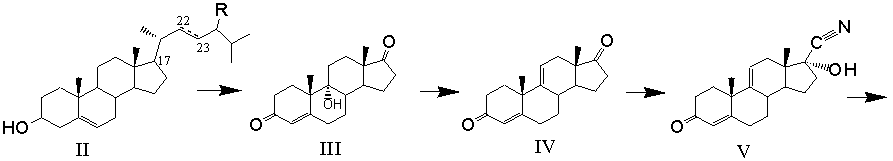
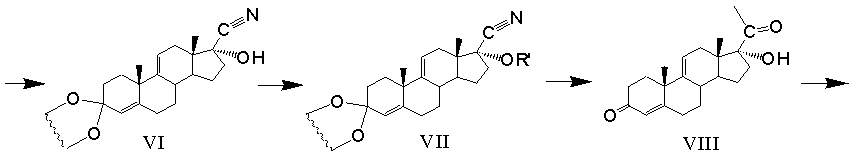
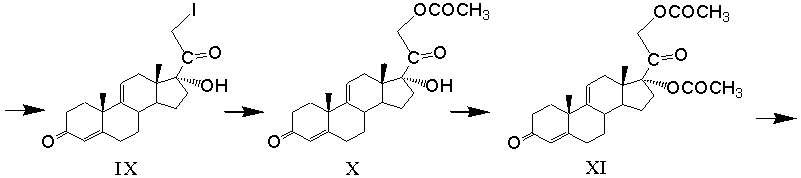
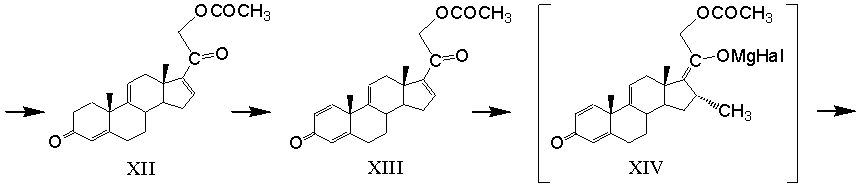
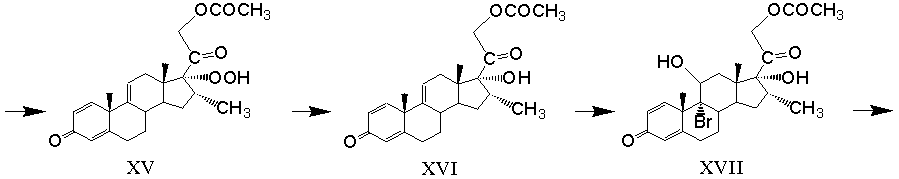
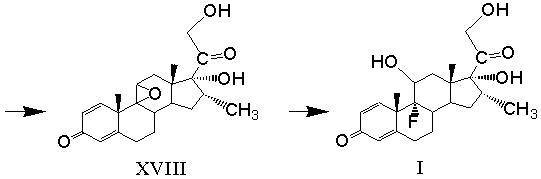
Рисунок 1- Синтез дексаметазона (I) из фитостерина (II) заявляемым способом, где: R=C2H5 - β-ситостерин; R=CH3 - кампестерин; R=C2H5 и ∆22(23) - стигмастерин, R=CH3 и ∆22(23) - брассикастерин; R'= СН(СН3)ОС2Н5 или СН(СН3)ОС4Н9
Ниже приводится в качестве примера подробное описание сущности изобретения.
Фитостерин общей формулы (II),
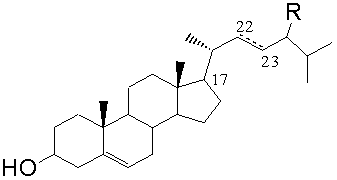
где R=C2H5 - β-ситостерин; R=CH3 - кампестерин; R=C2H5 и ∆22(23) - стигмастерин, R=CH3 и ∆22(23) - брассикастерин,
подвергают микробиологическому окислительному элиминированию боковой цепи при атоме С17 с одновременным образованием Δ4-3-кетосистемы и 9α-гидроксилированием культурой бактериальных клеток рода Mycobacterium (предпочтительно клеток бактерий Mycobacterium sp. ВКМ Ac-1817Д), при этом нагрузка исходного субстрата составляет от 4 до 20 г/л, а продолжительность ферментации - от 72 до 144 ч; процесс проводят в водной среде с селективностью превращения в 9-ОН-АД (III) до 72%, после чего клетки штамма-трансформанта отделяют. 9-ОН-АД (III) экстрагируют из полученной осветленной культуральной жидкости, практически не содержащей фитостерин, органическим растворителем (ароматическим углеводородом или хлорсодержащим углеводородом). Полученный 9-ОН-АД (III) с выделением в виде кристаллического порошка или без выделения из экстракта подвергают взаимодействию с дегидратирующим агентом с образованием Δ9(11)-АД (IV). Для этого в экстракт или в раствор кристаллического 9-ОН-АД (III) в апротонном органическом растворителе добавляют минеральную кислоту. При этом в качестве дегидратирующего агента используют минеральные кислоты, в том числе содержащие воду, а удаление избыточной воды может быть проведено в процессе реакции дегидратации. Селективность образования 9-ОН-АД (III) на стадии микробиологического окисления фитостерина (II) составляет 68-72%. Селективность образования Δ9(11)-АД (IV) на стадии дегидратации составляет 98-100%. Степень извлечения кристаллического Δ9(11)-АД из круда составляет 96-98%. Общий выход Δ9(11)-АД из фитостерина достигает 71%. Далее Δ9(11)-АД подвергают взаимодействию с ацетонциангидрином в присутствии едкого натра в водном метаноле, при этом получают циангидрин Δ9(11)-АД (V) с выходом более 94%. После этого проводят защиту 3-кетогруппы циангидрина Δ9(11)-АД (V) образованием циклического кеталя (пятичленного или шестичленного) взаимодействием с этиленгликолем или неопентилгликолем в присутствии кислого катализатора и агента, связывающего воду, например триэтилортоформиата. Затем проводят защиту 17α-гидроксигруппы образованием 17α-(1-алкокси)этилового эфира, после чего нитрильную группу соединения VII подвергают алкилированию действием метиллития или метилмагнийгалогенида. Кислым гидролизом защитных группировок получают Δ9(11)-ГПГ (VIII) с выходом от 89 до 92,4% в зависимости от природы защитных групп. Полученный Δ9(11)-ГПГ (VIII) превращают в 21-ацетокси-Δ9(11)-ГПГ (X) методом прямого C21-иодирования с последующим замещением атома иода на ацетоксигруппу действием ацетата калия, причем реакция замещения может быть проведена без выделения 21-иод-Δ9(11)-ГПГ (IX). 17α-Гидроксигруппу 21-ацетокси-Δ9(11)-ГПГ (X) ацетилируют методом смешанных ангидридов в среде хлористого метилена, используя ацетилирующую смесь, состоящую из уксусной кислоты, трифторуксусного ангидрида и каталитического количества п-ТСК, содержащую минимально необходимое количество трифторуксусного ангидрида, что позволяет полностью исключить образование нежелательного продукта енолацетилирования карбонильной группы при атоме С3. Полученное с выходом 92,54% соединение XI затем обрабатывают в среде диметилсульфоксида ацетатом калия с образованием 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII), который подвергают микробиологическому 1,2-дегидрированию с участием клеток N. simplex ВКМ Ас-2033Д, обладающих 3-кетостероид-∆1-дегидрогеназной активностью, при этом нагрузка исходного субстрата составляет от 10 до 20 г/л, а продолжительность ферментации - от 6 до 14 ч и процесс проводят при температуре 25°С в среде буферного раствора при рН ~5, содержащей до 8 об.% полярного растворителя, смешивающегося с водой (предпочтительно диметилсульфоксид или этанол), метил-β-циклодекстрин в количестве не более 2 молей на 1 моль исходного субстрата и экзогенный акцептор электронов - менадион - в эффективном количестве. Выделение 21-ацетата тетраена (XIII) из культуральной жидкости осуществляют экстракцией несмешивающимся с водой растворителем или экстракцией стероида из пасты биомассы любым подходящим растворителем после ее предварительного отделения от водной фазы. Селективность образования 21-ацетата тетраена (XIII) из 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) составляет 92-95%. Выход кристаллического 21-ацетата тетраена (XIII) после выделения составляет 83,5-84%. После этого проводят реакцию каталитического 1,4-присоединения метилмагнийбромида по Δ16-20-кетосистеме 21-ацетата тетраена (XIII) в среде апротонного растворителя (или смеси растворителей) с образованием магнийгалоидпроизводного 21-ацетокси-20-гидрокси-16α-метилпрегна-1,4,9(11),17(20)-тетраен-3-она (XIV), который гидролизуют действием водного раствора хлорида аммония в присутствии атмосферного воздуха. 17α-Гидропероксигруппу полученного самопроизвольным окислением 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XV) подвергают восстановлению в 17α-гидроксильную с образованием 1,4,9-триена (XVI), которое проводят действием иодида калия в среде смеси ароматического углеводорода и ацетона, причем указанная последовательность может быть осуществлена без выделения 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XV). Затем 1,4,9-триен (XVI) подвергают бромгидроксилированию действием гипобромной кислоты в среде апротонного полярного растворителя (например, тетрагидрофурана или диалкилкетона), содержащей воду в эффективном количестве, с образованием гипобромной кислоты in situ, причем в качестве источника гипобромной кислоты используют дибромантин (1,3-дибром-5,5-диметилгидантоин). Последующее 9β(11)-эпоксидирование 21-ацетокси-9α-бром-11β,17-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона (XVII) и удаление сложноэфирной защитной группировки при С21 проводят одновременно в водной среде, содержащей смесь апротонного полярного растворителя (например, тетрагидрофурана или диалкилкетона) и протонного растворителя (например, алифатического спирта), действием щелочного агента (например, гидроксида или карбоната щелочного металла). При этом последовательность реакций бромгидроксилирования 1,4,9-триена (XVI) и 9β(11)-эпоксидирования соединения XVII с одновременным удалением сложноэфирной защитной группировки при С21 могут быть осуществлены без выделения соединения XVII из реакционной смеси. Реакцию раскрытия 9β(11)-эпокисного кольца 17α,21-дигидрокси-16α-метил-9β(11)-эпоксипрегна-1,4-диен-3,20-диона (XVIII) проводят в среде 70% водной фтористоводородной кислоты, являющейся реагентом. Получают дексаметазон (I) с выходом 90,91% и содержанием основного вещества 91,1% (ВЭЖХ).
После очистки получают дексаметазон фармакопейного качества с содержанием основного вещества 97,8% (ВЭЖХ). Выход на очистке составляет 81,2%. Общий выход дексаметазона из фитостерина составляет 22,8%.
Заявленное изобретение иллюстрируется следующими примерами, не ограничивающими его.
Для трансформации фитостерина может быть использован соевый фитостерин с содержанием трансформируемых стеринов от 90 до 100%. При осуществлении изобретения-способа получения 9-ОН-АД (III) в описанном ниже примере использован фитостерин с содержанием трансформируемых стеринов 95,47%, в том числе (%): β-ситостерин 42,39; кампестерин 23,48; стигмастерин 26,08; брассикастерин 3,52.
Микробиологическое окисление фитостерина при осуществлении изобретения-способа получения дексаметазона из фитостерина может быть осуществлено клетками бактерий рода Mycobacterium, конкретно Mycobacterium sp. ВКМ Ac-1817Д.
В качестве органического растворителя при осуществлении изобретения-способа на операции экстракции 9-ОН-АД (III) и для получения Δ9(11)-АД (IV) в реакции последующей дегидратации могут быть использованы ароматические углеводороды (например, бензол или толуол) или хлорсодержащие углеводороды (например, дихлорметан, или хлороформ, или дихлорэтан).
В качестве дегидратирующего агента на стадии дегидратации 9-ОН-АД (III) могут быть использованы минеральные кислоты, в том числе содержащие воду, например хлорная кислота с содержанием воды до 43%, или серная кислота с содержанием воды до 30%, или ортофосфорная кислота с содержанием воды до 15%, а также ортофосфорная кислота с содержанием до 50% пирофосфорной кислоты.
Интенсификация процесса дегидратации при осуществлении изобретения-способа получения андроста-4,9(11)-диен-3,17-диона удалением избыточной воды в течение реакции дегидратации может быть проведена, например, методом азеотропной отгонки или проведением реакции в присутствии эффективного количества пирофосфорной кислоты, которая, реагируя с водой, превращается в условиях проведения реакции в ортофосфорную кислоту.
В таблице 1 представлены результаты дегидратации кристаллического 9-ОН-АД с образованием ∆9(11)-АД, а также результаты дегидратации в экстракте осветленной культуральной жидкости, в условиях по заявляемому способу и способами, описанными в литературных источниках.
Гидроцианирование Δ9(11)-АД (IV) проводят реакцией с ацетонциангидрином в качестве гидроцианирующего агента, а в качестве основного катализатора используют минеральное основание (например, гидроксид щелочного металла). Реакцию проводят в среде водного метанола.
Защиту 3-кетогруппы 17α-гидрокси-17β-цианандроста-4,9(11)-диен-3-она (V) осуществляют кетализацией реакцией с 1,2-диолом (например, этиленгликолем) с образованием диоксоланового кеталя или реакцией с 1,3 диолами (например, непентилгликолем), с образованием гексациклического (1,3-диоксанового) кеталя.
Защиту 17α-гидроксигруппы циклического кеталя общей формулы (VI) осуществляют этерификацией действием алкоксиэтилена (например, этоксиэтилена или бутоксиэтилена).
Алкилирование карбонитрильной группы 17α-этерифицированного производного циклического кеталя общей формулы (VII) проводят, используя метиллитий или галогениды метилмагния (например, метилмагния бромид или метилмагния хлорид) в качестве алкилирующего агента
С21-Иодирование Δ9(11)-ГПГ (VIII) проводят в среде смеси метанола и дихлорметана действием молекулярного иода в присутствии оксида кальция и хлорида кальция, а для проведения реакции последующего замещения атома иода на ацетоксигруппу в 21-иод-Δ9(11)-ГПГ (IX) могут быть использованы апротонные полярные органические растворители (например, диметилформамид, диметилсульфоксид, гексаметилфосфортриамид, ацетонитрил), а также диалкилкетоны (например, ацетон). Кроме того, последовательность процессов С21-иодирования Δ9(11)- ГПГ (VIII) и С21-ацетоксилирования 21-иод-Δ9(11)-ГПГ (IX) могут быть проведены без выделения 21-иод-Δ9(11)-ГПГ (IX) из реакционного раствора.
Этерификацию 17α-гидроксигруппы 21-ацетокси-Δ9(11)-ГПГ (X) проводят ацетилирующим агентом, в качестве которого могут быть использованы ангидрид, или хлорангидрид уксусной кислоты, или смесь уксусной кислоты и трифторуксусного ангидрида в эффективном соотношении.
Элиминирование 17α-ацетоксигруппы 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона (XI) проводят действием ацетата щелочного металла в среде апротонного органического растворителя (например, диметилформамида, диметилсульфоксида, гексаметилфосфортриамида и т.п.).
1,2-Дегидрирование 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона формулы (XII) проводят микробиологическим способом с применением клеток микроорганизма Nocardioides simplex ВКМ Ас-2033Д, обладающих 3-кетостероид-1-дегидрогеназной активностью. Основные параметры процесса микробиологического 1,2-дегидрирования 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона приведены в таблицах 2 и 3. Использован субстрат с содержанием массовой доли 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона от 80% до 95%. Нагрузка исходного субстрата дана в пересчете на содержание основного вещества.
Реакцию каталитического 1,4-присоединения метилмагнийбромида по Δ16-20-кетосистеме 21-ацетата тетраена (XIII) проводят при температуре от минус 70 до минус 10 ºС, а в качестве катализатора используют хлорид меди (I).
Аутоокисление магнийгалоидпроизводного 21-ацетокси-20-гидрокси-16α-метилпрегна-1,4,9(11),17(20)-тетраен-3-она (XIV) проводят при температуре от минус 10 до плюс 10 ºС молекулярным кислородом атмосферного воздуха в гетерогенной среде, состоящей из смешивающегося с водой апротонного растворителя (например, тетрагидрофурана, ацетонитрила, или их смеси) и не смешивающегося с водой апротонного растворителя (ароматического, например бензола, толуола) в эффективном соотношении.
Восстановление 17α-гидропероксигруппы 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона формулы (XV) в 17α-гидроксильную группу проводят действием восстанавливающего агента, в качестве которого используют йодид щелочного металла (натрия или калия) в эффективном количестве. Реакцию проводят в среде диалкилкетона (например, ацетона, метилэтилкетона) или в среде ароматического углеводорода (например, бензола, толуола) в присутствии диалкилкетона (например, ацетона) в эффективном количестве.
Кроме того, последовательность реакций, включающая каталитическое 1,4-присоединение метилмагнийгалогенида (например, матилмагнийбромида или метилмагнийхлорида) по Δ16-20-кетосистеме 21-ацетата тетраена (XIII), аутоокисление магнийгалоидпроизводного 21-ацетокси-20-гидрокси-16α-метилпрегна-1,4,9(11),17(20)-тетраен-3-она (XIV) и восстановление 17α-гидропероксигруппы 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XV) в 17α-гидроксильную группу, может быть осуществлена без выделения 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XV).
Реакцию бромгидроксилирования 1,4,9-триена (XVI) действием гипобромной кислоты проводят в среде апротонного полярного растворителя (например, тетрагидрофурана или диалкилкетона), содержащей воду в эффективном количестве, с образованием гипобромной кислоты in situ, причем источник гипобромной кислоты выбран из группы, содержащей 1,3-дибром-5,5-диметилгидантоин, N-бромсукцинимид, N-бромацетамид и т.п.
9β(11)-Эпоксидирование 21-ацетокси-9α-бром-11β,17-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона (XVII) и удаление сложноэфирной защитной группировки при С21 проводят одновременно в водной среде, содержащей смесь апротонного полярного растворителя (например, тетрагидрофурана или диалкилкетона) и протонного растворителя (например, алифатического спирта, выбранного из группы: метиловый, этиловый, изопропиловый) в эффективном количестве, действием щелочного агента (например, гидроксида или карбоната щелочного металла). Кроме того, последовательность реакций бромгидроксилирования 1,4,9-триена (XVI) и 9β(11)-эпоксидирования 21-ацетокси-9α-бром-11β,17-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона (XVII) с одновременным удалением сложноэфирной защитной группировки при С21 могут быть осуществлены без выделения 21-ацетокси-9α-бром-11β,17-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона (XVII) из реакционной смеси.
Примеры
Пример 1 Получение 9α-гидроксиандрост-4-ен-3,17-диона (9-ОН-АД, III)
А) Выращивание инокулята
Бактерии Mycobacterium sp. ВКМ Ac-1817Д культивируют в шейкере-термостате в течение 25 ч при температуре 30°C и частоте встряхивания 220 об/мин в качалочных колбах вместимостью 750 мл, содержащих по 100 мл жидкой питательной среды следующего состава (г/л): дрожжевой экстракт сухой 10; (NH3)2HPO4 1,5; KH2PO4 0,5; K2HPO4 x 3H2O 0,5; глицерин 10; полиэтиленгликоль сорбит моноолеат 3,0; MgSO4 x 7H2O 0,2; FeSO4 x 7H2O 0,005; ZnSO4 x 7H2O 0,002; вода дистиллированная до 1 л, pH 6,8 7,2.
Б) Проведение трансформации
Полученный посевной материал вносят в среду для трансформации в количестве 10% (по объему). Микробиологическую трансформацию фитостерина осуществляют в шейкере-термостате в течение 72 ч при температуре 30°C и частоте встряхивания 220 об/мин в качалочных колбах вместимостью 750 мл, содержащих по 100 мл среды для трансформации следующего состава (г/л): KH2PO4 0,8; K2HPO4 x 3H2O 4,2; (NH3)2SO4 3,0; глицерин 5,0; карбамид 0,13; MgSO4 x 7H2O 0,2; FeSO4 x 7H2O 0,01; ZnSO4 x 7H2O 0,002; полиэтиленгликоль сорбит моноолеат 1,0; фитостерин (в расчете на трансформируемые стерины) 4,0; вода дистиллированная до 1 л, pH 6,8 7,2. После стерилизации среды фитостерин гомогенизируют ультразвуком (42 кГц, 100 Вт) в течение 2 мин.
В) Получение экстракта
По окончании трансформации культуральную жидкость центрифугируют при 10 000 g в течение 40 мин при температуре 20-25°C. Получают осветленную культуральную жидкость (надосадочную жидкость), содержащую 9-ОН-АД (селективность трансформации 71,48%).
9-ОН-АД экстрагируют из надосадочной жидкости органическим растворителем. Получают экстракт, содержащий 9-ОН-АД (далее экстракт).
Г) выделение 9-ОН-АД
Растворитель упаривают в вакууме до начала кристаллизации стероидов, концентрат разбавляют н-гексаном в пропорции 1:1 по объему, перемешивают, охлаждают до 4°С и инкубируют в течение 2-3 ч. Образовавшиеся кристаллы отделяют фильтрованием и высушивают. Проводят две перекристаллизации из этанола, маточные растворы объединяют, упаривают под вакуумом до начала кристаллизации, охлаждают концентрат до 4°С и инкубируют в течение 4-6 ч. Образовавшиеся кристаллы отделяют фильтрованием, высушивают и перекристаллизовывают из этанола. Все полученные кристаллы объединяют, измельчают и высушивают под вакуумом. Получают 9-ОН-АД со степенью извлечения 82%, содержанием основного вещества ≥ 97%, содержанием примеси АД < 0,5% и температурой плавления 217-220°С (лит. 219-221°С [US4237220, 1980, пример 2]).
Пример 2 Получение андроста-4,9(11)-диен-3,17-диона (∆9(11)-АД, IV)
Приведено описание некоторых вариантов примера 2 получения ∆9(11)-АД (IV). Параметры и результаты других вариантов примера 2 приведены в таблице 1.
Вариант 1. Экстракт, полученный по п.В) примера 1 , помещают в колбу, снабженную обратным холодильником. К экстракту добавляют минеральную кислоту. Реакционную массу нагревают до кипения и выдерживают при перемешивании до окончания реакции. Затем смесь охлаждают до комнатной температуры и добавляют 5% водный раствор NaCl. Органический слой отделяют, водный слой экстрагируют тем же растворителем. Объединенные органические фазы промывают 5% раствором NaCl до нейтральной реакции и водой, затем сушат Na2SO4 и упаривают в вакууме досуха. Получают круд, содержащий ∆9(11)-АД и не содержащий 9-ОН-АД по данным ВЭЖХ анализа.
Вариант 2 (с азеотропной отгонкой воды). Экстракт, полученный по п.В) примера 1, помещают в колбу, снабженную насадкой Дина-Старка и обратным холодильником. Затем к экстракту добавляют минеральную кислоту. Реакционную массу нагревают до кипения и выдерживают при перемешивании до окончания реакции, собирая выделяющуюся воду в насадку. Затем смесь охлаждают до комнатной температуры. Обработку реакционной массы проводят, как описано в предыдущем варианте.
Селективность реакции дегидратации оценивают по данным ВЭЖХ анализа исходного экстракта (определение содержания 9-ОН-АД) и круда (содержание ∆9(11)-АД).
После выделения из круда получают ∆9(11)-АД со степенью извлечения 96-98%. Температура плавления от 203,5 до 206,5ºC (лит. 201-204.5ºC [US4127596, 1978, пример 7].
[α]D20 от+217 до +221 º (с=1.0, CHCl3) (лит. +220 º (CHCl3) [US4127596, 1978, пример 6]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,75 (д, J = 1,8 Гц, 1H, 4-CH); 5,55 (м, 1H, 11-CH); 1,35 (с, 3H, 19-CH3); 0,87 (с, 3H, 18-CH3).
Спектр ЯМР 13С (δ, м.д.; CDCl3): 221,0 (17-C(O)); 199,0 (3-C(O)); 168,9 (5-C); 145,1 (9-C); 124,2 (4-CH); 118,1 (11-CH); 48,0; 45,8; 41,1 36,8; 36,2; 34,2; 33,8; 33,4; 32,6; 31,1; 26,2; 22,6; 13,9 (19-СН3).
Вариант 3. К суспензии 5,0 г 9-OH-АД (III) в 60 мл сухого 1,2-дихлорэтана при перемешивании добавляют 10 мл 85% ортофосфорной кислоты. Реакционную смесь нагревают до кипения, при этом наблюдается полное растворение исходного вещества. Реакционную массу кипятят в течение 30 мин, затем охлаждают до комнатной температуры и добавляют 30 мл 5% водного раствора NaCl. Перемешивают в течение 3-5 мин. Органический слой отделяют, водный слой экстрагируют 1,2-дихлорэтаном. Объединенные органические экстракты промывают 5% водным раствором NaCl до нейтральной реакции, обрабатывают осветляющим углем.
Растворитель упаривают при атмосферном давлении до небольшого объема, после чего добавляют 5% водный раствор NaCl и отгонку продолжают. Полученную суспензию охлаждают до комнатной температуры, осадок отфильтровывают, промывают на фильтре водой и сушат до постоянной массы. Получают 4,46 г ∆9(11)-АД (IV) с выходом 95,15%.
Т. пл. 198-200ºC.
Вариант 4. К раствору 115 мг 9-OH-АД (III) в 1,5 мл абсолютного хлороформа добавляют 0,2 мл 80% H2SO4. Смесь выдерживают при кипении в течение 1,5 ч, затем разбавляют водой и переносят в делительную воронку. Водный слой отделяют, органический слой промывают водой до рН ~7. Растворитель упаривают в вакууме до прекращения погона, остаток кристаллизуют из этилацетата. Получают 97,34 мг соединения IV с выходом 90%. Т.пл. 198-202ºС.
Пример 3 Получение 17α-гидрокси-17β-цианоандроста-4,9(11)-диен-3-она (V)
К суспензии 50 г ∆9(11)-АД (IV) в 140 мл метанола в токе азота при комнатной температуре добавляют 45 мл ацетонциангидрина и 7 мл 2.68 н. раствора едкого натра в воде. Суспензию нагревают до температуры 35°С и выдерживают 50-60 мин при этой же температуре. Затем медленно добавляют 30 мл дистиллированной воды, реакционную массу охлаждают до комнатной температуры и выдерживают в течение 19-21 ч. После этого реакционную суспензию медленно разбавляют 130 мл воды. Осадок отфильтровывают, промывают сначала смесью воды и метанола, затем водой до рН ~7. Получают 43,84 г соединения V.
Т.пл. 225-229°С (лит. 211-220°С [DD281394, 1990, пример 1]);
[α]D20 +115 º (с=1.0, CHCl3) (лит. +118 º (с=1.0, CHCl3) [US4500461, 1985]).
Из маточного раствора дополнительно извлекают 7,81 г соединения V с т.пл. 214-218°С. Содержание примеси изомерного 17β-гидрокси-17α-цианандроста-4,9(11)-диен-3-она менее 1%.
Суммарно получают 51.65 г с выходом 94,2%.
Спектр 1H ЯМР (CDCl3) δ: 5.75 (м, 1Н, 4-Н), 5.57 (м, 1Н, 11-Н), 1.35 (с, 3Н, 19-СН3), 0.94 (с, 3Н, 18-СН3).
Спектр 13С ЯМР (CDCl3) δ: 199.7 (С-3), 169.8 (С-5), 144.3 (С-9), 124.0 (С-4), 120.7 (CN), 118.2 (С-11), 77.2 (С-17), 47.8, 45.3, 41.0, 38.5, 38.0, 34.2, 33.8, 32.8, 32.2 31.0, 26.7, 24.7 (19-СН3), 16.1 (18-СН3).
Пример 4 Получение 3.3-(2,2-диметилпропилен)-диокси-17α-гидрокси-17β-цианоандроста-5,9(11)-диена (VIa)
К суспензии 20 г 17α-гидрокси-17β-цианоандроста-4,9(11)-диен-3-она (V) в 40 мл сухого хлористого метилена в токе азота добавляют последовательно 20 г 2,2-диметилпропан-1,3-диола и 20 мл триэтилортоформиата. Реакционную массу охлаждают до 0-2°С, добавляют 2 г безводной п-ТСК и перемешивают в течение 7-8 ч. Затем добавляют 2 мл триэтиламина и 160 мл воды. Суспензию перемешивают в течение 30 мин, осадок отфильтровывают, промывают водой до рН 7, сушат до постоянной массы.
Получают 21,8 г первого кристаллизата соединения VIa.
Из маточного раствора дополнительно извлекают 3,47 г второго кристаллизата соединения VIa.
Суммарно получают 25,27 г соединения VIa с выходом 99%.
Т.пл. аналитического образца 253-255ºС (с разложением).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,51 (м, 1H, 11-CH); 5,40 (м, 1H, 6-CH); 3,50 (м, 4Н, 1'-ОСН2); 1,18 (с, 3H, 19-CH3); 0,98 (с, 3H, 18-CH3); 0,91 (д, J = 3.4 Гц, 6Н, 3'-СН3)
Спектр ЯМР 13С (δ, м.д.; CDCl3): 145,7 (9-C); 138,0 (5-C); 121,7 (CN); 120,8 (6-CH); 116,0 (11-С); 98,6 (3-CH); 70,5; 70,0; 48,6; 46,3; 44,0; 39,0; 38,9; 35,1; 33,0; 32,7; 31,3; 30,4; 28,1; 27,6; 25,1; 23,0; 22,9; 16,1 (19-СН3).
Найдено: C 75,40%, H 8,68%, N 3,51%. C25H35NO3. Вычислено C 75,53%, H 8,87%, N 3,52%.
Пример 5 Получение 3.3-этилендиокси-17α-гидрокси-17β-цианоанодроста-5,9(11)-диена (VIб)
К раствору 0,165 г безводной п-ТСК в 9,6 мл этиленгликоля добавляют 96 мл дихлорметана, 9,5 г 17α-гидрокси-17β-цианоандроста-4,9(11)-диен-3-она (V) и 4,7 мл триметилортоформиата. Реакционный раствор перемешивают в течение 2 ч при комнатной температуре. Затем из реакционной массы отгоняют растворитель при атмосферном давлении до половины первоначального объема, остаток охлаждают до комнатной температуры и добавляют 0,25 мл сухого триэтиламина.
Реакционный раствор переносят в делительную воронку и промывают водой. Растворитель упаривают досуха. К остатку добавляют 10 мл метанола, растирают, полученную суспензию охлаждают до 0°С и выдерживают не менее 6 ч при этой же температуре. Осадок отфильтровывают, промывают 1,6 мл метанола, сушат до постоянной массы. Получают 10,04 г соединения VIб с выходом 92,5%.
Т.пл. 201-203°С (лит. 202-205°C [Bull.Chem.Soc.Jpn. 1985, 58 (3), 978-980, соединение 10а]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,54 (м, 1H, 11-CH); 5,40 (м, 1H, 6-CH); 3,95 (м, 4Н, ОСН2-ОСН2); 1,20 (с, 3H, 19-CH3); 0,91 (с, 3H, 18-CH3).
Пример 6 Получение 3,3-(2,2-диметилпропилен)-диокси-17α-(1-этоксиэтокси)-17β-цианоадроста-5,9(11)-диена (VII)
К смеси 1 г 3,3(2,2-диметилпропилен)-диокси-17α-гидрокси-17β-цианоандроста-5,9(11)-диена (VIa) в 10 мл абсолютного ТГФ, охлажденной до 0°С, добавляют 1 мл этоксиэтилена и 100 мг безводной п-ТСК. Перемешивают 1,5 ч. По завершении реакции добавляют 0,1 мл триэтиламина и выдерживают 10 мин. Затем реакционную массу выливают в 100 мл воды. Выпавший осадок отфильтровывают, промывают на фильтре водой до нейтральной реакции и сушат до постоянной массы.
Получают 0,92 г соединения VII с выходом 78% (в виде смеси двух диастереомеров в соотношении 1:1).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,52 (м, 1H, 11-CH); 5,41 (м, 1H, 6-CH); 5,10, 5,00 (кв, J = 5.3 Гц, 1 Н, О2СН); 3,73-3.36 (м, 6Н, 1'-ОСН2, OCH2); 1.35, 1.31 (д, J = 5.3 Гц, 3H, CH3CH); 1.22, 1.18 (т, J = 7.1 Гц, 3H, CH3CH2O); 1,19 (с, 3H, 19-CH3); 0,97 (с, 3H, 18-CH3); 0,92 (м, 6Н, 3'-СН3).
Пример 7 Получение 17α-гидроксипрегна-4,9(11)-диен-3,20-диона (VIII)
Вариант 1. К раствору 90,65 г 3,3-(2,2-диметилпропилен)-диокси-17α-гидрокси-17β-цианоандроста-5,9(11)-диена (VIa) в 907 мл абсолютного диэтилового эфира, охлажденному до 0-2°С, в токе аргона добавляют 90,7 мл этоксиэтилена и 9,07 г безводной п-ТСК. Реакционную смесь перемешивают в течение 4 ч при температуре 0-2°С.
Затем реакционную массу охлаждают до температуры минус 25°С, добавляют 9 мл триэтиламина и перемешивают в течение 5 мин. После этого добавляют по каплям 384 мл 1,6 М раствора метиллития в диэтиловом эфире и охлаждение прекращают. Подогревают реакционную массу до комнатной температуры и выдерживают в течение 30 мин. Затем добавляют смесь 49,4 мл метанола, 2,7 мл воды и 2,7 мл концентрированной HCl. Через 5 мин реакционную массу разбавляют 181 мл метанола и добавляют 181 мл ~20-21% разбавленной HCl. Перемешивают в течение 18 ч. Эфир отгоняют при атмосферном давлении, добавляя постепенно в реакционную массу 1,36 л дистиллированной воды. Суспензию в воде охлаждают до температуры 0-2°С и выдерживают в течение 1 ч. Осадок отфильтровывают, промывают водой до нейтральной реакции и сушат до постоянной массы. Получают 79 г технического продукта, который обрабатывают осветляющим углем в дихлорметане, кристаллизуют из ацетона.
Получают 57,33 г первого кристаллизата соединения VIII с т. пл. 207-211°С (лит. т.пл. 206-211°С [US 4041055, 1977, пример 58].
Из маточного раствора дополнительно извлекают 9,47 г второго кристаллизата соединения VIII с т. пл. 207-209°С.
Всего получают 66,8 г соединения VIII с выходом 89,2%.
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,73 (д, J = 1,5 Гц, 1H, 4-CH); 5,53 (м, 1H, 11-CH); 2,27 (с, 3Н, 21-СН3); 1,33 (с, 3H, 19-CH3); 0,68 (с, 3H, 18-CH3).
Вариант 2. В условиях варианта 1 из 10 г 3.3-этилендиокси-17α-гидрокси-17β-цианандроста-5,9(11)-диена (VI) получают 8,54 г соединения VIII с выходом 92.4%.
Т.пл. 206-209°С.
Вариант 3. К реакционной массе, полученной в условиях варианта 1 из 10 г 3.3-этилендиокси-17α-гидрокси-17β-цианандроста-5,9(11)-диена (VIб) и 11 мл бутоксиэтилена в 40 мл толуола, при температуре минус 5°С добавляют 120 мл раствора CH3MgCl в толуоле, полученного из 44 мл раствора CH3MgCl в ТГФ (Мерк) отгонкой ТГФ. Реакционную массу перемешивают в течение 1,5 ч, затем добавляют 40 мл 10% водного раствора NH4Cl и 8 мл концентрированной HCl. Через 20 мин реакционный раствор переносят в делительную воронку, органический слой отделяют, водный экстрагируют толуолом. Раствор стероида в толуоле промывают водой до рН 7 и упаривают в вакууме досуха. К остатку добавляют 100 мл метанола. Суспензию нагревают до кипения, добавляют 1 мл концентрированной HCl и выдерживают в течение 10-15 мин. Затем к реакционной массе, охлажденной до комнатной температуры, добавляют 22 мл 12.5% водного раствора ацетата натрия и растворитель упаривают в вакууме. Полученную суспензию разбавляют 100 мл воды, осадок отфильтровывают, промывают водой. Осадок сушат до постоянной массы и перекристаллизовывают из метанола.
После очистки получают 6,25 г соединения VIII с т.пл. 210-212°С.
Пример 8 Получение 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-диона (IX)
К раствору 30 г 17α-гидроксипрегна-4,9(11)-диен-3,20-диона (VIII) в смеси 225 мл дихлорметана и 75 мл метанола добавляют 22,7 г оксида кальция, 1,5 мл дистиллированной воды и раствор 30 г иода и 5,1 г хлорида кальция в 75 мл метанола. Реакционную массу перемешивают при комнатной температуре в течение 1 ч и добавляют 300 мл воды. Перемешивают в течение 5 мин. Неорганический осадок отфильтровывают, промывают последовательно смесью хлористого метилена и метанола (4 : 1) и дихлорметаном. Водный слой отделяют и экстрагируют хлористым метиленом. Объединенный экстракт упаривают в вакууме до прекращения погона.
Остаток растирают с метанолом, осадок отфильтровывают. Получают 41,5 г соединения IX c количественным выходом.
Т.пл. 148-150°С (лит. 142-149°С [US3009935, 1954, пример 2]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,74 (д, J = 1,5 Гц, 1H, 4-CH); 5,53 (м, 1H, 11-CH); 4,20 (д, Jobs = 11,1 Гц (АВ-система), 1H, 21-CH2I); 3,98 (д, Jobs = 11,1 Гц (АВ-система), 1Н, 21-CH2I); 1,32 (с, 3H, 19-CH3); 0,65 (с, 3H, 18-CH3).
Пример 9 Получение 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона (X)
Вариант 1. К раствору 41,5 г 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-диона (IX) в 420 мл ацетона в токе инертного газа добавляют 41,5 г прокаленного ацетата калия. Суспензию кипятят в течение 2,5 ч, затем охлаждают до комнатной температуры и выливают в 4,3 л воды. Перемешивают в течение 30 мин. Осадок отфильтровывают, промывают на фильтре водой, сушат до постоянной массы. Получают 32.5 г соединения X с выходом 92%.
Т.пл. 225-230°С. (лит. 230-232°С [US 4041055, 1977, пример 66])
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,73 (с, 1H, 4-CH); 5,53 (м, 1H, 11-CH); 5,02 (д, Jobs = 17,4 Гц (АВ-система), 1H, 21-СН2О); 4,83 (д, Jobs = 17,4 Гц (АВ-система), 1H, 21-СН2О); 2,16 (с, 3Н, 21-ОС(О)СН3); 1,32 (с, 3H, 19-CH3); 0,64 (с, 3H, 18-CH3).
Вариант 2. К раствору 15 г 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-диона (IX) в 150 мл ацетонитрила в токе инертного газа добавляют 21 г прокаленного ацетата калия. Суспензию кипятят в течение 2,5 ч, затем охлаждают до комнатной температуры и выливают в 1,5 л воды. Перемешивают в течение 30 мин. Осадок отфильтровывают, промывают на фильтре водой, сушат до постоянной массы. Получают 12,41 г соединения X с выходом 97,2% и содержанием родственных соединений не более 5%.
Т.пл. 219 - 220°С.
Вариант 3. К остатку после упаривания растворителя, содержащему 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-дион (IX) и представляющему собой густое желтое масло, полученному по примеру 8 из 66.8 г 17α-гидроксипрегна-4,9(11)-диен-3,20-диона (VIII), добавляют 1 л ацетона и 84 г безводного ацетата калия. Реакционную массу нагревают до температуры 60-65°С и выдерживают в течение 2.5 ч. По окончании реакции реакционную массу охлаждают до комнатной температуры и выливают в 10 л воды. Осадок отфильтровывают, промывают водой. Получают 78 г соединения X с выходом 99,24%, считая на VIII.
После очистки получают 66,9 г соединения X с выходом 85,8% на очистке и содержанием родственных соединений не более 2%.
Т.пл. 229,1 - 229,8°С
Вариант 4. К остатку после отгонки растворителя, содержащему 17α-гидрокси-21-иодпрегна-4,9(11)-диен-3,20-дион (IX), полученному по примеру 8 из 33,1 г 17α-гидроксипрегна-4,9(11)-диен-3,20-диона (VIII), добавляют 330 мл диметилформамида и 48,3 г безводного ацетата калия. Реакционную массу нагревают до температуры 60-65°С, выдерживают в течение 2 ч, затем охлаждают до температуры 0°С. После выдержки в течение 3 ч осадок отфильтровывают, промывают диметилформамидом и водой.
Получают 34,2 г соединения X с выходом 87,8%, считая на VIII. Содержание родственных соединений 2.5%, содержание ДМФ 5%.
Т.пл. 207-209°С.
Пример 10 Получение 17α,21-диацетоксипрегна-4,9(11)-диен-3,20-диона (XI)
К ацетилирующей смеси, состоящей из 29,8 г безводной п-ТСК, 0,34 л ледяной уксусной кислоты и 0,125 л трифторуксусного ангидрида, в 1,34 л хлористого метилена добавляют 66,9 г 21-ацетокси-17α-гидроксипрегна-4,9(11)-диен-3,20-диона (X). Реакционную массу перемешивают при комнатной температуре в течение 2,5 ч, затем выливают в 3,24 л 5% водного раствора аммиака. Выдерживают 15 мин при температуре 10-15°С, после чего органический слой отделяют, водный слой экстрагируют хлористым метиленом. Объединенные экстракты промывают водой до нейтральной реакции. Растворитель упаривают при атмосферном давлении. Остаток кристаллизуют из воды. Осадок отфильтровывают, промывают водой, сушат до постоянной массы. Получают 68,6 г соединения XI с выходом 92,54%.
Т.пл. 220°C с разложением (лит. 215-220°C [Известия академии наук СССР, Серия химическая, 1991, 40(12), 2875-2878]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 5,75 (с, 1H, 4-CH); 5,55 (м, 1H, 11-CH); 4,88 (д, Jobs = 16,7 Гц (АВ-система), 1H, 21-СН2О); 4,65 (д, Jobs = 16,7 Гц (АВ-система), 1Н, 21-СН2О); 2,16 (с, 3Н, 21-ОС(О)СН3); 2,08 (с, 3Н, 17-ОС(О)СН3); 1,33 (с, 3H, 19-CH3); 0,70 (с, 3H, 18-CH3).
Пример 11 Получение 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII)
Реакционную суспензию, состоящую из 68,6 г 21,17α-диацетоксипрегна-4,9(11)-диен-3,20-диона (XI), 34,3 г прокаленного ацетата калия в 686 мл абсолютного диметилсульфоксида, нагревают в токе инертного газа до 80°С и выдерживают в течение 2 ч, после чего реакционную смесь охлаждают и медленно выливают в 6,9 л воды. Перемешивают в течение 30 мин. Осадок отфильтровывают, промывают водой и сушат до постоянной массы. Получают 58,87 г соединения XII с выходом 99,8%.
Т. пл. 114-117°С. По данным ТСХ и ПМР анализов примесь соединения XI отсутствует. Сумма посторонних (родственных) стероидов менее 5%.
После очистки т.пл. 128-130.5°С (лит. 128-129°С из эфира [US4290963, 1981, пример 15].
Спектр ЯМР 1H (δ, м.д.; CDCl3): 6,76 (м, 1Н, 16-СН); 5,73 (уш.с, 1H, 4-CH); 5,53 (м, 1H, 11-CH); 5,02 (д, Jobs = 16,1 Гц (АВ-система), 1H, 21-СН2О); 4,90 (д, Jobs = 16,1 Гц (АВ-система), 1Н, 21-СН2О); 2,16 (с, 3Н, 21-ОС(О)СН3); 1,33 (с, 3H, 19-CH3); 0,86 (с, 3H, 18-CH3).
Пример 12 Получение 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона (XIII)
В работе используют штамм Nocardioides simplex ВКМ Ас-2033Д, который поддерживают на скошенной агаризованной кукурузно-глюкозной среде следующего состава (г/л): кукурузный экстракт 10,0; глюкоза 10,0; водопроводная вода (до 1 литра), рН 7,0-7,2. Выращивание проводят в течение 3-5 сут при температуре 30°С. Культуру, выросшую на скошенной агаризованной среде, используют для получения первого инокулята или хранят в течение 1-2 месяцев в холодильнике при температуре 4°С.
А) Выращивание 1-го инокулята
Смыв культуры N. simplex с поверхности агаризованной среды проводят стерильной водопроводной водой; при этом при разведении в 10 раз значение оптической плотности полученной суспензии клеток должно составлять 0,35-0,40 (длина кюветы 0,5 см, длина волны 536 нм, Specord (Германия)). Полученную суспензию клеток (2-3 мл) вносят в колбы Эрленмейера вместимостью 750 мл, содержащие 50 мл среды (г/л): глюкоза 10, соевый пептон 4, дрожжевой экстракт 4, MgSO4 0,5, KH2PO4 2, K2HPO4 4, рН 7,0-7,2. Инкубируют на качалке (220 об/мин) при температуре 30ºС в течение 42-48 ч.
Б) Выращивание 2-го инокулята
В свежую среду того же состава вносят 5 об.% выросшего первого инокулята и инкубируют в течение 24-26 ч в тех же условиях.
В) Выращивание трансформирующей культуры
Для получения культуры с 1(2)-дегидрогеназной активностью клетки N. simplex выращивают в ферментере АНКУМ-2М (Россия) с рабочим объемом 5 л в условиях периодического культивирования с подпиткой. Посевной материал (2-й инокулят) вносят в количестве 5 об.% в среду следующего состава (г/л): дрожжевой экстракт 10, соевый пептон 20, глюкоза 20, MgSO4 0,5, KH2PO4 2, K2HPO4 4, рН 7,0. Среду без глюкозы стерилизуют в колбе при избыточном давлении в 1 атм в течение 30 мин. Раствор глюкозы (30 г в 150 мл воды) стерилизуют отдельно (0,5 атм, 30 мин). Через 6-8 ч культивирования вносят ацетат кортизона (в виде 1 % спиртового раствора) для индукции синтеза 3-кетостероид-1-дегидрогеназы. Режим выращивания культуры в ферментере: температура 29°С; рН 6,5-7,5; скорость вращения вала мешалки от 350 до 800 об/мин; исходный расход воздуха от 0,25 до 4 л/мин. Продолжительность выращивания 36-38 ч.
Полученную культуральную жидкость с клетками используют сразу либо замораживают и хранят при температуре минус 18ºС для последующего использования. Концентрация клеток в культуральной жидкости (сухой вес) составляет 13.4 г /л.
Клетки микроорганизма осаждают центрифугированием при 5000 х g в течение 30 мин и используют для проведения процесса 1,2-дегидрирования или хранят при
минус 18°С в течение месяца до использования.
Инкубирование проводят в аэрируемых условиях на роторной качалке при температуре 25°С
Стероиды экстрагируют этилацетатом. Основные параметры процесса и содержание стероидных соединений в экстракте в сравнении с известными аналогами представлены в таблице 2. Выход и параметры качества 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона (XIII) (по данным ВЭЖХ), полученного после выделения без дополнительной очистки в сравнении с известным аналогом, представлены в таблице 3.
Т.пл. 173-175°С (лит. 172-174°С [US2864834 , 1958, пример 1]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 7,21 (д, J = 10.1 Гц, 1H, 1-СН); 6,76 (м, 1Н, 16-СН); 6,28 (дд, J = 10.1 Гц, J = 1.8 Гц, 1H, 2-CH); 6,06 (т, J = 1.8 Гц, 1H, 4-CH); 5,55 (м, 1H, 11-CH); 5,03 (д, Jobs = 16,1 Гц (АВ-система), 1H, 21-СН2О); 4,89 (д, Jobs = 16,1 Гц (АВ-система), 1Н, 21-СН2О); 2,17 (с, 3Н, 21-ОС(О)СН3); 1,42 (с, 3H, 19-CH3); 0,91 (с, 3H, 18-CH3).
Вариант 1. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) проводят в среде, содержащей раствор статистически метилированного β-циклодекстрина (МЦД) в 0,05 М Na-ацетатном буфере (pH 5), с концентрацией 10,0 г/л. Стероидный субстрат (0,5 г в 100% исчислении основного вещества) вносят в 46,25 мл среды в виде раствора в диметилсульфоксиде (ДМСО), содержащего также 20 мг менадиона (4% от массы субстрата), при этом мольное соотношение МЦД: субстрат составляет 1.5:1, а концентрация ДМСО составляет 4 %об., считая на конечный объем трансформационной среды (50 мл). Затем добавляют 3,75 мл культуральной жидкости, весовое соотношение биомасса:субстрат составляет 1:10. Инкубирование проводят в течение 6 ч в аэрируемых условиях (220 об/мин). Результаты (по данным ТСХ-анализа) представлены в таблице 2.
Вариант 2. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20,0 г/л (1,0 г стероидного субстрата в 100% исчислении основного вещества) осуществляют, как описано в варианте 1, но используют 38.34 мл раствора МЦД в 0.02 М растворе NaH2PO4 (подкисляют до pH 5,0) и 10,66 мл культуральной жидкости, при этом мольное соотношение МЦД:субстрат составляет 2:1, весовое соотношение биомасса:субстрат составляет 1:7. Стероидный субстрат и менадион вносят в растворе этанола. Инкубирование проводят в течение 14 ч. Результаты (по данным ТСХ-анализа) представлены в таблице 2.
Вариант 3. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20.0 г/л (1 г стероидного субстрата в 100% исчислении основного вещества) осуществляют, как описано в варианте 2, но используют 38,34 мл раствора МЦД в 0,05 М Na-ацетатном буфере (подкисляют до pH 5,0) и 10,66 мл культуральной жидкости. При этом мольное соотношение МЦД:субстрат составляет 2:1, а весовое соотношение биомасса: субстрат равно 1:7. Инкубирование проводят в течение 9 ч. Результаты (по данным ВЭЖХ-анализа) представлены в таблице 2.
Вариант 4. Трансформацию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20,0 г/л (24 г стероидного субстрата в 100% исчислении основного вещества) в ферментере АНКУМ-2М с рабочим объемом 1,2 л проводят в условиях варианта 3, но используют 805,5 мл раствора МЦД в 0,05 М Na-ацетатном буфере (подкисляют до pH 5,0) и 298.5 мл культуральной жидкости. При этом мольное соотношение МЦД:субстрат составляет 2:1, а весовое соотношение биомасса:субстрат равно 1:6. Инкубирование проводят в течение 14 ч при оборотах вала мешалки 800 об/мин, уровень растворенного кислорода поддерживают выше 30% от уровня насыщения. Результаты (по данным ВЭЖХ-анализа) представлены в таблице 2.
Вариант 5. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20.0 г/л (1 г стероидного субстрата в 100% исчислении основного вещества) осуществляют, как описано в варианте 2, но используют раствор МЦД в 0,02 М Na-цитратном буфере (pH 5.0) и 670 мг влажной пасты биомассы, хранившейся в замороженном виде при температуре минус 18°С в течение 1 месяца (соответствует 142,8 мг сухого веса биомассы). При этом мольное соотношение МЦД:субстрат составляет 2:1, весовое соотношение биомасса:субстрат составляет 1:7, а концентрация этанола - 8% из расчета на конечный объем трансформационной среды (50 мл), Инкубирование проводят в течение 11 ч. Результаты (по данным ТСХ-анализа) представлены в таблице 2.
Вариант 6. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20.0 г/л (1 г стероидного субстрата в 100% исчислении основного вещества) осуществляют, как описано в варианте 5, но используют раствор МЦД в 0.02 М растворе NaH2PO4 (подкислен до pH 5,0). Инкубирование проводят в течение 14 ч. Результаты (по данным ТСХ-анализа) представлены в таблице 2.
Вариант 7. Биоконверсию 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) в концентрации 20,0 г/л (1 г стероидного субстрата в 100% исчислении основного вещества) осуществляют, как описано в варианте 5, но используют раствор МЦД в 0,05 М Na-ацетатном буфере (pH 5,0). Инкубирование проводят в течение 9 ч. Результаты (по данным ТСХ-анализа) представлены в таблице 2.
Пример 13 Получение 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XVI)
К раствору 1,05 г CuCl в 633 мл ацетонитрила добавляют 45,48 г 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона (XIII), 180 мл тетрагидрофурана и 690 мл толуола. Реакционную массу охлаждают до температуры минус 40ºС и добавляют 304 мл 1.4 М раствора CH3MgBr в смеси толуола и ТГФ (3:1 по объему). Затем реакционную массу выдерживают при температуре минус 40ºС в течение 15 мин. По окончании реакции реакционную массу медленно выливают в 2,28 л 20% водного раствора NH4Cl при температуре не выше 5ºС при интенсивном перемешивании. После дополнительной выдержки в течение 30 мин при температуре 0-5ºС реакционную массу, содержащую 21-ацетокси-17α-гидроперокси-16α-метилпрегна-1,4,9(11)-триен-3,20-дион (XV), переносят в делительную воронку. Органический слой отделяют, промывают 20% водным раствором NH4Cl и водой.
К полученному раствору соединения XV в толуоле в токе инертного газа добавляют 544 мл ацетона и 15,8 г иодистого калия. Суспензию перемешивают при комнатной температуре в течение 1 ч. Затем добавляют 10% водный раствор сульфита натрия до обесцвечивания реакционной массы и 20 мл воды. После этого реакционную массу, содержащую 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-дион (XVI), переносят в делительную воронку. Органический слой отделяют, промывают 10% водным раствором сульфита натрия и водой. Растворитель упаривают в вакууме до прекращения погона, остаток толуола выгоняют метанолом. Раствор соединения XVI в метаноле выливают в 10-кратное количество воды. Осадок отфильтровывают, промывают водой, сушат до постоянной массы.
Получают 46,7 г соединения с молярным выходом 94,4%, считая на соединение XIII. Т. пл. 188-192ºС. После кристаллизации из диэтилового эфира получают 35,63 г соединения XVI с выходом 76,3% на очистке.
Т. пл. 207-208,5ºС (лит. 206-211°С [US3947409, 1976, пример 1]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 7,17 (д, J = 10.1 Гц, 1H, 1-СН); 6,26 (дд, J = 10.1 Гц, J = 1.8 Гц, 1H, 2-CH); 6,05 (т, J = 1.8 Гц, 1H, 4-CH); 5,53 (м, 1H, 11-CH); 4,96 (д, Jobs = 17,5 Гц (АВ-система), 1H, 21-СН2О); 4,79 (д, Jobs = 17,5 Гц (АВ-система), 1Н, 21-СН2О); 2,16 (с, 3Н, 21-ОС(О)СН3); 1,38 (с, 3H, 19-CH3); 0,91 ( д, J = 7.1 Гц, 3Н, 16-CH3); 0,73 (с, 3H, 18-CH3).
Пример 14 Получение 21-ацетокси-9α-бром-11β,17α-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона (XVII)
К раствору 50 г 21-ацетокси-17α-гидрокси-16α-метилпрегна-1,4,9(11)-триен-3,20-диона (XVI) в 350 мл ацетона, охлажденному до температуры 10°С, добавляют 100 мл дистиллированной воды и 54 мл ~6,5% водного раствора HClO4. Затем к реакционной массе добавляют 25,24 г дибромантина порциями в течение 25-30 мин. Реакционную массу выдерживают в течение 45 мин, добавляют 10% раствор сульфита натрия в воде до обесцвечивания.
Затем к реакционной массе добавляют 560 мл воды. Осадок отфильтровывают, промывают водой. Получают 78 г влажной пасты соединения XVII, которую без высушивания используют для получения соединения XVIII.
Спектр ЯМР 1H высушенного в вакууме образца (δ, м.д.; DMSO-d6): 7,33 (д, J = 10.1 Гц, 1H, 1-СН); 6,22 (дд, J = 10.1 Гц, J = 1.8 Гц, 1H, 2-CH); 5,95 (уш. с, 1H, 4-CH); 5,57 (д, J = 5.4 Гц, 1H, 11-ОН); 5.22 (с, 1 Н, 17-OH); 5,03 (д, Jobs = 17,6 Гц (АВ-система), 1H, 21-СН2О); 4,78 (д, Jobs = 17,6 Гц (АВ-система), 1Н, 21-СН2О); 4.57 ( уш. с, 1 Н, 11-СН); 2,08 (с, 3Н, 21-ОС(О)СН3); 1,63 (с, 3H, 19-CH3); 0,88 (с, 3H, 18-CH3); 0,79 (д, J = 7.3 Гц, 3Н, 16-CH3).
Пример 15 Получение 17α,21-дигидрокси-16α-метил-9β(11)-эпоксипрегна-1,4-диен-3,20-диона (XVIII)
Вариант 1. К реакционной массе, полученной в примере 14 после обесцвечивания 10% раствором сульфита натрия, добавляют 300 мл метанола. Реакционную массу охлаждают до температуры 0°С, добавляют 200 мл 7,5% водного раствора NaOH и выдерживают при этой же температуре в течение 40 мин. Затем реакционную массу нейтрализуют уксусной кислотой. Растворитель упаривают до небольшого объема, к остатку добавляют 500 мл воды. Осадок отфильтровывают, промывают водой, сушат до постоянной массы. Получают 42,51 г соединения XVIII с выходом 90,97%, считая на соединение XVII, содержание примесей суммарно менее 1%.
Т.пл. 257-259°С (лит. 238-239,5°С [US4990612, 1991, пример 8]).
Спектр ЯМР 1H (δ, м.д.; CDCl3): 6,58 (д, J = 10.1 Гц, 1H, 1-СН); 6,18 (дд, J = 10.1 Гц, J = 1.8 Гц, 1H, 2-CH); 6,14 (уш. с, 1H, 4-CH); 4,60 (дд, Jobs = 20,0 Гц (АВ-система), J = 4.5 Гц, 1H, 21-СН2О); 4,22 (дд, Jobs = 20,0 Гц (АВ-система), J = 4.5 Гц, 1Н, 21-СН2О); 3.21 (уш. с, 1 Н, 11-СН); 3.06 (т, J = 4.5 Гц, 21-ОН); 1,42 (с, 3H, 19-CH3); 0,92 (с, 3H, 18-CH3); 0,89 (д, J = 7.2 Гц, 3Н, 16-CH3).
Вариант 2. 7 г влажной пасты 21-ацетокси-9α-бром-11β,17α-дигидрокси-16α-метилпрегна-1,4-диен-3,20-диона XVII, полученной по примеру 14 из 4,45 г соединения XVI, растворяют в 56 мл метанола и добавляют в токе инертного газа 2,57 г K2CO3. Реакционную массу выдерживают при комнатной температуре в течение 2 ч. По окончании реакции реакционную массу нейтрализуют уксусной кислотой и добавляют 24 мл воды. Осадок отфильтровывают, промывают водой. Получают 3.6 г соединения XVIII с выходом 86,5%.
Пример 16 Получение 11β,17α,21-тригидрокси-16α-метил-9α-фторпрегна-1,4-диен-3,20-диона (дексаметазона, I)
К 213 мл 70% водной фтористоводородной кислоты при температуре не выше минус 20°С добавляют равномерными порциями 42,51 г 17α,21-дигидрокси-16α-метил-9β(11)-эпоксипрегна-1,4-диен-3,20-диона (XVIII). Реакционную массу перемешивают в течение 1,5 ч при этой же температуре. По окончании реакции реакционную массу выливают в 4,68 л ~2% водного раствора аммиака при температуре не выше 10ºС. Суспензию выдерживают при температуре 0-2ºС в течение 2 ч. Осадок отфильтровывают, промывают водой, сушат до постоянной массы. Получают 40,71 г дексаметазона (I) с выходом 90,91% и содержанием основного вещества 91,1% (ВЭЖХ).
После очистки получают дексаметазон с содержанием основного вещества 97,8% (ВЭЖХ). Выход на очистке составляет 81,2%.
Т.пл. 264-265°С (лит. 263°С с разложением [US3947409, 1976, пример 7]).
Спектр ЯМР 1H (δ, м.д.; DMSO-d6): 7,28 (д, J = 10.1 Гц, 1H, 1-СН); 6,21 (дд, J = 10.1 Гц, J = 1.8 Гц, 1H, 2-CH); 5,99 (уш. с, 1H, 4-CH); 5,26 (уш. с, 1H, 11-ОН); 4.93 (с, 1Н, 17-OH); 4.65 (т, J = 5.9 Гц, 1Н, 21-ОН); 4,49 (дд, Jobs = 19,2 Гц (АВ-система), J = 5.9 Гц, 1H, 21-СН2О); 4.13 (м, 1Н, 11-СН);); 4,07 (дд, Jobs = 19,2 Гц (АВ-система), J = 5.9 Гц, 1Н, 21-СН2О); 1,47 (с, 3H, 19-CH3); 0,85 (с, 3H, 18-CH3); 0,77 (д, J = 7.3 Гц, 3Н, 16-CH3)
Таблица 1.
Основные параметры процесса получения андроста-4,9(11)-диен-3,17-диона (IV) заявляемым способом и известными аналогами
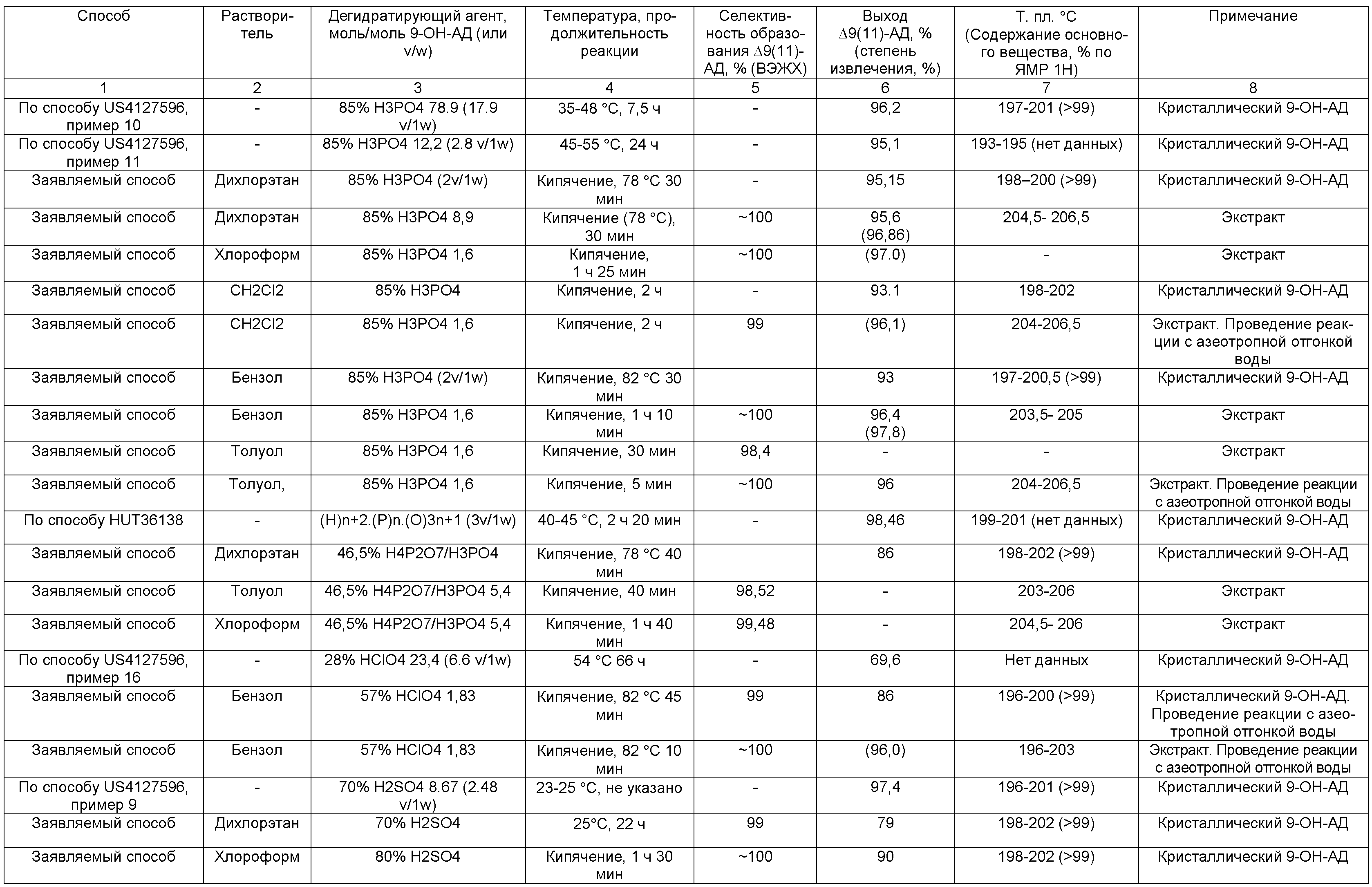
Таблица 2
Основные параметры процесса микробиологического 1,2-дегидрирования 21-ацетоксипрегна-4,9(11),16-триен-3,20-диона (XII) заявляемым способом и известными аналогами
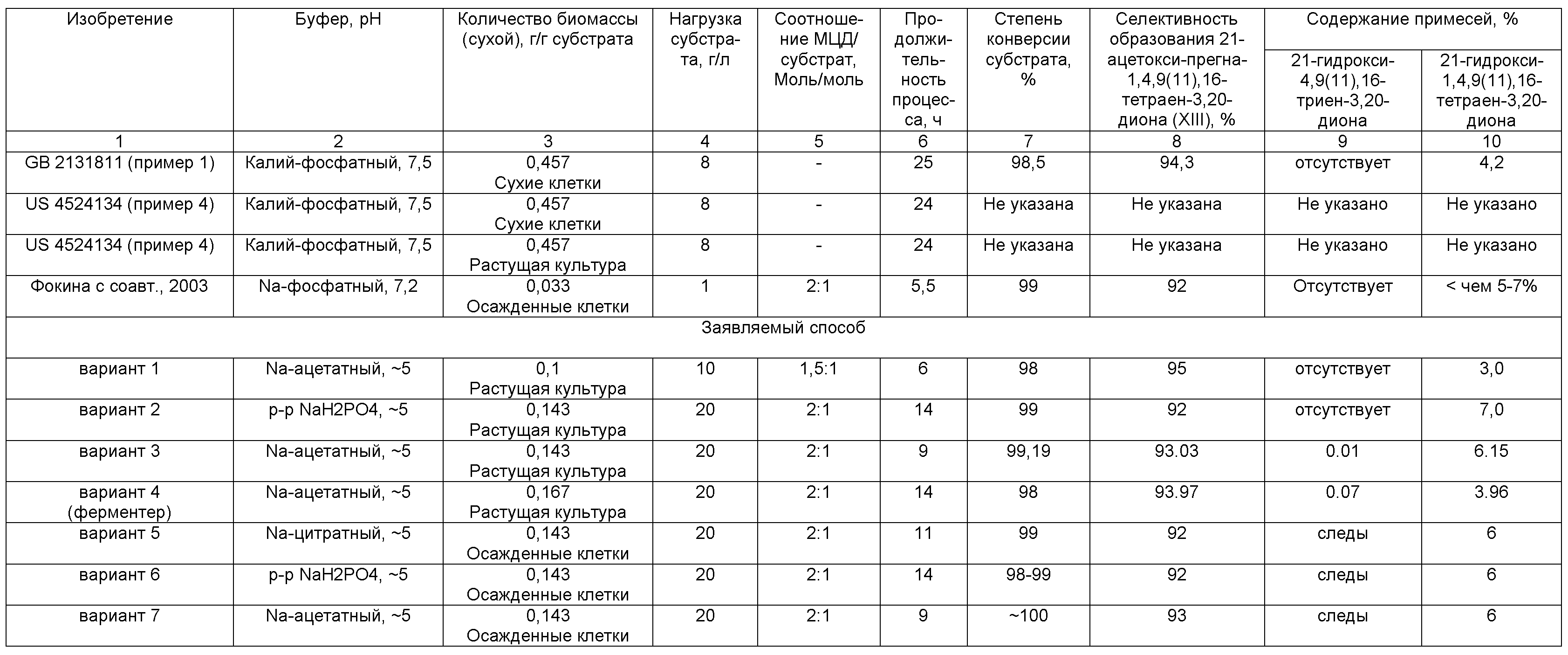
Таблица 3
Выход и параметры качества выделенного 21-ацетоксипрегна-1,4,9(11),16-тетраен-3,20-диона (XIII) по заявляемому способу и известному аналогу
|
Способ газолазерной резки крупногабаритных деталей из композиционных материалов и устройство для его осуществления
Полимерная композиция
Грунтовочная композиция
Способ адаптации рабочей части аэродинамической трубы для получения безындукционного обтекания моделей летательных аппаратов и устройство для его осуществления
Способ получения конъюгата нона-β-(1→3)-глюкозида с бычьим сывороточным альбумином скваратным методом
Износостойкий сплав на основе никеля для нанесения износо- и коррозионно-стойких покрытий на конструкционные элементы микроплазменным или сверхзвуковым газодинамическим напылением
Способ измерения температуры поверхности конструкции резистивным чувствительным элементом, устройство для его осуществления и способ изготовления устройства
Способ определения температурной характеристики резисторного чувствительного элемента, устройство для его осуществления и способ изготовления устройства
Устройство для измерения звукового давления
Устройство для измерения давления, температуры и теплового потока
Способ газолазерной резки крупногабаритных деталей из композиционных материалов и устройство для его осуществления
Полимерная композиция
Грунтовочная композиция
Способ адаптации рабочей части аэродинамической трубы для получения безындукционного обтекания моделей летательных аппаратов и устройство для его осуществления
Способ определения запасов устойчивости рулевого привода и устройство для его осуществления
Способ измерения температуры термопарами, измерительная информационная система для его осуществления и температурный переходник
Способ получения конъюгата нона-β-(1→3)-глюкозида с бычьим сывороточным альбумином скваратным методом
Износостойкий сплав на основе никеля для нанесения износо- и коррозионно-стойких покрытий на конструкционные элементы микроплазменным или сверхзвуковым газодинамическим напылением
Способ измерения температуры поверхности конструкции резистивным чувствительным элементом, устройство для его осуществления и способ изготовления устройства
Способ определения температурной характеристики резисторного чувствительного элемента, устройство для его осуществления и способ изготовления устройства

