Результат интеллектуальной деятельности: КОМБИНАЦИИ ИНГИБИТОРОВ ФОСФОИНОЗИТИД 3-КИНАЗЫ И ХИМИОТЕРАПЕВТИЧЕСКИХ АГЕНТОВ И СПОСОБЫ ПРИМЕНЕНИЯ
Вид РИД
Изобретение
ОТСЫЛКИ К РОДСТВЕННЫМ ЗАЯВКАМ
В данной безусловной заявке, поданной согласно 37 CFR §1.53(b), преимущество приоритета заявлено согласно 35 USC §119(е) в отношении предварительной заявки США с серийным №60/971773, поданной 12 сентября 2007 г., которая включена во всей своей полноте путем ссылки. ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение в целом относится к фармацевтическим комбинациям соединений, обладающих активностью против гиперпролиферативных расстройств, таких как рак, и включающих соединения, которые ингибируют активность Р13-киназы. Изобретение также относится к способам применения соединений для in vitro, in situ и in vivo диагностики или лечения клеток млекопитающих или ассоциированных патологических состояний.
ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ УРОВЕНЬ ТЕХНИКИ
Фосфатидилинозит является одним из ряда фосфолипидов, обнаруженных в клеточных мембранах и принимающих участие во внутриклеточной передаче сигнала. Передача сигнала клетками, опосредованная 3'-фосфорилированными фосфоинозитидами, вовлечена в ряд клеточных процессов, например, малигнизацию, передачу сигнала через ростовые факторы, воспаление и иммунитет (Rameh et al. (1999) J. Biol. Chem. 274: 8347-8350). Фермент, ответственный за образование таких фосфорилированных продуктов при передаче сигнала, фосфатидилинозитол-3-киназу (также обозначаемую как PI 3-киназа или Pl3K), изначально идентифицировали по активности, ассоциированной с вирусными онкобелками и тирозинкиназами - рецепторами ростовых факторов, которые фосфорилируют фосфатид ил инозит (PI) и его фосфорилированные производные по 3'-гидроксилу кольца инозита (Panayotou et al. (1992) Trends Cell Biol. 2: 358-60). Фосфоинозитид-3-киназы (Pl3K) представляют собой липидкиназы, которые фосфорилируют липиды по 3-гидроксильному остатку кольца инозита (Whitman et al. (1988) Nature, 332: 664). 3-Фосфорилированные фосфолипиды (PIP3), образованные с помощью Р13-киназ, действуют в качестве вторичных мессенджеров, вовлекающих киназы с липид-связывающими доменами (включая плекстрин-гомологичные (РН) участки), такие как Akt и PDK1, фосфоинозитидзависимая киназа-1 (Vivanco et al. (2002) Nature Rev. Cancer 2: 489; Phillips et al. (1998) Cancer 83: 41).
Семейство Pl3-киназ содержит по меньшей мере 15 разных ферментов, систематизированных по структурной гомологии и разделенных на 3 класса на основе гомологии последовательностей и продукта, образующегося в результате ферментативного катализа. Pl3-киназы I класса состоят из 2 субъединиц: каталитической субъединицы (110 кДа) и регуляторной субъединицы (85 кДа). Регуляторные субъединицы содержат SH2-домены (src-гомологичные домены 2) и связываются с остатками тирозина, фосфорилированными под действием рецепторов ростовых факторов, обладающих тирозинкиназной активностью, или продуктов онкогенов, тем самым запуская Pl3K-активность каталитической субъединицы р110, которая фосфорилирует свой липидный субстрат.Pl3-киназы 1 класса вовлечены в важные события передачи сигнала вниз по пути цитокинов, интегринов, ростовых факторов и иммунорецепторов, что предполагает, что регулирование этого пути может приводить к важным терапевтическим эффектам, таким как модулирование клеточной пролиферации и канцерогенеза. Pl3K I класса могут фосфорилировать фосфатидилинозит (Р1), фосфатидилинозит-4-фосфат и фосфатидилинозит-4,5-бифосфат (Р1Р2) с получением фосфатидилинозит-3-фосфата (PIP), фосфатидилинозит-3,4-бифосфата и фосфатидилинозит-3,4,5-трифосфата, соответственно. Pl3K II класса фосфорилируют Р1 и фосфатидилинозит-4-фосфат.Pl3K III класса могут фосфорилировать только Р1. Основной изоформой Pl3-киназы при раке является Pl3-киназа 1 класса - р110α (US 5824492; US 5846824; US 6274327). Другие изоформы вовлечены в сердечнососудистое и иммуновоспалительное заболевание (Workman P (2004) Biochem. Soc. Trans. 32: 393-396; Patel et al. (2004) Proc. Am. Assoc. of Cancer Res. (Abstract LB-247) 95th Annual Meeting, March 27-31, OrIando, Florida, USA; Ahmadi К and Waterfield MD (2004) "Phosphoinositide 3-Kinase: Function and Mechanisms" EncyClopedia of Biological Chemistry (Lennarz W J, Lane M D eds) Elsevier/Academic Press) и онкогенные мутации в Pl3-киназе (Samuels et al. (2004) Science 304: 554). Онкогенные мутации р110-альфа со значительной частотой обнаружены в солидных опухолях толстой кишки, молочной железы, головного мозга, печени, яичников, желудка, легких и головы и шеи. Нарушения в PTEN (гомологе фосфатазы и тензина с делецией на хромосоме 10) обнаруживаются при глиобластоме, меланоме, раке предстательной железы, эндометрия, яичников, молочной железы, легких, головы и шеи, гепатоцеллюлярном раке и раке щитовидной железы.
После первоначальной очистки и молекулярного клонирования Pl3-киназы было обнаружено, что она представляла собой гетеродимер, состоящий из субъединиц р85 и р110 (Otsu et al. (1991) Cell 65: 91-104; Hiles et al. (1992) Cell 70: 419-29). Впоследствии идентифицировано четыре разных Pl3K I класса, обозначенные как Pl3K-α (альфа), -β (бета), -δ (дельта) и -γ (гамма), каждая из которых состоит из разных каталитических субъединиц (110 кДа) и регуляторных субъединицы. Более конкретно, каждая из трех каталитических субъединиц, т.е. p110-альфа, p110-бета и р110-дельта, взаимодействует с одной и той же регуляторной субъединицей, р85; тогда как p110-гамма взаимодействует с другой регуляторной субъединицей, р101. Особенности экспрессии каждой из этих Pl3K в человеческих клетках и тканях также различны. В каждом из подтипов Pl3K-альфа, -бета и -дельта действие субъединицы р85 заключается в локализации Pl3-киназы на плазматической мембране благодаря взаимодействию ее SH2-домена с фосфорилированными тирозиновыми остатками (присутствующими в составе соответствующей последовательности) в белках-мишенях (Rameh et al. (1995) Cell, 83: 821-30; Volinia et al. (1992) Oncogene, 7: 789-93).
Путь Pl3-киназа/Ак1/РТЕМ представляет собой привлекательную мишень для разработки противораковых лекарственных средств, поскольку ожидается, что такие агенты будут ингибировать пролиферацию, реверсировать подавление апоптоза и преодолевать резистентность к цитотоксическим агентам в раковых клетках. Данные об ингибиторах Pl3-киназ опубликованы (Yaguchi et al. (2006) Jour. of the Nat. Cancer Inst. 98(8): 545-556; US 7173029; US 7037915; US 6608056; US 6608053; US 6838457; US 6770641; US 6653320; US 6403588; WO 2006/046031; WO 2006/046035; WO 2006/046040; WO 2007/042806; WO 2007/042810; WO 2004/017950; US 2004/092561; WO 2004/007491; WO 2004/006916; WO 2003/037886; US 2003/149074; WO 2003/035618; WO 2003/034997; US 2003/158212; EP 1417976; US 2004/053946; JP 2001247477; JP 08175990; JP 08176070). Аналоги вортманнина обладают Pl3-киназной активностью в организме млекопитающих (US 6703414; WO 97/15658).
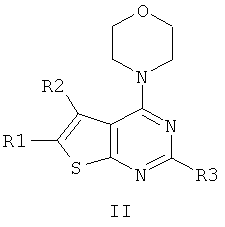
Соединения тиенопиримидинов формул I и II обладают p110-альфа-связывающей, Pl3-киназа-ингибирующей активностью и ингибируют рост раковых клеток (WO 2006/046031; US 2008/0039459; US 2008/0076768; US 2008/0076758; WO 2008/070740; WO 2008/073785.
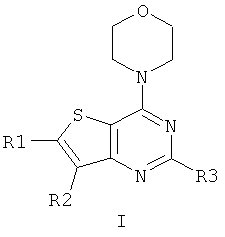
Соединение формулы I, GDC-0941 (Genentech Inc.), представляет собой селективный, перорально биодоступный ингибитор Pl3K с многообещающими фармакокинетическими и фармацевтическими свойствами (Belvin et al., American AssoClation for Cancer Research Annual Meeting 2008, 99th: April 15, Abstract 4004; Folkes et al., American AssoClation for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-146; Friedman et al., American AssoClation for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-110).
В настоящее время в лечении рака общепризнано использование комбинаций противораковых фармацевтических терапевтических средств, вводимых в режиме одновременного или последовательного дозирования. Успешная комбинированная терапия обеспечивает улучшенный и даже синергический эффект по сравнению с монотерапией, т.е. фармацевтическим лечением, ограниченным одним лекарственным средством. Проведено изучение комбинированной терапии для лечения таких гиперпролиферативных расстройств, как рак, включая противоопухолевую активность эрлотиниба в комбинации с капецитабином в моделях опухолевых ксенотрансплантатов человека (Ouchi et al. (2006) Cancer Chemother. Pharmacol. 57: 693-702) и эрлотиниба в комбинации с гемцитабином и цисплатином в моделях опухолевых ксенотрансплантатов немелкоклеточного рака легкого (NSClC) (Higgins et al. (2004) Anti-Cancer Drugs 15:503-512). Доклиническое изучение составляет основу для прогнозирования синергического действия противораковых фармацевтических терапевтических комбинаций на клинической стадии, включая капецитабин и таксаны для лечения рака молочной железы (Sawada et al. (1998) Clin. Cancer Res. 4: 1013-1019). Некоторые дозы и схемы проведения комбинированной терапии капецитабина и таксана могут улучшать безопасность без нарушения эффективности (O'Shaughnessy et al. (2006) Clin. Breast Cancer, Apr 7(1):42-50). Синергические эффекты противогрибковых комбинаций in vitro коррелируют с синергическим действием на клинической стадии (Steinbach et al. (2003) Clin. Inf. Dis. Oct 1; 37 Suppl. 3: S188-224).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение в целом относится к соединениям тиенопиримидинов формул I и II с противораковой активностью и более конкретно с Pl3-киназа-ингибирующей активностью, вводимым в комбинации с химиотерапевтическими агентами для ингибирования роста раковых клеток. Некоторые комбинации соединений формулы I и II с химиотерапевтическими агентами демонстрируют синергические эффекты в ингибировании роста раковых клеток in vitro и in vivo. Комбинации и способы по изобретению могут быть полезны в лечении гиперпролиферативных расстройств, таких как рак. Такие композиции могут ингибировать опухолевый рост у млекопитающих и могут быть полезны для лечения раковых пациентов-людей.
В одном из аспектов изобретение включает способ лечения гиперпролиферативного расстройства, включающий введение терапевтической комбинации млекопитающему в виде комбинированной композиции или путем чередования, при этом терапевтическая комбинация содержит терапевтически эффективное количество соединения, имеющего формулу I или II, и терапевтически эффективное количество химиотерапевтического агента, выбранного из эрлотиниба, доцетаксела, 5-FU (5-фторурацила), гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2 (ингибитора Akt-1/2), HPPD, рапамицина и лапатиниба.
Изобретение также относится к способам применения композиций для диагностики или лечения in vitro, in situ и in vivo клеток, организмов млекопитающих или ассоциированных патологических состояний.
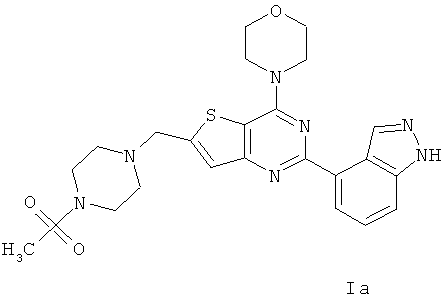
Согласно аспекту изобретения предложены терапевтические комбинации, содержащие 4-(2-(1Н-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин (US 2008/0076768; WO 2006/046031), также известный как GDC-0941 (Genentech, Inc.) и имеющий формулу Ia, и терапевтически эффективное количество химиотерапевтического агента, выбранного из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба.
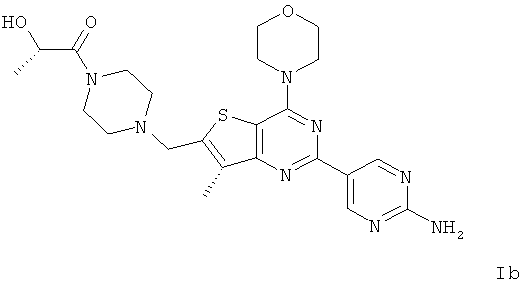
Согласно аспекту изобретения предложены терапевтические комбинации, содержащие (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1 -ил)-2-гидроксипропан-1-он (WO 2008/070740), имеющий формулу Ib, и терапевтически эффективное количество химиотерапевтического агента, выбранного из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба.
Согласно аспекту изобретения предложены терапевтические комбинации, содержащие 4-(2-(1Н-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[2,3-d]пиримидин-4-ил)морфолин (US 2008/0076758; WO 2006/046031), имеющий формулу IIa, и терапевтически эффективное количество химиотерапевтического агента, выбранного из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба.
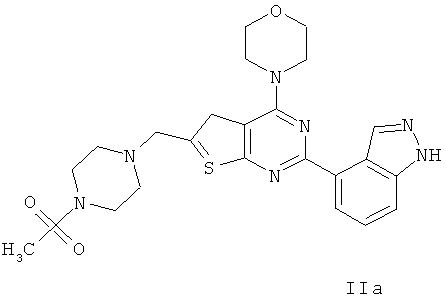
Соединения формулы Ia, Ib и IIa являются перорально биодоступными и в виде отдельных агентов обладают противоопухолевой активностью во многих моделях рака человека.
Соединения формулы I и II включают все стереоизомеры, геометрические изомеры, таутомеры, метаболиты и их фармацевтически приемлемые соли. Некоторые соединения формулы I и II являются сильнодействующими ингибиторами Pl3K, обладающими такими же физико-химическими и фармакокинетическими свойствами, как и лекарственные средства. Некоторые соединения формулы I и II демонстрируют повышенную селективность по отношению к Pl3K класса Ia по сравнению с классом Ib, в частности, к подтипу p110-альфа.
Фармацевтические композиции и терапевтические комбинации по изобретению содержат химиотерапевтический агент, выбранный из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба.
Кроме того, фармацевтические композиции по изобретению могут содержать фармацевтически приемлемый носитель.
Согласно другому аспекту изобретения предложены способы лечения гиперпролиферативного заболевания или расстройства, модулируемого PI3-киназами, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективных количеств соединения формулы I или II и химиотерапевтического агента. Соединение формулы I или II и химиотерапевтический агент могут быть изготовлены для совместного комбинированного введения в виде фармацевтической композиции, или они могут быть введены по отдельности путем чередования (последовательно) в виде терапевтической комбинации.
Согласно другому аспекту изобретения предложены способы лечения гиперпролиферативного расстройства, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективных количеств соединения формулы I или II и химиотерапевтического агента.
В следующем аспекте согласно настоящему изобретению предложен способ применения фармацевтической композиции по изобретению для лечения заболевания или состояния, модулируемого Pl3-киназой, у млекопитающего.
Дополнительный аспект изобретения относится к применению фармацевтической композиции по изобретению в изготовлении лекарства для лечения заболевания или состояния, модулируемого Pl3-киназой, у млекопитающего.
Другой аспект изобретения включает изделия или наборы, содержащие соединение формулы I или II, химиотерапевтический агент, контейнер и возможно инструкцию по медицинскому применению препарата или этикетку с показанием лечения.
Другой аспект изобретения относится к продукту, содержащему соединение, имеющее формулу I или II, и химиотерапевтический агент, выбранный из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба; в виде комбинированной композиции для раздельного, одновременного или последовательного применения в лечений гиперпролиферативного расстройства.
Другой аспект изобретения включает способ определения соединений, которые могут быть использованы в комбинации для лечения рака, включающий: а) введение терапевтической комбинации соединения, имеющего формулу I или II, и химиотерапевтического агента в НЕК2(рецептор-2 эпидермального фактора роста человека)-амплифицирующие раковые клетки молочной железы в ламинин-обогащенных, воссозданных содержащих базальные мембраны средах (basement membrame media), при этом химиотерапевтический агент направленно воздействует на рецептор HER2, связывается с ним или модулирует его, и б) измерение ингибирования клеточной пролиферации, где незлокачественные и злокачественные клетки молочной железы различают на основании одного или более чем одного фенотипического различия, выбранного из жизнеспособности клеток и ацинарного морфогенеза.
Другой аспект изобретения включает способ определения соединений, которые могут быть использованы в комбинации для лечения рака, включающий: а) введение терапевтической комбинации по пункту I формулы изобретения в линию опухолевых клеток in vitro с мутацией K-ras и б) измерение синергического или несинергического эффекта.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг.1-А показана сводная информация in vitro анализов клеточной пролиферации с использованием комбинаций соединения формулы Ia и различных химиотерапевтических агентов, вводимых одновременно. Клеточные линии охарактеризованы по типу опухоли и наличию известной мутации. Проведено сравнение индивидуальных измеренных величин EC50 для химиотерапевтического агента и соединения формулы Ia (GDC-0941) с величиной EC50 для комбинации и показатель в баллах для комбинации (combination index score) рассчитан по методу Chou и Talalay (Chou, Т. and Talalay, P. (1984) Adv. Enzyme Regul. 22: 27-55). Эффективность синергического действия оценена в баллах с использованием системы ранжирования и приведена в последней колонке.
На Фиг.1-В показана сводная информация in vitro анализов клеточной пролиферации с использованием комбинаций соединения формулы IIa и различных химиотерапевтических агентов. Клеточные линии охарактеризованы по типу опухоли и наличию мутации Ras. Проведено сравнение индивидуальных измеренных величин ЕС50 для химиотерапевтического агента и соединения формулы IIa с величиной ЕС50 для комбинации и показатель в баллах для комбинации рассчитан по методу Chou и Talalay (Chou, Т. and Talalay, P. (1984) Adv. Enzyme Regul. 22: 27-55). Эффективность синергического действия оценена в баллах с использованием системы ранжирования Chou и Talalay.
На Фиг.1-С показана сводная информация in vitro анализов клеточной пролиферации с использованием комбинаций соединения формулы Ib и различных химиотерапевтических агентов. Клеточные линии охарактеризованы по типу опухоли и наличию мутации Ras. Проведено сравнение индивидуальных измеренных величин EC50 для химиотерапевтического агента и соединения формулы Ib с величиной EC50 для комбинации и показатель в баллах для комбинации рассчитан по методу Chou и Talalay (Chou, Т. and Talalay, P. (1984) Adv. Enzyme Regul. 22: 27-55). Эффективность синергического действия оценена в баллах с использованием системы ранжирования Chou и Talalay.
На Фиг.2 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, 5-FU, соединения формулы Ia (GDC-0941) и комбинации 5-FU и соединения формулы Ia. Клетки MDA-MB-361 (тип опухоли молочной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением 5-FU (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения 5-FU (низ).
На Фиг.3 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, гемцитабина, соединения формулы Ia (GDC-0941) и комбинации гемцитабина и соединения формулы Ia. Клетки Cal-51 (тип опухоли молочной железы) обработаны одновременно (верх) и обработаны впоследствии соединением формулы Ia через 4 часа после введения гемцитабина (низ).
На Фиг.4 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, гемцитабина, соединения формулы Ia (GDC-0941) и комбинации гемцитабина и соединения формулы Ia. Клетки MDA-MB-361 (тип опухоли молочной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением гемцитабина (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения гемцитабина (низ).
На Фиг.5 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, эрлотиниба, соединения формулы Ia (GDC-0941) и комбинации эрлотиниба и соединения формулы Ia. Клетки А549 (тип опухоли легкого с K-ras G12C) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением эрлотиниба (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения эрлотиниба (низ).
На Фиг.6 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, эрлотиниба, соединения формулы Ia (GDC-0941) и комбинации эрлотиниба и соединения формулы Ia. Клетки Н23 (тип опухоли легкого с мутацией K-ras-G12C) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением эрлотиниба (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения эрлотиниба (низ).
На Фиг.7 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС5о), справа налево, темозоломида, соединения формулы Ia (GDC-0941) и комбинации темозоломида и соединения формулы Ia. Клетки U87 (тип опухоли - глиома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ).
На Фиг.8 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, темозоломида, соединения формулы Ia (GDC-0941) и комбинации темозоломида и соединения формулы Ia. Клетки А375 (тип опухоли - меланома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ).
На Фиг.9 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, темозоломида, соединения формулы Ia (GDC-0941) и комбинации темозоломида и соединения формулы Ia. Клетки MALME-3M (тип опухоли - меланома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ).
На Фиг.10 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, доксорубицина, соединения формулы Ia (GDC-0941) и комбинации доксорубицина и соединения формулы Ia. Клетки SKOV3 (тип опухоли яичников) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением доксорубицина (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения доксорубицина (низ).
На Фиг.11 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, доцетаксела, соединения формулы Ia (GDC-0941) и комбинации доцетаксела и соединения формулы Ia. Клетки РС3 (тип опухоли предстательной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением доцетаксела (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения доцетаксела (низ).
На Фиг.12 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки MDA-MB 361 (тип опухоли молочной железы), при варьировании концентраций (начиная от 4Х EC50), справа налево: (верх) 5-FU, соединения формулы IIa и комбинации одновременно вводимых 5-FU и соединения формулы IIa; (середина) доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa; и (низ) гемцитабина, соединения формулы IIa и комбинации одновременно вводимых гемцитабина и соединения формулы IIa.
На Фиг.13 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют: (верх) жизнеспособные клетки МТ3 (тип опухоли молочной железы) при варьировании концентраций (начиная от 4Х ЕС50), справа налево, доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa; и (низ) жизнеспособные клетки U87 (тип опухоли - глиома) при варьировании концентраций (начиная от 4Х ЕС50), справа налево, темозоломида, соединения формулы IIa и комбинации одновременно вводимых темозоломида и соединения формулы IIa.
На Фиг.14 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки ZR75-1 (тип опухоли молочной железы) при варьировании концентраций (начиная от 4Х ЕС50), справа налево: (верх) 5-FU, соединения формулы IIa и комбинации одновременно вводимых 5-FU и соединения формулы IIa; и (низ) доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa.
На Фиг.15 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) эрлотиниба и соединения формулы Ia (GDC-0941) из Фиг.1-А против линий опухолевых клеток без мутаций Ras (Ras WT (дикий тип), эксперименты 41, 42, 73-75, 77, 79-81, 83, 84, 86) и с мутациями Ras (мут. Ras, эксперименты 40, 69-72, 76, 78, 82, 144, 145).
На Фиг.16 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) PD-0325901 и соединения формулы Ia (GDC-0941) из Фиг.1-А против линий опухолевых клеток без мутаций Ras (Ras WT, эксперименты 22-24, 26-28, 31-33, 36-38, 55, 59, 61, 63-66, 85, 89-98, 149, 161, 162) и с мутациями Ras (мут. Ras, эксперименты 25, 30, 34, 35, 39, 56-58, 60,62,67,68, 146-148,150).
На Фиг.17 показаны результаты для синергидной линии опухолевых клеток MDA-MB-361 и не-синергидной линии опухолевых клеток МТ-3 в зависимости от времени обработки гемцитабином при уровнях дозировки, равных ЕС80. Уровни рАКТ (фосфо-АКТ) измеряли для момента времени Т=0 (без обработки, UT), 1 ч, 4 ч, 6 ч и 24 ч.
На Фиг.18 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) доцетаксела, 5-FU или гемцитабина и соединения формулы Ia (GDC-0941) из Фиг.1-А против линий опухолевых клеток, которые демонстрируют увеличение уровня pAkt или не демонстрируют никакого увеличения в уровне pAkt.
На Фиг.19 показаны результаты проточной цитометрии с использованием FACS (клеточный сортер с активацией флуоресценции): (верх) клеток МВ361 (слева направо), необработанных, обработанных соединением формулы Ia, 5FU и сначала 5FU, затем соединением формулы Ia (GDC-0941); (середина) клеток РС3 (слева направо), необработанных, обработанных соединением формулы Ia, доцетакселом, одновременно соединением формулы Ia и доцетакселом, сначала соединением формулы Ia, затем доцетакселом и сначала доцетакселом, затем соединением формулы Ia; и (низ) клеток МВ361 (слева направо), необработанных, обработанных соединением формулы Ia, гемцитабином и сначала гемцитабином, затем соединением формулы Ia.
На Фиг.20 показана обработка клеток ВТ474М1 в объемной (3D) культуре. Ацинусный рост (aClnni growth) и морфогенез коррелирует с продуцированием клеткой АТФ, выраженным в относительных световых единицах (RLU), в среде с добавлением 10% FBS (фетальная телячья сыворотка) в присутствии и в отсутствие 1 нМ херегулина-1 в результате обработки (слева направо): средой, DMSO (диметилсульфоксид), комбинацией 20 мкг/мл трастузумаба и 25 мкг/мл пертузумаба, 250 нМ соединения формулы Ia (GDC-0941) и комбинацией 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба и 250 нМ соединения формулы Ia.
На Фиг.21 показана обработка клеток ВТ474М1 в объемной (3D) культуре. Ацинусный рост и морфогенез коррелирует с продуцированием клеткой АТФ, выраженным в относительных световых единицах (RLU), в среде с добавлением 10% FBS в присутствии и в отсутствие 1 нМ херегулина-1 в результате обработки (слева направо): средой, DMSO, комбинацией 20 мкг/мл трастузумаба и 25 мкг/мл пертузумаба, 250 нМ соединения формулы IIa и комбинацией 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба и 250 нМ соединения формулы IIa.
На Фиг.21-А показана обработка клеток ВТ474М1 в объемной (3D) культуре. Ацинусный рост и морфогенез коррелирует с продуцированием клеткой АТФ, выраженным в относительных световых единицах (RLU), в среде с добавлением 10% FBS в присутствии и в отсутствие 1 нМ херегулина-1 в результате обработки (слева направо): DMSO, комбинацией 20 мкг/мл трастузумаба и 25 мкг/мл пертузумаба, 20 нМ соединения формулы Ib и комбинацией 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба и 20 нМ соединения формулы Ib.
На Фиг.22 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 (Charles River Labs) с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 150 мг/кг соединения формулы Ia (GDC-0941), 5 мг/кг доцетаксела и комбинацию соединения формулы Ia (150 мг/кг) и доцетаксела (5 мг/кг). Мышам вводили доцетаксел на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) внутривенно, тогда как соединение формулы Ia вводили ежесуточно в течение 21 суток путем перорального чреззондового кормления.
На Фиг.23 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 (Charles River Labs) с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 1-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 37,5 мг/кг соединения формулы IIa, 5 мг/кг доцетаксела и комбинацию 37,5 мг/кг соединения формулы IIa и 5 мг/кг доцетаксела. Мышам вводили доцетаксел на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) внутривенно, тогда как соединение формулы IIa вводили ежесуточно в течение 21 суток путем перорального чреззондового кормления.
На Фиг.24 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии NMRI с ксенотрансплантатами эксплантатов первичной опухоли молочной железы MAXF 401, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы Ia (GDC-0941), 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы Ia и 15 мг/кг доцетаксела. Мышам вводили внутривенно доцетаксел на 0-е сутки и 11-е сутки, тогда как соединение формулы Ia вводили на сутки 0-4, 11-17 и 21-28 путем перорального чреззондового кормления.
На Фиг.25 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами эксплантатов первичной опухоли молочной железы MAXF 401, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы IIa, 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы IIa и 15 мг/кг доцетаксела. Мышам вводили внутривенно доцетаксел на 0-е сутки и 11-е сутки, тогда как соединение формулы IIa вводили на сутки 0-3, 11-17 и 21-28 путем перорального чреззондового кормления.
На Фиг.26 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами эксплантатов первичной опухоли молочной железы MAXF 1162, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы Ia (GDC-0941), 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы Ia и 15 мг/кг доцетаксела. Мышам вводили внутривенно доцетаксел на 0-е, 11-е, 22-е и 44-е сутки, а соединение формулы Ia вводили на сутки 0-5, 11-16, 22-27, 30-32, 42 и 44 путем перорального чреззондового кормления.
На Фиг.27 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами первичной опухоли молочной железы MAXF 1162, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы IIa, 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы IIa и 15 мг/кг доцетаксела. Мышам вводили внутривенно доцетаксел на 0-е, 11-е, 22-е и 44-е сутки, а соединение формулы IIa вводили на сутки 0-5, 11-16, 22-23, 29-31 и 35-38 путем перорального чреззондового кормления.
На Фиг.28 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии CRL с ксенотрансплантатами опухоли немелкоклеточного рака легкого (NSClC) NCl-Н2122, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 50 мг/кг соединения формулы Ia (GDC-0941), 75 мг/кг эрлотиниба и комбинацию 50 мг/кг соединения формулы Ia и 75 мг/кг эрлотиниба. Мышам вводили ежесуточно эрлотиниб и соединение формулы Ia ежесуточно в течение 16 суток путем перорального чреззондового кормления.
На Фиг.29 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии CRL с ксенотрансплантатами опухоли немелкоклеточного рака легкого (NSClC) NCl-Н2122, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 50 мг/кг соединения формулы IIa, 75 мг/кг эрлотиниба и комбинацию 50 мг/кг соединения формулы IIa и 75 мг/кг эрлотиниба. Мышам вводили ежесуточно эрлотиниб и соединение формулы IIa в течение 14 суток (окончание исследования) путем перорального чреззондового кормления.
На Фиг.30 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии HRLN с ксенотрансплантатами клеток опухоли молочной железы MCF-7 (мутант по Pl3K), которым на 0-е сутки вводили: разбавитель МСТ и PBS (МСТ: 0,5% метилцеллюлозы/0,2% Твина 80, и PBS: забуференный фосфатом физиологический раствор), контроль IgG (иммуноглобулин G) (5 мг/кг), мышиное антитело против VEGF (сосудистый эндотелиальный ростовой фактор) mB20-4.1 (5 мг/кг), соединение формулы Ia (GDC-0941) (150 мг/кг) и комбинацию соединения формулы Ia (150 мг/кг) и мышиного антитела против VEGF mB20-4.1 (5 мг/кг). Животным вводили контроль IgG и mB20-4.1 внутрибрюшинно дважды в неделю в течение 3 недель, а соединение формулы Ia ежесуточно в течение 21 суток путем перорального чреззондового кормления, и мониторинг опухолевого роста проводили в течение дополнительных 41 суток (общее количество суток на одно исследование составляло 62). Соединение формулы Ia и mB20-4.1 вводили одновременно.
На Фиг.31 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии HRLN с ксенотрансплантатами клеток опухоли молочной железы MCF-7 (мутант по Pl3K), которым на 0-е сутки вводили: разбавитель МСТ и PBS (МСТ: 0,5% метилцеллюлозы/0,2% Твина 80, и PBS: забуференный фосфатом физиологический раствор), контроль IgG (5 мг/кг), мышиное антитело против VEGF mB20-4.1 (антиангиогенное средство) (5 мг/кг), соединение формулы IIa (100 мг/кг) и комбинацию соединения формулы IIa (100 мг/кг) и мышиного антитела против VEGF mB20-4.1 (5 мг/кг). Животным вводили контроль IgG и mB20-4.1 внугрибрюшинно дважды в неделю в течение 3 недель, а соединение формулы IIa перорально ежесуточно в течение 21 суток, и мониторинг опухолевого роста проводили в течение дополнительных 41 суток (общее количество суток на одно исследование составляло 62). Соединение формулы IIa и mB20-4.1 вводили одновременно.
На Фиг.32 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами опухолевых клеток глиомы U87MG, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (109 мг/кг), темозоломид (100 мг/кг) и комбинацию соединения формулы Ia (109 мг/кг) и темозоломида (100 мг/кг), а также у мышей, не получавших никакого лекарственного средства (нелеченная группа). Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток и темозоломид перорально ежесуточно в течение 5 суток.
На Фиг.33 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (150 мг/кг), гемцитабин (100 мг/кг) и комбинацию соединения формулы Ia (150 мг/кг) и гемцитабина (100 мг/кг), а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток, а гемцитабин внутрибрюшинно на 1-е, 4-е, 7-е и 10-е сутки (1х/3 сут х 4).
На Фиг.34 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы ВТ474, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) в дозах 18, 36 и 73 мг/кг, трастузумаб (20 мг/кг) и комбинации соединения формулы Ia в дозах 18, 36 и 73 мг/кг и трастузумаба (20 мг/кг), а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток, а трастузумаб внутривенно дважды в неделю в течение 3 недель.
На Фиг.35 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы ВТ474, которым на 0-е сутки вводили: соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 3 недель, соединение формулы Ib в дозе 2,5 мг/кг перорально дважды в неделю в течение 3 недель, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 3 недель, трастузумаб (15 мг/кг) внутривенно один раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель; соединения формулы Ib в дозе 2,5 мг/кг перорально дважды в неделю в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель; и соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.36 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы MCF-7, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, соединение формулы Ib в дозе 10 мг/кг перорально ежесуточно в течение 4 суток, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение суток 0-3, 10-26, соединение формулы Ib в дозе 2 мг/кг перорально ежесуточно в течение суток 0-4, 10-25 и комбинации: соединения формулы Ib в дозе 10 мг/кг перорально ежесуточно в течение 4 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение суток 0-3, 10-26 и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; и соединения формулы Ib в дозе 2 мг/кг перорально ежесуточно в течение суток 0-4, 10-25 и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.37 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы Fo5, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, соединение формулы Ia (GDC-0941) в дозах 36 и 73 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозах 2,5 и 5 мг/кг перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ia в дозе 73 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; и соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.38 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от Marian с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-231, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, соединение формулы Ia (GDC-0941) в дозах 36 и 73 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и соединение формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 8 суток и комбинации: соединения формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ia в дозе 73 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; и соединения формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 8 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 1,5 недели, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.39 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H1299, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ia (GDC-0941) в дозе 100 мг/кг перорально ежесуточно в течение 6 суток, соединение формулы Ia (50 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ia (25 мг/кг) перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia (100 мг/кг) перорально ежесуточно в течение 6 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 6 суток; соединения формулы Ia (50 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; и соединения формулы Ia (25 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.40 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H520, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ia (GDC-0941) в дозе 73 мг/кг перорально ежесуточно в течение 4 суток, соединение формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ia (18 мг/кг) перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 4 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 4 суток; соединения формулы Ia (36 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; и соединения формулы Ia (18 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.41 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H1299, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозе 5 мг/кг перорально два раза в неделю в течение 3 недель, соединение формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально два раза в неделю в течение 3 недель и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 3 недель; и соединения формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.42 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии NCR от Taconic с ксенотрансплантами клеток опухоли немелкоклеточного рака легкого (NSClC) NCl-Н2122, которым на 0-е сутки вводили: эрлотиниб (75 мг/кг) перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 16 суток и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 16 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально два раза в неделю в течение 3 недель и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 16 суток; и соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 16 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 16 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.43 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли меланомы человека А375, которым на 0-е сутки вводили: PD-0325901 (3 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (GDC-0941) в дозе 73 мг/кг перорально ежесуточно в течение 3 недель и комбинацию: PD-0325901 (3 мг/кг) перорально ежесуточно в течение 3 недель и соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.44 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли меланомы человека А375, которым на 0-е сутки вводили: темозоломид (100 мг/кг) перорально ежесуточно в течение 5 суток, соединение формулы Ib в дозе 10 мг/кг перорально один раз в неделю в течение 3 недель, соединение формулы Ib в дозе 5 мг/кг перорально раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 10 мг/кг перорально один раз в неделю в течение 3 недель и темозоломида (100 мг/кг) перорально ежесуточно в течение 5 суток; и соединения формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и темозоломида (100 мг/кг) перорально ежесуточно в течение 5 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.45 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (73 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (36 мг/кг) перорально ежесуточно в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; соединения формулы Ia (36 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ia (73 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.46 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ib (5 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ib (1 мг/кг) перорально ежесуточно в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ib (5 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ib (1 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.47 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ib (5 мг/кг) перорально раз в неделю в течение 3 недель, соединение формулы Ib (10 мг/кг) перорально раз в неделю в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ib (5 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ib (10 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.48 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии SClD/Beige с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека LuCap 35V, которым на 0-е сутки вводили: доцетаксел (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3), соединение формулы Ia (GDC-0941) (50 мг/кг) перорально ежесуточно в течение 18 суток, соединение формулы Ia (100 мг/кг) перорально ежесуточно в течение 18 суток и комбинации: доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ia (50 мг/кг) перорально ежесуточно в течение 18 суток; и доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ia (100 мг/кг) перорально ежесуточно в течение 18 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.49 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии SClD/Beige с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека LuCap 35V, которым на 0-е сутки вводили: доцетаксел (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3), соединение формулы Ib (2,5 мг/кг) перорально ежесуточно в течение 15 суток, соединение формулы Ib (5 мг/кг) перорально ежесуточно в течение 15 суток и комбинации: доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ib (2,5 мг/кг) перорально ежесуточно в течение 15 суток; и доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ib (5 мг/кг) перорально ежесуточно в течение 15 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.50 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии CRL с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека PC3-NCl, которым вводили на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4): доцетаксел (2,5 мг/кг) внутривенно, соединение формулы Ib (10 мг/кг) перорально на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4) и комбинацию: доцетаксела (2,5 мг/кг) внутривенно и соединения формулы Ib (10 мг/кг) перорально на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4), а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.51 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии CRL с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека PC3-NCl, которым на 0-е сутки вводили: гемцитабин (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза, соединение формулы Ia (GDC-0941) (150 мг/кг) перорально каждые 3 суток (1х/3 сут) за 4 раза, соединение формулы Ib (2,5 мг/кг) перорально каждые 3 суток (1х/3 сут) за 4 раза, соединение формулы Ib (5 мг/кг) перорально каждые 3 суток за 4 раза и комбинации: гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ia (150 мг/кг) перорально каждые 3 суток за 4 раза; гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (2,5 мг/кг) перорально каждые 3 суток за 4 раза; гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (5 мг/кг) перорально каждые 3 суток за 4 раза; и гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (10 мг/кг) перорально каждые 3 суток за 4 раза, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
На Фиг.52 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей от HarIan с ксенотрансплантатами клеток опухоли NSClC NCl-H2122 (K-ras), которым на 0-е сутки вводили: PD-0325901 (6,3 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ia (GDC-0941) в дозе 100 мг/кг перорально ежесуточно в течение 21 суток и комбинацию: PD-0325901 (6,3 мг/кг) перорально ежесуточно в течение 21 суток и соединения формулы Ia в дозе 100 мг/кг перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель).
ПОДРОБНОЕ ОПИСАНИЕ ТИПИЧНЫХ ВОПЛОЩЕНИЙ
Теперь будет приведена подробная ссылка на некоторые воплощения изобретения, примеры которых иллюстрируются сопровождающими структурами и формулами. Несмотря на то, что изобретение будет описано в сочетании с приведенными воплощениями, очевидно, что изобретение не предполагает ограничения этими воплощениями. Напротив, предполагается, что изобретение охватывает все альтернативные варианты, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, определенный формулой изобретения. Специалисту в данной области техники будут известны многие способы и вещества, аналогичные или эквивалентные изложенным в данном описании, которые могут быть использованы при практическом применении настоящего изобретения. Настоящее изобретение никоим образом не ограничено описанными способами и веществами. В том случае, если в одном или более чем одном из включенных литературы, патентов и аналогичных материалов имеется отличие от этой заявки или противоречие с этой заявкой, включая, но этим не ограничиваясь, определенные термины, употребление терминов, описанные методики или тому подобное, то эта заявка является контрольным документом.
ОПРЕДЕЛЕНИЯ
Слова "содержат", "содержащий", "включают", "включающий" и "включает", когда они используются в этом описании и формуле изобретения, предназначены для конкретизации наличия установленных признаков, нечто целого, компонентов или стадий, но они не исключают наличия или добавления одного или более чем одного их других признаков, нечто целого, компонентов, стадий или их групп.
Термин "алкил", как он использован в данном описании, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу из одного-двенадцати атомов углерода, причем этот алкильный радикал возможно может быть независимо замещен одним или более заместителями, описанными ниже. Примеры алкильных групп включают, но этим не ограничиваются, метил (Me, -СН3), этил (Et, -СН2СН3), 1-пропил (н-Pr, н-пропил, -СН2СН2СН3), 2-пропил (изо-Pr, изопропил, -СН(СН3)2), 1-бутил (н-Bu, н-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (изо-Bu, изобутил, -СН2СН(СН3)2), 2-бутил (втор-Bu, втор-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (трет-Bu, трет-бутил, -С(СН3)3), 1-пентил (н-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1 -бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3), 2-гексил (-СН(СН3)СН2СН2СН2СН3), 3-гексил (-СН(СН2СН3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С(СН3)3, 1-гептил, 1-октил и тому подобное.
Термин "алкенил" относится к линейному или разветвленному одновалентному углеводородному радикалу из двух-двенадцати атомов углерода, имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp2-двойную связь, причем этот алкенильный радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании, и включает радикалы, имеющие ориентации "цис" и "транс" или альтернативно, ориентации "Е" и "Z". Примеры включают, но этим не ограничиваются, этиленил или винил (-CH=CH2), аллил (-CH2CH=CH2) и тому подобное.
Термин "алкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу из двух-двенадцати атомов углерода, имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp-тройную связь, причем этот алкинильный радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании. Примеры включают, но этим не ограничиваются, этинил (-С=СН), пропинил (пропаргил, -CH2C≡CH) и тому подобное.
Термины "карбоцикл", "карбоциклил", "карбоциклическое кольцо" и "циклоалкил" относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему 3-12 атомов углерода в виде моноциклического кольца или 7-12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, имеющие 7-12 атомов, могут быть организованы, например, в виде бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-систем, а бициклические карбоциклы, имеющие 9 или 10 кольцевых атомов, могут быть организованы в виде бицикло-[5,6]- или -[6,6]-систем или в виде соединенных мостиковой связью систем, таких как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и тому подобное.
"Арил" обозначает одновалентный ароматический углеводородный радикал из 6-20 атомов углерода, образованный в результате удаления одного атома водорода от единственного атома углерода исходной ароматической кольцевой системы. Некоторые арильные группы представлены в приведенных в качестве примера структурах как "Ar". Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Типичные арильные группы включают, но этим не ограничиваются, радикалы, образованные из бензола (фенил), замещенных бензолов, нафталина, антрацена, бифенила, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и тому подобное. Арильные группы возможно замещены независимо одним или более заместителями, изложенными в данном описании.
Термины "гетероцикл", "гетероциклил" и "гетероциклическое кольцо" используются в данном описании взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (т.е. имеющему одну или более двойных и/или тройных связей в пределах кольца) карбоциклическому радикалу из 3-20 кольцевых атомов, в котором по меньшей мере один атом в кольце представляет собой гетероатом, выбранный из азота, кислорода и серы, при этом остальные кольцевые атомы представляют собой С, где один или более чем один кольцевой атом возможно независимо замещен одним или более заместителями, описанными ниже. Гетероцикл может представлять собой моноцикл, имеющий 3-7 кольцевых членов (2-6 атомов углерода и 1-4 гетероатома, выбранных из N, О, Р и S), или бицикл, имеющий 7-10 кольцевых членов (4-9 атомов углерода и 1-6 гетероатомов, выбранных из N, О, Р и S), например: бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-систему. Гетероциклы описаны в Paquette, Leo A.; "PrinClples of Modern HeterocyClic Chemistry" (W.A. Benjamin, New York, 1968), особенно в главах 1, 3, 4, 6, 7 и 9; "The Chemistry of HeterocyClic Compounds, A series of Monographs" (John Wiley & Sons, New York, 1950 до настоящего времени), в частности, в томах 13, 14, 16, 19 и 28; и в J. Am. Chem. Soc. (1960) 82: 5566. Термин "гетероцикл" включает гетероциклоалкокси. "Гетероциклил" также включает радикалы, при этом гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но этим не ограничиваются, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3Н-индолил, хинолизинил и N-пиридил-мочевины. Спиро-группировки также включены в объем этого определения. Примерами гетероциклической группы, где 2 кольцевых атома углерода замещены группировками оксо (=O), являются пиримидинонил и 1,1-диоксо-тиоморфолинил. Приведенные в данном описании гетероциклические группы возможно замещены независимо одним или более заместителями, изложенными в данном описании.
Термин "гетероарил" относится к одновалентному ароматическому радикалу из 5-, 6- или 7-членных колец и включает конденсированные кольцевые системы (по меньшей мере одно из которых является ароматическим) из 5-20 атомов, содержащие один или более гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Гетероарильные группы возможно замещены независимо одним или более заместителями, изложенными в данном описании.
Гетероциклические или гетероарильные группы могут быть присоединены по углероду (углерод-связанные), азоту (азот-связанные) или кислороду (кислород-связанные), если такое возможно. В качестве примера, а не ограничения, углерод-связанные гетероциклы или гетероарилы связываются по положению 2, 3, 4, 5 или 6 пиридина, по положению 3, 4, 5 или 6 пиридазина, по положению 2, 4, 5 или 6 пиримидина, по положению 2, 3, 5 или 6 пиразина, по положению 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, по положению 2, 4 или 5 оксазола, имидазола или тиазола, по положению 3, 4 или 5 изоксазола, пиразола или изотиазола, по положению 2 или 3 азиридина, по положению 2, 3 или 4 азетидина, по положению 2, 3, 4, 5, 6, 7 или 8 хинолина или по положению 1, 3, 4, 5, 6, 7 или 8 изохинолина.
В качестве примера, а не ограничения, азот-связанные гетероциклы или гетероарилы связываются по положению 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1Н-индазола, по положению 2 изоиндола или изоиндолина, по положению 4 морфолина и по положению 9 карбазола или β-карболина.
"Углерод-связанный моноциклический гетероарил" относится к пяти- или шестичленному, незамещенному или замещенному, моноциклическому гетероарильному радикалу, содержащему 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранных из N, О и S. Углерод-связанный моноциклический гетероарил присоединен по положению С-2 пиримидинового кольца в соответствии с формулами I и II по любому атому углерода моноциклической гетероарильной группы R3. Углерод-связанные моноциклические гетероарильные радикалы включают, но этим не ограничиваются: 2-пиридил, 3-пиридил, 4-пиридил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-имидазолил, 4-имидазолил, 3-пиразолил, 4-пиразолил, 2-пирролил, 3-пирролил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 3-пиридазинил, 4-пиридазинил, 5-пиридазинил, 2-пиримидинил, 5-пиримидинил, 6-пиримидинил, 2-пиразинил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 3-триазолил, 1-триазолил, 5-тетразолил, 1-тетразолил и 2-тетразолил. Углерод-связанные моноциклические гетероарилы возможно замещены независимо одним или более заместителями, изложенными в данном описании.
"Углерод-связанный конденсированный бициклический С3-С20гетероциклил" и "углерод-связанный конденсированный бициклический С1-С20гетероарил", содержащие один или более гетероатомов, независимо выбранных из азота, кислорода и серы, отличаются только своим ароматическим характером и имеют два кольца, конденсированные друг с другом, т.е. имеющие общую связь. Углерод-связанные конденсированные бициклические гетероциклильные и гетероарильные радикалы присоединены по положению С-2 пиримидинового кольца в соответствии с формулами I и II по любому атому углерода конденсированной бициклической С3-С20гетероциклильной или конденсированной бициклической С1-С20гетероарильной группы R3. Углерод-связанные конденсированные бициклические гетероциклильные и гетероарильные радикалы включают, но этим не ограничиваются: 1Н-индазол, 1Н-индол, индолин-2-он, 1-(индолин-1-ил)этанон, 1Н-бензо[d][1,2,3]триазол, 1Н-пиразоло[3,4-b]пиридин, 1Н-пиразоло[3,4-d]пиримидин, 1Н-бензо[d]имидазол, 1Н-бензо[d]имидазол-2(3Н)-он, 1Н-пиразоло[3,4-с]пиридин, 1Н-пирроло[2,3-с]пиридин, 3Н-имидазо[4,5-с]пиридин, 7Н-пирроло[2,3-d]пиримидин, 7Н-пурин, 1Н-пиразоло[4,3-d]пиримидин, 5Н-пирроло[3,2-d]пиримидин, 2-амино-1Н-пурин-6(9Н)-он, хинолин, хиназолин, хиноксалин, изохинолин, изохинолин-1(2Н)-он, 3,4-дигидроизохинолин-1(2Н)-он, 3,4-дигидрохинолин-2(1Н)-он, хиназолин-2(1Н)-он, хиноксалин-2(1Н)-он, 1,8-нафтиридин, пиридо[3,4-d]пиримидин и пиридо[3,2-b]пиразин. Конденсированные бициклические гетероциклы и конденсированные бициклические гетероарилы возможно замещены независимо одним или более заместителями, изложенными в данном описании.
Замещающие группы, которыми возможно замещены алкил, алкенил, алкинил, карбоциклил, гетероциклил, арил, гетероарил, конденсированный бициклический С4-С20гетероциклил и конденсированный бициклический С1-С20гетероарил, включают F, Cl, Br, I, CN, CF3, -NO2, оксо, R10, -C(=Y)R10, -C(=Y)OR10, -C(=Y)NR10R11, -(CR14R15)nNR10R11, -(CR14R15)nOR10, -NR10R11, -NR12C(=Y)R10, -NR12C(=Y)OR11, -NR12C(=Y)NR10R11, -NR12SO2R10,=NR12, OR10, -OC(=Y)R10, -OC(=Y)OR10, -OC(=Y)NR10R11, -OS(O)2(OR10), -OP(=Y)(OR10)(OR11), -OP(OR10)(OR11), SR10, -S(O)R10, -S(O)2R10, -S(O)2NR10R11, -S(O)(OR10), -S(O)2(OR10), -SC(=Y)R10, -SC(=Y)OR10, -SC(=Y)NR10R11, возможно замещенный С1-С12алкил, возможно замещенный С2-С8алкенил, возможно замещенный С2-С8алкинил, возможно замещенный С3-С12карбоциклил, возможно замещенный С2-С20гетероциклил, возможно замещенный С6-С20арил, возможно замещенный С1-С20гетероарил, -(CR14R15)t-NR12C(=O), (CR14R15)NR10R11 и (CR4R5)t-NR10R11.
Термины "лечить или обрабатывать" и "лечение или обработка" относятся как к терапевтическому лечению, так и к профилактическим или предохранительным мерам, при которых у объекта должно быть предотвращено или замедлено (снижено) нежелательное физиологическое изменение или расстройство, такое как рост, развитие или распространение рака. Для задач данного изобретения полезные или желаемые клинические результаты включают, но этим не ограничиваются, ослабление симптомов, уменьшение степени заболевания, состояние стабильности (т.е. без ухудшения) заболевания, задержку или замедление прогрессирования заболевания, уменьшение интенсивности или временное ослабление болезненного состояния и ремиссию (будь то частичную или полную), как детектируемые, так и недетектируемые. "Лечение" также может означать пролонгирование выживаемости по сравнению с ожидаемой выживаемостью в отсутствие получения лечения. Те, кто нуждается в таком лечении, включают уже имеющих данное состояние или расстройство, а также предрасположенных к данному состоянию или расстройству, или тех, у которых данное состояние или расстройство должно быть предотвращено.
Фраза "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (1) лечит конкретное заболевание, состояние или расстройство, (2) ослабляет, уменьшает интенсивность или устраняет один или более симптомов конкретного заболевания, состояния или расстройства или (3) предотвращает или задерживает начало развития одного или более симптомов конкретного заболевания, состояния или расстройства, изложенного в данном описании. В случае рака терапевтически эффективное количество лекарственного средства может уменьшать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) опухолевый метастаз; ингибировать, до некоторой степени, опухолевый рост; и/или ослаблять до некоторой степени один или более симптомов, ассоциированных с раком. В зависимости от возможности лекарственного средства предотвращать рост и/или уничтожать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксичным. Что касается терапии рака, то эффективность может быть измерена, например, путем оценки времени до прогрессирования заболевания (ТТР) и/или определения показателя ответной реакции (RR).
Термины "рак" и "раковый" относятся к физиологическому состоянию или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым клеточным ростом. "Опухоль" содержит одну или более раковых клеток. Примеры рака включают, но этим не ограничиваются, карциному, лимфому, бластому, саркому и лейкоз или лимфонеоплазии. Более конкретные примеры таких видов рака включают плоскоклеточный рак (например, рак плоскоклеточного эпителия), рак легкого, в том числе мелкоклеточный рак легкого, немелкоклеточный рак легкого ("NSClC"), аденокарциному легкого и плоскоклеточный рак легкого, рак брюшины, гепатоцеллюлярный рак, рак желудочно-кишечного тракта или желудка, в том числе гастроинтестинальный рак, рак поджелудочной железы, глиобластому, цервикальный рак, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстой кишки, рак прямой кишки, колоректальный рак, рак матки и эндометрия, рак слюнных желез, рак почки или почечный рак, рак предстательной железы, рак вульвы, рак щитовидной железы, печеночный рак, рак анального канала, рак полового члена, а также рак головы и шеи.
"Химиотерапевтический агент" представляет собой биологическое (высокомолекулярное) или химическое (низкомолекулярное) соединение, полезное в лечении рака, независимо от механизма действия. Классы химиотерапевтических агентов включают, но этим не ограничиваются: алкилирующие агенты, антиметаболиты, ядовитые алкалоиды веретеновидных растений, цитотоксичные/противоопухолевые антибиотики, ингибиторы топоизомеразы, белки, антитела, фотосенсибилизирующие вещества и ингибиторы киназ. Химиотерапевтические агенты включают соединения, используемые в "терапии с направленной доставкой" и традиционной химиотерапии без направленной доставки.
Примеры химиотерапевтических агентов включают: эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), доцетаксел (TAXOTERE®, Sanofi-Aventis), 5-FU (фторурацил, 5-фторурацил, №CAS (Chemical abstract cervice (идентификатор химических соединений)): 51-21-8), гемцитабин (GEMZAR®, Lilly), PD-0325901 (№CAS: 391210-10-9, Pfizer), цисплатин (цис-диамин, дихлорплатина(II), №CAS: 15663-27-1), карбоплатин (№CAS: 41575-94-4), паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), бевацизумаб (AVASTIN®, Genentech), трастузумаб (HERCEPTIN®, Genentech), пертузумаб (OMNITARG®, rhuMab (рекомбинантное человеческое моноклональное антитело) 2С4, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентазабицикло-[4.3.0]нона-2,7,9-триен-9-карбоксамид, №CAS: 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), тамоксифен ((Z)-2-[4-(1,2-бут-1 -енил)фенокси]-N,N-диметил-этанамин, NOLVADEX®, ISTUBAL®, VALODEX®), доксорубицин (ADRIAMYClN®), Akti-1/2, HPPD, рапамицин и лапатиниб (TYKERB®, GIaxo SmithKline).
Следующие примеры химиотерапевтических агентов включают: оксалиплатин (ELOXATIN®, Sanofi), бортезомид (VELCADE®, Millennium Pharm.), сутент (SUNITINIB®, SU11248, Pfizer), летрозол (FEMARA®, Novartis), иматиниба мезилат (GLEEVEC®, Novartis), XL-518 (ингибитор МЕК (митоген-активируемая киназа регулируемой внеклеточным сигналом киназы), Exelixis, WO 2007/044515), ARRY-886 (ингибитор МЕК, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор Pl3K, Semafore Pharmaceuticals), BEZ-235 (ингибитор Pl3K, Novartis), XL-147 (ингибитор Pl3K, Exelixis), ABT-869 (многоцелевого действия ингибитор рецепторных тирозинкиназ семейств VEGF и PDGF (фактор роста тромбоцитов), Abbott Laboratories и Genentech), АВТ-263 (ингибитор BCl-2/BCl-xL, Abbott Laboratories и Genentech), PTK787/ZK 222584 (Novartis), фулвестрант (FASLODEX®, AstraZeneca), лейковорин (фолиновая кислота), лонафарниб (SARASAR™, SCH 66336, Schering Plough), сорафениб (NEXAVAR®, BAY43-9006, Bayer Labs), гефитиниб (IRESSA®, AstraZeneca), иринотекан (CAMPTOSAR®, CPT-11, Pfizer), типифарниб (ZARNESTRA™, Johnson & Johnson), капецитабин (XELODA®, Roche), ABRAXANE™ (без кремофора), препараты наночастиц паклитаксела, сконструированных с использованием альбумина (American Pharmaceutical Partners, Schaumberg, II), валдетаниб (rINN (рекомендованное международное непатентованное наименование), ZD6474, ZACTIMA®, AstraZeneca), хлорамбуцил, AG1478, AG1571 (SU 5271; Sugen), темсиролимус (TORISEL®, Wyeth), пазопаниб (GIaxoSmithKline), канфосфамид (TELCYTA®, Telik), тиотепа и циклофосфамид (CYTOKCAN®, NEOSAR®); алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метипедопа и уредопа; этиленимины и метиламеламины, включающие алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (в том числе синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (в том числе его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности криптофицин 1 и криптофицин 8); доластатин; дуокармицин (в том числе синтетические аналоги, KW-2189 и СВ1-ТМ1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотные аналоги иприта, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид мехлоретаминоксида, мелфалан; новембихин, фенестерин, преднимустин, трофосфамид, урациловый аналог иприта; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, калихеамицин гамма 11, калихеамицин омега 11, динемицин, динемицин А; бифосфонаты, такие как клодронат; эсперамицин; а также неокарциностатиновый хромофор и родственные хромофоры хромопротеиновых энедииновых антибиотиков), алкациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофиллин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксо-рубицин, эпирубицин, эзорубицин, идарубицин, марселломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; антагонисты функции надпочечников (anti-adrenals), такие как аминоглутетимид, митотан, трилостан; вещество, являющееся источником восполнения (replenisher) фолиевой кислоты, такое как фролиновая кислота; ацеглатон; альдофосфамидгликозид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; эллиптиния ацетат; эпотилон; этоглуцид; галлия нитрат; гидроксимочевину; лентинан; лонидаинин; майтансиноиды, такие как майтансин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2"-трихлортриэтиламин; трихотецены (особенно, токсин Т-2, верракурин А, роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-С"); циклофосфамид; тиотепа; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорелбин (NAVELBINE®); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение "химиотерапевтического агента" также включены: (1) антигормональные агенты, действие которых заключается в регуляции или ингибировании воздействия гормонов на опухоли, как например, антиэстрогены и селективные модуляторы рецептора эстрогена (SERM), в том числе, например, тамоксифен (включая NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® (торемифина цитрат); (2) ингибиторы ароматазы, ингибирующие фермент ароматазу, которая регулирует продуцирование эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (эксеместан; Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARIMIDEX® (анастрозол; AstraZeneca); (3) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; а также троксацитабин (1,3-диоксолановый аналог нуклеозида цитозина); (4) ингибиторы протеинкиназ, такие как ингибиторы МЕК (WO 2007/044515); (5) ингибиторы липид-киназ; (6) антисмысловые олигонуклеотиды, в частности такие, которые ингибируют экспрессию генов в путях передачи сигналов, участвующих в аберрантной пролиферации клеток, например, РКС-альфа (протеинкиназа С), Raf и H-Ras, такие как облимерсен (GENASENSE®, Genta Inc.); (7) рибозимы, такие как ингибиторы экспрессии VEGF (например, ANGIOZYME®) и ингибиторы экспрессии HER2; (8) вакцины, как например, вакцины для генотерапии, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; rlL-2 (рекомбинантный интерлейкин-2) PROLEUKIN®; ингибиторы топоизомеразы 1, такие как LURTOTECAN®; rmRH ABARELIX®; (9) антиангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение "химиотерапевтического агента" также включены терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (AVASTIN®, Genentech); цетуксимаб (ERBITUX®, ImClone); панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idee), пертузумаб (OMNITARG™, 2C4, Genentech), трастузумаб (HERCEPTIN®, Genentech), тозитумомаб (Bexxar, Corixia) и конъюгат антитела и лекарственного средства, гемтузумаб-озогамицин (MYLOTARG®, Wyeth).
Гуманизированные моноклональные антитела с терапевтическим потенциалом в качестве химиотерапевтических агентов в комбинации с ингибиторами Pl3K по изобретению включают: алемтузумаб, аполизумаб, азелизумаб, атлизумаб, бипинеузумаб, бевацизумаб, биватузумаб-мертанзин, кантузумаб-мертанзин, цеделизумаб, цертолизумаб-пегол, сидфузизумаб, сидтузумаб, даклизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб-озогамицин, инотузумаб-озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфузитузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, ресивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб-тетраксетан, тадоцизумаб, тализумаб, тефибазумаб, тоцилизумаб, торализумаб, трастузумаб, тукотузумаб целмолейкин, тукуситузумаб, умавизумаб, уртоксазумаб и визилизумаб.
Термин "млекопитающее" включает, но этим не ограничивается, людей, мышей, крыс, морских свинок, обезьян, собак, кошек, лошадей, коров, свиней и овец и домашних птиц.
"Метаболит" представляет собой продукт, образуемый в процессе метаболизма конкретного соединения или его соли в организме. Метаболиты соединения могут быть идентифицированы с использованием общепринятых методик, известных в данной области техники, а их активности определены с использованием тестов, таких как тесты, изложенные в данном описании. Такие продукты могут получаться, например, в результате окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного вводимого соединения. Соответственно, изобретение включает метаболиты соединений по изобретению, в том числе соединения, получаемые способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Термин "инструкция по медицинскому применению препарата" используется в отношении инструкций, обычно вкладываемых в выпускаемые промышленностью упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях, касающихся применения таких терапевтических продуктов.
Фраза "фармацевтически приемлемая соль", как она использована в данном описании, относится к фармацевтически приемлемым органическим или неорганическим солям соединения по изобретению. Типичные соли включают, но этим не ограничиваются, соли сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат (мезилат), этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). При образовании фармацевтически приемлемой соли возможно включение другой молекулы, такой как ацетат-ион, сукцинат-ион или другой противоион. Противоион может представлять собой любую органическую или неорганическую группировку, которая стабилизирует заряд на исходном соединении. Кроме того, фармацевтически приемлемая соль может иметь более одного заряженного атома в своей структуре. В случаях, когда часть фармацевтически приемлемой соли представлена многими заряженными атомами, противоионов может быть много. Следовательно, фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более чем один противоион.
Если соединение по изобретению представляет собой основание, то желаемую фармацевтически приемлемую соль можно получить любым подходящим способом, доступным в данной области техники, например, обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидил-содержащая кислота (pyranosidyl aCld), такая как глюкуроновая кислота или галактуроновая кислота, альфа-оксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или тому подобное. Кислоты, которые, как правило, считаются подходящими для образования фармацевтически полезных или приемлемых солей из основных фармацевтических соединений рассмотрены, например, в Р. Stahl et al., Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al., Journal of Pharmaceutical Sciences (1977) 66(1) 119; P. Gould, International J. of Pharmaceutics (1986) 33 201 217; Anderson et al., The Practice of MediClnal Chemistry (1996), Academic Press, New York; Remington's Pharmaceutical Sciences, 18th ed., (1995) Mack Publishing Co., Easton PA; и в The Orange Book (Food & Drug Administration, Washington, D.C. на их веб-сайте). Эти описания включены в данное описание со ссылкой на них.
Если соединение по изобретению представляет собой кислоту, желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например, посредством обработки свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла или тому подобное. Иллюстративные примеры подходящих солей включают, но этим не ограничиваются, органические соли, происходящие из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, происходящие из натрия, кальция, калия, магния, марганца, железа, меди, цинка, аллюминия и лития.
Фраза "фармацевтически приемлемый" указывает на то, что вещество или композиция должны быть совместимы с химической и/или токсикологической точки зрения с другими ингредиентами, содержащимися в композиции, и/или с млекопитающим, подвергаемым лечению этой композицией.
"Сольват" относится к физической ассоциации или комплексу одной или более чем одной молекулы растворителя и соединения по изобретению. Соединения по изобретению могут существовать в несольватированных, а также в сольватированных формах. Примеры растворителей, которые образуют сольваты, включают, но этим не ограничиваются, воду, изопропанол, этанол, метанол, DMSO, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, молекулой растворителя в котором является вода. Эта физическая ассоциация включает в себя ионное и ковалентное связывание в различной степени, в том числе и связывание посредством водородных связей. В некоторых случаях сольват может быть выделен, например, когда одна или более чем одна молекула растворителя инкорпорирована в кристаллическую решетку кристаллического твердого вещества. Способы получения сольватов в общем известны, см. например, М. Caira et al., J. Pharmaceutical SCl., 93(3), 601-611 (2004). Аналогичные способы получения сольватов, полусольватов, гидратов и тому подобное описаны в Е. С.van Tender et al., AAPS Pharm. SCl. Tech., 5(1), artiCle 12 (2004); и A. L. Bingham et al., Chem. Commun., 603-604 (2001). Типичный неограничивающий способ включает растворение соединения по изобретению в желаемых количествах желаемого растворителя (органического растворителя или воды или их смесей) при температурах выше температуры окружающей среды и охлаждение раствора со скоростью, необходимой для образования кристаллов, которые далее выделяют стандартными способами. Аналитические методики, такие например, как ИК-спектроскопия, демонстрируют наличие растворителя (или воды) в кристаллах в виде сольвата (или гидрата).
Термин "синергический", как он использован в данном описании, относится к действию терапевтической комбинации, которое более эффективно, чем аддитивные эффекты двух или более отдельных агентов. В основе определения синергического взаимодействия между соединением формулы I или II и одним или более чем одним химиотерапевтическим агентом могут лежать результаты, полученные из анализов, изложенных в данном описании. Результаты этих исследований анализируют, используя метод Chou и Talalay для комбинации и анализ зависимости доза-эффект с применением программного обеспечения CalcuSyn с целью получения показателя для комбинации (Chou and Talalay, 1984, Adv. Enzyme Regul. 22: 27-55). Комбинации, предложенные согласно данному изобретению, оценены в нескольких аналитических системах, и данные могут быть проанализированы с использованием стандартной программы для количественного определения синергизма, аддитивизма и антагонизма по отношению к противораковым агентам. Предпочтительно используемой программой является программа, описанная Chou и Talalay в "New Avenues in Developmental Cancer Chemotherapy", Academic Press, 1987, часть 2. Величины показателя для комбинации менее 0,8 указывают на синергическое действие, величины выше 1,2 указывают на антагонизм, а величины от 0,8 до 1,2 указывают на аддитивные эффекты. Комбинированная терапия может обеспечивать "синергическое действие" и демонстрировать "синергический характер", т.е. эффект, достигаемый при совместном использовании активных ингредиентов, больше суммы эффектов, получаемых в результате использования соединений по отдельности. Синергический эффект может быть получен, когда активные ингредиенты: (1) изготавливают совместно и вводят или доставляют одновременно в комбинированной, стандартной лекарственной композиции; (2) доставляют путем чередования или параллельно в виде отдельных композиций; или (3) доставляют с использованием какой-либо другой схемы (введения). При доставке с использованием чередующейся терапии (alternation therapy) синергический эффект может быть получен, когда соединения вводят или доставляют последовательно, например, посредством разных инъекций в отдельных шприцах. В общем случае, при чередующейся терапии эффективную дозировку каждого активного ингредиента вводят последовательно, т.е. сериями (serially), тогда как при комбинированной терапии эффективные дозировки двух или более активных ингредиентов вводят совместно.
СОЕДИНЕНИЯ ФОРМУЛЫ I И II
Настоящее изобретение включает терапевтические комбинации, включающие соединения формулы I и II, которые имеют структуры:
или стереоизомеры, геометрические изомеры, таутомеры или их фармацевтически приемлемые соли, где:
R1 выбран из Н, F, Cl, Br, I, CN, -(CR14R15)mNR10R11, -C(R14R15)nNR12C(=Y)R10, -(CR14R15)nNR12S(O)2R10, -(CR14R15)mOR10, -(CR14R15)nS(O)2R10, -(CR14R15)nS(O)2NR10R11, -C(OR10)R11R14, -C(=Y)R10, -C(=Y)OR10, -C(=Y)NR10R11, -C(=Y)NR12OR10, -C(=O)NR12S(O)2R10, -C(=O)NR12(CR14R15)mNR10R11, -NO2, -NR12C(=Y)R11, -NR12C(=Y)OR11, -NR12C(=Y)NR10R11, -NR12S(O)2R10, -NR12SO2NR10R11, -SR10, -S(O)2R10, -S(O)2NR10R11, -SC(=Y)R10, -SC(=Y)OR10, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила и С1-С20гетероарила;
R2 выбран из Н, F, Cl, Br, I, CN, CF3, -NO2, -C(=Y)R10, -C(=Y)OR10, -C(=Y)NR10R11, -(CR14R15)mNR10R11, -(CR14R15)nOR10, -(CR14R15)t-NR12C(=O)(CR14R15)NR10R11, -NR12C(=Y)R10, -NR12C(=Y)OR10, -NR12C(=Y)NR10R11, -NR12SO2R10, OR10, -OC(=Y)R10, -OC(=Y)OR10, -OC(=Y)NR10R11, -OS(O)2(OR10), -OP(=Y)(OR10)(OR11), -OP(OR10)(OR11), SR10, -S(O)R10, -S(O)2R10, -S(O)2NR10R11, -S(O)(OR10), -S(O)2(OR10), -SC(=Y)R10, -SC(=Y)OR10, -SC(=Y)NR10R11, C1-C12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила и С1-С20гетероарила;
R3 представляет собой углерод-связанный моноциклический гетероарил, углерод-связанный конденсированный бициклический С3-С20гетероциклил или углерод-связанный конденсированный бициклический С1-С20гетероарил, где моноциклический гетероарил, конденсированный бициклический С3-С20гетероциклил и конденсированный бициклический С1-С20гетероарил возможно замещены одной или более группами, выбранными из F, Cl, Br, I, -CN, -NR10R11, -OR10, -C(O)R10, -NR10C(O)R11, -N(C(O)R11)2, -NR10C(O)NR10R11, -NR12S(O)2R10, -C(=O)OR10, -C(=O)NR10R11, С1-С12алкила и (С1-С12алкил)OR10;
R10, R11 и R12 независимо представляют собой Н, С1-С12алкил, С2-С8алкенил, С2-С8алкинил, С3-С12карбоциклил, С2-С20гетероциклил, С6-С20арил или С1-С20гетероарил,
или R10 и R11 вместе с азотом, к которому они присоединены, образуют С2-С20гетероциклическое кольцо, возможно замещенное одной или более группами, независимо выбранными из оксо, (CH2)mOR12, NR12R12, CF3, F, Cl, Br, I, SO2R12, C(=O)R12, NR12C(=Y)R12, NR12S(O)2R12, C(=Y)NR12R12, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила и С1-С20гетероарила;
R14 и R15 независимо выбраны из Н, С1-С12алкила или -(СН2)n-арила,
или R14 и R15 вместе с атомами, к которым они присоединены, образуют насыщенное или частично ненасыщенное С3-С12карбоциклическое кольцо;
где указанный алкил, алкенил, алкинил, карбоциклил, гетероциклил, арил и гетероарил возможно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, CN, CF3, -NO2, оксо, R10, -C(=Y)R10, -C(=Y)OR10, -C(=Y)NR10R11, -(CR14R15)nNR10R11, -(CR14R15)nOR10, -NR10R11, -NR12C(=Y)R10, -NR12C(=Y)OR11, -NR12C(=Y)NR10R11, -(CR14R15)mNR12SO2R10,=NR12, OR10, -OC(=Y)R10, -OC(=Y)OR10, -OC(=Y)NR10R11, -OS(O)2(OR10), -OP(=Y)(OR10)(OR11), -OP(OR10)(OR11), -SR10, -S(O)R10, -S(O)2R10, -S(O)2NR10R11, -S(O)(OR10), -S(O)2(OR10), -SC(=Y)R10, -SC(=Y)OR10, -SC(=Y)NR10R11, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила и С1-С20гетероарила;
Y представляет собой 0, S или NR12;
m равно 0, 1,2, 3, 4, 5 или б; и
n равно 1, 2, 3, 4, 5 или 6.
Типичные воплощения соединений формулы I и II включают таковые, где R1 представляет собой -(CR14R15)mNR10R11, где m равно 1, и R10 и R11 вместе с азотом, к которому они присоединены, образуют возможно замещенное С3-С20гетеро-циклическое кольцо. Это С3-С20гетероциклическое кольцо может представлять собой пиперазинил, возможно замещенный одной или более группами, выбранными из NR10R11, CF3, F, Cl, Br, I, SO2R10, C(=O)R10, NR12C(=Y)R11, NR12S(O)2R11, C(=Y)NR10R11 и С1-С12алкила.
Типичные воплощения соединений формулы I и II включают таковые, где R1 не является Н.
Типичные воплощения соединений формулы I и II включают таковые, где R2 представляет собой Н, СН3, С3-С12карбоцикпил, С2-С20гетероциклил, С6-С20арил или С1-С20гетероарил. Этот С1-С20гетероарил может представлять собой моноциклическую гетероарильную группу, выбранную из 2-пиридила, 3-пиридила, 4-пиридила, 3-изоксазолила, 4-изоксазолила, 5-изоксазолила, 2-имидазолила, 4-имидазолила, 3-пиразолила, 4-пиразолила, 2-пирролила, 3-пирролила, 2-тиазолила, 4-тиазолила, 5-тиазолила, 3-пиридазинила, 4-пиридазинила, 5-пиридазинила, 2-пиримидинила, 5-пиримидинила, 6-пиримидинила, 2-пиразинила, 2-оксазолила, 4-оксазолила, 5-оксазолила, 2-фуранила, 3-фуранила, 2-тиенила, 3-тиенила, 3-триазолила, 1-триазолила, 5-тетразолила, 1-тетразолила и 2-тетразолила.
Типичные воплощения соединений формулы I и II включают таковые, где R3 представляет собой 2-аминопиримидин-5-ил.
Типичные воплощения соединений формулы I и II включают таковые, где R3 представляет собой бициклическую гетероарильную группу, выбранную из 1Н-индазола, 1Н-индола, индолин-2-она, 1-(индолин-1-ил)этанона, 1Н-бензо[d][1, 2, 3]триазола, 1Н-пиразоло[3,4-b]пиридина, 1Н-пиразоло[3,4-d]пиримидина, 1Н-бензо[d]имидазола, 1Н-бензо[d]имидазол-2(3Н)-она, 1Н-пиразоло[3,4-с]пиридина, 1Н-пирроло[2,3-с]пиридина, 3Н-имидазо[4,5-с]пиридина, 7Н-пирроло[2,3-d]пиримидина, 7Н-пурина, 1Н-пиразоло[4,3-d]пиримидина, 5Н-пирроло[3,2-d]пиримидина, 2-амино-1Н-пурин-6(9Н)-она, хинолина, хиназолина, хиноксалина, изохинолина, изохинолин-1(2Н)-она, 3,4-дигидроизохинолин-1(2Н)-она, 3,4-дигидрохинолин-2(1Н)-она, хиназолин-2(1Н)-она, хиноксалин-2(1Н)-она, 1,8-нафтиридина, пиридо[3,4-d]пиримидина и пиридо[3,2-b]пиразина.
Типичные воплощения соединений формулы I и II включают таковые, где R3 представляет собой 1Н-индазол-4-ил.
Типичным соединением формулы I является 4-(2-(1Н-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин, имеющий формулу Ia:
Другим типичным соединением формулы I является (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)-пиперазин-1-ил)-2-гидроксипропан-1-он, имеющий формулу Ib:
Типичным соединением формулы II является 4-(2-(1Н-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[2,3-d]пиримидин-4-ил)морфолин, имеющий формулу IIa:
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ФОРМУЛЫ I И II
Соединения формулы I и II могут быть синтезированы способами синтеза, которые включают процессы, аналогичные хорошо известным в области химии, включая WO 2006/046031. Обычно, исходные соединения приобретают в коммерческих источниках, таких как Aldrich Chemicals (Milwaukee, WI), или легко получают, используя хорошо известные специалистам в данной области техники способы (например, получают способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-VerIag, Berlin, включая приложения (также доступные через онлайн-базу данных Beilstein).
Соединение формулы I и II может быть получено с использованием методик получения других тиофенов и пиримидинов (US 6608053; US 6492383; US 6232320; US 6187777; US 3763156; US 3661908; US 3475429; US 5075305; US 2003/220365; GB 1393161; WO 93/13664) и других гетероциклов, которые описаны в: Comprehensive HeterocyClic Chemistry, Editors Katritzky and Rees, Pergamon Press, 1984.
Соединения формулы I и II можно превратить в фармацевтически приемлемую соль, а соль можно превратить в свободное основание, используя традиционные способы. Соединения формулы I и II могут быть терапевтически эффективны в виде свободного основания или в виде фармацевтически приемлемой соли в зависимости от желаемых свойств, таких как растворимость, разложение (dissolution), гигроскопичная природа и фармакокинетические свойства. Примеры фармацевтически приемлемых солей включают соли с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; и органическими кислотами, такими как метансульфоновая кислота, бензолсульфоновая кислота, муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, этансульфоновая кислота, аспарагиновая кислота и глутаминовая кислота. Соль может представлять собой мезилат, гидрохлорид, фосфат, бензолсульфонат или сульфат. Соли могут представлять собой моно-соли или бис-соли. Например, соль мезилата может представлять собой моно-мезилат или бис-мезилат.
Соединения формулы I и II и соли также могут существовать в виде гидратов или сольватов.
При получении соединений формулы I и II может быть необходима защита функциональных групп (например, первичного или вторичного амина) промежуточных соединений. Необходимость в такой защите будет меняться в зависимости от природы отдаленных функциональных групп и условий способов получения. Подходящие амино-защитные группы включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-флуоренил-метиленоксикарбонил (Fmoc). Необходимость в такой защите легко определяется специалистом в данной области техники. Общее описание защитных групп и их использование приведено в Т. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
В иллюстративных целях на Схемах 1-7 показаны общие способы получения соединений по настоящему изобретению, а также ключевых промежуточных соединений. Для более подробного описания отдельных стадий реакций см. раздел Примеры, ниже. Специалистам в данной области техники очевидно, что для синтеза соединений по изобретению могут быть использованы и другие пути синтеза. Несмотря на то, что на приведенных ниже схемах изображены и рассмотрены конкретные исходные вещества и реагенты, они легко могут быть заменены на другие исходные вещества и реагенты для получения разнообразия производных и/или реакционных условий. К тому же, многие соединения, полученные описанными ниже способами, могут быть дополнительно модифицированы в свете данного описания с использованием общепринятых методов химии, хорошо известных специалистам в данной области техники.
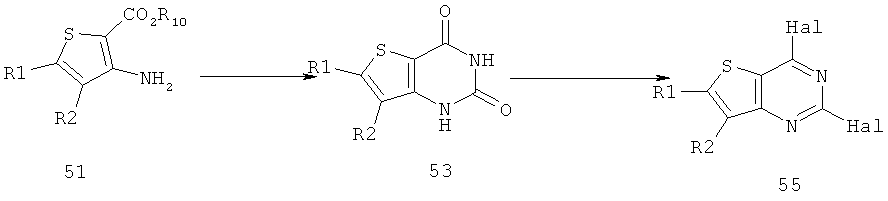
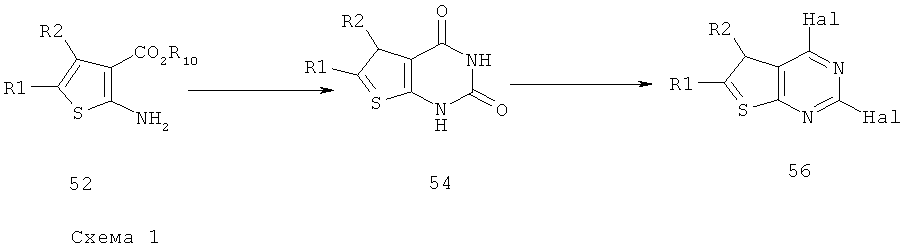
На Схеме 1 показан общий способ получения промежуточных соединений тиенопиримидинов 55 и 56 из реагентов 2-карбоксиэфира 3-аминотиофена и 2-амино-3-карбоксиэфира тиофена, соответственно, 51 и 52, где Hal представляет собой Cl, Br или I; и R1, R2 и R10 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств.
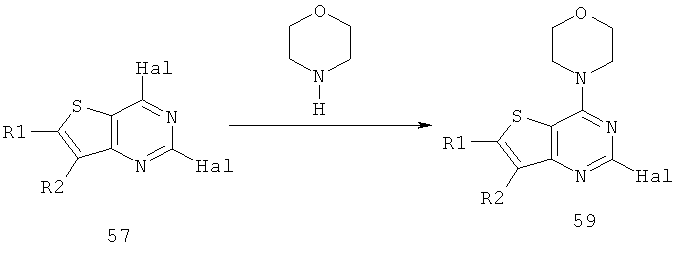
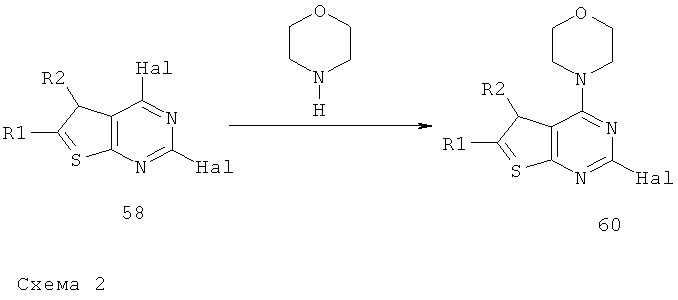
На Схеме 2 показан общий способ селективной замены 4-галогенида в промежуточных соединениях бис-галогенотиенопиримидинов 57 и 58 на морфолин в щелочных условиях в органическом растворителе с целью получения соединений 2-галогено-4-морфолино-тиенопиримидинов 59 и 60, соответственно, где Hal представляет собой Cl, Br или I; и R1 и R2 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств.
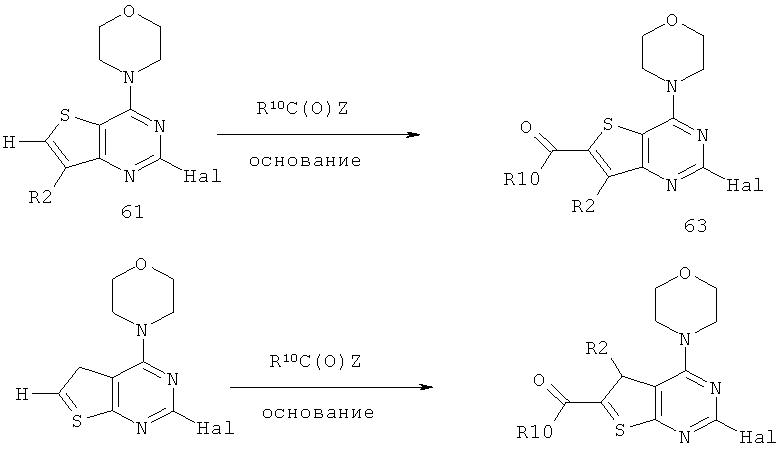
Схема 3
На Схеме 3 показан общий способ получения производных по положению 6 соединений 2-галогено-4-морфолино-6-водород-тиенопиримидинов 61 и 62, где R1 представляет собой Н. В результате обработки 61 или 62 реагентом для введения лития с целью удаления протона из положения 6, затем добавления ацилирующего реагента R10C(O)Z, где Z представляет собой уходящую группу, такую как галогенид, сложный эфир NHS (N-гидроксисукцинимид), карбоксилат или диалкиламино, получают соединения 2-галогено-4-морфолино-6-ацилтиенопиримидины 63 и 64, где Hal представляет собой Cl, Br или I; и R2 и R10 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств. Примером R10C(O)Z для получения 6-формил-соединений (R10 представляет собой Н) является N,N'-диметилформамид (DMF).
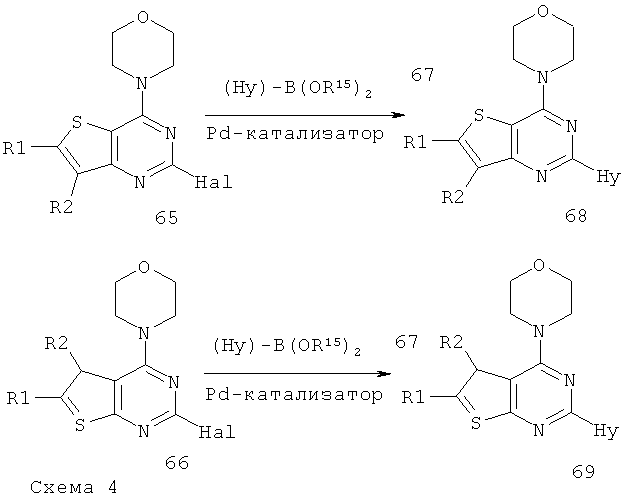
На Схеме 4 показан общий способ сочетания по Сузуки промежуточных соединений 2-галогено-пиримидинов (65 и 66) с реагентом (моноциклический гетероарил)-, (конденсированный бициклический гетероциклил)- или (конденсированный бициклический гетероарил)-бороновой кислотой (R15 представляет собой Н) или ее сложным эфиром (R15 представляет собой алкил) 67 с получением 2-замещенных (Ну) соединений 4-морфолино-тиенопиримидинов (68 и 69) формул I и II, где Hal представляет собой Cl, Br или I; и R1 и R2 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств. Для ознакомления с реакцией Сузуки см.: Miyaura et al. (1995) Chem. Rev. 95: 2457-2483; Suzuki, A. (1999) J. Organomet. Chem. 576: 147-168; Suzuki, А. в Metal-Catalyzed Cross-Coupling Reactions, Diederich, F., Stang, P.J., Eds., VCH, Weinheim, DE (1998), pp.49-97. Палладиевым катализатором может быть любой катализатор, обычно используемый для реакций перекрестного сочетания по Сузуки, такой как PdCl2(PPh3)2, Pd(PPh3)4, Pd(OAc)2, PdCl2(dppf)-DCM (бис(1,1′-бис(дифенилфосфанил)ферроцен-палладия(II) дихлорид/дихлорметан), Pd2(dba)3/P(t-Bu)3 (трис(дибензилиденацетон)дипалладий(0)/Р-(трет-Bu)3) (Owens et al. (2003) Bioorganic & Med. Chem. Letters 13: 4143-4145; MoIander et al. (2002) Organic Letters 4(11): 1867-1870; US 6448433).
На Схеме 5 показан общий способ синтеза алкинов 71, которые могут быть использованы для получения алкинилированных производных соединений 72 и 73. Пропаргиламины 71 могут быть получены в результате взаимодействия пропаргилбромида 70 с амином формулы R10R11NH (где R10 и R11 независимо выбраны из Н, алкила, арила и гетероарила, или R10 и R11 вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо) в присутствии соответствующего основания (Cs2CO3 или тому подобного). Для обзора синтезов алкиниламинов и родственных синтезов см. Booker-Milburn, K.I., Comprehensive Organic Functional Group Transformations (1995), 2: 1039-1074; и Viehe, H.G., (1967) Angew. Chem., Int. Ed. Eng., 6(9): 767-778. После этого алкины 71 могут быть приведены во взаимодействие с промежуточными соединениями 72 (X2 представляет собой бромо или иодо) или 73 (посредством сочетания Соногаширы) с получением соединений 74 и 75, соответственно, где R2 и R3 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств.
На Схеме 6 показан общий способ синтеза алкинов 77, которые могут быть использованы для получения алкинилированных производных соединений 72 и 73. Гем-диалкилпропаргиламины 77 могут быть получены с использованием способов, описанных Zaragoza et al. (2004) J. Med. Chem., 47: 2833. В соответствии со Схемой 6 гем-диалкилхлорид 76 (R14 и R15 независимо представляют собой метил, этил или другую алкильную группу) может быть приведен во взаимодействие с амином формулы R10R11NH (где R10 и R11 независимо выбраны из Н, алкила, арила и гетероарила, или R10 и R11 вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо) в присутствии CuCl и соответствующего основания (например, TEA (триэтиламин) или тому подобного) с получением алкина 77. Алкин 77 может быть приведен во взаимодействие с промежуточными соединениями 72 или 73 (посредством сочетания Соногаширы) с получением соединений 78 и 79, соответственно, где R2 и R3 такие, как они определены для соединений формулы и II или их предшественников или пролекарств.

На Схеме 7 показана общая схема синтеза алкинов 81, которые могут быть использованы для получения алкинилированных производных соединений 72 и 73. Бут-3-ин-1 -амины 81 (где R14R15 независимо представляют собой Н, алкил, арил, гетероарил, или R14 и R15 вместе с атомом углерода, к которому они присоединены, образуют карбоциклическое или гетероциклическое кольцо) могут быть получены в результате взаимодействия алкинов 80 (LG представляет собой тозилат или другую уходящую группу) с амином формулы R10R11NH (где R10 и R11 независимо выбраны из Н, алкила, арила и гетероарила, или R10 и R11 вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо) с использованием протокола, описанного Olomucki M. et al. (1960) Ann. Chim. 5: 845. Алкины 81 затем могут быть приведены во взаимодействие с промежуточными соединениями 72 или 73 (посредством сочетания Соногаширы) в соответствии с описаниями, представленными для Схем 5 и 6, с получением соединений 82 и 83, соответственно, где R2 и R3 такие, как они определены для соединений формулы I и II или их предшественников или пролекарств.
Фармацевтически приемлемую соль соединения тиенопиримидина формулы I или II можно получить, используя традиционные методики. Обычно такой способ включает обработку тиенопиримидина формулы I, который определен выше, подходящей кислотой в подходящем растворителе.
В способе по изобретению, который определен выше, как стадия аминирования, так и Pd-опосредованная стадия перекрестного сочетания протекают в традиционных условиях. Палладиевым катализатором может быть любой катализатор, обычно используемый для реакций перекрестного сочетания по Сузуки, такой как PdCl2(PPh3)2. Восстанавливающий агент обычно представляет собой боргидрид, такой как NaBH(ОАс)3, NaBH4 или NaCNBH4.
МЕТОДЫ РАЗДЕЛЕНИЯ
В способах получения соединений по данному изобретению предпочтительным может быть отделение продуктов реакции друг от друга и/или от исходных соединений. Желаемые продукты с каждой стадии или серий стадий разделяют и/или очищают (далее, разделяют) до желаемой степени гомогенности с использованием общеизвестных в данной области техники методик. Обычно такие методы разделения включают многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может включать в себя любое количество методов, в том числе, например: обращенно-фазовую и нормально-фазовую; стерическую эксклюзионную; ионообменную хроматографию; методы и устройство для жидкостной хроматографии высокого, среднего и низкого давления; мелкомасштабную аналитическую хроматографию; хроматографию с симулированным движением неподвижной фазы (SMB); и препаративную тонкослойную и толстослойную хроматографию, а также методики мелкомасштабной тонкослойной хроматографии и флэш-хроматографии.
Другой класс методов разделения включает обработку смеси реагентом, избирательно связывающимся или каким-либо другим образом взаимодействующим с отделяемым желаемым продуктом, непрореагировавшим исходным веществом, побочным продуктом реакции или тому подобное. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные среды или тому подобное. Альтернативно, данными реагентами могут быть кислоты в случае основного вещества, основания в случае кислотного вещества, связывающие реагенты, такие как антитела, связывающие белки, селективные хелатирующие агенты, такие как краун-эфиры, реагенты для жидкостной экстракции с использованием ионных жидкостей (LIX) или тому подобное.
Выбор соответствующих методов разделения зависит от природы рассматриваемых веществ. Например, от точки кипения и молекулярной массы при дистилляции и сублимации, наличия или отсутствия полярных функциональных групп при хроматографии, стабильности соединений в кислотных и щелочных средах при многофазной экстракции и тому подобного. Специалист в данной области техники будет применять методики, с наибольшей вероятностью обеспечивающие желаемое разделение.
Диастереомерные смеси могут быть разделены на их индивидуальные диастереомеры на основании различий их физико-химических свойств методами, хорошо известными специалисту в данной области техники, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены посредством превращения энантиомерной смеси в диастереомерную смесь в результате взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, в результате гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме этого, некоторые соединения по настоящему изобретению могут представлять собой атропизомеры (например, замещенные биарилы), и они рассматриваются как часть данного изобретения. Энантиомеры также могут быть разделены с использованием колонки для хиральной HPLC (высокоэффективной жидкостной хроматографии).
Индивидуальный стереоизомер, например энантиомер, по существу, не содержащий своего другого стереоизомера, может быть получен в результате разделения рацемической смеси с использованием такого метода, как образование диастереомеров с применением оптически активных разделяющих агентов (Eliel, E. and Wilen, S. "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994; Lochmuller, C. H., (1975) J. Chromatogr., 113(3): 283-302). Рацемические смеси хиральных соединений по изобретению могут быть разделены и выделены любым подходящим методом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение путем фракционной кристаллизации или другими методами, (2) образование диастереомерных соединений с хиральными дериватизирующими реагентами (chiral derivatizing reagents), разделение диастереомеров и превращение их в чистые стереоизомеры и (3) выделение, по существу, чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: "Drug Stereochemistry, Analytical Methods and Pharmacology", Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
В методе (1) диастереомерные соли могут быть образованы в результате взаимодействия энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, а-метил-р-фенилэтиламин (амфетамин) и тому подобное, с асимметрическими соединениями, несущими кислотную функциональную группировку, такими как карбоновая кислота и сульфоновая кислота. Такие диастереомерные соли могут быть подвергнуты разделению фракционной кристаллизацией или ионной хроматографией. При разделении оптических изомеров аминосоединений добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота, может приводить к образованию диастереомерных солей.
Альтернативно, согласно методу (2) подвергаемое разделению вещество приводят во взаимодействие с одним энантиомером хирального соединения для образования диастереомерной пары (Eliel, E. and Wilen, S. "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., 1994, p.322). Диастереомерные соединения могут быть образованы в результате взаимодействия ассиметрических соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментильные производные, с последующим разделением этих диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Метод определения оптической чистоты включает приготовление хиральных сложных эфиров рацемической смеси, таких как ментиловые эфиры, например, (-)-ментилхлорформиат, в присутствии основания или сложного эфира кислоты Мошера, α-метокси-α-(трифторметил)фенилацетата (Jacob III. J. Org. Chem., (1982) 47: 4165), и анализа 1ЯМР-спектра на предмет наличия двух атропизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропизомерных соединений могут быть разделены и выделены с использованием нормально-фазовой и обращенно-фазовой хроматографии, следуя методам разделения атропизомерных нафтил-изохинолинов (WO 96/15111). Согласно методу (3) рацемическая смесь двух энантиомеров может быть разделена хроматографией с использованием хиральной неподвижной фазы ("Chiral Liquid Chromatography" (1989) W. J. Lough, Ed., Chapman and Hall, New York; Okamoto, J. Chromatogr., (1990) 513: 375-378). Обогащенные или очищенные энантиомеры можно различить, используя методы идентификации других хиральных молекулы с ассиметрическими атомами углерода, такие как оптическое вращение и круговой дихроизм.
ХИМИОТЕРАПЕВТИЧЕСКИЕ АГЕНТЫ
Некоторые химиотерапевтические агенты продемонстрировали удивительные и неожидаемые свойства в комбинации с соединениями формулы I или II в отношении ингибирования клеточной пролиферации in vitro и in vivo. Такие химиотерапевтические агенты включают: эрлотиниб, доцетаксел, 5-FU, гемцитабин, PD-0325901, цисплатин, карбоплатин, паклитаксел, бевацизумаб, трастузумаб, пертузумаб, темозоломид, тамоксифен, доксорубицин, Akti-1/2, HPPD и рапамицин.
Эрлотиниб (TARCEVA®, OSI-774, Genentech) применяют для лечения немелкоклеточного рака легкого (NSClC), рака легкого, рака поджелудочной железы и некоторых других типов рака путем целенаправленного воздействия конкретно на тирозинкиназу - рецептор эпидермального ростового фактора (EGFR) (US 5747498; US 6900221; Moyer et al. (1997) Cancer Res. 57: 4838; PolIack et al. (1999) J. Pharmacol. Exp.Ther. 291: 739; Perez-Soler et al. (2004) J. Clin. Oncol. 22: 3238; Kirn et al. (2002) Curr. Opin. Invest. Drugs 3: 1385-1395; BIackhall et al. (2005) Expert. Opin. Pharmacother. 6: 995-1002). Эрлотинибом называют N-(3-этинилфенил)-6,7-бис(метоксиметокси)хиназолин-4-амин (регистрационный №CAS: 183321-74-6), и он имеет структуру:
Доцетаксел (TAXOTERE®, Sanofi-Aventis) применяют для лечения рака молочной железы, рака яичников и NSClC (US 4814470; US 5438072; US 5698582; US 5714512; US 5750561; Mangatal et al. (1989) Tetrahedron 45:4177; Ringel et al. (1991) J. Natl. Cancer Inst. 83: 288; Bissery et al. (1991) Cancer Res. 51: 4845; Herbst et al. (2003) Cancer Treat. Rev. 29: 407-415; Davies et al. (2003) Expert. Opin. Pharmacother. 4: 553-565). Доцетакселом называют (2R,3S)-N-карбокси-3-фенилизосерин, сложный N-трет-бутиловый эфир, сложный 13-иловый эфир, содержащий 5,20-эпокси-1,2,4,7,10,13-гексагидрокситакс-1-ен-9-он, 4-ацетат, 2-бензоат, тригидрат (US 4814470; ЕР 253738; регистрационный №CAS: 114977-28-5), и он имеет структуру:
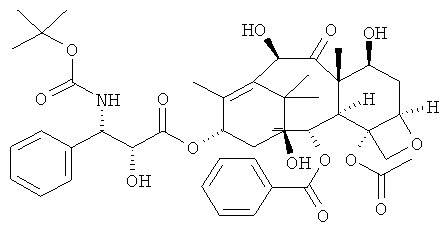
5-FU (фторурацил, 5-фторурацил, регистрационный №CAS: 51-21-8) представляет собой ингибитор тимидилатсинтазы и уже несколько десятилей используется в лечении рака, в том числе колоректального рака и рака поджелудочной железы (US 2802005; US 2885396; Duschinsky et al. (1957) J. Am. Chem. Soc. 79: 4559; Hansen, R.M. (1991) Cancer Invest. 9: 637-642). 5-FU называют 5-фтор-1H-пиримидин-2,4-дион, и он имеет структуру:
Гемцитабин (GEMZAR®, Lilly, регистрационный №CAS: 95058-81-4) представляет собой аналог нуклеозидов, который блокирует репликацию ДНК, и его применяют для лечения различных карцином, включая рак поджелудочной железы, рак молочной железы, NSClC, и лимфом (US 4808614; US 5464826; Hertel et al. (1988) J. Org. Chem. 53:2406; Hertel et al. (1990) Cancer Res. 50: 4417; Lund et al. (1993) Cancer Treat. Rev. 19: 45-55). Гемцитабином называют 4-амино-1-[3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-он, и он имеет структуру:
PD-0325901 (регистрационный №CAS: 391210-10-9, Pfizer) представляет собой аллостерический АТФ-неконкурентный ингибитор МЕК второго поколения для возможного лечения рака пероральными таблетками (US 6960614; US 6972298; US 2004/147478; US 2005/085550). Проведена фаза II клинических испытаний на предмет возможного лечения опухолей молочной железы, опухолей толстой кишки и меланомы. PD-0325901 называют (R)-N-(2,3-дигидроксипропокси)-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамид, и он имеет структуру:
Цисплатин (цис-диамин, дихлорплатина(II), регистрационный №CAS: 15663-27-1), представляет собой химиотерапевтическое лекарственное средство на основе платины, применяемое для лечения различных типов рака, включая саркомы, некоторые карциномы (например, мелкоклеточный рак легкого и рак яичников), лимфомы и эмбрионально-клеточные опухоли (Ochs et al. (1978) Cancer Treat. Rep.62: 239; Pinedo et al. (1978) Eur. J. Cancer 14: 1149; "Cysplatin, Current Status and New Developments", A.W. Prestayko et al., Eds., Academic Press, New York, 1980). Цисплатин (CDDP) был первым членом этого класса, который в настоящее время также включает карбоплатин и оксалиплатин.
Карбоплатин (регистрационный №CAS: 41575-94-4) представляет собой химиотерапевтическое лекарственное средство, применяемое против карциномы яичников, рака легкого, рака головы и шеи (US 4140707; Calvert et al. (1982) Cancer Chemother. Pharmacol. 9: 140; HarIand et al. (1984) Cancer Res. 44: 1693). Карбоплатином называют азанид, циклобутан-1,1-дикарбоновую кислоту, платину, и он имеет структуру:
Паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Princeton NJ, регистрационный №CAS: 33069-62-4) представляет собой соединение, выделенное из коры дерева тиса тихоокеанского, Taxus brevifolia, и применяется для лечения рака легкого, яичников, молочной железы и распространенных форм саркомы Калоши (Wani et al. (1971) J. Am. Chem. Soc. 93: 2325; Mekhail et al. (2002) Expert. Opin. Pharmacother. 3: 755-766). Паклитакселом называют β-(бензоиламино)-α-гидрокси-,6,12b-бис(ацетилокси)-12-(бензоилокси)-2а,3,4,4а,5,6,9,10,11,12,12а,12b-додекагидро-4,11-дигидрокси-4а,8,13,13-тетраметил-5-оксо-7,11-метано-1Н-циклодека(3,4)бенз(1,2-б)-оксет-9-иловый эфир (2aR-(2a-a,4-β,4a-β,6-β,9-a(a-R*,β-S*), 11-а, 12-а, 12а-а,2b-а))-бензолпропановой кислоты, и он имеет структуру:
Бевацизумаб (AVASTIN®, Genentech) представляет собой рекомбинантное гуманизированное моноклональное антитело против VEGF - сосудистого эндотелиального ростового фактора (US 6054297; Presta et al. (1997) Cancer Res. 57: 4593-4599). Его применяют в лечении рака, где он ингибирует опухолевый рост путем блокирования образования новых кровеносных сосудов. Бевацизумаб был первым доступным в клинике ингибитором ангиогенеза в Соединенных Штатах Америки, одобренным Управлением по контролю за качеством пищевых продуктов, медикаментов и косметических средств (FDA) в 2004 году к применению в комбинации со стандартной химиотерапией в лечении метастатического рака толстой кишки и большинства форм метастатического немелкоклеточного рака легкого. В настоящее время проводятся отдельные поздние стадии клинических исследований с целью определения его безопасности и эффективности для пациентов с: поддающимся лечению адъювантной терапией/неметастатическим раком толстой кишки, метастатическим раком молочной железы, метастатическим почечно-клеточным раком, метастатической полиморфной глиобластомой, метастатическим раком яичников, метастатической гормоно-резистентной формой рака предстательной железы и метастатическим или нерезектабельным локально распространенным раком поджелудочной железы (Ferrara et al. (2004) Nat. Rev. Drug Disc. 3: 391-400).
Бевацизумаб содержит подвергнутые мутации каркасные области человеческого IgG1 и антиген-связывающие гипервариабельные участки от мышиного моноклонального антитела против hVEGF A.4.6.1, которое блокирует связывание человеческого VEGF (hVEGF) с его рецепторами. Бевацизумаб имеет молекулярную массу примерно 149000 дальтон и гликозилирован. Бевацизумаб и другие гуманизированные анти-VEGF антитела также описаны в US 6884879. Дополнительные анти-VEGF антитела включают G6- или В20-серии антител, например, G6-31, В20-4.1 (WO 2005/012359; WO 2005/044853; US 7060269; US 6582959; US 6703020; US 6054297; WO 98/45332; WO 96/30046; WO 94/10202; EP 0666868B1; US 2006/009360; US 2005/0186208; US 2003/0206899; US 2003/0190317; US 2003/0203409; 20050112126; Popkov et al. (2004) Journal of Immunological methods, 288: 149-164. "Антитело В20-серии" представляет собой анти-VEGF антитело, которое происходит из последовательности антитела В20 или является происходящим из В20 антителом согласно любой из Фиг.27-29 в WO 2005/012359, полное описание которой конкретно включено в данное описание путем ссылки. В одном из воплощений антитело В20-серии связывается с функциональным эпитопом человеческого VEGF, содержащим остатки F17, М18, D19, Y21, Y25, Q89, 191, К101, Е103 и С104. Другие анти-VEGF антитела включают антитела, которые связываются с функциональным эпитопом человеческого VEGF, содержащим остатки F17, М18, D19, Y21, Y25, Q89, 191, К101, Е103 и С104, или, альтернативно, содержащим остатки F17, Y21, 022, Y25, D63, 183 и Q89.
Трастузумаб (HERCEPTIN®, huMab4D5-8, rhuMab HER2, Genentech) представляет собой происходящую из рекомбинантной ДНК гуманизированную, IgGI-каппа, версию моноклонального антитела - мышиного антитела против HER2, которое селективно связывается с высокой аффинностью в анализе с использованием клеток (Kd=5 нМ) с внеклеточным доменом белка - рецептора-2 эпидермального ростового фактора человека, HER2 (ErbB2) (US 5821337; US 6054297; US 6407213; US 6639055; Coussens L, et al. (1985) Science 230: 1132-9; SIamon DJ, et al. (1989) Science 244: 707-12). Трастузумаб содержит каркасные области антитела человека с гипервариабельными участками мышиного антитела (4D5), которые связываются с HER2. Трастузумаб связывается с антигеном HER2 и таким образом ингибирует рост раковых клеток. Как в анализах in vitro, так и на животных показано, что Трастузумаб ингибирует пролиферацию человеческих опухолевых клеток, которые сверхэкспрессируют HER2 (Hudziak RM, et al. (1989) Mol. Cell Biol. 9: 1165-72; Lewis GD, et al. (1993) Cancer Immunol. Immunother.; 37: 255-63; Baselga J, et al. (1998) Cancer Res. 58: 2825-2831). Трастузумаб является медиатором антитело-зависимой клеточной цитотоксичности, ADCC (Hotaling ТЕ, et al. (1996) [Abstract]. Proc. Annual Meeting Am. Assoc. Cancer Res.; 37: 471; Pegram MD, et al. (1997) [Abstract]. Proc. Am. Assoc. Cancer Res.; 38: 602; Sliwkowski et al. (1999) Seminars in Oncology 26(4), SuppI 12: 60-70; Yarden Y. and Sliwkowski, M. (2001) Nature Reviews: MolecuIar Cell Biology, MacmilIan Magazines, Ltd., Vol.2: 127-137). HERCEPTIN® был одобрен в 1998 г.для лечения пациентов с ErbB2-сверхэкспрессирующими метастатическими видами рака молочной железы (Baselga et al., (1996) J. Clin. Oncol. 14: 737-744). HERCEPTIN® одобрен FDA в 2006 г.в качестве части схемы лечения, состоящей из доксорубицина, циклофосфамида и паклитаксела, для адъювантного лечения пациентов с HER2-позитивным раком молочной железы с позитивными лимфатическими узлами. Имеется существенная клиническая необходимость в разработке дополнительных НЕР2-направленных терапий рака для таких пациентов с HER2-сверхэкспрессирующими опухолями или другими заболеваниями, ассоциированными с экспрессией HER2, которые не отвечают или слабо отвечают на лечение с использованием HERCEPTIN®.
Пертузумаб (OMNITARG™, rhuMab 2C4, Genentech) представляет собой находящееся на клинической стадии изучения гуманизированное антитело и первый агент в новом классе агентов, известных как ингибиторы димеризации HER (HDI), которые блокируют способность рецептора HER2 к совместному действию с другими членами семейства рецепторов HER, т.е. HER1/EGFR, HER3 и HER4 (US 6949245; Agus et al. (2002) Cancer Cell 2: 127-37; Jackson et al. (2004) Cancer Res. 64: 2601-9; Takai et al. (2005) Cancer 104: 2701-8). В раковых клетках, такое воздействие на способность HER2 к совместному действию с другими рецепторами HER-семейства блокирует передачу сигнала клеткой и в конечном счете может приводить к ингибированию роста раковых клеток и гибели раковой клетки. HDI, вследствие своего уникального способа действия, обладают способностью работать в большом разнообразии опухолей, включая опухоли, не экспрессирующие HER2 (Mullen et al. (2007) MolecuIar Cancer Therapeutics 6: 93-100).
Темозоломид (регистрационный №CAS: 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough) представляет собой пероральное химиотерапевтическое лекарственное средство, одобренное FDA для лечения анапластической астроцитомы, и проводились его исследования в отношении других типов опухолей головного мозга, таких как полиформная глиобластома (US 5260291; Stevens et al. (1984) J. Med. Chem. 27: 196; NewIands et al. (1997) Cancer Treat. Rev. 23: 35-61; Danson et al. (2001) Expert Rev. Anti Cancer Ther. 1: 13-19). Темозоломидом называют (4-метил-5-оксо-2,3,4,6,8-пентазабицикло-[4.3.0]-нона-2,7,9-триен-9-карбоксамид или 3,4-дигидро-3-метил-4-оксоимидазо-[5,1-d]-as-тетразин-8-карбоксамид (US 5260291, №CAS: 85622-93-1), и он имеет структуру:
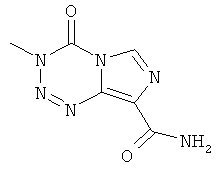
Тамоксифен (NOLVADEX®, ISTUBAL®, VALODEX®) представляет собой перорально активный селективный модулятор рецептора эстрогена (SERM), применяемый в лечении рака молочной железы и на сегодняшний день являющийся наиболее продаваемым в мире лекарственным средством для такого показания. Первоначально тамоксифен (Nolvadex®) был одобрен FDA (ICl Pharmaceuticals, в настоящее время AstraZeneca) в 1977 г.для лечения метастатического рака молочной железы (Jordan VC (2006) Br. J. Pharmacol. 147 (SuppI 1): S269-76). В настоящее время тамоксифен применяют для лечения как раннего, так и распространенного рака молочной железы, позитивного по эстрогеновому рецептору (ER), у женщин пред- и послеменопаузального периода (Jordan VC (1993) Br. J. Pharmacol. 110 (2): 507-17). Он также одобрен FDA для предупреждения рака молочной железы у женщин с высоким риском развития заболевания и для ослабления риска контралатерального (на противоположной молочной железе) рака молочной железы. Тамоксифеном называют (Z)-2-[4-(1,2-бут-1-енил)фенокси]-N,N-диметил-этанамин (регистрационный №CAS: 10540-29-1), и он имеет структуру:
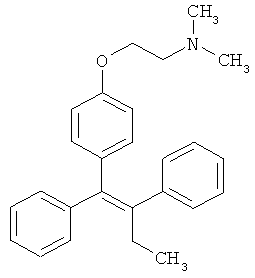
Доксорубицин (ADRIAMIClNE®, гидроксилдаунорубицин) представляет собой воздействующее на ДНК лекарственное средство, широко применяемое в химиотерапии с 1960-х годов. Он является антрациклиновым антибиотиком и по структуре родственен дауномицину, который также действует на ДНК как интеркалирующее средство. Доксорубицин обычно используют в лечении широкого диапазона типов рака. Доксорубицином называют (8S,10S)-10-(4-амино-5-гидрокси-6-метил-тетрагидро-2Н-пиран-2-илокси)-6,8,11-тригидрокси-8-(2-гидроксиацетил)-1-метокси-7,8,9,10-тетрагидротетрацен-5,12-дион (регистрационный №CAS: 23214-92-8), и он имеет структуру:
Akti-1/2 представляет собой способное проникать в клетку соединение хиноксалина, которое потенциально и селективно ингибирует активность Akt1/Akt2: IC50=58 нМ, 210 нМ и 2,12 мкМ для Akt1, Akt2 и Akt3, соответственно, в in vitro анализах киназ (Barnett et al. (2005) Biochem. J. 385: 399; DeFeo-Jones, et al. (2005) Mol. Cancer Ther. 4:271; Zhao et al. (2005) Bioorg. Med. Chem. Lett. 15: 905; Lindsley et al. (2005) Bioorg. Med. Chem. Lett. 15: 761; US 2006/142178; US 2005/159422; US 2004/102360). По-видимому, такое ингибирование является зависимым от плекстрин-гомологичного (РН) домена. Соединение не проявляет никакого ингибирующего действия против Akt, у которых отсутствует РН-домен, или других близкородственных киназ AGC-семейства, РКА (протеинкиназа А), РКС (протеинкиназа С) и SGK (сыворотка- и глюкокортикоид-индуцибельная киназа (serum- and glucocorticoid-induClble kinase)), даже в концентрации до 50 мкМ. Akt1-1/2 преодолевает Akt1/Akt2-опосредованную резистентность к химиотерапевтическим средствам в опухолевых клетках, и показано, что он блокирует основное/ую и стимулированное/ую фосфорилирование/активацию Akt1/Akt2 как в культивируемых клетках in vitro, так и у мышей in vivo. Akti-1/2 (EMD Biosciences Product, №124018) называют 1,3-дигидро-1-(1-((4-(6-фенил-1Н-имидазо[4,5-д]хиноксалин-7-ил)фенил)метил)-4-пиперидинил)-2Н-бензимидазол-2-он, и он имеет структуру:
HPPD представляет собой селективный ингибитор B-Raf-киназы (IC50 для В-Raf меньше 2 нМ, IC50 для pERK 87 нМ), проходящий доклинические исследования (US 2006/0189627). HPPD называют оксим 5-(1-(2-гидроксиэтил)-3-(пиридин-4-ил)-1Н-пиразол-4-ил)-2,3-дигидро-1Н-инден-1-она, он и имеет структуру:
Рапамицин (сиролимус, RAPAMUNE®) представляет собой иммуносупрессивное лекарственное средство, применяемое для предупреждения отторжения трансплантации органов и особенно полезное в трансплантатах почки. Рапамицин является макролидным антибиотиком ("мицин"), первоначально описанным как продукт бактерии Streptomyces hygroscopicus из образца почвы с острова Рапа-Нуи, более известного как остров Пасхи (Pritchard DI (2005). Drug Discovery Today 10 (10): 688-691). Рапамицин ингибирует ответ на интерлейкин-2 (IL-2) и тем самым блокирует активацию Т- и В-клеток. Принцип действия рапамицина заключается в связывании с цитозольным белком размером 12 кДа, связывающим FK (FKBP12). Комплекс рапамицин-РКВР12 ингибирует путь мишени рапамицина у млекопитающих (mTOR) путем прямого связывания с mTOR-комплексом-1 (mTORd). mTOR также называют как FRAP (FKBP-рапамицин-ассоциированный белок) или RAFT (мишень рапамицина и FKBP). Рапамицином называют (3S,6R,7E,9R, 10R, 12R, 14S, 15E, 17E, 19E,21 S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34а-гексадекагидро-9,27-дигидрокси-3-[(1R)-2-[(1S,3R,4R)-4-гидрокси-3-метоксициклогексил]-1-метилэтил]-10,21-диметокси-6,8,12,14,20,26-гексаметил-23,27-эпокси-3H-пиридо[2,1-с][1,4]-оксаазациклогентриаконтин-1,5,11,28,29(4H,6H,31Н)-пентон (регистрационный №CAS: 53123-88-9), и он имеет структуру:
Лапатиниб (TYKERB®, GW572016, GIaxo SmithKline) одобрен для применения в комбинации с капецитабином (XELODA®, Roche) для лечения пациентов с распространенным или метастатическим раком молочной железы, опухоли которого сверхэкспрессируют HER2 (ErbB2), которые ранее получали терапию, включающую антрациклин, таксан и трастузумаб. Лапатиниб представляет собой АТФ-конкурентный двойной тирозинкиназный ингибитор эпидермального ростового фактора (EGFR) и HER2/neu (ErbB-2) (US 6727256; US 6713485; US 7109333; US 6933299; US 7084147; US 7157466; US 7141576), который ингибирует аутофосфорилирование и активацию рецептора путем связывания с АТФ-связывающим карманом домена протеинкиназ EGFR/HER2. Лапатинибом называют N-(3-хлор-4-(3-фторбензилокси)фенил)-6-(5-((2-(метилсульфонил)-этиламино)-метил)фуран-2-ил)хиназолин-4-амин, и он имеет структуру:
БИОЛОГИЧЕСКАЯ ОЦЕНКА
Некоторые соединения формулы I и II специфически связываются с изоформами Pl3-киназы и ингибируют пролиферацию опухолевых клеток (WO 2006/046031; US 2008/0039459; US 2008/0076768; US 2008/0076758; WO 2008/070740; WO 2008/073785).
Некоторые соединения формулы Ia и IIa связываются с изоформой р110а с Юбо менее чем 1 микромоль/л и демонстрируют ингибирование in vivo опухолевого роста под действием одного агента в мышиных моделях ксенотрансплантатов. Соответственно, соединения формулы I и II могут быть использованы для лечения заболевания или расстройства, возникающего вследствие аномального клеточного роста, функционирования или поведения, в виде отдельных агентов или в комбинированной терапии с одним или более химиотерапевтическими агентами.
Некоторые типичные соединения формулы I и II, изложенные в данном описании, были получены, охарактеризованы и проанализированы на предмет их Pl3K-связывающей активности (Пример 13) и активности in vitro против опухолевых клеток (Пример 14). Диапазон Pl3K-связывающих активностей (IC50) составлял от менее 1 нМ (наномолярной концентрации, равной единице) до примерно 10 мкМ (микромолярной концентрации, равной десяти). Некоторые типичные соединения формулы I и II имеют величины IC50 для Pl3K-связывающей активности менее 10 нМ. Некоторые соединения формулы I и II имеют величины EC50 для ассоциированной с опухолевыми клетками активности менее 100 нМ.
Цитотоксическую или цитостатическую активность типичных соединений формулы I и II измеряли посредством: внедрения линии пролиферирующих опухолевых клеток млекопитающих в культуральную клеточную среду, добавления соединения формулы I или II, культивирования клеток в течение периода времени от примерно 6 часов до примерно 5 суток; и измерения жизнеспособности клеток (Пример 14). Анализы in vitro с использованием клеток применяли для измерения жизнеспособности, т.е. пролиферации (IC50), цитотоксичности (ЕС50) и индукции апоптоза (активации каспаз). Фармакодинамические и фармакокинетические свойства в отношении всасывания, распределения, метаболизма и экскреции (ADME) измеряли для некоторых типичных соединений, используя анализы, в том числе: анализы Сасо-2-проницаемости, выведения гепатоцитами, ингибирования цитохрома Р450, индукции цитохрома Р450, связывания белками плазмы и блокирования hERG-каналов (специфических калиевых каналов сердца человека (human ether-a-go-go)).
АНАЛИЗЫ IN VITRO КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ
Эффективность in vitro комбинаций соединений формулы I или II с химиотерапевтическими агентами измеряли в соответствии с анализом клеточной пролиферации из Примера 14 с использованием люминесцентного анализа жизнеспособности клеток CellTiter-Glo® от Promega Corp., Madison, WI. Этот гомогенный метод анализа основан на экспрессии рекомбинантной люциферазы Coleoptera (US 5583024; US 5674713; US 5700670), и в нем определяют количество жизнеспособных клеток в культуре на основании количества присутствующего АТФ, индикатора метаболически активных клеток (Crouch et al. (1993) J. Immunol. Meth. 160: 81-88; US 6602677). Анализ CellTiter-Glo® проводили в 96- или 384-луночном формате, что делает его подходящим для автоматического высокопроизводительного скрининга (HTS) (Сгее et al. (1995) AntiCancer Drugs 6: 398-404). Процедура данного гомогенного анализа включает добавление одного реагента (реагента CellTiter-Glo®) непосредственно в клетки, культивируемые в дополненной сывороткой среде. Стадий промывки клеток, удаления среды и многократного пипетирования не требуется. Система регистрирует минимально 15 клеток/лунка в 384-луночном формате через 10 минут после добавления реагента и перемешивания.
Гомогенный формат "добавить-перемешать-измерить" приводит к лизису клеток и генерации сигнала люминесценции, пропорционального количеству присутствующего АТФ. Количество АТФ прямо пропорционально количеству присутствующих в культуре клеток. В анализе CellTiter-Glo® генерируется люминесцентный сигнал типа свечения ("glow-type"), производимый под действием люциферазной реакции, который в общем случае характеризуется временем полужизни более пяти часов в зависимости от используемого типа клеток и используемой среды. Количество жизнеспособных клеток выражают в относительных световых единицах (RLU). Субстрат, люциферин жуков, подвергается окислительному декарбоксилированию под действием рекомбинантной люциферазы светлячков с сопровождающимся превращением АТФ в AMP и образованием фотонов. Продолжительное время полужизни снимает необходимость в применении инжекторов для реагентов и обеспечивает оперативность в отношении осуществления непрерывного или периодического режима обработки многочисленных планшетов. Этот анализ клеточной пролиферации может быть использован в различных многолуночных форматах, например 96- или 384-луночном формате. Данные можно регистрировать с помощью люминометра или визуализирующего устройства, CCD-камеры (камера, содержащая устройство с зарядовой связью). Выходной сигнал люминесценции приведен в виде относительных световых единиц (RLU), измеренных во времени.
Антипролиферативные эффекты типичных соединений формулы I и II и их комбинаций с химиотерапевтическими агентами измеряли в соответствии с анализом CellTiter-Glo® (Пример 14) против линий опухолевых клеток, приведенных на Фиг.1-А, 1-В и 1-С. Оценивали величины ЕС50 для тестируемых соединений и комбинаций. Диапазон in vitro активности в отношении пролиферативного потенциала клеток составлял от примерно 100 нМ до примерно 10 мкМ.
На Фиг.1-А показана сводная информация in vitro анализов клеточной пролиферации комбинаций соединения формулы Ia и различных химиотерапевтических агентов. На Фиг.1-В показана сводная информация in vitro анализов клеточной пролиферации комбинаций соединения формулы IIa и различных химиотерапевтических агентов. На Фиг.1-С показана сводная информация in vitro анализов клеточной пролиферации комбинаций соединения формулы Ib и различных химиотерапевтических агентов. Линии раковых клеток охарактеризованы по типу опухоли и наличию генной мутации.
Индивидуальные измеренные против конкретного типа клеток величины EC50 для соединений формулы Ia, Ib и IIa и химиотерапевтического агента сравнивают с величиной ЕСао для комбинации. Показатель для комбинации (combination index) (Cl) в баллах рассчитывают по методу Chou и Talalay (Chou, T. and Talalay, P. (1984) Adv. Enzyme Regul. 22: 27-55). Cl менее 0,8 указывает на синергическое действие. Cl от 0,8 до 1,2 указывает на аддитивность. Cl больше 1,2 указывает на антагонизм. Величины Cl на Фиг.1-А, 1-В и 1-С рассчитаны на основании концентраций ЕСьо (третья позиция справа). Эффективность синергического действия оценена согласно Chou и Talalay и приведена в последней колонке таблиц. Некоторые комбинации на Фиг.1-А, 1-В и 1-С демонстрируют удивительное и неожидаемое свойство синергического действия в анализах in vitro клеточной пролиферации в отношении линий клеток опухолевого типа, в том числе рака молочной железы, цервикального рака, рака толстой кишки, эндометрия, глиомы, рака легкого, меланомы, рака яичников, поджелудочной железы и предстательной железы. Другие комбинации на Фиг.1-А, 1-В и 1-С не демонстрируют никакого синергического действия; а демонстрируют только одну лишь аддитивность или антагонизм. Некоторые комбинации проявляют синергическое действие по отношению к опухолям одного или более типов, но не к остальным. Синергическое действие, продемонстрированное in vitro в анализах клеточной пролиферации, дает основание ожидать соответствующее синергическое действие в лечении видов рака, включая, но этим не ограничиваясь, рак молочной железы, цервикальный рак, рак толстой кишки, эндометрия, глиому, рак легкого, меланому, рак яичников, поджелудочной железы и предстательной железы, у пациентов-людей.
На Фиг.2 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, 5-FU, соединения формулы Ia и комбинации 5-FU и соединения формулы Ia. Клетки MDA-MB-361 (тип опухоли молочной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением 5-FU (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения 5-FU (низ). Наблюдают сильное синергическое действие в случае одновременного введения (Cl=0,11) и в случае последующего введения соединения формулы Ia (Cl=0,10). Наблюдают сильное влияние порядка (последовательности) введения. В случае предварительного введения соединения формулы Ia показано меньшее синергическое действие (Cl=0,67).
На Фиг.3 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, гемцитабина, соединения формулы Ia и комбинации гемцитабина и соединения формулы Ia. Клетки Cal-51 (тип опухоли молочной железы) обработаны одновременно (верх) и обработаны впоследствии соединением формулы Ia через 4 часа после введения гемцитабина (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,59) и сильное синергическое действие в случае последующего введения соединения формулы Ia (Cl=0,17).
На Фиг.4 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, гемцитабина, соединения формулы Ia и комбинации гемцитабина и соединения формулы Ia. Клетки MDA-MB-361 (тип опухоли молочной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением гемцитабина (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения гемцитабина (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,27), в случае предварительного введения соединения формулы Ia (Cl=0,46) и в случае последующего введения соединения формулы Ia (Cl=0,28).
На Фиг.5 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, эрлотиниба, соединения формулы Ia и комбинации эрлотиниба и соединения формулы Ia. Клетки А549 (тип опухоли легкого с K-ras G12C) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением эрлотиниба (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения эрлотиниба (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,17), в случае предварительного введения соединения формулы Ia (Cl=0,31) и в случае последующего введения соединения формулы Ia (Cl=0,33).
На Фиг.6 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, эрлотиниба, соединения формулы Ia и комбинации эрлотиниба и соединения формулы Ia. Клетки Н23 (тип опухоли легкого с мутацией K-ras-G12C) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением эрлотиниба (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения эрлотиниба (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,28), в случае предварительного введения соединения формулы Ia (Cl=0,39) и в случае последующего введения соединения формулы Ia (Cl=0,37).
На Фиг.7 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕСоо), справа налево, темозоломида, соединения формулы Ia и комбинации темозоломида и соединения формулы Ia. Клетки U87 (тип опухоли - глиома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,004), но не в случае предварительного введения соединения формулы Ia (Cl=1,13) и не в случае последующего введения соединения формулы Ia (Cl=1,41).
На Фиг.8 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, темозоломида, соединения формулы Ia и комбинации темозоломида и соединения формулы Ia. Клетки А375 (тип опухоли - меланома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,007), но не в случае предварительного введения соединения формулы Ia (Cl=0,99) и не в случае последующего введения соединения формулы Ia (Cl=1,02).
На Фиг.9 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, темозоломида, соединения формулы Ia и комбинации темозоломида и соединения формулы Ia. Клетки MALME-3M (тип опухоли - меланома) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением темозоломида (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения темозоломида (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,18), но не в случае предварительного введения соединения формулы Ia (Cl=1,46) и не в случае последующего введения соединения формулы Ia (Cl=1,22).
На Фиг.10 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х EC50), справа налево, доксорубицина, соединения формулы Ia и комбинации доксорубицина и соединения формулы Ia. Клетки SKOV3 (тип опухоли яичников) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением доксорубицина (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения доксорубицина (низ). Наблюдают синергическое действие в случае одновременного введения (С1=0,39) и в случае последующего введения соединения формулы Ia (Cl=0,18), но не в случае предварительного введения соединения формулы Ia (Cl=1,44).
На Фиг.11 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки, при варьировании концентраций (начиная от 4Х ЕС50), справа налево, доцетаксела, соединения формулы Ia и комбинации доцетаксела и соединения формулы Ia. Клетки PC3 (тип опухоли предстательной железы) обработаны одновременно (верх), предварительно обработаны соединением формулы Ia за 4 часа перед введением доцетаксела (середина) и обработаны впоследствии соединением формулы Ia через 4 часа после введения доцетаксела (низ). Наблюдают синергическое действие в случае одновременного введения (Cl=0,43) и в случае последующего введения соединения формулы Ia (Cl=0,30), но не в случае предварительного введения соединения формулы Ia (Cl=1,23).
На Фиг.12 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки MDA MD 361 (тип опухоли молочной железы) при варьировании концентраций (начиная от 4Х ЕС50), справа налево: (верх) 5-FU, соединения формулы IIa и комбинации одновременно вводимых 5-FU и соединения формулы IIa; (середина) доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa; и (низ) гемцитабина, соединения формулы IIa и комбинации одновременно вводимых гемцитабина и соединения формулы IIa. Наблюдают синергическое действие в случае одновременного введения 5-FU и соединения формулы IIa (Cl=0,34), доцетаксела и соединения формулы IIa (Cl=0,09) и гемцитабина и соединения формулы IIa (Cl=0,50).
На Фиг.13 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют: (верх) жизнеспособные клетки МТЗ (тип опухоли молочной железы) при варьировании концентраций (начиная от 4Х ЕС50), справа налево, доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa; и (низ) жизнеспособные клетки U87 (тип опухоли: глиома, негативная по PTEN мутация) при варьировании концентраций (начиная от 4Х EC50), справа налево, темозоломида, соединения формулы IIa и комбинации одновременно вводимых темозоломида и соединения формулы IIa. Наблюдают синергическое действие в случае одновременного введения в МТ3-клетки доцетаксела и соединения формулы IIa (Cl=0,69) и в и87-клетки темозоломида и соединения формулы IIa (Cl=0,67).
На Фиг.14 показаны результаты in vitro анализов клеточной пролиферации (Cell-Titer Glo, Promega), в которых измеряют жизнеспособные клетки ZR75-1 (тип опухоли молочной железы) при варьировании концентраций (начиная от 4Х EC50), справа налево: (верх) 5-FU, соединения формулы IIa и комбинации одновременно вводимых 5-FU и соединения формулы IIa; и (низ) доцетаксела, соединения формулы IIa и комбинации одновременно вводимых доцетаксела и соединения формулы IIa. Наблюдают синергическое действие в случае одновременного введения в ZR75-1-клетки 5-FU и соединения формулы IIa (Cl=0,47) и в ZR75-1 -клетки доцетаксела и соединения формулы IIa (Cl=0,46).
Корреляция между мутациями Ras и синергическими эффектами in vitro, приведенными на Фиг.1А (эксперименты 1-248), обусловленная комбинациями соединения формулы Ia и химиотерапевтических агентов, может быть продемонстрирована с помощью точечных диаграмм. Каждая точка на Фиг.15 и 16 соответствует эксперименту с Фиг.1А. Данные экспериментов объединяют в группы дикого типа Ras (Ras WT) или мутанта Ras (мут.Ras) с конкретными мутациями, отмеченными на Фиг.1А, строят график зависимости для синергического эффекта (показатель для комбинации, Cl), при этом синергическое действие возрастает при уменьшении Cl, который рассчитывают по методу Chou и Talalay (Chou, Т. and Talalay, P. (1984) Adv. Enzyme Regul. 22: 27-55).
На Фиг.15 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) эрлотиниба и соединения формулы Ia из Фиг.1А против линий опухолевых клеток с мутациями Ras и без них (эксперименты 71-73, 140-168, 230-231). Линии клеток с мутацией Ras показывают более сильное синергическое действие между эрлотинибом и соединением формулы Ia, чем линии клеток дикого типа Ras.
На Фиг.16 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) PD-0325901 и соединения формулы Ia из Фиг.1А против линий опухолевых клеток с мутациями Ras и без них (эксперименты 29-35, 74-83, 124-139, 175-184, 224-226, 232-236, 247, 248). Линии клеток с мутацией Ras показывают более сильное синергическое действие между PD-0325901 и соединением формулы Ia, чем линии клеток дикого типа Ras.
На Фиг.17 показаны результаты для синергидной линии опухолевых клеток MDA-MB-361 и не-синергидной линии опухолевых клеток МТ-3 в зависимости от времени обработки гемцитабином при уровнях дозировки, равных ЕС50. Уровни рАКТ измеряли для Т=0 (без обработки, UT), 1 ч, 4 ч, 6 ч и 24 ч. Известно, что конститутивная и индуцибельная активность Akt стимулирует резистентность к химиотерапии, трастузумабу или тамоксифену, в раковых клетках молочной железы (dark et al. (2002) Mol. Cancer Ther. 1(9): 707-17). Уровни фосфо-Akt (pAkt) можно измерить способом, описанным в Примере 18. Низкие величины Cl коррелируют с эффектом химиотерапевтического средства в отношении индуцирования увеличения уровней pAkt. Уровни pAkt (Ser473) определяли, используя наборы гранул от Biosource (Carlsbad, CA) и систему Luminex Bio-Plex (Bio-Rad, Hercules, CA). Обработка гемцитабином приводит к повышенным уровням рАКТ в синергидной клеточной линии (MDA-MB-361), но не в не-синергидной клеточной линии (МТ-3), демонстрируя, что увеличение уровней рАКТ в ответ на химиотерапию коррелирует с синергизмом и прогнозирует предрасположенность к синергизму под действием соединения формулы I или II и химиотерапевтического агента в лечении рака.
На Фиг.18 показана точечная диаграмма для экспериментов по синергическому действию (показатель для комбинации) доцетаксела, 5-FU или гемцитабина и соединения формулы Ia из Фиг.1А против линий опухолевых клеток, которые демонстрируют увеличение уровня pAkt или никакого увеличения уровня pAkt в ответ только лишь на химиотерапевтический агент. Клеточные линии, которые показывают увеличение уровней pAkt после обработки доцетакселом, 5-FU или гемцитабином, демонстрируют более сильный синергизм при использовании соединения формулы Ia, чем клеточные линии без pAkt-ответа.
Изобретение охватывает способ определения соединений, которые следует использовать в комбинации для лечения рака, включающий: а) введение терапевтической комбинации соединения, имеющего формулу I или II, и химиотерапевтического агента, выбранного из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба, в линию опухолевых клеток in vitro с мутацией K-ras и б) определение синергического или несинергического эффекта.
Несмотря на то, что в комбинациях, приведенных в качестве примера на Фиг.1А, 1 В, 1C и Фиг.2-14, могут быть задействованы и другие механизмы функционирования, результаты согласуются с тем, что ингибиторы Pl3K оказывают влияние на G1-фаза-специфический эффект в отношении ингибирования пролиферации опухолевых клеток, 5FU оказывает влияние на S-фаза-специфическое нарушение синтеза ДНК, гемцитабин оказывает влияние S-фаза-специфическое нарушение синтеза ДНК, а доцетаксел оказывает влияние на М-фаза-специфическую деполяризацию микротрубочек.
ПРОТОЧНАЯ ЦИТОМЕТРИЯ С ИСПОЛЬЗОВАНИЕМ FACS
Проводили проточную цитометрию для измерения влияния комбинированной терапии соединением формулы Ia и различными химиотерапевтическими агентами на клетки опухоли молочной железы МВЗ и клетки опухоли предстательной железы РС3. В анализе с использованием аннексина V/PI(иодид пропидия) детектируют ранние и поздние апоптотические события (Пример 15). Клетки, являющиеся позитивными по аннексину V, находятся на ранних стадиях апоптоза, а клетки, которые являются позитивными как по аннексину V, так и по PI, обозначаются как "мертвые" на диаграммных изображениях на Фиг.19. Остальные клетки образуют "живую" популяцию.
На Фиг.19 показаны результаты проточной цитометрии с использованием FACS (клеточный сортер с активацией флуоресценции): (верх) клеток МВ361 (слева направо), необработанных, обработанных соединением формулы Ia, 5FU и сначала 5FU, затем соединением формулы Ia; (середина) клеток PC3 (слева направо), необработанных, обработанных соединением формулы Ia, доцетакселом, одновременно соединением формулы Ia и доцетакселом, сначала соединением формулы Ia, затем доцетакселом и сначала доцетакселом, затем соединением формулы Ia; и (низ) клеток МВ361 (слева направо), необработанных, обработанных соединением формулы Ia, гемцитабином и сначала гемцитабином, затем соединением формулы Ia.
Соединение формулы Ia и генотоксичный химиотерапевтический агент вводили в ЕС50 через 24, 48, 72 часа. FACS (фиксация клеточного цикла (по PI), живые клетки по результатам окрашивания аннексином V и PI). Соединения добавляли либо одновременно, либо по отдельности с интервалом 4 часа перед введением и после введения соединения формулы Ia (Пример 19). Отмечали эффект вымывания, при этом максимального синергизма достигали через один час. Комбинации оставались синергическими, при этом оба лекарственных средства вымывались через один час после введения. Наблюдается усиление раннего и позднего апоптоза во всех трех комбинациях (Фиг.19), когда соединение формулы Ia комбинируют с химиотерапевтическим лекарственным средством. Низкие величины Cl, определенные по Chou и Talalay, позволяют предположить, что значительный результат в ингибировании опухоли вероятно имеет место тогда, когда эти лекарственные средства комбинируют in vivo.
ПРОСТРАНСТВЕННЫЙ (3D) АНАЛИЗ ДЕЙСТВИЯ КОМБИНАЦИЙ
В гландулярных тканях, таких как молочная железа, эпителий взаимодействует со специализированной формой внеклеточного матрикса, известной как базальная мембрана. Внеклеточный матрикс регулирует нормальное развитие молочной железы и патогенез. Применение реконструированной ламинин-обогащенной базальной мембраны (IrBM) к стандартной клеточной культуре может повторять основную ацинарную структуру молочной железы, и ее считают улучшенной моделью in vitro для имитации динамического состояния микроокружения опухоли (Debnath J, Brugge JS. (2005) Nat. Rev. Cancer 5: 675-88). Путем использования ингибиторов, обладающих широкой специфичностью, показано, что Pl3K-опосредованная передача сигнала вовлечена в ацинусное развитие клеток опухоли молочной железы человека НМТ-3522 Т4-2, растущих на IrBM, поэтому ингибирования Pl3K было достаточно для того, чтобы восстановить полярность апикально-базальной структуры и индуцировать остановку роста (Liu Н, Radisky DC, Wang F, Bissell MJ. (2004) J. Cell Biol., 164: 603-12). Принимая во внимание предполагаемый вклад Pl3K в инициацию и развитие рака молочной железы, культуральные 3D-cncTeMbi обеспечивают новое и универсальное средство для оценки эффективности низкомолекулярных ингибиторов Pl3K, таких как соединение формулы Ia.
Рецепторная тирозинкиназа HER2 (Neu/ErbB2) амплифицируется и сверхэкспрессируется приблизительно в 20% случаев рака молочной железы у человека и играет причинно обусловленную роль в канцерогенезе молочной железы (Yarden Y, Sliwkowski MX. (2001) Nat. Rev. Mol. Cell Biol. 2: 127-37). Такая повышенная экспрессия HER2, играющая роль при раке молочной железы у человека, демонстрируется посредством терапевтической эффективности моноклонального антитела против HER2 - трастузумаба (HERCEPTIN®, Genentech). В дополнение к гомодимеризации, HER2 также может действовать в качестве корецептора для других членов HER-семейства. Моноклональное антитело пертузумаб направленно воздействует на "плечо" (внеклеточный субдомен II) при димеризации HER2 и нарушает рекрутмент HER2 в комплексы HER-рецептор-лиганд (Franklin et al. (2004) Cancer Cell, 5: 317-28). Эффективность ингибирования Pl3K определяли с использованием соединений формулы Ia и IIa, комбинируя его с ингибированием терапевтическими антителами трастузумабом и пертузумабом передачи сигнала, обусловленной HER-семейством.
Изобретение охватывает способ определения соединений, которые могут быть использованы в комбинации для лечения рака, включающий: а) введение терапевтической комбинации по п.1 в НЕР2-амплифицирующие раковые клетки молочной железы в ламинин-обогащенных, воссозданных содержащих базальные мембраны средах, при этом химиотерапевтический агент направленно воздействует на рецептор HER2, связывается с ним или модулирует его, и б) измерение ингибирования клеточной пролиферации, где незлокачественные и злокачественные клетки молочной железы различают на основании одного или более чем одного фенотипического различия, выбранного из жизнеспособности клеток и ацинарного морфогенеза.
Проводили оценку комбинаций соединений формулы I и II и терапевтических антител в НЕК2-амплифицирующих клетках ВТ474М1 (Пример 16). Клетки культивировали в объемной (3D) ламинин-обогащенной, воссозданной базальной мембране, чтобы дать объяснение роли молекул внеклеточного матрикса в передаче сигнала с участием онкогенов и биологии онкогенов. Дополнительным следствием способности SD-культуры повторять онкогенное микроокружение является то, что она может быть более надежна для предсказания эффективности in vivo (по сравнению с двумерными (2D) культурами клеток на пластике) и может быть полезна для охарактеризования ингибиторов и генов-мишеней. SD-культуру использовали для оценки обусловленной HER-семейством передачи сигнала и измерения синергической эффективности ингибиторов. Жизнеспособность клеток и ацинарные фенотипы (морфогенез) использовали в качестве маркеров лекарственной эффективности (Пример 16).
НЕК2-амплифицирующие клетки ВТ474М1 применяли для детекции обусловленной HER-семейством передачи сигнала, используя новые фенотипы клеток 30-культуры и количественный показатель синергической эффективности, основанный на ацинарном морфогенезе (Пример 16). Одним из воплощений ламинин-обогащенных, воссозданных содержащих базальные мембраны сред является экстракт внеклеточного матрикса Engelbreth-Holm-Swarm(-capкомы мыши), поступающий в продажу под названием Матригель от BD (BD Matrigel™ (BD Biosciences)). Другим типичным воплощением внеклеточного матрикса (ЕСМ) для 30-культуры являются эпителиальные клетки почки собаки Madin-Darby. Характерное фенотипическое различие заключается в ацинарной структуре инвазивных (злокачественных) и неинвазивных (незлокачественных) клеток.
В качестве дополнительного анализа лекарственной эффективности можно провести оценку в баллах ацинарного морфогенеза. Каждый ингибитор титруют с целью исследования фармакодинамических (PD) маркеров данного пути, жизнеспособности клеток и ацинарного фенотипа. Устанавливали самую низкую ингибирующую концентрацию лекарственного средства, эффективно ингибирующую мишень. Подходящую концентрацию соединения формулы I и II для анализов в SD-культурах определяли путем введения возрастающих доз с учетом общей жизнеспособности клеток, фармакодинамики соответствующих маркеров, расположенных в данном пути ниже, (таких как фосфорилированная АКТ1) и изменений в ацинарном фенотипе. Для данных анализов выбирали самую низкую концентрацию лекарственного средства, которая эффективно ингибировала мишень. Концентрация соединения формулы Ia и На, равная 250 нМ, была выбрана в качестве конечной рабочей концентрации для анализов в SD-культурах.
Показано, что прямые ингибиторы HER2, такие как трастузумаб и пертузумаб, препятствуют активации некоторых нижележащих ключевых эффекторных путей, включая направление Pl3K-AKT. Совместное ингибирование Pl3K и обусловленной HER-семейством передачи сигнала в HER2-амплифицирующих раковых клетках молочной железы может приводить к эффективному ингибированию опухолевых клеток.
Посредством детектирования иммуноблотингом фосфорилированной АКТ1 (pAkt) было подтверждено, что соединение формулы Ia в концентрации 250 нМ эффективно ингибирует нисходящий Pl3K-сигналинг (Пример 16). Терапевтические антитела использовали в насыщающих концентрациях 20 мкг/мл и 25 мкг/мл для пертузумаба и трастузумаба, соответственно. Применение комбинации соединения формулы Ia и трастузумаба приводило к аддитивному подавлению 3D-роста ВТ474М1-ацинусов, культивируемых в Матригеле без лиганда HRG (херегулина). Для детекции лиганд-зависимой обусловленной гетеродимером HER2-HER3 передачи сигнала, которая считается приводящей к трансформации во многих клеточных линиях, в эти анализы добавляли 1 нМ HRG. Соединение формулы Ia и трастузумаб не оказывали никакого эффекта на HRG-индуцируемую пролиферацию. В сравнении с этим совместная обработка соединением формулы Ia и пертузумабом вызывала аддитивное уменьшение HRG-индуцируемого ацинусного роста и морфогенеза. Показано, что этот эффект был статистически значимым на протяжении многих клеточных делений. В отсутствие лиганда уменьшение 3D-роста ацинусов усиливалось после обработки соединением формулы Ia и трастузумабом по сравнению с соединением формулы Ia и пертузумабом. Комбинация трастузумаба и соединения формулы Ia ингибирует клеточную пролиферацию и ослабляет херегулин-индуцируемый морфогенез. Комбинация трастузумаба и соединения формулы Ia оказывает сильные и аддитивные эффекты на 3D-рост в нормальной сыворотке, однако при использовании данной комбинации в херегулин-обработанных средах никакой аддитивности в отношении ацинусного роста или морфогенеза не наблюдали. Комбинация пертузумаба и соединения формулы Ia эффективно, однако же аддитивно, ингибирует ВТ474М1-ацинусный рост и морфогенез. Тройная комбинация трастузумаба, пертузумаба и соединения формулы Ia синергически ингибирует рост и морфогенез "ацинусных бугорков" (aClnni bud) у ВТ474М1 (Фиг.20) как в HRG-дополненных, так и в стандартных средах.
Каждый ингибитор титровали с целью исследования релевантных фармакодинамических (PD) маркеров данного пути, жизнеспособности клеток и ацинарного фенотипа. Устанавливали самую низкую ингибирующую концентрацию лекарственного средства, которая эффективно ингибирует мишень, и использовали во всех 3D-анализах.
На Фиг.20 показано количественное определение роста клеток ВТ474 в объемной (3D) культуре. Жизнеспособность клеток определяли путем измерения уровней АТФ в клетках. Клетки ВТ474М1 культивировали либо в 10%-ной сыворотке, либо в 10%-ной сыворотке, содержащей 1 нМ херегулин, и обрабатывали ингибирующими комбинациями, как указано (слева направо): средами, DMSO, комбинацией 20 мкг/мл трастузумаба и 25 мкг/мл пертузумаба, 250 нМ соединения формулы Ia и комбинацией 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба и 250 нМ соединения формулы Ia. Ацинусный рост и морфогенез коррелирует с продуцированием клеткой АТФ, выраженным в относительных световых единицах (RLU), в среде с добавлением 10% FBS в присутствии и в отсутствие 1 нМ херегулина.
В стандартных средах (без херегулина) активность клеток сравнительно более низкая в присутствии трастузумаба, пертузумаба или соединения формулы Ia по отдельности, но не в HRG-обработанных средах. Комбинация трастузумаба и соединения формулы Ia ингибировала клеточную пролиферацию и ослабляла херегулин-индуцируемый морфогенез в нормальной сыворотке, но никакой аддитивности не наблюдали в отношении ацинусного роста или морфогенеза в херегулин-обработанных средах. Комбинация пертузумаба и соединения формулы Ia эффективно и аддитивно ингибировала ацинусный рост и морфогенез у ВТ474М1 как в стандартных, так и в херегулин-дополненных средах. Тройная комбинация трастузумаба, пертузумаба и соединения формулы Ia синергически ингибировала пролиферацию клеток ВТ474М1 и херегулин-индуцируемый морфогенез (Фиг.20) как в стандартных, так и в HRG-дополненных средах. Комбинация всех трех агентов синергически снижала жизнеспособность клеток как в стандартных, так и в HRG-дополненных средах. Херегулин-индуцируемый морфогенез клеток ВТ474М1 также прекращался под действием тройной комбинации, что определено посредством наблюдения под микроскопом. Эти данные позволяют предположить, что тройная комбинация соединения формулы Ia, трастузумаба и пертузумаба может обеспечивать улучшенную эффективность в отношении лечения НЕР2-амплифицирующего рака молочной железы у пациентов-людей.
Трастузумаб или соединение формулы Ia значительно уменьшают размер ацинусов в нормальных средах, но оказывают ограниченное влияние на HRG-индуцируемый морфогенез. Благодаря комбинированному действию Трастузумаб и соединение формулы Ia могут минимизировать размер ацинусов и морфогенез. Посредством анализа жизнеспособности клеток (Cell Titer-Glo) показано, что аддитивность трастузумаба и соединения формулы Ia приводит к уменьшению клеточного роста в нормальных средах, но никакого различия не было замечено при добавлении HRG.
Пертузумаб полностью ингибирует HRG-индуцируемый морфогенез, тогда как соединение формулы Ia только частично ослабляет данный фенотип. Совместно, пертузумаб и соединение формулы Ia ослабляют клеточный рост и морфогенез как в нормальных, так и в HRG-дополненных средах. Согласно оценке жизнеспособности клеток с использованием Cell Titer-Glo наблюдается уменьшение клеточной активности в присутствии пертузумаба и соединения формулы Ia как при применении в виде индивидуальных агентов, так и при комбинированной терапии в нормальных средах. HRG-обработанные ацинусы также демонстрируют похожую тенденцию, но несколько в меньшей степени. Согласно сравнительной оценке посредством Т-теста Дуннетта данных Cell Titer-Glo для повторов (n=8) комбинация пертузумаба и соединения формулы Ia значительно ингибирует клеточную активность (р=0,0054).
На Фиг.21 показан аналогичный эффект от добавления соединения формулы IIa к двойной обработке трастузумабом и пертузумабом. На Фиг.21 показан ВТ474-рост в клеточной 3D-культуре после обработки 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба или 250 нМ соединением формулы IIa, как указано. Соединение формулы IIa значительно уменьшало размер ацинусов в нормальных средах, однако в качестве отдельного агента оказывало ограниченное влияние на HRG-индуцируемый морфогенез. Благодаря комбинированному действию соединение формулы IIa, трастузумаб и пертузумаб значительно уменьшали размер ацинусов и морфогенез, как определено посредством измерения жизнеспособности клеток с использованием Cell Titer Glo. По сравнению с соединением формулы Ia соединение формулы IIa оказывается немного менее эффективным в концентрации 250 нМ как в виде отдельного агента (р=0,0001, Т-тест Дуннетта), так и в комбинации с трастузумабом и пертузумабом (р<0,0001, Т-тест Дуннетта).
На Фиг.21-А показан аналогичный эффект от добавления соединения формулы Ib к двойной обработке трастузумабом и пертузумабом. ВТ474-рост в клеточной ЗО-культуре измеряли после обработки 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба или 20 нМ соединением формулы Ib, как указано. Монотерапия соединением формулы Ib уменьшала размер ацинусов в нормальных и HRG-дополненных средах. Наиболее значительное уменьшение в размере ацинусов и морфогенезе, как определено посредством измерения жизнеспособности клеток с использованием Cell Titer Glo, обуславливалось обработкой комбинацией соединения формулы Ib, трастузумаба и пертузумаба.
ЭФФЕКТИВНОСТЬ IN VIVO ПРОТИВ ОПУХОЛЕВЫХ КСЕНОТРАНСПЛАНТАТОВ
Эффективность комбинаций по изобретению можно измерить in vivo посредством введения аллотрансплантатов или ксенотрансплантатов раковых клеток грызунам и подвергания животных, которым внесена опухоль, лечению данными комбинациями. В зависимости от клеточной линии, наличия или отсутствия некоторых мутаций в опухолевых клетках, последовательности введения соединения формулы I или II и химиотерапевтического агента, схемы введения и других факторов следует ожидать различные результаты. Испытуемых мышей обрабатывали лекарственным средством(ами) или контролем (разбавителем) и проводили мониторинг в течение нескольких недель или более с целью определения времени удвоения опухоли, порядка уменьшения числа клеток (log cell kill) и ингибирования (роста) опухоли (Пример 17).
На Фиг.22 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 (Charles River Labs) с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 150 мг/кг соединения формулы Ia, 5 мг/кг доцетаксела и комбинацию соединения формулы Ia (150 мг/кг) и доцетаксела (5 мг/кг). Мышам вводили доцетаксел на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) внутривенно, тогда как соединение формулы Ia вводили ежесуточно в течение 21 суток путем перорального чреззондового кормления. При введении в один и тот же день соединение формулы Ia вводили через 1 час после доцетаксела. Комбинация 150 мг/кг соединения формулы Ia с 5 мг/кг доцетаксела синергически ингибировала рост опухоли молочной железы MDA-MB-361.1 in vivo, больше, чем каждый отдельный агент сам по себе.
Группа из 11 животных, которым вводили соединение формулы Ia (150 мг/кг), показала 75% ингибирования через 21 сутки и 3 частичные регрессии и 66% ингибирования через 41 сутки. Группа из 10 животных, которым вводили доцетаксел (5 мг/кг), показала 78% ингибирования через 21 сутки и 2 частичные регрессии и 26% ингибирования через 41 сутки. Группа из 9 животных, которым вводили комбинацию соединения формулы Ia (150 мг/кг) и доцетаксела (5 мг/кг), показала 90% ингибирования через 21 сутки и 7 частичных регрессий и 83% ингибирования через 41 сутки. Комбинация показала более хорошую эффективность в ингибировании опухоли и статистическую значимость при сравнении с каждым используемым по отдельности лекарственным средством (р=0,0001 по сравнению с доцетакселом, и р=0,02 по сравнению с соединением формулы Ia).
На Фиг.23 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 (Charles River Labs) с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 1-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 37,5 мг/кг соединения формулы IIa, 5 мг/кг доцетаксела и комбинацию 37,5 мг/кг соединения формулы IIa и 5 мг/кг доцетаксела. Мышам вводили доцетаксел на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) внутривенно, тогда как соединение формулы IIa вводили ежесуточно в течение 21 суток путем перорального чреззондового кормления. При введении в один и тот же день соединение формулы IIa вводили через 1 час после доцетаксела. Группа из 10 животных, которым вводили 37,5 мг/кг соединения формулы IIa, показала 30% ингибирования и 2 частичные регрессии через 21 сутки. Группа из 10 животных, которым вводили 5 мг/кг доцетаксела, показала 35% ингибирования и 3 частичные регрессии через 21 сутки. Группа из 10 животных, которым вводили комбинацию соединения формулы IIa (37,5 мг/кг) и доцетаксел (5 мг/кг), показала 63% ингибирования. Комбинация показала более хорошую эффективность в ингибировании опухоли и статистическую значимость при сравнении с каждым используемым по отдельности лекарственным средством (р=0,0454 по сравнению с доцетакселом, и р=0,0174 по сравнению с соединением формулы IIa).
На Фиг.24 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии NMRI с ксенотрансплантатами эксплантатов первичной ("трижды негативной") опухоли молочной железы MAXF 401, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы Ia, 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы Ia и 15 мг/кг доцетаксела. MAXF 401 обозначает первичные опухоли молочной железы, взятые биопсией непосредственно у пациента, который давал ответ на доцетаксел, и имплантированные подкожно мышам. Это группа с раком молочной железы представляет собой субпопуляцию "трижды негативных" пациентов, негативных по HER2, негативных по ER (рецептору эстрогена) и негативных по PR (рецептору прогестерона). Чувствительность к доцетакселу сохраняется у мышей ex vivo. Мышам вводили внутривенно доцетаксел на 0-е сутки и 11-е сутки, тогда как соединение формулы Ia вводили на сутки 0-4, 11-17 и 21-28 путем перорального чреззондового кормления. Проводили мониторинг животных в течение дополнительных 22 суток на предмет опухолевого роста после последнего введения соединения формулы Ia (общее количество суток, в течение которых животных исследовали, составляло 50). При введении в один и тот же день соединение формулы Ia вводили через 1 час после доцетаксела. Группа из 10 животных, которым вводили 100 мг/кг соединения формулы Ia, показала 49% ингибирования на 28-е сутки. Группа из 10 животных, которым вводили 15 мг/кг доцетаксела, показала 95% ингибирования на 28-е сутки. Группа из 10 животных, которым вводили комбинацию соединения формулы Ia (150 мг/кг) и доцетаксела (15 мг/кг) показала более 90% ингибирования через 28 суток. В конце исследования (50-е сутки) у животных, которым вводили только доцетаксел и только соединение формулы Ia, отмечали повторный рост имеющихся у них опухолей и ингибирование (роста) опухоли уменьшалось с 95% до 68% и с 49% до 10%, соответственно. Однако, в конце исследования (50-е сутки) комбинация доцетаксела и соединения формулы Ia приводила к тому, что у всех десяти животных наблюдалась непрерывная опухолевая регрессия (более 90% ингибирования) первичных опухолей молочной железы MAXF 401, и она была статистически значимой при сравнении с каждым используемым по отдельности лекарственным средством (р=0,05 по сравнению с доцетаксела, и р<0,001 по сравнению с соединением формулы Ia). Принимая во внимание, что опухоли молочной железы MAXF 401 выделены из пациента, который давал ответ на терапию таксанами, наблюдаемая ex vivo улучшенная эффективность позволяет предположить, что комбинация на основе соединения формулы Ia может оказывать благоприятные клинические воздействия при раке молочной железы, когда его комбинируют с доцетакселом.
На Фиг.25 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами эксплантатов первичной опухоли молочной железы MAXF 401, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы IIa, 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы IIa и 15 мг/кг доцетаксела. MAXF 401 обозначает первичные опухоли молочной железы, взятые биопсией непосредственно у пациента, который давал ответ на доцетаксел, и имплантированные подкожно мышам. Чувствительность к доцетакселу сохраняется у мышей ех vivo. Мышам вводили внутривенно доцетаксел на 0-е сутки и 11-е сутки, тогда как соединение формулы IIa вводили на сутки 0-3, 11-17 и 21-28 путем перорального чреззондового кормления. Проводили мониторинг животных в течение дополнительных 22 суток на предмет опухолевого роста после последнего введения соединения формулы IIa (общее количество суток, в течение которых животных исследовали, составляло 50). При введении в один и тот же день соединение формулы IIa вводили через 1 час после доцетаксела. Группа из 10 животных, которым вводили 100 мг/кг соединения формулы IIa, показала 82% ингибирования (роста) опухоли на 28-е сутки. Группа из 10 животных, которым вводили 15 мг/кг доцетаксела, показала 95% ингибирования (роста) опухоли на 28-е сутки. Группа из 10 животных, которым вводили комбинацию соединения формулы IIa (150 мг/кг) и доцетаксела (15 мг/кг), показала 99% ингибирования на 28-е сутки. В конце исследования (50-е сутки) у животных, которым вводили только доцетаксел и только соединение формулы IIa, отмечали повторный рост имеющихся у них опухолей, и ингибирование (роста) опухоли уменьшалось до 68% и 51%, соответственно. Однако, в конце исследования (50-е сутки) при использовании комбинации 100 мг/кг соединения формулы IIa и 15 мг/кг доцетаксела наблюдалась непрерывная опухолевая регрессия (99% ингибирования) первичных опухолей молочной железы MAXF 401, и она была статистически значимой при сравнении с каждым используемым по отдельности лекарственным средством (р=0,0118 по сравнению с доцетакселом, и р=0,0005 по сравнению с соединением формулы IIa). Принимая во внимание, что опухоли молочной железы MAXF 401 выделены из пациента, который давал ответ на терапию таксанами, наблюдаемая ex vivo улучшенная эффективность позволяет предположить, что комбинация на основе соединения формулы IIa может оказывать благоприятные клинические воздействия при раке молочной железы, когда его комбинируют с доцетакселом.
На Фиг.26 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами эксплантатов первичной опухоли молочной железы MAXF 1162, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы Ia (GDC-0941), 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы Ia и 15 мг/кг доцетаксела. MAXF 1162 обозначает первичные опухоли молочной железы, взятые биопсией непосредственно у пациента, терапия которого доцетакселом была неудачной, и имплантированные подкожно мышам. Резистентность к доцетакселу сохраняется у этих мышей ех vivo. Мышам вводили внутривенно доцетаксел на 0-е, 11-е, 22-е и 44-е сутки, а соединение формулы Ia вводили на сутки 0-5, 11-16, 22-27, 30-32, 42 и 44 путем перорального чреззондового кормления. Проводили мониторинг животных в течение дополнительных 6 суток на предмет опухолевого роста после последнего введения соединения формулы Ia (общее количество суток, в течение которых животных исследовали, составляло 50). При введении в один и тот же день соединение формулы Ia вводили через 1 час после доцетаксела. Группа из 10 животных, которым вводили 100 мг/кг соединения формулы Ia, показала 54% ингибирования через 49 суток. Группа из 10 животных, которым вводили 15 мг/кг доцетаксела, показала 36% ингибирования через 49 суток. Группа из 10 животных, которым вводили комбинацию соединения формулы Ia (100 мг/кг) и доцетаксела (15 мг/кг), показала 87% ингибирования через 49 суток. В конце исследования (50-е сутки) при использовании комбинации наблюдалась непрерывная опухолевая регрессия (более 87% ингибирования), и она была статистически значимой при сравнении с каждым используемым по отдельности лекарственным средством (р=0,0005 по сравнению с доцетакселом, и р=0,0007 по сравнению с соединением формулы Ia). Принимая во внимание, что опухоли молочной железы MAXF 1162 выделены из пациента, терапия которого таксанами была неудачной, наблюдаемая ex vivo улучшенная эффективность позволяет предположить, что комбинация соединения формулы Ia с доцетакселом может оказывать благоприятные клинические воздействия в случаях таксан-резистентного рака молочной железы у людей.
На Фиг.27 показано изменение среднего объема опухоли во времени у самок nu/nu бестимусных и бесшерстных мышей линии NMRI с ксенотрансплантатами первичной опухоли молочной железы MAXF 1162, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 100 мг/кг соединения формулы IIa, 15 мг/кг доцетаксела и комбинацию 100 мг/кг соединения формулы IIa и 15 мг/кг доцетаксела. MAXF 1162 обозначает первичные опухоли молочной железы, взятые биопсией непосредственно у пациента, терапия которого доцетакселом была неудачной, и имплантированные подкожно мышам. Резистентность к доцетакселу сохраняется у этих мышей ех vivo. Мышам вводили внутривенно доцетаксел на 0-е, 11-е, 22-е и 44-е сутки, а соединение формулы IIa вводили на сутки 0-5, 11-16, 22-23, 29-31 и 35-38 путем перорального чреззондового кормления. Проводили мониторинг животных в течение дополнительных 12 суток на предмет опухолевого роста после последнего введения соединения формулы IIa (общее количество суток, в течение которых животных исследовали, составляло 50). При введении в один и тот же день соединение формулы IIa вводили через 1 час после доцетаксела. Группа из 10 животных, которым вводили 100 мг/кг соединения формулы IIa, показала 32% ингибирования через 49 суток. Группа из 10 животных, которым вводили 15 мг/кг доцетаксела, показала 36% ингибирования через 49 суток. Группа из 10 животных, которым вводили комбинацию соединения формулы IIa (100 мг/кг) и доцетаксела (15 мг/кг), показала 80% ингибирования через 49 суток. В конце исследования (50-е сутки) при использовании комбинации наблюдалась непрерывная опухолевая регрессия (более 80% ингибирования) первичных опухолей рака молочной железы MAXF 1162, и она была статистически значимой при сравнении с каждым используемым по отдельности лекарственным средством (р<0,0001 по сравнению с доцетакселом, и р=0,0166 по сравнению с соединением формулы IIa). Принимая во внимание, что опухоли молочной железы MAXF 1162 выделены из пациента, терапия которого таксанами была неудачной, наблюдаемая ex vivo улучшенная эффективность позволяет предположить, что комбинация соединения формулы IIa с доцетакселом может оказывать благоприятные клинические воздействия в случаях таксан-резистентного рака молочной железы у человека.
На Фиг.28 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии CRL с ксенотрансплантатами опухоли немелкоклеточного рака легкого (NSClC) NCl-1-12122, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 50 мг/кг соединения формулы Ia, 75 мг/кг эрлотиниба и комбинацию 50 мг/кг соединения формулы Ia и 75 мг/кг эрлотиниба. Мышам вводили ежесуточно эрлотиниб и соединение формулы Ia ежесуточно в течение 16 суток путем перорального чреззондового кормления. Проводили мониторинг животных на предмет опухолевого роста в течение дополнительных 5 суток (окончание исследования пришлось на 21-е сутки). И эрлотиниб, и соединение формулы Ia вводили одновременно. Группа из 8 животных, которым вводили 50 мг/кг соединения формулы Ia, показала 17% ингибирования (роста) опухоли через 20 суток. Группа из 8 животных, которым вводили 75 мг/кг эрлотиниба, показала 21% ингибирования через 20 суток. Группа из 8 животных, которым вводили комбинацию соединения формулы Ia (50 мг/кг) и эрлотиниба (75 мг/кг), показала 55% ингибирования через 20 суток. В конце исследования (21-е сутки) при использовании комбинации 50 мг/кг соединения формулы Ia и эрлотиниба наблюдалась аддитивность и значительная задержка опухолевого роста ксенотрансплантатов опухоли NSClC NCl-H2122 при сравнении с каждым используемым по отдельности лекарственным средством (р=0,032 по сравнению с эрлотинибом, и р=0,019 по сравнению с соединением формулы Ia).
На Фиг.29 показано изменение среднего объема опухоли во времени у самок nu/nu (бестимусных и бесшерстных) мышей линии CRL с ксенотрансплантатами опухоли немелкоклеточного рака легкого (NSClC) NCl-H2122, которым на 0-е сутки вводили: разбавитель МСТ (0,5% метилцеллюлозы/0,2% Твина 80), 50 мг/кг соединения формулы IIa, 75 мг/кг эрлотиниба и комбинацию 50 мг/кг соединения формулы IIa и 75 мг/кг эрлотиниба. Мышам вводили ежесуточно эрлотиниб и соединение формулы IIa в течение 14 суток (окончание исследования) путем перорального чреззондового кормления. И эрлотиниб, и соединение формулы IIa вводили одновременно. Группа из 9 животных, которым вводили 50 мг/кг соединения формулы IIa, показала 27% ингибирования (роста) опухоли на момент завершения исследования. Группа из 10 животных, которым вводили 75 мг/кг эрлотиниба, показала 34% ингибирования (роста) опухоли на момент завершения исследования. Группа из 9 животных, которым вводили комбинацию соединения формулы IIa (50 мг/кг) и эрлотиниба (75 мг/кг), показала 63% ингибирования (роста) опухоли на момент завершения исследования. В конце исследования (14-е сутки) при использовании комбинации 50 мг/кг соединения формулы IIa и 75 мг/кг эрлотиниба наблюдалась аддитивность и значительная задержка опухолевого роста ксенотрансплантатов опухоли NSClC при сравнении с каждым используемым по отдельности лекарственным средством (р=0,032 по сравнению с эрлотинибом, и р=0,029 по сравнению с соединением формулы IIa).
На Фиг.30 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии HRLN с ксенотрансплантатами клеток опухоли молочной железы MCF-7 (мутант по Pl3K), которым на 1-е сутки вводили: разбавитель МСТ и PBS (МСТ: 0,5% метилцеллюлозы/0,2% Твина 80, и PBS: забуференный фосфатом физиологический раствор), контроль IgG (5 мг/кг), мышиное антитело против VEGF mB20-4.1 (антиангиогенное средство) (5 мг/кг), соединение формулы Ia (150 мг/кг) и комбинацию соединения формулы Ia (150 мг/кг) и мышиного антитела против VEGF mB20-4.1 (5 мг/кг). Животным вводили контроль IgG и mB20-4.1 внутрибрюшинно дважды в неделю в течение 3 недель, а соединение формулы Ia ежесуточно в течение 21 суток путем перорального чреззондового кормления, и мониторинг опухолевого роста проводили в течение дополнительных 41 суток (общее количество суток на одно исследование составляло 62). Соединение формулы Ia и mB20-4.1 вводили одновременно. Группа из 13 животных при общем количестве 15 животных, которым вводили контроль IgG и исследовали, показала 19% ингибирования и 0 частичных регрессий через 21 сутки. Группа из 10 животных при общем количестве 15 животных, которым вводили мышиное антитело против VEGF mB20-4.1 и исследовали, показала 49% ингибирования и 0 частичных регрессий через 21 сутки. Группа из 13 животных, которым вводили 150 мг/кг соединения формулы Ia и исследовали, показала 36% ингибирования и 0 частичных регрессий через 21 сутки. Группа из 10 животных, которым вводили комбинацию соединения формулы Ia и мышиного антитела против VEGF mB20-4.1 и исследовали, показала 66% ингибирования и 4 полные регрессии через 21 сутки. Животные, которым вводили комбинацию соединения формулы Ia и мышиного антитела против VEGF mB20-4.1, показали значительное ингибирование и непрерывную регрессию опухолевого роста через 41 сутки после остановки введения (окончание исследования) при сравнении с каждым используемым по отдельности лекарственным средством (р<0,006 по сравнению с соединением формулы Ia, и р<0,01 по сравнению с mB20-4.1).
На Фиг.31 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии HRLN с ксенотрансплантатами клеток опухоли молочной железы MCF-7 (мутант по Pl3K), которым на 1-е сутки вводили: разбавитель МСТ и PBS (МСТ: 0,5% метилцеллюлозы/0,2% Твина 80, и PBS: забуференный фосфатом физиологический раствор), контроль IgG (5 мг/кг), мышиное антитело против VEGF mB20-4.1 (антиангиогенное средство) (5 мг/кг), соединение формулы IIa (100 мг/кг) и комбинацию соединения формулы IIa (100 мг/кг) и мышиного антитела против VEGF mB20-4.1 (5 мг/кг). Животным вводили контроль IgG и mB20-4.1 внугрибрюшинно дважды в неделю в течение 3 недель, а соединение формулы IIa перорально ежесуточно в течение 21 суток, и мониторинг опухолевого роста проводили в течение дополнительных 41 суток (общее количество суток на одно исследование составляло 62). Соединение формулы IIa и mB20-4.1 вводили одновременно. Группа из 12 животных при общем количестве 15 животных, которым вводили разбавитель и исследовали, показала 10% ингибирования и О частичных регрессий через 21 сутки. Группа из 10 животных при общем количестве 15 животных, которым вводили мышиное антитело против VEGF mB20-4.1 и исследовали, показала 49% ингибирования и 0 частичных регрессий через 21 сутки. Группа из 13 животных, которым вводили 100 мг/кг соединения формулы IIa и исследовали, показала 5% ингибирования и 0 частичных регрессий через 21 сутки. Группа из 10 животных при общем количестве 15 животных, которым вводили комбинацию соединения формулы IIa и мышиного антитела против VEGF mB20-4.1 и исследовали, показала 61% ингибирования и 1 полную регрессию через 21 сутки. Животные, которым вводили комбинацию соединения формулы IIa и мышиного антитела против VEGF mB20-4.1, показали значительное ингибирование и задержку опухолевого роста через 41 сутки после остановки введения при сравнении с каждым используемым по отдельности лекарственным средством (р<0,001 по сравнению с соединением формулы IIa, и р<0,01 по сравнению с mB20-4.1).
На Фиг.32 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами опухолевых клеток глиомы U87MG, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (109 мг/кг), темозоломид (100 мг/кг) и комбинацию соединения формулы Ia (109 мг/кг) и темозоломида (100 мг/кг), а также у мышей, не получавших никакого лекарственного средства (нелеченная группа). Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток и темозоломид перорально ежесуточно в течение 5 суток. Комбинация 109 мг/кг соединения формулы Ia с 100 мг/кг темозоломида синергически ингибировала опухолевый рост глиомы U87MG in vivo, больше, чем соединение формулы Ia или темозоломид по отдельности, и вызывала регрессию опухоли с последующей задержкой опухолевого роста.
На Фиг.33 показано изменение среднего объема опухоли во времени у бестимусных и бесшерстных мышей линии CD-1 от CR/Hollister с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-361.1, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (150 мг/кг), гемцитабин (100 мг/кг) и комбинацию соединения формулы Ia (150 мг/кг) и гемцитабина (100 мг/кг), а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток и гемцитабин внутрибрюшинно на 1-е, 4-е, 7-е и 10-е сутки (1х/3 сут х 4). Комбинация 150 мг/кг соединения формулы Ia со 100 мг/кг гемцитабина синергически ингибировала рост опухоли молочной железы MDA-MB-361.1 in vivo, больше, чем соединение формулы Ia или гемцитабин по отдельности, и вызывала регрессию опухоли с последующей задержкой опухолевого роста.
На Фиг.34 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы ВТ474, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) в дозах 18, 36 и 73 мг/кг, трастузумаб (20 мг/кг) и комбинации соединения формулы Ia в дозах 18, 36 и 73 мг/кг и трастузумаба (20 мг/кг), а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Животным вводили соединение формулы Ia перорально ежесуточно в течение 21 суток и трастузумаб внутрибрюшинно дважды в неделю в течение 3 недель. Комбинация 73 мг/кг соединения формулы Ia с 20 мг/кг трастузумаба синергически ингибировала рост опухоли молочной железы ВТ474 in vivo, больше, чем соединение формулы Ia или трастузумаб по отдельности, и вызывала регрессию опухоли с последующей задержкой опухолевого роста.
Обработка клеток ВТ474-М1 (перевиваемого in vivo субклона ВТ474) трастузумабом приводила к уменьшению уровня рАКТ, равному уменьшению, достигаемому при использовании соединения формулы Ia (GDC-0941). Добавление трастузумаба к 50 нМ соединению формулы Ia приводило к дозозависимому, возрастающему уменьшению уровня рАКТ. При максимальной дозе трастузумаба дополнительное снижение составляло 29-38% от такового, получаемого при обработке одним лишь GDC-0941. Усиление влияния комбинации (комбинаторный эффект) на ингибирование рАКТ не было кратковременным, и его еще можно было детектировать через 48 ч после обработки. Такой комбинаторный эффект, установленный для рАКТ, также отражался на компонентах передачи сигнала, расположенных ниже АКТ. Клетки ВТ474-М1 обрабатывали в течение четырех часов соединением формулы Ia в присутствии или в отсутствие трастузумаба. Добавление трастузумаба также уменьшало фосфорилирование прямого субстрата АКТ - PRAS40 (ТПг246) и отдаленного субстрата, фосфо-86-рибосомального белка (Ser235/236), позволяя предположить, что трастузумаб и соединение формулы Ia обладают усиленным комбинаторным эффектом на передачу сигнала ниже АКТ. То, что усиление ингибирования Pl3K/АКТ-пути приводит к снижению клеточной пролиферации/жизнеспособности, было продемонстрировано, когда клетки ВТ474-М1 обрабатывали в течение шести суток и измеряли жизнеспособность клеток. Один лишь трастузумаб уменьшал пролиферацию/жизнеспособность на 40%. В отсутствие трастузумаба величина IC50 для соединения формулы Ia составляла 296 нМ. Добавление трастузумаба приводило к дозозависимому, возрастающему снижению пролиферации/жизнеспособности. Максимальная доза трастузумаба 10 мкг/мл обеспечивала уменьшение на 64% концентрации соединения формулы Ia, которое необходимо для достижения соответствующей величины IC50, равной 106 нМ, также демонстрируя усиленный комбинаторный эффект в отношении уменьшения клеточной пролиферации/жизнеспособности. Похожий комбинаторный эффект на рАКТ и ингибирование пролиферации также наблюдали, когда обрабатывали клетки SKBR-3, но не когда обрабатывали нечувствительные к трастузумабу клетки KPL-4. Комбинация трастузумаба и соединения формулы Ia ингибирует синергически пролиферацию клеток ВТ474 и SKBR-3 почти во всем эффективном диапазоне лекарственного средства, как определено с использованием программного обеспечения CalcuSyn, показывая, что данная комбинация усиливает ингибирующий эффект на АКТ и на расположенные ниже его мишени, обеспечивая синергический эффект на пролиферацию трастузумаб-чувствительных раковых клеток молочной железы.
Комбинация трастузумаба и соединения формулы Ia аддитивно индуцирует апоптоз раковых клеток молочной железы ВТ474-М1, обработанных в течение 48 ч. Комбинация трастузумаба и соединения формулы Ia увеличивала аккумуляцию расщепленных каспазой-3 фрагментов, что является показателем активации этого ключевого эффектора - каспазы. Комбинирование трастузумаба и соединения формулы Ia также приводило к повышению уровня расщепленного фрагмента PARP (поли-(АДФ-рибоза)-полимеразы) массой 89 кДа, что является известным ответом на активацию каспазы-3. Активность каспаз-3 и -7 также увеличивалась, когда к обработке соединением формулы Ia добавляли трастузумаб. Комбинирование трастузумаба с 250 нМ соединением формулы Ia повышало активность каспаз-3 и -7 до уровня, аналогичного уровню, отмеченному при использовании в четыре раза более высокой дозы (1000 нМ) только лишь соединения формулы Ia. Важно, что такое повышение каспазной активности отражалось на апоптотическом индексе этих клеток. Добавление трастузумаба существенно уменьшало концентрацию соединения формулы Ia, необходимую для индукции апоптоза. Детектировали почти эквивалентный уровень апоптоза, когда клетки обрабатывали 100 нМ соединением формулы Ia и трастузумабом, по сравнению с тем, когда обрабатывали только 1000 нМ соединением формулы Ia. Как ожидалось, такое усиление апоптоза отражалось в уменьшении жизнеспособности клеток через 48 ч. Аналогичное увеличение каспазной активности и апоптоза наблюдали, когда клетки SKBR-3 обрабатывали ингибирующей комбинацией. Комбинация соединения формулы Ia с трастузумабом значительно снижает концентрацию соединения формулы Ia, необходимую для достижения порога активации каспазы и апоптоза в трастузумаб-чувствительных раковых клетках молочной железы. Следовательно, обработка трастузумабом могла бы быть полезна для повышения чувствительности HER2-амплифицирующих клеток к ингибированию Pl3K и, таким образом, для обеспечения дополнительного уровня опухолевой специфичности для Pl3K-ингибитора формулы Ia.
На Фиг.35 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы ВТ474, которым на 0-е сутки вводили: соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 3 недель, соединение формулы Ib в дозе 2,5 мг/кг перорально дважды в неделю в течение 3 недель, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 3 недель, трастузумаб (15 мг/кг) внутривенно один раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель; соединения формулы Ib в дозе 2,5 мг/кг перорально дважды в неделю в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель; и соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 3 недель и трастузумаба (15 мг/кг) внутривенно один раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 2,5 мг/кг соединения формулы Ib, вводимого ежесуточно, с 15 мг/кг трастузумаба, вводимого один раз в неделю, синергически ингибировала рост опухоли молочной железы ВТ474 in vivo, больше, чем соединение формулы Ia или трастузумаб по отдельности, и вызывала задержку опухолевого роста.
На Фиг.36 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы MCF-7, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель или соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение суток 0-3, 10-26 и комбинации: соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение суток 0-3, 10-26, и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 5,0 мг/кг соединения формулы Ib с 5 мг/кг В20-4.1 синергически ингибировала рост опухоли молочной железы MCF-7 in vivo, больше, чем соединение формулы Ib или В20-4.1 по отдельности, и вызывала регрессию опухоли.
На Фиг.37 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли молочной железы Fo5, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, соединение формулы Ia (GDC-0941) в дозах 36 и 73 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозах 2,5 и 5 мг/кг перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ia в дозе 73 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; и соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 36 мг/кг соединения формулы Ia с 5 мг/кг В20-4.1 синергически ингибировала рост опухоли молочной железы Роб in vivo, больше, чем соединение формулы Ia или В20-4.1 по отдельности, и вызывала задержку опухолевого роста. Комбинация 73 мг/кг соединения формулы Ia с 5 мг/кг В20-4.1 также синергически ингибировала рост опухоли молочной железы Fo5 in vivo, больше, чем соединение формулы Ia или В20-4.1 по отдельности, и вызывала задержку опухолевого роста. Комбинация 2,5 мг/кг соединения формулы Ib с 5 мг/кг В20-4.1 синергически ингибировала рост опухоли молочной железы Fo5 in vivo, больше, чем соединение формулы Ib или В20-4.1 по отдельности, и вызывала задержку опухолевого роста. Комбинация 5,0 мг/кг соединения формулы Ib с 5,0 мг/кг В20-4.1 синергически ингибировала рост опухоли молочной железы Fo5 in vivo, больше, чем соединение формулы Ib или В20-4.1 по отдельности, и вызывала регрессию опухолевого роста.
На Фиг.38 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от Marian с ксенотрансплантатами клеток опухоли молочной железы MDA-MB-231, которым на 0-е сутки вводили: мышиное антитело против VEGF В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель, соединение формулы Ia (GDC-0941) в дозах 36 и 73 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и соединение формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 8 суток и комбинации: соединения формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ia в дозе 73 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 21 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 3 недель; и соединения формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 8 суток и В20-4.1 (5 мг/кг) внутрибрюшинно дважды в неделю в течение 1,5 недели, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 5,0 мг/кг соединения формулы Ib с 5,0 мг/кг В20-4.1 синергически ингибировала рост опухоли молочной железы MDA-MB-231 in vivo, больше, чем соединение формулы Ib или В20-4.1 по отдельности, и вызывала задержку опухолевого роста.
На Фиг.39 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от Marian с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H1299, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (GDC-0941) в дозе 100 мг/кг перорально ежесуточно в течение 6 суток, соединение формулы Ia в дозе 50 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ia (25 мг/кг) перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia (100 мг/кг) перорально ежесуточно в течение 6 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 6 суток; соединения формулы Ia (50 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; и соединения формулы Ia (25 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 25 мг/кг соединения формулы Ia с 50 мг/кг эрлотиниба синергически ингибировала опухолевый рост немелкоклеточного рака легкого H1299 in vivo, больше, чем соединение формулы Ia или эрлотиниб по отдельности, и вызывала задержку опухолевого роста. Комбинация 50 мг/кг соединения формулы Ia с 50 мг/кг эрлотиниба также синергически ингибировала опухолевый рост немелкоклеточного рака легкого NCl-H1299 in vivo, больше, чем соединение формулы Ia или эрлотиниб по отдельности, и вызывала задержку опухолевого роста.
На Фиг.40 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H520, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (GDC-0941) в дозе 73 мг/кг перорально ежесуточно в течение 4 суток, соединение формулы Ia в дозе 36 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ia (18 мг/кг) перорально ежесуточно в течение 21 суток и комбинации: соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 4 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 4 суток; соединения формулы Ia (36 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; и соединения формулы Ia (18 мг/кг) перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 18 мг/кг соединения формулы Ia с 50 мг/кг эрлотиниба синергически ингибировала опухолевый рост немелкоклеточного рака легкого Н520 in vivo, больше, чем соединение формулы Ia или эрлотиниб по отдельности, и вызывала задержку опухолевого роста. Комбинация 36 мг/кг соединения формулы Ia с 50 мг/кг эрлотиниба также синергически ингибировала опухолевый рост немелкоклеточного рака легкого Н520 in vivo, больше, чем соединение формулы Ia или эрлотиниб по отдельности, и вызывала задержку опухолевого роста.
На Фиг.41 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток опухоли немелкоклеточного рака легкого (NSClC) H1299, которым на 0-е сутки вводили: эрлотиниб (50 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток, соединение формулы Ib в дозе 5 мг/кг перорально два раза в неделю в течение 21 суток, соединение формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 21 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; соединения формулы Ib в дозе 5 мг/кг перорально два раза в неделю в течение 3 недель и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток; и соединения формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 2,5 мг/кг соединения формулы Ib, вводимого перорально ежесуточно, с 50 мг/кг эрлотиниба, вводимого перорально ежесуточно, в течение 21 суток синергически ингибировала опухолевый рост немелкоклеточного рака легкого H1299 in vivo, больше, чем соединение формулы Ib или эрлотиниб по отдельности, и вызывала задержку опухолевого роста.
На Фиг.42 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии NCR от Taconic с ксенотрансплантами клеток опухоли немелкоклеточного рака легкого (NSClC) NCl-H2122, которым на 0-е сутки вводили: эрлотиниб (75 мг/кг) перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 16 суток, соединение формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 16 суток и комбинации: соединения формулы Ib в дозе 2,5 мг/кг перорально ежесуточно в течение 16 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 16 суток; соединения формулы Ib в дозе 5 мг/кг перорально ежесуточно в течение 16 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 16 суток; и соединения формулы Ib в дозе 7,5 мг/кг перорально ежесуточно в течение 16 суток и эрлотиниба (50 мг/кг) перорально ежесуточно в течение 16 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 2,5 мг/кг соединения формулы Ib с 75 мг/кг эрлотиниба синергически ингибировала опухолевый рост немелкоклеточного рака легкого NCl-H2122 in vivo, больше, чем соединение формулы Ib или эрлотиниб по отдельности, и вызывала остановку опухолевого роста.
На Фиг.43 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли меланомы человека А375, которым на 0-е сутки вводили: PD-0325901 (3 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (GDC-0941) в дозе 73 мг/кг перорально ежесуточно в течение 3 недель и комбинацию: PD-0325901 (3 мг/кг) перорально ежесуточно в течение 3 недель и соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 73 мг/кг соединения формулы Ia с 3 мг/кг PD-0325901 синергически ингибировала опухолевый рост меланомы человека А375 in vivo, больше, чем соединение формулы Ia или PD-0325901 по отдельности, и вызывала регрессию опухоли и задержку опухолевого роста.
На Фиг.44 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли меланомы человека А375, которым на 0-е сутки вводили: темозоломид (100 мг/кг) перорально ежесуточно в течение 5 суток, соединение формулы Ib в дозе 10 мг/кг перорально один раз в неделю в течение 3 недель, соединение формулы Ib в дозе 5 мг/кг перорально раз в неделю в течение 3 недель и комбинации: соединения формулы Ib в дозе 10 мг/кг перорально один раз в неделю в течение 3 недель и темозоломида (100 мг/кг) перорально ежесуточно в течение 5 суток; и соединения формулы Ib в дозе 5 мг/кг перорально один раз в неделю в течение 3 недель и темозоломида (100 мг/кг) перорально ежесуточно в течение 5 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 5 мг/кг соединения формулы Ib с 100 мг/кг темозоломида синергически ингибировала опухолевый рост меланомы человека А375 in vivo, больше, чем соединение формулы Ib или темозоломид по отдельности, и вызывала задержку опухолевого роста.
На Фиг.45 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ia (GDC-0941) (73 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ia (36 мг/кг) перорально ежесуточно в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ia (73 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; соединения формулы Ia (36 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ia (73 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 36 мг/кг соединения формулы Ia, вводимого ежесуточно, с 10 мг/кг доцетаксела синергически ингибировала рост опухоли яичников человека SKOV3 in vivo, больше, чем соединение формулы Ia или доцетаксел по отдельности, и вызывала задержку опухолевого роста. Комбинация 73 мг/кг соединения формулы Ia, вводимого ежесуточно, с 10 мг/кг доцетаксела также синергически ингибировала рост опухоли яичников человека SKOV3 in vivo, больше, чем соединение формулы Ia или доцетаксел по отдельности, и вызывала задержку опухолевого роста.
На Фиг.46 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ib (5 мг/кг) перорально ежесуточно в течение 3 недель, соединение формулы Ib (1 мг/кг) перорально ежесуточно в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ib (5 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ib (1 мг/кг) перорально ежесуточно в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 5 мг/кг соединения формулы Ib с 10 мг/кг доцетаксела синергически ингибировала рост опухоли яичников человека SKOV3 in vivo, больше, чем соединение формулы Ib или доцетаксел по отдельности, и вызывала остановку опухолевого роста.
На Фиг.47 показано изменение среднего объема опухоли во времени у самок nu/nu мышей от HarIan с ксенотрансплантатами клеток раковой опухоли яичников человека SKOV3, которым на 0-е сутки вводили: соединение формулы Ib (5 мг/кг) перорально раз в неделю в течение 3 недель, соединение формулы Ib (10 мг/кг) перорально раз в неделю в течение 3 недель, доцетаксел (10 мг/кг) внутривенно раз в неделю в течение 3 недель и комбинации: соединения формулы Ib (5 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель; и соединения формулы Ib (10 мг/кг) перорально раз в неделю в течение 3 недель и доцетаксела (10 мг/кг) внутривенно раз в неделю в течение 3 недель, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 10 мг/кг соединения формулы Ib, вводимого перорально раз в неделю, с 10 мг/кг доцетаксела синергически ингибировала рост опухоли яичников человека SKOV3 in vivo, больше, чем соединение формулы Ib или доцетаксел по отдельности, и вызывала задержку опухолевого роста.
На Фиг.48 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии SClD/Beige с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека LuCap 35V, которым на 0-е сутки вводили: доцетаксел (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3), соединение формулы Ia (GDC-0941) (50 мг/кг) перорально ежесуточно в течение 18 суток, соединение формулы Ia (100 мг/кг) перорально ежесуточно в течение 18 суток и комбинации: доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ia (50 мг/кг) перорально ежесуточно в течение 18 суток; и доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ia (100 мг/кг) перорально ежесуточно в течение 18 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 100 мг/кг соединения формулы Ia с 5 мг/кг доцетаксела синергически ингибировала рост первичной опухоли предстательной железы человека LuCap 35V in vivo, больше, чем соединение формулы Ia или доцетаксел по отдельности, и вызывала регрессию опухоли.
На Фиг.49 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей линии SClD/Beige с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека LuCap 35V, которым на 0-е сутки вводили: доцетаксел (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3), соединение формулы Ib (2,5 мг/кг) перорально ежесуточно в течение 15 суток, соединение формулы Ib (5 мг/кг) перорально ежесуточно в течение 15 суток и комбинации: доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ib (2,5 мг/кг) перорально ежесуточно в течение 15 суток; и доцетаксела (5 мг/кг) внутривенно на 1-е, 5-е и 9-е сутки (1х/4 сут х 3) и соединения формулы Ib (5 мг/кг) перорально ежесуточно в течение 15 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 2,5 мг/кг соединения формулы Ib с 5 мг/кг доцетаксела синергически ингибировала рост первичной опухоли предстательной железы человека LuCap 35V in vivo, больше, чем соединение формулы Ib или доцетаксел по отдельности, и вызывала регрессию опухоли. Комбинация 5,0 мг/кг соединения формулы Ib с 5 мг/кг доцетаксела также синергически ингибировала рост первичной опухоли предстательной железы человека LuCap 35V in vivo, больше, чем соединение формулы Ib или доцетаксел по отдельности, и вызывала регрессию опухоли.
На Фиг.50 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии CRL с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека PC3-NCl, которым вводили на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4) 2,5 мг/кг доцетаксела внутривенно, 2,5 мг/кг соединения формулы Ib на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4), соединение формулы Ib (10 мг/кг) перорально на 1-е, 5-е, 9-е и 13-е сутки (1х/4 сут х 4) и комбинацию: доцетаксела (2,5 мг/кг) внутривенно и соединения формулы Ib (10 мг/кг) перорально, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 10 мг/кг соединения формулы Ib, вводимого на 1-е, 5-е, 9-е и 13-е сутки, с 5 мг/кг доцетаксела синергически ингибировала рост первичной опухоли предстательной железы человека PC3-NCl in vivo, больше, чем соединение формулы Ib или доцетаксел по отдельности, и вызывала регрессию опухоли.
На Фиг.51 показано изменение среднего объема опухоли во времени у самок nu/nu мышей линии CRL с ксенотрансплантатами клеток опухоли первичного рака предстательной железы человека PC3-NCl, которым на 0-е сутки вводили: гемцитабин (100 мг/кг) внутрибрюшинно каждые 3 суток (1х/3 сут) за 4 раза, соединение формулы Ia (GDC-0941) (150 мг/кг) перорально каждые 3 суток (1х/3 сут) за 4 раза, соединение формулы Ib (2,5 мг/кг) перорально каждые 3 суток (1х/3 сут) за 4 раза, соединение формулы Ib (5 мг/кг) перорально каждые 3 суток за 4 раза и комбинации: гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ia (150 мг/кг) перорально каждые 3 суток за 4 раза; гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (2,5 мг/кг) перорально каждые 3 суток за 4 раза; гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (5 мг/кг) перорально каждые 3 суток за 4 раза; гемцитабина (100 мг/кг) внутрибрюшинно каждые 3 суток за 4 раза и соединения формулы Ib (10 мг/кг) перорально каждые 3 суток за 4 раза, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 150 мг/кг соединения формулы Ia, вводимого перорально каждые 3 суток (1х/3 сут) за 4 раза, с 100 мг/кг гемцитабина синергически ингибировала рост первичной опухоли предстательной железы человека PC3-NCl in vivo, больше, чем соединение формулы Ia или гемцитабин по отдельности, и вызывала регрессию опухоли и задержку опухолевого роста. Комбинация 2,5 мг/кг соединения формулы Ib, вводимого перорально каждые 3 суток (1х/3 сут) за 4 раза, с 100 мг/кг гемцитабина синергически ингибировала рост первичной опухоли предстательной железы человека PC3-NCl in vivo, больше, чем соединение формулы Ia или гемцитабин по отдельности, и вызывала регрессию опухоли и задержку опухолевого роста. Комбинация 5,0 мг/кг соединения формулы Ib, вводимого перорально каждые 3 суток (1х/3 сут) за 4 раза, с 100 мг/кг гемцитабина синергически ингибировала рост первичной опухоли предстательной железы человека PC3-NCl in vivo, больше, чем соединение формулы Ia или гемцитабин по отдельности, и вызывала регрессию опухоли и задержку опухолевого роста. Комбинация 10 мг/кг соединения формулы Ib, вводимого перорально каждые 3 суток (1х/3 сут) за 4 раза, с 100 мг/кг гемцитабина синергически ингибировала рост первичной опухоли предстательной железы человека PC3-NCl in vivo, больше, чем соединение формулы Ia или гемцитабин по отдельности, и вызывала регрессию опухоли и задержку опухолевого роста.
На Фиг.52 показано изменение среднего объема опухоли во времени у самок бестимусных и бесшерстных мышей от HarIan с ксенотрансплантатами опухолевых клеток немелкоклеточного рака легкого (NSClC) NCl-H2122 (K-ras), которым на 0-е сутки вводили: PD-0325901 (6,3 мг/кг) перорально ежесуточно в течение 21 суток, соединение формулы Ia (GDC-0941) в дозе 100 мг/кг перорально ежесуточно в течение 21 суток и комбинацию: PD-0325901 (6,3 мг/кг) перорально ежесуточно в течение 21 суток и соединения формулы Ia в дозе 100 мг/кг перорально ежесуточно в течение 21 суток, а также у мышей, не получавших никакого лекарственного средства (группа, получавшая разбавитель). Комбинация 100 мг/кг соединения формулы Ia с 6,3 мг/кг PD-0325901 синергически ингибировала опухолевый рост NSClC NCl-H2122 (K-ras) in vivo, больше, чем соединение формулы Ia или PD-0325901 по отдельности, и вызывала регрессию опухоли.
СОЗДАННАЯ ГЕННОИНЖЕНЕРНЫМ ПУТЕМ МЫШИНАЯ МОДЕЛЬ ДЛЯ ИЗУЧЕНИЯ ПРОТИВООПУХОЛЕВОЙ ЭФФЕКТИВНОСТИ
Комбинированная терапия соединениями формулы I и химиотерапевтическими агентами была эффективна в лечении мелкоклеточного рака легкого (SClC), немелкоклеточного рака легкого (NSClC) и аденокарциномы панкреатической железы (PDAC) в созданных генноинженерным путем мышиных моделях (GEMM), которые воспроизводят развитие человеческой опухоли в эндогенном тканевом микроокружении (Singh, M. and Johnson, L. (2006) Clin. Cancer Res. 12(18): 5312-5328; US 2007/0292948; оба документа включены во всей своей полноте путем ссылки). Удивительные и неожиданные результаты этих экспериментов могут предсказывать клинический ответ некоторых групп пациентов с отдельными типами опухолей и мутациями на такую комбинированную терапию соединениями формулы I и II и химиотерапевтическими агентами.
В модели SClC (Meuwissen et al. (2003), Cancer Cell 4(3): 181-189), несущего мутации, обычно обнаруживаемые в данной группе пациентов, само соединение формулы Ia не оказывало влияния на опухолевый рост по оценкам с использованием компьютерной микротомографии, но оказывало влияние (достоверное отношение рисков) на выживание по сравнению с контролями. Животные, которым вводили комбинацию соединения формулы Ia и мышиного антитела против VEGF-A mB20-4.1.1, показали значительное ингибирование и регрессию опухолевого роста по данным компьютерной микротомографии при сравнении с контролями и каждым лекарственным средством по отдельности. Кроме того, комбинация соединения формулы Ia и мышиного антитела против VEGF-A mB20-4.1 оказывала статистически значимое влияние на общую продолжительность выживания по сравнению с контролями. Животные, которым вводили тройную комбинацию соединения формулы Ia, карбоплатина и мышиного антитела против VEGF-A mB20-4.1.1, также показали измеряемый и устойчивый противоопухолевый ответ по сравнению с агентами по отдельности. Тройная комбинация существенно увеличивала продолжительность выживания по сравнению с агентами по отдельности, и соединение формулы Ia в этом тройном режиме усиливало эффект в плане выживания по сравнению с двойной комбинацией карбоплатина и мышиного антитела против VEGF-A mB20-4.1. К тому же, эти комбинаторные схемы значительно снижали частоту возникновения метастазов в отдаленных лимфатических узлах и печени по сравнению с контролями и каждым лекарственном средством по отдельности.
GEMM для рака поджелудочной железы (Aguirre et al. (2003) Genes & Development, 17: 3112-3126) охватывает мутации, обнаруживаемые у большей части пациентов с этим заболеванием. Такие мыши, при лечении их соединением формулы Ia, первоначально показали уменьшение опухолевого роста по данным ультразвуковой эхографии по сравнению с обработанными контролем мышами. Однако этот ответ не был устойчивым, а обработка агентами по отдельности оказывала умеренный (не являющийся статистически значимым) эффект на выживание у этих мышей. В отличие от этого, соединение формулы Ia в комбинации с мышиным антителом против VEGF-A mB20-4.1.1 показывало значительное уменьшение опухолевого роста по данным ультразвуковой эхографии (по сравнению с неподвергаемыми лечению мышами и обработкой агентами по отдельности), а также значительное влияние на выживание. Комбинированная терапия гемцитабином, либо вместе с мышиным антителом против VEGF-A mB20-4.1.1, либо без него, не показала значительных улучшений в ингибировании опухолевого роста (по данным ультразвуковой эхографии) или в выживании по сравнению с применением одного гемцитабина.
При использовании GEMM для K-ras-инициируемого NSClC (Johnson et al. (2001) Nature 410: 1111-1116; Jackson et al. (2001) Genes & Development 15: 3243-3248) обработка самим только соединением формулы Ia оказывала минимальное воздействие на опухолевый рост по измерениям с использованием компьютерной микротомографии и не оказывала никакого влияния на выживание по сравнению с контролями. Обработка соединением формулы Ia в комбинации с эрлотинибом продемонстрировала умеренное ингибирование опухолевого роста и эффект в плане выживания по сравнению с обработками этими агентами по отдельности. Обработка соединением формулы Ia в комбинации с мышиным антителом против VEGF-A mB20-4.1.1 вызывала значительное уменьшение опухолевого роста и увеличение продолжительности выживания по сравнению с контролями, хотя это несущественно превышало эффекты, наблюдаемые при использовании мышиного антитела против VEGF-A mB20-4.1.1 в качестве единственного агента. Животные, которым вводили тройную комбинацию соединения формулы Ia, карбоплатина и мышиного антитела против VEGF-A mB20-4.1.1, также показали измеряемый и устойчивый противоопухолевый ответ по сравнению с контролями, и также показано заметное воздействие на выживание в этой модели.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Фармацевтические композиции или композиции по настоящему изобретению включают комбинации соединений формулы I или II, химиотерапевтического агента и одного или более чем одного фармацевтически приемлемого носителя, глиданта, разбавителя или эксципиента.
Соединения формулы I или II и химиотерапевтические агенты по настоящему изобретению могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и подразумевается, что данное изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения формулы I или II и химиотерапевтические агенты по настоящему изобретению также могут сущестовать в разных таутомерных формах, и все такие формы включены в объем данного изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с разными энергиями, взаимое превращение которых происходит через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимные превращения посредством миграции протона, как например, при кето-енольной и имин-енаминовой изомеризациях. Валентные таутомеры включают взаимные превращения посредством реорганизации некоторых связанных электронов.
Фармацевтические композиции охватывают как нерасфасованную композицию, так и индивидуальные стандартные лекарственные формы, содержащие более одного (например, два) фармацевтически активных агента, включая соединение формулы I или II и химиотерапевтический агент, выбранный из перечня дополнительных агентов, изложенных в данном описании, вместе с любыми фармацевтически неактивными эксципиентами, разбавителями, носителями или глидантами. Нерасфасованная композиция и каждая индивидуальная стандартная лекарственная форма может содержать фиксированные количества вышеуказанных фармацевтически активных агентов. Нерасфасованная композиция представляет собой вещество, которое еще не прошло технологическую обработку для придания ей индивидуальной стандартной лекарственной формы. Иллюстративной стандартной лекарственной формой являются такие формы, как таблетки, пилюли, капсулы и тому подобное. Аналогично, также предполагается, что изложенный в данном описании способ лечения пациента путем введения фармацевтической композиции по настоящему изобретению охватывает введение нерасфасованной композиции и индивидуальных стандартных лекарственных форм.
Фармацевтические композиции также включают в себя меченные изотопом соединения по настоящему изобретению, которые идентичны соединениям, приведенным в данном описании, но при том лишь, что один или более чем один атом заменен на атом, имеющий атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Все изотопы любого указанного конкретного атома или элемента рассматриваются в объеме соединений по изобретению и их применений. Типичные изотопы, которые могут быть введены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2H, 3H, 11С, 13С, 14С, 13N, 15N, 15О, 17О. 18О, 32P, 33P, 35S, 18F, 36Cl, 123I и 125I. Некоторые меченные изотопом соединения по настоящему изобретению (например, 3H- и 14С-меченные) полезны в исследованиях по распределению соединения и/или субстрата в тканях. Изотопы тритий (3H) и углерод-14 (14С) полезны по причине легкости их получения и детекции. К тому же, замещение более тяжелыми изотопами, такими как дейтерий (2Н), может давать некоторые терапевтические преимущества, вытекающие из более высокой метаболической стабильности (например, увеличение времени полувыведения in vivo или понижение требований по дозировке) и, следовательно, может быть предпочтительно в некоторых случаях. Изотопы, испускающие позитроны, такие как 15О, 13N, 11С и 18F, полезны в исследованиях с использованием позитронной эмиссионной томографии (PET) степени занятости рецепторов субстратом.
Меченные изотопом соединения по настоящему изобретению в общем случае можно получить путем проведения процедур, аналогичных изложенным ниже на Схемах и/или в Примерах в данном описании, в результате замещения меченным изотопом реагентом немеченного изотопом реагента.
Соединения формулы I или II и химиотерапевтические агенты изготавливают в соответствии с установившейся фармацевтической практикой для применения в терапевтической комбинации для терапевтического лечения (включая профилактическое лечение) гиперпролиферативных расстройств у млекопитающих, в том числе людей. Согласно изобретению предложена фармацевтическая композиция, содержащая соединение формулы I или II вместе с одним или более чем одним фармацевтически приемлемым носителем, глидантом, разбавителем или эксципиентом.
Подходящие глиданты могут быть выбраны из диоксида кремния, порошкообразной целлюлозы, микрокристаллической целлюлозы, стеаратов металлов, алюмосиликата натрия, бензоата натрия, карбоната кальция, силиката кальция, кукурузного крахмала, карбоната магния, талька без примеси асбеста, стеаровета (stearowet) C, крахмала, крахмала 1500, лаурилсульфата магния, оксида магния и их комбинаций.
Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают такие вещества, как углеводы, воски, водорастворимые и/или набухаемые полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и тому подобное. Выбор используемого конкретного носителя, разбавителя или эксципиента будет зависеть от средств и цели, для решения которой будет применено соединение по настоящему изобретению. Обычно растворители выбирают из растворителей, известных специалистам в данной области техники как безопасные (GRAS (generally recognized/regarded as safe)) для введения млекопитающему. В общем случае, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или могут смешиваться с ней. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300) и т.д. и их смеси. Композиции также могут включать в себя одно или более из: буферов, стабилизирующих агентов, поверхностно-активных веществ, смачивающих агентов, смазывающих вещества, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, агентов, делающих композицию непрозрачной, глидантов, технологических добавок, красителей, подсластителей, отдушек, корригентов и других известных добавок для придания элегантного вида лекарственному средству (т.е. соединению по настоящему изобретению или его фармацевтической композиции) или для облегчения изготовления фармацевтического продукта (т.е. лекарственного средства).
Данные композиции могут быть изготовлены с использованием традиционных процедур растворения и смешивания. Например, нерасфасованное лекарственное вещество (т.е. соединение по настоящему изобретению или стабилизированную форму соединения, например, комплекс с производным циклодекстрина или другим известным комплексообразующим агентом) растворяют в подходящем растворителе в присутствии одного или более эксципиентов, описанных выше. Соединение по настоящему изобретению обычно изготавливают в фармацевтических лекарственных формах, чтобы обеспечить получение легко регулируемой дозировки лекарственного средства и облегчить пациенту соблюдение предписанной схемы (лечения).
Фармацевтическая композиция (или просто композиция) для применения может быть упакована разнообразно в зависимости от способа, используемого для введения лекарственного средства. В общем случае предназначенное для продажи изделие содержит контейнер с помещенной в него фармацевтической композицией в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники и включают такие материалы, как бутылки (пластиковые и стеклянные), саше, ампулы, пластиковые мешочки, металлические цилиндры и тому подобное. Кроме того, контейнер может содержать препятствующее вскрытию укупорочное средство (tamper-proof assemblage) для защиты от неосторожного проникновения к содержимому упаковки. К тому же, контейнер имеет помещенную на него этикетку, на которой описано содержимое контейнера. Этикетка также может содержать соответствующие предупреждения.
Фармацевтические композиции соединений по настоящему изобретению могут быть изготовлены для различных способов и типов введения. Например, соединение формулы I или II желаемой степени чистоты возможно может находиться в смеси с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington′s Pharmaceutical Sciences (1995) 18th edition, Mack Publ. Co., Easton, PA) в форме лиофилизированной композиции, размолотого порошка или водного раствора. Изготовление композиции может быть осуществлено путем смешивания при температуре окружающей среды, при соответствующем pH и желаемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов в используемых дозировках и концентрациях. Величина pH композиции зависит главным образом от конкретного применения и концентрации соединения, но может изменяться в диапазоне от примерно 3 до примерно 8.
Фармацевтическая композиция предпочтительно является стерильной. В частности, композиции, которые будут использованы для введения in vivo, должны быть стерильными. Такую стерилизацию легко осуществляют посредством фильтрования через мембраны для стерильной фильтрации.
Как правило, фармацевтическую композицию можно хранить в виде твердой композиции, лиофилизированной композиции или в виде водного раствора.
Фармацевтические композиции по изобретению будут дозированы и введены способом, т.е. в количествах, концентрациях, с соответствии со схемами, порядком, наличием разбавителей и путем введения, согласующимися с хорошей медицинской практикой,. В этом контексте рассматриваемые факторы включают конкретное подвергаемое лечению расстройство, конкретное подвергаемое лечению млекопитающее, клиническое состояние индивидуального пациента, причину расстройства, место доставки агента, способ введения, составление схемы введения и другие факторы, известные врачам. "Терапевтически эффективное количество" соединения, которое должно быть введено, будет обусловлено такого рода соображениями и представляет собой минимальное количество, необходимое для предупреждения, улучшения или лечения расстройства, опосредованного фактором свертывающей системы крови. Предпочтительно, чтобы такое количество было ниже количества, которое является токсичным для реципиента или которое делает реципиента значительно более подверженным кровоизлиянию.
В качестве общей нормы изначальное фармацевтически эффективное количество вводимого перорально или парентерально соединения формулы I или II на одну дозу будет находиться в диапазоне примерно 0,01-1000 мг/кг, а именно, примерно 0,1-20 мг/кг массы тела пациента в сутки, при этом типичный используемый начальный диапазон для соединения составит 0,3-15 мг/кг/сутки. Доза соединения формулы I или II и доза химиотерапевтического агента, которая должны быть введена, может находиться в диапазоне для каждого из них от примерно 1 мг до примерно 1000 мг на стандартную лекарственную форму или от примерно 10 мг до примерно 100 мг на стандартную лекарственную форму. Дозы соединения формулы I или II и химиотерапевтического агента можно вводить в соотношении от примерно 1:50 до примерно 50:1 по массе или в соотношении от примерно 1:10 до примерно 10:1 по массе.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы нетоксичны для реципиентов в применяемых дозировках и концентрациях и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметонийхлорид; бензалконийхлорид, бензетонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропил-парабен; катехол; резорцин; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем примерно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA (этилендиаминтетрауксусная кислота); сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы с металлами (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Кроме того, активные фармацевтические ингредиенты могут быть заключены в микрокапсулы, приготовленные, например, с использованием методик коацервации или путем межфазной полимеризации, например, в микрокапсулы из гидроксиметилцеллюлозы или желатина и микрокапсулы из поли-(метилметакрилата), соответственно, в коллоидные системы лекарственной доставки (например, липосомы, микросферы из альбумина, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методики описаны в Remington's Pharmaceutical Sciences, 18th edition, (1995) Mack Publ. Co., Easton, PA.
Можно изготовить композиции с непрерывным высвобождением соединений формулы I и II. Подходящие примеры композиций с непрерывным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащих соединение формулы I, причем эти матрицы существуют в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц с непрерывным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтил-метакрилат) или поливиниловый спирт)), полилактиды (US 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата, неразлагаемый этилен-винилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и ацетата лейпролида) и поли-0-(-)-3-гидроксимасляная кислота.
Фармацевтические композиции включают композиции, подходящие для способов введения, подробно изложенных в данном описании. Удобно возможное представление композиций в стандартной лекарственной форме, и они могут быть изготовлены любым из способов, хорошо известных в фармации. Методики и композиции обычно можно найти в Remington's Pharmaceutical Sciences 18th Ed. (1995) Mack Publishing Co., Easton, PA. Такие способы включают стадию приведения активного ингредиента в ассоциацию с носителем, который состоит из одного или более дополнительных ингредиентов. В общем случае композиции изготавливают путем непрерывного и равномерного приведения активного ингредиента в ассоциацию с жидкими носителями или тонкоизмельченными твердыми носителями или ими обоими и затем, если необходимо, придания формы продукту.
Композиции соединения формулы I или II и/или химиотерапевтического агента, подходящие для перорального введения, могут быть изготовлены в виде дискретных единиц, таких как пилюли, твердые или мягкие, например желатиновые, капсулы, облатки, пастилки, лепешки, водные или масляные суспензии, диспергируемые порошки или драже, эмульсии, сиропы или эликсиры, каждая из которых содержит предварительно определенное количество соединения формулы I или II и/или химиотерапевтического агента. Данное количество соединения формулы I или II и количество химиотерапевтического агента может быть включено в пилюлю, капсулу, раствор или суспензию в виде комбинированной композиции. Альтернативно, соединение формулы I или II и химиотерапевтический агент могут быть включены в пилюлю, капсулу, раствор или суспензию по отдельности для введения путем чередования.
Композиции могут быть изготовлены в соответствии с любым способом, известным в области изготовления фармацевтических композиций, и такие композиции могут содержать один или более агентов, включая подсластители, корригенты, красители и консерванты, с целью придания препарату приятного вкуса. Прессованные таблетки могут быть изготовлены в подходящей машине путем прессования активного ингредиента в свободно текучей форме, такой как порошок или драже, возможно в смеси со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены в подходящей машине путем формования смеси порошкообразного активного ингредиента, увлажненного инертным жидким разбавителем. Возможно, что таблетки могут иметь покрытие или риску и возможно могут быть изготовлены таким образом, чтобы обеспечить медленное или регулируемое высвобождение из него активного ингредиента.
Эксципиенты для таблеточной фармацевтической композиции по изобретению могут включать: наполнитель (или разбавитель) для увеличения общего объема порошкового лекарственного средства, из которого изготавливается таблетка; разрыхлители для способствования распаданию таблетки на небольшие фрагменты, в идеале на отдельные лекарственные частицы, при ее проглатывании и для стимулирования быстрого растворения и всасывания лекарственного средства; связующее вещество для обеспечения возможности образования драже и таблеток с необходимой механической прочностью и сохранения целостности таблетки после ее прессования, предотвращающее таблетки от распадания на составляющие их порошки в процессе упаковки, транспортировки и традиционного манипулирования; глидант для улучшения сыпучести порошка, составляющего таблетку, в процессе изготовления; смазывающее вещество для обеспечения того, чтобы в процессе изготовления таблетируемый порошок не прилипал к оборудованию, используемому для прессования таблетки (они улучшают сыпучесть порошковых смесей при прохождении через прессы и минимизируют истирание и разламывание при извлечении готовых таблеток из оборудования; антиагезивное вещество с функцией, аналогичной глиданту, уменьшающее в процессе изготовления прилипание порошка, составляющего таблетку, к машине, используемой для штампования формы таблетки; корригент, включаемый в таблетки для придания им более приятного вкуса или для маскировки неприятного вкуса; и краситель для облегчения идентификации и соблюдения больных режима и схемы лечени.
Приемлемы таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, подходящим для изготовления таблеток. Этими эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующие агенты и разрыхлители, такие как кукурузный крахмал или альгиновая кислота; связывающие вещества, такие как крахмал, желатин или аравийская камедь; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут быть покрыты с использованием известных методик, включая микроинкапсулирование, для задержки распадаемости и всасывания в желудочно-кишечном тракте и тем самым для обеспечения непрерывного действия в течение более длительного периода времени. Например, можно использовать такое вещество-замедлитель (time deIay material), как глицерилмоностеарат или глицерилдистеарат, сами по себе или с воском.
Для лечения глазных или других внешних тканей, например, рта и кожи, предпочтительно применяют композиции в виде мази или крема для местного применения, содержащих активный ингредиент(ы) в количестве, например, 0,075-20% масс./масс.При изготовлении в виде мази активные ингредиенты могут быть использованы либо с парафиновой, либо со смешивающейся с водой мазевой основой. Альтернативно, активные ингредиенты могут быть изготовлены в виде крема с использованием основы для крема типа масло-в-воде.
При желании водная фаза основы для крема может включать многоатомный спирт, т.е. спирт, имеющий две или более гидроксильные группы, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400) и их смеси. Композиции для местного применения при желании могут включать соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие зоны воздействия. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и родственные аналоги.
Масляная фаза эмульсий по данному изобретению может быть составлена известным образом и состоять из известных ингредиентов, включая смесь по меньшей мере одного эмульгатора с жиром или маслом либо и с жиром, и с маслом обоими. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует как стабилизатор. Совместно, эмульгатор(ы) со стабилизатором(ами) или без него(их) образуют эмульгирующий воск, и такой воск вместе с маслом и жиром составляет эмульгирующую мазевую основу, которая образует диспергируемую в масле фазу композиций в форме крема. Эмульгаторы и стабилизаторы эмульсий, подходящие для использования в композиции по изобретению, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии фармацевтических композиций по изобретению содержат активные вещества в смеси с эксципиентами, подходящими для изготовления водных суспензий. Такие эксципиенты включают суспендирующий агент, такой как карбоксиметилцеллюлозы натриевая соль, кроскармелоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие или смачивающие агенты, такие как фосфатид природного происхождения (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленокси-цетанол), продукт конденсации этиленоксида с неполным сложным эфиром, являющимся производным жирной кислоты и ангидрида гексита (например, полиоксиэтиленсорбитана моноолеат). Водная суспензия также может содержать один или более консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или более красителей, один или более корригентов и один или более подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции могут быть в форме стерильной инъецируемой композиции, такой как стерильная водная или масляная суспензия для инъекций. Эта суспензия может быть изготовлена согласно известным в данном области техники способам с использованием таких подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые упомянуты выше. Стерильная композиция для инъекций может представлять собой раствор или суспензию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, как например, раствор в 1,3-бутандиоле, или может быть приготовлена из лиофилизированного порошка. Среди приемлемых разбавителей и растворителей, которые могут быть использованы, находится вода, раствор Рингера и изотонический раствор хлорида натрия. К тому же, в качестве растворителя или суспендирующей среды традиционно могут быть использованы стерильные нелетучие масла. Для этой цели может быть использовано любое маловязкое нелетучее масло, включая синтетические моно- или диглицериды. В дополнение к этому, жирные кислоты, такие как олеиновая кислота, таким же образом могут быть использованы в изготовлении инъецируемых средств.
Количество активного ингредиента, которое может быть комбинировано с веществом-носителем для изготовления разовой лекарственной формы, будет варьировать в зависимости от подвергаемого лечению реципиента и конкретного способа введения. Например, композиция с пролонгированным высвобождением, предназначенная для перорального введения людям, может содержать приблизительно 1-1000 мг активного вещества в смеси с соответствующим и удобным количеством вещества-носителя, которое может варьировать от примерно 5 до примерно 95% от массы всей композиции (масса:масса). Фармацевтическая композиция может быть изготовлена таким образом, чтобы беспрепятственно обеспечить введение измеряемого количества. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать примерно 3-500 мкг активного ингредиента на один миллилитр раствора с тем, чтобы можно было осуществлять инфузию подходящего объема при скорости примерно 30 мл/ч.
Композиции, подходящие для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, делающие композицию изотоничной крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители.
Композиции, подходящие для местного введения в глаз, также включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в таких композициях в концентрации примерно 0,5-20% масс./масс., например примерно 0,5-10% масс./масс., например примерно 1,5% масс./масс.
Композиции, подходящие для местного введения в ротовую полость, включают лепешки, содержащие активный ингредиент в корригирующей основе, обычно сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Композиции для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например масло какао или салицилат.
Композиции, подходящие для введения внутрь легких или интраназального введения, имеют размер частиц, например, в диапазоне от 0,1 до 500 микрон (включая размеры частиц в диапазоне от 0,1 до 500 микрон с таким шагом (in increments), как 0,5, 1, 30 микрон, 35 микрон и т.д.), которые вводят посредством быстрой ингаляции через носовой проход или посредством ингаляции через рот, чтобы достичь альвеолярных мешочков. Подходящие композиции включают водные или масляные растворы активного ингредиента. Композиции, подходящие для введения аэрозоля или сухого порошка, могут быть изготовлены традиционными способами и могут быть доставлены вместе с другими терапевтическими агентами, как например, соединения используемые до сих пор в лечении или профилактике расстройств, которые описаны ниже.
Композиции, подходящие для вагинального введения, могут быть представлены в виде композиций в форме пессариев, тампонов, кремов, гелей, паст, пенок или спрея, содержащих в дополнение к активному ингредиенту такие носители, целесообразность использования которых в данной области техники известна.
Композиции могут быть упакованы в контейнеры для однократного приема или для многократного приема, например, герметично укупоренные ампулы и флаконы, и их можно хранить в высушенном сублимационной сушкой (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды, для инъекций непосредственно перед использованием. Экстемпоральные инъекционные растворы и суспензии готовят из стерильных порошков, драже и таблеток вида, описанного ранее. Предпочтительными стандартными лекарственными композициями являются композиции, содержащие суточную дозу или стандартную суточную субдозу, которые перечислены выше в данном описании, или соответствующую ее долю, активного ингредиента.
Согласно изобретению также предложены ветеринарные композиции, содержащие по меньшей мере один активный ингредиент, который определен выше, вместе с приемлемым в ветеринарии носителем, соответственно. Приемлемыми в ветеринарии носителями являются вещества, полезные для введения композиции, и они могут быть твердыми, жидкими или газообразными веществами, которые в других отношениях инертны или приемлемы в области ветеринарии и совместимы с активным ингредиентом. Эти ветеринарные композиции могут быть введены парентерально, перорально или любым другим желаемым способом.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединения формулы I и II могут быть применены в комбинации с другими химиотерапевтическими агентами для лечения гиперпролиферативного заболевания или расстройства, включая опухоли, виды рака и неопластическую ткань, вместе с предраковыми и не-неопластическими или нераковыми гиперпролиферативными расстройствами. В некоторых воплощениях соединение формулы I или II объединено в фармацевтической комбинированной композиции, или схеме введения в виде комбинированной терапии, со вторым соединением, которое обладает антигиперпролиферативными свойствами или которое полезно для лечения гиперпролиферативного расстройства. Второе соединение в такой фармацевтической комбинированной композиции или схеме введения предпочтительно обладает взаимодополняющими активностями с соединением формулы I или II и такими, что они не оказывают негативного влияния друг на друга. Такие соединения могут быть введены в количествах, эффективных для предполагаемой задачи. В одном из воплощений фармацевтическая композиция по данному изобретению содержит соединение формулы I или II или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль в комбинации с таким химиотерапевтическим агентом, который приведен в данном описании. В другом воплощении терапевтическую комбинацию вводят, используя схему введения, где терапевтически эффективное количество соединения, имеющего формулу 1 или II, вводят в диапазоне от двух раз в сутки (2х/сут) до одного раза каждые три недели (1х/3 нед) и терапевтически эффективное количество химиотерапевтического агента вводят в диапазоне от двух раз в сутки до одного раза каждые три недели.
Терапевтические комбинации по изобретению включают продукт, содержащий соединение, имеющее формулу I или II, и химиотерапевтический агент, выбранный из эрлотиниба, доцетаксела, 5-FU, гемцитабина, PD-0325901, цисплатина, карбоплатина, паклитаксела, бевацизумаба, трастузумаба, пертузумаба, темозоломида, тамоксифена, доксорубицина, Akti-1/2, HPPD, рапамицина и лапатиниба, в виде комбинированной композиции для раздельного, одновременного или последовательного применения в лечении гиперпролиферативного расстройства.
Комбинированная терапия может быть применена в соответствии со схемой одновременного или последовательного введения. При последовательном введении комбинация может быть введена за два или более введений.
Комбинированное введение включает совместное введение, при котором используют отдельные композиции или одну фармацевтическую композицию и последовательное введение в любом порядке, причем предпочтительно имеется период времени, в течение которого оба активных агента (или все) одновременно проявляют свои биологические активности.
Подходящими дозировками для любого из вышеупомянутых вводимых совместно агентов являются используемые в настоящее время дозировки, и они могут быть снижены благодаря комбинированному действию (синергическому действию) нового идентифицированного агента и других химиотерапевтических агентов или лечений, например, благодаря уменьшению терапевтического индекса или ослаблению токсичности или других побочных эффектов или последствий.
В конкретном воплощении противораковой терапии соединение формулы I или II или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемая соль могут быть объединены вместе с химиотерапевтическим агентом, включая гормональные средства или средства на основе антител, как например, изложенные в данном описании, а также объединены с хирургической терапией и лучевой терапией. Таким образом, комбинированные терапии по настоящему изобретению включают введение по меньшей мере одного соединения формулы I или II или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли и использование по меньшей мере одного другого способа лечения рака. Количества соединения(ий) формулы I или II и другого фармацевтически активного химиотерапевтического агента(ов) и соответственное хронометрирование введения будут выбраны с тем, чтобы достичь желаемого комбинированного терапевтического эффекта.
ВВЕДЕНИЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
Соединения по изобретению могут быть введены любым способом, соответствующим подвергаемому лечению состоянию. Подходящие способы включают пероральный, парентеральный (в том числе подкожный, внутримышечный, внутривенный, внутриартериальный, ингаляционный, интрадермальный, интратекальный, эпидуральный и инфузионные методики), трансдермальный, ректальный, назальный, местный (в том числе трансбуккальный и сублингвальный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Местное введение также может включать в себя использование трансдермального введения, как например, трансдермальных пластырей или устройств для ионофореза. Технология изготовления композиций лекарственных средств обсуждается в Remington's Pharmaceutical Sciences, 18th Ed., (1995) Mack Publishing Co., Easton, PA. Другие примеры лекарственных композиций можно найти в Liberman, H. A. and Lachman, L, Eds., Pharmaceutical Drugs, Marcel Decker, Vol 3, 2nd Ed., New York, NY. Для местного иммуносупрессивного лечения соединения могут быть введены внутрь пораженной ткани, включая перфузирование ингибитором или каким-либо иным способом приведение трансплантата в контакт с ингибитором перед трансплантацией. Очевидно, что предпочтительный способ может меняться, например, от состояния реципиента. Если соединение вводят перорально, оно может быть изготовлено в виде пилюли, капсулы, таблетки и т.д. вместе с фармацевтически приемлемым носителем, глидантом или эксципиентом. Если соединение вводят парентерально, оно может быть изготовлено вместе с фармацевтически приемлемым парентеральным напонителем или разбавителем и в стандартной лекарственной форме для инъекций, как подробно описано ниже.
Доза для лечения пациентов-людей может находиться в диапазоне от примерно 10 мг до примерно 1000 мг соединения формулы I или II. Типичная доза может составлять от примерно 100 мг до примерно 300 мг соединения. Доза может быть введена один раз в сутки (1х/сут), дважды в сутки (2х/сут) или более часто, в зависимости от фармакокинетических (РК) и фармакодинамических (PD) свойств, включая всасывание, распределение, метаболизм и экскрецию конкретного соединения. К тому же, на дозировку и схему введения лекарственного средства могут влиять факторы, определяющие токсичность. При введении перорально пилюля, капсула или таблетка могут приниматься путем проглатывания дважды в сутки, ежесуточно или с меньшей частотой, например, раз в неделю или один раз каждые две или три недели, в течение определенного периода времени. Эта схема может быть повторена для ряда циклов терапии.
СПОСОБЫ ЛЕЧЕНИЯ
Терапевтические комбинации: (1) соединения формулы I или II и (2) химиотерапевтического агента полезны для лечения заболеваний, состояний и/или расстройств, включая, но этим не ограничиваясь, таковые, характеризующиеся активацией Pl3-киназного пути. Соответственно, другой аспект этого изобретения включает способы лечения заболеваний или состояний, которые могут быть подвергнуты лечению посредством ингибирования липид-киназ, включая PI3. В одном из воплощений способ включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или II или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли. Терапевтические комбинации: (1) соединения формулы I или II и (2) химиотерапевтического агента могут быть применены для лечения гиперпролиферативного заболевания или расстройства, включая опухоли, виды рака и неопластическую ткань, вместе с предраковыми и не-неопластическими или нераковыми гиперпролиферативными расстройствами. В одном из воплощений человеческого пациента лечат терапевтической комбинацией и фармацевтически приемлемым носителем, адъювантом или разбавителем, где соединение формулы I или II или его метаболит в указанной терапевтической комбинации присутствует в количестве, необходимом для детектируемого ингибирования активности PIS-киназы.
Виды рака, которые могут быть подвергнуты лечению в соответствии со способами по данному изобретению, включают, но этим не ограничиваются, рак молочной железы, яичников, шейки матки, предстательной железы, яичка, мочеполового тракта, пищевода, гортани, глиобластому, нейробластому, рак желудка, кожи, кератокарциному, рак легкого, плоскоклеточный рак, большеклеточный рак, немелкоклеточный рак легкого (NSClC), мелкоклеточный рак, аденокарциному легкого, рак кости, рак толстой кишки, аденому, рак поджелудочной железы, аденокарциному, рак щитовидной железы, фолликулярную карциному, недифференцируемый рак, папиллярную карциному, семиному, меланому, саркому, рак мочевого пузыря, рак печени и желчных протоков, рак почки, ассоциированные с миелоидой тканью расстройства, ассоциированные с лимфоидной тканью расстройства, волосатоклеточный рак, рак ротовой полости и глотки (ее ротовой части), рак губы, языка, ротовой полости, ротоглотки, тонкого кишечника, ободочной и прямой кишки, толстого кишечника, прямой кишки, головного мозга и центральной нервной системы, болезнь Ходжкина и лейкоз.
Согласно другому аспекту этого изобретения предложена фармацевтическая композиция или терапевтическая комбинация для применения в лечении заболеваний или состояний, изложенных в данном описании, у млекопитающего, например, человека, страдающего от такого заболевания или состояния. Также предложено применение фармацевтической композиции в изготовлении лекарственного средства для лечения заболеваний и состояний, изложенных в данном описании, у теплокровного животного, такого как млекопитающее, например, человек, страдающего от такого расстройства.
МЕТАБОЛИТЫ СОЕДИНЕНИЙ ФОРМУЛЫ I и II
Кроме того, в объем данного изобретения попадают in vivo продукты метаболизма соединений формулы I и II, изложенных в данном описании. Такие продукты могут получаться, например, в результате окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного вводимого соединения. Соответственно, изобретение включает метаболиты соединений формулы I и II, в том числе соединения, получаемые способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Обычно продукты метаболизма идентифицируют посредством получения меченного радиоактивным (например, 14С или 3H изотопом соединения по изобретению, введения его парентерально в детектируемой дозе (например, больше приблизительно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, выдерживанию его в течение времени, достаточного для осуществления метаболизма (обычно от приблизительно 30 секунд до 30 часов) и выделения продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты легко выделить, поскольку они являются мечеными (другие выделяют путем использования антител, способных к связыванию эпитопов, оставшихся в метаболите). Структуры метаболитов определяют традиционным образом, например, анализом с использованием MS (масс-спектрометрия), LC/MS (жидкостная хроматография/масс-спектрометрия) или ЯМР (ядерный магнитный резонанс). В общем случае анализ метаболитов осуществляют таким же образом, что и традиционные исследования по метаболизму лекарственных средств, хорошо известные специалистам в данной области техники. Продукты метаболизма, поскольку по-другому они не обнаруживаются in vivo, полезны в диагностических исследованиях по терапевтическому дозированию соединений по изобретению.
ИЗДЕЛИЯ
В другом воплощении изобретения предложены изделие или "набор", содержащие соединения формулы I и II, полезные для лечения заболеваний и расстройств, описанных выше. В одном из воплощений набор включает в себя контейнер, содержащий соединение формулы I или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль. Кроме того, набор может содержать этикетку или инструкцию по медицинскому применению препарата, находящуюся на контейнере или вместе с ним. Термин "инструкция по медицинскому применению препарата" используется для отсылки к инструкциям, обычно вкладываемым в выпускаемые промышленностью упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях, касающихся применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку и т.д. Контейнер может быть изготовлен из ряда материалов, таких как стекло или пластик. Контейнер может содержать соединение формулы I или II или их композицию, которые эффективны для лечения данного состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешок для внутривенных растворов или флакон, имеющий пробку, которую можно проткнуть иглой для подкожных инъекций). По меньшей мере один активный агент в композиции представляет собой соединение формулы I или II. Этикетка или инструкция по медицинскому применению препарата может содержать указания, что композиция применяется для лечения состояния "выбора", такого как рак. В одном из воплощений этикетка или инструкция по медицинскому применению препарата содержит указания, что композиция, содержащая соединение формулы I или II, может быть использована для лечения расстройства, обусловленного аномальным клеточным ростом. Этикетка или инструкция по медицинскому применению препарата также может содержать указания, что композиция может быть использована для лечения других расстройств. Альтернативно или в дополнение к этому, изделие может дополнительно включать в себя второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он может дополнительно включать другие материалы, желательные с точки зрения продавца и потребителя, включая другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно содержать указания по введению соединения формулы I или II и, если она присутствует, то второй фармацевтической композиции. Например, если набор содержит первую композицию, содержащую соединение формулы I или II, и вторую фармацевтическую композицию, то набор может дополнительно содержать указания по одновременному, последовательному или раздельному введению первой и второй фармацевтических композиций нуждающемуся в этом пациенту.
В другом воплощении такие наборы подходят для доставки твердых пероральных форм соединения формулы I или II, таких как таблетки или капсулы. Такой набор предпочтительно включает ряд стандартных дозировок. Такой набор предпочтительно включает ряд стандартных дозировок. Такие наборы могут включать в себя карточку с дозировками, определенными с целью их предполагаемого применения. Примером такого набора является "блистерная упаковка". Блистерные упаковки хорошо известны в упаковочной индустрии и широко используются для упаковки фармацевтических стандартных лекарственных форм. При желании может быть придана памятка, например в виде чисел, букв или других маркировок либо с календарем-вкладышем, где указаны дни в схеме лечения, в которые могут быть введены дозировки.
Согласно одному из воплощений набор может содержать (а) первый контейнер с содержащимся в нем соединением формулы I или II; и возможно (б) второй контейнер с содержащейся в нем второй фармацевтической композицией, содержащей второе соединение, обладающее антигиперпролиферативной активностью. Альтернативно или в дополнение к этому, набор может дополнительно содержать третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он может дополнительно включать другие материалы, желательные с точки зрения продавца и потребителя, включая другие буферы, разбавители, фильтры, иглы и шприцы.
Если набор содержит композицию соединения формулы I или II и второй терапевтический агент, т.е. химиотерапевтический агент, то такой набор может содержать контейнер для помещения в него отдельных композиций, такой как разделенная бутылка или разделенный пакет из фольги, однако отдельные композиции также могут содержаться в одном неразделенном контейнере. Обычно, набор содержит указания по введению отдельных компонентов. Форма набора особенно полезна в тех случаях, когда отдельные компоненты предпочтительно вводят в разных лекарственных формах (например, пероральной и парентеральной), вводят с различными интервалами дозирования или в тех случаях, когда согласно предписанию врача желательно титрование индивидуальных компонентов комбинации.
ОБЩИЕ МЕТОДИКИ ПОЛУЧЕНИЯ
Общая методика А-1. Сочетание по Сузуки
Реакция сочетания по Сузуки используется для присоединения конденсированного бициклического гетероцикла или гетероарила в положение 2 пиримидинового кольца (см. Схему 4). Обычно, замещенный 2-хлор-4-морфолинотиено[3,2-d]пиримидин 5 или замещенный 2-хлор-4-морфолинотиено[2,3-d]пиримидин 6 могут быть объединены с 1,5 эквивалента 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазола 7 и растворены в 3 эквивалентах карбоната натрия в виде 1 молярного раствора в воде и равном объеме ацетонитрила. Добавляют низковалентный палладиевый реагент в каталитическом или превышающем каталитическое количестве, такой как бис(трифенилфосфин)палладия(И) дихлорид. Вместо указанного сложного эфира индазол-бороновой кислоты могут быть использованы разнообразные бороновые кислоты или сложные эфиры бороновых кислот. Кроме этого, альтернативно, азот индазола может быть защищен, например тетрагидропиранильной группой. См. соединение 40. В некоторых случаях вместо карбоната натрия для подведения рН водного слоя использовали ацетат калия. Реакционную смесь затем нагревали до примерно 140-150°С под давлением в микроволновом реакторе Biotage Optimizer (Biotage, Inc.) в течение 10-30 минут. Содержимое экстрагируют этилацетатом или другим органическим растворителем. После упаривания органического слоя продукт 8 или 9 может быть очищен на диоксиде кремния или обращенно-фазовой HPLC.
Общая методика А-2. Сочетание по Сузуки
Реакцию сочетания по Сузуки используют для присоединения моноциклического гетероарила в положение 2 пиримидинового кольца (см. Схему 4). Обычно, замещенный 2-хлор-4-морфолинотиено[3,2-d]пиримидин 5 или замещенный 2-хлор-4-морфолинотиено[2,3-d]пиримидин 6 могут быть объединены с 1,5 эквивалента 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-амина 7а и растворены в 3 эквивалентах карбоната натрия или калия в виде 1 молярного раствора в воде и равном объеме ацетонитрила. Добавляют низковалентный палладиевый реагент в каталитическом или превышающем каталитическое количестве, такой как бис(трифенилфосфин)палладия(II) дихлорид. Вместо указанного сложного эфира пинакол-бороновой кислоты могут быть использованы разнообразные бороновые кислоты или сложные эфиры бороновых кислот. Кроме этого, альтернативно, азот пиримидин-2-амина может быть защищен, например тетрагидропиранильной группой. В некоторых случаях вместо карбоната натрия для подведения рН водного слоя использовали ацетат калия. Реакционную смесь затем нагревали до примерно 100-150°С под давлением в микроволновом реакторе Biotage Optimizer (Biotage, Inc.) в течение 10-30 минут. Содержимое экстрагируют этилацетатом или другим органическим растворителем. После упаривания органического слоя продукт 8а или 9а может быть очищен на диоксиде кремния или обращенно-фазовой HPLC.
Общая методика В. Амидное сочетание
2-(1Н-Индазол-4-ил)-4-морфолинотиено[3,2-d]пиримидин-6-карбоновую кислоту 13 или 2-(1Н-индазол-4-ил)-4-морфолинотиено[2,3-d]пиримидин-6-карбоновую кислоту 14 обрабатывают 1,5 экв. HATU, 3 экв. алкиламина и 3 экв. DIPEA (диизопропилэтиламин) в DMF до концентрации приблизительно 0,1 М. Реакционную смесь перемешивают до завершения реакции и один раз экстрагируют в этилацетат с насыщенным раствором бикарбоната. Органический слой сушат, фильтруют и концентрируют, получая неочищенное промежуточное соединение. Это промежуточное соединение очищают обращенно-фазовой HPLC, получая продукт 15 или 16.
Общая методика В-1. Амидное сочетание
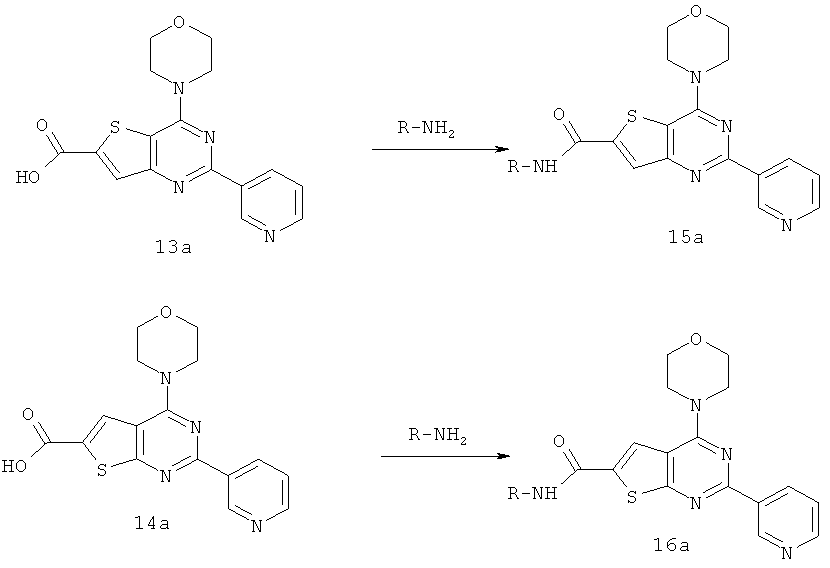
4-Морфолино-2-(пиридин-3-ил)тиено[3,2-d]пиримидин-6-карбоновую кислоту 13а или 4-морфолино-2-(пиридин-3-ил)тиено[2,3-d]пиримидин-6-карбоновую кислоту 14а обрабатывают 1,5 экв. HATU, 3 экв. алкиламина (R-NH2) и 3 экв. DIPEA в DMF до концентрации приблизительно 0,1 М. Реакционную смесь перемешивают до завершения реакции и один раз экстрагируют в этилацетат с насыщенным раствором бикарбоната. Органический слой сушат, фильтруют и концентрируют, получая неочищенное промежуточное соединение. Это промежуточное соединение очищают обращенно-фазовой HPLC, получая продукт 15а или 16а.
Общая методика В-2. Амидное сочетание
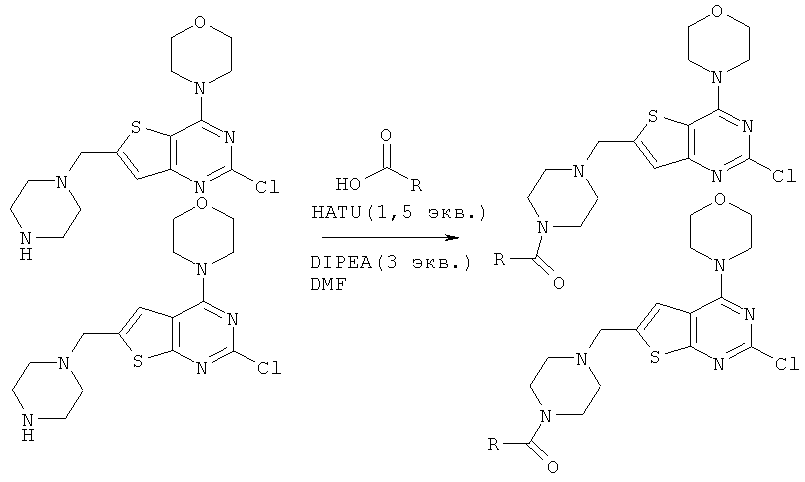
2-Хлор-4-морфолино-6-((пиперазин-1-ил)метил)тиено[3,2-d]пиримидин или 2-хлор-4-морфолино-6-((пиперазин-1-ил)метил)тиено[2,3-d]пиримидин обрабатывают 1,5 экв. HATU, 3 экв. карбоновой кислоты (RC02H) и 3 3KB.DIPEA в DMF до концентрации приблизительно 0,1 М. Реакционную смесь перемешивают до завершения реакции и один раз экстрагируют в этилацетат с насыщенным раствором бикарбоната. Органический слой сушат, фильтруют и концентрируют, получая неочищенное промежуточное соединение.
Общая методика В-3. Восстановительное аминирование
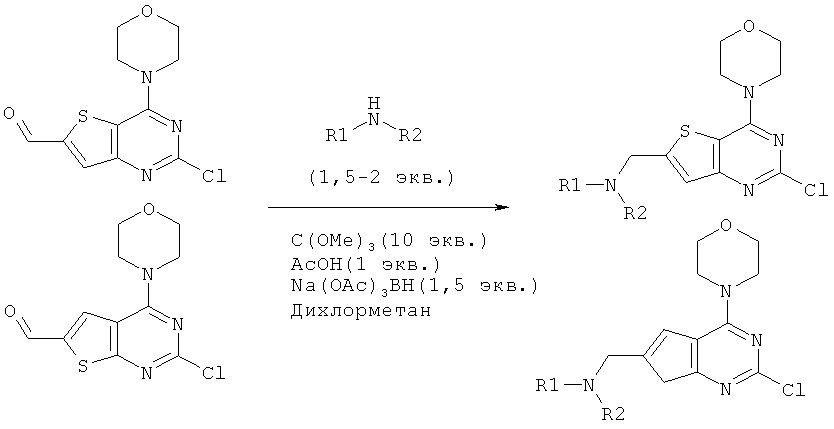
2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид 10 или 2-хлор-4-морфолинотиено[2,3-d]пиримидин-6-карбальдегид 33 растворяли в дихлорэтане до концентрации 0,2 М. К этому раствору добавляли 1,5-2,0 эквивалента амина (R1R2NH), 10 эквивалентов триметилортоформиата и 1 эквивалент уксусной кислоты. Смесь оставляли перемешиваться в течение 2-6 часов, затем добавляли 1,5 эквивалента триацетоксиборгидрида натрия. После перемешивания в течение 12-16 часов содержимое реакционной смеси выливали в насыщенный бикарбонат натрия и несколько раз экстрагировали этилацетатом, получая подвергнутое восстановительному аминированию промежуточное соединение, которое либо очищали на силикагеле, либо использовали в неочищенном виде в следующей реакции.
Общая методика С. Образование сульфонамида
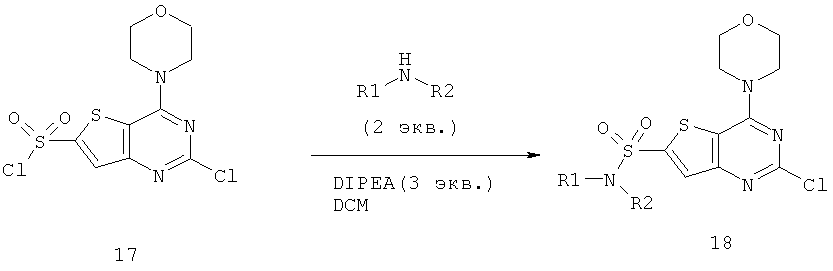 2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-сульфонилхлорид суспендировали в 1 мл DCM, затем добавляли 2 экв. амина и 3 экв. DIPEA. За ходом реакции до ее завершения следили с помощью LCMS. Неочищенные реакционные смеси разбавляли этилацетатом, экстрагировали насыщенным хлоридом аммония и один раз повторно экстрагировали этилацетатом. Органические слои объединяли и концентрировали досуха. Неочищенные сульфонамидные промежуточные соединения 18 использовали непосредственно в последующих реакциях сочетания по Сузуки.
2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-сульфонилхлорид суспендировали в 1 мл DCM, затем добавляли 2 экв. амина и 3 экв. DIPEA. За ходом реакции до ее завершения следили с помощью LCMS. Неочищенные реакционные смеси разбавляли этилацетатом, экстрагировали насыщенным хлоридом аммония и один раз повторно экстрагировали этилацетатом. Органические слои объединяли и концентрировали досуха. Неочищенные сульфонамидные промежуточные соединения 18 использовали непосредственно в последующих реакциях сочетания по Сузуки.
Общая методика Р. Синтез спирта
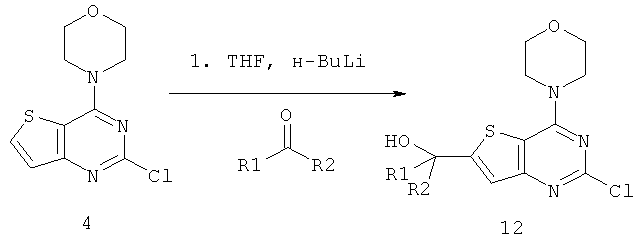
2-Хлор-4-морфолинотиено[3,2-d]пиримидин 4 суспендировали до 0,2 молярной концентрации в THF (тетрагидрофуран) и охлаждали до -50°С в бане сухой лед/ацетонитрил, затем добавляли 2 эквивалента 2,5 М н-BuLi в гексанах. Через 15 мин к раствору добавляли 3,0 молярных эквивалента циклического или ациклического кетона. Реакционную смесь продолжали перемешивать при -50°С в течение 1 ч и затем в большинстве случаев оставляли нагреваться до 0°С. Когда реакция завершалась по данным TLC (тонкослойной хроматографии) или масс-спектрометрии, ее гасили в насыщенном растворе хлориде аммония и содержимое реакционной смеси дважды экстрагировали EtOAc. Органический слой концентрировали и либо использовали в виде неочищенной смеси, либо очищенной на диоксиде кремния, либо продукт 12 растворяли в минимальном количестве ацетонитрила и фильтровали для удаления оставшегося исходного вещества 4.
Общая методика Е. Удаление трет-бутоксикарбонильной (ВОС) группы
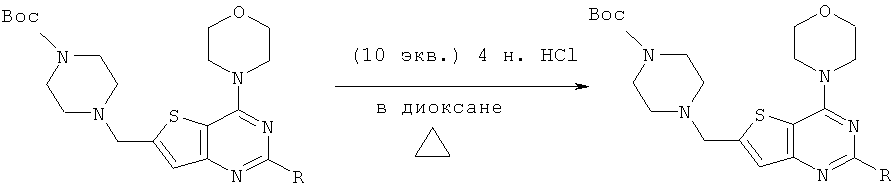
Десять или более эквивалентов 4 н. HCl в диоксане с дихлорметаном в качестве сорастворителя или без него добавляют к исходному соединению (приведенная выше общая схема, но также используются аналогичные структурные каркасы). Для удаления группы Вое обычно необходимо нагревание до 40°С включительно в течение нескольких часов. Реакционную смесь концентрируют досуха, и ее можно использовать неочищенной в последующих реакциях.
Общая методика F. Реакции сочетания по Сузуки "в одном реакторе"
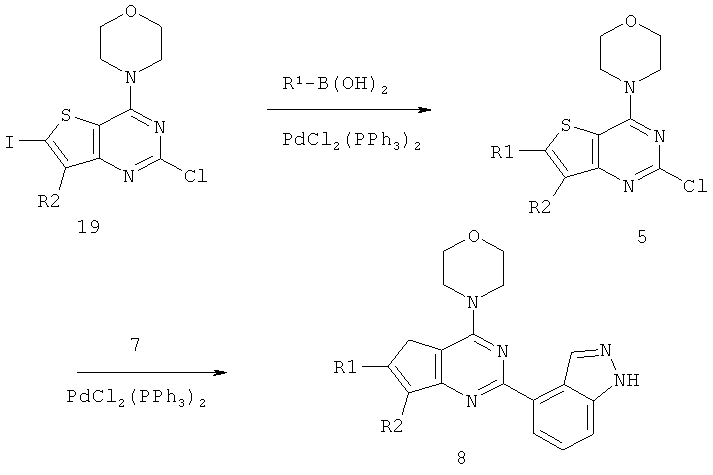
2-Хлор-6-иод-4-морфолинотиено[3,2-d]пиримидин 19 (1 экв.), фенилбороновую кислоту или гетероциклическую бороновую кислоту (R1-B(OH)2, 1,1 экв.) и бис(трифенилфосфин)палладия(II) дихлорид (0,1 экв.) в 1 М водном растворе Ма2СОз (3 экв.) и ацетонитриле (3 экв.) нагревали до 100°С в герметично закрытом микроволновом реакторе в течение 10-40 мин, получая соединение 5. После завершения реакции в тот же сосуд добавляли 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индазол 7 (1,3 экв.) и бис(трифенилфосфин)палладия(И) дихлорид (0,1 экв.). Реакционную смесь нагревали до 150°С в герметично закрытом микроволновом реакторе в течение 10-15 мин. Смесь экстрагировали этилацетатом (3×5 мл). Объединенные органические слои концентрировали, получая неочищенное соединение 8.
Общая методика G. Реакция амидного сочетания
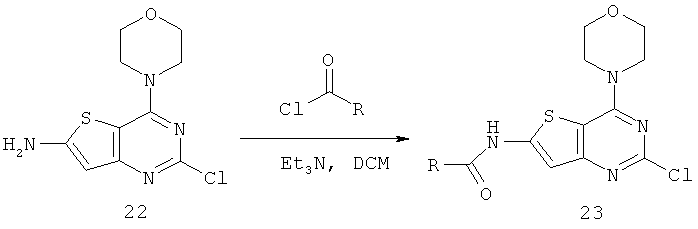
2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-амин 22 (1 экв.), хлорангидрид кислоты (1,5-2 экв.) и триэтиламин (2 экв.) перемешивали в дихлорметане. За ходом реакции до ее завершения следили с помощью LC/MS. Смесь упаривали, получая неочищенный амид 23, который использовали без очистки непосредственно на следующей стадии реакции.
Общая методика Н. Получение ацетамида, бензамидов и сульфонамидов
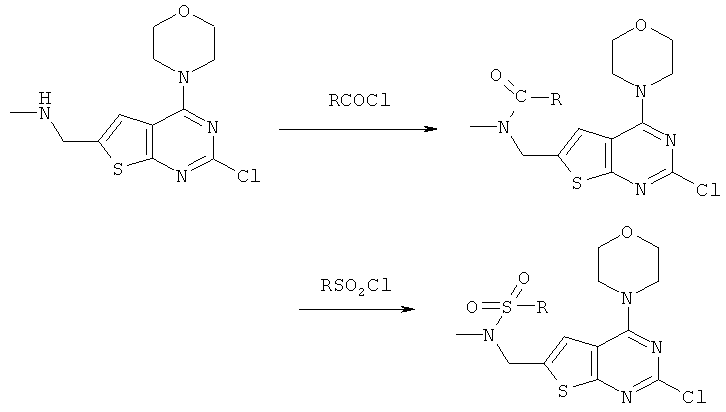
К 0,25-0,40 М раствору 1-(2-хлор-4-морфолинотиено[2,3-d]пиримидин-6-ил)-N-метилметанамина в DCM, охлажденному до 0°С, добавляли 1,5 экв. TEA с последующим добавлением по каплям 1,0-1,5 экв. хлорангидрида алкил- или арил-содержащей кислоты или сульфонилхлорида, растворенного в DCM. Реакционную смесь перемешивают при температуре окружающей среды и полноту прохождения реакции контролируют с использованием LCMS. По завершении реакции реакционный объем увеличивают, добавляя DCM, и к данному раствору добавляют разбавленный водный бикарбонат натрия. Органические и водные слои разделяют. В конце органический слой промывают рассолом и сушат (MgSO4). Обезвоженный органический раствор концентрируют в вакууме и продукт при необходимости очищают хроматографией на диоксиде кремния.
Общая методика I. Реакция амидного сочетания для бензоламина
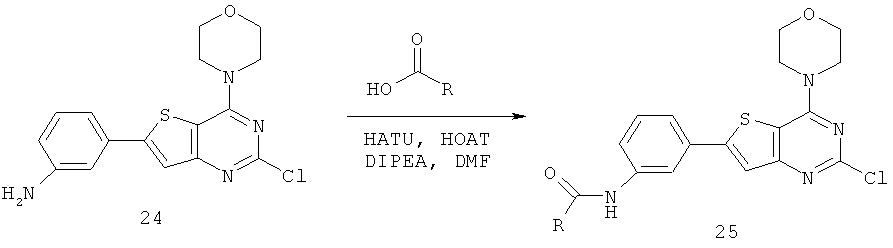
3-(2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-ил)бензоламин 24 (1 экв.), карбоновую кислоту (1,5 экв.), 1-гидрокси-7-азабензотриазол (НОАТ; 0,2 экв.), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU; 1,5 экв.) и N,N-диизопропилэтиламин (2,5 экв.) в DMF перемешивали при комнатной температуре. За ходом реакции до ее завершения следили с помощью LC/MS. Реакционную смесь разбавляли этилацетатом, промывали насыщенным бикарбонатом натрия и рассолом. Органический слой сушили над MgSO4, фильтровали и упаривали, получая продукт амид 25.
Общая методика J. Замещение йода в положении 6 и сочетание по Сузуки в положение 2
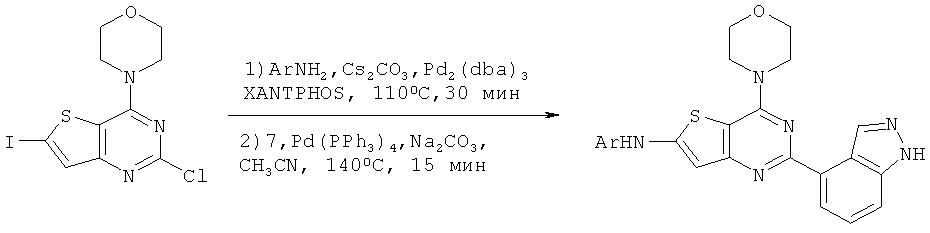
К раствору 2-хлор-6-йод-4-морфолинотиено[3,2-d]пиримидина 19 (0,05 г; 0,13 ммоль) в DMF (1,00 мл) добавляли соответствующий анилин (200 моль%), Cs2CO3 (50 моль%), Pd2(dba)s (5 моль%) и XANTPHOS (4,5-бис(дифенилфосфино)-9,9-диметилксантен; 10 моль%). Реакционную смесь нагревали до 110°С под давлением в микроволновом реакторе Biotage optimizer в течение 30 мин. Полученный раствор концентрировали в вакууме, получая соединение 26, следуя общей методике А.
Общая методика К. Аминоалкилацилирование по положению 6 и сочетание по Сузуки в положение 2
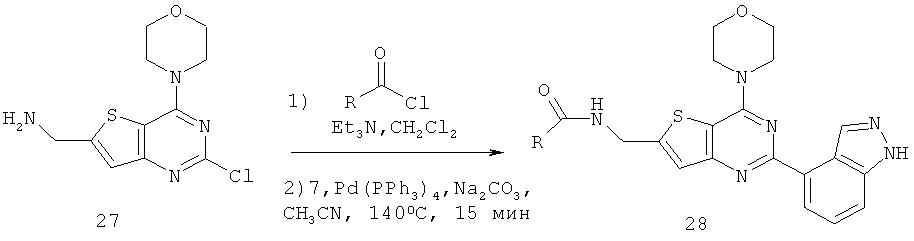
К раствору (2-хлор-4-морфолинотиено[3,2-d]пиримидин-6-ил)метанамина 27 (50 мг; 0,2 ммоль) в CH2Cl2 (4 мл) добавляли Et3N (84 мкл; 0,6 ммоль) и соответствующий хлорангидрид кислоты или его соль HCl (0,3 ммоль). Реакционную смесь перемешивали в течение 18-48 ч при комнатной температуре, затем гасили водой. Водный слой экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Неочищенный 2-хлорсодержащий продукт сочетали с боронатным реагентом 7 в присутствии палладиевого катализатора в соответствии с общей методикой А, получая соединение 28, которое очищали обращенно-фазовой HPLC.
Альтернативно, к раствору (2-хлор-4-морфолинотиено[3,2-d]пиримидин-6-ил)метанамина 27 (111 мг; 0,39 ммоль) в DMF (5 мл) добавляли 2,6-лутидин (48,2 мкл; 0,41 ммоль) и соответствующий хлорангидрид кислоты или его соль HCl (0,39 ммоль). Реакционную смесь перемешивали в течение 18-72 ч при комнатной температуре, затем гасили водой. Водный слой экстрагировали EtOAc. Объединенные органические слои сушили над Мд804 и концентрировали в вакууме. Неочищенный 2-хлорсодержащий продукт сочетали с боронатным реагентом 7 в присутствии палладиевого катализатора в соответствии с общей методикой А, получая 20 мг соединения 28, которое очищали обращенно-фазовой HPLC.
Общая методика L. Аминозамещение на фторпиридине
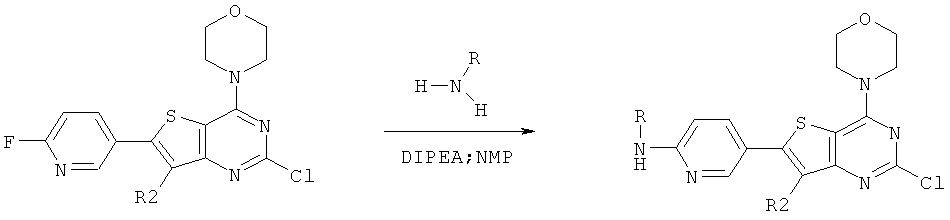
Смесь соединения 2-хлор-6-(6-фторпиридин-3-ил)-4-морфолинотиено[3,2-d]пиримидина или 2-хлор-6-(6-фторпиридин-3-ил)-4-морфолинотиено[2,3-d]пиримидина, примерно четырех эквивалентов первичного или вторичного амина (R представляет собой Н, С1-С12алкил, С2-С8алкенил, С2-С8алкинил, С3-С12карбоциклил, С2-С20гетероциклил, С6-С20арил или C1-С20гетероарил) и примерно двух эквивалентов диизопропилэтиламина в N-метилпирролидине (~0,1 М) нагревают до примерно 130-140°С в герметично закрытом микроволновом реакторе в течение 10~40 мин, затем удаляют летучие соединения в высоком вакууме. Неочищенную смесь очищают флэш-хроматографией, получая промежуточный 2-хлор-6-(6-аминопиридин-3-ил)-4-морфолинотиено[3,2-d]пиримидин или 2-хлор-6-(6-аминопиридин-3-ил)-4-морфолинотиено[2,3-d]пиримидин, который может быть подвергнут сочетанию по Сузуки с (моноциклический гетероарил)-, (конденсированный бициклический гетероцикл- или гетероарил)-боронатным реагентом, следуя общей методике А.
ПРИМЕРЫ
Следующие далее примеры включены с целью иллюстрации данного изобретения. Однако следует понимать, что эти примеры не ограничивают изобретение, а предназначены только для предложения способа практической реализации изобретения. Специалистам в данной области техники будет известно, что описанные химические реакции можно легко адаптировать для получения ряда других ингибиторов Pl3K по изобретению, и подразумевается, что альтернативные способы получения соединений по данному изобретению находятся в пределах объема изобретения. Например, синтез не приведенных в примерах соединений по изобретению может быть успешно проведен в результате модификаций, очевидных специалистам в данной области техники, например, посредством соответственной защиты мешающих групп (protecting interfering groups), путем использования других подходящих реагентов, известных в данной области техники как отличные от уже описанных и/или путем проведения общепринятых модификаций реакционных условий. Альтернативно, могут быть рассмотрены другие реакции, изложенные в данном описании или известные в данной области техники, как применимые для получения других соединений по изобретению.
Пример 1. 2,4-Дихлор-тиено[3,2-d]пиримидин 3
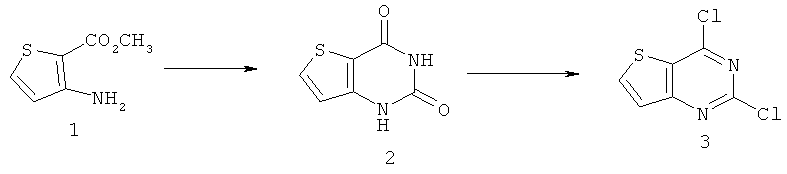
Смесь метил-3-амино-2-тиофенкарбоксилата 1 (13,48 г; 85,85 ммоль) и мочевины (29,75 г; 5 экв.) нагревали при 190°С в течение 2 часов. Горячую реакционную смесь выливали на раствор гидроксида натрия и какое-либо нерастворимое вещество удаляли фильтрованием. Далее смесь подкисляли (HCl, 2 н.), получая 1Н-тиено[3,2-d]пиримидин-2,4-дион 2 в виде белого осадка, который собирали фильтрованием и сушили на воздухе (9,49 г; 66%). 1H ЯМР (400 МГц, d6-DMSO)6.90(1H,d, J=5,2 Гц), 8.10(1H, d, J=5,2 Гц), 11.60-11.10 (2Н, br s).
Смесь 1Н-тиено[3,2-d]пиримидин-2,4-диона 2 (9,49 г; 56,49 ммоль) и оксихлорида фосфора (150 мл) нагревали при температуре дефлегмации в течение 6 ч. Затем реакционную смесь охлаждали и выливали на смесь лед/вода с энергичным перемешиванием, получая осадок. Затем смесь фильтровали, получая 2,4-дихлор-тиено[3,2-d]пиримидин 3 в виде белого твердого вещества (8,68 г; 75%). 1H ЯМР (400 МГц, CDCl3) 7.56 (1Н, d, J=5,5 Гц), 8.13 (1Н, d, J=5,5 Гц).
Пример 2. 2-Хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин 4
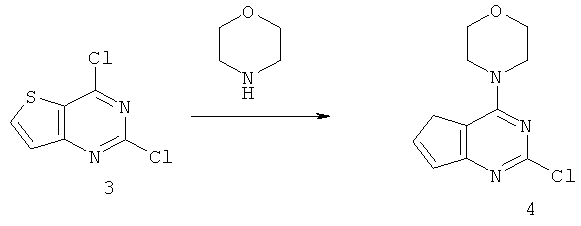
Смесь 2,4-дихлор-тиено[3,2-d]пиримидина 3 (8,68 г; 42,34 ммоль), морфолина (8,11 мл; 2,2 экв.) и МеОН (150 мл) перемешивали при комнатной температуре в течение 1 ч. Затем реакционную смесь фильтровали, промывали водой и МеОН, получая 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин 4 в виде белого твердого вещества (11,04 г; 100%). 1H ЯМР (400 МГц, d6-DMSO) 3.74 (4Н, t, J=4,9 Гц), 3.90 (4Н, t, J=4,9 Гц), 7.40 (1 H, d, J=5,6 Гц), 8.30 (1 H, d, J=5,6 Гц).
Пример 3. 2-Хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегид 10
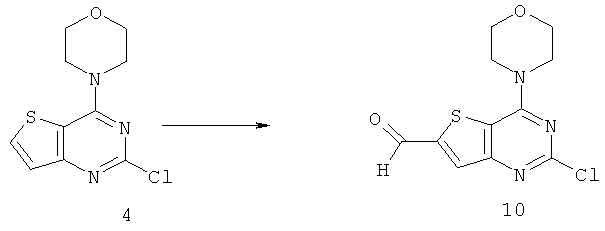
К суспензии 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидина 4 (1,75 г; 6,85 ммоль) в безводном THF (40 мл) при -78°С добавляли 2,5 М раствор н-бутиллития (н-BuLi) в гексане (3,3 мл; 1,2 экв.). После перемешивания в течение 1 ч добавляли безводный DMF (796 мкл; 1,5 экв.). Реакционную смесь перемешивали в течение 1 ч при -78°С и затем медленно нагревали до комнатной температуры. Еще через 2 ч при комнатной температуре реакционную смесь выливали на смесь лед/вода, получая желтый осадок. Его собирали фильтрованием и сушили на воздухе, получая 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегид 10 (1,50 г; 77%). 1H ЯМР (400 МГц, (d6-DMSO) 3.76 (4Н, t, J=4,9 Гц), 3.95 (4Н, t, J=4,9 Гц), 8.28 (1H, s), 10.20 (1H, s).
Пример 3а. 2-Хлор-4-морфолинотиено[2,3-d]пиримидин-6-карбальдегид 33
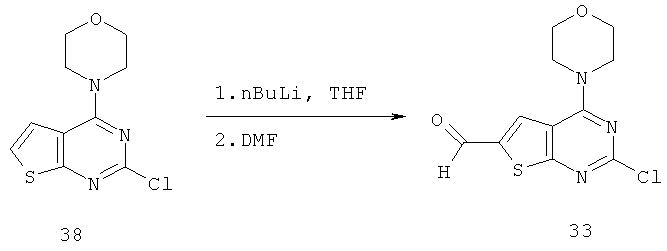
К суспензии 2-хлор-4-морфолинотиено[2,3-d]пиримидина 38 (1,75 г; 6,85 ммоль) в безводном THF (40 мл) при -78°С добавляли 2,5 М раствор н-бутиллития (н-BuLi) в гексане (3,3 мл; 1,2 экв.). После перемешивания в течение 1 ч добавляли безводный DMF (796 мкл; 1,5 экв.). Реакционную смесь перемешивали в течение 1 ч при -78°С и затем медленно нагревали до комнатной температуры. Еще через 2 ч при комнатной температуре реакционную смесь выливали на смесь лед/вода, получая желтый осадок, который собирали фильтрованием и сушили на воздухе, получая 2-хлор-4-морфолинотиено[2,3-d]пиримидин-6-карбальдегид 33 (1,50 г). MS (Q1 (первый квадруполь)) 284 (М+).
Пример 3b. 2-Хлор-6-иод-7-метил-4-морфолинотиено[3,2-d]пиримидин 41
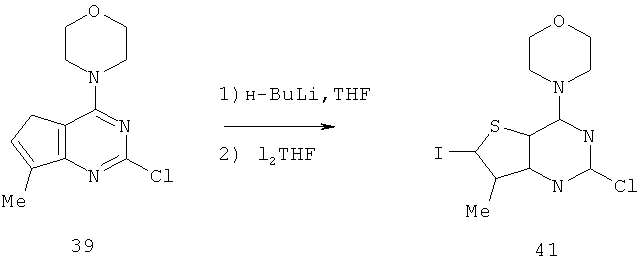
К раствору 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидина 39 (3,0 г; 11,1 ммоль; полученного в соответствии с методикой для синтеза 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидина, но начиная с этилового эфира 3-амино-4-метил-тиофен-2-карбоновой кислоты) в THF (60 мл) при -78°С добавляли н-BuLi (8,9 мл; 2,5 М в Et2O). Полученную суспензию нагревали до -40°С и перемешивали в течение 50 мин. Затем реакционную смесь охлаждали до -78°С и добавляли раствор l2 (5,6 г; 22,2 ммоль) в THF (30 мл). Раствор нагревали до комнатной температуры и перемешивали в течение 5 ч. Реакцию гасили добавлением воды. Органический слой отделяли и водный слой экстрагировали CH2Cl2. Объединенные органические слои промывали насыщенным водным Na2S2O3, сушили над Na2S2O4, фильтровали и концентрировали в вакууме, получая 2-хлор-6-иод-7-метил-4-морфолинотиено[3,2-d]пиримидин 41 (3,8 г; выход 84%).
Пример 3с. 4-(2-Хлор-6-(пиперазин-1 -илметил)тиено[3,2-d]пиримидин-4-ил)морфолин 30
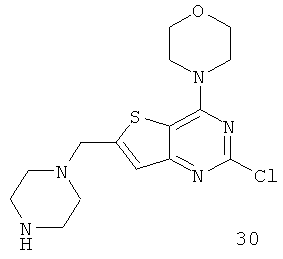
Смесь 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегида 10 (3,5 г), 1-ВОС-пиперазина (2,76 г) и триметилортоформиата (4,05 мл) перемешивали в 1,2-дихлорэтане (300 мл) в течение 1 ч при комнатной температуре. К ней добавляли триацетоксиборгидрид натрия (3,92 г) и реакционную смесь перемешивали в течение 24 часов при комнатной температуре. Затем смесь гасили рассолом, экстрагировали дихлорметаном, сушили (MgSO4) и растворитель удаляли в вакууме. Остаток очищали, используя флэш-хроматографию и получая трет-бутиловый эфир 4-(2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-илметил)-пиперазин-1-карбоновой кислоты (3,4 г). В результате обработки HCl в смеси дихлорметан/метанол получали 4-(2-хлор-6-(пиперазин-1-илметил)тиено[3,2-d]пиримидин-4-ил)морфолин 30.
Пример 3d. (2-Хпор-4-морфолинотиено[2,3-d]пиримидин-6-ил)-N-метил-метанамин 34
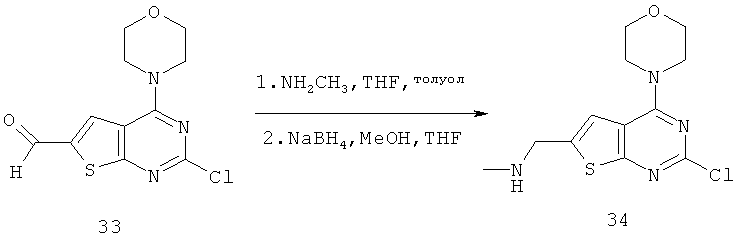
К 2-хлор-4-морфолинотиено[2,3-d]пиримидин-6-карбальдегиду 33 (2,0 г) в 50 мл толуола и 50 мл THF добавляли 20 мл 40%-ного метиламина в N2O. Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов. Растворители удаляли в вакууме, остаток растворяли в 50 мл МеОН и 50 мл THF и порциями добавляли NaBH4. Эту реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов и полноту реакции подтверждали, используя LCMS. Растворители удаляли в вакууме и неочищенный продукт очищали флэш-хроматографией (EtOAc/EtOH), получая 1,12 г соединения 34 (выход 53%). MS (Q1) 300 (М+).
Пример 3е. (2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-ил)-N-метил-метанамин 35
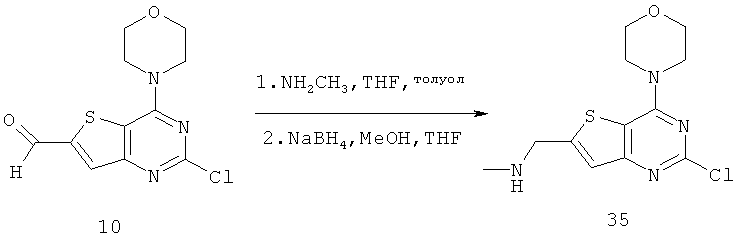
2-Хлор-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид 10 (2,0 г) растворяли в 50 мл толуола и 50 мл THF с последующим добавлением 20 мл 40%-ного метиламина в H2O. Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов. Растворители удаляли в вакууме, остаток растворяли в 50 мл метанола и 50 мл THF и порциями добавляли NaBH4. Эту реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов и полноту реакции подтверждали, используя LCMS. Растворители удаляли в вакууме и неочищенный продукт очищали флэш-хроматографией (EtOAc/EtOH), получая 1,12 г соединения 35 (выход 53%). MS (Q1) 300 (М+).
Пример 3f. (2-Хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)-N-метилметанамин 37
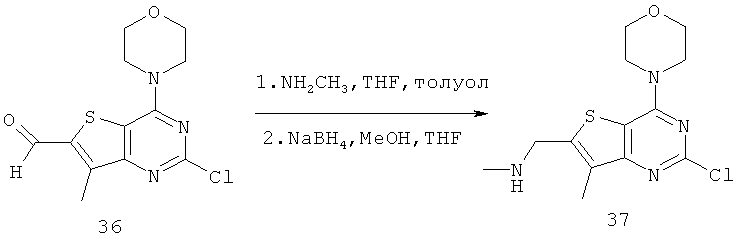
2-Хлор-7-метил-4-морфолинотиено-[3,2-d]пиримидин-6-карбальдегид 36 растворяли в 20 мл толуола и 20 мл THF с последующим добавлением 15 мл 40%-ного метиламина в H2O и реакционную смесь перемешивали в течение 24 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в 30 мл метанола и 30 мл THF с последующим добавлением NaBH4. Реакционную смесь перемешивали при комнатной температуре в течение по меньшей мере 24 часов и образование продукта подтверждали, используя LCMS. Растворители удаляли в вакууме и неочищенный продукт очищали флэш-хроматографией, получая 2,53 г (2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)-N-метилметанамина 37 (выход 70%). MS (Q1) 314 (М+).
Пример 4. 4-(2-Хлор-6-((4-(метилсульфонил)пиперазин-1-ил)метил)-5 тиено[3,2-d]пиримидин-4-ил)морфолин 31
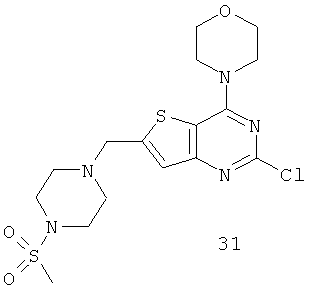
В результате взаимодействия между N-BOC-пиперазином и метансульфонилхлоридом в дихлорметане и триэтиламине получали трет-бутиловый эфир 4-метансульфонил-пиперазин-1-карбоновой кислоты. В результате отщепления защитной группы ВОС с использованием HCl (2 М) в дихлорметане получали 1-метансульфонил-пиперазина соль HCl.
Смесь 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегида 10 (1,00 г), 1-метансульфонил-пиперазина (750 мг) и триметилортоформиата (3,80 мл) перемешивали в 1,2-дихлорэтане (30 мл) в течение 6 ч при комнатной температуре. К ней добавляли триацетоксиборгидрид натрия (900 мг) и реакционную смесь перемешивали в течение 24 ч при комнатной температуре. Затем смесь гасили рассолом, экстрагировали дихлорметаном, сушили (MgSO4) и растворитель удаляли в вакууме. Остаток растирали с горячим этилацетатом, получая 4-(2-хлор-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин 31 в виде белого твердого вещества (1,01 г).
Пример 5. 2-Хлор-6-(4-метансульфонил-пиперазин-1-илметил)-4-морфолин-4-ил-тиено[2,3-d]пиримидин 32
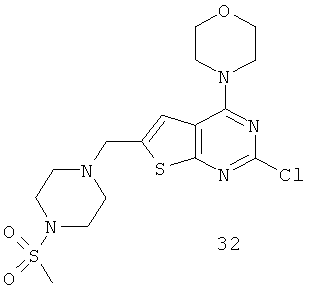
В результате взаимодействия между 1-метансульфонил-пиперазином, 5 солью HCl, и 2-хлор-4-морфолин-4-ил-тиено[2,3-d]пиримидин-6-карбальдегидом 33 с использованием общей методики С получали 2-хлор-6-(4-метансульфонил-пиперазин-1-илметил)-4-морфолин-4-ил-тиено[2,3-d]пиримидин.
Пример 6. 4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)-1 Н-индазол 7 - путь 1
Промежуточное соединение 7 получали в соответствии со способами из US 2008/0076768; US 2008/0076758; WO 2006/046031, включеными в данное описание путем ссылки.
Пример 8. 1-(Тетрагидро-2Н-пиран-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-15 диоксаборолан-2-ил)-1Н-индазол 40
Промежуточное соединение 40 получали в соответствии со способами из US 2008/0039459; US 2008/0076768; US 2008/0076758; WO 2006/046031, включенными в данное описание путем ссылки.
Пример 10. 2-(1 Н-Индазол-4-ил)-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегид 11
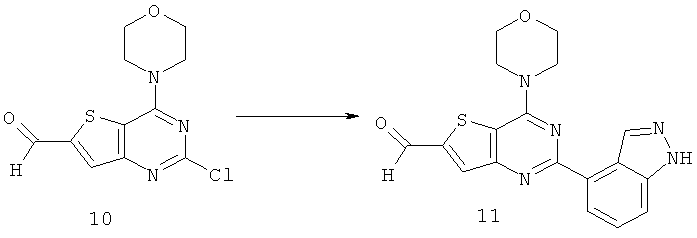
Смесь 2-хлор-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегида 10 (100 мг; 0,35 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индазола 7 (95 мг; 0,39 ммоль) и карбоната натрия (112 мг) суспендировали в толуоле (2,5 мл), этаноле (1,5 мл) и воде (0,7 мл). К ней добавляли бис(трифенилфосфин)палладия(И) хлорид (13,5 мг) и реакционный сосуд продували аргоном. Реакционную смесь облучали микроволнами при 120°С в течение 1 ч и затем распределяли между DCM и водой, органический слой промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный остаток очищали, используя флэш-хроматографию и получая 2-(1Н-индазол-4-ил)-4-морфолин-4-ил-тиено[3,2-d]пиримидин-6-карбальдегид 11 (97 мг).
Пример 11. 4-(2-(1Н-Индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин (соединение формулы Ia, GDC-0941):
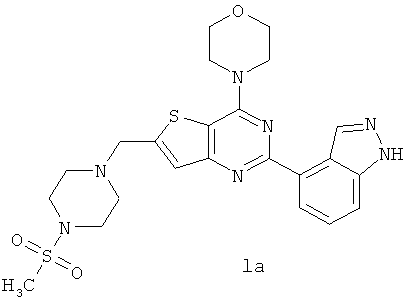
Смесь 4-(2-хлор-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолина 31 из Примера 4 (2,00 г), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индазола 7 (2,26 г), толуола (24 мл), этанола (12 мл), воды (6 мл), карбоната натрия (1,72 г) и PdCl2(PPh3)2 (325 мг) нагревали до 130°С в микроволновом реакторе в течение 90 минут (US 2008/0076768; WO 2006/046031, включенные в данное описание путем ссылки).
Реакционную смесь охлаждали, разбавляли хлороформом, промывали рассолом, сушили (MgSO4) и растворитель удаляли в вакууме. Остаток очищали, используя флэш-хроматографию (этилацетат, затем смесь 5% этилацетата/метанол) и затем в результате растирания с эфиром получали соединение формулы Ia, GDC-0941 (1,4 г). Данные MS: (ESI+(электрораспылительная ионизация, режим положительных ионов)): МН+514. Данные ЯМР: (CDCl3): 2.67-2.71 (4Н, m), 2.81 (3Н, s), 3.29-3.33 (4H, m), 3.89 (2H, s), 3.89-3.93 (4H, m), 4.08-4.12 (4H, m),7.41 (1H, s), 7.51 (1H, t, J=7,2 Гц), 7.60 (1Н, d, J=8,3 Гц), 8.28 (1H, d, J=7,5 Гц), 9.02 (1H, s), 10.10 (1H, br).
Пример 12. 4-(2-(1Н-Индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[2,3-d]пиримидин-4-ил)морфолин (соединение формулы IIa):
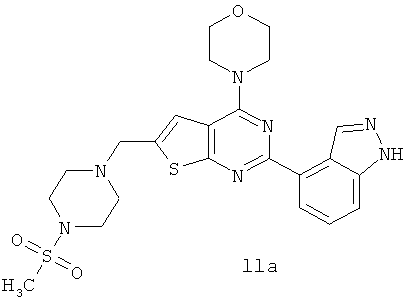
2-Хлор-6-(4-метансульфонил-пиперазин-1-илметил)-4-морфолин-4-ил-тиено[2,3-d]пиримидин 32 из Примера 5 приводили во взаимодействие с 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индазолом 7 и с использованием общей методики А получали соединение формулы IIa, которое очищали, используя флэш-хроматографию (US 2008/0076758; WO 2006/046031, включенные в данное описание путем ссылки). 1H ЯМР (400 МГц, CDCl3): 2.67 (m, 4H, 2×CH2), 2.81 (s, 3Н, СН3), 3.30 (m, 4H, 2×CH2), 3.83 (s, 2H, CH2), 3.92-3.94 (m, 4H, 2×CH2), 3.98-4.00 (m, 4H, 2×CH2), 7.17 (s, H, ArH), 7.50 (t, H, ArH, J=7,81 Гц), 7.59 (d, H, ArH, J=8,31 Гц), 8.31 (d, H, ArH, J=6,98 Гц), 10.12 (s br, H, NH). МН+=514,10.
Пример 12а. (S)-1-(4-((2-(2-Аминопиримидин-5-ил)-7-метил-4-морфолино-тиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он (соединение формулы Ib):
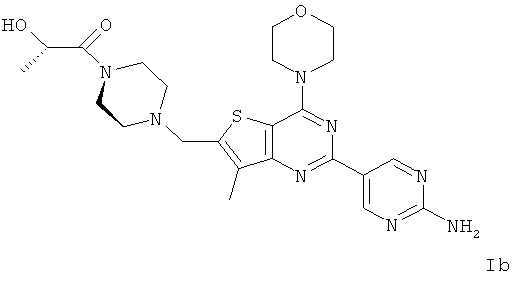
2-Хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид 36 (495 мг) приводили во взаимодействие с Вос-пиперазином с использованием общей методики В-3, получая трет-бутил-4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-карбоксилат.
трет-Бутил-4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)-метил)пиперазин-1-карбоксилат (777 мг) подвергали обработке с использованием общей методики Е, получая соль HCl 2-хлор-7-метил-4-морфолино-6-((пиперазин-1-ил)метил)тиено[3,2-d]пиримидина. Соль HCl 2-хлор-7-метил-4-морфолино-6-((пиперазин-1-ил)метил)тиено[3,2-d]пиримидина (590 мг) приводили во взаимодействие с молочной кислотой с использованием общей методики В-2, получая (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)-пиперазин-1-ил)-2-гидроксипропан-1-он.
(S)-1-(4-((2-Хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)-пиперазин-1-ил)-2-гидроксипропан-1-он (60 мг) приводили во взаимодействие с 50 мг 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-амина с использованием общей методики А-2, получая 10 мг соединения формулы Ib (WO 2008/070740, включено путем ссылки). MS (Q1) 499,3 (М)+.
Пример 13. Анализ связывания с р110α (альфа) Pl3K
Анализы связывания: начальные эксперименты по поляризации проводили на Analyst НТ 96-384 (Molecular Devices Corp, Sunnyvale, CA.). Образцы для измерения аффинности с использованием поляризации флуоресценции готовили, добавляя серийные разведения (1:3) раствора p110альфа Pl3K (Upstate Cell Signaling Solutions, Charlottesville, VA), начиная с конечной концентрации 20 мкг/мл в буфере для измерения поляризации (10 мМ Трис, рН 7,5; 50 мМ NaCl; 4 мМ MgCl2; 0,05% CHAPS (3-[(3-холамидопропил)-диметиламмонио]-1-пропан-сульфонат) и 1 мМ DTT (дитиотреит)), к PIP2 в конечной концентрации 10 мМ (Echelon-Inc., Salt Lake Clty, UT.). После инкубации в течение 30 минут при комнатной температуре реакции останавливали, добавляя GRP-1 и зонд PIP3-TAMRA (Echelon-lnc., Salt Lake City, UT.) в конечной концентрации 100 нМ и 5 нМ, соответственно. Прочитывание выполняли, используя стандартные фильтры с ограниченной полосой пропускания для родамина (Авозб.=530 нМ; Лисп.=590 нМ) в 384-луночных черных планшетах на малые объемы Proxiplates (PerkinElmer, Wellesley, MA.) Строили график зависимости величин поляризации флуоресценции от концентрации белка и с использованием аппроксимации данных к 4-х параметрическому уравнению с применением программного обеспечения KaleidaGraph (программное обеспечение для анализа синергического действия, Reading, PA) получали величины ЕС50. В этом эксперименте также устанавливали соответствующую концентрацию белка для использования в последующих экспериментах по конкуренции с ингибиторами.
Величины IC50 для ингибиторов определяли, добавляя 0,04 мг/мл p110альфа Pl3K (конечная концентрация) вместе с PIP2 (конечная концентрация 10 мМ) в лунки, содержащие серийные разведения (1:3) антагонистов в растворе АТФ в конечной концентрации 25 мМ (Cell Signaling Technology, Inc., Danvers, MA) в буфере для измерения поляризации. После инкубации в течение 30 минут при комнатной температуре реакции останавливали, добавляя GRP-1 и зонд PIP3-TAMRA (Echelon-Inc., Salt Lake City, UT.) в конечной концентрации 100 нМ и 5 нМ, соответственно. Прочитывание выполняли, используя стандартные фильтры с ограниченной полосой пропускания для родамина (Авозб.=530 нМ; Лисп.=590 нМ) в 384-луночных черных планшетах на малые объемы Proxiplates (PerkinElmer, Wellesley, MA.) Строили график зависимости величин поляризации флуоресценции от концентрации антагонистов и с использованием аппроксимации данных к 4-х параметрическому уравнению с применением программного обеспечения Assay Explorer (MDL, San Ramon, CA.) получали величины ЕС50.
Альтернативно, ингибирование Pl3K определяли в радиометрическом анализе, используя очищенный рекомбинантный фермент и АТФ в концентрации 1 мкМ. Выполняли серийные разведения соединения в 100%-ном DMSO. Реакционную смесь с киназой инкубировали в течение 1 ч при комнатной температуре и реакцию останавливали, добавляя PBS. Затем определяли величины IC50, используя аппроксимацию зависимости доза-ответ к сигмоидальной кривой (переменная крутизна).
Пример 14. Анализ клеточной пролиферации in vitro
Эффективность соединений формулы I или II измеряли в анализе клеточной пролиферации, применяя следующий протокол (Promega Corp.Technical Bulletin ТВ288; Mendoza et al. (2002) Cancer Res. 62: 5485-5488). Реагенты для анализа Cell-Titer Glo и протокол имеются в продаже (Promega). В анализе оценивается способность соединений проникать в клетки и ингибировать клеточную пролиферацию. Принцип анализа заключается в определении присутствующего количества жизнеспособных клеток посредством количественного определения присутствующего АТФ. Cell-Titer Glo представляет собой реагент, используемый для такого количественного определения. Это гомогенный анализ, в котором добавление Cell-Titer Glo приводит к лизису клеток и генерации люминесцентного сигнала в результате люциферазной реакции. Люминесцентный сигнал пропорционален количеству присутствующего АТФ.
Клетки: см. Фиг.1А-С для клеточных линий и типа опухоли.
Планшеты для DMSO и сред: 96-луночные полипропиленовые планшеты с коническим дном от Nunc (№по каталогу 249946).
Планшеты для клеток: 384-луночные черные ТС-планшеты (планшеты для тканевых культур) с прозрачным дном (для улучшенной микрофотосъемки) с крышкой от Falcon (353962).
Культуральная клеточная среда: из RPMI (Roswell Park Memorial Institute) или DMEM (модифицированная Дульбекко среда Игла) с высоким содержанием глюкозы, 10% фетальной сыворотки теленка, 2 мМ L-глутамин, P/S.
Cell Titer-Glo: Promega (№по каталогу G7572).
Методика
1-е сутки - посев клеток в планшеты для клеток, собрать клетки, провести посев РС3-клеток в количестве 1000 клеток в 54 мкл на лунку в 384-луночные планшеты для клеток для выполнения анализа через 3 суток. Инкубировать в течение ночи (O/N) при 37°С, 5% CO2.
2-е сутки - добавить к клеткам лекарственное средство в виде разведения соединений в планшетах для DMSO (серийные разведения 1:2 для 9 точек), а именно: добавить по 20 мкл соединений в концентрации 10 мМ во 2-ю колонку 96-луночного планшета. Выполнить серийные разведения 1:2 в поперечном направлении планшета (10 мкл+10 мкл 100%-ного DMSO) суммарно для 9 точек, используя Precision. В планшеты для сред (для получения разведения 1:50) добавить по 147 мкл сред во все лунки. Перенести по 3 мкл смеси DMSO+соединение из каждой лунки планшета для DMSO в каждую соответствующую лунку на планшете для сред, используя Rapidplate. Для исследований комбинации 2-х лекарственных средств (2 drug combo) перенести одно лекарственное средство в виде 1,5 мкл смеси DMSO+соединение из каждой лунки планшета для DMSO в каждую соответствующую лунку планшета для сред, используя Rapidplate. Затем перенести другое лекарственное средство (1,5 мкл) в планшет для сред.
Добавление лекарственного средства к клеткам в планшете для клеток (разведение 1:10), а именно: добавить по 6 мкл смеси среды+соединение непосредственно к клеткам (клетки уже находятся в 54 мкл среды). Инкубировать 3 суток при 37°С, 5% CO2 в инкубаторе, который не должен часто открываться.
5-е сутки - окраска планшетов: разморозить буфер Cell Titer Glo при комнатной температуре. Извлечь планшеты для клеток с 37°С и уравновесить до комнатной температуры в течение примерно 30 минут. Добавить буфер Cell Titer Glo к субстрату Cell Titer Glo (бутылка к бутылке). Добавить по 30 мкл реагента Cell Titer Glo в каждую лунку с клетками. Поместить на планшетный шейкер примерно на 30 минут. Снять показания люминесценции, используя планшетный ридер Analyst HT (полсекунды на лунку).
Анализы жизнеспособности клеток и анализы действия комбинаций: клетки высевали в количестве 1000-2000 клеток/лунка в 384-луночные планшеты на 16 ч. На вторые сутки делали девять серийных (1:2) разведении соединений в DMSO в 96-луночном планшете. Кроме того, соединения разводили в ростовых средах, используя робот Rapidplate (Zymark Corp., Hopkinton, MA). Разведенные соединения затем добавляли в четырех повторах в лунки 384-луночных планшетов для клеток и инкубировали при 37°С и 5% CO2. Через 4 суток измеряли относительные количества жизнеспособных клеток по люминесценции, используя Cell-Titer Glo (Promega) в соответствии с инструкциями производителя, и снимали показания на ридере Wallac Multilabel (PerkinElmer, Foster Clty). Величины ЕС50 рассчитывали, используя программное обеспечение Prism 4.0 (GraphPad, San Diego). Лекарственные средства при проведении анализов комбинаций вводили, начиная от концентраций 4Х EC50. В случаях, когда EC50 лекарственного средства составляла более 2,5 мкМ, самая высокая используемая концентрация составляла 10 мкМ. Во всех анализах ингибиторы Pl3K и химиотерапевтические агенты добавляли одновременно или по отдельности с интервалом 4 часа (одно перед другим).
Дополнительный типичный in vitro анализ клеточной пролиферации включает следующие стадии.
1. Аликвоту в 100 мкл клеточной культуры, содержащую примерно 104 клеток (см. Фиг.1А-С для клеточных линий и типа опухоли) в среде, помещали в каждую лунку 384-луночного планшета с непрозрачными лунками.
2. Готовили контрольные лунки, содержащие среду и не содержащие клеток.
3. В экспериментальные лунки добавляли соединение и инкубировали в течение 3-5 суток.
4. Планшеты уравновешивали до комнатной температуры в течение приблизительно 30 минут.
5. К объему культуральной клеточной среды, присутствующей в каждой лунке, добавляли равный объем реагента CellTiter-Glo.
6. Содержимое перемешивали в течение 2 минут на орбитальном встряхивателе с целью индукции лизиса клеток.
7. Планшет инкубировали при комнатной температуре в течение 10 минут для стабилизации сигнала люминесценции.
8. Люминесценцию регистрировали и представляли графические изображения в виде RLU (относительных световых единиц).
Альтернативно, клетки засевали при оптимальной плотности в 96-луночный планшет и инкубировали в течение 4 суток в присутствии тестируемого соединения. После этого в среду для анализа добавляли Alamar Blue™ и клетки инкубировали в течение 6 ч, после чего снимали показания при возбуждении при 544 нм, испускании при 590 нм. Величины ЕС50 расчитывали, используя аппроксимацию зависимости доза-ответ к сигмоидальной кривой.
Пример 15. FACS-анализ с использованием аннексина V/PI
Клетки (2 х 106) помещали в чашку (10 см) для культур тканей. Через 16 часов клетки подвергали воздействию 0,1%-ного DMSO (контроль) или соединения формулы Ia (содержащего 0,1% DMSO) в течение 48 часов. Затем клетки удаляли из чашки, используя трипсин, и промывали один раз PBS. Для детекции апоптоза клетки (МВ361, PC3) ресуспендировали в буфере для связывания (10 мМ Hepes(N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота)/МаОН [рН 7,4], 140 мМ NaCl и 2,5 мМ CaCl2) в концентрации 1 х 106 клеток/мл и непосредственно окрашивали 5 мкл смеси аннексии V/FITC (флуоресцеинизотиоцианат) (BD Pharmingen; Franklin Lakes, NJ) и 500 мкл раствора иодида пропидия (PI), содержащего PI (50 мкг/мл, Sigma), РНКазу (раствор 0,2 мг/мл, Sigma) и 0,1% Тритона-Х (Sigma) в PBS. Смесь инкубировали при комнатной температуре в течение 30 минут и клетки анализировали проточной цитометрией (BD Biosciences; San Jose, CA).
Пример 16. Ацинарный морфогенез HER2+ВТ474М1-клеток в ЗО-культуре
Биологическую активность ингибиторов Pl3K и большинства эффективных терапевтических комбинаций ингибиторов Pl3K и HER-семейства в HER2-амплифицирующих раковых клетках молочной железы определяли в 3D-культуре клеток, приготовленной с использованием "способа с наложением". Соединение формулы Ia использовали в виде суспензии в диметилсульфоксиде в концентрации 50 мМ. Херегулин-бета-1177-244 (далее обозначаемый как HRG) готовили для хранения в концентрации 225,8 мкМ. ВТ474М1-клетки обрабатывали 20 мкг/мл трастузумаба, 25 мкг/мл пертузумаба, 250 нМ соединением формулы Ia или 250 нМ соединением формулы IIa в 3D-культуре. Жизнеспособность клеток определяли путем измерения уровней АТФ в клетках, используя люминесцентный анализ жизнеспособности клеток Cell Titer-Glo (Promega). Снятие показаний основывалось на усреднении 3 повторов для каждого из условий анализа. Морфогенез оценивали количественно путем подсчета степени образования бугорков, используя фазово-контрастные изображения. 100 ацинусов для каждого из условий анализа продолжительностью обычно 9 суток подсчитывали на предмет количества бугорков, образованных на поверхности ацинусов. Культуральные среды обновляли каждые 3 суток.
При снятии кривой титрования ВТ474М1-клетки обрабатывали постепенно возрастающими дозами соединения формулы Ia для определения оптимальной концентрации, которая эффективно ингибирует маркеры, находящиеся ниже Pl3K (АКТ), и приводит к общему снижению жизнеспособности клеток (Cell Titer-Glo). ВТ474М1-клетки происходили из родительской ВТ474-клеточной линии, приобретенной через Американскую коллекцию типовых культур. Клетки пассировали на мышах для получения жизнеспособной эстроген-зависимой клеточной линии, подходящей для исследований in vitro и in vivo.
Все 3D-анализы проводили, используя "способ с наложением", как описано в (Lee et al. (2007) Nat. Methods. 4: 359-65). 48-луночные планшеты равномерно покрывали на льду 100 мкл Матригеля (BD Biosciences) с пониженным содержанием ростовых факторов. Затем планшеты переносили в инкубатор на 37°С на 20 минут, давая возможность матриксу заполимеризоваться. Собирали ВТ474М1-клетки и производили посев по 10000 клеток/лунка в покрытые Матригелем планшеты. Клетки культивировали в ростовой среде, дополненной 5% Матригеля и соответствующими лекарственными средствами или лигандом. Продолжительность анализов обычно составляла 9-10 суток, и ростовую среду заменяли каждые 3 суток. Фазово-контрастные изображения регистрировали с использованием цифровой камеры Sony (DXC-S500), приспособленной к микроскопу Leica DMIL. Жизнеспособность клеток определяли путем измерения уровней АТФ в клетках, используя люминесцентный анализ жизнеспособности клеток Cell Titer-Glo (Promega). Показания снимали с использованием люминометра и в расчет брали усреднение 3 повторов для каждого из условий анализа. Морфогенез оценивали количественно путем подсчета степени образования бугорков, используя фазово-контрастные изображения. Одну сотню ацинусов для каждого из условий анализа подсчитывали на предмет количества бугорков, образованных на поверхности ацинусов. Проводили подсчет баллов и группировали в категории: 0-1, 2-3 или ≥4 бугорка на все ацинусы.
Таблица 1. Комбинации ингибиторов Pl3K, оцененные в 3D-клеточной культуре
|
Пример 17. Опухолевый ксенотрансплантат in vivo
Животные, подходящие для трансгенных экспериментов, могут быть получены из стандартных коммерческих источников. Группам самок бестимусных и бесшерстных мышей линии CD-1 (Charles River Laboratory) подкожно в задний пах вводили имплантат, содержащий 20 миллионов раковых клеток молочной железы MDA-MB-361.1 (мутант по Pl3K) с Матригелем и 0,36 мг эстрогена на одну мышь. Группам самок nu/nu мышей линии NMRI (Janvier) вводили имплантат в виде фрагментов (150 мм3) первичной опухоли молочной железы MAXF 401 (Her2+/ER+/PR+) или MAXF 1162 (Her2+/ER+/PR+) (полученных биопсией непосредственно у двух отдельных пациентов с раком молочной железы) с Матригелем и 0,36 мг драже эстрогена на одну мышь. Группам самок nu/nu мышей линии NMRI (HarIan Labs) вводили имплантат в виде 10 миллионов раковых клеток молочной железы MCF-7 (мутант по Pl3K) с Матригелем и 0,36 мг драже эстрогена на одну мышь. Группам самок бестимусных nu/nu мышей (Charles River Laboratory) вводили имплантат в виде 15 миллионов клеток немелкоклеточного рака легкого NCl-H2122 (мутант K-ras) и Матригелем на одну мышь. В мышиные ксенотрансплантаты вводили на 1-е сутки лекарственное средство, лекарственную комбинацию или разбавитель в соответствии со схемой, определенной для каждой опухолевой модели. Доцетаксел вводили внутривенно, В20-1.4 вводили внутрибрюшинно и соединение формулы Ia и IIа доставляли внутрь путем перорального чреззондового кормления. Размер опухолей регистрировали дважды в неделю в ходе исследования. Массы мышей также регистрировали два раза в неделю и за мышами осуществляли регулярное наблюдение. Объем опухоли измеряли в двух направлениях (по длине и ширине), используя циркуль Ultra Cal-IV (модель 54-10-111; Fred V. Fowler Co., Inc.; Newton, MA), и анализировали, используя Excel, версия 11.2 (Microsoft Corporation; Redmond, WA). Графики, отражающие ингибирование опухоли, строили с использованием KaleidaGraph, версия 3.6 (Synergy Software; Reading, PA). Объем опухоли рассчитывали, используя формулу:
Размер опухоли (мм3)=(результат максимального измерения х результат минимального измерения2) × 0,5.
Массы тела животных измеряли, используя весы Adventurera Pro AV812 (Ohaus Corporation; Pine Brook, NJ). Графики строили с использованием KaleidaGraph, версия 3.6. Процентное изменение массы рассчитывали, используя формулу:
Процентное изменение массы в группе=(1 - (начальная масса/новая масса))×100.
Мышей, объем опухоли которых превышал 2000 мм3 или уменьшение массы тела которых составляло более 20% от их начальной массы, немедленно подвергали эвтаназии в соответствии с регулирующим руководством.
Процент ингибирования опухолевого роста (% INH) на момент завершения исследования (EOS) рассчитывали, используя формулу:
% INH=100×(средний объем опухолей на EOS у животных, получавших разбавитель - средний объем опухолей на EOS у животных, получавших лекарственное средство)/средний объем опухолей на EOS у животных, получавших разбавитель.
Частоту возникновения опухолей (Tl) определяли на основании количества измеряемых опухолей, оставшихся в каждой группе в конце исследования. Частичный ответ (PR) определяли как уменьшение в объеме опухоли, составляющее >50%, но <100% по сравнению с исходным объемом опухоли, наблюдаемое на какой-либо день исследования. Полный ответ (CR) определяли как 100% уменьшения в объеме опухоли по сравнению с начальным объемом опухоли, наблюдаемое на какой-либо день исследования. Данные анализировали и определяли р-величины, используя тест Дуннета со статистическим программным обеспечением JMP, версия 5.1.2 (SAS Institute; Cary, NC). Величины индивидуальных объемов опухолей на момент завершения исследования и среднего объема опухоли ± SEM (стандартная ошибка среднего) рассчитывали, используя статистическое программное обеспечение JMP, версия 5.1.2. Данные для массы тела наносили на график, основываясь на среднем процентном изменении от начальных масс тела ± SEM.
Пример 18. Анализ индукции фосфо-АКТ
Клетки высевали в количестве 5×105 клеток на лунку в 6-луночный планшет для тканевых культур и растили в течение ночи. Клетки обрабатывали химиотерапевтическим агентом в концентрации, равной ЕС80. После обработки клетки промывали один раз холодным PBS и подвергали лизису в 1Х буфере для экстракции клеток от Biosource (Carlsbad, СА), дополненном протеазными ингибиторами (Roche, Mannheim, Germany), 1 мМ PMSF (фенилметилсульфонил-фторид) и коктейлями 1 и 2 фосфатазных ингибиторов от Sigma (St. Louis, МО). Определение концентрации белка выполняли, используя набор для анализа белков Pierce BCA (Rockford, IL). Уровни pAkt (Ser473) и общей Akt оценивали, используя наборы гранул от Biosource (Carlsbad, СА) и систему Luminex Bio-Plex (Bio-Rad, Hercules, СА).
Приведенное выше описание рассматривается только в качестве иллюстрации принципов данного изобретения. Кроме того, поскольку многочисленные модификации и изменения будут легко очевидны специалистам в данной области техники, не желательно ограничивать изобретение точными показанными конструкцией и способом, которые описаны выше. Соответственно, все подходящие модификации и эквиваленты будут считаться попадающими в объем изобретения, который определен следующей ниже формулой изобретения.
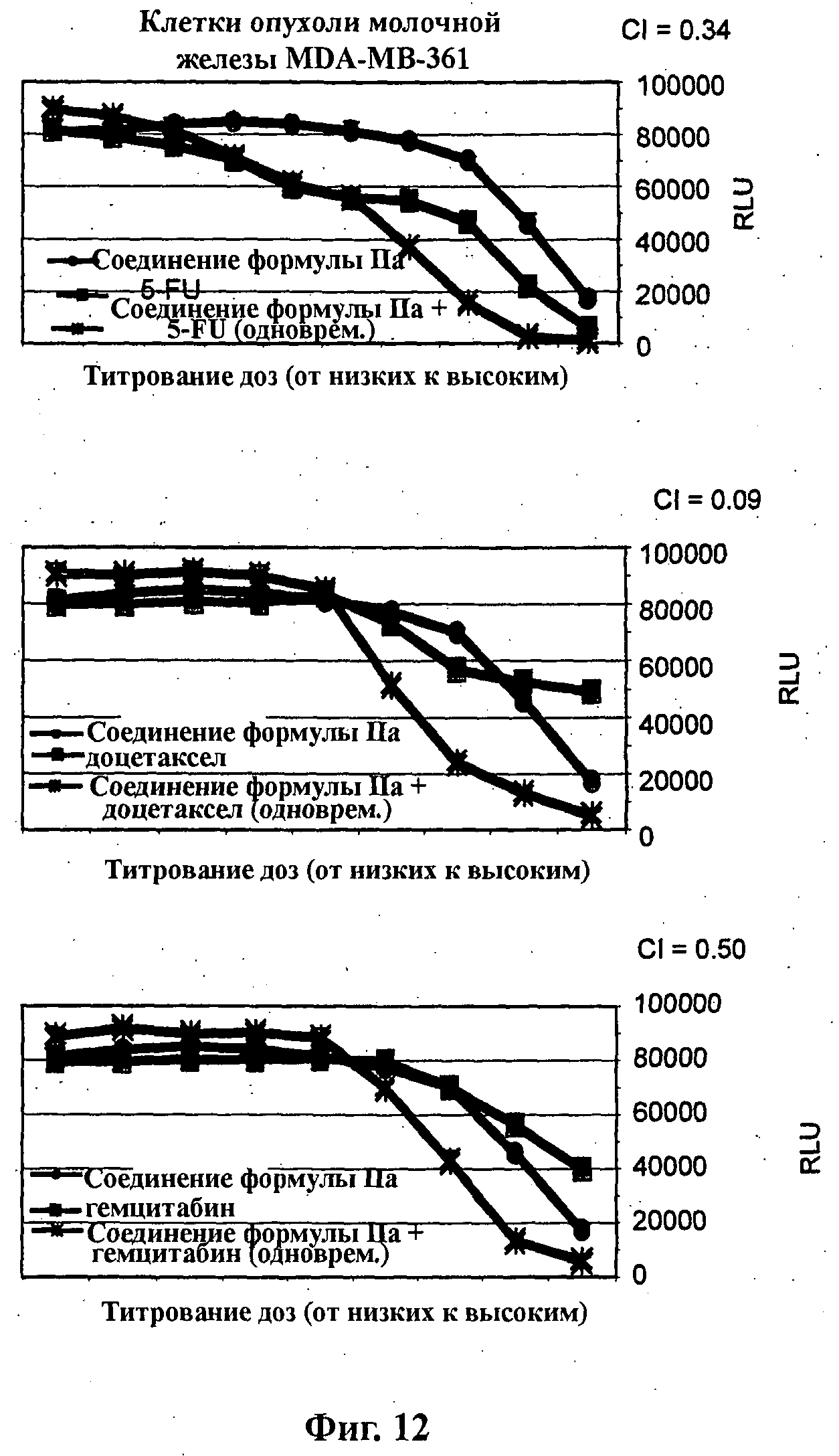
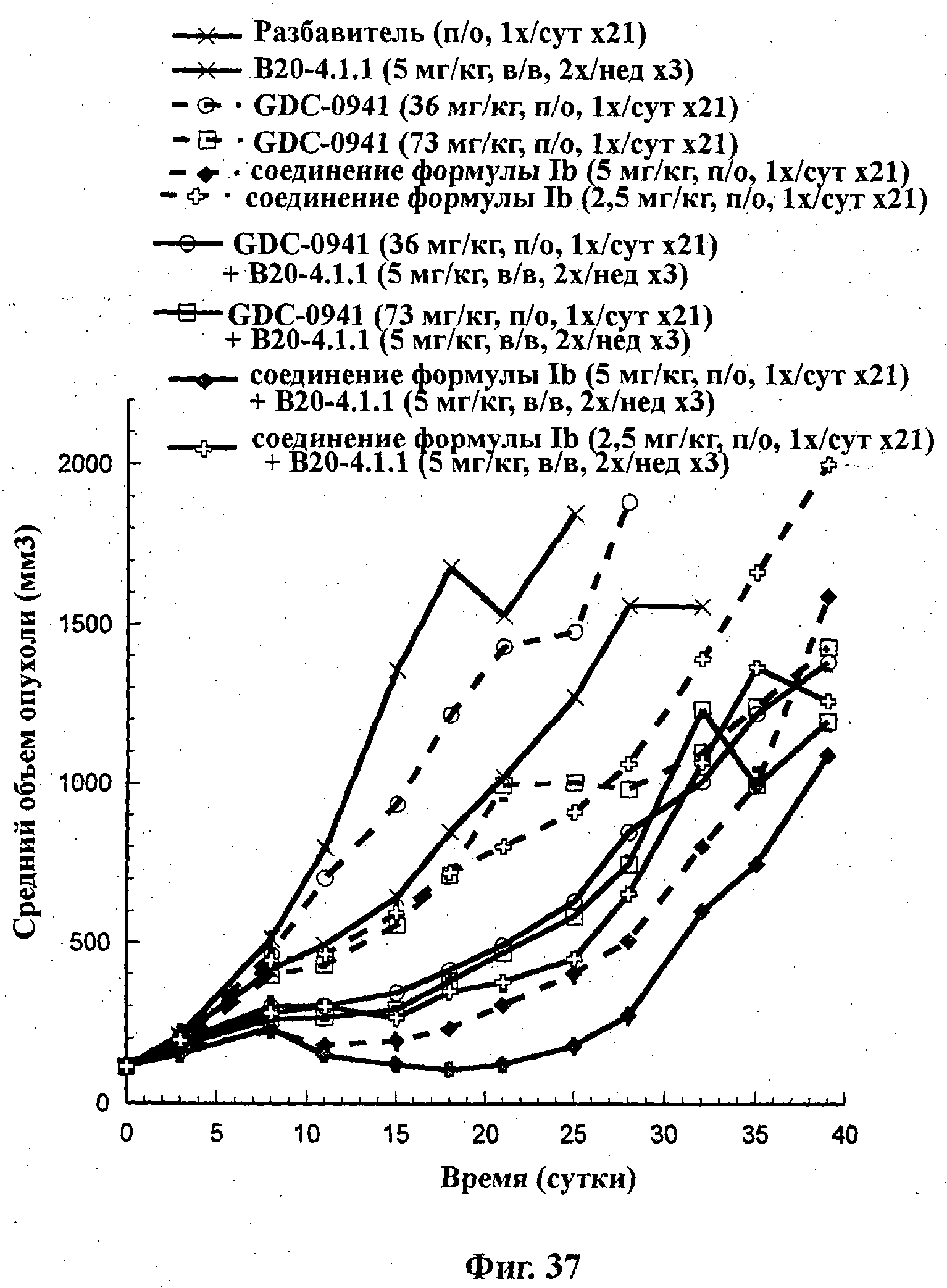
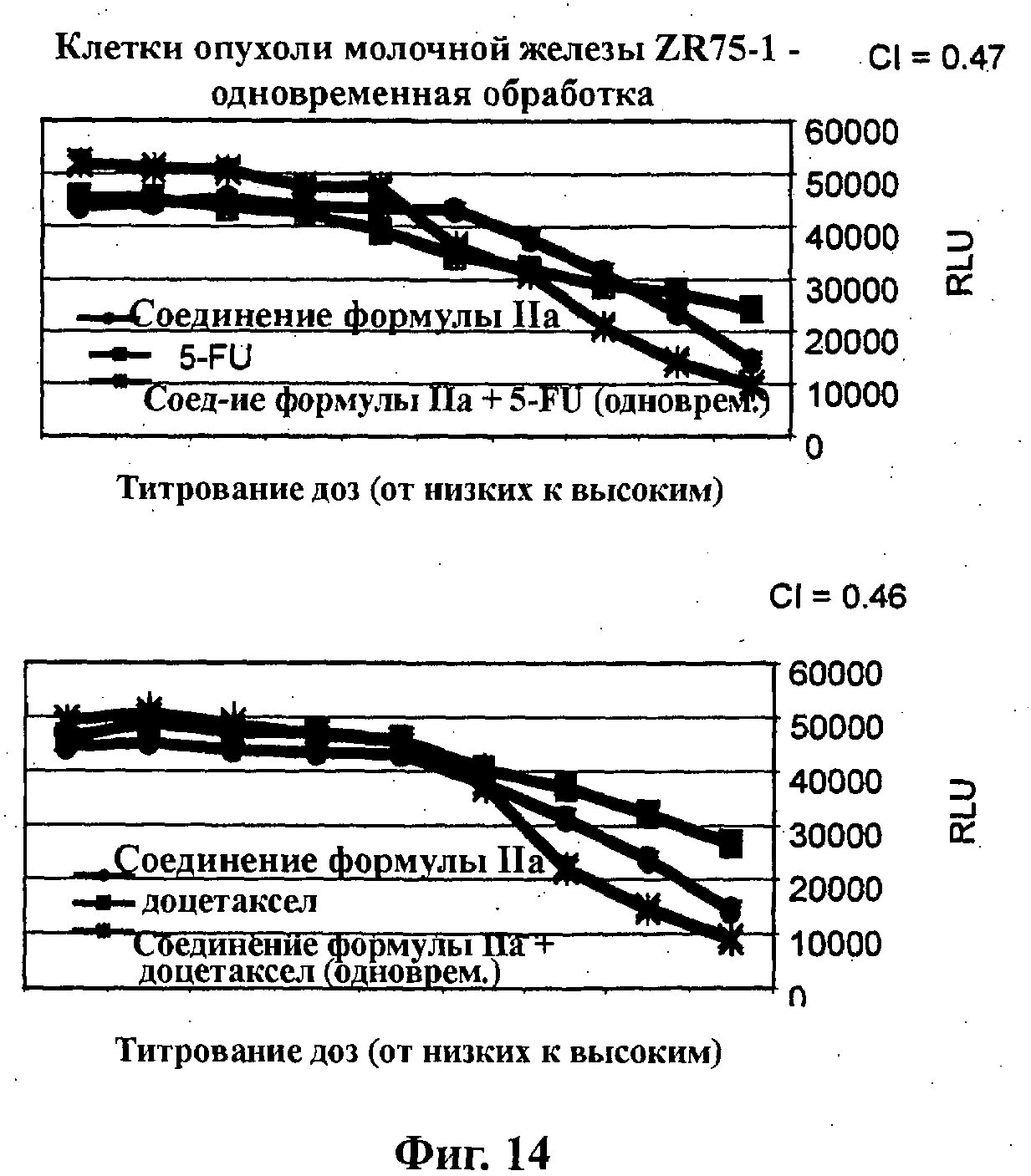
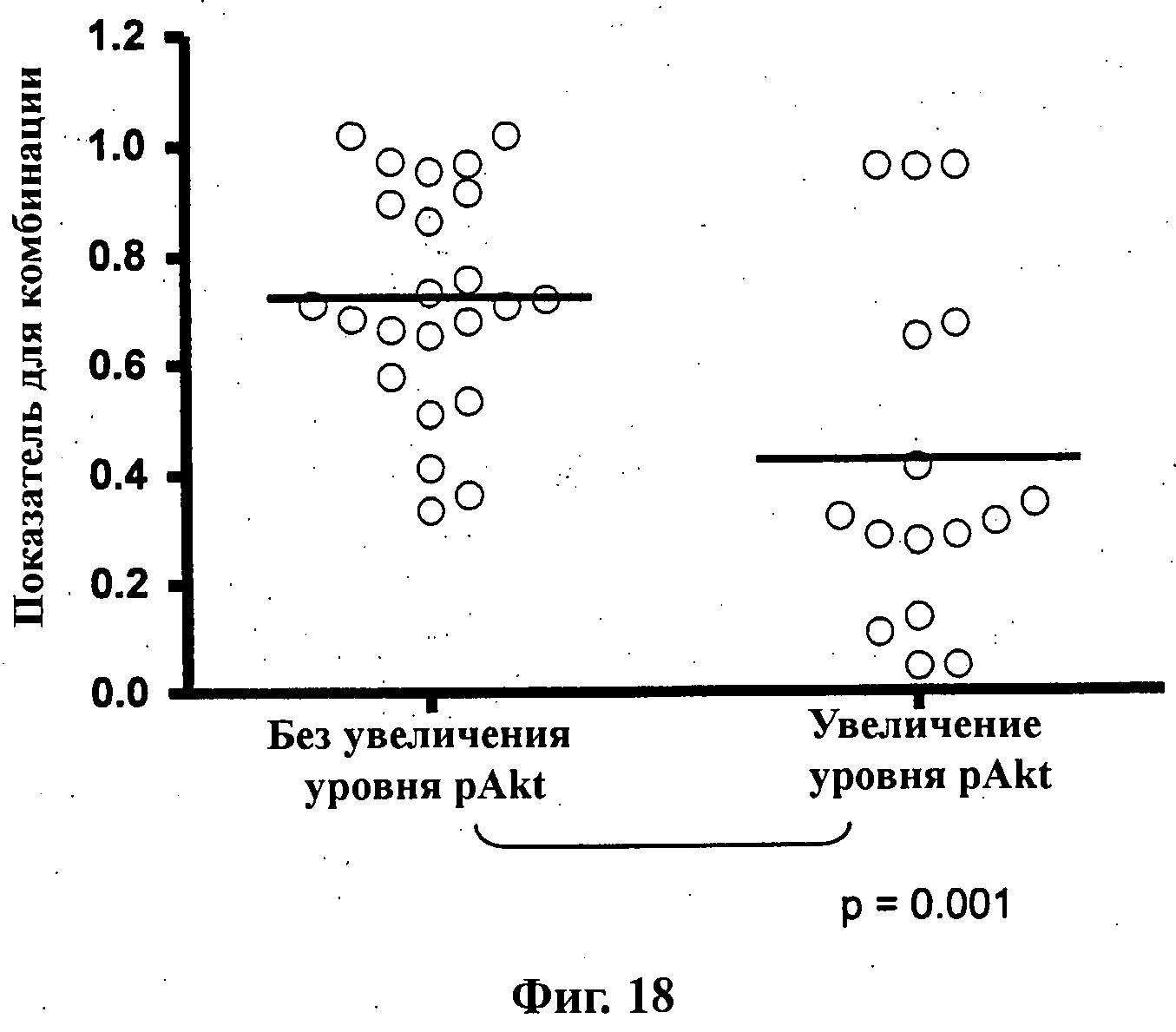
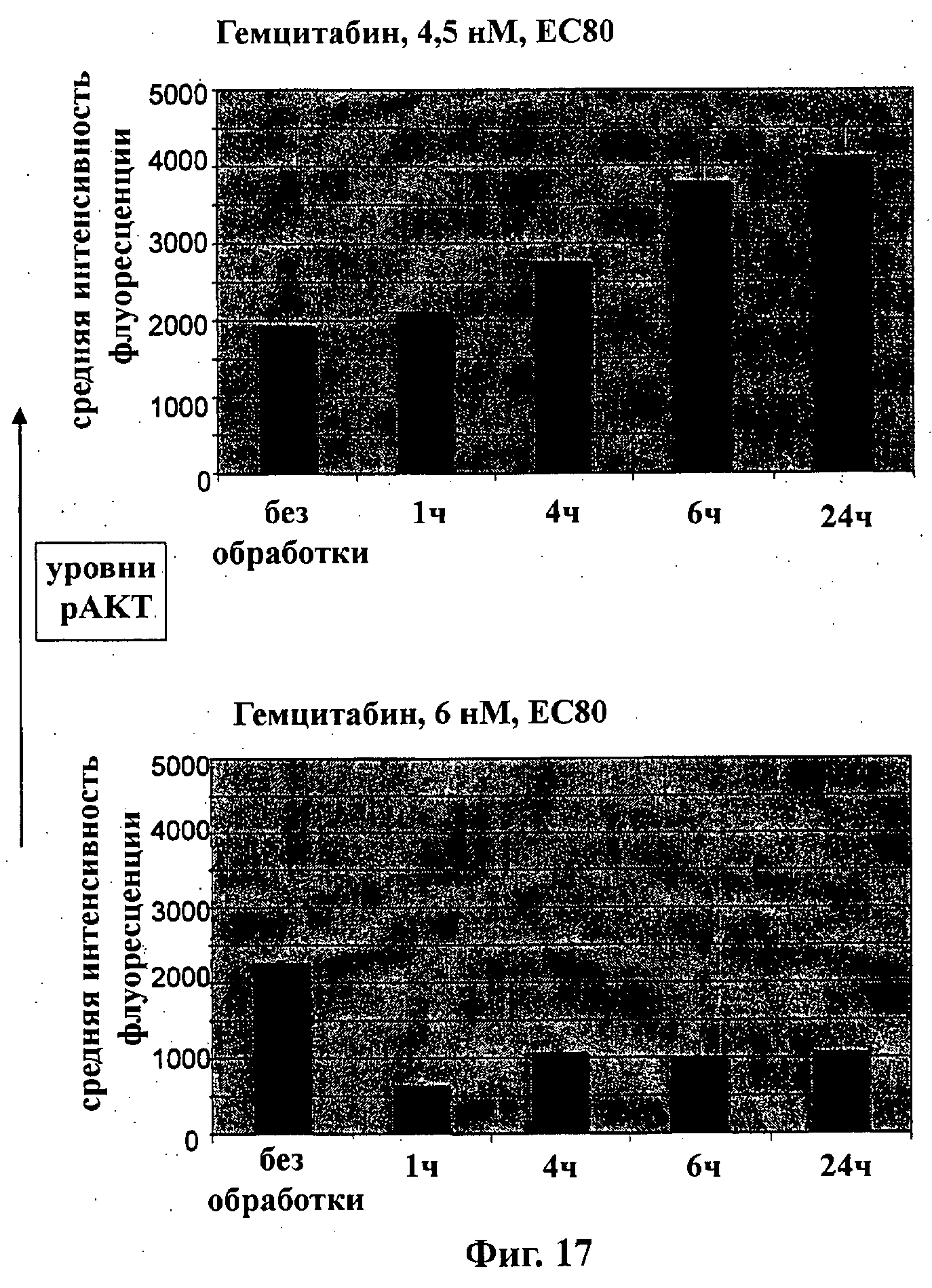
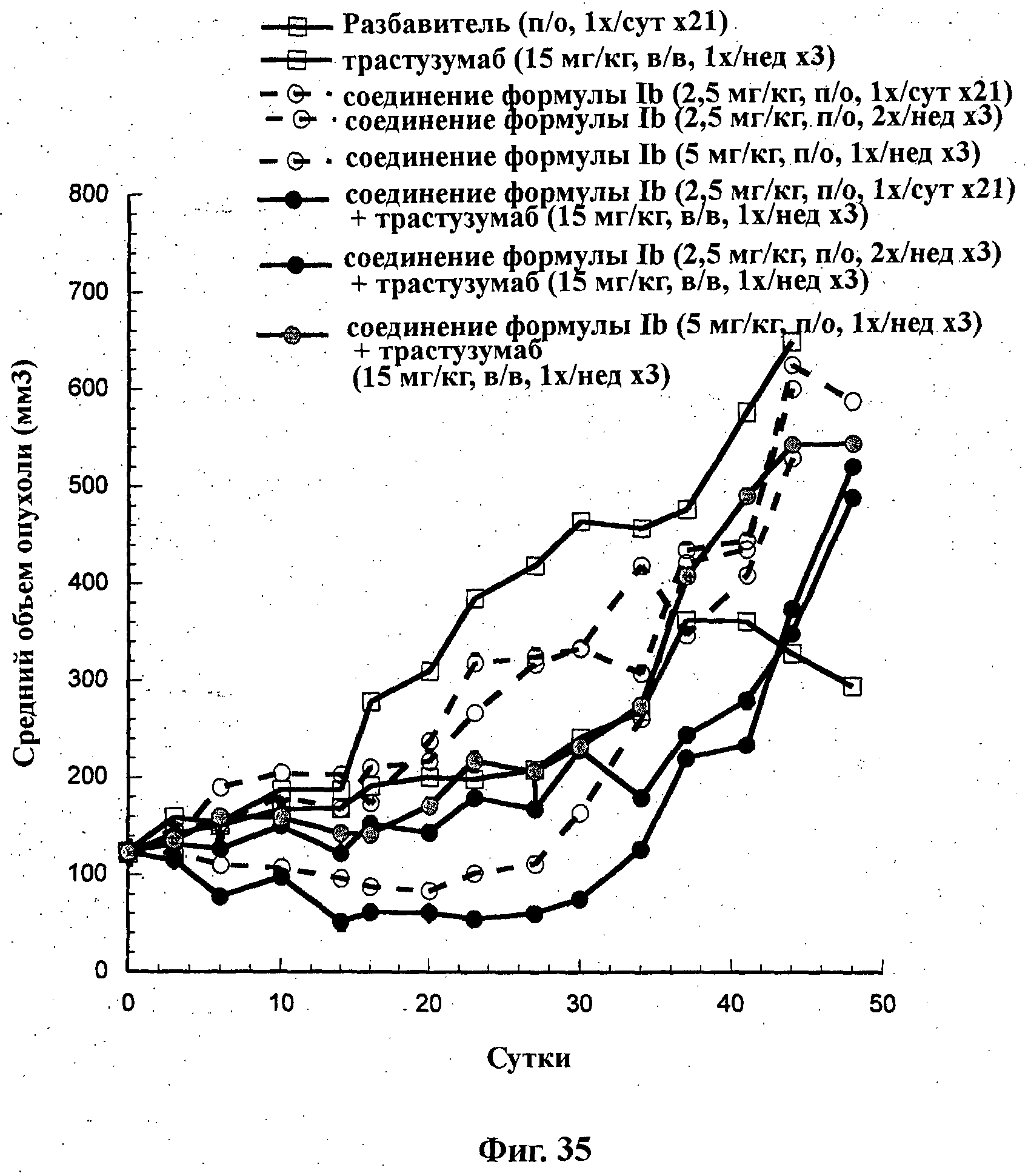
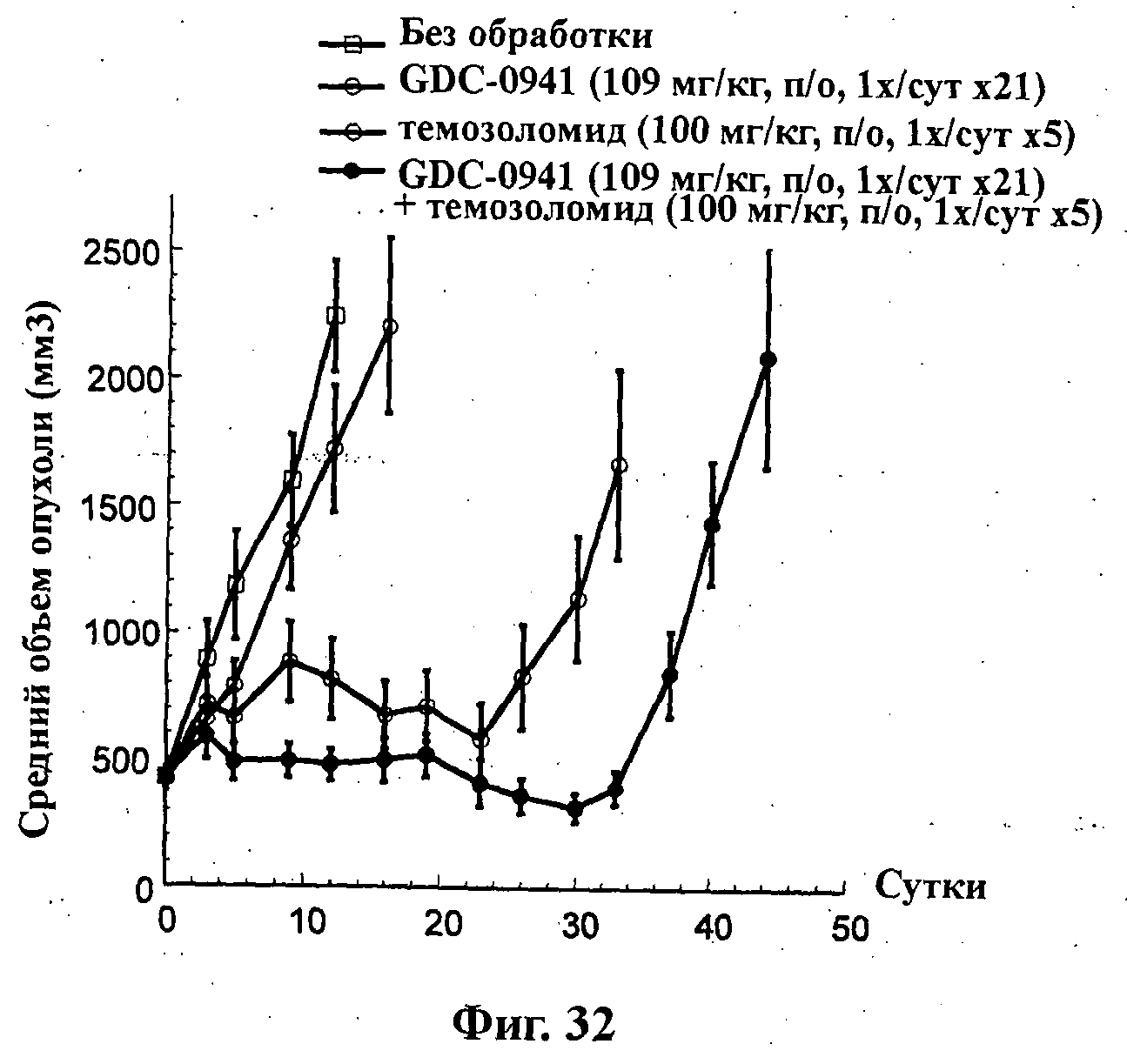
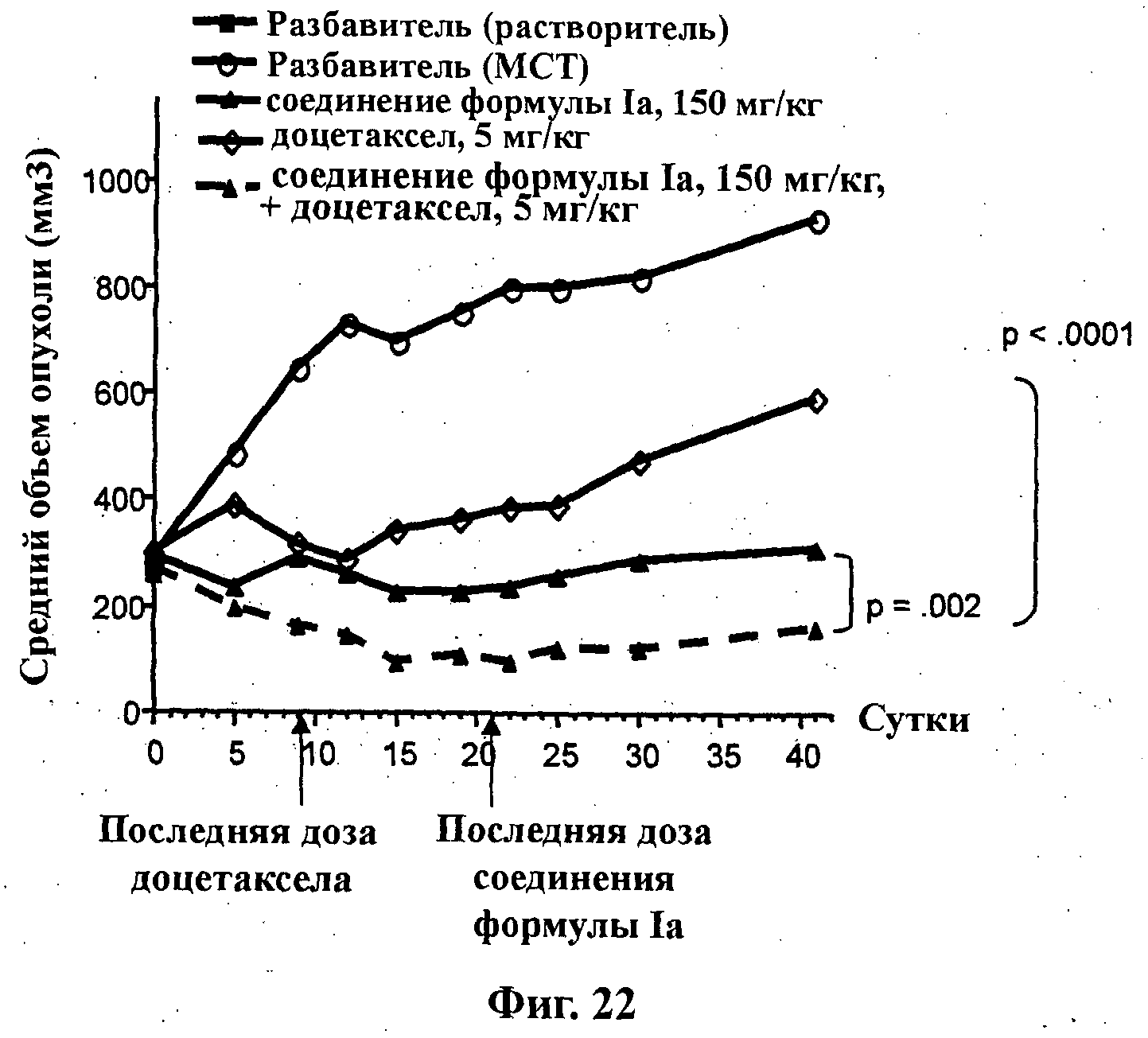
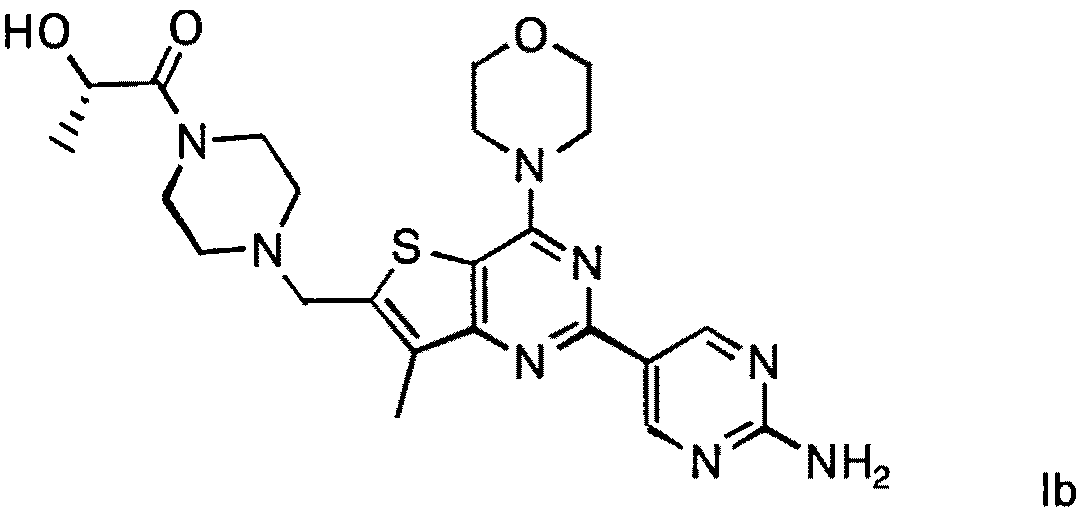
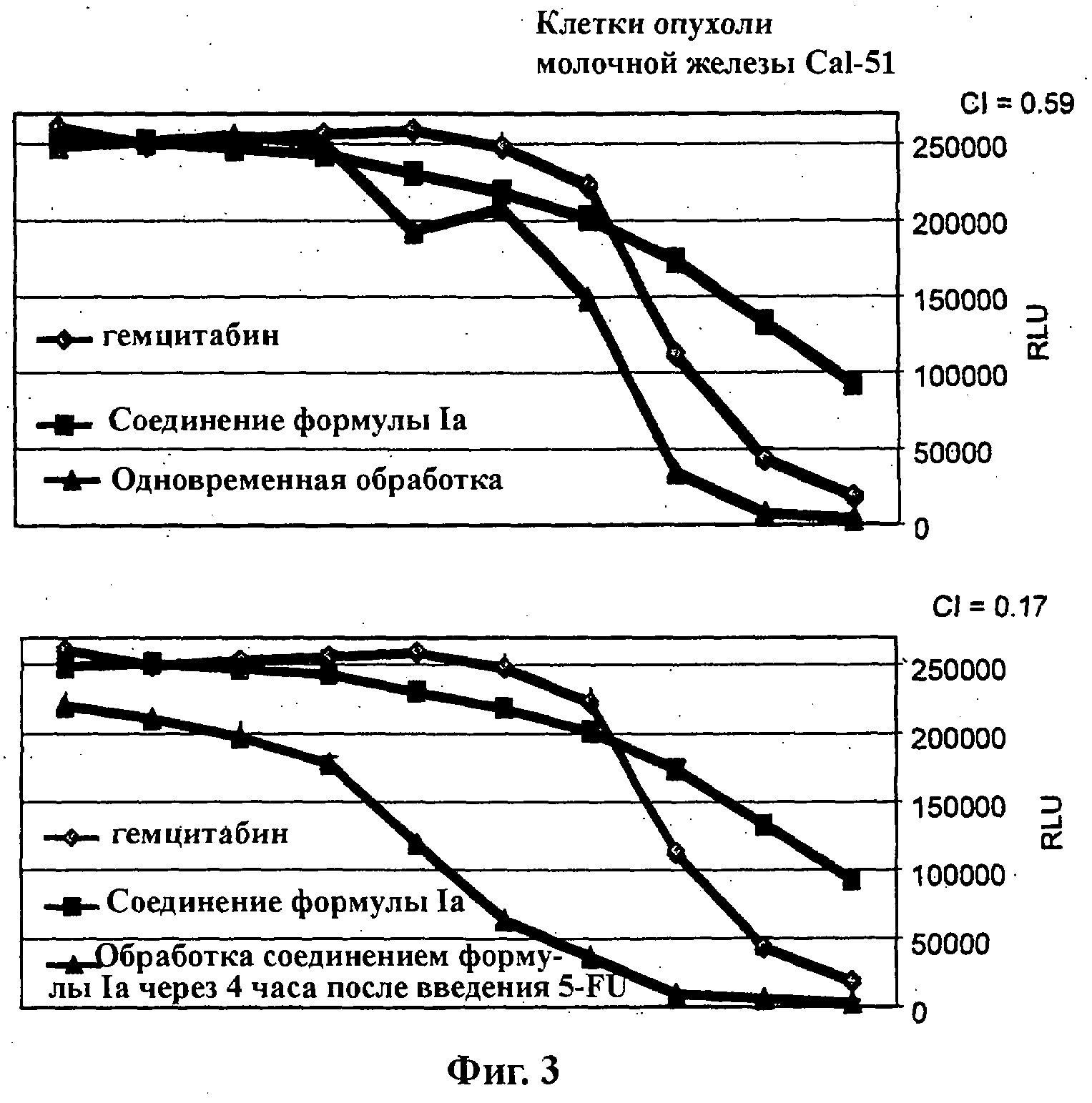
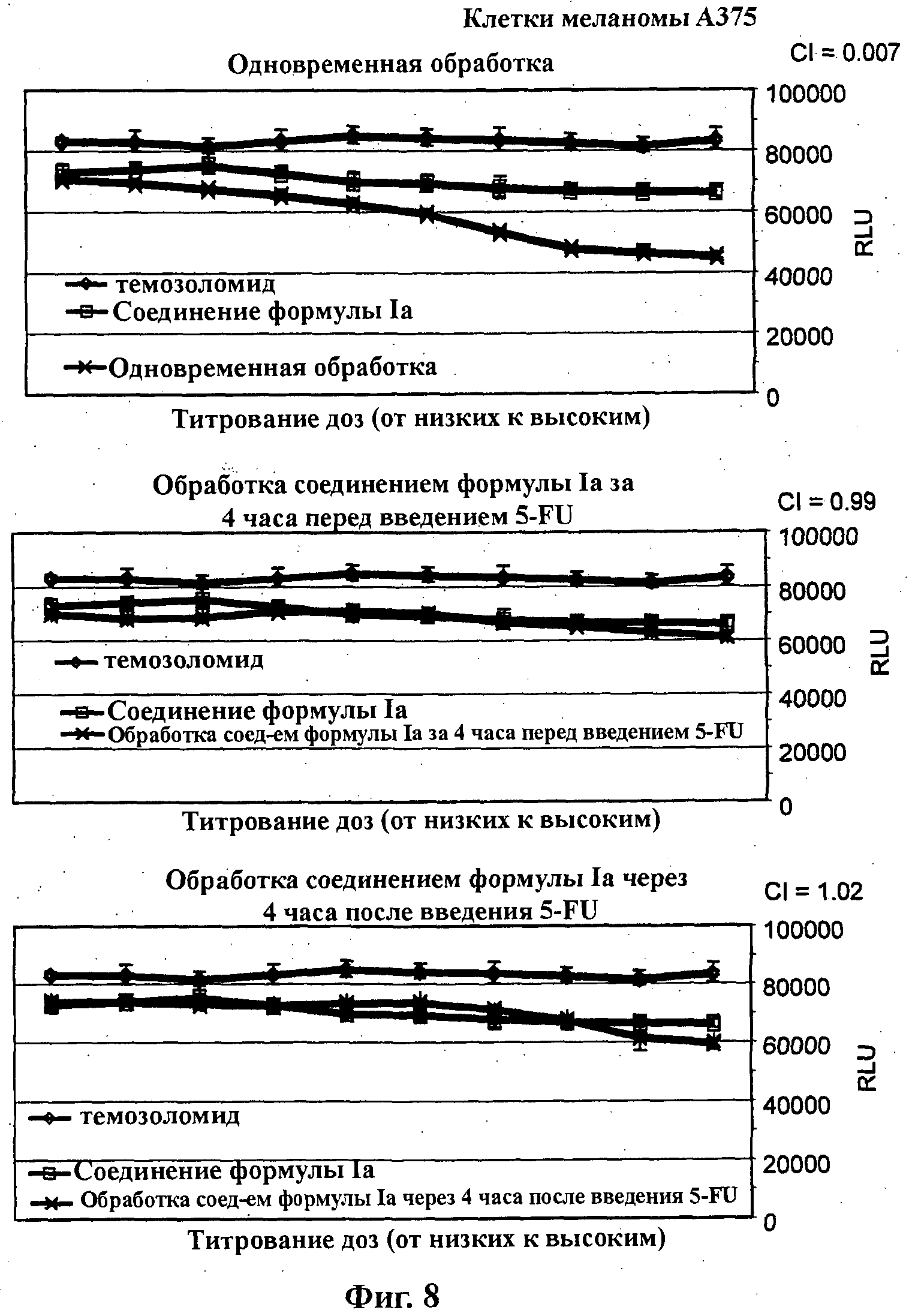
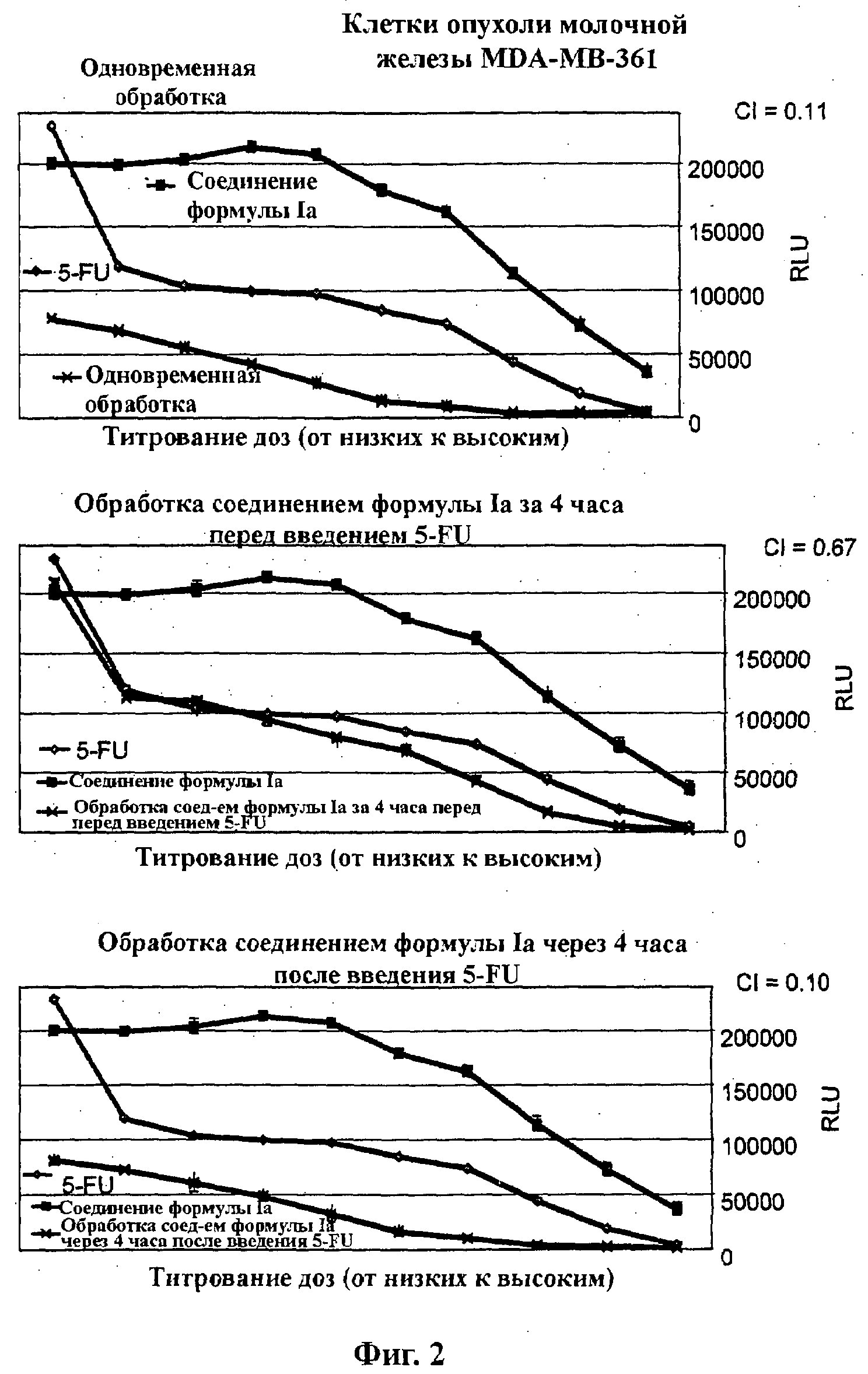

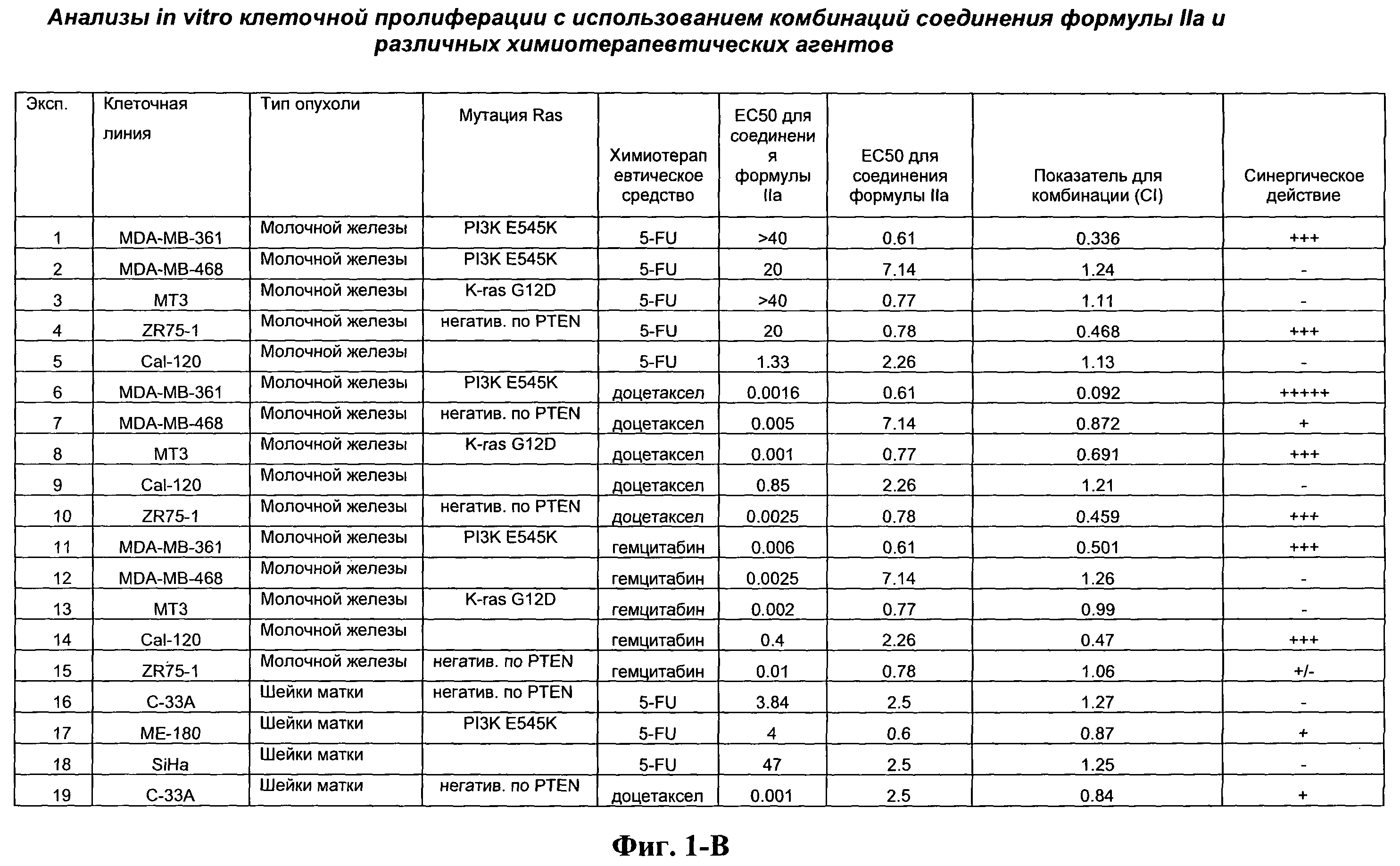
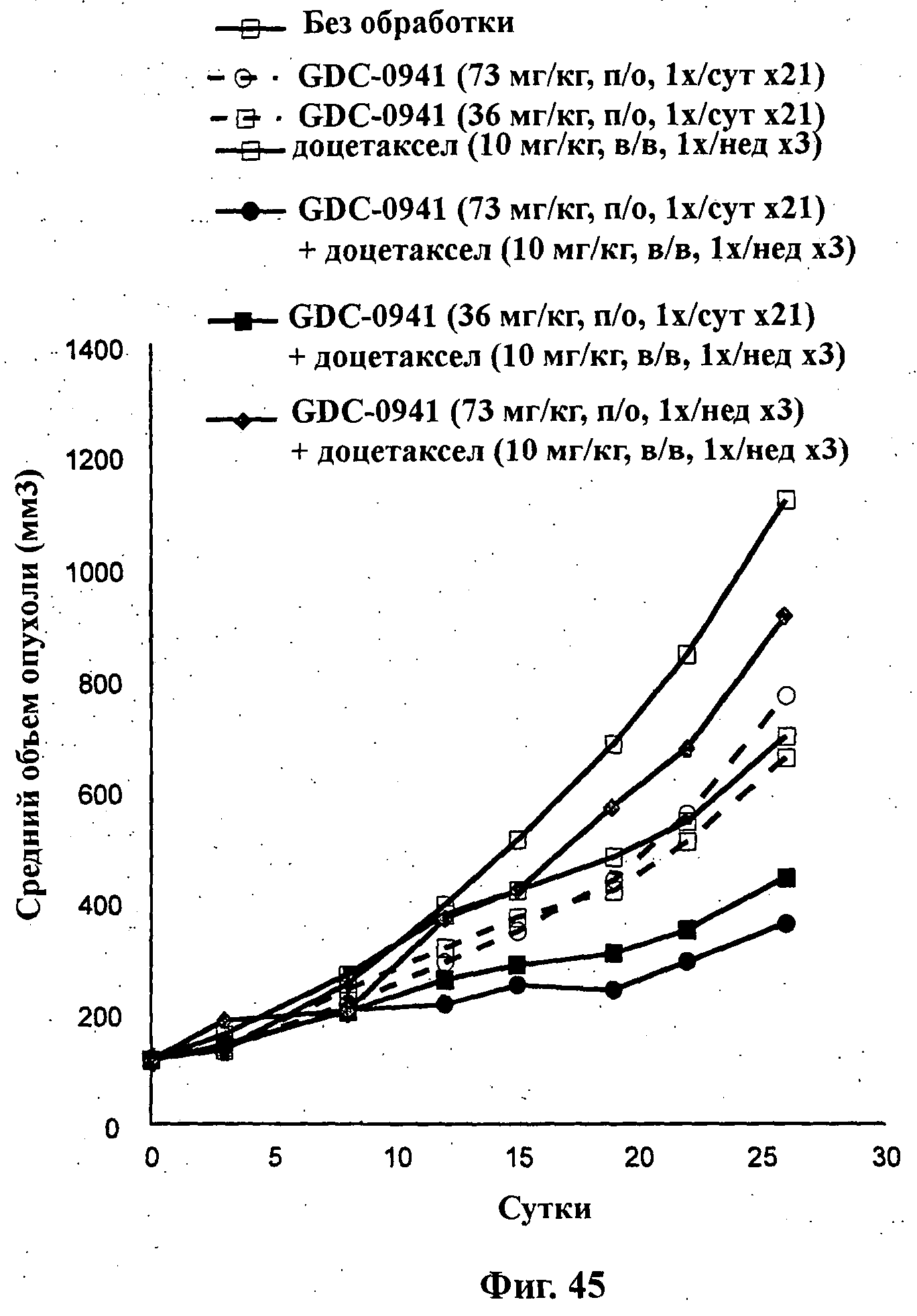
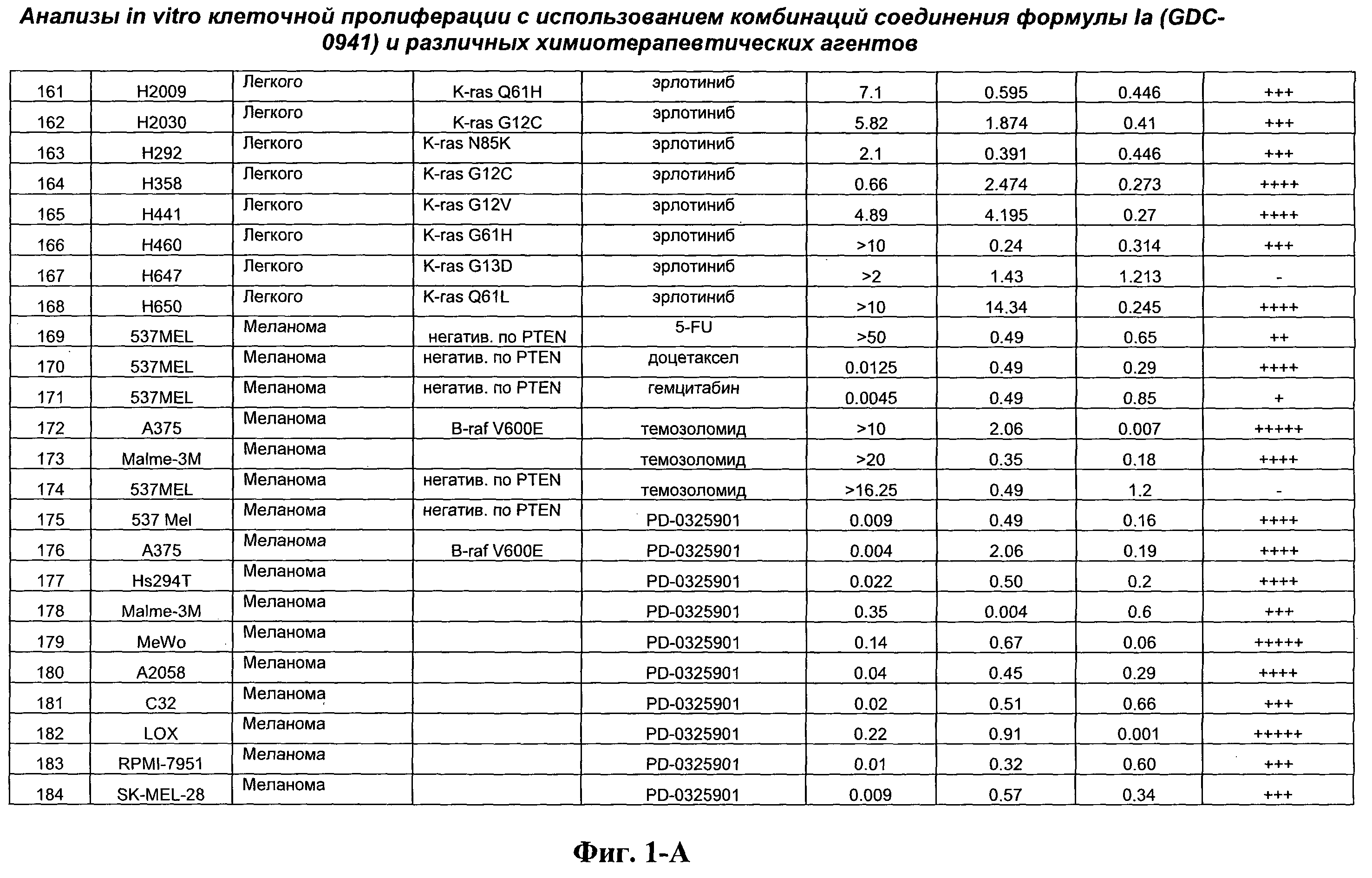
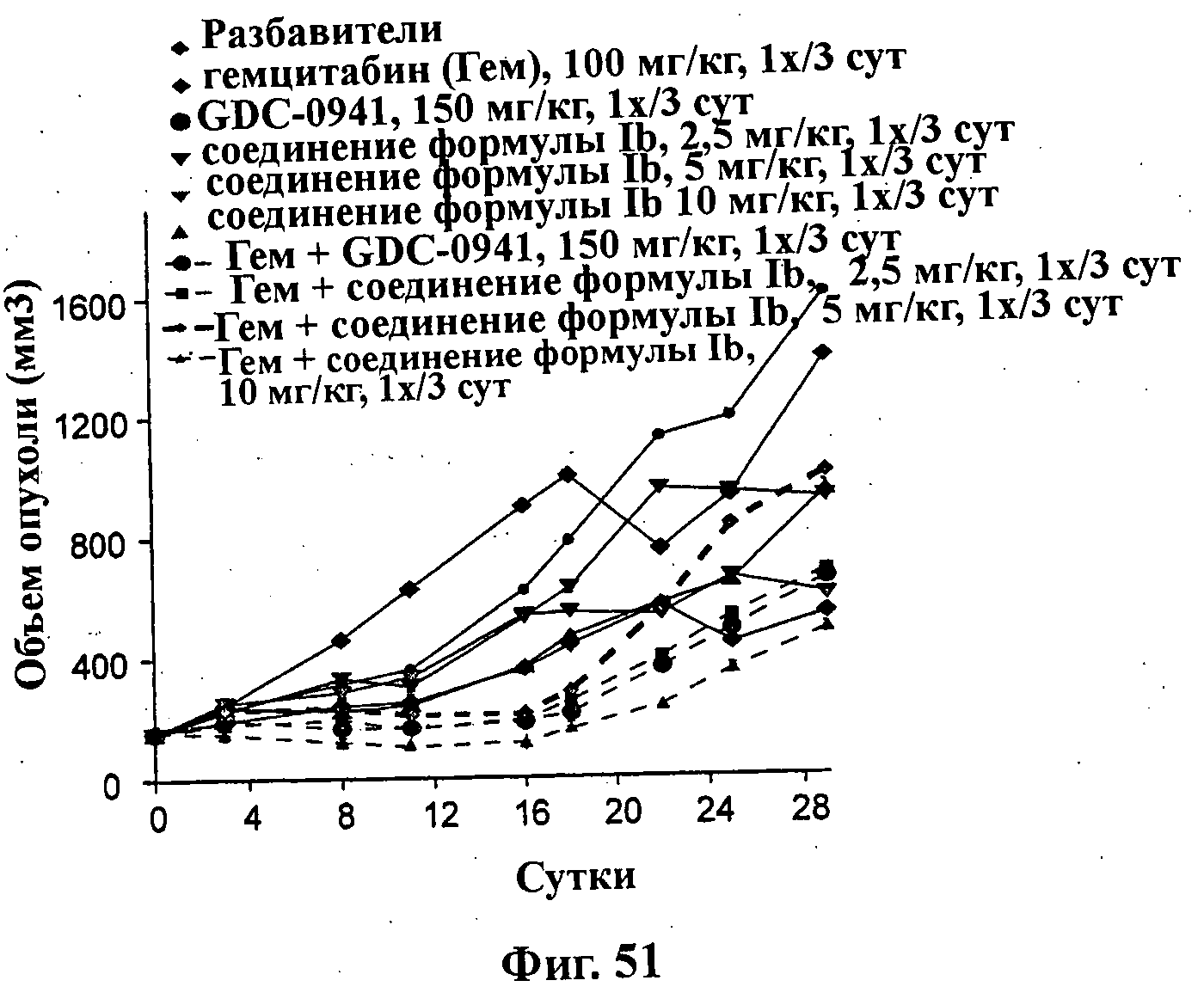
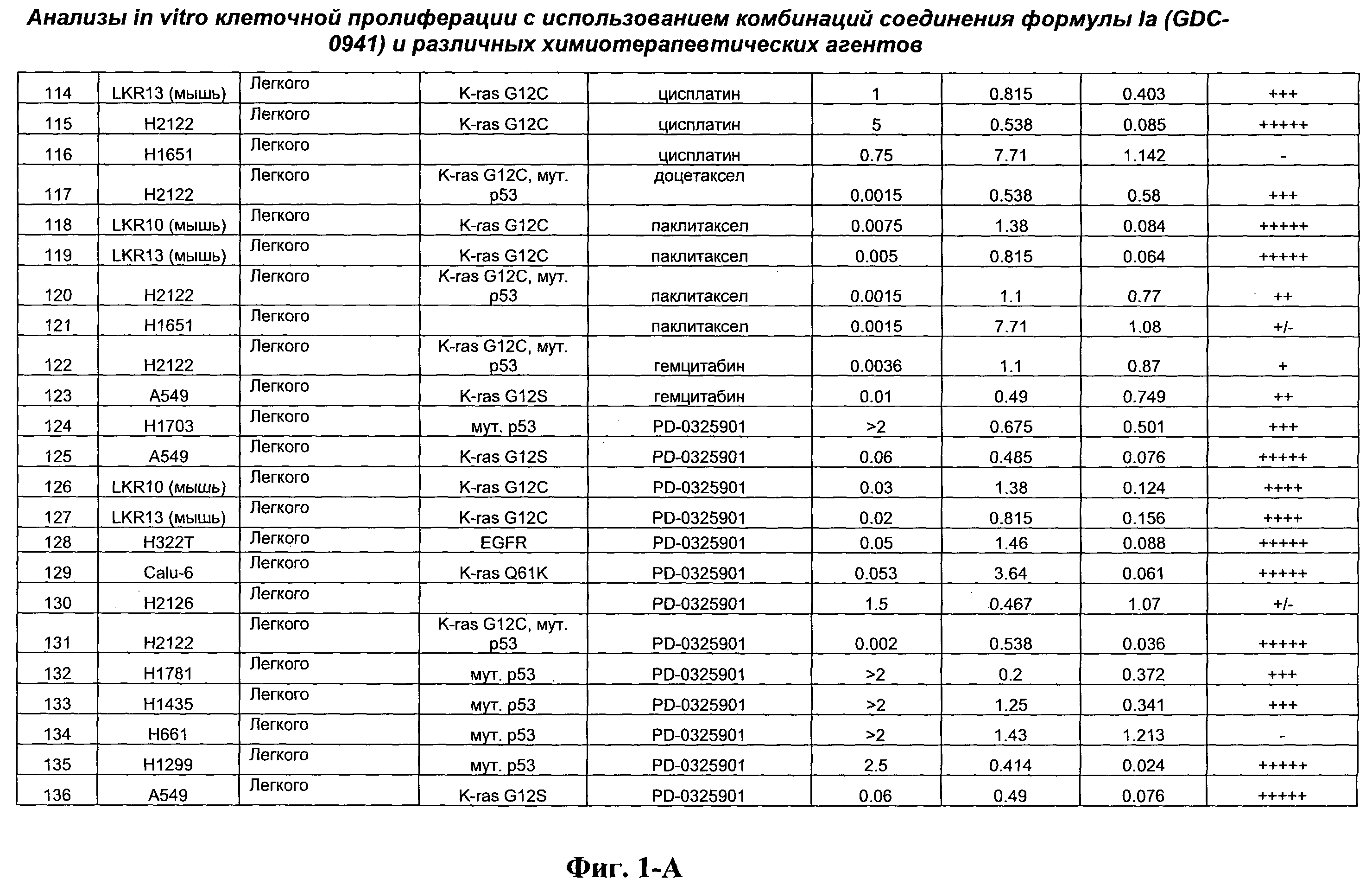
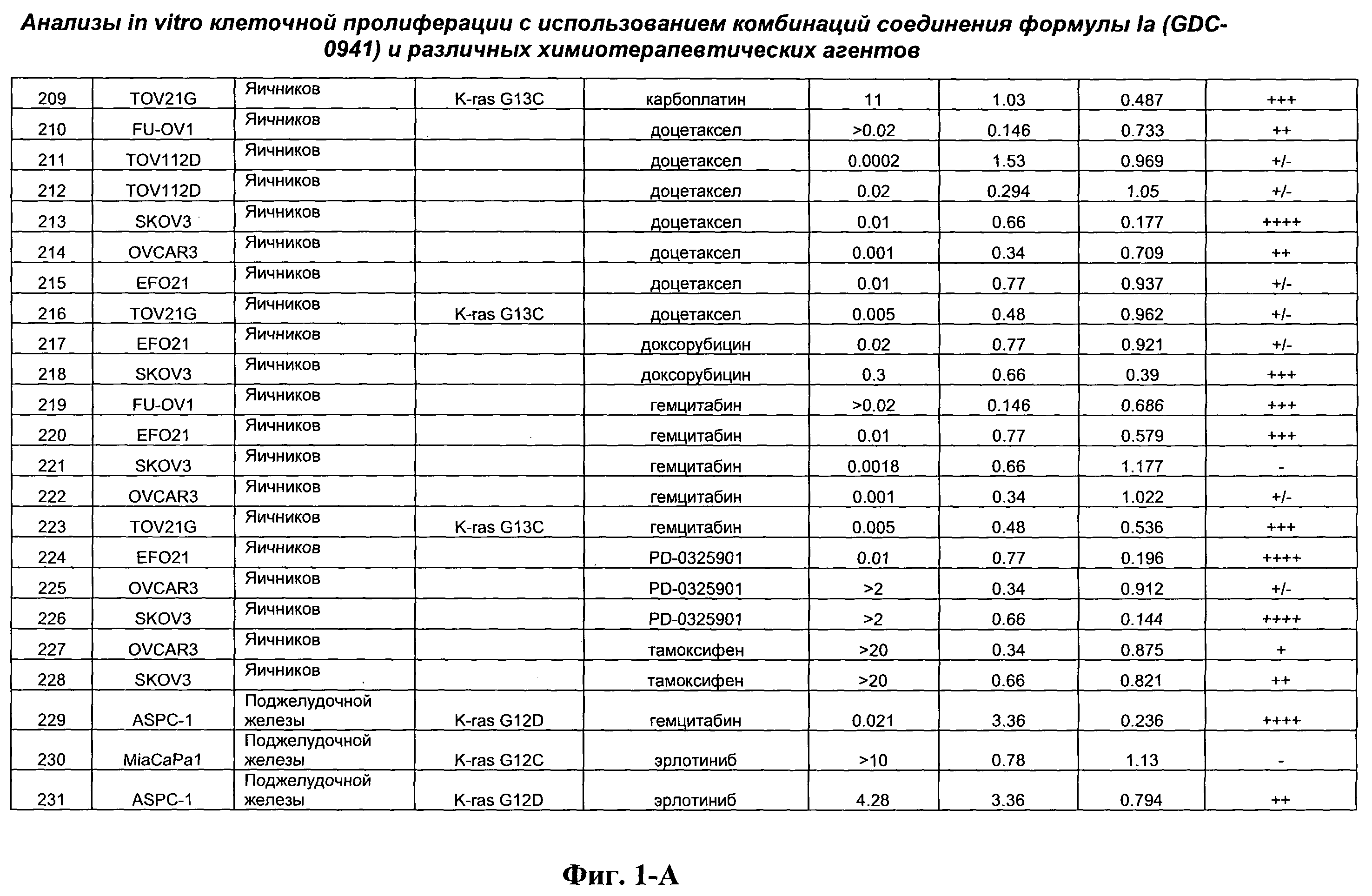
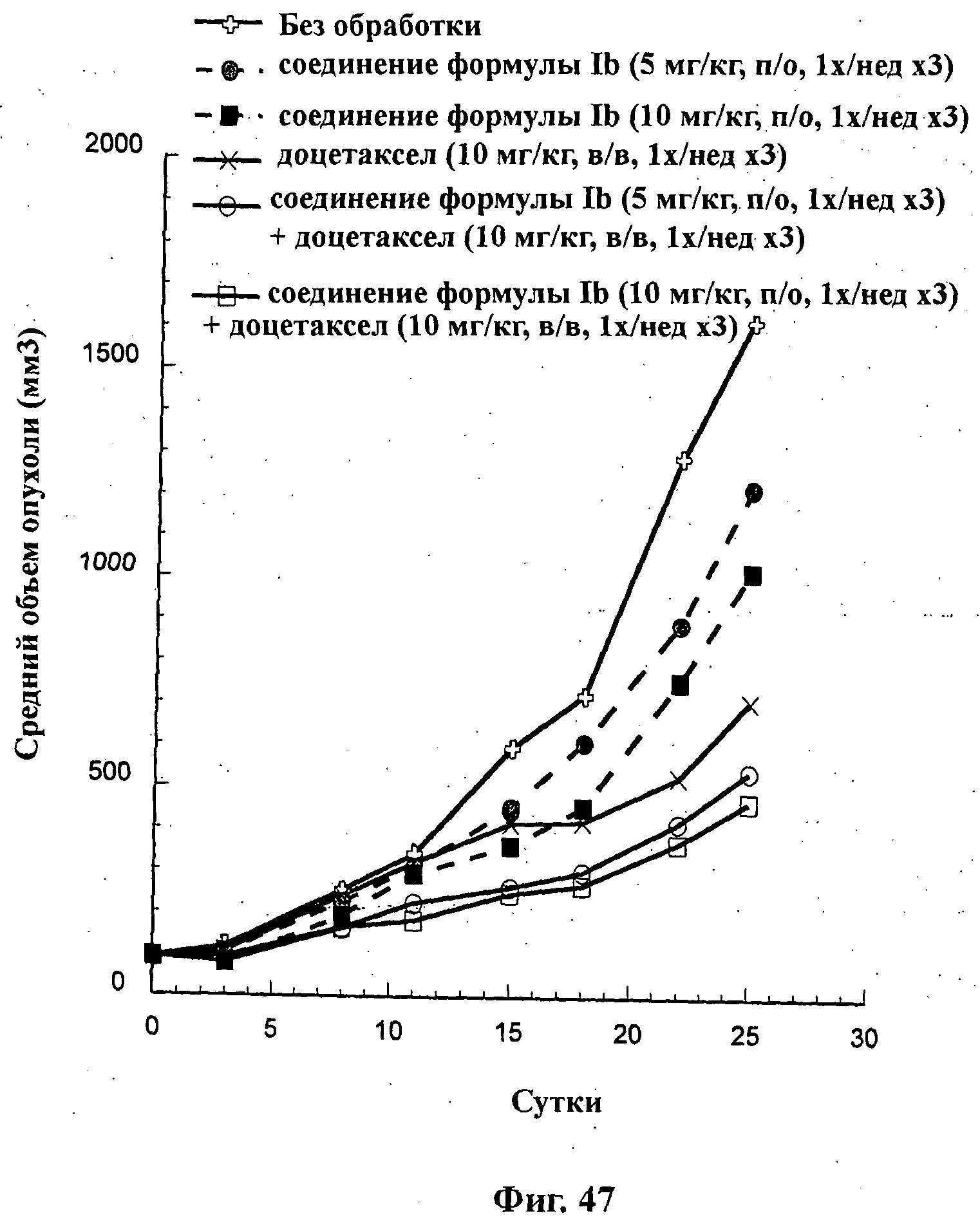
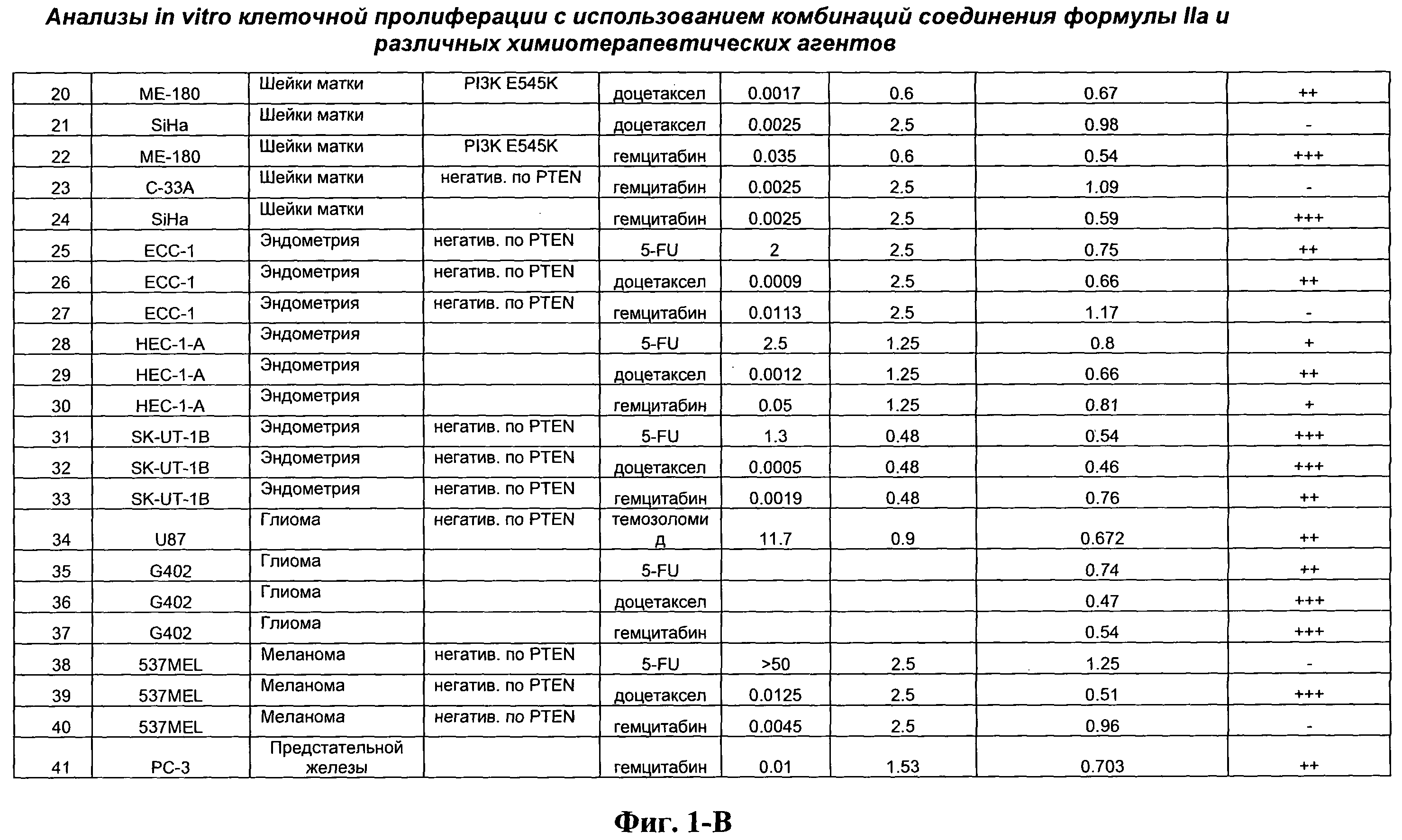
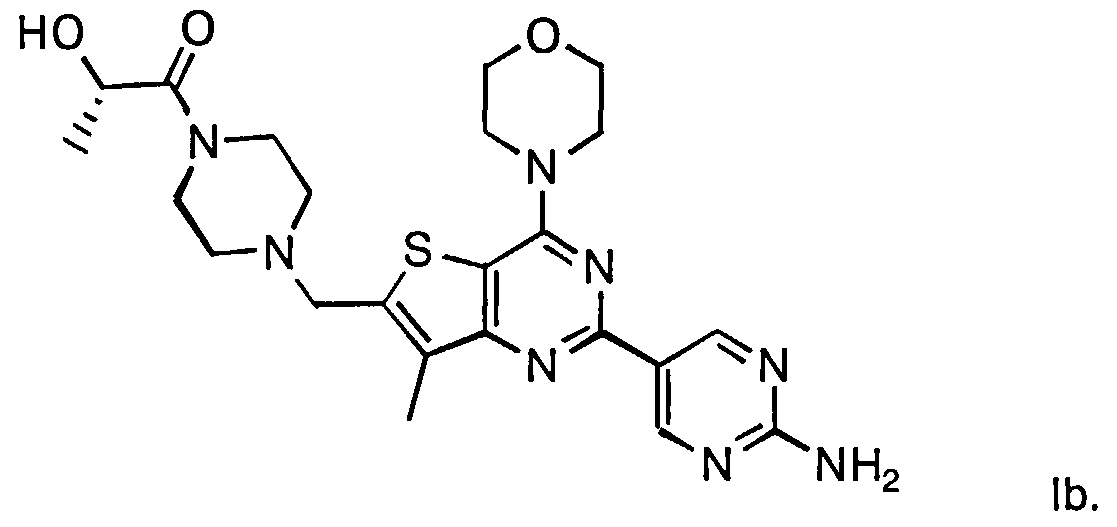
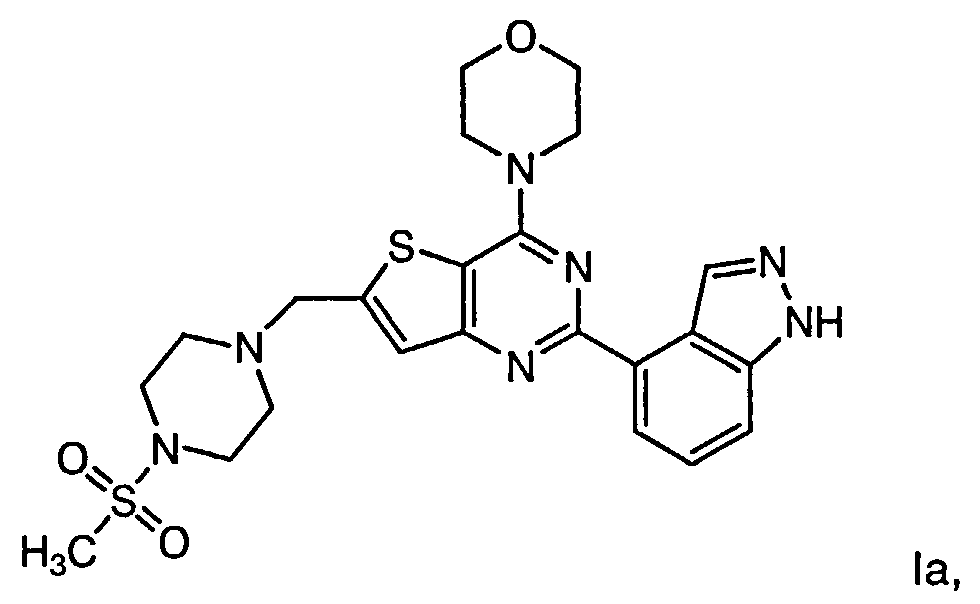
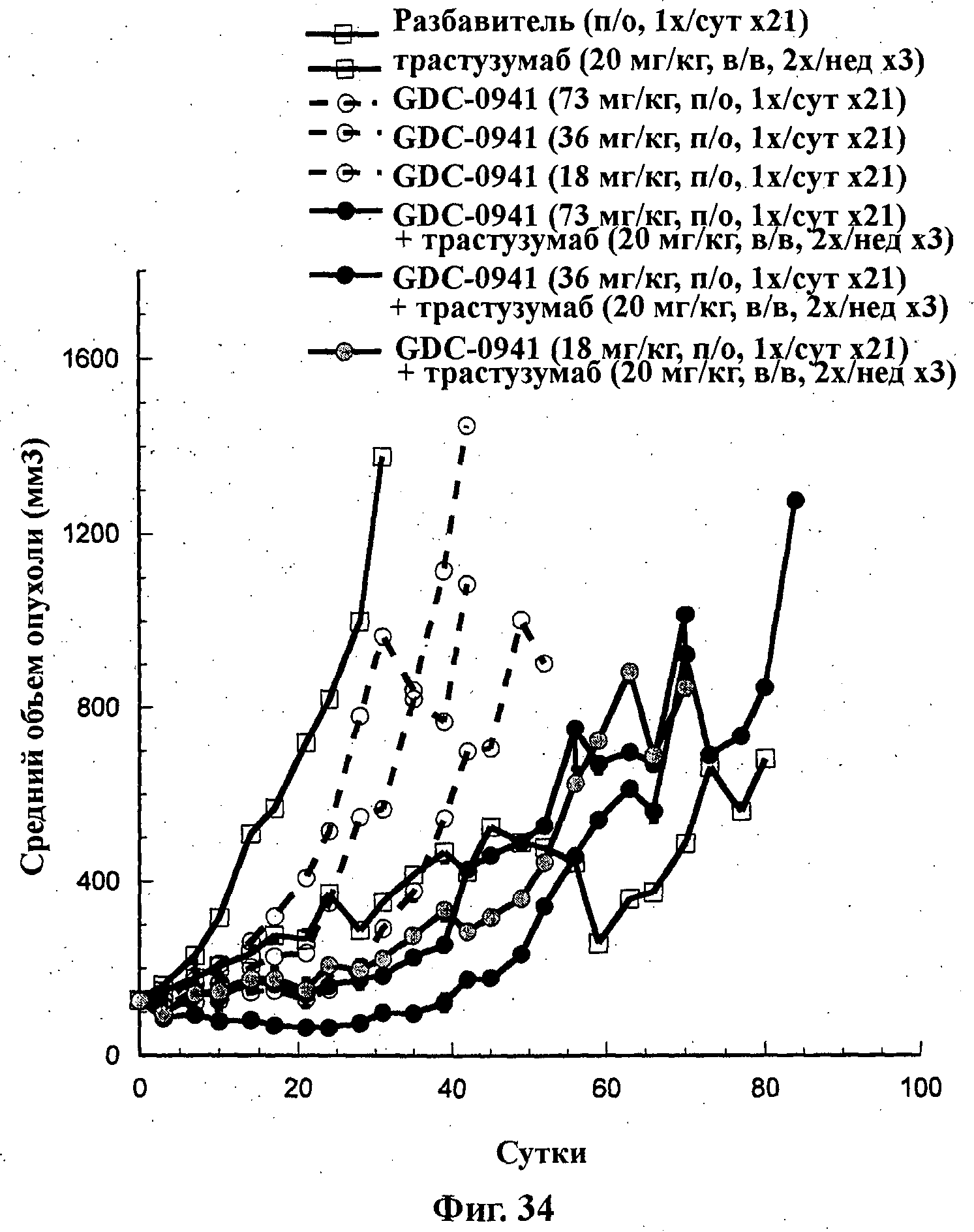
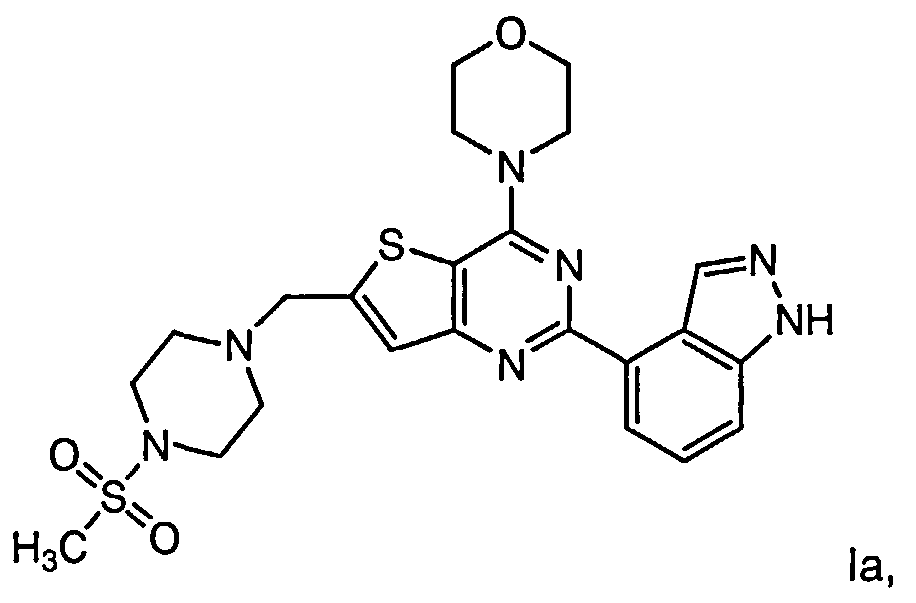
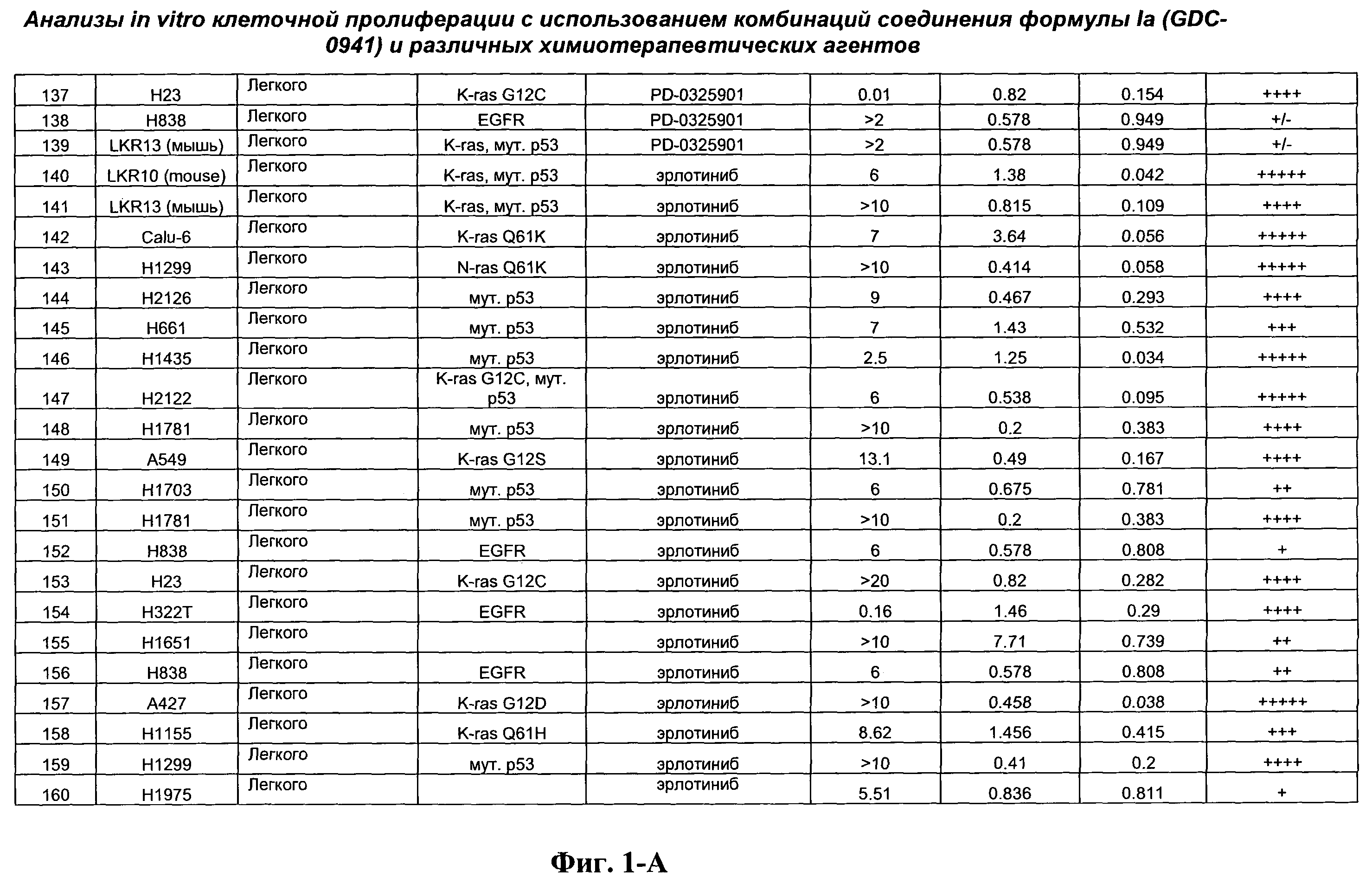
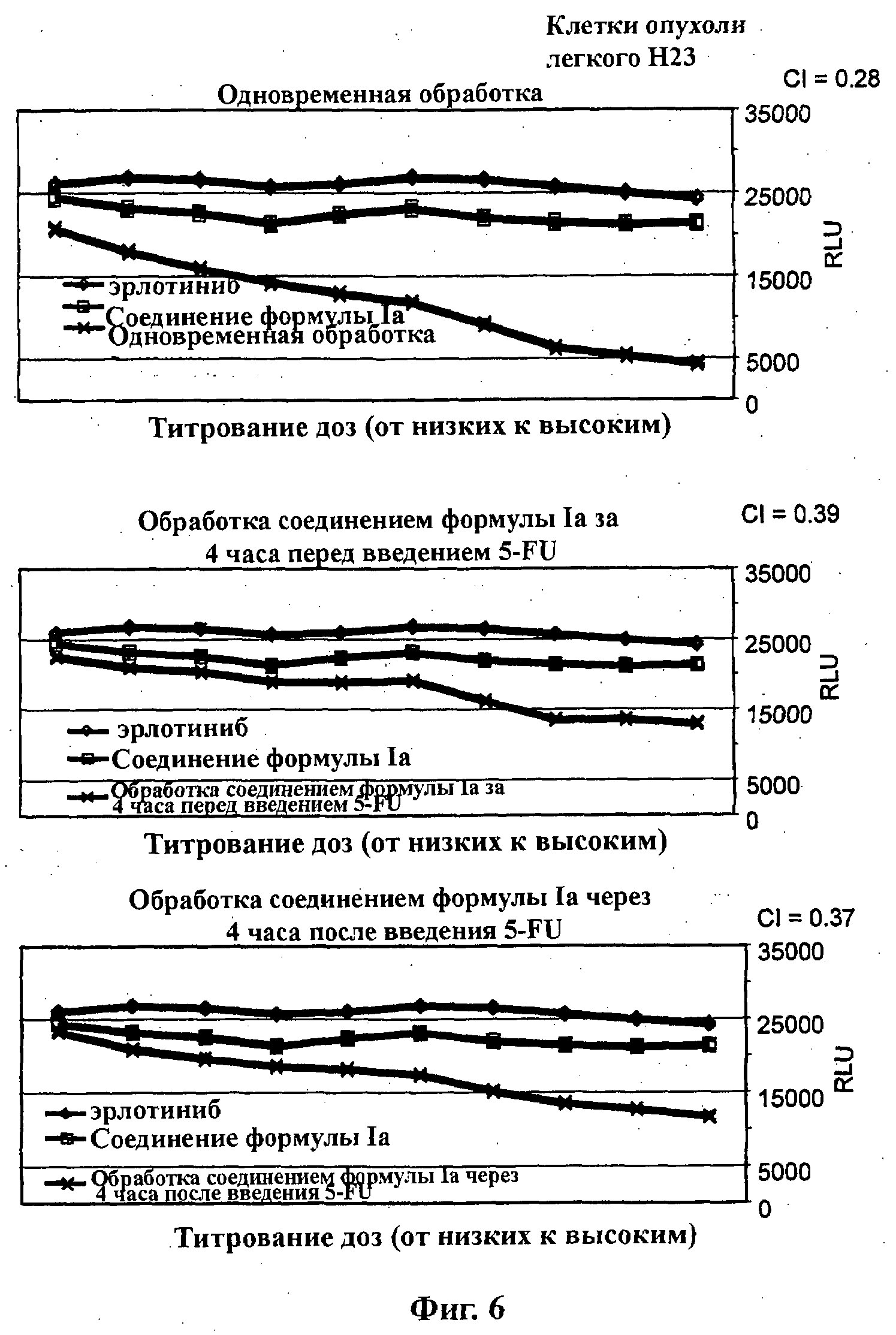

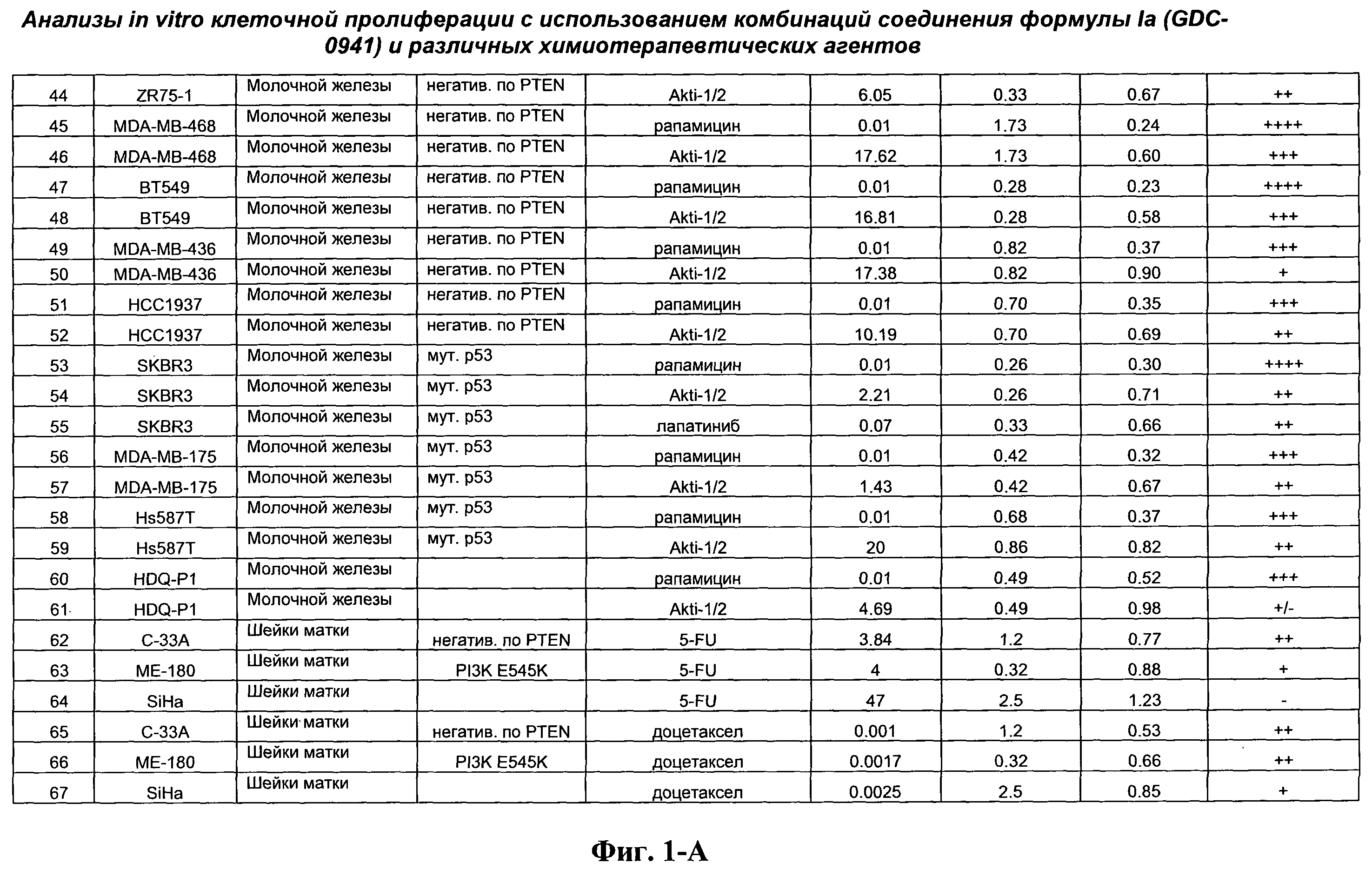
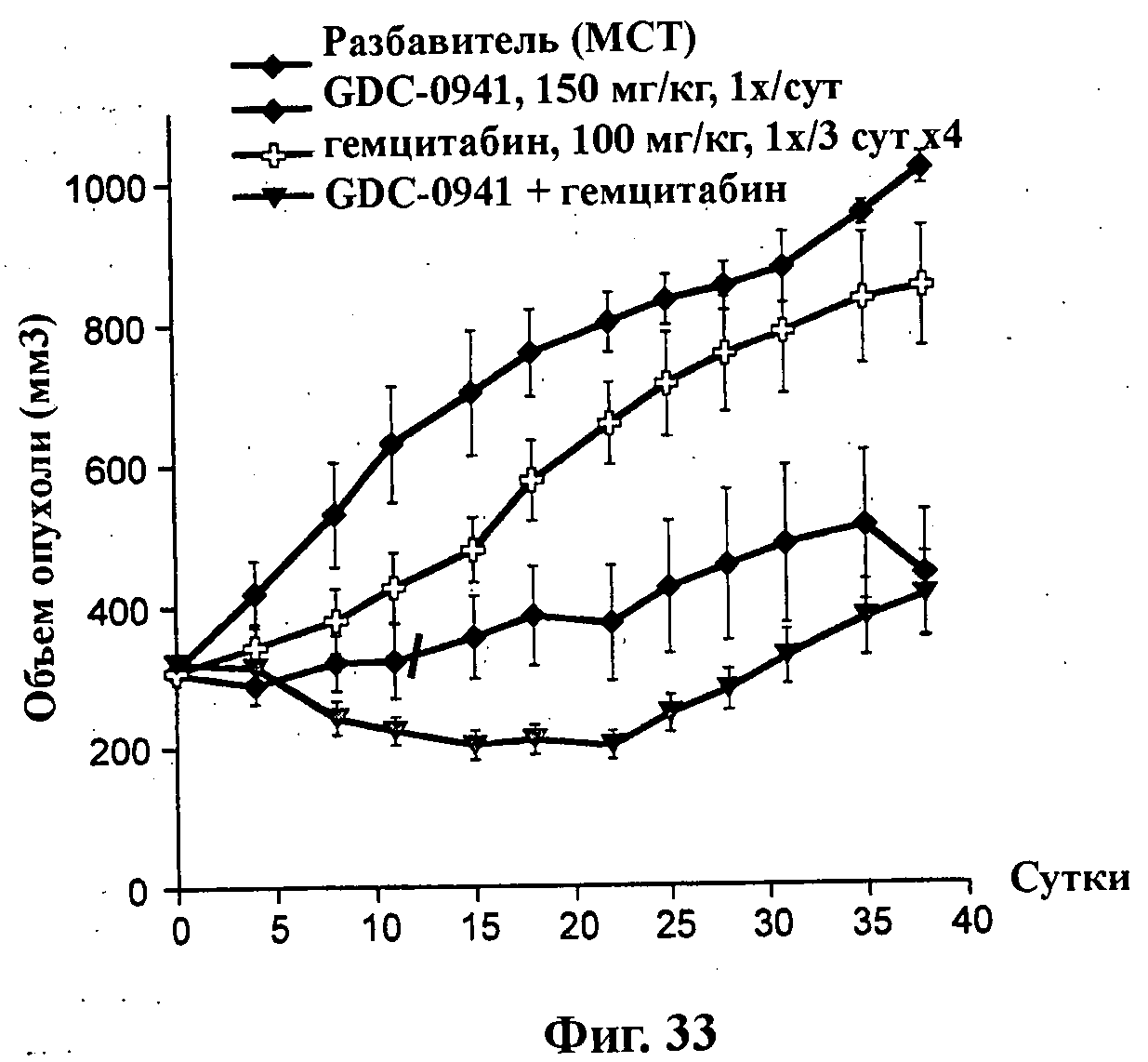
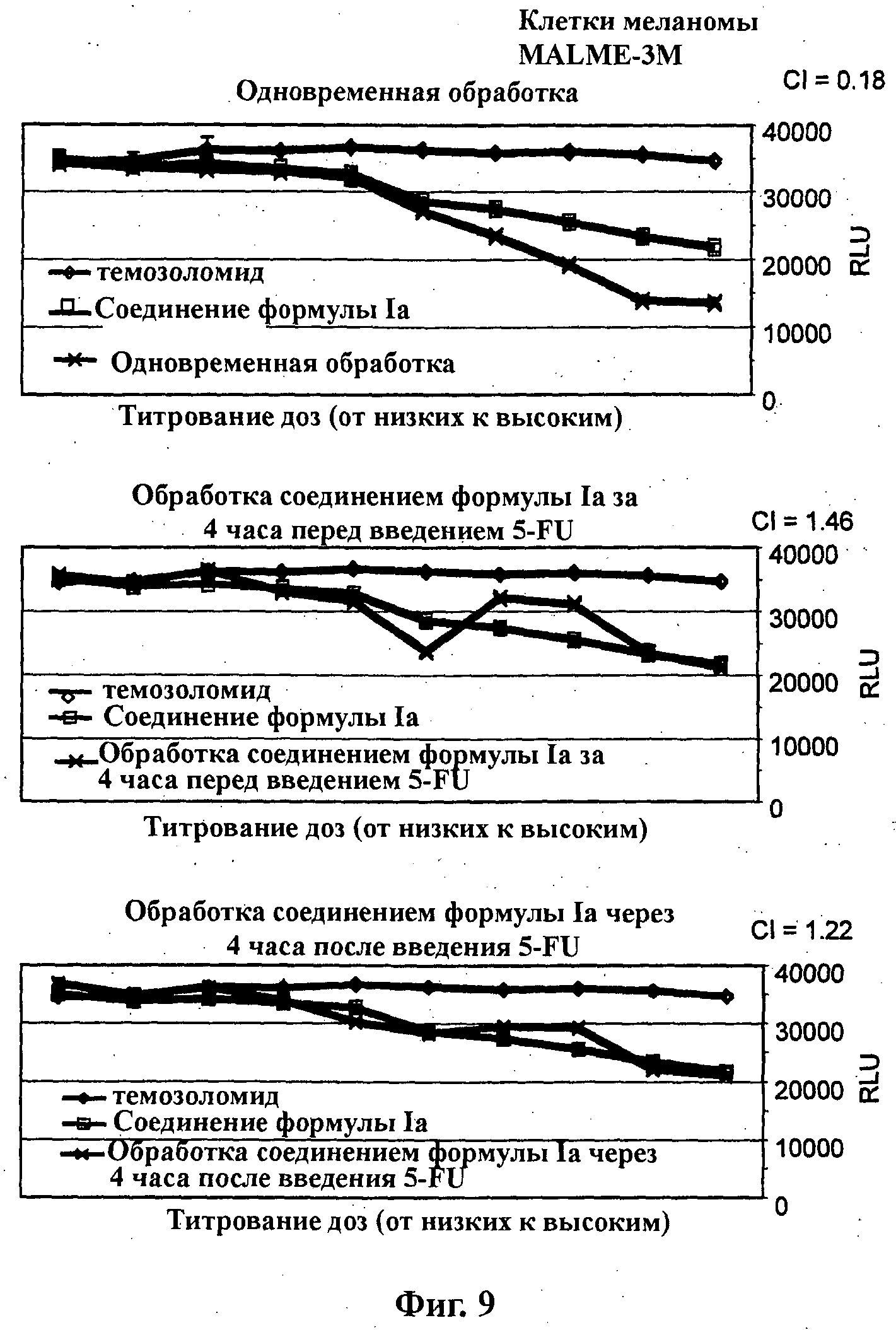
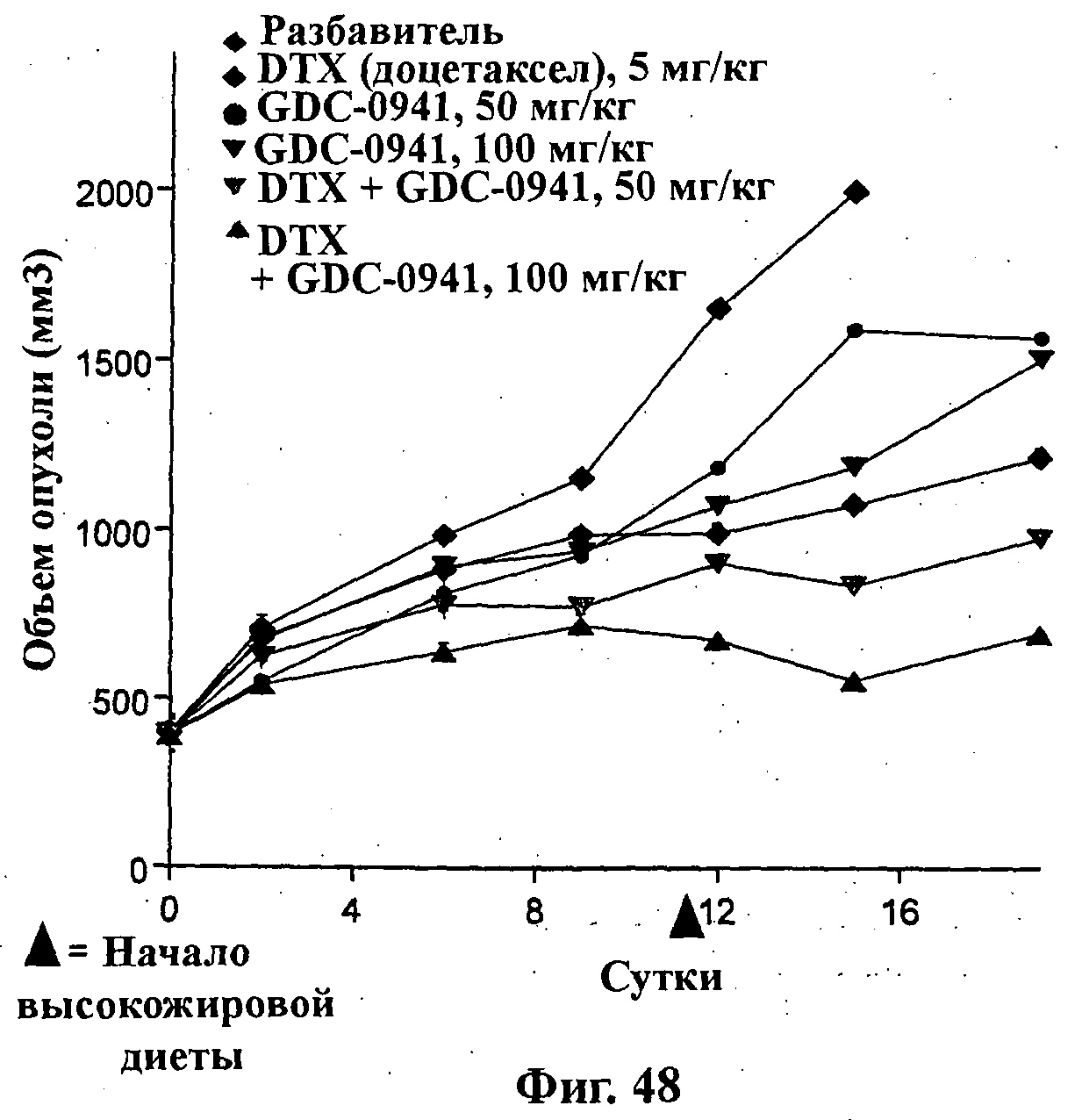
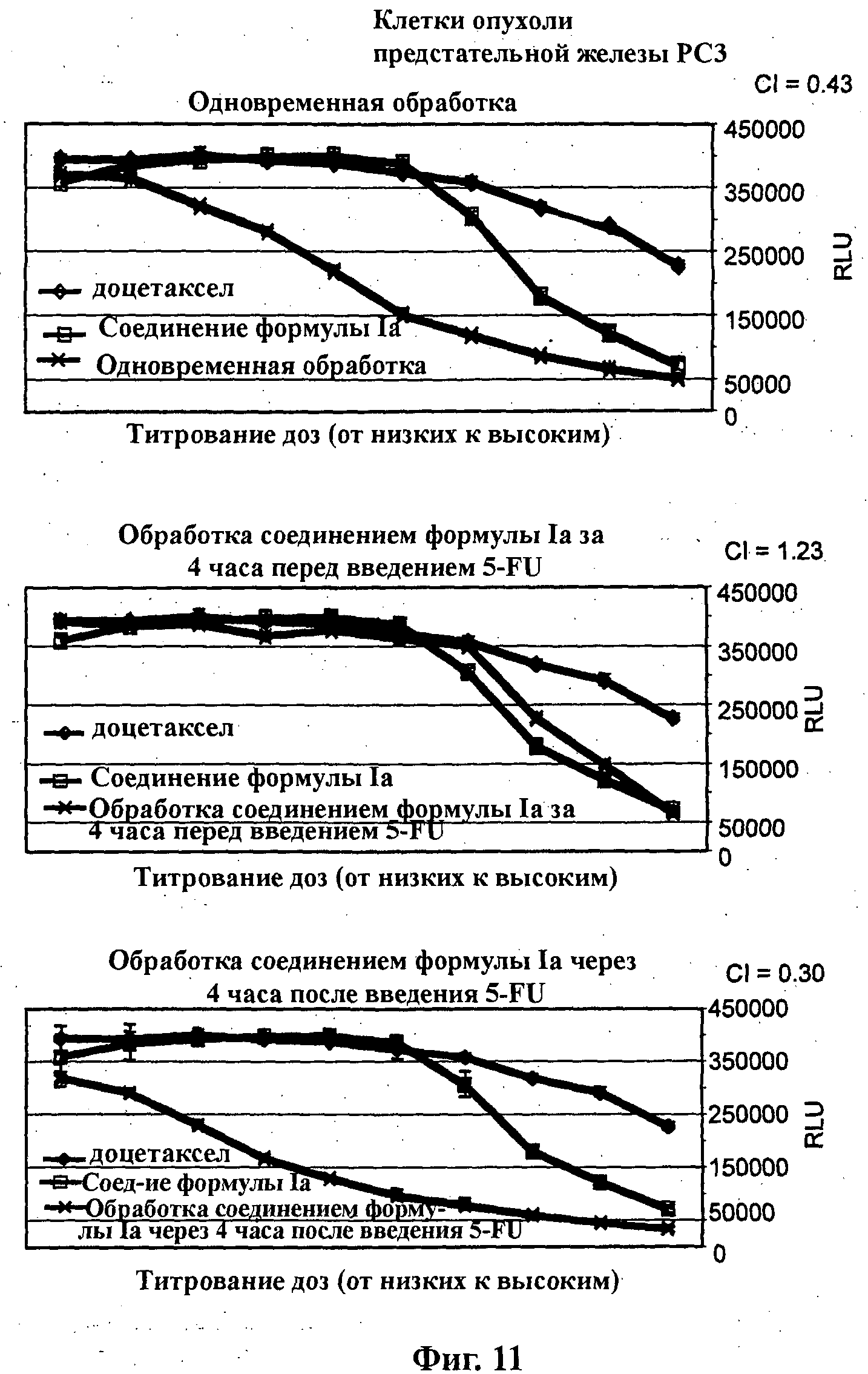
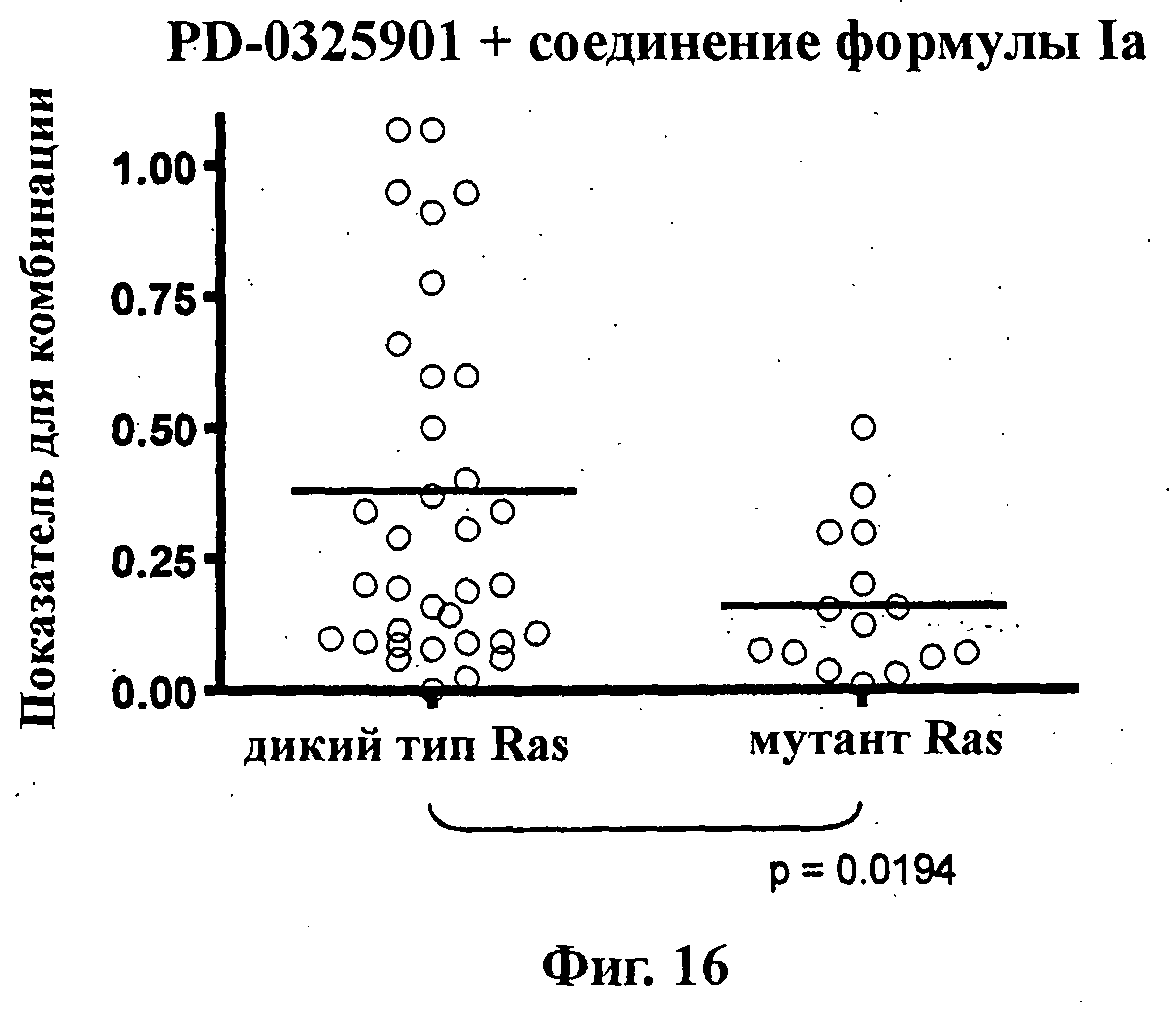
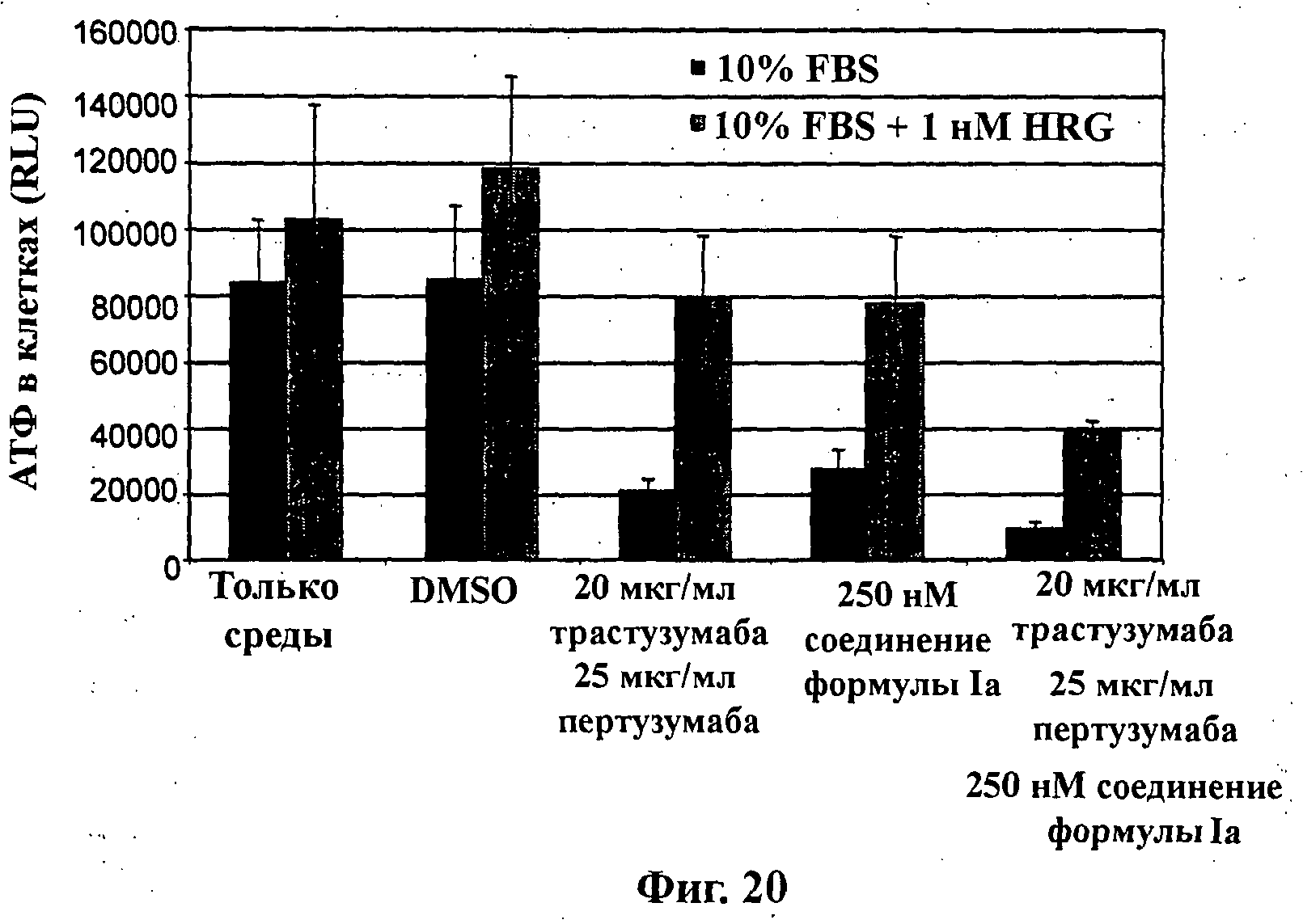
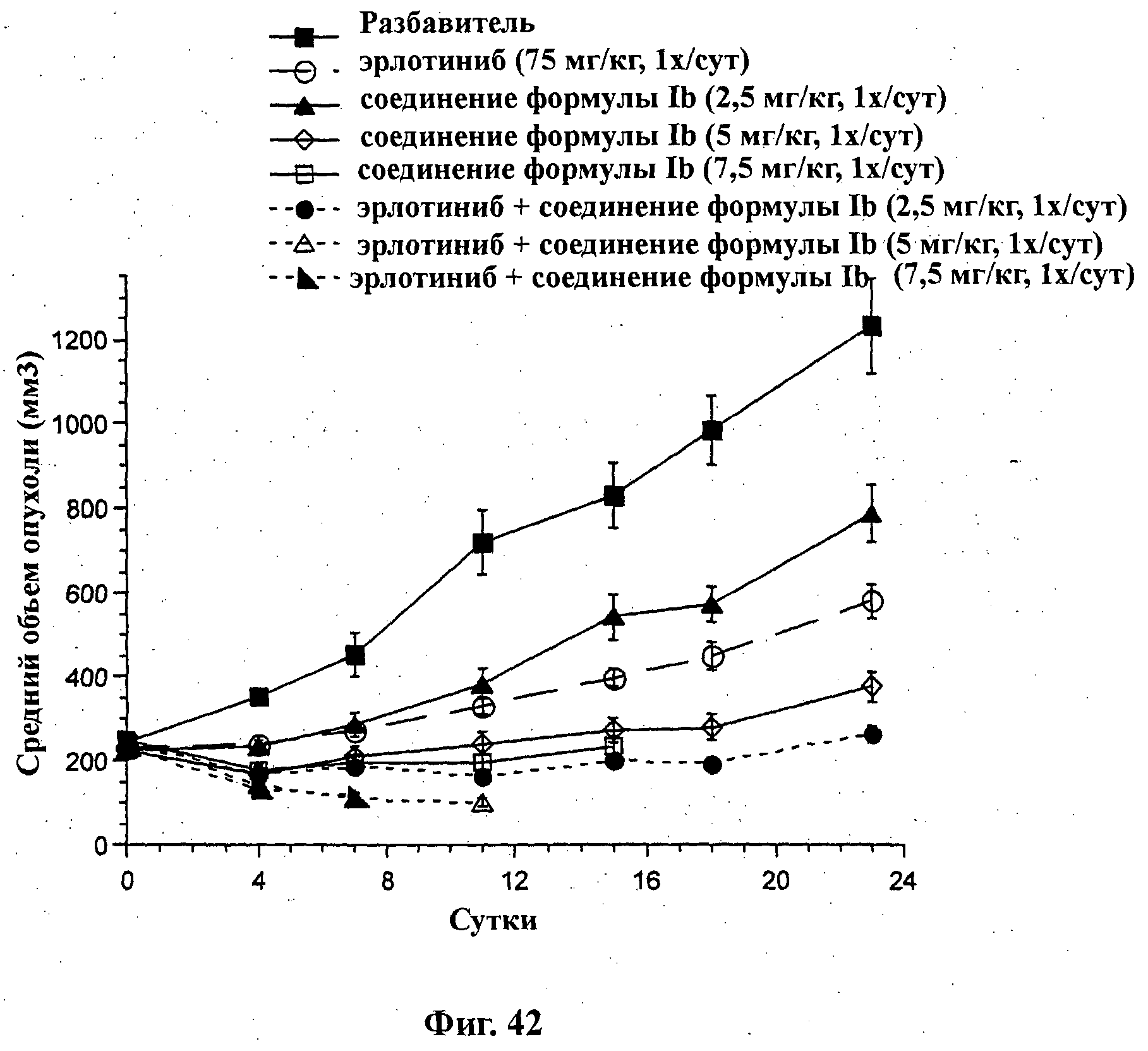
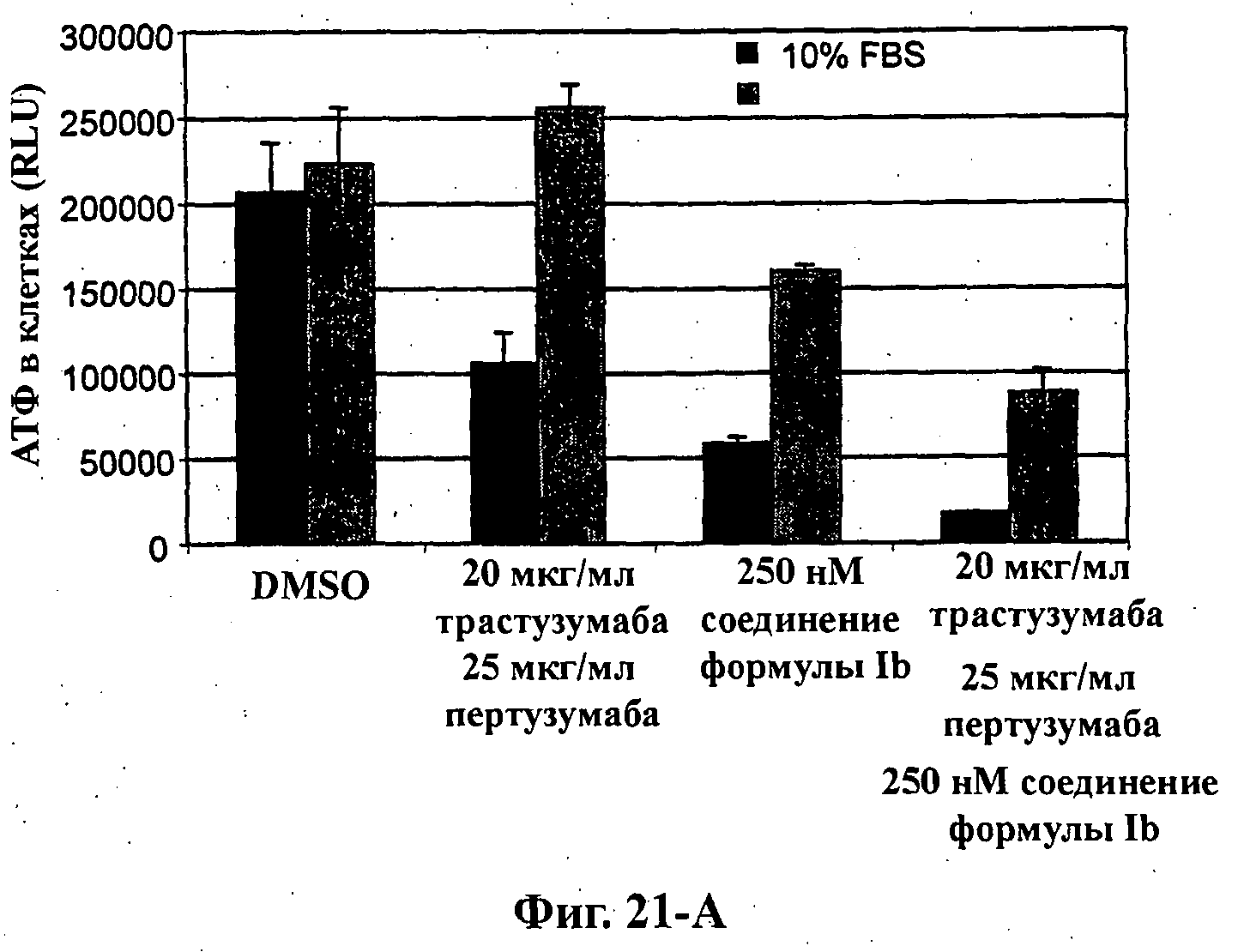
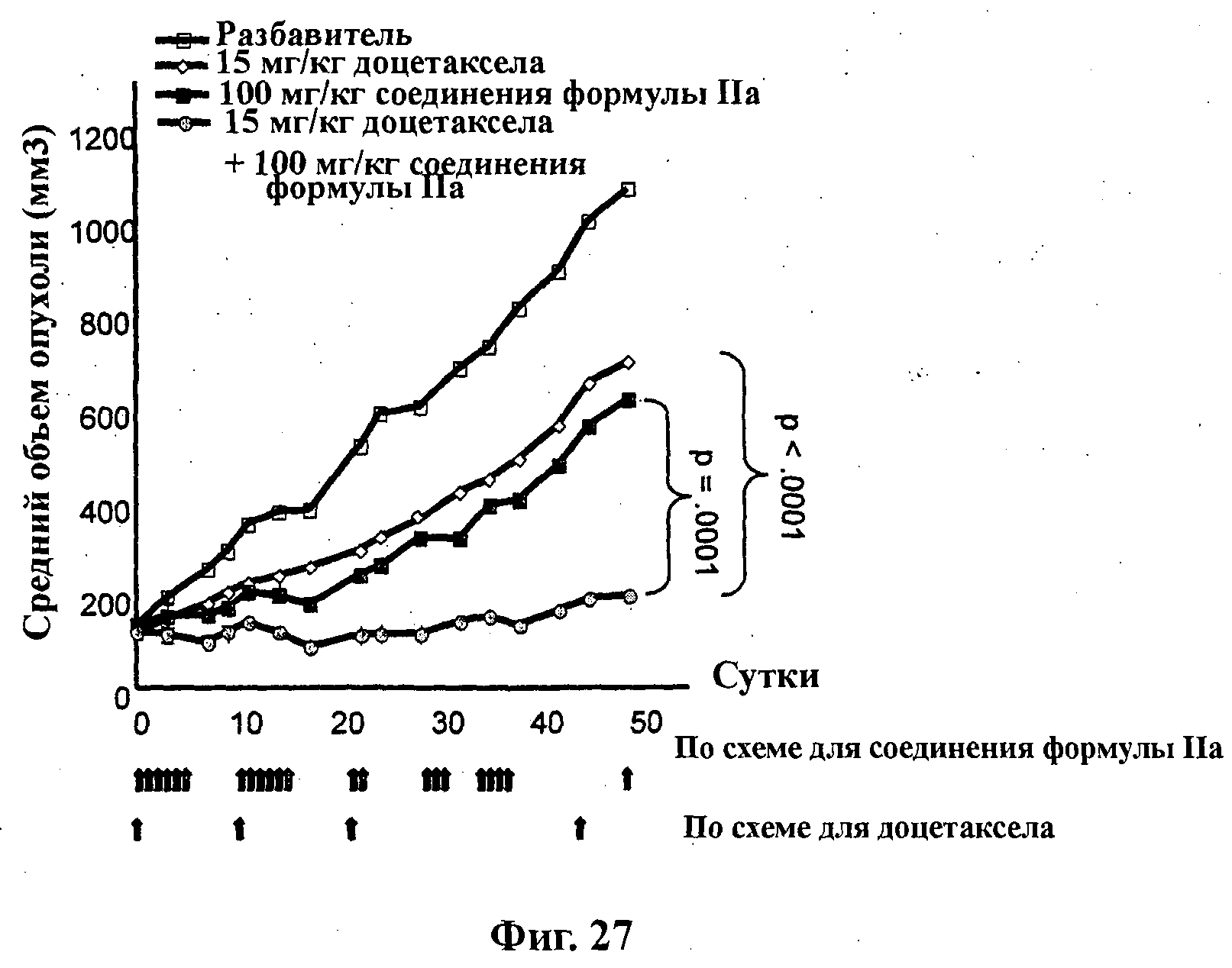
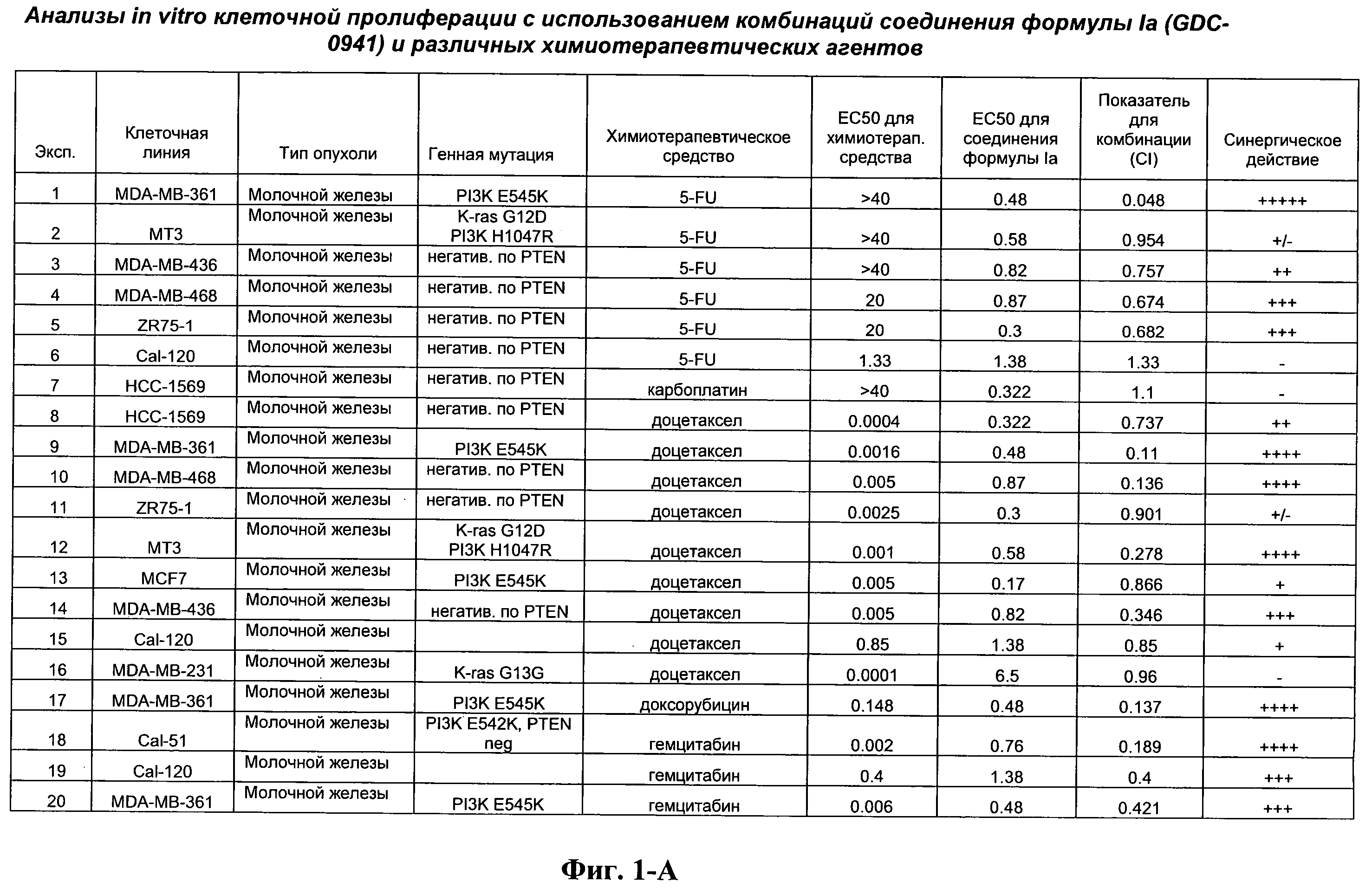
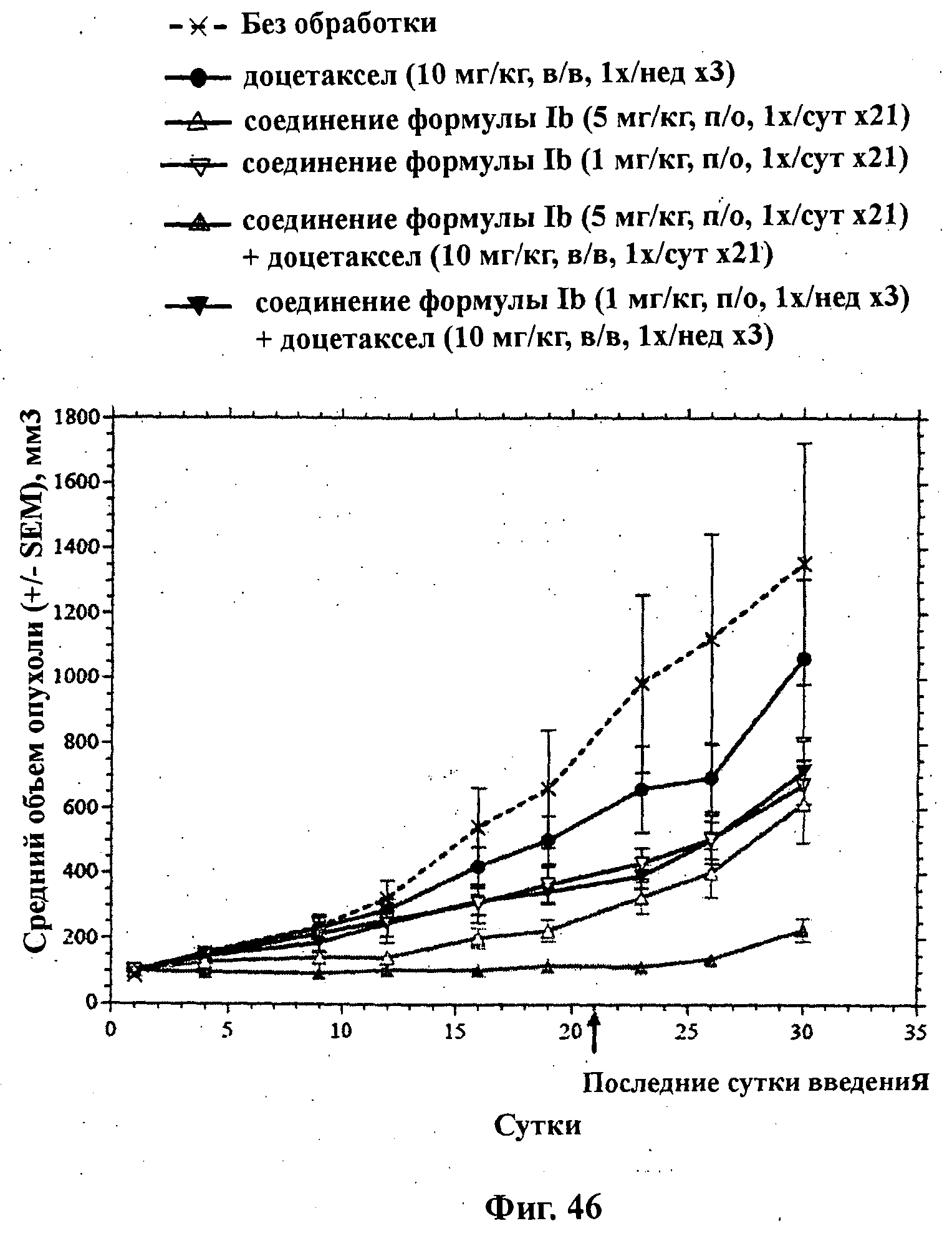
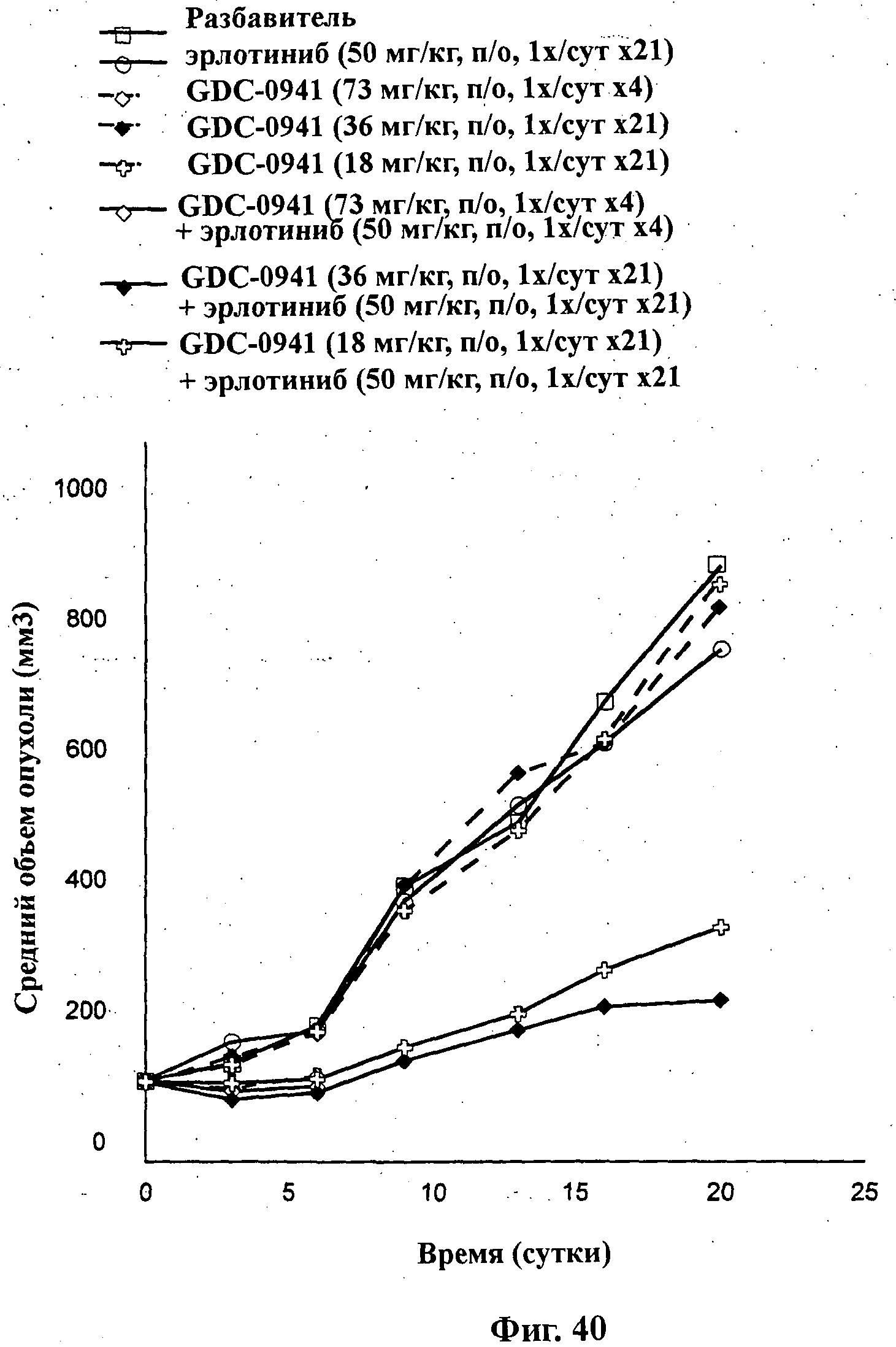
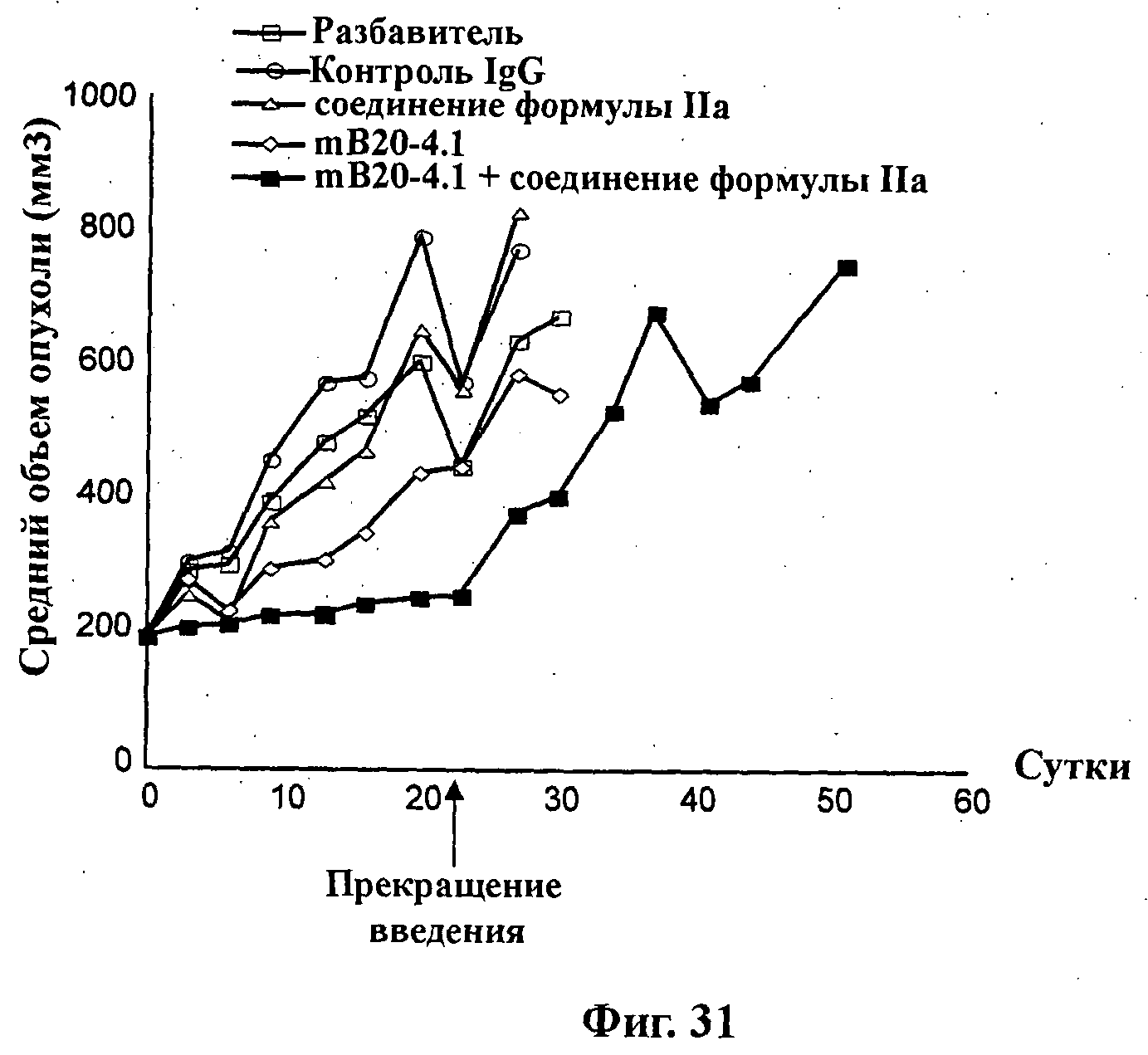
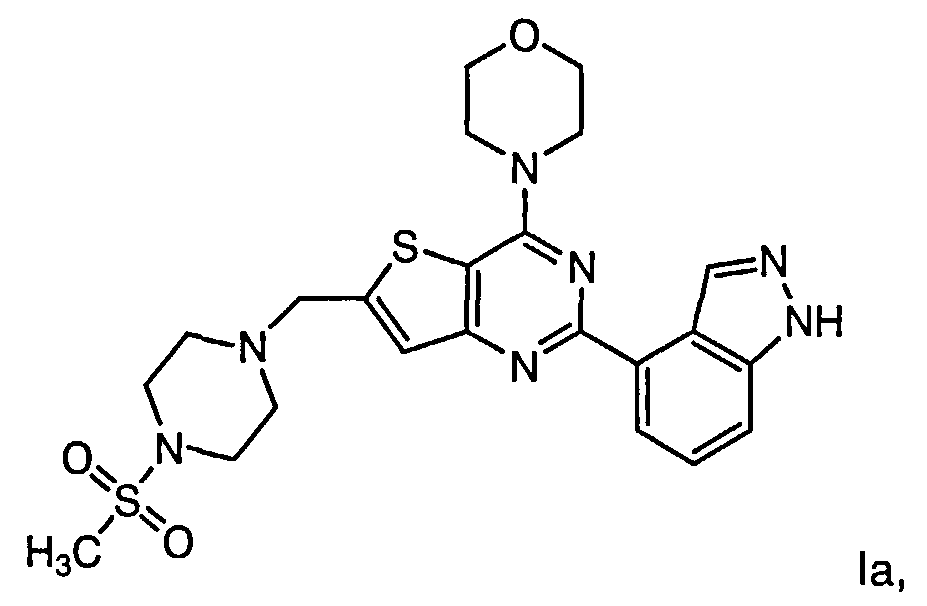
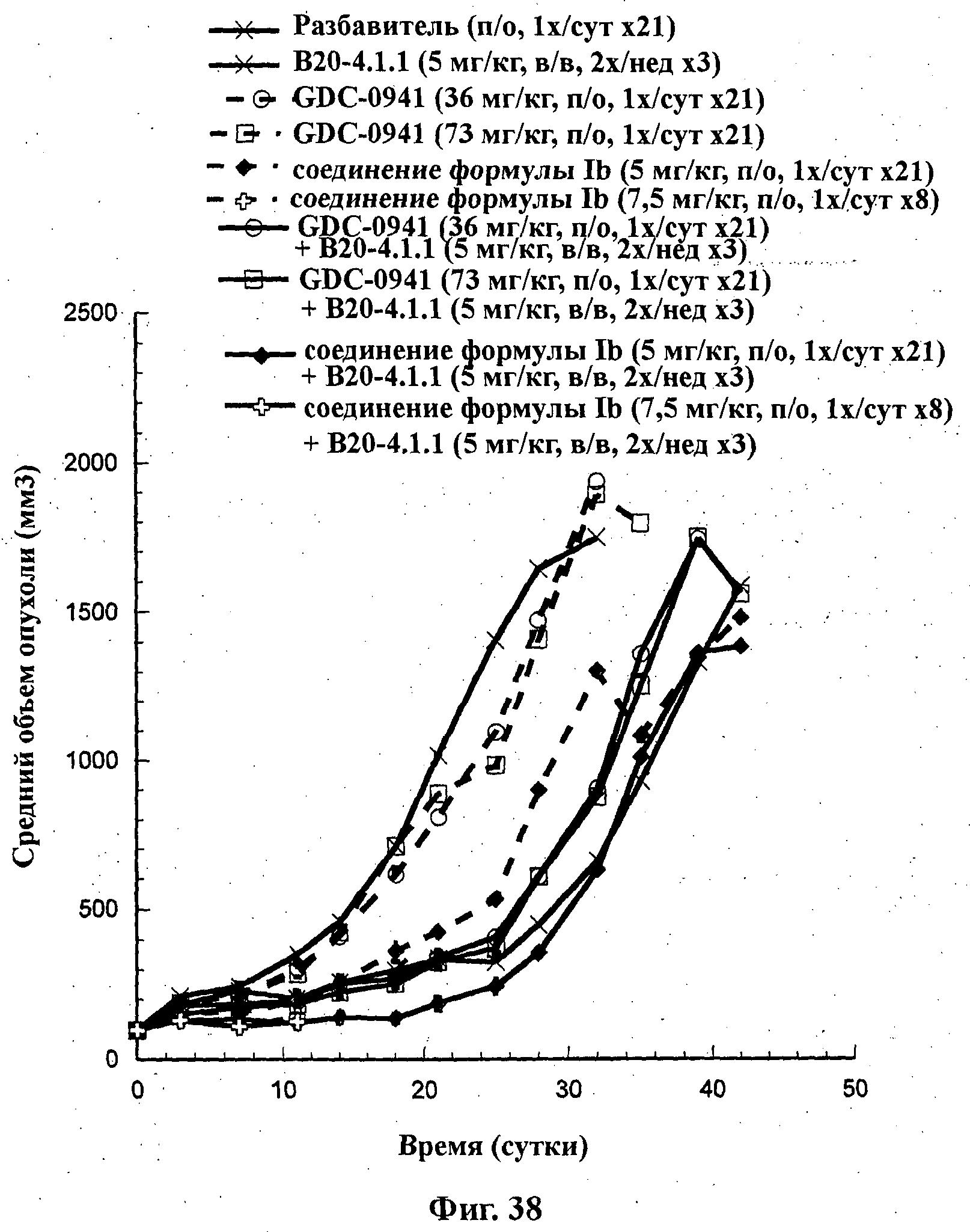
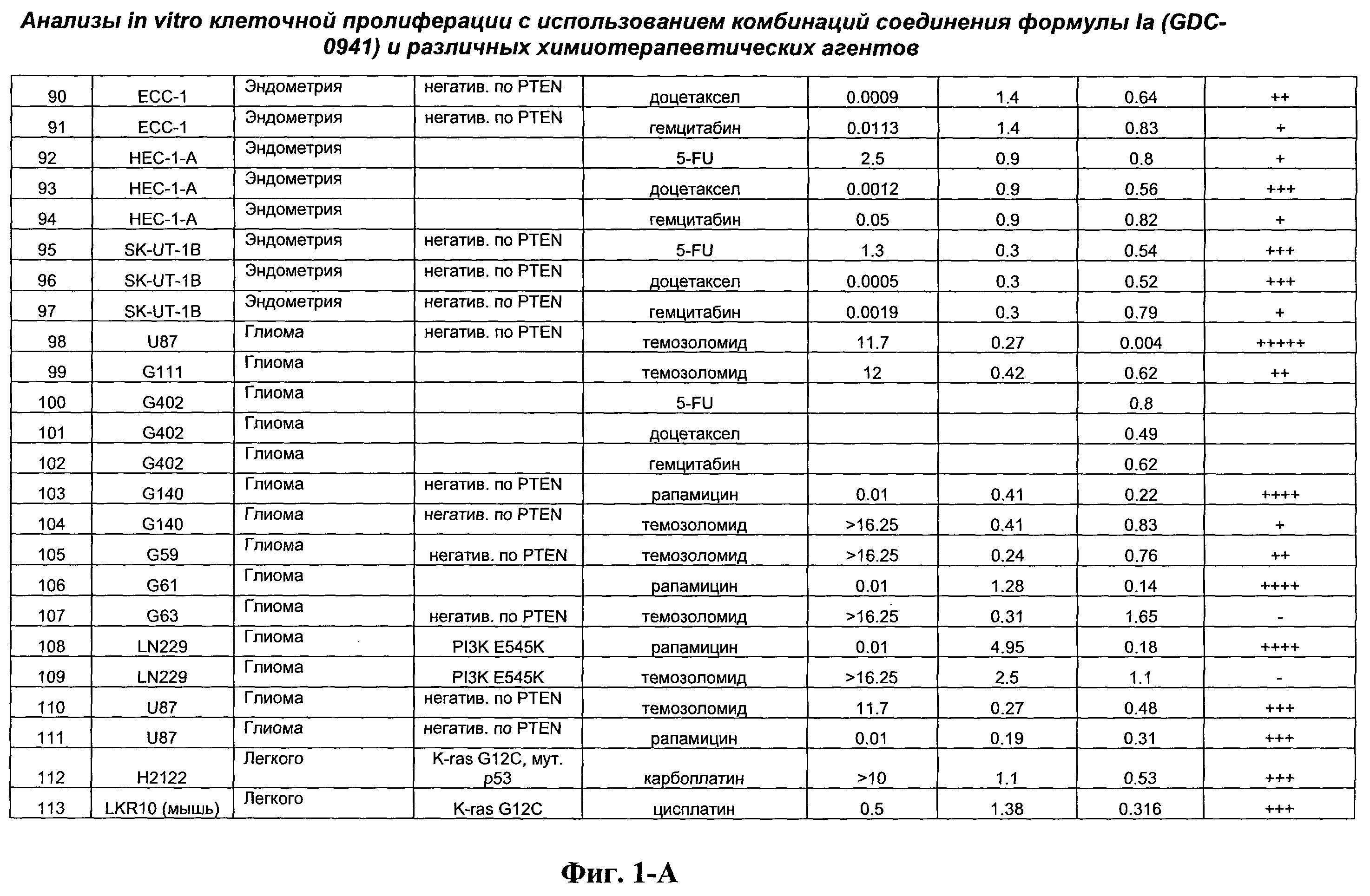
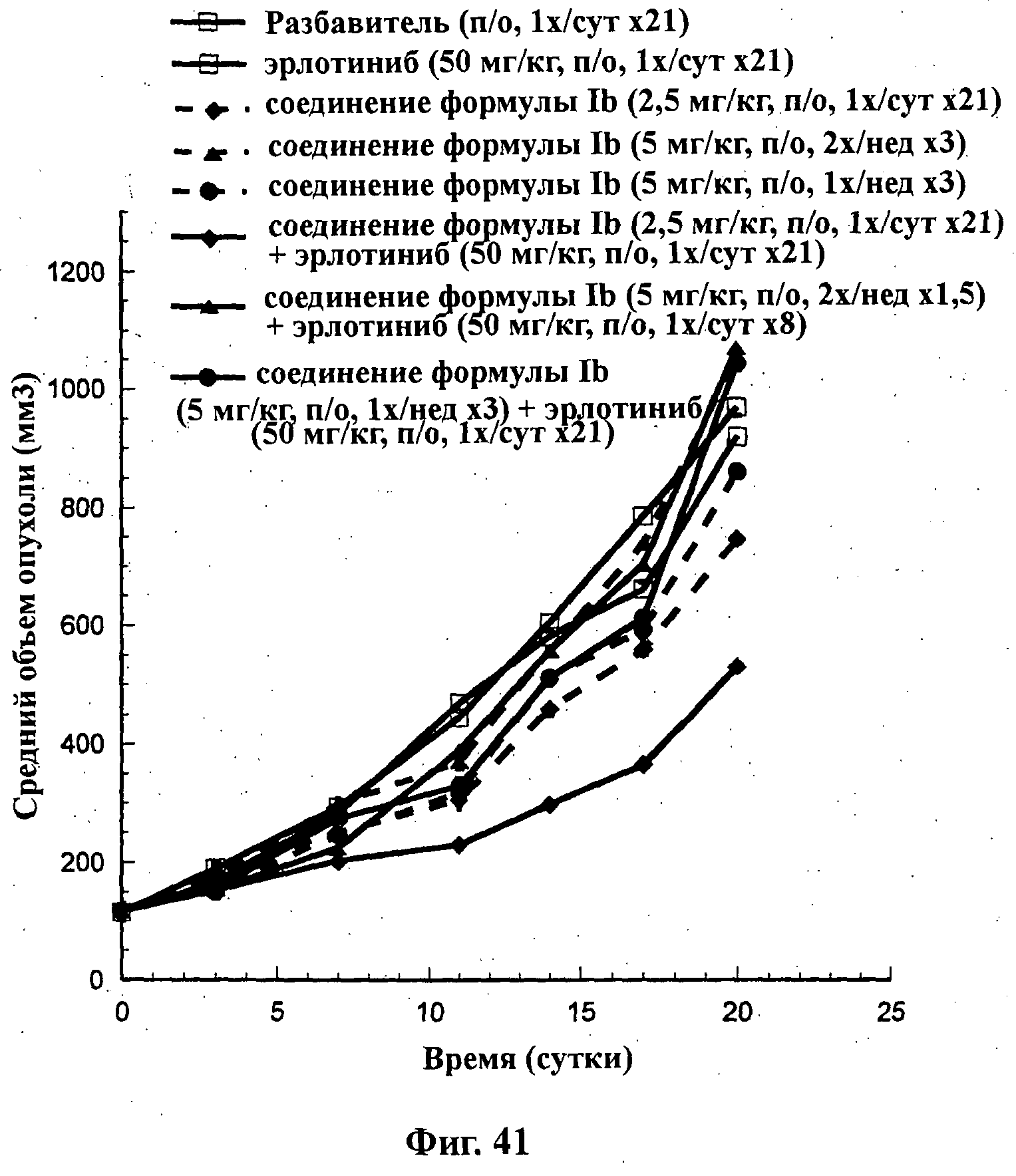
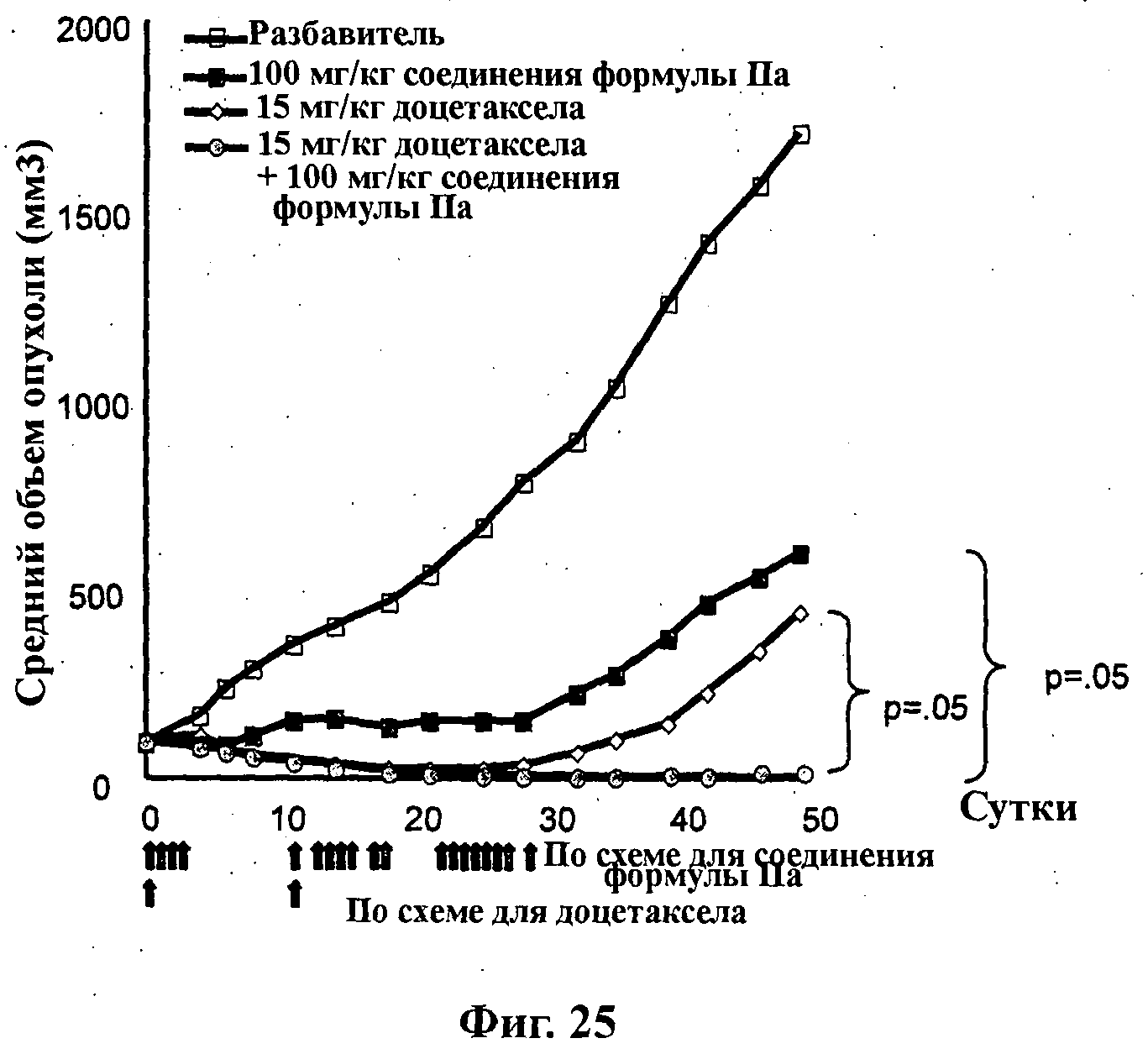
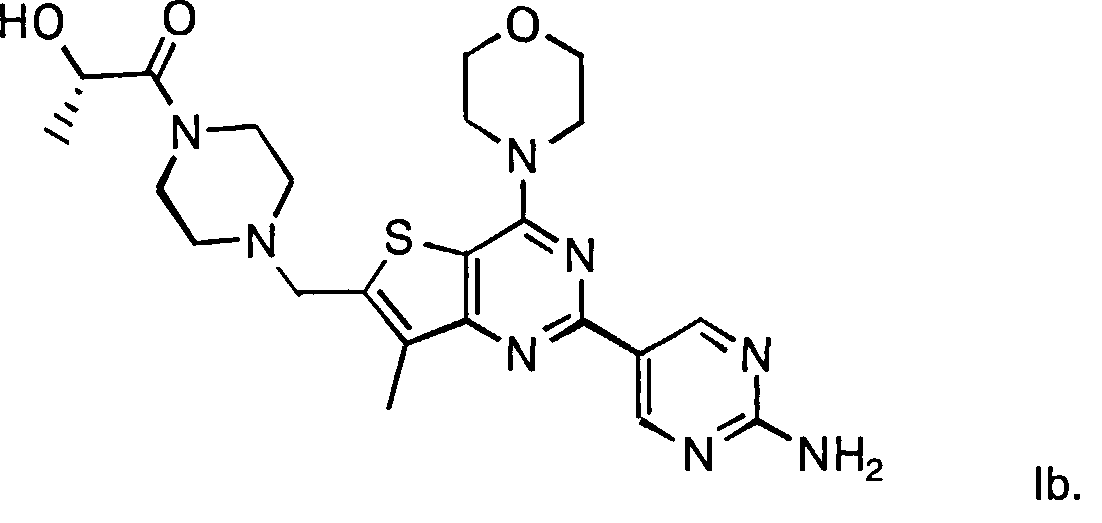
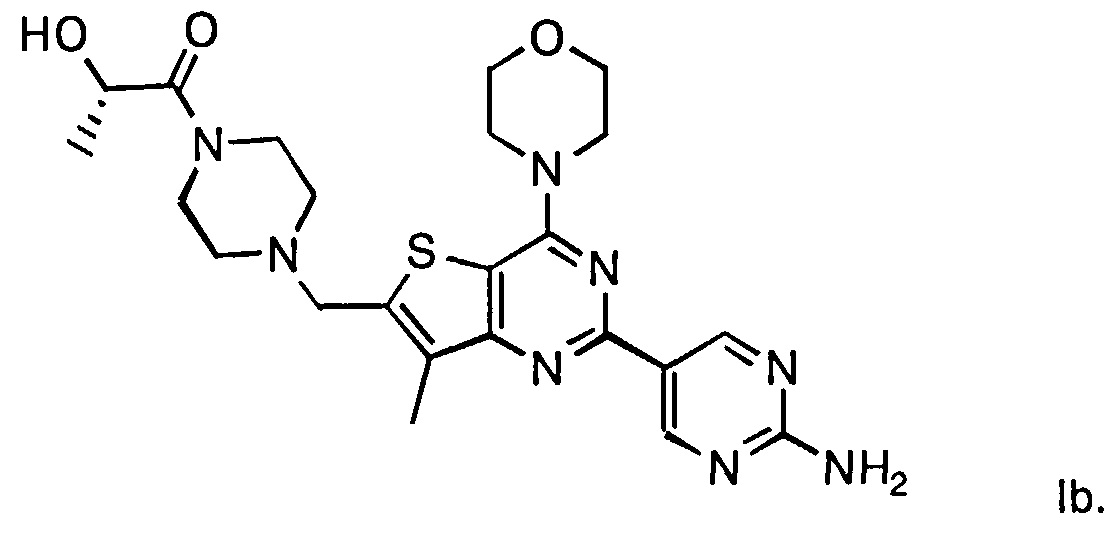
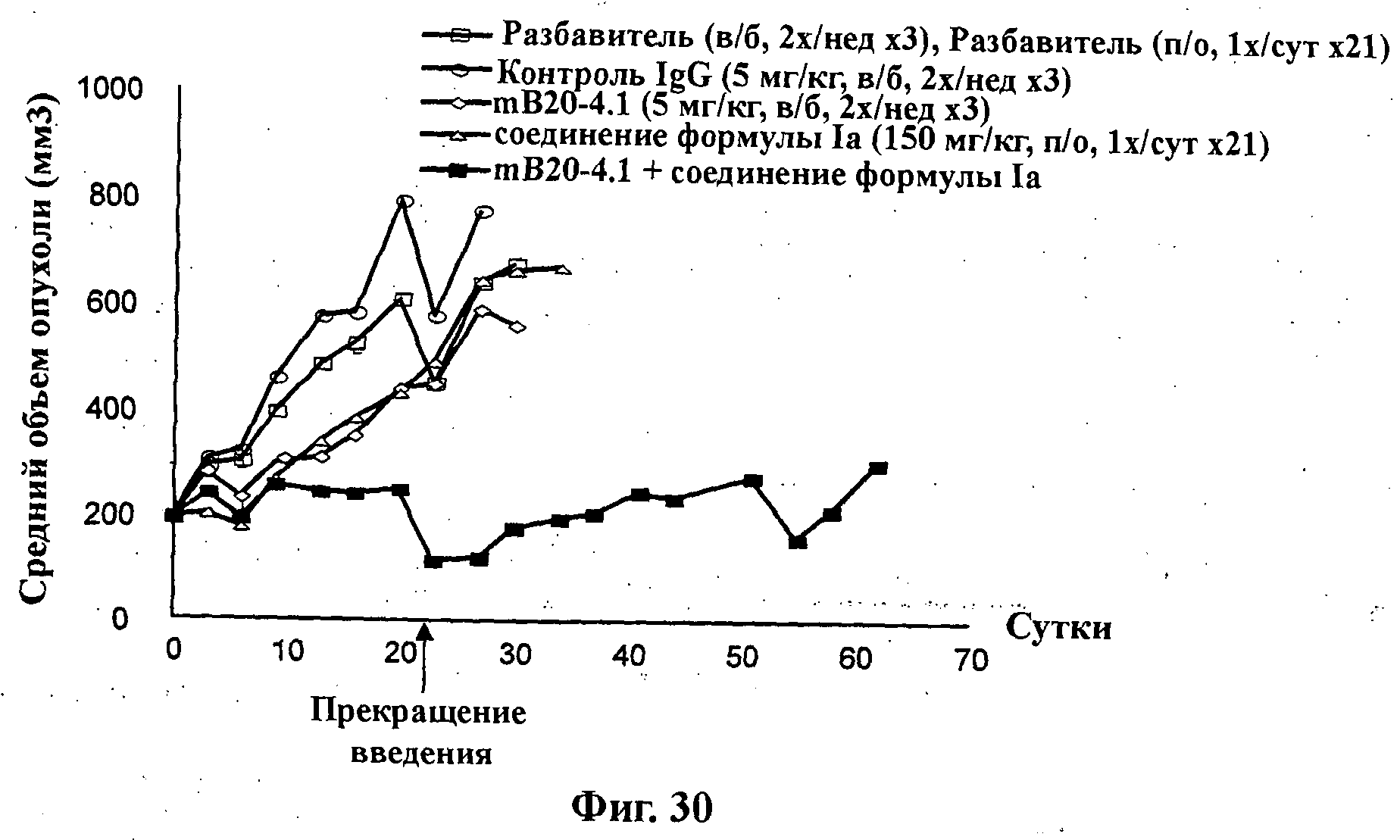
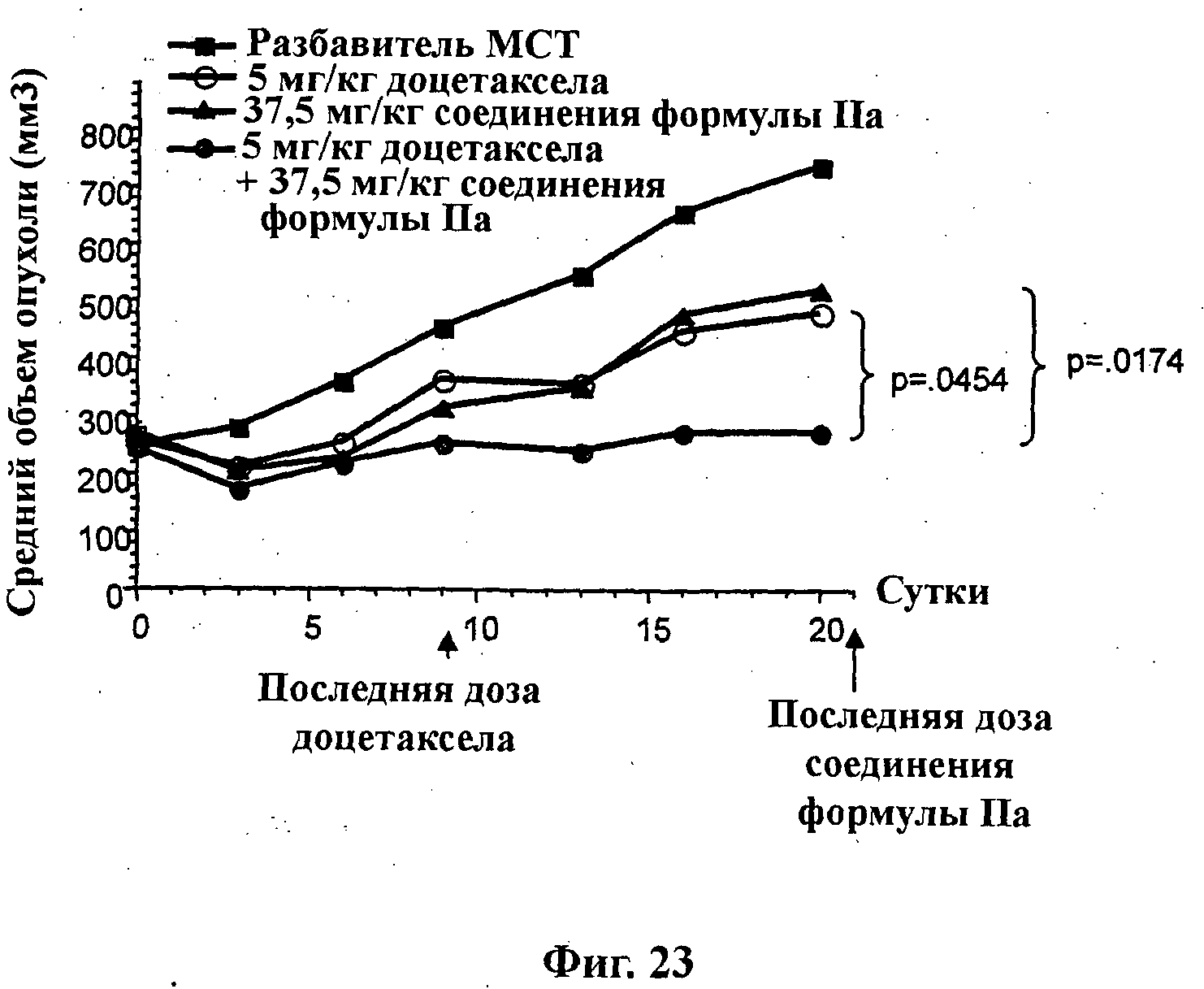
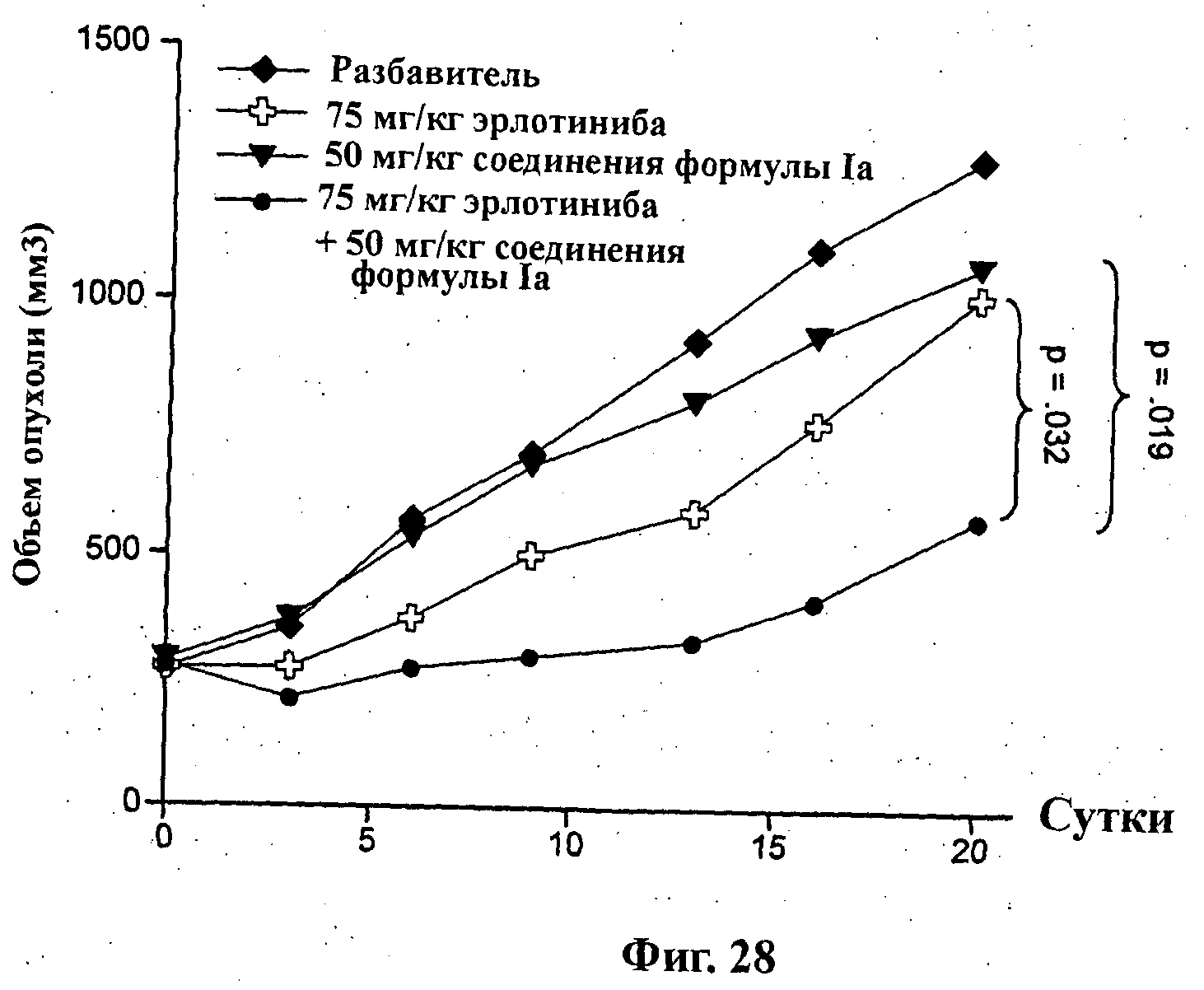
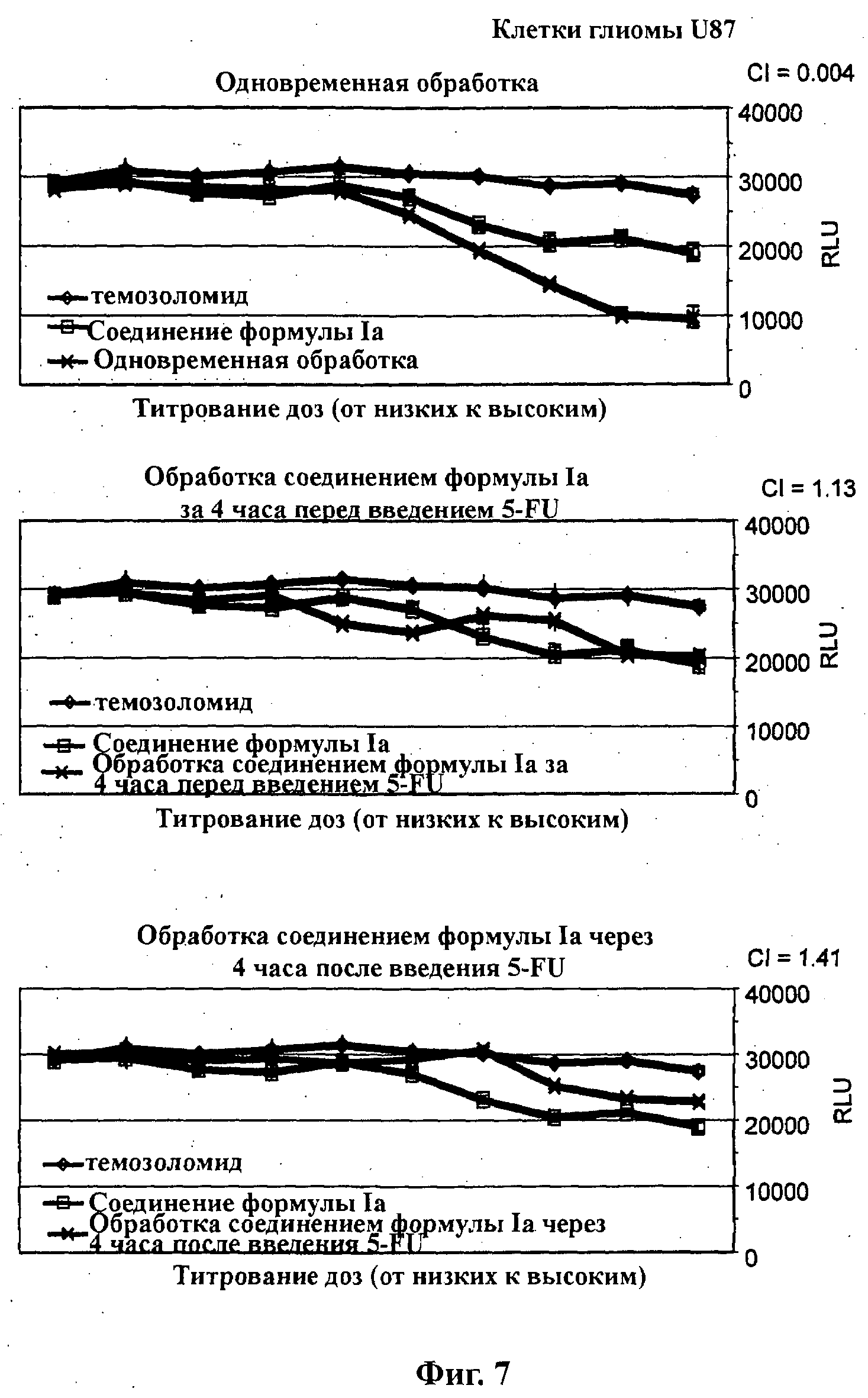
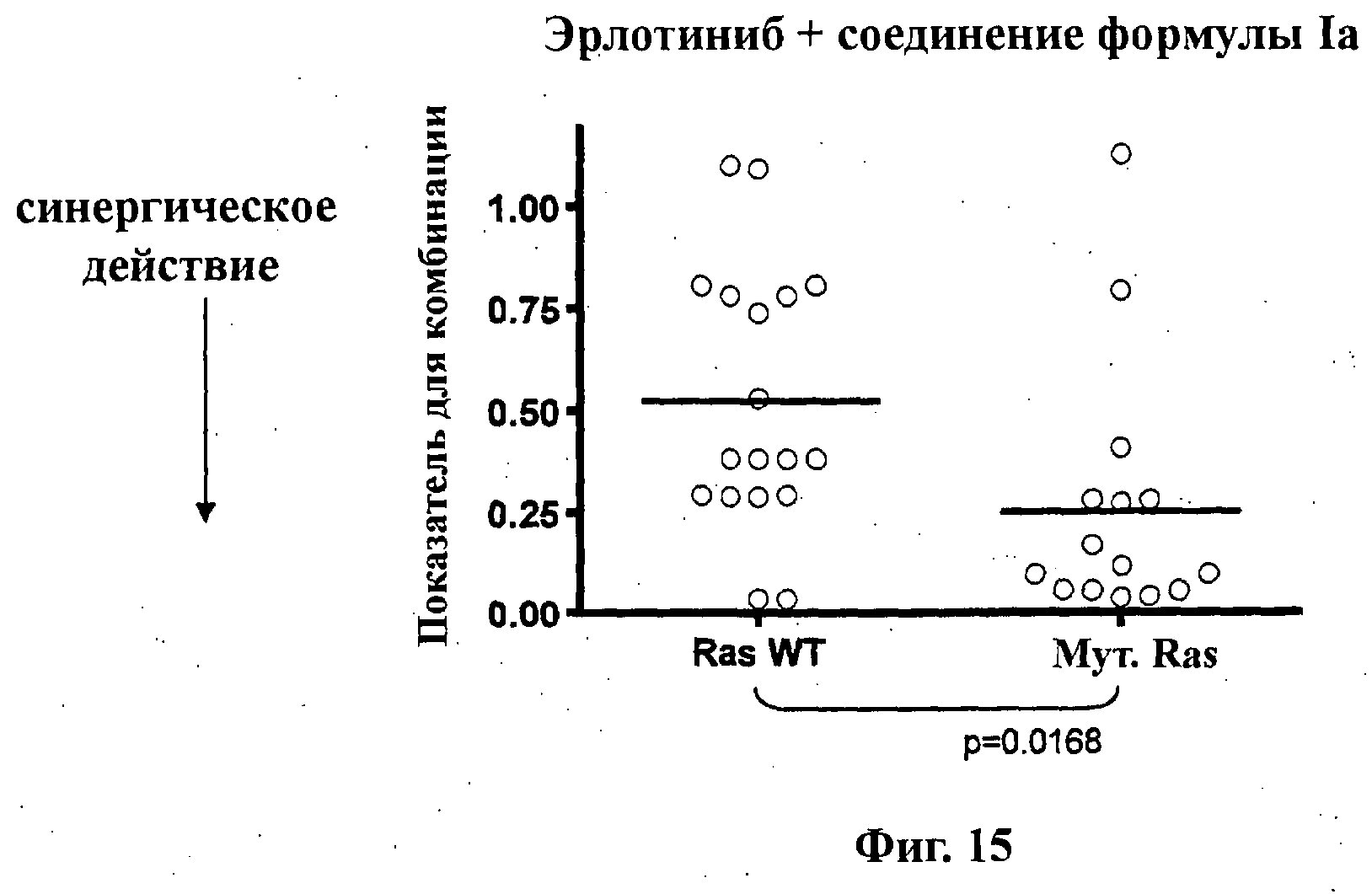
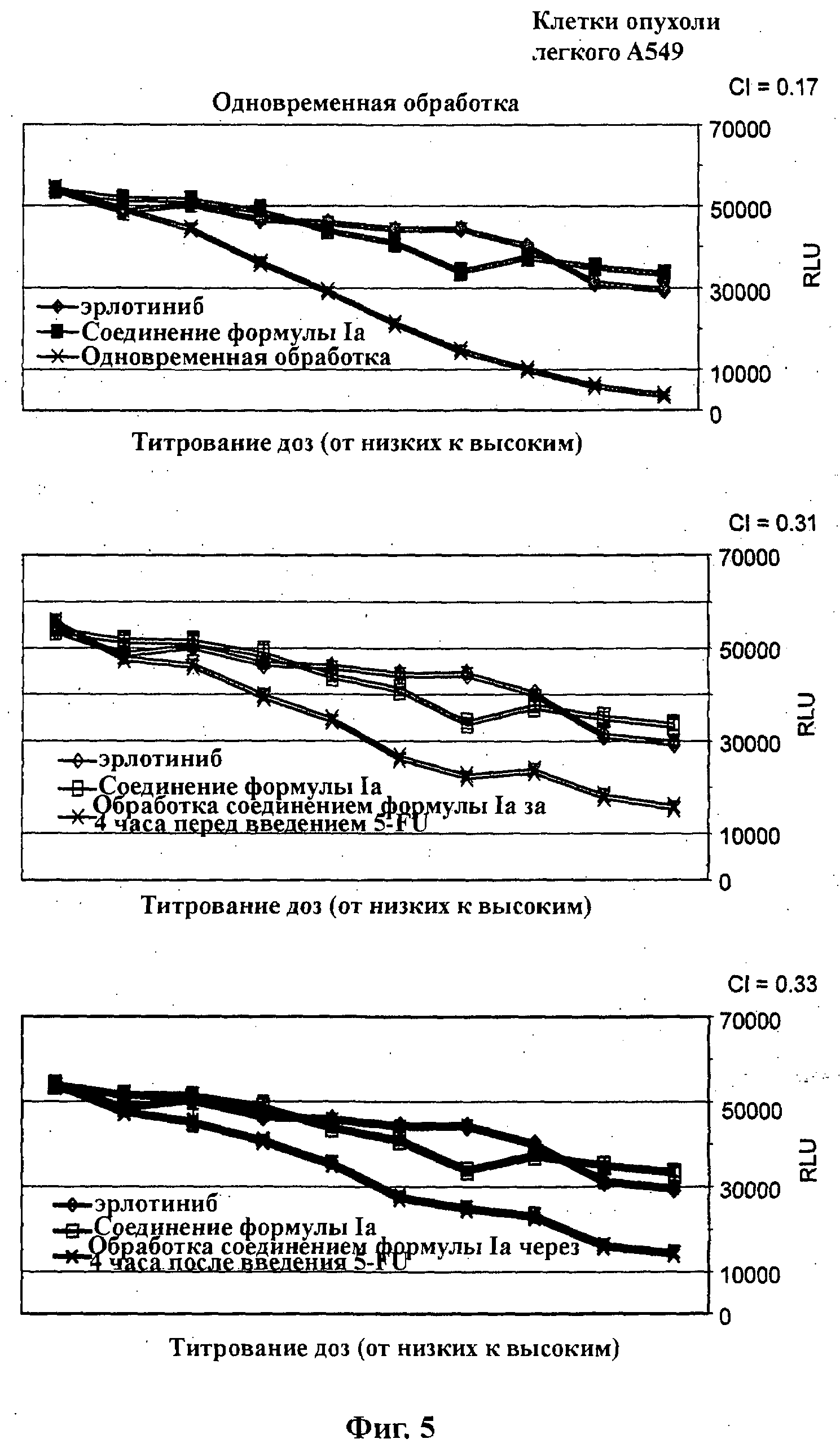
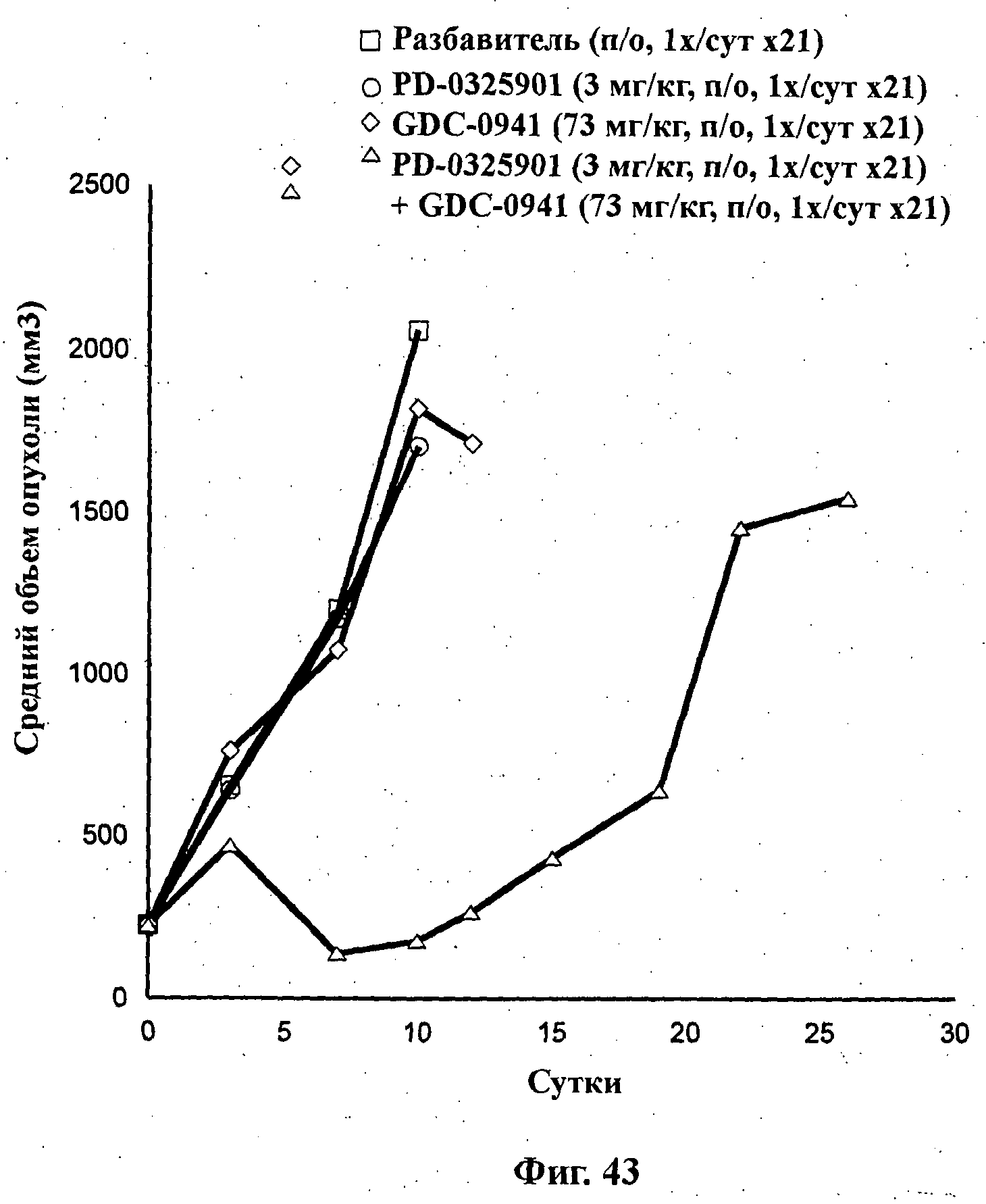
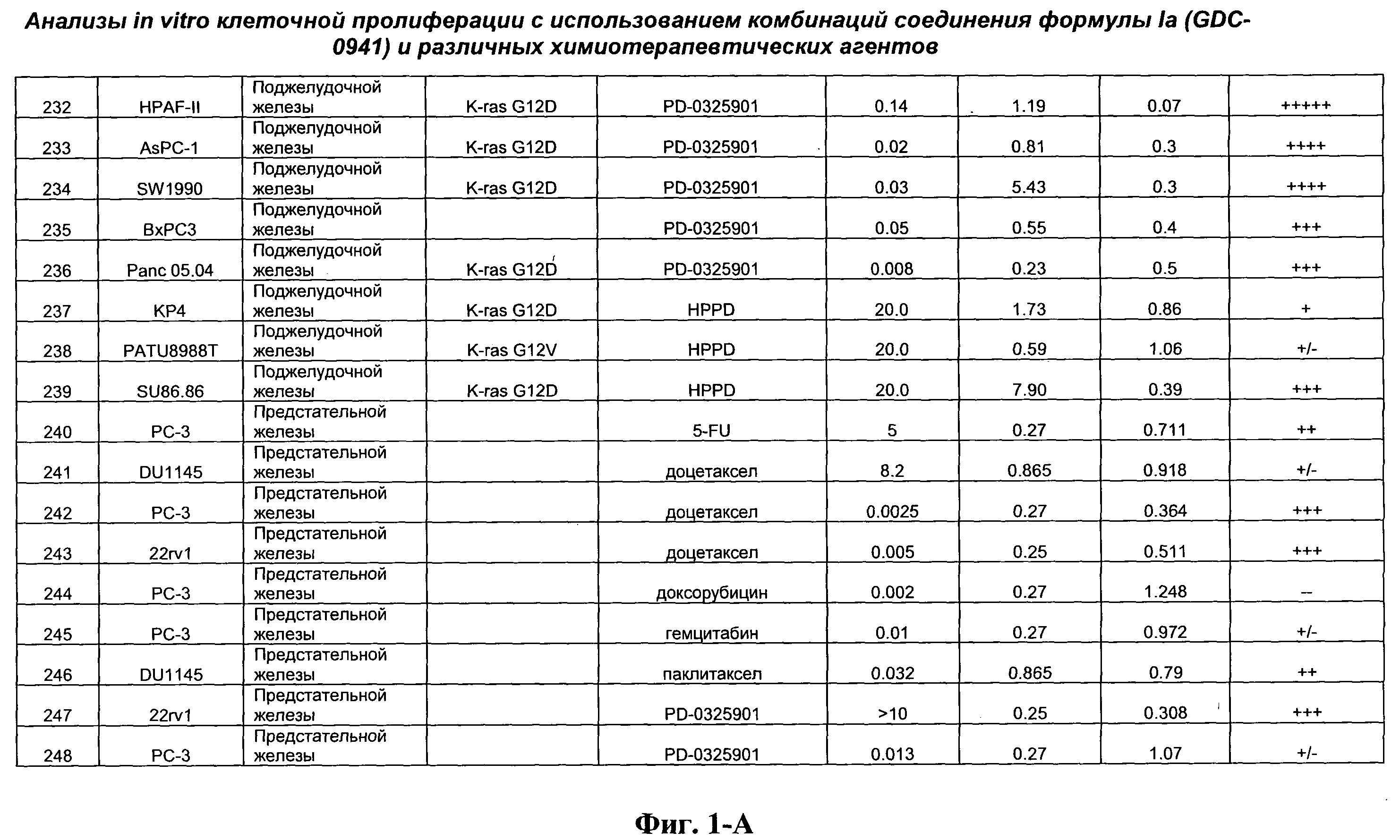
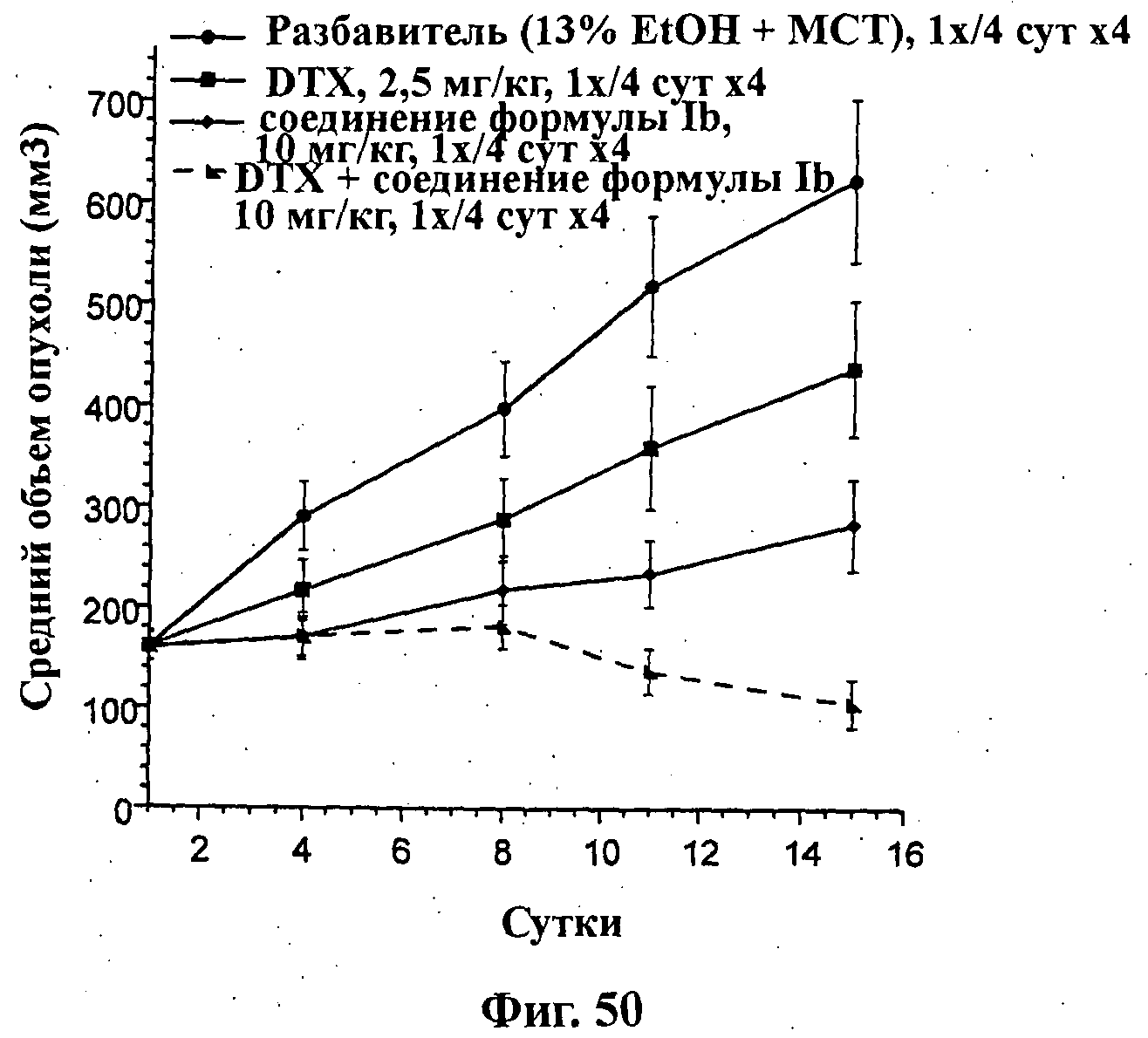
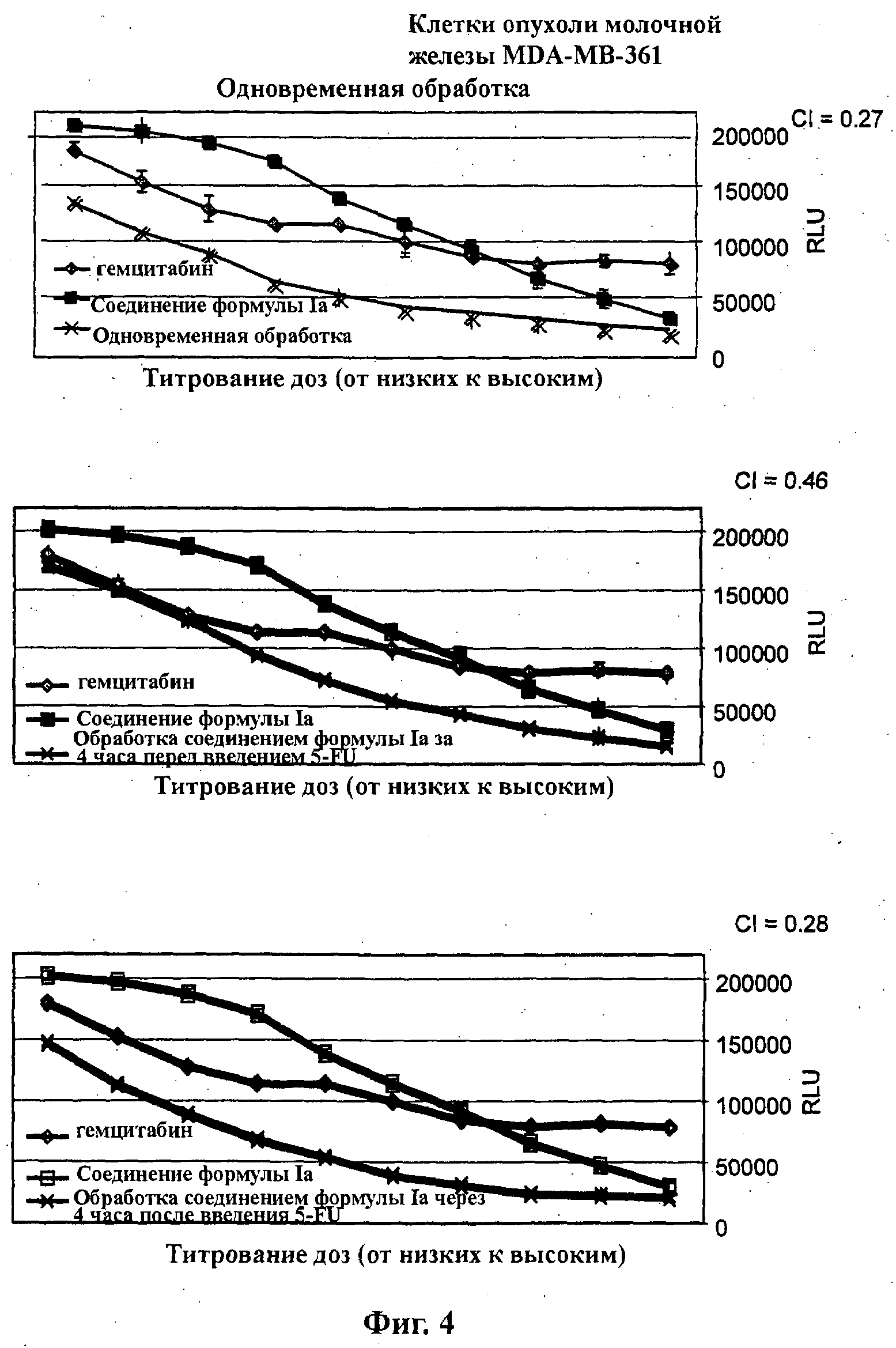
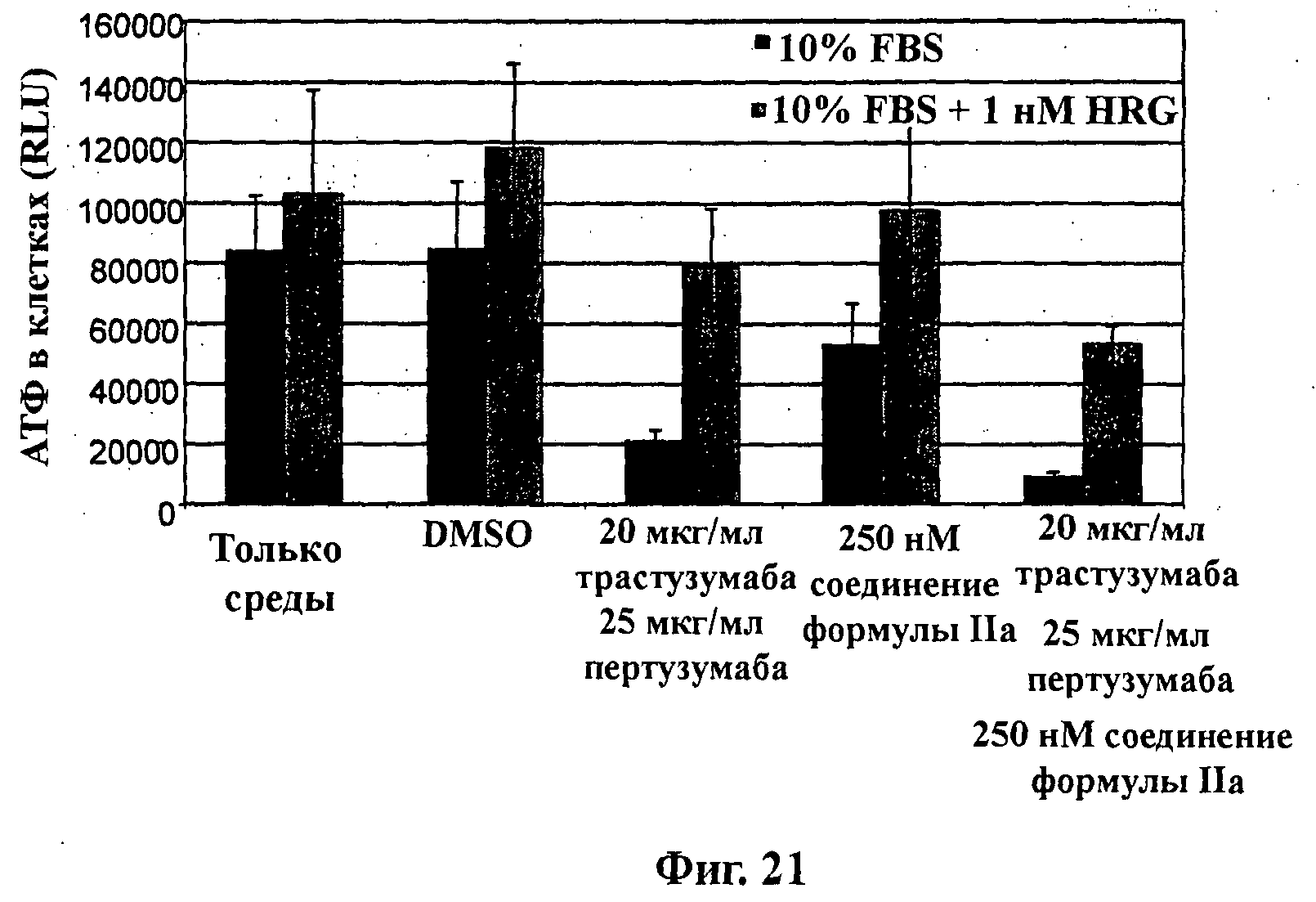
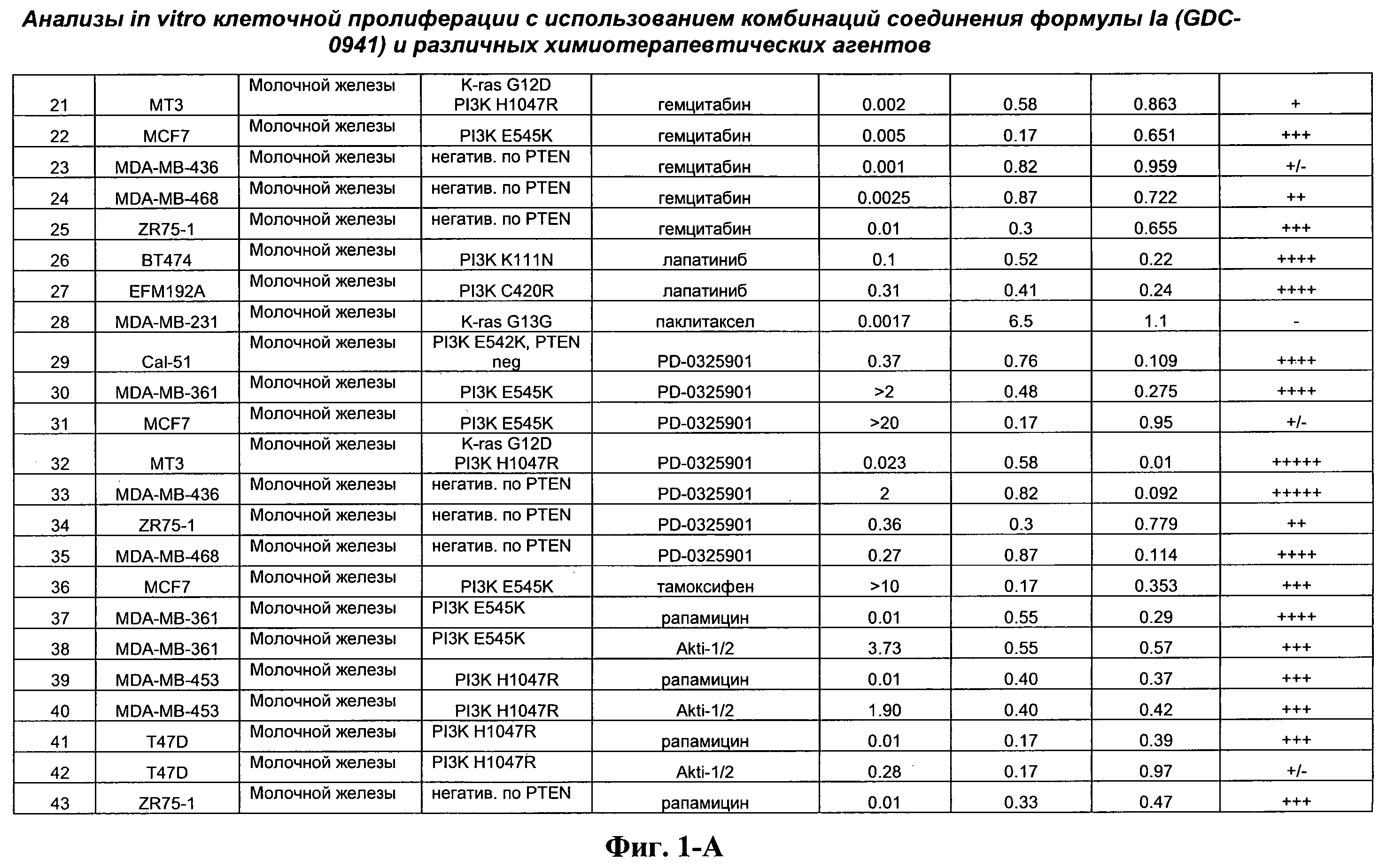
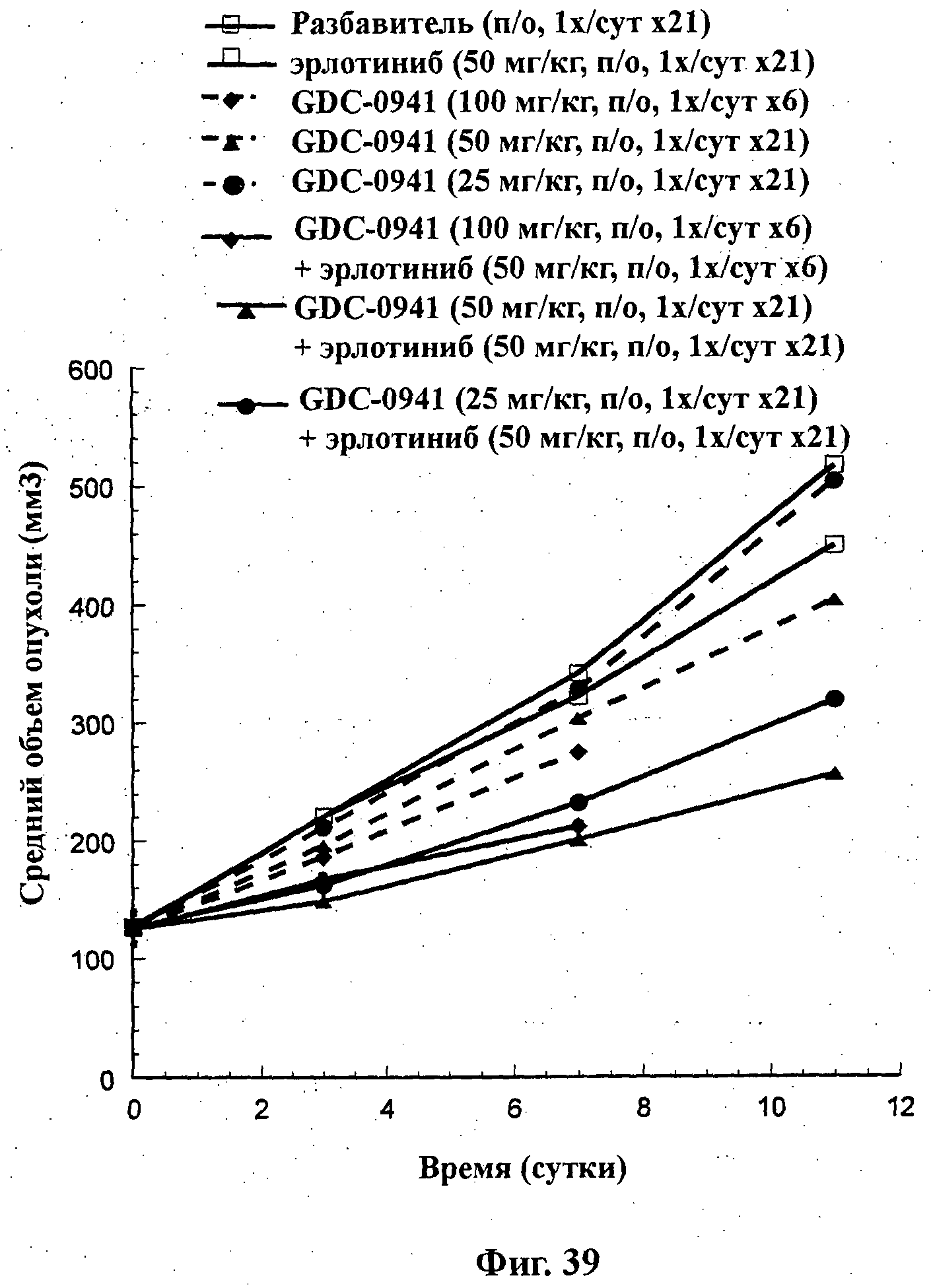
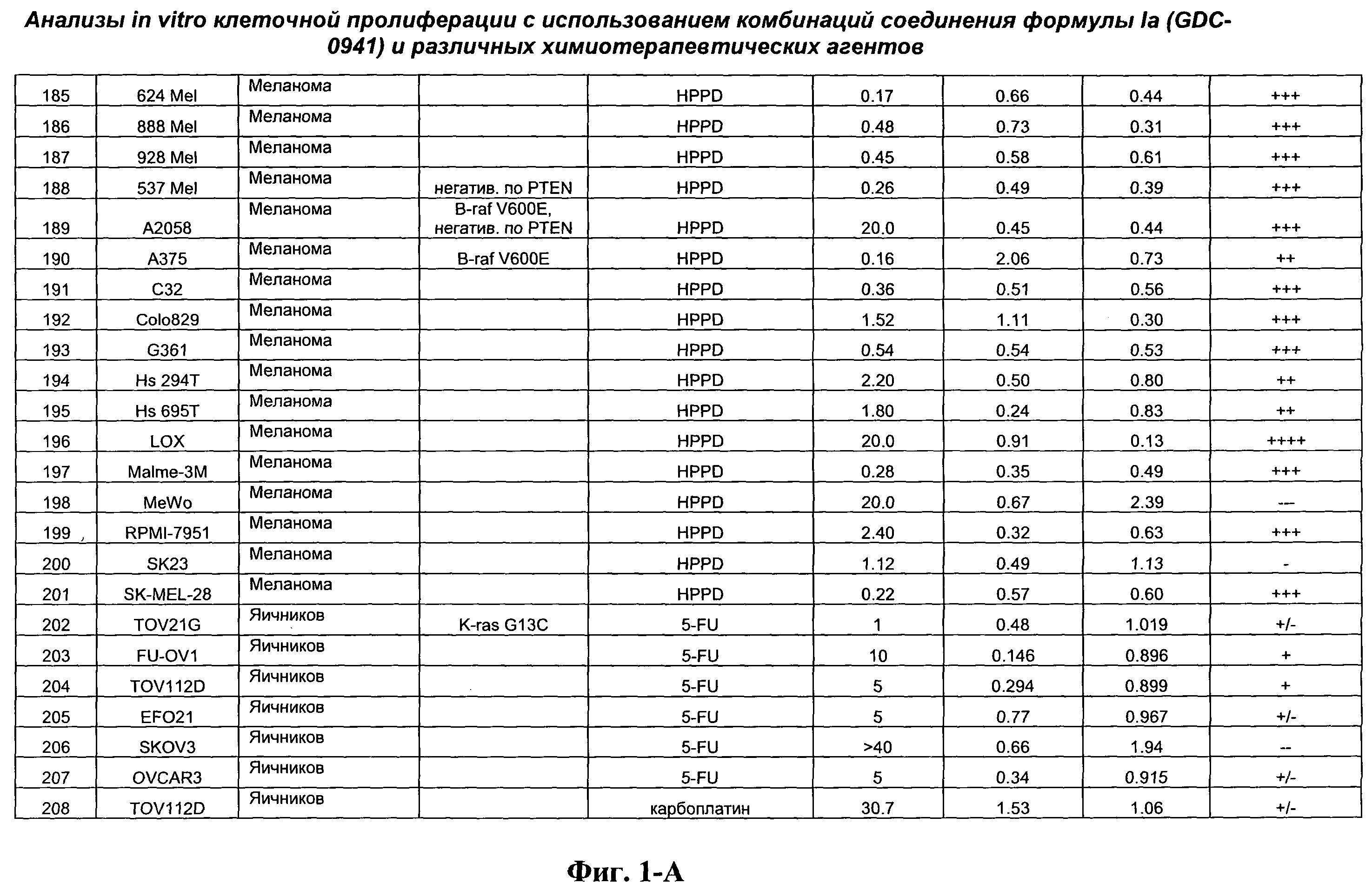
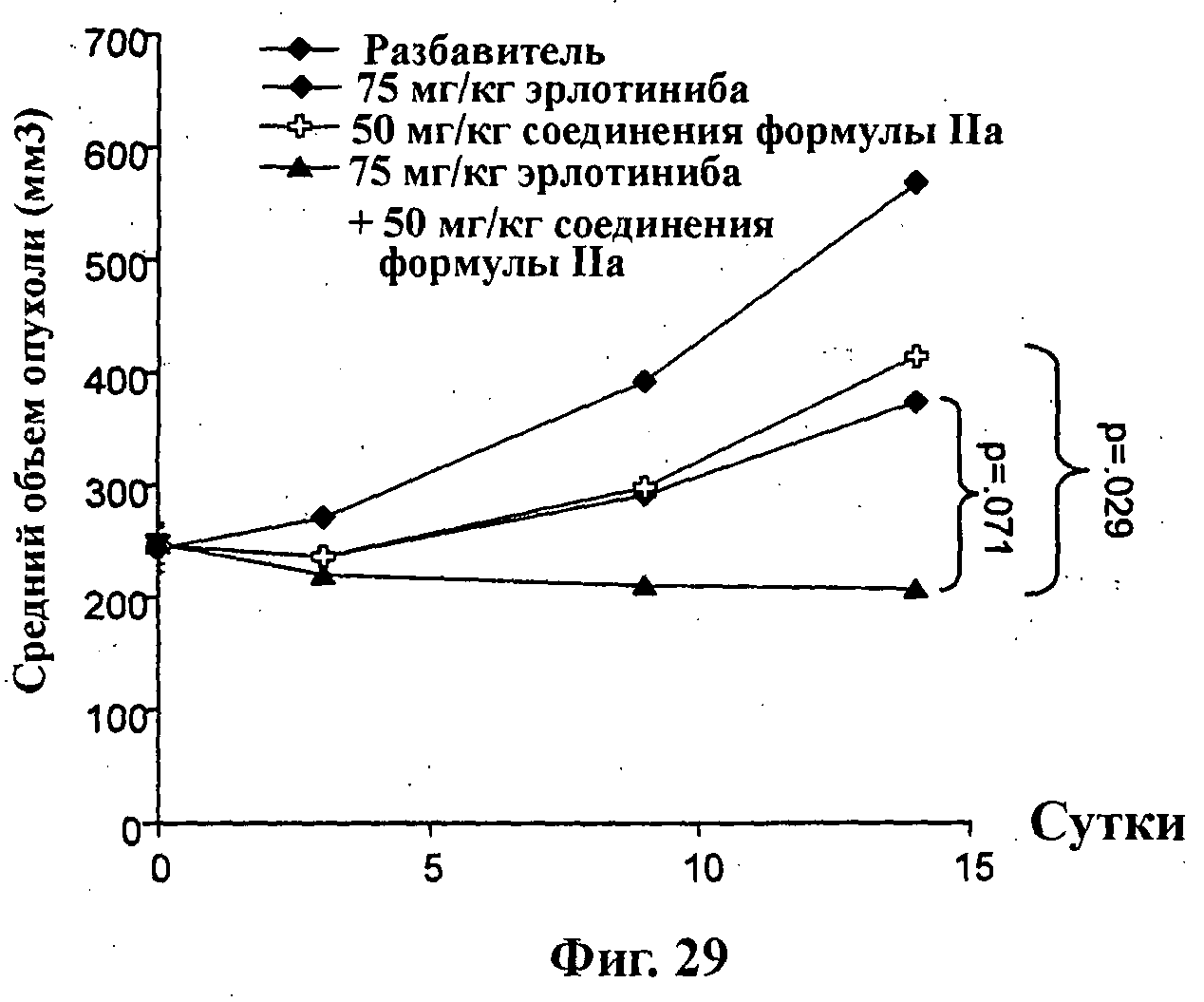
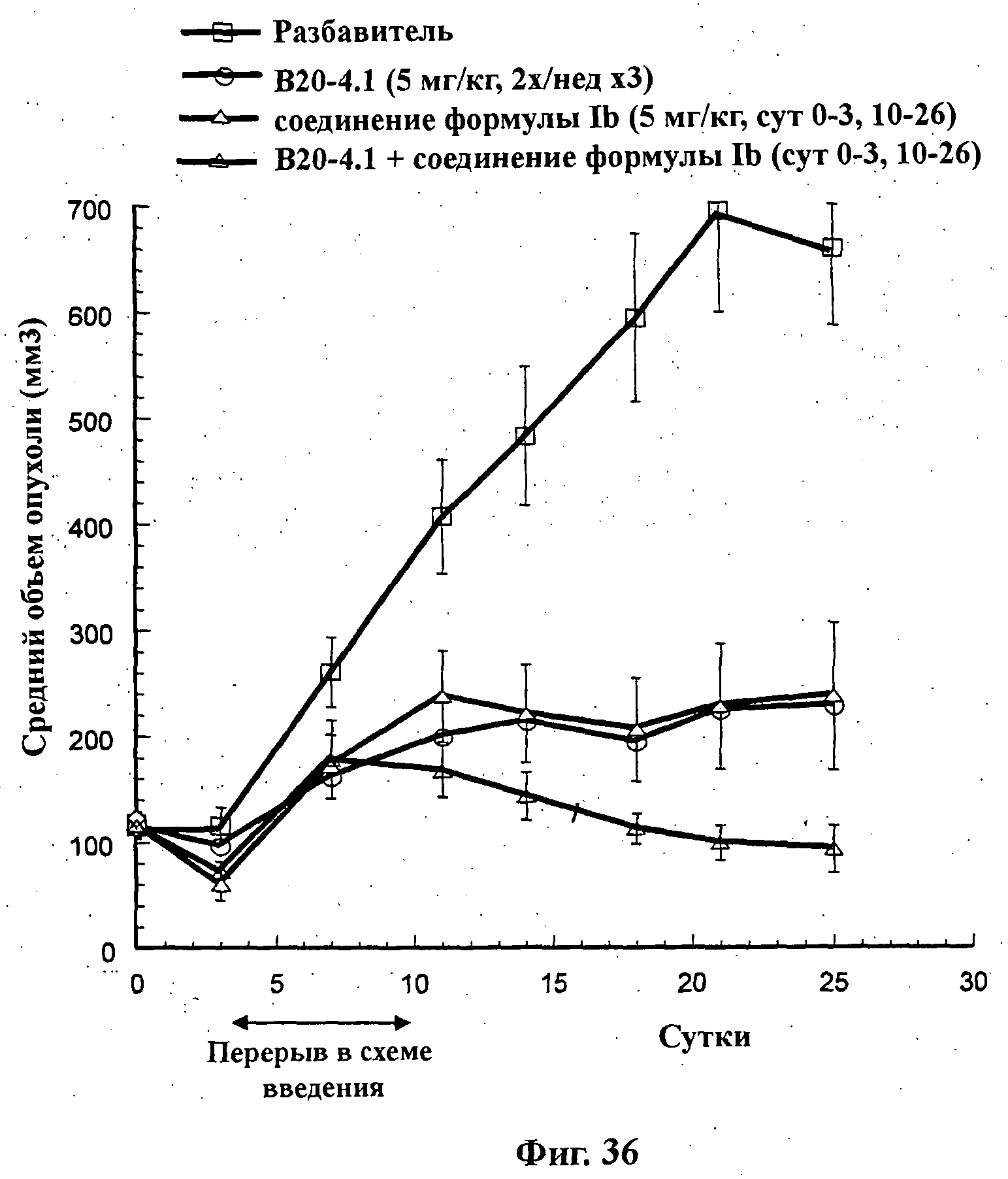
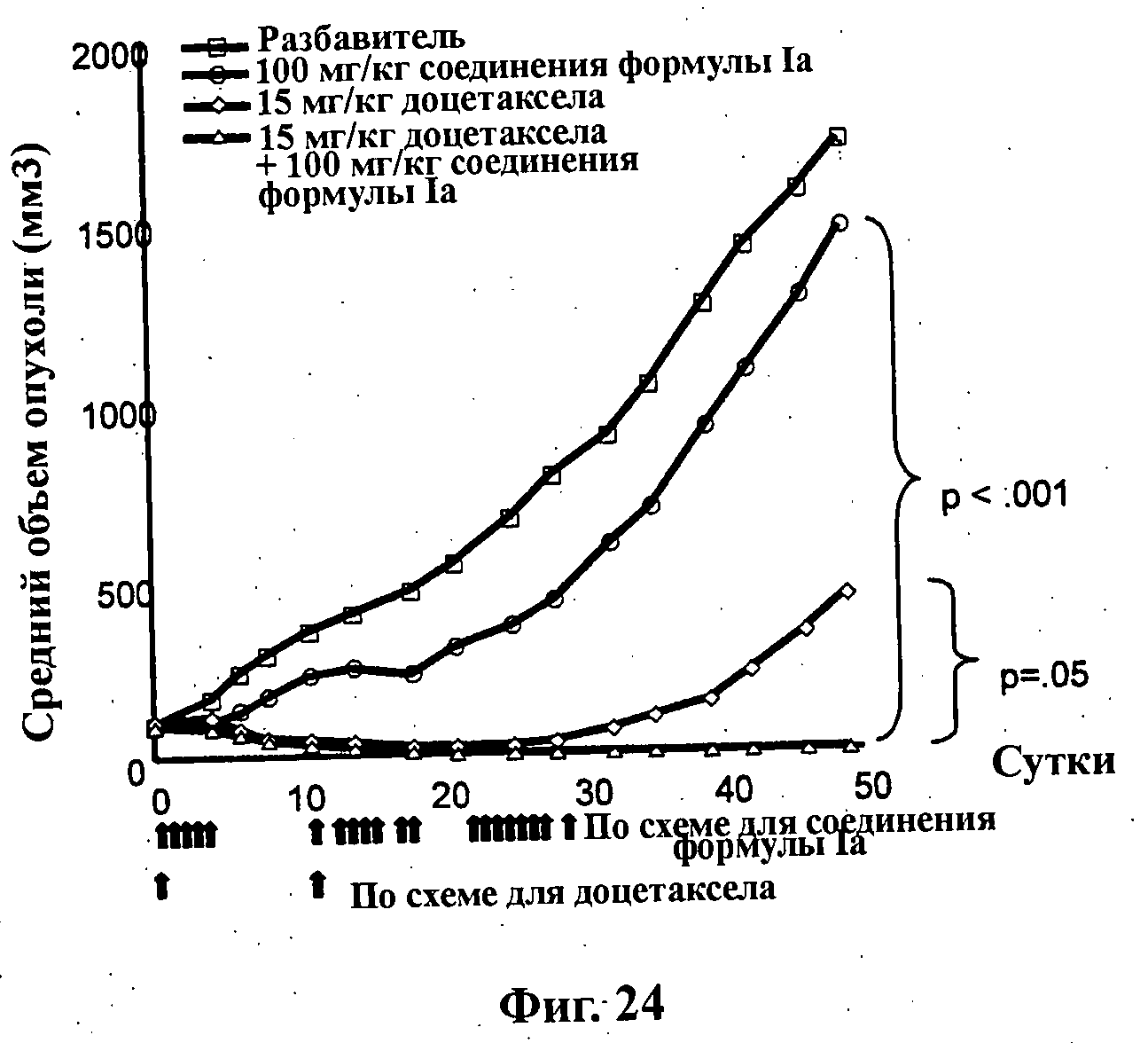
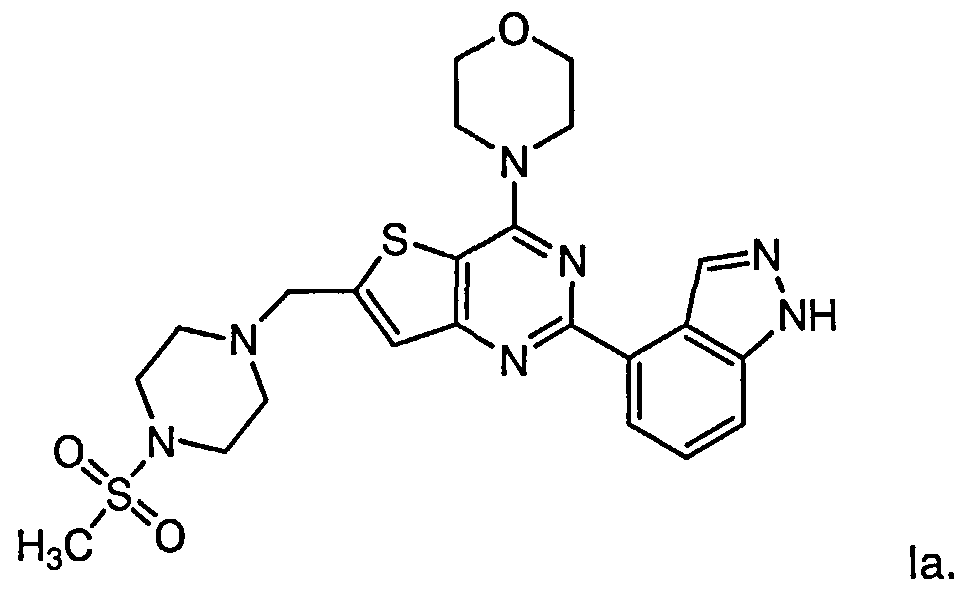
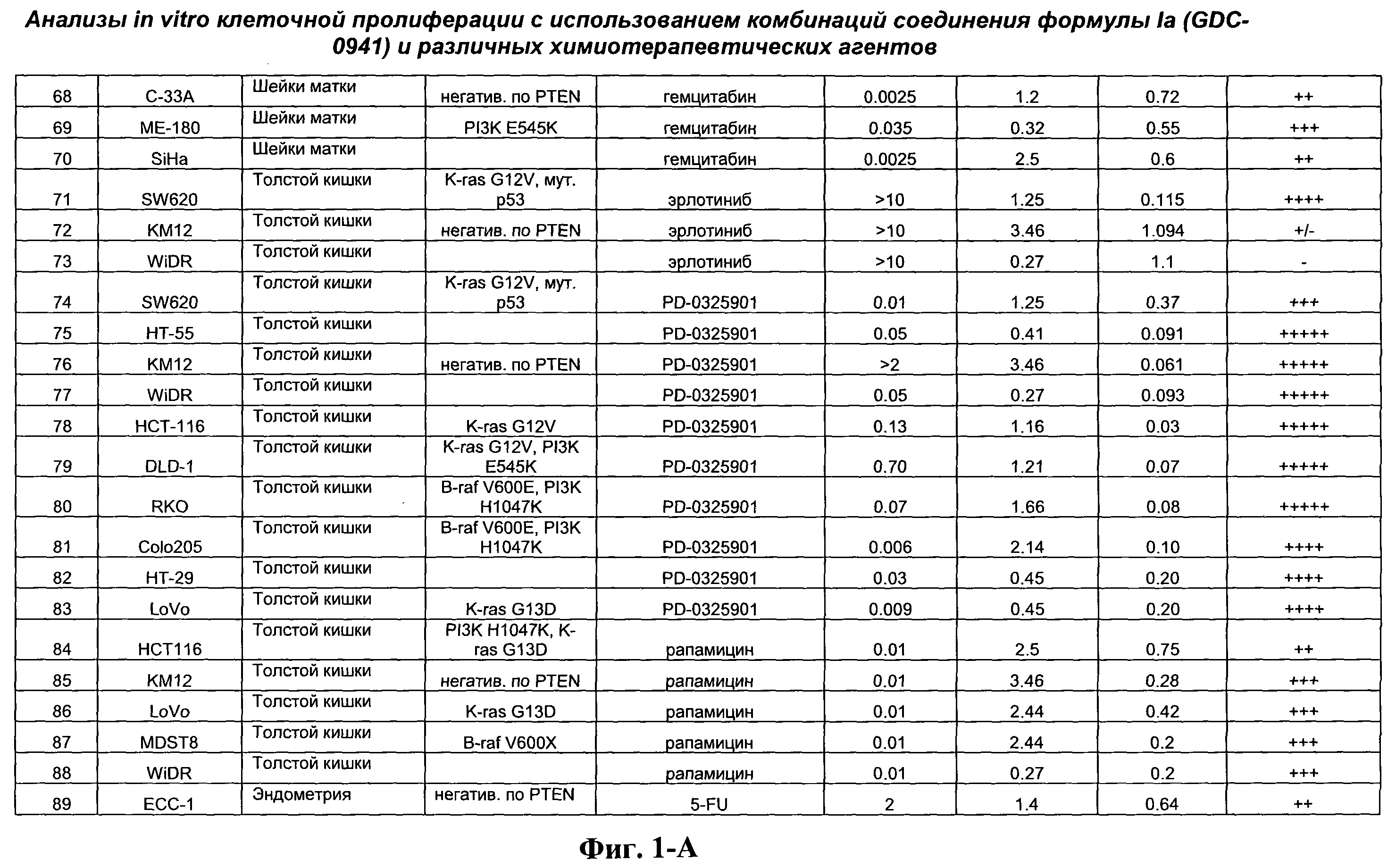
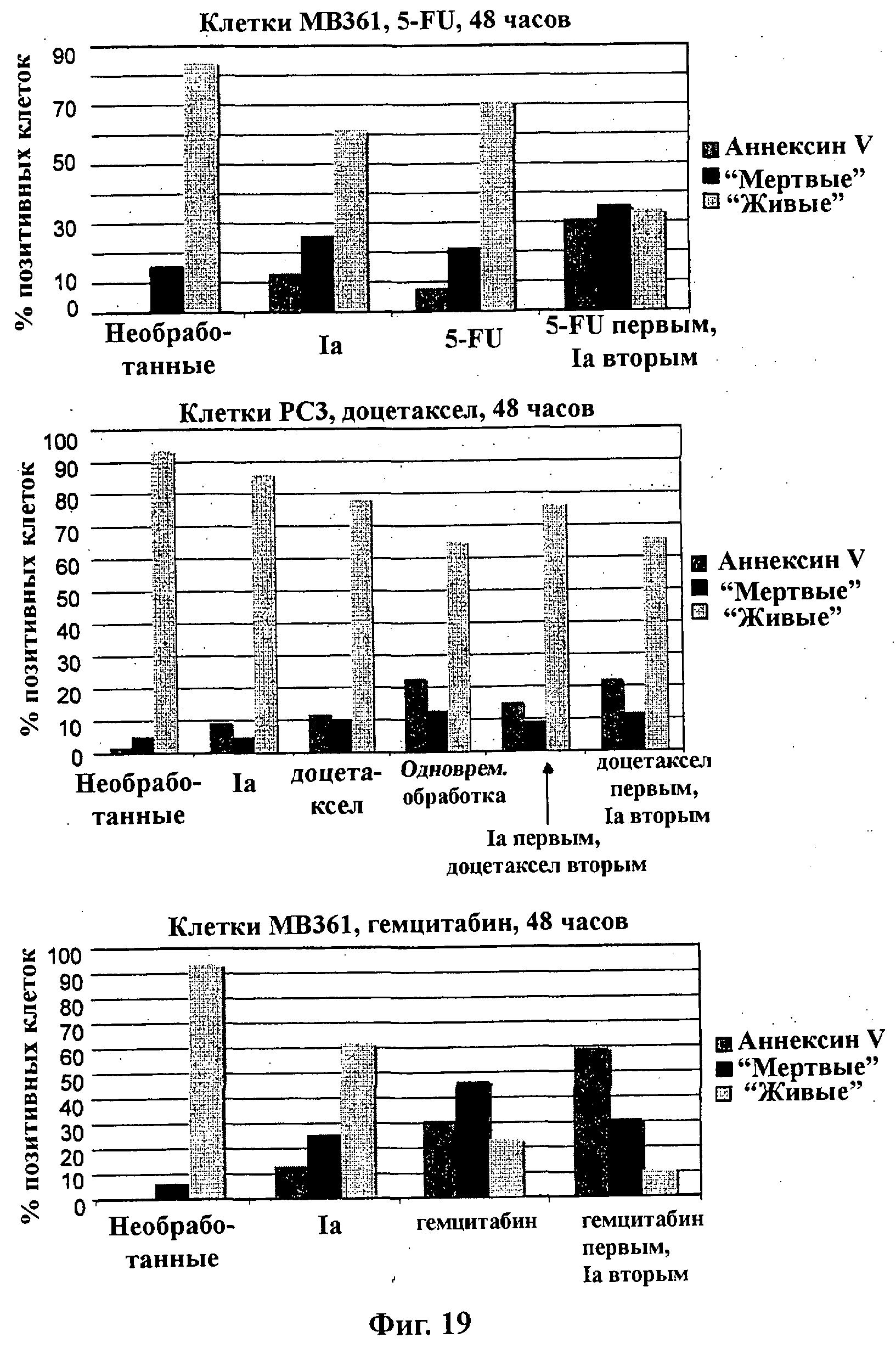
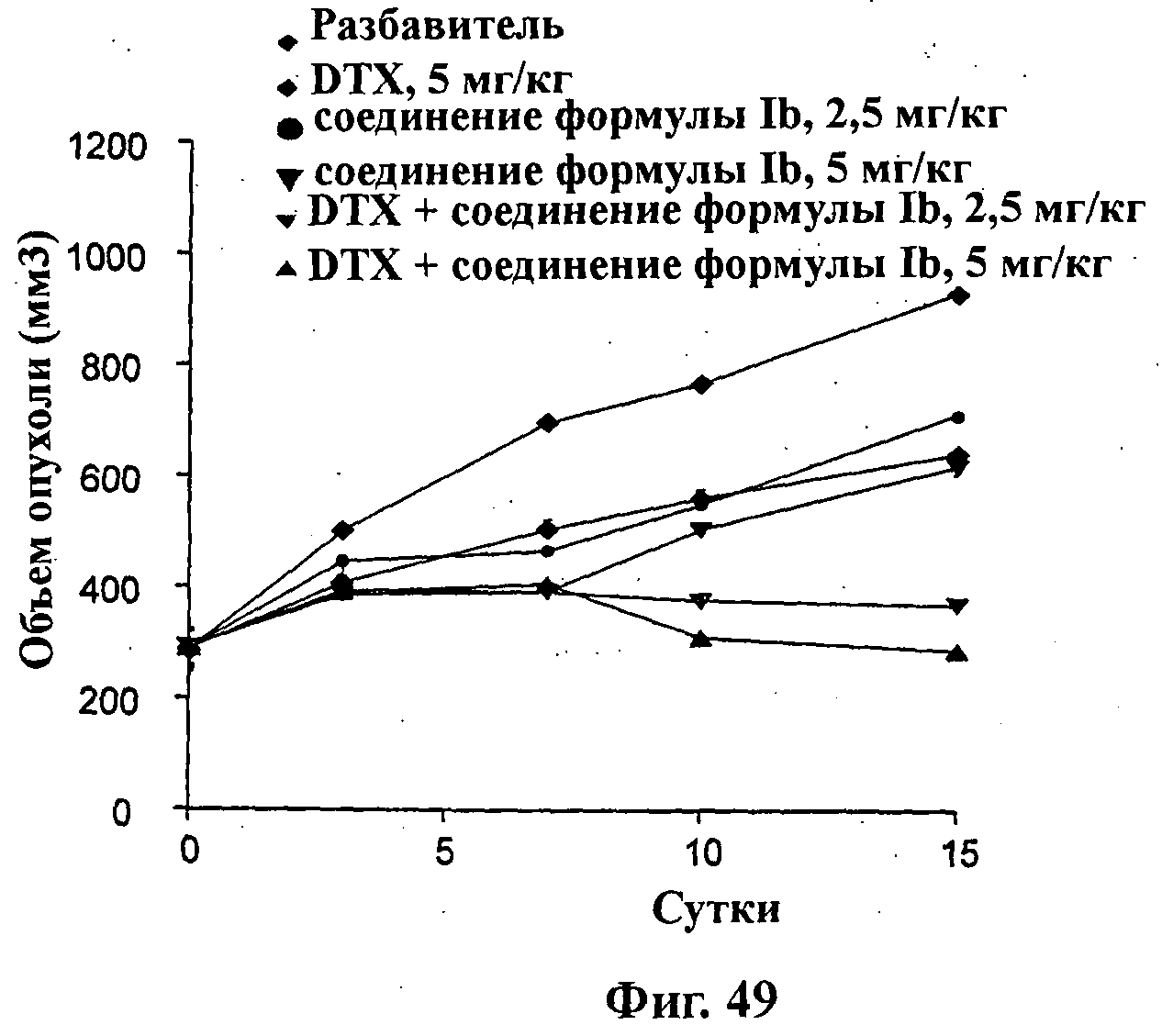
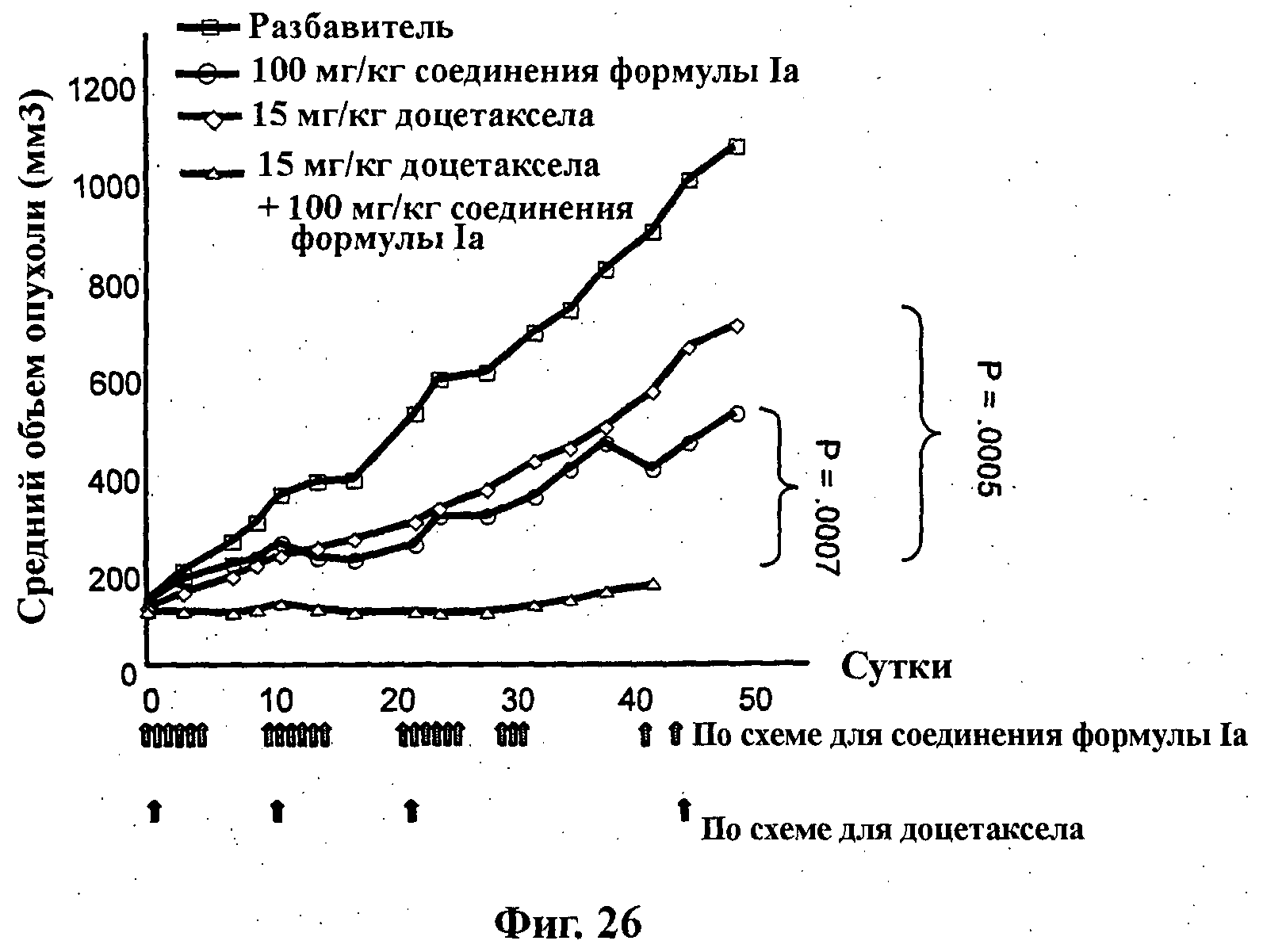
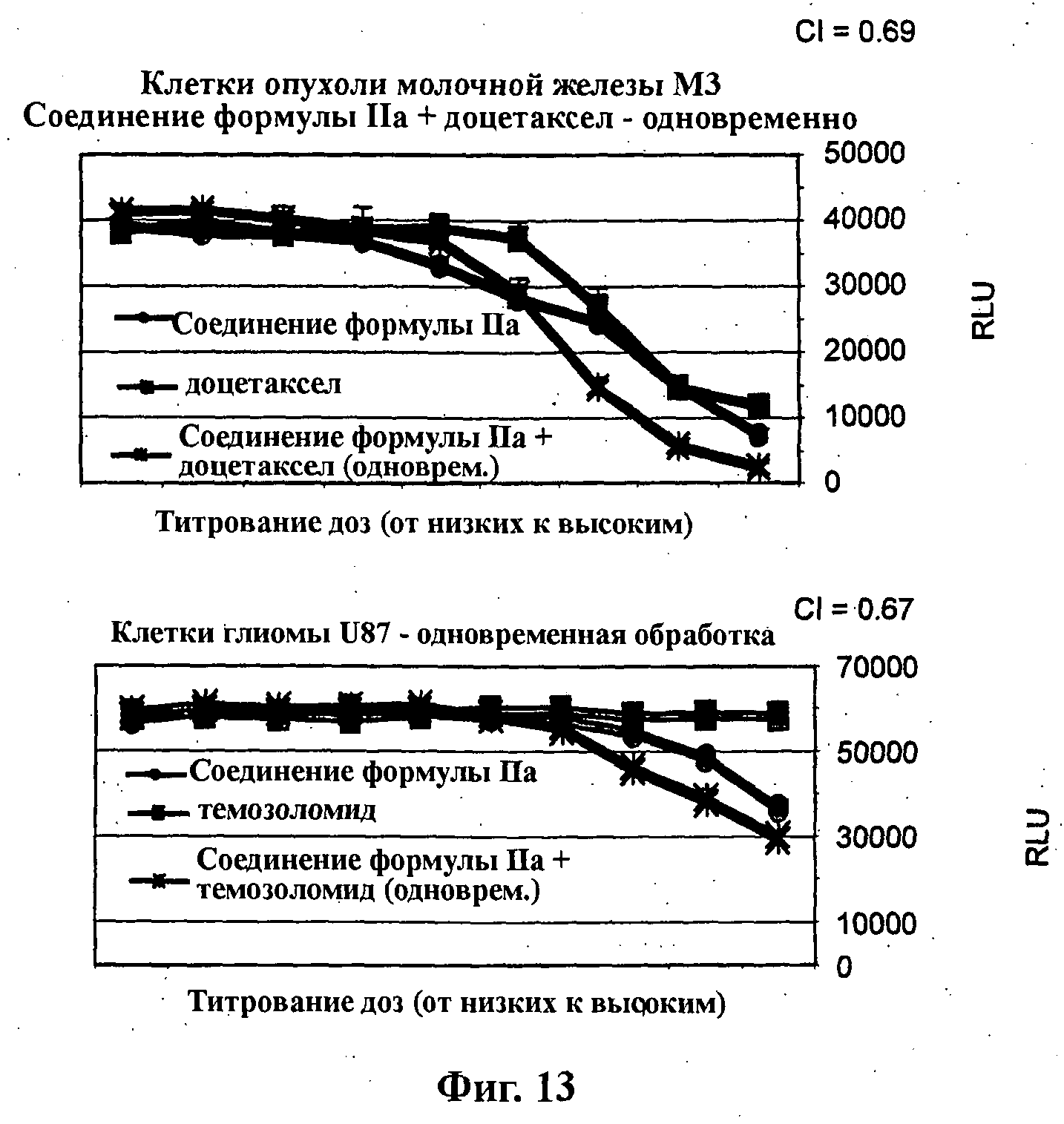
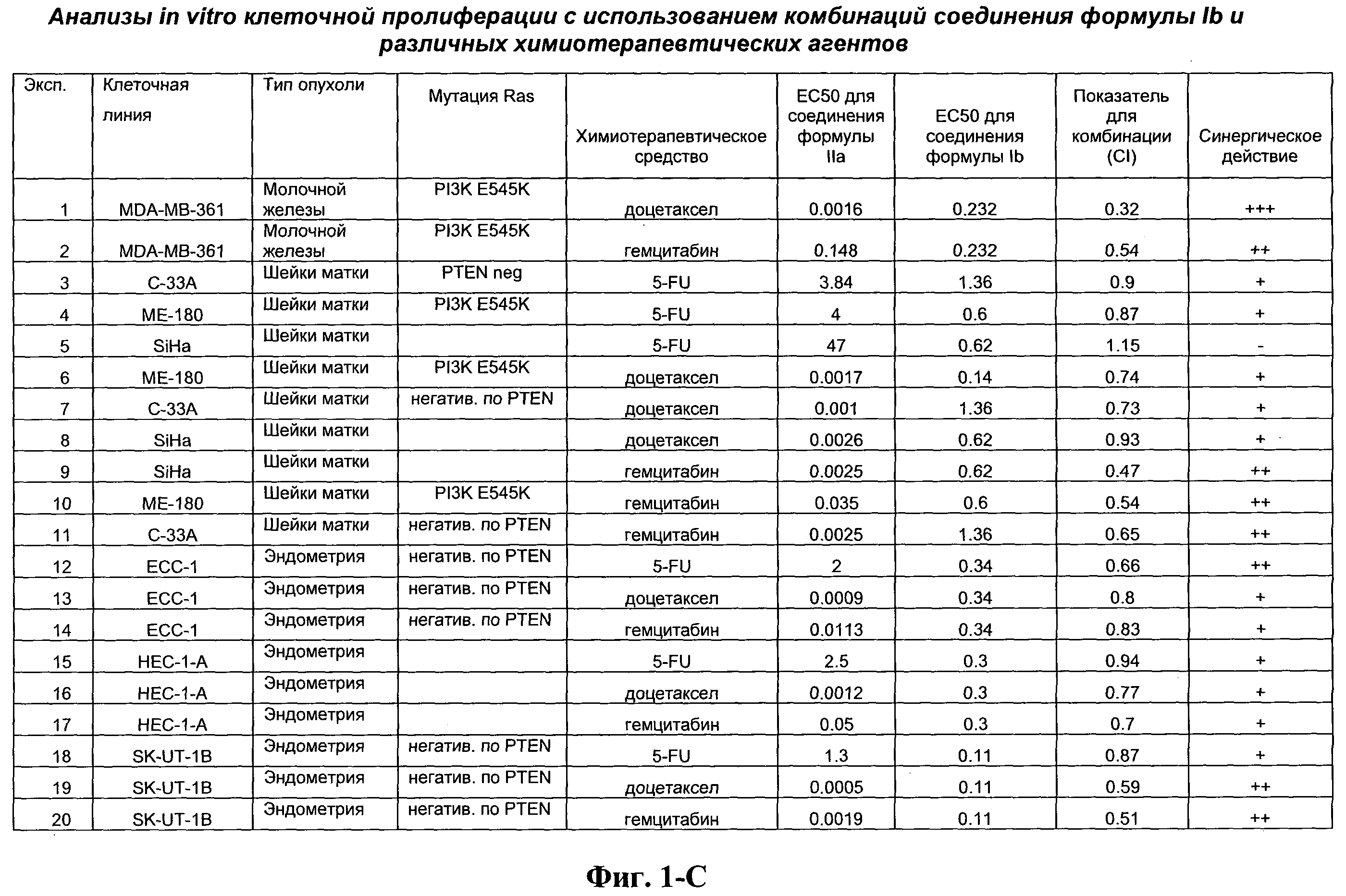
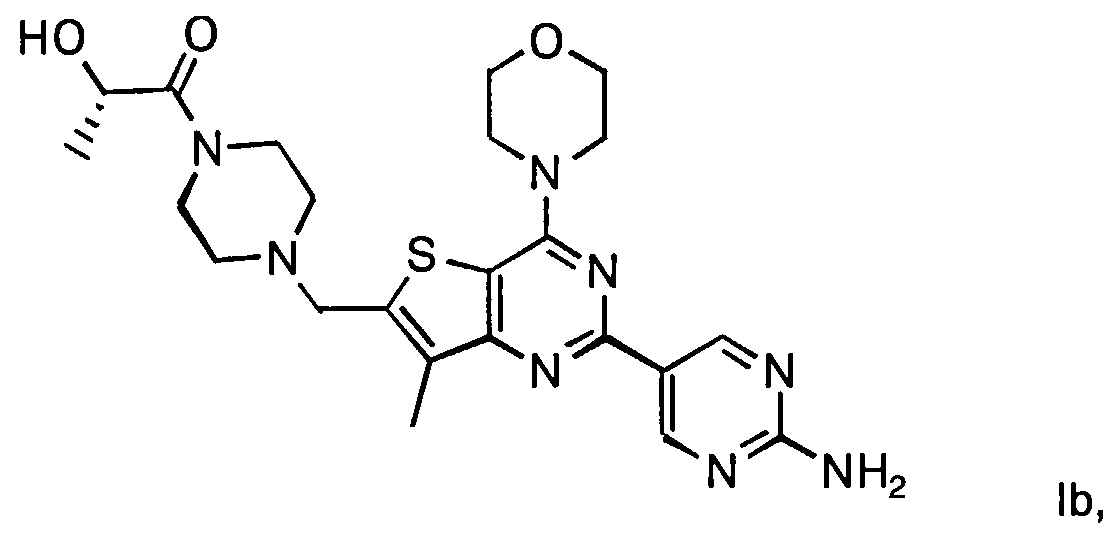
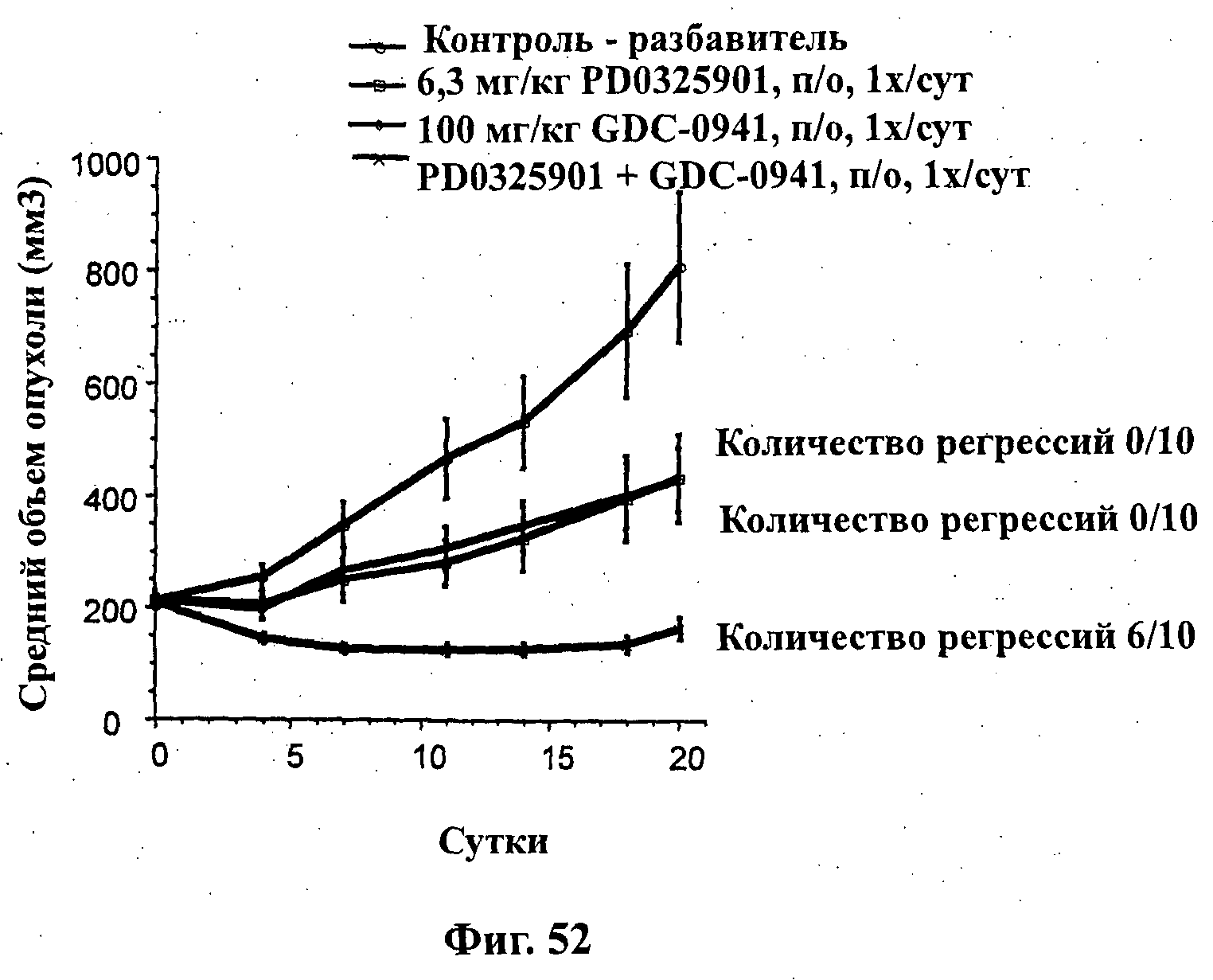
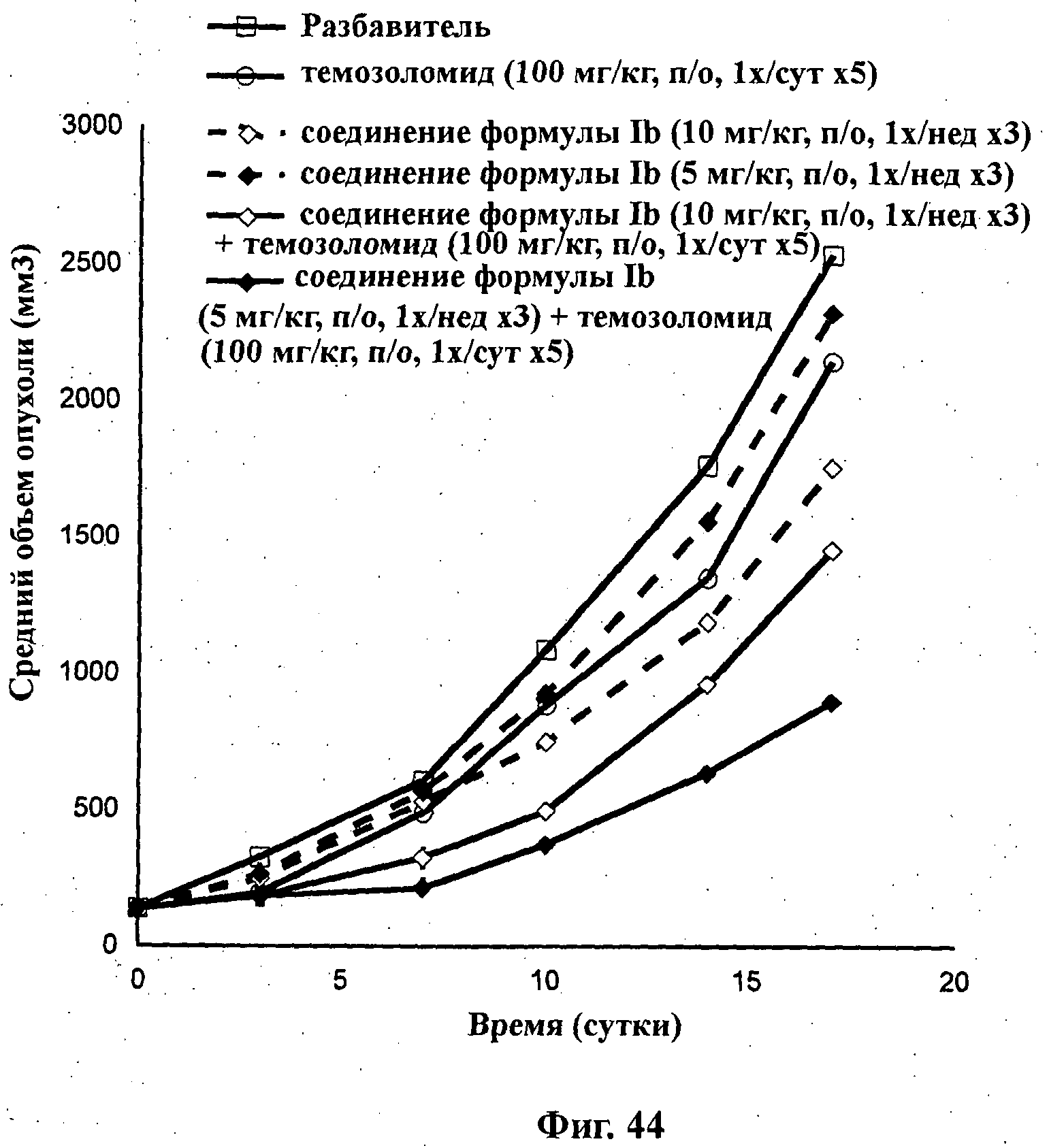
Ингибирование ангиогенеза
Новые 2-аминооксазолины в качестве лигандов taar1 для заболеваний цнс
Пиримидиновые соединения, композиции и способы применения
Антитела против c3b и способы профилактики и лечения связанных с комплементом нарушений
Производные 4-диметиламиномасляной кислоты
Осаждение и очистка белков полиэлектролитами
Гуманизированные антитела к фактору d и их применения
Очистка пегилированных полипептидов
Антитела против nrr notch1 и способы их применения
Гидроксилированные и метоксилированные циклопента[d]пиримидины в качестве ингибиторов акт протеинкиназ
Ингибирование ангиогенеза
Сульфонамидные производные как ингибиторы химазы
Новые 2-аминооксазолины в качестве лигандов taar1 для заболеваний цнс
Пиримидиновые соединения, композиции и способы применения
Антитела против c3b и способы профилактики и лечения связанных с комплементом нарушений
Производные 4-диметиламиномасляной кислоты
Осаждение и очистка белков полиэлектролитами
Гуманизированные антитела к фактору d и их применения
Очистка пегилированных полипептидов
Антитела против nrr notch1 и способы их применения

