Результат интеллектуальной деятельности: ТЕТРАЗОЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ СНИЖЕНИЯ КОНЦЕНТРАЦИИ МОЧЕВОЙ КИСЛОТЫ
Вид РИД
Изобретение
УРОВЕНЬ ТЕХНИКИ
Заболевания, вызванные повышенными концентрациями мочевой кислоты, относятся к двум основным категориям: заболевания, вызванные выпадением кристаллов мочевой кислоты, и заболевания, связанные с патологическим действием растворенной мочевой кислоты. Подагрический артрит представляет собой классический пример заболевания первой категории. Отложение кристаллов соли мочевой кислоты в почках также является основной причиной почечной дисфункции. Повышенные концентрации растворенной мочевой кислоты связаны с рядом заболеваний, включая сердечно-сосудистые и почечные заболевания.
Подагра обычно проявляется в виде воспаления одного или более суставов, приводя в результате к от слабой до сильной боли. Данные осложнения могут быть эпизодическими и/или хроническими. Со временем подагра может приводить к разрушению хрящей и костей, появлению отложений кристаллов мочевой кислоты, болям в почках и почечной дисфункции, а также почечным камням. Подагра также может влиять на другие органы.
Подагра вызывается гиперурикемией и последующим образованием и отложением кристаллов мочевой кислоты в тканях, суставах, почках и других органах. Мочевая кислота синтезируется в процессе метаболизма нормальных клеток или попадает в организм из некоторых видов пищи и напитков. Повышенная концентрация мочевой кислоты является результатом повышенного производства мочевой кислоты, нарушения почечного клиренса (или комбинацией избыточного производства и нарушенного клиренса), и также может быть результатом приема некоторых типов лекарственных средств, используемых для изменения состояния здоровья. (Примеры включают диуретики, пиразинамид, циклоспорин, низкие дозы аспирина, никотиновую кислоту и леводопу). Многие типы состояний здоровья также могут способствовать гиперурикемии и подагре, включая алкоголизм, лейкемию, лимфому, рак легких, синдром лизиса опухоли, курение, псориаз, ожирение, почечную дисфункцию, застойную сердечную недостаточность, голодание, анемию, высокое кровяное давление, диабет, отсутствие подвижности, синдром Леша-Найхана, синдром Дауна и дисфункцию щитовидной и паращитовидной железы.
Подагру обычно подразделяют на четыре категории, исходя из постепенного увеличения выраженности симптомов:
1) бессимптомная: повышенная концентрация мочевой кислоты в крови, но явные симптомы отсутствуют;
2) острый подагрический артрит: внезапное появление симптомов, часто в одном суставе (обычно в большом пальце), и затем появление симптомов в других суставах. Симптомы включают боль, отек, покраснение и повышение температуры;
3) периодическая подагра: бессимптомные фазы между приступами подагры;
4) хроническая узелковая подагра: хроническое состояние, которое может включать частые приступы, постоянную слабую боль и воспаление сосудов, разрушение хрящей и костей, появление отложений кристаллов мочевой кислоты, почечную дисфункцию и почечные камни.
Лекарственные средства, используемые в настоящее время для лечения острых симптомов подагры, включают нестероидные противовоспалительные лекарственные средства, колхицин и кортикостероиды. Все эти лекарственные средства могут оказывать от слабых до тяжелых побочных эффектов. Другие виды лечения данных острых симптомов исследуются, включая антитела и антагонисты воспалительных цитокинов, таких как интерлейкин 1.
Другие типы лекарственных средств используют для того, чтобы попытаться снизить частоту возникновения или тяжесть будущих приступов снижением концентрации мочевой кислоты. Тремя основными классами лекарственных средств являются ингибиторы ксантиноксидазы (например, аллопуринол), которые снижают производство мочевой кислоты из ксантина; средства, способствующее выведению мочевой кислоты (например, сульфинпиразон, пробенецид, бензбромарон и лозартан), которые имеют целью усилить выведение мочевой кислоты ингибированием обратного захвата секретированной мочевой кислоты в почечных канальцах посредством ингибирования переносчика мочевой кислоты (URAT1) (см. также опубликованную патентную заявку США № 2007/0010670, опубликованную 11 января 2007 (Japan Tobacco Inc.)), или других элементов обратного захвата мочевой кислоты; и уриказы, например пегилированную уриказу, такую как PURICASE (пегилированная рекомбинантная уриказа млекопитающего фирмы Savient). Данные лекарственные средства также часто приводят к значительным и нежелательным побочным эффектам. Например, сообщают, что аллопуринол является причиной, по меньшей мере, 100 случаев синдрома Стивенса - Джонсона/токсического эпидермального некролиза и приблизительно 30 случаев смерти каждый год в Европе (Halevy et al., Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 58(1):25-32, 2008). Пробенецид и бензбромарон запрещены для продажи в ряде стран из-за нежелательных побочных эффектов, таких как печеночная недостаточность в случае бензбромарона. Сообщают, что больные не очень хорошо соблюдают режим и следуют схеме лечения при приеме данных лекарственных средств (A. A. Reidel et al. "Compliance with Allopurinol Therapy among Managed Care Enrollees with Gout: A Retrospective Analysis of Administrative Claims." Journal of Rheumatology 2004: 31:1575-1581), вероятно из-за побочных эффектов и/или отсутствия улучшений.
Более 5 миллионов людей в США страдают от подагры (National Health and Nutrition Examination Survey 111, 1988-1994). В 1999 году сообщалось, что частота случаев гиперурикемии и подагры в США составляла 41 на 1000 и 14 на 1000 в Великобритании (T.R. Mikuls et al., "Gout Epidemiology: Results for the UK General Practice Research Database. 1990-1999." Annals of the Rheumatic Diseases 2005; 64:267-272). Последующие сообщения показывают, что частота случаев в США, Великобритании и других странах непрерывно растет. (K. L. Wallace et al., "Increasing Prevalence of Gout and Hyperuricemia over 10 Years Among Older Adults in a Managed Care Population." Journal of Rheumatology 2004; 31: 1582-1587). Новейшие данные предполагают, что у более чем 5 миллионов американцев в настоящее время можно диагностировать подагру. (E.Krishnan et al., "Gout in Ambulatory Care Settings in the United States." Journal of Rheumatology 2008; 35(3); 498-501).
Гиперурикемия и подагра являются особенно значительной проблемой у пациентов с пересаженными органами (Stamp, L., et al., "Gout in solid organ transplantation: a challenging clinical problem". Drugs (2005) 65(18); 2593-2611). Концентрация мочевой кислоты часто повышена у пациентов с трансплантатами почек, и стандартные иммунодепрессантные лекарственные средства, такие как циклоспорин, могут вызывать, в частности, тяжелую гиперурикемию. Аллопуринол противопоказан пациентам с трансплантатами из-за взаимодействия его с некоторыми иммунодепрессантами, такими как азатиоприн, и из-за повреждения костного мозга, вызванного их комбинацией. Кроме того, повышенная концентрация мочевой кислоты может способствовать отторжению трансплантата (Armstrong, K.A. et al., "Does Uric Acid Have a Pathogenetic Role in Graft Dysfunction and Hypertension in Renal Transplant Patients?" Transplantation (2005) 80(11); 1565-1571). Следовательно, существует особенно острая необходимость в безопасных агентах, которые ослабляют гиперурикемию у пациентов с трансплантатами.
Заболевания, связанные с повышенной концентрацией растворенной мочевой кислоты, часто сопровождаются проблемами с сосудами: гипертонией (Sundstrom et al., Relations of serum uric acid to longitudinal blood pressure tracking and Hypertension incidence. Hypertension. 45(1):28-33. 2005), прегипертонией (Syamela, S. et al. Association between serum uric acid and prehypertension among US adults. J Hypertens. 25 (8) 1583-1589, 2007), атеросклерозом (Ishizaka et al. Association between serum uric acid, metabolic syndrome, and carotid atherosclerosis in Japanese individuals. Arterioscler Thromb Vase Biol. (5); 1038-44, 2005), заболеванием периферических артерий (Shankar,A. et al. Association between serum uric acid level and peripheral artery disease. Atherosclerosis doi 10: 1016, 2007), воспалением сосудов (Zoccali et al., Uric acid and endothelial dysfunction in essential Hypertension. J Am Soc Nephrol. 17(5); 1466-71, 2006), сердечной недостаточностью (Strasak, A.M. et al. Serum uric acid and risk of cardiovascular mortality: A prospective, long-term study of 83,683 Austrian men. Clin Chem. 54 (2) 273-284, 2008; Pascual-Figal, Hyperuricaemia and long-term outcome after hospital discharge in acute heart failure patients. Eur J Heart Fail. 2006 Oct 23; [Epub ahead of print]; Cengel. A., et al., "Serum uric Acid Levels as a Predictor of In-hospital Death in Patients Hospitalized for Decompensated Heart Failure." Acta Cardiol. (Oct. 2005) 60(5); 489-492), инфарктом миокарда (Strasak, A.M. et al.; Bos et al., Uric acid is a risk factor for myocardial infarction и stroke: the Rotterdam study. Stroke. 2006 Jun: 37(6); 1503-7), почечной дисфункцией (Cirillo et al. Uric acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 17(12 Suppl 3):S165-8, 2006; Z. Avram and E. Krishnan, Hyperuricemia - where nephrology meets rheumatology. Rheumatology (Oxford), 47(7); 960-964. 2008), и инсультом (Bos et al., 2006). Мочевая кислота непосредственно вызывает эндотелиальную дисфункцию (Kanellis. et al. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 25(1):39-42, 2005: Khosla et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 67(5); 1739-42, 2005). У детей и подростков ранняя эссенциальная гипертензия связана с повышенной концентрацией мочевой кислоты в сыворотки крови, и снижение концентрации мочевой кислоты при использовании аллопуринола снижает кровяное давление у данных пациентов (Feig and Johnson, The role of uric acid in pediatric hypertension. J Ren Nutrition 17(1); 79-83, 2007; D.I. Feig et al., Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension. JAMA 300(8); 924-932. 2008). Фейг и др. также утверждают, что это является новым терапевтическим подходом, но что побочные эффекты существующих лекарственных средств для снижения концентраций мочевой кислоты могут ограничить или препятствовать их использованию. Гиперурикемия представляет собой независимый фактор риска во всех из данных состояний.
Повышенная концентрация растворенной мочевой кислоты также связана с или непосредственно вызывает воспалительную реакцию. Например, мочевая кислота переносится в гладкомышечные клетки сосудов посредством переносчика органических кислот, особенно уратным переносчиком URAT1, и затем стимулирует производство гладкомышечными клетками C-реактивного белка. MCP-1 и другие цитокины, посредством этого стимулирующие пролиферацию и другие изменения, связанные с атеросклерозом (Price et al., Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol. 17(7); 1791-5, 2006; Kang et al. Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005 25(5):425-33 (2005); Yamamoto et al., Allopurinol reduces neointimal hyperplasia in the carotid artery ligation model in spontaneously hypertensive rats. Hypertens. Res. 29 (11) 915-921. 2006), стимулирует производство мононуклеарными клетками человека IL-1β, IL-6 и TNF-α, вызывает заметное увеличение концентрации TNF-α при инфузии мышам, активирует эндотелиальные клетки и тромбоциты и увеличивает адгезию тромбоцитов (Coutinho et al. "Associations of Serum Uric Acid with Markers of Inflammation. Metabolic Syndrome, and Subclinical Coronary Atherosclerosis". Amer. J. Hypertens. (2007) 20: 83-89; Levya, F., et al., "Uric Acid in Chronic Heart Failure: A Marker of Chronic Inflammation", Eur. Heart J. (1998) 19(12); 1814-1822). Также показано, что мочевая кислота ингибирует биодоступность эндотелиального оксида азота и активирует ренин-ангиотензиновую систему (T.S. Perlstein et al. Uric Acid and the state of the intrarenal renin-angiotensin system in humans. Kidney International. 66: 1465-1470, 2004). Инокучи и др. показали, что интерлейкин 18 (IL-18) и другие отвечающие за воспаление агенты свидетельствуют о местном воспалении, связанном с подагрой, и что кристаллы солей мочевой кислоты ускоряют активацию IL-18 (T. Inokuchi et al. Plasma IL-18 and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 33(1):21-27, 206), который, как оказалось, является причиной почечной недостаточности. Концентрации IL-18 и других цитокинов также заметно повышены у людей, у которых еще нет подагры самой по себе, но у которых просто имеется повышенная концентрация мочевой кислоты (C. Ruggiero et al. Uric acid and inflammatory markers. European Heart Journal. 27: 1174-1181, 2006).
Гиперурикемия также связана с когнитивными нарушениями и другими формами дисфункции центральной нервной системы (Schretlen. D.J. et al. "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1); 136-140; Watanabe.S., et al., "Cerebral Oxidative Stress and Mitochondrial Dysfunction in Oxonate-Induced Hyperuricemic Mice", J. Health Science (2006) 52: 730-737).
Повышенные концентрации мочевой кислоты в плазме крови также связаны с повышенным риском рака и смертностью от рака (Strasak, A.M. et al. (2007) Serum Uric Acid and risk of cancer mortality in a large prospective male cohort. Cancer Causes Control 18 (9) 1021-1029: Strasak, A.M. et al. (2007) The role of serum uric acid as an antioxidant protecting against cancer: prospective study in more than 28000 older Austrian women. Annals Oncol 18 (11) 1893-1897; Jee, S.A. et al. (2004) Serum Uric Acid and risk of death from cancer, cardiovascular disease or all causes in men Eur. J. Cardiovascular Prev. Rehab. 11 (3) 185-191).
Повышенные концентрации мочевой кислоты связаны с предиабетом, резистентностью к инсулину, развитием диабета 2 типа и повышенной вероятностью возникновения различных нежелательных состояний у людей с диабетом, таких как заболевание периферических артерий, инсульт и повышенный риск смерти (Ioachimescu, A.G. et al. (2007) Serum uric acid, mortality and glucose control in patients with Type 2 diabetes mellitus: a PreCIS database study Diabet. Med. 24 (12) 1369-1374; Perry. I.J. et al (1995) Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men BMJ 310 (6979) 560-564: Chien. K-L et al. (2008) Plasma uric acid and the risk of Type 2 diabetes in a Chinese community Clin. Chem. 54 (2) 310-316; Sautin, Y. Y. et al. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress Am. J. Physiol. Cell Physiol, 293: C584-C596; Tseng, C.H. (2004) Independent association of uric acid levels with peripheral artery disease in Taiwanese patients with Type 2 diabetes Diabet. Med. 21 (7) 724-729: Lehto, S. et al. (1998) Serum uric acid is a strong predictor of stroke in patients with non-insulin dependent diabetes mellitus Stroke 29: 635-639).
Повышенные концентрации мочевой кислоты являются определяющей чертой синдрома Леша-Найхана. Люди с приступами апноэ во сне или нарушением дыхания во сне также имеют повышенные концентрации мочевой кислоты (Saito. H. et al. Tissue hypoxia in sleep apnea syndrome assessed by uric acid and adenosine. Chest 122: 1686-1694. 2002; Verhulst, S.L. et al. Sleep-disordered breathing and uric acid in overweight and obese children and adolescents. Chest 132: 76-80. 2007).
Повышенные концентрации мочевой кислоты связаны с преэклампсией (Bainbridge, S.A. and Roberts. J. M.. Uric Acid as a pathogenic factor in preeclampsia. Placenta Dec. 17 2007 epub ahead of print).
"Мочевая кислота является основным фактором воспалительной реакции, инициируемой P. falciparum в мононуклеарных клетках периферической крови человека…. Воспалительную реакцию, вызванную P. falciparum, считают главной причиной патогенеза малярии…" PLoS ONE 2009:4(4):e5194. Epub 2009 Apr 17.
В медицине существует большая необходимость в новых лекарственных средствах, с помощью которых можно безопасно, легко и эффективно лечить и предотвращать заболевания, связанные с повышением концентрации мочевой кислоты в крови, являются ли данные заболевания результатом кристаллизации мочевой кислоты или результатом действия повышенных (основанных ли на индивидуальном или среднестатистическом стандарте) концентраций растворенной мочевой кислоты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
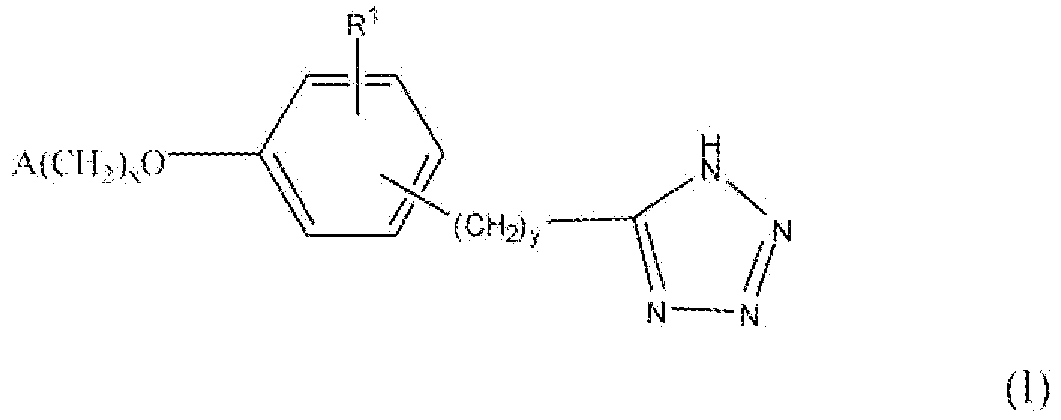
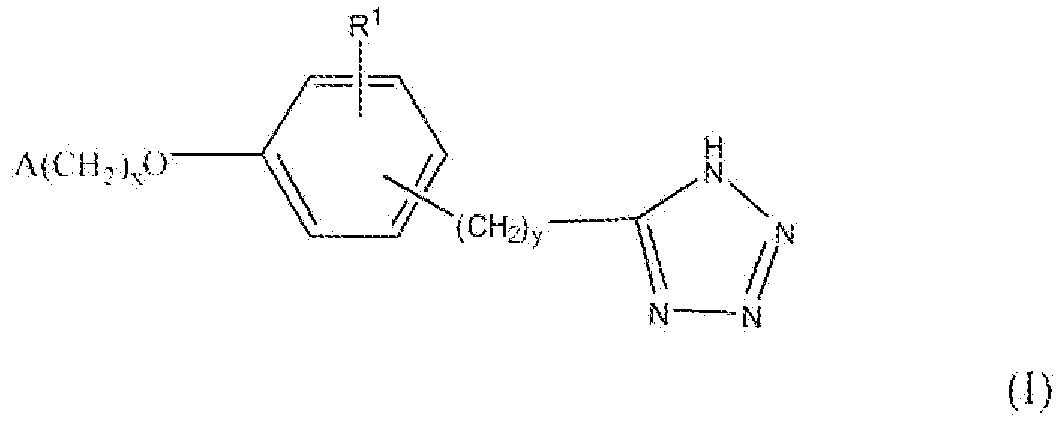
Настоящее изобретение относится к соединению, представленному формулой
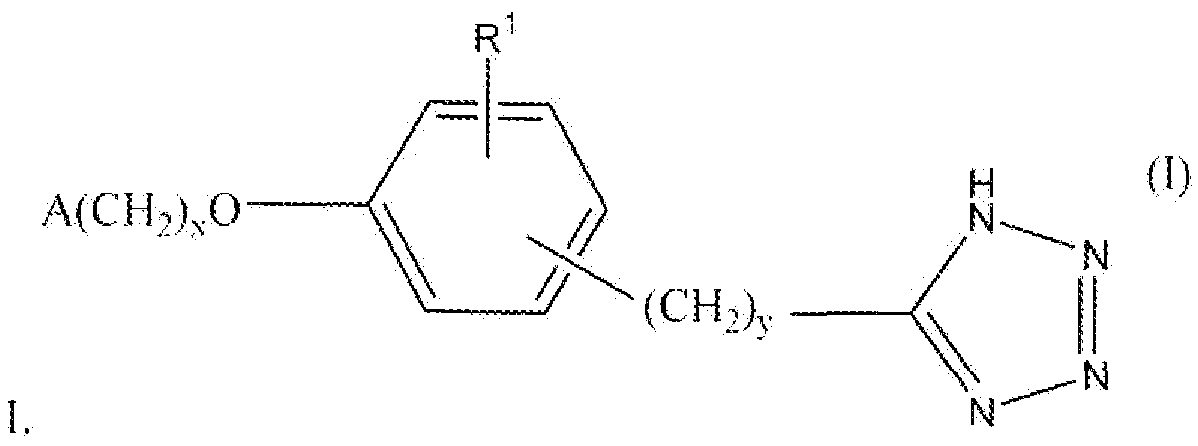
В формуле I x равно 1 или 2; y равно 0, 1, 2 или 3; R1 выбирают из группы, состоящей из водорода, алкила, содержащего 1 или 2 атома углерода, гидрокси, алкокси, содержащего 1 или 2 атома углерода, фтора, хлора, брома и амино. A представляет собой фенил, незамещенный или замещенный одним, двумя или тремя группами, выбранными из группы, состоящей из галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси; или циклоалкил, содержащий в кольце от 3 до 6 атомов, причем циклоалкил является незамещенным, или один или два атома углерода в кольце независимо монозамещают метилом или этилом; или 5- или 6-членное гетероароматическое кольцо, содержащее в кольце 1 или 2 гетероатома, выбранные из N, S и O, причем гетероароматическое кольцо ковалентно соединено с остальной частью соединения через атом углерода кольца.
Настоящее изобретение относится к способу снижения концентрации мочевой кислоты в крови или усилению выведения мочевой кислоты у субъекта, являющегося млекопитающим, включающему введение субъекту соединения настоящего изобретения в эффективном количестве для снижения концентрации мочевой кислоты в крови или усиления выведения мочевой кислоты у субъекта. Настоящее изобретение относится к соединению настоящего изобретения, применяемому для снижения концентрации мочевой кислоты в крови или усиления выведения мочевой кислоты у млекопитающего. Настоящее изобретение относится к применению соединения настоящего изобретения в получении лекарственного средства для снижения концентрации мочевой кислоты в крови или усиления выведения мочевой кислоты у млекопитающего. Настоящее изобретение относится к фармацевтической композиции для применения для снижения концентрации мочевой кислоты в крови или усиления выведения мочевой кислоты у субъекта, являющегося млекопитающим, содержащей соединение настоящего изобретения в эффективном количестве для снижения концентрации мочевой кислоты в крови или усиления выведения мочевой кислоты у субъекта. Настоящее изобретение относится к набору, содержащему одну или более однократных пероральных доз соединения настоящего изобретения, и инструкции для введения соединения для того, чтобы снизить концентрации мочевой кислоты в крови или усилить выведение мочевой кислоты у субъекта, являющегося млекопитающим.
Снижение концентрации мочевой кислоты, как описано в настоящем описании, можно применять для того, чтобы лечить или предотвращать ряд состояний, включая подагру (любую форму или все из: бессимптомной подагры, острого подагрического артрита, периодической подагры и хронической узелковой подагры), гиперурикемию, повышенную концентрацию мочевой кислоты, которая не соответствует концентрациям, при которых обычно ставят диагноз гиперурикемии, почечную дисфункцию, почечные камни, сердечно-сосудистые заболевания, риск развития сердечно-сосудистых заболеваний и другие последствия гиперурикемии, когнитивное нарушение, раннюю эссенциальную гипертензию и воспаление, вызванное Plastodium falciparum.
Настоящее изобретение основывается на наблюдениях, что соединения настоящего изобретения ингибируют URAT1 in vitro, как показано в примере 7. Ингибирование URAT1 представляет собой общепризнанную in vitro модель для снижения концентрации мочевой кислоты in vivo.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1: Зависящий от концентрации ингибирующий эффект соединения EB на поглощение 14C-урата в hURAT1-HEK клетках.
Фиг.2: Зависящий от концентрации ингибирующий эффект соединения EC на поглощение 14C-урата в hURAT1-HEK клетках.
Фиг.3: Концентрация соединения EB в плазме мышей.
Фиг.4: Калибровочная кривая для соединения EB в плазме крыс, LC-MS.
Фиг. 5: Концентрация соединения EB в плазме крыс.
Фиг.6: Калибровочная кривая для соединения EC в плазме крыс, LC-MS.
Фиг.7: Концентрация соединения EC в плазме крыс.
Фиг.8: Калибровочная кривая для соединения EG в плазме крыс, LC-MS.
Фиг.9: Концентрация соединения EG в плазме крыс.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Определения
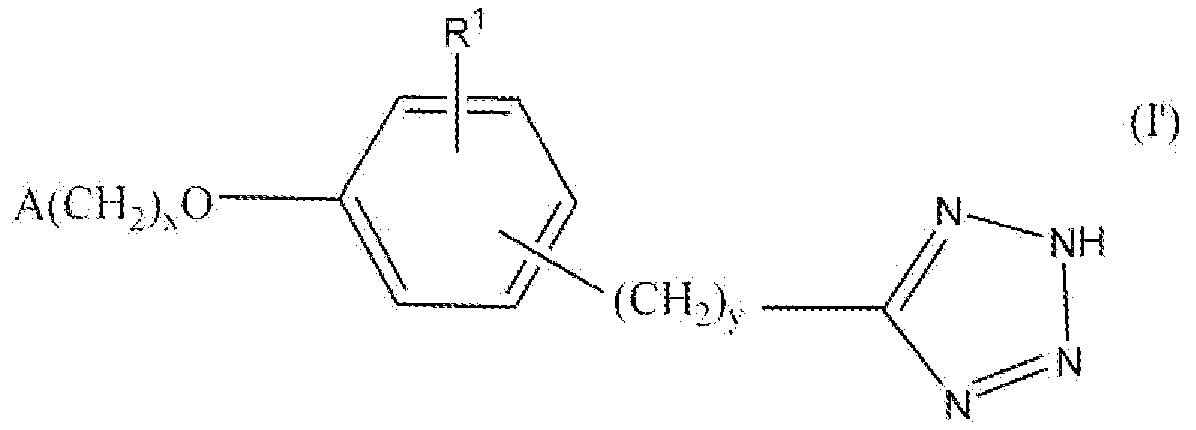
1H-Тетразолил-5-ильная группа и соответствующая 2H-тетразолил-5-ильная группа могут существовать в виде таутомеров. В настоящем описании соединения называют и структурные формулы изображают относительно 1H-таутомера. Все данные ссылки следует понимать как включающие обе таутомерные формы. Таким образом, например, "5-(3-(2,6-диметилбензилокси)фенил)-1H-тетразол" включает как 5-(3-(2,6-диметилбензилокси)фенил)-1H-тетразол, так и 5-(3-(2,6-диметилбензилокси)фенил)-2H-тетразол, и формула I включает как формулу I, как изображено выше, так и ее 2Η-тетразолил-5-ильную таутомерную форму, изображенную в формуле I' ниже.
Как используют в настоящем описании, термин "алкил" относится к алкильной группе с линейной или разветвленной цепью. Алкильная группа, идентифицированная как содержащая определенное число атомов углерода, относится к любой алкильной группе, имеющей конкретное число атомов углерода. Например, алкил, содержащий три атома углерода, может представлять собой пропил или изопропил; и алкил, содержащий четыре атома углерода, может представлять собой н-бутил, 1-метилпропил, 2-метилпропил или трет-бутил.
Как используют в настоящем описании, термин "галоген" относится к одному или более атомам фтора, хлора, брома и йода.
Как используют в настоящем описании, термин "перфтор", как в перфторметиле или перфторметокси, обозначает то, что рассматриваемая группа содержит атомы фтора вместо всех атомов водорода.
Определенные химические соединения называют в настоящем описании их химическим названием или двухбуквенным кодом, показанным ниже. Соединения EB-EI и соединение BD включены в объем формулы I, показанной выше.
|
Как используют в настоящем описании, неустановившийся термин "включающий" ничем не ограничен. Пункт формулы изобретения, в котором используется данный термин, может включать элементы в добавление к элементам, перечисленным в подобном пункте формулы изобретения.
Как используют в формуле изобретения, слово "или" обозначает "и/или", если такое прочтение имеет смысл в данном контексте. Так, например, фраза "снижение концентрации мочевой кислоты в крови или усиление выведения мочевой кислоты у субъекта, являющегося млекопитающим" является эквивалентным фразе "снижение концентрации мочевой кислоты в крови и/или усиление выведения мочевой кислоты у субъекта, являющегося млекопитающим".
СОЕДИНЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
В одном варианте осуществления соединения, способа, применения или фармацевтической композиции, описанных в сущности изобретения выше, соединение представлено формулой XLVI.
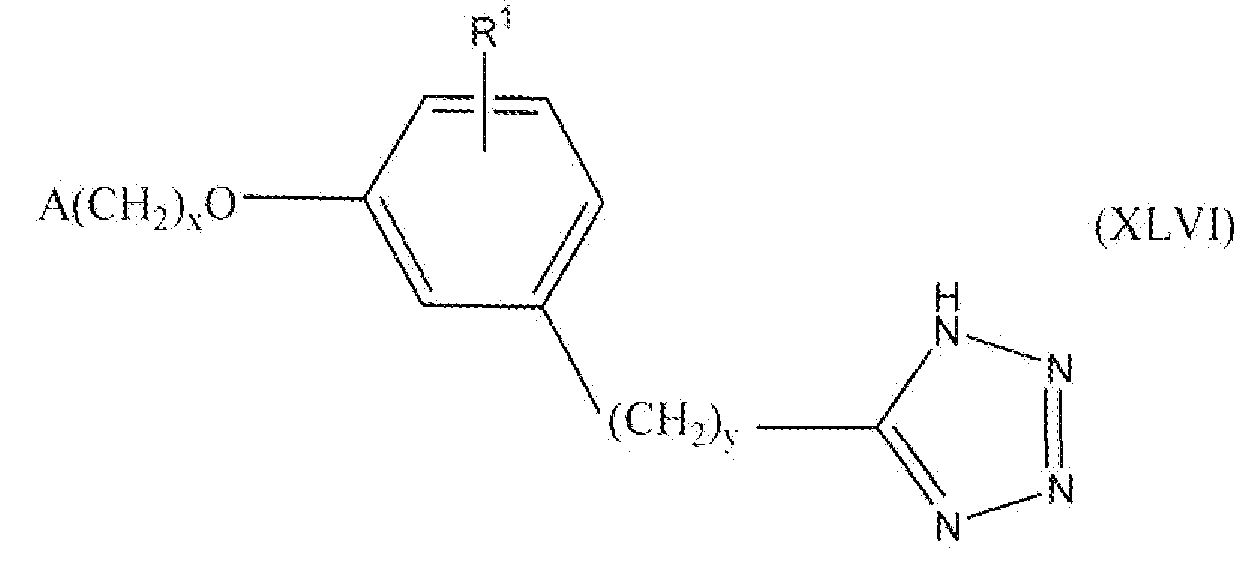
где x, y, R1 и A такие, как описано выше для формулы I.
В следующем варианте осуществления настоящего изобретения соединение представлено формулой XLVII
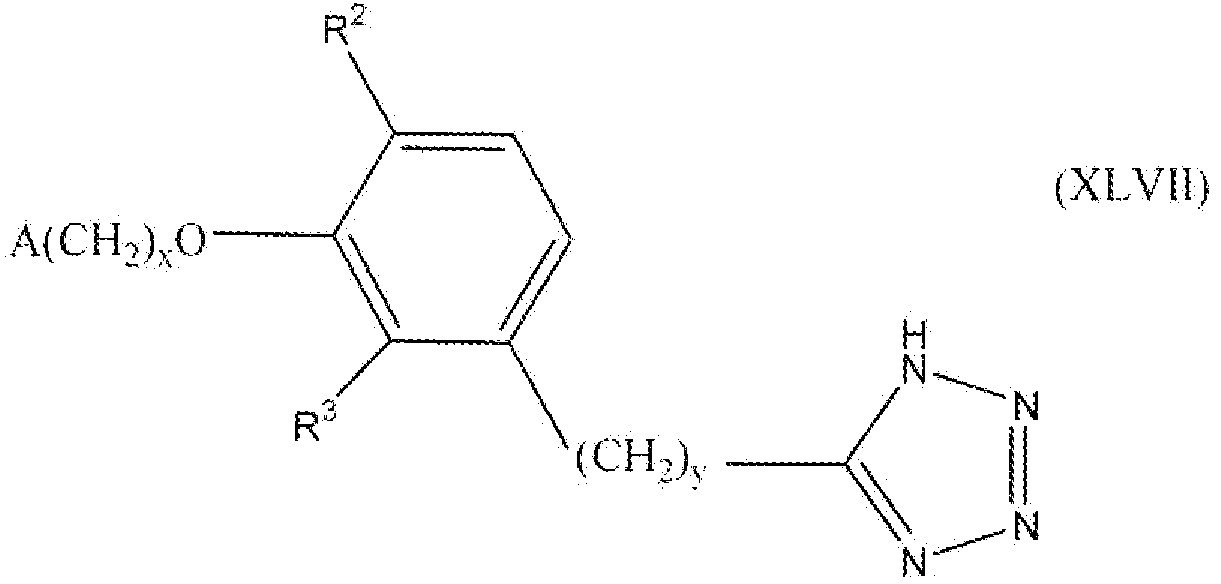
где x, y и A такие, как описано выше для формулы I; и один из R2 и R3 представляет собой водород, и другой выбирают из группы, состоящей из водорода, алкила, содержащего 1 или 2 атома углерода, гидрокси, алкокси, содержащего 1 или 2 атома углерода, фтора, хлора, брома и амино.
В одном варианте осуществления настоящего изобретения, в формуле I, XLVI или XLVII, x равно 1. В другом варианте осуществления A представляет собой фенил, незамещенный или замещенный одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, алкила, содержащего 1 или 2 атома углерода, перфторметила, алкокси, содержащего 1 или 2 атома углерода, и перфторметокси. В более конкретном варианте осуществления A представляет собой 2,6-диметилфенил или 2,6-дифторфенил. Предпочтительно A представляет собой 2,6-диметилфенил.
В одном варианте осуществления настоящего изобретения соединение представлено формулой XLVIII
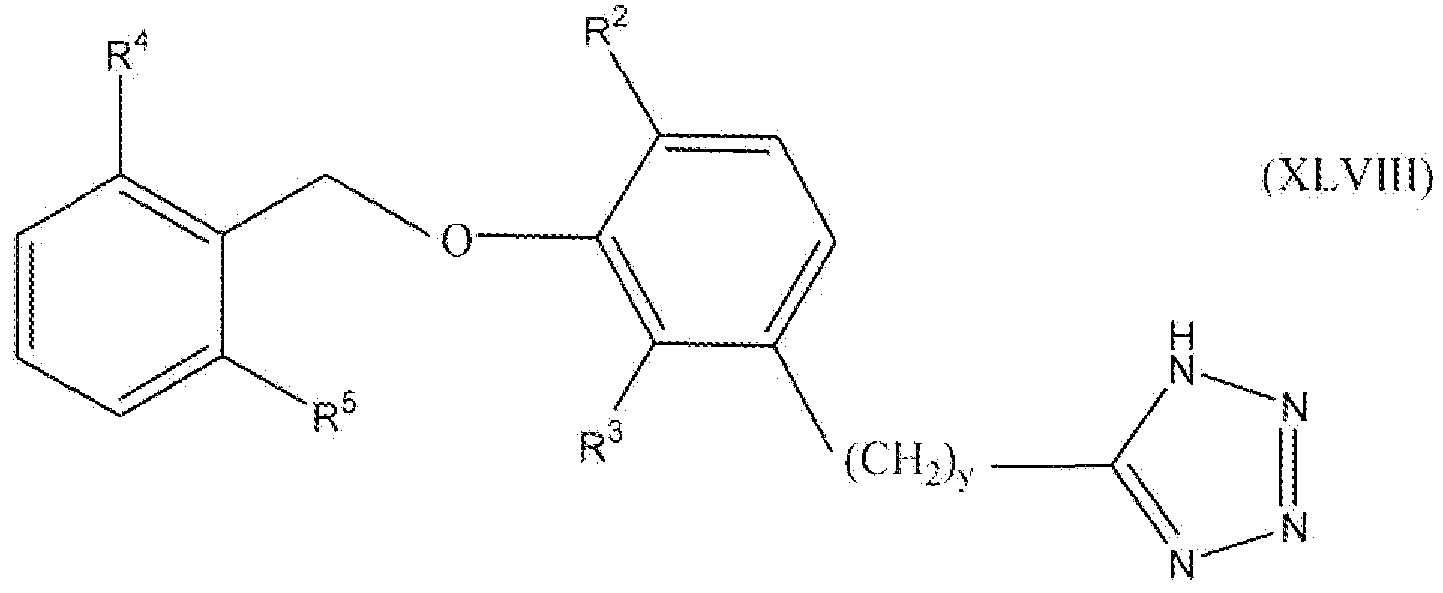
где y равно 0, 1, 2 или 3; один из R2 и R3 представляет собой водород, и другой выбирают из группы, состоящей из водорода, алкила, содержащего 1 или 2 атома углерода, гидрокси, алкокси, содержащего 1 или 2 атома углерода, фтора, хлора, брома и амино; и R4 и R5 независимо выбирают из группы, состоящей из метила, фтора и хлора.
В одном варианте осуществления настоящего изобретения, в формуле I, XLVI, XLVII или XLVIII, y равно 0, 1 или 2. В одном варианте осуществления настоящего изобретения в формуле XLVII или XLVIII R3 представляет собой водород, и R2 выбирают из группы, состоящей из водорода, алкила, содержащего 1 или 2 атома углерода, гидрокси, алкокси, содержащего 1 или 2 атома углерода, фтора, хлора, брома и амино. В более конкретном варианте осуществления R3 представляет собой водород, и R2 выбирают из группы, состоящей из водорода, метила и метокси. В другом варианте осуществления настоящего изобретения в формуле XLVII или XLVIII R2 представляет собой водород, и R3 выбирают из группы, состоящей из алкила, содержащего 1 или 2 атома углерода, гидрокси, алкокси, содержащего 1 или 2 атома углерода, фтора, хлора, брома и амино. В более конкретном варианте осуществления R2 представляет собой водород, и R3 выбирают из группы, состоящей из метила и метокси. В следующем варианте осуществления формулы XLVIII оба из R4 и R5 представляют собой метил или оба представляют собой фтор. Предпочтительно оба представляют собой метил.
В конкретных вариантах осуществления настоящего изобретения соединение выбирают из группы, состоящей из: 5-(3-(2,6-диметилбензилокси)фенил)-1H-тетразола; 5-(3-(2,6-диметилбензилокси)бензил)-1H-тетразола; 5-(3-(2,6-диметилбензилокси)-4-метоксибензил)-1H-тетразола; 5-(3-(2,6-диметилбензилокси)фенэтил)-1H-тетразола; 5-(3-(2,6-диметилбензилокси)-4-метилбензил)-1H-тетразола; 5-(4-(2,6-дифторбензилокси)бензил)-1H-тетразола; 5-(3-(2,6-диметилбензилокси)-2-метилбензил)-1H-тетразола и 5-(3-(2,6-диметилбензилокси)-2-метоксибензил)-1H-тетразола.
В одном варианте осуществления соединения настоящего изобретения, соединение находится в практически (по меньшей мере, 98%) чистой форме.
РЕАКЦИОННЫЕ СХЕМЫ
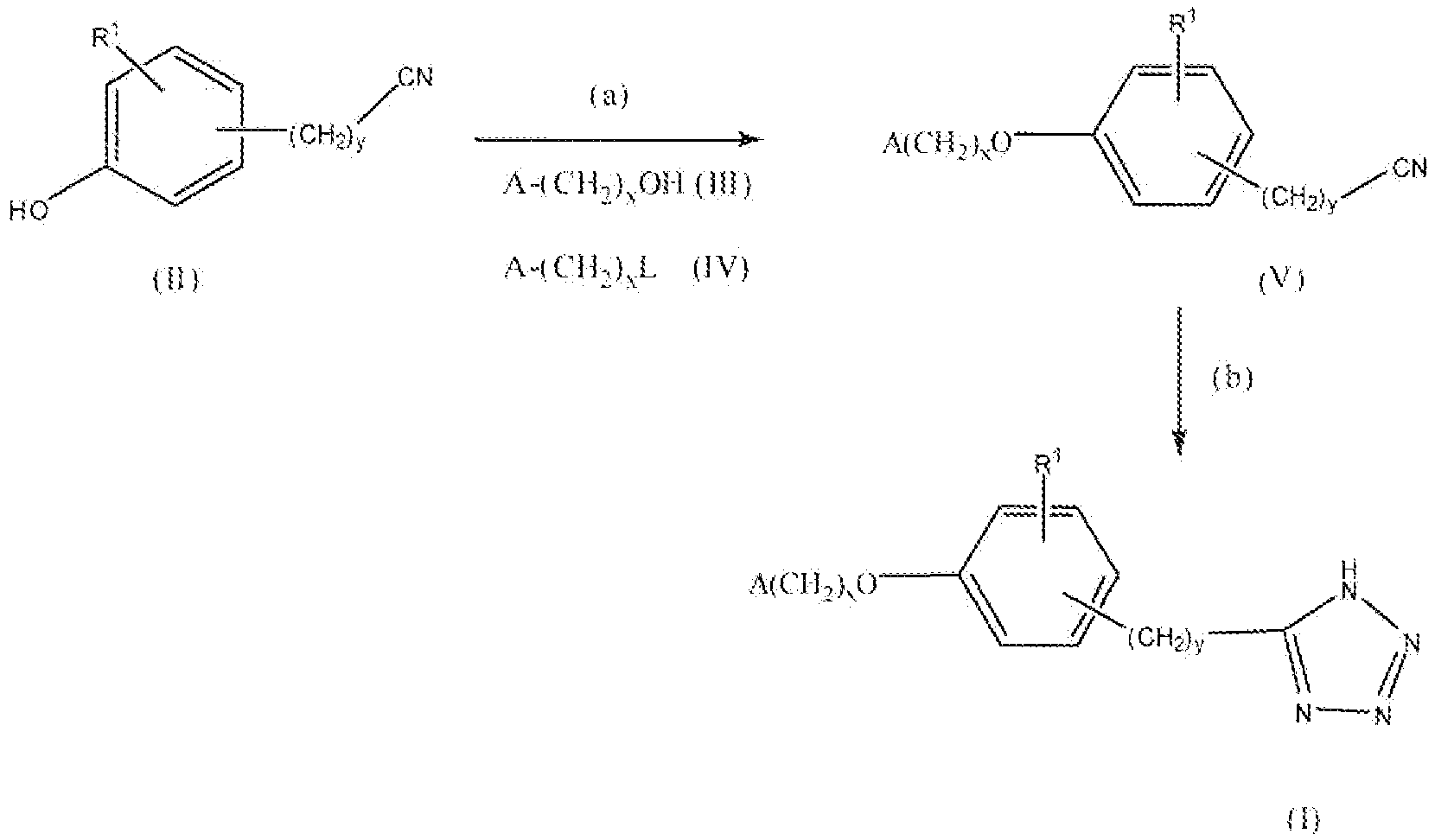
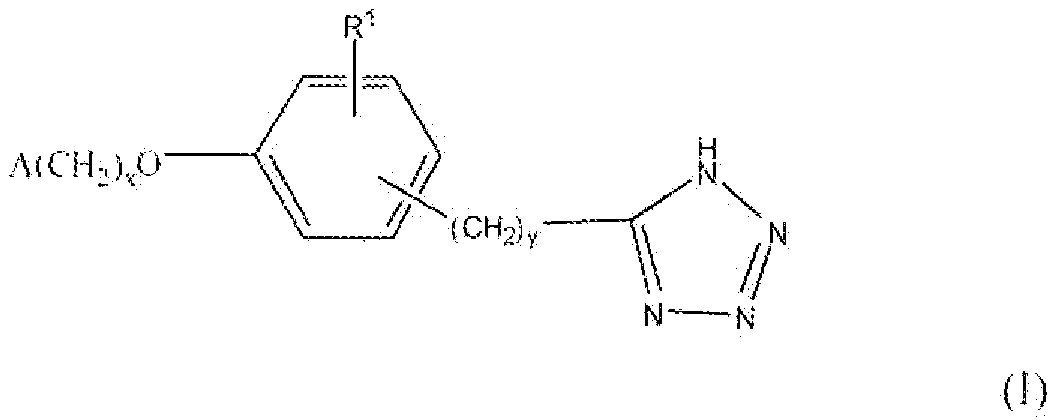
Соединение формулы I, где x равно 1 или 2, y равно 0-3, R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, т.е. соединения формулы:
где A такой, как описано выше, можно получить реакцией схемы 1. В реакции схемы 1 A, x, y и R1 такие, как определено выше. L представляет собой уходящую группу.
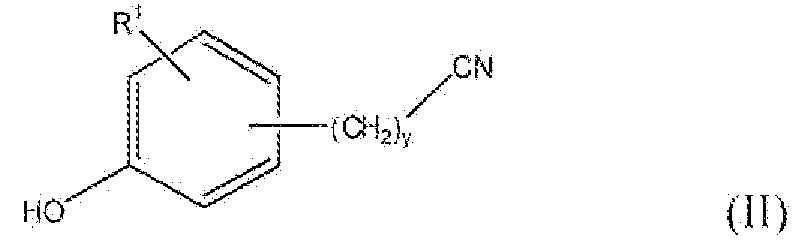
Соединение формулы II можно превратить в соединение формулы V реакцией стадии (a), применяя конденсацию по Мицунобу II c III, применяя трифенилфосфин и диэтилазодикарбоксилат или диизопропилазодикарбоксилат. Реакцию проводят в подходящем растворителе, например тетрагидрофуране. Можно применять любые из условий, обычно применяемые в реакциях Мицунобу, для проведения реакции стадии (a).
Соединение формулы V можно также получить этерификацией или алкилированием соединения формулы II соединением формулы IV, как в реакции стадии (a), применяя подходящее основание, например карбонат калия, триэтиламин, пиридин и подобные. Реакцию проводят в апротонных растворителях, например N,N-диметилформамиде, ацетонитриле, дихлорметане и подобном. В соединении формулы IV, L включает, но не ограничивается, мезилокси, тозилокси, хлор, бром, йод и подобные. Можно применять любой стандартный способ этерификации гидроксильной группы реакцией с галогенидом или уходящей группой для проведения реакции стадии (a).
Соединение формулы V можно превратить в соединение формулы I реакцией стадии (b) путем реакции нитрила с азидом, например триметилсилилазидом или азидом метала, например азидом натрия, азидом калия, азидом лития, причем предпочтительным азидом является азид натрия, в присутствии кислоты Льюиса, например хлорида цинка, хлорида магния, хлорида алюминия, тетрахлорида олова и подобных. Реакцию проводят в растворителе, например, N,N-диметилформамиде при температуре в диапазоне от 80°C до 145°C от 6 до 60 часов. В идеальном случае в реакции применяют взаимодействие нитрила со смесью азид натрия/хлорид аммония/N,N-диметилформамид при 120°C в течение 24 часов. Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Если A представляет собой фенил, замещенный 1 или 2 гидроксильными группами, обычно предпочтительно защищать гидроксильные группы. Подходящая защитная группа может быть описана в Protective Groups in Organic Synthesis у T. Greene. Защитную группу можно удалять после реакции стадии (b), применяя подходящие реагенты для деблокирования, такие как реагенты, описанные в Protective Groups in Organic Synthesis у T. Greene.
Реакционная схема 1
Соединение формулы I, где x равно 1 или 2, y равно 0-3, R1 представляет собой гидроксил, т.е. соединения формулы:
где A такой, как описано выше, можно получить реакцией схемы 2. В реакции схемы 2 A, x и y такие, как определено выше.
Соединение формулы V можно превратить в соединение формулы VI реакцией стадии (c) путем обработки соединения формулы V трибромидом бора или трихлоридом бора, применяя растворитель, например дихлорметан, в течение 4-48 часов при температуре в диапазоне от -72°C до 0°C. Можно применять любые условия, стандартные для данных реакций деметилирования, для проведения реакции стадии (c).
Соединение формулы VI можно превратить в соединение формулы I, где R1 представляет собой гидроксил, реакцией стадии (d) тем же способом, как описано в настоящем описании выше в реакции стадии (b). Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 2
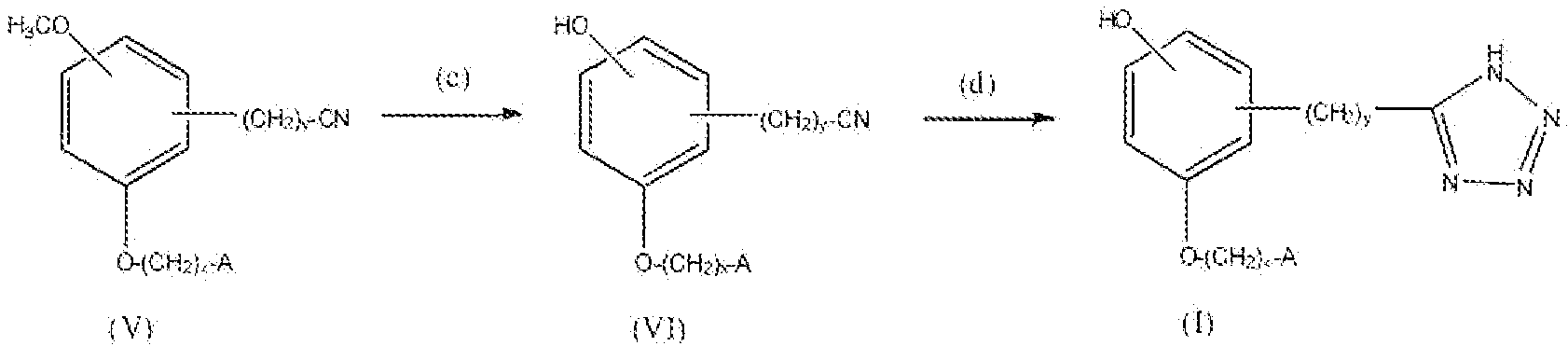
Соединение формулы I, где x равно 1 или 2, y равно 0-3, R1 представляет собой амино, т.е. соединения формулы:
где A такой, как описано выше, можно получить реакцией схемы 3. В реакции схемы 3 A, x и y такие, как определено выше. P представляет собой защитную группу.
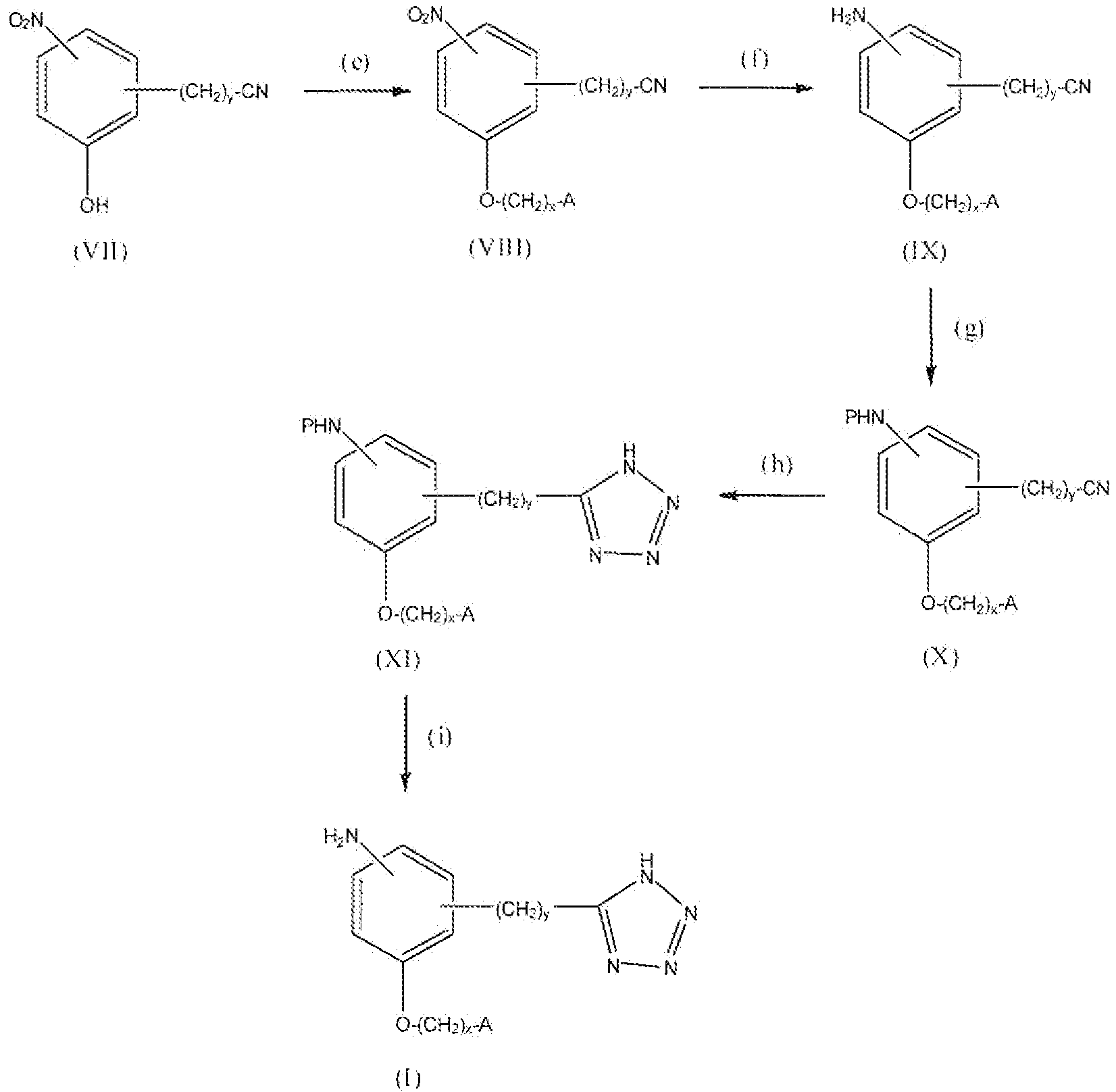
Соединение формулы VII можно превратить в соединение формулы VIII реакцией стадии (e) тем же способом, как описано в настоящем описании выше в реакции стадии (a). Соединение VIII можно превратить в соединение формулы IX реакцией стадии (f) путем восстановления нитрогруппы до амина. Восстанавливающие агенты могут представлять собой металлы, например Zn, Sn или Fe и подобные, и кислоту. Нитрогруппу можно также восстановить каталитическим гидрированием с получением амина. Предпочтительным способом восстановления является каталитическое гидрирование. Можно применять любые условия, стандартные для данных реакций восстановления, для проведения реакции стадии (f).
Соединение формулы IX можно превратить в соединение формулы X реакцией стадии (g) путем защиты аминогруппы. Подходящая защитная группа может быть описана в Protective Groups in Organic Synthesis у T. Greene. Соединение формулы X можно превратить в соединение формулы XI реакцией стадии (h) тем же способом, как описано в настоящем описании выше в реакции стадии (b). Соединение формулы XI можно превратить в соединение формулы I, где R1 представляет собой амино, реакцией стадии (i) путем удаления защитной группы аминофункции. Подходящие реагенты для деблокирования могут быть описаны в Protective Groups in Organic Synthesis у T. Greene. Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 3
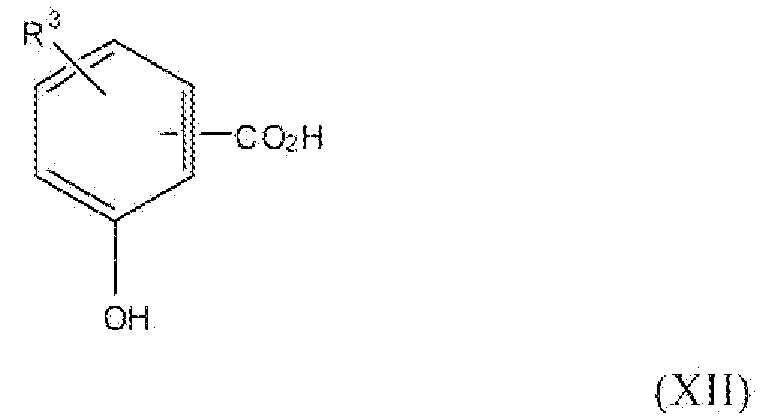
Соединение формулы II, где y равно 1-3, R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, т.е. соединения формулы:
и соединение формулы VII, где y равно 1-3, т.е. соединения формулы:
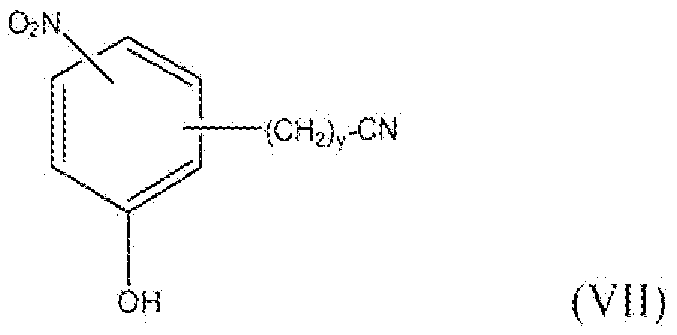
можно получить реакцией схемы 4. В реакции схемы 4 R3 представляет собой водород, фтор, бром, хлор, нитро, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода. P представляет собой защитную группу гидроксильной функции. R2 представляет собой алкильную группу, содержащую 1-2 атома углерода. Y представляет собой галоген.
Соединение формулы XII можно превратить в соединение формулы XIII реакцией стадии (j) путем предварительной защиты карбоксильной и гидроксильной групп, применяя подходящие защитные группы, такие как группы, описанные в Protective Groups in Organic Synthesis у T. Greene.
Соединение формулы XIII можно восстановить до соединения формулы XIV, где R3 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, применяя стандартный восстанавливающий реагент, который превращает сложноэфирную группу в спирт реакцией стадии (k). При проведении данной реакции предпочтительно, но не ограничиваясь этим, применять алюмогидрид лития. Реакцию проводят в подходящем растворителе, таком как тетрагидрофуран и подобном. Можно применять любые условия, стандартные для данных реакций восстановления, для проведения реакции стадии (k).
Соединение формулы XIII можно восстановить до соединения формулы XIV, где R3 представляет собой нитро, применяя восстанавливающий реагент, который превращает сложный эфир в спирт, но не восстанавливает нитрогруппу, например BH3-THF, NaBH4-AlCl3 и подобные. Можно применять любые условия, стандартные для данной реакции восстановления, для проведения реакции стадии (k). Соединение формулы XIV можно превратить в соединение формулы XV замещением гидроксильной группы галогеном, причем предпочтительным галогеном является бром или хлор. Подходящие галогенирующие агенты включают, но не ограничиваясь этим, тионилхлорид, оксалилхлорид, бром, трибромид фосфора, тетрабромид углерода и подобные. Можно применять любые условия, стандартные для данных реакций галогенирования, для проведения реакции стадии (l).
Соединение формулы XV можно превратить в соединение формулы XVI реакцией XV с цианидом щелочного металла, например цианидом натрия или калия или цианидом меди. Реакцию можно проводить в подходящем растворителе, например диметилсульфоксиде, N,N-диметилформамиде и подобном. Можно применять любые условия, обычно применяемые для получения нитрилов из галогенидов, для проведения реакции стадии (m).
Соединение формулы XVI можно превратить в соединение формулы XVII реакцией стадии (n) путем удаления защитной группы гидроксила, применяя подходящие реагенты для деблокирования, такие как реагенты, описанные в Protective Groups in Organic Synthesis у T. Greene. Соединение формулы XVII представляет собой соединение формулы II, где y равно 1, и R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода. Соединение формулы XVII также представляет собой соединение формулы VII, где y равно 1, и R3 представляет собой нитро. Соединение формулы XVI можно превратить в соединение формулы XVIII реакцией стадии (o) путем кислого или основного гидролиза. При проведении данной реакции предпочтительно применять основный гидролиз, например водный гидроксид натрия в этаноле и подобные. Для проведения реакции стадии (o) можно применять любые условия, обычно применяемые для гидролиза нитрилов до карбоновых кислот.
Соединение формулы XVIII можно восстановить для того, чтобы получить соединение формулы XIX реакцией стадии (p). Данную реакцию можно проводить тем же способом, как описано в настоящем описании выше в реакции стадии (k). Соединение формулы XIX можно превратить в соединение формулы XX реакцией стадии (q) тем же способом, как описано в настоящем описании выше в реакции стадии (l). Соединение формулы XX можно превратить в соединение формулы XXI реакцией стадии (r) тем же способом, как описано в настоящем описании выше в реакции стадии (m). Соединение формулы XXI можно превратить в соединение формулы XXII реакцией стадии (s) тем же способом, как описано в настоящем описании выше в реакции стадии (n).
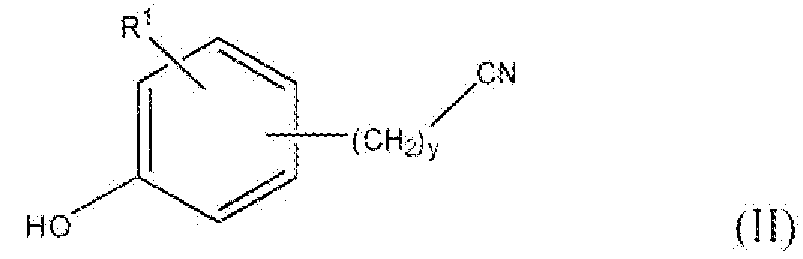
Соединение формулы XXII представляет собой соединение формулы II, в котором y равно 2, и R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода. Соединение формулы XVII также представляет собой соединение формулы VII, где y равно 2, и R3 представляет собой нитро.
Соединение формулы XXI можно гидролизовать тем же способом, как описано в настоящем описании выше в реакции стадии (o), для того, чтобы получить соединение формулы XXIII реакцией стадии (t).
Соединение формулы XXIII можно восстановить для того, чтобы получить соединение формулы XXIV реакцией стадии (u). Данную реакцию можно проводить тем же способом, как описано в настоящем описании выше в реакции стадии (k). Соединение формулы XXIV можно превратить в соединение формулы XXV реакцией стадии (v) тем же способом, как описано в настоящем описании выше в реакции стадии (l). Соединение формулы XXV можно превратить в соединение формулы XXVI реакцией стадии (w) тем же способом, как описано в настоящем описании выше в реакции стадии (m). Соединение формулы XXVI можно превратить в соединение формулы XXVII реакцией стадии (x) тем же способом, как описано в настоящем описании выше в реакции стадии (n). Соединение формулы XXVII представляет собой соединение формулы II, где y равно 3, и R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода. Соединение формулы XXVII также представляет собой соединение формулы VII, где y равно 3, и R3 представляет собой нитро. Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 4
Соединение формулы II, где y равно 0, R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, т.е. соединения формулы:
и соединение формулы VII, где y равно 0, т.е. соединения формулы:
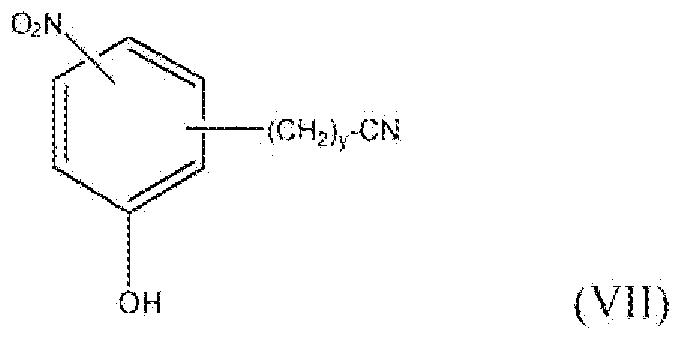
можно получить реакцией схемы 5. В реакции схемы 5 R3 представляет собой водород, нитро, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, P представляет собой защитную группу гидроксильной функции, R2 представляет собой алкильную группу, содержащую 1-2 атома углерода, R4 представляет собой H, хлор или бром.
Соединение формулы XXVIII можно превратить в соединение формулы XXIX реакцией стадии (y) путем предварительной защиты гидроксильной группы, применяя подходящие защитные группы, такие как группы, описанные в Protective Groups in Organic Synthesis у T. Greene, и затем гидролизуя сложный эфир для того, чтобы получить соединение формулы XXIX, где R4 представляет собой H.
Соединение формулы XXVIII можно превратить в соединение формулы XXIX, где R4 представляет собой хлор или бром, реакцией соединения формулы из стадии (y) с галогенирующим реагентом, например тионилхлоридом, пентахлоридом фосфора, трихлоридом фосфора, бромом, тетрабромидом углерода и подобным. Можно применять любые условия, обычно применяемые в данных реакциях галогенирования карбоновых кислот, для проведения реакции стадии (z).
Соединение формулы XXIX можно превратить в соединение формулы XXX реакцией (a') путем взаимодействия непосредственно с аммиаком или сначала обработкой соединения формулы XXIX конденсирующим реагентом, например дициклогексилкарбодиимидом, гексафторфосфатом бензотриазолилокси-трис(диметиламино)фосфония, и затем реакцией с аммиаком и подобным. Можно применять любые условия, обычно применяемые для ацилирования аммиака, для проведения реакции стадии (a'). Соединение формулы XXX можно превратить в соединение формулы XXXI реакцией стадии (b') путем дегидрирования, применяя реагенты, например тионилхлорид, пентаоксид фосфора, пентахлорид фосфора, оксихлорид фосфора, тетрахлорид углерода-трифенилфосфин, цианурхлорид и подобные. Реакцию проводят или без растворителей или в подходящем растворителе, например N,N-диметилформамиде и подобном. Можно применять любые условия, обычно применяемые для реакции дегидрирования, для проведения реакции стадии (b').
Соединение формулы XXXI можно превратить в соединение формулы XXXII реакцией стадии (c') путем удаления защитной группы с гидроксильной функции, применяя подходящие реагенты для деблокирования, такие как реагенты, описанные в Protective Groups in Organic Synthesis у T. Greene. Соединение формулы XXXII представляет собой соединение формулы II, где y равно 0, и R1 представляет собой водород, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода. Соединение формулы XXXII также представляет собой соединение формулы VII, где y равно 0, и R3 представляет собой нитро. Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 5
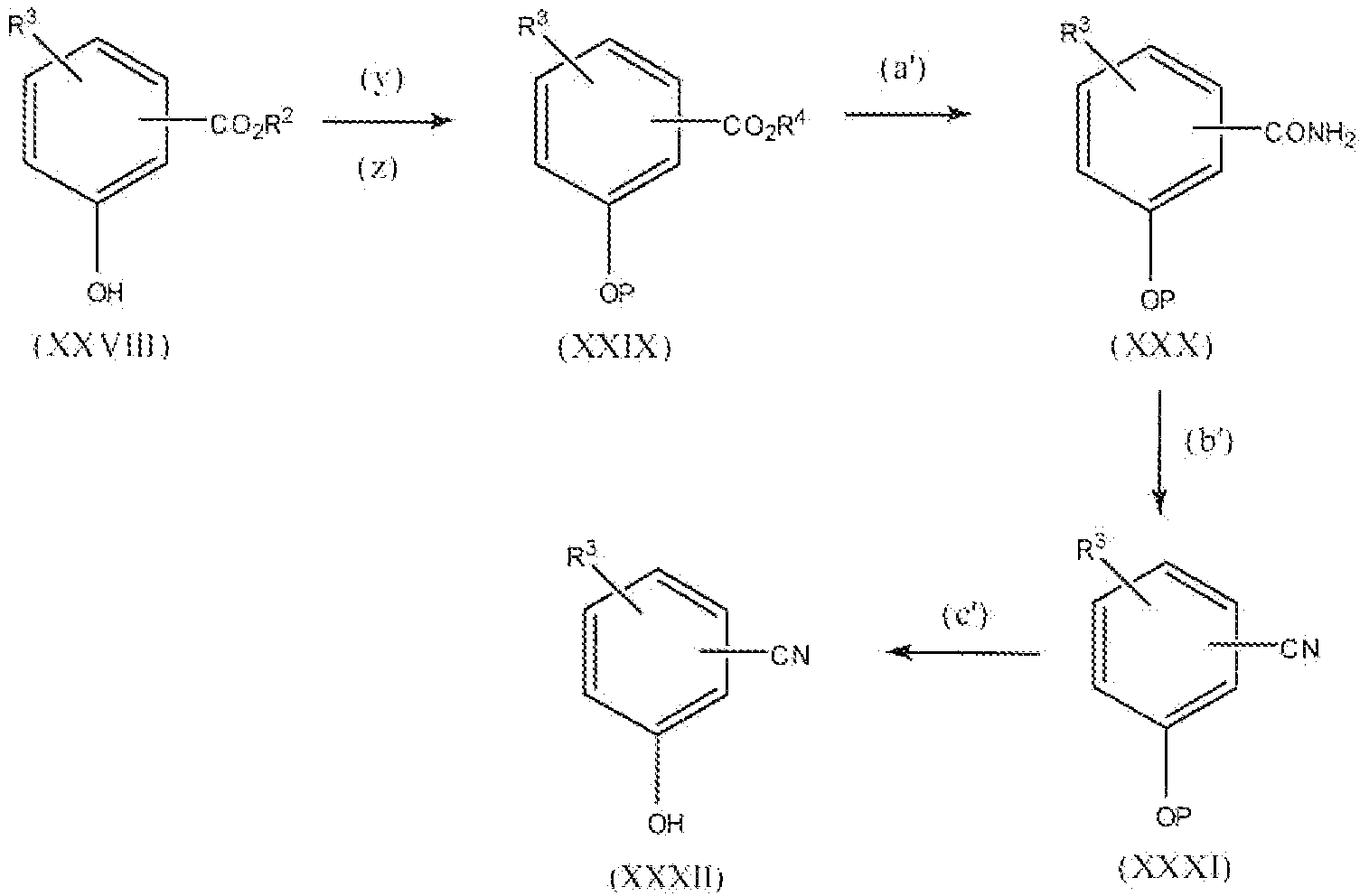
Соединение формулы III, где x равно 1 или 2, т.е. соединения формулы:
A-(CH2)xOH
и соединение формулы IV, где x равно 1 или 2, т.е. соединения формулы:
A-(CH2)xL
можно получить реакцией схемы 6. В реакции схемы 6 A такой, как описано выше. L представляет собой уходящую группу или галогенид. Соединение формулы XXXIII можно восстановить до соединения формулы XXXIV реакцией стадии (d'). Реакцию проводят, применяя стандартный восстанавливающий реагент, например гидрид щелочного металла, такой как алюмогидрид лития. Реакцию проводят в подходящем растворителе, таком как тетрагидрофуран. Можно применять любые условия, стандартные для данных реакций восстановления, для проведения реакции стадии (d'). Соединение формулы XXXIV представляет собой соединение формулы III, где x равно 1.
Соединение формулы XXXIV можно превратить в соединение формулы XXXV замещением гидроксильной группы уходящей группой или галогенидом, причем предпочтительной группой является бром или хлор. Подходящие реагенты для галогенирования включают, но не ограничиваясь этим, тионилхлорид, оксалилхлорид, бром, трибромид фосфора, тетрабромид углерода и подобные. Уходящие группы включают тозилат, мезилат и подобные. Можно применять любые условия, стандартные для данных реакций, для проведения реакции стадии (e'). Соединение формулы XXXV представляет собой соединение формулы IV, где x равно 1.
Соединение формулы XXXV можно превратить в соединение формулы XXXVI реакцией XXXV с цианидом щелочного металла, например цианидом натрия или калия. Реакцию проводят в подходящем растворителе, таком как этанол, диметилсульфоксид, N,N-диметилформамид и подобных. Можно применять любые условия, обычно применяемые при получении нитрилов, для проведения реакции стадии (f').
Соединение формулы XXXVI можно превратить в соединение формулы XXXVII реакцией стадии (g') кислотным или основным гидролизом. При проведении данной реакции предпочтительно применять основный гидролиз, например водный гидроксид натрия. Можно применять любые условия, обычно применяемые для гидролиза нитрилов, для проведения реакции стадии (g').
Соединение формулы XXXVII можно восстановить для того, чтобы получить соединение формулы XXXVIII реакцией стадии (h'). Данную реакцию можно проводить тем же способом, как описано в настоящем описании выше в реакции стадии (d'). Соединение формулы XXXVIII представляет собой соединение формулы III, где x равно 2.
Соединение формулы XXXVIII можно превратить в соединение формулы XXXIX реакцией стадии (i') тем же способом, как описано в настоящем описании выше в реакции стадии (e'). Соединение формулы XXXIX представляет собой соединение формулы IV, где x равно 2.
Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация. Если A представляет собой фенил, замещенный 1 или 2 группами гидроксила, обычно предпочтительно защищать гидроксильную группу соединения формулы XXXIII. Подходящая защитная группа может быть описана в Protective Groups in Organic Synthesis у T. Greene.
Реакционная схема 6
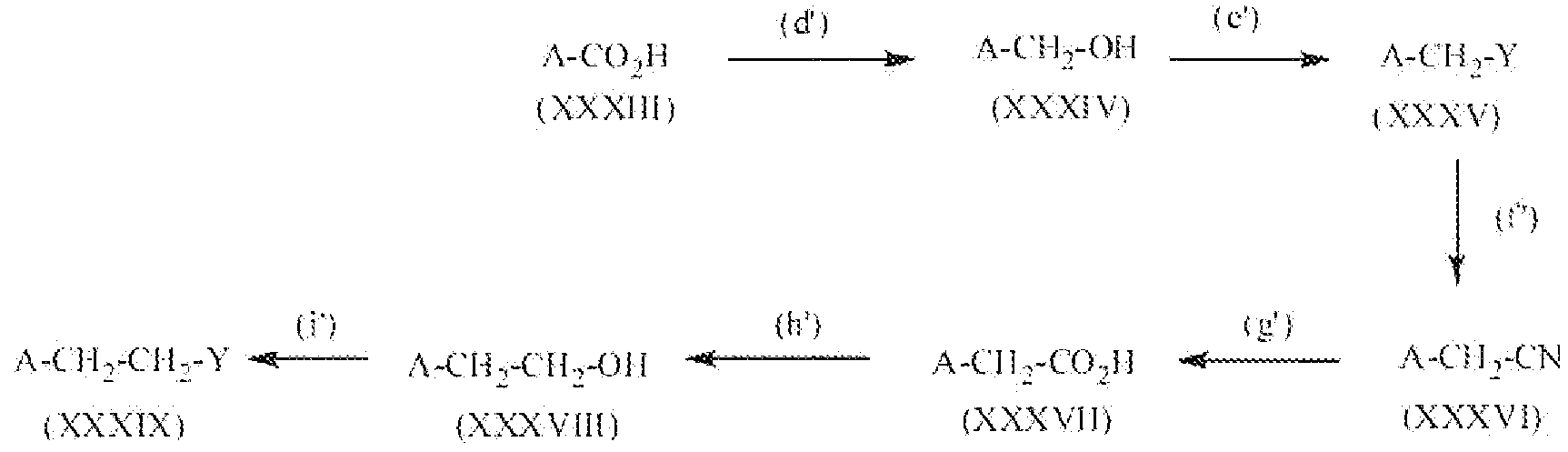
Соединение формулы XXVIII, где R3 представляет собой водород, нитро, фтор, бром, хлор, алкокси, содержащий от 1 до 2 атомов углерода, или алкил, содержащий от 1 до 2 атомов углерода, R2 представляет собой алкильную группу, содержащую 1-2 атома углерода, т.е. соединения формулы:
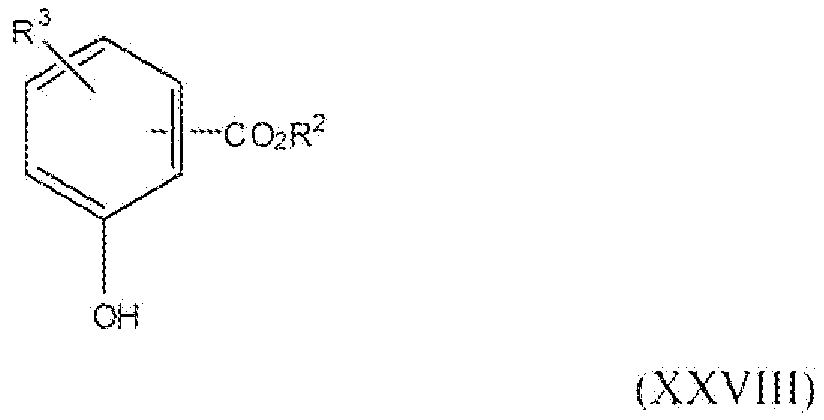
можно получить реакцией схемы 7. В реакции схемы 7 R2 и R3 такие, как определено выше.
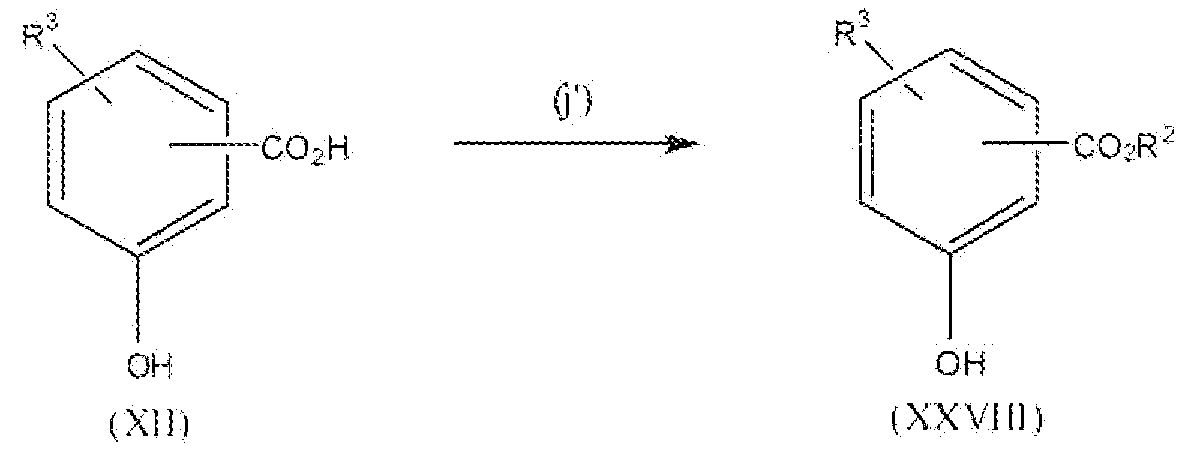
Соединение формулы XII можно превратить в соединение формулы XXVIII реакцией стадии (j') путем этерификации соединения формулы XII с метанолом или этанолом. Реакцию можно проводить или применяя катализаторы, например H2SO4, TsOH и подобные, или применяя дегидратирующий агент, например дициклогексилкарбодиимид и подобный. Можно применять любые из условий, стандартные в данных реакциях этерификации, для проведения реакции стадии (j'). Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 7
Соединение формулы XII, где R3 представляет собой хлор, бром или фтор, т.е. соединения формулы:
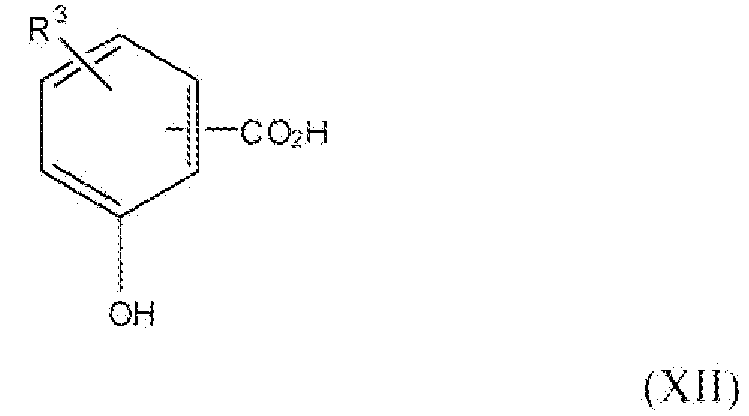
является или коммерчески доступным, или его можно получить согласно способам, описанным в следующей литературе:
1. 3-Br или F-2-OHC6H3CO2H
Canadian Journal of Chemistry (2001), 79(11) 1541-1545;
2. 4-Br-2-OHC6H3CO2H
WO 9916747 или JP 04154773;
3. 2-Br-6-OHC6H3CO2H
JP 47039101;
4. 2-Br-3-OHC6H3CO2H
WO 9628423;
5. 4-Br-3-OHC6H3CO2H
WO 2001002388;
6. 3-Br-5-OHC6H3CO2H
Journal of labelled Compounds and Radiopharmaceuticals (1992), 31 (3), 175-82;
7. 2-Br-5-OHC6H3CO2H и 3-Cl-4-OHC6H3CO2H
WO 9405153 и US 5519133;
8. 2-Br-4-OHC6H3CO2H и 3-Br-4-OHC6H3CO2H
WO 20022018323;
9. 2-Cl-6-OHC6H3CO2H
JP 06293700;
10. 2-Cl-3-OHC6H3CO2H
Proceedings of the Indiana Academy of Science (1983). Volume date 1982. 92, 145-51;
11. 3-Cl-5-OHC6H3CO2H
WO 2002000633 и WO 2002044145;
12. 2-Cl-5-OHC6H3CO2H
WO 9745400.
Соединение формулы XII, где R3 представляет собой алкокси, содержащий от 1 до 2 атомов углерода, т.е. соединения формулы:
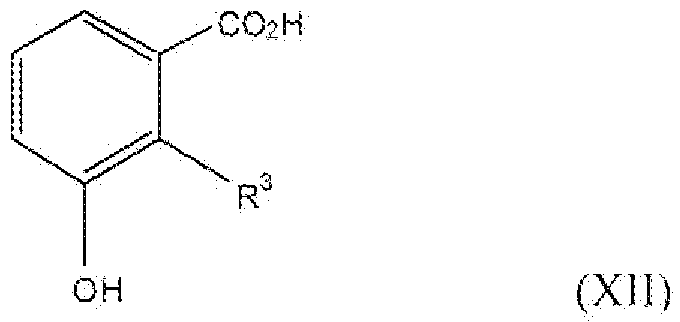
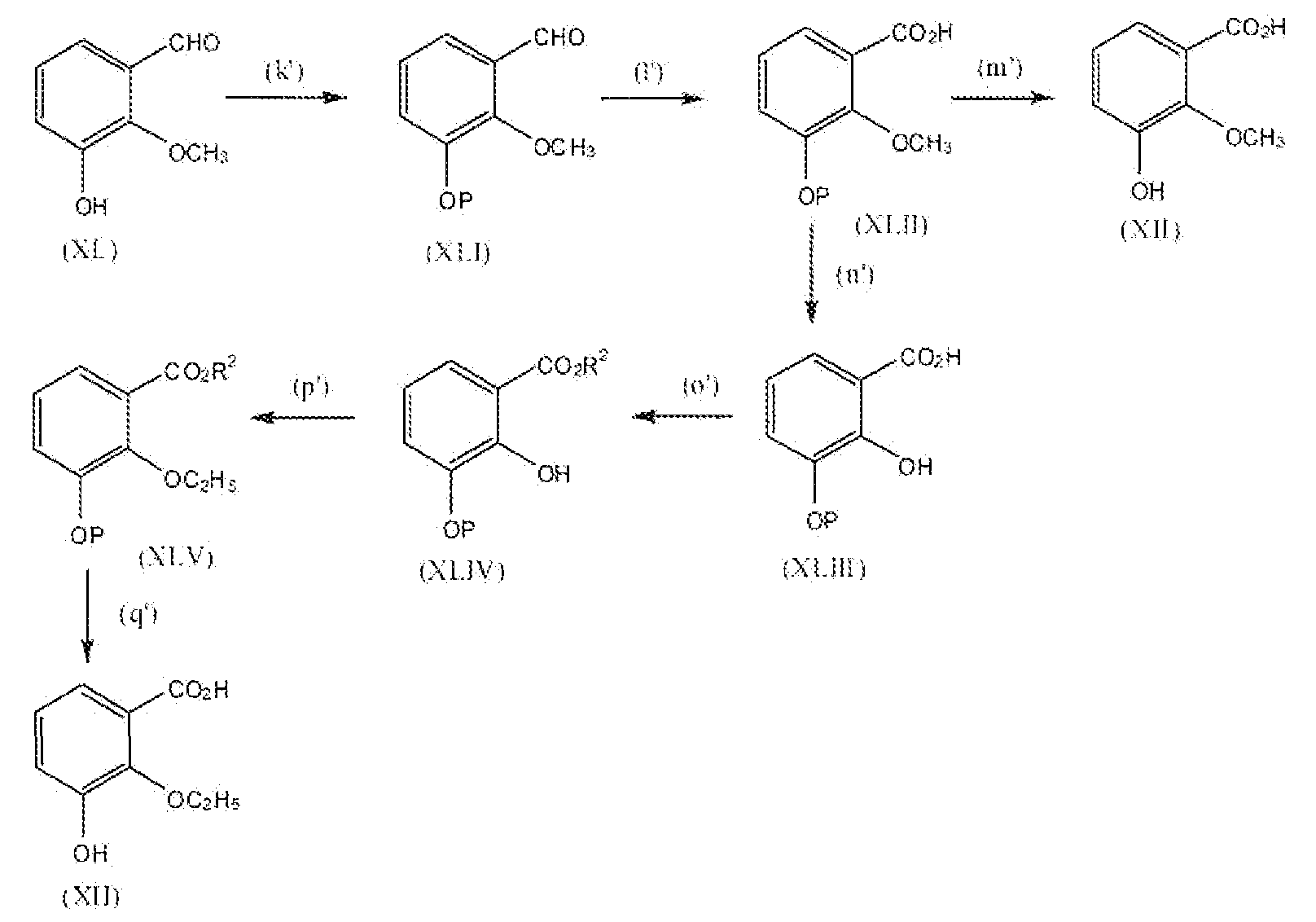
можно получить реакцией схемы 8. В реакции схемы 8 R2 представляет собой алкильную группу, содержащую от 1 до 2 атомов углерода. P представляет собой защитную группу гидроксила. Соединение формулы XL можно превратить в соединение формулы XLI реакцией стадии (k') путем защиты фенольной группы подходящей защитной группой. Подходящие условия для защитной группы могут быть описаны в Protective Groups in Organic Synthesis у T. Greene.
Соединение формулы XLI можно превратить в соединение формулы XLII окислением альдегида до карбоновой кислоты. Реакцию можно проводить, применяя подходящие окисляющие реагенты, например хлорхромат пиридиния, перманганат калия, перманганат натрия и подобные. Можно применять любые из условий, подходящие для данных реакций окисления, для проведения реакции стадии (l').
Соединение формулы XLII можно превратить в соединение формулы XII реакцией стадии (m'), где R3 представляет собой алкокси, содержащий 1 атом углерода, удалением защитной группы. Подходящие условия деблокирования могут быть описаны в Protective Groups in Organic Synthesis у T Greene.
Соединение формулы XLII можно превратить в соединение формулы XLIII обработкой соединения формулы XLII трибромидом бора или трихлоридом бора, применяя растворитель, например, дихлорметан, в течение 4-48 часов при температуре от -72°C до 0°C. Можно применять любые из условий, стандартные в данных реакциях деметилирования, для проведения реакции стадии (n').
Соединение формулы XLIII можно превратить в соединение формулы XLIV этерификацией соединения формулы XLIII с метанолом или этанолом. Реакцию можно проводить или применяя катализатор, например H2SO4, TsOH и подобный, или применяя дегидратирующий агент, например дициклогексилкарбодиимид и подобный. Можно применять любые из условий, стандартные для данных реакций этерификации, для проведения реакции стадии (o').
Соединение формулы XLIV можно превратить в соединение формулы XLV этерификацией или алкилированием соединения формулы XLIV этилгалогенидом, применяя подходящее основание, например карбонат калия, гидрид натрия, пиридин и подобные. Реакцию можно проводить в стандартных растворителях, таких как тетрагидрофуран, N,N-диметилформамид, дихлорметан и подобные. Реакцию обычно проводят при температурах от 0°C до 40°C. Можно применять любые из условий, подходящих для данных реакций алкилирования, для проведения реакции стадии (p').
Соединение формулы XLV можно превратить в соединение формулы XII реакцией стадии (q'), где R3 представляет собой алкокси, содержащий 2 атома углерода, удалением защитной группы. Подходящие условия деблокирования могут быть описаны в Protective Groups in Organic Synthesis у T Greene. Продукты можно выделить и очистить способами, такими как экстракция, упаривание, хроматография и перекристаллизация.
Реакционная схема 8
Соединение формулы XII, где R3 представляет собой алкокси, содержащий от 1 до 2 атомов углерода, т.е. соединения формулы:
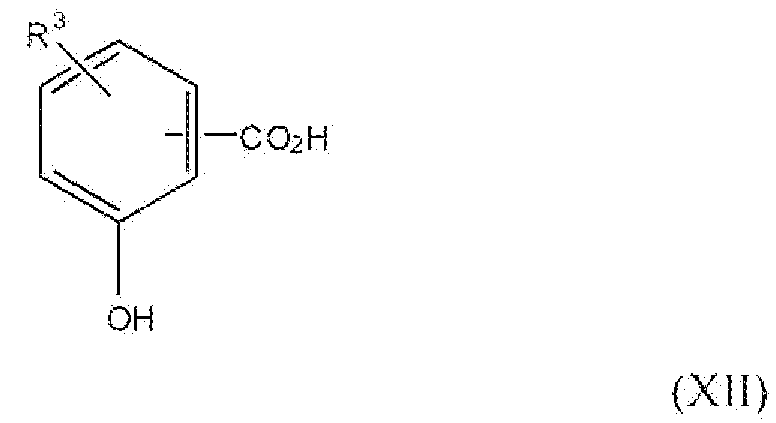
являются или коммерчески доступным, или их можно получить согласно способам, описанным в следующей литературе:
1. 2-OMe-4-OHC6H3CO2H
US 2001034343 или WO 9725992;
2. 5-OMe-3-OHC6H3CO2H
J.O.C (2001), 66(23), 7883-88;
3. 2-OMe-5-OHC6H3CO2H
US 6194406 (страница 96) и Journal of the American Chemical Society (1985), 107(8), 2571-3;
4. 3-OEt-5-OHC6H3CO2H
Taiwan Kexue (1996). 49(1), 51-56;
5. 4-OEt-3-OHC6H3CO2H
WO 9626176;
6. 2-OEt-4-OHC6H3CO2H
Takeda Kenkyusho Nempo (1965), 24, 221-8
JP 07070025;
7. 3-OEt-4-OHC6H3CO2H
WO 9626176.
Соединение формулы XII, где R3 представляет собой алкил, содержащий 1-2 атома углерода, т.е. соединения формулы:
являются или коммерчески доступный, или их можно получить согласно способам, описанным в следующей литературе:
1. 5-Me-3-OHC6H3CO2H и 2-Me-5-OHC6H3CO2H
WO 9619437
J.O.C. 2001, 66, 7883-88;
2. 2-Me-4-OHC6H3CO2H
WO 8503701;
3. 3-Et-2-OHC6H3CO2H и 5-Et-2-OHC6H3CO2H
J. Med. Chem. (1971), 14(3), 265;
4. 4-Et-2-OHC6H3CO2H
Yaoxue Xuebao (1998), 33(1), 67-71;
5. 2-Et-6-OHC6H3CO2H и 2-н-Pr-6-OHC6H3CO2H
J. Chem. Soc. Perkin Trans 1 (1979), (8), 2069-78;
6. 2-Et-3-OHC6H3CO2H
JP 10087489 и WO 9628423;
7. 4-Et-3-OHC6H3CO2H
J.O.C. 2001, 66, 7883-88
WO 9504046;
8. 2-Et-5-OHC6H3CO2H
J.A.C.S (1974), 96(7), 2121-9;
9. 2-Et-4-OHC6H3COH и 3-Et-4-OHC6H3CO2H
JP 04282345;
10. 1,3-Et-5-OHC6H3CO2H
Адаптированный синтез из J.O.C 2001, 66, 7883-88, применяя 2-этилакролеин.
ПРИМЕНЕНИЕ В СПОСОБАХ ЛЕЧЕНИЯ
Настоящее изобретение относится к способу снижения концентрации мочевой кислоты у субъекта, являющегося млекопитающим, или усиления выведения мочевой кислоты у субъекта, являющегося млекопитающим. Концентрацию мочевой кислоты у млекопитающего можно определить, применяя стандартные измерения. Обычно определяют концентрацию мочевой кислоты в крови. Мочевая кислота может также откладываться или осаждаться в тканях, приводя в результате к накоплению (например, тофус), на которые может влиять повышение или снижение концентрации мочевой кислоты в крови, и которые, наоборот, могут способствовать циркуляции мочевой кислоты. Способ настоящего изобретения для снижения концентрации мочевой кислоты можно применять для лечения или предотвращения ряда состояний, включая подагру, гиперурикемию, повышенную концентрацию мочевой кислоты, которая не соответствует концентрации, при которой обычно ставят диагноз гиперурикемии, почечные камни, почечную дисфункцию, сердечно-сосудистое заболевание, фактор риска возникновения сердечно-сосудистого заболевания и когнитивное нарушении. Снижая концентрацию мочевой кислоты, введение соединений настоящего изобретения замедляет развитие заболевания почек. Повышенная концентрация мочевой кислоты отождествляется с фактором риска развития сердечно-сосудистого заболевания. Показана характерная взаимосвязь между повышенной концентрацией мочевой кислоты и когнитивным нарушением у пожилых людей. (Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1); 136-140). Соответственно, способ настоящего изобретения для снижения концентрации мочевой кислоты можно применять для лечения или предотвращения когнитивного нарушения, включая когнитивное нарушение у пожилых людей. Хорошо известно, что люди с синдромом Леша-Найхана имеют повышенные концентрации мочевой кислоты и страдают от многочисленных последствий данной гиперурикемии, включая подагру. Таким образом, настоящее изобретение для снижения концентраций в крови и усиления выведения мочевой кислоты можно применять для лечения людей с синдромом Леша-Найхана.
Нормальный диапазон концентраций мочевой кислоты в крови составляет от 3,4 мг/дл до 7,0 мг/дл у мужчин, от 2,4 мг/дл до 6,0 мг/дл у женщин в пременопаузе, и от 2,5 мг/дл до 5,5 мг/дл у детей. Образование/осаждение кристаллов соли мочевой кислоты обычно возникает у мужчин при концентрациях 6,6 мг/дл или более и у женщин при концентрациях 6,0 мг/дл или более. Это показывает что концентрация мочевой кислоты, которая находится в так называемом нормальном диапазоне, может иметь нежелательные для здоровья последствия, вызывая даже подагру. Кроме того, величина, которая может находиться в нормальном диапазоне для популяции в целом, может быть повышенной для индивида. Сердечно-сосудистые и другие последствия повышенной концентрации мочевой кислоты могут возникать при концентрации в крови, находящейся в данных "нормальных" диапазонах. Следовательно, диагноз гиперурикемии необязательно является необходимым условием для благоприятного эффекта соединений настоящего изобретения.
Настоящее изобретение включает лечение гиперурикемии, связанной с подагрой, гипертензией, воспалением сосудов, сердечной недостаточностью, артерио-венозными заболеваниями, инфарктом миокарда, инсультом, преэклампсией, эклампсией, апноэ во сне, почечной дисфункции (включая почечную недостаточность, терминальную стадию почечной недостаточности [ESRD]), пересадкой органов, диуретиками, тиазидами, циклоспорином, аспирином, витамином C, никотиновой кислотой, леводопа (L-DOPA), цитотоксическими лекарственными средствами и определенными антибактериальными агентами (такими как пирозинамид), циррозом печени, дисфункцией щитовидной железы, дисфункцией паращитовидной железы, раком легких, анемией, лейкемией, лимфомой, множественной миеломой, синдромом лизиса опухоли, дисфункцией щитовидной и паращитовидной железы, синдромом Леша-Найхена, курением, употреблением алкоголя и псориазом. Настоящее изобретение включает лечение гиперурикемии, которая не приводит к подагре, образованию кристаллов солей мочевой кислоты, почечной дисфункции, отторжению трансплантата после пересадки органа, эндотелиальным заболеваниям (таким как воспаление), хронической сердечной недостаточности, артерио-венозным заболеваниям, преэклампсии, эклампсии, гипертензии и когнитивным нарушениям. В вариантах осуществления способа настоящего изобретения для лечения подагры снижают отложения в тканях мочевой кислоты, включая, но не ограничиваясь этим, тофус, и частота возникновения и тяжесть вспышек подагры также снижается.
Соединения настоящего изобретения можно вводить любым стандартным путем общего введения. Предпочтительно их вводят перорально. Соответственно, предпочтительно, чтобы лекарственные средства составлялись для перорального введения. Другие пути введения, которые можно применять согласно настоящему изобретению, включают ректальный, парентеральный, инъекцией (например, внутривенно, подкожно, внутримышечно или внутрибрюшинно) или назальный.
Следующие варианты осуществления каждого данного применения и способ лечения настоящего изобретения включают введение любого одного из вариантов осуществления соединений, описанный выше. Во избежание ненужного многословия, каждое данное соединение и группу соединений не повторяют, но их вводят в данное описание применений и способов лечения, если они повторяются.
И субъектов, являющихся людьми, и субъектов, являющихся другими млекопитающими, можно лечить согласно способу лечения настоящего изобретения. Оптимальная доза конкретного соединения настоящего изобретения для конкретного субъекта может быть определена в клинической ситуации опытным практикующим врачом. В случае перорального введение соединение настоящего изобретения обычно вводят взрослым в дневной дозе от 1 мг до 2500 мг, более предпочтительно от 1 мг до 1200 мг. В других вариантах осуществления настоящего изобретения соединение вводят в дозе от 400 мг до 1000 мг, от 600 мг до 800 мг, от 600 мг до 1000 мг или от 100 до 300 мг, вводимой один раз или дважды в день. Средний вес тела обычного взрослого составляет 60-70 килограмм, так что подходящий диапазон доз, выраженный в мг/кг, составляет приблизительно от 0,015 до 42 мг/кг, от 0,015 до 20 мг/кг, от 6,6 до 13 мг/кг, от 10 до 13 мг/кг, от 10 до 16 мг/кг или от 1,67 до 4,3 мг/кг, вводимой один раз или дважды в день. При лечении детей оптимальную дозу определяет лечащий врач. В случае перорального введения мышам соединение настоящего изобретения обычно вводят в дневной дозе от 1 до 300 мг соединения на килограмм веса тела.
Соединение настоящего изобретения можно вводить в комбинации с другими лекарственными средствами, снижающими концентрацию мочевой кислоты. В данном случае доза соединения настоящего изобретения является такой, как описано выше. Любое стандартное или исследуемое лекарственное средство, снижающее концентрацию мочевой кислоты, можно применять в комбинации с соединением настоящего изобретения. Примеры таких лекарственных средств включают ингибиторы ксантиноксидазы, такие как аллопуринол (от 100 мг/день до 1000 мг/день; более обычно от 100 мг/день до 300 мг/день), фебуксостат (от 40 мг/день до 120 мг/день: более конкретно от 60 мг/день до 80 мг/день) и оксипуринол: пуриказа/PEG-уриказа (от 4 мг до 12 мг каждые две недели инфузией); средства, способствующие выведению мочевой кислоты, такие как сульфинпиразон (от 100 мг/день до 800 мг/день), пробенецид (500 мг/день), лозартан (от 25 мг/день до 200 мг/день, более обычно от 50 мг/день до 100 мг/день), фенофибрат, JTT-552 (URAT-1 ингибитор), бензбромарон (от 70 мг/день до 150 мг/день) и статины, такие как аторвастатин (LIPITOR®). Другое лекарственное средство, снижающее концентрацию мочевой кислоты, можно вводить в обычно применяемом для него количестве или в количестве, которое является меньшим, чем обычно применяемое количество, введением ли меньших доз данного другого лекарственного средства или менее частым дозированием данного другого лекарственного средства.
Соединения настоящего изобретения можно вводить вместе с другими лекарственными средствами, применяемыми для снижения боли, связанной с приступами подагры, например нестероидными противовоспалительными агентами (NSAID), колхицином, кортикостероидами и другими анельгетиками.
В процессе снижения концентрации мочевой кислоты в крови ожидается, что соединения настоящего изобретения будут увеличивать концентрацию мочевой кислоты в моче. Для увеличения pH мочи и, таким образом увеличения растворимости мочевой кислоты, в комбинации с соединением настоящего изобретения можно вводить, например, цитрат или бикарбонат.
Можно вводить субъекту смесь соединения или соли настоящего изобретения с одним или более другим лекарственным средством, снижающим концентрацию мочевой кислоты, анальгетиками и агентами, увеличивающими pH. Альтернативно, соединение или соль настоящего изобретения и одно или более других лекарственных средств, снижающих концентрацию мочевой кислоты, анальгетики и агенты, увеличивающие pH, не смешивают вместе для получения смеси, но независимо вводят субъекту. Когда активные ингредиенты не смешивают вместе для получения однородной смеси или композиции, удобно предоставлять их в форме набора, содержащего одну или более однократных пероральных доз соединения настоящего изобретения, одну или более однократных пероральных доз одного или более других лекарственных средств, снижающих концентрацию мочевой кислоты, анальгетиков и агентов, повышающих pH, и инструкции для введения соединения настоящего изобретения в комбинации с другими активными ингредиентами. Предпочтительно компоненты набора упаковывают вместе, например в коробку или блистерную упаковку.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Настоящее изобретение относится к фармацевтической композиции, содержащий соединение настоящего изобретения, и необязательно фармацевтически приемлемый носитель. Следующие варианты осуществления фармацевтической композиции настоящего изобретения содержат любой один из вариантов осуществления соединений, описанных выше. Во избежание многословия каждое данное соединение и группу соединений не повторяют, но их вводят в данное описание фармацевтических композиций, если их повторяют.
Предпочтительно композицию адаптируют для перорального введения, например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсии или суспензии. Обычно пероральная композиция будет содержать от 1 мг до 2500 мг, более предпочтительно от 1 мг до 1200 мг соединения настоящего изобретения. В более конкретных вариантах осуществления настоящего изобретения пероральная композиция будет содержать от 400 мг до 1000 мг, от 600 мг до 800 мг, от 600 мг до 1000 мг или от 100 до 300 мг соединения настоящего изобретения. Удобно, чтобы пациент принимал одну или две таблетки; таблетки, покрытые оболочкой; драже или желатиновые капсулы в день. Однако композицию можно также адаптировать для введения любым другим стандартным способом общего введения, включая ректальный, например в форме суппозиториев, парентеральный, например в форме растворов для инъекций, или назальный.
Активные ингредиенты можно смешивать с фармацевтически инертным, неорганическим или органическим носителем для получения фармацевтических композиций. Можно применять лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и подобные, например, такие как носители для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул. Подходящие носители для мягких желатиновых капсул представляют собой, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и подобные. В зависимости от свойств активного ингредиента носители, однако, не требуются в случае мягких желатиновых капсул, отличных от самого мягкого желатина. Подходящие носители для получения растворов и сиропов представляют собой, например, воду, полиолы, глицерин, растительные масла и подобные. Подходящие носители для суппозиториев представляют собой, например, природные или гидрогенизированные масла, воски, жиры, полутвердые и жидкие полиолы и подобные.
Кроме того, фармацевтические композиции содержат консерванты, солюбилизаторы, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, агенты для покрытия или антиоксиданты.
Настоящее изобретение будет более понятно со ссылкой на следующие примеры, которые иллюстрируют, но не ограничивают настоящее изобретение, описанное в настоящем описании.
ПРИМЕРЫ
ПРИМЕР 1
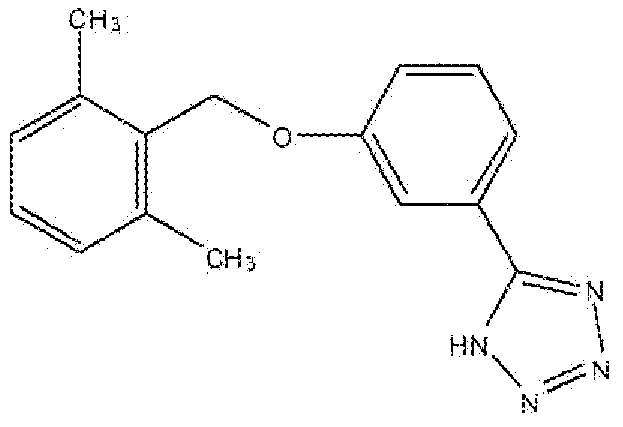
5-(3-(2,6-Диметилбензилокси)фенил)-1H-тетразол
Стадия A: Получение 3-(2,6-диметилбензилокси)бензонитрила
Раствор 2,6-диметилбензилового спирта (6,27 г, 46,1 ммоль) и диизопропилазодикарбоксилата (DIAD, 9,24 г, 45,7 ммоль) в сухом TΗF (30 мл) добавляли по каплям к раствору 3-гидроксибензонитрила (5 г, 37 ммоль) и трифенилфосфина (TPP, 11,99 г, 45,7 ммоль) в THF (100 мл) при 0°C. Реакционную смесь нагревали до комнатной температуры в течение 4 часов или до исчезновения всех исходных соединений, разбавляли простым эфиром и промывали водой (2×). Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия B: Получение 5-(3-(2,6-диметилбензилокси)фенил)-1H-тетразола
Смесь 3-(2,6-диметилбензилокси)бензонитрила (Стадия A, 3 г, 11,8 ммоль), азида натрия (0,847 г, 13 ммоль) и хлорида аммония (0,697 г, 13 ммоль) в сухом диметилформамиде (30 мл) нагревали в атмосфере аргона при 110°C в течение 14 часов или до исчезновения всех исходных соединений. К реакционной смеси добавляли воду для растворения всех твердых веществ; раствор обрабатывали соляным раствором и экстрагировали этилацетатом (2×). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (хлороформ-метанол 95:5) с получением указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,4 (с, 6H); 5,15 (с, 2H); 7,1 (д, 2H); 7,15 (м, 1H); 13 (дд, 1H); 7,5 (т, 1H); 7,65 (м, 1H); 7,7 (м, 1H).
ПРИМЕР 2
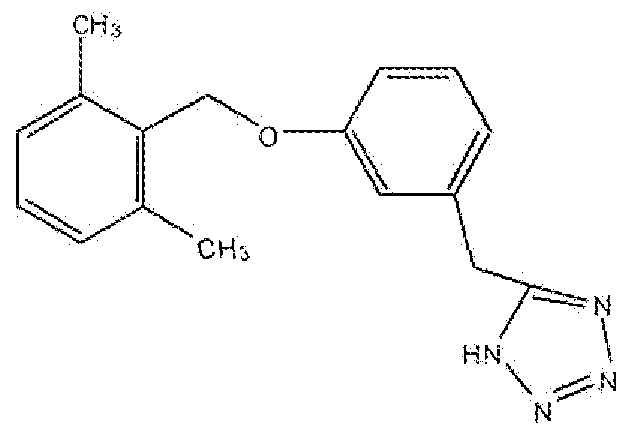
5-(3-(2,6-Диметилбензилокси)бензил)-1H-тетразол
Стадия A: Получение 2-(3-гидроксифенил)ацетонитрила
К раствору 2-(3-метоксифенил)ацетонитрила (3,6 г, 25,4 ммоль) в сухом метиленхлориде (20 мл) добавляли BBr3 (55 мл, 1M в CH2Cl2, 55 ммоль) при -78°C в атмосфере аргона. Реакционную смесь нагревали до температуры окружающей среды в течение 48 часов, гасили дробленым льдом и экстрагировали хлористым метиленом. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (CH2Cl2:этилацетат 4:1) с получением указанного в заголовке соединения в виде масла.
Стадия B: Получение 2-(3-(2,6-диметилбензилокси)фенил)ацетонитрила
Раствор 2-(3-гидроксифенил)ацетонитрила (Стадия A, 5 г, 37 ммоль) и диизопропилазодикарбоксилата (DlAD, 3,38 г, 16,7 ммоль) в сухом THF (20 мл) добавляли по каплям к раствору 2,6-диметилбензилового спирта (2,25 г, 16,5 ммоль) и трифенилфосфина (TPP, 4,3 г, 16,4 ммоль) в THF (30 мл) при 0°C в атмосфере аргона. Реакционная смесь перемешивали при комнатной температуре в течение 16 часов или до исчезновения всех исходных соединений. К смеси добавляли силикагель (25 г), растворители удаляли при пониженном давлении, наносили на колонку с силикагелем и элюировали смесью метиленхлорид:гексан (1:1) с получение светло-желтого кристаллического вещества.
Стадия C: Получение 5-(3-(2,6-диметилбензилокси)бензил)-1H-тетразола
Смесь 2-(3-(2,6-диметилбензилокси)фенил)ацетонитрила (Стадия B, 3,2 г, 12,7 ммоль), азида натрия (1,28 г, 16,7 ммоль) и хлорида аммония (1,08 г, 20,2 ммоль) в сухом диметилформамиде (30 мл) нагревали в атмосфере аргона при 90°C в течение 9 часов или до исчезновения всех исходных соединений и реакционную смесь концентрировали при пониженном давлении. Реакционную смесь выливали в этилацетат и промывали водой (2×), сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (метиленхлорид:метанол 9:1) с получением маслянистого продукта. Масло перемешивали со смесью 1:2 этилацетат:гексан в течение 10 минут и твердое вещество фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,3 (с, 6H); 4,25 (с, 2H); 5,15 (с, 2H); 6,84 (д, 1H); 6,96 (м, 2H); 7,08 (д, 2H); 7,18 (м, 1H); 7,28 (м, 1H).
ПРИМЕР 3
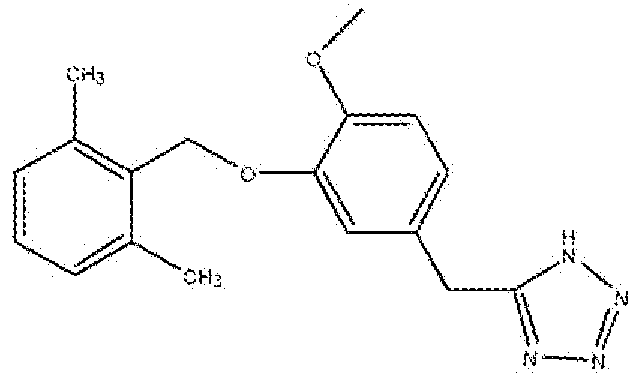
5-(3-(2,6-Диметилбензилокси)-4-метоксибензил)-1H-тетразол
Стадия A: Получение этил 3-гидрокси-4-метоксибензоата
Раствор 3-гидрокси-4-метоксибензойной кислоты (25 г, 148,67 ммоль) и моногидрата п-толуолсульфоновой кислоты (3,17 г, 16,66 ммоль) в абсолютном этаноле (300 мл) кипятили с обратным холодильником в течение 6 часов или до исчезновения всех исходных соединений. Реакционную смесь концентрировали, разбавляли EtOAc (60 мл) и промывали водой (20 мл). Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия B: Получение этил 3-(2,6-диметилбензилокси)-4-метоксибензоата
Раствор этил 3-гидрокси-4-метоксибензоата (Стадия A, 9,10 г, 46,4 ммоль) и диизопропилазодикарбоксилата (DIAD, 10,23 г, 50 ммоль) в сухом THF (20 мл) добавляли по каплям к раствору 2,6-диметилбензилового спирта (6,94 г, 51 ммоль) и трифенилфосфина (TPP, 13,27 г, 50 ммоль) в сухом THF (60 мл) при 0°C в атмосфере аргона. Реакционную смесь нагревали до комнатной температуры в течение 4 часов или до исчезновения всех исходных соединений, разбавляли эфиром и промывали водой (2×). Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия C: Получение (3-(2,6-диметилбензилокси)-4-метоксифенил)метанола
К раствору этил 3-(2,6-диметилбензилокси)-4-метоксибензоата (Стадия B, 6,04 г, 19,23 ммоль) в сухом THF (30 мл) добавляли по каплям LiAlH4 (1M в THF, 0,803 г, 21,16 ммоль) при 0°C в атмосфере аргона. Реакционную смесь перемешивали в течение 4 часов или до исчезновения всех исходных соединений, затем медленно гасили 0,1N HCl. К реакционной смеси добавляли EtOAc (20 мл). Реакционную смесь фильтровали и осадок промывали EtOAc (25 мл × 2). Объединенный органический слой промывали 0,1N HCl, солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1) с получением указанного в заголовке соединения.
Стадия D: Получение 2-((5-(бромметил)-2-метоксифенокси)метил)-1,3-диметилбензола
К раствору (3-(2,6-диметилбензилокси)-4-метоксифенил)метанола (Стадия C, 5,23 г, 20,4 ммоль) и CBr4 (10,16 г, 30,6 ммоль) в сухом CH2Cl2 (20 мл) добавляли порциями трифенилфосфин (8,03 г, 30,64 ммоль) при 0°C. Реакционную смесь перемешивали в течение 1,5 часов, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
1H ЯМР (400 МГц, (CDCl3): 2,43 (с, 6H); 3,83 (с, 3H); 4,53 (с, 2H); 5,08 (с, 2H); 6,84 (д, 1H); 7,0-7,03 (дд, 1H); 7,06-7,09 (м, 3H); 7,14-7,18 (м, 1H).
Стадия E: Получение 2-(3-(2,6-диметилбензилокси)-4-метоксифенил)ацетонитрила
Раствор 2-((5-(бромметил)-2-метоксифенокси)метил)-1,3-диметилбензола (Стадия D, 3,28 г, 9,7 ммоль) и NaCN (0,624 г, 12,7 ммоль) в сухом DMF (20 мл) нагревали при 120°C в течение 2,5 часов, затем охлаждали и разбавляли EtOAc (50 мл). Органический слой промывали водой (30 мл), солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия F: Получение 5-(3-(2,6-диметилбензилокси)-4-метоксибензил)-1H-тетразола
Смесь 2-(3-(2,6-диметилбензилокси)-4-метоксифенил)ацетонитрила (Стадия E, 2,17 г, 7,5 ммоль), азида натрия (0,590 г, 9,1 ммоль) и хлорида аммония (0,486 г, 9,1 ммоль) в сухом DMF (20 мл) нагревали в атмосфере аргона при 90°C в течение 16 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали, разбавляли водой и экстрагировали EtOAc (30 мл × 4). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5) с получением полутвердого продукта. Полутвердое вещество перемешивали со смесью 1:2 этилацетат:гексан (15 мл) в течение 10 минут и фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,3 (с, 6H); 3,68 (с, 3H); 4,22 (с, 2H); 4,98 (с, 2H); 6,78-6,81 (дд, 1H); 6,91-6,93 (д, 1H); 7,05-7,07 (д, 2H); 7,13-7,16 (м, 2H). МС: m/z 325,2 [M + H]+.
ПРИМЕР 4
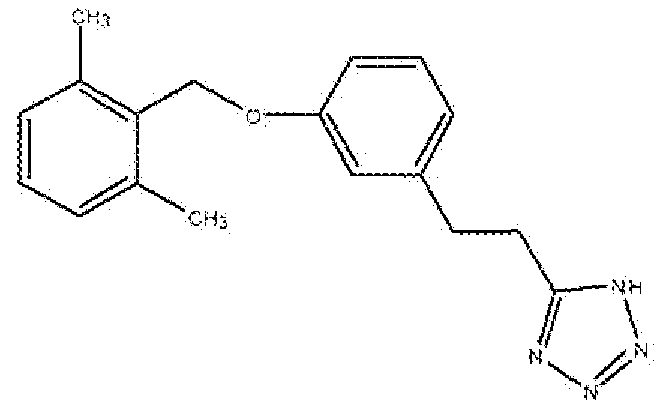
5-(3-(2,6-Диметилбензилокси)фенэтил])-1H-тетразол
Стадия A: Получение 3-(3-метоксифенил)пропаннитрила
Раствор 1-(2-бромэтил)-3-метоксибензола (10 г, 46,4 ммоль), NaCN (2,73 г, 55,8 ммоль) в сухом DMF (20 мл) нагревали при 90°C в течение 6 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали, разбавляли EtOAc (60 мл) и промывали водой (20 мл × 3), солевым раствором, органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения в виде масла.
Стадия B: Получение 3-(3-гидроксифенил)пропаннитрил
К перемешиваемому раствору 3-(3-метоксифенил)пропаннитрила (Стадия A, 1,71 г, 10,6 ммоль) в сухом CH2Cl2 (20 мл) добавляли BBr3 (1M в CH2Cl2, 5,32 г, 21,2 ммоль) при -78°C в атмосфере аргона. Реакционную смесь перемешивали при той же температуре в течение 2 часов и затем при 0°C в течение 4 часов или до исчезновения всех исходных соединений, гасили льдом, экстрагировали EtOAc (30 мл × 3). Объединенный органический слой тщательно промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия C: Получение 3-(3-(2,6-диметилбензилокси)фенил)пропаннитрила
Раствор 3-(3-гидроксифенил)пропаннитрила (Стадия B, 1,25 г, 8,5 ммоль) и диизопропилазодикарбоксилата (DIAD, 1,87 г, 9,26 ммоль) в сухом THF (10 мл) добавляли по каплям к раствору 2,6-диметилбензилового спирта (1,27 г, 9,3 ммоль) и трифенилфосфина (TPP, 2,43 г, 9,26 ммоль) в сухом THF (30 мл) при 0°C в атмосфере аргона. Реакционную смесь нагревали до комнатной температуры в течение 4 часов или до исчезновения всех исходных соединений, разбавляли простым эфиром и промывали водой (2×). Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия D: Получение 5-(3-(2,6-диметилбензилокси)фенэтил)-1H-тетразола
Смесь 3-(3-(2,6-диметилбензилокси)фенил)пропаннитрила (Стадия C, 2,62 г, 9,9 ммоль), азида натрия (0,899 г, 13,8 ммоль) и хлорида аммония (0,740 г, 13,8 ммоль) в сухом DMF (20 мл) нагревали в атмосфере аргона при 90°C в течение 16 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали, разбавляли водой и экстрагировали EtOAc (30 мл × 4). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5→92,5:7,5) с получением полутвердого продукта. Полутвердое вещество перемешивали со смесью 1:2 этилацетат:гексан (15 мл) в течение 10 минут и фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,48 (с, 6H); 3,02 (т, 2H); 3,19 (т, 2H); 4,98 (с, 2H); 6,80-6,81 (д, 1H); 6,86-6,89 (м, 2H); 7,05-7,07 (д,2H); 7,14-7,23 (м, 2H). МС: m/z 309,2 [M+H]+.
ПРИМЕР 5
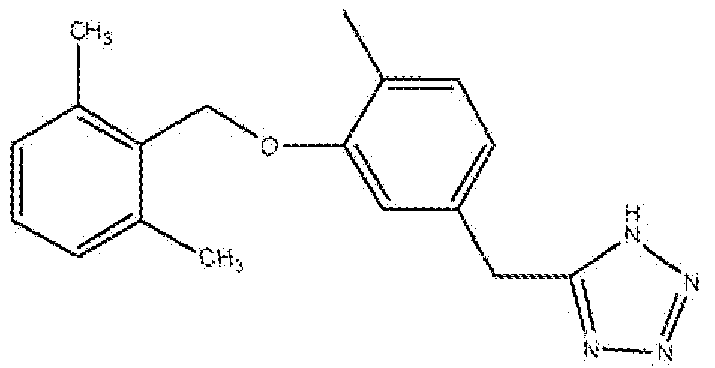
5-(3-(2,6-Диметилбензилокси)-4-метилбензил)-1H-тетразол
Стадия A: Получение 2-(3-гидрокси-4-метилфенил)ацетонитрила
К перемешиваемому раствору 2-(3-метокси-4-метилфенил)ацетонитрила (5 г, 31 ммоль) в сухом CH2Cl2 (20 мл) добавляли по каплям BBr3 (1M в CH2Cl2, 10,02 г, 40 ммоль) при -78°C в атмосфере аргона. Реакционную смесь перемешивали при той же температуре в течение 2 часов и затем при 0°C в течение 5 часов или до исчезновения всех исходных соединений, гасили льдом, экстрагировали EtOAc (30 мл × 3), объединенный органический слой тщательно промывали насыщенным NaHCO3, солевым раствором и сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1→CH2Cl2:гексан 1:1) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
Стадия B: Получение 2-(3-(2,6-диметилбензилокси)-4-метилфенил)ацетонитрила
К перемешиваемому раствору 2-(3-гидрокси-4-метилфенил)ацетонитрила (Стадия A, 2,18 г, 14,8 ммоль), K2CO3 (2,66 г, 19,2 ммоль) в сухом DMF (20 мл) добавляли 2,6-диметилбензилхлорид (2,97 г, 19,2 ммоль) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивали в течение 16 часов, разбавляли EtOAc (40 мл), промывали водой (20 мл) и солевым раствором. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия C: Получение 5-(3-(2,6-диметилбензилокси)-4-метилбензил)-1H-тетразола
Смесь 2-(3-(2,6-диметилбензилокси)-4-метилфенил)ацетонитрила (Стадия B, 1,12 г, 4,2 ммоль), азида натрия (0,400 г, 6,1 ммоль) и хлорида аммония (0,350 г, 6,5 ммоль) в сухом DMF (15 мл) нагревали в атмосфере аргона при 90°C в течение 16 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали, разбавляли водой и экстрагировали EtOAc (30 мл × 4). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (хлороформ:метанол 95:5→92,5:7,5) с получением полутвердого продукта. Полутвердое вещество перемешивали со смесью 1:2 этилацетат:гексан (15 мл) в течение 10 минут и фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,0 (с, 3H); 2,35 (с, 6H); 4,27 (с, 2H); 5,0 (с, 2H); 6,73-6,75 (дд, 1H); 7,08-7,1 (м, 3H); 7,15-7,19 (м, 2H). МС: m/z 309,2 [M+H]+.
ПРИМЕР 6
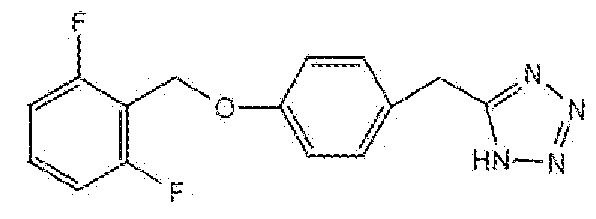
5-(4-(2,6-дифторбензилокси)бензил)-1H-тетразол
Стадия A: Получение 2-(4-(2,6-дифторбензилокси)фенил)ацетонитрила
К раствору 2-(4-гидроксифенил)ацетонитрила (5 г, 37,5 ммоль) и K2CO3 (6,74 г, 48,8 ммоль) в сухом DMF (20 мл) добавляли 2,6-дифторбензилбромид (7,77 г, 37,5 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре и концентрировали в вакууме. Неочищенный остаток растворяли в EtOAc и промывали водой и солевым раствором. Водный слой промывали еще один раз EtOAc. Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (270 МГц, CDCl3): 3,65 (с, 2H); 5,1 (с, 2H); 6,9-7,0 (м, 4H); 7,2-7,4 (м, 3H).
Стадия B: Получение 5-(4-(2,6-дифторбензилокси)бензил)-1H-тетразола
Смесь 2-(4-(2,6-дифторбензилокси)фенил)ацетонитрила (Стадия A, 5 г, 19,3 ммоль), азида натрия (1,3 г, 20 ммоль) и хлорида аммония (1,06 г, 20 ммоль) в сухом DMF (60 мл) нагревали при 90°C в течение 16 часов. Растворитель удаляли в вакууме и маслянистый остаток распределяли между EtOAc и водой (подкисляли до pH 1 конц. HCl). Органический слой промывали водой, сушили над Na2SO4, фильтровали и концентрировали до коричневого полутвердого вещества. Очистку осуществляли флеш-хроматографией на колонке с силикагелем (хлороформ:метанол, 9:1) с получением указанного в заголовке соединения в виде светло-кремового твердого вещества.
1H ЯМР (270 МГц, CDCl3): 4,0 (с, 2H); 5,1 (с, 2H); 6,7-6,9 (м, 4H); 7,0 (д, 2H); 7,2 (м, 1H).
ПРИМЕР 7
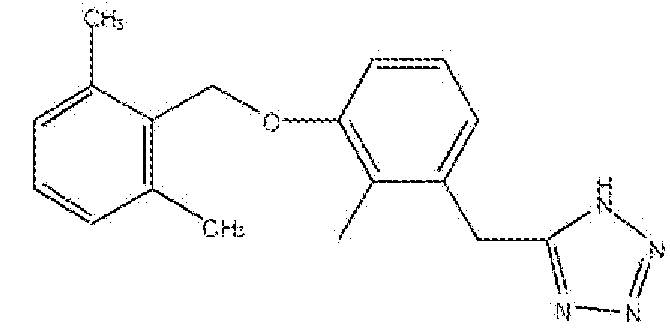
5-(3-(2,6-диметилбензилокси)-2-метилбензил)-1H-тетразол
Стадия A: Получение этил 3-гидрокси-2-метилбензоата
Раствор 3-гидрокси-2-метилбензойной кислоты (5,04 г, 33,12 ммоль) и моногидрата п-толуолсульфоновой кислоты (0,741 г, 3,89 ммоль) в абсолютном этаноле (150 мл) кипятили с обратным холодильником в течение 16 часов или до исчезновения всех исходных соединений. Реакционную смесь концентрировали, разбавляли EtOAc (30 мл) и промывали водой (20 мл). Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия B: Получение этил 3-(2,6-диметилбензилокси)-2-метилбензоата
К перемешиваемому раствору этил 3-гидрокси-2-метилбензоата (Стадия A, 3,1 г, 17,22 ммоль), K2CO3 (3,09 г, 22,38 ммоль) в сухом DMF добавляли 2,6-диметилбензилхлорид (3,19 г, 20,66 ммоль) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивали в течение 16 часов, разбавляли EtOAc (40 мл), промывали водой (20 мл) и солевым раствором. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия C: Получение (3-(2,6-диметилбензилокси)-2-метилфенил)метанола
К раствору этил 3-(2,6-диметилбензилокси)-2-метилбензоата (Стадия B, 5,94 г, 19,93 ммоль) в сухом THF (35 мл) добавляли по каплям LiAlH4 (1M в THF, 0,832 г, 21,92 ммоль) при 0°C в атмосфере аргона. Реакционную смесь перемешивали в течение 4 часов или до исчезновения всех исходных соединений, затем медленно гасили 0,1N HCl при 0°C, и добавляли к реакционной смеси EtOAc (20 мл). Реакционную смесь фильтровали и осадок промывали EtOAc (25 мл × 2). Объединенный органический слой промывали 0,1N HCl, солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 2:1) с получением указанного в заголовке соединения.
Стадия D: Получение 1-(бромметил)-3-(2,6-диметилбензилокси)-2-метилбензола
К раствору (3-(2,6-диметилбензилокси)-2-метилфенил)метанола (Стадия C, 3,68 г, 14,37 ммоль) и CBr4 (5,25 г, 15,8 ммоль) в сухом CH2Cl2 (20 мл) добавляли порциями трифенилфосфин (4,14 г, 15,8 ммоль) при 0°C. Реакционную смесь перемешивали в течение 4 часов, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1) с получением указанного в заголовке соединения. Твердое вещество дополнительно выдерживали в вакууме в течение 6 часов для высушивания.
Стадия E: Получение 2-(3-(2,6-диметилбензилокси)-2-метилфенил)ацетонитрила
Раствор 1-(бромметил)-3-(2,6-диметилбензилокси)-2-метилбензола (Стадия D, 4,28 г, 13,41 ммоль) и NaCN (0,789 г, 16,10 ммоль) в сухом DMF (20 мл) нагревали при 120°C в течение 3 часов, затем охлаждали и разбавляли EtOAc (50 мл). Органический слой промывали водой (30 мл), соляным раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1) с получением указанного в заголовке соединения.
Стадия F: Получение 5-(3-(2,6-диметилбензилокси)-2-метилбензил)-1H-тетразола
Смесь 2-(3-(2,6-диметилбензилокси)-2-метилфенил)ацетонитрила (Стадия E, 2,70 г, 10,19 ммоль), азида натрия (0,795 г, 12,23 ммоль) и хлорида аммония (0,653 г, 12,22 ммоль) в сухом DMF (20 мл) нагревали в атмосфере аргона при 90°C в течение 16 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали, разбавляли водой и экстрагировали EtOAc (30 мл × 4). Объединенный органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали флеш-хроматографией на колонке с силикагелем (хлороформ:метанол 92,5:7,5) с получением полутвердого продукта. Полутвердое вещество перемешивали со смесью 1:2 этилацетат:гексан (15 мл) в течение 10 минут и фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,01 (с, 3H); 2,32 (с, 6H); 4,24 (с, 2H); 5,01 (с, 2H); 6,78-6,79 (дд, 1H); 7,06-7,08 (д, 2H); 7,12-7,19 (м, 3H).
ПРИМЕР 8
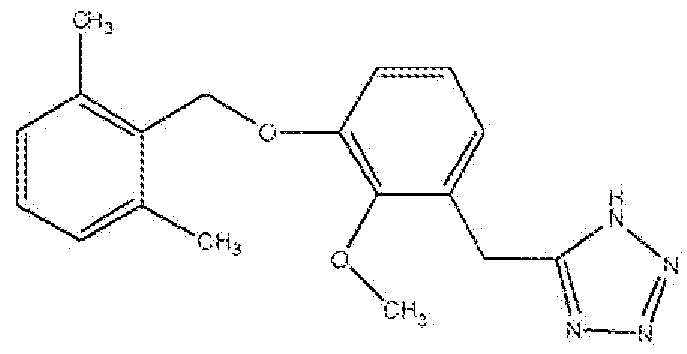
5-(3-(2,6-Диметилбензилокси)-2-метоксибензил)-1H-тетразол
Стадия A: Получение 3-(2,6-диметилбензилокси)-2-метоксибензальдегида
Раствор 3-гидрокси-2-метоксибензальдегида (5,06 г, 32,7 ммоль), 2,6-диметилбензилхлорида (5,04 г, 33,1 ммоль) и K2CO3 (4,78 г, 34,6 ммоль) в сухом DMF (15 мл) перемешивали при комнатной температуре в течение 16 часов в атмосфере аргона, затем разбавляли EtOAc (40 мл) и промывали водой (20 мл). Органический слой концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:этилацетат 4:1) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
Стадия B: Получение (3-(2,6-диметилбензилокси)-2-метоксифенил)метанола
К раствору 3-(2,6-диметилбензилокси)-2-метоксибензальдегида (Стадия B, 10,6 г, 32,7 ммоль) в сухом THF (40 мл) добавляли по каплям LiAlH4 (1M в THF, 0,95 г, 23,7 ммоль) при 0°C в атмосфере аргона. Реакционную смесь перемешивали в течение 1 часа или до исчезновения всех исходных соединений, затем гасили медленным добавлением воды, с последующим добавлением 1N HCl (5 мл), воды (10 мл), и добавляли к реакционной смеси EtOAc (20 мл). Реакционную смесь концентрировали и пропускали через колонку с тонким слоем силикагеля, применяя этилацетат, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
Стадия C: Получение 3-(2,6-диметилбензилокси)-2-метоксибензилметансульфоната
К раствору (3-(2,6-диметилбензилокси)-2-метоксифенил)метанола (Стадия B, 9,5 г, 32,7 ммоль) и триэтиламина (5,80 г, 57,4 ммоль) в сухом CH2Cl2 (100 мл) добавляли по каплям метансульфонилхлорид (3,5 мл, 45 ммоль) при 0°C в атмосфере аргона. Реакционную смесь нагревали до комнатной температуры в течение 6 часов или до исчезновения всех исходных соединений, нейтрализовали охлажденным 10% Na2SO4 и экстрагировали CH2Cl2. Органический слой концентрировали и очищали флеш-хроматографией на колонке с силикагелем (гексан:метиленхлорид 2:1) с получением указанного в заголовке соединения в виде светло-желтого масла.
Стадия D: Получение 2-(3-(2,6-диметилбензилокси)-2-метоксифенил)ацетонитрила
Раствор 3-(2,6-диметилбензилокси)-2-метоксибензилметансульфоната (Стадия C, 8,8 г, 30,26 ммоль) и NaCN (1,60 г, 32,6 ммоль) в сухом DMF (40 мл) нагревали при 85°C в течение 18 часов, затем охлаждали и разбавляли EtOAc (50 мл). Органический слой промывали водой (30 мл), концентрировали и пропускали через колонку с тонким слоем силикагеля, применяя метиленхлорид, с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия E: Получение 5-(3-(2,6-диметилбензилокси)-2-метоксибензил)-1H-тетразола
Смесь 2-(3-(2,6-диметилбензилокси)-2-метоксифенил)ацетонитрила (Стадия D, 3,2 г, 12,7 ммоль), азида натрия (0,86 г, 13,2 ммоль) и хлорида аммония (0,696 г, 13,0 ммоль) в сухом DMF (10 мл) нагревали в атмосфере аргона при 90°C в течение 16 часов или до исчезновения всех исходных соединений, реакционную смесь охлаждали и концентрировали при пониженном давлении и очищали флеш-хроматографией на колонке с силикагелем (хлороформ:метанол 9:1) с получением полутвердого продукта. Полутвердое вещество перемешивали со смесью 1:2 этилацетат:гексан (15 мл) в течение 10 минут и фильтровали с получением продукта в виде белого твердого вещества.
1H ЯМР (400 МГц, (CD3)2SO): 2,33 (с, 6H); 3,5 (с, 3H); 4,21 (с, 2H); 5,04 (с, 2H); 6,83-6,85 (дд, 1H); 7,05-7,09 (м, 3H); 7,15-7,23 (м, 2H).
ПРИМЕР 9: Анализ на ингибирование URAT1
URAT1 (Переносчик мочевой кислоты 1) экспрессируется на апикальной мембране почечных канальцев. Он опосредует обратный захват мочевой кислоты из мочи в кровь. Ингибирование URAT1 приводит к повышенному выведению мочевой кислоты с мочой и, следовательно, представляет собой потенциальный способ действия для лекарственных средств, которые снижают концентрацию мочевой кислоты в сыворотке крови. Пробенецид и бензбромарон, например, применяют в клинической практике для лечения подагры и гиперурикемии, и они оба действуют на URAT1, ослабляя обратный захват мочевой кислоты. Однако бензбромарон был изъят с рынка из-за токсичности для печени посредством независимых от URAT1 механизмов, и пробенецид действует на большое количество транспортных белков, приводя в результате к взаимодействию с рядом других лекарственных средств.
Анализ на ингибирование URAT1 in vitro является пригодным способом для обнаружения соединений с потенциальной активностью для снижения концентрации мочевой кислоты в сыворотки крови. Подходящий анализ включает трансфекцию клеток (например, клеток мезонефроса человека: "HEK") вектором, кодирующим человеческий URAT1, с последующим определением способности трансфицированных клеток поглощать меченную радиоактивностью мочевую кислоту. Активность соединений в качестве URAT1 ингибиторов оценивают, исходя из их способности блокировать поглощение мочевой кислоты трансфицированными клетками.
Испытуемые соединения и химические реагенты:
Бензбромарон (Sigma. Cat.No.B5774), Пробенецид (Sigma. Cat.No.P8761)), DMSO (Sigma. Cat.No.D-2650). [8-14C] урат (50-60 мКи/ммоль: American Radio Chemicals. Cat. No. ARC0513).
Субклонирование hURAT1 в вектор экспрессии:
Плазмидный вектор pCMV6-XL5, содержащий hURAT1 кДНК (Cat. No. SC 125624), и вектор экспрессии pCMV6-Neo (Cat. No.pCMVNEO) получали у OriGene Technologies, Inc. hURAT1 кДНК полной длины получали из вектора pCMV6-XL5 и субклонировали в вектор экспрессии pCMV6-Neo для получения hURAT1 экспрессионной плазмиды pCMV6-hURAT1. Последовательности проверяли автоматическим секвенатором ДНК.
Клеточная культура, трансфекция URAT1-экспрессирующих плазмид и обнаружение стабильно экспрессирующих HEK клеток hURAT1:
Клетки мезонефроса человека 293 (HEK) (ATTCC, Cat No. CRL-1573) выращивали в EMEM, снабженной 10% FBS и 2 мМ L-глутамином, и выдерживали при 37°C и 5% CO2. Для экспериментов по трансфекции клетки помещали на 60 мм чашки в 1 мл среды на чашку. После выдерживания в течение 18-24 часов клетки трансфицировали плазмидой pCMV6-hURAT1 или вектором экспрессии pCMV6-Neo, применяя в качестве агента трансфекции липофектин, следуя инструкциям изготовителя (Invitrogen, Cat. No. 18292). После выращивания клеток для трансфекции в среде EMEM в течение 72 часов и затем добавления 1 мг/мл генетицина (GIBCO. Cat. No 10131) отбирали стабильные трансфектанты. Стабильные трансфектанты, экспрессирующие hURAT1 (в настоящем описании ниже названы как hURAT1-HEK клетки), или клетки, содержащие только вектор экспрессии pCMV6-Neo (в настоящем описании ниже названы как mock-HEK клетки), проверяли, применяя способы полимеразной цепной реакции с обратной транскриптазой (RT-PCR).
Анализ на поглощение [8-14C]урата:
hURAT1 HEK клетки и mock-HEK клетки помещали в 24-луночные планшеты для клеточных культур с покрытием из поли-D-лизина (Becton Dickinson, Cat. No. 354414) при концентрации 3×10-1 в среду EMEM и выдерживали в течение ночи. Реакционные растворы, содержащие [8-14C]урат (55 мКи/ммоль), при конечной концентрации 50 мкм получали в присутствии или отсутствие испытуемых соединений в сбалансированном солевом растворе Хенкса (HBSS), содержащим 125 мМ глюконат натрия, 4,8 мМ глюконат калия, 1,3 мМ кальций, 5,6 мМ глюкозы, 1,2 мМ сульфат магния, 1,2 мМ KH2PO4 и 25 мМ HEPES (pH 7,4). Перед началом анализа по поглощению культуральную среду удаляли и клетки выдерживали в течение 5 минут в 0,6 мл HBSS. После удаления HBSS приготовленные реакционные растворы добавляли в каждую лунку и выдерживали в течение 5 минут при комнатной температуре. Затем реакционный раствор удаляли, клетки промывали дважды 0,6 мл охлажденным HBSS и лизировали 0,2 мл 0,1 M NaOH в течение 20 минут. Клеточные лизаты переносили в сцинтилляционные флаконы, содержащие 1 мл сцинтилляционной жидкости (Opti Phase SuperMIX. PerkinElmer. Cat No. 1200-439), и радиоактивность считали в Microbeta счетчике (1450, Wallac Jet, PerkinElmer). Испытуемые соединения растворяли в DMSO, и те же концентрации DMSO добавляли в лунки с mock-HEK клетками и hURAT1-HEK клетками, которые не содержали испытуемые соединения. Для каждого испытуемого соединения анализ на поглощение проводили 2 раза и осуществляли в трех экземплярах. Поглощение урата клетками для каждого испытуемого состояния представляли в виде среднего процента ингибирования по сравнению с DMSO контролем. Величины радиоактивности, полученные для лунок, которые содержали DMSO, принимали за 100% поглощение клетками. Данные по наблюдаемой концентрации - проценту ингибирования приводили к сигмоидальной модели концентрация-эффект, в которой:
% ингибирование = (100*концентрация^угловой коэффициент)/(IC50^угловой коэффициент + концентрация^угловой коэффициент)
Значения IC50 и углового коэффициента с границами доверительного интервала 95% определяли нелинейным, регрессионным способом наименьших квадратов, применяя Data Analysis ToolboxTM (MDL Information Systems, San Leandro. CA, USA).
Для оценки активности соединений в качестве URAT1 ингибиторов процент ингибирования поглощения мочевой кислоты обычно оценивали при концентрации лекарственного средства, равной 10 мкмоль (Таблица 1). Дополнительные концентрации лекарственного средства некоторых соединений испытывали для определения величин IC50 (Таблица 2). В данном примере соединения не обязательно испытывали одновременно.
|
|
ПРИМЕР 10: Фармакокинетические исследования с применением пероральной однократной дозы соединения EB на мышах
Определение концентрации соединения EB в плазме после принудительного введения однократной дозы самцам мышей:
Испытуемые соединения суспендировали в 1% HPMC, применяя гомогенизатор тканей для получения частиц минимального размера и максимальной однородности суспензии, и хранили при 4°C. Состав тщательно перемешивали непосредственно перед введением. Дозу 100 мг/кг соединения EB или среду (1% HPMC) вводили самцам мышей однократным пероральным принудительным введением. Мышей помещали в мочесборник после дозирования на 5 часов, и собирали суммарное количество мочи в течение 5 часов. Образцы замораживали при -80°C перед анализом, применяя LC/MS-MS.
Через 0, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения дозы собирали образцы крови (0,4 мл) кровотечением из орбитального синуса в K3EDTA пробирки. Образцы крови центрифугировали в течение 30 минут сбора при охлаждении (2-8°C) в течение 7 минут при 6000 об/мин. После центрифугирования плазму собирали в единичные пробирки для каждого животного в каждый момент времени и сразу замораживали при -80°C до анализа, применяя LC/MS-MS.
Данные подвергали фармакокинетическому анализу, применяя WinNonlin Standard (v2.1, Pharsight Corporation) и Microsoft EXCEL.
Протокол:
A. Плазма
1. Мышам принудительно вводили однократную дозу соединения EB, 100 мг/кг, и плазму собирали в определенные моменты времени.
2. Плазму хранили при -80°C до дня анализа.
3. Образцы размораживали в бане при 37°C в течение 5 минут и встряхивали с максимальной скоростью в течение 10 секунд.
4. Плазму мышей, 0,1 мл, смешивали с 0,2 мл ацетонитрила, встряхивали 1 минуту, центрифугировали при 14000 об/мин, 17000 g, при 4°C в течение 25 минут.
5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52) и 13 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 100 бар, 37°C температура колонки, способ 406975M1, серия 1029-08A, Agilent 1100 LC-MS.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
B. Калибровочная кривая
Стадия 1. Плазму животных с "пустышкой" (смешанная), 0,095 мл, смешивали с 0,005 мл 20× стокового раствора соединения EB в метаноле для получения 500 мкМ, 250 мкМ, 125 мкМ,..., концентраций соединения EB в плазме.
Например: 95 мкл плазмы + 5 мкл 10 мМ соединения EB в метаноле = 0,1 мл плазмы с 500 мкМ соединения EB.
Стадия 2. Образцы со стадии 1 встряхивали в течение 10 секунд при максимальной скорости.
Стадия 3. Добавляли ко всем образцам со стадии 2 0,2 мл ацетонитрила и все пробирки встряхивали при максимальной скорости в течение 1 минут.
Стадия 4. Все образцы со стадии 3 центрифугировали при 14000 об/мин, 17000 g, при 4°C, в течение 25 минут.
Стадия 5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52), 13 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 100 бар, 37°C температура колонки, способ 406975M1, серия 1029-08A, Agilent 1100 LC-MS.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
ВЭЖХ условия:
|
Результаты:
Соединение EB легко обнаруживали в плазме мышей. Время удержания и массу подтверждали в положительном и отрицательном режимах ионизации. AGILENT LC-MS серия 1029-08А.
“M-” = 279,2 100%,
“M+” = 281,2 100%
Молекулярный вес 280, среднее время удержания = 26,5 мин.
|
|
|
Линейные компоненты на графике в полулогарифмическом масштабе:
t1/2 (1-4 ч): 2,27
t1/2 (6-8 ч): 0,74
t1/2 (8-24 ч): 9,39
ПРИМЕР 11: Фармакокинетические исследования при введении пробной однократной пероральной дозы соединения EB крысам
Определение концентрации в плазме соединения EB после принудительного введения однократной дозы самцам крыс.
Испытуемые соединения суспендировали в 1% HPMC, применяя гомогенизатор тканей для получения частиц минимального размера и максимальной однородности суспензии, и хранили при 4°C. Состав тщательно перемешивали непосредственно перед введением. Дозу 100 мг/кг испытуемых соединений или среду (1% HPMC) вводили самцам крыс линии Sprague-Dawley однократным пероральным принудительным введением. Через 0, 1, 2, 4, 6, 8 и 24 часа после введения дозы собирали образцы крови (0,4 мл) кровотечением из орбитального синуса в K3EDTA пробирки. Образцы крови центрифугировали в течение 30 минут сбора при охлаждении (2-8°C) в течение 7 минут при 6000 об/мин. После центрифугирования плазму собирали в единичные пробирки для каждого животного в каждый момент времени и сразу замораживали при -80°C до анализа, применяя LC/MS-MS. Данные зависимости концентрации в плазме от времени подвергали фармакокинетическому анализу, применяя WinNonlin Standard (v2.1, Pharsight Corporation) и Microsoft EXCEL.
Протокол:
A. Плазма
1. Крысам принудительно вводили однократную дозу соединения EB, 100 мг/кг и плазму собирали в определенные моменты времени.
2. Плазму хранили при -80°C до дня анализа.
3. Образцы размораживали на бане при 37°C в течение 5 минут и встряхивали с максимальной скоростью в течение 10 секунд.
4. Плазму крыс, 0,1 мл, смешивали с 0,2 мл ацетонитрила, встряхивали 1 минуту, центрифугировали при 14000 об/мин, 17000 g, при 4°C в течение 25 минут.
5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52) и 13 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 100 бар, температура колонки 37°C, способ 406975M1, AGILENT серия 1029-08A, Agilent 1100 LC-MS.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
B. Калибровочная кривая
Стадия 1. Плазму животных с "пустышкой" (смешанная), 0,19 мл, смешивали с 0,01 мл 20× стокового раствора PN2107 в метаноле для получения 500 мкМ, 250 мкМ, 125 мкМ, ..., концентраций соединения EB в плазме.
Например: 190 мкл плазмы + 10 мкл 10 мМ соединения EB в метаноле = 0,2 мл плазмы с 500 мкМ соединения EB.
Стадия 2. Образцы со стадии 1 встряхивали в течение 10 секунд при максимальной скорости.
Стадия 3. Добавляли ко всем образцам со стадии 2 0,4 мл ацетонитрила и все пробирки встряхивали при максимальной скорости в течение 1 минут.
Стадия 4. Все образцы со стадии 3 центрифугировали при 14000 об/мин, 17000 g, при 4°C, в течение 25 минут.
Стадия 5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52), 13 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 100 бар, температура колонки 37°C.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
|
Результаты:
1. Калибровочную кривую строили с R^2 соответствием линейности = 0,9994 (фиг.4).
2. Соединение EB легко обнаруживали в плазме крыс. Время удержания и массу подтверждали в положительном и отрицательном режимах ионизации.
“M-” = 279,2 100%,
“M+” = 281,2 100%
Молекулярный вес 280, среднее время удержания = 26,5 мин.
Необработанные данные и расчеты приведены в таблице 7.
Концентрации соединений в плазме приведены в таблице 8.
|
|
|
ПРИМЕР 12: Фармакокинетические исследования при введении пробной однократной пероральной дозы соединения EC крысам
Определение концентрации в плазме соединения EC после принудительного введения однократной дозы самцам крыс.
Испытуемое соединение суспендировали в 1% HPMC, применяя гомогенизатор тканей для получения частиц минимального размера и максимальной однородности суспензии, и хранили при 4°C. Состав тщательно перемешивали непосредственно перед введением. Дозу 100 мг/кг испытуемых соединений или среду (1% HPMC) вводили самцам крыс линии Sprague-Dawley однократным пероральным принудительным введением. Через 0, 1, 2, 4, 6, 8 и 24 часа после введения дозы собирали образцы крови (0,4 мл) кровотечением из орбитального синуса в K3EDTA пробирки. Образцы крови центрифугировали в течение 30 минут сбора при охлаждении (2-8°C) в течение 7 минут при 6000 об/мин. После центрифугирования плазму собирали в единичные пробирки для каждого животного в каждый момент времени и сразу замораживали при -80°C до анализа, применяя LC/MS-MS. Данные зависимости концентрации в плазме от времени подвергали фармакокинетическому анализу, применяя WinNonlin Standard (v2.1, Pharsight Corporation) и Microsoft EXCEL.
Протокол:
A. Плазма
1. Крысам принудительно вводили однократную дозу соединения EC, 100 мг/кг, и плазму собирали в определенные моменты времени.
2. Плазму хранили при -80°C до дня анализа.
3. Образцы размораживали на бане при 37°C в течение 5 минут и встряхивали с максимальной скоростью в течение 10 секунд.
4. Плазму крыс, 0,1 мл, смешивали с 0,2 мл ацетонитрила, встряхивали 1 минуту, центрифугировали при 14000 об/мин, 17000 g, при 4°C в течение 25 минут.
5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52) и 15 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 107 бар, температура колонки 37°C, способ 406975M1, AGILENT серия 0226-09A, Agilent 1100 LC-MS.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
B. Калибровочная кривая
Стадия 1. Плазму животных с "пустышкой" (смешанная), 0,19 мл, смешивали с 0,01 мл 20× стокового соединения EC в метаноле для получения 500 мкМ, 250 мкМ, 125 мкМ концентраций соединения EC в плазме.
Например: 190 мкл плазмы + 10 мкл 10 мМ соединения EC в метаноле = 0,2 мл плазмы с 500 мкМ соединения EC.
Стадия 2. Образцы со стадии 1 встряхивали в течение 10 секунд при максимальной скорости.
Стадия 3. Добавляли ко всем образцам со стадии 2 0,4 мл ацетонитрила и все пробирки встряхивали при максимальной скорости в течение 1 минут.
Стадия 4. Все образцы со стадии 3 центрифугировали при 14000 об/мин, 17000 g, при 4°C, в течение 25 минут.
Стадия 5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex# AF0-3102-52), 15 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 107 бар, температура колонки 37°C.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
|
Результаты:
1. Калибровочную кривую строили с R^2 соответствием линейности = 0,9984 (см. фиг.6)
2. Соединение EC легко обнаруживали в плазме. Время удержания и массу подтверждали в положительном и отрицательном режимах ионизации (см. фиг.7).
“M-” = 293,2 100%, 527,2 65%
“M+” = 295,2 100%
Молекулярный вес 294, среднее время удержания = 23 мин.
|
|
|
ПРИМЕР 13: Фармакокинетические исследования при введении пробной однократной пероральной дозы соединения EG крысам
Определение концентрации в плазме соединения EG после принудительного введения однократной дозы самцам крыс.
Испытуемое соединение суспендировали в 1% HPMC, применяя гомогенизатор тканей для получения частиц минимального размера и максимальной однородности суспензии, и хранили при 4°C. Состав тщательно перемешивали непосредственно перед введением. Дозу 100 мг/кг испытуемых соединений или среду (1% HPMC) вводили самцам крыс линии Sprague-Dawley однократным пероральным принудительным введением. Через 0, 1, 2, 4, 6, 8 и 24 часа после введения дозы собирали образцы крови (0,4 мл) кровотечением из орбитального синуса в K3EDTA пробирки. Образцы крови центрифугировали в течение 30 минут сбора при охлаждении (2-8°C) в течение 7 минут при 6000 об/мин. После центрифугирования плазму собирали в единичные пробирки для каждого животного в каждый момент времени и сразу замораживали при -80°C до анализа, применяя LC/MS-MS. Данные зависимости концентрации в плазме от времени подвергали фармакокинетическому анализу, применяя WinNonlin Standard (v2.1, Pharsight Corporation) и Microsoft EXCEL.
Протокол:
A. Плазма
1. Крысам принудительно вводили однократную дозу соединения EG, 100 мг/кг и плазму собирали в определенные моменты времени.
2. Плазму хранили при -80°C до дня анализа.
3. Образцы размораживали на бане при 37°C в течение 5 минут и встряхивали с максимальной скоростью в течение 10 секунд.
4. Плазму крыс, 0,1 мл, смешивали с 0,2 мл ацетонитрила, встряхивали 1 минуту, центрифугировали при 14000 об/мин, 17000 g, при 4°C в течение 25 минут.
5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex # AF0-3102-52) и 15 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 107 бар, температура колонки 37°C, способ 406975M1, AGILENT серия 0226-09A, Agilent 1100 LC-MS.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
B. Калибровочная кривая
Стадия 1. Плазму животных с "пустышкой" (смешанная), 0,19 мл, смешивали с 0,01 мл 20× стокового соединения EG в метаноле для получения 500 мкМ, 250 мкМ, 125 мкМ, …, концентраций соединения EG в плазме.
Например: 190 мкл плазмы + 10 мкл 10 мМ соединения EG в метаноле = 0,2 мл плазмы с 500 мкМ соединения EG.
Стадия 2. Образцы со стадии 1 встряхивали в течение 10 секунд при максимальной скорости.
Стадия 3. Добавляли ко всем образцам со стадии 2 0,4 мл ацетонитрила и все пробирки встряхивали при максимальной скорости в течение 1 минут.
Стадия 4. Все образцы со стадии 3 центрифугировали при 14000 об/мин, 17000 g, при 4°C, в течение 25 минут.
Стадия 5. Надосадочные жидкости фильтровали через 0,45-микронный, 4 мм, PTFE мембранный шприцевой фильтр (Phenomenex# AF0-3102-52), 15 мкл впрыскивали и анализировали на Luna 3-микронной, 100A поры, C8(2), 150×3 мм, колонке с обращенной фазой (Phenomenex# 00F-4248-YO, SN#259151-7) в 50-минутном линейном градиенте от 40% до 69% (0,1% муравьиная кислота, 89,9% ацетонитрил, 10% метанол) при 0,25 мл/мин, 107 бар, температура колонки 37°C.
Все образцы прогоняли в двух экземплярах, 210 нм и 230 нм поглощение. Спектры регистрировали в режимах положительной и отрицательной ионизации.
|
Результаты:
1. Калибровочную кривую строили с R^2 соответствием линейности = 0,9997 (см. фиг.8).
2. Соединение EG легко обнаруживали в плазме крыс. Время удержания и массу подтверждали в положительном и отрицательном режимах ионизации (см. фиг.9).
“M-” = 307,2 100%
“M+” = 309,2 100%
Молекулярный вес 308, среднее время удержания = 30 мин.
Необработанные данные и расчеты
|
|
|
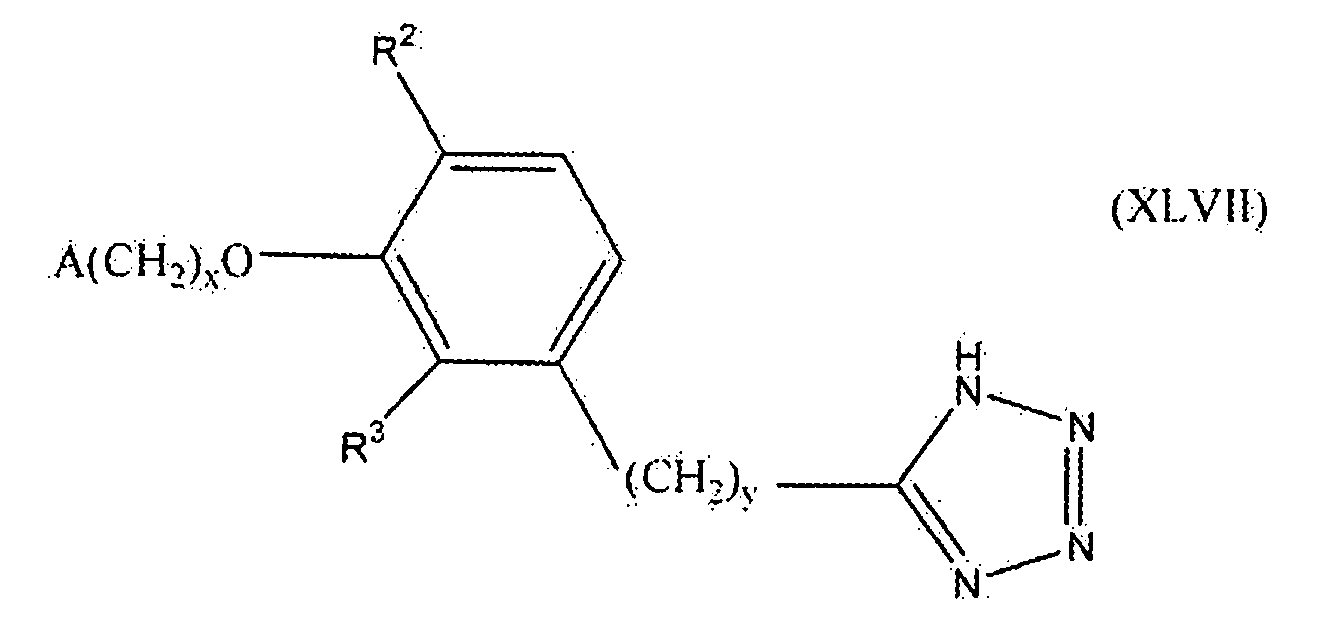
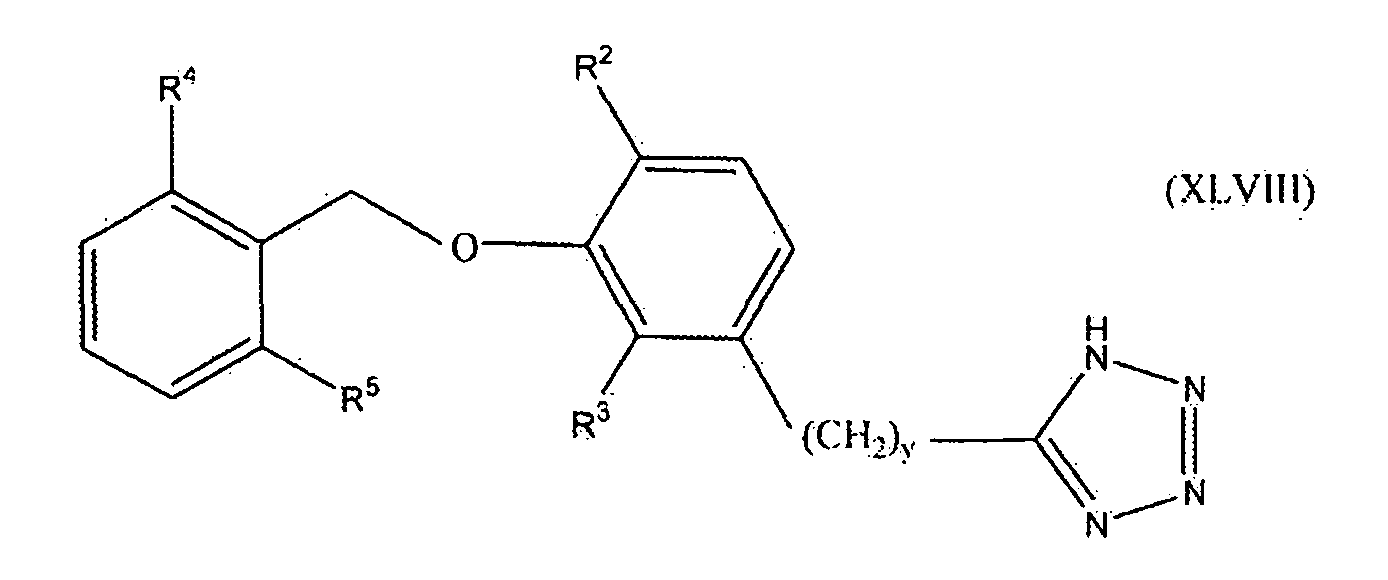
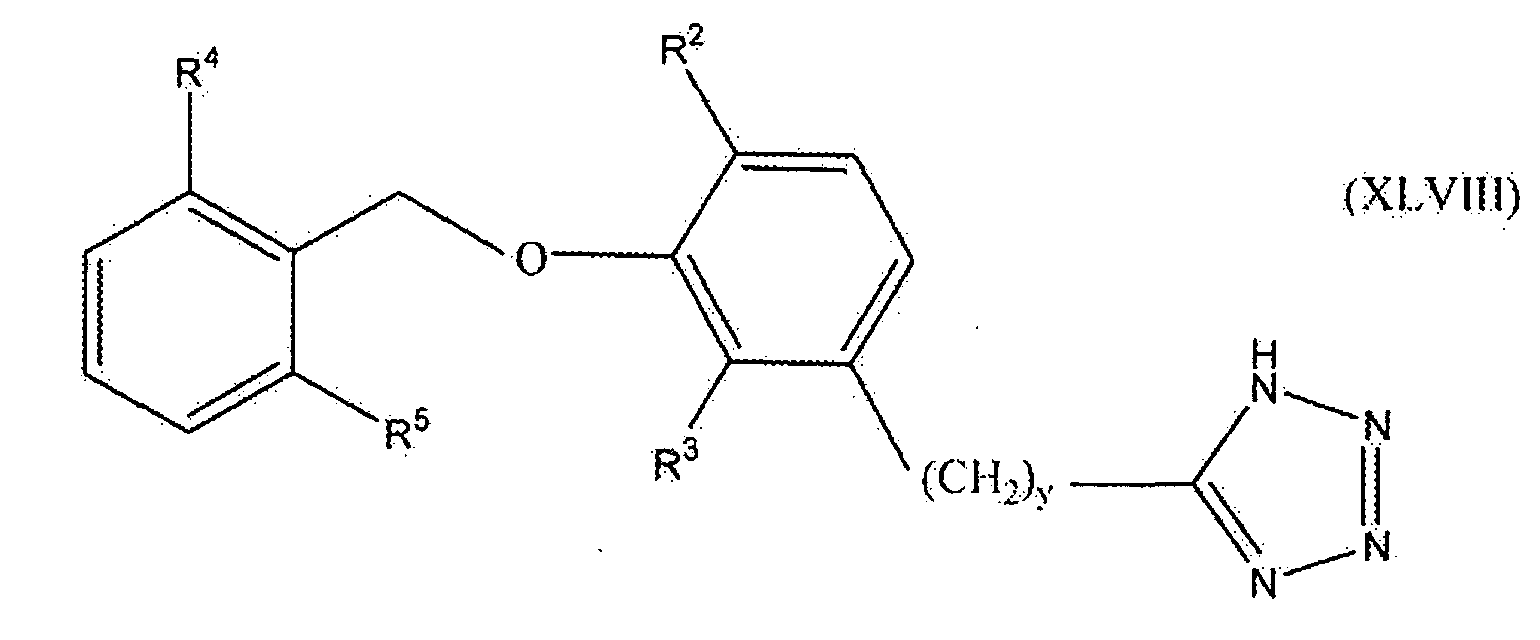
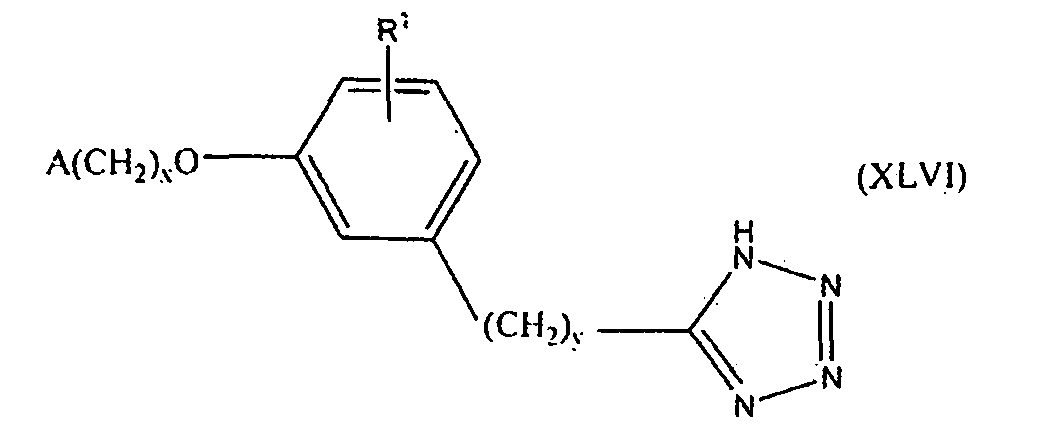
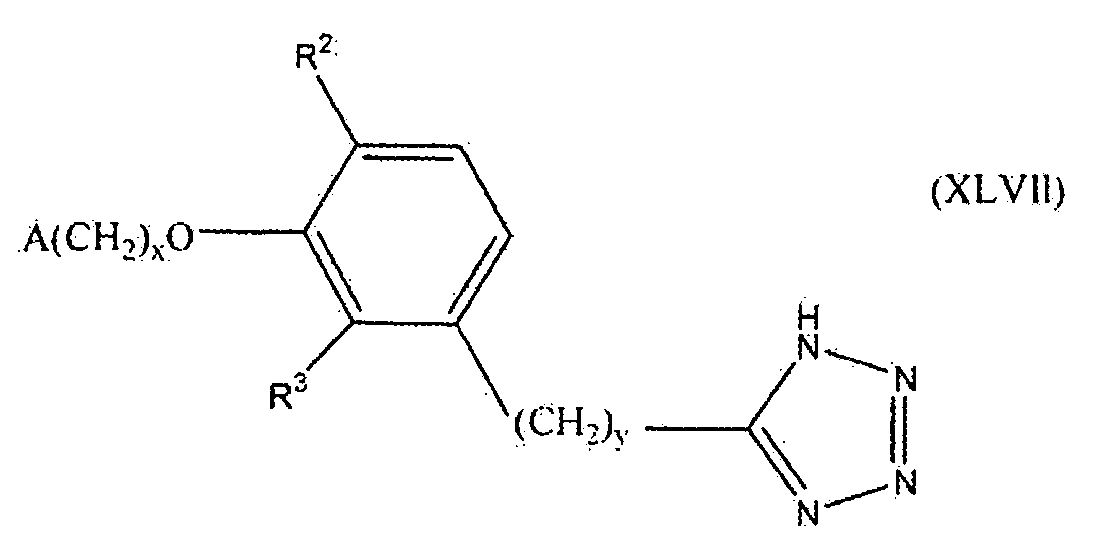
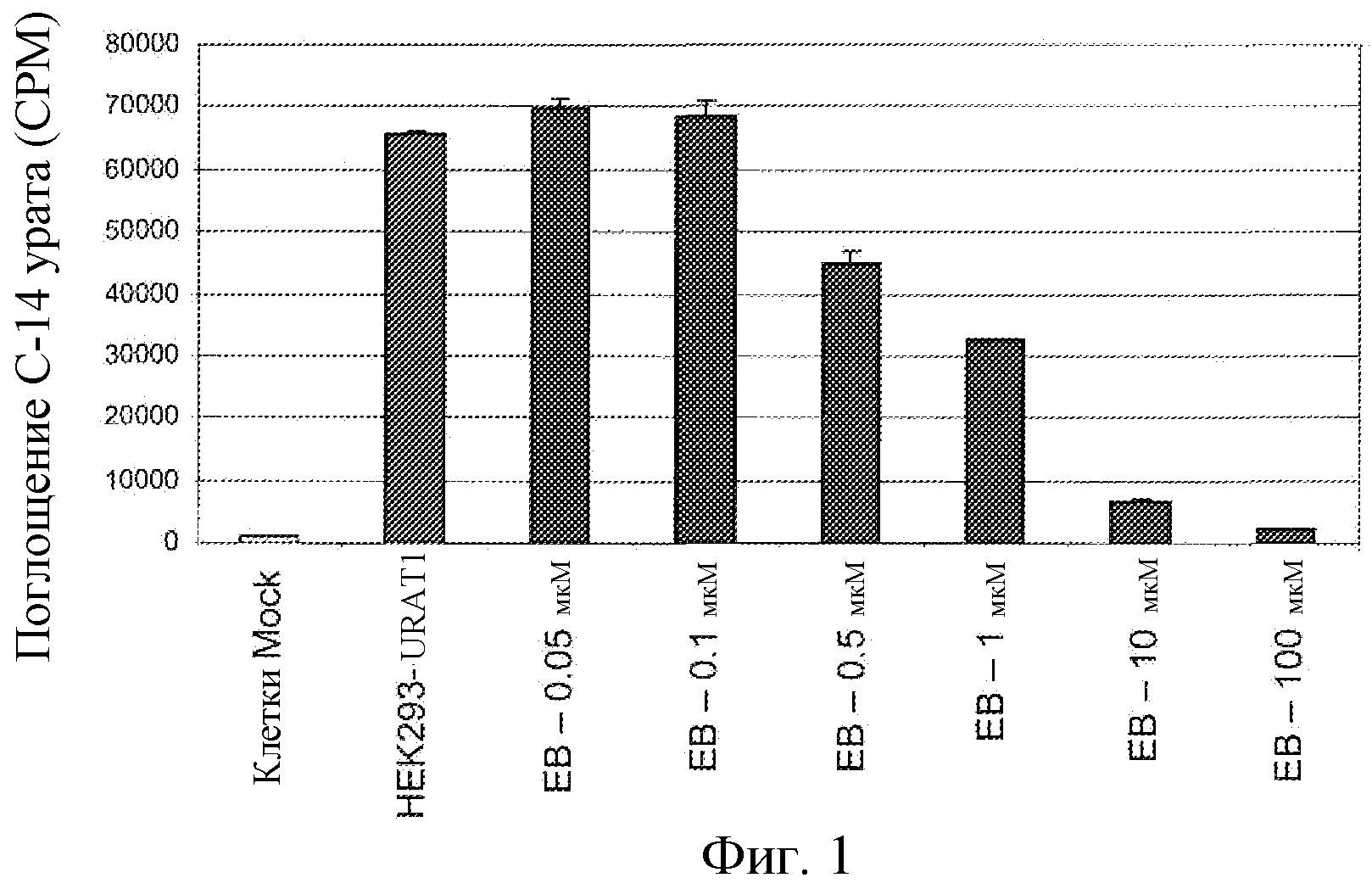
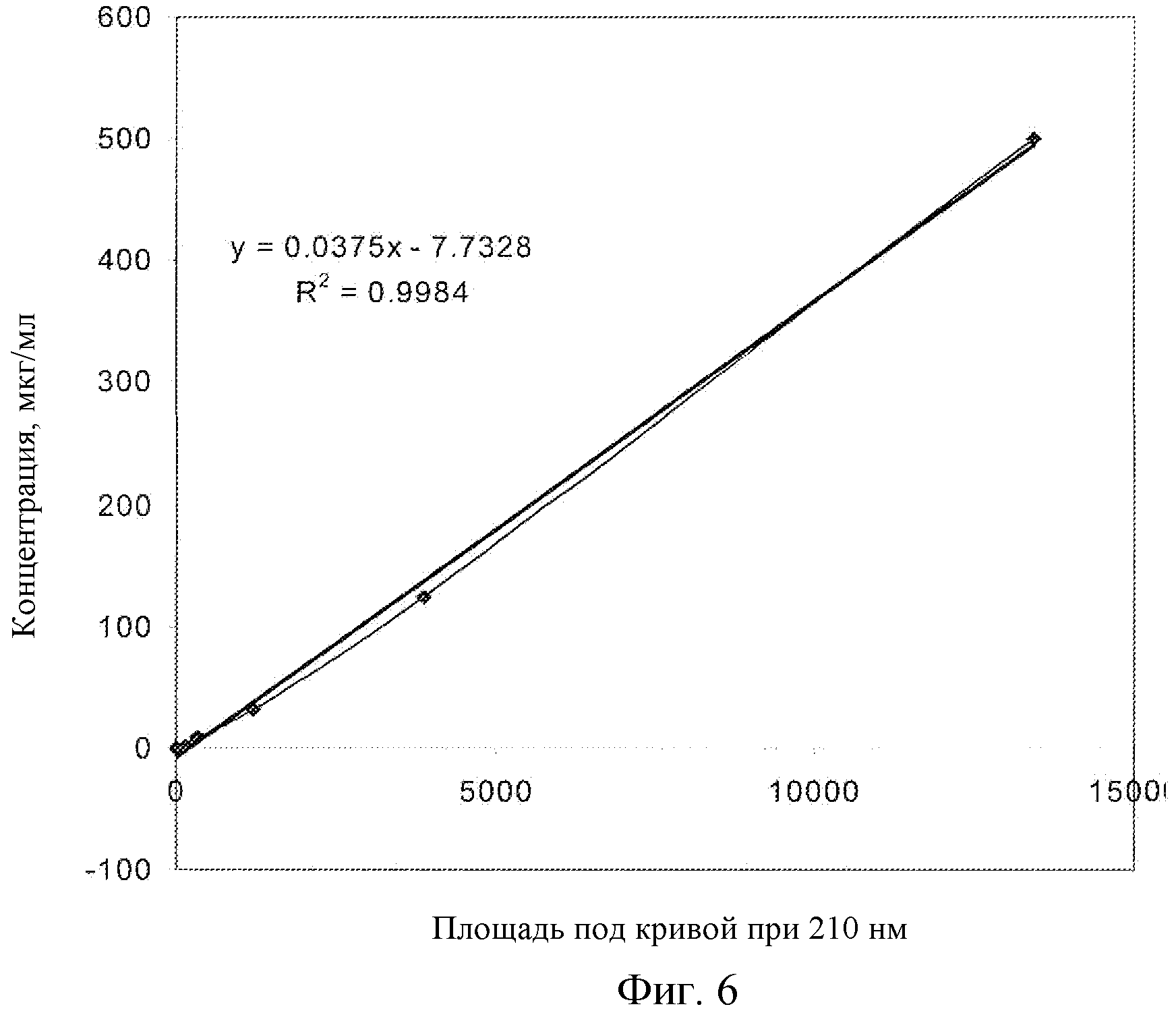
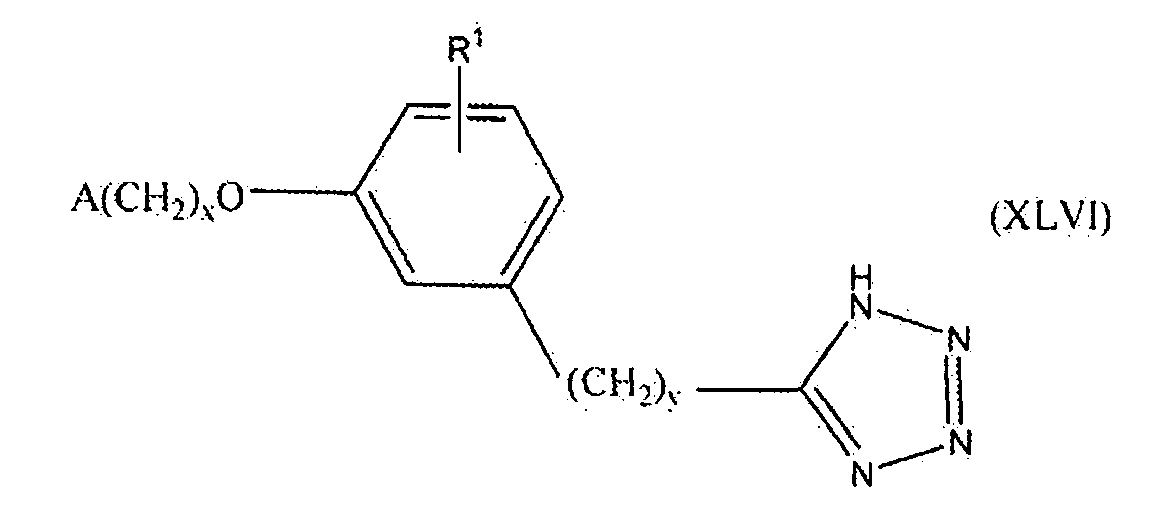
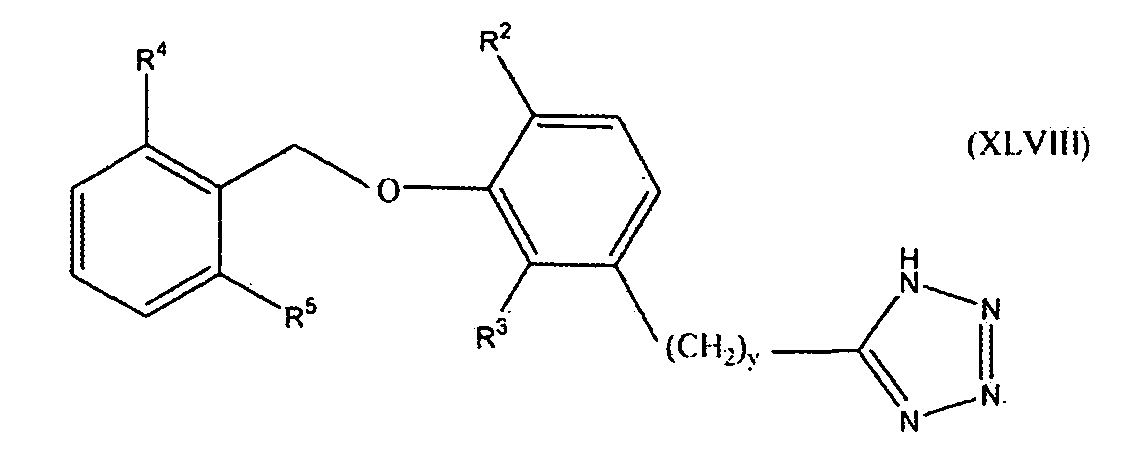
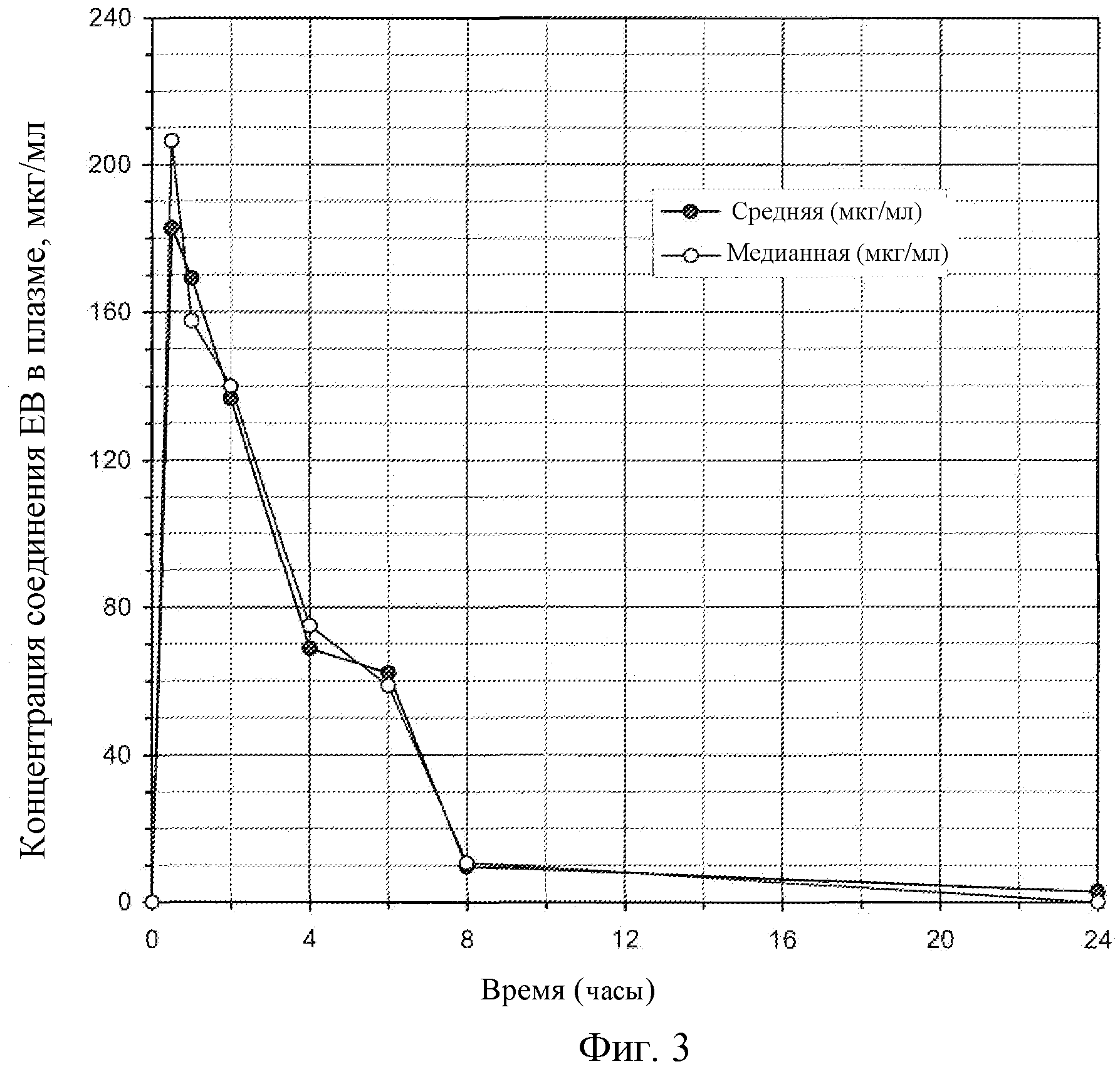
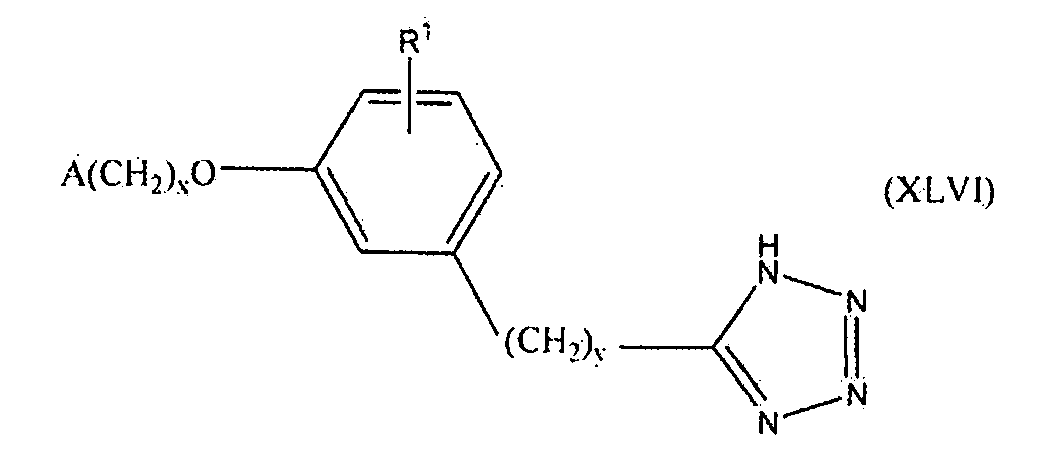
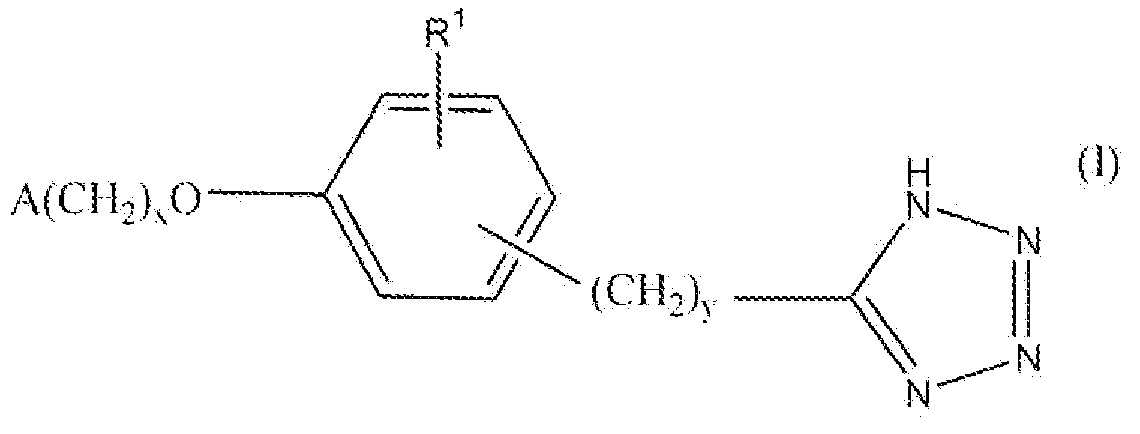
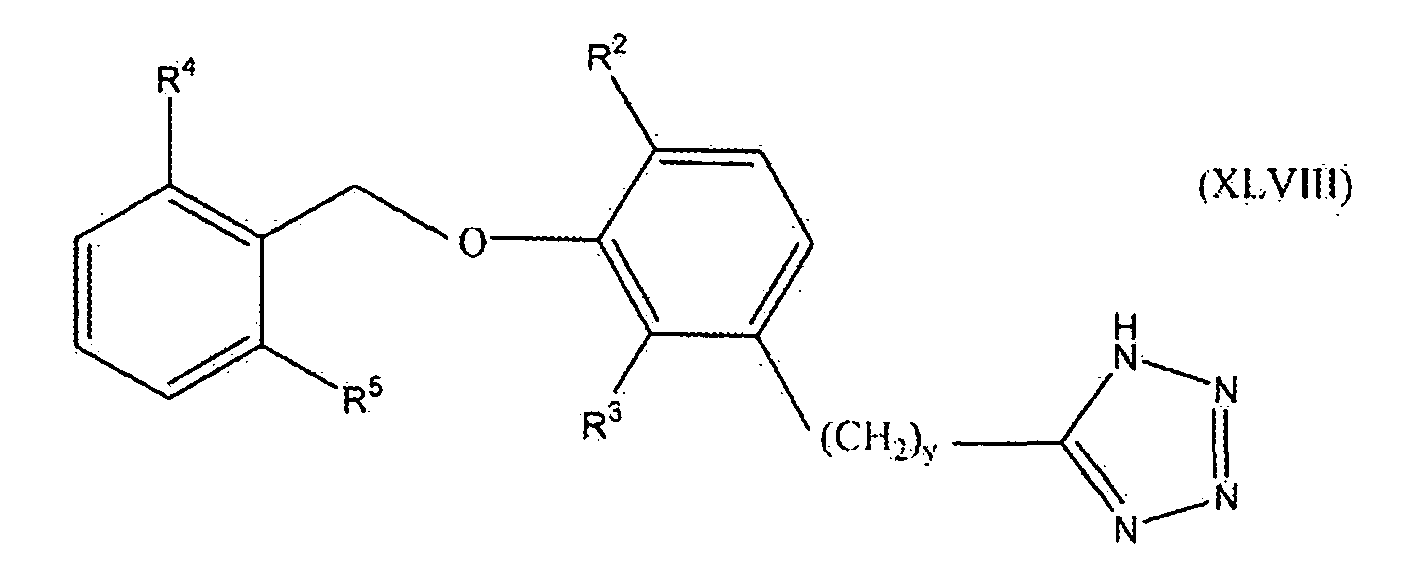
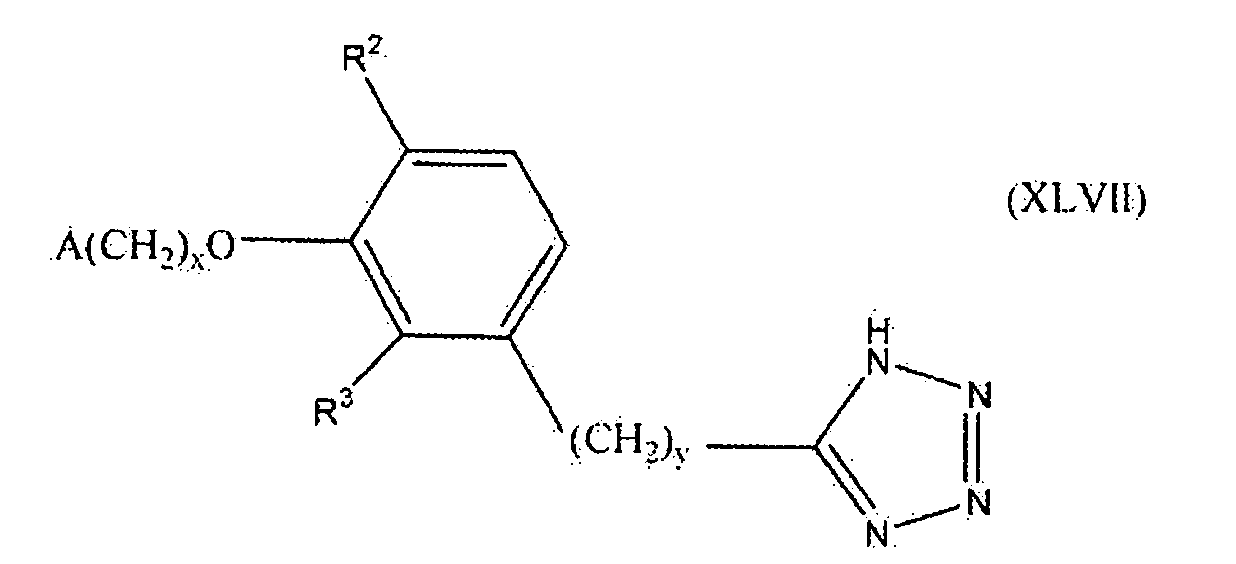
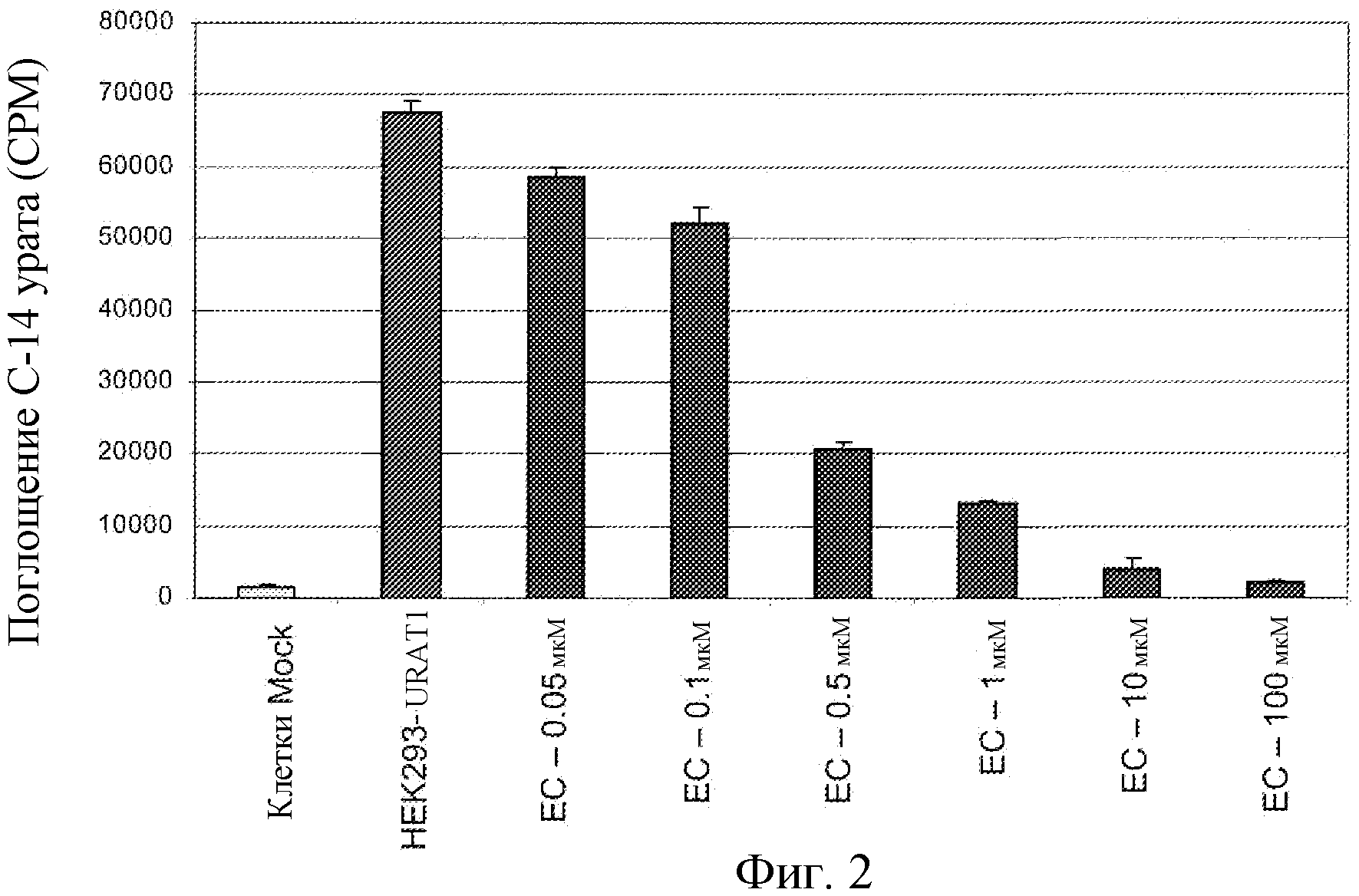
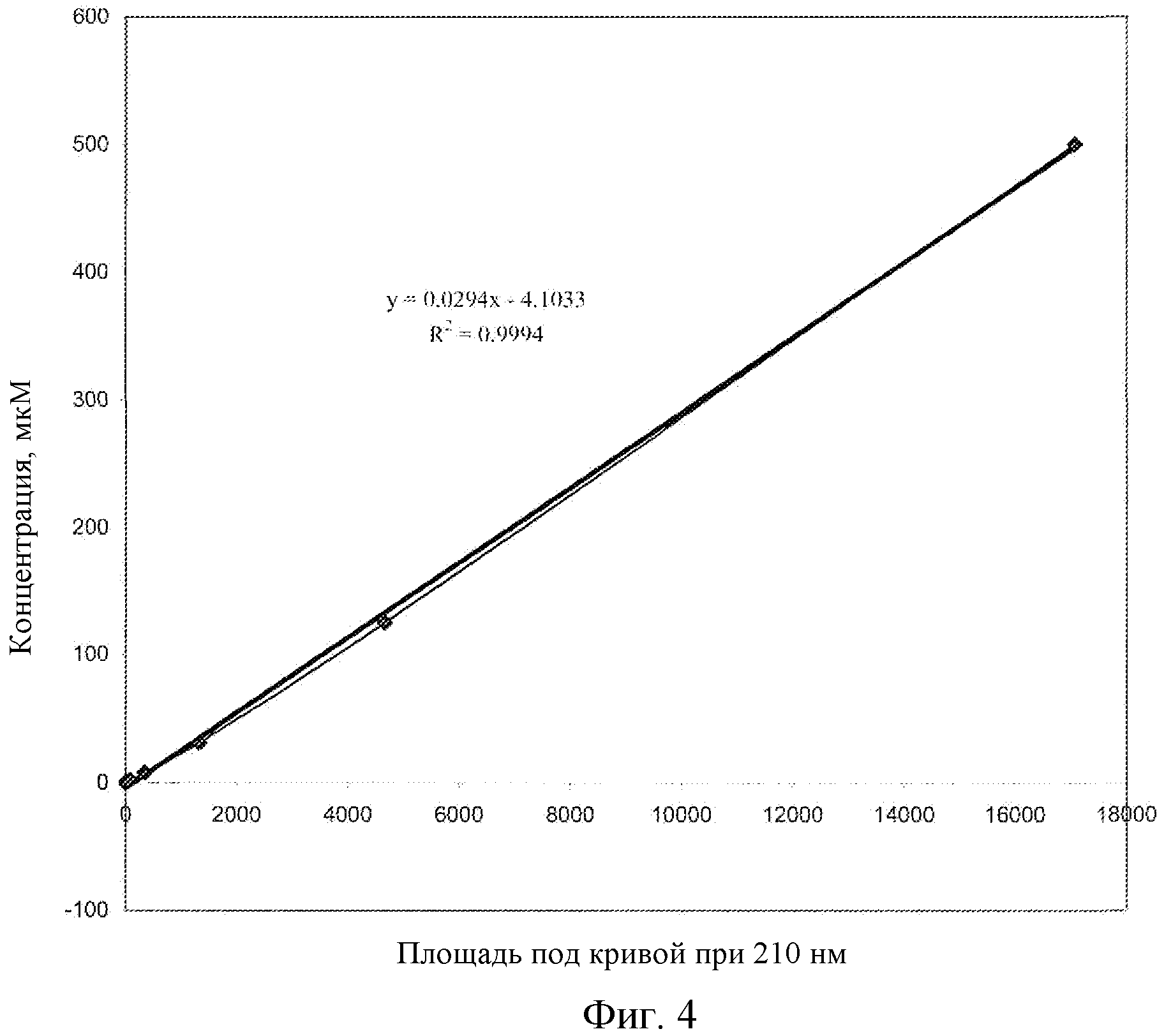
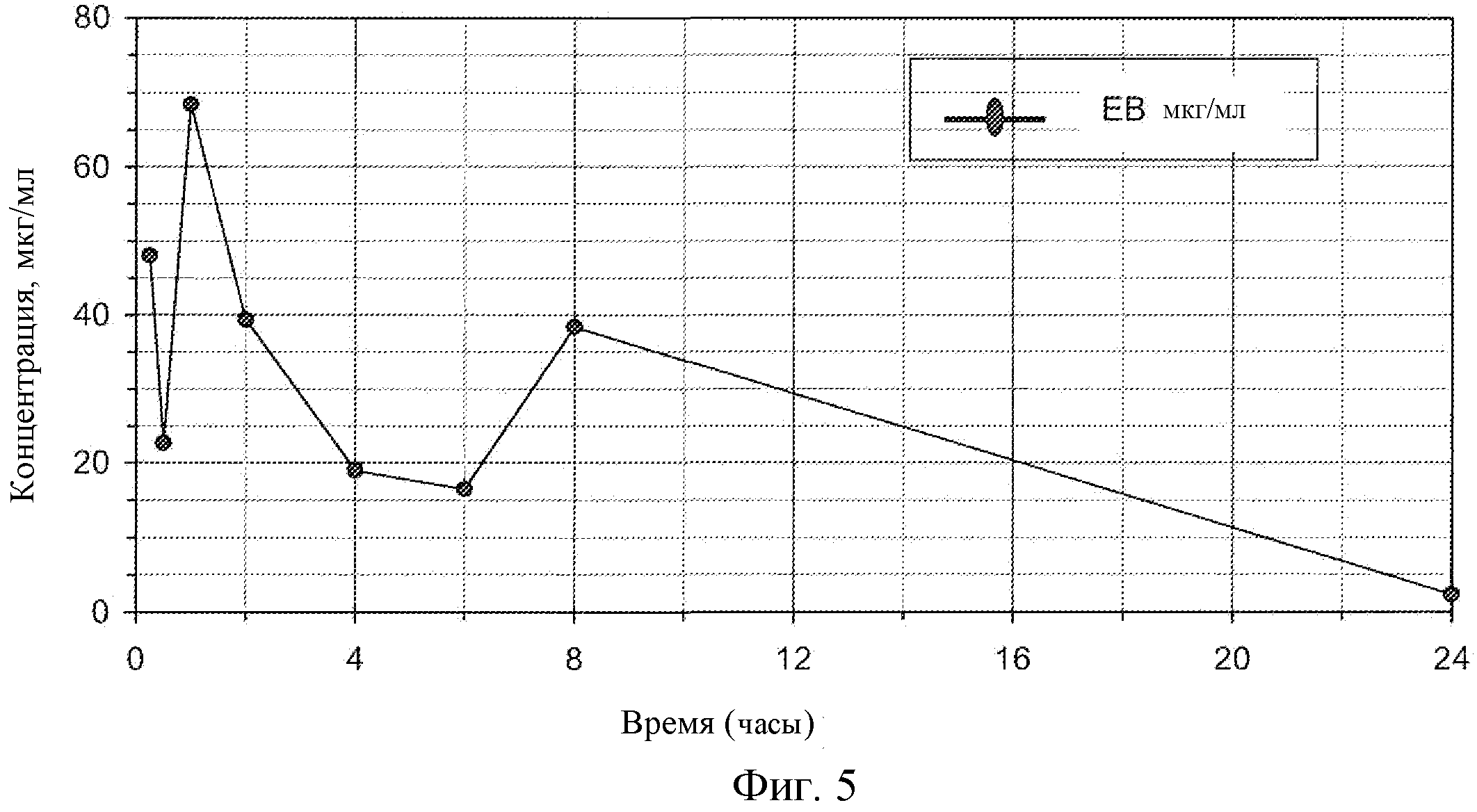
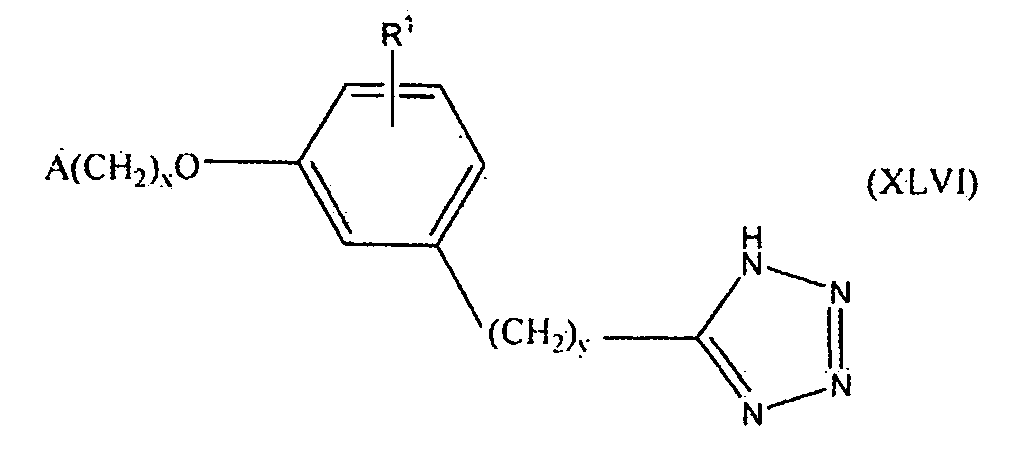
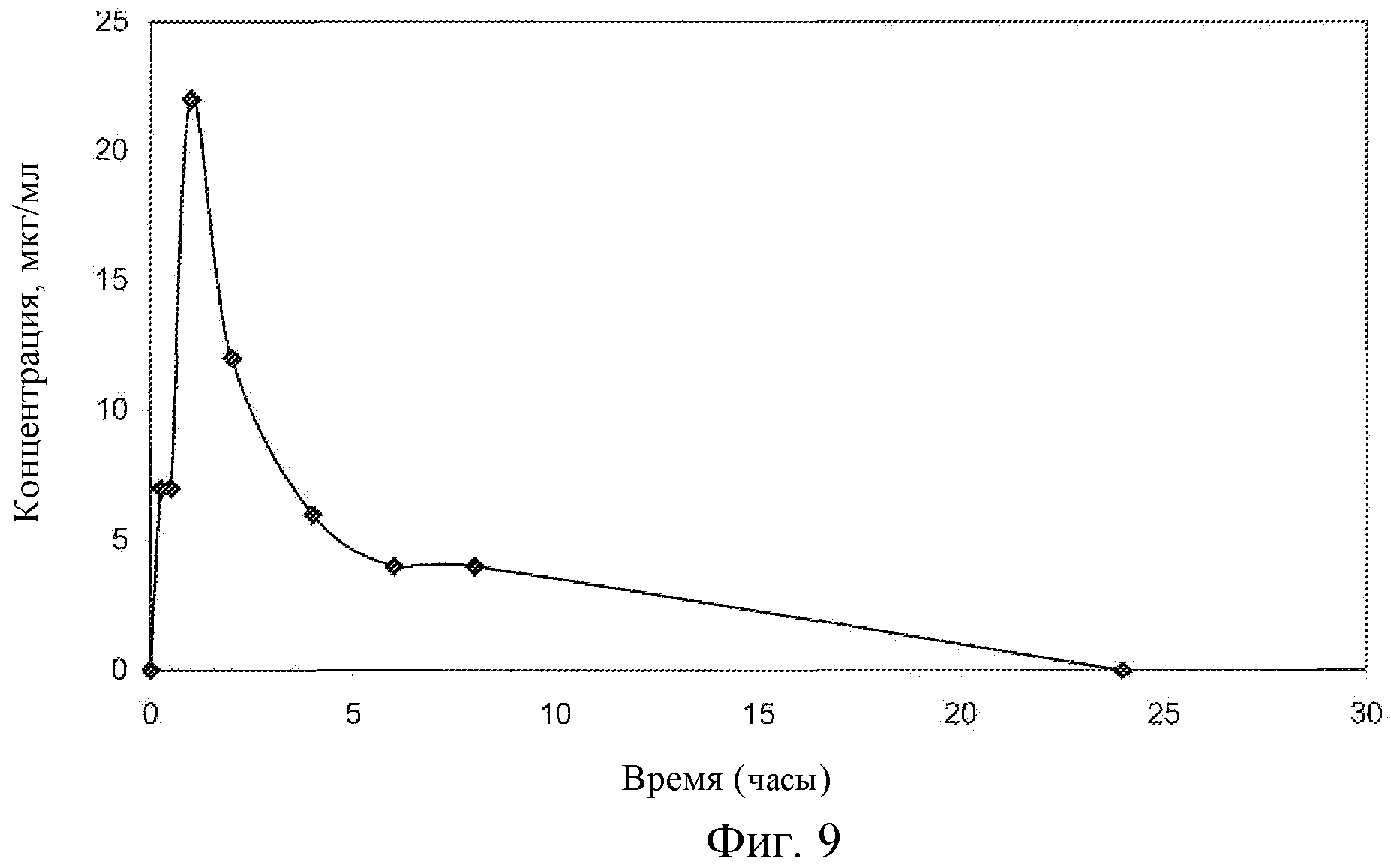
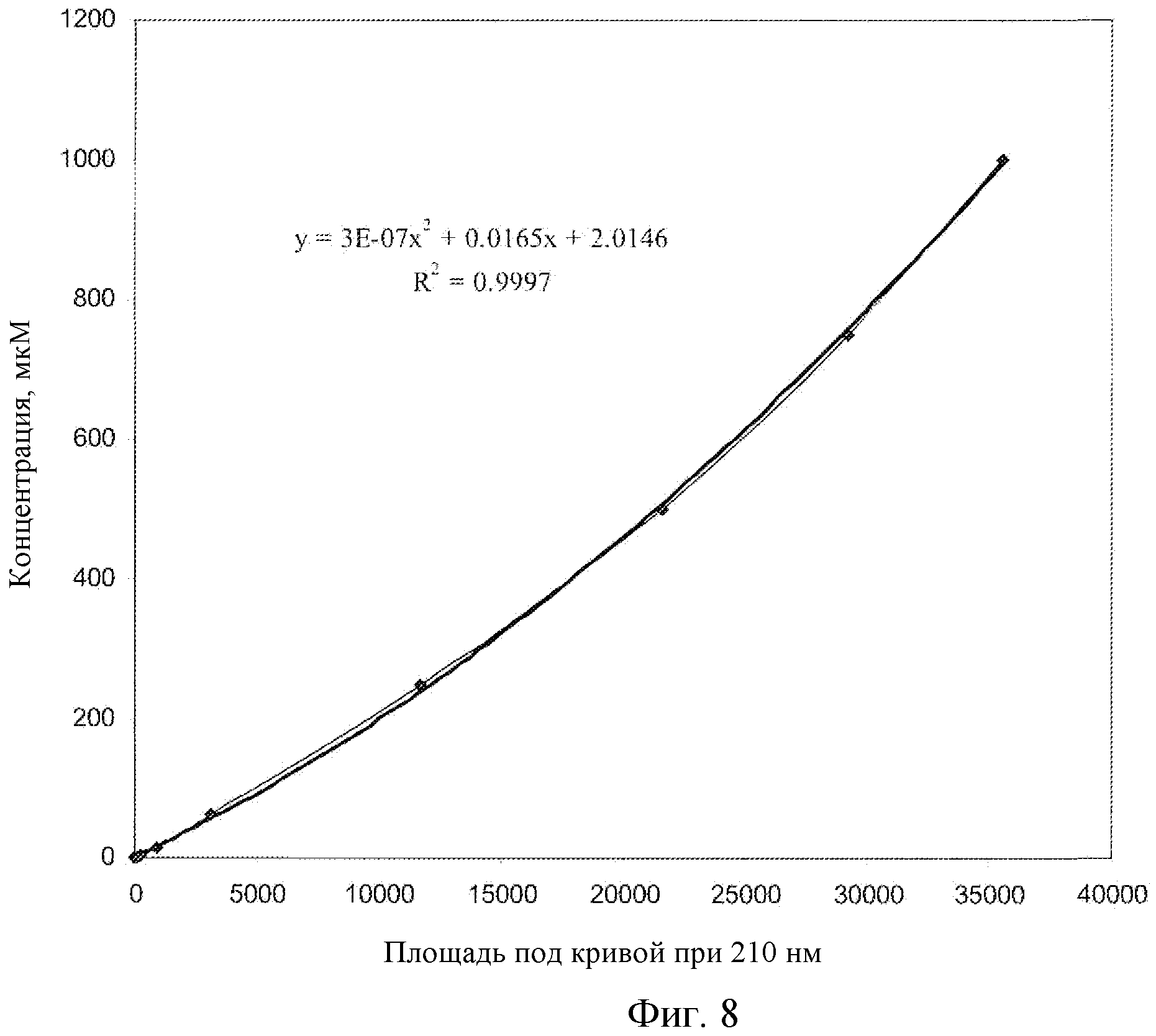
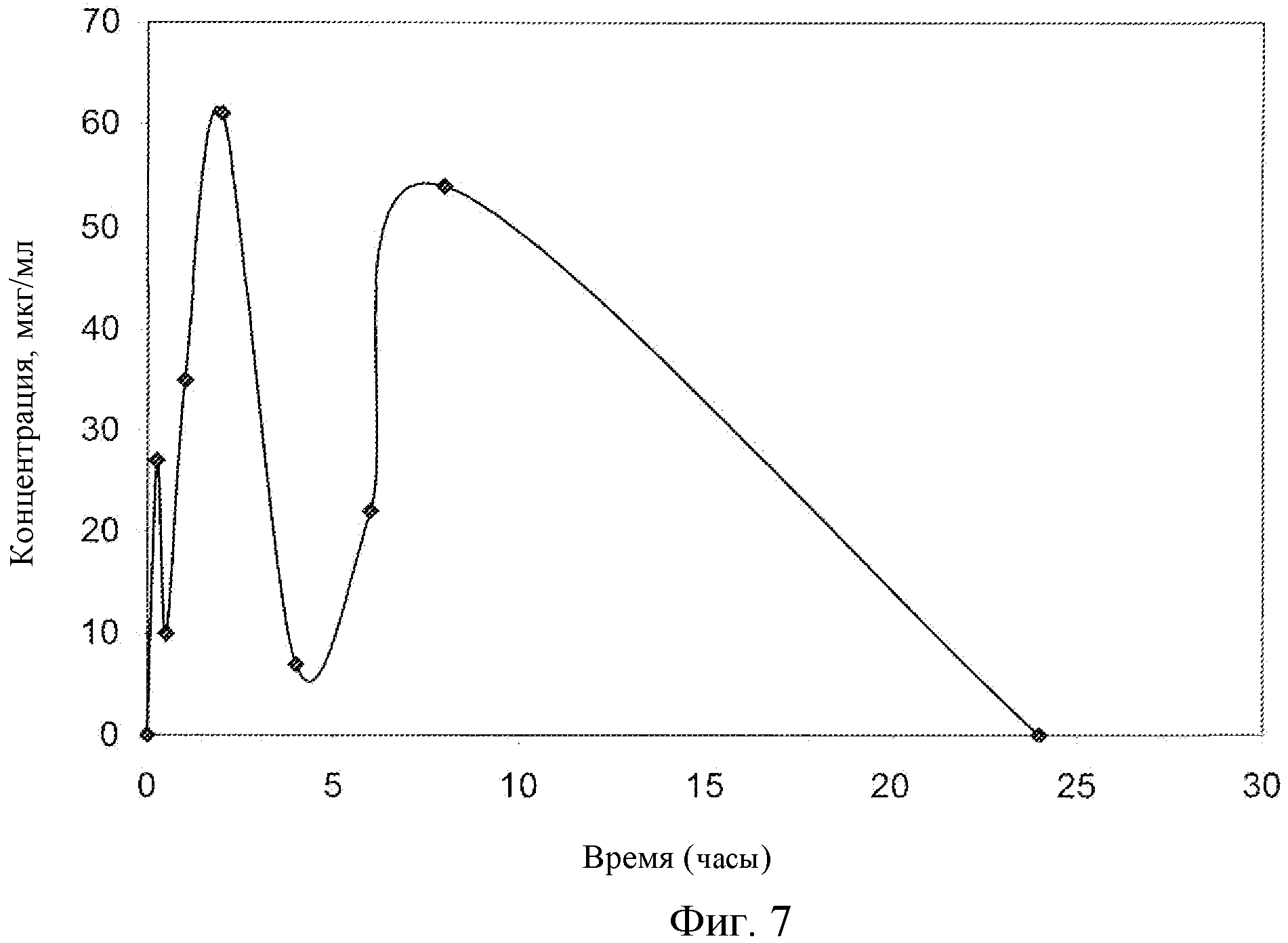
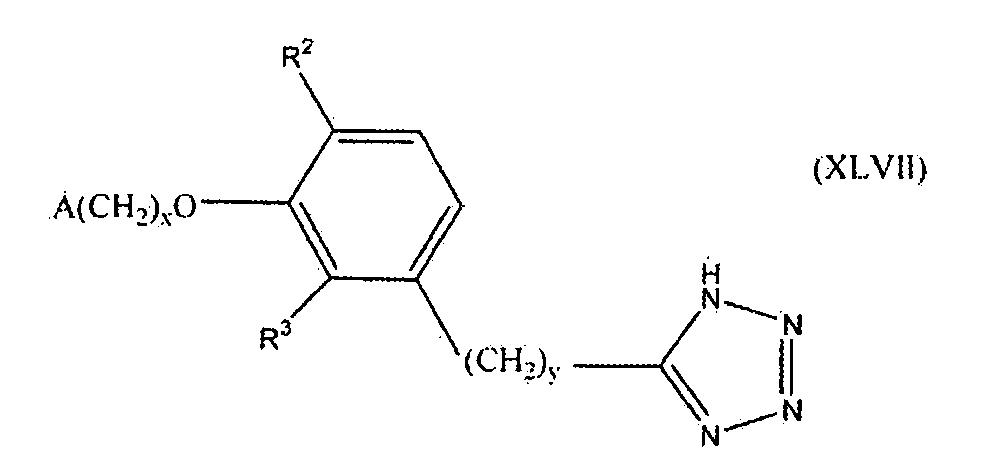
Соединения и способ снижения мочевой кислоты
Соединения и способ снижения мочевой кислоты
Соединения для лечения метаболических заболеваний
Соединение для лечения метаболических расстройств
Соединения амина, имеющие противовоспалительную, противогрибковую, противопаразитарную и противораковую активность
Композиции и способы для лечения митохондриальных заболеваний
Соединения амина, имеющие противовоспалительную, противогрибковую, противопаразитарную и противораковую активность
Соединения и способ снижения мочевой кислоты
Соединения и способ снижения мочевой кислоты
Соединения для лечения метаболических заболеваний
Соединение для лечения метаболических расстройств

