Результат интеллектуальной деятельности: РЕЖИМ ДОЗИРОВАНИЯ ИНГИБИТОРОВ КОМТ
Вид РИД
Изобретение
Данное изобретение относится к новым замещенным нитрокатехолам и их применению при лечении расстройств центральной и периферической нервной системы в соответствии с определенной схемой приема.
Основная причина применения ингибиторов КОМТ в качестве вспомогательных средств при L-ДОПА/ААОС терапии основывается на их способности уменьшать метаболическое 0-метилирование L-ДОПА до 3-0-метил-L-ДОПА (3-ОМД). Период вызванного L-ДОПА клинического улучшения непродолжителен, как результат короткого времени полужизни in vivo L-ДОПА в отличие от длительного времени полужизни 3-ОМД. Кроме того, 3-ОМД конкурирует с L-ДОПА за перенос через гематоэнцефалический барьер (ВВВ, от blood-brain barrier), что означает, что только очень ограниченное количество перорально введенной дозы L-ДОПА действительно достигает места действия, т.е. головного мозга. Обычно только через несколько лет после начальной L-ДОПА терапии с обычной схемой приема вызванное L-ДОПА клиническое улучшение снижается к концу каждого цикла дозирования, приводя к так называемому состоянию «изнашивания» двигательных флуктуации. Описана прямая взаимосвязь между феноменом «изнашивания» и накоплением 3-ОМД (Tohgi, H., et al., Neurosci. Letters, 132: 19-22, 1992). Предполагается, что это может быть следствием замедленного проникания L-ДОПА в мозг из-за конкуренции за транспортную систему через ВВВ с 3-ОМД (Reches, A. et al., Neurology, 32: 887-888, 1982) или, проще говоря, небольшое количество L-ДОПА способно достигнуть головного мозга (Nutt, J.G., Fellman, J.H., Clin. Neuropharmacol., 7: 35-49, 1984). В действительности ингибирование КОМТ защищает L-ДОПА от метаболического расщепления на периферии в ходе 0-метилирования, так что при повторных дозах L-ДОПА средняя величина концентрации L-ДОПА в плазме повышается. В дополнение к пониженной конкуренции за перенос к головному мозгу значительно больший процент перорально введенной дозы L-ДОПА способен достигнуть места действия. Таким образом, ингибирование КОМТ способствует увеличению биологической доступности L-ДОПА, и период антипаркинсонического действия удлиняется с однократными дозами L-ДОПА (Nutt, J.G., Lancet, 351: 1221-1222, 1998).
Наиболее эффективными ингибиторами КОМТ, описанными к настоящему времени, являются 3,4-дигидрокси-4'-метил-5-нитробензофенон (толкапон, Австралийский патент AU-B-69764/87) и (Е)-2-циано-N,N-диэтил-3-(3,4-дигидрокси-5-нитрофенил)акриламид (энтакапон, Германский патент DE 3740383 А1).
Имея по существу одинаковый фармакофор, толкапон отличается от энтакапона тем, что легко проникает в центральную нервную систему (ЦНС) и способен ингибировать церебральную КОМТ, а также периферическую КОМТ. Однако вскоре после его выпуска толкапон изъяли из продажи после того, как стало известно о нескольких случаях гепатотоксичности, включая три внезапных смерти от летального фульминантного гепатита. На сегодняшний день толкапон можно использовать только для больных паркинсонизмом, которые не восприимчивы к другим способам лечения, и строго только при постоянном наблюдении за функцией печени, что дорого и неудобно для пациента. Хотя фактические механистические причины печеночной токсичности, связанной с толкапоном, понятны неполностью, in vitro изучения показали, что толкапон можно уменьшить метаболически до реакционно-способных промежуточных соединений, и предполагается, что они могут образовывать ковалентные аддукты с белками печени, вызывая гепатоцеллюлярное повреждение (Smith, K.S. et al, Chem. Res. Toxicol., 16: 123-128,2003).
Энтакапон напротив, имея с толкапоном один и тот же нитрокатехоловый фармакофор, не связан с печеночной токсичностью и, как правило, рассматривается как безопасное лекарственное вещество. К сожалению, однако, энтакапон является значительно менее эффективным ингибитором КОМТ, чем толкапон, и обладает намного более коротким временем полужизни in-vivo. Это означает, что энтакапон имеет очень ограниченную продолжительность действия и, как следствие, лекарственно вещество следует вводить в очень больших дозах с каждой дозой L-ДОПА, принятой пациентом. В связи с этим клиническая эффективность энтакапона была подвергнута сомнению - действительно, недавнее исследование (Parashos, S.A. et al., Clin. Neuropharmacol., 27(3): 119-123, 2004) показало, что основной причиной для прекращения лечения энтакапоном пациентов с болезнью Паркинсона является ощутимая нехватка эффективности.
Кроме того, относительно короткое время полужизни in-vivo известных ингибиторов КОМТ требует продолжительных схем лечения, обычно включающих введение нескольких доз в день, что многие пациенты найдут обременительным. Например, толкапон следует вводить три раза в день. Следовательно, данное обстоятельство может мешать соблюдению пациентом схемы лечения и вредить качеству жизни.
Соответственно все еще существует потребность в ингибиторах КОМТ, проявляющих сбалансированные свойства биологической активности, биологической доступности и безопасности. А именно, существует потребность в ингибиторах КОМТ, имеющих длительно время полужизни in-vivo и, таким образом, пролонгированное действие на КОМТ, позволяющее, используя меньшее количество доз, получить требуемый лечебный эффект.
Нами в настоящее время неожиданно было обнаружено, что соединения общей формулы 1 представляют собой очень эффективные ингибиторы КОМТ, которые также обладают исключительно длительной продолжительностью действия по сравнению с ингибиторами КОМТ из известного уровня техники.
Кроме того, нами неожиданно было установлено, что соединения общей формулы 1 заметно усиливают биологическую доступность L-ДОПА и увеличивают доставку L-ДОПА к головному мозгу. Соединения значительно повышают уровни дофамина в головном мозге.
Даже более неожиданно повышенные уровни L-ДОПА остаются стабильными в течение двадцатичетырехчасового периода. Эти влияния как на активность КОМТ, так и биологическую доступность L-ДОПА через 24 часа после введения соединений общей формулы 1 заметно больше, чем те, что наблюдались с толкапоном, к настоящему времени единственным известным ингибитором КОМТ, обладающим довольно длительной продолжительностью действия. При коротких промежутках времени (2 и 7 часов) соединения общей формулы 1 дают увеличения при доставке L-ДОПА к головному мозгу, подобные тем, что наблюдались через 24 часа, и расходятся с теми, что наблюдались с толкапоном. Это приводит к более стабильной доставке L-ДОПА к головному мозгу после введения соединений общей формулы I, тогда как толкапон вызывает заметные колебания при доставке L-ДОПА к головному мозгу. Таким образом, соединения общей формулы 1 по всей вероятности обладают терапевтическими преимуществами вследствие длительного постоянного повышения уровней L-ДОПА, тогда как применение толкапона вероятно вызывает нежелательные побочные эффекты, такие как дискинезия, из-за внезапных увеличений и уменьшений уровней L-ДОПА.
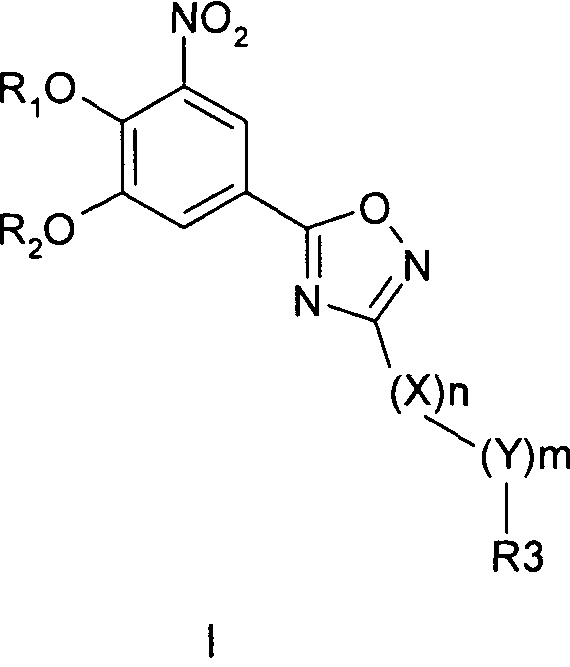
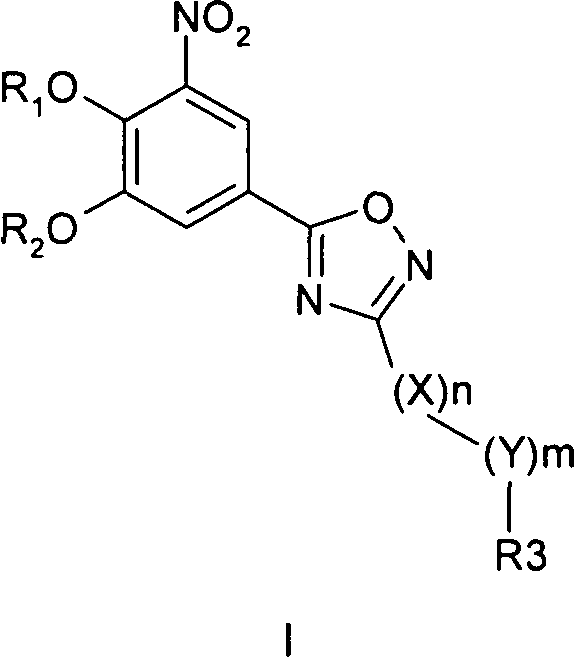
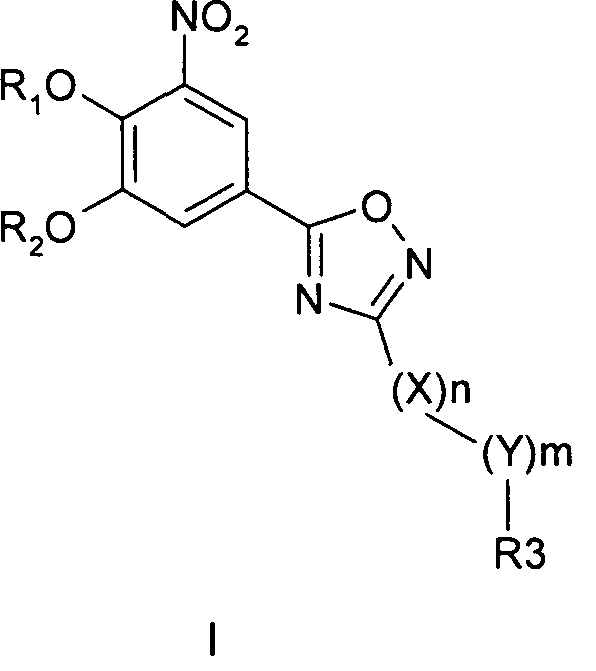
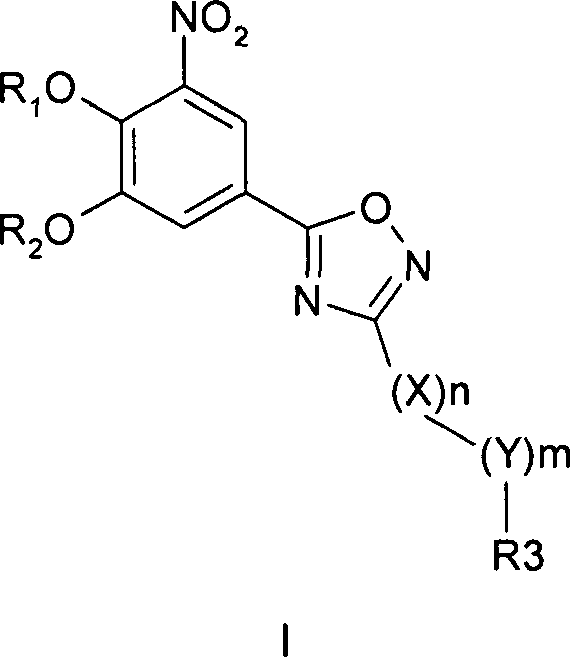
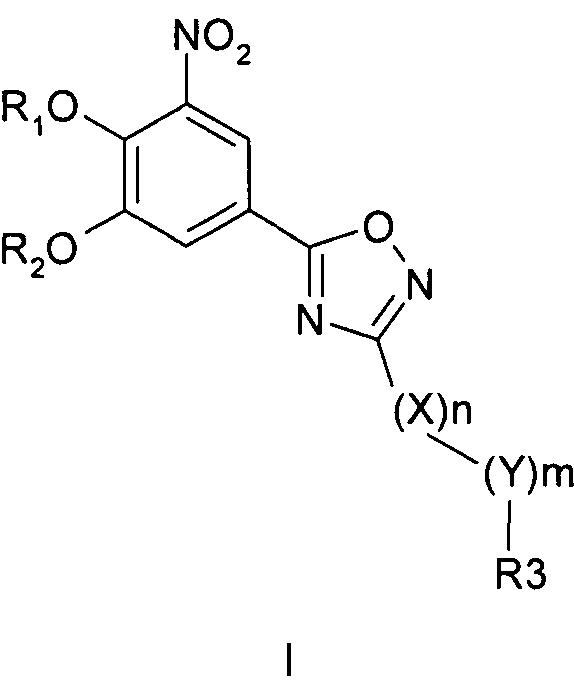
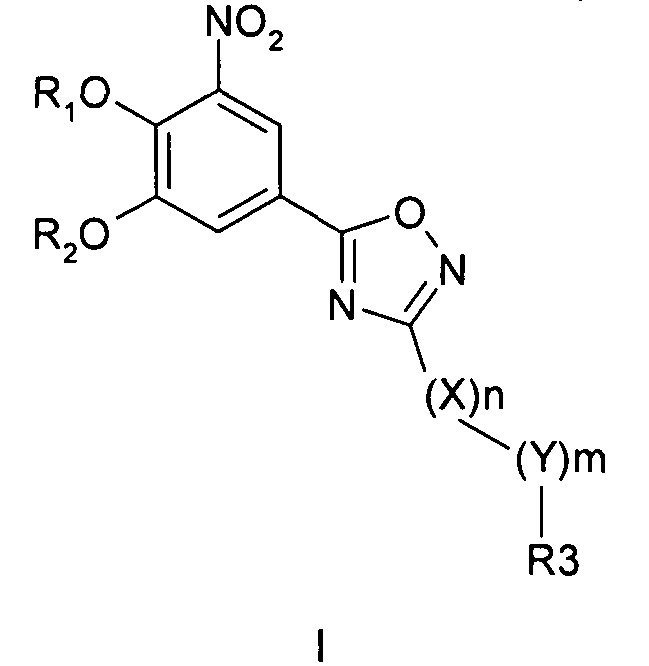
Соединения общей формулы 1 представляют собой соединения, имеющие следующую формулу
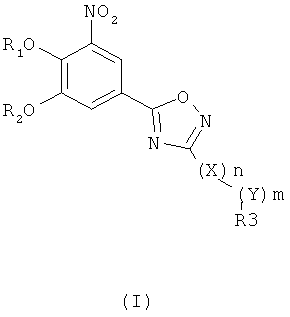
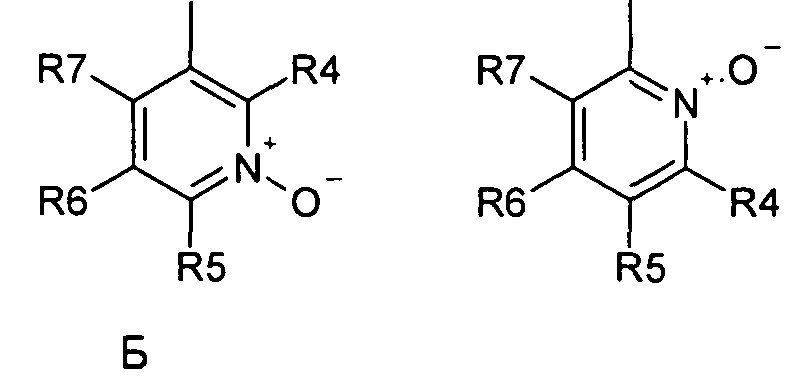
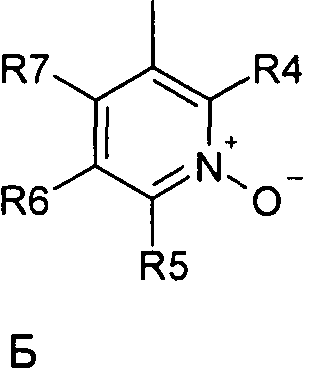
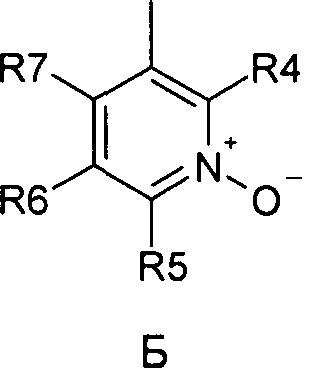
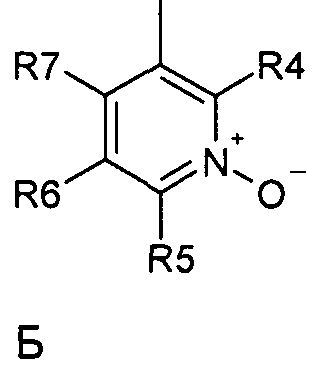
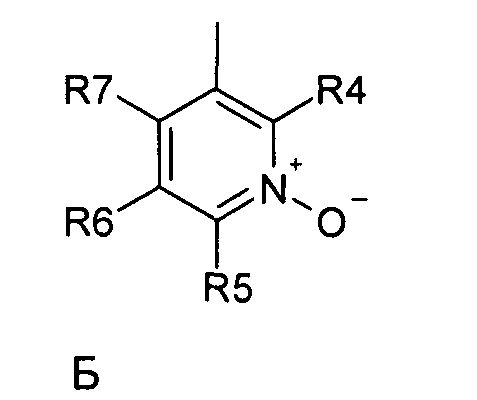
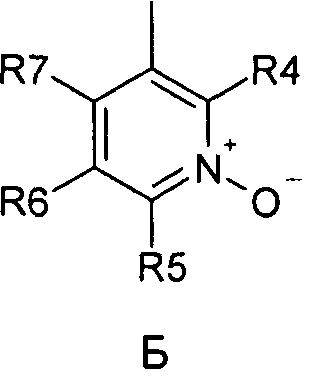
где R1 и R2 независимо друг от друга представляют собой водород или группу, которая гидролизуется при физиологических условиях, возможно замещенный 5 низший алканоил или ароил; Х представляет собой метиленовую группу; Y представляет собой атом кислорода, азота или серы; n представляет собой число 0, 1, 2 или 3, и m представляет собой число 0 или 1; R3 представляет собой группу N-оксида пиридина согласно формуле А, Б или В, которая присоединяется, как показано неотмеченной связью:
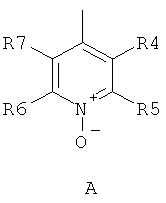
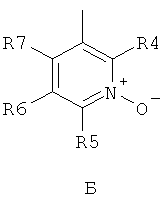
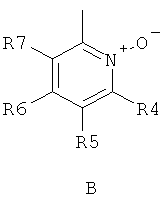
где R4, R5, R6 и R7 являются одинаковыми или разными и обозначают водород, низший алкил, низший тиоалкил, низший алкокси, арилокси или тиоарильную группу, низшую алканоильную или ароильную группу, возможно замещенную арильную группу, амино, низший алкиламино, низший диалкиламино, циклоалкиламино или гетероциклоалкиламино группу, низшую алкилсульфонильную или арилсульфонильную группу, галоген, галоалкил, трифторметил, циано, нитро или гетероарильную группу, или, взятые вместе, обозначают алифатические или гетероалифатические кольца, или ароматические или гетероароматические кольца; термин алкил означает углеродные цепи, неразветвленные или разветвленные, содержащие от одного до шести углеродных атомов; термин арил означает фенильную или нафтильную группу, возможно замещенную алкокси группами или галогенами; термин гетероциклоалкил представляет собой четырех - восьмичленное циклическое кольцо, возможно включающее дополнительные атомы кислорода, серы или азота; термин гетероарил представляет собой пяти- или шестичленное кольцо, включающее атом серы, кислорода или азота; термин галоген означает фтор, хлор, бром или йод.
В вышеприведенной формуле предпочтительно R4, R5, R6 и R7 независимо друг от друга представляют собой водород, C1-C6-алкил, C1-C6-тиоалкил, C1-C6-алкокси, С6-С12-арилокси или С6-С12-тиоарильную группу, C1-C6-алканоильную или С7-С13-ароильную группу, амино, C1-C6-алкиламино, C1-C6-диалкиламино, С7-С13-циклоалкиламино, С3-С12-гетероциклоалкиламино, С3-С12-алкилсульфонил, C3-C12-арилсульфонил, галоген, C1-C6-галоалкил, трифторметил, циано, нитро или гетероарильную группу; или два или более взятых вместе остатка R4, R5, R6 и R7 представляют собой алифатические или гетероалифатические кольца, или ароматические или гетероароматические кольца.
Предпочтительно С1-С6-алкильные остатки представляют собой метил, этил, н-пропил, изопропил, н-бутил, елюр-бутил, тpeт-бутил, гептил или гексил. Предпочтительно C1-C6-тиоалкильные остатки представляют собой тиометил, тиоэтил, тио-н-пропил, тио-изопропил и тио-н-бутил. Предпочтительно C1-C6-алкокси остатки представляют собой метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси и трет-бутокси. Предпочтительно С6-С12-арилокси остатки представляют собой фенокси или нафтокси, которые могут быть замещены. Предпочтительно С6-С12-тиоарильные остатки представляют собой тиофенил и тионафтил, которые могут быть замещены. Предпочтительно C1-C6-алканоильные остатки представляют собой метаноил, этаноил, пропаноил или бутаноил. Предпочтительно С7-С13-ароильные остатки представляют собой бензоил и нафтоил. Предпочтительно остатки C1-С6-алкиламино представляют собой метиламино, этиламино, н-пропиламино, изопропиламино и н-бутиламино. Предпочтительно остатки С1-С6-диалкиламино представляют собой диметиламино, диэтиламино, ди-н-пропиламино, ди-н-бутиламино, ди-изопропиламино, метилэтиламино, метилпропиламино и этилпропиламино. Предпочтительно остатки С3-С12-циклоалкиламино представляют собой пирролидино, пиперидино, циклогексиламино и дициклогексиламино. Предпочтительно остатки С3-С12-гетероциклоалкиламино представляют собой морфолино, 2,6-диметилморфолино, 3,5-диметилморфолино, пиперазино, N-метилпиперазино и N-этилпиперазино. Предпочтительно С1-С6-алкилсульфонильные или С6-С12-арилсульфонильные остатки представляют собой метилсульфонил, этилсульфонил, фенилсульфонил и толилсульфонил. Предпочтительно остатки галогенов представляют собой хлор, бром, йод и фтор. Предпочтительно C1-С6-галоалкил представляет собой хлорметил, фторметил, дихлорметил, дифторметил, трихлорметил и трифторметил. Предпочтительно гетероарильные остатки представляют собой пиридил, пиримидил, изоксазолил, оксазолил, изоксадиазолил, оксадиазолил, триазолил и тетразолил. В тех случаях, когда два или более взятых вместе остатка R4, R5, R6 и R7 представляют собой алифатические, или гетероалифатические кольца, или ароматические или гетероароматические кольца, предпочтительные объединенные остатки представляют собой индолизинил, изоиндолил, индолил, индазолил, пуринил, хинолизинил, нафтиридинил, изохинолил и хинолил. Предпочтительно каждый пит обозначает число 0 или 1, или оба означают 0 или 1.
В следующем описании медицинских показаний, методов лечения и схем приема фармацевтических композиций, содержащих соединения согласно общей формуле I по изобретению, наиболее предпочтительным примером соединения согласно общей формуле I является 5-[3-(2,5-дихлор-4,6-диметил-1-окси-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол, далее обозначаемый как соединение А, и его фармакологически приемлемые соли и эфиры.
Другие предпочтительные соединения вышеприведенной общей формулы (I) в следующих медицинских показаниях, методах лечения и схемах приема включают 3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-4-(трифторметил)пиридин-1 -оксид, 2-хлор-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-4,6-диметилпиридин-1-оксид, 3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-2-метил-6-(трифторметил)пиридин-1-оксид, 5-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-2-(трифторметил)пиридин-1 -оксид, 5-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-2-метил-4-(трифторметил)пиридин-1 -оксид, 3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-2,6-диметил-4-(трифторметил)пиридин-1-оксид, 3,5-дихлор-4-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)пиридин-1 -оксид, 3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-6-метил-2-фенил-4-(трифторметил)пиридин-1-оксид, 2-бром-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-4,5,6-триметилпиридин-1 -оксид, 2-хлор-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-4,5,6-триметилпиридин-1-оксид, 3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-2-(трифторметил)пиридин-1 -оксид, 2-хлор-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-6-метилпиридин-1-оксид, 2-бром-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-6-метилпиридин-1-оксид, 2-бром-5-хлор-3-(5-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-3-ил)-4,6-диметилпиридин-1-оксид, 5-[3-(2-хлор-1-окси-пиридин-4-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол- 1,2-диол, 5-[3-(2-морфолин-4-ил-1-окси-пиридин-4-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол-1,2-диол, 5-[3-(4-бром-1-окси-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол-1,2-диол, 5-[3-(2-морфолин-4-ил-1 -окси-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол-1,2-диол, 5-[3-(2-метил-1-окси-6-фенил-4-трифторметил-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол-1,2-диол и 5-[3-(2-бром-4,6-диметил-1-окси-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитро-бензол-1,2-диол, и их фармакологически приемлемые соли и эфиры.
Настоящее изобретение относится к применению соединений общей формулы I, их фармацевтически приемлемых солей или эфиров для предотвращения или лечения конкретных патологических состояний, особенно у людей (например расстройств центральной и периферической нервной системы), и к получению фармацевтических композиций, содержащих их.
Подвергающиеся лечению патологические состояния предпочтительно являются связанными с центральной и периферической нервной системой заболеваний людей. Предпочтительные заболевания включают двигательные расстройства и шизоаффективные расстройства. Двигательные расстройства характеризуются либо недостатком движения, либо чрезмерным движением. Двигательные расстройства, предпочтительно излечиваемые соединениями общей формулы I, включают болезнь Паркинсона, дистонию, дискинезию, экстрапирамидные синдромы, нарушение походки, тремор, хорею, дрожательный паралич, акатизию, атетоз, брадикинезию, обморожение, ригидность, постуральную неустойчивость, миоклонию и судорожные сокращения или синдром Туретта. Наиболее предпочтительным заболеванием является болезнь Паркинсона.
Как используется здесь, термин «лечение» и такие варианты, как «лечить» или «подвергающийся лечению», относятся к режиму, который может оказывать благоприятное воздействие на человека или животное. Лечение может относиться к существующему состоянию или может быть профилактическим (превентивным лечением). Лечение может включать целебное, облегчающее или профилактическое действия. Лечение может предотвращать или задерживать появление болезни, замедлять прогрессирование или улучшать симптомы заболевания или состояния.
Соединения общей формулы I предпочтительно используются для получения лекарства для предотвращения или лечения заболеваний, связанных с центральной и периферической нервной системой, согласно определенной схеме приема.
Подходящие схемы приема (режимы дозирования) включают режимы, имеющие периодичность дозирования в диапазоне от приблизительно двух раз в день до приблизительно одного раза через день.
Как используется здесь, термин «периодичность дозирования» относится к числу эффективных доз соединения общей формулы I, даваемых в определенный промежуток времени.
Предпочтительно периодичность дозирования выбрана из двух раз в день, одного раза в день и одного раза через день.
В случае периодичности дозирования два раза в день эффекты изобретения могут быть достигнуты при введении один раз в течение каждых 12 часов, даже когда время между введениями (или интервал дозирования) не составляет 12 часов. Дозы предпочтительно вводят с интервалами дозирования от 8 до 16 часов, более предпочтительно 12 часов, причем два интервала дозирования предпочтительно составляют приблизительно 24 часа. Подходящие неограниченные начальные точки для интервалов дозирования включают утро, середину дня, полдень, день, вечер и полночь. Например, схема приема два раза в день согласно изобретению может требовать введение дозы в 8.00 утра и другой дозы в 17.00 дня (в этом случае интервалы дозирования составляют 11 часов и 13 часов и сводятся приблизительно к 24 часам). Предпочтительно, чтобы временной интервал между двумя дозами составлял около 12 часов.
В случае периодичности дозирования один раз в день эффекты изобретения могут быть достигнуты при введении один раз в течение каждых 24 часов, даже когда время между введениями не составляет 24 часа. Дозы предпочтительно вводят с интервалами дозирования около 24 часов. Подходящие неограниченные начальные точки для интервалов дозирования включают утро, середину дня, полдень, день, вечер и полночь. Например, схема приема один раз в день согласно изобретению может требовать введение дозы в 8.00 утра и другой дозы в 8.00 утра следующего дня (в этом случае интервал дозирования составляет около 24 часов).
В случае периодичности дозирования один раз через день эффекты изобретения могут быть достигнуты при введении один раз в течение каждых 48 часов, даже когда время между введениями не составляет 48 часов. Дозы предпочтительно вводят с интервалами дозирования от 36 до 60 часов, причем интервалы дозирования предпочтительно в среднем составляют около 48 часов. Подходящие неограниченные начальные точки для интервалов дозирования включают утро, середину дня, полдень, день, вечер и полночь. Например, схема приема один раз через день согласно изобретению может требовать введение дозы в 8.00 утра первого дня и другой дозы в 13.00 третьего дня (в этом случае интервал дозирования составляет 53 часа). Предпочтительно время между введениями составляет около 48 часов.
В настоящем изобретении эффективные суточные дозы соединений общей формулы I находятся в диапазоне 1-1000 мг/день, более предпочтительно от 2 до 500 мг/день, даже более предпочтительно от 3 до 250 мг/день и наиболее предпочтительно 5-100 мг/день.
Предпочтительно, чтобы отдельные единицы дозирования соединений общей формулы I находились в интервале 1-500 мг, более предпочтительно от 2 до 300 мг/день, даже более предпочтительно от 3 до 100 мг/день и наиболее предпочтительно 5-50 мг, причем суточная доза может различаться в зависимости от времени введения. Например, при схеме приема два раза в день возможно вводить дозу, содержащую 11/24 суточной дозы соединения общей формулы I, в 8.00 утра и другую дозу, содержащую 13/24 суточной дозы соединения общей формулы I, в 17.00 дня.
Как используется здесь, термин «единица дозирования» относится к отдельному фармацевтическому препарату, например таблетке, содержащему соединение общей формулы I, который вводиться пациенту во время режима дозирования.
Предпочтительно, чтобы объект, подвергаемый лечению с соединением общей формулы I, также получал лечение с L-ДОПА и/или ингибитором декарбоксилазы ароматических L-аминокислот (AADC).
Подходящие AADC включают карбидопу и бенсеразид.
Соединения общей формулы I, L-ДОПА и AADC могут быть введены раздельно или в любом сочетании. Их можно вводить параллельно (например одновременно) или последовательно и с одинаковой или различающейся периодичностью дозирования. Например, соединения общей формулы I могут быть введены параллельно или последовательно с L-ДОПА. В случае одновременного введения также возможно объединять оба активных компонента в одном фармацевтическом препарате.
Согласно другому аспекту настоящего изобретения создан способ лечения по меньшей мере одного состояния или заболевания у пациента, нуждающегося в лечении, включающий стадию, на которой вводят пациенту приблизительно два раза в день - приблизительно один раз через день фармакологически эффективную дозу соединения общей формулы I, как определено выше.
Предпочтительно вводят один раз в день для всех воплощений изобретения.
Предпочтительно для всех способов по изобретению, чтобы объект, подвергаемый лечению с соединением общей формулы I, также получал лечение с L-ДОПА и/или ингибитором декарбоксилазы ароматических L-аминокислот (AADC).
Согласно другому аспекту изобретения создан способ снижения ингибирования КОМТ у объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ увеличения уровней L-ДОПА в головном мозге объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ увеличения уровней L-ДОПА в плазме объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ уменьшения уровней 3-0-метил-1--ДОПА (3-ОМД) в головном мозге объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ уменьшения уровней 3-ОМД в плазме объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ увеличения биологической доступности L-ДОПА в головном мозге объекта в течение 24-48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно другому аспекту изобретения создан способ увеличения биологической доступности L-ДОПА в плазме объекта в течение 24 - 48 часов, включающий стадию, на которой вводят объекту от приблизительно двух раз в день до приблизительно одного раза через день эффективную дозу соединения общей формулы I, как определено выше.
Согласно дополнительному аспекту изобретения создана фармацевтическая композиция, приспособленная к введению соединения общей формулы 1 от приблизительно двух раз в день до приблизительно одного раза через день.
Настоящее изобретение также относится к упаковке, включающей фармацевтическую композицию соединения общей формулы 1 в сочетании с инструкциями по вводу указанного препарата со схемой приема, имеющей периодичность дозирования в диапазоне от двух раз в день до приблизительно одного раза через день.
В одном воплощении соединения общей формулы 1 можно получить по способу, согласно которому соединение общей формулы IIА, IIБ или IIIВ,
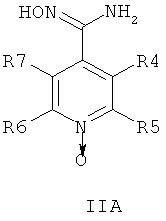
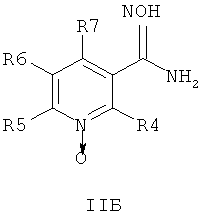
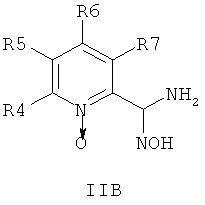
где R4, R5, R6 и R7 являются такими, как определено в общей формуле I, подвергают реакции циклизации, включающей конденсацию и дегидратацию, с соединением общей формулы III,
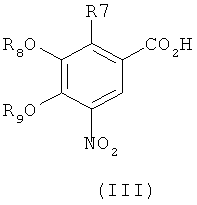
где R8 и R9 независимо друг от друга представляют собой водород или подходящие защитные группы для ароматических гидроксильных групп, при условиях, пригодных для получения оксадиазольных производных формулы IVA, IVБ или IVB,
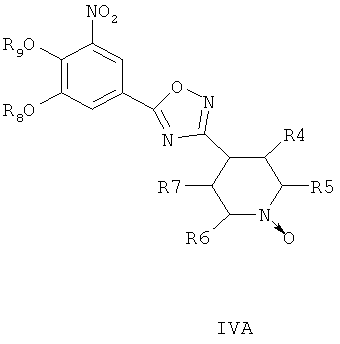
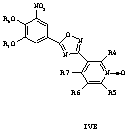
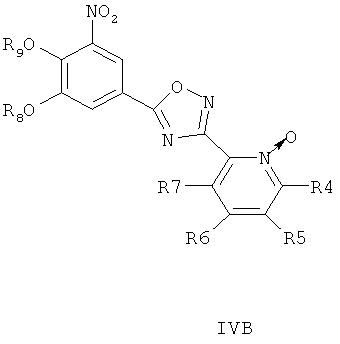
Затем при необходимости удаляют гидроксилзащищающие группы, чтобы получить соединения общей формулы I.
В другом воплощении соединения общей формулы 1 можно получить по способу, согласно которому соединение общей формулы VA, V5 или VB,
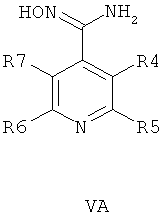
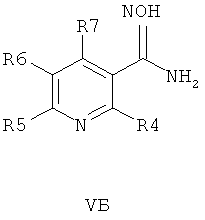
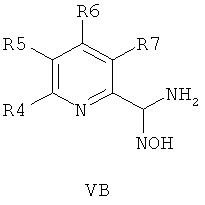
где R4, R5, R6 и R7 являются такими, как определено в общей формуле I, подвергают реакции циклизации, включающей конденсацию и дегидратацию, с соединением общей формулы III при условиях, пригодных для получения оксадиазольных производных формулы VIA, VIБ или VIB,
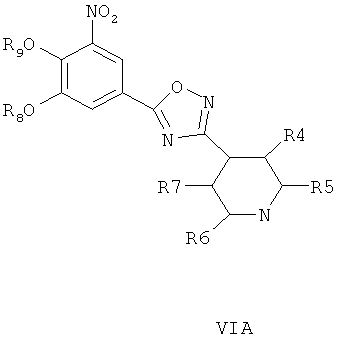
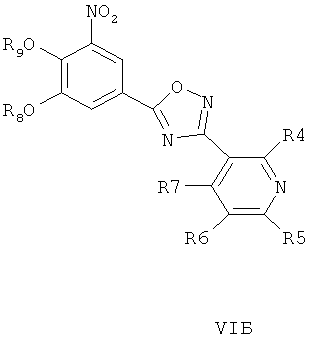
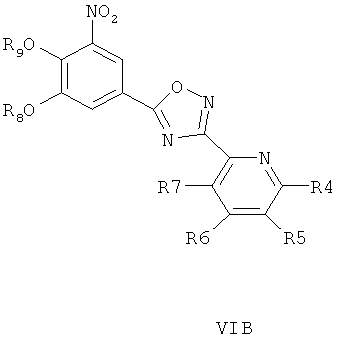
затем окисляют пиридиновый атом азота, что дает соединение согласно формуле IVA, IVB или IVB, как показано выше, и при необходимости удаляют гидроксилзащищающие группы, чтобы получить соединения общей формулы I.
Подходящие защитные группы для ароматических гидроксильных групп хорошо известны в данной области техники. Примеры подходящих защитных групп для ароматических гидроксильных групп включают метил, этил, изопропил, бензил, 4-метоксибензил, метоксиметил, бензилоксиметил, метоксиэтоксиметил, тетрагидропиранил, фенацил, аллил, триметилсилил, трет-бутилдиметилсилил, бензилоксикарбонил, трет-бутоксикарбонил, эфир, сульфонат, карбамат, фосфинат, ацетальные и кетальные производные.
В предпочтительном воплощении одна из групп R8 и R9 представляет собой водород, и другая является метилом. В особенно предпочтительном воплощении R8 представляет собой метил и R9 представляет собой водород.
В альтернативном предпочтительном воплощении защитные группы R8 и R9 заменяют на водород или группу, которая гидролизуется при физиологических условиях. Защитные группы R8 и R9 могут быть удалены независимо друг от друга в ходе отдельных реакционных стадий или могут быть удалены в ходе одной реакционной стадии. Таким же образом введение группы, которая гидролизуется при физиологических условиях, можно осуществлять либо на этой же, либо на последующей реакционной стадии.
В настоящем изобретении условия пригодные для получения оксадиазольных производных включают условия, которые дают оксадиазольное производное с высоким выходом и чистотой. Предпочтительно выход требуемого оксадиазольного производного составляет по меньшей мере 70%, более предпочтительно 75-99%, даже более предпочтительно 80-97% и наиболее предпочтительно 85-95%. Предпочтительно чистота требуемого оксадиазольного производного составляет по меньшей мере 90%, более предпочтительно по меньшей мере 95%, даже более предпочтительно по меньшей мере 99% и наиболее предпочтительно по меньшей мере 99,5%. Исходя из предмета настоящего изобретения квалифицированный специалист может обычным путем установить наиболее подходящие реакционные условия, чтобы оптимизировать выход и чистоту оксадиазола. Параметры, которые следует учитывать квалифицированному специалисту, включают, но не ограничиваются этим, реагенты, осуществляющие конденсацию, и дегидратирующие агенты, выбор защитных групп R8 и R9, систему растворителей, температуру реакции и время реакции, и растворимость реагентов.
Соединение общей формулы III требует активации перед реакцией конденсации с соединением формулы IIА-IIВ или VA-VB. Подходящие реагенты для активации соединения формулы III включают 1,1-карбонилдиимидазол, тионилхлорид, сульфонилхлорид, N,N'-дициклогексилкарбодиимид, 1-гидроксибензотриазол и М-(3-диметиламинопропил)-N'-этилкарбодиимид, фосген, РСl3, РОСl3, PCl5, ангидриды, трихлортриазин и хлордиметокситриазин и подобные. Особенно предпочтительны 1,1-карбонилдиимидазол и тионилхлорид. В некоторых случаях те же самые реагенты можно применять для выполнения стадии циклизации, которая состоит из конденсации и дегидратации. Альтернативные реагенты для осуществления конденсации и/или дегидратации включают пиридин и фторид тетрабутиламмония. Предпочтительно дегидратацию можно выполнить в ходе теплового нагревания реакционной смеси совместно с вышеупомянутыми реагентами.
Соединение общей формулы III можно активировать с избытком реагента, такого как тионилхлорид, в подходящем растворителе или без необходимости в дополнительном растворителе. Предпочтительно избыток реагента можно затем удалить, например, перегонкой, и заменить растворителем и другим реагентом, таким как пиридин, чтобы выполнить стадии конденсации и дегидратации. Предпочтительные системы растворителей для активации соединения общей формулы III и циклизации с соединениями общих формул IIА-IIВ или VA-VB представляют собой диполярные апротонные растворители, включающие диметилформамид, диметилсульфоксид, диметилацетамид и N-метилпирролидинон. Особенно предпочтительны диметилсульфоксид и диметилацетамид.
Подходящие температуры реакции и время реакции зависят от реакционной способности используемых для осуществления конденсации и дегидратации реагентов. Предпочтительно температура реакции находится в диапазоне от 0°С до точки кипения используемой системы растворителей, более предпочтительно в диапазоне 20-150°С и наиболее предпочтительно в диапазоне 25-120°С. Предпочтительно время реакции находится в интервале от 30 минут до 24 часов, более предпочтительно в интервале от 1 часа до 18 часов и наиболее предпочтительно 2-6 часов.
В альтернативном предпочтительном воплощении реакцию конденсации и дегидратации проводят в присутствии органического или неорганического основания. Подходящие предпочтительные основания включают триэтиламин, трибутиламин, 2,6-лутидин, N-метилморфолин, пиридин, имидазол, N-метилимидазол и 4-диметиламинопиридин. Особенно предпочтительные основания включают пиридин, N-метилимидазол и 4-диметиламинопиридин.
В предпочтительном воплощении настоящего изобретения конденсацию и дегидратацию проводят в ходе двух отдельных реакционных стадий. В этом конкретном воплощении разные агенты конденсации и дегидратации и системы растворителей можно использовать, чтобы оптимизировать выход и чистоту получаемого продукта.
В альтернативном предпочтительном воплощении настоящего изобретения конденсацию и дегидратацию проводят последовательно в одном и том же сосуде без выделения 0-ацилированных промежуточных соединений. В этом конкретном воплощении реагенты, осуществляющие конденсацию и дегидратацию, могут быть одинаковыми или разными, но предпочтительно идентичными.
Количество реагентов, осуществляющих конденсацию и дегидратацию, не является определяющим. Обычные количества реагентов, осуществляющих конденсацию и дегидратацию, включают по меньшей мере количество в 1 моль, предпочтительно от 2,1 моль до 5 моль, более предпочтительно 2,2-4 моль и наиболее предпочтительно от 2,3 моль до 3 моль на моль пиридинового производного. В тех случаях, когда реагенты, осуществляющие конденсацию и дегидратацию, также служат в качестве растворителя или сорастворителя, избыточное количество может быть намного выше.
Как упоминалось выше, в предпочтительных воплощениях изобретение включает стадию, на которой атом азота пиридиновой группировки VIA, VIБ или VIВ окисляют при подходящих условиях до соответствующего производного пиридил-N-оксида IVA, IVB или IVB после реакции циклизации.
В настоящем изобретении окислительные условия пригодные для получения пиридил-N-оксида включают условия, которые дают производное пиридил-N-оксида с высоким выходом и чистотой. Предпочтительно выход требуемого производного пиридил-N-оксида составляет по меньшей мере 90%, более предпочтительно 92-99%, даже более предпочтительно 94-98% и наиболее предпочтительно 95-97%. Предпочтительно чистота требуемого производного пиридил-N-оксида составляет по меньшей мере 90%, более предпочтительно по меньшей мере 95%, даже более предпочтительно по меньшей мере 99% и наиболее предпочтительно по меньшей мере 99,5%. Исходя из предмета настоящего изобретения квалифицированный специалист может обычным путем установить наиболее подходящие реакционные условия, чтобы оптимизировать выход и чистоту пиридил-N-оксида. Параметры, которые следует учитывать квалифицированному специалисту, включают, но не ограничиваются этим, окислитель, количество окислителя, выбор защитных групп, систему растворителей, температуру реакции и время реакции, и растворимость реагентов.
Предпочтительные окислители включают пероксид водорода, МnO2, надуксусную кислоту, трифторнадуксусную кислоту, трет-бутилгидропероксид, мета-хлорпероксибензойную кислоту, надсерные кислоты, Оксон®, комплекс мочевины с пероксидом водорода и трифторуксусный ангидрид, хлорхромат пиридиния и перманганат ионы. Особенно предпочтителен комплекс мочевины с пероксидом водорода и трифторуксусный ангидрид.
Предпочтительное количество окислителя находится в интервале эквимолярных количеств до 20-кратного избытка относительно пиридинового производного. Предпочтительно количество окислителя находится в интервале от 1,2-кратного до 10-кратного избытка, более предпочтительно от 1,5-кратного до 8-кратного избытка и наиболее предпочтительно от 2-кратного до 5-кратного избытка.
Предпочтительные системы растворителей для проведения окисления представляют собой растворители, которые инертны к окислителю. Особенно предпочтительны галогенированные растворители, такие как дихлорметан, хлороформ, хлорбензол и тетрахлорид углерода, ароматические растворители, такие как бензол и толуол, алканы, такие как циклогексан и гексан, и эфиры, такие как ТГФ, 1,4-диоксан и трет-бутилметиловый эфир.
Подходящие температуры реакции и время реакции зависят от реакционной способности используемого окислителя. Предпочтительно температура реакции находится в диапазоне от 0°С до точки кипения используемой системы растворителей, более предпочтительно в диапазоне 20-100°С и наиболее предпочтительно в диапазоне 40-80°С. Предпочтительно время реакции находится в интервале от 30 минут до 24 часов, более предпочтительно в интервале от 1 часа до 18 часов и наиболее предпочтительно 2-6 часов.
Окисление пиридинового атома азота можно выполнить на любой стадии процесса получения соединений согласно общей формуле I. Предпочтительно окисление проводят до образования соединений формул IIА-IIВ или альтернативно после образования оксадиазольного кольца как в соединениях формул VIA-VIB.
В другом аспекте изобретения соединения формулы НА, ИБ или ИВ получают в ходе взаимодействия соединений общей формулы VIIA, VIIB или VIIB,
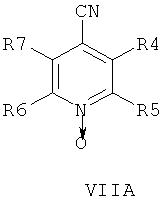
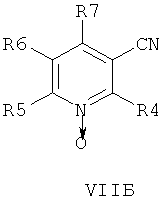
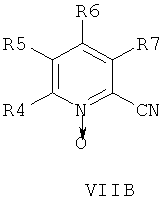
с гидроксиламином в присутствии хелатообразующего агента при подходящих условиях реакции.
В другом аспекте изобретения соединения формулы VA, VB или VB получают в ходе взаимодействия соединений общей формулы VIIIA, V111Б или VIIIB,
с гидроксиламином в присутствии хелатообразующего агента при подходящих условиях реакции.
В настоящем изобретении подходящие реакционные условия вышеприведенных реакций включают условия, которые дают производное амидоксима с высоким выходом и чистотой. Предпочтительно выход требуемого производного амидоксима составляет по меньшей мере 70%, более предпочтительно 72-95%, даже более предпочтительно 75-90% и наиболее предпочтительно 78-85%. Предпочтительно чистота требуемого производного амидоксима составляет по меньшей мере 90%, более предпочтительно по меньшей мере 95%, даже более предпочтительно по меньшей мере 96% и наиболее предпочтительно по меньшей мере 97%. Исходя из предмета настоящего изобретения квалифицированный специалист может обычным путем установить наиболее подходящие реакционные условия, чтобы оптимизировать выход и чистоту амидоксима. Параметры, которые следует учитывать квалифицированному специалисту, включают, но не ограничиваются этим, количество гидроксиламина, выбор катализатора, природу заместителей R4-R7, систему растворителей, температуру реакции и время реакции, и растворимость реагентов.
Предпочтительное количество гидроксиламина находится в интервале эквимолярных количеств до 50-кратного избытка относительно пиридинового производного. Предпочтительно количество гидроксиламина находится в интервале от 1,2-кратного до 20-кратного избытка, более предпочтительно от 1,5-кратного до 10-кратного избытка и наиболее предпочтительно от 3-кратного до 5-кратного избытка.
Предпочтительные хелатообразующие агенты включают 8-гидроксихинолин, орто-фенантролин и их гидраты и производные. Предпочтительное количество хелатообразующего агента находится в интервале 0,1-10 молярных %, более предпочтительно 0,5-5 молярных %, более предпочтительно 0,75-3 молярных % и наиболее предпочтительно 1-1,5 молярных %.
Система растворителей особенно не ограничена и включает воду, спирты, такие как метанол, этанол или изопропанол, эфиры, такие как ТГФ или 1,4-диоксан, и диполярные апротонные растворители, такие как диметилсульфоксид и тому подобные, или смеси этих растворителей.
Предпочтительно температура реакции находится в диапазоне от 0°С до точки кипения используемой системы растворителей, более предпочтительно в диапазоне 20-100°С и наиболее предпочтительно в диапазоне 40-80°С. Предпочтительно время реакции находится в интервале от 30 минут до 24 часов, более предпочтительно в интервале от 1 часа до 18 часов и наиболее предпочтительно 2-8 часов.
Биологическая доступность, биологическая активность, профиль безопасности и другие родственные свойства, известные в данной области техники (например проницаемость гематоэнцефалического барьера), соединений общей формулы I могут быть обычным способом оптимизированы квалифицированным специалистом на основе предмета настоящей заявки, изменяя заместители R1-R7 вышеприведенной общей формулы I, чтобы получить требуемую сбалансированную смесь свойств.
Соединения общей формулы I также могут находиться в форме их фармакологически приемлемых солей. Подходящие фармацевтически приемлемые противоионы известны в данной области техники.
Также возможно использовать пролекарства соединений общей формулы I, чтобы преобразовать терапевтический профиль активного соединения.
Материалы и способы
Анализ активности КОМТ
Печень самцов крыс Уистара в возрасте 60 дней и весом 240 - 260 г (Harlan-Interfauna Iberica, Барселона, Испания), содержащихся по две в клетке при регулируемых условиях окружающей среды (12 часовой цикл день / ночь и комнатная температура 24°С), использовали во всех экспериментах. После декапитации органы немедленно удаляли и гомогенизировали в 5 мМ фосфатном буфере с рН 7,8. Активность КОМТ оценивали по способности метилировать адреналин до метанефрина. Аликвоты по 0,5 мл гомогенатов печени предварительно инкубировали в течение 20 минут с 0,4 мл фосфатного буфера (5 мМ); затем реакционную смесь инкубировали в течение 15 минут с эпинефрином (2000 мкМ; 0,1 мл) в присутствии насыщенной концентрации 3-аденозил-L-метионина (500 мкМ), метильного донора; инкубационная среда также содержала паргилин (100 мкМ), MgCl2 (100 мкМ) и ЭГТА (1 мМ). Предварительную инкубацию и инкубацию проводили при 37°С в условиях защиты от света с непрерывным встряхиванием и без насыщения кислородом.
В экспериментах, предназначенных для оценки биологической доступности исследуемых соединений при пероральном введении, соединения по желудочному зонду вводили голодным с вечера крысам. Затем в определенные промежутки времени животных убивали декапитацией, печень удаляли и использовали для определения активности КОМТ, как описано выше. В конце периода инкубации (5 минут) пробирки переносили в лед и реакцию останавливали, добавляя 200 мкл 2 М перхлорной кислоты. Затем образцы центрифугировали (200 х д, 4 минуты, 4°С) и 500 мкл аликвот супернатанта, фильтрованного через фильтровальные трубки Spin-X с размером пор 0,22 мкм (Costar), использовали для анализа метанефрина. Анализ метанефрина проводили посредством высокоэффективной жидкостной хроматографии с электрохимическим детектированием. Нижние пределы детектирования метанефрина составляли от 350 до 500 фмоль (0,5 до 1,0 пмоль/мг белка/час).
Уровни L-ДОПА и его производных в целом головном мозге и плазме
Голодным с вечера крысам вводили перорально толкапон и соединения общей формулы I (3 мг/кг) или наполнитель (0,5% карбоксиметилцеллюлозу, 4 мл/кг). Спустя один, 6 или 23 часа крысам вводили перорально L-ДОПА (12 мг/кг) плюс бенсеразид (3 мг/кг) или наполнитель (0,5% карбоксиметилцеллюлозу, 4 мл/кг). Через один час крысам делали анестезию с пентобарбитоном натрия (60 мг/кг, интраперитонеально), кровь отбирали из полой вены и весь головной мозг быстро удаляли. Головной мозг хранили в 0,2 М перхлорной кислоте для последующего анализа L-ДОПА, 3-0-метил-L-ДОПА, дофамина, ДОФУК (3,4-дигидроксифенилуксусной кислоты) и ГВК (гомованилиновой кислоты). Пробы крови центрифугировали в течение 15 минут при 3000 g (4°С) и образцы плазмы хранили при -80°С до анализа L-ДОПА и 3-0-метил-L-ДОПА. Все эксперименты с животными осуществляли согласно Европейской Директиве номер 86/609 и правилам «Руководства по уходу и использованию лабораторных животных» (Guide for the Care and Use of Laboratory Animals), 7-е издание, 1996, Institute for Laboratory Animal Research (ILAR), Washington, DC.
Анализ L-ДОПА и производных катехола
L-ДОПА, 3-0-метил-L-ДОПА, дофамин и метаболиты (ДОФУК и ГВК) в диализированных пробах анализировали с помощью ВЭЖХ с электрохимическим детектированием, как описано прежде (Soares-da-Silva et al., Brain Res. 2000; 863: 293-297). Кратко аликвоты по 20 мкл вводили в хроматограф. Хроматографическая система состояла из насоса (Gilson 307) и 5 мкм колонки ODS2 из нержавеющей стали (Biophase; Bioanalytical Systems, West Lafayette, IN) длиной 25 см и диаметром 4,6 мм; пробы вводили посредством автоматического дозатора образца (Gilson 231), подключенного к дилютору Gilson (Gilson 401). Подвижная фаза представляла собой дегазированный раствор 0,1 мМ лимонной кислоты; 0,5 мМ октилсульфата натрия; 0,1 М ацетата натрия; 0,17 мМ Nа2ЭДТА; 1 мМ дибутиламина и метанола (10% об/об), доведенный до рН 3,5 с 2 М РСА (пирролидонкарбоновой кислотой) и подаваемый насосом со скоростью 1,0 мл мин'1. Детектирование проводили электрохимически со стеклоуглеродным электродом, электродом сравнения Ag/AgCl и амперометрическим детектором (Gilson 142); ячейкой детектора управляли при 0,75 В. Созданный ток обрабатывали, используя программное обеспечение для ВЭЖХ Gilson Unipoint. Нижний предел детектирования дофамина, ДОФУК и ГВК составлял от 350 до 1000 фмоль.
Описание фигур
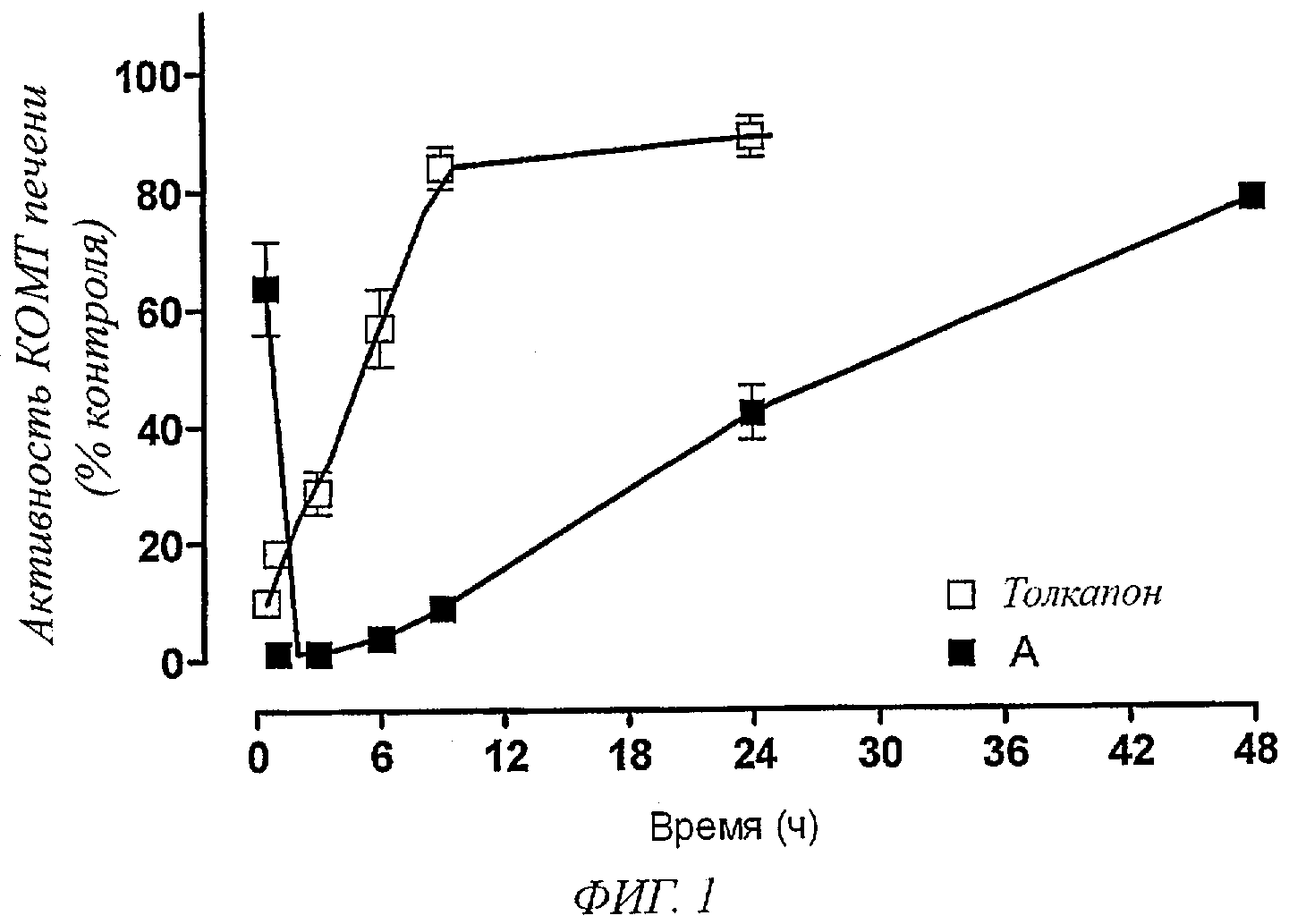
Фигура 1. Влияние соединения А и толкапона (3 мг/кг) на активность КОМТ печени через 0,5, 1, 3, 6, 9, 24 и 48 часов после введения ингибитора КОМТ. Символы представляют собой значения ±SEM (стандартная погрешность среднего) 5 экспериментов на группу. Значительно отличаются от соответствующих контрольных значений (*Р<0,05).
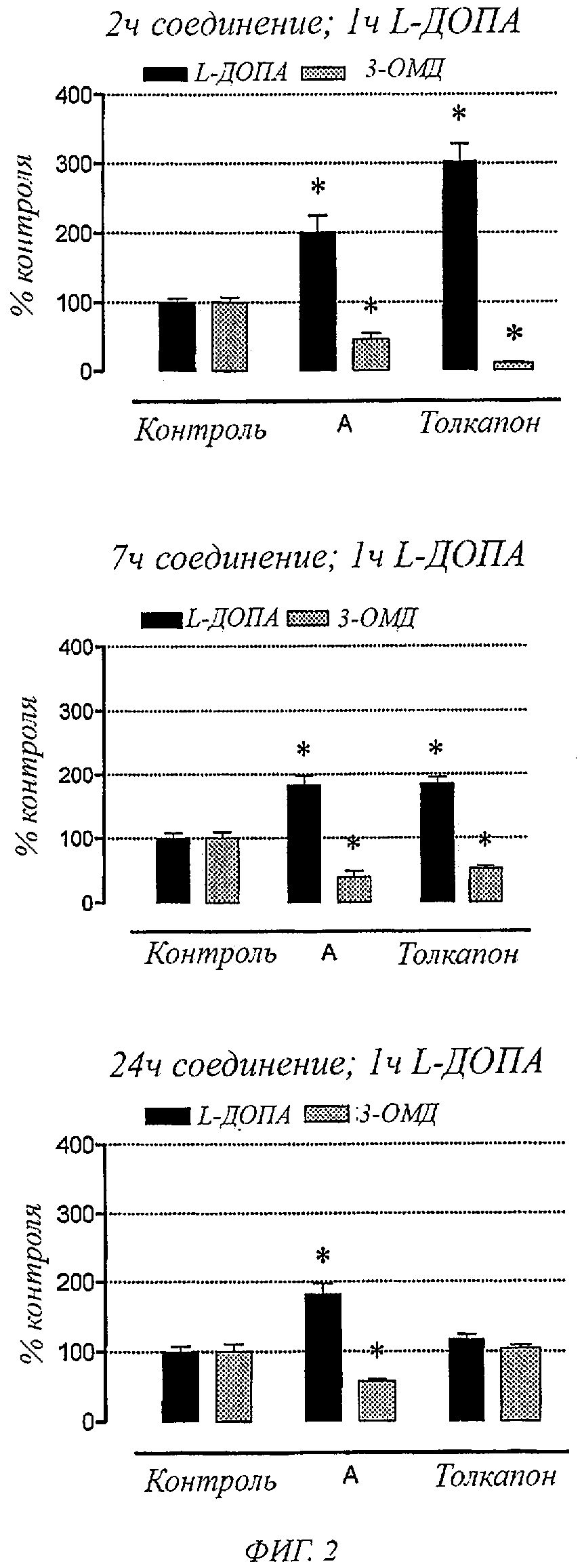
Фигура 2. Влияние соединения А и толкапона (3 мг/кг) на уровни в плазме L-ДОПА и 3-0-метил-b-ДОПА у крыс, обработанных с L-ДОПА (12 мг/кг) плюс бенсеразид (3 мг/кг), через 2, 7 и 24 часа после введения ингибитора КОМТ. В колонках представлены значения±SEM 5 экспериментов на группу. Значительно отличаются от соответствующих контрольных значений (*Р<0,05).
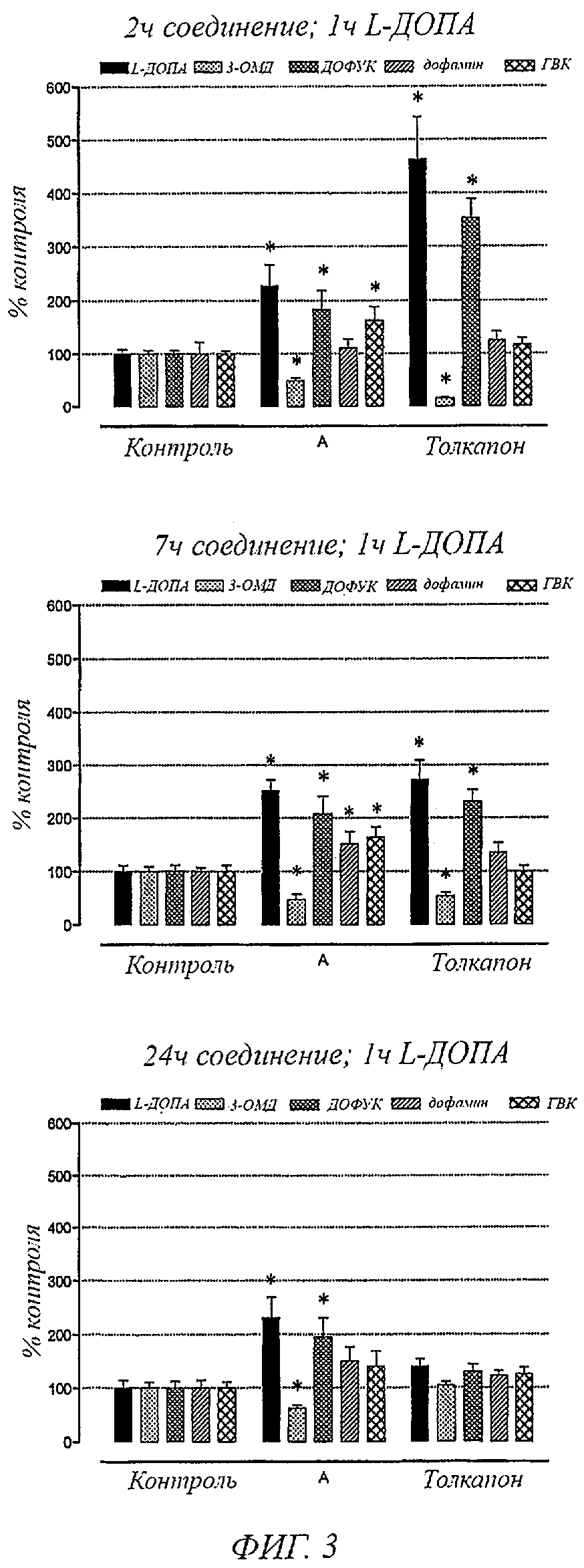
Фигура 3. Влияние соединения А и толкапона (3 мг/кг) на уровни в головном мозге L-ДОПА, 3-0-метил-L-ДОПА, дофамина, ДОФУК и ГВК у крыс, обработанных с L-ДОПА (12 мг/кг) плюс бенсеразид (3 мг/кг), через 2, 7 и 24 часа после введения ингибитора КОМТ. В колонках представлены значения ±SEM 5 экспериментов на группу. Значительно отличаются от соответствующих контрольных значений (*Р<0,05).
Результаты
Было установлено, что соединения общей формулы I, например соединение А, являются эффективными ингибиторами КОМТ печени, максимальный ингибирующий эффект достигается в течение 60 минут после их перорального введения (Фигура 1). Максимальный ингибирующий эффект толкапона наблюдался в пределах 30 минут после введения (Фигура 1). Через девять часов после введения толкапон проявляет минимальные ингибирующие эффекты, тогда как соединения общей формулы I, например соединение А, продолжают ингибировать активность КОМТ на 90% контрольных уровней (Фигура 1). Как показано на Фигуре 1, через 24 часа после введения соединения общей формулы I, например соединение А, способны ингибировать КОМТ печени на 60% контрольных уровней, тогда как толкапон опять почти лишен свойств ингибирования КОМТ.
Фигура 2 показывает уровни L-ДОПА и 3-0-метил-L-ДОПА в плазме крыс, обработанных с L-ДОПА плюс бенсеразид, через 2, 7 и 24 часа после введения толкапона и соединений общей формулы I, например соединения А, (3 мг/кг). L-ДОПА плюс бенсеразид вводили за 1 час до сбора проб крови. Данный момент времени был выбран, так как он представлял собой Тмакс для L-ДОПА. Как можно заметить, соединения общей формулы I, например соединение А, вызывали значительные увеличения L-ДОПА в плазме, сопровождаемые заметным уменьшением в циркуляции 3-0-метил-L-ДОПА, что было одинаковым для всех случаев предварительных обработок с соединениями общей формулы I, например соединением А, (1, 7 и 24 часа). Через 24 часа введение толкапона не влияло на уровни L-ДОПА и 3-0-метил-L-ДОПА в плазме. Значительные изменения уровней L-ДОПА и 3-0-метил-L-ДОПА в плазме под действием толкапона наблюдались только в короткие промежутки времени через 2 и 7 часов после введения соединения.
Фигура 3 показывает уровни L-ДОПА, 3-0-метил-L-ДОПА, ДОФУК, дофамина и ГВК в головном мозге крыс, обработанных с L-ДОПА плюс бенсеразид, через 2, 7 и 24 часа после введения толкапона и соединений общей формулы I, например соединения А, (3 мг/кг). L-ДОПА плюс бенсеразид вводили за 1 час до сбора проб головного мозга. Данный момент времени был выбран, так как он представлял собой Тмакс для L-ДОПА. Как можно заметить, соединения общей формулы I, например соединение А, вызывали значительные увеличения L-ДОПА, дофамина и ДОФУК в головном мозге, сопровождаемые заметным уменьшением 3-0-метил-L-ДОПА в головном мозге, что было одинаковым для всех случаев предварительных обработок с соединениями общей формулы I, например соединением А, (1, 7 и 24 часа). Через 24 часа введение толкапона не влияло на уровни L-ДОПА, дофамина, ДОФУК и 3-0-метил-L-ДОПА в головном мозге. Значительные изменения уровней L-ДОПА, дофамина, ДОФУК и 3-0-метил-L-ДОПА в головном мозге под действием толкапона наблюдались только через 2 и 7 часов после введения соединения.
Изобретение будет описано со ссылкой на следующий пример получения, который, как подразумевается, не ограничивают объем изобретения каким-либо образом.
Пример 1 - Получение соединения А
(5-[3-(2,5-Дихлор-4,6-диметил-1-окси-пиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол)
а) К перемешиваемому раствору 3,4-дибензилокси-5-нитробензойной кислоты (0,50 г, 1,318 ммоль) в диметилформамиде (5 мл) при комнатной температуре добавляли 1,1-карбонилдиимидазол (0,24 г, 1,48 ммоль) за один раз. После перемешивания в течение девяноста минут 2,5-дихлор-N'-гидрокси-4,6-диметилникотинимидамид (0,40 г, 1,71 ммоль) добавляли за один раз. Полученную в результате смесь перемешивали при 135°С в течение пяти часов и затем при комнатной температуре в течение ночи. Реакционную смесь выливали в лед - 2 н. HCl (100 мл) и полученный в результате осадок отфильтровывали, промывали водой и высушивали на воздухе. Перекристаллизация из изопропанола давала бледно-желтое твердое вещество (0,55 г, 72%).
б) К перемешиваемому раствору полученного выше твердого вещества (0,50 г, 0,866 ммоль) в дихлорметане (20 мл) добавляли комплекс присоединения мочевины и пероксида водорода (0,41 г, 4,36 ммоль) за один раз. Смесь охлаждали на бане с ледяной водой и по каплям добавляли трифторуксусный ангидрид (0,73 г, 3,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего отфильтровывали нерастворимое вещество. Фильтрат промывали водой и солевым раствором, высушивали над безводным сульфатом магния, фильтровали и выпаривали. Остаток перекристаллизовывали из изопропанола, что давало бледно-желтое твердое вещество (0,35 г, 68%).
в) К перемешиваемому раствору полученного выше твердого вещества (0,30 г, 0,5 ммоль) в дихлорметане (10 мл) при -78°С в атмосфере аргона добавляли трибромид бора (0,38 г, 1,5 ммоль) по каплям. Полученную в результате пурпурную суспензию перемешивали при комнатной температуре в течение одного часа, затем опять охлаждали до -78°С и осторожно гасили, добавляя воду. После перемешивания при комнатной температуре в течение одного часа осадок отфильтровывали, промывали водой и высушивали при 50°С в вакууме, что давало требуемое соединение в виде желтых кристаллов (0,18 г, 86%), Тпл 237-240°С.
Пример 2 - Фармацевтическая композиция
Подходящие характерные фармацевтические препараты получают согласно следующим спецификациям:
Капсула:
Соединение А 15,0% Моногидрат лактозы 43,0% Микрокристаллическая целлюлоза 30,0% Повидон 4,0% Кроскармеллоза натрия 5,0% Тальк 2,0% Стеарат магния 1,0%
Капсула;
Соединение А 15,0% Микрокристаллическая целлюлоза 72,5% Этилцеллюлоза 5,0% Натрия крахмала гликолят 6,0% Коллоидный диоксид кремния 0,5% Стеарат магния 1,0%
Таблетка:
Соединение А 20,0% Микрокристаллическая целлюлоза 25,0% Гидрофосфат кальция, дигидрат 40,0% Повидон 6,0% Кроскармеллоза натрия 6,0% Тальк 2,0% Стеарат магния 1,0%
Пример 3 - Схема приема
Больных, которые страдают двигательным расстройством и которые проходят L-ДОПА терапию, лечат таблетками, содержащими 50 мг соединения общей формулы I. Подтверждается значительное улучшение клинической картины.
Фармацевтические соединения
Режим введения нитрокатехолов
Режим введения для нитрокатехолов
Фармацевтические соединения
Режим введения нитрокатехолов
Режим введения для нитрокатехолов
Имидазолкарбоксамиды и их применение в качестве ингибиторов гидролазы амидов жирных кислот
Соединения мочевины и их применение в качестве ингибиторов ферментов

