Результат интеллектуальной деятельности: ПОГЛОТИТЕЛИ УФ/ВИДИМОГО СВЕТА ДЛЯ МАТЕРИАЛОВ ОФТАЛЬМОЛОГИЧЕСКИХ ЛИНЗ
Вид РИД
Изобретение
Область, к которой относится изобретение
Данное изобретение направлено на поглотители УФ/видимого света. В частности, данное изобретение относится к новым мономерам бензотриазола, особенно подходящим для использования в материалах имплантируемых офтальмологических линз.
Уровень техники
В качестве ингредиентов для полимерных материалов, используемых для изготовления офтальмологических линз, известны многие поглотители ультрафиолетового и видимого света. Такие поглотители, предпочтительно, ковалентно связаны с полимерной сеткой материала линзы, вместо простого физического включения в материал, для предотвращения их переноса, фазового разделения или вымывания из материала линзы. Такая стабильность является особенно важной для имплантируемых офтальмологических линз, где вымывание поглотителя может представлять как токсикологические проблемы, так и вести к потере активности блокирования УФ/видимого света в имплантате.
Известны многочисленные сополимеризующиеся поглотители на основе бензотриазола, бензофенона и триазина. Большинство из данных соединений известны как УФ-поглотители, хотя некоторые, как может быть известно, также поглощают некоторую часть видимого света. Многие поглотители содержат обычные олефиновые полимеризующиеся группы, такие как метакрилатные, акрилатные, метакриламидные, акриламидные или стирольные группы. Сополимеризация с другими ингредиентами в материалах линз, обычно с помощью радикального инициатора, включает поглотители в получаемую полимерную цепь. Включение дополнительных функциональных групп в поглотитель может повлиять на одно или несколько светопоглощающих свойств поглотителя, растворимость или реакционную способность. Если поглотитель не имеет достаточной растворимости в остальной части ингредиентов материала офтальмологической линзы или полимерного материала линзы, поглотитель может сращиваться в домены, которые могут взаимодействовать со светом и в результате приводить к сниженной оптической прозрачности линзы.
Примеры материалов полимерных офтальмологических линз, которые включают поглотители УФ света, можно найти в патентах США 5290892, 5331073 и 5693095.
Сущность изобретения
Настоящее изобретение предлагает поглощающие свет мономеры бензотриазола, которые поглощают как ультрафиолетовый свет, так и часть видимого света (″поглотители УФ/видимого света″). Данные поглотители подходят для использования в офтальмологических линзах, включая контактные линзы. Они особенно применимы в имплантируемых линзах, таких как интраокулярные линзы (IOLs).
Соединения-поглотители по настоящему изобретению поглощают свет с длинами волн между 400-450 нм в добавление к более высокоэнергетичным UVA лучам (ультрафиолетовым лучам спектра А) между 400-320 нм, UVB лучам (ультрафиолетовым лучам спектра Б) между 320-280 нм и UVC лучам (ультрафиолетовым лучам спектра С) ниже 280 нм. Они содержат реакционноспособные группы, которые дают возможность ковалентного присоединения поглотителей к материалам офтальмологических линз. Кроме того, поглотители по настоящему изобретению можно синтезировать приблизительно за 5 стадий из легко доступных исходных веществ.
Краткое описание чертежей
Фиг.1 показывает кривые процента пропускания для соединений-поглотителей УФ/видимого света WL-1-WL-7.
Фиг.2A-2J показывают кривые процента пропускания для материалов IOL, содержащих соединения-поглотители УФ/видимого света WL-1-WL-4, которые были подвергнуты тестированию светоустойчивости, давая эквивалент 10 или 20 лет воздействия света.
Подробное описание изобретения
Если не указано иным образом, все количества ингредиентов, выраженные в процентах, представлены в виде % масс./масс.
Поглотители УФ/видимого света по настоящему изобретению представлены формулой

в которой
R1=H, CH3, CH2CH3 или CH2OH;
R2=C1-C4 алкил или C1-C4 алкокси; и
R3=H, CH3, CH3O, F, Cl, Br, I или CF3.
Предпочтительные поглотители УФ/видимого света по настоящему изобретению представляют собой соединения, в которых R1=H или CH3, R2=C1-C4 алкокси и R3=H, CH3, CH3O, F, Cl или CF3.
Более предпочтительные поглотители по настоящему изобретению выбраны из группы, состоящей из:
2-гидрокси-5-метокси-3-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата;
3-(5-фтор-2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата;
3-(2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата;
3-(5-хлор-2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата;
2-гидрокси-5-метокси-3-(5-метокси-2H-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата;
2-гидрокси-5-метокси-3-(5-метил-2H-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата; и
2-гидрокси-5-метил-3-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата.
Наиболее предпочтительными поглотителями УФ/видимого света по настоящему изобретению являются 2-гидрокси-5-метокси-3-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)бензилметакрилат и 3-(5-хлор-2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилат.
Синтез поглотителей УФ/видимого света по настоящему изобретению описывается ниже.
Стадия 1: Производное фенола 1 синтезируют посредством гидроксиметилирования п-метоксифенола, как показано ниже.

На стадиях 2 и 3 синтезируют соль диазония производного 2-нитроанилина и затем осуществляют ее взаимодействие с 1, получая азокраситель.

На стадии 4 азокраситель обрабатывают восстановителем, например, формамидинсульфиновой кислотой, получая соответствующее соединение бензотриазола. Чистоту выделенного соединения бензотриазола можно повысить методиками, известными из уровня техники, включая отделение фильтрованием избытка восстановителя и побочных продуктов восстановителя перед добавлением протонных кислот и колоночную хроматографию.

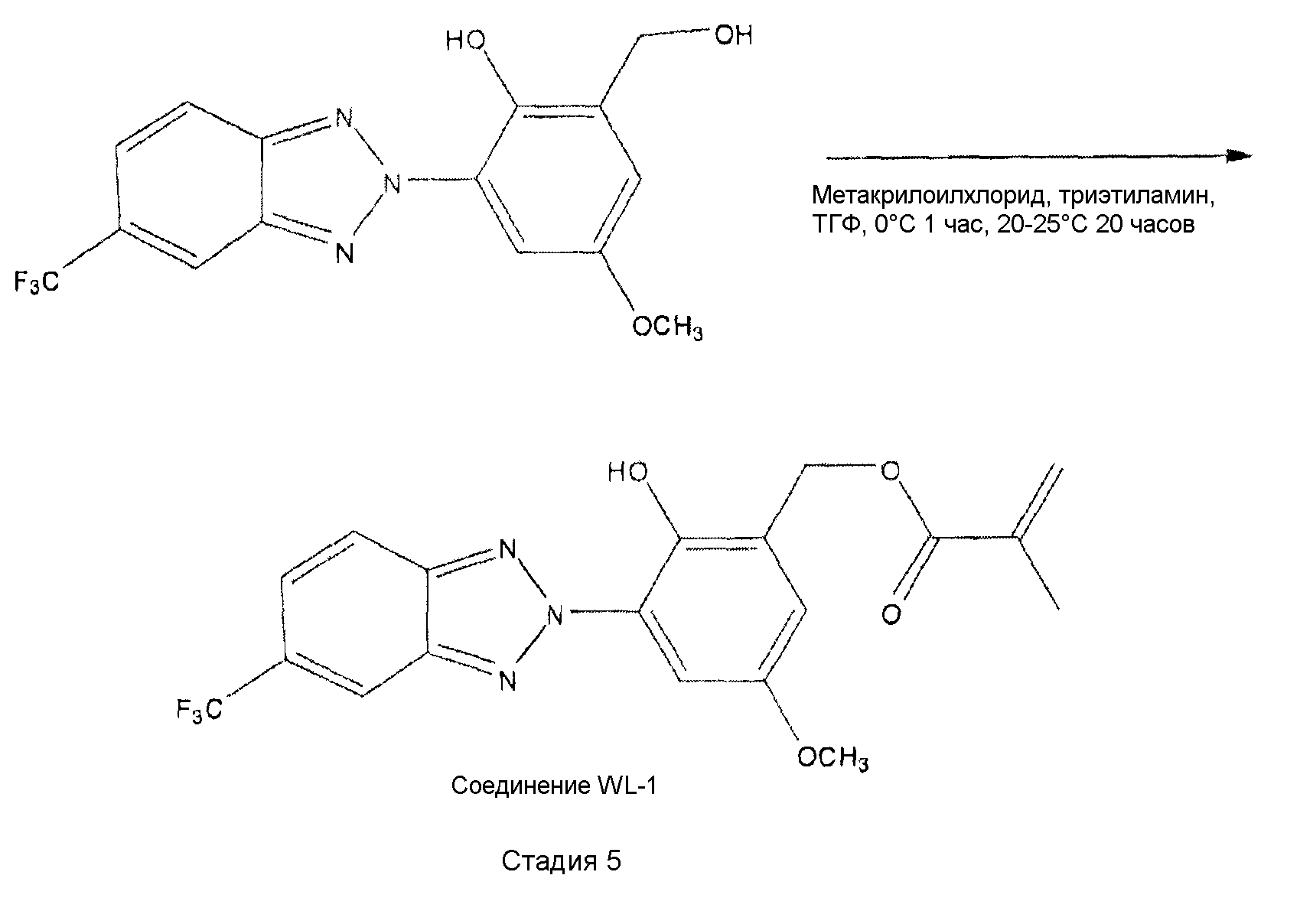
На стадии 5 бензотриазол из стадии 4 эстерифицируют, получая ″реакционноспособное″ соединение, которое содержит винильную группу. Под ″реакционноспособным″ соединением понимают, что винильная группа может полимеризоваться с образованием ковалентных связей при взаимодействии с виниловыми мономерами, со-мономерами, макромерами, сшивающими агентами и другими компонентами, обычно используемыми при изготовлении офтальмологических материалов на основе полимеров, особенно акриловых. Реакционноспособные группы, предпочтительно, представляют собой акрилатные или метакрилатные группы.
Поглотители УФ/видимого света по настоящему изобретению особенно подходят для использования в IOLs. Материалы IOL обычно будут содержать от 0,1 до 3% (масс./масс.) поглотителя УФ/видимого света по настоящему изобретению. Предпочтительно, материалы IOL будут содержать от 0,2 до 2,5% (масс./масс.) поглотителя по настоящему изобретению. Наиболее предпочтительно, материалы IOL будут содержать от 0,3 до 2% (масс./масс.) поглотителя по настоящему изобретению. Такие материалы устройства готовят сополимеризацией поглотителей по настоящему изобретению с другими ингредиентами, такими как формирующие устройство материалы, сшивающие агенты и, необязательно, хромофоры, блокирующие синий свет.
Многие мономеры, формирующие устройство, известны из уровня техники, и включают как акриловые, так и кремнийсодержащие мономеры, среди прочего. Смотри, например, патенты США № 7101949, 7067602, 7037954, 6872793, 6852793, 6846897, 6806337, 6528602 и 5693095. В случае IOLs, любой известный материал устройства IOL подходит для использования в композициях по настоящему изобретению. Предпочтительно, материалы офтальмологического устройства включают акриловый или метакриловый мономер, формирующий устройство. Более предпочтительно, формирующие устройство мономеры включают мономер формулы IV:

где в формуле IV:
A представляет собой H, CH3, CH2CH3 или CH2OH;
B представляет собой (CH2)m или [O(CH2)2]z;
C представляет собой (CH2)w;
m равно 2-6;
z равно 1-10;
Y представляет собой прямую связь либо O, S или NR' при условии, что, если Y представляет собой O, S или NR', то B представляет собой (CH2)m;
R' представляет собой H, CH3, Cn'H2n'+1 (n'=1-10), изо-OC3H7, C6H5 или CH2C6H5;
w равно 0-6 при условии, что m+w≤8; и
D представляет собой H, C1-C4 алкил, C1-C4 алкокси, C6H5, CH2C6H5 или галоген.
Предпочтительными мономерами формулы IV являются мономеры, в которых A представляет собой H или CH3, B представляет собой (CH2)m, m равно 2-5, Y представляет собой прямую связь или O, w равно 0-1 и D представляет собой H. Наиболее предпочтительными являются 2-фенилэтилметакрилат, 4-фенилбутилметакрилат, 5-фенилпентилметакрилат, 2-бензилоксиэтилметакрилат, 3-бензилоксипропилметакрилат и их соответствующие акрилаты.
Мономеры формулы IV известны, и их можно изготовить известными методами. Например, сопряженный спирт желаемого мономера можно объединить в реакционном сосуде с метилметакрилатом, тетрабутилтитанатом (катализатор) и ингибитором полимеризации, таким как 4-бензилоксифенол. Затем сосуд можно нагреть, чтобы содействовать реакции, и отогнать побочные продукты реакции, чтобы довести реакцию до завершения. Альтернативные схемы синтеза включают добавление метакриловой кислоты к сопряженному спирту и катализ карбодиимидом, или смешивание сопряженного спирта с метакрилоилхлоридом и основанием, таким как пиридин или триэтиламин.
Материалы устройства, как правило, включают всего, по меньшей мере, примерно 75%, предпочтительно, по меньшей мере, примерно 80% формирующих устройство мономеров.
Кроме поглотителя по настоящему изобретению и формирующего устройство мономера материалы устройства по настоящему изобретению обычно включают сшивающий агент. Сшивающий агент, используемый в материалах устройства согласно настоящему изобретению, может представлять собой любое ненасыщенное соединение с концевой этиленовой группой, имеющее более одной ненасыщенной группы. Подходящие сшивающие агенты включают, например, этиленгликольдиметакрилат, диэтиленгликольдиметакрилат, аллилметакрилат, 1,3-пропандиолдиметакрилат, 2,3-пропандиолдиметакрилат, 1,6-гександиолдиметакрилат, 1,4-бутандиолдиметакрилат, CH2=C(CH3)C(=O)O-(CH2CH2O)p-C(=O)C(CH3)=CH2, где p=1-50, и CH2=C(CH3)C(=O)O(CH2)tO-C(=O)C(CH3)=CH2, где t=3-20, и их соответствующие акрилаты. Предпочтительным сшивающим мономером является CH2=C(CH3)C(=O)O-(CH2CH2O)p-C(=O)C(CH3)=CH2, где p является таким, что среднечисленная молекулярная масса составляет примерно 400, примерно 600 или примерно 1000.
Как правило, общее количество сшивающего компонента составляет, по меньшей мере, 0,1% по массе и, в зависимости от особенностей и концентрации остальных компонентов и желаемых физических свойств, может находиться в диапазоне примерно до 20% по массе. Предпочтительный диапазон концентраций для сшивающего компонента составляет 1-5% для небольших, гидрофобных соединений с молекулярной массой в типичном случае менее чем 500 Дальтон, и 5-17% (масс./масс.) для более крупных гидрофильных соединений с молекулярными массами в типичном случае между 500 и 5000 Дальтон.
Подходящие инициаторы полимеризации для материалов устройства, содержащих поглотитель УФ/видимого света по настоящему изобретению, включают термические инициаторы и фотоинициаторы. Предпочтительные термические инициаторы включают пероксидные свободно-радикальные инициаторы, такие как трет-бутил(перокси-2-этил)гексаноат и ди(трет-бутилциклогексил)пероксидикарбонат (имеющийся в продаже в виде Perkadox® 16 от Akzo Chemicals Inc., Чикаго, Иллинойс). Инициаторы обычно присутствуют в количестве примерно 5% (масс./масс.) или менее. Поскольку свободно-радикальные инициаторы не становятся химически частью получаемого полимера, общее количество инициатора обычно не включается при определении количества других ингредиентов.
Материалы устройства, содержащие поглотитель УФ/видимого света по настоящему изобретению, необязательно также содержат реакционноспособное красящее вещество. Подходящие реакционноспособные соединения, поглощающие синий свет, включают соединения, описанные в патенте США № 5470932. Поглотители синего света обычно присутствуют в количестве примерно 0,01-0,5% (массовых).
IOLs, созданные из материалов по настоящему изобретению, могут быть любого исполнения, при котором их можно сворачивать или сгибать в профиль небольшого размера, который можно приспособить через относительно небольшой надрез. Например, IOLs могут иметь так называемую монолитную или сложную составную конструкцию и включать оптический и гаптический компонент. Оптический компонент представляет собой ту часть, которая служит в качестве линзы. Гаптический компонент присоединяется к оптическому и удерживает оптический компонент в надлежащем месте в глазу. Оптический и гаптический компоненты могут состоять из одного и того же материала или из различных материалов. Линзы называют сложными составными, когда оптический и гаптический компоненты изготавливают по отдельности и затем гаптический компонент присоединяют к оптическому. В монолитных линзах оптический и гаптический компонент формируют из одного куска материала. В зависимости от материала, гаптические компоненты затем вырезают, или вытачивают, из данного материала, получая IOL.
Кроме IOLs, материалы по настоящему изобретению также могут подходить для использования в других офтальмологических устройствах, таких как контактные линзы, кератопротезы и роговичные имплантаты или кольца.
Далее изобретение будет иллюстрировано следующими ниже примерами, которые предназначены быть иллюстративными, а не ограничивающими.
ПРИМЕР 1
Синтез (2-гидрокси-5-метокси-1,3-фенилен)диметанола. В 2 литровой реакционной колбе, оборудованной магнитной мешалкой, суспендировали 200 г п-метоксифенола в 1300 мл воды. К перемешиваемому раствору добавляли раствор формальдегида (335 мл, 37% в воде), после чего следовало добавление 55,8 г оксида кальция. Реакционную смесь накрывали алюминиевой фольгой и выдерживали при комнатной температуре в течение 10 дней. Твердое вещество отфильтровывали и затем суспендировали в 1 л деионизированной воды. К перемешиваемой суспензии при комнатной температуре добавляли 130 мл ледяной уксусной кислоты. Серовато-белое твердое вещество отфильтровывали, промывали гексаном и затем сушили под высоким вакуумом в течение 72 часов, получая 118,9 г (37,8%). 1H ЯМР (ДМФА-D7) дельта: 3,75 (с, 3H, OCH3), 4,73 (с, 4H, CH2O), 5,39 (ушир. с, 2H, CH2OH), 6,85 (с, 2H, Ar-H). 13C ЯМР (ДМФА-D7) дельта: 55,12 (1С, ОСН3), 59,84 (2C, CH2OH), 111,18 (2C, Ar-CH), 129,51 (2C, Ar-CCH2), 146,30 (1С, Ar-COH), 152,95 (1С, Ar-COCH3).

ПРИМЕР 2
Синтез 2-(гидроксиметил)-4-метокси-6-((2-нитро-4-(трифторметил)фенил)диазенил)фенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 19,9 г 2-нитро-4-(трифторметил)анилина (96,5 ммоль) (Aldrich, Милуоки, Висконсин), 41 мл концентрированной HCl (водн.), 100 мл деионизированной воды, 100 мл этанола и 80 мл ТГФ. Суспензию перемешивали в течение 30 минут и в течение 60 минут по каплям добавляли 7,07 г нитрита натрия (102 ммоль) в 30 мл воды, поддерживая температуру реакционной смеси при 0°C. Реакционную смесь перемешивали в течение дополнительного 1 часа. Добавляли 300 мг сульфаминовой кислоты, чтобы разрушить избыток нитрита. Смесь фильтровали, чтобы удалить нерастворенное твердое вещество. Отфильтрованную смесь диазония вместе с раствором гидроксида натрия (15,1 г в 100 мл воды) по каплям добавляли к раствору, содержащему 19,7 г (2-гидрокси-5-метокси-1,3-фенилен)диметанола (107 ммоль), 4,3 гидроксида натрия, 200 мл деионизированной воды и 50 мл ТГФ при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 4 часов. Смесь выливали в 3 л воды. Смесь подкисляли до рН 3-4. Темный неочищенный продукт отфильтровывали и промывали несколькими литрами воды, получая темное твердое вещество, которое сушили под высоким вакуумом при 55°C в течение 72 часов, получая 15,2 г (выход 42%) продукта.
Синтез 2-(гидроксиметил)-4-метокси-6-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)фенола. В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 7,70 г (20,7 ммоль) 2-(гидроксиметил)-4-метокси-6-((2-нитро-4-(трифторметил)фенил)диазенил)фенола из примера 2, часть 1, 25 мл деионизированной воды, 1,85 г гидроксида натрия и 80 мл 1-пропанола. Смесь нагревали до 80°C и медленно добавляли 6,55 г (60,6 ммоль) формамидинсульфиновой кислоты (Aldrich) одновременно с раствором 3,0 г NaOH в 50 мл деионизированной воды. Реакционную смесь нагревали при 80°C в течение 2 часов. Реакционную смесь охлаждали при -20°C в течение 2 часов и фильтровали. Твердое вещество растворяли в 2,5 л деионизированной воды, содержащей 4 грамма NaOH. рН корректировали до 2,0, используя 1н. HCl. Полученное в результате твердое вещество отфильтровывали, промывали обильным количеством деионизированной воды, фильтровали и сушили, получая 2,2 г (31%) желтого твердого вещества. 1H ЯМР (CDCl3) дельта: 11,03 (с, 1H, Ar-OH), 8,30 (с, 1H, Ar-H бензотриазол 4-положение), 8,07 (д, 1H, Ar-H бензотриазол 6-положение), 7,88 (с, 1H, Ar-H фенол), 7,69 (д, 1H, Ar-H бензотриазол), 7,08 (с, 1H, Ar-H фенол), 4,84 (с, 2H, Ar-CH2), 3,90 (с, 3H, Ar-OCH3).
ПРИМЕР 3 (Соединение WL-1)
Синтез 2-гидрокси-5-метокси-3-(5-(трифторметил)-2Н-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата. В 250 мл круглодонной 3-горлой колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 1,15 г (3,39 ммоль) 2-(гидроксиметил)-4-метокси-6-(5-(трифторметил)-2Н-бензо[d][1,2,3]триазол-2-ил)фенола из примера 2 в 50 мл безводного ТГФ, содержащего ингибитор ВНТ (Aldrich). Добавляли триэтиламин (1,4 мл, 11 ммоль) и смесь охлаждали до -10°C. По каплям добавляли метакрилоилхлорид (0,436 г, 4,17 ммоль) и смесь перемешивали в течение 1 часа при -10°C, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Твердое вещество отфильтровывали и промывали 100 мл диэтилового эфира. Фильтрат выливали в 100 мл диэтилового эфира и промывали 0,5н HCl и водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя, получая желаемый продукт в виде темно-желтого маслянистого вещества, которое перекристаллизовывали из метанола, получая 0,35 г продукта (25%). [М+Н+]=408,1. 1H ЯМР (CDCl3) дельта: 10,97 (с, 1Н, Ar-OH), 8,30 (с, 1Н, Аг-Н бензотриазол), 8,07 (д, 1Н, Аг-Н бензотриазол), 7,91 (с, 1Н, Ar-H фенол), 7,69 (д, 1Н, Ar-Н бензотриазол), 7,10 (с, 1Н, Ar-Н фенол), 6,21 (с, 1Н, С=С-Н транс), 5,62 (с, 1Н, С=С-Н цис), 5,40 (с, 2Н, Ar-СН2), 3,90 (с, 3Н, Ar-ОСН3), 2,01 (с, 3Н, С=С-СН3). Элементный анализ: вычислено %С (56,02), %Н (3,96), %N (10,32), %F (13,99); найдено %С (56,11), %Н (3,96), %N (10,24), %F (14,37).

ПРИМЕР 4
Синтез 2-((4-фтор-2-нитрофенил)диазенил)-6-(гидроксиметил)-4-метоксифенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 25,1 г (161 ммоль) 4-фтор-2-нитроанилина (Aldrich), HCl (водн.) (J.T. Baker), 100 мл деионизированной воды и 100 мл абсолютного этанола. Суспензию охлаждали до 0°C. В течение 60 минут по каплям добавляли 11,8 г (171 ммоль) нитрита натрия (Sigma-Aldrich) в 50 мл воды, поддерживая температуру реакционной смеси при 0°C. Реакционную смесь перемешивали в течение дополнительного 1 часа. Добавляли 420 мг сульфаминовой кислоты (Aldrich) и реакционную смесь перемешивали в течение дополнительных 30 минут и затем фильтровали. В 1 л круглодонной колбе объединяли 32,5 г (176 ммоль) (2-гидрокси-5-метокси-1,3-фенилен)диметанола, 200 мл деионизированной воды и 200 мл этанола. 32,3 г (807 ммоль) NaOH (Aldrich) растворяли в 100 мл воды и приблизительно одну четвертую часть данного количества добавляли к раствору, содержащему производное фенола при 0°C. Раствор соли диазония и остающийся раствор гидроксида натрия по каплям добавляли к реакционной смеси при 0°C в течение 1 часа и затем перемешивали в течение дополнительного 1 часа при 0°C и в течение 3 часов при комнатной температуре. Реакционную смесь выливали в 3 л воды и pH подкисляли до 4, используя 1н. HCl. Твердое вещество собирали фильтрованием, промывали несколькими литрами воды и сушили под вакуумом (0,1 мм Hg (13,33 Па)) при 55°C в течение 72 часов, используя P2O5 в качестве осушителя, получая 29,9 г (68%) темного твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
Синтез 2-(5-фтор-2Н-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 2-((4-фтор-2-нитрофенил)диазенил)-6-(гидроксиметил)-4-метоксифенол из примера 4, часть I и 200 мл этанола. Гранулы NaOH (21,7 г, 542 ммоль) (97+%, реагент A.C.S., Aldrich) растворяли в 100 мл деионизированной воды и приблизительно одну четвертую часть раствора по каплям добавляли к реакционной смеси. Затем смесь нагревали до 80°C и к реакционной смеси в течение 30 минут одновременно добавляли 29,3 г (271 ммоль) формамидинсульфиновой кислоты (Aldrich) и остающуюся часть раствора гидроксида натрия. Смесь перемешивали при 80°C в течение 3 часов, затем содержимое выливали в 3 л деионизированной воды и подкисляли до pH 4, используя 1н. HCl. Твердое вещество собирали фильтрованием, суспендировали в 300 мл метанола и перемешивали в течение 30 минут, в то же время охлаждая ниже -20°C. Твердое вещество собирали фильтрованием, затем суспендировали в 300 мл этанола, снова охлаждали ниже -20°C, фильтровали и затем сушили, получая 7,8 г (30%) желтого твердого вещества, которое использовали в следующей стадии эстерификации.
ПРИМЕР 5 (Соединение WL-2)
Синтез 3-(5-фтор-2Н-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата. Эстерификацию осуществляли, используя продукт из примера 4, часть II. В 250 мл круглодонной колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 5,06 г (17,5 ммоль) 2-(5-фтор-2Н-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола в 60 мл безводного ТГФ. Добавляли триэтиламин (8,7 мл, 62 ммоль) и смесь охлаждали до -10-0°C. По каплям добавляли метакрилоилхлорид (2,26 г, 21,6 ммоль) и смесь перемешивали в течение 1 часа при -10°C, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Соли отфильтровывали и промывали 100 мл ТГФ (≥99,9%, безводный, содержащий ингибитор, Aldrich). К фильтрату добавляли диэтиловый эфир (100 мл), промывали 1н. HCl и водой, сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя, получая желаемый продукт, который перекристаллизовывали из этанола, получая 2,5 г (40%) желтого твердого вещества. 1H ЯМР (CDCl3) дельта: 11,03 (с, 1Н, фенол ОН), 7,94 (м, 1Н, Ar-Н бензотриазольное кольцо), 7,87 (с, 1Н, Ar-Н фенольное кольцо), 7,53 (м, 1Н, Ar-Н бензотриазольное кольцо), 7,29 (м, 1Н, Ar-Н бензотриазольное кольцо), 7,05 (с, 1Н, Ar-Н фенольное кольцо), 6,21 (с, 1Н, С=С-Н транс), 5,61 (с, 1Н, С=С-Н цис), 5,39 (с, 2Н, Ar-СН2), 3,89 (с, 3Н, Ar-ОСН3), 2,01 (с, 3Н, С=С-CH3).

ПРИМЕР 6
Синтез 2-(гидроксиметил)-4-метокси-6-((2-нитрофенил)-диазенил)фенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 19,9 г (144 ммоль) 2-нитроанилина (Aldrich), HCl (водн.), 100 мл деионизированной воды и 100 мл этанола. Смесь охлаждали до 0°C и в течение 60 минут по каплям добавляли 10,6 г (153 ммоль) нитрита натрия в 50 мл воды, поддерживая температуру реакции при -10-0°C. Реакционную смесь перемешивали в течение дополнительного 1 часа. Добавляли 300 мг сульфаминовой кислоты, чтобы разрушить избыток нитрита, и смесь перемешивали в течение дополнительных 20 минут. Твердое вещество отфильтровывали, фильтрат, содержащий соль диазония, отделяли и хранили холодным при -10°C. NaOH (29,3 г, 731 ммоль) растворяли в 100 мл воды и приблизительно одну четвертую часть добавляли к раствору (2-гидрокси-5-метокси-1,3-фенилен)диметанола в 100 мл деионизированной воды и 200 мл этанола. Смесь соли диазония и остающийся раствор гидроксида натрия добавляли к реакционной смеси, содержащей производное фенола, в течение 1 часа при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 3 часов. Содержимое выливали в 3 л воды и рН подкисляли до 6, используя 1н. HCl. Полученное в результате твердое вещество промывали несколькими литрами воды и затем сушили при 55°C в течение 40 часов, используя P2O5 в качестве осушителя, получая 24,4 г (55,8%) темного твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
Синтез 2-(2H-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, стандартной капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 23,6 г (77,7 ммоль) 2-(гидроксиметил)-4-метокси-6-((2-нитрофенил)диазенил)фенола и 200 мл этанола. NaOH (18,8 г, 470 ммоль) растворяли в 100 мл деионизированной воды и приблизительно одну четвертую часть раствора по каплям добавляли к реакционной смеси. Смесь нагревали до 80°C и к реакционной смеси в течение 30 минут одновременно по каплям добавляли 25,1 г (232 ммоль) формамидинсульфиновой кислоты и остающуюся часть раствора гидроксида натрия. Смесь перемешивали при 80°C в течение 3 часов, выливали в 3,5 л воды и затем подкисляли до рН 4, используя 1н. HCl. Полученное в результате твердое вещество собирали фильтрованием и обрабатывали, как в примере 5, получая светло-желтое твердое вещество (4,3 г, 20,5%).
ПРИМЕР 7 (Соединение WL-3)
Синтез 3-(2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата. В 250 мл круглодонной колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 4,03 г (14,9 ммоль) 2-(2H-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола из примера 6 в 50 мл безводного ТГФ. Добавляли триэтиламин (7,4 мл, 53 ммоль) и смесь охлаждали до -10°C. По каплям добавляли метакрилоилхлорид (1,99 г, 19,0 ммоль) и смесь перемешивали в течение 1 часа при 0°C, после чего следовало перемешивание в течение 6 часов при комнатной температуре. Смесь выливали в 200 мл диэтилового эфира и промывали 0,5н HCl и водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя, получая желаемый продукт в виде темно-желтого маслянистого вещества, которое перекристаллизовывали из метанола, получая 1,73 г светло-желтого твердого вещества (34%). 1H ЯМР (CDCl3) дельта: 11,26 (с, 1Н, Ar-ОН), 7,92 (д, 2Н, Ar-Н бензотриазол), 7,91 (с, 1Н, Ar-Н метаположение относительно фенола), 7,49 (д, 2Н, Ar-Н бензотриазол, 5,6-положение), 7,05 (с, 1Н, Ar-Н метаположение относительно фенола), 6,21 (с, 1Н, С=С-Н транс), 5,61 (с, 1Н, С=С-Н цис), 5,40 (с, 2Н, Ar-СН2), 3,90 (с, 3Н, Ar-ОСН3), 2,01 (с, 3Н, С=С-CH3).

ПРИМЕР 8
Синтез 2-((5-хлор-2-нитрофенил)диазенил)-6-(гидроксиметил)-4-метоксифенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 30,0 г (174 ммоль) 5-хлор-2-нитроанилина (Aldrich), конц. HCl (водн.) (J.T. Baker), 100 мл деионизированной воды и 100 мл абсолютного этанола. Суспензию охлаждали до 0°C и в течение 30 минут по каплям добавляли 12,7 г (184 ммоль) нитрита натрия в 50 мл воды, поддерживая температуру реакционной смеси при 0°C. Реакционную смесь перемешивали в течение дополнительного 1 часа. Добавляли сульфаминовую кислоту (430 мг), чтобы разрушить избыток нитрита, и реакционную смесь перемешивали в течение дополнительных 20 минут. Твердое вещество отфильтровывали и фильтрат, содержащий соль диазония, отделяли и хранили холодным при -10°C. NaOH (34,9 г, 873 ммоль) растворяли в 100 мл воды и приблизительно одну четвертую часть добавляли к раствору (2-гидрокси-5-метокси-1,3-фенилен)диметанола в 100 мл деионизированной воды и 200 мл этанола. Смесь соли диазония и остающийся раствор гидроксида натрия добавляли к реакционной смеси, содержащей производное фенола, в течение 1 часа при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 2 часов. Содержимое выливали в 3 л деионизированной воды и рН корректировали до 5, используя 1н. HCl. Полученное в результате твердое вещество промывали несколькими литрами воды и затем сушили под вакуумом (0,1 мм Hg (13,33 Па)) при 55°C в течение 40 часов, используя P2O5 в качестве осушителя, получая 28,0 г (48%) темного твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
Синтез 2-(5-хлор-2H-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 27,4 г (81,0 ммоль) 2-((5-хлор-2-нитрофенил)диазенил)-6-(гидроксиметил)-4-метоксифенола и 200 мл этанола. NaOH (19,7 г, 493 ммоль) растворяли в 100 мл деионизированной воды и приблизительно одну четвертую часть по каплям добавляли к реакционной смеси. Смесь нагревали до 80°C и к реакционной смеси одновременно по каплям добавляли 26,5 г (245 ммоль) формамидинсульфиновой кислоты и остающуюся часть раствора гидроксида натрия. Смесь перемешивали при 80°C в течение 2 часов и затем выливали в 3 л деионизированной воды. Смесь подкисляли до рН 3, используя 1н. HCl, и твердое вещество собирали фильтрованием, промывали обильным количеством воды и затем обрабатывали как в примере 5, получая 7,4 г (30%) твердого вещества, которое использовали в следующей стадии эстерификации.
ПРИМЕР 9 (Соединение WL-4)
Синтез 3-(5-хлор-2H-бензо[d][1,2,3]триазол-2-ил)-2-гидрокси-5-метоксибензилметакрилата. В 250 мл круглодонной колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 3,98 г (13,0 ммоль) 2-(5-хлор-2H-бензо[d][1,2,3]триазол-2-ил)-6-(гидроксиметил)-4-метоксифенола в 60 мл безводного ТГФ. Добавляли триэтиламин (6,4 мл) и смесь охлаждали до -10-0°C. По каплям добавляли метакрилоилхлорид (1,62 г, 15,5 ммоль) и смесь перемешивали в течение 1 часа при -10-0°C, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Твердое вещество отфильтровывали и промывали 100 мл диэтилового эфира. Органический слой промывали 1н. HCl и водой, сушили над сульфатом магния, отфильтровывали и концентрировали с помощью роторного испарителя, получая желтое маслянистое вещество, которое перекристаллизовывали из этанола, получая 1,73 г (34%) светло-желтого твердого вещества. 1H ЯМР (CDCl3) дельта: 11,00 (с, 1H, Ar-OH), 7,92 (с, 1H, Ar-H бензотриазол), 7,88 (с, 1H, Ar-H фенол), 7,87 (д, 1H, Ar-H бензотриазол), 7,45 (д, 1H, Ar-H бензотриазол), 7,06 (с, 1H, Ar-H фенол), 6,21 (с, 1H, C=C-H транс), 5,62 (с, 1H, C=C-H цис), 5,38 (с, 2H, Ar-CH2), 3,89 (с, 3Н, Ar-ОСН3), 2,01 (с, 3H, C=C-CH3).

ПРИМЕР 10
Синтез 2-(2-(гидроксиметил)-4-метокси-6-(5-метокси-2H-бензо[d][1,2,3]триазол-2-ил)фенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 24,8 г (148 ммоль) 4-метокси-2-нитроанилина (Aldrich), конц. HCl (водн.) (J.T. Baker), 150 мл воды и 150 мл абсолютного этанола. Смесь охлаждали до -20°C и в течение 30 минут по каплям добавляли раствор, включающий 10,8 г (156 ммоль) нитрита натрия в 40 мл воды. Реакционную смесь перемешивали в течение дополнительного 1 часа и затем добавляли сульфаминовую кислоту (315 мг), чтобы разрушить избыток нитрита. Нерастворимое твердое вещество отфильтровывали и фильтрат, содержащий соль диазония, отделяли и хранили холодным при -10°C. NaOH (29,5 г, 739 ммоль) растворяли в 100 мл воды и приблизительно одну четвертую часть добавляли к раствору, включающему (2-гидрокси-5-метокси-1,3-фенилен)диметанол в 100 мл деионизированной воды и 200 мл этанола. Смесь соли диазония и остающийся раствор гидроксида натрия добавляли к реакционной смеси, содержащей производное фенола, в течение 1 часа при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 3 часов. Содержимое выливали в 3 л воды и pH корректировали до 4,5, используя 1н. НС1. Полученное в результате твердое вещество отфильтровывали, промывали несколькими литрами воды и затем сушили под вакуумом (0,1 мм Hg (13,33 Па)) при 65°C в течение 20 часов, используя P2O5 в качестве осушителя, получая 33,5 г (68%) темного твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
Синтез 2-(гидроксиметил)-4-метокси-6-(5-метокси-2Н-бензо[d][1,2,3]триазол-2-ил)фенола. В 500 мл круглодонную 3-горлую колбу, оборудованную магнитной мешалкой, капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 2-(гидроксиметил)-4-метокси-6-((4-метокси-2-нитрофенил)диазенил)фенол из примера 10, часть I и 200 мл абсолютного этанола (Pharmco Products, Брукфилд, Коннектикут). Гранулы NaOH, 97% (Aldrich, 16,8 г, 420 ммоль) растворяли в 80 мл деионизированной воды и приблизительно одну четвертую часть раствора по каплям добавляли к азосмеси. Реакционную смесь нагревали до 80°C и к азосмеси в течение 0,5 часа одновременно по каплям добавляли 22,6 г (209 ммоль) формамидинсульфиновой кислоты (Aldrich) и остающуюся часть раствора гидроксида натрия. Смесь нагревали при 80°C в течение 3 часов, затем выливали в 3,5 л воды и подкисляли до pH 4-5 конц. НС1 (J.T. Baker). Неочищенный продукт подвергали перекристаллизации из этанола, затем собирали и сушили под вакуумом (0,1 мм Hg (13,33 Па)) при 50°C в течение 72 часов, получая 7 г (27%) светло-желтого твердого вещества.
ПРИМЕР 11 (Соединение WL-5)
Синтез 2-гидрокси-5-метокси-3-(5-метокси-2Н-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата. Эстерификацию осуществляли, используя продукт из примера 10, часть II. В 100 мл круглодонной 3-горлой колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 3,24 г (10,8 ммоль) 2-(гидроксиметил)-4-метокси-6-(5-метокси-2Н-бензо[d][1,2,3]триазол-2-ил)фенола в 60 мл безводного ТГФ. Добавляли триэтиламин (1,2 мл) и смесь охлаждали до -10-0°С. По каплям добавляли метакрилоилхлорид (1,40 г, 13,4 ммоль) и смесь перемешивали в течение 2 часов при 0°С, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Соли отфильтровывали, фильтрат выливали в 100 мл диэтилового эфира и промывали 0,5н HCl и водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя, получая желаемый продукт в виде темно-желтого маслянистого вещества, которое перекристаллизовывали из этанола, получая светло-желтое твердое вещество. 1H ЯМР (CDCl3) дельта: 11,20 (с, 1H, фенол OH), 7,80 (м, 1H, Ar-H бензотриазол), 7,77 (с, 1H, Ar-H фенол), 7,17 (м, 1H, Ar-H бензотриазол), 7,14 (с, 1H, Ar-H фенол), 7,10 (м, 1H, Ar-H бензотриазол), 6,21 (с, 1H, C=C-H транс), 5,61 (с, 1H, C=C-H цис), 5,38 (с, 2H, Ar-CH2), 3,92 (с, 3Н, Ar-ОСН3, фенол), 3,88 (с, 3H, Ar-OCH3, бензотриазол), 2,01 (с, 3H, C=C-CH3).

ПРИМЕР 12
Синтез 2-(гидроксиметил)-4-метокси-6-((4-метил-2-нитрофенил)диазенил)фенола. В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, добавляли 24,3 г (160 ммоль) 4-метил-2-нитроанилина, 98% (Aldrich), 67 мл конц. HCl (водн.), 100 мл воды и 100 мл абсолютного этанола. Смесь охлаждали до -10-0°C и в течение 30 минут при температуре -10-0°C по каплям добавляли 11,6 г (169 ммоль) нитрита натрия в 40 мл воды. Реакционную смесь перемешивали в течение дополнительного 1 часа и добавляли 315 мг сульфаминовой кислоты, чтобы разрушить избыток нитрита. После дополнительных 20 минут перемешивания реакционную смесь фильтровали и отделяли холодный фильтрат. В 1 л колбе суспендировали (2-гидрокси-5-метокси-1,3-фенилен)диметанол в 200 мл деионизированной воды и 100 мл этанола. Готовили раствор 32,2 г (805 ммоль) гидроксида натрия и приблизительно одну четвертую часть добавляли к производному фенола. Производное фенола охлаждали до 0°C и смесь соли диазония и остающийся раствор гидроксида натрия одновременно добавляли к производному фенола в течение 1 часа при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 3 часов. Смесь выливали в 3 л воды и рН корректировали до 4,5, используя 1н. HCl. Неочищенный продукт сушили при 65°C в течение 20 часов под высоким вакуумом (0,1 мм Hg (13,33 Па)), используя P2O5 в качестве осушителя, получая 30,8 г (61%). Продукт использовали в следующей стадии без дополнительной очистки.
Синтез 2-(гидроксиметил)-4-метокси-6-(5-метил-2H-бензо[d][1,2,3]триазол-2-ил)фенола. В 500 мл круглодонную 3-горлую колбу, оборудованную магнитной мешалкой, стандартной капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 30,0 г (94,6 ммоль) азосоединения из части 1 и 200 мл этанола. NaOH (22,9 г, 573 ммоль) растворяли в 100 мл деионизированной воды и приблизительно одну четвертую часть раствора по каплям добавляли к реакционной смеси. Реакционную смесь нагревали до 80°C. К азосмеси в течение 30 минут одновременно добавляли формамидинсульфиновую кислоту (30,7 г, 284 ммоль) и остающуюся часть раствора гидроксида натрия. Смесь нагревали при 80°C в течение 3 часов. Реакционную смесь выливали в 3 л воды и затем подкисляли до рН 4-5 конц. 1н. HCl. Твердое вещество отфильтровывали, затем растворяли в 3 л воды, содержащей 5 г NaOH, и подкисляли до pH 2 с помощью 1н. HCl. Твердое вещество снова отфильтровывали и затем сушили при 55°C под высоким вакуумом (0,1 мм Hg (13,33 Па)) в течение 40 часов, получая 20 г (74%), которые использовали в следующей стадии без дополнительной очистки.
ПРИМЕР 13 (Соединение WL-6)
Синтез 2-гидрокси-5-метокси-3-(5-метил-2Н-бензо[d][1,2,3]триазол-2-ил)бензилметакрилата. В 1000 мл круглодонной 3-горлой колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 19,5 г (68,3 ммоль) 2-(гидроксиметил)-4-метокси-6-(5-метил-2Н-бензо[d][1,2,3]триазол-2-ил)фенола в 200 мл безводного ТГФ. Добавляли триэтиламин (34 мл, 240 ммоль) и смесь охлаждали до -10°C. По каплям добавляли метакрилоилхлорид (8,55 г, 81,8 ммоль) и смесь перемешивали в течение 2 часов при 0°C, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Твердое вещество отфильтровывали, промывали диэтиловым эфиром, фильтрат выливали в 100 мл диэтилового эфира и промывали 0,5н HCl и водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя. Неочищенный продукт перекристаллизовывали из этанола, получая 7,2 г (30%) светло-желтого твердого вещества. 1H ЯМР (CDCl3) дельта: 11,28 (с, 1Н, Ar-ОН), 7,89 (с, 1Н, Ar-Н бензотриазол 4-положение), 7,79 (д, 1Н, Ar-Н бензотриазол 6-положение), 7,66 (с, 1Н, Ar-Н фенол 6-положение), 7,26 (д, 1Н, Ar-Н бензотриазол 7-положение), 7,03 (с, 1Н, Ar-Н фенол 4-положение), 6,21 (с, 1Н, С=С-Н транс), 5,61 (с, 1H, C=C-H цис), 5,39 (с, 2H, Ar-CH2), 3,89 (с, 3Н, Ar-ОСН3), 2,54 (с, 3H, Ar-CH3), 2,01 (с, 3H, C=C-CH3).

ПРИМЕР 14
Синтез 2-(гидроксиметил)-4-метил-6-((2-нитро-4-(трифторметил)фенил)диазенил)фенола. В 500 мл круглодонной колбе, оборудованной магнитной мешалкой, объединяли 26,0 г (126 ммоль) 2-нитро-4-(трифторметил)анилина, 53 мл конц. HCl, 100 мл деионизированной воды и 150 мл абсолютного этанола. Смесь охлаждали до 0°C и в течение 60 минут по каплям добавляли нитрит натрия в 30 мл воды. Реакционную смесь перемешивали в течение дополнительного 1 часа и добавляли 300 мг сульфаминовой кислоты, чтобы разрушить избыток нитрита. Твердое вещество отфильтровывали и холодный фильтрат отделяли. В 1 л колбе суспендировали (2-гидрокси-5-метокси-1,3-фенилен)диметанол в 200 мл деионизированной воды и 100 мл этанола. Приблизительно одну четвертую часть раствора гидроксида натрия (25,4 г, 635 ммоль) в 100 мл воды добавляли к фенольной смеси при 0°C. Смесь соли диазония и остающийся раствор гидроксида натрия одновременно добавляли к фенольной смеси в течение 1 часа. Реакционную смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 4 часов. Смесь выливали в 3 л воды и рН корректировали до 3-4, используя 1н. HCl. Твердое вещество отфильтровывали, промывали обильным количеством воды и затем сушили под вакуумом (0,1 мм Hg (13,33 Па)) при 55°C в течение 40 часов, получая 27,4 г (76%) твердого вещества красного цвета.
Синтез 2-(гидроксиметил)-4-метил-6-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)фенола. В 1 л круглодонную колбу, оборудованную магнитной мешалкой, капельной воронкой, воронкой для добавления порошка и вводом для азота, добавляли 26,0 г (73,2 ммоль) 2-(гидроксиметил)-4-метил-6-((2-нитро-4-(трифторметил)фенил)диазенил)фенола и 300 мл этанола. Гидроксид натрия (17,6 г, 441 ммоль) растворяли в 100 мл деионизированной воды и приблизительно одну четвертую часть раствора по каплям добавляли к азосмеси. Реакционную смесь нагревали до 80°C и к азосмеси в течение 30 минут добавляли 23,9 г (221 ммоль) формамидинсульфиновой кислоты и остающуюся часть раствора гидроксида натрия. Реакционную смесь нагревали при 80°C в течение 3 часов, выливали в 3 л деионизированной воды, подкисляли до рН 4, используя 1н. HCl. Твердое вещество собирали фильтрованием под вакуумом, промывали обильным количеством воды и затем сушили в течение 20 часов под вакуумом (0,1 мм Hg (13,33 Па)) при 42°C, получая 18,6 г (79%) серовато-белого твердого вещества.
ПРИМЕР 15 (Соединение WL-7)
Синтез 2-гидрокси-5-метил-3-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)метакрилата. В 100 мл круглодонной 3-горлой колбе, оборудованной магнитной мешалкой и вводом для азота, растворяли 3,58 г (11,1 ммоль) 2-(гидроксиметил)-4-метил-6-(5-(трифторметил)-2H-бензо[d][1,2,3]триазол-2-ил)фенола в 60 мл безводного ТГФ. Добавляли триэтиламин (5,6 мл, 40 ммоль) и смесь охлаждали до -10°C. По каплям добавляли метакрилоилхлорид (1,373 г, 13,1 ммоль) и смесь перемешивали в течение 1 часа при -10°C, после чего следовало перемешивание в течение 20 часов при комнатной температуре. Твердое вещество отфильтровывали, промывали диэтиловым эфиром, полученный в результате фильтрат выливали в 100 мл диэтилового эфира и промывали 0,5н HCl и водой. Органический слой сушили над сульфатом магния, фильтровали и перекристаллизовывали из диэтилового эфира, получая 1,90 г (44%) белого твердого вещества. 1H ЯМР (CDCl3) дельта: 11,17 (с, 1H, Ar-OH), 8,29 (с, 1H, Ar-H бензотриазол 4-положение), 8,21 (с, 1H, Ar-H фенол 6-положение), 8,06 (д, 1H, Ar-H бензотриазол 6-положение), 7,69 (д, 1H, Ar-H бензотриазол 7-положение), 7,29 (с, 1H, Ar-H фенол 4-положение), 6,20 (с, 1H, C=C-H транс), 5,61 (с, 1H, C=C-H цис), 5,39 (с, 2H, Ar-CH2), 2,42 (с, 3Н, Ar-СН3), 2,00 (с, 3H, C=C-CH3).

ПРИМЕР 16
Кривые пропускания для соединений WL-1-WL-7 получали спектроскопией ультрафиолетовой и видимой части спектра. Каждое соединение растворяли в хлороформе и оценивали на УФ-спектрометре Perkin Elmer Lambda 35. Результаты показаны на фигуре 1, и результаты 1% Т и 10% Т показаны в таблице 1.
|
ПРИМЕР 17
Рецептуры акриловой IOL
Из соединений WL-1-WL-4 составляли рецептуры IOL материалов, как показано в таблицах 2-6. Все компоненты были перемешаны вихревой мешалкой в 30 мл стеклянном сосуде, дегазированы азотом и затем профильтрованы фильтрующим шприцом с использованием 0,2-микронного тефлонового фильтра в прямоугольные полипропиленовые формы глубиной ~1 мм. Образцы термически отверждали при 70°C в течение 1 часа и при 110°C в течение 2 часов, затем экстрагировали в ацетоне при 50°C в течение 6 часов с заменой на свежий растворитель каждые 90 минут.
|
PEA=2-фенилэтилакрилат
PEMA=2-фенилэтилметакрилат
BzA=бензилакрилат
BzMA=бензилметакрилат
BDDA=1,4-бутандиолдиакрилат
Этоксилат вторичного спирта, сложный эфир метакриловой кислоты = сложный эфир метакриловой кислоты и поверхностно-активного вещества Tergitol™ 15-S-30 (Dow/Union Carbide)
AIBN=2,2'-азобис(2-метилпропионитрил)
|
|
|
|
ПРИМЕР 18
Светоустойчивость
Образцы рецептур 17A, 17B, 17C, 17D, 17E, 17F, 17G, 17H, 17I и 17J подвергали УФ облучению от 300 до 800 нм, используя тестовую камеру Atlas Suntest CPS+ (Atlas Electric Devices Company, Чикаго, Иллинойс), использующую ксеноновую дуговую лампу с интенсивностью света приблизительно 8-10 мВт/см2 на высоте тестируемого образца. Температура среды фосфатно-солевого буфера составляла 35°C. Спектры УФ/видимого диапазона срезов образцов толщиной 0,9 мм регистрировали, используя УФ-спектрометр Perkin Elmer Lambda 35. Результаты воздействия света, эквивалентные 20 годам (примеры 17A-17H) или 10 годам (примеры 17I и 17J), показаны на фиг.2A-2J.
Данное изобретение было описано посредством ссылки на конкретные предпочтительные варианты осуществления; однако необходимо понимать, что его можно осуществить на практике в его других конкретных формах или вариантах без отклонения от его характерных или существенных характеристик. Варианты осуществления, описанные выше, поэтому рассматриваются как иллюстративные во всех аспектах и не ограничивающие, причем объем изобретения указывается прилагаемой формулой изобретения, а не предшествующим описанием.













Система для доставки линзы
Выравнивание торических линз с использованием предоперационных изображений
Хирургическая кассета с акустическим воздушным отражателем
Гидрогелевая интраокулярная линза и способ ее формирования
Фармацевтическая композиция, содержащая фторхинолоновое антибиотическое лекарственное средство
Асферическая тороидальная внутриглазная линза
Поверхность линзы с комбинированными дифракционными, торическими и асферическими компонентами
Аккомодирующая система искусственного хрусталика
Зональные дифракционные мультифокальные внутриглазные линзы
Линза с увеличенной глубиной фокуса (edof) для усиления псевдоаккомодации с использованием динамики зрачка
Система для доставки линзы
Выравнивание торических линз с использованием предоперационных изображений
Хирургическая кассета с акустическим воздушным отражателем
Гидрогелевая интраокулярная линза и способ ее формирования
Фармацевтическая композиция, содержащая фторхинолоновое антибиотическое лекарственное средство
Асферическая тороидальная внутриглазная линза
Поверхность линзы с комбинированными дифракционными, торическими и асферическими компонентами
Аккомодационная интраокулярная линза (иол) с торическим оптическим элементом и увеличенной глубиной фокуса
Аккомодирующая система искусственного хрусталика
Зональные дифракционные мультифокальные внутриглазные линзы

