2,6-ДИНИТРОСОДЕРЖАЩИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПУРИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЕ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ. К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармацевтической химии, в частности, к 2, 6-динитросодержащим замещенным производным пурина, способу их получения и их использованию.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Злокачественные опухоли (рак) являются одним из самых распространенных заболеваний, оказывающих в настоящее время пагубное воздействие на здоровье человека и угрожающих жизни человека. Ежегодно во всем мире от рака умирает более 5 миллионов человек. Несмотря на то, что в настоящее время разработаны и применяются определенные терапевтические средства и методы, такие как хирургия, радиотерапия, химиотерапия и т.д., показатель эффективности лечения в общем не является высоким. В настоящее время метод химиотерапии в основном характеризуется рядом недостатков, таких как низкая селективность, тяжелые побочные эффекты и т.д. Таким образом, организации, занимающиеся разработками в области фармацевтики в различных странах мира, сосредотачивают свои усилия на создании противоопухолевых лекарственных средств, которые обладали бы более низкой токсичностью, слабым побочным эффектом, высокой противораковой активностью, высокой стабильностью и т.д.
В соответствии с имеющейся информацией ряд производных пурина характеризуется определенной противовирусной и противоопухолевой активностью. См. соответствующие документы: ЕР 0353955, WO 9201968, JP 10120682, KR 9100441 и т.д.
В известных технических решениях в данной области раскрывается ряд яд замещенных производных пурина, например, Незамещенные производные пурина, использовавшиеся для лечения аллергических заболеваний, раскрыты в патенте US 4853386; производные N6-циклопропиламино-9Н-пурина, обладающие противовирусной активностью, раскрыты в патентах JP 2003-55377 A и JP 2003-119197 А. Гликолизированные производные пурина, обладающие противовоспалительным эффектом раскрыты в J. Org. Chem. (стр.3212~3215. том 69, 2004). Производные N2-бутилфенил-2-дезоксипурина, обладающие активностью ДНК-полимеразы а эукариотических клеток, раскрыты в J. Med. Chem. (стр.175-181, том. 27, 1984). 2,6,9-тризамещенные производные пурина раскрыты в Tetraheron Letters (1827~1830, том. 39, 1998). Кроме того, ряд соединений, обладающих противоопухолевым эффектом, раскрыт в патенте CN 200510026846. Перед специалистами в данной области техники стоит задача разработать N2,N6-дизамещенные производные пурина, характеризующиеся более высокой противоопухолевой активностью, при проведении дальнейших исследований с целью усиления противоопухолевой активности N2,N6-дизамещенного соединения пурина.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩЕСТВА ИЗОБРЕТЕНИЯ
Техническая проблема настоящего изобретения заключается в проведении исследований по разработке N2,N6-дизамещенных производных пурина, обладающих более низкой токсичностью, более широким противораковым диапазоном, более высокой противораковой активностью и высокой стабильностью. Настоящее изобретение предусматривает создание соединений 2, 6-динитросодержащего замещенного пурина по формуле (А) либо их солей или сольватов, либо сольватов их солей:
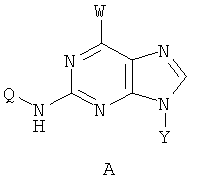
Где W обозначает выборочно монозамещенный C1~C6алкиламино с прямой или разветвленной цепью, выборочно монозамещенный С3~С6алкил, либо алкенил, либо алкиниламино с прямой или разветвленной цепью, выборочно дизамещенный C1~C6алкиламино с прямой или разветвленной цепью, выборочно дизамещенный С3~С6алкил, либо алкенил, либо алкиниламино с прямой или разветвленной цепью, W также может обозначать амино, замещенный двумя различными С1~С6алканами с прямой или разветвленной цепью, или обозначать амино, замещенный двумя различными С3~С6олефинами с прямой или разветвленной цепью, либо амино, один конец которого замещен С1~С6алканом, а другой конец замещен С3~С6олефином, либо выборочно замещенный гетероциклсодержащий вторичный азот, такой как пирролидин, пиперидин, морфин или пиперазидин; заместитель обозначает C1~C6алкил, либо галоген, либо гидроксил с прямой или разветвленной цепью;
Y обозначает Н или фармацевтически приемлемый сахарид, в котором сахарид обозначает предпочтительно любую из нижеприведенных формул:
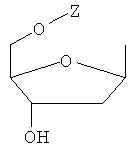 ,
, 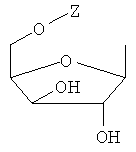 ,
, 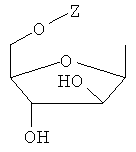 ,
, 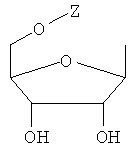
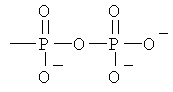
Z обозначает Н или любую из нижеприведенных формул:
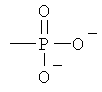 ,
, 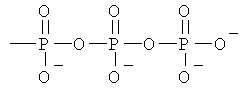 ,
, 
Q обозначает Н или любую из нижеприведенных формул:
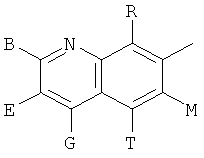 ,
, 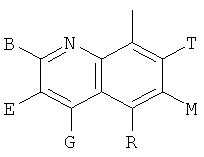 ,
, 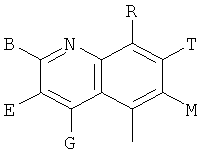 ,
,
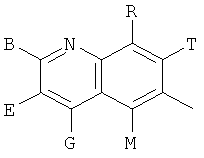 ,
, 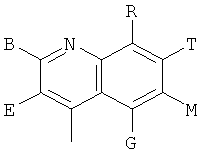 ,
, 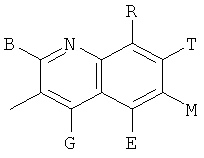 ,
,
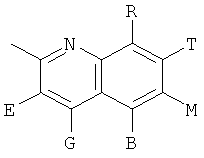 ,
, 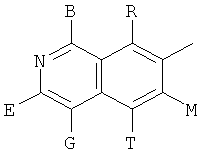 ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  ,
, 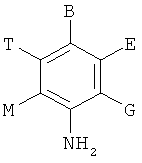
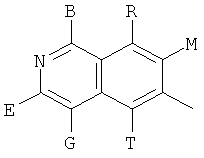
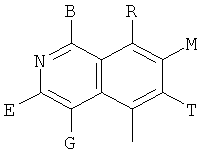
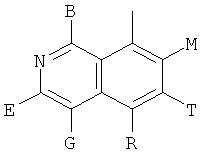
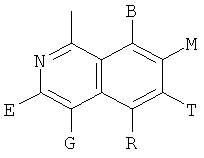
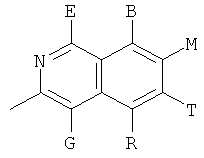
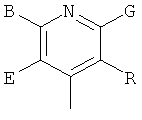
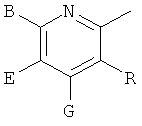
в которых В, Е, G, R, Т, М каждый независимо обозначает Н или C1~C6алкил либо галоалкил с прямой или разветвленной цепью, С3~С6циклоалкил, галоген, CN, NH2, метоксил, этиоксил или нитро.
W предпочтительно обозначает амино, циклопропиламино, циклобутиламино, метиламино, этиламино, пропиламино, изопропиламино, диметиламино, диэтиламино, метилэтиламино, аллиламино, метилаллиламино, этилаллиламино, пропилаллиламино, диаллиламино, этаноламино или любую из нижеприведенных формул:
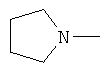 ,
, 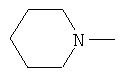 ,
, 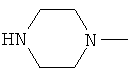 ,
, 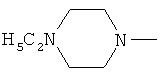 ,
, 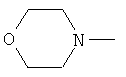 ,
,
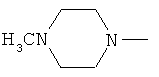 ,
,  ,
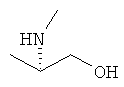
, 
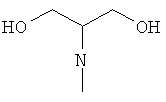
W предпочтительно обозначает циклопропиламино, диметиламино, диэтиламино, метилэтиламино, аллиламино, диаллиламино или любую из нижеприведенных формул:
, , ,  , ,
, ,
,
Q предпочтительно обозначает любую из нижеприведенных формул:
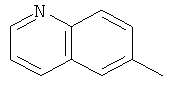 ,
, 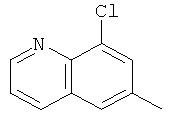 ,
, 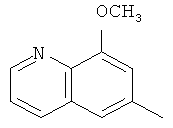 ,
, 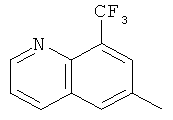 ,
,
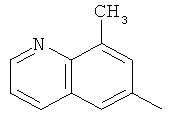 ,
, 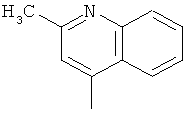 ,
, 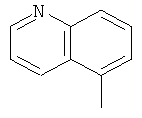 ,
, 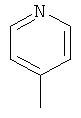 ,
,
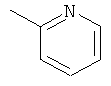 ,
, 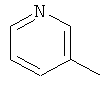 ,
, 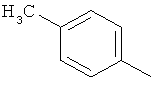 ,
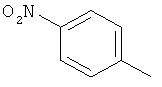
, 
в которых Y является Н.
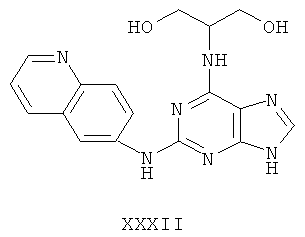
Целью настоящего изобретения является создание нижеприведенных соединений:
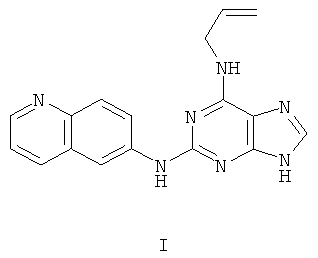
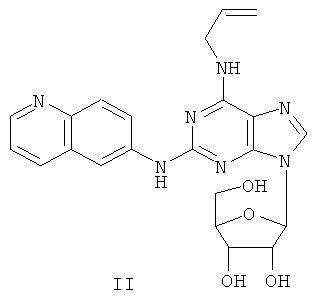
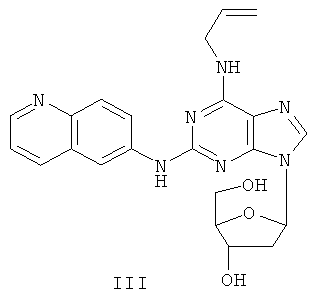



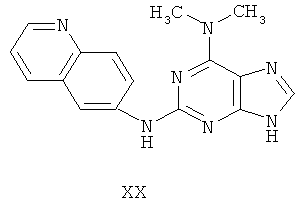
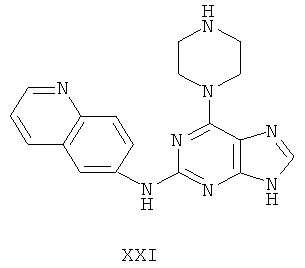
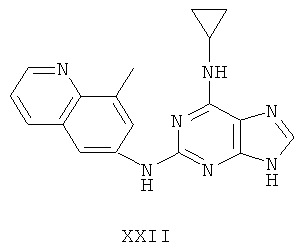
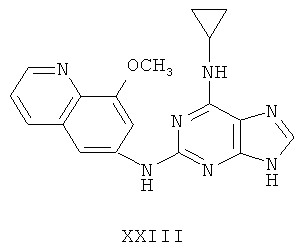
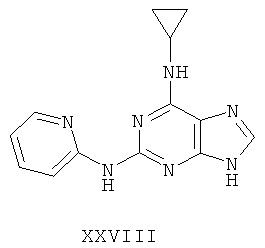
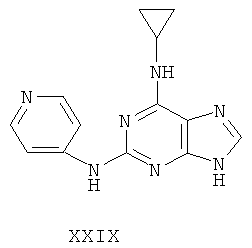
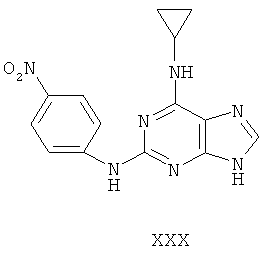
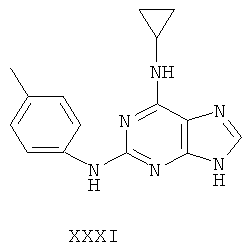
Другой целью настоящего изобретения является создание способа получения вышеприведенных соединений по формуле (А) либо их солей или сольватов, либо сольватов их солей, и способ проиллюстрирован нижеприведенной формулой:
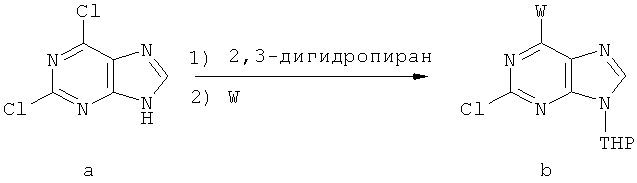
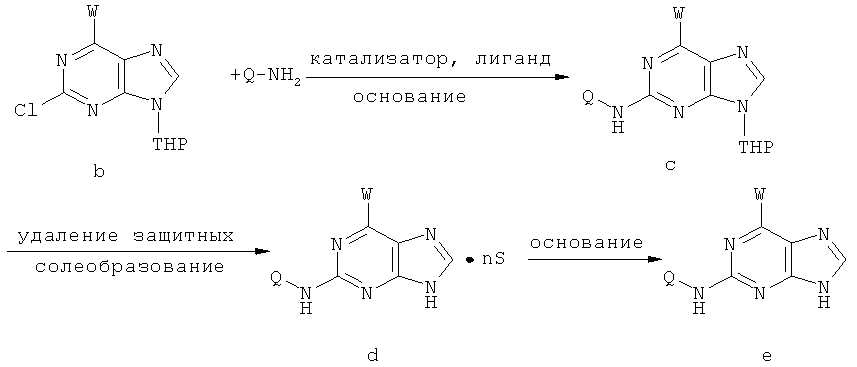
Способ получения соединений включает следующие этапы:
1) на первом этапе соединение (а) реагирует с 2,3-дигидропираном в присутствии таких катализаторов, как паратолуолсульфоновая кислота, соль пиридиния паратолуолсульфоновой кислоты или смоляная кислота, либо иного катализатора для защиты 9-го атома азота пурина; при этом в процессе реакции молярное отношение соединения (а) к 2,3-дигидропирану составляет приблизительно 1:1~5; далее в присутствии нетравящего растворителя, такого как триэтиламин, карбонат натрия, карбонат калия или бикарбонат натрия происходит конденсирование с W и получают соединение (b); при этом молярное отношение соединения (а) к W составляет приблизительно 1:1~5, температура реакции конденсирования с W составляет приблизительно 20~100°С, предпочтительно приблизительно 40~60°С.
2) далее протекает каталитическая реакция сочетания и реакция удаления защитных групп и солеобразования между соединением (b) и Q-NH2, в результате чего получают соединение (d); при этом молярное отношение соединения (b) к Q-NH2 составляет приблизительно 1:0,5~2.
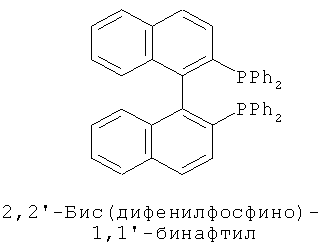
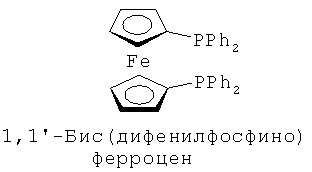
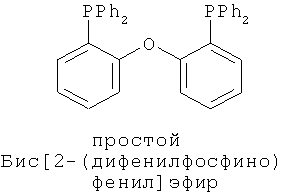
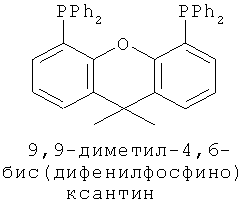
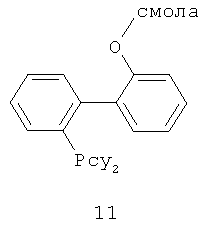
В каталитической реакции сочетания лиганд включает три-о-толилфосфин, три-трет-бутилфосфин, 2,2′-дифенилфосфин -1,1′-бинафталин, 1,1′-дифенилфосфин-ферроцен, простой эфир бис(2-дифенилфосфинофенил), 9,9-диметил-4,5-дифенилфосфин ксантен, либо лиганд представляет собой соединения по формуле 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11; катализатор является катализатором на основе переходных металлов - палладия или никеля - например, PdCl2, Pd(OAc)2, Pd2(dba)3, Ni(OAc)2 или Ni/C; основанием является трет-бутоксид натрия, трет-бутоксид калия, карбонат калия, карбонат цезия или фосфат трикалия. Растворителем является апротонный растворитель, такой как тетрагидрофуран, простой изопропиловый эфир, простой этиленгликольдиметиловый эфир, диоксан, пиридин, 1-метил-2-пирролидон (NMP), 1,3-диметилтриметиленмочевина (DMPU), толуол или ксилол, либо смеси растворителей, содержащие один или несколько растворителей, выбранных из вышеприведенных.
При протекании каталитической реакции сочетания температура реакции составляет приблизительно 15~150°С, предпочтительно приблизительно 55~120°С, либо реакция протекает при воздействии микроволнового нагрева. Реакция снятия защитной группы и солеобразования на этапе 2 могла бы протекать в кислотной среде, такой как соляная кислота, серная кислота, бромистоводородная кислота, метансульфоновая кислота, бензолсульфоновая кислота, паратолуолсульфоновая кислота, малеионовая кислота, фумаровая кислота, молочная кислота или лимонная кислота и т.д. При этом молярное отношение соединения (с) к соляной кислоте, серной кислоте, бромистоводородной кислоте, метансульфоновой кислоте, бензолсульфоновой кислоте, паратолуолсульфоновой кислоте, малеионовой кислоте, фумаровой кислоте, молочной кислоте или лимонной кислоте может составлять 1:1~10 соответственно.
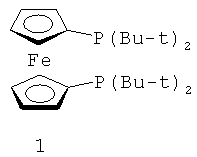
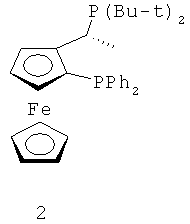
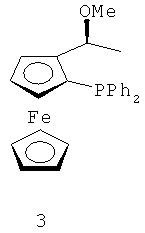

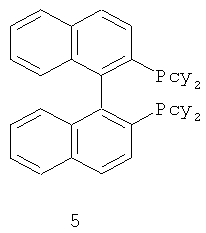
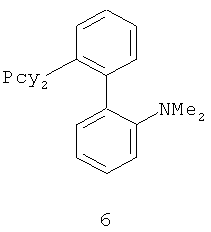
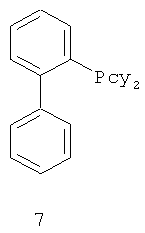
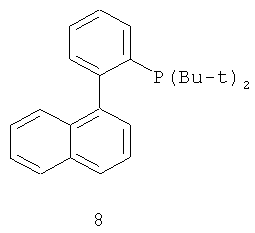
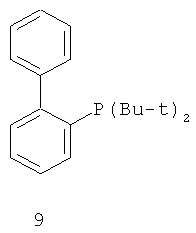
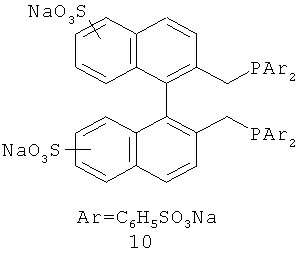
3) Нейтрализация соединения (d) карбонатом натрия, карбонатом калия, гидроксидом натрия или гидроксидом калия для получения соединения (е).
Дополнительной целью настоящего изобретения является создание фармацевтической композиции, в которой фармацевтическая композиция состоит из соединений по формуле (А) либо их солей или сольватов, либо сольватов их солей и фармацевтически приемлемого вспомогательного вещества. Соль является солью присоединения кислоты, полученной с помощью органической или неорганической кислоты, при этом кислота предпочтительно является соляной кислотой, серной кислотой, бромистоводородной кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, паратолуолсульфоновой кислотой, малеионовой кислотой, фумаровой кислотой, молочной кислотой, лимонной кислотой, либо соль является солью присоединения основания, полученной с помощью органического или неорганического основания. Фармацевтическая композиция выполнена в форме таблетки, капсулы, пилюли, жидкого препарата для перорального применения, гранулы, порошка, инъекции, имплантата или препарата для внешнего применения.
Испытания противоопухолевой активности in vitro и in vivo показывают, что соединения А в соответствии с настоящим изобретением обладают противоопухолевой активностью. Соединения оказывают ингибирующее действие на рост клеток рака (Colon 26) и саркомы S180 у мышей. Соединения А либо соли или их сольваты, либо сольваты их солей могли бы быть использованы для изготовления лекарственного средства для лечения или профилактики опухолевых заболеваний. Опухолевые заболевания включают рак легких, рак печени, лейкемию, остеокарциному, рак поджелудочной железы, рак кожи, меланому, метрокарциному, оофорому, ректальную карциному, желудочную карциному, рак толстой кишки, карциному груди, карциному придатков матки, карциному эндометрия, рак шейки матки, карциному влагалища, карциному наружных половых органов, карциному пищевода, карциному тонкого кишечника, карциному эндокраниума, карциному мягких тканей, карциному мочеиспускательного канала, рак предстательной железы, лимфоцитому, рак мочевого пузыря, рак почек или рак мочеиспускательного канала, опухоли позвоночника, нейроглиальные опухоли мозга и аденому гипофиза.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ И ЕГО ПРЕДПОЧТИТЕЛЬНЫХ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ
Ниже, в частности, приведено описание настоящего изобретения со ссылкой на примеры. Указанные примеры приведены в целях иллюстрации технического решения настоящего изобретения и не должны рассматриваться как ограничивающие настоящее изобретение.
Примеры 1-3 Получение соединений формулы I, II, III
Пример 1 Получение соединения I
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл), и далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) при дефлегмировании, в течение 15 минут к нему добавляли аллиламин (7 мл), и далее в смеси протекала реакция в течение 0,5 ч. Завершение реакции проверяли методом тонкослойной хроматографии, и далее смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило отделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12 г). Выход составлял приблизительно 77,3%.
2. В 250 мл трехгорловую колбу поочередно добавляли твердый пурин (10,4 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат белого цвета (11,5 г). Выход составлял приблизительно 82,6% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества полностью растворяли и получали прозрачный раствор оранжевого цвета. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,5 г).
1H-NMR (DMSO-d6+D2O, ppm) δ: 4,29 (2Н, s), 5,21 (1Н, dd, J=2,0, 10,4 Hz), 5,32 (1H, dd, J=2,0, 17,2 Hz), 6,10 (1Н, т), 7,98 (1Н, dd, J=5,2, 8,4 Hz), 8,20 (1H, d, J=9,2 Hz), 8,34 (перекрыты), 8,84 (2Н, перекрыты), 8,92 (1Н, d, J=8,4 Hz), 9,08 (1Н, dd, J=5,2, 1,2 Hz).
13C-NMF (DMSO-d6, ppm) δ: 43,0, 106,0, 113,2, 116,4, 121,6, 122,4, 128,7, 129,8, 134,2, 134,4, 138,9, 141,1,142,5, 144,5, 149,3, 151,5, 155,4.
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество светло-желтого цвета. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение I (5,4 г).
(+)-ESI MS m/z: 318 [M+H]+.
Пример 2 Получение соединения II
Соединение I (2,5 г) и бис (триметилсилил) ацетамид (3,5 мл) смешивали в безводном ацетонитриле (10 мл). Вышеуказанную смесь перемешивали при комнатной температуре в течение 1 часа. Далее к смеси добавляли раствор тетраацетил робофуранозы (3,5 г), растворенной в ацетонитриле (8 мл), и тиметилсилилтрифлата (0,6 мл) и нагревали при дефлегмировании в течение 5 часов. К смеси добавляли бычий сывороточный альбумин (0,7 мл) и далее перемешивали в течение 24 часов. Завершение реакции проверяли методом тонкопленочной хроматографии. Растворитель концентрировали при пониженном давлении, и осадок растворяли в метаноле (15 мл). Через вышеуказанную смесь пропускали газообразный аммиак в течение 1,5 часа. Растворитель удаляли при пониженном давлении, остаток очищали методом колончатой хроматографии на силикагеле и получали соединение II (2,6 г).
(+)-ESI MS m/z: 450 [M+H]+.
Пример 3 Получение соединения III
1. 60% NaH (0,4 г) и безводный ацетонитрил (50 мл) смешивали с соединением I (2,5 г). Вышеуказанную смесь перемешивали в защитной атмосфере азота в течение 30 минут. К смеси в течение 10 минут добавляли по порциям 3,5-ди-пара-толуолсульфонил-2-дезокси-β-D-рибофураноза-1-хлорид (3 г). После протекания реакции при комнатной температуре в течение 2 часов и последующего фильтрования фильтрат концентрировали до сухого состояния и получали масляное вещество. Далее масляное вещество очищали методом колончатой хроматографии и получали твердое вещество (2,5 г).
2. Вышеуказанное твердое вещество, 50% метоксид натрия (0,6 г) и метанол (100 мл) смешивали, и между ними протекала реакция при комнатной температуре в течение 5 часов при постоянном перемешивании. Значение pH вышеуказанной смеси регулировали до нейтрального значения с помощью уксусной кислоты. Растворитель отгоняли, остаток очищали методом колончатой хроматографии и получали соединение III (1,3 г).
(+)-ESI MS m/z: 434 [M+H]+.
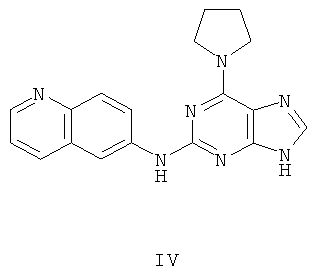
Пример 4-6 Получение соединений формулы IV, V, VI
Пример 4 Получение соединения IV
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (7,9 мл), далее в течение 15 минут к нему добавляли пирролидин (7,8 мл) при этой температуре, и далее в смеси протекала реакция при этой температуре в течение 0,5 ч. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,3 г). Выход составил приблизительно 75,6%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,9 г). Выход составил приблизительно 82,6% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (12,0 г).
1H-NMR (DMSO-d6, ppm) δ: 2,06 (4Н, s), 2,41 (6H, s), 3,93(4H, brs), 7,95 (1H, dd,J=5,2 Hz, J=8,4 Hz), 8,17(1H, d, J=9,2 Hz), 8,31(1H, dd, J=2,4 Hz, J=9,4 Hz), 8,45(1H, s), 8,85(1H, d, J=2,0 Hz), 8,90 (1H, d, J=8,4 Hz), 9,04 (1H, d, J=4 Hz), 10,03 (замена 1Н, s, D2O исчезла).
4. В 100 мл колбе перемешивали соль метансульфоновой кислоты (10 г), полученную на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение IV (5,4 г).
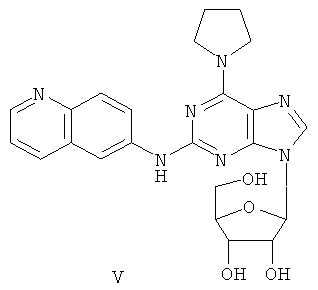
Пример 5 Получение соединения V
Соединение IV (2,5 г) и бис (триметилсилил) ацетамид (3,5 мл) смешивали в безводном ацетонитриле (10 мл). Вышеуказанную смесь перемешивали при комнатной температуре в течение 1 часа. Далее к смеси добавляли раствор тетраацетил робофуранозы (3,5 г), растворенной в ацетонитриле (8 мл), и тиметилсилилтрифлата (0,6 мл) и нагревали при дефлегмировании в течение 5 часов. К смеси добавляли бычий сывороточный альбумин (0,7 мл) и далее перемешивали в течение 24 часов. Завершение реакции проверяли методом тонкопленочной хроматографии. Растворитель концентрировали при пониженном давлении, и осадок растворяли в метаноле (15 мл). Через вышеуказанную смесь пропускали газообразный аммиак в течение 1,5 часа. Растворитель удаляли при пониженном давлении, остаток очищали методом колончатой хроматографии на силикагеле и получали соединение V (2,3 г).
(+)-ESI MS m/z: 464 [М+Н]+.
Пример 6 Получение соединения VI
1. 60% NaH (0,4 г) и безводный ацетонитрил (50 мл) смешивали с соединением IV (2,5 г). Вышеуказанную смесь перемешивали в защитной атмосфере азота в течение 30 минут. К смеси в течение 10 минут добавляли по порциям 3,5-ди-пара-толуолсульфонил-2-дезокси-β-D-рибофураноза-1-хлорид (2,9 г). В смеси протекала реакция дополнительно в течение 2 часов. После фильтрования фильтрат концентрировали до сухого состояния и получали масляное вещество. Далее масляное вещество очищали методом колончатой хроматографии и получали твердое вещество (2,5 г).
2. Вышеуказанное твердое вещество, 50% метоксид натрия (0,7 г) и метанол (100 мл) смешивали, и между ними протекала реакция при комнатной температуре в течение 5 часов при постоянном перемешивании. Значение pH вышеуказанной смеси регулировали до нейтрального значения с помощью уксусной кислоты. Растворитель отгоняли, остаток очищали методом колончатой хроматографии и получали соединение VI (1,4 г).
(+)-ESI MS m/z: 448 [М+Н]+.
Пример 7-9 Получение соединений формулы VII, VIII, IX
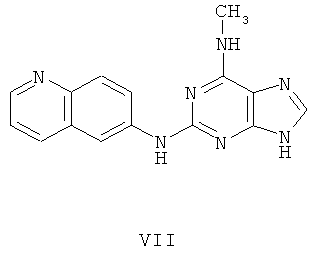
Пример 7 Получение соединения VII
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли метиламингидрохлорид (4,6 г), далее в течение 30 минут к нему добавляли триэтиламин (21 мл) при этой температуре, и далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (11,0 г). Выход составил приблизительно 77,7%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (10,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (10,7 г). Выход составил приблизительно 82,2% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,3 г).
1H-NMR (DMSO-d6, ppm) δ: 2,44 (6Н, s), 3,15 (3H, s), 7,95 (1H, dd, J=5,2 Hz, J=8,0 Hz), 8,18 (1H, d, J=8,8 Hz), 8,33 (1H, dd, J=2,4 Hz, J=9,4 Hz), 8,70 (1H, s), 8,86 (1H, d, J=2,0 Hz), 8,91 (1H, d, J=8,4 Hz), 9,05 (1H, dd, J=1,2 Hz, J=5,2 Hz), 10,14 (замена 1H, s, D2O исчезала).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение VII (5,2 г).
Пример 8 Получение соединения VIII
Свободное основание соединения VII (2,5 г) и бис (триметилсилил) ацетамид (3,7 мл) смешивали в безводном ацетонитриле (10 мл). Вышеуказанную смесь перемешивали при комнатной температуре в течение 1 часа. Далее к смеси добавляли раствор тетраацетил робофуранозы (3,5 г), растворенной в ацетонитриле (8 мл), и тиметилсилилтрифлата (0,6 мл) и нагревали при дефлегмировании в течение 5 часов. К смеси добавляли бычий сывороточный альбумин (0,7 мл) и далее перемешивали в течение 24 часов. Завершение реакции проверяли методом тонкопленочной хроматографии. Растворитель концентрировали при пониженном давлении, и осадок растворяли в метаноле (15 мл). Через вышеуказанную смесь пропускали газообразный аммиак в течение 1,5 часа. Растворитель удаляли при пониженном давлении, остаток очищали методом колончатой хроматографии на силикагеле и получали соединение VIII (2,7 г).
(+)-ESI MS m/z: 424 [M+H]+.
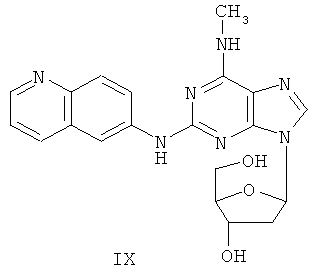
Пример 9 Получение соединения IX
1. 60% NaH (0,42 г) и безводный ацетонитрил (50 мл) смешивали с соединением VII (2,5 г). Вышеуказанную смесь перемешивали в защитной атмосфере азота в течение 30 минут. К смеси в течение 10 минут добавляли по порциям 3,5-ди-пара-толуолсульфонил-2-дезокси-β-D-рибофураноза-1-хлорид (3,5 г). В смеси протекала реакция в течение 2 часов. После фильтрования фильтрат концентрировали до сухого состояния и получали масляное вещество. Далее масляное вещество очищали методом колончатой хроматографии и получали твердое вещество (2,3 г).
2. Вышеуказанное твердое вещество, 50% метоксид натрия (0,75 г) и метанол (100 мл) смешивали, и между ними протекала реакция при комнатной температуре в течение 5 часов при постоянном перемешивании. Значение pH вышеуказанной смеси регулировали до нейтрального значения с помощью уксусной кислоты. Растворитель отгоняли, остаток очищали методом колончатой хроматографии и получали соединение IX (1,2 г).
(+)-ESI MS m/z: 408 [M+H]+.
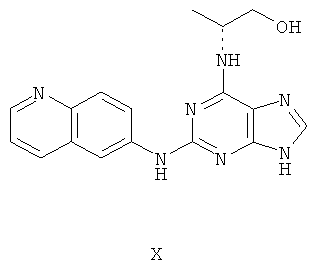
Пример 10 Получение соединения Х
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (7,9 г), и далее к нему добавляли DL-аминопропанол (7,0 мл) при этой температуре. Далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (11,6 г). Выход составил приблизительно 70,4%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,6 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,5 г). Выход составил приблизительно 79,0% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после Завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (12,3 г).
1H-NMR (DMSO-d6, ppm) δ: 1,32 (3H, d, J=6,4 Hz), 2,44 (6H, s), 3,63 (2H, m), 4,43 (1H, brs), 7,96 (1H, dd, J=5,2 Hz, J=8,4 Hz), 8,00 (1H, brs), 8,19 (1H, d, J=9,2 Hz), 8,29 (1H, dd, J=2,4 Hz, J=9,4 Hz), 8,82 (1H, s), 8,85 (1H, d, J=2,0 Hz), 8,90 (1H, d, J=8,4 Hz), 9,05 (1H, dd, J=1,2 Hz, J=5,2 Hz), 10,18 (1H, s).
4. В 100 мл колбе перемешивали метансульфонат (12 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение Х (6,5 г).
(+)-ESI MS m/z: 335 [M+H]+.
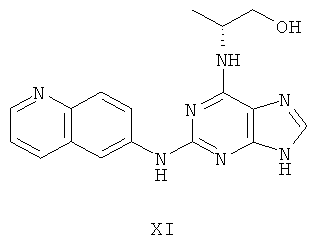
Пример 11 Получение соединения XI
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (7,9 г), и далее к нему добавляли L-аминопропанол (7,0 мл). Далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,2 г). Выход составил приблизительно 74,0%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,6 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,1 г). Выход составил приблизительно 83,2% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,9 г).
1H-NMR (DMSO-d6, ppm) δ: 1,32 (3H, d, J=6,8 Hz), 2,44 (6H, s), 3,63 (2H, m), 4,43 (1H, brs), 7,96 (1H, dd, J=5,2 Hz, J=8,4 Hz), 8,01 (1H, brs), 8,19 (1H, d, J=9,2 Hz), 8,29 (1H, dd, J=2,4 Hz, J=9,4 Hz), 8,82 (1H, s), 8,85 (1H, d, J=2,4 Hz), 8,90 (1H, d, J=8,8 Hz), 9,05 (1H, d, J=1,2 Hz, J=5,2 Hz), 10,19 (1H, s).
4. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XI (5,8 г).
Пример 12 Получение соединения XII
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли метилпиперазин (9,0 г) и триэтиламин (8 мл). Далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,0 г). Выход составил приблизительно 67,4%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,8 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,0 г). Выход составил приблизительно 77,8% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали соединение (10,0 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,5 г).
4. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество светло-желтого цвета. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XII (5,9 г).
(+)-ESI MS m/z: 361 [M+H]+.
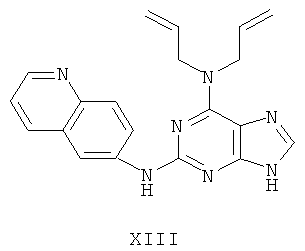
Пример 13 Получение соединения XIII
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли диаллиламин (11,4 мл) при этой температуре в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,9 г). Выход составил приблизительно 73,1%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (12,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,2 г). Выход составил приблизительно 79,7% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (10,8 г).
1H-NMR (DMSO-d6, ppm) δ: 2,44 (6H, s), 4,61 (4H, s), 5,19-5,27 (4H, m), 6,01 (2H, m), 7,98 (1Н, dd, J=5,6 Hz, J=8,6 Hz), 8,14-8,18 (2H, m), 8,32 (1H, dd, J=2,4 Hz, J=9,2 Hz), 8,82 (1H, d, J=2,0 Hz), 8,86 (1H, d, J=8,8 Hz), 9,04 (1H, dd, J=1,6 Hz, J=5,4 Hz), 9,91 (замена 1H, s, D2O исчезла).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XIII (5,6 г).
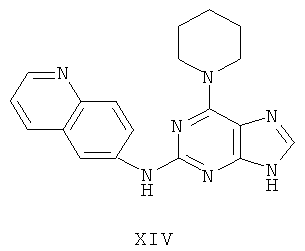
Пример 14 Получение соединения XIV
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли пиперидин (9,2 мл) при этой температуре в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,8 г). Выход составил приблизительно 75,2%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,5 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,3 г). Выход составил приблизительно 75,9% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,7 г).
1H-NMR (DMSO-d6, ppm) δ: 1,70 (6Н, m), 2,44 (6H, s), 4,19 (4H, s), 7,99 (1H, dd, J=5,2 Hz, J=8,2 Hz), 8,19 (2H, m), 8,32(1H, dd, J=2,0 Hz, J=9,4 Hz), 8,79(1H, d, J=2,0 Hz), 8,91(1H,d,J=8,0 Hz), 9,05 (1H, dd, J=1,2 Hz, J=5,2 Hz), 9,95 (замена 1H, s, D2O исчезала).
4. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XIV (6,0 г).
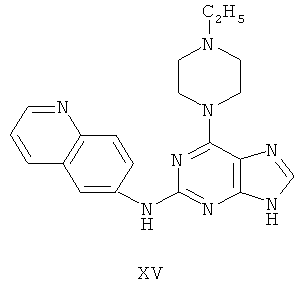
Пример 15 Получение соединения XV
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли N-этилпиперазин (10,3 г) при этой температуре в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (13,0 г). Выход составил приблизительно 70,0%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (12,2 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (13,2 г). Выход составил приблизительно 83,0% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (10,9 г).
1H-NMR (DMSO-d6, ppm) δ: 1,30 (3H, d, J=7,4 Hz), 2,43 (6H, s),3,21 (4H, m), 3,59 (2H, m), 3,70 (2H, m), 5,45 (2H, m), 7,99 (1H, dd, J=5,2 Hz, J=8,4 Hz), 8,08 (1H, brs), 8,20 (1H, d, J=9,2 Hz), 8,34 (1H, dd, J=2,0 Hz, J=9,4 Hz), 8,82 (1H, d, J=2,4 Hz), 8,98 (1H, d, J=8,4 Hz), 9,05 (1H, dd, J=1,2 Hz, J=5,6 Hz), 9,77 (1H, brs), 9,93 (1H, s).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XV (5,8 г).
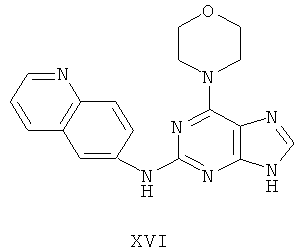
Пример 16 Получение соединения XVI
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (7,9 мл) и к нему добавляли морфолин (7,9 мл) при этой температуре. Далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (11,2 г). Выход составил приблизительно 65,4%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,6 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,5 г). Выход составил приблизительно 76,8% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали соединение (10,0 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,7 г).
1H-NMR (DMSO-d6, ppm) δ: 2,42 (6Н, s), 3,78 (4Н, m), 4,22 (4H, s), 7,98 (1H, dd, J=5,2 Hz, J=8,4 Hz), 8,10 (1H, s), 8,17 (1H, d, J=9,2 Hz), 8,32 (1H, dd, J=2,0 Hz, J=9,4 Hz), 8,80 (1H, d, J=1,6 Hz), 8,94 (1H, d, J=8,8 Hz), 9,04 (1H, d, J=5,2 Hz), 9,89 (1H, s).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XVI (5,4 г).
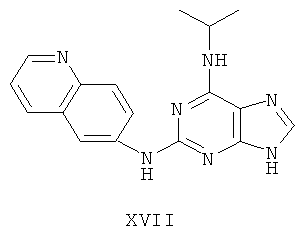
Пример 17 Получение соединения XVII
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли изопропиламин (7,7 мл) при дефлегмировании в течение 15 минут. Далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (10,8 г). Выход составил приблизительно 70,0%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,7 г). Выход составил приблизительно 83,6% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали соединение (10,0 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (9,7 г).
1H-NMR (DMSO-d6, ppm) δ: 1,35 (6H, d, J=6,4 Hz), 2,47 (6H, s), 4,35 (1H, brs), 7,97 (1H, dd, J=5,2 Hz, J=8,6 Hz), 8,09 (1H, brs), 8,20 (1H, d, J=9,2 Hz), 8,31(1H, dd, J=2,0 Hz, J=9,4 Hz), 8,83(2H, наложенный), 8,89 (1H, d, J=8,4Hz), 9,06 (1H, dd, J=1,2 Hz, J=5,2 Hz), 10,20 (1H, s).
(+)-ESI MS m/z: 320 [M+H]+.
4. В 100 мл колбе перемешивали метансульфонат (9 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество светло-желтого цвета. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XVII (4,8 г).
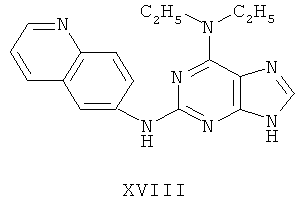
Пример 18 Получение соединения XVIII
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли диэтиламин (9,6 мл) при этой температуре в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,3 г). Выход составил приблизительно 75,1%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (11,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,9 г). Выход составил приблизительно 82,2% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,3 г).
1H-NMR (DMSO-d6, ppm) δ: 1,30 (6Н, t, J=7,0 Hz), 2,44 (6H, s), 3,97 (4Н, brs), 8,02 (1H, dd, J=5,6 Hz, J=8,4 Hz), 8,21 (1H, d, J=9,2 Hz), 8,31-8,35 (2H, m), 8,85 (1H, d, J=2,0 Hz), 8,94 (1H, d, J=8,4 Hz), 9,09 (1H, d, J=5,2 Hz).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XVIII (5,5 г).
(+)-ESI MS m/z: 334 [M+H]+.
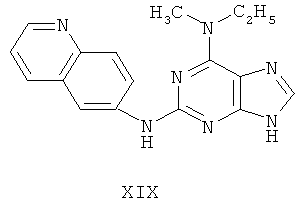
Пример 19 Получение соединения XIX
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и к нему добавляли метилэтиламин (7,7 мл) при дефлегмировании в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердое вещество (10,3 г). Выход составил приблизительно 65,9%.
2. В 250 мл трехгорловую колбу поочередно добавляли 6-аминохинолин (5,0 г), 2-хлор-N-метил-N-этил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (10,3 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (10,5 г). Выход составил приблизительно 75,0% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали соединение (10,0 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,2 г).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XIX (5,4 г).
Соединение XIX: (+)-ESI MS m/z: 320 [M+H]+.
Пример 20 Получение соединения XX
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли диметиламин гидрохлорид (7,3 мл) и к нему добавляли триэтиламин (22 мл) при этой температуре в течение 30 минут, далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (8,9 г). Выход составил приблизительно 59,8%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (8,9 г), полученный на предыдущем этапе, 6-аминохинолин (4,5 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (10,5 г). Выход составил приблизительно 86,4% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (9,8 г).
4. В 100 мл колбе перемешивали метансульфонат (9 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XX (4,9 г).
(+)-ESI MS m/z: 306 [M+H]+.
Пример 21 Получение соединения XXI
1. В 100 мл трехгорловой колбе смешивали 2,6-дихлорпурин (10 г), этилацетат (50 мл), соль пиридиния паратолуолсульфоновой кислоты (0,2 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 5 минут к ней добавляли 2,3-дигидропиран (12 мл). Далее в смеси протекала реакция при 50~60°С в течение 3 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (8 мл) и пиперазин (7,3 мл) в течение 20 минут, далее в смеси протекала реакция при этой температуре в течение 1 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердый пурин (12,4 г). Выход составил приблизительно 72,7%.
2. В 250 мл трехгорловую колбу поочередно добавляли пурин (12,0 г), полученный на предыдущем этапе, 6-аминохинолин (5,0 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,4 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,7 г). Выход составил приблизительно 85,0% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (12,1 г).
1H-NMR (DMSO-d6, ppm) δ: 2,43 (9H, s), 3,22 (2H, m),3,54-3,66 (4Н, m), 5,41 (2H, m), 8,01 (1H, dd, J=5,2 Hz, J=8,6 Hz), 8,12 (1H, s), 8,21 (1H, d, J=9,6 Hz), 8,34 (1H, dd, J=2,0 Hz, J=9,0 Hz), 8,83 (1H, d, J=2,0 Hz), 8,99 (1H, d, J=8,8 Hz), 9,06 (1H, d, J=4,2 Hz), 9,98 (замена 2Н, brs, D2O исчезала).
4. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл). К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXI (5,3 г).
Пример 22 Получение соединения XXII
1. В 3000 мл трехгорловой колбе смешивали 2,6-дихлорпурин (300 г), этилацетат (1500 мл) и соль пиридиния паратолуолсульфоновой кислоты (3 г). Вышеуказанную смесь перемешивали и нагревали до температуры 35°С, в течение 30 минут к ней добавляли 2,3-дигидропиран (360 мл). Далее в смеси протекала реакция при 50~60°С в течение 5 ч. Завершение реакции проверяли методом тонкослойной хроматографии. В колбу добавляли триэтиламин (240 мл) и к нему добавляли циклопропиламин (204 мл) при дефлегмировании в течение 30 минут. Далее в смеси протекала реакция при этой температуре в течение 0,5 часа. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали этилацетатом, а фильтрат промывали водой 3 раза и расслаивали. Органический слой концентрировали до тех пор, пока не происходило выделение большого количества твердых частиц. После фильтрования осадок промывали этилацетатом 3 раза и далее высушивали в вакууме при 50°С в течение 5 часов и получали твердое соединение 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (364 г).
2. В 250 мл трехгорловую колбу поочередно добавляли 6-амино-8-метилхинолин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (10,2 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,2 г). Выход составил приблизительно 85,3% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10,0 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,3 г).
1H-NMR (DMSO-d6, ppm) δ: 0,75 (2Н, s), 0,95 (2Н, m), 2,41 (6Н, s), 2,76 (3Н, s), 3,12 (1Н, brs), 7,84 (1H, dd, J=4,8 Hz, J=8,2 Hz), 8,12 (1H, s), 8,50 (замена 1Н, brs, D2O исчезала), 8,71 (3Н, m), 8,93 (1H, d, J=4,0 Hz), 10,05 (замена 1H, s, D2O исчезала).
4. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXII (5,3 г).
Пример 23 Получение соединения XXIII
1. В 250 мл трехгорловую колбу поочередно добавляли 6-амино-8-метоксиилхинолин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (9,3 г), катализатор Pd(OAc)2 (0,25 г), лиганд 7 (0,25 г), трет-бутоксид натрия (4,5 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,0 г). Выход составил приблизительно 88,8% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали соединение (10,0 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,8 г).
1H-NMR (DMSO-d6, ppm) δ: 0,73 (2H, s), 0,93 (2H, m),2,37 (6Н, s), 3,14 (1H, brs), 4,13 (3H, s), 7,96 (2H, m), 8,30 (замена 1H, s, D2O исчезала), 8,64 (2H, brs), 8,86 (2H, m), 10,01 (замена 1H, s, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXIII (6,1 г).
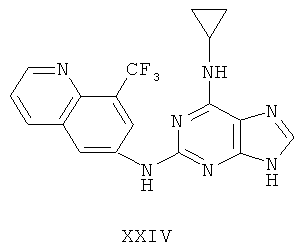
Пример 24 Получение соединения XXIV
1. В 250 мл трехгорловую колбу поочередно добавляли 6-амино-8-трифторметилхинолин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (7,6 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (4,0 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (8,5 г). Выход составил приблизительно 76,8% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (8 г), полученный на предыдущем этапе, ацетон (50 мл) и воду (40 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (3,8 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (8,7 г).
1H-NMR (DMSO-d6, ppm) δ: 0,69 (2H, s), 0,76 (2H, brs), 2,40 (6H, s), 2,97 (1H, brs), 7,77 (1H, dd, J=4,0 Hz, J=8,2 Hz), 8,46 (3H, m), 8,53 (1H, d, J=8,4 Hz), 8,86 (замена 1Н, brs, D2O исчезала), 9,10 (1H, d, J=4,0 Hz), 9,46 (замена 1H, brs, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (8 г), полученный на предыдущем этапе, и воду (40 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXIV (4,5 г).
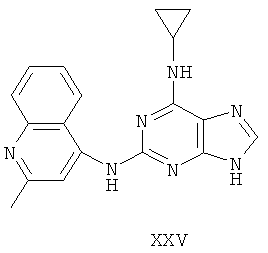
Пример 25 Получение соединения XXV
1. В 250 мл трехгорловую колбу поочередно добавляли 2-метил-4-аминохинолин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (10,2 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,0 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (11,8 г). Выход составил приблизительно 89,9% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали твердый метансульфонат (10,9 г).
1H-NMR (DMSO-d6, ppm) δ: 0,75 (2H, m), 0,87 (2H, m), 2,37 (6H, s), 2,79 (3H, s), 3,10 (1H, brs), 7,78 (1H, m), 8,02 (2H, m), 8,35 (замена 1Н, brs, D2O исчезала), 8,60 (1Н, m), 8,91 (2H, m), 10,68 (замена 1H, s, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXV (5,2 г).
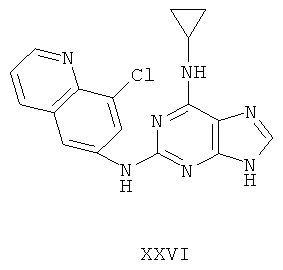
Пример 26 Получение соединения XXVI
1. В 250 мл трехгорловую колбу поочередно добавляли 8-хлор-6-аминохинолин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (9,0 г), катализатор Pd(OAc)2 (0,25 г), лиганд 7 (0,25 г), трет-бутоксид натрия (4,5 г) и простой этиленгликольдиметиловый эфир (100 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (4,6 г). Выход составил приблизительно 37,7% в расчете на аминохинолин.
3. В 250 мл одногорловой колбе смешивали конъюгат (4,5 г), полученный на предыдущем этапе, ацетон (30 мл) и воду (30 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (2 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали твердый метансульфонат (4,5 г).
1H-NMR (DMSO-d6, ppm) δ: 0,74 (2H, m), 0,98 (2H, m), 2,42 (6H, s), 3,07 (1H, s), 7,63 (1H, m), 8,32 (1H, d, J=8,4 Hz), 8,47-8,54 (2H, m), 8,74-8,87 (2H, m), 10,04 (замена 1H, brs, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (4 г), полученный на предыдущем этапе, и воду (25 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXVI (2,3 г).
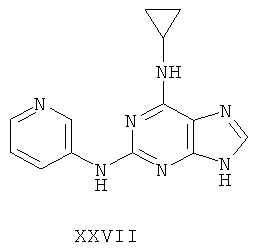
Пример 27 Получение соединения XXVII
1. В 250 мл трехгорловую колбу поочередно добавляли 3-аминопиридин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (16,0 г), катализатор Pd(OAc)2 (0,4 г), лиганд 7 (0,4 г), трет-бутоксид натрия (7,5 г) и простой этиленгликольдиметиловый эфир (130 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,9 г). Выход составил приблизительно 69,1% в расчете на аминопиридин.
3. В 250 мл одногорловой колбе смешивали соединение (10 г), полученное на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (6 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (12 г).
1H-NMR (DMSO-d6, ppm) δ: 0,68 (2H, m), 0,93 (2H, m), 2,38 (6H, s), 3,01 (1H, brs), 7,97 (1H, dd, J=5,6 Hz, J=8,8 Hz), 8,24 (замена of 1H, brs, D2O исчезала), 8,46 (1H, d, J=5,2 Hz), 8,54 (1H, brs), 8,69 (1H, d, J=8,4 Hz), 9,66 (1H, s), 10,25 (замена 1Н, brs, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXVII (5,3 г).
Пример 28 Получение соединения XXVIII
1. В 250 мл трехгорловую колбу поочередно добавляли 2-аминопиридин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (16,0 г), катализатор Pd(OAc)2 (0,4 г), лиганд 7 (0,4 г), трет-бутоксид натрия (7,5 г) и простой этиленгликольдиметиловый эфир (130 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (10,5 г). Выход составил приблизительно 56,0% в расчете на аминопиридин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (6 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (11,7 г).
1H-NMR (DMSO-d6, ppm) δ: 0,78 (2Н, m), 0,98 (2Н, m), 2,39 (6H, s), 3,06 (1H, brs), 7,30 (1H, m), 7,47 (1H, d, J=8,8 Hz), 8,14 (1H, m), 8,30 (1H, s), 8,47 (1H, s), 9,17 (замена 1H, brs, D2O исчезала), 11,71 (замена 1H, brs, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (11 г), полученный на предыдущем этапе, и воду (60 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXVIII (5,0 г).
Пример 29 Получение соединения XXIX
1. В 250 мл трехгорловую колбу поочередно добавляли 4-аминопиридин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (16,0 г), катализатор Pd(OAc)2 (0,4 г), лиганд 7 (0,4 г), трет-бутоксид натрия (7,5 г) и простой этиленгликольдиметиловый эфир (130 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (13,7 г). Выход составил приблизительно 73,4% в расчете на аминопиридин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (6 мл). Все твердые вещества растворяли до получения прозрачного раствора. Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (10,9 г).
1H-NMR (DMSO-d6, ppm) δ: 0,71 (2H, m), 0,92 (2H, m), 2,42 (6H, s), 3,05 (1H, brs), 8,38 (2H, brs), 8,54 (2H, m), 8,75 (1H, s), 11,03 (замена 1H, brs, D2O исчезала).
3. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXIX (5,0 г).
Пример 30 Получение соединения XXX
1. В 250 мл трехгорловую колбу поочередно добавляли паранитроанилин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (10,9 г), катализатор Pd(OAc)2 (0,3 г), лиганд 7 (0,3 г), трет-бутоксид натрия (5,8 г) и простой этиленгликольдиметиловый эфир (130 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,5 г). Выход составил приблизительно 85,6% в расчете на аминопиридин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (5 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (10,3 г).
1H-NMR (DMSO-d6, ppm) δ: 0,70 (2H, m), 0,95 (2H, m), 2,48 (6H, s), 3,06 (1H, brs), 8,13 (2H, m), 8,19 (2H, m), 8,49 (1H, brs), 8,99 (1H, s), 10,26 (1H, s).
3. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXX (5,1 г).
Пример 31 Получение соединения XXXI
1. В 250 мл трехгорловую колбу поочередно добавляли пара-толуидин (5,0 г), 2-хлор-N-циклопропил-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амин (13,7 г), катализатор Pd(OAc)2 (0,4 г), лиганд 7 (0,4 г), трет-бутоксид натрия (7,5 г) и простой этиленгликольдиметиловый эфир (130 мл). Вышеуказанную смесь перемешивали и нагревали до дефлегмирования, и реакция в смеси протекала в течение 2,5 часов при дефлегмировании. Завершение реакции проверяли методом тонкослойной хроматографии. Реакционную смесь охлаждали до комнатной температуры. После фильтрования осадок полностью промывали простым этиленгликольдиметиловым эфиром 2 раза, и фильтрат концентрировали до сухого состояния, остаток очищали методом колончатой хроматографии на силикагеле и получали конъюгат (12,3 г). Выход составил приблизительно 72,3% в расчете на аминопиридин.
3. В 250 мл одногорловой колбе смешивали конъюгат (10 г), полученный на предыдущем этапе, ацетон (60 мл) и воду (60 мл). Вышеуказанную смесь перемешивали и нагревали и затем добавляли метансульфоновую кислоту (6 мл). Далее в вышеуказанной смеси протекала реакция в течение 1 часа при дефлегмировании, и происходило естественное охлаждение смеси до комнатной температуры после завершения перемешивания. После фильтрования осадок промывали ацетоном 3 раза, затем высушивали в вакууме при 40°С в течение 6 часов и получали метансульфонат (10,8 г).
1H-NMR (DMSO-d6, ppm) δ: 0,79 (2H, m), 0,97 (2H, m), 2,34 (3H, s), 2,44 (6H, s), 7,21 (2H, d, J=8,4 Hz), 3,07 (1H, brs), 7,67 (2H, d, J=8,4 Hz), 8,49 (1H, s), 9,56 (1H, brs).
3. В 100 мл колбе перемешивали метансульфонат (10 г), полученный на предыдущем этапе, и воду (50 мл) и нагревали при постоянном перемешивании до его растворения. К смеси добавляли 10% раствор карбоната калия, значения pH регулировали до приблизительно 10, и затем выделялось твердое вещество. Вышеуказанную смесь охлаждали и фильтровали, осадок промывали ацетоном, высушивали в вакууме и получали соединение XXXI (5,0 г).
Были проведены испытания на противоопухолевую активность in vitro и in vivo с использованием соединений, полученных в вышеприведенных примерах. При этом использовали in vitro метод анализа с сульфородамином В и метод анализа МТТ, и время действия составило приблизительно 72 часа. Конкретные данные об активности приведены в таблице 1. Данные об ингибирующем действии соединений на рост рака Conlon26 у мышей приведены в таблице 2, и об ингибирующем действии соединений на рост саркомы S180 у мышей приведены в таблице 3.
|
|
Объяснение: перорально означает введение препарата через ротовой зонд; интраперитонеально означает введение препарата посредством абдоминальной инъекции. СТХ означает циклофосфамид для инъекции.
|
Объяснение: перорально означает введение препарата через ротовой зонд; интраперитонеально означает введение препарата посредством абдоминальной инъекции. СТХ означает циклофосфамид для инъекции.
Данные об активности in vitro в таблице 1 показывают, что большинство соединений обладают определенной противоопухолевой активностью, при этом соединения I, VII, XIII, XIV, XV, XXI и XXVI проявляют более высокую противоопухолевую активность по отношению к четырем различным раковым клеткам, в частности, активность соединения IV является максимальной. В соответствии с определенным результатом активности in vivo, при введении дозы 100 мг/кг соединение I обладает более сильным ингибирующим действием на рак Colon26 и саркому S180 у мышей. По сравнению с соединением I соединение XX обладает более сильным противоопухолевым действием. При введении дозы 100 мг/кг процент ингибирования соединения XX в отношении рака Colon26 у мышей достигает 72,31%, и его процент ингибирования саркомы S180 у мышей - 72,19%.
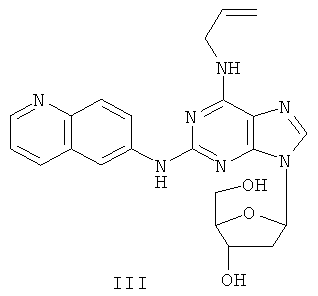
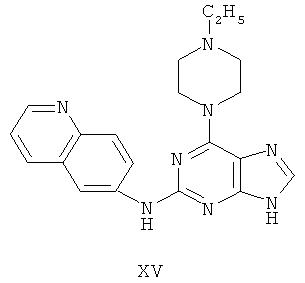
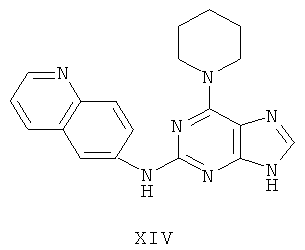
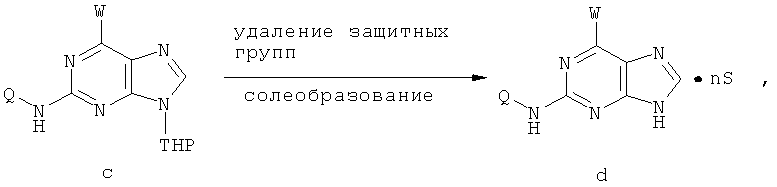
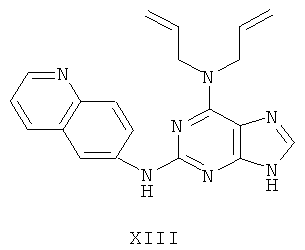
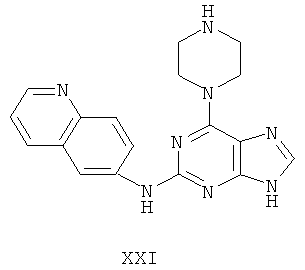

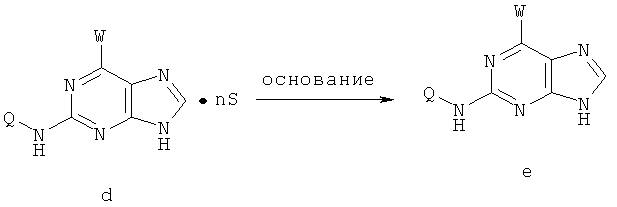
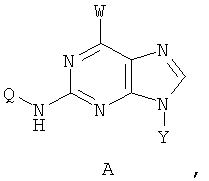
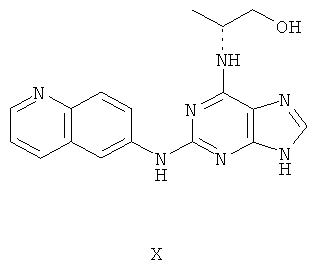

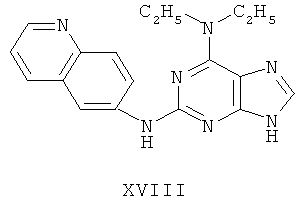
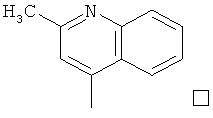
Соединения тиенопиридазина, их получение, содержащие их фармацевтические композиции, и их применение
Соединения тиенопиридазина, их получение, содержащие их фармацевтические композиции, и их применение

