Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ФТОРПОЛИМЕРОВ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к способу получения дисперсий фторполимеров, к дисперсиям фторполимеров, получаемым указанным способом, и к циклическим фторсодержащим поверхностно-активным веществам, применимым в указанном способе.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Фторполимеры, т.е. полимеры, имеющие фторированную основную цепь, давно известны и благодаря ряду их полезных свойств, таких как стойкость против теплового старения, химическая стойкость, устойчивость против атмосферных воздействий, стойкость к УФ-облучению и т.д., их используют в разнообразных областях применения.
Часто применяемый способ производства фторполимеров использует водно-эмульсионную полимеризацию одного или более фторированных мономеров, обычно с использованием фторированных поверхностно-активных веществ. Часто применяемые фторированные поверхностно-активные вещества включают перфтороктановые кислоты и их соли, в частности, аммониевую соль перфтороктановой кислоты.
В последнее время перфторалкановые кислоты, имеющие 8 или более атомов углерода, вызывают озабоченность с точки зрения экологической безопасности. Например, было найдено, что перфторалкановые кислоты демонстрируют способность к бионакоплению. В связи с этим такие соединения пытаются снять с производства и разрабатываются способы производства полимерных продуктов с применением альтернативных поверхностно-активных веществ, обладающих более благоприятным токсикологическим профилем.
С этой целью в последнее время применяют несколько различных подходов, обычно с участием фторсодержащих поверхностно-активных веществ, имеющих перфторалкильную цепь, прерываемую одним или более атомов кислорода, включенных в основную цепь, причем указанная цепь содержит ионную карбоксилатную группу на одном из ее концов.
Примеры этих соединений, которым придан улучшенный биоаккумуляционный профиль по сравнению с перфторалкановыми кислотами, имеющими 8 или более атомами углерода, можно найти, в частности, в US 2007276103 (3M INNOVATIVE PROPERTIES CO) 29.11.2007, US 2007015864 (3M INNOVATIVE PROPERTIES CO) 18.01.2007, US 2007015865 (3M INNOVATIVE PROPERTIES CO) 18.01.2007, US 2007015866 (3M INNOVATIVE PROPERTIES CO) 18.01.2007.
Таким образом, было бы желательно найти альтернативные фторированные поверхностно-активные вещества, которые можно применять при эмульсионной полимеризации фторированных мономеров, которые, желательно, показывают более низкое биологическое накопление и более низкую биологическую устойчивость, чем перфторалкановые кислоты, имеющие 8 или более атомов углерода.
Кроме того, было бы желательно, чтобы поверхностно-активные свойства указанных альтернативных фторированных поверхностно-активных веществ были бы такими, чтобы указанную полимеризацию можно было проводить традиционным и экономичным образом, используя оборудование, обычно применяемое для водно-эмульсионной полимеризации фторированных мономеров с традиционными поверхностно-активными веществами.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
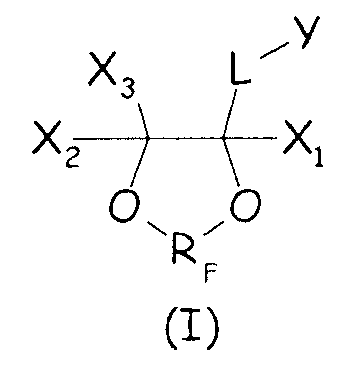
Было найдено, что циклические фторсодержащие соединения следующей формулы (I):
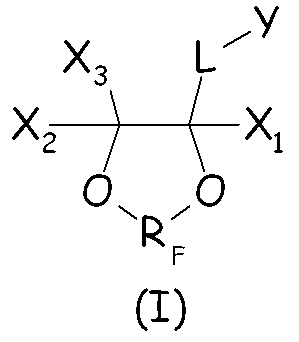
как подробно описано ниже, являются эффективными при водно-эмульсионной полимеризации, даже когда их применяют без добавления других поверхностно-активных веществ, таких как перфторалкановые кислоты и их соли.
Более того, авторы настоящего изобретения неожиданно обнаружили, что вышеуказанные циклические фторсодержащие соединения (I) обладают значительно улучшенными свойствами в отношении биологической устойчивости по сравнению с производными перфторалкановых кислот, что намного улучшает их токсикологический профиль.
И, наконец, эти циклические фторсодержащие соединения (I) обладают более высокой летучестью, чем производные перфторалкановых кислот, так что можно значительно понизить их остаточное содержание в конечных продуктах, получаемых из содержащих их дисперсий фторполимеров.
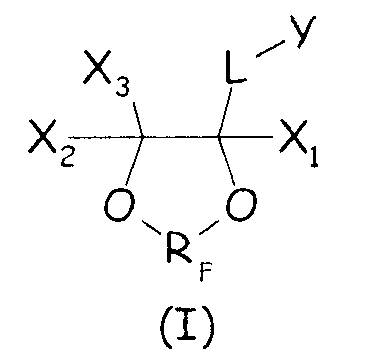
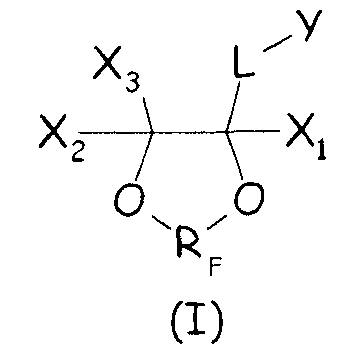
Таким образом, в одном аспекте настоящее изобретение относится к способу получения фторполимера, включающему водно-эмульсионную полимеризацию одного или более фторированных мономеров, при котором указанную водно-эмульсионную полимеризацию проводят в присутствии по меньшей мере одного циклического фторсодержащего соединения следующей формулы (I):
где X1, X2, X3 - одинаковые или отличные один от другого, являются независимо выбранными из H, F и C1-6-(пер)фторалкильных групп, необязательно содержащих один или более атомов кислорода, включенных или не включенных в цепь; L представляет собой связь или двухвалентную группу; RF представляет собой двухвалентную фторированную мостиковую С1-3-группу; Y представляет собой гидрофильную функциональную группу, выбранную из анионных функциональных групп, катионных функциональных групп и неионных функциональных групп.
Гидрофильную функциональную группу Y можно предпочтительно выбирать из неионных функциональных групп формулы -(ORH)n-OH, где RH представляет собой двухвалентную углеводородную группу, и n представляет собой целое число от 1 до 15.
В качестве альтернативы гидрофильную функциональную группу можно предпочтительно выбирать из катионных функциональных групп формул:
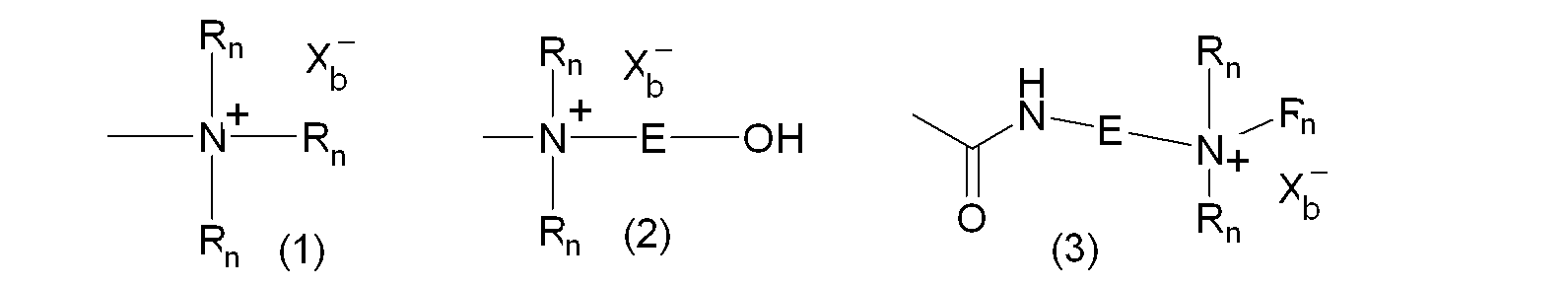
где Rn является одинаковым или различным в каждом случае его применения и представляет собой атом водорода или С1-6-углеводородную группу (предпочтительно алкильную группу), E представляет собой двухвалентную С1-3-углеводородную группу, и Xb - представляет собой анион, выбранный из OH-, Cl-, Br-, I-.
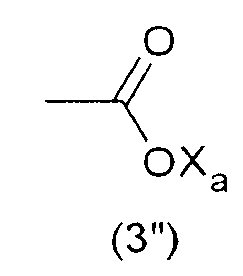
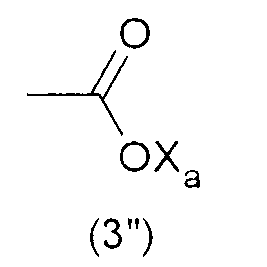
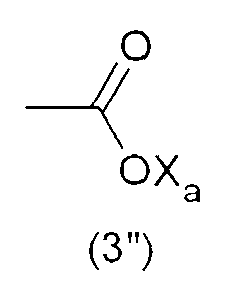
Тем не менее, гидрофильную функциональную группу Y предпочтительно выбирают из анионных функциональных групп, в частности, из функциональных групп формул:
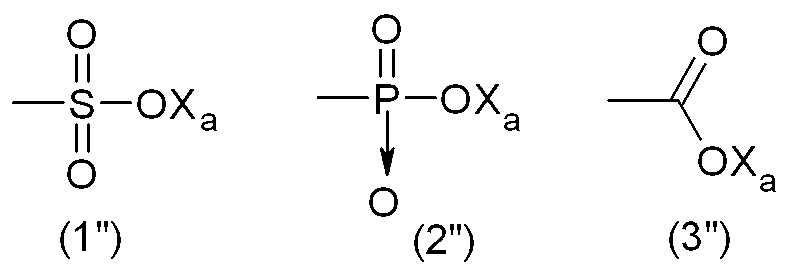
где Xa представляет собой Н, одновалентный металл, предпочтительно щелочной металл, или аммониевую группу формулы -N(R'n)4, где R'n - одинаковый или различный в каждом случае его применения, представляет собой атом водорода или С1-6-углеводородную группу (предпочтительно алкильную группу).
Наиболее предпочтительно, гидрофильная функциональная группа Y представляет собой карбоксилат формулы (3"), как подробно описано выше.
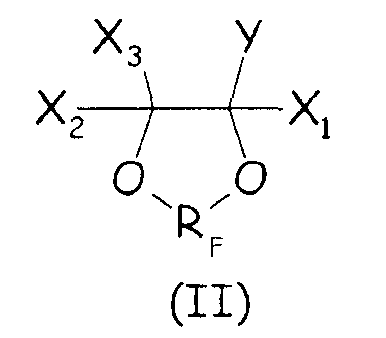
Согласно первому варианту осуществления настоящего изобретения циклическое фторсодержащее соединение соответствует формуле (II), представленной ниже:
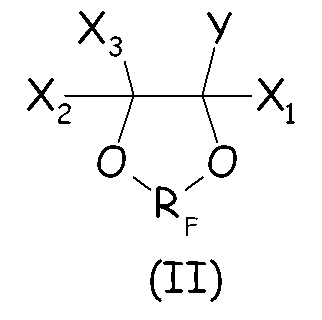
где X1, X2, X3, Y и RF имеют те же значения, что определено выше.
Более предпочтительно, циклическое фторсодержащее соединение соответствует формуле (III), представленной ниже:
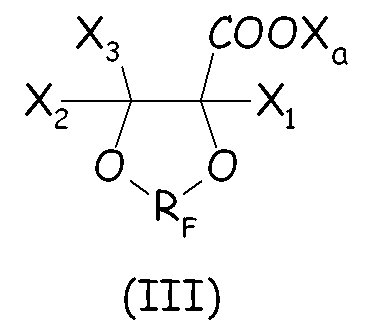
где RF, X1, X2, X3 и Xa имеют те же значения, что определено выше.
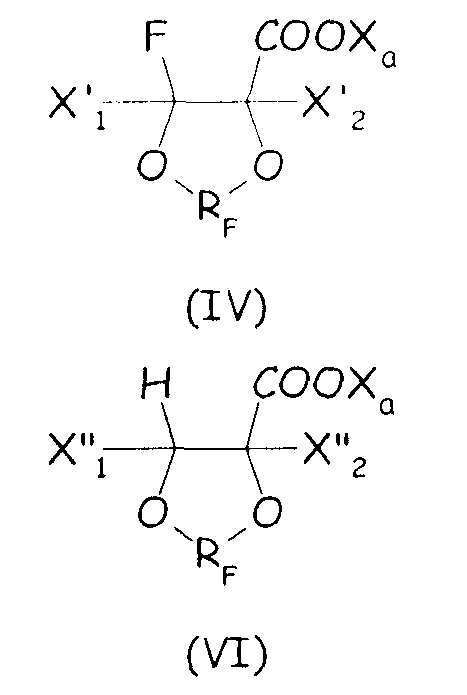
Согласно первому варианту этого предпочтительного способа осуществления циклическое фторсодержащее соединение соответствует формуле (IV):
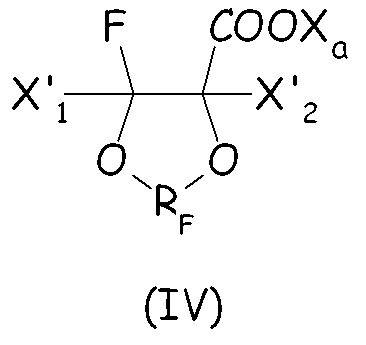
где X'1 и X'2, одинаковые или отличные один от другого, независимо представляют собой атом фтора, группу -R'f или группу -OR'f, где R'f представляет собой С1-3-перфторалкильную группу, предпочтительно при условии, что по меньшей мере один из X'1 и X'2 является отличным от фтора, и RF, и X3 имеют те же значения, что определены выше.
Соединения формулы (IV) можно предпочтительно получать по реакции перфтораллилфторсульфатных производных, имеющих формулу:
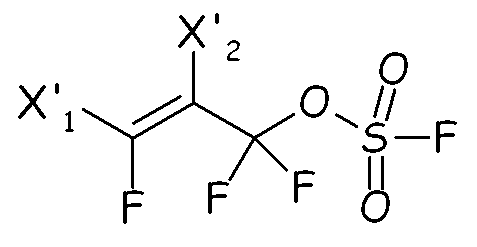
с бис-гипофторитом формулы:
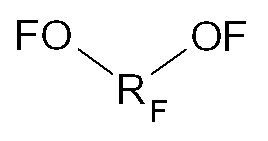
так, чтобы получать соответствующий аддукт, имеющий формулу:
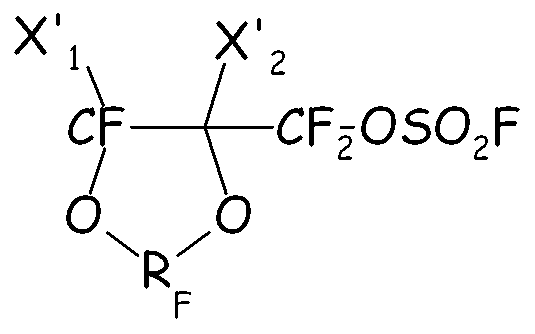
из которого посредством гидролиза получают желаемое соединение (IV).
Гидролиз вышеуказанного аддукта предпочтительно осуществляют посредством щелочного гидролиза водным раствором неорганического основания, например, водным раствором КОН, необязательно с последующей обработкой водным кислотным раствором (например, HClaq) для получения карбоновых кислот и/или дальнейшей нейтрализации для введения требующегося противокатиона в карбоксильной группу.
В альтернативном способе получения циклических фторсодержащих соединений формулы (IV), указанных выше в настоящем документе, проводят реакцию циклического фторолефина с карбонилфторидом в присутствии фторидов, как представлено на схеме, представленной ниже:
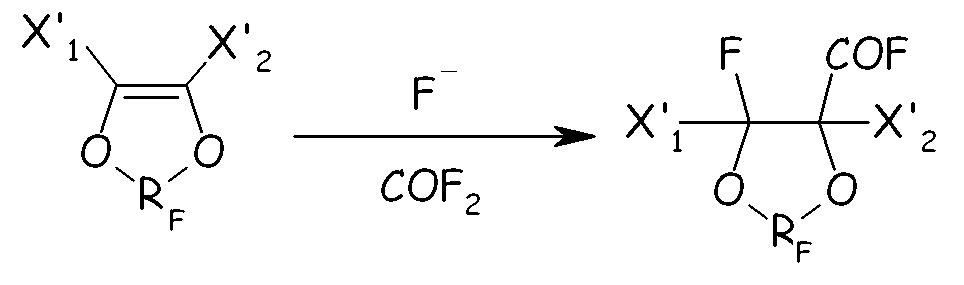
где X'1, X'2, RF имеют значения, как определено выше.
Карбонилфторидные производные, полученные таким образом, можно легко гидролизовать с получением желаемого соединения (IV).
В еще одном альтернативном способе циклическое фторсодержащее соединение формулы (IV) можно получать, добавляя метанол к циклическому фторолефину для получения циклического фторированного метанольного производного, как представлено на схеме, представленной ниже в настоящем документе:
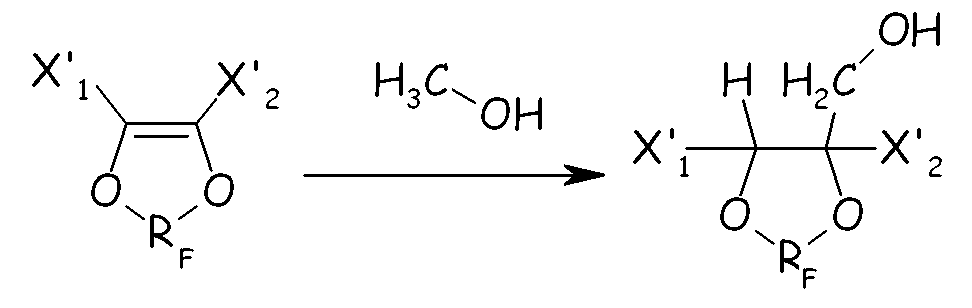
где X'1, X'2 и RF имеют значения, как определено выше. Циклическое спиртовое производное можно далее преобразовывать в соединение (IV) по следующим стадиям:
(i) этерификация циклического спирта фторированным ацилфторидом с получением соответствующего сложного эфира:
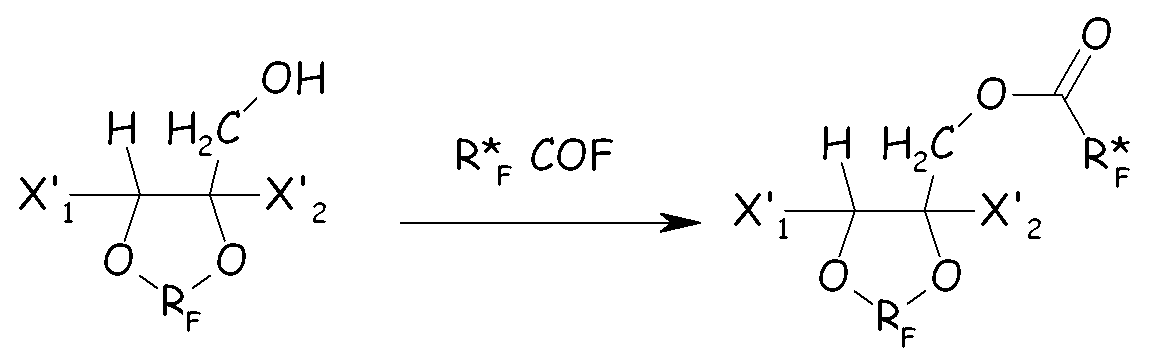
(ii) полное фторирование всех связей С-Н, преобразуя их в связи С-F этого последнего, с получением соответствующего перфторированного сложноэфирного соединения:
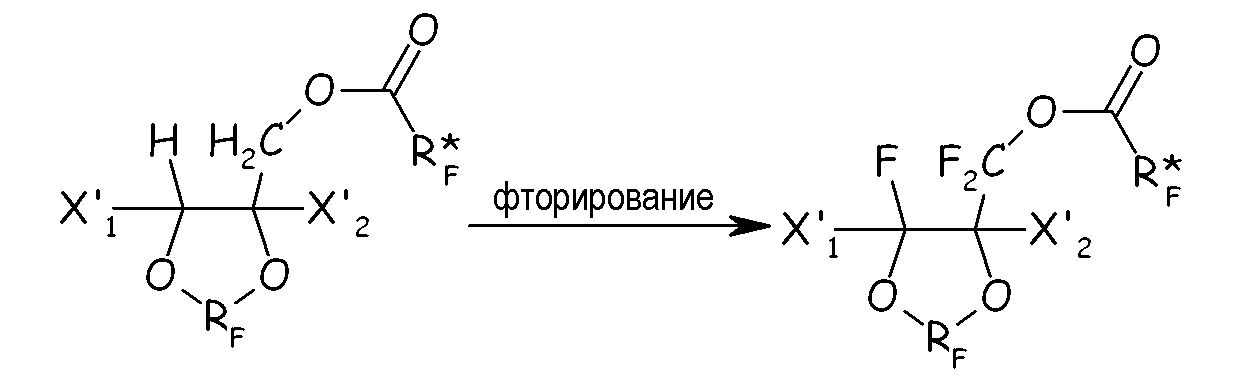
(iii) разложение перфторированного сложного эфира с получением соответствующего перфторацильного соединения:
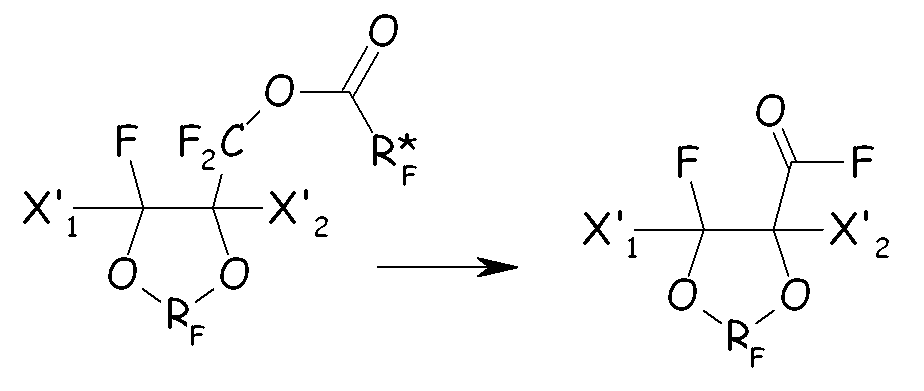
(iv) гидролиз и обработка основанием с получением соответствующего карбоксилатного производного (IV):
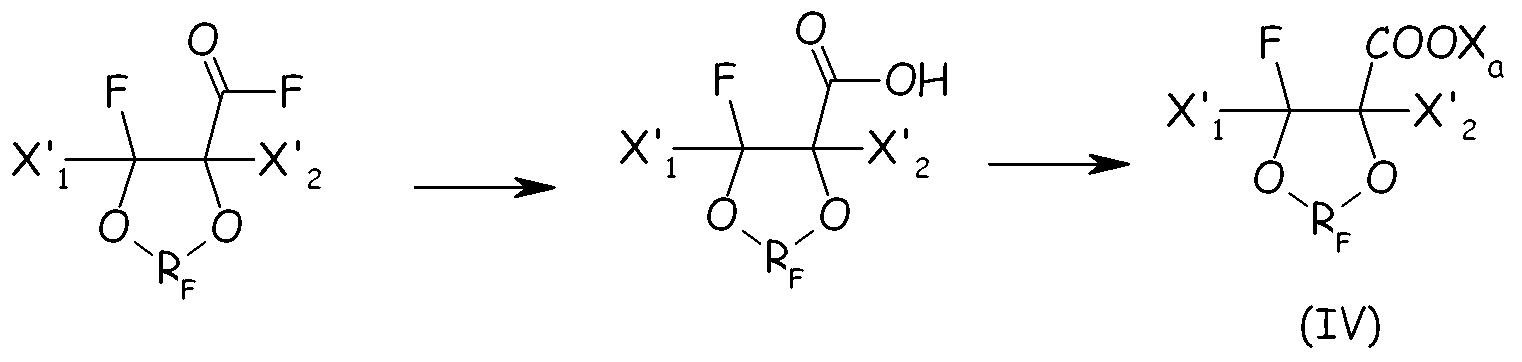
где во всех формулах выше в настоящем документе X'1, X'2, RF и Xa имеют значения, как определено выше; R* F представляет собой (пер)фторуглеродную группу.
Любой другой способ, обеспечивающий полное фторирование связей С-Н, но сохраняющий спиртовую/карбоксильную функциональную группу в защищенной форме, также может подходить для преобразования вышеуказанного циклического фторированного метанольного производного в соединение (IV).
Циклическое фторсодержащее соединение (IV) первого варианта этого предпочтительного способа осуществления настоящего изобретения более предпочтительно соответствует формуле (V):
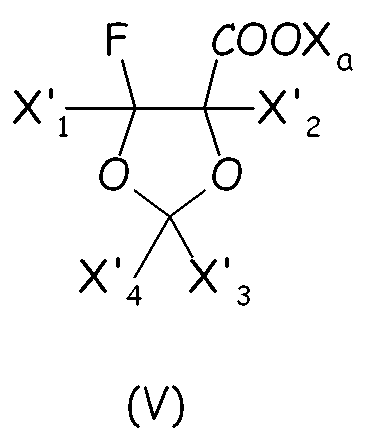
где X'1, X'2, X'3, X'4, одинаковые или отличные один от другого, независимо представляют собой атом фтора, -R'f или -OR'f, где R'f представляет собой С1-3-перфторалкильную группу.
Неограничивающие примеры циклических фторсодержащих соединений формулы (V) предпочтительно представляют собой:
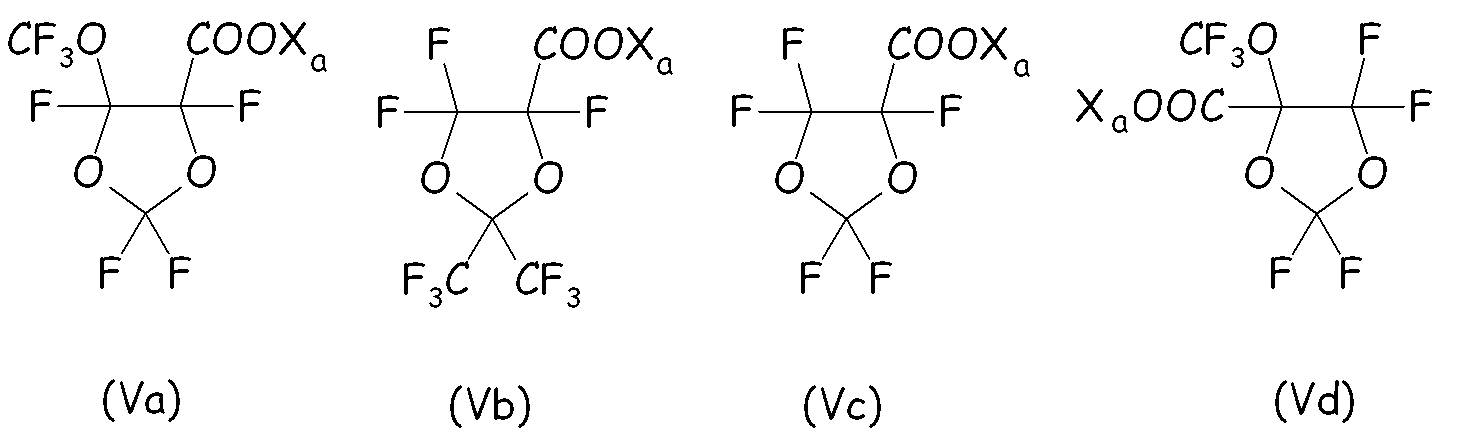
Согласно второму варианту этого предпочтительного способа осуществления настоящего изобретения циклическое фторсодержащее соединение соответствует формуле (VI), представленной ниже:
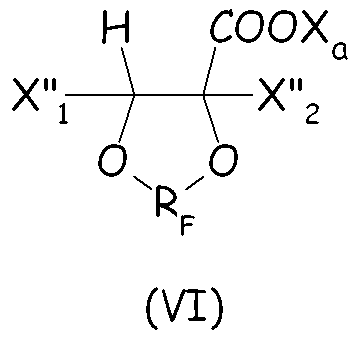
где X"1 и X"2, одинаковые или отличные один от другого, независимо представляют собой атом фтора, группу -R'f или группу -OR'f, где R'f представляет собой С1-3-перфторалкильную группу, и RF и Xa имеют те же значения, что определены выше.
Циклическое фторсодержащее соединение формулы (VI) можно получать, добавляя углеводородный первичный спирт к циклическому фторолефину для получения циклического фторированного спиртового производного, как представлено на схеме, представленной ниже:
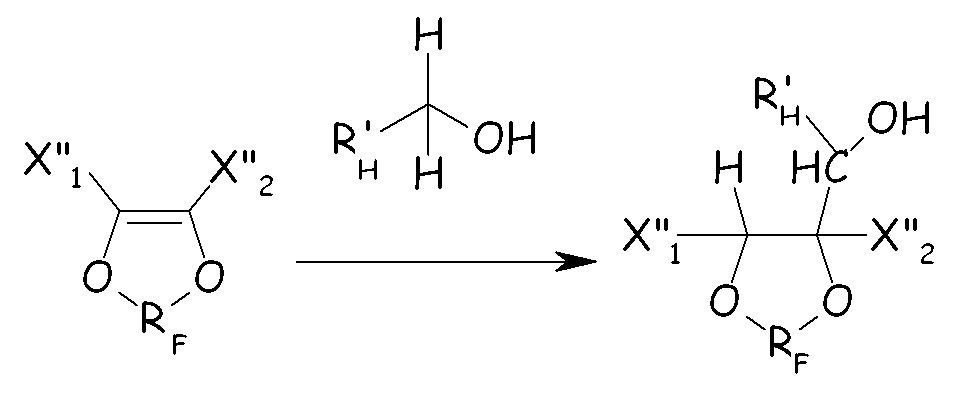
где X"1, X"2 и RF имеют значения, как определено выше, и R'H представляет собой Н или С1-6-углеводородную группу.
Подходящие углеводородные спирты включают алифатические спирты, такие как низшие первичные алканолы, имеющие от 1 до 4 атомов углерода. Конкретные примеры включают метанол, этанол, пропанол и бутанол, причем метанол является особо предпочтительным.
Реакцию фторированного олефина со спиртом можно проводить, как описано в CHAMBERS, R. D. Fluorine in Organic Chemistry. Oxford (UK): Blackwell Publishing, 2004. ISBN 0849317908. p.199 and ss.
Получаемое в результате циклическое фторированное спиртовое производное можно химически окислять некоторым окислителем до соответствующего производного карбоновой кислоты (необязательно с последующими подходящими стадиями гидролиза/нейтрализации), как представлено ниже:
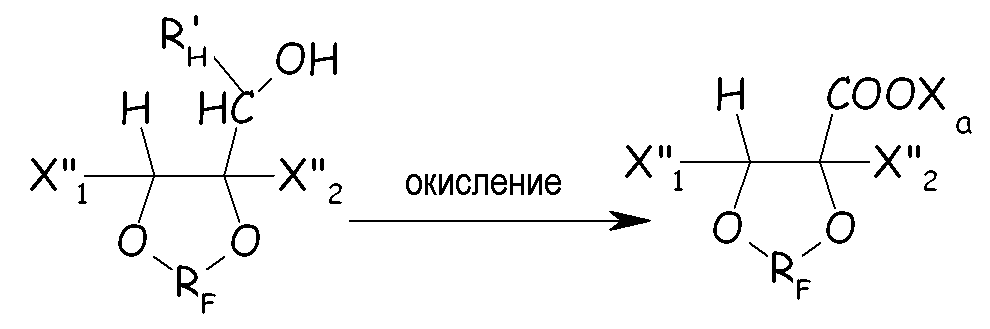
где X"1, X”2, R'H, RF и Xa имеют те же значения, что определены выше.
Примеры окислителей включают, например, перманганат калия, оксид хрома (VI), RuO4 или OsO4 необязательно в присутствии NaOCl, азотной кислоты с железом в качестве катализатора, четырехокиси азота. Обычно окисление проводят в кислых или основных условиях, предпочтительно в основных условиях, при температуре между 10° и 100°C. Кроме химического окисления можно также применять электрохимическое окисление.
Циклическое фторсодержащее соединение (VI) второго варианта этого предпочтительного способа осуществления настоящего изобретения более предпочтительно соответствует формуле (VII):
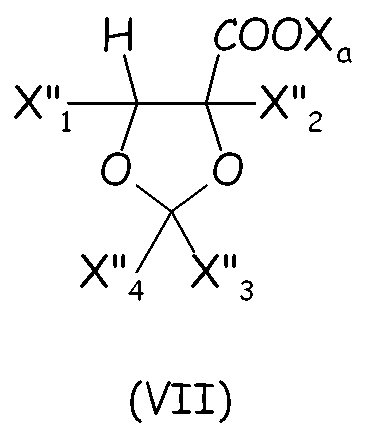
где X"1, X"2, X"3, X"4, одинаковые или отличные один от другого, независимо представляют собой атом фтора, -R'f или -OR'f, где R'f представляет собой С1-3-перфторалкильную группу.
Неограничивающие примеры циклического фторсодержащего соединения формулы (VII) предпочтительно представляют собой:
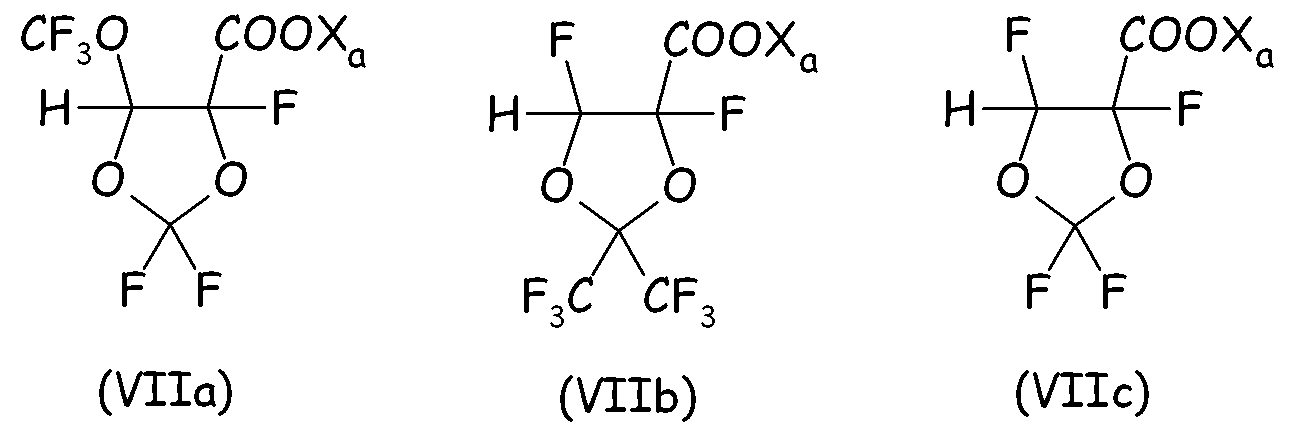
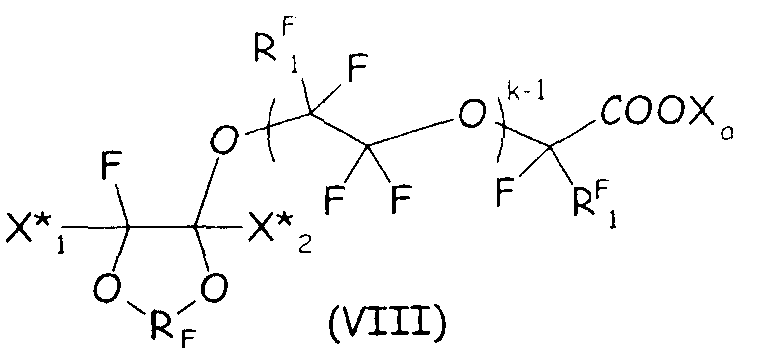
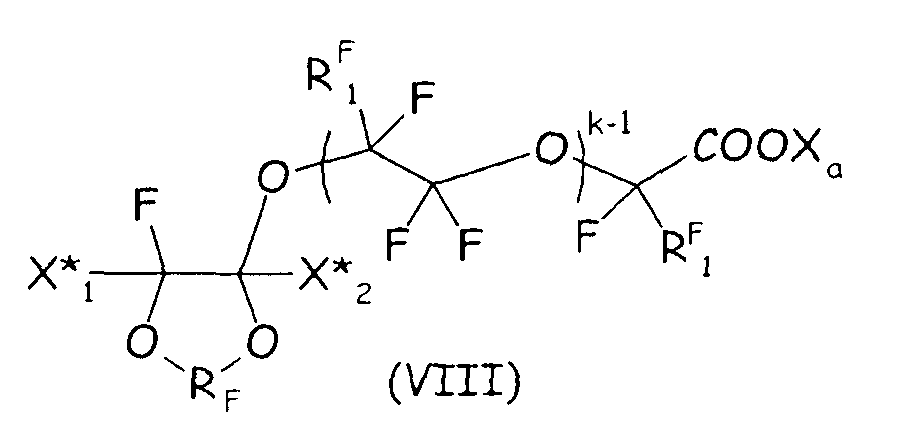
Согласно второму варианту осуществления настоящего изобретения циклическое фторсодержащее соединение соответствует формуле (VIII), представленной ниже:
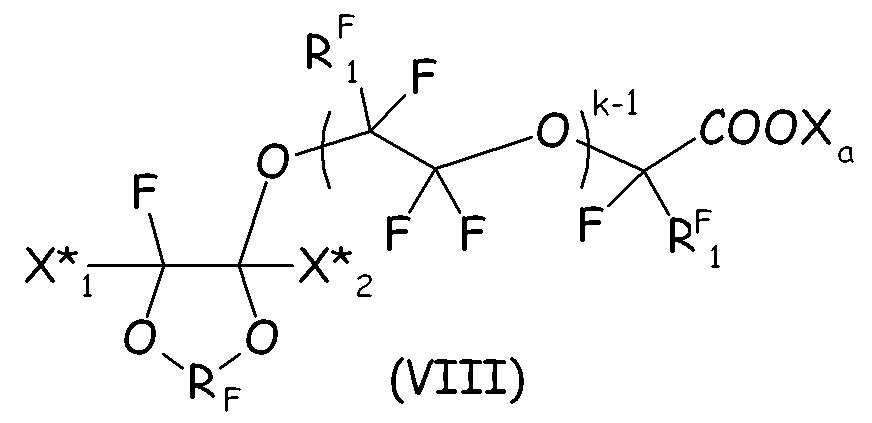
где RF и Xa имеют те же значения, что подробно описаны выше; X* 1, X* 2, одинаковые или отличные один от другого, независимо представляют собой атом фтора, -R'f или -OR'f, где R'f представляет собой С1-3-перфторалкильную группу; RF 1 представляет собой F или CF3, k представляет собой целое число от 1 до 3.
Соединения формулы (VIII) предпочтительно можно получать посредством реакции ненасыщенного фтордиоксола с гидрогенизированным производным гликоля, как представлено ниже, так чтобы получать соединение формулы (X), являющееся продуктом однократного присоединения:
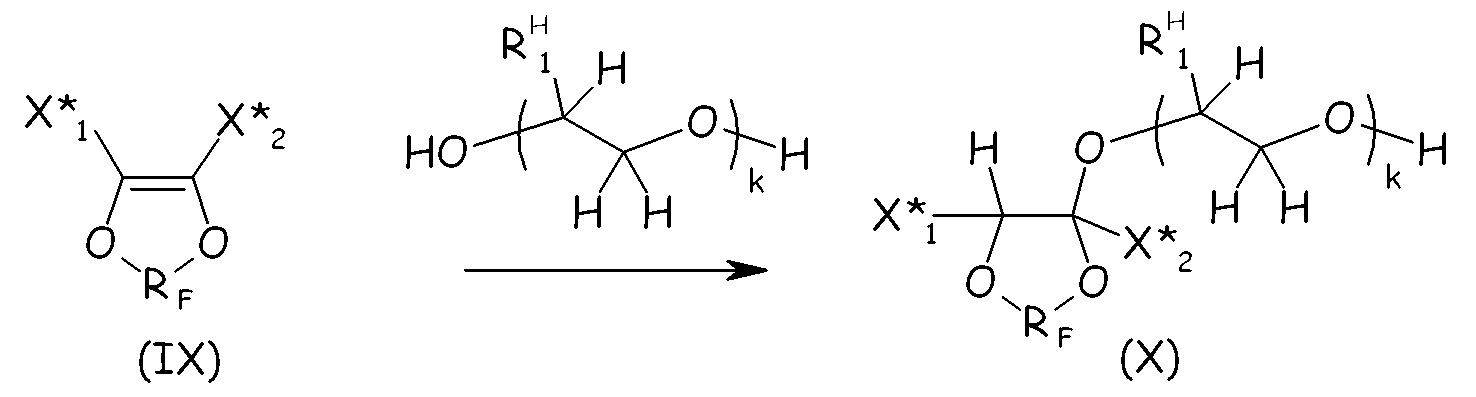
где X* 1, X* 2, RF, k имеют те же значения, что определено выше; RH 1 представляет собой Н или -CH3.
Основный катализ обычно принимают для создания условий, благоприятствующих этой реакции. Добавление гидрогенизированного производного гликоля обычно проводят, применяя 1 экв. указанного ненасыщенного диоксола (IX) на эквивалент основания в указанном гликоле, так чтобы максимизировать выход желаемого соединения (X), являющегося продуктом однократного присоединения. Поскольку гидроксильная функциональная группа обычно нестабильна в условиях фторирования, свободную гидроксильную группу продукта присоединения (Х) обычно защищают перед проведением полного фторирования:
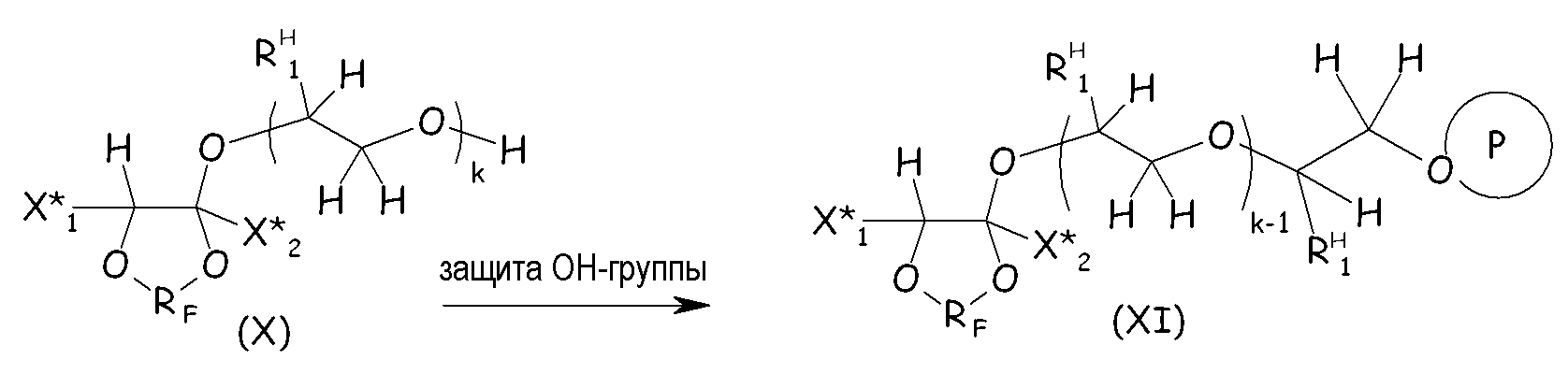
где X*1, X*2, RF, RH 1, k имеют значения, определенные выше; и Р, обведенное кружком, обозначает защитную группу.
Выбор защитного средства не ограничен конкретно, при условии что эта группа является стабильной в условиях фторирования. Обычно, предпочтительной будет этерификация (пер)фторированным ацилфторидом. В качестве альтернативы можно проводить реакцию соединения (Х) с любым карбонилдифторидом, карбонилфторидбромидом и карбонилфторидхлоридом (предпочтительно с карбонилдифторидом), чтобы защитить гидроксильную группу в виде фторформиатной группы, которая преимущественно является стабильной при фторировании.
Защищенный продукт присоединения (XI) (например, в форме сложного эфира или фторформиата) затем фторируют в соответствии со стандартными процедурами, обычно применяя элементарный фтор, для получения соответствующего перфторированного соединения (XII):
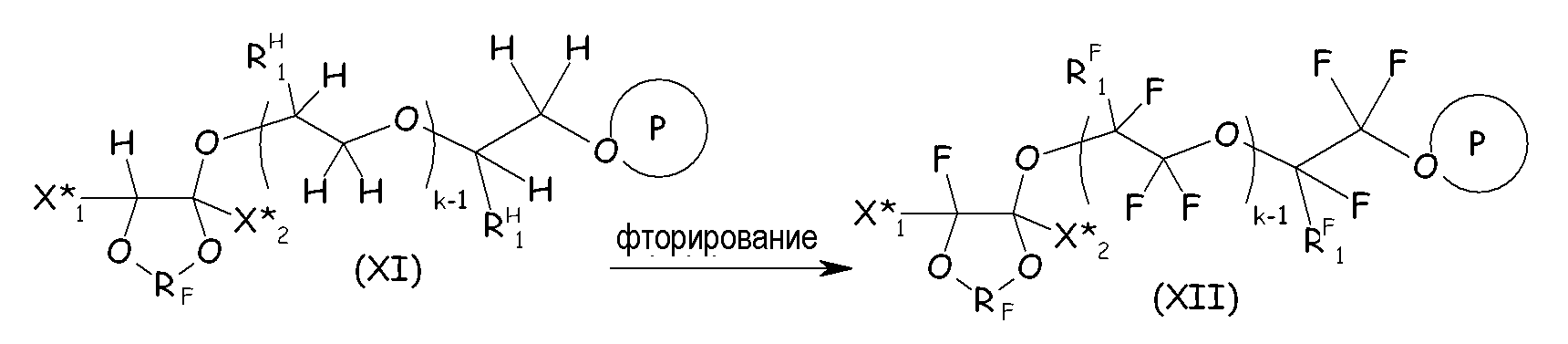
где X* 1, X* 2, RF, RH 1, RF 1, k и Р, обведенное кружком, имеют те же значения, как подробно описано выше.
Затем на указанное перфторированное соединение (XII) воздействуют реакционными условиями, соответствующими разложению/гидролизу защитной группы гидроксильной функциональной группы, получая соответствующий ацилфторид, который затем посредством гидролиза/нейтрализации преобразуют в желаемое соединение (VIII):
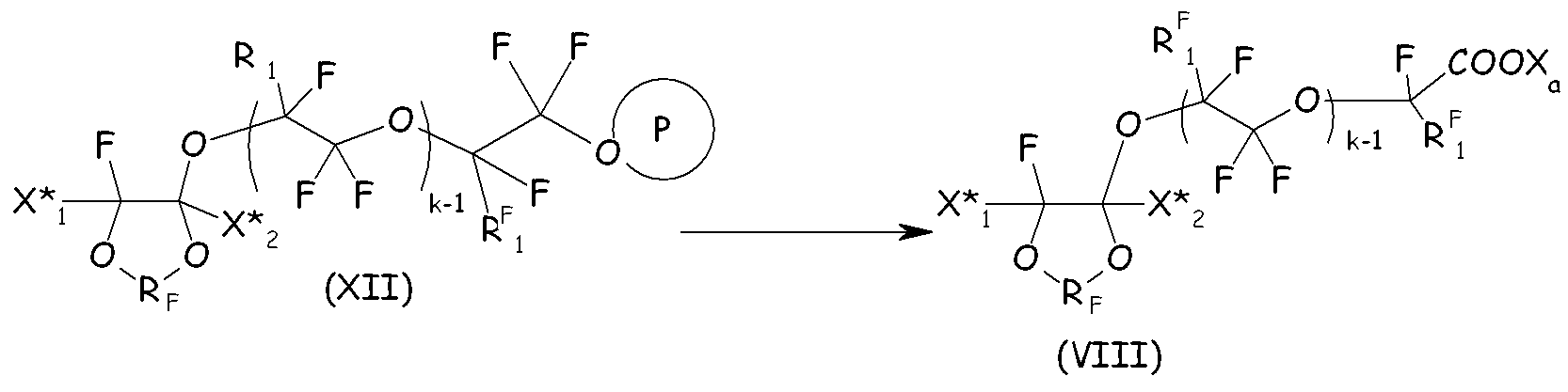
где X*1, X*2, RF, RF 1, Xa, k и Р, обведенное кружком, имеют те же значения, что подробно описано выше.
Этот путь синтеза предпочтительно можно с успехом применять для преобразования ненасыщенных перфтордиоксолов формул:
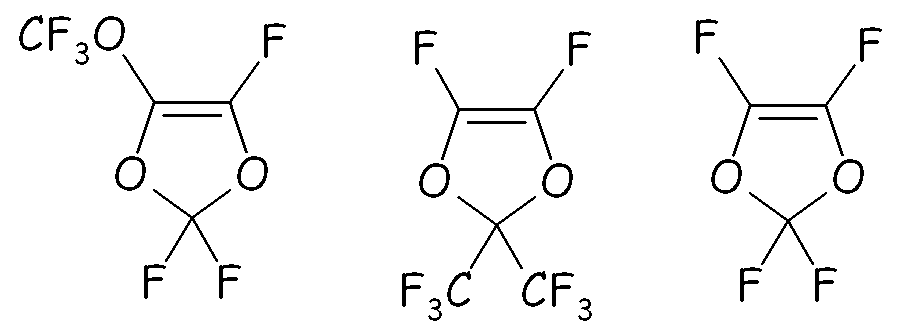
в соответствующие циклические фторсодержащие соединения (VIII) по реакции с этиленгликолем, по реакции с фторацильным соединением (например, (CF3)2-CF-COF), получая соответствующий сложный эфир, или по реакции с карбонилфторидом (например, COF2), получая соответствующий фторформиат, посредством фторирования, получая соответствующий перфторированный сложный эфир или перфторформиат, разложения указанных перфторированного сложного эфира или перфторформиата и конечного гидролиза/нейтрализации, как представлено на следующей схеме:
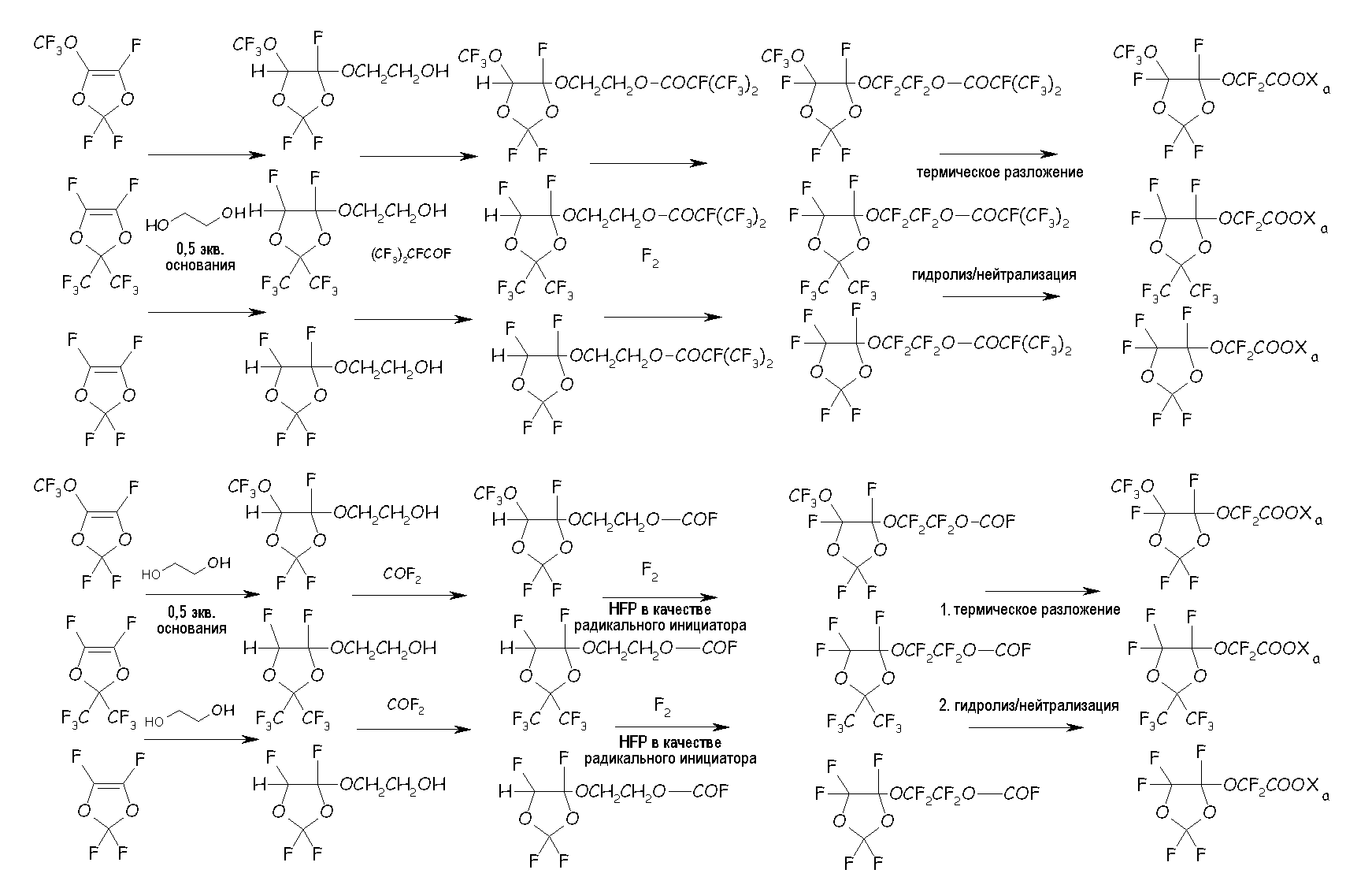
где Xa имеет то же значение, что определено выше. Кроме того, понятно, что для защиты гидроксильного фрагмента можно применять и другие ацилфториды, отличные от (CF3)2CFCOF, или другие карбонилфториды, отличные от COF2, такие как, например, CF3COF.
Предпочтительно, перфторированный сложный эфир и/или перфторформиат можно расщеплять до соответствующего ацилфторида посредством термического разложения в присутствии нуклеофила или электрофила, обычно в присутствии фторида металла формулы MeFy, причем металл имеет валентность, обозначаемую символом y, равную 1 или 2, в частности, в присутствии NaF, CaF2, AgF, CsF, KF, предпочтительно, KF.
В ином случае, перфторированный сложный эфир и/или перфторформиат можно гидролизовать в водной среде, обычно в присутствии подходящего поглотителя HF, например, KF, который, как известно, в водной среде улавливает HF, образуя KHF2.
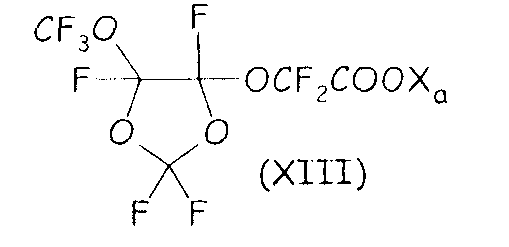
Было найдено, что среди этих соединений циклическое фторсодержащее соединение формулы (XIII), представленное ниже:
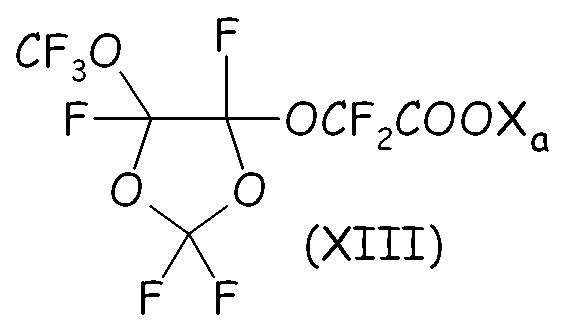
где Xa имеет значение, определенное выше, является особо полезным в способе согласно настоящему изобретению.
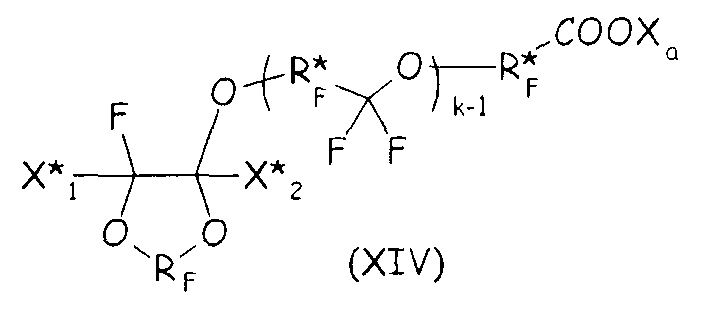
В более общем случае синтетический подход, подробно описанный выше в настоящем документе для второго варианта осуществления настоящего изобретения, можно с успехом применять для получения циклических фторсодержащих соединений, соответствующих формуле (XIV), представленной ниже, что составляет следующий вариант осуществления настоящего изобретения:
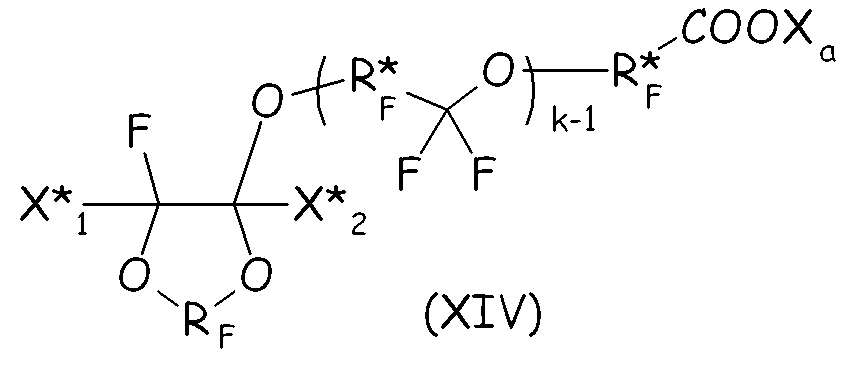
где RF и Xa имеют те же значения, что подробно описаны выше; X* 1, X* 2, одинаковые или отличные один от другого, независимо представляют собой атом фтора, -R'f или -OR'f, где R'f представляет собой С1-3-перфторалкильную группу; R* F представляет собой двухвалентную фторированную группу, k представляет собой целое число от 1 до 3.
Соединения формулы (XIV) можно получать, следуя путем, похожим на тот, что подробно описан выше для соединений формулы (VIII), по реакции ненасыщенного фтордиоксола с гидрогенизированным производным двухатомного спирта, при условии что такой двухатомный спирт содержит по меньшей мере один фрагмент -CH2OH, как представлено ниже, так, чтобы получать соединение формулы (XV), являющееся продуктом однократного присоединения:
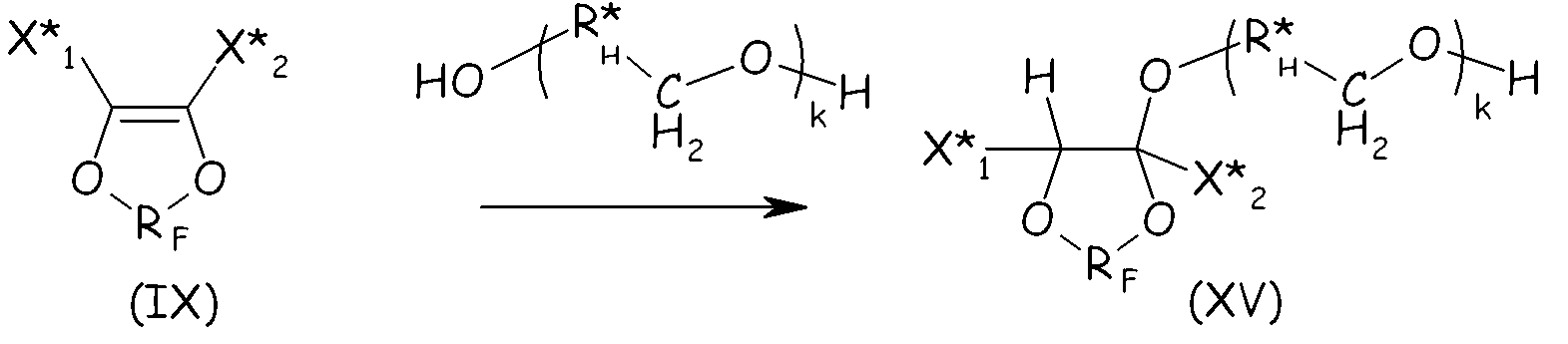
где X* 1, X* 2, RF, k имеют те же значения, что определено выше; R*H представляет собой двухвалентную гидрогенизированную группу.
Защиту гидроксильной функциональной группы и фторирование с последующим разложением/гидролизом группы, защищающей гидроксильную функциональную группу, для получения соответствующего ацилфторида и конечный гидролиз/нейтрализацию до конечного желаемого соединения (XIV) можно проводить так, как подробно описано выше для соединений (VIII):
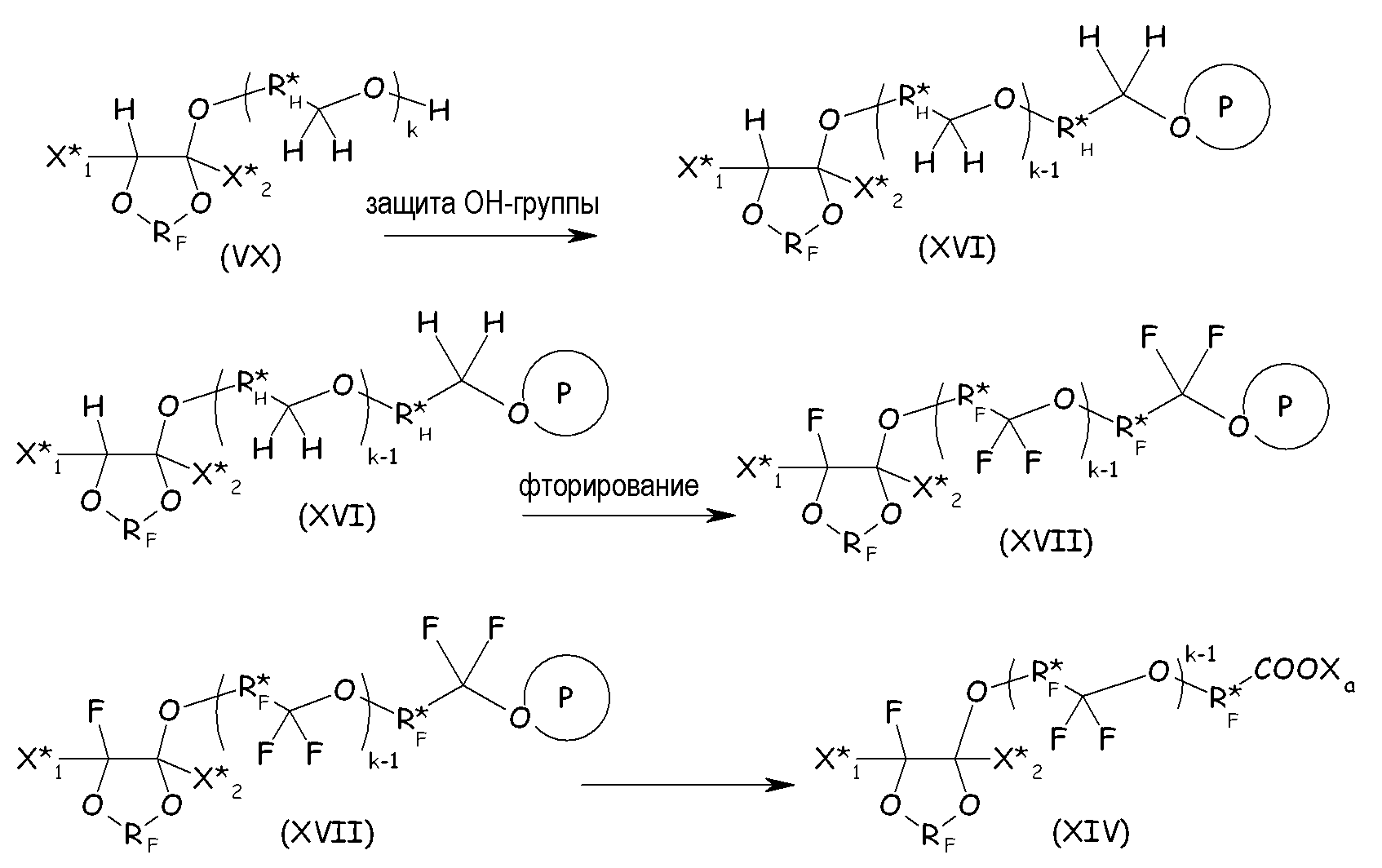
где X* 1, X* 2, RF, R*F, R*H, k имеют значения, определенные выше; и Р, обведенное кружком, обозначает защитную группу.
Выбор защитного средства не ограничен конкретно, при условии, что эта группа стабильна в условиях фторирования. Обычно предпочтительными путями будут этерификация (пер)фторированным ацилбромидом или образование фторформиата действием карбонилфторида.
Этот путь синтеза можно предпочтительно с успехом применять для преобразования ненасыщенного перфтордиоксола, имеющего формулы:
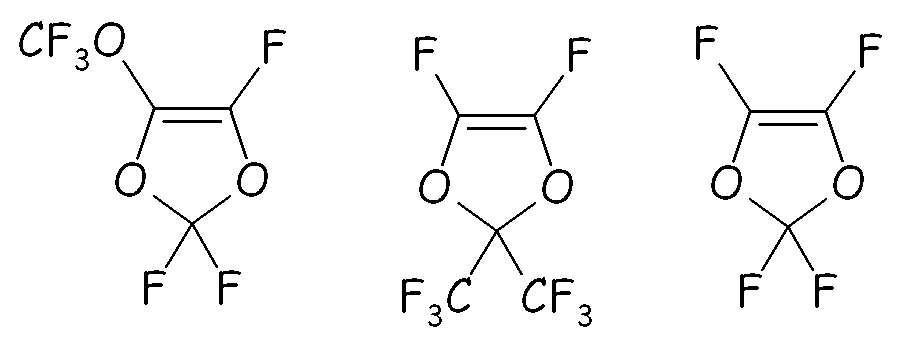
в соответствующие циклические фторсодержащие соединения (XIV) путем реакции с разными двухатомными спиртами, подобно, предпочтительно, реакции с участием пропиленгликоля, по реакции с фторацильным соединением (например, CF3-COF), получая соответствующий сложный эфир, или по реакции с карбонилфторидом (например, COF2), получая соответствующий фторформиат, посредством фторирования, получая соответствующий перфторированный сложный эфир, разложения указанного перфторированного сложного эфира или перфторформиата и конечного гидролиза/нейтрализации, как представлено на следующей схеме:
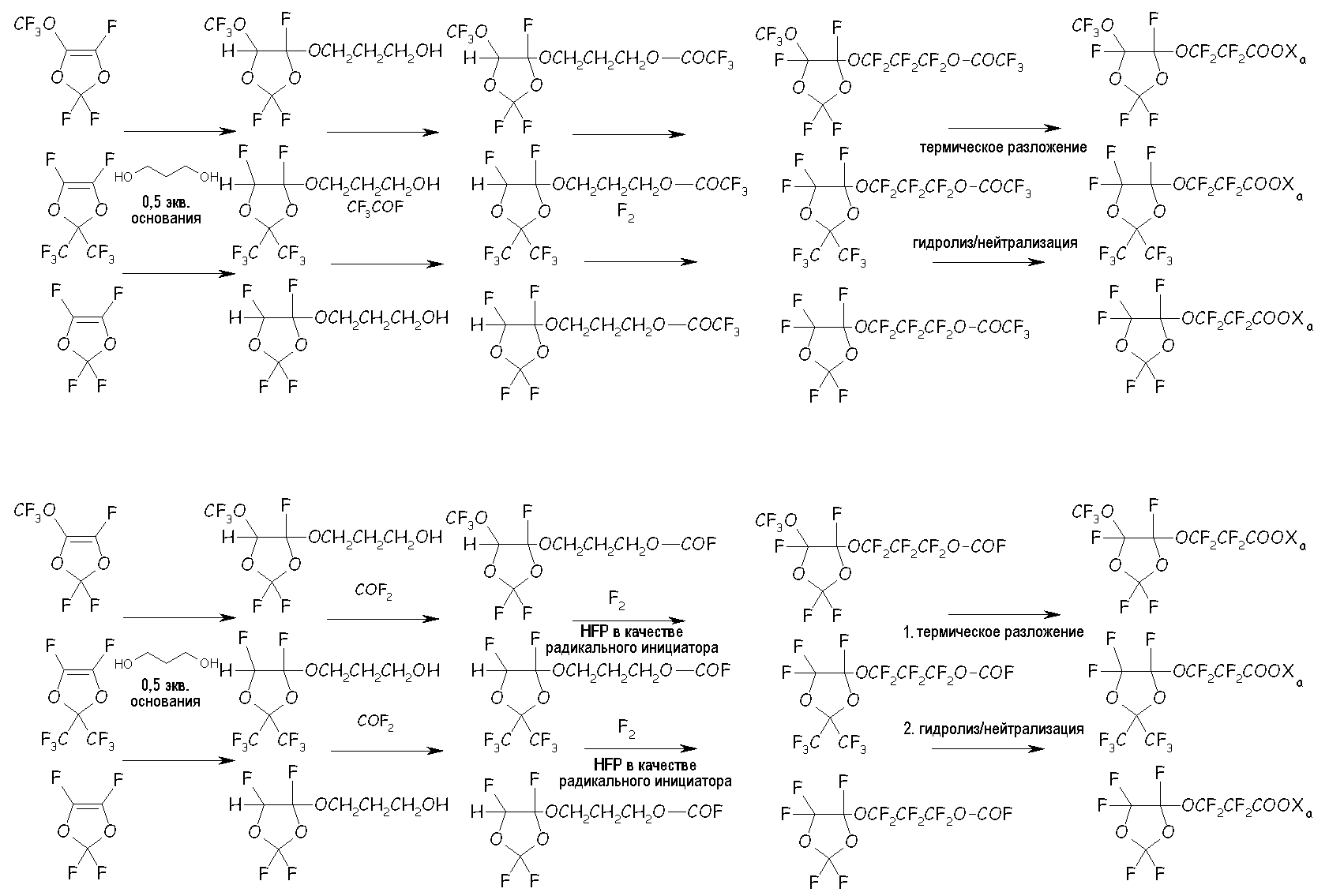
Среди этих соединений особо полезным в способе согласно настоящему изобретению было найдено циклическое фторсодержащее соединение формулы (XVIII), представленное ниже:
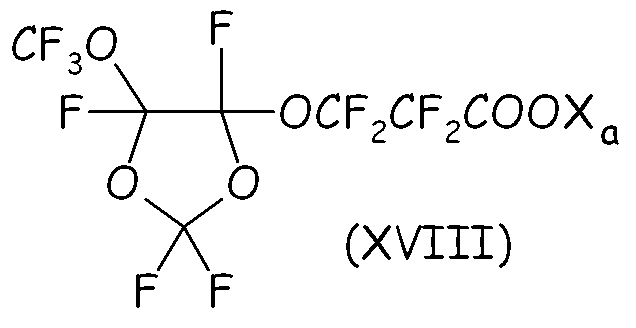
где Xa имеет значение, определенное выше.
В способе согласно настоящему изобретению одно или более из циклических фторсодержащих соединений формулы (I) применяют при водно-эмульсионной полимеризации одного или более фторированных мономеров, в частности мономеров, фторированных в газовой фазе.
Под мономерами, фторированными в газовой фазе, понимают мономеры, которые в условиях полимеризации присутствуют в газообразной форме. В конкретном варианте осуществления настоящего изобретения полимеризацию фторированных мономеров начинают в присутствии циклического фторсодержащего соединения формулы (I), т.е. полимеризацию инициируют в присутствии тех же самых соединений. Применяемое количество циклического фторсодержащего соединения формулы (I) может варьировать в зависимости от желаемых свойств, таких как количество твердых веществ, размер частиц и т.д. Обычно количество циклического фторсодержащего соединения формулы (I) будет составлять от 0,001% по массе в расчете на массу воды при полимеризации до 5% по массе. Практический диапазон составляет от 0,05% по массе до 1% по массе.
Хотя полимеризацию обычно инициируют в присутствии циклического фторсодержащего соединения формулы (I), не исключено дополнительное добавление фторсодержащего соединения формулы (I) в процессе полимеризации, хотя обычно это не потребуется.
Тем не менее, при полимеризации может быть желательным добавление определенного мономера в форме водной эмульсии. Например, фторированные мономеры и, в частности, перфторированные сомономеры, которые являются жидкими в условиях полимеризации, можно преимущественно добавлять в форме водной эмульсии. Такую эмульсию таких сомономеров предпочтительно получают, применяя в качестве эмульгатора циклическое фторсодержащее соединение формулы (I).
Водно-эмульсионную полимеризацию можно проводить при температуре от 10 до 150°С, предпочтительно от 20°C до 130°C и обычно при давлении от 2 до 50 бар, в частности, от 5 до 35 бар.
Реакционную температуру можно изменять в течение полимеризации, например, для воздействия на распределение по молекулярной массе, т.е. для получения широкого распределения по молекулярной массе или для получения бимодального или мультимодального распределения по молекулярной массе.
Значение рН полимеризационной среды может быть в диапазоне рН 2-11, предпочтительно 3-10, наиболее предпочтительно 4-10.
Водно-эмульсионную полимеризацию обычно инициируют посредством некоторого инициатора, включая любой из известных инициаторов, применяемых для инициирования свободно-радикальной полимеризации фторированных мономеров. Подходящие инициаторы включают пероксиды и азо-соединения и инициаторы на окислительно-восстановительной основе. Конкретные примеры пероксидных инициаторов включают пероксид водорода, пероксид натрия или бария, диацилпероксиды, такие как диацетилпероксид, дисукцинилпероксид, дипропионилпероксид, дибутирилпероксид, дибензоилпероксид, ди-трет-бутилпероксид, бензоилацетилпероксид, пероксид диглутаровой кислоты и дилаурилпероксид и другие надкислоты и их соли, такие как, например, соли аммония, натрия или калия. Примеры надкислот включают надуксусную кислоту. Можно также применять сложные эфиры надкислоты, и их примеры включают трет-бутилпероксиацетат и трет-бутилпероксипивалат. Примеры неорганических инициаторов включают, например, соли аммония, щелочных металлов или щелочноземельных металлов с персульфатами, марганцевой или марганцовистой кислотой или марганцовистыми кислотами. Персульфатный инициатор, например персульфат аммония (APS), можно применять отдельно или в комбинации с восстановителем. Подходящие восстановители включают бисульфиты, такие как, например, бисульфит аммония или метабисульфит натрия, тиосульфаты, такие как, например, тиосульфат аммония, калия или натрия, гидразины, азодикарбоксилаты и азодикарбоксилдиамид (ADA). Дополнительные восстановители, которые можно применять, включают формальдегидсульфоксилат натрия (Rongalite) или фторалкилсульфинаты, как раскрыто в патенте США № 5285002. Восстановитель обычно уменьшает период полужизни персульфатного инициатора. Кроме того, можно добавлять катализатор, представляющий собой соль металла, например, соли меди, железа или серебра. Количество инициатора может составлять от 0,01% по массе (в расчете на твердое вещество получаемого фторполимера) до 1% по массе. В одном варианте осуществления настоящего изобретения количество инициатора составляет от 0,05 до 0,5% по массе. В другом варианте осуществления настоящего изобретения это количество может составлять от 0,05 до 0,3% по массе.
Водно-эмульсионную полимеризацию можно проводить в присутствии других материалов, таких как, предпочтительно, буферные вещества и, если требуется, комплексообразователи и переносчики цепи.
Примеры переносчиков цепи, которые можно применять, включают диметиловый простой эфир, метил-трет-бутиловый простой эфир, алканы, имеющие от 1 до 5 атомов углерода, такие как этан, пропан и н-пентан, галогенированные углеводороды, такие как CCl4, CHCl3 и CH2Cl2, и гидрофторуглеродные соединения, такие как CH2F-CF3 (R134a). Кроме того, в способе согласно настоящему изобретению эффективными переносчиками цепи могут быть сложные эфиры, такие как этилацетат и малоновые эфиры.
Примеры фторированных мономеров, которые можно полимеризовать с применением циклического фторсодержащего соединения согласно формуле (I) в качестве эмульгатора в способе согласно настоящему изобретению, включают частично или полностью фторированные газообразные мономеры, включая фторированные олефины, такие как тетрафторэтилен (TFE), хлортрифторэтилен (CTFE), гексафторпропилен (HFP), винилфторид (VF), винилиденфторид (VDF), частично или полностью фторированные аллильные простые эфиры и частично или полностью фторированные алкильные или алкоксивинильные простые эфиры.
Кроме того, водно-эмульсионную полимеризацию можно проводить в присутствии фторированных жидкостей, обычно делающих возможным образование наноразмерных капель (со средним размером менее 50 нм, предпочтительно менее 30 нм), стабилизированных в водной дисперсии посредством присутствия циклического фторсодержащего соединения формулы (I).
Если способ согласно настоящему изобретению требуется проводить в присутствии фторированной жидкости, как подробно описано выше, может оказаться предпочтительным сначала однородно смешать циклическое соединение и указанную жидкость в водной фазе, возможно в водной среде, и затем ввести водную смесь соединения (I) и указанной жидкости в полимеризационную среду. Эта методика особо выгодна, поскольку эта предварительная смесь может дать возможность выгодно получать эмульсию указанной жидкости в водной фазе, содержащей циклическое соединение, где эта эмульсия содержит диспергированные капли указанной жидкости, имеющие средний размер, равный, предпочтительно менее 50 нм, более предпочтительно менее 40 нм, еще более предпочтительно менее 30 нм.
Жидкости, которые можно применять согласно этому варианту осуществления, предпочтительно представляют собой (пер)фторированные простые полиэфиры, содержащие повторяющиеся звенья (R1), причем указанные повторяющиеся звенья содержат по меньшей мере одну простоэфирную связь в главной цепи и по меньшей мере один атом фтора (фторполиоксиалкеновая цепь). Предпочтительно, повторяющиеся звенья R1 (пер)фторированного простого полиэфира выбирают из группы, состоящей из:
(I) -CFX-O-, где X представляет собой -F или -CF3; и
(II) -CF2-CFX-O-, где X представляет собой -F или -CF3; и
(III) -CF2-CF2-CF2-O-; и
(IV) -CF2-CF2-CF2-CF2-O-; и
(V) -(CF2)j-CFZ-O-, где j представляет собой целое число, выбранное из 0 и 1, и Z представляет собой фторполиоксиалкеновую цепь, содержащую от 1 до 10 повторяющихся звеньев, выбранных из классов (I)-(IV), указанных выше; и их смесей.
Если требуется, чтобы (пер)фторированный простой полиэфир содержал повторяющиеся звенья R1 разных типов, то указанные повторяющиеся звенья преимущественно оказываются распределенными произвольно вдоль фторполиоксиалкеновой цепи.
Предпочтительно, (пер)фторированный простой полиэфир представляет собой соединение, соответствующее формуле (I-p), представленной ниже:
T1-(CFX)p-O-Rf-(CFX)p'-T2 (I-p)
где:
- каждый из Х независимо представляет собой F или CF3;
- p и p', одинаковые или отличные один от другого, представляют собой целые числа от 0 до 3;
- Rf представляет собой фторполиоксиалкеновую цепь, содержащую повторяющиеся звенья R°, причем указанные повторяющиеся звенья являются выбранными из группы, состоящей из:
(i) -CFXO-, где X представляет собой F или CF3,
(ii) -CF2CFXO-, где X представляет собой F или CF3,
(iii) -CF2CF2CF2O-,
(iv) -CF2CF2CF2CF2O-,
(v) -(CF2)j-CFZ-O-, где j представляет собой целое число, выбранное из 0 и 1, и Z представляет собой группу общей формулы -ORf'T3, где Rf' представляет собой фторполиоксиалкеновую цепь, содержащую повторяющиеся звенья числом от 0 до10, причем указанные повторяющиеся звенья являются выбранными из следующих групп: -CFXO-, -CF2CFXO-, -CF2CF2CF2O-, -CF2CF2CF2CF2O-, причем каждый из Х независимо представляет собой F или CF3; и T3 представляет собой С1-C3-перфторалкильную группу, и их смесей;
- T1 и T2, одинаковые или отличные один от другого, представляют собой Н, атомы галогенов, C1-C3-фторалкильные группы, необязательно содержащие один или более атомов Н или атомов галогенов, отличных от фтора.
В полимеризации также могут участвовать нефторированные мономеры, такие как этилен и пропилен.
Дополнительные примеры фторированного мономера, который можно применять в водно-эмульсионной полимеризации согласно настоящему изобретению, включают фторированные мономеры, соответствующие формуле: CF2=CF-O-Rf, где Rf представляет собой перфторированную алифатическую группу, которая может содержать один или более атомов кислорода.
Кроме того, в полимеризации могут участвовать и сомономеры, которые имеют функциональную группу, такую как, например, группа, способная участвовать в реакции пероксидной вулканизации (пероксидного отверждения). Такие функциональные группы включают галогены, такие как Br или I, а также нитрильные группы.
Водно-эмульсионную полимеризацию можно применять для получения разнообразных фторполимеров, включая перфторполимеры, которые имеют полностью фторированную основную цепь, а также частично фторированные фторполимеры. Кроме того, водно-эмульсионная полимеризация может в результате давать фторполимеры, перерабатываемые в расплаве, а также фторполимеры, которые не являются перерабатываемыми в расплаве, такие как, например, политетрафторэтилен и так называемый модифицированный политетрафторэтилен. Данный способ полимеризации может дополнительно давать фторполимеры, которые можно вулканизировать с получением фторэластомеров, а также фтортермопластов. Фтортермопласты обычно представляют собой фторполимеры, которые имеют явную и хорошо различимую точку плавления, обычно в диапазоне от 60 до 320°С или от 100 до 320°С. Таким образом, они имеют существенную кристаллическую фазу. Фторполимеры, которые применяют для получения фторэластомеров, обычно являются аморфными и/или имеют незначительное количество кристалличности, так что у этих фторполимеров нельзя различить никакой или почти никакой точки плавления.
Согласно варианту осуществления способа согласно настоящему изобретению данный способ включает полимеризацию в водной эмульсии в присутствии смеси циклического фторсодержащего соединения формулы (I) и по меньшей мере одного дополнительного эмульгатора, отличного от циклического фторсодержащего соединения формулы (I).
Выбор указанного дополнительного эмульгатора не является ограниченным конкретно. Обычно фторированные эмульгаторы будут применять в комбинации с циклическим фторсодержащим соединением формулы (I).
Более конкретно, фторированный эмульгатор [поверхностно-активное вещество (FS)], имеющий формулу:
Rf§(X-)j(M+)j
где Rf§ представляет собой С3-C30-(пер)фторалкильную цепь, (пер)фтор(поли)оксиалкиленовую цепь, X- представляет собой -COO-, -PO3 - или -SO3 -, M+ выбран из H+, NH4 + и иона щелочного металла, и j может представлять собой 1 или 2.
В качестве неограничивающего примера поверхностно-активных веществ (FS) можно назвать перфторкарбоксилаты аммония и/или натрия, и/или (пер)фторполиоксиалкилены, имеющие одну или более концевую карбоксильную группу.
Другими примерами фторированных поверхностно-активных веществ являются (пер)фтороксиалкиленовые поверхностно-активные вещества, описанные в US 2007015864 (3M INNOVATIVE PROPERTIES) 08.01.2007, US 2007015865 (3M INNOVATIVE PROPERTIES CO) 18.01.2007, US 2007015866 (3M INNOVATIVE PROPERTIES CO) 18.01.2007, US 2007025902 (3M INNOVATIVE PROPERTIES CO) 01.02.2007.
Более предпочтительно, фторированный эмульгатор [поверхностно-активное вещество (FS)] является выбранным из:
- CF3(CF2)n1COOM', в котором n1 представляет собой целое число в диапазоне от 4 до 10, предпочтительно от 5 до 7, и более предпочтительно равное 6; M' представляет собой H, NH4, Na, Li или K, предпочтительно NH4;
- T(C3F6O)n0(CFXO)m0CF2COOM", в котором T представляет собой Cl или перфторалкоксидную группу формулы CkF2k+1O, где k представляет собой целое число от 1 до 3, один атом F необязательно заменен на атом Cl; n0 представляет собой целое число в диапазоне от 1 до 6; m0 представляет собой целое число в диапазоне от 0 до 6; M" представляет собой H, NH4, Na, Li или K; X представляет собой F или CF3;
- F-(CF2-CF2)n2-CH2-CH2-RO3M"', в котором R представляет собой P или S, предпочтительно S, M'" представляет собой H, NH4, Na, Li или K, предпочтительно H; n2 представляет собой целое число в диапазоне от 2 до 5, предпочтительно n2=3;
- бифункциональные фторированные поверхностно-активные вещества A-Rf-B, в которых A и B, одинаковые или отличные один от другого, представляют собой -(O)pCFX-COOM*; M* представляет собой H, NH4, Na, Li или K, предпочтительно M* представляет собой NH4; X = F или CF3; p представляет собой целое число, равное 0 или 1; Rf представляет собой линейную или разветвленную перфторалкильную цепь или цепь (пер)фторированного простого полиэфира, такие, чтобы среднечисленная молекулярная масса A-Rf-В была в диапазоне от 300 до 3000, предпочтительно от 500 до 2000;
- R'f-O-(CF2)r-O-L-COOM', где R'f представляет собой линейную или разветвленную перфторалкильную цепь, необязательно содержащую атомы кислорода в цепи, M' представляет собой Н, NH4, Na, Li или K, предпочтительно M' представляет собой NH4; r равно от 1 до 3; L представляет собой двухвалентную фторированную мостиковую группу, предпочтительно -CF2CF2- или -CFX-, X = F или CF3;
- R"f-(OCF2)u-O-(CF2)v-COOM", где R"f представляет собой линейную или разветвленную перфторалкильную цепь, необязательно содержащую атомы кислорода в цепи, M" представляет собой Н, NH4, Na, Li или K, предпочтительно M" представляет собой NH4; u и v представляют собой целые числа от 1 до 3;
- R"'f-(O)t-CHQ-L-COOM'", где R'"f представляет собой линейную или разветвленную перфторалкильную цепь, необязательно содержащую атомы кислорода в цепи, Q = F или CF3, t равно 0 или 1, M"' представляет собой Н, NH4, Na, Li или K, предпочтительно M"' представляет собой NH4; L представляет собой двухвалентную фторированную мостиковую группу, предпочтительно -CF2CF2- или -CFX-, X = F или CF3;
- и их смесей.
Особо хорошие результаты были получены со смесями соединения (I) с бифункциональными фторированными поверхностно-активными веществами A-Rf-B; указанное бифункциональное поверхностно-активное вещество A-Rf-B предпочтительно соответствует формуле MzOOC-CFXz-O-Rfz-CFXz-COOMz', где Mz представляет собой Н, NH4, Na, Li или K, предпочтительно Mz представляет собой NH4; Xz = F, -CF3; Rfz представляет собой цепь (пер)фторированного простого полиэфира, содержащего повторяющиеся звенья, соответствующие одной или более из формул: -(C3F6O)-; -(CF2CF2O)-; -(CFL0O)-, где L0 = F, -CF3; -(CF2(CF2)Z'CF2O)-, где z' равно 1 или 2; -(CH2CF2CF2O)-.
Rfz предпочтительно имеет одну из следующих структур:
1) -(CF2O)a-(CF2CF2O)b-, где a и b ≥ 0; если a и b одновременно >0, тогда отношение b/a обычно составляет от 0,01 до 10 включительно;
2) -(CF2-(CF2)Z'-CF2O)b'-, где b' > 0 и z' равно 1 или 2;
3) -(C3F6O)r-(C2F4O)b-(CFL0O)t-, где r, b и t ≥ 0, L0 = F, -CF3; если r, b и t одновременно > 0, тогда отношение r/b обычно находится в диапазоне 0,5-2,0 и (r+b)/t находится в диапазоне 10-30;
4) -(OC3F6)r-(OCFL0)t-OCF2-R*f-CF2O-(C3F6O)r-(CFL0O)t-, где R*f представляет собой фторалкиленовую группу, содержащую от 1 до 4 атомов углерода; L0 = F, -CF3; r, t ≥ 0.
Наиболее предпочтительное бифункциональное фторированное поверхностно-активное вещество A-Rf-B соответствует формуле MzOOC-CFXz-O-(CF2O)a-(CF2CF2O)b-CFXz-COOMz, где Mz представляет собой Н, NH4, Na, Li или K, предпочтительно Mz представляет собой NH4; Xz = F, -CF3; и оба a и b > 0 и являются выбранными так, чтобы b/a составляло от 0,3 до 10, и молекулярная масса данного поверхностно-активного вещества составляла от 500 до 2000.
Если способ согласно настоящему изобретению требуется осуществлять в присутствии смеси циклического соединения и дополнительного фторированного эмульгатора, как подробно описано выше, может оказаться предпочтительным сначала однородно смешать циклическое соединение и дополнительный эмульгатор в водной фазе, а затем ввести водную смесь соединения (I) и указанного эмульгатора в полимеризационную среду. Эта методика особенно выгодна, когда дополнительный фторированный эмульгатор является слаборастворимым в воде. Таким образом, эта предварительная смесь может сделать возможным выгодное получение эмульсии указанного фторированного эмульгатора в водной фазе, содержащей циклическое соединение, где эта эмульсия содержит диспергированные капли указанного фторированного эмульгатора, имеющие средний размер предпочтительно менее 50 нм, предпочтительно менее 40 нм, более предпочтительно менее 30 нм.
Кроме того, в дополнение к этому водно-эмульсионную полимеризацию в этом варианте осуществления можно проводить в присутствии фторированных жидкостей, как указано выше, как правило, делая возможным образование наноразмерных капель (со средним размером менее 50 нм, предпочтительно менее 30 нм) стабилизированных в водной дисперсии присутствием смеси циклического фторсодержащего соединения формулы (I) и по меньшей мере одного дополнительного эмульгатора, отличного от циклического фторсодержащего соединения формулы (I).
Фторированные жидкости, которые можно применять в комбинации с указанной смесью соединения (I) и эмульгатора, являются теми, которые указаны выше и которые подходят для применения в комбинации с циклическим фторсодержащим соединением формулы (I).
Результатом процесса водно-эмульсионной полимеризации согласно настоящему изобретению является дисперсия фторполимера в воде, содержащая циклическое фторсодержащее соединение формулы (I). Обычно количество твердых веществ фторполимера в дисперсии, которое является прямым результатом полимеризации, будет варьировать между 3% по массе и примерно 40% по массе, в зависимости от условий полимеризации. Типичный диапазон находится между 5 и 30% по массе, например, между 10 и 25% по массе.
Размер частиц (среднеобъемный диаметр) фторполимера обычно находится между 40 нм и 400 нм, причем предпочтителен типичный размер частиц между 60 нм и примерно 350 нм. Общее количество циклического фторсодержащего соединения формулы (I) в дисперсии, получаемой в результате, обычно находится между 0,001 и 5% по массе, в расчете на количество твердых веществ фторполимера в дисперсии. Типичное количество может составлять от 0,01 до 2% по массе или от 0,02 до 1% по массе.
Если желательным является полимер в твердой форме, этот фторполимер можно выделять из дисперсии посредством коагуляции. Также, в зависимости от требований той области применения, в которой фторполимер должен использоваться, этот фторполимер можно фторировать впоследствии, чтобы преобразовать любые термически нестабильные концевые группы в стабильные концевые группы CF3-.
Для применения в качестве наносимых покрытий желательна водная дисперсия фторполимера, и поэтому отделять или коагулировать фторполимер из дисперсии не потребуется. Для получения дисперсии фторполимеров, подходящей для применения в качестве наносимых покрытий, как например, при импрегнировании тканей или при нанесении покрытий на металлические подложки, например, при изготовлении кухонной утвари, обычно будет желательным добавление дополнительных стабилизирующих поверхностно-активных веществ и/или дополнительное увеличение содержания твердых веществ фторполимеров. Например, к дисперсии фторполимеров можно добавлять неионные стабилизирующие поверхностно-активные вещества. Обычно эти вещества будут добавлять в количестве от 1 до 12% по массе в расчете на твердые вещества фторполимеров. Примеры неионных поверхностно-активных веществ, которые можно добавлять таким образом, включают R1-O-[CH2CH2O]n-[R2O]m-R3 (NS), где R1 представляет собой ароматическую или алифатическую углеводородную группу, имеющую от 6 до 18 атомов углерода, R2 представляет собой алкилен, имеющий 3 атома углерода, R3 представляет собой водород или C1-3-алкильную группу, n имеет значение от 0 до 40, m имеет значение от 0 до 40, а сумма n+m составляет по меньшей мере 2. Следует понимать, что звенья, обозначенные в вышеуказанной формуле (NS) как n и m, могут появляться в виде блоков или могут присутствовать в чередующейся или неупорядоченной конфигурации. Примеры неионных поверхностно-активных веществ согласно формуле (VI), указанной выше, включают алкилфенолоксиэтилаты, такие как этоксилированный п-изооктилфенол, коммерчески доступный под фирменным названием TRITON™, как, например, TRITON™ Х 100, где число этокси-звеньев равно примерно 10, или TRITON™ X 114, где число этокси-звеньев равно примерно 7-8. Дальнейшие примеры включают вещества, в которых R1 из вышеприведенной формулы (NS) представляет собой алкильную группу из 4-20 атомов углерода, m равно 0, и R3 представляет собой водород. Их пример включает изотридеканол, этоксилированный примерно восемью этоксигруппами, и который коммерчески доступен как GENAPOL® X080 от Clariant GmbH. Можно также применять неионные поверхностно-активные вещества согласно формуле (NS), в которых гидрофильная часть содержит блок-сополимер этоксигрупп и пропоксигрупп. Такие неионные поверхностно-активные вещества коммерчески доступны от Clariant GmbH под фирменным названием GENAPOL® PF 40 и GENAPOL® PF 80.
Если нужно или если требуется, количество твердых веществ фторполимера в дисперсии можно сконцентрировать до количества, составляющего 30-70% по массе. Можно применять любые известные процедуры концентрирования, включая ультрафильтрацию и термическое концентрирование.
Еще одним объектом настоящего изобретения являются дисперсии фторполимеров, содержащие по меньшей мере одно циклическое фторсодержащее соединение формулы (I), как описано выше.
Указанные дисперсии фторполимеров обычно получают способом согласно настоящему изобретению.
Концентрацию циклического фторсодержащего соединения формулы (I) в дисперсиях фторполимеров согласно настоящему изобретению можно при необходимости уменьшать, следуя традиционным процедурам. В этой связи можно назвать ультрафильтрацию, комбинированную с рециркуляцией фильтрата, как описано в US 4369266 (HOECHST AG) 18.01.1983, обработку ионообменными смолами в присутствии неионного поверхностно-активного вещества (как описано в EP 1155055 A (DYNEON GMBH) 21.11.2001), анионного поверхностно-активного вещества (как продемонстрировано в EP 1676868 A (SOLVAY SOLEXIS SPA) 05.07.2006) или полиэлектролита (как указано в EP 1676867 A (SOLVAY SOLEXIS SPA) 05.07.2006).
Таким образом, настоящее изобретение также относится к способу выделения циклического фторсодержащего соединения формулы (I) из содержащей его дисперсии фторполимера. Этот способ предпочтительно включает контактирование дисперсии фторполимера с твердым адсорбирующим материалом, обычно с ионообменной смолой, предпочтительно анионообменной смолой: циклическое фторсодержащее соединение формулы (I) преимущественно адсорбируют (по меньшей мере частично) на этом твердом адсорбирующем материале. Циклическое фторсодержащее соединение формулы (I) можно эффективно выделять с твердого адсорбирующего материала посредством стандартных процедур, включая элюцию, тепловую десорбцию и т.п. В случае элюции, в частности, с анионообменной смолы, циклическое фторсодержащее соединение формулы (I) можно выделять посредством элюции кислым раствором. Обычно для этой цели можно применять водную среду, содержащую кислоту и органический растворитель, смешивающийся с водой. Особенно эффективны смеси неорганической кислоты и спирта в воде. Циклическое фторсодержащее соединение (I) можно предпочтительно выделять из таких жидких фаз стандартными способами, включая предпочтительно кристаллизацию, дистилляцию (например, в форме сложного эфира) и т.п.
Другими объектами настоящего изобретения также являются циклическое фторсодержащее соединение (I), как подробно описано выше, и способы его получения.
Далее в настоящем описании будет дано подробное объяснение настоящего изобретения со ссылкой на следующие примеры, цель которых является исключительно иллюстративной и не предполагает ограничения объема настоящего изобретения.
Препаративный пример 1
Синтез соединения VIIa (где Xa = NH4)
Пример 1a. Реакция между перфтор-5-метокси-1,3-диоксолом (MDO, (A) на схеме, представленной ниже) и метанолом
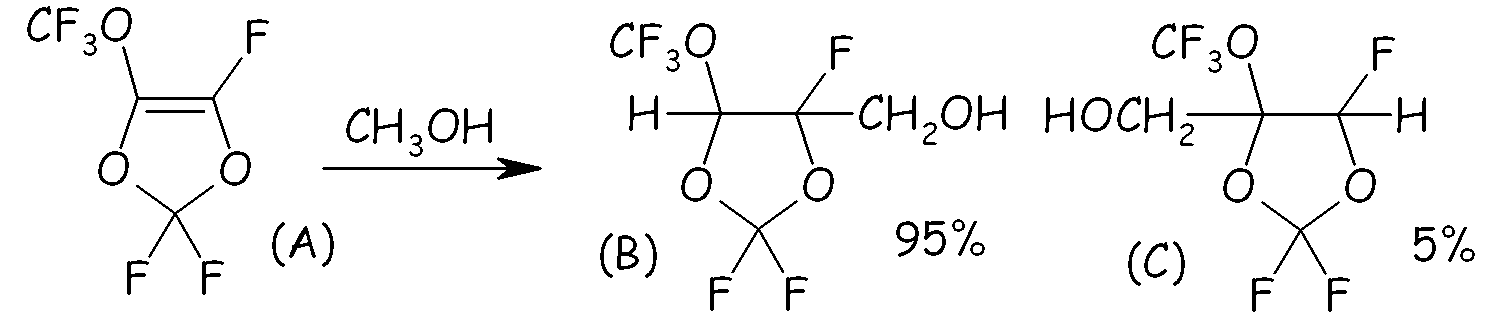
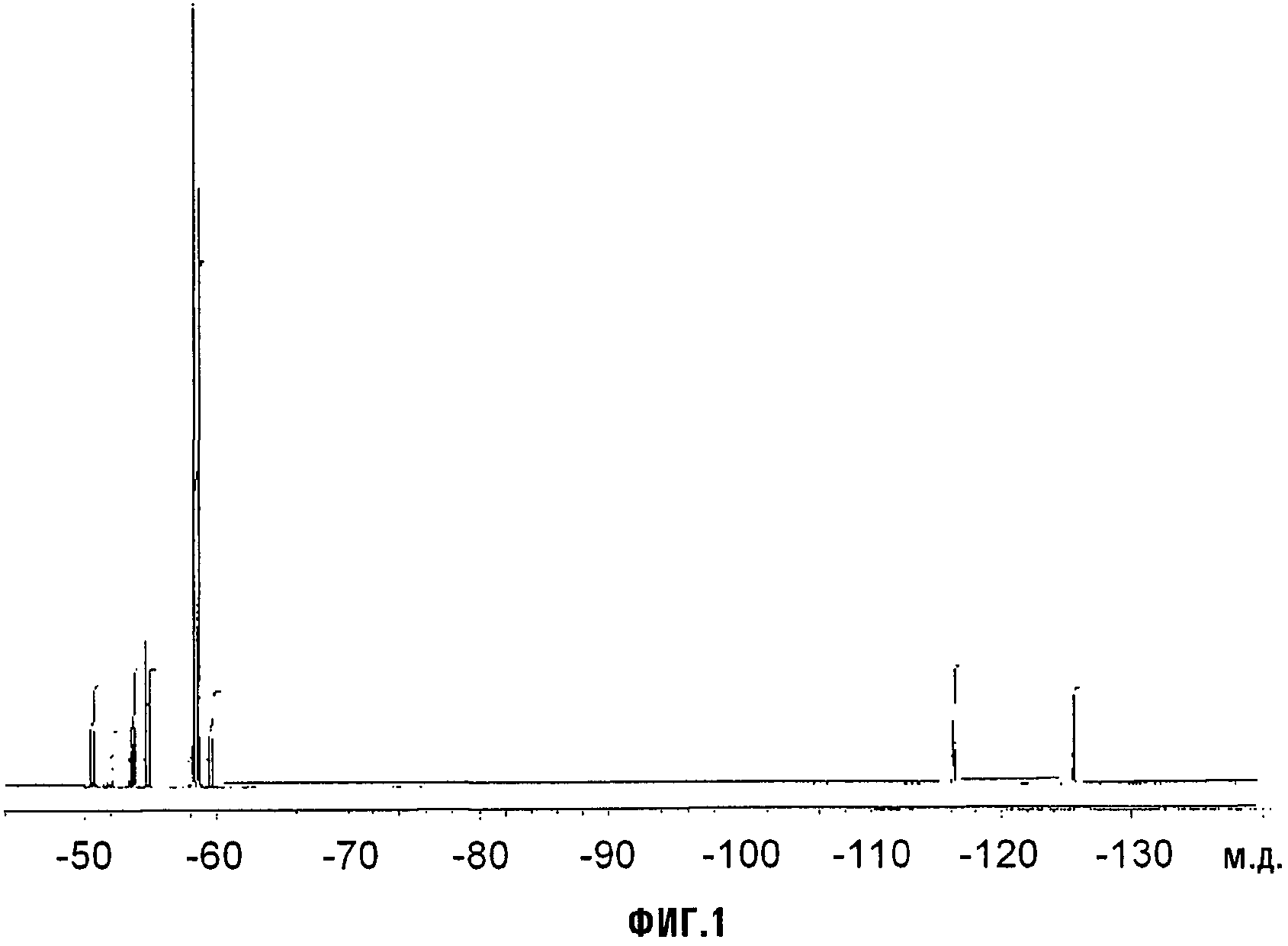
В сосуд высокого давления, изготовленный из нержавеющей стали, оснащенный цифровым манометром и магнитной мешалкой, вносили CaCO3 (7,14 ммоль, 0,714 г). После осторожного вакуумирования при комнатной температуре в сосуд вносили смесь, состоявшую из CH3OH (3,57 моль, 114 г), ди-трет-бутилпероксида (DTBP; 71,4 ммоль, 10,5 г) и MDO (0,714 моль, 150 г). Затем при интенсивном перемешивании сосуд нагревали при 134°C в течение 21 часа, проводя мониторинг внутреннего давления. После завершения реакции сосуд охлаждали до комнатной температуры, и неочищенную реакционную смесь извлекали и промывали несколько раз дистиллированной водой. Органическую (нижнюю) фазу сначала сушили над MgSO4, фильтровали и окончательно перегоняли. Выход выделенного продукта составлял 56% по отношению к исходному MDO (A), т.кип. = 142°C. Селективность составляла 95% для желаемого изомера (B); 5% для альтернативного изомера (C). Фиг.1 представляет спектр 19F-ЯМР, записанный с соединением (B).
Пример 1b. Окисление спиртового промежуточного соединения (B)
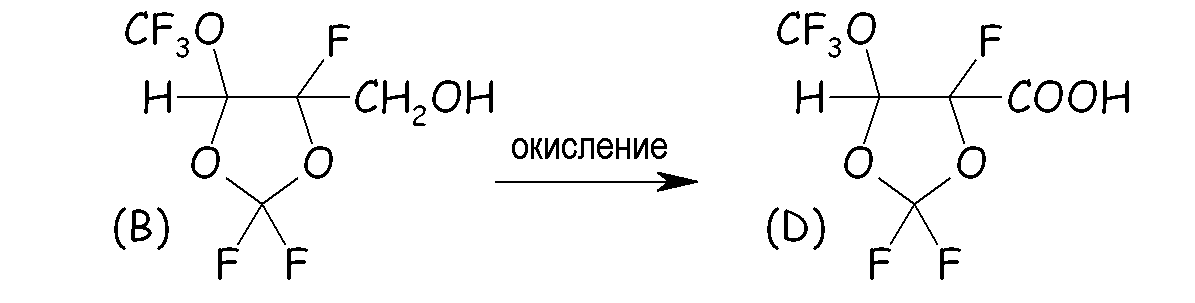
Водный раствор, состоявший из KMnO4 (238 ммоль, 37,6 г), NaOH (238 ммоль, 9,52 г) в 200 мл дистиллированной H2O вносили в 3-горлую стеклянную круглодонную колбу, оснащенную магнитной мешалкой, капельной воронкой, термометром и охлаждающей колонкой с водопроводной водой (обратным холодильником). Колбу нагревали с интенсивным перемешиванием до 80°C и затем в основной окисляющий раствор медленно, по каплям, добавляли 238 ммоль (50 г) продукта, полученного со стадии 1a. Наблюдали непосредственное экзотермическое высвобождение (+15°C) вместе с образованием осадка MnO2. После завершения этого добавления раствор дополнительно перемешивали при 80°C в течение 40 мин. Затем неочищенную реакционную смесь охлаждали до комнатной температуры, фильтровали, подкисляли до pH 1 концентрированной HCl (37% масс./масс.) и несколько раз экстрагировали CH2Cl2. Органический слой сушили над MgSO4, фильтровали и затем выпаривали CH2Cl2. Выход выделенного продукта составлял 55%, преобразование продукта из 1a составляло 100%. pKa (D) = 2,8.
Пример 1c. Синтез VIIa посредством основного гидролиза кислоты (D)
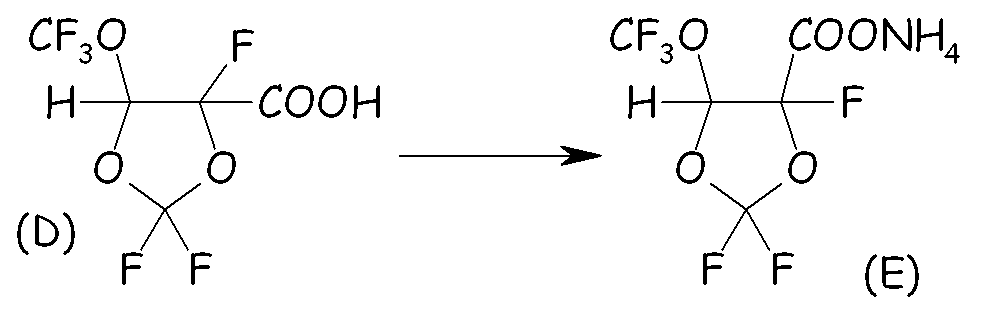
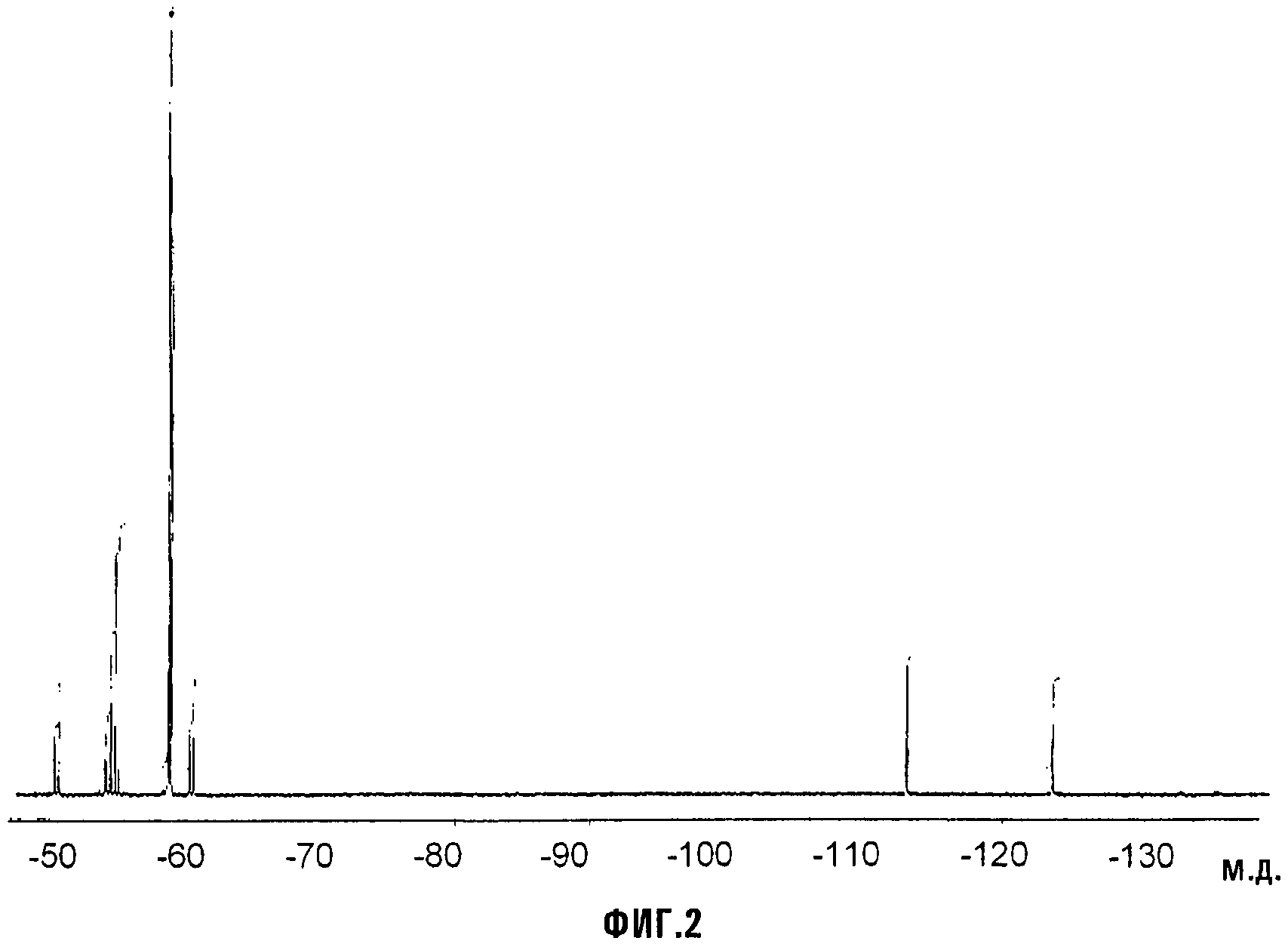
Органический раствор, состоявший из (D) (127 ммоль; 30,6 г) и 200 мл CH2Cl2, вносили в 2-горлую стеклянную круглодонную колбу, оснащенную магнитной мешалкой, охлаждающей колонкой с водопроводной водой (обратным холодильником) и трубкой-барботером. Смесь охлаждали до 0°C с интенсивным перемешиванием и через органическую смесь барботировали большой избыток газообразного NH3. Барботирование NH3 (газ) продолжали до завершения осаждения аммониевой соли. Неочищенную смесь фильтровали, твердое вещество сушили в вакуумном сушильном шкафу при 40°C и пониженном давлении (20 мм Hg) в течение 2 часов. Получали хлопьевидное твердое вещество белого цвета. Выход выделенного соединения (E) (VIIa, с Xa, представляющим собой NH4) составлял 100%. Термогравиметрический анализ (TGA), проведенный на воздухе, показывал уменьшение массы на 10% при 148°C и на 50% при 182°C. Фиг.2 представляет спектр 19F-ЯМР соединения (E). LC-MS показал сильный пик при m/z = 255 (соответствующий карбоксилатному фрагменту соединения (E)). Острую токсичность при пероральном приеме соединения VIIa оценивали согласно стандартной практике; было найдено, что значение LD50 превышало 2000 мг/кг.
Препаративный пример 2
Синтез соединения Va (где Xa = NH4)
Пример 2b. Этерификация спиртового промежуточного соединения (B)
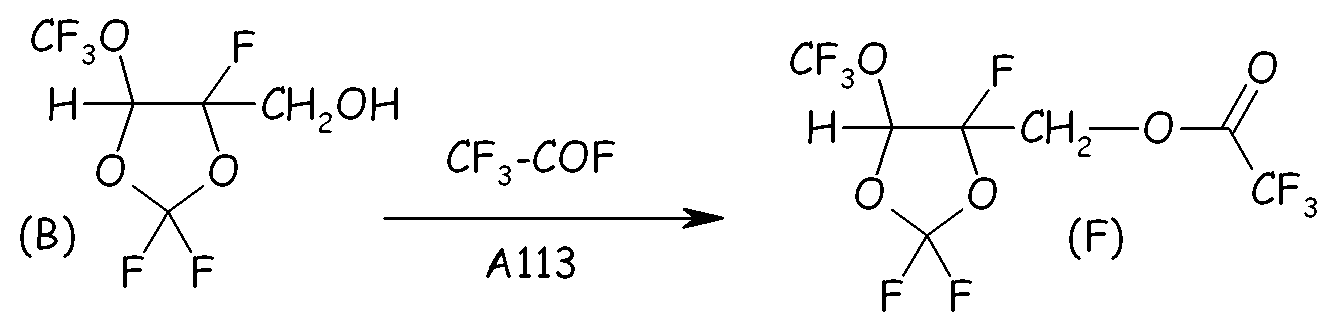
Спиртовое промежуточное соединение (B) (333 ммоль, 70 г), полученное из Примера 1a, реагировало с CF3COF (350 ммоль, 40,5 г) в 200 мл A113 при T=0°C. Растворитель и непрореагировавший CF3COF удаляли посредством отгонки при 40°C/600 мм Hg.
Пример 2c. Фторирование сложного эфира (F)
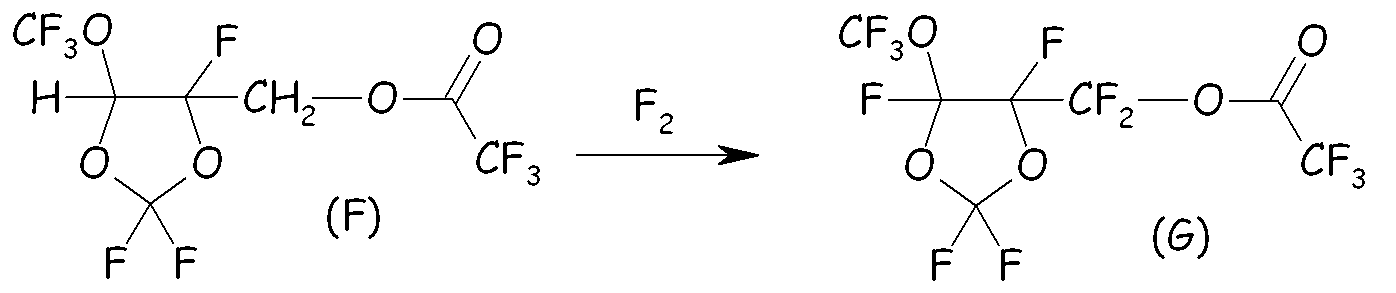
Сложный эфир (F) разбавляли в A113 (200 мл) и фторировали смесью F2/N2 (20/80) при температуре от 0 до 10°C. Мониторинг реакции осуществляли посредством газовой хроматографии. После завершения фторирования остаточный F2 выдували посредством барботирования потока азота. Перфторированный сложный эфир (G) получали после удаления растворителя при пониженном давлении.
Пример 2d. Гидролиз перфторированного сложного эфира (G)
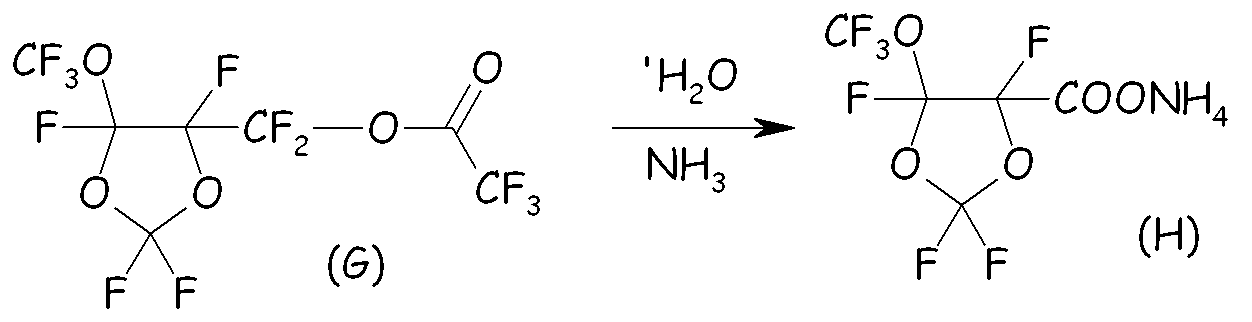
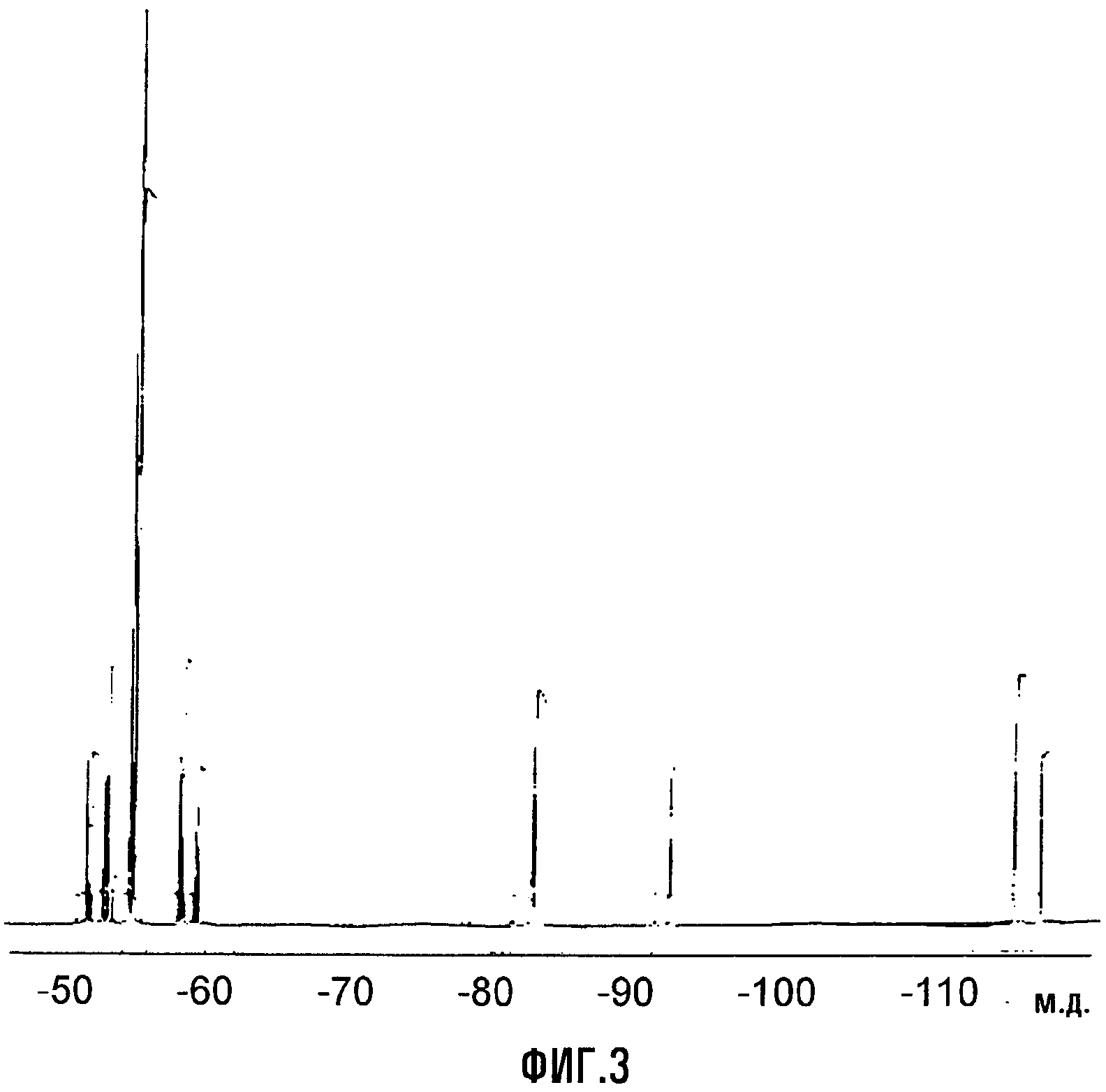
Перфторированный сложный эфир (G) гидролизовали в воде при 0°C, получая соответствующую кислоту с количественным выходом. Выделявшийся HF нейтрализовывали 1,5 моль-экв. KF, получая твердый KHF2, который отделяли фильтрованием. После удаления растворителя свободную кислоту (т.кип. = 160°C) и CF3COOH (т.кип. = 72°C) разделяли фракционной перегонкой. После этого в раствор кислоты в CH2Cl2 (200 мл) барботировали газообразный NH3; затем извлекали аммониевую соль (H) (формула Va, где Xa = NH4) с выходом 75% моль (по отношению к спиртовому промежуточному соединению (B)). Термогравиметрический анализ (TGA), проведенный на воздухе, показывал уменьшение массы на 10% при 145°C и на 50% при 182°C. Фиг.3 представляет спектр 19F-ЯМР, записанный с соединением (H).
Пример 2e. Получение фторформиата спиртового промежуточного соединения (B)
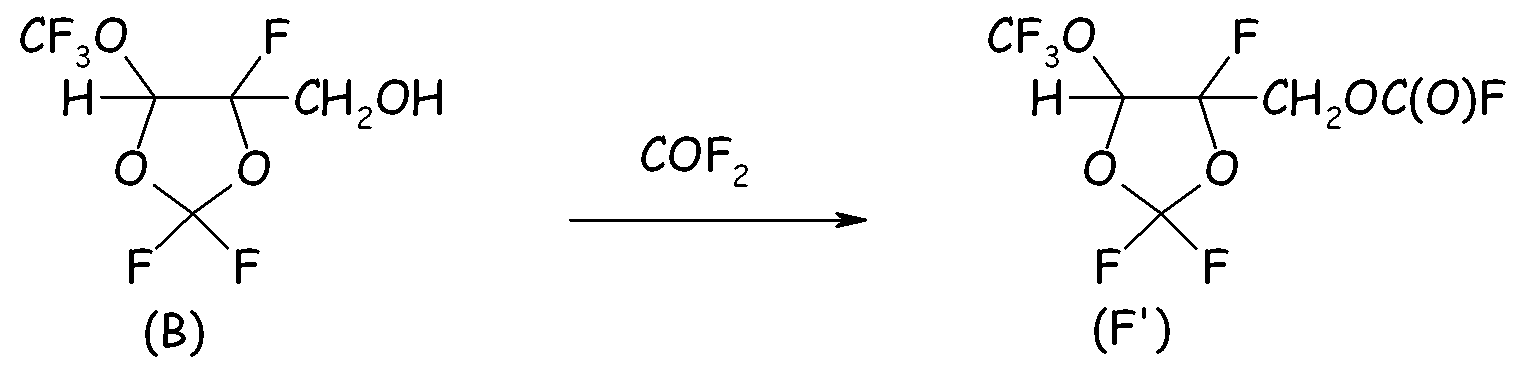
В 250-мл реактор, изготовленный из нержавеющей стали и оснащенный механической мешалкой, устройством для впуска газа, устройством для выпуска газа, термопарой для контроля за внутренней температурой и внешней охлаждающей баней, загружали 99 г спирта вышеуказанной формулы (B) и 34 г порошкообразного NaF и устанавливали внешнюю температуру при 15°C. Затем, поддерживая в реакторе интенсивное перемешивание, в реактор вводили COF2 (2,0 нормо-л/ч, полученный по реакции между CO (2,5 нормо-л/ч) и F2 (2,0 нормо-л/ч)), разбавленный He (1,0 нормо-л/ч). Выходящие газы анализировали посредством системы ГХ для оценки степени превращения COF2. Спустя 6,0 часов введение реагентов прекращали, и неочищенную смесь фильтровали для отделения неорганических солей. Жидкий продукт анализировали посредством 19F-ЯМР, показывавшим почти полное и селективное преобразование исходного спирта в желаемый фторформиат.
Пример 2f. Фторирование фторформиата (F')
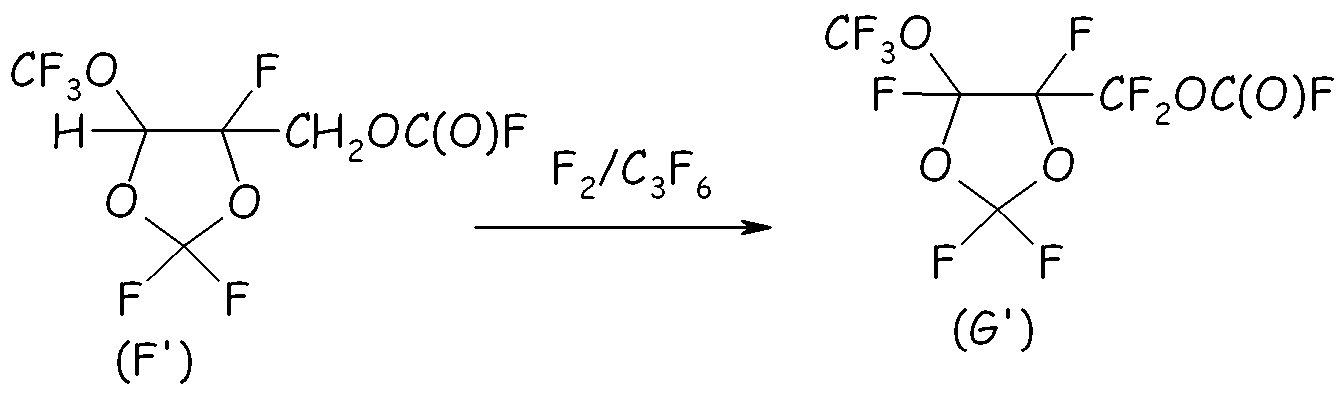
В 250-мл реактор, изготовленный из нержавеющей стали и оснащенный механической мешалкой, двумя устройствами для впуска газа, одним устройством для выпуска газа, термопарой для контроля за внутренней температурой и внешней охлаждающей баней, водили 81 г фторформиата формулы (F'), который фторировали согласно такой же процедуре, что и в Примере 1, за исключением того, что F2 вводили при 8 нормо-л/ч, разбавляя его Не (3,0 нормо-л/ч). Спустя 15 часов внутренняя температура быстро падала от 5°C до 0°C и не наблюдалось никакого дополнительного преобразования F2. Неочищенную смесь собирали и анализировали посредством ГХ и 19F-ЯМР. Желаемый перфторформиат (G') получали с выходом, составлявшим примерно 96%.
Пример 2g. Гидролиз перфторформиата (G')
Следовали той же процедуре, что подробно описана в Примере 2d.
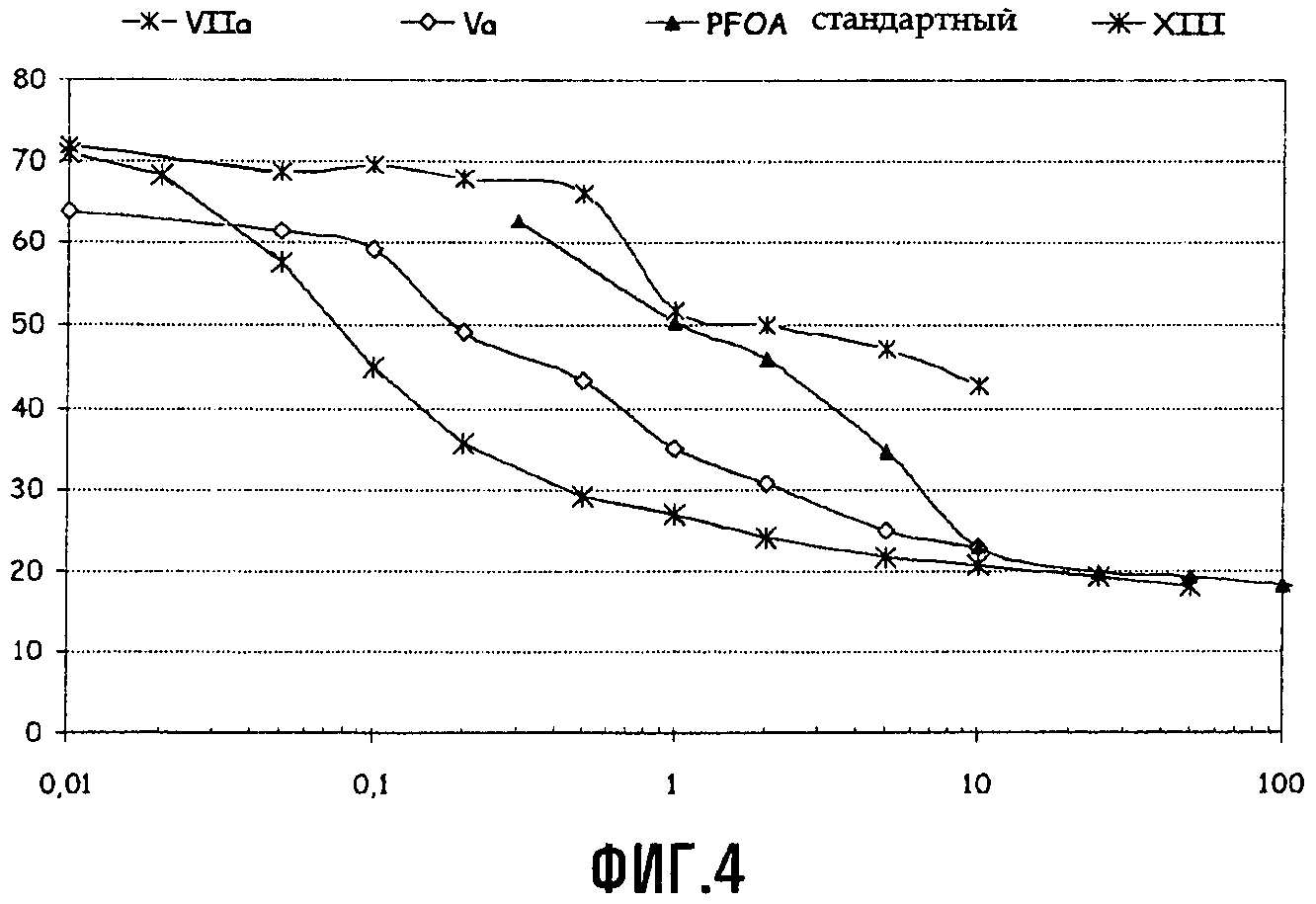
Определение поверхностного натяжения водного раствора соединений Va и VIIa (с Xa = NH4)
Измерения поверхностного натяжения проводили в разбавленных растворах аммониевых солей соединений (Va) и (VIIa) в воде при температуре 25°C, используя тензиометр LAUDA TE1C, оснащенный платиновым кольцом; первичные данные обрабатывали по методике Hun-Mason. Для целей сравнения при тех же условиях определяли также поверхностное натяжение водного раствора перфтороктаноата аммония (APFO). График для поверхностного натяжения (в мН/м) как функции концентрации (в г/л) для соединений Va, VIIa и APFO представлен на Фиг.4.
Моделирование, проведенное с применением теории функционала плотности, для предсказания биологической устойчивости соединения Va
Применяя теорию функционала плотности, определяли конформационную структуру минимальной энергии для карбоксилатного аниона соединения (Va) или в вакууме, или в водном растворе; в частности, определяли объем и поверхность молекулы в растворе, свободную энергию сольватации, длину основной цепи молекулы, колебательную энтропию, эквивалентный диаметр, электрические заряды при атомах кислорода и углерода карбоксильных групп, дипольный момент.
Кроме того, рассчитывали структурные и энергетические данные, как описано выше, для нескольких фторсодержащих поверхностно-активных веществ, таких как перфтороктаноат, и других соединений, имеющих атомы кислорода в цепи, для которых были доступны данные о биологической устойчивости, чтобы установить соответствующие корреляции между указанными данными и профилем биологической устойчивости. В частности, было найдено, что отношение между свободной энергией сольватации и длиной молекулы прямо коррелирует с долей (%) соединения, элиминированного из организмов живых крыс через 96 часов после введения, как определено по анализу мочи.
На основании указанной взаимосвязи оказалось возможным определить экскрецию/элиминацию для циклического соединения формулы (Va), которые через 96 часов после введения превышали 95%, тогда как ожидаемая экскреция перфтороктаноата из живого организма на основании аналогичных расчетов, составляла только 5%. Эти данные хорошо демонстрируют, что циклические соединения согласно настоящему изобретению действительно обладают более благоприятным профилем биологической устойчивости, чем традиционные фторсодержащие поверхностно-активные вещества.
Уровни в крови и моче и фармакокинетические параметры соединений Va и VIIa (Xa = NH4)
Соединения Va и VIIa (Xa = NH4) вводили в единичной пероральной дозе (через желудочный зонд) трем самцам крыс Wistar (SPF) при уровнях доз, составлявших примерно 70 мкмоль/кг для сухой аммониевой соли, что соответствовало 21,2 мг/кг для соединения Va и 19,9 мг/кг для соединения VIIa. Образцы крови отбирали за 15 минут до введения и через 4, 8, 12, 24, 72 и 168 часов после введения. Для соединения Va максимальную концентрацию в плазме (Cmax) наблюдали через 4 ч (tmax) после перорального введения с достоверным периодом полуэлиминации (полувыведения) из плазмы, равным 3,1-4,5 часов. Для соединения VIIa максимальную концентрацию в плазме (Cmax) наблюдали также через 4 ч (tmax) с достоверным периодом полуэлиминации из плазмы, равным 8,0-8,9 часов. ФК-параметры этих соединений после однократной пероральной дозы (через желудочный зонд) самцам крыс приведены в таблице, представленной ниже:
|
данные относятся к трем животным, которым вводили дозу препарата.
Индивидуальные и средние уровни в моче у крыс после однократного перорального введения соответствовали рассчитанным по моче периодам полуэлиминации, равным 9, 13, 10 часов с кумулятивной экскрецией за 168 часов после введения, составившей 97-107% для соединения VIIa; соединение Va представило рассчитанные по моче периоды полуэлиминации, равные 15, 11 и 28 часов, с кумулятивной экскрецией за 168 часов после введения, составившей 80-83%.
Соединение XIII вводили трем самцам крыс Wistar в однократной пероральной дозе 73 мкмоль/кг, что соответствовало 26,05 мг/кг. Образцы крови отбирали за 15 минут до введения и через 4, 8, 12, 24, 72 и 168 часов после введения. Образцы мочи получали для временных интервалов 0-12, 12-24, 24-72, 72-96, 96-168 часов после введения дозы. Концентрацию соединения XIII в плазме и моче определяли утвержденным аналитическим способом. Максимальную концентрация в плазме (Cmax) наблюдали через 4 часа (tmax). Средняя кумулятивная экскреция с мочой за 168 часов после введения составляла около 82%.
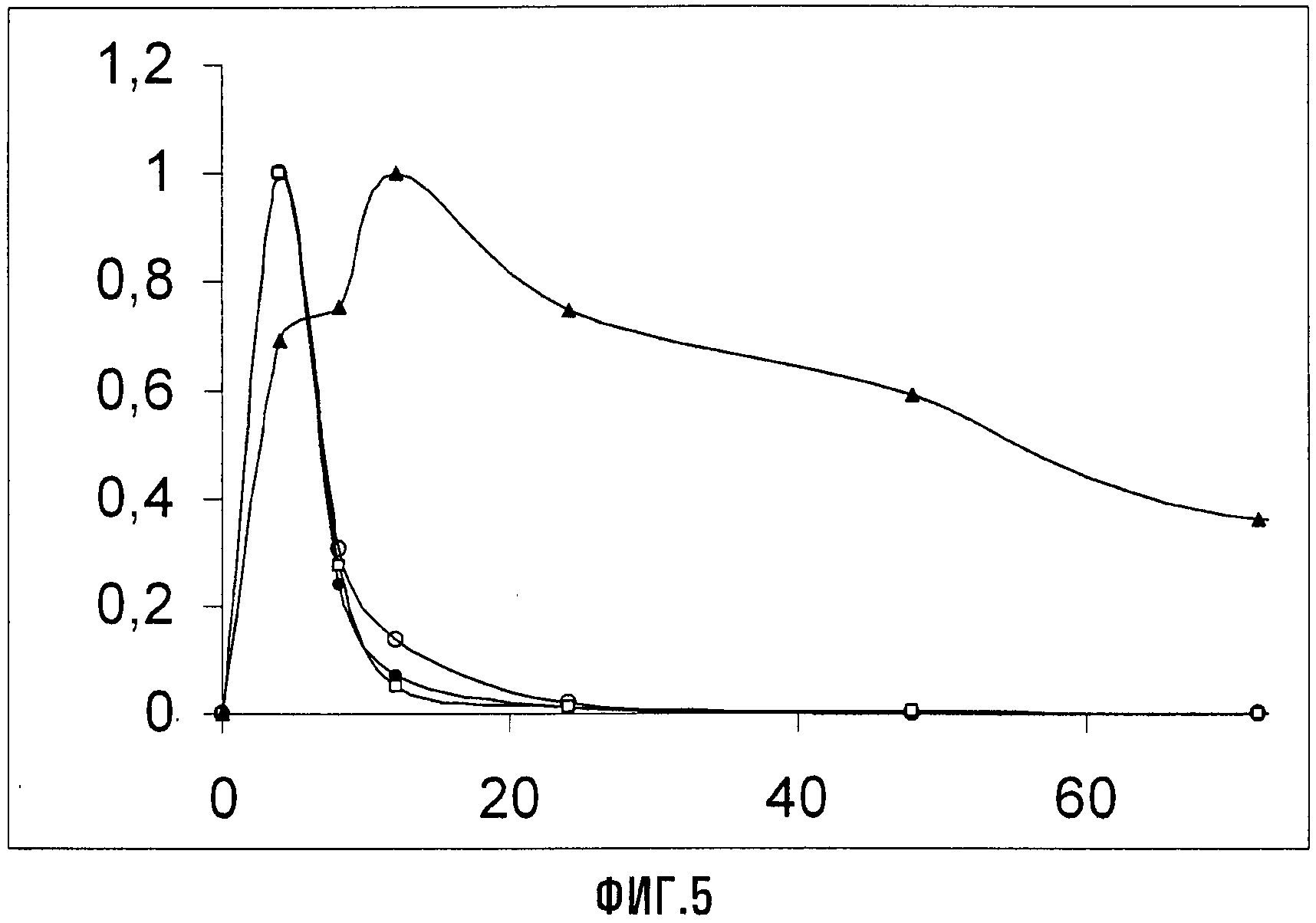
Результаты, относящиеся к концентрациям в плазме для соединений VIIa (Xa = NH4), Va (Xa = NH4), XIII (Xa = NH4) и APFO, как функции времени представлены на Фиг.5. Этот график изображает отношение C/Cmax для соединения Va (Xa = NH4) ( ), соединения VIIa (Xa = NH4) (O), для соединения XIII (Xa = NH4) (□) и APFO (▲), где C представляет собой текущую концентрацию в плазме, и Cmax представляет собой максимальную концентрацию в плазме в виде функции от времени (в часах). Экспериментальные данные показывают значительно более быструю элиминацию соединений Va, VIIa и XIII (Xa = NH4) из крови после однократного перорального введения, чем то, что наблюдали для APFO. Кумулятивная экскреция с мочой всех трех соединений за 96 и 168 часов после введения всегда была больше 80%.
), соединения VIIa (Xa = NH4) (O), для соединения XIII (Xa = NH4) (□) и APFO (▲), где C представляет собой текущую концентрацию в плазме, и Cmax представляет собой максимальную концентрацию в плазме в виде функции от времени (в часах). Экспериментальные данные показывают значительно более быструю элиминацию соединений Va, VIIa и XIII (Xa = NH4) из крови после однократного перорального введения, чем то, что наблюдали для APFO. Кумулятивная экскреция с мочой всех трех соединений за 96 и 168 часов после введения всегда была больше 80%.
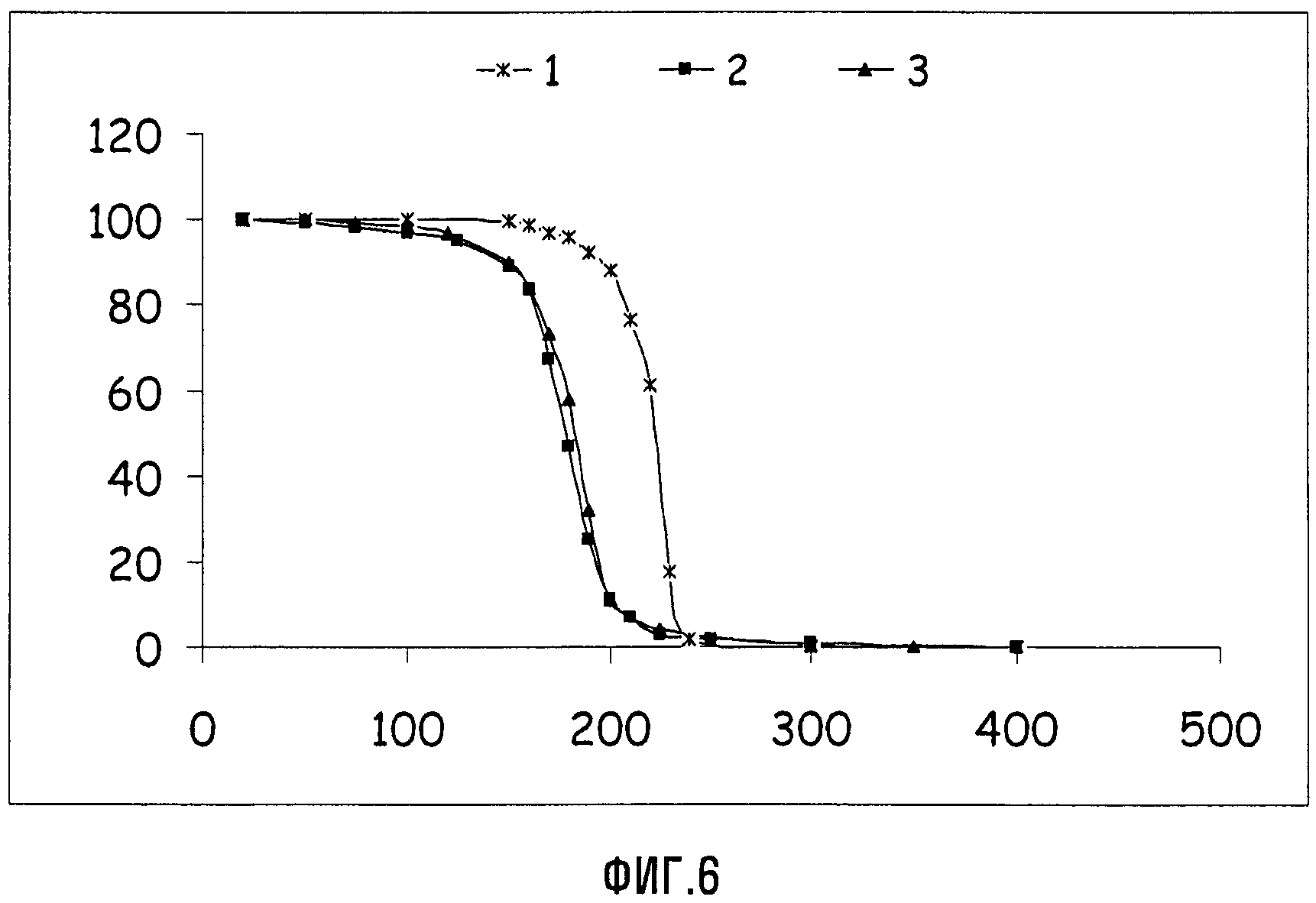
TGA-анализы соединений Va и VIIa (Xa = NH4) в сравнении с APFO
Фиг.6 представляет графики TGA в виде уменьшения массы (в %) как функции температуры (в °С) для APFO (1), соединений Va (2) и VIIa (Xa = NH4) (3). Эти данные хорошо демонстрируют, что циклические соединения являются более летучими, чем перфторалкановые кислоты, и поэтому можно ожидать, что они оставляют более низкие остаточные уровни в конечных образцах, полученных из содержащих их дисперсий.
Пример полимеризации 3: Полимеризация PTFE в присутствии соединения Va (Xa = NH4)
В полимеризационный реактор с общим объемом 100 см3, оснащенный механической мешалкой, загружали 60 см3 деионизированной воды, 0,12 г соединения Va (Xa = NH4) и 1,0 г твердого парафина с точкой размягчения при 52-58°С. Реактор вакуумировали и нагревали до 70°C. В реакторе поддерживали механическое перемешивание, и в него загружали газообразный TFE до достижения избыточного давления 20 бар. Полимеризацию инициировали раствором, содержавшим 0,5 мг пероксодисульфата аммония (NH4)2S2O8 (APS) и 9,6 мг пероксида диянтарной кислоты (DSAP). Реакционное давление поддерживали при установленном значении 20 бар изб.д. посредством введения газообразного TFE. Реакционную температуру повышали до 80°C со скоростью 0,5°C/мин. Спустя 80 мин введение TFE прекращали, реактор продували и охлаждали. Получали стабильную дисперсию PTFE, содержавшую 20% по массе твердого вещества, без образования коагулята в реакторе в процессе полимеризации. По измерениям рассеяния лазерного излучения (LLS) было найдено, что диаметр частиц латекса составлял 235 нм.
Препаративный пример 4
Синтез соединения XIII (с Xa = NH4)
Пример 4a: Реакция между перфтор-5-метокси-1,3-диоксолом (MDO, (A) на схеме, представленной ниже) и этиленгликолем
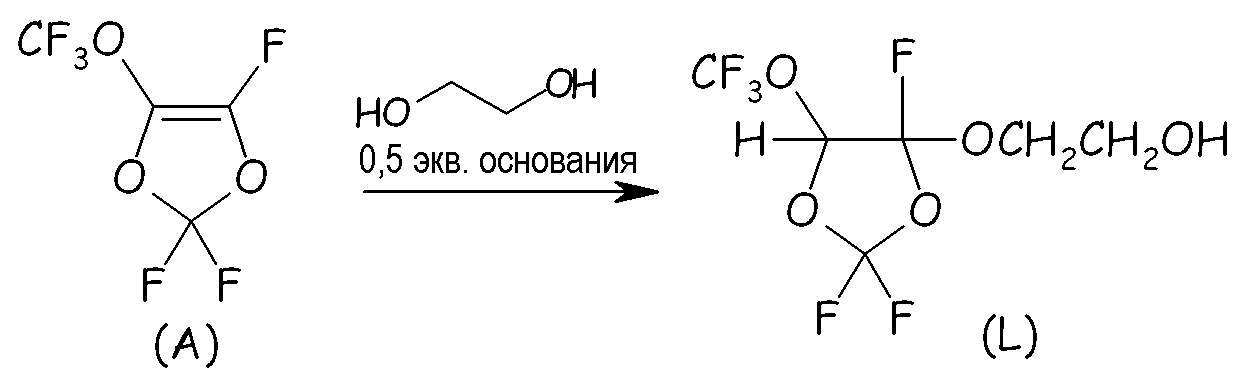
В четырехгорлый круглодонный стеклянный реактор, оснащенный магнитной мешалкой, термометром, холодильником, поддерживаемым при -75°С (сухой лед с изопропиловым спиртом) и двумя капельными воронками, вносили 450 г этиленгликоля; реактор охлаждали на ледяной бане до 0°C; затем, спустя полчаса, добавляли раствор 11,4 г (285 мэкв.) NaOH (тв.) в 60 мл дистиллированной воды H2O. После небольшого экзотермического эффекта смесь нагревалась до 80°C; при этом медленно добавляли 150 г (714 ммоль) MDO. Закончив это добавление, реакционную смесь перемешивали еще в течение 2 часов. После охлаждения до 20°С добавляли 250 мл дихлорметана, и смесь, полученную в результате этого, дважды промывали раствором соли. Органическую (нижнюю) фазу сначала сушили над MgSO4, фильтровали и затем выпаривали CH2Cl2. Было найдено, что выход выделенного продукта соединения (I) составлял 74%.
Пример 4b. Этерификация спиртового промежуточного соединения (L)
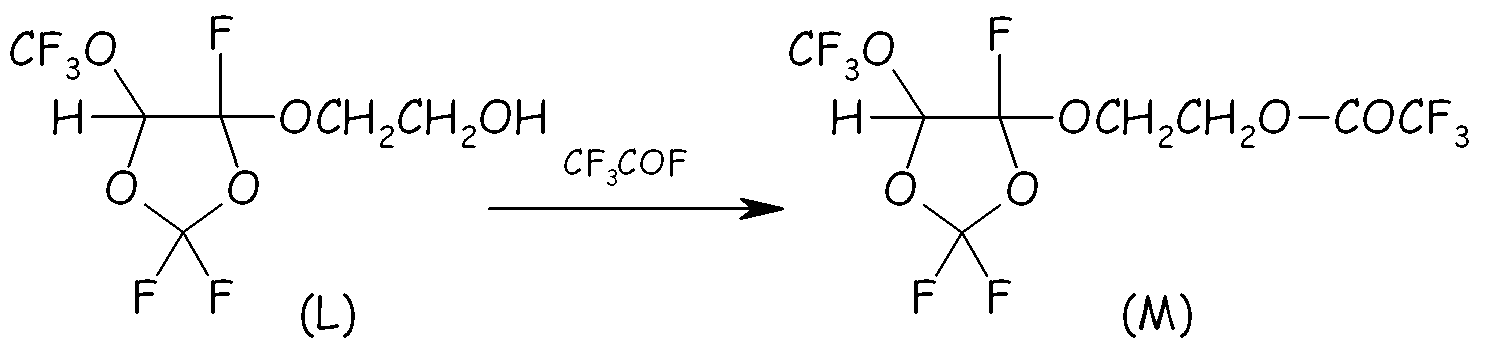
Спиртовое промежуточное соединение (L) (184 ммоль, 50 г), полученное из Примера 4a, реагировало с CF3COF (200 ммоль, 23,2 г) в 150 мл A113 при T=0°C. Растворитель и непрореагировавший CF3COF удаляли посредством отгонки при 40°C/600 мм Hg.
Пример 4c. Фторирование сложного эфира (M)
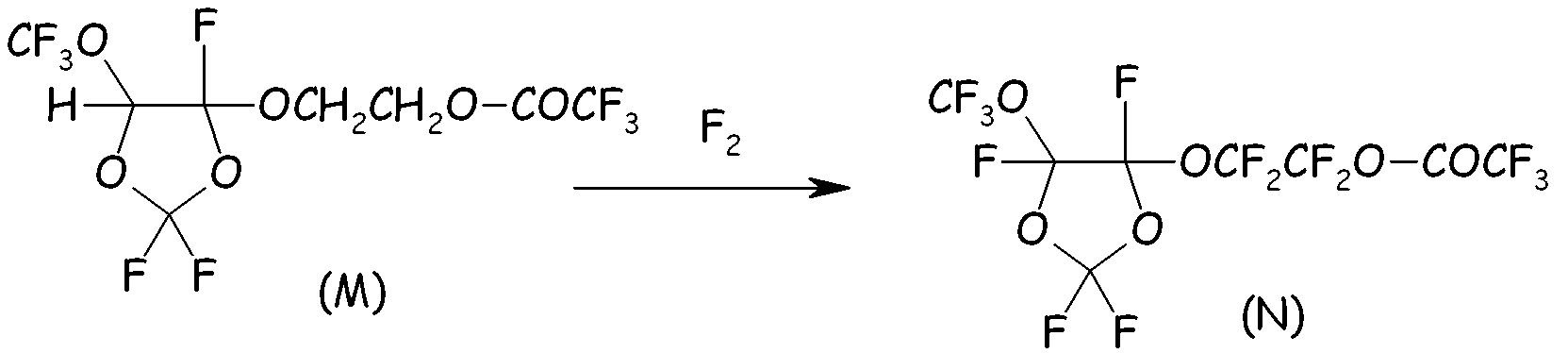
Сложный эфир (M) разбавляли в A113 (150 мл) и фторировали смесью F2/N2 (20/80) при температуре от 0 до 30°C. Мониторинг реакции осуществляли посредством газовой хроматографии. После завершения фторирования остаточный F2 выдували барботированием потока азота. Перфторированный сложный эфир (N) извлекали после удаления растворителя посредством фракционной перегонки.
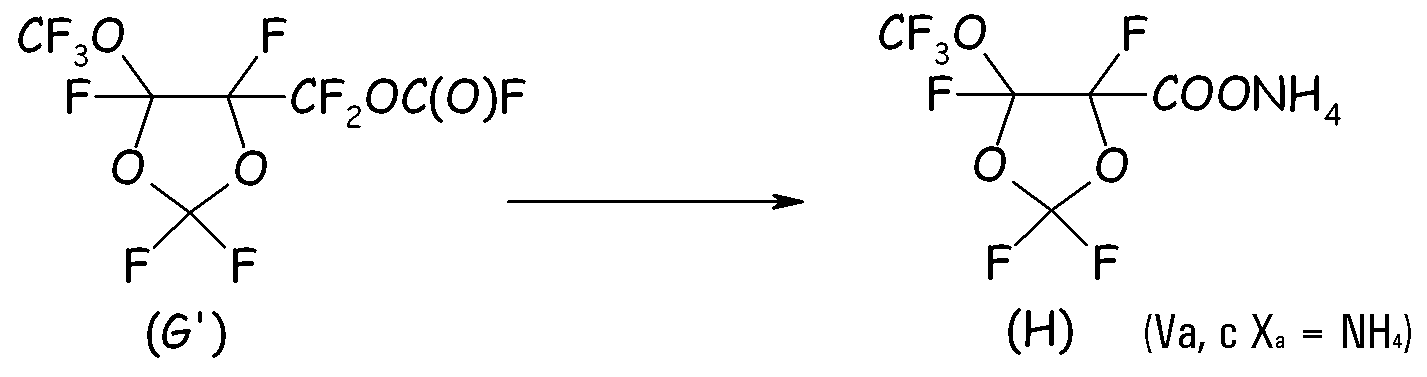
Пример 4d. Гидролиз перфторированного сложного эфира (N)
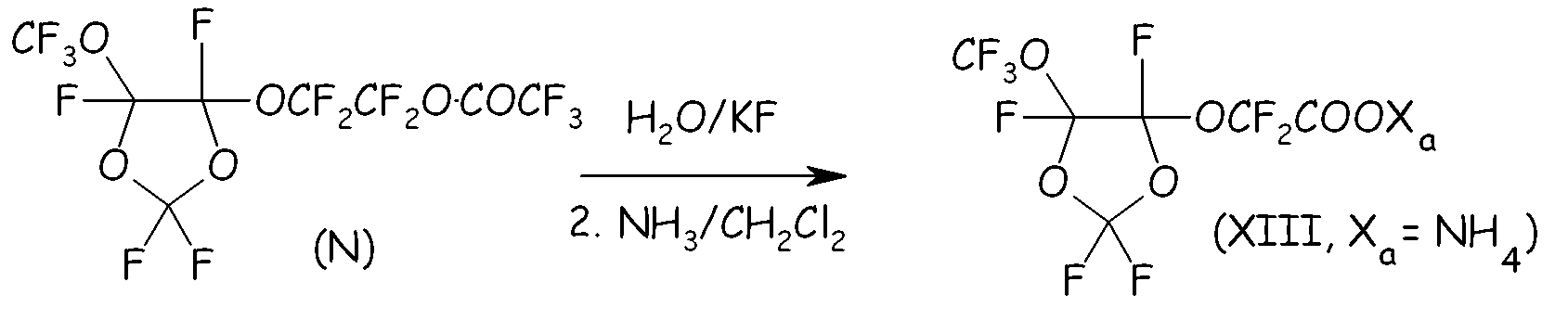
Перфторированный сложный эфир (N) гидролизовали в воде при 0°C, получая соответствующую кислоту с количественным выходом. Выделявшуюся HF нейтрализовывали 1,5 моль экв. KF, получая твердый KHF2, который отделяли фильтрованием. После удаления растворителя свободную кислоту (XIII, Xa= H) и CF3-COOH (т.кип.=72°C) отделяли друг от друга посредством фракционной перегонки.
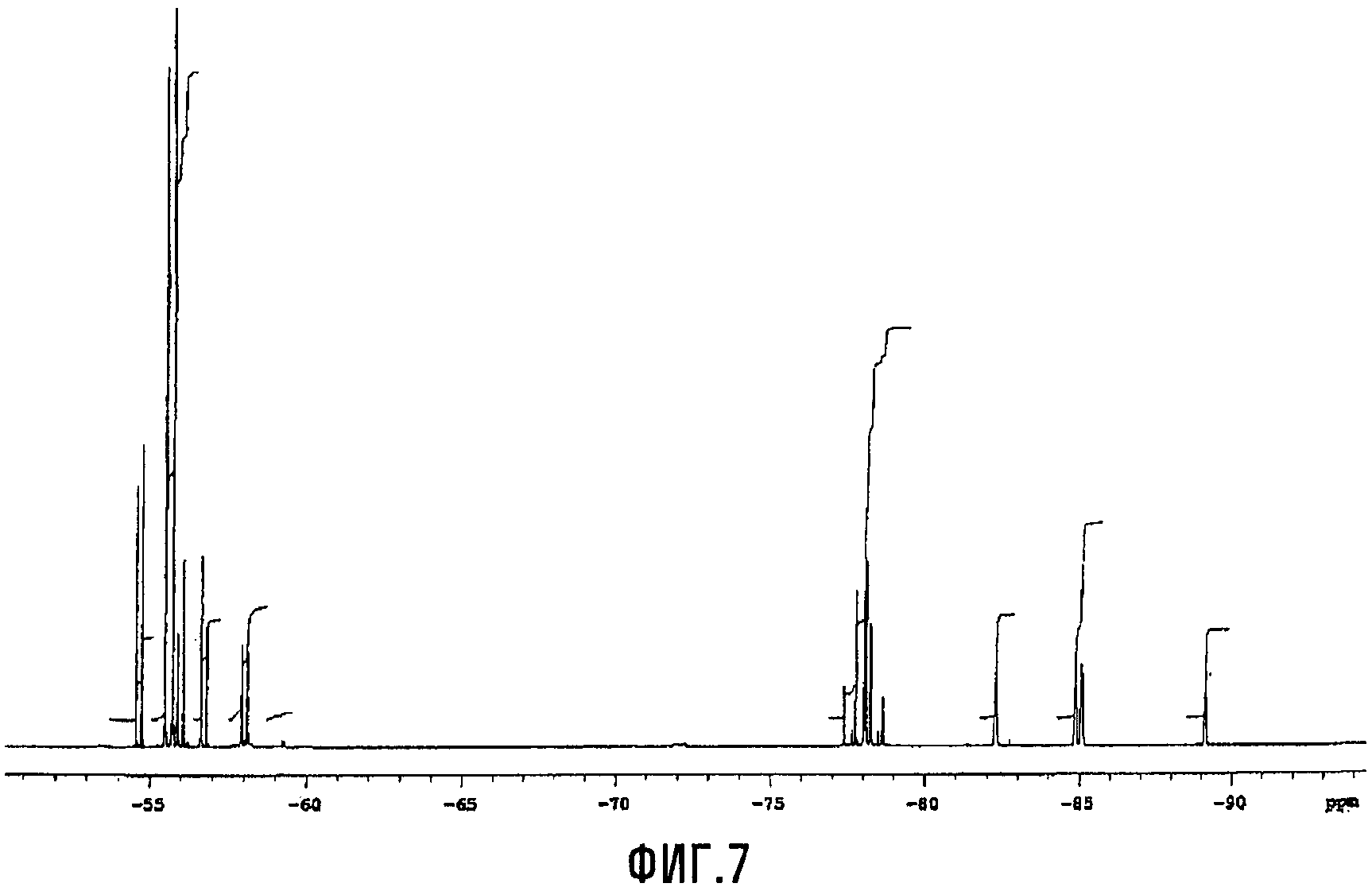
Кислоту (XIII, Xa = H) солюбилизировали в CH2Cl2 (200 мл); затем в указанный раствор барботировали газообразный NH3; после этого получали аммониевую соль (XIII, Xa = NH4) с выходом 75% моль (в отношении к спиртовому промежуточному соединению (L)). Термогравиметрический анализ (TGA), проведенный на воздухе, показывал уменьшение массы на 10% при 159°C и на 50% при 191°C. Фиг.7 представляет спектр 19F-ЯМР, записанный с аммониевой солью (XIII, Xa = NH4).
Пример 4e. Получение фторформиата из спиртового промежуточного соединения (L)
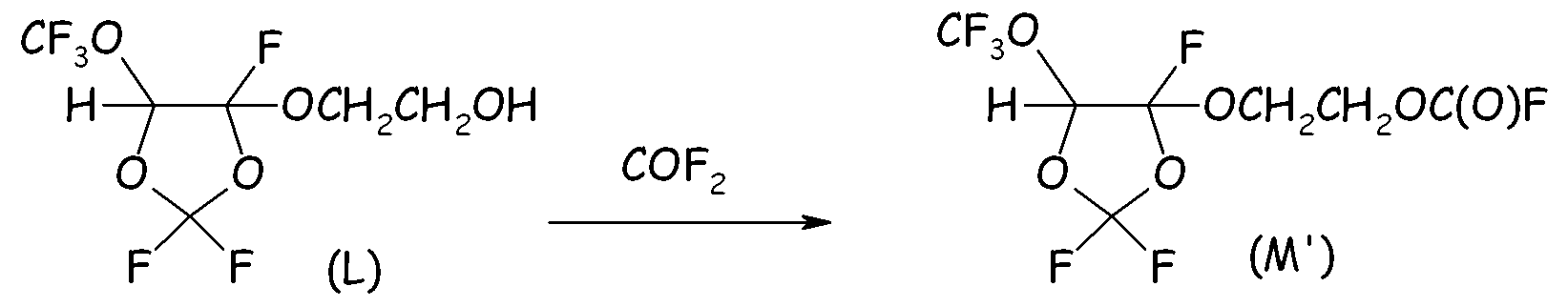
В 500-мл реактор, изготовленный из нержавеющей стали, оснащенный механической мешалкой, устройством для впуска газа, устройством для выпуска газа, термопарой для контроля за внутренней температурой и внешней охлаждающей баней, вносили 393 г спирта вышеуказанной формулы (L) и 92 г порошообразного NaF и внешнюю температуру устанавливали при 15°C.
Затем, поддерживая интенсивное перемешивание реакционной среды, в реактор вводили COF2 (6,0 нормо-л/ч, полученный по реакции между CO (7,0 нормо-л/ч) и F2 (6,0 нормо-л/ч)) разбавленный Не (2,0 нормо-л/ч). Выходящие газы анализировали посредством системы ГХ для оценки степени преобразования COF2. Спустя 6,75 часа введение реагентов прекращали, и неочищенную смесь фильтровали для отделения неорганических солей. Жидкий продукт анализировали посредством 19F-ЯМР, который показывал почти полное и селективное преобразование исходного спирта в желаемый фторформиат (M').
Пример 4f. Фторирование фторформиата (M')
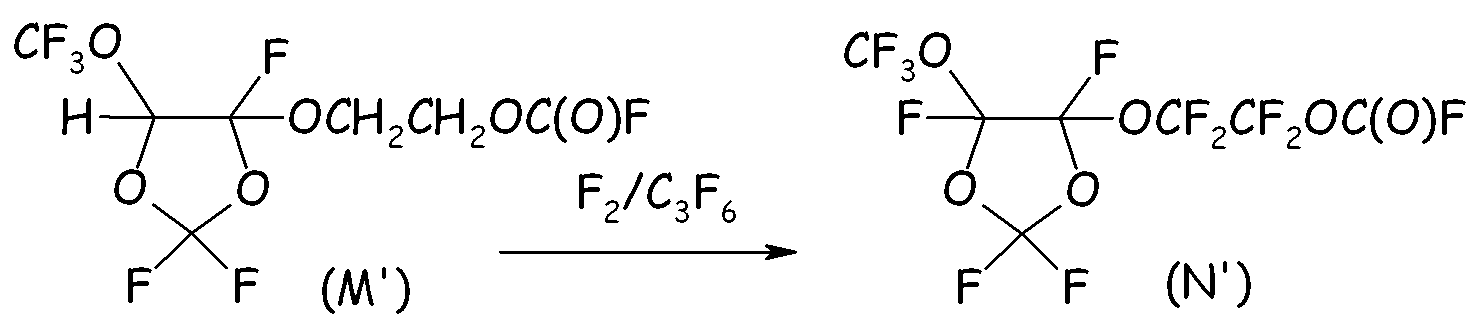
В 500-мл реактор, изготовленный из нержавеющей стали, оснащенный механической мешалкой, двумя устройствами для впуска газа, одним устройством для выпуска газа, термопарой для контроля за внутренней температурой и внешней охлаждающей баней, загружали 278 г фторформиата вышеуказанной формулы (M'), и внешнюю температуру устанавливали при 0°C. Затем, поддерживая в реакторе интенсивное перемешивание, через впускные устройства в реактор вводили два разных потока газов: F2 (2,3 нормо-л/ч), разбавленный Не (4,5 нормо-л/ч), и C3F6 (0,3 нормо-л/ч), разбавленный Не (1,5 нормо-л/ч). Выходящие газы пропускали через ловушку с NaF и анализировали посредством ГХ для оценки степени преобразования F2, тем самым оценивая степень преобразования C-H в C-F. Внутреннюю температуру поддерживали постоянной при +5°C. Спустя 57 часов происходило быстрое снижение внутренней температуры с 5°C до 0°C без наблюдаемого дополнительного преобразования F2. Введение реагентов прекращали, и остаточный HF удаляли инертным газом. Неочищенную смесь собирали и анализировали посредством ГХ и 19F-ЯМР. Желаемый перфторфторформиат (N') получали с выходом, составлявшим приблизительно 95%.
Пример 4g. Гидролиз перфторформиата (N')
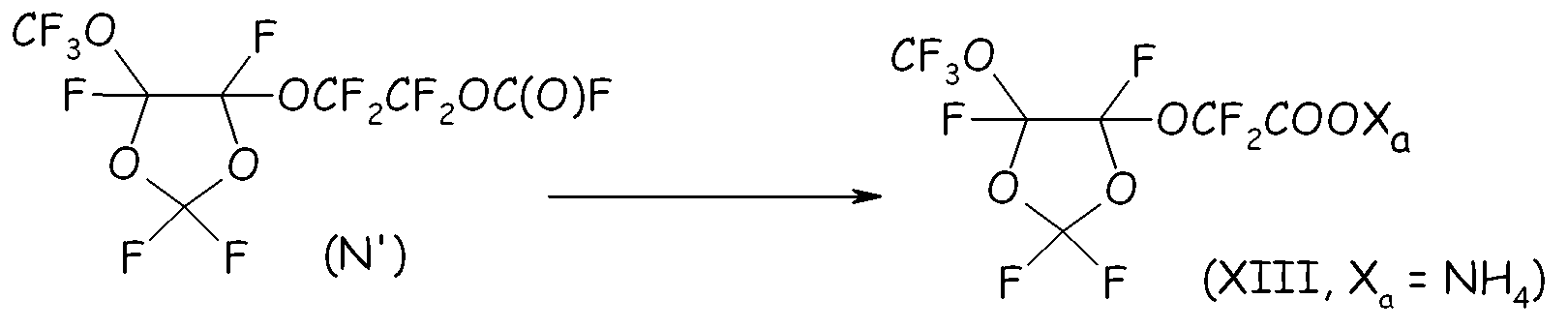
Следовали той же процедуре, что подробно описана в стадии 4d.
Определение поверхностного натяжения водного раствора соединения XIII (с Xa = NH4)
Измерения поверхностного натяжения проводили в разбавленных растворах аммониевых солей соединения (XIII), как подробно описано выше для (Va) и (VIIa). График, демонстрирующий поверхностное натяжение (в мН/м) в виде функции концентрации (в г/л), для соединения (XIII) представлен на Фиг.4.
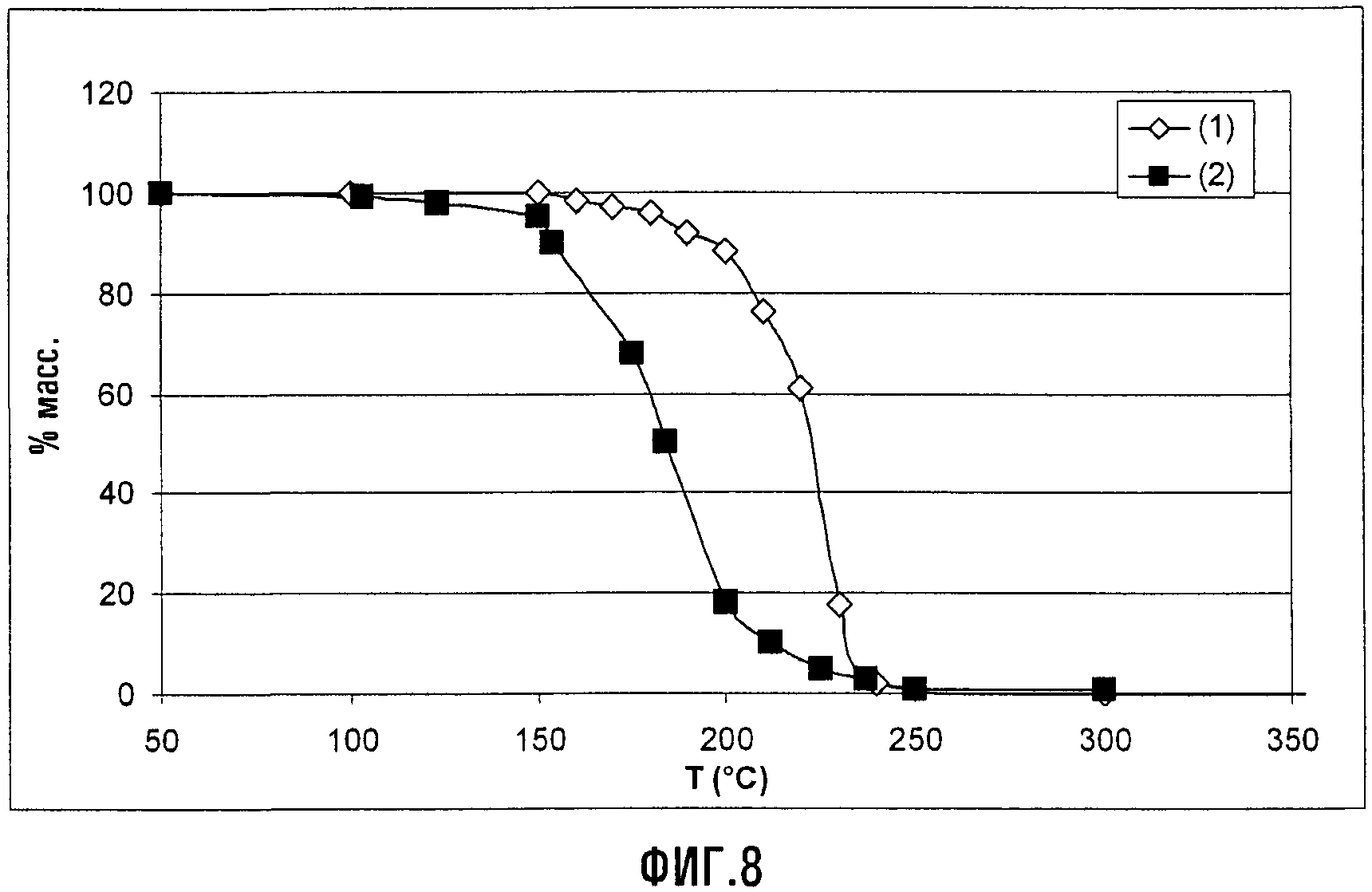
TGA-анализы соединения XIIIa (Xa = NH4) в сравнении с APFO
Фиг.8 представляет графики TGA в виде уменьшения массы (в %) как функции температуры (в °С) для APFO (1) и соединения XIII (Xa = NH4) (2). Эти данные хорошо демонстрируют, что циклическое соединение является более летучим, чем перфторалкановые кислоты, возможно вследствие явлений декарбоксилирования, и, таким образом, можно ожидать, что оно оставляет более низкие уровни остаточных веществ в конечных продуктах, полученных из содержащих их дисперсий.
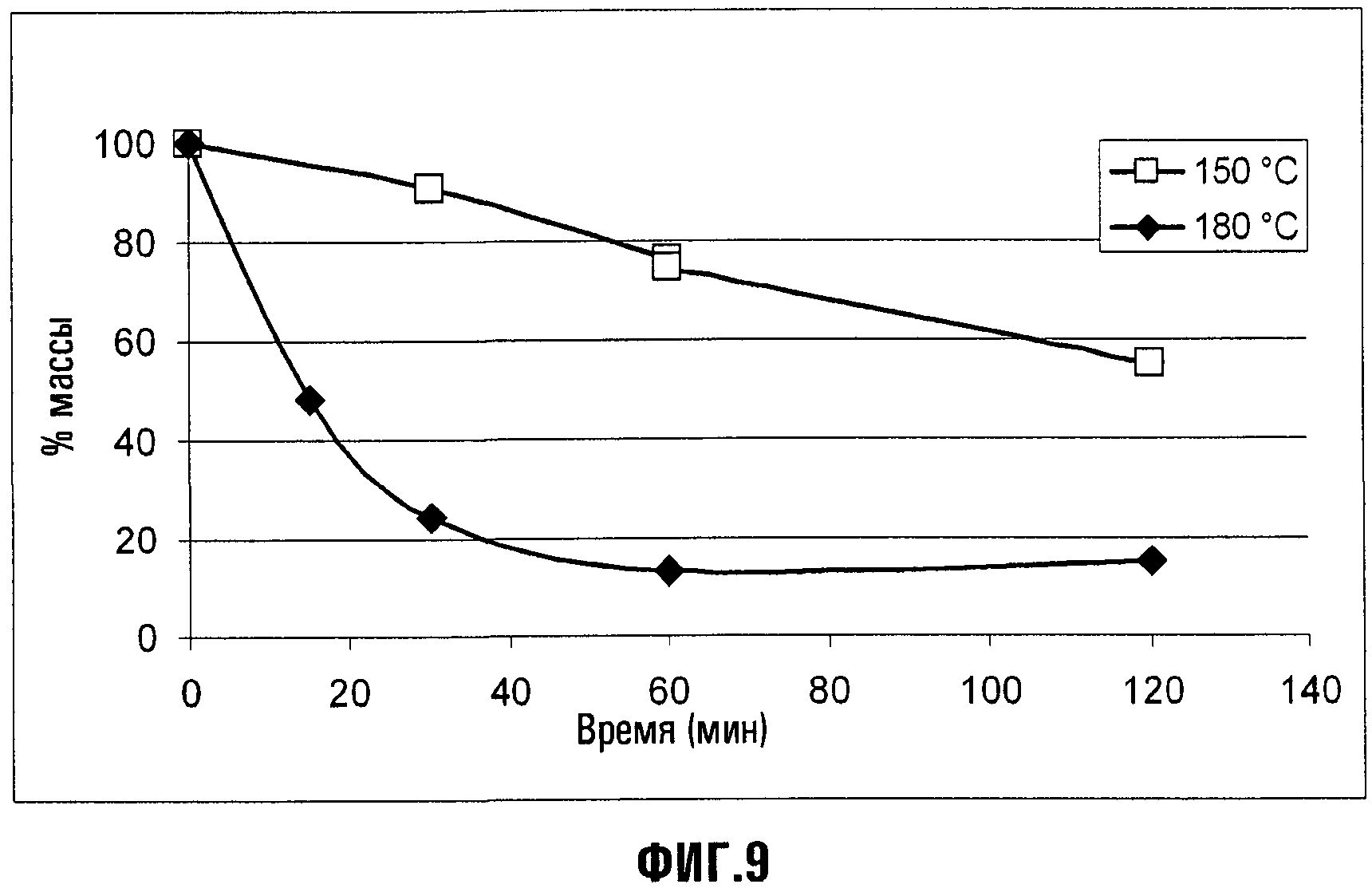
Кроме того, для оценки кинетики декарбоксилирования было выполнено изотермическое сканирование в вакууме для соединения XIII (Xa = NH4) при T = 150 и 180°C; эти графики представлены на Фиг.9, где на абсциссе дано время (в минутах), а другая ось показывает % массы относительно начальной массы. ГХ, сопряженная с масс-спектрометрией, сделала возможной идентификацию в циклическом C5O4F9H (т.е. соответствующем декарбоксилированном соединении) детектированного значительно преобладающего летучего материала.
Пример полимеризации 5: Полимеризация PTFE в присутствии соединения XIII (Xa = NH4) и выделение соединения XIII (Xa = NH4) посредством ионного обмена
В полимеризационный реактор с общим объемом 5000 мл, оснащенный механической мешалкой (500 об/мин), загружали 3 л деионизированной воды; реактор нагревали до 60°С и дополнительно загружали 60 г 10%-ного по массе водного раствора соединения XIII (Xa = NH4) и 60 г твердого парафина с точкой размягчения при 52-58°С. Реактор вакуумировали и нагревали до 70°С. Поддерживая в реакторе механическое перемешивание, в реактор вводили газообразный TFE до достижения избыточного давления 20 бар. Полимеризацию инициировали, вводя 30 мл раствора, содержавшего 4 г/л пероксодисульфата аммония (NH4)2S2O8 (APS), и устанавливая температуру при 80°С. Реакционное давление поддерживали при установленном значении 20 бар изб.д. посредством введения газообразного TFE. После введения 1450 г TFE реактор продували и охлаждали. Получали стабильную дисперсию PTFE, содержащую примерно 32% по массе твердого вещества, без образования коагулята в реакторе в процессе полимеризации. По измерениям рассеяния лазерного излучения (LLS) было найдено, что диаметр частиц латекса составлял 230 нм.
Дисперсию PTFE, полученную так, как подробно описано выше, стабилизировали, добавляя 4,5% по массе (в расчете на твердые вещества) неионного поверхностно-активного вещества Tergitol® TMN 100X. Дисперсию разбавляли до содержания твердых веществ, равного 9%, и очищали, обрабатывая анионообменными смолами Amberjet® 4400 OH. Было найдено, что очищенная дисперсия содержала менее 5 м.д. соединения XIII (в расчете на твердые вещества). Никакой коагулят в процессе очистки не образовывался.
Пример полимеризации 6: Сополимеризация TFE и перфторпропилвинилового простого эфира в присутствии соединения VIIa (Xa = NH4) и перфторированного простого полиэфира в качестве поверхностно-активного вещества
В полимеризационный реактор с общим объемом 5000 мл, оснащенный механической мешалкой (470 об/мин), загружали 2550 г деионизированной воды; реактор нагревали до 60°С и в него дополнительно загружали 150 г 5%-ного по массе водного раствора соединения XIII (Xa = NH4) и 200 г 1%-ного по массе водного раствора аммониевой соли перфторированного простого полиэфира с двумя концевыми карбоксильными группами формулы: XaOOC-CF2O-(CF2O)n(CF2CF2O)m-CF2-COOXa (Xa = NH4, n, m являются такими, чтобы средняя молекулярная масса составляла 1800). Реактор вакуумировали и нагревали до 80°С. Поддерживая в реакторе механическое перемешивание, в реактор нагнетали газообразный TFE до достижения избыточного давления 20 бар и вводили начальную порцию (20 г) перфторпропилвинилового простого эфира (PPVE). Полимеризацию инициировали, вводя 35 мл раствора, содержавшего 6 г/л пероксодисульфата аммония (NH4)2S2O8 (APS), и устанавливая температуру при 80°С. Реакционное давление поддерживали при установленном значении 20 бар изб.д. посредством введения газообразного TFE. После введения 100 г TFE вводили дополнительный перфторпропилвиниловый простой эфир (PPVE) в 5 последовательных порциях, соответствовавших общей загрузке 45 г. После введения 1300 г TFE реактор продували и охлаждали. Получали стабильную дисперсию сополимера TFE/PPVE, содержащую примерно 30% по массе твердого вещества, без образования коагулята в реакторе в процессе полимеризации. По измерениям рассеяния лазерного излучения (LLS) было найдено, что диаметр частиц латекса составлял 97 нм.
Пример полимеризации 7: Полимеризация TFE в присутствии соединения XIIIa (Xa = NH4) и последующее концентрирование с помутнением
В полимеризационный реактор с общим объемом 5 л, оснащенный импеллерной мешалкой, загружали 3 л деионизированной воды. Реактор, свободный от кислорода, нагревали до 65°C и систему перемешивания устанавливали на 500 об/мин. В реактор загружали 60 г твердого парафина, 9 г соединения XIII (с Xa = NH4) и TFE до избыточного давления 20 бар. Полимеризацию инициировали, вводя 30 см3 раствора, содержащего 120 мг пероксодисульфата аммония (NH4)2S2O8 (APS) и 15 мг соли Мора (NH4)2Fe(SO4)2·6H2O. После начала реакции реакционное давление поддерживали при 20 бар изб.д., посредством введения TFE в газовую фазу. Реакционная температура повышалась до 80°С. Спустя 130 мин завершали введение 1600 г TFE, закрывали клапаны мономера и останавливали перемешивание. Реактор разгерметизировали, продували и охлаждали.
Полученная таким образом дисперсия полимера была стабильной и содержала 33% масс./масс. твердого вещества, в реакторе не детектировали никакого коагулята. Согласно рассеянию лазерного излучения (LLS), диаметр частиц латекса составлял 200 нм, а по результатам DSC-анализа (дифференциальной сканирующей калориметрии), точка первого плавления была при 335°C и теплота кристаллизации составляла -42 Дж/г. Указанную дисперсию концентрировали до помутнения в пирексовом реакторе, получая конечную композицию с концентрацией, равной 74,3% масс./масс., и затем составляли образец массой 600 г, содержащий 60% PTFE, 5,8% неионного эмульгатора Triton® X-100 и обладающий следующими свойствами: pH 10,7; вязкость (20°C) = 31,5 сП; вязкость (35°C) = 22,5 сП; проводимость = 1132 мСм/см; устойчивость к напряжению сдвига (61°C) = 627 сек.
Сравнительная дисперсия, полимеризованная таким же образом, но с применением APFO в качестве поверхностно-активного вещества, обычно обладала свойствами в следующем диапазоне: PTFE = 59-61%; эмульгатор Triton® X-100 = 5-7%; pH 9,5-11; вязкость (20°C) = 35 сП максимально; вязкость (35°C) = 50 сП максимально; проводимость = 800-1300 мСм/см; устойчивость к напряжению сдвига (61°C) = 300-350 сек.
Пример полимеризации 8: Полимеризация TFE в присутствии соединения XIIIa (Xa = NH4) и выделение его полимера в виде сухого порошка
Стадия 8a - Полимеризация
В полимеризационный реактор с общим объемом 5 л, оснащенный импеллерной мешалкой, загружали 3 л деионизированной воды. Реактор, свободный от кислорода, нагревали до 70°C, и систему перемешивания устанавливали на 500 об/мин. В реактор загружали 60 г твердого парафина, 9 г соединения XIII (с Xa = NH4), из которых 5,5 г распределяли в течение реакции, и TFE до избыточного давления 20 бар. Полимеризацию инициировали, вводя 16 см3 раствора, содержащего 8 мг (NH4)2S2O8 (APS) и 160 мг пероксида диянтарной кислоты (DSAP). После начала реакции реакционное давление поддерживали при 20 бар изб.д., посредством введения TFE в газовую фазу. Реакционная температура повышалась до 85°С. Спустя 146 мин завершали введение 1400 г TFE, закрывали клапаны для введения мономера и останавливали перемешивание. Реактор разгерметизировали, продували и охлаждали. Полученная таким образом дисперсия полимера была стабильной и содержала 29% масс./масс. твердого вещества, в реакторе не обнаруживали никакого коагулята. Согласно рассеянию лазерного излучения (LLS), диаметр частиц латекса составлял 227 нм, а по результатам DSC-анализа, точка первого плавления была при 338,4°C и теплота кристаллизации составляла -33,3 Дж/г.
Стадия 8b - Получение продукта в виде сухого порошка
Дисперсию со стадии 8а коагулировали, промывали и сушили в течение 32 часов, соответственно при 140-160-180°C. Согласно ГХ-анализу, остаточное количество соединения (XIII) на сухом порошке составляло < 20 м.д. (предел анализа) во всех трех случаях.
Препаративный пример 9: Адсорбция/десорбция соединения XIII (Xa = NH4) на ионообменных смолах
4 г анионообменной смолы Dowex MSA, предварительно промытой деминерализированной водой и высушенной, контактировали в течение 24 часов со 100 г 1,3%-ного масс./масс. раствора соединения XIII (с Xa = NH4). Было найдено, что насыщение смолы составляло 24,5%.
Полученную таким образом израсходованную/насыщенную смолу промывали деминерализированной водой с вакуумным отсосом досуха. Часть этой смолы, 3,5 г, после дополнительной стадии промывки (30 мл воды) экстрагировали 60 мл раствора, содержщего 70% метанола и 30% серной кислоты, и опять промывали 30 мл воды. Кислый раствор и промывную воду собирали, разбавляли водой, омыляли NaOH до pH 11,2 и окончательно разбавляли водой до 250 г. ГХ-анализ показал 70%-ный выход соединения (XIII).
Пример полимеризации 10: Получение латекса PVDF в присутствии смеси поверхностно-активного вещества
В реактор с внутренним объемом 7,57 л загружали 5241 г деионизированной воды, 134 г 10%-ного масс./масс. водного раствора соединения XIII (Xa = NH4) и 5,4 мг аммониевой соли перфторированного простого полиэфира с двумя концевыми карбоксильными кислотными группами формулы: XaOOC-CF2O-(CF2O)n(CF2CF2O)m-CF2-COOXa (Xa = NH4, n, m являются такими, чтобы средняя молекулярная масса составляла 1800) и 4 г воска. Реактор нагревали до 100°C и продували в течение 2 мин. Температура повышалась до 122,5°C, и в реактор нагнетали под давлением винилиденфторид (VDF) до 650 psi (44,8 бар). Для инициирования полимеризации в реактор добавляли 24,4 мл ди-трет-бутилпероксида и в течение всей полимеризации поддерживали давление 650 psi (44,8 бар). После достижения желаемой степени преобразования (2298 г израсходованного мономера) прекращали введение мономера и останавливали перемешивание, реактор охлаждали и из реактора собирали полимерный латекс, содержавший 28% по массе твердого вещества, со средним размером частиц диспергированного полимера, равным 282 нм. Латекс фильтровали для сбора возможного коагулята и реактор осматривали для определения количества наростов (например, полимера, налипшего на лопасти мешалки и стенки реактора).
Пример полимеризации 11: Сополимеризация TFE с перфторпропилвиниловым простым эфиром в присутствии соединения VIIa (Xa = NH4) и перфторированного простого полиэфира, применяемого в качестве поверхностно-активного вещества, предварительно смешанных в форме микроэмульсии
Стадия 10a - Получение стабильной дисперсии в воде соединения XIII (с Xa = NH4) и дополнительно фторированного эмульгатора
В стеклянной колбе, оснащенной мешалкой, при осторожном перемешивании смешивали 24,00 г поверхностно-активного вещества формулы XIII (с Xa = NH4), 24,00 г деминерализированной H2O; 12,00 г полимерного перфторированного простого эфира с двумя концевыми карбоксильными кислотными группами формулы: HOOC-CF2O-(CF2O)n(CF2CF2O)m-CF2-COOH (n, m являются такими, чтобы средняя молекулярная масса составляла 1800). Система самопроизвольно образовывала микроэмульсию, которая имеет вид прозрачной, термодинамически стабильной дисперсии. Было найдено, что капли, измеренные по рассеянию лазерного излучения (LLS), имели средний диаметр, равный 11,7 нм.
Стадия 10b - Полимеризация тетрафторэтилена (TFE) и перфторпропилвинилового простого эфира (PPVE)
В реактор с внутренним объемом 5 л загружали 3 л воды и 33 мл вышеуказанной микроэмульсии. Температуру повышали до 75°С; в реактор загружали 50 г перфторпропилвинилового простого эфира и нагнетали этан, повышая давление до 470 мбар, и окончательно повышали давление до 20 бар, нагнетая TFE. Полимеризацию инициировали, добавляя персульфат аммония (0,48 г вводили в начале процесса, в дальнейшем пятью порциями вводили еще 0,30 г в комбинации с дополнительными порциями перфторпропилвинилового простого эфира). Полимеризацию продолжали 76 мин до достижения общего потребления 1500 г мономеров. Получали латекс, содержавший 31% масс. твердых веществ, с частицами со средним диаметром 60 нм (как определено посредством LLS) сополимера TFE/PPVE (PPVE: 3,1% масс.), имевшего MFI 30 г/10 мин (372°C/5 кг, измерено согласно ASTM D 1238), точку плавления при 305,7°C и теплоту кристаллизации, равную -27,3 Дж/г (измерено согласно ASTM D 3418).
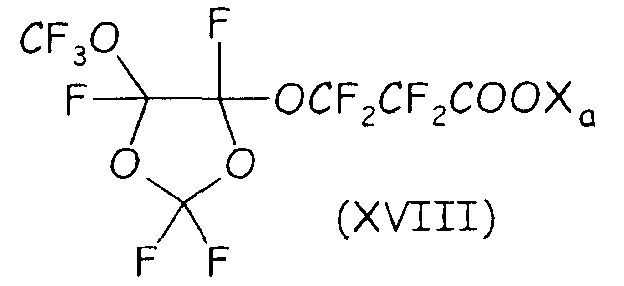
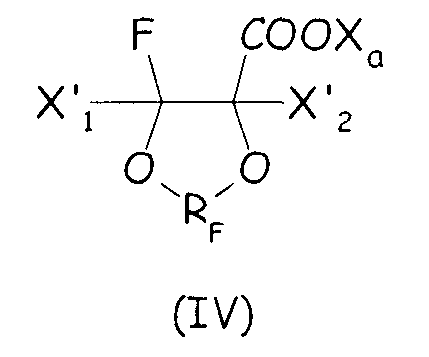
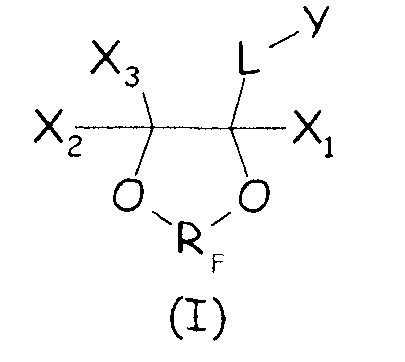
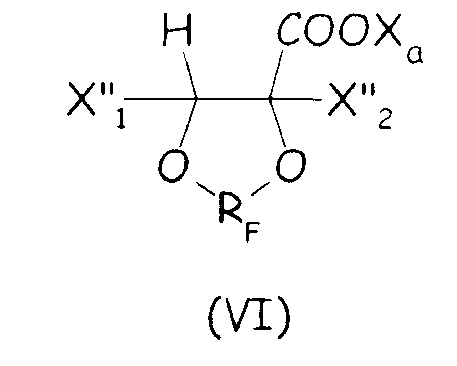
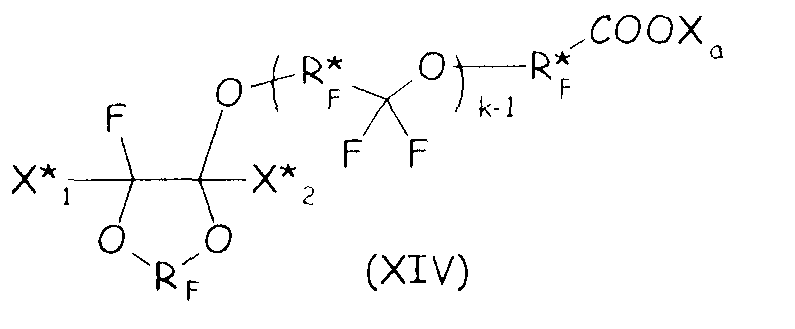
Способ смазывания коробки передач ветряной турбины
Способ и катализатор для синтеза трифторэтилена
Способ фторирования галоолефинов
Водные композиции для маслоотталкивающей обработки бумаги, содержащие соли перфторполиэфиров дикарбоновых кислот
Способ смазывания коробки передач ветряной турбины
Способ и катализатор для синтеза трифторэтилена
Способ фторирования галоолефинов
Способ получения альфа-йодперфторалканов и альфа, омега-дийодперфторалканов
Фторэластомеры
Перфторэластомеры

