НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ СУХИХ ФАРМАЦЕВТИЧЕСКИХ ФОРМ, ДИСПЕРГИРУЕМЫХ В ВОДЕ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ПОЛУЧЕННЫЕ ТАКИМ СПОСОБОМ
Вид РИД
Изобретение
Давно известно, что в отношении традиционного исследования фармакологических и токсикологических свойств лекарственных средств одним из факторов, определяющих активность, является количественная характеристика всасывания активного компонента. Кинетика все чаще становится объектом систематических исследований с учетом того, что интенсивность ответа и, часто, его природа являются функцией концентрации, получаемой на уровне воспринимающего органа. В значительном числе таких исследований фармакокинетики и, следовательно, биодоступности показана существенная полезность модификаций, в частности галеновых, осуществляемых в ходе получения фармацевтических форм, главным образом, с целью получения форм, наиболее приспособленных для введения через пищеварительный тракт.
Наибольшая проблема при слабом кишечном всасывании лекарственных средств, видимо, связана с тем, что всасывание происходит в течение достаточно короткого времени на малой площади поверхности кишечника. Таким образом, для увеличения всасывания важно, чтобы действующее вещество быстро и в полном объеме было направлено к месту всасывания в растворимой форме.
Лекарственные средства обладают низкой растворимостью в воде или в низкой степени переходят в солевую форму при прохождении в желудок, так что при этом они всасываются только частично. В литературных источниках предшествующего уровня техники указано, что всасывание в пищеварительном тракте может быть благоприятным образом изменено за счет подбора размера частиц, за счет прибавления, в частности, неионогенных поверхностно-активных веществ, а также за счет прибавления солюбилизирующего агента.
С одной стороны, тонкое измельчение действующего вещества увеличивает соответственным образом внешнюю удельную поверхность порошкообразного вещества и представляет собой подход к решению этой проблемы. А с другой стороны, тонкое измельчение приемлемо только для некоторых фармацевтических форм, таких, как дисперсии или наполненные желатиновые капсулы. Измельчение не может быть общим решением проблемы всасывания, поскольку некоторые действующие вещества трудно тонко измельчать, так как они являются легкоплавкими или имеют слишком неустойчивую химическую структуру.
Прибавление поверхностно-активных веществ может увеличивать растворимость некоторых действующих веществ и за счет этого улучшать кинетику всасывания, но не позволяет систематически получать повышенные концентрации в крови. Кроме того, для получения существенного результата часто требуется прибавлять поверхностно-активные вещества в значительном количестве (от 25 до 50%). Такое улучшение при прохождении по пищеварительному тракту, видимо, является результатом уменьшения поверхностного натяжения, вызывающего увеличение проницаемости слизистой оболочки пищеварительного тракта. Однако большое количество поверхностно-активных веществ часто оказывает послабляющее действие, что не способствует хорошему всасыванию.
Другой подход состоит в прибавлении эмульгатора, в частности сложного эфира жирной кислоты и углевода (сложного эфира сахарозы), и представляет собой иной принцип. Такой сложный эфир увеличивает липофильность соединения и, таким образом, облегчает переход через кишечный барьер. Однако процесс такого типа дает приемлемые результаты только в случае соединений с большой липофильностью и требует высоких концентраций этого сложного эфира.
Недавно для улучшения всасывания действующих веществ, имеющих низкую растворимость в биологических жидкостях, была предпринята попытка увеличить скорость растворения за счет образования твердых дисперсий. Такие твердые дисперсии, описанные Chiou и соавт. в J. Pharm. Sci. 60 (194) 1281-1302, представляют собой систему, которая в зависимости от способа, используемого для ее получения, может иметь различные структуры (см. Fort et al. Pharm. Acta Helv. 61 (3) (1986) 69-88 или Bloc et al. Pharm. Acta Helv. 62 (1987) 23-27), соответствующие различным кристаллическим состояниям. Стекловидное состояние, притом, что его можно рассматривать как вариант твердого состояния, характеризуется жидкой фазой, способствующей его структурной неупорядоченности. Такое стекловидное состояние является малоупорядоченным и легко может быть разрушено. Оно в существенной степени способствуют увеличению скорости растворения, в частности, малорастворимых веществ в водных средах. Однако, несмотря на большое число публикаций, касающихся получения твердых дисперсий особенно на основе макроголов или полоксамеров, такая техника известна только в ограниченной степени по причине трудности обобщения. В некоторых случаях скорость растворения является большой. В других случаях скорость растворения значительно меньше и имеет тенденцию к достижению предельного значения.
Так, например, для одного и того же действующего вещества в сравнимых концентрациях были зафиксированы очень значительные различия в скорости перевода в раствор в зависимости от совместно сплавленного агента. В некоторых случаях была зафиксирована также невозможность достижения полной солюбилизации действующего вещества даже после длительного времени контакта или, в частности, тенденция к перекристаллизации.
Другой подход к решению этой проблемы заключается в получении твердых дисперсий агента с терапевтическим действием в гидрофильном наполнителе, имеющих повышенную растворимость в водной среде. Этот подход состоит, прежде всего, в растворении активного вещества с терапевтическим действием в очень легко испаряющемся органическом растворителе, к которому прибавляют полимер с высокой гидрофильностью, такой, как поливинилпирролидон. Затем растворитель выпаривают досуха для получения совместного осадка агента с терапевтическим действием и гидрофильного полимера.
Такая технология позволяет получить заметное улучшение кинетики всасывания, но она не может быть адаптирована к любому типу действующих веществ. Кроме того, при такой технологии часто требуется прибавлять к раствору поверхностно-активное вещество, которое увеличивает способность к смачиванию средами пищеварительного тракта и при необходимости ограничивает процесс образования кристаллов, спонтанно происходящий при хранении твердых дисперсий. Образование кристаллических продуктов способствует уменьшению скорости растворения в зависимости от времени (см. Kigudin et al. Chem. Pharma. Bull. 9 (1961) 866-872, Duchène D. Pharma 1 (11) (1985) 1064-1073).
В то же время, при такой технологии требуется образование совместных осадков в лактаме с высокой гидрофильностью, таком, как поливинилпирролидон, имеющем молекулярную массу в интервале от 10000 до 5000, и кислород- или хлорсодержащем растворителе или их смесях (см. US-5776495).
Последний по времени подход представлен в WO 2005/034920 на имя Life Cycle Pharma, в которой описана технология Melt Dose. Такая технология обеспечивает лучшее оральное всасывание соединений, имеющих низкую растворимость в воде. Такая технология состоит в растворении соединений в растворителе, облегчающем проникновение в эпителий кишечника для обеспечения перехода в поток крови.
Растворитель, описанный в этом документе, представляет собой наполнитель, который может быть гидрофобным или гидрофильным, смешивается с водой и имеет температуру плавления меньше 250°C. Предпочтительный растворитель представляет собой полиэтиленгликоль, необязательно дополненный полоксамером 188.
В этом документе полиэтиленстеарат 32 не упомянут, а инертный материал, требующийся для распыления наполнителя, представляет собой не микрокристаллическую целлюлозу, а лактозу.
В настоящем изобретении приведено более простое и более удовлетворительное решение проблемы перевода в раствор и кишечного всасывания действующих веществ, которые являются малорастворимыми или нерастворимыми в воде или не могут быть переведены в солевую форму в желудочном соке.
Способ по настоящему изобретению состоит в получении дисперсии одного или нескольких действующих веществ в легкоплавком сложном эфире жирной кислоты и полиоксиэтилена 32. Данную дисперсию напыляют в горячем состоянии на зернистый эксципиент в псевдоожиженном слое. Полученную таким образом порошкообразную смесь распределяют в фармацевтических композициях после разбавления при необходимости фармацевтически приемлемым нетоксичным инертным эксципиентом. Выражение "легкоплавкий" в этом случае означает, что такой сложный эфир плавится при температуре ниже 80°C и более предпочтительно в интервале от 40 до 50°C.
В предпочтительном варианте осуществления настоящего изобретения в качестве сложного эфира жирной кислоты и полиоксиэтилена 32 используют полиоксиэтилендистеарат 32, плавление которого происходит в интервале приблизительно от 50 до 60°C. Полиоксиэтилендистеарат 32 представляет собой коммерчески доступный продукт. Один из продуктов, относящихся к той же группе, продается под маркой Kessco® PEG 1540 DS (Stepan).
В другом предпочтительном варианте осуществления настоящего изобретения зернистый эксципиент представляет собой инертный материал, такой, как целлюлоза, декстран, коллоидный диоксид кремния, полимеры или сополимеры винилпирролидона, поливинилпирролидоны, акриловые полимеры, такие, как поликарбофил, и аналогичные соединения.
Предпочтительный зернистый материал представляет собой микрокристаллическую целлюлозу и предпочтительно микрокристаллическую целлюлозу фармацевтического качества, реализуемую под названием AVICEL PH, и более предпочтительно микрокристаллическую целлюлозу, продаваемую под названием AVICEL PH 105.
Содержание действующего вещества, диспергированного в сложном эфире жирной кислоты и полиоксиэтилена 32, может изменяться в широком интервале, поскольку такие сложные эфиры являются очень хорошими растворителями. Можно одинаково хорошо получать как разбавленные растворы, так и концентрированные растворы.
Концентрация действующего вещества предпочтительно находится в интервале от 30 до 50% действующего вещества в сложном эфире жирной кислоты. Такие значения позволяют легко переходить в раствор. Предпочтительная концентрация находится в интервале от 40 до 50% действующего вещества.
Среди действующих веществ, которые можно вводить в композиции по настоящему изобретению, можно назвать, в частности:
• противовоспалительные и болеутоляющие средства:
салсалат;
бенорилат;
оксаметацин;
индометацин;
парацетамол;
пироксикам;
тиениловая кислота;
этензамид;
трамадол;
• иммуносуппрессоры:
циклоспорин;
такролимус;
• антигистаминные средства:
терфенадин;
бромфенирамин;
хлорфенирамин;
• противогрибковые и противотрихомонадные средства:
метронидазол;
орнидазол;
дапсон;
итраконазол;
тербинафин;
• противовирусные средства:
цитарабин;
ганцикловир;
ацикловир;
• антипсихотические средства:
сульпирид;
сультоприд;
амисульприд;
• гормональные средства:
эстрадиол и его сложные эфиры (17β-валерат);
эстрон;
эстриол;
прогестерон и его производные;
• сердечно-сосудистые и сосудорасширяющие средства:
добутамин;
дилтиазем;
нифедипин и аналогичные (нитрендипин, нисолдипин и т.п.);
• противоязвенные средства:
пирензепин;
ранитидин;
омепразол;
лансопразол;
• антибактериальные средства:
эритромицин;
флумеквин;
окситетрациклин;
пиперациллин;
цефуроксим;
амфотерицин;
• антиаритмические средства:
пропафенон;
амиодарон;
кордарон;
флекаинид;
галлопамил;
верапамил;
дипиридамол;
диизопирамид;
• урикозурические средства:
бензбромарон;
пробенецид;
сульфинпиразон;
аллопуринол;
• антимигреневые средства:
флунаризин;
производные эрготамина;
суматриптан;
• антидепрессанты:
флувоксамин;
флуанизон;
флуоксетин;
пароксетин;
• антигормональные средства:
флутамид;
• бронходилататоры:
тулобутерол;
талинолол;
преналтерол;
• анксиолитические средства:
тиотиксен;
тразодон;
доксепин;
карбамазепин;
• коронародилататоры:
этаверин;
пентоксифиллин;
эбурнамонин;
• диуретические средства:
фуросемид;
триамтерен;
торасемид;
гидрохлортиазид;
• антиспазматические средства:
флавоксат;
тримебутин;
флорогусинол;
• средства, ингибирующие экскрецию кальция:
клодронат;
памидронат;
алендронат;
• антикоагулянты:
пиндион;
тромексан;
• болеутоляющие средства:
фентанил;
декстропропоксифен;
суфентанил;
• опиатные производные:
налбуфин;
налтрексон;
дигидрокодеинон;
бупренорфин;
метадон.
Особенно предпочтительным применением настоящего изобретения является получение фармацевтических форм, биодоступность которых улучшена в значительной степени, а действующее вещество представляет собой антилипидемическое и/или гипохолестеринемическое средство. Более конкретно можно упомянуть композиции на основе производных клофибровой или фенофибровой кислоты, таких, как, например, клофибрат, фенофибрат, гемфиброзил, безафибрат, ципрофибрат, пирифибрат или симфибрат. Можно упомянуть также ингибиторы HMG-coA-редуктаз (статины), такие, как аторвастатин, церивастатин, флувастатин, правастатин и его натриевая соль или симвастатин, или также триптаны, такие, как суматриптан или фроватриптан.
В особом случае производных клофибровой кислоты интерес в растворителе, таком, как сложный эфир жирной кислоты и полиоксиэтилена 32, состоит в том, что этот продукт не способен благоприятствовать или вызывать переэтерификацию или увеличивать токсичность действующего вещества.
Другая особенность настоящего изобретения состоит в возможности получать биодоступные формы гормональных средств, которые не могут всасываться или всасываются в малой степени в пищеварительном тракте, таких, как прогестерон, андростерон, ацетат хлормадинона или меленгестрол. Традиционно используют производные, алкилированные в положении 17α или 6α, для получения активных соединений, вводимых через пищеварительный тракт (ципротерон, демегестон, промегестон, ацетат норэтинодиола, этинилэстрадиол). Недостатком такого замещения является индуцирование вторичных осложняющих явлений (андрогенное или антиандрогенное действие, антиэстрогенное действие и, главным образом, гепатотоксические эффекты), которых особенно следует избегать. Именно поэтому часто используют природный прогестерон или его производные (дигидропрогестерон, 17α-гидроксипрогестерон), поскольку они проявляют активность при введении через пищеварительный тракт, малотоксичны, но при этом менее активны, чем собственно прогестерон.
Дисперсии по настоящему изобретению позволяют получать фармацевтические композиции, содержащие помимо действующего вещества, диспергированного в инертном носителе, один или несколько фармацевтически приемлемых нетоксичных инертных эксципиентов.
Также возможно к композициям, полученным после введения в инертный материал и последующего высушивания в псевдоожиженном слое, прибавлять разбавитель, наполнитель, ароматизатор, краситель, антиагрегант, гелеобразователь или пленкообразователь.
Содержание действующего вещества рассчитывают так, чтобы конечный фармацевтический препарат содержал действующее вещество в эффективном и не имеющем токсического эффекта количестве. Количество эксципиента рассчитывают так, чтобы концентрация активной фракции составляла около 50%, т.е. чтобы оно не превосходило 50% и находилось предпочтительно в интервале от 20 до 40%.
Особый вариант представляет собой получение фармацевтических композиций на основе абсорбата фенофибрата в микрокристаллической целлюлозе. Содержание действующего вещества в таких композициях находится в интервале от 50 до 150 мг на единичную дозу и предпочтительно составляет 60, 90 или 130 мг фенофибрата. Содержание микрокристаллической целлюлозы находится в интервале от 40 до 60 мг на единичную дозу.
В случае прогестерона можно получать фармацевтические композиции, в которых содержание действующего вещества находится в интервале от 5 до 50 мг и предпочтительно в интервале от 25 до 40 мг на единичную дозу. Содержание полиоксиэтилендистеарата 32 может находиться в интервале от 5 до 100 мг на единичную дозу, а содержание эксципиента может находиться также в интервале от 5 до 100 мг.
Композиции на основе фенофибрата, на основе прогестерона или на основе амиодарона приведены далее в качестве примеров и не налагают ограничений на настоящее изобретение.
Пример I
Фенофибрат 25 г
Полиоксиэтилендистеарат 32 26 г
Микрокристаллическая целлюлоза 44 г
Стеарат магния 0,5 г
Полученная таким образом смесь была распределена в 1000 желатиновых капсул, содержащих по 25 мг фенофибрата на единичную дозу.
Пример II
Прогестерон 30 г
Полиоксиэтилендистеарат 32 25 г
Микрокристаллическая целлюлоза 55 г
Мальтодекстрин 12 г
Карбонат кальция 5 г
Тальк 5 г
В расчете на 1000 желатиновых капсул, содержащих по 30 мг прогестерона.
Пример III
Амиодарон 150 г
Полиоксиэтилендистеарат 32 180 г
Поливинилпирролидон 20 г
Коллоидный диоксид кремния 45 г
Рисовый крахмал 105 г
В расчете на 1000 таблеток, содержащих по 150 мг амиодарона.
Объектом настоящего изобретения является также получение сублингвальных форм или буккальных таблеток. Они предназначены для помещения под язык в случае сублингвальных таблеток или для приклеивания к небу в случае буккальных таблеток. Таблетки таких типов, получаемые способом по настоящему изобретению, обладают еще более высокой биодоступностью.
Такие сублингвальные или буккальные таблетки получают способом по настоящему изобретению, при этом порошкообразный зернистый продукт превращают затем в таблетки, прибавляя связующее вещество, таблетирующий агент и скользящее вещество.
На фигурах 1-4 показаны результаты фармакокинетических исследований, осуществленных с композициями по настоящему изобретению для различных действующих веществ. Минимальная эффективная концентрация (CEM) указана для каждого из соединений. В приведенных далее примерах термины "средство" и "препарат" эквиваленты термину "композиция".
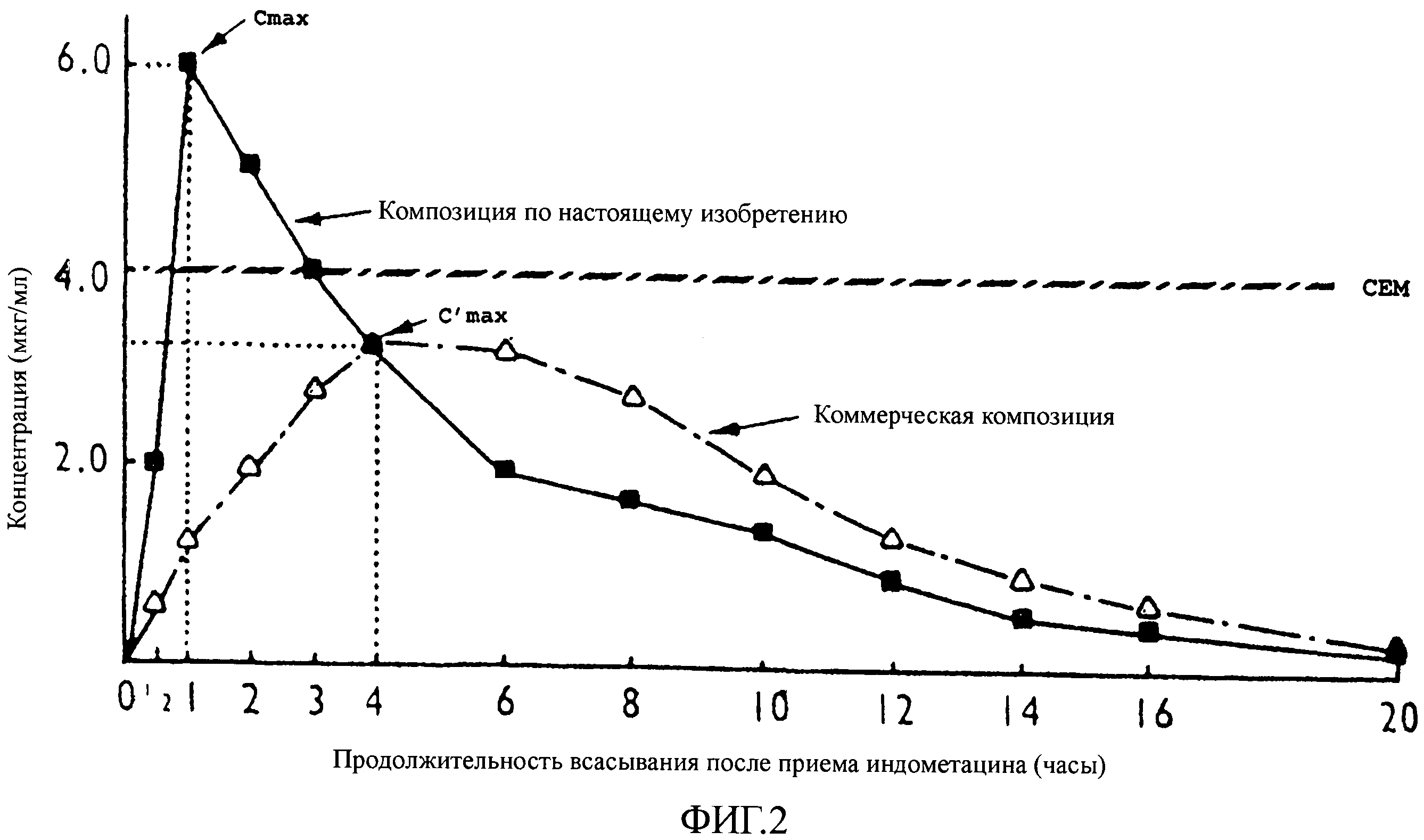
На фиг.1 представлен график, показывающий среднюю концентрацию в крови (в мкг/мл) в зависимости от продолжительности всасывания после приема таблеток парацетамола (в часах). Для средства по настоящему изобретению результаты получены с препаратом на основе 0,350 г парацетамола, полученным способом по настоящему изобретению. Было произведено сравнение данных результатов с результатами, полученными с коммерчески доступными таблетками, содержащими такое же количество парацетамола.
На этой фигуре можно видеть, что в случае средства по настоящему изобретению пик концентрации достигается только через 1 час после приема таблеток парацетамола, а максимальная концентрация (Cmax) составляет при этом 6,0 мкг/мл. В случае коммерчески доступного средства подобный пик концентрации в крови появляется только через 4 часа и при этом максимальная концентрация (C'max) составляет меньше 4,0 мкг/мл. В то же время, в случае композиции парацетамола по настоящему изобретению CEM парацетамола достигается всего за 1/2 часа, тогда как в случае коммерчески доступного средства аналогичная концентрация достигается только через 2 часа.
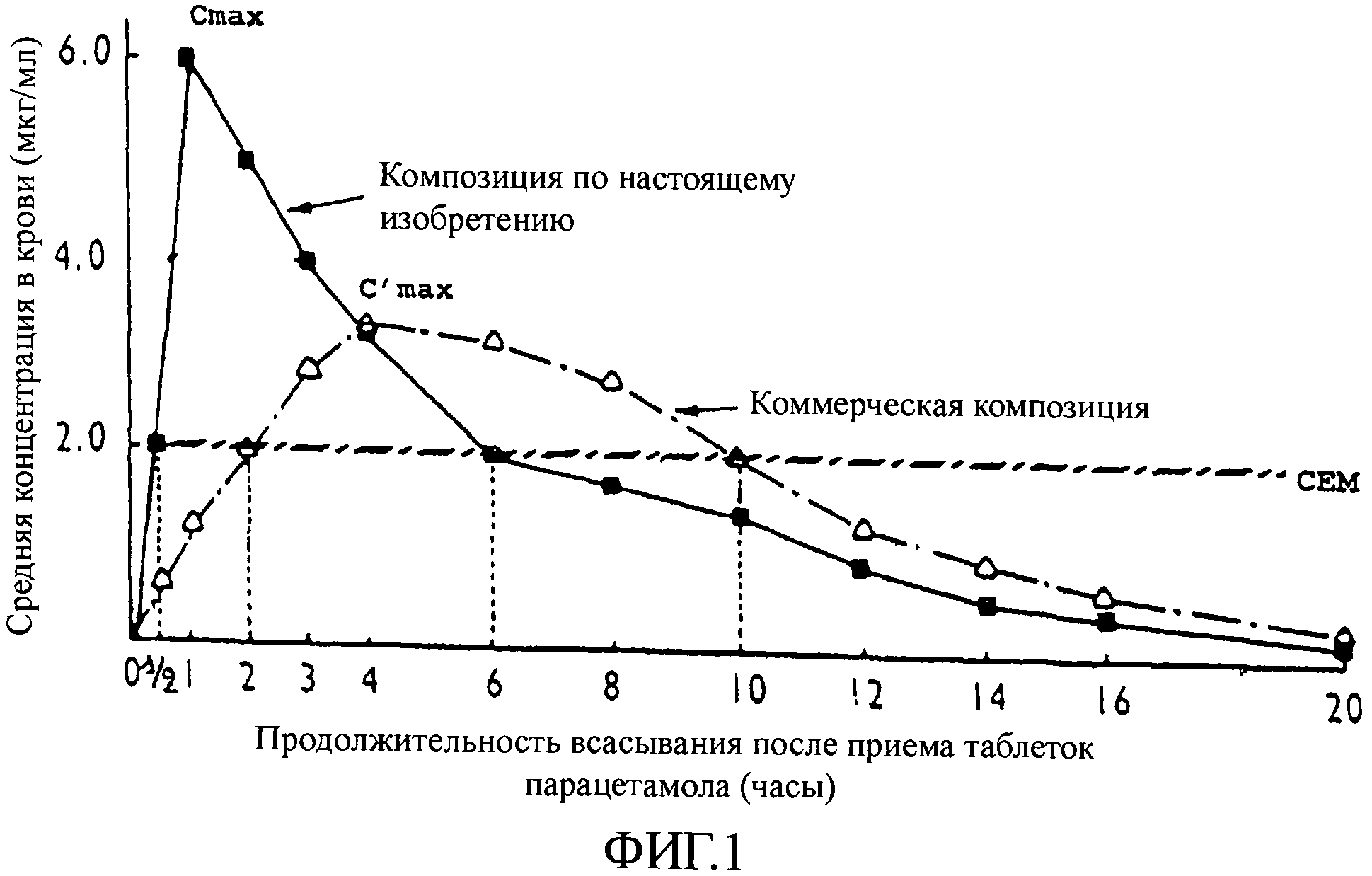
На фиг.2 представлен график, показывающий концентрацию в крови (в мкг/мл) в зависимости от продолжительности всасывания после приема индометацина (в часах). На этой фигуре представлены результаты, полученные со средством, которое приготовлено способом по настоящему изобретению и которое в качестве действующего вещества содержит индометацин, по сравнению с результатами, полученными с коммерчески доступным средством, содержащим индометацин.
Согласно представленным кривым пик концентрации в крови для средства по настоящему изобретению появляется быстро, всего лишь через 1 час после приема индометацина. К тому же, в случае средства по настоящему изобретению максимальная концентрация (Cmax), фиксируемая для этого пика, составляет 6 мкг/мл. В случае коммерчески доступного средства необходимо ожидать 4 часа после приема для того, чтобы был зафиксирован пик концентрации в крови, при этом максимальная концентрация (C'max) для коммерчески доступного средства составляет меньше 4 мкг/мл. К тому же, CEM индометацина достигается через 3 часа после приема средства по настоящему изобретению, тогда как в случае коммерчески доступного средства аналогичная концентрация не достигается совсем.
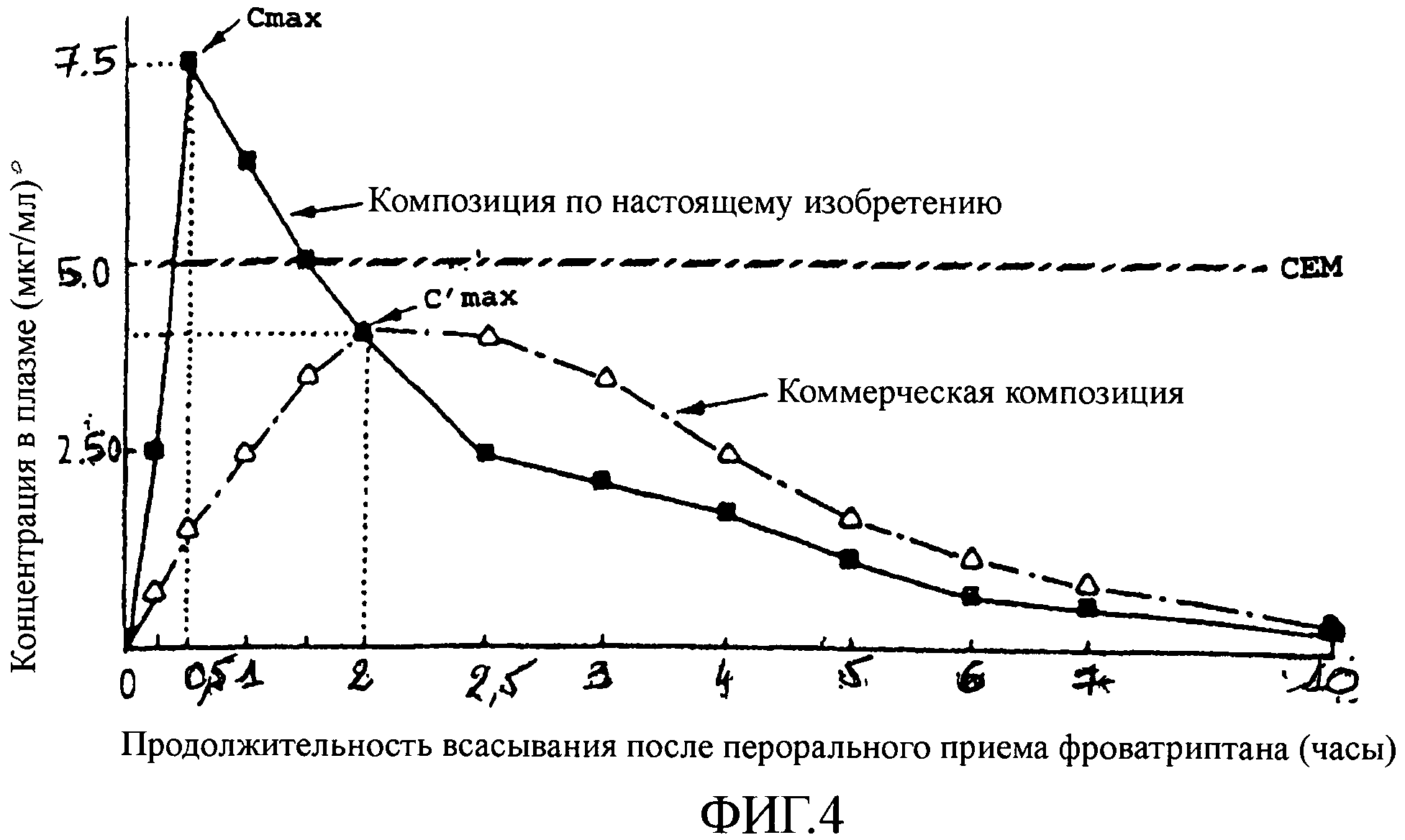
На фиг.3 представлен график, показывающий концентрацию в крови (в мкг/мл) в зависимости от продолжительности всасывания после приема суматриптана (в часах). Кривые отображают результаты, полученные с композицией по настоящему изобретению, действующим веществом которой является суматриптан, по сравнению с результатами, полученными с коммерчески доступным продуктом.
На этом графике можно видеть, что пик концентрации в крови достигается всего лишь через 1 час после приема композиции по настоящему изобретению, а максимальная концентрация (Cmax) составляет при этом 3 мкг/мл. В случае коммерчески доступного продукта пик концентрации в крови появляется только через 4 часа после приема, а максимальная концентрация (C'max) для данного коммерчески доступного продукта составляет меньше 2 мкг/мл. К тому же, CEM для суматриптана достигается через 3 часа после приема композиции по настоящему изобретению, тогда как для коммерчески доступного продукта такая концентрация не достигается совсем.
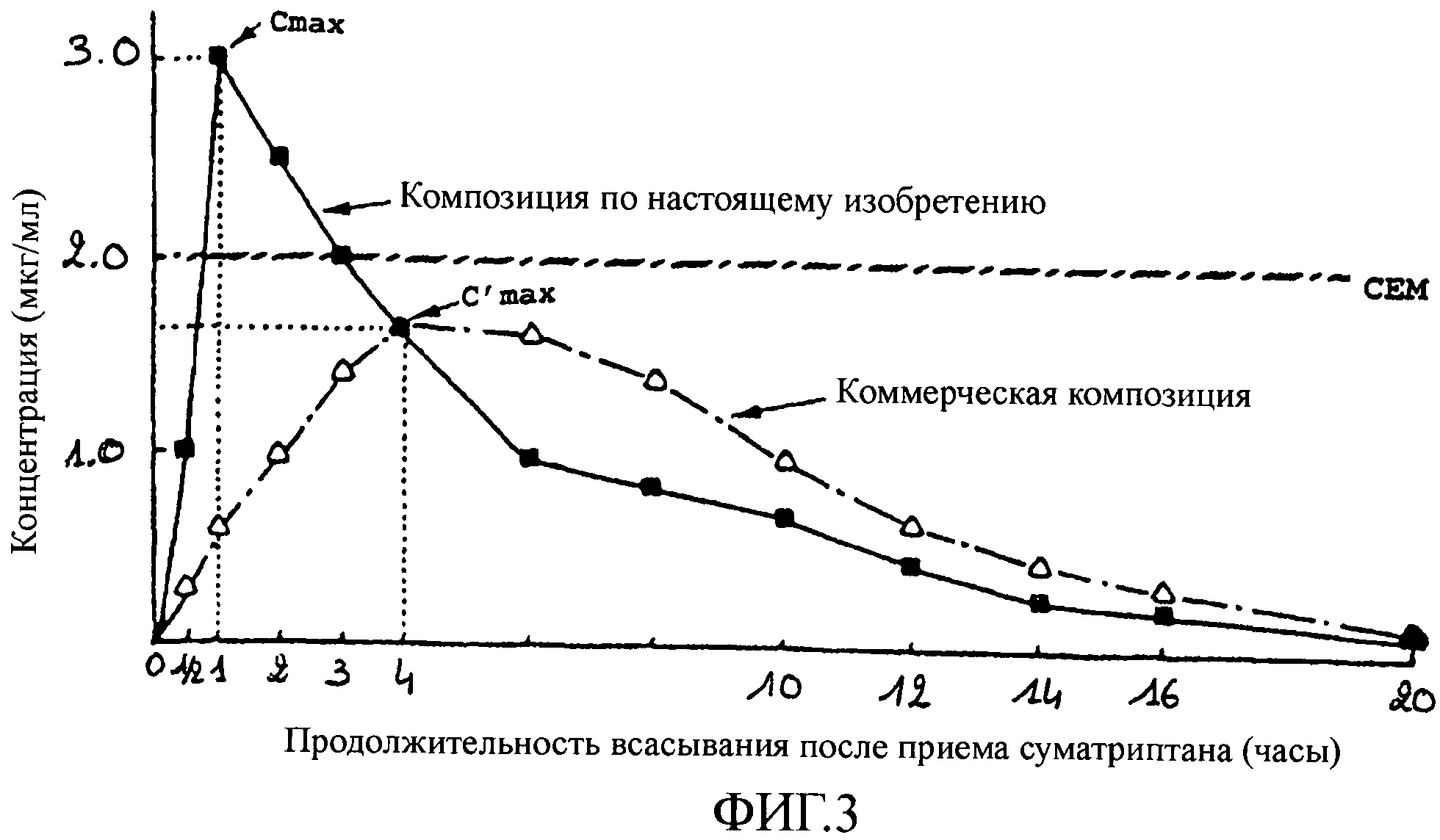
На фиг.4 представлен график, показывающий концентрацию в плазме (в мкг/мл) в зависимости от продолжительности всасывания после перорального приема фроватриптана (в часах). Результаты получены исходя из препарата по настоящему изобретению, действующим веществом которого является фроватриптан. Было произведено сравнение данных результатов с результатами, полученными с коммерчески доступным препаратом данного лекарственного средства.
Из этой фигуры видно, что пик концентрации достигается всего лишь через 1/2 часа после приема препарата по настоящему изобретению, а максимальная концентрация (Cmax) составляет при этом 7,5 мкг/мл. В случае коммерчески доступного средства необходимо ожидать 2 часа до появления пика концентрации, при этом максимальная концентрация (C'max) составляет меньше 5 мкг/мл. Кроме того, CEM фроватриптана достигается через 1 час 30 мин после приема препарата по настоящему изобретению, тогда как для коммерчески доступного средства такая концентрация не достигается совсем.
Полученные результаты показывают, что в случае композиций по настоящему изобретению действующие вещества всасываются организмом более быстро и в значительно большем количестве. Действительно, всасывание действующих веществ заметно облегчено за счет композиций по настоящему изобретению, а концентрации Cmax, полученные с композициями по настоящему изобретению, выше концентраций C'max, полученных с аналогичными коммерчески доступными продуктами.
К тому же, в случае индометацина, суматриптана и фроватриптана концентрации CEM для коммерчески доступных композиций не были достигнуты совсем. При этом в случае композиций по настоящему изобретению концентрации CEM были достигнуты для каждого из действующих веществ, использованных в данных примерах. К тому же, данные концентрации были достигнуты приблизительно не более чем через 3 часа после приема композиций по настоящему изобретению. Таким образом, композиции по настоящему изобретению обеспечивают не только более быстрое всасывание, но позволяют также достигать эффективных концентраций за малое время в случаях, когда с коммерческими доступными композициями такие концентрации не достигаются.