Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ОМЕГА-ИОДАЛИФАТИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ
Вид РИД
Изобретение
Изобретение относится к области органической химии, в частности к способам получения ω-иодалифатических карбоновых кислот и их эфиров на основе реакции расщепления алифатических циклических кетонов действием пероксида водорода в присутствии иода и солей меди (I), соответствующей основным принципам «зеленой» химии (Green Chemistry) (фиг.1).
ω-Иодалифатические карбоновые кислоты и их эфиры могут применяться в различных областях техники, в том числе в органической и фармацевтической химии, биохимии и в медицине (в частности, в качестве радиофармпрепаратов).
Способов получения иодалифатических карбоновых кислот и их эфиров, отвечающих принципам «зеленой» химии, известно немного.
Известен способ получения иодалифатических карбоновых кислот, описанный в немецком патенте [1]. Способ заключается в получении ω-иодалифатических карбоновых кислот из ω-бромалифатических карбоновых кислот путем обмена галогенов при нагревании в тонком слое инертного носителя (фиг.1).
Главный недостаток этого способа - это малодоступность ω-бромалифатических карбоновых кислот, кроме того, реализация способа происходит при нагревании (80-150°С), необходимо отделение полученного продукта от исходного субстрата и выходы продуктов недостаточно высокие.
Известен другой способ, где в условиях классической реакции нуклеофильного замещения при действии на этил-6-бромгексаноат натрий иодидом в среде ацетона с невысоким выходом получили соответствующую ω-иодалифатическую кислоту [2] (фиг.2).
И в данном случае основным недостатком является то, что используемые субстраты являются малодоступными реагентами. Кроме того, способ получения включает в себя стадию хроматографического разделения ω-иодалифатических карбоновых кислот от ω-бромалифатических карбоновых кислот, необходимо нагревание до 80-150°С, также не допускается наличие каких-либо количеств воды, поскольку могут образовываться гидрокси-алифатические карбоновые кислоты. Выходы целевых продуктов достигаются не более 75%.
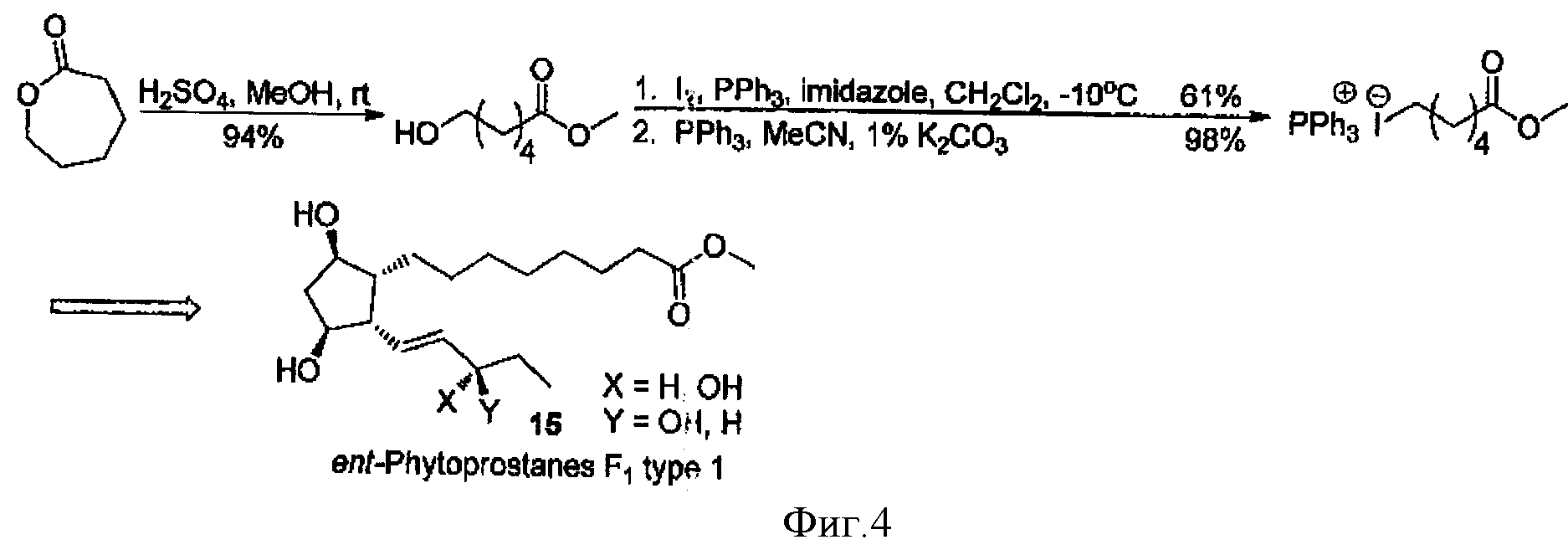
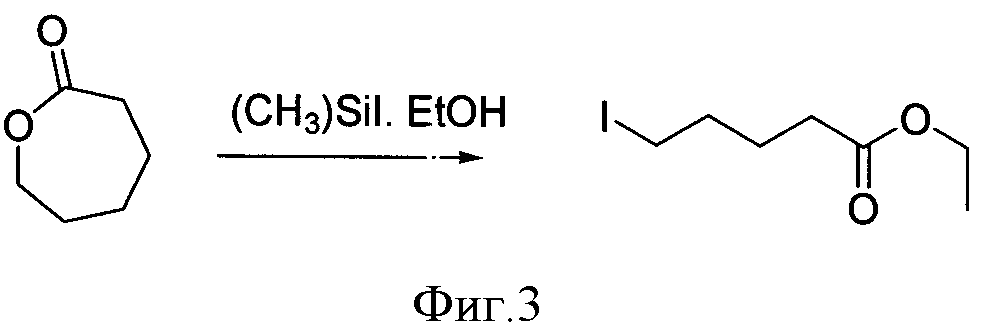
Другим, также наиболее общим методом получения ω-иодалифатических кислот, является реакция раскрытия лактонов в присутствии различных источников иода (фиг.3). Так, этиловый эфир 6-иодгексановой кислоты был получен с очень низким выходом (46%) действием триметилсилил иодида на валеролактон в этаноле [3].
В другой работе подобное соединение (метиловый эфир 6-иодгексановой кислоты) было получено через реакцию раскрытия до метилового эфира гидроксикислоты, и последующей обработкой иодом и трифенилфосфином. Полученный трифенилфосфоний иодид используется в синтезе простагландинов (фиг.4) [4].
Однако эти методы получения эфиров 6-иодгексановой кислоты также требуют использование малодоступных и экологически небезопасных реактивов.
Наиболее близким к предлагаемому можно считать метод расщепления гидропероксидов циклоалкана действием ионов Cu (II) (фиг. 5) [5]. Гидропероксиды авторы получают из соответствующих кетонов действием перекиси водорода в присутствии Сu2+ - ионов.
Главный недостаток этого способа - это невысокие выходы целевых продуктов (составляли всего 41-72%). Низкие выходы обусловлены тем, что используемый катализатор меди (II) хлорид и соотношения реагентов не позволяют полностью окислить циклические кетоны. Кроме того, для введения иода в карбоновые кислоты авторы статьи предлагают использовать соли галогенидов при комнатной температуре, в данных условиях галогенирование протекает очень медленно, не полностью и с образованием побочных продуктов - ω-гидроксикарбоновых кислот.
Новая техническая задача - упрощение способа, повышение выхода и чистоты целевых продуктов при расширении их ассортимента.
Для решения поставленной задачи в способе получения ω-иодалифатических карбоновых кислот и их эфиров, включающем расщепление алифатических циклических кетонов под действием пероксида водорода в присутствии катализатора ионов меди и соединений иода, при комнатной температуре, подвергают расщеплению такие циклическте кетоны, как циклогексанон, или циклогептанон, или 4-метилциклогексанон, также в качестве катализатора используют меди (I) хлорид; получение проводят при следующем соотношении компонентов: циклические кетоны - пероксид водорода - меди (I) хлорид - 1:5:0,1, при перемешивании, в течение 10-20 часов, в присутствии метанольных или этанольных растворов иода, также количество иода берут в следующем соотношении: циклические кетоны - иод - 1:0,5. Также для выделения и разделения целевых продуктов в реакционную массу добавляют насыщенный раствор натрия гидрокарбоната, при этом ω-иодалифатические карбоновые кислоты переходят в водный слой в виде натриевых солей, а их эфиры отделяют путем экстракции водного слоя этилацетатом, после этого этилацетатное извлечение осушают с помощью натрия сульфата безводного, этилацетат отгоняют и получают эфиры ω-иодалифатических карбоновых кислот. Также, для выделения только ω-иодалифатических карбоновых кислот, реакционную массу, содержащую смесь кислот и их эфиров, экстрагируют метиленхлоридом, добавляют воду и трифторуксусную кислоту, перемешивают при комнатной температуре в течение 20-24 часов. Затем растворитель отгоняют и получают ω-иодалифатические карбоновые кислоты, не требующие дополнительной очистки.
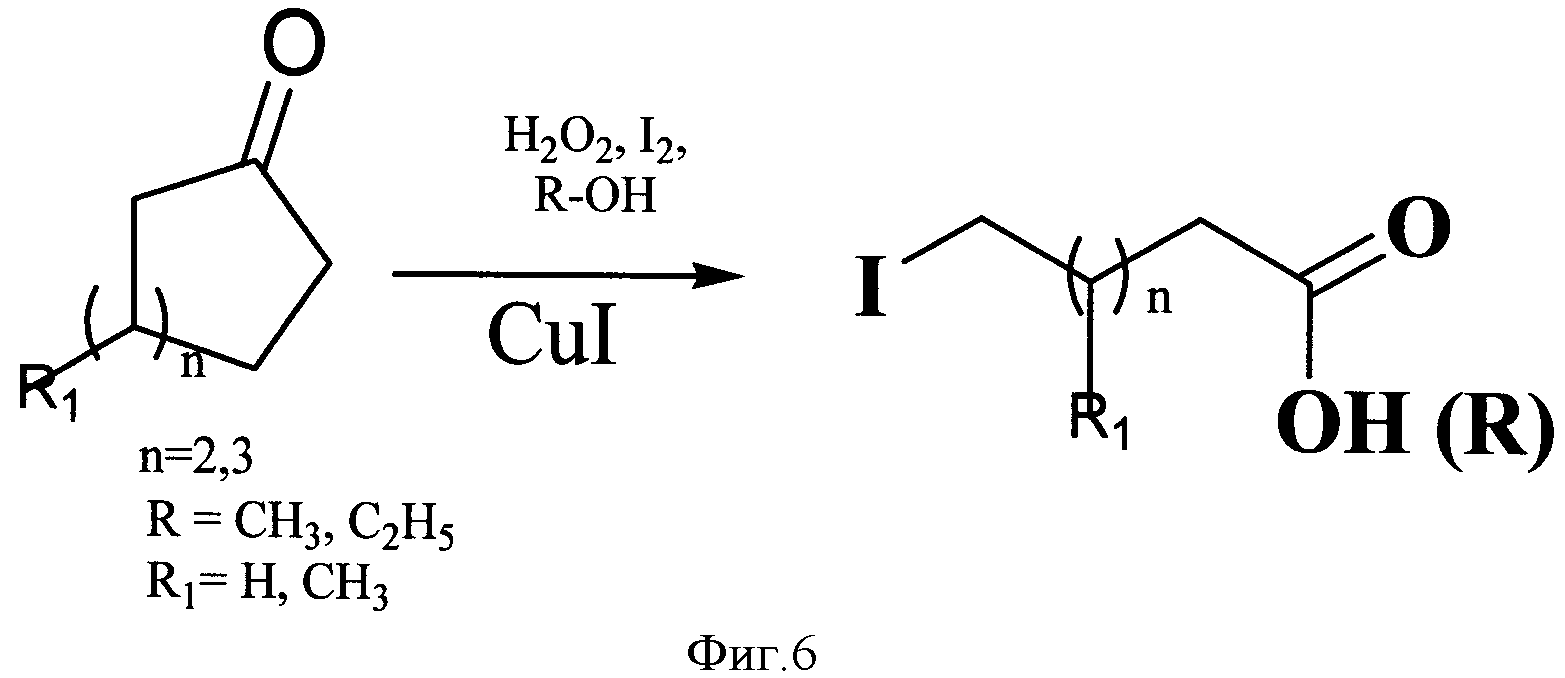
Предлагается использование реакции расщепления алифатических циклических кетонов действием пероксида водорода в присутствии иода и солей меди (I), соответствующей основным принципам «зеленой» химии (Green Chemistry), с образованием ω-иодалифатических карбоновых кислот и их эфиров (фиг.6).
В предлагаемом способе получения ω-иодалифатических карбоновых кислот и их эфиров алифатические циклические кетоны под действием пероксида водорода и солей меди (I) при соотношении циклические кетоны - пероксид водорода - меди (I) хлорид - 1:5:0,1 при перемешивании при комнатной температуре в течение 10-20 часов образуют гидроперекиси, которые в присутствии спиртовых растворов иода (при соотношении субстрат - иод - 1:0,5) раскрываются, образуя ω-иодалифатические карбоновые кислоты и их эфиры. Подобранные условия, а именно соотношения реагентов, время реакции и комнатная температура, позволяют получить продукты с суммарным выходом 80-98%.
Для выделения и разделения целевых продуктов предложен максимально простой способ. При добавлении насыщенного раствора натрия гидрокарбоната в реакционную массу ω-иодалифатические карбоновые кислоты переходят в водный слой в виде натриевых солей, а их эфиры легко отделяют путем экстракции водного слоя этилацетатом. Затем этилацетатное извлечение осушают с помощью натрия сульфата безводного, этилацетат отгоняют и получают эфиры ω-иодалифатических карбоновых кислот, не требующие дополнительной очистки. Для выделения только ω-иодалифатических карбоновых кислот реакционную массу, содержащую смесь кислот и их эфиров, экстрагируют метиленхлоридом, добавляют воду и трифторуксусную кислоту, перемешивают при комнатной температуре в течение 20-24 часов. Затем растворитель и трифторуксусную кислоту отгоняют и получают ω-иодалифатические карбоновые кислоты, не требующие дополнительной очистки.
Отличительные признаки проявили в заявляемой методике совокупности новые свойства, явным образом не вытекающие из уровня техники в данной области и не очевидные для специалиста.
Предлагаемая совокупность признаков не описана в патентной и научно-технической литературе.
Примеры конкретных способов получения ω-иодалифатических карбоновых кислот
и их эфиров из циклических кетонов
Пример 1. Получение метилового эфира ω-иодгексановой кислоты при 20-28°С.
К раствору циклогексанона в 10 мл метанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2О2, ρ=1,125 г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
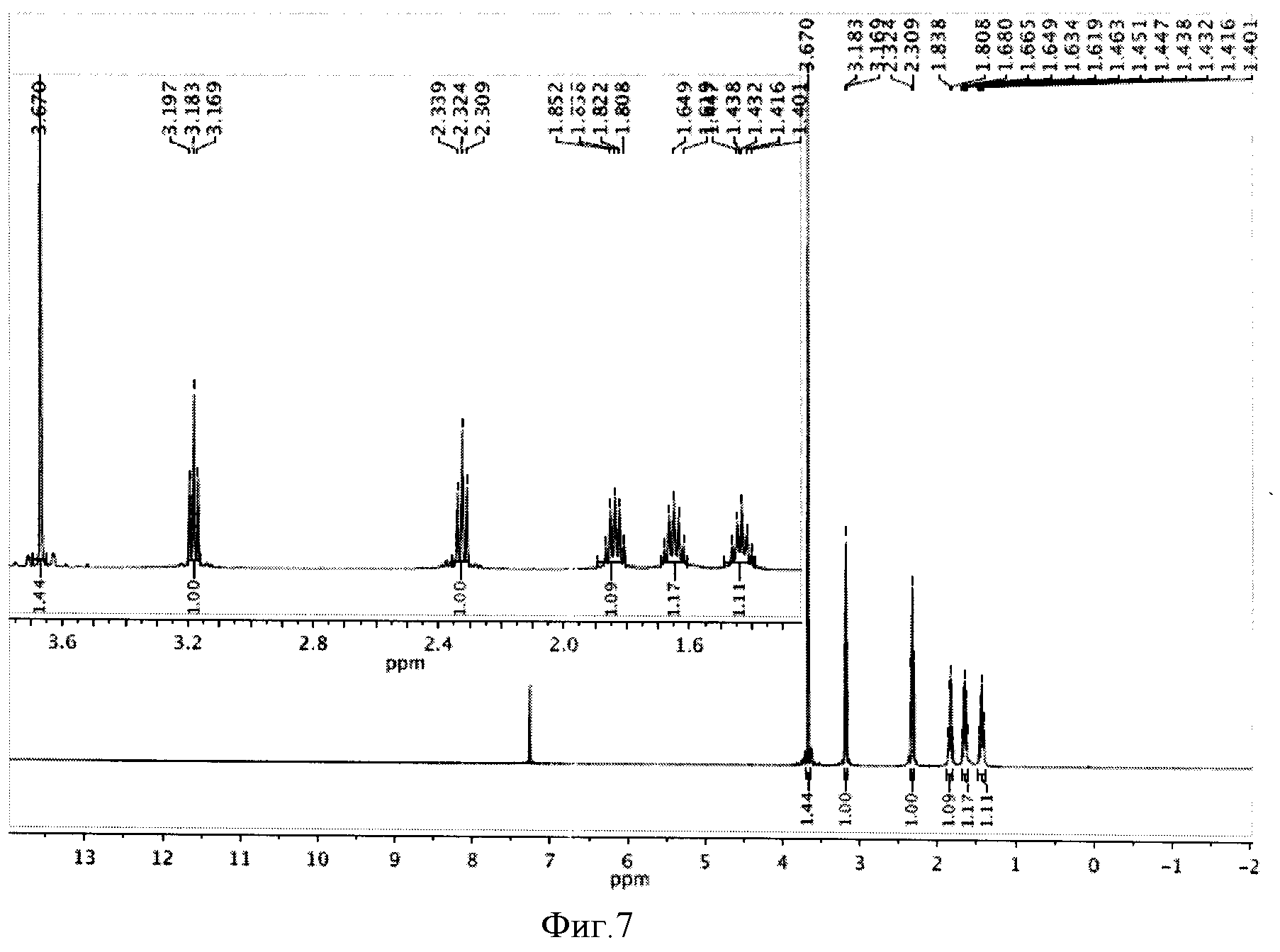
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа и натрия сульфит для окисления остатка иода. Далее реакционную смесь фильтровали, отбрасывая осадок, содержащий соли меди (I) хлориды и иодиды. Полученный фильтрат экстрагировали этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,248 г (81%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,67 (с, 3Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (кв, 2Н), 1,62 (кв, 2Н), 1,43 (кв, 2Н) (фиг.7).
Пример 2. Получение метилового эфира ω-иодгексановой кислоты при 29-35°С.
К раствору циклогексанона в 10 мл метанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2O2, ρ=1,125г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа и натрия сульфит для окисления остатка иода. Далее реакционную смесь фильтровали, отбрасывая осадок, содержащий соли меди (I) хлориды и иодиды. Полученный фильтрат экстрагировали этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,124 г (73%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,67 (с, 3Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (кв, 2Н), 1,62 (кв, 2Н), 1,43 (кв, 2Н) (фиг.7).
Пример 3. Получение метилового эфира ω-иодгексановой кислоты при 36-39°С.
К раствору циклогексанона в 10 мл метанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2О2, р=1,125 г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа и натрия сульфит для окисления остатка иода. Далее реакционную смесь фильтровали, отбрасывая осадок, содержащий соли меди (I) хлориды и иодиды. Полученный фильтрат экстрагировали этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,063 г (69%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,67 (с, 3Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (кв, 2Н), 1,62 (кв, 2Н), 1,43 (кв, 2Н) (фиг.7).
Пример 4. Получение ω-иодгексановой кислоты при 20-28°С.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
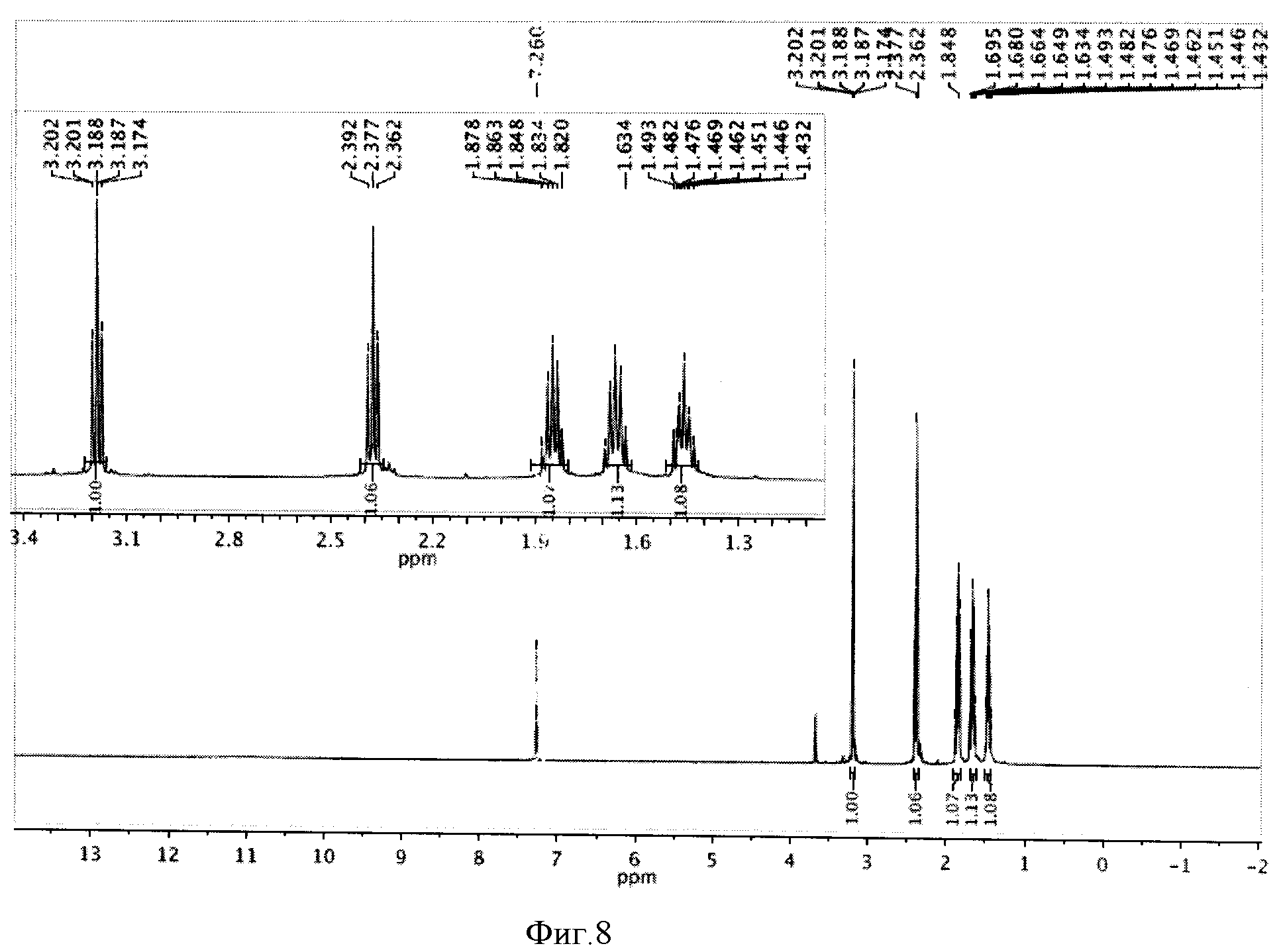
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,708 г (98%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 5. Получение ω-иодгексановой кислоты при 29-35°С.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2О2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,614 г (85%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 6. Получение ω-иодгексановой кислоты при температуре гидролиза - 29-35°С.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при температуре 29-35°С в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,578 г (80%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 7. Получение ω-иодгексановой кислоты при температуре гидролиза - 36-45°С.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при температуре 29-35°С в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,434 г (60%). Спектр ЯМР 1H (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 8. Получение ω-иодгексановой кислоты при времени гидролиза 12 ч.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 12 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,390 г (54%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47(м, 2Н) (фиг.8).
Пример 9. Получение ω-иодгексановой кислоты при времени гидролиза 16 ч.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 16 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,433 г (60%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 10. Получение ω-иодгексановой кислоты при времени гидролиза 20 ч.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2О2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 20 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,693 г (96%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 11. Получение ω-иодгексановой кислоты при времени гидролиза 28 ч.
К раствору циклогексанона в 10 мл метанола (3 ммоль, 0,294 г, ρ=0,946 г/см3) добавляли иод (1,5 ммоль, 0,381 г), катализатор меди (I) хлорида (0,3 ммоль, 0,03 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (6 ммоль, 0,638 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 6 ммоль (0,638 г) пергидроля и меди (I) хлорида (0,15 ммоль, 0,015 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 28 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выделенная ω-иодгексановая кислота не требовала дополнительной очистки. Выход 0,708 г (98%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,19 (т, 2Н), 2,38 (т, 2Н), 1,85 (м, 2Н), 1,63 (м, 2Н), 1,47 (м, 2Н) (фиг.8).
Пример 12. Получение метилового эфира ω-иодгексановой кислоты при соотношение компонентов в реакционной смеси циклогексанон - пероксид водорода - меди (I) хлорид - 1:3:0,1.
К раствору циклогексанона в 10 мл метанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2О2, ρ=1,125 г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 3 ммоль (0,319 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 3 ммоль (0,319 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа и натрия сульфит для окисления остатка иода. Далее реакционную смесь фильтровали, отбрасывая осадок, содержащий соли меди (I) хлориды и иодиды. Полученный фильтрат экстрагировали этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 0,893 г (58%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,67 (с, 3Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (кв, 2Н), 1,62 (кв, 2Н), 1,43 (кв, 2Н) (фиг.7).
Пример 13. Получение метилового эфира ω-иодгексановой кислоты при соотношение компонентов в реакционной смеси циклогексанон - пероксид водорода - меди (I) хлорид - 1:6:0,1.
К раствору циклогексанона в 10 мл метанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2O2, ρ=1,125г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 12 ммоль (1,275 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа и натрия сульфит для окисления остатка иода. Далее реакционную смесь фильтровали, отбрасывая осадок, содержащий соли меди (I) хлориды и иодиды. Полученный фильтрат экстрагировали этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,263 г (82%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,67 (с, 3Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (кв, 2Н), 1,62 (кв, 2Н), 1,43 (кв, 2Н) (фиг.7).
Пример 14. Получение этилового эфира ω-иодгексановой кислоты.
К раствору циклогексанона в 10 мл этанола (6 ммоль, 0,588 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в этаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 5 мл этанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 6 часов в тех же условиях.
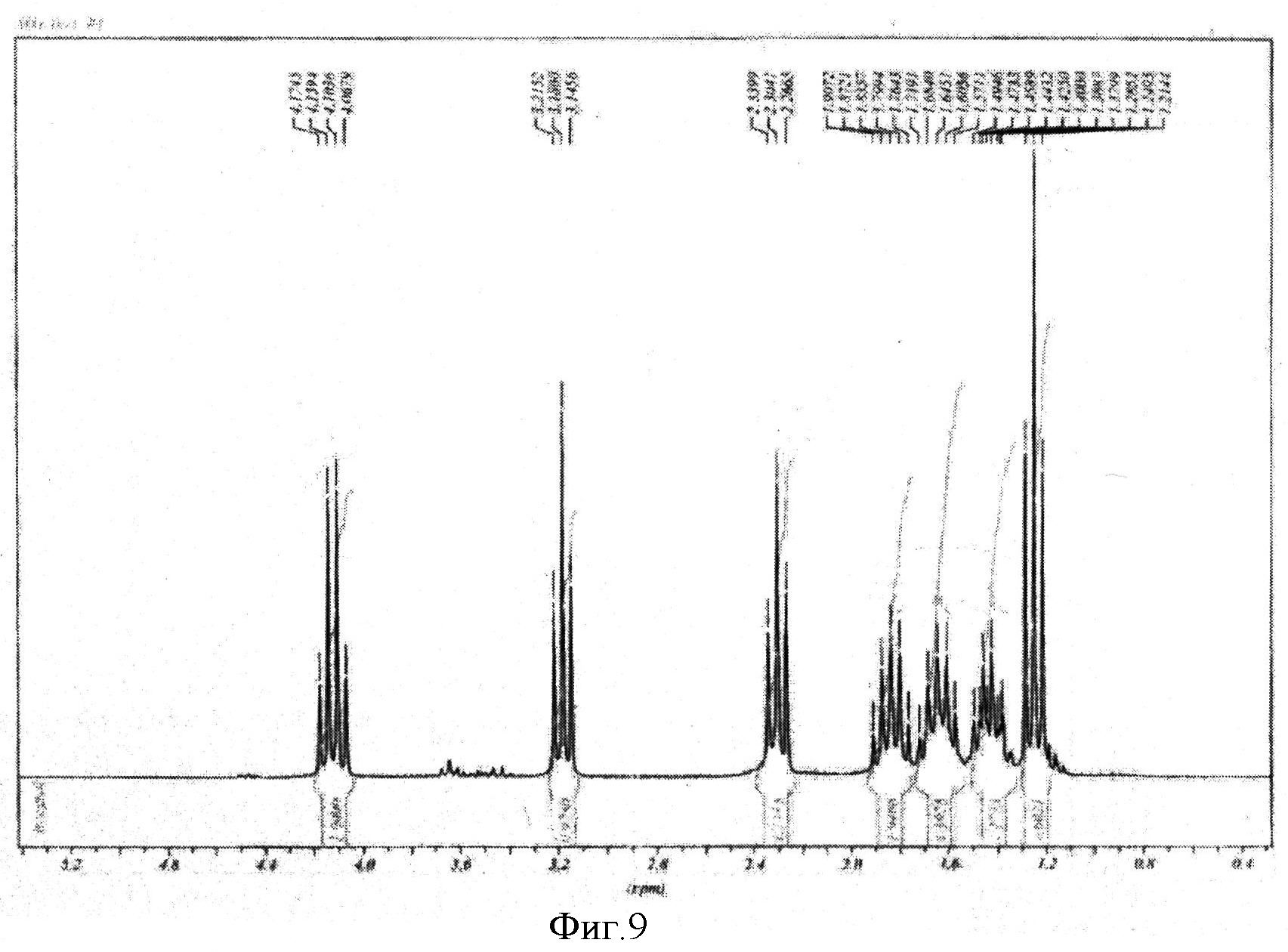
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа. Затем проводили экстракцию этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,332 г (82%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 4,14 (кв, 2Н), 3,18 (т, 2Н), 2,32 (т, 2Н), 1,84 (м, 2Н), 1,62 (м, 2Н), 1,43 (м, 2Н), 1,25 (т, 3Н) (фиг.9).
Пример 15. Получение ω-иодгептановой кислоты.
К раствору циклогептанона в 10 мл метанола (2 ммоль, 0,228 г, ρ=0,956 г/см3) добавляли иод (1 ммоль, 0,254 г), катализатор меди (I) хлорида (0,2 ммоль, 0,02 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (4 ммоль, 0,425 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 4 мл метанола) в течение 4 часов. Далее добавляли 4 ммоль (0,425 г) пергидроля и меди (I) хлорида (0,1 ммоль, 0,01 г), перемешивали при комнатной температуре 10 часов и после этого добавили еще 2 ммоль (0,212 г) пергидроля при перемешивании еще 8 часов в тех же условиях.
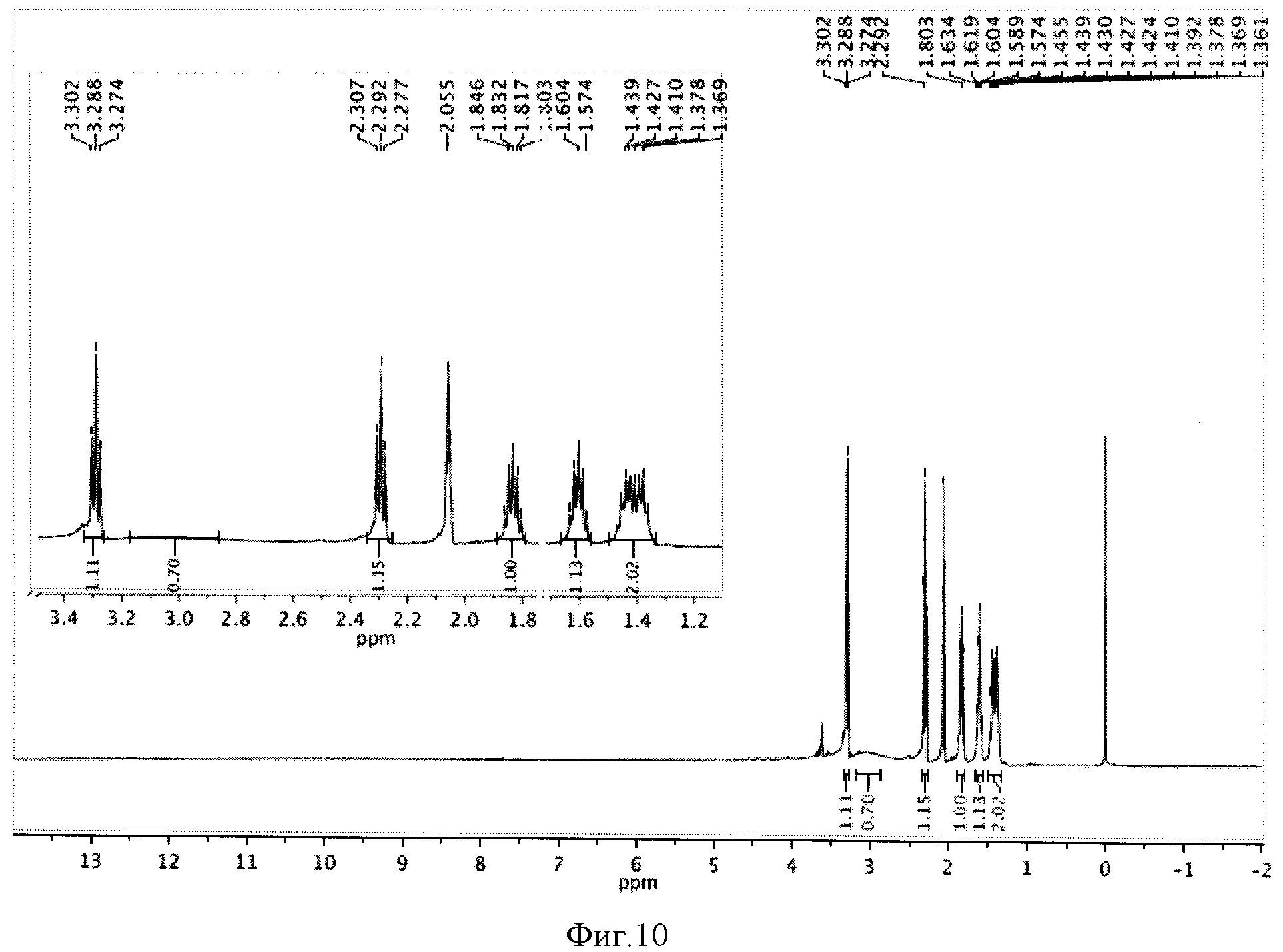
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 0,504 г (98%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): 3,29 (т, 2Н), 2,29 (т, 2Н), 2,05 (м, 2Н), 1,82 (м, 2Н), 1,59 (м, 2Н), 1,41 (м, 2Н) (фиг.10).
Пример 16. Получение метилового эфира ω-иод-γ-метилгексановой кислоты.
К раствору 4-метилциклогексанона в 10 мл метанола (6 ммоль, 0,672 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2O2, ρ=1,125 г/см3) в 5 мл метанола) в течение 6 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 8 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 8 часа в тех же условиях.
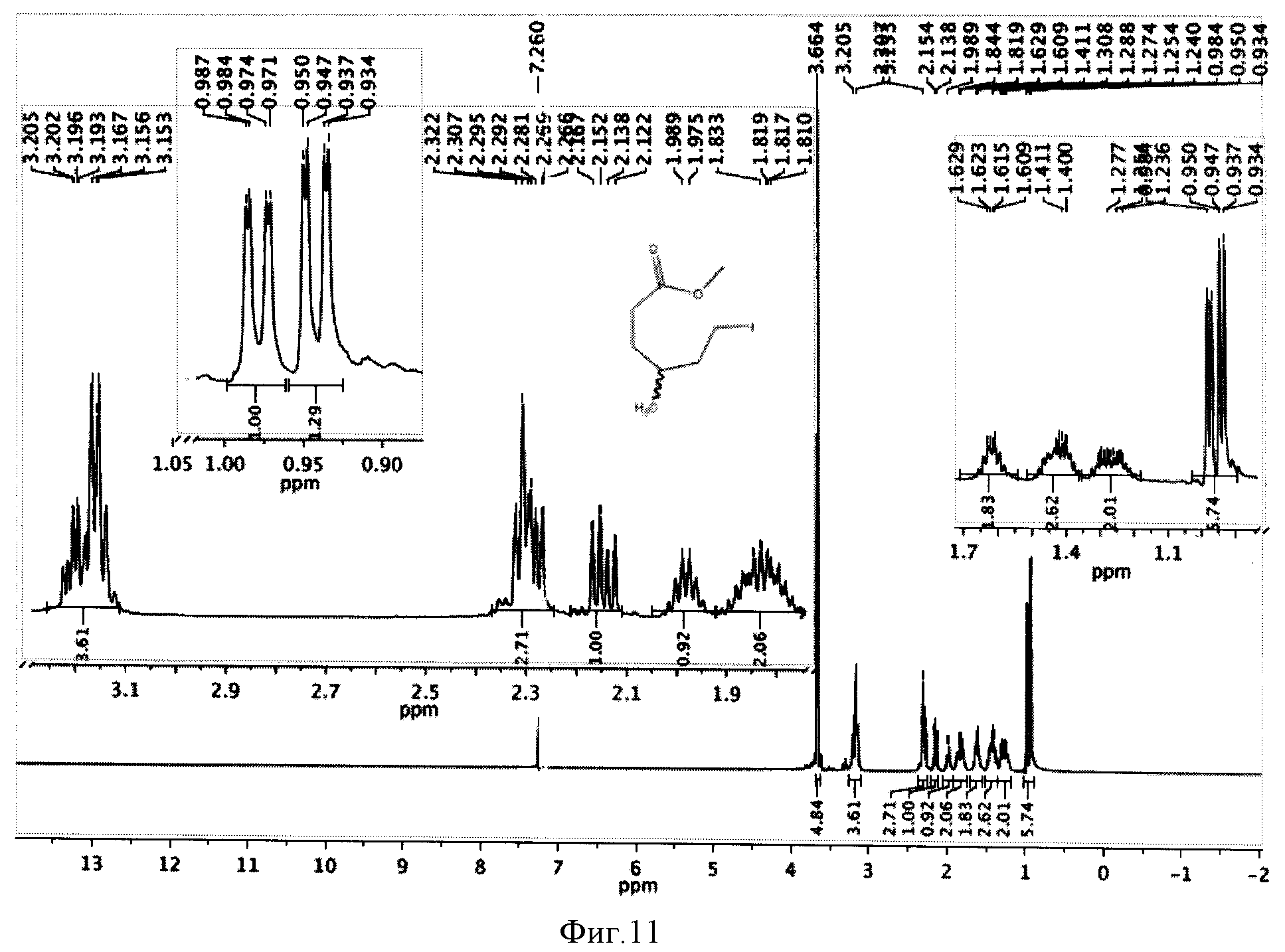
К реакционной смеси добавляли насыщенный раствор натрия гидрокарбоната до прекращения выделения углекислого газа, натрия сульфит до обесцвечивания иода и фильтровали, осадок отбрасывая. Затем проводили экстракцию этилацетатом (2×10 мл). Этилацетатное извлечение осушали путем пропускания через натрий сульфат безводный и растворитель отгоняли. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,344 г (83%). Спектр ЯМР 1Н (рацемическая смесь) (500 MHz, CDCl3, δ, м.д.): 3,66 (с, 3Н), 3,20 (т, 2Н), 2,31 (м, 2Н), 2,15 (м, 2Н), 1,98 (м, 2Н), 1,82 (м, 2Н), 1,62 (м, 2Н), 1,40 (м, 2Н), 1,26 (м, 2Н), 0,95 (д, 3 Н), 0,94 (д, 3 Н) (фиг.11).
Пример 17. Получение ω-иод-γ-метилгексановой кислоты.
К раствору 4-метилциклогексанона в 10 мл метанола (6 ммоль, 0,672 г, ρ=0,946 г/см3) добавляли иод (3 ммоль, 0,762 г), катализатор меди (I) хлорида (0,6 ммоль, 0,06 г). Затем при перемешивании при комнатной температуре по каплям вносили раствор пероксида водорода в метаноле (12 ммоль, 1,275 г пергидроля (32%-ного Н2О2, ρ=1,125 г/см3) в 5 мл метанола) в течение 4 часов. Далее добавляли 12 ммоль (1,275 г) пергидроля и меди (I) хлорида (0,3 ммоль, 0,03 г), перемешивали при комнатной температуре 8 часов и после этого добавили еще 6 ммоль (0,638 г) пергидроля при перемешивании еще 8 часа в тех же условиях.
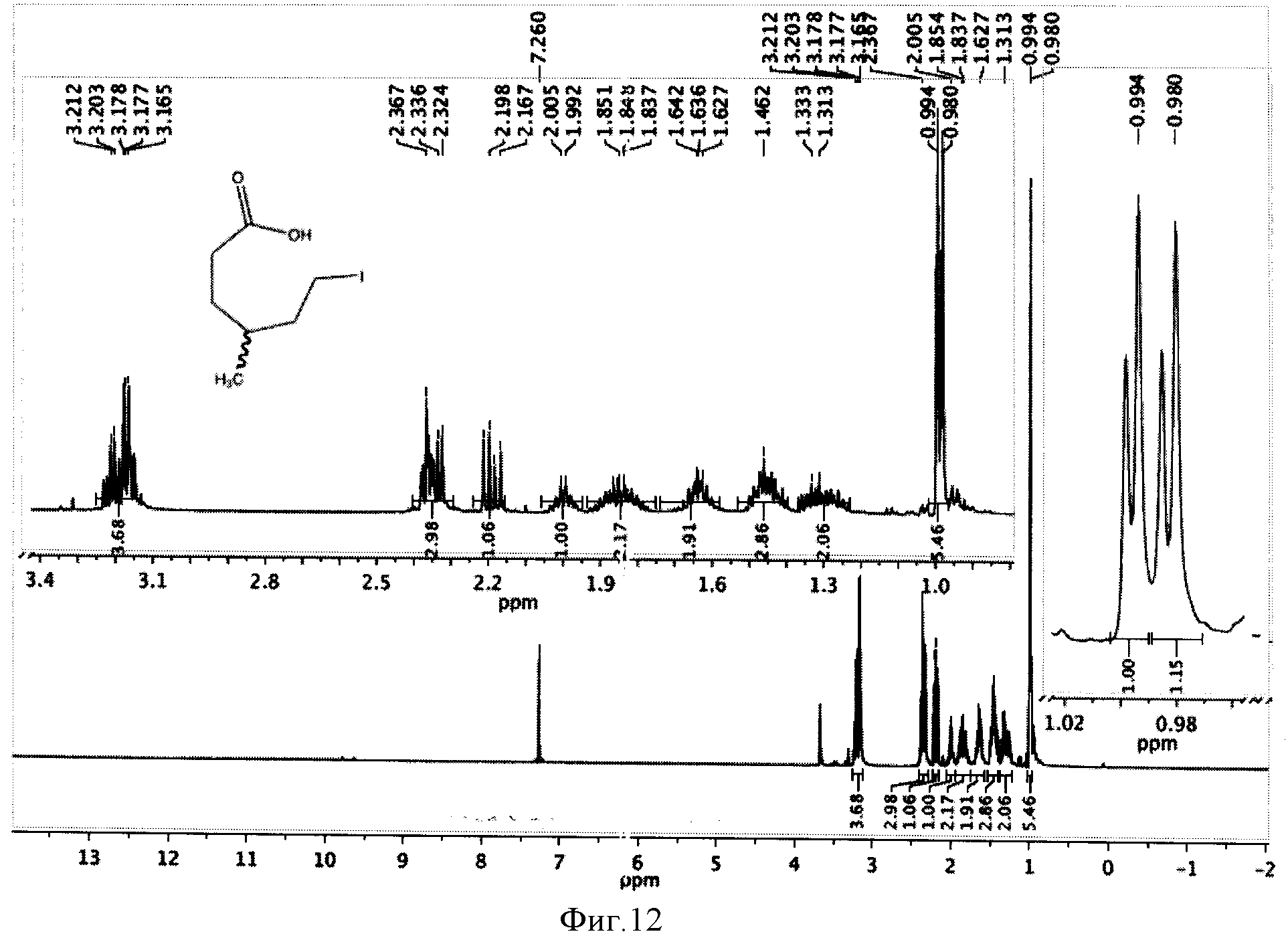
К реакционной смеси добавляли натрия сульфит для окисления остатка иода, отфильтровывали и отбрасывали осадок. Затем проводили экстракцию фильтрата метиленхлоридом (2×5 мл). К полученному извлечению (10 мл) добавляли 5 мл воды и 1 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 24 часов и отгоняли растворители. Полученную светло-желтую маслообразную массу сушили под вакуумом. Выход 1,49 г (97%). Спектр ЯМР 1Н (500 MHz, CDCl3, δ, м.д.): (рацемическая смесь) 3,66 (с, 3Н), 3,20 (т, 2Н), 2,34 (м, 2Н), 2,18 (м, 2Н), 2,00 (м, 2Н), 1,85 (м, 2Н), 1,64 (м, 2Н), 1,46 (м, 2Н), 1,33 (м, 2Н), 0,99 (д, 3Н), 0,98 (д, 3Н) (фиг.12).
Предложен способ получения ω-иодалифатических карбоновых кислот и их эфиров, заключающийся в том, что алифатические циклические кетоны под действием пероксида водорода и солей меди (I) при соотношении циклические кетоны - пероксид водорода - меди (I) хлорид - 1:5:0,1 при перемешивании при комнатной температуре в течение 10-20 часов образуют гидроперекиси, которые в присутствии спиртовых растворов иода (соотношение субстрата и иода - 1:0,5) раскрываются, образуя ω-иодалифатические карбоновые кислоты и их эфиры. Подобранные условия, а именно соотношения реагентов, время реакции и комнатная температура, позволяют получить продукты с суммарным выходом 80-98%.
Для выделения и разделения целевых продуктов предложен максимально простой способ. При добавлении насыщенного раствора натрия гидрокарбоната в реакционную массу ω-иодалифатические карбоновые кислоты переходят в водный слой в виде натриевых солей, а их эфиры легко отделяются путем экстракции водного слоя этилацетатом. Затем этилацетатное извлечение осушают с помощью натрия сульфата безводного, этилацетат отгоняют и получают эфиры ω-иодалифатических карбоновых кислот, не требующие дополнительной очистки. Для выделения только ω-иодалифатических карбоновых кислот реакционную массу, содержащую смесь кислот и их эфиров, экстрагируют метиленхлоридом, добавляют воду и трифторуксусную кислоту, перемешивают при комнатной температуре в течение 20-24 часов. Затем растворитель отгоняют и получают ω-иодалифатические карбоновые кислоты, не требующие дополнительной очистки.
Таким образом, заявляемое изобретение по сравнению с известными аналогами позволяет посредством одной стадии получить смесь ω-иодалифатических карбоновых кислот и их эфиров и простым способом отделить кислоту от ее эфира с суммарным выходом продуктов 80-98%.
Как видно из экспериментальной части, предлагаемый способ получения не предполагает использование дорогих реагентов и использование сложных манипуляций и позволяет получить ω-иодалифатические карбоновые кислоты и их эфиры в две стадии, при том что суммарный выход продуктов составляет 80-98%. Также при получении ω-иодалифатических карбоновых кислот и их эфиров не требуется стадия очистки продуктов и максимально упрощена процедура их выделения.
Таким образом, предлагаемый способ имеет следующие преимущества перед другими способами получения иодалифатических карбоновых кислот и их эфиров:
1) алифатические циклические кетоны - это дешевые, коммерчески доступные субстраты, не обладающие токсичностью;
2) высокие выходы целевых продуктов;
3) применение малотоксических реагентов и растворителей;
4) проведение реакции без нагревания;
5) без образования побочных продуктов;
6) отсутствует необходимость дополнительной очистки целевых продуктов;
7) простые способы выделения целевых продуктов;
8) получение одним способом кислот и их эфиров (метиловых и этиловых);
9) отсутствие многостадийности, простота получения.
Обоснование режима
Экспериментальным путем подобран оптимальный температурный режим получения ω-иодалифатических карбоновых кислот и их эфиров (примеры 1-5). Установлено, что реакцию необходимо проводить при температуре 20-28°С, при этом суммарный выход продуктов составляет 80-98%, а увеличение температуры до 29-35°С приводит к снижению выхода на 8-14%.
Оптимальным является соотношение компонентов в реакционной смеси циклические кетоны - пероксид водорода - меди (I) хлорид - 1:5:0,1. Такое соотношение позволяет практически полностью окислить циклические кетоны, что необходимо для достижения высоких выходов целевых продуктов (примеры 1, 12-13).
Для стадии гидролиза принципиальными оказались температурный режим и время проведения гидролиза. Оптимальным является проведение гидролиза при комнатной температуре, при этом выход ω-иодалифатических карбоновых кислот свыше 95%. Так, при более высокой температуре (более 30°С) выходы существенно снижались (на 10-15%), так как помимо гидролиза происходило нуклеофильное замещение иода на гидроксильную группу или атом водорода в целевых продуктах, что приводило к образованию побочных продуктов (примеры 4, 6, 7).
Выход ω-иодалифатических карбоновых кислот достигается наибольшим при проведении гидролиза в течение 20-24 часов (примеры 4, 8-11). Уменьшение времени реакции (менее 20 ч) приводит к неполному гидролизу эфиров и, следовательно, к снижению выхода целевых кислот.
Источники информации, принятые во внимание при составлении описания:
1. LAUFER; PETER MACHULLA; YURGEN STOCKLIN; GERHARD.
METHOD OF MAKING 1. SUP. 123 LABELED FATTY ACIDS. Publication number: 4290965, Publication date: 1981-22-09.
2. C.Hardouin, M.J.Kelso, F.A.Romero, T.J.Rayl, D.Leung, I.Hwang, B.F.Cravatt, D.L.Boger // J.Med. Chem. 2007, 50, P.3359.
3. P.Kraft, R.Cadalbert. The Thia-Analog of Ambrettolide. Synthesis and Odor of 1,8-Oxathiacyclohexadecan-2-one // Synlett 1997; №5: P 600-602.
4. S. El. Fangour, A.Guy, V. Despres, J.-P. Vidal, J.-C. Rossi, T. Durand. Total Synthesis of the Eight Diastereomers of the Syn-Anti-Syn Phytoprostanes Fl Types I and II // J. Org. Chem. 2010, 69, №7. P.2498-2503.
5. Г.И. Никишин, A.B. Александров, A.B. Игнатенко, E.K. Старостина. Синтез ω-галогеналифатических кислот каталитическим разложением циклоалкан гидропероксидов ионами меди // Известия АН СССР, Серия химическая. 1984. №11. С.2628-2630.
ПРИЛОЖЕНИЕ
Фигура 1 - Схема получения 6-иодгексановой кислоты.
Фигура 2 - Схема получения этилового эфира 6-иодгексановой кислоты из этилового эфира 6-бромгексановой кислоты.
Фигура 3 - Схема получения этилового эфира 6-иодгексановой кислоты из лактона.
Фигура 4 - Схема получения метилового эфира 6-иодгексановой кислоты из лактона.
Фигура 5 - Схема получения ω-производных карбоновых кислот из циклических кетонов.
Фигура 6 - Схема получения ω-иодалифатических карбоновых кислот и их эфиров из циклических кетонов.
Фигура 7 - Спектр ЯМР 1Н метиловый эфир ω-иодгексановой кислоты (500 МГц, растворитель CDCl3).
Фигура 8 - Спектр ЯМР 1Н ω-иодгексановой кислоты (500 МГц, растворитель CDCl3).
Фигура 9 - Спектр ЯМР 1Н этилового эфира ω-иодгексановой кислоты (500 МГц, растворитель CDCl3).
Фигура 10 - Спектр ЯМР 1Н ω-иодгептановой кислоты (500 МГц, растворитель CDCl3).
Фигура 11 - Спектр ЯМР 1Н метилового эфира ω-иод-γ-метилгексановой кислоты (500 МГц, растворитель CDCl3).
Фигура 12 - Спектр ЯМР 1Н ω-иод-γ-метилгексановой кислоты (500 МГц, растворитель CDCl3).
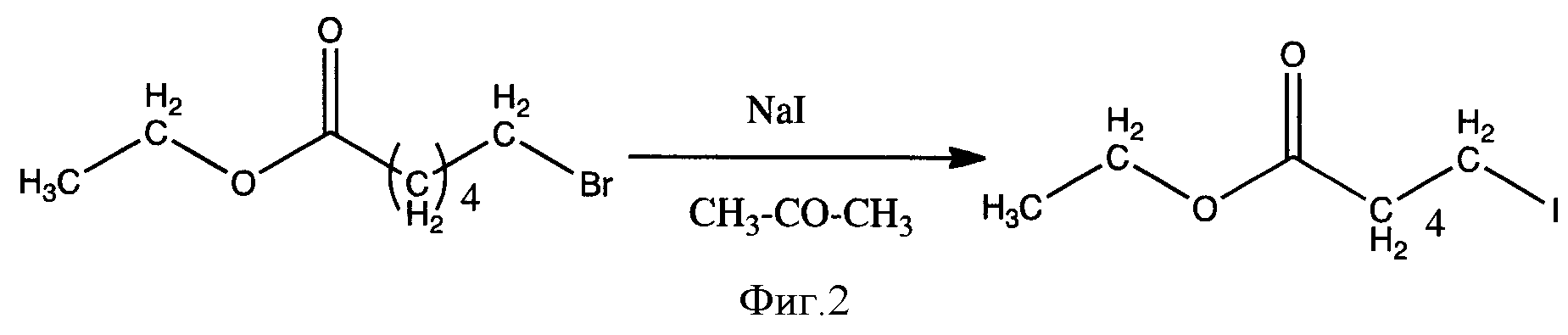
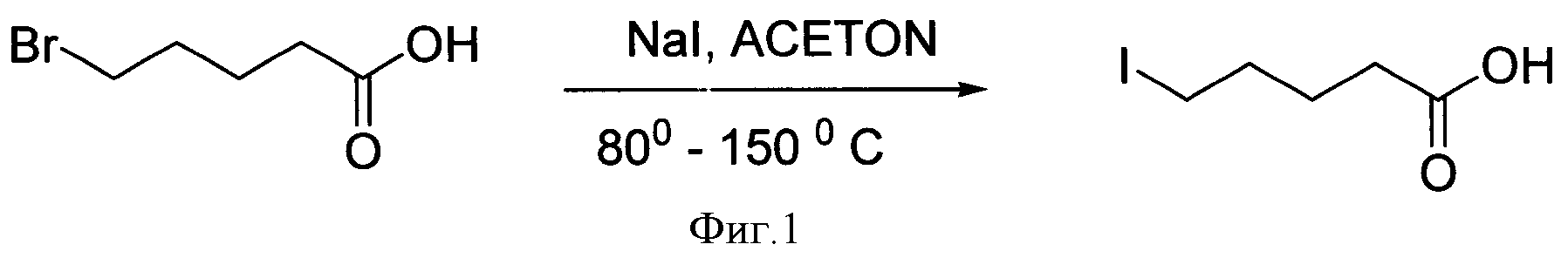
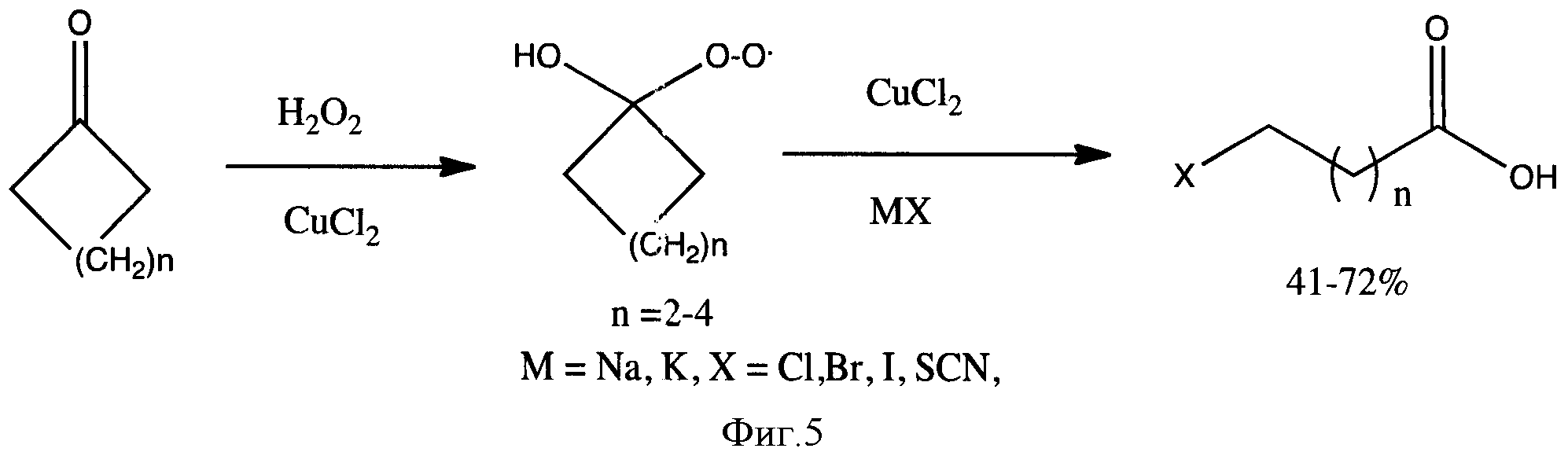
Средство, повышающее продукцию оксида азота макрофагами in vitro, на основе гуминовых кислот из торфа болот томской области и способ его получения
Способ получения ω-(бис(пиридин-2-илметил)амино)алифатических кислот - прекурсоров с хелатными центрами для связывания металлов
Газосодержащее контрастное средство для ультразвуковой диагностики
Способ прогнозирования туберкулеза легких с множественной лекарственной устойчивостью
Средство для инъекционной терапии гастроэзофагеальной рефлюксной болезни
Средство, обладающее противоописторхозным действием и способ его получения
Способ получения п-иодфенилжирных кислот
Водорастворимый реагент для органического синтеза и способ его получения
Способ прогнозирования риска развития тяжелого поражения нервной системы у новорожденных детей с различным сроком гестации в неонатальном периоде
Способ лечения подошвенных бородавок
Способ получения α(1,2)-l-рамно-α(1,4)-d-галактопиранозилуронана из корневищ acorus calamus l.
Способ профилактики канцерогенного действия диэтилнитрозамина у экспериментальных животных
Средство, повышающее продукцию оксида азота макрофагами in vitro, на основе гуминовых кислот из торфа болот томской области и способ его получения

