Результат интеллектуальной деятельности: НОВЫЕ СОЕДИНЕНИЯ, ПРИМЕНЕНИЕ И ПОЛУЧЕНИЕ ИХ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к соединениям пиразина, которые действуют как ингибиторы протеинкиназ, особенно типа FLT3-тирозинкиназы. Изобретение также относится к фармацевтическим средствам, включающим эти соединения, и к применению соединений для изготовления лекарственного средства для лечения гематологических злокачественных новообразований, таких как AML, MLL, T-ALL, B-ALL и CMML, миелопролиферативных заболеваний, других пролиферативных заболеваний, таких как злокачественное новообразование, аутоиммунных заболеваний и заболеваний кожи, таких как псориаз и атопический дерматит.
Уровень техники
Протеинкиназы задействованы в регуляции клеточного метаболизма, пролиферации, дифференциации и жизнеспособности. Протеинкиназы фосфорилируют белки по сериновым/треониновым или тирозиновым остаткам. Активация одного класса киназ обычно приводит к активации более чем одного сигнального пути через посредство сигнального перекрестного действия. Рецепторы тирозинкиназ (RTK) представляют собой основной тип рецепторов клеточной поверхности, у которых внутриклеточная часть рецептора имеет киназный домен. Активирующие лиганды представляют собой пептидные/белковые гормоны, такие как FL-лиганд, сосудистый эндотелиальный ростовой фактор (VEGF), эпидермальный ростовой фактор (EGF), фибробластный ростовой фактор (FGF), нервный ростовой фактор (NGF), тромбоцитами произведенный ростовой фактор (PDGF), инсулин и т.п. Связывание лиганда с внеклеточным доменом RTK в результате приводит к димеризации рецептора и к конформационному изменению, которое активирует киназный сайт внутриклеточного домена. Киназная активность приводит к каскаду сигнальной трансдукции посредством фосфорилирования других белков, что и регулирует клеточную физиологию и системы генной экспрессии (в качестве обзора см. Schlessinger, J. (2000) Cell 103: 211-225; и Blume-Jensen P. & Hunter T. (2001) Nature 411: 355-365). Внутриклеточные сигнальные белки, активируемые в сигнальном каскаде, могут быть другими киназами и/или белками, задействованными в транскрипции и трансляции. Существует несколько семейств внутриклеточных киназ. Янус-киназное (JAK) семейство тирозиновых киназ (JAK l, 2, 3 и Thy 1) активируется посредством взаимодействия с другими белками (см. O'Shea, J.J. et al. (2002) Cell 109 (Suppl.) 121-131 и приведенные там ссылки). Сериновые/треониновые киназы, такие как протеинкиназное C (PKC) семейство изоферментов и митоген-активируемые киназы (MAP-киназное семейство), также задействованы в регуляции клеточной жизнеспособности, пролиферации и дифференциации. PKC-изоферменты активируются кальцием, и диацилглицерин представляет собой аллостерический активатор некоторых членов семейства PKC (альфа, бета, гамма). Внутриклеточные киназы взаимодействуют с другими белками и часто перемещаются к другим компартментам при активации (см. Manning, G. et al. (2002) Science 298: 1912- 1934; Martin. P.M. & Hussaini L.M. (2005) Expert Opin. Ther. Targets 9(2) 299-313 и приведенные там ссылки). Мембранная ассоциация может регулироваться путем меристоилирования, как в случае PKC изоферментов. Ядерная ассоциация описана для нескольких различных классов киназ. MAP-киназы активируются другими белками и способны к перемещению в ядро, где белки, задействованые в транскрипции, и регуляторы клеточного цикла и дифференциации оказываются фосфорилированными.
В процессе нормального развития и дифференциации как активация, так и дезактивация киназы строго регулируются. Онкогенные мутации, приводящие к конститутивно активным киназам, могут трансформировать нормальные клетки в злокачественные опухолевые клетки. Активированная мутация может в результате предсталять собой хромосомную транслокацию, проводящую к возрастанию выработки белка слияния, например, как в случае хронической миеломной лейкемии, при которой ABL-тирозинкиназный домен объединяется с BCR белком (в качестве обзора см. Ostman, A. (2007) Helix Review Series Oncology 2: 2-9; и Deininger, M. et al. (2005) Blood 105: 2640-2653).
При нормальном гематопоэзе, FLT3 активна на стадии миелобласта, но FLT3 активность затем угнетается при нормальной гематопоэтической дифференциации в зрелые кровяные клетки (Gilliand, D.G, & Griffin, J.D. (2002) Blood 100: 1532-1542; Weisel, K.C. et al. (2007) Ann. N.Y. Acad. Sci. 1106: 190-196). При острой миеломной лейкемии, (AML) экспрессия FLT3 высока у большинства пациентов (70-90%) (Carow, C.E. et al. (1996) Blood 87 (3): 1089-1096; и Rosnet, O. et al. (1993) Crit. Rev. Oncogenesis 4: 595-613). Кроме того, FLT3 киназная активность повышена у одной трети пациентов из-за внутренней тандемной дупликации в околомембранной позиции (FLT3-ITD), в результате приводящей к димеризации лиганд-независимого рецептора и к конститутивной активной киназе. FLT3-ITD представляет собой предвещающий симптомный маркер со статистически достоверным снижением выживаемости обследуемой группы пациентов, имеющих мутацию, особенно в том случае, когда затронуты обе аллели. Существуют также активирующие точечные мутации (FLT3-PM) для FLT3, описанные у AML пациентов. Эти активирующие мутации могут быть обнаружены в активационной петле киназного домена (AL-мутации) или в околомембранном домене (JM-мутации). В качестве обзора см. Carow, C.E. et al. (1996) Blood 87 (3): 1089-1096; Tickenbrock, L. et al. (2006) Expert Opin. Emerging Drugs 11(1): 153-165; Anjali S. & Advani, A.S. (2005) Current Pharmaceutical Design 11: 3449-3457; Lee B.H. et al. (2007) Cancer Cell 12: 367-380); Stam, R.W. et al. (2005) Blood 106(7): 2484-2490; и приведенные там ссылки. Кроме того, FLT3-ITD или FLT3-PM обнаружены у подгруппы пациентов с другими лимфоидными или миелоидными злокачественными опухолями, такими как MLL, T-ALL и CMML, и высокая FLT3-активность была описана в случае B-ALL (в качестве обзора см. Lee, B.H. et al. (2007) Cancer Cell 12: 367-380.
Однако, FLT3 активность является составной частью нормального гематопоэза. В том случае, когда пролиферация незрелых бластных клеток костного мозга нерегулируема, путем сверхстимуляции киназ, таких как FLT3, в результате можно получить уменьшение количества других гематопоэтических клеток. Бластные клетки затем поступают в кровоток вместо зрелых дифференцированных клеток. Состояние острой лейкемии в результате приводит к анемии и нейтропении. Таким образом, блокирование вредной киназной активности может уменьшить пролиферацию бластных клеток и снизить вероятность лейкемического состояния. Различные ингибиторы FLT3 киназы тестировали на моделях AML и при клинических симптомах, в которых задействована FLT3 (Cheng, Y. & Paz, K. (2008) I Drugs 11(1): 46-56; Kiyoi, H. et al. (2007) Clin. Cancer Res. 13(15): 4575-4582; Roboz, G.J. et al. (2006) Leukemia 20: 952-957; Tse, K-F. et al. (2002) Leukemia 16: 2027-2036; Smith, B.D. et al. (2004) Blood 103: 3669-3676; Knapper, S. et al. (2006) Blood 108 (10): 3494-3503; и Furukawa, Y. et al. (2007) Leukemia 21: 1005-1014). AML клеточная линия MV4-11 несет FLT3-ITD. Эта клеточная линия очень чувствительная в испытаниях жизнеспособности/пролиферации для ингибиторов FLT3 активности. Однако, в случае ex-vivo клеток пациента имеет место также столкновение между сигнальными путями, молекулы, активируемые после FLT3 рецептора, могут также быть активированы другими киназами. Knapper et al. 2006 показал, что даже если аутофосфорилирование FLT3 было подавлено в клетках пациента после воздействия ингибиторов FLT3, уровень фосфорилирования после эффекторов подавления STAT и ERK не сокращался, возможно из-за дисрегуляции других сигнальных путей, независимо от FLT3-фосфорилирования.
Активность FLT3 и других RTK регулируется аутофосфорилированием и компенсационным процессом, фосфорилирование рецептора затем устраняется специфическими фосфатазами, которые также подвержены регуляции. Дисрегуляция компенсационного процесса и дефосфорилирования с участием фосфатаз может также иметь воздействие на RTK-активность и, таким образом, вносить изменения в жизнеспособность и пролиферацию клеток. Поскольку существует несколько направлений регуляции, ингибитору киназы необходимо иметь определенную форму, касающуюся его избирательности взаимодействия с мишенью и механизма действия для эффективного ингибирования пролиферации и жизнеспособности злокачественного новообразования или пролиферативного заболевания.
Раскрытие изобретения
Это изобретение относится в целом к определенным соединениям пиразина, которые могут действовать как ингибиторы рецептора тирозинкиназы FLT3, и относится к фармацевтическим композициям и способам.
Несмотря на то, что не возлагаются надежды, связанные с теорией, верится, что соединения, описанные в этом документе, могут применяться, например, для лечения или предотвращения гематологических злокачественных новообразований, таких как острая миеломная лейкемия (AML); недифференцированная лейкемия (MLL); T-клеточного типа острая лимфоцитарная лейкемия (T-ALL); B-клеточного типа острая лимфоцитарная лейкемия (B-ALL); хроническая миеломоноцитарная лейкемия (CMML); миелопролиферативные заболевания; другие пролиферативные заболевания, такие как злокачественное новообразование; аутоиммунные заболевания; и заболевания кожи, такие как псориаз и атопический дерматит.
Соединения, кроме того, могут применяться в сочетании с молекулярно направленными агентами, такими как традиционный цитотоксический агент или соединение, применяемое в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии.
В первом аспекте это изобретение предоставляет соединение формулы (I) и геометрические изомеры, рацематы, таутомеры и оптические изомеры его, а также и фармацевтически приемлемые соли, гидраты, N-оксиды и физиологически гидролизуемые и приемлемые сложные эфиры и любые формы их пролекарств:
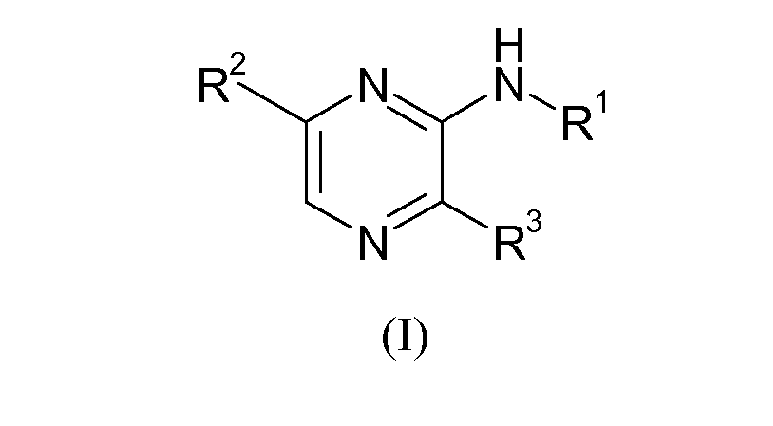
где
R1 выбирают из группы, состоящей из
(a) индолилэтила,
(b) циклогексила,
(c) гидроксициклогексила,
(d) 1,3-бензотиазолила,
(e) C1-3-алкил-1,3-бензотиазолила,
(f) бензотиенила,
(g) индолила,
(h) индазолила,
(i) C1-3-алкилиндолила,
(j) карбоксииндолила,
(k) C1-3-алкоксикарбонилиндолила,
(l) карбамоилиндолила,
(m) 4-метилпиперазин-1-илкарбонилиндолила,
(n) карбоксиметилиндолила,
(o) ацетиламинофенила и
(p) C1-3-алкилбензимидазолила;
R2 выбирают из группы, состоящей из
(a) пиридинила,
(b) фторпиридинила,
(c) хлорпиридинила,
(d) C1-3-алкоксипиридинила,
(e) тиенила,
(f) фурила,
(g) фенила,
(h) фторфенила,
(i) гидроксифенила,
(j) цианофенила,
(k) гидроксиметилфенила,
(l) аминофенила,
(m) карбамоилфенила,
(n) C1-3-алкиламинокарбонилфенила,
(o) диметиламинокарбонилфенила,
(p) (C1-2-алкокси-C2-3-алкиламинокарбонил)фенила,
(q) (циано-C2-3-алкиламинокарбонил)фенила,
(r) (диметиламино-C2-3-алкиламинокарбонил)фенила,
(s) N-метокси-N-метиламинокарбонилфенила,
(t) морфолин-4-илкарбонилфенила,
(u) пиперидин-1-илкарбонилфенила и
(v) хинолинила;
R3 представляет собой водород или NH2;
при условии, что соединение не представляет собой
4-(6-{[2-(1H-индол-3-ил)этил]амино}пиразин-2-ил)бензамид;
N'-(1H-индол-5-ил)-5-(хинолин-5-ил)пиразин-2,3-диамин;
5-(3-аминофенил)-N'-(1H-индол-5-ил)пиразин-2,3-диамин;
3-[5-амино-6-(1H-индол-5-иламино)пиразинил]фенол;
4-[5-амино-6-(1H-индол-5-иламино)пиразинил]фенол; или
1-метил-N-[6-(2-пиридинил)пиразинил]-1H-бензимидазол-2-амин.
Предпочтительной группой соединений изобретения являются соединения формулы (I), где R предствляет собой H, образующие соединения формулы (Ia):
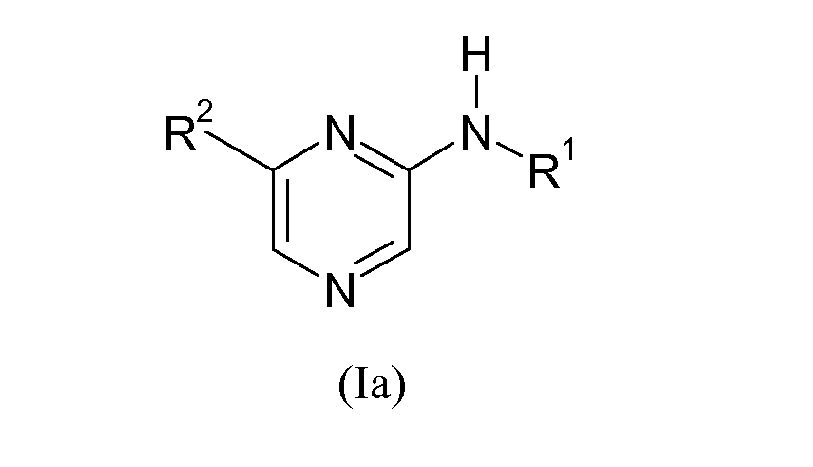
где
R1 выбирают из группы, состоящей из
(a) гидроксициклогексила,
(b) C1-3-алкил-1,3-бензотиазол-5-ила,
(c) 1,3-бензотиазолила,
(d) бензотиенила,
(e) индолила,
(f) C1-3-алкилиндол-5-ила,
(g) карбоксииндолила,
(h) C1-3-алкоксикарбонилиндолила и
R2 выбирают из группы, состоящей из
(a) пиридинила,
(b) фторпиридинила и
(c) карбамоилфенила.
Более предпочтительной группой соединений формулы (Ia) являются такие, в которых R1 выбирают из группы, состоящей из
(a) 4-гидроксициклогексила,
(b) 2-метил-1,3-бензотиазол-5-ила,
(c) 1,3-бензотиазол-5-ила,
(d) индол-5-ила,
(e) индол-6-ила, и
R2 выбирают из группы, состоящей из
(a) 4-пиридинила,
(b) 2-фтор-4-пиридинила и
(c) 4-карбамоилфенила.
Предпочтительные соединения формулы (Ia) представляют собой
N-(6-пиридин-4-илпиразин-2-ил)-1H-индол-5-амин,
N-[6-(2-фторпиридин-4-ил)пиразин-2-ил]-1H-индол-5-амин,
N-(6-пиридин-4-илпиразин-2-ил)-1H-индол-6-амин,
N-(6-пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин,
2-метил-N-(6-пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин,
4-[6-(1H-индол-5-иламино)пиразин-2-ил]бензамид и
4-{6-[(4-гидроксициклогексил)амино]пиразин-2-ил}бензамид.
Предпочтительной группой соединений изобретения являются соединения формулы (I), где R3 представляет собой NH2, образующие соединения формулы (Ib)
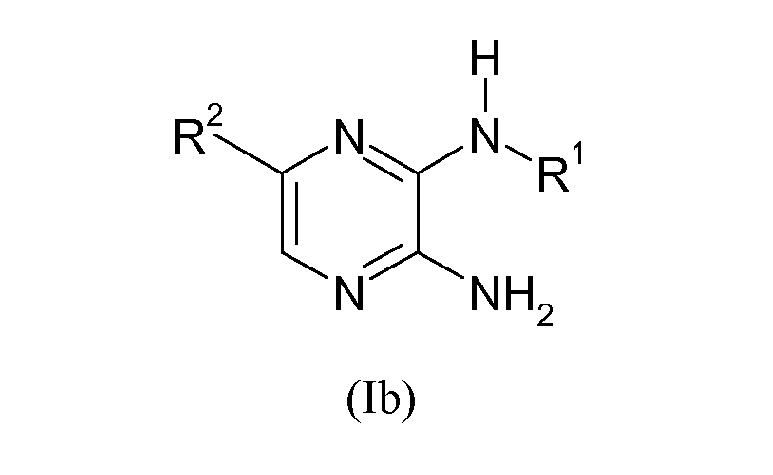
где
R1 выбирают из группы, состоящей из
(a) индолэтила,
(b) циклогексила,
(c) гидроксициклогексила,
(d) C1-3-алкил-1,3-бензотиазолила,
(e) бензотиенила,
(f) индолила,
(g) индазолила,
(h) C1-3-алкилиндол-5-ила и
(i) карбамоилиндолила;
R2 выбирают из группы, состоящей из
(a) пиридинила,
(b) хлорпиридинила,
(c) фторпиридинила,
(d) C1-3-алкоксипиридинила,
(e) тиенила,
(f) фурила,
(g) фенила,
(h) фторфенила,
(i) гидроксифенила,
(j) цианофенила,
(k) гидроксиметилфенила,
(l) аминофенила,
(m) карбамоилфенила,
(n) C1-3-алкиламинокарбонилфенила,
(o) диметиламинокарбонилфенила,
(p) (C1-2-алкокси-C2-3-алкиламинокарбонил)фенила,
(q) циано-C2-3-алкиламинокарбонил)фенила,
(r) (диметиламино-C2-3-алкиламинокарбонил)фенила,
(s) (N-метокси-N-метиламинокарбонилфенила,
(t) (пиперидин-1-илкарбонил)фенила,
(u) (морфолин-4-илкарбонил)фенила,
(v) хинолинила.
Более предпочтительной группой соединений Формулы (Ib) является такая, в которой R1 выбирают из группы, состоящей из
(a) 2-(индол-3-ил)этила,
(b) 4-гидроксициклогексила,
(c) индол-5-ила,
(d) индол-4-ила,
(e) индазол-5-ила,
(f) 2-метилиндол-5-ила, и
R2 выбирают из группы, состоящей из
(a) 3-пиридинила,
(b) 4-пиридинила,
(c) 2-хлорпиридин-4-ила,
(d) 3-тиенила,
(e) 3-фурила,
(f) 3 -фторфенила,
(g) 3-гидроксифенила,
(h) 4-цианофенила,
(i) 4-аминофенила,
(j) 4-карбамоилфенила,
(k) 3-карбамоилфенила,
(l) 4-диметиламинокарбонилфенила,
(m) 4-[(2-метоксиэтил)аминокарбонил]фенила,
Предпочтительные соединения формулы (Ib) представляют собой
N3-1H-индол-5-ил-5-пиридин-4-илпиразин-2,3-диамин,
N3-1H-индол-5-ил-5-пиридин-3-илпиразин-2,3-диамин,
5-(2-хлорпиридин-4-ил)-N3-1H-индол-5-илпиразин-2,3-диамин,
N3-(2-метил-1H-индол-5-ил)-5-пиридин-4-илпиразин-2,3-диамин,
N3-(2-метил-1H-индол-5-ил)-5-пиридин-3-илпиразин-2,3-диамин,
N3-1H-индол-4-ил-5-пиридин-4-илпиразин-2,3-диамин,
N3-1H-индол-5-ил-5-(3-тиенил)пиразин-2,3-диамин,
5-(3-фурил)-N3-1H-индол-5-илпиразин-2,3-диамин,
N3-1H-индол-5-ил-5-фенилпиразин-2,3-диамин,
5-(3-фторфенил)-N3-1H-индол-5-илпиразин-2,3-диамин,
3-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]бензамид,
4-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]бензамид,
4-{5-амино-6-[(2-метил-1H-индол-5-ил)амино]пиразин-2-ил}бензамид,
4-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]-N-(2-метоксиэтил)бензамид,
4-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]-N-(2-цианоэтил)бензамид,
4-[5-амино-6-(1H-индол-4-иламино)пиразин-2-ил]бензамид,
N3-[2-(1H-индол-3-ил)этил]-5-пиридин-4-илпиразин-2,3-диамин,
N3-[2-(1H-индол-3-ил)этил]-5-пиридин-3-илпиразин-2,3-диамин,
4-(5-амино-6-{[2-(1H-индол-3-ил)этил]амино}пиразин-2-ил)бензамид,
4-(5-амино-6-{[2-(1H-индол-3-ил)этил]амино}пиразин-2-ил)-N,N-диметилбензамид,
5-(4-аминофенил)-N3-[2-(1H-индол-3-ил)этил]пиразин-2,3-диамин,
транс-4-[(3-амино-6-пиридин-4-илпиразин-2-ил)амино]циклогексанол,
3-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]фенол,
N3-1H-индазол-5-ил-5-пиридин-4-илпиразин-2,3-диамин,
4-[5-амино-6-(1H-индазол-5-иламино)пиразин-2-ил]-N-(2-метоксиэтил)бензамид и
4-[5-амино-6-(1H-индазол-5-иламино)пиразин-2-ил]бензамид.
В одном аспекте настоящее изобретение относится к соединению формулы (I) для применения в терапии, особенно для применения при лечении или профилактике связанных с FLT3 заболеваний. Примеры связанных с FLT3 заболеваний включают острую миеломную лейкемию (AML); недифференцированную лейкемию (MLL); T-клеточного типа острую лимфоцитарную лейкемию (T-ALL); B-клеточного типа острую лимфоцитарную лейкемию (B-ALL); хроническую миеломоноцитарную лейкемию (CMML). Настоящее изобретение также относится к соединению формулы (I) для применения при лечении или профилактике гематологических заболеваний, связанных с нерегулируемой киназной активностью, таких как миелопролиферативные заболевания; другие пролиферативные заболевания, такие как злокачественное новообразование; аутоиммунные заболевания; и заболевания кожи, такие как псориаз и атопический дерматит.
В другом аспекте настоящее изобретение относится к фармацевтическому составу, включающему соединение формулы (I) в качестве активного ингредиента, в комбинации с фармацевтически приемлемым разбавителем или носителем, особенно для применения при лечении или профилактике связанного с FLT3 заболевания.
В одном аспекте настоящее изобретение относится к способу лечения пациента, человека или животного, страдающего от связанного с FLT3 заболевания. В следующем аспекте настоящее изобретение относится к способу лечения пациента, человека или животного, страдающего от гематологических злокачественных новообразований, таких как острая миеломная лейкемия (AML); недифференцированная лейкемия (MLL); T-клеточного типа острая лимфоцитарная лейкемия (T-ALL); B-клеточного типа острая лимфоцитарная лейкемия (B-ALL); хроническая миеломоноцитарная лейкемия (CMML), и других гематологических заболеваний, таких как миелопролиферативные заболевания; других пролиферативных заболеваний, таких как злокачественное новообразование; аутоиммунных заболеваний; и заболеваний кожи, таких как псориаз и атопический дерматит. Способ может включать введение пациенту (например, человеку или животному, собаке, кошке, лошади, корове), нуждающемуся в этом, эффективного количества одного или нескольких соединений формулы (I), их солей или композиций, содержащих соединения или соли.
Способы, описанные в этом документе, включают такие, в которых пациент рассматривается как нуждающийся в индивидуально назначенном лечении. Определение пациента как нуждающегося в таком лечении может быть на основании оценки пациента или специалиста в области здравоохранения и может быть субъективным (например, точка зрения) или объективным (например, определяемое тестом или диагностическим методом).
В других аспектах изобретение относится к способу лечения пациента, страдающего от связанного с FLT3 заболевания или нарушения или подверженного связанному с FLT3 заболеванию или нарушению, включающему введение указанному пациенту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что указанного пациента лечат от упомянутого заболевания или нарушения.
В следующем аспекте настоящее изобретение относится к применению соединения формулы (I) (например, в качестве лекарственного средства) для лечения заболевания, нарушения или патологического состояния, связанного с нежелательной активностью FLT3 киназы, как описано в этом документе.
В другом аспекте настоящее изобретение относится к применению соединения формулы (I) в производстве лекарственного средства, содержащего соединение формулы I для лечения заболевания, нарушения или патологического состояния, связанного с нежелательной активностью FLT3 киназы, как описано в этом документе.
Один аспект настоящего изобретения относится к фармацевтической композиции, включающей эффективное количество комбинации ингибитора рецептора тирозинкиназы FLT3, в соответствии с формулой (I), и другой молекулярно направленный агент, предпочтительно, традиционный цитотоксический агент или соединение, применяемое в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии; и, необязательно, фармацевтически приемлемый носитель.
Другой аспект изобретение предоставляет способ предотвращения или лечения гематологических злокачественных новообразований, миелопролиферативного нарушения, других пролиферативных нарушений, аутоиммунных нарушений и заболеваний кожи, включающий введение пациенту, человеку или животному, нуждающемуся в этом, ингибитора рецептора тирозинкиназы FLT3, в соответствии с формулой (I), одновременно или последовательно с другим молекулярно направленным агентом, предпочтительно, с традиционным цитотоксическим агентом, или соединением, применяемым в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии; в количествах, достаточных для обеспечения терапевтического эффекта.
И еще другой аспект изобретение относится к ингибитору рецептора тирозинкиназы FLT3, в соответствии с формулой (I), совместно с другим молекулярно направленным агентом, таким как традиционный цитотоксический агент, или соединение, применяемое в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии; для производства лекарственного средства для лечения гематологических злокачественных новообразований, миелопролиферативного нарушения, других пролиферативных заболеваний, аутоиммунных нарушений и заболеваний кожи.
Другой аспект изобретения относится к способу получения фармацевтической композиции, в которой ингибитор рецептора тирозинкиназы FLT3, в соответствии с формулой (I), и другой молекулярно направленный агент, такой как традиционный цитотоксический агент, или соединение, применяемое в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии; в суммарном терапевтическом количестве смешивают до однородной массы с фармацевтически приемлемым носителем.
И еще в другом аспекте изобретение относится к продукту, содержащему ингибитор рецептора тирозинкиназы FLT3, в соответствии с формулой (I), дополнительно включающему другой молекулярно направленный агент, такой как традиционный цитотоксический агент, или соединение, применяемое в постхимиотерапии, терапии, ориентированной на сохранение стволовых клеток, и в MLL-реаранжированной детской острой лимфобластной лейкемии; в качестве комбинированного лекарственного средства для одновременного, раздельного или последовательного применения в терапии гематологических злокачественных новообразований, миелопролиферативного нарушения, других пролиферативных заболеваний, аутоиммунных нарушений и заболеваний кожи.
Другой аспект настоящего изобретения относится к способу для получения соединения, в соответствии с формулой (I) изобретения, включающий взаимодействие 2-амино-3,5-дибромпиразина с соответствующим амином, с последующей реакцией сочетания Сузуки. А именно, способ получения соединения, в соответствии с формулой (I) изобретения, включающий одну или несколько нижеуказанных стадий: 2-амино-3,5-дибромпиразин (3 экв.) и соответствующий амин растворяют в 4 мл воды и полученную в результате смесь нагревают до 195°C в течение 1 часа. Добавляют воду и этилацетат и фазы разделяют. Водную фазу экстрагируют еще раз этилацетатом. Объединенную органическую фазу промывают (водой или солевым раствором) и концентрируют, получая неочищенную смесь продукта и непрореагировавшего амина или спирта. Неочищенную смесь используют без дополнительной очистки или определения характеристик в последующей реакции Сузуки, которую осуществляют в соответствии с типовыми протоколами Сузуки, опубликованными в литературе.
Химические реактивы, применяемые в синтетических методах, описанных в этом документе, могут включать, например, растворители, реагенты, катализаторы, реагенты введения защитных групп и удаления защитных групп. Способы, описанные выше, могут также дополнительно включать стадии либо перед, либо после стадий, описанных конкретно в этом документе, для добавления или удаления подходящих защитных групп с целью, прежде всего, обеспечить синтез соединений. Кроме того, различные синтетические стадии могут осуществляться в альтернативной последовательности или в другом порядке с получением целевых соединений. Синтетические химические процессы превращения, применимые для синтеза соответствующих соединений, являются известными в данной области техники и включают, например, такие, которые описаны в изданиях R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); L. Fieser, M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) и более поздние их издания.
Способы осуществления реакций, описанных выше, хорошо известны специалисту в данной области техники. Необходимые исходные вещества для получения соединений формулы (I) представляют собой либо известные, либо могут быть получены по аналогии с получением известных соединений. Соединения формулы (I) могут содержать один или несколько хиральных атомов углерода, и вследствие этого они могут быть получены в форме оптических изомеров, например, в виде чистого энантиомера, или в виде смеси энантиомеров (рацематов), или в виде смеси, содержащей диастереомеры. Разделение смесей оптических изомеров с получением чистых энантиомеров хорошо известно в данной области техники и, например, может быть достигнуто фракционной кристаллизацией солей с оптически активными (хиральными) кислотами или с помощью хроматографического разделения на хиральных колонках. Все изомерные формы, возможные (чистые энантиомеры, диастереомеры, таутомеры, рацемические смеси и неравномерные смеси двух энантиомеров) для описанных соединений, подпадают под действие изобретения. В том случае когда соединения, описанные в этом документе, содержат олефиновые двойные связи с геометрической асимметрией, предполагают, что они включают как транс так и цис (E и Z) геометрические изомеры.
Соединения формулы (I) могут применяться сами по себе или могут быть выделены в виде их фармакологически приемлемых солей (соли, полученные присоединением кислоты или основания). Под фармакологически приемлемыми аддитивными солями, упомянутыми выше, подразумевают такие, которые включают терапевтически активные, нетоксичные, образованные присоединением кислоты или основания формы солей, которые соединения способны образовать. Соединения, которые имеют основные свойства, могут быть превращены в их фармацевтически приемлемые кислотно-аддитивные соли путем обработки основной формы соответствующей кислотой. Типичные кислоты включают неорганические кислоты, такие как хлористоводородная, бромистоводородная, йодоводородная, серная кислота, фосфорная кислота; и органические кислоты, такие как муравьиная кислота, уксусная кислота, пропановая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, щавелевая кислота, бензолсульфокислота, толуолсульфокислота, метансульфокислота, трифторуксусная кислота, фумаровая кислота, янтарная кислота, яблочная кислота, виноградная кислота, лимонная кислота, салициловая кислота, пара-аминосалициловая кислота, памовая кислота, бензойная кислота, аскорбиновая кислота и им подобные. Типичные основно-аддитивные солевые формы представляют собой натриевые, калиевые, кальциевые соли и соли с фармацевтически приемлемыми аминами, такими как, например, аммиак, алкиламины, бензатин, и аминокислоты, такие как, например, аргинин и лизин. Термин «аддитивная соль», как он применяется в этом документе, также включает сольваты, которые соединения и их соли способны образовать, такие как, например, гидраты, алкоголяты и им подобные.
Для клинического применения соединения изобретения включаются в рецептуру фармацевтических составов для перорального, ректального, парентерального или других способов введения. Фармацевтические составы обычно получают путем смешивания активного вещества или его фармацевтически приемлемой соли со стандартными фармацевтическими эксципиентами. Примеры эксципиентов представляют собой воду, желатин, аравийскую камедь, лактозу, микрокристаллическую целлюлозу, крахмал, натриевую соль гликолята крахмала, гидрофосфат кальция, стеарат магния, тальк, коллоидный диоксид кремния и им подобные. Такие препаративные формы могут также содержать другие фармакологически активные агенты и стандартные вспомогательные вещества, такие как стабилизаторы, увлажняющие агенты, эмульгаторы, вкусоароматизирующие вещества, буферные компоненты и им подобные. Как правило, количество активного соединения составляет в диапазоне 0,1-95% от массы лекарственного средства, предпочтительно, в диапазоне 0,2-20% масс. в лекарственных средствах для парентерального применения, и более предпочтительно, в диапазоне 1-50% масс. в лекарственных средствах для перорадьного введения.
Лекарственные формы далее могут быть получены известными способами, такими как грануляция, прессование, микрокапсулирование, нанесение покрывающей оболочки методом распыления и т.п. Лекарственные формы могут быть получены путем стандартных методов в виде дозированных лекарствнных форм таблеток, капсул, гранул, порошков, сиропов, суспензий, суппозиториев или инъекционных форм. Жидкие лекарственные формы могут быть получены путем растворения или суспендирования активного вещества в воде или в других подходящих средах. Таблетки и гранулы могут быть покрыты оболочкой стандартного вида изготовления.
Уровень содержания дозы и периодичность приема дозировки определенного соединения меняют в зависимости от различных факторов, включающих активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст пациента, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость выведения, комбинирование с лекарственными средствами, тяжесть подлежащего лечению патологического состояния, переносимость терапии пациентом. Дневная доза может, например, изменяться в диапазоне от приблизительно 0,001 мг до приблизительно 100 мг на кг массы тела, вводиться одноразово или множественными дозировками, например, от приблизительно 0,01 мг до приблизительно 1000 мг каждая. Обычно такая дозировка дается перорально, но и парентеральное введение может быть выбрано.
Определения
Нижеприведенные определения следует использовать на всем протяжении описания изобретения и приведенной в завершение формулы изобретения.
Термины "связанное с FLT3 заболевание" и "заболевание или патологическое состояние, связанное с нежелательной активностью FLT3" применяют взаимозаменяемо в этом документе для обозначения какого-либо нарушения или симптома, в которые вовлечена FLT3 в процессе представления заболевания или симптома. Связанные с FLT3 заболевания таким образом включают, например, но не ограничиваются только ими, гематологические злокачественные новообразования, такие как острая миеломная лейкемия (AML); недифференцированная лейкемия (MLL); T-клеточного типа острая лимфоцитарная лейкемия (T-ALL); B-клеточного типа острая лимфоцитарная лейкемия (B-ALL) и хроническая миеломоноцитарная лейкемия (CMML).
Если не указано иначе термин "C1-6-алкил" обозначает алкильную группу с неразветвленной или разветвленной цепью, имеющей от 1 до 6 атомов углерода. Примеры указанного C1-6-алкила включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил и пентил и гексил с неразветвленной и разветвленной цепью. В качестве составных частей в диапазоне "C1-6-алкил" все его подгруппы рассматриваются, так как C1-5-алкил, C1-4-алкил, C1-3-алкил, C1-2-алкил, C2-6-алкил, C2-5-алкил, C2-4-алкил, C2-3-алкил, C3-6-алкил, C4-5-алкил и т.п. Аналогично, "арил-C1-6-алкил" обозначает C1-6-алкильную группу, замещенную арильной группой. Примеры включают бензил, 2-фенилэтил, 1-фенилэтил и 1-нафтилметил.
Если не указано иначе, термин "C1-3-алкокси" обозначает алкоксигруппу с неразветвленной или разветвленной цепью, имеющей от 1 до 3 атомов углерода. Примеры указанных C1-3-алкокси включают метокси, этокси, н-пропокси, изопропокси. В качестве составных частей в диапазоне "C1-3-алкокси" все его подгруппы рассматриваются, так как C1-2-алкокси и C2-3-алкокси.
Если не указано иначе, термин "C1-3-алкоксикарбонил" обозначает алкоксигруппу с неразветвленной или разветвленной цепью, имеющей от 1 до 3 атомов углерода, связанных с карбонильной группой. Примеры указанных C1-3-алкоксикарбонилов включают метоксикарбонил, этоксикарбонил, изопропоксикарбонил. В качестве составных частей в диапазоне "C1-3-алкоксикарбонил" все его подгруппы рассматриваются, так как C1-2-алкоксикарбонил и C2-3-алкоксикарбонил.
"Фармацевтически приемлемый" обозначает пригодный в получении фармацевтической композиции, который полностью безвреден, нетоксичен и как биологически, так и как-либо иначе не может быть неприемлемым, и включает применимый для ветеринарного использования, а также и для людей, для фармацевтического использования.
"Лечение", как применяют в этом документе, включает профилактику названного заболевания или патологического состояния, или улучшение, или устранение заболевания, некогда установленного.
"Эффективное количество" обозначает количество соединения, которое дает терапевтический эффект для субъекта, подвергаемого лечению. Терапевтический эффект может быть объективным (например, измеряемым с помощью какого-либо теста или маркера) или субъективным (например, пациент появляет признаки терапевтического эффекта или он чувствует эффект).
Термин "пролекарственные формы" обозначает фармакологически приемлемое производное, такое как сложный эфир или амид, такое производное, которое является биотрансформируемым в организме с образованием активного лекарственного средства. Ссылка делается на Goodman and Gilman's, The Pharmacological basis of Therapeutics, 8th ed., Mc-Graw-Hill, Int. Ed. 1992, "Biotransformation of Drugs", p. 13-15; и "The Organic Chemistry of Drug Design and Drug Action" by Richard B. Silverman. Chapter 8, p. 352. (Academic Press, Inc. 1992. ISBN 0-12-643730-0).
Комбинации заместителей и переменных характеристик, предусмотренных этим изобретением, представляют собой только такие, которые в результате приводят к образованию стабильных соединений. Термин "стабильные", как он применяется в этом документе, относится к соединениям, которые проявляют стабильность, достаточную для обеспечения промышленного производства, и которые обеспечивают целостность соединения в течение соответствующего периода времени для применения их в целях, подробно описанных в этом документе (например, терапевтическое введение пациенту для лечения связанного с FLT3 нарушения или заболевания (включая те, которые описаны в этом документе), например, гематологические злокачественные новообразования, такие как острая миеломная лейкемия (AML); недифференцированная лейкемия (MLL); T-клеточного типа острая лимфоцитарная лейкемия (T-ALL); B-клеточного типа острая лимфоцитарная лейкемия (B-ALL) и хроническая миеломоноцитарная лейкемия (CMML)).
Изложение перечня химических групп в каких-либо определениях переменных характеристик этого документа включает определения таких переменных характеристик, как какая-либо одиночная группа или комбинация перечисленных групп. Изложение примера осуществления изобретения для переменной характеристики этого документа включает такой пример осуществления изобретения, который представляет собой какой-либо один пример осуществления изобретения, или в комбинации с какими-либо другими примерами осуществления изобретения или их частями.
Изобретение будет теперь дополнительно проиллюстрировано нижеприведенными, не ограничивающими его примерами. Конкретные нижеприведенные примеры представляют собой всего лишь иллюстративные и не ограничивают остальное раскрытие изобретения каким бы то ни было другим путем. Без дополнительных исследований очевидно, что средний специалист в данной области техники способен, основываясь на описании этого документа, применять настоящее изобретение в полном объеме. Все процитированные в этом документе публикации таким образом включены в качестве ссылок во всей полноте.
Структуры, описанные в этом документе, могут содержать определенные -NH-, -NH2 (амино) и -OH (гидроксил) группы, в которых соответствующий(е) водородный(е) атом(ы) не присутствует(ют) явным образом; хотя их надо понимать как -NH-, -NH2 или -OH в зависимости от конкретного случая.
Методы
1H ядерный магнитный резонанс (ЯМР) и 13C ЯМР регистрировали на Bruker Advance DPX 400 спектрометре при 400,1 МГц и 100,6 МГц, соответственно. Все спектры регистрировали, используя остаточный растворитель или тетраметилсилан (ТМС) в качестве внутреннего стандарта.
Низкого разрешения электрораспылительной ионизации масс-спектры (LRESIMS) получали, используя Agilent MSD масс-спектрометр или Waters ZQ масс-спектрометр. Высокого разрешения электрораспылительной ионизации масс-спектры (HRESIMS) получали на Agilent LC/MSD TOF, соединенном с Agilent 1100 LC-системой, ионный источник: ESI (ионизация методом электрораспыления), ионная полярность: пол, представление данных: в виде графического изображения, диапазон сканирования: 100-1100 Да, масс-спектрометра параметры: фрагментор 215 В, скиммер 560 В och OCT RF (octpole rods) 250 В.; рекомендуемые массы 121,050873 и 922,009798 (Agilent reference Mix); ЖХ (LC): A 15 мМ ацетата аммония; B 100 MeCN; скорость потока 400 мкл/мин, изократический режим. Флэш-хроматографию проводили на силикагеле Merck silica gel 60 (230-400 mesh). Микроволновое облучение проводили, применяя Smith Creator или Optimizer (Personal Chemistry), используя 0,5-2 мл или 2-5 мл ампулы Smith Process vials, снабженные алюминиевыми капсулами и перегородками. Соединения назывались автоматически с использованием ACD/NAME 6.0 (Advanced Chemistry Development, Inc., Toronto, Canada).
Результаты аналитической ЖХМС (LCMS) получали, используя:
Система A: Agilent MSD масс-спектрометр; Agilent 1100 система; ACE 3 C8 колонка (50Ч3,0 мм); водную 0,1% трифторуксусную кислоту (TFA) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 1 мл/мин градиентное время 3,0 мин (градиент 10-97% ацетонитрила); или
Система B: Agilent MSD масс-спектрометр; Agilent 1100 система; YMC ODS-AQ колонка (33Ч3,0 мм); водную 0,1% трифторуксусную кислоту (TFA) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 1 мл/мин градиентное время 3,0 мин (градиент 10-97% ацетонитрила); или
Система C: Waters ZQ масс-спектрометр; Waters 996 PDA детектор (DAD 215-395 нм); ACE C8 (3 мкм) колонка (30Ч3,0 мм) (от ACT); водный 10 мМ ацетат аммония (pH=7) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 1 мл/мин градиентное время 3,2 мин (градиент 5-100% ацетонитрила).
Препаративную ВЭЖХ (HPLC) проводили на системе Gilson, снабженной следующим:
Система D: ACE C8 5 мкм (21,2Ч50 мм) колонка. Водную 0,1% трифторуксусную кислоту (TFA) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 25 мл/мин градиентное время 6 мин.; или
Система E: XTerra Prep MS C18 5 мкм (19Ч50 мм) колонка. Водный 50 мМ NH4HCO3 (pH=10) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 25 мл/мин градиентное время 6 мин; или Xterra MS C18 5 мкм (30Ч100 мм) колонка.
Водный 50 мМ NH4HCO3 (pH=10) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 40 мл/мин градиентное время 8,5 мин; или
Система F: YMC ODS-AQ 10 мкМ (30Ч150 мм) колонка. Водную 0,1% трифторуксусную кислоту (TFA) и ацетонитрил применяли в качестве подвижных фаз при скорости потока элюента 45 мл/мин градиентное время 8,5 мин.
Нижеприведенные сокращения использовали
ДМСО (DMSO) обозначает диметилсульфоксид,
ВЭЖХ (HPLC) обозначает высокоэффективная жидкостная хроматография,
ТФУК (TFA) обозначает трифторуксусную кислоту.
МСВР (HRMS) обозначает масс-спектрометрия высокого разрешения
ПРИМЕРЫ
Методика A:
Общая методика для SNAr для 2-амино-3,5-дибромпиразина
2-Амино-3,5-дибромпиразин, триэтиламин (3 экв.) и соответствующий амин или спирт (3 экв.) растворяли в 4 мл воды и полученную в результате смесь нагревали до 195°C в течение 1 часа. Воду и этилацетат добавляли и фазы разделяли. Водную фазу экстрагировали еще раз этилацетатом. Объединенные органические фазы промывали (водой и солевым раствором) и концентрировали с получением неочищенной смеси продукта и непрореагировавшего амина или спирта. Эту неочищенную смесь использовали без дополнительной очистки или определения характеристик в последующей реакции Сузуки.
Методика B:
Общая методика для реакции сочетания Сузуки
Смесь пиразинилбромида из методики A (1 экв.), соответствующей бороновой кислоты (1 экв.), K2CO3 (3 экв.) и Pd(dppf)Cl2*CH2Cl2 (0,1 экв.) в 4 мл диоксан/вода (4:1) нагревали до 150°C в течение 15 мин. Смесь фильтровали через небольшой слой силикагеля и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ (ACE C8 колонка; подвижная фаза: 0,1% TFA - CH3CN), получая указанное в заголовке соединение в виде белого твердого вещества в форме его соответствующей трифторуксусной соли.
Промежуточное соединение 1
5-бром-N3-1H-индол-5-илпиразин-2,3-диамин
Используя методику A и 2-амино-3,5-дибромпиразин (100 мг) и 5-аминоиндол (200 мг), получали 150 мг смеси, содержащей в соотношении 1:1 5-аминоиндола и целевого продукта. Масс-спектр m/z 303 [M+H]+, который использовали без дополнительной очистки или регистрации его характеристик.
ПРИМЕР 1
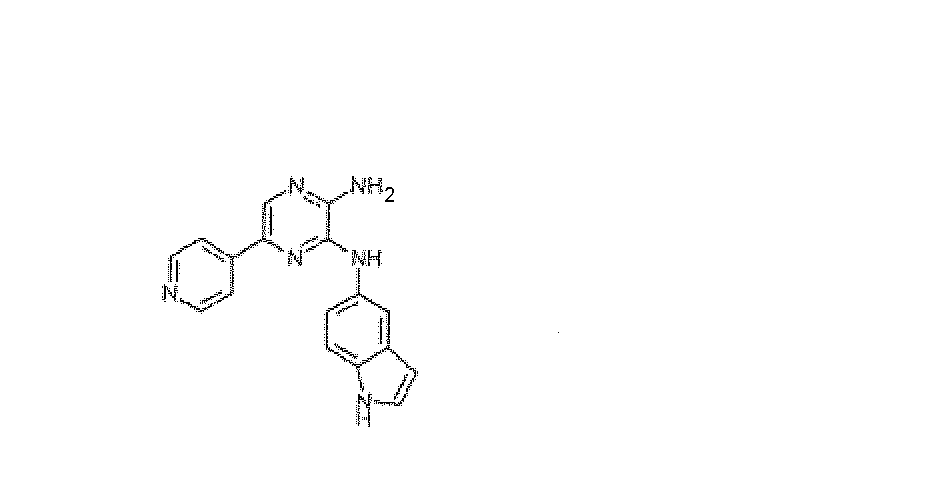
N3-1H-Индол-5-ил-5-пиридин-4-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (20 мг) и 4-пиридилбороновую кислоту (12 мг), получали 1,7 мг указанного в заголовке соединения. Масс-спектр m/z 303 [M+H]+.
1H ЯМР (400 МГц, CD3OD) д м.д. 6,48 (д, J=3,01 Гц, 1H), 7,26-7,33 (м, 1H), 7,33-7,50 (м, 2H), 7,92 (д, J=1,25 Гц, 1H), 8,37 (с, 1H), 8,45 (д, J=7,03 Гц, 2H), 8,64 (д, J=7,03 Гц, 2H).
ПРИМЕР 2
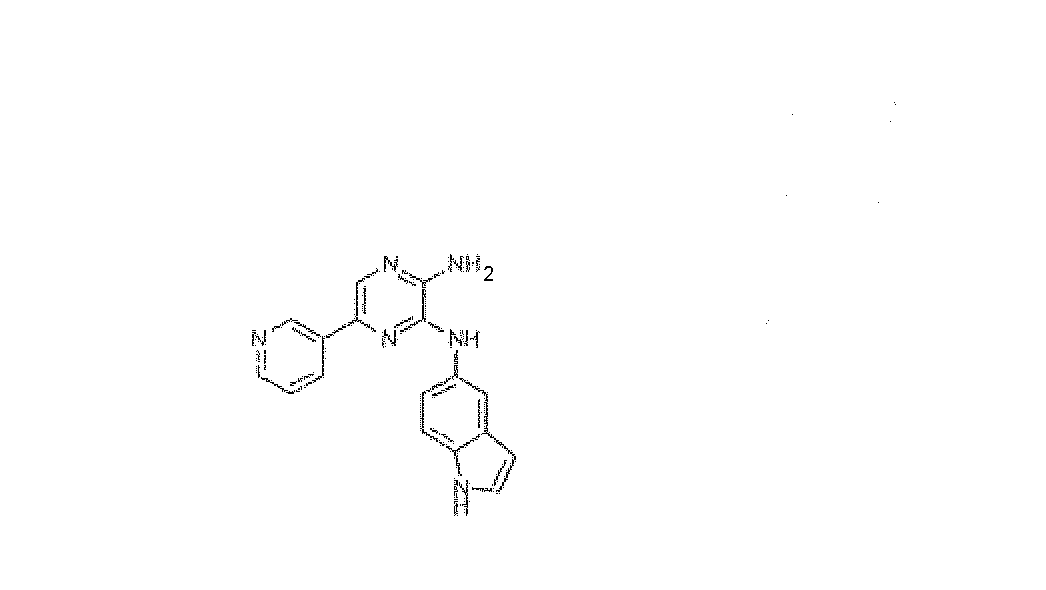
N3-1H-Индол-5-ил-5-пиридин-3-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (20 мг) и 3-пиридилбороновую кислоту (12 мг), получали 1,3 мг указанного в заголовке соединения. HRMS, рассчитано для C17H14N6: 302,1280, получено: 302,1279.
1H ЯМР (400 МГц, CD3OD): 6,49 (д, 1H, J=4 Гц), 7,29-7,46 (м, 3H), 7,88-7,94 (м, 2H), 8,02 (с, 1H), 8,64 (д, 1H, J=8 Гц), 8,84 (д, 1H, J=8 Гц), 9,19 (с, 1H).
ПРИМЕР 3
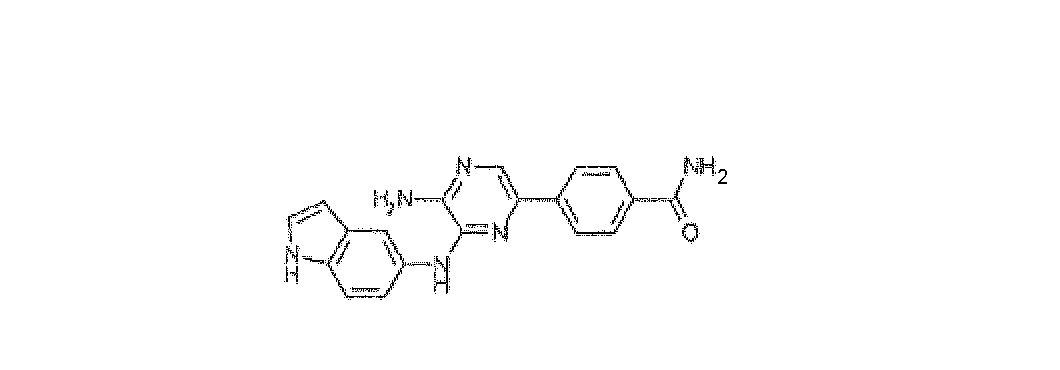
4-[5-Амино-6-(1H-индол-5-иламино)пиразин-2-ил]бензамид, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (20 мг) и 4-бензамид бороновой кислоты (16 мг), получали 0,9 мг указанного в заголовке соединения. HRMS рассчитано для C19H16N6O: 344,1386, найдено: 344,1381.
1H ЯМР (400 МГц, CD3OD): д 6,49 (д, 1H, J=4 Гц), 7,30 (д, 1H, J=4 Гц), 7,42-7,47 (м, 2H), 7,81 (с, 1H), 7,94 (д, 2H, J=8 Гц), 8,01-8,06 (м, 3H).
ПРИМЕР 4
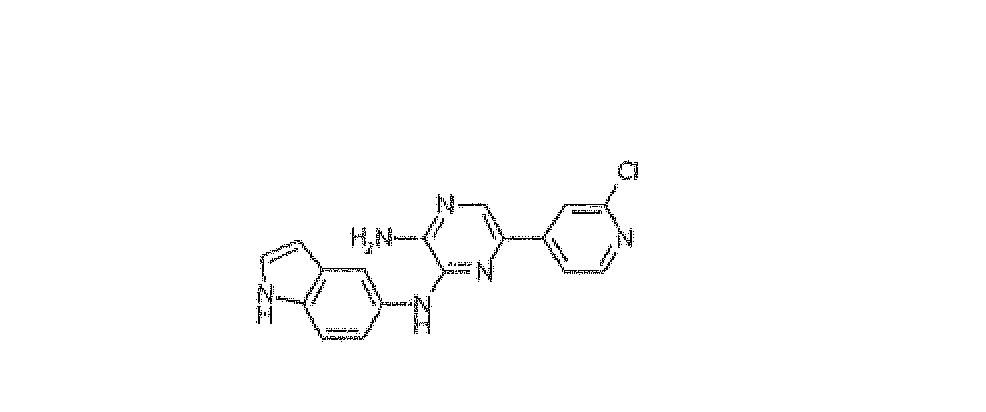
5-(2-Хлорпиридин-4-ил)-N3-1H-индол-5-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (20 мг) и 2-хлорпиридин-4-илбороновую кислоту (20 мг), получали 4,0 мг указанного в заголовке соединения.
ПРИМЕР 5
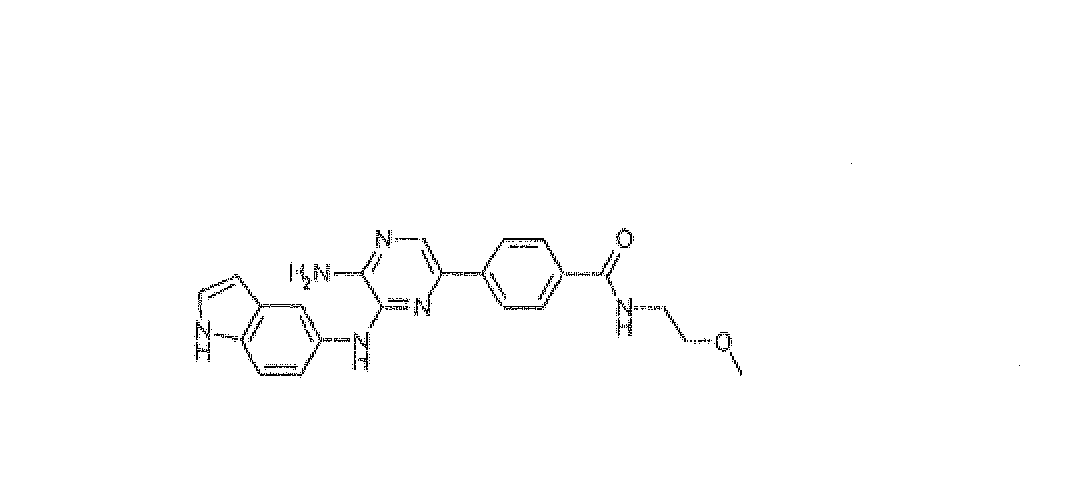
4-[5-Амино-6-(1H-индол-5-иламино)пиразин-2-ил]-N-(2-метоксиэтил)бензамид, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (25 мг) и [4-[[(2-метоксиэтил)амино]карбонил]фенил]бороновую кислоту (27 мг), получали 4,2 мг указанного в заголовке соединения. Масс-спектр m/z 403 [M+H]+.
ПРИМЕР 6
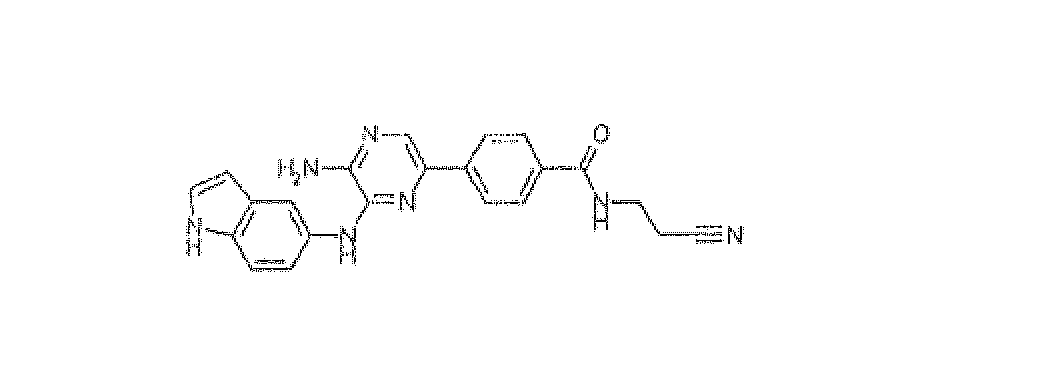
4-[5-Амино-6-(1H-индол-5-иламино)пиразин-2-ил]-N-(2-цианоэтил)бензамид, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-5-илпиразин-2,3-диамин (25 мг) и [4-(2-цианоэтиламинокарбонил)фенил]бороновую кислоту (27 мг), получали 3,2 мг указанного в заголовке соединения. Масс-спектр m/z 398 [M+H]+.
1H ЯМР (500 МГц, ДМСО-d6) д м.д. 2,78 (т, J=6,70 Гц, 2H), 3,51 (кв., J=6,09 Гц, 2H), 6,42 (с, 1H), 7,27-7,47 (м, 3H), 7,76-8,18 (м, 6H), 8,38 (с, 1H), 8,63-9,11 (м, 1H), 10,98 (с, 1H).
Промежуточное соединение 2
5-Бром-Н3-1H-индол-4-илпиразин-2,3-диамин
Используя методику A и 2-амино-3,5-дибромпиразин (300 мг) и 4-аминоиндол (470 мг), получали 700 мг смеси, содержащей в соотношении 1:1 4-аминоиндол и целевой продукт. Масс-спектр m/z 303 [M+H]+, который использовали без дополнительной очистки или регистрации его характеристик.
ПРИМЕР 7
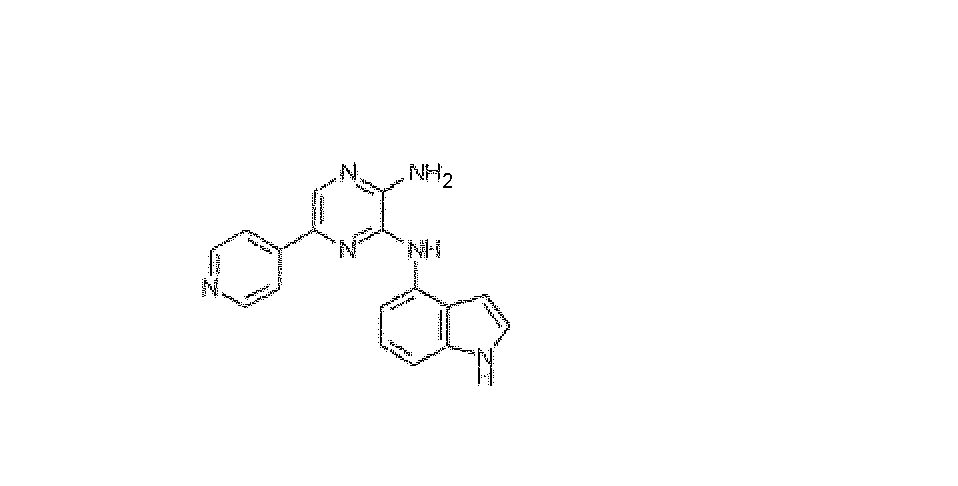
N3-1H-Индол-4-ил-5-пиридин-4-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-4-илпиразин-2,3-диамин (25 мг) и 4-пиридинилбороновую кислоту (15 мг), получали 1,2 мг указанного в заголовке соединения. HRMS рассчитано для C17H14N6: 302,1280, найдено: 302,1278.
1H ЯМР (400 МГц, CD3OD) м.д. 6,41 (д, J=3 Гц, 1H), 7,19 (д, J=7 Гц, 1H), 7,21-7,32 (м, 2H), 7,38 (д, J=7 Гц, 1H), 8,33 (д, J=6 Гц, 2H), 8,44 (с, 1H), 8,57 (д, J=6 Гц, 2H).
ПРИМЕР 8
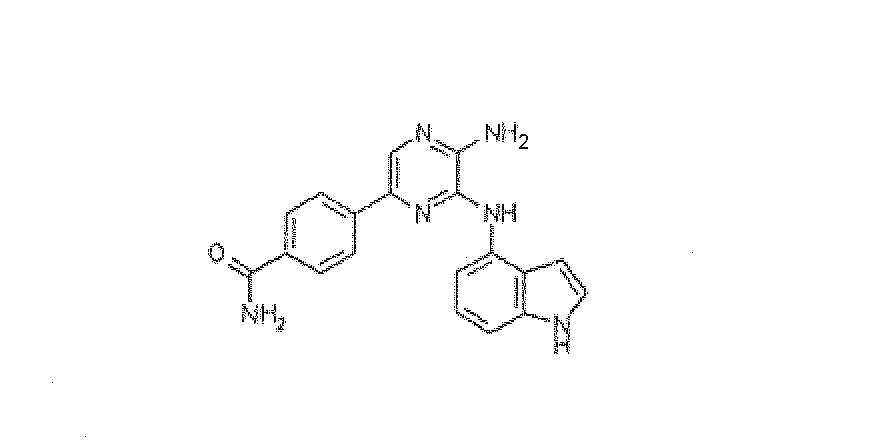
4-[5-Амино-6-(1H-индол-4-иламино)пиразин-2-ил]бензамид, трифторацетат
Используя методику B и 5-бром-N3-1H-индол-4-илпиразин-2,3-диамин (25 мг) и 4-бензамид бороновой кислоты (20 мг), получали 1,1 мг указанного в заголовке соединения. HRMS рассчитано для C19H16N6O: 344,1386, найдено: 344,1384.
1H ЯМР (400 МГц, CD3OD) д 6,50 (д, J=2 Гц, 1H), 7,20 (т, J=7 Гц, 1H), 7,28 (д, J=3 Гц, 1H), 7,33 (д, J=8 Гц, 1H), 7,49 (д, J=7 Гц, 1H), 7,82-7,96 (м, 5H).
Промежуточное соединение 3
5-Бром-N3-(2-метил-1H-индол-5-ил)пиразин-2,3-диамин
Используя методику A и 2-амино-3,5-дибромпиразин (300 мг) и 5-амино-2-метилиндол (520 мг), получали 400 мг смеси, содержащей в соотношении 1:1 5-амино-2-метилиндол и целевой продукт. Масс-спектр m/z 319 [M+H]+, который использовали без дополнительной очистки или регистрации его характеристик.
ПРИМЕР 9
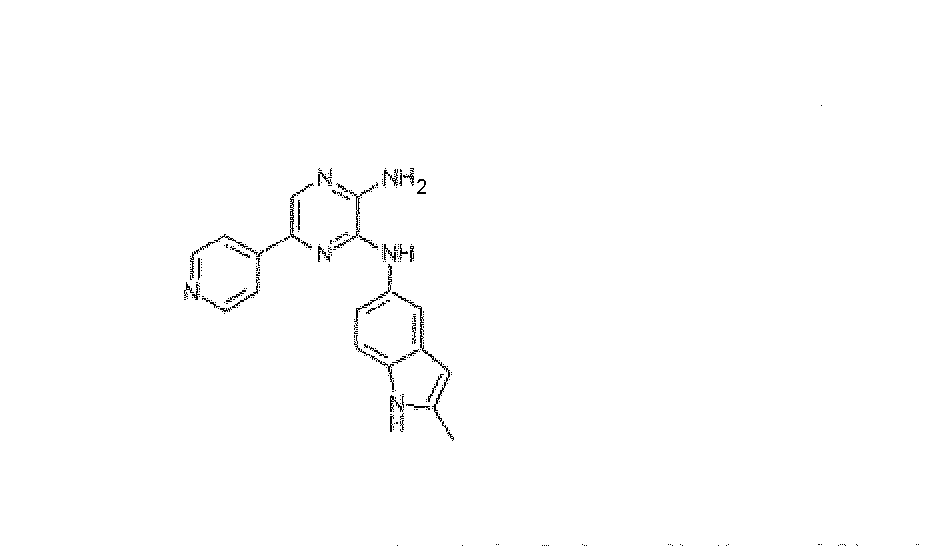
N3-(2-Метил-1H-индол-5-ил)-5-пиридин-4-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-(2-метил-1H-индол-5-ил)пиразин-2,3-диамин (26 мг) и 4-пиридинилбороновую кислоту (14 мг), получали 3,0 мг указанного в заголовке соединения. HRMS рассчитано для C18H16N6: 316,1436, найдено: 316,1437.
1H ЯМР (400 МГц, CD3OD) д м.д. 2,45 (с, 3H), 7,16-7,47 (м, 3H), 7,76 (с, 1H), 8,35 (с, 1H), 8,45 (д, J=6 Гц, 2H), 8,65 (д, J=6 Гц, 2H).
ПРИМЕР 10
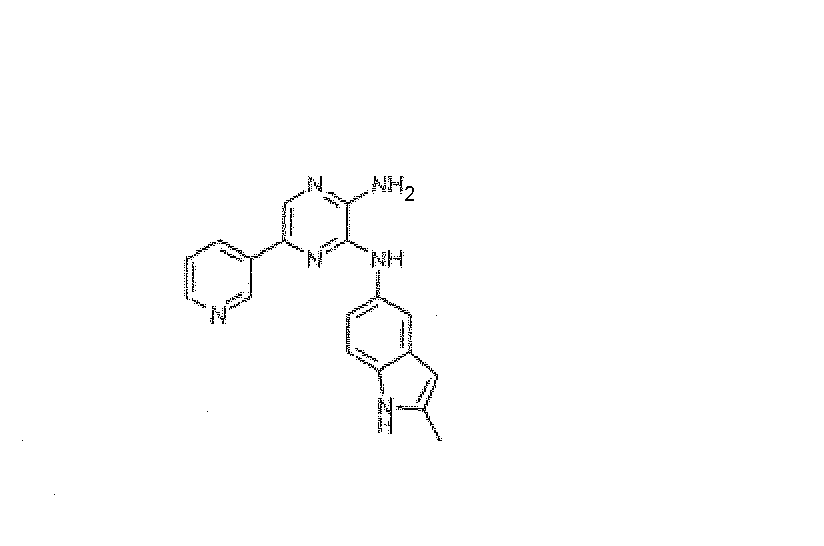
N3-(2-Метил-1H-индол-5-ил)-5-пиридин-3-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-(2-метил-1H-индол-5-ил)пиразин-2,3-диамин (26 мг) и 3-пиридинилбороновую кислоту (14 мг), получали 3,4 мг указанного в заголовке соединения. HRMS рассчитано для C18H16N6: 316,1436, найдено: 316,1434.
ПРИМЕР 11
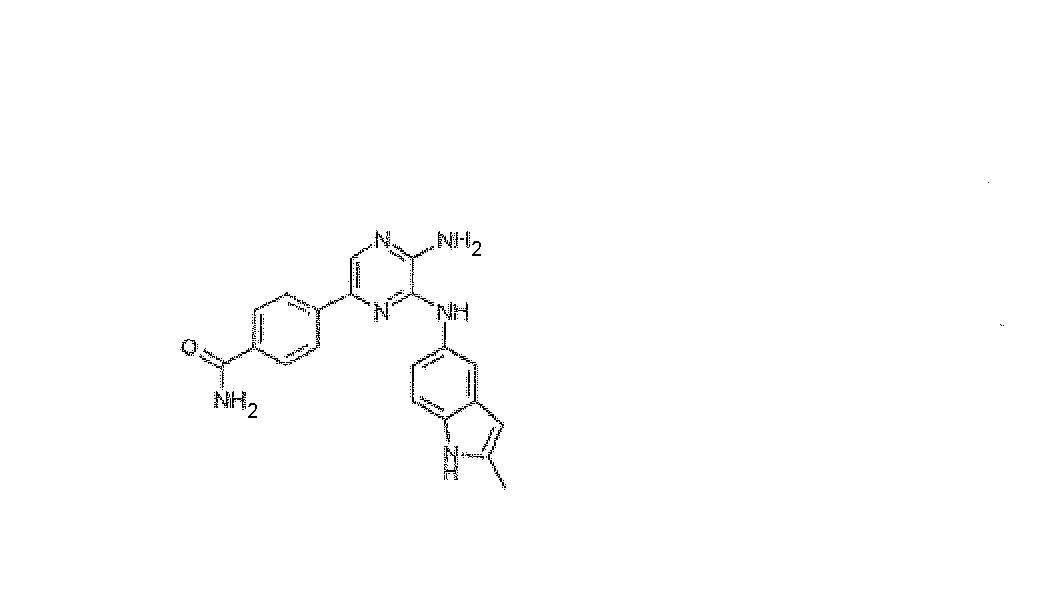
4-{5-Амино-6-[(2-метил-1H-индол-5-ил)амино]пиразин-2-ил}бензамид, трифторацетат
Используя методику B и 5-бром-N3-(2-метил-1H-индол-5-ил)пиразин-2,3-диамин (26 мг) и 4-бензамид бороновой кислоты (19 мг), получали 2,2 мг указанного в заголовке соединения. HRMS рассчитано для C20H18N6O: 358,1542, найдено: 358,1542.
1H ЯМР (400 МГц, CD3OD) д 2,46 (с, 3H), 7,15-7,48 (м, 3H), 7,77 (с, 1H), 7,90 (с, 1H), 7,91-7,96 (м, 2H), 8,00-8,12 (м, 2H).
Промежуточное соединение 4
5-Бром-N3-(1H-индазол-5-ил)пиразин-2,3-диамин
Используя методику A и 2-амино-3,5-дибромпиразин (300 мг) и 5-аминоиндазол (470 мг), получали 320 мг смеси, содержащей в соотношении 1:3 5-аминоиндазол и целевой продукт. Масс-спектр m/z 306 [M+H]+, который использовали без дополнительной очистки или регистрации его характеристик.
ПРИМЕР 12
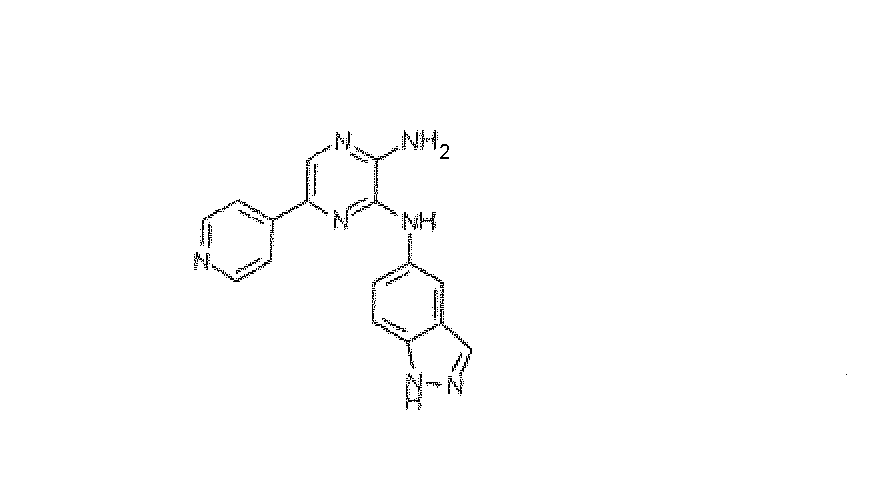
N3-1H-Индазол-5-ил-5-пиридин-4-илпиразин-2,3-диамин, трифторацетат
Используя методику B и 5-бром-N3-(1H-индазол-5-ил)пиразин-2,3-диамин (15 мг) и 4-пиридилбороновую кислоту (9 мг), получали 1,3 мг указанного в заголовке соединения. HRMS рассчитано для C16H13N7: 303,1232, найдено: 303,1231.
ПРИМЕР 13
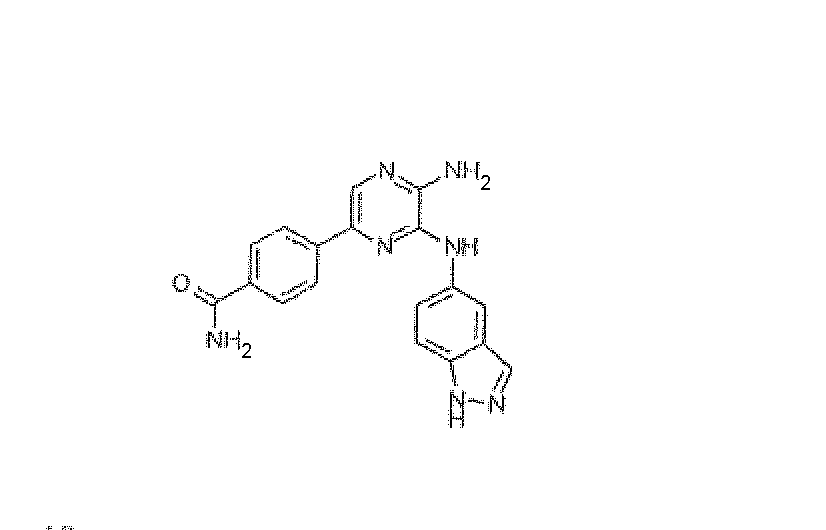
4-[5-Амино-6-(1H-индазол-5-иламино)пиразин-2-ил]бензамид, трифторацетат
Используя методику B и 5-бром-N3-(1H-индазол-5-ил)пиразин-2,3-диамин (15 мг) и 4-бензамид бороновой кислоты (12 мг), получали 1,5 мг указанного в заголовке соединения. HRMS рассчитано для C18H15N7O: 345,1338, найдено: 345,1335.
ПРИМЕР 14
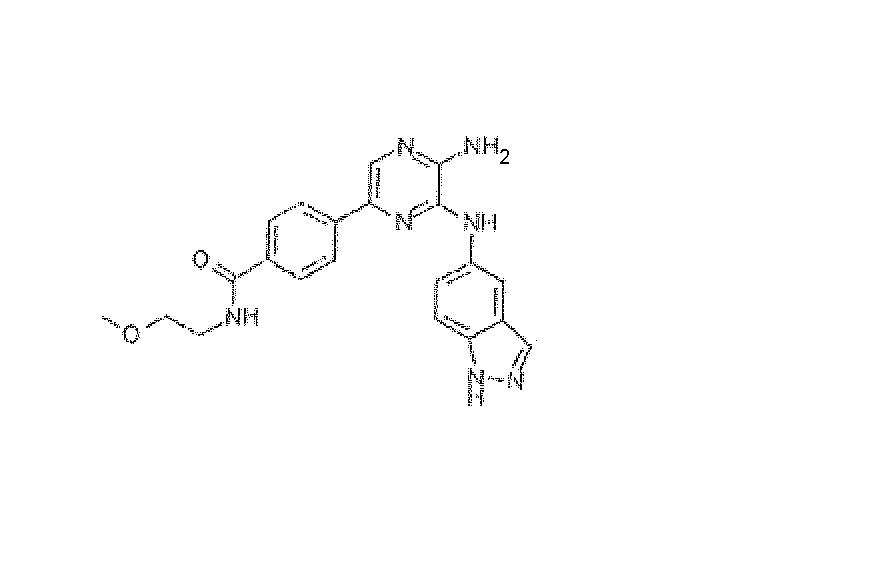
4-[5-Амино-6-(1H-индазол-5-иламино)пиразин-2-ил]-N-(2-метоксиэтил)бензамид, трифторацетат
Используя методику B и 5-бром-N3-(1H-индазол-5-ил)пиразин-2,3-диамин (15 мг) и [4-[[(2-метоксиэтил)амино]карбонил]фенил]бороновую кислоту (16 мг), получали 2,5 мг указанного в заголовке соединения. HRMS рассчитано для C21H21N7O2: 403,1757, найдено: 403,1751.
Промежуточное соединение 5
5-Бром-N3-[2-(1H-индол-3-ил)этил]пиразин-2,3-диамин
Используя методику A: 2-Амино-3,5-дибромпиразин (300 мг) и триптамин (570 мг), получали 600 мг смеси, содержащей в соотношении 1:1 триптамин и целевой продукт. Масс-спектр m/z 333 [M+H]+, который использовали без дополнительной очистки или регистрации его характеристик.
ПРИМЕР 15
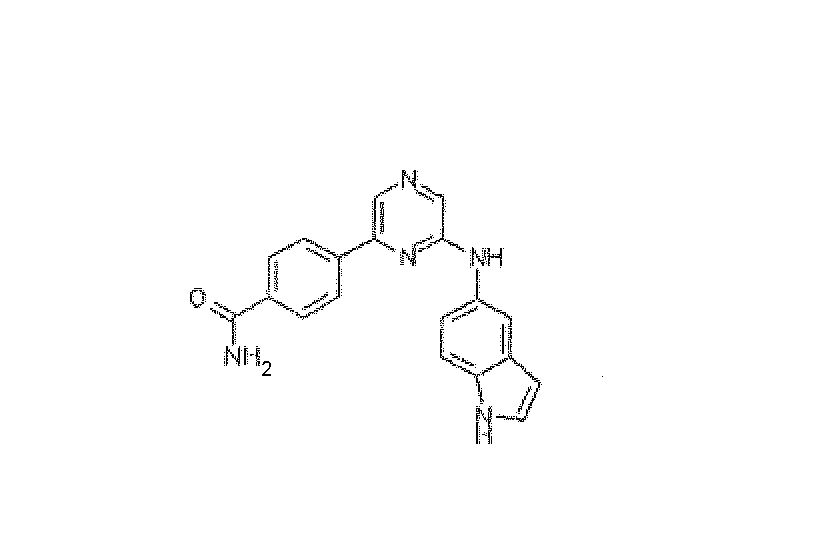
4-[6-(1H-Индол-5-иламино)пиразин-2-ил]бензамид, трифторацетат
5-Аминоиндол (100 мг), 2,6-дихлорпиразин (100 мг) и триэтиламин (135 мг) смешивали в 4 мл ацетонитрила и нагревали до 150°C в течение 1 ч. Водный насыщенный раствор NaHCO3 и дихлорметан добавляли к реакционной смеси и фазы разделяли. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы промывали солевым раствором и концентрировали. Неочищенное промежуточное соединение, 6-хлор-N-(1H-индол-5-ил)пиразин-2-амин, (4-аминокарбонилфенил)бороновую кислоту (121 мг), K2CO3 (275 мг) и Pd(тетракис(трифенилфосфин)) (38 мг) растворяли в 4 мл диоксана и 1 мл H2O, и реакционную смесь нагревали до 100°C в течение ночи. 1M NaOH(водн.) и дихлорметан добавляли к смеси, и фазы разделяли. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы промывали солевым раствором и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ (ACE C8 колонка; подвижная фаза: 0,1% TFA - CH3CN), получая указанное в заголовке соединение (85 мг) в виде белого твердого вещества в форме его соответствующей трифторацетатной соли. HRMS рассчитано для C19H15N5O: 329,1277, найдено: 329,1279. 1H ЯМР (400 МГц, CD3OD) д 7,27 (д, J=3,01 Гц, 1H), 7,31-7,37 (м, 1H), 7,40-7,43 (м, 1H), 7,47-7,51 (м, 1H), 7,85-7,93 (м, 1H), 7,95-8,08 (м, 3H), 8,19-8,26 (м, 2H), 8,38 (с, 1H).
ПРИМЕР 16
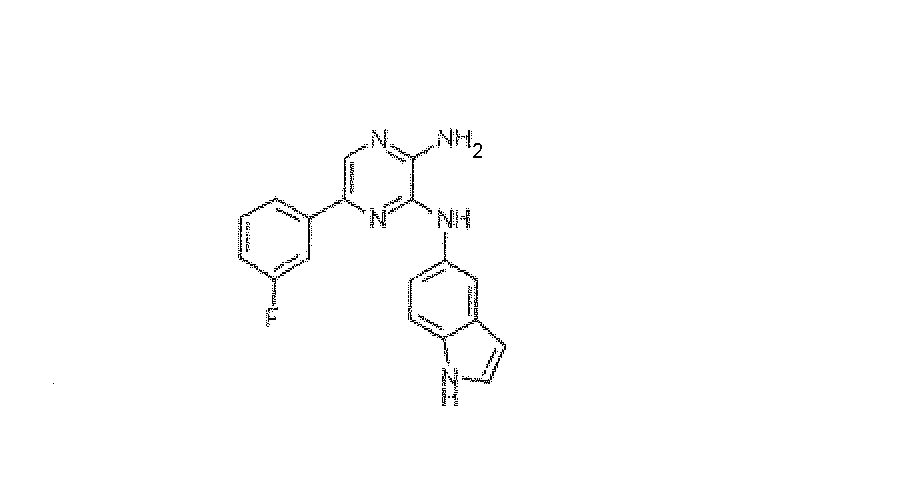
5-(3-фторфенил)-N~3-1H-индол-5-илпиразин-2,3-диамин. Приобретали у BioFocus DPI: HRMS рассчитано для C18H14FN5: 319,123324, найдено: 319,123684. Масс-спектр m/z 320 [M+H]+.
ПРИМЕР 17
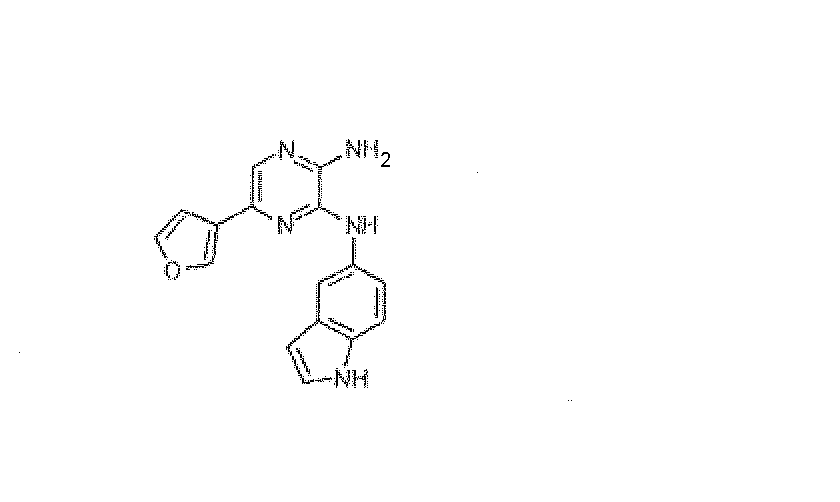
5-(3-фурил)-N~3-1H-индол-5-илпиразин-2,3-диамин.
Приобретали у BioFocus DPI: HRMS рассчитано для C16H13N5O: 291,112010, найдено: 291,112130. Масс-спектр m/z 292 [M+H]+.
ПРИМЕР 18
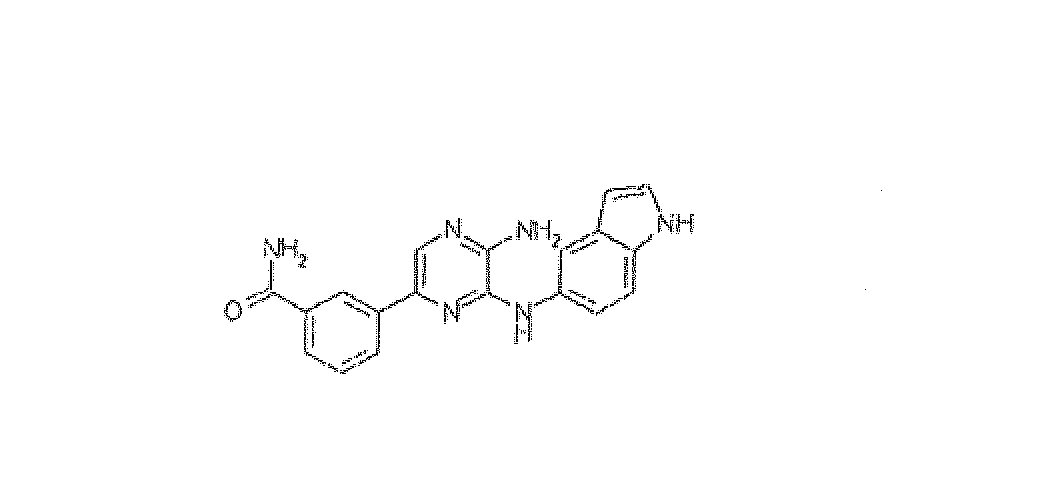
3-[5-амино-6-(1H-индол-5-иламино)пиразин-2-ил]бензамид. Приобретали у BioFocus DPI: HRMS рассчитано для C19H16N6O: 344,138559, найдено: 344,138509. Масс-спектр m/z 345 [M+H]+.
ПРИМЕР 19
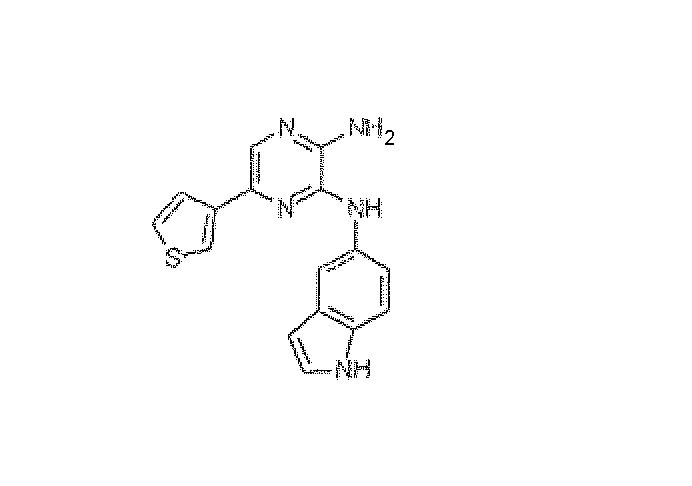
N~3-1H-индол-5-ил-5-(3-тиенил)пиразин-2,3-диамин. Приобретали у BioFocus DPI: HRMS рассчитано для C16H13N5S: 307,089166, найдено: 307,089106. Масс-спектр m/z 308 [M+H]+.
ПРИМЕР 20
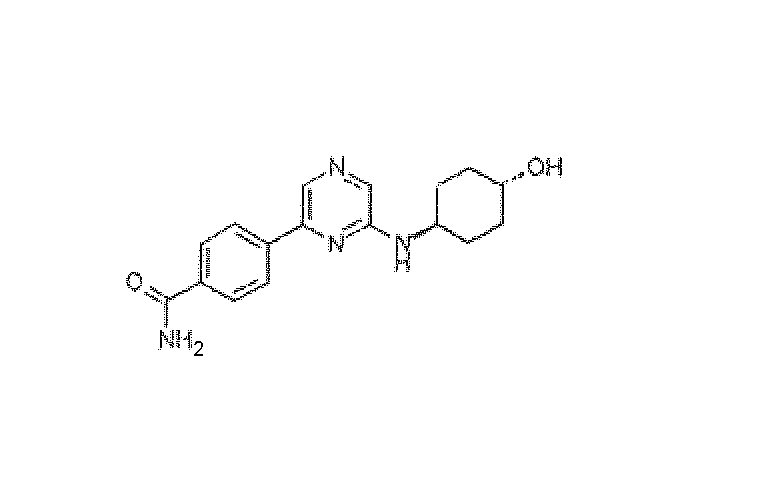
4-{6-[(транс-4-Гидроксициклогексил)амино]пиразин-2-ил}бензамид, трифторацетат
2,6-Дихлорпиразин (500 мг), транс-4-аминоциклогексанол (380 мг) и триэтиламин (500 мг) растворяли в 4 мл ацетонитрила/1 мл воды, и реакционную смесь нагревали до 150°C в течение 15 мин. Воду и дихлорметан добавляли к смеси и фазы разделяли. Водную фазу еще раз экстрагировали дихлорметаном. Объединенные органические фазы промывали (водой и солевым раствором) и упаривали, получая 750 мг промежуточного соединения 6-хлор-N-(транс-4-гидроксициклогексил)пиразин-2-амина с чистотой 85%. Часть этого вещества (30 мг), карбонат калия (55 мг), 4-бензамид бороновой кислоты (26 мг) и Pd(тетракис(трифенилфосфин)) (5 мг) растворяли в 4 мл диоксана и 1 мл H2O, и реакционную смесь нагревали до 100°C в течение ночи. 1M NaOH(водн.) и дихлорметан добавляли к смеси, и фазы разделяли. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы промывали солевым раствором и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ (ACE C8 колонка; подвижная фаза: 0,1% TFA - CH3CN), получая указанное в заголовке соединение (5,0 мг) в виде белого твердого вещества в форме его соответствующей трифторацетатной соли. HRMS рассчитано для C17H20N4O2: 312,1586, найдено: 312,1585.
ПРИМЕР 21
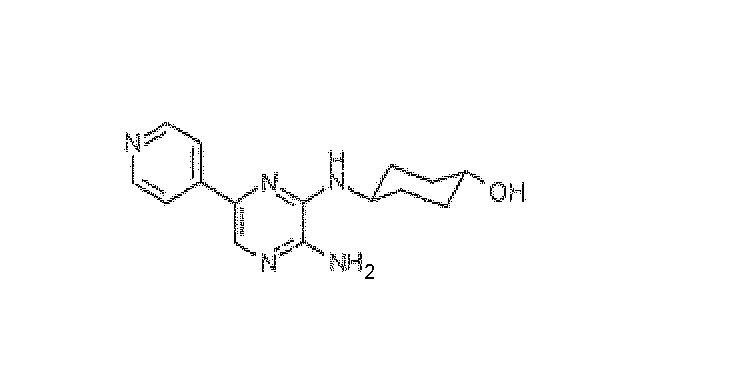
транс-4-[(3-Амино-6-пиридин-4-илпиразин-2-ил)амино]циклогексанол
Суспензию 2,6-дибром-3-аминопиразина (6,44 г, 0,0255 моль), K2CO3 (6,9 г, 0,05 моль) и транс-4-аминоциклогексанола (HCl соль) (7,55 г, 0,05 моль) в H2O (10,0 мл) нагревали до кипения с обратным холодильником в течение 72 ч (гомогенный раствор быстро образовывался и спустя приблизительно 30 ч твердое вещество медленно выпадало в осадок. Смесь охлаждали и нерастворимое твердое вещество отделяли и промывали водой, что давало возможность получить 4,336 г (59%) промежуточного соединения, транс-4-[(3-амино-6-бромпиразин-2-ил)амино]циклогексанола. К раствору неочищенного вещества (4,336 г, 0,0151 моль), 4-пиридилбороновой кислоты (1,84 г, 0,0151 моль) тетракис(трифенилфосфина)палладия(0) (870 мг, 0,7 ммоль; 5% моль) в PhMe (200 мл) добавляли водный раствор 2M карбоната натрия (40 мл), и этанол (40 мл). Смесь нагревали до кипения с обратным холодильником в течение ночи. Смесь концентрировали упариванием и нерастворимое темно-окрашенное твердое вещество отделяли фильтрованием. Это вещество затем растворяли в MeOH и проводили флэш-хроматографию на диоксиде кремния с EtOAc-MeOH (9:1), получая бледно-желтое твердое вещество (2,2 г). Последующее элюирование с EtOAc-MeOH (7:1) давало дополнительную массу бледно-желтого твердого вещества (930 мг), которое было достаточно сильно засорено силикагелем. Обе массы твердого вещества объединяли и очищали препаративной ВЭЖХ (ACE C8 колонка; подвижная фаза: 0,1% TFA-CH3CN), что давало возможность получить 2,2 г указанного в заголовке продукта. Чистота по данным ВЭЖХ 100%; 1H ЯМР (400 МГц, ДМСО-d6) д м.д. 1,33-1,40 (м, 4H), 1,89-1,92 (м, 2H), 2,01-2,04 (м, 2H), 3,47-3,49 (м, 1H), 3,93-3,97 (м, 1H), 8,29 (с, 1H), 8,41 (д, 2H, J=5,0 Гц), 8,80 (д, 2H, J=5,0 Гц). Масс-спектр (API-ES/Positive); m/z: 286 (M+H)+.
ПРИМЕР 22
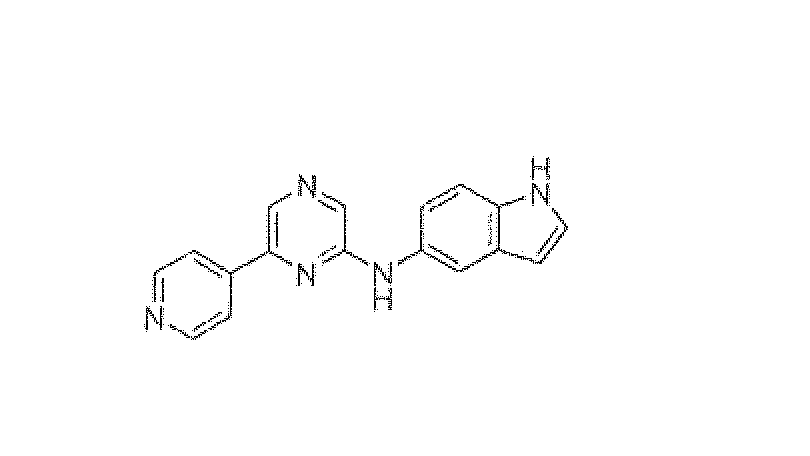
N-(6-Пиридин-4-илпиразин-2-ил)-1H-индол-5-амин
Смесь 2,6-дихлорпиразина (0,845 г, 5,67 ммоль), 5-аминоиндола (0,5 г, 3,78 ммоль), BINAP (0,051 г, 0,0831 ммоль), трет-бутоксида натрия (0,51 г, 5,29 ммоль) и ацетата палладия (0,0186 г, 0,0831 ммоль) в толуоле (25 мл) нагревали до 85°C в течение 22 ч в атмосфере азота. CH2Cl2 добавляли, реакционную смесь фильтровали через Celite, и растворитель упаривали. Остаток очищали колоночной хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая 0,180 г (13%) промежуточное соединение N-(6-хлорпиразин-2-ил)-1H-индол-5-амин.
1H ЯМР (CD3OD) д 7,97 (с, 1H), 7,84 (с, 1H), 7,77 (с, 1H), 7,42-7,39 (д, J=8,63 Гц, 1H), 7,28-7,20 (м, 2H), 6,47-6,46 (д, J=2,83 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 245 (M+H)+.
Смесь N-(6-хлорпиразин-2-ил)-1H-индол-5-амина (0,030 г, 0,123 ммоль), пиридин-4-бороновой кислоты (0,018 г, 0,147 ммоль), карбоната натрия (0,067 г, 0,615 ммоль) и тетракис(трифенилфосфин)палладия(0) (0,007 г, 0,006 ммоль) в DME:вода (3:2, 5 мл) нагревали до кипения с обратным холодильником в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном. Органический слой промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали флэш-хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая N-(6-пиридин-4-илпиразин-2-ил)-1H-индол-5-амин (0,011 г, 31%) в виде желтого твердого вещества.
1H ЯМР (CD3OD) д 8,70-8,68 (д, J=6,17 Гц, 2H), 8,47 (с, 1H), 8,15 (с, 1H), 8,14 (д, J=1,50 Гц, 2H), 7,96 (д, J=1,70 Гц, 1H), 7,45-7,43 (д, J=8,64 Гц, 1H), 7,37-7,35 (дд, J=10,46, 1,83 Гц, 1H), 7,29-7,28 (д, J=3,06 Гц, 1H), 6,49-6,48 (д, J=3,02 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 288 (M+H)+.
ПРИМЕР 23
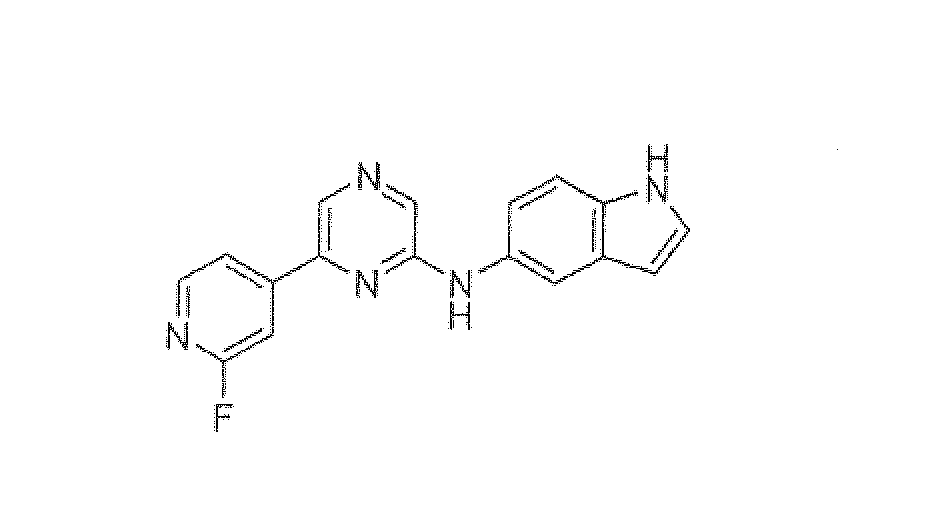
N-[6-(2-фторпиридин-4-ил)пиразин-2-ил]-1H-индол-5-амин
Смесь N-(6-хлорпиразин-2-ил)-1H-индол-5-амина (0,05 г, 0,205 ммоль), 2-фторпиридин-4-бороновой кислоты (0,057 г, 0,4 ммоль), карбоната натрия (0,112 г, 1,025 ммоль) и татракис(трифенилфосфин)палладия(0) (0,012 г, 0,01 ммоль) в DME:вода (3:2, 3 мл) нагревали до кипения с обратным холодильником в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном. Органический слой промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали флэш-хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая указанное в заголовке соединение (0,015 г, 24%) в виде желтого твердого вещества.
1H ЯМР (CD3OD) д 8,47 (с, 1H), 8,35-8,33 (д, J=5,29 Гц, 1H), 8,16 (с, 1H), 8,01-8,00 (д, J=5,13 Гц, 1H), 7,94 (с, 1H), 7,78 (с, 1H), 7,45-7,43 (д, J=8,64 Гц, 1H), 7,35-7,33 (дд, J=10,33, 1,72 Гц, 1H), 7,29 (д, J=2,95 Гц, 1H), 6,48-6,47 (д, J=2,58 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 306 (M+H)+.
ПРИМЕР 24
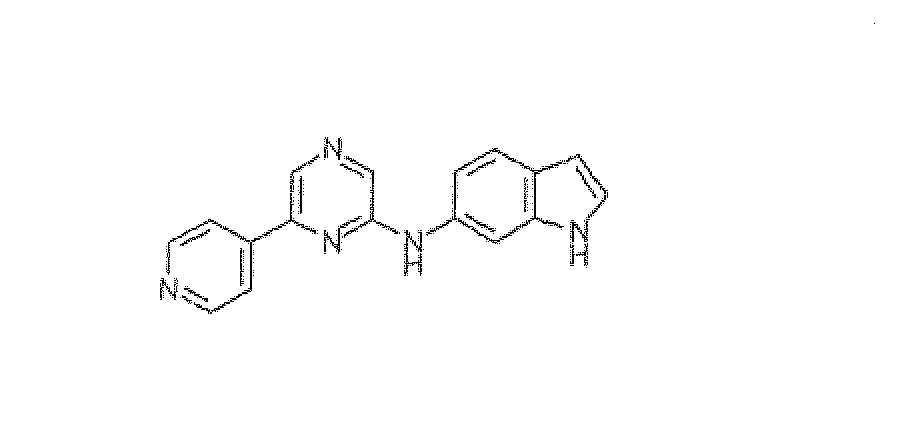
N-(6-Пиридин-4-илпиразин-2-ил)-1H-индол-6-амин
Смесь 2,6-дихлорпиразина (0,150 г, 1,006 ммоль), 6-аминоиндола (0,200 г, 1,51 ммоль), BINAP (0,0137 г, 0,02215 ммоль), трет-бутоксида натрия (0,136 г, 1,409 ммоль) и ацетата палладия (0,005 г, 0,02215 ммоль) в толуоле (8 мл) нагревали при 85°C в течение 16 ч в атмосфере азота. CH2Cl2 добавляли, реакционную смесь фильтровали через Celite и растворитель упаривали. Остаток очищали колоночной хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая 0,070 г (33%) промежуточного соединения (6-хлорпиразин-2-ил)-(1H-индол-6-ил)амин.
1H NMR (CDCl3) д 8,36 (ушир.с, 1H, NH), 8,08 (с, 1H), 7,92 (с, 1H), 7,64-7,59 (м, 2H), 7,23 (с, 1H), 7,01-6,98 (д, J=8,37 Гц, 1H), 6,87 (с, 1H, NH), 6,56 (с, 1H). Масс-спектр (API-ES/Positive); m/z: 245 (M+H)+.
Смесь (6-хлорпиразин-2-ил)-(1H-индол-6-ил)амина (0,070 г, 0,2868 ммоль), пиридин-4-бороновой кислоты (0,042 г, 0,344 ммоль), карбоната натрия (0,150 г, 1,43 ммоль) и тетракис(трифенилфосфин)палладия(0) (0,0165 г, 0,0143 ммоль) в DME:вода (3:2, 5 мл) нагревали при кипении с обратным холодильником в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном. Органический слой промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали флэш-хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая N-(6-пиридин-4-илпиразин-2-ил)-1H-индол-6-амин (0,030 г, 36,5%) в виде желтого твердого вещества.
1H ЯМР (CD3OD) д 8,73-8,72 (д, J=5,68 Гц, 2H), 8,51 (с, 1H), 8,23-8,20 (м, 4H), 7,56-7,54 (д, J=8,46 Гц, 1H), 7,22 (д, J=2,99 Гц, 1H), 7,14 (дд, J=10,18, 1,74 Гц, 1H), 6,45 (д, J=2,75 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 288 (M+H)+.
ПРИМЕР 25
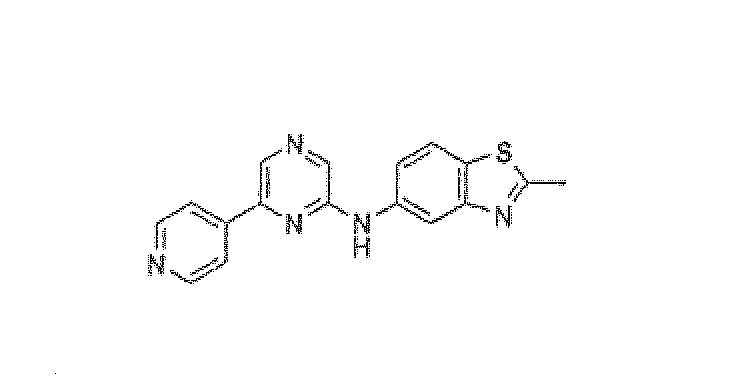
2-Метил-N-(6-пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин
Смесь 2,6-дихлорпиразина (0,150 г, 1,006 ммоль), 5-амино-2-метилбензотиазола (0,250 г, 1,51 ммоль), BINAP (0,0137 г, 0,02215 ммоль), трет-бутоксида натрия (0,136 г, 1,409 ммоль) и ацетата палладия (0,005 г, 0,02215 ммоль) в толуоле (8 мл) нагревали при 85°C в течение 16 ч в атмосфере аргона. CH2Cl2 добавляли, реакционную смесь фильтровали через Celite и растворитель упаривали. Остаток очищали колоночной хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая 0,180 г (65%) промежуточного соединения, (6-хлорпиразин-2-ил)-(2-метилбензотиазол-5-ил)амина.
1H ЯМР (CDCl3) д 7,59-7,54 (д, J=15,6 Гц, 2H), 7,38 (с, 1H), 7,19-7,16 (д, J=8,65 Гц, 1H), 6,98-6,95 (д, J=8,41 Гц, 1H), 6,71 (ушир.с, 1H, NH), 2,31 (с, 3H, CH3). Масс-спектр (API-ES/Positive); m/z: 277 (M+H)+. Смесь (6-хлорпиразин-2-ил)-(2-метилбензотиазол-5-ил)амина (0,075 г, 0,271 ммоль), пиридин-4-бороновой кислоты (0,040 г, 0,326 ммоль), карбоната натрия (0,143 г, 1,35 ммоль) и тетракис(трифенилфосфин)палладия(0) (0,0156 г, 0,0135 ммоль) в DME:вода (3:2, 5 мл) нагревали до кипения с обратным холодильником в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном. Органический слой промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали флэш-хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая 2-метил-N-(6-пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин (0,075 г, 86,5%) в виде желтого твердого вещества.
1H ЯМР (CD3OD) д 8,82 (м, 3H), 8,72 (с, 1H), 8,45-8,44 (м, 2H), 8,35 (с, 1H), 7,92-7,90 (д, J=8,67 Гц, 1H), 7,61-7,59 (дд, J=10,63, 1,94 Гц, 1H), 2,89 (с, 3H, CH3). Масс-спектр (API-ES/Positive); m/z: 320 (M+H)+.
ПРИМЕР 26
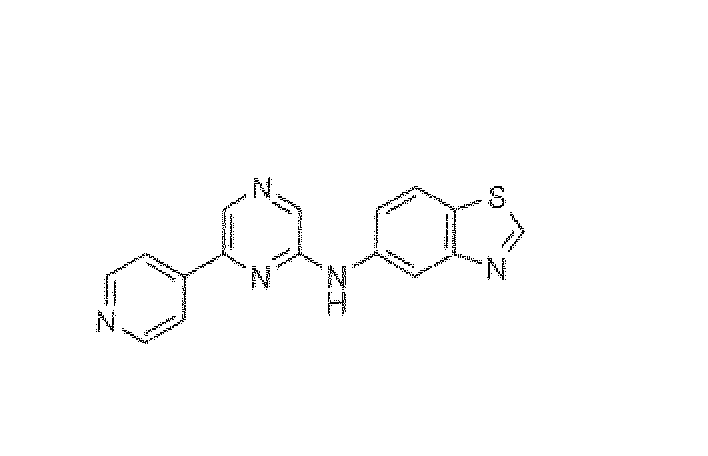
N-(6-Пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин
Смесь 2,6-дихлорпиразина (0,150 г, 1,006 ммоль), 5-аминобензотиазола (0,151 г, 1,006 ммоль), BINAP (0,0137 г, 0,02215 ммоль), трет-бутоксида натрия (0,136 г, 1,409 ммоль) и ацетата палладия (0,005 г, 0,02215 ммоль) в толуоле (8 мл) нагревали при 85°C в течение 16 ч в атмосфере азота. CH2Cl2 добавляли, реакционную смесь фильтровали через Celite и растворитель упаривали. Остаток очищали колоночной хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая 0,140 г (53%) промежуточного соединения, бензотиазол-5-ил-(6-хлорпиразин-2-ил)амина.
1H ЯМР (CDCl3) д 10,12 (с, 1H), 9,38 (с, 1H), 8,59 (с, 1H), 8,22 (с, 1H), 8,11-8,08 (д, J=8,67 Гц, 1H), 8,02 (с, 1H), 7,6-7,57 (д, J=8,67 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 263 (M+H)+.
Смесь бензотиазол-5-ил-(6-хлорпиразин-2-ил)амина (0,06 г, 0,228 ммоль), пиридин-4-бороновой кислоты (0,043 г, 0,342 ммоль), карбоната натрия (0,124 г, 1,14 ммоль) и тетракис(трифенилфосфина)палладия(0) (0,013 г, 0,0114 ммоль) в DME:вода (3:2, 5 мл) нагревали при кипении с обратным холодильником в течение 22 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном. Органический слой промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали флэш-хроматографией (5% метанол в CH2Cl2 в качестве элюента), получая N-(6-пиридин-4-илпиразин-2-ил)-1,3-бензотиазол-5-амин (0,035 г, 50%) в виде желтого твердого вещества.
1H ЯМР (CD3OD) д 9,31 (с, 1H), 8,98 (д, J=2,02 Гц, 1H), 8,75-8,74 (д, J=5,31 Гц, 2H), 8,64 (с, 1H), 8,31 (с, 1H), 8,24-8,22 (д, J=5,99 Гц, 2H), 8,06-8,04 (д, J=8,77 Гц, 1H), 7,74-7,71 (дд, J=10,73, 2,01 Гц, 1H). Масс-спектр (API-ES/Positive); m/z: 306 (M+H)+.
Биологические методы
Способность соединения изобретения ингибировать FLT3 может быть определена посредством использования испытаний in vitro и in vivo, известных из уровня техники. Некоторые испытания киназы in vitro по ингибированию FLT3 описаны в литературе и предусматривают применение клонированного киназного домена и оценку фосфорилирования субстратного пептида. Кроме того, клеточные линии, экспрессирующие FLT3, применяют для оценки воздействия на жизнеспособность и пролиферацию при клеточных испытаниях.
Испытание ингибирования фермента
Соединения, в соответствие с изобретением, характеризовали по ингибированию FLT3 ими с помощью нижеприведенных методов:
Испытание in vitro на FLT3 киназе
Испытание по ингибированию фермента для тирозинкиназного домена FLT3 осуществляли с помощью метода поляризации флуоресценции, Immobilized Metal Ion Affinity-Based Fluorescence Polarization (IMAP) из Molecular Devices.
Краткое изложение: киназную активность оценивали при инкубировании флуоресцирующего пептидного субстрата с киназным доменом. После завершения киназной реакции добавляли связывающий буфер. После фосфорилирования субстрата флуоресцирующий пептид приобретает способность связываться с покрытыми металлом наночастицами. В том случае когда субстрат связан с наночастицей, скорость вращения пептида снижена, и поэтому поляризация флуоресценции (fluorescence polarization (fp)) становится высокой. Соединения, ингибирующие киназную активность фермента, в результате будут приводить к низкой степени фосфорилирования субстрата и к низкому значению fp-сигнала.
Реагенты
Комплект буферных растворов IMAP с Progressive Binding System (Molecular Devices, #R8124):
Реакционный буфер: 10 мМ Трис-HCl pH 7,2 с 10 мМ MgCl2, 0,05% NaN3 и 0,01% Tween 20. Перед использованием DTT доводили до 1 мМ DTT конечной концентрации (готовый реакционный буфер).
Связывающий раствор готовили из комплекта буферных растворов в соответствие с рекомендациями производителя. Связывающий реагент разбавляли 1:1500 с использованием 40% связывающего буфера A и 60% связывающего буфера B.
Применяемый фермент FLT3 представлял собой рекомбинантную человеческую FLT3 от фирмы Upstate (#14-500) 7,2 Ед./мл, по N-концу меченный GST, аминокислоты 564 - конца.
Применяемый субстратный пептид: FAM-CSKtide от фирмы Molecular Devices (#R7269) 20 мкМ, 5FAM-KKKKEEIYFFFG-NH2.
ATP базовый раствор 10 мМ
DTT базовый раствор 100 мМ
Разбавители соединения: 0,01% Tween 20+1% DMSO в реакционном буфере. Реагенты разбавляли в готовом реакционном буфере до рабочих растворов.
Условия испытания
Конечные концентрации:
FLT3: 0,0125 Ед./мл (зависит от партии)
FAM-CSKtide: 100 нМ
ATP: 100 мкМ
Чувствительная доза соединения: одинадцать ступенчатых разбавлений 1:3, диапазон концентраций 25000-0,42 нМ, 5000-0,085 нМ, соответственно 500-0,0085 нМ в зависимости от эффективности соединения.
Протокол
I. Проведение киназной реакции в объеме 20 мкл в течение 1 ч:
Внесите пипеткой в 96-луночный планшет, черный на Ѕ площади:
5 мкл разбавленного соединения или среды
5 мкл субстратного пептида (400 нМ)
5 мкл фермента (0,05 Ед./мл) или готового реакционного буфера для неспецифического фонового значения (NSB)
5 мкл ATP (400 мкМ)
Закройте планшет и инкубируйте при комнатной температуре с легким перемешиванием
II. Инкубация для связывания в течение 2 ч (минимальное время):
Добавьте 60 мкл связывающего раствора.
Закройте планшет и инкубируйте при комнатной температуре с легким перемешиванием
III. Анализ поляризации флуоресценции:
Проведите измерения для флуоресцеина, используя устройство планшет-ридер (Analyst AD), длина волны возбуждающего излучения 485 и длина волны эмиссии 530, считывающее время интегрирования 0,1 сек (Alternatively Victor2 V Wallac 485/535 нм)
Исходные концентрации тестируемых соединений составляли 10 мМ в 100% DMSO. При испытаниях соединения тестировали в одной точке при 10 и 1 микромолярных концентрациях, разбавляли в реакционном буфере, как описано выше. Соединения с ингибирующей активностью более, чем 60% ингибированием при 1 микромолярной концентрации далее были протестированы в зависимости от дозы для определения IC50, используя одинадцать ступенчатых разбавлений 1:3 (обычный диапазон от 25000 нМ до 0,42 нМ, более эффективные соединения испытывали от 500 нМ до 0,0085 нМ). Значения IC50 расчитывали по уравнению (А+((В-А)/(1+((C/x)ΛD)))), в котором A равно min, B равно max, C равно IC50 и D равно тангенсу угла наклона графической кривой.
Соединения, в соответствии с изобретением, могут проявлять значения IC50 между 1 нМ и 2 мкМ (например, между 1 нМ и 1 мкМ, между 1 нМ и 500 нМ, между 1 нМ и 100 нМ, между 1 нМ и 25 нМ, между 1 нМ и 10 нМ).
В частности, IC50 N3-1H-индол-5-ил-5-пиридин-4-илпиразин-2,3-диамина (AKN-028) составляет 6 нМ.
Клеточные исследования
AML клеточная линия MV4-11 имеет FLT3-внутреннюю тандемную дупликацию. Эта клеточная линия широко используется для оценки воздействия ингибиторов FLT3-киназы на жизнеспособность и пролиферацию.
Клетки высевали с низкой плотностью в 96-луночные планшеты. Добавляли последовательные разведения соединений и клетки инкубировали в течение 72 часов. Общее количество жизнеспособных клеток определяли, используя проточную цитометрию, в конце обработки, и воздействие соединений расчитывали как % ингибирования в сравнении с обработанными средой клетками.
Условия культивирования клеток и культуры
Все клетки культивировали при стандартных условиях культивирования клеточной культуры, при 37°C и в атмосфере 5% CO2 при 90% влажности.
AML-клеточную линию MV4-11 культивировали в среде с высоким содержанием глюкозы DMEM Glutamax (4500 г/л глюкозы), дополненной 10% фетальной бычьей сывороткой (FBS) от Invitrogen. Клетки пересевали дважды в неделю, доращивая до плотности примерно 2 миллиона клеток на мл объема перед пересевом.
Исследования жизнеспособности и пролиферации
Для определения жизнеспособности 3000-5000 клеток высевали в 50 микролитров культуральной среды, в 96-луночный планшет. Последовательные разведения 1:3 растворов соединений от 10 мМ ДМСО (DMSO) базового раствора осуществляли в бессывороточной культуральной среде, дополненной пенициллином и стрептомицином. 50 микролитров растворов последовательных разведении добавляли к клеточной суспензии. Конечная концентрация соединений была от 5-микромолярной до 0,8 нМ, или от 500 нМ до 0,08 нМ, соответственно. Концентрацию ДМСО поддерживали постоянной, равной 0,05%.
В конце обработки 100 микролитров реагента для определения жизнеспособности (Guava ViaCount) добавляли в каждую лунку, и количество клеток, и их жизнеспособность определяли, используя проточную цитометрию (Guava 96-well ViaCount assay). Как правило, опыты с обработанными средой (0,05% ДМСО) клетками клеточной линии проводили в трехкратной повторности на протяжении всего эксперимента.
% выживаемости рассчитывали в сравнении с обработанными средой клетками в конце эксперимента.
Величины EC50 рассчитывали, используя уравнение (A+((B-A)/(1+((C/x)ΛD)))), в котором A равно min, B равно max, C равно IC50 и D равно тангенсу угла наклона графической кривой.
Результаты
|
|
Исследование in vitro при комбинации FLT3-ингибитора и химиотерапии
Последовательный ряд зависимости синергических активностей соединений формулы (I) и стандартных химиотерапевтических агентов, применяемых при лечении AML, устанавливали, как описано у Brown et al. (2006) Leukemia 20: 1368-1376, и результаты анализировали с использованием программного обеспечения Calcusyn Software, в соответствии с правилами Спои и Talalay (1981) Eur. J. Biochem.
Лиофилизированный препарат цитотоксических дипептидов
Лиофилизированные препараты мелфалана флуфенамида
Лиофилизированный препарат цитотоксических дипептидов
Лиофилизированные препараты мелфалана флуфенамида
Модифицированная сульфамидаза и способ её получения
С5-связующие полипептиды

