Результат интеллектуальной деятельности: СОЕДИНЕНИЯ, ИМЕЮЩИЕ АРИЛСУЛЬФОНАМИДНУЮ СТРУКТУРУ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ
Вид РИД
Изобретение
Область изобретения, к которой относится изобретение
Настоящее изобретение находит применение в области терапии, и, более конкретно, оно относится к арилсульфонамидосоединениям, способу их получения, содержащим их фармацевтическим композициям и их применению в качестве терапевтических агентов, особенно при лечении дегенеративных нарушений.
Уровень техники
Известно, что многие физиологические и патологические процессы характеризуются как значительной гиперпролиферацией, так и подвижностью клеток. Среди них имеются физиологические процессы, подобные, например, эмбриогенезу или развитию и дифференциации ткани, а также патологические процессы, среди которых имеются опухоли или, более обычно, нарушения, поражающие различные участки или органы тела: легкие, мышцы, кости, кожу, а также нервную, лимфатическую, желудочно-кишечную, почечную, макуло-глазную, сердечно-сосудистую систему и тому подобное.
При патологических или непатологических условиях высокая клеточная пролиферация и подвижность в основном зависит от активности металлопротеаз цинка, класса каталитических протеаз, присутствующих в организме людей (называемых также протеиназами), которые, как известно, координируют ионы цинка в их каталитическом сайте и которые способны гидролизовать амидные связи в пептидной цепи белков.
Среди металлопротеаз цинка имеются металлопротеазы внеклеточного матрикса (далее обозначаемые ММР), ADAMS (дизинтегрин и металлопротеазы) и ADAMT (дизинтегрин и металлопротеаза с повторами тромбоспондина типа I).
После продуцирования эти протеазы остаются прикрепленными на клеточных мембранах или экскретируются во внеклеточный матрикс (ЕСМ), важную физиологическую структуру, содержащую организованную трехмерную сетчатую структуру клеток окружающих тканей, которые электрически, химически и физически соединяются друг с другом.
Как таковые, они могут играть ключевую роль в некоторых внеклеточных процессах, включающих взаимодействия клетка-клетка и клетка-ЕСМ, а также в физиологических внутриклеточных процессах, например, росте, развитии и ремоделировании тканей, трансдукции внутри- и внеклеточных сигналов и феномене адгезии.
Протеолитическая активность этих металлопротеаз цинка в физиологических условиях значительно и тонко регулируется эндогенными ингибиторами, известными как тканевые ингибиторы металлопротеаз (TIMP), которые, как было обнаружено, играют фундаментальную роль также в регуляции активности ADAM и ADAMT.
Так, тонкое равновесие между ММР и их ингибиторами делает возможным надлежащее функционирование всех из физиологических процессов, в которых принимает участие ММР, таких как, например, эмбрионный рост и развитие, морфогенез тканей, миграция клеток и ремоделирование матриксов, репродуктивные процессы, т.е. менструальный цикл и овуляция, образование костей, адипогенез, заживление ран и ангиогенез или даже высвобождение и процессинг биоактивных молекул, как например, внутри- или межклеточные сигналы пептидов.
Вследствие их диффузии в теле человека и их роли в оказании усилия, таким образом очевидно, что любое изменение в регуляции даже одного из указанных выше процессов, например, из-за патологий, подобных опухолям, развитие которых может определить либо сверхэкспрессию, либо недостаточную экспрессию ММР, может привести, почти неизбежно, к возникновению дегенеративных процессов, приводящих к аномальному изменению и/или развитию тканей.
Примеры указанных выше патологий, в которых могут участвовать сверхэкспрессия или недостаточная экспрессия ММР, таким образом приводящая к измененной тканевой морфологии с нерегулируемой пролиферацией клеток, могут включать артрит и нарушения соединительных тканей; нейродегенеративные нарушения, такие как рассеянный склероз, болезнь Альцгеймера, удар и ALS (боковой амиотрофический склероз); сердечно-сосудистые нарушения, такие как атеросклероз, аневризма, сердечная недостаточность, застойная кардиомиопатия; легочные заболевания, такие как эмфизема или муковисцидоз; язвы желудка; сепсис и аутоиммунные нарушения.
Помимо этого, во время тканевой дегенерации измененная экспрессия этих протеаз цинка может также зависеть, например, от типа клеток, активации их проферментативных форм, путей генной транскрипции, а также механизмов экскреции и эндоцитоза. Внеклеточная и внутриклеточная пороговая величина активных металлопротеаз цинка часто регулируется на поверхности клеточной мембраны посредством устранения их каталитической активности другими металлопротеазами, подобными ММР, закрепленными на клеточной мембране, известными как ММР мембранного типа, альфа-конвертазой фактора некроза опухоли, более известной как ТАСЕ (и соответствующей ADAM-17), или даже другими ADAM или ADAMT.
Поэтому для терапевтических целей, когда имеют место патологические нарушения, характеризующиеся значительной активностью металлопротеаз на клеточной поверхности инвазивных и гиперпролиферирующих клеток, может быть желательным ингибирование этих МТ-ММР или некоторых других ADAM или ADAMT.
До сих пор по меньшей мере 23 разных фермента, которые, как известно, принадлежат к семейству ММР, были классифицированы на подклассы согласно их специфичности к субстратам. Известно, что среди них, например, ММР-1, ММР-8 и ММР-13 действуют на коллагеназу; с другой стороны, известно, что ММР-2 и ММР-9 действуют на желатиназу как на мишень, и известно, что ММР-3, ММР-10 и ММР-11 действуют на стромелизин как на мишень.
Помимо этого, к настоящему времени идентифицирована и характеризована четвертая подгруппа мембранного типа ММР, названная МТ-ММР, а именно, МТ1-ММР (ММР-14), МТ2-ММР (ММР-15), МТ3-ММР (ММР-16), МТ4-ММР (ММР-17), МТ5-ММР (ММР-24) и МТ6-ММР (ММР-26); оказываемое действие, однако, было выяснено только для некоторых из них (см. для ссылки HG Munshi et al., Cancer Metastasis Rev., 2006, 25, 45-56; и VS Golubkov et al., J. Biol. Chem, 2005, 280, 25079-25086).
Известно, например, что ММР-14 является ответственной за активацию про-ММР-2 на наружной поверхности некоторых типов клеток, например, клеток гладких мышц васкулярной ткани в ангиогенетических процессах (см. для ссылки N. Koshikawa et al., J. Cell. Biol. 2000, 148, 615-624; и Y. Itoh, H. Nagase, Essays in Biochemistry, 2002, 38, 21-36). Кроме того, известно, что ММР-14 гиперэкспрессируется на мембранах некоторых типов опухолевых клеток, таких как в меланомах (см. в качестве ссылки NE Sounni et al., Int. J. Cancer, 2002, 98, 23-28), аденокарциноме молочной железы (см. в качестве ссылки NE Sounni, et al., FASEB J 2002, 16, 555-564) и в глиомах (AT Belien, et al., J. Cell Biol 1999, 144, 373-384; и EI Deryugina, et al., Cancer Res, 2002; 62: 580-588).
ММР-14 может также активировать другие про-ММР, подобные про-ММР-13, гиперэкспрессия которых, как известно, коррелирует в некоторых типах клеток с опухолями, воспалением или сердечно-сосудистыми и нейродегенеративными нарушениями (см., например, AR Folgueras et al., Int. J. Dev. Biol., 2004, 48, 411-424; и JO Degushi, et al., Circulation, 2005, 2708-2715).
Известно, что даже другие ММР способствуют активации про-ММР-2, и/или про-ММР-13, и/или про-ММР-9. Известно, что, например, ММР-15, ММР-16, ММР-17 и ММР-24 активируют про-ММР-2 и про-ММР-13; ММР-17 действует только для активации про-ММР-2, тогда как ММР-26 активирует про-ММР-2 и про-ММР-9; см. в качестве ссылки AR Folgueras et al. (Int. J. Dev. Biol., 2004, 48, 411-424).
Кроме того, ММР-2 и ММР-13 продуцируются из пролиферирующих и инвазивных клеток и активируются или так или иначе становятся активируемыми в ЕСМ каталитической активностью ММ-ММР мембранной поверхности; поэтому они представляют собой предварительное средство для движения клеток через дигестивную проницаемую поверхность ЕСМ.
На основании предыдущих исследований по так называемым “деградомикам” (см. в качестве ссылки C Lopez-Otin et al., Nature Rev. 2002, 3, 509-519; и CM Overall et al., Nature Review Cancer, 2006, 6, 227-239) возможность нацеливания на некоторые ММР в качестве кандидатов для доставки лекарственного средства при раковых и других патологиях, но при исключении препятствий физиологическим функциям, вызываемых некоторыми другими ММР, в настоящее время широко подтверждается.
По существу исследования продолжают проводить для разработки ингибиторов ММР, которые можно применять в терапии и которые способны селективно воздействовать на патологические поражения без указанных выше недостатков.
Следовательно, когда активность некоторых ММР должна быть ингибирована, чтобы ограничить имеющий место дегенеративный процесс и противодействовать ему, активность некоторых других типов ММР, регулирующих физиологические процессы развития и морфогенеза, не должна быть ослаблена, что, как указано выше, может привести к нежелательным побочным действиям.
Известно, что среди них, например, мышечно-скелетный синдром с фибропролиферативными действиями в суставной капсуле колена имеет место при недостаточности нормальной активности ремоделирования ткани, вызванной ММР-1. Таким же образом, ингибирование некоторых ММР, принимающих участие в регуляции онкогенеза, таких как, например, ММР-3, может вызвать увеличение клеточной пролиферации и инвазии.
Поэтому в терапии отсутствие ингибирования ММР-1 и ММР-3, рассматриваемых как не являющихся мишенями ММР, представляется очень желательным.
В противоположность этому, обнаружено, что при явной селективности по отношению к ММР-2 и ММР-9 имеют место проапоптические действия на опухолевые клеточные культуры без проявления значительных побочных эффектов.
Следовательно, поиск молекул, способных специфически регулировать активность определенных ММР, когда утрачены нормальные механизмы, может обеспечить пригодные соединения для лечения некоторых заболеваний.
В данной области известны различные ингибиторы металлопротеаз, некоторые из которых относятся к сульфонамидопроизводным.
Среди них имеются, например, содержащие карбоциклическую боковую цепь N-замещенные ингибиторы металлопротеаз, описанные в WO 01/70720, и их фармацевтические композиции.
В US 6686355 описаны производные бифенила, содержащие циклическую азотсодержащую сульфонамидогруппу, в качестве ингибиторов ММР.
В WO 2004/069365 описаны диагностические агенты для получения изображения, содержащие ингибиторы металлопротеаз матрикса, имеющие сульфонамидогруппы, замещенные N,N-диалкильной цепью и должным образом меченные γ-испускающим радионуклидом.
В WO 98/39329 описаны производные сульфонамидогидроксамовой кислоты, специфически поражающие как мишени ММР-2, ММР-9 и ММР-13; приведенные в ней в качестве примеров несколько соединений содержат сульфонамидогруппы, замещенные боковой N,N-диалкильной цепью.
В US 7067670 описаны производные алкилсульфонамидогидроксамовых кислот, обладающие ингибирующей активностью в отношении ММР-2 и ММР-13.
В US 6500948 описаны пиридилокси- и пиридилтиоарилсульфонамидопроизводные, обладающие ингибирующей ММР активностью, у которых N-атом сульфонамидогруппы является частью шестичленного гетероцикла, содержащего атомы углерода, соседние с указанным выше N-атомом.
В US 6495568 описаны производные алкил- или циклоалкилсульфонамидогидроксамовых кислот в качестве ингибиторов металлопротеаз матрикса.
В US 5985900 описаны сульфонамидопроизводные, характеризующиеся фенил- или фенилен-SO2NH-частью, обладающие ингибирующий ММР активностью.
В US 99/42443 описаны производные сульфониламиногидроксамовой кислоты, содержащие группу арил-SO2NH-, которые охарактеризованы как разрушающие матрикс металлопротеиназы.
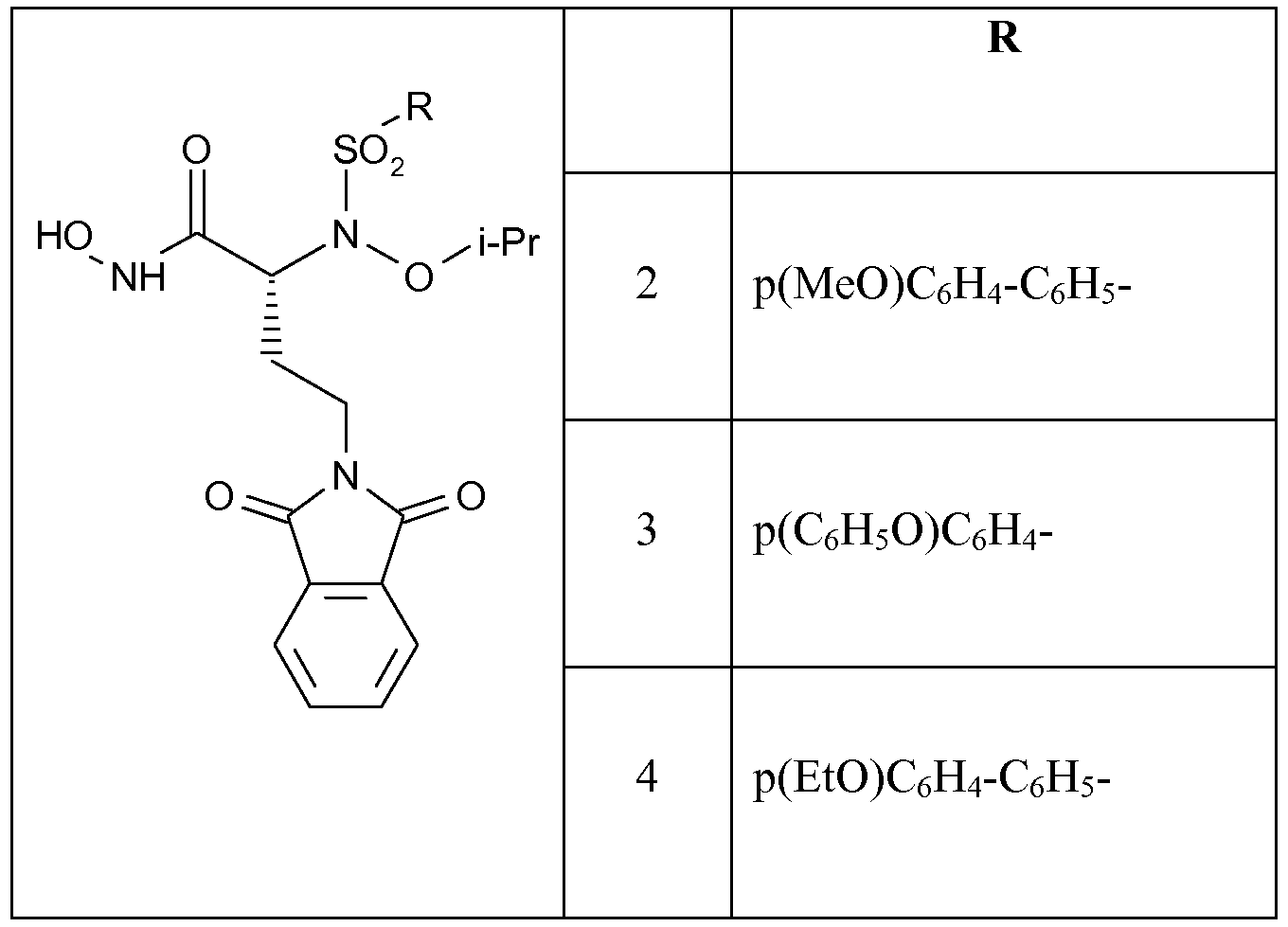
В данной области известны другие сульфонамидоингибиторы ММР. Среди них имеются селективные ингибиторы желатиназы А (ММР-2), описанные Rossello et al. в Bioorg. & Med. Chem. 12 (2004) 2441-2450 и имеющие указанную ниже формулу (А), где R представляет собой группу, выбранную из изопропила, аллила или п-(бензилокси)бензила.
Кроме того, ингибиторы ММР-2, проявляющие хорошую селективность в отношении ММР-2/ММР-1, были также описаны Tuccinardi et al. (Bioorg. & Med. Chem. 14 (2006) 4260-4276); среди описанных в этой публикации в качестве примеров соединений имеется производное указанной ниже формулы (В)
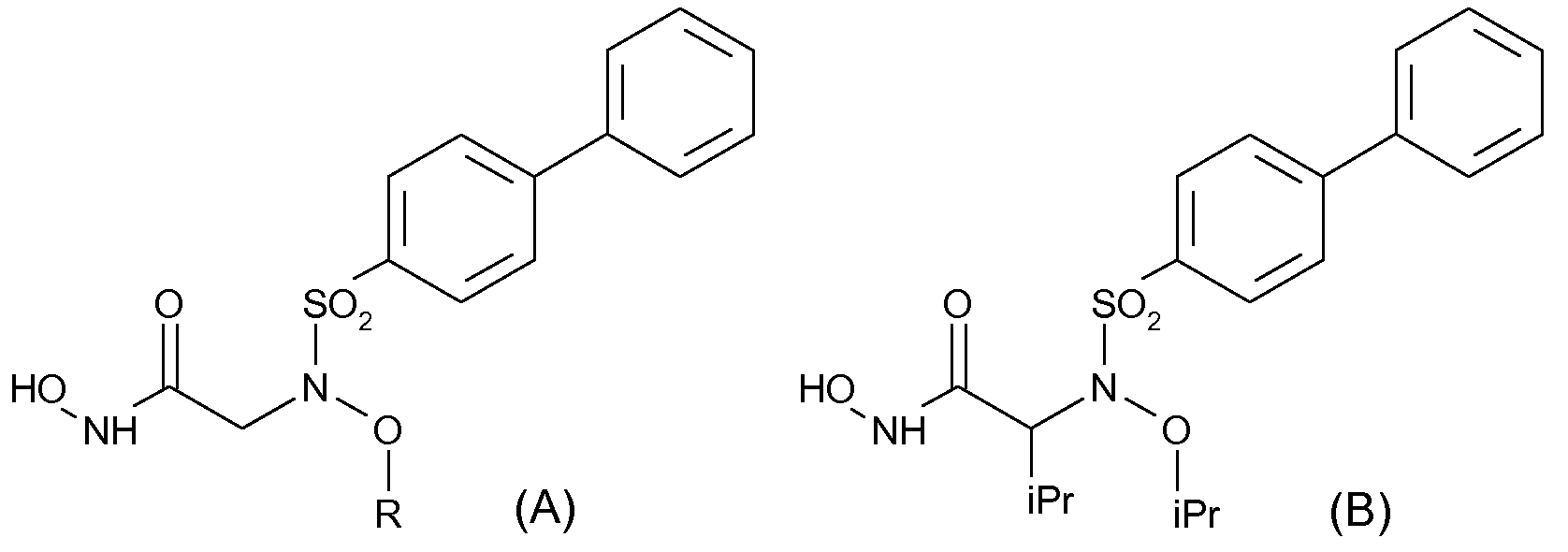
Помимо указанных выше публикаций, в предыдущих работах в данной области указывается, что применение селективных ингибиторов ММР-2, которые не действуют на некоторые ММР, такие как ММР-1 и ММР-3, позволяет блокировать прорастание (инвазию) клеток НТ1080 (очень инвазивной фибросаркомы) и клеток HUVEC (эндотелия вены пупка человека) на моделях хемоинвазии и ангиогенеза (A. Rossello, et al., Bioorg & Med. Chem., 2004, 12, 2441-2450; и A. Rossello, et al., Bioorg & Med. Chem. Lett., 2005, 15, 1321-1326).
Как указывалось в вышеупомянутой публикации Bioorg & Med. Chem. Lett., (2005), была разработана возможная модель антиангиогенеза и были синтезированы определенные соединения, обозначаемые в ней (5b) и (5с), формулы которых представлены ниже
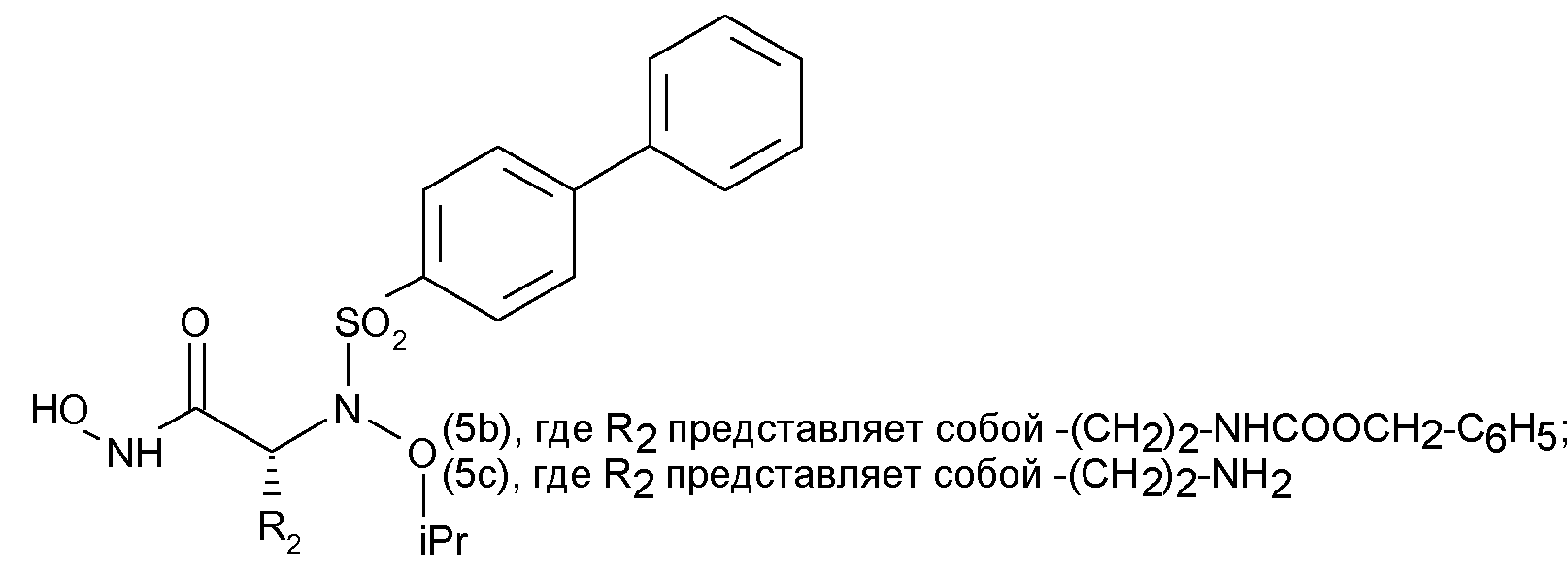
На основании результатов, полученных на выделенных ферментах и согласно способу, описанному CG Knight et al. (см. в качестве ссылки FEBS Lett. 1992, 296, 263; и Methods Enzymol. 1995, 248, 470), причем указанный способ включает применение флуорогенного субстрата FS-1 (например, Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2), как указывается Rossello et al., in Bioorg. Med. Chem. Lett. 12 (2004), 2441-2450, показано, что соединение (5b) является двойным ингибитором ММР-2 (величина IC50, соответствующая 0,41 нМ) и ММР-14 (величина IC50, соответствующая 7,7 нМ).
В этом отношении, если не оговорено особо, в нижеследующих фармакологических и экспериментальных разделах, вышеуказанное соединение (5b) известного уровня техники, здесь называется “ссылочным соединением (5b)”.
Кроме того, в вышеуказанных публикациях Bioorg. Med. Chem. Lett. 15 (2005), 1321-1326, и Bioorg. Med. Chem. Lett. 15 (2005), 2311-2314 описаны химические синтетические промежуточные продукты следующей формулы
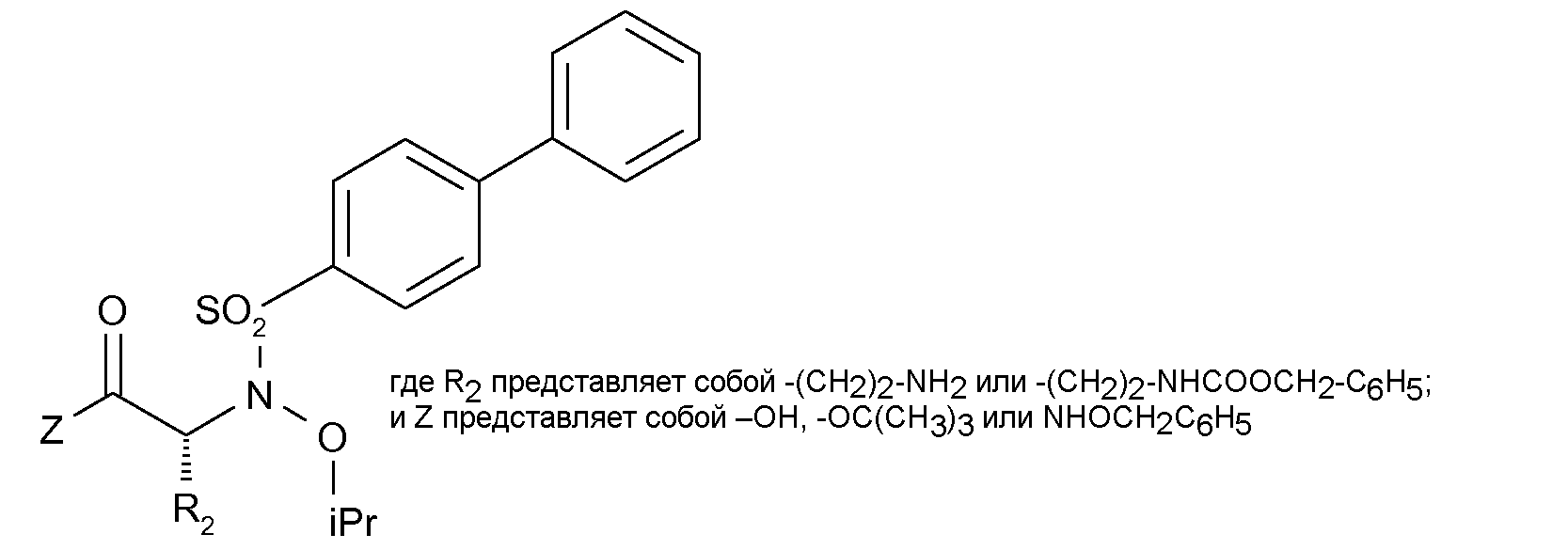
Авторами изобретения теперь обнаружен новый класс ингибиторов металлопротеазы Zn, имеющих арилсульфонамидную структуру, которые на основании испытаний на ингибирование имеют IC50 в молярном диапазоне нано/субнановеличин и в результате являются особенно эффективными в отношении мишеней-ферментов, в частности ММР-2, ММР-13 и ММР-14.
Задача изобретения
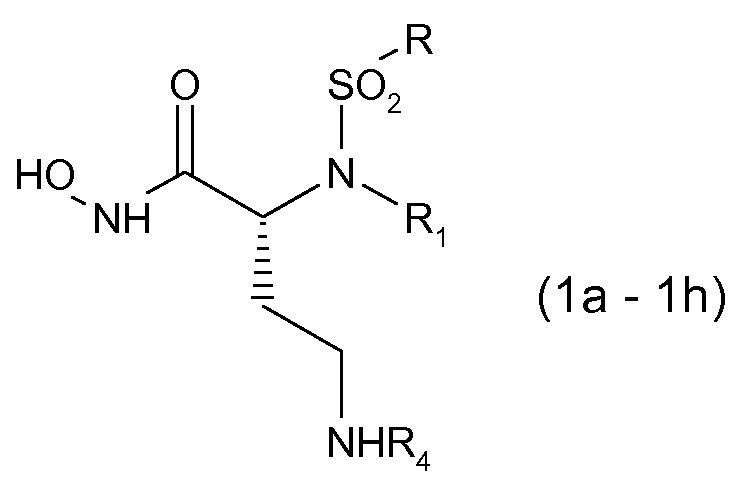
Таким образом, первой задачей настоящего изобретения является соединение, имеющее следующую общую формулу (I)
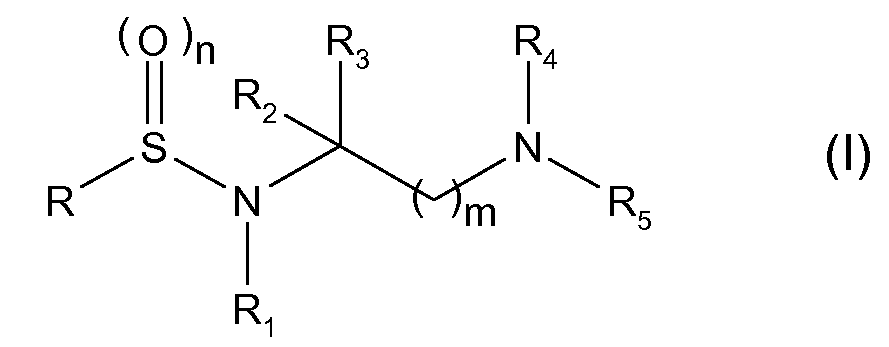
где
R представляет собой группу формулы -Ar-X-Ar' (II), где Ar представляет собой ариленовую, гетероариленовую, арильную или гетероарильную группу и Ar' является таким же или другим и независимо от Ar представляет собой арильную или гетероарильную группу или Н; причем указанные Ar и Ar' необязательно замещены одной или более группами, выбранными из
(i) неразветвленного или разветвленного алкила, алкокси, гидроксиалкила, алкоксиалкила, аминоалкила, алкиламино, аминоацила, ациламино или перфторированного алкила, каждый из которых имеет от 1 до 4 атомов углерода в алкильной цепи;
(ii) неразветвленной или разветвленной С2-С6алкенильной или алкинильной группы;
(iii) галогена или цианогруппы (-CN);
Х представляет собой одинарную связь или представляет собой двухвалентный линкер (соединяющую группу), выбранный из неразветвленной или разветвленной С1-С4алкиленовой цепи, -О-, -S-, -S(O)2-, -CO-, -NR'-, -NR'CO- или -CONR'-, где R' представляет собой Н или неразветвленную или разветвленную С1-С4алкильную группу;
R1 представляет собой -ОН или группу -ORa, где Ra выбран из неразветвленных или разветвленных С1-С4алкильных или С2-С4алкенильных групп; или Ra представляет собой группу формулы (III)
-(CH2)p-Z-(CH2)r-W (III)
где р равно нулю или целому числу от 1 до 4; Z представляет собой одинарную связь или двухвалентный линкер, выбранный из -О-, -NR'-, -NR'CO- или -CONR'-, где R' имеет значения, указанные выше; r равно нулю или целому числу от 1 до 4 и W представляет собой фенил или 5- или 6-членный гетероцикл, каждый из которых необязательно замещен одной или более группами, выбранными из -NH2, -COR', -CONHR', -COOR' или -SO2NHR', где R' имеет значения, указанные выше, арилом или гетероарилом, или одной или более вышеуказанными группами от (i) до (ii);
R2 и R3 являются одинаковыми или разными и каждый независимо представляет собой Н, неразветвленную или разветвленную С1-С4алкильную группу, необязательно замещенную гидроксилом или С1-С4алкоксигруппами, или связывающую цинк группу, выбранную из -COOH, -COORb, -CONHOH, -CONHORb, -CONRbOH, -CONHS(O)2Rb, -CONH2, -CONHRb или -Р(О)(ОН)2, где Rb представляет собой неразветвленную или разветвленную алкильную, арилалкильную или гетероарилалкильную группу, имеющую от 1 до 4 атомов углерода в алкильной цепи; или любая из вышеуказанных групп R2 или R3 связывается с R1 так, что образовывалось 5-7-членное гетероциклическое кольцо, содержащее по меньшей мере два соседних гетероатома N-O, необязательно замещенных одной или более оксогруппами (=О);
R4 представляет собой Н или группу, выбранную из -CORc, -COORc, -S(O)2Rc, -CONHRc или -S(O)2NHRc, где Rc представляет собой группу, выбранную из С3-С6циклоалкила, неразветвленного или разветвленного алкила, арила, гетероарила, арилалкила, гетероарилалкила, алкиларила, алкилгетероарила, 5- или 6-членного гетероциклила, алкилгетероциклила или гетероциклоалкила, имеющего от 1 до 4 атомов углерода в алкильной цепи;
R5 представляет собой Н или R4 и R5 вместе с атомом N, к которому они присоединены, образуют необязательно бензоконденсированный 4-6-членный гетероцикл, необязательно замещенный группой Ra, которая имеет значения, указанные выше, и/или одной или более оксогруппами (=О);
n равно 1 или 2;
m равно целому числу от 1 до 6;
при условии, что, когда R представляет собой бифенил-4-ил, R1 представляет собой изопропокси, m и n, оба, равны 2, R5 представляет собой Н, один из R2 или R3 представляет собой -COOH, -COOC(CH3)3, -CONHOH или -CONHOCH2C6H5 и другой из R2 или R3 представляет собой Н, то R4 не является Н или бензилоксикарбонилом;
и его фармацевтически приемлемые соли.
Соединения формулы (I) могут иметь один или более асимметричных атомов углерода, иначе называемых хиральными атомами углерода, и поэтому могут существовать в форме индивидуальных энантиомеров, рацематов, диастереоизомеров и любой их смеси, предполагается, что все они включены в объем настоящего изобретения.
Как указано выше, в соединениях формулы (I) изобретения R представляет собой группу формулы -Ar-X-Ar' (II), где Ar представляет собой ариленовую или гетероариленовую группу, связанную с -Х-Ar', или когда Х представляет собой одинарную связь и Ar' представляет собой Н, арильную или гетероарильную группу.
В данном контексте, несмотря на тот факт, что в настоящее время предполагается, что арилен и гетероарилен обозначают двухвалентную группу (например, фенилен-С6Н4-), оба термина «арил» и «арилен» (и, таким образом, «гетероарил» и «гетероарилен») используют в описании взаимозаменяемым образом, если не оговорено особо.
В настоящем описании, если не оговорено иначе, авторы изобретения предполагают, что термин «арильная группа» (и, таким образом, «ариленовая группа») означает карбоциклическую ароматическую группу.
Авторы изобретения предполагают, что термин «гетероарил» (и, таким образом, «гетероарилен») означает 5- или 6-членный ароматический гетероцикл с 1-3 гетероатомами или гетероатомными группами, выбранными из N, NH, O или S.
Подходящие примеры арильных или гетероарильных групп, относящихся к Ar и Ar', а также к любой другой арильной или гетероарильной группе, присутствующей в соединениях формулы (I), могут таким образом включать фенил, пирролил, фурил, тиенил, пиразолил, имидазолил, оксазолил, изоксазолил, пиридил, пиримидинил, тиазолил и тому подобное.
Что касается формулы (II), специалисту в данной области ясно, что как Ar, так и Ar', могут быть непосредственно соединены друг с другом, когда Х представляет собой одинарную связь, так что образуется группа -Ar-Ar'-, или в альтернативном случае они могут быть связаны друг с другом через любой подходящий двухвалентный линкер среди линкеров, указанных выше, таким образом образуя, например группы R, соответствующие -Ar-O-Ar'-, Ar-S-Ar', -Ar-CONR'-Ar' и тому подобное.
Помимо этого, при указании на Ar', как соответствующую атому водорода Н, любую из вышеуказанных групп R можно идентифицировать соответственно самим -Ar (когда Х представляет собой связь) или группой -Ar-OH, -Ar-SH, -Ar-CONR'H и тому подобное.
Как ранее указывалось, любая из вышеуказанных групп Ar и/или Ar' может быть необязательно дополнительно замещена в любом свободном положении одной или более группами, указываемыми в пунктах от (i) до (iii).
Среди необязательных заместителей, если не оговорено особо, авторы изобретения предполагают, что неразветвленным или разветвленным алкилом с 1-4 атомами углерода в цепи является любая из С1-С4алкильных групп, таким образом включающая метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
Таким же образом, при указании на алкокси авторы изобретения предполагают любые соответствующие алкилоксигруппы, такие как метокси, этокси, н-пропокси, изопропокси и тому подобное.
Как указано выше, алкильные группы могут быть дополнительно замещены гидроксилом (-ОН), амино (-NH2) или даже указанными выше алкоксигруппами (-OAlk), так что образуется гидроксиалкил (НО-Alk-), аминоалкил (H2N-Alk-) или алкоксиалкильные группы (Alk-O-Alk-), соответственно.
По аналогии, термины «алкиламино» авторы изобретения относят к аминогруппе, которая дополнительно замещена любой из вышеуказанных алкильных групп, так что образуются группы Alk-NH-.
При применении термина «ацил», если не оговорено особо, авторы изобретения предполагают любую из групп, обычно идентифицируемых как группы Alk(CO)-, где остаток Alk представляет собой только любую неразветвленную или разветвленную С1-С4алкильную группу.
Подходящие примеры ацильных групп могут таким образом включать ацетил (CH3CO-), пропионил (CH3CH2CO-), бутирил [СН3(СН2)2СО-], изобутирил [(CH3)2CHCO-], валерил [CH3(CH2)3CO-] и тому подобное.
На основании вышеуказанного, аминоацильные группы могут таким образом включать H2NCO-, а также любую из вышеуказанных ацильных групп, у которых алкильная цепь должным образом замещена амино, таких как, например, аминоацетил (H2NCH2CO-), аминопропионил [H2NCH2CH2CO- или CH3CH(NH2)CO-] и тому подобное.
По аналогии, если не оговорено особо, ациламиногруппы могут быть подходящим образом представлены карбоксамидогруппами, в которых любая из вышеуказанных ацильных групп связана с -NH-, такими как, например, ацетамидо (CH3CONH-), пропионамидо (CH3CH2CONH-), бутирамидо [CH3(CH2)2CONH-] и тому подобное.
При применении термина «перфторированный алкил» авторы изобретения предполагают любые из вышеуказанных алкильных групп, у которых все атомы водорода заменены атомами фтора, подобные, например, группам трифторметил, -C2F5, C3F7 или C4F9.
При применении термина «неразветвленная или разветвленная С2-С6алкенильная или алкинильная группа», авторы изобретения предполагают любые из С2-С6углеводородных цепей, содержащих по меньшей мере одну двойную связь или тройную связь, соответственно.
Подходящие примеры алкенильных или алкинильных групп согласно изобретению таким образом включают винил, аллил, пропенил, изопропенил, бутенил, изобутенил, гексенил, этинил, пропинил, бутинил и тому подобное.
Наконец, при применении термина «атом галогена» авторы изобретения предполагают любой из атомов фтора, хлора, брома или йода.
Согласно первому варианту осуществления в соединениях формулы (I) R представляет собой группу формулы (II), где Ar представляет собой необязательно замещенную фениленовую группу, Х представляет собой одинарную связь или двухвалентный линкер, выбранный из -О-, -S- или -NH-, и Ar' представляет собой Н или необязательно замещенную фенильную группу.
Предпочтительными заместителями в этом классе являются неразветвленные или разветвленные С1-С4алкильные группы или алкоксигруппы или атомы галогена.
Еще более предпочтительно, в соединениях формулы (I) R представляет собой группу формулы (II), в которой Ar представляет собой фениленовую группу, Х представляет собой одинарную связь или -О- и Ar' представляет собой Н или фенильную группу, причем фениленовая и фенильная группы необязательно замещены неразветвленными или разветвленными С1-С4алкильными группами или алкоксигруппами или атомами галогена.
Еще более предпочтительными в этом классе являются соединения формулы (I), в которой R представляет собой группу, выбранную из бифенил-4-ила, 4-бромфенила, 4-(4'-метоксифенил)фенила, 4-(4'-этоксифенил)фенила, 4-феноксифенила, 4-(4'-метоксифенокси)фенила и 4-(4'-этоксифенокси)фенила.
Согласно другому аспекту изобретения, в соединениях формулы (I) R1 представляет собой группу -ОН или -ORa, где Ra представляет собой алкил или алкенил, или R1 представляет собой группу формулы (III), где p, Z и r имеют значения, указанные выше, и W представляет собой фенил или 5- или 6-членный гетероцикл, каждый из которых необязательно дополнительно замещен, как указано выше.
В настоящем изобретении, если не оговорено особо, при применении термина «5- или 6-членный гетероцикл» или «гетероциклическая группа» авторы изобретения предполагают любой 5- или 6-членный ароматический или неароматический гетероцикл, потому включающий насыщенные, частично ненасыщенные или даже полностью ненасыщенные кольца с 1-3 гетероатомами или гетероатомными группами, выбранными из N, NH, O или S.
Из вышеуказанного специалисту в данной области ясно, что указанное выше определение гетероцикла или гетероциклической группы включает также любой полностью ненасыщенный гетероцикл, известный также как гетероарильная группа.
Подходящие примеры гетероциклических групп, не включающие гетероциклические группы, уже указанные как находящиеся в пределах определения гетероарила, могут таким образом содержать тетрагидрофуран, пирролин, пирролидин, морфолин, тиоморфолин, пиперидин, пиперазин и тому подобное.
Согласно предпочтительному варианту осуществления изобретения, в соединениях формулы (I) R1 представляет собой группу -ORa, где Ra представляет собой алкильную или алкенильную группу, указываемую выше, или R1 представляет собой группу формулы (III), где р равно 1 или 2, Z представляет собой одинарную связь или двухвалентную группу, выбранную из -О- или -NH-, r равно 0, 1 или 2 и W представляет собой необязательно замещенную фенильную или гетероциклическую группу, которая имеет указанные выше значения.
Еще более предпочтительными в данном классе являются соединения формулы (I), в которой R1 выбран из изопропокси, бензилокси, 4-фенилбензилокси, аллилокси, 2-[2-(пиперазинил-1-ил)этокси]этокси или 2-[2-(4-(этилкарбонил)пиперазинил-1-ил)этокси]этокси.
Что касается R2 и R3 в формуле (I), они независимо представляют собой Н, необязательно замещенную алкильную группу или связывающую цинк группу среди групп, указанных ранее.
Альтернативно, R2 или R3 может быть связан с R1 так, чтобы образовывать 5-7-членное гетероциклическое кольцо, содержащее по меньшей мере два соседних гетероатома N-O, причем, например, N-атом связан с S в формуле (I) и атом О является частью самого R1.
В качестве неограничивающего примера подходящие соединения формулы (I), в которой один из R2 или R3 (например, R2) связан с R1 так, чтобы образовывать вышеуказанные гетероциклические структуры, могут таким образом содержать
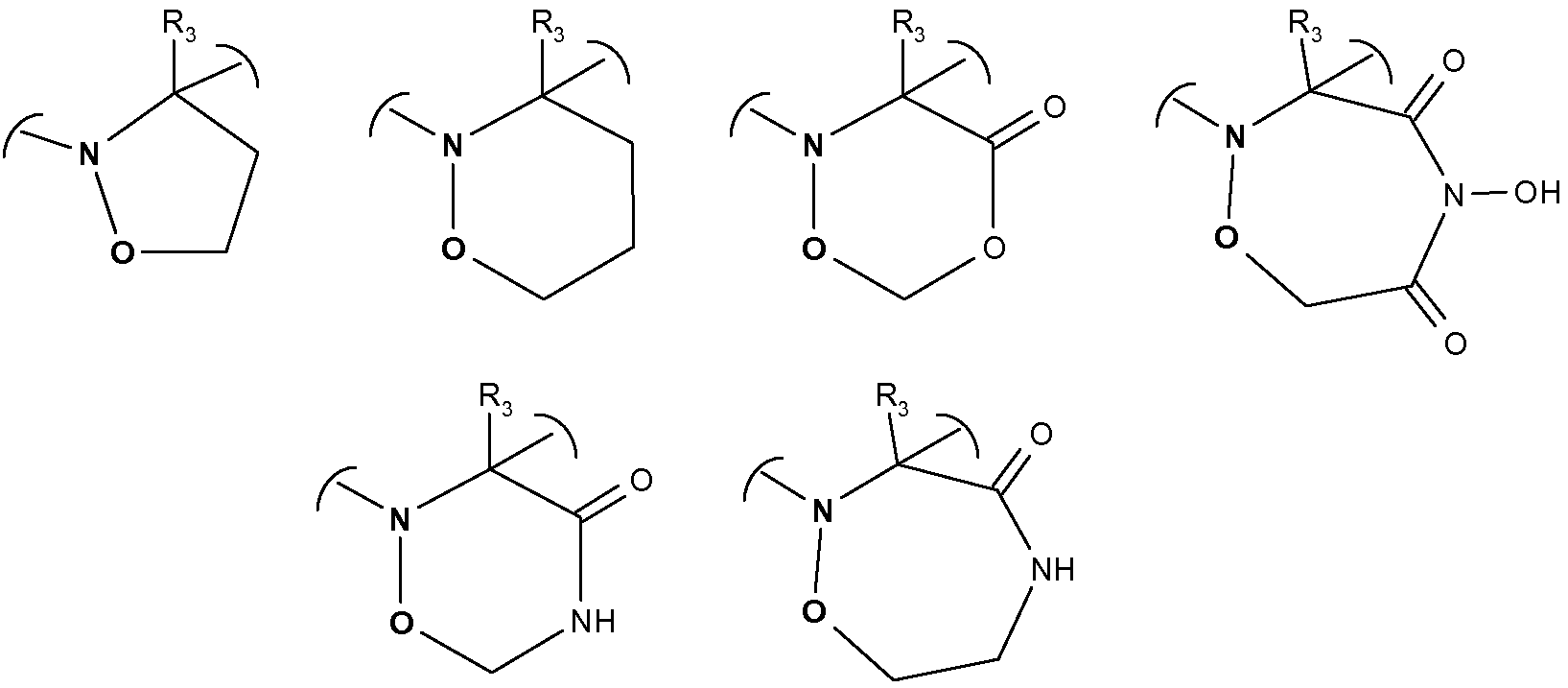
где вышеуказанные соседние гетероатомы N-O представлены отчетливо.
Согласно предпочтительному варианту осуществления, R2 и R3, каждый независимо, выбраны из Н, неразветвленного или разветвленного С1-С4алкила, -СООН, -COORb, -CONHOH или -CONHORb, где Rb представляет собой неразветвленную или разветвленную алкильную, арилалкильную или гетероарилалкильную группу, имеющую от 1 до 4 атомов углерода в алкильной цепи.
Еще более предпочтительно, в этом классе соединений R2 и R3 представляют собой, оба, атомы Н или один из них представляют собой Н и другой из R2 или R3 представляет собой -СООН или -CONHOH.
Что касается значений R4, указанная группа может представлять собой атом водорода Н или карбонил, карбоксил, сульфонил, карбоксамидо- или сульфонамидогруппу, дополнительно превращенную в производное посредством вышеуказанных групп Rc.
Что касается значений Rc, если не указано особо, при применении термина «С3-С6циклоалкил» авторы изобретения предполагают любое 3-6-членное циклоалифатическое кольцо, такое как циклопропил, циклобутил, циклопентил и циклогексил.
Из указанных выше определений значений алкила, арила, гетероарила и гетероцикла (или гетероциклила) любая группа с комбинированным названием, такая как арилалкил, алкиларил, гетероарилалкил, алкилгетероарил, алкилгетероциклил или гетероциклилалкил, должна быть понятна специалисту в данной области.
Просто в качестве примера, если не оговорено особо, при применении термина «алкиларил» авторы изобретения предполагают любую арильную группу, дополнительно замещенную алкилом, например, п-этилфенил (пС2Н5-C6H4-); термин «арилалкил» авторы изобретения вместо этого относят к алкильной группе, дополнительно замещенной арилом, например, 2-фенилэтил (С6Н5-СН2-СН2-) и тому подобное.
Из всего указанного выше специалисту в данной области должно быть понятно, что аналогичное рассмотрение можно применять для гетероарилалкильных, алкилгетероарильных, гетероциклоалкильных или алкилгетероциклических групп.
Что касается R5 в формуле (I), он представляет собой Н, или, альтернативно, R4 и R5 вместе с атомом N, с которым они связаны, образуют необязательно бензоконденсированный 4-6-членный гетероцикл, который имеет указанные выше значения.
Подходящие примеры указанных гетероциклов, например, замещенных Ra и оксогруппами, могут таким образом включать
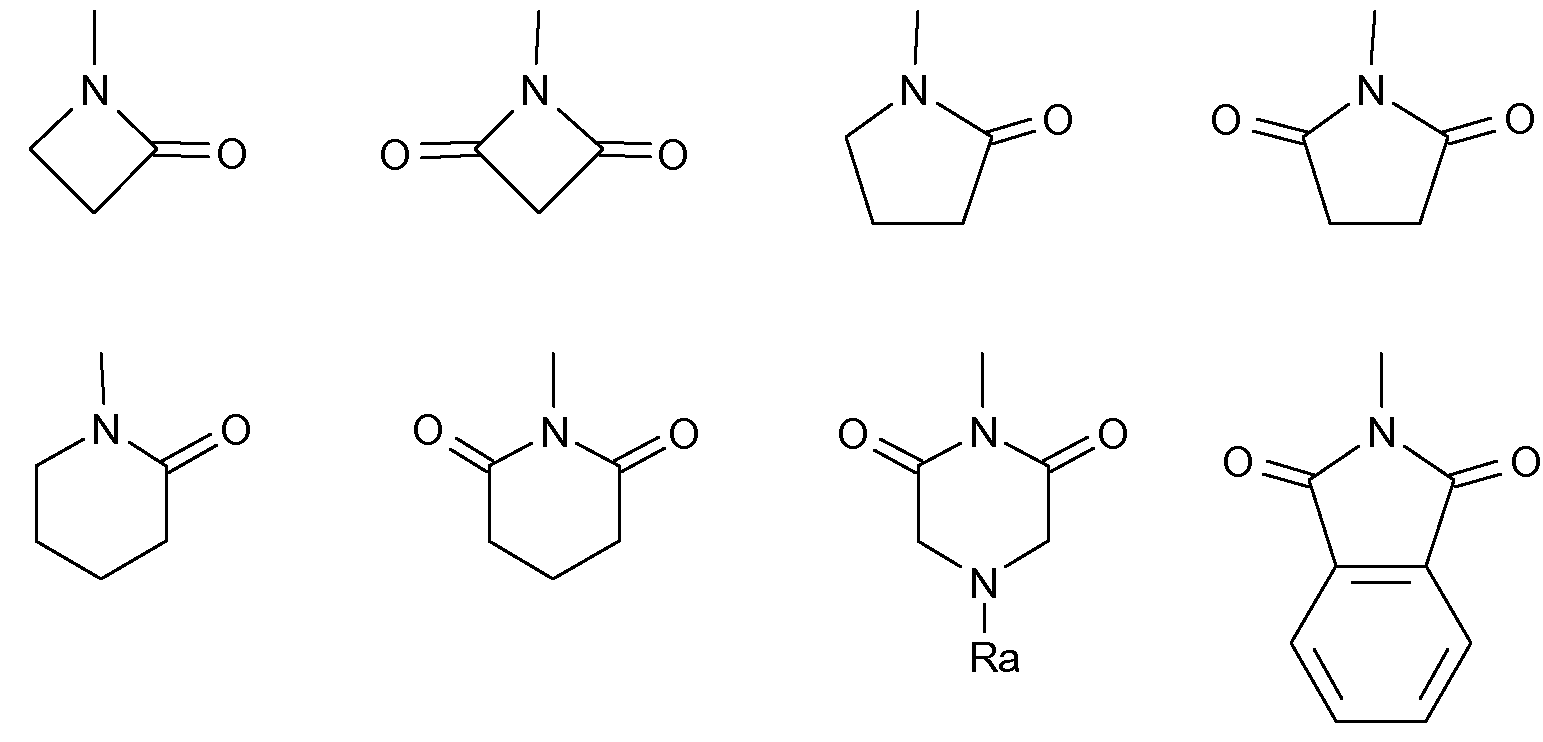
Согласно дополнительному предпочтительному варианту осуществления изобретения, R4 выбран из -CORc, -COORc или -S(O)2Rc, где Rc представляет собой арильную, неразветвленную или разветвленную алкильную или арилалкильную группу, имеющую от 1 до 4 атомов углерода в алкильной цепи, и R5 представляет собой Н.
Еще более предпочтительными в данном классе являются соединения формулы (I), в которой R4 выбран из ацетила, бензоила, фенацетила, 4-фенилбутаноила, бензилоксикарбонила, метансульфонила, фенилсульфонила или бензилсульфонила и R5 представляет собой Н.
Согласно еще одному другому варианту осуществления изобретения, R4 и R5 вместе с атомом N, к которому они присоединены, образуют N-фталимидогруппу.
Наконец, согласно дополнительному предпочтительному варианту осуществления изобретения, в соединениях формулы (I) n и m, оба, равны 2.
Как указывалось ранее, соединения формулы (I) изобретения могут также присутствовать в форме фармацевтически приемлемой соли.
Термин “фармацевтически приемлемая соль”, применяемый в настоящем описании, относится к производным соединений изобретения, у которых исходное соединение подходящим образом модифицировано превращением любой из свободных кислотных или основных групп, если она присутствует, в соответствующую аддитивную соль с любым основанием или кислотой, которая обычно предполагается как фармацевтически приемлемая.
Не говоря о том, что соответствующие соли соединений формулы (I) изобретения являются нетоксичными, они характеризуются также высокой стабильностью, включающей физиологическую стабильность при применении и введении.
Подходящие примеры упомянутых солей могут таким образом включать минеральные или органические кислотно-аддитивные соли основных остатков, таких как аминогруппы, а также минеральные или органические основно-аддитивные соли кислотных остатков, таких как карбоксильные, фосфоновые или серные группы.
Предпочтительные катионы неорганических оснований, которые можно подходящим образом применять для получения солей в изобретении, содержат ионы щелочных или щелочноземельных металлов, таких как калий, натрий, кальций или магний. Предпочтительные катионы органических оснований включают, среди прочих, катионы первичных, вторичных и третичных аминов, таких как этаноламин, диэтаноламин, морфолин, глюкамин, N-метилглюкамин, N,N-диметилглюкамин.
Предпочтительные анионы неорганических кислот, которые можно подходящим образом применять для получения солей соединений изобретения, включают ионы галогенов, такие как хлориды, бромиды, иодиды или другие подходящие ионы, такие как сульфаты.
Предпочтительные анионы органических кислот включают анионы, обычно применяемые в фармацевтических технологиях для получения солей основных веществ, таких как, например, ацетат, трифторацетат, сукцинат, цитрат, фумарат, малеат или оксалат.
Предпочтительные аминокислоты, которые можно подходящим образом применять для получения солей соединений изобретения, могут включать также, например, таурин, глицин, лизин, аргинин, орнитин, аспарагиновую и глутаминовую кислоты.
Определенные примеры соединений формулы (I) изобретения вместе со способом их получения описаны в нижеследующем экспериментальном разделе. Способ получения соединений изобретения можно проводить согласно общепринятым способам, хорошо известным специалистам в области методик синтетической органической химии.
Следовательно, дополнительной задачей изобретения является способ получения соединений формулы (I), который включает
а) реакцию соединения формулы (IV) с соединением формулы (V)

где R, R1, R2, R3, R4, R5, n и m имеют значения, указанные выше, так чтобы получить соединение формулы (I) изобретения, и необязательно
b) превращение соединения формулы (I), полученного на стадии (а), в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
Указанный выше способ является особенно подходящим, когда он является восприимчивым к модуляции должным образом посредством любого подходящего варианта, так чтобы получить любые требуемые соединения формулы (I).
На стадии (а) способа реакцию между соединениями формулы (IV) и (V) проводят согласно общепринятым способам для образования сульфонамидо- или сульфинамидогрупп с n, равным 2 или 1, соответственно.
Реакцию обычно проводят в известных рабочих условиях конденсации Мицунобу, в присутствии подходящих растворителей, включающих, среди других растворителей, тетрагидрофуран, дихлорметан, ацетонитрил, N-метилпирролидон, бензол, толуол, м-ксилол и их смеси.
В этом отношении указанную выше реакцию можно проводить в присутствии подходящего агента конденсации, либо как такового, либо подходящим образом нанесенного на полимерные смолы, включающего, например, диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD), 1,1'-азодикарбонилдипиперидин (ADDP), N,N,N',N'-тетраметилазодикарбоксамид (TMAD), трифенилфосфин (PPh3), трибутилфосфин (PBu3) и тому подобное.
Из всего указанного выше специалисту в данной области должно быть ясно, что на стадии (а) способа любая из групп R, R1, R2, R3, R4 и R5, в дальнейшем коротко обозначаемых группами “R”, может присутствовать как таковая или же может присутствовать в любой нужным образом защищенной форме.
Более конкретно, функциональные группы, которые присутствуют в любом из соединений формулы (IV) или (V) и которые могут вызывать нежелательные побочные реакции и образование побочных продуктов, необходимо нужным образом защитить перед проведением реакции конденсации. Кроме того, после завершения упомянутых реакций может следовать последующее удаление тех же защитных групп.
В настоящем изобретении, если не оговорено особо, термин “защитная группа” означает защитную группу, адаптированную для защиты функциональной группы, с которой она связана. В частности, защитные группы применяют для сохранения аминогрупп, гидроксильных или карбоксильных функциональных групп. Подходящие защитные группы поэтому могут включать, например, бензил, бензилоксикарбонил, алкиловые или бензиловые сложные эфиры или другие заместители, обычно применяемые для защиты таких функциональных групп, которые все хорошо известны специалистам в данной области [см. для общей ссылки T. W. Green; Protective Groups in Organic Synthesis (Wiley, N.Y. 1981)].
Кроме того, селективную защиту или снятие защиты у любой из этих групп, например, включающих карбоксил, гидроксил и аминогруппы, можно все выполнить согласно очень хорошо известным способам, обычно применяемым в органической синтетической химии.
Помимо указанного выше, как в стадии (b) способа, любую из групп “R” в конечных соединениях формулы (I), которую можно легко идентифицировать как превращенную в производное группу, например, любой сложный эфир или амид, можно получить из функциональной группы, производным которой она является, обработкой согласно общепринятым способам.
В качестве неограничивающего примера, например, в случае получения соединения формулы (I), в которой R2 представляет собой группу -COORb, Rb представляет собой алкильную группу и R3 представляет собой Н, такое же соединение можно получить согласно настоящему способу: (i) исходя из соединения формулы (V), где R2 и R3 имеют значения, указанные выше, как в стадии (а); или, альтернативно, (ii) исходя из соответствующего соединения формулы (I), где R2 представляет собой -СООН, превращением должным образом карбоксильной группы в требуемую группу -COORb, как в стадии (b) способа.
Условия вышеуказанных реакций являются хорошо известными в области получения эфиров карбоновых кислот.
Аналогичные соображения можно применять, например, при получении карбоксамидов реакцией должным образом соответствующего карбоксильного производного с любым подходящим амином и обработкой согласно хорошо известным рабочим условиям.
Таким же образом, например при получении соединения формулы (I), у которого R2 представляет собой гидроксамовую группу -CONHOH, способ можно проводить сначала путем взаимодействия соединения формулы (I), где R2 представляет собой карбоксил, полученного на стадии (а), в присутствии подходящих реагентов, подобных, например, О-(трет-бутилдиметилсилил)гидроксиламину, О-(тетрагидро-2Н-пиран-2-ил)гидроксиламину, О-тритилгидроксиламину или О-бензилгидроксиламину, в зависимости от того, какой может быть случай.
Последующее снятие защиты у полученного промежуточного производного, например кислотным гидролизом в присутствии трифторуксусной кислоты или обработкой триметилсилилтрифлатом, или каталитическим гидрированием в случае О-бензилгидроксиламина, может привести к получению требуемого соединения с R2, представляющим собой -CONHOH.
В данной области известно несколько дополнительных способов, позволяющих превратить данную группу в соединение формулы (I) в другую группу. Они могут включать, например, превращение аминогруппы (-NH2) или карбоксамидогруппы (-CONH2) в соответствующее N-замещенное производное; превращение карбоксильной группы в соответствующее производное бензилового сложного эфира реакцией с бензилбромидом в присутствии карбоната цезия в хорошо известных рабочих условиях; превращение карбоксильной группы (-СООН) в соответствующую группу (-CONHSO3H) сначала превращением ее в (-CONH2) с последующей реакцией с хлорсульфоновой кислотой в присутствии 2-пиколина; превращение карбоксильной группы (-СООН) в соответствующую группу [-CH2PO(OH)2] сначала восстановлением ее в гидроксиметил (-СН2ОН), последующим превращением в (-CH2Cl) при помощи тионилхлорида с последующей реакцией с триэтилфосфитом с образованием соответствующей группы [-CH2P(OH)2] и, наконец, гидролизом ее в [-CH2PO(OH)2].
Все из указанных выше реакций и их рабочие условия являются хорошо известными в данной области и позволяют получить различные соединения формулы (I).
Ясно, что также в стадии (b) способа любую функциональную группу в соединениях формулы (I), получаемых на стадии (а), которая может привести к образованию нежелательных побочных продуктов, необходимо должным образом защитить до того, как будет протекать реакция, согласно известным способам.
Для ссылки на определенные рабочие условия, применяемые при получении соединений формулы (I) см. в качестве примера нижеследующую схему 1, которая изображает синтетические пути получения репрезентативных соединений изобретения. Особые подробности, однако, можно найти в экспериментальном разделе.
На приведенной ниже схеме 1 показано получение некоторых репрезентативных соединений формулы (I) изобретения, у которых n и m, оба, равны 2; R, R1 и R4 имеют указанные здесь значения, один из R2 или R3 представляет собой водород и другой из R2 или R3 представляет собой гидроксильную или гидроксамовую группу (-СООН или -CONHOH) и R5 представляет собой Н.
Схема 1
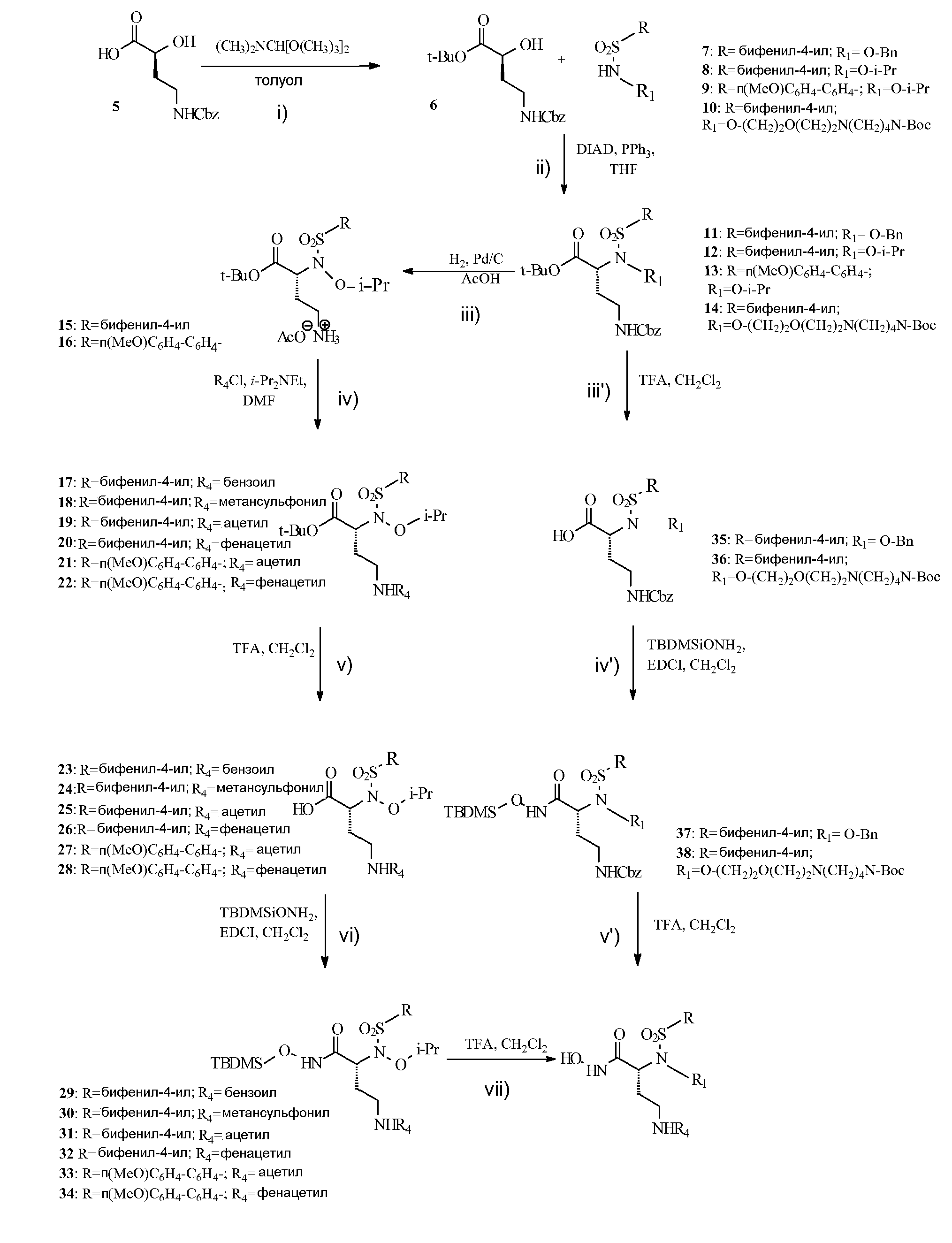
Указанный выше способ получения по существу содержит следующие стадии:
i) защита карбоксильной группы соединения (5) любой подходящей защитной группой, например, ди-трет-бутилацеталем N,N-диметилформамида в толуоле (см. в качестве общей ссылки Rossello et al. Bioorg. Med. Chem. Lett, 2005, 15, 1321), с получением таким образом соединения формулы (6):
ii) реакция любого из соединений (7-10) с соединением формулы (6) проведением ее согласно хорошо известным рабочим условиям Мицунобу, например, в присутствии диизопропилазодикарбоксилата (DIAD) и трифенилфосфина (PPh3) в качестве конденсирующих агентов, в подходящем растворителе, подобном тетрагидрофурану, с получением таким образом соответствующих соединений формулы (11-14) и обработку их, как в указанных ниже альтернативных путях:
iii) снятие защиты у аминогруппы соединений (12-13) согласно общепринятым способам, включающим, например, каталитическое гидрирование с палладиевыми или платиновыми катализаторами в уксусной кислоте, так чтобы получить соединения (15-16);
iv) функционализация должным образом аминогруппы соединений (15-16), так чтобы образовывалась любая требуемая группа -NHR4, как в соединениях (17-22), например, посредством реакции с любым подходящим ацилирующим агентом;
v) снятие защиты у карбоксильной функциональной группы, так чтобы получить соответствующие соединения (23-28), в подходящих условиях гидролиза, например, в присутствии трифторуксусной кислоты и в подходящем растворителе, подобном дихлорметану;
vi) превращение той же карбоксильной группы соединений (23-28) в подходящее силилпроизводное (29-34) согласно известным способам, например, при помощи О-(трет-бутилдиметилсилил)гидроксиламина в дихлорметане с последующим добавлением 1-[3-(диметиламино)пропил]-3-этилкарбодиимида;
vii) и гидролиз соединений (29-34), например, трифторуксусной кислотой в дихлорметане, так чтобы получить требуемые соединения формулы (I);
или же
iii') снятие защиты у карбоксильной функциональной группы у соединений (11 и 14), так чтобы получить соединения (35 и 36), например, обработкой как на стадии (v);
iv') превращение таким образом полученных соединений в соответствующие силилпроизводные (37-38), например, обработкой, как на стадии (vi);
v') и гидролиз соединений (37-38), например, обработкой, как на стадии (vii), с получением таким образом требуемых соединений формулы (I).
Наконец, необязательное получение солей соединений формулы (I) можно проводить превращением должным образом свободных кислотных групп (например, карбоновых, сульфоновых, фосфоновых и тому подобное) или свободных аминогрупп в соответствующие фармацевтически приемлемые соли. В этом случае рабочие условия, применяемые для необязательного получения солей соединений изобретения, также находятся в пределах обычных знаний специалиста в данной области.
Из всего указанного выше специалисту в данной области должно быть ясно, что указанный выше способ, подходящий для любого из его вариантов для получения подходящих соединений формулы (I) изобретения, можно подходящим образом модифицировать, чтобы адаптировать условия реакции для определенных требований, например, выбором подходящих агентов конденсации, растворителей и защитных групп, в зависимости от того, какой может быть случай.
Соединения формул (IV) и (V) в качестве исходных веществ настоящего способа являются известными или их можно легко получить согласно известным способам.
Сульфонамидопроизводные формулы (IV), например, можно получить реакцией любого подходящего аминосоединения с подходящим производным сульфонилхлорида по существу следующим образом:
R1-NH2 + R-S(O)2Cl → R-S(O)2NHR1
Кроме того, если как вышеуказанный амин, так и производное сульфонилхлорида, являются по существу неизвестными, их можно легко получить согласно известным способам из коммерчески доступных соединений.
Аналогичное рассмотрение можно применять для соединений формулы (V), которые, если они не являются коммерчески доступными сами по себе, можно получить пригодным образом согласно общепринятым способам, хорошо известным в данной области.
Фармакология
Ингибирующая активность
Согласно настоящему изобретению, соединения формулы (I) обладают ингибирующей активностью против металлопротеиназ матрикса и поэтому являются применимыми в терапии при лечении патологий, при которых изменена регуляция указанных ферментов.
Более конкретно, соединения испытывали для доказательства их активности против ММР-2, ММР-13 и ММР-14 согласно способу, описанному в примере 16.
Как описано в нем, ингибирующую активность соединений изобретения оценивали на известном флуорогенном субстрате (Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2), кратко обозначенном как FS-6 (см. в качестве ссылки U. Neumann, Analytical Biochem. 2004, 328, 166-173).
Указанный флуорогенный субстрат получали из аналогичного FS-1 (см. в качестве ссылки вышеуказанную публикацию CG Knight et al., в Febs Lett. 1992, 296, 263-266) посредством инсерции остатка лизина между остатками МСА и пролина.
Поскольку элонгация пептидной цепи в FS-6 была описана как повышение способности субстрата гидролизоваться ММР, предположительно вследствие стерически затрудненной части Мса, которая может быть ответственной за пониженную аффинность субстрата для некоторых ММР, настоящие соединения испытывали согласно этому более точному и очень чувствительному способу, основанному на FS-6.
Следовательно, в результате таким образом полученных экспериментальных данных и комментариев к ним (см. пример 16), ясно, что соединения изобретения обладают ингибирующей активностью против ММР-2, ММР-13 и ММ-14, заметно превосходящей ингибирующую активность структурно близкого соединения известного уровня техники, в описании идентифицированного как ссылочное соединение (5b). Вследствие их неожиданного профиля активности, следовательно, соединения изобретения можно преимущественно применять в терапии при лечении патологий, в общем называемых дегенеративными нарушениями, в которых принимают участие указанные выше ферменты.
Согласно этому последнему аспекту, соединениями формулы (I) для терапевтической цели являются соединения, ранее описанные, и такие соединения включают также вышеуказанные соединения известного уровня техники, описанные только как химические промежуточные продукты.
Следовательно, дополнительным вариантом осуществления изобретения является соединение формулы (I), где R, R1, R2, R3, R4, R5, n и m имеют значения, указанные выше, с условием, что когда R представляет собой бифенил-4-ил, R1 представляет собой изопропокси, m и n, оба, равны 2, R5 представляет собой Н, один из R2 или R3 представляет собой -CONHOH и другой из R2 и R3 представляет собой Н, то R4 не является Н или бензилоксикарбонилом, и его фармацевтически приемлемые соли, для применения в качестве лекарственных средств.
Кроме того, в объем изобретения включено применение указанных выше соединений формулы (I), где R, R1, R2, R3, R4, R5, n и m имеют значения, указанные выше, с условием, что когда R представляет собой бифенил-4-ил, R1 представляет собой изопропокси, m и n, оба, равны 2, R5 представляет собой Н, один из R2 или R3 представляет собой -CONHOH и другой из R2 и R3 представляет собой Н, то R4 не является Н или бензилоксикарбонилом, и его фармацевтически приемлемых солей при изготовлении лекарственного средства для лечения дегенеративных нарушений.
Упомянутые дегенеративные нарушения включают опухоли и в общем патологии, приводящие к измененной тканевой морфологии с нерегулируемой пролиферацией клеток. Такие патологии могут таким образом включать артрит и нарушения соединительной ткани; нейродегенеративные нарушения, такие как рассеянный склероз, болезнь Альцгеймера, удар и ALS (боковой амиотрофический склероз), сердечно-сосудистые нарушения, такие как атеросклероз, аневризма, сердечная недостаточность, застойная кардиомиопатия; легочные заболевания, такие как эмфизема или муковисцидоз; язвы желудка; сепсис и аутоиммунные нарушения.
Еще в одном варианте осуществления изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента фармацевтически эффективное количество соединения формулы (I), где R, R1, R2, R3, R4, R5, n и m имеют значения, указанные выше, с условием, что когда R представляет собой бифенил-4-ил, R1 представляет собой изопропокси, m и n, оба, равны 2, R5 представляет собой Н, один из R2 или R3 представляет собой -CONHOH и другой из R2 и R3 представляет собой Н, то R4 не является Н или бензилоксикарбонилом, и его фармацевтически приемлемые соли в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Композиции изобретения можно целесообразно получать согласно общепринятым способам, широко известным в области получения фармацевтических форм, они могут содержать любой из носителей, разбавителей или эксципиентов, известных в данной области для предполагаемой цели.
В еще одном аспекте изобретение относится к способу лечения вышеуказанных дегенеративных нарушений, который включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), где R, R1, R2, R3, R4, R5, n и m имеют значения, указанные выше, с условием, что когда R представляет собой бифенил-4-ил, R1 представляет собой изопропокси, m и n, оба, равны 2, R5 представляет собой Н, один из R2 или R3 представляет собой -CONHOH и другой из R2 и R3 представляет собой Н, то R4 не является Н или бензилоксикарбонилом, и его фармацевтически приемлемых солей.
Из всего указанного выше можно легко предположить, что соединения данного изобретения могут иметь широкий диапазон применений в терапии и поэтому могут быть должным образом изготовлены в составе препарата согласно общепринятым способам для предназначенного пути введения, т.е. местного, перорального или энтерального введения.
С целью лучшей иллюстрации настоящего изобретения, без цели любого его ограничения, теперь приводятся следующие примеры. В этом отношении дополнительные применения, включающие возможные варианты к препаративному способу, которые будут очевидны специалисту в данной области, таким образом должны рассматриваться как включенные в объем настоящего изобретения.
Экспериментальный раздел
Некоторые репрезентативные соединения изобретения получали согласно следующим схемам 1-6; если не оговорено особо, ссылка на нумерацию соединений, указываемую в следующих схемах, будет сохраняться.
Соединения, здесь пронумерованные как соединения (1а-1h), получали способом, указанным на схеме 1.
|
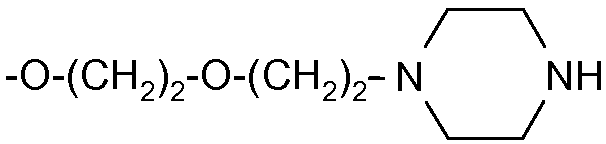
Оптически активный α-гидрокси-трет-бутиловый сложный эфир (6) синтезировали непосредственной этерификацией коммерчески доступной α-гидроксикислоты (5) ди-трет-бутилацеталем N,N-диметилформамидом (см. Rosello, A. et al.; Bioorg. Med. Chem. Lett, 2005, 15, 1321). Реакция конденсация Мицунобу сульфонамидов (7-10) с α-гидрокси-трет-бутиловым сложным эфиром (6) давала трет-бутиловые сложные эфиры (11-14). Расщепление кислотой сложных эфиров (11, 14) давало карбоновые кислоты (35, 36), которые превращали в их О-силилат (37, 38) при обработке О-(трет-бутилдиметилсилил)гидроксиламином. Гидролиз трифторуксусной кислотой трет-бутил-О-силилата (37, 38) давал гидроксамовые кислоты (1а, 1h). трет-Бутиловые сложные эфиры (17-22) получали Pd-катализируемым гидрированием (12, 13) с последующим ацилированием коммерческими ацилхлоридами. Расщепление кислотой сложных эфиров (17-22) давало карбоксилаты (23-28), которые затем превращали в их соответствующий трет-бутил-О-силилат (29, 34). Гидроксамовые кислоты (1b-1g) затем получали обработкой (29, 34) трифторуксусной кислотой.
Дополнительные соединения изобретения (2-4) также получали согласно схеме 2.
(S)-α-Гидрокси-трет-бутиловый сложный эфир (40) получали прямой этерификацией коммерчески доступной α-гидроксикислоты (39) с применением ди-трет-бутилацеталя N,N-диметилформамида. Сочетание по Мицунобу сульфонамидов (9, 41 и 42) с α-гидрокси-трет-бутиловым сложным эфиром (40) давало трет-бутиловые сложные эфиры (43-45). Расщепление кислотой (43, 44) давало карбоновые кислоты (46, 47), которые превращали в их О-силилат (48, 49) при обработке О-(трет-бутилдиметилсилил)гидроксиламином. Гидролиз трифторуксусной кислотой трет-бутил-О-силилата (48, 49) давал гидроксамовые кислоты (2 и 3). Сочетание по Сузуки коммерчески доступной 4-этоксифенилбороновой кислоты со сложным эфиром (45) давало бифениловый сложный эфир (50), который превращали в гидроксамовую кислоту (4) с применением методики, описанной выше.
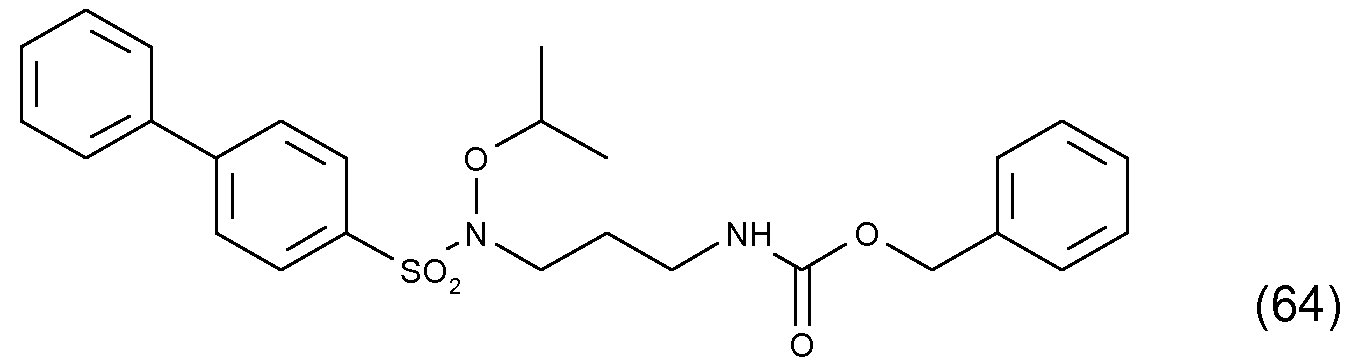
Дополнительное соединение изобретения, в описании указанное как соединение (64), у которого оба из R2 и R3 в формуле (I) представляют собой атомы Н, получали, как показано ниже на схеме 5, посредством реакции конденсации Мицунобу между коммерчески доступным спиртом (63) и сульфонамидом (8).
Сульфонамиды (7-10 и 41, 42), применяемые на схемах 1 и 2, получали, как показано на схеме 3. Коммерчески доступные арилсульфонилхлориды (56-59) сочетали с подходящими О-алкилгидроксиламинами (53-55) при обработке N-метилморфолином (см. Rossello, A. et al.; Bioorg. Med. Chem. 2004, 12, 2441). Тогда как О-алкилгидроксиламины (53 и 54) были коммерчески доступными, (55) получали, как описано на схеме 4. Реакция N-защищенного коммерческого аминоспирта (60) с ди-трет-бутилдикарбонатом давала производное пиперазина (61). Реакция Мицунобу с участием N-гидроксифталимида давала (62), который превращали в (55) гидразинолизом гидратом гидразина в этаноле.
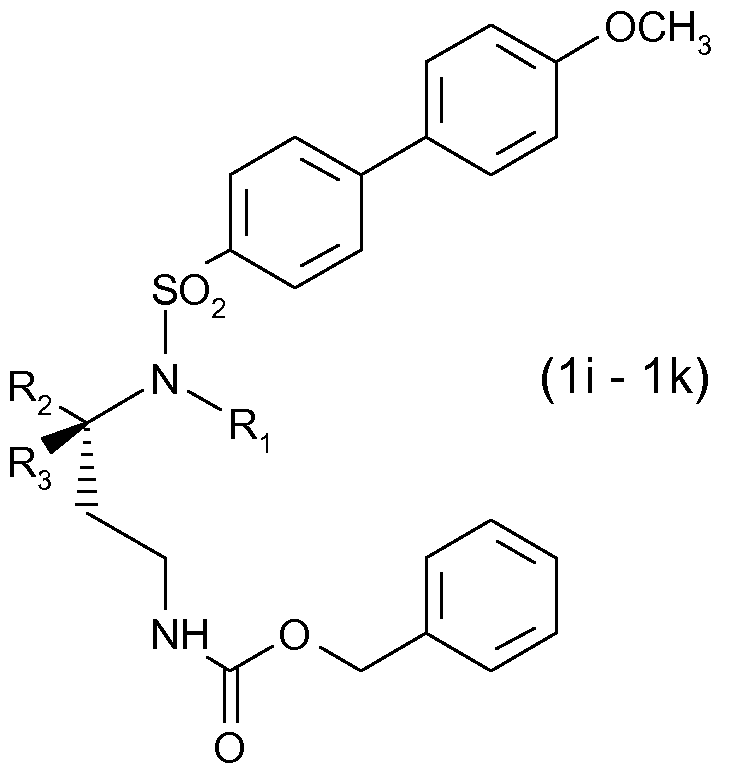
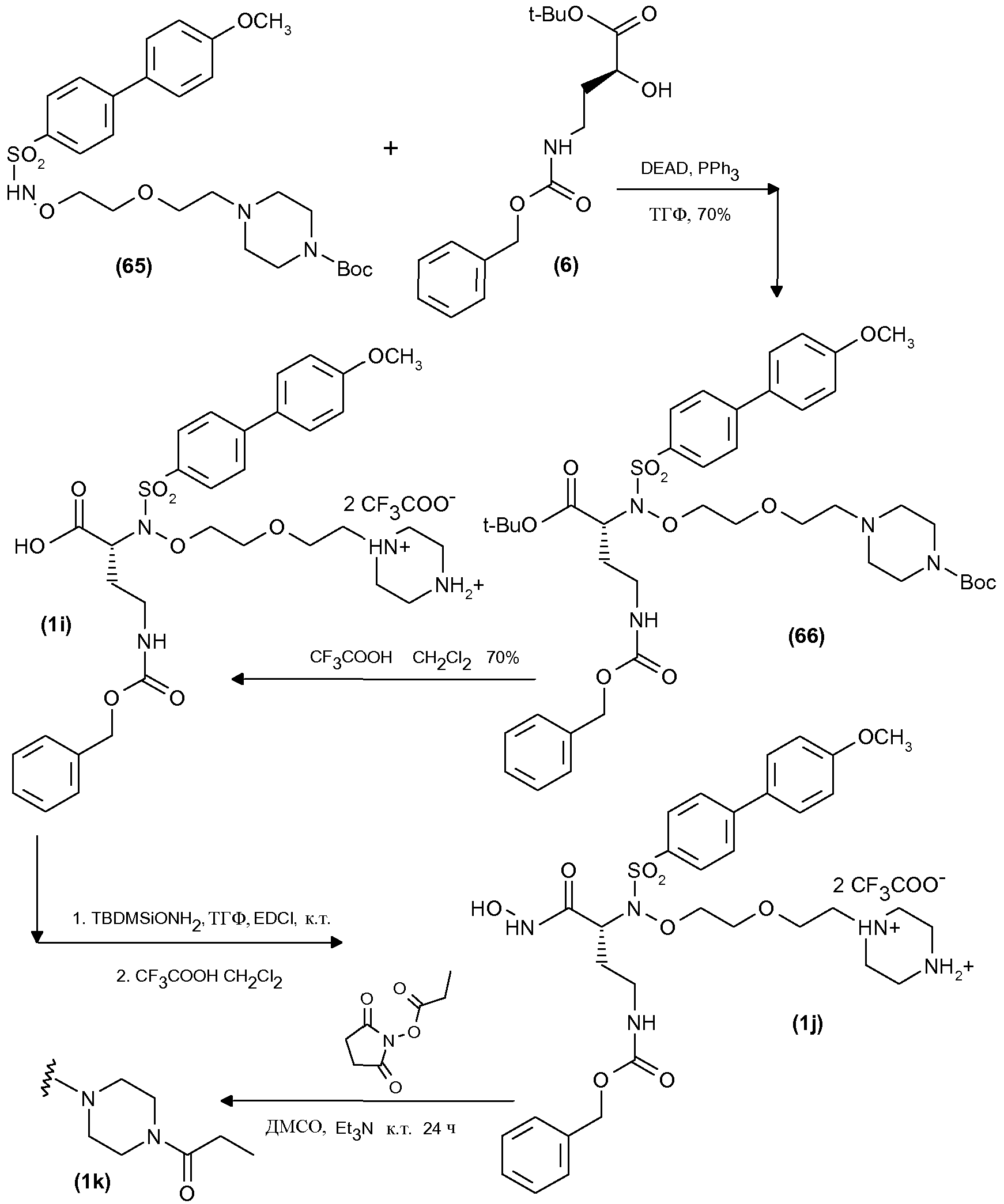
Получены также дополнительные соединения изобретения (1i - 1k), имеющие следующую формулу
|
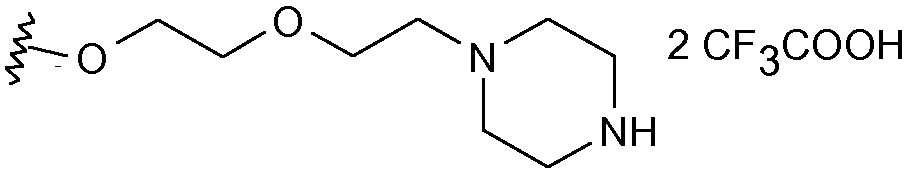
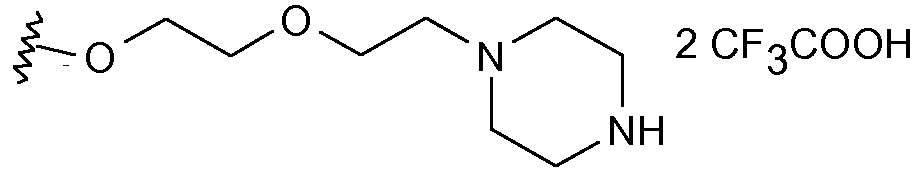
Соединения (1i) и (1j) получали, как показано на схеме 6, соответствующей реакцией, как ранее указано в условиях сочетания Мицунобу, сульфонамидопроизводного (65) с соединением (6) схемы 1. Полученное соединение (66) затем освобождали от защитной группы трифторуксусной кислотой, так чтобы получить соединение (1i) в виде дитрифторацетатной соли.
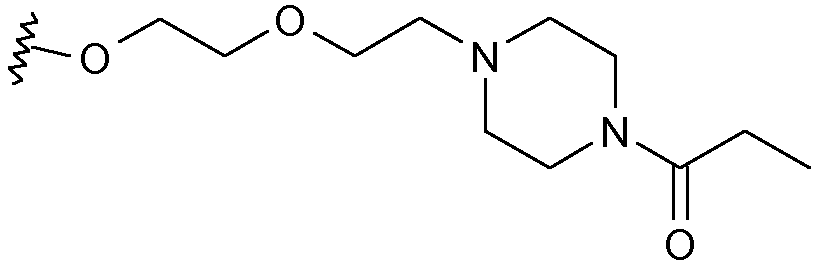
Эту последнюю карбоновую кислоту затем превращали в дитрифторацетат соответствующего производного гидроксамовой кислоты (1j), как указано ранее.
Соединение (1j) затем ацилировали у атома N пиперазина 2,5-диоксопирролидин-1-илпропионатом обработкой согласно общепринятым способам в присутствии триэтиламина, так чтобы получить соответствующее соединение (1k).
Исходное соединение (65) получали аналогично тому, как показано на схемах 3 и 4 для получения соединения (10).
Спектры 1Н ЯМР регистрировали на Varian Gemini 200 (200 МГц) с применением CDCl3 или ДМСО-d6 в качестве растворителей.
Схема 1
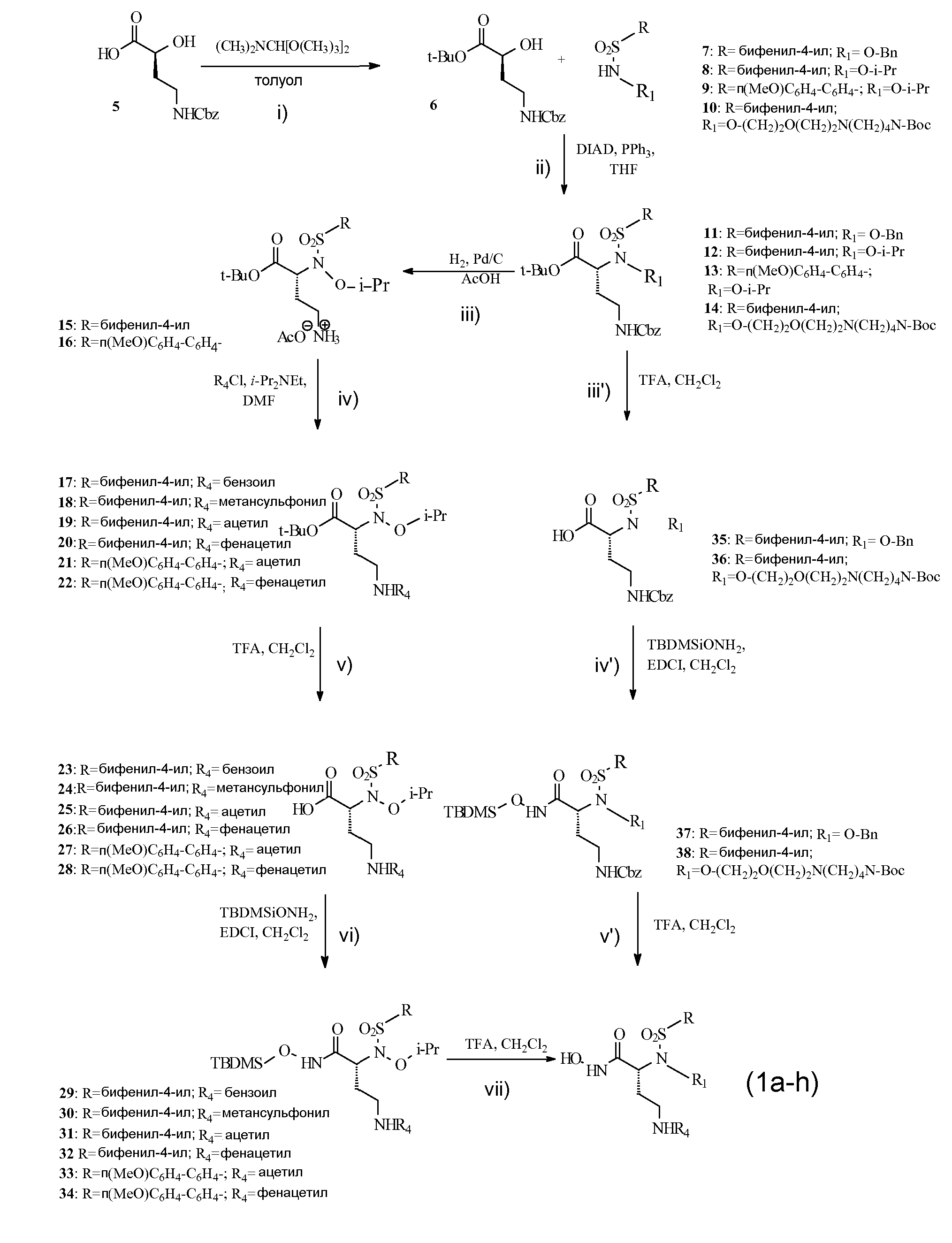
Схема 2
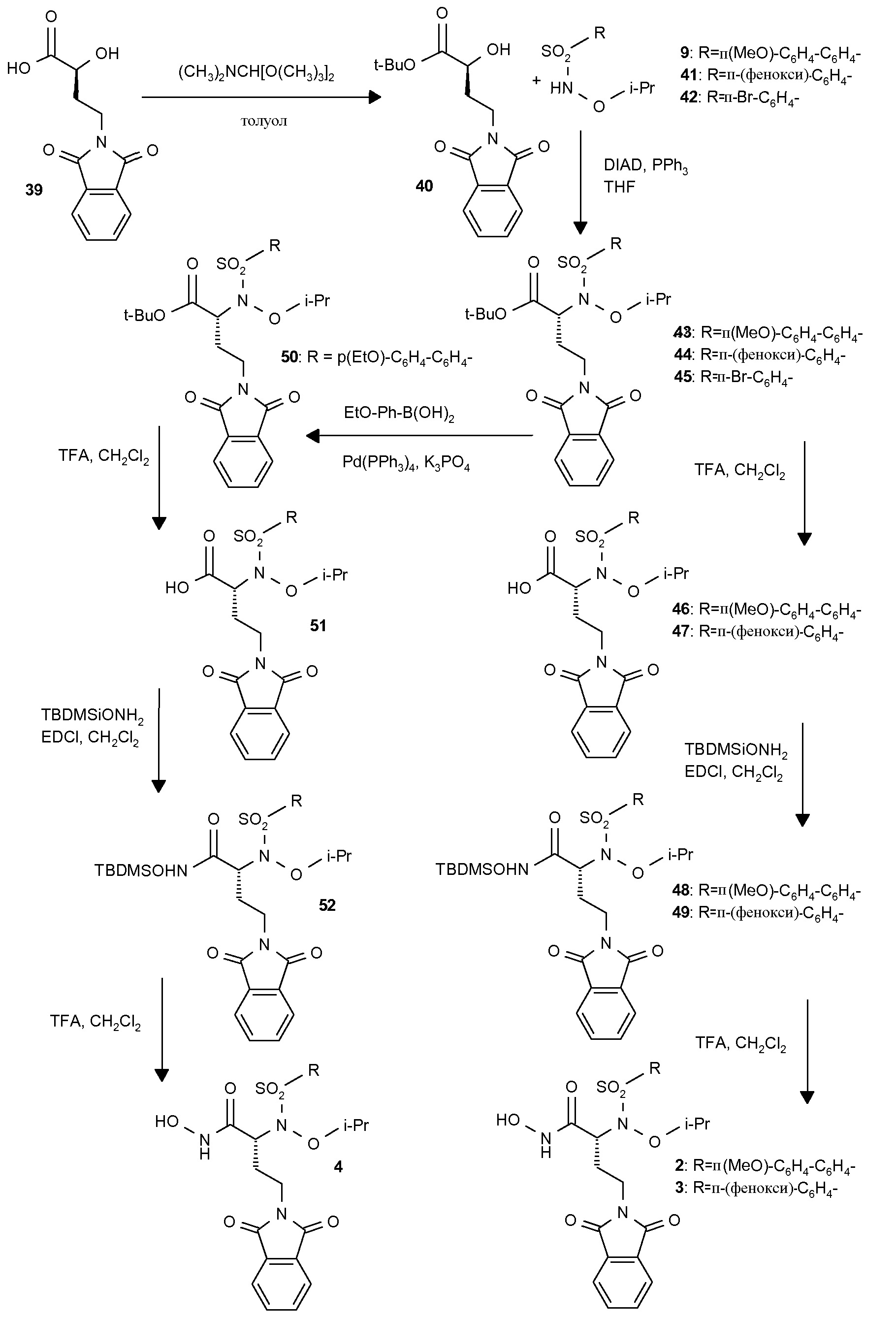
Схема 3
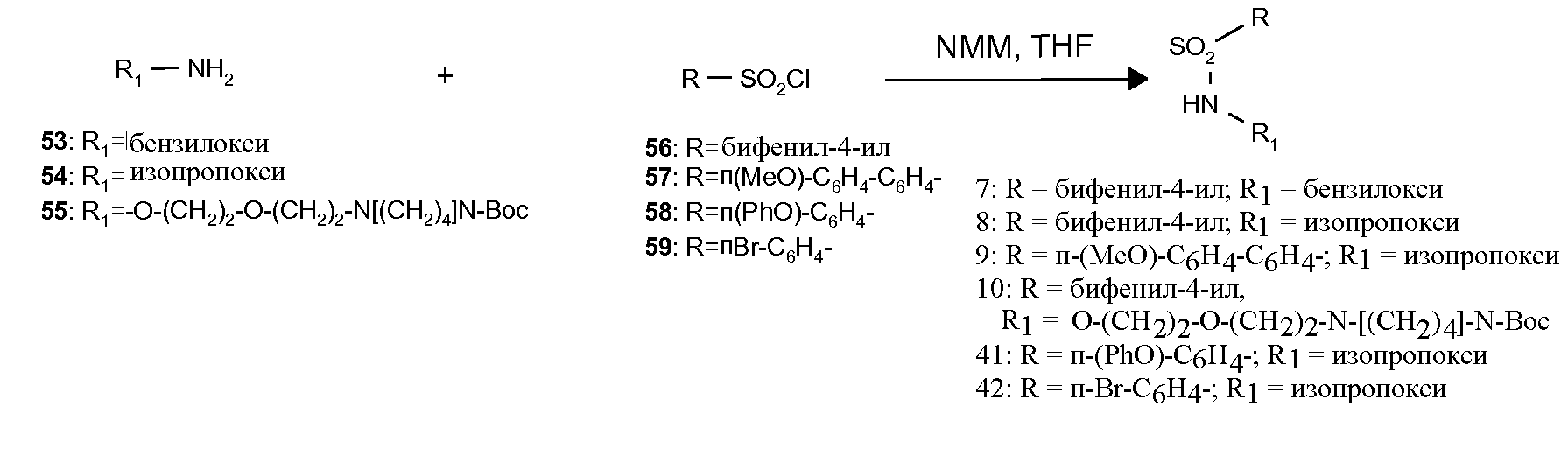
Схема 4
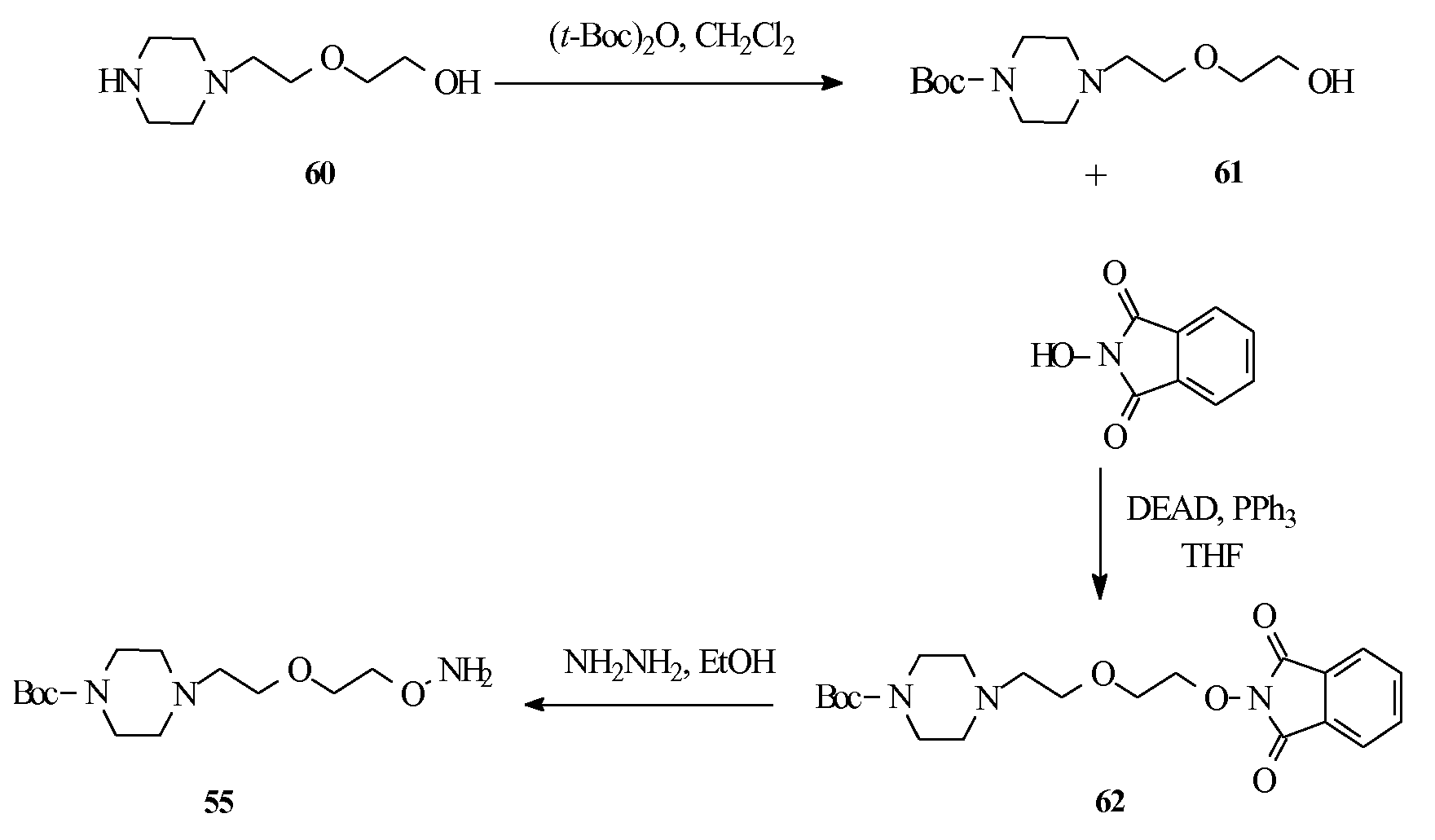
Схема 5
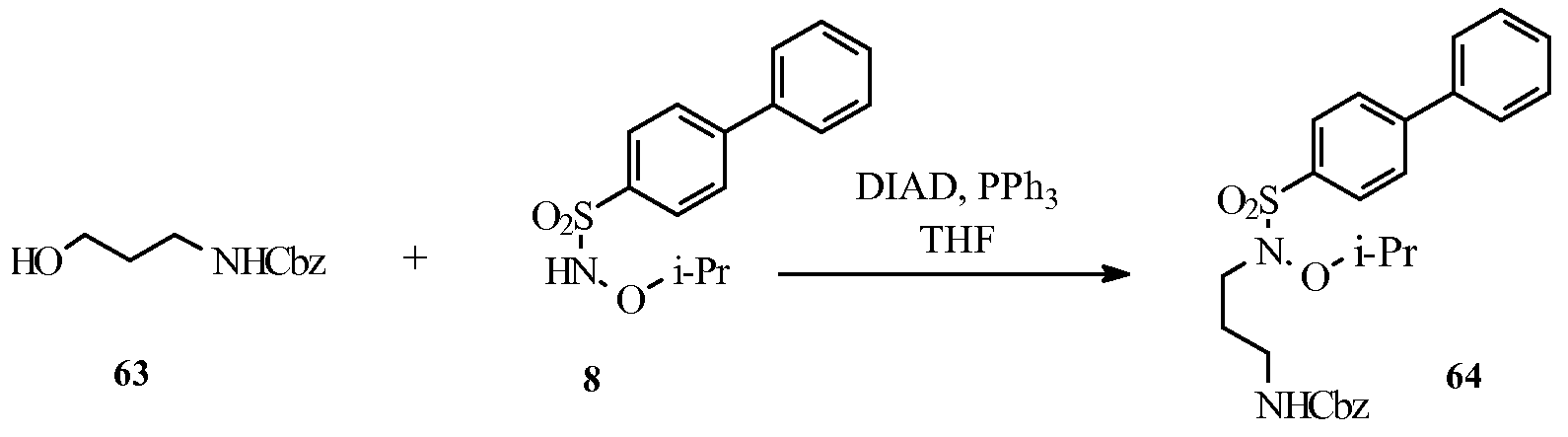
Схема 6
Пример 1
Получение соединения (1а): (R)-бензил-3-(N-(бензилокси)бифенил-4-илсульфонамидо)-4-(гидроксиамино)-4-оксобутилкарбамат
Получение соединения (6)
Раствор (S)-(+)-Z-4-амино-2-гидроксимасляной кислоты (5) (5 г, 19,74 ммоль) в толуоле (38 мл), содержащий ди-трет-бутилацеталь N,N-диметилформамида (18,92 мл, 78,96 ммоль) нагревали до 95°С в течение 3 ч. Растворитель затем выпаривали и сырой продукт очищали флэш-хроматографией на силикагеле (н-гексан/EtOAc = 7:4), получая при этом (6) (3,4 г, выход 55,7%) в виде желтого твердого вещества.
Т. пл. 42-44°С; [α]20 D = -5,9° (с = 10,1 мг/мл, CHCl3).
1H ЯМР (CDCl3) δ: 1,47 (с, 9H); 1,76-1,85 (м, 1H); 1,94-2,09 (м, 1H); 2,74 (ушир. с, 1H); 3,36 (дд, J=6,04 Гц, J=11,99 Гц, 2H); 4,10 (дд, J=4,02 Гц, J=8,05 Гц, 1H); 5,09 (с, 2H); 5,21 (ушир. с, 1H); 7,31-7,37 (м, 5H).
Анал. Вычисл. для C16H23NO5: C, 62,12; H, 7,49; N, 4,53. Найдено: C, 62,22; H, 7,48; N, 4,53.
Получение соединения (7)
Раствор бифенил-4-сульфонилхлорида (56) (3,17 г, 12,53 ммоль) в безводном ТГФ (32 мл) добавляли по каплям к перемешиваемому и охлажденному (0°С) раствору гидрохлорида О-бензилгидроксиламина (53) (2 г, 12,53 ммоль) и N-метилморфолина (2,75 мл, 25,06 ммоль) в безводном ТГФ (32 мл). Спустя 30 мин при этих условиях реакционную смесь перемешивали при комнатной температуре (к.т.) в течение 3 дней, затем разбавляли AcOEt и промывали Н2О, получая после обработки сульфонамид (7) (3,62 г, 85%) в виде белого твердого вещества.
Т. пл. 130-132°С.
1H ЯМР (CDCl3) δ: 5,01 (с, 2H); 7,01 (с, 1H); 7,35 (м, 5H); 7,44-7,51 (м, 3H); 7,57-7,62 (м, 2H); 7,70-7,75 (м, 2H); 7,97-8,01 (м, 2H).
Получение соединения (11)
Диизопропилазодикарбоксилат (DIAD) (1,28 мл, 6,52 ммоль) добавляли по каплям к раствору, содержащему вторичный спирт (6) (0,8 г, 2,61 ммоль), сульфонамид (7) (1,3 г, 3,91 ммоль) и трифенилфосфин (2,05 г, 7,83 ммоль) в безводном ТГФ (45 мл) в атмосфере азота при 0°С. Образовавшийся раствор перемешивали в течение 5 час при к.т. и выпаривали при пониженном давлении с получением сырого продукта, который очищали флэш-хроматографией на силикагеле (н-гексан/EtOAc = 3:1), получая при этом соединение (11) (0,95 г, выход 58%) в виде чистого желтого масла.
1H ЯМР (CDCl3) δ: 1,25 (с, 9H); 1,90-2,04 (м, 2H); 3,16-3,40 (м, 2H); 4,16 (т, J=7,1 Гц, 1H); 4,93-5,00 (м, 1H); 5,07-5,19 (м, 4H); 7,33-7,37 (м, 10H); 7,41-7,59 (м, 5H); 7,66-7,70 (м, 2H); 7,92-7,97 (м, 2H).
13C ЯМР (CDCl3) δ: 22,09; 27,87; 37,59; 62,75; 66,76; 80,87; 82,56; 127,43; 127,67; 128,16; 128,56; 128,72; 128,92; 129,14; 129,89; 129,94; 133,89; 134,80; 139,17; 146,84; 156,27.
Получение соединения (35)
Трифторуксусную кислоту (0,9 мл, 57,00 ммоль) добавляли по каплям к перемешиваемому раствору трет-бутилового сложного эфира (11) (136 мг, 0,21 ммоль) в свежеперегнанном CH2Cl2 (1,0 мл), охлажденному до 0°С. Раствор перемешивали в течение 5 час при 0°С и растворитель удаляли в вакууме, получая при этом соединение (35) (128 мг, выход 100%) в виде масла.
1H ЯМР (CDCl3) δ: 1,60-1,70 (м, 1H); 1,88-2,08 (м, 1H); 3,19 (м, 2H); 4,30 (т, J=6,9 Гц, 1H); 5,02-5,17 (м, 4H); 7,28-7,34 (м, 10H); 7,40-7,51 (м, 3H); 7,55-7,59 (м, 2H); 7,65-7,70 (м, 2H); 7,91-7,95 (м, 2H).
Получение соединения (37)
К раствору карбоновой кислоты (35) (117 мг, 0,2 ммоль) и О-(трет-бутилдиметилсилил)гидроксиламина (44 мг, 0,3 ммоль) в свежеперегнанном CH2Cl2 (3,6 мл), охлажденному до 0°С, добавляли в виде порций гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI) (57,5 мг, 0,3 ммоль). После перемешивания при к.т. в течение 20 час смесь промывали Н2О и органическую фазу сушили и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (н-гексан/EtOAc = 2,5:1), получая при этом соединение (37) (28 мг, выход 20%) в виде желтого масла.
1H ЯМР (CDCl3) δ: 0,13 (с, 6H); 0,90 (с, 9H); 1,90-2,11 (м, 2H); 2,80-3,40 (м, 2H); 4,13-4,22 (м, 1H); 5,02-5,27 (м, 4H); 7,31-7,46 (м, 13H); 7,53-7,58 (м, 2H); 7,64-7,68 (м, 2H); 7,84-7,88 (м, 2H).
Получение указанного в заголовке соединения (1a)
Трифторуксусную кислоту (0,15 мл, 1,85 ммоль) добавляли по каплям к перемешиваемому раствору соединения (37) (23 мг, 0,03 ммоль) в свежеперегнанном CH2Cl2 (1 мл), охлажденному до 0°С. Раствор перемешивали в течение 5 час при 0°С и растворитель удаляли в вакууме с получением сырого продукта, который перекристаллизовывали из Et2O и н-гексана, получая при этом (1а) (10 мг, выход 53%) в виде твердого вещества.
1H ЯМР (CDCl3) δ: 1,99 (м, 2H); 3,00-3,35 (м, 2H); 4,29 (м, 1H); 5,08-5,26 (м, 4H); 7,34-7,45 (м, 13H); 7,53-7,58 (м, 2H); 7,66-7,70 (м, 2H); 7,87-7,91 (м, 2H).
Пример 2
Получение соединения (1b): (R)-N-(4-(гидроксиамино)-3-(N-изопропоксибифенил-4-илсульфонамидо)-4-оксобутил)бензамида
Получение соединения (8)
N-Изопропокси-1,1'-бифенил-4-сульфонамид получали, как ранее описано Rossello, A. et al. (Bioorg. Med. Chem. 2004, 12, 2441).
Получение соединения (12)
трет-Бутиловый сложный эфир (12) получали из производного сульфонамида (8) (1,08 г, 3,72 ммоль) и спирта (6) (0,76 г, 2,48 ммоль) по методике, ранее описанной для получения соединения (11), как описано в примере 1. Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 5:1), получая при этом (12) (1,14 г, выход 79%) в виде желтого масла.
[α]20 D = +55о (с = 9,1 мг/л, CHCl3).
1H ЯМР (CDCl3) δ: 1,22-1,25 (м, 15H); 2,04 (м, 2H); 3,22-3,37 (м, 2H); 4,12 (дд, J=7,14 Гц, J=14,29 Гц, 1H); 4,43 (септет, J=6,2 Гц, 1H); 5,08 (с, 2H); 7,34 (м, 5H); 7,42-7,53 (м, 3H); 7,55-7,61 (м, 2H); 7,7-7,74 (м, 2H); 7,94-7,98 (м, 2H).
Получение соединения (15)
Раствор соединения (12) (0,74 г, 1,27 ммоль) в МеОН (80 мл) перемешивали в атмосфере водорода в присутствии 10% Pd-C (0,20 г) и ледяной уксусной кислоты (80 мл) в течение 17 час при комнатной температуре. Образовавшуюся смесь фильтровали на целите и фильтрат упаривали при пониженном давлении, получая при этом (15) (0,60 г, выход 93%) в виде коричневатого масла.
1H ЯМР (CDCl3) δ: 1,10 (ушир. с, 9H); 1,20 (т, J=4,4 Гц, 6H); 2,16-2,30 (м, 2H); 3,16 (м, 2H); 4,34-4,46 (м, 2H); 7,40-7,51 (м, 3H); 7,56-7,59 (м, 2H); 7,71-7,75 (м, 2H); 8,00-8,04 (м, 2H).
13C ЯМР (CDCl3) δ: 21,15; 21,22; 27,72; 36,70; 62,97; 80,03; 82,49; 127,03; 127,45; 127,59; 127,76; 128,67; 129,14; 130,49; 133,60; 139,30; 146,89.
Получение соединения (17)
Раствор соединения (15) (0,30 г, 0,59 ммоль) в сухом ДМФА (6 мл) обрабатывали бензоилхлоридом (0,08 мл, 0,70 ммоль) и i-Pr2NEt (0,20 мл, 1,18 ммоль). Реакционную смесь перемешивали при к.т. в течение 17 час, затем разбавляли этилацетатом, промывали Н2О, сушили над Na2SO4 и упаривали. Сырой продукт очищали флэш-хроматографией (н-гексан/AcOEt = 2,5:1), получая при этом (17) (112 мг, выход 34%) в виде желтого масла.
1H ЯМР (CDCl3) δ: 1,13 (ушир. с, 9H); 1,23 (д, J=5,1 Гц, 3H); 1,26 (д, J=4,4 Гц, 3H); 2,10-2,21 (м, 2H); 3,34-3,50 (м, 1H); 3,80-3,92 (м, 1H); 4,23 (т, J=7,1 Гц, 1H); 4,45 (септет, 1H); 7,39-7,59 (м, 10H); 7,69-7,73 (м, 2H); 7,93-7,98 (м, 2H).
Получение соединения (23)
Карбоновую кислоту (23) (89 мг, выход 100%) получали из производного сложного эфира (17) (0,10 г, 0,18 ммоль) по методике, ранее описанной для получения соединения (35), как в примере 1.
1H ЯМР (CDCl3) δ: 1,20 (т, J=5,1 Гц, 6Н); 1,80-2,28 (м, 2Н); 3,40-3,55 (м, 1Н); 3,60-3,82 (м, 1Н); 4,30-4,50 (м, 2Н); 6,56 (ушир. с, 1Н); 7,43-7,58 (м, 10Н); 7,66-7,69 (м, 2Н); 7,91-7,94 (м, 2Н).
Получение соединения (29)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (23) (90 мг, 0,18 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 2:1) давала требуемый продукт (30 мг, выход 27%).
1H ЯМР (CDCl3) δ: 0,16 (с, 6Н); 0,93 (c, 9H); 1,22 (д, J=6,2 Гц, 3Н); 1,28 (д, J=6,2 Гц, 3Н); 2,02-2,16 (м, 2Н); 3,20 (м, 1Н); 3,43 (м, 1Н); 4,06-4,20 (м, 1Н); 4,46 (септет, 1Н); 6,76 (ушир. с, 1Н); 7,35-7,65 (м, 10Н); 7,73-7,77 (м, 2Н); 7,87-7,91 (м, 2Н); 9,01 (ушир. с, 1Н).
Получение указанного в заголовке соединения (1b)
По методике, аналогичной методике, применяемой для получения соединения (1а) в примере 1, трет-бутил-О-силилат (29) (30 мг, 0,05 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (15 мг, выход 60,5%) после перекристаллизации из Et2O.
1H ЯМР (CDCl3) δ: 1,24 (т, J=6,5 Гц, 6Н); 1,44-1,69 (м, 1Н); 2,05-2,21 (м, 1Н); 3,10-3,50 (м, 2Н); 4,20-4,50 (м, 2Н); 7,10 (ушир. с, 1Н); 7,30-7,55 (м, 10Н); 7,59-7,63 (м, 2Н); 7,86-7,90 (м, 2Н).
Пример 3
Получение соединения (1с): (R)-N-гидрокси-2-(N-изопропоксибифенил-4-илсульфонамидо)-4-(метилсульфонамидо)бутанамида
Получение соединения (18)
Раствор соединения (15), полученного согласно примеру 2 (0,30 г, 0,60 ммоль) в сухом ТГФ (3 мл), обрабатывали метансульфонилхлоридом (0,05 мл, 0,60 ммоль) и N-метилморфолином (0,13 мл, 1,2 ммоль). Реакционную смесь перемешивали при комнатной температуре на протяжении ночи, затем разбавляли AcOEt, промывали Н2О, сушили над Na2SO4 и упаривали. Сырой продукт очищали флэш-хроматографией (н-гексан/AcOEt = 3:2), получая при этом (18) (100 мг, выход 32%) в виде желтого масла.
1H ЯМР (CDCl3) δ: 1,14 (ушир. с, 9Н); 1,20-1,26 (м, 6H); 2,04 (ушир. с, 2Н); 2,95 (с, 3Н); 3,29 (ушир. с, 2Н); 4,25 (т, J=7,1 Гц, 1Н); 4,42 (септет, 1Н); 7,43-7,54 (м, 3Н); 7,58-7,62 (м, 2Н); 7,74-7,78 (м, 2Н); 7,96-8,00 (м, 2Н).
Получение соединения (24)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, производное сложного эфира (18) (100 мг, 0,19 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (24) (73 мг, выход 79%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,22 (д, J=6,2 Гц, 6Н); 2,06-2,17 (м, 2H); 2,91 (с, 3Н); 3,21 (ушир. с, 2Н); 4,34-4,44 (м, 2Н); 7,42-7,53 (м, 3Н); 7,61-7,66 (м, 2Н); 7,74-7,78 (м, 2Н); 7,95-7,99 (м, 2Н).
Получение соединения (30)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (24) (70 мг, 0,15 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 1:1) давала требуемый продукт (32 мг, выход 33%).
1H ЯМР (CDCl3) δ: 0,15 (с, 6Н); 0,93 (c, 9H); 1,20 (д, J=6,2 Гц, 3Н); 1,25 (д, J=5,8 Гц, 3Н); 2,00-2,15 (м, 2Н); 2,83 (с, 3Н); 2,89-2,99 (м, 2Н); 4,23-4,29 (м, 1Н); 4,40 (септет, 1Н); 7,42-7,52 (м, 3Н); 7,61-7,66 (м, 2Н); 7,79-7,83 (м, 2Н); 7,96-8,00 (м, 2Н); 8,64 (ушир. с, 1Н).
Получение указанного в заголовке соединения (1с)
По методике, аналогичной методике, применяемой для получения соединения (1а) в примере 1, трет-бутил-О-силилат (30) (30 мг, 0,05 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (15 мг, выход 60%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,24 (т, J=6,4 Гц, 6Н); 2,01-2,18 (м, 2H); 2,88 (с, 3Н); 3,00-3,20 (м, 2Н); 4,40-4,48 (м, 2Н); 4,88 (ушир. с, 1Н); 7,42-7,53 (м, 3Н); 7,62-7,66 (м, 2Н); 7,78-7,83 (м, 2Н); 7,95-7,99 (м, 2Н).
Пример 4
Получение соединения (1d): (R)-4-ацетамидо-N-гидрокси-2-(N-изопропоксибифенил-4-илсульфонамидо)бутанамида
Получение соединения (19)
По методике, аналогичной методике, применяемой для получения соединения (17) в примере 2, производное сложного эфира (15) (0,30 г, 0,59 ммоль) ацилировали ацетилхлоридом. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 1:1) давала требуемый продукт (19) (60 мг, выход 22%).
1H ЯМР (CDCl3) δ: 1,19-1,24 (м, 15Н); 1,90-2,01 (м, 5H); 3,13-3,23 (м,1Н); 3,53 (м, 1Н); 4,12 (м, 1Н); 4,40 (септет, 1Н); 6,11 (ушир. с, 1Н); 7,41-7,52 (м, 3Н); 7,57-7,61 (м, 2Н); 7,72-7,76 (м, 2Н); 7,94-7,98 (м, 2Н).
13С ЯМР (CDCl3) δ: 21,17; 23,48; 27,76; 36,17; 63,59; 79,80; 82,25; 127,39; 127,52; 128,72; 129,16; 130,14; 133,80; 139,19; 146,84; 170,22.
Получение соединения (25)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, производное сложного эфира (19) (60 мг, 0,12 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (25) (60 мг, выход 100%).
1H ЯМР (CDCl3) δ: 1,16 (д, J=2,01, 3Н); 1,19 (д, J=2,01 Гц, 3H); 1,92-2,10 (м, 5Н); 3,15-3,60 (м, 2Н); 4,20-4,39 (м, 2Н); 6,73 (ушир. с, 1Н); 7,42-7,52 (м, 3Н); 7,59-7,63 (м, 2Н); 7,73-7,77 (м, 2Н); 7,93-7,97 (м, 2Н); 10,24 (ушир. с, 1Н).
Получение соединения (31)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (25) (60 мг, 0,14 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 1:2) давала требуемый продукт (12 мг, выход 16%).
1H ЯМР (CDCl3) δ: 0,15 (с, 6Н); 0,93 (c, 9H); 1,21 (д, J=6,2 Гц, 3Н); 1,25 (д, J=6,2 Гц, 3Н); 1,90-2,05 (м, 5Н); 2,80-3,26 (м, 2Н); 4,06-4,16 (м, 1Н); 4,44 (септет, 1Н); 5,95 (ушир. с, 1Н); 7,42-7,53 (м, 3Н); 7,58-7,65 (м, 2Н); 7,76-7,80 (м, 2Н); 7,92-7,96 (м, 2Н); 8,90 (ушир. с, 1Н).
Получение указанного в заголовке соединения (1d)
По методике, аналогичной методике, применяемой для получения соединения (1а) в примере 1, трет-бутил-О-силилат (31) (12 мг, 0,02 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (11 мг, выход 90%).
1H ЯМР (CDCl3) δ: 1,23 (д, J=6,7 Гц, 6Н); 1,90-2,08 (м, 5H); 3,04-3,35 (м, 2Н); 4,22 (м, 1Н); 4,40 (септет, 1Н); 6,73 (ушир. с, 1Н); 7,42-7,52 (м, 3Н); 7,59-7,63 (м, 2Н); 7,76-7,80 (м, 2Н); 7,92-7,96 (м, 2Н).
Пример 5
Получение соединения (1е): (R)-N-гидрокси-2-(N-изопропоксибифенил-4-илсульфонамидо)-4-(2-фенилацетамидо)бутанамид
Получение соединения (20)
По методике, аналогичной методике, применяемой для получения соединения (17) в примере 2, производное сложного эфира (15) (0,30 г, 0,59 ммоль) ацилировали фенилацетилхлоридом. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 2:1) давала требуемый продукт (55 мг, выход 16%).
1H ЯМР (CDCl3) δ: 1,10-1,22 (м, 15Н); 1,81-2,05 (м, 2H); 3,00-3,21 (м, 1Н); 3,50-3,60 (м, 3Н); 3,96 (т, J=7,5 Гц, 1Н); 4,38 (септет, 1Н); 7,28-7,37 (м, 5Н); 7,43-7,54 (м, 3Н); 7,57-7,62 (м, 2Н); 7,68-7,73 (м, 2Н); 7,80-7,84 (м, 2Н).
Получение соединения (26)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, производное сложного эфира (20) (55 мг, 0,09 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (26) (53 мг, выход 100%).
1H ЯМР (CDCl3) δ: 1,14 (д, J=3,4 Гц, 3Н); 1,17 (д, J=3,4 Гц, 3H); 1,93-2,10 (м, 2Н); 3,14 (м, 1Н); 3,47 (м, 1Н); 3,61 (с, 2Н); 4,13 (т, J=7,3 Гц, 1Н); 4,33 (септет, 1Н); 5,32 (ушир. с, 1Н); 6,14 (ушир. с, 1Н); 7,22-7,26 (м, 1Н); 7,30-7,37 (м, 4Н); 7,42-7,53 (м, 3Н); 7,58-7,63 (м, 2Н); 7,67-7,73 (м, 2Н); 7,82-7,86 (м, 2Н).
Получение соединения (32)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (26) (53 мг, 0,10 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином, получая при этом соединение (32) (54 мг, выход 80%).
Получение указанного в заголовке соединения (1е)
По методике, аналогичной методике, применяемой для получения соединения (1а) в примере 1, трет-бутил-О-силилат (32) (54 мг, 0,08 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (30 мг, выход 68%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,18-1,32 (м, 6Н); 1,83-2,22 (м, 2H); 3,10-3,28 (м, 2Н); 3,48 (с, 2Н); 4,11 (м, 1Н); 4,39 (септет, 1Н); 6,23 (ушир. с, 1Н); 7,16-7,36 (м, 5Н); 7,43-7,47 (м, 3Н); 7,57-7,61 (м, 2Н); 7,67-7,72 (м, 2Н); 7,86-7,90 (м, 2Н).
Пример 6
Получение соединения (1f): (R)-4-ацетамидо-N-гидрокси-2-(N-изопропокси-4'-метоксибифенил-4-илсульфонамидо)бутанамида
Получение соединения (9)
Раствор коммерчески доступного 4'-метоксибифенил-4-илсульфонилхлорида (1 г, 3,53 ммоль) в безводном ТГФ (8 мл) добавляли по каплям к перемешиваемому и охлажденному (0°С) раствору гидрохлорида О-изопропилгидроксиламина (0,4 г, 3,53 ммоль) и N-метилморфолина (0,77 мл, 7,06 ммоль) в безводном ТГФ (8 мл). После выдерживания 30 мин при этих условиях реакционную смесь перемешивали при комнатной температуре в течение 3 дней, затем разбавляли AcOEt и промывали Н2О, получая при этом после обработки сульфонамид (9) (0,94 г, выход 83%) в виде белого твердого вещества.
Т. пл. = 162-163°С.
1H ЯМР (CDCl3) δ: 1,20 (д, J=6,2 Гц, 6Н); 3,86 (c, 3H); 4,28 (септет, J=6,0 Гц, 1Н); 6,80 (с, 1Н); 6,97-7,04 (м, 2Н); 7,53-7,60 (м, 2Н); 7,68-7,72 (м, 2Н); 7,93-7,97 (м, 2Н).
Получение соединения (13)
трет-Бутиловый сложный эфир (13) получали из сульфонамида (9) (482 мг, 1,5 ммоль) и спирта (6) (310 мг, 1,0 ммоль) по методике, предварительно описанной для получения соединения (11) в примере 1. Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 5:2) с получением (13) (455 мг, выход 74%) в виде желтого масла.
1H ЯМР (CDCl3) δ: 1,21-1,32 (м, 15Н); 1,90-2,10 (м, 2H); 3,10-3,50 (м, 2Н); 3,86 (с, 3Н); 4,06-4,16 (м, 1Н); 4,43 (септет, J=6,2 Гц, 1Н); 5,08 (с, 2Н); 6,97-7,02 (м, 2Н); 7,34 (м, 5Н); 7,51-7,56 (м, 2Н); 7,65-7,70 (м, 2Н); 7,90-7,94 (м, 2Н).
Получение соединения (16)
По методике, аналогичной методике, применяемой для получения соединения (15), сложный эфир (13) (450 мг, 0,73 ммоль) гидрировали в присутствии 10% Pd-C, получая при этом (16) (428 мг, выход 100%).
1H ЯМР (CDCl3) δ: 1,15-1,27 (м, 15Н); 2,04-2,10 (м, 2H); 3,06 (м, 2Н); 3,86 (с, 3Н); 4,27 (м, 1Н); 4,41 (септет, J=6,2 Гц, 1Н); 6,98-7,02 (м, 2Н); 7,52-7,56 (м, 2Н); 7,68-7,72 (м, 2Н); 7,95-7,99 (м, 2Н).
Получение соединения (21)
По методике, аналогичной методике, применяемой для получения соединения (17) в примере 2, сложный эфир (16) (215 мг, 0,40 ммоль) ацилировали ацетилхлоридом. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 2:3) давала требуемый продукт (86 мг, выход 42%).
1H ЯМР (CDCl3) δ: 1,19-1,25 (м, 15Н); 1,90-2,04 (м, 5H); 3,13-3,50 (м, 2Н); 3,87 (с, 3Н); 4,12 (м, 1Н); 4,41 (септет, 1Н); 6,99-7,03 (м, 2Н); 7,53-7,57 (м, 2Н); 7,69-7,73 (м, 2Н); 7,91-7,95 (м, 2Н).
Получение соединения (27)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, сложный эфир (21) (81 мг, 0,15 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (27) (50 мг, выход 67%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,15-1,22 (м, 6Н); 1,88-2,05 (м, 5H); 3,19-3,35 (м, 2Н); 3,84 (с, 3Н); 4,24 (т, 1Н); 4,37 (септет, 1Н); 6,30 (ушир. с, 1Н); 6,96-7,00 (м, 2Н); 7,53-7,58 (м, 2Н); 7,67-7,71 (м, 2Н); 7,89-7,94 (м, 2Н).
Получение соединения (33)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (27) (50 мг, 0,10 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 1:2) давала требуемый продукт (45 мг, выход 71%).
1H ЯМР (CDCl3) δ: 0,15 (с, 6Н); 0,92 (c, 9H); 1,22-1,27 (м, 6Н); 1,91-2,00 (м, 5Н); 3,00-3,20 (м, 2Н); 3,87 (с, 3Н); 4,06-4,13 (м, 1Н); 4,43 (септет, 1Н); 5,89 (ушир. с, 1Н); 6,98-7,03 (м, 2Н); 7,56-7,60 (м, 2Н); 7,72-7,76 (м, 2Н); 7,88-7,93 (м, 2Н); 8,80 (ушир. с, 1Н).
Получение указанного в заголовке соединения (1f)
По методике, аналогичной методике, применяемой для получения соединения (1а), трет-бутил-О-силилат (33) (43 мг, 0,07 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (31 мг, выход 90%) после перекристаллизации из Et2O и н-гексана.
Т. пл. = 83-85°С;
1H ЯМР (CDCl3) δ: 1,20-1,26 (м, 6Н); 1,94-2,04 (м, 5H); 3,02-3,40 (м, 2Н); 3,86 (с, 3Н); 4,22 (м, 1Н); 4,45 (септет, 1Н); 6,09 (ушир. с, 1Н); 6,98-7,02 (м, 2Н); 7,55-7,59 (м, 2Н); 7,71-7,75 (м, 2Н); 7,89-7,94 (м, 2Н).
Пример 7
Получение соединения (1g): (R)-N-гидрокси-2-(N-изопропокси-4'-метоксибифенил-4-илсульфонамидо)-4-(2-фенилацетамидо)бутанамида
Получение соединения (22)
По методике, аналогичной методике, применяемой для получения соединения (17) в примере 2, сложный эфир (16) (200 мг, 0,37 ммоль) ацилировали фенилацетилхлоридом. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 3:2) давала требуемый продукт (53 мг, выход 24%).
1H ЯМР (CDCl3) δ: 1,16-1,25 (м, 15Н); 1,90-2,05 (м, 2H); 3,12 (м, 2Н); 3,57 (с, 2Н); 3,87 (с, 3Н); 3,94 (т, J=7,5 Гц, 1Н); 4,37 (септет, 1Н); 6,98-7,04 (м, 2Н); 7,31-7,37 (м, 5Н); 7,52-7,57 (м, 2Н); 7,64-7,68 (м, 2Н); 7,77-7,81 (м, 2Н).
Получение соединения (28)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, сложный эфир (22) (48 мг, 0,08 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (28) (46 мг, выход 100%).
1H ЯМР (CDCl3) δ: 1,15 (д, J=1,8 Гц, 3Н); 1,18 (д, J=1,8 Гц, 3H); 1,93-2,10 (м, 2Н); 3,10 (м, 2Н); 3,56 (с, 2Н); 3,86 (с, 3Н); 4,15 (м, 1Н); 4,37 (септет, 1Н); 6,98-7,02 (м, 2Н); 7,30-7,39 (м, 5Н); 7,54-7,58 (м, 2Н); 7,63-7,67 (м, 2Н); 7,80-7,84 (м, 2Н).
Получение соединения (34)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (28) (42 мг, 0,07 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 2:1) давала требуемый продукт (17 мг, выход 32%).
1H ЯМР (CDCl3) δ: 0,15 (с, 6Н); 0,92 (c, 9H); 1,17-1,25 (м, 6Н); 1,91-1,95 (м, 2Н); 3,00-3,20 (м, 2Н); 3,50 (с, 2Н); 3,87 (с, 3Н); 4,01-4,06 (м, 1Н); 4,39 (септет, 1Н); 5,75 (ушир. с, 1Н); 6,98-7,02 (м, 2Н); 7,21-7,34 (м, 5Н); 7,54-7,59 (м, 2Н); 7,67-7,71 (м, 2Н), 7,82-7,87 (м, 2Н).
Получение указанного в заголовке соединения (1g)
По методике, аналогичной методике, применяемой для получения соединения (1а), трет-бутил-О-силилат (34) (15 мг, 0,02 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (10 мг, выход 77%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,19-1,22 (м, 6Н); 1,94-1,98 (м, 2H); 3,00-3,20 (м, 2Н); 3,51 (с, 2Н); 3,86 (с, 3Н); 4,10-4,16 (м, 1Н); 4,41 (септет, 1Н); 5,89 (ушир. с, 1Н); 6,97-7,01 (м, 2Н); 7,21-7,32 (м, 5Н); 7,53-7,57 (м, 2Н); 7,65-7,69 (м, 2Н); 7,83-7,87 (м, 2Н).
Пример 8
Получение соединения (1h): (R)-бензил-4-(гидроксиамино)-4-оксо-3-(N-(2-(2-(пиперазин-1-ил)этокси)этокси)бифенил-4-илсульфонамидо)бутилкарбамата
Получение соединения (61)
К раствору 1-[2-(2-гидроксиэтокси)этил]пиперазина (60) (5,0 г, 28,7 ммоль) в CH2Cl2 (20 мл) добавляли по каплям раствор ди-трет-бутилдикарбоната (6,9 г, 31,5 ммоль) в CH2Cl2 (20 мл) при 0°С. После перемешивания при комнатной температуре в течение 12 час раствор разбавляли Et2O, промывали насыщенным раствором NaHCO3, насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая при этом Вос-защищенное производное (61) (7,2 г, выход 92%).
1H ЯМР (CDCl3) δ: 1,43 (с, 9Н); 2,45 (т, J=4,9 Гц, 4Н); 2,57 (т, J=5,3 Гц, 2Н); 3,44 (т, J=5,1 Гц, 4Н); 3,56-3,67 (м, 6Н); 4,17 (т, J=5,6 Гц, 1Н).
13С ЯМР (CDCl3) δ: 28,47; 43,46; 53,16; 57,97; 61,86; 67,69; 72,44; 79,69; 155,00.
Получение соединения (62)
Диэтилазодикарбоксилат (DEAD) (2,15 мл, 13,65 ммоль) по каплям добавляли к раствору, содержащему спирт (61) (2,50 г, 9,10 ммоль), N-гидроксифталимид (1,48 г, 9,10 ммоль) и трифенилфосфин (3,58 г, 13,6 ммоль) в безводном ТГФ (100 мл), в атмосфере азота. Образовавшийся раствор перемешивали на протяжении ночи при к.т. и упаривали при пониженном давлении с получением сырого продукта, который очищали флэш-хроматографией на силикагеле (н-гексан/AcOEt = 4:1), получая при этом (62) (4,30 г, выход 98%) в виде чистого желтого масла.
1H ЯМР (CDCl3) δ: 1,44 (с, 9Н); 2,38 (т, J=4,9 Гц, 4Н); 2,50 (т, J=5,6 Гц, 2Н); 3,39 (т, J=5,3 Гц, 4Н); 3,64 (т, J=5,6 Гц, 2Н); 3,82 (т, J=4,2 Гц, 2Н); 4,37 (т, J=4,3 Гц, 2Н); 7,72-7,86 (м, 4Н).
Получение соединения (55)
Гидрат гидразина (1,73 мл, 35,87 ммоль) добавляли к раствору соединения (62) (4,30 г, 10,25 ммоль) в этаноле (210 мл). После перемешивания при комнатной температуре в течение 14 час смесь фильтровали и фильтрат концентрировали. Остаток разбавляли AcOEt, осадок удаляли фильтрованием и фильтрат упаривали, получая при этом требуемый О-алкилгидроксиламин (55) (2,15 г, выход 72,6%).
1H ЯМР (CDCl3) δ: 1,45 (с, 9Н); 2,45 (т, J=4,9 Гц, 4Н); 2,61 (т, J=5,8 Гц, 2Н); 3,43 (т, J=5,1 Гц, 4Н); 3,58-3,65 (м, 4Н); 3,80-3,85 (м, 2Н).
Получение соединения (10)
По методике, аналогичной методике, применяемой для получения соединения (7) в примере 1, О-алкилгидроксиламин (55) (2,85 г, 9,86 ммоль) подвергают реакции с бифенил-4-сульфонилхлоридом (56). Колоночная хроматография на силикагеле (AcOEt) продукта давала требуемый сульфонамид (3,05 г, выход 61%).
1H ЯМР (CDCl3) δ: 1,44 (с, 9Н); 2,44 (т, J=4,9 Гц, 4Н); 2,58 (т, J=5,6 Гц, 2Н); 3,43 (т, J=5,3 Гц, 4Н); 3,61 (т, J=5,5 Гц, 2Н); 3,67-3,71 (м, 2Н); 4,10-4,15 (м, 2Н); 7,41-7,53 (м, 3Н); 7,58-7,63 (м, 2Н); 7,71-7,75 (м, 2Н); 7,97-8,01 (м, 2Н).
Получение соединения (14)
трет-Бутиловый сложный эфир (14) получали из сульфонамида (10) (1,18 г, 2,33 ммоль) и спирта (6) по методике, ранее описанной для получения соединения (11) в примере 1. Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 1:5), получая при этом требуемый продукт (1,39 г, выход 75%) в виде желтого масла.
1H ЯМР (CDCl3) δ: 1,33 (с, 9Н); 1,43 (c, 9H); 1,74-1,98 (м, 2Н); 2,42 (м, 4Н); 2,58 (т, J=5,3 Гц, 2Н); 3,12-3,26 (м, 2Н); 3,40 (м, 4Н); 3,56-3,65 (м, 4Н); 4,09-4,41 (м, 3Н); 5,05 (с, 2Н); 5,22 (ушир. с, 1Н); 7,32 (м, 5Н); 7,40-7,74 (м, 7Н); 7,96-8,00 (м, 2Н).
Получение соединения (36)
По методике, аналогичной методике, применяемой для получения соединения (35) в примере 1, сложный эфир (14) (1,39 г, 1,74 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (36) (1,30 г, выход 86%) после перекристаллизации из Et2O.
1H ЯМР (CDCl3) δ: 1,80-2,05 (м, 2Н); 3,19-3,29 (м, 6H); 3,40 (м, 4Н); 3,62 (м, 10Н); 4,15 (м, 2Н); 4,29 (т, J=6,5 Гц, 1Н); 5,01 (с, 2Н); 5,35 (ушир. с, 1Н); 7,29 (м, 5Н); 7,43-7,52 (м, 3Н); 7,57-7,63 (м, 2Н); 7,70-7,74 (м, 2Н); 7,90-7,94 (м, 2Н).
Получение указанного в заголовке соединения (1h)
По методике, аналогичной методике, применяемой для получения соединения (37) в примере 1, карбоновую кислоту (36) (0,14 г, 0,16 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (CHCl3/МеОН = 9:1), получая при этом требуемую гидроксамовую кислоту (13 мг, выход 12%).
1H ЯМР (CDCl3) δ: 1,80-2,00 (м, 2Н); 2,47-3,17 (м, 16H); 4,13 (м, 3Н); 4,92 (с, 2Н); 6,25 (ушир. с, 1Н); 7,18 (м, 5Н); 7,38-7,64 (м, 7Н); 7,96 (м, 2Н).
Пример 9
Получение соединения (2): (R)-4-(1,3-диоксоизоиндолин-2-ил)-N-гидрокси-2-(N-изопропокси-4'-метоксибифенил-4-илсульфонамидо)бутанамида
Получение соединения (40)
По методике, аналогичной методике, применяемой для получения соединения (6) в примере 1, (S)-α-гидрокси-1,3-диоксо-2-изоиндолинмасляную кислоту (39) (1,0 г, 4,0 ммоль) обрабатывали ди-трет-бутилацеталем N,N-диметилформамида. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 3:2) продукта дала требуемый сложный эфир (530 мг, выход 43%) в виде белого твердого вещества.
Т. пл. = 122-123°С.
1H ЯМР (CDCl3) δ: 1,46 (с, 9Н); 1,87-2,23 (м, 2H); 3,02 (д, J=5,3 Гц, 1Н); 3,86 (т, J=7,3 Гц, 2Н); 4,07-4,16 (м, 1Н); 7,69-7,75 (м, 2Н); 7,80-7,87 (м, 2Н).
Получение соединения (43)
трет-Бутиловый сложный эфир (43) получали из сульфонамида (9) (400 мг, 1,24 ммоль) и спирта (40) (250 мг, 0,82 ммоль) по методике, ранее описанной для получения соединения (11). Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 2:1), получая при этом требуемый продукт (217 мг, выход 43%).
1H ЯМР (CDCl3) δ: 1,24-1,29 (м, 15Н); 2,10-2,30 (м, 2H); 3,51-3,74 (м, 2Н); 3,87 (с, 3Н); 4,06-4,23 (м, 1Н); 4,46 (септет, 1Н); 6,98-7,03 (м, 2Н); 7,52-7,88 (м, 10Н).
Получение соединения (46)
По методике, аналогичной методике, применяемой для соединения (35), сложный эфир (43) (205 мг, 0,33 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (46) (135 мг, выход 74%) после перекристаллизации из Et2O.
Т. пл. = 185-187°С.
1H ЯМР (CDCl3) δ: 1,21 (д, J=6,2 Гц, 3Н); 1,27 (д, J=6,2 Гц, 3Н); 2,10-2,28 (м, 2Н); 3,61 (м, 2Н); 3,87 (с, 3Н); 4,33 (м, 1Н); 4,46 (септет, 1Н); 6,98-7,02 (м, 2Н); 7,50-7,54 (м, 4Н); 7,65-7,85 (м, 6Н).
Получение соединения (48)
По методике, аналогичной методике, применяемой для получения соединения (37), карбоновую кислоту (46) (130 мг, 0,23 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 3:2) давала требуемый продукт (110 мг, выход 70%).
1H ЯМР (CDCl3) δ: 0,26 (с, 6Н); 1,00 (c, 9H); 1,23 (д, J=6,2 Гц, 3Н); 1,30 (д, J=6,2 Гц, 3Н); 2,15-2,30 (м, 2Н); 3,30-3,60 (м, 2Н); 3,89 (с, 3Н); 4,03 (м, 1Н); 4,43 (септет, 1Н); 6,98-7,02 (м, 2Н); 7,16 (м, 2Н); 7,36-7,41 (м, 2Н); 7,61 (м, 4Н); 7,68-7,72 (м, 2Н); 8,72 (ушир. с, 1Н).
Получение соединения (2)
По методике, аналогичной методике, применяемой для соединения (1а), трет-бутил-О-силилат (48) (100 мг, 0,14 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (61 мг, 77%) после перекристаллизации из Et2O и н-гексана.
Т. пл. = 75-76°С.
1H ЯМР (CDCl3) δ: 1,23 (д, J=6,2 Гц, 3Н); 1,30 (д, J=6,2 Гц, 3Н); 2,12-2,32 (м, 2Н); 3,40-3,53 (м, 2Н); 3,88 (с, 3Н); 4,02-4,20 (м, 1Н); 4,46 (септет, 1Н); 6,98-7,02 (м, 2Н); 7,29-7,39 (м, 2Н); 7,42-7,46 (м, 2Н); 7,63 (м, 4Н); 7,72-7,76 (м, 2Н).
Пример 10
Получение соединения (3): (R)-4-(1,3-диоксоизоиндолин-2-ил)-N-гидрокси-2-(N-изопропокси-4-феноксифенилсульфонамидо)бутанамида
Получение соединения (41)
По методике, аналогичной методике, применяемой для получения соединения (7), О-изопропилгидроксиламин (54) (415 мг, 3,72 ммоль) подвергают реакции с 4-феноксибензолсульфонилхлоридом (58), получая при этом требуемый сульфонамид (41) (989 мг, выход 86%).
1H ЯМР (CDCl3) δ: 1,19 (д, J=6,2 Гц, 6Н); 4,25 (септет, J=6,2 Гц, 1Н); 6,71 (с, 1Н); 7,03-7,10 (м, 4Н); 7,19-7,27 (м, 1Н); 7,38-7,46 (м, 2Н); 7,83-7,88 (м, 2Н).
Получение соединения (44)
трет-Бутиловый сложный эфир (44) получали из сульфонамида (41) (378 мг, 1,12 ммоль) и спирта (40) по методике, ранее описанной для соединения (11). Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 7:2), получая при этом требуемый продукт (253 мг, выход 51%).
1H ЯМР (CDCl3) δ: 1,18-1,45 (м, 15Н); 1,99-2,38 (м, 2H); 3,56-3,74 (м, 2Н); 4,06-4,15 (м, 1Н); 4,43 (септет, J=6,2 Гц, 1Н); 6,94-6,98 (м, 2Н); 7,09-7,12 (м, 2Н); 7,21-7,28 (м, 2Н); 7,39-7,47 (м, 2Н); 7,67-7,83 (м, 5Н).
Получение соединения (47)
По методике, аналогичной методике, применяемой для получения соединения (35), сложный эфир (44) (250 мг, 0,42 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (47) (164 мг, 71%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,20 (д, J=6,2 Гц, 3Н); 1,25 (д, J=6,2 Гц, 3Н); 2,10-2,30 (м, 2Н); 3,55-3,75 (м, 2Н); 4,25-4,35 (м, 1Н); 4,44 (септет, J=6,2 Гц, 1Н); 6,88-6,92 (м, 2Н); 7,09-7,13 (м, 2Н); 7,21-7,28 (м, 2Н); 7,39-7,47 (м, 2Н); 7,69-7,84 (м, 5Н).
Получение соединения (49)
По методике, аналогичной методике, применяемой для получения соединения (37), карбоновую кислоту (47) (160 мг, 0,30 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином, получая при этом требуемый продукт (178 мг, выход 87%).
1H ЯМР (CDCl3) δ: 0,26 (с, 6Н); 1,00 (с, 9Н); 1,21 (д, J=6,2 Гц, 3Н); 1,26 (д, J=6,2 Гц, 3Н); 2,10-2,31 (м, 2Н); 3,40-3,65 (м, 2Н); 3,90-4,10 (м, 1Н); 4,40 (септет, J=6,2 Гц, 1Н); 6,59 (ушир. с, 1Н); 7,08-7,13 (м, 2Н); 7,22-7,30 (м, 2Н); 7,41-7,49 (м, 2Н); 7,58-7,62 (м, 2Н); 7,69-7,83 (м, 5Н).
Получение указанного в заголовке соединения (3)
По методике, аналогичной методике, применяемой для получения соединения (1а), трет-бутил-О-силилат (49) (170 мг, 0,25 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (3) (97 мг, выход 68%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,22 (д, J=6,2 Гц, 3Н); 1,26 (д, J=6,2 Гц, 3Н); 2,07-2,28 (м, 2Н); 3,43-3,65 (м, 2Н); 4,05-4,20 (м, 1Н); 4,44 (септет, J=6,2 Гц, 1Н); 6,70-6,83 (м, 2Н); 7,09-7,13 (м, 2Н); 7,22-7,29 (м, 2Н); 7,40-7,48 (м, 2Н); 7,64-7,83 (м, 5Н).
Пример 11
Получение соединения (4): (R)-4-(1,3-диоксоизоиндолин-2-ил)-2-(4'-этокси-N-изопропоксибифенил-4-илсульфонамидо)-N-гидроксибутанамид
Получение соединения (42)
По методике, аналогичной методике, применяемой для получения соединения (7), О-изопропилгидроксиламин (54) (436 мг, 3,91 ммоль) подвергают реакции с 4-бромбензолсульфонилхлоридом (59), получая при этом требуемый сульфонамид (42) (870 мг, 75%).
1H ЯМР (CDCl3) δ: 1,18 (д, J=6,2 Гц, 6Н); 4,25 (септет, J=6,2 Гц, 1Н); 6,80 (с, 1Н); 7,66-7,70 (м, 2Н); 7,75-7,80 (м, 2Н).
Получение соединения (45)
трет-Бутиловый сложный эфир (45) получали из сульфонамида (42) (864 мг, 2,94 ммоль) и спирта (40) по методике, ранее описанной для получения соединения (11). Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 5:1), получая при этом требуемый продукт (720 мг, выход 63%).
1H ЯМР (CDCl3) δ: 1,19-1,40 (м, 15Н); 2,05-2,20 (м, 2Н); 3,55-3,80 (м, 2Н); 4,02-4,20 (м, 1Н); 4,43 (септет, J=6,2 Гц, 1Н); 7,60-7,76 (м, 6Н); 7,80-7,87 (м, 2Н).
Получение соединения (50)
Смесь сложного эфира (45) (200 мг, 0,34 ммоль), 4-этоксифенилбороновой кислоты (96 мг, 0,58 ммоль), Pd(PPh3)4 (20 мг, 0,017 ммоль) и К3РО4 (166 мг, 0,78 ммоль) в 4,5 мл смеси 5:1 диоксан/Н2О нагревали до 85°С в атмосфере азота. Спустя 2 час реакционную смесь разбавляли насыщенным раствором NaHCO3, экстрагировали AcOEt и сушили над Na2SO4. Органический слой концентрировали в вакууме и сырой продукт очищали флэш-хроматографией на силикагеле (н-гексан/AcOEt = 4:1), получая при этом (50) (193 мг, выход 88%).
1H ЯМР (CDCl3) δ: 1,20-1,39 (м, 15Н); 1,45 (т, J=6,9 Гц, 3Н); 2,04-2,35 (м, 2Н); 3,50-3,80 (м, 2Н); 4,05-4,15 (м, 3Н); 4,46 (септет, J=6,2 Гц, 1Н); 6,97-7,01 (м, 2Н); 7,51-7,88 (м, 10Н).
Получение соединения (51)
По методике, аналогичной методике, применяемой для получение соединения (35), сложный эфир (50) (193 мг, 0,30 ммоль) обрабатывали TFA, получая при этом требуемую карбоновую кислоту (51) (150 мг, 87%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,21 (д, J=6,2 Гц, 3Н); 1,24 (д, J=6,2 Гц, 3Н); 1,46 (т, J=6,9 Гц, 3Н); 2,10-2,30 (м, 2Н); 3,50-3,75 (м, 2Н); 4,10 (кв, J=6,9 Гц, 2Н); 4,22-4,50 (м, 2Н); 6,97-7,01 (м, 2Н); 7,48-7,53 (м, 4Н); 7,65-7,85 (м, 6Н).
Получение соединения (52)
По методике, аналогичной методике, применяемой для получения соединения (37), карбоновую кислоту (51) (140 мг, 0,25 ммоль) сочетали с О-(трет-бутилдиметилсилил)гидроксиламином. Колоночная хроматография на силикагеле (н-гексан/AcOEt = 2:1) давала требуемый продукт (120 мг, выход 71%).
1H ЯМР (CDCl3) δ: 0,26 (с, 6Н); 1,00 (с, 9Н); 1,23 (д, J=6,2 Гц, 3Н); 1,30 (д, J=6,2 Гц, 3Н); 1,47 (т, J=6,9 Гц, 3Н); 2,10-2,30 (м, 2Н); 3,30-3,60 (м, 2Н); 3,90-4,05 (м, 1Н); 4,11 (кв., J=6,9 Гц, 2Н); 4,43 (септет, J=6,2 Гц, 1Н); 6,99-7,01 (м, 2Н); 7,10-7,22 (м, 2Н); 7,35-7,39 (м, 2Н); 7,60-7,71 (м, 6Н); 8,70 (ушир. с, 1Н).
Получение указанного в заголовке соединения (4)
По методике, аналогичной методике, применяемой для получения соединения (1а), трет-бутил-О-силилат (52) (120 мг, 0,17 ммоль) обрабатывали TFA, получая при этом требуемую гидроксамовую кислоту (4) (83 мг, 82%) после перекристаллизации из Et2O и н-гексана.
1H ЯМР (CDCl3) δ: 1,24 (д, J=6,2 Гц, 3Н); 1,30 (д, J=6,2 Гц, 3Н); 1,46 (т, J=6,9 Гц, 3Н); 2,10-2,35 (м, 2Н); 3,40-3,60 (м, 2Н); 4,05-4,16 (м, 3Н); 4,46 (септет, J=6,2 Гц, 1Н); 6,96-7,01 (м, 2Н); 7,31-7,44 (м, 4Н); 7,63-7,75 (м, 6Н).
Пример 12
Получение соединения (64): бензил-3-(N-изопропоксибифенил-4-илсульфонамидо)пропилкарбамата
Карбамат (64) получали из сульфонамида (8) (400 мг, 1,37 ммоль) и коммерческого спирта (63) по методике, ранее описанной для получения соединения (11). Сырую реакционную смесь очищали флэш-хроматографией (н-гексан/AcOEt = 3:1), получая при этом требуемый продукт (400 мг, выход 87%).
1H ЯМР (CDCl3) δ: 1,23-1,27 (м, 8Н); 1,76-1,89 (м, 2Н); 3,30 (кв., J=6,2 Гц, 2Н); 4,54 (септет, J=6,2 Гц, 1Н); 5,08 (с, 2Н); 7,34 (м, 5Н); 7,41-7,53 (м, 3Н); 7,59-7,63 (м, 2Н); 7,72-7,76 (м, 2Н); 7,88-7,93 (м, 2Н).
Пример 13
Получение соединения (1i)
Получение соединения (65)
К раствору соединения (55) примера 8 (1,76 г; 6 ммоль) в ТГФ (15 мл) при 0°С добавляли N-метилморфолин (0,66 мл, 6 ммоль) и раствор 4'-метокси-[1,1'-бифенил]-4-сульфонилхлорида (1,69 г, 6 ммоль) в ТГФ (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней, затем концентрировали. Остаток растворяли в этилацетате, промывали водой и органическую фазу концентрировали с получением оранжевого масла. Сырой продукт очищали флэш-хроматографией (EtOAc), получая при этом соединение (65) в виде желтого масла с выходом 75%.
1H ЯМР (CDCl3, 200 МГц): 7,95 (д, 2Н); 7,68 (д, 2Н); 7,55 (д, 2Н); 7,0 (д, 2Н); 4,12 (м, 2Н); 3,86 (с, 3Н); 3,67 (м, 4Н); 3,6 (м, 4Н); 3,42 (т, J=5,3 Гц, 4Н); 2,56 (т, J=5,6 Гц, 2Н); 2,42 (т, J=4,95 Гц, 4Н); 1,44 (с, 9Н).
Получение соединения (66)
DEAD (0,153 мл, 0,97 ммоль) добавляли по каплям при 0°С к раствору сульфонамида (65) (458,8 мг; 0,97 ммоль), соединения (6) примера 1 (300 мг, 0,97 ммоль) и трифенилфосфина (255 мг; 0,97 ммоль) в сухом ТГФ (16 мл).
Смесь перемешивали при комнатной температуре на протяжении ночи, затем концентрировали и очищали флэш-хроматографией (гексан/этилацетат, 1/5), получая при этом соединение (66) в виде масла с выходом 70%.
1H ЯМР (CDCl3, 200 МГц): 7,95 (д, J=8,6 Гц, 2Н); 7,57 (м, 4Н); 7,26 (с, 5Н); 7,0 (д, J=8,8 Гц, 2Н); 5,18 (м, 1Н); 5,06 (с, 2Н); 4,33 (м, 1Н); 4,14 (м, 2Н); 3,87 (с, 3Н); 3,62 (м, 4Н); 3,39 (м, 4Н); 3,21 (м, 2Н); 2,56 (т, J=5,4 Гц, 2Н); 2,4 (м, 4Н); 1,92 (м, 2Н); 1,44 (с, 9Н); 1,34 (с, 9Н).
Получение соединения (1i)
К раствору трет-бутилового сложного эфира (66) (800 мг, 0,966 ммоль) в сухом CH2Cl2 (12 мл) по каплям при 0°С добавляли TFA (7,4 мл, 96,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 час и затем 4 час при комнатной температуре. Растворитель и TFA удаляли в вакууме, получая при этом карбоновую кислоту (1i) как дитрифторацетатную соль в виде белой пены с выходом 79%.
1H ЯМР (CDCl3, 200 МГц): 9,45 (ушир. с, +NH2, +NH); 8,0 (д, J=7,8 Гц, 2Н); 7,68 (м, 4Н); 7,4 (с, 5Н); 7,1 (д, J=8,2 Гц, 2Н); 5,6 (с, 1Н); 5,13 (с, 2Н); 4,27 (м, 3Н); 3,97 (с, 3Н); 3,78 (м, 10Н); 3,44 (м, 4Н); 1,98 (м, 2Н).
Пример 14
Получение соединения (1j)
К раствору кислоты (1i) (150 мг, 0,167 ммоль) в сухом CH2Cl2 (4 мл) добавляли О-(трет-бутилдиметилсилил)гидроксиламин (79 мг, 0,534 ммоль) и EDCI (96 мг, 0,501 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 час, затем концентрировали и остаток растворяли в EtOAc и промывали водой. Органическую фазу сушили (Na2SO4) и упаривали в вакууме. Полученное таким образом белое твердое вещество (140 мг) растворяли в сухом CH2Cl2 (3 мл) и обрабатывали TFA (0,8 мл). Реакционную смесь перемешивали при 0°С в течение 1 час, затем давали ей возможность достичь комнатной температуры и перемешивали в течение 4 час. Раствор концентрировали с получением коричневого масла, которое растирали со смесью CH2Cl2/Et2O, получая при этом дитрифторацетат гидроксамовой кислоты (1j) в виде коричневого твердого вещества с выходом 50%.
1H ЯМР (CDCl3, 200 МГц): 7,86 (д, J=7,2 Гц, 2Н); 7,56 (м, 4Н); 7,24 (с, 5Н); 6,94 (д, J=8,2 Гц, 2Н); 5,73 (с, 1Н), 5,1 (с, 2Н); 4,1 (м, 4Н); 3,82 (с, 3Н); 3,53 (м, 8Н); 3,25 (м, 4Н), 2,05 (м, 2Н).
Пример 15
Получение соединения (1k)
К раствору (1j) (40 мг, 0,04 ммоль) в сухом ДМСО (1 мл) добавляли 2,5-диоксипирролидин-1-илпропионат (10 мг, 0,534 ммоль) и ТЕА (10 капель, рН=8). Реакционную смесь перемешивали при комнатной температуре в течение 24 час, затем добавляли 10 мл Et2O. Реакцию останавливали и раствор охлаждали до -15°С, затем оставляли до достижения 0°С. Слой этилового простого эфира декантировали и оставшееся желтое масло концентрировали в вакууме. Чистое соединение (1k) получали после хроматографии (CH2Cl2/МеОН) в виде бесцветного масла с выходом 24%.
1H ЯМР (CDCl3, 200 МГц): 7,88 (д, J=8,2 Гц, 2Н); 7,6 (м, 4Н); 7,26 (с, 5Н); 6,96 (д, J=8,8 Гц, 2Н); 5,09 (с, 2Н), 4,34 (м, 3Н); 3,87 (с, 3Н); 3,62 (м, 6Н); 3,45 (м, 4Н), 2,59 (м, 2Н); 2,46 (м, 4Н); 2,28 (кв, J=7,5 Гц, 2Н), 1,68 (м, 2Н); 1,11 (т, J=7,5 Гц, 3Н).
Пример 16
Анализы ингибирования ММР
Рекомбинантная про-ММР-2 человека была поставлена Prof. Gillian Murphy (Department of Oncology, University of Cambridge, UK), тогда как про-ММР-13 и про-ММР-14 были куплены у Calbiochem. Проферменты активировали непосредственно перед применением ацетатом п-аминофенилртути(II) (АРМА: 2 мМ в течение 1 час при 37°С для про-ММР-2 и 1 мМ в течение 30 мин при 37°С для про-ММР-13). Про-ММР-14 активировали трипсином 5 мкг/мл в течение 15 мин при 37°С с последующей обработкой ингибитором соевого трипсина (SBTI) 23 мкг/мл. Для измерений величин анализа исходные растворы ингибитора (ДМСО, 100 мМ) дополнительно разбавляли для получения семи разных концентраций (0,01 нМ - 100 мкМ) для каждой ММР в буфере для флуориметрического анализа (FAB: 50 мМ Трис, рН=7,5, 150 мМ NaCl, 10 мМ CaCl2, 0,05% Brij 35 и 1% ДМСО). Активированный фермент (конечная концентрация 2,9 нМ для ММР-2, 1 нМ для ММР-14, 0,33 нМ для ММР-13) и растворы ингибитора инкубировали в буфере для анализа в течение 4 час при 25°С. После добавления 200 мкМ раствора флуорогенного субстрата Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (Bachem) (для ссылки см. Neumann, U.; Kubota, H.; Frei, K.; Ganu, V.; Leppert, D. Anal. Biochem. 2004, 328, 166) для всех ферментов в ДМСО (конечная концентрация 2 мкМ), мониторинг гидролиза проводили каждые 15 сек в течение 20 мин, регистрируя повышение во флуоресценции (λex=325 нм, λem=395 нм) с применением аппарата для прочтения планшетов Molecular Device SpectraMax Gemini XS.
Анализы проводили в трех повторностях в общем объеме 200 мкл на лунку в 96-луночных титрационных микропланшетах (Corning, Black, NBS). В контрольных лунках отсутствовал ингибитор. Активность ингибирования ММР выражали в относительных единицах флуоресценции (RFU). Процент ингибирования вычисляли из контрольных реакций без ингибитора. IC50 определяли с применением формулы: Vi/Vo=1/(1+[I]/IC50), где Vi представляет собой начальную скорость расщепления субстрата в присутствии ингибитора при концентрации [I] и V0 представляет собой начальную скорость в отсутствие ингибитора. Результаты анализировали с применением программного обеспечения SoftMax Pro и программного обеспечения GraFit.
|
Как ясно показано в приведенной выше таблице 1, соединения формулы (I) изобретения обладают значительной ингибирующей активностью против ММР-2, ММР-13 и ММР-14 и поэтому могут быть применимыми в терапии для лечения дегенеративных нарушений, в которых принимают участие те же самые ферменты.
Как ни странно, на основании этого очень чувствительного способа оценки ингибирующей активности против испытуемых ферментов можно прийти к выводу, что соединения изобретения обладают ингибирующей активностью, заметно более высокой, чем проявляло структурно близкое ссылочное соединение (5b) известного уровня техники. Интересно, что ингибирующая активность соединений изобретения была значительно выше, чем ингибирующая активность ссылочного соединения (5b), по меньшей мере на один порядок выше его величины, для ММР-2; сравнимый профиль ингибирования можно было также наблюдать при рассмотрении ингибирования ММР-13 и ММР-14, хотя и в меньшей степени. Кроме того, такой профиль постоянно имел общие характеристики и постоянно наблюдался при испытании всего класса репрезентативных соединений изобретения. Замечательным является тот факт, что помимо того, что соединения являются особенно активными при ингибировании вышеуказанных металлопротеаз ММР-2, ММР-13 и ММР-14, когда каждая рассматривается отдельно, соединения изобретения являются эффективными при ингибировании всех этих ферментов, что является особенно благоприятным в терапии, где известно, что все они играют роль в дегенеративных процессах.
Пример 17
Эксперимент по ингибированию in vivo
Соединение (2) как репрезентативное соединение изобретения испытывали в анализе in vivo для оценки его ингибирующей активности на раковых клетках. В частности, культуры HUVEC, купленные у Cascade Biologies (Portland, OR), культивировали на покрытых желатином колбах с применением М199, 10% FCS (фетальная бычья сыворотка, Seromed, Milan, Italy), дополненной FGF (фибробластный фактор роста, PeproTech, Inc., Rocky Hill, NJ) (1 мкг aFGF (кислотный фибробластный фактор роста) + 1 мкг bFGF (основный фибробластный фактор роста)/100 мл среды), EGF (эпидермальный фактор роста) (1 мкг/100 мл среды), гепарином (10 мг/100 мл среды), (ICN Pharmaceuticals, Inc., Casta Mesa, CA), гидрокортизоном (Sigma Chemical Co.) (0,1 мг/100 мл среды).
Анализ роста
На день 0 клетки засевали в 96-луночных микропланшетах при плотности 1000 клеток в 200 мкл полной культуральной среды. К клеткам добавляли ингибитор при различных концентрациях (10, 100 мкМ). HUVEC количественно определяли через 24, 48 и 72 ч. Непрямую количественную оценку оптической плотности (OD) получали фиксацией/окрашиванием клеток в течение 20 минут раствором кристалла фиолетового [см. для ссылки Fassina, G. et al., Clin Cancer Res, (2004), 10, 4865-73].
Хемоинвазия
Инвазию HUVEC анализировали в камерах Boyden (Costar) с применением не содержащей сыворотку фибробластной кондиционированной среды (FB-CM) в качестве хемоаттрактантов. Клетки обрабатывали ингибитором при указанных дозах и инкубировали при 37°С в течение 5 часов в увлажненной атмосфере. Инвазию клеток анализировали денситометрическим сканированием всей поверхности фильтра. Каждый тест проводили в трех повторностях и повторяли три раза [см. для ссылки Albini, A., et al., Int. J. Dev. Biol., (2004), 48, 563-71].
Морфогенез на матригеле
Матригель оттаивали при 4°С на бане лед-вода и 200 мкл концентрированного раствора (10 мг/мл) пипетировали в лунки для тканевых культур диаметром 13 мм, избегая появления даже маленьких пузырьков. Матригель затем полимеризовали в течение 1 час при 37°С. После протекания полимеризации 7×104 клеток в 1 мл полной среды осторожно пипетировали на верх геля. Планшеты затем инкубировали при 37°С в увлажненной атмосфере с 5% СО. Проводили мониторинг анализов и для фотографирования применяли инвертированный микроскоп.
Эндотелиальные клетки образовывали взаимосвязанные сетки в матригеле, которые уменьшались антиангиогенными соединениями (см. для ссылки вышеуказанную публикацию Albini, A. et al.).
Результаты
Испытуемое соединение (2) проявляло зависимое как от времени, так и от дозы ингибирование роста клеток по сравнению с необработанными контролями. При испытании с более высокой дозой (100 мкМ) авторы изобретения наблюдали 100% ингибирование, начиная от 48 часов обработки. При анализе инвазии соединение (2) при испытании с более высокой дозой (100 мкМ) обнаружило значительную ингибирующую активность в отношении миграции эндотелиальных клеток через реконструированную базальную мембрану. При высокой дозе (100 мкМ) соединение (2) начинало проявлять некоторую проапоптотическую активность. Через 48 ч соединение проявляло способность индуцировать программированную гибель клеток. Согласно этим данным, соединение (2), по-видимому, является особенно активным в качестве ингибитора активности раковых клеток.
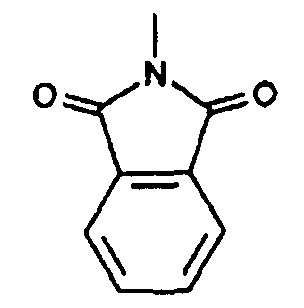
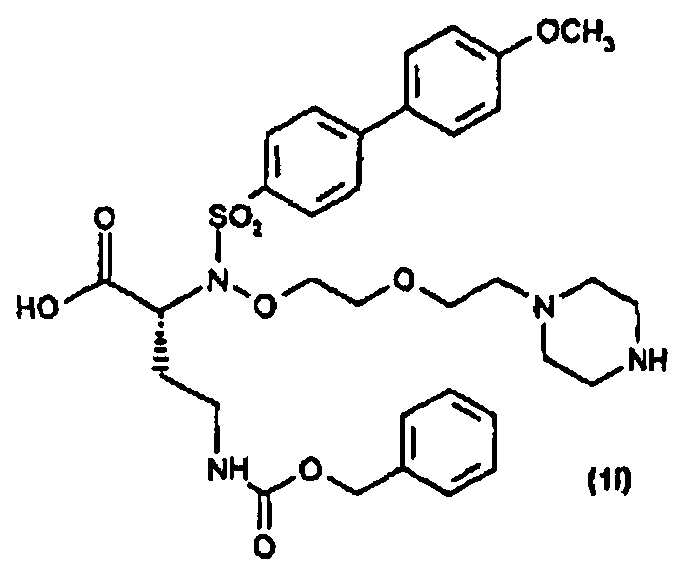
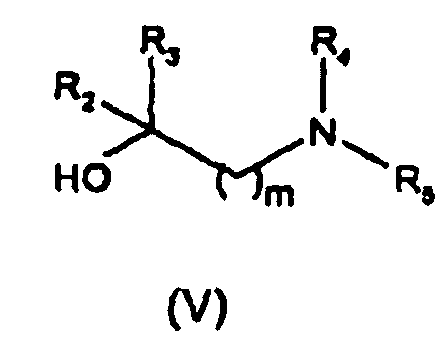
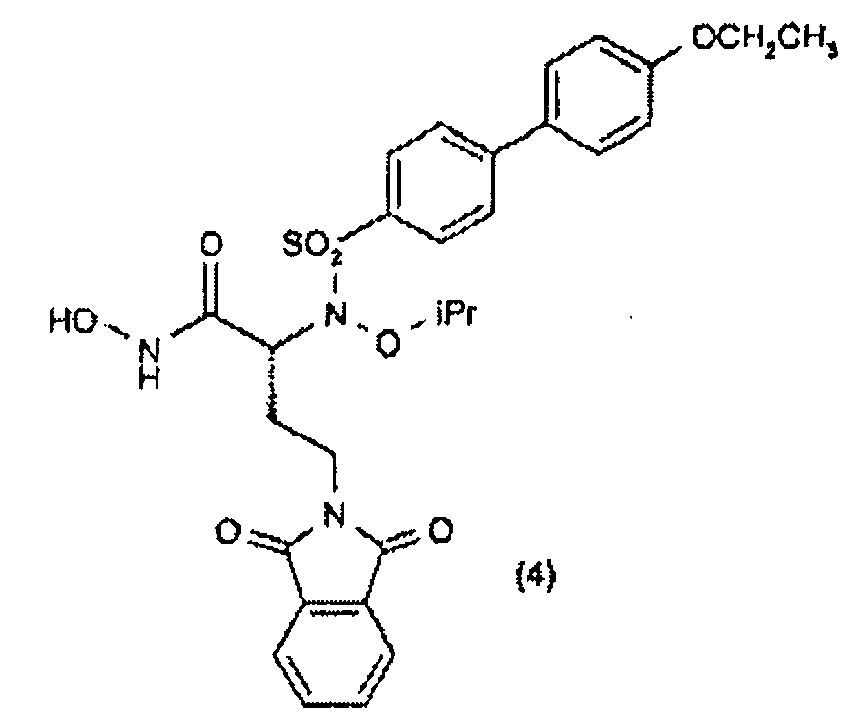
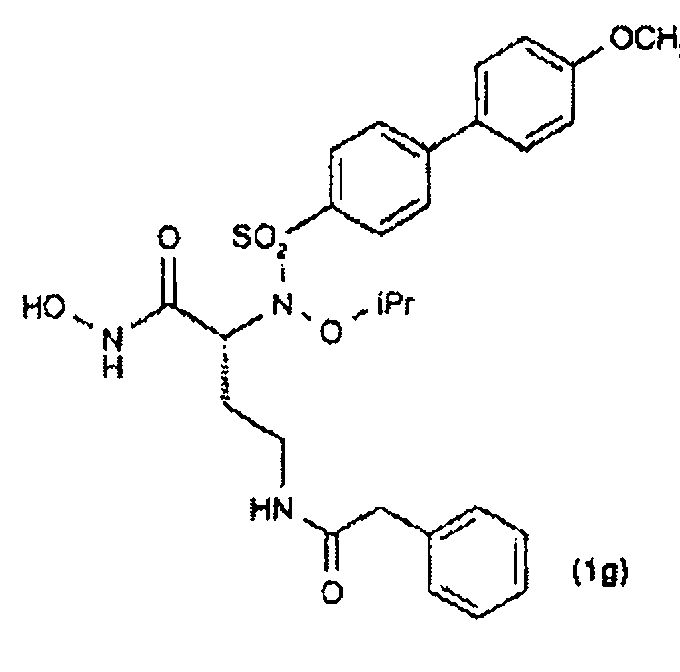
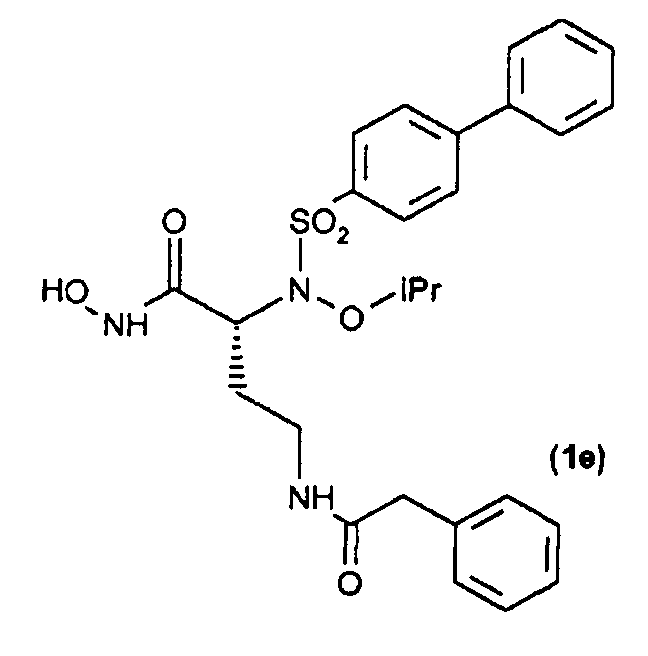
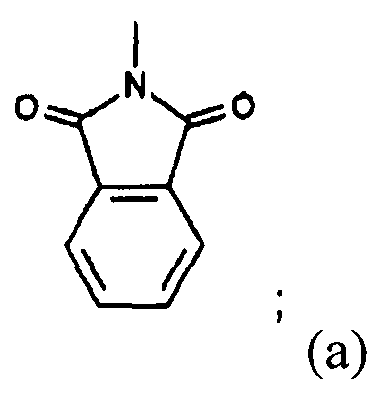
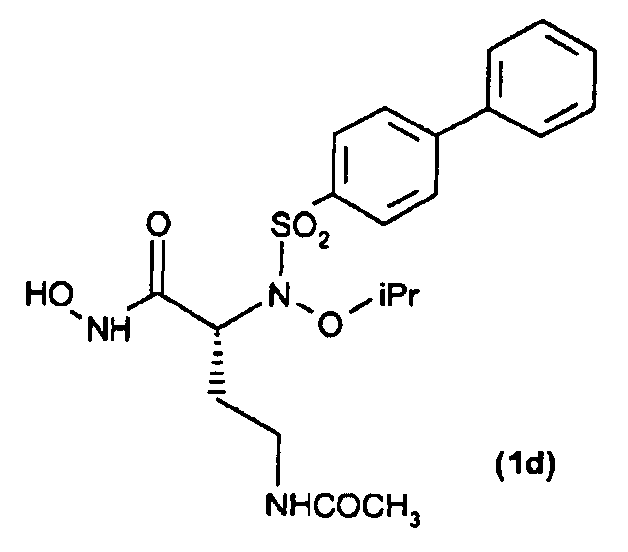
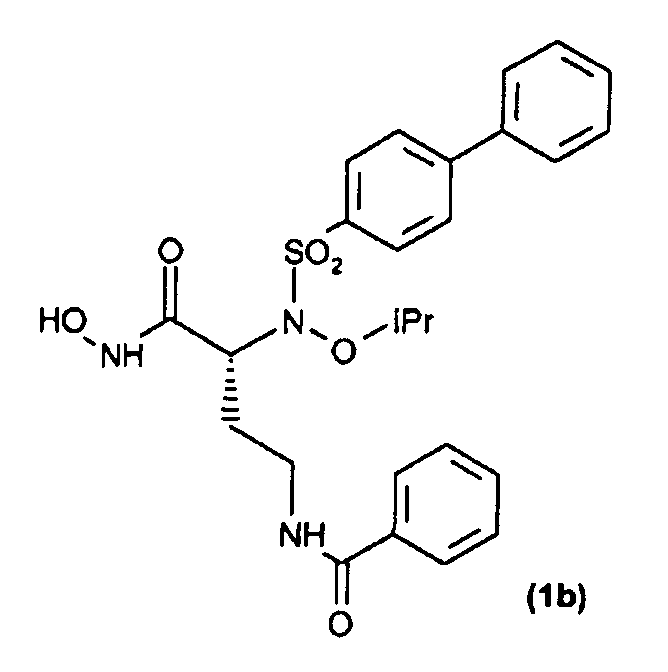
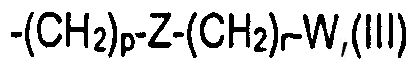
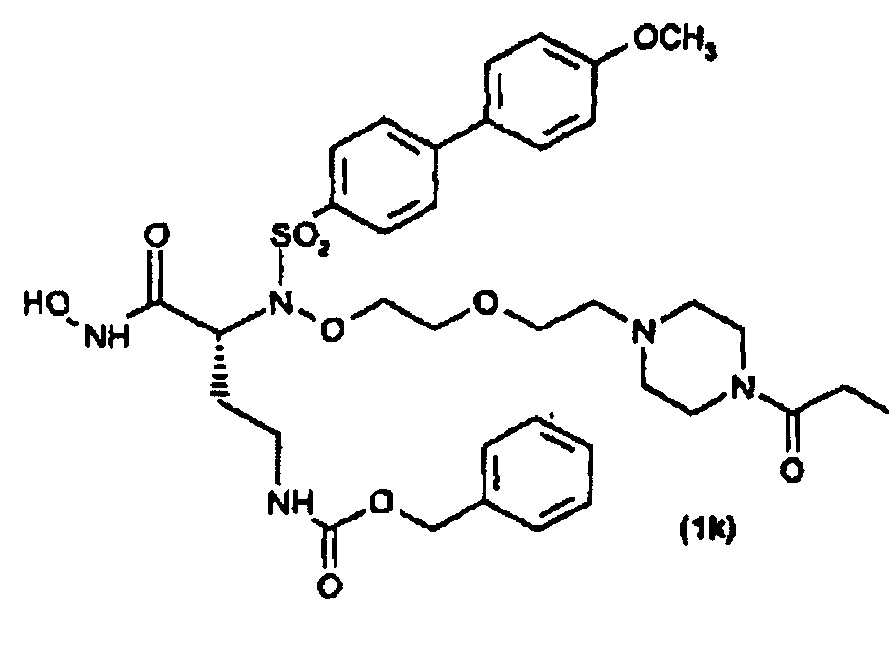
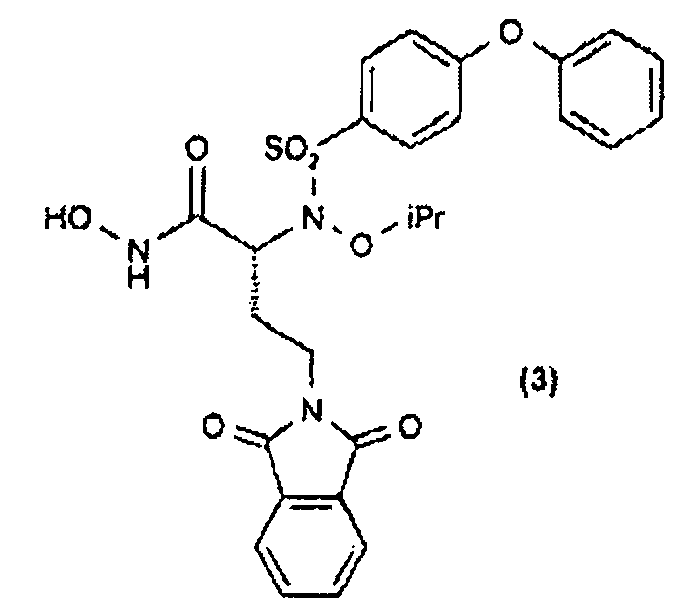
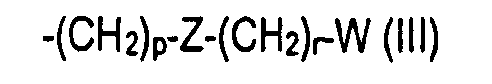
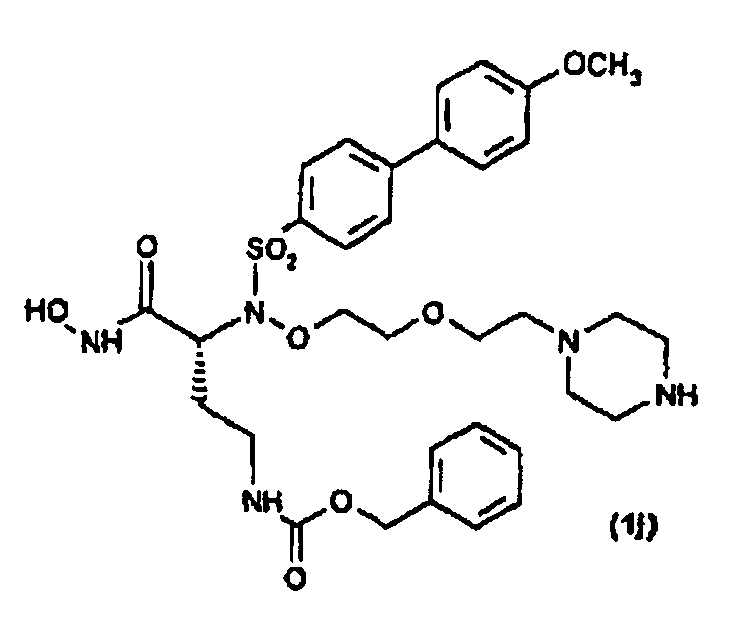
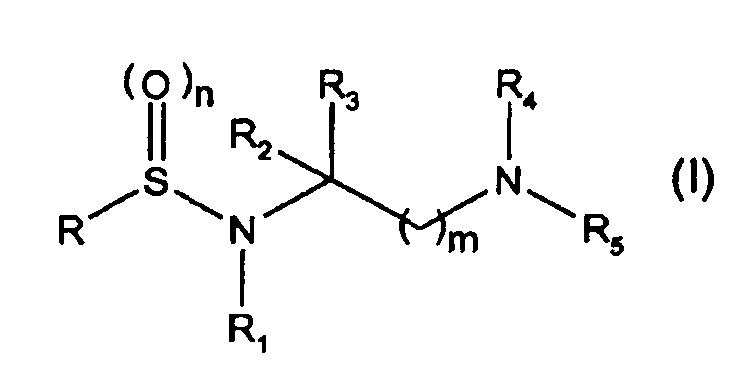
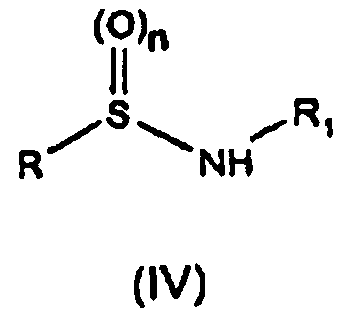
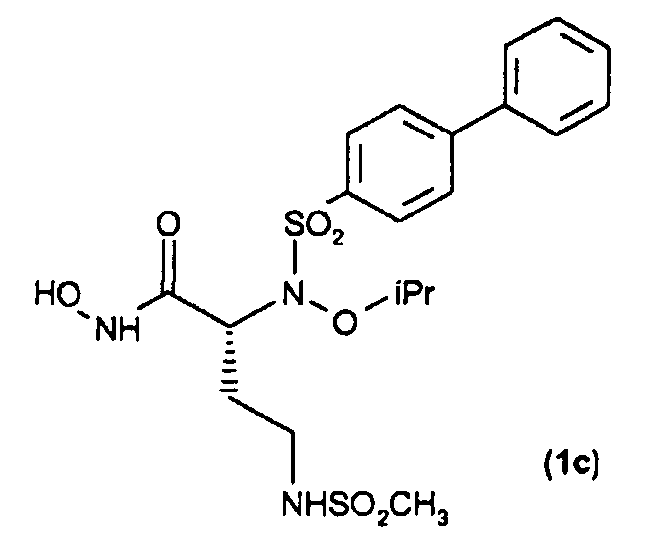
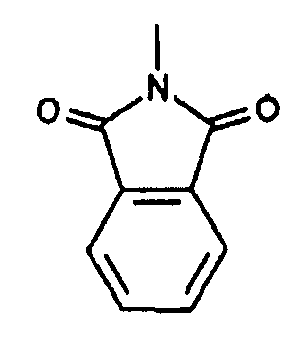
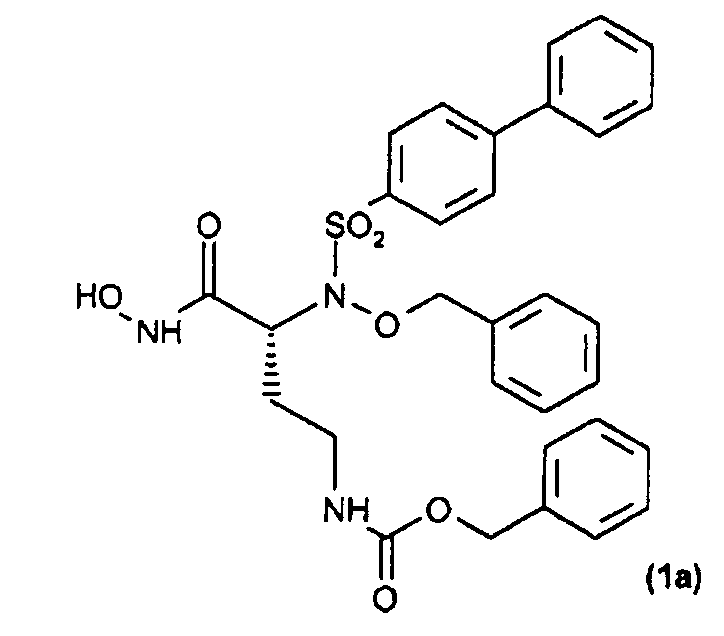
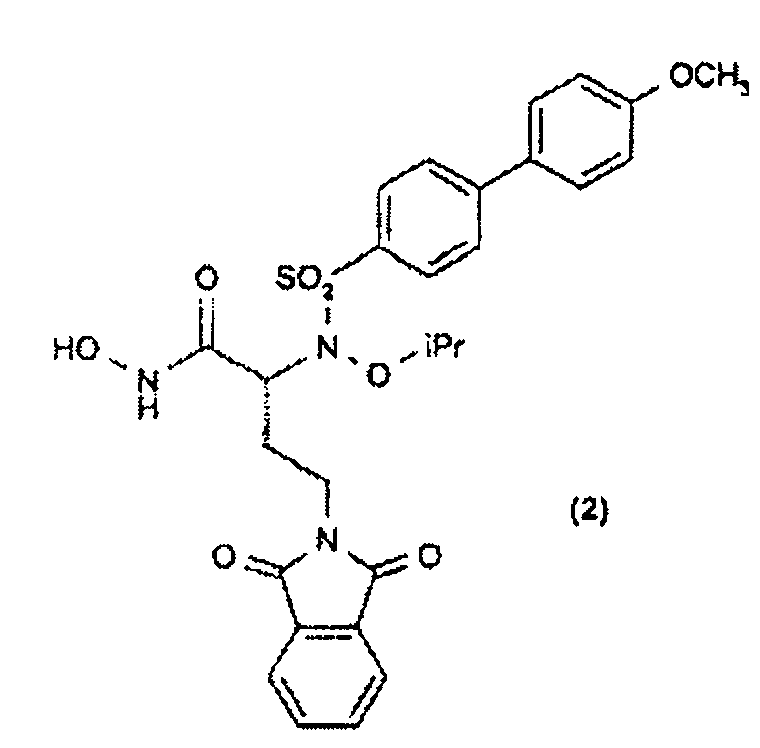
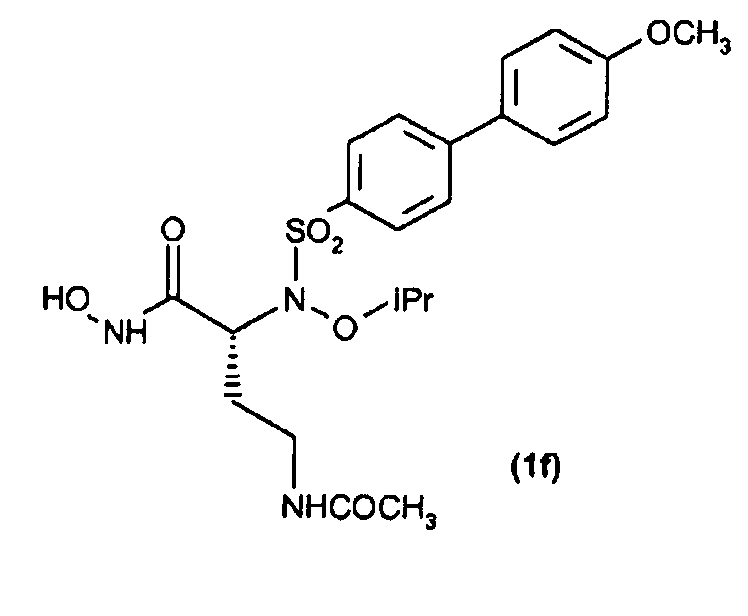
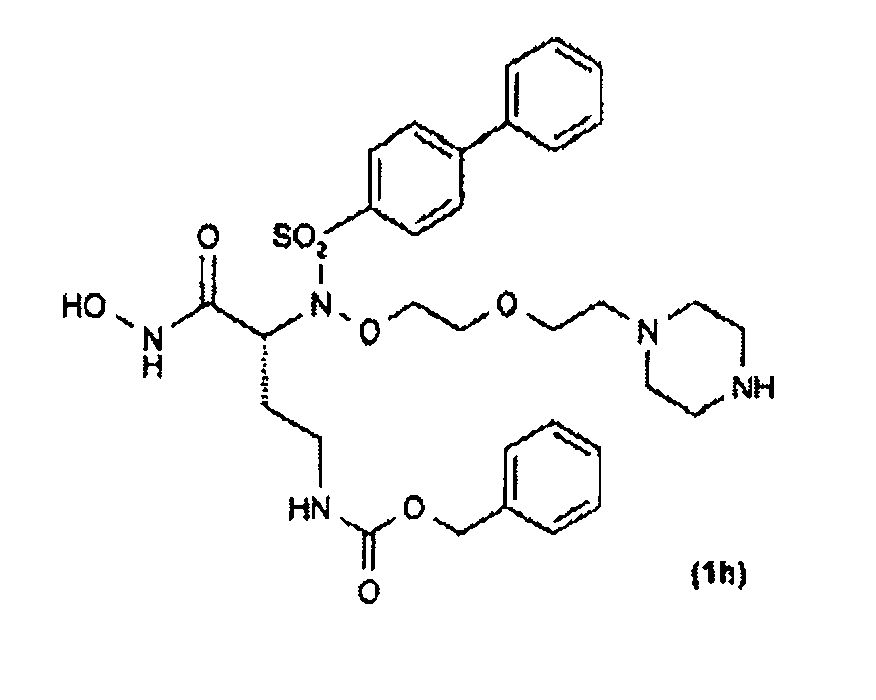
Способ получения контрастных агентов

