Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ
Вид РИД
Изобретение
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка связана с предварительной заявкой на патент США №60/864566, поданной 6 ноября 2006, предварительной заявкой на патент США №60/892523, поданной 1 марта 2007 и предварительной заявкой на патент США №60/957988, поданной 24 августа 2007, причем каждая из перечисленных заявок включена в настоящую заявку с помощью ссылки во всей своей полноте.
Предпосылки изобретения
Область техники, к которой относится изобретение
Настоящее изобретение в основном относится к соединениям, которые ингибируют активность протеинкиназ, а также к композициям и способам, связанным с этими соединениями.
Уровень техники
Рак (а также другие гиперпролиферативные заболевания) характеризуется неуправляемой пролиферацией клеток. Потеря нормального управления клеточной пролиферацией, по-видимому, часто является результатом генетического повреждения путей передачи сигналов в клетке, которые регулируют развитие клеточного цикла. Клеточный цикл состоит из синтеза ДНК (S- фазы), деления клетки или митоза (M-фазы) и кажущихся пауз в активности клетки, именуемых пауза 1 (G1) и пауза 2 (G2). M-фаза состоит из митоза и цитокинеза (разделения на две клетки). Все стадии клеточного цикла регулируются строго организованным каскадом фосфорилирования белков, и в осуществление этих стадий фосфорилирования вовлечено несколько семейств протеинкиназ. Кроме того, активность многих протеинкиназ у человека увеличивается в опухолях, по сравнению с нормальными тканями, и это увеличение активности может являться следствием многих факторов, в том числе повышенных уровней киназ или изменения экспрессии коактиваторов или ингибирующих белков.
В клетках присутствуют белки, которые управляют переходом от одной фазы клеточного цикла к другой. Например, циклины представляют собой семейство белков, концентрации которых увеличиваются и уменьшаются во время клеточного цикла. В соответствующий момент времени циклины запускают различные циклинзависимые протеинкиназы (CDK), которые фосфорилируют субстраты, необходимые для продвижения по клеточному циклу. Активность определенных CDK в определенные моменты времени является существенной как для инициирования клеточного цикла, так и для его координированного развития. Например, CDK1 представляет собой наиболее известный регулятор клеточного цикла, который управляет развитием M-фазы. Однако был идентифицирован ряд других митотических протеинкиназ, принимающих участие в M-фазе, которые включают членов семейств polo, aurora и NIMA (Never-In-Mitosis-A), и киназ, вовлеченных в контрольные точки митоза, окончание митоза и цитокинез.
Киназы Pim (например, Pim-1 киназа, Pim-2 киназа, Pim-3 киназа) являются семейством онкогенных серин/треонин киназ. Известно, что киназа Pim-1 вовлечена в ряд сигнальных путей цитокинов в качестве эффектора нисходящего направления. Будучи активированной, киназа Pim-1 вызывает развитие клеточного цикла, ингибирование апоптоза и модулирование других путей передачи сигнала, включая ее собственный. Кроме того, известно, что киназа Pim-1 вызывает активацию таких факторов транскрипции, как NFAT, p100, c-Myb и pap-1 и ингибирование других факторов, например, HP1. Нормальная экспрессия киназы Pim-1 наблюдается в клетках гематопоэтического происхождения, например, печени, тимусе, селезенке и костном мозге телят. Кроме того, экспрессия наблюдается в простате и эпителиальных клетках ротовой полости. Полагают, что киназа Pim-1 вовлечена в инициирование или развитие злокачественного перерождения, ведущего, в том числе, к злокачественным заболеваниям, в том числе лимфоме Беркитта, раку простаты, раку ротовой полости и диффузным крупноклеточным лимфомам.
Поскольку Pim-киназы вовлечены в развитие ряда злокачественных заболеваний человека, существует необходимость в рациональной разработке специфичных и селективных ингибиторов для лечения рака и других состояний, которые опосредованы и/или связаны с белками Pim-киназ. Настоящее изобретение решает эту задачу и обеспечивает другие аналогичные преимущества.
Сущность изобретения
Настоящее изобретение в основном направлено на соединения, а также на фармацевтические композиции, включающие указанные соединения, где соединения имеют общие структуры, представленные следующими формулами (I) и (II):
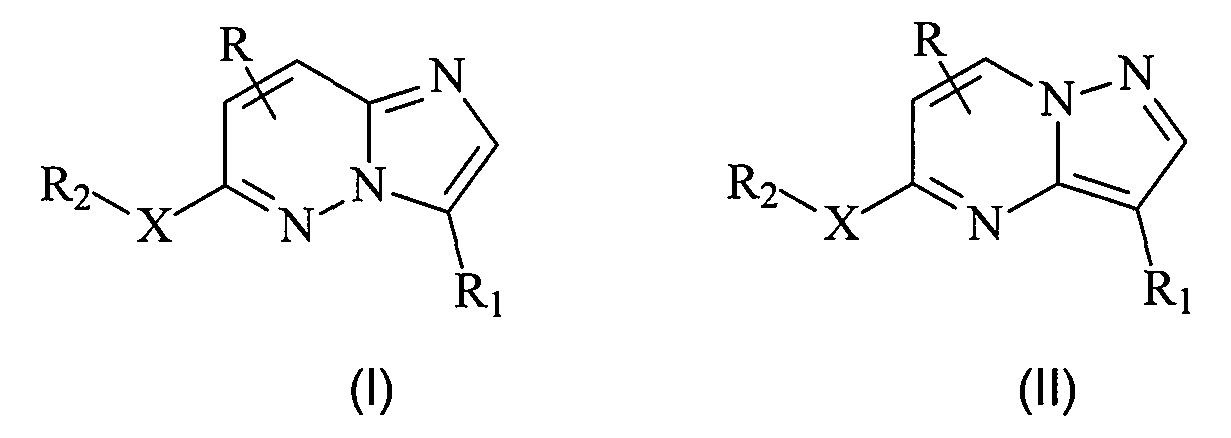
включая их стереоизомеры, пролекарства и фармацевтически приемлемые соли, где R, R1, R2 и X соответствуют данным в настоящей заявке определениям.
Эти соединения по настоящему изобретению применимы в широком диапазоне терапевтических приложений и могут использоваться для лечения таких заболеваний как рак, который хотя бы частично опосредован активностью протеинкиназы. Соответственно, в одном из аспектов настоящего изобретения, описанные в заявке соединения входят в состав фармацевтически приемлемых композиций для введения субъекту при наличии такой необходимости.
В другом аспекте в изобретении разработан способ лечения или предотвращения заболеваний, опосредованных протеинкиназой, например, рака, причем способ включает введение пациенту, у которого имеется необходимость в таком лечении, терапевтически эффективного количества описанного в заявке соединения или фармацевтически приемлемой композиции, включающей такое соединение. В некоторых вариантах осуществления заболевание, опосредованное протеинкиназой, представляет собой заболевание, опосредованное Pim-киназой, например, раковую опухоль, экспрессирующую Pim-1 киназу.
Другой аспект настоящего изобретения относится к способу ингибирования активности протеинкиназы в биологическом образце, причем этот способ включает приведение биологического образца в контакт с соединением, описанным в настоящей заявке, или фармацевтически приемлемой композицией, включающей указанное соединение. В некоторых вариантах осуществления протеинкиназа является Pim-киназой.
Другой аспект настоящего изобретения относится к способу ингибирования активности протеинкиназы у пациента, где способ включает введение пациенту соединения, описанного в настоящей заявке, или фармацевтически приемлемой композиции, включающей такое соединение. В некоторых вариантах осуществления протеинкиназа является Pim-киназой.
Эти и другие аспекты настоящего изобретения станут ясны при ознакомлении с приведенным ниже подробным описанием и приложенным иллюстративным материалом. С этой целью в заявке цитируются некоторые патенты и другие документы для более конкретного изложения различных аспектов настоящего изобретения. Каждый из этих документов включен в настоящую заявку с помощью ссылки во всей полноте.
Краткое описание иллюстративного материала
На фиг.1 показана ингибирующая активность типовых соединений в отношении Pim-1 киназы.
На фиг.2 показаны результаты скрининга соединения 7-29 (таблица VII) с точки зрения селективности в отношении панели серин/треонин и тирозинкиназ по данным радиометрического анализа.
На фиг.3-5 показаны результаты фосфо-Bad окрашивания клеток MV-4-11, обработанных соединениями 7-19, 7-29 и 7-31, соответственно.
Подробное описание изобретения
В соответствии с основным аспектом настоящего изобретения разработаны соединения, применимые в качестве ингибиторов протеинкиназ, а также композиции и способы, относящиеся к этим соединениям. Соединения по настоящему изобретению имеют структуры, соответствующие приведенным ниже формулам (I) или (II):
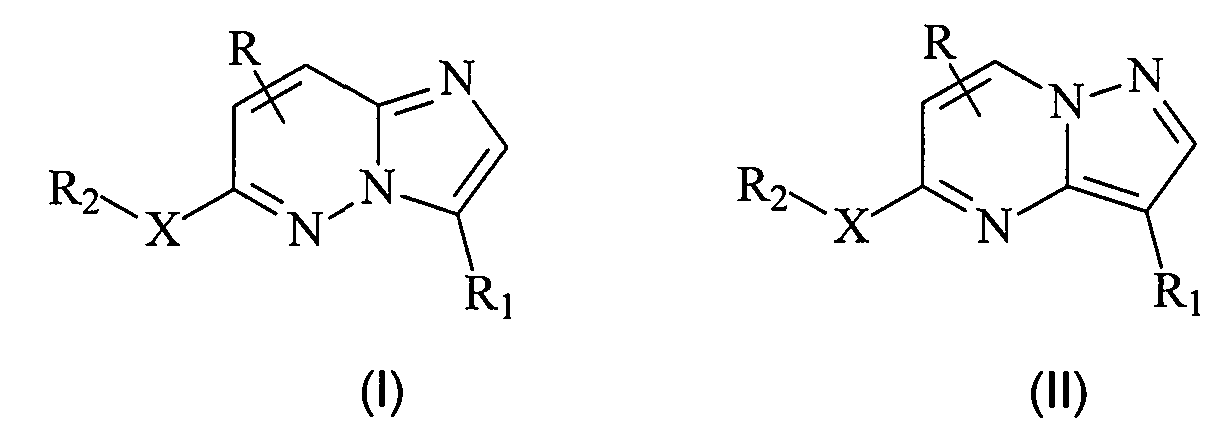
включая их стереоизомеры, пролекарства и фармацевтически приемлемые соли, где
X представляет собой NH, S, O, SO или SO2;
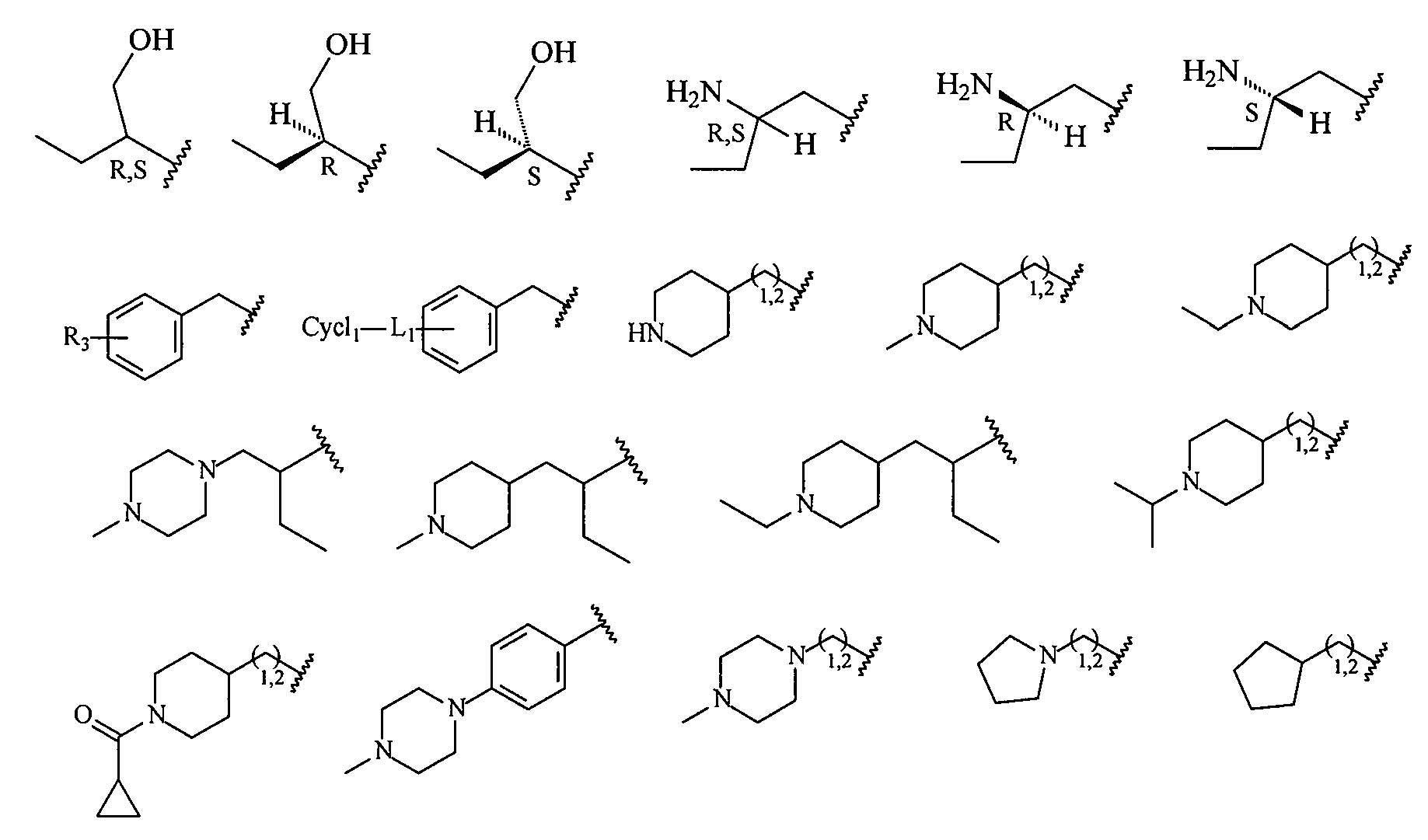
R представляет собой H, -OH, галоген, алкил, галогеналкил, алкокси или галогеналкокси;
R1 представляет собой карбоцикл, замещенный карбоцикл, гетероцикл или замещенный гетероцикл; или структуру, выбранную из
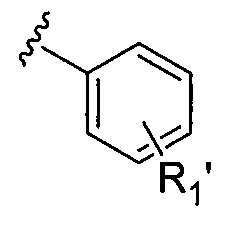
где R1' означает замещение в п-, о- или м-положения одним или несколькими заместителями из числа галогена, -OCF3, -OCHF2, -CF3, -OCH3, -NH2, -NO2, -OH, -COCH3, -NHSO2CH3 или -N(CH3)2.
R2 представляет собой -(CH2)n-циклопропил, -(CH2)n-циклопентил, -(CH2)n-циклогексил, -SO2-CH3, -SO2-(CH2)nCH3, -(CH2)n-пиперонил, -(CH2)n-пиперидил, -(CH2)n-пиперазинил, -(CH2)n-фурил, -(CH2)n-тиофен, -(CH2)n-пиридил, -(CH2)n-пиримидил, -(CH2)nOCH3, -(CH2)nOH или -(CH2)nN(CH3)2, где n означает 0,1,2,3 или 4 и каждый из перечисленных фрагментов необязательно замещен одним или несколькими заместителями; или структуру выбранную из
где заместитель L является необязательным и, в случае своего присутствия, представляет собой NH, S, O, SO или SO2; R3 означает один или несколько необязательных заместителей; и Cycl1 представляет собой карбоцикл, замещенный карбоцикл, гетероцикл или замещенный гетероцикл.
Если не указано иное, следующие термины, приведенные в описании и формуле изобретения, имеют значения, которые обсуждаются ниже:
Термин «алкил» относится к насыщенному линейному или разветвленному углеводородному радикалу, включающему от одного до шести атомов углерода, предпочтительно от одного до четырех атомов углерода, например, метилу, этилу, пропилу, 2-пропилу, н-бутилу, изобутилу, трет-бутилу, пентилу, гексилу и т.п., предпочтительно метилу, этилу, пропилу или 2-пропилу. Типовые насыщенные алкилы с линейной цепью включают метил, этил, н-пропил, н-бутил, н-пентил, н-гексил и т.п.; в то же время насыщенные разветвленные алкилы включают изопропил, втор-бутил, изобутил, трет-бутил, изопентил и т.п. Типовые насыщенные циклические алкилы включают циклопропил, циклобутил, циклопентил, циклогексил, -CH2-циклогексил, и т.п.; в то же время ненасыщенные циклические алкилы включают циклопентенил, циклогексенил, -CH2-циклогексенил и т.п. В отношении циклических алкилов употребляется также термин «циклоалкил». Ненасыщенные алкилы содержат, по меньшей мере, одну двойную или тройную связь между соседними атомами углерода (их именуют также терминами «алкенил» или «алкинил» соответственно). Типовые линейные или разветвленные алкенилы включают этиленил, пропиленил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил и т.п.; тогда как типовые линейные и разветвленные алкинилы включают ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-бутинил и т.п.
Термин «алкилен» означает линейный насыщенный двухвалентный углеводородный радикал, включающий от одного до шести атомов углерода, или разветвленный насыщенный двухвалентный углеводородный радикал, включающий от трех до шести атомов углерода, например, метилен, этилен, 2,2-диметилэтилен, пропилен, 2-метилпропилен, бутилен, пентилен и т.п., предпочтительно метилен, этилен или пропилен.
Термин «циклоалкил» относится к насыщенному циклическому углеводородному радикалу, включающему от трех до восьми атомов углерода, например, циклопропилу, циклобутилу, циклопентилу или циклогексилу.
Термин «алкокси» означает радикал -ORa, где Ra представляет собой алкил, соответствующий данному выше определению, например, метокси, этокси, пропокси, бутокси и т.п.
Термин «галоген» означает фтор, хлор, бром или йод, предпочтительно фтор или хлор.
Термин «галогеналкил» означает алкил, замещенный одним или несколькими, предпочтительно, одним, двумя и тремя одинаковыми или различными атомами галогенов, например, -CH2Cl, -CF3, -CH2CF3, -CH2CCl3 и т.п.
Термин «галогеналкокси» означает радикал -ORb, где Rb означает определенный выше галогеналкил, например, трифторметокси, трихлорэтокси, 2,2-дихлорпропокси и т.п.
Термин «ацил» означает радикал -C(O)Rc, где Rc означает водород, алкил или галогеналкил, соответствующий данному выше определению, например, формил, ацетил, трифторацетил, бутаноил и т.п.
Термин «арил» относится к моноциклической или полициклической группе с конденсированными циклами (т.е. два соседних атома углерода входят одновременно в оба цикла), где указанные циклы состоят только из атомов углерода в количестве от 6 до 12, имеющей полностью сопряженную пи-электронную систему. Примерами арильных групп, не ограничиваясь перечисленными, являются фенил, нафтил и антраценил. Арильная группа может быть замещенной или незамещенной. Если арильная группа является замещенной, она может быть замещена одним или несколькими заместителями, которые определены ниже, более предпочтительно одним, двумя или тремя, еще более предпочтительно одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила (где алкил может быть необязательно замещен одним или двумя заместителями), галогеналкила, галогена, гидрокси, алкокси, меркапто, алкилтио, циано, ацила, нитро, фенокси, гетероарила, гетероарилокси, галогеналкила, галогеналкокси, карбокси, алкоксикарбонила, амино, алкиламино, диалкиламино, арила, гетероарила, карбоцикла или гетероцикла (где арил, гетероарил, карбоцикл или гетероцикл могут быть необязательно замещенными).
Термин «гетероарил» относится к моноциклической группе или группе с конденсированными циклами (т.е. два соседних атома углерода входят одновременно в оба цикла), содержащей от 5 до 12 циклических атомов, в число которых входят один, два, три или четыре циклических гетероатома, выбранных из N, O или S, причем остальные циклические атомы являются атомами углерода, и, кроме того, имеющей полностью сопряженную пи-электронную систему. Примерами незамещенных гетероарильных групп, не ограничиваясь указанными, являются пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиридин, пиримидин, хинолин, изохинолин, пурин, триазол, тетразол, триазин и карбазол. Гетероарильные группы могут быть замещенными или незамещенными. Если гетероарильная группа является замещенной, она может быть замещена одним или несколькими заместителями, которые определены ниже, более предпочтительно одним, двумя или тремя, еще более предпочтительно одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила (где алкил может быть необязательно замещен одним или двумя заместителями), галогеналкила, галогена, гидрокси, алкокси, меркапто, алкилтио, циано, ацила, нитро, галогеналкила, галогеналкокси, карбокси, алкоксикарбонила, амино, алкиламино, диалкиламино, арила, гетероарила, карбоцикла или гетероцикла (где арил, гетероарил, карбоцикл или гетероцикл могут быть необязательно замещенными).
Термин «карбоцикл» относится к насыщенной, ненасыщенной или ароматической циклической системе, включающей от 3 до 14 циклических атомов углерода. Термин «карбоцикл», независимо от того, является ли данный цикл насыщенным или частично ненасыщенным, относится также к циклам, которые являются необязательно замещенными. Термин «карбоцикл» включает арил. Термин «карбоцикл» включает также алифатические циклы, которые конденсированы с одним или несколькими ароматическими или неароматическими циклами, например, декагидронафтил или тетрагидронафтил, где радикал или точка присоединения находятся на алифатическом цикле. Карбоциклическая группа может быть замещенной или незамещенной. Если карбоциклическая группа является замещенной, она может быть замещена одним или несколькими заместителями, которые определены ниже, более предпочтительно одним, двумя или тремя, еще более предпочтительно одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила (где алкил может быть необязательно замещен одним или двумя заместителями), галогеналкила, галогена, гидрокси, алкокси, меркапто, алкилтио, циано, ацила, нитро, галогеналкила, галогеналкокси, карбокси, алкоксикарбонила, амино, алкиламино, диалкиламино, арила, гетероарила, карбоцикла или гетероцикла (где арил, гетероарил, карбоцикл или гетероцикл могут быть необязательно замещенными).
Термин «гетероцикл» относится к насыщенной, ненасыщенной или ароматической циклической системе, включающей от 3 до 14 циклических атомов, в которой один, два или три циклических атома являются гетероатомами, выбранными из N, O или S(O)m (где m представляет собой целое число от 0 до 2), причем оставшиеся циклические атомы являются атомами углерода, где один или два атома углерода необязательно могут быть заменены карбонильными группами. Термин «гетероцикл» включает гетероарил. Гетероцикл может быть необязательно замещен независимо одним или несколькими заместителями, которые определены ниже, предпочтительно одним, двумя или тремя заместителями, выбранными из алкила (где алкил может быть необязательно замещен одним или двумя заместителями), галогеналкила, циклоалкиламино, циклоалкилалкила, циклоалкиламиноалкила, циклоалкилалкиламиноалкила, цианоалкила, галогена, нитро, циано, гидрокси, алкокси, амино, алкиламино, диалкиламино, гидроксиалкила, карбоксиалкила, аминоалкила, алкиламиноалкила, диалкиламиноалкила, аралкила, гетероаралкила, арила, гетероарила, карбоцикла, гетероцикла (где арил, гетероарил, карбоцикл или гетероцикл могут быть необязательно замещенным), аралкила, гетероаралкила, насыщенного или ненасыщенного гетероциклоамино, насыщенного или ненасыщенного гетероциклоаминоалкила и -CORd (где Rd представляет собой алкил). Более конкретно термин гетероциклил включает, не ограничиваясь перечисленными, тетрагидропиранил, 2,2-диметил-1,3-диоксолан, пиперидино, N-метилпиперидин-3-ил, пиперазино, N-метилпирролидин-3-ил, пирролидино, морфолино, 4-циклопропилметилпиперазино, тиоморфолино, тиоморфолино-1-оксид, тиоморфолино-1,1-диоксид, 4-этилоксикарбонилпиперазино, 3-оксопиперазино, 2-имидазолидон, 2-пирролидинон, 2-оксогомопиперазино, тетрагидропиримидин-2-он, а также их производные. В некоторых вариантах осуществления гетероциклическая группа необязательно замещена одним или двумя заместителями, независимо выбранными из галогена, алкила, алкила, замещенного карбокси, сложного эфира, гидрокси, алкиламино, насыщенного или ненасыщенного гетероциклоамино, насыщенного или ненасыщенного гетероциклоаминоалкила или диалкиламино.
Термины «необязательный» или «необязательно» означают, что описанное вслед за ними событие или обстоятельство может иметь место, но необязательно, и что данное описание включает случаи, в которых событие или обстоятельство имеет место и случаи, в которых оно не имеет места. Например, фраза «гетероциклическая группа, необязательно замещенная алкильной группой» означает, что алкильная группа может присутствовать, но может и не присутствовать, и данное описание включает ситуации, в которых гетероциклическая группа замещена алкильной группой, и ситуации, в которых гетероциклическая группа не замещена алкильной группой.
Наконец, термин «замещенный» в настоящем описании означает любую из перечисленных выше групп (например, алкил, арил, гетероарил, карбоцикл, гетероцикл и т.д.), в которой хотя бы один атом водорода заменен на заместитель. В случае оксозаместителя (“=O”) заменены два атома водорода. В контексте настоящего изобретения «заместители» включают галоген, гидрокси, оксо, циано, нитро, амино, алкиламино, диалкиламино, алкил, алкокси, тиоалкил, галогеналкил, гидроксиалкил, арил, замещенный арил, арилалкил, замещенный арилалкил, гетероарил, замещенный гетероарил, гетероарилалкил, замещенный гетероарилалкил, гетероцикл, замещенный гетероцикл, гетероциклалкил, замещенный гетероциклалкил, -NReRf, -NReC(=O)Rf, -NReC(=O)NReRf, -NReC(=O)ORf, -NReSO2Rf, -ORe, -C(=O)Re, -C(=O)ORe, -C(=O)NReRf, -OC(=O)NReRf, -SH, -SRe, -SORe, -S(=O)NH2, -S(=O)2Re, -OS(=O)2Re, -S(=O)2ORe, где Re и Rf являются одинаковыми или различными и независимо представляют собой водород, алкил, галогеналкил, замещенный алкил, арил, замещенный арил, арилалкил, замещенный арилалкил, гетероарил, замещенный гетероарил, гетероарилалкил, замещенный гетероарилалкил, гетероцикл, замещенный гетероцикл, гетероциклалкил или замещенный гетероциклалкил.
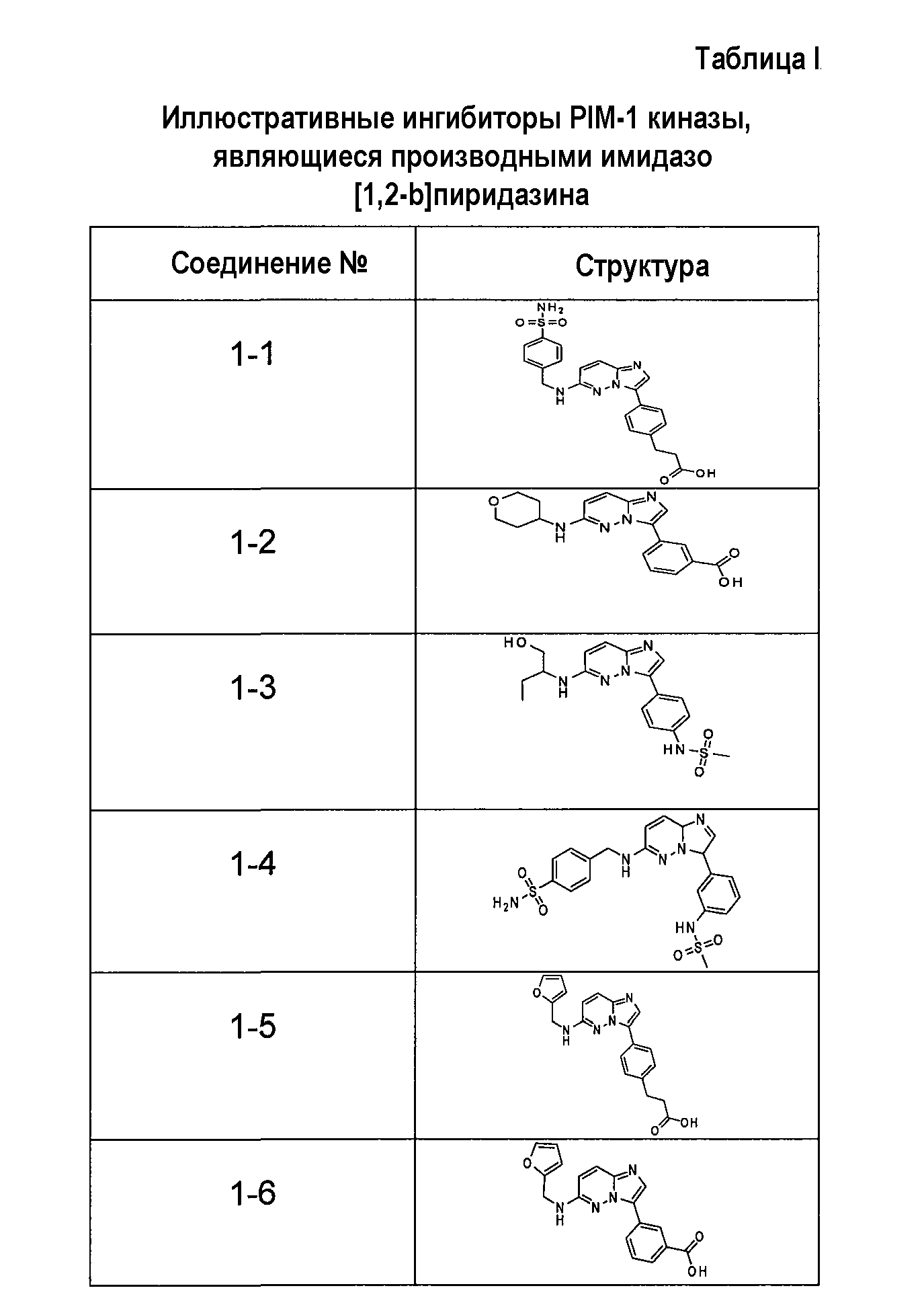
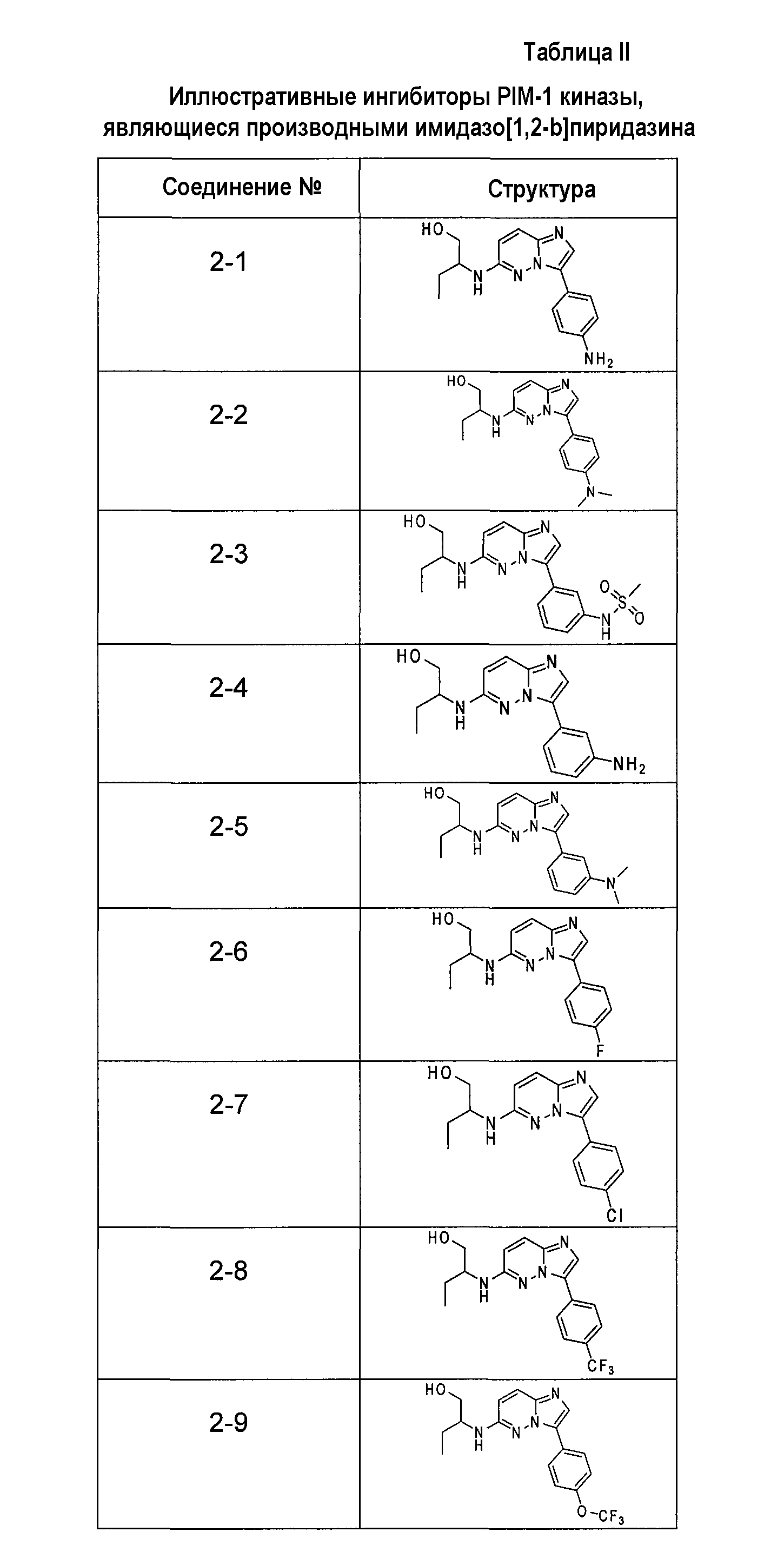
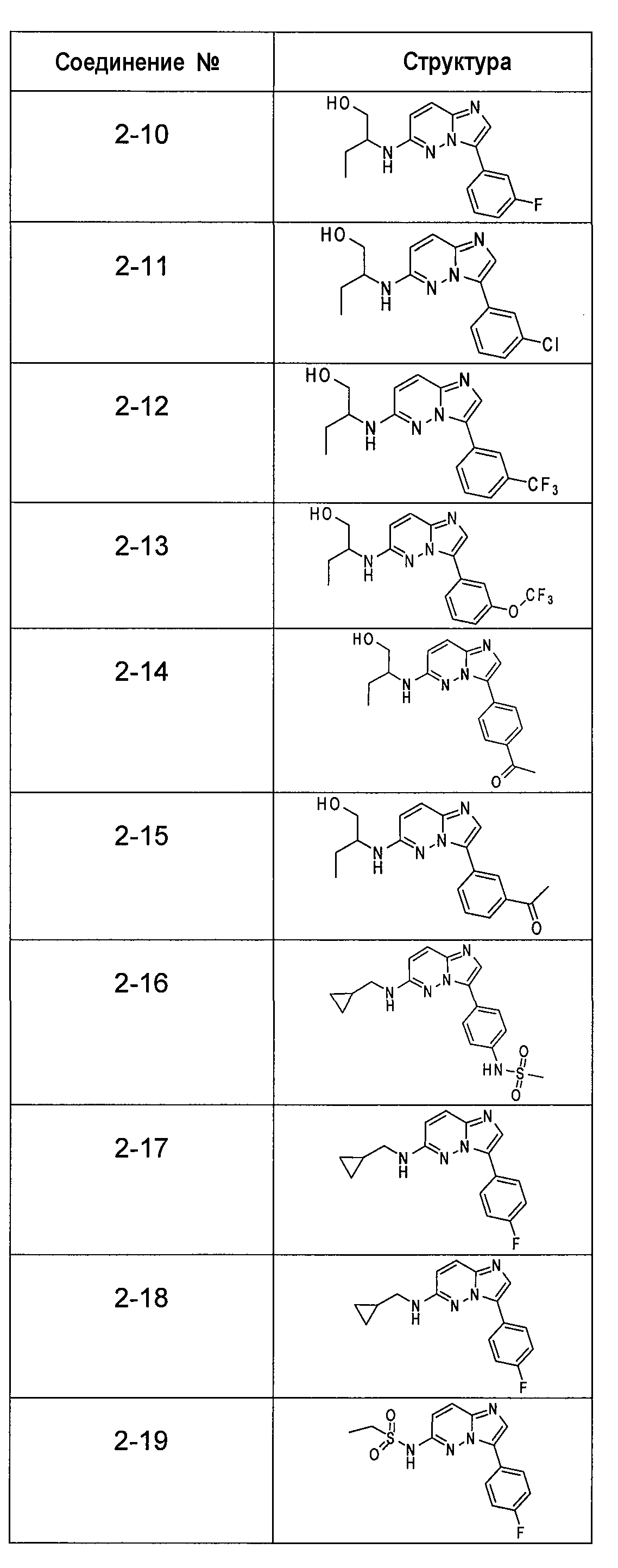
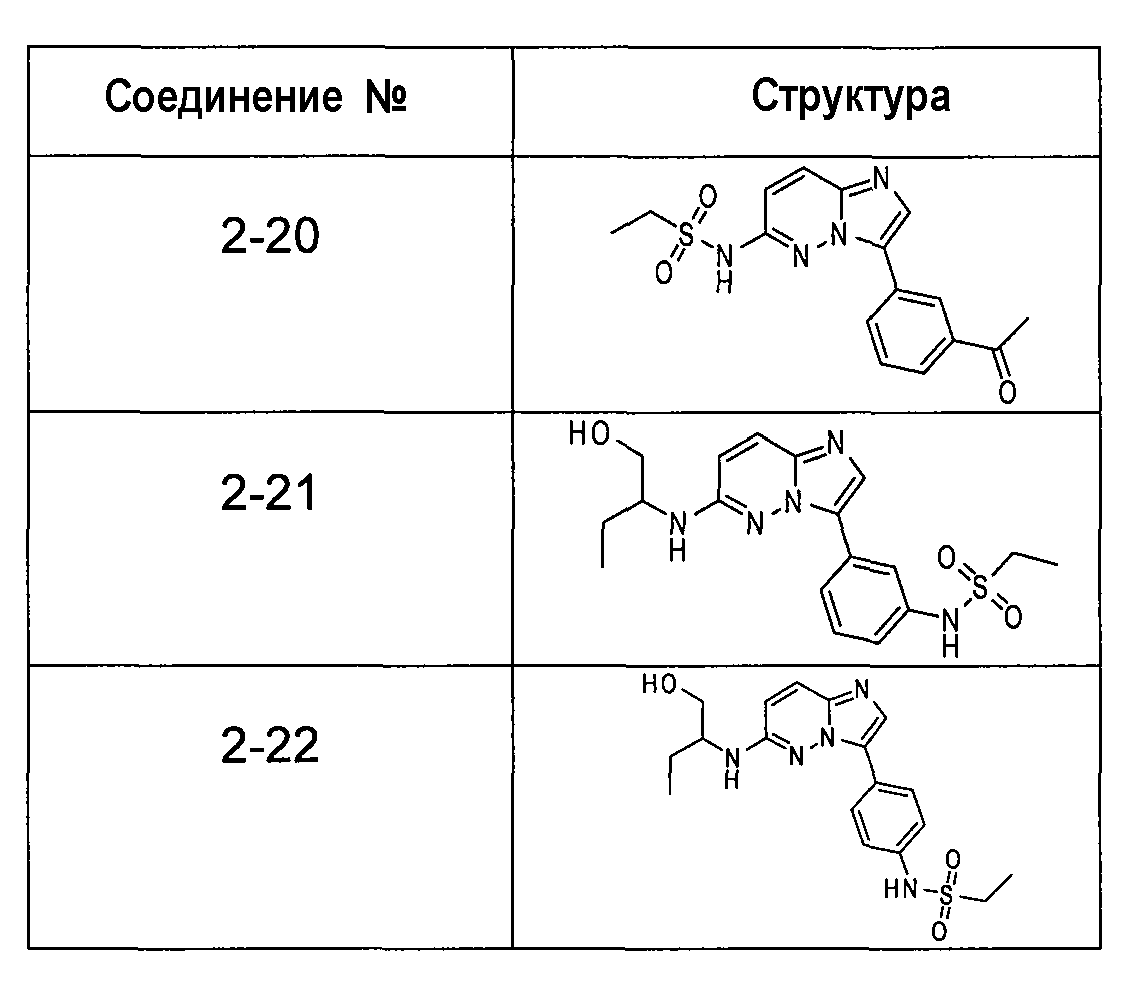
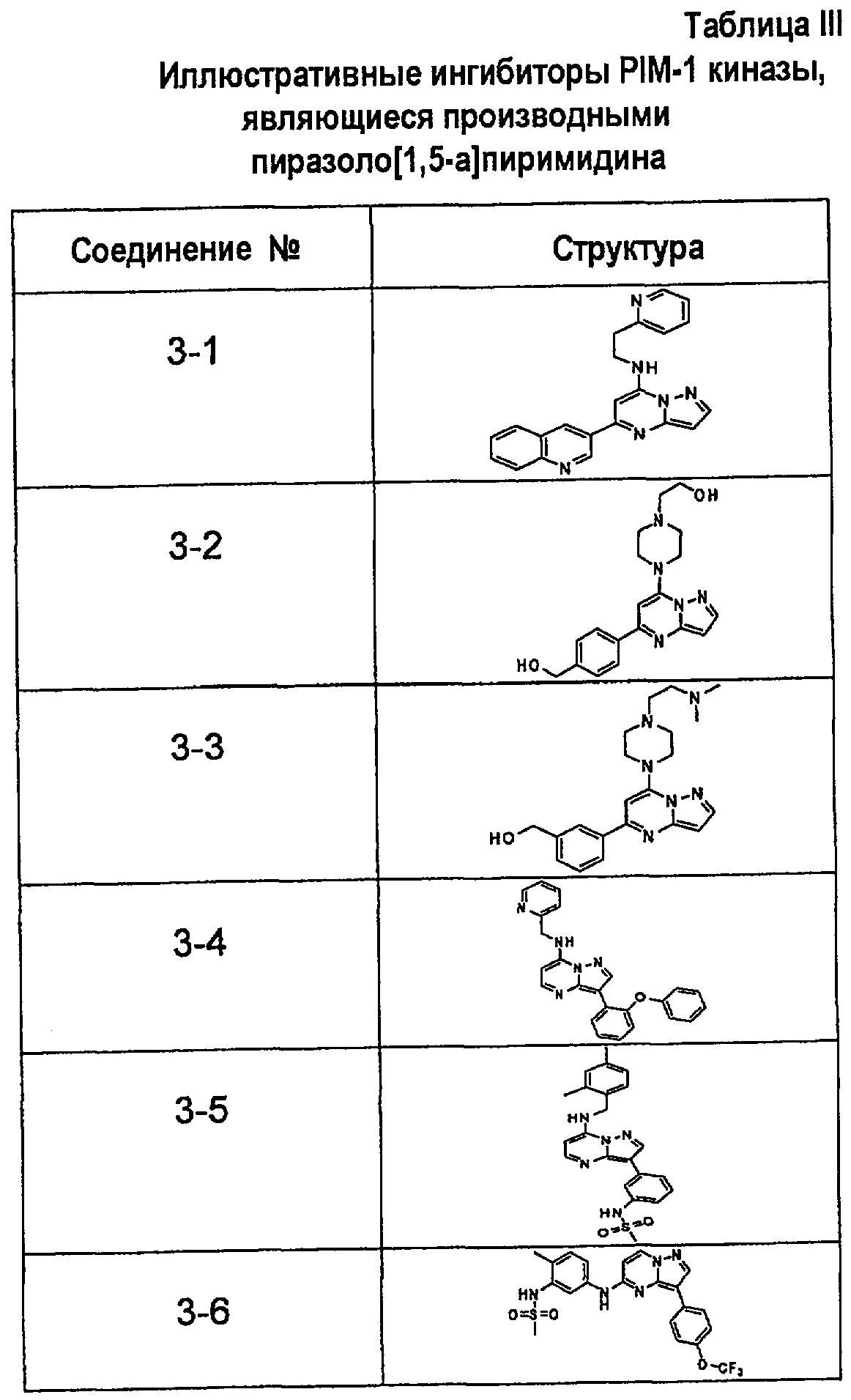
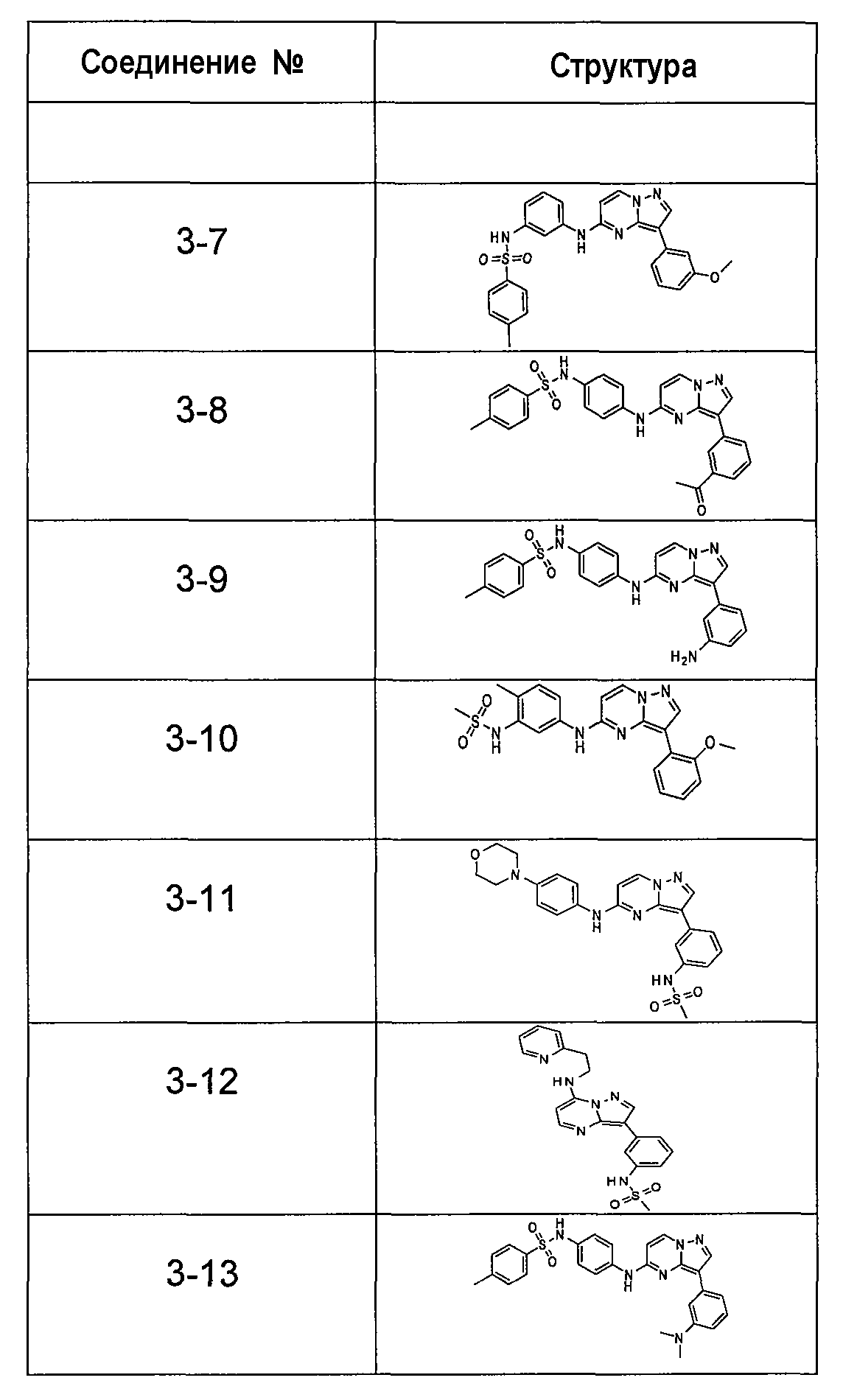
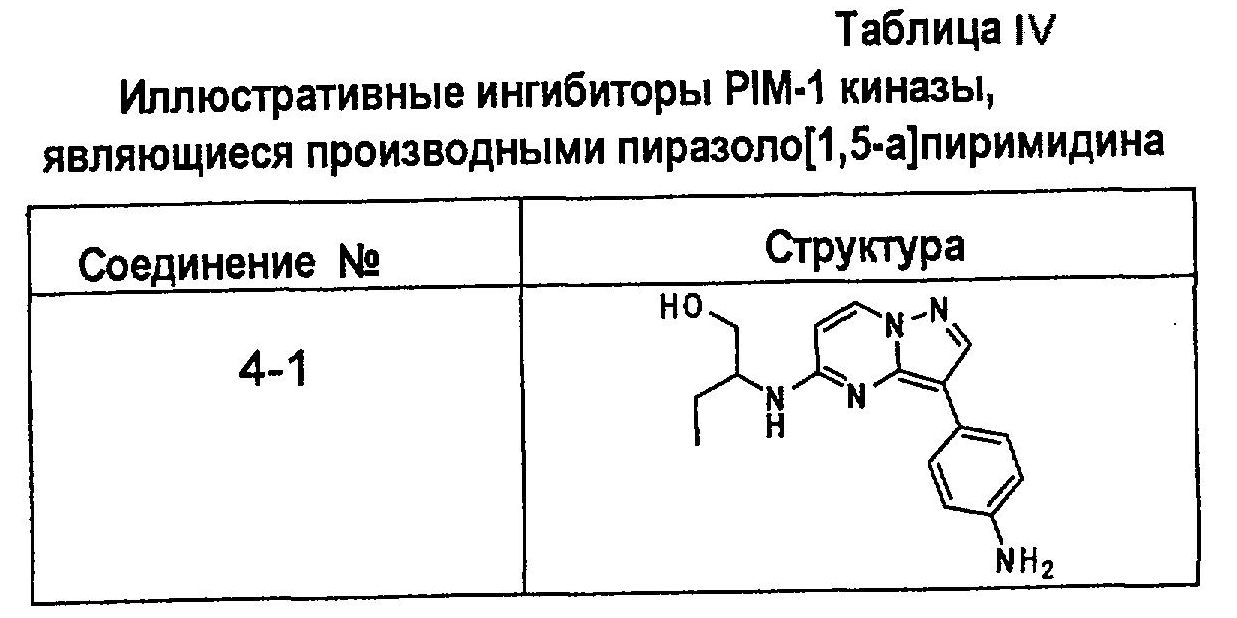
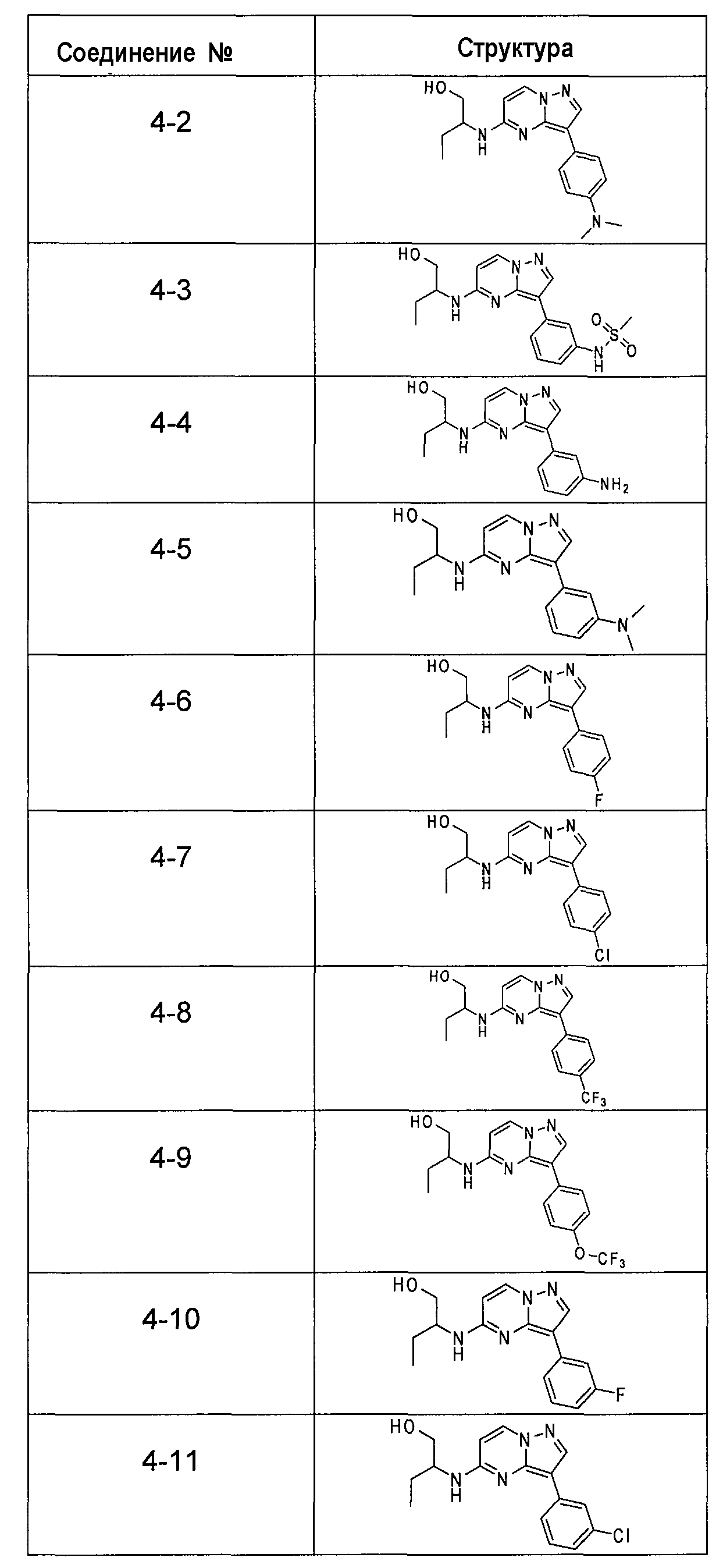
Ниже приведены некоторые иллюстративные соединения, соответствующие структурам (I) и (II), предназначенные для применения, описанного в настоящей заявке.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R1 представляет собой 5-6-членную насыщенную, частично ненасыщенную или полностью ненасыщенную моноциклическую структуру, включающую 0-3 гетероатома, где гетероатомы выбраны из азота, кислорода и серы.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R1 представляет собой п-, о- или м-замещенный фенил с одним или несколькими заместителями, выбранными из -F, -Cl, -CF3, -OCF3, -OCH3, -CH3, -NO2, -N(CH3)2, -NH2, -NHSO2CH3, -NHSO2CH2CH3, -COCH3, -COOH, -CH2NH2, -OH, -SO2NH2, -SCH3, пиперазина или морфолина.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R1 представляет собой необязательно замещенный пиразолил, фурил, тиофен, пиридил, пиримидил или индолильную группу.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R1 имеет структуру
где R1' означает один или несколько необязательных заместителей или, в более конкретном варианте осуществления, о-, м- или п-замещение одним или несколькими заместителями из числа галогена.
-OCF3, -CF3, -OCH3, -OCHF2, -NH2, -NO2, -OH, -COCH3, -NHSO2CH3 или -N(CH3)2.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R2 представляет собой 2-бутан-1-ол, -SO2CH3, -SO2CH2CH3, -CH2CH2OCH3, -CH2CH2CH2OH или -CH2CH2N(CH3)2.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R2 представляет собой необязательно замещенный -(CH2)n-циклопропил, -(CH2)n-циклопентил, -(CH2)n-циклогексил, -(CH2)n-пиперонил, -(CH2)n-пиперидил, -(CH2)n-пиперазинил, -(CH2)n-фурил, -(CH2)n-тиофен, -(CH2)n-пиридил или -(CH2)n-пиримидил.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R2 имеет структуру, выбранную из
где заместитель L является необязательным и в случае наличия представляет собой NH, S, O, SO или SO2; R3 означает один или несколько необязательных заместителей; и Cycl1 означает карбоцикл, замещенный карбоцикл, гетероцикл или замещенный гетероцикл.
В более конкретном аспекте, в показанных выше структурах (I) и (II) R2 имеет следующую структуру:
где заместитель L является необязательным и в случае наличия представляет собой NH, S, O, SO или SO2; и Cycl1 означает карбоцикл, замещенный карбоцикл, гетероцикл или замещенный гетероцикл, и в более конкретном варианте осуществления Cycl1 представляет собой 5-6-членный насыщенный, частично ненасыщенный или полностью ненасыщенный моноцикл, включающий 0-3 гетероатома, где гетероатомы выбраны из азота, кислорода и серы.
В других конкретных вариантах осуществления структур (I) и (II), X представляет собой NH и R1 является замещенным или незамещенным фенилом (где R соответствует данному выше определению и R1' означает отсутствие заместителя или наличие одного или нескольких заместителей), и соединения имеют следующие структуры (I-A) и (II-A), соответственно:
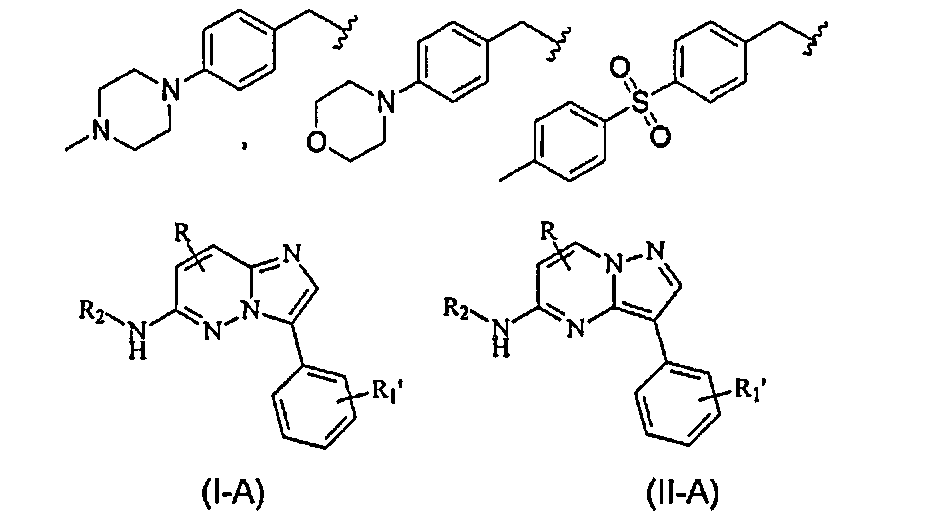
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
В более конкретных вариантах осуществления структур (I-A) или (II-A) R представляет собой алкил, например метил, и соединения имеют следующие структуры (I-Aa) и (II-Aa):
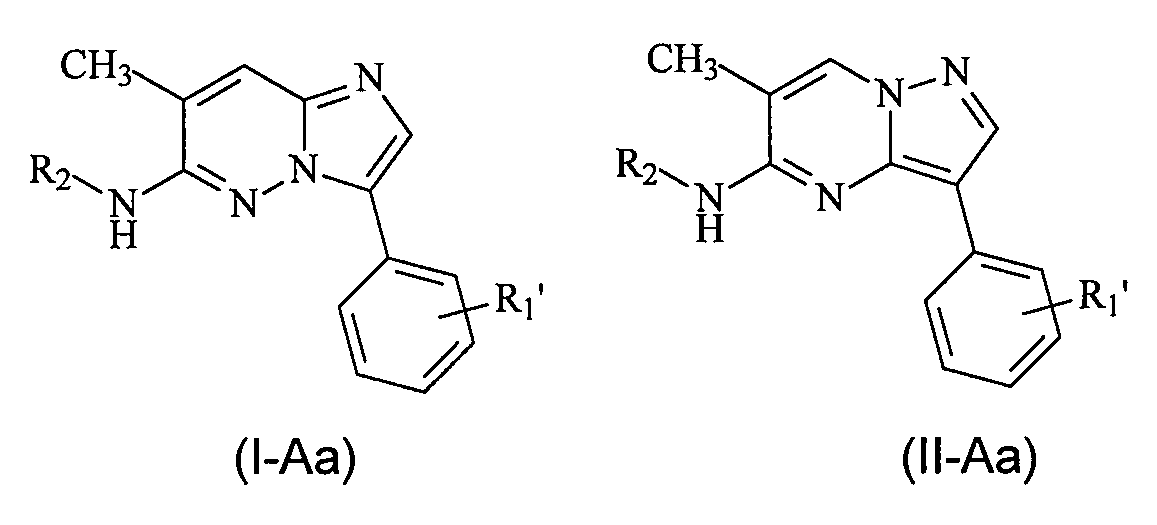
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
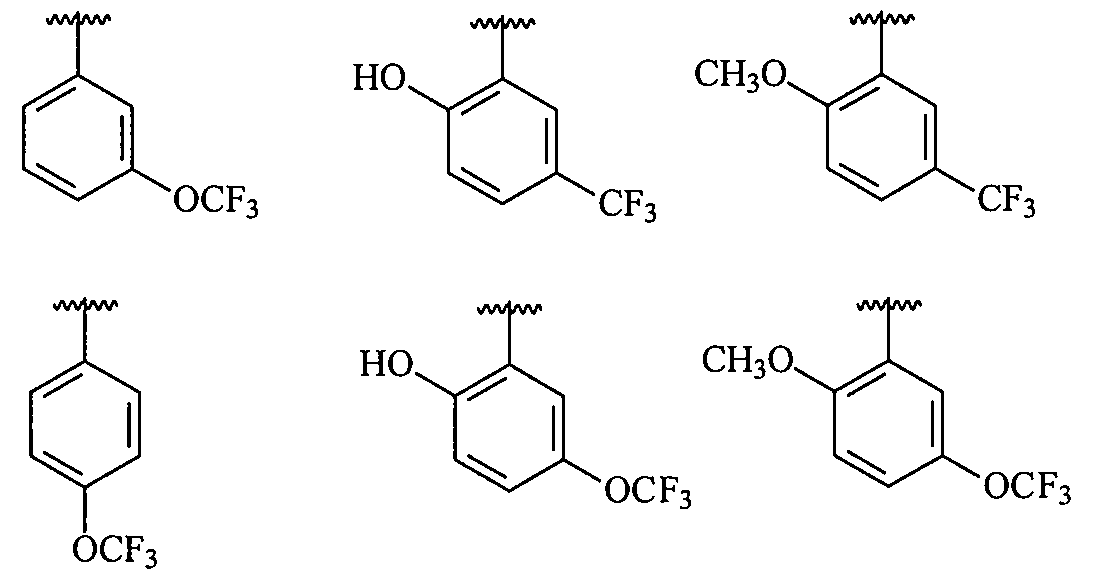
В более конкретных вариантах осуществления структур (I-A), (II-A), (I-Aa) и (II-Aa) R1 представляет собой замещенный фенил, имеющий, по меньшей мере, один п-, о- или м-заместитель, выбранный из галогена, -OCF3, -OCHF2, -CF3, -OCH3, -NH2, -NO2, -OH, -COCH3, -NHSO2CH3 или -N(CH3)2, и в более конкретном варианте осуществления R1 представляет собой замещенный фенил, имеющий, по меньшей мере, один п-, о- или м-заместитель, выбранный из -OCF3, -OCHF2, -CF3, -OCH3 и -OH, и в еще более конкретном варианте осуществления заместитель R1 выбран из
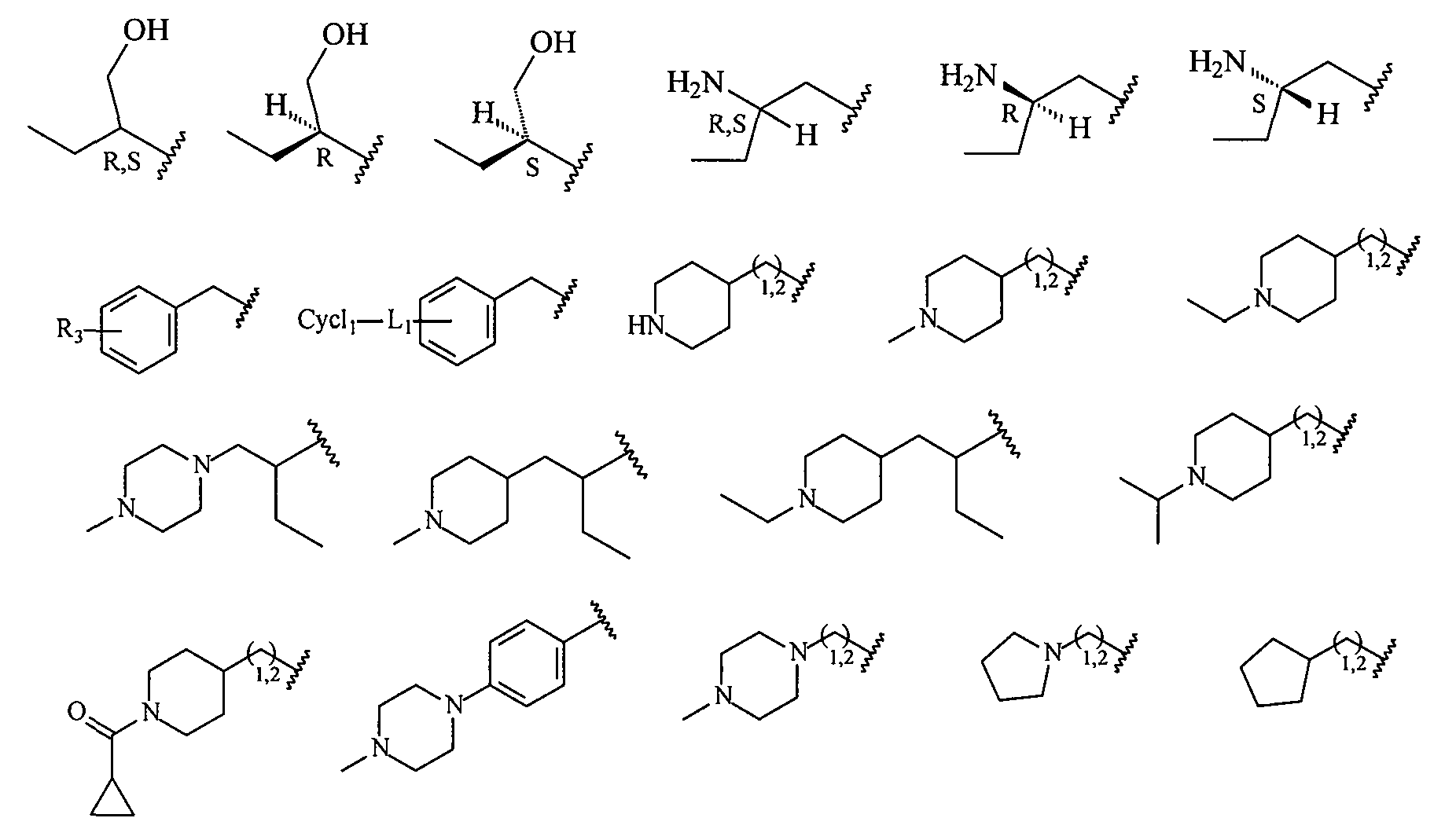
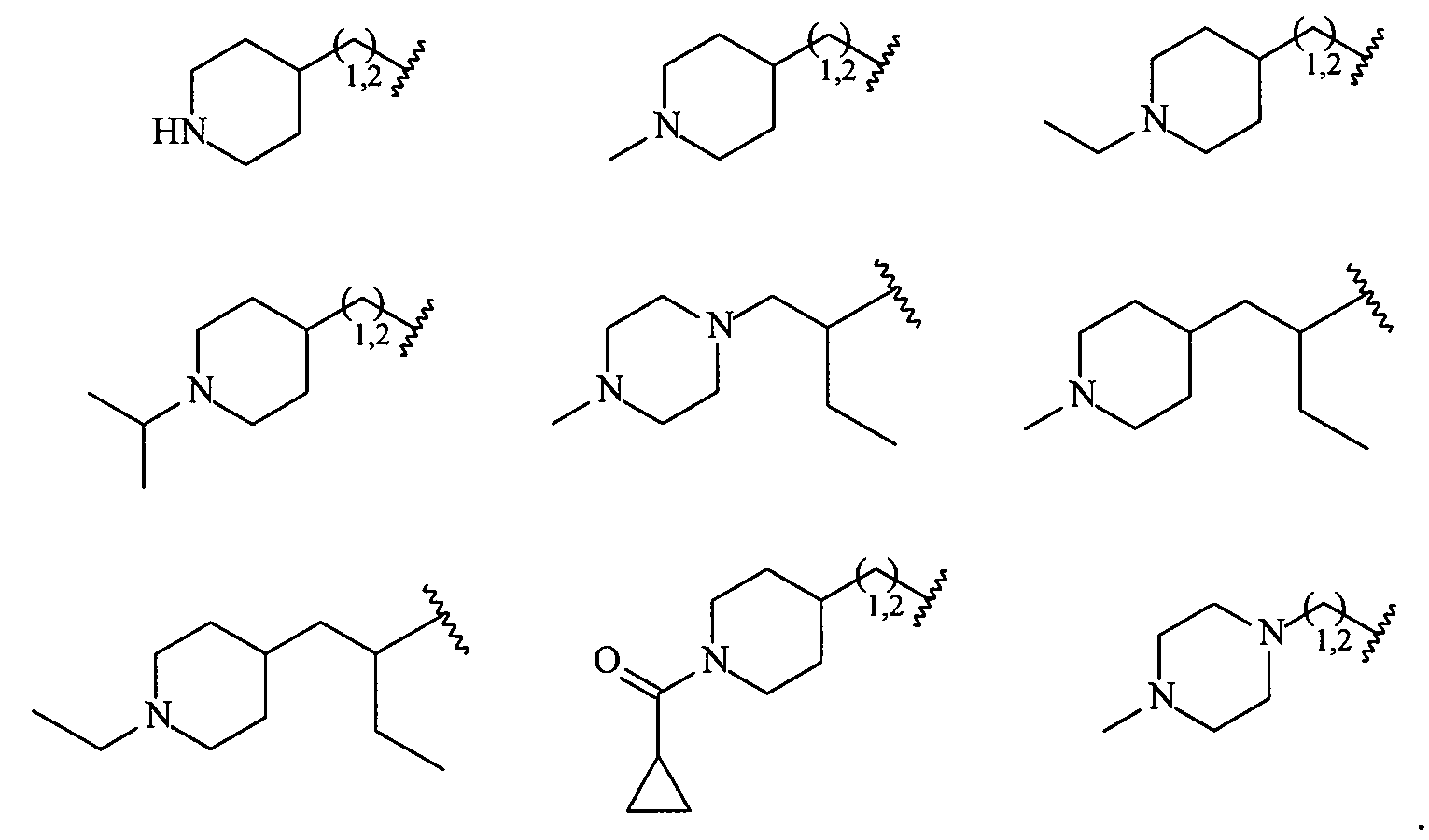
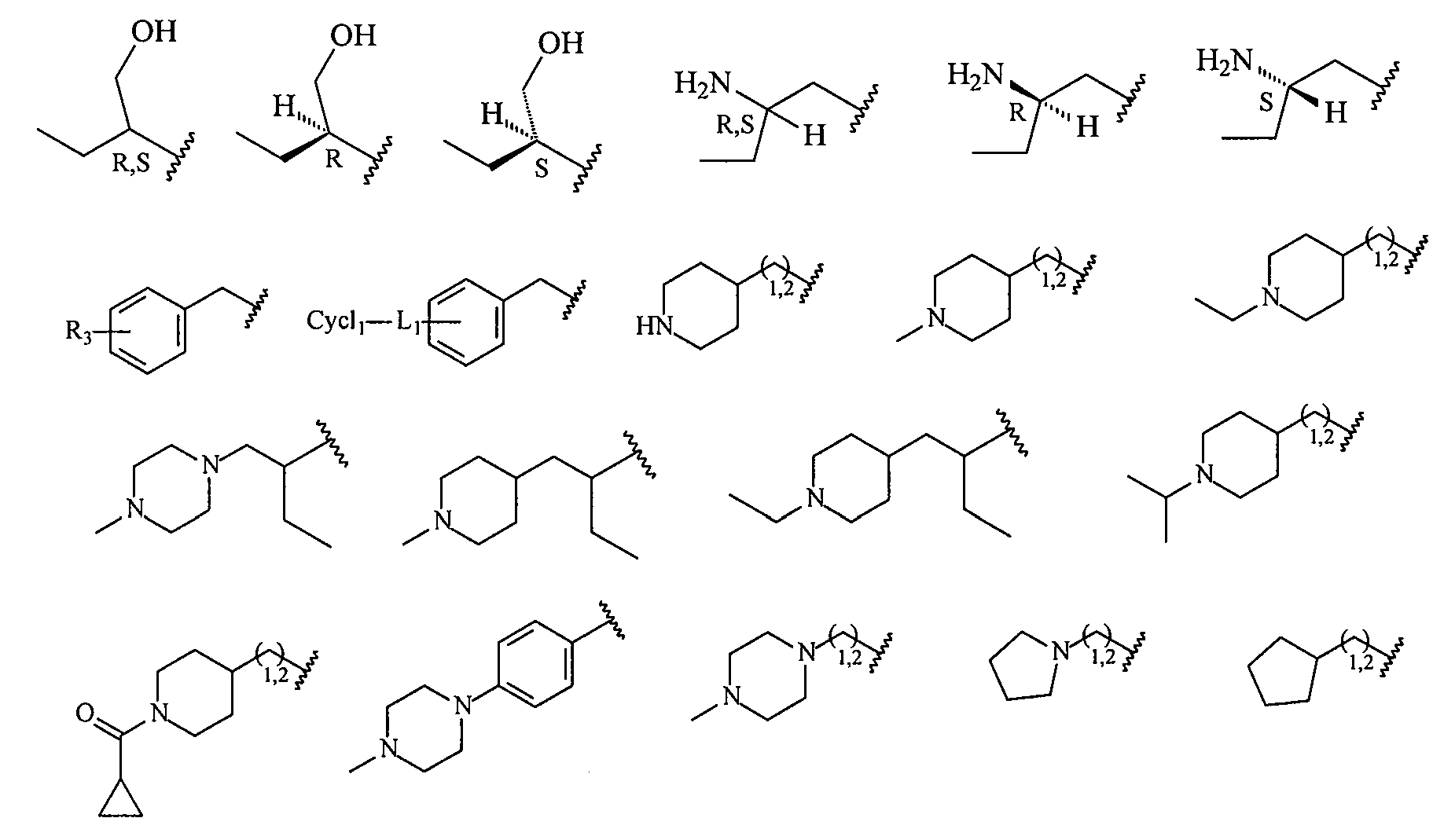
В более конкретных вариантах осуществления структур (I-A), (II-A), (I-Aa) и (II-Aa) R2 представляет собой -(CH2)1,2-пиперид-4-ил, замещенный -(CH2)1,2-пиперид-4-ил, -(CH2)1,2-пиперазин-1-ил или замещенный -(CH2)1,2-пиперазин-1-ил, например, фрагмент, выбранный из
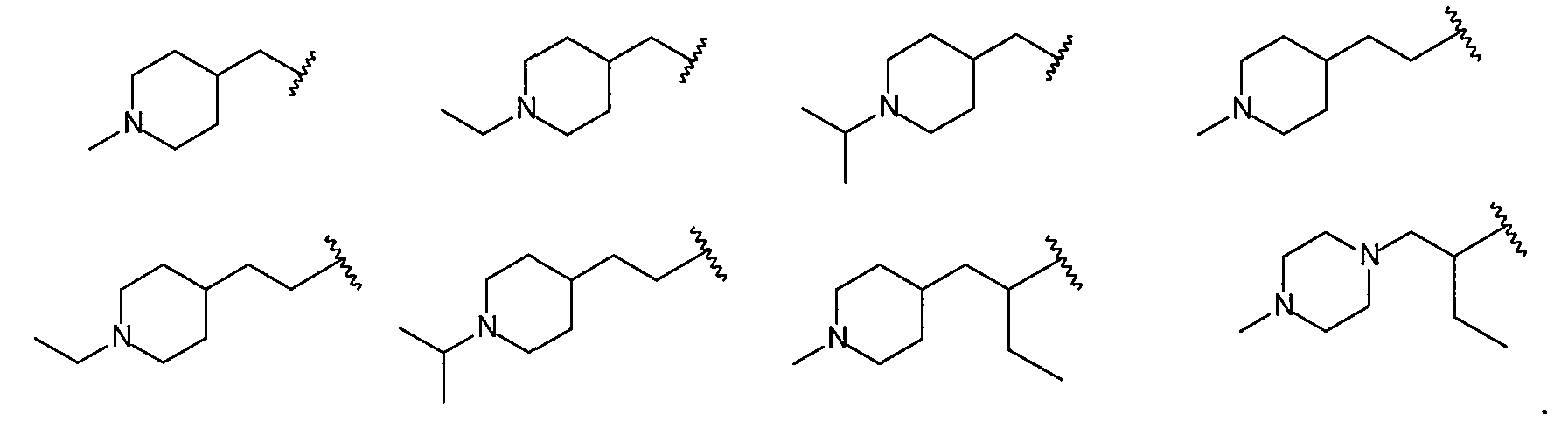
Более конкретно R2 выбран из
В еще одном конкретном варианте осуществления структур (I) и (II), X представляет собой O, и R1 является замещенным или незамещенным фенилом (где R соответствует данным выше определениям, и R1' означает отсутствие заместителя или наличие одного или нескольких заместителей) и соединения имеют следующие структуры (I-B) и (II-B), соответственно:
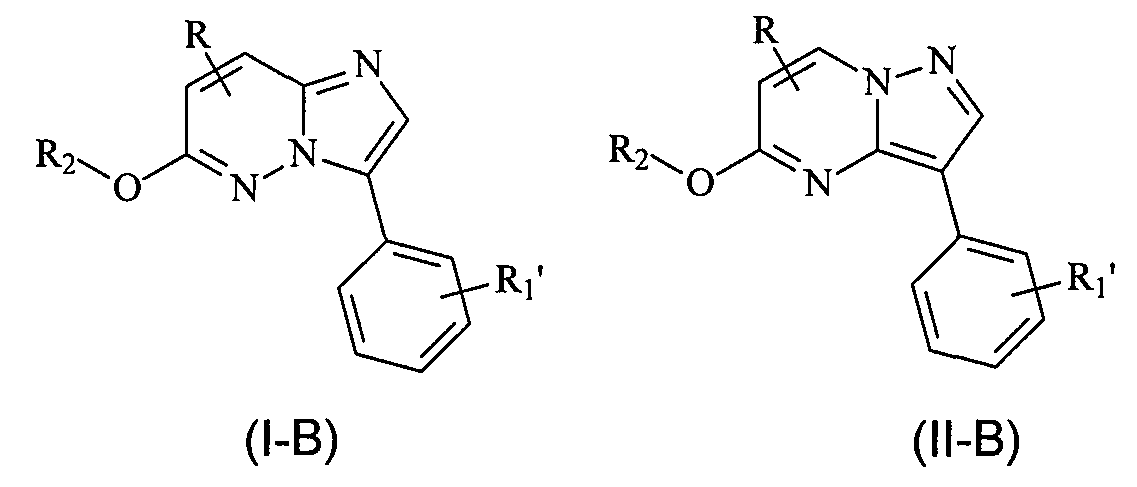
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
В более конкретных вариантах осуществления структур (I-B) или (II-B) R представляет собой алкил, например метил, и соединения имеют следующие структуры (I-Bb) и (II-Bb):
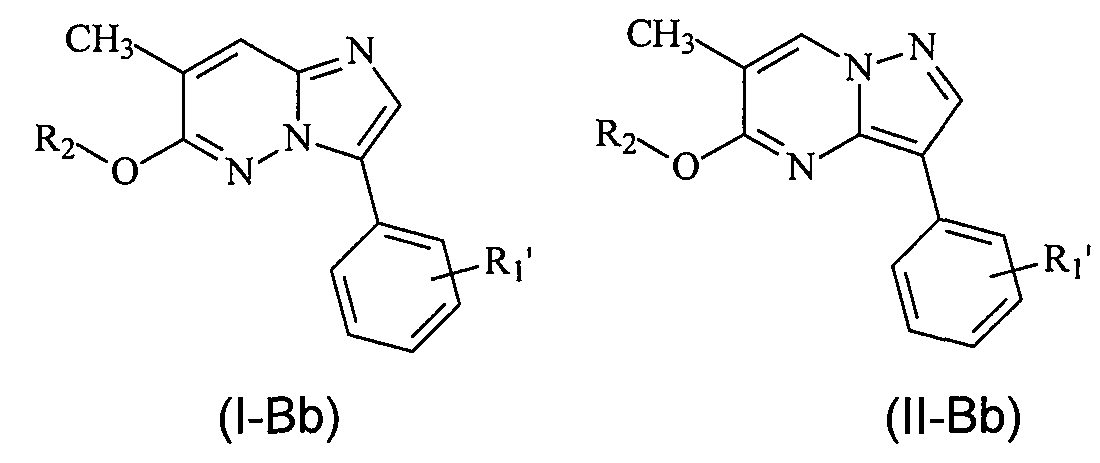
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
В более конкретных вариантах осуществления структур (I-B), (II-B), (I-Bb) и (II-Bb) R1 представляет собой замещенный фенил, имеющий, по меньшей мере, один п-, о- или м-заместитель, выбранный из галогена, -OCF3, -OCHF2, -CF3, -OCH3, -NH2, -NO2, -OH, -COCH3, -NHSO2CH3 или -N(CH3)2, и в более конкретном варианте осуществления R1 представляет собой замещенный фенил, имеющий, по меньшей мере, один п-, о- или м-заместитель, выбранный из -OCF3, -OCHF2, -CF3, -OCH3 и -OH, и в еще более конкретном варианте осуществления заместитель R1 выбран из
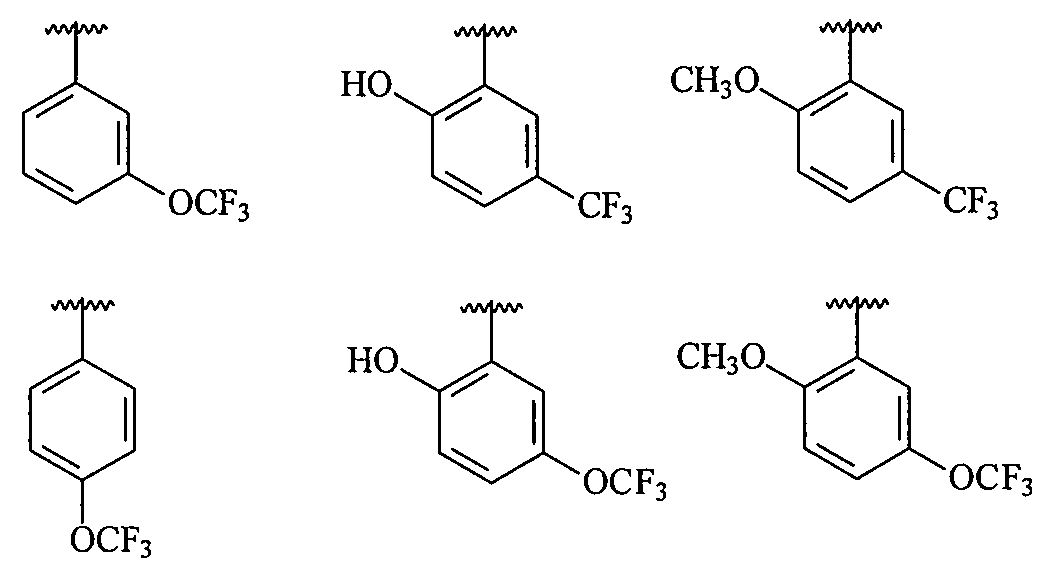
В более конкретных вариантах осуществления структур (I-B), (II-B), (I-Bb) и (II-Bb) R2 представляет собой -(CH2)n-циклопропил, -(CH2)n-циклопентил, -(CH2)n-циклогексил, -SO2-CH3, -SO2-(CH2)nCH3, -(CH2)n-пиперонил, -(CH2)n-пиперидил, -(CH2)n-пиперазинил, -(CH2)n-фурил, -(CH2)n-тиофен, -(CH2)n-пиридил, -(CH2)n-пиримидил, -(CH2)nOCH3, -(CH2)nOH или -(CH2)nN(CH3)2, где n означает 0, 1, 2, 3 или 4 и каждый из приведенных выше фрагментов необязательно замещен одним или несколькими заместителями; или же R2 является структурой, выбранной из
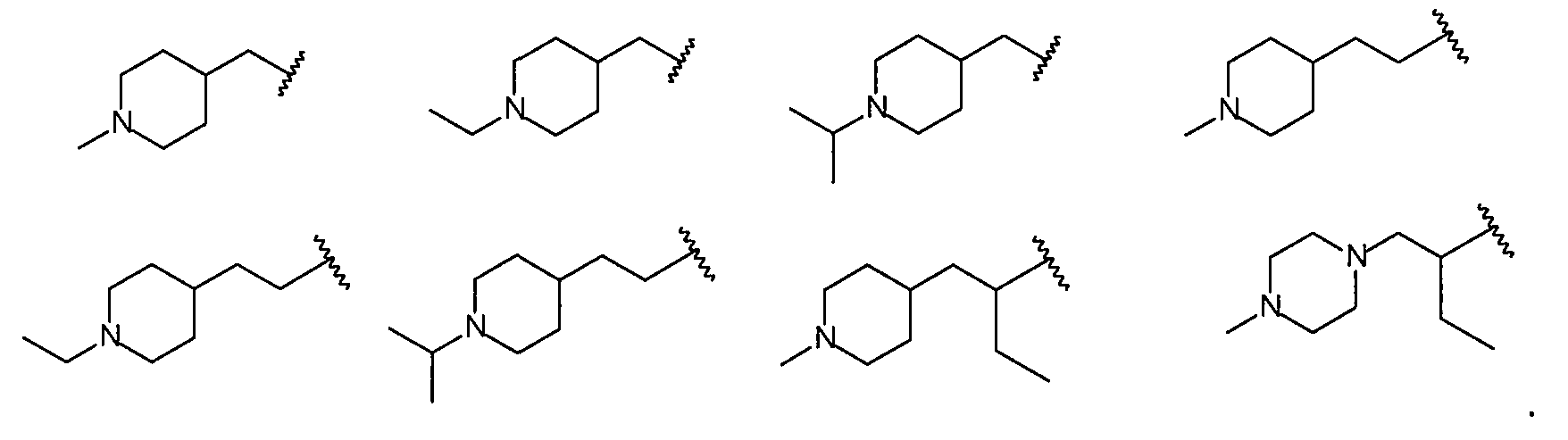
В более конкретных вариантах осуществления структур (I-B), (II-B), (I-Bb) и (II-Bb) R2 представляет собой -(CH2)1,2-пиперид-4-ил, замещенный -(CH2)1,2-пиперид-4-ил, -(CH2)1,2-пиперазин-1-ил или замещенный -(CH2)1,2-пиперазин-1-ил, например, фрагмент, выбранный из
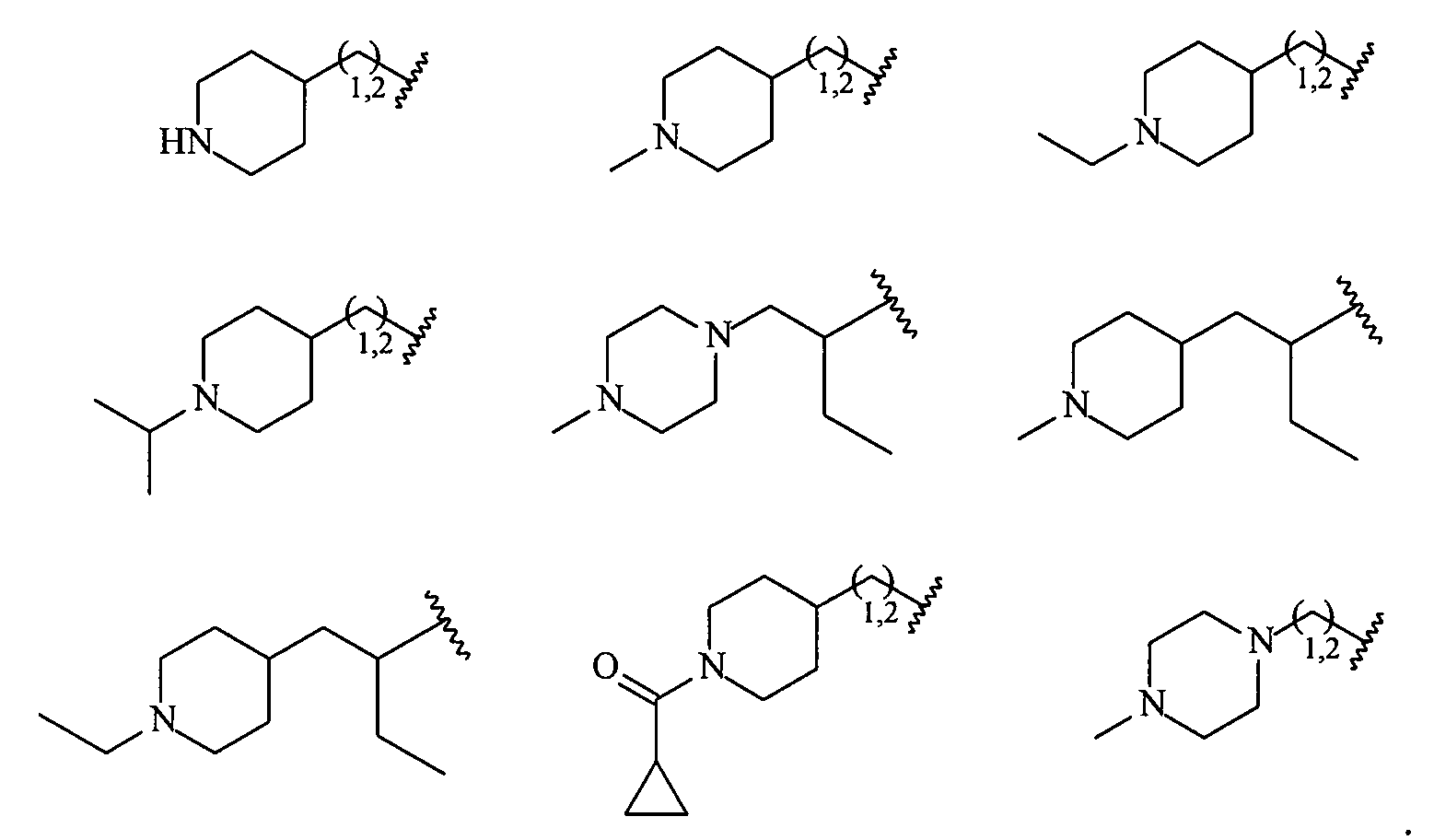
Более конкретно R2 выбран из
В еще одном конкретном варианте осуществления структур (I) и (II) X представляет собой S, SO или SO2, и R1 является замещенным или незамещенным фенилом (где в приведенных ниже формулах R1' означает отсутствие заместителя или наличие одного или нескольких заместителей) и соединения имеют следующие структуры (I-C) и (II-C), соответственно:
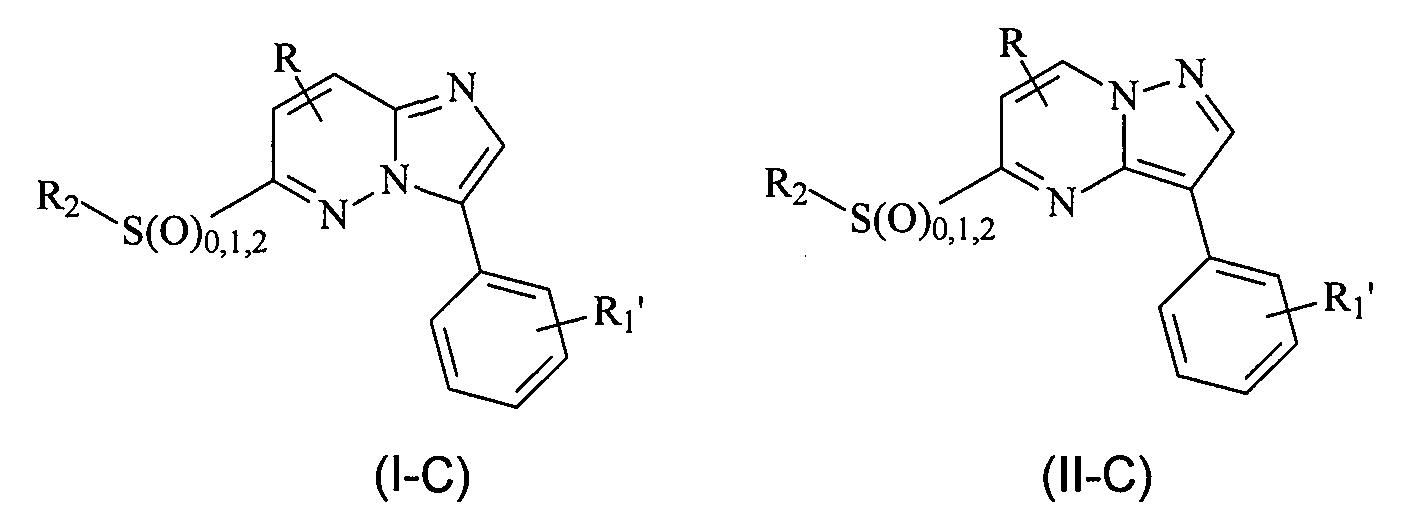
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
В более конкретных вариантах осуществления структур (I-C) или (II-C), R представляет собой алкил, например, метил, и соединения имеют следующие структуры (I-Cc) и (II-Cc):
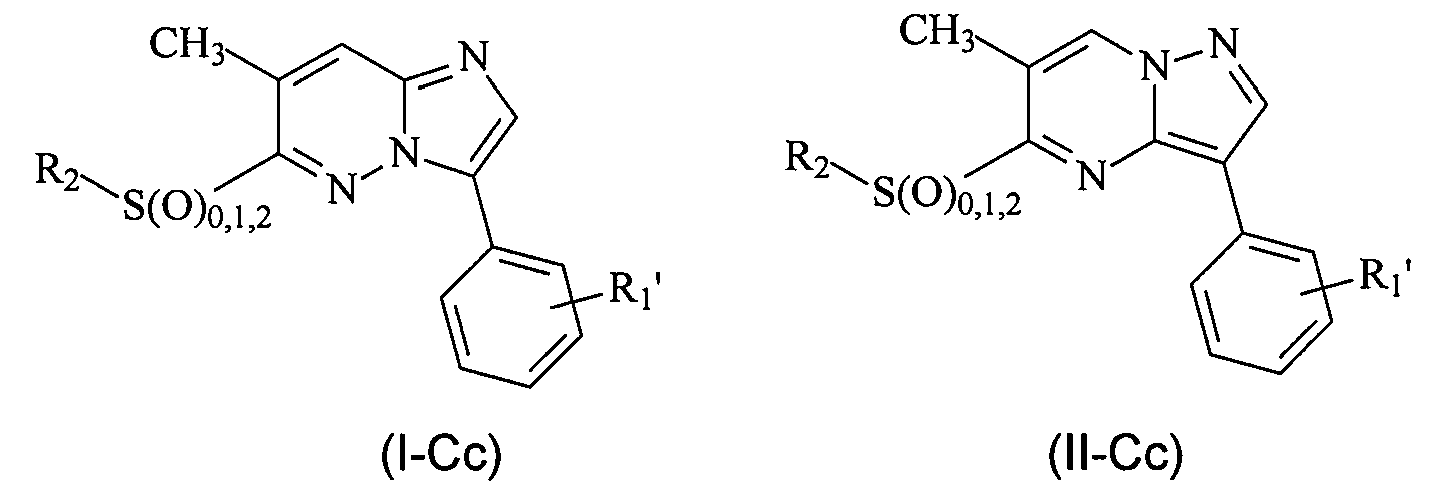
или являются их стереоизомерами, пролекарствами или фармацевтически приемлемыми солями.
В более конкретных вариантах осуществления структур (I-C), (II-C), (I-Cc) и (II-Cc), R1 представляет собой замещенный фенил, имеющий, по меньшей мере, п-, о- или м-заместитель, выбранный из галогена, -OCF3, -OCHF2, -CF3, -OCH3, -NH2, -NO2, -OH, -COCH3, -NHSO2CH3 и -N(CH3)2, и в более конкретном варианте осуществления R1 представляет собой замещенный фенил, имеющий, по меньшей мере, один п-, о- или м-заместитель, выбранный из -OCF3, -OCHF2, -CF3, -OCH3 и -OH, и в еще более конкретном варианте осуществления заместитель R1 выбран из
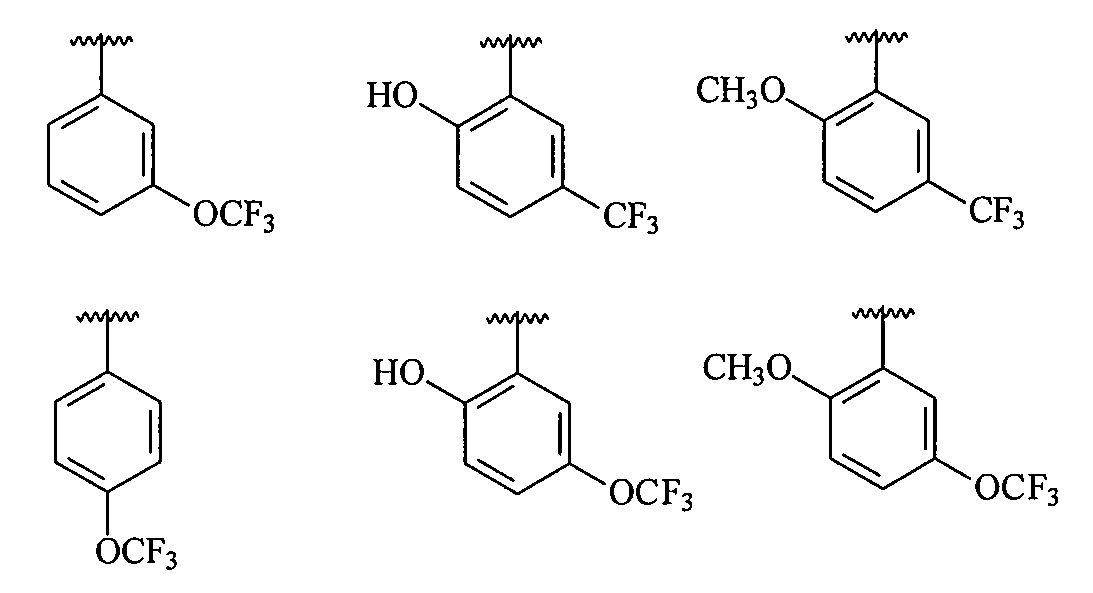
В более конкретных вариантах осуществления структур (I-C), (II-C), (I-Cc) и (II-Cc) R2 представляет собой -(CH2)n-циклопропил, -(CH2)n-циклопентил, -(CH2)n-циклогексил, -SO2-CH3, -SO2-(CH2)nCH3, -(CH2)n-пиперонил, -(CH2)n-пиперидил, -(CH2)n-пиперазинил, -(CH2)n-фурил, -(CH2)n-тиофен, -(CH2)n-пиридил, -(CH2)n-пиримидил, -(CH2)nOCH3, -(CH2)nOH или -(CH2)nN(CH3)2, где n означает 0, 1, 2, 3 или 4 и каждый из приведенных выше фрагментов необязательно замещен одним или несколькими заместителями; или же R2 является структурой, выбранной из
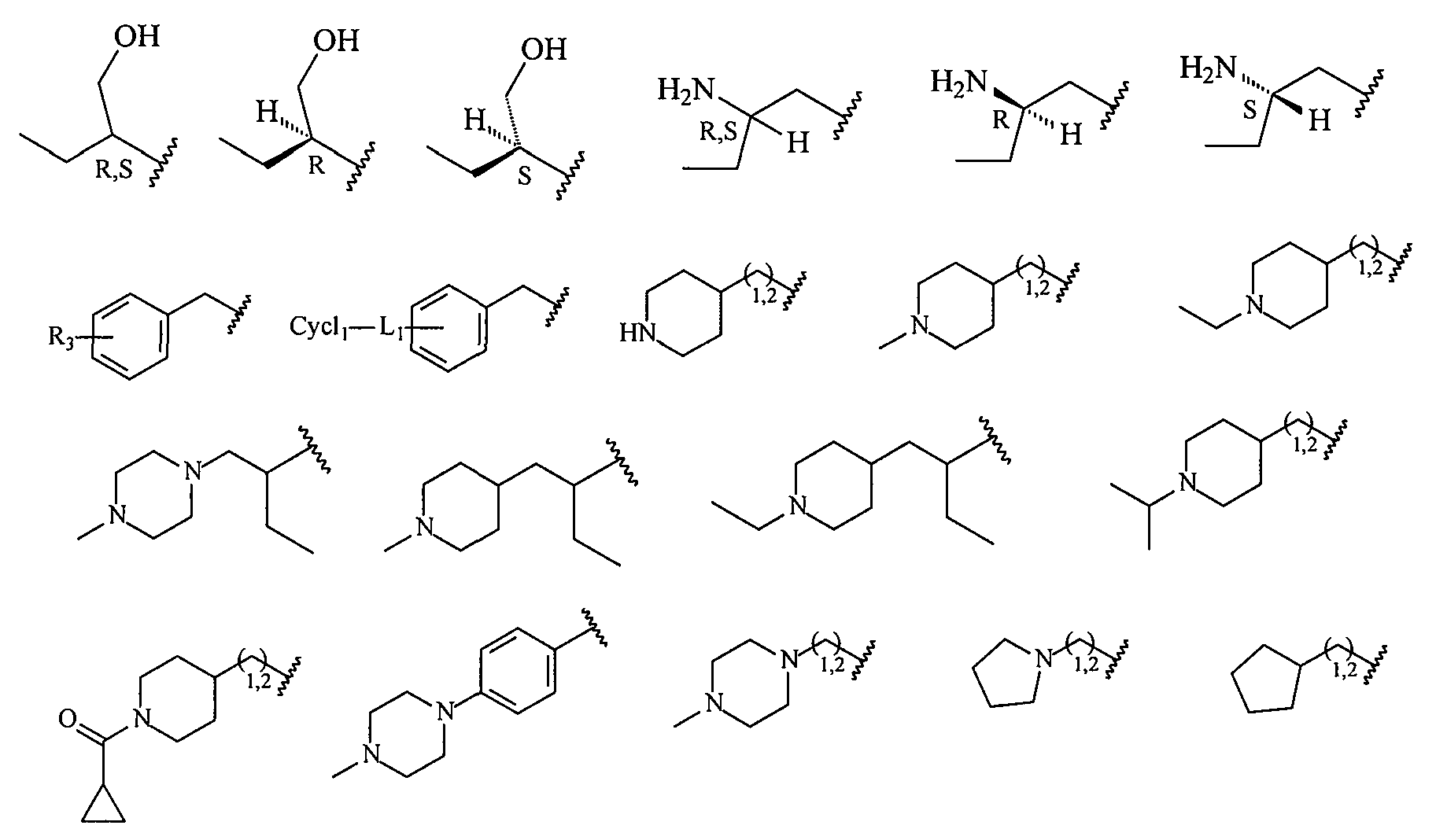
В более конкретных вариантах осуществления структур (I-C), (II-C), (I-Cc) и (II-Cc) R2 представляет собой -(CH2)1,2-пиперид-4-ил, замещенный -(CH2)1,2-пиперид-4-ил, -(CH2)1,2-пиперазин-1-ил или замещенный -(CH2)1,2-пиперазин-1-ил, например, фрагмент, выбранный из
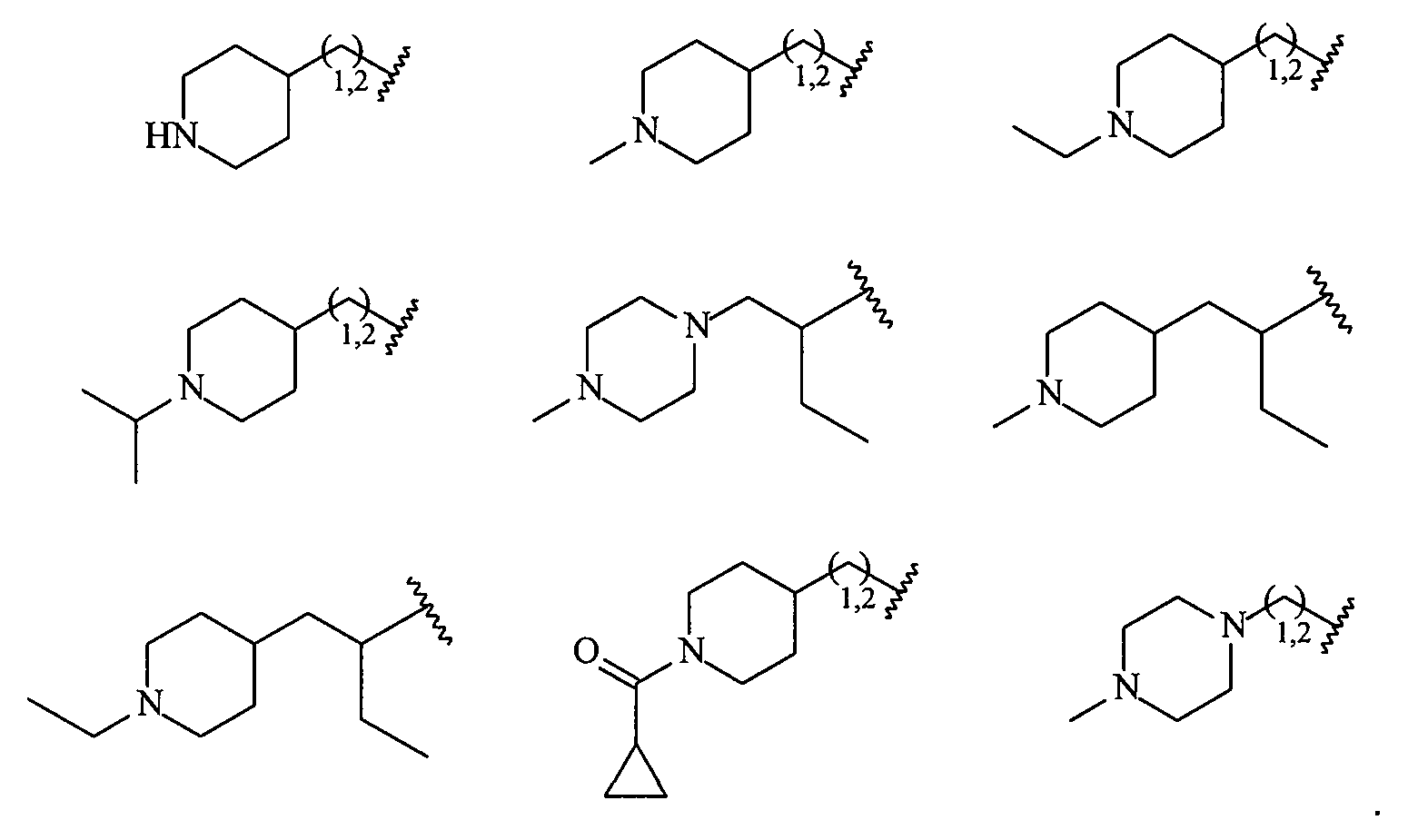
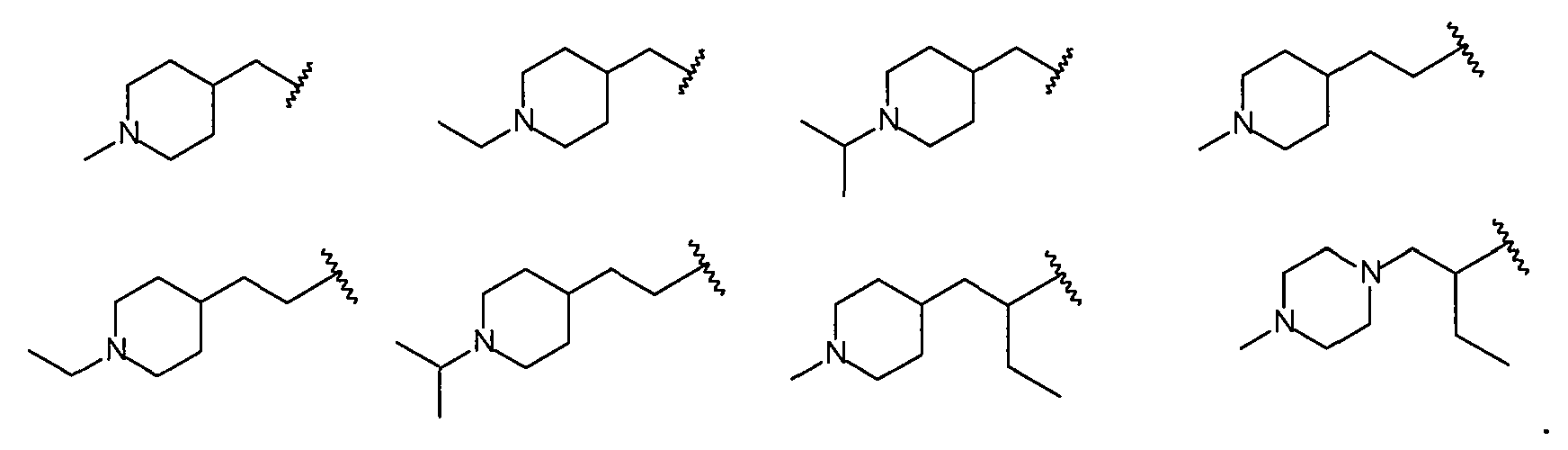
Более конкретно R2 выбран из
В более конкретных аспектах соединения показанной выше структуры (I) имеют структурные формулы, приведенные в таблице II (соединения от 2-1 до 2-22).
В более конкретных аспектах соединения показанной выше структуры (II) имеют структурные формулы, приведенные в таблице III (соединения от 3-1 до 3-13).
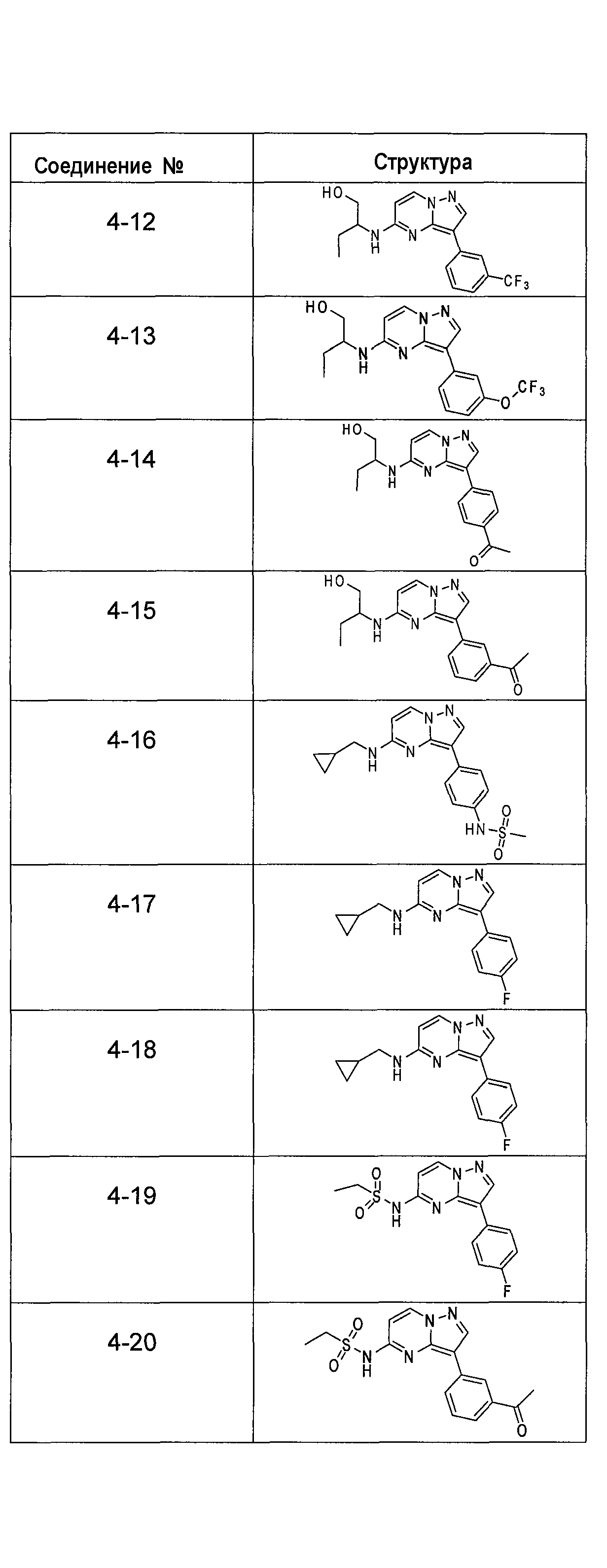
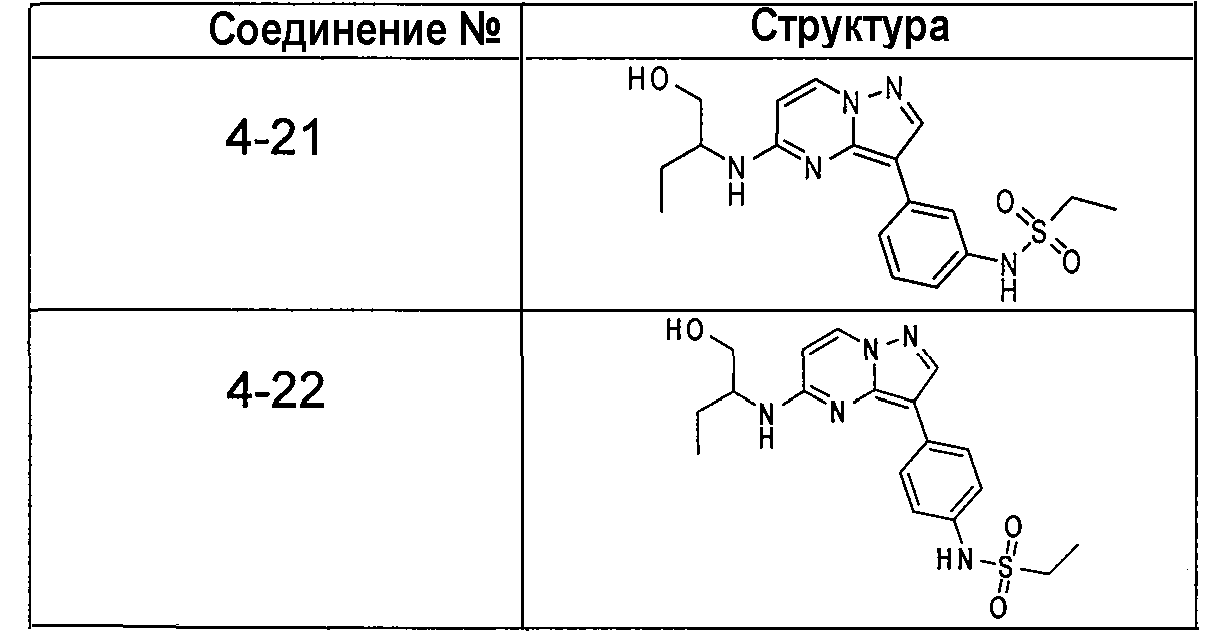
В более конкретных аспектах разработаны соединения показанной выше структуры (II), имеющие структурные формулы, приведенные в таблице IV (соединения от 4-1 до 4-22).
В более конкретных аспектах разработаны соединения приведенной выше структуры (I), имеющие структурные формулы, приведенные в таблице VII (соединения от 7-1 до 7-51).
Соединения, которые имеют одинаковую молекулярную формулу, но различаются природой или последовательностью связей между атомами или расположением атомов в пространстве, называют «изомерами». Изомеры, которые отличаются расположением атомов в пространстве, называют «стереоизомерами». Стереоизомеры, которые не являются зеркальными отображениями друг друга, называют «диастереомерами», и стереоизомеры, которые являются несовместимыми зеркальными отображениями друг друга, именуют «энантиомерами». Если в соединении имеется асимметрический центр, например, связанный с четырьмя различными группами, возможно существование пары энантиомеров. Энантиомер может быть охарактеризован абсолютной конфигурацией асимметрического центра и описан с помощью R- и S- номенклатуры Кана и Прелога (Cahn, R., Ingold, C., and Prelog, V. Angew. Chem. 78:413-47, 1966; Angew. Chem. Internat. Ed. Eng. 5:385-415, 511, 1966) или направлением, в котором молекула вращает плоскость поляризованного света, и назван правовращающим или левовращающим (т.е. соответственно (+)- или (-)-изомером, соответственно). Хиральное соединение может существовать либо в виде индивидуального энантиомера, либо в виде их смеси. Смесь, содержащую равные количества энантиомеров, именуют «рацемической смесью».
Соединения по настоящему изобретению могут иметь один или несколько асимметрических центров; следовательно такие соединения могут быть получены в виде индивидуальных (R)- или (S)-стереоизомеров или их смесей. Если нет иных указаний, описание или наименование конкретного соединения в данной заявке и формуле изобретения подразумевает включение обоих индивидуальных энантиомеров и их смесей, рацемических или иных. Способы установления пространственного строения и разделения стереоизомеров хорошо известны в технике (см. обсуждение в главе 4 Advanced Organic Chemistry, 4th edition, March, J., John Wiley and Sons, New York City, 1992).
Соединения по настоящему изобретению могут демонстрировать явления таутомерии и структурной изомерии. Например, описанные в настоящей заявке соединения могут иметь E- или Z-конфигурацию относительно двойной связи, соединяющей фрагмент 2-индолинона и фрагмент пиррола, или же соединения могут являться смесью E- и Z-изомеров. Настоящее изобретение охватывает любые таутомерные или структурно-изомерные формы и их смеси, которые обладают способностью модулировать активность aurora-2 киназы, и оно не ограничено какой-либо одной таутомерной или структурно-изомерной формой.
Предполагается, что соединение по настоящему изобретению могло бы подвергаться метаболизму под действием ферментов в организме, например, в организме человека, с образованием метаболитов, которые могут модулировать активность протеинкиназ. Такие метаболиты входят в объем настоящего изобретения.
Соединение по настоящему изобретению или его фармацевтически приемлемая соль могут вводиться пациенту, в частности человеку, в чистом виде или в составе фармацевтической композиции, в которой упомянутые соединения смешаны с подходящими носителями или наполнителями (наполнителем). Методики приготовления и введения лекарственных средств могут быть найдены, например, в Remington's Pharmacological Sciences, Mack Publishing Co., Easton, PA, последнее издание.
Термин «фармацевтическая композиция» относится к смеси одного или нескольких соединений, описанных в настоящей заявке, или их фармацевтически приемлемых солей или пролекарств, с другими химическими компонентами, например, фармацевтически приемлемыми наполнителями. Цель получения фармацевтической композиции заключается в облегчении введения соединения в организм.
Термин «фармацевтически приемлемый наполнитель» относится к инертному веществу, добавленному в фармацевтическую композицию для дополнительного облегчения введения соединения. Примеры наполнителей включают, не ограничиваясь указанными, карбонат кальция, фосфат кальция, различные сахара и различные типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли.
Термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Такие соли могут включать (1) кислотно-аддитивные соли, которые получены взаимодействием свободного основания исходного соединения с неорганическими кислотами, например, хлористоводородной кислотой, бромистоводородной кислотой, азотной кислотой, фосфорной кислотой, серной кислотой, перхлорной кислотой и т.п., или с органическими кислотами, например, уксусной кислотой, щавелевой кислотой, (D)- или (L)-яблочными кислотами, малеиновой кислотой, метансульфоновой кислотой, этансульфоновой кислотой, п-толуолсульфоновой кислотой, салициловой кислотой, винной кислотой, лимонной кислотой, янтарной кислотой, малоновой кислотой и т.п., предпочтительно хлористоводородной кислотой или (L)-яблочной кислотой; или (2) соли, образующиеся в том случае, когда кислотный протон, присутствующий в исходном соединении, замещен либо ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла либо ионом алюминия; или координирован с органическим основанием, например, этаноламином, диэтаноламином, триэтаноламином, трометамином, N-метилглутамином и т.п.
Соединение по настоящему изобретению может также действовать в качестве пролекарства или быть предназначенным для действия в качестве пролекарства. Термин «пролекарство» относится к агенту, который превращается в необходимое лекарственное средство in vivo. Пролекарства часто находят применение, поскольку в определенных ситуациях их можно вводить легче, чем само лекарственное средство. Они могут, например, быть биодоступными при пероральном введении, в то время как само лекарственное средство не является биодоступным. Пролекарство может также обладать лучшей растворимостью в фармацевтической композиции по сравнению с самим лекарственным средством. Примерами пролекарств, не ограничиваясь перечисленными, могли бы являться соединения по настоящему изобретению, которые вводятся в форме сложных эфиров («пролекарства»), фосфатов, амидов, карбаматов или мочевин.
Термин «терапевтически эффективное количество» относится к такому количеству вводимого соединения, которое до определенной степени облегчает один или несколько симптомов подвергаемого лечению расстройства. Применительно к лечению рака, термин «терапевтически эффективное количество» относится к такому количеству, которое приводит к одному из следующих результатов: (1) уменьшению размеров опухоли; (2) подавлению опухолевых метастазов; (3) торможению роста опухоли и/или (4) облегчению одного или нескольких симптомов, связанных с раком.
Термины «заболевание» или «состояние, опосредованное протеинкиназой» в настоящем описании означают любое заболевание или опасное состояние, о котором известно, что протеинкиназа играет определенную роль в его развитии. Термины «заболевание» или «состояние, опосредованное протеинкиназой» также означают такие заболевания или состояния, которые облегчаются при лечении ингибитором протеинкиназы. Такие состояния включают, не ограничиваясь указанными, раковые опухоли, которые экспрессируют Pim-киназы, в частности Pim-1 киназу, а также другие гиперпролиферативные расстройства, связанные с экспрессией Pim-киназы. В некоторых вариантах осуществления раковое заболевание является раком ободочной кишки, груди, желудка, простаты, поджелудочной железы или ткани яичников.
Термины «заболевание» или «состояние, опосредованное Pim-киназой» в настоящем описании означают любое заболевание или опасное состояние, о котором известно, что Pim 1 киназа, Pim 2 киназа и/или Pim 3 киназа экспрессируются и/или играют определенную роль в его развитии. Кроме того, термины «заболевание» или «состояние, опосредованное Pim-киназой» также означают те заболевания или состояния, которые облегчаются при лечении ингибитором Pim-киназы.
В настоящем описании термины «вводить» или «введение» относятся к доставке соединения по настоящему изобретению или его фармацевтически приемлемой соли, или же фармацевтической композиции, содержащей соединение по настоящему изобретению или его фрамацевтически приемлемую соль по настоящему изобретению, в организм пациента, с целью профилактики или лечения расстройства, связанного с протеинкиназой.
Подходящие пути введения могут включать, не ограничиваясь этим, пероральное, ректальное, трансмукозальное или интестинальное введение, или внутримышечные, подкожные, интрамедуллярные, интратекальные прямые интравентрикулярные, внутривенные, интравитреальные, интраперитонеальные, интраназальные или внутриглазные инъекции. В некоторых вариантах осуществления предпочтительными путями введения являются пероральный и внутривенный.
С другой стороны, можно вводить соединения местным, а не системным путем, например, с помощью инъекции соединения непосредственно в солидную опухоль, часто в виде состава с пролонгированным или отсроченным высвобождением.
Кроме того, лекарственное средство может вводиться в составе системы нацеленной доставки, например, в липосомах, покрытых опухоль-специфичным антителом. В этом способе липосомы могут быть нацелены на опухоль и селективно поглощаться ей.
Фармацевтические композиции по настоящему изобретению могут производиться способами, хорошо известными в технике, например, с помощью обычных методик смешивания, растворения, гранулирования, изготовления драже, растирания, эмульгирования, инкапсулирования, улавливания или лиофилизации.
Фармацевтические композиции, предназначенные для применения по настоящему изобретению, могут быть получены любым обычным способом с применением одного или нескольких физиологически приемлемых носителей, в том числе наполнителей и вспомогательных веществ, которые облегчают превращение действующих соединений в препараты, которые могут найти фармацевтическое применение. Подходящий состав фармацевтического препарата зависит от выбранного пути введения.
Для введения путем инъекции соединения по настоящему изобретению могут включаться в состав водных растворов, предпочтительно в физиологически совместимых буферах, например, растворе Хенкса, растворе Рингера или физиологическом солевом буферном растворе. Для трансмукозального введения в составах применяют проникающие вещества, подходящие для прохождения барьера, который необходимо преодолеть. Такие проникающие вещества хорошо известны в технике.
В случае перорального введения составы на основе соединений по настоящему изобретению могут быть получены смешиванием действующих соединений с фармацевтически приемлемыми носителями, хорошо известными в технике. Такие носители дают возможность вводить соединения по настоящему изобретению в состав таблеток, пилюль, ромбических таблеток, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального приема пациентом. Фармацевтические препараты для перорального применения могут быть изготовлены с применением твердого наполнителя, необязательного измельчения полученной смеси и переработкой смеси в гранулы после добавления других подходящих вспомогательных компонентов, если это желательно, с получением таблеток или ядер драже. Применимыми наполнителями являются, в частности, такие наполнители как сахара, в том числе лактоза, сахароза, маннит или сорбит, препараты целлюлозы, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал и картофельный крахмал, а также другие материалы, такие как желатин, камедь трагаканта, метилцеллюлоза, гидроксипропилметилцлеллюлоза, натрий карбоксиметилцлеллюлоза и/или поливинилпирролидон (PVP). Если это желательно, могут быть добавлены дезинтегрирующие средства, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота. Также могут применяться соли, например, альгинат натрия.
Ядра драже снабжают соответствующим покрытием. С этой целью можно применять концентрированные растворы сахаров, которые могут необязательно содержать гуммиарабик, тальк, поливинилпирролидон, карбопол гель, полиэтиленгликоль и/или диоксид титана, лаковые растворы и подходящие органические растворители или смеси растворителей. В покрытие таблеток или драже можно добавлять красители или пигменты для идентификации или для характеристики различных комбинаций дозировки действующих компонентов.
Фармацевтические композиции, которые могут применяться перорально, включают плотно закрытые капсулы, изготовленные из желатина, а также мягкие, запечатанные капсулы, изготовленные из желатина и пластификатора, например, глицерина или сорбита. Плотно закрытые капсулы могут содержать действующие ингредиенты в смеси с наполнителем, например, лактозой, связующим веществом, например, крахмалом, и/или смазывающим средством, например, тальком или стеаратом магния, и, необязательно, стабилизаторами. В мягких капсулах действующие соединения могут быть растворены или суспендированы в подходящих жидкостях, например, жирных маслах, жидких парафинах или жидких полиэтиленгликолях. В эти составы также могут быть добавлены стабилизаторы. Фармацевтические составы, которые могут применяться помимо этого, включают твердые желатиновые капсулы. Капсулы или пилюли могут быть упакованы в бутылки из коричневого стекла или пластика для защиты действующего соединения от света. Контейнеры, содержащие капсулы с действующим соединением, предпочтительно хранят при регулируемой температуре, близкой к комнатной (15-30°C).
Для введения с помощью ингаляции, соединения, применяемые по настоящему изобретению, удобно поставлять в форме аэрозольных спреев, применяя упаковки под давлением или распылитель и подходящий пропеллент, например, не ограничиваясь перечисленным, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан или диоксид углерода. В случае аэрозоля под давлением, единица дозировки может регулироваться применением клапана, выпускающего отмеренное количество препарата. Капсулы и картриджи, изготовленные, например, из желатина, для применения в ингаляторах или инсуффляторах, могут содержать смесь порошка соединения с подходящей порошковой основой, например, лактозой или крахмалом.
Соединения по настоящему изобретению могут также быть включены в состав для парентерального введения, например, в виде болюсной инъекции или продолжительной инфузии. Составы для инъекции могут быть представлены в виде дозированных лекарственных форм, например, в ампулах или в контейнерах, содержащих большое количество доз, с добавлением консервантов. Эти композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать вспомогательные добавки, например, суспендирующие, стабилизирующие и/или диспергирующие средства.
Фармацевтические композиции для парентерального введения включают водные растворы или растворимые в воде формы, например, не ограничиваясь перечисленным, соль действующего соединения. Кроме того, могут быть приготовлены суспензии действующих соединений в липофильном носителе. Подходящие липофильные носители включают жирные масла, например, кунжутное масло, синтетические сложные эфиры жирных кислот, например, этилолеат и триглицериды, или такие носители, как липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, например, натрий-карбоксиметилцеллюлозу, сорбит или декстран. Суспензия может также необязательно содержать подходящие стабилизаторы и/или средства, которые увеличивают растворимость соединений, чтобы сделать возможным получение растворов с высокой концентрацией.
С другой стороны, действующий ингредиент может иметь форму порошка для восстановления жидкого препарата перед применением с использованием подходящего носителя, например, стерильной апирогенной воды.
Соединения по настоящему изобретению можно также включать в состав композиций для ректального введения, например, суппозиториев или удерживающих клизм, применяя, например, обычные основы суппозиториев, такие как масло какао или другие глицериды.
Помимо описанных выше составов, соединения по настоящему изобретению можно включать в составы препаратов с пролонгированным высвобождением. Такие составы длительного действия можно вводить при помощи имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Для такого пути введения соединение по настоящему изобретению можно включать в подходящие полимерные или гидрофобные вещества (например, в состав эмульсии с фармакологически приемлемым маслом), в ионообменные смолы или вводить в виде плохо растворимых производных, например, не ограничиваясь этим, плохо растворимых солей.
Неограничивающим примером фармацевтического носителя для гидрофобных соединений по настоящему изобретению является система сорастворителей, например, система сорастворителей VPD, содержащая бензиловый спирт, неполярное ПАВ, смешивающийся с водой органический полимер и водную фазу. VPD представляет собой смесь 3% (масса/объем) бензилового спирта, 8% (масса/объем) неполярного ПАВ полисорбат 80 и 65% (масса/объем) полиэтиленгликоля 300, доведенную до необходимого объема добавлением абсолютного этанола. Система сорастворителей VPD (VPD:D5W) состоит из VPD, разбавленного 1:1 5% раствором декстрозы в воде. Данная система сорастворителей хорошо растворяет гидрофобные соединения и демонстрирует низкую токсичность при системном введении. В действительности пропорции компонентов в такой системе сорастворителей могут значительно варьироваться без нарушения ее характеристик по растворимости и токсичности. Кроме того, можно менять природу сорастворителей: например, вместо полисорбата 80 можно применять другие неполярные растворители с низкой токсичностью, можно варьировать молекулярную массу полиэтиленгликоля и заменять его другими биосовместимыми полимерами, например, поливинилпирролидоном, и, наконец, декстрозу можно менять на другие сахара или полисахариды.
В качестве альтернативы можно применять другие системы доставки гидрофобных фармацевтических соединений. Липосомы и эмульсии являются хорошо известными примерами носителей для доставки гидрофобных лекарственных средств. Кроме того, можно применять некоторые органические растворители, например, диметилсульфоксид, хотя часто ценой более высокой токсичности.
Кроме того, соединения по настоящему изобретению могут доставляться с применением системы с отсроченным высвобождением, например полупроницаемой матрицы из твердых гидрофобных полимеров, содержащей терапевтическое средство. Были выявлены различные материалы, обеспечивающие отсроченное высвобождение, и они хорошо известны специалисту в данной области техники. Капсулы с отсроченным высвобождением, в зависимости от их химической природы, могут высвобождать соединения за срок от нескольких недель до более чем 100 дней. В зависимости от химической природы и биологической стабильности терапевтического средства могут применяться дополнительные стратегии стабилизации белка.
Фармацевтические композиции по настоящему изобретению также могут включать подходящие твердые или гелевые носители или наполнители. Примеры таких носителей или наполнителей включают, не ограничиваясь перечисленным, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли.
Многие из соединений по настоящему изобретению, модулирующих активность протеинкиназы, могут применяться в форме физиологически приемлемых солей, в которых заявленные соединения могут образовывать отрицательно или положительно заряженные частицы. Примеры солей, в которых соединение образует положительно заряженный фрагмент, включают, не ограничиваясь перечисленными, четвертичные аммониевые соли (определенные в других местах настоящей заявки), как, например, гидрохлорид, сульфат, карбонат, лактат, тартрат, малат, малеат, сукцинат, где атом азота четвертичной аммониевой группы является атомом, входящим в некоторые из соединений по настоящему изобретению, который вступил во взаимодействие с подходящей кислотой. Соли, в которых соединение по настоящему изобретению образует отрицательно заряженные частицы, включают, не ограничиваясь указанными, соли натрия, калия, кальция и магния, полученные взаимодействием карбоксильной кислотной группы соединения с подходящим основанием (например, гидроксидом натрия (NaOH), гидроксидом калия (KOH), гидроксидом кальция (Ca(OH)2) и т.д.).
Фармацевтические композиции, которые подходят для применения по настоящему изобретению, включают композиции, в которых действующие ингредиенты содержатся в количествах, достаточных для достижения намеченной цели, например, модулирования активности протеинкиназы и/или лечения или предотвращения расстройства, связанного с протеинкиназой.
Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предотвращения, облегчения или улучшения симптомов заболевания или увеличения продолжительности жизни субъекта, подвергаемого лечению.
Определение терапевтически эффективного количества находится полностью в рамках компетенции специалиста в данной области, особенно в свете подробного раскрытия изобретения, приведенного в настоящей заявке.
Для любого соединения, применяемого в способах по настоящему изобретению, терапевтически эффективное количество или доза могут быть на первом этапе определены из данных исследований на клеточных культурах. Затем может быть определена дозировка для применения у животных моделей, так, чтобы достичь диапазона концентрации в крови, который включает значение IC50, определенное для клеточной культуры (т.е. концентрации тестируемого соединения, при которой достигается половина максимально возможного ингибирования активности протеинкиназы). После этого полученная информация может быть использована для более точного определения дозировок, применимых у людей.
Токсичность и терапевтическая эффективность соединений, описанных в настоящей заявке, может быть определена по стандартным фармацевтическим методикам на клеточных культурах или экспериментальных животных, например, путем определения IC50 и LD50 (оба параметра обсуждаются в другом месте настоящей заявки) для данного соединения. Данные, полученные в результате описанного исследования клеточных культур и экспериментов на животных, могут использоваться при определении диапазона дозировок для применения у людей. Дозировка может меняться в зависимости от применяемой лекарственной формы и пути введения. Точный состав, путь введения и дозировка могут быть выбраны лечащим врачом в зависимости от состояния пациента. (См., например, Goodman & Gilman's The Pharmacological Basis of Therapeutics, Ch.3, 9th ed., Ed. by Hardman, J., and Limbard, L., McGraw-Hill, New York City, 1996, p.46).
Вводимые количества и промежутки между введениями могут быть индивидуально скорректированы для обеспечения уровня активных частиц в плазме, который является достаточным для поддержания эффекта модулирования киназ. Эти уровни концентрации в плазме именуются минимальными эффективными концентрациями (MEC). MEC будет меняться для каждого соединения, но его можно определить из данных in vitro, например, концентрацию, необходимую для достижения 50-90% ингибирования киназы, можно установить с использованием анализов, описанных в настоящей заявке. Дозировки, необходимые для достижения MEC, будут зависеть от индивидуальных характеристик и пути введения. Для определения концентрации в плазме можно применять ВЭЖХ анализ или биологические анализы.
Диапазоны дозировок также можно определять с использованием значений MEC. Для этого следует вводить соединения в режиме, который поддерживает уровни в плазме, превышающие MEC в течение 10-90% времени, предпочтительно 30-90% и наиболее предпочтительно 50-90%.
По имеющимся данным, терапевтически эффективные количества соединений по настоящему изобретению могут меняться от примерно 2,5 мг/м2 до 1500 мг/м2 в день. Дополнительные иллюстративные количества находятся в пределах 0,2-1000 мг/qid (четыре раза в день), 2-500 мг/qid и 20-250 мг/qid.
В случае местного введения или селективного поглощения эффективная местная концентрация лекарственного препарата может быть не связана с концентрацией в плазме, и можно применять другие известные в технике методики для определения правильных дозировок и интервалов между ними.
Вводимое количество композиции безусловно будет зависеть от подвергаемого лечению субъекта, тяжести заболевания, способа введения, мнения лечащего врача и т.д.
Если это желательно, композиции можно помещать в упаковку или дозирующее устройство, например, комплект, одобренный FDA, который может включать одну или несколько единиц дозированной лекарственной формы, содержащей действующий ингредиент. Эта упаковка может включать, например, металлическую или пластиковую фольгу, такую как блистерная упаковка. Упаковка или дозирующее устройство могут быть снабжены инструкциями по введению. Упаковка или дозирующее устройство могут быть также снабжены уведомлением, связанным с контейнером, имеющим форму, предписанную государственным ведомством, регулирующим производство, применение или продажу фармацевтических препаратов, причем в этом уведомлении отражено одобрение данным ведомством формы композиции или возможность введения человеку или животным. Такое уведомление, например, может заключаться в нанесении маркировки, одобренной Управлением по надзору за продуктами питания и лекарственными средствами США для лекарств, отпускаемых по рецепту, или представлять собой вкладыш в продукт, одобренной формы. Композиции, включающие соединения по настоящему изобретению в сочетании с совместимым фармацевтическим носителем могут также быть изготовлены, помещены в подходящий контейнер и помечены как применимые для лечения обозначенного заболевания. Соответствующие условия применения, указанные на этикетке, могут включать лечение опухолей, ингибирование ангиогенеза, лечение фиброза, диабета и т.п.
Как упоминалось выше, соединения и композиции по настоящему изобретению должны найти применение для лечения большого числа заболеваний и состояний, опосредованных протеинкиназами, включая заболевания и состояния, опосредованные aurora-2 киназой. Такие заболевания могут включать, в качестве примера, а не ограничения, раковые заболевания, например, рак легких, NSCLC (немелкоклеточный рак легких), овсяно-клеточный рак, рак кости, рак поджелудочной железы, рак кожи, дерматофибросаркому выбухающую, рак головы и шеи, кожную или внутриглазную меланому, рак матки, рак яичников, колоректальный рак, рак анальной области, рак желудка, рак ободочной кишки, рак груди, гинекологические опухоли (например, саркомы матки, карциному фаллопиевых труб, карциному эндометрия, карциному шейки матки, карциному вагины или карциному вульвы), болезнь Ходжкина, гепатоклеточный рак, рак пищевода, рак тонкой кишки, раковые заболевания эндокринной системы (например, раковые заболевания щитовидной железы, поджелудочной железы, околощитовидной железы или надпочечников), саркомы мягких тканей, рак уретры, рак пениса, рак простаты (особенно устойчивый к лечению гормонами), хроническую или острую лейкемию, солидные опухоли, возникающие в детском возрасте, гиперэозинофилию, лимфоцитарные лимфомы, рак мочевого пузыря, рак почек или мочеточника (например, карциному почечных клеток, карциному почечной лоханки), педиатрические злокачественные заболевания, новообразования центральной нервной системы (например, первичную лимфому ЦНС, опухоли спинного мозга, медулобластому, глиомы ствола головного мозга или аденомы гипофиза), пищевод Барретта (предраковый синдром), неопластические заболевания кожи, псориаз, фунгоидный микоз, а также доброкачественную гипертрофию простаты, заболевания, связанные с диабетом, такие как диабетическая ретинопатия, ишемия ретины и неоваскуляризация ретины, цирроз печени, ангиогенез, сердечно-сосудистые заболевания, такие как атеросклероз, иммунологические заболевания, например, аутоиммунные заболевания и почечные заболевания.
Соединения по настоящему изобретению могут применяться в комбинации с одним или несколькими другими химиотерапевтическими средствами. Дозировка соединения по настоящему изобретению может быть скорректирована с учетом взаимодействия между лекарственными средствами. В одном из вариантов осуществления химиотерапевтическое средство выбирают из группы, состоящей из ингибиторов митоза, алкилирующих средств, антиметаболитов, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, например, CAMPTOSAR (иринотекана), модификаторов биологической реакции, антигормонов, антиангиогенных средств, таких как MMP-2, MMP-9 и ингибиторов COX-2, антиандрогенов, координационных комплексов платины (цисплатина и т.д.), замещенных мочевин, например, гидроксимочевины; производных метилгидразина, например, прокарбазина; ингибиторов коры надпочечников, например, митотана, аминоглютетимида, гормонов и антагонистов гормонов, например, адренокортикостероидов (например, преднизона), прогестинов (например, гидроксипрогестерона капроата), эстрогенов (например, диэтилстильбэстрола), антиэстрогенов, например, тамоксифена, андрогенов, например, тестостерона пропионата и ингибиторов ароматазы, таких как анастрозола и AROMASIN (экземестана).
Примеры алкилирующих средств, которые могут применяться в комбинациях по упомянутым выше способам, включают, не ограничиваясь указанными, фторурацил (5-FU) сам по себе или в комбинации с лейковорином; другие аналоги пиримидина, такие как UFT, капецитабин, гемцитабин и цитарабин, алкилсульфонаты, например, бусульфан (применяемый при лечении хронической гранулоцитарной лейкемии), импросульфан и пипосульфан; азиридины, например, бензодепа, карбоквон, метуредепа и уредепа; этиленимины и метилмеламины, например, алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилолмеламин; а также азотистые иприты, например, хлорамбуцил (применяемый при лечении хронической лимфоцитарной лейкемии, первичной макроглобулинемии и не Ходжкинских лимфом), циклофосфамид (применяемый при лечении болезни Ходжкина, множественной миеломы, нейробластомы, рака груди, рака яичников, рака легких, опухоли Вилмса и рабдомиосаркомы), эстрамустин, ифосфамид, новембрихин, преднимустин и урацил иприт (применяемый при лечении первичного тромбоцитоза, не Ходжкинской лимфомы, болезни Ходжкина и рака яичников); и триазины, например, дакарбазин (применяемый при лечении саркомы мягких тканей).
Примеры антиметаболитических химиотерапевтических средств, которые могут применяться в комбинациях по упомянутым выше способам, включают, не ограничиваясь указанными, аналоги фолиевой кислоты, например, метотрексат (применяемый при лечении острой лимфоцитарной лейкемии, хориокарциномы, фунгоидного микоза, рака груди, рака головы и шеи, и остеогенной саркомы) и птероптерин; а также аналоги пурина, например, меркаптопурин и тиогуанин, который находит применение в лечении острой гранулоцитарной, острой лимфоцитарной и хронической гранулоцитарной лейкемии.
Примеры химиотерапевтических средств на основе природных продуктов, которые могут применяться в комбинациях по упомянутым выше способам, включают, не ограничиваясь указанными, алкалоиды барвинка, например, винбластин (применяемый при лечении рака груди и яичек), винкристин и виндезин; эпиподофиллотоксины, например, этопозид и тенипозид, которые применяются при лечении рака яичек и саркомы Капоши; антибиотические химиотерапевтические средств, например, даунорубицин, доксорубицин, эпирубицин, митомицин (применяемый для лечения рака желудка, шейки матки, ободочной кишки, груди, мочевого пузыря и поджелудочной железы), дактиномицин, темозоломид, пликамицин, блеомицин (применяемый при лечении рака кожи, пищевода и мочеполового тракта); и ферментные химиотерапевтические средства, например, L-аспарагиназа.
Примеры применимых ингибиторов COX-II включают Vioxx, CELEBREX (целекоксиб), валдекоксиб, паракоксиб, рофекоксиб и Cox 189.
Примеры применимых ингибиторов матриксной металлопротеиназы описаны в WO 96/33172 (опубликованной 24 октября 1996), WO 96/27583 (опубликованной 7 марта 1996), европейской заявке на патент № 97304971.1 (поданной 8 июля 1997), европейской заявке на патент № 99308617.2 (поданной 29 октября 1999), WO 98/07697 (опубликованной 26 февраля 1998), WO 98/03516 (опубликованной 29 января 1998), WO 98/34918 (опубликованной 13 августа 1998), WO 98/34915 (опубликованной 13 августа 1998), WO 98/33768 (опубликованной 6 августа 1998), WO 98/30566 (опубликованной 16 июля 1998), европейской патентной публикации 606046 (опубликованной 13 июля 1994), европейской патентной публикации 931788 (опубликованной 28 июля 1999), WO 90/05719 (опубликованной 31 мая 1990), WO 99/52910 (опубликованной 21 октября 1999), WO 99/52889 (опубликованной 21 октября 1999), WO 99/29667 (опубликованной 17 июня 1999), международной патентной заявке PCT № PCT/IB98/01113 (поданной 21 июля 1998), европейской патентной заявке № 99302232.1 (поданной 25 марта 1999), патентной заявке Великобритании № 9912961.1 (поданной 3 июня 1999), патенте США № 5863949 (выданном 26 января 1999), патенте США № 5861510 (выданном 19 января 1999) и европейской патентной публикации 780386 (опубликованной 25 июня 1997), причем все публикации включены в настоящую заявку во всей полноте с помощью ссылки. Предпочтительными ингибиторами MMP-2 и MMP-9 являются такие ингибиторы, которые не имеют активности или имеют небольшую активность по ингибированию MMP-1. Более предпочтительны те ингибиторы, которые селективно ингибируют MMP-2 и/или MMP-9 по отношению к другим матриксным металлопротеиназам (т.е. MMP-1, MMP-3, MMP-4, MMP-5, MMP-6, MMP-7, MMP-8, MMP-10, MMP-11, MMP-12 и MMP-13).
Некоторыми конкретными примерами ингибиторов MMP, применимых по настоящему изобретению, являются AG-3340, RO 32-3555, RS 13-0830 и соединения, выбранные из: 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты; гидроксамида 3-экзо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты; гидроксамида (2R,3R) 1-[4-(2-хлор-4-фторбензилокси)бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; гидроксамида 4-[4-(4-фторфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты; 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклобутил)амино]пропионовой кислоты; гидроксамида 4-[4-(4-хлорфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты; гидроксамида (R) 3-[4-(4-хлорфенокси)бензолсульфониламино]тетрагидропиран-3-карбоновой кислоты; гидроксамида (2R,3R) 1-[4-(4-фтор-2-метилбензилокси)бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; 3-[[(4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]пропионовой кислоты; 3-[[(4-(4-фторфенокси)бензолсульфонил]-(4-гидроксикарбамоилтетрагидропиран-4-ил)амино]пропионовой кислоты; гидроксамида 3-экзо-3-[4-(4-хлорфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты; гидроксамида 3-эндо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты; и гидроксамида (R) 3-[4-(4-фторфенокси)бензолсульфониламино]тетрагидрофуран-3-карбоновой кислоты; а также фармацевтически приемлемых солей и сольватов этих соединений.
Другие антиангиогенные средства, другие ингибиторы COX-II и другие ингибиторы MMP также могут применяться в рамках настоящего изобретения.
Кроме того, соединения по настоящему изобретению можно применять в сочетании с другими ингибиторами трансдукции сигнала, например, средствами, которые ингибируют реакцию на EGFR (рецептор эпидермального фактора роста), например, антителами против EGFR, антителами против EGF и молекулами, которые являются ингибиторами EGFR; ингибиторами VEGF (сосудистого эндотелиального фактора роста); и ингибиторами рецептора erbB2, например, органическими молекулами и антителами, которые связываются с рецептором erbB2, такими как HERCEPTIN (Genentech, Inc., South San Francisco, CA). Ингибиторы EGFR описаны, например, в WO 95/19970 (опубликованной 27 июля 1995), WO 98/14451 (опубликованной 9 апреля 1998), WO 98/02434 (опубликованной 22 января 1998) и патенте США №5747498 (выданном 5 мая 1998), и эти вещества могут применяться в настоящем изобретении, как описано в настоящей заявке.
Средства, ингибирующие EGFR, включают, не ограничиваясь перечисленными, моноклональные антитела C225 и анти-EGFR 22MAb (ImClone Systems, Inc., New York, NY), соединения ZD-1839 (AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDX-447 (Medarex Inc., Annandale, NJ) и OLX-103 (Merck & Co., Whitehouse Station, NJ) и EGF-слитый токсин (Seragen Inc., Hopkinton, MA).
Эти и другие EGFR-ингибирующие средства могут применяться в настоящем изобретении. Ингибиторы VEGF, например, SU-5416 и SU-6668 (Sugen Inc., South San Francisco, CA) также могут применяться в комбинации с соединениями по настоящему изобретению. Ингибиторы VEGF описаны, например, в WO 01/60814 A3 (опубликованной 23 августа 2001), WO 99/24440 (опубликованной 20 мая 1999), международной заявке PCT PCT/IB99/00797 (поданной 3 мая 1999), WO 95/21613 (опубликованной 17 августа 1995), WO 99/61422 (опубликованной 2 декабря 1999), патенте США №5834504 (выданном 10 ноября 1998), WO 01/60814, WO 98/50356 (опубликованной 12 ноября 1998), патенте США №5883113 (выданном 16 марта 1999), патенте США №5886020 (выданном 23 марта 1999), патенте США №5792783 (выданном 11 августа 1998), WO 99/10349 (опубликованной 4 марта 1999), WO 97/32856 (опубликованной 12 сентября 1997), WO 97/22596 (опубликованной 26 июня 1997), WO 98/54093 (опубликованной 3 декабря 1998), WO 98/02438 (опубликованной 22 января 1998), WO 99/16755 (опубликованной 8 апреля 1999) и WO 98/02437 (опубликованной 22 января 1998), причем все упомянутые источники включены в настоящую заявку во всей своей полноте с помощью ссылки. Другими примерами некоторых специфичных ингибиторов VEGF, применимых в настоящем изобретении, являются IM862 (Cytran Inc., Kirkland, WA); моноклональное антитело против VEGF, разработанное Genentech, Inc.; и ангиозим, т.е. синтетический рибозим компаний Ribozyme (Boulder, CO) и Chiron (Emeryville, CA). Эти и другие ингибиторы VEGF могут применяться в настоящем изобретении, как описано в тексте заявки. Кроме того, в комбинациях с соединениями по настоящему изобретению могут применяться ингибиторы рецептора pErbB2, например, GW-282974 (Glaxo Wellcome plc) и моноклональные антитела AR-209 (Aronex Pharmaceuticals Inc., The Woodlands, TX) и 2B-1 (Chiron), например, описанные в WO 98/02434 (опубликованной 22 января 1998), WO 99/35146 (опубликованной 15 июля 1999), WO 99/35132 (опубликованной 15 июля 1999), WO 98/02437 (опубликованной 22 января 1998), WO 97/13760 (опубликованной 17 апреля 1997), WO 95/19970 (опубликованной 27 июля 1995), патенте США № 5587458 (выданном 24 декабря 1996) и патенте США № 5877305 (выданном 2 марта 1999), которые включены в настоящую заявку во всей их полноте с помощью ссылки. Ингибиторы рецептора ErbB2, применимые в настоящем изобретении, также описаны в патенте США №6284764 (выданном 4 сентября 2001), включенном в настоящую заявку во всей полноте с помощью ссылки. Соединения, являющиеся ингибиторами рецептора erbB2, и соединения, описанные в упомянутых выше заявках PCT, патентах США и предварительных заявках на патент США, а также другие соединения и вещества, которые ингибируют рецептор erbB2, могут применяться в сочетании с соединениями по настоящему изобретению в соответствии с настоящим изобретением.
Соединения по настоящему изобретению могут также применяться в сочетании с другими средствами, применимыми при лечении рака, включая, но не ограничиваясь перечисленным, средства, способные улучшать противоопухолевый иммунный ответ, например, антитела против CTLA4 (цитотоксичного лимфоцитарного антигена 4) и другие агенты, способные блокировать CTLA4; а также антипролиферативные средства, например другие ингибиторы фарнезил протеин трансферазы, например, ингибиторы фарнезил протеин трансферазы, описанные в источниках, цитированных в разделе «предпосылки изобретения» патента США №6258824 B1.
Описанный выше способ можно осуществлять в комбинации с радиационной терапией, где количество соединения по настоящему изобретению, применяемого в сочетании с радиационной терапией, является эффективным для лечения указанных выше заболеваний.
Методики осуществления радиационной терапии известны в технике, и эти методики могут применяться в комбинационной терапии, описанной в настоящей заявке. Особенности введения соединения по настоящему изобретению в этой комбинированной терапии могут быть определены, как описано в тексте заявки.
Изобретение может быть понято более глубоко при рассмотрении следующих неограничивающих примеров.
ПРИМЕРЫ
Пример 1
Выявление целевых соединений на основе компьютерного анализа
Расчеты по виртуальному скринингу (Friesner et al., J. Med. Chem. 47, 1739-1749, 2004; Schrodinger. L. L. C., New York (http://www.schrodinger.com); Schrodinger LLC. FirstDiscovery Technical Notes; Schrodinger Press: Portland, 2003) осуществляли на основе кристаллической структуры PIM-1 киназы в комплексе с AMP-PNP в качестве шаблона (Qian et al., J. Biol. Chem. 280, 6130-6137, 2005; Jacobs et al., J. Biol. Chem. 280, 13728, 2005; Kumar et al., J. Mol. Biol. 348, 183, 2005; Bullock et al., J. Med. Chem. 48, 7604-7614, 2005; Ryan et al., PCT Publication WO 2004/024895; Jeremy et al., PCT Publication WO 2004/058769). Компьютерный скрининг ~1,5 миллиона сфокусированных и диверсифицированных соединений, подобных лекарствам из библиотек Life Chemicals, Maybridge, TimTec, BioFocus ComGenex и Ambinter, привел к отбору 63 соединений-кандидатов исключительно из библиотеки BioFocus (BioFocus, 2464, Massachusetts Avenue, Cambridge, MA 02140, USA, www.biofocus.com). Было обнаружено, что девять соединений проявляют активность в нижней части микромолярного диапазона (8-10 мкМ) и 2 из них, как было обнаружено, демонстрируют активность при концентрации <8 мкМ в анализе прямого связывания с PIM-1 киназой. При выявлении областей молекул, значимых для специфической активности по отношению к Pim-1 киназе, наиболее активные соединения принадлежали к классам имидазо[1,2-b]пиридазинов (Beswick et al., PCT Publication WO 1996/9631509; Raboisson et al., Tetrahedron 59, 5869-5878, 2003) и пиразоло[1,5-a]пиримидинов (Williamson et al., Bioorganic & Med. Chem. Letters 15, 863-867, 2005). Как указано ниже, соединения, выбранные в результате виртуального скрининга, были отфильтрованы на основании характера связывания, QikProp (Schrodinger. L. L. C., New York (http://www.schrodinger.com); Schrodinger LLC. QikProp Technical Notes; Schrodinger Press: Portland, 2003) (растворимости, проникающей способности) критериев Липинского (CA Lipinski, Adv. Drug Del. Rev. 23, 3, 1997) и наличия желаемых фармакофорных групп. Эти классы соединений служили в качестве структурных шаблонов для оптимизации, синтеза и скрининга целевых соединений по взаимодействию с PIM-1 киназой.
1. Предварительная обработка трехмерной базы данных лигандов
Использовали базу данных из внешнего источника в форме файлов формата sdf от Life Chemicals (16173), Maybridge (Hitfinder и screening collection; 16000, 58855), TimTec (Actimol Collection; 82000), BioFocus (45842), ComGenex (4573) и Ambinter (5534) и генерировали их трехмерные координаты в формате mae для каждого из sdf файлов, используя модуль LigPrep, входящий в состав пакета программ Schrodinger. Полученные координаты сохраняли в файле multi mae. В модуле LigPrep используется специальная предосторожность, касающаяся состояния протонирования ионизируемых групп (например, аминов, амидинов, карбоновых кислот) и заключающаяся в предположении, что все выбранные лиганды ионизированы при физиологическом значении pH 7,4. Генерировали отдельные файлы формата multi mae, подходящие для программ виртуального скрининга QikProp и Glide. Каждую из баз данных вместе с конечной библиотекой, состоящей из 154122 молекул, использовали для виртуального скрининга.
2. Получение координат белков и определение активных сайтов
Опорные координаты белков, использованные для программы виртуального скрининга Glide, получали из результатов рентгеноструктурного анализа PIM-1 киназы в комплексе с AMP-PNP (код регистрации в pdb: 1XR1). Затем удаляли молекулы воды и редактировали порядок и геометрию недостающих связей. Добавляли атомы водорода и на основе объединенной структуры комплекса проводили расчеты по компоновке структуры белка. Полностью уточненную структуру с учетом связи с молекулой AMP-PNP вводили далее в расчеты Grid для определения активного сайта, как совокупности аминокислот, расположенных в сфере радиусом 12Å с центром на связанном лиганде.
3. Виртуальный скрининг библиотек, отфильтрованных программой QikProp, с применением программы Glide
Как правило, молекулы каждого файла multi mae обсчитывали с помощью программы QikProp. В качестве критерия для выбора подобных соединений в базах данных использовали коэффициент Танимото, и это привело к окончательному выбору молекул 9267, 26593, 16394, 29394, 13258 и 3964 из всех баз данных для виртуального скрининга.
4. Заключительная обработка и критерии выбора соединений
Соединения, имеющие требуемые суммы баллов в программе Glide, образующие желаемые водородные связи и гидрофобные взаимодействия, оценивали с точки зрения межатомных расстояний для дальнейшего анализа. Конформационную стабильность каждого соединения-кандидата также оценивали по разности энергии силового поля конформации соединения в комплексе и конформации свободного соединения с минимальной энергией, и соединения-кандидаты с наибольшей суммой баллов из этой категории отбирали для дальнейшего анализа. Соединения в каждой из трех категорий просматривали визуально для того, чтобы удалить соединения-кандидаты, не имеющие идеальной геометрии водородных связей, гидрофобной поверхности молекул или торсионных углов. Полученные 236 структур анализировали далее с использованием QikProp для расчета log S, проникающей способности, MW и критериев Липинского. В результате число соединений уменьшилось до 69. Эти соединения-кандидаты объединяли в группы, и сходные соединения с подобной структурой представляли в виде одной позиции. Отбирали шесть производных имидазо[1,2-b]пиридазина и 13 производных пиразоло[1,5-a]пиримидина и оценивали их с точки зрения способности ингибировать активность PIM-1 киназы в анализе in vitro.
5. Результаты
Имидазо[1,2-b]пиридазины и пиразоло[1,5-a]пиримидины из библиотеки BioFocus приведены в таблице I и таблице III. Характер связывания этих структур на основании предсказаний докинга показал, что фрагмент имидазо[1,2-b]пиридазина располагается подобно фрагменту аденина и взаимодействует с остатками шарнирной области Glu121, Arg122 и Pro123. Ароматические группы в положении R1 с различными заместителями в 3-м положении представляются более предпочтительными и демонстрируют более стабильную конформацию в кармане PIM-1 киназы. Заместители в положении C-8 является более предпочтительными, чем заместители в положении C-6. Данные расчетов для этих двух молекулярных структур наводили на мысль, что заместители R1 и R2 демонстрируют сильную энергию связей по сравнению с пиразоло[1,5-a]пиримидином. На основе этих анализов авторы изобретения оптимизировали соединения, выбранные в библиотеке BioFocus и разработали новые соединения, приведенные в таблицах II и IV.
Пример 2
Синтез производных имидазо[1,2-b]пиридазина
Некоторые иллюстративные соединения по настоящему изобретению получали, как показано на приведенных ниже схемах реакций и описано в подробных синтетических примерах.
Схема 1
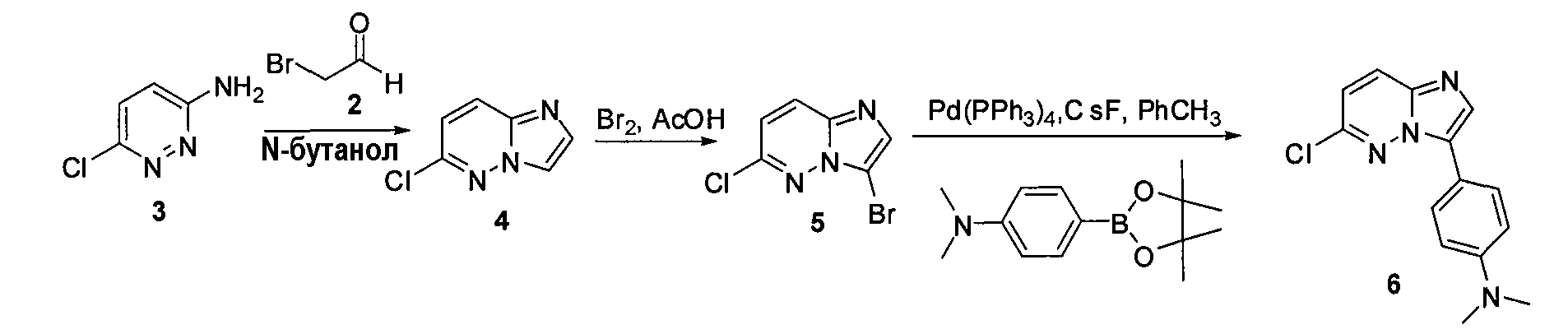
Схема 2
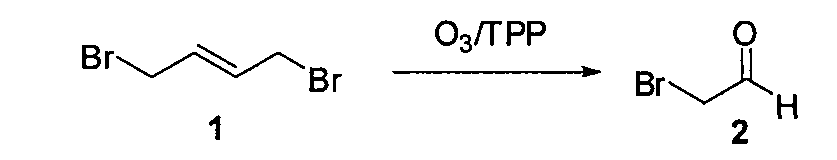
1. Получение бромацетальдегида (2)
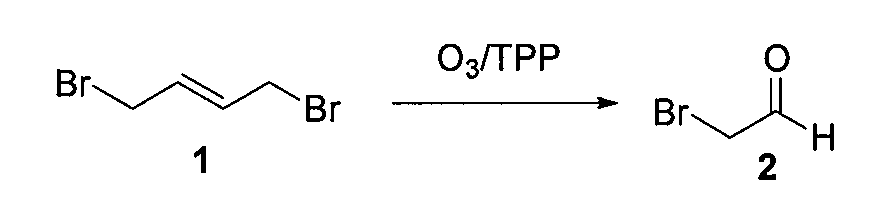
1,4-дибром-транс-2-бутен (1) (10 г, 0,046 моль) растворяли в сухом CH2Cl2 (100 мл), охлаждали до -78°C и барботировали газообразный озон до появления устойчивой синей окраски (~30 мин). Через раствор пропускали поток газообразного азота до исчезновения синей окраски, получая бесцветный раствор. Порциями в течение 1 часа добавляли трифенилфосфин (12,9 г, 0,046 моль). Температуру реакционной смеси доводили до 0°C и выдерживали смесь в холодильнике в течение 15 часов. Растворитель (CH2Cl2) удаляли из реакционной смеси (без применения вакуума) и густой осадок перегоняли при 40°C в вакууме (1 мм Hg), поддерживая температуру в приемной колбе на уровне -78°C (при перегонке нужно уделить особое внимание поддержанию циркуляции холодной воды (~0-5°C)). Бромацетальдегид (2) (2,8 г, выход=50%) получали в виде светло-желтой жидкости, обладающей сильным слезоточивым действием.
2. 6-хлоримидазо[1,2-b]пиридазин (4)
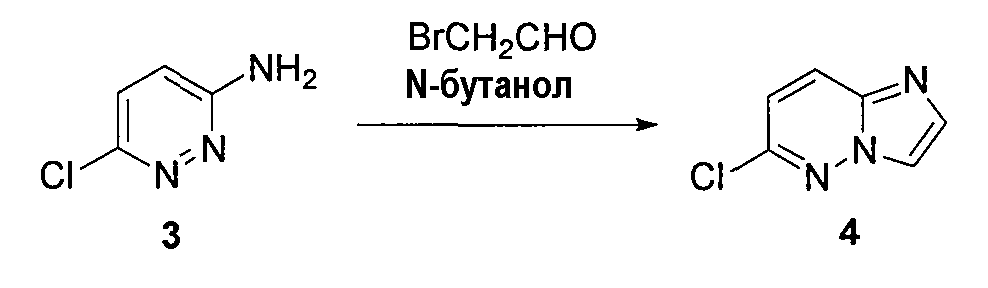
3-амино-6-хлорпиридазин (1,5 г, 0,0116 моль) растворяли в н-бутаноле (12 мл), охлаждали до 0°C и добавляли бромацетальдегид (2,8 г, 0,023 моль). Реакционную смесь кипятили с обратным холодильником в течение 20 часов и удаляли бутанол при пониженном давлении. К реакционной смеси добавляли воду и экстрагировали EtOAc (5×20 мл). Объединенные органические слои высушивали (Na2SO4), концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (EtOAc/гексан), получая 6-хлоримидазо[1,2-b]пиридазин (4) (690 мг, выход 40%).
3. 3-бром-6-хлоримидазо[1,2-b]пиридазин (5)
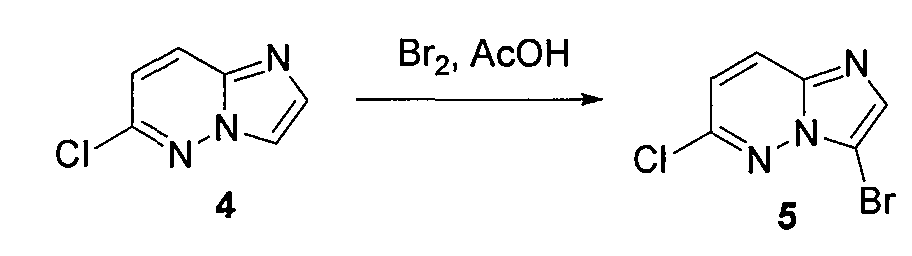
6-хлоримидазо[1,2-b]пиридазин (4) (500 мг, 0,0032 моль) смешивали с ледяной уксусной кислотой (5 мл) и медленно при комнатной температуре добавляли бром (0,4 мл). Через 20 минут осаждалось твердое вещество, которое отделяли фильтрованием. Это твердое вещество промывали эфиром (3×15 мл) и высушивали на воздухе, получая 3-бром-6-хлоримидазо[1,2-b]пиридазин (5) (400 мг, выход=60%).
4. [4-(6-хлоримидазо[1,2-b]пиридазин-3-ил)фенил]диметиламин (6)
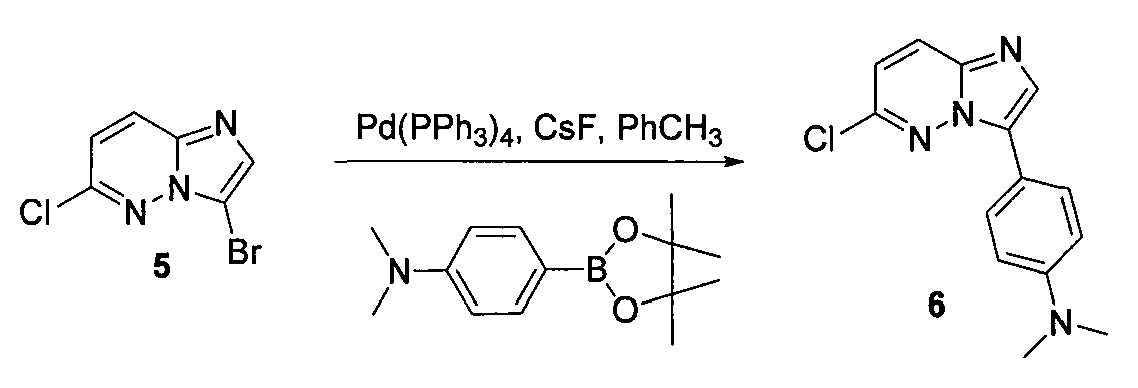
В двугорлой круглодонной колбе смешивали Pd(PPh3)4 (0,07 г, 0,068 ммоль) и CsF (0,31 г, 0,0020 моль) с безводным PhCH3 (1,6 мл). Через реакционную смесь в течение 10 минут барботировали аргон, после чего добавляли 3-бром-6-хлоримидазо[1,2-b]пиридазин (5) и затем бороновую кислоту. Реакционную смесь кипятили с обратным холодильником в течение 20 часов. Удаляли из реакционной смеси растворитель при пониженном давлении. Остаток смешивали с EtOAc (20 мл) и фильтровали через целит. Фильтрат концентрировали при пониженном давлении, получая остаток, который очищали колоночной хроматографией (MeOH/CH2Cl2), получая [4-(6-хлоримидазо[1,2-b]пиридазин-3-ил)фенил]диметиламин (6) (50 мг, выход=30%).
5. 2-[3-(4-диметиламинофенил)имидазо[1,2-b]пиридазин-6-ил(±)-2-амино]бутан-1-ол (7)
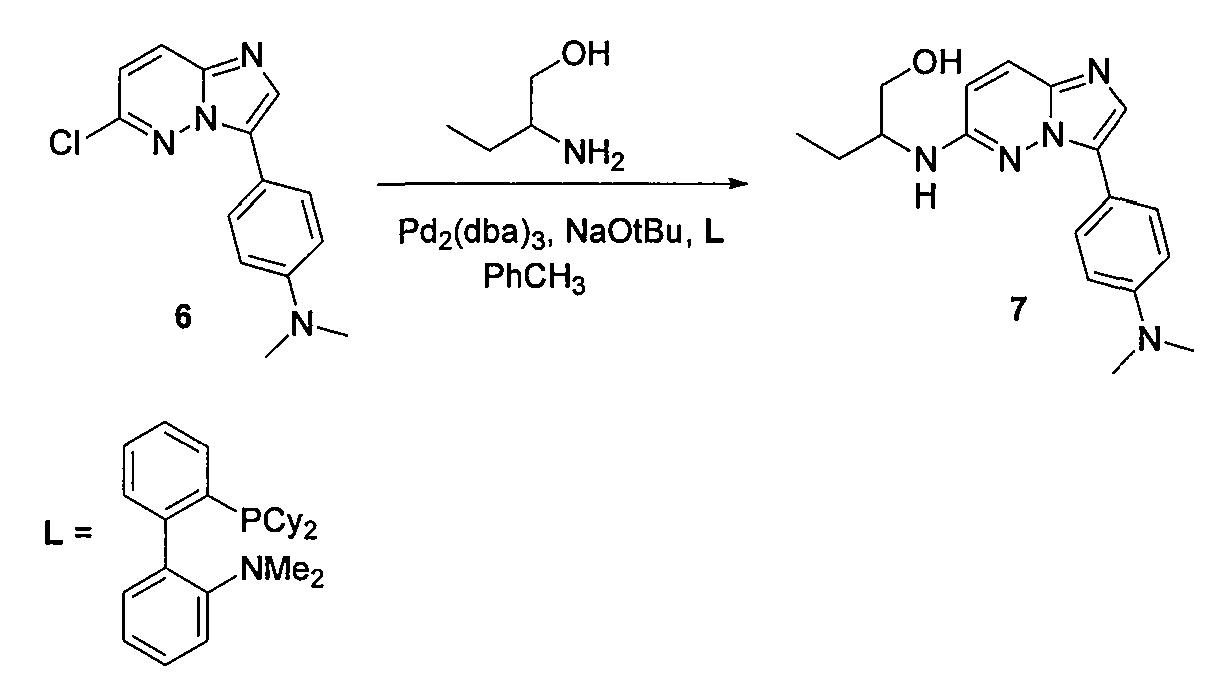
В двугорлой круглодонной колбе смешивали Pd2(dba)3 (0,001 г, 0,0009 ммоль), трет-бутоксид натрия (0,02 г, 0,256 ммоль) и лиганд (0,001 г, 0,0027 ммоль) с безводным PhCH3. Через реакционную смесь в течение 10 минут пропускали аргон и затем добавляли [4-(6-хлоримидазо[1,2-b]пиридазин-3-ил)фенил]диметиламин (6) (0,05 г, 0,18 ммоль) и (±)-2-амино-1-бутанол (0,02 г, 0,22 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 24 часов. Из реакционной смеси под пониженным давлением удаляли толуол, остаток смешивали с EtOAc (25 мл) и фильтровали через целит. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (MeOH/CH2Cl2), получая 2-[3-(4-диметиламинофенил)имидазо[1,2-b]пиридазин-6-ил(±)-2-амино]бутан-1-ол (7) (15 мг, выход=31%, чистота по данным ВЭЖХ=97%).
6. Общая схема и методики синтеза соединений 9 и 10
Схема 3
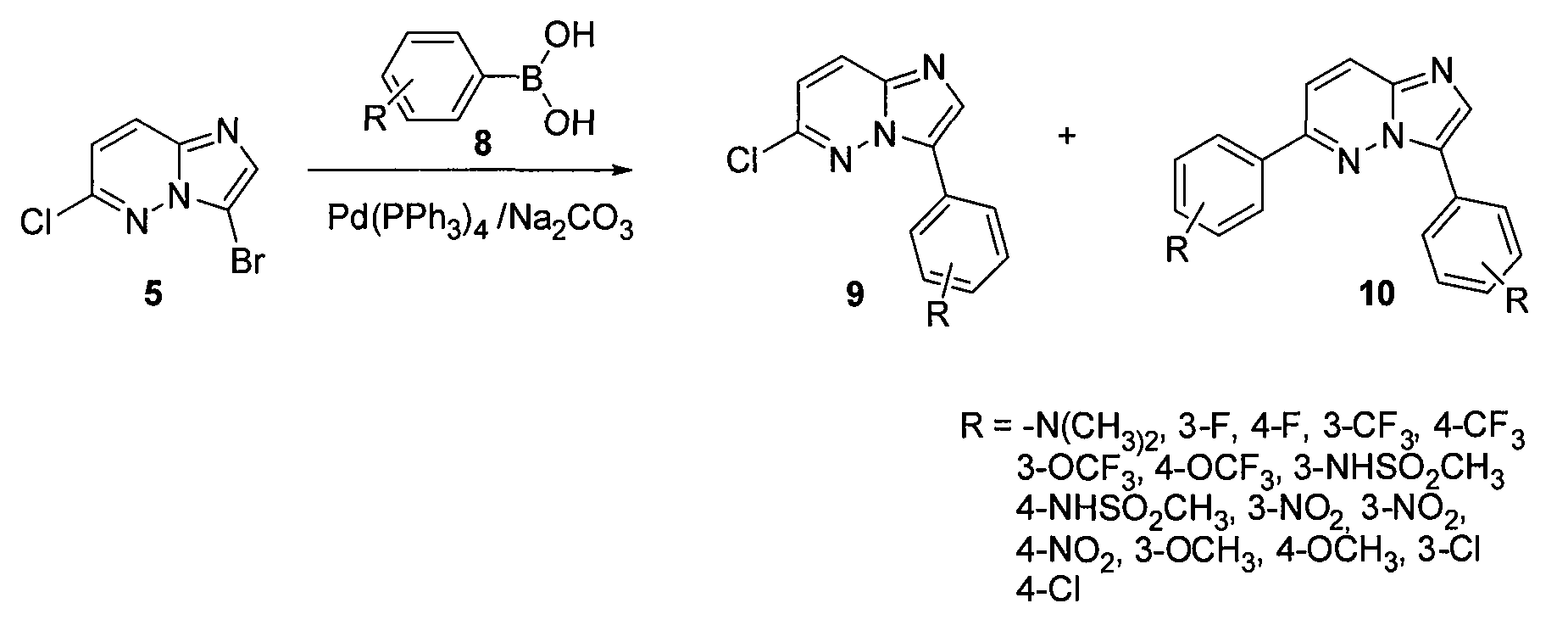
В двугорлой круглодонной колбе смешивали Pd(PPh3)4 (0,068 ммоль) и CsF (0,0020 моль) с безводным PhCH3 (1,6 мл). Через реакционную смесь в течение 10 минут барботировали аргон, после чего добавляли 3-бром-6-хлоримидазо[1,2-b]пиридазин (5) и затем 3- или 4-замещенную бороновую кислоту. Реакционную смесь кипятили с обратным холодильником в течение 20 часов. Удаляли из реакционной смеси растворитель при пониженном давлении. Остаток смешивали с EtOAc (20 мл) и фильтровали через целит. Фильтрат концентрировали при пониженном давлении, получая остаток, который очищали колоночной хроматографией (MeOH/CH2Cl2), получая [3- или 4-(замещенный)-(6-хлоримидазо[1,2-b]пиридазин-3-ил)фенил]диметиламин (9) и (10) в соотношении 6:4.
7. Схема синтеза 2-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-ил(±)-2-амино)бутан-1-ола (соединение 7-17)
Схема 4
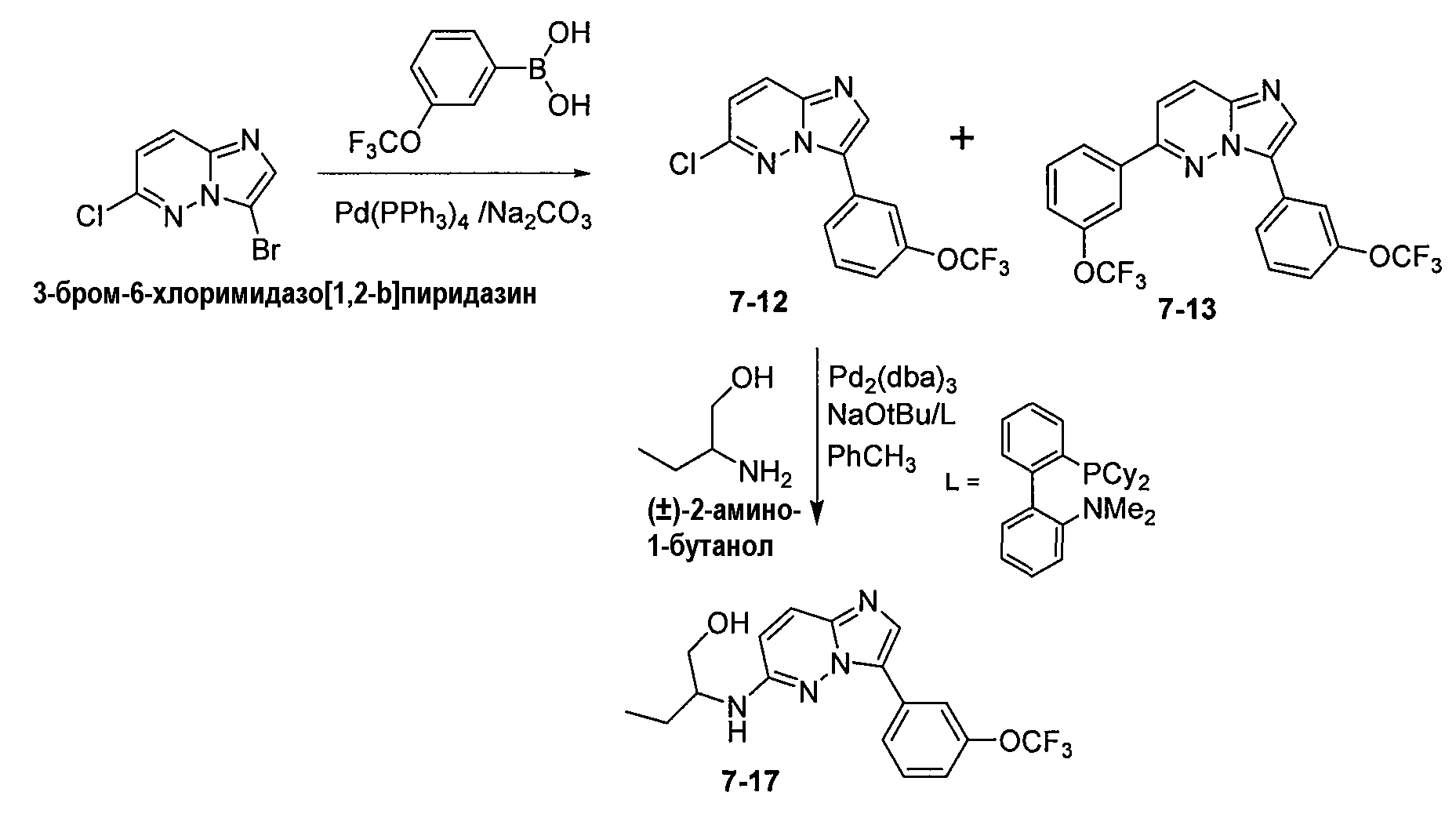
a. Синтез 6-хлор-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазина (соединения 7-12, 7-13).
К дегазированной смеси растворителей MeOH/толуол (1:4, 5 мл) и 2М Na2CO3 (0,215 мл, 0,430 ммоль) в атмосфере аргона добавляли 3-бром-6-хлоримидазо[1,2-b]пиридазин (100 мг, 0,430 ммоль), 3-фторметоксифенилбороновую кислоту (98 мг, 0,430 ммоль) и Pd(PPh3)4 (8,95 мг, 7,74 мкМ, 0,018 экв). Полученную реакционную смесь нагревали до кипения с обратным холодильником в течение ночи (12 ч). ТСХ (5% MeOH/DCM, Rf=0,2) показала присутствие исходного соединения 3-бром-6-хлоримидазо[1,2-b]пиридазина и образование пятен двух новых соединений с сильной флуоресценцией. Реакционную смесь концентрировали и неочищенный продукт очищали с помощью CombiFlash Companion, используя систему растворителей 0%-70% EtOAc/гексан 40 мин (4 г колонка RediSep Flash с нормальной фазой при скорости потока 18 мл/мин), что приводило к разделению двух продуктов, соединения 7-12 63 мг (46,7%) и соединения 7-13 13 мг (6,88%).
b. Синтез 2-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ола (соединения 7-17)
К толуолу, использованному в качестве растворителя, добавляли соединение 7-12 (30 мг), 2-амино-1-бутанол (18,08 мкМ, 2 экв.), лиганд (5,65 мг, 0,15 экв.), Pd2(dba)3 (6,57 мг, 0,05 экв.) и NaOt-Bu (13,05 мг, 1,42 экв.). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение ночи (12 ч). Неочищенный продукт концентрировали и проводили препаративную ТСХ в системе растворителей 10% MeOH/DCM, получая 10 мг рацемата соединения 7-17 (28,5%).
9. Схема синтеза соединений 7-27 и 7-28
Схема 5
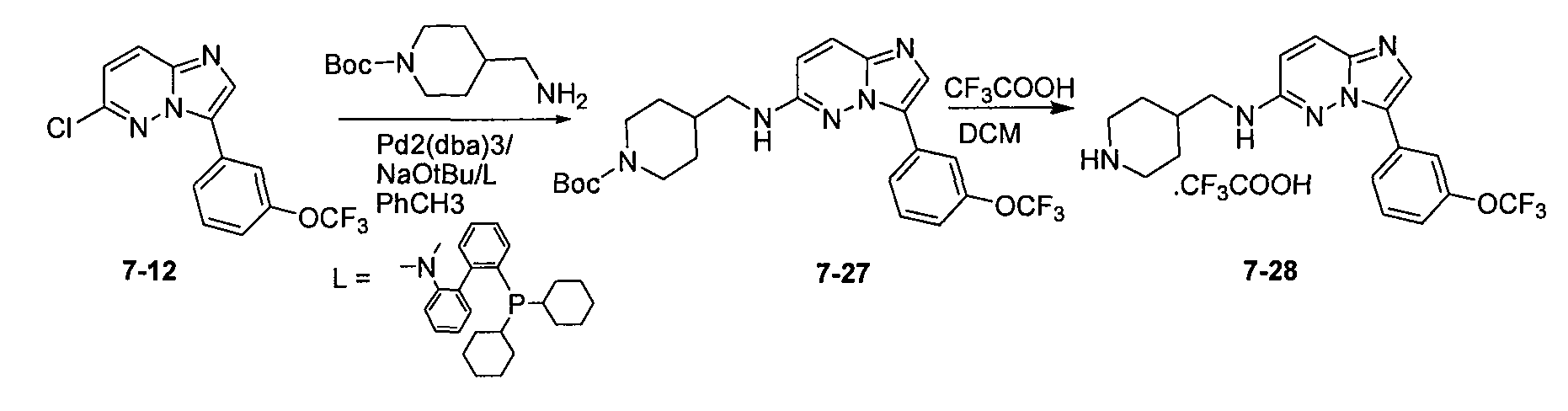
10. Синтез трет-бутил 4-((3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)метил)пиперидин-1-карбоксилата (соединения 7-27)
К толуолу добавляли соединение 7-12 (100 мг, 0,319 ммоль), 4-аминометил-1-Boc-пиперидин (68,3 мг, 0,319 ммоль), лиганд (18,8 мг, 0,048 ммоль), Pd2(dba)3 (0,05 экв.) и NaOt-Bu (1,5 экв.). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение ночи (12 ч). Неочищенный продукт концентрировали и проводили препаративную ТСХ в системе растворителей 10% MeOH/DCM, получая 84 мг соединения 7-27 (53,6%).
11. Синтез 2,2,2-трифторацетата N-(пиперидин-4-илметил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амина (соединения 7-28)
1 мл DCM и 1 мл ТФУ (0,098 ммоль) последовательно добавляли к соединению 7-27 (48 мг, 0,0). Реакция завершалась в течение 1 ч при комнатной температуре по данным ТСХ (20% MeOH/DCM) Rf=0,1. Концентрирование для полного удаления ТФУ приводило к получению неочищенной соли 7-28 и ТФУ. Препаративная ТСХ (20% MeOH/DCM) позволила получить 48 мг (97%) бесцветного твердого вещества.
12. Схема синтеза 7-29
Схема 6
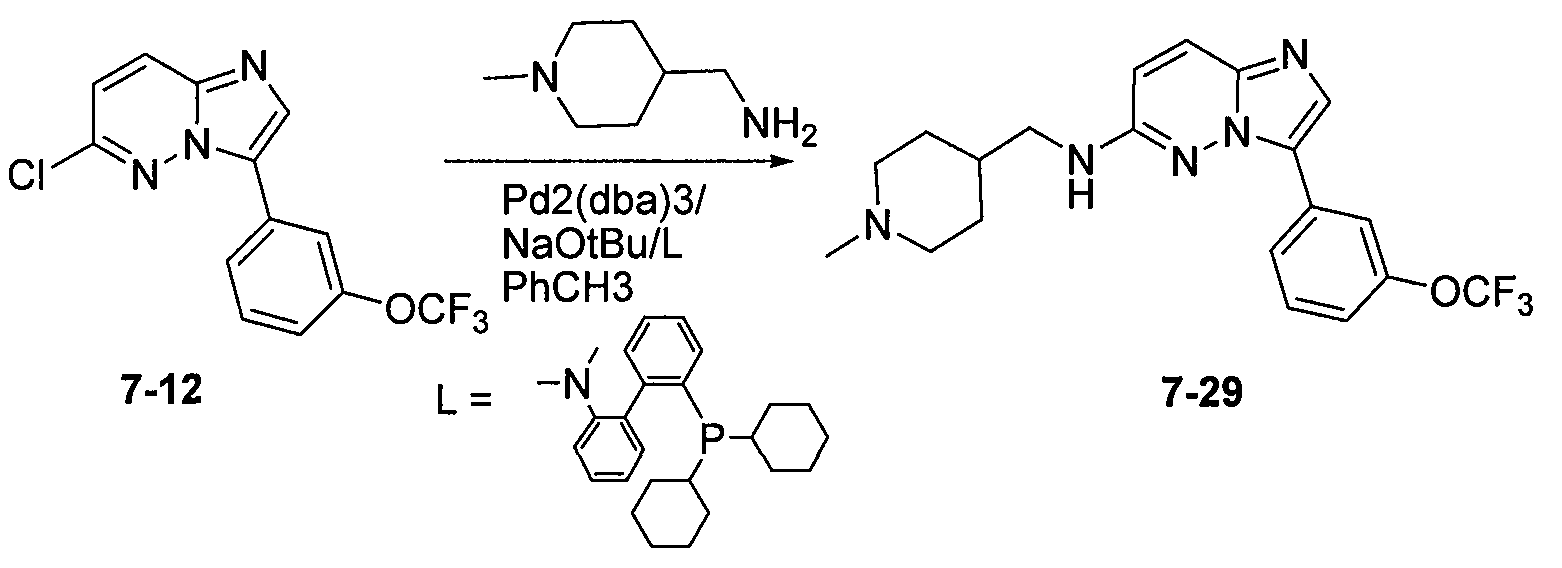
13. Синтез N-((1-метилпиперидин-4-ил)метил)-3-(3-(трифторметокси)фенилимидазо[1,2-b]пиридазин-6-амина (соединения 7-29):
К толуолу (5 мл) добавляли соединение 7-12 (40 мг, 0,128 ммоль), (1-метилпиперидин-4-ил)метанамин (24,53 мг, 0,191 ммоль), лиганд (7,53 мг, 0,019 ммоль), Pd2(dba)3 (8,76 мг, 9,56 ммоль) и NaOt-Bu (17,40 мг, 0,181 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение ночи (12 ч). Неочищенный продукт концентрировали и проводили препаративную ТСХ в системе растворителей 10% MeOH/DCM, получая 17,6 мг соединения 7-23 (50%).
14. Схема синтеза соединения 7-31
Схема 7
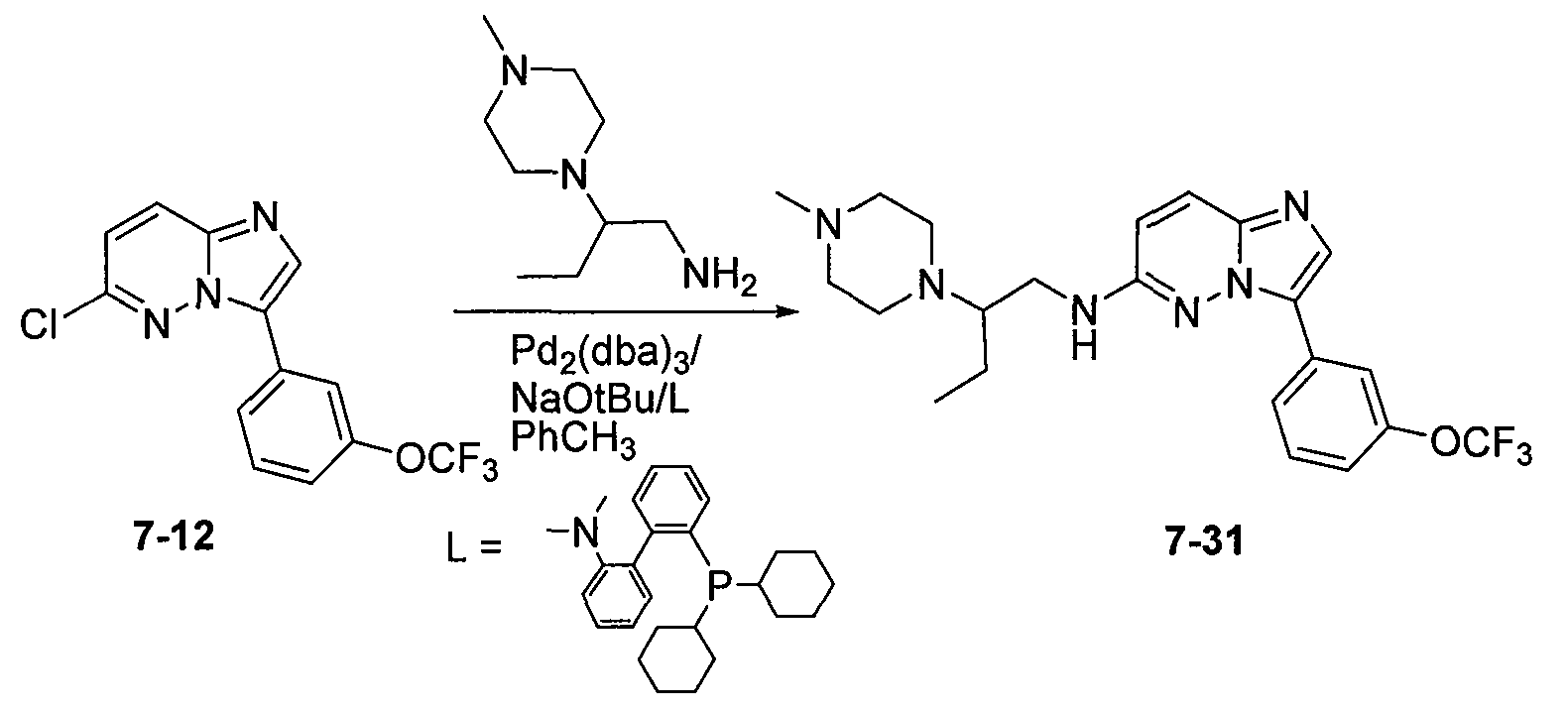
15. ±N-(2-(4-метилпиперазин-1-ил)бутил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-31)
К толуолу (5 мл) добавляли соединение 7-12 (40 мг, 0,128 ммоль), добавляли (±)2-(4-метилпиперазин-1-ил)бутан-1-амин (0,191 ммоль), лиганд (0,019 ммоль), Pd2(dba)3 (9,56 ммоль) и NaOt-Bu (0,181 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение ночи (12 ч). Неочищенный продукт концентрировали и проводили препаративную ТСХ в системе растворителей 10% MeOH/DCM, получая 22 мг соединения 7-31.
Пример 3
Исследование активности PIM-1 киназы
A. Исследование ингибирования Pim-1 киназы
Один из иллюстративных способов, которым можно определить активность Pim-1 киназы, заключается в установлении количества АТФ, оставшейся в растворе после взаимодействия с Pim-1 киназой in vitro. Такую возможность предоставляет набор Kinase-Glo Assay Kit (Promega, Inc., Madison, WI). АТФ, оставшаяся в растворе после взаимодействия с киназой, служит субстратом для люциферазы, которая катализирует превращение люциферина в оксилюциферин с испусканием одного фотона света. Таким образом, люминесцентный сигнал, регистрируемый прибором Luminoskan Ascent Instrument (Thermo Electron Corp., Milford, MA), коррелирует с количеством АТФ, оставшейся после взаимодействия с киназой, и находится в обратной зависимости от активности киназы. Этот анализ эффективен при определении значений IC50 ингибиторов киназы при ингибировании Pim-1 киназы. Анализы проводят в двух экземплярах в объеме 50 мкл в белых плоскодонных 96-луночных планшетах. Ингибиторы добавляют к 1× раствору буфера киназы, 10 мкМ АТФ, 100 мкМ Pim-1 специфичного субстрата, 50 нг активного фермента Pim-1 и воды в последовательных разбавлениях, в диапазоне концентраций от микромолярных до наномолярных. Эти растворы инкубируют при 30°С при 360 об/мин в течение 2 часов. После инкубирования в каждую лунку добавляют 50 мкл реагента Kinase-Glo, включая все ячейки с положительным и отрицательным контролем, и инкубируют при комнатной температуре в течение 15 минут. Затем планшет считывают на приборе Luminoskan Ascent, и результаты отображают с помощью программного обеспечения Ascent Software версии 2.6. Затем можно рассчитать значения IC50 для каждого исследованного ингибитора.
В качестве альтернативы активность Pim-1 киназы можно определить в другом анализе in vitro количественным измерением фосфорилирования известного субстрата Pim-1. Осуществить это позволяет набор Z-Lyte Protein Kinase Assay Kit (Invitrogen, Madison WI) с использованием методики Fluorescent Resonance Energy Transfer (FRET) (флуоресцентного резонансного переноса энергии). Вкратце известный Pim-1 субстрат (серин-треониновый субстрат 7 от Invitrogen), который несет два флуорофора на противоположных концах (кумарин и флуоресцеин), инкубируют с ферментом Pim-1 и потенциальным ингибитором. Затем реакцию с киназой останавливают и добавляют обрабатывающий реагент. Этот реагент, а именно протеаза, будет расщеплять только нефосфорилированный субстрат, разделяя два упомянутых флуорофора и уменьшая величину переноса FRET, который может наблюдаться между ними. Затем FRET может быть измерен с использованием спектрофотометра, например, Gemini EM (Molecular Devices). Уменьшение FRET является указанием на активность ингибитора.
B. Исследование ингибиторов Pim-1 киназы на основе клеточной культуры
Исследования на основе клеточных культур могут применяться для оценки способности соединений по настоящему изобретению ингибировать один или несколько видов клеточной активности, например, рост и/или выживание раковых клеток. Раковые клетки многих линий могут быть получены из American Type Culture Collection (ATCC) (Американская Коллекция Типовых культур) и других источников. Вкратце, клетки высевают в 96-луночные белые непрозрачные планшеты, обработанные культурами тканей (Thermo Electron, Vantaa, Finland) в количестве от 5000 до 10000 клеток на лунку в зависимости от скорости пролиферации клеток в 100 мкл подходящей ростовой среды (определенной ATCC). Затем клетки подвергают действию исследуемого препарата в подходящей концентрации или такого же количества ДМСО (разбавитель препаратов) и дают расти в их присутствии в течение 96 часов. После этого в каждую лунку добавляют 100 мкл реагента Cell-Titer-Glo (Promega, Inc., Madison, WI). Затем планшеты встряхивают в течение 2 минут при комнатной температуре, чтобы провести лизис клеток и инкубируют в течение 10 минут при комнатной температуре для стабилизации люминесцентного сигнала. Подобно аналитическому реагенту Kinase-Glo от Promega, этот реагент содержит как фермент люциферазу, так и его субстрат люциферин. Люцифераза, активированная действием АТФ в клеточном лизате, катализирует превращение люциферина в оксилюциферин, т.е. реакцию, которая приводит к излучению света. Количество излученного света пропорционально количеству АТФ в клеточном лизате, которое, в свою очередь, пропорционально количеству клеток и позволяет получить коэффициент клеточной пролиферации.
Кроме того, для определения специфического ингибирования фермента Pim-1 в клеточной культуре выполняют анализ с применением Вестерн-блоттинга. С этой целью клетки, обработанные потенциальным ингибитором Pim-1, подвергают лизису под действием буфера, подходящего для изоляции и консервации белков (1% Nonidet P-40, 150 мМ NaCl, 50 мМ Tris pH 8,0, 5 мМ ЭДТУ, коктейль III ингибиторов протеазы 1:500 [Calbiochem], 100 мМ NaF, 100 мМ ортованадат натрия). Затем количественно определяют концентрацию белка в полученном лизате с использованием набора BCA Protein Assay Kit (Pierce). Известные количества белка, например, 10 мкг, помещают на 12% SDS-полиакриламидный гель и подвергают восстановлению, денатурируя SDS-PAGE. Подвергшиеся электрофорезу белки переносят на нитроцеллюлозную мембрану, которую затем исследуют антителами к p-21 и фосфо(Thr 145) p-21. Поскольку треонин-145 белка p-21 является субстратом для Pim-1, количественное определение фосфорилирования белка на этом участке в обработанных клетках должно дать средство для оценки эффективности исследуемых ингибиторов Pim-1.
C. Данные о специфической активности Pim-1 киназы:
Применяя методики, в основном совпадающие с описанными выше, тестировали иллюстративные соединения на ингибирование активности Pim-1 киназы. На фиг.1 показаны результаты для иллюстративных соединений, исследованных в концентрации 10 мкМ, с применением аналитического набора Z-LYTE. Величины приведены в процентах от контроля, не обработанного тестируемыми соединениями. Как показано на фиг.1, соединения проявляли эффективность при ингибировании активности Pim-1 киназы в данном анализе.
В дополнение к этому, определяли значения IC50 в отношении Pim-1 киназы для иллюстративных соединений, используя аналитический набор Promega Kinase-Glo, причем результаты исследования суммированы ниже в таблице V. Помимо этого, иллюстративные соединения подвергали оценке по действию на культуры клеток, экспрессирующих киназу Pim-1. Значения IC50, представляющие собой концентрации, необходимые для ингибирования роста клеток на 50% от необработанного контроля, приведены в мкМ в таблице VI ниже. Таким образом, по данным различных исследований, соединения являются активными ингибиторами Pim-1 киназы и способны ингибировать рост клеток.
|
|
Пример 4
Синтез производных имидазо[1,2-b]пиридазина
Другую группу соединений по настоящему изобретению, включающую иллюстративные соединения, приведенные в таблице VII, получали в соответствии со следующими синтетическими примерами.
1. Получение бромацетальдегида (2)
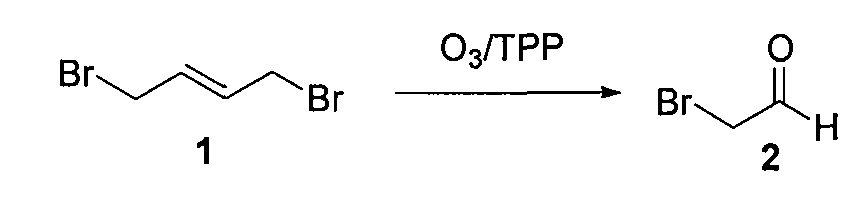
1,4-дибром-транс-2-бутен (1) (10 г, 0,046 моль) растворяли в сухом CH2Cl2 (100 мл), охлаждали до -78°C и барботировали газообразный озон до появления устойчивой синей окраски (~30 мин). Через раствор пропускали поток газообразного азота до исчезновения синей окраски, получая бесцветный раствор. Порциями в течение 1 часа добавляли трифенилфосфин (12,9 г, 0,046 моль). Температуру реакционной смеси доводили до 0°C и выдерживали смесь в холодильнике в течение 15 часов. Растворитель (CH2Cl2) удаляли из реакционной смеси (без применения вакуума) и густой осадок перегоняли при 40°C в вакууме (1 мм Hg), поддерживая температуру в приемной колбе на уровне -78°C [Примечание: при перегонке нужно уделить особое внимание поддержанию циркуляции холодной воды (~0-5°C)]. Бромацетальдегид (2) (2,8 г, выход=50%) получали в виде светло-желтой жидкости, обладающей сильным слезоточивым действием.
2. 6-хлоримидазо[1,2-b]пиридазин (4)
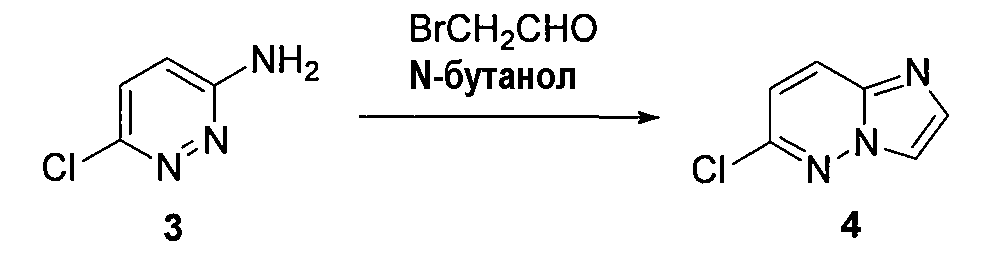
3-амино-6-хлорпиридазин (1,5 г, 0,0116 моль) растворяли в н-бутаноле (12 мл), охлаждали до 0°C и добавляли бромацетальдегид (2,8 г, 0,023 моль). Реакционную смесь кипятили с обратным холодильником в течение 20 часов и удаляли бутанол при пониженном давлении. К реакционной смеси добавляли воду и экстрагировали EtOAc (5×20 мл). Объединенные органические слои высушивали (Na2SO4), концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (EtOAc/гексан), получая 6-хлоримидазо[1,2-b]пиридазин (4) (690 мг, выход 40%).
3. 3-бром-6-хлоримидазо[1,2-b]пиридазин (5)
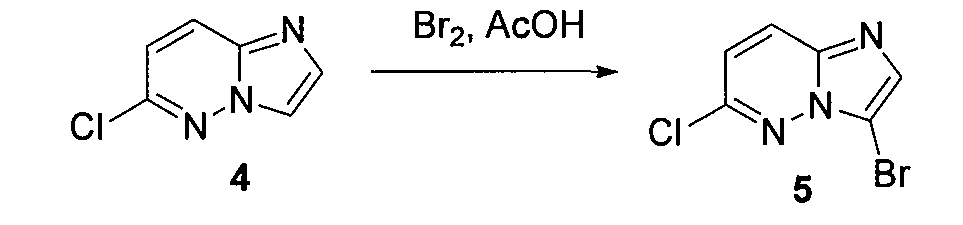
6-хлоримидазо[1,2-b]пиридазин (4) (500 мг, 0,0032 моль) смешивали с ледяной уксусной кислотой (5 мл) и медленно при комнатной температуре добавляли бром (0,4 мл). Через 20 минут осаждалось твердое вещество, которое отделяли фильтрованием. Это твердое вещество промывали эфиром (3×15 мл) и высушивали на воздухе, получая 3-бром-6-хлоримидазо[1,2-b]пиридазин (5) (400 мг, выход=60%).
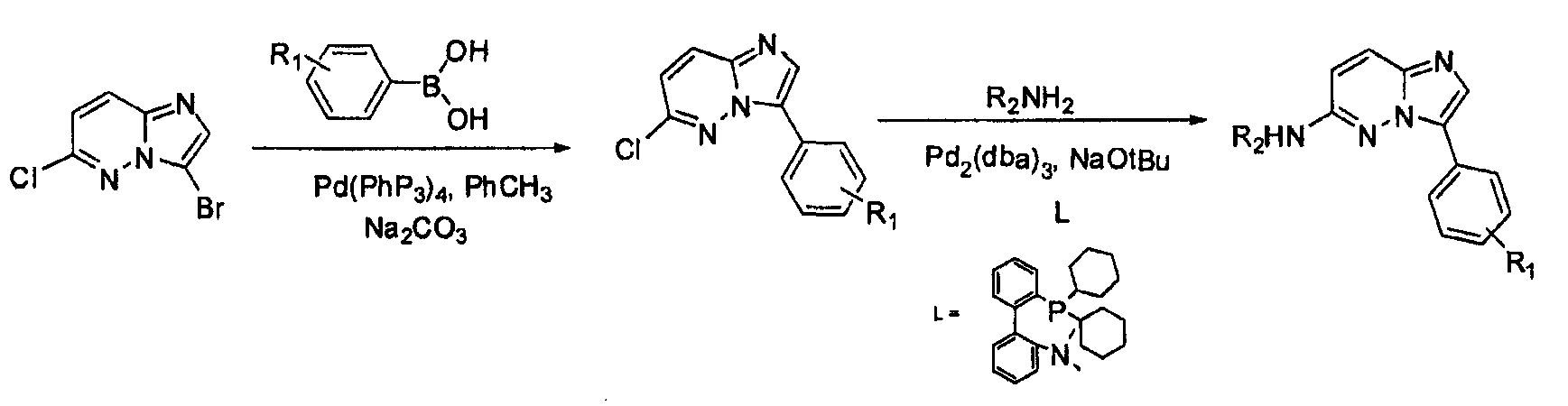
4. Общая методика получения 6-хлор-3-замещенных-имидазо[1,2-b]пиридазинов
Реакционную смесь, содержащую 3-бром-6-хлоримидазо[1,2-b]пиридазин (0,43 ммоль), бороновую кислоту (0,43 ммоль), Pd(PPh3)4 (7,74 мкмоль, 0,018 экв.) и Na2CO3 (2М, 0,43 ммоль) в 5 мл смеси толуол/MeOH (4:1) дегазировали аргоном в течение 10 мин. Реакционную смесь кипятили с обратным холодильником в течение ночи. Полученную смесь фильтровали через MgSO4 и концентрировали в вакууме. Остаток очищали хроматографией с помощью системы combiflash (от 0% до 70% EtOAc/гексан), получая желаемые соединения.
5. Общая методика получения 3,6-дизамещенных-имидазо[1,2-b]пиридазинов
Реакционную смесь, содержащую 6-хлор-3-замещенный-имидазо[1,2-b]пиридазин (0,096 ммоль), амин (0,191 ммоль), лиганд (0,014 ммоль, 0,15 экв.), Pd2(dba)3 (7,17 мкмоль, 0,075 экв.) и NaOt-Bu (0,136 ммоль, 1,4 экв.) в 5 мл толуола, дегазировали Ar в течение 10 мин. Полученную смесь кипятили с обратным холодильником в течение ночи. Концентрирование и очистка препаративной ТСХ позволяли получить желаемые продукты.
12-(3-(3-(диметиламино)фенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-4)
1H-ЯМР: (400 МГц, CD3OD) 7,92 (м, 2H), 7,65 (с, 1H), 7,30 (м, 2H), 6,94 (д, J=10 Гц, 1H), 6,81 (м, 1H), 4,26 (м, 1H), 4,20 (м, 1H), 3,01 (с, 6H), 1,04 (т, J=7,6 Гц, 3H). МС m/z: 326,1, 255,2.
2-(3-(4-фторфенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-10)
1H-ЯМР (400 МГц, CD3OD) 8,09 (м, 2H), 7,92 (м, 2H), 7,25 (т, J=8,5 Гц, 2H), 6,97 (д, J=9,4 Гц, 1H), 4,45 (м, 1H), 4,21 (м, 1H), 3,17 (м, 1H), 1,68 (м, 1H), 1,52 (м, 1H), 1,06 (т, J=7,5 Гц, 3H).
2-(3-(3-фторфенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-11)
1H-ЯМР (400 МГц, CD3OD) 7,96 (м, 3H), 7,88 (д, J=7,8 Гц, 1H), 7,50 (м, 1H), 7,13 (м, 1H), 7,00 (д, J=8,6 Гц, 1H), 4,50 (м, 1H), 4,23 (м, 1H), 3,21 (м, 1H), 1,70 (м, 1H), 1,54 (м, 1H), 1,06 (т, J=7,6 Гц, 3H).
N-циклопентил-3-(3-фторфенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-15)
1H-ЯМР (400 МГц, CD3OD) 8,17 (д, J=11,3 Гц, 1H), 7,90 (д, J=6,8 Гц, 1H), 7,82 (с, 1H), 7,61 (д, J=8,5 Гц, 1H), 7,4 (м, 1H), 7,03 (м, 1H), 6,72 (д, J=9,5 Гц, 1H), 4,16 (м, 1H), 1,77 (м, 4H), 1,66 (м, 4H).
2-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-17)
1H-ЯМР (400 МГц, CD3OD) 8,24 (с, 1H), 8,04 (с, 1H), 7,97 (м, 2H), 7,57 (т, J=6,2 Гц, 1H), 7,27 (д, J=8,2 Гц, 1H), 7,02 (дд, J1=1,4 Гц, J2=9,6 Гц, 1H), 4,78 (д, J=10,9 Гц, 1H), 4,25 (т, J=8,9 Гц, 1H), 1,64 (м, 2H), 1,08 (т, J=7,6 Гц, 3H).
(R)-1-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-илокси)бутан-2-амин (соединение 7-18)
1H-ЯМР (400 МГц, CD3OD) 8,24 (с, 1H), 8,02 (с, 1H), 7,98 (т, J=9,57 Гц, 2H), 7,58 (т, J=7,8 Гц, 1H), 7,27 (д, J=9,57 Гц, 1H), 7,02 (д, J=9,57 Гц, 1H), 4,48 (м, 1H), 4,25 (м, 1H), 1,71~1,56 (м, 2H), 1,08 (м, 3H).
(S)-1-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-илокси)бутан-2-амин (соединение 7-19)
1H-ЯМР (400 МГц, CD3OD) 8,22 (с, 1H), 8,02 (с, 1H), 7,95 (м, 2H), 7,55 (т, J=8,2 Гц, 1H), 7,27 (м, 1H), 7,00 (дд, J1=9,9 Гц, 1H), 4,42 (м, 1H), 4,18 (м, 1H), 1,67~1,51 (м, 2H), 1,06 (м, 3H).
N-(циклопропилметил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-20)
1H-ЯМР (400 МГц, CD3OD) 8,43 (с, 1H), 7,97 (д, J=7,8 Гц, 1H), 7,85 (с, 1H), 7,62 (д, J=9,5 Гц, 1H), 7,52 (т, J=8,2 Гц, 1H), 7,20 (д, J=7,5 Гц, 1H), 6,75 (д, J=9,3 Гц, 1H), 3,21 (д, J=6,8 Гц, 2H), 1,2 (м, 1H), 0,55 (м, 2H), 0,28 (м, 2H).
N-(3-(6-(1-гидроксибутан-2-иламино)имидазо[1,2-b]пиридазин-3-ил)фенил)метансульфонамид (соединение 7-23)
1H-ЯМР (400 МГц, CD3OD) 8,29 (с, 1H), 7,95 (с, 1H), 7,92 (м, 1H), 7,74 (д, J=7,8 Гц, 1H), 7,44 (т, J=7,8 Гц, 1H), 7,18 (м, 1H), 6,96 (м, 1H), 4,51 (м, 1H), 4,36 (м, 1H), 3,24 (м, 1H), 3,00 (с, 3H), 1,74 (м, 1H), 1,60 (м, 1H), 1,05 (т, J=7,5 Гц, 3H).
2-(3-(3-(трифторметил)фенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-24)
1H-ЯМР (400 МГц, CD3OD) 8,64 (с, 1H), 8,22 (с, 1H), 8,06 (с, 1H), 7,94 (м, 1H), 7,65 (с, 2H), 7,00 (д, J=9,9 Гц, 1H), 4,45 (м, 1H), 4,19 (т, J=8,2 Гц, 1H), 3,20 (м, 1H), 1,60 (м, 2H), 1,04 (т, J=8,5 Гц, 3H).
N-(циклопропилметил)-3-(3-(трифторметил)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-25)
1H-ЯМР (400 МГц, CD3OD) 8,82 (с, 1H), 8,19 (с, 1H), 7,86 (с, 1H), 7,60 (м, 3H), 6,74 (м, 1H), 3,20 (м, 2H), 1,18 (м, 1H), 0,55 (м, 2H), 0,26 (м, 2H).
трет-бутил 4-((3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)метил)пиперидин-1-карбоксилат (соединение 7-27)
1H-ЯМР (400 МГц, CD3OD) 8,39 (с, 1H), 7,96 (д, J=8,2 Гц, 1H), 7,85 (с, 1H), 7,62 (дд, J1=2,0 Гц, J2=9,9 Гц, 1H), 7,49 (м, 1H), 7,21 (д, J=8,2 Гц, 1H), 6,71 (дд, J1=2,0 Гц, J2=9,57 Гц, 1H), 4,07 (м, 4H), 3,26 (м, 4H), 1,82 (д, J=12,7 Гц, 2H), 1,42 (с, 9H), 1,57 (м, 1H).
N-(пиперидин-4-илметил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-28)
1H-ЯМР (400 МГц, CD3OD) 8,37 (с, 1H), 7,99 (д, J=8,2 Гц, 1H), 7,88 (с, 1H), 7,66 (д, J=9,2 Гц, 1H), 7,54 (т, J=8,2 Гц, 1H), 7,24 (д, J=7,24 Гц, 1H), 6,75 (д, J=9,5 Гц, 1H), 2,94 (м, 4H), 2,04 (м, 4H), 1,44 (м, 1H).
N-((1-метилпиперидин-4-ил)метил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-29)
1H-ЯМР (400 МГц, CD3OD) 8,38 (с, 1H), 8,00 (д, J=7,9 Гц, 1H), 7,86 (с, 1H), 7,64 (м, 1H), 7,53 (т, J=8,2H, 1H), 7,22 (д, J=7,5 Гц, 1H), 6,74 (д, J=9,9 Гц, 1H), 3,00 (д, J=12 Гц, 2H), 2,30 (с, 3H), 1,90 (д, J=12,6 Гц, 3H), 1,38 (м, 2H), 2,20 (т, J=11,6 Гц, 2H).
N-(2-(пирролидин-1-ил)этил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-30)
1H-ЯМР (400 МГц, CD3OD) 8,29 (с, 1H), 8,02 (д, J=7,6 Гц, 1H), 7,84 (с, 1H), 7,64 (д, J=9,6 Гц, 1H), 7,53 (т, J=8 Гц, 1H), 7,31 (м, 1H), 7,22 (д, J=8,4 Гц, 1H), 6,74 (д, J=9,6 Гц, 1H), 3,56 (т, J=6,8 Гц, 2H), 2,81 (т, J=6,8 Гц, 2H), 2,63 (с, 4H), 1,81 (м, 4H).
Пример 5
Синтез производных имидазо[1,2-b]пиридазина и производных 7-метилимидазо[1,2-b]пиридазина
Дополнительные соединения по настоящему изобретению, включая иллюстративные соединения, приведенные в таблице VII, получали в соответствии со следующими синтетическими примерами. В приведенных примерах соединение 7-12 получали, как описано выше в примере 2, в то время как соединение 11 получали следующим способом:
6-хлор-5-метилпиридазин-3-амин 7:
3,6-дихлор-4-метилпиридазин 6 (1 г) растворяли в 5 мл этанола и добавляли гидроксид аммония (10 мл). Полученную реакционную смесь закупоривали в емкость высокого давления и нагревали до 100°C в течение 48 часов. Реакционную смесь охлаждали, выпаривали растворители и очищали с помощью CombiFlash Companion, используя систему растворителей гексан/DCM 40:60 (4 г колонка RediSep Flash с нормальной фазой, со временем прохождения мин при потоке 18 мл/мин), получая 0,640 г (72,7%) соединения 7 в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CD3OD) 7,08 (с, 1H), 4,72 (с, 2H), 2,27 (с, 3H). ESI-MC m/z 143,9 (M+H)+.
6-хлор-7-метилимидазо[1,2-b]пиридазин 8:
6-хлор-5-метилпиридазин-3-амин 7 (0,6 г, 4,18 ммоль) растворяли в н-бутаноле (10 мл) и добавляли хлорацетальдегид (0,328 г, 4,18 ммоль). Реакционную смесь кипятили в течение 6 часов и удаляли н-бутанол при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (DCM/гексан, 70:30), получая соединение 8 (0,234 г, выход=33,4%).
1H-ЯМР (300 МГц, CD3OD) 8,66 (с, 1H), 8,05 (д, J=1,7 Гц, 1H), 7,96 (д, J=8,2 Гц, 1H), 2,58 (с, 3H). ESI-МС m/z 167,8 (М+H)+.
3-бром-6-хлор-7-метилимидазо[1,2-b]пиридазин 9:
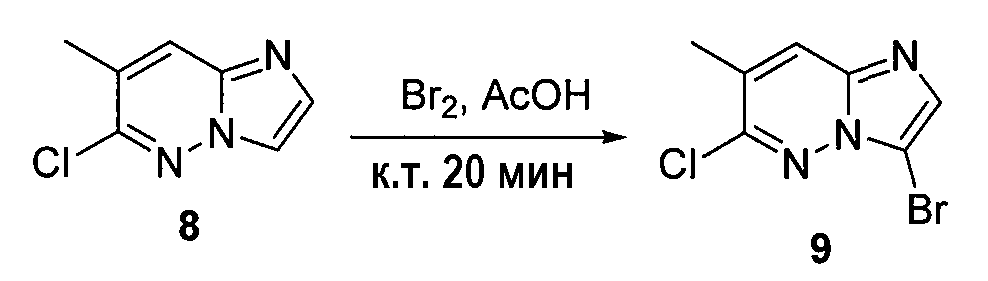
6-хлор-7-метилимидазо[1,2-b]пиридазин 8 (0,230 г, 1,372 ммоль) смешивали с ледяной уксусной кислотой (10 мл) и медленно при комнатной температуре добавляли бром (0,070 мл, 1,372 ммоль). Через 20 минут растворители выпаривали, полученное твердое коричневое вещество промывали эфиром (3×15 мл) и высушивали на воздухе, получая соединение 9 (0,236 г, выход 69,8%).
1H-ЯМР (300 МГц, CD3OD) 7,79 (д, J=7,32 Гц, 1H), 6,93 (с, 1H), 2,64 (с, 3H). ESI-МС m/z 247,9 (М+H)+.
6-хлор-7-метил-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин 11:
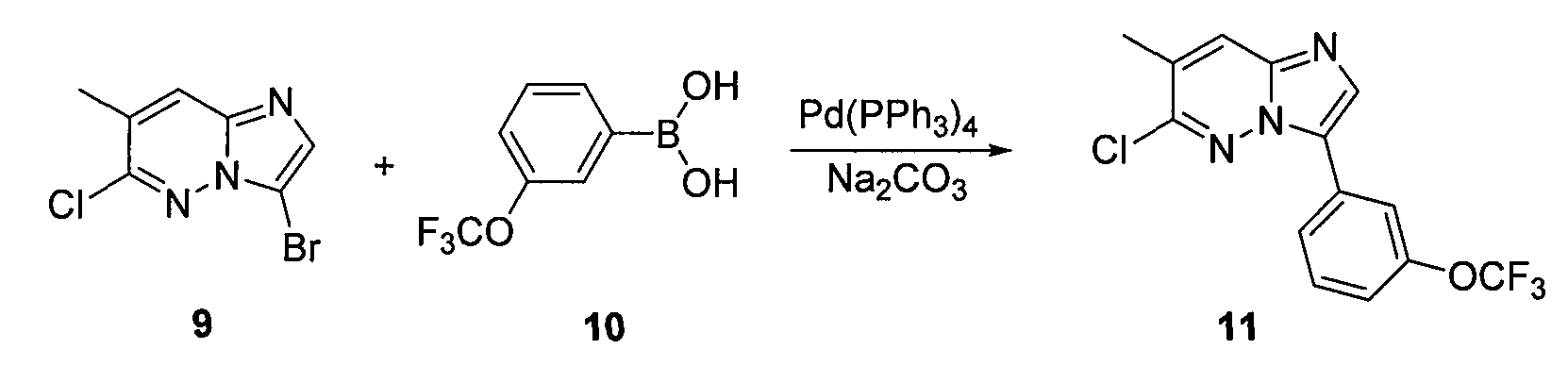
3-бром-6-хлор-7-метилимидазо[1,2-b]пиридазин 9 (100 мг, 0,406 ммоль) растворяли в 1,4-диоксане (10 мл) и добавляли 3-фторметоксибороновую кислоту 10 (84 мг, 0,406 ммоль), Pd(PPh3)4 (9,38 мг, 8,11 мкмоль) и Na2CO3 (47,3 мг, 0,446 ммоль). Реакционную смесь нагревали микроволновым излучением при 150°C в течение 30 минут. ТСХ (6% MeOH/DCM) показала завершение реакции. Концентрирование и препаративная ТСХ (6% MeOH/DCM) позволили получить соединение 11.
1H-ЯМР (300 МГц, CDCl3) 8,01 (д, J=1,2 Гц, 1H), 7,95 (м, 2H), 7,51 (т, J=8,1 Гц, 1H), 7,21 (м, 1H), 6,96 (м, 1H), 2,69 (с, 3H), 1,54 (с, 3H). 19F-ЯМР (300 МГц, CDCl3) -59,08.
(S)-2-(3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)бутан-1-ол (соединение 7-32):
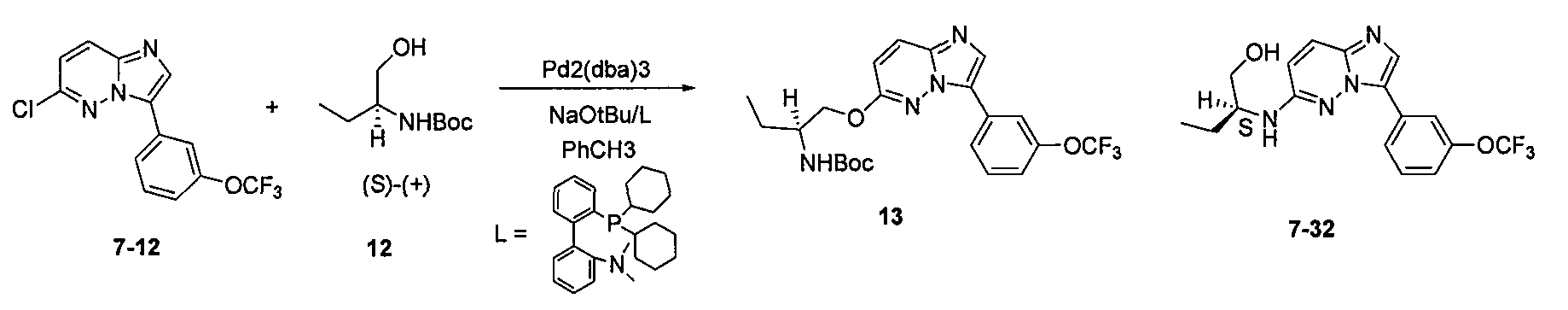
К толуолу (10 мл) добавляли соединение 7-12 (100 мг, 0,319 ммоль), N-Boc-(S)-(+)-2-амино-1-бутанол 12 (121 мг, 0,638 ммоль), лиганд (18,82 мг, 0,048 ммоль), NaOt-Bu (43,5 мг, 0,453 ммоль) и Pd2(dba)3 (21,90, 0,024 ммоль). Полученную реакционную смесь дегазировали в течение 10 минут в атмосфере аргона и затем обрабатывали микроволновым излучением в течение 1 ч при 165°C. Концентрирование и очистка препаративной ТСХ (10% MeOH/DCM), позволяли получить соединение 13 и соединение 7-32. По данным ЯМР соединение 7-32 представляло собой продукт с удаленной группой Boc. Неочищенный продукт концентрировали, проводили препаративную ТСХ, используя систему растворителей 10% MeOH/DCM, и получали 27 мг 7-32 в виде твердого желтого вещества (23,12%).
1H-ЯМР (400 МГц, CD3OD) 8,30 (с, 1H), 8,02 (д, J=7,8 Гц, 1H), 7,84 (с, 1H), 7,62 (д, J=9,6 Гц, 1H), 7,53 (т, J=8,2 Гц, 1H), 7,21 (д, J=8,5 Гц, 1H), 6,78 (д, J=9,6 Гц, 1H), 3,93 (т, J=5,8 Гц, 1H), 3,76 (м, 1H), 3,65 (м, 1H), 1,73 (м, 2H), 1,03 (т, J=7,5 Гц, 3H). ESI-МС m/z 367,13 (М+H)+.
6-((1-метилпиперидин-4-ил)метокси)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин (соединение 7-33):
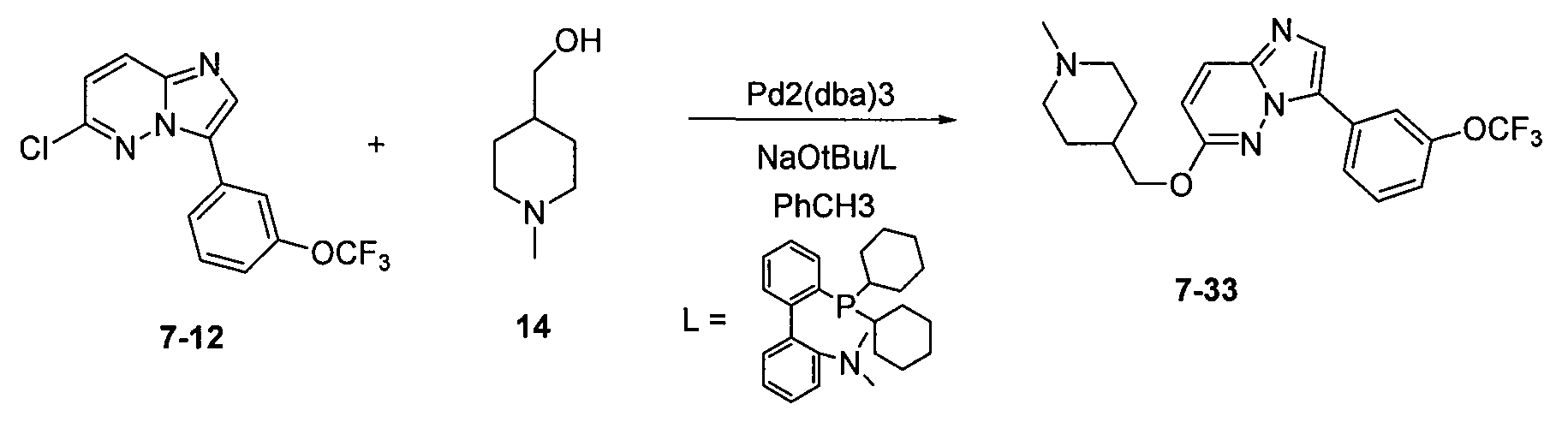
К толуолу (5 мл) добавляли соединение 7-12 (50 мг, 0,159 ммоль), (1-метилпиперидин-4-ил)метанол 14 (30,9 мг, 0,239 ммоль), лиганд (9,41 мг, 0,024 ммоль), NaOt-Bu (21,75 мг, 0,226 ммоль) и Pd2(dba)3 (10,95 мг, 0,012 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение 12 ч. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 17,6 мг соединения 7-33.
1H-ЯМР (400 МГц, CD3OD) 8,30 (с, 1H), 8,05 (с, 1H), 7,97 (м, 3H), 7,58 (т, J=8,2 Гц, 1H), 6,96 (д, J=9,9 Гц, 1H), 4,29 (д, J=6,5 Гц, 2H), 3,94 (д, J=6,15 Гц, 1H), 3,07 (т, J=12,64 Гц, 4H), 2,33 (т, J=12,30 Гц, 4H), 2,02 (с, 3H). ESI-МС m/z 407,18 (М+H)+.
7-метил-6-((1-метилпиперидин-4-ил)метокси)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин (соединение 7-34):
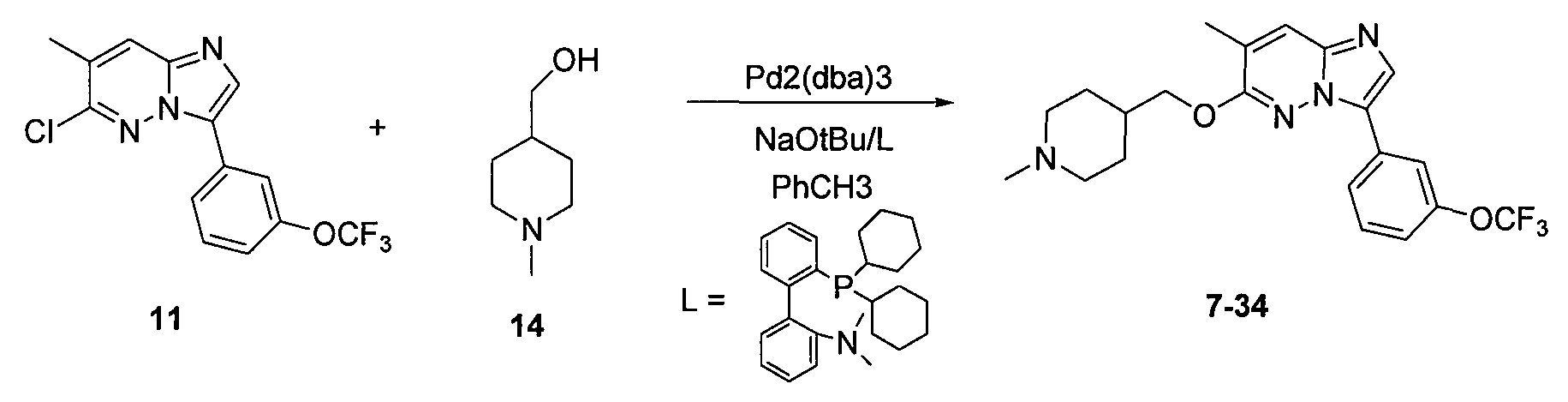
К толуолу (5 мл) добавляли соединение 11 (80 мг, 0,224 ммоль), (1-метилпиперидин-4-ил)метанол 14 (47,3 мг, 0,366 ммоль), лиганд (14,41 мг, 0,037 ммоль), NaOt-Bu (33,3 мг, 0,347 ммоль) и Pd2(dba)3 (16,77 мг, 0,018 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали действием микроволнового излучения при 165°C. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 111,5 мг (11,2%) соединения 7-34.
1H-ЯМР (300 МГц, CD3OD) 8,27 (с, 1H), 7,96 (м, 2H), 7,55 (м, 1H), 7,23 (д, J=8,1 Гц, 1H), 6,76 (с, 1H), 4,18 (с, 2H), 2,96 (д, J=11,7 Гц, 1H), 2,56 (с, 3H), 2,31 (с, 3H), 2,11 (т, J=12,3 Гц, 2H), 1,88 (д, J=12,3 Гц, 4H), 1,44 (д, J=12,6 Гц, 2H). ESI-МС m/z 421,18 (М+H)+.
N-((1-изопропилпиперидин-4-ил)метил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-35):
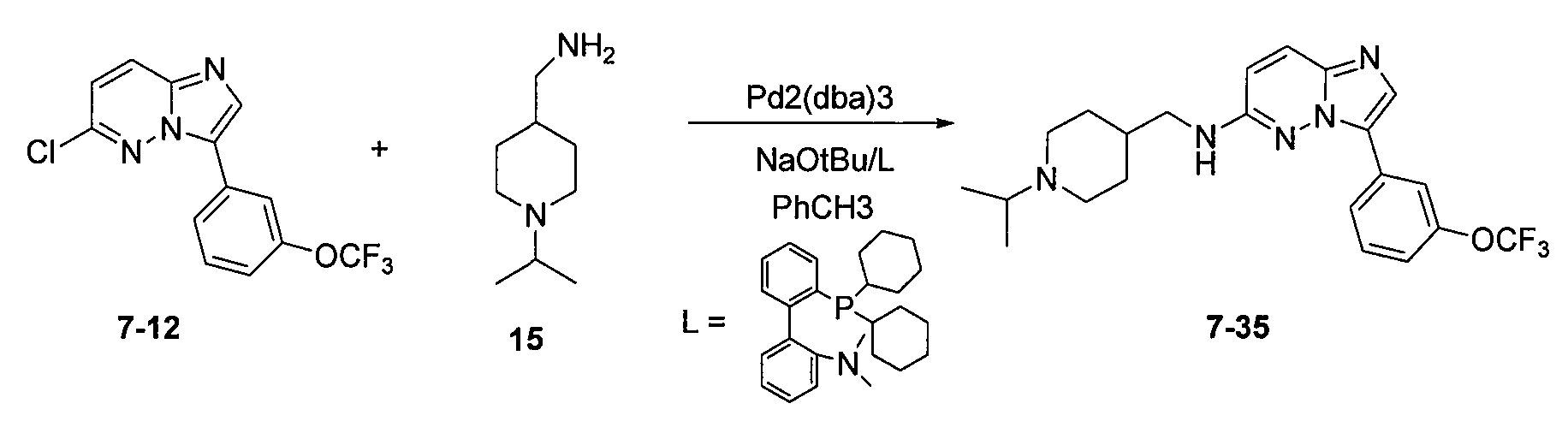
К толуолу (10 мл) добавляли соединение 7-12 (10 мг, 0,319 ммоль), (1-изопропилпиперидин-4-ил)метанамин 15 (74,9 мг, 0,478 ммоль), лиганд (18,82 мг, 0,048 ммоль), NaOH (43,5 мг, 0,453 ммоль) и Pd2(dba)3 (21,90 мг, 0,024 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения в течение 12 ч. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 11 мг (7,96%) соединения 7-35.
1H-ЯМР (300 МГц, CD3OD) 7,60 (c, 1H), 7,18 (д, J=9,3 Гц, 1H), 7,07 (с, 1H), 6,84 (д, J=9,3 Гц, 1H), 6,73 (т, J=8,1 Гц, 1H), 6,43 (д, J=6,3 Гц, 1H), 5,94 (д, J=9,9 Гц, 1H), 2,85 (с, 1H), 2,14 (м, 3H), 1,93 (м, 2H), 1,42 (м, 3H), 1,07 (м, 4H), 0,286 (2с, 2CH3). ESI-МС m/z 434,23 (М+H)+.
Циклопропил(4-((3-(3-трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-иламино)метил)пиперидин-1-ил)метанон (соединение 7-36):
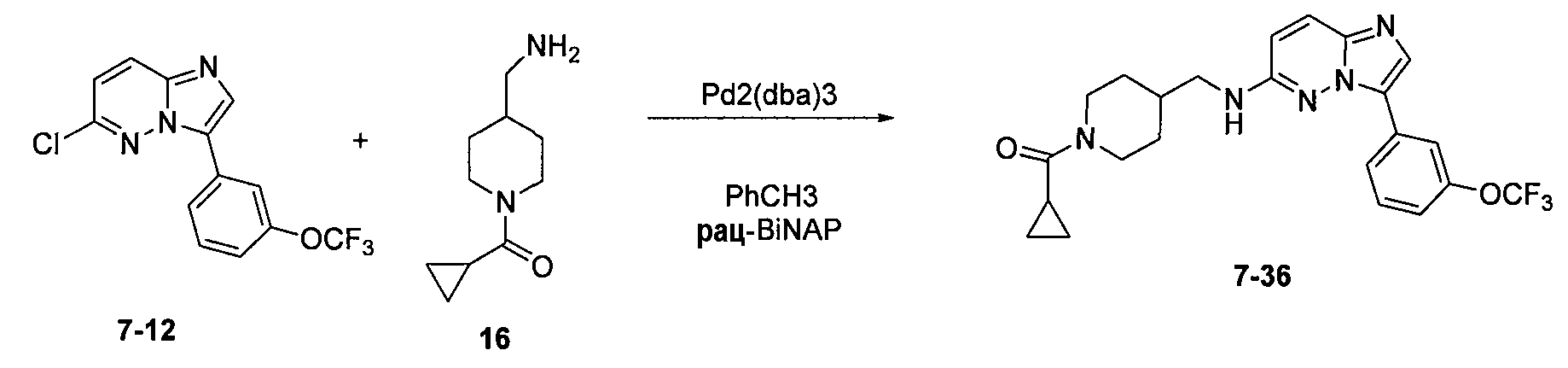
К толуолу (10 мл) добавляли соединение 7-12 (10 мг, 0,319 ммоль), (1-циклопропилкарбонилперидин-4-ил)метанамин 16 (69,7 мг, 0,383 ммоль), рац-BINAP (7,94 мг, 0,013 ммоль) и Pd2(dba)3 (5,84 мг, 6,38 мкмоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали до кипения с обратным холодильником в течение 12 ч. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 29 мг (19,8%) соединения 7-36.
1H-ЯМР (300 МГц, CD3OD) 7,59 (с, 1H), 7,16 (д, J=7,2 Гц, 1H), 7,04 (д, J=6,3 Гц, 1H), 6,81 (м, 1H), 6,70 (м, 1H), 6,40 (д, J=6,3 Гц, 1H), 5,90 (м, 1H), 3,70 (м, 1H), 3,54 (м, 1H), 2,36 (м, 1H), 1,83 (м, 1H), 1,28 (м, 1H), 1,12 (м, 3H), 0,4 (м, 2H), 0,027 (м, 4H). ESI-МС m/z 460,20 (M+H)+.
7-метил-N-((1-метилпиперидин-4-ил)метил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-37):
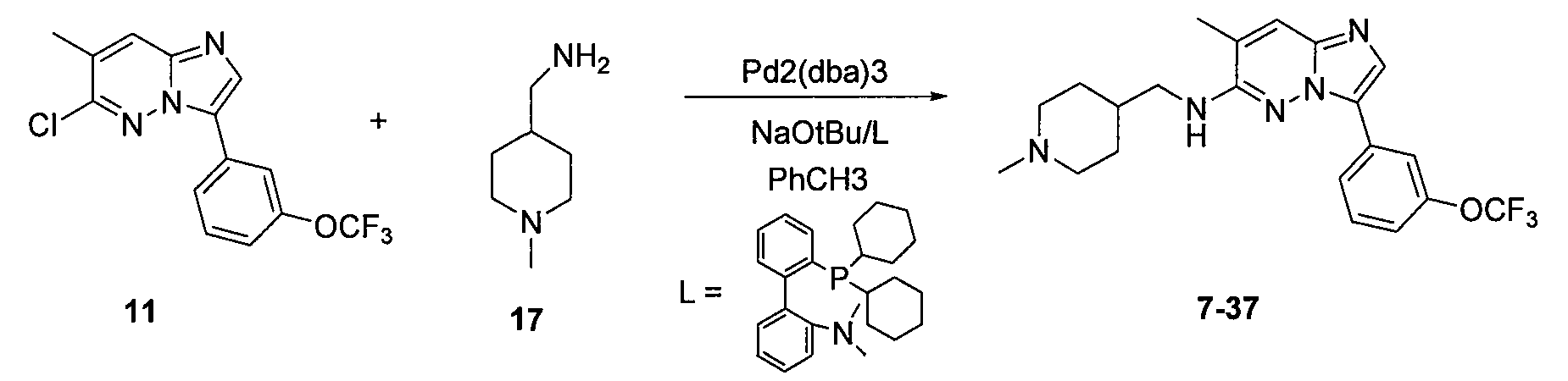
К толуолу (5 мл) добавляли соединение 11 (57 мг, 0,174 ммоль), (1-метилпиперидин-4-ил)метанамин 17 (26,8 мг, 0,209 ммоль), лиганд (10,27 мг, 0,026 ммоль), NaOt-Bu (23,40 мг, 0,244 ммоль) и Pd2(dba)3 (11,95 мг, 0,013 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали с помощью микроволнового излучения при 150°C в течение 1 ч. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 5,3 мг (7,26%) соединения 7-37.
1H-ЯМР (300 МГц, CD3OD) 8,35 (с, 1H), 8,01 (д, J=8,4 Гц, 1H), 7,83 (с, 1H), 7,51 (т, J=7,8 Гц, 2H), 7,21 (д, J=8,1 Гц, 2H), 3,65 (м, 2H), 2,36 (м, 3H), 2,98 (д, J=11,4 Гц, 2H), 2,48 (м, 2H), 2,26 (д, J=0,9 Гц, 3H), 2,24 (с, 3H), 1,91 (м, 4H), 1,32 (м, 1H). 19F-ЯМР (300 МГц, CD3OD) -56,456.
FTMC+p MALDI: 420,20113 (M+H)+, точное теоретическое значение массы: 420,20112.
N-((1-этилпиперидин-4-ил)метил)-7-метил-3-((трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-38):
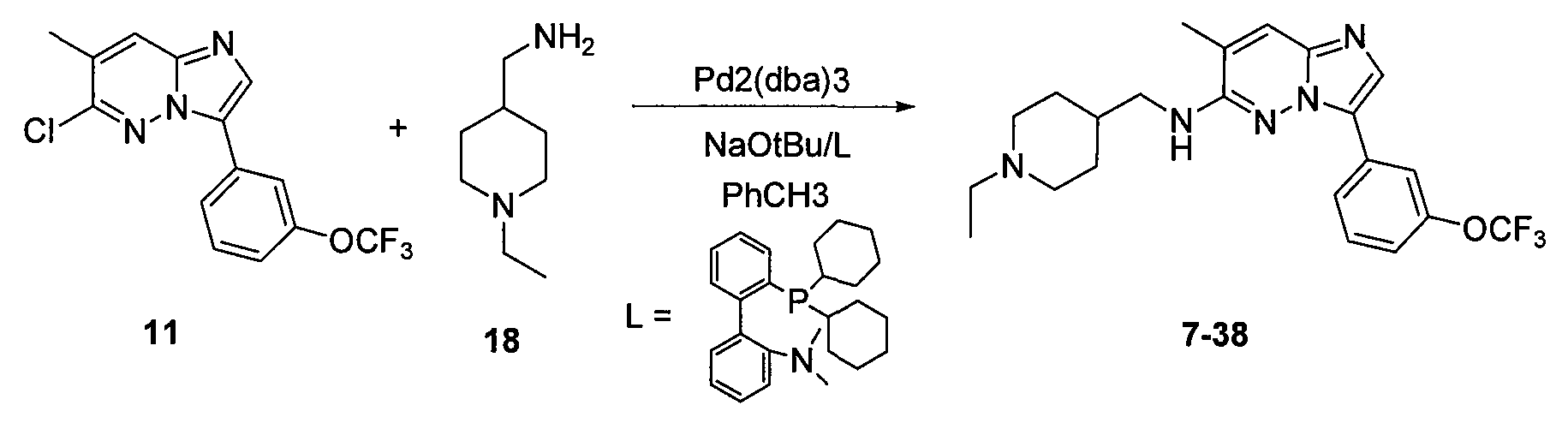
К толуолу (5 мл) добавляли соединение 11 (50 мг, 0,153 ммоль), (1-этилпиперидин-4-ил)метанамин 18 (26 мг, 0,183 ммоль), лиганд (9,01 мг, 0,023 ммоль), NaOt-Bu (20,53 мг, 0,214 ммоль) и Pd2(dba)3 (10,48 мг, 0,011 ммоль). Полученную реакционную смесь дегазировали в течение 10 мин в атмосфере аргона и затем нагревали с помощью микроволнового излучения при 150°C в течение 1,5 ч. Концентрирование и препаративная ТСХ (10% MeOH/DCM) позволяли получить 11,9 мг (17,99%) соединения 7-38.
1H-ЯМР (300 МГц, CD3OD) 8,335 (с, 1H), 7,99 (д, J=7,8 Гц, 1H), 7,81 (с, 1H), 7,49 (т, J=7,8 Гц, 2H), 7,21 (м, 1H), 3,34 (м, 2H), 3,00 (д, J=11,1 Гц, 2H), 2,44 (м, 4H), 2,26 (с, 3H), 1,91 (м, 4H), 1,32 (м, 1H), 1,08 (т, J=6,9 Гц, 3H). 19F-ЯМР (300 МГц, CD3OD) -56,423.
FTMC+p MALDI: 434,36365 (M+H)+, точное теоретическое значение массы: 433,20895.
N-((1-этилпиперидин-4-ил)метил)-3-(3-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-39):
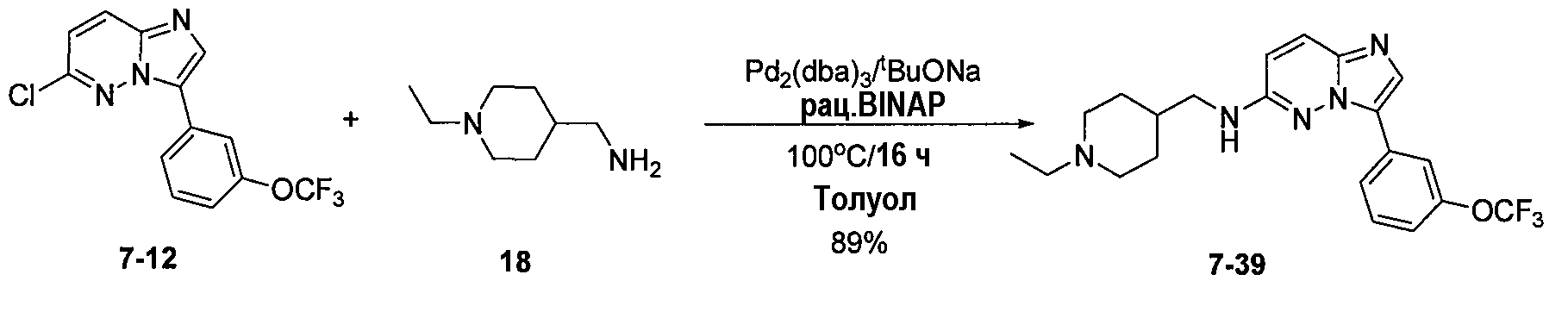
К раствору соединения 7-12 (0,250 г, 0,797 ммоль) и (1-этилпиперидин-4-ил)метанамин 18 (0,113 г, 0,797 ммоль) в толуоле (10 мл) добавляли трет-бутоксид натрия (0,138 г, 1,435 ммоль), рац.-BINAP (0,030 г, 0,048 ммоль) и Pd2(dba)3 (0,022 г, 0,024 ммоль), и эту смесь нагревали до 100°C в течение ночи. Через 16 ч полученный темно-коричневый раствор охлаждали и концентрировали при пониженном давлении. Твердое вещество дополнительно очищали с использованием хроматографии CombiFlash (6 г колонка), элюент: 5% TEA в смеси этилацетат/гексан (10-100) для удаления загрязнений и 5% TEA в смеси этилацетат/CH3OH (90:10) для получения соединения 7-39 (89%).
1H-ЯМР (ДМСО-d6/300 МГц): 8,50 (с, 1H), 8,06 (д, J=13,2 Гц, 1H), 7,77 (д, J=9,9 Гц, 1H), 7,57 (т, J=7,8 Гц, 1H), 7,28 (м, 2H), 6,76 (д, J=9,9 Гц, 1H), 3,35 (м, 2H), 3,16 (м, 2H), 2,87 (д, J=9,9 Гц, 2H), 2,27 (м, 2H), 1,76 (м, 4H), 1,22 (м, 1H), 0,97 (т, J=6,9 Гц, 3H).
Соединения 7-40-7-51 получали в соответствии со следующей общей методикой:
К раствору 6-хлор-3-(2-метокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазина или 6-хлор-3-(2-метокси-4-(трифторметокси)фенил)имидазо[1,2-b]пиридазина (0,149 г, 0,434 ммоль) и амина (0,434 ммоль) в толуоле (5 мл) добавляли трет-бутоксид натрия (0,075 г, 0,780 ммоль), рац.-BINAP (0,012 г, 0,013 ммоль) и Pd2(dba)3 (0,016 г, 0,026 ммоль), и эту смесь нагревали до 100°C в течение ночи. Через 16 ч полученный темно-коричневый раствор охлаждали и концентрировали при пониженном давлении. Твердое вещество дополнительно очищали с использованием хроматографа CombiFlash (6 г колонка), элюент: 5% TEA в смеси этилацетат/гексан (10-100) для удаления загрязнений и 5% TEA в смеси этилацетат/CH3OH (90:10) для желаемых метоксипродуктов.
Метоксигруппу удаляли путем растворения метоксисоединения (0,222 ммоль) в безводном дихлорметане (DCM) (3 мл) и добавления BBr3 (1,0 М в DCM, 0,667 мл) при -78°C и перемешивания при комнатной температуре в течение ночи. Через 16 ч полученный темно-коричневый раствор гасили NaHCO3. ВЭЖХ анализ показывал завершение реакции. После экстрагирования DCM и высушивания, остаток очищали с использованием хроматографии combiFlash, элюент метанол/этилацетат (5% TEA), соотношение 5-50%. Rf=0,23, 50% этилацетат (5% TEA)/CH3OH. Структуры соединений подтверждали 1H-ЯМР и масс-спектрометрией (МС).
3-(2-метокси-5-(трифторметил)фенил)-N-(4-(4-метилпиперазин-1-ил)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-40):
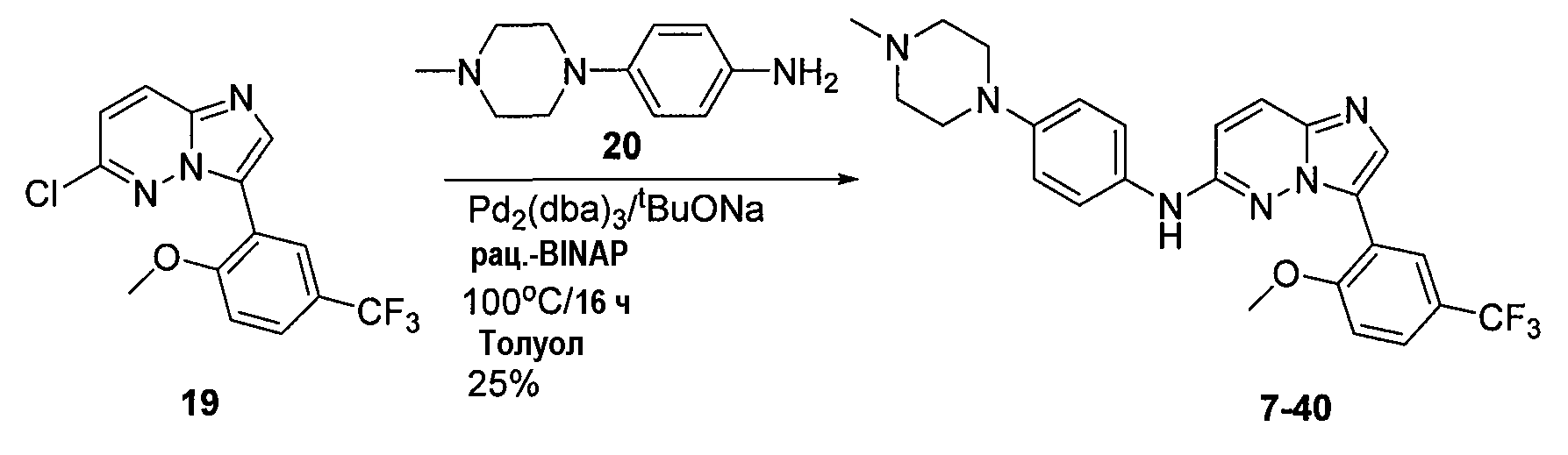
1H-ЯМР (CD3OD/400 МГц): 8,36 (д, J=2,1 Гц, 1H), 7,79 (с, 1H), 7,68 (с, 1H), 7,64 (с, 1H), 7,54 (дд, J=8,5, 1,7 Гц, 2H), 6,87 (м, 3H), 3,92 (с, 3H), 3,12 (т, J=18,3 Гц, 4H), 2,62 (т, J=18,3 Гц, 4H), 2,33 (с, 3H). ESI-МС (ES+, m/z): 483,2 (M++1, 100,0).
3-(2-гидрокси-5-(трифторметил)фенил)-N-(4-(4-метилпиперазин-1-ил)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-41):
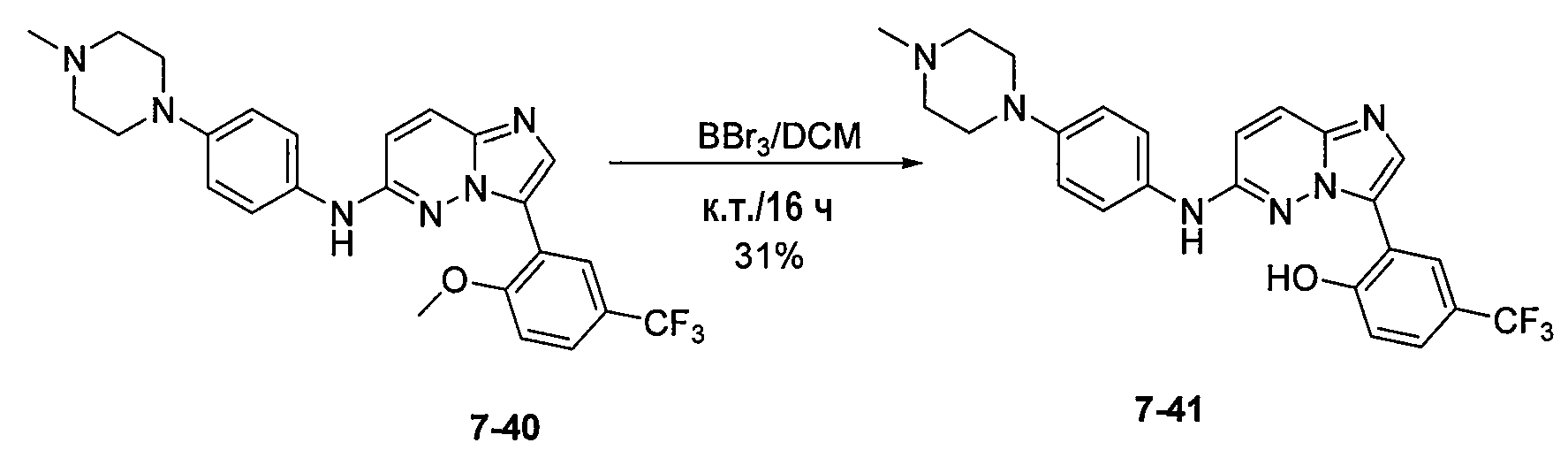
1H-ЯМР (ДМСО-d6/400 МГц): 9,28 (с, 1H), 8,29 (д, J=2,1 Гц, 1H), 7,90 (д, J=9,9 Гц, 1H), 7,81 (с, 1H), 7,77 (дд, J=8,6, 2,1 Гц, 1H), 7,55 (д, J=8,9 Гц, 2H), 7,40 (д, J=8,6 Гц, 1H), 6,90 (т, J=9,9 Гц, 3H), 3,64 (д, J=12,3 Гц, 4H), 3,50 (д, J=13,3 Гц, 4H), 2,33 (с, 3H). ESI-МС (ES+, m/z): 469,3 (M++1, 10,0).
3-(2-метокси-5-(трифторметокси)фенил)-N-((1-метилпиперидин-4-ил)метил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-42):
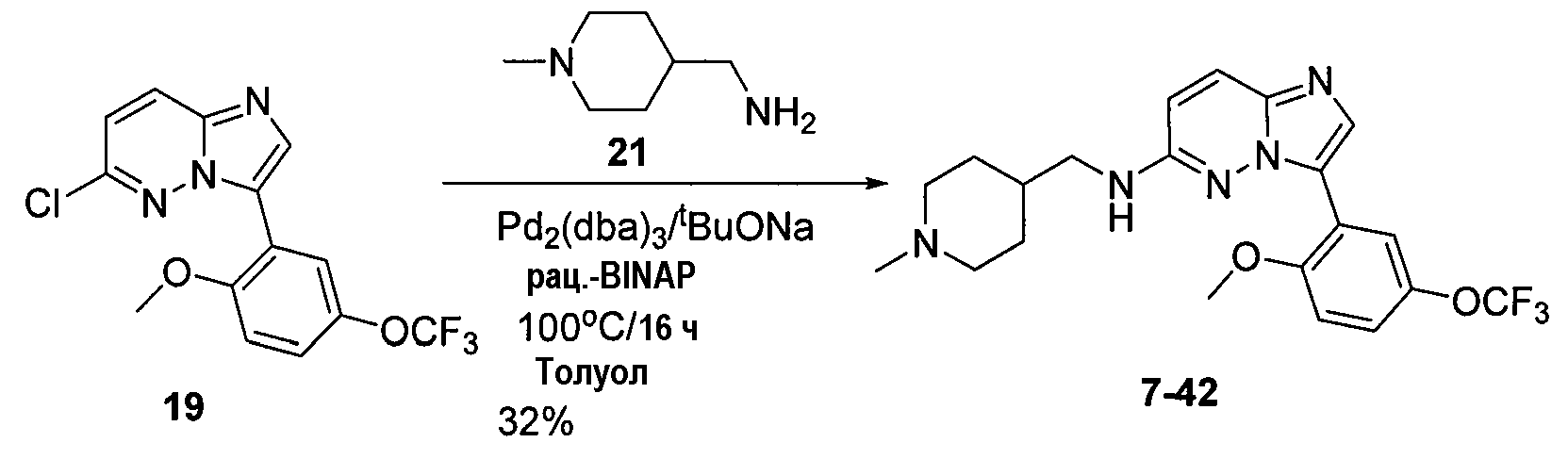
1H-ЯМР (CD3OD/400 МГц): 8,46 (д, J=2,4 Гц, 1H), 7,94 (с, 1H), 7,64 (д, J=9,6 Гц, 1H), 7,23 (дт, J=9,2, 2,0 Гц, 1H), 7,18 (д, J=9,3 Гц, 1H), 6,73 (д, J=9,6 Гц, 1H), 3,95 (с, 3H), 3,27 (м, J=18,3 Гц, 2H), 3,15 (м, J=18,3 Гц, 2H), 2,48 (с, 3H), 2,41 (т, J=18,3 Гц, 2H), 1,90 (м, J=18,3 Гц, 3H), 1,29 (м, J=18,3 Гц, 2H). ESI-МС (ES+, m/z): 436,2 (M++1, 100,0).
3-(2-гидрокси-5-(трифторметокси)фенил)-N-((1-метилпиперидин-4-ил)метил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-43):
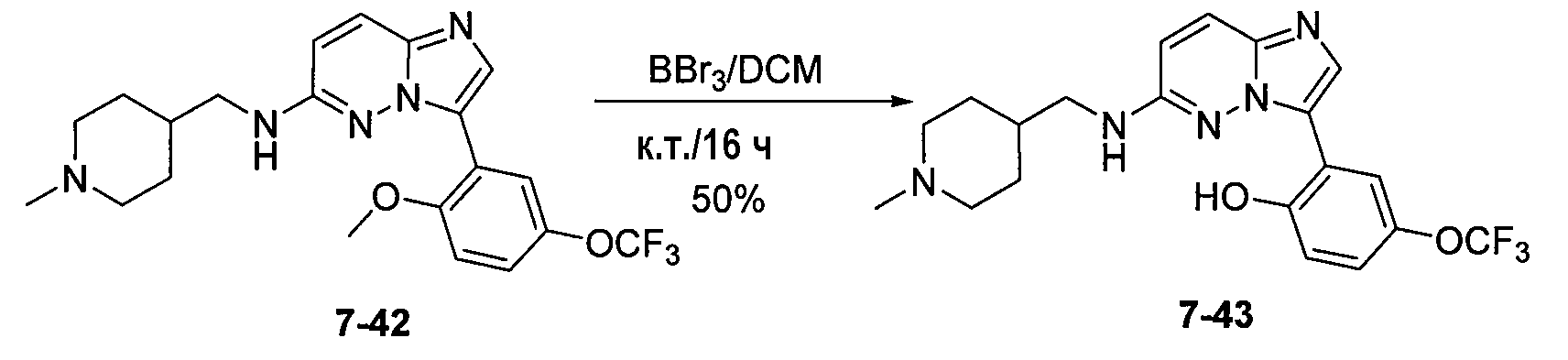
1H-ЯМР (CD3OD/400 МГц): 8,60 (д, J=2,4 Гц, 1H), 8,04 (с, 1H), 7,74 (д, J=9,9 Гц, 1H), 7,13 (м, J=9,2, 2,0 Гц, 2H), 7,04 (д, J=8,9 Гц, 1H), 6,72 (д, J=9,6 Гц, 1H), 3,13 (т, J=6,5 Гц, 4H), 2,75 (д, J=11,3 Гц, 2H), 2,48 (с, 3H), 1,81 (т, J=12,0 Гц, 2H), 1,72 (д, J=12,0 Гц, 2H), 1,60 (м, J=18,3 Гц, 1H). ESI-МС (ES+, m/z): 422,2 (M++1, 100,0).
3-(2-метокси-5-(трифторметокси)фенил)-N-(2-(1-метилпиперидин-4-ил)этил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-44):
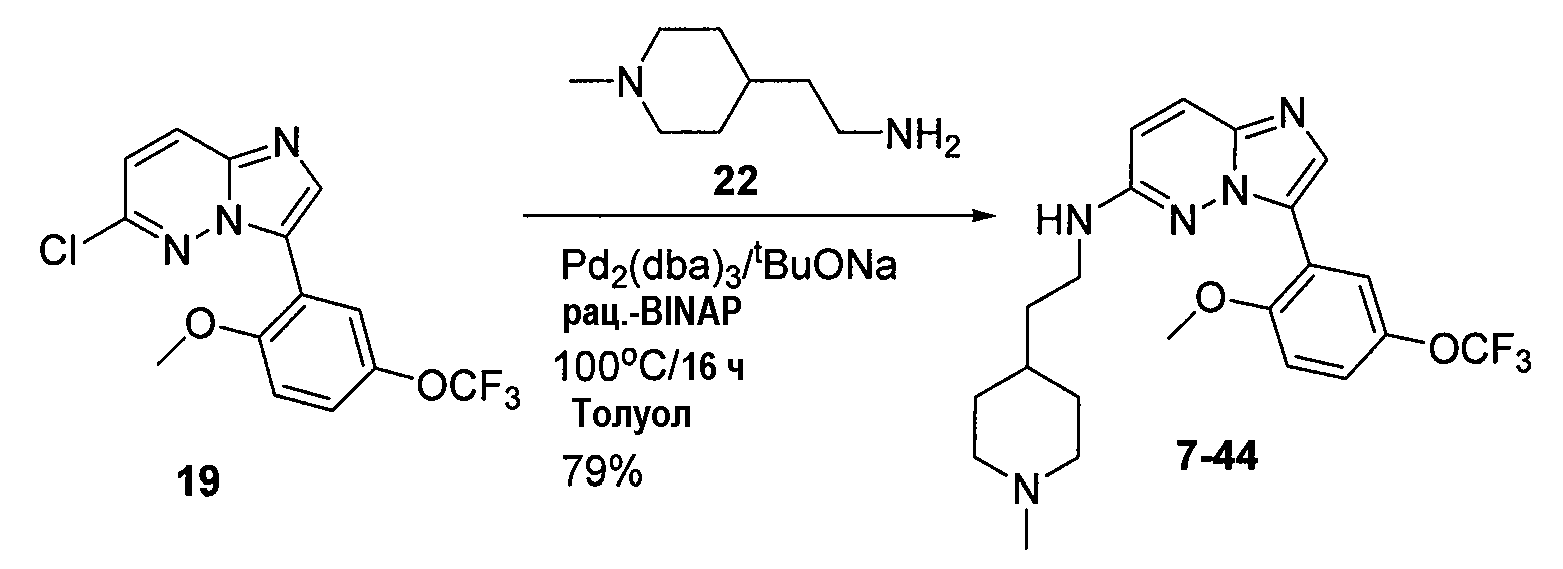
1H-ЯМР (ДМСО-d6/300 МГц): 8,53 (д, J=2,7 Гц, 1H), 7,93 (с, 1H), 7,75 (д, J=9,6 Гц, 1H), 7,30 (дд, J=8,7, 3,0 Гц, 1H), 7,22 (д, J=9,3 Гц, 1H), 6,97 (т, J=5,1 Гц, 1H), 6,77 (д, J=9,3 Гц, 1H), 3,91 (с, 3H), 2,81 (д, J=11,7 Гц, 2H), 2,48 (с, 3H), 1,89 (т, J=11,7 Гц, 2H), 1,52 (д, J=12,3 Гц, 2H), 1,27 (м, 1H), 1,17 (м, 2H), 0,84 (м, 4H). ESI-МС (ES+, m/z): 450,2 (M++1, 20,0).
3-(2-гидрокси-5-(трифторметокси)фенил)-N-(2-(1-метилпиперидин-4-ил)этил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-45):
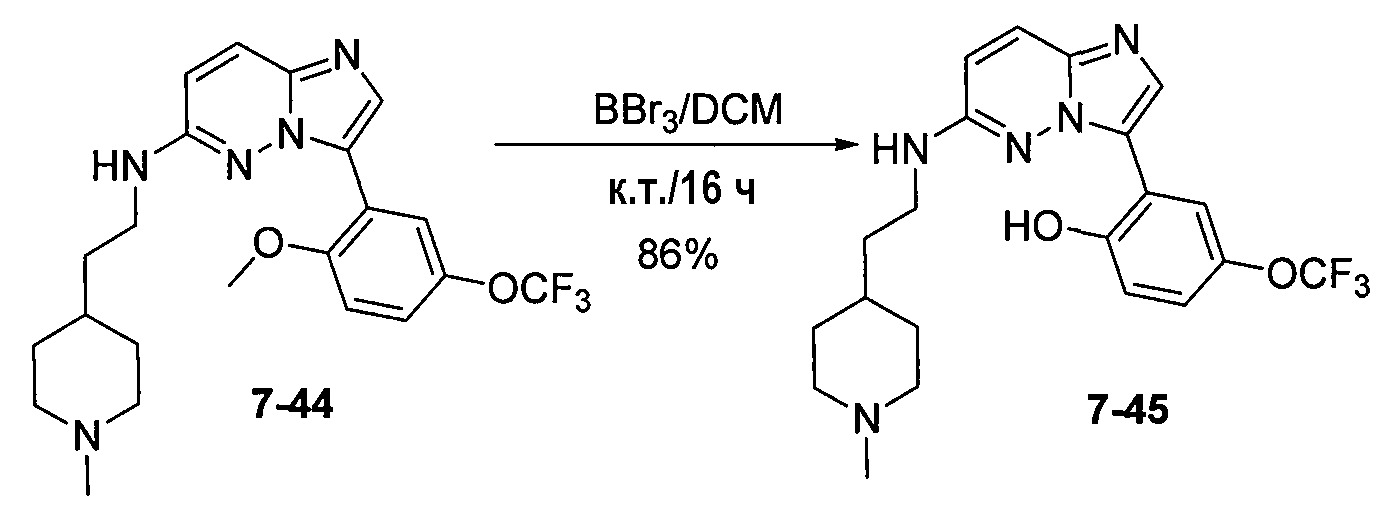
1H-ЯМР (CD3OD+CDCl3/300 МГц): 8,01 (д, J=3,1 Гц, 1H), 7,88 (с, 1H), 7,63 (д, J=9,6 Гц, 1H), 7,07 (дд, J=8,9, 1,7 Гц, 1H), 6,97 (д, J=8,7 Гц, 1H), 6,70 (д, J=9,6 Гц, 1H), 3,34 (с, 3H), 2,90 (т, J=6,5 Гц, 2H), 2,37 (т, J=11,2 Гц, 2H), 1,69 (д, J=12,3 Гц, 2H), 1,45 (м, 1H), 1,28 (м, 2H), 0,93 (м, 4H). ESI-МС (ES+, m/z): 436,3 (M++1, 20,0).
N-((1-этилпиперидин-4-ил)метил)-3-(2-метокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-46):
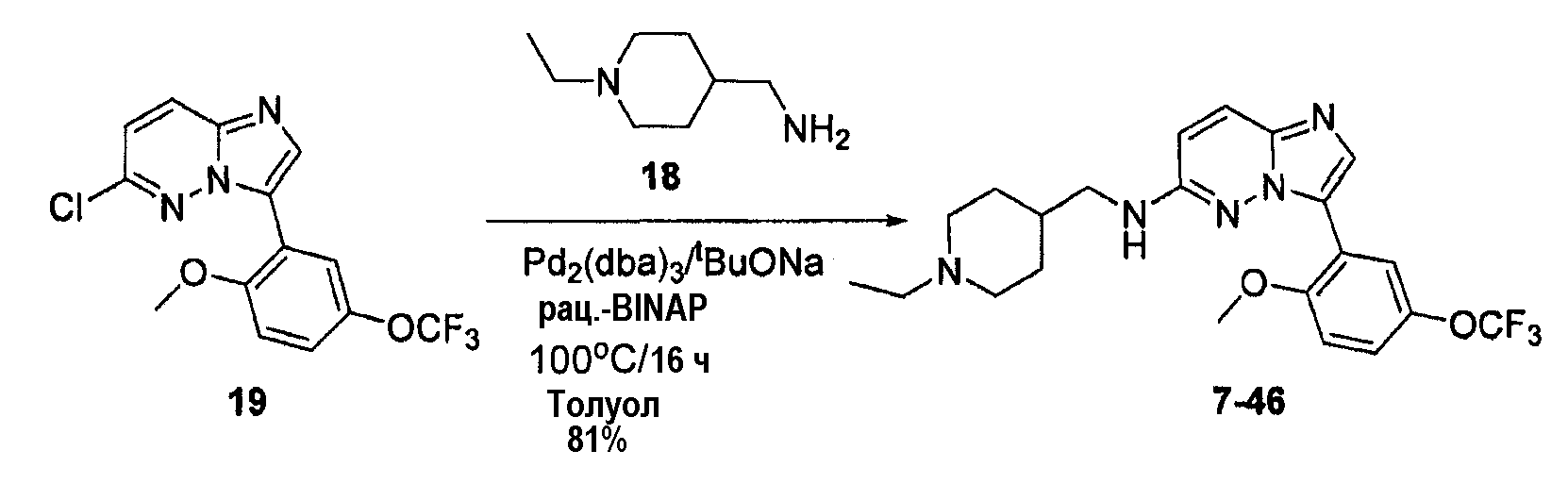
1H-ЯМР (ДМСО-d6/400 МГц): 8,27 (с, 1H), 7,98 (с, 1H), 7,78 (д, J=9,9 Гц, 1H), 7,33 (д, J=8,9 Гц, 1H), 7,25 (д, J=9,2 Гц, 1H), 7,15 (т, J=5,2 Гц, 1H), 6,77 (д, J=9,6 Гц, 1H), 3,95 (с, 3H), 3,15 (т, J=5,8 Гц, 4H), 2,88 (д, J=11,3 Гц, 2H), 2,30 (кв, J=7,2 Гц, 2H), 1,82 (т, J=10,9 Гц, 2H), 1,75 (д, J=13,3 Гц, 2H), 1,65 (м, 1H), 0,99 (т, J=7,2 Гц, 3H). ESI-МС (ES+, m/z): 450,2 (M++1, 20,0).
N-((1-этилпиперидин-4-ил)метил)-3-(2-гидрокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-47):
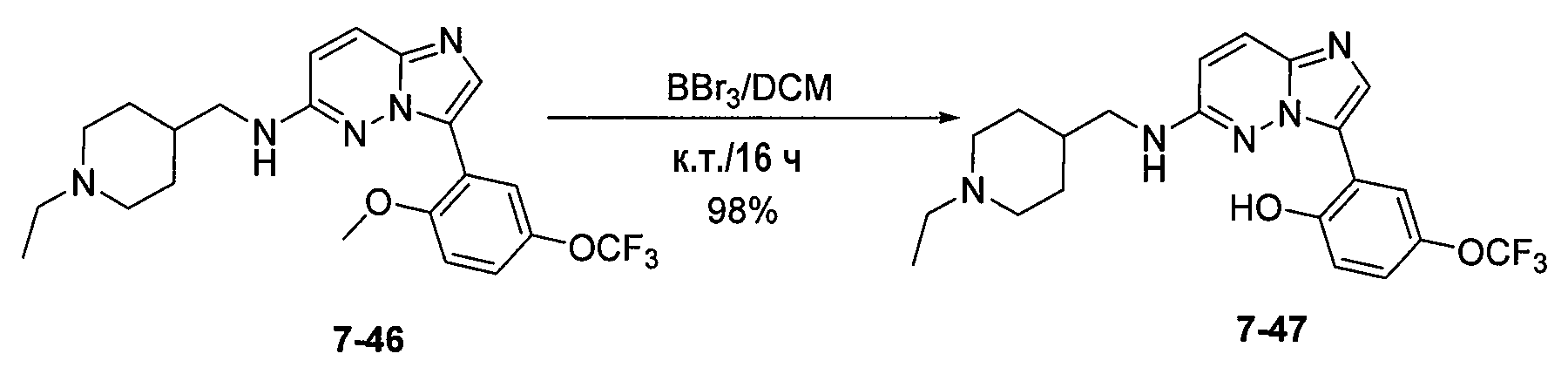
1H-ЯМР (ДМСО-d6/300 МГц): 8,60 (с, 1H), 7,96 (д, J=3,6 Гц, 1H), 7,75 (дд, J=9,9, 3,6 Гц, 1H), 7,30 (д, J=8,7 Гц, 1H), 7,23 (д, J=9,0 Гц, 1H), 7,14 (с, 1H), 6,76 (д, J=9,9 Гц, 1H), 3,16 (м, 4H), 2,78 (м, 2H), 2,30 (кв, J=7,2 Гц, 2H), 1,82 (м, 2H), 1,76 (м, 2H), 1,67 (м, 1H), 0,99 (т, J=7,2 Гц, 3H). ESI-МС (ES+, m/z): 436,2 (M++1, 100,0).
N-((1-изопропилпиперидин-4-ил)метил)-3-(2-метокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-48):
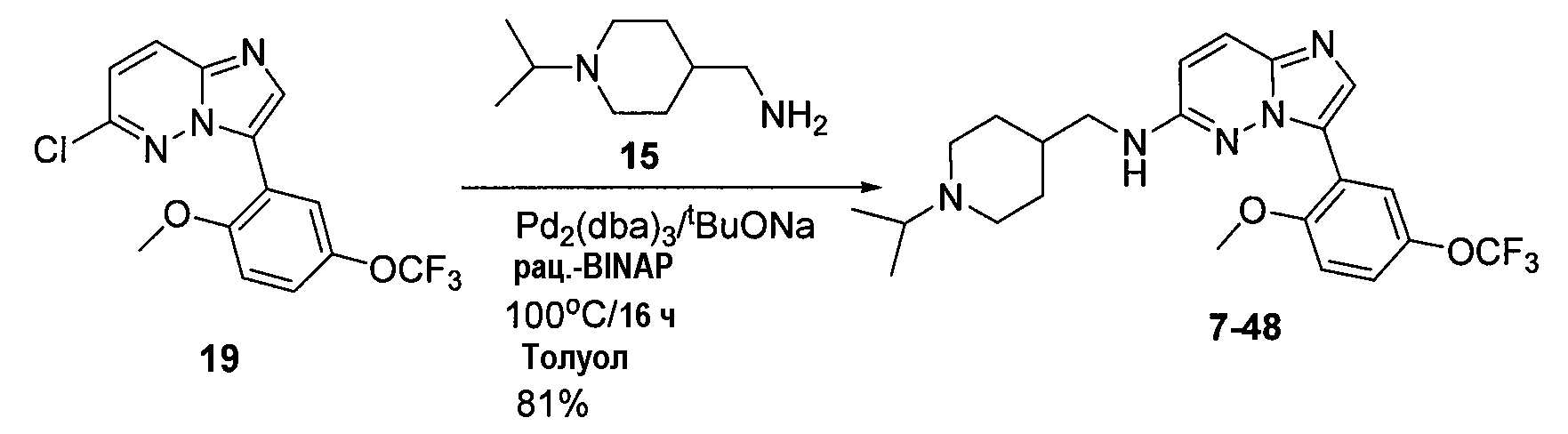
1H-ЯМР (ДМСО-d6/300 МГц): 8,52 (д, J=2,1 Гц, 1H), 7,88 (с, 1H), 7,66 (д, J=10,7 Гц, 1H), 7,24 (д, J=9,0 Гц, 1H), 7,14 (д, J=13,7 Гц, 1H), 7,04 (т, J=5,2 Гц, 1H), 6,68 (д, J=9,4 Гц, 1H), 3,84 (с, 3H), 3,06 (т, J=5,9 Гц, 4H), 2,71 (д, J=8,9 Гц, 2H), 2,55 (т, J=6,5 Гц, 2H), 1,99 (т, J=9,2 Гц, 2H), 1,68 (д, J=13,3 Гц, 1H), 1,53 (м, 1H), 0,84 (д, J=7,2 Гц, 6H). ESI-МС (ES+, m/z): 464,2 (M++1, 50,0).
N-((1-изопропилпиперидин-4-ил)метил)-3-(2-гидрокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-49):
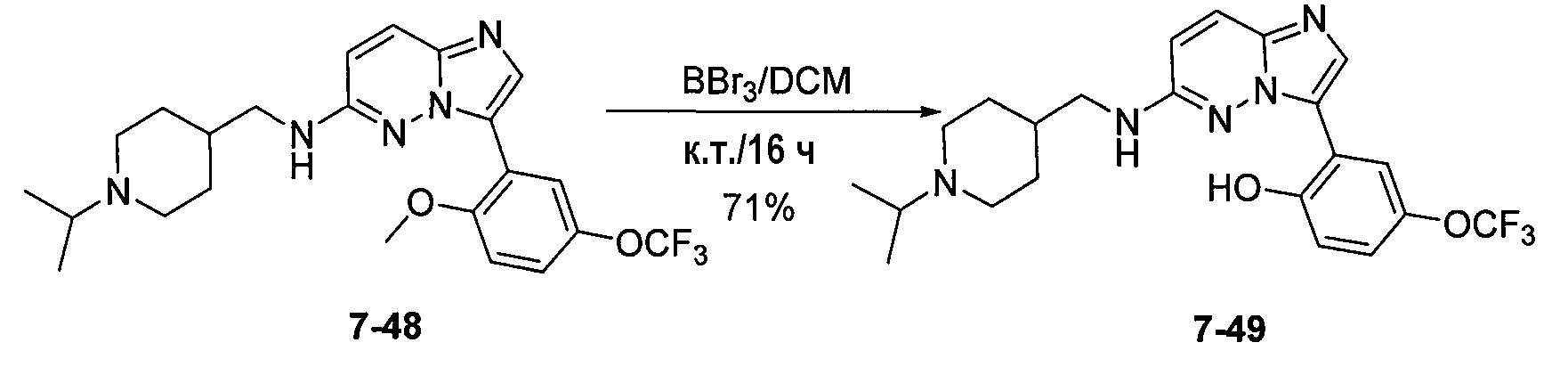
1H-ЯМР (ДМСО-d6/300 МГц): 8,59 (с, 1H), 8,04 (д, J=2,4 Гц, 1H), 7,75 (дд, J=9,8, 2,0 Гц, 1H), 7,13 (д, J=8,3 Гц, 2H), 7,03 (д, J=6,8 Гц, 1H), 6,73 (д, J=7,8 Гц, 1H), 3,13 (м, 4H), 2,87 (м, 2H), 2,25 (м, 2H), 1,74 (м, 4H), 0,98 (д, J=6,8 Гц, 6H). ESI-МС (ES+, m/z): 450,2 (M++1, 40,0).
N-(2-(1-этилпиперидин-4-ил)этил)-3-(2-метокси-5-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-50):
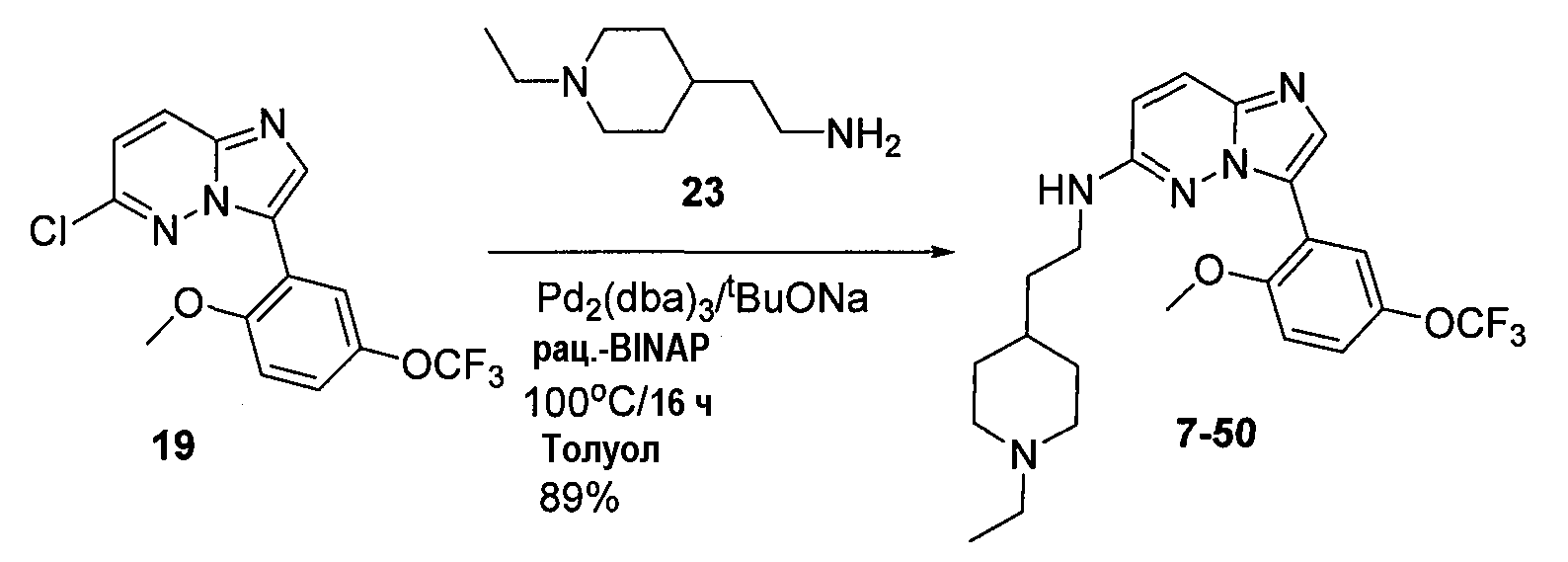
1H-ЯМР (ДМСО-d6/300 МГц): 8,58 (д, J=2,7 Гц, 1H), 7,95 (с, 1H), 7,76 (д, J=9,7 Гц, 1H), 7,30 (дд, J=8,5, 2,0 Гц, 1H), 7,22 (д, J=9,3 Гц, 1H), 6,98 (т, J=5,2 Гц, 1H), 6,77 (д, J=9,3 Гц, 1H), 3,91 (с, 3H), 3,21 (м, 2H), 2,82 (м, 2H), 2,29 (т, J=7,4 Гц, 2H), 1,78 (м, 2H), 1,66 (м, 2H), 1,50 (м, 2H), 1,28 (м, 1H), 1,15 (м, 2H), 0,94 (т, J=7,2 Гц, 3H). ESI-МС (ES+, m/z): 464,2 (M++1, 20,0).
N-((1-изопропилпиперидин-4-ил)метил)-3-(4-(трифторметокси)фенил)имидазо[1,2-b]пиридазин-6-амин (соединение 7-51):
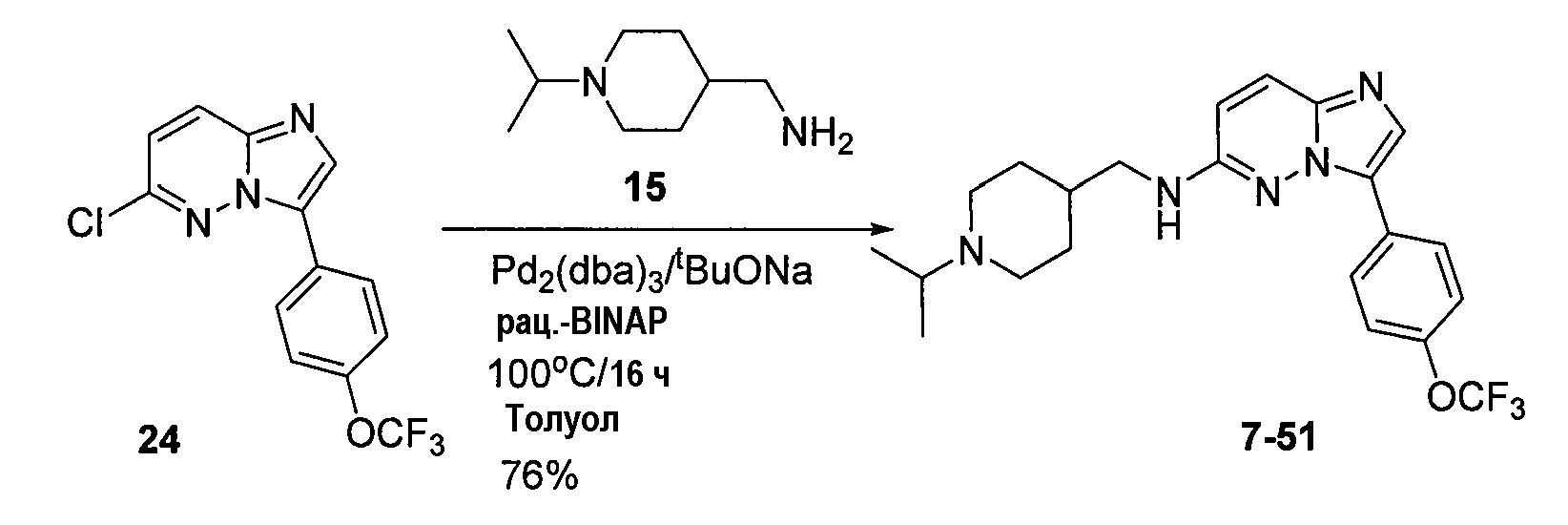
1H-ЯМР (ДМСО-d6/300 МГц): 8,32 (дт, J=9,8, 2,4 Гц, 2H), 7,93 (с, 1H), 7,75 (д, J=9,3 Гц, 1H), 7,44 (д, J=8,3 Гц, 2H), 7,19 (т, J=5,4 Гц, 1H), 6,74 (д, J=9,8 Гц, 1H), 3,14 (т, J=4,9 Гц, 4H), 2,79 (д, J=11,2 Гц, 2H), 2,63 (м, 1H), 2,04 (т, J=10,3 Гц, 2H), 1,74 (м, 2H), 1,23 (м, 1H), 0,91 (д, J=6,3 Гц, 6H). ESI-МС (ES+, m/z): 434,3 (M++1, 40,0).
Пример 6
Иллюстративные ингибиторы PIM-1 киназы, являющиеся производными имидазо[1,2-b]пиридазина
Структуры дополнительных ингибиторов Pim-1 киназы, выявленных в настоящем изобретении и синтезированных согласно методикам, подробно описанным в настоящей заявке, приведены ниже в таблице VII.
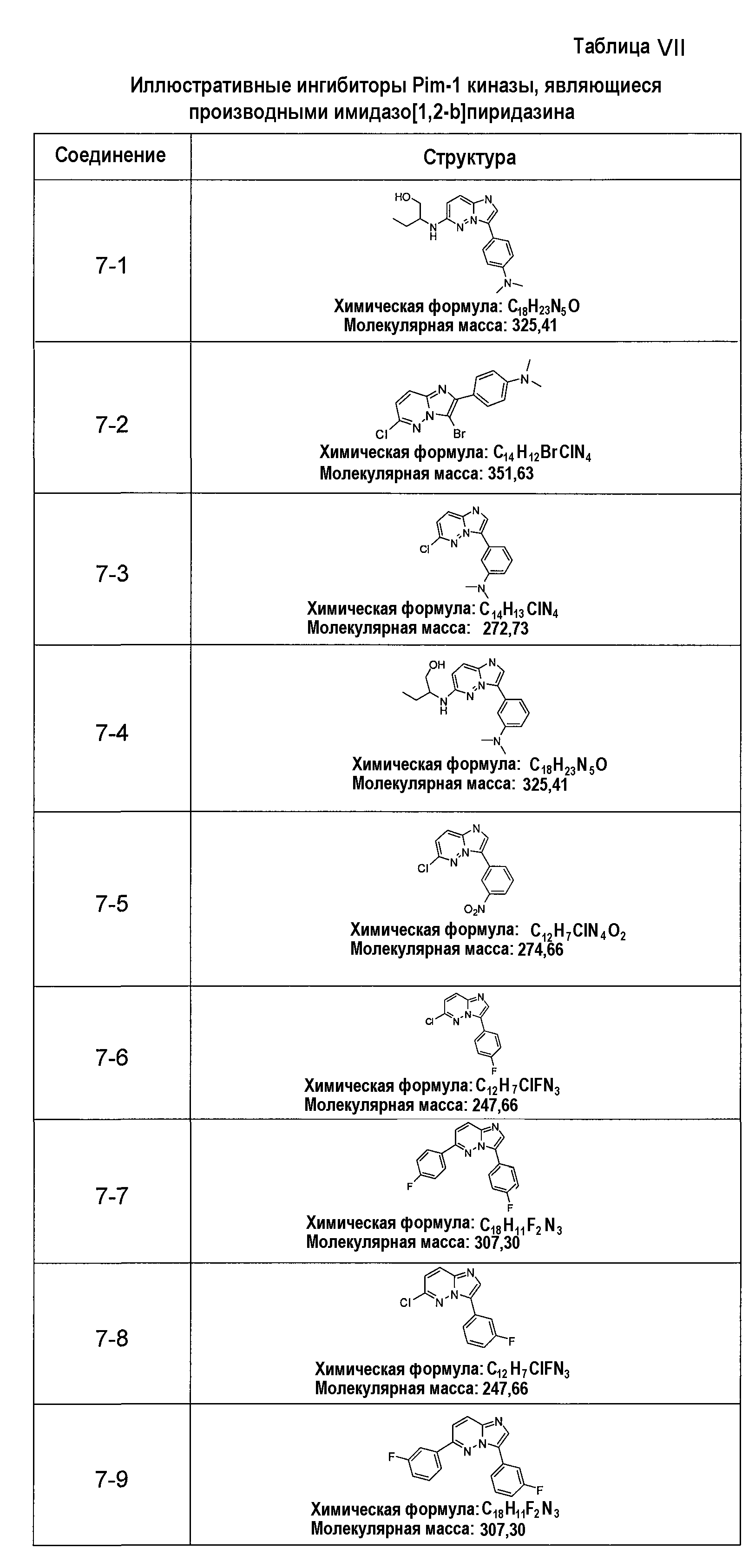
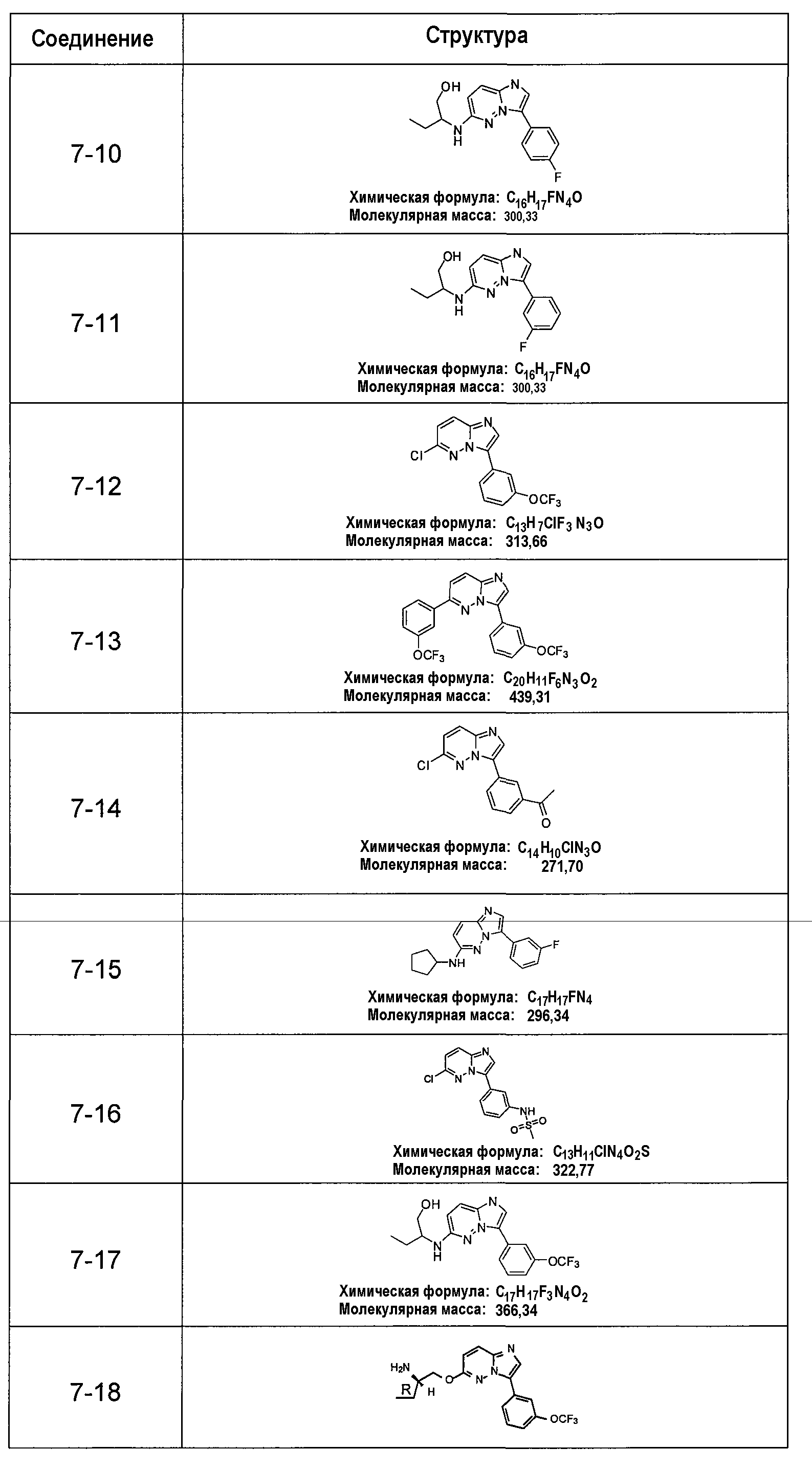
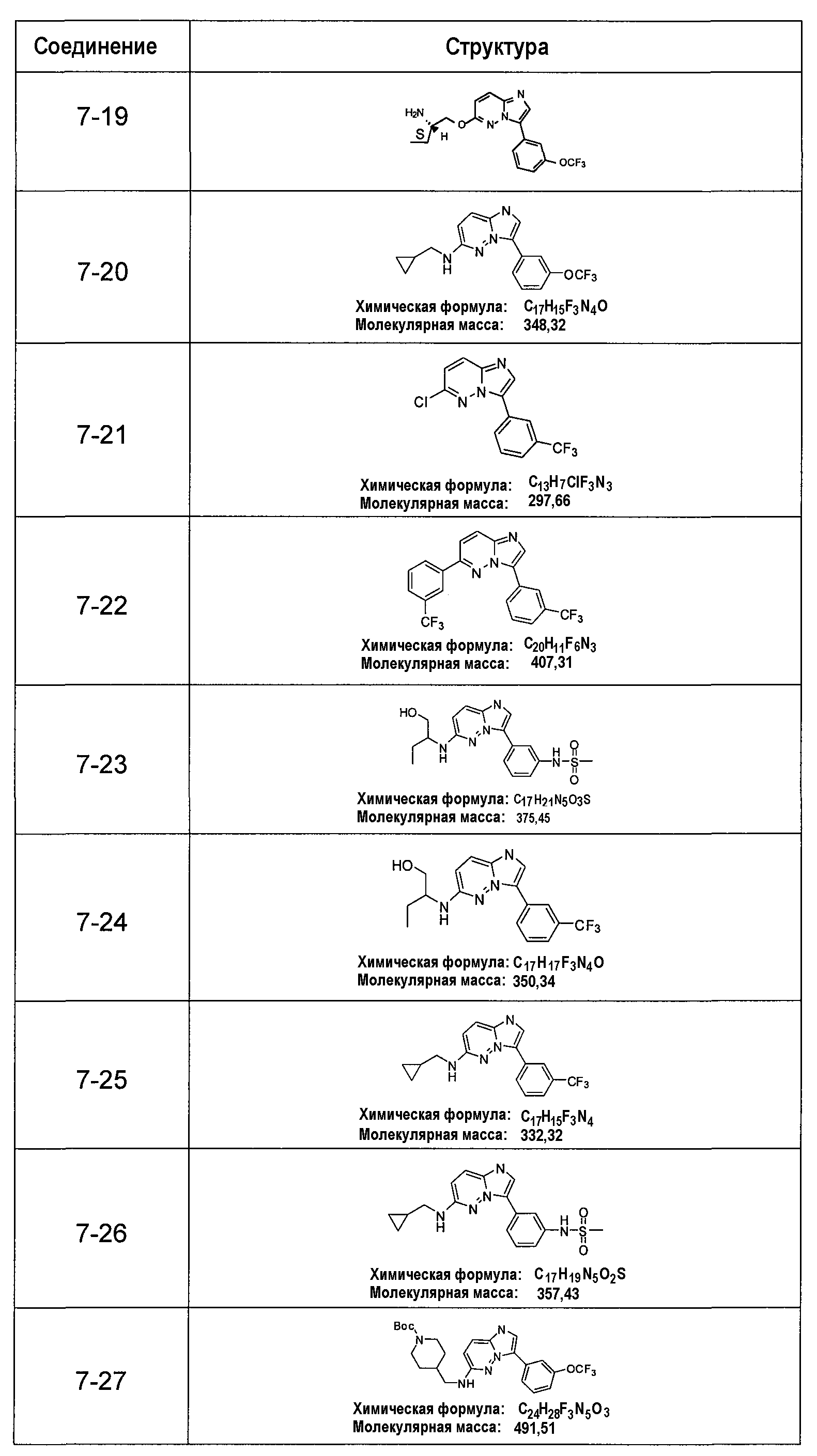
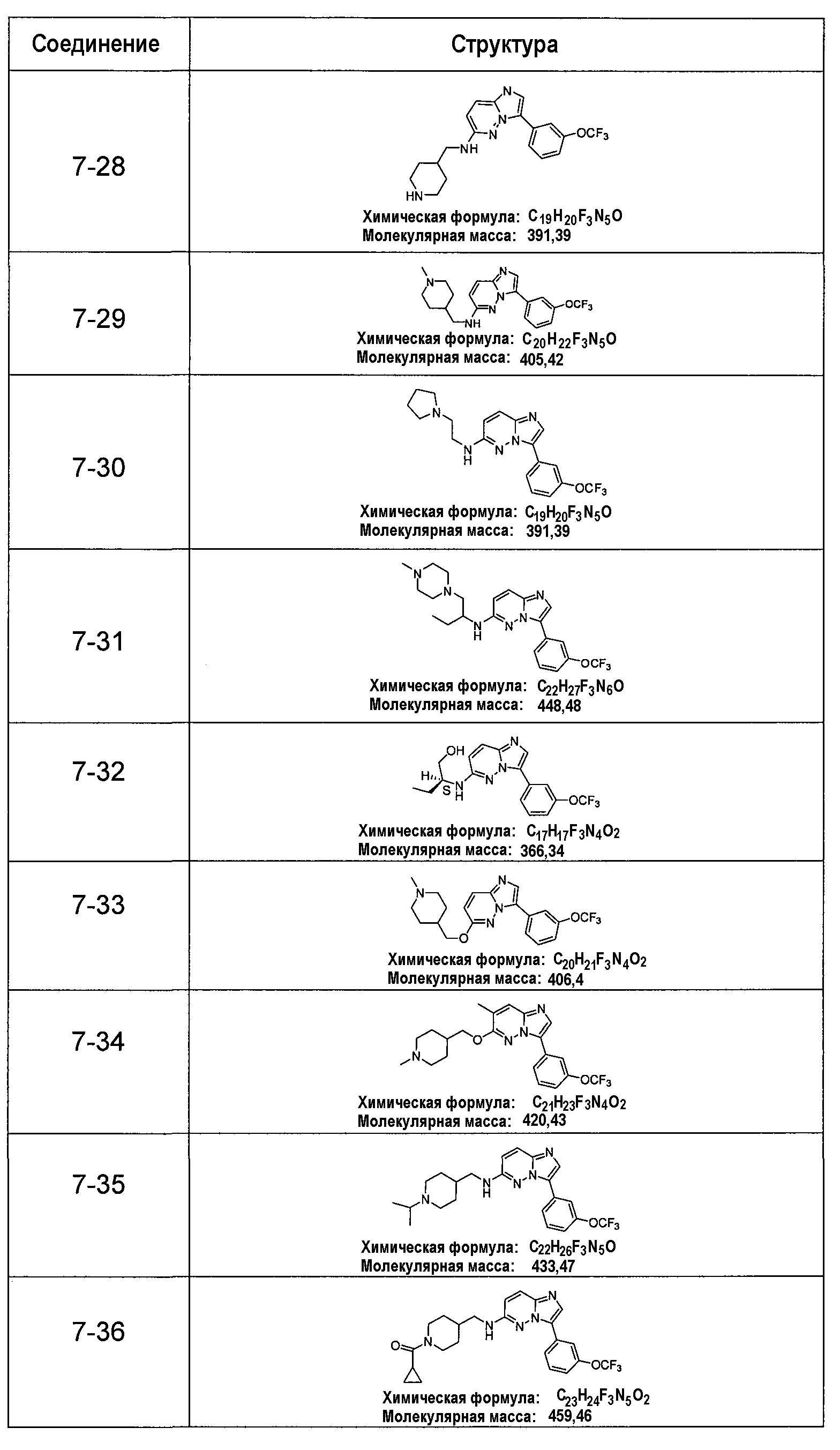
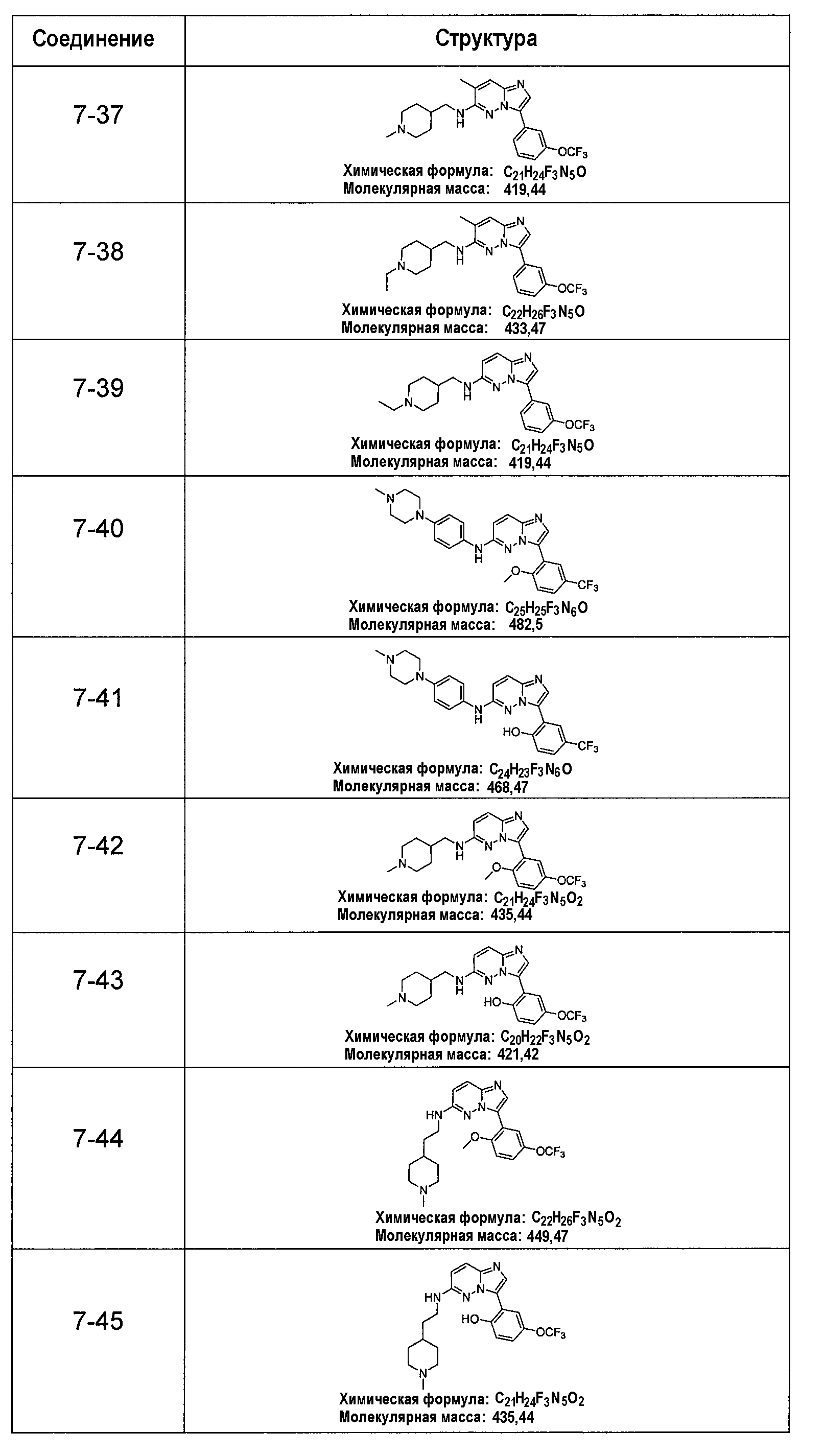
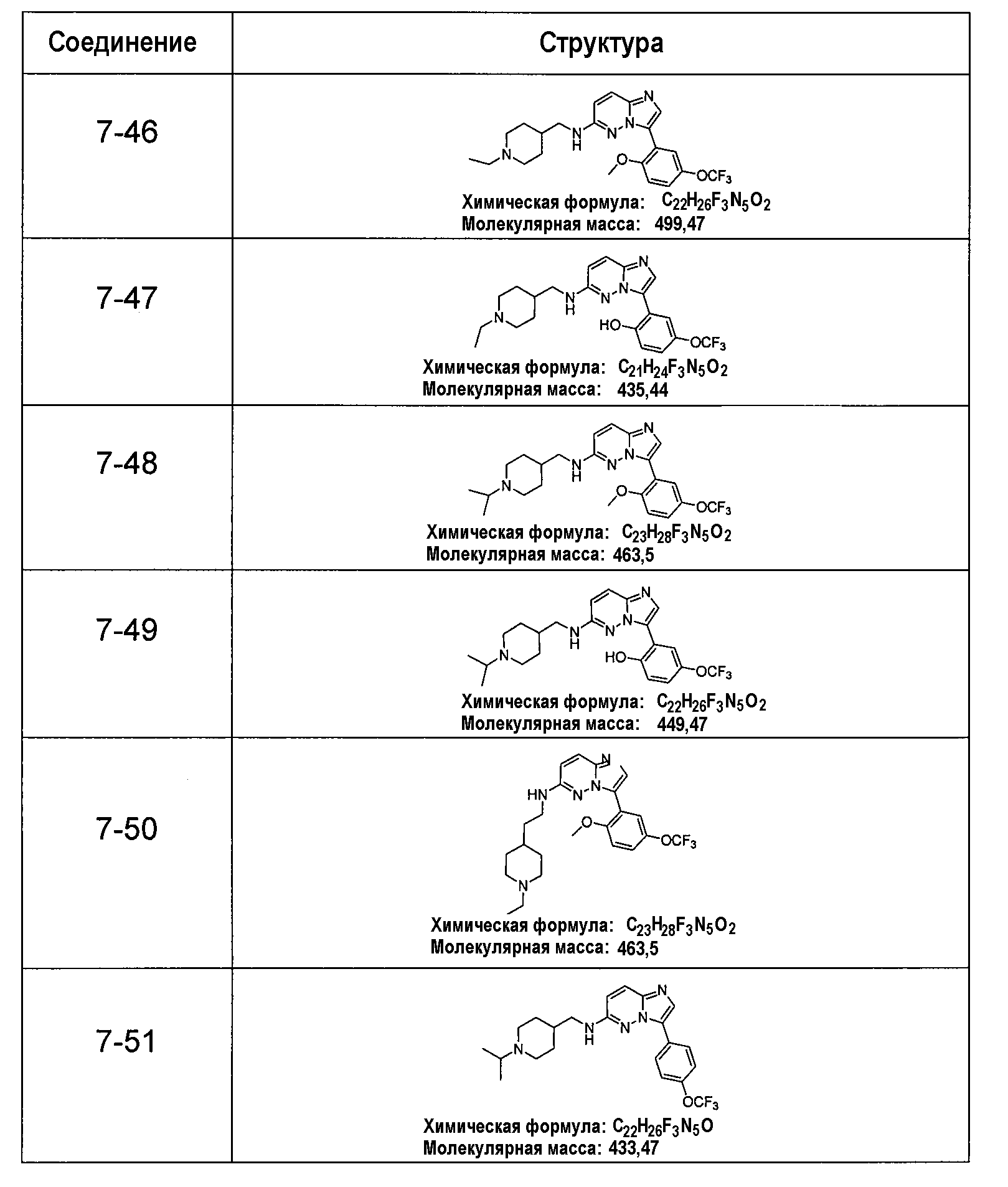
Пример 7
Исследование активности PIM-1 киназы
Значения IC50 определяли для иллюстративных соединений (например, показанных в таблице VII), используя аналитический набор Promega Kinase-Glo, и результаты анализов приведены ниже в таблице VIII. Кроме того, иллюстративные соединения исследовали на активность в отношении клеточных культур, экспрессирующих Pim-1 киназу. Значения IC50, соответствующие концентрациям, требуемым для ингибирования роста клеток на 50% по сравнению с необработанным контролем, приведены в мкМ ниже в таблице IX. Таким образом, по данным нескольких методик исследования данные иллюстративные соединения являются активными ингибиторами Pim-1 киназы и способны ингибировать рост опухолевых клеток.

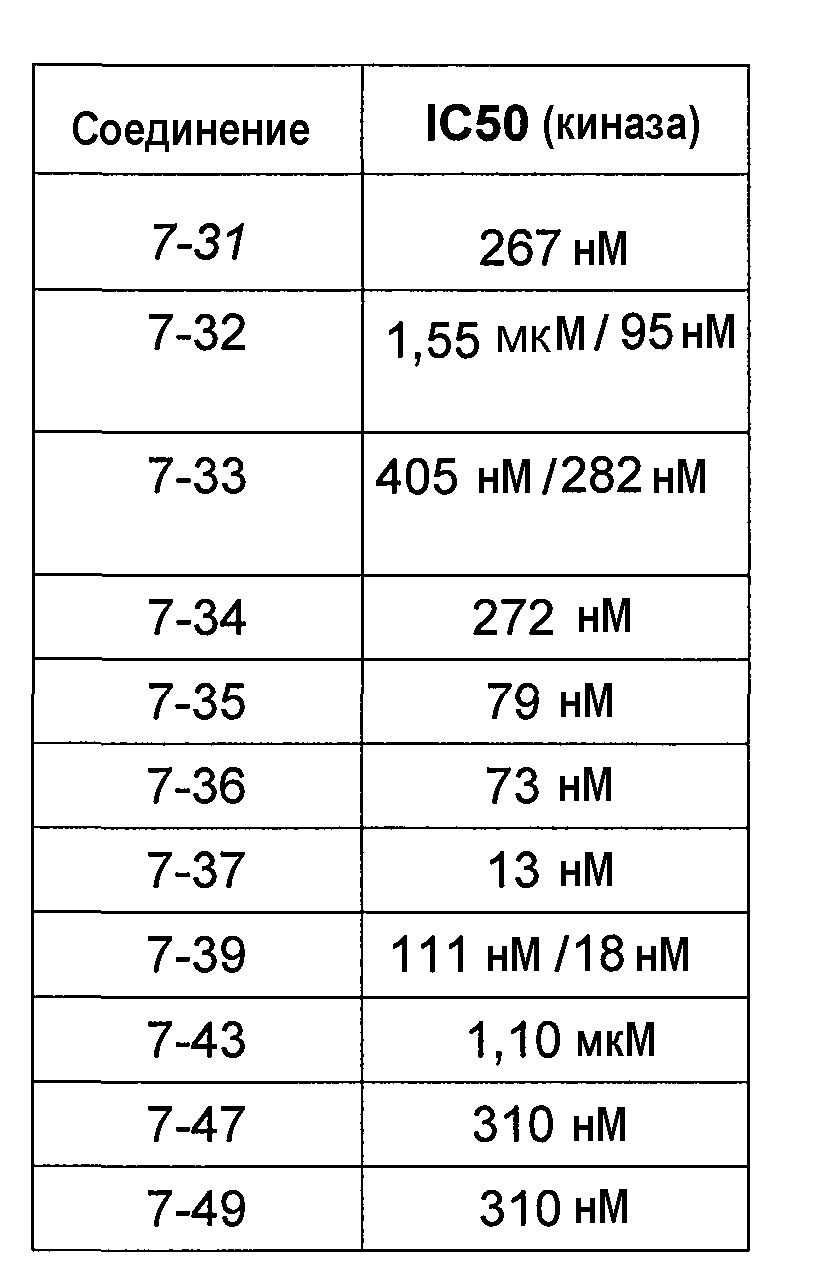
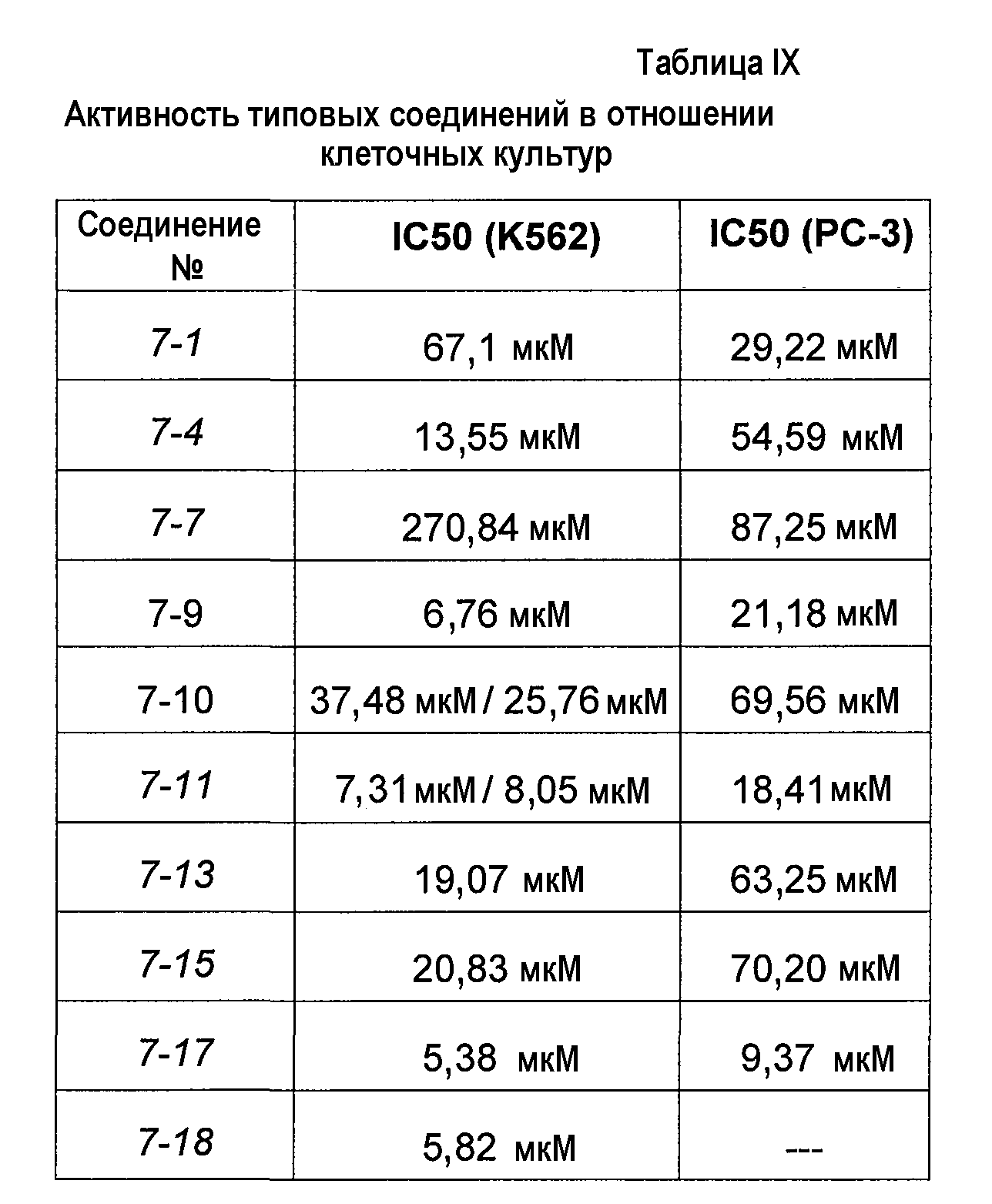
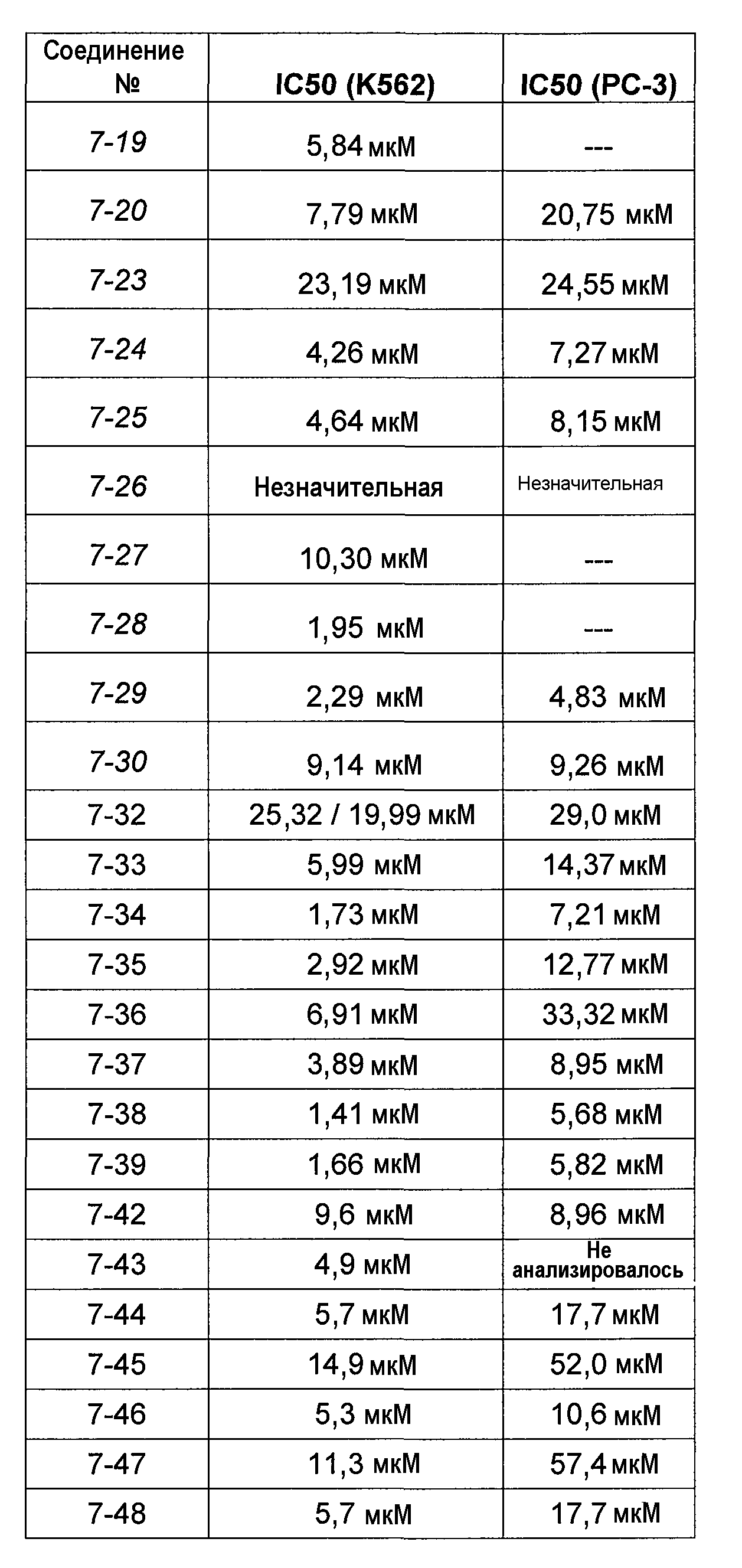
Пример 8
Селективность соединения 7-29 в отношении отдельных протеинкиназ
Соединение 7-29 (таблица VII) оценивали с точки зрения селективности в отношении панели Ser/Thr и тирозинкиназ в радиометрическом анализе при концентрации 1 мМ. Результаты показаны на фиг.2. Что касается протестированных Ser/Thr киназ, соединение 7-29 продемонстрировало >100-кратную селективность в отношении Pim-1 киназы, по сравнению с другими протестированными киназами. Однако, кроме того, соединение 7-29 продемонстрировало селективность в отношении Flt3, Mek1 и TrkA. Это соединение не продемонстрировало значительной активности в отношении панели других Ser/Thr киназ, включая Aurora-A, CDK1, CDK2, Plk3 и Nek2 и тирозинкиназ, включая Able, c-Kit, EGFR и Jak2.
Пример 9
Этот пример демонстрирует ингибиторную активность в отношении Pim-1 киназы для гидрохлоридов иллюстративных соединений 7-19, 7-29 и 7-31.
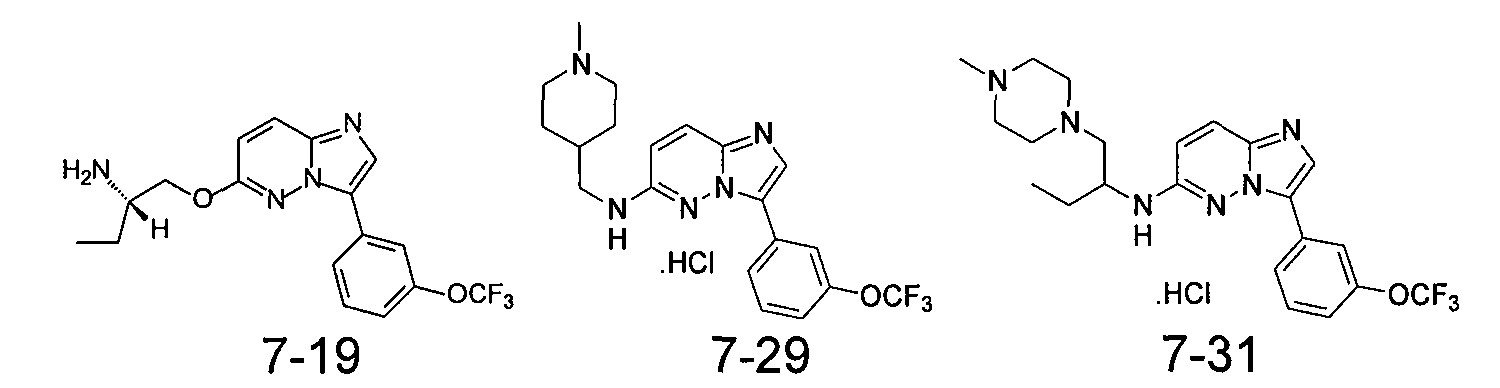
Один иллюстративный способ определения влияния ингибиторов Pim-1 на активность киназы Pim-1 заключается в измерении уровней фосфорилирования белка Bad по остатку серина 112 (S112) (фосфо-Bad или pBad). Известно, что Pim-1 вызывает фосфорилирование Bad по остатку S112 для того, чтобы инактивировать белок и ингибировать его связывание с членами антиапоптического семейства Bcl-2, тем самым позволяя этим членам семейства Bcl-2 дополнительно ингибировать апоптические сигналы.
Вкратце, клетки MV-4-11 (бифенотипической миеломоцитарной лейкемии B) высевали в колбы T25 в не содержащей сыворотку среде (SFM) в количестве 1,5×105 клеток/мл и давали расти в течение 24 часов. Через 24 часа в отдельные колбы добавляли ингибиторы Pim-1 в концентрациях 10, 5, 1, 0,5, 0,1 или 0,01 мкМ. Обработку ингибиторами Pim-1 продолжали в течение 1 часа. После часовой обработки клетки собирали и из каждого образца получали клеточные лизаты. Эквивалентные общие количества белка из лизатов наносили на 10% трис-глициновый гель (Invitrogen) для анализа SDS-PAGE. После разделения белков с помощью SDS-PAGE их переносили на мембраны из нитроцеллюлозы для вестерн-блоттинга. Первичные антитела для фосфо-Bad (S112) (Cell Signaling Technologies) применяли для определения уровней фосфо-Bad (S112). Для расчета EC50 ингибиторов фосфорилирования Bad (S112), определяли общее содержание белка Bad. Для этого из исходного вестерн-блота удаляли антитела и вводили новые зонды, используя антитела, которые распознают белок Bad (Cell Signaling Technologies), в независимости от статуса фосфорилирования белка. Для количественного определения содержания каждой полосы вестерн-блота применяли денситометрию и полученные данные использовали для определения EC50 ингибиторов Pim-1 по изменению содержания белка фосфо-Bad.
На фиг.3-5 показаны результаты фосфо-Bad окрашивания клеток MV-4-11, обработанных соединениями 7-19, 7-29 и 7-31 соответственно. После 1 ч обработки ингибиторами Pim-1 уровни pBad уменьшались в зависимости от дозы, демонстрируя почти полное отсутствие pBad при наиболее высоком содержании ингибитора. Общее содержание Bad было сходным во всех тестовых группах. Было определено, что значения EC50 для соединений 7-19, 7-29 и 7-31 составляют 635 нМ, 7,9 нМ и 57,4 нМ, соответственно.
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/4067eaa9435cbf1b2bcd08285445560d.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/0e58d99d8790b3818fdf94fc78c3fdc4.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/bf0a09dc0bde6e083556376179479ae6.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/223cb1d4e74fdd2e9ecda7732d66a8a8.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/5608c2d0c4d0eb1e87197d43c1aaa1d7.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/8e9b0c87ffef2081c0bd5cc99b7e9372.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/cd0045c7cf727de718b5a0f276a65c13.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/700809fbaeb83c2e159c871b6256130c.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/1ba007ae7193a8914738e7dabc041ec1.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/9ba6bf08e37c1821a60aa96f9d14fa90.png)
![ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ](https://fips.edrid.ru/images/rid/5f/39/be/d7f4abc9aac1045a1b9fd27f1feac5b3.png)
Композиция для ухода за полостью рта
Композиция для ухода за полостью рта
Ингибиторы jak2 и alk2 и способы их использования
Пролекарства альвоцидиба, имеющие повышенную биодоступность

