Результат интеллектуальной деятельности: СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ
Вид РИД
Изобретение
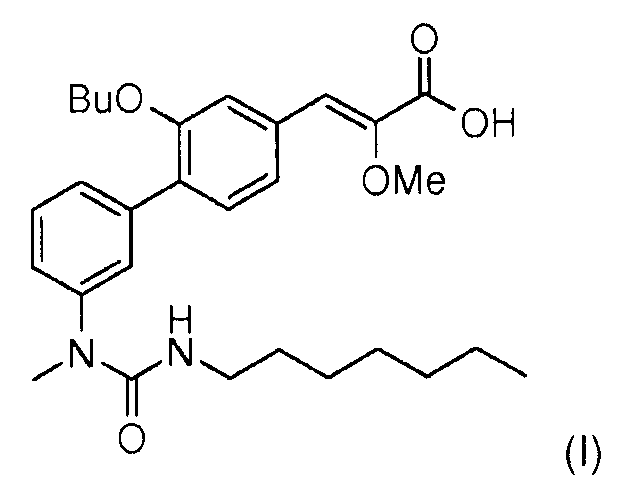
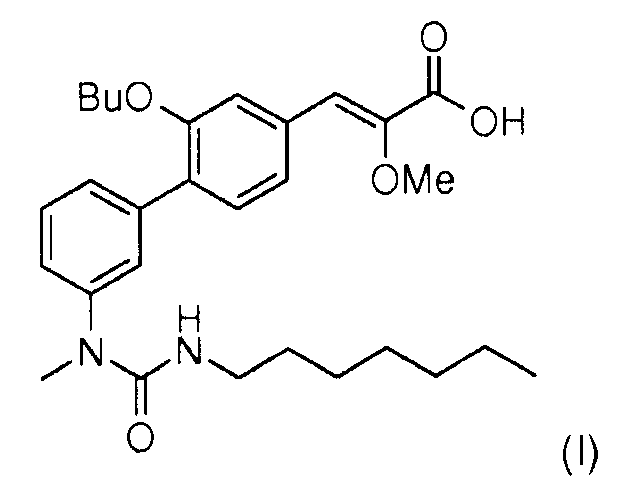
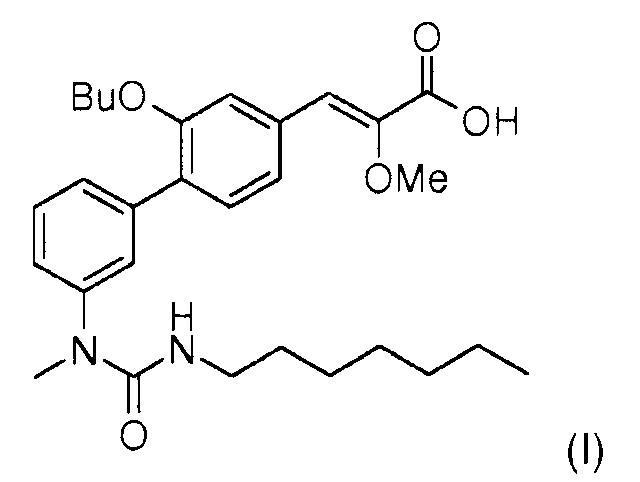
Настоящее изобретение относится к новому способу синтеза (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты формулы (I),
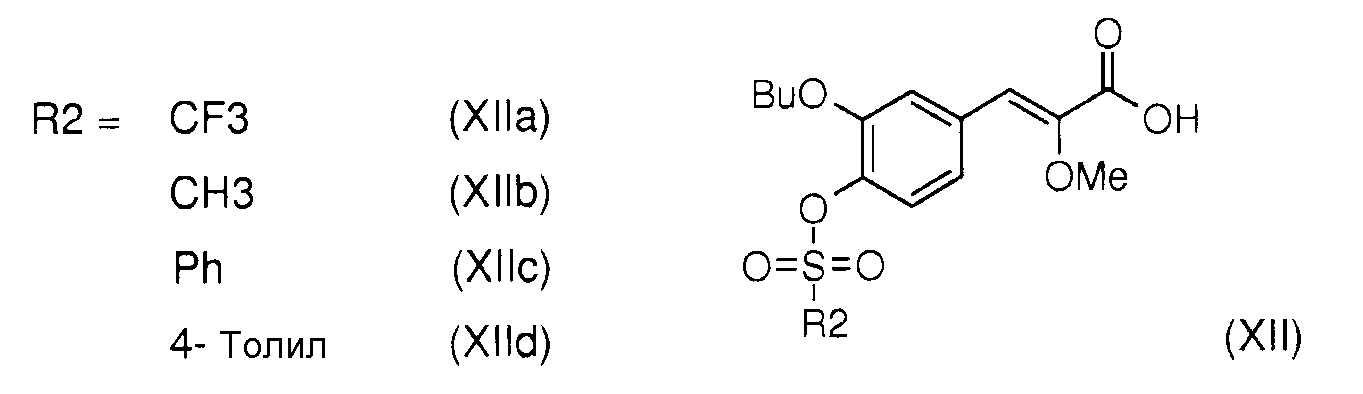
через промежуточный продукт реакции общей формулы (XII),
(Z)-3-[2-Бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловая кислота формулы (I) является модулятором рецепторов PPARs (рецепторы, активирующие пролиферацию пероксисом) (англ. Peroxisome proliferating activated receptors).
Синтез соединения формулы (I) описан 8 стадиями в международной заявке WO2007/049158, представленной на фиг.1:
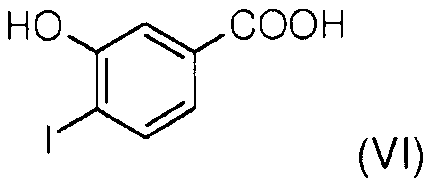
Стадия 1: 3-гидроксибензойную кислоту йодируют в присутствии йодида натрия и гипохлорита натрия, получая 4-йодо-3-гидроксибензойную кислоту (VI).
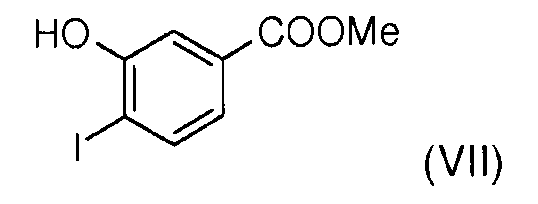
Стадия 2: 4-йодо-3-гидроксибензойную кислоту (VI) подвергают этерификации метанолом в присутствии серной кислоты для получения метил 3-гидрокси-4-йодобензоата (VII).
Стадия 3: Взаимодействие метил 3-гидрокси-4-йодобензоата (VII) с бутилйодидом в присутствии карбоната калия позволяет получить метил 3-бутокси-4-йодобензоат (VIII).
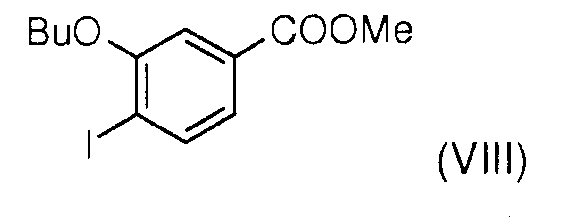
Стадия 4: Соединение (VIII) далее восстанавливают боргидридом лития для получения (3-бутокси-4-йодофенил)метанола (IX).
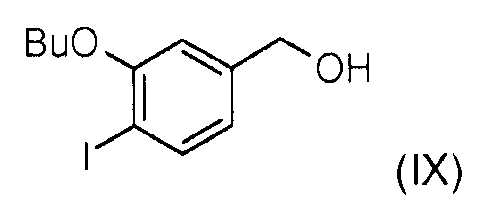
Стадия 5: Окисление (3-бутокси-4-йодофенил)метанола (IX) диоксидом марганца позволяет получить 3-бутокси-4-йодобензальдегид (Х).
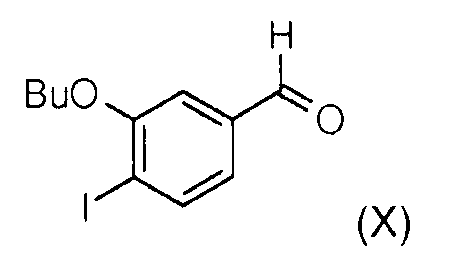
Стадия 6: Реакция Виттига между 3-бутокси-4-йодобензальдегидом (Х) и (диэтоксифосфорил)метоксиметилацетатом (XI) в присутствии гидрида натрия позволяет получить смесь метил (Z)-3-(3-бутокси-4-йодофенил)-2-метоксиакрилата (II) и метил (Е)-3-(3-бутокси-4-йодофенил)-2-метоксиакрилата (II') в соотношении 60/40. Чистый метил (Z)-3-(3-бутокси-4-йодофенил)-2-метоксиакрилат (II) получают после хроматографии на колонке с силикагелем.
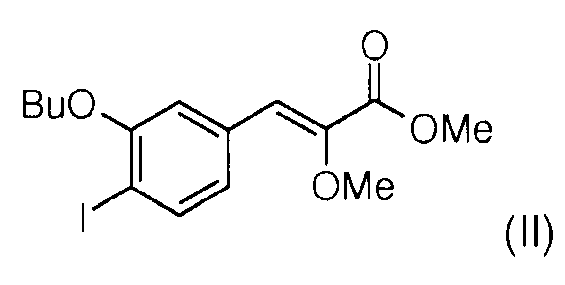
Стадия 7: Реакция сочетания Сузуки, катализируемая палладием, проводилось между метил (Z)-3-(3-бутокси-4-йодофенил)2-метоксиакрилатом (II) и 3-(1-метил-3-пентилуреидо)фенилбороновой кислотой (III) для получения метил (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакрилата (IV)
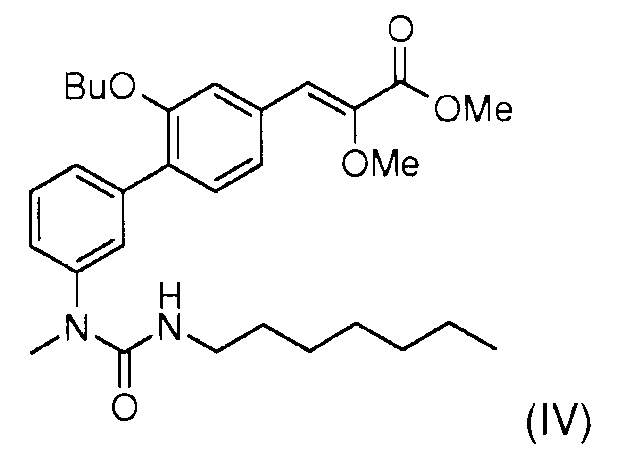
Стадия 8: Омыление метил (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакрилата (IV) гидроксидом натрия позволяет получить (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловую кислоту (I).
Первым недостатком данного способа синтеза является использование длинной линейной последовательности реакций, состоящей из 8 стадий, между коммерческим соединением (V) и (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислотой (I). Линейный синтез всегда невыгоден при проведении его в промышленном масштабе, где предпочтителен конвергентный синтез, в частности, из-за стремления добиться большего выхода.
С другой стороны, использование диоксида марганца также является недостатком из-за промышленных отходов, выделяемых им.
Наконец, разделение двух изомеров (Z) и (Е) метил 3-(3-бутокси-4-йодофенил)-2-метоксиакрилата на стадии 6, при котором используется хроматография на колонке с силикагелем, трудно осуществлять в промышленном масштабе.
Кроме того, метил (Z)-3-(3-бутокси-4-йодофенил)-2-метоксиакрилат (II), промежуточный продукт в данном способе, представляет собой бесцветное масло, что может создавать проблемы разделения при осуществлении в промышленном масштабе.
Наконец очистка (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты (I) в вышеописанном способе синтеза делает необходимым осуществление трех хроматографических разделений на колонке с силикагелем.
Настоящее изобретение нацелено на решение вышеперечисленных проблем, и предоставляет упрощенный способ синтеза (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты формулы (I), содержащий меньше стадий, более экономичный и подходящий для применения в промышленном масштабе.
Действительно, новый способ синтеза (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты (I), объект настоящего изобретения, является конвергентным синтезом, содержащим максимум одну линейную последовательность из 4 стадий, как показано на фиг.2.
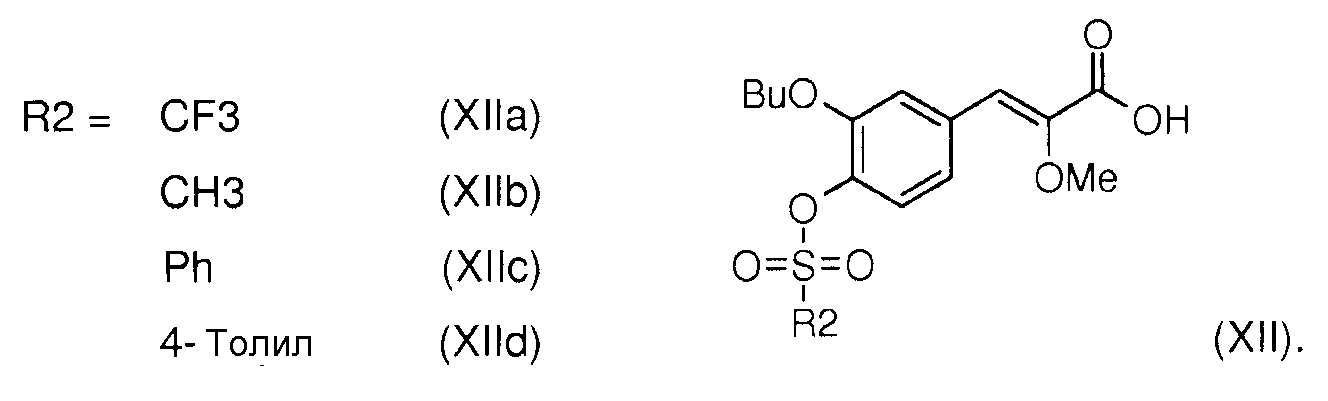
Данный новый способ позволяет также получить в 3 стадии (Z)-3-(-3-бутокси-4-трифторметансульфонилоксифенил)2-метоксиакриловую кислоту формулы (XIIa) как промежуточный продукт реакции получения (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты (I).
Стадии способа по изобретению являются следующими:
Стадия 1 способа по изобретению:
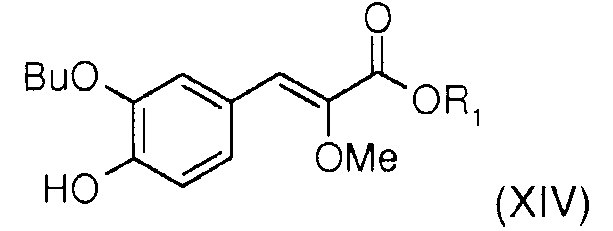
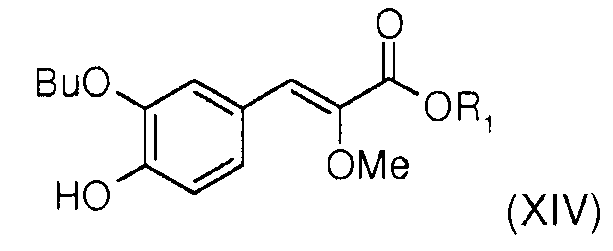
На первой стадии в новом способе синтеза прибегают к реакции Виттига между (метоксиалкоксикарбонилметил)трифенилфосфонийгалогенидом (XVII) [полученным способом, описанным в Tetrahedron 1994, 50, 7543-7556] и 3-бутокси-4-гидроксибензальдегидом (XV) [полученным сочетанием коммерческого 3-бром-4-гидроксибензальдегида (XVI) и бутаноатом щелочного металла, такого как натрий, например, в присутствии соли меди (I) или комплекса палладия в апротонном полярном растворителе, таком как диметилформамид (ДМФА), например, при температуре, составляющей от 60°С до 150°С (в издании SVnth Commun 1990, 20, 2659 описаны подобные реакции)] в присутствии органического амина, такого как тетрагидрофуран (ТГФ), например, что позволяет получить алкил (Z)-3-(3-бутокси-4-гидроксифенил)-2-метоксиакрилат общей формулы (XIV), где R1 представляет собой алкильный радикал. Этот способ позволяет получить в основном изомер (Z) (>95%), благодаря чему, в отличие от способа синтеза, описанного в международной заявке WO2007/049158, можно полностью избавиться от операции разделения изомеров Z/Е на колонке с силикагелем.
Стадия 2 способа по изобретению:
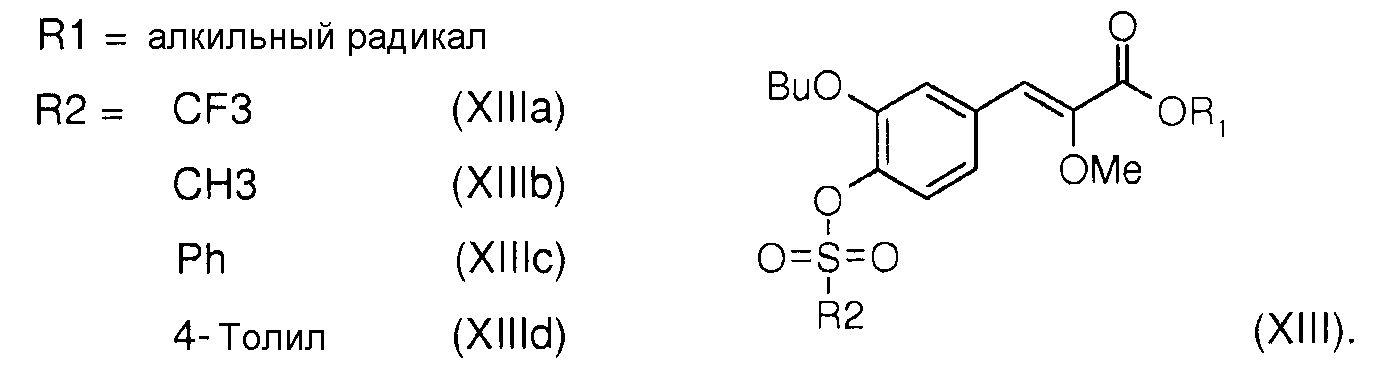
Алкил (Z)-3-(3-бутокси-4-гидроксифенил)-2-метоксиакрилат (XIV) подвергают реакции с трифлик ангидридом, например, или мезилхлоридом, или бензолсульфонилхлоридом, или тозилхлоридом в присутствии органического основания, такого, например, как дихлорметан, для получения, соответственно, алкил (Z)-2-метокси-3-(3-бутокси-4-фторметансульфонилоксифенил)акрилата (XIIIa), или алкил (Z)-2-метокси-3-(3-бутокси-4-метансульфонилоксифенил)акрилата (XIIIb), или алкил (Z)-2-метокси-3-(3-бутокси-4-фенилсульфонилоксифенил)акрилата (XIIIc), или алкил (Z)-2-метокси-3-(3-бутокси-4-толилсульфонилоксифенил)акрилата (XIIId):
Стадия 3 способа по изобретению:
Омыление сложного эфира (XIII), например, алкил (Z)-2-метокси-3-(3-бутокси-4-трифторметансульфонилоксифенил)акрилата (XIIIa) основанием, таким как гидроксид лития, например, в растворителе, таком как ТГФ, например, в присутствии воды позволяет получить (Z)-3-(3-бутокси-4-трифторметансульфонилоксифенил)-2-метоксиакриловую кислоту (XIIa).
Таким же образом омыление сложных эфиров XIIIb, XIIIc или XIIId приводит, соответственно, к кислотам (XIIb), (XIIc) или (XIId).
Стадия 4 способа по изобретению:
Стадия 4 способа, являющегося объектом настоящего изобретения, состоит в синтезе (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты (I) путем сочетания, предпочтительно катализируемого палладием или никелем, например, (Z)-3-(3-бутокси-4-трифторметансульфонилоксифенил)-2-метоксиакриловой кислоты (XIIa) с 3-гептил-1-метил-1-[3-(4,4,5,5-тетраметил-[1,3,2]-диоксаборолан-2-ил)-фенил]мочевиной.
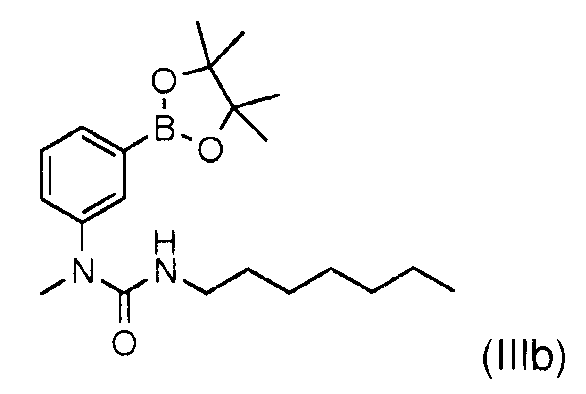
или с соответствующей бороновой кислотой (IIIa)
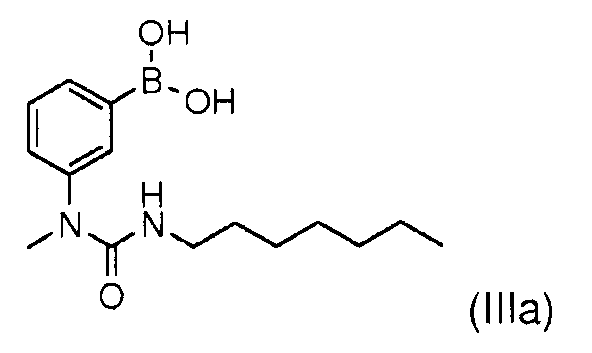
Соединение (IIIb) может быть получено, в частности, как описано в международной заявке WO 2007/049158, а именно:
а- 1-(3-бромфенил)-3-гептил-1-метилмочевина.
Получают (3-бромфенил)1-метиламин следующим образом:
150 г (500 ммоль) трет-бутил (3-бромфенил)-N-метилкарбамата вносят в 600 мл дихлорметана и 383 мл (5 моль) трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, затем обрабатывают насыщенным водным раствором карбоната натрия и экстрагируют дихлорметаном. Органическую фазу промывают водой, сушат над сульфатом магния, фильтруют и упаривают. Получают 99 г (100%) (3-бромфенил)метиламина.
Затем:
3,2 мл (20 ммоль) гептилизоцианата добавляют к раствору 2,5 г (13 ммоль) (3-бромфенил)метиламина, полученного вышеуказанным способом, в 10 мл тетрагидрофурана в присутствии 2 мл триэтиламина. Реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Реакцию останавливают путем добавления 2 мл воды, затем смесь экстрагируют этилацетатом. Органические фазы собирают и сушат над сульфатом натрия. Растворители выпаривают, затем осадок очищают хроматографией на колонке с силикагелем, элюируя смесью гептан/этилацетат 70/30. Получают 3,4 г (77%) 1-(3-бромфенил)-3-гептил-1-метилмочевины в виде твердого вещества.
b- 3-гептил-1-метил-1-[3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]мочевина:
4,0 г (15,5 ммоль) бис-пинакон диборана добавляют к смеси 3,4 г (10 ммоль) 1-(3-бромфенил)-3-гептил-1-метилмочевины, 3,0 г (31 ммоль) ацетата калия в присутствии 380 мг (0,5 ммоль) дифенилфосфино-ферроцена дихлорида палладия в 15 мл диметилформамида. Смесь перемешивают в течение 3 часов при 90°С. Реакцию останавливают путем добавления 50 мл воды, затем экстрагируют этилацетатом. Органические фазы собирают и сушат над сульфатом натрия. Растворители выпаривают, затем остаток очищают хроматографией на колонке с силикагелем, элюируя смесью гептан/этилацетат 70/30. Получают 2,5 г (64%) 3-гептил-1-метил-1-[3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]мочевины (IIIb) в виде масла.
Само соединение (IIIa) получают так, как описано в международной заявке WO2007/049158, а именно:
а- 1-(3-бромфенил)-3-гептил-1-метилмочевина.
Методика идентична описанной выше.
b- 3-(3-гептил-1-метилмочевина)фенилбороновая кислота.
Взаимодействием 113 г (345 ммоль) 1-(3-бромфенил)-3-гептил-1-метилмочевины в 1,1 л тетрагидрофурана, 127 мл (380 ммоль) раствора метиллития 1,6М в диэтиловом эфире, 530 мл (760 ммоль) раствора 1,7М трет-бутиллития в пентане и 97 мл (904 ммоль) триметилборана, получают 36 г (36%) 3-(3-гептил-1-уреидо)фенилбороновой кислоты (IIIa) в виде розового порошка после очистки сырого осадка хроматографией на колонке с силикагелем, элюируя смесью гептан/этилацетат 50/50 и перекристаллизации из смеси этилацетат/гептан.
Таким же образом реакция сочетания кислот (XIIb), (XIIc) или (XIId) (кислоты формулы (XII), в которых R2 представляет собой метил, фенил или толил) приводит к получению (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты формулы (I).
(Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловую кислоту формулы (I) очищают перекристаллизацией из диизопропилового эфира способом, используемом в промышленном масштабе.
Условия сочетания на данной последней стадии известны специалисту в данной области. В рамках настоящего изобретения специалист может использовать как обычные условия сочетания (см. A.Suzuki et al., SVnth. Commun. 1981, 11, 513 или Sharp, M.J. Tet. Lett. 1985, 26, 5997), так и оптимизированные условия (см., например, Littke, A.F. et al, J. Am. Chem. Soc. 2000, 122(17), 4020-4028). Способ реализации, описанный ниже, включает в себя не ограниченный выбор условий.
Согласно настоящему изобретению под алкилом подразумевают насыщенную углеводородную цепь, линейную или разветвленную, содержащую от 1 до 4 атомов углерода. Предпочтительно такой радикал выбирают из радикалов метил, этил, пропил, бутил, изо-пропил и трет-бутил.
Согласно настоящему изобретению под алкокси подразумевают атом кислорода, замещенный насыщенной углеводородной цепью, линейной или разветвленной, содержащей от 1 до 4 атомов углерода.
Согласно настоящему изобретению под алкоксакарбонилом подразумевают карбонил, замещенный алкокси, значение которого определено ранее.
Согласно настоящему изобретению под галогеном подразумевают атом хлора, фтора, брома или йода.
Настоящее изобретение относится также к различным промежуточным продуктам реакции, используемым в способе по изобретению, а именно:
- сульфонаты общей формулы (XII), в которых R2 выбирают из метила, трифторметила, фенила и толила, представленные (Z)-3-(3-бутокси-4-трифторметансульфонилоксифенил)-2-метоксиакриловой кислотой (XIIa), (Z)-3-(3-бутокси-4-метансульфонилоксифенил)-2-метоксиакриловой кислотой (XIIb), (Z)-3-(3-бутокси-4-фенилсульфонилоксифенил)-2-метоксиакриловой кислотой (XIIc) и (Z)-3-(3-бутокси-4-толилсульфонилоксифенил)-2-метоксиакриловой кислотой (XIId).
- сульфонаты общей формулы (XIII), в которых R1 означает алкил, а R2 означает метил, трифторметил, фенил или толил, представленные алкил (Z)-2-метокси-3-(3-бутокси-4-трифторметансульфонилоксифенил)акрилатом (XIIIa), алкил (Z)-2-метокси-3-(3-бутокси-4-метансульфонилоксифенил)акрилатом (XIIIb), алкил (Z)-2-метокси-3-(3-бутокси-4-фенилсульфонилоксифенил)акрилатом (XIIIc) и алкил (Z)-2-метокси-3-(3-бутокси-4-толилсульфонилоксифенил)акрилатом (XIIId).
- алкил (Z)-3-(3-бутокси-4-гидроксифенил)-2-метоксиакрилат (XIVc), где R1 означает алкил:
Другие характеристики и преимущества настоящего изобретения будут выявлены при знакомстве с методиками, приведенными ниже для иллюстрации и не имеющими ограничительный характер.
ПОЛУЧЕНИЕ ИСХОДНЫХ СОЕДИНЕНИЙ
3-бутокси-4-гидроксибензальдегид (XV)
2,16 г (93,9 ммоль) натрия растворяют в 15 мл н-бутанола при 110°С в течение 3 часов. Добавляют 20 мл диметилформамида при комнатной температуре, затем реакционную среду несколько раз дегазируют. Добавляют 3,4 г (34,1 ммоль) хлорида меди при комнатной температуре, затем реакционную среду перемешивают в течение 10 минут. Добавляют 6,2 г (31 ммоль) 3-бром-4-гидроксибензальдегида, затем реакционную смесь перемешивают в течение 2 часов при 120°С. Реакционную смесь охлаждают до комнатной температуры, затем реакцию останавливают путем добавления 50 мл раствора соляной кислоты 2М. Реакционную смесь экстрагируют 250 мл этилацетата. Растворители выпаривают, затем осадок фильтруют через слой силикагеля (2 см): элюент (гептан/этилацетат 8/2). После выпаривания растворителей получают 5,5 г оранжевого масла. Данное масло перекристаллизовывают из пентана. После фильтрации получают 4,8 г 3-бутокси-4-гидроксибензальдегида в виде твердого коричневого осадка.
Выход=80%.
ЯМР 1Н (400 МГц, CDCl3): 0,99 (т, J=8Гц, 3H, CH3); 1,52 (гекс, J=8 Гц, 2H, CH2); 1,85 (пент, J=8Гц, 2H, CH2); 4,14 (т, J=8 Гц, 2H, CH2); 6,24 (с, 1H, ArH); 7,05 (д, J=8 Гц, 1H, ArH); 7,42 (м, 2H, ArH+OH); 9,83 (с, 1H, CHO).
(Метоксиметоксикарбонилметил)трифенилфосфонийхлорид (XVII)
25 г (0,186 моль) метил-диметоксиацетата добавляют к 27 мл (0,215 моль) ацетилхлорида при комнатной температуре. Добавляют 0,1 г (0,2 моль%) дийода, затем реакционную смесь перемешивают в течение 16 часов при 55°С. Избыток ацетилхлорида выпаривают в вакууме, затем осадок растворяют в 100 мл дихлорметана. Добавляют 49 г (0,204 моль) трифенилфосфина, далее реакционную смесь перемешивают при 37°С в течение 3 часов. Растворители выпаривают, затем осадок перекристаллизовывают из диизопропилового эфира. Получают 70 г (метоксиметоксикарбонилметил)трифенилфосфонийхлорида.
Выход = 88%.
ЯМР 1Н (400 МГц, CDCl3): 3,61 (с, 3H, OCH3); 3,90 (с, 3H, OCH3); 7,66 (м, 6H, ArH); 7,77 (м, 3H, ArH); 8,01 (м, 6H, ArH); 8,71 (д, J=13 Гц, 1H, CH).
СТАДИИ СПОСОБА
СТАДИЯ 1: Алкил (Z)-3-(3-бутокси-4-гидроксифенил)-2-метоксиакрилат
4,0 г (20,6 ммоль) 3-бутокси-4-гидроксибензальдегида и 13,25 г (30,9 ммоль) (метоксиметоксикарбонилметил)трифенилфосфония хлорида суспендируют в 40 мл тетрагидрофурана. Добавляют 6 мл (42 ммоль) триэтиламина, затем реакционную смесь перемешивают в течение 16 часов при 40°С. Реакционную смесь фильтруют, затем выпаривают растворители. Добавляют 50 мл диэтилового эфира, смесь перемешивают в течение 10 минут при комнатной температуре, далее фильтруют. После выпаривания растворителей выделяют осадок из смеси гептан/этилацетат 8/2, затем фильтруют через слой силикагеля (2 см). После выпаривания растворителей получают 5,3 г метил (Z)-3-(3-бутокси-4-гидроксифенил)-2-метоксиацетата.
Выход = 92%.
ЯМР 1Н (400 МГц, CDCl3): 0,91 (т, J=8 Гц, 3H, CH3); 1,42 (гекс, J=8 Гц, 2H, CH2); 1,75 (пент, J=8Гц, 2H, CH2); 3,66 (с, 3H, OCH3); 3,78 (с, 3H, OCH3); 4,00 (т, J=8Гц, 2H, CH2); 5,79 (с, 1H, CH=); 6,84 (д, J=8 Гц, 1H, ArH); 6,88 (с, 1H, ArH); 7,13 (д, J=8 Гц, 1H, ArH); 7,39 (с, 1H, OH).
СТАДИЯ 2: Метил (Z)-2-метокси-3-(3-бутокси-4-трифторметансульфонилоксифенил)акрилат (XIIIa)
2,6 г (9,27 ммоль) метил (Z)-3-(3-бутокси-4-гидроксифенил)2-метоксиакрилата растворяют в 25 мл дихлорметана, затем добавляют 2 мл триэтиламина. Медленно добавляют 1,67 мл трифлат-ангидрида (10,2 ммоль) при 0°С. Реакционную смесь перемешивают в течение 2 часов при 0°С. Реакцию останавливают добавлением насыщенного раствора гидрокарбоната натрия. Смесь экстрагируют этилацетатом. Органические фазы объединяют, сушат над сульфатом натрия, затем фильтруют через слой силикагеля (1 см), растворители выпаривают. Получают 3,57 г метил (Z)-2-метокси-3-(3-бутокси-4-трифторметансульфонилоксифенил)акрилата.
Выход = 93%.
ЯМР 1Н (400 МГц, CDCl3): 0,91 (т, J=7Гц, 3H, CH3); 1,46 (гекс, J=7 Гц, 2H, CH2); 1,76 (пент, J=7Гц, 2H, CH2); 3,73 (с, 3H, OCH3); 3,80 (с, 3H, OCH3); 4,00 (т, J=7Гц, 2H, CH2); 6,83 (с, 1H, CH=); 7,11 (д, J=8 Гц, 1H, ArH); 7,20 (д, J=8 Гц, 1H, ArH); 7,45 (с, 1H, ArH).
СТАДИЯ 3: (Z)-3-(3-бутокси-4-трифторметансульфонилоксифенил)-2-метоксиакриловая кислота (XIIa)
1,53 г (36,3 ммоль) гидроксида лития добавляют в раствор 7,5 г (18,2 ммоль) метил (Z)-2-метокси-3-(3-бутокси-4-трифторметансульфонилоксифенил)акрилата в 50 мл смеси 10/1 тетрагидрофуран/вода. Реакционную смесь перемешивают в течение 5 часов при 68°С. Реакционную смесь подкисляют раствором соляной кислоты 2N до рН=1. Реакционную смесь экстрагируют два раза 100 мл смеси гептан/этилацетат 1/2. Органические фазы собирают, затем сушат над сульфатом натрия. Растворители выпаривают, затем осадок выделяют из 30 мл пентана. Получают 4,7 г (Z)-3-(3-бутокси-4-трифторметансульфонилоксифефенил-2-метоксиакриловой кислоты в виде белого твердого вещества.
Выход = 63%.
ЯМР 1Н (400 МГц, CDCl3): 1,00 (т, J=7Гц, 3H, CH3); 1,56 (гекс, J=7 Гц, 2H, CH2); 1,86 (пент, J=7Гц, 2H, CH2); 3,86 (с, 3H, OCH3); 4,10 (т, J=7Гц, 2H, CH2); 7,07 (с, 1H, CH=); 7,23 (д, J=8 Гц, 1H, ArH); 7,33 (д, J=8 Гц, 1H, ArH); 7,56 (с, 1H, ArH).
СТАДИЯ 4: (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловая кислота (I)
870 мг (2,2 ммоль) (Z)-3-(3-бутокси-4-трифторметансульфонилоксифенил)-2-метоксиакриловой кислоты (XII), 981 мг (2,62 ммоль) (3-гептил-1-метил-1-[3-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]мочевины (III) [полученной согласно международной заявке WO2007/049158], 14 мг (3 моль%) ацетата палладия и 46 мг (6 моль%) дициклогексилбифенилфосфина растворяют в 8 мл диетилформамида, добавляют 1,5 мл раствора 2М фосфата калия, и смесь дегазируют несколько раз. Реакционную смесь перемешивают в течение 2 часов при 90°С. Реакцию останавливают путем добавления раствора соляной кислоты 2М, затем смесь экстрагируют этилацетатом. Органические фазы промывают раствором хлорида натрия, затем сушат над Na2SO4. Растворители выпаривают, затем осадок выделяют из 10 мл смеси гептан/AcOEt 1/1. Фильтруют через слой силикагеля (1 см). Выпаривание растворителей. Осадок выпадает из пентана. Твердое вещество перекристаллизовывают из смеси изопропиловый эфир/гептан. Получают 510 мг (Z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты в виде твердого вещества.
Выход = 47%.
Точка плавления = 99°С.
ЯМР 1Н (400 МГц, CDCl3): 0,85 (т, J=7Гц, 3H, CH3); 0,95 (т, J=7 Гц, 3H, CH3); 1,24 (м, J=8Гц, 2H, CH2); 1,42 (м, 4H, CH2); 1,78 (м, 2H, CH2); 3,18 (тд, J=7Гц, J=6Гц, 2H, NCH2); 3,33 (с, 3H, NCH3); 3,89 (с, 3H, OCH3); 4,05 (т J=7 Гц, 2H, OCH2); 4,46 (т, J=6 Гц, 1H, NH); 7,17 (с, 1H, =CH); 7,23 (д, J=8Гц, 1H, ArH); 7,35-7,54 (м, 6H, ArH).
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/55d13cc6cccee331be54618cb0bb598f.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/8bacb13550faa95992860361a2c5d0c6.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/ec5c8d19ebcb7f7d48f65a0418d5e6ad.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/263fb4a51ebefdd2a8cf6e9abd5ed2eb.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/e73a5f7b1ffb21a03859c4615043c906.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/4516e947601be9f057cfdcf65af43fd2.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/7c9fe35780b397ed7615b0418696eb3c.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/4a1897e8bbb0d99a1a79daf5a7ec7a60.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/25eeee4a9df26610d36ae1fb87e6587a.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/a5f27e444d7ad13a9d463108a8598dad.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/4eb17dbd102687a3bd7d25c896b9c8ea.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/b0e037de99ff548b9403a5f774307943.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/cdd0eb7493232f6bb3a70401466ca2be.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/71e04e4563c8376b1282204cf2232b10.jpg)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/95181c6a2adec6dc2c7a2a6e05dd511a.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/8e7659f5dc0c5e9d04b73120e27e232f.png)
![СПОСОБ СИНТЕЗА(Z)-3-[2-БУТОКСИ-3'-(3-ГЕПТИЛ-1-МЕТИЛУРЕИДО)БИФЕНИЛ-4-ИЛ]-2-МЕТОКСИАКРИЛОВОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/88/d5/41/94689ff1609ada077ff470b292727226.png)
Новые 4-(гетероциклоалкил)бензол-1,3-диольные соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека, а также в косметических средствах
Новые 4-(азациклоалкил)бензол-1,3-диоловые соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека и в косметических средствах
Способ получения адапаленовых гелей
Комбинация, включающая пирролидон-5-карбоновую кислоту и, по меньшей мере, одно соединение из цитруллина, аргинина и аспарагина, и их применение при лечении атопического дерматита
Применение адапалена и пероксида бензоила для продолжительного лечения угрей обыкновенных
Новые 4-(гетероциклоалкил)бензол-1,3-диольные соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека, а также в косметических средствах
Схема лечения заболеваний, связанных с акне
Новые 4-(азациклоалкил)бензол-1,3-диоловые соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека и в косметических средствах
Усовершенствованные способы и композиции для безопасного и эффективного лечения эритемы
Увлажняющий агент для кожи и его применение
Композиции, включающие по меньшей мере одно производное нафтойной кислоты, бензоилпероксид и по меньшей мере один пленкообразующий компонент, способы их получения и их применения
Новые бензолсульфонамидные соединения, способ их получения и применение в терапии и косметике

