СПОСОБ СИНТЕЗА ПОЛИГИДРОКСИСТИЛЬБЕНОВЫХ СОЕДИНЕНИЙ
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Объектом настоящего изобретения является новый способ синтеза полигидроксистильбеновых соединений.
Более конкретно, изобретение относится к способу синтеза ресвератрола и пицеатаннола.
Уровень техники
Полигидроксистильбены представляют собой соединения, которые встречаются в различных растениях и которые привлекли особое внимание, т.к. они проявляют многообразие терапевтических свойств.
Эти производные включают ресвератрол (E-3,5,4'-тригидроксистильбен) и пицеатаннол (E-3,5,3',4'-тетрагидроксистильбен) с формулами:
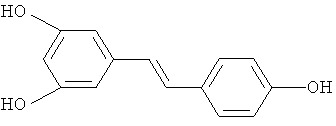
ресвератрол
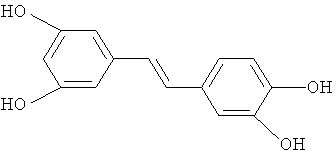
пицеатаннол
Ресвератрол и пицеатаннол являются соединениями, принадлежащими к классу полифенолов, которые, как известно, проявляют антиоксидантный эффект, допускающий предотвращение или замедление вредоносного воздействия окислительного стресса.
В терапевтической области ресвератрол числится как антиагрегант тромбоцитов, противовоспалительное или сосудорасширяющее средство или как ингибитор клеточной пролиферации.
Большинство путей, используемых для получения полигидроксистильбенов, требуют защиты фенольных функциональных групп в форме эфирных производных. Защиту наиболее часто получают с помощью метильной, метиленовой, изопропильной или бензильной групп. Для синтеза полигидроксистильбенов требуется, наконец, стадия снятия защиты для того, чтобы освободить фенольные функциональные группы. Стадию снятия эфирной защиты с фенолов обычно легко осуществляют в случае простых производных, таких как анизол, с помощью хлорида алюминия, как в J.Chem.Soc. (1944), 330. Однако эта реакция является сложной в случае, в частности, производных стильбена, благодаря присутствию двойной связи, и в особенности, если молекула имеет активирующие группы (иными словами, электронодонорные группы) в ароматических кольцах молекулы, такие как эфирные группы. Применение сильных кислот, таких как бромистоводородная кислота или хлорид алюминия, широко распространенная и наиболее дешевая из кислот Льюиса, приводит к значительному разложению молекулы во время этих реакций снятия защиты. В более конкретном случае бензиловых эфиров, снятие защиты осуществляют путем гидрогенолиза, который приводит, в конкретном случае стильбеновых производных, к гидрированию двойной связи.
Для того чтобы преодолеть эти недостатки, в случае этих стильбеновых производных в реакциях O-деметилирования или O-дебензилирования обычно используют трибромид бора, как в WO2003/086414, или трихлорид бора в присутствии тетрабутиламмонийиодида, как в Tetrahedron Lett., 44, 1 (2003), 193-98, и, в конкретном случае изопропильной группы, трихлорид бора в чистом виде, как в Tetrahedron, 59 (2003), 3315-21. Однако BCI3, а также BBr3 являются дорогими реагентами, которые опасно применять в промышленности.
В случае других реагентов, таких как метилмагнийиодид, как в J. Org. Chem., 62, 2 (1997), 417-21, или гидрохлорид пиридина, как в J. Agric. Food Chem., 47, 10 (1999), 3974-77, применяют большие количества реагентов и жесткие условия реакции (высокая температура) для выходов, которые являются обычно весьма средними.
Некоторые авторы используют хлорид алюминия в пиридине в качестве реагента и растворителя для того, чтобы получить ресвератрол при температурах 165-170°C, как в Патенте CN 1663939. Однако, в дополнение к весьма специфическим условиям реакции, этот растворитель является токсичным, и необходимо избегать его применения в промышленности. То же самое относится к реакции Akiyama et al., (конкретно, к реакциям O-дебензилирования), в которой используют хлорид алюминия и N,N-диметиланилин в качестве реагентов и которая описана, например, в J. Med. Chem. (2003), 46 (16), 3547 для синтеза, в частности, ресвератрола и пицеатаннола. Однако ароматический амин является дорогим, высокотоксичным и трудноудалимым, что делает этот способ малоинтересным для промышленности.
Учитывая, что ни одно из решений, описанных выше, не является истинно удовлетворительным с промышленной точки зрения, Компания-Заявитель подыскивала способ, который является более подходящим для снятия защиты с алкоксистильбеновых или аралкоксистильбеновых производных, более конкретно, для цели синтеза ресвератрола и пицеатаннола.
Раскрытие изобретения
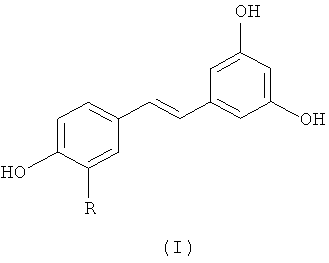
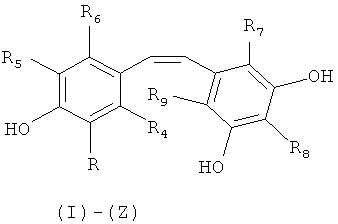
Объектом настоящего изобретения является, таким образом, способ синтеза производного (E)-стильбена формулы (I)-(E) или производного (Z)-стильбена формулы (I)-(Z):
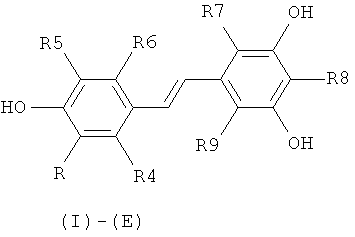
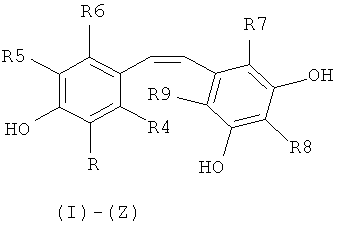
в которой:
R представляет собой водород или OH-группу, R4, R5, R6, R7, R8 и R9, независимо, представляют собой водород или заместитель, выбранный из:
- галогена;
- нитро-группы;
- линейной или разветвленной C1-C6-алкильной группы;
- линейной или разветвленной C2-C6-алкенильной группы;
- C3-C10-циклоалкильной группы;
- циклоалкилалкильной группы, в которой циклоалкильная и алкильная группы являются такими, как определено выше;
- моноциклической, бициклической или трициклической C6-C14-арильной группы;
- C7-C16-аралкильной группы;
- группы C(=O)R10, в которой R10 представляет собой линейную или разветвленную C1-C6-алкильную группу, C3-C10-циклоалкильную группу, циклоалкилалкильную группу, в которой циклоалкильная и алкильная группы являются такими, как определено выше, моноциклическую, бициклическую или трициклическую C6-C14-арильные группы, C7-C16-аралкильную группу, группу OR11, в которой R11 представляет собой водород, линейную или разветвленную C1-C6-алкильную группу, C3-C10 циклоалкильную группу, циклоалкилалкильную группу, в которой циклоалкильная и алкильная группы являются такими, как определено выше, моноциклическую, бициклическую или трициклическую C6-C16-арильную группу или C7-C16-аралкильную группу, или группу NR12R13, в которой R12 и R13, независимо, представляют собой водород, линейную или разветвленную C1-C6-алкильную группу, C3-C10 циклоалкильную группу, циклоалкилалкильную группу, в которой циклоалкильная и алкильная группы являются такими, как определено выше, моноциклическую, бициклическую или трициклическую C6-C14-арильную группу или C7-C16-аралкильную группу;
причем все вышеупомянутые алкильные, алкенильные, циклоалкильные, циклоалкилалкильные, арильные или аралкильные радикалы являются незамещенными или замещенными,
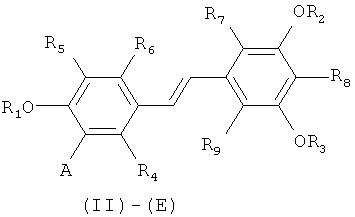
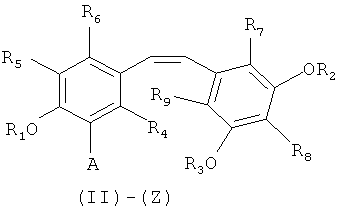
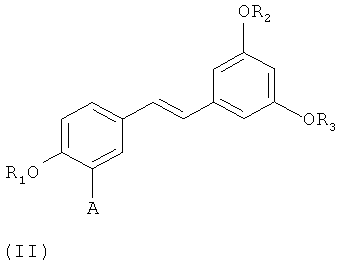
путем снятия защиты с соединения формулы (II)-(E) или (II)-(Z):
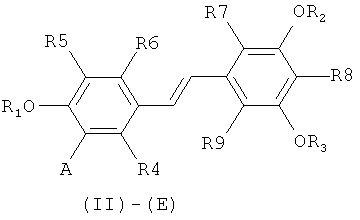
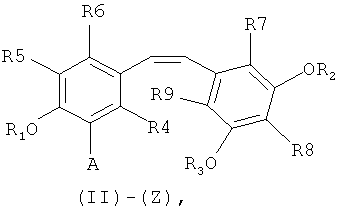
в которой A представляет собой водород или группу 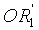 , и R1, R2, R3 и
, и R1, R2, R3 и 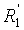 независимо представляют собой линейную или разветвленную C1-C6-алкильную группу или C7-C16-аралкильную группу, необязательно, замещенную по арильной части одной или более C1-C4-алкоксигруппами или галогеновыми группами,
независимо представляют собой линейную или разветвленную C1-C6-алкильную группу или C7-C16-аралкильную группу, необязательно, замещенную по арильной части одной или более C1-C4-алкоксигруппами или галогеновыми группами,
характеризующийся тем, что снятие защиты осуществляют путем применения галогенида алюминия и третичного амина формулы NRaRbRc, где Ra, Rb и Rc, независимо, представляют собой линейную или разветвленную C1-C4-алкильную группу.
По предпочтительному объекту изобретение также относится к способу синтеза производного (E)-стильбена формулы (I):
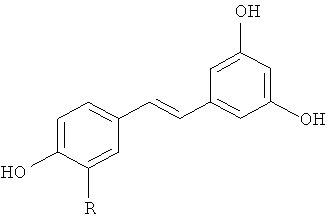
(I),
в которой R представляет собой водород или OH-группу,
путем снятия защиты с соединения формулы (II):
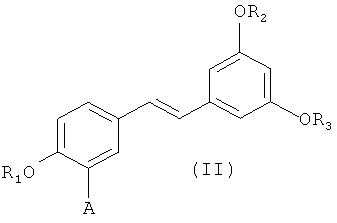
(II),
в которой A представляет собой водород или группу , и R1, R2, R3 и 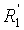 независимо представляют собой линейную или разветвленную C1-C6-алкильную группу или C7-C16-аралкильную группу, необязательно, замещенную по арильной части одной или более C1-C4-алкоксигруппой или галогеновой группой,
независимо представляют собой линейную или разветвленную C1-C6-алкильную группу или C7-C16-аралкильную группу, необязательно, замещенную по арильной части одной или более C1-C4-алкоксигруппой или галогеновой группой,
характеризующийся тем, что снятие защиты осуществляют путем применения галогенида алюминия и третичного амина формулы NRaRbRc, в котором Ra, Rb и Rc, независимо, представляют собой линейную или разветвленную C1-C4-алкильную группу.
В настоящем описании подразумевают, что термин “линейная или разветвленная C1-C6-алкильная группа” означает, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группу.
Галогеновый радикал означает Cl, Br, F или I.
Подразумевают, что термин “линейная или разветвленная C1-C4 алкильная группа” означает, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную или трет-бутильную группу.
Подразумевают, что термин "линейная или разветвленная C2-C6-алкенильная группа" означает, например, этенильную или винильную, пропенильную или аллильную, 1-пропенильную, н-бутенильную, изобутенильную, 3-метилбут-2-енильную, н-пентенильную или гексенильную группу.
Термин “C1-C4-алкоксигруппа” обозначает, например, метокси-, этокси-, пропокси- или бутоксигруппу.
Подразумевают, что термин “C3-C10-циклоалкильная группа” означает, например, циклопропильную, циклобутильную, циклопентильную или циклогексильную группу.
Подразумевают, что термин “циклоалкилалкильная группа” означает, например, циклопропилметильную, циклобутилметильную, циклопентилметильную, циклогексилметильную, циклогептилметильную, циклопропилэтильную или циклогексилэтильную группу.
Подразумевают, что термин “моноциклическая, бициклическая или трициклическая C6-C14-арильная группа” означает, например, фенильную, нафтильную, инденильную или антраценильную группу.
Подразумевают, что термин “C7-C16-аралкильная группа” означает, например, бензильную, 1-фенилэтильную, нафталенилметильную или 1-нафталенилэтильную группу.
Способ изобретения распространяется, в частности, на применение соединений формулы (II)-(E) или (II)-(Z), где R1 R2, R3 и , независимо, представляют собой линейную или разветвленную C1-C3-алкильную группу или бензильную группу, необязательно, замещенную по фенильной части одной или более C1-C4-алкоксигруппами и, в особенности, к применению соединений формулы (II)-(E) или (II)-(Z), где R1, R2, R3 и 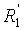 , независимо, представляют собой метильную группу или бензильную группу, a R4, R5, R6, R7, R3 и R9 являются такими, как определено выше.
, независимо, представляют собой метильную группу или бензильную группу, a R4, R5, R6, R7, R3 и R9 являются такими, как определено выше.
По предпочтительному объекту, способ изобретения распространяется на применение соединений формулы (II), где R1, R2, R3 и 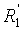 , независимо, представляют собой линейную или разветвленную C1-C3-алкильную группу или бензильную группу, необязательно, замещенную по фенильной части одной или более C1-C4-алкоксигруппами, и, в особенности, к применению соединений формулы (II), где R1, R2, R3 и , независимо, представляют собой метильную группу или бензильную группу.
, независимо, представляют собой линейную или разветвленную C1-C3-алкильную группу или бензильную группу, необязательно, замещенную по фенильной части одной или более C1-C4-алкоксигруппами, и, в особенности, к применению соединений формулы (II), где R1, R2, R3 и , независимо, представляют собой метильную группу или бензильную группу.
В настоящем изобретении применяют галогенид алюминия, который может быть выбран из хлорида алюминия, бромида алюминия и иодида алюминия, причем хлорид алюминия является предпочтительным.
По предпочтительному объекту изобретения применяют третичный амин, который может быть выбран из триэтиламина, трипропиламина, трибутиламина, N,N-диметилэтиламина, N,N-диэтилметиламина и N,N-диметилбутиламина, причем триэтиламин является предпочтительным.
Молярное соотношение реагентов галогенид алюминия:третичный амин, используемое в способе по изобретению, можно варьировать между 1:1 и 1:4, предпочтительно между 1:1 и 1:2 и в особенности между 1:1,5 и 1:1,6.
Обычно применяют 1-10 мольных эквивалентов галогенида алюминия и 1-20 мольных эквивалентов третичного амина на эфирную группу, подлежащую снятию в соединениях формулы (II)-(E), (II)-(Z) или (II), предпочтительно 1-4 мольных эквивалента галогенида алюминия и 1-6 мольных эквивалентов третичного амина на эфирную группу, подлежащую снятию, и в особенности, 2-2,2 мольных эквивалента галогенида алюминия и 3-3,5 мольных эквивалента третичного амина на эфирную группу, подлежащую снятию.
В соответствии с изобретением способ по изобретению может быть осуществлен без растворителя или с использованием растворителей, не содержащих гидроксильных групп и не содержащих кислородных атомов. Предпочтительно используют растворитель или смесь растворителей. Более конкретно, в качестве примеров растворителей, подходящих для настоящего изобретения, можно упомянуть галогенированные алифатические углеводороды или галогенированные или негалогенированные ароматические углеводороды. Более конкретно, в качестве примеров галогенированных алифатических углеводородов можно упомянуть дихлорометан или 1,2-дихлороэтан.
Более конкретно, в качестве примеров галогенированных или негалогенированных ароматических углеводородов можно упомянуть толуол или хлорбензол.
Предпочтительными растворителями являются толуол и хлорбензол, в частности хлорбензол.
Согласно первой альтернативной форме изобретения нет никаких ограничений по использованию галогенида алюминия и третичного амина. Галогенид алюминия и третичный амин можно вводить в любом порядке.
Согласно предпочтительному воплощению изобретения галогенид алюминия прибавляют к третичному амину, а затем вводят соединения формулы (II)-(E), (II)-(Z) или (II).
Согласно другому предпочтительному воплощению изобретения соединения формулы (II)-(E), (II)-(Z) или (II) прибавляют к третичному амину, а затем вводят галогенид алюминия.
Согласно второй альтернативной форме изобретения предварительно получают комплекс галогенид алюминия/третичный амин и его, необязательно, выделяют перед введением соединения формулы (II)-(E), (II)-(Z) или (II). Предпочтительно проводят реакцию галогенида алюминия и третичного амина при температуре между 50°C-60°C в течение 1-4 ч, необязательно, в растворителе, таком как описанный выше.
Температура, при которой осуществляют способ изобретения, составляет обычно между 50°C и 120°C. Предпочтительно используют диапазон температур между 80°C и 100°C.
Время реакции можно варьировать в соответствии с условиями реакции, в частности температурой, и составляющими. Анализ реакционной среды с помощью ВЭЖХ позволяет подтвердить исчезновение соединений формулы (II)-(E), (II)-(Z) или (II).
Соединения формулы (II)-(E), (II)-(Z) или (II) могут быть получены с помощью способов, известных на существующем уровне техники, например в EP 1466884, WO 2003/086414 и US 2004/0015020.
Следующие примеры иллюстрируют изобретение и не имеют ограничивающего характера.
Осуществление изобретения
Пример 1
56,2 г триэтиламина (555,4 ммоль) вводят в 20 мл хлорбензола в трехгорлой круглодонной колбе. Используют атмосферу азота, реакционную среду охлаждают до 0-5°C и прибавляют 45 г безводного хлорида алюминия (337,5 ммоль) мелкими порциями на протяжении 30 мин. Среду выдерживают при комнатной температуре в течение 30 мин при перемешивании, а затем доводят до 60°C, причем эту температуру поддерживают в течение 1 ч. Затем в течение 1 ч добавляют 10 г (E)-3,5,4'-триметоксистильбена (37 ммоль), растворенного в 20 мл хлорбензола. Смесь выдерживают при 60°C при перемешивании в течение 4 ч, а затем при 80°C в течение 4 ч. Ее доводят обратно до комнатной температуры, проводят разделение путем отстаивания и верхнюю хлорбензольную фазу отделяют. Нижнюю фазу медленно прибавляют к 100 г смеси лед/вода (50/50). Среду выдерживают при перемешивании в течение 1 ч и экстрагируют несколько раз этилацетатом.
Объединенные органические фазы промывают водой и концентрируют с образованием 7,6 г (E)-ресвератрола, т.е. с выходом сырого продукта 90%. Сырой продукт растворяют в этаноле при 60°C и ресвератрол осаждают путем прибавления воды для того, чтобы получить 6 г осадка, показывающего точку плавления 262-264°C.
Протонные и 13C спектры ЯМР согласуются со структурой (E)-ресвератрола.
Пример 2
4,5 г безводного хлорида алюминия (33,7 ммоль) вводят в атмосфере азота при комнатной температуре и при перемешивании в 10,37 г трибутиламина (56 ммоль) в трехгорлой круглодонной колбе. Среду доводят до 60°C и выдерживают на этом уровне в течение 4 ч. Затем вводят 1 г (E)-3,5,4'-триметоксистильбена (3,7 ммоль) и реакционную среду доводят до 80°C в течение 2 ч и до 100°C в течение 2 ч. Ее доводят обратно до комнатной температуры, а затем прибавляют 10 г смеси вода/лед (50/50). Ее выдерживают при 0-5°C при перемешивании в течение 3 ч и экстрагируют 4-5 раз 10 мл метилэтилкетона. Объединенные органические фазы промывают 10 мл насыщенного водного раствора бикарбоната натрия, а затем 10 мл воды. После концентрирования органической фазы количественное определение с помощью ВЭЖХ (внешняя калибровка) дает выход (E)-ресвератрола 75%.
Пример 3
4 мл хлорбензола и 6 г триэтиламина (59,3 ммоль) вводят в трехгорлую круглодонную колбу. Используют атмосферу азота, реакционную среду охлаждают до 0-5°C и 4,9 г безводного хлорида алюминия (36,7 ммоль) вводят мелкими порциями при этой температуре. Среду доводят до 50°C и выдерживают при этой температуре в течение 1 ч, а затем 2 г (E)-3,5,4'-трибензилоксистильбена (4 ммоль), растворенного в 5 мл хлорбензола, прибавляют в течение 1 ч при этой температуре. Эту температуру поддерживают в течение 4 ч и затем в течение 4 ч поддерживают температуру 80°C. Среду доводят обратно до комнатной температуры, проводят разделение путем отстаивания, тяжелую фазу отделяют и выливают на 20 мл смеси лед/вода (50/50). Среду выдерживают при перемешивании в течение 2 ч и экстрагируют этилацетатом. Органическую фазу промывают насыщенным водным раствором бикарбоната натрия, а затем водой и затем концентрируют с образованием 0,93 г сырого (E)-ресвератрола, т.е. практически с количественным выходом по отношению к исходному трибензилресвератролу.
Пример 4
4,5 мл хлорбензола и 15,7 г триэтиламина (155,1 ммоль; 21 мол. экв.) вводят в трехгорлую круглодонную колбу. Используют атмосферу азота, реакционную среду охлаждают до 0-5°C и 12,8 г безводного хлорида алюминия (95 ммоль; 13 мол. экв.) прибавляют в течение 30 мин. Среду доводят до 60°C в течение 1 ч. При этой температуре в течение 1 ч вводят 2,2 г тетраметилпицеатаннола (7,3 ммоль), растворенного в 4,5 мл хлорбензола, и реакционную среду выдерживают при этой температуре в течение 4 ч, а затем при 80°C в течение 4 ч. Ее доводят обратно до комнатной температуры, проводят разделение путем отстаивания, тяжелую фазу отделяют и гидролизуют путем выливания по каплям на 40 г смеси вода/лед (50/50). Смесь выдерживают при перемешивании при этой температуре в течение 1 ч 30 мин. Среду экстрагируют 4 раза по 25 мл метилэтилкетона и органические фазы промывают насыщенным раствором бикарбоната натрия, а затем водой. 1,62 г сырого (E)-пицеатаннола выделяют в форме коричневого твердого вещества.
Продукт очищают из смеси (5/95) метанол/вода для того, чтобы получить пицеатаннол, показывающий точку плавления 233-234°C.
Протонные и 13C спектры ЯМР согласуются со структурой (E)- пицеатаннола.
Пример 5
5 г (E)-3,5,4'-триметоксистильбена (18,5 ммоль) вводят в 20 г триэтиламина (197,6 ммоль) в трехгорлой круглодонной колбе объемом 100 мл. Смесь нагревают до 50°C в атмосфере азота и 16 г безводного хлорида алюминия (120 ммоль) прибавляют мелкими порциями в течение 30 мин при этой температуре. Реакционную среду затем доводят до 80°C в течение 2 ч, а затем до 100°C в течение 2 ч. Ее охлаждают до приблизительно 75°C, медленно прибавляют 10 мл безводного этанола, а затем при этой температуре 75°C прибавляют 50 мл воды в течение 30 мин. Реакционную среду затем охлаждают до комнатной температуры, при которой ее выдерживают в течение 3 ч, и среду экстрагируют 1 раз 35 мл и 3 раза 30 мл метилэтилкетона. Объединенные органические фазы промывают 30 мл воды, затем 30 мл насыщенного раствора бикарбоната натрия и 30 мл воды. После концентрирования органических фаз прибавляют 15 мл абсолютного этанола, смесь нагревают с обратным холодильником, а затем прибавляют 46 г воды в течение приблизительно 1 ч также при кипячении с обратным холодильником. Смесь охлаждают до комнатной температуры и оставляют при перемешивании в течение 3 ч. Осадок отфильтровывают и промывают на фильтре 9 г смеси вода/этанол (80/20 по массе).
После высушивания при 40°C в вакууме в течение 24 ч получают 3,1 г (E)-ресвератрола, т.е. выход 73,4%.
Анализы методами ВЭЖХ и ЯМР согласуются со структурой (E)-ресвератрола.
Пример 6
5,6 г триэтиламина (55,5 ммоль) вводят в 4 мл метиленхлорида. Среду охлаждают до 0-5°C и 4,5 г безводного хлорида алюминия (33,5 ммоль) прибавляют мелкими порциями в течение 15 мин при перемешивании и в атмосфере азота. Реакционную среду затем доводят до 50°C в течение 4 ч. Ее охлаждают, и растворитель, и избыток амина концентрируют при комнатной температуре для того, чтобы получить приблизительно 8,5 г слегка дымящего розоватого твердого вещества, образованного комплексом AICI3/триэтиламин. К твердому веществу прибавляют 2 мл хлорбензола и смесь доводят до 60°C. При этой температуре в течение одного часа прибавляют 1 г 3,5,4'-триметоксистильбена (3,7 ммоль), растворенного в 2 мл хлорбензола. Смесь выдерживают при 60°C в течение 4 ч, а затем при 80°C в течение 2 ч. Реакционную среду доводят обратно до комнатной температуры, а затем гидролизуют путем прибавления 10 г смеси вода/лед (50/50). Выдерживают температуру 0-5°C в течение 1 ч и среду экстрагируют несколько раз этилацетатом. После концентрирования органической фазы полученный осадок промывают 6 мл хлорбензола и высушивают в вакууме с образованием 0,6 г сырого (E)-ресвератрола.
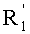
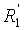
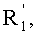
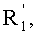
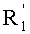
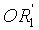
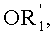
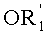
Способ деацетилирования α-аминоацеталей
Способ получения водного раствора глиоксиловой кислоты
Способ получения оптически активных альфа-аминоацеталей
Способ деацетилирования α-аминоацеталей
Способ получения водного раствора глиоксиловой кислоты
Способ выделения глиоксиловой кислоты из водной реакционной среды, содержащей глиоксиловую кислоту и соляную кислоту
Способ получения оптически активных альфа-аминоацеталей

