Результат интеллектуальной деятельности: СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R
Вид РИД
Изобретение
РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет по предварительным заявкам на патент США №№ 62/579120, поданной 30 октября 2017 года; 62/710381, поданной 16 февраля 2018 года; 62/647368, поданной 23 марта 2018 года; 62/742736, поданной 8 октября 2018 года, и заявке на Европейский патент № EP18305566.4, поданной 4 мая 2018 года, каждая из которых включена в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к лечению и/или предупреждению астмы и родственных состояний. В частности, настоящее изобретение относится к введению антагониста рецепторов интерлейкина-4 (IL-4R) для лечения или предупреждения астмы у нуждающегося в этом пациента.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Астма представляет собой хроническое воспалительное заболевание дыхательных путей, характеризующееся гиперчувствительностью дыхательных путей, острым и хроническим бронхоспазмом, отеком и закупоркой дыхательных путей слизью. Считается, что воспалительный компонент астмы охватывает много типов клеток, в том числе тучные клетки, эозинофилы, T-лимфоциты, нейтрофилы и эпителиальные клетки, а также их биологические продукты. Пациенты с астмой наиболее часто проявляют симптомы свистящего дыхания, одышки, кашля и сдавленности в груди. Для большинства пациентов с астмой схема терапии контролирующими препаратами и терапии бронходилататорами обеспечивает соответствующий продолжительный контроль. Ингаляционные кортикостероиды (ICS) считаются "золотым стандартом" в контроле симптомов астмы, а ингаляционные бета-2-агонисты являются наиболее эффективными бронходилататорами, доступными в настоящее время. Исследования показали, что комбинированная терапия ICS ингаляционным бета-2-агонистом длительного действия (LABA) обеспечивает лучший контроль над астмой, чем высокие дозы ICS в отдельности. Следовательно, комбинированная терапия представляла собой рекомендованное лечение для субъектов, у которых не удавалось достичь контроля низкими дозами ICS в отдельности.
Тем не менее, по оценкам у 5% - 10% популяции астма имеет клиническое проявление, несмотря на максимальное рекомендованное лечение комбинациями противовоспалительных лекарственных средств и бронходилататоров. Кроме того, на эту популяцию с тяжелой астмой приходится до 50% всех затрат на поддержание здоровья при госпитализации, использование услуг неотложной помощи и внеплановые визиты врачей. Существует неудовлетворенная потребность в новом терапевтическом средстве в этой популяции с тяжелой астмой, поскольку многие из этих пациентов слабо реагируют на ICS вследствие ряда клеточных и молекулярных механизмов. Кроме того, отдаленные побочные эффекты системных и ингаляционных кортикостероидов в отношении метаболизма костной ткани, функции надпочечников и роста у детей приводят к попыткам свести к минимуму количество потребления кортикостероидов. Несмотря на то, что состояние большей части пациентов с астмой контролируют надлежащим образом с помощью существующих средств лечения, у пациентов с тяжелой неконтролируемой астмой (например, с тяжелой не поддающейся лечению кортикостероидами астмой или не переносящей стероиды астмой) существует немного вариантов терапевтических средств лечения, которые могут контролировать заболевание на должном уровне. Следствием невосприимчивости к терапии или отсутствия соблюдения режима терапии является потеря контроля астмы и в конечном итоге обострение астмы.
По оценкам, 45% пациентов с тяжелой астмой нуждаются в системных глюкокортикоидах, чтобы контролировать свое заболевание и предотвращать опасные для жизни обострения, связанные с повышенным риском необратимого повреждения ткани легких, прогрессирующей стойкой обструкцией дыхательных путей и ускоренным снижением функции легких. Однако системные глюкокортикоиды действуют неселективно и ассоциированы со значительной полиорганной токсичностью и широкой иммуносупрессией. Существует потребность в более безопасных и более эффективных целенаправленно воздействующих средствах терапии, которые предотвращают обострения и нарушение функции легких, улучшают симптомы астмы и контроль, а также снижают или устраняют потребность в пероральных глюкокортикоидах.
Примерно 20% пациентов с астмой имеют неконтролируемое заболевание от умеренной до тяжелой степени с рецидивирующими обострениями и постоянными симптомами, несмотря на максимальное стандартное лечение контролирующими препаратами. Эта популяция населения подвержена повышенному риску заболеваемости (особенно обострений) и является причиной издержек значительных ресурсов здравоохранения. Эти пациенты характеризуются значимо сниженной функцией легких, несмотря на максимальное лечение, и им суждено неизбежно потерять функцию легких в дальнейшем. Ни одно из утвержденных в настоящее время средств лечения не продемонстрировало замедления этого неизбежного снижения у данных пациентов или последовательного и значительного повышения функции легких.
Соответственно, в данной области существует необходимость в новых целенаправленно воздействующих средствах терапии для лечения и/или предупреждения астмы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно одному аспекту предусматривается способ лечения субъекта, страдающего тяжелой неконтролируемой астмой (например, тяжелой стероидозависимой астмой), предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где множество поддерживающих доз вводят во время фазы лечения, предусматривающей фазу индукции, фазу снижения перорального кортикостероида (OCS) и фазу поддержания OCS.
В определенных иллюстративных вариантах осуществления поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят один раз в две недели (q2w). В определенных иллюстративных вариантах осуществления поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят раз в четыре недели (q4w).
В определенных вариантах осуществления субъекту вводят нагрузочную дозу и введение поддерживающей дозы осуществляют согласно схеме дозирования 500 мг q4w или 750 мг q4w.
В определенных вариантах осуществления нагрузочная доза исключается. В определенных вариантах осуществления введение дозы субъекту осуществляют согласно схеме дозирования 500 мг q4w или 750 мг q4w.
В определенных иллюстративных вариантах осуществления нагрузочная доза составляет приблизительно 600 мг антитела или его антигенсвязывающего фрагмента и/или каждая поддерживающая доза антитела или его антигенсвязывающего фрагмента составляет приблизительно 300 мг антитела или его антигенсвязывающего фрагмента.
В определенных иллюстративных вариантах осуществления поддерживающие дозы антитела или его антигенсвязывающего фрагмента вводят в течение по меньшей мере 24 недель.
В определенных иллюстративных вариантах осуществления первую поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят через две недели после нагрузочной дозы антитела или его антигенсвязывающего фрагмента.
В определенных иллюстративных вариантах осуществления продолжительность фазы снижения OCS составляет приблизительно 16 недель.
В определенных иллюстративных вариантах осуществления применение OCS субъектом снижается в ходе фазы снижения OCS. В определенных иллюстративных вариантах осуществления субъект использует 50% или меньше, 75% или меньше или 90% или меньше OCS в фазе поддержания по сравнению с фазой индукции. В определенных иллюстративных вариантах осуществления применение OCS субъектом в фазе поддержания снижается до приблизительно 5 мг/сутки или меньше. В других иллюстративных вариантах осуществления OCS снижается и/или исключается, например, у субъекта постепенно снижают предыдущую дозу OCS. В определенных иллюстративных вариантах осуществления введение OCS полностью исключают из схемы лечения.
В определенных иллюстративных вариантах осуществления субъект характеризуется содержанием эозинофилов в крови, составляющим приблизительно 150 клеток/мкл или меньше. В определенных иллюстративных вариантах осуществления субъект характеризуется содержанием эозинофилов в крови более приблизительно 150 клеток/мкл. В определенных иллюстративных вариантах осуществления субъект характеризуется содержанием эозинофилов в крови более приблизительно 300 клеток/мкл.
В определенных иллюстративных вариантах осуществления у субъекта наблюдается снижение тяжелых обострений астмы в годовом исчислении. В определенных иллюстративных вариантах осуществления у субъекта наблюдается улучшение функции легких, измеряемой по объему форсированного выдоха (FEV1). В других вариантах осуществления у субъекта наблюдается улучшение функции легких в мелких дыхательных путях и/или уменьшение воспаления в мелких дыхательных путях. В определенных вариантах осуществления улучшение функции легких и уменьшение воспаления измеряют по скорости форсированного выдоха при 25-75% от объема легких (FEF25-75).
В определенных иллюстративных вариантах осуществления применение OCS субъектом оптимизируют до лечения антителом или его антигенсвязывающим фрагментом. В определенных иллюстративных вариантах осуществления OCS представляет собой преднизон или преднизолон.
В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат последовательности определяющих комплементарность участков (CDR) тяжелой и легкой цепей из пары последовательностей вариабельного участка тяжелой цепи (HCVR)/вариабельного участка легкой цепи (LCVR), предусматривающей SEQ ID NO: 1 и 2. В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат три последовательности CDR тяжелой цепи, предусматривающие SEQ ID NO: 3, 4 и 5 соответственно, и три последовательности CDR легкой цепи, предусматривающие SEQ ID NO: 6, 7 и 8 соответственно. В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат HCVR, содержащий аминокислотную последовательность под SEQ ID NO: 1, и LCVR, содержащий аминокислотную последовательность под SEQ ID NO: 2.
В определенных иллюстративных вариантах осуществления субъектом является взрослый человек. В определенных иллюстративных вариантах осуществления субъектом является подросток. В определенных иллюстративных вариантах осуществления субъектом является взрослый человек или подросток, например, возрастом 12 лет или старше.
В одном аспекте предусматривается способ лечения субъекта, страдающего тяжелой неконтролируемой астмой (например, тяжелой стероидозависимой астмой), предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где множество поддерживающих доз вводят во время фазы лечения, предусматривающей фазу индукции, фазу снижения перорального кортикостероида (OCS) и фазу поддержания, и где антитело или его антигенсвязывающий фрагмент содержат последовательности CDR тяжелой и легкой цепей из пары последовательностей HCVR/LCVR, предусматривающей SEQ ID NO: 1 и 2.
В другом аспекте предусматривается способ лечения субъекта, страдающего тяжелой неконтролируемой астмой, например, тяжелой стероидозависимой астмой, предусматривающий введение субъекту нагрузочной дозы приблизительно 600 мг антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где каждая поддерживающая доза составляет приблизительно 300 мг антитела или его антигенсвязывающего фрагмента, где множество поддерживающих доз вводят во время фазы лечения, предусматривающей фазу индукции, фазу снижения перорального кортикостероида (OCS) и фазу поддержания, и где антитело или его антигенсвязывающий фрагмент содержат последовательности CDR тяжелой и легкой цепей из пары последовательностей HCVR/LCVR, предусматривающей SEQ ID NO: 1 и 2.
В другом аспекте предусматривается способ снижения частоты тяжелого обострения в годовом исчислении у субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени, предусматривающий введение субъекту q2w или q4w антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R.
В определенных иллюстративных вариантах осуществления доза составляет 200 мг q2w или 300 мг q2w.
В определенных иллюстративных вариантах осуществления поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят раз в четыре недели (q4w).
В определенных вариантах осуществления субъекту вводят нагрузочную дозу и поддерживающие дозы и введение дозы субъекту осуществляют согласно схеме дозирования 500 мг q4w или 750 мг q4w.
В определенных вариантах осуществления нагрузочная доза исключается. В определенных вариантах осуществления введение дозы субъекту осуществляют согласно схеме дозирования 500 мг q4w или 750 мг q4w.
В определенных иллюстративных вариантах осуществления субъект характеризуется содержанием эозинофилов в крови, составляющим менее приблизительно 150 клеток/мкл, составляющим приблизительно 150 клеток/мкл или больше или составляющим более приблизительно 300 клеток/мкл.
В определенных иллюстративных вариантах осуществления субъект характеризуется уровнем фракции оксида азота в выдыхаемом воздухе (FeNO), составляющим приблизительно 25 частей на миллиард (ppb) или больше, характеризуется уровнем FeNO, составляющим приблизительно 50 ppb или больше, или характеризуется уровнем FeNO, составляющим от приблизительно 25 ppb или больше до приблизительно 50 ppb.
В другом аспекте предусматривается способ улучшения оценки FEV1 у субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени, предусматривающий введение субъекту q2w или q4w антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R.
В определенных иллюстративных вариантах осуществления вводят дозу 200 мг q2w или 300 мг q2w. В определенных иллюстративных вариантах осуществления вводят дозу 500 мг q4w или 750 мг q4w.
В определенных иллюстративных вариантах осуществления субъект характеризуется содержанием эозинофилов в крови, составляющим менее приблизительно 150 клеток/мкл, составляющим приблизительно 150 клеток/мкл или больше или составляющим более приблизительно 300 клеток/мкл.
В определенных иллюстративных вариантах осуществления субъект характеризуется уровнем фракции оксида азота в выдыхаемом воздухе (FeNO), составляющим приблизительно 25 частей на миллиард (ppb) или больше, характеризуется уровнем FeNO, составляющим приблизительно 50 ppb или больше, или характеризуется уровнем FeNO, составляющим от приблизительно 25 ppb или больше до приблизительно 50 ppb.
В другом варианте осуществления у субъекта наблюдается снижение на по меньшей мере 10%, 15%, 20% или 25% уровня биомаркера, выбранного из группы, состоящей из FeNO, эотаксина-3, общего IgE, периостина и хемокина, регулируемого тимусом и активацией (TARC), на неделе 4, неделе 12 или неделе 24 после введения антитела к IL-4R или его фрагмента.
В определенных иллюстративных вариантах осуществления субъектом является взрослый человек. В определенных иллюстративных вариантах осуществления субъектом является подросток. В определенных иллюстративных вариантах осуществления субъектом является взрослый человек или подросток, например, возрастом 12 лет или старше.
В других аспектах в настоящем изобретении предусматривается способ улучшения показателя скорости форсированного выдоха при 25-75% от объема легких (FEF25-75) у субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени, предусматривающий введение субъекту q2w или q4w антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R.
В одном варианте осуществления доза составляет 200 мг q2w или 300 мг q2w. В одном варианте осуществления доза составляет 500 мг q4w или 750 мг q4w.
В одном варианте осуществления субъект характеризуется содержанием эозинофилов в крови менее приблизительно 150 клеток/мкл. В одном варианте осуществления субъект характеризуется содержанием эозинофилов в крови, составляющим приблизительно 150 клеток/мкл или больше. В одном варианте осуществления субъект характеризуется содержанием эозинофилов в крови, составляющим приблизительно 300 клеток/мкл или больше.
В другом варианте осуществления субъект характеризуется уровнем FeNO, составляющим приблизительно 25 ppb или больше. В другом варианте осуществления субъект характеризуется уровнем FeNO, составляющим приблизительно 50 ppb или больше. В другом варианте осуществления субъект характеризуется уровнем FeNO, составляющим от приблизительно 25 ppb или больше до приблизительно 50 ppb.
В другом варианте осуществления у субъекта наблюдается снижение на по меньшей мере 10%, по меньшей мере 15%, по меньшей мере 20% или по меньшей мере 25% уровня биомаркера, выбранного из группы, состоящей из FeNO, эотаксина-3, общего IgE, периостина и хемокина, регулируемого тимусом и активацией (TARC), на неделе 4, неделе 12 или 24 после введения антитела к IL4R или его фрагмента.
В определенных иллюстративных вариантах осуществления субъектом является взрослый человек. В определенных иллюстративных вариантах осуществления субъектом является подросток. В определенных иллюстративных вариантах осуществления субъектом является взрослый человек или подросток, например, возрастом 12 лет или старше.
В другом аспекте настоящее изобретение относится к способу снижения или устранения применения OCS у субъекта, страдающего тяжелой астмой, зависимой от стероидов, при этом способ предусматривает введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R; и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где снижение применения OCS на по меньшей мере 50% или больше, по меньшей мере 75% или больше или по меньшей мере 90% или больше достигается на неделе 24 после введения нагрузочной дозы.
В одном варианте осуществления применение OCS снижается до менее 5 мг в сутки на неделе 24 после введения нагрузочной дозы. В другом варианте осуществления OCS практически устраняется через некоторый период времени (например, 1 год) после введения нагрузочной дозы. В определенных вариантах осуществления OCS практически устраняется через 40 недель, 45 недель, 50 недель, 52 недели или больше после первой дозы, следующей за введением нагрузочной дозы.
В одном варианте осуществления поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят один раз в две недели (q2w). В одном варианте осуществления нагрузочная доза составляет приблизительно 600 мг антитела или его антигенсвязывающего фрагмента. В одном варианте осуществления каждая поддерживающая доза антитела или его антигенсвязывающего фрагмента составляет приблизительно 300 мг антитела или его антигенсвязывающего фрагмента. В другом варианте осуществления поддерживающие дозы антитела или его антигенсвязывающего фрагмента вводят в течение по меньшей мере 24 недель. В одном варианте осуществления первую поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят через две недели после нагрузочной дозы антитела или его антигенсвязывающего фрагмента. В одном варианте осуществления OCS представляет собой преднизон или преднизолон.
В одном варианте осуществления антитело или его антигенсвязывающий фрагмент содержат последовательности определяющих комплементарность участков (CDR) тяжелой и легкой цепей из пары последовательностей вариабельного участка тяжелой цепи (HCVR)/вариабельного участка легкой цепи (LCVR), предусматривающей SEQ ID NO: 1 и 2. В одном варианте осуществления антитело или его антигенсвязывающий фрагмент содержат три последовательности CDR тяжелой цепи, предусматривающие SEQ ID NO: 3, 4 и 5 соответственно, и три последовательности CDR легкой цепи, предусматривающие SEQ ID NO: 6, 7 и 8 соответственно. В одном варианте осуществления антитело или его антигенсвязывающий фрагмент содержат HCVR, содержащий аминокислотную последовательность под SEQ ID NO: 1, и LCVR, содержащий аминокислотную последовательность под SEQ ID NO: 2.
В определенных иллюстративных вариантах осуществления поддерживающую дозу антитела или его антигенсвязывающего фрагмента вводят раз в четыре недели (q4w).
В определенных вариантах осуществления субъекту вводят нагрузочную дозу и введение дозы субъекту осуществляют согласно схеме дозирования 500 мг q4w или 750 мг q4w.
В определенных вариантах осуществления нагрузочная доза исключается. В определенных вариантах осуществления введение дозы субъекту осуществляют согласно схеме дозирования 500 мг q4w и 750 мг q4w.
В определенных иллюстративных вариантах осуществления субъектом является взрослый человек. В определенных иллюстративных вариантах осуществления субъектом является подросток. В определенных иллюстративных вариантах осуществления субъектом является взрослый человек или подросток, например, возрастом 12 лет или старше.
В другом аспекте предусматривается способ лечения субъекта, страдающего зависимой от перорального кортикостероида (OCS) астмой от умеренной до тяжелой степени, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат последовательности определяющих комплементарность участков (CDR) тяжелой и легкой цепей из пары последовательностей вариабельного участка тяжелой цепи (HCVR)/вариабельного участка легкой цепи (LCVR), предусматривающей SEQ ID NO: 1 и 2. В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат три последовательности CDR тяжелой цепи, предусматривающие SEQ ID NO: 3, 4 и 5 соответственно, и три последовательности CDR легкой цепи, предусматривающие SEQ ID NO: 6, 7 и 8 соответственно. В определенных иллюстративных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат HCVR, содержащий аминокислотную последовательность под SEQ ID NO: 1, и LCVR, содержащий аминокислотную последовательность под SEQ ID NO: 2.
В определенных иллюстративных вариантах осуществления нагрузочная доза составляет приблизительно 600 мг антитела или его антигенсвязывающего фрагмента. В определенных иллюстративных вариантах осуществления каждая поддерживающая доза антитела или его антигенсвязывающего фрагмента составляет приблизительно 300 мг антитела или его антигенсвязывающего фрагмента.
В определенных иллюстративных вариантах осуществления нагрузочная доза составляет приблизительно 400 мг антитела или его антигенсвязывающего фрагмента. В определенных иллюстративных вариантах осуществления каждая поддерживающая доза антитела или его антигенсвязывающего фрагмента составляет приблизительно 200 мг антитела или его антигенсвязывающего фрагмента.
В определенных иллюстративных вариантах осуществления возраст субъекта составляет 12 лет или старше.
В определенных иллюстративных вариантах осуществления OCS представляет собой преднизон или преднизолон.
В другом аспекте предусматривается способ лечения субъекта, страдающего астмой от умеренной до тяжелой степени и сопутствующим атопическим дерматитом от умеренной до тяжелой степени, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В другом аспекте предусматривается способ лечения субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени, где манифестация астмы произошла, когда возраст субъекта был старше 40 лет, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В другом аспекте предусматривается способ лечения субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени и одним или обоими из сопутствующего хронического риносинусита и назального полипоза, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В другом аспекте предусматривается способ лечения субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени и сопутствующим аллергическим ринитом, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В другом аспекте предусматривается способ улучшения качества жизни, связанного с аллергическим ринитом, у субъекта, страдающего неконтролируемой астмой от умеренной до тяжелой степени и сопутствующим аллергическим ринитом, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В другом аспекте предусматривается способ улучшения качества жизни, связанного с аллергическим ринитом, у субъекта, страдающего зависимой от перорального кортикостероида астмой, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В определенных иллюстративных вариантах осуществления наблюдается улучшение в отношении ежедневных показателей симптомов астмы в утреннее и вечернее время.
В определенных иллюстративных вариантах осуществления зависимая от перорального кортикостероида астма представляет собой зависимую от перорального кортикостероида тяжелую астму.
В другом аспекте предусматривается способ улучшения контроля астмы у субъекта, страдающего зависимой от перорального кортикостероида астмой, предусматривающий введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где нагрузочную дозу и множество поддерживающих доз вводят в качестве дополнительной поддерживающей терапии астмы.
В определенных иллюстративных вариантах осуществления связанное с состоянием здоровья качество жизни улучшается.
В определенных иллюстративных вариантах осуществления зависимая от перорального кортикостероида астма представляет собой зависимую от перорального кортикостероида тяжелую астму.
Другие варианты осуществления будут очевидны из обзора последующего подробного описания, графических материалов, таблиц и прилагаемой формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Вышеизложенные и другие признаки и преимущества настоящего изобретения будут в большей степени понятны из следующего подробного описания иллюстративных вариантов осуществления во взаимосвязи с прилагаемыми графическими материалами. В файле настоящего патента содержится по меньшей мере один графический материал/фотография, выполненные в цвете. Копии настоящего патента с цветным(цветными) графическим(графическими) материалом(материалами)/фотографией(фотографиями) будет представлены патентным ведомством по запросу и при оплате необходимого взноса.
На фиг. 1 изображена схема исследования Venture (EFC13691) для испытания фазы 3. EOS обозначает конец исследования, EOT - конец лечения, OCS - пероральный глюкокортикоид, q2w раз в 2 недели, R - визит рандомизации. анагрузочная доза 600 мг (или сопоставимое плацебо) в день 1; bво время этого визита происходит рандомизация и первое введение исследуемого медицинского продукта; cпериод скрининга может быть увеличен до 10 недель для пациентов, у которых наблюдается обострение астмы, которое требует изменения дозы глюкокортикоидов для обеспечения стабилизации в течение 2 недель до рандомизации.
На фиг. 2 изображена диаграмма CONSORT, показывающая распределение пациентов для исследования Venture (EFC13691).
Фиг. 3 представляет собой диаграмму исходных демографических данных для популяции пациентов.
На фиг. 4A - фиг. 4D графически изображены первичные и вторичные конечные точки на протяжении 24-недельного периода лечения в популяции, сформированной согласно назначенному лечению (ITT). На фиг. 4A изображены первичные и вторичные конечные точки для перорального глюкокортикоида на неделе 24. На фиг. 4B изображена частота тяжелых обострений в годовом исчислении. На фиг. 4C изображено изменение FEV1 (в литрах) до введения бронхолитического средства. На фиг. 4D изображено изменение FeNO (в ppb).
На фиг. 5A - фиг. 5B изображены результаты на неделе 24 по подгруппам на основе исходного уровня эозинофилов в крови. На фиг. 5A изображены данные первичной конечной точки. На фиг. 5B изображены данные вторичной конечной точки для перорального глюкокортикоида.
На фиг. 6A - фиг. 6B изображены тяжелые обострения астмы (Фиг. 6A) и FEV1 (л) до введения бронхолитического средства (фиг. 6B) на протяжении 24-недельного периода лечения по подгруппам на основе исходного уровня эозинофилов в крови.
На фиг. 7 изображена схема исследования Quest для испытания фазы 3.
Фиг. 8 представляет распределение пациентов для исследования Quest.
На фиг. 9A - фиг. 9B графически изображены тяжелые обострения астмы в популяции ITT и в подгруппах, определенных на основе исходного содержания эозинофилов в крови ≥ 150 и ≥ 300 клеток/мкл (фиг. 9A), и в подгруппах, определенных на основе исходных уровней FeNO < 25 ppb, от ≥ 25 до 50 ppb и ≥ 50 ppb (фиг. B).
На фиг. 10A - фиг. 10C изображены изменения относительно исходного уровня FEV1 с течением времени в популяции ITT (фиг. 10A) на неделе 12 в подгруппах, определенных на основе исходного содержания эозинофилов в крови ≥ 150 и ≥ 300 клеток/мкл (фиг. 10B), и в подгруппах, определенных на основе исходных уровней FeNO < 25 ppb, от ≥ 25 до 50 ppb и ≥ 50 ppb (фиг. 10C).
На фиг. 11A - фиг. 11B графически изображен ретроспективный анализ тяжелых обострений астмы (фиг. 11A) и изменения от исходного уровня FEV1 (фиг. 11B) у пациентов с высокими (≥ 25 ppb) или низкими (< 25 ppb) исходными уровнями FeNO и высоким (≥ 150 клеток/мкл) или низким (< 150 клеток/мкл) исходным содержанием эозинофилов в крови.
На фиг. 12 изображены исходные демографические данные и клинические характеристики подростков (n=107) и взрослых людей (n=1795). Жирным текстом выделены ключевые различия между подгруппами. FeNO - фракция оксида азота в выдыхаемом воздухе; LABA - β-агонист длительного действия; SD - стандартное отклонение.
На фиг. 13A - фиг. 13B графически изображено снижение тяжелых обострений и улучшение FEV1 в общей популяции, сформированной согласно назначенному лечению(ITT). Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; треугольники - 200 мг дупилумаба q2w; Xs - 300 мг дупилумаба q2w. ***P < 0,001 относительно плацебо. CI - доверительный интервал; LS - метод наименьших квадратов; SE - стандартная ошибка; стрелка - первичная конечная точка.
На фиг. 14A - фиг. 14B графически изображено снижение частоты тяжелого обострения у подростков и взрослых людей. Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; оранжевый - 200 мг дупилумаба q2w; синий - 300 мг дупилумаба q2w. ***P < 0,001 относительно плацебо; NS - недостоверно.
На фиг. 15A - фиг. 15B графически изображено улучшение FEV1 на неделе 12 и на неделе 52 у подростков и взрослых людей. Несмотря на более высокие исходные уровни, подростки характеризовались большим увеличением FEV1. *P < 0,05, **P < 0,01, ***P < 0,001 относительно плацебо.
На фиг. 16A - фиг. 16B графически изображено улучшение FEV1 на протяжении 52-недельного периода лечения у подростков и взрослыхлюдей. *P < 0,05, **P < 0,01 относительно плацебо.
На фиг. 17 изображено, что профили нежелательных явлений были сопоставимы между подгруппами (популяция по оценке безопасности). Эозинофилию идентифицируют как AE с HLT как эозинофильные нарушения, или PT как повышенное содержание эозинофилов. HLT - термин высокого уровня; PT - предпочтительный термин; SAE - тяжелое нежелательное явление; TEAE - появившееся во время лечения нежелательное явление.
На фиг. 18A - фиг. 18B графически изображено улучшение в процентах прогнозируемого FEV1 на протяжении 52-недельного периода лечения у подростков и взрослых людей. Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; треугольники - 200 мг дупилумаба q2w; Xs - 300 мг дупилумаба q2w. *P < 0,05, **P < 0,01 относительно плацебо.
На фиг. 19A - фиг. 19B графически изображены уровни FeNO на протяжении 52-недельного периода лечения у подростков и взрослых людей. Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; треугольники - 200 мг дупилумаба q2w; Xs - 300 мг дупилумаба q2w. *P < 0,05, **P < 0,01, ***P < 0,001 относительно плацебо.
На фиг. 20A - фиг. 20B графически изображены показатели ACQ-5 на протяжении 52-недельного периода лечения у подростков и взрослых людей. Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; треугольники - 200 мг дупилумаба q2w; Xs - 300 мг дупилумаба q2w. *P < 0,05, **P < 0,01, ***P < 0,001 относительно плацебо.
На фиг. 21A - фиг. 21B графически изображены показатели AQLQ на протяжении 52-недельного периода лечения у подростков и взрослых людей. Светло-серые кружки - 1,14 мл плацебо; темно-серые кружки - 2 мл плацебо; треугольники - 200 мг дупилумаба q2w; Xs - 300 мг дупилумаба q2w. *P < 0,05, **P < 0,01, ***P < 0,001 относительно плацебо.
На фиг. 22 изображены TEAE (PT), встречающиеся у ≥ 10% пациентов, в соответствии с подгруппами подростков и взрослых людей (популяция по оценке безопасности).
На фиг. 23 изображена информация о TEAE, представляющем собой конъюнктивит (популяция по оценке безопасности).
На фиг. 24 изображена информация о TEAE, представляющем собой эозинофилию (популяция по оценке безопасности). Эозинофилию идентифицируют как AE с HLT как эозинофильные нарушения, или PT как повышенное содержание эозинофилов.
На фиг. 25A - фиг. 25D изображен эффект дупилумаба на протяжении 24-недельного периода лечения в отношении показателей симптома до полудня в популяции ITT (фиг. 25A) и в подгруппе пациентов, у которых снижено применение OCS на 100% к неделе 24 (фиг. 25B), а также в отношении показателей симптомов после полудня в популяции ITT (фиг. 25C) и в подгруппе пациентов, у которых снижено применение OCS на 100% к неделе 24 (фиг. 25D). *P< 0,05, ***P< 0,001. SE - стандартная ошибка. Треугольники - плацебо; кружки - 300 мг дупилумаба q2w.
На фиг. 26A - фиг. 26B изображен эффект дупилумаба в отношении контроля над астмой и HRQoL у пациентов с OCS-зависимой тяжелой астмой, демонстрирующее показатель ACQ-5 (фиг. 26A) и общий показатель AQLQ (фиг. 26B). *P< 0,05, *P< 0,01, ***P< 0,001. SE - стандартная ошибка. Минимальное клинически значимое различие составляет 0,5 для всех шкал. Треугольники - плацебо; кружки - 300 мг дупилумаба q2w.
На фиг. 27 изображен эффект дупилумаба в отношении частоты тяжелого обострения, FEV1 и соотношения FEV1/FVC у пациентов с неконтролируемой астмой от умеренной до тяжелой степени, которым было > 40 лет на момент манифестации астмы, и характеризующихся исходным FEV1/FVC до введения бронхолитического средства < 0,7 или ≥ 0,7.
ПОДРОБНОЕ ОПИСАНИЕ
Перед описанием настоящего изобретения необходимо понимать, что настоящее изобретение не ограничивается конкретными описанными способами и условиями экспериментов, поскольку такие способы и условия могут варьировать. Также необходимо понимать, что терминология, используемая в данном документе, предназначена лишь с целью описания конкретных вариантов осуществления, и не предполагает ограничительный характер, поскольку объем настоящего изобретения будет ограничиваться лишь прилагаемой формулой изобретения.
Если не определено иное, все технические и научные термины, используемые в данном документе, имеют такое же значение, которое обычно понимается специалистом в данной области техники, к которой принадлежит настоящее изобретение.
Используемый в данном документе термин "приблизительно", при использовании со ссылкой на конкретное описываемое числовое значение, означает, что значение может отличаться от описываемого значения на не более чем 1%. Например, используемое в данном документе выражение "приблизительно 100" включает 99 и 101 и все значения между ними (например, 99,1, 99,2, 99,3, 99,4 и т.д.).
Используемые в данном документе термины "лечить", "лечение" или т.п. означают облегчать симптомы, устранять причинность симптомов, либо на временной, либо на постоянной основе, или предупреждать или замедлять проявление симптомов указанного нарушения или состояния.
Хотя любые способы и материалы, подобные или эквивалентные тем, которые описаны в данном документе, можно использовать при осуществлении настоящего изобретения на практике, ниже описаны типичные способы и материалы. Все публикации, упомянутые в данном документе, включены в данный документ посредством ссылки в полном объеме.
Способы снижения частоты возникновения обострений астмы
Настоящее изобретение включает способы снижения частоты возникновения обострений астмы у нуждающегося в этом субъекта, предусматривающие введение субъекту фармацевтической композиции, содержащей антагонист IL-4R. Согласно определенным вариантам осуществления антагонист IL-4R представляет собой антитело или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R. Иллюстративные антитела к IL-4R, которые можно использовать в контексте способов, представленных в настоящем изобретении, описаны в других частях данного документа. Используемое в данном документе выражение "обострение астмы" означает повышение тяжести и/или частоты возникновения и/или продолжительности одного или нескольких симптомов или проявлений астмы. Выражение "обострение астмы" также включает любое ухудшение состояния дыхательной системы субъекта, которое требует терапевтического вмешательства или подлежит лечению с помощью терапевтического вмешательства по поводу астмы (такого как, например, лечение стероидами, лечение ингаляционными кортикостероидами, госпитализация и т.д.). Существует два типа явлений обострения астмы: явление потери контроля астмы (LOAC) и явление тяжелого обострения.
Согласно определенным вариантам осуществления явление потери контроля астмы (LOAC) определяют как одно или несколько из следующего: (a) 6 или более дополнительных облегчающих дыхание впрыскиваний сальбутамола/альбутерола или левосальбутамола/левалбутерола в течение 24-часового периода (по сравнению с исходным уровнем) в течение 2 последовательных дней; (b) повышения дозы ICS в 4 раза или больше по отношению к дозе на момент 2 визита и (c) применения системных кортикостероидов в течение 3 дней или больше или (d) госпитализации или визита в отделение неотложной помощи вследствие астмы, требующей применения системных кортикостероидов.
В определенных случаях обострение астмы можно обозначить как "явление тяжелого обострения астмы". Явление тяжелого обострения астмы означает случай, требующий незамедлительного вмешательства в форме лечения либо системными кортикостероидами, либо ингаляционными кортикостероидами в дозе, превышающей в четыре раза или больше дозу, принимаемую до этого случая. Согласно определенным вариантам осуществления явление тяжелого обострения астмы определяют как ухудшение астмы, требующее следующего: применения системных кортикостероидов в течение 3 дней или больше или госпитализации или визита в отделение неотложной помощи вследствие астмы, требующей применения системных кортикостероидов. Таким образом, общее выражение "обострение астмы" включает и охватывает более специфическую подкатегорию "тяжелых обострений астмы". Соответственно, включены способы снижения частоты возникновения тяжелых обострений астмы у нуждающегося в этом пациента.
"Снижение частоты" обострения астмы означает, что у субъекта, который получал фармацевтическую композицию, содержащую антагонист IL-4R, наблюдается меньше обострений астмы (т.е. меньше на по меньшей мере одно обострение астмы) после лечения, чем до лечения, или не наблюдаются обострения астмы в течение по меньшей мере 4 недель (например, 4, 6, 8, 12, 14 или более недель) после начала лечения фармацевтической композицией. "Снижение частоты" обострения астмы альтернативно означает, что после введения фармацевтической композиции, вероятность того, что у субъекта наблюдается обострение астмы снижается на по меньшей мере 10% (например, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% или больше) по сравнению с субъектом, который не получал фармацевтической композиции.
Настоящее изобретение включает способы снижения частоты обострений астмы у нуждающегося в этом субъекта, предусматривающие введение субъекту фармацевтической композиции, содержащей антагонист IL-4R, а также введение субъекту одной или нескольких поддерживающих доз ингаляционного кортикостероида (ICS) и/или одной или нескольких поддерживающих доз второго контролирующего препарата, например, бета-агониста длительного действия (LABA) или антагониста лейкотриеновых рецепторов (LTA). Подходящие ICS включают без ограничения флутиказон (например, флутиказона пропионат, например, Flovent™), будесонид, мометазон (например, мометазона фуроат, например, Asmanex™), флунизолид (например, Aerobid™), дексаметазона ацетат/фенобарбитал/теофиллин (например, Azmacort™), беклометазона дипропионат HFA (Qvar™) и т.п. Подходящие LABA включают без ограничения сальметерол (например, Serevent™), формотерол (например, Foradil™) и т.п. Подходящие LTA включают без ограничения монтелукаст (например, Singulaire™), зафирлукаст (например, Accolate™) и т.п.
Настоящее изобретение включает способы снижения частоты обострений астмы у нуждающегося в этом субъекта, предусматривающие введение фармацевтической композиции, содержащей антагонист IL-4R, субъекту, а также введение субъекту одного или нескольких лекарственных препаратов, облегчающих дыхание, для устранения или снижения одного или нескольких ассоциированных с астмой симптомов. Подходящие лекарственные препараты, облегчающие дыхание, включают без ограничения агонисты бета2-адренергических рецепторов длительного действия, такие как, например, альбутерол (например, сальбутамол, например, Proventil™, Ventolin™, Xopenex™ и т.п.), пирбутерол (например, Maxair™), метапротеренол (например, Alupent™) и т.п.
Способы улучшения ассоциированных с астмой параметров
Настоящее изобретение также включает способы улучшения одного или нескольких ассоциированных с астмой параметров у нуждающегося в этом субъекта, где способы предусматривают введение субъекту фармацевтической композиции, содержащей антагонист IL-4R. Снижение частоты обострения астмы (как описано выше) может быть взаимосвязано с улучшением одного или нескольких ассоциированных с астмой параметров; однако такая взаимосвязь необязательно наблюдается во всех классах.
Примеры "ассоциированных с астмой параметров" включают (1) относительное процентное изменение от исходного уровня (например, на неделе 12) объема форсированного выдоха за 1 секунду (FEV1); (2) относительное процентное изменение от исходного уровня (например, на неделе 12), измеренное с помощью скорости форсированного выдоха при 25-75% от объема легких (FEF25-75); (3) частоту в годовом исчислении явлений потери контроля астмы, на протяжении периода лечения; (4) частоту в годовом исчислении явлений тяжелого обострения на протяжении периода лечения; (5) время до наступления явлений потери контроля астмы на протяжении периода лечения; (6) время до наступления явлений тяжелого обострения на протяжении периода исследования; (7) время до наступления явлений потери контроля астмы на протяжении всего периода исследования; (8) время до наступления явлений тяжелого обострения на протяжении всего периода исследования; (9) использование медицинских услуг; (10) изменение от исходного уровня до недели 12 i) показателей симптомов астмы в утреннее и вечернее время, ii) показателя ACQ-5, iii) показателя AQLQ, iv) PEF в утреннее и вечернее время, v) числа ингаляций/день сальбутамола/альбутерола или левосальбутамола/левалбутерола для облегчения симптомов, vi) ночных пробуждений; (11) изменение от исходного уровня до недели 12 и недели 24 i) показателя теста для оценки результатов лечения заболеваний носа и придаточных пазух носа из 22 вопросов (SNOT-22), ii) показателя по госпитальной шкале тревоги и депрессии (HADS), iii) показателя по опроснику EuroQual (EQ-5D-3L или EQ-5D-5L). "Улучшение ассоциированного с астмой параметра" означает повышение от исходного уровня одного или нескольких из FEV1, PEF до полудня или PEF после полудня и/или снижение от исходного уровня одного или нескольких из суточного применения альбутерола/левалбутерола, показателя ACQ5, среднего числа пробуждений в ночное время или показателя SNOT-22. Используемый в данном документе термин "исходный уровень" по отношению к ассоциированному с астмой параметру означает числовую величину ассоциированного с астмой параметра для пациента до или в течение всего периода введения фармацевтической композиции, содержащей антагонист IL-4R.
Чтобы определить, "улучшился ли" ассоциированный с астмой параметр, параметр измеряют количественно на исходном уровне и во временной точке после введения фармацевтической композиции, описанной в данном документе. Например, ассоциированный с астмой параметр можно измерять в день 1, день 2, день 3, день 4, день 5, день 6, день 7, день 8, день 9, день 10, день 11, день 12, день 14 или на неделе 3, неделе 4, неделе 5, неделе 6, неделе 7, неделе 8, неделе 9, неделе 10, неделе 11, неделе 12, неделе 13, неделе 14, неделе 15, неделе 16, неделе 17, неделе 18, неделе 19, неделе 20, неделе 21, неделе 22, неделе 23, неделе 24 или дольше после первичного лечения фармацевтической композицией. Различие между величиной параметра в определенной временной точке после начала лечения и величиной параметра на исходном уровне используют для установления того, произошло ли "улучшение" ассоциированного с астмой параметра (например, повышение или снижение, в зависимости от ситуации, в зависимости от конкретного параметра, измерение которого осуществляют).
Термины "приобретать" или "приобретение", используемые в данном документе, относятся к получению во владение физического объекта или величины, например, числовой величины, "прямым приобретением" или "непрямым приобретением" физического объекта или величины, таких как ассоциированный с астмой параметр. "Прямое приобретение" означает осуществление способа (например, осуществление синтетического или аналитического способа) для получения физического объекта или величины. "Непрямое приобретение" относится к получению физического объекта или величины от другой стороны или источника (например, лаборатории третьей стороны, которая непосредственно приобрела физический объект или величину). Прямое приобретение физического объекта включает осуществление способа, который предусматривает физическое изменение физического вещества, например, исходного материала. Иллюстративные изменения включают получение физического объекта из двух или более исходных материалов, разрезание или фрагментацию вещества, отделение или очистку вещества, объединение двух или более отдельных объектов в смесь, осуществление химической реакции, которая включает разрыв или образование ковалентной или нековалентной связи. Непосредственное приобретение величины включает осуществление способа, который предусматривает физическое изменение образца или другого вещества, например, осуществление аналитического способа, который предусматривает физическое изменение вещества, например, образца, аналита или реагента (иногда обозначаемое в данном документе как "физический анализ").
Информация, которую приобретают непосредственно, может быть представлена в форме отчета, например, представленного в бумажной или электронной форме, например, из базы данных или приложения в онлайн режиме ("App"). Отчет или информация может быть представлена, например, учреждением здравоохранения, таким как больница или клиника; или медицинским работником, таким как врач или медицинская сестра.
Объем форсированного выдоха за 1 секунду (FEV1). Согласно определенным вариантам осуществления введение пациенту антагониста IL-4R в результате приводит к повышению от исходного уровня объема форсированного выдоха за 1 секунду (FEV1). Способы измерения FEV1 известны из уровня техники. Например, для измерения FEV1 у пациента можно использовать спирометр, который соответствует рекомендациям Американского торакального общества (ATS)/Европейского респираторного общества (ERS) 2005 года. В качестве руководства можно использовать стандартизацию спирометрии согласно ATS/ERS. Спирометрию, как правило, проводят с 6 до 10 часов утра после воздержания от альбутерола в течение по меньшей мере 6 часов. Легочные функциональные пробы, как правило, проводят в положении сидя, а наибольшую величину записывают в качестве FEV1 (в литрах).
Настоящее изобретение включает терапевтические способы, которые в результате приводят к повышению FEV1 от исходного уровня на по меньшей мере 0,05 л на неделе 12 после начала лечения фармацевтической композицией, содержащей антагонист IL-4R. Например, введение антагониста IL-4R нуждающемуся в этом субъекту вызывает увеличение FEV1 от исходного уровня на приблизительно 0,05Л, 0,10Л, 0,12Л, 0,14Л, 0,16Л, 0,18Л, 0,20Л, 0,22Л, 0,24Л, 0,26Л, 0,28Л, 0,30Л, 0,32Л, 0,34Л, 0,36Л, 0,38Л, 0,40Л, 0,42Л, 0,44Л, 0,46Л, 0,48Л, 0,50Л или больше на неделе 12.
FEF25-75%. Согласно определенным вариантам осуществления введение пациенту антагониста IL-4R в результате приводит к повышению FEF25-75% от исходного уровня. Способы измерения FEF известны из уровня техники. Например, для измерения FEV1 у пациента можно использовать спирометр, который соответствует рекомендациям Американского торакального общества (ATS)/Европейского респираторного общества (ERS) 2005 года. FEF25-75 (скорость форсированного выдоха от 25% до 75%) представляет собой скорость (в литрах в секунду), с которой человек может высвобождать среднюю половину своего воздуха во время максимального выдоха (т.e. форсированная жизненная емкость или FVC). Параметр относится к среднему потоку от точки, в которой 25 процентов FVC было выдохнуто, до точки, в которой 75 процентов FVC было выдохнуто. FEF25-75% субъекта предоставляет информацию о функции мелких дыхательных путей, такую как степень заболевания и/или воспаления мелких дыхательных путей. Изменение FEF25-75 представляет собой ранний показатель обструктивного заболевания легких. В определенных вариантах осуществления улучшение и/или увеличение параметра FEF25-75% представляет собой улучшение на по меньшей мере 10%, 25%, 50% или больше по сравнению с исходным уровнем. В определенных вариантах осуществления способы по настоящему изобретению приводят к нормальным значениям FEF25-75% у субъекта (например, значениям в диапазоне от 50-60% и до 130% от среднего значения).
Максимальная скорость выдоха в утреннее и вечернее время (PEF до полудня и PEF после полудня). Согласно определенным вариантам осуществления введение антагониста IL-4R пациенту в результате приводит к повышению от исходного уровня максимальной скорости выдоха в утреннее (до полудня) и/или вечернее время (после полудня) (PEF до полудня и/или PEF после полудня). Способы измерения PEF известны из уровня техники. Например, согласно одному способу измерения PEF пациентам выдают электронный измеритель PEF для записи PEF в утреннее (до полудня) и вечернее время (после полудня) (а также суточного применения альбутерола, показателей симптомов астмы в утреннее и вечернее время и числа пробуждений в ночное время вследствие симптомов астмы, которые требуют экстренного приема лекарственных препаратов). Пациентов инструктируют относительно применения устройства и предоставляют пациентам инструкции по применению электронного измерителя PEF в письменном виде. Кроме того, медицинский работник может инструктировать пациентов в отношении того, как записывать соответствующие переменные в электронный измеритель PEF. PEF до полудня, как правило, определяют в течение 15 минут после подъема (с 6 до 10 часов утра) перед приемом альбутерола в любой дозе. PEF после полудня, как правило, определяют в вечернее время (с 6 до 10 часов вечера) перед приемом альбутерола в любой дозе. Субъекты должны стараться отказаться от приема альбутерола в течение по меньшей мере 6 часов до измерения своей PEF. Пациент предпринимает три попытки измерения PEF, и все 3 величины записывают с помощью электронного измерителя PEF. Обычно для оценки используют максимальную величину. Исходную PEF до полудня можно рассчитать в виде среднего измерения до полудня, записанного в течение 7 дней до введения первой дозы фармацевтической композиции, содержащей антагонист IL-4R, а исходную PEF после полудня можно рассчитать в виде среднего измерения после полудня, записанного в течение 7 дней до введения первой дозы фармацевтической композиции, содержащей антагонист IL-4R.
Настоящее изобретение включает терапевтические способы, которые в результате приводят к повышению PEF до полудня и/или PEF после полудня от исходного уровня на по меньшей мере 1,0 л/мин. на неделе 12 после начала лечения фармацевтической композицией, содержащей антагонист IL-4R. Например, согласно настоящему изобретению введение антагониста IL-4R нуждающемуся в этом субъекту вызывает повышение PEF от исходного уровня на приблизительно 0,5 л/мин., 1,0 л/мин., 1,5 л/мин., 2,0 л/мин., 2,5 л/мин., 3,0 л/мин., 3,5 л/мин., 4,0 л/мин., 4,5 л/мин., 5,0 л/мин., 5,5 л/мин., 6,0 л/мин., 6,5 л/мин., 7,0 л/мин., 7,5 л/мин., 8,0 л/мин., 8,5 л/мин., 9,0 л/мин., 9,5 л/мин., 10,0 л/мин., 10,5 л/мин., 11,0 л/мин., 12,0 л/мин., 15 л/мин., 20 л/мин. или больше на неделе 12.
Применение альбутерола/левалбутерола. Согласно определенным вариантам осуществления введение пациенту антагониста IL-4R в результате приводит к снижению от исходного уровня суточного применения альбутерола или левалбутерола. Число ингаляций альбутерола/левалбутерола может записываться пациентами ежедневно в дневнике, измерителе PEF или другом записывающем устройстве. На протяжении лечения фармацевтической композицией, описанной в данном документе, применение альбутерола/левалбутерола, как правило, можно осуществлять при необходимости в зависимости от симптомов, а не на постоянной основе или профилактически. Исходное число ингаляций альбутерола/левалбутерола в сутки можно рассчитать на основе средней величины в течение 7 дней до введения первой дозы фармацевтической композиции, содержащей антагонист IL-4R.
Настоящее изобретение включает терапевтические способы, которые в результате приводят к снижению применения альбутерола/левалбутерола от исходного уровня на по меньшей мере 0,25 впрыскивания в сутки на неделе 12 после начала лечения фармацевтической композицией, содержащей антагонист IL-4R. Например, введение антагониста IL-4R нуждающемуся в этом субъекту вызывает снижение применения альбутерола/левалбутерола от исходного уровня на приблизительно 0,25 впрыскивания в сутки, 0,50 впрыскивания в сутки, 0,75 впрыскивания в сутки, 1,00 впрыскивание в сутки, 1,25 впрыскивания в сутки, 1,5 впрыскивания в сутки, 1,75 впрыскивания в сутки, 2,00 впрыскивания в сутки, 2,25 впрыскивания в сутки, 2,5 впрыскивания в сутки, 2,75 впрыскивания в сутки, 3,00 впрыскивания в сутки или больше на неделе 12.
Применение OCS. Согласно определенным вариантам осуществления введение антагониста IL-4R пациенту можно применять в сочетании с OCS, таким как преднизон для перорального применения. Количество введений OCS может ежедневно записываться пациентами в дневнике, измерителе PEF или другом записывающем устройстве. На протяжении лечения фармацевтической композицией, описанной в данном документе, периодическое кратковременное применение преднизона, как правило, можно использовать для контроля острых эпизодов астмы, например, эпизодов, при которых с помощью бронходилататоров и других противовоспалительных средств не удается контролировать симптомы. В других аспектах преднизон используют параллельно с другим ICS или в качестве замены другого ICS. Преднизон для перорального применения можно вводить в дозах приблизительно 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 35 мг или 40 мг. OCS можно необязательно вводить один раз в сутки или несколько раз в сутки (например, два раза в сутки, три раза в сутки, четыре раза в сутки и т.п.)
В определенных иллюстративных вариантах осуществления в настоящем изобретении предусматриваются способы снижения или устранения зависимости субъекта от применения OCS. Снижение или устранение зависимости от стероидов очень преимущественно и необходимо. В определенных вариантах осуществления снижение на 50% или больше (например, 50%, 60%, 70%, 80%, 90% или больше) дозы OCS достигается после введения средства терапии на основе антитела к IL-4R в течение определенного периода времени (например, на неделе 24). В определенных вариантах осуществления OCS практически исключается через 40 недель, 45 недель, 50 недель, 52 недели или больше после первой дозы, следующей за введением нагрузочной дозы. В других вариантах осуществления уровень применения OCS снижается до менее 5 мг в сутки (например, менее 5 мг, 4 мг, 3 мг, 2 мг или меньше в сутки). В других вариантах осуществления зависимость от применения OCS по существу устраняется через 3 месяца, 6 месяцев, 9 месяцев или 1 год после лечения антителом к IL4R или его фрагментом.
Показатель согласно опроснику по контролю астмы из 5 пунктов (ACQ). Согласно определенным вариантам осуществления введение антагониста IL-4R пациенту в результате приводит к снижению от исходного уровня показателя согласно опроснику по контролю астмы из пяти пунктов (ACQ5). ACQ5 представляет собой утвержденный опросник для оценки контроля астмы.
Настоящее изобретение включает терапевтические способы, которые в результате приводят к снижению показателя ACQ5 от исходного уровня на по меньшей мере 0,10 балла на неделе 12 после начала лечения фармацевтической композицией, содержащей антагонист IL-4R. Например, введение антагониста IL-4R нуждающемуся в этом субъекту вызывает снижение показателя ACQ от исходного уровня на приблизительно 0,10 балла, 0,15 балла, 0,20 балла, 0,25 балла, 0,30 балла, 0,35 балла, 0,40 балла, 0,45 балла, 0,50 балла, 0,55 балла, 0,60 балла, 0,65 балла, 0,70 балла, 0,75 балла, 0,80 балла, 0,85 балла или больше на неделе 12.
Пробуждения в ночное время. Согласно определенным вариантам осуществления введение антагониста IL-4R пациенту в результате приводит к снижению от исходного уровня среднего числа пробуждений в ночное время.
В определенных вариантах осуществления способы обеспечивают снижение среднего числа пробуждений в ночное время от исходного уровня в по меньшей мере приблизительно 0,10 раза за ночь на неделе 12 после начала лечения. Например, введение антагониста IL-4R нуждающемуся в этом субъекту может вызвать снижение среднего числа пробуждений в ночное время от исходного уровня в приблизительно 0,10 раза за ночь, 0,15 раза за ночь, 0,20 раза за ночь, 0,25 раза за ночь, 0,30 раза за ночь, 0,35 раза за ночь, 0,40 раза за ночь, 0,45 раза за ночь, 0,50 раза за ночь, 0,55 раза за ночь, 0,60 раза за ночь, 0,65 раза за ночь, 0,70 раза за ночь, 0,75 раза за ночь, 0,80 раза за ночь, 0,85 раза за ночь, 0,90 раза за ночь, 0,95 раза за ночь, 1,0 раз за ночь, 2,0 раза за ночь или больше на неделе 12.
Показатель теста для оценки результатов лечения заболеваний носа и придаточных пазух носа из 22 пунктов (SNOT-22). Согласно определенным вариантам осуществления введение пациенту антагониста IL-4R в результате приводит к снижению от исходного уровня показателя теста для оценки результатов лечения заболевания носа и придаточных пазух носа из 22 пунктов (SNOT-22). SNOT-22 представляет собой утвержденный опросник для определения влияния хронического риносинусита на качество жизни (Hopkins et al. 2009, Clin. Otolaryngol. 34: 447-454).
Настоящее изобретение включает терапевтические способы, которые в результате приводят к снижению показателя SNOT-22 от исходного уровня на по меньшей мере 1 балл на неделе 12 после начала лечения фармацевтической композицией, содержащей антагонист IL-4R. Например, введение антагониста IL-4R нуждающемуся в этом субъекту может вызывать снижение показателя SNOT-22 от исходного уровня на приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 баллов или больше на неделе 12.
Биомаркеры. В определенных вариантах осуществления у субъекта наблюдается улучшение функции легких, измеряемое с помощью биомаркера, например, биомаркера, связанного с тяжелой стероидозависимой астмой или тяжелой неконтролируемой астмой. Например, биомаркером может быть фракция оксида азота в выдыхаемом воздухе (FeNO), эотаксин-3, общий IgE, периостин или хемокин, регулируемый тимусом и активацией (TARC). В определенных вариантах осуществления на улучшение функции легких указывает снижение или увеличение (в зависимости от ситуации) на неделе 4, неделе 12 или неделе 24 после лечения.
Способы лечения астмы
В некоторых вариантах осуществления в настоящем изобретении предусматриваются способы лечения астмы, в том числе, например, неконтролируемой астмы от умеренной до тяжелой степени или в недостаточной степени контролируемой астмы, у нуждающегося в этом субъекта, где способы предусматривают введение субъекту фармацевтической композиции, содержащей антагонист IL-4R. В определенных вариантах осуществления способы пригодны для лечения неконтролируемой астмы от умеренной до тяжелой степени у субъекта.
Используемый в данном документе термин "астма" можно использовать взаимозаменяемо с "рецидивирующей астмой" или "бронхиальной астмой". "Астма", "бронхиальная астма" и "рецицивирующая астма" относятся к астме, для которой справедлива одна или любая комбинация из следующих: симптомы возникают в течение 2 дней в неделю или реже; симптомы не нарушают повседневной деятельности; симптомы в ночное время появляются реже 2 дней в месяц; или одно или несколько исследований легочных функций (например, объем форсированного выдоха за одну секунду (FEV1) и/или максимальная скорость выдоха (PEF) более 80%) являются нормальными, когда субъект не страдает от приступа астмы.
Используемый в данном документе термин "персистирующая астма" или "персистирующая бронхиальная астма" относится к астме, которая протекает более тяжело, чем (бронхиальная) астма/рецидивирующая (бронхиальная) астма. У субъекта, страдающего персистирующей астмой или персистирующей бронхиальной астмой, наблюдается одно или несколько из следующего: симптомы чаще 2 дней в неделю; симптомы, которые не нарушают повседневной деятельности; симптомы в ночное время, которые появляются более 2 дней в месяц; или одно или несколько исследований легочных функций (например, объем форсированного выдоха за одну секунду (FEV1) и/или максимальная скорость выдоха (PEF) менее 80%), которые не являются нормальными, когда субъект не страдает от приступов астмы; субъект зависит от ежедневного применения лекарственного препарата для контроля астмы; субъект принимал системный стероид чаще одного раза за последний год после тяжелого приступа астмы или применение бета-2-агониста кратковременного действия более двух дней в неделю для облегчения симптомов астмы.
Астма/рецидивирующая астма, бронхиальная астма/рецидивирующая бронхиальная астма и персистирующая астма/персистирующая бронхиальная астма могут быть классифицированы как "слабая", "умеренная", "тяжелая" или "степень от умеренной до тяжелой". "Слабую рецидивирующую астму" или "слабую рецидивирующую бронхиальную астму" определяют по наличию симптомов менее одного раза в неделю и объему форсированного выдоха за одну секунду (FEV1) или максимальной скорости выдоха (PEF) ≥ 80%. "Слабая персистирующая астма" или "слабая персистирующая бронхиальная астма" отличается тем, что частота возникновения симптомов составляет более одного раза в неделю, но реже одного раза в день, а вариабельность FEV1 или PEF составляет < 20% - 30%. "Умеренную рецидивирующую астму" или "умеренную рецидивирующую бронхиальную астму" определяют по наличию симптомов менее одного раза в неделю и объему форсированного выдоха за одну секунду (FEV1) или максимальной скорости выдоха (PEF) на уровне 60-80%. "Умеренную персистирующую астму" или "умеренную персистирующую бронхиальную астму" определяют по наличию ежедневных симптомов, обострений, которые могут влиять на деятельность и/или сон, ночных симптомов более одного раза в неделю, ежедневному применению ингаляционного бета-2-агониста кратковременного действия и объему форсированного выдоха за одну секунду (FEV1) или максимальной скорости выдоха (PEF) на уровне 60-80%. "Тяжелую рецидивирующую астму" или "тяжелую рецидивирующую бронхиальную астму" определяют по наличию симптомов менее одного раза в неделю и объему форсированного выдоха за одну секунду (FEV1) или максимальной скорости выдоха (PEF) на уровне 60%. "Тяжелую персистирующую астму" или "тяжелую персистирующую бронхиальную астму" определяют по наличию ежедневных симптомов, частых обострений, которые могут влиять на деятельность и/или сон, частых ночных симптомов, ограничению физической активности, ежедневному применению ингаляционного бета-2-агониста кратковременного действия и объему форсированного выдоха за одну секунду (FEV1) или максимальной скорости выдоха (PEF) на уровне 60%. "Рецидивирующую астму от умеренной до тяжелой степени" или "рецидивирующую бронхиальную астму от умеренной до тяжелой степени" определяют по наличию симптомов между симптомами умеренной рецидивирующей астмы/умеренной рецидивирующей бронхиальной астмы и тяжелой рецидивирующей астмы/тяжелой рецидивирующей бронхиальной астмы. "Персистирующую астму от умеренной до тяжелой степени" или "персистирующую бронхиальную астму от умеренной до тяжелой степени" определяют по наличию симптомов между симптомами умеренной персистирующей астмы/умеренной персистирующей бронхиальной астмы и тяжелой персистирующей астмы/тяжелой персистирующей бронхиальной астмы.
Используемый в данном документе термин "в недостаточной степени контролируемая астма" относится к пациентам, у которых астма либо "не контролируется надлежащим образом" либо "очень слабо контролируется", как определено в "Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma," National Heart, Blood and Lung Institute, NIH, Aug. 28, 2007. "Не контролируемую надлежащим образом астму" определяют по наличию симптомов более двух дней в неделю, пробуждений в ночное время от одного до трех раз в неделю, некоторых ограничений повседневной деятельности, применению бета2-агониста кратковременного действия для контроля симптомов более двух раз в неделю, FEV1 на уровне 60-80% от прогнозируемой величины и/или личного рекорда, показателю ATAQ, составляющему 1-2, показателю ACQ, составляющему 1,5 или больше, и показателю ACT, составляющему 16-19. "Очень слабо контролируемую астму" определяют по наличию симптомов в течение дня, пробуждений в ночное время четыре раза или больше в неделю, существенных ограничений повседневной деятельности, применению бета2-агониста кратковременного действия для контроля симптомов несколько раз в день, FEV1 менее 60% от прогнозируемой величины и/или личного рекорда, показателю ATAQ, составляющему 3-4, отсутствию данных о показателе ACQ и показателю ACT, составляющему 15 или меньше.
В некоторых вариантах осуществления субъекта определяют как имеющего "неконтролируемую астму от умеренной до тяжелой степени", если субъект получает такой диагноз от врача на основании руководств 2009 года Глобальной инициативы по бронхиальной астме (GINA) и одного или нескольких из следующих критериев: i) текущее лечение умеренными или высокими дозами ICS/LABA (2 введения 250 мкг флутиказона пропионата два раза в сутки или эквипотентная суточная доза ICS) со стабильной дозой ICS/LABA в течение периода, составляющего 1 месяц или больше, до введения нагрузочной дозы антагониста IL-4R; ii) FEV1, составляющий 40-80% от прогнозируемой нормы до введения нагрузочной дозы антагониста IL-4R; iii) показатель ACQ-5, составляющий 1,5 или больше, до введения нагрузочной дозы антагониста IL-4R; iv) обратимость на по меньшей мере 12% и 200 мл FEV1 после 200-400 мкг (2-4 ингаляции) сальбутамола/альбутерола до введения нагрузочной дозы антагониста IL-4R; или v) в течение 1 года до введения нагрузочной дозы антагониста IL-4R наблюдалось любое из следующих явлений: (a) лечение с применением 1 или более системной пульс-терапии стероидами (пероральной или парентеральной) по причине обострения астмы, (b) госпитализация или визит в отделение неотложной/скорой медицинской помощи по причине обострения астмы.
Выражение "тяжелая астма" относится к астме, при которой соответствующий контроль не может быть достигнут путем лечения высокими дозами ингаляционных кортикостероидов и дополнительных контролирующих препаратов (например, ингаляционных бета 2-агонистов длительного действия, монтелукаста и/или теофиллина) или терапией пероральным кортикостероидом (например, в течение по меньшей мере шести месяцев в году), или утрачивается, когда лечение снижается. В определенных вариантах осуществления тяжелая астма включает астму, лечение которой осуществляют высокими дозами ICS и по меньшей мере одним дополнительным контролирующим препаратом (например, LABA, монтелукаст или теофиллин) или пероральными кортикостероидами > 6 месяцев/год, где по меньшей мере одно из следующего происходит или может произойти, если лечение снижается: ACT < 20 или ACQ > 1,5; по меньшей мере 2 обострения за последние 12 месяцев; по меньшей мере 1 обострение, лечение которого осуществляли в больнице или требующее искусственной вентиляции легких за последние 12 месяцев; или FEV1<80% (если FEV1/FVC ниже нижней границы нормы).
Выражение "стероидозависимая астма" относится к астме, которая требует одного или нескольких из следующих способов лечения: лечение в виде частой кратковременной пульс-терапии пероральными кортикостероидами в течение последних 12 месяцев; регулярное применение высоких доз ингаляционных кортикостероидов в течение последних 12 месяцев; регулярное применение инъекционных кортикостероидов длительного действия; ежедневное применение пероральных кортикостероидов; пероральные кортикостероиды через день или длительное применение пероральных кортикостероидов в прошлом году.
Выражение "зависимая от перорального кортикостероида астма" относится к субъекту, имеющему ≥ 3 30-дневных курсов приема перорального кортикостероида (OCS) в течение 12-месячного периода и первичный диагноз астмы в течение 12 месяцев после первого курса приема OCS. У субъектов с OCS-зависимой астмой могут также наблюдаться одно или любая комбинацию из следующего: получили прописанные врачом LABA и высокую дозу ICS (общая суточная доза > 500 мкг эквивалента сухого порошкообразного состава на основе флутиказона пропионата) в течение по меньшей мере 3 месяцев (ICS и LABA могут быть частями комбинированного продукта или предоставлены в отдельных ингаляторах); получили дополнительные поддерживающие контролирующие астму лекарственные препараты в соответствии со стандартной практикой лечения, например, антагонисты лейкотриеновых рецепторов (LTRA), теофиллин, антагонисты мускариновых рецепторов длительного действия (LAMA), вторичные ICS и кромоны; получили OCS для лечения астмы в дозе от ≥ 7,5 до ≤ 30 мг (преднизон или эквивалент преднизолона); получили дозу OCS, вводимую через день (или разные дозы через день); FEV1 в утреннее время до введения бронхолитического средства (BD) составляет < 80% от прогнозируемой нормы; наличие признаков астмы, подтверждаемых обратимостью FEV1 ≥ 12% и ≥ 200 мл после BD (альбутерол/сальбутатомол) (15-30 мин. после введения 4 впрыскиваний альбутерола/сальбутатомола); или имеют в анамнезе по меньшей мере одно явление обострения астмы в течение 12 месяцев.
В одном аспекте предусматриваются способы лечения астмы, предусматривающие (a) отбор пациента, который характеризуется содержанием эозинофилов в крови, составляющим по меньшей мере 300 клеток на микролитр; и (b) введение пациенту фармацевтической композиции, содержащей антагонист IL-4R.
В другом аспекте предусматриваются способы лечения астмы, предусматривающие (a) отбор пациента, который характеризуется содержанием эозинофилов в крови, составляющим 200-299 клеток на микролитр; и (b) введение пациенту фармацевтической композиции, содержащей антагонист IL-4R.
В другом аспекте предусматриваются способы лечения астмы, предусматривающие (a) отбор пациента, который характеризуется содержанием эозинофилов в крови, составляющим менее 200 клеток на микролитр; и (b) введение пациенту фармацевтической композиции, содержащей антагонист IL-4R.
В родственном аспекте предусматриваются способы лечения астмы, предусматривающие дополнительную терапию к фоновой терапии. В определенных вариантах осуществления антагонист IL-4R вводят в виде дополнительной терапии пациенту с астмой, который получает фоновую терапию в течение определенного периода времени (например, 1 недели, 2 недель, 3 недель, 1 месяца, 2 месяцев, 5 месяцев, 12 месяцев, 18 месяцев, 24 месяцев или дольше) (также называемого "стабильной фазой"). В некоторых вариантах осуществления фоновая терапия предусматривает ICS и/или LABA.
В некоторых вариантах осуществления настоящее изобретение включает способ снижения зависимости пациента с астмой от ICS и/или LABA для лечения одного или нескольких обострений астмы, предусматривающий (a) отбор пациента, у которого имеется астма от умеренной до тяжелой степени, которая не контролируется фоновой терапией астмы, предусматривающей ICS, LABA или их комбинацию; и введение пациенту фармацевтической композиции, содержащей антагонист IL-4R.
В некоторых вариантах осуществления настоящее изобретение охватывает способы лечения или облегчения состояний или осложнений, ассоциированных с астмой, таких как хронический риносинусит, аллергический ринит, аллергический грибковый риносинусит, аллергический бронхолегочный аспергиллез, объединенное заболевание дыхательных путей, синдром Черджа-Стросса, васкулит, хроническое обструктивное заболевание легких (COPD) и бронхоспазм, вызванный физической нагрузкой.
Настоящее изобретение также включает способы лечения персистирующей астмы. Используемый в данном документе термин "персистирующая астма" означает, что у субъекта возникают симптомы по меньшей мере один раз в неделю днем и/или ночью, при этом симптомы длятся от нескольких часов до нескольких дней. В определенных альтернативных вариантах осуществления персистирующая астма является "слабо персистирующей" (например, более двух раз в неделю, но менее одного раза в день с симптомами, которые являются достаточно тяжелыми, чтобы нарушать повседневную деятельность или сон, и/или если дыхательная функция является нормальной или обратимой при ингаляции бронходилататора), "умеренно персистирующей" (например, симптомы появляются ежедневно, при этом сон прерывается по меньшей мере раз в неделю и/или при этом дыхательная функция является умеренно нарушенной) или "тяжело персистирующей" (например, непрерывные симптомы, несмотря на корректное применение утвержденных лекарственных препаратов, и/или если дыхательная функция сильно нарушена).
Антагонисты рецепторов интерлейкина-4
Способы, представленные в настоящем изобретении, предусматривают введение нуждающемуся в этом субъекту терапевтической композиции, содержащей антагонист IL-4R. Используемый в данном документе термин "антагонист IL-4R" означает любое средство, которое связывается или взаимодействует с IL-4R человека и ингибирует нормальную биологическую сигнальную функцию IL-4R при экспрессии IL-4R на клетке in vitro или in vivo. Неограничивающие примеры категорий антагонистов IL-4R включают малые молекулы антагонистов IL-4R, аптамеры к IL-4R, пептидные антагонисты IL-4R (например, молекулы "пептид-ассоциированных антител"), а также антитела или антигенсвязывающие фрагменты антител, которые специфически связываются с IL-4R человека. Согласно определенным вариантам осуществления антагонист IL-4R предусматривает антитело к IL-4R, которое можно использовать в контексте способов, представленных в настоящем изобретении, описанное в других частях данного документа. Например, согласно одному варианту осуществления антагонистом IL-4R является антитело или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R и содержат последовательности CDR (определяющего комплементарность участка) тяжелой цепи и легкой цепи из вариабельного участка тяжелой цепи (HCVR) и вариабельного участка легкой цепи (LCVR) под SEQ ID NO:1 и 2 соответственно.
Термин "IL4R человека" (hIL-4R) относится к рецептору цитокинов человека, который специфически связывается с интерлейкином-4 (IL-4), таким как IL-4Rα.
Термин "антитело" относится к молекулам иммуноглобулинов, содержащим четыре полипептидные цепи, две тяжелые (H) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями, а также их мультимерам (например, IgM). Каждая тяжелая цепь содержит вариабельный участок тяжелой цепи (в данном документе обозначен аббревиатурой HCVR или VH) и константный участок тяжелой цепи. Константный участок тяжелой цепи содержит три домена: CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельный участок легкой цепи (в данном документе обозначен аббревиатурой LCVR или VL) и константный участок легкой цепи. Константный участок легкой цепи содержит один домен (CL1). Участки VH и VL можно дополнительно подразделять на участки гипервариабельности, называемые участками, определяющими комплементарность (CDR), чередующиеся с более консервативными участками, называемыми каркасными участками (FR). Каждый VH и VL состоит из трех CDR и четырех FR, расположенных от аминоконца к карбоксиконцу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. В различных вариантах осуществления FR антитела к IL-4R (или его антигенсвязывающего участка) могут быть идентичны последовательностям иммуноглобулина зародышевой линии человека или могут быть модифицированы естественным или искусственным путем. Аминокислотная консенсусная последовательность может быть определена на основании анализа "бок-о-бок" двух или более CDR.
Термин "антитело" также включает антигенсвязывающие фрагменты целых молекул антител. Термины "антигенсвязывающий участок" антитела, "антигенсвязывающий фрагмент" антитела и т.п., используемые в данном документе, включают любые встречающиеся в природе, получаемые ферментативным путем, синтетические или получаемые методиками генной инженерии полипептид или гликопротеин, специфически связывающиеся с антигеном с образованием комплекса. Антигенсвязывающие фрагменты антитела могут быть получены, например, из целых молекул антител с помощью любых подходящих стандартных методик, таких как протеолитическое расщепление или рекомбинантные методики генной инженерии, включающие манипуляцию с ДНК, кодирующей вариабельные и необязательно константные домены антитела, и ее экспрессию. Такая ДНК известна и/или легкодоступна, например, из коммерческих источников, библиотек ДНК (в том числе, например, библиотек "фаг-антитело"), или может быть синтезирована. ДНК можно секвенировать и с ней можно проводить химические манипуляции или манипуляции с помощью методик молекулярной биологии, например, для расположения одного или нескольких вариабельных и/или константных доменов в подходящей конфигурации или для введения кодонов, создания цистеиновых остатков, модификации, добавления или удаления аминокислот и т.д.
Неограничивающие примеры антигенсвязывающих фрагментов включают (i) Fab-фрагменты; (ii) F(ab')2-фрагменты; (iii) Fd-фрагменты; (iv) Fv-фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb-фрагменты и (vii) минимальные распознающие единицы, состоящие из аминокислотных остатков, имитирующих гипервариабельный участок антитела (например, выделенный участок, определяющий комплементарность (CDR), такой как пептид CDR3), или пептид c ограниченной конформационной свободой FR3-CDR3-FR4. Другие сконструированные молекулы, такие как домен-специфические антитела, однодоменные антитела, антитела с удаленным доменом, химерные антитела, CDR-привитые антитела, диатела, триатела, тетратела, минитела, нанотела (например, моновалентные антитела, бивалентные антитела и т.д.), иммунофармацевтические средства на основе модульного белка малого размера (SMIP) и вариабельные домены IgNAR акулы, также включены в выражение "антигенсвязывающий фрагмент".
Антигенсвязывающий фрагмент антитела, как правило, будет содержать по меньшей мере один вариабельный домен. Вариабельный домен может иметь любой размер или аминокислотный состав и будет, как правило, содержать по меньшей мере один CDR, который прилегает к одной или нескольким каркасным последовательностям или находится в рамке считывания с ними. В антигенсвязывающих фрагментах, имеющих домен VH, связанный с доменом VL, домены VH и VL могут располагаться относительно другу друга в любом подходящем порядке. Например, вариабельный участок может быть димерным и содержать димеры VH-VH, VH-VL или VL-VL. Альтернативно, антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
В определенных вариантах осуществления антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный с по меньшей мере одним константным доменом. Неограничивающие иллюстративные конфигурации вариабельных и константных доменов, которые можно выявить в антигенсвязывающем фрагменте антитела, описанного в данном документе, включают (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3 и (xiv) VL-CL. В любой конфигурации вариабельных и константных доменов, в том числе любых иллюстративных конфигурациях, перечисленных выше, вариабельные и константные домены могут быть либо непосредственно связаны друг с другом, либо могут быть связаны с помощью целого или частичного шарнирного или линкерного участка. Шарнирный участок может состоять из по меньшей мере 2 (например, 5, 10, 15, 20, 40, 60 или больше) аминокислот, которые в результате приводят к образованию гибкой или полугибкой связи между прилегающими вариабельными и/или константными доменами в одной молекуле полипептида, как правило, шарнирный участок может состоять из 2-60 аминокислот, как правило, 5-50 или, как правило, 10-40 аминокислот. Кроме того, антигенсвязывающий фрагмент антитела, описанного в данном документе, может содержать гомодимер или гетеродимер (или другой мультимер) из любых конфигураций вариабельных и константных доменов, изложенных выше, в нековалентной ассоциации друг с другом и/или с одним или несколькими мономерными доменами VH или VL (например, с помощью дисульфидной связи(связей)).
Как и в случае с целыми молекулами антител антигенсвязывающие фрагменты могут быть моноспецифическими или мультиспецифическими (например, биспецифическими). Мультиспецифический антигенсвязывающий фрагмент антитела будет, как правило, содержать по меньшей мере два различных вариабельных домена, где каждый вариабельный домен способен специфически связываться с отдельным антигеном или с другим эпитопом того же антигена. Любой формат мультиспецифических антител может быть адаптирован для применения в контексте антигенсвязывающего фрагмента антитела, описанного в данном документе, с помощью стандартных методик, доступных в данной области техники.
Константный участок антитела важен с точки зрения способности антитела связывать комплемент и опосредовать клеточнозависимую цитотоксичность. Таким образом, изотип антитела может быть выбран на основании того, требуется ли антителу опосредовать цитотоксичность.
Термин "антитело человека" включает антитела, имеющие вариабельные и константные участки, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Антитела человека, представленные в настоящем изобретении, при этом могут включать аминокислотные остатки, не кодируемые последовательностями иммуноглобулинов зародышевой линии человека (например, мутации, вводимые случайным или сайт-специфичным мутагенезом in vitro или соматической мутацией in vivo), например, в CDR и, в частности, CDR3. Однако термин "антитело человека" не включает антитела, в которых последовательности CDR, полученные из зародышевой линии другого вида млекопитающего, такого как мышь, привиты на последовательности каркасных участков человека.
Термин "рекомбинантное антитело человека" включает все антитела человека, получаемые, экспрессируемые, создаваемые или выделяемые рекомбинантными методиками, такие как антитела, экспрессируемые с помощью рекомбинантного вектора экспрессии, трансфицированного в клетку-хозяина (описанные дополнительно далее), антитела, выделяемые из комбинаторной библиотеки рекомбинантных антител человека (описанные дополнительно далее), антитела, выделяемые из животного (например, мыши), которое является трансгенным по генам иммуноглобулинов человека (см., например, Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295), или антитела, полученные, экспрессированные, созданные или выделенные посредством любых других способов, которые предусматривают сплайсинг последовательностей генов иммуноглобулинов человека с другими последовательностями ДНК. Такие рекомбинантные антитела человека имеют вариабельные и константные участки, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Однако в определенных вариантах осуществления такие рекомбинантные антитела человека подвергают мутагенезу in vitro (или в случае использования животного, трансгенного по последовательностям Ig человека, соматическому мутагенезу in vivo) и, таким образом, аминокислотные последовательности участков VH и VL рекомбинантных антител представляют собой последовательности, которые, будучи полученными из последовательностей VH и VL зародышевой линии человека и родственными им, могут не встречаться в природе в репертуаре антител зародышевой линии человека in vivo.
Антитела человека могут встречаться в двух формах, что связано с гетерогенностью шарнирных участков. В одной форме молекула иммуноглобулина содержит стабильную четырехцепочечную конструкцию массой примерно 150-160 кДа, в которой димеры удерживаются вместе посредством межцепочечной дисульфидной связи, которая связывает тяжелые цепи. Во второй форме димеры не соединены межцепочечными дисульфидными связями, и образуется молекула массой приблизительно 75-80 кДа, состоящая из ковалентно связанных легкой и тяжелой цепей (полуантитело). Эти формы крайне сложно разделить даже после аффинной очистки.
Частота появления второй формы в различных изотипах интактных IgG обусловлена без ограничения структурными различиями, связанными с изотипом шарнирного участка антитела. Единственная аминокислотная замена в шарнирном участке шарнира IgG4 человека может значительно снизить появление второй формы (Angal et al. (1993) Molecular Immunology 30:105) до уровней, обычно наблюдаемых при использовании шарнира IgG1 человека. Настоящее изобретение охватывает антитела с одной или несколькими мутациями в шарнире, участке CH2 или CH3, что может быть предпочтительным, например, в условиях получения, для повышения выхода предпочтительной формы антитела.
Термин "выделенное антитело" означает антитело, которое было идентифицировано и отделено и/или извлечено из по меньшей мере одного компонента своего естественного окружения. Например, антитело, которое было отделено или удалено из по меньшей мере одного компонента организма, или из ткани или клетки, в которой антитело изначально присутствует или продуцируется естественным путем, представляет собой "выделенное антитело". Выделенное антитело также включает антитело in situ в рекомбинантной клетке. Выделенные антитела представляют собой антитела, которые были подвергнуты по меньшей мере одной стадии очистки или выделения. Согласно определенным вариантам осуществления выделенное антитело может практически не содержать другого клеточного материала и/или других химических соединений.
Термин "специфически связывает" и т.п. означает, что антитело или его антигенсвязывающий фрагмент образуют комплекс с антигеном, который является сравнительно устойчивым в физиологических условиях. Способы определения наличия специфического связывания антитела с антигеном хорошо известны из уровня техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс и т.п. Например, антитело, которое "специфически связывается" с IL-4R, как представлено в настоящем изобретении, включает антитела, которые связываются с IL-4R или его частью с KD менее приблизительно 1000 нМ, менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, измеренном в анализе поверхностного плазмонного резонанса. Однако выделенное антитело, которое специфически связывается с IL-4R человека, характеризуется перекрестной реактивностью по отношению к другим антигенам, таким как молекулы IL-4R из других (отличных от человека) видов.
Антитела к IL-4R, пригодные для способов по настоящему изобретению, могут содержать одну или несколько аминокислотных замен, вставок и/или делеций (например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 замен и/или 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 вставок и/или 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 замен) в каркасных участках и/или участках CDR вариабельных доменов тяжелой и легкой цепей по сравнению с соответствующими последовательностями зародышевой линии, из которых были получены антитела. Такие мутации можно легко определить посредством сравнения аминокислотных последовательностей, раскрытых в данном документе, с последовательностями зародышевой линии, доступными, например, из публичных баз данных последовательностей антител. Настоящее изобретение включает способы, предусматривающие применение антител и их антигенсвязывающих фрагментов, которые получены из любых аминокислотных последовательностей, раскрытых в данном документе, где одна или несколько аминокислот (например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 аминокислот) в одном или нескольких каркасных участках и/или одном или нескольких (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 по отношению к тетрамерному антителу или 1, 2, 3, 4, 5 или 6 по отношению к HCVR и LCVR антитела) участках CDR подвергнуты мутации в соответствующий остаток(остатки) последовательности зародышевой линии, из которой антитело получено, или в соответствующий остаток(остатки) последовательности другой зародышевой линии человека, или в консервативную аминокислотную замену соответствующего остатка(остатков) зародышевой линии (такие изменения последовательностей называются в данном документе собирательно "мутациями зародышевой линии"). Специалист в данной области техники, начиная с последовательностей вариабельных участков тяжелой и легкой цепи, раскрытых в данном документе, может легко получить множество антител и антигенсвязывающих фрагментов, которые содержат одну или несколько отдельных мутаций зародышевой линии или их комбинации. В определенных вариантах осуществления все остатки каркасных участков и/или CDR в доменах VH и/или VL подвергнуты обратной мутации с получением остатков, встречающихся в исходной последовательности зародышевой линии, из которой было получено антитело. В других вариантах осуществления лишь определенные остатки подвергнуты обратной мутации в исходную последовательность зародышевой линии, к примеру лишь подвергнутые мутации остатки, обнаруженные в первых 8 аминокислотах FR1 или в последних 8 аминокислотах FR4, или лишь подвергнутые мутации остатки, обнаруженные в CDR1, CDR2 или CDR3. В других вариантах осуществления один или несколько остатков каркасных участков и/или CDR подвергнуты мутации в соответствующий остаток(остатки) последовательности другой зародышевой линии (т.е. последовательности зародышевой линии, которая отличается от последовательности зародышевой линии, из которой изначально получено антитело). Кроме того, антитела могут содержать любую комбинацию из двух или более мутаций зародышевых линий в каркасных участках и/или участках CDR, например, где определенные отдельные остатки подвергнуты мутации в соответствующий остаток последовательности определенной зародышевой линии, в то время как определенные другие остатки, которые отличаются от последовательности исходной зародышевой линии, сохраняются или подвергнуты мутации в соответствующий остаток последовательности другой зародышевой линии. Сразу после получения, антитела и антигенсвязывающие фрагменты, которые содержат одну или несколько мутаций зародышевых линий, могут быть легко исследованы в отношении одного или нескольких предпочтительных свойств, таких как улучшенная специфичность связывания, повышенная аффинность связывания, улучшенные или усиленные биологические антагонистические или агонистические свойства (в зависимости от ситуации), сниженная иммуногенность и т.п. Применение антител и антигенсвязывающих фрагментов, полученных с помощью этого общего способа, охвачено в настоящем изобретении.
Настоящее изобретение также включает способы, предусматривающие применение антител к IL-4R, содержащих варианты любых из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе, с одной или несколькими консервативными заменами. Например, настоящее изобретение включает применение антител к IL-4R с аминокислотными последовательностями HCVR, LCVR и/или CDR, например, с 10 или менее, 8 или менее, 6 или менее, 4 или менее и т.п. консервативными аминокислотными заменами по отношению к любой из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе.
Термин "поверхностный плазмонный резонанс" относится к оптическому феномену, который обеспечивает анализ взаимодействий в реальном времени посредством выявления изменений концентраций белков в биосенсорной матрице, например, с помощью системы BIAcore™ (подразделение Biacore Life Sciences в GE Healthcare, Пискатауэй, Нью-Джерси).
Термин "KD" относится к равновесной константе диссоциации взаимодействия определенного антитела и антигена.
Термин "эпитоп" относится к антигенной детерминанте, которая взаимодействует со специфическим антигенсвязывающим сайтом в вариабельном участке молекулы антитела, известном как паратоп. Один антиген может иметь более одного эпитопа. Таким образом, различные антитела могут связываться с различными областями на антигене и могут оказывать различные биологические эффекты. Эпитопы могут быть конформационными или линейными. Конформационный эпитоп образован пространственно сближенными аминокислотами из различных сегментов линейной полипептидной цепи. Линейный эпитоп образован смежными аминокислотными остатками в полипептидной цепи. При определенных условиях эпитоп может включать фрагменты сахаридов, фосфорильных групп или сульфонильных групп антигена.
Получение антител человека
Из уровня техники известны способы получения антител человека в трансгенных мышах. Любые такие известные способы можно использовать для создания антител человека, которые специфически связываются с IL-4R человека.
С помощью технологии VELOCIMMUNE® (см., например, US 6596541, Regeneron Pharmaceuticals) или любого другого известного способа получения моноклональных антител изначально выделяют высокоаффинные химерные антитела к IL-4R с вариабельным участком человека и константным участком мыши. Технология VELOCIMMUNE® включает получение трансгенной мыши с геномом, содержащим вариабельные участки тяжелой и легкой цепей человека, функционально связанные с эндогенными локусами константного участка мыши так, что мышь продуцирует антитело, содержащее вариабельный участок человека и константный участок мыши в ответ на антигенную стимуляцию. ДНК, кодирующую вариабельные участки тяжелой и легкой цепей антитела, выделяют и функционально связывают с ДНК, кодирующей константные участки тяжелой и легкой цепей человека. Затем ДНК экспрессируют в клетке, способной экспрессировать полностью человеческое антитело.
Как правило, на мышь, полученную с помощью VELOCIMMUNE®, воздействуют антигеном, представляющим интерес, и из мышей, которые экспрессируют антитела, извлекают лимфатические клетки (такие как B-клетки). Лимфатические клетки можно сливать с линией клеток миеломы с получением иммортализированных линий клеток гибридомы, и такие линии клеток гибридомы подвергают скринингу и отбирают для идентификации линий клеток гибридомы, которые продуцируют антитела, специфические в отношении антигена, представляющего интерес. ДНК, кодирующую вариабельные участки тяжелой цепи и легкой цепи, можно выделить и связать с константными участками требуемых изотипов тяжелой цепи и легкой цепи. Такой белок-антитело можно получать в клетке, такой как клетка CHO. Альтернативно, ДНК, кодирующую антиген-специфические химерные антитела или вариабельные домены легкой и тяжелой цепей, можно выделить непосредственно из антиген-специфических лимфоцитов.
Изначально выделяют высокоаффинные химерные антитела с вариабельным участком человека и константным участком мыши. Антитела характеризуют и отбирают по предпочтительным характеристикам, в том числе по аффиности, селективности, эпитопу и т.д., с помощью стандартных процедур, известных специалистам в данной области техники. Константные участки мыши заменяют требуемым константным участком человека с получением полностью человеческого антитела, представленного в настоящем изобретении, например, IgG1 или IgG4 дикого типа или модифицированных IgG1 или IgG. В то время как выбранный константный участок может варьировать в зависимости от конкретного применения, у вариабельного участка сохраняются характеристики высокоаффинного связывания с антигеном и специфичности в отношении мишени.
Как правило, антитела, которые можно использовать в способах, характеризуются высокой аффинностью, как описано выше, при измерении связывания с антигеном как при иммобилизации на твердой фазе, так и в жидкой фазе. Константные участки мыши заменяют требуемыми константными участками человека с получением полностью человеческих антител, представленных в настоящем изобретении. В то время как выбранный константный участок может варьировать в зависимости от конкретного применения, у вариабельного участка сохраняются характеристики высокоаффинного связывания с антигеном и специфичности в отношении мишени.
В одном варианте осуществления антитело человека или его антигенсвязывающий фрагмент, которые специфически связываются с IL-4R, которые можно использовать в контексте способов, представленных в настоящем изобретении, содержат три CDR тяжелой цепи (HCDR1, HCDR2 и HCDR3), содержащихся в вариабельном участке тяжелой цепи (HCVR) с аминокислотной последовательностью под SEQ ID NO: 1. Антитело или антигенсвязывающий фрагмент могут содержать три CDR легкой цепи (LCVR1, LCVR2, LCVR3), содержащиеся в вариабельном участке легкой цепи (LCVR) с аминокислотной последовательностью под SEQ ID NO: 2. Способы и методики идентификации CDR в аминокислотных последовательностях HCVR и LCVR хорошо известны из уровня техники и могут быть использованы для идентификации CDR в определенных аминокислотных последовательностях HCVR и/или LCVR, раскрытых в данном документе. К иллюстративным общепринятым способам, которые можно применять для идентификации границ CDR, относятся, например, определение по Kabat, определение по Chothia и определение по AbM. В целом, определение по Kabat основано на вариабельности последовательностей, определение по Chothia основано на положении участков структурных петель и определение по AbM является компромиссным решением между подходами по Kabat и Chothia. См., например, "Kabat, Sequences of Proteins of Immunological Interest", National Institutes of Health, Bethesda, Md. (1991); Al-Lazikani et al., J. Mol. Biol. 273:927-948 (1997); и Martin et al., Proc. Natl. Acad. Sci. USA 86:9268-9272 (1989). Общедоступные базы данных также доступны для идентификации последовательностей CDR в антителе.
В определенных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат шесть CDR (HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 и LCDR3) из пар аминокислотных последовательностей вариабельного участка тяжелой и легкой цепей (HCVR/LCVR) под SEQ ID NO: 1 и 2.
В определенных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат шесть CDR (HCDR1/HCDR2/HCDR3/LCDR1/LCDR2/LCDR3) с аминокислотными последовательностями под SEQ ID NO: 3/4/5/6/7/8.
В определенных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат пары аминокислотных последовательностей HCVR/LCVR под SEQ ID NO: 1 и 2.
В одном варианте осуществления антителом является дупилумаб, который содержит пары аминокислотных последовательностей HCVR/LCVR под SEQ ID NO: 1 и 2.
Фармацевтические композиции
Настоящее изобретение включает способы, которые предусматривают введение пациенту антагониста IL-4R, где антагонист IL-4R содержится в фармацевтической композиции. Фармацевтические композиции, представленные в настоящем изобретении, составляют с подходящими носителями, вспомогательными средствами и другими средствами, которые обеспечивают подходящий перенос, доставку, переносимость и т.п. Множество подходящих составов можно найти в справочнике, известном всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания. Эти составы включают, например, порошки, пасты, мази, желе, воска, масла, липиды, везикулы, содержащие (катионные или анионные) липиды (такие как LIPOFECTIN™), конъюгаты ДНК, безводные абсорбционные пасты, эмульсии типа "масло в воде" и "вода в масле", эмульсии карбовакс (полиэтиленгликоли с различной молекулярной массой), полужидкие гели и полужидкие смеси, содержащие карбовакс. См. также Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
Доза антитела, вводимого пациенту, может варьироваться в зависимости от возраста и размерных характеристик пациента, симптомов, состояний, пути введения и т.п. Дозу обычно рассчитывают в соответствии с весом тела или площадью поверхности тела. В зависимости от тяжести состояния частоту и продолжительность лечения можно регулировать. Эффективные дозы и схемы введения фармацевтических композиций, содержащих антитела к IL-4R, можно определить эмпирически; например, динамику состояния пациента можно контролировать с помощью периодической оценки и, соответственно, регулировать дозу. Кроме того, межвидовой расчет дозировок можно осуществлять с помощью хорошо известных из уровня техники способов (например, Mordenti et al., 1991, Pharmaceut. Res. 8:1351).
Известны различные системы доставки и их можно применять для введения фармацевтических композиций, представленных в настоящем изобретении, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, рекомбинантные клетки, способные экспрессировать мутантные вирусы, опосредованный рецепторами эндоцитоз (см., например, Wu et al., 1987, J. Biol. Chem. 262:4429-4432). Способы введения включают без ограничения внутрикожные, внутримышечные, интраперитонеальные, внутривенные, подкожные, интраназальные, интратрахеальные, эпидуральные и пероральные пути. Композицию можно вводить любым удобным способом, например инфузией или болюсной инъекцией, абсорбцией через эпителиальные или кожно-слизистые покровы (например, слизистую ротовой полости, слизистую прямой кишки и кишечника и т.д.), и можно вводить совместно с другими биологически активными средствами.
Фармацевтическую композицию, представленную в настоящем изобретении, можно доставлять подкожно или внутривенно с помощью стандартной иглы и шприца. Кроме того, в отношении подкожной доставки при доставке фармацевтической композиции, представленной в настоящем изобретении, широко используется устройство для доставки в виде шприца-ручки (например, шприц-ручка для автоинъекции). Такое устройство для доставки в виде шприца-ручки может быть многоразового и одноразового использования. В устройстве для доставки в виде шприца-ручки многоразового использования, как правило, используется заменяемый картридж, который содержит фармацевтическую композицию. После введения всей фармацевтической композиции из картриджа и после опустошения картриджа пустой картридж можно легко утилизировать и заменить новым картриджем, который содержит фармацевтическую композицию. Затем устройство для доставки в виде шприца-ручки можно использовать повторно. В устройстве для доставки в виде шприца-ручки одноразового использования заменяемый картридж отсутствует. Вместо этого устройство для доставки в виде шприца-ручки одноразового использования выпускают предварительно заполненным фармацевтической композицией, содержащейся в резервуаре устройства. После того как резервуар опустошается от фармацевтической композиции, устройство полностью утилизируют.
Многочисленные многоразовые устройства для доставки в виде шприца-ручки и автоинжектора используются в подкожной доставке фармацевтической композиции. Примеры включают без ограничения AUTOPEN™ (Owen Mumford, Inc., Вудсток, Великобритания), шприц-ручку DISETRONIC™ (Disetronic Medical Systems, Бургдорф, Швейцария), шприц-ручку HUMALOG MIX 75/25™, шприц-ручку HUMALOG™, шприц-ручку HUMALIN 70/30™ (Eli Lilly and Co., Индианаполис, Индиана), NOVOPEN™ I, II и III (Novo Nordisk, Копенгаген, Дания), NOVOPEN JUNIOR™ (Novo Nordisk, Копенгаген, Дания), шприц-ручку BD™ (Becton Dickinson, Франклин-Лэйкс, Нью-Джерси), OPTIPEN™, OPTIPEN PRO™, OPTIPEN STARLET™ и OPTICLIK™ (Sanofi-Aventis, Франкфурт, Германия), при этом упомянуты лишь некоторые из них. Примеры многоразовых устройств для доставки в виде шприца-ручки, используемых при подкожном введении фармацевтической композиции, представленной в настоящем изобретении, включают без ограничения шприц-ручку SOLOSTAR™ (Sanofi-Aventis), FLEXPEN™ (Novo Nordisk) и KWIKPEN™ (Eli Lilly), автоинжектор SURECLICKTM (Amgen, Таузанд-Окс, Калифорния), PENLETTM (Haselmeier, Штутгарт, Германия), EPIPEN (Dey, L.P.) и шприц-ручку HUMIRATM (Abbott Labs, Эббот-Парк, Иллинойс), при этом упомянуты лишь некоторые из них. Примеры устройств для доставки большого объема (например, инжекторов большого объема) включают без ограничения болюсные инжекторы, такие как, например, BD Libertas West SmartDose, Enable Injections, SteadyMed PatchPump, Sensile SenseTrial, YPsomed YpsoDose, Bespak Lapas и т.п.
Для непосредственного введения в пазухи носа фармацевтические композиции, представленные в настоящем изобретении, можно вводить с помощью, например, микрокатетера (например, эндоскопа и микрокатетера), распылителя аэрозоля, дозатора порошка, небулайзера или ингалятора. Способы предусматривают введение антагониста IL-4R нуждающемуся в этом субъекту в аэрозольном составе. Например, антитела к IL-4R в виде аэрозоля можно вводить пациенту для лечения астмы. Аэрозольные антитела можно получать, как описано, например, в US 8178098, включенном в данный документ посредством ссылки во всей своей полноте.
В определенных ситуациях фармацевтическую композицию можно доставлять в системе с контролируемым высвобождением. В одном варианте осуществления можно использовать насос (см. Langer, выше; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201). В другом варианте осуществления можно использовать полимерные материалы; см. Medical Applications of Controlled Release, Langer and Wise (eds.), 1974, CRC Pres., Boca Raton, Florida. В еще одном другом варианте осуществления систему с контролируемым высвобождением можно поместить вблизи мишени для композиции, при этом требуется лишь часть системной дозы (см., например, Goodson, 1984, в Medical Applications of Controlled Release, выше, том. 2, стр. 115-138). Другие системы с контролируемым высвобождением описаны в обзоре Langer, 1990, Science 249:1527-1533.
Инъекционные препараты могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, капельных инфузий и т.д. Эти инъекционные препараты можно получать с помощью известных способов. Например, инъекционные препараты можно получать, например, растворением, суспендированием или эмульгированием антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, обычно используемых для инъекций. Водной средой для инъекций является, например, физиологический солевой раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства и т.д., которые можно использовать в комбинации с подходящим солюбилизирующим средством, таким как спирт (например, этанол), многоатомный спирт (например, пропиленгликоль, полиэтиленгликоль), неионогенное поверхностно-активное вещество (например, полисорбат 80, HCO-50 (полиоксиэтиленовый (50 моль) аддукт гидрогенизированного касторового масла)) и т.д. В качестве масляной среды используют, например, кунжутное масло, соевое масло и т.д., которые можно использовать в комбинации с солюбилизирующим средством, таким как бензилбензоат, бензиловый спирт и т.д. Полученной таким способом инъекционной формой обычно наполняют подходящую ампулу.
Преимущественно фармацевтические композиции для перорального или парентерального применения, описанные выше, получают в лекарственных формах в стандартной дозе, подходящей для подбора дозы активных ингредиентов. Такие лекарственные формы в стандартной дозе включают, например, таблетки, пилюли, капсулы, инъекционные формы (ампулы), суппозитории и т.д.
Иллюстративные фармацевтические композиции, содержащие антитело к IL-4R, которые могут быть использованы в настоящем изобретении, раскрыты, например, в публикации заявки на патент США № 2012/0097565.
Доза
Количество антагониста IL-4R (например, антитела к IL-4R), вводимого субъекту согласно способам, представленным в настоящем изобретении, как правило, представляет собой терапевтически эффективное количество. Используемое в данном документе выражение "терапевтически эффективное количество" означает количество антагониста IL-4R, которое в результате приводит к одному или нескольким из следующего: (a) снижения частоты обострений астмы; (b) улучшения одного или нескольких ассоциированных с астмой параметров (как определено в других частях данного документа) и/или (c) выявляемого улучшения одного или нескольких симптомов или проявлений воспалительного состояния верхних дыхательных путей. Выражение "терапевтически эффективное количество" также включает количество антагониста IL-4R, которое ингибирует, предупреждает, снижает или задерживает прогрессирование астмы у субъекта.
В случае антитела к IL-4R терапевтически эффективное количество может составлять от приблизительно 0,05 мг до приблизительно 700 мг, например, приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 1,0 мг, приблизительно 1,5 мг, приблизительно 2,0 мг, приблизительно 3,0 мг, приблизительно 5,0 мг, приблизительно 7,0 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, приблизительно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг, приблизительно 240 мг, приблизительно 250 мг, приблизительно 260 мг, приблизительно 270 мг, приблизительно 280 мг, приблизительно 290 мг, приблизительно 300 мг, приблизительно 310 мг, приблизительно 320 мг, приблизительно 330 мг, приблизительно 340 мг, приблизительно 350 мг, приблизительно 360 мг, приблизительно 370 мг, приблизительно 380 мг, приблизительно 390 мг, приблизительно 400 мг, приблизительно 410 мг, приблизительно 420 мг, приблизительно 430 мг, приблизительно 440 мг, приблизительно 450 мг, приблизительно 460 мг, приблизительно 470 мг, приблизительно 480 мг, приблизительно 490 мг, приблизительно 500 мг, приблизительно 510 мг, приблизительно 520 мг, приблизительно 530 мг, приблизительно 540 мг, приблизительно 550 мг, приблизительно 560 мг, приблизительно 570 мг, приблизительно 580 мг, приблизительно 590 мг, приблизительно 600 мг, приблизительно 610 мг, приблизительно 620 мг, приблизительно 630 мг, приблизительно 640 мг, приблизительно 650 мг, приблизительно 660 мг, приблизительно 670 мг, приблизительно 680 мг, приблизительно 690 мг или приблизительно 700 мг антитела к IL-4R. В определенных вариантах осуществления вводят 300 мг антитела к IL-4R.
Количество антагониста к IL-4R, содержащегося в отдельных дозах, можно выражать в миллиграммах антитела на килограмм веса тела пациента (т.е. мг/кг). Например, антагонист IL-4R можно вводить пациенту в дозе от приблизительно 0,0001 до приблизительно 10 мг/кг веса тела пациента. Например, антагонист IL-4R можно вводить в дозе 1 мг/кг, 2 мг/кг, 3 мг/кг или 4 мг/кг.
В некоторых вариантах осуществления доза антагониста IL-4R может варьировать в соответствии с содержанием эозинофилов. Например, субъект может характеризоваться содержанием эозинофилов в крови (высокое содержание эозинофилов в крови), составляющим ≥ 300 клеток/мкл, или 300-499 клеток/мкл, или ≥ 500 клеток/мкл (HEo); содержанием эозинофилов в крови, составляющим 200-299 клеток/мкл (умеренное содержание эозинофилов в крови); или содержанием эозинофилов в крови, составляющим < 200 клеток/мкл (низкое содержание эозинофилов в крови).
В определенных вариантах осуществления способы предусматривают нагрузочную дозу от приблизительно 400 до приблизительно 600 мг антагониста IL-4R.
В определенных вариантах осуществления способы предусматривают одну или несколько поддерживающих доз от приблизительно 200 до приблизительно 300 мг антагониста IL-4R.
В определенных вариантах осуществления ICS и LABA вводят в течение всего периода введения антагониста IL-4R.
В определенных вариантах осуществления нагрузочная доза предусматривает 600 мг антитела к IL-4R или его антигенсвязывающего фрагмента, a одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В определенных вариантах осуществления нагрузочная доза предусматривает 400 мг антитела к IL-4R или его антигенсвязывающего фрагмента, a одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В определенных вариантах осуществления нагрузочная доза предусматривает 400 мг антитела к IL-4R или его антигенсвязывающего фрагмента, a одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели, которые могут быть повышены до 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В других вариантах осуществления нагрузочная доза предусматривает 600 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В других вариантах осуществления нагрузочная доза предусматривает 400 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В других вариантах осуществления нагрузочная доза предусматривает 600 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят один раз в неделю.
В других вариантах осуществления нагрузочная доза предусматривает 400 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят один раз в неделю.
В других вариантах осуществления нагрузочная доза предусматривает 600 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в три недели.
В других вариантах осуществления нагрузочная доза предусматривает 400 мг антитела к IL-4R или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в три недели.
В одном варианте осуществления возраст субъекта составляет от 6 до < 18 лет, и антитело к IL-4R или его антигенсвязывающий фрагмент вводят в дозе 2 мг/кг или 4 мг/кг.
В другом варианте осуществления возраст субъекта составляет от 12 до < 18 лет, и антитело к IL-4R или его антигенсвязывающий фрагмент вводят в дозе 2 мг/кг или 4 мг/кг.
В другом варианте осуществления возраст субъекта составляет от 6 до < 12 лет, и антитело к IL-4R или его антигенсвязывающий фрагмент вводят в дозе 2 мг/кг или 4 мг/кг.
В другом варианте осуществления возраст субъекта составляет от 2 до < 6 лет, и антитело к IL-4R или его антигенсвязывающий фрагмент вводят в дозе 2 мг/кг или 4 мг/кг.
В еще одном другом варианте осуществления возраст субъекта составляет < 2 лет, и антитело к IL-4R или его антигенсвязывающий фрагмент вводят в дозе 2 мг/кг или 4 мг/кг.
Средства комбинированной терапии
Определенные варианты осуществления способов, представленных в настоящем изобретении, включают введение субъекту одного или нескольких терапевтических средств в комбинации с антагонистом IL-4R. Используемое в данном документе выражение "в комбинации с" означает, что дополнительные терапевтические средства вводят до, после содержащей антагонист IL-4R фармацевтической композиции или параллельно с ней. В некоторых вариантах осуществления термин "в комбинации с" включает последовательное или сопутствующее введение антагониста IL-4R и второго терапевтического средства. Настоящее изобретение также включает способы лечения астмы или ассоциированного состояния или осложнения с целью снижения частоты возникновения по меньшей мере одного обострения, предусматривающие введение антагониста IL-4R в комбинации со вторым терапевтическим средством для дополнительной или синергетической активности.
Например, при введении "до" фармацевтической композиции, содержащей антагонист IL-4R, дополнительное терапевтическое средство можно вводить за приблизительно 72 часа, приблизительно 60 часов, приблизительно 48 часов, приблизительно 36 часов, приблизительно 24 часа, приблизительно 12 часов, приблизительно 10 часов, приблизительно 8 часов, приблизительно 6 часов, приблизительно 4 часа, приблизительно 2 часа, приблизительно 1 час, приблизительно 30 минут, приблизительно 15 минут или приблизительно 10 минут до введения фармацевтической композиции, содержащей антагонист IL-4R. При введении "после" фармацевтической композиции, содержащей антагонист IL-4R, дополнительное терапевтическое средство можно вводить через приблизительно 10 минут, приблизительно 15 минут, приблизительно 30 минут, приблизительно 1 час, приблизительно 2 часа, приблизительно 4 часа, приблизительно 6 часов, приблизительно 8 часов, приблизительно 10 часов, приблизительно 12 часов, приблизительно 24 часа, приблизительно 36 часов, приблизительно 48 часов, приблизительно 60 часов или приблизительно 72 часа после введения фармацевтической композиции, содержащей антагонист IL-4R. Введение "совместно" с фармацевтической композицией, содержащей антагонист IL-4R, означает, что дополнительное терапевтическое средство вводят субъекту в отдельной лекарственной форме в течение менее 5 минут (до, после или в то же время) от введения фармацевтической композиции, содержащей антагонист IL-4R, или вводят субъекту в одном комбинированном дозированном составе, содержащем как дополнительное терапевтическое средство, так и антагонист IL-4R.
Дополнительным терапевтическим средством может быть, например, другой антагонист IL-4R, антагонист IL-1 (в том числе, например, антагонист IL-1, как изложено в патенте США № 6927044), антагонист IL-6, антагонист IL-6R (в том числе, например, антитело к IL-6R, как изложено в патенте США № 7582298), антагонист TNF, антагонист IL-8, антагонист IL-9, антагонист IL-17, антагонист IL-5, антагонист IgE, антагонист CD48, ингибитор лейкотриена, противогрибковое средство, NSAID, бета2-агонист длительного действия (например, сальметерол или формотерол), ингаляционный кортикостероид (например, флутиказон или будесонид), системный кортикостероид (например, пероральный или внутривенный), метилксантин, недокромил натрия, кромолин натрия или их комбинации. Например, в определенных вариантах осуществления фармацевтическую композицию, содержащую антагонист IL-4R, вводят в комбинации с комбинацией, содержащей бета2-агонист длительного действия и ингаляционный кортикостероид (например, флутиказон+сальметерол [например, Advair® (GlaxoSmithKline)] или будесонид+формотерол [например, SYMBICORT® (Astra Zeneca)]).
Схемы введения
Согласно определенным вариантам осуществления несколько доз антагониста IL-4R можно вводить субъекту в течение определенного промежутка времени. Данные способы предусматривают последовательное введение субъекту нескольких доз антагониста IL-4R. Используемое в данном документе выражение "последовательное введение" означает, что каждую дозу антагониста IL-4R вводят субъекту в различный момент времени, например, в различные дни, разделенные предварительно определенным интервалом (например, часами, днями, неделями или месяцами). Настоящее изобретение включает способы, которые предусматривают последовательное введение пациенту одной начальной дозы антагониста IL-4R, затем одной или нескольких вторичных доз антагониста IL-4R, а затем необязательно одной или нескольких третичных доз антагониста IL-4R.
Настоящее изобретение включает способы, предусматривающие введение субъекту фармацевтической композиции, содержащей антагонист IL-4R, с частотой введения дозы приблизительно четыре раза в неделю, два раза в неделю, один раз в неделю (q1w), один раз в две недели (раз в две недели или q2w), один раз в три недели (раз в три недели или q3w), один раз в четыре недели (ежемесячно или q4w), один раз в пять недель (q5w), один раз в шесть недель (q6w), один раз в восемь недель (q8w), один раз в двенадцать недель (q12w) или с меньшей частотой до тех пор, пока не будет достигнут терапевтический ответ. В определенных вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в неделю. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в две недели. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в три недели. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в четыре недели (ежемесячное введение дозы). В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в пять недель. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в шесть недель. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в восемь недель. В других вариантах осуществления, включающих введение фармацевтической композиции, содержащей антитело к IL-4R, можно использовать введение дозы в количестве приблизительно 75 мг, 100 мг, 150 мг, 200 мг или 300 мг один раз в двенадцать недель. В других вариантах осуществления путь введения может быть подкожным.
Термин "неделя" или "недели" относится к периоду (n x 7 дней) ± 2 дня, например, (n x 7 дней) ± 1 день или (n x 7 дней), где "n" означает количество недель, например, 1, 2, 3, 4, 5, 6, 8, 12 или больше.
Термины "начальная доза", "вторичные дозы" и "третичные дозы" относятся к временной последовательности введения антагонистa IL-4R. Таким образом, "начальная доза" представляет собой дозу, которую вводят в начале схемы лечения (также обозначается как "исходная доза"); "вторичные дозы" представляют собой дозы, которые вводят после введения начальной дозы; и "третичные дозы" представляют собой дозы, которые вводят после вторичных доз. Все из начальных, вторичных и третичных доз могут содержать одинаковое количество антагониста IL-4R, но, как правило, могут отличаться друг от друга по частоте введения. Однако в определенных вариантах осуществления количество антагониcта IL-4R, содержащегося в начальных, вторичных и/или третичных дозах, отличается друг от друга (например, при необходимости регулируется в сторону повышения или понижения) на протяжении курса лечения. В определенных вариантах осуществления две или более (например, 2, 3, 4 или 5) доз вводят в начале схемы лечения в виде "нагрузочных доз", затем последующие дозы вводят с меньшей частотой (например, "поддерживающие дозы"). В одном варианте осуществления поддерживающая доза может быть ниже нагрузочной дозы. Например, одну или несколько нагрузочных доз, составляющих 600 мг антагониста IL-4R можно вводить после поддерживающих доз, составляющих от приблизительно 75 мг до приблизительно 300 мг.
В определенных вариантах осуществления нагрузочная доза антагониста IL-4R составляет от приблизительно 400 до приблизительно 600 мг. В одном варианте осуществления нагрузочная доза антагониста IL-4R составляет 400 мг. В другом варианте осуществления нагрузочная доза антагониста IL-4R составляет 600 мг.
В определенных вариантах осуществления поддерживающая доза антагониста IL-4R составляет от приблизительно 200 до приблизительно 300 мг. В одном варианте осуществления поддерживающая доза антагониста IL-4R составляет 200 мг. В другом варианте осуществления поддерживающая доза антагониста IL-4R составляет 300 мг.
В определенных вариантах осуществления нагрузочная доза в два раза больше, чем поддерживающая доза.
В некоторых вариантах осуществления нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления у субъекта имеется OCS-зависимая астма, и нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления у субъекта имеется сопутствующий атопический дерматит от умеренной до тяжелой степени, и нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления у субъекта имеется OCS-зависимая астма, и нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления у субъекта имеется сопутствующий атопический дерматит от умеренной до тяжелой степени, и нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в две недели.
В некоторых вариантах осуществления нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В некоторых вариантах осуществления у субъекта имеется OCS-зависимая астма, и нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В некоторых вариантах осуществления у субъекта имеется сопутствующий атопический дерматит от умеренной до тяжелой степени, и нагрузочная доза предусматривает 600 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 300 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В некоторых вариантах осуществления нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В некоторых вариантах осуществления у субъекта имеется OCS-зависимая астма, и нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В некоторых вариантах осуществления у субъекта имеется сопутствующий атопический дерматит от умеренной до тяжелой степени, и нагрузочная доза предусматривает 400 мг антитела или его антигенсвязывающего фрагмента, а одна или несколько поддерживающих доз предусматривают 200 мг антитела или его антигенсвязывающего фрагмента, которые вводят раз в четыре недели.
В одном иллюстративном варианте осуществления каждую вторичную и/или третичную дозу вводят через 1-14 (например, 1, 1½, 2, 2½, 3, 3½, 4, 4½, 5, 5½, 6, 6½, 7, 7½, 8, 8½, 9, 9½, 10, 10½, 11, 11½, 12, 12½, 13, 13½, 14, 14½ или больше) недель после непосредственно предшествующей дозы. Выражение "непосредственно предшествующая доза" означает в последовательности нескольких введений дозу антагониста IL-4R, которую вводят пациенту до введения ближайшей следующей дозы в последовательности без промежуточных доз.
Способы могут включать введение пациенту любого количества вторичных и/или третичных доз антагониста IL-4R. Например, в определенных вариантах осуществления пациенту вводят только одну вторичную дозу. В других вариантах осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или больше) вторичных доз. Аналогично, в определенных вариантах осуществления пациенту вводят только одну третичную дозу. В других вариантах осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или больше) третичных доз.
В вариантах осуществления, включающих несколько вторичных доз, каждую вторичную дозу можно вводить с той же частотой, что и другие вторичные дозы. Например, каждую вторичную дозу можно вводить пациенту через 1-2 недели после непосредственно предшествующей дозы. Аналогично, в вариантах осуществления, включающих несколько третичных доз, каждую третичную дозу можно вводить с той же частотой, что и остальные третичные дозы. Например, каждую третичную дозу можно вводить пациенту через 2-4 недели после непосредственно предшествующей дозы. Альтернативно, частота, с которой вторичные и/или третичные дозы вводят пациенту, может варьироваться на протяжении схемы лечения. Частота введения может регулироваться врачом на протяжении курса лечения в зависимости от потребностей отдельного пациента после клинического обследования.
Настоящее изобретение включает способы, предусматривающие последовательное введение пациенту антагониста IL-4R и второго терапевтического средства для лечения астмы или ассоциированного состояния. В некоторых вариантах осуществления способы предусматривают введение одной или нескольких доз антагониста IL-4R, после чего следует введение одной или нескольких доз (например, 2, 3, 4, 5, 6, 7, 8 или больше) второго терапевтического средства. Например, можно вводить одну или несколько доз антагониста IL-4R, составляющих от приблизительно 75 мг до приблизительно 300 мг, после которых можно вводить одну или несколько доз (например, 2, 3, 4, 5, 6, 7, 8 или больше) второго терапевтического средства (например, ингаляционного кортикостероида или любого бета2-агониста или любого другого терапевтического средства, описанного в других частях данного документа) для лечения, облегчения, снижения интенсивности или уменьшения тяжести одного или нескольких симптомов астмы. В некоторых вариантах осуществления антагонист IL-4R вводят в одной или нескольких дозах (например, 2, 3, 4, 5, 6, 7, 8 или больше), что приводит к улучшению одного или нескольких ассоциированных с астмой параметров, после чего следует введение второго терапевтического средства для предупреждения повторного возникновения по меньшей мере одного симптома астмы. Альтернативные варианты осуществления относятся к сопутствующему введению антагониста IL-4R и второго терапевтического средства. Например, вводят одну или несколько доз (например, ,2, 3, 4, 5, 6, 7, 8 или больше) антагониста IL-4R и вводят второе терапевтическое средство в отдельной дозировке с одинаковой или различной частотой по отношению к антагонисту IL-4R. В некоторых вариантах осуществления второе терапевтическое средство вводят до, после антагониста IL-4R или совместно с ним.
В определенных вариантах осуществления антагонист IL-4R вводят раз в две недели в течение 12 недель, 14 недель, 16 недель, 18 недель, 20 недель, 22 недель, 24 недель, 26 недель, 28 недель, 30 недель, 32 недель, 34 недель, 36 недель, 38 недель, 40 недель, 42 недель, 44 недель, 46 недель, 48 недель или больше. В других вариантах осуществления антагонист IL-4R вводят раз в четыре недели в течение 12 недель, 16 недель, 20 недель, 24 недель, 28 недель, 32 недель, 36 недель, 40 недель, 44 недель, 48 недель или больше. В конкретных вариантах осуществления антагонист IL-4R вводят в течение по меньшей мере 24 недель.
Настоящее изобретение включает способы лечения субъекта, страдающего тяжелой неконтролируемой астмой (например, тяжелой стероидозависимой астмой), предусматривающие введение субъекту нагрузочной дозы антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R. В определенных вариантах осуществления способы предусматривают введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента, где множество поддерживающих доз вводят на протяжении фазы лечения. Фаза лечения включает фазу индукции, фазу снижения OCS и фазу поддержания OCS.
В определенных иллюстративных вариантах осуществления фаза индукции включает период, в течение которого субъекты непрерывно получают свою(свои) дозу(дозы) OCS. В определенных иллюстративных вариантах осуществления фаза снижения включает период, в течение которого субъекты получают более низкую дозу OCS по сравнению с дозой, полученной во время фазы индукции. В определенных иллюстративных вариантах осуществления фаза поддержания включает период, в течение которого субъект получает определенное стабильное количество или дозу(дозы) OCS. Альтернативно, фаза поддержания включает период, в течение которого терапия/введение OCS снижается или устраняется. В определенных вариантах осуществления применение OCS пациентом полностью исключается, и пациент не получает стероидов в течение менее 1 года лечения антителом к IL4R или его фрагментом (например, в течение 1 года, 6 месяцев, 3 месяцев или 1 месяца после первичного лечения).
В другом аспекте способ лечения субъекта, страдающего тяжелой стероидозависимой астмой и/или тяжелой неконтролируемой астмой, предусматривает введение субъекту нагрузочной дозы, составляющей приблизительно 600 мг антитела или его антигенсвязывающего фрагмента, которые специфически связываются с рецептором интерлейкина-4 (IL-4R), и введение субъекту множества поддерживающих доз антитела или его антигенсвязывающего фрагмента. Каждая поддерживающая доза составляет приблизительно 300 мг антитела или его антигенсвязывающего фрагмента, где множество поддерживающих доз вводят на протяжении фазы лечения, предусматривающей фазу индукции, фазу снижения перорального кортикостероида (OCS) и фазу поддержания, где антитело или его антигенсвязывающий фрагмент содержат последовательности CDR тяжелой и легкой цепей из пары последовательностей HCVR/LCVR, предусматривающей SEQ ID NO: 1 и 2.
Популяции лечения
Способы, представленные в настоящем изобретении, предусматривают введение нуждающемуся в этом субъекту терапевтической композиции, содержащей антагонист IL-4R. Выражение "нуждающийся в этом субъект" означает человека или не относящегося к человеку животного, у которых наблюдаются один или несколько симптомов или проявлений астмы (например, неконтролируемой астмы от умеренной до тяжелой степени), или которым поставили диагноз астмы. Например, "нуждающийся в этом субъект" может включать, например, субъектов, которые до лечения характеризуются (или характеризовались) одним или несколькими ассоциированными с астмой параметрами, такими как, например, нарушенный FEV1 (например, менее 2,0 л), нарушенная FEF 25-75%; нарушенная PEF до полудня (например, менее 400 л/мин.), нарушенная PEF после полудня (например, менее 400 л/мин.), показатель ACQ5, составляющий по меньшей мере 2,5, по меньшей мере 1 пробуждение в ночное время в неделю и/или показатель SNOT-22, составляющий по меньшей мере 20. В различных вариантах осуществления данные способы можно использовать для лечения слабой, от умеренной до тяжелой или тяжелой астмы у нуждающихся в этом пациентов. В определенных вариантах осуществления данные способы можно применять для лечения слабой, от умеренной до тяжелой или тяжелой астмы у нуждающихся в этом пациентов, где у пациентов дополнительно наблюдается сопутствующий атопический дерматит от умеренной до тяжелой степени.
В родственном варианте осуществления "нуждающимся в этом субъектом" может быть субъект, которому до получения антагониста IL-4R назначали комбинацию ICS/LABA или он принимает ее в настоящее время. Примеры ICS включают мометазона фуроат, будесонид и флутиказона пропионат. Примеры LABA включают формотерол и сальметерол. Примеры средств терапии ICS/LABA включают средство комбинированной терапии флутиказон/сальметерол и средство комбинированной терапии будесонид/формотерол. Например, настоящее изобретение включает способы, которые предусматривают введение антагониста IL-4R пациенту, который проходил регулярный курс ICS/LABA в течение двух или более недель непосредственно до введения антагониста IL-4R (такие средства предшествующего лечения обозначены в данном документе как "средства фонового лечения"). Настоящее изобретение включает терапевтические способы, в которых средства фонового лечения продолжают принимать в комбинации с введением антагониста IL-4R. В еще одних других вариантах осуществления количество компонента ICS, компонента LABA или как того, так и другого постепенно снижают до или после начала введения антагониста IL-4R. В некоторых вариантах осуществления настоящее изобретение включает способы лечения пациентов с персистирующей астмой в течение по меньшей мере ≥ 12 месяцев. В одном варианте осуществления пациент с персистирующей астмой может быть устойчивым к лечению терапевтическим средством, таким как кортикостероид, и ему можно вводить антагонист IL-4R согласно способам по настоящему изобретению.
В некоторых вариантах осуществления "нуждающимся в этом субъектом" может быть субъект с повышенными уровнями ассоциированного с астмой биомаркера. Примеры ассоциированных с астмой биомаркеров включают без ограничения IgE, хемокин, регулируемый тимусом и активацией (TARC), эотаксин-3, CEA, YKL-40 и периостин. В некоторых вариантах осуществления "нуждающимся в этом субъектом" может быть субъект с содержанием эозинофилов в крови ≥ 300 клеток/мкл, 200-299 клеток/мкл или < 200 клеток/мкл. В одном варианте осуществления "нуждающимся в этом субъектом" может быть субъект с повышенным уровнем воспаления в бронхах или дыхательных путях, измеренным по фракции выдыхаемого оксида азота (FeNO).
В некоторых вариантах осуществления "нуждающийся в этом субъект" выбран из группы, состоящей из субъекта возрастом 18 лет или старше, субъекта возрастом 12 лет или старше, субъекта возрастом от 12 до 17 лет (от 12 до < 18 лет), субъекта возрастом от 6 до 11 лет (от 6 до < 12 лет) и субъекта возрастом от 2 до 5 лет (от 2 до < 6 лет). В некоторых вариантах осуществления "нуждающийся в этом субъект" выбран из группы, состоящей из взрослого человека, подростка и ребенка. В некоторых вариантах осуществления "нуждающийся в этом субъект" выбран из группы, состоящей из взрослого человека возрастом от 18 лет или старше, подростка возрастом от 12 до 17 лет (от 12 до < 18 лет), ребенка возрастом от 6 до 11 лет (от 6 до < 12 лет) и ребенка возрастом от 2 до 5 лет (от 2 до < 6 лет). Возраст субъекта может быть менее 2 лет, например, от 12 до 23 месяцев или от 6 до 11 месяцев.
Нормальный уровень IgE у здоровых субъектов составляет менее приблизительно 100 кЕ/л (например, измеренный с помощью анализа IMMUNOCAP® [Phadia Inc., Портедж, Мичиган]). Таким образом, настоящее изобретение включает способы, предусматривающие отбор субъекта, который характеризуется повышенным уровнем IgE в сыворотке крови, при этом уровень IgE в сыворотке крови составляет более приблизительно 100 кЕ/л, более приблизительно 150 кЕ/л, более приблизительно 500 кЕ/л, более приблизительно 1000 кЕ/л, более приблизительно 1500 кЕ/л, более приблизительно 2000 кЕ/л, более приблизительно 2500 кЕ/л, более приблизительно 3000 кЕ/л, более приблизительно 3500 кЕ/л, более приблизительно 4000 кЕ/л, более приблизительно 4500 кЕ/л или более приблизительно 5000 кЕ/л, и введение субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-4R.
Уровни TARC у здоровых субъектов находятся в диапазоне от 106 нг/л до 431 нг/л, при этом среднее значение составляет приблизительно 239 нг/л. (Иллюстративной аналитической системой для измерения уровня TARC является набор ELISA для количественного определения TARC, предлагаемый под № по кат. DDN00 от R&D Systems, Миннеаполис, Миннесота.) Таким образом, настоящее изобретение включает способы, предусматривающие отбор субъекта, который характеризуется повышенным уровнем TARC в сыворотке крови, при этом уровень TARC в сыворотке крови составляет более приблизительно 431 нг/л, более приблизительно 500 нг/л, более приблизительно 1000 нг/л, более приблизительно 1500 нг/л, более приблизительно 2000 нг/л, более приблизительно 2500 нг/л, более приблизительно 3000 нг/л, более приблизительно 3500 нг/л, более приблизительно 4000 нг/л, более приблизительно 4500 нг/л или более приблизительно 5000 нг/л, и введение субъекту фармацевтической композиции, содержащей терапевтически эффективное количество антагониста IL-4R.
Эотаксин-3 принадлежит к группе хемокинов, высвобождаемых эпителиальными клетками дыхательных путей, который положительно регулируется цитокинами Th2 IL-4 и IL-13 (Lilly et al 1999, J. Allergy Clin. Immunol. 104: 786-790). Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R для лечения пациентов с повышенными уровнями эотаксина-3, например, более приблизительно 100 пг/мл, более приблизительно 150 пг/мл, более приблизительно 200 пг/мл, более приблизительно 300 пг/мл или более приблизительно 350 пг/мл. Уровни эотаксина-3 в сыворотке крови можно измерить, например, с помощью ELISA.
Периостин представляет собой белок внеклеточного матрикса, вовлеченный в Th2-опосредованные воспалительные процессы. Обнаружено, что уровни периостина повышены у пациентов с астмой (Jia et al 2012 J Allergy Clin Immunol. 130:647-654.e10. doi: 10.1016/j.jaci.2012.06.025. электронная публикация от 1 августа 2012 г.). Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R для лечения пациентов с повышенными уровнями периостина.
Фракционный выдыхаемый NO (FeNO) представляет собой биомаркер воспаления бронхов или дыхательных путей. FeNO продуцируется эпителиальными клетками дыхательных путей в ответ на воспалительные цитокины, в том числе IL-4 и IL-13 (Alwing et al 1993, Eur. Respir. J. 6: 1368-1370). Уровни FeNO у здоровых взрослых людей находятся в диапазоне от 2 до 30 частей на миллиард (ppb). Иллюстративным анализом для измерения FeNO является использование устройства NIOX Aerocrine AB, Сольна, Швеция. Оценку можно проводить до спирометрии или после воздержания от пищи в течение по меньшей мере часа. Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R пациентам с повышенными уровнями выдыхаемого NO (FeNO), например, более приблизительно 30 ppb, более приблизительно 31 ppb, более приблизительно 32 ppb, более приблизительно 33 ppb, более приблизительно 34 ppb или более приблизительно 35 ppb.
Раково-эмбриональный антиген (CEA) (также известный как молекула клеточной адгезии 5 CEA [CEACAM5]) представляет собой опухолевый маркер, который, как было обнаружено, связан с заболеваниями легкого неопухолевой природы (Marechal et al 1988, Anticancer Res. 8: 677-680). Уровни CEA в сыворотке крови можно измерить с помощью ELISA. Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R пациентам с повышенными уровнями CEA, например, более приблизительно 1,0 нг/мл, более приблизительно 1,5 нг/мл, более приблизительно 2,0 нг/мл, более приблизительно 2,5 нг/мл, более приблизительно 3,0 нг/мл, более приблизительно 4,0 нг/мл или более приблизительно 5,0 нг/мл.
YKL-40 (названный по своим N-концевым аминокислотам тирозину (Y), лизину (K) и лейцину (L) и своей молекулярной массе 40 кДа) представляет собой хитиназа-подобный белок, который, как обнаружено, положительно регулируется и связан с обострением астмы, IgE и эозинофилами (Tang et al 2010 Eur. Respir. J. 35: 757-760). Уровни YKL-40 в сыворотке крови измеряют, например, с помощью ELISA. Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R пациентам с повышенными уровнями YKL-40, составляющими, например, более приблизительно 40 нг/мл, более приблизительно 50 нг/мл, более приблизительно 100 нг/мл, более приблизительно 150 нг/мл, более приблизительно 200 нг/мл или более приблизительно 250 нг/мл.
Периостин представляет собой секретируемый белок внеклеточного матрикса, ассоциированный с фиброзом, а его экспрессия положительно регулируется рекомбинантными IL-4 и IL-13 в культивируемых эпителиальных клетках бронхов и фибробластах бронхов (Jia et al. (2012) J. Allergy Clin. Immunol.130:647). У пациентов-людей с астмой уровни экспрессии периостина связаны с толщиной ретикулярной базальной мембраны, показателем субэпителиального фиброза. Там же. Настоящее изобретение включает способы, предусматривающие введение антагониста IL-4R пациентам с повышенными уровнями периостина.
Эозинофилы и нейтрофилы индуцированной мокроты являются общепринятыми прямыми маркерами воспаления дыхательных путей (Djukanovic et al 2002, Eur. Respire. J. 37: 1S-2S). Отделение мокроты индуцируют ингаляцией гипертонического солевого раствора и подсчитывают количество клеток в соответствии со способами, известными из уровня техники, например, в соответствии с руководствами Европейского респираторного общества.
В некоторых вариантах осуществления субъектов разделяют на следующие группы: содержание эозинофилов в крови (высокое содержание эозинофилов в крови) ≥ 300 клеток/мкл (HEo), или 300-499 клеток/мкл, или ≥ 500 клеток/мкл, содержание эозинофилов в крови 200-299 клеток/мкл (умеренное содержание эозинофилов), или содержание эозинофилов в крови < 200 клеток/мкл (низкое содержание эозинофилов), и введение антитела к IL-4R или его антигенсвязывающего фрагмента осуществляют в дозе или согласно схеме введения на основании уровня эозинофилов.
В некоторых вариантах осуществления субъектов разделяют на следующие группы: содержание эозинофилов в крови ≥ 300 клеток/мкл, 300-499 клеток/мкл или ≥ 500 клеток/мкл (высокое содержание эозинофилов в крови); содержание эозинофилов в крови ≥ 150 клеток/мкл (умеренное содержание эозинофилов); или содержание эозинофилов в крови < 150 клеток/мкл (низкое содержание эозинофилов), и ведение антитела к IL-4R или его антигенсвязывающего фрагмента осуществляют в дозе или согласно схеме введения на основании уровня эозинофилов.
В некоторых вариантах осуществления субъект имеет астму "эозинофильного фенотипа", определяемую по содержанию эозинофилов в крови ≥ 150 клеток/мкл, содержанию эозинофилов в крови ≥ 300 клеток/мкл, содержанию эозинофилов в крови 300-499 клеток/мкл или содержанию эозинофилов в крови ≥ 500 клеток/мкл, и ему вводят антитело к IL-4R или его антигенсвязывающий фрагмент.
Способы определения ассоциированных с астмой фармакодинамических параметров
Настоящее изобретение включает способы определения одного или нескольких ассоциированных с астмой фармакодинамических параметров у нуждающегося в этом субъекта, вызванных введением фармацевтической композиции, содержащей антагонист IL-4R. Снижение частоты обострения астмы (как описано выше) или улучшения одного или нескольких ассоциированных с астмой параметров (как описано выше) может быть связано с улучшением одного или нескольких фармакодинамических ассоциированных с астмой параметров; однако такая связь не обязательно наблюдается во всех случаях.
Примеры "ассоциированных с астмой фармакодинамических параметров" включают, например, следующие: (a) уровни экспрессии биомаркеров; (b) анализ сывороточных белков и РНК; (c) уровни эозинофилов и нейтрофилов индуцированной мокроты; (d) выдыхаемый оксид азота (FeNO) и (e) содержание эозинофилов в крови. "Улучшение ассоциированного с астмой фармакодинамического параметра" означает, например, снижение от исходного уровня одного или нескольких биомаркеров, таких как TARC, эотаксин-3 или IgE, снижение эозинофилов или нейтрофилов в мокроте, FeNO, периостина или содержания эозинофилов в крови. Используемый в данном документе термин "исходный уровень" по отношению к ассоциированному с астмой фармакодинамическому параметру означает числовую величину ассоциированного с астмой фармакодинамического параметра для пациента до или в течение всего периода введения фармацевтической композиции, описанной в данном документе.
Для оценки ассоциированного с астмой фармакодинамического параметра параметр измеряют количественно на исходном уровне и во временной точке после введения фармацевтической композиции. Например, ассоциированный с астмой фармакодинамический параметр можно измерять в день 1, день 2, день 3, день 4, день 5, день 6, день 7, день 8, день 9, день 10, день 11, день 12, день 14 или на неделе 3, неделе 4, неделе 5, неделе 6, неделе 7, неделе 8, неделе 9, неделе 10, неделе 11, неделе 12, неделе 13, неделе 14, неделе 15, неделе 16, неделе 17, неделе 18, неделе 19, неделе 20, неделе 21, неделе 22, неделе 23, неделе 24 или дольше после первичного лечения фармацевтической композицией. Различие между величиной параметра в определенной временной точке после начала лечения и величиной параметра на исходном уровне используют для установления того, произошло ли изменение, такое как "улучшение" ассоциированного с астмой фармакодинамического параметра (например, повышение или снижение, в зависимости от ситуации, в зависимости от конкретного параметра, измерение которого осуществляют).
В определенных вариантах осуществления введение антагониста IL-4R пациенту вызывает изменение, такое как снижение или повышение экспрессии определенного биомаркера. Ассоциированные с астмой биомаркеры включают без ограничения следующие: (a) общий IgE; (b) хемокин, регулируемый тимусом и активацией (TARC); (c) YKL-40; (d) раково-эмбриональный антиген в сыворотке крови; (e) эотаксин-3 в плазме крови и (f) периостин в сыворотке крови. Например, введение антагониста IL-4R пациенту с астмой может вызвать одно или несколько из снижения уровней TARC или эотаксина-3 или снижения уровней общего IgE в сыворотке крови. Снижение можно обнаружить на неделе 1, неделе 2, неделе 3, неделе 4, неделе 5 или позже после введения антагониста IL-4R. Экспрессию биомаркеров можно определить способами, известными из уровня техники. Например, уровни белка можно измерить с помощью ELISA (твердофазного иммуноферментного анализа). Уровни РНК можно измерить, например, с помощью обратной транскрипции, связанной с полимеразной цепной реакцией (RT-PCR).
Экспрессию биомаркеров, описанных выше, можно определить по выявлению белка или РНК в сыворотке крови. Образцы сыворотки крови также можно использовать для контроля дополнительных биомаркеров, представляющих собой белок или РНК, связанных с ответом на лечение антагонистом IL-4R, передачей сигнала IL-4/IL-13, астмой, атопией или эозинофильными заболеваниями (например, посредством измерения растворимых IL-4Rα, IL-4, IL-13, периостина). В некоторых вариантах осуществления образцы РНК используют для определения уровней РНК (негенетический анализ), например, уровней РНК биомаркеров; и в других вариантах осуществления образцы РНК используют для секвенирования транскриптомов (например, генетического анализа).
Составы
В некоторых вариантах осуществления антитело или его антигенсвязывающий фрагмент составляют в композиции, содержащей i) приблизительно 150 мг/мл антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R, ii) приблизительно 20 мМ гистидина, iii) приблизительно 12,5 мМ ацетата, iv) приблизительно 5% (вес/об.) сахарозы, v) приблизительно 25 мМ гидрохлорида аргинина, vi) приблизительно 0,2% (вес/об.) полисорбата 80, где значение pH состава составляет приблизительно 5,9, и где вязкость состава составляет приблизительно 8,5 сантипуазы.
В альтернативных вариантах осуществления антитело или его антигенсвязывающий фрагмент составляют в композиции, содержащей i) приблизительно 175 мг/мл антитела или его антигенсвязывающего фрагмента, которые специфически связываются с IL-4R, ii) приблизительно 20 мМ гистидина, iii) приблизительно 12,5 мМ ацетата, iv) приблизительно 5% (вес/об.) сахарозы, v) приблизительно 50 мМ гидрохлорида аргинина, vi) приблизительно 0,2% (вес/об.) полисорбата 80, где значение pH состава составляет приблизительно 5,9, и где вязкость состава составляет приблизительно 8,5 сантипуазы.
В конкретных вариантах осуществления антитело или его антигенсвязывающий фрагмент содержат HCVR, содержащий аминокислотную последовательность под SEQ ID NO: 1, и LCVR, содержащий аминокислотную последовательность под SEQ ID NO: 2.
Настоящее изобретение дополнительно иллюстрируют следующие примеры, которые не должны рассматриваться как дополнительное ограничение. Содержание графических материалов и всех ссылок, патентов и опубликованных заявок на патенты, цитируемых в настоящей заявке, полностью включено в данный документ посредством ссылки для всех целей.
Кроме того, в соответствии с настоящим изобретением можно использовать общепринятые методики молекулярной биологии, микробиологии и рекомбинантной ДНК в пределах компетенции специалиста в данной области техники. Такие методики в полном объеме объясняются в литературе. См., например, Green & Sambrook, Molecular Cloning: A Laboratory Manual, Fourth Edition (2012) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York; DNA Cloning: A Practical Approach, Volumes I and II (D.N. Glover ed. 1985); Oligonucleotide Synthesis (M.J. Gait ed. 1984); Nucleic Acid Hybridization [B.D. Hames & S.J. Higgins eds. (1985)]; Transcription And Translation [B.D. Hames & S.J. Higgins, eds. (1984)]; Animal Cell Culture [R.I. Freshney, ed. (1986)]; Immobilized Cells And Enzymes [IRL Press, (1986)]; B. Perbal, A Practical Guide To Molecular Cloning (1984); F.M. Ausubel et al. (eds.), Current Protocols in Molecular Biology, John Wiley & Sons, Inc. (1994).
ПРИМЕРЫ
Следующие примеры изложены для того, чтобы обеспечить специалистов в данной области полным раскрытием и описанием получения и применения способов и композиций, описанных в настоящем изобретении, и они не предполагают ограничения объема того, что авторы настоящего изобретения рассматривают в качестве своего изобретения. Были приложены усилия для обеспечения точности в отношении используемых чисел (например, количеств, температуры и т.п.), однако, необходимо учитывать некоторые экспериментальные ошибки и отклонения. Если не указано иное, то части являются частями по весу, молекулярная масса является средней молекулярной массой, температура представлена в градусах Цельсия, а давление является атмосферным или близким к атмосферному.
Иллюстративный антагонист IL-4R, используемый в следующих примерах, представляет собой антитело человека к IL-4R под названием дупилумаб (также упоминаемое в данном документе как "mAb1").
Пример 1. Испытание фазы III исследования VENTURE (NCT02528214)
Тяжелая неконтролируемая астма может привести к зависимости от пероральных кортикостероидов с системным воздействием стероидов. Это может потенциально привести к серьезным краткосрочным и долгосрочным неблагоприятным эффектам, включая увеличение веса, диабет, остеопороз, глаукому, тревожность, депрессию, сердечно-сосудистые заболевания и иммуносупрессию. Пациенты с тяжелой хронической астмой живут с глубоким снижением функции легких, примерно 52 процента от прогнозируемого нормального уровня у тех, кто в данном исследовании находится на исходном уровне. Снижение функции легких влияет на их способность нормально дышать и может привести к частым обострениям, требующим неотложного лечения и госпитализации. Эти проблемы возникают даже у пациентов, лечение которых осуществляют с помощью систематического введения OCS.
Испытание/исследование фазы 3 проводили для оценки исследуемого дупилумаба у взрослых людей и подростков с тяжелой стероидозависимой астмой без требований к минимальному содержанию эозинофилов в крови с получением дополнительных 300 мг дупилумаба или плацебо раз в 2 недели в течение 24 недель. Испытание фазы 3 (VENTURE) включало 210 пациентов (203 завершили рандомизированный период лечения, 101 в группе дупилумаба и 102 в группе плацебо) с тяжелой астмой и регулярным применением поддерживающих OCS в течение шести месяцев до исследования (фиг. 1). В данном исследовании предписанным OCS был преднизон или преднизолон. Пациентов рандомизировали с использованием соотношения рандомизации 1:1 и лечили либо дупилумабом, 300 мг раз в две недели с нагрузочной дозой 600 мг, либо плацебо (фиг. 2). Срединное исходное содержание эозинофилов в данном исследовании составляло 260 эозинофилов/микролитр. Первичной конечной точкой было снижение дозы глюкокортикоидов на неделе 24. Ключевые вторичные конечные точки включали долю пациентов, достигших снижения дозы глюкокортикоидов на 50% и снижения дозы глюкокортикоидов до 5 мг/сутки на неделе 24. Частоту тяжелого обострения и объем форсированного выдоха за 1 секунду (FEV1) до введения бронхолитического средства оценивали в общей популяции и у пациентов с содержанием эозинофилов в крови ≥ 300 клеток/мкл. Безопасность оценивали в целом.
Критерии включения для данного исследования приведены ниже в таблице 1. Исходные демографические данные для исследования показаны на фиг. 3.
Таблица 1. Критерии включения.
|
Также применяли критерии исключения для исследования Venture; при этом критерием исключения для пациентов было EOS < 150, ограниченное не более чем 25% от общей популяции.
Анализируемой первичной конечной точкой было процентное снижение дозы OCS на неделе 24. Анализируемые ключевые вторичные конечные точки включали снижение дозы OCS на 50% или больше и снижение дозы OCS до < 5 мг/сутки. Другие исследуемые вторичные конечные точки включали достижение пациентом максимально возможного снижения в соответствии с протоколом и дальнейшее отсутствие потребности в OCS. Использовали критерии эффективности, специфические в отношении заболевания. Критериями было снижение тяжелого обострения в годовом исчислении и улучшение функции легких (FEV1). На фиг. 2 показано общее распределение пациентов в исследовании.
Первичный исход
В популяции, сформированной согласно назначенному лечению (ITT), лечение дупилумабом значительно снижало дозу перорального глюкокортикоида по сравнению с плацебо, сохраняя при этом контроль астмы: процентное изменение среднего значения (стандартная ошибка [SE]) методом наименьших квадратов (LS) от исходного уровня до недели 24 (−70,1% (4,90) относительно −41,9% (4,57) от исходного уровня соответственно (P < 0,001; фиг. 4A; таблица 2)). Наблюдаемое срединное изменение от исходного уровня до недели 24 у пациентов, лечение которых осуществляли дупилумабом, составило 100% (межквартильный диапазон (IQR) от 62,5% до 100%) относительно 50% (IQR от 0% до 100%) в группе плацебо.
Таблица 2. Краткое изложение исходов на неделе 24 - популяция ITT.
|
CI обозначает доверительный интервал, FeNO - фракция оксида азота в выдыхаемом воздухе, FEV1 - объем форсированного выдоха за 1 секунду, IQR - межквартильный интервал, LS - метод наименьших квадратов, ppb - части на миллиард, q2w - раз в 2 недели, SD - стандартное отклонение, и SE - стандартная ошибка. aРассчитано только по наблюдаемым данным. bFeNO тестировали непараметрически.
Вторичные исходы
Исходы снижения глюкокортикоидов
В отношении первичной конечной точки, через 24 недели в общей популяции добавление дупилумаба к стандартным средствам терапии значительно снижало применение поддерживающих пероральных глюкокортикоидов (OCS) на 70,1% (срединное значение 100 процентов) по сравнению с 41,9% с плацебо (срединное значение 50 процентов) (p < 0,001).
В предварительно определенных анализах пациентов с исходным содержанием эозинофилов, составляющим 300 клеток/микролитр или больше, добавление дупилумаба значительно снижало применение OCS в среднем на 80 процентов (срединное значение 100 процентов) по сравнению с 43 процентами для плацебо (срединное значение 50 процентов).
Доля пациентов, достигших ≥ 50% снижения дозы перорального глюкокортикоида относительно исходного уровня на неделе 24, была значительно выше при применении дупилумаба по сравнению с плацебо (80% относительно 50%; P < 0,001; наблюдаемые значения: 80% для дупилумаба, 53% для плацебо) (фиг. 4A; таблица 2). Анализы чувствительности также показали большую долю пациентов с 50%, 75% и 90% снижением пероральных глюкокортикоидов при применении дупилумаба (таблица 3). Значительно большее количество пациентов, получавших дупилумаб, по сравнению с плацебо, достигло снижения дозы перорального глюкокортикоида до < 5 мг/сутки (69% относительно 33%; P < 0,001; наблюдаемые значения: 72% для дупилумаба, 37% для плацебо) (фиг. 4A; таблица 2).
Таблица 3. Анализы чувствительности: процентное снижение дозы перорального глюкокортикоида (мг/сутки) на неделе 24, проанализированное с помощью модели пропорциональных рисков - популяция ITT.
|
Данные представляют собой среднее значение (SD) или кол-во (%). CI обозначает доверительный интервал, q2w - раз в 2 недели, и ITT - назначенное лечение. Процентное снижение дозы перорального глюкокортикоида на неделе 24 классифицировали по пяти порядковым категориям (от 90% до 100%, от 75% до < 90%, от 50% до < 75%, от > 0% до < 50%, отсутствие снижения или какого-либо увеличения дозы перорального глюкокортикоида, или выбыл из исследования). В модели использовали категорию конечной точки в качестве зависимой переменной и группы лечения, оптимизированную на исходном уровне дозу перорального глюкокортикоида, области и подгруппы на основании исходного уровня эозинофилов в крови (≥ 150 клеток/мкл или < 150 клеток/мкл) в качестве ковариат.
Примечательно, что 48% пациентов, лечение которых осуществляли дупилумабом, по сравнению с 26% пациентов, лечение которых осуществляли плацебо, достигли максимально возможного снижения дозы глюкокортикоидов на неделе 24 (Р=0,002; наблюдаемые значения: 52% для дупилумаба, 30% для плацебо) (фиг. 4A; таблица 2). Подобным образом, 48% пациентов, лечение которых осуществляли дупилумабом, по сравнению с 25% пациентов, лечение которых осуществляли плацебо, больше не нуждались в пероральных глюкокортикоидах на неделе 24 (Р=0,002; наблюдаемые значения: 52% для дупилумаба, 29% для плацебо) (фиг. 4A; таблица 2). Дупилумаб обеспечивал последовательное снижение итоговых показателей для перорального глюкокортикоида независимо от исходного содержания эозинофилов в крови (фиг. 5A и фиг. 5B; таблица 4).
Таблица 4. Анализы конечных точек для перорального глюкокортикоида в подгруппах на неделе 24 по подгруппам на основании исходного содержания эозинофилов крови.
|
CI обозначает доверительный интервал, LS - метод наименьших квадратов, SE - стандартную ошибку, и q2w - раз в 2 недели.
Хотя улучшения наблюдали во всех подгруппах на основании исходного содержания эозинофилов в крови, величина эффекта лечения была наибольшей у индивидуумов с более высоким исходным содержанием эозинофилов (например, отношение рисков по сравнению с плацебо у пациентов с ≥ 50% снижением дозы перорального глюкокортикоида составило 6,59 (95% CI от 2,1 до 20,4) для пациентов с ≥ 300 клеток/мкл и 2,91 (95% CI от 1,3 до 6,6) для пациентов с < 300 клеток/мкл на исходном уровне). В общей популяции 69 процентов среди пациентов, получавших дупилумаб, смогли снизить свою дозу OCS до менее 5 мг в сутки при сохранении контроля астмы по сравнению с 33 процентами пациентов, получавших плацебо (p менее 0,0001); в группе с высоким EOS 84 процента среди пациентов с применением дупилумаба смогли снизить свою дозу OCS до менее 5 мг в сутки по сравнению с 40 процентами для плацебо. (p равняется 0,0002.) Половина пациентов полностью исключила применение перорального глюкокортикоида. Несмотря на снижение уровня глюкокортикоидов, дупилумаб по сравнению с плацебо в общей популяции и в подгруппе EOS ≥ 300 клеток/мкл обеспечил снижение тяжелых обострений на 59,3% (P < 0,001) и 71,1% и улучшение FEV1 на 0,22 л (P < 0,001) и 0,32 л соответственно.
Обострения и FEV1
В дополнение к значительному снижению применения перорального глюкокортикоида в течение 24-недельного периода лечения дупилумаб обеспечил значительное (P < 0,001) снижение тяжелых обострений астмы по сравнению с плацебо на 59,3% в общей популяции (фиг. 4B и таблица 7), а также обеспечил улучшение FEV1 на среднее значение, определенное LS (SE), составляющее 0,22 л (0,05) (относительно 0,01 л [0,05] в группе плацебо, P < 0,001) на неделе 24 в общей популяции. Хотя дупилумаб обеспечивал снижение частоты тяжелых обострений астмы в годовом исчислении и улучшение FEV1 по сравнению с плацебо независимо от исходного содержания эозинофилов (фиг. 6A и фиг. 6B, таблица 5), эти преимущества были более выраженными у пациентов с более высоким исходным содержанием эозинофилов в крови. Например, дупилумаб обеспечивал уменьшение тяжелых обострений на 71,1% и улучшение FEV1 на среднее значение, определенное LS (SE), составляющее 0,32 л (95% CI от 0,10 до 0,54) (оба P < 0,001 относительно плацебо) у пациентов с исходным содержанием эозинофилов в крови ≥ 300 клеток/мкл.
Таблица 5. Анализы обострений и изменения FEV1 до введения бронхолитического средства в подгруппах на неделе 24 по подгруппам на основании исходного содержания эозинофилов в крови.
|
CI обозначает доверительный интервал, FEV1 - объем форсированного выдоха за 1 секунду, LS - метод наименьших квадратов, q2w - раз в 2 недели, SD - стандартное отклонение, и SE - стандартную ошибку.
Улучшения FEV1 были быстрыми и устойчивыми уже на второй неделе (изменение среднего значения, определенного LS, составило 0,15 л; 95% CI от 0,04 до 0,26) и дополнительно увеличивались к неделе 24 (P < 0,05 во все моменты времени) (фиг. 4C и таблица 2). Через 24 недели дупилумаб обеспечил улучшение функции легких, что оценивали по увеличению объема форсированного выдоха за одну секунду (FEV1) на 220 мл (15 процентов) в общей популяции (p=0,0007) по сравнению с 10 мл для плацебо, и на 320 мл (25 процентов) по сравнению с 120 мл для плацебо у пациентов с содержанием эозинофилов, составляющим 300 клеток/мкл или больше (p=0,0049).
Другие вторичные и исследуемые исходы
В исследование фазы 3 включали пациентов со стероидозависимой тяжелой астмой, независимо от уровня эозинофилов или других биомаркеров и результаты показали улучшения по сравнению с плацебо в отношении функции легких и обострений в подгруппах пациентов: у пациентов с исходным содержанием эозинофилов более 300 клеток/мкл; более 150 клеток/мкл и менее 150 клеток/мкл. Дупилумаб продемонстрировал последовательное улучшение функции легких в рамках программы лечения астмы у пациентов с тяжелой астмой, испытывающих затруднение в связи со снижением своей повседневной способности к дыханию.
Показатели ACQ-5 на неделе 24 указали на значительное улучшение (P=0,002) контроля астмы при применении дупилумаба по сравнению с плацебо (различие среднего значения, определенного LS, изменения от исходного уровня: −0,47 [95% CI от −0,76 до −0,18]). При применении дупилумаба среднее значение, определенное методом наименьших квадратов, улучшения от исходного уровня (−1,05) на неделе 24 составляло две минимальных клинически значимых разницы 0,5 для прибора ACQ-5.
Лечение дупилумабом обеспечивало подавление FeNO на второй неделе и было устойчивым в течение 24-недельного периода лечения (P < 0,001 по сравнению с плацебо во все моменты времени; фиг. 4D). Процент пациентов с FeNO < 25 ppb (верхняя граница нормы) (таблица 6) увеличивался с 43,6% на исходном уровне до 84,4% в группе дупилумаба, тогда как изменений в группе плацебо не наблюдалось (с 44,7% до 45,1%).
Таблица 6. Доля пациентов, достигающих подавления FeNO до < 25 ppb на исходном уровне и неделе 24.
|
FeNO обозначает фракцию оксида азота в выдыхаемом воздухе, IQR - межквартильный интервал, ppb - части на миллиард, q2w - раз в 2 недели, и SD - стандартное отклонение.
Ежедневные показатели симптомов астмы в утреннее и вечернее время у пациентов с зависимой от перорального кортикостероида тяжелой астмой
Показатели симптомов астмы у пациентов записывали в виде баллов в электронном дневнике в утреннее время для симптомов в течение ночи (симптомы до полудня) и в вечернее время (симптомы после полудня) для симптомов в течение дня с оценкой их тяжести от 0 (самая легкая) до 4 (самая тяжелая). Изменения от исходного уровня показателей симптомов астмы в течение 24-недельного периода лечения анализировали с использованием моделей со смешанным эффектом с повторными измерениями.
Средние значения показателей симптомов до полудня/после полудня в группах дупилумаба и плацебо соответственно составляли 1,37/1,37 и 1,50/1,52 в популяции ITT (n=210) и 1,45/1,49 и 1,50/1,52 у пациентов, которые снизили применение OCS на 100% к неделе 24 (40,5%). В группе дупилумаба показатели симптомов быстро улучшились (изменение среднего значения, определенного LS, от исходного уровня показателей симптомов до полудня/после полудня на неделе 2 ─0,18/─0,23; оба P < 0,05 по сравнению с плацебо) с продолжающимся улучшением на 16 неделе (─0,47/─0,47, оба P < 0,05 по сравнению с плацебо) и сохраняли положительный эффект до недели 24 (фиг. 25A и фиг. 25C). Пациенты в группе дупилумаба, которые снизили применение OCS на 100% к неделе 24, продемонстрировали схожий характер ответа с большей величиной улучшения показателей симптомов (фиг. 25B и фиг. 25D). В целом, наиболее частыми появившимися во время лечения нежелательными явлениями, возникающими у пациентов, лечение которых осуществляли дупилумабом и плацебо, была эозинофилия (14% относительно 1%). Реакции в месте инъекции возникали у 9% пациентов, лечение которых осуществляли дупилумабом, и у 4% пациентов, лечение которых осуществляли плацебо.
Дупилумаб обеспечивал быстрое и устойчивое улучшение показателей ежедневных симптомом астмы в утреннее и вечернее время у пациентов с OCS-зависимой тяжелой астмой, несмотря на отмену OCS. Улучшение показателей симптомов было наибольшим у пациентов, которые снизили применение OCS на 100% к неделе 24. Дупилумаб в целом хорошо переносился.
Популяция: ITT; подгруппа 100% снижения OCS. Конечные точки: изменение среднего значения, определенного LS, от исходного уровня показателей симптомов до полудня/после полудня на протяжении периода лечения. Группы лечения: 300 мг дупилумаба q2w; плацебо.
Контроль астмы и связанное с состоянием здоровья качество жизни
Контроль астмы оценивали путем еженедельной записи в электронном дневнике утвержденного опросника по контролю астмы из 5 пунктов (ACQ-5), по которому более высокий показатель (диапазон 0-6) указывал на меньший контроль. Связанное с состоянием здоровья качество жизни (HRQoL) оценивали с помощью самостоятельно заполняемого опросника оценки качества жизни у больных астмой из 7 пунктов (AQLQ), по которому более высокие общие показатели (диапазон 0-7) указывали на лучшее HRQoL. Изменения от исходного уровня показателей ACQ-5 и AQLQ в течение 24-недельного периода лечения анализировали с использованием моделей со смешанным эффектом с повторными измерениями.
В группах дупилумаба и плацебо соответственно средние исходные показатели ACQ-5 составляли 2,42 и 2,58, а средние исходные показатели AQLQ составляли 4,38 и 4,31. В группе дупилумаба контроль астмы быстро улучшился (неделя 2, изменение среднего значения, определенного LS, от исходного уровня показателя ACQ-5, 0,57; P=0,002 по сравнению с плацебо), дополнительно улучшился на неделе 12 (1,01; P=0,001 по сравнению с плацебо) и оставался стабильным до 24 недели (1,05; P=0,002 по сравнению с плацебо)(фиг. 26A). У пациентов, получавших лечение дупилумабом, наблюдали улучшение среднего значения, определенного LS, от исходного уровня показателя AQLQ на 0,76 на неделе 12 (P=0,14 по сравнению с плацебо), которое дополнительно улучшилось до 0,89 на неделе 24 (P=0,008 по сравнению с плацебо) (фиг. 26B). В целом, наиболее частыми появившимися во время лечения нежелательными явлениями, возникающими у пациентов, лечение которых осуществляли дупилумабом и плацебо, была эозинофилия (14% относительно 1%). Реакции в месте инъекции возникали у 9% пациентов, лечение которых осуществляли дупилумабом, и у 4% пациентов, лечение которых осуществляли плацебо.
Добавление дупилумаба по сравнению с плацебо обеспечивало значительное улучшение контроля астмы и улучшение HRQoL у пациентов с OCS-зависимой тяжелой астмой. Улучшение контроля астмы произошло уже на неделе 2 и сохранялось в течение 24 недель. Дупилумаб в целом хорошо переносился.
Популяция: ITT. Конечные точки: Изменение среднего значения, определенного LS, от исходного уровня ACQ-5 на неделях 2, 12 и 24; изменение среднего значения, определенного LS, от исходного уровня AQLQ на неделях 12 и 24; безопасность в течение периода лечения. Группы лечения: 300 мг дупилумаба q2w; плацебо.
Безопасность
Частота TEAE была одинаковой во всех группах лечения (62,1% относительно 64,5% для дупилумаба относительно плацебо) в популяции по оценке безопасности. TEAE согласно предпочтительным терминам Медицинского словаря для нормативно-правовой деятельности (MedDRA), наиболее часто встречающимися у ≥ 5% пациентов, лечение которых осуществляли дупилумабом, по сравнению с плацебо, были вирусные инфекции верхних дыхательных путей (8,7% относительно 17,8%), бронхит (6,8% относительно 5,6%), синусит (6,8% относительно 3,7%), грипп (2,9% относительно 5,6%), реакции в месте инъекции (8,7% относительно 3,7%) и лабораторные показатели эозинофилии (сгруппированные как предпочтительные термины "увеличение содержания эозинофилов" и "эозинофилия") (13,6% относительно 0,9%). Согласно протоколу исследования, все случаи содержания эозинофилов > 3000 клеток/мкл при лечении должны были регистрироваться как AE и встречались у 12,6% пациентов, лечение которых осуществляли дупилумабом, относительно 0,9% в группе плацебо. Зарегистрированные TEAE эозинофилии были исключительно лабораторными данными без каких-либо клинических последствий или ассоциированных AE.
Серьезные TEAE были зарегистрированы у 9 (8,7%) пациентов, лечение которых осуществляли дупилумабом, и у 6 (5,6%), лечение которых осуществляли плацебо; серьезные TEAE не были связаны с исследуемым медицинским продуктом. В данном исследовании не было случаев летального исхода. Появившиеся во время лечения гуморальные иммунные ответы на лекарственное средство наблюдались у 5 пациентов в каждой группе (дупилумаб 5,0%; плацебо 4,7%) и не оказывали значительного влияния на эффективность или безопасность.
Таблица 7. Изменения FEV1 и биомаркеров воспаления на неделях 4 и 24 у пациентов, лечение которых осуществляли дупилумабом, по сравнению с плацебо.
|
*P < 0,05, **P < 0,01, ***P < 0,001 относительно плацебо.
CI - доверительный интервал, SE - стандартная ошибка, SD - стандартное отклонение, FEV1 - объем форсированного выдоха за 1 секунду, FeNO - фракция оксида азота в выдыхаемом воздухе, TARC - хемокин, регулируемый тимусом и активацией.
Способы
Схема исследования и надзор
В этом многонациональном рандомизированном двойном слепом плацебо-контролируемом исследовании фазы 3 оценивали эффективность и безопасность дупилумаба у пациентов с зависимой от перорального глюкокортикоида тяжелой астмой. Пациенты завершили 8-10-недельный период оптимизации дозы перорального глюкокортикоида с последующей рандомизацией 1:1 для получения дупилумаба или плацебо в течение 24-недельного периода лечения. Этот период лечения состоял из 4-недельного периода индукции, в течение которого продолжалась оптимизированная доза перорального глюкокортикоида; 16-недельного периода снижения перорального глюкокортикоида (недели 4-20), в течение которого доза глюкокортикоидов снижалась каждые 4 недели в соответствии с заранее определенным алгоритмом протокола; 4-недельного поддерживающего периода, в течение которого пациенты оставались на дозе глюкокортикоидов, установленной на неделе 20; и 12-недельного периода оценки после лечения. Удовлетворяющих критериям включения пациентов, которые завершили лечение, допустили к долгосрочному открытому расширенному исследованию.
Исследование проводили в соответствии с Хельсинкской декларацией, руководящими принципами Международной конференции по гармонизации надлежащей клинической практики и применимыми нормативно-правовыми требованиями. Независимый комитет по мониторингу данных и безопасности проводил слепой мониторинг данных по безопасности пациентов. Местный институциональный наблюдательный совет или комитет по этике в каждом исследовательском центре следили за проведением испытаний и документацией. Все пациенты дали письменное информированное согласие, прежде чем принимать участие в испытании.
Пациенты
К участию допускались пациенты в возрасте ≥ 12 лет с диагностированной врачом астмой в течение ≥ 12 месяцев, исходя из Руководящих принципов Глобальной инициативы по астме 2014 года. Пациенты должны были регулярно принимать системные глюкокортикоиды в течение предыдущих 6 месяцев (от 5 до 35 мг/сутки преднизона или преднизолона или эквивалента) в течение 4 недель до скрининга, а также высокие дозы ингаляционного глюкокортикоида (общая суточная доза флутиказона пропионата или равносильного эквивалента > 500 мкг) в комбинации с не более двумя контролирующими препаратами (например, β2-агонист длительного действия или антагонист лейкотриеновых рецепторов) в течение ≥ 3 месяцев. Подходящие пациенты должны были характеризоваться объемом форсированного выдоха за 1 секунду (FEV1) до введения бронхолитического средства ≤ 80% от прогнозируемой нормы (≤ 90% для подростков), обратимостью 17 FEV1 ≥ 12% и 200 мл, или гиперреактивностью дыхательных путей, задокументированной в течение 12 месяцев до скринингового визита 1. Пациентов набирали без минимальных требований к исходному содержанию эозинофилов в крови или мокроте или любых других биомаркеров типа 2 (например, FeNO или IgE). Ключевые критерии исключения включали заболевания легких, кроме астмы, ухудшение астмы, требующее неотложного лечения или госпитализации в течение 4 недель после визита 1, а также курильщики в настоящий момент или курильщики, которые бросили курить в течение 6 месяцев до скрининга или имели курение в анамнезе > 10 пачко-лет
Лечение и процедуры
Пациенты были рандомизированы (1:1) для получения подкожно 300 мг дупилумаба (после нагрузочной дозы 600 мг в день 1) в качестве дополнительной терапии или соответствующего плацебо раз в 2 недели (q2w). Рандомизация проводилась с помощью интерактивной технологии ответа с помощью голосовой/цифровой связи, и пациентов стратифицировали в соответствии с оптимизированной дозой перорального глюкокортикоида (≤ 10 мг/сутки или > 10 мг/сутки преднизона/преднизолона) и страной. Пациенты, использующие другие пероральные глюкокортикоиды, были переведены на клинически сопоставимую дозу преднизона или преднизолона в течение периода скрининга.
Оптимизированная доза перорального глюкокортикоида была определена как самая низкая доза, которую пациент мог переносить без увеличения на ≥ 0,5 показателя по опроснику по контролю астмы из 5 пунктов (ACQ-5), тяжелого обострения или любого клинически значимого явления, требующего регуляции дозы перорального глюкокортикоида. Во время фазы снижения дозы дозу перорального глюкокортикоида снижали каждые 4 недели, чтобы свести к минимуму риск клинически значимых явлений и эффектов предыдущей дозы. Регуляция дозы не допускалась после недели 20, за исключением соображений безопасности. Прием фоновых контролирующих препаратов от астмы был продолжен в стабильной дозе, и применение β2-агонистов кратковременного действия было разрешено по мере необходимости для симптомов астмы.
Конечные точки
Первичной конечной точкой эффективности было процентное снижение дозы перорального глюкокортикоида от исходного уровня к неделе 24 при сохранении контроля астмы. Пациент считался поддерживающим контроль астмы между неделями 20 и 24 , если ни одно клинически значимое явление (на основании суждения исследователя) не требовало регуляции дозы перорального глюкокортикоида. Для пациентов, у которых наблюдалось обострение, конечной дозой перорального глюкокортикоида считалась доза на одну ступень выше, чем та, которую они получали во время обострения.
Ключевыми вторичными конечными точками эффективности, оцененными у пациентов, поддерживающих контроль астмы, были доля пациентов, достигших ≥ 50% снижения от исходного уровня дозы перорального глюкокортикоида, и доля пациентов, достигших снижения дозы перорального глюкокортикоида до < 5 мг/сутки. Другие вторичные конечные точки включали абсолютное снижение дозы перорального глюкокортикоида, долю пациентов, достигших максимально возможного снижения дозы перорального глюкокортикоида, и долю пациентов, которым больше не требуются пероральные глюкокортикоиды.
Дополнительные конечные точки эффективности включали частоту в годовом исчислении явлений тяжелого обострения в течение периода лечения (определяется как необходимость госпитализации, визита в отделение неотложной помощи или лечения в течение ≥ 3 дней системными глюкокортикоидами в количестве, превышающем текущую дозу в по меньшей мере 2 раза); абсолютное изменение от исходного уровня FEV1 до введения бронхолитического средства на неделях 2, 4, 8, 12, 16, 20 и 24; и изменение от исходного уровня показателя ACQ-5 на неделе 24.
Исследуемую конечную точку абсолютного изменения от исходного уровня FeNO (ppb) оценивали с использованием прибора NIOX (Aerocrine AB, Солна, Швеция) на неделях 2, 4, 8, 12, 16, 20 и 24.
Статистический анализ
Было установлено, что 90 рандомизированных пациентов на группу лечения дали бы в исследовании 94% мощности (2-выборочный тест при α=0,05), чтобы выявить различие в лечении в 27% в суточной дозе глюкокортикоида 18 при условии, что общее стандартное отклонение составляет 50%.
Первичную конечную точку анализировали с использованием модели ковариационного анализа (ANCOVA). Модель включала процентное снижение дозы перорального глюкокортикоида на неделе 24 в качестве зависимой переменной и группы лечения, оптимизированную на исходном уровне дозу перорального глюкокортикоида, области (объединенные страны) и подгруппы на основании исходного содержания эозинофилов (≥ 150 клеток/мкл, < 150 клеток/мкл) в качестве ковариат. Различие в лечении проверяли при 2-выборочном уровне значимости α=0,05. Для пациентов, которые прекратили исследование или у которых отсутствовали данные о дозе перорального глюкокортикоида на неделе 24 (2 пациента в группе дупилумаба и 1 в группе плацебо), основным подходом к обработке отсутствующих данных была модель комбинации закономерностей методом множественного восстановления (PMM с помощью MI).
Ключевые вторичные и другие комбинированные вторичные конечные точки анализировали с использованием моделей логистической регрессии. Частоту в годовом исчислении явлений тяжелого обострения в течение 24-недельного периода лечения анализировали с помощью модели отрицательной биномиальной регрессии. Подход с применением моделей со смешанными эффектами с повторными измерениями использовали для анализа изменений от исходного уровня FEV1 до введения бронхолитического средства в различные моменты времени в течение 24-недельного периода лечения и изменений от исходного уровня показателя по опроснику по контролю астмы из 5 пунктов (ACQ-5) на неделе 24.
Анализ эффективности проводили на популяции, сформированной согласно назначенному лечению (ITT), определяемой как все рандомизированные пациенты, проанализированные в соответствии с назначенным лечением, независимо от полученного лечения. Первичные и ключевые вторичные конечные точки, FEV1 и частоту тяжелого обострения астмы также анализировали в подгруппах пациентов, определяемых на основании исходных уровней эозинофилов в крови (≥ 300 клеток/мкл, < 300 клеток/мкл, ≥ 150 клеток/мкл и < 150 клеток/мкл). Популяция по оценке безопасности включала всех пациентов, которые получили ≥ 1 дозу или частичную дозу исследуемого средства лечения, проанализированных в соответствии с полученным лечением.
Все анализы проводили с использованием программного обеспечения SAS, версии 9.4 (SAS Institute).
Вывод
Это исследование продемонстрировало, что дупилумаб в качестве дополнительной терапии обеспечивал значительное уменьшение применения перорального глюкокортикоида у пациентов с зависимой от перорального глюкокортикоида тяжелой астмой, уменьшение тяжелых обострений астмы на 59,3% и улучшение FEV1 на 0,22 л в общей популяции со снижением на 71% обострений и улучшением на 0,32 л FEV1 у пациентов с "эозинофилией" с исходным содержанием эозинофилов в крови ≥ 300 клеток/мкл. Лечение дупилумабом также обеспечивало улучшение контроля астмы и снижение уровней FeNO, маркера воспаления дыхательных путей 2 типа.
Добавление 300 мг дупилумаба раз в 2 недели (q2w) (по сравнению с плацебо) обеспечивало значительное снижение применения перорального кортикостероида (OCS) на неделе 24 (среднее значение, определенное методом наименьших квадратов [LS] составило 70,1% относительно 41,9%, срединное значение 100% относительно 50%) при одновременном снижении частоты тяжелого обострения астмы в течение 24-недельного периода лечения (59%) и улучшении объема форсированного выдоха за 1 секунду (FEV1) на неделе 24 (различие средних значений, определенных LS, составляет 0,22 л), и в целом он хорошо переносился пациентами с OCS-зависимой тяжелой астмой.
Дупилумаб является первым биологическим веществом, продемонстрировавшим положительную эффективность на основе множества итоговых показателей в отношении астмы в общей популяции исследования, независимо от исходного содержания эозинофилов в крови (т.е. ≥ 300, < 300, ≥ 150 и < 150 клеток/мкл). В самом деле, 28,6% включенных пациентов характеризовались исходным содержанием эозинофилов в крови < 150 клеток/мкл. В этой подгруппе у 75% пациентов, лечение которых осуществляли дупилумабом, дозы перорального глюкокортикоида снизились на 50%, а у 62% пациентов дозы перорального глюкокортикоида снизились до < 5 мг/сутки. Эти данные противоречат предыдущим исследованиям с моноклональными антителами к интерлейкину-5, включая меполизумаб и бенрализумаб, которые показали эффект лечения исключительно у пациентов с высоким исходным содержанием эозинофилов в крови.
В этом исследовании у пациентов, лечение которых осуществляли плацебо, также отмечалось снижение зависимости от перорального глюкокортикоида на 41,9%. Лучшее соблюдение схем приема лекарственных средств в условиях клинического исследования, возможно, способствовало этому наблюдению. Однако ближе к концу исследования у этих пациентов, лечение которых осуществляли плацебо, наблюдалось умеренное ухудшение функции легких (FEV1), что дополнительно подчеркивает необходимость лечения, которое улучшает функцию легких у пациентов с зависимой от перорального глюкокортикоида тяжелой астмой. Способность дупилумаба повышать функцию легких так же заметно, как это было в данном исследовании, даже несмотря на отмену приема глюкокортикоида, указывает на то, что он, по-видимому, ингибирует ключевые факторы воспаления легких, которые приводят к снижению функции легких.
Дупилумаб обеспечивал снижение уровней FeNO в условиях значительной отмены перорального глюкокортикоида в популяции исследования с персистирующим воспалением 2 типа (определяется по повышенному FeNO), несмотря на систематическое применение глюкокортикоида.
Дупилумаб обеспечивал уменьшение дозы перорального глюкокортикоида на наблюдаемое среднее значение 74% (наблюдаемое срединное значение 100%) в более широкой популяции без требований к минимальному исходному содержанию эозинофилов в крови. Не желая быть связанными научной теорией, эти данные указывают на то, что дупилумаб с его двойной блокадой сигнальных путей интерлейкина-4 и интерлейкина-13 посредством блокады альфа-рецептора интерлейкина-4 ингибирует воспаление 2 типа более широко, чем целенаправленное воздействие только на эозинофилы. В то время как интерлейкин-4 играет центральную роль в дифференцировке и пролиферации клеток T-хелперов 2, индуцируя продуцирование цитокинов и синтез IgE, интерлейкин-13 играет ключевую роль в патологических особенностях заболевания, таких как гиперплазия бокаловидных клеток, выработка слизи, сократительная способность гладких мышц и гиперчувствительность дыхательных путей.
У пациентов с зависимой от глюкокортикоида тяжелой астмой дупилумаб обычно хорошо переносился, а профиль безопасности соответствовал предыдущим исследованиям по астме и другим показаниям, таким как эозинофильный эзофагит, назальный полипоз и атопический дерматит. У пациентов, лечение которых осуществляли дупилумабом, наблюдалось большее среднее кратковременное увеличение содержания эозинофилов в крови от исходного уровня по сравнению с плацебо с увеличением доли пациентов (12,6%) с содержанием эозинофилов > 3000 клеток/мкл. У пациентов с кратковременным повышением содержания эозинофилов в крови не было сопутствующих клинических AE или последствий. Увеличение содержания эозинофилов в крови согласуется с гипотезой о том, что дупилумаб блокирует функцию интерлейкина-4 и интерлейкина-13 в отношении выживаемости, активации и рекрутинга эозинофилов в ткани, но не возврата из костного мозга, что приводит к кратковременному увеличению содержания циркулирующих эозинофилов. Поскольку глюкокортикоиды подавляют циркулирующие эозинофилы, большее снижение перорального глюкокортикоида в группе дупилумаба также может способствовать повышению содержания эозинофилов. Между группами дупилумаба и плацебо не наблюдалось связанных с лечением AE конъюнктивита, в отличие от исследований дупилумаба при атопическом дерматите.
В заключение следует отметить, что дополнительная терапия дупилумабом обеспечивала значительное снижение потребности в пероральных глюкокортикоидах, одновременно уменьшая тяжелые обострения и улучшая функцию легких (FEV1) у пациентов с зависимой от глюкокортикоидов тяжелой астмой вне зависимости от исходного содержания эозинофилов в крови, и в целом хорошо переносилась.
Пример 2. Испытание фазы III исследования QUEST (NCT02414854)
Способы
Пациентов с астмой, возрастом ≥12 лет, с астмой от умеренной до тяжелой степени, неконтролируемой с применением ICS и одного или двух контролирующих препаратов, рандомизировали 2:1 для добавления подкожно 200 или 300 мг дупилумаба раз в 2 недели (q2w) или сопоставимого плацебо в течение 52 недель в двойном слепом плацебо-контролируемом исследовании фазы 3 (NCT02414854). Первичными конечными точками были частота в годовом исчислении тяжелых обострений астмы и абсолютное изменение от исходного уровня к неделе 12 объема форсированного выдоха за 1 секунду (FEV1) до введения бронхолитического средства в общей популяции исследования. Вторичные конечные точки включали обострения и FEV1 у пациентов с ≥ 300 эозинофилов/мкл. Также оценивали контроль астмы и безопасность дупилумаба. Комбинированными первичными конечными точками были частота в годовом исчислении тяжелого обострения в течение 52 недель и изменение FEV1 (л) от исходного уровня к неделе 12.
Конкретные детали исследования описаны ниже. В этом рандомизированном двойном слепом плацебо-контролируемом испытании в параллельных группах оценивали эффективность дупилумаба у пациентов с неконтролируемой астмой от умеренной до тяжелой степени. Пациенты завершили период скрининга 4 ± 1 неделя с последующей рандомизацией на дупилумаб и плацебо сопоставимого объема, 52-недельным рандомизированным периодом лечения и 12-недельным периодом последующего наблюдения после окончания лечения (см. фиг. 7).
Пациенты
Пациенты в возрасте ≥ 12 лет с диагностированной врачом на основании Руководящих принципов Глобальной инициативы по бронхиальной астме 2014 года персистирующей астмой в течение ≥ 12 месяцев допускались к участию, отвечая следующим ключевым критериям: текущее лечение ингаляционным глюкокортикоидом в дозе от средней до высокой (общая суточная доза флутиказона пропионата или равносильного эквивалента > 500 мкг) плюс не более двух дополнительных контролирующих препаратов (например, β2-агонист длительного действия или антагонист лейкотриеновых рецепторов); объем форсированного выдоха за 1 секунду (FEV1) до введения бронхолитического средства (BD) ≤ 80% от прогнозируемой нормы (≤ 90% для лиц в возрасте от 12 до 17 лет); обратимость FEV1 ≥ 12% и 200 мл; показатель по опроснику по контролю астмы из 5 пунктов (ACQ-5) ≥ 1,5; ухудшение астмы в течение предшествующего года, которое требовало госпитализации, неотложной медицинской помощи или лечения системными глюкокортикоидами в течение ≥ 3 суток. Пациентов набирали независимо от исходного содержания эозинофилов или биомаркеров 2 типа в крови. (См. фиг. 8).
Лечение и процедуры
Пациентов рандомизировали (2:2:1:1) для получения в течение 52 недель дополнительной терапии подкожно 200 мг (нагрузочная доза 400 мг) или 300 мг дупилумаба (нагрузочная доза 600 мг) раз в 2 недели (q2w) или плацебо сопоставимого объема для каждой активной дозы (поставляется в предварительно заполненных шприцах, 1,14 мл для 200 мг дупилумаба и 2,0 мл для 300 мг дупилумаба). Рандомизацию проводили с помощью интерактивной технологии ответа с помощью голосовой/цифровой связи и стратифицировали по возрасту (< 18 лет, ≥ 18 лет), содержанию эозинофилов в периферической крови (< 300 клеток/мкл, ≥ 300 клеток/мкл) при скрининге, уровню дозы ингаляционного глюкокортикоида (средний/высокий) и стране. Прием фоновых лекарственных препаратов для контроля астмы продолжался в стабильной дозе на протяжении всего исследования и ежедневно записывался пациентами в электронном дневнике. Было разрешено применение ингаляционных глюкокортикоидов, β2-агонистов длительного действия, антагонистов мускариновых рецепторов длительного действия, антагонистов лейкотриеновых рецепторов и метилксантинов. На протяжении всего исследования пациентам разрешалось применение агонистов β2-адренергических рецепторов кратковременного действия, необходимых для облегчения симптомов. Измеряли биомаркеры 2 типа; биомаркеры включали содержание эозинофилов в крови, FeNO, IgE в сыворотке крови, периостин, TARC и эотаксин-3 в плазме крови.
Конечные точки
Первичными конечными точками эффективности были частота в годовом исчислении явлений тяжелого обострения в течение 52-недельного периода лечения и абсолютное изменение от исходного уровня FEV1 до BD на неделе 12 в общей популяции исследования. Эти конечные точки также включали в качестве вторичных конечных точек исследования для индивидуумов с содержанием эозинофилов в крови ≥ 300 эозинофилов/мкл. Дополнительные вторичные конечные точки исследования обобщены в таблице 8. Тяжелое обострение астмы определяли как ухудшение астмы, требующее лечения в течение ≥ 3 дней системными глюкокортикоидами или госпитализации или визита в отделение неотложной помощи, требующих системных глюкокортикоидов. Безопасность и переносимость регистрировали в соответствии с частотой появившихся во время лечения нежелательных явлений (TEAE) и серьезных TEAE.
Таблица 8. Краткое изложение итоговых показателей исследования согласно многоуровневой процедуре тестирования.
|
ACQ-5 обозначает версию опросника по контролю астмы из 5 пунктов, а AQLQ (S) - опросник по оценке качества жизни у больных астмой (стандартизированную версию).
Статистический анализ
Было установлено, что размер выборки из примерно 1638 пациентов даст в исследовании 99% мощность (2-выборочный тест при α=0,05) для выявления снижения на 55% относительного риска частоты в годовом исчислении тяжелых обострений (т.e. частота в годовом исчислении 0,6 и 0,27 для групп плацебо и дупилумаба соответственно). Предполагалось также, что этот размер выборки обеспечит 98% мощность для обнаружения различия в лечении в изменении FEV1 до BD от исходного уровня, составляющей 0,15 л к неделе 12. Анализ эффективности проводили на популяции, сформированной согласно назначенному лечению (ITT), определяемой как все рандомизированные пациенты, проанализированные в соответствии с назначенным лечением, независимо от того получали ли лечение. Частоту в годовом исчислении тяжелых обострений анализировали с использованием модели отрицательной биномиальной регрессии, включающей четыре группы лечения, возраст, область, исходные уровни эозинофилов, исходный уровень дозы ингаляционного глюкокортикоида и предшествующие обострения за 1 год до этого в качестве ковариат. Изменение от исходного уровня в непрерывных конечных точках, таких как FEV1 и исходы по оценке пациентов, анализировали с использованием модели со смешанными эффектами с повторными измерениями (MMRM), включающей лечение, возраст, исходные уровни эозинофилов, исходный уровень дозы ингаляционного глюкокортикоида, визит, взаимодействие с эффектом фактора лечения в зависимости от визита, исходное значение и взаимодействие с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат. Пол и исходный рост включали в качестве ковариат только в модели для параметров спирометрии.
Результаты
Исходные демографические данные и клинические характеристики популяции ITT показаны в таблице 4 и в целом были сопоставимыми во всех четырех группах лечения (таблица 10). У 1902 пациентов 200/300 мг дупилумаба q2w по сравнению с плацебо обеспечивали снижение частоты в годовом исчислении тяжелого обострения в течение 52-недельного периода лечения на 48%/46% (оба P < 0,0001) (фиг. 9A). Улучшение FEV1 наблюдали на неделе 12 (различие средних значений, определенных LS, по сравнению с плацебо 0,14 л/0,13 л; оба P < 0,0001) в общей популяции.
Анализы в предварительно определенных на основании исходного содержания эозинофилов в крови подгруппах показали значительное снижение частоты обострений (P < 0,001) при применении 200 и 300 мг дупилумаба по сравнению с плацебо сопоставимого объема у пациентов с ≥ 300 эозинофилов/мкл (снижение на 65,8% и 67,4% по сравнению с плацебо) и пациентов с ≥ 150 эозинофилов/мкл (снижение на 55,8% и 59,8% по сравнению с плацебо). Имели место устойчивые тенденции, но отсутствие значимости в отношении конечных точек по обострениям и FEV1 у пациентов с <300 эозинофилов/мкл. Анализы в предварительно определенных на основании исходных уровней FeNO подгруппах показали аналогичный эффект (P < 0,001). (См. фиг. 9B и таблицу 9)
Таблица 9. Краткое изложение первичной конечной точки эффективности и вторичных конечных точек.
|
В общей популяции исследования 200 и 300 мг q2w обеспечивали улучшение FEV1 до BD на неделе 12 на 0,32 л и 0,34 л соответственно (различие в 0,14 и 0,13 л по сравнению с сопоставимыми плацебо, P < 0,001) (фиг. 10A). У пациентов с ≥ 300 эозинофилов/мкл улучшения FEV были более значительными: дупилумаб обеспечивал улучшение FEV1 на неделе 12 на 0,43 л и 0,47 л соответственно (различие в 0,21 и 0,24 л по сравнению с сопоставимыми плацебо, Р < 0,001). (См. фиг. 10B.) Улучшение FEV1 было быстрым (со значительными различиями по сравнению с плацебо, очевидными при первой оценке на неделе 2 для обеих схем) и поддерживалось в течение 52-недельного периода лечения (P < 0,001 для обеих схем на неделе 52). Кроме того, анализ углового коэффициента для FEV1 после введения бронхолитического средства между неделями 8 и 52 показал потерю функции легких при приеме плацебо на 0,04 л/год без потери на любой из доз дупилумаба (P < 0,05).
Улучшения FEV1 на неделе 12 (P < 0,05) при обеих схемах дозирования были более значительными в подгруппе пациентов с более высокими исходными уровнями FeNO (0,19 и 0,12 л для FeNO от ≥ 25 до 50; 0,30 и 0,39 л для FeNO ≥ 50 ppb). (См. фиг. 10C и таблицу 8.)
Кроме того, 200 и 300 мг дупилумаба обеспечивали значительное улучшение процентного изменения FEV1 до введения бронхолитического средства от исходного уровня к неделе 12 по сравнению с плацебо: 21,34% относительно 12,11% и 23,08% относительно 13,67% соответственно (P < 0,001). Частота явлений тяжелого обострения, приводящих к госпитализации или визиту в отделение неотложной помощи в течение 52-недельного периода лечения, составила 0,035 относительно 0,065 (Р=0,004) при сравнении комбинации пациентов, лечение которых осуществляли дупилумабом, с комбинацией пациентов, лечение которых осуществляли плацебо. Это приводило к снижению относительного риска для дупилумаба по сравнению с плацебо на 46,8%. (См. таблицу 9.)
Дупилумаб обеспечивал значительное улучшение ACQ-5 уже на неделе 2, и эффект сохранялся в течение курса лечения (P < 0,01). Аналогичным образом, показатель по стандартизированной версии опросника по оценке качества жизни у больных астмой, показатели симптомов астмы до полудня и после полудня, а также максимальная скорость выдоха до полудня и после полудня, улучшались на неделе 24 и неделе 52. (См. таблицу 9.)
Пациенты, лечение которых осуществляли дупилумабом, показывали более значительное снижение от исходного уровня FeNO, общего IgE, периостина, эотаксина-3 и TARC в течение курса лечения по сравнению с плацебо (таблица 13). В обеих группах лечения наблюдалось временное повышение содержания эозинофилов в крови, которое к неделе 52 снижалось до уровней, близких к исходному.
Чтобы лучше понять эффект дупилумаба в отношении пациентов с признаками воспаления 2 типа, проводили анализы для оценки взаимосвязей биомаркеров эффективности. Каждый биомаркер тестировали в сплайн-модели без штрафов на взаимодействия с эффектом фактора биомаркера в зависимости от лечения в отношении обострений и FEV1. В этих анализах взаимодействия для эозинофилов и FeNO были значимыми (P < 0,05), когда итоговым показателем были обострения, в то время как эозинофилы, FeNO, периостин, ECP, IgE и эотаксин-3 были значимыми для FEV1 на неделе 12 (таблица 11). Эффект дупилумаба в отношении обострений был аналогичным для уровней IgE выше и ниже срединного значения на исходном уровне (167 МЕ/мл) и более значительным для улучшения FEV1 при уровнях IgE выше срединного значения.
У пациентов, лечение которых осуществляли дупилумабом, с исходным содержанием эозинофилов в крови ≥ 150 клеток/мкл и FeNO ≥ 25 ppb (2 тип - высокий) наблюдалась большая польза лечения по сравнению с плацебо как в отношении снижения частоты тяжелого обострения, так и в отношении улучшения FEV1. (См. фиг. 11A и фиг. 11B.) У пациентов с исходным содержанием эозинофилов < 150 клеток/мкл и FeNO < 25 ppb не наблюдалось никакого эффекта лечения (2 тип - низкий). Тем не менее, у пациентов, лечение которых осуществляли дупилумабом, с исходным содержанием эозинофилов в крови < 150 клеток/мкл и FeNO ≥ 25 ppb или ≥ 150 клеток/мкл и FeNO < 25 ppb наблюдалось численное снижение частоты тяжелого обострения.
Наиболее частым нежелательным явлением в группах, лечение которых осуществляли дупилумабом, по сравнению с плацебо были реакции в месте инъекции (15%/18% относительно 5%/10% соответственно). В отличие от исследований дупилумаба при атопическом дерматите частота конъюнктивита была схожей между дупилумабом и плацебо.
Таблица 10. Исходные демографические параметры и клинические характеристики (популяция ITT).
|
ACQ-5 обозначает опросник по контролю астмы из 5 пунктов, AQLQ (S) - опросник по оценке качества жизни у больных астмой (стандартизированная версия), BMI - индекс массы тела, FeNO - фракцию оксида азота в выдыхаемом воздухе, FEV1 - объем форсированного выдоха за 1 секунду, LABA - β2-агонист длительного действия, min - max - от минимального значения до максимального, ppb - части на миллиард, q2w - раз в 2 недели, q4w - раз в 4 недели, SD - стандартное отклонение, и TARC - хемокин, регулируемый тимусом и активацией.
†ACQ-5 является сообщаемой пациентом оценкой достаточности контроля астмы и изменения контроля астмы, что происходит либо спонтанно, либо в результате лечения. Более высокие показатели указывают на меньший контроль; рассчитан общий показатель от 0 до 6.
‡AQLQ (S) является сообщаемой пациентом оценкой влияния астмы на качество жизни. Более высокие показатели указывают на лучшее качество жизни; рассчитан общий показатель от 1 до 7.
§Показатели симптомов астмы представляют собой сообщаемые пациентом наблюдаемые при пробуждении и в вечернее время оценки симптомов астмы и их влияния на деятельность (после полудня) и сон (до полудня). Более высокие показатели указывают на большее нарушение; симптомы оценивают в диапазоне от 0 до 4.
Таблица 11. Анализ первичных конечных точек в подгруппах по исходному содержанию эозинофилов в крови и уровням FeNO.
|
*CI обозначает доверительный интервал, EOS - эозинофилы, FeNO - фракцию оксида азота в выдыхаемом воздухе, FEV1 - объем форсированного выдоха за 1 секунду, LS - метод наименьших квадратов, NA - не применимо, ppb - части на миллиард, q2w - раз в 2 недели, q4w - раз в 4 недели, и SE - стандартную ошибка.
†Получено с использованием отрицательной биномиальной модели с общим числом явлений, начиная от рандомизации до визита 18 или до даты последнего контакта в качестве зависимой переменной; четыре группы лечения, возраст, область (страна суммарно), исходный уровень эозинофилов, исходный уровень дозы ICS и количество тяжелых обострений за год до исследования в качестве ковариат и логарифмически преобразованная стандартизированная продолжительность наблюдения в качестве переменной смещения.
‡Изменения на неделе 12 от исходного уровня получали с использованием подхода MMRM с изменением от исходного уровня значений FEV1 до введения бронхолитического средства на неделе 12 в качестве зависимой переменной, а также лечения, возраста, пола, исходного роста, области (страна суммарно), исходного уровня эозинофилов, исходного уровня дозы ICS, визита, взаимодействия с эффектом фактора лечения в зависимости от визита, исходного значения FEV1 до введения бронхолитического средства и взаимодействия с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат.
Таблица 12. Краткое изложение дополнительных вторичных конечных точек.
|
* ACQ-5 обозначает версию опросника по контролю астмы из 5 пунктов, AQLQ - опросник по оценке качества жизни у больных астмой, CI - доверительный интервал, ER - отделение неотложной помощи, LS - метод наименьших квадратов, PEF - максимальную скорость выдоха, q2w - раз в 2 недели, q4w - раз в 4 недели, и SE - стандартную ошибку.
†Получено исходя из модели MMRM с изменением от исходного уровня общего показателя AQLQ до недели 24 или 52 в качестве зависимой переменной и четырьмя группами лечения, возрастом, областью (страна суммарно), исходным уровнем эозинофилов, исходным уровнем дозы ICS, визитом, взаимодействием с эффектом фактора лечения в зависимости от визита, исходным общим показателем AQLQ и взаимодействием с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат.
‡Получено исходя из модели MMRM с изменением от исходного уровня ACQ-5 до недели 24 или 52 в качестве зависимой переменной и четырьмя группами лечения, возрастом, областью (страна суммарно), исходным уровнем эозинофилов, исходным уровнем дозы ICS, визитом, взаимодействием с эффектом фактора лечения в зависимости от визита, исходным ACQ-5 и взаимодействием с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат.
§Получено исходя из модели MMRM с изменением от исходного уровня показателя симптомов до полудня/после полудня (периодическое среднее значение) до недели 52 в качестве зависимой переменной и лечением, возрастом, областью (страна суммарно), исходным уровнем эозинофилов, исходным уровнем дозы ICS, визитом, взаимодействием с эффектом фактора лечения в зависимости от визита, исходным показателем симптомов до полудня/после полудня и взаимодействием с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат.
¶Получено исходя из модели MMRM с изменением от исходного уровня значений PEF до полудня/после полудня (периодическое среднее значение) до недели 52 в качестве зависимой переменной и лечением, возрастом, полом, исходным ростом, областью (страна суммарно), исходным уровнем эозинофилов, исходным уровнем дозы ICS, визитом, взаимодействием с эффектом фактора лечения в зависимости от визита, исходным значением PEF до полудня/после полудня и взаимодействием с эффектом фактора исходного уровня в зависимости от визита в качестве ковариат.
#Получено с использованием отрицательной биномиальной модели с общим числом явлений, начиная от рандомизации до визита 18 или до даты последнего контакта в качестве зависимой переменной; две объединенные группы лечения, возраст, область (страна суммарно), исходный уровень эозинофилов, исходный уровень дозы ICS и число тяжелых обострений за год до исследования в качестве ковариат и логарифмически преобразованная стандартизированная продолжительность наблюдения в качестве переменной смещения.
Таблица 13. Краткое изложение изменения от исходного уровней биомаркера 2 типа.
|
*min - max обозначает от минимального значения до максимального, ppb - части на миллиард, q2w - раз в две недели, и SD - стандартное отклонение.
Таблица 14. Краткое изложение теста взаимодействия на эффективность.
|
* P-значения для тестирования эффектов взаимодействия фактора лечения в зависимости от биомаркера на основании сплайн-моделей отрицательной биномиальной регрессии без штрафов в популяции ITT.
† P-значения для тестирования эффектов взаимодействия фактора лечения в зависимости от биомаркера на основании регрессионных сплайн-моделей без штрафов в популяции ITT.
Обсуждение
Дупилумаб обеспечивал значительное снижение частоты в годовом обобщении тяжелого обострения в популяции ITT, причем наблюдались более сильные эффекты лечения при повышении исходных уровней эозинофилов в крови и FeNO. Дупилумаб также обеспечивал значительное снижение частоты наиболее тяжелых обострений астмы, требующих госпитализации или визита в отделение неотложной помощи. Оценка FEV1 и контроля астмы с течением времени показала, что эффективность дупилумаба была быстрой, причем значительные различия по сравнению с плацебо проявились уже при первой оценке на неделе 2 и сохранялись в течение 52-недельного периода лечения для обеих схем дозирования. Существенные и клинически значимые улучшения FEV1 от 0,32 до 0,34 л наблюдались на неделе 12 независимо от исходного содержания эозинофилов в крови с еще большим увеличением от 0,43 до 0,47 л у пациентов с исходным содержанием эозинофилов в крови ≥ 300 эозинофилов/мкл.
Кроме того, анализ углового коэффициента для FEV1 после введения бронхолитического средства показал, что по сравнению с потерей функции легких, наблюдаемой у пациентов, получавших плацебо, никакой потери у пациентов, лечение которых осуществляли дупилумабом, не наблюдалось, что указывает на потенциальный эффект дупилумаба в отношении ремоделирования дыхательных путей. Анализ углового коэффициента показал, что пациенты, принимавшие плацебо, теряли в среднем приблизительно 40 мл в год, что согласуется с данными из других когорт с астмой. Кроме того, поскольку IL-4Rα экспрессируется на гладкомышечных клетках, возможно, что существует прямой бронхолитический эффект лекарственного средства в дополнение к эффектам в отношении воспалительных факторов 2 типа.
Последовательное и глубокое улучшение, наблюдаемое при применении дупилумаба, вероятно, связано с его уникальным механизмом действия. В связи с тем, что в последнее время все большее внимание сообщества по изучению астмы уделяется обострениям, что продиктовано опасениями по поводу экономической эффективности, сместился акцент с серьезных вопросов заболеваемости и качества жизни, связанных со значительной потерей функции легких, наблюдаемой у пациентов с астмой от умеренной до тяжелой степени. Несмотря на современные средства терапии, этим пациентам с астмой от умеренной до тяжелой степени суждено продолжать терять функцию легких и испытывать прогрессирование заболевания со временем. Таким образом, возможность того, что новое лечение может обеспечить существенное восстановление клинически значимых уровней функции легких и, возможно, даже предотвратить дальнейшее ухудшение состояния, может обеспечить огромную пользу этим пациентам.
Результаты этого исследования подтверждают, что интерлейкин-4 и интерлейкин-13 являются ключевыми проксимальными факторами воспаления 2 типа при астме. Дупилумаб является первым биологическим веществом, которое значительно снижает уровни FeNO, в дополнение к другим системным биомаркерам 2 типа, таким как IgE, что подтверждает его биологическую активность при воспалении дыхательных путей. Не желая быть связанными научной теорией, уникальный механизм действия дупилумаба с двойной блокадой передачи сигналов интерлейкина-4 и интерлейкина-13 может объяснить, почему дупилумаб демонстрирует значительный лечебный эффект в более широкой популяции пациентов и беспрецедентный эффект в отношении улучшения функции легких, предполагая потенциальный прямой бронхолитический эффект в дополнение к его противовоспалительному эффекту. Следует отметить, что это исследование показывает наиболее заметную связь пользы с исходными уровнями эозинофилов в крови по сравнению с двумя другими базовыми исследованиями с применением дупилумаба. Хотя нет четкого объяснения, почему эта связь была более заметной в этом исследовании, содержание эозинофилов в крови может быть недостаточным критерием воспаления 2 типа, указывая на то, что другие биомаркеры воспаления 2 типа, такие как FeNO, могут быть важны. Тем не менее, в целом во всех трех исследованиях дупилумаб, по-видимому, направлен на более широкую популяцию с астмой, чем популяции, определяемые только повышенными уровнями эозинофилов или IgE в крови, что требуется для других утвержденных биологических веществ.
Активность дупилумаба была продемонстрирована в отношении нескольких атопических/аллергических состояний, которые часто являются сопутствующими у пациентов с астмой. В этом исследовании более 80% пациентов страдали сопутствующими атопическими или аллергическими состояниями, включая атопический дерматит (приблизительно 10% популяции), назальный полипоз (приблизительно 20% популяции) и аллергический ринит (более 65% популяции). Высокий уровень сопутствующих атопических/аллергических состояний позволяет предположить, что эти пациенты страдают от системной избыточной активности оси воспаления 2 типа и, таким образом, лечение астмы с применением дупилумаба может одновременно помочь в облегчении этих связанных состояний.
Дупилумаб в большинстве случаев хорошо переносился и имел приемлемый профиль безопасности. За исключением реакций в месте инъекции, частота TEAE была схожей в группах лечения. В соответствии с механизмом действия и аналогично тому, что наблюдалось в испытаниях с атопическим дерматитом, у пациентов, лечение которых осуществляли дупилумабом, наблюдалось большее среднее временное увеличение по сравнению с исходным содержанием эозинофилов в крови по сравнению с плацебо. Согласно протоколу исследования, все случаи содержания эозинофилов > 3000 клеток/мкл при лечении должны были регистрироваться как AE в этом исследовании. Большинство наблюдаемых случаев повышения содержания эозинофилов были лабораторными данными без каких-либо клинических последствий или ассоциированных AE. Увеличение содержания эозинофилов в крови согласуется с гипотезой о том, что дупилумаб блокирует функцию интерлейкина-4 и интерлейкина-13 в отношении выживаемости, активации и рекрутинга эозинофилов в ткани, но не возврата из костного мозга, на который влияет IL-5. В результате первичное лечение дупилумабом может привести к временному повышению содержания циркулирующих эозинофилов в крови. Между группами дупилумаба и плацебо не наблюдалось связанных с лечением AE конъюнктивита, в отличие от исследований дупилумаба при атопическом дерматите.
В заключение, в крупнейшем на сегодняшний день исследовании дупилумаба у пациентов с неконтролируемой астмой от умеренной до тяжелой степени здесь продемонстрировано, что двойная блокада интерлейкина-4 и интерлейкина-13 дупилумабом оказывает эффективное лечение широкой популяции с астмой, обеспечивая значительное снижение частоты тяжелых обострений, быстрое и устойчивое улучшение функции легких и контроля астмы, а также облегчение симптомов. Наиболее достоверные результаты наблюдались у пациентов с повышенными иммунными характеристиками 2 типа, включая содержание эозинофилов и FeNO. Дупилумаб является единственным биологическим веществом, демонстрирующим эффективность в многочисленных исследованиях пациентов с астмой от умеренной до тяжелой степени, независимо от исходных уровней биомаркеров 2 типа. Дупилумаб в большинстве случаев хорошо переносился и имел приемлемый профиль безопасности. Эти данные подтверждают целесообразность применения дупилумаба в качестве эффективной дополнительной терапии для этой популяции пациентов с астмой с высокой неудовлетворенной потребностью.
Пример 3. Испытание фазы III исследования QUEST. Дупилумаб обеспечивает снижение частоты тяжелого обострения и улучшение функции легких у пациентов подросткового возраста с неконтролируемой астмой от умеренной до тяжелой степени
Распространенность астмы у детей и подростков возросла за последние 30 лет (Asher (2014) Int. J. Tuberc. Lung Dis.). В 2011 году примерно 11,4% подростков (в возрасте 12-17 лет) в США сообщили, что в настоящее время страдают астмой (Bloom (2011) Vital and Health Statistics Series).
Уровень заболеваемости астмой у подростков такой же (или часто выше), как и у детей младшего возраста, однако подростки реже обращаются за медицинской помощью (Couriel (2003) J. Paediatric Resp. Rev.). Многие подростки недооценивают тяжесть своей астмы и переоценивают свою восприимчивость бронхолитических средств (Rhee (2008) J. Asthma; Andersson (2013) Pediatrics). Астма оказывает глубокое влияние на физическое, психологическое и социальное здоровье подростков и отрицательно влияет на связанное со здоровьем качество жизни (Cui (2016) J. Pediatrics).
В этом исследовании оценивали эффективность и безопасность дупилумаба по подгруппам подростков (в возрасте 12-17 лет) и взрослых людей (в возрасте ≥ 18 лет) с неконтролируемой астмой от умеренной до тяжелой степени. Конечными точками, оцененными в течение 52-недельного периода лечения, были годовая частота тяжелых обострений и изменение от исходного уровня FEV1 до введения бронхолитического средства (л). Исходные демографические данные и клинические характеристики показаны на фиг. 12.
Критерии включения. Возраст ≥ 12 лет с диагностированной врачом неконтролируемой астмой в течение ≥ 12 месяцев (Глобальная инициатива по бронхиальной астме (GINA) 2014 г.); проходит лечение с помощью ICS (ингаляционные кортикостероиды) в дозе от средней до высокой плюс не более 2 дополнительных контролирующих препаратов; FEV1 (объем форсированного выдоха за 1 секунду) до введения бронхолитического средства ≤ 80% от прогнозируемой нормы (взрослые люди) и ≤ 90% (подростки) при скрининге и на исходном уровне; обратимость после бронхолитического средства ≥ 12% и 200 мл; показатель ACQ-5 (опросник по контролю астмы из 5 пунктов) ≥ 1,5 при скрининге и на исходном уровне; ≥ 1 обострение в течение предшествующего года; отсутствие минимальных требований к исходному содержанию эозинофилов в крови или любому другому биомаркеру 2 типа.
Критерии исключения. Хроническое обструктивное заболевание легких или другие заболевания легких, которые могут нарушать функцию легких; тяжелое обострение астмы в течение 1 месяца после визита включения или в течение периода скрининга; активный курильщик, курильщик, который бросил курить в течение 6 месяцев до скрининга или с курением в анамнезе >10 пачко-лет; сопутствующее заболевание, которое может помешать оценке исследуемого лекарственного средства.
Статистический анализ. Анализ эффективности проводили на популяции ITT, определяемой как все рандомизированные пациенты, проанализированные в соответствии с назначенным лечением, независимо от полученного лечения.
Частоту в годовом исчислении тяжелых обострений астмы в течение 52-недельного периода лечения анализировали с помощью модели отрицательной биномиальной регрессии. Изменения от исходного уровня FEV1 в различные моменты времени в течение 52-недельного периода лечения анализировали с использованием модели со смешанными эффектами с повторными измерениями.
Первичные конечные точки, частоту тяжелого обострения астмы и FEV1, также анализировали в подгруппах пациентов, определенных по возрасту (< 18 лет и > 18 лет). Популяция по оценке безопасности включала всех пациентов, которые получили ≥ 1 дозу или часть дозы исследуемого средства лечения, проанализированных в соответствии с полученным лечением.
Дупилумаб обеспечивал уменьшение тяжелых обострений и улучшение FEV1 в общей популяции ITT (фиг. 13A и фиг. 13B), снижение частоты тяжелого обострения у подростков и взрослых людей (фиг. 14A и фиг. 14B) и улучшение FEV1 на неделе 12 (фиг. 15A) и неделе 52 (фиг. 15B), а также в течение 52-недельного периода лечения (фиг. 16A и фиг. 16B) у подростков и взрослых людей.
Дупилумаб обеспечивал улучшение прогнозируемого процента FEV1 в течение 52-недельного периода лечения у подростков и взрослых людей (фиг 18A и фиг. 18B). Оценивали уровни FeNO (фиг. 19A и фиг. 19B), показатели ACQ-5 (фиг. 20A и фиг. 20B) и показатели AQLQ (фиг. 21A и фиг. 21B).
Подростки составляли 107/1902 включенных пациентов (34 в группах дупилумаба, 21/18 в группах с сопоставимыми плацебо); 35,5% составляли женщины, среднее исходное значение FEV1 2,33 л, среднее значение прогнозируемого % FEV1 70,45%, среднее количество тяжелых обострений за предшествующий год 1,91. У подростков, получавших плацебо, наблюдалось меньше тяжелых обострений (0,36/0,33), чем у взрослых людей (0,89/1,00). У подростков 200 мг дупилумаба обеспечивало снижение частоты в годовом исчислении обострения на 46,4%, в то время как 300 мг дупилумаба не оказывало лечебного эффекта по сравнению с плацебо (не желая быть связанными с научной теорией, это могло иметь место из-за небольшого размера выборки и несбалансированного числа предшествующих явлений (среднее значение 1,53 относительно 2,22 соответственно)). Нескорректированная частота обострения составила 0,46 (300 мг дупилумаба) и 0,76 (плацебо). Значительные улучшения изменения от исходного значения FEV1 (л) по сравнению с плацебо наблюдали у подростков (200 мг дупилумаба: среднее значение, определенное методом наименьших квадратов, 0,36 [95% CI 0,12-0,61]; 300 мг: 0,27 [0,02-0,52]) (P < 0,05), и они были численно выше по сравнению со взрослыми людьми (200 и 300 мг: 0,12 [0,07-0,18]).
Профиль нежелательных явлений был сопоставим между подгруппами (фиг. 17, фиг. 22, фиг. 23 и фиг. 24). Наиболее частыми появившимися во время лечения нежелательными явлениями (TEAE), чаще встречающимися в объединенной группе дупилумаба, были следующие: у подростков - вирусные инфекции дыхательных путей (плацебо 2 [5,1%]); дупилумаб 7 [10,3%]); у взрослых людей - эритема в месте инъекции (плацебо 34 [5,7%]; дупилумаб 168 [14,1%]). Эозинофилия наблюдалась только в популяции взрослых людей.
Дупилумаб обеспечивал значительное снижение частоты в годовом исчислении тяжелого обострения и улучшение функции легких у взрослых людей с неконтролируемой астмой от умеренной до тяжелой степени. Улучшение FEV1 было быстрым и устойчивым в течение 52-недельного периода лечения. Дупилумаб также обеспечивал значительное улучшение функции легких у подростков с неконтролируемой астмой от умеренной до тяжелой степени, при этом наблюдалось численное снижение тяжелых обострений.
Как и у взрослых людей, у подростков улучшение FEV1 было быстрым и устойчивым в течение 52-недельного периода лечения. Величина улучшения FEV1 была выше у подростков. Дупилумаб в целом хорошо переносился.
Пример 4. Испытание фазы III исследования QUEST. Дупилумаб обеспечивает улучшение связанного со здоровьем качества жизни при риноконъюнктивите, улучшение функции легких и снижение частоты тяжелых обострений у пациентов с астмой от умеренной до тяжелой степени
Связанное со здоровьем качество жизни у пациентов с сопутствующим аллергическим ринитом
Аллергический ринит (AR), распространенное сопутствующее заболевание 2 типа у пациентов с астмой, способствует увеличению общего бремени заболеваний. В этом анализе исследования фазы 3 LIBERTY ASTHMA QUEST (NCT02414854) у пациентов с неконтролируемой астмой от умеренной до тяжелой степени оценивали эффект дупилумаба в отношении показателя по стандартизированному опроснику качества жизни при риноконъюнктивите [RQLQ(S)+12] у пациентов с собственной оценкой сопутствующего AR.
Пациенты с астмой возрастом ≥ 12 лет, неконтролируемые с ICS в дозах от средней до высокой плюс ≤ 2 дополнительных контролирующих препаратов, получали дополнительно 200/300 мг дупилумаба или сопоставимое плацебо раз в 2 недели (q2w) в течение 52 недель. Пациенты с AR в анамнезе согласно собственной оценке (63,5%; n/N=1207/1902) заполняли утвержденный RQLQ(S) + 12 на неделях 12 и 52. Не регистрировали клинический диагноз AR.
Общий показатель RQLQ (S) + 12 (исходное среднее значение [SD] 1,90 [1,12] - 2,01 [1,16]) значительно улучшался при применении 200/300 мг дупилумаба q2w по сравнению с плацебо на неделе 52 (различие для среднего значения, определенного методом наименьших квадратов, [95% CI] −0,42 [−0,61, −0,24]/−0,39 [−0,56, −0,21]; P < 0,0001). 200/300 мг дупилумаба обеспечивали значительное (P < 0,001) улучшение показателей в доменах деятельности (0,44 [0,68, 0,21] / 0,39 [0,61, 0,16]), сна (0,47 [0,69, 0,25] / 0,38 [0,59, 0,17]) и глазных симптомов (0,37 [0,58, 0,16] / 0,39 [0,59, 0,19]) от исходного уровня к неделе 52 по сравнению с плацебо; и к неделе 12 для 300 мг дупилумаба (0,23 [0,42, 0,04], 0,26 [0,45, 0,07], 0,26 [0,45, 0,08] соответственно; P < 0,05). Показатели для домена назальных симптомов значительно улучшились при применении 200/300 мг дупилумаба по сравнению с плацебо к неделе 12 (0,36 [0,56, 0,16] / 0,32 [0,51, 0,13]; P<0,001) и к неделе 52 (0,61 [0,84, 0,39] / 0,55 [0,76, 0,33]; P < 0,0001). Наиболее частым нежелательным явлением с более высокой частотой при применении дупилумаба по сравнению с плацебо была реакция в месте инъекции (15%/18% относительно 5%/10%).
Дупилумаб обеспечивал значительное улучшение специфического для риноконъюнктивита связанного со здоровьем качества жизни у пациентов с неконтролируемой астмой от умеренной до тяжелой степени и сопутствующим AR и в целом хорошо переносился.
Популяция: пациенты с сопутствующим AR. Конечные точки/визит: изменение среднего значения, определенного LS, от исходного уровня в течение 52-недельного периода лечения для доменов RQLQ (назальные симптомы, глазные симптомы, деятельность, сон); безопасность (ITT). Группы лечения: 200 мг и 300 мг дупилумаба q2w и сопоставимое плацебо.
Улучшение функции легких и снижение тяжелого обострения у пациентов с сопутствующим аллергическим ринитом или без него
В ретроспективном анализе исследования фазы 3 LIBERTY ASTHMA QUEST (NCT02414854) у пациентов с астмой (≥ 12 лет, неконтролируемая с ICS в дозе от средней до высокой плюс ≤ 2 дополнительных контролирующих препаратов) с сопутствующим AR в анамнезе согласно собственной оценке (63,5%; n/N=1207/1902) или без сопутствующего AR оценивали эффект добавления 200 мг или 300 мг дупилумаба или сопоставимого плацебо раз в 2 недели (q2w) в отношении частоты в годовом исчислении тяжелых обострений и объема форсированного выдоха за 1 секунду (FEV1). Не регистрировали клинический диагноз AR.
Исходные характеристики пациентов с AR и без него были в целом схожими. Частота в годовом исчислении тяжелых обострений снижалась по сравнению с плацебо при применении 200 мг дупилумаба q2w (относительный риск с AR: 0,606 [95% CI, 0,451-0,814]; P=0,0009; без AR: 0,406 [95% CI, 0,273-0,605]; P < 0,0001) с аналогичными результатами для 300 мг q2w. FEV1 улучшался на неделе 12 при применении 200 мг дупилумаба q2w (различие среднего значения, определенного LS, по сравнению с плацебо с AR: 0,14 л [95% CI, 0,07-0,21]; P < 0,0001; без AR: 0,13 л [95% CI, 0,05-0,22]; P=0,0023) и сохранялся до недели 52 (как с AR, так и без него: P < 0,0001) со схожими результатами на неделе 52 для 300 мг q2w. Наиболее частым нежелательным явлением в группах, лечение которых осуществляли дупилумабом (по сравнению с плацебо), была реакция в месте инъекции (200 мг/300 мг по сравнению с сопоставимым плацебо: 15%/18% по сравнению с 5%/10%).
Дупилумаб обеспечивал существенное улучшение FEV1 и снижение частоты в годовом исчислении тяжелого обострения в этой популяции с трудно контролируемой астмой с сопутствующим AR, а также у пациентов без сопутствующего AR.
Популяция: пациенты с сопутствующим AR и без него (AR определен в соответствии с CSR). Конечные точки: изменение среднего значения, определенного LS, от исходного уровня FEV1 на неделях 12 и 52; тяжелые обострения в течение 52-недельного периода лечения. Безопасность: ITT.
Пример 5. Испытание фазы III исследования QUEST - Дупилумаб обеспечивает подавление биомаркеров 2 типа у пациентов с астмой с сопутствующим хроническим риносинуситом и назальным полипозом (CRS+NP) или без них, или с хроническим риносинуситом без назального полипоза (CRS - NP) у пациентов с астмой от умеренной до тяжелой степени
В исследовании фазе 3 LIBERTY ASTHMA QUEST (NCT02414854) 200/300 мг дупилумаба раз в 2 недели по сравнению с сопоставимым плацебо обеспечивали подавление биомаркеров 2 типа у пациентов с неконтролируемой астмой от умеренной до тяжелой степени и улучшение связанного со здоровьем качества жизни, оцениваемого с помощью SNOT-22, в трудно поддающейся лечению подгруппе с сопутствующим хроническим риносинуситом с назальным полипозом (CRS+NP) или (CRSwNP) и с сопутствующим хроническим риносинуситом без назального полипоза (CRS - NP). В данном ретроспективном анализе оценивали эффект дупилумаба в отношении биомаркеров 2 типа в этой подгруппе.
Исходный уровень/изменение от исходного уровня с течением времени оценивали для фракции оксида азота в выдыхаемом воздухе (FeNO), общего IgE и эотаксина-3. CRS с NP или без него был согласно собственной оценке у 20,1% (n/N=382/1897) пациентов.
Исходные значения FeNO и эотаксина-3 были численно выше у пациентов с CRS - NP или CRS+NP, чем у пациентов без них. Подавление биомаркеров было явным у всех пациентов, лечение которых осуществляли дупилумабом, к неделе 12. На неделе 52 значительное подавление биомаркеров наблюдали у пациентов с CRS - NP или CRS+NP и без них, о чем свидетельствуют срединное значение процентного изменения от исходного уровня (200/300 мг дупилумаба по сравнению с сопоставимым плацебо) с CRS - NP или CRS+NP: FeNO 46,2/37,7 относительно 5,5/6,4, IgE 74,8/76,8 относительно 0,0/2,0, эотаксин-3 47,7/50,9 относительно 1,5/5,4 (все P ≤ 0,0001); без CRS - NP или CRS+NP: FeNO 31,0/35,9 относительно 5,9/10,1, IgE 67,3/67,7 относительно 3,3/6,6, эотаксин-3 31,8/37,2 относительно 0,0/0,8 (все P < 0,0001). Наиболее частым нежелательным явлением с более высокой частотой при применении дупилумаба по сравнению с плацебо была реакция в месте инъекции (15%/18% относительно 5%/10%).
Дупилумаб обеспечивал подавление местных и системных биомаркеров 2 типа у пациентов с CRS+/-NP и без них.
Популяция: пациенты с сопутствующими CRS или NP, или без них. Конечные точки: процентное изменение от исходного уровня общего IgE в сыворотке крови, эотаксина-3 в плазме крови и FeNO в течение 52-недельного периода лечения. Безопасность: ITT. Группы лечения: 200 и 300 мг дупилумаба q2w и сопоставимое плацебо.
Пример 6. Испытание фазы III исследования QUEST. Дупилумаб обеспечивает снижение тяжелых обострений и улучшение функции легких у пациентов с неконтролируемой астмой от умеренной до тяжелой степени с поздней манифестацией
В исследовании фазы 3 LIBERTY ASTHMA QUEST (NCT02414854) в данном ретроспективном анализе оценивали эффективность дупилумаба у пациентов с поздней манифестацией астмы (возраст > 40 лет) и исходным соотношением FEV1 после введения бронхолитического средства/форсированная жизненная емкость легких [FVC] < 0,7 (что предполагает стойкую обструкцию дыхательных путей) или ≥ 0,7.
Частоту в годовом исчислении тяжелых обострений в течение 52-недельного периода лечения оценивали с использованием моделей отрицательной биномиальной регрессии. Изменения от исходного уровня в отношении FEV1 до и после введения бронхолитического средства (л) и соотношения FEV1 до введения бронхолитического средства/FVC на неделях 12 и 52 анализировали с использованием моделей со смешанными эффектами с повторными измерениями.
200 мг и 300 мг дупилумаба q2w по сравнению с плацебо обеспечивали значительное снижение частоты в годовом исчислении тяжелых обострений у пациентов с поздней манифестацией астмы и стойкой обструкцией дыхательных путей (68,8% и 75,7% соответственно, P < 0,0001) и у пациентов без стойкой обструкции дыхательных путей (55,1% и 50,7% соответственно, оба Р < 0,05) (фиг. 27). На неделе 12 FEV1 до и после введения бронхолитического средства и соотношение FEV1/FVC улучшалось у пациентов, лечение которых осуществляли дупилумабом, с поздней манифестацией астмы и стойкой обструкцией дыхательных путей (P < 0,05 по сравнению с плацебо, в одной либо в обеих дозах). Аналогичные улучшения наблюдали на неделе 52 (200 мг дупилумаба q2w Р < 0,05 для FEV1 до и после введения бронхолитического средства; 300 мг дупилумаба q2w FEV1 до введения бронхолитического средства P=0,09, FEV1 после введения бронхолитического средства P=0,06). У пациентов с поздней манифестацией астмы без стойкой обструкции дыхательных путей было более умеренное улучшение по сравнению с плацебо в отношении FEV1 до введения бронхолитического средства на неделях 12 и 52, чем у пациентов со стойкой обструкцией дыхательных путей (P ≥ 0,05). Наиболее частым нежелательным явлением в группах, лечение которых осуществляли дупилумабом, по сравнению с сопоставимым плацебо были реакции в месте инъекции (15%/18% относительно 5%/10%).
У пациентов с поздней манифестацией астмы со стойкой обструкцией дыхательных путей или без нее дупилумаб обеспечивал значительное снижение частоты тяжелого обострения. Кроме того, улучшения функции легких наблюдались на неделях 12 и 52 у пациентов с поздней манифестацией астмы и стойкой обструкцией дыхательных путей, у которых обычно наблюдаются худшие исходы астмы, чем у пациентов без стойкой обструкции дыхательных путей.
Популяция: популяция ITT с возрастом манифестации астмы > 40 лет и FEV1 после BD/FVC < 0,7; популяция ITT с возрастом манифестации астмы > 40 лет и FEV1 после BD/FVC ≥ 0,7.
Конечные точки/визит (данные для включения в реферат: тяжелые обострения в течение 52-недельного периода лечения; изменение среднего значения, определенного LS, от исходного уровня для FEV1 до BD (л) на неделях 12 и 52; изменение среднего значения, определенного LS, от исходного уровня для FEV1 после BD (л) на неделях 12 и 52; изменение среднего значения, определенного LS, от исходного уровня для соотношения FEV1/FVC на неделях 12 и 52; безопасность.
Группы лечения: 200 мг дупилумаба q2w, 300 мг дупилумаба q2w и группы сопоставимого плацебо.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110>
<120> СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R
<130> 601407: SA9-217-4
<140>
<141>
<150>
<151>
<150>
<151>
<160> 8
<170> PatentIn версия 3.5
<210> 1
<211> 124
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический полипептид HCVR
<400> 1
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Glu Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Gly Ser Gly Phe Thr Phe Arg Asp Tyr
20 25 30
Ala Met Thr Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Ser Ile Ser Gly Ser Gly Gly Asn Thr Tyr Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Lys Asp Arg Leu Ser Ile Thr Ile Arg Pro Arg Tyr Tyr Gly Leu
100 105 110
Asp Val Trp Gly Gln Gly Thr Thr Val Thr Val Ser
115 120
<210> 2
<211> 112
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический полипептид LCVR
<400> 2
Asp Ile Val Met Thr Gln Ser Pro Leu Ser Leu Pro Val Thr Pro Gly
1 5 10 15
Glu Pro Ala Ser Ile Ser Cys Arg Ser Ser Gln Ser Leu Leu Tyr Ser
20 25 30
Ile Gly Tyr Asn Tyr Leu Asp Trp Tyr Leu Gln Lys Ser Gly Gln Ser
35 40 45
Pro Gln Leu Leu Ile Tyr Leu Gly Ser Asn Arg Ala Ser Gly Val Pro
50 55 60
Asp Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Lys Ile
65 70 75 80
Ser Arg Val Glu Ala Glu Asp Val Gly Phe Tyr Tyr Cys Met Gln Ala
85 90 95
Leu Gln Thr Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys
100 105 110
<210> 3
<211> 8
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид HCDR1
<400> 3
Gly Phe Thr Phe Arg Asp Tyr Ala
1 5
<210> 4
<211> 8
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид HCDR2
<400> 4
Ile Ser Gly Ser Gly Gly Asn Thr
1 5
<210> 5
<211> 18
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид HCDR3
<400> 5
Ala Lys Asp Arg Leu Ser Ile Thr Ile Arg Pro Arg Tyr Tyr Gly Leu
1 5 10 15
Asp Val
<210> 6
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид LCDR1
<400> 6
Gln Ser Leu Leu Tyr Ser Ile Gly Tyr Asn Tyr
1 5 10
<210> 7
<211> 3
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид LCDR2
<400> 7
Leu Gly Ser
1
<210> 8
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид LCDR3
<400> 8
Met Gln Ala Leu Gln Thr Pro Tyr Thr
1 5
<---

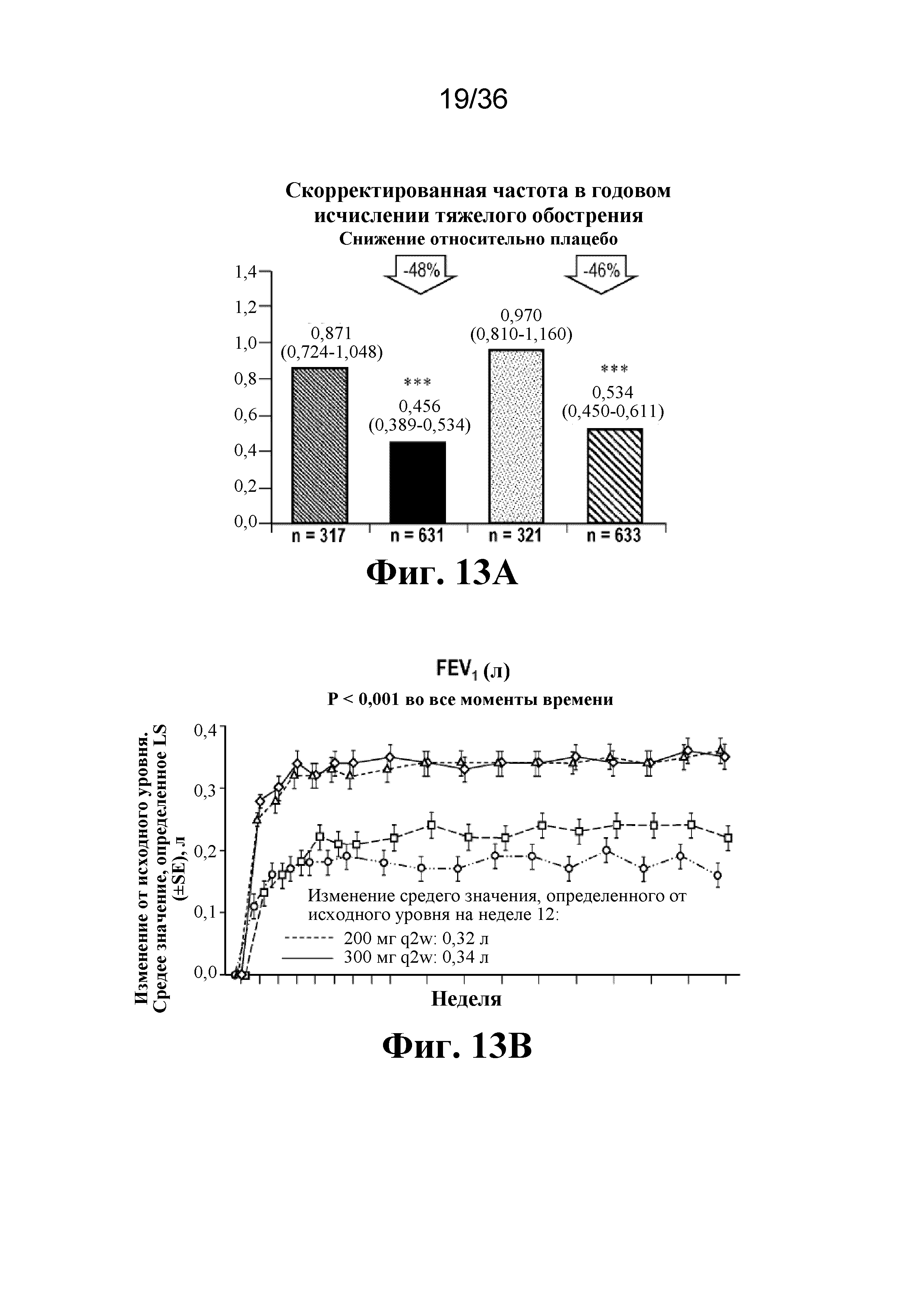
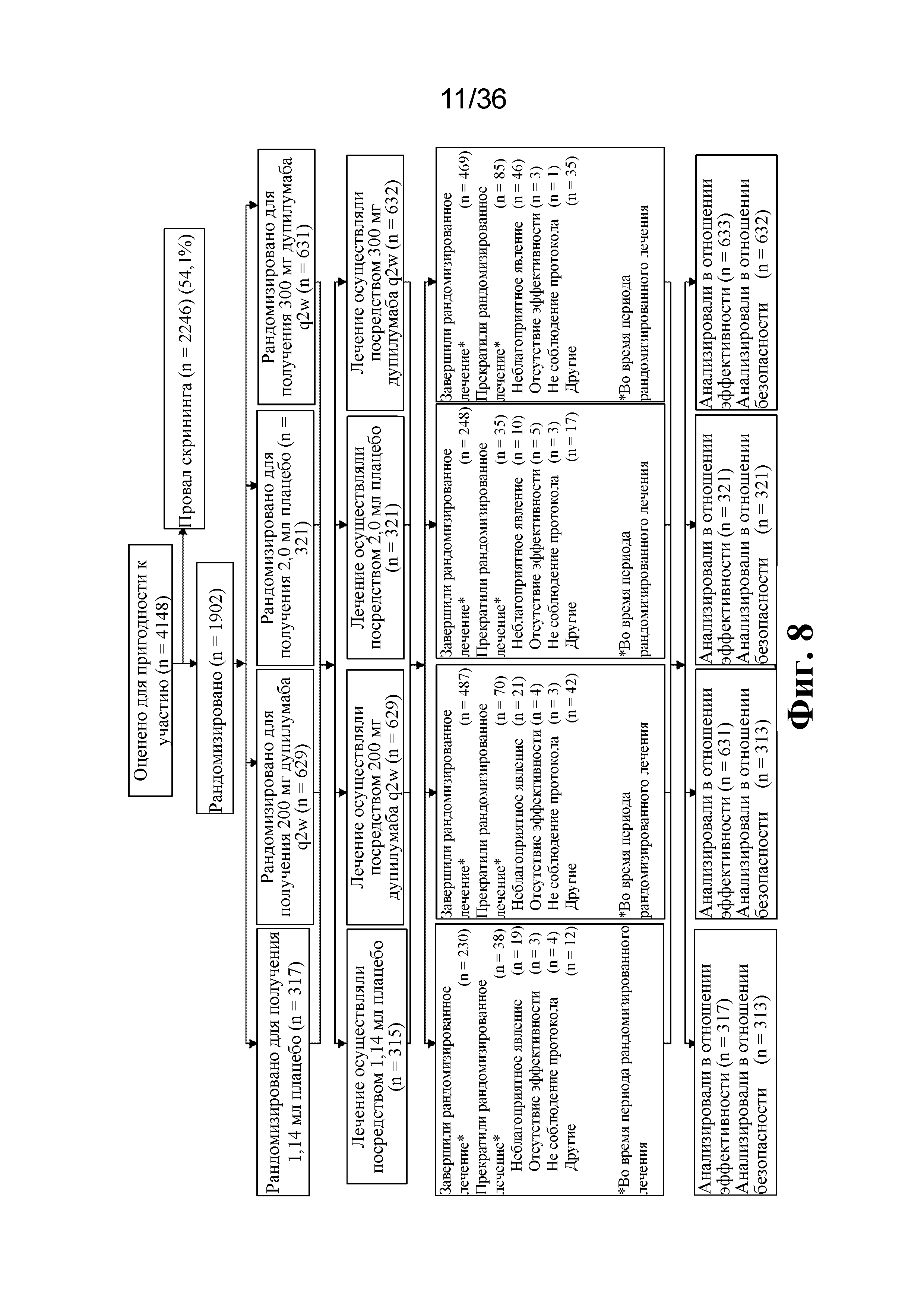
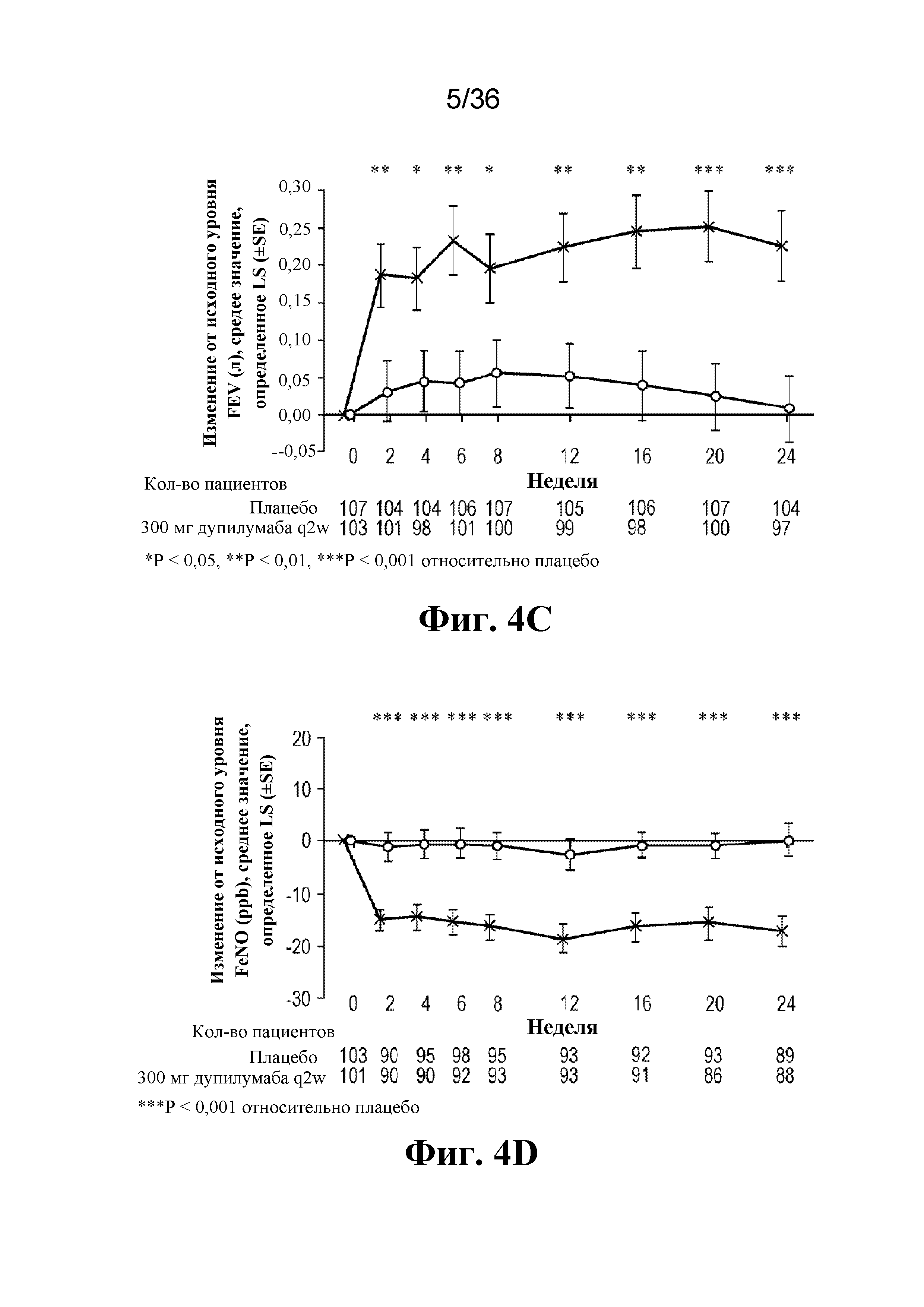
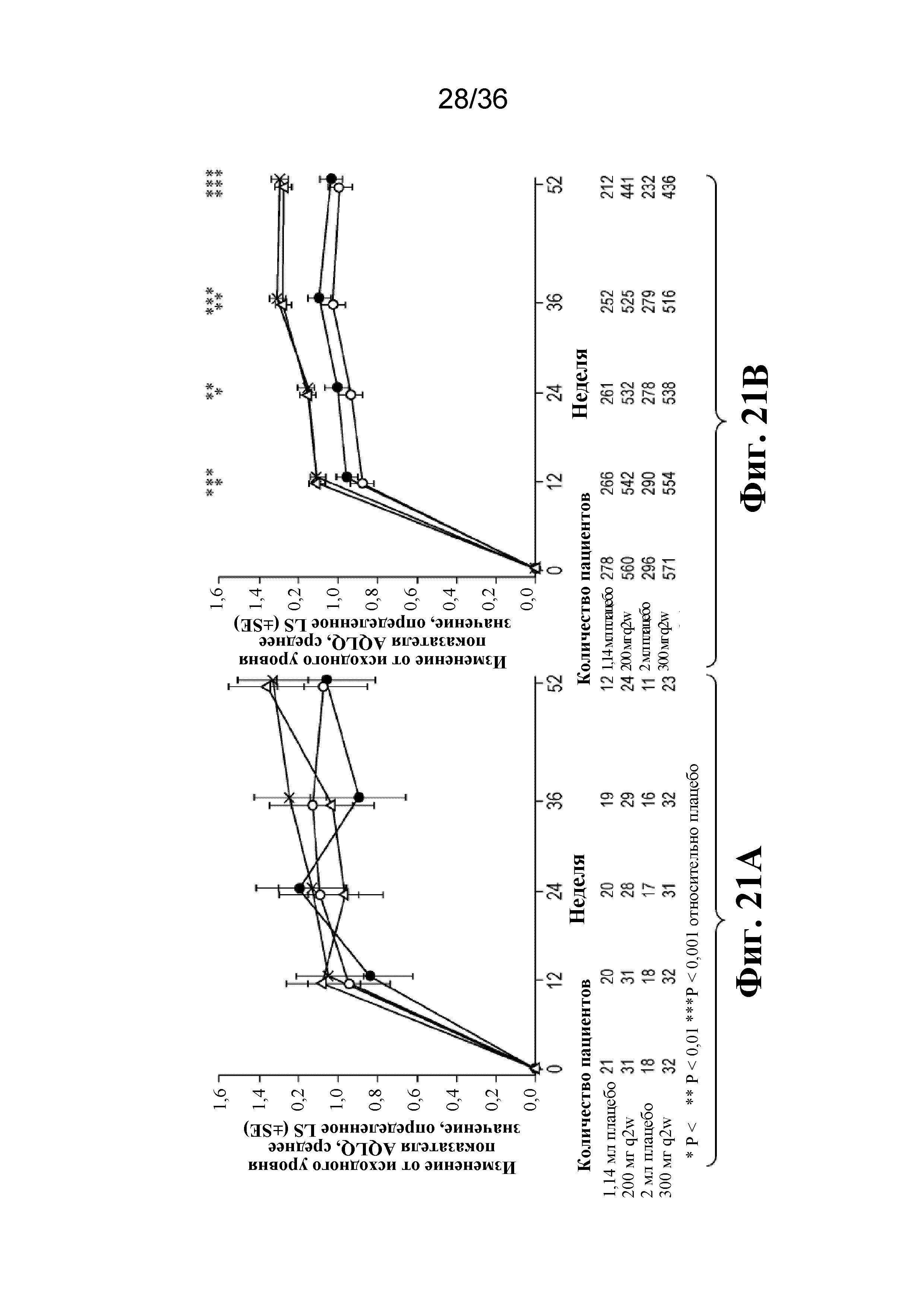
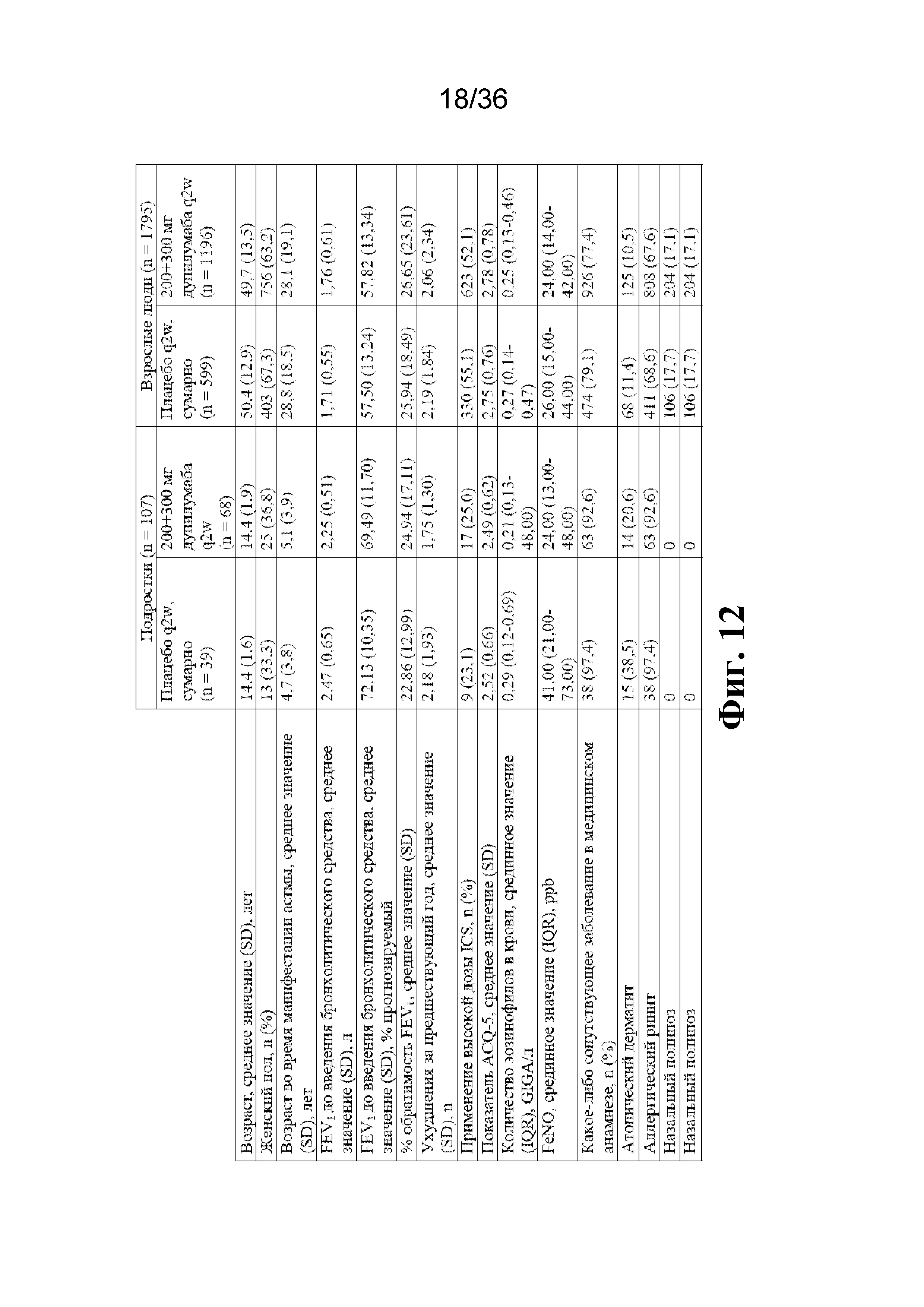
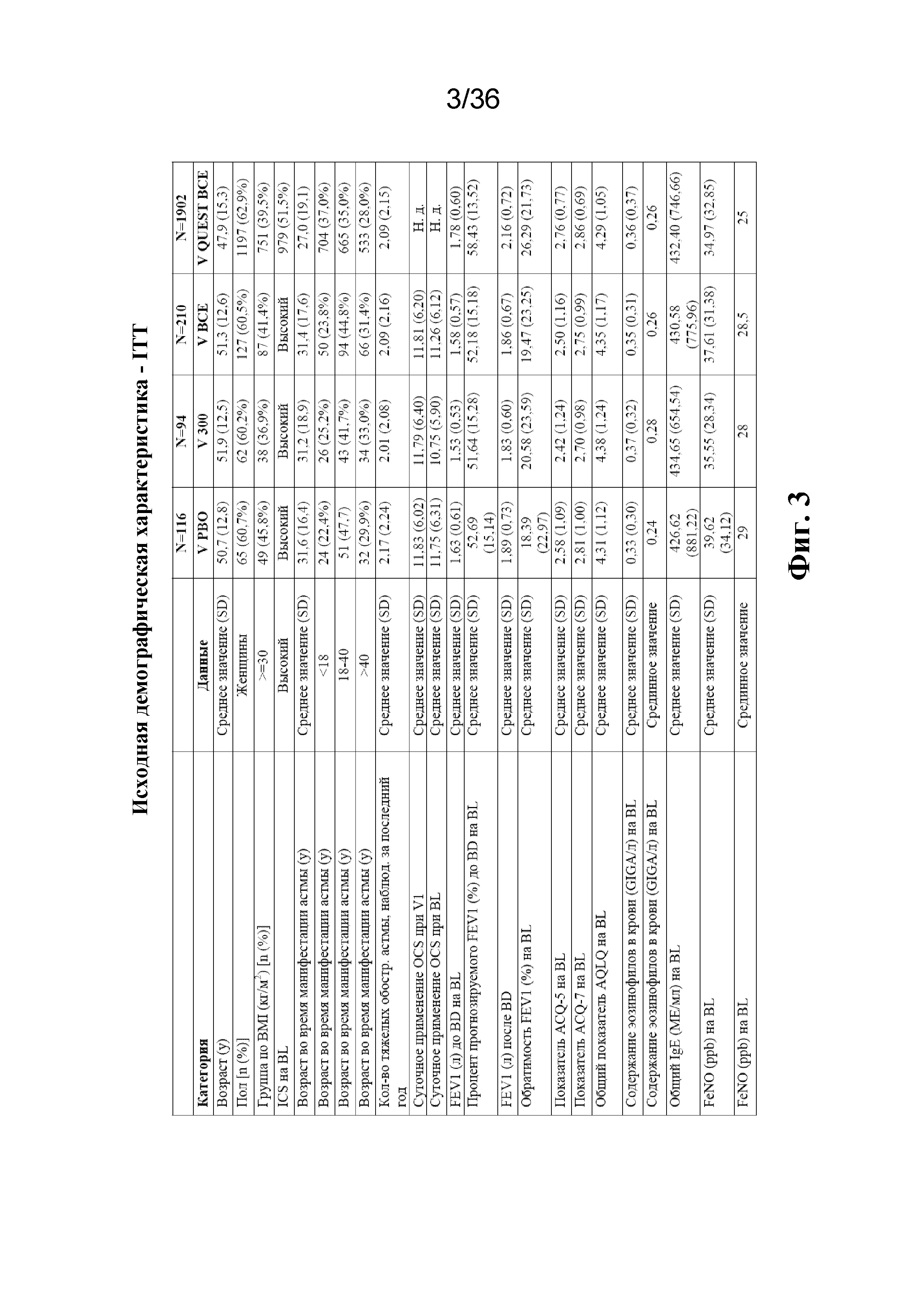
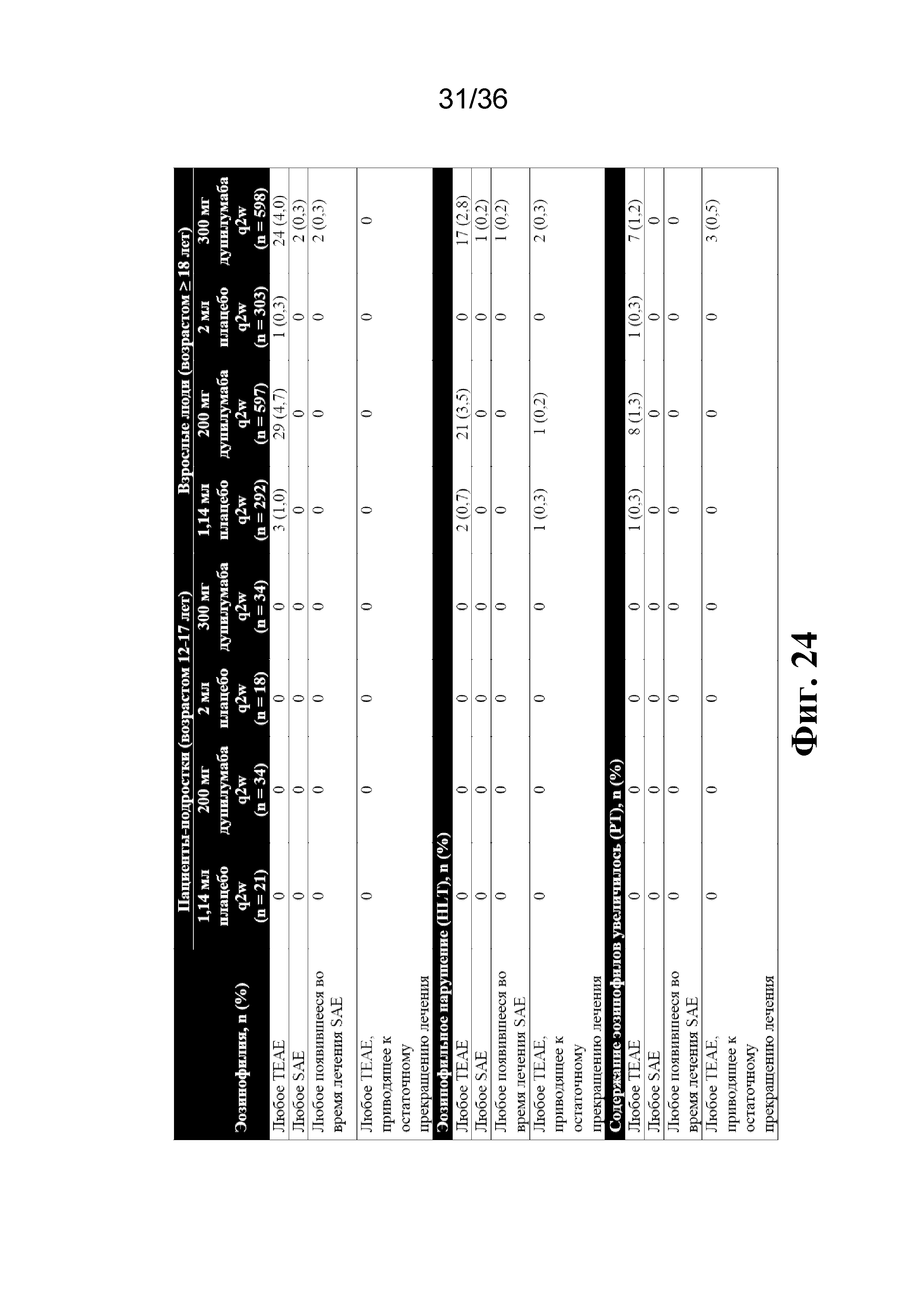
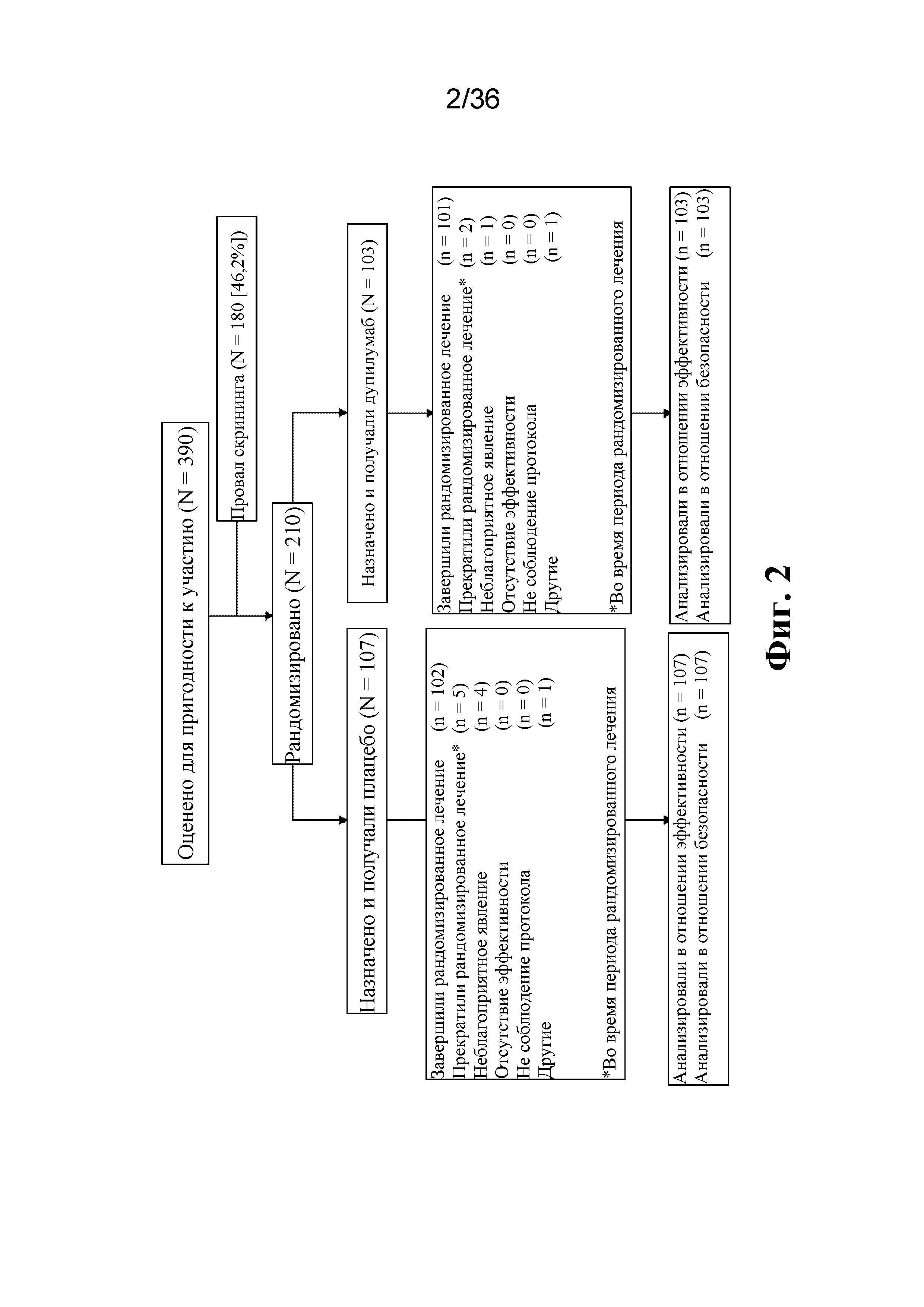
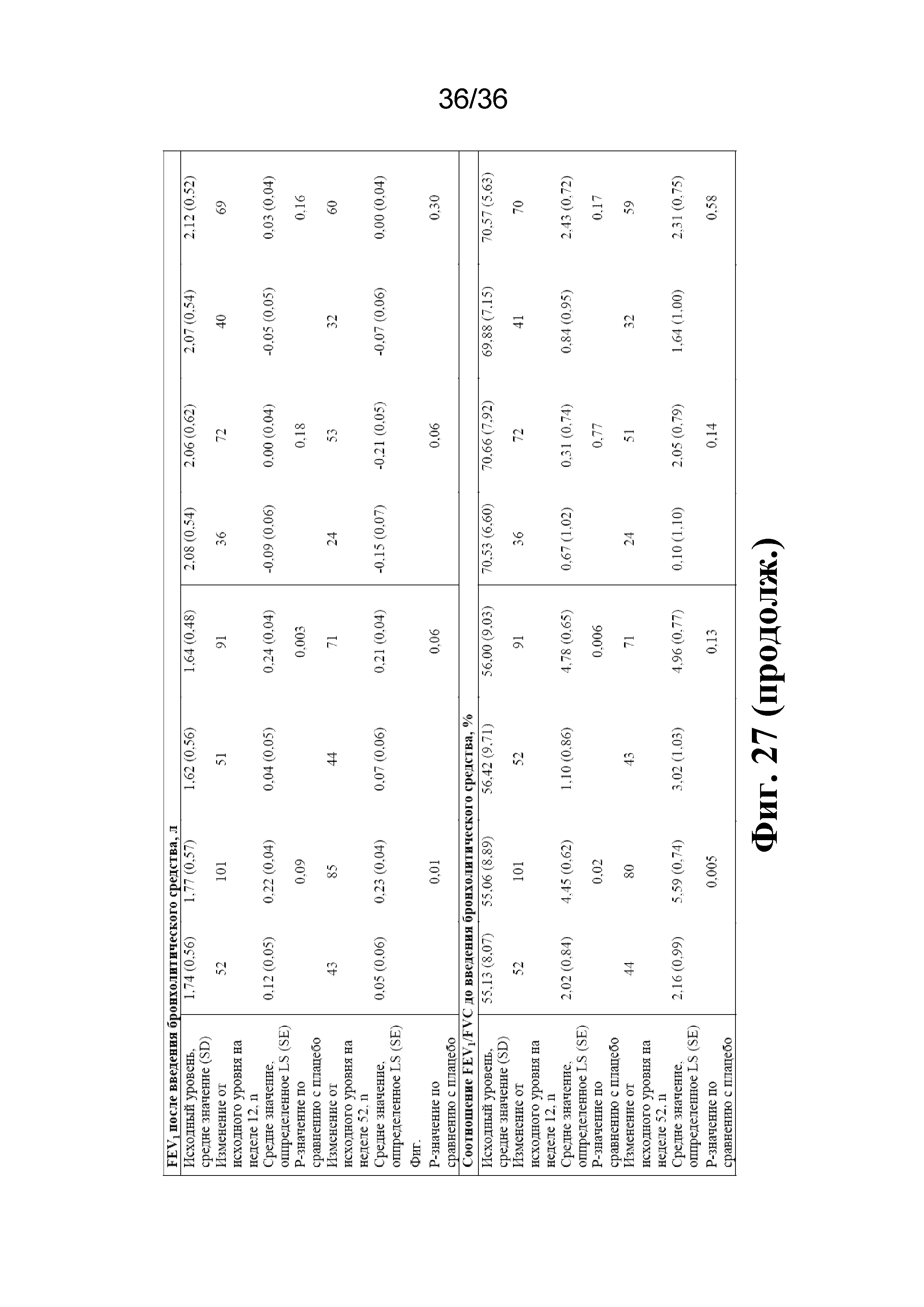
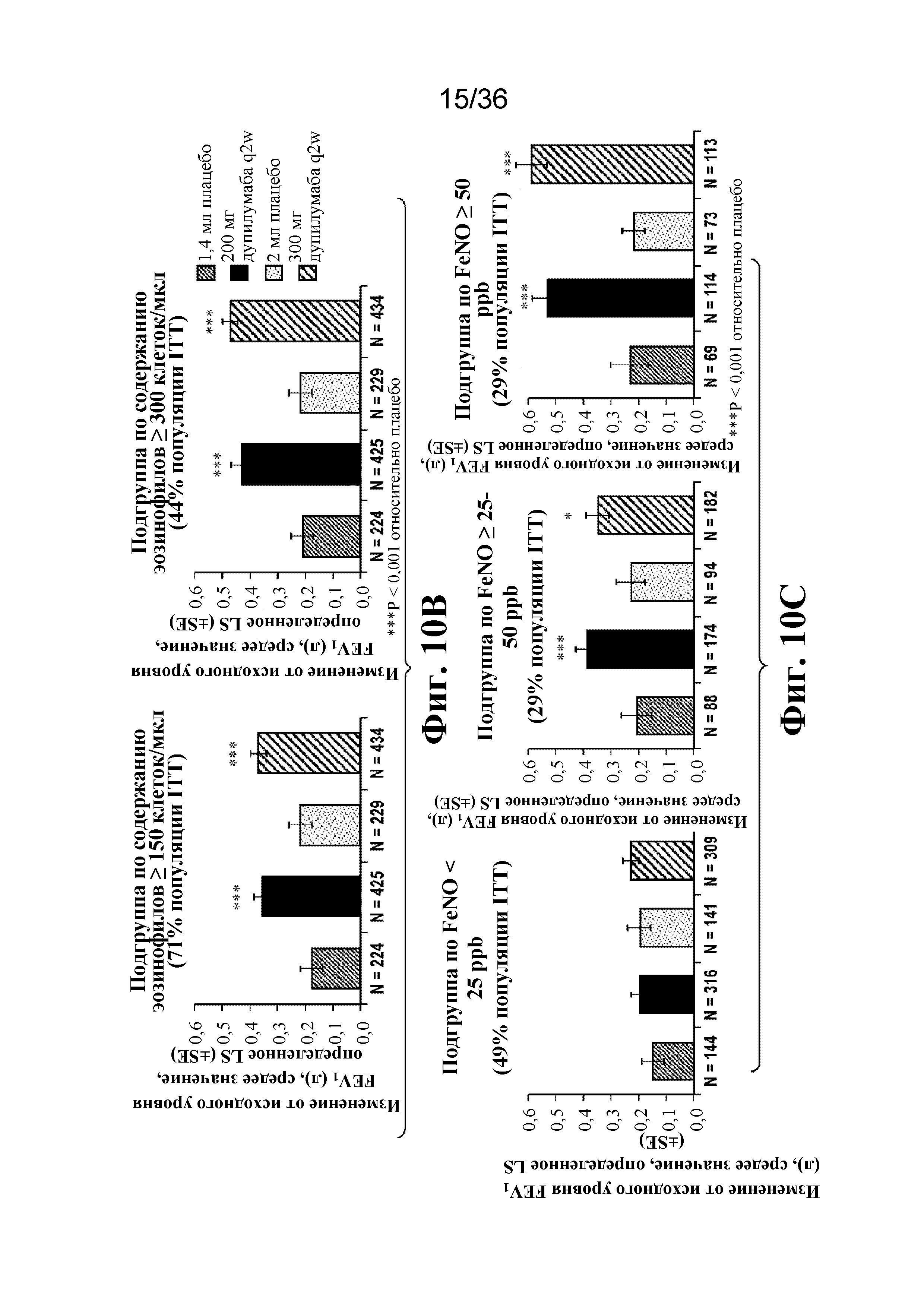
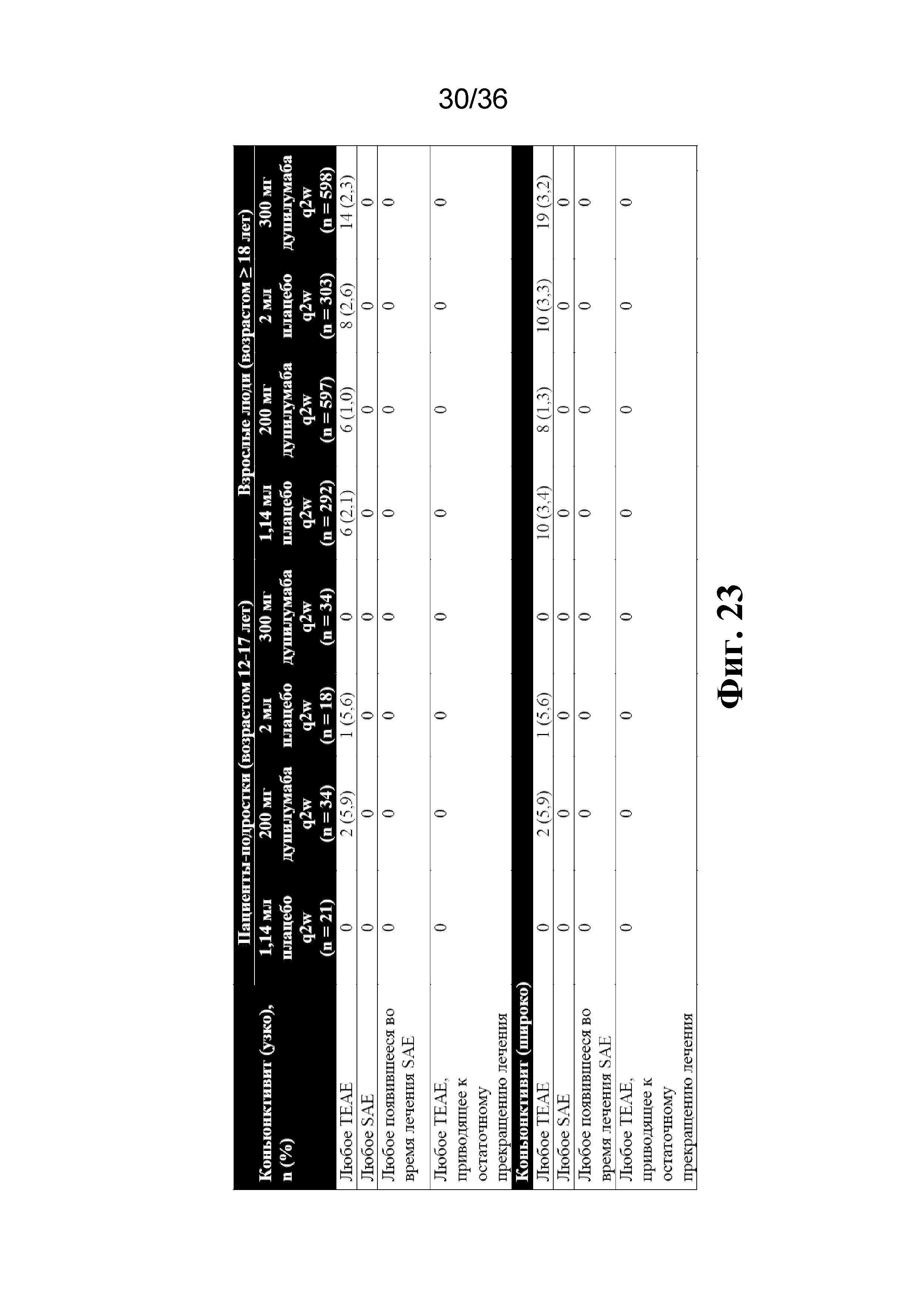
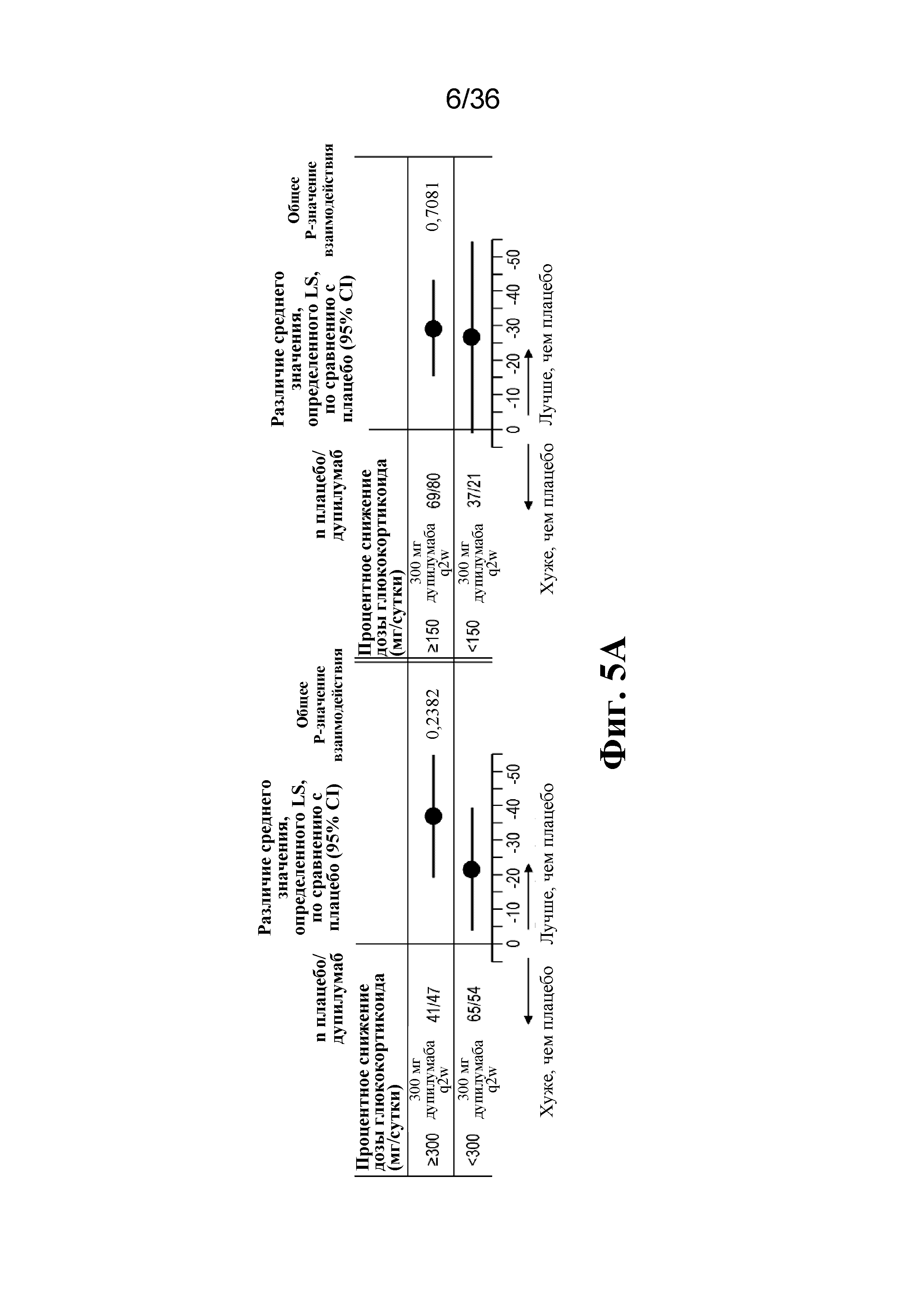
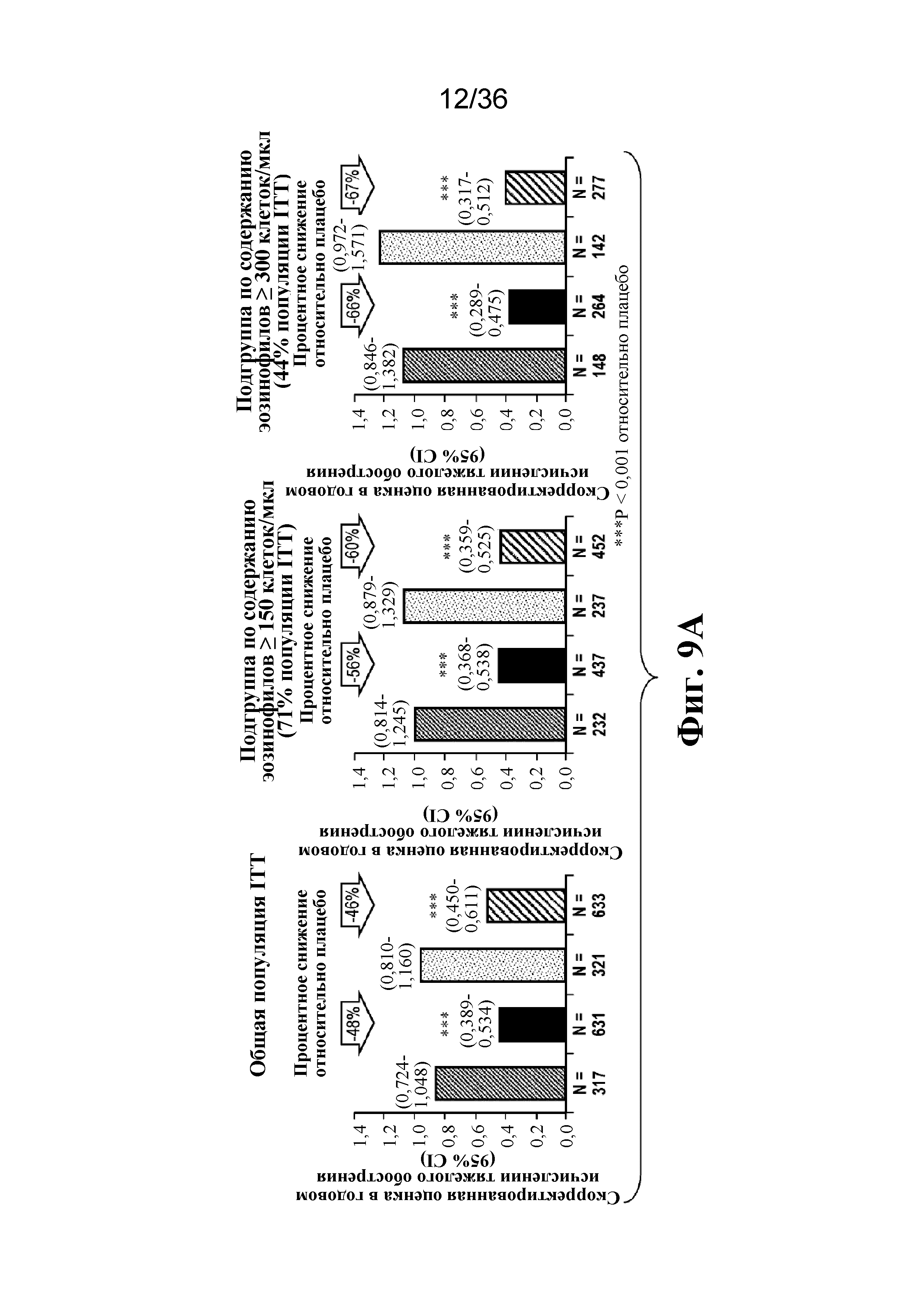
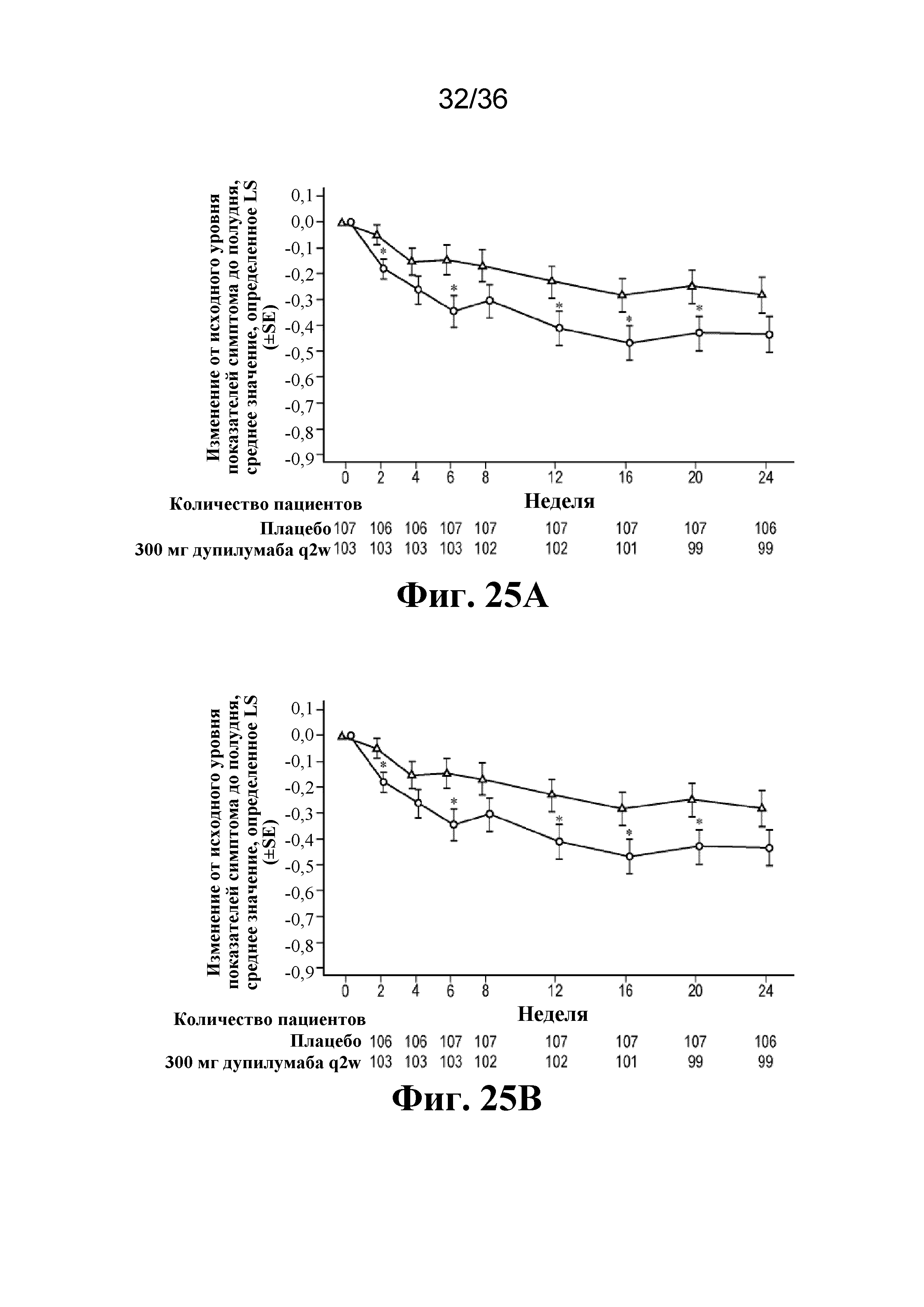


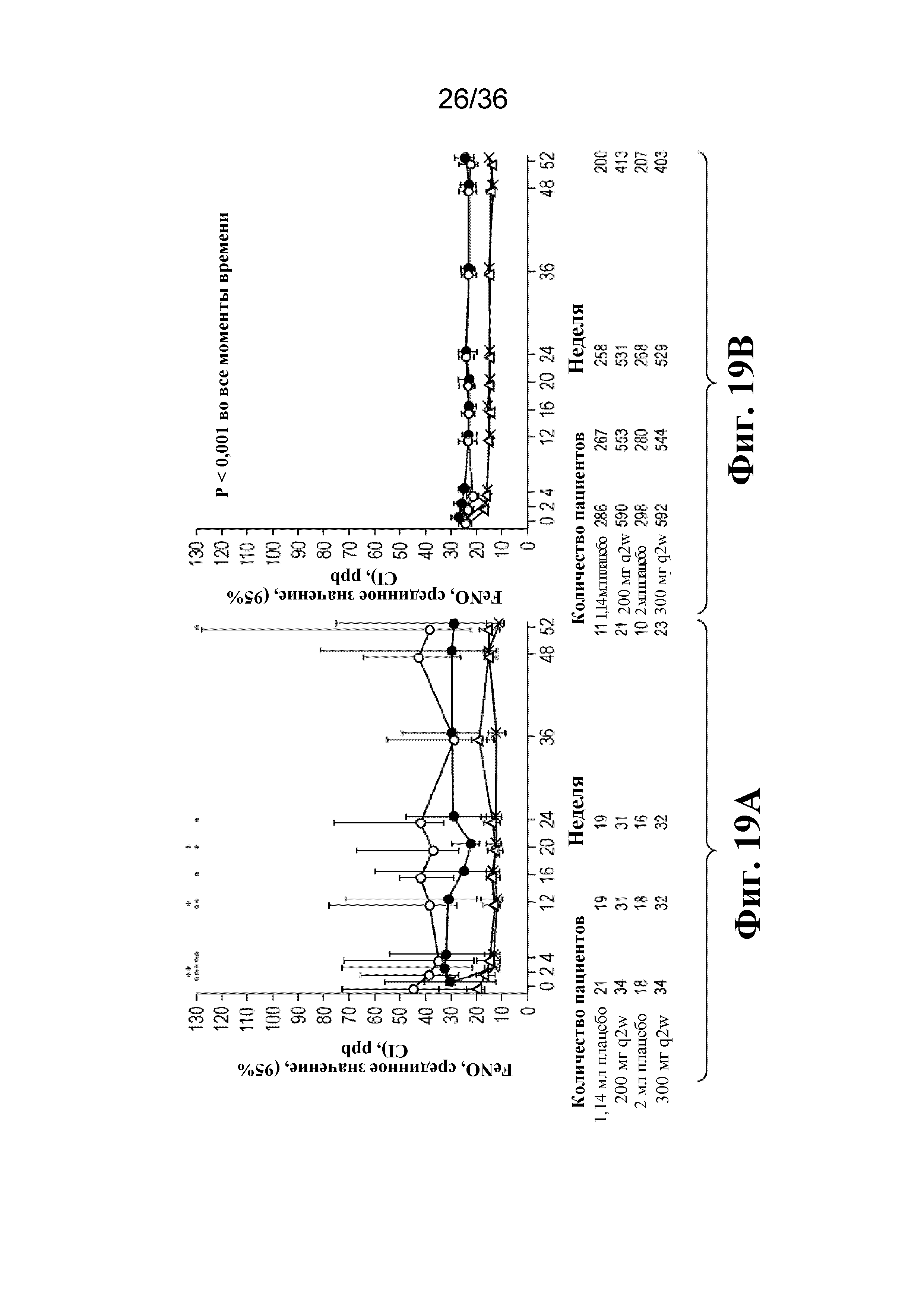
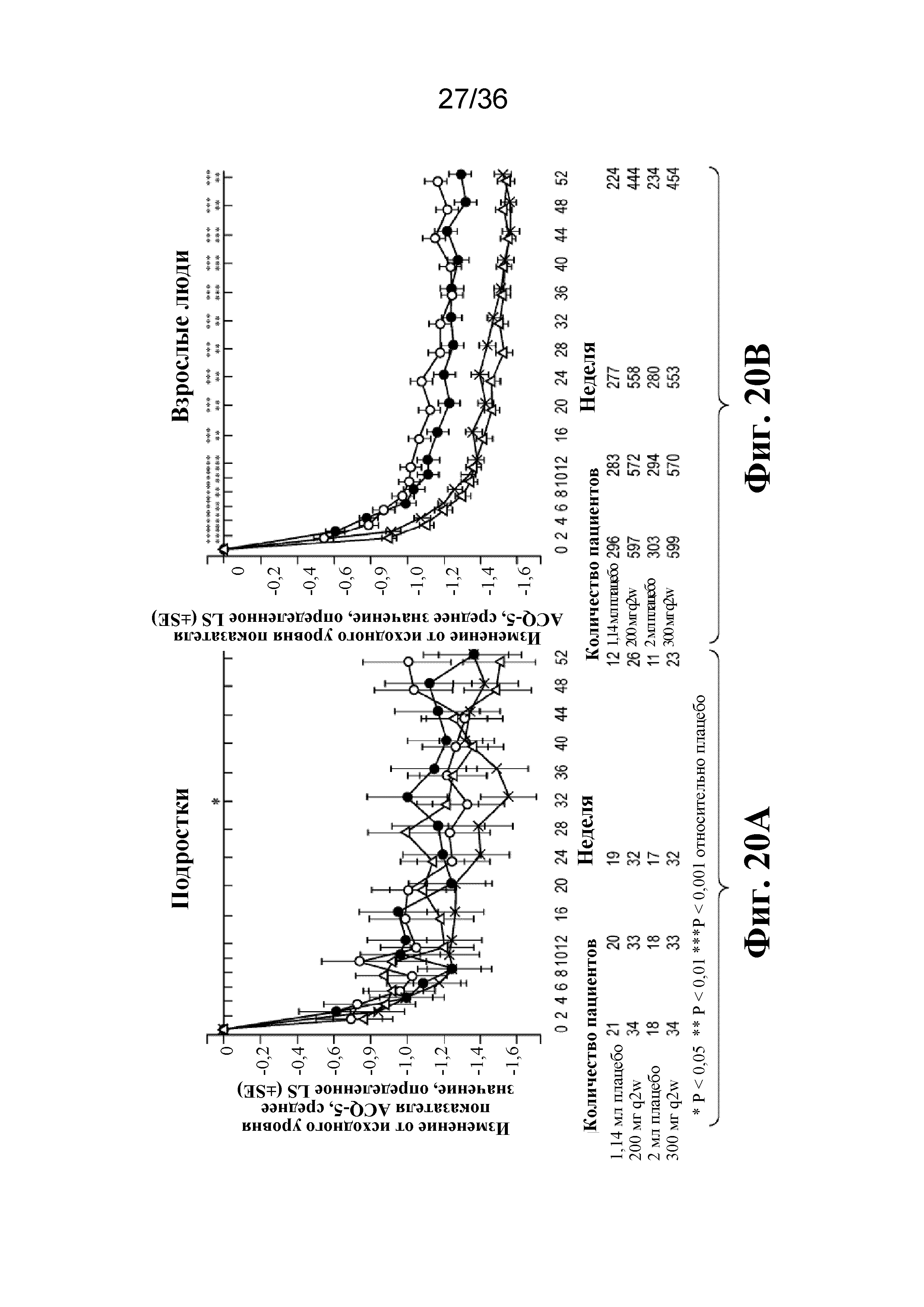

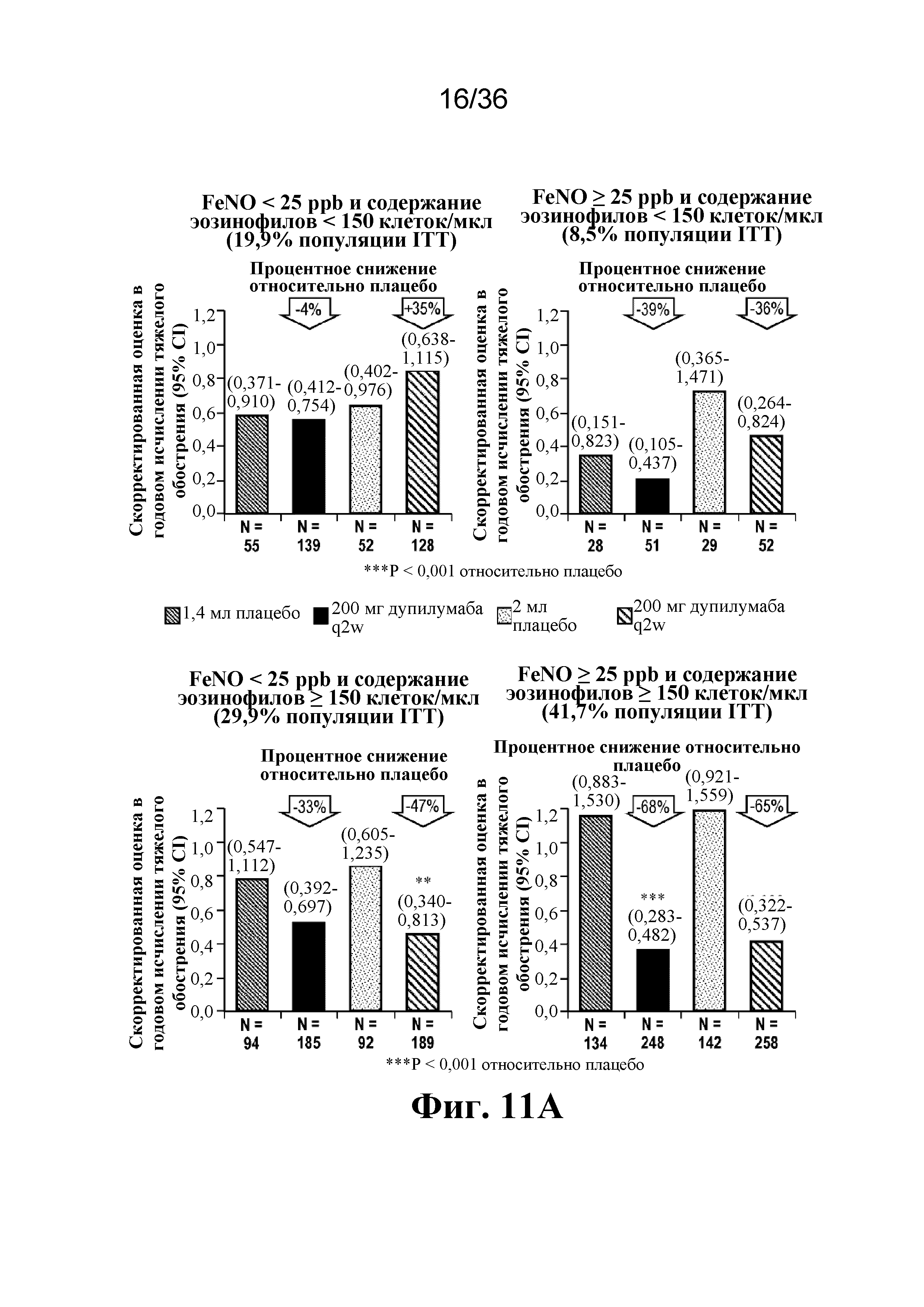
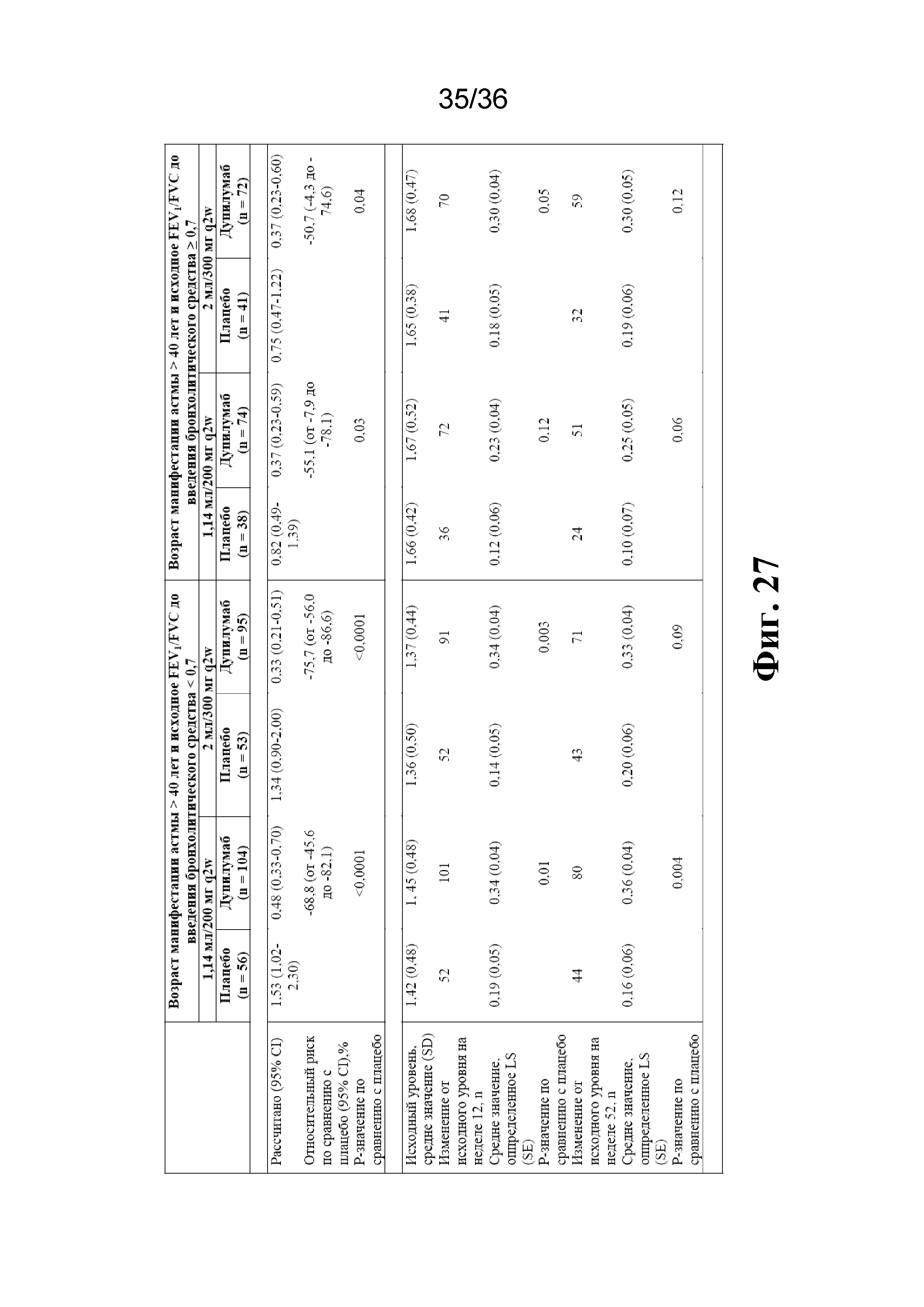

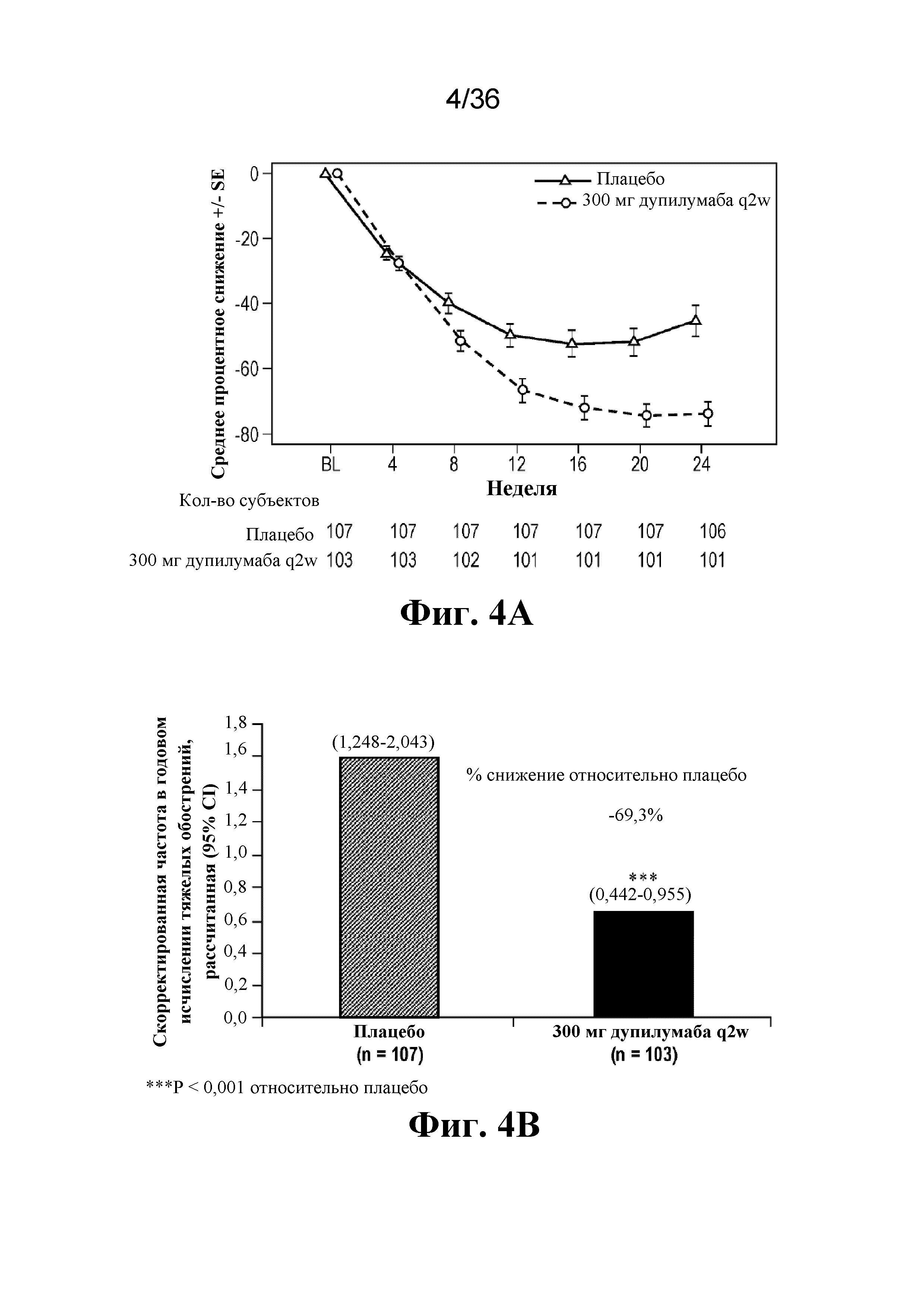
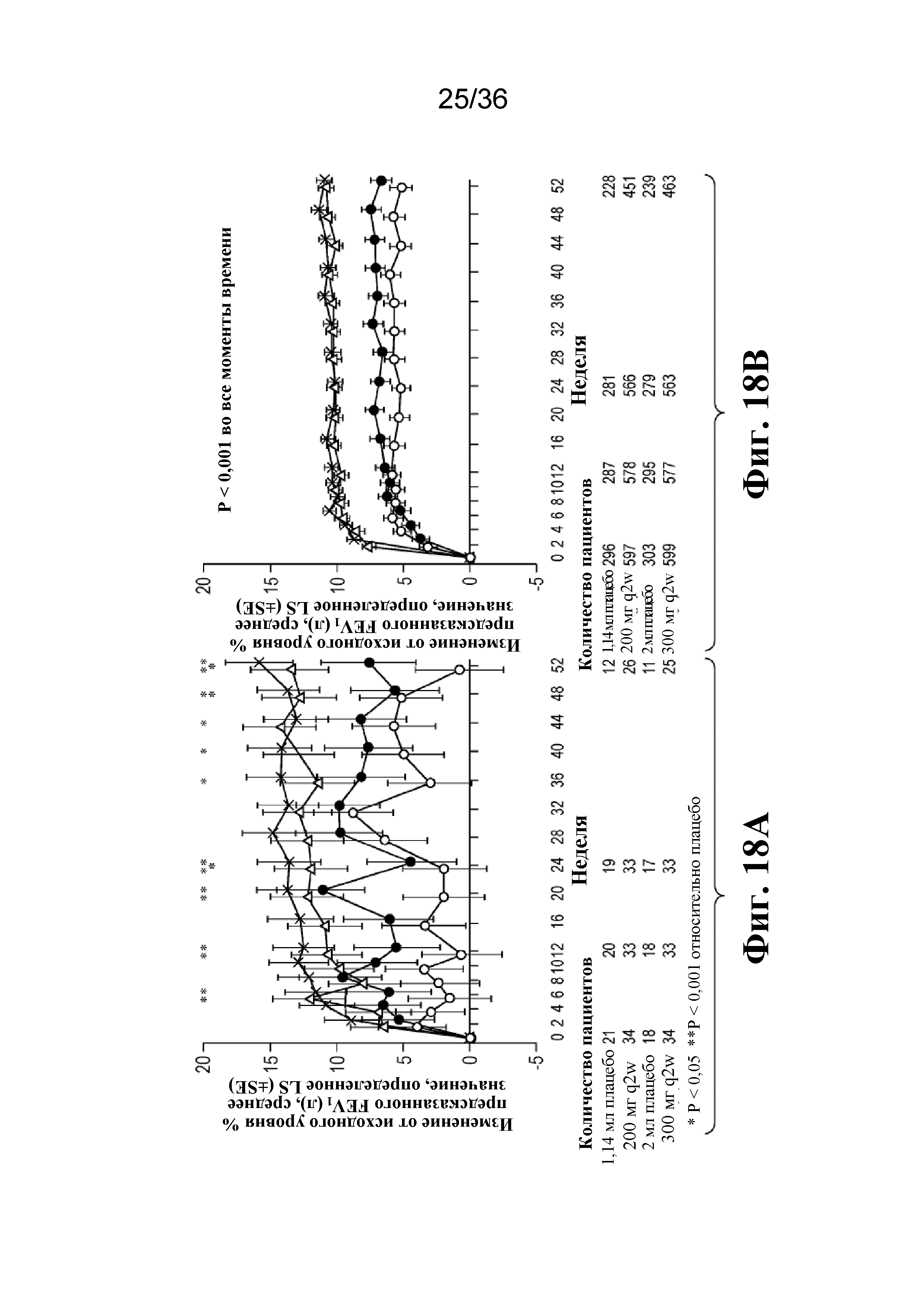
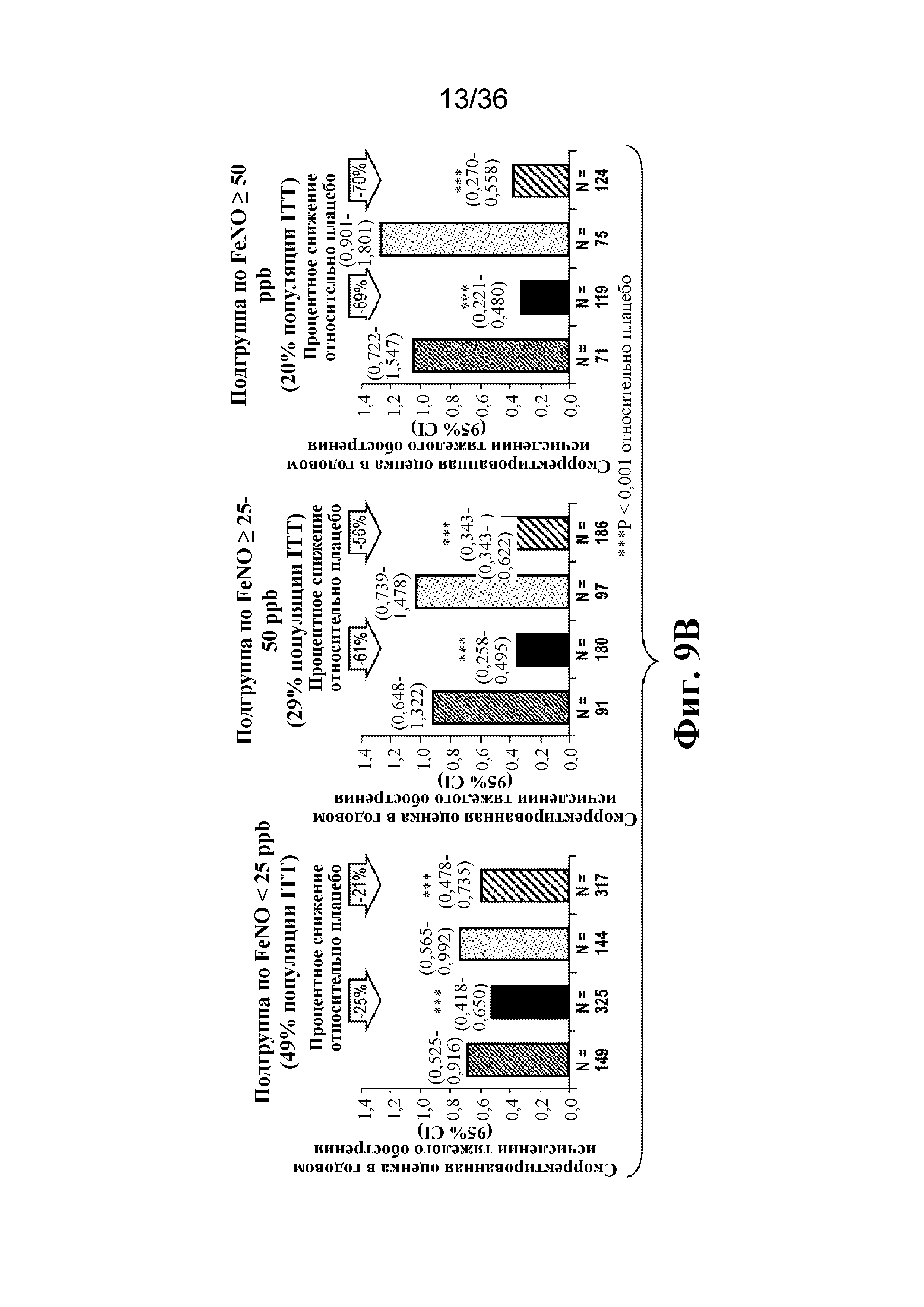
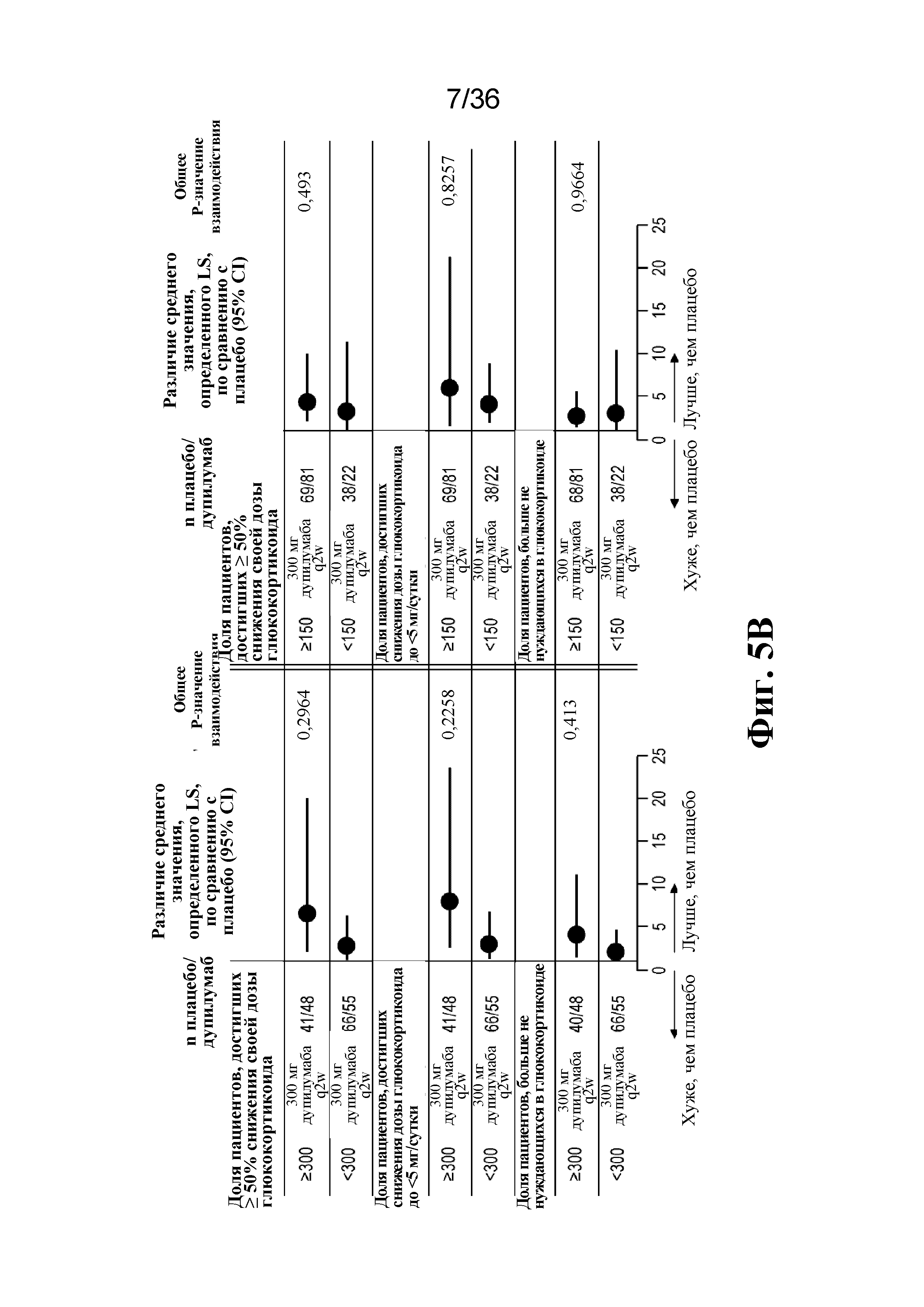
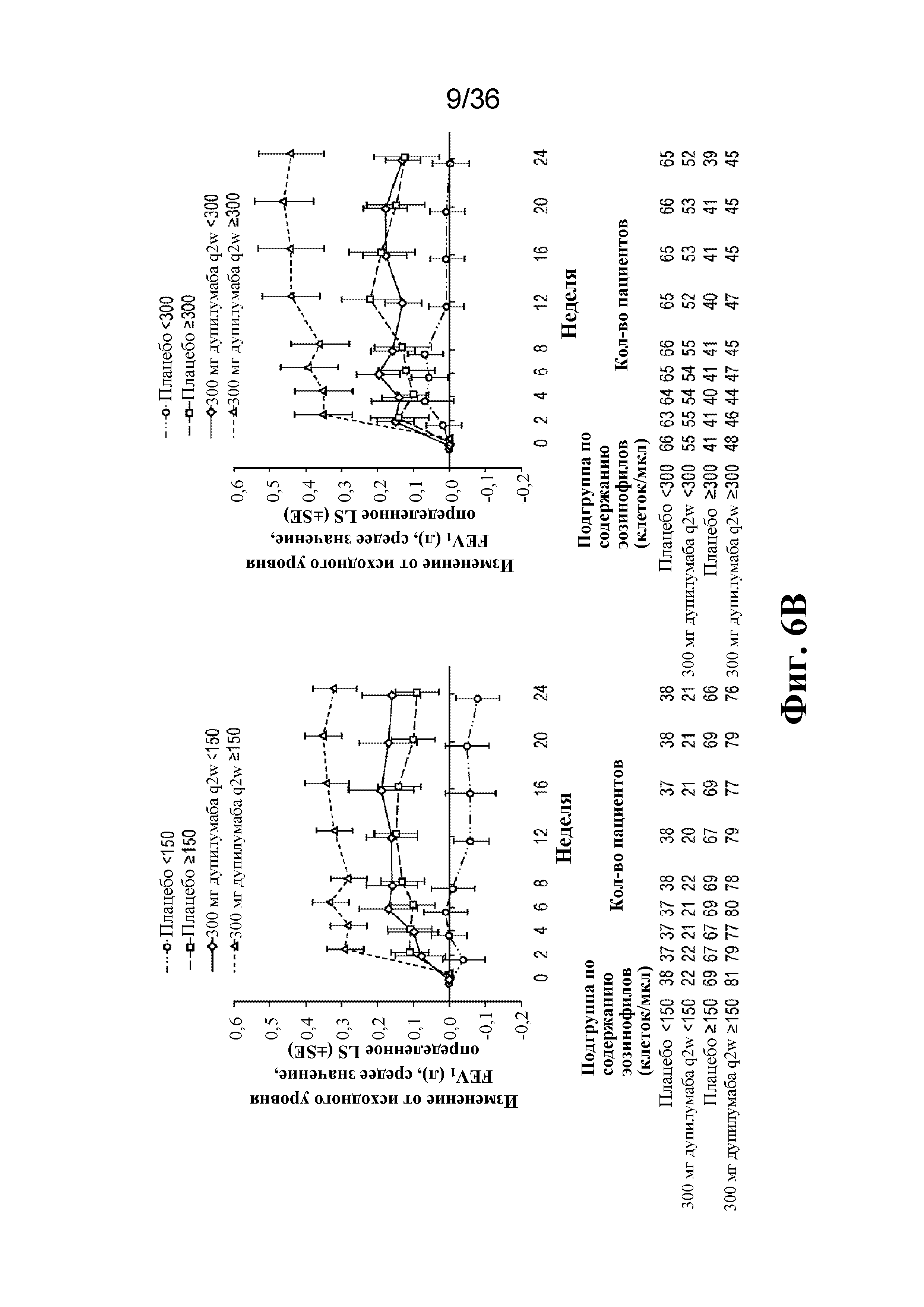
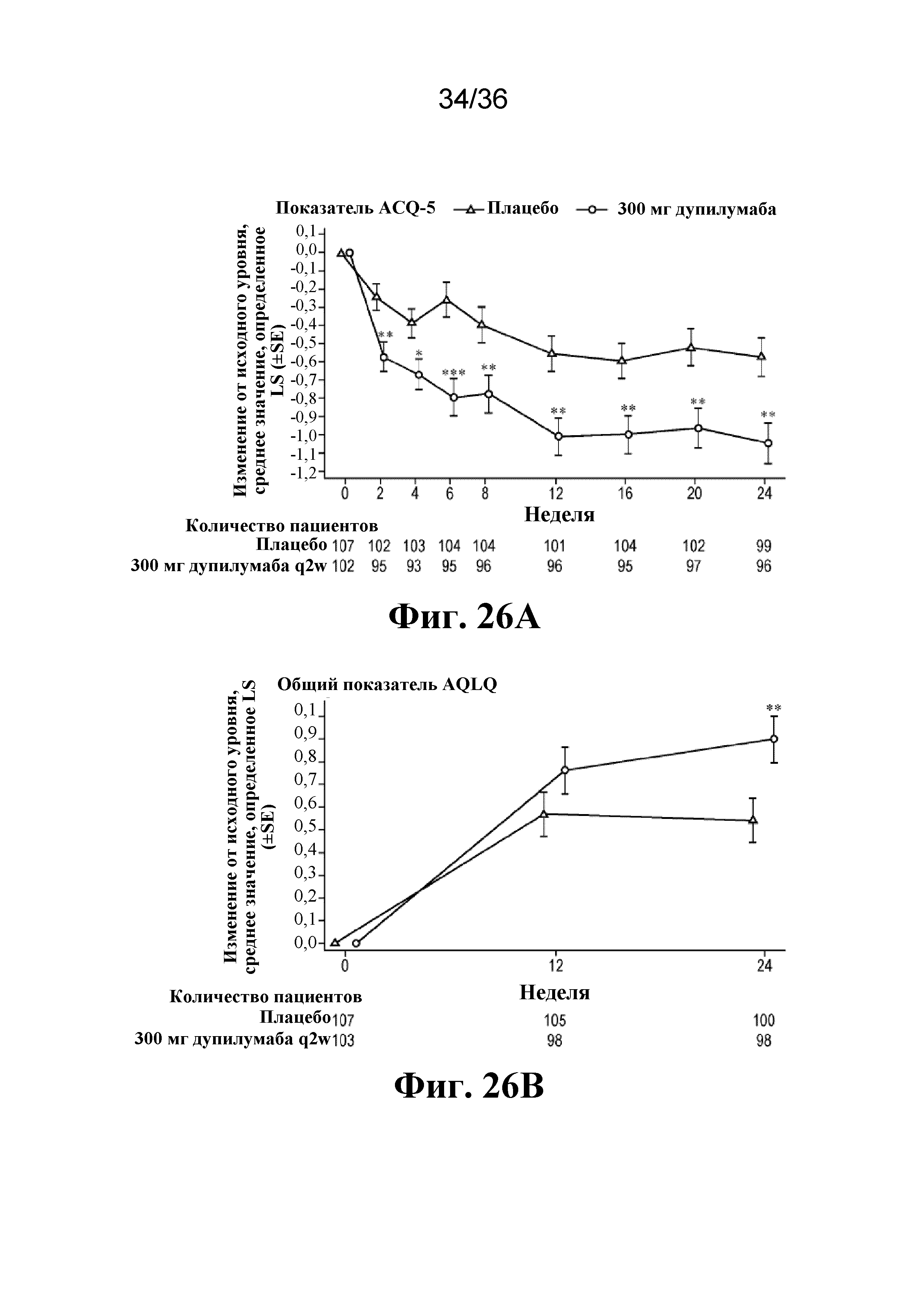

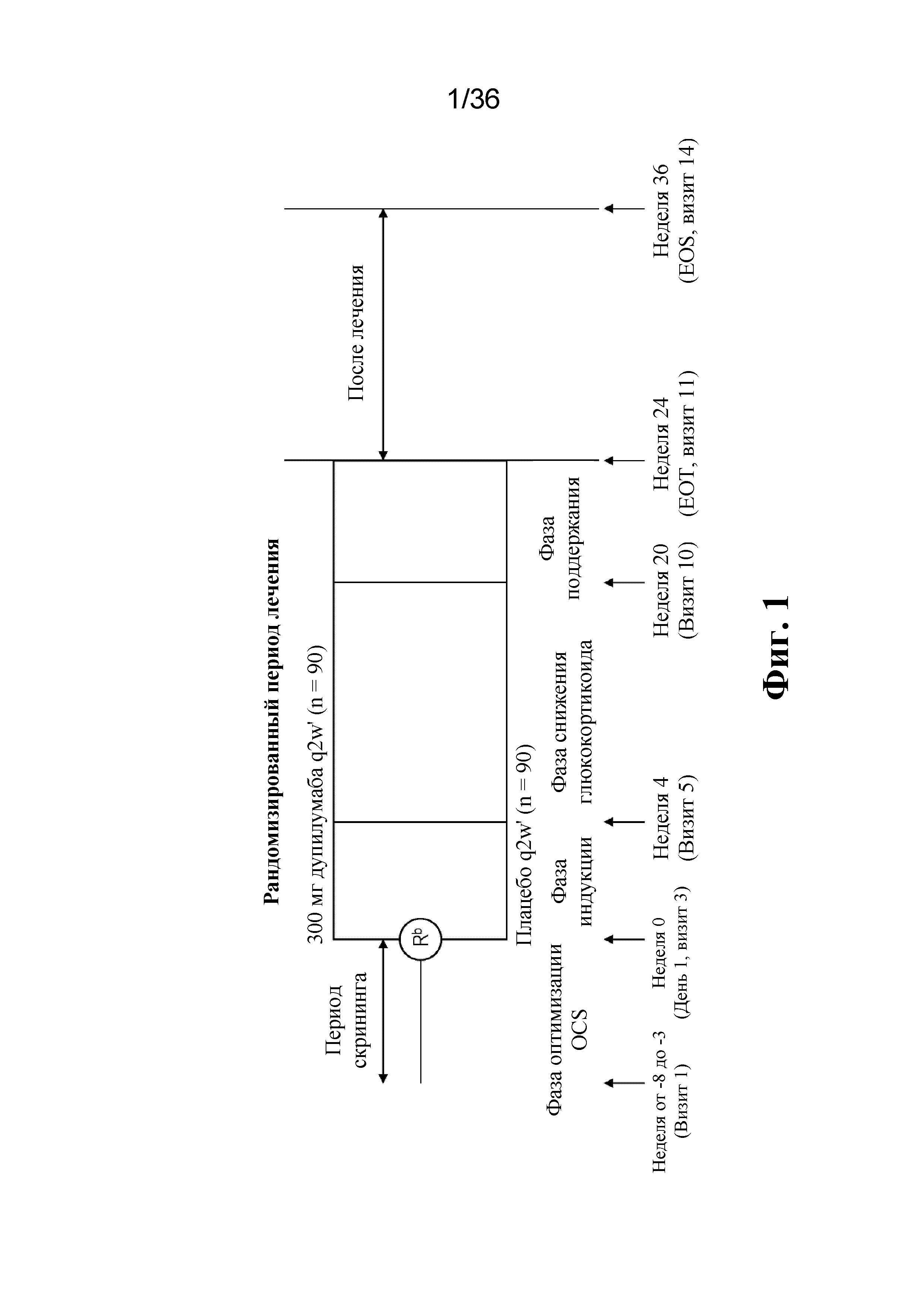
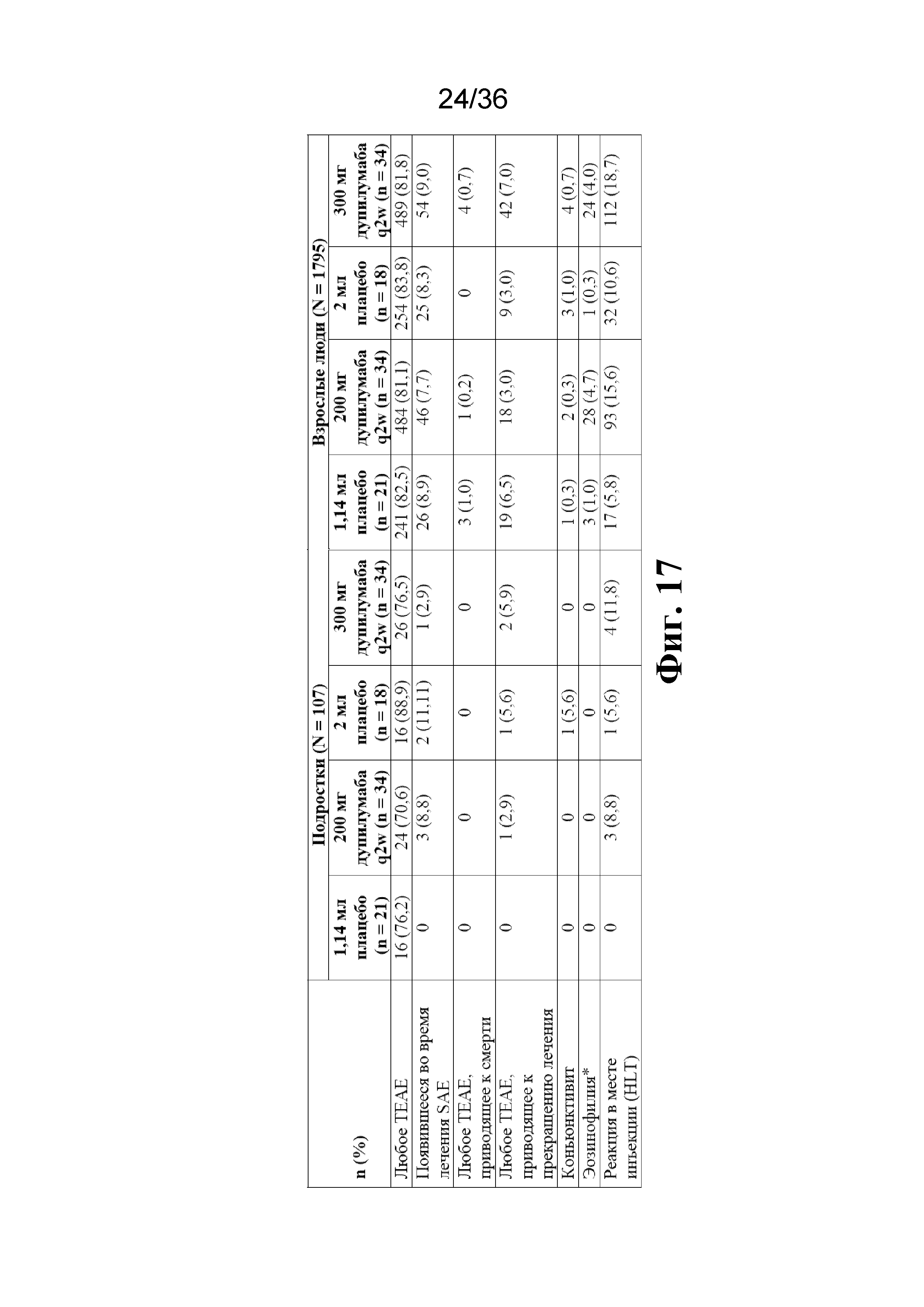
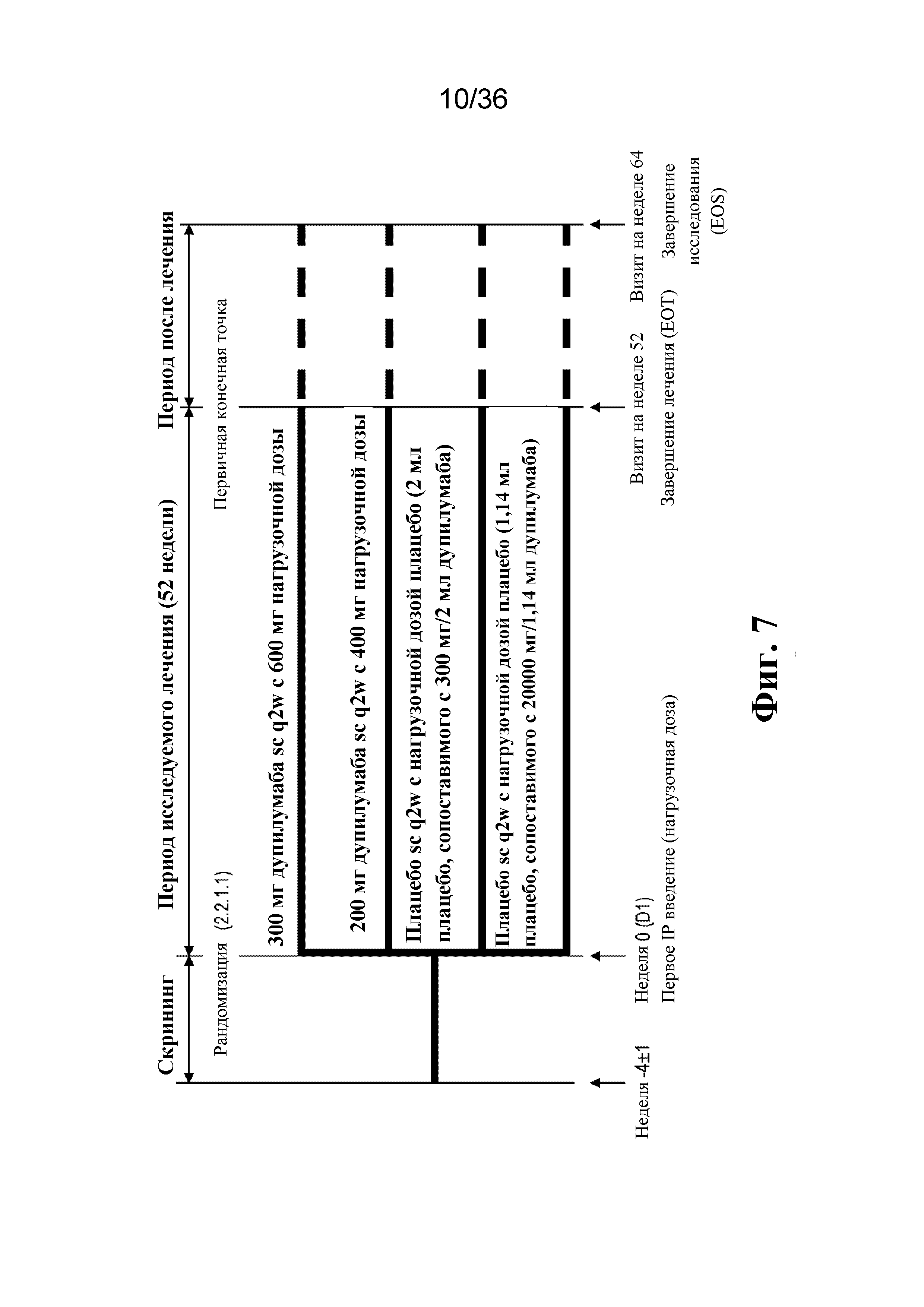
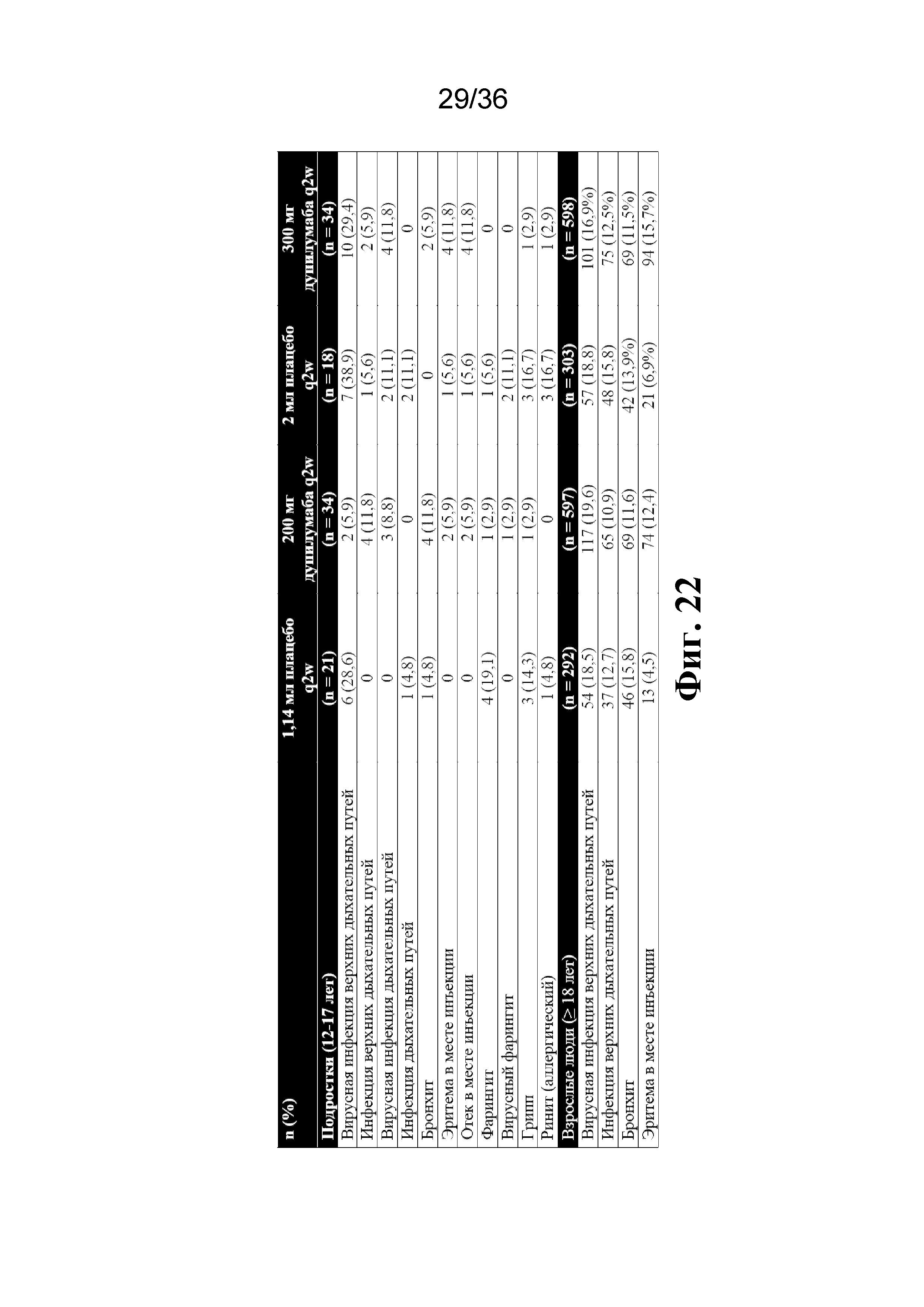
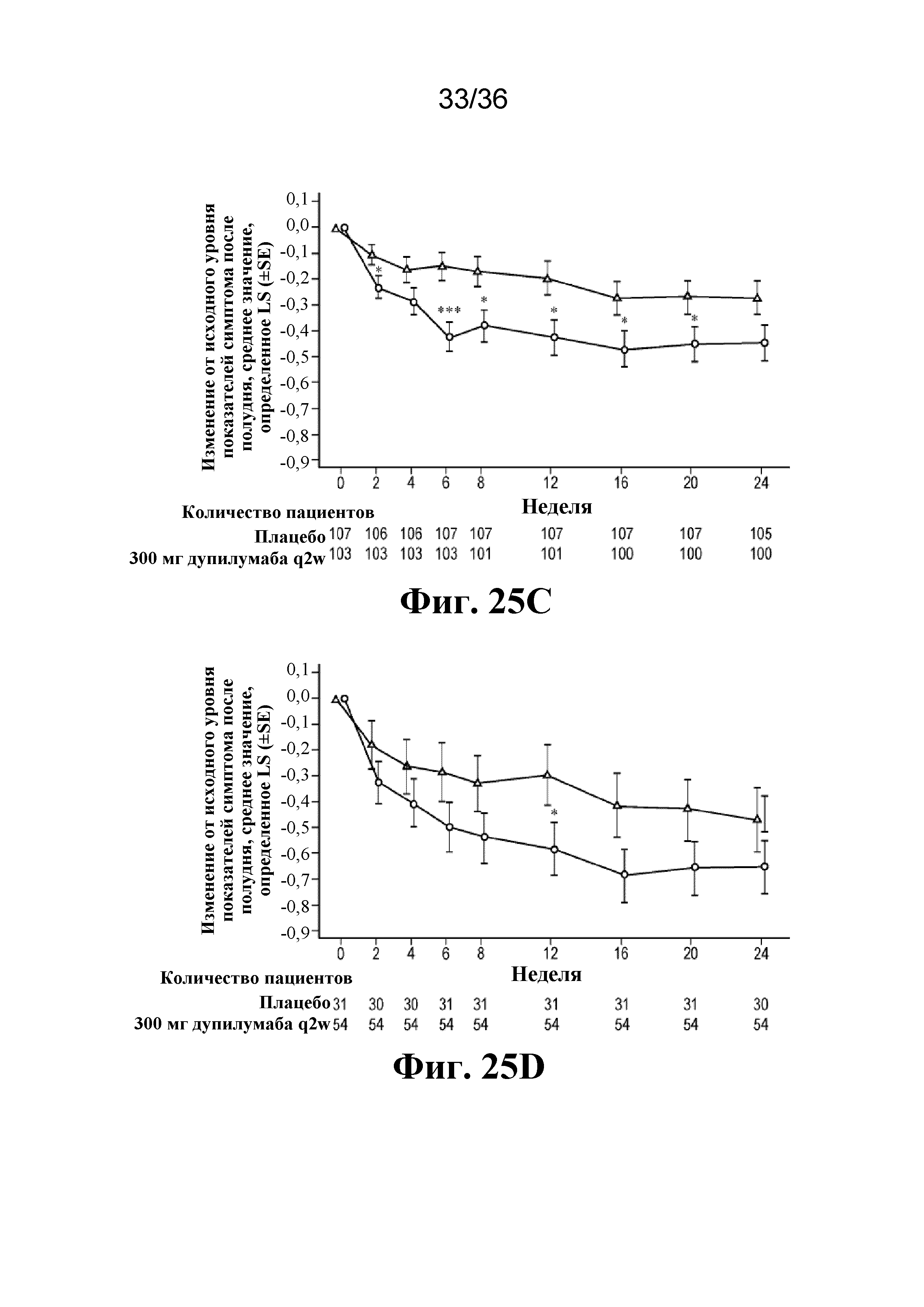

Антитела человека с высокой аффинностью к фактору роста нервов человека
Антитела человека к cd20 человека и способ их использования
Легковыделяемые биспецифические антитела с природным иммуноглобулиновым форматом
Человеческие антитела с высокой аффинностью к рецепторам il-4 человека
Высокоаффинные человеческие антитела к человеческому ангиопоэтину-2
Гуманизированные fcγr мыши
Высокоаффинные человеческие антитела к pcsk9
Стабилизированные составы, содержащие антитела к рецептору интерлейкина 6 (il-6r)
Антитела против gdf8 человека
Легковыделяемые биспецифические антитела с природным иммуноглобулиновым форматом

