Результат интеллектуальной деятельности: СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА НА ОСНОВЕ ТИТАН-СОДЕРЖАЩЕГО ЦЕОЛИТА ДЛЯ ЭПОКСИДИРОВАНИЯ ПРОПИЛЕНА
Вид РИД
Изобретение
Настоящее изобретение относится к способу регенерации катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала, к регенерированному катализатору, и к непрерывному способу получения пропиленоксида.
В последние годы были разработаны различные титансодержащие цеолиты, которые полезны при катализе органических реакций, таких как превращение олефинов в эпоксиды. Например, в WO 98/55229 А1 раскрывается производство и далее использование цеолита, содержащий титан, для эпоксидирования олефинов.
Титансодержащие цеолиты представляют большой промышленный интерес, и в этом контексте экономические и экологические соображения имеют большое значение. Эффективная регенерация таких цеолитов для последующего повторного использования при катализе органических реакций была бы очень предпочтительной по сравнению с их заменой свежим катализатором.
В ЕР 1221442 А1 раскрыта регенерация катализатора на основе содержащего титан цеолит, используемого при эпоксидировании олефинов с пероксидом водорода, причем способ включает проведение реакции эпоксидирования, где регенерация отработанного катализатора проводится пероксидом водорода в присутствии олефина, посредством чего реакция эпоксидирования продолжается, и где регенерация достигается путем изменения направления подачи пероксида водорода. Пероксид водорода также является ценным исходным материалом и как таковой трудно обрабатывается из-за его склонности к спонтанному разложению. В WO 2005/000827 А1 раскрывается регенерация катализатора на основе силиката титана после процесса непрерывного эпоксидирования пропена с пероксидом водорода. Катализатор периодически регенерируется с помощью метанольного растворителя при температуре по меньшей мере 100°. Метанол представляет собой ценное органическое соединение, которое требует дорогостоящего и трудоемкого восстановления. После регенерации эпоксидирование необходимо перезапустить при более высокой температуре по сравнению со свежим катализатором в WO 2005/000827 А1.
В WO 2007/013739 А1 раскрывается регенерация катализатора на основе титан-осодержащего молекулярного сита, где после предварительной обработки отработанного катализатора водой или спиртом предварительно обработанный катализатор приводится в контакт со смесью, которая включает пероксид водорода, воду и спирт. Таким образом, этот процесс включает две обязательные и последующие стадии, в которых отработанный катализатор приводится в контакт с двумя различными растворами. Кроме того, требуется обработка смеси, содержащей пероксид водорода, воду и спирт.
Поэтому задача настоящего изобретения состоит в обеспечении простого и экономичного способа регенерации катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала, используемого при эпоксидировании олефинов. Еще одна задача настоящего изобретения состоит в обеспечении регенерированного катализатора, содержащего цеолит, содержащего титан, в качестве каталитически активного материала, который может быть легко повторно использован при эпоксидировании олефинов.
Таким образом, настоящее изобретение относится к способу регенерации катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала, включающему
(i) стадию, включающую
(a) непрерывное получение пропиленоксида, включающее
(a1) введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, в реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2) воздействие на поток поступающего материала согласно (a1) в реакторе условиями эпоксидирования в присутствии указанного катализатора, с получением реакционной смеси, содержащей пропиленоксид и указанный органический растворитель;
(а3) удаление потока продукта, содержащего пропиленоксид и органический растворитель, из реактора;
(b) прекращение введения потока поступающего материала в реактор;
(c) промывку катализатора жидкой водной системой;
причем стадия (i) дополнительно включает повторение последовательности этапов (а)-(с) n раз, причем n представляет собой целое число и равно по меньшей мере 1; причем указанный способ регенерации дополнительно включает
(ii) стадию, включающую кальцинирование катализатора, полученного на этапе (с), после повторения последовательности этапов (а)-(с) n раз;
где последовательность стадий (i)-(ii) необязательно повторяют m раз, причем m представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (i)-(ii), n является одинаковым или различным. Неожиданным образом, согласно способу в соответствии с настоящим изобретением, регенерация катализатора, содержащего цеолит, содержащий титан в качестве каталитически активного материала, применяемого при эпоксидировании олефинов, может осуществляться посредством промывки катализатора жидкой водной системой согласно этапу (с) согласно стадии (i) по меньшей мере один раз без последующего кальцинирования, где после указанной промывки, катализатор проявляет такую же или по существу такую же каталитическую активность как соответствующий свежий катализатор уже без стадии кальцинирования, и где результаты регенерации могут даже быть усилены посредством кальцинирования катализатора согласно стадии (ii) после одной или более следующих стадий промывки.
Стадия (i)
Стадия (i) согласно настоящему изобретению включает
(a) непрерывное получение пропиленоксида, включающее
(a1) введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, в реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(a1) воздействие на поток поступающего материала согласно (a1) в реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3) удаление потока продукта, содержащего пропиленоксид и органический растворитель из реактора;
(b) прекращение введения потока поступающего материала в реактор;
(с) промывку катализатора жидкой водной системой;
причем стадия (i) дополнительно включает повторение последовательности этапов (а)-(с) n раз, причем п представляет собой целое число и равно по меньшей мере 1.
Стадия (a1)
Поток поступающего материала согласно (a1) содержит пропен, пероксид водорода или источник пероксида водорода, и органический растворитель.
Пероксид водорода или источник пероксида водорода
Как пероксид водорода, так и источник пероксида водорода могут применяться в качестве эпоксидирующего средства в потоке поступающего материала согласно (a1).
В случае применения пероксида водорода, предпочтительно, что пероксид водорода представляет собой водный раствор пероксида водорода, где раствор содержит предпочтительно от 30 до 50 мас. % пероксида водорода относительно общего количества воды.
Возможно также, что пероксид водорода образуется in situ в реакционной смеси из водорода и кислорода в присутствии подходящего катализатора или каталитической системы, например, в присутствии цеолита, содержащего титан, дополнительно содержащего один или более благородных металлов, или цеолита, содержащего титан, и дополнительного катализатора, содержащего один или более благородных металлов, например, нанесенного на подходящую подложку, такую как древесный уголь или подходящий неорганический оксид или смесь неорганических оксидов. Для получения пероксида водорода в потоке поступающего материала согласно (a1) может быть использован антрахиноновый процесс. Этот процесс основан на каталитическом гидрировании антрахинонового соединения с образованием соответствующего антрагидрохинонового соединения, последующей его реакции с кислородом с образованием пероксида водорода и последующей экстракции образованного пероксида водорода. Цикл завершается повторным гидрированием соединения антрахинона, которое снова образуется при окислении. Обзор антрахинонового процесса приведен в “Ullmann’s Encyclopedia of Industrial Chemistry”, 5th edition, volume 13, pages 447 to 456. В качестве альтернативы пероксид водорода может быть получен путем анодного окисления серной кислоты с одновременным выделением водорода на катоде с получением пероксодисульфоновой кислоты. Гидролиз пероксодисульфоновой кислоты образует сначала пероксосульфовую кислоту, а затем пероксид водорода и серную кислоту, которая, таким образом, восстанавливается. В еще одном варианте пероксид водорода может быть получен непосредственно из элементов водорода и кислорода.
Предпочтительно, пероксид водорода применяют в потоке поступающего материала согласно (a1).
Органический растворитель
Органические растворители, применяемые в (a1), в действительности представляют собой все растворители, известные для этой цели. Предпочтительными являются органические растворители, такие как спирты, нитрилы, и их смеси, необязательно также вода. Предпочтительно, органический растворитель выбирают из группы, состоящей из метанола и ацетонитрила. Более предпочтительно, органическим растворителем является ацетонитрил.
Поток поступающего материала согласно (a1)
В общем, поток поступающего материала согласно (a1) может быть получен любым стандартным способом. Таким образом, поток поступающего материала в (a1) может быть получен посредством смешивания потока, содержащего пропен, потока, содержащего пероксид водорода или источник пероксида водорода, и потока, содержащего органический растворитель, которое охватывает последовательное перемешивание, то есть смешивание потока, содержащего пропен, с потоком, содержащим органический растворитель, и смешивание полученного потока с потоком, содержащим пероксид водорода или источник пероксида водорода.
В общем, поток поступающего материала согласно (a1) не ограничен относительно молярного отношения пропена и пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом источника пероксида водорода. Предпочтительно, пропен присутствует в молярном избытке в потоке поступающего материала согласно (a1) относительно пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом источника пероксида водорода. Предпочтительно, молярное отношение пропена и пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом источника пероксида водорода, в потоке поступающего материала согласно (a1) составляет от 1 до 1,6, более предпочтительно от 1,1 до 1,55, более предпочтительно от 1,2 до 1,5, более предпочтительно от 1,40 до 1,45.
В общем, поток поступающего материала согласно (a1), содержащий пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, не ограничен в отношении дополнительных компонентов содержащихся потоком поступающего материала. Таким образом, поток поступающего материала согласно (a1) может дополнительно содержать по меньшей мере один дополнительный компонент, например, по меньшей мере одну соль, содержащую калий.
Соль, содержащая калий
Что касается химической природы по меньшей мере одной калиевой соли, то никаких конкретных ограничений не существует. Предпочтительно по меньшей мере одну калиевую соль выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, по меньшей мере одной органической калиевой соли и комбинаций по меньшей мере одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли.
Предпочтительные неорганические соли, содержащие калий, включают, но без ограничения к этому, галогениды калия, такие как хлорид калия или бромид калия, нитрат калия, сульфат калия, гидросульфат калия, гидроксид калия, перхлорат калия, соли калия, содержащие фосфор, такие как дигидрофосфат калия или гидрофосфат дикалия или фосфат калия, или пирофосфаты калия, такие как одноосновный пирофосфат калия или двухосновный пирофосфат калия или трехосновный пирофосфат калия или четырехосновный пирофосфат калия, или этидронаты калия, такие как одноосновный этидронат калий или двухосновный этидронат калия, или трехосновный этидронат калия, или четырехосновный этидронат калия, цианат калия, оксиды калия, такие как оксид калия (K2О) или супероксид калия (KО2) или пероксид калия (K2О2).
Предпочтительные органические соли, содержащие калий включают, но без ограничения к этому, карбонат калия (К2СО3), гидрокарбонат калия, калиевые соли алифатических насыщенных карбоновых кислот, таких как монокарбоновые кислоты, предпочтительно имеющие от 1 до 6, более предпочтительно от 1 до 5, более предпочтительно от 1 до 4, более предпочтительно от 1 до 3 атомов углерода, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, дикарбоновые кислоты, предпочтительно имеющие от 2 до 6, более предпочтительно от 2 до 4 атомов углерода, такие как щавелевая кислота, малоновая кислота, янтарная кислота, винная кислота, трикарбновые кислоты, предпочтительно имеющие от 6 до 10 атомов углерода, такие как лимонная кислота или изолимонная кислота или пропан-1,2,3-трикарбоновая кислота, или тетракарбоновые кислоты. Предпочтительно, органическую калиевую соль выбирают из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновые кислоты, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия. Более предпочтительно, органическую калиевую соль выбирают из группы, состоящей из формиата калия, ацетата калия, пропионата калия, карбоната калия и гидрокарбоната калия. Более предпочтительно, органическую калиевую соль выбирают из группы, состоящей из формиата калия, ацетата калия, карбоната калия и гидрокарбоната калия.
Поэтому, соль, содержащая калий, предпочтительно выбирается из группы, состоящей из по меньшей мере одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, дигидрофосфата калия или гидрофосфата дикалия или фосфата калия или пирофосфатов калия, таких как одноосновный пирофосфат калия или двухосновный пирофосфат калия или трехосновный пирофосфат калия или четырехосновный пирофосфат калия, или этидронатов калия, таких как одноосновный этидронат калия или двухосновный этидронат калия или трехосновный калийный этидронат или четырехосновный этидронат калия, по меньшей мере одной органической калиевой соли, выбранной из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбонат калия, а также комбинации по меньшей мере одной из по меньшей мере одной неорганической соли калия и по меньшей мере одной из по меньшей мере одной органической соли.
Более предпочтительно, соль, содержащую калий выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, выбранной из группы, состоящей из дигидрофосфата калия или гидрофосфата дикалия или фосфата калия, гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, по меньшей мере одной органической калиевой соли, выбранной из группы, состоящей из формиата калия, ацетата калия, пропионата калия, карбоната калия и гидрокарбоната калия, а также комбинации по меньшей мере одной из по меньшей мере одной неорганической калиевой соли и по меньшей мере одной из по меньшей мере одной органической калиевой соли.
Особенно предпочтительно, соль, содержащая калий, представляет собой дигидрофосфат калия, гидрофосфат дикалия или формиат калия. Поэтому, если используется одна калиевая соль, солью, содержащей калий, является наиболее предпочтительной дигидрофосфат калия, гидрофосфат дикалия или формиат калия. Если используются две или более солей, содержащих калий, одна калиевая соль представляет собой дигидрофосфат калия, гидрофосфат дикалия или формиат калия.
Что касается концентрации соли, содержащей калий в потоке поступающего материала, конкретных ограничений не существует. Предпочтительно, концентрация соли, содержащей калий, в потоке поступающего материала составляет по меньшей мере 10%, предпочтительно в интервале от 10 до 100%, предпочтительно от 20 до 100%, более предпочтительно от 30 до 100%, более предпочтительно от 40 до 100% от предела растворимости соли, содержащей калий в потоке поступающего материала. Термин “предел растворимости по меньшей мере одной калиевой соли в потоке поступающего материала”, как применяется в контексте настоящего изобретения, относится к концентрации насыщения соли, содержащей калий в жидком потоке поступающего материала, де, при добавлении еще соль, содержащую калий, концентрация соли, содержащей калий, в растворенном в смеси виде, не увеличивается, и соль, содержащая калий начнет осаждаться. Предел растворимости соли, содержащей калий, в потоке поступающего материала будет зависеть от состава смеси и условий, таких как температура и давление, при которых обеспечивается поток поступающего материала. Определение предела растворимости соли, содержащей калий, в смеси, является простой и прямой задачей для специалиста, знающего указанные условия и указанный состав данной смеси. Простая процедура оценки того, является ли количество добавляемой соли, содержащей калий, выше предела растворимости, состоит в пропускании потока поступающего материала перед тем, как подвергнуть его условия эпоксидирования в (а2), через фильтр и измерении падение давления через фильтр. Если перепад давления через фильтр со временем возрастает по потоку, а соль, содержащая калий, обнаруживается на фильтре, когда он отключен, количество добавляемой соли, содержащей калий, уже превышает предел растворимости.
Предпочтительно на стадии (a1), молярное отношение калия, содержащегося в соли, содержащей калий, относительно пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом источника пероксида водорода, содержащегося в потоке поступающего материала, находится в интервале от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1. Молярное количество калия, содержащегося в соли, содержащей калий, относится к суммарному молярному количеству калия, содержащегося во всех солях, содержащих калий, применяемых в (a1), если используются две или более соли, содержащие калий.
Далее предпочтительно в (a1), молярное отношение калия относительно пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом источника пероксида водорода, содержащегося в потоке поступающего материала, находится в интервале от 10×l0-6:l до 5000×10-6:1, предпочтительно от 15×10-6:1 до 2500×10-6:1, более предпочтительно от 20×10-6:1 до 1,500×10-6:1, более предпочтительно от 25×10-6:1 до 1,300×10-6:1, более предпочтительно от 30×10-6:1 до 1,000×10-6:1.
Предпочтительно, поток поступающего материала согласно (a1) свободен от дигидрофосфата аммония. Более предпочтительно, поток поступающего материала согласно (a1) свободен от фосфата аммония, гидрофосфата аммония и дигидрофосфата аммония. Более предпочтительно, поток поступающего материала согласно (a1) свободен от карбоната аммония, гидрокарбоната аммония, дигидрофосфата аммония, гидрофосфата аммония, фосфата аммония, гидропирофосфата аммония, пирофосфата аммония, хлорида аммония, нитрата аммония и ацетата аммония. Более предпочтительно, поток поступающего материала согласно (a1) свободен от аммониевой соли. Термин “свободен от”, как применяется в этом контексте, относится к концентрации соответствующего соединения, равной не более 2 мас. частей на миллион, предпочтительно не более 1 мас. частей на миллион, на основе общей массы потока поступающего материала.
Предпочтительно, поток поступающего материала согласно (a1) содержит натрий при молярном отношении натрия относительно пероксида водорода или одного эквивалента пероксида водорода, являющегося результатом пероксида водорода, находится в интервале от 1×10-6:1 до 250×10-6:1, предпочтительно от 5×10-6:1 до 50×10-6:1. Предпочтительно, смесь, предпочтительно поток поступающего материала согласно (al) не содержит растворенный дигидрофосфат натрия (NaH2PO4), более предпочтительно ни растворенный дигидрофосфат натрия, ни растворенный динатрия гидрофосфат (Na2HPO4), более предпочтительно ни растворенный дигидрофосфат натрия, ни растворенный динатрия гидрофосфат, ни растворенный фосфат натрия (Na3PO4).
В случае, когда поток поступающего материала согласно (a1) дополнительно содержит по меньшей мере одну соль, содержащую калий, жидкий поток поступающего материала предпочтительно получают путем объединения по меньшей мере четырех отдельных потоков. Предпочтительно, водный поток, содержащий по меньшей мере одну растворенную соль, содержащую калий, объединяют с потоком, содержащим пропен, или с потоком, содержащим пероксид водорода или источник пероксида водорода, или с потоком, содержащим органический растворитель, или со смешанным потоком из двух или трех из этих потоков, предпочтительно с потоком, содержащим пероксид водорода или источник пероксида водорода, или с потоком, содержащим органический растворитель, или с их смешанным потоком.
Поток, содержащий пропен, может дополнительно содержать пропан, где предпочтительно по меньшей мере 98 мас. %, более предпочтительно меньшей мере 99 мас. %, более предпочтительно меньшей мере 99,5 мас. %, более предпочтительно меньшей мере 99,9 мас. % потока состоит из пропена и пропана. Предпочтительно, массовое отношение пропена относительно пропана в потоке составляет по меньшей мере 7:3. Например, может использоваться коммерчески доступный пропен, который может быть либо пропеном полимерного назначения, либо пропеном химического назначения. Как правило, пропен полимерного назначения имеет содержание пропена в интервале от 99 до 99,8 мас. % и содержание пропана в интервале от 0,2 до 1 мас. %. Пропен химического назначения обычно имеет содержание пропена в интервале от 92 до 98 мас. % и содержание пропана в интервале от 2 до 8 мас. %. Предпочтительно используется поток с содержанием пропена в интервале от 99 до 99,8 мас. % и содержанием пропана в интервале от 0,2 до 1 мас. %.
Поток, содержащий пероксид водорода или источник пероксида водорода, может быть получен любым обычным способом. Вполне возможно получить поток, содержащий пероксид водорода путем превращения серной кислоты в пероксодисерную кислоту путем анодного окисления с одновременным выделением водорода на катоде. Затем гидролиз пероксодисерной кислоты приводит через пероксимоносерную кислоту к пероксиду водорода и серной кислоте, которую, таким образом, получают обратно. Возможно также получение пероксида водорода из элементов. В зависимости от конкретного способа получения поток, содержащий пероксид водорода, может быть, например, водным или водным/метанольным потоком пероксида водорода, предпочтительно водным потоком пероксид водорода. В случае применения водного потока пероксида водорода, содержание потока относительно пероксида водорода находится, как правило, в интервале от 3 до 85 мас. %, предпочтительно от 25 до 75 мас. %, более предпочтительно от 30 до 50 мас. %, как например от 30 до 40 мас. % или от 35 до 45 мас. %, от 40 до 50 мас. %. Предпочтительно, по меньшей мере 25 мас. %, более предпочтительно по меньшей мере 30 мас. %, более предпочтительно по меньшей мере 35 мас. % потока, содержащего пероксид водорода состоит из воды и пероксида водорода. Предпочтительные диапазоны составляют от 30 до 80 мас. % или от 35 до 75 мас. % или от 40 до 70 мас. %.
Чтобы обеспечить достаточную стабильность пероксида водорода во время экстракции водой, предпочтительно по существу чистой водой, к воде обычно добавляют подходящие стабилизирующие агенты, предпочтительно применяют по существу чистую воду. В частности, следует упомянуть сильные неорганические кислоты и/или хелатирующие агенты. Согласно предпочтительным способам экстракции небольшие количества нитратов и/или фосфатов и пирофосфатов соответственно добавляют в качестве стабилизирующих агентов либо в виде кислот, либо в виде солей натрия. Эти стабилизирующие агенты обычно добавляют в количествах, таких что неочищенный водный раствор пероксида водорода содержит от 50 до 400 мас. частей на миллион катионов натрия, от 100 до 700 мас. частей на миллион фосфора, вычисленного как фосфат (РО43-), и от 50 до 400 мас. частей на миллион нитратных анионов, в каждом случае вычисленных относительно пероксида водорода, содержащегося в неочищенном водном растворе пероксида водорода. Предпочтительные диапазоны составляют, например, от 50 до 200 мас. частей на миллион или от 50 до 100 мас. частей на миллион катионов натрия, от 100 до 500 мас. частей на миллион или от 100 до 300 мас. частей на миллион фосфора и от 50 до 200 мас. частей на миллион или от 50 до 100 мас. частей на миллион нитрата. Кроме того, возможно применение других стабилизирующих агентов, такие как станниты, такие как станнит натрия (Na2SnO2) и/или органические фосфоновые кислоты, в частности органические дифосфоновые кислоты, такие как этидроновая кислота. Предпочтительно, поток водного пероксида водорода содержит натрий с молярным отношением натрия относительно пероксида водорода в интервале от 1×10-6:1 до 250×10-6:1, более предпочтительно от 5×10-6:1 до 50×10-6:1.
В общем, молярное отношение воды относительно органического растворителя в потоке поступающего материала, обеспечиваемого в (a1), не имеет специальных ограничений. Предпочтительно, в частности в случае, когда органический растворитель представляет собой ацетонитрил, молярное отношение воды относительно органического растворителя не составит не более 1:4, более предпочтительно в интервале от 1:50 до 1:4, предпочтительно от 1:15 до 1:4.1, более предпочтительно от 1:10 до 1:4.2.
Катализатор согласно (a1)
В общем, цеолит, содержащий титан, применяемый в качестве каталитически активного материала, может иметь каркасную структуру цеолита согласно следующим трехзначным обозначениям: ABW, АСО, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFX, AFY, АНТ, ANA, АРС, APD, AST, ASV, ATN, ATO, ATS, ATT, ATV, AWO, AWW, ВСТ, BEA, ВЕС, BIK, BOG, BPH, BRE, CAN, CAS, CDO, CFI, CGF, CGS, СНА, CHI, CLO, CON, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EMT, EPI, ERI, ESV, ETR, EUO, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFR, ISV, ITE, ITH, ITW, IWR, IWW, JBW, KFI, LAU, LEV, LIO, LOS, LOV, LTA, LTL, LTN, MAR, MAZ, MEI, MEL, МЕР, MER, MMFI, MFS, MON, MOR, MSO, MTF, MTN, MTT, MTW, MWW, NAB, NAT, NEES, NON, NPO, OBW, OFF, OSI, OSO, PAR, PAU, PHI, PON, RHO, RON, RRO, RSN, RTE, RTH, RUT, RWR, RWY, SAO, SAS, SAT, SAV, SBE, SBS, SBT, SFE, SFF, SFG, SFH, SFN SFO, SGT, SOD, SSY, STF, STI, STT, TER, THO, TON, TSC, UEI, UFI, UOZ, USI, UTL, VET, VFI, VNI, VSV, WEI, WEN, YUG, ZON, или смешанную структуру двух или более из этих каркасных структур. В отношении трехкодовых обозначений и их определений делается ссылка “Atlas of Zeolite Framework Types”, 5th edition, Elsevier, London, England (2001).
Предпочтительно цеолит, содержащий титан, имеет MFI каркасную структуру, MEL каркасную структуру, MWW каркасную структуру, каркасную структуру MWW-типа, или смешанную структуру двух или более из этих каркасных структур, предпочтительно MFI каркасную структуру, MWW каркасную структуру или каркасную структуру MWW-типа, более предпочтительно MWW каркасную структуру или каркасную структуру MWW-типа. Более предпочтительно, цеолит, содержащий титан, представляет собой цеолит, известный как “TS-1” (силикат титана-1) или “TiMWW”.
Предпочтительно, в частности в случае, когда цеолит, содержащий титан, представляет собой TiMWW, цеолит, содержащий титан, содержит одно или более из А1, В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Со, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd, предпочтительно одно или более из В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd, более предпочтительно Zn.
Термин “содержащий титан цеолит с каркасной структурой типа MWW”, как применяется в контексте настоящего изобретения, также обозначается как “TiMWW”, относительно цеолитов с каркасной структурой MWW, который содержит титан в качестве изоморфного элемента замещения в цеолитной каркасной структуре. Предпочтительно, цеолитная каркасная структура по существу свободна от алюминия и состоит по существу из кремния, титана и кислорода. Предпочтительно, по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % цеолитной каркасной структуры состоит из кремния, титана и кислорода. Необязательно, содержащий титан цеолит с каркасной структурой типа MWW может содержать экстра-каркасный титан, под котором понимают каждый вид титана, который не является частью MWW цеолитной каркасной структуры. Получение TiMWW катализаторов описано, например, в US 2007043226 A1, в частности в Примерах 3 и 5.
Содержание титана в содержащем титан цеолите с каркасной структурой типа MWW не имеет конкретных ограничений. Предпочтительно, содержащий титан цеолит с каркасной структурой типа MWW, содержащийся в катализаторе в (a1), содержит титан, вычисленный как элементарный титан, в количестве в интервале от 0.1 до 5 мас. %, более предпочтительно от 0.2 до 4 мас. %, более предпочтительно от 0.5 до 3 мас. %, более предпочтительно от 1 до 2 мас. %, на основе общей массы содержащего титан цеолита с каркасной структурой типа MWW. Поэтому настоящее изобретение относится к способу, как описано выше, где содержащий титан цеолит с каркасной структурой типа MWW, содержащийся в катализаторе в (a1), содержит титан, вычисленный как элементарный титан, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы содержащего титан цеолита с каркасной структурой типа MWW.
В дополнение к титану, содержащий титан цеолит с каркасной структурой типа MWW может содержать по меньшей мере один дополнительный элемент, отличный от титана, кремния и кислорода. В общем, вполне возможно, что этот по меньшей мере один дополнительный элемент является изоморфным элементом замещения, который является частью цеолитной каркасной структуры MWW. Предпочтительно, этот по меньшей мере один дополнительный элемент не является изоморфным элементом замещения. Такой дополнительный элемент, который не является изоморфным элементом замещения, может быть добавлен к цеолиту, например, способом распыления, способом влажной пропитки, способом пропитки по влагоемкости или любым другим подходящим способом. Предпочтительно, по меньшей мере один дополнительный элемент выбирают из группы, состоящей из А1, В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd и комбинации двух или более из них, предпочтительно из группы, состоящей из В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd и комбинации двух или более из них. Более предпочтительно, содержащий титан цеолит с каркасной структурой типа MWW содержит цинк в качестве дополнительного элемента в дополнение к титану, кремнию и кислороду. Более предпочтительно, содержащий титан цеолит с каркасной структурой типа MWW содержит цинк в качестве единственного дополнительного элемента в дополнение к титану, кремнию и кислороду. Более предпочтительно, содержащий титан цеолит с каркасной структурой типа MWW одержит цинк в качестве единственного дополнительного элемента в дополнение к титану, кремнию и кислороду, где по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % цеолитной каркасной структуры состоит из кремния, титана и кислорода. Более предпочтительно, в случае, когда содержащий титан цеолит с каркасной структурой типа MWW содержит цинк в качестве единственного дополнительного элемента, по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % содержащего титан цеолита с каркасной структурой типа MWW состоит из цинка, титана, кремния и кислорода; этот содержащий титан цеолит с каркасной структурой типа MWW, который содержит цинк в качестве единственного дополнительного элемента, также обозначается как “ZnTiMWW”.
ZnTiMWW катализатор
Содержание цинка в содержащем титан цеолите с каркасной структурой типа MWW не имеет конкретных ограничений. Предпочтительно, содержащий титан цеолит с каркасной структурой типа MWW, содержащийся в катализаторе в (a1), содержит цинк, вычисленный как элементарный цинк, в количестве в интервале от 0.1 до 5 мас. %, более предпочтительно от 0.2 до 4 мас. %, более предпочтительно от 0.5 до 3 мас. %, более предпочтительно от 1 до 2 мас. %, на основе общей массы содержащего титан цеолита с каркасной структурой типа MWW. Поэтому, настоящее изобретение относится к способу, как описано выше, где содержащий титан цеолит с каркасной структурой типа MWW, содержащийся в катализаторе в (a1), содержит цинк, вычисленный как элементарный цинк, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы содержащего титан цеолита с каркасной структурой типа MWW.
Таким образом, предпочтительно цеолит, содержащий титан, который содержится в катализаторе в (a1), имеет MWW каркасную структуру, содержит цинк и содержит титан, вычисленный как элементарный титан, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан, и содержит цинк, вычисленный как элементарный цинк, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан.
Катализатор согласно (a1), включающий содержащий титан цеолит с каркасной структурой типа MWW, может состоять из содержащего титан цеолита с каркасной структурой типа MWW, предпочтительно состоящего из TiMWW или ZnTiMWW, как описано. В таких случаях катализатором может быть содержащий титан цеолит с каркасной структурой типа MWW в виде цеолитного порошка, который может быть формованным, например, в виде гранул, микросферы, такой как микросфера, полученная распылительной сушкой или грануляцией распылением, формованного тела, имеющего, например, форму гранулы, таблетки, цилиндра, колеса, звезды или сферы.
Предпочтительно, катализатор согласно (a1), содержащий содержащий титан цеолит с каркасной структурой типа MWW, предпочтительно TiMWW или ZnTiMWW, получают в виде формованного изделия, содержащего содержащий титан цеолит с каркасной структурой типа MWW, предпочтительно TiMWW или ZnTiMWW, посредством надлежащего смешивания содержащего титан цеолита с каркасной структурой типа MWW с по меньшей мере одним связующим и/или с по меньшей мере одним предшественником связующего, и необязательно по меньшей мере одним порообразующим агентом и/или по меньшей мере одним пластифицирующим агентом. Формованные изделия могут быть сформированы в любой мыслимой геометрии, такой как нити, например, имеющие прямоугольное, треугольное шестиугольное, квадратное, овальное или круговое поперечное сечение, звезды, таблетки, сферы, полые цилиндры и тому подобное. Примерами таких связующих являются оксиды металлов, такие как, например, SiO2, Al2O3, ТiO2, ZrO2 или MgO или глины или смеси двух или более из этих оксидов или смешанные оксиды по меньшей мере двух из Si, Al, Ti, Zr, и Mg, причем SiO2 является предпочтительным. Порообразующие агенты, такие как мезопора-образующие агенты, включают полимерные виниловые соединения, такие как полиалкиленоксиды, такие как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры. Пастирующие агенты включают органические, в частности гидрофильные полимеры, такие как углеводы, такие как целлюлоза, производные целлюлозы, такие как метилцеллюлоза, и крахмал, такой как картофельный крахмал, обойная штукатурка, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутен или политетрагидрофуран. Применение воды, спиртов или гликолей или их смесей, таких как смеси воды и спирта, или воды и гликоля, как например, вода и метанол, или вода и этанол, или вода и пропанол, или вода и пропиленгликоль, в качестве пастирующих агентов может быть указано. Предпочтительно, катализатор согласно (a1) применяется в форме формованного изделия, имеющего форму экструдатов, предпочтительно экструдатов, имеющих длину предпочтительно от 1 до 10 мм, более предпочтительно от 1 до 7 мм, более предпочтительно от 1 до 5 мм, и диаметр предпочтительно от 0.1 до 5 мм, более предпочтительно от 0.2 до 4 мм, более предпочтительно от 0.5 до 2 мм. В частности, что касается предпочтительного катализатора согласно (a1), включающего ZnTiMWW, то предпочтительно использовать этот катализатор в форме микропорошка или в форме формованного изделия, где формованное изделие предпочтительно содержит указанный микропорошок.
Указанный катализатор согласно (a1) в форме микропорошока, содержащего ZnTiMWW, предпочтительно характеризуется следующими признаками и вариантами выполнения настоящего изобретения, включая комбинации вариантов выполнения настоящего изобретения согласно данным зависимостям:
1. микропорошок, частицы которого имеют значение Dv10 по меньшей мере 2 микрометра, причем указанный микропорошок содержит мезопоры, имеющие средний диаметр поры (4V/A) в интервале от 2 до 50 нм, как определено посредством Hg порозиметрии согласно DIN 66133, и содержащий на основе массы микропорошка, по меньшей мере 95 мас. % микропорошкового свободного от алюминия цеолитного материала со структурой типа MWW, содержащего титан и цинк, (ZnTiMWW). Значение Dv10 понимают как определяемое согласно ссылочному примеру 8 из WO 2013/117536.
2. Микропорошок согласно варианту выполнения настоящего изобретения 1, имеющий значение Dv10 в интервале от 2 до 5.5 микрометра, предпочтительно от 3 до 5.5 микрометра.
3. Микропорошок согласно варианту выполнения настоящего изобретения 1 или 2, имеющий значение Dv50 в интервале от 7 до 25 микрометра и необязательно значение Dv90 в интервале от 26 до 85 микрометра. Значения Dv50 и Dv90 понимают как определяемые согласно ссылочному примеру 8 из WO 2013/117536.
4. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-3, где мезопоры имеют средний диаметр пор (4V/A) в интервале от 10 до 50 нм, предпочтительно от 15 до 40 нм, более предпочтительно от 20 до 30 нм, как определено посредством Hg порозиметрии согласно DIN 66133.
5. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-4, дополнительно содержащий макропоры, имеющие средний диаметр поры (4V/A) в интервале от более 50 нм, причем указанные макропоры предпочтительно имеют средний диаметр поры в интервале от 0.05 до 3 микрометра, как определено посредством Hg порозиметрии согласно DIN 66133.
6. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-5, где микропоры в ZnTiMWW имеют средний диаметр поры в интервале от 1.0 до 1.2 нанометров, как определено посредством адсорбции азота согласно DIN 66135.
7. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-6, содержащий, на основе массы микропорошка, по меньшей мере 99 мас. %, предпочтительно по меньшей мере 99.7 мас. % ZnTiMWW.
8. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-7, где ZnTiMWW содержит цинк в количестве от 1.0 до 2.0 мас. %, предпочтительно от 1.2 до 1.9 мас. %, вычисленный как Zn и на основе массы ZnTiMWW.
9. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-8, где ZnTiMWW содержит титан в количестве от 1.0 до 2.0 мас. %, предпочтительно от 1.2 до 1.8 мас. %, вычисленный как Ti и на основе массы ZnTiMWW.
10. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-9, имеющий кристалличность, как определено посредством анализа дифракции рентгеновских лучей (XRD), по меньшей мере (80+/-10)%, предпочтительно по меньшей мере (85+/-10)%. Кристалличность понимают как определенную согласно ссылочному примеру 10 в WO 2013/117536.
11. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-10, содержащий, на основе массы микропоршка и при вычислении в виде элемента, менее 0.001 мас. %, предпочтительно менее 0.0001 мас. % благородного металла, предпочтительно выбранного из группы, состоящей из золота, серебра, платины, палладия, иридия, рутения, осмия и смеси двух или более из них, более предпочтительно выбранного из группы, состоящей из золота, платины, золота и смеси двух или более из них.
12. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-11, содержащий, на основе массы микропоршка и при вычислении в виде элемента, менее 0.1 мас. %, предпочтительно менее 0.01 мас. % бора.
13. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-12, имеющий насыпную плотность в интервале от 80 до 100 г/мл.
14. Микропорошок согласно любому из вариантов выполнения настоящего изобретения 1-13, представляющий собой распылительный порошок, предпочтительно получаемый или полученный распылительной сушкой.
Далее, указанный катализатор согласно (a1) в форме формованного изделия, содержащего ZnTiMWW, предпочтительно характеризуется следующими признаками и вариантами выполнения настоящего изобретения, включая комбинации вариантов выполнения настоящего изобретения согласно данным зависимостям:
1. Формованное изделие, содержащее микропорошковый свободный от алюминия цеолитный материал со структурой типа MWW, содержащий титан и цинк, (ZnTiMWW), причем указанное формованное изделие предпочтительно содержит микропорошок содержащий, на основе массы микропорошка, по меньшей мере 95 мас. % микропорошкового свободного от алюминия цеолитного материала со структурой типа MWW, содержащего титан и цинк, (ZnTiMWW), причем указанное формованное изделие более предпочтительно содержит микропорошок согласно любому из вариантов выполнения микропорошка 1-14, как определено выше, причем формованное изделие предпочтительно дополнительно содержит по меньшей мере одно связующее, предпочтительно связующее на основе диоксида кремния.
2. Формованное изделие согласно варианту выполнения настоящего изобретения 1, содержащее мезопоры, имеющие средний диаметр поры в интервале от 4 до 40 нм, предпочтительно от 20 до 30 нм, как определено посредством Hg порозиметрии согласно DIN 66133.
3. Формованное изделие согласно варианту выполнения настоящего изобретения 1 или 2, имеющий кристалличность, как определено посредством анализа XRD, по меньшей мере (55+/-10)%, предпочтительно в интервале ((55-75)+/-10)%. Кристалличность понимают как определенную согласно ссылочному примеру 10 в WO 2013/117536.
4. Формованное изделие согласно любому из вариантов выполнения настоящего изобретения 1-3, содержащий микропорошок в количестве в интервале от 70 до 80 мас. % и связующее на основе диоксида кремния в количестве от 30 до 20 мас. %, причем микропорошок вместе с связующим на основе диоксида кремния составляет по меньшей мере 99 мас. % от формованного изделия, где формованное изделие имеет концентрацию силанольных групп относительно общего числа атомов Si не более 6%, предпочтительно не более 3%, как определено согласно 29Si MAS ЯМР. Под концентрацией силанольных групп понимают определенную согласно ссылочному примеру 3 в WO 2013/117536.
5. Формованное изделие согласно любому из вариантов выполнения настоящего изобретения 1-4, представляющее собой нить с круглым поперечным сечением и диаметром в диапазоне от 1,5 до 1,7 мм и имеющую прочность на раздавливание по меньшей мере 5 Н, предпочтительно в интервале от 5 до 20 Н, более предпочтительно в интервале от 12 до 20 Н, причем прочность на раздавливание определяется испытательной машиной Z2.5/TS1S для испытания прочности на раздавливание согласно способу, описанному в ссылочном примере 2 в WO 2013/117536.
6. Формованное изделие согласно любому из вариантов выполнения настоящего изобретения 1-5, причем 29Si-ЯMP спектр указанного формованного изделия включает шесть пиков в следующих положениях
пик 1 при -98 +/- х ррm,
пик 2 при -104 +/- х ррm,
пик 3 при -110 +/- х ррm,
пик 4 при -113 +/- х ррm,
пик 5 при -115 +/- х ррm,
пик 6 при -118 +/- х ррm,
где х в любом из пиков равно 1.5, предпочтительно 1.0, более предпочтительно 0.5,
где Q, которое определяется как
Q=100*{[a1+a2]/[а4+а5+а6]}/а3
равно не более 2.5, предпочтительно не более 1.6, предпочтительно не более 1.4, причем [a1+а2] составляет сумму площадей пиков для пиков 1 и 2, и [а4+а5+а6] составляет сумму площадей пиков для пиков 4, 5, и 6, и аз представляет собой площадь пика 3. Под этими 29Si-ЯМР характеристиками понимаются полученные согласно ссылочному примеру 4 в WO 2013/117536.
7. Формованное изделие согласно любому из вариантов выполнения настоящего изобретения 1-6, имеющее поглощение воды в интервале от 3 до 8 мас. %, предпочтительно от 4 до 7 мас. %. Под поглощением воды понимают определенное согласно ссылочному примеру 6 в WO 2013/117536.
8. Формованное изделие согласно любому из вариантов выполнения настоящего изобретения 1-7, причем инфракрасный спектр указанного формованного изделия содержит полосу в области (3700-3750)+/-20 см-1 и полосу в области (3670-3690)+/-20 см-1, где отношение интенсивностей полосы в области (3700-3750)+/-20 см-1 относительно полосы в области (3670-3690)+/-20 см-1 не составит не более 1.5, предпочтительно не более 1.4. Под этими IR характеристиками понимаются определенные согласно ссылочному примеру 5 в WO 2013/117536.
Далее, предпочтительный способ получения указанного катализатора согласно (a1) в форме микропорошка и/или формованного изделия, содержащего ZnTiMWW, характеризуется следующими признаками и вариантами выполнения, включая комбинации вариантов выполнения настоящего изобретения согласно данным зависимостям:
1. Способ, включающий
(a) обеспечение суспензии, содержащей микропорошковый свободный от алюминия цеолитный материал со структурой типа MWW, содержащий титан и цинк, (ZnTiMWW);
(b) воздействие на суспензию, полученную в (а), распылительной сушкой с получением микропорошка;
(c) необязательно кальцинирование микропорошка, полученного в (b),
где микропорошок, полученный в (b) или (с), предпочтительно в (с), предпочтительно представляет собой микропорошок согласно любому из указанных вариантов выполнения микропорошка 1-14, как описано выше.
2. Способ согласно варианту выполнения настоящего изобретения 1, где суспензия, полученная на этапе (а), имеет содержание твердого вещества в интервале от 5 до 25 мас. %, предпочтительно от 10 до 20 мас. %, причем суспензия предпочтительно представляет собой водную суспензию.
3. Способ согласно варианту выполнения настоящего изобретения 1 или 2, где ZnTiMWW согласно (а) содержит цинк в количестве от 1.0 до 2.0 мас. %, предпочтительно от 1.2 до 1.9 мас. %, вычисленный как Zn, и титан в количестве от 1.0 до 2.0 мас. %, предпочтительно от 1.2 до 1.8 мас. %, вычисленный как Ti и на основе массы ZnTiMWW.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1-3, где на этапе (b), распылительное устройство, предпочтительно оросительная колонная применяется для распылительной сушки суспензии, причем указанное устройство имеет по меньшей мере одну распылительную форсунку, предпочтительно по меньшей мере одну двухкомпонентную форсунку, причем указанная форсунка имеет диаметр в интервале от 3.5 до 4.5 мм.
5. Способ согласно любому из вариантов выполнения настоящего изобретения 1-4, где на этапе (b), распылительное устройство, предпочтительно оросительная колонная применяется для распылительной сушки суспензии, причем указанное устройство работает с форсуночным газом, имеющим температуру в интервале от 20 до 50°С, предпочтительно от 20 до 30°С, и сушильным газом, имеющим температуру в интервале от 250 до 350°С, предпочтительно от 275 до 325°С, причем указанный форсуночный газ предпочтительно представляет собой инертный газ, более предпочтительно технический азот, и указанный сушильный газ предпочтительно представляет собой инертный газ, более предпочтительно технический азот.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1-5, где на этапе (с), микропорошок кальцинируют при температуре в интервале от 600 до 700°С в течение времени в интервале от 0.5 до 6 ч.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1-6, дополнительно включающий
(d) формование микропорошка, полученного на этапе (b) или (с) с получением формованного изделия;
(e) необязательно сушку и/или кальцинирование формованного изделия, полученного на этапе (d).
8. Способ согласно варианту выполнения настоящего изобретения 7, где формование согласно (d) включает
(аа) смешивание микропорошка со связующим или предшественником связующего, предпочтительно связующим на основе диоксида кремния или предшественником связующего на основе диоксида кремния, где массовое отношение ZnTiMWW, содержащегося в микропорошке, относительно диоксида кремния, содержащегося в или происходящего из связующего на основе диоксида кремния, находится в интервале от 3:7 до 1:4, с получением смеси;
(bb) формование смеси, полученной на этапе (аа), с получением формованного изделия, причем указанное формование предпочтительно включает подвергание смеси, полученной на этапе (аа), экструзии в результате которой предпочтительно получают нити, имеющие диаметр предпочтительно в интервале от 1.0 до 2.0 мм, более предпочтительно от 1.5 до 1.7 мм.
9. Способ согласно варианту выполнения настоящего изобретения 8, где на этапе (аа), углевод и/или воду добавляют в качестве пастирующего агента.
10. Способ согласно варианту выполнения настоящего изобретения 8 или 9, где смешивание на этапе (аа) проводят в течение времени в интервале от 15 до 60 мин, предпочтительно от 30 до 55 мин, более предпочтительно от 40 до 50 мин.
11. Способ согласно любому из вариантов выполнения настоящего изобретения 7-10, где на этапе (d), никакой мезопорообразующий агент, выбранный из группы, состоящей из полиалкиленоксидов, таких как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры не добавляется.
12. Способ согласно любому из вариантов выполнения настоящего изобретения 7-11, где на этапе (е), формованное изделие сушат при температуре в интервале от 100 до 150°С в течение времени в интервале от 10 до 20 ч и кальцинируют при температуре в интервале от 500 до 600°С в течение времени в интервале от 0.5 до 2 ч.
13. Способ согласно любому из вариантов выполнения настоящего изобретения 7-12, дополнительно включающий
(f) воздействие на формованное изделие, полученное на этапе (d) или (е), предпочтительно на этапе (е), обработкой водой;
(g) необязательно сушку и/или кальцинирование обработанного водой формованного изделия,
где формованное изделие полученное на этапе (f) или (g), предпочтительно на этапе (g), предпочтительно представляет собой формованное изделие согласно любому из указанных вариантов выполнения формованного изделия 1-8, как описано выше.
14. Способ согласно варианту выполнения настоящего изобретения 13, где на этапе (f), обработка водой включает обработку формованного изделия жидкой водой в автоклаве при автогенном давлении при температуре в интервале от 100 до 200°С, предпочтительно от 125 до 175°С, более предпочтительно от 140 до 150°С в течение периода времени от 2 до 24 часов, предпочтительно от 6 до 10 ч.
15. Способ согласно варианту выполнения настоящего изобретения 13 или 14, где на этапе (f), массовое отношение формованного изделия относительно воды находится в интервале от 0.02 до 0.08, предпочтительно от 0.03 до 0.07, более предпочтительно от 0.04 до 0.06.
16. Способ согласно любому из вариантов выполнения настоящего изобретения 13-15, где на этапе (g), обработанное водой формованное изделие сушат при температуре в интервале от 100 до 150°С в течение времени в интервале от 10 до 20 ч и кальцинируют при температуре в интервале от 400 до 500°С в течение времени в интервале от 1 до 3 ч.
17. Способ согласно любому из вариантов выполнения настоящего изобретения 7-16, где формованное изделие не подвергают обработке паром.
Что касается указанного предпочтительного способа получения указанного катализатора согласно (a1) в виде микропорошка и/или формованного изделия, содержащего ZnTiMWW, описанного выше в вариантах выполнения настоящего изобретения 1-17, ZnTiMWW, на основе которого суспензия в варианте выполнения настоящего изобретения 1.(а), может быть получена согласно всеми стандартными методам. Например, можно получить микропорошковый свободный от алюминия цеолитный материал со структурой типа MWW, содержащий титан (TiMWW), и подвергнуть TiMWW подходящей обработке для получения ZnTiMWW. Кроме того, можно получить свободный от алюминия цеолитный материал со структурой типа MWW (MWW) и подвергнуть MWW подходящей обработке для получения ZnTiMWW где, например, оба Zn и Ti соответствующим образом включены в MWW. Кроме того, возможно получить свободный от алюминия цеолитный материал со структурой типа MWW, где во время синтеза каркасной структуры типа MWW вводится Ti, и полученный материал подвергается соответствующей обработке с включением Zn, или Zn вводится, и полученный материал подвергают соответствующей обработке для включения Ti или вводят оба Zn и Ti. В качестве возможных способов получения TiMWW можно применять способы, как описано, например, в US 6,114,551, или в Wu et al., “Hydrothermal Synthesis of a novel Titanosilicate with MWW Topology”, Chemistry Letters (2000), pp. 774-775. Предпочтительно, свободный от алюминия цеолитный материал со структурой типа MWW, содержащий Ti (TiMWW), получают на первой стадии и на второй стадии, TiMWW подвергают подходящей обработке с получением ZnTiMWW. Более предпочтительно, ZnTiMWW получают согласно способу, включающему
(I) получение свободного от алюминия цеолитного материала со структурой типа MWW, содержащего бор (B-MWW);
(II) деборирование B-MWW с получением свободного от алюминия цеолитного материала со структурой типа MWW (MWW);
(III) включение титана (Ti) в MWW с получением свободного от алюминия цеолитного материала со структурой типа MWW, содержащего Ti (TiMWW);
(IV) предпочтительно кислотную обработку TiMWW;
(V) включение цинка (Zn) в TiMWW с получением ZnTiMWW.
Предпочтительно на стадии (I) B-MWW получают способом, предпочтительные стадии и условия которого определены в следующих вариантах выполнения 1-28 и при соответствующих зависимостях, как указано:
1. Способ получения свободного от алюминия содержащего бор цеолитного материала, содержащего каркасную структуру MWW (B-MWW), включающий
(a) гидротермальный синтез предшественника B-MWW из смеси для синтеза, содержащей воду, источник кремния, источник бора и матричное соединение MWW, получая предшественник B-MWW в его маточном растворе, причем маточный раствор имеет значение рН выше 9;
(b) регулирование значения рН маточного раствора, полученного на этапе (а), и содержащего предшественник B-MWW, до значения в интервале от 6 до 9;
(c) отделение предшественника B-MWW от маточного раствора, с установленным значением рН, полученного на этапе (b), путем фильтрации в фильтровальном устройстве.
2. Способ согласно варианту выполнения настоящего изобретения 1, где на этапе (а), по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. % смеси для синтеза состоит из воды вода, источника кремния, источника бора и матричного соединения.
3. Способ согласно варианту выполнения настоящего изобретения 1 или 2, где на этапе (а), источник кремния выбирают из группы, состоящей из пирогенного диоксида кремния, коллоидного диоксида кремния и их смеси, причем источником кремния предпочтительно является коллоидный диоксид кремния, более предпочтительно стабилизированный аммиаком диоксид кремния, источник бора выбирают из группы, состоящей из борной кислоты, боратов, оксида бора и смеси двух или более из них, причем источником бора предпочтительно является борная кислота, и матричное соединение MWW выбирают из группы, состоящей из пиперидина, гексаметиленамина, N,N,N,N’,N’,N’-гексаметил-1,5-пентандиаммониевого иона, 1,4-бис(N-метилпирролидиния)бутана, гидроксида октилтриметиламмония, гидроксида гептилтриметиламмония, гидроксида гексилтриметиламмония, гидроксида N,N,N-триметил-1-адамантиламмония и смеси двух или более из них, предпочтительно матричным соединением MWW является пиперидин.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1-3, где на этапе (а), смесь для синтеза состоит из источника бора, вычисленного как элементарный бор, относительно источника кремния, вычисленного как элементарный кремний, при молярном отношении в интервале от 0.4:1 до 2.0:1, предпочтительно от 0.6:1 до 1.9:1, более предпочтительно от 0.9:1 до 1.4:1, воды относительно источника кремния, вычисленного как элементарный кремний, при молярном отношении в интервале от 1:1 до 30:1, предпочтительно от 3:1 до 25:1, более предпочтительно от 6:1 до 20:1; и матричного соединения относительно источника кремния, вычисленного как элементарный кремний, при молярном отношении в интервале от 0.4:1 до 2.0:1, предпочтительно от 0.6:1 до 1.9:1, более предпочтительно от 0.9:1 до 1.4:1.
5. Способ согласно любому из вариантов выполнения настоящего изобретения 1-4, где на этапе (а) гидротермальный синтез проводится при температуре в интервале от 160 до менее 180°С, предпочтительно от 170 до 175°С, в течение периода времени в интервале от 1 до 72 ч, предпочтительно от 6 до 60 ч, более предпочтительно от 12 до 50 ч.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1-5, где на этапе (а), гидротермальный синтез проводится по меньшей мере частично при перемешивании.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1-6, где на этапе (а), смесь для синтеза дополнительно содержит затравочный материал, предпочтительно цеолитный материал, содержащий каркасную структуру MWW, более предпочтительно содержащий бор цеолитный материал, содержащий каркасную структуру MWW.
8. Способ согласно варианту выполнения настоящего изобретения 7, где смесь для синтеза содержит затравочный материал относительно источника кремния при массовом соотношении в интервале от 0.01:1 до 1:1, предпочтительно от 0.02:1 до 0.5:1, более предпочтительно от 0.03:1 до 0.1:1, вычисленном как количество затравочного материала в кг относительно кремния, содержащегося в источнике кремния, вычисленного как диоксид кремния, в кг.
9. Способ согласно любому из вариантов выполнения настоящего изобретения 1-8, где значение рН матричного раствора, полученного на этапе (а), выше 10, предпочтительно в интервале от 10.5 до 12, более предпочтительно от 11 до 11.5.
10. Способ согласно любому из вариантов выполнения настоящего изобретения 1-9, где на этапе (b), значение рН матричного раствора, полученного на этапе (а), доводят до значения в интервале от 6.5 до 8.5, предпочтительно от 7 до 8.
11. Способ согласно любому из вариантов выполнения настоящего изобретения 1-10, где на этапе (b), значение рН регулируют посредством способа, включающего
(аа) добавление кислоты в матричный раствор, полученный на этапе (а), содержащий предшественник B-MWW, где добавление предпочтительно осуществляют по меньшей мере частично при перемешивании.
12. Способ согласно варианту выполнения настоящего изобретения 11, где на этапе (аа), добавление проводится при температуре в интервале от 20 до 70°С, предпочтительно от 30 до 65°С, более предпочтительно от 40 до 60°С.
13. Способ согласно варианту выполнения настоящего изобретения 11 или 12, где на этапе (аа) кислотой является неорганическая кислота, предпочтительно водный раствор, содержащий неорганическую кислоту.
14. Способ согласно варианту выполнения настоящего изобретения 13, где неорганическую кислоту выбирают из группы, состоящей из фосфорной кислоты, серной кислоты, соляной кислоты, азотной кислоты и смеси двух или более из них, причем неорганическая кислота предпочтительно представляет собой азотную кислоту
15. Способ согласно любому из вариантов выполнения настоящего изобретения 11-14, где способ дополнительно включает
(bb) перемешивание маточного раствора, к которому добавили кислоту согласно (аа), где в ходе (bb) кислота не добавляется к маточному раствору.
16. Способ согласно варианту выполнения настоящего изобретения 15, где на этапе (bb), перемешивание проводится при температуре в интервале от 20 до 70°С, предпочтительно от 25 до 65°С, более предпочтительно от 30 до 60°С.
17. Способ согласно любому из вариантов выполнения настоящего изобретения 1-16, где на этапе (b), размер частиц, содержащихся в матричном растворе, выраженный как соответствующие значения Dv10, Dv50, и Dv90, увеличивается на по меньшей мере 2%, предпочтительно по меньшей мере 3%, более предпочтительно по меньшей мере 4.5% относительно Dv10, на по меньшей мере 2%, предпочтительно по меньшей мере 3%, более предпочтительно по меньшей мере 4.5% относительно Dv50, и на по меньшей мере 5%, предпочтительно по меньшей мере 6%, более предпочтительно по меньшей мере 7% относительно Dv90.
18. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 17, где матричный раствор с урегулированным значением рН, полученный на этапе (b), имеет содержание твердых веществ в интервале от 1 до 10 мас. %, предпочтительно от 4 до 9 мас. %, более предпочтительно от 7 до 8 мас. %, на основе общей массы матричного раствора с урегулированным значением рН, полученного на этапе (b).
19. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 18, где матричный раствор с урегулированным значением рН, полученный на этапе (b), имеет сопротивление фильтрации в интервале от 10 до 50 мПа*с/м2, предпочтительно от 15 до 45 мПа*с/м2, более предпочтительно от 20 до 40 мПа*с/м2.
20. Способ согласно любому из вариантов выполнения настоящего изобретения 1-19, дополнительно включающий
(d) промывку предшественника B-MWW, полученного на этапе (с), предпочтительно остатка на фильтре, полученного на этапе (с), где промывку предпочтительно проводят с применением воды в качестве промывочного средства.
21. Способ согласно варианту выполнения настоящего изобретения 20, где на этапе (d) остаток на фильтре, полученный на этапе (с) имеет стойкость к вымыванию в интервале от 10 до 50 мПа*с/м2, предпочтительно от 15 до 45 мПа*с/м2, более предпочтительно от 20 до 40 мПа*с/м2.
22. Способ согласно варианту выполнения настоящего изобретения 20 или 21, где промывку проводят до достижения проводимости фильтрата не более 300 микросименс/см, предпочтительно не более 250 микросименс/см, более предпочтительно не более 200 микросименс/см.
23. Способ согласно любому из вариантов выполнения настоящего изобретения 1-22, дополнительно включающий
(e) сушку предшественника B-MWW, полученного на этапе (с), предпочтительно на этапе (d), при температуре в интервале от 20 до 50°С, предпочтительно от 20 до 40°С, более предпочтительно от 20 до 30°С, где сушка предпочтительно проводится посредством воздействия на B-MWW потока газа, предпочтительно потока азота.
24. Способ согласно любому из вариантов выполнения настоящего изобретения 1-23, где остаточная влажность предшественника B-MWW, полученного на этапе (с), предпочтительно на этапе (d), более предпочтительно на этапе (е), находится в интервале от 80 до 90 мас. %, предпочтительно от 80 до 85 мас. %.
25. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 24, дополнительно содержащий
(f) получение суспензии, предпочтительно водной суспензии, содержащей предшественник B-MWW, полученный на этапе (с), предпочтительно на этапе (d), более предпочтительно на этапе (е), и имеющей содержание твердых веществ в интервале от 10 до 20 мас. %, предпочтительно от 12 до 18 мас. %, более предпочтительно от 14 до 16 мас. %;
(g) распылительную сушку суспензии, полученной на этапе (f), содержащей предшественник B-MWW, получая распылительный порошок;
(h) кальцинирование распылительного порошка, полученного на этапе (g), содержащего предшественник B-MWW, предпочтительно при температуре в интервале от 500 до 700°С, более предпочтительно от 550 до 650°С, более предпочтительно от 575 до 625°С в течение периода времени в интервале от 1 до 24 ч, предпочтительно от 2 до 18 ч, более предпочтительно от 6 до 12 ч, получая распылительный порошок, который на по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.5 мас. % состоит из B-MWW.
26. Способ согласно варианту выполнения настоящего изобретения 25, где на этапе (h), кальцинирование проводится в непрерывном режиме, предпочтительно во вращающейся обжиговой печи, предпочтительно при производительности в интервале от 0.5 до 20 кг распылительного порошка в час.
27. Способ согласно варианту выполнения настоящего изобретения 25 или 26, где степень кристаллизации B-MWW, содержащегося в распылительном порошке, полученном на этапе (h), составляет по меньшей мере (75±5) %, предпочтительно по меньшей мере (80±5) %, как определено согласно XRD.
28. Способ согласно любому из вариантов выполнения настоящего изобретения 25-27, где удельная площадь поверхности BET B-MWW, содержащегося в распылительном порошке, полученном на этапе (h), составляет по меньшей мере 300 м2/г, предпочтительно в интервале от 300 до 500 м2/г, как определено согласно DIN 66131.
Предпочтительно, стадию (II) осуществляют способом, предпочтительные стадии и условия которого определены в следующих вариантах выполнения настоящего изобретения 1-7 и соответствующими зависимостями, как указано:
1. Способ получения цеолитного материала, включающий
(a) обеспечение содержащего бор цеолитного материала со структурой типа MWW (B-MWW), полученного согласно стадии (I);
(b) деборирование B-MWW посредством обработки B-MWW жидкой системой растворителей, с получением, таким образом, деборированного B-MWW (MWW);
где жидкая система растворителей выбирается из группы, состоящей из воды, одноатомных спиртов, многоатомных спиртов и смесей двух или более из них, и где указанная жидкая система растворителей не содержит неорганическую или органическую кислоту или ее соль, причем кислота выбирается из группы, состоящей из соляной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, муравьиной кислоты, уксусной кислоты, пропионовой кислоты, щавелевой кислоты и винной кислоты.
2. Способ согласно варианту выполнения настоящего изобретения 1, где жидкая система растворителей не содержит неорганические или органические кислоты или их соли.
3. Способ согласно варианту выполнения настоящего изобретения 1 или 2, где жидкую систему растворителей выбирают из группы, состоящей из воды, метанола, этанола, пропанола, этан-1,2-диола, пропан-1,2-диола, пропан-1,3-диола, пропан-1,2,3-триола, и смесей двух или более из них, предпочтительно воды.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1-3, где обработка согласно (b) проводится при температуре в интервале от 50 до 125°С.
5. Способ согласно любому из вариантов выполнения настоящего изобретения 1-4, где обработка согласно (b) проводится в течение периода времени в интервале от 6 до 20 ч.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1-5, где обработка согласно (b) проводится в ходе по меньшей мере 2 отдельных стадий, где между по меньшей мере 2 стадиями обработки, MWW сушат, предпочтительно при температуре в интервале от 100 до 150°С.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1-6, дополнительно включающий
(с) последующую обработку MWW, полученного на этапе (b), способом, включающим
(с. 1) отделение MWW от жидкой системы растворителей;
(с. 2) предпочтительно сушку отделенного MWW, предпочтительно посредством распылительной сушки;
(с. 3) необязательно кальцинирование MWW, полученного на этапе (с. 1) или (с. 2), предпочтительно при температурах в интервале от 500 до 700°С.
Что касается стадии (III), то предпочтительно подходящую исходную смесь, предпочтительно водную смесь, содержащую MWW и содержащий Ti, и предпочтительно содержащую по меньшей мере один подходящий микропоры-образующий агент, подвергают гидротермальной кристаллизации при автогенном давлении. Можно применять по меньшей мере один подходящий затравочный материал. В качестве подходящего предшественника, содержащего Ti, в качестве примера можно упомянуть тетраалкилортотитанаты, такие как тетрабутилортотитанат. В качестве подходящего микропоры-образующего агента в качестве примера можно упомянуть пиперидин, гексаметиленимин или смеси пиперидина и гексаметиленимина. Предпочтительно, время кристаллизации находится в интервале от 4 до 8 дней, более предпочтительно от 4 до 6 дней. В ходе гидротермального синтеза смесь для кристаллизации можно перемешивать. Температуры, применяемые во время кристаллизации, предпочтительно находятся в интервале от 160 до 200°С, более предпочтительно от 160 до 180°С. После гидротермального синтеза полученный кристаллический цеолитный материал TiMWW предпочтительно подходящим образом отделяют от маточного раствора. Возможны все способы отделения TiMWW от его маточного раствора. Эти методы включают, например, фильтрацию, ультрафильтрацию, диафильтрацию и методы центрифугирования или, например, процессы распылительной сушки и процессы грануляции распылением. Может применяться комбинация двух или более этих методов. Согласно настоящему изобретению TiMWW предпочтительно отделяют от его материнской жидкости путем фильтрации с получением осадка на фильтре, который предпочтительно подвергают промывке, предпочтительно водой. Затем осадок на фильтре, необязательно далее обработанный до получения подходящей суспензии, подвергают распылительной сушке или ультрафильтрации. Перед отделением TiMWW от маточного раствора можно увеличить содержание TiMWW в маточном растворе, концентрируя суспензию. Если применяется промывка, то предпочтительно продолжать процесс промывки до тех пор, пока промывная вода не будет иметь проводимость менее 1000 микросименс/см, более предпочтительно менее 900 микросименс/см, более предпочтительно менее 800 микросименс/см, более предпочтительно менее 700 микросименс/см. После отделения TiMWW от его маточного раствора, предпочтительно достигаемого путем фильтрации, и после промывки промытый осадок на фильтре, содержащий TiMWW, предпочтительно подвергают предварительной сушке, например, воздействуя на осадок на фильтре подходящим газовым потоком, предпочтительно потоком азота, в течение периода времени предпочтительно в интервале от 4 до 10 ч, более предпочтительно от 5 до 8 часов. Затем предварительно высушенный осадок на фильтре предпочтительно сушат при температурах в интервале от 100 до 300°С, более предпочтительно от 150 до 275°С, более предпочтительно от 200 до 250°С в подходящей атмосфере, такой как технический азот, воздух или обедненным воздухом, предпочтительно в воздухе или обедненном воздухе. Такая сушка может осуществляться, например, посредством распылительной сушки. После сушки TiMWW может подвергаться кальцинированию при температурах в интервале от 500 до 700°С, более предпочтительно от 550 до 675°С, более предпочтительно от 600 до 675°С в подходящей атмосфере, такой как технический азот, воздух или обедненным воздухом, предпочтительно в воздухе или обедненном воздухе. Предпочтительно кальцинирование не проводится согласно (III).
Предпочтительно, стадии (III) и (IV) осуществляются способом, предпочтительные стадии и условия которого определены в следующих вариантах выполнения настоящего изобретения 1-27 и при соответствующих зависимостях, как указано:
1. Способ получения содержащего титан цеолитного материала, имеющего MWW каркасную структуру, включающий
(a) обеспечение деборонированного кристаллического цеолитного материала MWW, полученного согласно стадии (II);
(b) включения титана в цеолитный материал, полученный согласно этапу (а), включающее
(b. 1) обеспечение водной смеси для синтеза, содержащей цеолитный материал, полученный на стадии (i), MWW матричное соединение и источник титана, где молярное отношение MWW матричного соединения относительно Si, вычисленного как SiO2 и содержащегося в цеолитном материале, полученном на этапе (а), находится в интервале от 0.5:1 до 1.4:1;
(b. 2) гидротермальный синтез титансодержащего цеолитного материала, имеющего MWW каркасную структуру, из водной смеси для синтеза, полученной на этапе (b. 1), с получением матричного раствора, содержащего титансодержащий цеолитный материал, имеющий MWW каркасную структуру;
(c) распылительную сушку матричного раствора, полученного на этапе (b. 2), содержащего титансодержащий цеолитный материал, имеющий MWW каркасную структуру.
2. Способ согласно варианту выполнения настоящего изобретения 1, где на этапе (b. l), MWW матричное соединение выбирают из группы, состоящей из пиперидина, гексаметиленимина, N,N,N,N’,N’,N’-гексаметил-l,5-пентадиаммониевого иона, 1,4-бис(N-метилпирролидиния)бутана, октилтриметиламмония гидроксида, гептилтриметиламмония гидроксида, гексилтриметиламмония гидроксида, и смеси двух или более из них, причем MWW матричное соединение предпочтительно представляет собой пиперидин.
3. Способ согласно варианту выполнения настоящего изобретения 1 или 2, где на этапе (b. 1), источник титана выбирают из группы, состоящей из тетрабутилортотитаната, тетраизопропилортотитаната, тетраэтилортотитаната, диоксида титана, тетрахлорида титана, трет-бутоксида титана, и смеси двух или более из них, где источник титана предпочтительно представляет собой тетрабутилортотитанат.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1-3, где в водной смеси для синтеза на этапе (b. 1), молярное отношение Ti, вычисленного как ТiO2 и содержащегося в источнике титана, относительно Si, вычисленного как SiO2 и содержащегося в цеолитном материале, имеющем молярное отношение В2O3: SiO2 не более 0.02:1, находится в интервале от 0.005:1 до 0.1:1, предпочтительно от 0.01:1 до 0.08:1, более предпочтительно от 0.02:1 до 0.06:1.
5. Способ согласно любому из вариантов выполнения настоящего изобретения 1-4, где в водной смеси для синтеза на этапе (b. 1), молярное отношение Н2O относительно Si, вычисленного как SiO2 и содержащегося в цеолитном материале, имеющем молярное отношение В2O3:SiO2 не более 0.02:1, находится в интервале от 8:1 до 20:1, предпочтительно от 10:1 до 18:1, более предпочтительно от 12:1 до 16:1.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1-5, где в водной смеси для синтеза на этапе (b. 1), молярное отношение MWW матричного соединения относительно Si, вычисленного как SiO2 и содержащегося в цеолитном материале, полученном на стадии (i), находится в интервале от 0.5:1 до 1.7:1, предпочтительно от 0.8:1 до 1.5:1, более предпочтительно от 1.0:1 до 1.3:1.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1-6, где на этапе (b. 2), гидротермальный синтез проводится при температуре в интервале от 80 до 250°С, предпочтительно от 120 до 200°С, более предпочтительно от 160 до 180°С.
8. Способ согласно любому из вариантов выполнения настоящего изобретения 1-7, где на этапе (b. 2), гидротермальный синтез проводится в течение периода времени в интервале от 10 до 100 ч, более предпочтительно от 20 до 80 ч, более предпочтительно от 40 до 60 ч.
9. Способ согласно любому из вариантов выполнения настоящего изобретения 1-8, где на этапе (b. 2), гидротермальный синтез проводится в закрытой системе при автогенном давлении.
10. Способ согласно любому из вариантов выполнения настоящего изобретения 1-9, где ни в ходе (b. 2), ни после (b. 2) и перед (с), титансодержащий цеолитный материал, имеющий MWW каркасную структуру, не отделяется от его матричного раствора.
11. Способ согласно любому из вариантов выполнения настоящего изобретения 1-10, где матричный раствор, подвергнутый этапу (с), содержащий титансодержащий цеолитный материал, имеющий MWW каркасную структуру, имеет содержание твердых веществ, необязательно после концентрирования или разбавления, в интервале от 5 до 25 мас. %, более предпочтительно от 10 до 20 мас. %, на основе общей массы матричного раствора, содержащего титансодержащий цеолитный материал.
12. Способ согласно любому из вариантов выполнения настоящего изобретения 1-11, где в ходе распылительной сушки на этапе (с), температура входа сушильного газа находится в интервале от 200 до 350°С, и температура выхода сушильного газа находится в интервале от 70 до 190°С.
13. Способ согласно любому из вариантов выполнения настоящего изобретения 1-12, где цеолитный материал, имеющий MWW каркасную структуру, полученный на этапе (с), имеет содержание Si в интервале от 30 до 40 мас. %, вычисленный как элементарный Si, где общее содержание органического углерода (ТОС) находится в интервале от 0 до 14 мас. %, и содержание Ti составляет от 2.1 до 2.8 мас. %, вычисленный как элементарный Ti, в каждом случае на основе общей массы цеолитного материала.
14. Способ согласно любому из вариантов выполнения настоящего изобретения 1-13, дополнительно включающий
(d) обработку титансодержащего цеолитного материала, имеющего MWW каркасную структуру, полученного на стадии (iii), водным раствором, имеющим рН не более 5.
15. Способ согласно варианту выполнения настоящего изобретения 14, где после (с) и до (d), высушенный распылением титансодержащий цеолитный материал, имеющий MWW каркасную структуру, полученный на этапе (с), не подвергается кальцинированию.
16. Способ согласно варианту выполнения настоящего изобретения 14 или 15, где на этапе (d), массовое отношение водного раствора относительно титансодержащего цеолитного материала, имеющего MWW каркасную структуру, находится в интервале от 10:1 до 30:1, предпочтительно от 15:1 до 25:1, более предпочтительно от 18:1 до 22:1.
17. Способ согласно любому из вариантов выполнения настоящего изобретения 14-16, где на этапе (d) водный раствор содержит неорганическую кислоту, предпочтительно выбранную из группы, состоящей из фосфорной кислоты, серной кислоты, соляной кислоты, азотной кислоты и смеси двух или более из них, причем водный раствор предпочтительно содержит азотную кислоту.
18. Способ согласно любому из вариантов выполнения настоящего изобретения 14-17, где на этапе (d), водный раствор имеет значение рН в интервале от 0 до 5, предпочтительно от 0 до 3, более предпочтительно от 0 до 2.
19. Способ согласно любому из вариантов выполнения настоящего изобретения 14-18, где на этапе (d), титан-содержащий цеолитный материал, имеющий MWW каркасную структуру, обрабатывают водным раствором при температуре в интервале от 50 до 175°С, предпочтительно от 70 до 125°С, более предпочтительно от 95 до 105°С.
20. Способ согласно любому из вариантов выполнения настоящего изобретения 14-19, где на этапе (d), титансодержащий цеолитный материал, имеющий MWW каркасную структуру, обрабатывают водным раствором в течение периода времени в интервале от 0.1 до 6 ч, предпочтительно от 0.3 до 2 ч, более предпочтительно от 0.5 до 1.5 ч.
21. Способ согласно любому из вариантов выполнения настоящего изобретения 14-20, где обработка согласно (d) проводится в закрытой системе при автогенном давлении.
22. Способ согласно любому из вариантов выполнения настоящего изобретения 14-21, дополнительно содержащий
(е) отделение титансодержащего цеолитного материала, имеющего MWW каркасную структуру, полученного на этапе (d), от водного раствора, необязательно с последующей промывкой титансодержащего цеолитного материала, имеющего MWW каркасную структуру.
23. Способ согласно варианту выполнения настоящего изобретения 22, где (е) включает сушку отделенного и необязательно промытого титансодержащего цеолитного материала, имеющего MWW каркасную структуру.
24. Способ согласно любому из вариантов выполнения настоящего изобретения 14-23, дополнительно включающий
(f) получение суспензии, предпочтительно водной суспензии, содержащей титансодержащий цеолитный материал, имеющий MWW каркасную структуру, полученный на этапе (d), предпочтительно на этапе (е), причем указанная суспензия имеет содержание твердых веществ предпочтительно в интервале от 5 до 25 мас. %, более предпочтительно от 10 до 20 мас. %, на основе общей массы суспензии, и воздействие на суспензию распылительной сушкой.
25. Способ согласно варианту выполнения настоящего изобретения 24, где в ходе распылительной сушки температура входа сушильного газа находится в интервале от 200 до 330°С, и температура выхода сушильного газа находится в интервале от 120 до 180°С.
26. Способ согласно любому из вариантов выполнения настоящего изобретения 14-25, дополнительно включающий
(g) кальцинирование титан-содержащего цеолитного материала, имеющего MWW каркасную структуру, полученного на этапе (d), предпочтительно на этапе (е), более предпочтительно на этапе (f), где кальцинирование предпочтительно проводят при температуре в интервале от 400 до 800°С, более предпочтительно от 600 до 700°С.
27. Способ согласно варианту выполнения настоящего изобретения 26, где на стадии (vii), кальцинирование проводится в непрерывном режиме, предпочтительно со скоростью в интервале от 0.2 до 2.0 кг цеолитного материала в час, более предпочтительно от 0.5 до 1.5 кг цеолитного материала в час.
Согласно стадии (V), TiMWW, полученный согласно стадии (IV), подвергают соответствующей обработке Zn для получения ZnTiMWW, используемого для получения суспензии согласно (а). В общем, что касается (V), особых ограничений не существует, при условии, что может быть получен вышеописанный предпочтительный ZnTiMWW с предпочтительным содержанием Zn и Ti. Наиболее предпочтительно, стадия (V) включает по меньшей мере одну подходящую стадию пропитки, более предпочтительно по меньшей мере одну стадию влажной пропитки. Что касается этой стадии пропитки, предпочтительно проводить контакт TiMWW, предпочтительно полученного согласно (IV), с по меньшей мере одним подходящим Zn-содержащим предшественником в по меньшей мере одном подходящем растворителе (влажная пропитка), наиболее предпочтительно воде. В качестве подходящего Zn-содержащего предшественника особенно предпочтительны водорастворимые соли Zn, особенно предпочтительными являются дигидрат ацетата цинка. Кроме того, предпочтительно получить раствор Zn-содержащего предшественника, предпочтительно водный раствор, и суспендировать TiMWW в этом растворе. Далее предпочтительно пропитка проводится при повышенных температурах по сравнению с комнатной температурой, предпочтительно в интервале от 75 до 125°С, более предпочтительно от 85 до 115°С, в течение периода времени предпочтительно в интервале от 3,5 до 5 ч, более предпочтительно от 3 до 6 часов. Предпочтительным является перемешивание суспензии во время пропитки. После пропитки полученный ZnTiMWW предпочтительно отделяют от суспензии. Возможны все способы отделения ZnTiMWW от суспензии. Особенно предпочтительно, отделение проводится посредством фильтрации, ультрафильтрации, диафильтрации или центрифугирования. Может применяться комбинация двух или более этих методов. Согласно настоящему изобретению ZnTiMWW предпочтительно отделяют от суспензии фильтрованием с получением осадка на фильтре, который предпочтительно подвергают промывке, предпочтительно водой. Если промывка применяется, может быть предпочтительнее продолжать промывку до тех пор, пока промывочная воды не будет иметь проводимость менее 1000 микросименс/см, более предпочтительно менее 900 микросименс/см, более предпочтительно менее 800 микросименс/см, более предпочтительно менее 700 микросименс/см. Затем промытый осадок на фильтре подвергают предварительной сушке, например, подвергая осадок на фильтре подходящему потоку газа, предпочтительно потоку азота, в течение периода времени предпочтительно в интервале от 5 до 15 ч, более предпочтительно от 8 до 12 ч.
Если TiMWW или ZnTiMWW применяется в качестве каталитически активного материала согласно настоящему изобретению, органический растворитель предпочтительно представляет собой ацетонитрил.
В общем, предпочтительный ZnTiMWW катализатор может быть получен согласно конкретной методике, как описано в WO 2013/117536 А2, которая включена в настоящую заявку в полном объеме посредством ссылки.
TS-1 катализатор
Согласно настоящему изобретению, катализатор силикат титана-1, предпочтительно катализатор силикат титана-1 в виде неподвижного слоя, может применяться в качестве катализатора. Силикат титана-1 представляет собой микропористый цеолит со структурой типа MFI, который не содержит алюминий, и в котором Si(IV) в решетке силиката частично замещен на титан, как например Ti(IV). Термин “микропоры”, как применяется в контексте настоящего изобретения, относится к порам, имеющим размер поры менее 2 нм, определенный согласно DIN 66134.
Цеолит сииликат титана-1 катализатора может в принципе быть получен любым возможным методом. Как правило, синтез по меньшей мере одного содержащего титан цеолита согласно настоящему проводится в гидротермальных системах, включающих комбинацию активного источника оксида кремния и источника титана, такого как оксид титана, с по меньшей мере одним матричным соединением, способных образовывать желаемый содержащий титан цеолит в водной суспензии, например, в основной суспензии. Как правило, используются органические шаблоны. Предпочтительно, синтез проводится при повышенных температурах, например, при температурах в интервале от 150 до 200°С, предпочтительно от 160 до 180°С. В принципе, любое подходящее соединение может быть использовано в качестве источника оксида кремния. Типичные источники оксида кремния (SiO2) включают силикаты, гидрогель диоксида кремния, кремниевую кислоту, коллоидный диоксид кремния, пирогенный диоксид кремния, тетраалкоксисиланы, гидроксиды кремния, осажденный диоксид кремния и глины. Могут использоваться как так называемый диоксид кремния, полученный «влажной» технологией, так и диоксид кремния, полученный «сухой» технологией. В этих случаях диоксид кремния особенно предпочтительно аморфен, где размер частиц диоксида кремния находится, например, в интервале от 5 до 100 нм, а площадь поверхности частиц диоксида кремния находится, например, в интервале от 50 До 500 м2/г. Коллоидный диоксид кремния, среди прочего, коммерчески доступен как Ludox®, Syton®, Nalco®, или Snowtex®. Полученный “влажной” технологией диоксид кремния, среди прочего, коммерчески доступен как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Полученный “сухой” технологией диоксид кремния, среди прочего, коммерчески доступен как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. В рамках настоящего изобретения также предполагается использовать соединение-предшественник диоксида кремния в качестве источника оксида кремния. Например, тетраалкоксисиланы, такие как, например, тетраэтоксисилан или тетрапропоксисилан, можно упомянуть в качестве соединения-предшественника.
В качестве матрицы можно использовать любую матрицу, подходящую для обеспечения желательной цеолитной структуры MFI. В частности, используют гидроксид тетрапропиламмония, более предпочтительно тетра-н-пропиламмонийгидроксид. В предпочтительном варианте выполнения способа согласно изобретению, по меньшей мере один порообразующий агент удаляется в более поздней стадии кальцинирования, как описано ниже.
Как правило, синтез силиката титана-1 проходит в автоклаве периодическим образом, так что реакционная суспензия подвергается аутогенному давлению в течение нескольких часов или нескольких дней до получения цеолита силикат титана-1. Согласно предпочтительному варианту осуществления настоящего изобретения синтез в общем протекает при повышенных температурах, где температуры во время стадии гидротермальной кристаллизации обычно находятся в интервале от 150 до 200°С, предпочтительно в интервале от 160 до 180°С. Обычно реакция протекает в течение периода времени в диапазоне от нескольких часов до нескольких дней, предпочтительно в течение периода времени в интервале от 12 ч до 48 ч, более предпочтительно от 20 до 30 ч. Кроме того, возможно добавлять затравочные кристаллы к партиям синтеза.
Согласно варианту выполнения настоящего изобретения, полученный кристаллический силикат титана-1 отделяют от реакционной суспензии, т.е. от матричного раствора, необязательно промывают и сушат.
Можно использовать все методы, известные для отделения кристаллического силиката титана-1 от суспензии. Среди прочего следует упомянуть фильтрацию, ультрафильтрацию, диафильтрацию и методы центрифугирования.
В случае промывки кристаллического силиката титана-1 стадия промывки может быть проведена с использованием любого подходящего промывочного средства, такого как, например, вода, спирты, такие как, например, метанол, этанол или метанол и пропанол, или этанол и пропанол, или метанол и этанол и пропанол, или смеси воды и по меньшей мере одного спирта, такие как, например, вода и этанол или вода и метанол, или вода и этанол, или вода и пропанол, или вода и метанол и этанол, или вода и метанол и пропанол, или вода и этанол и пропанол или вода и этанол и метанол и пропанол. В качестве промывочного средства используются вода или смесь воды и по меньшей мере одного спирта, предпочтительно воды и этанола.
Сушку кристаллического силиката титана-1 осуществляют при температурах, в общем, в интервале от 80 до 160°С, предпочтительно от 90 до 145°С, особенно предпочтительно от 100 до 130°С.
Вместо вышеупомянутых способов разделения, таких как, среди прочего, фильтрации, ультрафильтрации, диафильтрации и способов центрифугирования, суспензия может, согласно альтернативному варианту выполнения настоящего изобретения, также быть подвергнута распылительным методам, например, грануляции распылением и распылительной сушке.
Если отделение кристаллического силиката титана-1 проводится распылительным методом, стадия отделения и стадия сушки могут быть объединены в одну стадию. В этом случае может быть использована либо реакционная суспензия как таковая, либо концентрированная реакционная суспензия. Кроме того, можно добавить подходящую добавку, как например, по меньшей мере одно подходящее связующее и/или по меньшей мере один порообразующий агент к суспензии, либо к реакционной суспензии как таковой, либо к концентрированной суспензии, до распылительной сушки или гранулирования распылением. Подходящие связующие описаны подробно ниже. В качестве порообразующего агента могут быть использованы все порообразующие агенты, описанные выше. В случае, если суспензия высушивается распылением, порообразующий агент - если он добавлен - может быть добавлен двумя способами. Во-первых, порообразующий агент может быть добавлен в реакционную смесь до распылительной сушки. Однако также можно добавить часть порообразующего агента в реакционную смесь до распылительной сушки, при этом оставшуюся часть порообразующего агента добавляют к высушенному распылением материалу.
В случае, когда суспензию сначала концентрируют для повышения содержания силиката титана-1 в суспензии, можно достичь концентрирования, например, путем выпаривания, например, выпаривания при пониженном давлении или путем фильтрации в перекрестном потоке. Подобным образом, суспензию можно концентрировать путем разделения указанной суспензии на две фракции, где твердое вещество, содержащееся в одной из двух фракций, отделяют фильтрованием, диафильтрацией, ультрафильтрацией или методами центрифугирования и суспендируют после необязательной стадии промывки и/или стадии сушки в другой фракции суспензии. Полученную таким образом концентрированную суспензию затем можно подвергать распылительным способам, например, гранулированию распылением и распылительной сушке.
Согласно альтернативному варианту выполнения настоящего изобретения, концентрирование достигается путем отделения по меньшей мере одного содержащего титан цеолита от суспензии и повторного суспендирования содержащего титан цеолита, необязательно вместе с по меньшей мере одной подходящей добавкой, как уже было описано выше, где содержащий титан цеолит может быть подвергнут по меньшей мере одной стадии промывки и/или по меньшей мере одной стадии сушки перед повторным суспендированием. Затем повторно суспендированный содержащий титан цеолит можно использовать для распылительных способов, предпочтительно распылительной сушки.
Сушка распылением является прямым методом сушки взвесей, суспензий или растворов путем подачи хорошо диспергированной взвеси, суспензии или раствора текучая среда - твердое вещество, часто необязательно содержащих связующий агент, в распылитель с последующей термической сушкой в потоке горячего воздуха. Распылитель может быть одного из нескольких типов. Наиболее распространенным является барабанное распыление, в котором применяется высокоскоростное вращение колеса или диска для разбивания суспензии на капли, которые отлетают с колеса в камеру и подвергаются термической сушке до того, как удариться о стенки камеры. Распыление также может быть достигнуто с применением форсунки для однокомпонентной текучей среды, которая работает на основе гидростатического давления, проталкивая суспензию через небольшую форсунку. Также применяют форсунки для многокомпонентных текучих сред, где давление газа применяют для проталкивания суспензии через форсунку. Распыленный материал, полученный сушкой распылением и гранулированием распылением, как например, сушка в псевдоожиженном слое, может содержать твердое вещество и/или полые сферы, и может практически полностью состоять из таких сфер, которые имеют, например, диаметр в интервале от 5 до 500 мкм или от 5 до 300 мкм. Могут применяться однокомпонентные или мультикомпонентные форсунки. Также возможно использование вращающегося распылителя. Возможные температуры на входе для используемого газа-носителя составляют, например, от 200 до 600°С, предпочтительно от 300 до 500°С. Температура на выходе газа-носителя составляет, например, от 50 до 200°С. Воздух, обедненный воздух или кислородно-азотные смеси с содержанием кислорода вплоть до 10% по объему, предпочтительно, вплоть до 5% по объему, более предпочтительно, менее 5% по объему, например, до 2% по объему, могут быть указаны в качестве газа-носителя. Распылительные способы могут проводиться в противотоке или в прямотоке.
Предпочтительно, силикат титана-1 отделяют от реакционной суспензии стандартными фильтрацией или центрифугированием, необязательно сушат и/или кальцинируют, и повторно суспендируют, предпочтительно в смеси, предпочтительно водной смеси по меньшей мере одного связующего материала и/или одного порообразующего агента. Полеченную суспензию затем предпочтительно подвергают распылительной сушке или грануляции распылением. Полученный распыленный материал можно подвергнуть дополнительной стадии промывки, причем указанная стадия промывки осуществляется, как описано выше. Необязательно промытый распыленный материал затем сушат и кальцинируют, где сушка и кальцинирование предпочтительно осуществляются, как описано выше.
Согласно альтернативному варианту выполнения настоящего изобретения, кристаллизация силиката титана-1 не осуществляется до того, как вышеописанная суспензия была высушена распылением. Поэтому, образуется первая суспензия, содержащая источник диоксида кремния, предпочтительно диоксид кремния, источник оксида титана, и матричное соединение, способная образовывать силикат титана-1. Затем суспензию подвергают распылительной сушке, где затем, необязательно добавляется дополнительный порообразующий агент к высушенному распылительной сушкой силикату титана-1.
Высушенный распылением силикат титана-1, полученный согласно вышеуказанным способам, может, необязательно, подвергаться по меньшей мере одному процессу промывки. Если по меньшей мере один процесс промывки проводится, предпочтительно по меньшей мере одна стадия сушки и/или по меньшей мере одна стадия кальцинирования следует.
Силикат титана-1, необязательно полученный распылительными методами, может далее подвергаться по меньшей мере одной стадии кальцинирования, которая проводится согласно предпочтительному варианту выполнения настоящего изобретения после стадии сушки, или вместо стадии сушки. По меньшей мере одна стадия кальцинирования проводится при температурах в общем в интервале от 350 до 750°С, предпочтительно от 400 до 700°С, особенно предпочтительно от 450 до 650°С.
Кальцинирование силиката титана-1 может осуществляться в атмосфере подходящего газа, где воздух и/или обедненный воздух является предпочтительным. Кроме того, кальцинирование предпочтительно осуществляется в муфельной печи, вращающейся печи и/или ленточной печи для обжига, где кальцинирование в общем осуществляется в течение одного часа или более, например, в течение периода времени в интервале от 1 до 24 ч или в интервале от 4 до 12 ч. В способе согласно настоящему изобретению соответственно возможно, например, кальцинировать силикат титана-1 один раз, два раза или более часто в течение в каждом случае по меньшей мере 1 часа, как например, в каждом случае в интервале от 4 до 12 ч, предпочтительно от 4 до 8 часов, где возможно, чтобы температуры во время стадии кальцинирования оставались постоянными или менялись непрерывно или прерывисто. Если кальцинирование производится дважды или более часто, температуры кальцинирования на отдельных стадиях могут быть разными или идентичными.
Таким образом, предпочтительный вариант выполнения настоящего изобретения относится к способу, как описано выше, где силикат титана-1, отделенный от суспензии, например, фильтрацией или распылительной сушкой, промывают подходящим промывочным веществом, и затем подвергают по меньшей мере одной стадии сушки. Сушка осуществляется при температурах, в общем, в интервале от 80 до 160°С, предпочтительно от 90 до 145°С, особенно предпочтительно от 100 до 130°С. Наиболее предпочтительно, после сушки, проводится стадия кальцинирования. Эта стадия проводится при температурах в общем в интервале от 350 до 750°С, предпочтительно от 400 до 700°С, особенно предпочтительно от 450 до 650°С.
Силикат титана-1, полученный, как описано выше, в общем может непосредственно применяться в качестве катализатора на стадиях (i) и (iii). Однако особенно предпочтительно применять катализатор в виде неподвижного слоя на обеих стадиях (i) и (iii), т.е. не применять кристаллический цеолитный материал per se в качестве катализатора, а применять кристаллический материал, обработанный с получением формованного изделия, содержащего силикат титана-1. Таким образом, согласно предпочтительному варианту выполнения настоящего изобретения, формованное изделие, содержащее силикат титана-1, как описано выше, применяется в качестве катализатора.
В общем, в случае, когда формованное изделие применяется в качестве катализатора, указанный катализатор может содержать все возможные дополнительные соединения в дополнение к силикату титана-1 согласно настоящему изобретению, например, среди прочего, по меньшей мере одно связующее и/или по меньшей мере один порообразующий агент. Кроме того, катализатор может содержать по меньшей мере один пастирующий агент вместо по меньшей мере одного связующего и/или по меньшей мере одного порообразующего агента или в дополнение к по меньшей мере одному связующему и/или по меньшей мере одному порообразующему агенту.
В качестве связующего являются подходящими все соединения, которые обеспечивают адгезию и/или когезию между силикатом титана-1, подлежащим формованию, которые выходят за пределы физической сорбции, которая может присутствовать без связующего. Примерами таких связующих являются оксиды металлов, такие как, например, SiO2, Аl2O3, TiO2, ZrO2 или MgO или глины или смеси двух или более из этих соединений. Глинистые минералы и природные или синтетически полученные оксиды алюминия, такие как, например, альфа-, бета-, гамма-, дельта-, эта-, каппа-, хи- или тета-оксид алюминия и их неорганические или металлоорганические соединения-предшественники, такие как, например, гиббсит, байерит, бемит или псевдобемит, или триалкоксиалюминаты, такие как, например, триизопропилат алюминия, особенно предпочтительны в качестве связующих А12O3. Другими предпочтительными связующими являются амфифильные соединения, имеющие полярную и неполярную часть и графит.
Далее связующие представляют собой, например, глины, такие как, например, монтмориллониты, каолины, метакаолин, гекторит, бентониты, галлоизиты, дикиты, накриты или анакситы.
Эти связующие могут использоваться как таковые. Также в рамках настоящего изобретения можно использовать соединения, из которых связующее образуется за по меньшей мере одну дополнительную стадию при получении формованных изделий. Примерами таких предшественников связующего являются тетраалкоксисиланы, тетраалкоксититанаты, тетраалкоксицирконаты или смесь двух или более различных тетраалкоксисиланов или смеси двух или более различных тетраалкоксититанатов или смеси двух или более различных тетраалкоксицирконатов или смеси по меньшей мере одного тетраалкоксисилана и по меньшей мере одного тетраалкоксититаната или по меньшей мере одного тетраалкоксисилана и по меньшей мере одного тетраалкоксицирконата или по меньшей мере одного тетраалкоксититаната и по меньшей мере одного тетраалкоксицирконата или смесь по меньшей мере одного тетраалкоксисилана и по меньшей мере одного тетраалкоксититаната и по меньшей мере одного тетраалкоксицирконата.
В контексте настоящего изобретения связующие, которые либо полностью, либо частично содержат SiO2, или которые представляют собой предшественник SiO2, из которого SiO2 образуется за по меньшей мере одну дополнительную стадию, являются особенно предпочтительными. В этом контексте как так называемый диоксид кремния, полученный «влажной» технологией, так и диоксид кремния, полученный «сухой» технологией. В этих случаях диоксид кремния особенно предпочтительно аморфен, где размер частиц диоксида кремния находится, например, в интервале от 5 до 100 нм, а площадь поверхности частиц диоксида кремния находится, например, в интервале от 50 До 500 м2/г
Коллоидный диоксид кремния, предпочтительно в виде щелочного и/или аммиачного раствора, более предпочтительно в виде аммиачного раствора, коммерчески доступен как Ludox®, Syton®, Nalco® or Snowtex®. Диоксид кремния, полученный “влажной” технологией является коммерчески доступным, среди прочего, например, как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Диоксид кремния, полученный “сухой” технологией является коммерчески доступным, среди прочего, например, как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. Среди прочего, аммиачный раствор коллоидного диоксида кремния является предпочтительным в настоящем изобретении. Соответственно, настоящее изобретение также описывает катализатор, содержащий формованное изделие, как описано выше, причем указанное формованное изделие содержит силикат титана-1, как описано выше, и дополнительно SiO2 в качестве связующего материала, где связующее, используемое согласно (I), является связующим, содержащим или образующим SiO2. В общем, содержащий титан цеолит также может быть формован без использования связующего.
Таким образом, настоящее изобретение также относится к способу, в котором на стадиях (i) и (iii), катализатор на основе силиката титана-1 получают путем формования силиката титана-1 с получением формованного изделия, содержащего силикат титана-1 и предпочтительно по меньшей мере одно связующее, в частности связующее на основе диоксида кремния.
При желании, по меньшей мере один порообразующий агент можно добавить в смесь силиката титана-1 и по меньшей мере одного связующего или по меньшей мере предшественника связующего, для дальнейшей обработки и для формования катализатора в виде формованного тела, который будет использоваться в качестве катализатора в виде неподвижного слоя. Порообразующими агентами, которые могут быть использованы, являются все соединения, которые в отношении формованного изделия обеспечивают удельный размер пор и/или удельное распределение пор по размерам и/или определенные объемы пор. В частности, порообразующие агенты, которые обеспечивают в отношении формованного продукта микропоры и/или микропоры, в частности мезопоры и микропоры.
Таким образом, настоящее изобретение также относится к способу, в котором на стадиях (i) и (iii), катализатор на основе силиката титана-1 получают путем формования силиката титана-1 с получением формованного изделия, содержащего силикат титана-1 и предпочтительно по меньшей мере одно связующее, в частности связующее на основе диоксида кремния, причем формованное изделие в частности имеет микропоры и мезопоры.
Что касается примеров для порообразующих агентов, которые могут быть использованы, делается ссылка на порообразующие агенты, уже упомянутые выше. Предпочтительно, порообразующие агенты, используемые в способе формования по изобретению, представляют собой полимеры, которые являются диспергируемыми, суспендируемыми или эмульгируемыми в воде или в смеси водных растворителей. Особенно предпочтительными полимерами являются полимерные виниловые соединения, такие как, например, полиалкиленоксиды, такие как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры, углеводы, такие как, например, целлюлоза или производные целлюлозы, такие как, например, метилцеллюлоза, сахара или натуральные волокна. Другими подходящими порообразующими агентами являются, например, пульпа или графит.
Если желательно, чтобы распределение пор по размеру достигалось, можно использовать смесь двух или более порообразующих агентов. В особенно предпочтительном варианте осуществления способа согласно изобретению, как описано ниже, порообразующие агенты удаляют путем кальцинирования с получением пористого катализатора в виде формованного тела. Предпочтительно, порообразующие агенты, которые обеспечивают мезопоры и/или микропоры, особенно предпочтительно мезопоры, добавляют к смеси по меньшей мере одного связующего и силиката титана-1 для формования силиката титана-1. В общем, силикат титана-1 также может быть сформирован с получением катализатора в виде формованного тела без использования порообразующего агента.
Помимо связующего необязательно порообразующего агента также возможно добавлять дополнительные компоненты, например, по меньшей мере один пастирующий агент, к смеси, которая формуется с получением катализатора в виде формованного тела.
Если по меньшей мере используется один приклеивающий агент, указанный приклеивающий агент используется либо вместо, либо в дополнение к по меньшей мере одному порообразующему агенту. В частности, в качестве приклеивающего агента могут быть использованы соединения, которые также действуют как порообразующие агенты. Приклеивающими агентами, которые могут быть использованы, являются все соединения, которые, как известно, подходят для этой цели. Это предпочтительно органические, в частности гидрофильные полимеры, такие как, например, целлюлоза, производные целлюлозы, такие как, например, метилцеллюлоза и крахмал, такие как, например, картофельный крахмал, обойная штукатурка, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутен или политетрагидрофуран. Использование воды, спиртов или гликолей или их смесей, таких как смеси воды и спирта, или воды и гликоля, такие как, например, вода и метанол, или вода и этанол, или вода и пропанол, или вода и пропиленгликоль, в качестве пастирующих агентов может быть упомянуто. Предпочтительно, в качестве приклеивающего агента используют целлюлозу, производные целлюлозы, воду и смеси двух или более этих соединений, такие как вода и целлюлоза или вода и производные целлюлозы. В особенно предпочтительном варианте осуществления способа согласно изобретению по меньшей мере один приклеивающий агент удаляют путем кальцинирования, как дополнительно описано ниже, для получения формованного изделия.
Согласно еще одному варианту выполнения настоящего изобретения, по меньшей мере одна кислотная добавка может быть добавлена к смеси, которая формуется с получением формованного изделия. Если используется кислотная добавка, предпочтительными являются органические кислотные соединения, которые могут быть удалены путем кальцинирования. В этом контексте могут быть упомянуты карбоновые кислоты, такие как, например, муравьиная кислота, щавелевая кислота и/или лимонная кислота. Также возможно использовать два или более этих кислотных соединений.
Порядок добавления компонентов к смеси, которая формуется с получением формованного изделия, не является критическим. Если используется, например, комбинация связующего, порообразующего агента, пастирующего агента и необязательно по меньшей мере одного кислотного соединения, можно как сначала добавить по меньшей мере одно связующее, а затем по меньшей мере один порообразующий агент, по меньшей мере одно кислотное соединение и, наконец, по меньшей мере один приклеивающий агент, так и поменять последовательность в отношении по меньшей мере одного связующего, по меньшей мере одного порообразующего агента, по меньшей мере один кислотного соединения и по меньшей мере один приклеивающего агента.
После добавления по меньшей мере одного связующего и/или по меньшей мере одного пастирующего агента и/или по меньшей мере одного порообразующего агента и/или по меньшей мере одной кислотной добавки к смеси, содержащей силикат титана-1, смесь обычно гомогенизируют от 10 до 180 минут. В частности, для гомогенизации особенно предпочтительны месильные машины, бегуны или экструдеры. Смесь предпочтительно замешивают. В промышленном масштабе измельчение в бегунах является предпочтительным для гомогенизации. Гомогенизация, как правило, осуществляется при температурах в интервале от около 10°С до точки кипения приклеивающего агента и атмосферном давлении или слегка надатмосферном давлении. Необязательно, по меньшей мере одно из соединений, описанных выше, можно добавить. Смесь, полученную таким образом, гомогенизируют, предпочтительно замешивают, до образования экструдируемого пластического материала.
Затем гомогенизированную смесь формуют для получения формованного изделия. Могут быть использованы все известные способы формования, такие как экструзия, распылительная сушка, гранулирование распылением, брикетирование, то есть механическое сжатие с добавлением или без добавления дополнительного связующего, или гранулирование, то есть уплотнение круговыми и/или вращательными движениями.
Предпочтительными способами формования являются способы, в которых используются обычные экструдеры для формования смеси, содержащей силикат титана-1. Таким образом, например, получают экструдаты диаметром от 1 до 10 мм и предпочтительно от 2 до 5 мм. В дополнение к использованию экструдера экструзионный пресс также может быть использован для получения формованных изделий. Форма формованных изделий, изготовленных согласно изобретению, может быть выбрана по желанию. В частности, возможны, среди прочего, сферы, овальные формы, цилиндры или таблетки. Аналогичным образом могут быть упомянуты полые структуры, например, полые цилиндры или сотовые структуры или также звездообразные геометрические формы.
Формование может происходить при атмосферном давлении или при давлении выше атмосферного, например, в диапазоне давлений от 1 бар до нескольких сотен бар. Кроме того, уплотнение может иметь место при температуре окружающей среды или при температуре выше, чем температура окружающей среды, например, в диапазоне температур от 20 до 300°С. Если сушка и/или кальцинирование являются частью стадии формования, возможны температуры до 600°С. Наконец, уплотнение может происходить в атмосфере окружающей среды или в контролируемой атмосфере. Контролируемые атмосферы представляют собой, например, атмосферы инертного газа, восстанавливающие атмосферы и/или окислительные атмосферы.
Стадия формования предпочтительно сопровождается по меньшей мере одной стадией сушки. Эта по меньшей мере одна стадия сушки проводится при температурах в диапазоне в общем от 80 до 160°С, предпочтительно от 90 до 145°С и особенно предпочтительно от 100 до 130°С, как правило в течение 6 ч или более, например, в интервале от 6 до 24 ч. Однако в зависимости от содержания влаги в материале, подлежащем сушке, также возможны более короткие времена сушки, такие как, например, около 1, 2, 3, 4 или 5 часов.
До и/или после стадии сушки, предпочтительно полученный экструдат может, например, быть измельчен. Предпочтительно, таким образом, получают гранулы или стружки, имеющие диаметр частиц от 0,1 до 5 мм, в частности от 0,5 до 2 мм.
Согласно предпочтительному варианту выполнения настоящего изобретения, сушка формованных изделий, соответственно, предпочтительно сопровождается по меньшей мере одной стадией кальцинирования. Кальцинирование проводится при температурах в общем в интервале от 350 до 750°С, предпочтительно от 400 до 700°С, особенно предпочтительно от 450 до 650°С. Кальцинирование может быть осуществлено в любой подходящей газовой атмосфере, где предпочтительным является воздух и/или обедненный воздух. Кроме того, кальцинирование предпочтительно проводят в муфельной печи, вращающейся печи и/или ленточной печи для обжига, где продолжительность кальцинирования обычно составляет 1 час или более, например, в интервале от 1 до 24 ч или в интервале от 3 до 12 ч. В способе согласно настоящему изобретению соответственно возможно, например, кальцинировать катализатор в виде формованного тела один раз, два раза или более часто в течение в каждом случае по меньшей мере 1 часа, как например, в каждом случае в интервале от 3 до 12 ч, где возможно, чтобы температуры во время стадии кальцинирования оставались постоянными или менялись непрерывно или прерывисто. Если кальцинирование производится дважды или более часто, температуры кальцинирования на отдельных стадиях могут быть разными или идентичными.
Согласно особенно предпочтительному варианту выполнения настоящего изобретения, катализатор в виде формованного тела подвергают гидротермальной обработке. Гидротермальная обработка может быть проведена с использованием любого подходящего метода, известного специалистам в данной области техники. Таким образом, катализатор или формованный катализатор, в общем, контактирует с водой или парами воды. Как правило, указанная гидротермальная обработка проводится путем загрузки катализатора или согласно изобретению вместе с водой в автоклав, нагревания суспензии до температуры в интервале от 100 до 200°С, предпочтительно в интервале от 120 до 150°С при давлении в интервале от 1,5 до 5 бар, предпочтительно в интервале от 2 до 3 бар, в течение периода времени в интервале от 1 до 48 часов, предпочтительно в интервале от 24 до 48 часов. Как правило, по меньшей мере следует одна стадия промывки, предпочтительно с водой в качестве моющего средства. После обработки водой катализатор сушат и/или кальцинируют, где сушка и кальцинирование проводятся, как уже описано выше. Согласно предпочтительному варианту выполнения настоящего изобретения гидротермальная обработка проводится путем перемешивания катализатора в виде формованного тела в автоклаве, причем скорость перемешивания доводится до скорости перемешивания, так чтобы избежать истирания, насколько это возможно. Однако, если катализатор используется в форме цилиндрических экструдатов, для достижения цилиндрических экструдатов с закругленными краями требуется некоторое истирание. С такими экструдатами, имеющими закругленные края, может быть достигнута более высокая объемная плотность, например, для использования экструдатов в качестве катализатора в виде неподвижного слоя в трубчатом реакторе R1 и/или в реакторе шахтного типа R2. Кроме того, пылеобразование указанных катализаторов в процессе эпоксидирования на стадиях (i) и (iii) снижается.
Кроме того, в процессе эпоксидирования согласно настоящему изобретению, применяется катализатор силикат титана - 1, как описано выше, имеющий микропоры м мезопоры, содержащий от 49.5 до 80%, предпочтительно от 69.5 до 80 мас. % силиката титана-1, на основе общей массы катализатора, и от 19.5 до 50%, предпочтительно от 19.5 до 30 мас. % по меньшей мере одного связующего, предпочтительно связующего на основе диоксида кремния, на основе общей массы катализатора в виде формованного тела.
Если TS-1 применяется в качестве каталитически активного материала согласно настоящему изобретению, органическим растворителем предпочтительно является метанол.
Этап (а2)
Согласно (а2), поток поступающего материала согласно (al) подвергают в реакторе условиям эпоксидирования в присутствии катализатора, и получают реакционную смесь, содержащую пропиленоксид и органический растворитель.
Реактор, используемый на этапе (а2), может работать в изотермическом или адиабатическом режиме, где предпочтительно, что реактор на этапе (а2) является изотермическим реактором.
Удобно, что скорость конверсии исходных материалов может регулироваться путем регулирования температуры, давления, WHSV исходных материалов и тому подобного. В качестве примера, температуру реакции можно регулировать так, чтобы по меньшей мере превращалось 90% эпоксидирующего средства. Количество исходного материала, присутствующего в реакционной смеси до и после реакции эпоксидирования, может быть проанализировано с помощью любого подходящего способа, например, хроматографии.
В общем, непрерывная реакция эпоксидирования на этапе (а2) может быть проведена в любом подходящем сосуде или реакторе. Предпочтительно, реакция на этапе (а2) проводится в по меньшей мере одном непрерывно работающем реакторе, таком как трубчатый реактор или реактор с пучком труб, который предпочтительно содержит по меньшей мере одну охлаждающую рубашку, окружающей по меньшей мере одну трубу, где температуру реакционной смеси контролируют изотермически с использованием теплоносителя, предпочтительно путем пропускания теплоносителя через рубашку реактора. Если реакция на этапе (а2) проводится в таком реакторе, содержащем по меньшей мере одну охлаждающую рубашку, термин «температура реакции эпоксидирования», как используется здесь, определяется как температура теплоносителя до контроля температуры реакции смеси, предпочтительно как температура теплоносителя на входе в рубашку изотермического реактора.
Предпочтительно, по меньшей мере один из реакторов, в которых проводится реакция согласно (а2), является трубчатым реактором или реактором с пучком труб.
Катализатор, содержащий цеолит, содержащий титан, может применяться в любой подходящей форме, описанной выше, включая порошок, микропорошок, предпочтительно распылительный порошок, как например формованное изделие, содержащее порошок, или как, например, формованное изделие, содержащее микропорошок, предпочтительно распылительный порошок. Предпочтительно, катализатор, содержащий цеолит, содержащий титан, применяется в виде формованного изделия, содержащего порошок или микропорошок, предпочтительно распылительный порошок, более предпочтительно в виде формованного изделия, содержащего микропорошок, предпочтительно распылительный порошок.
Катализатор, используемый на этапе (а2) согласно настоящему изобретению, может быть расположен в реакторе любым возможным способом. Предпочтительно, катализатор расположен в виде псевдоожиженного слоя или как неподвижного слоя, более предпочтительно в виде неподвижного слоя.
Предпочтительно, условия эпоксидирования на этапе (а2) содержат температуру реакции эпоксидирования в интервале от 20 до 100°С, более предпочтительно от 25 до 80°С, более предпочтительно от 25 до 60°С, более предпочтительно от 30 до 60°С.
Предпочтительно, условия эпоксидирования на этапе (а2) содержат давление реакции эпоксидирования в интервале от 5 до 100 бар, более предпочтительно от 10 до 32 бар, более предпочтительно от 15 до 25 бар, где давление реакции эпоксидирования определяется как давление на выходе изотермического реактора
Таким образом, условия эпоксидирования на этапе (а2) предпочтительно содержат температуру реакции эпоксидирования в интервале от 20 до 100°С, и давление реакции эпоксидирования находится в интервале от 5 до 100 бар, более предпочтительно температура реакции эпоксидирования находится в интервале от 25 до 60°С, и давление реакции эпоксидирования находится в интервале от 10 до 32 бар, более предпочтительно температура реакции эпоксидирования находится в интервале от 30 до 60°С, и давление реакции эпоксидирования находится в интервале от 15 до 25 бар.
Термин “условия эпоксидирования”, как применяется в контексте настоящего изобретения, относится к реакции эпоксидирования на этапе (а2), где в течение по меньшей мере 98%, предпочтительно по меньшей мере 99%, более предпочтительно по меньшей мере 99.9% от общего времени реакции температура реакции и давление находятся в указанных выше диапазонах.
Термин «общее время реакции», как применяется в контексте настоящего изобретения, относится к времени реакции, когда данный слой катализатора применяется, прежде чем он либо будет списан, либо подвергнут регенерации. В частности, в начале реакции эпоксидирования на этапе (а2), когда катализатор является свежим, то есть при запуске реакции эпоксидирования на этапе (а2), температура реакции или давление или и то и другое могут находиться за пределами указанных выше диапазонов на короткий период времени. Предпочтительно, скорость потока теплоносителя выбирается таким образом, что разность температуры между температурой на входе и температурой на выходе не составит не более 3 K, более предпочтительно не более 2 K, более предпочтительно не более 1 K.
Предпочтительно, реакция эпоксидирования согласно этапе (а2) по настоящему изобретению проводится при практически постоянном превращении эпоксидирующего средства, предпочтительно пероксида водорода. Предпочтительно, чтобы определить конверсию пероксида водорода, молярную скорость потока пероксида водорода в отходящем потоке, удаляемом на стадии (а3), называемый здесь mout, сравнивают с молярным расходом пероксида водорода в потоке поступающего материала на стадии (a1), обозначаемой здесь как min, и где конверсия пероксида водорода определяется как 100×(1-mout/min). Предпочтительно, температура на входе теплоносителя, описанного выше, регулируется в вышеупомянутых предпочтительных диапазонах для того, чтобы конверсия пероксида водорода была по существу постоянной в пределах от 80 до 100%, более предпочтительно от 90 до 100%, более предпочтительно от 95 до 100%, более предпочтительно от 99 до 100%, более предпочтительно от 99,5 до 100%, более предпочтительно от 99,9 до 100%. Термин “условия эпоксидирования содержат”, как применяется в контексте настоящего изобретения, относится к реакции эпоксидирования на этапе (а2), где в течение по меньшей мере 98%, предпочтительно по меньшей мере 99%, более предпочтительно по меньшей мере 99.9% от общего времени реакции, превращение пероксида водорода находится в определенных выше диапазонах. Термин «общее время реакции», как применяется в контексте настоящего изобретения, относится к времени реакции, когда данный слой катализатора применяется, прежде чем он либо будет списан, либо подвергнут регенерации. В частности, в начале реакции эпоксидирования на этапе (а2), когда катализатор является свежим, то есть при запуске реакции эпоксидирования на этапе (а2), конверсия пероксида водорода может находиться за пределами указанных выше диапазонов на короткий период времени. Предпочтительно, температура реакции не поддерживается постоянной во время реакции, но регулируется непрерывно или постадийно, чтобы обеспечить постоянную конверсию пероксида водорода. В общем, из-за определенной дезактивации катализатора, температура реакции непрерывно или постадийно увеличивается. Предпочтительно, температура реакции непрерывно или постадийно увеличивается не более чем на 1 K/д (Кельвин/день), более предпочтительно менее чем на 1 K/д.
Предпочтительно, реакционная смесь, которая присутствует в реакторе на этапе (а2), является жидкой в условиях эпоксидирования. Предпочтительно реакционная смесь состоит из одной жидкой фазы, двух жидких фаз или трех или более жидких фаз. Предпочтительно реакционная смесь в реакторе на этапе (а2) состоит из одной жидкой фазы или двух жидких фаз, более предпочтительно одной жидкой фазы.
В общем, реактор, используемый на этапе (а2) согласно настоящему изобретению, может быть расположен горизонтально или вертикально. Предпочтительно, реактор расположен вертикально. В предпочтительном вертикально размещенном реакторе поток поступающего материала, обеспеченный на этапе (a1), может проходить в режиме восходящего потока или в режиме нисходящего потока, причем предпочтительным является режим восходящего потока. Предпочтительно, по сравнению с направлением хода потока поступающего материала, теплоноситель пропускается через рубашку в виде сопутствующего потока.
В общем, реакция эпоксидирования на этапе (а2) может быть проведена в одном или более реакторах, где эти реакторы могут быть расположены параллельно или последовательно. Предпочтительно, реакция на этапе (а2) проводится в одном реакторе или в по меньшей мере двух реакторах, предпочтительно двух реакторах, которые расположены последовательно, где между двумя расположенными последовательно реакторами может быть проведена соответствующая промежуточная обработка. Если реакция проводится в двух реакторах, расположенных последовательно, предпочтительно, что работает первый реактор, как описано выше, то есть в качестве изотермического реактора, а второй реактор, то есть нижестоящий реактор, работает как адиабатический или по существу адиабатический реактор. Используемый здесь термин «реактор» также охватывает два или более реактора, расположенных параллельно, где пропускаемый поток поступающего материала разделяется на два или более подпотока, каждый подпоток пропускается в реактор, и выходящие потоки, удаляемые из реактора, объединяются с получением общего выходящего потока. Следовательно, реакцию эпоксидирования можно проводить в одном первом реакторе, как например двух или более первых реакторах, например, 2, 3, 4 первых реакторах, которые расположены параллельно и которые представляют собой изотермические реакторы, и в по меньшей мере одном втором реакторе, как например двух или более вторых реакторах, например, 2, 3, 4 вторых реакторах, которые расположены параллельно и которые являются адиабатическими или по существу адиабатными реакторами.
Если реакция эпоксидирования согласно (а2) проводится в двух реакторах, расположенных последовательно, предпочтительно, чтобы в первом реакторе, который является предпочтительно изотермическим реактором, конверсия пероксида водорода сохранялась по существу постоянной в диапазоне от 80 до 99%, предпочтительно от 85 до 98%, более предпочтительно от 90 до 97%, а во втором реакторе, который спроектирован как адиабатический или по существу адиабатический реактор, общая конверсия пероксида водорода, то есть конверсия пероксида водорода с учетом конверсии в первом и втором реакторе, доводится до значения более 99%, предпочтительно по меньшей мере 99,5%, более предпочтительно по меньшей мере 99,9%.
Этап (а3)
Согласно этапу (а3) из реактора удаляется поток продукта, содержащий пропиленоксид и органический растворитель. Указанный поток продукта обычно подвергают по меньшей мере одной стадии обработки для выделения пропиленоксида из потока продукта. Кроме того, органический растворитель, который обычно включает побочные продукты реакции эпоксидирования, предпочтительно подвергают одной или более стадиям обработки, чтобы обеспечить рециркуляцию органического растворителя, предпочтительно ацетонитрила, предпочтительно после одной или более стадий очистки, на этап (a1). Предпочтительная последовательность стадий обработки описана в ссылочном примере 2 и на Фиг. 1 и 2 ниже.
Этап (b)
Согласно (b) введение потока поступающего материала в реактор согласно (a1) останавливают.
Предпочтительно, этап (b) дополнительно содержит отделение реакционной смеси, содержащей пропиленоксид и органический растворитель, от катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала. Это отделение реакционной смеси от отработанного катализатора, содержащего цеолит, содержащий титан, может быть достигнуто любым подходящим способом, таким как прокачка, дренирование, декантирование, фильтрация и тому подобное. Необязательно, этап (b) дополнительно содержит сушку катализатора до проведения промывки на этапе (с), где предпочтительно катализатор не сушат перед проведением промывки на этапе (с). Если сушка проводится, предпочтительно указанная сушка включает приведение катализатора в контакт с потоком газа содержащим азот, предпочтительно при температуре указанного потока газа в диапазоне от 40 до 200°С, более предпочтительно в интервале от 55 до 150°С, более предпочтительно в интервале от 70 до 100°С. Указанное разделение может быть достигнуто также путем промывки реактора с помощью органического растворителя, предпочтительно ацетонитрила.
Таким образом, в предпочтительном варианте выполнения настоящего изобретения, реактор промывается от пероксида водорода, пропилена и пропиленоксида, заменяя поток поступающего материала согласно (a1) потоком, по существу состоящим из органического растворителя, предпочтительно из ацетонитрила, до тех пор, пока реактор практически не содержит пероксид водорода, пропилен и пропиленоксид, и необязательно проводится сброс давления и опустошение реактора. Например, поток, по существу состоящий из органического растворителя, предпочтительно ацетонитрила, содержит по меньшей мере 80 мас. %, предпочтительно по меньшей мере 75 мас. %, более предпочтительно по меньшей мере 80 мас. % органического растворителя и необязательно содержит по меньшей мере еще одну жидкость, предпочтительно содержащую воду. Поэтому предпочтительно по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 98 мас. %, более предпочтительно по меньшей мере 99 мас. % потока, по существу состоящего из органического растворителя, предпочтительно из ацетонитрила, состоит из органического растворителя, предпочтительно ацетонитрила, и воды, где по меньшей мере 60 мас. %, предпочтительно по меньшей мере 70 мас. %, более предпочтительно по меньшей мере 75 мас. %, более предпочтительно по меньшей мере 80 мас. % потока состоит из органического растворителя, предпочтительно ацетонитрила. Указанное промывание предпочтительно проводится при температуре указанного потока, по существу состоящего из органического растворителя, предпочтительно ацетонитрила, в интервале от 20 до 90°С, более предпочтительно в интервале от 25 до 80°С, более предпочтительно в интервале от 30 до 70°С.
В альтернативном предпочтительном варианте выполнения настоящего изобретения, способ согласно настоящему изобретению дополнительно содержит получение потока поступающего материала на этапе (a1) посредством смешивания потока, содержащего пропен, потока, содержащего пероксид водорода или источник пероксида водорода, и потока, содержащего органический растворитель, где этап (b) включает
(b1) остановку потока, содержащего пероксид водорода или источник пероксида водорода;
(b2) остановку потока, содержащего пропен, и потока, содержащего органический растворитель;
(b3) необязательно сброс давления и опустошение реактора,
где этап (b2) проводится предпочтительно по меньшей мере 30 мин, более предпочтительно по меньшей мере 60 мин, после стадии (b1).
Этап (с)
Согласно этапу (с), катализатор промывают жидкой водной системой.
Возможные компоненты жидкой водной системы согласно этапу (с) и их соответствующие количества, как описано ниже, следует понимать как относящиеся к жидкой водной системе, применяемой на этапе (с), перед добавлением к катализатору, содержащему цеолит, содержащий титан.
В общем, жидкая водная система согласно (с) не ограничена в отношении воды, содержащейся в ней. Жидкая водная система может, таким образом, содержать например, по меньшей мере 50 мас. %, или по меньшей мере 75 мас. %, или по меньшей мере 85 мас. % вода, на основе общей массы жидкой водной системы. Предпочтительно, жидкая водная система согласно (с) содержит по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды на основе общей массы жидкой водной системы.
Вода, применяемая на этапе (с), может быть получена из любого подходящего источника. Предпочтительно, вода, применяемая на этапе (с), представляет собой деминерализованную воду или конденсат.
Предпочтительно, жидкая водная система согласно (с) содержит пероксид водорода только в незначительных количествах, тогда как она предпочтительно свободна от или по меньшей мере по существу свободна от метанола. Предпочтительно, жидкая водная система согласно (с) содержит не более 5 мас. %, более предпочтительно 1 мас. %, более предпочтительно 0.1 мас. %, более предпочтительно 0.01 мас. %, более предпочтительно 0.001 мас. %, более предпочтительно 0.0001 мас. % метанола, на основе общей массы жидкой водной системы.
Предпочтительно, жидкая водная система согласно (с) содержит пероксид водорода только в незначительных количествах, тогда как она предпочтительно свободна от или по меньшей мере по существу свободна от пероксида водорода. Предпочтительно, жидкая водная система согласно (с) содержит не более 0.1 мас. %, предпочтительно 0.01 мас. %, более предпочтительно 0.001 мас. %, более предпочтительно 0.0001 мас. % пероксида водорода, на основе общей массы жидкой водной системы.
Предпочтительно, жидкая водная система согласно (с) имеет значение рН от 4 до 10, предпочтительно от 7 до 9 как определено с помощью рН электрода.
Особенно предпочтительно, жидкая водная система согласно (с) состоит из воды.
Промывка на этапе (с) может быть выполнена в любом возможном контейнере. Таким образом, промывка может быть проведена, например, в реакторе, содержащем катализатор, или в любом другом подходящем сосуде, в случае удаления катализатора из реактора для промывки на этапе (с). Предпочтительно, промывка на этапе (с) проводится в реакторе, содержащем катализатор.
Таким образом, промывка на этапе (с) предпочтительно проводится в реакторе, содержащем катализатор, где жидкая водная система согласно (с) содержит по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы.
Промывка на этапе (с) может проводиться либо периодическим, либо в непрерывном режиме. В случае, если промывка проводится периодическим образом, катализатор контактирует один или несколько раз с определенным количеством жидкой водной системы. Промывка может, таким образом, быть выполнена путем погружения катализатора в жидкую водную систему. Во время промывки можно подвергнуть жидкую водную систему вместе с катализатором перемешиванию. Возможно, что когда промывка на этапе (с) проводится периодическим образом, жидкая водная система может быть заменена один или более раз.
В случае, если промывка проводится в непрерывном режиме, катализатор непрерывно контактирует с потоком жидкой водной системы. В случае, если реактор, содержащий катализатор, расположен вертикально, и промывка проводится в реакторе или промывка проводится вне реактора в любом другом подходящем вертикально размещенном сосуде, промывка может выполняться либо в восходящем, либо в нисходящем режиме, предпочтительно в режиме нисходящего потока. Предпочтительно, промывка на этапе (с) проводится в непрерывном режиме. Более предпочтительно, промывка на этапе (с) проводится в непрерывном режиме в реакторе, содержащем катализатор.
Таким образом, промывка на этапе (с) предпочтительно проводится в непрерывном режиме в реакторе, содержащем катализатор, где жидкая водная система согласно (с) содержит по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы.
Предпочтительно, промывка в непрерывном режиме проводится в трубчатом реакторе или реакторе с пучком труб с жидкой водной системой, имеющей часовую объемную скорость жидкости (LHSV) в диапазоне от 1 до 20 м/ч, более предпочтительно от 5 до 15 м/ч, более предпочтительно от 5 до 10 м/ч. Часовая объемная скорость жидкости LHSV на этапе (с) определяется как объемный скорость потока жидкой водной системы в кубических метрах в час, поделенный на полное поперечное сечение в квадратных метрах пустой трубы реактора или пустых труб реактора в случае использования реактора с пучком труб, где труба имеет постоянный диаметр по всей ее длине, а трубки имеют постоянные диаметры по всей их длине, соответственно.
Таким образом, промывка на этапе (с) предпочтительно проводится в непрерывном режиме в трубчатом реакторе или реакторе с пучком труб, содержащем катализатор, где жидкая водная система содержит по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы, и она проводится с жидкой водной системой, имеющей LHSV в интервале от 1 до 20 м/ч, более предпочтительно от 5 до 15 м/ч, более предпочтительно от 5 до 10 м/ч.
Промывка на этапе (с) может проводиться при любой подходящей температуре жидкой водной системы. Предпочтительно, промывка на этапе (с) проводится при температуре жидкой водной системы в интервале от 30 до 90°С, предпочтительно от 40 до 80°С.
Промывка на этапе (с) может проводиться в течение любого периода времени, необходимого для достижения достаточной промывки катализатора, применяемого на этапе (а2). Предпочтительно, промывка на этапе (с) проводится в течение от 1 до 25 часов, предпочтительно от 1,5 до 20 часов, более предпочтительно от 2 до 12 часов, более предпочтительно от 2,5 до 9 часов, более предпочтительно от 3 до 6 часов.
Таким образом, промывка на этапе (с) предпочтительно осуществляется при температуре жидкой водной системы в интервале от 30 до 90°С в течение от 2,5 до 9 часов, более предпочтительно при температуре жидкой водной системы в интервале от 40 до 80°С в течение от 3 до 6 часов.
Таким образом, промывка на этапе (с) предпочтительно проводится в непрерывном режиме в трубчатом реакторе или реакторе с пучком труб, содержащем катализатор, где жидкая водная система содержит по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы, и она осуществляется с жидкой водной системой, имеющей LHSV в интервале от 5 до 15 м/ч при температуре жидкой водной системы в интервале от 30 до 90°С в течение от 2,5 до 9 часов, более предпочтительно от 5 до 10 ч/ч при температуре жидкой водной системы в интервале от 40 до 80°С в течение от 3 до 6 часов.
Согласно способу согласно настоящему изобретению, промывка на этапе (с) обычно контролируется посредством общей концентрации органического углерода в жидкой водной системе после контакта с катализатором, и промывка на этапе (с) проводится до тех пор, пока не достигается определенная концентрация общего органического углерода жидкой водной системы после контакта с катализатором. Поскольку общая концентрация органического углерода проходит через максимум в ходе промывки на этапе (с), определенная концентрация, которая предпочтительно достигается, относится к значению, достигнутому после прохождения максимума.
Предпочтительно промывка на этапе (с) выполняется до тех пор, пока общая концентрация органического углерода в жидкой водной системе после контакта с катализатором не составит 25% или менее, предпочтительно 20% или менее, предпочтительно 15% или менее, предпочтительно 10% или менее, Более предпочтительно 5% или менее, более предпочтительно 4% или менее, более предпочтительно 3% или менее, более предпочтительно 2% или менее, более предпочтительно 1% или менее от максимальной общей концентрации органического углерода, обнаруженной в ходе промывки на этапе (с), где общий объем жидкой водной системы равен или больше внутреннего объема реактора. Под общей концентрацией органического углерода понимают определенную согласно DIN EN 1484.
Момент времени, когда этап (b) проводится, и введение потока поступающего материала в реактор, останавливается, чтобы позволить промывку на этапе (с), выбирается предпочтительно на основе уменьшения селективности относительно образования пропиленоксида на этапе (а2) относительного средней селективности реакции на этапе (а2) в течение первых 100 ч проведения этапа (а). Селективность в отношении образования пропиленоксида на этапе (а2) в контексте настоящего изобретения понимается как селективность в отношении образования пропиленоксида на этапе (а2) на основе пероксида водорода и рассчитывается, как показано в сравнительном примере 1. Таким образом, этап (b) может быть проведена, например, когда селективность в отношении образования пропиленоксида на этапе (а2) уменьшилась на 15% или менее, или на 10% или менее, или на 6% или менее относительно средней селективности реакции на этапе (а2) в течение первых 100 ч проведения этапа (а). Предпочтительно, стадия (b) проводится, когда селективность в отношении образования пропиленоксида на этапе (а2) уменьшилась на 4% или менее, предпочтительно на 3% или менее, более предпочтительно на 2% или менее по сравнению со средней селективностью реакции на этапе (а2) в течение первых 100 ч проведения стадия (а).
Стадия (i) дополнительно включает повторение последовательности этапов (а)-(c) n раз, причем n представляет собой целое число и равно по меньшей мере 1. Таким образом, последовательность этапов (а)-(с) повторяют по меньшей мере один раз перед осуществлением стадии (ii). Предпочтительно, n находится в интервале от 1-10 и, таким образом, равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10. Более предпочтительно, n находится в интервале от 1 до 6 и, таким образом, равно 1, 2, 3, 4, 5, или 6. Более предпочтительно, n находится в интервале от 1-4 и, таким образом, равно 1, 2, 3, или 4.
Согласно способу согласно настоящему изобретению, катализатор, полученный на этапе (b) предпочтительно промывают только одной жидкой системой, содержащей по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы. Эта промывка предпочтительно осуществляется в непрерывном режиме. Таким образом, предпочтительно, что катализатор не обрабатывают никакой другой жидкой системой между этапом (b) и стадией (ii), за исключением жидкой системы, содержащей по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы. Таким образом, предпочтительно, ни предварительная, ни последующая обработка отличной жидкой водной системой, предпочтительно любой другой жидкой водной системой, не проводится, где промывка реактора органическим растворителем, предпочтительно ацетонитрилом на этапе (b) не исключается этим предпочтительным вариантом выполнения настоящего изобретения.
Согласно способу согласно настоящему изобретению, катализатор, полученный на этапе (b), предпочтительно не обрабатывают потоком газа состоящим из, предпочтительно содержащим озон.
Необязательно, этап (с) дополнительно включает сушку промытого катализатора. Если проводится сушка, указанная сушка предпочтительно включает приведение катализатора в контакт с потоком газа, содержащим азот, предпочтительно при температуре указанного потока газа в интервале от 40 до 200°С, более предпочтительно в интервале от 55 до 150°С, более предпочтительно в интервале от 70 до 100°С. Согласно настоящему изобретению, указанная сушка предпочтительно не проводится после этапу (с) промывки, после которой кальцинирование не проводится, т.е. проводится после каждой этапу (с) промывки, после которой стадия (ii) проводится.
Стадия (ii)
Способ регенерации согласно настоящему изобретению дополнительно содержит
(ii) стадию, включающую кальцинирование катализатора, полученного на этапе (с), после повторения последовательности этапов (а)-(с) n раз.
Предпочтительно, кальцинирование на стадии (ii) проводят при температуре слоя катализатора в интервале от 300 до 600°С, предпочтительно от 350 до 550°С, более предпочтительно от 400 до 500°С.
Кальцинирование на стадии (ii) может осуществляться в статической окружающей газовой атмосфере или в газовом потоке, с которым катализатор приводится в контакт. Предпочтительно, кальцинирование на стадии (ii) проводят в газовом потоке. В общем, указанный газовый поток не ограничен в отношении присутствующих в нем компонентов. Предпочтительно, указанный газовый поток содержит кислород, предпочтительно кислород и азот. В общем, указанный газовый поток, содержащий кислород, предпочтительно кислород и азот, не ограничен в отношении источника, используемого для кислорода, предпочтительно кислорода и азота. Предпочтительно воздух, обедненный, или их смесь, более предпочтительно воздух или обедненный применяется в качестве источника. Таким образом, кальцинирование на стадии (ii) предпочтительно осуществляют в потоке газа, содержащем кислород, предпочтительно кислород и азот, где поток газа, содержащий кислород, предпочтительно кислород и азот, предпочтительно представляет собой воздух, обедненный воздух, или их смесь, более предпочтительно воздух или обедненный воздух.
Таким образом, кальцинирование на стадии (ii) предпочтительно проводят при температуре слоя катализатора в интервале от 300 до 600°С, предпочтительно от 350 до 550°С, более предпочтительно от 400 до 500°С в потоке газа, содержащем кислород, предпочтительно кислород и азот, где поток газа, содержащий кислород, предпочтительно кислород и азот, предпочтительно представляет собой воздух, обедненный воздух, или их смесь, более предпочтительно воздух или обедненный воздух.
Кальцинирование на стадии (ii) может проводиться в течение любого периода времени, необходимого для достижения достаточного кальцинирования катализатора в способе регенерации катализатора согласно настоящему изобретению. Предпочтительно, кальцинирование на стадии (ii) проводится в течение от 1 до 15 часов, предпочтительно от 2 до 10 часов, более предпочтительно от 3 до 7 часов.
Таким образом, кальцинирование на стадии (ii) предпочтительно осуществляется при температуре слоя катализатора в интервале от 300 до 600°С в течение от 1 до 15 часов, более предпочтительно от 350 до 550°С в течение от 2 до 10 часов, более предпочтительно от 400 до 500°С в течение от 3 до 7 часов, в потоке газа, содержащем кислород, предпочтительно кислород и азот, где поток газа, содержащий кислород, предпочтительно кислород и азот, предпочтительно представляет собой воздух, обедненный воздух, или их смесь, более предпочтительно воздух или обедненный воздух.
Кальцинирование на стадии (ii) может проводиться в любом возможном контейнере. Таким образом, кальцинирование на стадии (ii) может проводиться, например, в реакторе, содержащем катализатор, или в любом другом подходящем сосуде, в случае, когда катализатор удаляется из реактора для кальцинирования на стадии (ii). Предпочтительно, кальцинирование на стадии (ii) проводится в реакторе, содержащем катализатор.
Последовательность стадий (i) и (ii) согласно настоящему изобретению для регенерации катализатора может быть повторена m раз прежде чем катализатор перестают использовать, причем m представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (i) и (ii), n является одинаковым или различным. Таким образом, последовательность стадий (i) и (ii) повторяют по меньшей мере один раз, где при каждом повторении последовательности стадий (i) и (ii), n является одинаковым или различным. Предпочтительно, m находится в интервале от 1 до 10 и, таким образом, равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10. Более предпочтительно, m находится в интервале от 1 до 6 и, таким образом, равно 1, 2, 3, 4, 5, или 6. Более предпочтительно, m находится в интервале от 1 до 4 и, таким образом, равно 1, 2, 3, или 4.
Предпочтительно, m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10, тогда как n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10, более предпочтительно 1, 2, 3, 4, 5, или 6, более предпочтительно 1, 2, 3, или 4. Более предпочтительно, m равно 1, 2, 3, 4, 5, или 6, тогда как n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10, более предпочтительно 1, 2, 3, 4, 5, или 6, более предпочтительно 1, 2, 3, или 4. Более предпочтительно, m равно 1, 2, 3, или 4, тогда как n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10, более предпочтительно 1, 2, 3, 4, 5, или 6, более предпочтительно 1, 2, 3, или 4.
Регенерированный катализатор
Настоящее изобретение также относится к регенерированному катализатору, содержащему цеолит, содержащий титан, в качестве каталитически активного материала, получаемому или полученному способом согласно настоящему изобретению и как описано выше.
Предпочтительно, катализатор, регенерированный согласно настоящему изобретению, проявляет в способе получения пропиленоксида дифференциальную температуру превращения, равную не более 5 K, предпочтительно не более 2 K, более предпочтительно не более 1 K, где дифференциальная температура превращения определена как абсолютная разность между
(А1) температурой, при которой достигнуто предопределенное превращение пероксида водорода в указанном способе получения пропиленоксида, в котором использован указанный регенерированный катализатор в качестве катализатора, и
(В1) температурой, при которой указанное предопределенное превращение пероксида водорода достигнуто в указанном способе получения пропиленоксида, в котором соответствующий свежий катализатор использован в качестве катализатора при остальных идентичных условиях реакции эпоксидирования.
После определенного периода времени в реакции эпоксидирования наблюдается снижение каталитической активности катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала. Сниженная каталитическая активность непосредственно связана с уменьшенной скоростью превращения для по меньшей мере одного из исходных материалов, то есть олефина и/или эпоксидирующего средства, где уменьшенная скорость превращения может быть скомпенсирована за счет увеличения общей температуры реакции. Это означает, что при продолжении работы катализатора требуется постепенное повышение температуры реакции относительно начальной температуры, что делает процесс эпоксидирования все более неэффективным.
Однако подвергая катализатор, содержащий цеолит, содержащий титан, прошедший процесс эпоксидирования, способу регенерации согласно настоящему изобретению, его исходная каталитическая активность может быть восстановлена. Первоначальная каталитическая активность здесь относится к каталитической активности свежеприготовленного катализатора. Поскольку каталитическая активность обычно непосредственно связана с температурой реакции при остальных идентичных условиях реакции, эффективность регенерации отработанного катализатора может быть получена на основе температуры реакции, необходимой для поддержания заданной скорости превращения. В данном случае, катализатор, регенерированный согласно настоящему изобретению, благоприятным образом проявляет в способе получения оксида олефина, температуру превращения, которая отклоняется не более чем на 5 K, предпочтительно не более 2 K, более предпочтительно не более 1 K от температуры превращения свежего катализатора при остальных идентичных условиях эпоксидирования.
Кроме того, предпочтительно, чтобы катализатор, регенерированный согласно способу согласно настоящему изобретению, проявляет в способе получения пропиленоксида дифференциальную селективность, равную не более 1, где дифференциальная селективность определена как абсолютная разница в % между
(А2) селективностью на основе пероксида водорода в указанном способе получения пропиленоксида, в котором регенерированный катализатор
(В2) селективностью на основе пероксида водорода в указанном способе получения пропиленоксида, в котором соответствующий свежий катализатор использован в качестве катализатора при остальных идентичных условиях реакции эпоксидирования, где селективность, основанная на пероксиде водорода, определена как моли полученного пропиленоксида, поделенные на моли превращенного пероксида водорода, ×100.
Качество катализатора, содержащего цеолит, содержащий титан, регенерированного согласно способу согласно настоящему изобретению, может быть также количественно оценено путем сравнения селективности регенерированного катализатора с селективностью свежего катализатора при остальных идентичных условиях эпоксидирования. При продолжительном использовании также наблюдается снижение селективности катализатора. Благоприятно то, что в данном случае после подачи в процесс регенерации согласно настоящему изобретению, катализатор, содержащий цеолит, содержащий титан, имеет селективность, которая отклоняется не более чем на 1 процентную единицу от селективности свежего катализатора при одинаковой реакции эпоксидирования условия.
Непрерывный способ получения пропиленоксида
Согласно настоящему изобретению, предпочтительно, что реакция эпоксидирования проводится в более чем одном реакторе, например, в по меньшей мере двух реакторах, трех реакторах или четырех реакторах, которые работают параллельно. Согласно этому способу предпочтительно, чтобы в ходе регенерации катализатора в данном реакторе реакция эпоксидирования проводилась в одном или более оставшихся реакторах. Таким образом, пропиленоксид может быть получен в непрерывном режиме и одновременно может проводиться регенерация согласно настоящему изобретению. Может быть предпочтительным, что в соответствии с этим способом, реакция эпоксидирования в по меньшей мере двух реакторах запускается последовательно с подходящей задержкой между соответствующими пусками.
Настоящее изобретение относится к непрерывному способу получения пропиленоксида, включающему
(i') стадию, включающую
(а') непрерывное получение пропиленоксида в первом реакторе, включающее
(а1') введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, в первый реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2') воздействие на поток поступающего материала согласно (а1') в первом реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3') удаление потока продукта, содержащего пропиленоксид и органический растворитель из первого реактора;
(b') прекращение введения потока поступающего материала в первый реактор;
(с') промывку катализатора жидкой водной системой;
причем стадия (i') дополнительно включает повторение последовательности этапов (а')-(с') n' раз, причем n' представляет собой целое число и равно по меньшей мере 1; причем способ получения пропиленоксида дополнительно включает
(ii') стадию, включающую кальцинирование катализатора, полученного на этапе (с'), после повторения последовательности этапов (а')-(с') n' раз;
где последовательность стадий (i')-(ii') необязательно повторяют m' раз, где m' представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (i')-(ii'), n' является одинаковым или различным;
причем способ получения пропиленоксида дополнительно включает
(iʺ) стадию, включающую
(аʺ) непрерывное получение пропиленоксида во втором реакторе, включающее
(a1ʺ) введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, во второй реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2ʺ) воздействие на поток поступающего материала согласно (a1ʺ) во втором реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3ʺ) удаление потока продукта, содержащего пропиленоксид и органический растворитель, из второго реактора;
(bʺ) прекращение введения потока поступающего материала во второй реактор;
(сʺ) промывку катализатора жидкой водной системой;
стадия (iʺ) дополнительно включает повторение последовательности этапов (аʺ)-(сʺ) nʺ раз, причем nʺ представляет собой целое число и равно по меньшей мере 1; причем способ получения пропиленоксида дополнительно содержит
(iiʺ) стадию, включающую кальцинирование катализатора, полученного на этапе (сʺ), после повторения последовательности этапов (аʺ)-(сʺ) nʺ раз;
где последовательность стадий (iʺ)-(iiʺ) необязательно повторяют mʺ раз, причем mʺ представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (iʺ)-(iiʺ), nʺ является одинаковым или различным;
где в ходе по меньшей мере одной последовательности этапов (b') и (с') или по меньшей мере одной последовательности этапов (b'), (с') и (ii'), пропиленоксид получают согласно (аʺ).
Настоящее изобретение далее охарактеризовано следующими вариантами выполнения настоящего изобретения и комбинациями вариантов выполнения настоящего изобретения, указанных посредством соответствующих зависимостей:
1. Способ регенерации катализатора, содержащего цеолит, содержащий титан, в качестве каталитически активного материала, включающий
(i) стадию, включающую
(а) непрерывное получение пропиленоксида, включающее
(a1) введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, в реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2) воздействие на поток поступающего материала согласно (a1) в реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3) удаление потока продукта, содержащего пропиленоксид и органический растворитель, из реактора;
(b) прекращение введения потока поступающего материала в реактор;
(c) промывку катализатора жидкой водной системой;
причем стадия (i) дополнительно включает повторение последовательности этапов (а)-(с) n раз, причем n представляет собой целое число и равно по меньшей мере 1; причем способ регенерации дополнительно включает
(ii) стадию, включающую кальцинирование катализатора, полученного на этапе (с), после повторения последовательности этапов (а)-(с) n раз;
где последовательность стадий (i)-(ii) необязательно повторяют m раз, причем m представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (i)-(ii), n является одинаковым или различным.
2. Способ согласно варианту выполнения настоящего изобретения 1, включающий получение потока поступающего материала согласно (a1) посредством смешивания потока, содержащего пропен, потока, содержащего пероксид водорода или источник пероксида водорода, и потока, содержащего органический растворитель.
3. Способ согласно варианту выполнения настоящего изобретения 2, в котором поток поступающего материала согласно (a1) дополнительно содержит по меньшей мере одну соль, содержащую калий.
4. Способ согласно варианту выполнения настоящего изобретения 2, в котором по меньшей мере одна соль, содержащая калий, представляет собой одну или более из дигидрофосфата калия, гидрофосфата дикалия, и формиата калия.
5. Способ согласно варианту выполнения настоящего изобретения 3 или 4, в котором получение потока поступающего материала согласно (a1) включает объединение водного потока, содержащего по меньшей мере одну растворенную соль, содержащую калий, с потоком, содержащим пропен, или с потоком, содержащим пероксид водорода или источник пероксида водорода, или с потоком, содержащим органический растворитель, или со смешанным потоком двух или трех из этих потоков, предпочтительно с потоком, содержащим пероксид водорода или источник пероксида водорода, или с потоком, содержащим органический растворитель, или с их смешанным потоком.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1-5, где органический растворитель представляет собой метанол или ацетонитрил, предпочтительно ацетонитрил.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1-6, где молярное отношение пропена относительно пероксида водорода или относительно пероксида водорода, происходящего из источника пероксида водорода, в потоке поступающего материала согласно (a1) находится в интервале от 1:1 до 1.6:1, предпочтительно от 1.1:1 до 1.55:1, более предпочтительно от 1.2:1 до 1.5:1, более предпочтительно от 1.30:1 до 1.45:1.
8. Способ согласно любому из вариантов выполнения настоящего изобретения 1-7, где реактор согласно (а2) представляет собой изотермически работающий реактор.
9. Способ согласно любому из вариантов выполнения настоящего изобретения 1-8, где согласно (а2) температуру реакционной смеси контролируют изотермически, применяя теплоноситель, предпочтительно пропуская теплоноситель через рубашку реактора.
10. Способ согласно варианту выполнения настоящего изобретения 9, где условия эпоксидирования согласно (а2) включают температуру реакции эпоксидирования в интервале от 20 до 100°С, предпочтительно от 25 до 80°С, более предпочтительно от 30 до 60°С, где температура реакции эпоксидирования определяется как температура теплоносителя до контроля температуры реакционной смеси, предпочтительно как температура теплоносителя на входе в рубашку изотермически работающего реактора.
11. Способ согласно любому из вариантов выполнения настоящего изобретения 1-10, где условия эпоксидирования на этапе (а2) содержат давление реакции эпоксидирования в интервале от 5 до 100 бар, предпочтительно от 10 до 32 бар, более предпочтительно от 15 до 25 бар, где давление реакции эпоксидирования определяется как давление при выходе из изотермически работающего реактора.
12. Способ согласно любому из вариантов выполнения настоящего изобретения 1-11, где цеолит, содержащий титан, имеет MFI каркасную структуру, MEL каркасную структуру, MWW каркасную структуру, каркасную структуру MWW-типа, или смешанную структуру двух или более из этих каркасных структур, предпочтительно MFI каркасную структуру, MWW каркасную структуру или каркасную структуру MWW-типа, более предпочтительно MWW каркасную структуру или каркасную структуру MWW-типа.
13. Способ согласно любому из вариантов выполнения настоящего изобретения 1-12, где по меньшей мере 99 мас. %, предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % каркасной структуры цеолита, содержащего титан, состоит из кремния, титана и кислорода.
14. Способ согласно любому из вариантов выполнения настоящего изобретения 1-13, где цеолит, содержащий титан, содержит одно или более из А1, В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd, предпочтительно одно или более из В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, Cd, более предпочтительно Zn.
15. Способ согласно варианту выполнения настоящего изобретения 14, где цеолит, содержащий титан, который содержится в катализаторе согласно (а2) имеет MWW каркасную структуру, содержит цинк и содержит титан, вычисленный как элементарный титан, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан, и содержит цинк, вычисленный как элементарный цинк, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан.
16. Способ согласно любому из вариантов выполнения настоящего изобретения 1-15, где катализатор, содержащий цеолит, содержащий титан, присутствует в реакторе в виде катализатора в виде неподвижного слоя.
17. Способ согласно любому из вариантов выполнения настоящего изобретения 1-16, где катализатор, содержащий цеолит, содержащий титан, присутствует в реакторе в виде формованного изделия, содержащего цеолит, содержащий титан, где формованное изделие предпочтительно содержит цеолит, содержащий титан, в количестве в интервале от 70 до 80 мас. % и связующее в количестве в интервале от 30 до 20 мас. %, на основе общей массы формованное изделие, где связующим предпочтительно является связующее на основе диоксида кремния.
18. Способ согласно любому из вариантов выполнения настоящего изобретения 1-17, включающий получение потока поступающего материала согласно (a1) посредством смешивания потока, содержащего пропен, потока, содержащего пероксид водорода или источник пероксида водорода, и потока, содержащего органический растворитель, где для прекращения введения потока поступающего материала в реактор согласно (b), получение потока поступающего материала согласно (a1) включает
(a11) остановку потока, содержащего пероксид водорода или источник пероксида водорода, и поддержание смешивания потока, содержащего пропен, и потока, содержащего органический растворитель, с получением потока поступающего материала;
(a12) остановку потока, содержащего пропен, и потока, содержащего органический растворитель;
где (a11) проводится предпочтительно по меньшей мере 30 мин, более предпочтительно по меньшей мере 60 мин, после (а12), и где, после (а12), реактор необязательно подвергают сбросу давления и опустошению.
19. Способ согласно любому из вариантов выполнения настоящего изобретения 1-18, где жидкая водная система согласно (с) содержит по меньшей мере 95 мас. %, предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.9 мас. %, более предпочтительно по меньшей мере 99.99 мас. % воды, на основе общей массы жидкой водной системы.
20. Способ согласно любому из вариантов выполнения настоящего изобретения 1-19, где жидкая водная система согласно (с) содержит не более 5 мас. %, предпочтительно не более 1 мас. %, более предпочтительно не более 0.1 мас. %, более предпочтительно не более 0.01 мас. %, более предпочтительно не более 0.001 мас. %, более предпочтительно не более 0.0001 мас. % метанола, на основе общей массы жидкой водной системы.
21. Способ согласно любому из вариантов выполнения настоящего изобретения 1-20, где жидкая водная система согласно (с) содержит не более 0.1 мас. %, предпочтительно не более 0.01 мас. %, более предпочтительно не более 0.001 мас. %, более предпочтительно не более 0.0001 мас. % пероксида водорода, на основе общей массы жидкой водной системы.
22. Способ согласно любому из вариантов выполнения настоящего изобретения 1-21, где согласно (с), промывку катализатора проводят в реакторе, содержащем катализатор.
23. Способ согласно любому из вариантов выполнения настоящего изобретения 1-22, где общий объем жидкой водной системы, применяемой согласно (с), равен или больше, чем внутренний объем реактора.
24. Способ согласно любому из вариантов выполнения настоящего изобретения 1-23, где промывку согласно (с) проводят в непрерывном режиме.
25. Способ согласно варианту выполнения настоящего изобретения 24, где реактор согласно (а) представляет собой трубчатый реактор или реакторе с пучком труб, и промывку согласно (с) проводят жидкой водной системой при часовой объемной скорости жидкости (LHSV) в интервале от 1 до 20 м/ч, предпочтительно от 5 до 15 м/ч, более предпочтительно от 5 до 10 м/ч.
26. Способ согласно любому из вариантов выполнения настоящего изобретения 1-25, где промывку согласно (с) проводят при температуре жидкой водной системы в интервале от 30 до 90°С, предпочтительно от 40 до 80°С.
27. Способ согласно любому из вариантов выполнения настоящего изобретения 1-26, где промывку согласно (с) проводят в течение от 1 до 25 часов, предпочтительно в течение от 1.5 до 20 часов, более предпочтительно в течение от 2 до 12 часов, более предпочтительно в течение от 2.5 до 9 часов, более предпочтительно в течение от 3 до 6 часов.
28. Способ согласно любому из вариантов выполнения настоящего изобретения 1-27, где промывку согласно (с) проводят до тех пор, пока общая концентрация органического углерода жидкой водной системы после контакта с катализатором не составит не более 25%, предпочтительно не более 20%, предпочтительно не более 15%, предпочтительно не более 10%, более предпочтительно не более 5%, более предпочтительно не более 1% от максимальной общей концентрации органического углерода, обнаруженной в ходе промывки на этапе (с).
29. Способ согласно любому из вариантов выполнения настоящего изобретения 1-28, где последовательность этапов (b) и (с) проводится, когда селективность реакции эпоксидирования согласно (а2) уменьшилась на 4%, предпочтительно на 3%, более предпочтительно на 2%, относительно средней селективности реакции эпоксидирования согласно (а2) в ходе первых 100 ч проведения этапа (а), где селективность реакции эпоксидирования определяется как молярное количество пропиленоксида, полученного на этапе (а2), относительно молярного количества пероксида водорода, взаимодействовавшего на этапе (а2).
30. Способ согласно любому из вариантов выполнения настоящего изобретения 1-29, где n находится в интервале от 1 до 10, предпочтительно от 1 до 6, более предпочтительно от 1 до 4.
31. Способ согласно любому из вариантов выполнения настоящего изобретения 1-30, где кальцинирование согласно стадии (ii) проводят при температуре катализатора в интервале от 300 до 600°С, предпочтительно от 350 до 550°С, более предпочтительно от 400 до 500°С.
32. Способ согласно любому из вариантов выполнения настоящего изобретения 1-31, где кальцинирование согласно стадии (ii) проводят, применяя поток газа, содержащий кислород, предпочтительно кислород и азот, где поток газа предпочтительно представляет собой воздух, обедненный воздух, или их смесь, более предпочтительно воздух или обедненный воздух.
33. Способ согласно любому из вариантов выполнения настоящего изобретения 1-32, где кальцинирование согласно стадии (ii) проводят в течение от 1 до 15 часов, предпочтительно от 2 до 10 часов, более предпочтительно от 3 до 7 часов.
34. Способ согласно любому из вариантов выполнения настоящего изобретения 1-33, где кальцинирование согласно стадии (ii) проводят в реакторе, содержащем катализатор.
35. Способ согласно любому из вариантов выполнения настоящего изобретения 1-34, где последовательность стадий (i)-(ii) повторяют m раз, причем m представляет собой целое число в интервале от 1 до 10, предпочтительно от 1 до 6, более предпочтительно от 1 до 4.
36. Регенерированный катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала, получаемый или полученный способом согласно любому из вариантов выполнения настоящего изобретения 1-35.
37. Регенерированный катализатор согласно варианту выполнения настоящего изобретения 36, где цеолит, содержащий титан, имеет MWW каркасную структуру, где по меньшей мере 99 мас. %, предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % каркасной структуры цеолита, содержащего титан, состоит из кремния, титана и кислорода, где цеолит, содержащий титан, предпочтительно содержит Zn, и где цеолит, содержащий титан, содержит титан, вычисленный как элементарный титан, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан, и предпочтительно содержит цинк, вычисленный как элементарный цинк, в количестве в интервале от 0.1 до 5 мас. %, предпочтительно от 1 до 2 мас. %, на основе общей массы цеолита, содержащего титан.
38. Регенерированный катализатор согласно варианту выполнения настоящего изобретения 36 или 37, где регенерированный катализатор представляет собой формованное изделие, содержащее цеолит, содержащий титан, где формованное изделие предпочтительно содержит цеолит, содержащий титан, в количестве в интервале от 70 до 80 мас. %, и связующее в количестве в интервале от 30 до 20 мас. %, на основе общей массы формованного изделия, где связующим предпочтительно является связующее на основе диоксида кремния.
39. Регенерированный катализатор согласно любому из вариантов выполнения настоящего изобретения 36-38, проявляющий, в способе получения пропиленоксида, дифференциальную температуру превращения, равную не более 5 K, предпочтительно не более 2 K, более предпочтительно не более 1 K, где дифференциальная температура превращения определена как абсолютная разность между
(А1) температурой, при которой достигнуто предопределенное превращение пероксида водорода в указанном способе получения пропиленоксида, в котором использован указанный регенерированный катализатор в качестве катализатора, и
(В1) температурой, при которой указанное предопределенное превращение пероксида водорода достигнуто в указанном способе получения пропиленоксида, в котором соответствующий свежий катализатор использован в качестве катализатора при остальных идентичных условиях реакции эпоксидирования.
40. Регенерированный катализатор согласно любому из вариантов выполнения настоящего изобретения 36-39, проявляющий, в способе получения пропиленоксида, дифференциальную селективность, равную не более 1, где дифференциальная селективность определена как абсолютная разность в % между
(А2) селективностью на основе пероксида водорода в указанном способе получения пропиленоксида, в котором использован указанный регенерированный катализатор в качестве катализатора, и
(В2) селективностью на основе пероксида водорода в указанном способе получения пропиленоксида, в котором соответствующий свежий катализатор использован в качестве катализатора при остальных идентичных условиях реакции эпоксидирования, где селективность, основанная на пероксиде водорода, определена как моли полученного пропиленоксида, поделенные на моли превращенного пероксида водорода, ×100.
41. Непрерывный способ получения пропиленоксида, включающий
(i') стадию, включающую
(а') непрерывное получение пропиленоксида в первом реакторе, включающее
(а1') введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, в первый реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2') воздействие на поток поступающего материала согласно (а1') в первом реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3') удаление потока продукта, содержащего пропиленоксид и органический растворитель, из первого реактора;
(b') прекращение введения потока поступающего материала в первый реактор;
(с') промывку катализатора жидкой водной системой;
стадия (i') дополнительно включает повторение последовательности этапов (а')-(с') n' раз, причем n' представляет собой целое число и равно по меньшей мере 1; причем способ получения пропиленоксида дополнительно включает
(ii') стадию, включающую кальцинирование катализатора, полученного на этапе (с'), после повторения последовательности этапов (а')-(с') n' раз;
где последовательность стадий (i')-(ii') необязательно повторяют m' раз, причем m' представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (i')-(ii'), n' является одинаковым или различным;
причем способ получения пропиленоксида дополнительно включает
(iʺ) стадию, включающую
(аʺ) непрерывное получение пропиленоксида во втором реакторе, включающее
(a1") введение потока поступающего материала, содержащего пропен, пероксид водорода или источник пероксида водорода, и органический растворитель, во второй реактор, содержащий катализатор, содержащий цеолит, содержащий титан, в качестве каталитически активного материала;
(а2ʺ) воздействие на поток поступающего материала согласно (a1ʺ) во втором реакторе условиями эпоксидирования в присутствии катализатора, с получением реакционной смеси, содержащей пропиленоксид и органический растворитель;
(а3ʺ) удаление потока продукта, содержащего пропиленоксид и органический растворитель из второго реактора;
(bʺ) прекращение введения потока поступающего материала во второй реактор;
(cʺ) промывку катализатора жидкой водной системой;
стадия (iʺ) дополнительно включает повторение последовательности этапов (аʺ)-(сʺ) nʺ раз, причем nʺ представляет собой целое число и равно по меньшей мере 1; причем способ получения пропиленоксида дополнительно включает
(iiʺ) стадию, включающую кальцинирование катализатора, полученного на этапе (сʺ), после повторения последовательности этапов (аʺ)-(сʺ) nʺ раз;
где последовательность стадий (iʺ)-(iiʺ) необязательно повторяют mʺ раз, причем mʺ представляет собой целое число и равно по меньшей мере 1, где при каждом повторении последовательности стадий (iʺ)-(iiʺ) nʺ является одинаковым или различным;
где в ходе по меньшей мере одной последовательности этапов (b') и (с') или по меньшей мере одной последовательности этапов (b'), (с') и (ii'), пропиленоксид получают согласно (аʺ).
Настоящее изобретение далее проиллюстрировано посредством следующих чертежей, ссылочных примеров, примеров и сравнительных примеров.
Краткое описание чертежей
Фиг. 1 показывает блок-схему способа согласно ссылочному Примеру 2. На Фиг. 1 буквы и числа имеют следующие значения:
А узел эпоксидирования А
В узел эпоксидирования В
С узел дистилляции
D узел дистилляции
Е узел дистилляции
F узел частичной дистилляции потока
G узел смесителя-отстойника
Н узел выделения ацетонитрила
I узел рециркуляции ацетонитрила
(1)-(20) потоки согласно особенно предпочтительному способу, как описано в примерах
S0, S01, S02, S1, S2, S3, S4, S4b, S5, L1, L2, TL1, TL2, TL2, BL2
потоки согласно предпочтительному способу, как описано в общем описании и примерах.
Фиг. 2 подробно показывает блок-схему узла частичной дистилляции потока F на Фиг. 1 и на Фиг. 2 буквы и цифры имеют следующие значения:
F1 первый узел фракционирования узла частичной дистилляции потока F
F2 второй узел фракционирования узла частичной дистилляции потока F
(13), (13а), (14), (15), (15а), (15b), (15 с), (16), (19), (20)
потоки согласно особенно предпочтительному способу, как описано в примерах
S1, S2, S3, S4, S4a, S4b, S4c, S5, TL2
потоки согласно предпочтительному способу, как описано в общем описании и примерах.
Примеры
Ссылочный пример 1: Получение формованного изделия, содержащего ZnTiMWW распылительный порошок
Катализатор в форме формованного изделия получили, как описано в WO 2013/117536 А2, в примере 5, в частности в Примерах 5.1-5.6, на стр. 83-99. Способы характеризации указанного формованного изделия, упомянутого в упомянутых примерах 5.1-5.6, описаны в Ссылочных примерах 2-10 на стр. 66-71 документа WO 2013/117536 А2. Характерные свойства катализатора показаны на Фиг. 20-27 в WO 2013/117536 А2 и соответствующем описании указанного чертежа на стр. 104 документа WO 2013/117536 А2.
Ссылочный пример 2: установка для реакции эпоксидирования
Что касается аббревиатур, делается ссылка на схемы на Фиг. 1 и 2, в общем описанные в разделе «Краткое описание чертежей». Все приведенные давления являются абсолютными давлениями.
1.1 Получение потока S0 (этап (а))
а) Эпоксидирование в основном реакторе эпоксидирования (узел эпоксидирования А)
Основным реактором А был вертикально установленный трубчатый реактор с 5 трубами (длина труб: 12 м, внутренний диаметр трубы: 38 мм), причем каждая труба была оборудована аксиально размещенной многоточечной термопарой с 10 равноотстоящими точками измерения, заключенными в подходящую защитную гильзу диаметром 18 мм. В каждую пробирку было загружено 17,5 кг формованных изделий ZnTiMWW катализатора, полученных согласно Ссылочному примеру 1 (пост-обработанные формованные изделия). Свободное пространство, в результате оставшееся, было заполненными стеатитными сферами (диаметр 3 мм). Теплота реакции удалялась путем циркуляции термостатируемого теплоносителя (смесь вода/гликоль) во межтрубном пространстве прямотоком по отношению к подаче. Скорость потока теплоносителя регулировали таким образом, чтобы разница температур между входом и выходом не превышала 1°С. Температура реакции, указанная ниже, определялась как температура теплоносителя, поступающего в оболочку реактора. На выходе из реактора давление контролировали регулятором давления и поддерживали постоянным при 20 бар.
Реактор подпитывали снизу жидким монофазным потоком (1). Поток (1) получили путем смешивания четырех потоков (2), (3), (3а) и (4). Температура потока (1) не контролировалась активным образом, но обычно находилась в диапазоне от 20 до 40°С:
- Поток (2), имеющий скорость потока 85 кг/ч. По меньшей мере 99,5 мас. % потока (2) состояли из ацетонитрила, пропена и воды. Этот поток (2) приходит со дна ацетонитрил рециркулирующего узла дистилляции рецикла ацетонитрила (I).
- Поток (3), имеющий скорость потока 15 кг/ч, представлял собой водный раствор пероксида водорода, имеющий концентрацию пероксида водорода 40 мас. % (марка «неочищенный/промытый» от Solvay с ТОС в диапазоне от 100 до 400 мг/кг. Водный раствор пероксида водорода подавали из резервуара для хранения, обеспечивая непрерывную подачу, и подавали с использованием подходящего дозирующего насоса.
- Поток (3а) представлял собой водный поток, включающий растворенный формиат калия. Дальнейший поток подавался из резервуара для хранения, что обеспечивало непрерывную подачу, и подавался с использованием подходящего дозирующего насоса. Концентрация формиата калия составляла 2,5 мас. %, скорость подачи потока (S3a) составляла 370 г/ч. Поток (3а) тщательно смешивали с потоком (3) до того, как объединенный поток был смешан с потоком, возникающим в результате смешивания потока (2) и (4).
- Поток (4) представлял собой подпитывающий поток чистого ацетонитрила (химическая марка, от Ineos, чистота около 99,9%, содержащий от 70 до 180 мас. частей на миллион пропионитрила, 5-20 мас. частей на миллион ацетамида и менее 100 мас. частей на миллион воды в качестве примесей). Достаточно свежий ацетонитрил был добавлен для компенсации потерь в способе. В обычных условиях добавляли в среднем от 100 до 150 г/ч подпитывающего ацетонитрила.
Выходящий поток (5), покидающий узел эпоксидирования А, отбирали каждые 20 минут для определения концентрации пероксида водорода с использованием метода на основе титанилсульфата и для расчета превращения пероксида водорода. Превращение пероксида водорода определялось как 1100×(1-mout/min) где min представляет собой молярный скорость потока потока Н2О2 при подпитке реактора, и mout представляет собой молярный скорость потока потока Н2О2 на выходе из реактора. На основе полученных значений превращения пероксида водорода температура на входе теплоносителя была отрегулирована таким образом, чтобы конверсия пероксида водорода была по существу постоянной в интервале от 90 до 92%. Температура на входе теплоносителя была установлена при 30°С в начале данного цикла со свежей порцией катализатора эпоксидирования и при необходимости увеличивалась для поддержания превращения пероксида водорода в указанном диапазоне. Требуемое повышение температуры обычно составляло менее 1 K/д.
Выходящий поток (5), выходящий из узла эпоксидирования А, пропускался через узел теплообмена. Поток, выходящий из узла теплообмена (поток (6), S0), подавался в узел эпоксидирования В.
b) Эпоксидирование в конечном реакторе (узел эпоксидирования В)
Конечный реактором В служил адиабатический реактор с неподвижным слоем. В этом контексте термин «адиабатический» относится к режиму работы, согласно которому никакое активное охлаждение не проводится, и согласно которому конечный реактор соответствующим образом изолирован для минимизации потерь тепла. Конечный реактор В имел длину 4 м и диаметр 100 мм. Реактор заполняли 9 кг того же катализатора эпоксидирования, который использовался в основном реакторе эпоксидирования А. Оставшееся пространство заполнялось стеатитными сферами (диаметром 3 мм). Рабочее давление конечного реактора В составляло 10 бар, которое поддерживалось постоянным подходящим регулятором давления на выходе из реактора. Выходящий поток из конечного реактора В отбирали каждые 20 мин для определения концентрации пероксида водорода с использованием метода на основе титанилсульфата.
Выходящий поток конечного реактора В, поток (6), был сброшен под давлением в испарительную емкость, и как жидкость, так и газ из этой емкости подавались в разделительную колонну для легкокипящих соединений (узел дистилляции С).
Поток (6) (поток S0) имел среднее содержание ацетонитрила от 69 до 70 мас. %, и содержание пропиленоксида 9-11 мас. %, как например 9.8 мас. %, содержание воды 17 мас. %, содержание пропена около 3 мас. %, содержание пропана около 0.05 мас. %, содержание пероксида водорода около 250 мас. частей на миллион, содержание пропиленгликоля около 0.1 мас. % и содержание кислорода около 150 мас. частей на миллион.
1.2 Отделение пропиленоксида от потока S0 с получением потока S1 (этап (b))
а) Отделение легкокипящих соединений от потока (6) (поток S0) с получением потока (8) (поток S01)
Поток (6) был направлен в разделительную колонну легкокипящих соединений (узел дистилляции С), работающую при 1,1 бар. Дистилляционная колонна имела длину 8,5 м, диаметр 170 мм и была оборудована 40 барботажными тарелками, испарителем на дне и конденсатором наверху. Колонна работала как смешанная промывочная/дистилляционная колонна. В качестве промывочного средства отводили часть нижнего потока узла дистилляции D (поток 11, около 20-30 кг/ч), охлаждали до 10°С и вводили в верхнюю часть колонны. Жидкие и газовые входящие потоки вводили в колонну в разных точках. Точка подачи жидкой части потока (6) была выше барботажной тарелки 37; газовую часть потока (6) вводили в колонну выше барботажной тарелки 28 (считая сверху). Газовый поток (7), выходящий из охлаждающего средства в верхней части колонны, содержал в основном пропен, пропан (который содержался в качестве примеси в используемом пропене полимерной марки), кислород, образованный как побочный продукт, и небольшое количество других легкокипящих соединений (ацетонитрил (1-2 об. %), пропиональдегид (около 200 об. частей на миллион), ацетон (около 100 об. частей на миллион, Н2 (около 400 об. частей на миллион), СO2 (около 400 об. частей на миллион) и ацетальдегид (около 100 об. частей на миллион)) и по существу не содержал пропиленоксид (менее 300 об. частей на миллион). Этот верхний поток был направлен на сгорание для утилизации. Нижний поток разделительной колонны легкокипящих соединений (поток (8), то есть поток S01), имеющий температуру 70°С, имел содержание пропена от 100 до 200 мас. частей на миллион.
b) Отделение пропиленоксида от потока (8) (поток S01) с получением потока S02
Поток S01, полученный согласно части 1.2 а) выше, вводили в дистилляционную колонну (узел дистилляции D), чтобы отделить пропиленоксид от потока S01. Колонна имела высоту 50 м и диаметр 220 мм и была оснащена уплотнителем (Sulzer ВХ64) с общей длиной уплотнителя 27,5 м, разделенным на 8 слоев длиной 3060 мм каждый и двумя слоями с длиной 1530 мм каждый. Между каждым слоем установлены распределители промежуточного потока. Колонна работала при верхнем давлении 750 мбар. Точка подачи потока S01 располагалась ниже четвертого слоя уплотнителя, считая сверху. Верхний поток колонны конденсировали и частично возвращали в колонну в виде флегмы (коэффициент обратного потока 5:1). Остаток (поток (9)), имеющий скорость потока 10,1 кг/ч, был взят в качестве верхнего продукта и по существу состоял из пропиленоксида, имеющего чистоту более 99,9 мас. %. Нижний испаритель работал таким образом, что концентрация пропиленоксида в кубовых потоках была ниже 100 мас. частей на миллион. Конечная температура кубового потока составляла около 69°С. Поток SO2 был затем разделен на две части. Основная его часть (поток (10), скорость потока около 85 кг/ч) была отправлена в следующую дистилляционную колонну (узел дистилляции Е). Остаток (поток (11), 20-30 кг/ч) охладили и рециркулировали на вершину колонны легкокипящих соединений (узел дистилляции С) в качестве промывочного средства, как описано выше в части 1.2 а). Этот поток S02 имел содержание ацетонитрила около 80 мас. %, содержание пропиленоксида менее 100 мас. частей на миллион, содержание воды около 20 мас. %, содержание пропиленгликоля около 0.1 мас. % и содержание гидроксипропанола около 0.1 мас. %.
с) Отделение легкокипящих соединений от потока (10) (поток S02) с получением потока(13)(поток S1)
Поток S02, полученный согласно части 1.2 б) выше, был введен в колонну для разделения легкокипящих соединений (узел дистилляции Е). Эта колонна для разделения легкокипящих соединений имела высоту 8 м и номинальный диаметр 150 мм и была оснащена 35 барботажными тарелками. Колонка работали при верхнем давлении 2 бар, и поток S02 вводили выше барботажной тарелки 7 (считая снизу). Полученный поток, отводимый сверху колонны (поток (12), скорость потока около 1 кг/ч), покидал колонну с температурой от 40 до 45°С и не конденсировался, так как колонна работала без внутреннего потока флегмы. Помимо ацетонитрила (6500 об. частей на миллион), этот поток, отводимый сверху колонны, содержал в основном азот, который использовался для поддержания рабочего давления колонны при значении 2 бар, и небольшие количества легкокипящих соединений (ацетальдегид (900 об. частей на миллион), кислород (300 об. частей на миллион) и пропиональдегид (320 об. частей на миллион). Этот верхний поток был направлен на сгорание для утилизации. Нижний испаритель работал с постоянной подачей в него количества (5 кг/ч) насыщенного пара при давлении 16 бар. Температура нижней части колонны составляла 100°С. Нижний поток, поток S1, в основном состоял из ацетонитрила и воды, причем оставшуюся часть составляли высококипящие соединения. Этот поток S1 имел содержание ацетонитрила около 80 мас. % и содержание воды около 20 мас. %
1.3 Разделение потока S1 на потоки S2 и S3 (этап (с))
Стадия (с), поток S1, скорость потока 86 кг/ч, полученный согласно части 1.2 с) выше, разделялся на два потока, потоки S2 (поток (13а согласно Фиг. 1) и S3 (поток 14 согласно Фиг. 1). Поток S2 имел скорость потока 84 кг/ч, и поток S3 имел скорость потока 2 кг/ч. Поток S3, 2.3% потока S1, подвергали узлу частичной дистилляции потока F (колонны частичной дистилляции потока).
1.4 Частичная дистилляция потока S1 (этап (d))
Первый узел фракционирования, т.е. первая дистилляционная колонна F1, имела высоту 9,5 м и диаметр 85 мм и была оборудована 6,5 метрами металлическим структурированным уплотнением Rombopak 9М, установленным в трех одинаковых слоях. Над первым слоем структурированного уплотнения, отсчитывая сверху, вводили поток S3 ((поток 14)) в первую дистилляционную колонну. Температура подачи потока S3 составляла 60±3°С. Первая дистилляционная колонна работала при верхнем давлении около 1,4 бар и кубовой температуре 92±5°С. Возврат флегмы не применялся. Количество пара, подаваемого в нижний испаритель первого узла фракционирования, контролировалось таким образом, чтобы концентрация ацетонитрила на дне составляла от 10 до 25 мас. %. Кубовый поток S4b (поток (15b), около 3% потока S3) был удален. Этот поток состоял в основном из воды (72-85 мас. %) и ацетонитрила (10-24 мас. %). Сумма всех анализируемых высококипящих компонентов (27 компонентов) варьировалась в диапазоне 2-10 мас. %. Верхний поток, поток S4a паровой фракции (поток 15а), имеющий температуру 85±3°С, не конденсировался и пропускался на дно второго узла фракционирования, то есть второй дистилляционной колонны F2. S4a вошел в F2 ниже последнего слоя структурированного уплотнителя, считая сверху. F2 имел высоту 9,5 м и диаметр 85 мм и был оснащен 6,5 м металлическим структурированным уплотнением Rombopak 9М, установленным в 3 одинаковых слоях. Вторая дистилляционная колонна работала при верхнем давлении около 1,25 бар и кубовой температуре 85±5°С. Верхний поток, поток S4c паровой фракции (поток (15 с), не более 1% потока S4a), полностью конденсировался внешним конденсатором на верху колонны (не показан на Фиг. 2) и практически полностью применялся для использования конденсированного жидкого потока в виде флегмы во вторую дистилляционную колонну. Жидкий кубовый поток S4 (поток 15) был удален и проходил на следующую стадию (рециркуляция потока S4). Поток S4 имел содержание ацетонитрила около 80 мас. % и содержание воды около 20 мас. %.
1.5 Рециркуляция потока S4 (этап (4))
а) Получение жидкого потока S5
Поток S4 (поток 15 согласно Фиг. 1 и Фиг. 2) смешивался с потоком S2 (поток (13а) согласно Фиг. 1 и Фиг. 2). Таким образом, поток S4 откачивался обратно в поток растворителя ацетонитрила. Смешивание происходило в точке ниже по ходу потока, где поток S3 отделялся от потока S1. Этот объединенный поток, имеющий скорость потока 86 кг/ч, смешивали с жидким потоком Р (обозначенным как поток (20) на Фиг. 1 и Фиг. 2) с получением потока S5. Поток Р представлял собой свежий поток пропена, содержащий пропан (полимерная марка, чистота >96 мас. %, сжиженный под давлением, скорость подачи: 10.9 кг/ч). Чтобы получить поток S5, объединенный поток S2 и S4 дополнительно смешивали с двумя другими потоками: первым из этих потоков является поток (16) согласно Фиг. 1, причем указанный поток получают из верхней части узла дистилляции Н. Второй из этих потоков представляет собой поток (19) согласно Фиг. 1, причем указанный поток получают из узла выделения ацетонитрила I. Оба потока (16) и (19) подробно описаны ниже.
b) Регулирование температуры потока S5 и отделение жидких фаз L1 и L2
Поток S5, имеющий скорость потока 130 кг/ч±10 кг/ч, затем подавали в смеситель-отстойник, работающий при 18 бар и температуре в диапазоне 15±5°С. Отстойник имел объем 5,3 литра. Получены две жидкие фазы L1 и L2, водная фаза L2 и органическая фаза L1. Верхнюю органическую фазу L1 удаляли из резервуара отстойника в качестве потока (17), кубовая водная фаза L2 удалялась из резервуара отстойника в качестве потока (18). Поток (17) имел скорость потока 110 кг/ч±11 кг/ч. Поток (17) затем пропустили через узел рециркулирования ацетонитрила I, поток (18) пропустили через узел выделения ацетонитрила Н, из которого поток (16), упомянутый выше, был получен. Поток (17), полученный таким образом, имел содержание ацетонитрила около 45-51 мас. %, содержание пропена около 49-55 мас. % и содержание воды около 2-5 мас. %. Поток (18), полученный таким образом, имел содержание ацетонитрила около 19-21 мас. %, содержание воды около 79-81 мас. % и содержание пропена менее 0.5 мас. %.
c) Выделение ацетонитрила (узел выделения ацетонитрила Н)
Для того чтобы рециркулировать как можно больше растворителя и чтобы минимизировать потери ацетонитрила, поток (18) вводили в дистилляционную колонну, из которой поток (16), также называемый как поток TL2, был получен как верхний поток, который в свою очередь, рециркулировали в поток растворителя, как описано выше. Для этой цели была использована дистилляционная колонна высотой 9,5 м и диаметром 100 мм, оборудованная 50 барботажными тарелками. Колонка работала при верхнем давлении 1,5 бар с коэффициентом обратного потока 1:4. Поток (18) подавали в колонну выше барботажной тарелки 26 (считая снизу). Температура на дне составляла около 113°С, а нижний продукт состоял в основном из воды, содержащей высококипящие побочные продукты. Типичный состав нижнего потока был следующим (мас. %, приводятся в скобках): вода (>99,0), пропенгликоль (0,5), ацетонитрил (не более 0,001), дипропиленгликоль (0,06), ацетамид (0,01), уксусная кислота (0,03), ТОС (2.4)). После необязательного количественного анализа этот поток был отброшен. Головной продукт (поток (16)=поток TL2) имел следующие типичные диапазоны состава (мас. %, приводятся в скобках): ацетонитрил (75-80), вода (15-20), низкокипящие вещества (например, пропен, 1). Как описано выше поток (16) перерабатывается в поток поступающего материала, который передается в узел смесителя-отстойника.
d) Рециркуляция ацетонитрила (узел рециркуляции ацетонитрила I)
Для рециркуляции ацетонитрила поток (17), полученный из узла смесителя-отстойника G, вводили в дистилляционную колонну высотой 10 м и номинальным диаметром 200 мм, снабженную 40 барботажными тарелками. Колонка работала при верхнем давлении 18 бар и коэффициенте обратного потока 1:4. Поток (17) подавали в колонну выше барботажной тарелки 26 (считая сверху). Верхний продукт (поток (19)), также называемый как поток TL1, содержащий в основном пропен (около 97 об. %) с небольшими количествами пропана (около 1-3 об. %) возвращался в сырье узла смесителя-отстойника G, как описано выше. Таким образом, избыток пропена удаляли из пара (17) и рециркулировали. Кубовый поток (поток (2), также называемый как поток BL1), имел температуру в интервале от 106 до 100°С. Точные рабочие параметры колонки, такие как затраты энергии в сборнике, регулируются таким образом, что количество пропена, возвращаемое в реактор с потоком (2), находится в таком диапазоне, что молярное отношение пропена к пероксиду водорода в потоке (1) составляет около 1:1,43. Для вышеупомянутой скорости подачи 15 кг/ч водного пероксида водорода это означает, что условия, которые необходимо отрегулировать, такие что скорость потока пропена в потоке (2) составляет около 9,7 кг/ч. До подачи потока (2) в основной реактор эпоксидирования А, ацетонитрил (поток (4), химический марка, от Ineos, чистота около 99,9%, содержащий от 70 до 180 мас. частей на миллион пропионитрила, 5-20 мас. частей на миллион ацетамида и <100 мас. частей на миллион воды в качестве примесей) необязательно добавляли для компенсации возможных потерь растворителя.
Точное количество дополнительно добавленного ацетонитрила зависело от потерь в выходящих потоках и побочных продуктах, но также от количества образцов, взятых для анализа. Типичное количество дополнительно добавленного ацетонитрила для вышеописанной технологической схемы может быть в интервале от 100 до 150 г/ч.
Пример 1: Разовая регенерация ZnTiMWW катализатора
Частичная регенерация ZnTiMWW катализатора на основе реакционной установки для эпоксидирования, как описано в ссылочном примере 2 выше. Реактор загрузили ZnTiMWW катализатором согласно Ссылочному примеру 1. Реакция эпоксидирования непрерывно проводилась до тех пор, пока селективность относительно образования пропиленоксида на этапе (а2) на основе пероксида водорода не снизилась на 2% относительно средней селективности относительно образования пропиленоксида на этапе (а2) на основе пероксида водорода в течение первых 1000 ч проведения этапа (а). Затем реакцию останавливали, и реактор промывали от Н2О2, пропилена и пропиленоксида. После сброса давления и опорожнения реактор промывали при 70°С деминерализованной водой сверху вниз с LHSV около 7 м/ч (LHSV, на основе поперечного сечения пустых трубок) в течение по меньшей мере 3 часов. По истечении этого времени жидкая водная система, покидающая реактор, показала общую концентрацию органического углерода менее 0,1% от максимального значения, и общий объем жидкой водной системы, полученный из реактора в процессе промывки, был больше объема реактора. Затем реактор был опустошен и перезапущен. Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1500 часов до тех пор, пока температура реакции не достигла значения, которое она имела до регенерации.
Сравнительный пример 1: Непрерывная работа согласно ссылочному примеру 2 без регенерации
В качестве сравнительного примера, способ согласно Ссылочному примеру 2 проводили в течение 2,500 ч без регенерации катализатора.
Результаты для Примера 1 и Сравнительного примера 1
Результаты для примера 1 и сравнительного примера 1 обсуждаются ниже и показаны в таблице 1. Температура на входе теплоносителя узла эпоксидирования А, как в примере 1, так и сравнительном примере 1, была отрегулирована в ходе непрерывной работы согласно ссылочному примеру 2 для того, чтобы сохранить превращение пероксида водорода по существу постоянным в интервале от 90 до 92% (см. Ссылочный пример 2, раздел 1.1 а). Конечная температура теплоносителя (охлаждающая вода) узла эпоксидирования А после общего рабочего времени 2500 часа показана в таблице 1 как для сравнительного примера 1, так и для примера 1. Кроме того, скорость дезактивации приводится как отношение разности температуры теплоносителя на входе в конце эксперимента и в начале эксперимента, поделенная на общее рабочее время 2500 ч, выраженная как °С/день. Кроме того, в Таблице 1 представлена селективность S реакции эпоксидирования на основе пероксида водорода после общего рабочего времени 2500 ч. Селективность S/%=(n(PO)/n(Н2O2))×100, где n(РО) представляет собой молярное количество пропиленоксида, обнаруженное непосредственно вниз по ходу потока к узлу В, и n(Н2O2) представляет собой молярное количество пероксида водорода, превращенное в ходе реакции эпоксидирования.
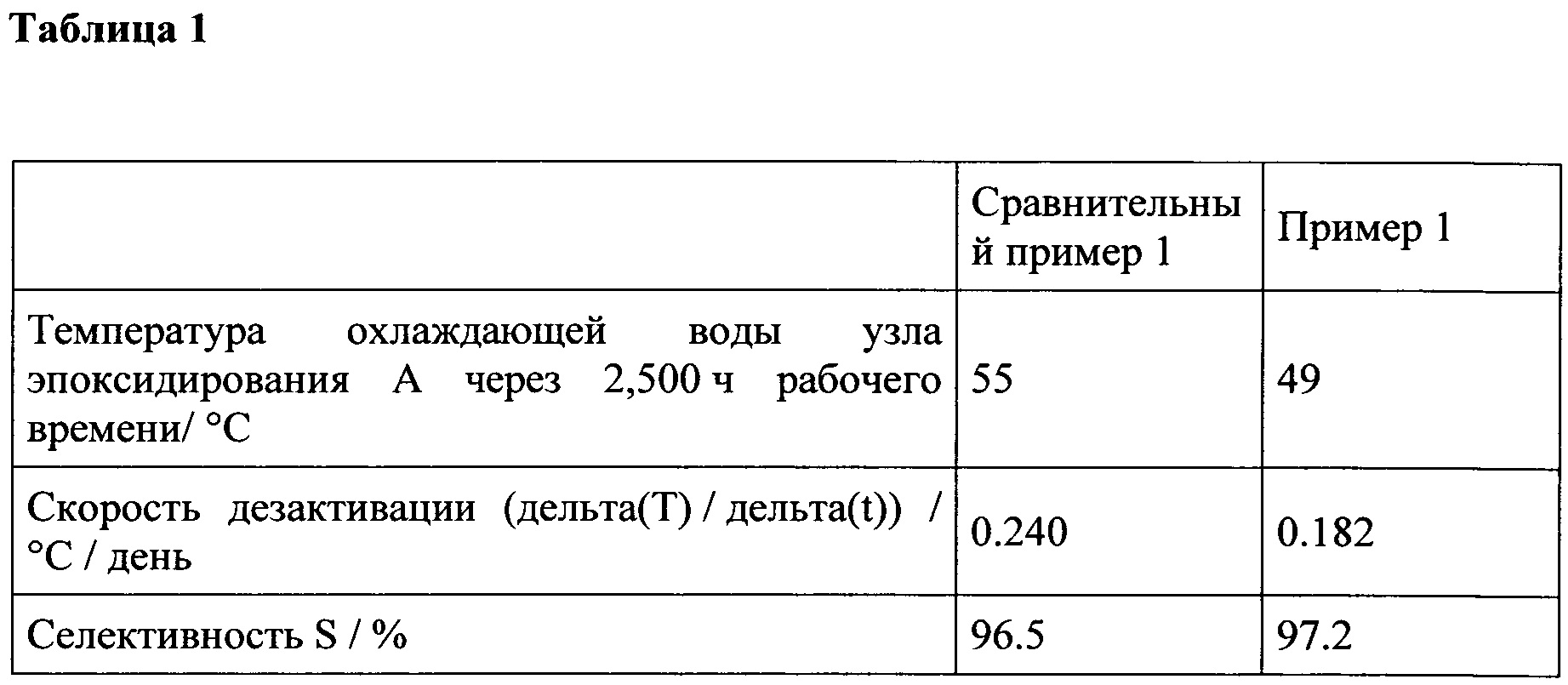
Как ясно показано в таблице 1, способ согласно настоящему изобретению позволяет пройти узел эпоксидирования А после общего рабочего времени 2500 часа при значительно более низкой температуре охлаждающей воды, то есть катализатор обладает значительно более высокой производительностью, чем в сравнительный пример 1. Далее, также скорость дезактивации (дельта (Т) / дельта (t)) отличается для двух экспериментов и значительно ниже в отношении примера согласно настоящему изобретению. Тем не менее способ согласно настоящему изобретению обеспечивает, после общего рабочего времени 2500 часа, значительно более высокую селективность, чем сравнительный пример 1.
Регенерированный катализатор согласно примеру 1 показал дифференциальную температуру превращения 1 K, где дифференциальная температура превращения определена как абсолютная разность между (А1) температурой, при которой превращение пероксида водорода 90-92% было достигнуто при использовании регенерированного катализатора в качестве катализатора, и (В1) температурой, при которой указанное определенное превращение пероксида водорода было достигнуто, когда соответствующий свежий катализатор использовался при остальных идентичных условиях реакции эпоксидирования. Далее, регенерированный катализатор согласно примеру 1 показал дифференциальную селективность 0,3%, где дифференциальная селективность определена как абсолютная разность в % между (А2) селективностью на основе пероксида водорода в процессе эпоксидирования, в котором использован указанный регенерированный катализатор в качестве катализатора, и (В2) селективностью на основе пероксида водорода в указанном процессе эпоксидирования, в котором соответствующий свежий катализатор использован в качестве катализатора при остальных идентичных условиях реакции эпоксидирования.
Пример 2.1: регенерация ZnTiMWW катализатора согласно настоящему изобретению, включающая три стадии промывки
Частичная регенерация ZnTiMWW катализатора на основе реакционной установки для эпоксидирования, как описано в ссылочном примере 2 выше.
Эпоксидирование
Частичная регенерация ZnTiMWW катализатора на основе реакционной установки для эпоксидирования, как описано в ссылочном примере 2 выше. В основной реактор загрузили ZnTiMWW катализатор согласно Ссылочному примеру 1. Реакция эпоксидирования проводилась в непрерывном режиме в течение около 1300 часов, до тех пор, пока “температура реакции эпоксидирования”, определенная как температура теплоносителя на входе в оболочку основного реактора, начальное значение которой в начале реакции эпоксидирования составляло 30°С, не достигла значения 50°С. В этот момент времени реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Первая стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1400 ч, до тех пор, пока температура реакции снова не достигала значения 50°С. В этот момент времени, реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Вторая стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1200 ч, до тех пор, пока температура реакции снова не достигала значение 50°С. В этот момент времени, реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Остаточные количества ацетонитрила и воды удалялись из реактора путем пропускания азот через слой катализатора при температуре азота 70°С.
Третья стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора. Стадия кальцинирования
Затем азот пропускали через слой катализатора в течение около 60 часов при начальной температуре азота 70°С, где эта температура увеличивалась до значения 100°С при 10 К/ч. Затем катализатор кальцинировали путем пропускания газовой смеси воздуха и азота через слой катализатора при расходе 0.58 кг/с. Температуру указанной смеси дополнительно непрерывно изменяли при 10 К/ч от начального значения 100°С до конечного значения 450°С и затем сохраняли по существу постоянной при значении 450°С в течение 6 часов. В начале кальцинирования, газовая смесь, проходящая в реактор, имела содержание кислорода 4 об. %. Как только достигали 450°С, указанное содержание кислорода доводили до значения 21 об. %. После кальцинирования при 450°С, реактор охлаждали до температуры около 30°С.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1000 ч.
Пример 2.2: регенерация ZnTiMWW катализатора согласно, включающая две стадии промывки
Частичная регенерация ZnTiMWW катализатора на основе реакционной установки для эпоксидирования, как описано в ссылочном примере 2 выше.
Эпоксидирование
Основной реактор загрузили ZnTiMWW катализатор согласно Ссылочный пример
1. Реакция эпоксидирования проводилась в непрерывном режиме в течение около 1200 часов, до тех пор, пока ʺтемпература реакции эпоксидированияʺ, определенная как температура теплоносителя на входе в оболочку основного реактора, начальное значение которой в начале реакции эпоксидирования составляло 30°С, не достигла значения 50°С. В этот момент времени, реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Первая стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1300 ч, до тех пор, пока температура реакции снова не достигала значение 50°С. В этот момент времени, реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Остаточные количества ацетонитрила и воды удалялись из реактора путем пропускания азот через слой катализатора при температуре азота 70°С.
Вторая стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора.
Стадия кальцинирования
Затем азот пропускали через слой катализатора в течение около 60 часов при начальной температуре азота 70°С, где эта температура увеличивалась до значения 100°С при 10 K/ч. Затем катализатор кальцинировали путем пропускания газовой смеси воздуха и азота через слой катализатора при расходе 0.58 кг/с. Температуру указанной смеси дополнительно непрерывно изменяли при 10 K/ч от начального значения 100°С до конечного значения 450°С и затем сохраняли по существу постоянной при значении 450°С в течение 6 часов. В начале кальцинирования, газовая смесь, проходящая в реактор, имела содержание кислорода 4 об. %. Как только достигали 450°С, указанное содержание кислорода доводили до значения 21 об. %. После кальцинирования при 450°С, реактор охлаждали до температуры около 30°С.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1000 ч.
Сравнительный пример 2: регенерация ZnTiMWW катализатора, включающая одну стадию промывки
Эпоксидирование
Регенерацию ZnTiMWW катализатора на основе реакционной установки для эпоксидирования, как описано в ссылочном примере 2 выше. Основной реактор загрузили ZnTiMWW катализатором согласно Ссылочный пример 1. Реакция эпоксидирования проводилась в непрерывном режиме в течение около 2200 часов, до тех пор, пока ʺтемпература реакции эпоксидированияʺ, определенная как температура теплоносителя на входе в оболочку основного реактора, начальное значение которой в начале реакции эпоксидирования составляло 30°С, не достигла значения 50°С. В этот момент времени, реакцию эпоксидирования остановили путем остановки потока пероксида водорода; течение ацетонитрила и пропена продолжалось до тех пор, пока эпоксидирование в трубках не было завершено. Затем реактор очистили от Н2О2, пропилена и пропиленоксида, пропуская смесь ацетонитрила и воды через реактор (80 мас. % ацетонитрила, 20 мас. % воды), где температура этой смеси составляла 50°С, с последующим опустошением реактора от основной доли смеси ацетонитрила и воды.
Остаточные количества ацетонитрила и воды удалялись из реактора путем пропускания азот через слой катализатора при температуре азота 70°С.
Стадия промывки
Затем реакционные трубки заполняли снизу водой, имеющей температуру 70°С, и воду, имеющую температура 70°С, затем пропускали сверху вниз через трубки в течение около 6 ч. Промывка водой прекращалась, как только проводимость выходящей воды была ниже 200 микросименс. Затем воду сливали из реактора.
Стадия кальцинирования
Затем азот пропускали через слой катализатора в течение около 60 часов при начальной температуре азота 70°С, где эта температура увеличивалась до значения 100°С при 10 К/ч. Затем катализатор кальцинировали путем пропускания газовой смеси воздуха и азота через слой катализатора при расходе 0.58 кг/с. Температуру указанной смеси дополнительно непрерывно изменяли при 10 К/ч от начального значения 100°С до конечного значения 450°С и затем сохраняли по существу постоянной при значении 450°С в течение 6 часов. В начале кальцинирования, газовая смесь, проходящая в реактор, имела содержание кислорода 4 об. %. Как только достигали 450°С, указанное содержание кислорода доводили до значения 21 об. %. После кальцинирования при 450°С, реактор охлаждали до температуры около 30°С.
Эпоксидирование
Непрерывная работа согласно Ссылочному примеру 2 возобновлялась и проводилась в течение примерно 1000 часов.
Результаты примеров 2.1 и 2.2 и сравнительного примера 2
Результаты примеров 2.1 и 2.2 и сравнительного примера 2 обсуждаются ниже и показаны в таблице 2 ниже. Температура на входе теплоносителя узла эпоксидирования А как в примерах 2.1 и 2.2, так и в сравнительном примере 2 была отрегулирована во время непрерывной работы согласно Ссылочному примеру 2, чтобы сохранить превращение пероксида водорода практически постоянным при около 96%. Селективность S реакции эпоксидирования на основе пероксида водорода, как указано в таблице 2, определяется как (n(РО)/n(H2O2))×100, где n(РО) означает молярное количество пропиленоксида, обнаруженное непосредственно ниже по ходу потока к узлу В, и n(H2O2) представляет собой молярное количество пероксида водорода, превращенное в ходе реакции эпоксидирования.
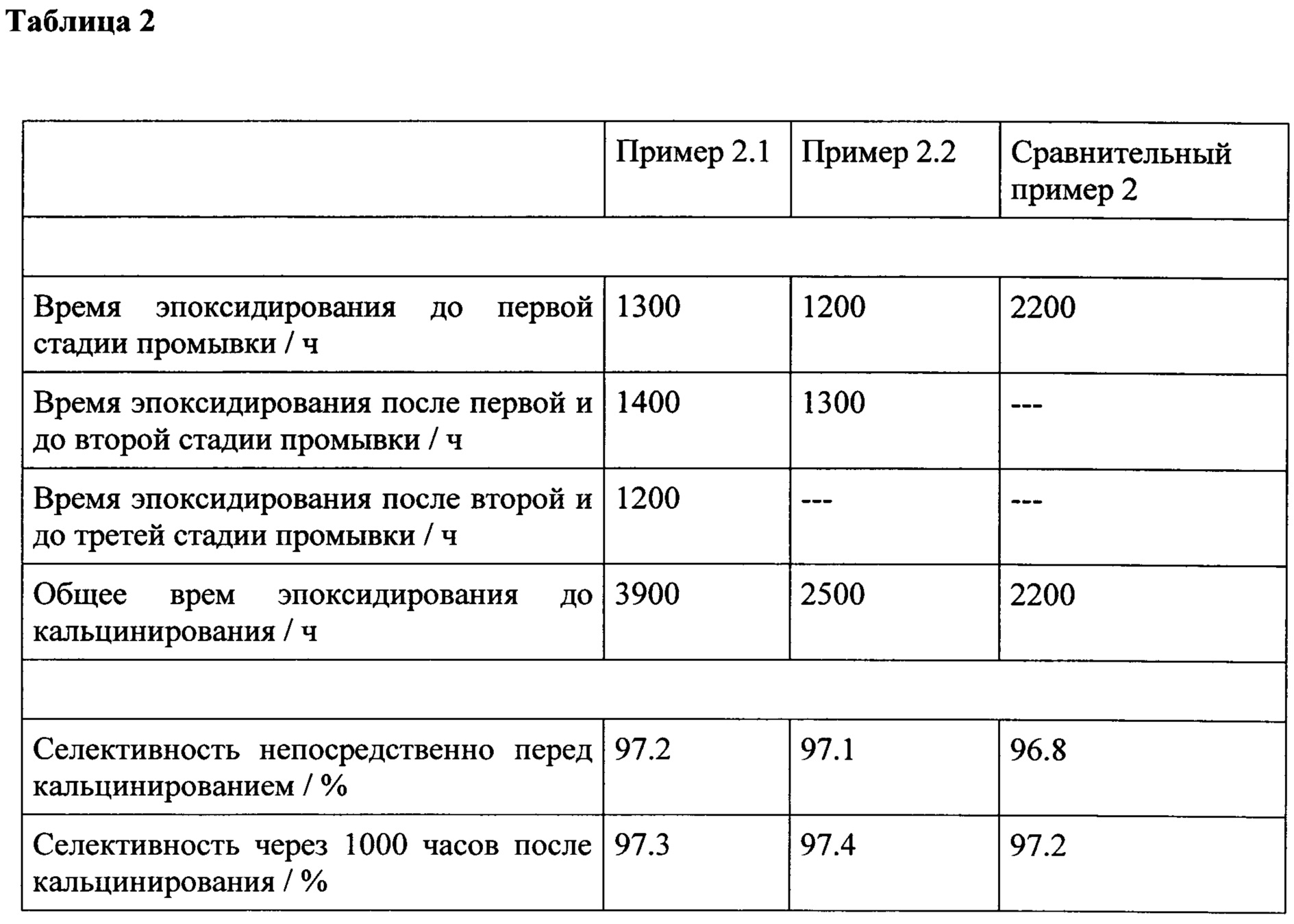
Как ясно показано в таблице 2, способ согласно изобретению, где одна стадия промывки (Пример 2.2) и две стадии промывки (Пример 2.1) выполняются без последующей стадии кальцинирования, приводят к небольшому, но тем не менее существующему улучшению селективности (1000 часов после кальцинирования) по сравнению с обычным способом (сравнительный пример 2), согласно которому кальцинирование проводится непосредственно после (только) стадии промывки. Этот заметный результат достигается, хотя общее время эпоксидирования до стадия кальцинирования выше (плюс 300 часов по сравнению с примером 2.2) или даже значительно выше (плюс 1700 часов по сравнению с примером 2.1). Таким образом, достоверно показано, что настоящее изобретение на основе частичной регенерации согласно стадии (i), избегая стадии кальцинирования после каждой стадии промывки, таким образом, представляет собой значительно менее сложный способ, и неожиданным образом приводит к еще более улучшенным результатам в отношении наиболее важного параметра, в частности для проведения способа в промышленном масштабе, т.е. селективности в отношении ценного продукта.
Список ссылочных источников
- WO 98/55229 А1
- ЕР 1221442 А1
- WO 2005/000827 А1
- WO 2007/013739 А1
- Ullmann’s Encyclopedia of Industrial Chemistry, 5th edition, volume 13, pp. 447-456
- Atlas of Zeolite Framework Types, 5th edition, Elsevier, London, England (2001)
- US 2007043226 Al
- US 6,114,551
- Wu et al., “Hydrothermal Synthesis of a novel Titanosilicate with MWW Topology”, Chemistry Letters (2000), pp. 774-775
- WO 2013/117536 A2.
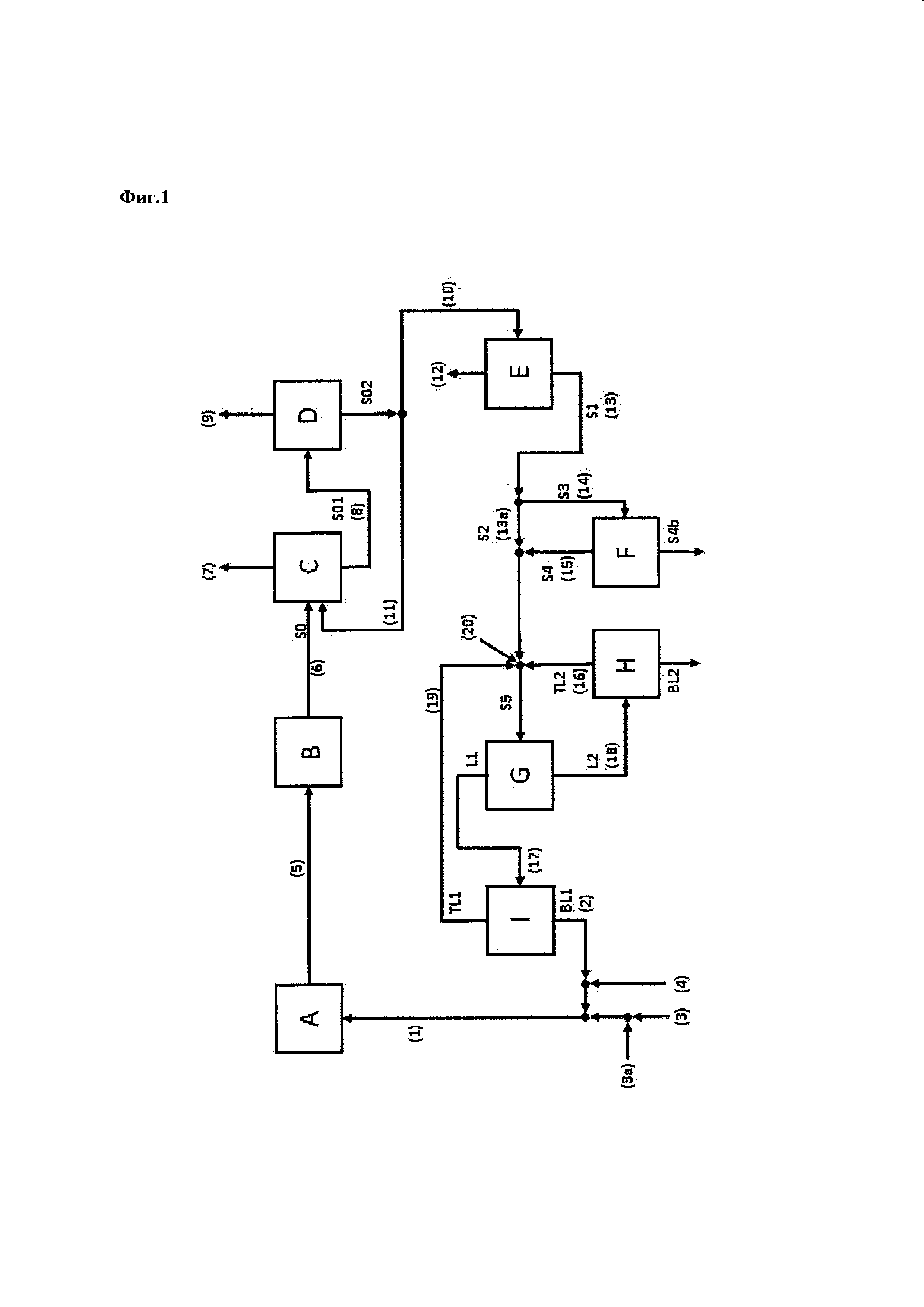
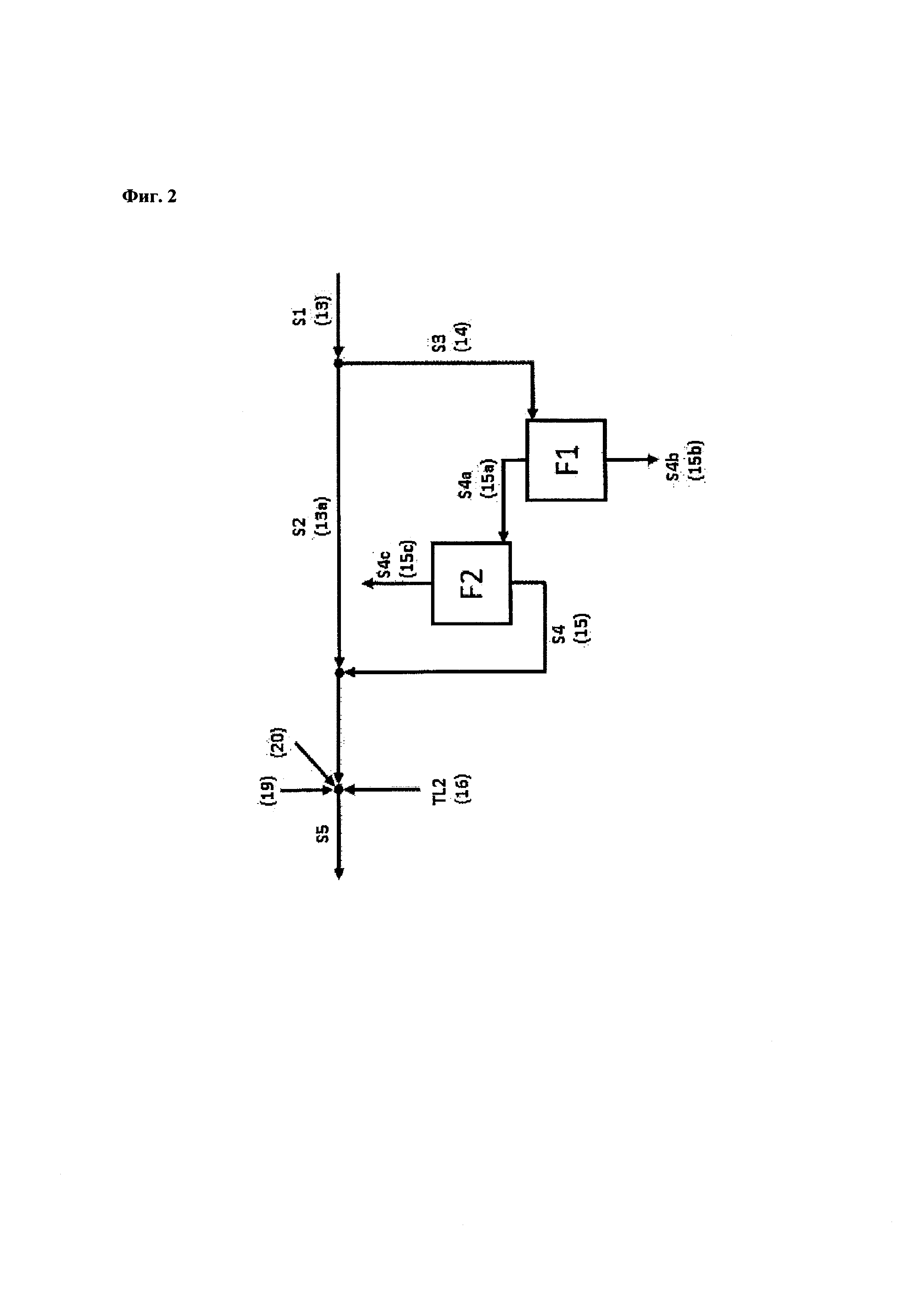
Способ хранения жидкой в условиях хранения мономерной фазы
Способ транспортировки жидкой мономерной фазы, извлеченной из резервуара для хранения, в резервуаре автозаправщика или танкера
Способ получения механически стабильных водопоглощающих полимерных частиц
Способ получения акриловой кислоты
Способ получения триэтилентетрамина (тэта) через этилендиаминдиацетонитрил (эддн)
Способ получения смеси этиленаминов
Средство для нанесения покрытий на вспенивающиеся частицы стирольного полимеризата
Способ для обнаружения и подсчета жизнеспособных микроорганизмов вида legionella pneumophila и набор для его осуществления
Способ получения смесей этиленаминов
Эластичный пеноматериал из частиц на основе смесей полиолефина/полимера стирола
Извлечение пропена посредством очистки в скруббере со смесью растворитель/вода

