Результат интеллектуальной деятельности: ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к замещенным 3-азабицикло[3,1,0]гексанам в качестве ингибиторов кетогексокиназы, способам получения указанных соединений, а также способам, включающим введение указанных соединений млекопитающему, которое нуждается в этом.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Диабет является одной из основных проблем общественного здравоохранения из-за его растущей распространенности и связанных с ним рисков для здоровья. Заболевание характеризуется высокими уровнями глюкозы в крови, возникающими вследствие нарушений продуцирования инсулина, действия инсулина или и того, и другого. Известны две основные формы диабета, 1 типа и 2 типа. Диабет 1 типа (Д1Т) развивается, когда иммунная система организма разрушает бета-клетки поджелудочной железы, единственные клетки в организме, производящие гормон инсулин, который регулирует уровень глюкозы в крови. Для выживания людям с диабетом 1 типа необходимо вводить инсулин инъекцией или при помощи насоса. Сахарный диабет 2 типа (обычно называемый Д2Т), как правило, начинается либо с устойчивости к инсулину, либо когда в организме продуцируется недостаточно инсулина для поддержания приемлемого уровня глюкозы в крови.
Хотя Д2Т чаще всего ассоциирован с гипергликемией и устойчивостью к инсулину, другие заболевания, ассоциированные с Д2Т, включают устойчивость к инсулину в печени, нарушение толерантности к глюкозе, диабетическую невропатию, диабетическую нефропатию, диабетическую ретинопатию, ожирение, дислипидемию, гипертензию, гиперинсулинемию и неалкогольную жировую болезнь печени (НАЖБП).
НАЖБП является проявлением метаболического синдрома в печени и включает целый спектр патологических состояний печени, в том числе стеатоз, неалкогольный стеатогепатит (НАСГ), фиброз, цирроз и, в конечном итоге, печеночно-клеточную карциному. НАЖБП и НАСГ считаются основными формами жировой болезни печени, поскольку ими страдают большинство людей с повышенным уровнем липидов в печени. Степень тяжести НАЖБП/НАСГ определяют на основании присутствия липидов, инфильтратов воспалительных клеток, баллонирования гепатоцитов и степени фиброза. Хотя не у всех людей, страдающих стеатозом, он прогрессирует до НАСГ, это происходит у значительной части пациентов.
Данные, полученные недавно в исследовании с участием людей, свидетельствуют о том, что употребление фруктозы может способствовать развитию НАЖБП/НАСГ (Vos, M. B., and Lavine, J. E. (2013, Hepatology 57, 2525-2531). В сравнении с глюкозой, фруктоза способствует значительному возрастанию de novo синтеза липидов (Stanhope, K. L., Schwarz, et al., (2009), J Clin Invest 119, 1322-1334), что является характерным для пациентов с НАЖБП (Lambert, J. E., et al., (2014), Gastroenterology 146, 726-735). Исследования с участием людей показали, что кратковременное употребление в пищу фруктозы вызывает повышение уровня триглицеридов в печени, и что прекращение употребления фруктозы может приводить к обращению вспять процесса накопления триглицеридов в печени (Schwarz, J. M., Noworolski, et al., (2015), J Clin Endocrinol Metab 100, 2434-2442). Кроме того, у подростков, страдающих НАЖБП, сокращение на 50% употребления сахара в течение 10 дней приводило к сокращению уровня печеночных триглицеридов на 20% (Schwarz, J. M., Noworolski, et al., (2015) PP07-3: Isocaloric Fructose Restriction for 10 Days Reduces Hepatic De Novo Lipogenesis and Liver Fat in Obese Latino and African American Children. http://press.endocrine.org.proxy1.athensams.net/doi/abs/10.1210/endo-meetings.2015.OABA.6.PP07-3).
Широкая распространенность Д2Т, ожирения и НАЖБП/НАСГ, а также сопутствующих заболеваний, таких как сердечно-сосудистые заболевания и инсульт, привела к повышению необходимости как в превентивных мерах, так и в методах терапевтического вмешательства. Диапазон стратегий в современных методах фармакологического лечения Д2Т включает использование средств, повышающих секрецию инсулина, влияющих на действие инсулина (тиазолидиндионы (TZD), бигуаниды), изменяющих метаболизм липидов (TZD, фибраты), влияющих на определяемое центральной нервной системой пищевое поведение, стимулирующих экскрецию глюкозы с мочой (ингибиторы SGLT2) и уменьшающих абсорбцию питательных вещества (ингибиторы липазы). Ингибирование KHK метаболизма фруктозы представляет собой новую альтернативу современным стратегиям лечения.
Кетогексокиназа (KHK) является основным ферментом в метаболизме фруктозы и катализирует превращение фруктозы в фруктоза-1-фосфат (F1P). KHK экспрессируется в виде двух альтернативно сплайсированных вариантов мРНК, называемых KHKa и KHKc, которые возникают в результате альтернативного сплайсинга третьего экзона. Аффинность и эффективность KHKc для фосфорилирования фруктозы намного выше, чем у KHKa, о чем свидетельствует намного более низкое значение Km (Ishimoto, Lanaspa et al., PNAS 109, 4320-4325, 2012). В то время как KHKa экспрессируется повсеместно, наиболее высокая экспрессия KHKc имеет место в печени, почках и кишечнике, основных зонах метаболизма фруктозы в организме (Diggle CP, et al. (2009) J Histochem Cytochem 57: 763-774; Ishimoto, Lanaspa, et al., PNAS 109, 4320-4325, 2012). Кроме того, сообщалось о мутациях потери функции фермента у человека, которые не приводили к неблагоприятным эффектам, за исключением появления фруктозы в моче после употребления в пищу сахара.
Более тяжелым состоянием, связанным с метаболизмом фруктозы, является врожденная непереносимость фруктозы (ВНФ, OMIM №229600), вызываемая дефектами в гене альдолазы B (GENE: ALDOB), которая представляет собой фермент, ответственный за разрушение F1P, и действует сразу после этапа KHK в метаболическом пути (Bouteldja N, et. al, J. Inherit. Metab. Dis. 2010 Apr; 33(2): 105-12; Tolan, DR, Hum Mutat. 1995; 6(3): 210-8; http://www.omim.org/entry/229600). Это редкое заболевание, которым, по оценкам, страдает 1 из 20000 людей, и мутации приводят к накоплению F1P, истощению АТФ и повышению уровня мочевой кислоты, сочетание этих признаков вызывает гипогликемию, гиперурикемию и молочный ацидоз, в числе прочих метаболических расстройств. При ВНФ нарушается способность организма метаболизировать пищевую фруктозу, что приводит к появлению острых симптомов, таких как рвота, тяжелая гипогликемия, диарея и расстройство желудка, что, в свою очередь, ведет к долгосрочным нарушениям роста, повреждению печени и почек, и, потенциально, к смерти (Ali M et al., J. Med. Genet. 1998 May: 35(5): 353-65). Пациенты обычно страдают на протяжении первых лет жизни до постановки диагноза, и единственным методом лечения является избегание фруктозы в диете. Это является проблематичным из-за присутствия данного макронутриента в большинстве продуктов питания. Помимо физических симптомов, многие пациенты испытывают эмоциональную и социальную изоляцию вследствие их необычной диеты, и постоянно борются за соблюдение строгих диетических ограничений (HFI-INFO Discussion Board, http://hfiinfo.proboards.com.; дата просмотра 14 декабря 2015 г.). У некоторых пациентов, даже при внешнем отсутствии симптомов, развивается НАЖБП и заболевание почек, что подчеркивает неадекватность самостоятельно соблюдаемого диетического ограничения как единственного варианта лечения, и наличие высокой неудовлетворенной потребности в методах лечения данного состояния.
При гипергликемических состояниях эндогенное продуцирование фруктозы происходит по полиоловому пути, пути, в котором глюкоза превращается в фруктозу с сорбитом в качестве промежуточного соединения. Активность данного пути возрастает при гипергликемии. В посвященных этому исследованиях авторы продемонстрировали, что мыши с «нулевой» мутацией KHK были защищены от вызываемого глюкозой увеличения массы тела, устойчивости к инсулину и от стеатоза печени, это свидетельствует о том, что в условиях гипергликемии эндогенно продуцируемая фруктоза может вносить вклад в развитие устойчивости к инсулину и стеатоза печени (Lanaspa, M.A., et al., Nature Comm. 4, 2434, 2013). Таким образом, ожидается, что ингибирование KHK может приносить пользу при многих заболеваниях, связанных с изменениями метаболизма любой, или обеих, из эндогенной или употребленной в пищу фруктозы.
Сохраняется потребность в легко вводимом терапевтическом препарате для лечения кардиометаболических и ассоциированных заболеваний, включая диабет (Д1Т и/или Д2Т), идиопатический Д1Т (типа 1b), латентный аутоиммунный диабет взрослых (ЛАДВ), юношеский Д2Т (ЮД), юношеский атипичный диабет (ЮАД), диабет взрослого типа у молодых (ДВТМ), связанный с недостаточностью питания диабет, гестационный диабет, гипергликемию, устойчивость к инсулину, устойчивость к инсулину в печени, нарушение толерантности к глюкозе, диабетическую невропатию, диабетическую нефропатию, заболевание почек (например, острую почечную недостаточность, дисфункцию канальцев, провоспалительные изменения в проксимальных канальцах), диабетическую ретинопатию, дисфункцию адипоцитов, висцеральное отложение жировой ткани, ожирение, нарушения пищевого поведения, чрезмерное потребление сахара, дислипидемию (в том числе гиперлипидемию, гипертриглицеридемию, повышенные уровни общего холестерина, высокие уровни ЛНП-холестерина и низкие уровни ЛВП-холестерина), гиперинсулинемию, НАЖБП (в том числе связанные заболевания, такие как стеатоз, НАСГ, фиброз, цирроз и печеночно-клеточная карцинома), ВНФ, болезнь коронарных артерий, заболевание периферических сосудов, гипертензию, эндотелиальную дисфункцию, нарушение податливости сосудов, застойную сердечную недостаточность, инфаркт миокарда (например, некроз и апоптоз), инсульт, геморрагический инсульт, ишемический инсульт, легочную гипертензию, рестеноз после ангиопластики, перемежающуюся хромоту, постпрандиальную липемию, метаболический ацидоз, кетоз, артрит, остеопороз, гипертрофию левого желудочка, заболевание периферических артерий, дегенерацию желтого пятна, катаракту, гломерулосклероз, хроническую почечную недостаточность, метаболический синдром, синдром X, предменструальный синдром, стенокардию, тромбоз, атеросклероз, транзиторные ишемические атаки, рестеноз сосудов, нарушение метаболизма глюкозы, состояния с нарушением уровней глюкозы в плазме натощак, гиперурикемию, подагру, эректильную дисфункцию, заболевания кожи и соединительной ткани, язвы стопы, язвенный колит, гиперлипопротеинемию (apo B), болезнь Альцгеймера, шизофрению, когнитивное расстройство, воспалительное заболевание кишечника, язвенный колит, болезнь Крона и синдром раздраженной кишки.
ЧЕРТЕЖИ
На Фигуре 1 приведена ПРД картина кристаллической свободной кислоты примера 4.
На Фигуре 2 приведена ПРД картина кристаллической натриевой соли примера 5.
На Фигуре 3 приведены структуры полученных в примерах соединений из таблицы 4.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
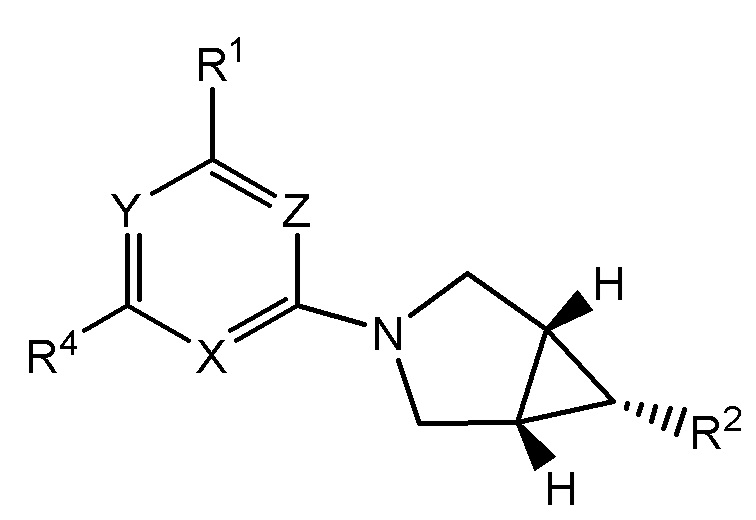
Настоящее изобретение относится к соединениям формулы I
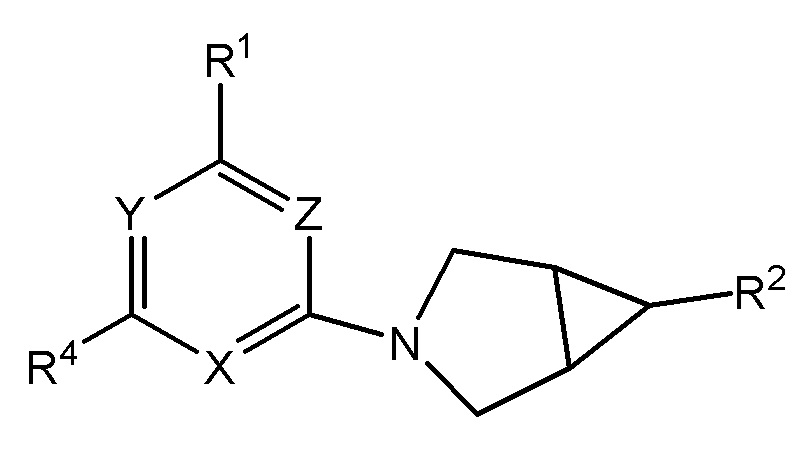 (I)
(I)
или их фармацевтически приемлемой соли, где
Y представляет собой N или C-CN;
Z представляет собой N или CH;
X представляет собой N или CR3;
при условии, что по меньшей мере одно из Y, Z или X представляет собой N;
R1 представляет собой C3-7 циклоалкил или 4-7-членный гетероциклический фрагмент, при этом гетероциклический фрагмент содержит 1-2 атома, независимо выбранных из азота, кислорода и серы, и при этом циклоалкил или гетероциклический фрагмент имеет 0-3 заместителей, независимо выбранных из -C1-3 алкила и -OH, причем -C1-3 алкил замещен 0-3 атомами галогена, и при условии, что присутствует не более одного заместителя -OH; или
N(C1-3 алкил)2, NH(C1-3 алкил) или NH(C3-4 циклоалкил), при этом каждый C1-3 алкил замещен 0-1 OH;
R2 представляет собой -(L)m-CON(RN)2, -(L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, -L-(CH2)nC(O)RC, L-(CH2)nCONHSO2RS, -L-(CH2)nSO2NHCORS, -L-(CH2)nSO2NHCONH2 или -L- (CH2)n-тетразол-5-ил;
m равно 0 или 1;
n равно 0 или 1;
RN представляет собой H или -C1-3 алкил;
RS представляет собой H или -C1-3 алкил;
L представляет собой CH2, CHF или CF2;
RC представляет собой -C1-4 алкилокси, -C1-4 алкилоксикарбонилокси-C1-4 алкилокси или -C1-4 алкилкарбонилокси-C1-4 алкилокси;
R3 представляет собой H, галоген, -CN, -C1-3 алкил, -OC1-3 алкил, -C1-3 алкил, замещенный 1-3 атомами галогена, или -C3-4 циклоалкил; и
R4 представляет собой циклопропил, циклобутил или -C1-3 алкил, замещенный 0-5 атомами галогена, насколько позволяет валентность.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где X, Y и Z представляют собой любое из следующих:
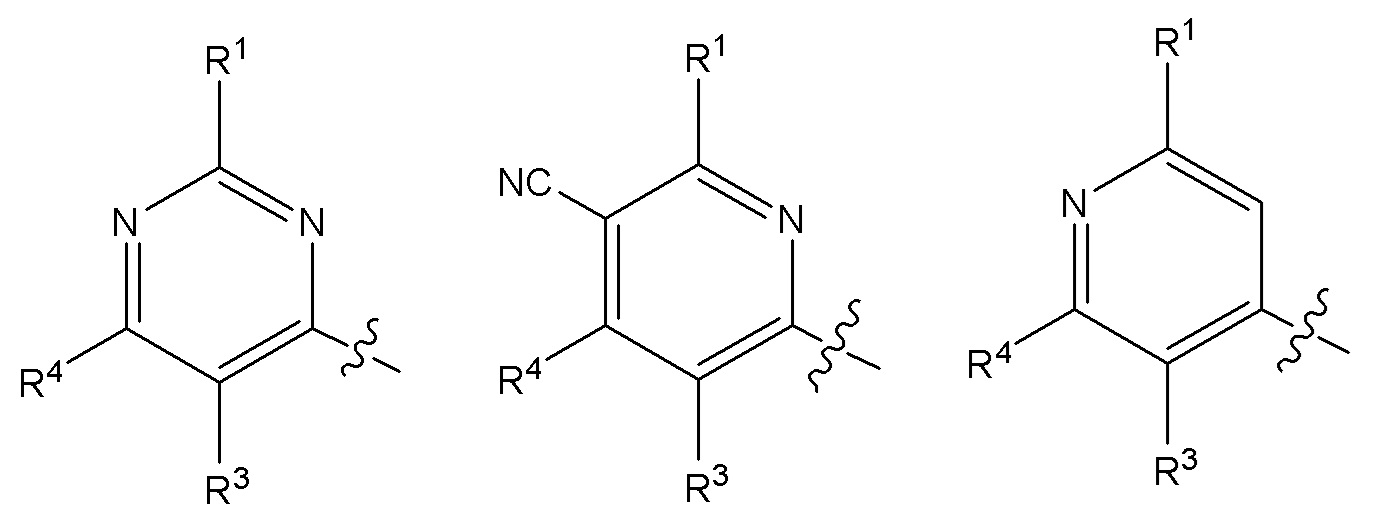
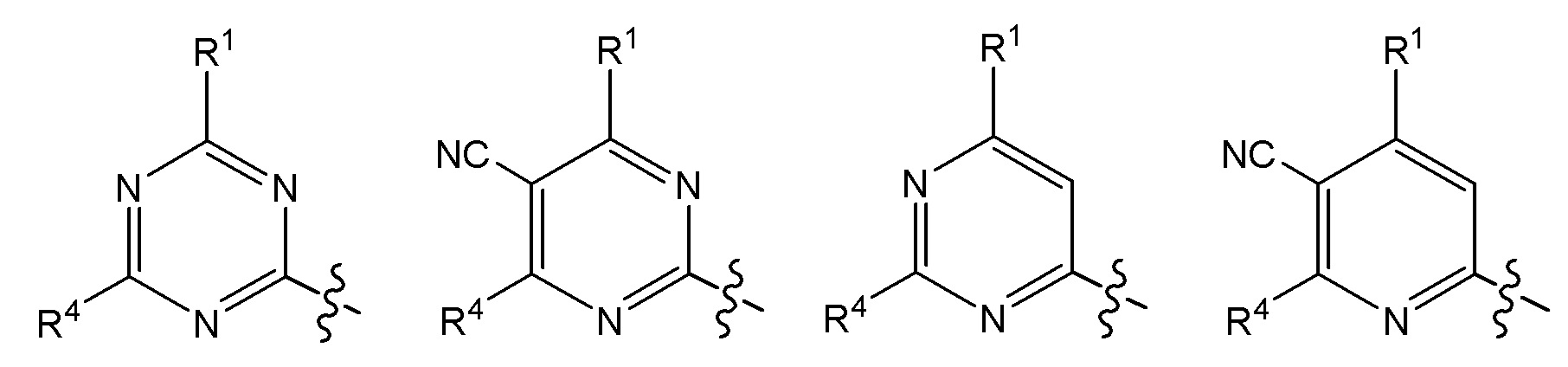
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где
Y представляет собой N или C-CN;
Z представляет собой N или CH;
X представляет собой CR3;
при условии, что по меньшей мере одно из Y или Z представляет собой N;
R1 представляет собой C3-7 циклоалкил или 4-7-членный гетероциклический фрагмент, при этом гетероциклический фрагмент содержит 1-2 атома, независимо выбранных из азота, кислорода и серы, и при этом циклоалкил или гетероциклический фрагмент имеет 0-3 заместителей, независимо выбранных из -C1-3 алкила и -OH, причем -C1-3 алкил замещен 0-3 атомами F (где галоген представляет собой F), при условии, что присутствует не более одного заместителя -OH; или
N(C1-3 алкил)2, NH(C1-3 алкил) или NH(C3-4 циклоалкил), при этом каждый C1-3 алкил замещен 0-1 OH;
R2 представляет собой -(L)m-CON(RN)2, -(L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, -L-(CH2)nC(O)RC, L-(CH2)nCONHSO2RS, -L-(CH2)nSO2NHCORS, -L-(CH2)nSO2NHCONH2 или -L- (CH2)n-тетразол-5-ил;
m равно 0 или 1;
n равно 0 или 1;
RN представляет собой H или -C1-3 алкил;
RS представляет собой H или -C1-3 алкил;
L представляет собой CH2, CHF или CF2;
RC представляет собой -C1-4 алкилокси, -C1-4 алкилоксикарбонилокси-C1-4 алкилокси или -C1-4 алкилкарбонилокси-C1-4 алкилокси;
R3 представляет собой H, галоген, -CN, -C1-3 алкил, -OC1-3 алкил, -C1-3 алкил, замещенный 1-3 атомами галогена, или -C3-4 циклоалкил; и
R4 представляет собой -C1-3 алкил, замещенный 0-5 атомами галогена, насколько позволяет валентность.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где
Y представляет собой C-CN;
Z представляет собой N;
X представляет собой CR3;
R1 представляет собой C3-7 циклоалкил или 4-7-членный гетероциклический фрагмент, при этом гетероциклический фрагмент содержит 1-2 атома, независимо выбранных из азота, кислорода и серы, и при этом циклоалкил или гетероциклический фрагмент имеет 0-3 заместителей, независимо выбранных из -C1-3 алкила и -OH, при условии, что присутствует не более одного заместителя -OH;
R2 представляет собой -(L)m-CON(RN)2, -(L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, -L-(CH2)nC(O)RC, L-(CH2)nCONHSO2RS, -L-(CH2)nSO2NHCORS или -L- (CH2)n-тетразол-5-ил;
m равно 0 или 1;
n равно 0 или 1;
RN представляет собой H или -C1-3 алкил;
RS представляет собой H или -C1-3 алкил;
L представляет собой CH2, CHF, или CF2;
RC представляет собой -C1-4 алкилокси, -C1-4 алкилоксикарбонилокси-C1-4 алкилокси или -C1-4 алкилкарбонилокси-C1-4 алкилокси;
R3 представляет собой H, галоген, -CN, -C1-3 алкил, -OC1-3 алкил, -C1-3 алкил, замещенный 1-3 атомами галогена, или -C3-4 циклоалкил; и
R4 представляет собой -C1-3 алкил, замещенный 0-5 атомами галогена, насколько позволяет валентность.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где
Y представляет собой N;
Z представляет собой N;
X представляет собой CR3;
R1 представляет собой C3-7 циклоалкил или 4-7-членный гетероциклический фрагмент, при этом гетероциклический фрагмент содержит 1-2 атома, независимо выбранных из азота, кислорода и серы, и при этом циклоалкил или гетероциклический фрагмент имеет 0-3 заместителей, независимо выбранных из -C1-3 алкила и -OH, при условии, что присутствует не более одного заместителя -OH;
R2 представляет собой -(L)m-CON(RN)2, -(L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, -L-(CH2)nC(O)RC, L-(CH2)nCONHSO2RS, -L-(CH2)nSO2NHCORS или -L- (CH2)n-тетразол-5-ил;
m равно 0 или 1;
n равно 0 или 1;
RN представляет собой H или -C1-3 алкил;
RS представляет собой H или -C1-3 алкил;
L представляет собой CH2, CHF или CF2;
RC представляет собой -C1-4 алкилокси, -C1-4 алкилоксикарбонилокси-C1-4 алкилокси или -C1-4 алкилкарбонилокси-C1-4 алкилокси;
R3 представляет собой H, галоген, -CN, -C1-3 алкил, -OC1-3 алкил, -C1-3 алкил, замещенный 1-3 атомами галогена, или -C3-4 циклоалкил; и
R4 представляет собой -C1-3 алкил, замещенный 0-5 атомами галогена, насколько позволяет валентность.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где
Y представляет собой N или C-CN;
Z представляет собой N или CH;
X представляет собой CR3;
при условии, что по меньшей мере одно из Y или Z представляет собой N;
R1 представляет собой C3-7 циклоалкил или 4-7-членный гетероциклический фрагмент, при этом гетероциклический фрагмент содержит 1-2 атома, независимо выбранных из азота, кислорода и серы, и при этом циклоалкил или гетероциклический фрагмент имеет 0-3 заместителей, независимо выбранных из -C1-3 алкила и -OH, при условии, что присутствует не более одного заместителя -OH;
R2 представляет собой -(L)m-CON(RN)2, -(L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, L (CH2)nC(O)RC, L-(CH2)nCONHSO2RS, -L-(CH2)nSO2NHCORS или -L-(CH2)n-тетразол-5-ил;
m равно 0 или 1;
n равно 0 или 1;
RN представляет собой H или -CH3;
RS представляет собой H или -CH3;
L представляет собой CH2, CHF или CF2;
RC представляет собой -C1-4 алкилокси, -C1-4 алкилоксикарбонилокси-C1-4 алкилокси или -C1-4 алкилкарбонилокси-C1-4 алкилокси;
R3 представляет собой H, -Cl, -CH3, -CH2CH3, -O-CH3, циклопропил или CN; и
R4 представляет собой -CF3, -CHF2 или -CF2CH3.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где RN представляет собой H или -CH3.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где RS представляет собой H или -CH3.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R2 представляет собой -CH2CO2H (n равно 0 и L представляет собой CH2). Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R2 представляет собой -CH2CO2H, CH2CO2CH3 или -CH2CO2CH2CH3 (n равно 0, Rc представляет собой CH3 или CH2CH3, когда присутствует, и L представляет собой CH2). Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R2 представляет собой CH2CH2CO2H, -CH2CH2CO2CH3 или -CH2CH2CO2CH2CH3 (n равно 1, Rc представляет собой CH3 или CH2CH3, когда присутствует, и L представляет собой CH2).
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R2 представляет собой -(L)m-CON(RN)2, (L)m-SO2RS, -L-(CH2)nSO2RS, -L-(CH2)nCO2H, L (CH2)nC(O)RC, L-(CH2)nCONHSO2RS, L (CH2)nSO2NHCORS или -L-(CH2)n-тетразол-5-ил.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R3 представляет собой H, -Cl, -CH3, CH2CH3, -O-CH3, циклопропил или CN.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R4 представляет собой -CF3, -CHF2 или CF2CH3.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R1 представляет собой циклобутил (C4 циклоалкил), имеющий 0-3 заместителей, независимо выбранных из -CH3 и -OH, при условии, что присутствует не более одного заместителя -OH.
Другой вариант осуществления относится к любому другому варианту осуществления, обсуждаемому в настоящем документе применительно к соединениям формулы I, или их фармацевтически приемлемой соли, где R1 представляет собой 4-7-членный гетероциклический фрагмент, выбранный из азетидин-1-ила, пирролидин-1-ила и пиперидин-1-ила (R1 представляет собой 4-7-членный гетероциклический фрагмент), имеющий 0-3 заместителей, независимо выбранных из CH3 и -OH, при условии, что присутствует не более одного заместителя -OH.
Предпочтительный вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где X, R2, m, n, RN, RS, L, RC, R3 и R4 представляют собой любой вариант осуществления, описанный в настоящем документе, при этом R1 представляет собой азетидин-1-ил, пирролидин-1-ил и пиперидин-1-ил, имеющий 0-2 заместителей -CH3 и имеющий 0-1 заместителей -OH, и при этом Y представляет собой C-CN и Z представляет собой N, или каждый из Y и Z представляет собой N.
Другой предпочтительный вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где X, R2, m, n, RN, RS, L, RC, R3 и R4 представляют собой любой вариант осуществления, описанный в настоящем документе, при этом R1 представляет собой азетидин-1-ил, имеющий 1-2 заместителей -CH3 и имеющий 0-1 заместителей -OH, и при этом Y представляет собой C-CN и Z представляет собой N, или каждый из Y и Z представляет собой N.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, где Y представляет собой C-CN и Z представляет собой N, или каждый из Y и Z представляет собой N.
Другой вариант осуществления изобретения относится к соединениям формулы I(a)
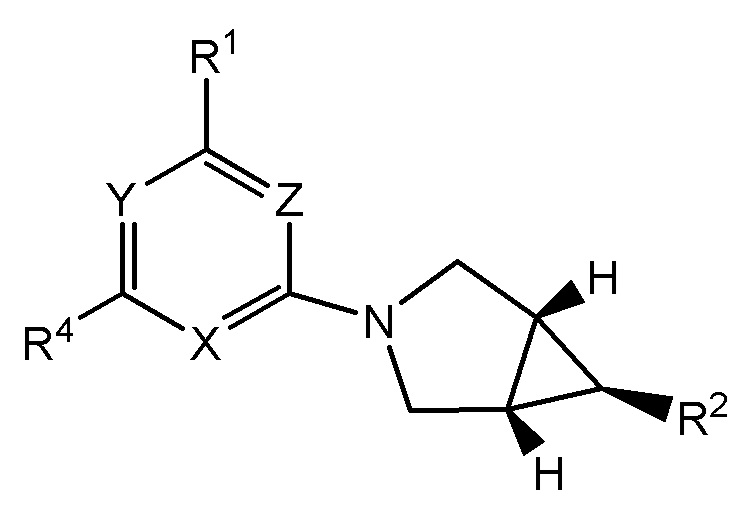 (Ia),
(Ia),
или их фармацевтически приемлемой соли, где заместитель R2 на азабицикло[3,1,0]гекс-6-иле и атомы H на атомах углерода моста находятся в одной плоскости, и при этом X, Y, Z, R2, m, n, RN, RS, L, RC, R3 и R4 представляют собой любой вариант осуществления, описанный в настоящем документе.
Другой вариант осуществления изобретения относится к соединениям формулы I(b)
 (Ib),
(Ib),
или их фармацевтически приемлемой соли, где заместитель R2 на азабицикло[3,1,0]гекс-6-иле и атомы H на атомах углерода моста находятся в одной плоскости, и при этом X, Y, Z, R2, m, n, RN, RS, L, RC, R3 и R4 представляют собой любой вариант осуществления, описанный в настоящем документе.
Используемый в настоящем документе термин «алкил» означает одновалентную углеводородную группу с прямой или разветвленной цепью формулы -CnH(2n+1). Неограничивающие примеры включают метил, этил, пропил, бутил, 2-метилпропил, 1,1-диметилэтил, пентил и гексил.
Используемый в настоящем документе термин «циклоалкил» означает циклическую одновалентную углеводородную группу формулы -CnH(2n-1), содержащую по меньшей мере три атома углерода. Неограничивающие примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Используемый в настоящем документе термин «алкилокси» означает алкильный заместитель, присоединеный через атом кислорода. Неограничивающие примеры включают метокси, этокси, пропокси и бутокси.
Используемый в настоящем документе термин «алкилоксикарбонилокси» означает алкокси-группу, присоединенную через карбонильную группу (-CO-). Неограничивающие примеры включают метоксикарбонил, этоксикарбонил и пропоксикарбонил.
Используемый в настоящем документе термин «алкилкарбонилокси» означает алкильную группу, присоединенную через карбонилокси-группу (-C(=O)-O-). Репрезентативные примеры включают метилкарбонилокси, этилкарбонилокси и трет-бутилкарбонилокси.
Используемый в настоящем документе термин «алкилоксикарбонилоксиалкилокси» означает алкилоксикарбонилокси-группу, присоединенную через алкилокси-группу.
Используемый в настоящем документе термин «галоген» означает F, Cl, Br, I.
Используемый в настоящем документе термин «гетероциклический фрагмент» означает циклоалкильную группу, имеющую 4-7 атомов углерода, в которой одна или более кольцевых метиленовых групп (-CH2-) были заменены группой, выбранной из -O-, -S- или -N-, где требования валентности для -N- удовлетворяются на счет H или создается точка присоединения.
Общепринятые сокращения, используемые в настоящем документе:
АДФ означает аденозиндифосфат;
АТФ означает аденозинтрифосфат;
CDCl3 означает дейтерохлороформ;
CO2Et означает этилкарбоксилат;
DCM означает дихлорметан;
DIPEA означает N,N-диизопропилэтиламин;
DMF означает диметилформамид;
ДМСО означает диметилсульфоксид;
EtOAc означает этилацетат;
Ч или ч или час означает час(ы);
HEPES означает 4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота;
KCl означает хлорид калия;
Мин означает минута(ы);
MgCl2 означает хлорид магния;
NaHCO3 означает бикарбонат натрия;
Na2SO4 означает сульфат натрия;
НАДH означает никотинамидадениндинуклеотид (восстановленная форма)
НАД+ означает никотинамидадениндинуклеотид (окисленная форма)
PEP означает фосфоенолпируват;
RT или rt означает комнатная температура;
TCEP означает трис(2-карбоксиэтил)фосфин;
TFA означает трифторуксусная кислота;
THF означает тетрагидрофуран.
Другой вариант осуществления относится к соединениям формулы I, или их фармацевтически приемлемой соли, при этом каждое соединение независимо выбирают из любого одного или более полученных в примерах соединений, предложенных в настоящем документе.
Одним из способов осуществления изобретения является введение соединения формулы (I) в форме пролекарства. Таким образом, некоторые производные соединения формулы (I), которые сами могут иметь небольшую, или совсем не иметь, фармакологическую активность, при введении в, или на, тело могут превращаться в соединение формулы (I), имеющее желаемую активность, например, за счет гидролитического расщепления, в частности, гидролитического расщепления, катализируемого ферментом эстеразой или пептидазой. Такие производные называют «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в сборниках «Pro-drugs as Novel Delivery Systems», Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association). Также можно сделать ссылку на Nature Reviews/Drug Discovery, 2008, 7, 355 и Current Opinion in Drug Discovery and Development, 2007, 10, 550.
Пролекарства по изобретению могут, например, быть получены путем замены соответствующих функциональных групп, присутствующих в соединениях формулы (I), определенными фрагментами, известными специалистам в данной области как «профрагменты», описанные, например, в «Design of Prodrugs» автора H. Bundgaard (Elsevier, 1985).
Таким образом, пролекарство по изобретению представляет собой (a) сложноэфирное или амидное производное карбоновой кислоты в соединении формулы (I); (b) сложноэфирное, карбонатное, карбаматное, фосфатное или эфирное производное гидроксильной группы в соединении формулы (I); (c) амидное, иминовое, карбаматное или аминопроизводное аминогруппы в соединении формулы (I); (d) тиоэфирные, тиокарбонатные, тиокарбаматные или сульфидные производные тиоловой группы в соединении формулы (I); или (e) оксимовое или иминовое производное карбонильной группы в соединении формулы (I).
Некоторые конкретные примеры пролекарств по изобретению включают соединения, в которых R2 представляет собой -L-(CH2)nC(O)RC. Ниже приводится более общее руководство в отношении пролекарств по данному изобретению:
(i) где соединение формулы (I) содержит функциональную группу карбоновой кислоты (-COOH), ее сложный эфир, например, соединение, в котором атом водорода функциональной группы карбоновой кислоты соединения формулы (I) заменен C1-8 алкилом (например, этилом) или (C1-8 алкил)C(=O)OCH2- (например, tBuC(=O)OCH2-);
(ii) где соединение формулы (I) содержит спиртовую функциональную группу (-OH), ее сложный эфир, например, соединение, в котором атом водорода спиртовой функциональной группы соединения формулы (I) заменен -CO(C1-8 алкилом) (например, метилкарбонилом) или спирт эстерифицирован аминокислотой;
(iii) где соединение формулы (I) содержит спиртовую функциональную группу (-OH), ее эфир, например, соединение, в котором атом водорода спиртовой функциональной группы соединения формулы (I) заменен (C1-8 алкил)C(=O)OCH2- или -CH2OP(=O)(OH)2;
(iv) где соединение формулы (I) содержит спиртовую функциональную группу (-OH), ее фосфат, например, соединение, в котором атом водорода спиртовой функциональной группы соединения формулы (I) заменен -P(=O)(OH)2 или -P(=O)(ONa)2 или -P(=O)(O-)2Ca2+;
(v) где соединение формулы (I) содержит первичную или вторичную функциональную аминогруппу (-NH2 или -NHR, где R ≠ H), ее амид, например, соединение, в котором, в зависимости от обстоятельств, один или оба атома водорода функциональной аминогруппы соединения формулы (I) заменен/заменены (C1-10) алканоилом, -COCH2NH2, или аминогруппа дериватизирована аминокислотой;
(vi) где соединение формулы (I) содержит первичную или вторичную функциональную аминогруппу (-NH2 или -NHR, где R ≠ H), ее амин, например, соединение, в котором, в зависимости от обстоятельств, один или оба атома водорода функциональной аминогруппы соединения формулы (I) заменен/заменены -CH2OP(=O)(OH)2.
Некоторые соединения формулы (I) могут сами действовать как пролекарства других соединений формулы (I). Также возможно соединять вместе два соединения формулы (I) в форме пролекарства. В определенных обстоятельствах пролекарство соединения формулы (I) может быть образовано путем внутреннего связывания двух функциональных групп в соединении формулы (I), например, путем образования лактона.
При использовании в настоящем документе «формула I» также может иметь названия «соединение(я) по изобретению», «соединение(я) по настоящему изобретению», «изобретение» и «соединение формулы I». Такие термины используют взаимозаменяемо. Кроме того, подразумевают, что варианты осуществления, описанные в настоящем документе со ссылкой на формулу I, также относятся к соединениям формулы I(a) или формулы I(b). Такие термины также должны включать все формы соединения формулы I, в том числе его гидраты, сольваты, клатраты, изомеры, кристаллические (включая сокристаллы) и некристаллические формы, изоморфы, полиморфы, таутомеры и метаболиты. Например, соединения по изобретению, или их фармацевтически приемлемые соли, могут существовать в не сольватированной и сольватированной формах. Если растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, независимо от содержания влаги. Однако если растворитель или вода связаны слабо, как в канальных сольватах и гигроскопических соединениях, содержание воды/растворителя будет зависеть от содержания влаги и условий высушивания. В таких случаях нестехиометрия будет нормой.
В соответствии с принятой в настоящее время системой классификации органические гидраты подразделяют на гидраты с изолированным расположением молекул воды, канальные гидраты или гидраты с молекулами воды, координированными вокруг иона металла, смотри сборник Polymorphism in Pharmaceutical Solids автора K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Гидраты с изолированным расположением молекул воды представляют собой гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом из-за находящихся между ними органических молекул. В канальных гидратах молекулы воды находятся в каналах решетки, рядом с другими молекулами воды. В гидратах с молекулами воды, координированными вокруг иона металла, молекулы воды связаны с ионом металла.
Если растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, независимо от содержания влаги. Однако если растворитель или вода связаны слабо, как в канальных сольватах и гигроскопических соединениях, содержание воды/растворителя будет зависеть от содержания влаги и условий высушивания. В таких случаях нестехиометрия будет нормой.
В объем изобретения также входят многокомпонентные комплексы (отличные от солей и сольватов), в которых лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы такого типа включают клатраты (комплексы включения лекарственного средства в молекулы «хозяина») и сокристаллы. Последние, как правило, определяют как кристаллические комплексы нейтральных молекулярных компонентов, которые связаны друг с другом посредством нековалентных взаимодействий, но это также может быть комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителей или путем физического совместного измельчения компонентов, смотри Chem Commun, 17, 1889-1896, авторов O. Almarsson и M. J. Zaworotko (2004). Обзор многокомпонентных комплексов можно найти в J Pharm Sci, 64 (8), 1269-1288, автора Haleblian (август 1975 г.).
Соединения по изобретению могут содержать ассиметричные или хиральные центры и, таким образом, существовать в разных стереоизомерных формах. Если нет иных указаний, подразумевают, что все стереоизомерные формы соединений по изобретению, а также их смеси, включая рацемические смеси, являются частью настоящего изобретения. Кроме того, изобретение включает все геометрические и позиционные изомеры. Например, если соединение по изобретению содержит двойную связь или конденсированное кольцо, как цис-, так и транс-формы, а также смеси, входят в объем изобретения.
Диастереомерные смеси можно разделять на отдельные диастереоизомеры на основании их физико-химических различий методами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделять путем превращения энантиомерной смеси в диастереомерную смесь за счет реакции с соответствующим оптически активным соединением (например, хиральным вспомогательным соединением, таким как хиральный спирт или хлорид кислоты Мошера), разделения диастереоизомеров и превращения (например, за счет гидролиза) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также можно разделять с использованием колонки для хиральной ВЭЖХ. Альтернативно, конкретные стереоизомеры можно синтезировать, используя оптически активный исходный материал, методом ассиметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, либо путем превращения одного стереоизомера в другой методом ассиметричной трансформации.
Если соединения по изобретению обладают одним или более стереогенными центрами и в названии или структуре отсутствует указание на стереохимию, понятно, что название или структура должны включать все формы соединения, в том числе рацемическую форму. Если соединения по изобретению обладают двумя или более стереогенными центрами и в названии или структуре имеется указание на абсолютную или относительную стереохимию, обозначения R и S относятся, соответственно, к каждому стереогенному центру в восходящем числовом порядке (1, 2, 3 и так далее) согласно общепринятый системе нумерации IUPAC для каждой молекулы. Стереогенные центры молекул могут быть представлены несколькими альтернативными комбинациями сплошных и пунктирных клиньев. Многие полученные в примерах соединения, приведенные в настоящем документе, могут содержать 3,1,0 кольцевую систему с мезо-стереохимией в соответствии с правилами наименования химических соединений IUPAC или правилами Кана-Ингольда-Прелонга, которые были использованы для присвоения названий полученным в примерах и промежуточным соединениям, наряду с использованием программ ChemBioDraw Ultra 14.0.0.117 и/или ACD/Name v. 12.0. Следует отметить, что связи могут быть обозначены клиньями или пунктиром, и при этом представлять одну и ту же стереохимию, например, сравните примеры 1 и 54, вследствие вращения вокруг связи между атомом азота фрагмента 3.1.0 и центральным фрагментом, и которое также может иметь место вокруг связи между центральным фрагментом и R1, где центральный фрагмент представляет собой пиридинил, пиримидинил, или триазинил, в зависимости от определений X, Y, и Z.
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение формулы I, или его фармацевтически приемлемую соль, определенное в любом из вариантов осуществления, описанных в настоящем документе, в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом.
Настоящее изобретение также относится к любому одному или сочетанию из следующего:
способ лечения заболевания, в случае которого предписано введение ингибитора KHK, у субъекта, который нуждается в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли;
применение соединения формулы I, или его фармацевтически приемлемой соли, в производстве медикамента для лечения заболевания, в случае которого предписано введение ингибитора KHK;
соединение формулы I, или его фармацевтически приемлемая соль, для применения в качестве медикамента;
соединение формулы I, или его фармацевтически приемлемая соль, для применения в лечении заболевания, в случае которого предписано введение ингибитора KHK;
фармацевтическая композиция, содержащая соединение формулы I, или его фармацевтически приемлемую соль, и фармацевтически приемлемый эксципиент;
фармацевтическая композиция для лечения заболевания, в случае которого предписано введение ингибитора KHK, содержащая соединение формулы I, или его фармацевтически приемлемую соль.
При использовании в настоящем документе, лечение заболевания, в случае которого предписано введение ингибитора KHK, означает, что по меньшей мере одно соединение формулы I, или его фармацевтически приемлемую соль, вводят пациенту, который нуждается в этом, для лечения, или используют в производстве медикамента для лечения пациента, который нуждается в этом, путем ингибирования KHK и последующего метаболизма фруктозы, для лечения заболевания, нарушения, состояния или сопутствующего заболевания (как правило, называемого в настоящем документе заболеванием), выбранного из любого одного или более из: Д1Т, Д2Т, идиопатического Д1Т, ЛАДВ, ЮД, ЮАД, ДВТМ, связанного с недостаточностью питания диабета, гестационного диабета, гипергликемии, устойчивости к инсулину, устойчивости к инсулину в печени, нарушения толерантности к глюкозе, диабетической невропатии, диабетической нефропатии, заболевания почек, острой почечной недостаточности, дисфункции канальцев, провоспалительных изменений в проксимальных канальцах, диабетической ретинопатии, дисфункции адипоцитов, висцерального отложения жировой ткани, ожирения, нарушения пищевого поведения, чрезмерного потребления сахара, дислипидемии, гиперлипидемии, гипертриглицеридемии, повышенных уровней общего холестерина, высоких уровней ЛНП-холестерина, низких уровней ЛВП-холестерина, гиперинсулинемии, НАЖБП, стеатоза, НАСГ, фиброза, цирроза, печеночно-клеточной карциномы, ВНФ, болезни коронарных артерий, заболевания периферических сосудов, гипертензии, эндотелиальной дисфункции, нарушения податливости сосудов, застойной сердечной недостаточности, инфаркта миокарда, инсульта, геморрагического инсульта, ишемического инсульта, легочной гипертензии, рестеноза после ангиопластики, перемежающейся хромоты, постпрандиальной липемии, метаболического ацидоза, кетоза, артрита, остеопороза, гипертрофии левого желудочка, заболевания периферических артерий, дегенерации желтого пятна, катаракты, гломерулосклероза, хронической почечной недостаточности, метаболического синдрома, синдрома X, предменструального синдрома, стенокардии, тромбоза, атеросклероза, транзиторных ишемических атак, рестеноза сосудов, нарушения метаболизма глюкозы, состояний с нарушением уровней глюкозы в плазме натощак, гиперурикемии, подагры, эректильной дисфункции, заболеваний кожи и соединительной ткани, язв стопы, язвенного колита, гиперлипопротеинемии (apo B), болезни Альцгеймера, шизофрении, когнитивного расстройства, воспалительного заболевания кишечника, язвенного колита, болезни Крона и синдрома раздраженной кишки.
В другом варианте осуществления изобретение относится к способу лечения заболевания, выбранного из любого одного или сочетания из: Д1Т, Д2Т, устойчивости к инсулину, заболевания почек, острой почечной недостаточности, дисфункции канальцев, провоспалительных изменений в проксимальных канальцах, дисфункции адипоцитов, висцерального отложения жировой ткани, ожирения, нарушения пищевого поведения, чрезмерного потребления сахара, дислипидемии, гиперлипидемии, гипертриглицеридемии, повышенных уровней общего холестерина, высоких уровней ЛНП-холестерина, низких уровней ЛВП-холестерина, НАЖБП, стеатоза, НАСГ, фиброза, цирроза, печеночно-клеточной карциномы, ВНФ, гипертензии, эндотелиальной дисфункции, метаболического синдрома, гиперурикемии и подагры.
Изобретение также относится к фармацевтической композиции, содержащей соединение формулы I, или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, для использования в лечении любого одного или более заболеваний, обсуждаемых в настоящем документе.
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение формулы I, или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом.
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение формулы I, или его фармацевтически приемлемую соль, как определено в любом из вариантов осуществления, описанных в настоящем документе, в смеси с по меньшей мере одним другим терапевтическим средством, описанным в настоящем документе.
Выражение «терапевтически эффективное количество» означает количество соединения по изобретению, которое (i) лечит или предотвращает конкретное заболевание, (ii) ослабляет, уменьшает или устраняет один или более симптомов конкретного заболевания, или (iii) предотвращает или отсрочивает возникновение одного или более симптомов конкретного заболевания, описанного в настоящем документе.
Термин «млекопитающее» относится к теплокровным животным, включая людей (мужчин или женщин) и животных-компаньонов (например, собак, кошек, лошадей и так далее), а также других животных, включая морских свинок, мышей, крыс, песчанок, крупный рогатый скот, коз, овец, обезьян и шимпанзе.
Термин «пациент» является альтернативой термину «млекопитающее».
Выражение «фармацевтически приемлемые» указывает на то, что вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, входящими в состав препарата, и/или с организмом млекопитающего, которого лечат этим препаратом.
Термины «лечение», «лечить» или «терапия» включают как превентивное, то есть, профилактическое, так и паллиативное воздействие, то есть, облегчение, ослабление или замедление прогрессирования заболевания пациента, либо любого повреждения ткани, связанного с заболеванием.
Настоящее изобретение включает все фармацевтически приемлемые меченые изотопами соединения формулы I, в которых один или более атомов заменены атомами, имеющими то же атомное число, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O.
Некоторые меченые изотопами соединения формулы I, например, те, которые содержат радиоактивный изотоп, полезны в исследованиях распределения в тканях лекарственного средства и/или субстрата. Радиоактивные изотопы тритий, то есть, 3H, и углерод-14, то есть, 14C, являются особенно полезными для этой цели ввиду легкости их включения в соединения и доступности средств для их обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, то есть, 2H, может иметь определенные терапевтические преимущества, возникающие вследствие большей метаболической стабильности, например, повышенного времени полураспада in vivo, или необходимости в меньшей дозе, и, таким образом, в некоторых обстоятельствах может быть предпочтительным.
Замещение позитронно-активными изотопами, такими как 11C, 18F, 15O и 13N, может быть полезным для исследований методом позитронно-эмиссионной томографии (ПЭТ) с целью изучения занятости рецептора субстратом.
Меченые изотопами соединения формулы I, как правило, могут быть получены общепринятыми методами, известными специалистам в данной области, или методами, аналогичными тем, которые описаны в сопроводительных разделах «Примеры» и «Получение», с использованием соответствующих меченых изотопами реагентов вместо ранее используемых немеченых реагентов.
Комбинированные средства
Соединения по настоящему изобретению могут быть использованы, отдельно или в сочетании с другими терапевтическими средствами, для лечения различных состояний или заболеваний. Соединение(я) по настоящему изобретению и другое терапевтическое средство(а) можно вводить одновременно (либо в одной и той же лекарственной форме, либо в разных лекарственных формах) или последовательно.
Введение двух или более соединений «в сочетании» означает, что два соединения вводят достаточно близко по времени, чтобы присутствие одного изменяло биологические эффекты другого. Два или более соединений можно вводить одновременно, параллельно или последовательно. Кроме того, одновременное введение можно выполнять путем смешивания соединений перед введением или путем введения соединений в один и тот же момент времени, но в разных лекарственных формах, в одни и те же, или разные, зоны введения.
В другом варианте осуществления соединения по данному изобретению вводят совместно с любым одним или более дополнительными терапевтическими средствами, описанными в настоящем документе. Комбинированные средства вводят млекопитающему в терапевтически эффективном количестве для лечения заболеваний, описанных в настоящем документе.
Выражения «параллельное введение», «совместное введение», «одновременное введение» и «введение в одно и то же время» означают, что соединения вводят в сочетании.
В другом варианте осуществления настоящего изобретения соединение формулы I можно вводить совместно со средством против ожирения, которое выбирают из группы, состоящей из действующих избирательно в кишечнике ингибиторов MTP (например, дирлотапида, митратапида и имплитапида, R56918 (CAS № 403987 и CAS № 913541-47-6)), агонистов холицистокинина-A (CCK-A) (например, N-бензил-2-[4-(1H-индол-3-илметил)-5-оксо-1-фенил-4,5-дигидро-2,3,6,10b-тетраазабензо[e]азулен-6-ил]-N-изопропилацетамида, описанного в PCT публикации № WO 2005/116034 или публикации патента США № 2005-0267100 A1), агонистов 5HT2c (например, лоркасерина), агониста MCR4 (например, соединений, описанных в US 6818658), ингибитора липазы (например, цетилистата), PYY3-36 (при использовании в настоящем документе «PYY3-36» включает аналоги, такие как пегилированные PYY3-36, например, те, которые описаны в публикации патента США 2006/0178501), антагонистов опиоидов (например, налтрексона), сочетания налтрексона с бупропионом, олеоил-эстрона (CAS № 180003-17-2), обинепитида (TM30338), прамлинтида (симлин®), тезофензина (NS2330), лептина, лираглутида, бромкриптина, ингибиторов липазы (таких как тетрагидролипстатин, то есть орлистат), эксенатида (баета®), AOD-9604 (CAS № 221231-10-3) и сибутрамина.
Другие средства против ожирения включают ингибиторы 11b-гидроксистероид-дегидрогеназы 1 (11β-HSD типа 1), ингибитор стеароил-CoA-десатуразы 1 (SCD-1), ингибиторы обратного захвата моноаминов (такие как сибутрамин), симпатомиметики, агонисты β3-адренорецепторов, агонисты дофамина (такие как бромкриптин), аналоги меланоцит-стимулирующего гормона, антагонисты меланин-концентрирующего гормона, лептин (белок OB), аналоги лептина, агонисты лептина, антагонисты галанина, анорексигенные средства (такие как агонист бомбезина), антагонисты нейропептида Y (например, антагонисты NPY Y5), тиреомиметики, дегидроэпиандростерон или его аналог, агонисты или антагонисты рецепторов глюкокортикоидов, антагонисты рецепторов орексина, агонисты рецепторов глюкагоноподобного пептида 1, цилиарные нейротрофические факторы (такие как аксокин™, поставляемые компанией Regeneron Pharmaceuticals, Inc., Tarrytown, NY и компанией Procter & Gamble, Cincinnati, OH), ингибиторы человеческого агути-связанного белка (AGRP), антагонисты рецепторов грелина, антагонисты или обратные агонисты рецепторов гистамина 3, агонисты рецепторов нейромедина U, ингибиторы MTP/ApoB (например, действующие избирательно в кишечнике ингибиторы MTP), антагонисты рецепторов орексина, сочетание налтрексона с бупропионом и тому подобное.
В другом варианте осуществления настоящего изобретения соединение формулы I можно вводить совместно с антидиабетическим средством, выбранным из группы, состоящей из ингибиторов ацетил-CoA-карбоксилазы (ACC) (например, тех, которые описаны в WO 2009144554, WO 2003072197, WO 2009144555 и WO 2008065508), ингибиторов диацилглицерин-O-ацилтрансферазы 1 (DGAT-1) (например, тех, которые описаны в WO 09016462 или WO 2010086820, AZD7687 или LCQ908), ингибиторов моноацилглицерин-O-ацилтрансферазы, ингибиторов фосфодиэстеразы (PDE)-10, активаторов AMPK, препаратов сульфонилмочевины (например, ацетогексамида, хлорпропамида, диабинеса, глибенкламида, глипизида, глибурида, глимепирида, гликлазида, глипентида, гликидона, глисоламида, толазамида и толбутамида), меглитинида, ингибиторов α-амилазы (например, тендамистата, трестатина и AL-3688), ингибиторов α-глюкозид-гидролазы (например, акарбозы), ингибиторов α-глюкозидазы (например, адипозина, камиглибоза, эмиглитата, миглитола, воглибоза, прадимицина Q и сальбостатина), агонистов PPARγ (например, балаглитазона, циглитазона, дарглитазона, энглитазона, изаглитазона, пиоглитазона и розиглитазона), агонистов PPAR α/γ (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, MK-0767, SB-219994 и сароглитазара), бигуанидов (например, метформина), антагонистов рецепторов глюкагона, модуляторов, таких как агонисты, глюкагоноподобного пептида 1 (GLP-1) (например, эксендина-3, эксендина-4, ZYOG-1 и TTP273), лираглутида (виктоза®), албиглутида, экзенатида (баета®, бидуреон®), албиглутида, ликсисенатида, дулаглутида, семаглутида (NN-9924), TTP-054, ингибиторов протеинтирозинфосфатазы 1B (PTP-1B) (например, тродусквемина, экстракта Hyrtios и соединений, раскрытых в Zhang, S., et al., Drug Discovery Today, 12(9/10), 373-381 (2007)), активаторов SIRT-1 (например, ресвератрола, GSK2245840 или GSK184072), ингибиторов дипептидилпептидазы IV (DPP-IV) (например, тех, которые описаны в WO 2005116014, ситаглиптина, вилдаглиптина, алоглиптина, дутоглиптина, линаглиптина и саксаглиптина), секретагогов инсулина, ингибиторов окисления жирных кислот, антагонистов A2, ингибиторов аминоконцевой киназы c-jun (JNK), активаторов глюкокиназы (GKa) (например, тех, которые описаны в WO 2010103437, WO 2010103438, WO 2010013161, WO 2007122482, TTP-399, TTP-355, TTP-547, AZD1656, ARRY403, MK-0599, TAK-329, AZD5658 или GKM-001), инсулина и аналогов инсулина, миметиков инсулина, ингибиторов гликогенфосфорилaзы (например, GSK1362885), агонистов рецепторов VPAC2, ингибиторов SGLT2 (например, тех, которые описаны в E.C. Chao et al. Nature Reviews Drug Discovery 9, 551-559 (июль 2010 г.), включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин (CSG452), ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также те, которые описаны в WO 2010023594), модуляторов рецепторов глюкагона, таких как те, которые описаны в Demong, D.E. et al. Annual Reports in Medicinal Chemistry 2008, 43, 119-137; модуляторов GPR119 (например, особенно агонистов, таких как те, которые описаны в WO 2010140092, WO 2010128425, WO 2010128414, WO 2010106457, Jones, R.M. et al. в: Medicinal Chemistry 2009, 44, 149-170 (например, MBX-2982, GSK1292263, APD597 и PSN821)), производных или аналогов FGF21 (например, тех, которые описаны в Kharitonenkov, A. et al., Current Opinion in Investigational Drugs 2009, 10(4)359-364), модуляторов рецепторов TGR5 (также называемых GPBAR1) (например, INT777 и агонистов, таких как те, которые описаны в Zhong, M., Current Topics in Medicinal Chemistry, 2010, 10(4), 386-396), агонистов GPR40 (например, тех, которые описаны в Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая, но без ограничения, модуляторы, особенно агонисты, TAK-875, GPR120, активаторов высокоаффинных рецепторов никотиновой кислоты (HM74A) и ингибиторов SGLT1, таких как GSK1614235 (список антидиабетических средств, например, в WO 2011005611, в частности, тех, которые приведены на странице 28, строке 35 до страницы 30, строки 19), ингибиторов или модуляторов ферментов карнитин-пальмитоилтрансфераз, ингибиторов фруктоза-1,6-дифосфатазы, ингибиторов альдозоредуктазы, ингибиторов минералкортикоидных рецепторов, ингибиторов TORC2, ингибиторов CCR2 и/или CCR5, ингибиторов изоформ PKC (например, PKCα, PKCβ, PKCγ), ингибиторов синтетазы жирных кислот, ингибиторов серин-пальмитоилтрансферазы, модуляторов GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающего белка 4, глюкокортикоидных рецепторов, рецепторов соматостатина (например, SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторов или модуляторов PDHK2 или PDHK4, ингибиторов MAP4K4, модуляторов представителей семейства IL1, включая IL1-бета, модуляторов RXR-альфа; подходящие антидиабетические средства включают средства с механизмами действия, перечисленными в Carpino, P.A., Goodwin, B. Expert Opin. Ther. Pat, 2010, 20(12), 1627-51.
В другом варианте осуществления настоящего изобретения соединение формулы I можно вводить совместно со средствами, обычно используемыми пациентами с диабетом, например, гормоном щитовидной железы (таким как синтроид), любым средством против диабетической невропатии (например, габапентином, амитриптилином), либо средством или средствами для лечения депрессии любого типа (например, флуоксетином, сертралином, пароксетином, эсциталопрамом, циталопрамом, дулоксетином, левомилнаципраном, венлафаксином, десвенлафаксином, бупропионом, трициклическими антидепрессантами, включая имипрамин, нортриптилин, протриптилин, амитриптилин, доксепин, тримипрамин и дезипрамин).
В другом варианте осуществления настоящего изобретения соединение формулы I можно вводить совместно со средством, модулирующим уровни холестерина/липидов, при этом средство, модулирующее уровни холестерина/липидов, выбирают из группы, состоящей из ингибиторов HMG-CoA-редуктазы (например, правастатина, ловастатина, аторвастатина, симвастатина, флувастатина, NK-104 (другое название итавастатин или нисвастатин или нисбастатин) и ZD-4522 (другое название розувастатин или атавастатин или визастатин)), ингибиторов экспрессии гена HMG-CoA-редуктазы, ингибиторов сквален-синтетазы, ингибиторов сквален-эпоксидазы, ингибиторов сквален-циклазы, комбинированных ингибиторов сквален-эпоксидазы/сквален-циклазы или ингибиторов CETP, фибратов, ниацина, ионообменной смолы, антиоксидантов, секвестрантов желчных кислот (таких как квестран), ингибиторов ACAT, ингибиторов секреции MTP/APO β, ингибиторов липооксигеназы, ингибиторов поглощения холестерина, ингибиторов белка-переносчика сложных эфиров холестерина, такого средства, как мипомерсен, и/или антиатеросклеротических средств, включая модуляторы PCSK9.
В другом варианте осуществления соединение формулы I можно вводить совместно со средствами для лечения НАСГ и/или НАЖБП, такими как обетихолевая кислота (OCA, интерсепт), GFT505 (элафибранор), ингибиторы каспазы (например, эмрикасан), индукторы глутатионтрансферазы (например, олтипраз), ингибиторы аденозилметионин-декарбоксилазы (например, SAMe), конъюгат жирной кислоты/желчной кислоты (FABAC), такой как арамхол, аналоги FGF21, включая пегилированный FGF-21 пролонгированного действия (BMS-986036), двойной антагонист рецепторов CCR2/CCR5 (например, ценикривирок или TAK652), ингибитор галектина-3 (например, GR-MD-02), ингибитор апоптоз-стимулирующей киназы 1 (например, GS-4997), ингибитор 5-липоксигеназы (например, типелукаст), киРНК против HSP 47 (например, ND-L02-s0201), орлистат, TZD и другие сенсибилизаторы инсулина, метформин, сложные этиловые эфиры омега-3 кислот (например, ловаза), фибраты, ингибиторы HMG-CoA-редуктазы, эзетимиб, пробукол, урсодезоксихолевая кислота, агонисты TGR5, агонисты FXR, витамин E, бетаин, пентоксифиллин, антагонисты CB1, карнитин, N-ацетилцистеин, восстановленный глутатион, лоркасерин, сочетание налтрексона с бупропионом, ингибиторы SGLT2, фентермин, топирамат, аналоги инкретинов (GLP и GIP) и блокаторы рецепторов ангиотензина.
Дополнительные терапевтические средства включают антикоагулянты или средства, ингибирующие коагуляцию, антитромбоцитарные средства или средства, препятствующие образованию тромбов, ингибиторы тромбина, тромболитические или фибринoлитические средства, антиаритмические средства, антигипертензивные средства, блокаторы кальциевых каналов (L-типа и T-типа), сердечные гликозиды, диуретики, антагонисты минералкортикоидных рецепторов, средства-доноры NO, такие как органонитраты, средства, стимулирующие высвобождение NO, такие как ингибиторы фосфодиэстеразы, средства, снижающие уровни холестерина/липидов, и средства, способствующие улучшению липидного профиля, противовоспалительные средства (стероидные и нестероидные), средства против остеопороза, средства заместительной гормональной терапии, пероральные контрацептивы, средства против тревожности, антипролиферативные средства, противоопухолевые средства, противоязвенные средства и средства против гастроэзофагеальной рефлюксной болезни, гормон роста и/или секретагоги гормона роста, тиреомиметики (включая антагонист рецептора тиреоидного гормона), противоинфекционные средства, противовирусные средства, антибактериальные средства и противогрибковые средства.
Также включены средства, используемые в отделении интенсивной терапии, например, добутамин, дофамин, адреналин, нитроглицерин, нитропруссид и так далее.
Также включены комбинированные препараты, используемые для лечения васкулита, например, азатиоприн, циклофосфамид, микофенолят, мофетил, ритуксимаб и так далее.
В другом варианте осуществления настоящее изобретение относится к комбинации, в которой второе средство представляет собой по меньшей мере одно средство, выбранное из ингибитора фактора Xa, антикоагулянта, антитромбоцитарного средства, средства, препятствующего образованию тромбов, тромболитического средства и фибринолитического средства. Иллюстративные ингибиторы фактора Xa включают апиксабан и ривароксабан. Примеры подходящих антикоагулянтов для использования в сочетании с соединениями по изобретению включают варфарин, синтетический пентасахарид и гепарины (например, нефракционированные и низкомолекулярные гепарины, такие как эноксапарин и дальтепарин).
Используемый в настоящем документе термин «антитромбоцитарные средства» (или средства, препятствующие образованию тромбов) означает средства, ингибирующие функцию тромбоцитов, например, за счет ингибирования агрегации, адгезии или гранулярной секреции тромбоцитов. Средства включают, но не ограничиваются ими, различные известные нестероидные противовоспалительные лекарственные средства (НСПВЛС), такие как аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам, а также их фармацевтически приемлемые соли или пролекарства. Из НСПВЛС предпочтительными являются аспирин (ацетилсалициловая кислота или ASA) и ингибиторы COX-2, такие как целебрекс или пироксикам. Другие подходящие средства, препятствующие образованию тромбов, включают антагонисты IIb/IIIa (например, тирофибан, эптифибатид и абциксимаб), антагонисты рецептора тромбоксана A2 (например, ифетробан), ингибиторы синтетазы тромбоксана A2, ингибиторы PDE-III (например, цилостазол, дипиридамол), а также их фармацевтически приемлемые соли или пролекарства.
Используемый в настоящем документе термин «антитромбоцитарные средства» (или средства, препятствующие образованию тромбов) также включает антагонисты рецепторов АДФ, предпочтительно антагонисты пуринергических рецепторов P2Y1 и P2Y12, более предпочтительно P2Y12. Предпочтительные антагонисты рецепторов P2Y12 включают тикагрелор, прасугрел, тиклопидин и клопидогрел, включая их фармацевтически приемлемые соли или пролекарства. Клопидогрел является более предпочтительным средством. Тиклопидин и клопидогрел также являются предпочтительными соединениями, поскольку известно, что они мягко действуют в желудочно-кишечном тракте.
Используемый в настоящем документе термин «ингибиторы тромбина» (или антитромбиновые средства) означает ингибиторы сериновой протеазы тромбина. При ингибировании тромбина нарушаются различные опосредуемые тромбином процессы, такие как опосредуемая тромбином активация тромбоцитов (то есть, например, агрегация тромбоцитов и/или гранулярная секреция ингибитора активатора плазминогена 1 и/или серотонина) и/или образование фибрина. Многие ингибиторы тромбина известны специалисту в данной области и эти ингибиторы предусмотрены для использования в сочетании с соединениями по настоящему изобретению. Такие ингибиторы включают, но не ограничиваются ими, аргатробан, бороаргининовые производные, боропептиды, дабигатран, гепарины (нефракционированные и индивидуальные низкомолекулярные), гирудин, аргатробан и мелагатран, включая их фармацевтически приемлемые соли и пролекарства. Бороаргининовые производные и боропептиды включают N-ацетильные и пептидные производные бороновой кислоты, например, C-концевые производные альфа-аминобороновой кислоты, лизина, орнитина, аргинина, гомоаргинина и их соответствующие изотиоурониевые аналоги. Используемый в настоящем документе термин «гирудин» включает соответствующие производные или аналоги гирудина, называемые в настоящем документе гирулогами, например, дисульфатогирудин. Используемый в настоящем документе термин «тромболитические или фибринолитические средства» (или тромболитики или фибринолитики) означают средства, лизирующие сгустки крови (тромбы). Такие средства включают тканевой активатор плазминогена (природный или рекомбинантный) и его модифицированные формы, анистреплазу, урокиназу, стрептокиназу, тенектеплазу (TNK), ланотеплазу (nPA), ингибиторы фактора VIIa, ингибиторы PAI-1 (то есть, ингибиторы инактиваторов тканевого активатора плазминогена), ингибиторы альфа2-антиплазмина и анизоилированного активаторного комплекса плазминогена и стрептокиназы, включая их фармацевтически приемлемые соли или пролекарства. Используемый в настоящем документе термин «анистреплаза» означает анизоилированный активаторный комплекс плазминогена и стрептокиназы, описанный, например, в EP 028 489, содержание которого включено в настоящий документ посредством ссылки. Используемый в настоящем документе термин «урокиназа» означает как двух-, так и одноцепочечную урокиназу, в настоящем документе последнюю также называют проурокиназой.
Неограничивающие примеры подходящих антиаритмических средств включают: средства I класса (такие как пропафенон), средства II класса (такие как метопролол, атенолол, карвадиол и пропранолол), средства III класса (такие как соталол, дофетилид, амиодарон, азимилид и ибутилид), средства IV класса (такие как дитиазем и верапамил), средства, открывающие K+ каналы, такие как ингибиторы IAch и ингибиторы IKur (например, такие соединения, которые описаны в WO 01/40231).
Соединения по изобретению можно использовать в сочетании с антигипертензивными средствами, и такую антигипертензивную активность могут с легкостью определять специалисты в данной области стандартными методами (например, путем измерения кровяного давления). Примеры подходящих антигипертензивных средств включают: блокаторы альфа-адренорецепторов, блокаторы бета-адренорецепторов, блокаторы кальциевых каналов (например, дилтиазем, верапамил, нифедипин и амлодипин), сосудорасширяющие средства (например, гидралазин), диуретики (например, хлортиазид, гидрохлортиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид, бензтиазид, этакриновую кислоту, трикринафен, хлорталидон, торсемид, фуросемид, музолимин, буметанид, триамтренен, амилорид, спиронолактон), ингибиторы ренина, ингибиторы ACE (например, каптоприл, зофеноприл, фосиноприл, эналаприл, цераноприл, цилазоприл, делаприл, пентоприл, квинаприл, рамиприл, лизиноприл), антагонисты рецепторов AT-1 (например, лозартан, ирбесартан, валсартан), антагонисты рецепторов ET (например, ситаксентан, атрасентан и соединения, раскрытые в патентах США №№ 5612359 и 6043265), двойные антагонисты ET/AII (например, соединения, раскрытые в WO 00/01389), ингибиторы нейтральной эндопептидазы (NEP), ингибиторы вазопептидазы (двойные ингибиторы NEP-ACE) (например, гемопатрилат и нитраты). Иллюстративным антиангинальным средством является ивабрадин.
Примеры подходящих блокаторов кальциевых каналов (L-типа или T-типа) включают дилтиазем, верапамил, нифедипин, амлодипин и мибефрадил.
Примеры подходящих сердечных гликозидов включают дигиталис и уабаин.
В другом варианте осуществления соединение формулы I можно вводить совместно с одним или более диуретиками. Примеры подходящих диуретиков включают (a) петлевые диуретики, такие как фуросемид (например, лазикс™), торсемид (например, демадекс™), беметанид (например, бумекс™) и этакриновая кислота (например, эдекрин™); (b) тиазидные диуретики, такие как хлортиазид (например, диурил™, эсидрикс™ или гидродиурил™), гидрохлортиазид (например, микрозид™ или оретик™), бензтиазид, гидрофлуметиазид (например, салурон™), бендрофлуметиазид, метилхлортиазид, политиазид, трихлорметиазид и индапамид (например, лозол™); (c) фталимидиновые диуретики, такие как хлорталидон (например, гигротон™) и метолазон (например, зароксолин™); (d) хиназолиновые диуретики, такие как хинетазон и (e) калийсберегающие диуретики, такие как триамтерен (например, дирениум™) и амилорид (например, мидамор™ или модуретик™).
В другом варианте осуществления соединение формулы I можно вводить совместно с петлевым диуретиком. В другом варианте осуществления петлевой диуретик выбирают из фуросемида и торсемида. В другом варианте осуществления одно или более соединений формулы I можно вводить совместно с фуросемидом. В другом варианте осуществления одно или более соединений формулы I можно вводить совместно с торсемидом, который, необязательно, может представлять собой торсемид в форме с контролируемым или модифицированным высвобождением.
В другом варианте осуществления соединение формулы I можно вводить совместно с тиазидным диуретиком. В другом варианте осуществления тиазидный диуретик выбирают из группы, состоящей из хлортиазида и гидрохлортиазида. В другом варианте осуществления одно или более соединений формулы I можно вводить совместно с хлортиазидом. В другом варианте осуществления одно или более соединений формулы I можно вводить совместно с гидрохлортиазидом.
В другом варианте осуществления одно или более соединений формулы I можно вводить совместно с фталимидиновым диуретиком. В другом варианте осуществления фталимидиновый диуретик представляет собой хлорталидон.
Примеры подходящих антагонистов минералкортикоидных рецепторов включают сприонолактон и эплеренон.
Примеры подходящих ингибиторы фосфодиэстеразы включают: ингибиторы PDE III (такие как цилостазол) и ингибиторы PDE V (такие как силденафил).
Специалисты в данной области понимают, что соединения по данному изобретению также можно использовать в сочетании с другими видами терапии сердечно-сосудистых или цереброваскулярных заболеваний, включая ЧКВ, стентирование, применение высвобождающих лекарственное средство стентов, терапию стволовыми клетками, применение таких медицинских устройств, как имплантированные кардиостимуляторы, дефибрилляторы, или ресинхронизирующую терапию сердца.
В другом варианте осуществления изобретение относится к комбинированной терапии, в которой соединения по данному изобретению также могут быть использованы в сочетании с другими фармацевтическими средствами для лечения заболеваний, состояний и/или нарушений, описанных в настоящем документе. Таким образом, также предложены способы лечения, включающие введение соединений по изобретению в сочетании с другими фармацевтическими средствами.
Введение и дозы
Как правило, соединение по изобретению вводят в количестве, эффективном для лечения заболевания, описанного в настоящем документе. Соединения по изобретению вводят любым подходящим путем введения в форме фармацевтической композиции, адаптированной для такого пути введения, и в дозе, эффективной для запланированного лечения. Терапевтически эффективные дозы соединений, необходимые для лечения прогрессирующего заболевания, могут быть с легкостью определены специалистом в данной области с использованием доклинического и клинического подходов, известных в области медицины.
Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, в результате чего соединение попадает в желудочно-кишечный тракт, либо можно использовать трансбуккальное или подъязычное введение, при котором соединение попадает из ротовой полости непосредственно в кровоток.
В другом варианте осуществления соединения по изобретению также можно вводить непосредственно в кровоток, в мышцы или во внутренний орган. Подходящие способы парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, интравентрикулярный, интрауретральный, интрастернальный, интракраниальный, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольные (в то числе микроигольные) инъекторы, безыгольные инъекторы и инфузионные устройства.
В другом варианте осуществления соединения по изобретению также можно вводить топически на кожу или слизистую оболочку, то есть, методом дермального или чрескожного введения. В другом варианте осуществления соединения по изобретению также можно вводить интраназально или путем ингаляции. В другом варианте осуществления соединения по изобретению можно вводить ректально или вагинально. В другом варианте осуществления соединения по изобретению также можно вводить непосредственно в глаз или ухо.
Режим дозирования для соединений и/или композиций, содержащих соединения, основан на различных факторах, включая тип, возраст, массу тела, пол и медицинское состояние пациента; степень тяжести состояния; путь введения и активность конкретного используемого соединения. Таким образом, режим дозирования может варьироваться в широких пределах. Для лечения вышеуказанных состояний полезными являются уровни доз в диапазоне от примерно 0,01 мг до примерно 100 мг на килограмм массы тела в сутки. В одном варианте осуществления общая суточная доза соединения по изобретению (вводимая одной или разделенными дозами), как правило, составляет от примерно 0,01 до примерно 100 мг/кг. В другом варианте осуществления общая суточная доза соединения по изобретению составляет от примерно 0,1 до примерно 50 мг/кг, и в другом варианте осуществления от примерно 0,5 до примерно 30 мг/кг (то есть, мг соединения по изобретению на кг массы тела). В одном варианте осуществления доза составляет от 0,01 до 10 мг/кг/сутки. В другом варианте осуществления доза составляет от 0,1 до 1,0 мг/кг/сутки. Единичная доза композиций может содержать такие количества или их доли, которые суммарно составляют суточную дозу. Во многих случаях введение соединения будет повторено несколько раз в сутки (как правило, не более 4 раз). Введение несколько раз в сутки, как правило, можно использовать для увеличения общей суточной дозы, при необходимости.
Для перорального введения композиции могут быть предоставлены в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 миллиграмм активного ингредиента для симптоматической корректировки дозы для пациента. Медикамент, как правило, содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента или, в другом варианте осуществления, от примерно 1 мг до примерно 100 мг активного ингредиента. При внутривенном введении дозы могут находиться в диапазоне от примерно 0,01 до примерно 10 мг/кг/минуту при инфузии с постоянной скоростью.
Подходящие субъекты в соответствии с настоящим изобретением включают субъектов-млекопитающих. Млекопитающие в соответствии с изобретением включают собак, кошек, крупный рогатый скот, коз, лошадей, овец, свиней, грызунов, зайцеобразных, приматов и тому подобное, и включают млекопитающих in utero. В одном варианте осуществления подходящими субъектами являются люди. Субъекты-люди могут быть любого пола и находиться на любой стадии развития.
Фармацевтические композиции
Для лечения заболеваний, описанных в настоящем документе, соединения по изобретению можно вводить в виде соединения, как такового. Альтернативно, для медицинского применения подходят фармацевтически приемлемые соли, поскольку они могут иметь более высокую растворимость в воде в сравнении с исходным соединением.
В другом варианте осуществления настоящее изобретение относится к фармацевтическим композициям. Такие фармацевтические композиции содержат соединение по изобретению в сочетании с фармацевтически приемлемым носителем. Носитель может быть твердым веществом, жидкостью, или и тем, и другим, и может быть сформулирован с соединением в виде композиции со стандартной дозой, например, таблетки, которая может содержать от 0,05% до 95% по массе активного соединения. Соединение по изобретению может быть связано с подходящими полимерами в качестве направляющих носителей для лекарственного средства. Другие фармакологически активные вещества также могут присутствовать.
Соединения по изобретению можно вводить любым подходящим путем введения, предпочтительно, в форме фармацевтической композиции, адаптированной для такого пути введения, и в дозе, эффективной для запланированного лечения. Активные соединения и композиции, например, можно вводить перорально, ректально, парентерально или топически.
В случае перорального введения твердая лекарственная форма может, например, быть предоставлена в дискретных единицах, таких как твердые или мягкие капсулы, пилюли, саше, пастилки или таблетки, каждая из которых содержит заранее определенное количество по меньшей мере одного соединения по настоящему изобретению. В другом варианте осуществления препарат для перорального введения может быть в порошковой или гранулированной форме. В другом варианте осуществления пероральную лекарственную форму вводят сублингвально, в виде, например, пастилки. В таких твердых лекарственных формах соединения формулы I обычно находятся в сочетании с одним или более вспомогательными веществами. Такие капсулы или таблетки могут содержать препарат с контролируемым высвобождением. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные средства или могут быть изготовлены с энтеросолюбильными покрытиями.
В другом варианте осуществления препарат для перорального введения может быть в жидкой лекарственной форме. Жидкие лекарственные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области (то есть, воду). Такие композиции также могут содержать вспомогательные вещества, такие как увлажняющие, эмульгирующие, суспендирующие, вкусовые (например, подслащивающие) и/или ароматизирующие средства.
В другом варианте осуществления настоящее изобретение относится к парентеральной лекарственной форме. «Парентеральное введение» включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинные, внутримышечные инъекции, интрастернальные инъекции и инфузии. Инъекционные препараты (то есть, стерильные инъекционные водные или масляные суспензии) могут быть сформулированы способом, известным в данной области, с использованием соответствующих диспергирующих, увлажняющих средств и/или суспендирующих средств.
В другом варианте осуществления настоящее изобретение относится к топической лекарственной форме. «Топическое введение» включает, например, чрескожное введение, например, при помощи чрескожных пластырей или устройств для ионофореза, внутриглазное введение, либо интраназальное введение или ингаляцию. Композиции для топического введения также включают, например, топические гели, спреи, мази и кремы. Топический препарат может содержать соединение, усиливающее абсорбцию или проникновение активного ингредиента через кожу или другие пораженные области. Если соединения по данному изобретению вводят при помощи устройства для чрескожного введения, введение будет выполнено с использованием либо пластыря в виде резервуара с пористой мембраной, либо пластыря с твердой матрицей. Типичные препараты для данной цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, перевязочные материалы, пены, пленки, кожные пластыри, слоистые пластинки, имплантаты, губки, волокна, повязки и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, медицинский вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. В препарат можно включать усилители проникновения, смотри, например, B. C. Finnin and T. M. Morgan, J. Pharm. Sci., vol. 88, pp. 955-958, 1999.
Препараты, подходящие для топического введения в глаз, включают, например, глазные капли, в которых соединение по данному изобретению растворено или суспендировано в соответствующем носителе. Типичный препарат, подходящий для введения в глаз или в ухо, может быть в форме капель микронизированной суспензии или раствора в изотоническом, pH-скорректированном, стерильном солевом растворе. Другие препараты, подходящие для введения в глаз или в ухо, включают мази, биоразлагаемые (то есть, абсорбируемые гелевые губки, коллаген) и не биоразлагаемые (то есть, силиконовые) имплантаты, слоистые пластинки, линзы и суспензии из частиц или везикул, таких как ниосомы или липосомы. Можно включать полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, либо гетерополисахаридный полимер, например, геллановая камедь, наряду с консервантом, таким как бензалкония хлорид. Такие препараты также могут быть доставлены с помощью ионофореза.
В случае интраназального введения или введения путем ингаляции активные соединения по изобретению удобно доставлять в форме раствора или суспензии из контейнера с распылителем, при этом пациент сдавливает контейнер или нажимает на головку распылителя, либо в виде аэрозоля из находящегося под давлением контейнера или небулайзера, с использованием соответствующего пропеллента. Препараты, подходящие для интраназального введения, как правило, вводят в виде сухого порошка (либо отдельно, либо в виде смеси, например, в сухой смеси с лактозой, либо в виде смешанных частиц компонентов, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка или в виде аэрозоля из находящегося под давлением контейнера, насоса, спрея, пульверизатора (предпочтительно, пульверизатора, в котором используются электрогидродинамические силы для создания мелкодисперсного тумана) или небулайзера, с использованием или без использования соответствующего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. В случае интраназального использования порошок может содержать биоадгезивное средство, например, хитозан или циклодекстрин.
В другом варианте осуществления настоящее изобретение относится к ректальной лекарственной форме. Такая ректальная лекарственная форма может представлять собой, например, суппозиторий. Масло какао является традиционной основой суппозиториев, однако различные альтернативные варианты можно использовать в зависимости от обстоятельств.
Можно использовать также другие материалы носителя и способы введения, известные в области фармацевтики. Фармацевтические композиции по изобретению можно получать любым из известных методов фармацевтики, таких как методы создания эффективных препаратов и методы введения. Вышеприведенные соображения относительно эффективных препаратов и методов введения хорошо известны в данной области и описаны в стандартных руководствах. Формулирование лекарственных средств описано, например, в Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
ПОЛУЧЕНИЕ
Следует отметить, что при получении соединений формулы I в некоторых способах получения, описанных в настоящем документе, может потребоваться защита удаленной функциональной группы (например, первичного амина, вторичного амина, карбоксила в предшественниках соединения формулы I). Необходимость в такой защите будет варьироваться в зависимости от природы удаленной функциональной группы и от условий способов получения. Необходимость в такой защите может с легкостью определять специалист в данной области. Использование таких методов защиты/снятия защиты также находится в пределах компетенции специалиста в данной области. Для получения общей информации о защитных группах и их использовании смотри T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, некоторые соединения содержат первичные амино- или карбоксильные функциональные группы, которые, если их оставить без защиты, могут препятствовать протеканию реакций в других участках молекулы. Соответственно, такие функциональные группы можно защищать соответствующими защитными группами, которые можно удалять на последующих этапах. Подходящие защитные группы для защиты амино- и карбоксильных групп включают защитные группы, обычно используемые в пептидном синтезе (например, N-t-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметиленоксикарбонил (Fmoc) для аминов и низших алкиловых или бензиловых эфиров карбоновых кислот), которые, как правило, химически неактивны в описанных условиях и, как правило, могут быть удалены без химического изменения других функциональных групп в соединениях формулы I.
Схемы реакций, описанные ниже, предназначены для общего описания методологии, используемой в получении соединений по настоящему изобретению. Некоторые из соединений по настоящему изобретению содержат один хиральный центр со стереохимическим обозначением (R). Специалисту в данной области понятно, что все синтетические превращения можно осуществлять аналогичным образом, являются ли материалы богатыми энантиомерами или рацемическими. Кроме того, получение желательного оптически активного материала можно выполнять в любой желательный момент времени в последовательности операций с использованием хорошо известных методов, таких как описанные в настоящем документе и в химической литературе.
В приведенных ниже схемах реакций переменные X, Y, Z, R1, R2, R3, R4, RC, RN, RS, L, m и n являются такими, как указано в настоящем документе для соединений формулы (I), если нет иных указаний. В приведенных ниже схемах реакций некоторые уходящие группы обозначены LG1 или LG2, каждая из которых независимо может представлять собой галоген, SO2-алкил, SO2-арил, S-алкил, S-арил, S(O)-алкил, S(O)-арил, либо атом кислорода, связанный с фосфоросодержащим фрагментом. Каждая группа LG3 независимо может представлять собой уходящую группу, такую как любой алкил- или арилсульфонат (например, мезилат, тозилат или трифлат), или галоген, или любая другая группа, которая может быть замещена амином. Каждый «алкил» является независимым от другого и, как правило, содержит 1-6 атомов углерода. Арил, как правило, представляет собой фенил. Если защитная группа обозначена PG1, она может представлять собой алкильную аминозащитную группу, такую как бензил, бензгидрил или тому подобное; защитную группу карбамата, такую как Boc, Cbz или тому подобное; или амидную защитную группу, такую как трифторацетамид.
Пиримидинильные и цианопиридинильные кольца могут быть получены, как показано на схеме 1. Промежуточные соединения формулы 6 могут быть приобретены или обычным образом синтезированы в реакциях конденсации, как показано на схеме 1. Сложные эфиры 1 (где R3 может представлять собой F, Cl, Br, алкил и тому подобное) могут быть депротонированы за счет действия основания, такого как трет-бутоксид калия, диизопропиламид лития, гидрид натрия и тому подобное, и вступать в реакцию со сложными эфирами 2, с получением бета-кетоэфиров 3. Альтернативно, кетоны общей формулы 7 могут быть обработаны аналогичными основаниями и вступать в реакцию с хлорформиатами 8, с получением аналогичных бета-кетоэфиров 3.
Сложные эфиры 3 затем можно конденсировать с такими реагентами, как мочевина, с образованием пиримидинов 5, с нагреванием или без нагревания, или, альтернативно, с катализом кислотой или основанием. Активацию гидроксила уходящей группы можно осуществлять такими реагентами, как оксигалогенид фосфора, пентагалогенид фосфора, алкил- или арилтиолы, а также их соли (с последующим окислением или без него), BOP, PyBOP или другие аналогичные активирующие реагенты, с получением соединений общей формулы 6.
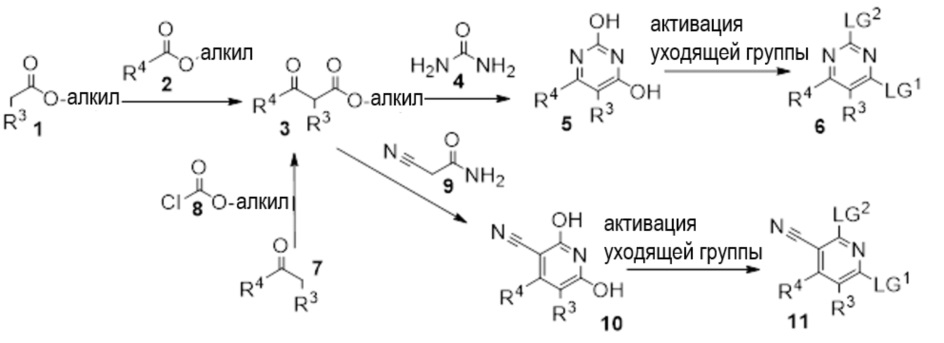
Схема 1
Соединения общей формулы 11 можно приобретать или синтезировать, начиная с бета-кетоэфиров 3, которые могут вступать в реакцию с цианоацетамидом 9, с получением соединений общей формулы 10. Их можно превращать в соединения общей формулы 11 методом, аналогичным превращению 5 в 6.
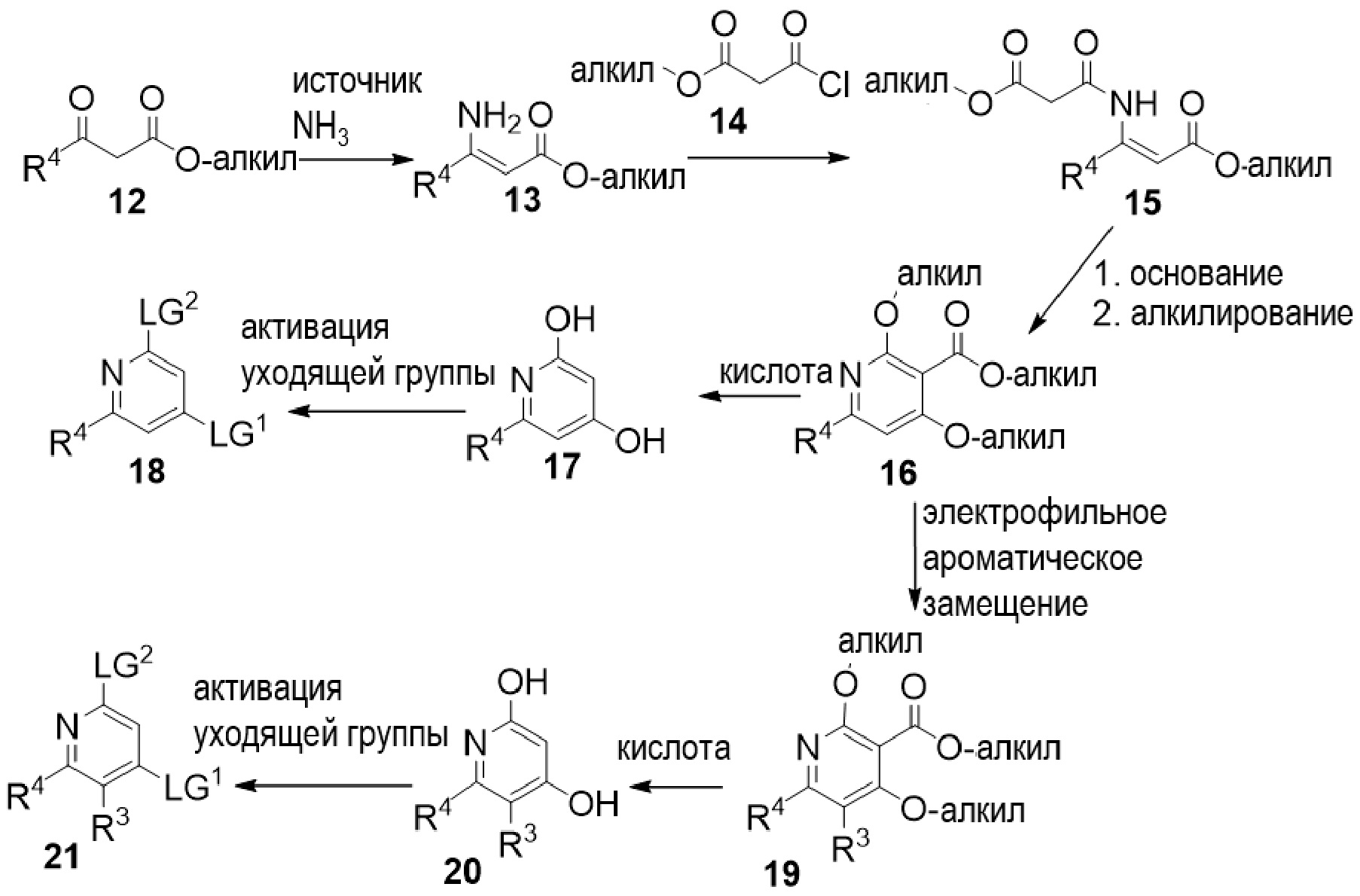
Схема 2
Промежуточные соединения 18 можно синтезировать, как показано на схеме 2. Начиная с бета-кетоэфиров 12, проводят обработку источником аммиака, таким как ацетат аммония, хлорид аммония, гидроксид аммония, раствор аммиака в растворителе и тому подобное, в разных условиях, в то числе с нагреванием или без нагревания, или, альтернативно, с катализом кислотой или основанием, с получением соединений общей формулы 13. Обработка хлоридами кислот 14 затем может приводить к получению соединений общей формулы 15. Обработка основанием может приводить к циклизации пиридина, и алкилирование полученных гидроксильных групп может приводить к получению пиридинов 16. Обработка кислотой, такой как фтористоводородная, хлористоводородная, бромистоводородная, йодистоводородная, или различные кислоты Льюиса, с нагреванием или без нагревания может приводить к получению соединений общей формулы 17. Активация гидроксильных функциональных групп уходящих групп, с образованием промежуточных соединений общей формулы 18, может происходить аналогично тому, как это происходит в условиях, описанных для превращения 5 в 6 на схеме 1. Альтернативно, может быть получен пиридин с заменой (где R3 представляет собой F, Cl, Br и алкилы, которые могут быть введены путем электрофильного ароматического замещения такими методами, как алкилирование по Фриделю-Крафтсу) в результате реакции соединений формулы 16 с одним из различных соединений, обеспечивающих электрофильное ароматическое замещение, таких как газообразный хлор, бром, selectFluor™, N-фторбензолсульфонимид, N-галосукцинимиды или любые другие известные источники электрофильного галогенида или алкилгалогенидов, в присутствии алюминиевых катализаторов, с образованием соединений общей формулы 19. Затем это соединение может быть превращено в промежуточные соединения общей формулы 21 методами, аналогичными тем, которые описаны для превращения 16 в 18.
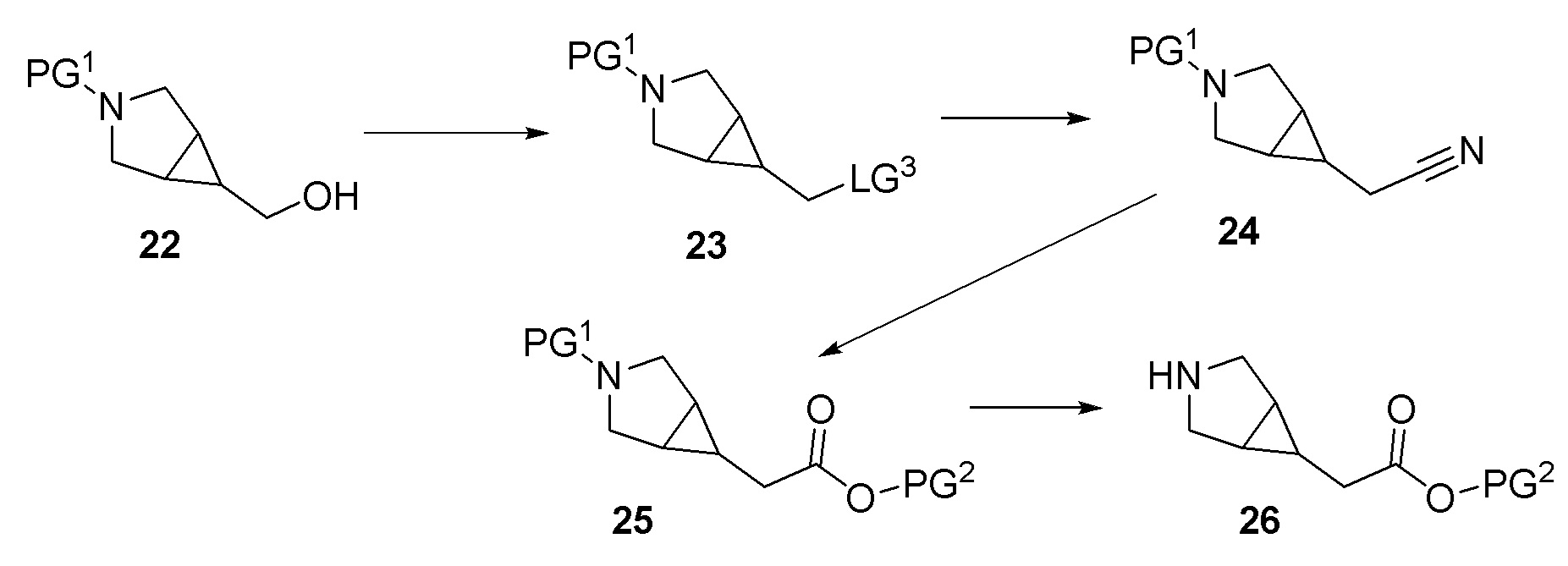
Схема 3A
Амины общей формулы 26 можно приобретать или обычным образом синтезировать, как показано на схемах 3A-3D. Начиная с защищенных [3,1,0]азабициклогексанов 22 (приобретенных или синтезированных методом, аналогичным тому, который описан в Berliner, M. A. et al. Org. Process Res. Dev. 2011, 15, 1052-1062), гидроксильный фрагмент может быть превращен в LG3 стандартными методами и замещен с помощью известных углеродсодержащих реагентов гомологизации, таких как цианиды натрия или калия, с получением нитрилов 24. Затем нитрильный фрагмент может быть гидролизован до сложных эфиров 25 (или карбоновой кислоты) в различных стандартных условиях для катализа кислотой или основанием, где PG2 представляет собой алкил (например, C1-6 алкил) или бензил. Удаление PG1 можно осуществлять разными способами, описанными в литературе, с получением сложных аминоэфиров 26.
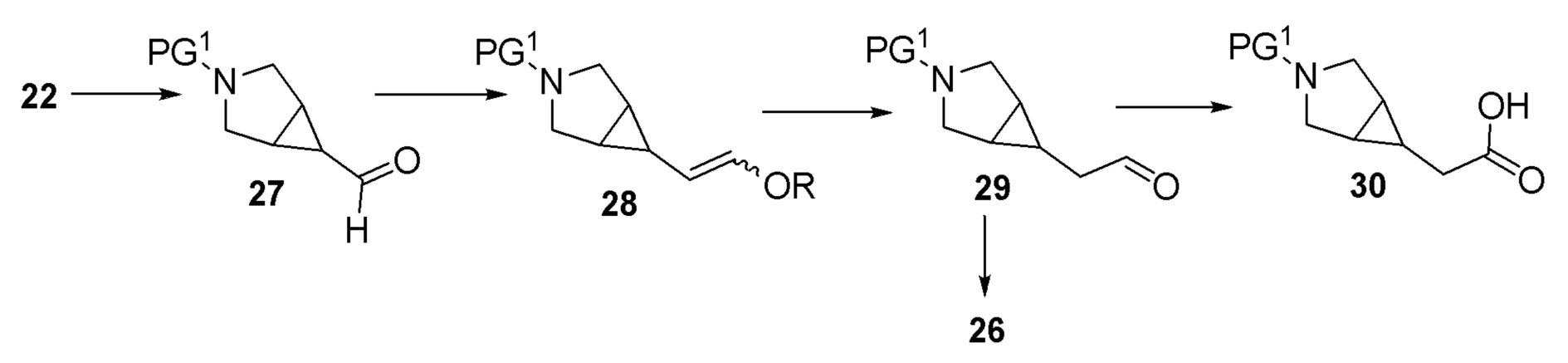
Схема 3B
Альтернативно, как показано на схеме 3B, гидроксильный фрагмент в 22 может быть окислен до альдегида 27 и гомологизирован с использованием реакции Виттинга и гидролиза, с получением гомологизированных альдегидов 29. Дальнейшее окисление с использованием различных окислителей, таких как хлорит натрия, хлорная известь, перманганат калия или другие, может приводить к образованию карбоновых кислот 30 или сложных эфиров 26.
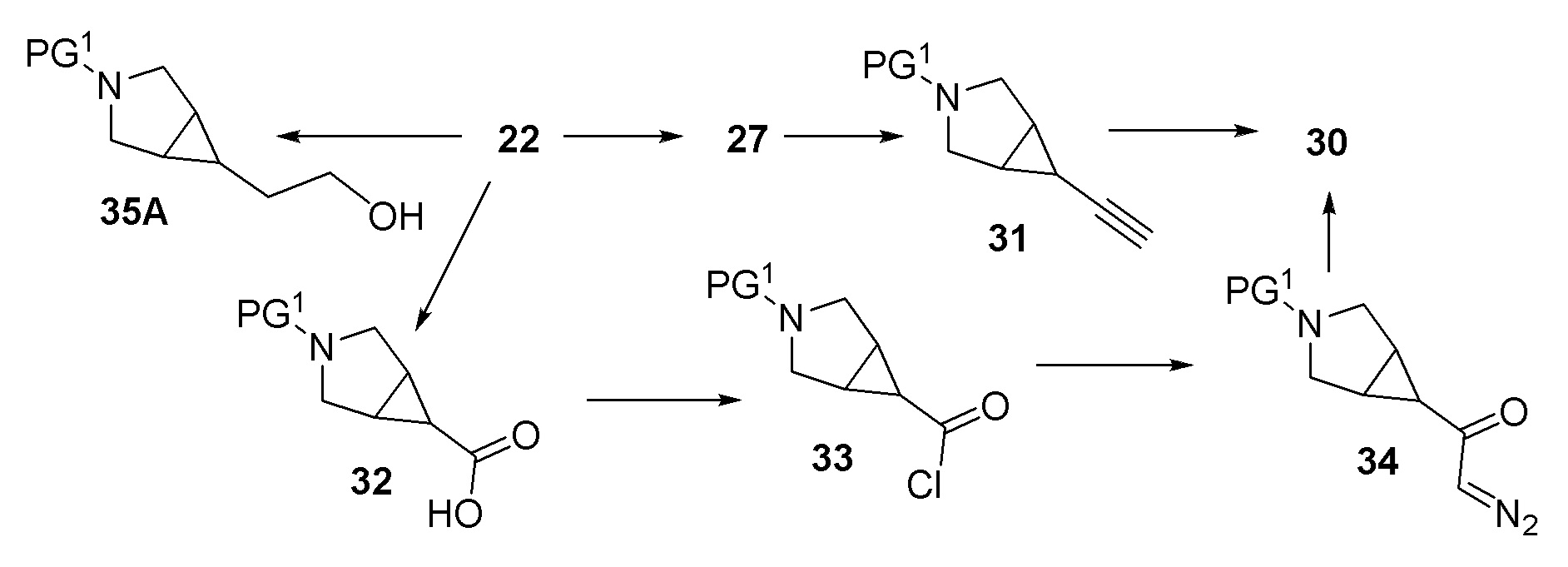
Схема 3C
Альтернативно, как показано на схеме 3C, альдегиды 27 могут быть превращены в алкины 31 с использованием различных условий, таких как реагент Гилберта-Сейферта, Охира-Бестмана, CBr4 с PPh3 или другие. Алкин затем может быть превращен в карбоновые кислоты 30 с использованием кислот Бренстеда или Льюиса, или при катализе металлом, например, катализе золотом. Альтернативно, гидроксильная группа может быть окислена до кислот 32 и обработана в условиях реакции гомологизации Арндта-Эйстерта (32 в 33 в 34 в 30), с получением гомологизированных кислот 30. Альтернативно, соединения общей формулы для промежуточного соединения 35A могут быть синтезированы путем функционализации спиртов 22 в различных условиях, описанных в литературе (смотри, например, WO 2010116328).
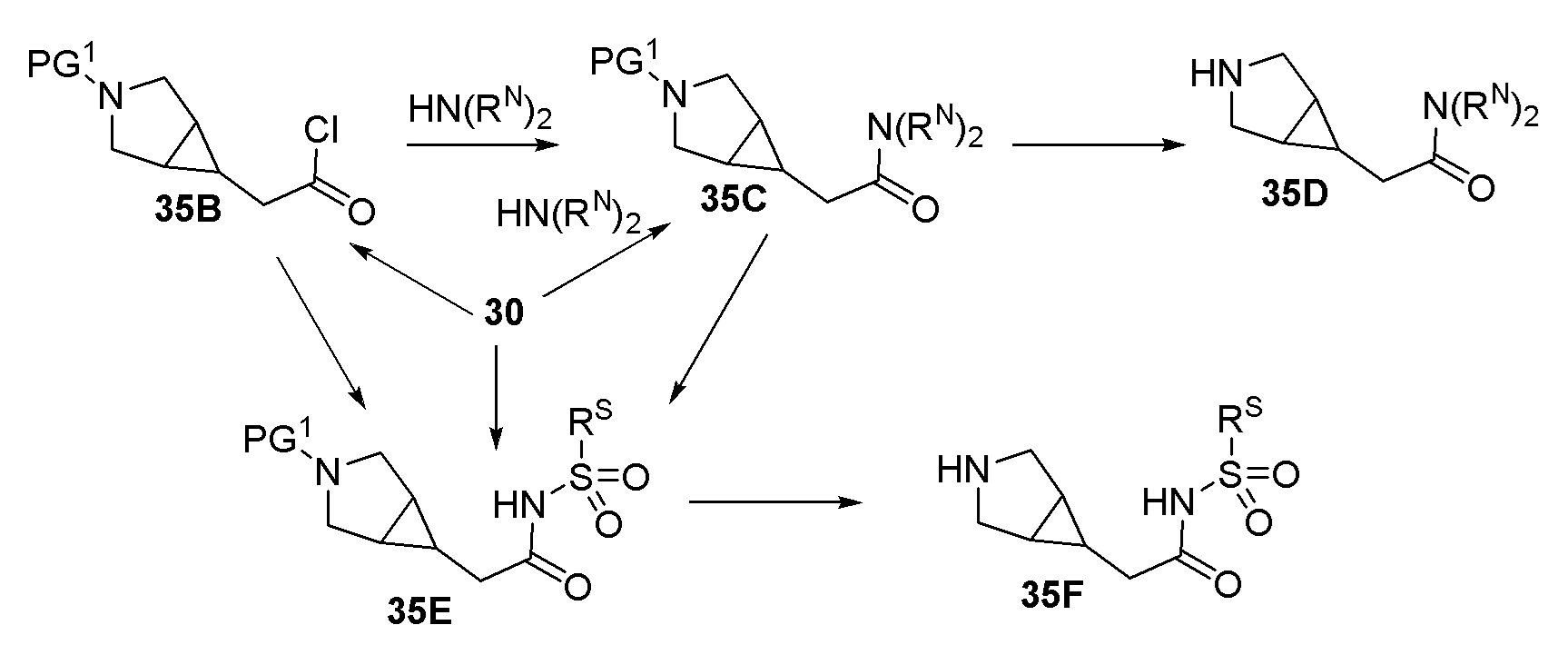
Схема 3D
Амины формулы 35D, 35F, 35H, 35K, 35N могут быть синтезированы, как описано в литературе, или синтезированы, как указано на схемах 3D-3F. Начиная с 30, обработка реагентом, который замещает гидроксил хлоридом (таким как оксихлорид фосфора, оксалилхлорид, пентахлорид фосфора, тионилхлорид, сульфурилхлорид и другие в присутствии или в отсутствие DMF) может приводить к получению хлорангидридов 35B. Последующая обработка амином, HN(RN)2, в присутствии любого основания, такого как DIPEA, TEA, DBU, K2CO3, NaHCO3 или любые другие, может приводить к получению амидов 35C. Альтернативно, 30 можно непосредственно связывать с амином, используя любой реагент, связывающий амиды, для активации карбоновой кислоты (например, EDC, HATU, T3P, COMU, DCC и многие другие, описанные в литературе), с получением 35C. Хлорангидрид 35B может быть обработан сульфонамидом, H2NS(O)2RS, с получением ацилсульфонамидов 35E. Альтернативно, 30 можно превращать в 35E с использованием сульфонамида, H2NS(O)2RS, и условий, аналогичных тем, которые описаны для превращения 30 в 35C.
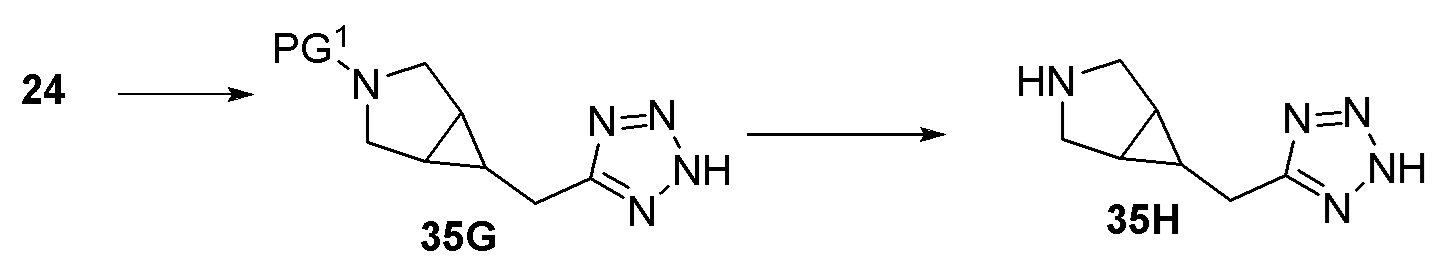
Схема 3E
Промежуточное соединение 24 можно превращать в тетразол 35G, добавляя азид, такой как азид натрия, калия, триметилсилилазид, трибутилтиназид или другие, при нагревании или с добавлением катализатора для ускорения реакции. Затем получают тетразол 35H с использованием стандартных методов для удаления PG1.
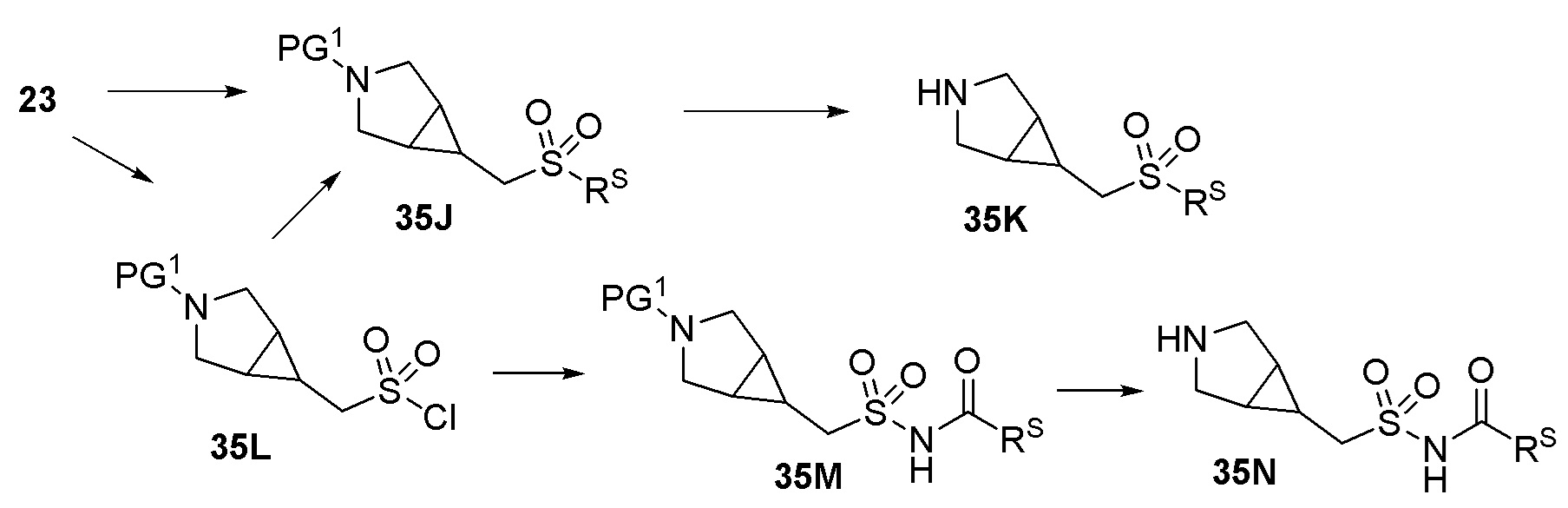
Схема 3F
Промежуточное соединение 23 можно превращать в сульфон 35J различными способами, такими как замещение уходящей группы сульфиновой кислотой, либо натриевой, калиевой или другой солью сульфиновой кислоты, HOS(O)RS, в нейтральных или щелочных условиях. Альтернативно, промежуточное соединение 23 можно превращать в 35J в процессе, состоящем из замещения уходящей группы на промежуточном соединении 23 тиолом, либо натриевой, калиевой или другой солью тиола, с получением тиоэфира, который затем может быть окислен до сульфона с использованием окислителя, такого как мета-хлорпербензойная кислота, пероксид водорода, перманганат калия или многие другие окислители. Альтернативно, промежуточное соединение 23 можно превращать в сульфонилхлорид 35L путем обработки тиомочевиной, а затем хлорной известью; или за счет обмена металл-галоген при использовании такого реагента, как магний или бутиллитий, с последующей обработкой диоксидом серы или источником диоксида серы, таким как DABCO-SO2, и последующим хлорированием с использованием NCS, тионилхлорида, оксихлорида фосфора или других хлорирующих реагентов; либо другими способами, известными из литературы. Промежуточное соединение 35L можно превращать в 35J путем обработки алкилирующим реагентом, таким как алкиллитий, алкилмагний-галогенид, триалкилалюминий или любые другие нуклеофильные источники алкильных групп. Промежуточное соединение 35L можно превращать в ацилсульфонамид 35M путем обработки амидом, H2NC(O)RS, в присутствии основания, такого как гидрид натрия, литийдиизопропиламид, карбонат калия, DBU или другие основания. Удаление защитных групп 35C, 35E, 35G, 35J и 35M можно осуществлять в кислых, щелочных, гидрогенолитических или других условиях, известных из литературы для удаления конкретной защитной группы, с получением 35D, 35F, 35H, 35K и 35N, соответственно.
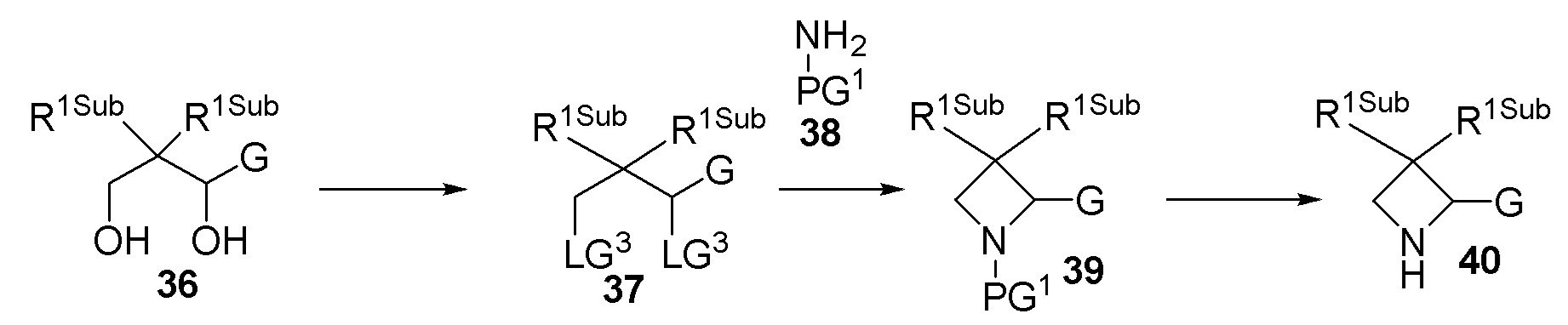
Схема 4A
Азетидины 40 (где G может представлять собой H или любой C1-3 алкил) можно приобретать, синтезировать, как описано в литературе (например, в J. Med. Chem. 1994, 37, 4195), или синтезировать, как указано на схемах 4A-4C. Промежуточные диолы 36 можно превращать в промежуточные соединения 37 путем активации мезилхлоридом или ангидридом, трифлатным ангидридом и другими сульфонат-образующими реагентами или превращать в галогенидную уходящую группу с использованием тионилхлорида, четырехбромистого углерода с трифенилфосфином, йода с трифенилфосфином или имидазолом, или различных других реагентов. Обработка 37 амином 38 может приводить к получению азетидинов 39. Стандартные методы снятия защиты позволяют получать промежуточные соединения общей формулы 40, которые в конечном итоге становятся R1 в соединениях формулы (I), таким образом, R1Sub представляет собой H, если группа R1 не замещена, или R1Sub представляет собой -C1-3 алкил и -OH, как определено в любом варианте осуществления соединений формулы (I) для заместителей R1.
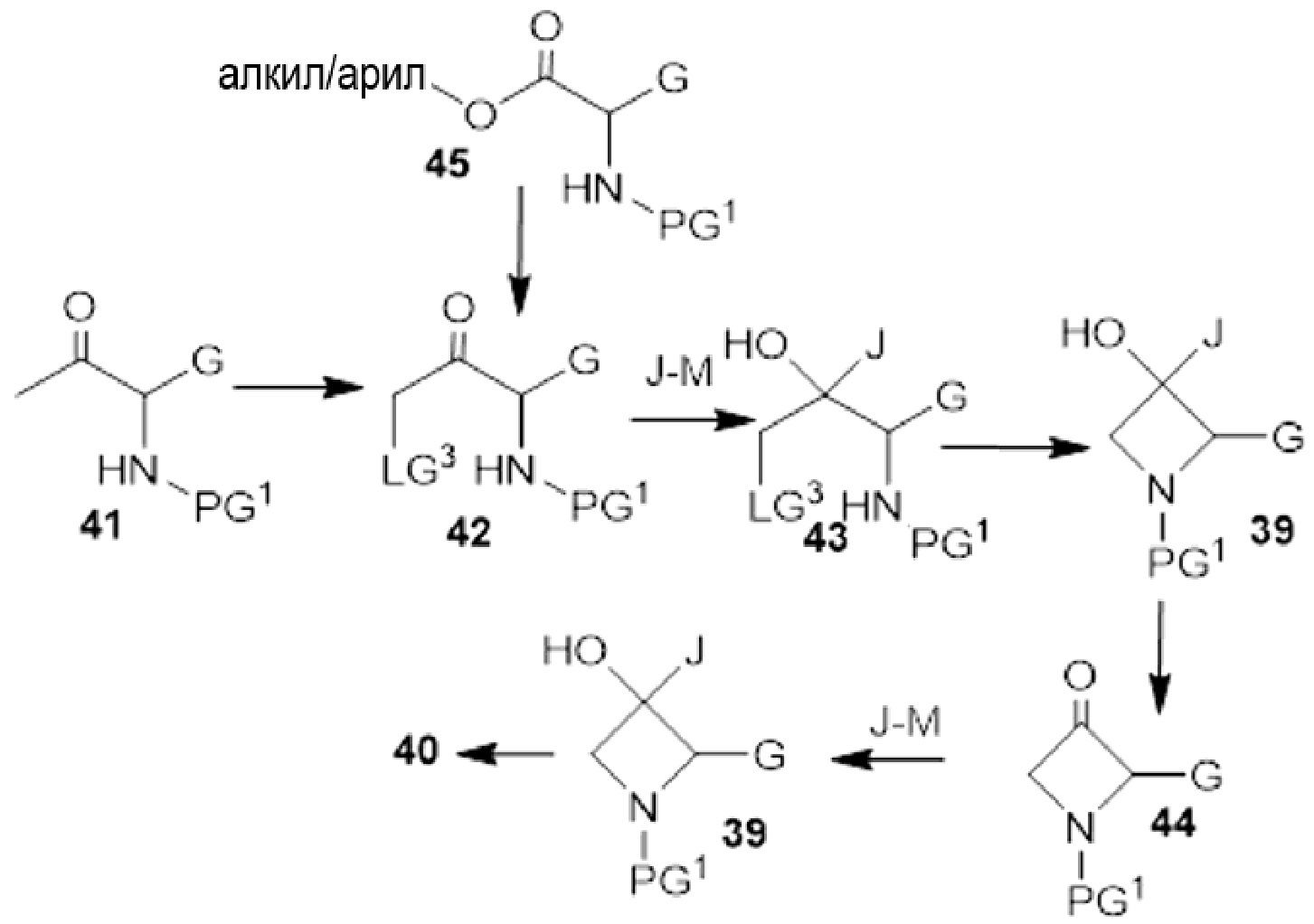
Схема 4B
Альтернативно, как показано на схеме 4B, если J представляет собой водород, может иметь место окисление, с получением кетонов 44 (где G может представлять собой H или любой C1-3 алкил). Обработка любым известным источником гидрида металла (J-M, где J представляет собой водород и M представляет собой металлический противоион, например, литий, магний, цинк, алюминий, бор или другие) может приводить к получению азетидинильных промежуточных соединений 40 для R1, с оказанием влияния на желательный с точки зрения стереохимии результат за счет выбора реагентов. Альтернативно, кетоны 44 можно обрабатывать металл-алкилирующими агентами (J-M, где J представляет собой любой C1-3 алкил и M представляет собой металлический противоион, например, литий, магний, цинк, алюминий, бор или другие), такими как алкилмагний-галогениды, алкиллитий или многие другие источники нуклеофильных алкильных групп, с получением соединений общей формулы 39, где J представляет собой алкил. Их можно переводить в азетидины 40, как описано ранее. Альтернативно, кетоны 41 можно активировать с уходящей группой (LG3) путем обработки основанием и источником электрофильного галогена, с получением кетонов 42. Затем можно проводить дериватизацию аналогично превращению 44 до 39, с получением соединений 43. Их можно подвергать воздействию щелочных условий, с получением азетидинов 39, где J представляет собой алкил или водород, которые можно переводить в промежуточные соединения 40, как описано ранее. Альтернативно, сложные эфиры 45 можно превращать в кетоны 42 в реакции гомологизации с включением уходящей группы, используя такие реагенты, как хлоруксусная кислота или дигалометан, оба реагента в присутствии сильного основания или с сульфоний-илидом, а также многие другие реагенты, описанные в литературе. Промежуточное соединение 42 затем можно переводить в 40, как описано ранее.
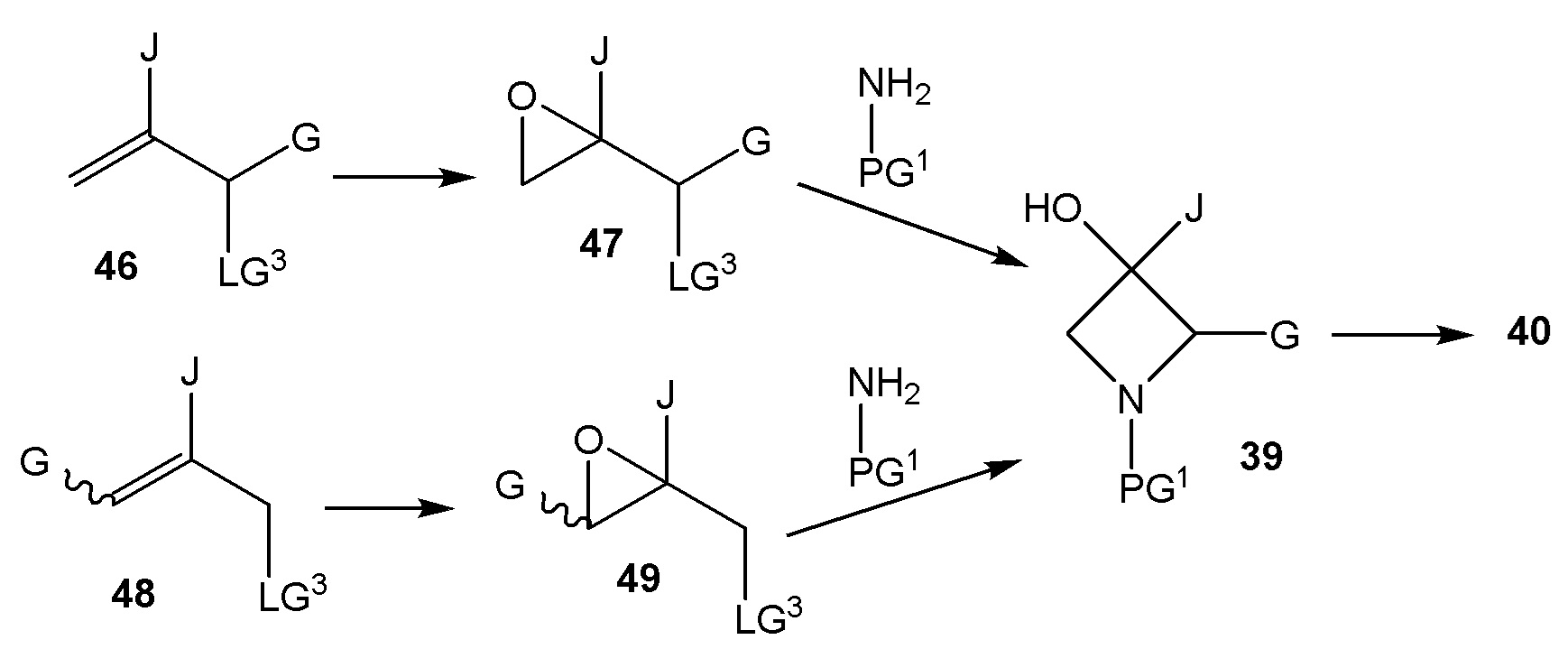
Схема 4C
Альтернативно, алкены 46 или 48 (где J может представлять собой любой алкил или водород; G может представлять собой H или любой C1-3 алкил) можно обрабатывать различными окислителями, такими как м-CPBA (мета-хлорпербензойная кислота), пероксид водорода, трет-бутилгидропероксид, создавать условия эпоксидирования по Шарплессу, условия эпоксидирования по Ши или множество других условий, известных в литературе, с получением эпоксидов 47 или 49, соответственно. Эпоксиды 47 или 49 можно обрабатывать амином аналогично превращению 37 в 39, с получением азетидинов 39, которые можно переводить в промежуточные соединения 40.
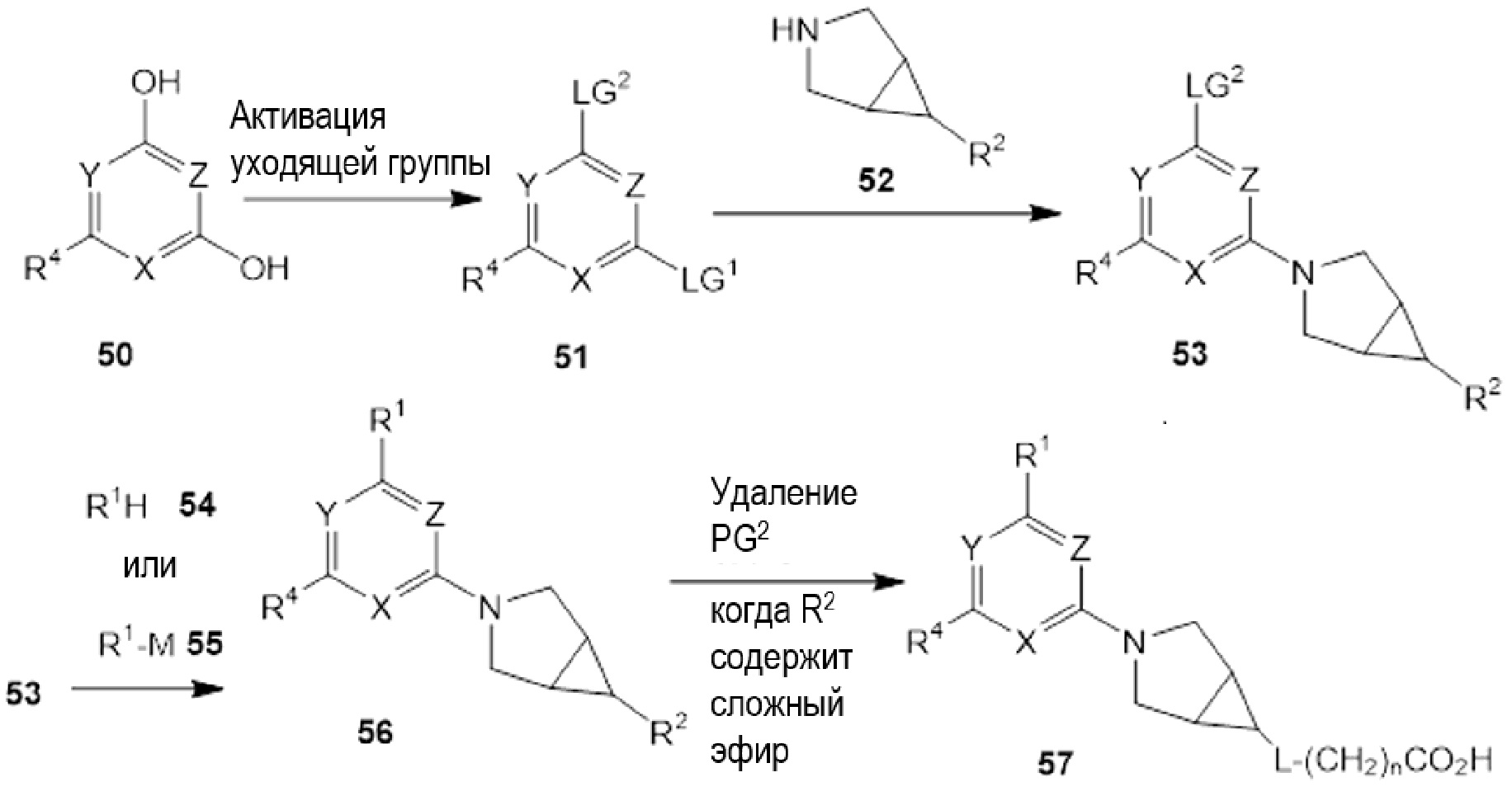
Схема 5
Промежуточные соединения формулы 56 и 57, в целом, можно синтезировать, как показано на схеме 5. Начиная с бис-гидроксигетероарилов общей формулы 50 (приобретенных, известных из литературы или описанных в предыдущих схемах), превращение в промежуточные соединения общей формулы 51 можно выполнять методом, аналогичным методу, описанному для превращения промежуточного соединения 5 в 6 на схеме 1. Амины общей формулы 52 (приобретенные, известные из литературы или описанные в предыдущих схемах, например, 30 или 25, у которых сначала должна быть снята защита в кислых, щелочных, гидрогенолитических или других условиях, известных из литературы для удаления конкретной защитной группы,) могут быть связаны с 51 в щелочных или кислых условиях посредством реакции SNAr в присутствии таких оснований, как натрия, калия или цезия карбонат, бикарбонат, гидроксид, ацетат, или основание органический амин, например, триэтиламин, диизопропилэтиламин, DBU и тому подобное, или при катализе палладием с различными источниками, лигандами и основаниями палладия, с получением промежуточных соединений 53. Впоследствии их можно связывать с аминами общей формулы 54 (приобретенными, известными из литературы или описанными в предыдущих схемах, например, 40) аналогично предыдущей стадии, но часто при более высоких температурах, с получением промежуточных соединений 56. Альтернативно, обработка соединений 53 комплексами алкил-металл или металлоид 55, такими как алкил-цинк, алкилбороновая кислота, соли боронат, трифторборат и тому подобное, при катализе палладием может приводить к получению промежуточных соединений 56. Когда R2 содержит сложный эфир (смотри схему 3A), карбоновая кислота может быть демаскирована с использованием различных условий, известных из литературы, с получением промежуточных соединений 57.
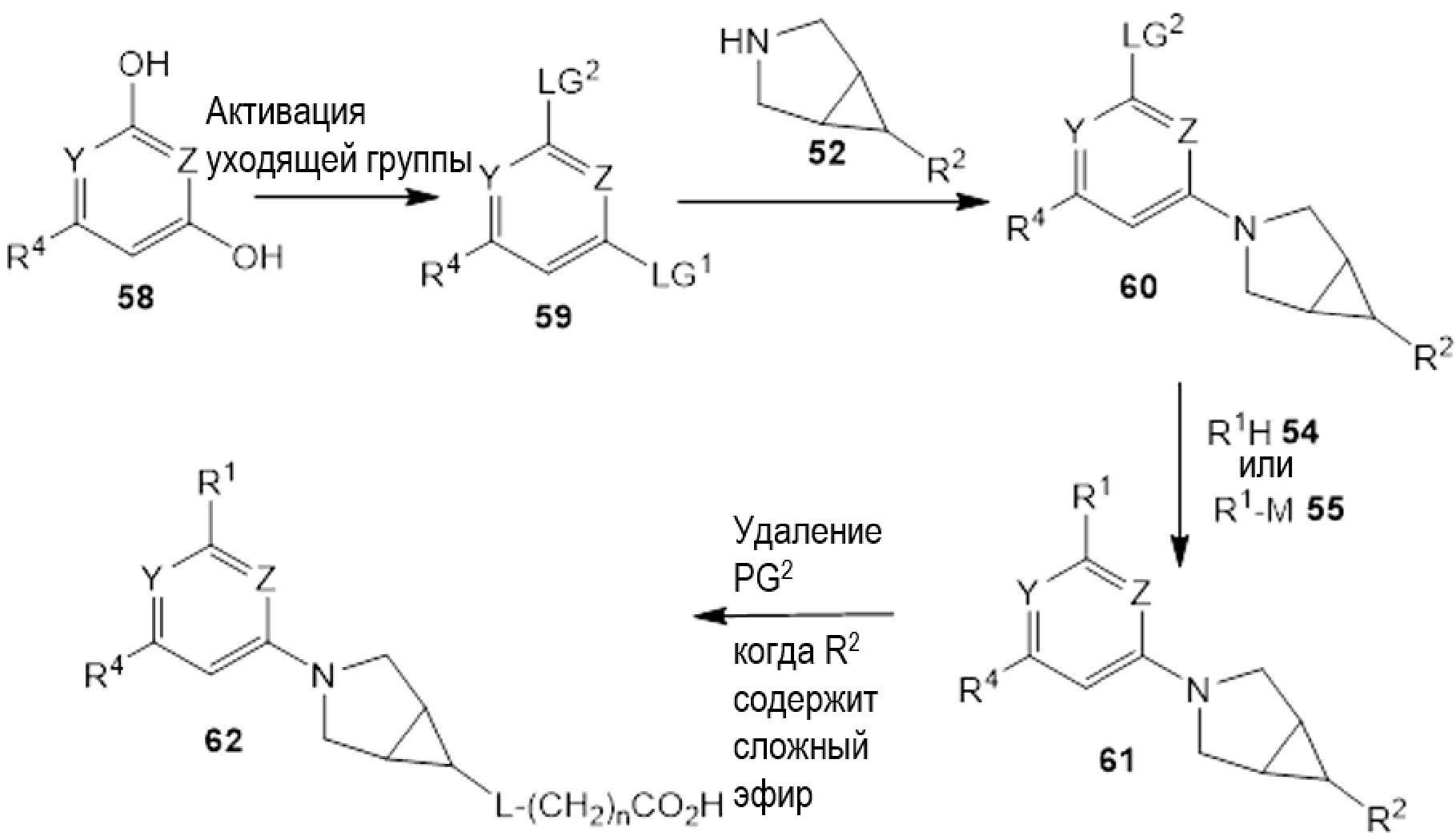
Схема 6A
Альтернативно, промежуточные соединения 60, 61 и 62 (схема 6A) можно синтезировать методом, аналогичным методам, описанным для промежуточных соединений 53, 56 и 57, соответственно, как показано на схеме 5.
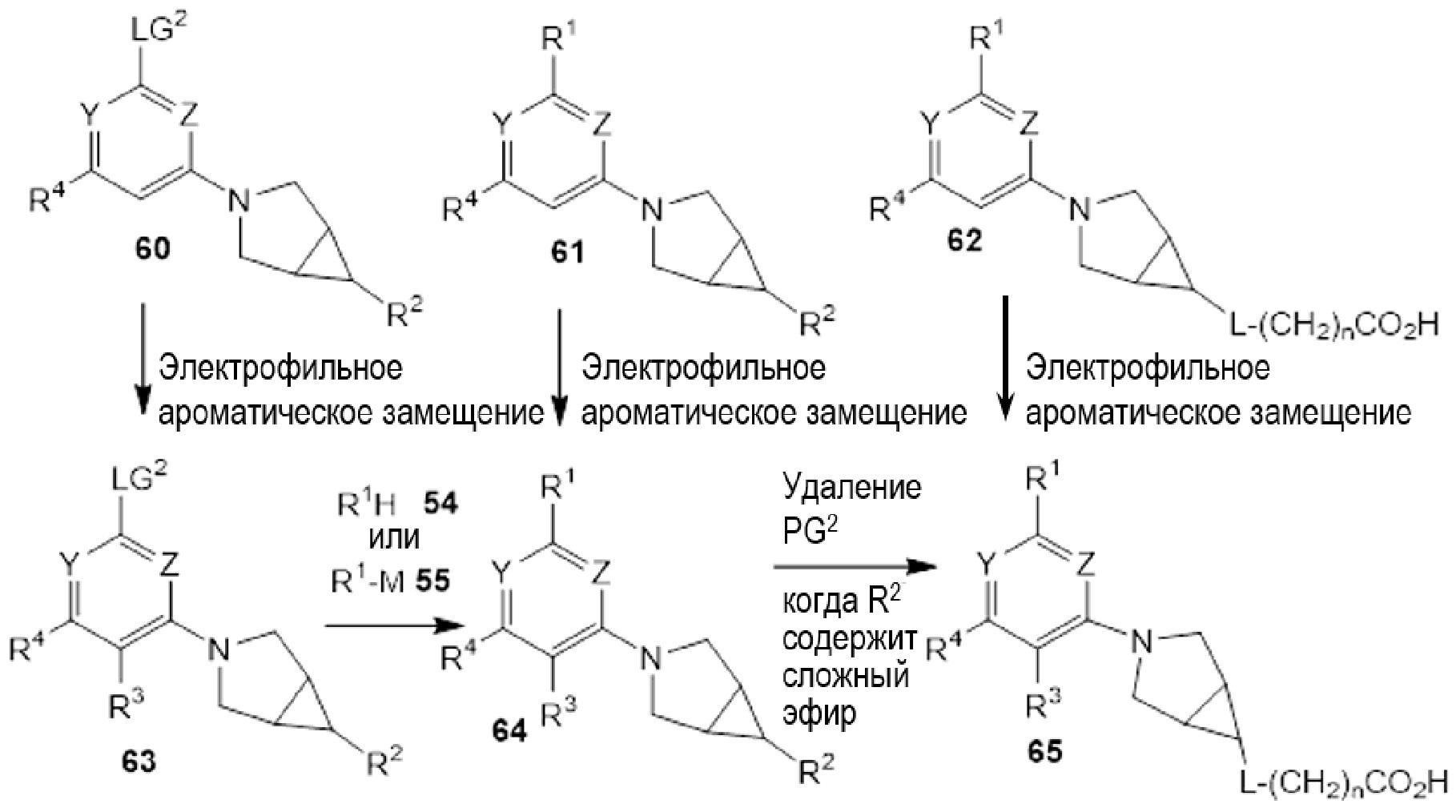
Схема 6B
Промежуточные соединения 60, 61 и 62 можно подвергать реакциям электрофильного ароматического замещения аналогично методам, описанным для превращения промежуточного соединения 16 в 19 на схеме 2, с получением промежуточных соединений 63, 64 и 65, соответственно, где R3=F, Cl, Br, I или алкилы, которые могут быть введены посредством электрофильного ароматического замещения с использованием таких методов, как алкилирование по Фриделю-Крафтсу. Промежуточные соединения 63 и 64 затем могут быть переведены в соединения формулы 65 методами, аналогичными уже описанным методам.
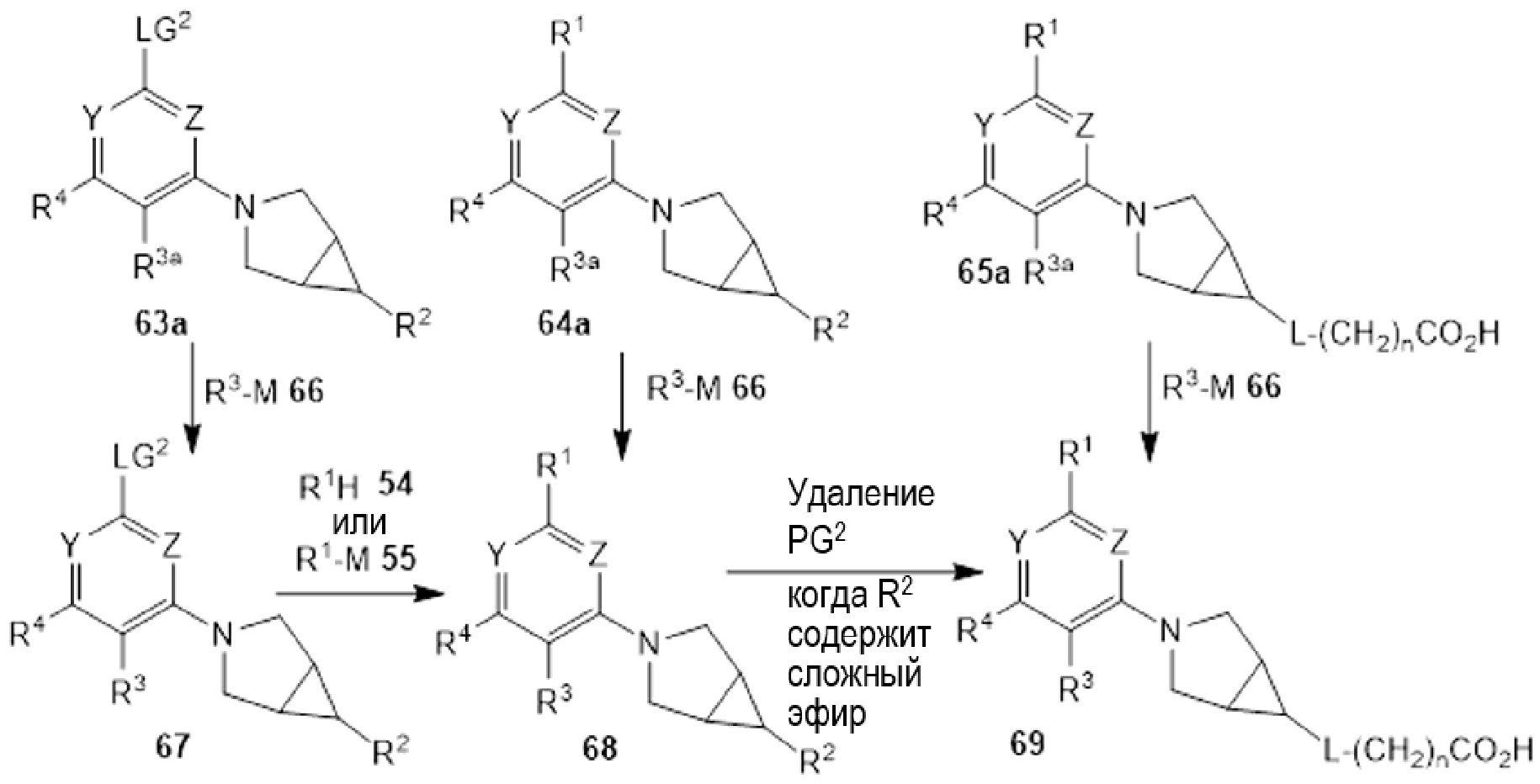
Схема 6C
Альтернативно, как показано на схеме 6C, соединения 63a, 64a и 65a (где R3a=галоген) можно превращать в соединения общей формулы 67, 68 и 69, соответственно (где R3=Me, Et, iPr, cPr и OMe) обработкой R3M (реагент 66, где M может представлять собой металл или металлоид, такой как натрий, калий, цинк, олово, бор, алюминий, магний или другие) с катализом палладием или медью аналогично описанному связыванию 53 с 55, с получением соединений 56 (схема 5).
Иллюстративные промежуточные соединения
2,4-дихлор-6-(дифторметил)пиримидин
Раствор этилдифторацетата (250 г, 2,01 моль) и EtOAc (1070 г, 12,10 моль) нагревали до 70°C и обрабатывали раствором этоксида натрия (151 г, 2,22 моль) в безводном этаноле (2500 мл) в течение 2 ч. Полученную желтую смесь перемешивали при 70°C в течение 14 ч. Охлажденную реакционную смесь подкисляли до pH=2-3 раствором 4M HCl в EtOAc, что приводило к осаждению твердых веществ. Смесь фильтровали через слой целита®, и отфильтрованный осадок промывали EtOAc (4×30 мл). Фильтрат концентрировали, с получением сырого этил-4,4-дифтор-3-оксобутаноата (200 г, 59,8%) в виде желтого масла, которое использовали в следующей стадии без дополнительной очистки.
В раствор этил-4,4-дифтор-3-оксобутаноата (100 г, 602 ммоль) в безводном толуоле (1000 мл) по каплям добавляли мочевину (43,4 г, 722 ммоль) и 2M этоксид натрия в этаноле (81,7 г, 1,20 моль). Полученный желтый раствор перемешивали при rt в течение 30 мин, а затем перемешивали при 120°C в течение 16 ч. Затем желтую суспензию дополнительно перемешивали при 130°C в течение 16 ч. Желтую суспензию охлаждали до rt и концентрировали, с получением 6-(дифторметил)пиримидин-2,4-диола в виде желтого твердого вещества (100 г, колич.), которое непосредственно использовали в следующей стадии без дополнительной очистки.
В двух отдельных партиях, коричневую суспензию 6-(дифторметил)пиримидин-2,4-диола (97,6 г, 602 ммоль) и N,N-диметиланилина (67,8 г, 560 ммоль) в ацетонитриле (1000 мл) охлаждали до 0°C и добавляли по каплям оксихлорид фосфора (231 мл, 2,48 моль). После завершения добавления полученную смесь нагревали до 95°C в течение 16 ч. Затем реакционную смесь охлаждали до 25°C, гасили ледяной водой (1000 мл) и экстрагировали метил-трет-бутиловым эфиром (8×500 мл). Объединенные органические слои промывали рассолом (200 мл), сушили над безводным Na2SO4, фильтровали и концентрировали, с получением коричневого масла (100 г). Две партии объединяли и очищали хроматографией на колонке (соотношение петролейный эфир/EtOAc от 100:0 до 98:2), с получением 2,4-дихлор-6-(дифторметил)пиримидина (92,0 г) в виде светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ: 7,87 (с, 1H), 6,72 (т, 1H).
2,4-дихлор-6-(дифторметил)-5-метилпиримидин
Раствор этилпропионата (200 г, 1,96 моль) в THF (1250 мл) порционно обрабатывали гидридом натрия (60% в минеральном масле, 78,3 г, 1,96 моль). Затем полученную суспензию обрабатывали этилдифторацетатом (486 г, 3,92 моль) по каплям в течение 2 ч. Суспензию нагревали при 50°C в течение 19 ч. Затем охлажденную реакционную смесь обрабатывали 10% серной кислотой (600 мл) и экстрагировали EtOAc (4×500 мл). Объединенные органические слои промывали рассолом (1000 мл), сушили над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали хроматографией на колонке, элюируя смесью петролейный эфир/EtOAc (от 100:0 до 5:1), с получением этил-4,4-дифтор-2-метил-3-оксобутаноата (260 г, 74%) в виде красного масла, которое использовали непосредственно в следующей стадии.
В двух отдельных партиях, в раствор 4,4-дифтор-2-метил-3-оксобутаноата (130 г, 722 ммоль) в безводном толуоле (1,44 л) по каплям добавляли мочевину (52,0 г, 866 ммоль) и 2M этоксид натрия в этаноле (98,2 г, 1,44 моль). Полученный желтый раствор перемешивали при rt в течение 30 мин, а затем перемешивали при 130°C в течение 16 ч. Охлажденные реакционные смеси объединяли и концентрировали, с получением 6-(дифторметил)-5-метилпиримидин-2,4-диола (254 г) в виде светло-желтого твердого вещества, которое использовали непосредственно в следующей стадии.
Смесь 6-(дифторметил)-5-метилпиримидин-2,4-диола (84,7 г, 481 ммоль) и пентахлорида фосфора (401 г, 1,92 моль) перемешивали при 140°C в течение 16 ч. Охлажденную реакционную смесь вливали в ледяную воду (5000 мл) и экстрагировали метил-трет-бутиловым эфиром (8×1000 мл). Органическую фазу промывали рассолом (3000 мл), сушили над Na2SO4, фильтровали и концентрировали, с получением темно-коричневого масла (300 г, сырое). Сырой продукт разделяли на три партии и очищали хроматографией на колонке, элюируя смесью петролейный эфир/EtOAc (от 100:0 до 98:2), с получением 2,4-дихлор-6-(дифторметил)-5-метилпиримидина в виде красного масла (92 г, 30%).
1H ЯМР (400 МГц, CD3OD) δ: 6,83 (т, 1H), 2,49 (с, 3H).
2,4-дихлор-5-метил-6-(трифторметил)пиримидин
В раствор этилпропионата (35,0 г, 340 ммоль) в THF (350 мл) при 25°C добавляли гидрид натрия (60% в минеральном масле, 13,7 г, 343 ммоль). Серую суспензию нагревали до 50°C и этилтрифторацетат (97,4 г, 685 ммоль) добавляли в смесь по каплям в течение 15 мин. Реакционную смесь перемешивали при 50°C в течение 16 ч. Охлажденную реакционную смесь медленно добавляли в 10% серную кислоту при 0°C. Полученную желтую смесь экстрагировали EtOAc (3×500 мл), объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, с получением этил-4,4,4-трифтор-2-метил-3-оксобутаноата (60 г), который использовали непосредственно в следующей стадии.
В раствор этил-4,4,4-трифтор-2-метил-3-оксобутаноата (60,0 г, 303 ммоль) в безводном толуоле (500 мл) порционно добавляли мочевину (21,8 г, 363 ммоль) и свежеприготовленный 2M этоксид натрия в этаноле (41,2 г, 606 ммоль). Полученный желтый раствор перемешивали при rt в течение 15 мин, а затем нагревали до 130°C в течение 48 ч. Реакционную смесь концентрировали и растворитель удаляли, с получением сырого 5-метил-6-(трифторметил)пиримидин-2,4-диола (60 г) в виде смолы, которую использовали в следующей стадии без дополнительной очистки.
5-метил-6-(трифторметил)пиримидин-2,4-диол (120 г, 480 ммоль) добавляли в оксихлорид фосфора (371,0 г, 2,420 ммоль) при 0°C и по каплям обрабатывали N,N-диметиланилином (54,6 г, 451 ммоль). Полученную смесь нагревали до 100°C в течение 16 ч. Темную реакционную смесь охлаждали до rt и вливали в ледяную воду. Водный слой экстрагировали метил-трет-бутиловым эфиром (3×1000 мл), объединенные органические слои сушили над Na2SO4 и концентрировали, с получением темно-желтого масла (80 г). Сырой продукт растворяли в н-гексане и небольшое количество образовавшегося нерастворимого материала удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении, с получением 2,4-дихлор-5-метил-6-(трифторметил)пиримидина (40 г, 36%) в виде желтого масла с примесью остаточного н-гексана.
1H ЯМР (400 МГц, CD3OD) δ: 2,53 (с, 3H).
2,4-дихлор-6-(1,1-дифторэтил)пиримидин
Стадия 1: 6-(1,1-дифторэтил)пиримидин-2,4-диол
Раствор гексаметилдисилазида лития (217 мл, 1M раствор в THF, 217 ммоль) в сухом THF (400 мл) охлаждали в атмосфере аргона до -78°C и по каплям обрабатывали EtOAc (19,1 г, 217 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 ч, затем по каплям обрабатывали этил-2,2-дифторпропионатом (15,0 г, 110 ммоль). Перемешивание продолжали в течение 4 ч при -78°C. По каплям добавляли насыщенный раствор хлорида аммония (150 мл). Смесь нагревали до rt, подкисляли 1M HCl (150 мл) и оставляли стоять на 2 ч. Фазы разделяли, водную фазу экстрагировали EtOAc, и объединенные органические фазы промывали 1M HCl, рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя смесью петролейный эфир/EtOAc (от 100:0 до 7:3), с получением этил-4,4-дифтор-3-оксопентаноата (27 г) в виде желтого масла, которое использовали непосредственно в следующей стадии.
В раствор этил-4,4-дифтор-3-оксопентаноата (20,0 г, 111 ммоль) и мочевины (8,00 мг, 133 ммоль) в безводном толуоле (400 мл) и этаноле (30 мл) добавляли твердый этоксид натрия (30200 мг, 222 ммоль) при rt. Затем смесь нагревали до 125°C с обратным холодильником, оснащенным ловушкой Дина-Старка. Реакционную смесь охлаждали до rt и растворитель удаляли при пониженном давлении. Осадок подкисляли до pH=4 4Н HCl в EtOAc и экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали, с получением сырого продукта (20,0 г) в виде желтого масла. Сырой продукт очищали смесью EtOH:петролейный эфир (1:1), собирая указанное в заголовке соединение (11,6 г, 59%) в виде твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 5,71 (с, 1H), 1,93 (т, 3H).
Стадия 2
В раствор 6-(1,1-дифторэтил)пиримидин-2,4-диола (9,60 г, 54,5 ммоль) в ацетонитриле (120 мл) добавляли оксихлорид фосфора (41,8 г, 273 ммоль), а затем N,N-диизопропиламин (704 мг, 5,45 ммоль). Смесь перемешивали при 80°C в течение 16 ч. Реакционную смесь охлаждали до rt и вливали в ледяную воду (60 мл). Смесь подщелачивали до pH=7-8 насыщенным водным раствором карбоната натрия и экстрагировали EtOAc (3×30 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали, с получением коричневого масла. Сырой продукт очищали хроматографией на колонке, элюируя смесью DCM/петролейный эфир, с получением 2,4-дихлор-6-(1,1-дифторэтил)пиримидина (6,5 г, 56%) в виде прозрачного масла.
1H ЯМР (400 МГц, CD3OD) δ: 7,85 (с, 1H), 1,97 (т, 3H).
2,4-дихлор-6-(1,1-дифторэтил)-5-метилпиримидин
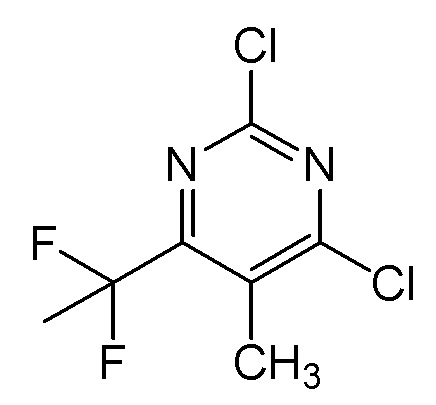
Стадия 1: этил-4,4-дифтор-2-метил-3-оксопентаноат
В раствор этилпропионата (15,0 г, 147 ммоль) в THF (70 мл) порционно добавляли гидрид натрия (60% в минеральном масле, 5,87 г, 147 ммоль). Затем полученную серую суспензию по каплям обрабатывали этил-2,2-дифторпропионатом (24,3 г, 176 ммоль) в течение 15 мин. Суспензию нагревали до 50°C в течение 4 ч, затем перемешивали при 16°C в течение 60 ч. Смесь медленно вливали в 10% раствор серной кислоты (60 мл) и экстрагировали EtOAc (2×50 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя смесью EtOAc:петролейный эфир (1:10), с получением указанного в заголовке соединения (18 г) в виде коричневого масла.
1H ЯМР (400 МГц, CD3OD) δ: 3,76 (к, 2H), 3,52 (к, 1H), 1,32 (т, 3H), 0,98 (д, 3H), 0,83 (т, 3H).
Стадия 2
В раствор этил-4,4-дифтор-2-метил-3-оксопентаноата (18 г, 93 ммоль) и мочевины (6,68 г, 111 ммоль) в толуоле (270 мл) добавляли раствор этоксида натрия (12,6 г,185 ммоль) в этаноле (90 мл). Раствор перемешивали при 130°C в течение 16 ч. Охлажденную реакционную смесь концентрировали, с получением 6-(1,1-дифторэтил)-5-метилпиримидин-2,4-диола (19 г) в виде серого твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
Смесь 6-(1,1-дифторэтил)-5-метилпиримидин-2,4-диола (7,5 г, 39 ммоль) в оксихлориде фосфора (50 мл) и DMF (8 мл) перемешивали при 100°C в течение 5 ч. Охлажденную реакционную смесь осторожно вливали в ледяную воду (150 мл) и экстрагировали EtOAc (3×80 мл). Объединенные органические слои промывали рассолом (2×100 мл), сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, с получением 2,4-дихлор-6-(1,1-дифторэтил)-5-метилпиримидина в виде желтого масла (6,0 г, 67%).
1H ЯМР (400 МГц, CDCl3) δ: 2,59 (с, 3H), 2,01 (т, 3H).
(2S,3R)-3-гидрокси-2-метилазетидин-1-ий [(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат
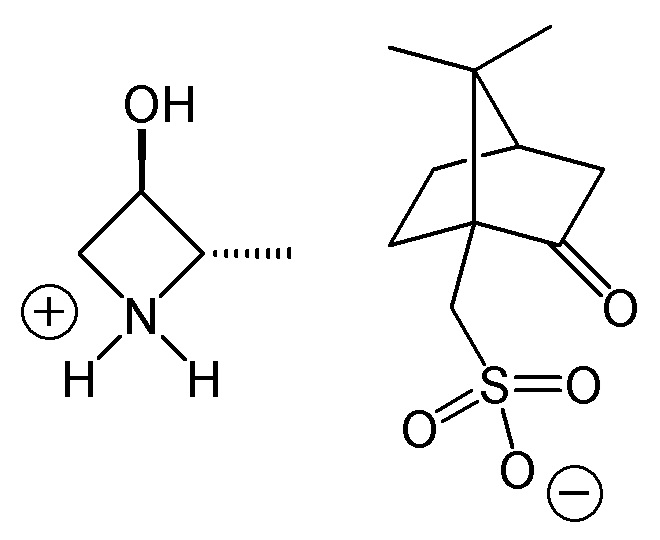
Стадия 1: (2R)-2-[(1R)-1-бромэтил]оксиран
В трех отдельных реакционных сосудах раствор (2E)-бут-2-ен-1-ола (967 г, 13,4 моль) в хлороформе (10 л) обрабатывали бромом (2,15 кг, 13,4 моль) на протяжении 2 ч при 0°C. Смесь перемешивали при 15°C в течение 30 мин. Смеси гасили насыщенным раствором тиосульфата натрия (500 мл) при 15°C. Три реакционные смеси объединяли и экстрагировали DCM (3×5 л). Объединенные органические слои концентрировали в вакууме, с получением транс-2,3-дибромбутан-1-ола (10,5 кг, колич.) в виде желтого масла, которое переносили в следующую стадию без дополнительной очистки. В трех отдельных реакционных сосудах раствор KOH (711 г, 12,7 моль) в воде (6 л) по каплям добавляли в раствор транс-2,3-дибромбутан-1-ола (3,33 кг, 12,7 моль) в THF (9 л) при 15°C. Реакционную смесь перемешивали при rt в течение 2 ч. Три реакционные смеси объединяли и органический слой отделяли. Водную фазу экстрагировали EtOAc (3×5 л). Объединенные органические слои промывали рассолом (5 л x 3), сушили Na2SO4, фильтровали и концентрировали в вакууме, с получением указанного в заголовке соединения (6,5 кг, колич.) в виде желтого масла, которое переносили в следующую стадию без дополнительной очистки.
1H ЯМР (600 МГц, CD3OD) δ: 3,86 (квин., 1H), 3,19-3,22 (м, 1H), 2,94 (т, 1H), 2,76-2,78 (м, 1H), 1,73 (д, 3H).
Стадия 2: (2S,3R)-1-(дифенилметил)-2-метилазетидин-3-ол
В двух отдельных реакционных сосудах раствор (2R)-2-[(1R)-1-бромэтил]оксирана (3,28 кг, 16,2 моль) и бензгидриламина (2,97 кг, 16,2 моль) в безводном этаноле (5,41 л) обрабатывали NaHCO3 (2,07 кг, 24,34 моль) и смесь перемешивали при rt в течение 80 ч. Затем смесь дополнительно перемешивали при 65°C в течение 24 ч. Две реакционные смеси охлаждали до rt, объединяли и фильтровали. Фильтрат концентрировали. Осадок растворяли в DCM (10 л), промывали насыщенным водным раствором хлорида аммония (2×5 л), сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир/EtOAc (от 50:1 до 1:1), с получением указанного в заголовке соединения (3,18 кг, ~80% чистоты, выход 36,5%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ: 7,16-7,46 (м, 10H), 4,34 (с, 1H), 3,93 (к, 1H), 3,66 (т, 1H), 3,03 (к, 1H), 2,58 (т, 1H), 0,76 (д, 3H).
Стадия 3: (2S,3R)-1-(дифенилметил)-3-гидрокси-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат
В раствор [(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоновой кислоты (2,7 кг, 12 моль) в этаноле (8 л) добавляли раствор (2S,3R)-1-(дифенилметил)-2-метилазетидин-3-ола (3,18 кг, 11,7 моль) в этаноле (2 л). Полученный раствор выпаривали для удаления EtOH. Осадок обрабатывали метил-трет-бутиловым эфиром (5 л) и выпаривали до того, как оставался ~1 л растворителя. Осадок дополнительно обрабатывали метил-трет-бутиловым эфиром (5 л) и фильтровали. Отфильтрованный осадок сушили в вакууме, с получением белого твердого вещества (3,5 кг), которое растворяли в DCM (7,6 л), и добавляли EtOAc (10,9 л). Смесь перемешивали при rt в течение 30 мин, что приводило к осаждению белого твердого вещества, которое собирали фильтрованием. Отфильтрованный осадок суспендировали в DCM (10,6 л), перемешивали при rt в течение 10 мин, а затем в раствор добавляли EtOAc (10,6 л). Смесь перемешивали при rt в течение 30 мин и полученный белый осадок собирали фильтрованием. Отфильтрованный осадок растворяли в DCM (10,6 л), перемешивали при rt в течение 10 мин, затем добавляли EtOAc (10,6 л). Реакционную смесь перемешивали при rt в течение 30 мин, и осажденное твердое вещество собирали фильтрованием, с получением белого твердого вещества (1,3 кг, э.и.=95,2% методом хиральной СЖХ). Этот материал растворяли в DCM (7 л) и нагревали с обратным холодильником в течение 40 мин. Добавляли EtOAc (3,5 л) и смесь дополнительно перемешивали при 40°C в течение 20 мин, при этом осаждалось белое твердое вещество. Твердое вещество собирали фильтрованием. Отфильтрованный осадок сушили в вакууме, с получением указанного в заголовке соединения (1,1 кг, э.и.=98,2% методом хиральной СЖХ, выход хирального разделения 62,9%) в виде белого твердого вещества.
1H ЯМР (600 МГц, CD3OD) δ: 7,44-7,59 (м, 10H), 5,66 (с, 1H), 4,35-4,41 (м, 1H), 4,25-4,30 (м, 2H), 3,73-3,78 (м, 1H), 3,37 (д, 1H), 2,80 (д, 1H), 2,68-2,74 (м, 1H), 2,36 (дт, 1H), 2,02-2,09 (м, 2H), 1,91 (д, 1H), 1,60-1,66 (м, 1H), 1,40-1,45 (м, 1H), 1,16 (с, 3H), 1,09 (д, 3H), 0,88 (с, 3H).
Стадия 4
Частичный раствор (2S,3R)-1-(дифенилметил)-3-гидрокси-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (18,96 г, 39,04 ммоль) в метаноле (60 мл) обрабатывали 10% гидроксидом палладия на угле (1,11 г) в реакционном сосуде из нержавеющей стали. Реакционный сосуд продували газообразным азотом, затем заполняли газообразным водородом (60 psi; 4,083 атм.). Реакционную смесь перемешивали при rt в течение 17 ч, затем повторно создавали давление газообразным водородом (55 psi; 3,743 атм.). Еще через 24 ч реакционную смесь продували газообразным азотом и фильтровали через слой целита®, элюируя метанолом (4×80 мл). Объединенные фильтраты выпаривали, с получением белого маслянистого полутвердого вещества. Этот материал суспендировали в гептане (100 мл), бока колбы очищали шпателем и гептаны декантировали. Этот процесс повторяли два раза, твердое вещество суспендировали в гептанах (200 мл) и перемешивали при rt в течение 2,5 ч. Твердое вещество собирали фильтрованием, суспендировали в гептанах (100 мл) и перемешивали при rt в течение 1 ч. Твердое вещество собирали фильтрованием, суспендировали в гептанах (120 мл) и интенсивно перемешивали в течение 24 ч. Твердое вещество собирали фильтрованием, с получением (2S,3R)-3-гидрокси-2-метилазетидин-1-ий[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (11,8 г, 95%) в виде белого твердого вещества.
1H ЯМР (600 МГц, CD3OD) δ: 4,27-4,34 (м, 2H), 4,04-4,09 (м, 1H), 3,76-3,80 (м, 1H), 3,31 (д, 1H), 2,80 (д, 1H), 2,62-2,69 (м, 1H), 2,34-2,39 (м, 1H), 2,04-2,09 (м, 2H), 1,92 (д, 1H), 1,63-1,68 (м, 1H), 1,54 (д, 3H), 1,41-1,47 (м, 1H), 1,13 (с, 3H), 0,88 (с, 3H).
(2S,3R)-3-гидрокси-2,3-диметилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат
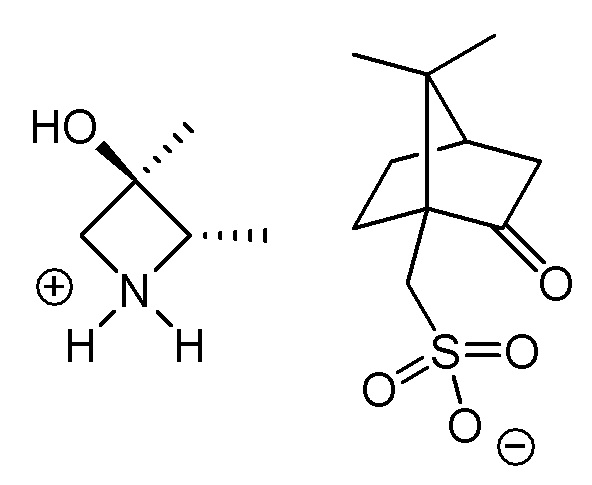
Стадия 1: трет-бутил[(2S)-4-хлор-3-оксобутан-2-ил]карбамат
Магниевую стружку (120 г, 4,90 моль) и йод (50 мг) объединяли в трехгорлой 250-мл круглодонной колбе, оснащенной обратным холодильником. Добавляли раствор трет-бутилхлорида (22,5 г, 245 ммоль) в THF (80 мл), а затем этилбромид (5 мл). Реакционную смесь нагревали до 60°C и наблюдали интенсивное кипение. Дополнительное количество трет-бутилхлорида (428 г, 4,65 моль) в THF (1,52 л) добавляли по каплям через дополнительную воронку с такой скоростью, чтобы поддерживать умеренное стекание флегмы. После завершения добавления темный раствор с Mg стружкой нагревали при 60°C в течение 30 мин, затем охлаждали до 0°C. К охлажденному раствору Гриньяра добавляли триэтиламин (120 г, 1,19 моль) и твердый хлорацетат натрия (139 г, 1,19 моль). Затем по каплям добавляли раствор метилового эфира Boc-L-аланина (157 г, 0,77 моль) в толуоле (900 мл). Реакционную смесь нагревали до rt и перемешивали в течение 16 ч. Затем реакционную смесь охлаждали до 0°C и по каплям добавляли уксусную кислоту (320 г, 5,50 моль) в воде (640 мл). Добавляли водный 2M раствор HCl (70 мл), доводя значение pH водного слоя до ~4-5. Реакционную смесь перемешивали при rt в течение 45 мин до прекращения выделения газа. Слои разделяли и водный слой экстрагировали EtOAc (500 мл). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (60 мл) и рассолом (30 мл). Органические слои сушили над безводным Na2SO4 и концентрировали, с получением желтого масла. В масло добавляли гептан (300 мл) и перемешивали при rt в течение 30 мин. Полученное твердое вещество фильтровали и промывали гептаном, с получением указанного в заголовке соединения (105 г, 61%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 5,08 (уш.с, 1H), 4,50-4,57 (м, 1H), 4,23-4,32 (м, 2H), 1,44 (с, 9H), 1,36 (д, 3H).
Стадия 2: трет-бутил[(2S,3S)-4-хлор-3-гидрокси-3-метилбутан-2-ил]карбамат
В раствор трет-бутил[(2S)-4-хлор-3-оксобутан-2-ил]карбамата (90 г, 0,40 моль) в DCM (2,0 л), охлажденный до -70°C, по каплям добавляли метилбромид магния (460 мл, 1,38 моль, 3M в диэтиловом эфире). Смесь перемешивали при -70°C в течение 1 ч, затем нагревали до ~-5°C и перемешивали в течение 5 ч. Реакционную смесь по каплям гасили насыщенным водным раствором хлорида аммония (500 мл) с такой скоростью, чтобы внутренняя температура не поднималась выше 10°C. Серая суспензия становилась молочно-белой, затем значение pH доводили до ~2 2Н водным раствором HCl. Органический слой отделяли и водный слой экстрагировали DCM (3×800 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Сырой продукт растворяли в смеси гексан/EtOAc (10/1, 200 мл). Желтую смесь нагревали до 50°C, перемешивали в течение 10 мин и затем медленно охлаждали до 0°C. Образовавшееся твердое вещество фильтровали, с получением указанного в заголовке соединения (45 г, 47%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 4,72 (уш.с, 1H), 3,77-3,87 (м, 1H), 3,60 (д, 1H), 3,52 (д, 1H), 1,46 (с, 9H), 1,30 (с, 3H), 1,21 (д, 3H).
Стадия 3
В раствор трет-бутил[(2S,3S)-4-хлор-3-гидрокси-3-метилбутан-2-ил]карбамата (55 г, 0,23 ммоль) в DCM (20 мл) и метаноле (100 мл) добавляли 4Н HCl в диоксане (150 мл) при 0°C. Коричневую смесь нагревали до 20°C и перемешивали в течение 2,5 ч. Коричневую смесь концентрировали, с получением коричневого масла (40 г, 100%), которое растворяли в CH3CN (300 мл) и обрабатывали твердым NaHCO3 (146 г, 1,74 моль). Белую суспензию перемешивали при 70°C в течение 4 часов, охлаждали до rt, фильтровали через целит® и промывали ацетонитрилом. Желтый фильтрат концентрировали в вакууме, с получением (2S,3R)-2,3-диметилазетидин-3-ола (22 г, 75%) в виде коричневого масла. Соединение использовали в следующей стадии без дополнительной очистки.
В желтый раствор (2S,3R)-2,3-диметилазетидин-3-ола (23,4 г, 0,23 моль) в ацетонитриле (130 мл) добавляли [(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоновую кислоту (48 г, 0,21 моль) и перемешивали при 15°C в течение 4 ч. Образовавшийся преципитат собирали фильтрованием, с получением (2S,3R)-3-гидрокси-2,3-диметилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (50 г, 65%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ: 4,36 (к, 1H), 3,89 (д, 1H), 3,76 (д, 1H), 3,32 (д, 1H), 2,80 (д, 1H), 2,63-2,72 (м, 1H), 2,36 (дт, 1H), 2,02-2,10 (м, 2H), 1,93 (д, 1H), 1,60-1,68 (м, 1H), 1,42-1,48 (м, 7H), 1,16 (с, 3H), 0,88 (с, 3H).
(2S)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат
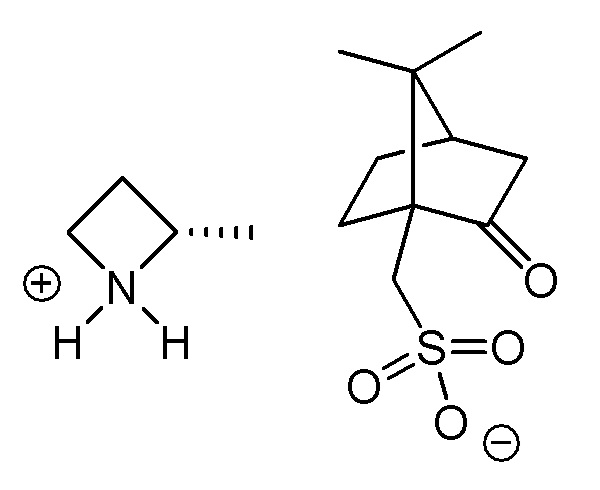
Стадия 1: (2S)-1-(дифенилметил)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат
Раствор R-(-)-1,3-бутандиола (20,0 г, 222 ммоль) и DIPEA (101,5 мл, 585,0 ммоль) в ацетонитриле (444 мл) охлаждали до -30°C и по каплям обрабатывали трифторметансульфоновым ангидридом (81,2 мл, 480 ммоль) через дополнительную воронку в течение 90 мин, поддерживая внутреннюю температуру реакционной смеси в диапазоне от -30 до -35°C. После завершения добавления реакционную смесь перемешивали в течение 10 мин при -30°C, затем дополнительно по каплям обрабатывали трифторметансульфоновым ангидридом (1,5 мл) и дополнительно перемешивали при -30°C в течение 15 мин. Затем реакционную смесь дополнительно обрабатывали DIPEA (101,5 мл, 585,0 ммоль) на протяжении 15 мин, поддерживая внутреннюю температуру на уровне -30°C. Еще через 10 мин при -30°C реакционную смесь по каплям обрабатывали раствором бензгидриламина (38 мл) в ацетонитриле (40 мл) в течение 30 мин через дополнительную воронку, поддерживая внутреннюю температуру реакционной смеси ниже -30°C. Реакционную смесь перемешивали при -30°C в течение 20 мин, затем помещали в ледяную баню на 30 мин. Затем реакционную смесь перемешивали при rt в течение 30 мин, с последующим нагреванием при 45°C в течение 30 мин. Реакционную смесь охлаждали до rt, вливали в деионизированную воду (900 мл) и экстрагировали толуолом (1 л). Водную фазу обратно экстрагировали толуолом (300 мл), объединенные органические слои промывали водой (2×250 мл), сушили над Na2SO4, фильтровали и выпаривали. Сырой продукт растворяли в DCM (300 мл) и наносили на слой силикагеля (300 мл SiO2, предварительно промытый 1:1 смесью гептан/EtOAc). Слой промывали смесью 1:1 гептан/EtOAc (1,2 л) и фильтрат выпаривали, с получением красного масла (50,2 г). Сырой продукт растворяли в метаноле (200 мл), помещали в водяную баню при 10°C, и партиями обрабатывали [(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоновой кислотой (49 г) в течение 5 минут. Раствор перемешивали при rt в течение 2 ч, растворитель выпаривали и твердые вещества сушили в высоком вакууме в течение 15 ч, с получением твердого вещества (99,2 г). Твердое вещество растворяли в DCM (100 мл) и перемешивали при rt в течение 10 мин, с получением темного раствора. EtOAc (850 мл) медленно добавляли при перемешивании и через ~5 мин из раствора осаждалось твердое вещество. Суспензию перемешивали при rt в течение 2 ч, твердое вещество собирали фильтрованием и промывали EtOAc (50 мл). Твердое вещество растворяли в DCM (100 мл) и добавляли EtOAc (700 мл). Смесь перемешивали при rt, и из раствора немедленно осаждалось твердое вещество. Суспензию перемешивали при rt в течение 15 ч, затем твердое вещество собирали фильтрованием, промывали EtOAc (50 мл) и сушили при пониженном давлении, с получением указанного в заголовке соединения (66,7 г, выход 65%) в виде белого твердого вещества.
1H ЯМР (500 МГц, CD3OD) δ: 7,54-7,59 (м, 4H), 7,43-7,53 (м, 6H), 5,67 (с, 1H), 4,69-4,76 (м, 1H), 3,97-4,02 (м, 2H), 3,36 (д, 1H), 2,81 (д, 1H), 2,70-2,75 (м, 1H), 2,58-2,64 (м, 1H), 2,31-2,39 (м, 2H), 2,03-2,09 (м, 2H), 1,91 (д, 1H), 1,62-1,66 (м, 1H), 1,41-1,47 (м, 1H), 1,16 (с, 3H), 1,11 (д, 3H), 0,88 (с, 3H); Элементный анализ: Рассчитано для C27H35NO4S: C=69,05%, H=7,51%, N=2,98%; Найдено: C=68,90%, H=7,59%, N=2,91%.
Стадия 2
В 300-мл реактор из нержавеющей стали наливали раствор (2S)-1-(дифенилметил)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (29,4 г, 62,6 ммоль) в метаноле (125 мл) и 20% Pd(OH)2/C (1,78 г). Реактор продували азотом три раза, затем водородом три раза, затем создавали давление водорода 60 psi (4,083 атм) и перемешивали при rt в течение 16 ч. Водород выпускали и реактор продували азотом. Реакционную смесь фильтровали через слой целита®, элюируя метанолом (100 мл), и фильтрат концентрировали в вакууме, с получением белого твердого вещества. Белое твердое вещество суспендировали в смеси EtOAc/метил-трет-бутиловый эфир (1:1, 200 мл) и перемешивали в течение 1 ч при 60°C. После охлаждения до rt суспензию дополнительно перемешивали в течение часа и твердое вещество собирали фильтрованием. Полученное твердое вещество суспендировали в метил-трет-бутиловом эфире (100 мл) и перемешивали при rt в течение 16 часов. Твердое вещество собирали фильтрованием, промывали метил-трет-бутиловым эфиром (25 мл) и сушили при пониженном давлении, с получением (2S)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (18,1 г, 95%) в виде белого твердого вещества.
1H ЯМР (500 МГц, CD3OD) δ: 4,59-4,66 (м, 1H), 4,05 (к, 1H), 3,92 (тд, 1H), 3,32 (м, 1H), 2,80 (д, 1H), 2,59-2,70 (м, 2H), 2,36 (дт, 1H), 2,25-2,32 (м, 1H), 2,03-2,10 (м, 2H), 1,92 (д, 1H), 1,62-1,68 (м, 1H), 1,57 (д, 3H), 1,41-1,47 (м, 1H), 1,15 (с, 3H), 0,89 (с, 3H); Элементный анализ: Рассчитано для C14H25NO4S: C=55,42%, H=8,31%, N=4,62%; Найдено: C=55,59%, H=8,41%, N=4,49%.
(2S)-2-метилазетидин гидрохлорид
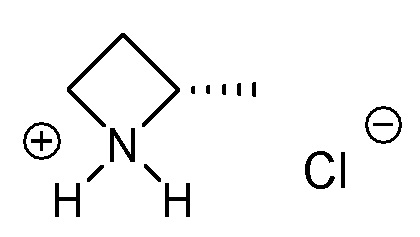
Стадия 1: (2R)-4-[(метилсульфонил)окси]бутан-2-илметансульфонат
Раствор (3R)-бутан-1,3-диола (3 г, 30 ммоль) и триэтиламина (10,1 г, 99,9 ммоль) в DCM (60 мл) охлаждали до 0°C и по каплям обрабатывали метансульфонилхлоридом (11,4 г, 99,9 ммоль) при 0°C. Через 15 мин ледяную баню удаляли и смесь перемешивали при rt в течение 2 ч. Смесь разбавляли насыщенным водным раствором хлорида аммония (80 мл) и экстрагировали DCM (3×50 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали, с получением осадка. Осадок очищали хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (от 1:4 до 3:2), с получением указанного в заголовке соединения (7,3 г, 89%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ: 5,00 (с, 1H), 4,35 (т, 2H), 3,07 (с, 3H), 3,06 (с, 3H), 2,05-2,12 (м, 2H), 1,50 (д, 3H).
Стадия 2
(2R)-4-[(метилсульфонил)окси]бутан-2-илметансульфонат (7,20 г, 29,2 ммоль) растворяли в бензиламине (19,2 мл, 175 ммоль) и перемешивали при 45°C в течение 16 ч. Реакционную смесь охлаждали до rt и добавляли смесь циклогексан/метил-трет-бутиловый эфир (1:1), что приводило к осаждению белого вещества. Осадок удаляли фильтрованием, фильтрат выпаривали при пониженном давлении и очищали хроматографией на колонке, элюируя DCM и 1% смесью гидроксид аммония/метанол, (от 100:0 до 99,5:0,5), с получением светло-желтого масла (2,5 г, 53%). Это желтое масло (2,28 г, 14,1 ммоль) растворяли в метаноле (50 мл) и обрабатывали 10% гидроксидом палладия на угле (500 мг). Полученную суспензию нагревали до 50°C в атмосфере газообразного водорода (30 PSI; 2,041 атм) в течение 20 ч, затем нагревали до 60°C и дополнительно перемешивали в атмосфере водорода (30 PSI; 2,041 атм) в течение 40 ч. Охлажденную реакционную смесь фильтровали, фильтрат обрабатывали 4Н HCl в EtOAc (15 мл) и перемешивали при rt в течение 30 мин. Смесь концентрировали, с получением (2S)-2-метилазетидин гидрохлорида (1,47 г, 96,6%) в виде белой смолы.
1H ЯМР (400 МГц, CDCl3) δ: 4,50-4,60 (м, 1H), 3,97-4,04 (м, 1H), 3,75-3,90 (м, 1H), 2,58-2,65 (м, 1H), 2,26-2,35 (м, 1H), 1,54 (д, 3H).
этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат
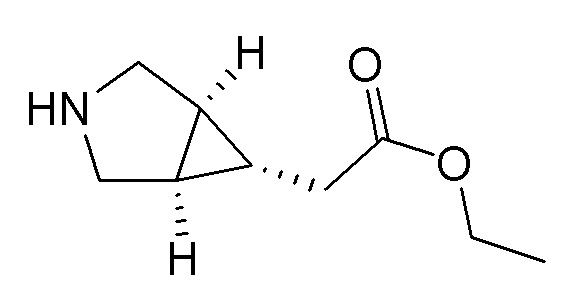
Стадия 1: [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метилметансульфонат
Получение [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанола описано в Berliner, M. A. et al., Org. Process Res. Dev. 2011, 15, 1052-1062.
В раствор [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанола (95,0 г, 396 ммоль) в сухом THF (1230 мл) и DMF (95 мл) добавляли триэтиламин (241 г, 2,38 моль) при 0°C. Смесь перемешивали при 0°C в течение 5 мин и по каплям обрабатывали метансульфонилхлоридом (82,22 г, 717,8 ммоль) в течение 5 мин. Смесь перемешивали при 10°C в течение 16 ч. Реакционную смесь гасили добавлением насыщенного раствора NaHCO3 (1000 мл) и затем смесь экстрагировали метил-трет-бутиловым эфиром (5×500 мл). Органическую фазу концентрировали в вакууме, с получением указанного в заголовке соединения (99 г, 89%) в виде коричневого масла.
МС(ЭС+): 281,9 (M+H).
Стадия 2: [(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетонитрил
В раствор [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метилметансульфоната (99 г, 352 ммоль) в DMF (700 мл) добавляли цианид натрия (18,49 г, 377,3 ммоль) при 20°C. Смесь перемешивали при 20°C в течение 16 ч. В реакционную смесь добавляли насыщенный водный раствор NaHCO3 (200 мл) и смесь экстрагировали метил-трет-бутиловым эфиром (2×150 мл). Органические слои сушили над Na2SO4, фильтровали и концентрировали, с получением коричневого масла (50 г). Коричневое масло очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир/EtOAc (от 10:1 до 5:1), с получением указанного в заголовке соединения (37 г, 50%) в виде желтого масла.
МС(ХИАД): 213,1 (M+H).
Стадия 3: этил[(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетат
В этанол (215 мл) добавляли концентрированную серную кислоту (108 мл) при 0°C. Смесь перемешивали при 10°C в течение 5 мин, затем повторно охлаждали до 0°C. Раствор [(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетонитрила (37 г, 170 ммоль) в EtOH (95 мл) добавляли в смесь EtOH и серной кислоты при 0°C. Смесь перемешивали при 80°C в течение 16 ч. Значение pH смеси доводили до 9 5M NaOH при 0°C, и продукт экстрагировали EtOAc (5×500 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, с получением желтого масла (45 г). Желтое масло очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир/EtOAc (от 10:1 до 5:1), с получением указанного в заголовке соединения (37 г, 82%) в виде желтого масла.
МС(ХИАД): 260,1 (M+H).
Стадия 4: этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат
В раствор этил[(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетата (37 г, 140 ммоль) в EtOH (1500 мл) добавляли 10% гидроксид палладия на угле (5 г, 4 ммоль). Смесь дегазировали, три раза продували азотом и дегазировали, а затем три раза продували газообразным водородом. Смесь перемешивали в атмосфере водорода (50 PSI; 3,402 атм) при 50°C в течение 16 ч. Охлажденную реакционную смесь продували азотом, фильтровали и отфильтрованный осадок промывали MeOH (500 мл). Фильтрат концентрировали в вакууме, с получением этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетата (22 г, 91%) в виде желтого масла.
МС(ЭС+): 170,1 (M+H).
этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат, соль трифторуксусной кислоты
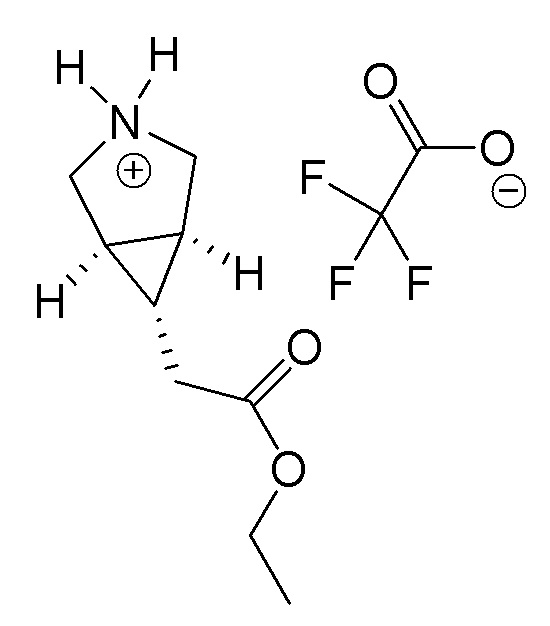
Стадия 1: трет-бутил(1R,5S,6s)-6-(2-этокси-2-оксоэтил)-3-азабицикло[3,1,0]гексан-3-карбоксилат
В раствор [(1R,5S,6s)-3-(трет-бутоксикарбонил)-3-азабицикло[3,1,0]гекс-6-ил]уксусной кислоты (400 мг, 1,66 ммоль, MFCD12198681) в DCM (12 мл) добавляли этанол (0,4 мл), 4-диметиламинопиридин (203 мг, 1,66 ммоль) и N,N'-дициклогексилкарбодиимид (342 мг, 1,66 ммоль) при rt. Полученную бесцветную суспензию перемешивали при rt в течение 16 ч. Смесь разбавляли водой (15 мл) и водным раствором хлорида аммония (10 мл). Продукт экстрагировали DCM (3×25 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали, с получением осадка (650 мг) в виде белого твердого вещества, которое очищали флэш-хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (от 1% до 11% EtOAc), с получением указанного в заголовке соединения (350 мг, 78%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 4,15 (к, 2H), 3,53-3,64 (м, 2H), 3,29-3,37 (м, 2H), 2,17-2,32 (м, 2H), 1,44 (с, 9H), 1,35-1,38 (м, 2H), 1,27 (т, 3H), 0,88-0,92 (м, 1H).
Стадия 2
В раствор трет-бутил(1R,5S,6s)-6-(2-этокси-2-оксоэтил)-3-азабицикло[3,1,0]гексан-3-карбоксилата (340 мг, 1,26 ммоль) в DCM (6 мл) добавляли TFA (5 мл). Смесь перемешивали при rt в течение 1 ч. Смесь концентрировали до сухого состояния, с получением этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетата, соли трифторуксусной кислоты (400 мг, 99%), в виде коричневой жидкости.
1H ЯМР (400 МГц, CD3OD) δ 4,13 (к, 2H), 3,37-3,45 (м, 4H), 2,35 (д, 2H), 1,72-1,77 (м, 2H), 1,25 (т, 3H), 1,06-1,12 (м, 1H).
этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат гидрохлорид
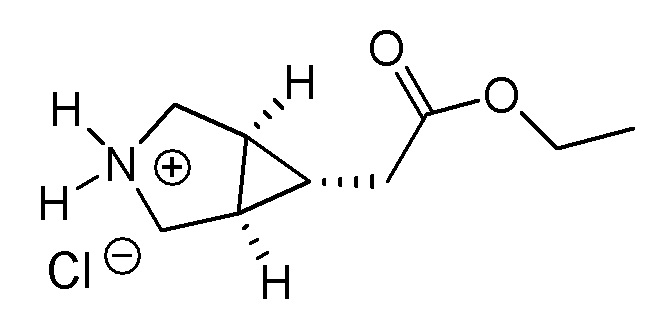
Стадия 1: трет-бутил(1R,5S,6r)-6-(бромметил)-3-азабицикло[3,1,0]гексан-3-карбоксилат
В раствор трет-бутил(1R,5S,6r)-6-(гидроксиметил)-3-азабицикло[3,1,0]гексан-3-карбоксилата (5,1 г, 23,91 ммоль, MFCD14525755) в DCM (180 мл) добавляли тетрабромид углерода (11,9 г, 35,9 ммоль) и трифенилфосфин (9,41 г, 35,9 ммоль) при 5°C. Реакционную смесь нагревали до rt и перемешивали в течение 12 ч. Реакционную смесь выпаривали до сухого состояния и очищали хроматографией на колонке, элюируя смесью петролейный эфир/EtOAc (от 100:1 до 10:1), с получением указанного в заголовке соединения (5,6 г, 85%) в виде желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 3,54 (д, 2H), 3,32-3,43 (м, 4H), 1,61-1,64 (м, 2H), 1,46 (с, 9H), 1,03-1,05 (м, 1H).
Стадия 2: трет-бутил(1R,5S,6s)-6-(цианометил)-3-азабицикло[3,1,0]гексан-3-карбоксилат
В раствор трет-бутил(1R,5S,6r)-6-(бромметил)-3-азабицикло[3,1,0]гексан-3-карбоксилата (6000 мг, 21,73 ммоль) в DMF (150 мл) добавляли цианид натрия (1600 мг, 32,6 ммоль) при rt и реакционную смесь перемешивали в течение 16 ч при rt. Желтую смесь разбавляли EtOAc (100 мл) и промывали рассолом (100 мл). Органический слой сушили над Na2SO4, фильтровали и выпаривали, с получением желтого масла, которое очищали хроматографией на колонке, элюируя смесью петролейный эфир/EtOAc (от 100:1 до 5:1), с получением указанного в заголовке соединения (4,0 г, 83%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 3,58 (дд, 2H), 3,30-3,35 (м, 2H), 2,45-2,51 (м, 1H), 2,31-2,36 (м, 1H), 1,49-1,52 (м, 2H), 1,41 (с, 9H), 0,88-0,91 (м, 1H).
Стадия 3
Ацетилхлорид (300 мг, 3,82 ммоль) добавляли в сухой этанол (2,5 мл) при 0°C и перемешивали при rt в течение 1 ч в запаянной колбе. Трет-бутил(1R,5S,6s)-6-(цианометил)-3-азабицикло[3,1,0]гексан-3-карбоксилат (85 мг, 0,38 ммоль) добавляли в раствор, и смесь перемешивали при 70°C в течение 68 ч. Раствор охлаждали до rt и концентрировали, с получением этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат гидрохлорида (80 мг, >99%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 4,15-4,18 (м, 2H), 3,44-3,47 (м, 4H), 2,36-2,38 (м, 2H), 1,74-1,78 (м, 2H), 1,25-1,30 (м, 3H), 1,14-1,17 (м, 1H).
метил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат гидрохлорид
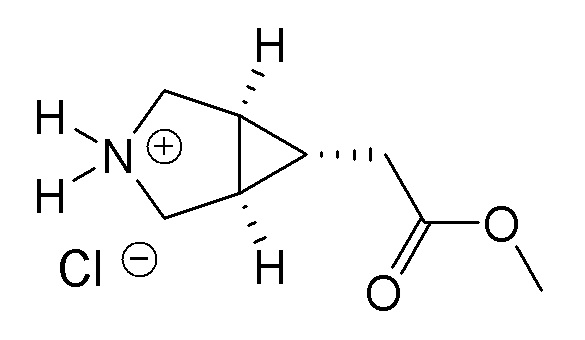
Стадия 1: (1R,5S,6r)-3-бензил-6-(хлорметил)-3-азабицикло[3,1,0]гексан
Получение [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанола описано в Berliner, M. A. et al., Org. Process Res. Dev. 2011, 15, 1052-1062.
В перемешиваемый раствор [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанола (620 г, 3,05 моль) в метаноле (600 мл) добавляли 4M HCl в метаноле (6,2 л) при 10°C на протяжении 45 мин, и смесь перемешивали в течение 15 мин. Реакционную смесь медленно нагревали до 25-30°C в течение 2 ч. Растворитель выпаривали при пониженном давлении, с получением сырого продукта. Сырой продукт растирали с эфиром (1,5 л), с получением [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанол гидрохлорида (703 г, выход 96%) в виде бледно-коричневого твердого вещества, которое использовали непосредственно в следующей стадии.
В перемешиваемый раствор [(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метанол гидрохлорида (699 г, 2,91 моль) в толуоле (1,4 л) добавляли тионилхлорид (693 г, 5,83 моль) при 5-10°C на протяжении 30 мин и перемешивали в течение 15 мин. Температура реакционной смеси медленно повышалась до 45°C, смесь перемешивали в течение 30 мин. Реакционную смесь охлаждали до rt и концентрировали при пониженном давлении. Сырой продукт растворяли в EtOAc (5 л) и насыщенном растворе NaHCO3 (3 л, pH=~8) и перемешивали в течение 1 ч, затем слои разделяли. Водный слой затем экстрагировали EtOAc (2×2 л). Объединенные органические слои промывали рассолом (2,0 л), сушили над безводным Na2SO4 и выпаривали при пониженном давлении, с получением указанного в заголовке соединения (611 г, 95%) в виде жидкости коричневого цвета.
1H ЯМР (600 МГц, ДМСО-d6) δ 7,30 (т, 2H), 7,20-7,25 (м, 3H), 3,51-3,56 (м, 4H), 2,87 (д, 2H), 2,29 (д, 2H), 1,54-1,57 (м, 1H), 1,43 (с, 2H).
Стадия 2: [(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетонитрил
В перемешиваемый раствор (1R,5S,6r)-3-бензил-6-(хлорметил)-3-азабицикло[3,1,0]гексана (664 г, 2,99 моль) в DMF (2,9 л) добавляли цианид натрия (191 г, 3,89 моль) при rt и смесь медленно нагревали до 50°C в течение 48 ч. Реакционную смесь охлаждали до rt, гасили водой (10 л) и экстрагировали EtOAc (3×4 л). Объединенные органические слои промывали водой (5 л), рассолом (3 л), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Сырой продукт очищали хроматографией на колонке, элюируя 20% EtOAc в петролейном эфире, с получением указанного в заголовке соединения (593 г, 93,2%) в виде жидкости коричневого цвета.
1H ЯМР (600 МГц, ДМСО-d6) δ 7,27-7,32 (м, 2H), 7,19-7,26 (м, 3H), 3,54 (с, 2H), 2,87 (д, 2H), 2,45 (д, 2H), 2,28 (д, 2H), 1,33-1,41 (м, 3H).
Стадия 3: метил[(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетат
Ацетилхлорид (2,21 кг, 28,3 моль) добавляли в метанол (3,77 л) при 0°C на протяжении 1 ч. Температура реакционной смеси медленно повышалась до 45°C в течение 30 мин. Реакционную смесь вновь охлаждали до 0°C и добавляли раствор [(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетонитрила (400 г, 1,88 моль) в метаноле (700 мл) на протяжении 2 ч при 0°C. Полученный раствор медленно нагревали до 65°C в течение 4 ч. Реакционную смесь охлаждали до rt и концентрировали при пониженном давлении. Сырой продукт растворяли в EtOAc (6 л) и насыщенном растворе NaHCO3 (4 л, pH ~8) и перемешивали в течение 1 ч. Слои разделяли, и водный слой затем экстрагировали EtOAc (2×1 л). Объединенные органические слои промывали рассолом (2,0 л), сушили над безводным Na2SO4 и выпаривали при пониженном давлении, с получением указанного в заголовке соединения (377 г, 82%) в виде жидкости коричневого цвета.
1H ЯМР (600 МГц, CDCl3) δ 7,19-7,31 (м, 5H), 3,67 (с, 3H), 3,56 (с, 2H), 2,99 (д, 2H), 2,34 (д, 2H), 2,18 (д, 2H), 1,50-1,54 (м, 1H), 1,23 (с, 2H).
Стадия 4
В раствор метил[(1R,5S,6s)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]ацетата (542 г, 2,21 моль) в метаноле (550 мл) добавляли 4M HCl в метаноле (5,4 л) при 10°C на протяжении 30 мин. Реакционную смесь нагревали до rt и перемешивали в течение 2 ч. Растворитель выпаривали при пониженном давлении. Сырой продукт растирали с эфиром (1,5 л), с получением не совсем белого твердого вещества (545 г, выход 87,7%), которое использовали непосредственно в следующей стадии. Сырой продукт (420 г, 149 моль) растворяли в метаноле (4 л) в автоклаве и обрабатывали 10% Pd(OH)2/C (41,4 г, 50% влаги) в атмосфере азота, автоклав дважды продували азотом, помещали в атмосферу газообразного водорода (100 psi; 6,805 атм) и нагревали до 70°C в течение 8 ч. Реакционную смесь охлаждали до rt и перемешивали в течение 4 ч. Реакционную смесь фильтровали через слой целита® и промывали метанолом (2×1 л). Фильтрат выпаривали при пониженном давлении. Сырой продукт растирали с эфиром (1 л) и твердое вещество собирали фильтрованием, с получением метил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат гидрохлорида (345 г, выход 99%) в виде не совсем белого твердого вещества.
1H ЯМР (600 МГц, ДМСО-d6) δ: 9,25-9,80 (уш.с, 2H), 4,05-4,44 (уш.с, 1H), 3,2-3,4 (уш.с, 1H), 3,21 (с, 3H), 3,15 (с, 2H), 2,30 (д, 2H), 1,60 (с, 2H), 1,20-1,27 (м, 1H).
2,6-дихлор-4-(1,1-дифторэтил)-5-фторпиридин-3-карбонитрил
Стадия 1: 4-(1,1-дифторэтил)-5-фтор-2,6-дигидроксипиридин-3-карбонитрил
В раствор этил-2,2-дифторпропаноата (10,0 г, 72,4 ммоль) в THF (10,0 мл) добавляли гидрид натрия (60% в минеральном масле, 3,19 г, 79,6 ммоль) и смесь нагревали до 50°C. Этилфторацетат (15,4 г, 145 ммоль) добавляли по каплям в течение 1 мин и реакционную смесь перемешивали при 50°C в течение 2 ч. Раствор вливали в водный раствор хлорида аммония (100 мл) при 0°C. Смесь экстрагировали EtOAc (3×150 мл), промывали рассолом (100 мл), сушили над Na2SO4, фильтровали и концентрировали, с получением желтого масла (13 г). Сырой продукт растворяли в этаноле (200 мл) и обрабатывали 2-цианоацетамидом (5,52 г, 65,6 ммоль) и пиперидином (5,59 г, 65,6 ммоль). Полученный бесцветный раствор перемешивали при 50°C в течение 16 ч. Продукт осаждали из раствора и собирали фильтрованием, с получением указанного в заголовке соединения (10 г, 70%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 8,20 (уш.с, 2H), 1,89 (т, 3H).
Стадия 2
Смесь 4-(1,1-дифторэтил)-5-фтор-2,6-дигидроксипиридин-3-карбонитрила (10,0 г, 45,8 ммоль) и пентахлорида фосфора (95,5 г, 458 ммоль) перемешивали при 130°C в течение 32 ч. Реакционную смесь охлаждали до rt и вливали в водный раствор NaHCO3 (750 мл) при 0°C. Продукт экстрагировали EtOAc (3×150 мл), промывали рассолом (150 мл), сушили над Na2SO4, фильтровали и концентрировали, с получением желтого масла. Сырой продукт очищали колоночной хроматографией (смесь EtOAc/петролейный эфир от 0:100 до 3:97), с получением 2,6-дихлор-4-(1,1-дифторэтил)-5-фторпиридин-3-карбонитрила (6,0 г, 51%) в виде желтого масла.
1H ЯМР (400 МГц, ДМСО-d6) δ: 2,10 (т, 3H).
2,4-дихлор-6-(1,1-дифторэтил)пиридин
Стадия 1: 6-(1,1-дифторэтил)пиридин-2,4-диол
Суспензию этил-6-(1,1-дифторэтил)-2,4-дигидроксипиридин-3-карбоксилата (10,5 г, 42,5 ммоль) в 6Н HCl (100 мл) перемешивали при 100°C в течение 16 ч. Реакционную смесь охлаждали до rt и выпаривали при пониженном давлении, с получением указанного в заголовке соединения (8,0 г, 90%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 7,9-8,6 (м, 2H), 6,62 (с, 1H), 6,27 (с, 1H), 1,95 (т, 3H).
Стадия 2: 2,4-дихлор-6-(1,1-дифторэтил)пиридин
Смесь 6-(1,1-дифторэтил)пиридин-2,4-диола (7,0 г, 33 ммоль) и пентахлорида фосфора (34,4 г, 165 ммоль) перемешивали при 125°C в течение 20 ч. Смесь гасили ледяной водой (200 мл) и экстрагировали EtOAc (2×100 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя петролейным эфиром, с получением 2,4-дихлор-6-(1,1-дифторэтил)пиридина (2,5 г, выход 36%) в виде светло-желтого масла.
МС(ЭС+): 211,6 (M+H).
Примеры
Пример 1: [(1R,5S,6R)-3-{5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
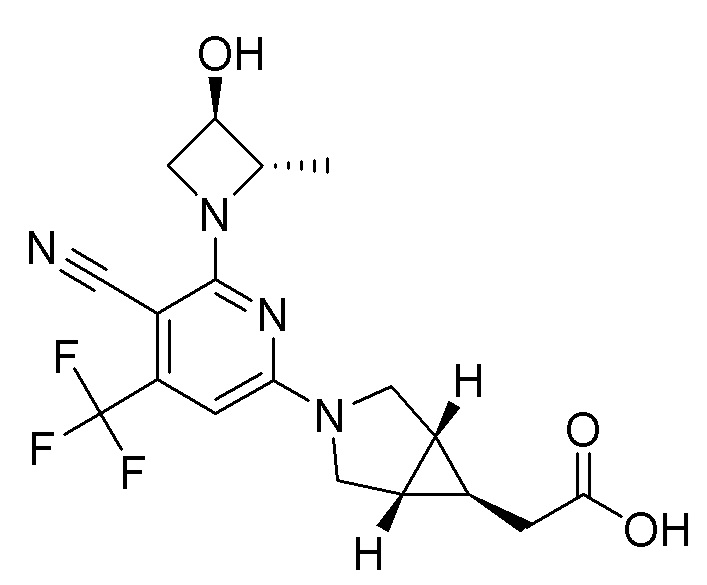
Стадия 1: этил{(1R,5S,6s)-3-[6-хлор-5-циано-4-(трифторметил)пиридин-2-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат
Суспензию 2,6-дихлор-4-(трифторметил)пиридин-3-карбонитрила (2,4 г, 9,8 ммоль), этил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетата (1,7 г, 9,8 ммоль) и NaHCO3 (2,6 г, 31 ммоль) в этаноле (25 мл) перемешивали при rt в течение ночи. Реакционную смесь концентрировали, разбавляли насыщенным водным раствором NaHCO3 и экстрагировали EtOAc (3×25 мл). Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле (10-35% EtOAc в н-гептане), с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (2,2 г, 57%).
МС(ЭС+): 374,2 (M+H), 1H ЯМР (600 МГц, ДМСО-d6) δ: 6,90 (с, 1H), 4,07 (к, 2H), 3,79 (м, 2H), 3,67-3,53 (м, 2H), 2,43-2,21 (м, 2H), 1,75-1,57 (м, 2H), 1,19 (т, 3H), 0,81 (дт, 1H).
Стадия 2: этил[(1R,5S,6R)-3-{5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
Этил{(1R,5S,6s)-3-[6-хлор-5-циано-4-(трифторметил)пиридин-2-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат (2,1 г, 5,7 ммоль), (2S,3R)-3-гидрокси-2-метилазетидин-1-ий[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат (2,0 г, 6,2 ммоль), NaHCO3 (1,7 г, 20 ммоль) суспендировали в этаноле и перемешивали при 80°C в течение 18 ч. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (200 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Полученное белое твердое вещество переносили в следующую стадию без очистки.
МС(ЭС+): 447,0 (M+Na).
Стадия 3
Гидроксид натрия (40 мл, 1M водн.) добавляли в суспензию этил[(1R,5S,6R)-3-{5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (2,5 г, 5,9 ммоль) в этаноле (80 мл), и реакционную смесь перемешивали при rt в течение 1 ч. Реакционную смесь концентрировали, разбавляли водой (25 мл), подкисляли 1Н HCl до pH=2 и экстрагировали EtOAc (3×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, с получением белого твердого вещества. Белое твердое вещество объединяли с продуктом другого процесса получения в тех же условиях, с получением 1,1 г для очистки. Белое твердое вещество суспендировали при кипячении с обратным холодильником в течение 3 ч в MTBE/н-Hep и затем при rt в течение 5 дней. Затем суспензию фильтровали и отфильтрованный осадок промывали н-гептаном, с получением соединения Примера 1 в виде белого твердого вещества (2,4 г, 73%). (ТП=193,2-195,8°C). Твердое вещество затем растворяли в EtOAc при кипячении с обратным холодильником и фильтровали при высокой температуре. Фильтрат концентрировали и перекристаллизовывали из смеси этил ацетат/н-гептан. Твердое вещество собирали вакуумным фильтрованием и сушили в вакуумной печи при 50°C в течение 2 ч, с получением соединения Примера 1 в виде белого твердого вещества (1,4 г, 44%).
ТП=189,9-196,8°C. МС(ЭС+): 397,1 (M+H). 1H ЯМР (600 МГц, ДМСО-d6) δ: 12,10 (уш.с, 1H), 6,22 (с, 1H), 5,63 (уш.с, 1H), 4,54 (т, 1H), 4,20 (квин, 1H), 4,06 (уш.с, 1H), 3,94-3,60 (м, 3H), 3,51 (уш.с, 2H), 2,24 (д, 2H), 1,60 (уш.д, 2H), 1,40 (д, 3H), 0,74 (уш.с, 1H).
Пример 2: [(1R,5S,6R)-3-{3-хлор-5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
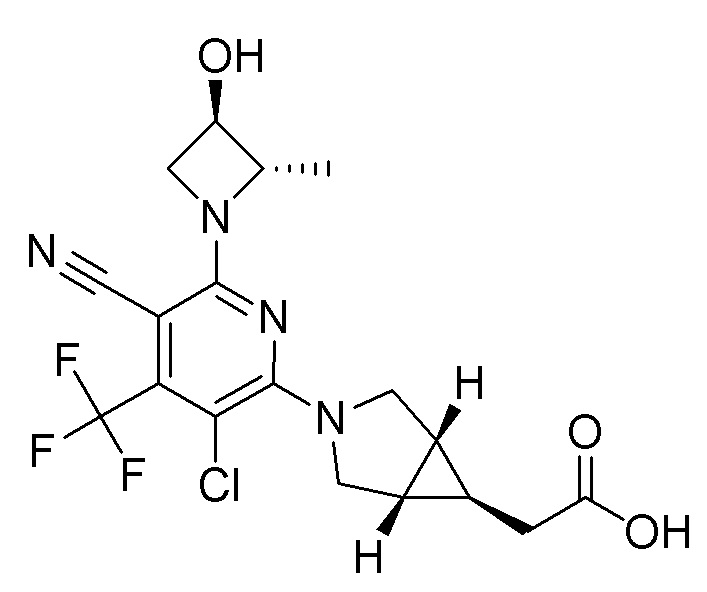
Стадия 1
Этил[(1R,5S,6R)-3-{3-хлор-5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
Этил[(1R,5S,6R)-3-{5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат (60 мг, 0,14 ммоль) в DMF (2,5 мл) обрабатывали N-хлорсукцинимидом (28,3 мг, 0,212 ммоль) при rt, и смесь перемешивали в течение 16 ч при 25°C. Смесь разбавляли водой (15 мл) и насыщенным водным раствором хлорида аммония (5 мл), затем экстрагировали EtOAc (15 мл x 3). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме, с получением указанного в заголовке соединения (80 мг, колич.) в виде не совсем желтого твердого вещества, которое непосредственно использовали в следующей стадии.
Стадия 2
Соединение Примера 2 получали по методике, аналогичной методике Примера 1, стадии 3, с использованием этил[(1R,5S,6R)-3-{3-хлор-5-циано-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-4-(трифторметил)пиридин-2-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата и очищали препаративной обращенно-фазовой ВЭЖХ, с получением соединения Примера 2 (30 мг, 49%) в виде белого твердого вещества.
МС(ЭС+): 431,1 (M+H). 1H ЯМР (400 МГц, CD3OD) δ 4,70 (дд, 1H), 4,39-4,25 (м, 2H), 4,24-4,08 (м, 2H), 3,86-3,67 (м, 3H), 2,30 (д, 2H), 1,59 (уш.с, 2H), 1,48 (д, 3H), 0,90-0,74 (м, 1H).
Пример 3: Метил[(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
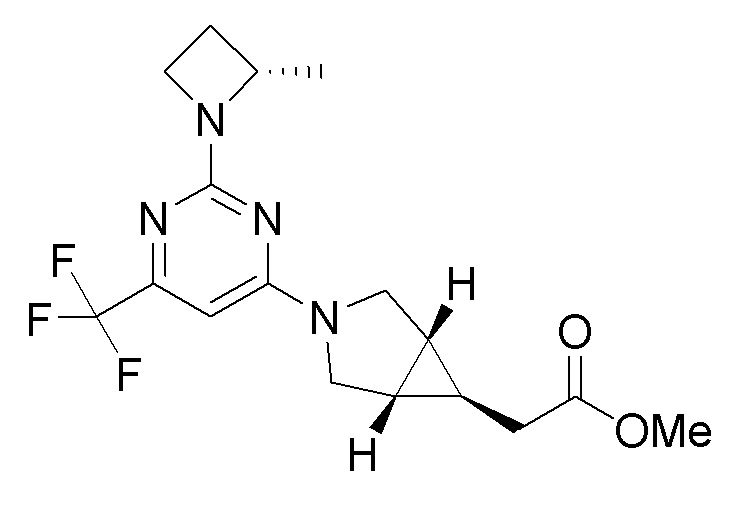
Стадия 1:
Метил{(1R,5S,6s)-3-[2-хлор-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат
В раствор метил(1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетат гидрохлорида (120,2 г, 627,2 ммоль) в DCM (1250 мл) по каплям добавляли 2,4-дихлор-6-(трифторметил)пиримидин (145,7 г, 671,5 ммоль) в DCM (50 мл) при -72°C; дополнительную воронку промывали DCM (50 мл) и смыв добавляли в реакционную колбу. DIPEA (273 мл, 1570 ммоль) добавляли в течение 10 мин, поддерживая температуру реакционной среды от -70°C до -60°C. Смесь перемешивали при температуре от -65°C до -63°C в течение 1 ч и затем нагревали до 25°C в течение 3 ч. Полученный прозрачный раствор концентрировали до ~1/5 исходного объема. К полученной густой суспензии добавляли MTBE (700 мл) и гептан (700 мл), и полученную суспензию перемешивали при 25°C в течение 10 мин, затем твердое вещество отфильтровывали и промывали смесью MTBE-гептан (4:1). Объединенный маточный раствор концентрировали в вакууме до консистенции масла, которое объединяли с гептаном (1200 мл). Полученную гетерогенную смесь перемешивали при 25°C в течение 2,5 дней. Образовывалось белое твердое вещество. Жидкость декантировали, твердое вещество промывали гептаном (200 мл) и сушили в потоке азота. Полученный указанный в заголовке продукт использовали в следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 6,47 (с, 1H), 4,07 (д, 1H), 3,71 (с, 3H), 3,53-3,68 (м, 3H), 2,36-2,49 (м, 1H), 2,21-2,34 (м, 1H), 1,60-1,73 (м, 2H), 0,88-0,97 (м, 1H).
Стадия 2
Метил{(1R,5S,6s)-3-[2-хлор-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат из Стадии 1 растворяли в ацетонитриле (1500 мл) и добавляли (2S)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат (223,0 г, 735 ммоль). Смесь перемешивали при 60°C и добавляли DIPEA (77,0 мл, 442 ммоль) в течение 3 ч. Смесь перемешивали в течение 3 ч, затем добавляли DIPEA (180 мл, 1,03 моль) в течение 3 ч и смесь перемешивали при 60°C в течение 18 ч. Вновь добавляли (2S)-2-метилазетидиний[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат (18,0 г, 59 ммоль) и смесь дополнительно перемешивали при 60°C в течение 18 ч. Смесь концентрировали до ~1/4 исходного объема и полученное желтое масло распределяли между 500 мл воды, 400 мл гептана и 400 мл MTBE. Водную фазу отделяли и вновь экстрагировали смесью MTBE-гептан (1:1) (2×150 мл). Объединенный органический экстракт промывали 120 мл насыщенного раствора NaHCO3 (120 мл), затем перемешивали с SiO2 (70 г) и безводным MgSO4 (70 г). Твердое вещество отфильтровывали, и прозрачный раствор концентрировали, с получением 216,6 г соединения Примера 3 в виде бесцветного масла.
МС(ЭС+): 371,1 (M+H). 1H ЯМР (400 МГц, CDCl3) δ: 5,91 (с, 1H), 4,37-4,48 (м, 1H), 3,87-4,05 (м, 3H), 3,70 (с, 3H), 3,50-3,64 (м, 1H), 3,41-3,50 (м, 2H), 2,33-2,42 (м, 1H), 2,31 (д, 2H), 1,88-1,99 (м, 1H), 1,52-1,59 (м, 2H), 1,49 (д, 3H), 0,88-0,96 (м, 1H).
Пример 4: [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
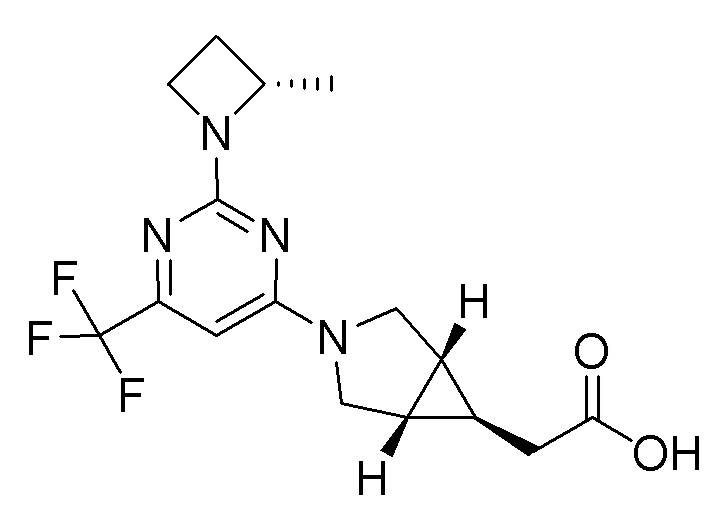
В перемешиваемый раствор неочищенного метил[(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата в метаноле (650 мл) добавляли раствор гидроксида натрия (35,1 г, 877 ммоль) в воде (70 мл) небольшими порциями с перемешиванием при температуре от 5°C до 15°C. Смесь становилась прозрачной через 30 мин. Прозрачный раствор перемешивали при rt в течение 3 ч, затем концентрировали до ~1/3 исходного объема и осадок разбавляли водой (750 мл) и рассолом (250 мл), затем промывали смесью MTBE (260 мл) и гептана (130 мл). Органический смыв отбрасывали. Водную фазу промывали смесью MTBE-гептан (2:1) (2×300 мл) и органические слои отбрасывали. Водный слой затем объединяли с MTBE (250 мл) и гептаном (250 мл) и охлаждали до 0°C. С перемешиванием при температуре от 0°C до 4°C медленно добавляли 6 M водный раствор HCl (130 мл), а затем 1 M водный раствор KHSO4 (150 мл), и полученную смесь перемешивали в течение 15 мин. Органическую фазу отделяли и водную фазу дополнительно экстрагировали смесью MTBE (170 мл) и гептана (170 мл). Объединенный органический экстракт промывали смесью вода-рассол (1:1) (150 мл), сушили над безводным MgSO4 (60 г) и SiO2 (60 г), фильтровали и концентрировали, с получением бесцветного масла. Его объединяли (в виде концентрированного раствора в MTBE) с другой партией соединения, которую получали в идентичных условиях в том же масштабе. Объединенный раствор в MTBE концентрировали в вакууме, затем добавляли гептан (2000 мл) и суспензию вновь концентрировали, постепенно увеличивая вакуум, с получением желаемого продукта (406,0 г). Часть данного материала (196 г) растворяли в MTBE (220 мл) при температуре от 60°C до 63°C, медленно перемешивали и добавляли гептан (1500 мл) при температуре от 55°C до 60°C. В смесь вводили затравку кристаллического указанного в заголовке соединения (50 мг). Смесь перемешивали при 60°C в течение 30 мин, затем вновь добавляли гептан (1700 мл) в течение 20 мин. Гетерогенную смесь перемешивали при 60°C в течение 2 ч, затем медленно охлаждали до 25°C и перемешивали в течение 20 ч. Небольшое количество твердого вещества прилипало к стенкам колбы и было с легкостью перенесено шпателем в жидкую фазу, после чего смесь дополнительно перемешивали при 25°C в течение 24 ч. Твердое вещество отфильтровывали, промывали 5% MTBE в гептане и сушили в вакууме при 50°C в течение 48 ч, с получением соединения Примера 4 в виде белого кристаллического вещества (178,2 г, 73% за 3 стадии). Кристаллическое твердое вещество Примера 4 также было получено с использованием аналогичных условий очистки без затравки.
ТП: 122-123°C, [α]D +86,3° (CDCl3, c=1,37). МС(ЭС+): 357,3 (M+H). 1H ЯМР (400 МГц, CDCl3) δ: 10,84 (уш.с, 1H), 5,92 (с, 1H), 4,38-4,51 (м, 1H), 3,89-4,10 (м, 3H), 3,53-3,66 (м, 1H), 3,41-3,53 (м, 2H), 2,30-2,46 (м, 3H), 1,94 (ддт, 1H), 1,55-1,63 (м, 2H), 1,50 (д, 3H), 0,94 (м, 1H).
Анализ методом порошковой рентгеновской дифракции проводили с использованием дифрактометра Bruker AXS D4 Endeavor, оснащенного Cu источником излучения. Щель расходимости устанавливали на 0,6 мм, при этом для вторичной оптики использовали переменные щели. Дифрагированное излучение обнаруживали детектором PSD-Lynx Eye. Напряжение и силу тока рентгеновской трубки устанавливали на 40 кВ и 40 мА, соответственно. Данные получали в тэта-2тэта гониометре при длине волны Cu Kα1 =1,54056 Å от 3,0 до 40,0 градусов 2-тэта, используя размер шага 0,020 градусов и время шага 0,3 секунды. Образцы готовили, помещая их в силиконовый держатель для образцов с низким уровнем фона, и вращали во время сбора. Данные получали с использованием программы Bruker DIFFRAC Plus и анализировали с помощью программы EVA diffract plus.
Файл с данными ПРД не обрабатывали до поиска пиков. Используя алгоритм поиска пиков в программе EVA, выбирали пики с пороговым значением 1, и значение ширины 0,3 использовали для предварительных отнесений пиков. Результат автоматизированных отнесений визуально проверяли для гарантии обоснованности и при необходимости проводили корректировку вручную. Как правило, выбирали пики с относительной интенсивностью ≥3%. Пики, которые не были разрешены или соответствовали шуму, также были отброшены. Типичная ошибка, связанная с положением пика при ПРД, указанная в Фармакопее США и Фармакопее Японии, составляет до +/- 0,2°.
Характерные пики для кристаллической свободной кислоты Примера 4 включают значения угла 2θ (°) примерно 9,0, 10,4, 15,0 и 21,4 +/- 0,2°. В другом варианте осуществления кристаллической свободной кислоты Примера 4 характерные пики включают значения угла 2θ (°) примерно 9,0, 15,0 19,6, 21,4 и 26,5 +/- 0,2°. В другом варианте осуществления кристаллической свободной кислоты Примера 4 характерные пики включают значения угла 2θ (°) примерно 9,0, 10,4, 11,5, 15,0, 16,5, 19,6, 21,4 и 26,5 +/- 0,2°. В другом варианте осуществления кристаллической свободной кислоты Примера 4 характерные пики включают значения угла 2θ (°) примерно 10,4, 11,5, 15,0, 19,6 и 26,5 +/- 0,2°. В Таблице 1 приведен список пиков ПРД для кристаллической свободной кислоты Примера 4, к указанным пикам применимо значение +/- 0,2°. На Фигуре 1 приведена картина ПРД кристаллической свободной кислоты Примера 4.
Таблица 1: Список пиков ПРД для кристаллической свободной кислоты Примера 4
|
Пример 5: [(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
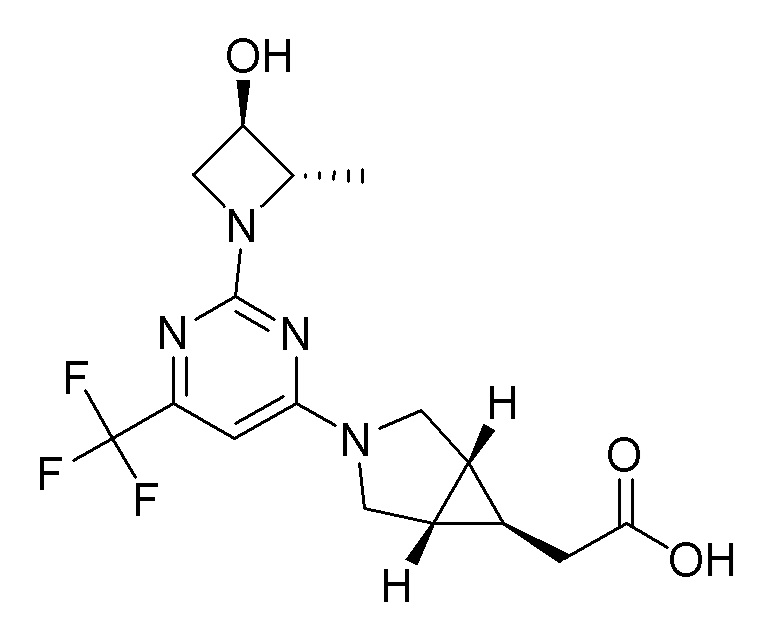
Стадия 1: метил[(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
Раствор метил{(1R,5S,6s)-3-[2-хлор-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетата (1,55 г, 4,60 ммоль), (2S,3R)-3-гидрокси-2-метилазетидин-1-ий[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфоната (1,62 г, 5,10 ммоль), триэтиламина (1,6 мл, 12,0 ммоль) и ацетонитрила (15,4 мл) нагревали при 60°C в течение 16 ч. Реакционную смесь охлаждали до rt и концентрировали. Добавляли воду (15 мл) и реакционную смесь экстрагировали EtOAc (10 мл x 3). Объединенные органические слои концентрировали и очищали флэш-хроматографией (смесь EtOAc/гептан, от 0% до 100%) на колонке с силикагелем, с получением указанного в заголовке соединения (1,3 г, 73%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 5,98 (с, 1H), 4,31 (ддд, 1H), 4,23 (т, 1H), 4,21-4,11 (м, 1H), 4,09-3,89 (м, 1H), 3,76 (дд, 1H), 3,72 (с, 3H), 3,67-3,54 (м, 1H), 3,53-3,41 (м, 2H), 2,34 (д, 2H), 1,61-1,58 (м, 2H), 1,54 (д, 3H), 0,98-0,88 (м, 1H).
Стадия 2
В раствор метил[(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (1,30 г, 3,36 ммоль) в метаноле (5 мл) добавляли 2M водный раствор NaOH (4,2 мл, 8,4 ммоль). Через 3 ч при rt реакционную смесь гасили 1M водным раствором гидросульфата калия (10 мл), экстрагировали трет-бутил-метиловым эфиром (10 мл x 3) и концентрировали, с получением соединения Примера 5 (1,2 г, 96%). Кристаллическую форму натриевой соли получали, смешивая соединение Примера 5 (500 мг, 1,34 ммоль) с 1M NaOH (1,34 мл, 1,34 ммоль). Раствор перемешивали при rt в течение 5 минут, затем сушили при пониженном давлении, с получениемa белого твердого вещества. Добавляли EtOAc (3 мл), гептан (0,5 мл) и воду (0,1 мл) и суспензию перемешивали при rt в течение 16 ч. Полученное белое твердое вещество выделяли и сушили, с получением соединения Примера 5 в виде кристаллической натриевой соли.
МС(АД+): 373,4 (M+H). 1H ЯМР (400 МГц, CDCl3) δ: 5,96 (с, 1H), 4,34-4,25 (м, 1H), 4,25-4,17 (м, 1H), 4,17-4,10 (м, 1H), 4,06-3,88 (м, 1H), 3,74 (дд, 1H), 3,65-3,53 (с, 1H), 3,53-3,43 (м, 2H), 2,45-2,27 (м, 2H), 1,62-1,55 (м, 2H), 1,52 (д, 3H), 0,97-0,87 (м, 1H).
Анализ методом порошковой рентгеновской дифракции проводили с использованием дифрактометра Bruker AXS D4 Endeavor, оснащенного Cu источником излучения. Щель расходимости устанавливали на 0,6 мм, при этом для вторичной оптики использовали переменные щели. Дифрагированное излучение обнаруживали детектором PSD-Lynx Eye. Напряжение и силу тока рентгеновской трубки устанавливали на 40 кВ и 40 мА, соответственно. Данные получали в тэта-2тэта гониометре при длине волны Cu Kα1=1,54056 Å от 3,0 до 40,0 градусов 2-тэта, используя размер шага 0,020 градусов и время шага 0,3 секунды. Образцы готовили, помещая их в силиконовый держатель для образцов с низким уровнем фона, и вращали во время сбора. Данные получали с использованием программы Bruker DIFFRAC Plus и анализировали с помощью программы EVA diffract plus.
Файл с данными ПРД не обрабатывали до поиска пиков. Используя алгоритм поиска пиков в программе EVA, выбирали пики с пороговым значением 1, и значение ширины 0,3 использовали для предварительных отнесений пиков. Результат автоматизированных отнесений визуально проверяли для гарантии обоснованности и при необходимости проводили корректировку вручную. Как правило, выбирали пики с относительной интенсивностью ≥3%. Пики, которые не были разрешены или соответствовали шуму, также были отброшены. Типичная ошибка, связанная с положением пика при ПРД, указанная в Фармакопее США и Фармакопее Японии, составляет до +/- 0,2°.
Характерные пики для кристаллической натриевой соли Примера 5 включают значения угла 2θ (°) примерно 5,9, 11,5, 11,8, 13,3, 21,5 +/- 0,2°. В другом варианте осуществления кристаллической натриевой соли Примера 5 характерные пики включают значения угла 2θ (°) примерно 5,9, 10,3, 11,5, 11,8, 13,3, 16,5, 21,5 и 22,6 +/- 0,2°. В другом варианте осуществления кристаллической натриевой соли Примера 5 характерные пики включают значения угла 2θ (°) примерно 5,9, 10,3, 11,8, 16,5 и 21,5 +/- 0,2°. В Таблице 2 приведен список пиков ПРД для кристаллической натриевой соли Примера 5, к указанным пикам применимо значение +/- 0,2°. На Фигуре 2 приведена картина ПРД кристаллической натриевой соли Примера 5.
Таблица 2: Список пиков ПРД для кристаллической натриевой соли Примера 5
|
Пример 6: {(1R,5S,6s)-3-[2-циклобутил-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}уксусная кислота
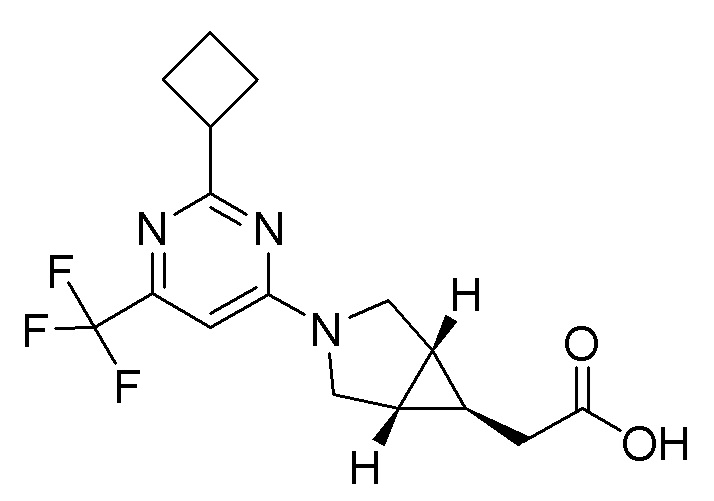
Стадия 1: Этил{(1R,5S,6s)-3-[2-циклобутил-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат
В раствор этил{(1R,5S,6s)-3-[2-хлор-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетата (50 мг, 0,14 ммоль; получен по методике, аналогичной методике получения соединения стадии 1 Примера 3) в сухом DMF (3 мл) добавляли (tBu3P)2Pd (7,3 мг, 0,014 ммоль). Смесь продували азотом и добавляли 0,5 M раствор бромида циклобутилцинка в THF (0,86 мл, 0,43 ммоль). Полученную серую суспензию продували азотом и затем перемешивали в закрытом сосуде при 100°C в течение 1 ч. Смесь вливали в насыщенный водный раствор NH4Cl (15 мл) и экстрагировали EtOAc (3×15 мл). Объединенный органический экстракт промывали рассолом, сушили над Na2SO4 и концентрировали. Осадок очищали препаративной ТСХ, элюируя смесью EtOAc-петролейный эфир (1:5), с получением указанного в заголовке соединения в виде бесцветной смолы (45 мг, выход 85%).
МС(ЭС+): 369,9 (M+H).
Стадия 2
Соединение Примера 6 синтезировали по методике, аналогичной методике Примера 1, стадии 3, с использованием этил{(1R,5S,6s)-3-[2-циклобутил-6-(трифторметил)пиримидин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетата и очищали препаративной обращенно-фазовой ВЭЖХ, с получением 15 мг соединения (выход 36%) в виде белого твердого вещества.
МС(ЭС+): 342,1 (M+H). 1H ЯМР (400 МГц, CD3OD) δ: 6,57 (с, 1H), 4,04-4,18 (м, 1H), 3,46-3,76 (м, 4H), 2,20-2,50 (м, 6H), 1,98-2,14 (м, 1H), 1,85-1,96 (м, 1H), 1,57-1,76 (м, 2H), 0,79-0,94 (м, 1H).
Пример 7: [(1R,5S,6R)-3-{5-циклопропил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
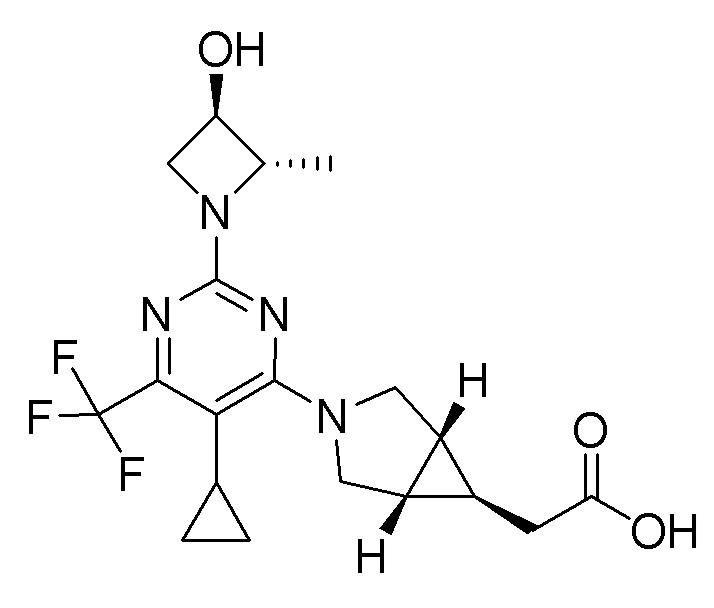
Стадия 1: Этил[(1R,5S,6R)-3-{5-циклопропил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
В смесь этил[(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (100 мг, 0,250 ммоль; синтезирован по методике, аналогичной методике получения соединения стадии 1 Примера 5), добавляли циклопропилтрифторборат калия (185 мг, 1,25 ммоль), AgNO3 (8,5 мг, 0,050 ммоль), K2S2O8 (338 мг, 1,25 ммоль), DCE (5,0 мл) и воду (5,0 мл). Затем добавляли TFA (57 мг, 0,50 ммоль). Реакционный сосуд закрывали и реакционную смесь перемешивали при 50°C в течение 16 ч. Реакционную смесь разбавляли водным раствором хлорида аммония (10 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали, с получением сырого продукта в виде желтого масла, которое очищали препаративной ТСХ с 10% MeOH в DCM, с получением указанного в заголовке соединения (30 мг, 27%) в виде бесцветного масла.
МС(ЭС+): 441,1 (M+H).
Стадия 2
Соединение Примера 7 синтезировали по методике, аналогичной методике Примера 1, стадии 3, с использованием этил[(1R,5S,6R)-3-{5-циклопропил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата и очищали препаративной обращенно-фазовой ВЭЖХ, с получением 10 мг соединения (выход 36%) в виде белого твердого вещества.
МС(ЭС+): 413,1 (M+H). 1H ЯМР (400 МГц, CD3OD) δ: 4,30-4,19 (м, 3H), 4,15-4,07 (м, 2H), 3,70-3,56 (м, 3H), 2,31 (д, 2H), 1,90-1,81 (м, 1H), 1,58-1,53 (м, 2H), 1,47 (д, 3H), 1,02-0,95 (м, 2H), 0,93-0,84 (м, 1H), 0,49-0,41 (м, 2H).
Пример 8: [(1R,5S,6R)-3-{5-этил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
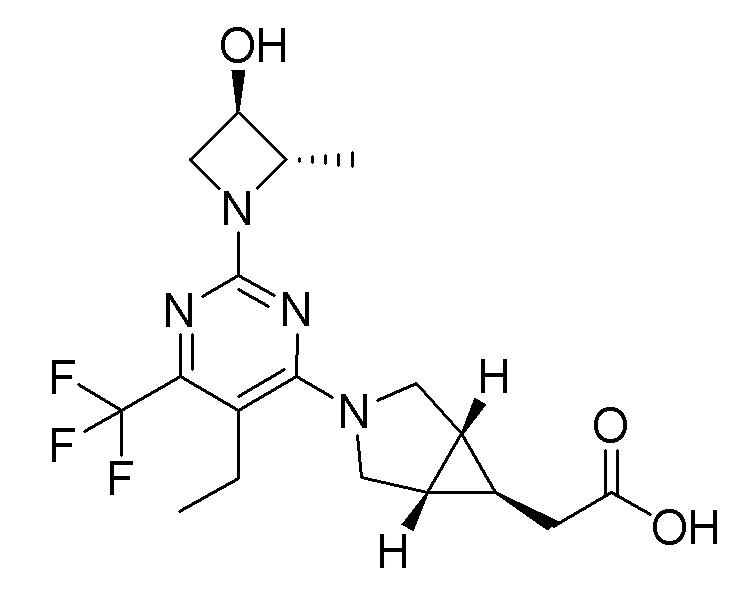
Стадия 1: этил[(1R,5S,6R)-3-{5-бром-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
В находящийся при температуре 0°C раствор этил[(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (100 мг, 0,259 ммоль; получен по методике, аналогичной методике получения метил[(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата) в сухом ацетонитриле (10 мл) добавляли N-бромсукцинимид (60 мг, 0,29 ммоль, 85% чистоты) и реакционную смесь перемешивали в течение 1 ч при 0°C. Смесь разбавляли водным раствором бикарбоната натрия, экстрагировали EtOAc (30 мл x 3) и объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали, с получением сырого материала, который очищали хроматографией на силикагеле (EtOAc в петролейном эфире, от 0% до 40%), с получением указанного в заголовке соединения (110 мг, выход 91%) в виде светло-желтого твердого вещества.
Стадия 2: этил[(1R,5S,6R)-3-{5-этенил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
В смесь этил[(1R,5S,6R)-3-{5-бром-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (50 мг, 0,11 ммоль), трибутилвинилолова (51 мг, 0,16 ммоль) и Pd(PPh3)2Cl2 (11 мг, 0,015 ммоль) в сухом диоксане (5,0 мл) добавляли тетрабутиламмоний бромид (35 мг, 0,11 ммоль). Красную реакционную смесь перемешивали при 50°C в течение 16 ч. Черную реакционную смесь разбавляли водным раствором NH4Cl и экстрагировали EtOAc (20 мл) три раза. Объединенный органический слой промывали рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали, с получением сырого продукта в виде красного масла. Осадок очищали препаративной ТСХ (смесь петролейный эфир:EtOAc=1:1), с получением сырого продукта, и повторно очищали в тех же условиях препаративной ТСХ (смесь петролейный эфир:EtOAc=1:1), с получением указанного в заголовке соединения в виде белого твердого вещества (15 мг). МС(ЭС+): 427,1 (M+H).
Стадия 3: этил[(1R,5S,6R)-3-{5-этенил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетат
В смесь этил[(1R,5S,6R)-3-{5-этенил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (50 мг, 0,020 ммоль, 17% чистоты) в сухом этаноле (10,0 мл) добавляли Pd/C (2,1 мг, 0,0020 ммоль). Черную суспензию перемешивали при 25°C в течение 16,0 часов в атмосфере водорода (30 Psi; 2,041 атм.). Катализатор отфильтровывали и фильтрат концентрировали, с получением 35 мг желаемого продукта в виде белого твердого вещества.
Стадия 4
Соединение Примера 8 синтезировали по методике, аналогичной методике Примера 1, стадии 3, с использованием этил[(1R,5S,6R)-3-{5-этенил-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата и очищали препаративной обращенно-фазовой ВЭЖХ, с получением 6 мг соединения в виде белого твердого вещества.
МС(ЭС+): 401,0 (M+H). 1H ЯМР (400 МГц, CD3OD) δ 4,22 (дд, 1H), 4,16-4,08 (м, 2H), 4,03 (т, 2H), 3,75-3,64 (м, 2H), 3,45-3,48 (м, 1H), 2,73 (к, 2H), 2,36-2,29 (д, 2H), 1,63-1,59 (уш.м 2H), 1,49 (д, 3H), 1,03 (т, 3H), 0,91-0,83 (м, 1H).
Пример 9: (2S,3R)-2,3-диметил-1-[4-{(1R,5S,6S)-6-[(метилсульфонил)метил]-3-азабицикло[3,1,0]гекс-3-ил}-6-(трифторметил)пиримидин-2-ил]азетидин-3-ол
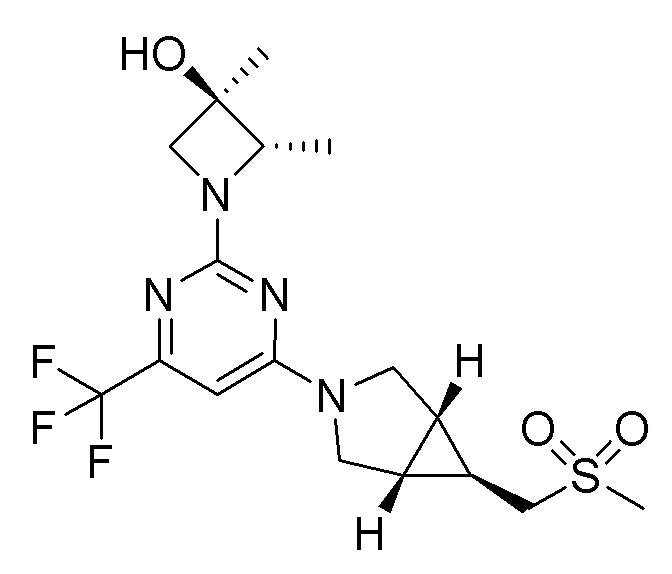
Стадия 1: (1R,5S,6r)-3-бензил-6-(иодметил)-3-азабицикло[3,1,0]гексан
[(1R,5S,6r)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил]метилметансульфонат (600 мг, 2,13 ммоль) и NaI (639, 4,26 ммоль) суспендировали в MeCN (5 мл) и перемешивали в течение 16 ч. Белую суспензию разбавляли NH4Cl (20 мл) и экстрагировали EtOAc (30 мл x 3). Объединенные органические слои концентрировали, с получением красного масла, которое очищали флэш-хроматографией (смесь петролейный эфир/EtOAc, от 0 до 40%) на колонке с силикагелем, с получением указанного в заголовке соединения (500 мг, 75%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,36-7,18 (м, 5H), 3,57 (с, 2H), 3,12 (д, 2H), 2,98 (д, 2H), 2,37-2,24 (м, 2H), 1,87-1,76 (м, 1H), 1,36-1,29 (м, 2H)
Стадия 2: (1R,5S,6r)-3-бензил-6-[(метилсульфонил)метил]-3-азабицикло[3,1,0]гексан
(1R,5S,6r)-3-бензил-6-(иодметил)-3-азабицикло[3,1,0]гексан (500 мг, 1,60 ммоль) растворяли в EtOH (10 мл). Порционно добавляли метансульфинат натрия (489 мг, 4,79 ммоль). Желтый раствор перемешивали при 80°C в течение 16 ч, затем при rt в течение 48 ч. Реакционную смесь разбавляли водой (50 мл) и экстрагировали EtOAc (30 мл x 3). Объединенные органические слои концентрировали, с получениемa бесцветного масла, которое очищали флэш-хроматографией (смесь петролейный эфир/EtOAc, от 50 до 80%) на колонке с силикагелем, с получением указанного в заголовке соединения (310 мг, 73%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,41-7,10 (м, 5H), 3,59 (с, 2H), 3,04 (д, 2H), 2,96-2,86 (м, 5H), 2,44-2,33 (м, 2H), 1,75-1,65 (м, 1H), 1,53-1,41 (м, 2H).
Стадия 3: (1R,5S,6r)-6-[(метилсульфонил)метил]-3-азабицикло[3,1,0]гексан
(1R,5S,6r)-3-бензил-6-[(метилсульфонил)метил]-3-азабицикло[3,1,0]гексан (310 мг, 1,17 ммоль) растворяли в EtOH (10 мл) и добавляли 10 масс% Pd/C (249 мг, 0,234 ммоль). Суспензию перемешивали при 50 psi (3,402 атм.) H2 в течение 48 ч. Реакционную смесь фильтровали и фильтрат концентрировали, с получением белого твердого вещества (200 мг, 98%), которое непосредственно использовали в следующей стадии без очистки.
Стадия 4
Соединение Примера 9 получали по методике, аналогичной методике Примера 1, стадии 1-2, начиная с (1R,5S,6r)-6-[(метилсульфонил)метил]-3-азабицикло[3,1,0]гексана (40 мг, 0,11 ммоль). По завершении, реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали EtOAc. Органические слои концентрировали и очищали препаративной ВЭЖХ (Phenomenex Gemini C18 250*50 10 мкм, от 26% MeCN в воде (0,225% муравьиной кислоты) до 46% MeCN в воде (0,225% муравьиной кислоты)), с получением соединения Примера 9 (30 мг, выход 25% за две стадии) в виде белого твердого вещества.
МС(ЭС+): 420,9 (M+H). 1H ЯМР (400 МГц, CD3OD) δ 6,13 (с, 1H), 4,16 (к, 1H), 4,09-3,87 (м, 1H), 3,87-3,75 (м, 2H), 3,75-3,62 (м, 1H), 3,61-3,46 (м, 2H), 3,16 (д, 2H), 2,99 (с, 3H), 1,87 (с, 2H), 1,42 (д, 3H), 1,39 (с, 3H), 0,98 (тт, 1H)
Пример 10: 2-[(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]-N-(метилсульфонил)ацетамид
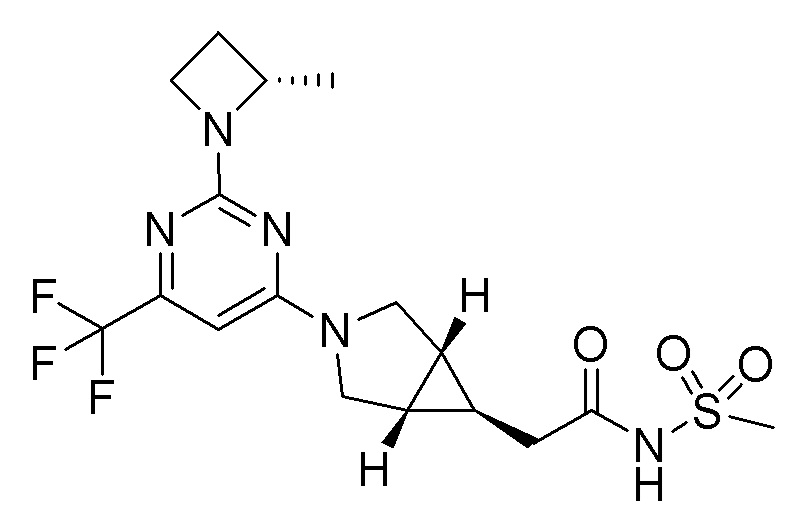
[(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусную кислоту (75 мг, 0,21 ммоль) растворяли в DCM (6 мл). Добавляли карбонилдиимидазол (34 мг, 0,21 ммоль). Через 2 ч добавляли метансульфонамид (22 мг, 0,23 ммоль) и 1,8-диазабициклоундец-7-ен (38 мг, 0,25 ммоль). После перемешивания в течение 16 ч реакционную смесь разбавляли раствором NH4Cl (15 мл) и экстрагировали DCM (15 мл x 3). Объединенные органические слои концентрировали, и сырой материал очищали препаративной ВЭЖХ (Agela Durashell C18 150*25 5 мкм, подвижная фаза: от 43% MeCN в воде (0,225% муравьиной кислоты) до 63% MeCN в воде (0,225% МК), с получением соединения Примера 10 в виде белого твердого вещества (32 мг, 35%).
МС(ЭС+): 434,0 (M+H). 1H ЯМР (400 МГц, CD3OD) δ 6,05 (с, 1H), 4,50-4,35 (м, 1H), 4,04-3,83 (м, 3H), 3,71-3,40 (м, 3H), 3,24 (с, 3H), 2,45-2,36 (м, 1H), 2,34 (д, 2H), 2,00-1,86 (м, 1H), 1,69-1,57 (м, 2H), 1,48 (д, 3H), 0,90-0,79 (м, 1H).
Пример 11: (1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-6-(1H-тетразол-5-илметил)-3-азабицикло[3,1,0]гексан
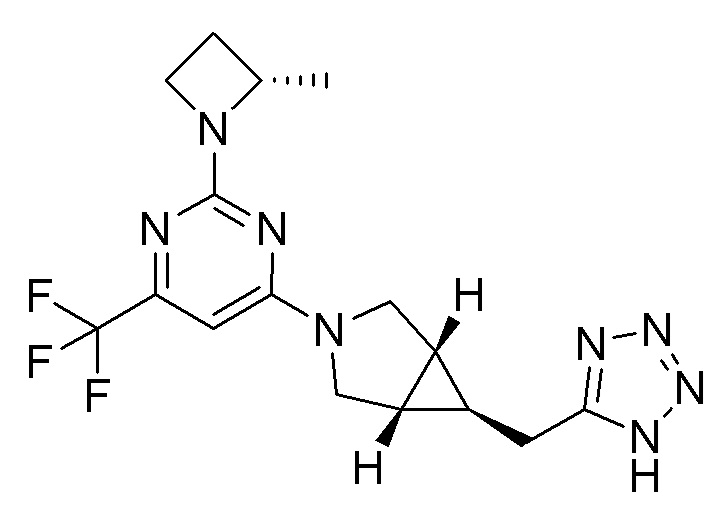
Стадия 1: трет-бутил(1R,5S,6s)-6-(1H-тетразол-5-илметил)-3-азабицикло[3,1,0]гексан-3-карбоксилат
трет-бутил(1R,5S,6s)-6-(цианометил)-3-азабицикло[3,1,0]гексан-3-карбоксилат (50 мг, 0,22 ммоль) растворяли в толуоле (2 мл) и добавляли азид трибутилолова (224 мг, 0,675 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 16 ч, затем охлаждали до rt. Реакционную смесь разбавляли насыщенным водным раствором Na2CO3 (5 мл) и водой (5 мл), и промывали DCM (15 мл x 2). Затем водный слой подкисляли до pH=5 и экстрагировали 10:1 смесью CH2Cl2:MeOH (15 мл x 3). Объединенные органические слои концентрировали, с получением указанного в заголовке соединения (50 мг, 84%) в виде бесцветного масла. Данное соединение использовали без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 3,60-3,48 (м, 2H), 3,46-3,29 (м, 2H), 3,08 (дд, 1H), 2,90 (дд, 1H), 1,65-1,50 (м, 2H), 1,43 (с, 9H), 1,13-1,02 (м, 1H).
Стадия 2: (1R,5S,6s)-6-(1H-тетразол-5-илметил)-3-азабицикло[3,1,0]гексан, соль трифторуксусной кислоты
Трет-бутил(1R,5S,6s)-6-(1H-тетразол-5-илметил)-3-азабицикло[3,1,0]гексан-3-карбоксилат (45 мг, 0,10 ммоль) растворяли в DCM (3 мл) и добавляли TFA (1,5 мл). Через 2 ч при rt реакционную смесь концентрировали до сухого состояния, с получением указанного в заголовке соединения, которое использовали без дополнительной очистки.
Стадия 3
Соединение Примера 11 получали по методике, аналогичной методике Примера 1, стадии 1-2, начиная с (1R,5S,6s)-6-(1H-тетразол-5-илметил)-3-азабицикло[3,1,0]гексана (соли трифторуксусной кислоты, 70 мг, 0,12 ммоль). По завершении, реакционную смесь гасили насыщенным водным раствором NH4Cl (20 мл), подкисляли до pH=5 и экстрагировали EtOAc (20 мл x 3). Органические слои концентрировали и очищали препаративной ВЭЖХ (Daiso 150*25 5 мкм, от 36% MeCN в воде (0,225% муравьиной кислоты) до 66% MeCN в воде (0,225% муравьиной кислоты)), с получением соединения Примера 11 (9 мг, 19%) в виде белого твердого вещества.
МС(ЭС+): 381,0 (M+H). 1H ЯМР (400 МГц, CD3OD) δ 6,07 (с, 1H), 4,50-4,35 (м, 1H), 4,09-3,84 (м, 3H), 3,72-3,56 (м, 1H), 3,56-3,44 (м, 2H), 3,00 (д, 2H), 2,46-2,34 (м, 1H), 2,02-1,86 (м, 1H), 1,82-1,67 (м, 2H), 1,49 (д, 3H), 1,04-0,94 (м, 1H).
Пример 12: [(1R,5S,6R)-3-{3-хлор-2-(1,1-дифторэтил)-6-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]пиридин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
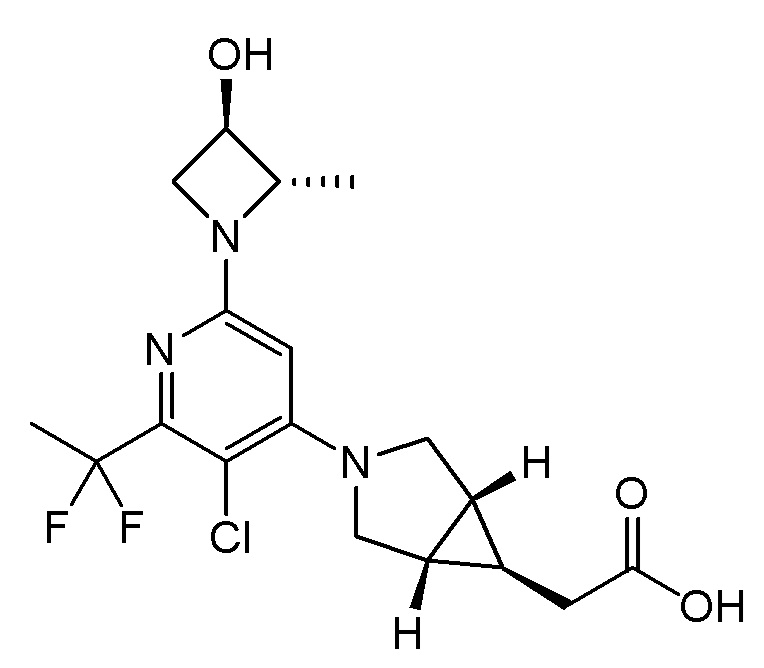
Стадия 1: этил-3-амино-4,4-дифторпент-2-еноат
В две отдельные партии раствора EtOAc (6,0 г, 70 ммоль) в THF (50 мл) порционно добавляли гидрид натрия (60% в минеральном масле, 2,72 г, 68,1 ммоль). После завершения добавления по каплям добавляли этил-2,2-дифторпропаноат (11,3 г, 81,7 ммоль) в течение 15 мин. Реакционную смесь нагревали при 50°C в течение 4 ч, затем перемешивали при rt в течение 16 ч. Реакционную смесь вливали в 10% раствор серной кислоты (50 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Сырые продукты объединяли и очищали хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (1:5), с получением желтого масла. Продукт растворяли в этаноле (150 мл) и обрабатывали ацетатом аммония (42,8 г, 555 ммоль). Смесь нагревали при 80°C в течение 16 ч. Растворитель удаляли при пониженном давлении, осадок разбавляли водным раствором NaHCO3 (100 мл) и экстрагировали DCM (2×100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали, с получением указанного в заголовке соединения (22,5 г, 90,5%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3) δ: 4,89 (с, 1H), 4,16 (к, 2H), 1,81 (т, 3H), 1,28 (т, 3H).
Стадия 2: этил-3-[(3-этокси-3-оксопропаноил)амино]-4,4-дифторпент-2-еноат
В раствор этил-3-амино-4,4-дифторпент-2-еноата (22,5 г, 126 ммоль) и пиридина (11,9 г, 151 ммоль) в DCM (250 мл) по каплям добавляли этил-3-хлор-3-оксопропаноат (18,9 г, 126 ммоль) при 0°C. Раствор перемешивали при rt в течение 16 ч. Реакционную смесь промывали 1Н HCl (250 мл) и насыщенным водным раствором NaHCO3 (250 мл), сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (1:10), с получением указанного в заголовке соединения (18,9 г, выход 51,3%) в виде светло-желтого масла.
1H ЯМР (400 МГц, CDCl3) δ: 10,53 (уш.с, 1H), 5,77 (с, 1H), 4,18-4,28 (м, 4H), 3,45 (с, 2H), 1,99 (т, 3H), 1,23-1,39 (м, 6H).
Стадия 3: этил-6-(1,1-дифторэтил)-2,4-дигидроксипиридин-3-карбоксилат
Суспензию этил-3-[(3-этокси-3-оксопропаноил)амино]-4,4-дифторпент-2-еноата (18,9 г, 64,4 ммоль) и трет-бутоксида калия (8,68 г, 77,3 ммоль) в EtOH (100 мл) перемешивали при 80°C в течение 4 ч, а затем при 10°C в течение 16 ч. Растворитель удаляли при пониженном давлении, и осадок вливали в ледяную воду (150 мл). Полученный раствор подкисляли до pH=2 водным 2Н раствором HCl. Продукт экстрагировали EtOAc (2×200 мл). Объединенные органические слои фильтровали, и собирали белый осадок (13,0 г). Фильтрат концентрировали до сухого состояния и промывали MeOH, с получением дополнительного белого твердого вещества (1,5 г), которое объединяли с отфильтрованным твердым веществом, с получением указанного в заголовке соединения (14,5 г, выход 91%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,82 (уш.с, 2H), 6,30 (с, 1H), 4,23 (к, 2H), 1,92 (т, 3H), 1,28 (т, 3H).
Стадия 4: этил-6-(1,1-дифторэтил)-2,4-диэтоксипиридин-3-карбоксилат
В смесь этил-6-(1,1-дифторэтил)-2,4-дигидроксипиридин-3-карбоксилата (2,00 г, 8,09 ммоль) и твердого карбоната калия (2,80 г, 20,2 ммоль) в DMF (35 мл) по каплям добавляли йодэтан (2,52 г, 16,2 ммоль) при 0°C. Смесь перемешивали при 30°C в течение 16 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3×35 мл). Объединенные органические слои промывали рассолом (2×40 мл), сушили над Na2SO4, фильтровали и концентрировали, с получением сырого продукта (2,51 г, >100%) в виде желтого масла, которое непосредственно использовали в следующей стадии.
1H ЯМР (400 МГц, CDCl3) δ: 6,87 (с, 1H), 4,36-4,44 (м, 4H), 4,15 (к, 2H), 1,94 (т, 3H), 1,41 (т, 3H), 1,33-1,39 (м, 6H).
Стадия 5: этил-5-хлор-6-(1,1-дифторэтил)-2,4-диэтоксипиридин-3-карбоксилат
В раствор этил-6-(1,1-дифторэтил)-2,4-дигидроксипиридин-3-карбоксилата (2,50 г, 8,24 ммоль) в ацетонитриле (30 мл) добавляли N-хлорсукцинимид (2,20 г, 16,5 ммоль). Бесцветную реакционную смесь перемешивали при 100°C в течение 16 ч. Реакционную смесь разбавляли водой (120 мл) и насыщенным водным раствором NaHCO3 (30 мл). Продукт экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали колоночной хроматографией, элюируя смесью EtOAc/петролейный эфир (от 0:100 до 96:4), с получением указанного в заголовке соединения (2,1 г, 75%) в виде светло-желтого масла.
1H ЯМР (400 МГц, CDCl3) δ: 4,33-4,34 (м, 4H), 4,21 (к, 2H), 2,02 (т, 3H), 1,35-1,46 (м, 9H).
Стадия 6: 5-хлор-6-(1,1-дифторэтил)пиридин-2,4-диол
Раствор этил-5-хлор-6-(1,1-дифторэтил)-2,4-диэтоксипиридин-3-карбоксилата (2,10 г, 6,21 ммоль) в 48% водном растворе бромистоводородной кислоты (25 мл) перемешивали при 110°C в течение 48 ч. Реакционную смесь концентрировали и обрабатывали гидроксидом аммония (6 мл). Реакционную смесь концентрировали, с получением указанного в заголовке соединения (3,1 г, >100%, 30% чистоты) в виде светло-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 6,36 (с, 1H), 1,93 (т, 3H).
Стадия 7: 3,4,6-трихлор-2-(1,1-дифторэтил)пиридин
Смесь 5-хлор-6-(1,1-дифторэтил)пиридин-2,4-диола (1,80 г, 2,6 ммоль, 30% чистоты) в оксихлориде фосфора (18 мл) и DMF (4,5 мл) перемешивали при 100°C в течение 16 ч. Реакционную смесь вливали в ледяную воду (80 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали рассолом (2×50 мл), сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (от 0:100 до 0,5:99,5), с получением указанного в заголовке соединения (540 мг, 85%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ: 7,56 (с, 1H), 2,08 (т, 3H).
Стадия 8: этил{(1R,5S,6s)-3-[3,6-дихлор-2-(1,1-дифторэтил)пиридин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетат
Смесь 3,4,6-трихлор-2-(1,1-дифторэтил)пиридина (50 мг, 0,2 ммоль), этила (1R,5S,6s)-3-азабицикло[3,1,0]гекс-6-илацетата (34 мг, 0,20 ммоль) и триэтиламина (62 мг, 0,61 ммоль) в DMF (2 мл) перемешивали при 60°C в течение 16 ч. Смесь разбавляли водой (15 мл) и водным раствором хлорида аммония (10 мл), и экстрагировали EtOAc (3×15 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на колонке, элюируя смесью EtOAc/петролейный эфир (от 0:100 до 7:93), с получением указанного в заголовке соединения (70 мг) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ: 6,84 (с, 1H), 4,13 (к, 2H), 4,02-4,08 (м, 2H), 3,45-3,54 (м, 2H), 2,33 (д, 2H), 1,97 (т, 3H), 1,57-1,62 (м, 2H), 1,25 (т, 3H), 0,99-1,06 (м, 1H).
Стадия 9
В раствор этил{(1R,5S,6s)-3-[3,6-дихлор-2-(1,1-дифторэтил)пиридин-4-ил]-3-азабицикло[3,1,0]гекс-6-ил}ацетата (70 мг, 0,18 ммоль) в диоксане (5 мл) добавляли (2S,3R)-3-гидрокси-2-метилазетидин-1-ий[(1R,4S)-7,7-диметил-2-оксобицикло[2,2,1]гепт-1-ил]метансульфонат (64,3 мг, 0,200 ммоль), трет-бутоксид натрия (71,0 мг, 0,738 ммоль), хлор(2-дициклогексилфосфино-2',6'-ди-и-пропокси-1,1'-бифенил)[2-(2-аминоэтилфенил)]палладий(II) (6,73 мг, 0,00923 ммоль) и 2-дициклогексилфосфино-2′,6′-диизопропоксибифенил (4,31 мг, 0,00923 ммоль) в атмосфере азота. Реакционную смесь перемешивали при 70°C в течение 16 ч, затем при 80°C в течение 40 ч. Охлажденную реакционную смесь разбавляли водой, подкисляли до pH=5 2Н HCl и экстрагировали EtOAc (3×25 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали препаративной тонкослойной хроматографией и затем очищали препаративной ВЭЖХ, с получением соединения Примера 12 (10,5 мг, 14%) в виде белого твердого вещества.
МС(ЭС+): 401,9 (M+H). 1H ЯМР (400 МГц, CD3OD) δ: 5,78 (с, 1H), 4,09-4,15 (м, 2H), 3,88-3,96 (м, 3H), 3,41-3,47 (м, 1H), 3,18-3,24 (м, 2H), 2,26 (д, 2H), 1,92 (т, 3H), 1,49-1,51 (м, 2H), 1,46 (д, 3H), 1,14-1,18 (м, 1H).
Пример 13: [(1R,5S,6R)-3-{2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-5-метокси-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]уксусная кислота
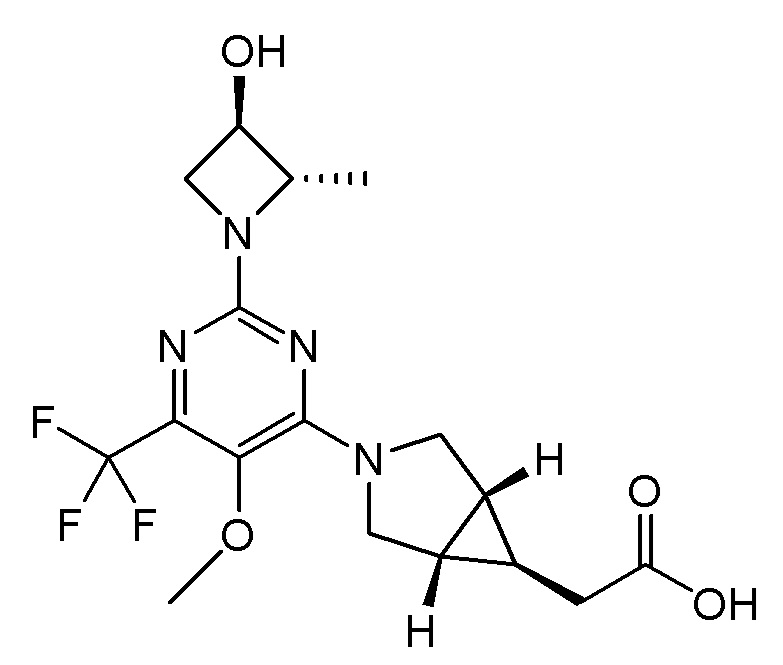
В раствор этил[(1R,5S,6R)-3-{5-бром-2-[(2S,3R)-3-гидрокси-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3,1,0]гекс-6-ил]ацетата (50 мг, 0,13 ммоль) в метаноле (5,0 мл) добавляли метоксид натрия (16,9 мг, 0,313 ммоль) и бромид меди (I) (2,2 мг, 0,016 ммоль) и нагревали до 60°C в течение 16 ч. Дополнительно добавляли бромид меди (I) (2,2 мг, 0,016 ммоль) и реакционную смесь нагревали при 60°C в течение 16 ч. Реакционную смесь разбавляли водным раствором хлорида аммония и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали, с получением сырого продукта, который очищали препаративной обращенно-фазовой ВЭЖХ, с получением 20 мг (выход 48%) соединения Примера 13 в виде белого твердого вещества.
МС(ЭС+): 403,0 (M+H). 1H ЯМР (400 МГц, CD3OD) δ: 4,19-3,97 (м, 5H), 3,64-3,57 (м, 3H), 3,57 (с, 3H), 2,30 (уш.д, 2H), 1,57 (уш.с, 2H), 1,46 (с, 3H), 0,91-0,81 (м, 1H).
Биологические данные
Был разработан скрининг-анализ для KHK в связанной ферментной системе, в котором используется продукт реакции KHK для управления сигналом поглощения в кинетическом режиме. KHK превращает фруктозу и АТФ в F1P и АДФ. АДФ затем служит субстратом для пируваткиназы, которая превращает PEP в пируват, который затем восстанавливается до лактата ферментом лактатдегидрогеназой с параллельным окислением НАДH до НАД+. Происходящее в результате истощение НАДH контролируют путем измерения абсорбции при 340 нм.
Рекомбинантные KHK-C и KHK-A человека экспрессировали в E. coli в виде слитого белка с His-меткой и очищали методом хроматографии на Ni-NTA. Синтезировали кДНК на основании эталонной последовательности NCBI NP_006479.1, наряду с последовательностями для N-концевой His-метки и сайта расщепления тромбином, и клонировали в вектор pET28a(+). Белок экспрессировали в BL-21 (DE3) с использованием индукции IPTG и очищали на колонке с Ni-NTA, а затем на колонке с Superdex 75. Очищенные KHK-C и KHK-A обрабатывали тромбином для удаления His-метки и окончательную очистку проводили методом аффинной очистки с помощью Ni-NTA/стрептавидина. Препарат белка имел чистоту ~95% при оценке методом SDS-ПААГ, и молекулярная масса, подтвержденная методом масс-спектрометрии, составляла 32663 Да (ожидаемая масса 32667 Да).
В одном анализе, называемом анализом A, используют аналитический 384-луночный планшет Corning 3653 и получают данные методом УФ-спектроскопии в непрерывном режиме при rt. Соединения готовили в ДМСО в виде 4 мМ маточных растворов, разбавляли, используя схему полулогарифмического разбавления с 11 точками, на Biomek FX (Beckman Coulter) и инкубировали при rt в течение 30 минут с реакционной смесью, содержащей 50 мМ HEPES, pH 7,4, 140 мМ KCl, 3,5 мМ MgCl2, 0,8 мМ фруктозу, 2 мМ TCEP, 0,8 мМ PEP, 0,7 мМ НАДH, 0,01% Triton X-100, 30 Ед/мл пируваткиназы-лактатдегидрогеназы и 10 нМ очищенную KHK-C. Концентрация соединения в каждой лунке находилась в диапазоне от 1 нМ до 100 мкМ. Реакцию запускали добавлением 0,2 мМ АТФ. Поглощение измеряли в течение 30 минут на ридере SpectraMax (Molecular Devices) после добавления АТФ. Полученные концентрации основаны на конечном объеме смеси 40 мкл (так называемая конечная концентрация).
Контроли: N8-(циклопропилметил)-N4-(2-(метилтио)фенил)-2-(пиперазин-1-ил)пиримидо[5,4-d]пиримидин-4,8-диамин в конечной концентрации 2 мкМ использовали в качестве контроля с высоким процентом эффективности (HPE) и 2,5% раствор ДМСО, присутствующий во всех реакционных лунках, использовали в качестве контроля с нулевым процентом эффективности (ZPE). Скорость реакции измеряли во временном окне 300-1800 секунд в единицах измерения 1000*ОЕ/мин (оптическая единица в минуту), и рассчитывали средние значения из 16 лунок для каждого из контролей ZPE и HPE, СредZPE и СредHPE, соответственно.
Процент ингибирования (% ингибирования) рассчитывали для каждой лунки, используя уравнение:
100-100 x (Величина поглощения соединения - СредHPE)
(СредZPE - СредHPE)
Строили график зависимости % ингибирования от log концентрации соединения с использованием GraphPad Prism, данные подставляли в уравнение «log[соединение] против ответа - вариабельный наклон», используя нелинейный регрессионный анализ для получения величин IC50. Для каждого тестируемого соединения полученная величина IC50 представляет собой среднюю величину из по меньшей мере двух отдельных анализов, проведенных в разные дни.
Соединения, имеющие величину IC50 менее 20 нМ, исследовали во втором анализе KHK, называемом анализом B, с использованием в 10 раз меньше фермента и измерением поглощения в течение 3 часов, получая величины IC50 ниже, чем нижний предел 10 нМ анализа A. Соединения готовили в ДМСО в виде 4 мМ маточных растворов, разбавляли, используя схему 2-кратного разбавления с 11 точками, на Biomek FX, охватывая диапазон концентраций от 97 пМ до 100 нМ, и инкубировали с реакционной смесью, полученной так же, как в анализе A, но содержащей 1 нМ KHK-C. Реакцию запускали добавлением 0,2 мМ АТФ, и поглощение контролировали в течение 3 часов при 340 нм. Скорости реакции и величины IC50 рассчитывали, как описано выше.
Третий анализ KHK, называемый анализом C, проводили при высоких концентрациях фруктозы и АТФ, условиях, которые более соответствуют физиологическим концентрациям природных субстратов фермента KHK. Анализ C проводили, как описано выше для анализа B, за исключением использования 8 мМ фруктозы и 2 мМ АТФ, и диапазона концентраций соединения от 10 пМ до 1 мкМ или от 50 пМ до 5 мкМ, используя схему полулогарифмического разбавления.
Четвертый анализ, называемый анализом D, проводили с использованием KHK-A человека для оценки эффективности соединений в ингибировании активности этого фермента. Соединения готовили в ДМСО в виде 4 мМ маточных растворов, разбавляли, используя схему 2-кратного разбавления с 11 точками, на Biomek FX, охватывая диапазон конечных концентраций от 0,25 до 250 нМ, и инкубировали с реакционной смесью, полученной так же, как в анализе A, но содержащей 8 мМ фруктозу и 1 нМ KHK-A. Реакцию запускали добавлением 0,2 мМ АТФ, и поглощение контролировали в течение 3 часов при 340 нм. Скорости реакции и величины IC50 рассчитывали, как описано выше.
Таблица 3. Биологические данные для анализов A, B, C и D+
|
+ Среднее значение IC50 для (#) количества экспериментов на каждый Пример.
Соединения следующих Примеров, представленные в Таблице 4, были получены с использованием условий, аналогичных условиям для соединений справочных Примеров, приведенных в колонке, обозначенной «Справ. Пример №», с использованием некритических, рутинных изменений. Таблица 4 также содержит биологические данные из анализа A для соединений этих Примеров. Структуры соединений этих Примеров из Таблицы 4 приведены на Фигуре 3.
Таблица 4. Примеры и биологические данные для анализа A
|
+ Среднее значение IC50 для (#) количества экспериментов на каждый Пример.
* В Примерах 37 и 38 использован Способ 1: Колонка: Xbridge C18 2,1×50 мм, 5мкм. Температура: 40°C. Подвижная фаза A: 0,0375% TFA в H2O. Подвижная фаза B: 0,01875% TFA в ацетонитриле. Начальные условия: B: 1%, A: 99%. Градиент: B: 1%, A: 99% до B: 5%, A: 95% от t=0,00 мин до 0,60 мин, затем до B: 100% от t=0,60 мин до 4,00 мин, затем до B: 1%, A: 99% от t=4,00 мин до 4,30 мин, удержание до t=4,70 мин. Скорость потока=0,8 мл/мин, объем впрыска 2 мкл.
**В Примерах 39 и 51 использован Способ 2: Колонка: Xbridge C18 2,1×50 мм, 5 мкм. Температура: 40°C. Подвижная фаза A: 0,0375% TFA в H2O. Подвижная фаза B: 0,01875% TFA в ацетонитриле. Начальные условия: B: 10%, A: 90%. Удержание от t=0,00 мин до 0,50 мин. Градиент: B: 10%, A: 90% до B: 100%, A: 0% от t=0,50 мин до 4,00 мин, затем до B: 10%, A: 90% от t=4,00 мин до 4,30 мин, удержание до t=4,70 мин. Скорость потока=0,8 мл/мин, объем впрыска 2 мкл.
***В Примерах 53 и 54 использован Способ 3: Колонка: OJ-H 4,6×100 мм, 5 мкм; Подвижная фаза A: метанол (по объему); Подвижная фаза B: CO2 (по объему). Градиент: 80,0% CO2/20,0% метанол, изократический в течение 5 мин. Поток: 1,5 мл/мин. Обратное давление: 100 бар.
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/010f582b4b1b9caba67247e18129cc50.png)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/0e0efac417305051c8dddf04f3172307.jpg)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/1890cc05509d1235b9de3d1a52de6f93.jpg)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/2f8cabbbeaa38e86856635e95b4bafa4.png)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/95caedc5dd32ce823a71c5423d0e2b98.png)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/7eeeecc9d8d523fe0023fd39e15d044b.png)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/e31b17b286ff4e7313f4df822c42d983.png)
![ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ](https://fips.edrid.ru/images/rid/21/a2/d5/a419e356c41cbbeb5ca9f661852417b1.jpg)
Устройство для манипулирования капсулами и оборудование для манипулирования капсулами, содержащее такое устройство
Производные пирроло[2,3-d]пиримидина
Антагонисты pcsk9
Соли и полиморфы 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6н-азепино[5,4,3-cd]индол-6-она
Твердые формы ингибитора диссоциации транстиретина
Простатоассоциированные антигены и иммунотерапевтические схемы на основе вакцин
Комбинация инотузумаба озогамицина и торизела для лечения рака
Антагонисты pcsk9
Улучшенные антитела-антагонисты против gdf-8 и их применения
Жидкие лекарственные композиции апиксабана
Замещенные-6,8-диоксабицикло[3.2.1.]октан-2,3-диольные соединения в качестве агентов, нацеленных на asgpr

