Результат интеллектуальной деятельности: ХЕМОЭНЗИМАТИЧЕСКИЙ СИНТЕЗ ПРОИЗВОДНЫХ 2,5-ДИКЕТОМОРФОЛИНА
Вид РИД
Изобретение
Область техники
Изобретение относится к синтезу производных 2,5-дикетоморфолина, представляющих собой мономеры для получения биоразлагаемых полимеров полидепсипептидов (полиэфирамидов). Полимеры этого типа находят применение в медицине и могут быть использованы при хирургическом сшивании тканей, в качестве костных пластин, а также носителей для контролируемого высвобождения лекарственных веществ. Производные 2,5-дикетоморфолина представляют интерес как биологически активные соединения: они обладают антибиотической, антиоксидантной, антикоагулянтной, а также иммуностимулирующей активностью, проявляют ингибиторное действие по отношению к различным ферментам.
Уровень техники
В научной и патентной литературе известны способы получения производных 2,5-дикетоморфолина методами классического органического многостадийного синтеза, включающие стадии постановки и снятия защитных групп и использующие активированные производные амино- и оксикислот. Все известные способы можно свести к двум основным подходам: первоначальное образование амидной связи, а затем циклизация в лактон, и наоборот, этерификация с последующей циклизацией в лактам [Smelcerovic A. et al. J. of Amino Acids. 2014, 46, P. 825.]. Чаще всего авторами используются альфа-галогензамещенные производные галогенангидридов карбоновых кислот [CN 108299332, 20-07-2018; Cook A. et al. J. Chem. Soc. 1949, P. 2347.; Smelcerovic A et al. J. Mol. Struct. 2011, 985, P. 397.], N-защищенные производные аминокислот [Pedras M. et al. Org. Lett. 2004, 6, P. 4615.; Kagamizono T. et al. J. Antibiot. 1995, 48, P. 1407.], O-защищенные производные аминокислот [Hughes A. et al. J. Org. Chem. 2005, 70, P. 3079.]. В патентном документе [US 005817751, 06-10-1998] сообщается о применении для синтеза предшественника производного 2,5-дикетоморфолина реакции Уги, заключающейся в создании N-замещенной амидной связи путем конденсации четырех компонентов: оксикислоты, аминокислоты, альдегида или кетона и изонитрила, причем аминокислота ковалентно связана сложноэфирной связью через карбоксигруппу с твердой подложкой, содержащей гидроксильные группы. Последующая циклизация предшественника приводит к образованию целевого продукта с регенерацией подложки.
Известные способы получения производных 2,5-дикетоморфолина имеют существенные ограничения. При необходимости получения энантиомерно чистых соединений нужно использовать оптически чистые исходные вещества. Для региоселективного превращения необходимы активированные реагенты. В связи с полифункциональностью исходных реагентов возникает необходимость использования защитных групп, что значительно усложняет синтез и делает его многостадийным, вследствие чего общий выход конечного продукта не превышает 50%, несмотря на довольно высокие выходы конкретных стадий синтеза. Применение биокатализа для синтеза подобных соединений позволяет преодолеть указанные ограничения и получить конечный продукт со значительно более высоким выходом и энантиомерной чистотой более 99,9%.
Наиболее близким к заявленному решению является способ, описанный в патентном документе [CN 108299332, 20-07-2018], авторы которого синтезировали (3R,6R и 3S,6S)-3-(бензилоксикарбонил-этилен)-6-метил-морфолин-2,5-дион в две стадии исходя из 2-бромпропионилхлорида и гамма-бензил-глутаминовой кислоты, причем в зависимости от способа очистки общий выход продукта оказался невелик и составил 3,4-34,8%. Кроме того, получение 2-бромпропионилхлорида также потребует проведения реакций с участием опасных и агрессивных реагентов. Способ синтеза 2,5-дикетоморфолинов, указанный в патентном документе [US 005817751, 06-10-1998], с применением реакции Уги описан выше и имеет сходные ограничения.
Раскрытие изобретения
Технической проблемой, на решение которой направлено изобретение, является разработка простого в исполнении способа синтеза производных 2,5-дикетоморфолина, обеспечивающего высокий выход целевого продукта и его энантиомерную чистоту.
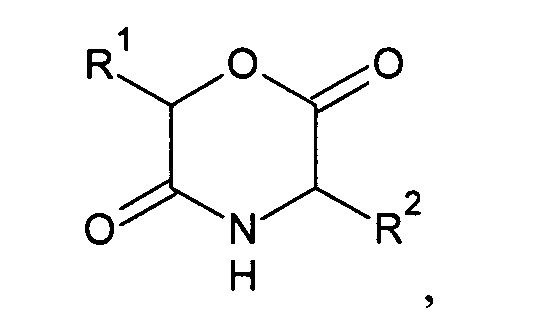
Заявлен способ синтеза производных 2,5-дикетоморфолина, общей формулы

где R1=Н, СН3, арил; R2=Н, СН3, арил, CH2-арил, изопропил, пропил, бутил, оксиметил, меткаптометил, 1-оксиэтил, 1-метилпропил, 2-метилпропил, индол-3-ил-метил, карбамоилметил, карбамоилэтил, карбоксиметил, карбоксиэтил, 4-аминобутил, 3-гуанидилпропил, 2-(метилсульфанил)-этил исходя из аминокислот и амидов оксикислот, включающий стадию образования амидной связи с использованием пенициллинацилазы и последующую циклизацию под действием активирующего агента, например, N,N'-дициклогексилкарбодиимида. Способ заключается в том, что готовят насыщенные водные растворы аминокислоты, являющейся нуклеофилом, и амида оксикислоты, являющегося ацильным донором, в мольном соотношении компонентов от 5:1 до 1:3, доводят рН до 9-11, добавляют биокатализатор - растворимую, иммобилизованную или мутантную форму пенициллинацилазы для обеспечения необходимого превращения, выдерживают реакционную смесь при 10-35°С до достижения максимального выхода N-оксиацил-аминокислоты, доводят рН до 2-3, полученную N-оксиацил-аминокислоту экстрагируют органическим растворителем, добавляют эквимолярное количество N,N'-дициклогексилкарбодиимида, смесь выдерживают до образования осадка N,N'-дициклогексилмочевины, осадок отделяют, оставшийся раствор упаривают и выделяют твердый остаток целевого продукта. При этом количество биокатализатора для оптимального превращения определяют эмпирически при отборе проб реакционной смеси в модельном эксперименте, проводимом в малом объеме с соблюдением аналогичных экспериментальных условий и концентраций, и слежении за накоплением целевого продукта. Время, необходимое для достижения максимального выхода N-оксиацил-аминокислоты определяют эмпирически в модельном эксперименте, проводимом в малом объеме с соблюдением аналогичных экспериментальных условий и концентраций, при слежении за накоплением целевого продукта. Для осуществления синтеза в качестве органического растворителя используют этилацетат, бутилацетат, изобутилацетат, гептан, четыреххлористый углерод, хлороформ, дихлорметан, ацетонитрил, циклопентилметиловый эфир или их смеси, при этом удаление растворителя осуществляют отгонкой или в вакууме. Отделение осадка проводят фильтрацией через фильтровальную бумагу, ткань или пористый фильтр, а также декантацией.
В качестве биокатализатора могут быть использованы различные формы (растворимые, иммобилизованные, мутантные) бактериального фермента пенициллинацилазы из Escherichia coli, Alcaligenes faecalis, Kluyvera citrophila, Providencia rettgeri, Arthrobacter viscosus, Bacillus megaterium и других источников сходные по свойствам и способные катализировать образование амидной связи между аминогруппой аминокислоты и карбоксильной группой оксикислоты (схема 1). Известно, что многочисленные методы иммобилизации пенициллинацилаз, как и других ферментов, позволяют сохранить каталитические свойства фермента, а также делают возможным повторное применение биокатализатора, часто приводят к его стабилизации, а также позволяют улучшить эффективность процесса и его экономические показатели [R.A. Sheldon et al. Chem. Soc. Rev. 2013, 42, P. 6223; A.I. Kallenberg et al. Adv. Synth. Catal. 2005, 347, P. 905; N.A. Pchelintsev et al. J. Mol. Catal. B: Enzymatic 2009, 56, Р. 202]. Биокаталитическая реакция ацильного переноса протекает стереоселективно: ацилированию преимущественно подвергается L-энантиомер аминокислоты, что позволяет включать в реакцию рацемическую смесь аминокислоты, а не индивидуальный энантиомер. Кроме того, биокаталитическая реакция протекает в диапазоне температур от 10 до 35°С в мягких условиях в водной среде, без использования органических растворителей и образования токсичных отходов. В качестве ацильного донора используется амид оксикислоты в виде рацемической смеси или индивидуального энантиомера, в качестве нуклеофила - аминокислота в виде рацемической смеси или индивидуального энантиомера, мольное соотношение ацильного донора и нуклеофила варьируется от 1:5 до 3:1, соответственно. Значение рН реакционной смеси варьируется от 9 до 11. Время проведения реакции пропорционально содержанию фермента в реакционной смеси и обеспечивается использованием определенного количества биокатализатора, которое подбирается эмпирически для каждой пары реагентов исходя из потребности провести реакцию за необходимый промежуток времени по технологическим, экономическим или иным соображениям, например, по результатам предварительного кинетического эксперимента, проводимого в тех же условиях, что и препаративный синтез, но в небольшом масштабе, например, в 1-2 мл реакционной смеси со слежением за кинетикой реакции, заключающимся в отборе проб и определении концентрации компонентов в каждый момент времени, например, с использованием высокоэффективной жидкостной хроматографии. Выбор оптимальных условий, концентраций реагентов и количества биокатализатора возможен также при использовании других подходов, например, микрореакторов или расчетов, основанных на фундаментальном понимании кинетических закономерностей процесса [R. Wohlgemuth et al. Trends Biotechnol. 2015, 33, P. 302; J.L. van Roon et al. Biotechnol. Adv. 2007, 25, Р. 137]. Применение биокатализа позволяет достичь очень высокого выхода N-оксиацил-аминокислоты. Продукт реакции выделяется из реакционной смеси экстракцией органическим растворителем, например, этилацетатом в кислой среде, например, при рН 2-3. При необходимости отделения непрореагировавшего ацильного донора после биокаталитического превращения, например, в случае проведения процесса при соотношениях ацильного донора по отношению к нуклеофилу больше чем 1:1, рН реакционной смеси доводят до 12±1 и извлекают непрореагировавший ацильный донор экстракцией органическим растворителем.
На второй стадии N-оксиацил-аминокислота подвергается реакции внутримолекулярной этерификации с образованием производного 2,5-дикетоморфолина. Циклизация протекает в органическом растворителе при добавлении активирующего реагента, например, N,N'-дициклогексилкарбодиимида в количестве эквимолярном N-оксиацил-аминокислоте. В процессе реакции образуется N,N'-дициклогексилмочевина, выпадающая в осадок. Выход реакции циклизации также очень высокий, поскольку образуется термодинамически устойчивый шестичленный цикл. Очистка целевого продукта заключается в отделении осадка N,N'-дициклогексилмочевины, например, декантацией, центрифугированием или фильтрованием через фильтровальную бумагу, ткань или пористый фильтр и последующем удалении (отгонкой) растворителя, например, в вакууме или с помощью роторного испарителя.
Структура и состав всех синтезированных соединений подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота подтверждена методом высокоэффективной жидкостной хроматографии.

Техническим результатом заявленного способа является производное 2,5-дикетоморфолина, синтезированное с участием пенициллинацилазы как высокоэффективного биокатализатора, позволяющего стереоселективно создавать амидную связь в одну стадию. Последующая циклизация приводит к получению целевого продукта с общим выходом не менее 80 масс. % по отношению к исходному компоненту (нуклеофилу или ацильному донору), взятому в меньшем количестве. Прикладным аспектом настоящего изобретения является то, что производные 2,5-дикетоморфолина являются мономерами для получения полидепсипептидов - перспективных биоразлагаемых полимеров биомедицинского назначения [Feng Y. et al. Int. J. Mol. Sci. 2009, 10, P. 589], а также обладают биологической активностью.
Осуществление изобретения
Ниже представлено более детальное описание заявляемого способа, которое не ограничивает объем притязаний заявляемого изобретения, а демонстрирует возможность осуществления изобретения с достижением заявляемого технического результата.
Все используемые реагенты являются коммерчески доступными, все процедуры, если не оговорено особо, осуществляли в диапазоне температур от 10 до 35°С.
Пример 1. Синтез (3S,6S)-3-бензил-6-фенил-морфолин-2,5-диона.
Амид (S)-миндальной кислоты и D,L-фенилаланин (до конечных концентраций 0,2 и 0,1 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 11 и начали реакцию добавлением аликвоты исходного концентрированного раствора пенициллинацилазы из Alcaligenes faecalis, создавая концентрацию активных центров фермента в реакционной смеси 1 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 15°С. По истечении 25 минут рН реакционной смеси довели 5 М раствором KOH до 12, экстрагировали три раза этилацетатом непрореагировавший амид (S)-миндальной кислоты, при этом объем этилацетата при каждой экстракции был равен объему реакционной смеси. Для извлечения N-(S)-манделил-(S)-фенилаланина концентрированной HCl рН довели до 2, продут экстрагировали три раза этилацетатом, при этом объем этилацетата при каждой экстракции был равен объему реакционной смеси. Этилацетат упарили на роторном испарителе до образования 0,5 М раствора, добавили N,N'-дициклогексилкарбодиимид до конечной концентрации 0,5 М. Осадок N,N'-дициклогексилмочевины отфильтровали через пористый фильтр с диаметром пор 40 мкм, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (3S,6S)-3-бензил-6-фенил-морфолин-2,5-диона составил 94% в пересчете на L-энантиомер фенилаланина. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99,1%.
Пример 2. Синтез (3S,6S)-3-бензил-6-фенил-морфолин-2,5-диона.
Амид (S)-миндальной кислоты и D,L-фенилаланин (до конечных концентраций 0,1 и 0,3 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 10 и начали реакцию добавлением аликвоты исходного концентрированного раствора пенициллинацилазы из Alcaligenes faecalis, создавая концентрацию активных центров фермента в реакционной смеси 1 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 20°С. По истечении 30 минут рН реакционной смеси довели до 2, экстрагировали продукт бути л ацетатом, при этом объем бутилацетата был равен одной трети объема реакционной смеси, добавили эквимолярное N-(S)-манделил-(S)-фенилаланину количество N,N'-дициклогексилкарбодиимида, после образования осадка N,N'-дициклогексилмочевины его отделили при помощи пористого фильтра, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (3S,6S)-3-бензил-6-фенил-морфолин-2,5-диона составил 91% в пересчете на амид (S)-миндальной кислоты. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99%.
Пример 3. Синтез (3S,6R)-3-метил-6-фенил-морфолин-2,5-диона
Амид (R)-миндальной кислоты и D,L-аланин (до конечных концентраций 0,2 и 0,5 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 9,0 и начали реакцию добавлением аликвоты исходного концентрированного раствора мутантной формы бeтaD484N пенициллинацилазы из Escherichia coli, создавая концентрацию активных центров фермента в реакционной смеси 1 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 10°С. По истечении 40 минут рН реакционной смеси концентрированной HCl довели до 2 и экстрагировали два раза этилацетатом продукт, при этом объем этилацетата при каждой экстракции был равен объему реакционной смеси. Этил ацетат упарили на роторном испарителе до образования 0,5 М раствора, добавили N,N'-дициклогексилкарбодиимид до конечной концентрации 0,5 М. Осадок N,N'-дициклогексилмочевины отфильтровали через стеклянный пористый фильтр с диаметром пор 40 мкм, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (3S,6R)-3-метил-6-фенил-морфолин-2,5-диона составил 86% в пересчете на амид (R)-миндальной кислоты. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99,5%.
Пример 4. Синтез (3S,6S)-3-(индол-3-ил-метил)-6-фенил-морфолин-2,5-диона.
Амид (S)-миндальной кислоты и D,L-триптофан (до конечных концентраций 0,2 и 0,1 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 10,6 и начали реакцию добавлением аликвоты исходного концентрированного раствора пенициллинацилазы из Alcaligenes faecalis, создавая концентрацию активных центров фермента в реакционной смеси 1 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 25°С. По истечении 25 минут рН реакционной смеси довели 5 М раствором KOH до 12, экстрагировали два раза бутилацетатом непрореагировавший амид (S)-миндальной кислоты, при этом объем бутилацетата при каждой экстракции был равен объему реакционной смеси. Для извлечения N-(S)-манделил-(S)-триптофана концентрированной HCl рН довели до 2, выпавшее масло центрифугировали 5 мин при 5000 об/мин, декантировали, растворили в ацетонитриле до образования 0,5 М раствора, добавили N,N'-дициклогексилкарбодиимид до конечной концентрации 0,5 М. Осадок N,N'-дициклогексилмочевины отфильтровали через стеклянный пористый фильтр с диаметром пор 40 мкм, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (3S,6S)-3-(индол-3-ил-метил)-6-фенил-морфолин-2,5-диона составил 91% в пересчете на L-энантиомер триптофана. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99,2%.
Пример 5. Синтез (3S,6S)-3-карбамоилметил-6-фенил-морфолин-2,5-диона.
Амид (S)-миндальной кислоты и D,L-аспарагин моногидрат (до конечных концентраций 0,2 и 0,1 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 10 и начали реакцию добавлением аликвоты исходного иммобилизованного препарата пенициллинацилазы из Alcaligenes faecalis, чтобы обеспечить необходимую скорость реакции, создавая концентрацию активных центров фермента в реакционной смеси 1 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 30°С. По истечении 30 минут фильтрованием отделили препарат иммобилизованного фермента, рН реакционной смеси (супернатанта) довели 5 М раствором KOH до 12, экстрагировали три раза этилацетатом не прореагировавший амид (S)-миндальной кислоты, при этом объем этилацетата при каждой экстракции был равен объему реакционной смеси. Для извлечения N-(S)-манделил-(S)-аспарагина концентрированной HCl рН довели до 7, упарили в вакууме водную фазу до сухого остатка, который промыли 2 раза этилацетатом. Продукт извлекли из твердого остатка ацетонитрилом до образования 0,5 М раствора, добавили N,N'-дициклогексилкарбодиимид до конечной концентрации 0,5 М. Осадок N,N'-дициклогексилмочевины отфильтровали через стеклянный пористый фильтр с диаметром пор 40 мкм, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (3S,6S)-3-карбамоилметил-6-фенил-морфолин-2,5-диона составил 82% в пересчете на L-энантиомер аспарагина. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99,0%.
Пример 6. Синтез (6S)-фенил-морфолин-2,5-диона.
Амид хлоруксусной кислоты и D,L-фенилаланин (до конечных концентраций 0,3 и 0,1 М соответственно) растворили в дистиллированной воде, 5 М раствором гидроксида калия довели рН до 10 и начали реакцию добавлением аликвоты исходного концентрированного раствора пенициллинацилазы из Alcaligenes faecalis, создавая концентрацию активных центров фермента в реакционной смеси 8 мкМ. Реакцию проводили в термостатируемой ячейке рН-стата Titrino 719 (Metrohm, Швейцария) при 35°С. По истечении 40 минут рН реакционной смеси довели 5 М раствором KOH до 12, экстрагировали три раза бутилацетатом не прореагировавший амид хлоруксусной кислоты, при этом объем бутилацетата при каждой экстракции был равен объему реакционной смеси. Для извлечения N-хлорацетил-(S)-фенилаланина концентрированной HCl значение рН довели до 2 и экстрагировали три раза бутилацетатом продукт, при этом объем бутилацетата при каждой экстракции был равен объему реакционной смеси. Бутилацетат упарили на роторном испарителе, остаток растворили в 5 М KOH и нагревали в течение 1 часа при 80°С. Концентрированной HCl значение рН довели до 2, экстрагировали три раза бутилацетатом N-оксиацетил-(S)-фенилаланин. Бутилацетат упарили на роторном испарителе до образования 0,5 М раствора, добавили N,N'-дициклогексилкарбодиимид до конечной концентрации 0,5 М. Осадок N,N'-дициклогексилмочевины отфильтровали через стеклянный пористый фильтр с диаметром пор 40 мкм, фильтрат упарили на роторном испарителе, твердый остаток высушили в вакууме до постоянной массы. Выход (6S)-фенил-морфолин-2,5-диона составил 87% в пересчете на L-энантиомер фенилаланина. Структура и состав синтезированного соединения подтверждены методами 1Н ЯМР и масс-спектрометрии, химическая чистота по результатам анализа методом высокоэффективной жидкостной хроматографии составила 99,6%.
Средство для ингибирования фермента поли(адф-рибозо) полимеразы
Способ синтеза пептидов, в том числе бета-лактамных антибиотиков, при использовании варианта пенициллинацилазы
Мутант пенициллинацилазы из e.coli с улучшенными свойствами
Применение по новому назначению бензилбензоата в качестве антигрибкового средства, получение и применение антимикозных фармацевтических композиций
Способ улучшения каталитических свойств пенициллинацилазы из escherichia coli и применение мутантной пенициллинацилазы
Способ улучшения каталитических свойств пенициллинацилазы
Селективные ингибиторы глицеральдегид-3-фосфатдегидрогеназы микобактерий
Применение фурансульфонатов для ингибирования транскетолазы патогена mycobacterium tuberculosis
Способ синтеза альфа-кетометилселенобутирата

