Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ ЦИНКОВОГО МЕТАЛЛОКОМПЛЕКСА ХЛОРИНА-e И ЕГО ПРИМЕНЕНИЕ
Вид РИД
Изобретение
Предлагаемая группа изобретений относится к области биомедицины, касается производного цинкового металлокомплекса хлорина-е6, которое может быть использовано в качестве агента, применяемого в фотодинамической терапии (ФДТ) для диагностики и лечения онкологических заболеваний.
Онкологические заболевания в настоящее время являются одной из острейших медико-социальных проблем человечества. Одним из основных нехирургических методов лечения различных форм опухолевых заболеваний является метод фотодинамической терапии. Однако существующие противоопухолевые препараты имеют ряд серьезных недостатков, среди которых низкая селективность накопления в опухолевых тканях и недостаточная водорастворимость, что приводит к возрастанию токсичности и снижению эффективности ФДТ. Таким образом, создание новых агентов ФДТ с улучшенными фармакокинетическими показателями - актуальная задача здравоохранения. На сегодняшний день известен ряд соединений, способных проявлять световую противоопухолевую токсичность по отношению к опухолевых клеткам. Терапевтическое действие данных молекул-фотосенсибилизаторов обусловлено генерацией активных форм кислорода и радикалов под действием светового излучения, что приводит к активации различных механизмов гибели опухолевых тканей [P. Mroz, A. Yaroslavsky, G.B. Kharkwal, M.R. Hamblin, Cell Death Pathways in Photodynamic Therapy of Cancer, Cancers 3 (2011) 2516-2539].
Известно большое количество фотоактивных соединений на основе хлориновых фотосенсибилизаторов. Некоторые из них успешно применяются в клинической практике.
Например, ФОТОЛОН (RU 2152790 С1, кл. A61K 31/79, G01N 33/52, опубл. 20.07.2000 г.), основное действующее вещество которого - хлорин-е6, представляющий собой 18-карбокси-20-(карбоксиметил)-8-этенил-13-этил-2,3-дигидро-3,7,12,17-тетраметил-21Н,23Н-порфин-2-пропионовую кислоту. В лекарственной форме хлорин-e6 содержится в виде тринатриевой соли вместе с низкомолекулярным медицинским поливинлпирролидоном.
Другой препарат на основе фотосенсибилизатора - РАДАХЛОРИН (RU 2183956 С1, кл. A61K 31/409, А61Р 35/00, опубл. 27.06.2002 г.), являющийся смесью хлорина-е6, пурпурина-5, а также хлорина-р6.
Еще один известный препарат ФОТОДИТАЗИН (RU 2144538 С1, кл. C07D 487/22, A61K 31/40, А61Р 35/00, опубл. 20.01.1998 г.) представляет собой бис-N-метил-D-глюкаминовую соль хлорина-е6 в виде водного раствора, содержащего поливинилпирролидон. Выпускается в виде концентрата и геля-пенетратора.
Кроме этого, эффективным агентом ФДТ является японский препарат NPe6 (Talaporfin) (US 2008254570 А1, кл. H01L 21/00, опубл. 16.10.2008 г.), представляющий собой натриевую соль N-аспартил-хлорина-e6.
Одной из важнейших характеристик фотосенсибилизатора является однородность состава. Это связано с тем, что наибольшими квантовыми выходами синглетного кислорода обладают соединения на основе хлорина-е6, а примесные компоненты зачастую менее активны.
Препараты ФОТОЛОН, ФОТОДИТАЗИН, РАДАХЛОРИН и NPe6 в настоящее время активно применяются для лечения различных онкологических заболеваний, однако они обладают рядом недостатков, такими как неоднородность состава и низкая селективность накопления в опухолевой ткани, что заметно снижает их общую эффективность. Препараты ФОТОЛОН, ФОТОДИТАЗИН, РАДАХЛОРИН и NPe6 являются водными растворами, состоящими из основного действующего вещества на основе хлорина-е6, а также ряда различных добавок. Количество и состав добавок может варьироваться в различных пределах. Наиболее химически однородный агент - ФОТОДИТАЗИН, однако, он имеет ограниченный срок хранения (порядка 1 года) и только в холодильной камере. Поэтому создание однородного по составу агента ФДТ является перспективным направлением создания новых лекарственных препаратов. Также препараты ФОТОЛОН, ФОТОДИТАЗИН, РАДАХЛОРИН и NPe6 представляют собой фотосенсибилизирующие агенты 2 поколения, главным недостатком которых служит низкая селективность распределения между здоровой и опухолевой тканями [Materials 2015, 8(7), 4421-4456].
Для повышения селективности накопления фотосенсибилизатора в опухолях создаются фотоактивные гибридные молекулы-конъюгаты с адресными (направляющими) фрагментами.
Например, показано, что молекулы, имеющие в своем составе адресные молекулы, такие как антитела, сиалиловые кислоты, биотин, некоторые пептиды, селективно связывающиеся с опухолевыми клетками, могут значительно снизить токсичность и повысить активность препарата на их основе за счет избирательного связывания с онкоэкспрессируемыми рецепторами на поверхности клеток [J. Pharm. Sci. 103 (2014) 71-77; J. Mater. Chem. В 4 (2016) 6758-6772].
Известен препарат на основе конъюгата хлорина-е6, связанного с белком трансферрином, рецепторы которого сверхэкспрессируются опухолевыми тканями (US 20020137901 А1, кл. C07K 14/79, A61K 38/00, A61K 41/00, C07K 14/435,опубл. 26.09.2002).
Также известен препарат на основе конъюгата хлорина-е6, связанного с фолиевой кислотой, которая позволяет провести доставку фотоактивной части непосредственно к опухолевым клеткам за счет связывания с фолатными рецепторами на их поверхности (US 2012059018 A1, A61K 31/519, A61P 35/00,C07D 487/22, опубл. 08.03.2012 г.).
Также разработано производное хлорина-е6, ковалентно соединенное с олигонуклеотидами, показывающее высокую эффективность связывания со злокачественными новообразованиями (US 20060105974 A1, кл. A61K 48/00, A61K 38/00, A61K 41/00, A61K 47/48, С07Н 21/00, С07Н 21/04, C12N 15/113, опубл. 18.05.2006).
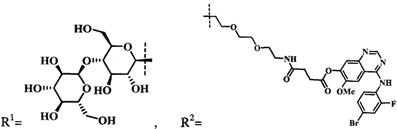
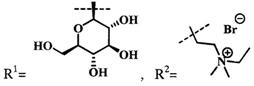
Среди существующих конъюгатов ФДТ наиболее близким к предлагаемому изобретению является конъюгат металлокомплекса пурпуринимида с моно- и дисахаридами (US 2002198157 A1, кл. А61Р 35/00, С07Н 15/26, С07Н 23/00, опубл. 26.12.2002 г.). Данные конъюгаты состоят из фотоактивного ядра на основе металлокомплекса пурпуринимида, связанных с углеводными фрагментами (фиг. 1). Вышеописанные конъюгаты были разработаны в Roswell Park Cancer Institute в группе доктора Pandey. Структуры данных молекулы были выбраны исходя из нацеленности на терапевтическую мишень в виде Галектина-3, экспрессируемого некоторыми опухолевыми клетками. Был синтезирован ряд соединений с различными молекулами углеводов, соединенных с пурпуринимидом, также выбраны несколько типов линкеров для их связи. Показано, что биологическая активность сильно зависит от природы углевода и типа линкера. Например, молекулы, не содержащие углеводных остатков демонстрируют уменьшение активности по сравнению с аналогами с галактозой и лактозой.
Соединение 4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7-ол является активной частью лиганда рецепторов факторов роста эпидермиса (EGFR) и эндотелия сосудов (VEGFR), а так же эффективным ингибитором тирозинкиназ, что делает его потенциально удобным агентом доставки и самостоятельным химиотерапевтическим агентам [J. Med. Chem. 2012, 55, 10797; Clin. Ther. 2011, 33, 315; Clin. Ther. 2012, 34, 221].
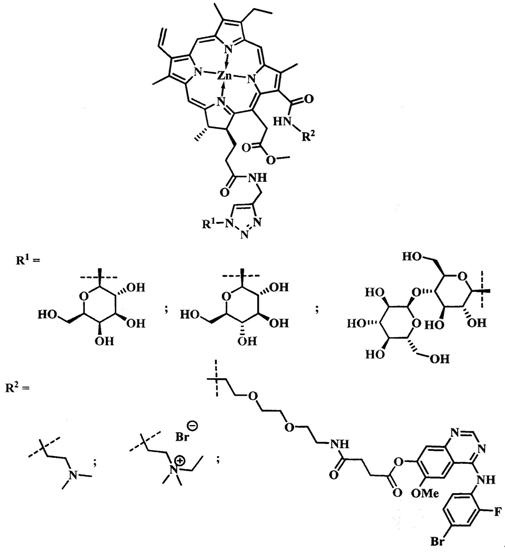
В задачу изобретения положено создание нового производного цинкового металлокомплекса хлорина-е6, связанного посредством химических связей с 4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7-олом, улучшающего биораспределение фото-сенсибилизатора за счет связывания с рецепторами EGFR/VEGFR на поверхности опухолевых клеток, а также его применение в качестве агента для фотодинамической терапии.
Техническим результатом от использования предлагаемой группы изобретений является повышение однородности, улучшение водорастворимости, увеличение селективности накопления опухолевыми тканями по сравнению со здоровыми, снижение системной токсичности.
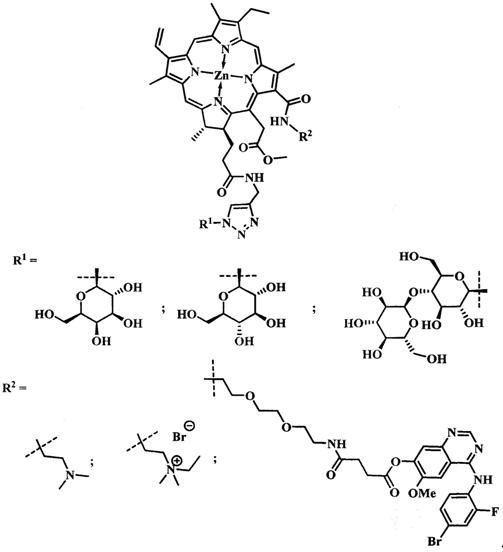
Поставленная задача достигается тем, что производное цинкового металлокомплекса хлорина-е6 имеет общую формулу:
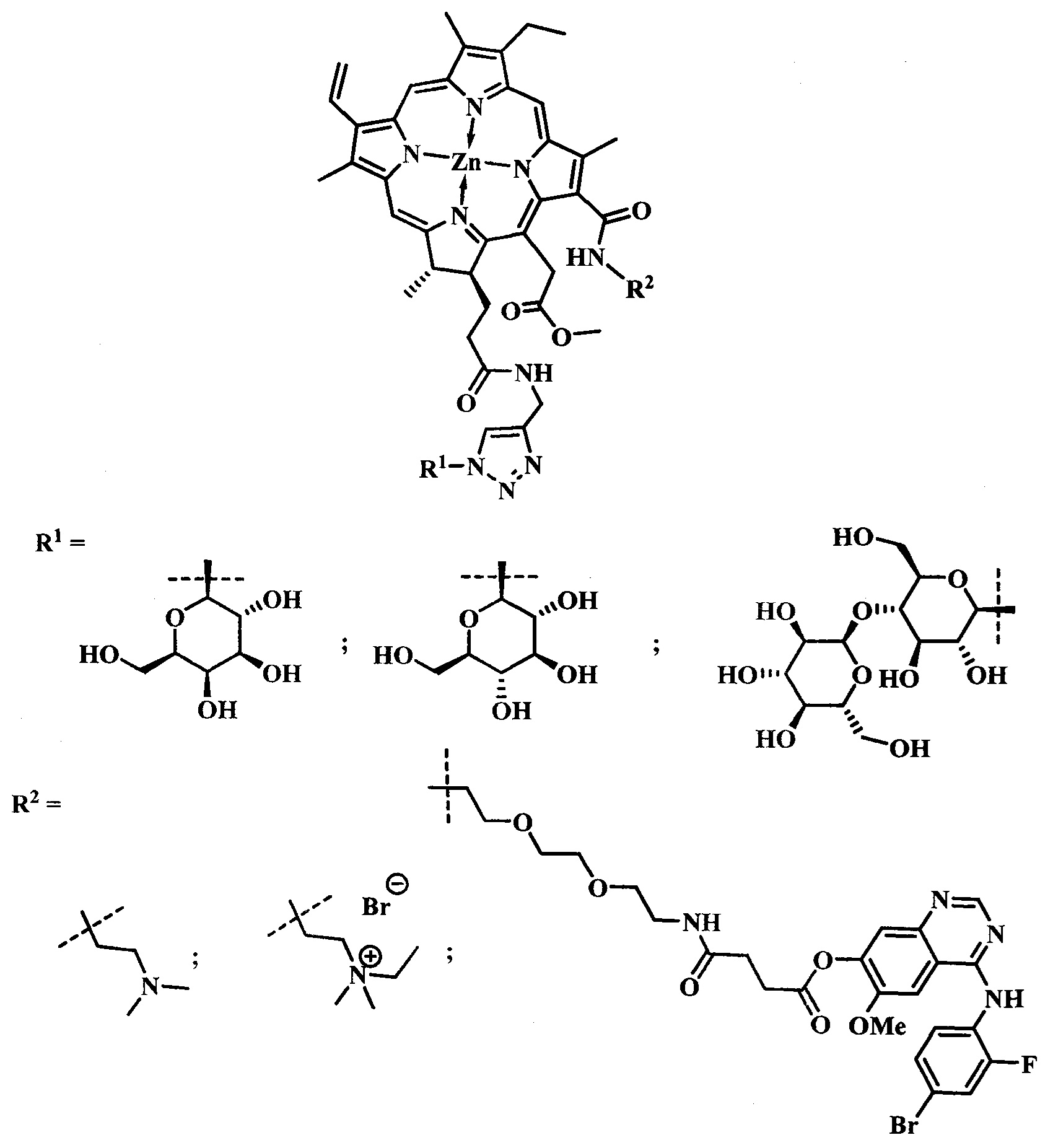
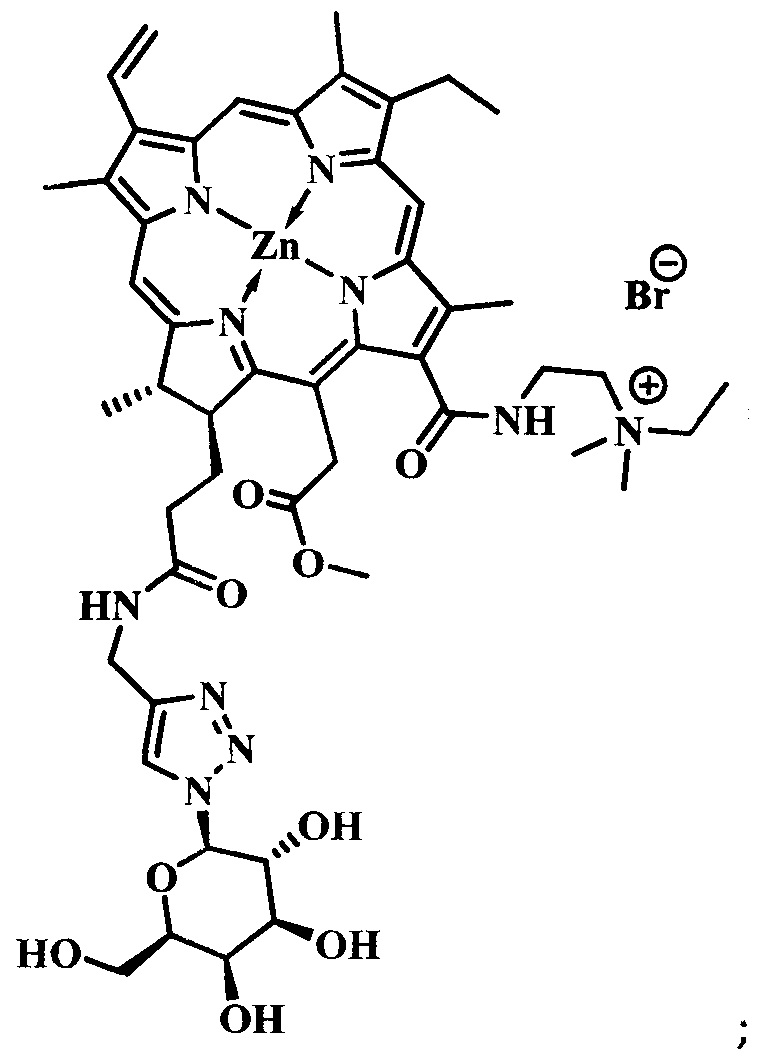
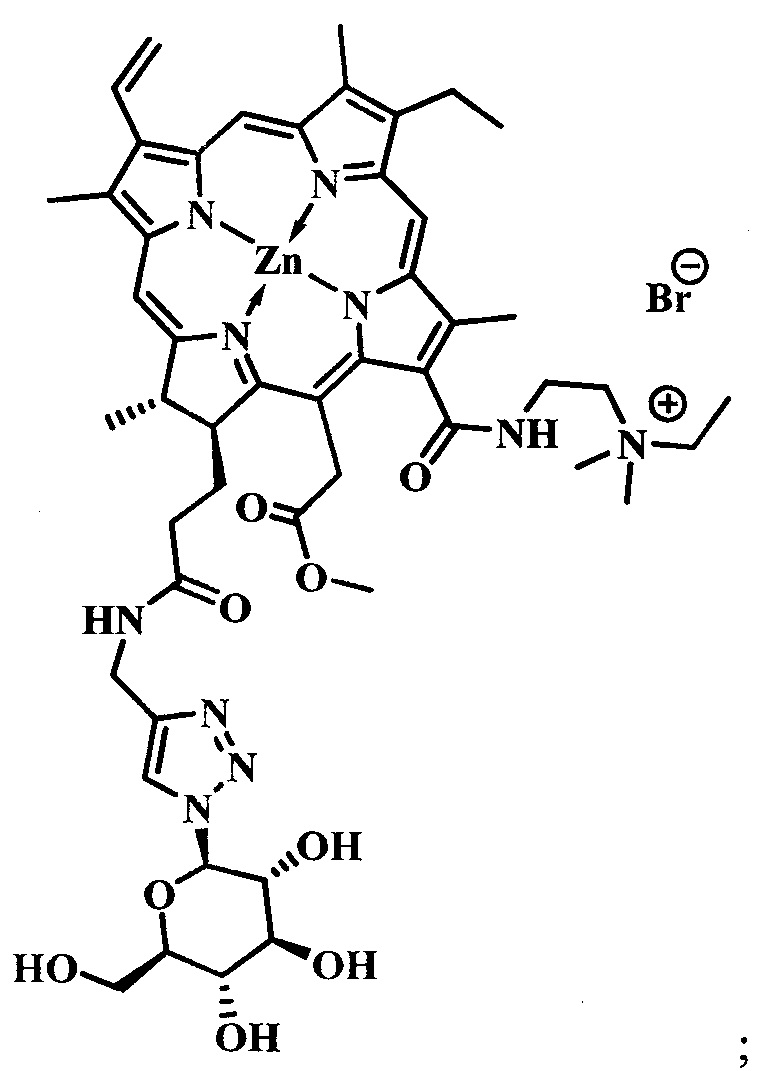
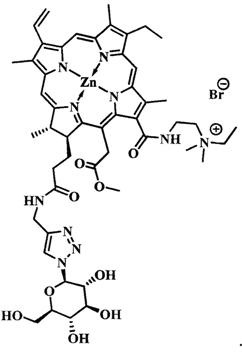
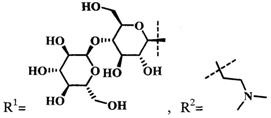
при 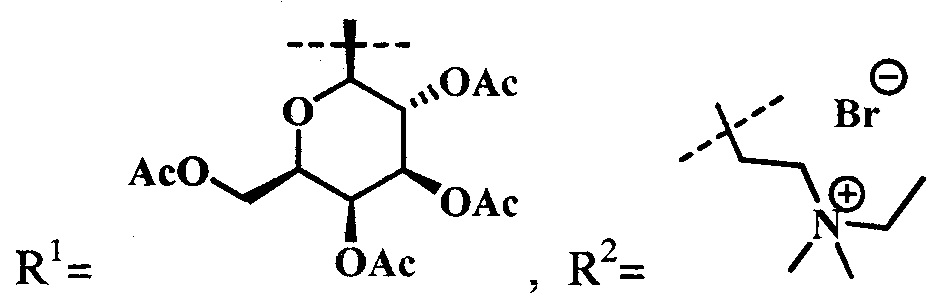 производное представляет собой бромид цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6, общей формулы:
производное представляет собой бромид цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6, общей формулы:
при 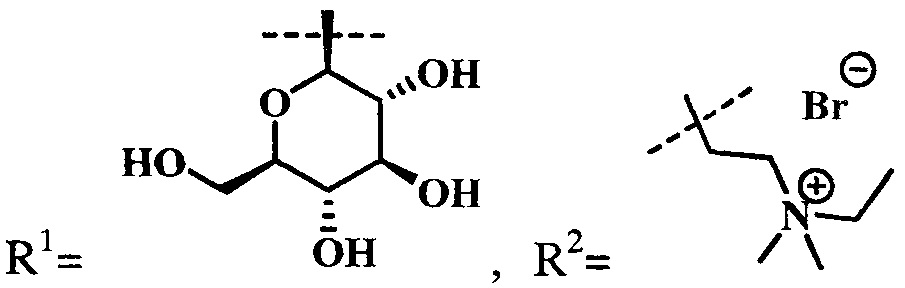 производное представляет собой бромид цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 общей формулы:
производное представляет собой бромид цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 общей формулы:
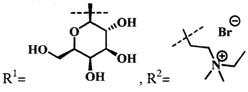
при 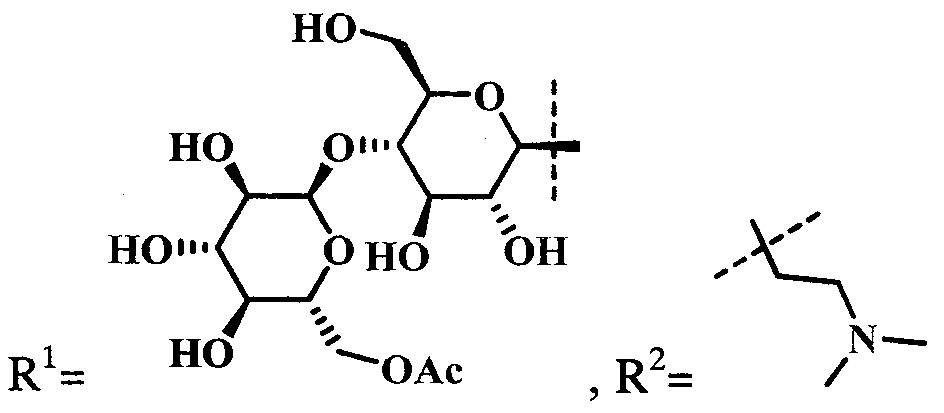 производное представляет собой цинковый комплекс 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 общей формулы:
производное представляет собой цинковый комплекс 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 общей формулы:
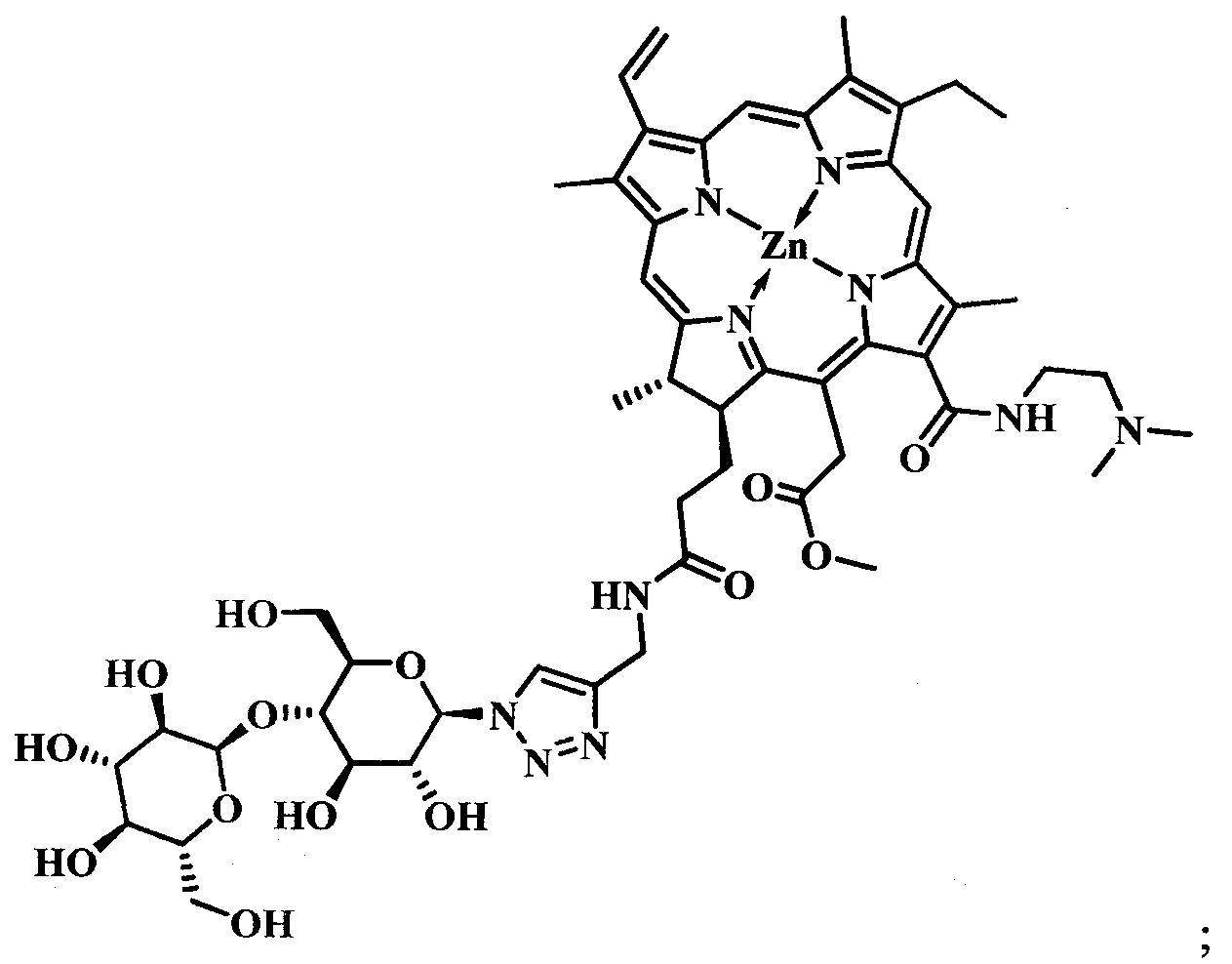
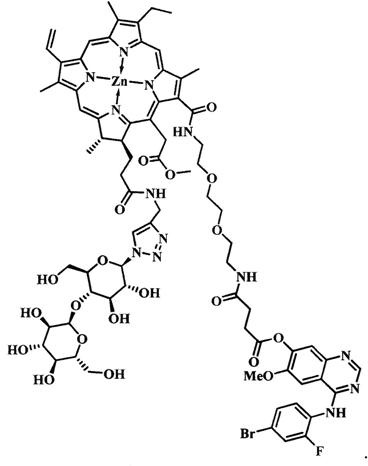
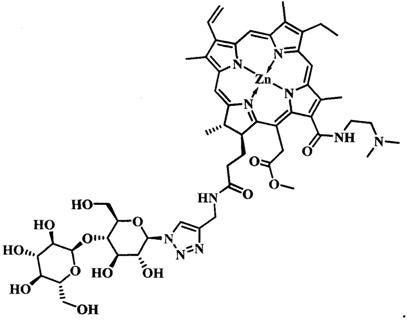
при 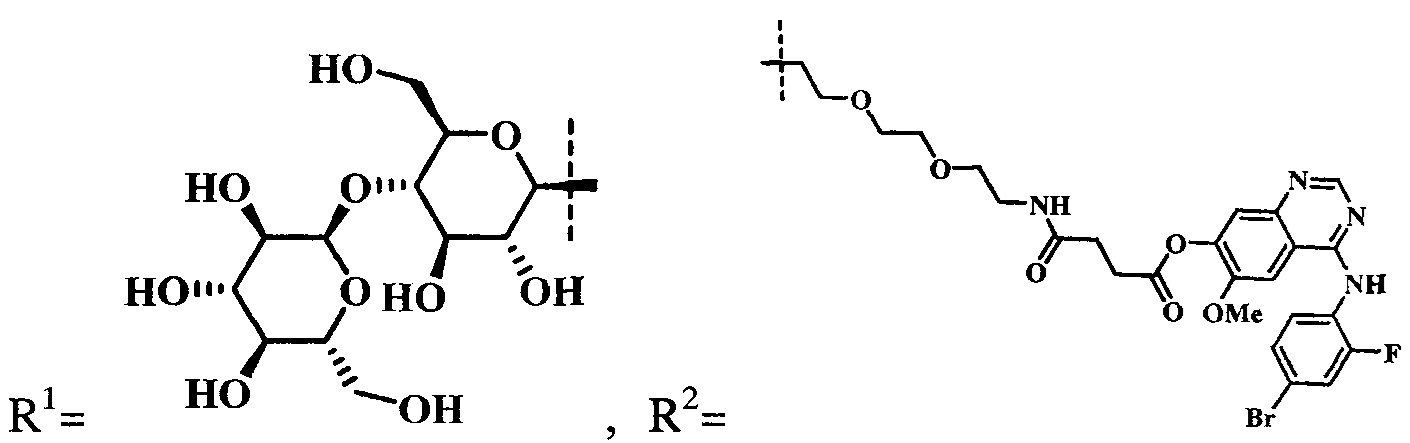 представляет собой цинковый комплекс 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)этил))амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-e6 общей формулы:
представляет собой цинковый комплекс 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)этил))амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-e6 общей формулы:
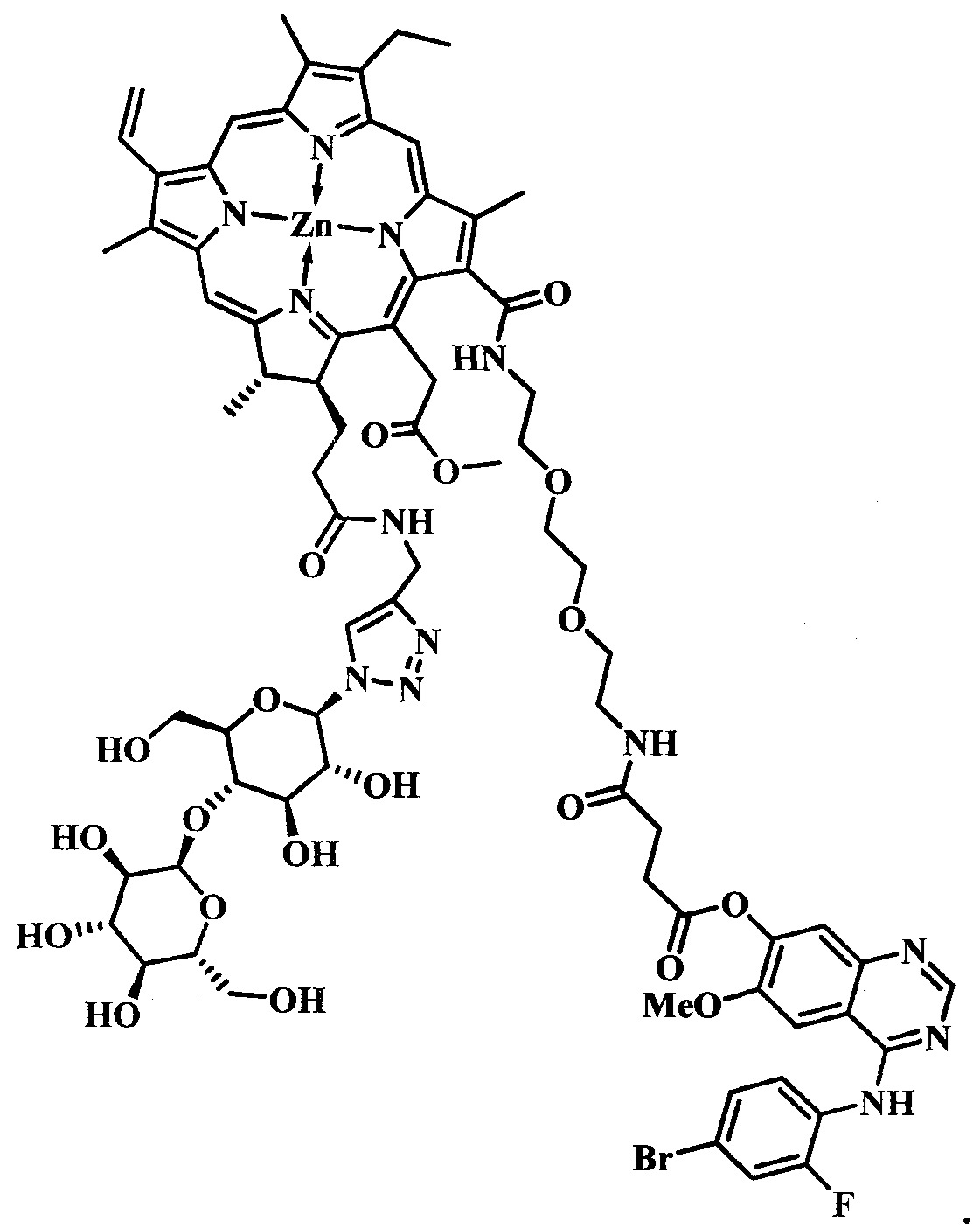
Поставленная задача достигается также тем, что полученное производное применяют в качестве агента для фотодинамической терапии.
На фиг. 1 представлены примеры синтезированных конъюгатов пурпуринимида-р6 с моно- и ди-сахарами.
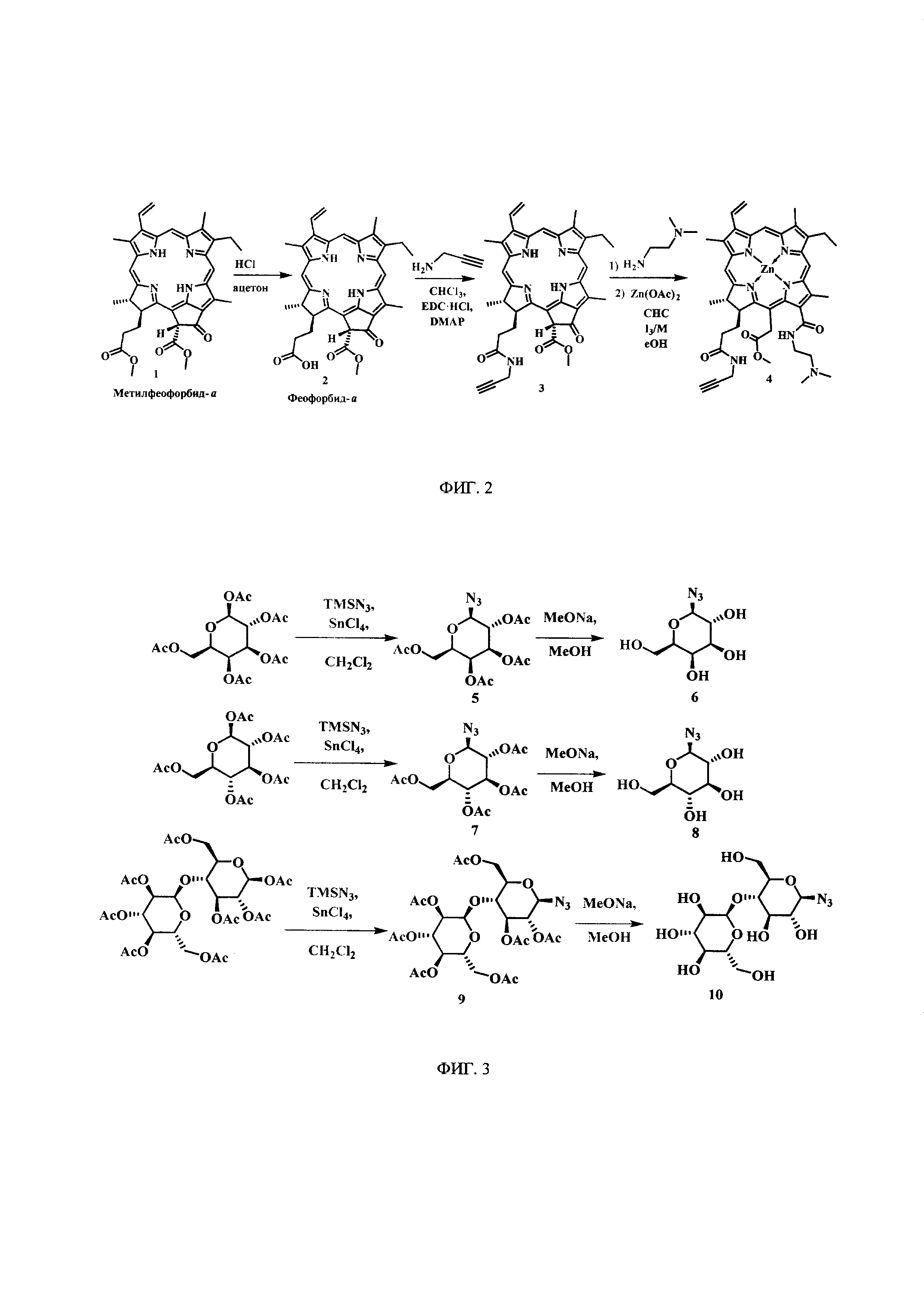
На фиг. 2 представлены схемы синтеза Феофорбид-а (2), пропаргиламидфеофорбида-а (3), цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-пропаргиламидхлорина-е6 (4).
На фиг. 3 представлены схемы синтеза 2,3,4,6-тетра-O-ацетил-β-D-галактопиранозил азида (5), β-D-галактопиранозил азида (6), 2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил азида (7), β-D-глюкопиранозил азида (8), гепта-О-ацетил-β-мальтозазида (9), β-мальтозазида (10).
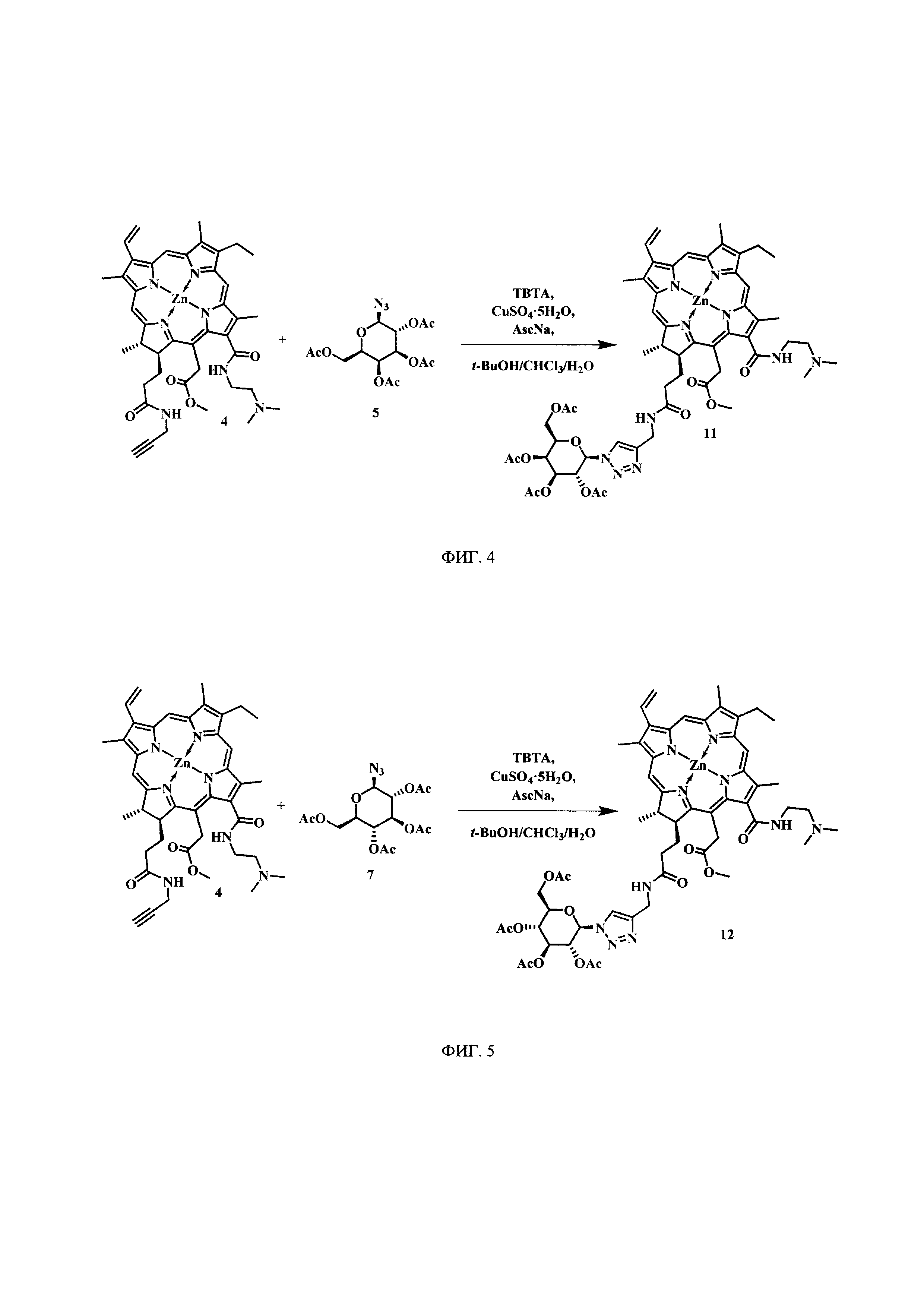
На фиг. 4 представлена схема синтеза цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(тетра-O-ацетил-1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (11).
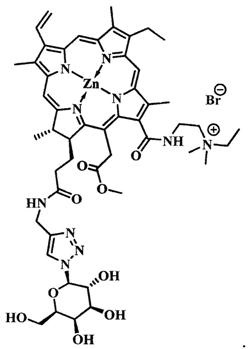
На фиг. 5 представлена схема синтеза цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(тетра-O-ацетил-1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (12).
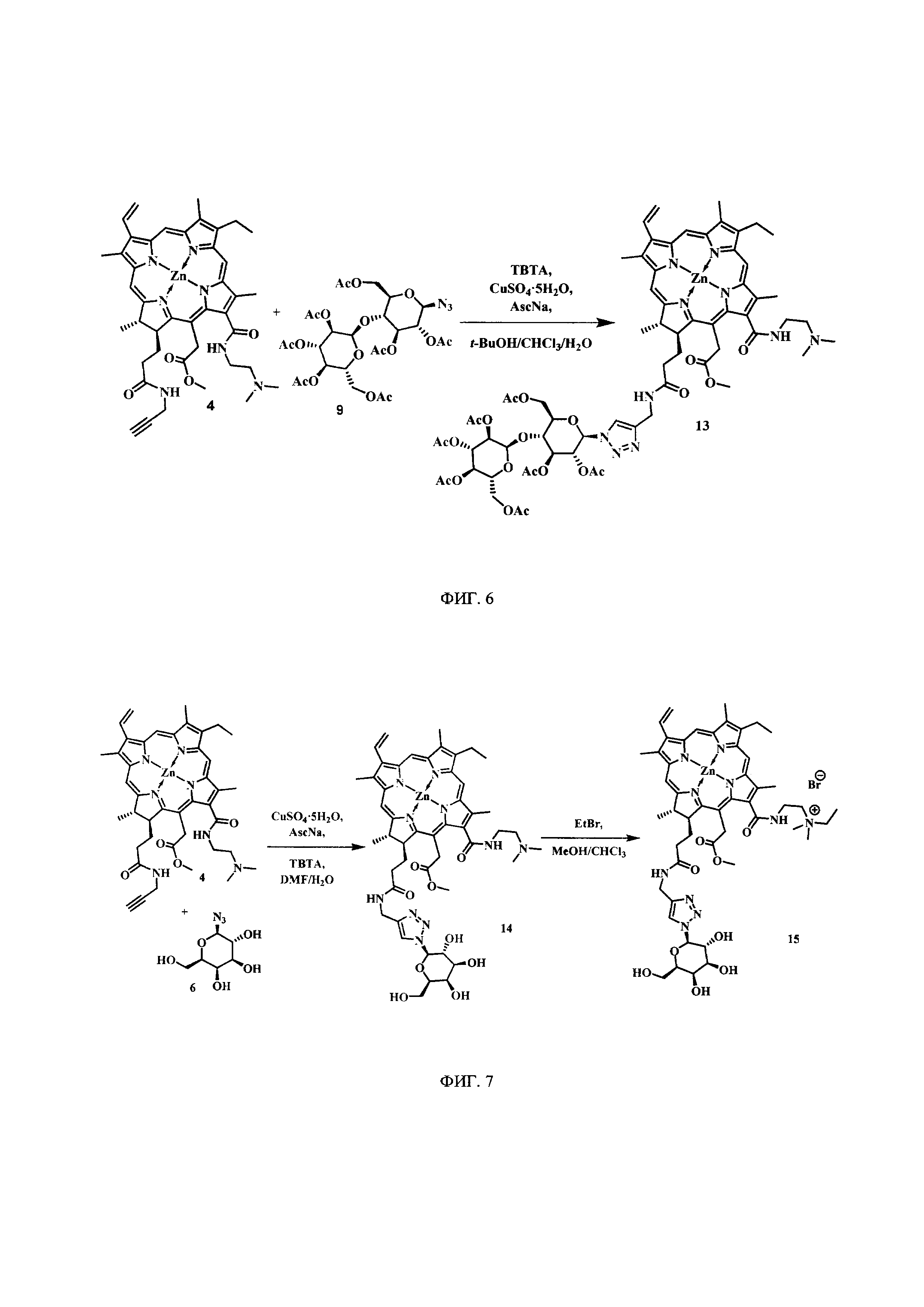
На фиг. 6 представлена схема синтеза цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(гепта-O-ацетил-1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (13).
На фиг. 7 представлена схема синтеза цинкового комплекса 13-(диметил аминоэтил)амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (14), бромида цинкового комплекса 13-(триметиламмонийэтил) амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (15).
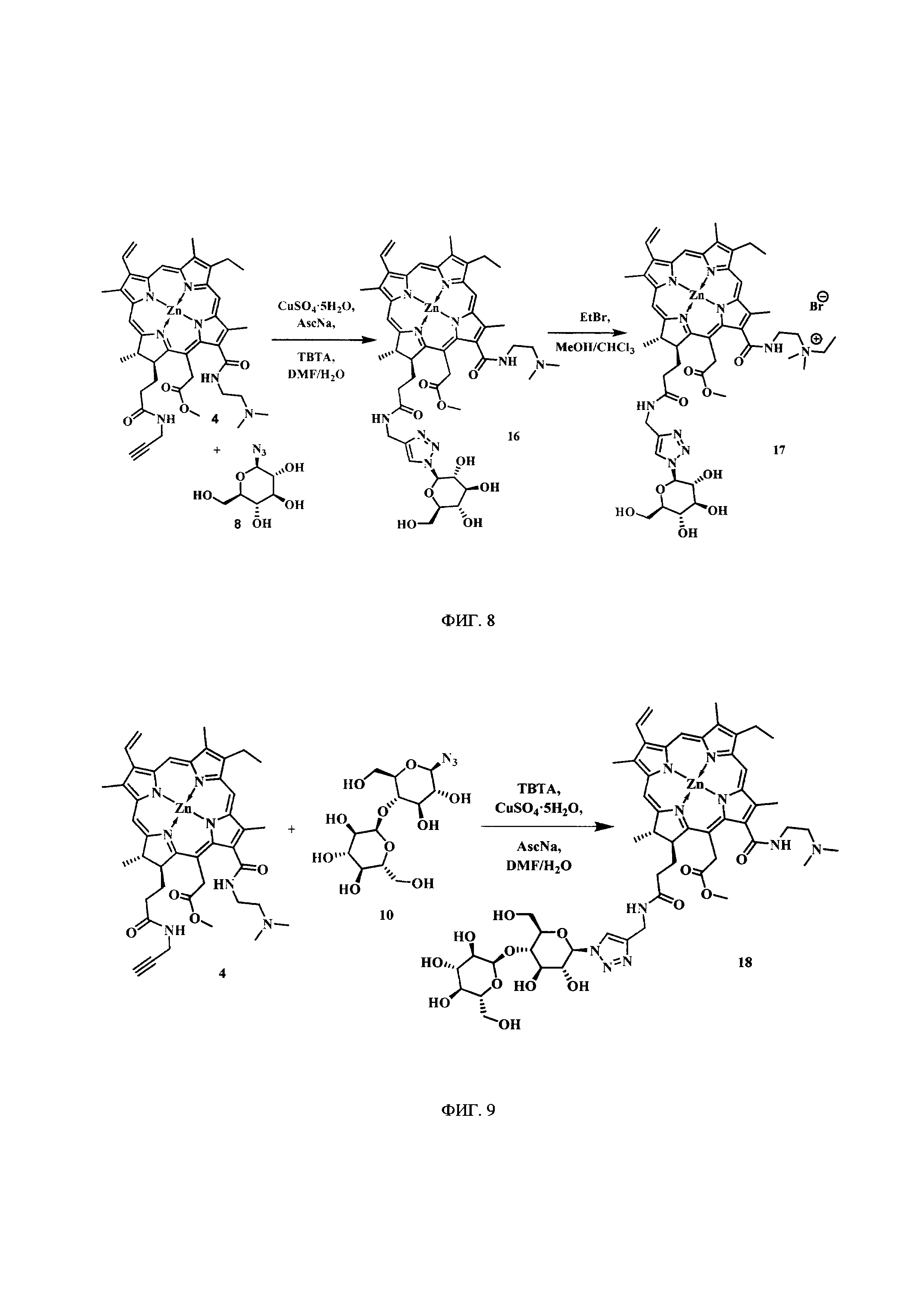
На фиг. 8 представлена схема синтеза цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (16), бромида цинкового комплекса 13-(диметилэтиламмоний этил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (17).
На фиг. 9 представлена схема синтеза цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (18).
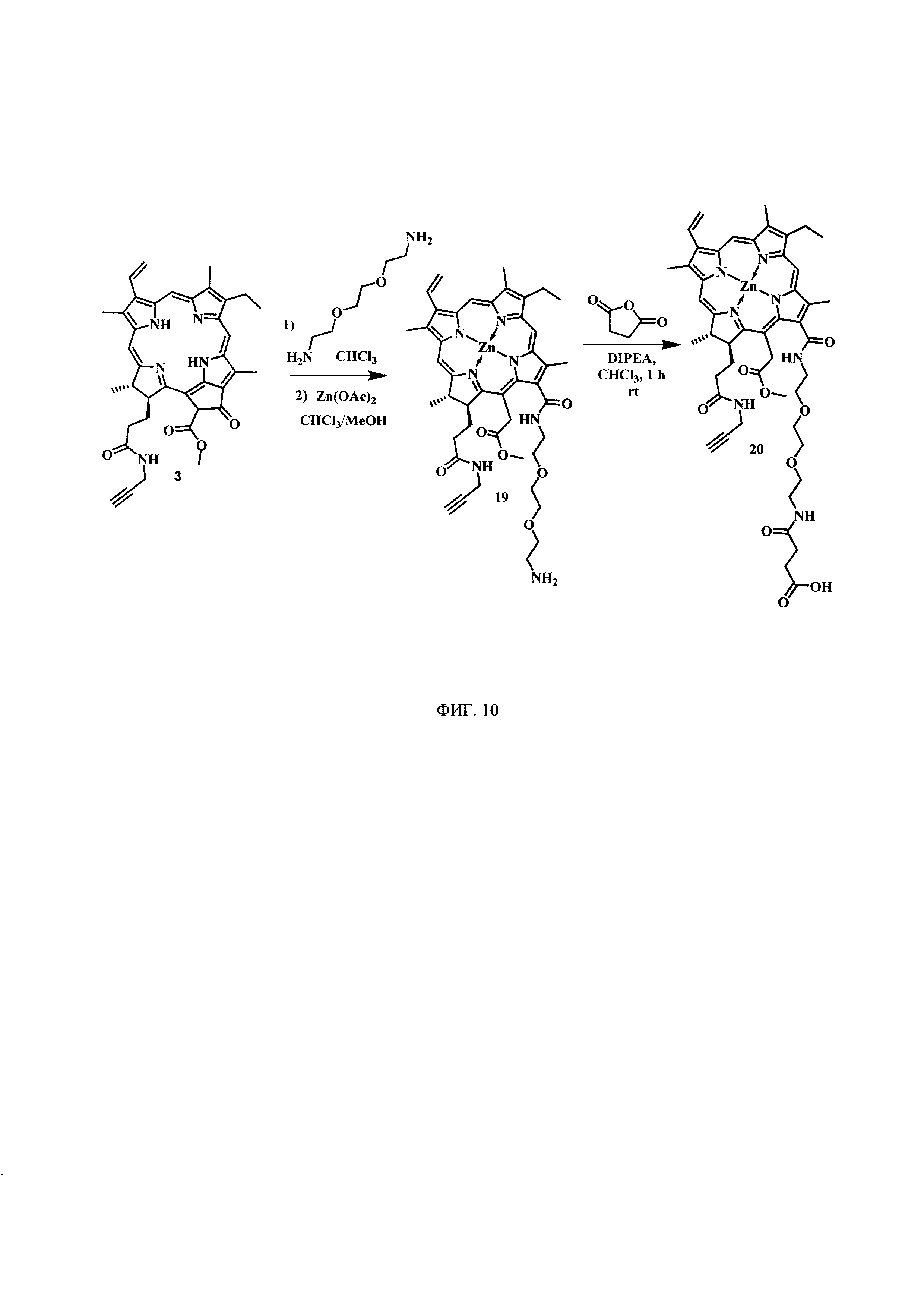
На фиг. 10 представлена схема синтеза 13-(N-(2-(2-(2-аминоэтокси)этокси)этил))амид-17(3)-пропаргиламидхлорина-е6 (19), цинкового комплекса 13-(N-(2-(2-(2-N-(3-карбокси-1-оксопропил)-аминоэтокси)этокси)этил))амид-17(3)-пропаргиламидхлорина-e6 (20).
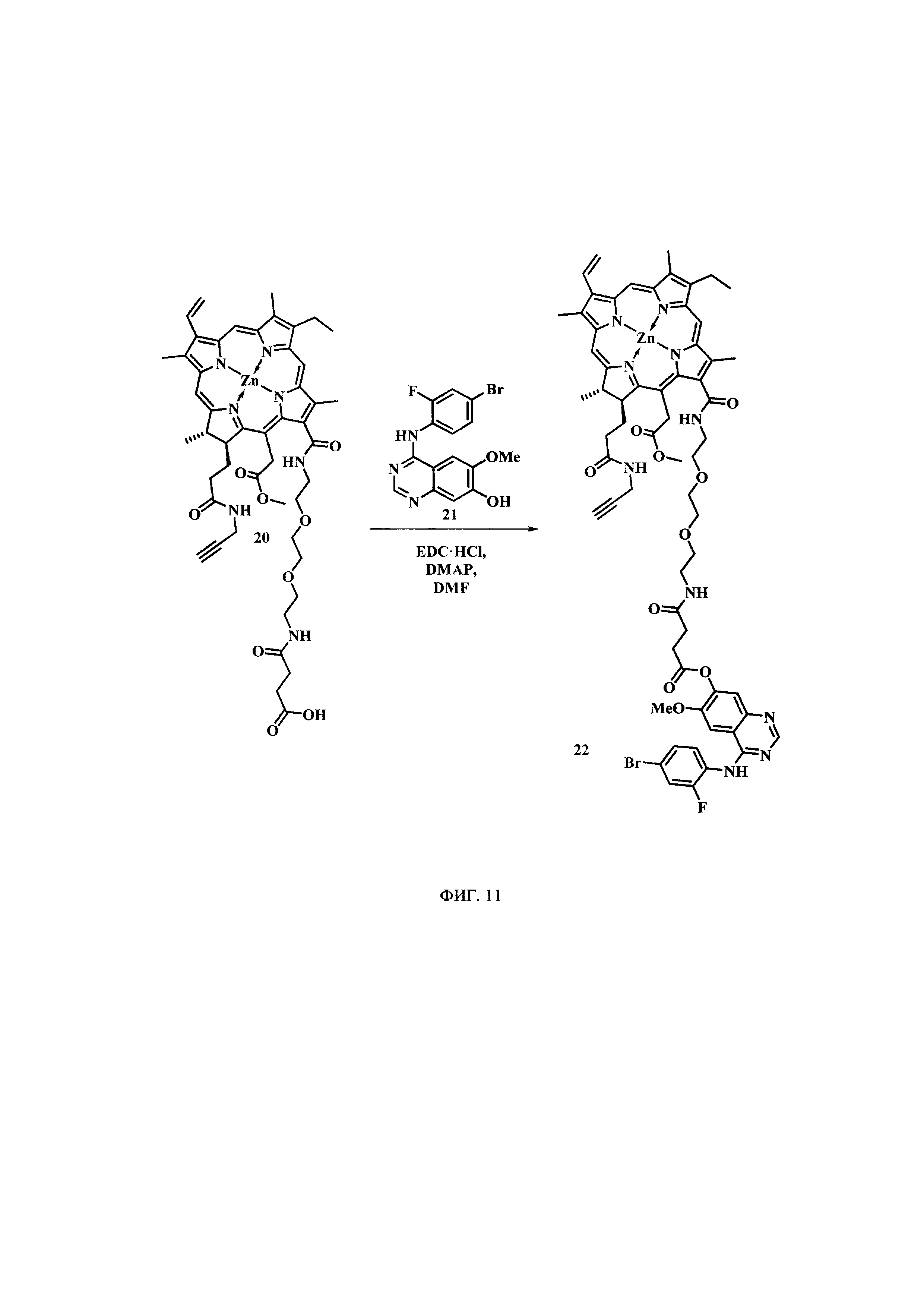
На фиг. 11 представлена схема синтеза цинкового комплекса 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)-этил))амид-17(3)-пропаргиламидхлорина-е6 (22).
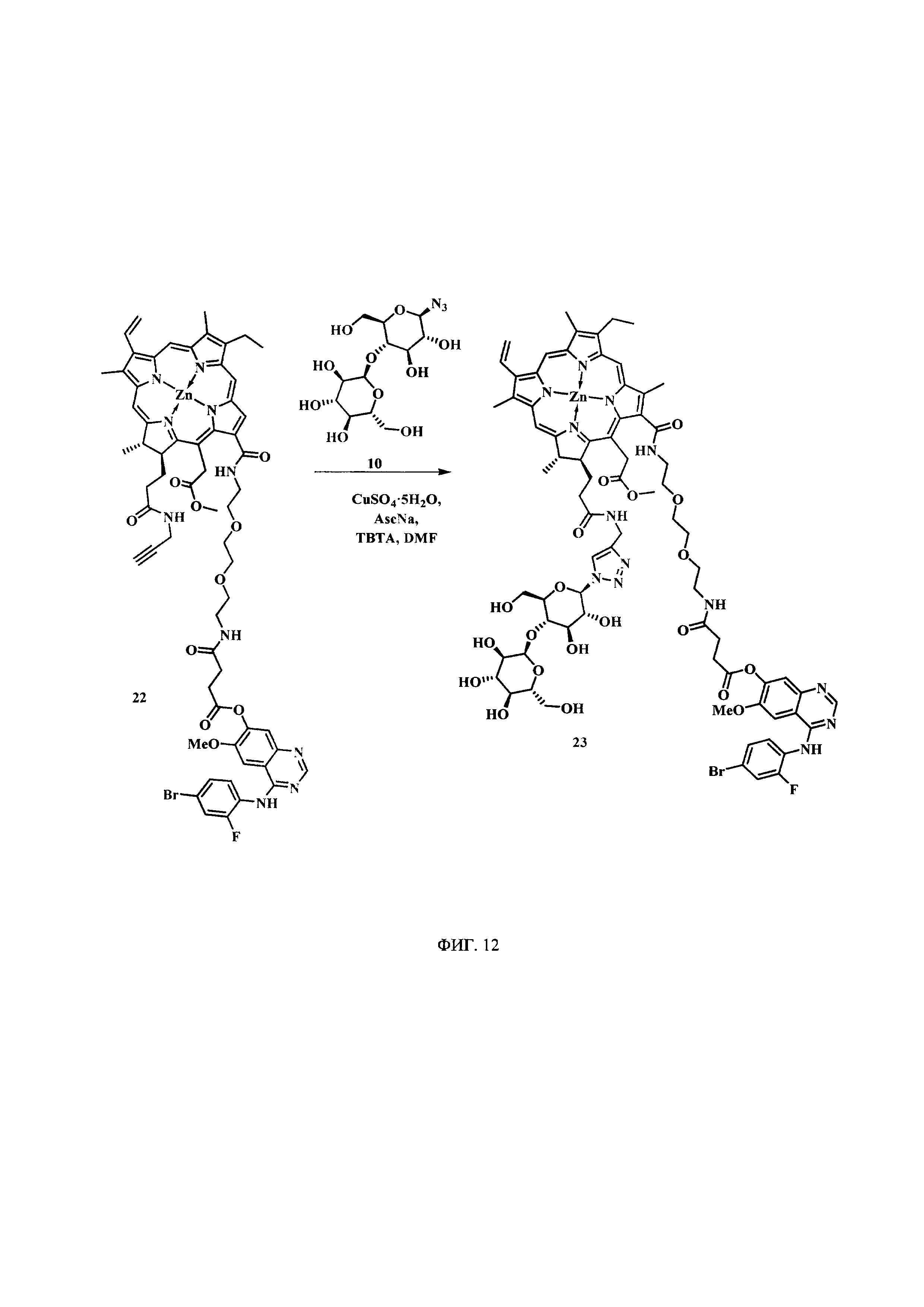
На фиг. 12 представлена схема синтеза цинкового комплекса 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)этил))амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (23).
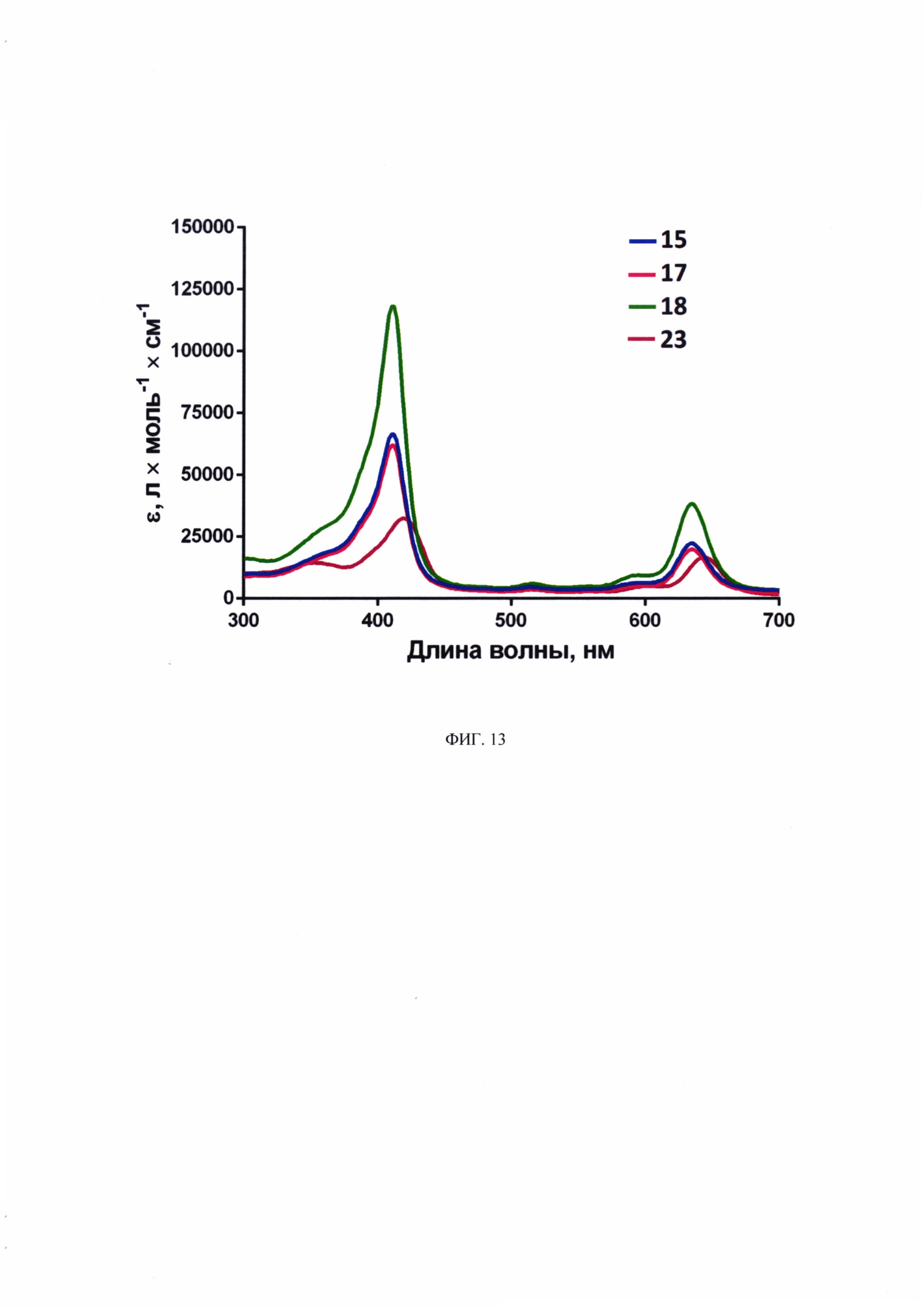
На фиг. 13 представлены представлены спектры поглощения соединений 15, 17, 18, 23 в воде, концентрация 5 мкМ.
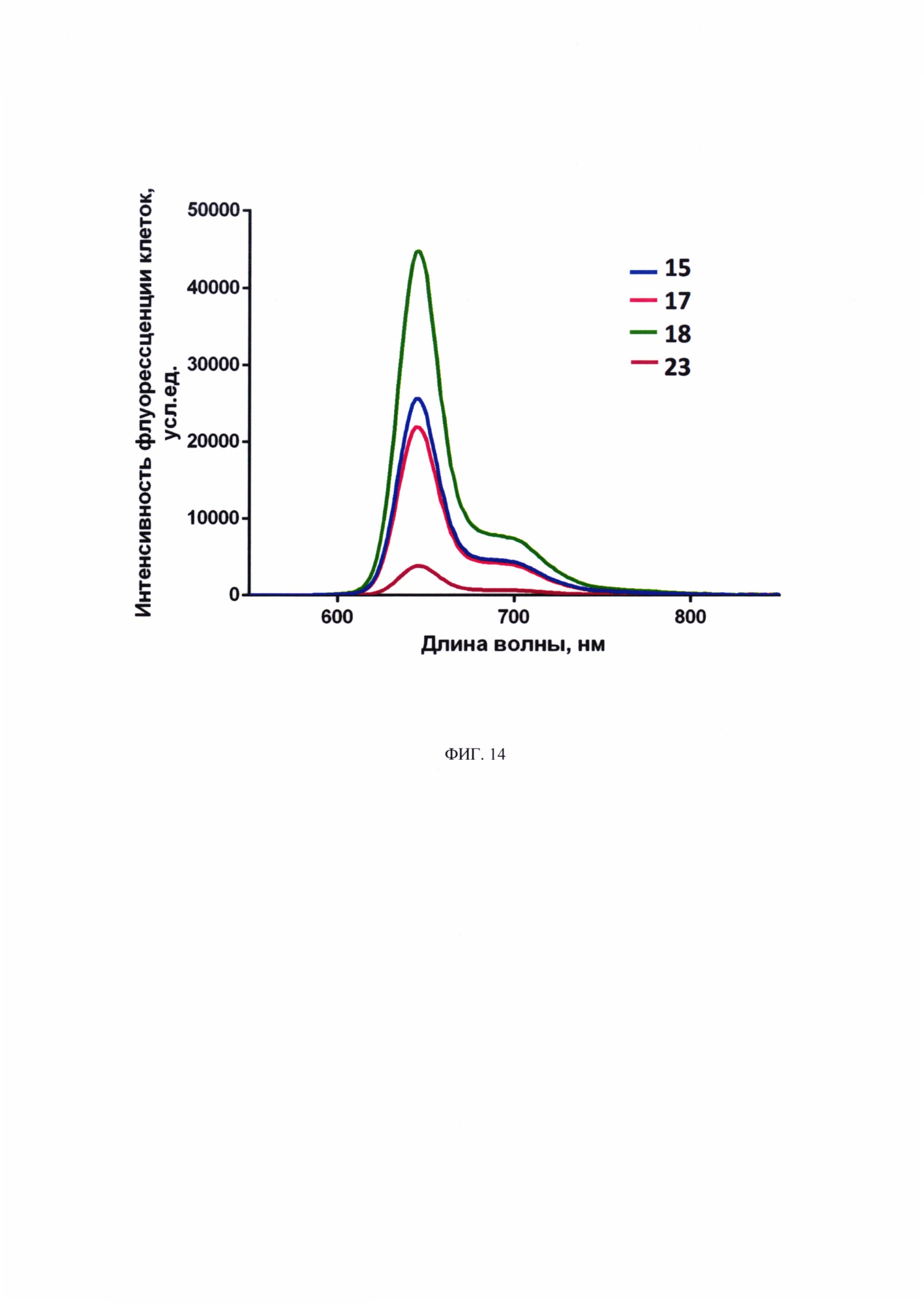
На фиг. 14 представлены спектры флуоресценции соединений 15, 17, 18, 23 в воде, концентрация 5 мкМ.
На фиг. 15 представлена внутриклеточная локализация соединений 15, 17, 18, 23 в клетках линий А431 и СНО. Концентрация 5 мкМ, инкубация 4 часа. λEx.=450 нм; λEm.=600-740 нм.
На фиг. 16 представлена интенсивность флуоресценции клеток после инкубации с соединениями 15, 17, 18, 23 в концентрации 5 мкМ в течение 4 часов.
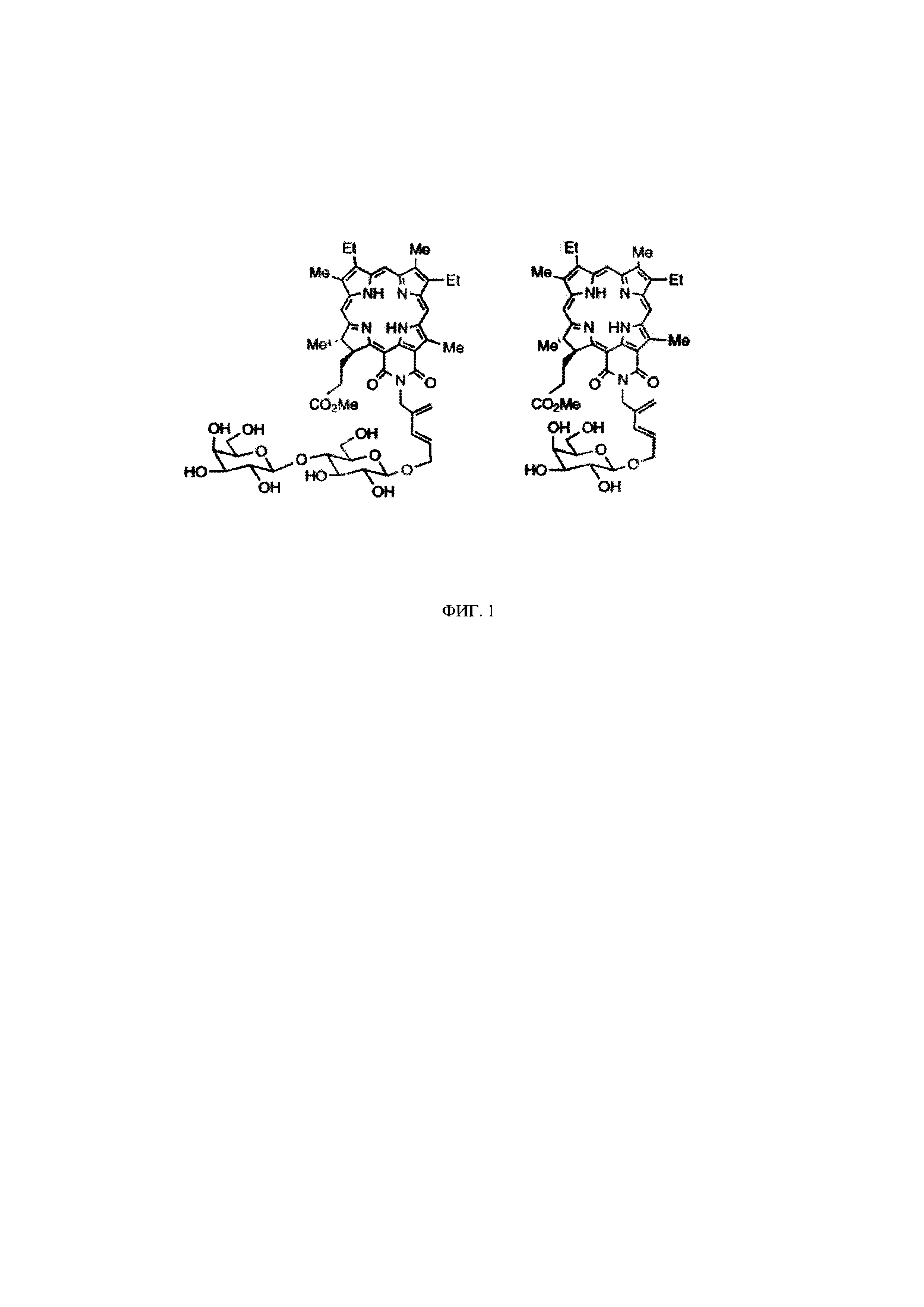
На фиг. 17 представлена зависимость жизнеспособности культур клеток А431 и СНО от концентрации соединений 15, 17, 18 в темноте и при облучении в дозе 20 Дж/см2.
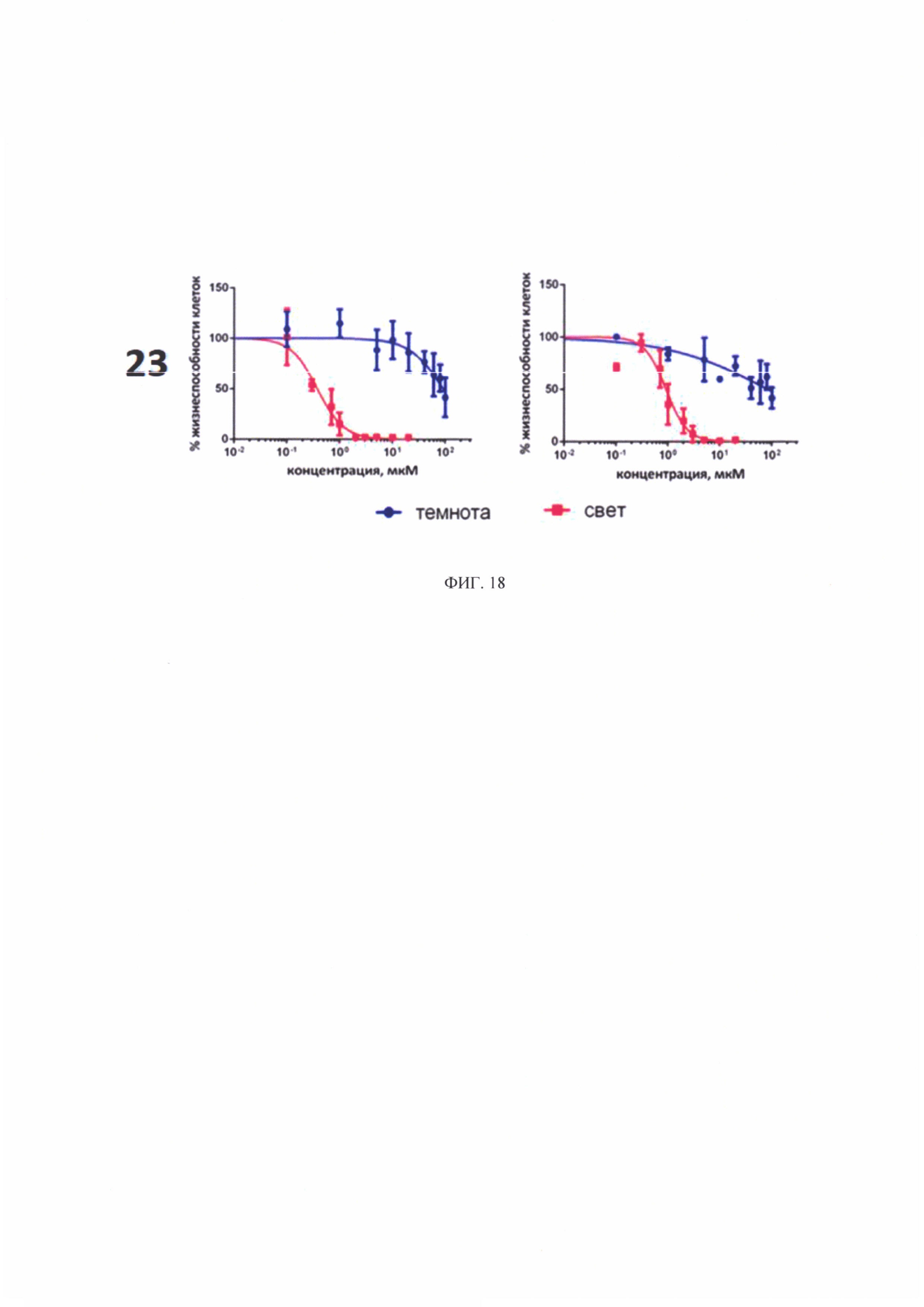
На фиг. 18 представлена зависимость жизнеспособности культур клеток А431 и СНО от концентрации соединений 23 в темноте и при облучении в дозе 20 Дж/см2.
Синтез предлагаемого производного цинкового металлокомплекса хлорина-е6 осуществляют следующим образом.
Синтез Феофорбид-а (2) (фиг. 2). В круглодонную колбу с мешалкой помещали 0,500 г метилфеофорбида-а 1 и добавляли 15 мл ацетона. Далее по каплям приливали 0,7 мл концентрированной соляной кислоты. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 48 часов. Растворитель удаляли при пониженном давлении. Остаток растворяли в 30 мл хлороформа, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 5% МеОН - 95% CHCl3) феофорбид-а (2) получают в виде темно-зеленого порошка (0,430 г, 89%). Соединение известно [J. Org. Chem. 45 (1980) 1969-1674].
Синтез пропаргиламидфеофорбида-а (3) (фиг. 2). В круглодонную колбу «А» с мешалкой в атмосфере аргона поместили 0,430 г (1 эквивалент) феофорбида-а 2, 0,284 г (2 эквивалента) гидрохлорида N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDC⋅HCl) и 8 мл CHCl3. Перемешивали при 0°С в течение 30 мин. В круглодонной колбе «Б» с мешалкой смешали 0,081 г (2 эквивалента) пропаргиламина, 0,045 г (0,5 эквивалента) DMAP и 8 мл хлороформа, смесь перемешивали 10 минут при 0°С. Затем шприцем перенесли содержимое колбы «Б» в колбу «А» и перемешивали в течение 18 часов. Растворитель удаляли при пониженном давлении. Остаток растворили в 30 мл хлороформа, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 2% МеОН - 98% CHCl3) пропаргиламид феофорбида-а (3) выделен в виде темно-зеленого порошка (0,395 г, 87%).
Спектр 1Н ЯМР (400 MHz, CDCl3, δ, м.д.): 9.49 (br.s, 2Н), 8.63 (s, 1H), 7.98 (dd, J=17.5, 11.5 Hz, 1H), 6.30 (d, J=18.2 Hz, 1H), 6.27 (s, 1H), 6.20 (d, J=19.9 Hz), 5.36 (br. s, 1H), 4.53 (br. s, 1H), 4.27 (br. s, 1H), 3.87 (s, 3H), 3.78-3.55 (m, 7H), 3.41 (s, 3H), 3.25 (s, 3H), 2.70 (br.s, 1H), 2.46 (br. s, 1H), 2.33-2.15 (m, 2H), 1.94 (s, 1H), 1.83 (d, J=6.6 Hz, 3H), 1.68 (t, J=7.6 Hz, 3H), -1.66 (br. s, 2H).
Спектр 13C ЯМР (101 MHz, CDCl3, δ, м.д.): 189.36, 172.27, 171.76, 169.56, 137.93, 128.86, 104.64, 104.26, 97.59, 79.16, 64.77, 52.97, 51.52, 50.25, 29.69, 28.83, 23.31, 19.55, 17.19, 12.16, 11.26.
MS (MALDI): m/z 630.1 [M]+.
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-пропаргиламидхлорина-е6 (4) (фиг. 2). В круглодонную колбу с мешалкой поместили 0,395 г соединения 3 и добавили 16 мл хлороформа, затем к смеси по каплям прибавляли 2 мл N,N-диметилэтилендиамина. Перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении. Остаток растворяли в 20 мл хлороформа, экстрагировали водой (3×50 мл), органическую фазу сушили над Na2SO4. Полученную органическую фазу растворили в 10 мл хлороформа и добавили по каплям диацетат цинка (0,433 г, 5 эквивалентов), растворенный в 4 мл метанола. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем растворитель удаляли при пониженном давлении. Остаток растворили в 30 мл хлороформа, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале 5% МеОН - 95% CHCl3, затем 15% МеОН - 85% CHCl3). Цинковый комплекс функционализированного хлорина-e6 (4) выделен в виде темно-зеленого порошка (0,402 г, 82%).
Спектр 1Н ЯМР (400 MHz, CDCl3, δ, м.д.): 9.51 (s, 2Н), 8.96 (br.s, 1H), 8.66 (s, 1H), 8.34 (t, J=5.4 Hz, 1H), 8.22 (dd, J=17.8, 11.6 Hz, 1H), 6.22 (d, J=17.9 Hz, 1H), 6.00 (d, J=11.6 Hz, 1H), 5.41 (d, J=19.1 Hz, 1H), 5.07 (d, J=18.9 Hz, 1H), 4.42 (q, J=7.0 Hz, 1H), 4.20 (d, J=9.9 Hz, 1H), 3.91-3.75 (m, 6H), 3.69 (br.s, 4H), 3.37 (s, 3H), 3.35 (s, 3H), 3.26-3.16 (m, 3H), 3.05 (t, J=2.3 Hz, 1H), 2.73 (br.s, 6H), 2.47-2.38 (m, 1H), 2.07 (d, J=8.8 Hz, 2H), 1.67 (t, J=7.5 Hz, 3H), 1.61 (d, J=7.0 Hz, 3H).
Спектр 13C ЯМР (101 MHz, CDCl3, δ, м.д.): 173.24, 171.72, 170.35, 165.14, 162.77, 151.66, 147.96, 146.14, 144.07, 143.10, 141.20, 140.74, 138.58, 137.23, 133.04, 132.29, 130.66, 119.20, 101.79, 101.75, 99.87, 93.16, 81.19, 72.81, 52.24, 51.65, 46.26, 43.88, 37.46, 32.02, 29.99, 27.78, 22.80, 18.87, 17.89, 12.30, 11.76, 10.93.
MS (MALDI): m/z 779.0 [M]+.
Синтез 2,3,4,6-тетра-O-ацетил-β-D-галактопиранозил азида (5) (фиг. 3). В колбу Шлейка с мешалкой и заполненную аргоном поместили 2 г пентаацетат β-D-галактозы и добавили 20 мл безводного дихлорметана, затем к смеси по каплям прибавляли 1 Моль/л раствор четыреххлористого олова в дихлорметане (2,56 мл, 0,5 эквивалента), затем триметилсилилазид (0,82 мл, 1,2 эквивалента). Перемешивали при 0°С в течение 3 ч. Затем реакционная смесь была разбавлена 80 мл дихлорметана и промыта насыщенным раствором NaHCO3 (70 мл), водой (50 мл) и насыщенным раствором поваренной соли (50 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: 50% ПЭ - 50% EtOAc) 2,3,4,6-тетра-O-ацетил-β-D-галактопиранозил азид (5) выделен в виде белого кристаллического порошка (1,8 г, 95%).
Спектр 1Н ЯМР (300 MHz, CDCl3, δ, м.д.): 5.42 (dd, J=3.3, 1.0 Hz, 1H), 5.16 (dd, J=10.4, 8.7 Hz, 1H), 5.03 (dd, J=10.3, 3.3 Hz, 1H), 4.59 (d, J=8.7 Hz, 1H), 4.17 (dd, J=6.5, 2.4 Hz, 2H), 4.05-3.98 (m, 1H), 2.17 (s, 3H), 2.09 (s, 3H), 2.06 (s, 3H), 1.99 (s, 3H).
Спектр 13C ЯМР (75 MHz, CDCl3, δ, м.д.): 170.51, 170.25, 170.13, 169.51, 88.44, 72.99, 70.85, 68.17, 66.97, 61.36, 20.82.
Синтез β-D-галактопиранозил азида (6) (фиг. 3). В круглодонную колбу с магнитной мешалкой помещали 0,2 г 2,3,4,6-тетра-O-ацетил-β-D-галактопиранозил азида и растворяли его в 4 мл метанола. Далее по каплям прибавляли метилат натрия до образования сильнощелочной среды (рН=11), перемешивали 30 минут. Затем по каплям прибавляли в колбу концентрированную соляную кислоту до нейтральной реакции. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 20% СН3ОН - 80% CHCl3, затем 50% СН3ОН - 50% CHCl3) β-D-галактопиранозил азид (5) выделен в виде белого кристаллического порошка (0,094 г, 95%).
ИК: 2100 см-1 (полоса азидной группы).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 4.45 (d, J=6.5 Hz, 1H), 3.89 (s, 1H), 3.84-3.71 (m, 2H), 3.65 (d, J=5.8 Hz, 1H), 3.56-3.48 (m, 2H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 92.61, 78.93, 74.97, 71.98, 70.23, 62.46.
Синтез 2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил азида (7) (фиг. 3). В заполненную аргоном колбу Шленка с мешалкой поместили 2 г β-D-пентаацетат глюкозы и добавили 20 мл безводного дихлорметана, затем к смеси по каплям прибавляли 1 Моль/л раствор четыреххлористого олова в дихлорметане (2,56 мл, 0,5 эквивалента), затем триметилсилилазид (0,82 мл, 1,2 эквивалента). Перемешивали при 0°С в течение 3 ч. Затем реакционная смесь была разбавлена 80 мл дихлорметана и промыта насыщенным раствором NaHCO3 (70 мл), водой (50 мл) и насыщенным раствором поваренной соли (50 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: 50% петролейный эфир - 50% EtOAc) 2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозил азид (7) был выделен в виде белого некристаллического порошка (1,66 г, 87%).
Спектр 1Н ЯМР (300 MHz, CDCl3, δ, м.д.): 5.22 (t, J=9.4 Hz, 1H), 5.10 (t, J=9.7 Hz, 1H), 4.96 (t, J=9.2 Hz, 1H), 4.65 (d, J=8.8 Hz, 1H), 4.22 (ddd, J=14.8, 12.5, 3.5 Hz, 2H), 3.79 (ddd, J=9.9, 4.7, 2.3 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H).
Спектр 13С ЯМР (75 MHz, CDCl3, δ, м.д.): 169.29, 169.19, 87.91, 74.01, 72.58, 70.61, 67.84, 61.64, 20.57.
Синтез β-D-глюкопиранозил азида (8) (фиг. 3). В круглодонную колбу с магнитной мешалкой помещали 0,3 г 2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил азида и растворяли его в 4 мл метанола. Далее по каплям прибавляли метилат натрия до образования сильнощелочной среды (рН=11), перемешивали 30 минут. Затем по каплям прибавляли в колбу концентрированную соляную кислоту до нейтральной реакции. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 20% СН3ОН - 80% CHCl3, затем 50% СН3ОН - 50% CHCl3) β-D-глюкопиранозил азид (8) выделен в виде белого некристаллического порошка (0,165 г, количественно).
ИК: 2098 см-1 (полоса азидной группы).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 4.50 (d, J=8.5 Hz, 1H), 3.90 (d, J=11.9 Hz, 1H), 3.70 (dd, J=12.0, 5.3 Hz, 1H), 3.43-3.35 (m, 2H), 3.31 (d, J=17.6 Hz, 2H), 3.16 (t, J=8.7 Hz, 1H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 90.67, 78.76, 76.68, 73.35, 69.70, 61.12.
Синтез гепта-О-ацетил-β-мальтозазида (9) (фиг. 3). В колбу Шленка с мешалкой и заполненную аргоном поместили 1,5 г β-D-октоацетат мальтозы и добавили 20 мл безводного дихлорметана, затем к смеси по каплям прибавляли 1 Моль/л раствор четыреххлористого олова в дихлорметане (1,1 мл, 0,5 эквивалента), затем триметилсилилазид (0,35 мл, 1,2 эквивалента). Перемешивали при 0°С в течение 3 ч. Затем реакционная смесь была разбавлена 80 мл дихлорметана и промыта насыщенным раствором NaHCO3 (70 мл), водой (50 мл) и насыщенным раствором поваренной соли (50 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент 50% петролейный эфир - 50% EtOAc) гепта-О-ацетил-β-мальтозазид (9) выделен в виде белого кристаллического порошка (1,66 г, 98%).
Спектр 1Н ЯМР (300 MHz, CDCl3, δ, м.д.): 5.39 (dd, J=12.3, 7.0 Hz, 1H), 5.26 (t, J=8.9 Hz, 1H), 5.05 (t, J=9.9 Hz, 1H), 4.85 (dd, J=10.5, 4.0 Hz, 1H), 4.82-4.68 (m, 2H), 4.50 (dd, J=12.3, 2.5 Hz, 1H), 4.28-4.19 (m, 2H), 4.08-3.91 (m, 2H), 3.78 (ddd, J=9.7, 4.3, 2.6 Hz, 1H), 2.15 (s, 3H), 2.10 (s, 3H), 2.06-1.98 (m, 15H).
Спектр 13C ЯМР (75 MHz, CDCl3, δ, м.д.): 170.50, 170.39, 170.08, 169.92, 169.48, 169.39, 95.69, 87.46, 75.08, 74.23, 72.31, 71.47, 69.97, 69.24, 68.62, 67.92, 62.51, 61.44, 20.85, 20.78, 20.69, 20.58.
Синтез β-мальтозазида (10) (фиг. 3). В круглодонную колбу с магнитной мешалкой помещали 0.3 г гепта-О-ацетил-β-мальтозил азида и растворяли его в 4 мл метанола. Далее по каплям прибавляли метилат натрия до образования сильнощелочной среды (рН=11), перемешивали 30 минут. Затем по каплям прибавляли в колбу концентрированную соляную кислоту до нейтральной реакции. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: 50% СН3ОН - 50% CHCl3) (3-мальтозазид (10) выделен в виде белого некристаллического порошка (0,165 г, 89%).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 5.19 (d, J=3.8 Hz, 1H), 4.55 (d, J=8.6 Hz, 1H), 3.93 (dd, J=12.3, 1.6 Hz, 1H), 3.85 (dd, J=12.6, 3.9 Hz, 2H), 3.74-3.55 (m, 5H), 3.53-3.43 (m, 2H), 3.37 (s, 1H), 3.28 (t, J=9.3 Hz, 1H), 3.22 (t, J=8.8 Hz, 1H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 102.90, 92.00, 80.65, 78.74, 77.87, 75.05, 74.82, 74.37, 74.14, 71.50, 62.73, 61.96.
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(тетра-O-ацетил-1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхло-рина-е6 (11) (фиг. 4). В круглодонную колбу, снабженную дефлегматором, помещали 0,06 г соединения 4 и 0,034 г (1,2 эквивалента) 2,3,4,6-тетра-O-ацетил-β-D-галактопиранозил азида 5. Смесь растворяли в 6 мл t-BuOH и 3 мл CHCl3. В отдельной колбе смешивали 0,004 г CuSO4⋅5H2O, 0,006 г (0,4 эквивалента) аскорбата натрия (AscNa), 0,008 г (0,2 эквивалента) трис(1-бензил-1Н-1,2,3-триазол-4-ил)метил)амина (ТВТА) и 6 мл воды; не растворившиеся частицы твердой фазы разбивали ультразвуком. После этого приготовленный раствор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток растворяли в хлороформе, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 20% МеОН - 80% CHCl3) целевой конъюгат (11) выделили в виде темно-зеленого кристаллического порошка (0,063 г, 71%).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): δ 9.52 (s, 1Н), 9.50 (s, 1H), 8.80 (s, 1H), 8.65 (s, 1H), 8.39 (t, 1H), 8.22 (dd, J=17.8, 11.6 Hz, 1H), 8.06 (s, 1H), 6.25-6.17 (m, 2H), 6.00 (d, 1H), 5.56 (t, 2H), 5.51-5.34 (m, 2H), 5.09 (d, J=18.5 Hz, 1H), 4.52 (t, 1H), 4.42 (dd, J=14.3, 7.1 Hz, 1H), 4.31 (d, J=5.6 Hz, 2H), 4.21 (d, J=9.5 Hz, 1H), 4.08 (dd, J=11.6, 5.1 Hz, 1H), 3.95 (dd, J=11.5, 7.2 Hz, 1H), 3.80 (q, J=15.4, 7.8 Hz, 3H), 3.65 (s, 3H), 2.86 (s, 2H), 2.17-2.04 (m, 6H), 1.92 (d, J=1.9 Hz, 6H), 1.77 (s, 3H), 1.66 (t, J=14.2, 6.7 Hz, 3H), 1.60 (d, J=7.8 Hz, 3H).
Спектр 1Н ЯМР (101 MHz, DMSO-d6, δ, м.д.): 173.58, 172.24, 170.20, 170.13, 169.69, 168.74, 165.31, 163.04, 151.80, 148.37, 146.26, 145.70, 144.20,143.37, 141.60, 140.92, 138.79, 137.41, 133.27, 132.45, 130.92, 128.99, 127.97, 122.35, 119.41, 102.14, 100.12, 93.38, 84.38, 79.41, 73.11, 70.67, 69.18, 67.84, 67.53, 61.75, 52.60, 51.82, 45.18, 34.22, 28.99, 23.07, 20.66, 20.60, 20.54, 20.22, 19.13, 18.14, 12.54, 11.94, 11.17.
MS (MALDI): m/z 1152.4 [М+].
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(тетра-O-ацетил-1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлори-на-е6 (12) (фиг. 5). В круглодонную колбу, снабженную дефлегматором, помещали 0,06 г соединения 4, 0,034 г (1,2 эквивалента) 2,3,4,6-тетра-O-ацетил-β-D-глюкогшранозил азида 7. Смесь растворяли в 6 мл t-BuOH и 3 мл CHCl3. В отдельной колбе смешивали 0,004 г (2 эквивалента) CuSO4⋅5H2O, 0,006 г (0,4 эквивалента) аскорбата натрия, 0,008 г (0,2 эквивалента) ТВТА и 6 мл воды; не растворившиеся частицы твердой фазы разбивали ультразвуком. После этого приготовленный раствор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток растворили в хлороформе, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 20% МеОН - 80% CHCl3.) целевой конъюгат 12 выделили в виде темно-зеленого кристаллического порошка (0,06 г, 67%).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): 9.51 (d, J=5.6 Hz, 2H), 8.83 (s, 1H), 8.65 (s, 1H), 8.38 (t, J=5.6 Hz, 1H), 8.22 (dd, J=17.8, 11.5 Hz, 1H), 8.13 (s, 1H), 6.30-6.17 (m, 2H), 6.00 (dd, J=11.5, 1.6 Hz, 1H), 5.64-5.37 (m, 4H), 5.18-5.02 (m, 2H), 4.42 (q, J=7.1 Hz, 1H), 4.35-4.18 (m, 4H), 4.13-3.95 (m, 2H), 3.81 (dd, J=15.3, 7.6 Hz, 3H), 3.68 (s, 3H), 3.53 (d, J=8.5 Hz, 1H), 2.92 (s, 1H), 2.00 (s, 6H), 1.92 (d, J=6.6 Hz, 6H), 1.74 (s, 3H), 1.67 (t, J=8.5 Hz, 3H), 1.59 (d, J=7.1 Hz, 3H).
Спектр 1H ЯМР (101 MHz, DMSO-d6, δ, м.д.): 173.29, 171.90, 169.95, 169.52, 169.31, 168.41, 162.76, 151.93, 151.56, 145.62, 143.96, 143.11, 140.66, 138.54, 137.15, 133.01, 132.21, 130.68, 121.78, 101.89, 83.70, 79.19, 73.17, 72.19, 70.00, 67.46, 61.78, 51.60, 33.98, 22.81, 20.42, 20.36, 20.22, 19.89, 18.88, 17.89, 12.30, 11.71, 10.93.
MS (MALDI): m/z 1152.1 [М+].
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(гепта-O-ацетил-1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (13) (фиг. 6). В круглодонную колбу, снабженную дефлегматором, помещали 0,06 г соединения 4 и 0,061 г. (1,2 эквивалента) гепта-О-ацетил-β-мальтозазида 9. Смесь растворяли в 6 мл t-BuOH и 3 мл CHCl3. Смесь растворяли в 6 мл t-BuOH и 3 мл CHCl3. В отдельной колбе смешивали 0,004 г (2 эквивалента) CuSO4⋅5H2O, 0,006 г (0,4 эквивалента) аскорбата натрия, 0,008 г (0,2 эквивалента) ТВТА и 6 мл воды; не растворившиеся частицы твердой фазы разбивали ультразвуком. После этого приготовленный раствор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток растворили в хлороформе, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 20% МеОН - 80% CHCl3) целевой конъюгат (13) выделили в виде темно-зеленого кристаллического порошка (0,07 г, 64%).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): 9.51 (d, J=6.1 Hz, 2H), 8.80 (s, 1H), 8.65 (s, 1H), 8.37 (t, J=5.3 Hz, 1H), 8.33 (s, 1H), 8.22 (dd, J=17.7, 11.6 Hz, 1H), 8.04 (s, 1H), 6.28-6.16 (m, 2H), 6.00 (d, J=11.5 Hz, 1H), 5.62-5.41 (m, 3H), 5.37-5.18 (m, 2H), 5.14-4.85 (m, 3H), 4.47-3.94 (m, 10H), 3.81 (d, J=7.5 Hz, 2H), 3.68 (s, 3H), 3.53 (d, J=7.9 Hz, 2H), 2.86 (s, 2H), 2.03-1.87 (m, 21H), 1.76-1.52 (m, 12H).
Спектр 1Н ЯМР (101 MHz, DMSO-d6, δ, м.д.): 173.30, 171.92, 170.05, 169.96, 169.82, 169.66, 169.50, 169.15, 168.63, 165.05, 162.75, 151.55, 148.11, 146.01, 145.54, 143.95, 143.12, 141.34, 140.65, 138.54, 137.15, 136.22, 133.80, 133.01, 132.19, 130.68, 128.73, 127.72, 121.83, 119.12, 101.90, 101.62, 99.88, 95.72, 93.11, 83.29, 79.19, 74.43, 73.77, 73.33, 70.54, 69.39, 68.90, 68.13, 67.67, 62.82, 61.35, 57.64, 52.33, 51.58, 46.35, 44.79, 37.40, 33.97, 32.13, 30.08, 22.82, 20.49, 20.42, 20.35, 20.31, 20.27, 19.90, 18.88, 17.89, 12.30, 11.70, 10.93.
MS (MALDI): m/z 1442.3 [М+].
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (14) (фиг. 7). В круглодонную колбу, снабженную дефлегматором, помещали 0,1 г соединения 4 и 0,031 г (1,2 эквивалента) β-D-галактопиранозил азида 6, смесь растворяли в 5 мл диметилформамида. В другой круглодонной колбе смешивали 0,010 г (2 эквивалента) CuSO4⋅5H2O, 0,010 г (0,4 эквивалента) аскорбат натрия, 0,014 г (0,2 эквивалента) ТВТА и 5 мл воды, для ускорения растворения использовали ультразвук. После этого приготовленный раствор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 20% СН3ОН - 80% CHCl3, затем 50% СН3ОН - 50% CHCl3) выделяли целевой конъюгат (14) в виде темно-зеленого кристаллического порошка (0,104 г, 83%).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 9.56 (d, J=12.7 Hz, 2H), 8.64 (s, 1H), 8.12 (dd, J=17.8, 11.5 Hz, 1H), 7.90 (d, J=6.1 Hz, 1H), 6.18 (d, J=18.3 Hz, 1H), 6.00 (d, J=11.4 Hz, 1H), 5.51-5.36 (m, 2H), 5.19 (d, J=18.8 Hz, 1H), 4.43 (d, J=23.5, 11.6 Hz, 1H), 4.31 (q, J=15.4 Hz, 3H), 4.01 (t, J=9.4 Hz, 1H), 3.91-3.79 (m, 3H), 3.75 (s, 3H), 3.73 (d, J=7.6 Hz, 1H), 3.70-3.65 (m, 2H), 3.65-3.56 (m, 6H), 3.40 (s, 3H), 3.37 (s, 3H), 3.34 (s, 3H), 3.33 (s, J=5.0 Hz, 3H), 2.63 (s, 2H), 2.49-2.39 (m, 1H), 2.25 (s, 6H), 2.11-2.00 (m, 1H), 1.98-1.83 (m, 1H), 1.73 (s, 3H), 1.66 (d, J=7.1 Hz, 3H).
Спектр 1H ЯМР (101 MHz, CD3OD, δ, м.д.): 175.06, 174.12, 165.70, 161.99, 152.80, 147.34, 145.21, 144.92, 143.60, 141.34, 141.24, 138.88, 138.12, 132.98, 132.64, 130.43, 121.43, 119.14, 118.42, 102.25, 101.36, 99.88, 92.71, 88.70, 78.36, 73.80, 69.87, 68.83, 60.83, 52.59, 51.35, 44.10, 37.83, 37.48, 34.13, 32.11, 30.21, 29.24, 22.04, 18.84, 16.75, 10.96, 10.58, 9.71.
MS (MALDI): m/z 983,6. [М+].
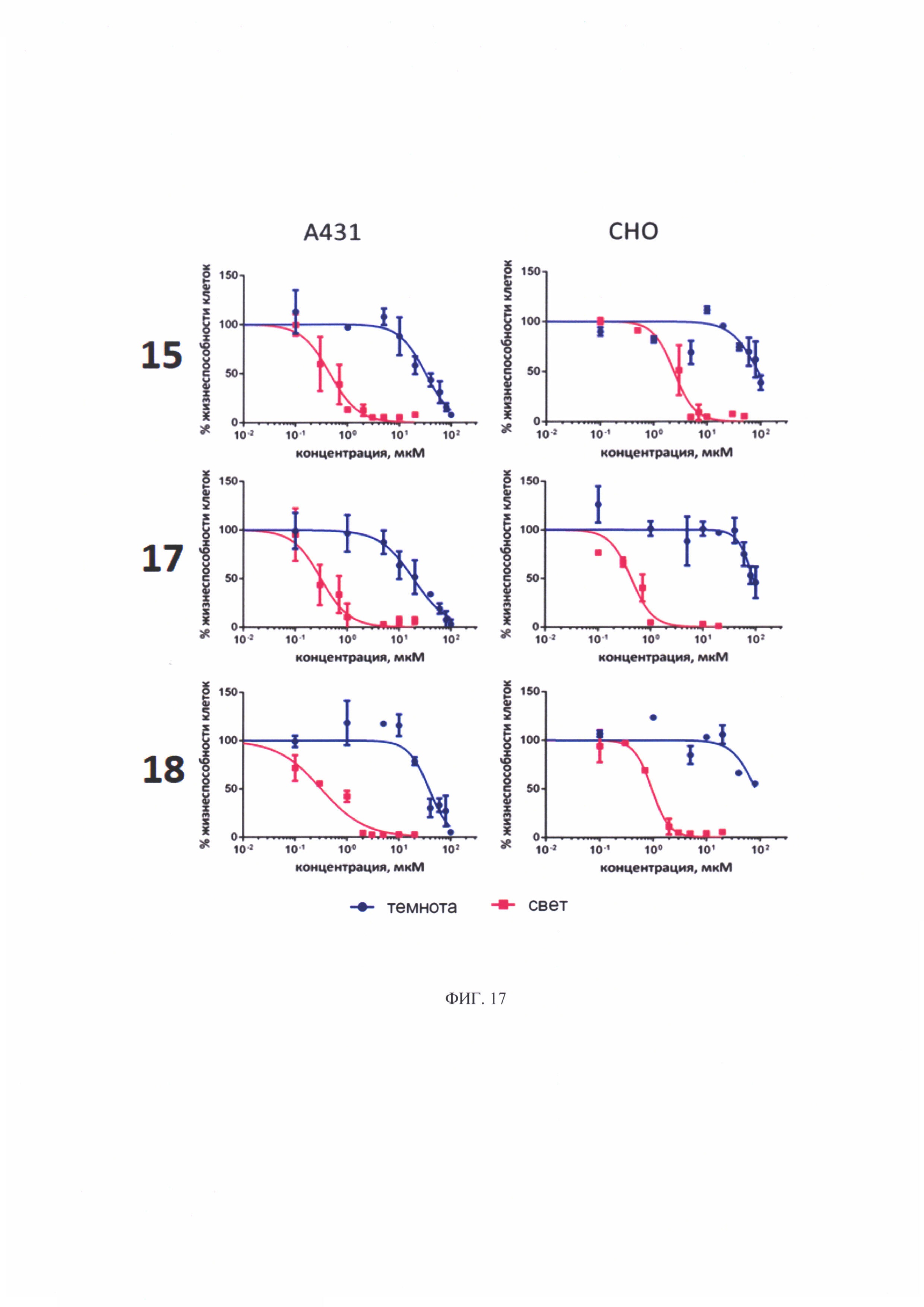
Синтез бромида цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-галактопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6, (15) (фиг. 7). В круглодонную колбу с магнитной мешалкой поместили 0,050 г соединения 14, растворили в смеси из 2 мл хлороформа и 3 мл метанола, добавили 2 мл бромистого этила. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Растворитель удаляли при пониженном давлении. Выделено 0,051 г (количественно) аммонийной соли 15 темно-зеленого твердого кристаллического порошка.
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 9.60 (d, J=24.9 Hz, 2H), 8.69 (s, J=8.0 Hz, 1H), 8.21-8.06 (m, 1H), 7.97 (s, 1H), 6.20 (d, J=17.6 Hz, 1H), 6.03 (d, J=10.3 Hz, 1H), 5.46 (d, J=9.2 Hz, 1H), 5.42-5.07 (m, 2H), 4.53-4.44 (m, 1H), 4.39-4.27 (m, 3H), 4.07 (t, J=9.3 Hz, 1H), 3.95-3.78 (m, 5H), 3.73 (d, J=4.3 Hz, 5H), 3.64 (d, J=4.0 Hz, 4H), 3.10-2.87 (m, 6H), 2.60-2.48 (m, 1H), 2.32-2.21 (m, 1H), 2.21-2.10 (m, 1H), 1.97-1.86 (m, 1H), 1.77 (q, J=7.3 Hz, 3H), 1.71 (d, J=7.0 Hz, 3H).
Спектр 1Н ЯМР (101 MHz, CD3OD, δ, м.д.): δ 175.95, 175.52, 172.99, 167.43, 163.64, 154.51, 149.15, 146.95, 146.37, 145.06, 143.00, 140.31, 139.75, 134.61, 134.39, 131.98, 131.74, 122.89, 120.09, 103.98, 102.59, 101.34, 94.33, 90.12, 79.81, 75.21, 71.33, 70.29, 62.31, 52.88, 51.01, 44.49, 39.81, 35.61, 33.52, 31.49, 23.44, 20.86, 20.28, 18.20, 14.46, 12.38, 12.19, 11.14, 8.38.
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (16) (фиг. 8). В круглодонную колбу, снабженную дефлегматором, помещали 0,123 г соединения 4 и 0,039 г (1,2 эквивалента) β-D-глюкопиранозил азида 8, смесь растворяли в 5 мл диметилформамида. В другой круглодонной колбе смешивали 0,008 г (2 эквивалента) CuSO4⋅5H2O, 0,012 г (0,4 эквивалента) аскорбата натрия, 0,016 г (0,2 эквивалента) ТВТА и 5 мл воды, для ускорения растворения использовали ультразвук. После этого приготовленный раствор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 20% СН3ОН - 80% CHCl3, затем 50% СН3ОН - 50% CHCl3) целевой конъюгат (16) выделен в виде темно-зеленого кристаллического порошка (0,106 г, 69%).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): δ 9.58 (d, J=13.4 Hz, 2H), 8.66 (s, 1H), 8.14 (dd, J=17.9, 11.5 Hz, 1H), 7.86 (s, 1H), 6.20 (d, J=17.9 Hz, 1H), 6.02 (d, J=10.2 Hz, 1H), 5.52-5.41 (m, 2H), 5.22 (d, J=19.0 Hz, 1H), 4.46 (d, J=7.2 Hz, 1H), 4.32 (d, J=2.7 Hz, 3H), 3.90-3.74 (m, 8H), 3.52-3.34 (m, 12H), 2.72 (s, 2H), 2.52-2.39 (m, 1H), 2.33 (s, 6H), 2.31-2.20 (m, 1H), 2.13-2.03 (m, 1H), 1.98-1.87 (m, 1H), 1.75 (t, J=7.5 Hz, 3H), 1.69 (d, J=7.2 Hz, 3H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 176.70, 175.75, 174.42, 167.35, 163.65, 154.44, 149.33, 148.99, 146.86, 146.37, 145.25, 142.98, 142.91, 140.54, 139.77, 134.65, 134.30, 133.43, 132.07, 123.55, 120.07, 103.90, 103.01, 101.53, 94.38, 89.69, 81.23, 78.62, 74.11, 71.03, 62.51, 59.21, 54.21, 53.00, 45.70, 39.48, 39.05, 35.72, 33.73, 31.82, 23.70, 20.49, 18.40, 12.61, 12.23, 11.36.
MS (MALDI): m/z 984,3. [М+].
Синтез бромида цинкового комплекса 13-(диметилэтиламмонийэтил)амид-17(3)-(1ʺ-дезокси-β-D-глюкопиранозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлори-на-е6 (17) (фиг. 8). В круглодонную колбу с магнитной мешалкой поместили 0,050 г соединения 16, растворили в смеси из 2 мл хлороформа и 3 мл метанола, добавили 2 мл бромистого этила. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Растворитель удаляли при пониженном давлении. Выделено 0,051 г (количественно) аммонийной соли 17 в виде темно-зеленого твердого кристаллического порошка.
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 9.59 (d, J=23.1 Hz, 2H), 8.67 (s, J=5.3 Hz, 1H), 8.13 (dd, J=17.9, 11.6 Hz, 1H), 7.91 (s, 1H), 6.20 (d, J=17.3 Hz, 1H), 6.03 (d, J=11.5 Hz, 1H), 5.50 (d, J=9.2 Hz, 1H), 5.42-5.09 (m, 2H), 4.48 (d, J=7.1 Hz, 1H), 4.34 (s, 3H), 3.90-3.56 (m, 13H), 3.16 (s, 4H), 3.04 (s, J=15.4 Hz, 2H), 2.59-2.47 (m, 1H), 2.33-2.21 (m, 1H), 2.16-2.07 (m, 1H), 1.97-1.85 (m, 1H), 1.76 (t, J=7.5 Hz, 3H), 1.70 (d, J=7.1 Hz, 3H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 175.89, 175.43, 170.66, 167.30, 163.50, 154.40, 146.10, 142.87, 140.21, 134.26, 131.64, 123.23, 119.96,108.20,103.84, 102.48, 101.20, 93.86, 89.39, 80.94, 78.31, 73.83, 70.76, 62.21, 53.78, 52.78, 51.01, 44.17, 42.92, 39.95, 39.70, 37.06, 35.46, 33.39, 31.35, 23.36, 20.17, 18.09, 12.28, 12.08, 11.04, 8.35.
Синтез цинкового комплекса 13-(диметиламиноэтил)амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (18) (фиг. 9). В круглодонную колбу, снабженную дефлегматором, помещали 0,1 г соединения 4 и 0,055 г (1,2 эквивалента) β-D-глюкопиранозил азида 10. Смесь растворяли в 5 мл DMF. Пока смесь перемешивалась, в другой круглодонной колбе смешивали CuSO4⋅5H2O 0,007 г. (2 экв.), 0,010 г (0,4 эквивалента) аскорбата натрия, 0,014 г (0,2 эквивалента) ТВТА и 5 мл воды, для ускорения растворения использовали ультразвук. После этого приготовленный катализатор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали с помощью магнитной мешалки при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 1% триэтиламина - 10% МеОН - 89% CHCl3, затем 3% триэтиламина -20% МеОН - 77% CHCl3.) целевой конъюгат (18) выделен в виде темно-зеленого кристаллического порошка (0,074 г, 50%).
Спектр 1Н ЯМР (400 MHz, CD3OD, δ, м.д.): 9.58 (d, J=19.1 Hz, 2H), 8.66 (s, 1H), 8.14 (dd, J=17.8,11.6 Hz, 1H), 7.89 (s, 1H), 6.21 (dd, J=17.9, 1.6 Hz, 1H), 6.04 (dd, J=11.5, 1.6 Hz, 1H), 5.56-5.40 (m, 2H), 5.32-5.15 (m, 2H), 4.48 (q, J=7.0 Hz, 1H), 4.33 (s, 3H), 4.09-3.99 (m, 1H), 3.97-3.80 (m, 6H), 3.75 (s, 7H), 3.72-3.54 (m, 8H), 3.46 (s, 3H), 3.40-3.38 (m, 3H), 3.36 (s, J=5.8 Hz, 3H), 2.96 (s, 6H), 2.57-2.43 (m, 1H), 2.37-2.22 (m, 1H), 2.17-2.05 (m, 1H), 2.01-1.86 (m, 1H), 1.76 (t, J=7.5 Hz, 3H), 1.70 (d, J=7.1 Hz, 3H).
Спектр 13C ЯМР (101 MHz, CD3OD, δ, м.д.): 176.70, 175.75, 167.50, 154.60, 149.20, 147.04, 146.39, 145.26, 143.04, 142.81, 140.65, 139.87,134.71, 134.44, 132.02, 123.51, 120.18, 104.06, 103.12, 102.94, 101.54, 94.42, 89.47, 80.48, 79.69, 78.34, 75.26, 75.06, 74.35, 73.68, 71.69, 62.93, 61.92, 59.21, 53.06, 45.17, 39.82, 38.34, 35.72, 33.66, 32.97, 31.76, 23.92, 23.71, 20.49, 18.40, 12.61, 12.31, 11.36, 9.43.
MS (MALDI): m/z 1145,8. [М+].
Синтез цинкового комплекса 13-(N-(2-(2-(2-аминоэтокси)этокси)этил))амид-17(3)-пропаргиламидхлорина-e6 (19) (фиг. 10). В круглодонную колбу с мешалкой поместили 0,208 г соединения 3 и добавили 8 мл хлороформа, затем к смеси по каплям прибавляли 1 мл 1,2-бис(2-аминоэтокси)этана и перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении. Остаток растворили в 20 мл хлороформа, экстрагировали водой (3×50 мл), органическую фазу сушили над Na2SO4. Полученную органическую фазу растворили в 7 мл хлороформа и добавили по каплям раствор 0,325 г диацетат цинка в 4 мл метанола. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении. Остаток растворили в 30 мл хлороформа, экстрагировали водой (3×30 мл). Органическую фазу сушили над Na2SO4. Целевой аминохлорин (19) выделен в виде темно-зеленого порошка (0,200 г, 72%). Использовали далее без дополнительной очистки.
Синтез цинкового комплекса 13-(N-(2-(2-(2-N-(3-карбокси-1-оксопропил)-аминоэтокси)этокси)этил))амид-17(3)-пропаргиламидхлорина-e6 (20) (фиг. 10). В круглодонную колбу с мешалкой поместили 0,170 г соединения 19 и добавили 8 мл хлороформа, затем к смеси прибавили 0,026 г (1,3 эквивалента) янтарного ангидрида и 0,026 г (1 эквивалент) диизопропилэтиламина. Перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли при пониженном давлении. Остаток растворили в 20 мл хлороформа, экстрагировали водой (3×50 мл), органическую фазу сушили над Na2SO4. После проведения колоночной хроматографии на силикагеле (элюент: 30% МеОН - 70% CHCl3) выделен целевой функционализированный хлорин (20) в виде темно-зеленого кристаллического порошка (0,190 г, количественно).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): 9.50 (d, J=6.3 Hz, 2H), 8.84 (s, 1H), 8.33 (t, J=5.4 Hz, 1H), 8.22 (dd, J=17.8, 11.6 Hz, 1H), 7.95 (t, J=5.3 Hz, 1H), 6.21 (d, J=17.9 Hz, 1H), 6.00 (d, J=11.6 Hz, 1H), 5.43 (d, J=18.7 Hz, 1H), 5.07 (d, J=19.0 Hz, 1H), 4.42 (q, J=7.2 Hz, 1H), 4.20 (d, J=9.9 Hz, 1H), 3.88-3.77 (m, 6H), 3.72 (s, J=4.8 Hz, 3H), 3.69-3.60 (m, 7H), 3.48 (t, J=5.9 Hz, 2H), 3.04 (s, 1H), 2.39 (d, J=6.2 Hz, 3H), 2.33 (d, J=6.1 Hz, 3H), 2.07 (d, J=8.6 Hz, 2H), 1.67 (t, J=7.5 Hz, 3H), 1.60 (d, J=7.0 Hz, 3H).
Спектр 13C ЯМР (400 MHz, DMSO-d6, δ, м.д.): 206.52, 173.30, 171.74, 171.15, 170.10, 165.01, 162.77, 151.53, 148.17, 145.98, 143.92, 143.13, 141.40, 140.64, 138.64, 137.15, 133.93, 133.00, 132.17, 130.70, 119.13, 101.94, 101.63, 99.90, 93.12, 81.20, 72.81, 69.73, 69.66, 69.22, 68.95, 52.29, 51.60, 46.26, 37.32, 32.07, 30.71, 30.05, 29.31, 27.79, 22.84, 18.89, 17.91, 12.32, 11.65, 10.94.
MS (MALDI): m/z 939,8. [М+].
Синтез цинкового комплекса 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)-этил))амид-17(3)-пропаргиламидхлорина-е6 (22) (фиг. 11). В первую круглодонную колбу с мешалкой поместили 0,095 г соединения 20, 0,038 г (2 эквивалента) EDC⋅HCl и 4 мл диметилформамида. Перемешивали при 0°С в течение 30 мин. Во вторую колбу с мешалкой добавили 0,073 г (2 эквивалента) 4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7-ола (21), который ранее был синтезирован в соответствии с методикой, известной из патента WO 2010/028254 А2, 0,006 г (0,5 эквивалента) DMAP и 3 мл хлороформа, смесь перемешивали 10 минут при 0°С. Затем шприцем перенесли содержимое второй колбы в первую и перемешивали 18 часов. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале CHCl3, затем 4% МеОН - 96% CHCl3) целевой конъюгат (22) выделен в виде темно-зеленого порошка (0,075 г, 58%).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): δ 9.75 (s, 1H), 9.51 (d, J=5.0 Hz, 2H), 8.84 (s, 1H), 8.65 (s, 1H), 8.32 (t, J=5.4 Hz, 1H), 8.27-8.17 (m, 2H), 8.09 (t, J=5.5 Hz, 1H), 7.96 (s, 1H), 7.71-7.65 (m, 1H), 7.54-7.43 (m, 2H), 7.36 (s, 1H), 6.21 (d, J=19.1 Hz, 1H), 6.00 (d, J=12.7 Hz, 1H), 5.43 (d, J=19.1 Hz, 1H), 5.07 (d, J=18.7 Hz, 1H), 4.41 (q, J=7.2 Hz, 1H), 4.20 (d, J=9.6 Hz, 1H), 3.92 (s, 3H), 3.88-3.77 (m, 5H), 3.75-3.70 (m, 2H), 3.67 (s, 5H), 3.52 (t, J=5.8 Hz, 2H), 3.37 (s, 3H), 3.05 (t, J=2.4 Hz, 1H), 2.86 (t, J=6.9 Hz, 2H), 2.55 (t, J=7.0 Hz, 2H), 2.06 (d, J=8.9 Hz, 2H), 1.66 (t, J=7.5 Hz, 3H), 1.60 (d, J=7.0 Hz, 3H).
Спектр 13C ЯМР (400 MHz, DMSO-d6, δ, м.д.): δ 173.28, 171.70, 170.49, 170.46, 170.07, 165.00, 162.77, 157.95, 157.31, 155.45, 153.16, 151.52, 150.23, 148.17, 145.97, 145.18, 145.13, 143.91, 143.12, 141.40, 140.64, 138.67, 137.14, 133.93, 132.99, 132.15, 130.69,129.65, 127.58, 126.01, 125.89, 120.63, 119.53, 119.29, 119.12, 118.10, 118.01, 113.10, 103.47,101.94, 101.62, 99.90, 93.11, 81.18, 79.17, 72.80, 69.74, 69.65, 69.22, 68.94, 56.56, 52.28, 51.57, 46.23, 37.29, 32.07, 30.03, 29.82, 28.94, 27.78, 22.82, 18.86, 17.88, 12.31, 11.63, 10.92.
MS (MALDI): m/z 1286,8. [М+].
Синтез цинкового комплекса 13-(N-(2-(2-(2-N-(4-((4'-бром-2'-фторфенил)амино)-6-метоксихиназолин-7)-1-оксобутиратоаминоэтокси)этокси)этил))амид-17(3)-(1ʺ-дезокси-β-мальтозил-1ʺ-(1',2',3'-триазол-4'-ил)-метил)амидхлорина-е6 (23) (фиг. 12). В круглодонную колбу, снабженную дефлегматором, помещали 0,058 г соединения 22, 0,02 г (1,2 эквивалента) β-мальтозазида 10, смесь растворяли в 6 мл диметилформамида. В другой круглодонной колбе смешивали 0,002 г (0,2 эквивалента) CuSO4⋅5H2O, 0,003 г (0,4 эквивалента) аскорбата натрия, 0,005 г (0,2 эквивалента) ТВТА и 6 мл воды, для ускорения растворения использовали ультразвук. После этого приготовленный катализатор вносили по каплям в реакционную смесь. Реакционную смесь перемешивали при 50°С в течение 1 часа. Растворитель удаляли при пониженном давлении. После проведения колоночной хроматографии на силикагеле (элюент: вначале 20% МеОН - 80% CHCl3, затем 40% МеОН - 60% CHCl3) выделяли целевой конъюгат (23) в виде темно-зеленого кристаллического порошка (0,066 г, 88%).
Спектр 1Н ЯМР (400 MHz, DMSO-d6, δ, м.д.): δ 9.93 (s, 1Н), 9.50 (d, J=4.8 Hz, 2H), 8.84 (s, 1H), 8.65 (s, 1H), 8.38-8.31 (m, 1H), 8.26-8.17 (m, 2H), 8.11 (t, J=5.6 Hz, 1H), 8.05 (s, 1H), 8.00 (s, 1H), 7.66 (dd, J=9.9, 1.9 Hz, 1H), 7.52-7.43 (m, 2H), 7.36 (s, 1H), 6.21 (d, J=19.4 Hz, 1H), 5.99 (d, J=11.5 Hz, 1H), 5.72 (d, J=2.9 Hz, 1H), 5.66-5.38 (m, 5H), 5.16-4.88 (m, 5H), 4.59-4.49 (m, 3H), 4.42 (q, J=6.7 Hz, 1H), 4.36-4.18 (m, 3H), 3.93 (s, 3H), 3.86-3.41 (m, 30Н), 3.13-3.03 (m, 2H), 2.86 (t, J=6.9 Hz, 2H), 2.55 (t, J=7.0 Hz, 2H), 1.66 (t, J=7.5 Hz, 3H), 1.59 (d, J=7.0 Hz, 3H).
Спектр 13C ЯМР (400 MHz, DMSO-d6, δ, м.д.): δ 173.33, 171.92, 170.53, 170.47, 170.11, 165.08, 162.75,157.35, 153.18, 151.54, 150.21, 148.16, 145.98, 145.18, 145.12, 144.84, 143.91, 143.12, 141.41, 140.64, 138.66, 137.14, 133.91, 132.98, 132.17, 130.70, 129.67, 127.55, 121.86, 120.58, 119.50, 113.15, 103.77, 101.95, 100.91, 87.06, 79.11, 77.92, 76.65, 76.25, 73.53, 73.22, 72.41, 71.52, 69.85, 69.74, 69.66, 69.22, 68.96, 60.74, 60.23, 56.67, 51.62, 34.07, 32.12, 29.84, 28.96, 22.86,18.88, 17.89, 12.32, 11.64, 10.93.
MS (MALDI): m/z 1656,1. [М+].
Анализ спектральных свойств полученных соединений.
Спектры поглощения и флуоресценции регистрировали с помощью планшетного спектрофотометра-спектрофлуориметра Synergy Мх (BioTek, США). Для анализа использовали растворы производных хлорина-e6 в концентрации 5 мкМ. В качестве растворителя использовали деионизированную воду. Спектры поглощения регистрировали в диапазоне длин волн 300-700 нм с шагом 2 нм. Спектры флуоресценции регистрировали при возбуждении на 410 нм, шаг - 2 нм, диапазон регистрации - 550-850 нм.
Коэффициент молярной экстинкции (ε) определяли по формуле:
ε=D/cl, где:
D - оптическая плотность, с - концентрация вещества, l - длина пути.
Квантовый выход флуоресценции исследуемых соединений рассчитывали относительно родамина В (Sigma, США) в воде (0,31). Расчеты производились по формуле:
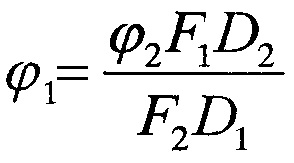
где:
ϕ1 - квантовый выход флуоресценции исследуемого соединения,
ϕ2 - квантовый выход флуоресценции стандартного соединения;
D1 и D2 - оптические плотности эталонного и исследуемого растворов, соответственно.
Показано, что исследуемые соединения 15, 17, 18, 23 характеризуются интенсивным поглощением в коротковолновой области (полоса Соре) и в длинноволновой области (Q-полоса) (фиг. 13). Максимумы поглощения составили 410 нм и 635 нм соответственно. Коэффициент молярной экстинкции в коротковолновой части спектра значительно выше, что характерно для хромофоров хлоринового ряда (Ferreira et al., 2008; Ormond, Freeman, 2013). Для соединения 23 (с дигалактозилом и вандетанибом) наблюдается смещение пиков в длинноволновую часть спектра (420 нм и 645 нм).
Флуоресценция производных хлорина-e6 регистрируется в длинноволновой области 620-700 нм спектра (фиг. 14), характерной для хлориновых соединений. Пик интенсивности флуоресценции находится на 645 нм.
На основании зарегистрированных спектров флуоресценции и значений экстинкции растворов были рассчитаны значения относительного квантового выхода флуоресценции, представленные в таблице 1. В качестве соединения с известным квантовым выходом флуоресценции использован родамин Б.
Для соединения 23 показано смещение пиков в спектре поглощения, наименьшая интенсивность флуоресценции и низкий уровень квантового выхода флуоресценции. Это может быть связано с агрегацией молекул соединения в водном растворе.
Клеточные культуры.
Исследования внутриклеточной локализации и фотодинамической активности соединений проводились на клеточных культурах:
Линия А-431 - эпидермальная карцинома кожи человека (АТСС® CRL-1555™), обладающая высоким уровнем экспрессии рецептора эпидермального фактора роста (EGFR);
Линия СНО - клетки яичника китайского хомячка (АТСС® CRL-9618TM). Клетки неопухолевой природы, экспрессия EGFR отсутствует.
Клетки выращивали на питательной среде ИГЛА MEM с 10% эмбриональной телячьей сывороткой (HyClone, США) и 2 мМ L-глутамина (ПанЭко, Россия) при 37°С и атмосфере 5% CO2. Культивирование проводили в культуральных флаконах площадью 25 см2 (Corning, США). Клетки пересаживали 3 раза в неделю в соотношении 1:7. Снятие клеток с культурального флакона осуществляли раствором трипсин : ЭДТА (1:1) (ПанЭко, Россия), для отмывки клеток использовали 10 мМ фосфатно-солевой буфер (PBS). Центрифугирование клеток проводилось на центрифуге 541 OR (Eppendorf, Германия). Состояние клеток оценивали с помощью инвертированного микроскопа Axiovert 200 (Carl Zeiss, Германия).
Анализ внутриклеточной локализации соединений.
Для исследования внутриклеточной локализации исследуемых соединений суспензию клеток высаживали в тонкодонные 96-луночные планшеты (Corning, США) в концентрации 9×103 клеток на лунку и инкубировали в течение 24 часов при 37°С и атмосфере 5% CO2. Затем питательную среду заменяли раствором исследуемого соединения в бессывороточной среде в концентрации 5 мкМ, 200 мкл/лунку, и инкубировали 4 часа. По завершении инкубации клетки трижды отмывали от фотосенсибилизатора и фиксировали 4% раствором формальдегида в течение 30 минут.
Изображения клеток получали с помощью системы лазерной сканирующей конфокальной микроскопии Axio Observer Z1 LSM 710 (Carl Zeiss, Германия). Флуоресценцию возбуждали на длине волны 405 нм и регистрировали в диапазоне 600-740 нм.
На основании полученных изображений производился анализ поступления соединений в клетки. Количественная оценка интенсивности флуоресценции клеток проводилась с использованием программы ZEN 2012 (Carl Zeiss, Германия).
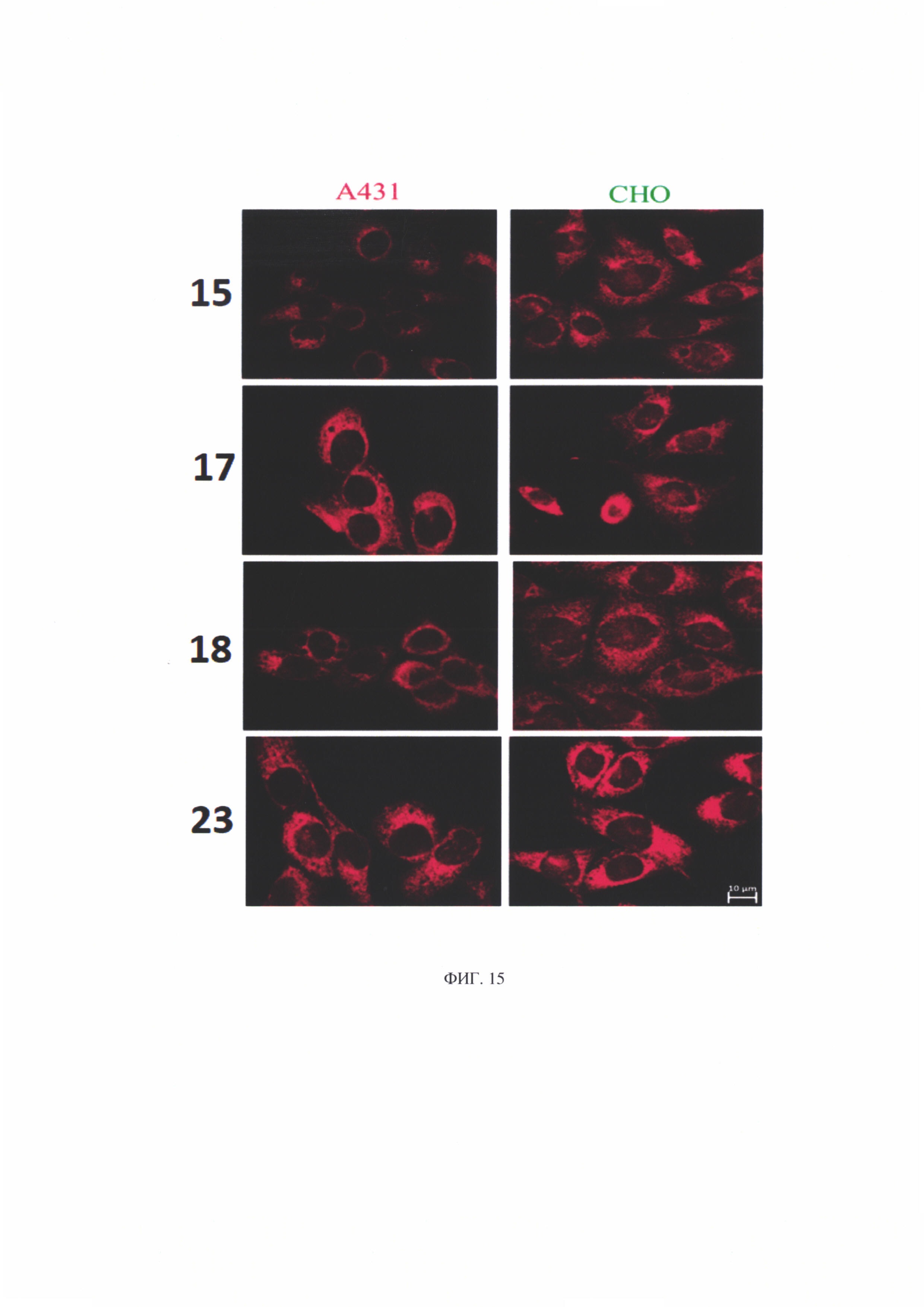
Исследование внутриклеточной локализации соединений проводилось на клетках с разным уровнем экспрессии рецептора эпидермального фактора роста (EGFR). Было предположено, что соединение 23 (с дигалактозилом и вандетанибом) может избирательно накапливаться в клетках с высоким уровнем экспрессии EGFR. С этой целью для исследования были выбраны линия СНО, клетки которой не экспрессируют ни EGFR, ни VEGFR (Colagar et al., 2013) и линия А431, клетки которой характеризуются гиперэкспрессией EGFR (Stanton et al., 1994). Уровень экспрессии EGFR в культурах клеток, используемых в лаборатории, был ранее проверен методом проточной цитометрии с окраской антителами, специфичными к данному рецептору (Otvagin et al., 2018).
Анализ флуоресценции клеток после их инкубации с исследуемыми соединениями показал, что соединения 15, 17, 18, 23 быстро накапливаются в клетках (фиг. 15). Характер распределения свидетельствует о том, что все соединения преимущественно локализуются в цитоплазме во внутриклеточных мембранах.
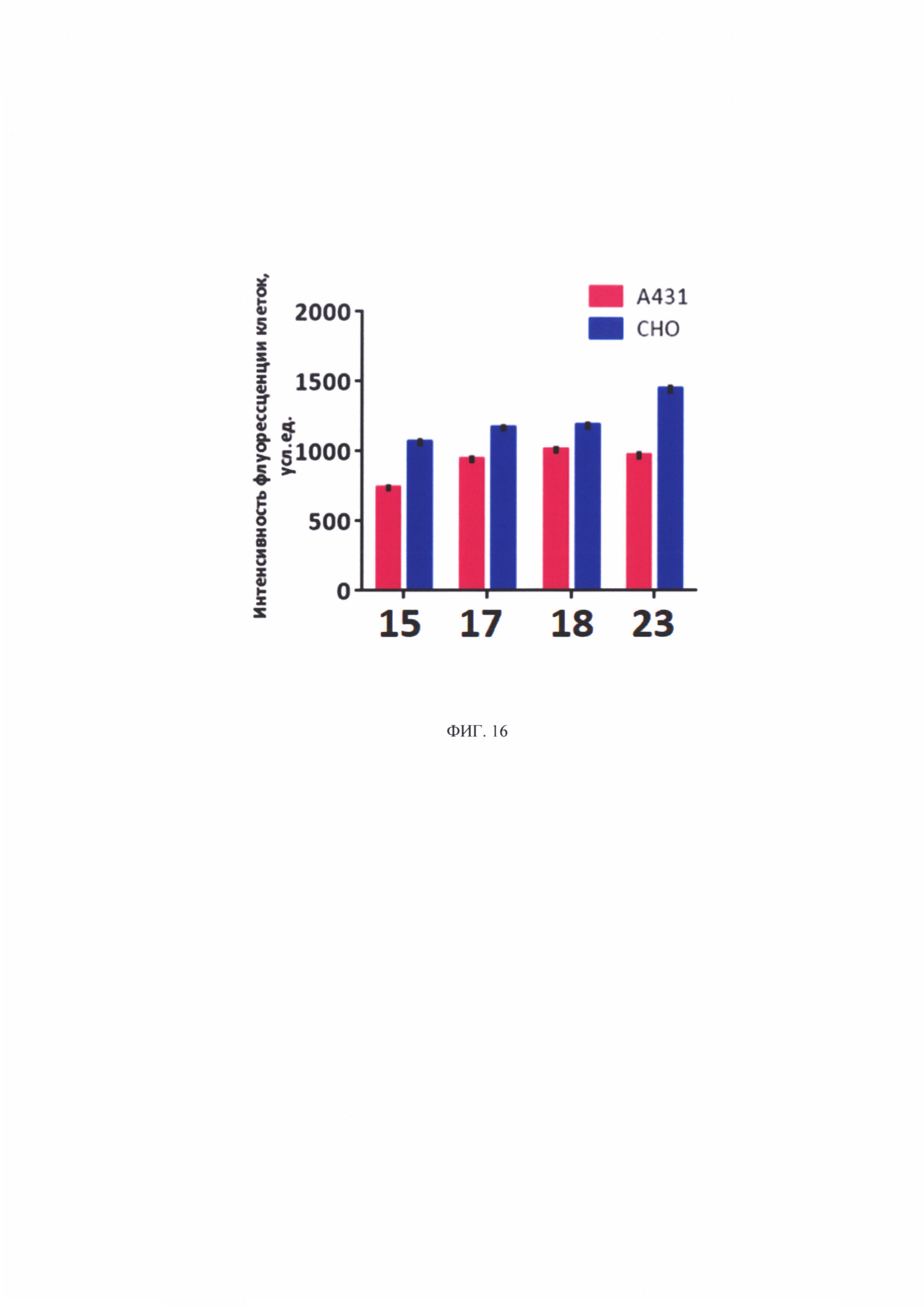
Результаты полуколичественной оценки накопления соединений в клеточных линиях представлены на фиг. 16. На интенсивность флуоресценции клеток существенно влияет лиганд, присоединенный к хлорину-е6.
Показано, что исследуемые соединения больше накапливаются в клетках неопухолевой природы (СНО). Это может говорить о неспецифическом накопления конъюгатов в клетках - проникновении посредством диффузии. Однако данный вопрос требует дальнейшего изучения.
Анализ фотодинамической активности соединений в отношении клеток опухолевой и неопухолевой природы.
Фотодинамическую активность оценивали in vitro. Для этого клетки высаживали на 96-луночный планшет в количестве 4×103 на лунку и инкубировали при 37°С и атмосфере 5% СО2 в течение 24 часов. Затем питательную среду заменяли 200 мкл бессывороточной среды с фотосенсибилизатором в различных концентрациях и инкубировали клетки в течение 4 часов. После окончания инкубации среду с соединением в лунках планшета заменяли соответствующей ростовой питательной средой. Далее клетки облучали с помощью светодиодного излучателя (Шилягина и др., 2014) в термостатируемых условиях (37°С) на TermoStat plus (Eppendorf, Германия).
Доза облучения составляла 20 Дж/см2 на длине волны 670 нм, мощность светового потока - 32 мВт/см2, время облучения - 10 минут 25 секунд. Жизнеспособность клеточной культуры оценивали через 24 часа после облучения при помощи МТТ-теста.
МТТ-тест.
Для образования водонерастворимых окрашенных кристаллов формазана в ростовую среду вносили МТТ-реагент (Alfa Aesar, Великобритания) в конечной концентрации 0,5 мг/мл и инкубировали клетки в течение 4 часов. После отбирали инкубационную среду и добавляли 200 мкл диметилсульфоксида (ДМСО). Измерение оптической плотности полученных в лунке растворов проводили на планшетном спектрофотометре Synergy MX (BioTek, США) на длине волны 570 нм. Жизнеспособность клеток оценивали по отношению значения оптической плотности раствора формазана в каждой пробе к контролю (клетки без фотосенсибилизатора). Относительную жизнеспособность клеток рассчитывали по формуле:
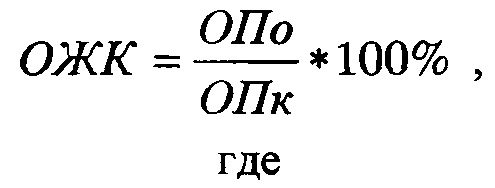
ОПо - усредненная оптическая плотность в лунках с обработанными клетками (за вычетом «бланка»), ОПк - усредненная оптическая плотность в лунках с необработанными клетками (за вычетом «бланка»).
По полученным результатам были построены кривые зависимости «доза-эффект» и рассчитано значение IC50 для каждого соединения (концентрация фотосенсибилизатора, снижающая жизнеспособность клеток на 50%).
Показано, что все исследуемые соединения способны вызывать фотоиндуцированную гибель клеток, однако фотодинамическая активность существенно отличалась в зависимости от присоединенного лиганда.
На рисунках 16 и 17 представлены графики зависимости жизнеспособности клеток А431 и СНО от концентрации соединений 15, 17, 18, 23 в темноте и при освещении.
Для соединении 15, 17, 18 (с углеводными лигандами) темновая токсичность наблюдалась при концентрациях 15-35 мкМ в отношении клеток А431 и 80-90 мкМ в отношении клеток СНО. При облучении в дозе 20 Дж/см2 токсичность существенно возрастала. IC50 (концентрация соединений, при которой наблюдалось снижение жизнеспособности культуры на 50%) во всех случаях не превышала 2,5 мкМ (фиг. 17).
В случае добавления к соединению 18 (с дигалактозилом) в качестве второго лиганда, вандетаниба, наблюдаются изменения цитотоксичности соединения (фиг. 18). У соединения 23 увеличивается фотодинамическая активность и уменьшается темновая токсичность по сравнению с соединением 18. Значения ингибирующей концентрации IC50 всех исследованных соединений представлены в таблице 2.
Минимальной темновой токсичностью обладает соединение 23. Максимальная световая токсичность наблюдается у соединений - 17 и 23. Оптимальным отношением темнота/свет обладает соединение 23 (конъюгат цинкового комплекса хлорина-е6 с дигалактозилом и вандетанибом).


Представленные данные показывают, что наличие гидрофильного производного мальтозы, присоединенного к хлориновому фрагменту, в предлагаемом производном обеспечивает достаточную водорастворимость, позволяет полученным соединениям избирательно накапливаться в опухолевых клетках, что обеспечивает их селективную доставку к опухолевой ткани и снижает их системную токсичность.
Устройство для управления сходимостью рентгеновского пучка и способ изготовления дифракционного блока в составе указанного устройства (варианты)
Способ каталитического пиролиза углеводородной смеси c-c в низшие олефины c-c
Катализатор для пиролиза углеводородной смеси с1-с4 и способ его получения
Солнечный оптический телескоп космического базирования (варианты)
Композиция для визуализации и повреждения клеток-мишеней
Способ получения наногидроксиапатита
Способ контроля наличия глубоких дефектов матрицы gaas, связанных с встраиванием в неё слоя квантовых точек inas
Способ получения загущающей присадки к смазочным маслам
Способ управления работой гибкого ротора на электромагнитных подшипниках и система для его осуществления
Применение вакуумного осаждения германия из газовой среды германа в качестве способа удаления диоксида кремния с рабочей поверхности кремниевой подложки и способ изготовления монокристаллической плёнки германия на кремниевой подложке, включающий указанное применение
Производное 1',2',3'-триметоксибензо[4',5':4,5]-6,7-дигидроциклогепта-[2,3-e]-1h-1-метилиндола и его применение
Производное 1',2',3'-триметоксибензо[4',5':4,5]-6,7-дигидроциклогепта-[3,2-f]-1h-1-метилиндола и его применение
Производное n-(1s)-1',2',3'-триметокси-6,7-дигидро-1н-бензо[5',6':5,4]циклогепта-[3,2-f]бензофуран-1-ил)ацетамида и его применение
Керамообразующая огнестойкая силиконовая резина
Способ оценки функционального состояния коллагеносодержащей ткани
Рекомбинантный иммунотоксин, специфичный к клеткам, экспрессирующим рецептор her2
Производное 1', 2', 3'-триметоксибензо[5', 6:5, 4]-1h-6, 7-дигидроциклогепта[3, 2-f]бензофурана и его применение
Композиция для визуализации и повреждения клеток-мишеней
Винтовая опора с решетчатой направляющей
Порфиразин, порфиразиновый комплекс гадолиния и их применение

