Результат интеллектуальной деятельности: РЕГЕНЕРАЦИЯ ТИТАНСОДЕРЖАЩЕГО ЦЕОЛИТА
Вид РИД
Изобретение
Настоящее изобретение относится к способу регенерации катализатора, содержащего титансодержащий цеолит, причем указанный катализатор использовался в способе получения оксида олефина и содержит осажденную на нем калиевую соль, причем указанный способ регенерации включает в себя стадии: (а) отделения реакционной смеси от катализатора, (b) промывки катализатора, полученного на стадии (а), с помощью жидкой водной системы; (с) необязательно сушки катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре менее 300°С; (d) обжига катализатора, полученного на стадии (с), в газовом потоке, содержащем кислород, при температуре, по меньшей мере, 300°С. Кроме того, настоящее изобретение относится к регенерированному катализатору, содержащему титансодержащий цеолит в качестве каталитически активного материала, получаемому или полученному посредством способа настоящего изобретения.
В последние годы были разработаны различные титансодержащие цеолиты, которые являются полезными в качестве катализаторов органических реакций, таких как конверсия олефинов в эпоксиды. Например, патенты WO-A 98/55229 и WO-A 2011/064191 описывают получение и дальнейшее использование гетерогенных титансодержащих цеолитов при эпоксидировании.
Гетерогенные титансодержащие цеолиты представляют собой значительный промышленный интерес и в данном контексте экономические и экологические соображения имеют существенное значение. Эффективная регенерация таких цеолитов для последующего повторного использования в катализе органических реакций будет иметь значительное предпочтение перед их заменой свежим катализатором.
Патент ЕР-А 0.934.116 описывает способ регенерации отработанного катализатора, состоящего из силиката титана, в результате синтеза эпоксида посредством реакции между олефином и перекисью водорода. Обработка отработанного катализатора включает в себя промывку метанолом, с последующей сушкой в потоке газообразного азота при 75°С и далее с последующей стадией действительной регенерации, которая предусматривает нагрев при 300°С в течение 7 часов. Метанол, который должен быть обеспечен в больших количествах и с достаточно высокой чистотой, является ценным органическим соединением и требует для своего повторного использования дорогостоящего и трудоемкого восстановления.
Патент ЕР-А 1.371.414 описывает способ регенерации катализатора на основе диоксида кремния, содержащего титан, после эпоксидирования кумола, включающий в себя пропускание жидкого пропена через отработанный катализатор при температуре не ниже, чем максимальная температура реакции эпоксидирования. Пропен также представляет собой ценное органическое соединение, и использовать его в больших количествах в промышленном масштабе было бы экономически невыгодно.
Патент ЕР-А 1.221.442 описывает регенерацию титансодержащего цеолитного катализатора, используемого при эпоксидировании олефинов с перекисью водорода, способ, включающий в себя выполнение реакции эпоксидирования, который отличается тем, что регенерацию отработанного катализатора осуществляют с перекисью водорода в присутствии олефина, при этом реакция эпоксидирования продолжается и отличается тем, что регенерация достигается посредством изменения направлении загрузки перекиси водорода. Перекись водорода также является ценным исходным продуктом, который как сам по себе сложно поддается обработке из-за его склонности к спонтанному разложению.
Патент WO-A 2005/000827 описывает регенерацию катализатора на основе силикалита титана после осуществления способа непрерывного эпоксидирования пропена с перекисью водорода. Катализатор периодически регенерируют с помощью метанолового растворителя при температуре, по меньшей мере, 100°С. Вышеуказанный метанол является ценным органическим соединением, которое требует дорогостоящего и затратного по времени восстановления. Кроме того, после регенерации, эпоксидирование требует перезапуска при более высокой температуре по сравнению со свежим катализатором согласно патенту WO-A 2005/000827.
Патент WO-A 2007/013739 описывает регенерацию титансодержащего молекулярного сита, которая отличается тем, что, после предварительной обработки отработанного катализатора водой или спиртом, полученный таким способом, предварительно обработанный катализатор подвергают контакту со смесью, которая содержит перекись водорода, воду и спирт. Таким образом, данный способ включает в себя две обязательных и последовательных стадии, на которых отработанный катализатор подвергают контакту с двумя различными растворами.
Патент US 2003/0187284 А1 описывает способ получения эпоксида в присутствии цеолитного катализатора и регенерации катализатора посредством его обработки раствором, содержащим кислоту с величиной рКа менее 6.
Патент US 2012/142950 А1 описывает непрерывный способ получения пропиленоксида, который включает в себя реагирование пропена с перекисью водорода в метаноловом растворе в присутствии катализатора на основе силикалита титана-1 с получением пропиленоксида.
Патент WO 2011/115234 А1 описывает способ регенерации титаносиликатных катализаторов.
Патент US 2004/058798 А1 описывает способ регенерации титансодержащих катализаторов на основе диоксида кремния посредством нагревания отработанных катализаторов при температуре, по меньшей мере, 400°С в присутствии кислородсодержащего газового потока.
Патент US 5.916.835 описывает способ регенерации отработанных нецеолитных гетерогенных катализаторов.
Таким образом, целью настоящего изобретения является создание простого и экономически эффективного способа регенерации катализатора, содержащего титансодержащий цеолит, в качестве каталитически активного материала, используемого при эпоксидировании олефинов. Еще одной целью настоящего изобретения является обеспечение регенерированного катализатора, содержащего титансодержащий цеолит, в качестве каталитически активного материала, который может быть легко использован повторно в катализе при эпоксидировании олефинов.
Таким образом, настоящее изобретение относится к способу регенерации катализатора, содержащего титансодержащий цеолит в качестве каталитически активного материала, причем указанный катализатор использовался в способе получения оксида олефина, который включает в себя:
(i) обеспечение смеси, содержащей органический растворитель, олефин, эпоксидирующий агент и, по меньшей мере, частично растворенную калийсодержащую соль;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей органический растворитель и оксид олефина, и с получением катализатора, содержащего осажденную на нем калиевую соль;
причем указанный способ регенерации включает в себя:
(a) отделение смеси, полученной на стадии (ii), от катализатора;
(b) промывку катализатора, полученного на стадии (а), с помощью жидкой водной системы;
(c) необязательно сушку катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре менее 300°С;
(d) обжиг катализатора, полученного на стадии (b) или (с), в газовом потоке, содержащем кислород, при температуре, по меньшей мере, 300°С.
Неожиданно, в соответствии со способом регенерации согласно настоящему изобретению, который включает в себя промывку отработанного катализатора, содержащего титансодержащий цеолит, с помощью жидкой водной системы, в сочетании с необязательной сушкой и дальнейшим обжигом, был получен регенерированный катализатор, содержащий титансодержащий цеолит, с отличными каталитическими свойствами, который может быть легко использован повторно, например, в способе получения оксида олефина.
В связи с этим, авторы обнаружили, что после воздействия на отработанный катализатор, содержащий титансодержащий цеолит в качестве каталитически активного материала, посредством регенерации согласно настоящему изобретению, его активность и селективность в течение длительного срока сравнима с активностью соответствующего свежего катализатора, содержащего титансодержащий цеолит. Такой благоприятный результат может быть получен после выполнения только одного цикла стадий регенерации (а)-(d).
Кроме того, неожиданно было обнаружено, что даже повторяющиеся циклы стадий (a)-(b) не оказывают неблагоприятного влияния на активность и селективность катализатора, содержащего титансодержащий цеолит в качестве каталитически активного материала. Регенерация согласно стадиям (а)-(b) настоящего изобретения, таким образом, зарекомендовала себя как ''мягкий'' способ, который может применяться к одному и тому же каталитическому материалу многократно, поскольку не наблюдалось какого-либо отрицательного действия на каталитическую активность и, таким образом, предположительно, на цеолитную структуру, после нескольких повторений стадий (а)-(b).
Стадия (a)
Первая стадия регенерации (а) требует отделения реакционной смеси, полученной в результате реакции эпоксидирования олефина, от катализатора, который содержит титансодержащий цеолит в качестве каталитически активного материала.
Данное отделение реакционной смеси от отработанного катализатора, содержащего титансодержащий цеолит, может быть достигнуто любым подходящим способом, таким как откачивание, слив, декантация, фильтрация и тому подобное. Предпочтительно, в случае, если реакцию эпоксидирования осуществляют в периодическом режиме, предпочтительно отделять смесь, полученную на стадии (ii), от отработанного катализатора посредством фильтрации. В случае, если реакцию эпоксидирования осуществляют в непрерывном режиме, предпочтительно отделять смесь, полученную на стадии (ii), от отработанного катализатора посредством прекращения воздействия на смесь, обеспеченную на стадии (i), с помощью условий эпоксидирования согласно стадии (ii), и посредством воздействия на отработанный катализатор с помощью стадии регенерации (b), как только вся смесь, полученная на стадии (ii) была удалена из реактора, в котором осуществлялось эпоксидирование, либо в реакторе, либо в любом другом подходящем сосуде после извлечения отработанного катализатора из реактора.
Если отработанный катализатор, содержащий титансодержащий цеолит, удаляют из реактора после стадий (i) и (ii) и регенерируют в отдельном сосуде, может быть обеспечен только короткий перерыв в производственном процессе, поскольку реактор может быть быстро наполнен загрузкой второго катализатора, что позволяет немедленное возобновление реакции эпоксидирования.
Стадия (b)
После отделения на стадии (а), отработанный катализатор, содержащий титансодержащий цеолит, промывают с помощью жидкой водной системы согласно стадии (b).
Жидкая водная система, используемая на стадии (b), предпочтительно содержит, по меньшей мере, 75 вес. %, предпочтительно, по меньшей мере, 90 вес. %, более предпочтительно, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы. В соответствии с вариантом осуществления настоящего изобретения, жидкая водная система, используемая на стадии (b), представляет собой воду, предпочтительно деионизированную воду.
В настоящем способе, условия температуры и давления на стадии (b) выбирают таким образом, чтобы водная система поддерживалась в жидком состоянии в течение, по меньшей мере, 90%, предпочтительно, по меньшей мере, 95%, более предпочтительно, по меньшей мере, 99% времени промывки. Предпочтительно, водная система находится в жидком состоянии во время промывки.
Предпочтительно, промывку на стадии (b) с помощью жидкой водной системы осуществляют при давлении в диапазоне от 0,5 до 2 бар, более предпочтительно от 0,8 до 1,5 бар, более предпочтительно от 1,0 до 1,4 бар. Предпочтительно, промывку на стадии (b) с помощью жидкой водной системы осуществляют при температуре жидкой водной системы в диапазоне от 25 до 95°С, более предпочтительно от 40 до 90°С, более предпочтительно от 60 до 80°С. Более предпочтительно, промывку на стадии (b) с помощью жидкой водной системы осуществляют при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С. Более предпочтительно, промывку на стадии (b) с помощью жидкой водной системы осуществляют при давлении в диапазоне от 1,0 до 1,4 бар и температуре в диапазоне от 60 до 80°С.
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают жидкой водной системой, содержащей, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С.
Как правило, значение рН жидкой водной системы согласно стадии (b) не имеет особых ограничений. В зависимости от предпочтительного содержания воды в жидкой водной системе, значение рН может находиться в диапазоне от 4 до 10, предпочтительно в диапазоне от 5 до 9, более предпочтительно в диапазоне от 6 до 8. Предпочтительно, рН находится в диапазоне от 6,5 до 7,5, более предпочтительно от 6,6 до 7,4, более предпочтительно от 6,7 до 7,3, более предпочтительно от 6,8 до 7,2. Значение рН следует понимать согласно определению с помощью рН-чувствительного стеклянного электрода, где жидкая водная система находится в инертной атмосфере, чтобы не допустить, например, чтобы жидкая водная система вступала в контакт с атмосферной двуокисью углерода, который, если поглощается жидкой водной системой, приведет к снижению рН.
Предпочтительно, кислотную обработку катализатора не проводят на стадии (b). Таким образом, предпочтительно, чтобы жидкая водная система не содержала соединений со значением рКа 8 или менее, предпочтительно 6 или менее. ''Не содержала соединений со значением рКа'' следует понимать в контексте настоящего изобретения таким образом, что жидкая водная система содержит менее чем 0,1 вес. % таких соединений, предпочтительно менее 0,01 вес. %, предпочтительно менее 0,001 вес. %, более предпочтительно менее 0,0001 вес. %, более предпочтительно менее 0,00001 вес. % и более предпочтительно менее 0,000001 вес. %.
Более предпочтительно, кислотную обработку катализатора не проводят в течение всего способа регенерации согласно настоящему изобретению. Таким образом, предпочтительно, чтобы соединения со значением рКа 8 или менее, предпочтительно 6 или менее, не использовались в течение всего способа регенерации согласно настоящему изобретению.
Предпочтительно, жидкая водная система на стадии (b) содержит менее 10 вес. % метанола, более предпочтительно менее 5 вес. % метанола, более предпочтительно менее 1 вес. % метанола, предпочтительно менее 0,1 вес. % метанола, более предпочтительно менее 0,01 вес. % метанола, и более предпочтительно менее 0,001 вес. % метанола, в расчете на общий вес жидкой водной системы.
Неожиданно, было обнаружено, что в этих условиях на стадии (b), промывка катализатора, содержащего титансодержащий цеолит, водной системой практически не приводит к изменениям цеолитной структуры титансодержащего цеолита. Таким образом, было обнаружено, что контактирование согласно стадии (b) не оказывает неблагоприятного воздействия на каталитическую активность катализатора, содержащего титансодержащий цеолит.
Непрерывный режим
В соответствии с предпочтительным вариантом осуществления настоящего изобретения, промывку на стадии (b) осуществляют в непрерывном режиме, которая отличается тем, что катализатор непрерывно контактирует с потоком жидкой водной системы, которую пропускают через катализатор.
Предпочтительно, промывку в непрерывном режиме проводят при среднечасовой скорости подачи (WHSV) в диапазоне от 1 до 20 ч-1, более предпочтительно от 5 до 15 ч-1, более предпочтительно от 5 до 10 ч-1. Среднечасовая скорость подачи на стадии (b) определяется массовой скоростью потока жидкой водной системы, разделенной на массу катализатора, содержащего титансодержащий цеолит, который подвергают регенерации.
Согласно этому варианту, можно проводить промывку катализатора в реакторе, в котором осуществляли реакцию эпоксидирования согласно стадии (ii). В этом случае, как уже упоминалось выше, предпочтительно прекратить воздействие на смесь, обеспеченную на стадии (i), с помощью условий эпоксидирования согласно стадии (ii), и подвергнуть отработанный катализатор стадии регенерации (b) в непрерывном режиме в реакторе, как только вся смесь, полученная на стадии (ii), была удалена из реактора, в котором осуществлялось эпоксидирование. Также можно удалить отработанный катализатор из реактора, как только вся смесь, полученная на стадии (ii), была удалена из реактора, загрузить катализатор в другой подходящий сосуд, в котором может быть проведена непрерывная промывка, и подвергать катализатор воздействию непрерывной промывки согласно стадии (b).
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают в непрерывном режиме с помощью жидкой водной системы, содержащей, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что промывку в непрерывном режиме осуществляют в реакторе согласно стадии (ii).
Периодический режим
Согласно другому варианту настоящего изобретения, промывку на стадии (b) осуществляют в периодическом режиме, который отличается тем, что катализатор контактирует один раз или несколько раз с определенным количеством жидкой системы. Например, предпочтительно, чтобы промывку на стадии (b) осуществляли посредством погружения катализатора в жидкую водную систему. Во время регенерации, можно подвергнуть смесь, полученную на стадии (ii), включая или исключая катализатор, перемешиванию. Возможно, когда промывка на стадии (b) выполняется в периодическом режиме, то жидкая водная система может быть заменена один или более раз.
Согласно этому варианту, можно проводить промывку катализатора в реакторе, в котором осуществляли реакцию эпоксидирования согласно стадии (ii). В этом случае, как уже упоминалось выше, предпочтительно, прекратить воздействие на смесь, обеспеченную на стадии (i), с помощью условий эпоксидирования согласно стадии (ii), и подвергнуть отработанный катализатор стадии регенерации (b) в периодическом режиме в реакторе, как только вся смесь, полученная на стадии (ii), была удалена из реактора, в котором осуществлялось эпоксидирование. Также можно удалить отработанный катализатор из реактора, как только вся смесь, полученная на стадии (ii), была удалена из реактора, загрузить катализатор в другой подходящий сосуд, в котором может быть проведена периодическая промывка, и подвергать катализатор воздействию периодической промывки согласно стадии (b).
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают в периодическом режиме с помощью жидкой водной системы, содержащей, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что промывку в периодическом режиме осуществляют в реакторе согласно стадии (ii).
Предпочтительно, промывку согласно стадии (b) осуществляют до тех пор, пока содержание калия в жидкой водной системе после контакта с катализатором не будет составлять не более 1000 весовых частей на миллион (вес. ч.н.м.), предпочтительно не более 250 вес. ч.н.м., более предпочтительно не более 25 вес. ч.н.м.
Предпочтительно, промывку согласно стадии (b) осуществляют до тех пор, пока содержание калия в жидкой водной системе после контакта с катализатором по отношению к содержанию калия в жидкой водной системе до контакта с катализатором не будет составлять не более 333:1, предпочтительно не более 100:1, более предпочтительно не более 10:1, более предпочтительно 1,2:1.
Как правило, если деионизированная вода используется в качестве жидкой водной системы, предпочтительно подвергать катализатор промывке согласно стадии (b) до тех пор, пока электрическая проводимость жидкой водной системы после контакта с катализатором, содержащим титансодержащий цеолит в качестве каталитически активного материала, не будет составлять не более 500 микросименс, предпочтительно не более 400 микросименс, более предпочтительно не более 300 микросименс.
Стадия (с)
После промывки согласно стадии (b), полученный катализатор, содержащий титансодержащий цеолит, может быть необязательно высушен в стадии (с) в газовом потоке, содержащем инертный газ, при температуре ниже 300°С.
Предпочтительно, температура находится в диапазоне от 20 до 200°С, предпочтительно от 25 до 100°С, более предпочтительно от 30 до 50°С.
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (с), сушку проводят, предпочтительно при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С. Кроме того, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают жидкой водной системой, которая содержит, по меньшей мере 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что на стадии (с), катализатор, полученный на стадии (b), сушат в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С.
Продолжительность сушки на стадии (с) зависит от количества катализатора, содержащего титансодержащий цеолит, который необходимо высушить в газовом потоке, содержащем инертный газ, при повышенных температурах. Возможно, что большие количества катализатора, содержащего титансодержащий цеолит, потребуют более длительного периода времени по сравнению с небольшим количеством катализатора, содержащего титансодержащий цеолит. Предпочтительно, чтобы сушка на стадии (с) осуществлялась в течение периода времени в диапазоне от 5 до 350 часов, предпочтительно от 10 до 250 часов, более предпочтительно от 12 до 100 часов.
Среднечасовая скорость подачи (WHSV) газового потока, содержащего инертный газ на стадии (с), не является предметом определенных ограничений и, как правило, находится в диапазоне от 100 до 2000 ч-1, предпочтительно от 500 до 1500 ч-1, более предпочтительно от 500 до 1000 ч-1. Среднечасовая скорость подачи на стадии (с) определяется массовой скоростью газового потока, содержащего инертный газ, разделенной на массу катализатора, содержащего титансодержащий цеолит, в реакторе.
Предпочтительно, по меньшей мере, 90 об. %, предпочтительно, по меньшей мере, 95 об. %, более предпочтительно, по меньшей мере, 99 об. % газового потока, содержащего инертный газ, согласно стадии (с), состоят из, по меньшей мере, одного инертного газа. Предпочтительно, по меньшей мере, один инертный газ выбирают из группы, состоящей из азота, гелия, аргона и смеси двух, трех или более из них. Предпочтительно, по меньшей мере, 90 об. %, предпочтительно, по меньшей мере, 95 об. %, более предпочтительно, по меньшей мере, 99 об. %, более предпочтительно, по меньшей мере, 99,9 об. % газового потока, содержащего инертный газ, согласно стадии (с), состоят из азота, предпочтительно технического азота.
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (с), сушку проводят, предпочтительно при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С. Кроме того, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают жидкой водной системой, которая содержит, по меньшей мере 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что на стадии (с), катализатор, полученный на стадии (b), сушат в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С, и который отличается тем, что, по меньшей мере, 99 об. % предпочтительно, по меньшей мере, 99,9 об. % газового потока состоят из азота, предпочтительно, технического азота.
Для получения удовлетворительных результатов, предпочтительно выполнять сушку согласно стадии (с) до тех пор, пока содержание воды в газом потоке, содержащем инертный газ, после контакта с катализатором, содержащим титансодержащий цеолит, не будет аналогичным содержанию воды в газовом потоке, содержащем инертный газ, до его контакта с катализатором. Предпочтительно, сушку на стадии (с) выполняют до тех пор, пока содержание воды в газовом потоке, содержащем инертный газ, после контакта с катализатором по отношению к содержанию воды в газовом потоке, содержащем инертный газ, до контакта с катализатором, не будет составлять не более 1,10:1, предпочтительно не более 1,08:1, более предпочтительно не более 1,05:1, более предпочтительно не более 1,03:1.
В качестве альтернативы, сушку на стадии (с) могут предпочтительно выполнять до тех пор, пока объемная доля воды в газовом потоке, содержащем инертный газ, после контакта с катализатором, содержащим титансодержащий цеолит, не будет составлять не более 500 объемных частей на миллион (об. ч.н.м.), предпочтительно не более 400 об. ч.н.м., предпочтительно не более 300 об. ч.н.м., более предпочтительно не более 250 об. ч.н.м., по отношению к общему объему газового потока, содержащего инертный газ.
Стадия (d)
Согласно стадии (d), катализатор, полученный на стадии (b) или (с), предпочтительно на стадии (с), подвергают обжигу в газовом потоке, содержащем кислород, при температуре, по меньшей мере, 300°С.
Предпочтительно, обжиг согласно стадии (d) проводят при температуре в диапазоне от 300 до 600°С, предпочтительно от 325 до 575°С, более предпочтительно от 350 до 550°С, более предпочтительно от 375 до 525°С, более предпочтительно от 400 до 500°С.
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (с), сушку проводят, предпочтительно при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С. Кроме того, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают жидкой водной системой, которая содержит, по меньшей мере 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что на стадии (с), катализатор, полученный на стадии (b), сушат в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С, и который отличается тем, что на стадии (d), катализатор, полученный на стадии (с), подвергают обжигу в газовом потоке, содержащем кислород, при температуре в диапазоне от 375 до 525°С, предпочтительно от 400 до 500°С.
Предпочтительно, газовый поток, содержащий кислород, используемый на стадии (d), имеет содержание кислорода, по меньшей мере, 1 об. %, например, по меньшей мере, 5 об. %, по меньшей мере, 10 об. %, по меньшей мере, 15 об. % или, по меньшей мере, 20 об. %. Более предпочтительно, газовый поток, содержащий кислород, используемый на стадии (d), имеет содержание кислорода в диапазоне от 1 до 50 об. %, более предпочтительно от 3 до 40 об. %, более предпочтительно от 5 до 30 об. %. Если газовый поток, содержащий кислород, используемый на стадии (d), имеет содержание кислорода менее 100 об. %, газовый поток может содержать один или более дополнительных газов, таких как азот, аргон, гелий, диоксид углерода, водяной пар или смесь двух или более из них. Более предпочтительно, газовый поток, содержащий кислород, используемый для обжига катализатора, содержащего титансодержащий цеолит, на стадии (d) представляет собой воздух или обедненный воздух.
Таким образом, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (с), сушку проводят, предпочтительно при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С. Кроме того, настоящее изобретение предпочтительно относится к способу, как описано выше, который отличается тем, что на стадии (b), катализатор, полученный на стадии (а), промывают жидкой водной системой, которая содержит, по меньшей мере 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С, и который отличается тем, что на стадии (с), катализатор, полученный на стадии (b), сушат в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С, и который отличается тем, что на стадии (d), катализатор, полученный на стадии (с), подвергают обжигу в газовом потоке, содержащем кислород, при температуре в диапазоне от 375 до 525°С, предпочтительно от 400 до 500°С, и который отличается тем, что газовой поток, содержащий кислород, используемый на стадии (d), содержит кислород в диапазоне от 3 до 40 об. %, предпочтительно от 5 до 50 об. %.
Предпочтительно, чтобы среднечасовая скорость подачи (WHSV) газового потока, содержащего кислород, на стадии (d) находилась в диапазоне от 100 до 2000 ч-1, предпочтительно от 500 до 1500 ч-1, более предпочтительно от 500 до 1000 ч-1. Среднечасовая скорость подачи на стадии (d) определяется массовой скоростью газового потока, содержащего кислород, разделенной на массу катализатора, содержащего титансодержащий цеолит, в реакторе.
Предпочтительно, согласно стадии (d) катализатор, полученный на стадии (с) или (d), предпочтительно на стадии (с), нагревают до температуры обжига со скоростью в диапазоне от 0,5 до 5 К/мин, предпочтительно от 1 до 4 К/мин, более предпочтительно от 2 до 3 К/мин.
Предпочтительно, обжиг на стадии (d) осуществляют в течение периода времени в диапазоне от 1 до 15 часов, более предпочтительно от 2 до 10 часов, более предпочтительно от 3 до 7 часов.
Сушка согласно стадии (с), а также обжиг согласно стадии (d), могут осуществляться либо в реакторе, согласно стадии (ii), либо вне реактора согласно стадии (ii). Если промывку согласно стадии (b) осуществляют в реакторе согласно стадии (ii), может быть целесообразно осуществлять сушку согласно стадии (с), если она выполняется, также в реакторе согласно стадии (ii). Что касается обжига согласно стадии (d), может быть целесообразно осуществлять его в реакторе согласно стадии (ii), если промывку согласно стадии (b) и сушку согласно стадии (с), если выполняются, также осуществляют в реакторе согласно стадии (ii), возможно, в зависимости от материала и компоновки реактора.
В соответствии с настоящим изобретением, стадии (b)-(d) могут повторяться, по меньшей мере, один раз. Таким образом, после обжига согласно стадии (d), обожженный катализатор может быть подвергнут стадии (b), повторно еще одной последовательностью стадий (b), необязательно (с), и (d). В данном цикле, соответствующие условия стадий могут быть изменены по сравнению с другим циклом. Таким образом, например, в данной последовательности (b)-(d), сушку согласно стадии (с) выполняют несмотря на то, что в другой последовательности (b)-(d), указанную сушку согласно стадии (с) не выполняли. В соответствии с настоящим изобретением, последовательность стадий (b)-(d) может повторяться от 1 до 5 раз, например, один, два, три раза, четыре раза, пять раз, при таких же или различных условиях на соответствующих стадиях (b)-(d). Принимая во внимание ''мягкие'' условия регенерации в соответствии с настоящим изобретением, было обнаружено, что даже повторение последовательности стадий (b)-(d) несколько раз не оказывает негативного воздействия на цеолитную структуру катализатора, и такое повторение может привести к очень эффективному удалению калия из катализатора.
В соответствии с настоящим изобретением, предпочтительно, чтобы в ходе последовательности стадий (а)-(d), отработанный катализатор промывали жидкой водной системой согласно стадии (b), причем данная промывка на стадии (b) является единственной обработкой с помощью жидкой системы. По сравнению с патентом WO-A 2007/013739, отсутствует сочетание стадии предварительной обработки и последующей обработки с использованием другой жидкой смеси. В частности, в соответствии с предпочтительным способом настоящего изобретения, жидкая водная система, используемая на стадии (b), по существу, состоит из воды, и по сравнению со способом согласно патенту WO-A 2007/013739, обработка водой в качестве единственной обработки с помощью жидкой смеси представляет собой чрезвычайно более ''мягкую'' регенерацию, чем обработка перекисью водорода.
Таким образом, настоящее изобретение относится к способу, как описано выше, который отличается тем, что промывка согласно стадии (b) является единственной обработкой с помощью жидкой системы во время способа регенерации, который включает в себя стадии (а), (b), необязательно (с) и (d).
Стадия (i)
В соответствии с настоящим изобретением, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), получают посредством способа получения оксида олефина, который включает в себя:
(i) обеспечение смеси, содержащей органический растворитель, олефин, эпоксидирующий агент и, по меньшей мере, частично растворенную калийсодержащую соль;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей органический растворитель и оксид олефина, и с получением катализатора, содержащего осажденную на нем калиевую соль.
Органическими растворителями, которые используются на стадии (i), в принципе, являются все растворители, известные для этой цели. Предпочтение отдается использованию органических растворителей, таких как спирты, нитрилы и их смеси, необязательно также вода. Особенно предпочтительно, чтобы органический растворитель выбирали из группы, состоящей из метанола и ацетонитрила.
Количества используемого органического растворителя могут варьироваться в широких пределах. Возможные количества используемого органического растворителя составляют от 5 до 25 г органического растворителя на грамм используемого эпоксидирующего агента. Например, органический растворитель используют в количестве от 8 до 16 г органического растворителя на грамм используемого эпоксидирующего агента, или от 10 до 14 г органического растворителя на грамм используемого эпоксидирующего агента.
Олефин, используемый на стадии (i), предпочтительно выбирают из группы, состоящей из этана, пропена, 1-бутена, 2-бутена, изобутена, бутадиена, пенденов, пиперилена, гексенов, гексадиенов, гептенов, октенов, диизобутена, триметилпентена, ноненов, додецена, тридецена, тетрадецена, эйконсенов, трипропена, тетрапропена, полибутадиенов, полиизобутенена, изопренов, терпенов, гераниола, линалоола, линалилацетата, метиленциклопропана, циклопентена, циклогексена, норборнена, циклогептена, винилциклогексана, винилоксирана, винилциклогексена, стирола, циклооктена, циклооктадиена, винилнорборнена, индена, тетрагидроиндена, метилстирола, дициклопентадиена, дивинилбензола, циклододецена, циклододекатриена, стильбена, дифеилбутадиена, витамина А, бета-каротина, винилиденфторида, аллилгалогенидов, хлористого бутилена, металлилхлорида, дихлорбутена, аллилового спирта, металлилового спирта, бутенолов, бутендиолов, циклопентендиолов, пентенолов, октадиенолов, тридеценолов, ненасыщенных стероидов, этоксиэтена, изоэвгенола, анетола, ненасыщенных карбоциклических кислот, таких как акриловая кислота, метакриловая кислота, кротоновая кислота, малеиновая кислота, винилуксусная кислота, ненасыщенных жирных кислот, таких как олеиновая кислота, линолевая кислота, пальмитиновая кислота, жиров и масел природного происхождения и их смесей. Особенно предпочтительно, чтобы олефин представлял собой пропен.
Предпочтительно, чтобы эпоксидирующим агентом, используемым на стадии (i), являлась перекись водорода. Кроме того, предпочтительно, чтобы перекись водорода представляла собой водный раствор перекиси водорода, который отличается тем, что раствор содержит предпочтительно от 30 до 50 вес. % перекиси водорода по отношению к общему количеству воды. Возможно также, что перекись водорода образуется in situ (на месте) в реакционной смеси из водорода и кислорода в присутствии подходящего катализатора или каталитической системы, например, в присутствии титансодержащего цеолита, дополнительно содержащего один или более благородных металлов, или титансодержащего цеолита и дополнительного катализатора, содержащего один или более благородных металлов, например, нанесенного на подходящий носитель, такой как уголь или подходящий неорганический оксид или смесь неорганических оксидов.
Для получения перекиси водорода, используемой на стадии (i), может быть использован способ на основе антрахинона. Данный способ основан на каталитической гидрогенизации соединения антрахинона с образованием соответствующего соединения антрагидрохинона, его последующей реакцией с кислородом с образованием перекиси водорода и последующей экстракцией образовавшейся перекиси водорода. Цикл завершается повторной гидрогенизацией соединения антрахинона, которое повторно образуется при окислении. Обзор способа с использованием антрахинона представлен в ''Ullmann's Encyclopedia of Industrial Chemistry'', 5th edition, volume 13, pages 447 to 456.
В качестве альтернативы, перекись водорода можно получить посредством анодного окисления серной кислоты с одновременным выделением водорода на катоде с получением пероксодисерной кислоты. Гидролиз пероксодисерной кислоты сперва образует пероксосерную кислоту, а затем перекись водорода и серную кислоту, которая, таким образом, восстанавливается.
В качестве дополнительной альтернативы, перекись водорода может быть получена непосредственно из элементов водорода и кислорода.
Таким образом, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение смеси, содержащей органический растворитель, пропен, перекись водорода и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что органический растворитель выбирают из группы, состоящей из метанола и ацетонитрила;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей органический растворитель и пропиленоксид, и с получением катализатора, содержащего осажденную на нем калиевую соль.
Калиевая соль
Что касается химической природы, по меньшей мере, одной калиевой соли, не существует каких-либо особых ограничений. Предпочтительно, по меньшей мере, одну калиевую соль выбирают из группы, состоящей, по меньшей мере, из одной неорганической калиевой соли, по меньшей мере, одной органической калиевой соли и комбинации, по меньшей мере, одной неорганической калиевой соли и, по меньшей мере, одной органической калиевой соли.
Предпочтительные неорганические калийсодержащие соли включают в себя, но не ограничиваются ими, галогениды калия, такие как хлорид калия или бромид калия, нитрат калия, сульфат калия, гидросульфат калия, гидроксид калия, перхлорат калия, калиевые соли, содержащие фосфор, такие как дигидрофосфат калия или гидроортофосфат калия, или фосфат калия, или пирофосфаты калия, такие как одноосновный пирофосфат калия или двухосновный пирофосфат калия, или трехосновный пирофосфат калия, или четырехосновный пирофосфат калия, или этидронаты калия, такие как одноосновный этидронат калия или двухосновный этидронат калия, или трехосновный этидронат калия, или четырехосновный этидронат калия, цианат калия, оксиды калия, такие как оксид калия (K2O) или супероксид калия (KO2) или перекись калия (K2O2).
Предпочтительные органические калийсодержащие соли включают в себя, но не ограничиваются ими, карбонат калия (K2CO3), гидрокарбонат калия, калиевые соли алифатических насыщенных карбоновых кислот, такие как монокарбоновые кислоты, предпочтительно имеющие от 1 до 6, более предпочтительно от 1 до 5, более предпочтительно от 1 до 4, более предпочтительно от 1 до 3 атомов углерода, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, дикарбоновые кислоты, предпочтительно имеющие от 2 до 6, более предпочтительно от 2 до 4 атомов углерода, такие как щавелевая кислота, малоновая кислота, янтарная кислота, винные кислоты, трикарбоновые кислоты, предпочтительно имеющие от 6 до 10 атомов углерода, такие как лимонная кислота или изолимонная кислота или пропан-1,2,3-трикарбоновая кислота, или тетракарбоновые кислоты. Предпочтительно, органическую калиевую соль выбирают из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия. Более предпочтительно, органическую калиевую соль выбирают из группы, состоящей из формиата калия, ацетата калия, пропионата калия, карбоната калия и гидрокарбоната калия. Более предпочтительно, органическую калиевую соль выбирают из группы, состоящей из формиата калия, ацетата калия, карбоната калия и гидрокарбоната калия.
Таким образом, калийсодержащую соль предпочтительно выбирают из группы, состоящей, по меньшей мере, из одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, дигидрофосфата калия или гидроортофосфата калия, или фосфата калия, или пирофосфатов калия, таких как одноосновный пирофосфат калия или двухосновный пирофосфат калия, или трехосновный пирофосфат калия, или четырехосновный пирофосфат калия, или этидронатов калия, таких как одноосновный этидронат калия или двухосновный этидронат калия, или трехосновный этидронат калия, или четырехосновный этидронат калия, по меньшей мере, одну органическую калиевую соль выбирают из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия, и комбинации, по меньшей мере, одной из, по меньшей мере, одной из неорганических калиевых солей и, по меньшей мере, одной из, по меньшей мере, одной из органических калиевых солей.
Более предпочтительно, калийсодержащую соль выбирают из группы, состоящей, по меньшей мере, из одной неорганической калиевой соли, выбранной из группы, состоящей из дигидрофосфата калия или гидроортофосфата калия, или фосфата калия, гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, по меньшей мере, одну органическую калиевую соль выбирают из группы, состоящей из формиата калия, ацетата калия, пропионата калия, карбоната калия и гидрокарбоната калия, и комбинации, по меньшей мере, одной из, по меньшей мере, одной из неорганических калиевых солей и, по меньшей мере, одной из, по меньшей мере, одной из органических калиевых солей.
Особенно предпочтительно, калийсодержащая соль согласно стадии (i) представляет собой дигидрофосфат калия, гидроортофосфат калия или формиат калия. Таким образом, если согласно стадии (i), используют одну единственную калиевую соль, калийсодержащая соль наиболее предпочтительно представляет собой дигидрофосфат калия, гидроортофосфат калия или формиат калия. Если согласно стадии (i) используют две или более калийсодержащие соли, одна калиевая соль представляет собой дигидрофосфат калия, гидроортофосфат калия или формиат калия.
Таким образом, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение смеси, содержащей органический растворитель, пропен, перекись водорода и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что органический растворитель выбирают из группы, состоящей из метанола и ацетонитрила, и который отличается тем, что калийсодержащую соль выбирают из группы, состоящей из дигидрофосфата калия, гидроортофосфата калия, формиата калия и смеси двух или более из них;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей органический растворитель и пропиленоксид, и с получением катализатора, содержащего осажденную на нем калиевую соль.
Согласно стадии (i), обеспечивается смесь, содержащая калийсодержащую соль. Что касается концентрации калийсодержащей соли в жидком потоке поступающего материала, не существует никаких особых ограничений. Предпочтительно, концентрация калийсодержащей соли в смеси, обеспеченной на стадии (i), составляет, по меньшей мере, 10%, предпочтительно находится в диапазоне от 10 до 100%, предпочтительно от 20 до 100%, более предпочтительно от 30 до 100%, более предпочтительно от 40 до 100% предела растворимости калийсодержащей соли в жидком потоке поступающего материала, обеспеченном на стадии (i). Термин ''предел растворимости, по меньшей мере, одной калиевой соли в жидком потоке поступающего материала'', как используется в контексте настоящего изобретения, относится к концентрации насыщения калийсодержащей солью жидкого потока поступающего материала, где посредством добавления большего количества калийсодержащей соли, концентрация калийсодержащей соли в качестве растворенного вещества в смеси не увеличивается и калийсодержащая соль начнет выпадать в осадок. Предел растворимости калийсодержащей соли в смеси будет зависеть от состава смеси и от условий, таких как температура и давление, при которых смесь обеспечивают на стадии (i). Определение предела растворимости калийсодержащей соли в смеси является простой и прямолинейной задачей для специалиста в данной области техники, владеющего информацией об указанных условиях и указанном составе данной смеси. Простой процедурой для оценки того, превышает ли количество добавляемой калийсодержащей соли предел растворимости, является пропускание смеси, перед воздействием на нее с помощью условий эпоксидирования на стадии (ii), через фильтр и измерение перепада давления на фильтре. Если перепад давления на фильтре увеличивается по мере того, как пропускают поток, и калийсодержащую соль находят на фильтре, когда его выводят из работы, количество добавляемой калийсодержащей соли уже превышает предел растворимости.
Предпочтительно, на стадии (i), молярное соотношение калия, содержащегося в калийсодержащей соли, по отношению к эпоксидирующему агенту, предпочтительно перекиси водорода, содержащейся в смеси, находится в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1. Молярное количество калия, содержащегося в калийсодержащей соли, относится к общему молярному количеству калия, содержащемуся во всех калийсодержащих солях, используемых на стадии (i), если используются две или более калийсодержащих соли.
Далее предпочтительно, на стадии (i), молярное соотношение калия по отношению к эпоксидирующему агенту, предпочтительно перекиси водорода, в смеси, находится в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1.
Таким образом, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение смеси, содержащей органический растворитель, пропен, перекись водорода и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что органический растворитель выбирают из группы, состоящей из метанола и ацетонитрила, и который отличается тем, что калийсодержащую соль выбирают из группы, состоящей из дигидрофосфата калия, гидроортофосфата калия, формиата калия и смеси двух или более из них;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей органический растворитель и пропиленоксид, и с получением катализатора, содержащего осажденную на нем калиевую соль,
который отличается тем, что смесь согласно стадии (i) содержит калийсодержащую соль с молярным соотношением калия, содержащегося в калийсодержащей соли, по отношению к перекиси водорода в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1.
Предпочтительно, способ получения оксида олефина согласно настоящему изобретению представляет собой непрерывный способ. Таким образом, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение жидкого потока поступающего материала, содержащего органический растворитель, олефин, эпоксидирующий агент и, по меньшей мере, частично растворенную калийсодержащую соль;
(ii) подачу потока поступающего материала, обеспеченного на стадии (i), в реактор эпоксидирования, содержащий катализатор, содержащий титансодержащий цеолит в качестве каталитически активного материала, и воздействие на поток поступающего материала с помощью условий реакции эпоксидирования в реакторе эпоксидирования, с получением реакционной смеси, содержащей органический растворитель и оксид олефина, и с получением катализатора, который содержит осажденную на нем калиевую соль.
Более предпочтительно, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение жидкого потока поступающего материала, содержащего органический растворитель, пропен, перекись водорода и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что органический растворитель выбирают из группы, состоящей из метанола и ацетонитрила и, который отличается тем, что калийсодержащую соль выбирают из группы, состоящей из дигидрофосфата, гидроортофосфата калия, формиата калия, и смеси двух или более из них;
(ii) подачу потока поступающего материала, обеспеченного на стадии (i), в реактор эпоксидирования, содержащий катализатор, содержащий титансодержащий цеолит в качестве каталитически активного материала, и воздействие на поток поступающего материала с помощью условий реакции эпоксидирования в реакторе эпоксидирования, с получением смеси, содержащей органический растворитель и пропиленоксид, и с получением катализатора, который содержит осажденную на нем калиевую соль,
который отличается тем, что смесь согласно стадии (i) содержит калийсодержащую соль с молярным соотношением калия, содержащегося в калийсодержащей соли, по отношению к перекиси водорода в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1.
Предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), не содержит дигидрофосфата аммония. Более предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), не содержит фосфата аммония, гидрофосфата аммония и дигидрофосфата аммония. Более предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), не содержит карбоната аммония, гидрокарбоната аммония, дигидрофосфата аммония, гидрофосфата аммония, фосфата аммония, гидропирофосфата аммония, пирофосфата аммония, хлорида аммония, нитрата аммония и ацетата аммония. Более предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), не содержит аммониевой соли. Термин ''не содержит'', как используется в данном контексте настоящего изобретения, относится к концентрации соответствующего соединения не более 2 вес. ч.н.м., предпочтительно не более 1 вес. ч.н.м., в расчете на общий вес смеси, предпочтительно жидкого потока поступающего материала.
Предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), содержит натрий в молярном соотношении натрия по отношению к эпоксидирующему агенту, предпочтительно перекиси водорода, в диапазоне от 1×10-6:1 до 250×10-6:1, предпочтительно от 5×10-6:1 до 50×10-6:1. Предпочтительно, смесь, предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), не содержит растворенного дигидрофосфата натрия (NaH2PO4), более предпочтительно ни растворенного дигидрофосфата натрия, ни растворенного гидроортофосфата натрия (Na2HPO4), более предпочтительно ни растворенного дигидрофосфата натрия, ни растворенного гидроортофосфата натрия, ни растворенного фосфата натрия (Na3PO4).
Жидкий поток поступающего материала
Как правило, жидкий поток поступающего материала может быть обеспечен на стадии (i) в соответствии с любым возможным способом. Предпочтительно, жидкий поток поступающего материала обеспечивают на стадии (i) посредством объединения, по меньшей мере, четырех отдельных потоков, где первый поток содержит эпоксидирующий агент, предпочтительно перекись водорода, второй поток содержит олефин, предпочтительно пропен и необязательно пропан, третий поток содержит органический растворитель, предпочтительно выбранный из группы, состоящей из метанола и ацетонитрила, и необязательно воды, а четвертый поток содержит калийсодержащую соль.
Эти, по меньшей мере, четыре отдельных потока могут быть объединены в любом подходящем порядке. Предпочтительно, поток, содержащий калийсодержащую соль, объединяют с потоком, содержащим эпоксидирующий агент, и полученный объединенный поток объединяют с потоком, который получают после объединения потока, содержащего органический растворитель, и потока, содержащего олефин. Таким образом полученный поток представляет собой жидкий поток, обеспеченный на стадии (i).
Предпочтительно, поток, содержащий пропен, дополнительно содержит пропан и отличается тем, что предпочтительно, по меньшей мере, 98 вес. %, более предпочтительно, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,5 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % потока состоит из пропена и пропана. Предпочтительно, весовое соотношение пропена и пропана в потоке составляет, по меньшей мере, 7:3. Например, может использоваться коммерчески доступный пропен, который может представлять собой либо пропен полимерного сорта, либо пропен химического сорта. Как правило, пропен полимерного сорта имеет содержание пропена в диапазоне от 99 до 99,8 вес. % и содержание пропана в диапазоне от 0,2 до 1 вес. %. Пропен химического сорта, как правило, имеет содержание пропена в диапазоне от 92 до 98 вес. % и содержание пропана в диапазоне от 2 до 8 вес. %. Предпочтительно, используется поток, имеющий содержание пропена в диапазоне от 99 до 99,8 вес. %, и содержание пропана в диапазоне от 0,2 до 1 вес. %.
Предпочтительно, поток, содержащий олефин, предпочтительно пропен и необязательно пропан не содержит катионов калия (K+) и фосфора (Р) в виде анионов, по меньшей мере, одной фосфорной оксикислоты. Термин ''не содержит катионов калия (K+)'', как используется в данном контексте настоящего изобретения, относится к потоку, содержащему олефин, содержащий катионы калия (K+) в количестве менее 1 вес. ч.н.м., предпочтительно менее 0,1 вес. ч.н.м., в расчете на общий вес потока. Термин ''не содержит фосфора (Р) в виде анионов, по меньшей мере, одной фосфорной оксикислоты'', как используемый в данном контексте настоящего изобретения, относится к потоку, содержащему олефин, который содержит фосфор (Р) в виде анионов, по меньшей мере, одной фосфорной оксикислоты в количестве менее 1 вес. ч.н.м., предпочтительно менее 0,1 вес. ч.н.м., в расчете на общий вес потока.
Поток, содержащий перекись водорода, может быть получен в соответствии с любым возможным способом. Можно получить поток, содержащий перекись водорода, посредством преобразования серной кислоты в пероксопиросерную кислоту посредством анодного окисления с одновременным выделением водорода на катоде. Гидролиз пероксопиросерной кислоты затем приводит к получению с помощью пероксомоносерной кислоты перекиси водорода и серной кислоты, которую, таким образом, получают обратно. Получение перекиси водорода из элементов также возможно. В зависимости от конкретного способа получения, поток, содержащий перекись водорода, может представлять собой, например, водный или водный/метанольный поток перекиси водорода, предпочтительно водный поток перекиси водорода. В случае использования водного питающего потока перекиси водорода, содержимое потока по отношению к перекиси водорода, как правило, находится в диапазоне от 3 до 85 вес. %, предпочтительно от 25 до 75 вес. %, более предпочтительно от 30 до 50 вес. %, например, от 30 до 40 вес. % или от 35 до 45 вес. %, или от 40 до 50 вес. %. Предпочтительно, по меньшей мере, 25 вес. %, более предпочтительно, по меньшей мере, 30 вес. %, более предпочтительно, по меньшей мере, 35 вес. % потока, содержащего перекись водорода, состоят из воды и перекиси водорода. Предпочтительные диапазоны составляют от 30 до 80 вес. % или от 35 до 75 вес. %, или от 40 до 70 вес. %.
Согласно настоящему изобретению, предпочтительно использовать поток, содержащий перекись водорода, который был получен в виде неочищенного (сырого) раствора перекиси водорода посредством экстракции смеси, полученной согласно способу, известному как способ с использованием антрахинона, с помощью которого производится практически вся продукция перекиси водорода в мире (см, например, Ullmann's Encyclopedia of Industrial Chemistry, 5th edition, volume A 13 (1989) pages 443-466), который отличается тем, что используется раствор антрахинона, содержащий алкильную группу, предпочтительно, имеющую от 2 до 10 атомов углерода, более предпочтительно, по меньшей мере, 5 атомов углерода, например, 5 атомов углерода или 6 атомов углерода, и в котором используемый растворитель обычно состоит из смеси двух различных растворителей. Данный раствор антрахинона обычно называют рабочим раствором. В данном способе, перекись водорода, которая образуется с помощью способа с использованием антрахинона, обычно отделяют посредством экстракции из соответствующего рабочего раствора после цикла гидрогенизации/повторного окисления. Указанная экстракции может быть выполнена предпочтительно, по существу, с чистой водой, при этом получают неочищенный водный раствор перекиси водорода. Хотя, как правило, можно дополнительно очистить полученный таким образом неочищенный водный раствор перекиси водорода посредством перегонки, предпочтительно, согласно настоящему изобретению, использовать такой неочищенный водный раствор перекиси водорода, который не был подвергнут очистке с помощью перегонки. Кроме того, как правило, можно подвергнуть неочищенный водный раствор перекиси водорода дальнейшей стадии экстракции, которая отличается тем, что используется подходящий экстракционный агент, предпочтительно органический растворитель. Более предпочтительно, органический растворитель, используемый для данной дополнительной стадии экстракции, является тем же самым растворителем, который используют в способе на основе антрахинона. Предпочтительно, экстракцию проводили с использованием только одного из растворителей в рабочем растворе и наиболее предпочтительно с использованием только наиболее неполярного растворителя рабочего раствора. В случае, если неочищенный водный раствор перекиси водорода подвергают такой дополнительной стадии экстракции, получают так называемый неочищенный промытый раствор перекиси водорода. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, неочищенный промытый раствор перекиси водорода используется в качестве питающего потока перекиси водорода. Получение неочищенного раствора описывается, например, в европейской патентной заявке ЕР 1.122.249 А1. Что касается термина ''по существу чистой воды'', сделана ссылка на параграф 10, стр. 3 заявки ЕР 1.122.249 А1, которая включена в виде ссылки.
Для того, чтобы обеспечить достаточную стабильность перекиси водорода во время экстракции водой, предпочтительно, по существу, чистой водой, подходящие стабилизирующие агенты обычно добавляют в воду, предпочтительно используемую, по существу, чистую воду. В частности, следует упомянуть сильные неорганические кислоты и/или хелатообразующие агенты. В соответствии с предпочтительными способами экстракции, небольшие количества нитратов и/или фосфатов и пирофосфатов, соответственно, добавляют в качестве стабилизирующих агентов, либо в виде кислот или натриевых солей. Данные стабилизирующие агенты обычно добавляют в таких количествах, чтобы неочищенный водный раствор перекиси водорода содержал от 50 до 400 вес. ч.н.м. катионов натрия, от 100 до 700 вес. ч.н.м. фосфора в пересчете на фосфат (РО43-), и от 50 до 400 вес. ч.н.м. нитратных анионов, в каждом случае в пересчете по отношению к перекиси водорода, содержащейся в неочищенном водном растворе перекиси водорода. Предпочтительные диапазоны составляют, например, от 50 до 200 вес. ч.н.м. или от 50 до 100 вес. ч.н.м. катионов натрия, от 100 до 500 вес. ч.н.м. или от 100 до 300 вес. ч.н.м. фосфора, и от 50 до 200 вес. ч.н.м. или от 50 до 100 вес. ч.н.м. нитрата. Кроме того, можно использовать другие стабилизирующие агенты, такие как станниты, например, станнит натрия (Na2SnO2), и/или органические фосфоновые кислоты, в частности органические дифосфоновые кислоты, такие как этидроновая кислота. Предпочтительно, водный поток перекиси водорода содержит натрий с молярным соотношением натрия по отношению к перекиси водорода в диапазоне от 1×10-6:1 до 250×10-6:1, более предпочтительно от 5×10-6:1 до 50×10-6:1.
Как правило, молярное соотношение воды по отношению к органическому растворителю в жидком потоке поступающего материала, обеспеченном на стадии (i), не является предметом определенных ограничений. Предпочтительно, в частности, в случае, если органическим растворителем является ацетонитрил, то молярное соотношение воды по отношению к органическому растворителю составляет не более 1:4, более предпочтительно находится в диапазоне от 1:50 до 1:4, предпочтительно от 1:15 до 1:4,1, более предпочтительно от 1:10 до 1:4,2.
Таким образом, отработанный катализатор, содержащий титансодержащий цеолит, который подвергается стадиям регенерации (а)-(d), предпочтительно получают посредством способа получения пропиленоксида, который включает в себя:
(i) обеспечение жидкого потока поступающего материала, содержащего органический растворитель, пропен, необязательно пропан, перекись водорода, воду, а также, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что органический растворитель предпочтительно выбирают из группы, состоящей из метанола и ацетонитрила и, который отличается тем, что калийсодержащую соль предпочтительно выбирают из группы, состоящей из дигидрофосфата, гидроортофосфата калия, формиата калия, и смеси двух или более из них;
(ii) подачу потока поступающего материала, обеспеченного на стадии (i), в реактор эпоксидирования, содержащий катализатор, содержащий титансодержащий цеолит в качестве каталитически активного материала, и воздействие на поток поступающего материала с помощью условий реакции эпоксидирования в реакторе эпоксидирования, с получением смеси, содержащей органический растворитель, пропиленоксид, воду, необязательно пропен, необязательно пропан, и с получением катализатора, который содержит осажденную на нем калиевую соль,
который отличается тем, что смесь согласно стадии (i) содержит калийсодержащую соль с молярным соотношением калия, содержащегося в калийсодержащей соли, по отношению к перекиси водорода в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1.
В случае, если ацетонитрил используется в качестве растворителя, смесь, обеспеченная на стадии (i), предпочтительно жидкий поток поступающего материала, обеспеченный на стадии (i), предпочтительно содержит:
ацетонитрил в количестве от 60 до 75 вес. %, предпочтительно от 60 до 65 вес. %, в расчете на общий вес жидкого потока поступающего материала;
перекись водорода в количестве от 6 до 10 вес. %, предпочтительно от 7 до 9 вес. %, в расчете на общий вес жидкого потока поступающего материала;
воду с молярным соотношением воды по отношению к ацетонитрилу не более 1:4, предпочтительно в диапазоне от 1:50 до 1:4, предпочтительно от 1:15 до 1:4,1, более предпочтительно от 1:10 до 1:4,2;
пропен с молярным соотношением пропена по отношению к перекиси водорода, содержащейся в жидком потоке поступающего материала, в диапазоне от 1:1 до 1,5:1, предпочтительно от 1,1:1 до 1,4:1; а также
необязательно пропан с молярным соотношением пропана по отношению к сумме пропена и пропана в диапазоне от 0,0001:1 до 0,15:1, предпочтительно от 0,001:1 до 0,05:1;
и отличается тем, что, по меньшей мере, 95 вес. %, предпочтительно от 95 до 100 вес. %, более предпочтительно от 98 до 100 вес. % жидкого потока поступающего материала, обеспеченного на стадии (i), состоят из пропена, перекиси водорода, ацетонитрила, воды, калийсодержащей соли и необязательно пропана.
Стадия (ii)
Смесь, обеспеченную на стадии (i), подвергают на стадии (ii) в подходящем реакторе воздействию подходящих условий эпоксидирования в присутствии катализатора, содержащего титансодержащий цеолит, в качестве каталитически активного материала.
Катализатор, содержащий титансодержащий цеолит в качестве каталитически активного материала
Как правило, титансодержащий цеолит, используемый в качестве каталитически активного материала, может иметь тип каркасной структуры в соответствии со следующими трехбуквенными кодами: ABW, АСО, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFX, AFY, АНТ, ANA, АРС, APD, AST, ASV, ATN, ATO, ATS, ATT, ATV, AWO, AWW, ВСТ, BEA, ВЕС, BIK, BOG, BPH, BRE, CAN, CAS, CDO, CFI, CGF, CGS, СНА, CHI, CLO, CON, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EMT, EPI, ERI, ESV, ETR, EUO, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFR, ISV, ITE, ITH, ITW, IWR, IWW, JBW, KFI, LAU, LEV, LIO, LOS, LOV, LTA, LTL, LTN, MAR, MAZ, MEI, MEL, МЕР, MER, MMFI, MFS, MON, MOR, MSO, MTF, MTN, MTT, MTW, MWW, NAB, NAT, NEES, NON, NPO, OBW, OFF, OSI, OSO, PAR, PAU, PHI, PON, RHO, RON, RRO, RSN, RTE, RTH, RUT, RWR, RWY, SAO, SAS, SAT, SAV, SBE, SBS, SBT, SFE, SFF, SFG, SFH, SFN SFO, SGT, SOD, SSY, STF, STI, STT, TER, THO, TON, TSC, UEI, UFI, UOZ, USI, UTL, VET, VFI, VNI, VSV, WEI, WEN, YUG, ZON, или смешанную структуру из двух или более из этих каркасных структур. Что касается трехбуквенных кодов и их определений, делается ссылка на ''Atlas of Zeolite Framework Types'', 5th edition, Elsevier, London, England (2001).
Кроме того, предпочтительно, чтобы титансодержащий цеолит имел каркасную структуру MFI, каркасную структуру MEL, каркасную структуру MWW, каркасную структуру типа MWW, каркасную структуру ITQ, каркасную структуру BEA, каркасную структуру MOR, или смешанную структуру из двух или более из этих каркасных структур, предпочтительно каркасную структуру MFI, каркасную структуру MWW или каркасную структуру типа MWW. Более предпочтительно, чтобы титансодержащий цеолит представляет собой цеолит, известный как ''TS-1'' (силикалит титана-1) или TiMWW.
Предпочтительно, в частности, в случае, если титансодержащий цеолит представляет собой TiMWW, чтобы титансодержащий цеолит содержал, по меньшей мере, один элемент, который выбирают из группы, состоящей из Al, В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Со, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, предпочтительно из группы, состоящей из В, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Со, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, более предпочтительно из группы, состоящей из Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Со, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au. Более предпочтительно, чтобы титансодержащий цеолит содержал дополнительно Zn.
Термин ''титановый цеолит с каркасной структурой типа MWW'', как используется в контексте настоящего изобретения, также упоминаемый как ''TiMWW'', относится к цеолиту с каркасной структурой MWW, который содержит титан в качестве элемента изоморфного замещения в цеолитной каркасной структуре. Предпочтительно, цеолитная каркасная структура, по существу, не содержит алюминия и, по существу, состоит из кремния, титана и кислорода. Предпочтительно, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,5 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % цеолитной каркасной структуры состоят из кремния, титана и кислорода. Необязательно, титановый цеолит с каркасной структурой типа MWW может содержать внекаркасный титан, который следует понимать как любой вид титана, который не является частью цеолитной каркасной структуры MWW. Получение катализаторов TiMWW описано, например, в патенте US 2007043226 А1, в частности в Примерах 3 и 5 патента US 2007043226 А1.
Содержание титана в титановом цеолите с каркасной структурой типа MWW не имеет особых ограничений. Предпочтительно, титановый цеолит с каркасной структурой типа MWW, содержащийся в катализаторе на стадии (ii), содержит титан, в пересчете на элементарный титан, в количестве в диапазоне от 0,1 до 5 вес. %, более предпочтительно от 0,2 до 4 вес. %, более предпочтительно от 0,5 до 3 вес. %, более предпочтительно от 1 до 2 вес. %, в расчете на общий вес титанового цеолита с каркасной структурой типа MWW. Таким образом, настоящее изобретение относится к способу, как описано выше, который отличается тем, что титановый цеолит с каркасной структурой типа MWW, содержащийся в катализаторе на стадии (ii), содержит титан, в пересчете на элементарный титан, в количестве в диапазоне от 0,1 до 5 вес. %, предпочтительно от 1 до 2 вес. %, кремний, в расчете на общий вес на титанового цеолита с каркасной структурой типа MWW.
В дополнение к титану, титановый цеолит с каркасной структурой типа MWW может содержать, по меньшей мере, один дополнительный элемент, иной, нежели титан, кремний и кислород. Как правило, возможно, что данный, по меньшей мере, один дополнительный элемент представляет собой элемент изоморфного замещения, который является частью цеолитной каркасной структуры MWW. Предпочтительно, данный, по меньшей мере, один дополнительный элемент не является элементом изоморфного замещения. Такой дополнительный элемент, который не является элементом изоморфного замещения, может быть применен к цеолиту, например, с помощью способа распыления, мокрой пропитки, например, посредством пропитки по влагоемкости, или любым другим подходящим способом. Предпочтительно, по меньшей мере, один дополнительный элемент выбирают из группы, состоящей из Al, Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, а также комбинации двух или более из них, предпочтительно из группы, состоящей из Zr, V, Nb, Та, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, a также комбинации двух или более из них. Более предпочтительно, титановый цеолит с каркасной структурой типа MWW содержит цинк в качестве дополнительного элемента в дополнение к титану, кремнию и кислороду. Более предпочтительно, титановый цеолит с каркасной структурой типа MWW содержит цинк в качестве единственного дополнительного элемента в дополнение к титану, кремнию и кислороду. Более предпочтительно, титановый цеолит с каркасной структурой типа MWW содержит цинк в качестве единственного дополнительного элемента в дополнение к титану, кремнию и кислороду, и отличается тем, что, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,5 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % цеолитной каркасной структуры состоят из кремния, титана и кислорода. Более предпочтительно, в случае, если титановый цеолит с каркасной структурой типа MWW содержит цинк в качестве единственного дополнительного элемента, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,5 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % титанового цеолита с каркасной структурой типа MWW состоят из цинка, титана, кремния и кислорода; такой титановый цеолит с каркасной структурой типа MWW, который содержит цинк в качестве единственного дополнительного элемента, также называют ''ZnTiMWW''.
ZnTiMWW катализатор
Содержание цинка в титановом цеолите с каркасной структурой типа MWW не имеет особых ограничений. Предпочтительно, титановый цеолит с каркасной структурой типа MWW, содержащийся в катализаторе на стадии (ii), содержит цинк, в пересчете на элементарный цинк, в количестве в диапазоне от 0,1 до 5 вес. %, более предпочтительно от 0,2 до 4 вес. %, более предпочтительно от 0,5 до 3 вес. %, более предпочтительно от 1 до 2 вес. %, в расчете на общий вес титанового цеолита с каркасной структурой типа MWW. Таким образом, настоящее изобретение относится к способу, как описано выше, который отличается тем, что титановый цеолит с каркасной структурой типа MWW, содержащийся в катализаторе на стадии (ii), содержит цинк, в пересчете на элементарный цинк, в количестве в диапазоне от 0,1 до 5 вес. %, предпочтительно от 1 до 2 вес. %, в расчете на общий вес титанового цеолита с каркасной структурой типа MWW.
Катализатор согласно стадии (ii), содержащий титановый цеолит с каркасной структурой типа MWW, может состоять из титанового цеолита с каркасной структурой типа MWW, предпочтительно состоит из TiMWW или ZnTiMWW, как описано. В таких случаях, катализатор может представлять собой титановый цеолит с каркасной структурой типа MWW в виде цеолитного порошка, который может подвергаться формованию, например, в виде гранул, микросфер, таких как микросферы, полученные посредством распылительной сушки или посредством распылительной грануляции, при этом формованное тело имеет, например, форму гранулы, таблетки, цилиндра, колеса, звезды, сферы и так далее.
Предпочтительно, катализатор согласно стадии (ii), содержащий титановый цеолит с каркасной структурой типа MWW, предпочтительно TiMWW или ZnTiMWW, получают в виде формованного изделия содержащего титановый цеолит с каркасной структурой типа MWW, предпочтительно TiMWW или ZnTiMWW, посредством подходящего смешивания титанового цеолита с каркасной структурой типа MWW, по меньшей мере, с одним связующим веществом и/или, по меньшей мере, с одним предшественником связующего вещества, и необязательно, по меньшей мере, с одним порообразующим агентом и/или, по меньшей мере, с одним пластификатором. Формованные изделия могут иметь форму любой возможной геометрии, например, форму заготовок, имеющих, например, прямоугольное, треугольное, гексагональное, квадратное, овальные или круглое поперечное сечение, в виде звезд, таблеток, сфер, полых цилиндров, и тому подобное. Примерами таких связующих веществ являются оксиды металлов, такие как, например, SiO2, Al2O3, TiO2, ZrO2 или MgO или глины или смеси двух или более из этих оксидов или смеси оксидов, по меньшей мере, двух из Si, Al, Ti, Zr и Mg с SiO2, которые является предпочтительным. Порообразующие агенты, такие как образующие мезопоры агенты, включают в себя полимерные виниловые соединения, такие как полиалкиленоксиды, например, полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и сложные полиэфиры. Клеящие агенты включают в себя органические вещества, в частности, гидрофильные полимеры, например, углеводы, такие как целлюлоза, производные целлюлозы, такие как метилцеллюлоза, и крахмал, такой как картофельный крахмал, обойный клей, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутилен или политетрагидрофуран. Следует упомянуть использование воды, спиртов или гликолей, или их смесей, таких как смеси воды и спирта, или воды и гликоля, таких как, например, воды и метанола, или воды и этанола, или воды и пропанола, или воды и пропиленгликоля, в качестве клеящих агентов. Предпочтительно, катализатор согласно стадии (ii) используют в виде формованного изделия, имеющего форму экструдатов, предпочтительно экструдатов, имеющих длину предпочтительно от 1 до 10 мм, более предпочтительно от 1 до 7 мм, более предпочтительно от 1 до 5 мм, и диаметр предпочтительно от 0,1 до 5 мм, более предпочтительно от 0,2 до 4 мм, более предпочтительно от 0,5 до 2 мм. В частности, что касается предпочтительного катализатора согласно стадии (ii), содержащего ZnTiMWW, предпочтительно использовать данный катализатор в виде микропорошка или в виде формованного изделия, отличающегося тем, что формованное изделие предпочтительно содержит указанный микропорошок.
Указанный катализатор согласно стадии (ii) настоящего изобретения в виде микропорошка, содержащего ZnTiMWW, предпочтительно отличается следующими особенностями и вариантами, включая комбинации вариантов в соответствии с указанными зависимостями:
1. Микропорошок, частицы которого имеют значение Dv10, по меньшей мере, 2 мкм, причем указанный микропорошок содержит мезопоры, имеющие средний диаметр пор (4V/A) в диапазоне от 2 до 50 нм, как определено методом ртутной порометрии в соответствии с DIN 66133, и содержит, в расчете на вес микропорошка, по меньшей мере, 95 вес. % микропористого цеолитного материала без содержания алюминия структурного типа MWW, содержащего титан и цинк (ZnTiMWW). Значение Dv10 следует понимать согласно определению в соответствии со Справочным примером 5.1 настоящего изобретения.
2. Микропорошок согласно варианту 1, имеющий значение Dv10 в диапазоне от 2 до 5,5 мкм, предпочтительно от 3 до 5,5 мкм.
3. Микропорошок согласно варианту 1 или 2, имеющий значение Dv50 в диапазоне от 7 до 25 мкм и значение Dv90 в диапазоне от 26 до 85 мкм. Значения Dv50 и Dv90 следует понимать согласно определению в соответствии со Справочным примером 5.1 настоящего изобретения.
4. Микропорошок согласно любому из вариантов 1-3, в котором мезопоры имеют средний диаметр пор (4V/A) в диапазоне от 10 до 50 нм, предпочтительно от 15 до 40 нм, более предпочтительно от 20 до 30 нм, как определено методом ртутной порометрии в соответствии с DIN 66133.
5. Микропорошок согласно любому из вариантов 1-4, который дополнительно содержит макропоры, имеющие средний диаметр пор (4V/A) в диапазоне от более чем 50 нм, причем указанные макропоры предпочтительно имеют средний диаметр пор в диапазоне от 0,05 до 3 мкм, как определено методом ртутной порометрии в соответствии с DIN 66133.
6. Микропорошок согласно любому из вариантов 1-5, в котором микропоры ZnTiMWW имеют средний диаметр пор в диапазоне от 1,0 до 1,2 нм, как определено методом адсорбции азота в соответствии с DIN 66135.
7. Микропорошок согласно любому из вариантов 1-6, содержащий, в расчете на вес микропорошка, по меньшей мере, 99 вес. %, предпочтительно, по меньшей мере, 99,7 вес. % ZnTiMWW.
8. Микропорошок согласно любому из вариантов 1-7, в котором ZnTiMWW содержит цинк в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,9 вес. %, в пересчете на Zn и в расчете на вес ZnTiMWW.
9. Микропорошок согласно любому из вариантов 1-8, в котором ZnTiMWW содержит титан в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,8 вес. %, в пересчете на Ti и в расчете на вес ZnTiMWW.
10. Микропорошок согласно любому из вариантов 1-9, имеющий степень кристалличности, как определено рентгенодифракционным (XRD) анализом, по меньшей мере, (80 +/- 10) %, предпочтительно, по меньшей мере, (85 +/- 10) %. Степень кристалличности следует понимать согласно определению в соответствии со Справочным примером 5.7 настоящего изобретения.
11. Микропорошок согласно любому из вариантов 1-10, содержащий, в расчете на общий вес микропорошка и в пересчете на элемент, менее 0,001 вес. %, предпочтительно менее 0,0001 вес. % благородного металла, предпочтительно выбранного из группы, состоящей из золота, серебра, платины, палладия, иридия, рутения, осмия и смеси двух или более из них, более предпочтительно выбранного из группы, состоящей из золота, платины, золота и смеси двух или более из них.
12. Микропорошок согласно любому из вариантов 1-11, содержащий, в расчете на общий вес микропорошка и в пересчете на элемент, менее 0,1 вес. %, предпочтительно менее 0,01 вес. % бора.
13. Микропорошок согласно любому из вариантов 1-12, имеющий объемную плотность в диапазоне от 80 до 100 г/мл.
14. Микропорошок согласно любому из вариантов 1-13, который представляет собой распылительный порошок, предпочтительно получаемый или полученный посредством распылительной сушки.
Кроме того, указанный катализатор согласно стадии (ii) настоящего изобретения в виде формованного изделия, содержащего ZnTiMWW, предпочтительно отличается следующими особенностями и вариантами, включая комбинации вариантов в соответствии с указанными зависимостями:
1. Формованное изделие, содержащее микропористый цеолитный материал без содержания алюминия структурного типа MWW, содержащее титан и цинк (ZnTiMWW), причем указанное формованное изделие предпочтительно содержит микропорошок, содержащий, в расчете на вес микропорошка, по меньшей мере, 95 вес. % микропористого цеолитного материала без содержания алюминия структурного типа MWW, содержащего титан и цинк (ZnTiMWW), причем указанное формованное изделие более предпочтительно содержит микропорошок согласно любому из вариантов микропорошка 1-14, как описано выше, формованное изделие предпочтительно дополнительно содержит, по меньшей мере, одно связующее вещество, предпочтительно связующее вещество на основе диоксида кремния.
2. Формованное изделие согласно варианту 1, содержащее мезопоры, имеющие средний диаметр пор в диапазоне от 4 до 40 нм, предпочтительно от 20 до 30 нм, как определено методом ртутной порометрии в соответствии с DIN 66133.
3. Формованное изделие согласно варианту 1 или 2, имеющее степень кристалличности, как определено рентгенодифракционным (XRD) анализом, по меньшей мере, (55 +/- 10) %, предпочтительно в диапазоне от ((55 до 75) +/- 10) %. Степень кристалличности следует понимать согласно определению в соответствии со Справочным примером 5.7 настоящего изобретения.
4. Формованное изделие согласно любому из вариантов 1-3, содержащее микропорошок в количестве в диапазоне от 70 до 80 вес. % и связующее вещество на основе диоксида кремния в количестве от 30 до 20 вес. %, причем микропорошок вместе со связующим веществом на основе диоксида кремния составляют, по меньшей мере, 99 вес. % формованного изделия, и которое отличается тем, что формованное изделие имеет концентрацию силанольных групп по отношению к общему числу атомов Si не более 6%, предпочтительно не более 3%, как определено методом ядерного магнитного резонанса при вращении образца под магическим углом 29Si MAS ЯМР. Концентрацию силанольных групп следует понимать согласно определению в соответствии со Справочным примером 5.2 настоящего изобретения.
5. Формованное изделие согласно любому из вариантов 1-4, представляющее собой заготовку, имеющую круглое поперечное сечение и диаметр в диапазоне от 1,5 до 1,7 мм, и имеющую прочность на раздавливание, по меньшей мере, 5 Н, предпочтительно в диапазоне от 5 до 20 Н, более предпочтительно в диапазоне от 12 до 20 Н, причем прочность на раздавливание определяется с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS1S в соответствии с методом, как описано в Справочном примере 5.3 настоящего изобретения.
6. Формованное изделие согласно любому из вариантов 1-5, причем 29Si-ЯМР-спектр указанного формованного изделия содержит шесть пиков в следующих положениях
пик 1 при -98 +/- х частей на миллион (ч.н.м.),
пик 2 при -104 +/- х ч.н.м.,
пик 3 при -110 +/- х ч.н.м.,
пик 4 при -113 +/- х ч.н.м.,
пик 5 при -115 +/- х ч.н.м.,
пик 6 при -118 +/- х ч.н.м.,
где х в любом из пиков составляет 1,5, предпочтительно 1,0, более предпочтительно 0,5,
где Q, которое определено как
Q=100*{[a1+a2]/[а4+а5+а6]}/а3
составляет не более 2,5, предпочтительно не более 1,6, предпочтительно не более 1,4, где [a1+а2] представляет собой сумму площадей пиков 1 и 2, и [а4+a5+а6] представляет собой сумму площадей пиков 4, 5 и 6, и а3 представляет собой площадь пика 3. Данные характеристики 29Si-ЯМР следует понимать согласно определению в соответствии со Справочным примером 5.4 настоящего изобретения.
7. Формованное изделие согласно любому из вариантов 1-6, имеющее водопоглощение в диапазоне от 3 до 8 вес. %, предпочтительно от 4 до 7 вес. %. Водопоглощение следует понимать согласно определению в соответствии со Справочным примером 5.5 настоящего изобретения.
8. Формованное изделие согласно любому из вариантов 1-7, причем инфракрасный спектр указанного формованного изделия содержит полосу в области (3700-3750) см-1 +/- 20 см-1 и полосу в области (3670-3690) см-1 +/- 20 см-1, где соотношение интенсивности полосы в области (3700-3750) см-1 +/- 20 см-1 по отношению к полосе в области (3670-3690) см-1 +/- 20 см-1, составляет не более 1,5, предпочтительно не более 1,4. Данные ИК характеристики следует понимать согласно определению в соответствии со Справочным примером 5.6 настоящего изобретения.
Кроме того, предпочтительный способ получения указанного катализатора согласно стадии (ii) в виде микропорошка и/или формованного изделия, содержащего ZnTiMWW, отличается следующими особенностями и вариантами, включая комбинации вариантов в соответствии с указанными зависимостями:
1. Способ, который включает в себя:
(a) получение суспензии, содержащей микропористый цеолитный материал без содержания алюминия структурного типа MWW, содержащий титан и цинк (ZnTiMWW);
(b) воздействие на суспензию, полученную на стадии (а), посредством распылительной сушки для получения микропорошка;
(c) необязательно обжиг микропорошка, полученного на стадии (b),
который отличается тем, что микропорошок, полученный на стадии (b) или (с), предпочтительно на стадии (с), представляет собой предпочтительно микропорошок согласно любому из указанных вариантов микропорошка 1-14, как описано выше.
2. Способ согласно варианту 1, который отличается тем, что суспензия, полученная на стадии (а) имеет содержание твердых веществ в диапазоне от 5 до 25 вес. %, предпочтительно от 10 до 20 вес. %, причем суспензия предпочтительно является водной суспензией.
3. Способ согласно варианту 1 или 2, который отличается тем, что ZnTiMWW согласно стадии (а) содержит цинк в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,9 вес. %, в пересчете на Zn, и титан в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,8 вес. %, в пересчете на Ti и в расчете на вес ZnTiMWW.
4. Способ согласно любому из вариантов 1-3, который отличается тем, что на стадии (b), распылительный аппарат, предпочтительно распылительная башня, используется для распылительной сушки суспензии, причем указанный аппарат имеет, по меньшей мере, одно распылительное сопло, предпочтительно, по меньшей мере, одно двухкомпонентное сопло, причем указанное сопло имеет диаметр в диапазоне от 3,5 до 4,5 мм.
5. Способ согласно любому из вариантов 1-4, который отличается тем, что на стадии (b), распылительный аппарат, предпочтительно распылительная башня используется для распылительной сушки суспензии, причем указанный аппарат работает с распылительным газом, имеющим температуру в диапазоне от 20 до 50°С, предпочтительно от 20 до 30°С, и сушильным газом, имеющим температуру в диапазоне от 250 до 350°С, предпочтительно от 275 до 325°С, причем указанный распылительный газ предпочтительно представляет собой инертный газ, более предпочтительно технический азот, и указанный сушильный газ предпочтительно представляет собой инертный газ, более предпочтительно технический азот.
6. Способ согласно любому из вариантов 1-5, который отличается тем, что на стадии (с) микропорошок подвергают обжигу при температуре в диапазоне от 600 до 700°С в течение времени в диапазоне от 0,5 до 6 часов.
7. Способ согласно любому из вариантов 1-6, который дополнительно включает в себя:
(d) формование микропорошка, полученного на стадии (b) или (с), с получением формованного изделия;
(e) необязательно сушку и/или обжиг формованного изделия, полученного на стадии (d).
8. Способ согласно варианту 7, который отличается тем, что формование согласно стадии (d) включает в себя:
(аа) смешивание микропорошка со связующим веществом или предшественником связующего вещества, предпочтительно связующим веществом на основе диоксида кремния или предшественником связующего вещества на основе диоксида кремния, которое отличается тем, что весовое соотношение ZnTiMWW, содержащегося в микропорошке, и диоксида кремния, содержащегося или получаемого вследствие связующего вещества, находится в диапазоне от 3:7 до 1:4, для получения смеси;
(bb) формование смеси, полученной на стадии (аа), для получения формованного изделия, причем указанное формование предпочтительно включает в себя воздействие на смесь, полученную на стадии (аа), посредством экструзии, в результате которой предпочтительно получают заготовки, имеющие диаметр предпочтительно в диапазоне от 1,0 до 2,0 мм, более предпочтительно от 1,5 до 1,7 мм.
9. Способ согласно варианту 8, который отличается тем, что на стадии (аа) добавляют углевод и/или воду в качестве клеящего вещества.
10. Способ согласно варианту 8 или 9, который отличается тем, что смешивание на стадии (аа) осуществляют в течение времени в диапазоне от 15 до 60 минут, предпочтительно от 30 до 55 минут, более предпочтительно от 40 до 50 минут.
11. Способ согласно любому из вариантов 7-10, который отличается тем, что на стадии (d) не добавляют образующий мезопоры агент, предпочтительно выбранный из группы, состоящей из полиалкиленоксидов, таких как полиэтиленоксид, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и полиэфиры.
12. Способ согласно любому из вариантов 7-11, который отличается тем, что на стадии (е), формованное изделие сушат при температуре в диапазоне от 100 до 150°С в течение времени в диапазоне от 10 до 20 часов и обжигают при температуре в диапазоне от 500 до 600°С в течение времени в диапазоне от 0,5 до 2 часов.
13. Способ согласно любому из вариантов 7-12, который дополнительно включает в себя:
(f) воздействие на формованное изделие, полученное на стадии (d) или (е), предпочтительно на стадии (е), посредством обработки водой;
(g) необязательно сушку и/или обжиг обработанного водой формованного изделия,
который отличается тем, что формованное изделие, полученное на стадии (f) или (g), предпочтительно на стадии (g), представляет собой предпочтительно формованное изделие согласно любому из указанных вариантов формованного изделия 1-8, как описано выше.
14. Способ согласно варианту 13, который отличается тем, что на стадии (f), обработка водой включает в себя обработку формованного изделия жидкой водой в автоклаве при автогенном давлении при температуре в диапазоне от 100 до 200°С, предпочтительно от 125 до 175°С, более предпочтительно от 140 от 150°С в течение периода времени от 2 до 24 часов, предпочтительно от 6 до 10 часов.
15. Способ согласно варианту 13 или 14, который отличается тем, что на стадии (f), весовое соотношение формованного изделия и воды находится в диапазоне от 0,02 до 0,08, предпочтительно от 0,03 до 0,07, более предпочтительно от 0,04 до 0,06.
16. Способ согласно любому из вариантов 13-15, который отличается тем, что на стадии (g), обработанное водой формованное изделие сушат при температуре в диапазоне от 100 до 150°С в течение времени в диапазоне от 10 до 20 часов и обжигают при температуре в диапазоне от 400 до 500°С в течение времени в диапазоне от 1 до 3 часов.
17. Способ согласно любому из вариантов 7-16, который отличается тем, что формованное изделие не подвергают обработке паром.
Что касается указанного предпочтительного способа получения указанного катализатора согласно стадии (b) в виде микропорошка и/или формованного изделия, содержащего ZnTiMWW, описанного выше в вариантах 1-17, цеолит ZnTiMWW, на основе которого получают суспензию в варианте 1.(а), может быть получен в соответствии со всеми возможными методами. Например, можно получить микропористый цеолитный материал без содержания алюминия структурного типа MWW, содержащий титан (TiMWW), и подвергнуть TiMWW подходящей обработке для получения ZnTiMWW. Кроме того, можно получить цеолитный материал без содержания алюминия структурного типа MWW (MWW) и подвергнуть MWW подходящей обработке для получения ZnTiMWW, который отличается тем, что, например, как Zn, так и Ti, соответствующим образом включены в MWW. Кроме того, возможно получить цеолитный материал без содержания алюминия структурного типа MWW, который отличается тем, что во время синтеза каркасной структуры типа MWW, вводят Ti и полученный материал подвергают подходящей обработке для введения Zn, или вводят Zn и полученный материал подвергают подходящей обработке для введения Ti, или вводятся оба материала, Zn и Ti. В качестве возможных методов получения TiMWW, могут быть упомянуты способы, как описано, например, в патенте US 6.114.551 или в Wu et al., ''Hydrothermal Synthesis of a novel Titanosilicate with MWW Topology'', Chemistry Letters (2000), pp. 774-775. Предпочтительно, цеолитный материал без содержания алюминия структурного типа MWW, содержащий Ti (TiMWW) получают на первой стадии, а на второй стадии TiMWW подвергают подходящий обработке для получения ZnTiMWW. Более предпочтительно, ZnTiMWW получают согласно способу, который включает в себя:
(I) получение цеолитного материала без содержания алюминия структурного типа MWW, содержащего бор (B-MWW);
(II) деборирование B-MWW для получения цеолитного материала без содержания алюминия структурного типа MWW (MWW);
(III) введение титана (Ti) в MWW для получения цеолитного материала без содержания алюминия структурного типа MWW, содержащего Ti (TiMWW);
(IV) предпочтительно кислотную обработку TiMWW;
(V) воздействие на TiMWW посредством пропитки цинком (Zn) для получения ZnTiMWW.
Предпочтительно, на стадии (I), B-MWW получают посредством способа, предпочтительные стадии и условия которого определены в соответствии со следующими вариантами 1-28 и соответствующими зависимостями, как указано:
1. Способ получения борсодержащего цеолитного материала без содержания алюминия, имеющего каркасную структуру MWW (B-MWW), который включает в себя:
(a) гидротермальный синтез предшественника B-MWW из смеси для синтеза, содержащей воду, источник кремния, источник бора, а также соединение-шаблон MWW, с получением предшественника B-MWW в своем маточном растворе, причем маточный раствор имеет рН выше 9;
(b) регулирование рН маточного раствора, полученного на стадии (а), и содержащего предшественник B-MWW, до величины в диапазоне от 6 до 9;
(c) отделение предшественника B-MWW от маточного раствора с отрегулированным рН, полученного на стадии (b), посредством фильтрации в фильтрующем устройстве.
2. Способ согласно варианту 1, который отличается тем, что на стадии (а), по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 99 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. % смеси для синтеза состоят из воды, источника кремния, источника бора и соединения-шаблона.
3. Способ согласно варианту 1 или 2, который отличается тем, что на стадии (а), источник кремния предпочтительно выбирают из группы, состоящей из пирогенного диоксида кремния, коллоидного диоксида кремния, и их смеси, причем источником кремния предпочтительно является коллоидный диоксид кремния, более предпочтительно диоксид кремния, стабилизированный аммиаком, источник бора предпочтительно выбирают из группы, состоящей из борной кислоты, боратов, оксида бора и смеси двух или более из них, причем источником бора предпочтительно является борная кислота, и соединение-шаблон MWW предпочтительно выбирают из группы, состоящей из пиперидина, гексаметиленимина, ионов N,N,N,N',N',N'-гексаметил-1,5-пентандиаммония, 1,4-бис(N-метилпирролидиний)бутана, гидроксида октилтриметиламмонийхлорида, гидроксида гептилтриметиламмония, гидроксида гексилтриметиламмония, гидроксида N,N,N-триметил-1-адамантиламмония и смеси двух или более из них, причем соединение-шаблон MWW предпочтительно представляет собой пиперидин.
4. Способ согласно любому из вариантов 1-3, который отличается тем, что на стадии (а), смесь для синтеза содержит источник бора, в пересчете на элементарный бор, по отношению к источнику кремния, в пересчете на элементарный кремний, в молярном соотношении в диапазоне от 0,4:1 до 2,0:1, предпочтительно от 0,6:1 до 1,9:1, более предпочтительно от 0,9:1 до 1,4:1, воду по отношению к источнику кремния, в пересчете на элементарный кремний, в молярном соотношении в диапазоне от 1:1 до 30:1, предпочтительно от 3:1 до 25:1, более предпочтительно от 6:1 до 20:1; и соединение-шаблон по отношению к источнику кремния, в пересчете на элементарный кремний, в молярном соотношении в диапазоне от 0,4:1 до 2,0:1, предпочтительно от 0,6:1 до 1,9:1, более предпочтительно от 0,9:1 до 1,4:1.
5. Способ согласно любому из вариантов 1-4, который отличается тем, что на стадии (а), гидротермальный синтез осуществляют при температуре в диапазоне от 160 до менее чем 180°С, предпочтительно от 170 до 175°С, в течение периода времени в диапазоне от 1 до 72 часов, предпочтительно от 6 до 60 часов, более предпочтительно от 12 до 50 часов.
6. Способ согласно любому из вариантов 1-5, который отличается тем, что на стадии (а), гидротермальный синтез осуществляют, по меньшей мере, частично при перемешивании.
7. Способ согласно любому из вариантов 1-6, который отличается тем, что на стадии (а), смесь для синтеза дополнительно содержит затравочный материал, предпочтительно цеолитный материал, имеющий каркасную структуру MWW, более предпочтительно борсодержащий цеолитный материал, имеющий каркасную структуру MWW.
8. Способ согласно варианту 7, который отличается тем, что смесь для синтеза содержит затравочный материал, по отношению к источнику кремния, в весовом соотношении в диапазоне от 0,01:1 до 1:1, предпочтительно от 0,02:1 до 0,5:1, более предпочтительно от 0,03:1 до 0,1:1, в пересчете на количество затравочного материала в кг по отношению к кремнию, содержащемуся в источнике кремния, в пересчете на диоксид кремния в кг.
9. Способ согласно любому из вариантов 1-8, который отличается тем, что рН маточного раствора, полученного на стадии (а), составляет выше 10, предпочтительно находится в диапазоне от 10,5 до 12, более предпочтительно от 11 до 11,5.
10. Способ согласно любому из вариантов 1-9, который отличается тем, что на стадии (b), рН маточного раствора, полученного на стадии (а), доводят до значения в диапазоне от 6,5 до 8,5, предпочтительно от 7 до 8.
11. Способ согласно любому из вариантов 1-10, который отличается тем, что на стадии (b), рН регулируют с помощью метода, который включает в себя:
(аа) добавление кислоты к маточному раствору, полученному на стадии (а), содержащему предшественник B-MWW, и который отличается тем, что добавление предпочтительно осуществляют, по меньшей мере, частично при перемешивании.
12. Способ согласно варианту 11, который отличается тем, что на стадии (аа), добавление осуществляют при температуре в диапазоне от 20 до 70°С, предпочтительно от 30 до 65°С, более предпочтительно от 40 до 60°С.
13. Способ согласно варианту 11 или 12, который отличается тем, что на стадии (аа), кислота представляет собой неорганическую кислоту, предпочтительно водный раствор, содержащий неорганическую кислоту.
14. Способ согласно варианту 13, который отличается тем, что неорганическую кислоту предпочтительно выбирают из группы, состоящей из фосфорной кислоты, серной кислоты, соляной кислоты, азотной кислоты, а также смеси двух или более из них, причем неорганическая кислота предпочтительно представляет собой азотную кислоту.
15. Способ согласно любому из вариантов 11-14, при этом способ дополнительно включает в себя:
(bb) перемешивание маточного раствора, к которому была добавлена кислота согласно стадии (аа), и который отличается тем, что во время стадии (bb) кислоту не добавляют к маточному раствору.
16. Способ согласно варианту 15, который отличается тем, что на стадии (bb), перемешивание осуществляют при температуре в диапазоне от 20 до 70°С, предпочтительно от 25 до 65°С, более предпочтительно от 30 до 60°С.
17. Способ согласно любому из вариантов 1-16, который отличается тем, что на стадии (b), размер частиц, содержащихся в маточном растворе, выраженный соответствующим значением Dv10, Dv50 и Dv90, увеличивается, по меньшей мере, на 2%, предпочтительно, по меньшей мере, на 3%, более предпочтительно, по меньшей мере, на 4,5% в отношении Dv10, по меньшей мере, на 2%, предпочтительно, по меньшей мере, на 3%, более предпочтительно, по меньшей мере, на 4,5% в отношении Dv50, и, по меньшей мере, на 5%, предпочтительно, по меньшей мере, на 6%, более предпочтительно, по меньшей мере, на 7% в отношении Dv90.
18. Способ согласно любому из вариантов 1-17, который отличается тем, что маточный раствор с отрегулированным рН, полученный на стадии (b), имеет содержание твердых веществ в диапазоне от 1 до 10 вес. %, предпочтительно от 4 до 9 вес. %, более предпочтительно от 7 до 8 вес. %, в расчете на общий вес маточного раствора с отрегулированным рН, полученного на стадии (b).
19. Способ согласно любому из вариантов 1-18, который отличается тем, что маточный раствор с отрегулированным рН, полученный на стадии (b), имеет фильтрационное сопротивление в диапазоне от 10 до 50 мПа*с/м2, предпочтительно от 15 до 45 мПа*с/м2, более предпочтительно от 20 до 40 мПа*с/м2.
20. Способ согласно любому из вариантов 1-19, который дополнительно включает в себя:
(d) промывку предшественника B-MWW, полученного на стадии (с), предпочтительно фильтровального осадка, полученного на стадии (с), и который отличается тем, что промывку предпочтительно выполняют с использованием воды в качестве промывочного агента.
21. Способ согласно варианту 20, который отличается тем, что на стадии (d), фильтровальный осадок, полученный на стадии (с), имеет стойкость к вымыванию в диапазоне от 10 до 50 мПа*с/м2, предпочтительно от 15 до 45 мПа*с/м2, более предпочтительно от 20 до 40 мПа*с/м2.
22. Способ согласно варианту 20 или 21, который отличается тем, что промывку осуществляют до тех пор, пока электрическая проводимость фильтрата не будет составлять не более 300 мкСм/см, предпочтительно не более 250 мкСм/см, более предпочтительно не более 200 мкСм/см.
23. Способ согласно любому из вариантов 1-22, который дополнительно включает в себя:
(e) сушку предшественника B-MWW, полученного на стадии (с), предпочтительно на стадии (d), при температуре в диапазоне от 20 до 50°С, предпочтительно от 20 до 40°С, более предпочтительно от 20 до 30°С, и который отличается тем, что сушку предпочтительно осуществляют посредством воздействия на B-MWW газовым потоком, предпочтительно потоком азота.
24. Способ согласно любому из вариантов 1-23, который отличается тем, что остаточная влажность предшественника B-MWW, полученного на стадии (с), предпочтительно на стадии (d), более предпочтительно на стадии (е), находится в диапазоне от 80 до 90 вес. %, предпочтительно от 80 до 85 вес. %.
25. Способ согласно любому из вариантов 1-24, который дополнительно включает в себя:
(f) получение суспензии, предпочтительно водной суспензии, содержащей предшественник B-MWW, полученный на стадии (с), предпочтительно на стадии (d), более предпочтительно на стадии (е), и имеющей содержание твердых веществ в диапазоне от 10 до 20 вес. %, предпочтительно от 12 до 18 вес. %, более предпочтительно от 14 до 16 вес. %.
(g) распылительную сушку суспензии, полученной на стадии (f), содержащей предшественник B-MWW, с получением распылительного порошка;
(h) обжиг распылительного порошка, полученного на стадии (g), содержащего предшественник B-MWW, предпочтительно при температуре в диапазоне от 500 до 700°С, более предпочтительно от 550 до 650°С, более предпочтительно от 575 до 625°С в течение периода времени в диапазоне от 1 до 24 часов, предпочтительно от 2 до 18 часов, более предпочтительно от 6 до 12 часов, с получением распылительного порошка, который, по меньшей мере, на 99 вес. %, более предпочтительно, по меньшей мере, на 99,5 вес. % состоит из B-MWW.
26. Способ согласно варианту 25, который отличается тем, что на стадии (h), обжиг осуществляют в непрерывном режиме, предпочтительно во вращающейся обжиговой печи, предпочтительно с пропускной способностью в диапазоне от 0,5 до 20 кг распылительного порошка в час.
27. Способ согласно варианту 25 или 26, который отличается тем, что степень кристалличности B-MWW, содержащегося в распылительном порошке, полученном на стадии (h), составляет, по меньшей мере, (75±5)%, предпочтительно, по меньшей мере, (80±5)%, как определено с помощью рентгенодифракционного (XRD) анализа.
28. Способ согласно любому из вариантов 25-27, который отличается тем, что удельная площадь поверхности по методу BET B-MWW, содержащегося в распылительном порошке, полученном на стадии (h), составляет, по меньшей мере, 300 м2/г, предпочтительно находится в диапазоне от 300 до 500 м2/г, как определено в соответствии с DIN 66131.
Предпочтительно, стадию (II) осуществляют с помощью способа, предпочтительные стадии и условия которого определены в соответствии со следующими вариантами 1-7 и соответствующими зависимостями, как указано:
1. Способ получения цеолитного материала, который включает в себя:
(a) обеспечение борсодержащего цеолитного материала структурного типа MWW (B-MWW), полученного в соответствии со стадией (I);
(b) деборирование B-MWW посредством обработки B-MWW системой жидких растворителей с получением не содержащего бора B-MWW (MWW);
который отличается тем, что систему жидких растворителей предпочтительно выбирают из группы, состоящей из воды, одноатомных спиртов, многоатомных спиртов и смеси двух или более из них, и который отличается тем, что указанная система жидких растворителей не содержит неорганическую или органическую кислоту или ее соль, причем кислоту предпочтительно выбирают из группы, состоящей из соляной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, муравьиной кислоты, уксусной кислоты, пропионовой кислоты, щавелевой кислоты и винной кислоты.
2. Способ согласно варианту 1, который отличается тем, что система жидких растворителей не содержит неорганическую или органическую кислоту или ее соль.
3. Способ согласно варианту 1 или 2, который отличается тем, что систему жидких растворителей предпочтительно выбирают из группы, состоящей из воды, метанола, этанола, пропанола, этан-1,2-диола, пропан-1,2-диола, пропан-1,3-диола, пропан-1,2,3-триола, а также смеси двух или более из них, предпочтительно воды.
4. Способ согласно любому из вариантов 1-3, который отличается тем, что обработку согласно стадии (b) осуществляют при температуре в диапазоне от 50 до 125°С.
5. Способ согласно любому из вариантов 1-4, который отличается тем, что обработку согласно стадии (b) осуществляют в течение времени в диапазоне от 6 до 20 часов.
6. Способ согласно любому из вариантов 1-5, который отличается тем, что обработку согласно стадии (b) осуществляют, по меньшей мере, с помощью 2 отдельных стадий, и который отличается тем, что между, по меньшей мере, двумя стадиями обработки, MWW сушат, предпочтительно при температуре в диапазоне от 100 до 150°С.
7. Способ согласно любому из вариантов 1-6, который дополнительно включает в себя:
(с) последующую обработку MWW, полученного на стадии (b) с помощью способа, включающего в себя:
(с.1) отделение MWW от системы жидких растворителей;
(с.2) предпочтительно сушку отделенного MWW, предпочтительно посредством распылительной сушки;
(с.3) необязательно обжиг MWW, полученного на стадии (с.1) или (с.2), предпочтительно при температурах в диапазоне от 500 до 700°С.
Что касается стадии (III), предпочтительно подходящую исходную смесь, предпочтительно водную смесь, содержащую MWW и содержащий Ti предшественник, и предпочтительно содержащую, по меньшей мере, один подходящий образующий микропоры агент, подвергают гидротермальной кристаллизации при автогенном давлении. Можно использовать, по меньшей мере, один подходящий затравочный материал. В качестве подходящего Ti содержащего предшественника могут быть упомянуты, в качестве примера, тетраалкилортотитанаты, такие как тетрабутилортотитанат. В качестве подходящего образующего микропоры агента могут быть упомянуты, в качестве примера, пиперидин, гексаметиленимин или смеси пиперидина и гексаметиленимина. Предпочтительно, время кристаллизации находится в диапазоне от 4 до 8 дней, более предпочтительно от 4 до 6 дней. В течение гидротермального синтеза, смесь для кристаллизации могут перемешивать. Температуры, применяемые в процессе кристаллизации, находятся предпочтительно в диапазоне от 160 до 200°С, более предпочтительно от 160 до 180°С. После гидротермального синтеза, полученный кристаллический цеолитный материал TiMWW предпочтительно, с помощью соответствующего метода, отделяют от маточного раствора. Все методы отделения TiMWW от его маточного раствора являются возможными. Эти методы включают, например, фильтрацию, ультрафильтрацию, диафильтрацию и методы центрифугирования или, например, способы распылительной сушки и способы распылительного гранулирования. Сочетание двух или более из этих методов может быть применено. Согласно настоящему изобретению, TiMWW предпочтительно отделяют от его маточного раствора посредством фильтрации с получением фильтровального осадка, который предпочтительно подвергают промывке, предпочтительно водой. Впоследствии, осадок на фильтре, необязательно дополнительно обрабатывают для получения подходящей суспензии, подвергают распылительной сушке или ультрафильтрации. Перед отделением TiMWW от его маточного раствора, можно повысить содержание TiMWW в маточном растворе посредством концентрирования суспензии. Если используется промывочный агент, предпочтительно продолжать процесс промывки до тех пор, пока промывочная вода не будет иметь электрическую проводимость менее 1000 мкСм/см, более предпочтительно менее 900 мкСм/см, более предпочтительно менее 800 мкСм/см, более предпочтительно менее 700 мкСм/см. После отделения TiMWW от его маточного раствора, которое предпочтительно достигается с помощью фильтрации, и после промывки, промытый фильтровальный осадок, содержащий TiMWW, предпочтительно подвергают предварительной сушке, например, посредством воздействия на фильтровальный осадок подходящим потоком газа, предпочтительно потоком азота, в течение времени предпочтительно в диапазоне от 4 до 10 часов, более предпочтительно от 5 до 8 часов. Затем, предварительно высушенный фильтровальный осадок предпочтительно сушат при температурах в диапазоне от 100 до 300°С, более предпочтительно от 150 до 275°С, более предпочтительно от 200 до 250°С в соответствующей атмосфере, такой как технический азот, воздух или обедненный воздух, предпочтительно воздух или обедненный воздух. Такая сушка может осуществляться, например, посредством распылительной сушки. После сушки, TiMWW могут подвергать обжигу при температурах в диапазоне от 500 до 700°С, более предпочтительно от 550 до 675°С, более предпочтительно от 600 до 675°С в соответствующей атмосфере, такой как технический азот, воздух или обедненный воздух, предпочтительно воздух или обедненный воздух. Предпочтительно, обжиг не проводят в соответствии со стадией (III).
Предпочтительно, стадии (III) и (IV) осуществляют с помощью способа, предпочтительные стадии и условия которого определены в соответствии со следующими вариантами 1-27 и соответствующими зависимостями, как указано:
1. Способ получения титансодержащего цеолитного материала, имеющего каркасную структуру MWW, который включает в себя:
(a) обеспечение не содержащего бора кристаллического цеолитного материала MWW, полученного в соответствии со стадией (II);
(b) введение титана в цеолитный материал, обеспеченный на стадии (а), которое включает в себя:
(b.1) получение водной смеси для синтеза, содержащей цеолитный материал, обеспеченный на стадии (i), соединение-шаблон MWW и источник титана, и которое отличается тем, что молярное соотношение соединения-шаблона MWW по отношению к Si, в пересчете на SiO2, и содержащегося в цеолитном материале, обеспеченном на стадии (а), находится в диапазоне от 0,5:1 до 1,4:1;
(b.2) гидротермальный синтез титансодержащего цеолитного материала, имеющего каркасную структуру MWW, из водной смеси для синтеза, полученной на стадии (b.1), с получением маточного раствора, содержащего титансодержащий цеолитный материал, имеющий каркасную структуру MWW;
(c) распылительную сушку маточного раствора, полученного на стадии (b.2), содержащего титансодержащий цеолитный материал, имеющий каркасную структуру MWW.
2. Способ согласно варианту 1, который отличается тем, что на стадии (b.1), соединение-шаблон MWW выбирают из группы, состоящей из пиперидина, гексаметиленимина, ионов N,N,N,N',N',N'-гексаметил-1,5-пентандиаммония, 1,4-бис(N-метилпирролидиний)бутана, гидроксида октилтриметиламмонийхлорида, гидроксида гептилтриметиламмония, гидроксида гексилтриметиламмония и смеси двух или более из них, причем соединение-шаблон MWW предпочтительно представляет собой пиперидин.
3. Способ согласно варианту 1 или 2, который отличается тем, что на стадии (b.1), источник титана предпочтительно выбирают из группы, состоящей из тетрабутилортотитаната, тетраизопропилортотитаната, тетраэтилортотитаната, диоксида титана, тетрахлорида титана, трет-бутоксида титана и смеси двух или более из них, источником титана предпочтительно является тетрабутилортотитанат.
4. Способ согласно любому из вариантов 1-3, который отличается тем, что в водной смеси для синтеза на стадии (b.1), молярное соотношение Ti, в пересчете на TiO2, и содержащегося в источнике титана, по отношению к Si, в пересчете на SiO2, и содержащегося в цеолитном материале, имеющем молярное соотношение В2О3:SiO2 не более 0,02:1, находится в диапазоне от 0,005:1 до 0,1:1, предпочтительно от 0,01:1 до 0,08:1, более предпочтительно от 0,02:1 до 0,06:1.
5. Способ согласно любому из вариантов 1-4, который отличается тем, что в водной смеси для синтеза на стадии (b.1), молярное соотношение Н2О по отношению к Si, в пересчете на SiO2, и содержащегося в цеолитном материале, имеющем молярное соотношение В2О3:SiO2 не более 0,02:1, находится в диапазоне от 8:1 до 20:1, предпочтительно от 10:1 до 18:1, более предпочтительно от 12:1 до 16:1.
6. Способ согласно любому из вариантов 1-5, который отличается тем, что в водной смеси для синтеза на стадии (b.1), молярное соотношение соединения-шаблона MWW по отношению к Si, в пересчете на SiO2, и содержащегося в цеолитном материале, обеспеченном на стадии (i), находится в диапазоне от 0,5:1 до 1,7:1, предпочтительно от 0,8:1 до 1,5:1, более предпочтительно от 1,0:1 до 1,3:1.
7. Способ согласно любому из вариантов 1-6, который отличается тем, что на стадии (b.2), гидротермальный синтез осуществляют при температуре в диапазоне от 80 до 250°С, предпочтительно от 120 до 200°С, более предпочтительно от 160 до 180°С.
8. Способ согласно любому из вариантов 1-7, который отличается тем, что на стадии (b.2), гидротермальный синтез осуществляют в течение периода времени в диапазоне от 10 до 100 часов, более предпочтительно от 20 до 80 часов, более предпочтительно от 40 до 60 часов.
9. Способ согласно любому из вариантов 1-8, который отличается тем, что на стадии (b.2), гидротермальный синтез осуществляют в замкнутой системе при автогенном давлении.
10. Способ согласно любому из вариантов 1-9, который отличается тем, что ни во время стадии (b.2), ни после стадии (b.2) и перед стадией (с), титансодержащий цеолитный материал, имеющий каркасную структуру MWW, отделяют от своего маточного раствора.
11. Способ согласно любому из вариантов 1-10, который отличается тем, что маточный раствор, подвергаемый стадии (с), и содержащий титансодержащий цеолитный материал, имеющий каркасную структуру MWW, имеет содержание твердых веществ, необязательно после концентрирования или разбавления, в диапазоне от 5 до 25 вес. %, более предпочтительно от 10 до 20 вес. %, в расчете на общий вес маточного раствора, содержащего титансодержащий цеолитный материал.
12. Способ согласно любому из вариантов 1-11, который отличается тем, что во время распылительной сушки на стадии (с), температура сушильного газа на входе находится в диапазоне от 200 до 350°С, а температура сушильного газа на выходе находится в диапазоне от 70 до 190°С.
13. Способ согласно любому из вариантов 1-12, который отличается тем, что цеолитный материал, имеющий каркасную структуру MWW, полученный на стадии (с), имеет содержание Si в диапазоне от 30 до 40 вес. %, в пересчете на элементарный Si, общее содержание органического углерода (ТОС) в диапазоне от 0 до 14 вес. % и содержание Ti в диапазоне от 2,1 до 2,8 вес. %, в пересчете на элементарный титан, в каждом случае в расчете на общий вес цеолитного материала.
14. Способ согласно любому из вариантов 1-13, который дополнительно включает в себя:
(d) обработку титансодержащего цеолитного материала, имеющего каркасную структуру MWW, полученного на стадии (iii) водным раствором, имеющим рН не более 5.
15. Способ согласно варианту 14, который отличается тем, что после стадии (с) и перед стадией (d), высушенный распылением титансодержащий цеолитный материал, имеющий каркасную структуру MWW, полученный на стадии (с), не подвергают обжигу.
16. Способ согласно варианту 14 или 15, который отличается тем, что на стадии (d), весовое соотношение водного раствора по отношению к титансодержащему цеолитному материалу, имеющему каркасную структуру MWW, находится в диапазоне от 10:1 до 30:1, предпочтительно от 15:1 до 25:1, более предпочтительно от 18:1 до 22:1.
17. Способ согласно любому из вариантов 14-16, который отличается тем, что на стадии (d), водный раствор содержит неорганическую кислоту, предпочтительно выбранную из группы, состоящей из фосфорной кислоты, серной кислоты, соляной кислоты, азотной кислоты и смеси двух или более из них, водный раствор предпочтительно содержит азотную кислоту.
18. Способ согласно любому из вариантов 14-17, который отличается тем, что на стадии (d), водный раствор имеет значение рН в диапазоне от 0 до 5, предпочтительно от 0 до 3, более предпочтительно от 0 до 2.
19. Способ согласно любому из вариантов 14-18, который отличается тем, что на стадии (d), титансодержащий цеолитный материал, имеющий каркасную структуру MWW, обрабатывают водным раствором при температуре в диапазоне от 50 до 175°С, предпочтительно от 70 до 125°С, более предпочтительно от 95 до 105°С.
20. Способ согласно любому из вариантов от 14-19, который отличается тем, что на стадии (d), титансодержащий цеолитный материал, имеющий каркасную структуру MWW, обрабатывают водным раствором в течение периода времени в диапазоне от 0,1 до 6 часов, предпочтительно от 0,3 до 2 часов, более предпочтительно от 0,5 до 1,5 часов.
21. Способ согласно любому из вариантов 14-20, который отличается тем, что обработку согласно (d) осуществляют в замкнутой системе при автогенном давлении.
22. Способ согласно любому из вариантов 14-21, который дополнительно включает в себя:
(e) отделение титансодержащего цеолитного материала, имеющего каркасную структуру MWW, полученного на стадии (d) из водного раствора, необязательно с последующей промывкой отделенного титансодержащего цеолитного материала, имеющего каркасную структуру MWW.
23. Способ согласно варианту 22, который отличается тем, что стадия (е) включает в себя сушку отделенного и необязательно промытого титансодержащего цеолитного материала, имеющего каркасную структуру MWW.
24. Способ согласно любому из вариантов 14-23, который дополнительно включает в себя:
(f) получение суспензии, предпочтительно водной суспензии, содержащей титансодержащий цеолитный материал, имеющий каркасную структуру MWW, полученный на стадии (d), предпочтительно на стадии (е), причем указанная суспензия имеет содержание твердых веществ предпочтительно в диапазоне от 5 до 25 вес. %, более предпочтительно от 10 до 20 вес. %, в расчете на общий вес суспензии, и воздействие на суспензию распылительной сушкой.
25. Способ согласно варианту 24, который отличается тем, что во время распылительной сушки, температура сушильного газа на входе находится в диапазоне от 200 до 330°С, а температура сушильного газа на выходе находится в диапазоне от 120 до 180°С.
26. Способ согласно любому из вариантов 14-25, который дополнительно включает в себя:
(g) обжиг титансодержащего цеолитного материала, имеющего каркасную структуру MWW, полученного на стадии (d), предпочтительно на стадии (е), более предпочтительно на стадии (f), который отличается тем, что обжиг предпочтительно проводят при температуре в диапазоне от 400 до 800°С, более предпочтительно от 600 до 700°С.
27. Способ согласно варианту 26, который отличается тем, что на стадии (vii), обжиг осуществляют в непрерывном режиме, предпочтительно с расходом в диапазоне от 0,2 до 2,0 кг цеолитного материала в час, более предпочтительно от 0,5 до 1,5 кг цеолитного материала в час.
Согласно стадии (V), TiMWW, предпочтительно полученный в соответствии со стадией (IV), подвергают подходящей обработке цинком (Zn) для получения ZnTiMWW, используемого для приготовления суспензии в соответствии со стадией (а). В целом, что касается стадии (V), то не существует никаких особых ограничений при условии, что может быть получен вышеуказанный предпочтительный ZnTiMWW, имеющий предпочтительное содержание Zn и Ti. Наиболее предпочтительно, стадия (V) включает в себя, по меньшей мере, одну подходящую стадию пропитки, более предпочтительно, по меньшей мере, одну стадию мокрой пропитки. Что касается данной стадии пропитки, предпочтительно, чтобы TiMWW, предпочтительно полученный согласно стадии (IV), взаимодействовал с, по меньшей мере, одним подходящим Zn-содержащим предшественником в, по меньшей мере, одном подходящем растворителе (мокрая пропитка), наиболее предпочтительно воде. В качестве подходящего Zn-содержащего предшественника, особенно предпочтительными являются водорастворимые соли цинка, причем дигидрат ацетата цинка является особенно предпочтительным. Кроме того, предпочтительно приготовить раствор цинксодержащего предшественника, предпочтительно водный раствор, и суспендировать TiMWW в этом растворе. Далее предпочтительно, осуществляют пропитку при повышенных температурах, по отношению к комнатной температуре, предпочтительно в диапазоне от 75 до 125°С, более предпочтительно от 85 до 115°С, в течение времени, предпочтительно в диапазоне от 3,5 до 5 часов, более предпочтительно от 3 до 6 часов. Перемешивание суспензии в процессе пропитки является предпочтительным. После пропитки, полученный ZnTiMWW предпочтительно соответствующим способом отделяют от суспензии. Все способы отделения ZnTiMWW от суспензии являются возможными. Особенно предпочтительно, отделение осуществляют с помощью способов фильтрации, ультрафильтрации, диафильтрации или центрифугирования. Сочетание двух или более из этих способов может быть применено. В соответствии с настоящим изобретением, ZnTiMWW предпочтительно отделяют от суспензии посредством фильтрования с получением осадка на фильтре, который предпочтительно подвергают промывке, предпочтительно водой. Если используется промывка, предпочтительно продолжать процесс промывки до тех пор, пока промывочная вода не будет иметь электрическую проводимость менее 1000 мкСм/см, более предпочтительно менее 900 мкСм/см, более предпочтительно менее 800 мкСм/см, более предпочтительно менее 700 мкСм/см. Затем предпочтительно промытый фильтровальный осадок подвергают предварительной сушке, например, посредством воздействия на фильтровальный осадок подходящим газовым потоком, предпочтительно потоком азота, в течение времени, предпочтительно в диапазоне от 5 до 15 часов, более предпочтительно от 8 до 12 часов.
Если TiMWW или ZnTiMWW используется в качестве каталитически активного материала согласно настоящему изобретению, предпочтительно, чтобы органический растворитель содержал, предпочтительно, по существу, состоял из ацетонитрила.
Таким образом, настоящее изобретение относится к способу регенерации катализатора, содержащего титансодержащий цеолит, имеющий каркасную структуру MWW, необязательно содержащего цинк, в качестве каталитически активного материала, причем указанный катализатор использовался в способе получения оксида олефина, который включает в себя:
(i) обеспечение смеси, содержащей ацетонитрил, олефин, эпоксидирующий агент и, по меньшей мере, частично растворенную калийсодержащую соль;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей ацетонитрил и оксид олефина, и с получением катализатора, содержащего осажденную на нем калиевую соль;
причем указанный способ регенерации включает в себя:
(a) отделение смеси, полученной на стадии (ii), от катализатора;
(b) промывку катализатора, полученного на стадии (а), с помощью жидкой водной системы;
(c) необязательно сушку катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре менее 300°С;
(d) обжиг катализатора, полученного на стадии (с), в газовом потоке, содержащем кислород, при температуре, по меньшей мере, 300°С.
Особенно предпочтительно, настоящее изобретение предпочтительно относится к способу регенерации катализатора, содержащего титансодержащий цеолит, имеющий каркасную структуру MWW, необязательно содержащего цинк, в качестве каталитически активного материала, причем указанный катализатор использовался в непрерывном способе получения пропиленоксида, который включает в себя:
(i) обеспечение смеси, содержащей ацетонитрил, пропен, перекись водорода, воду, необязательно пропан и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что калийсодержащую соль выбирают из группы, состоящей из дигидрофосфата калия, гидроортофосфата калия, формиата калия и смеси двух или более из них;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей ацетонитрил и пропиленоксид, воду, необязательно пропен, необязательно пропан, и с получением катализатора, содержащего осажденную на нем калиевую соль,
который отличается тем, что смесь согласно стадии (i) содержит калийсодержащую соль с молярным соотношением калия, содержащегося в калийсодержащей соли, по отношению к перекиси водорода в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1,
причем указанный способ регенерации включает в себя:
(a) отделение смеси, полученной на стадии (ii), от катализатора;
(b) промывку катализатора, полученного на стадии (а), с помощью жидкой водной системы, которая содержит, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С;
(c) необязательно сушку катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С;
(d) обжиг катализатора, полученного на стадии (b) или (с), предпочтительно на стадии (с), в газовом потоке, содержащем кислород, используемом на стадии (d), содержит кислород в диапазоне от 3 до 40 об. %, предпочтительно от 5 до 50 об. % в расчете на общий объем газового потока при температуре в диапазоне от 375 до 525°С, предпочтительно от 400 до 500°С.
Катализатор TS-1
В соответствии с настоящим изобретением, катализатор на основе силикалита титана-1, предпочтительно катализатор на основе силикалита титана-1 с неподвижным слоем, может быть использован в качестве катализатора. Силикалит титана-1 представляет собой микропористый цеолит структурного типа MFI, который не содержит алюминия, и в котором Si(IV) в силикатной решетке частично заменен титаном в качестве Ti(IV). Термин ''микропоры'', как используется в контексте настоящего изобретения, относится к порам, имеющим размер пор менее 2 нм, как определено в соответствии с DIN 66134.
Цеолит - силикалит титана-1 катализатора может быть, в принципе, получен любым возможным способом. Как правило, синтез, по меньшей мере, одного титанового цеолита согласно настоящему изобретению проводят в гидротермальных системах, которые предполагают объединение активного источника оксида кремния и источника титана, например, оксида титана, с, по меньшей мере, одним соединением-шаблоном, способным образовывать необходимый титановый цеолит в водной суспензии, например, в основной суспензии. Как правило, используются органические шаблоны. Предпочтительно, синтез осуществляют при повышенных температурах, например, температурах в диапазоне от 150 до 200°С, предпочтительно от 160 до 180°С.
В принципе, любое подходящее соединение может использоваться в качестве источника оксида кремния. Типичные источники оксида кремния (SiO2) включают силикаты, гидрогель диоксида кремния, кремниевую кислоту, коллоидный диоксид кремния, пирогенный диоксид кремния, тетраалкоксисиланы, гидроксиды кремния, осажденный диоксид кремния и глины. Могут быть использованы оба, так называемый диоксид кремния по ''мокрому способу'', и так называемый диоксид кремния по ''сухому способу''. В этих случаях, диоксид кремния особенно предпочтительно является аморфным и отличается тем, что размер частиц диоксида кремния находится, например, в диапазоне от 5 до 100 нм, а площадь поверхности частиц диоксида кремния находится, например, в диапазоне от 50 до 500 м2/г. Коллоидный диоксид кремния является, среди прочего, коммерчески доступным как Ludox®, Syton®, Nalco® или Snowtex®. Диоксид кремния по ''мокрому способу'' является, среди прочего, коммерчески доступным как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Диоксид кремния по ''сухому способу'' является, среди прочего, коммерчески доступным как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. Также в объеме настоящего изобретения можно использовать соединение-предшественник диоксида кремния в качестве источника оксида кремния. Например, тетраалкоксисиланы, такие как, например, тетраэтоксисилан или тетрапропоксисилан, могут быть упомянуты в качестве соединения-предшественника.
В качестве шаблона может быть использован любой шаблон, подходящий для обеспечения необходимой цеолитной структуры MFI. В частности, используют гидроксид тетрапропиламмония, более предпочтительно гидроксид тетра-н-пропиламмония. В предпочтительном варианте осуществления способа согласно изобретению, по меньшей мере, один порообразующий агент удаляют на более поздней стадии посредством обжига, как описано ниже.
Как правило, синтез силикалита титана-1 осуществляют в периодическом режиме в автоклаве таким образом, чтобы реакционная суспензия подвергалась воздействию автогенного давления в течение нескольких часов или нескольких дней до тех пор, пока не будет получен цеолит силикалит титана-1. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, синтез обычно протекает при повышенных температурах и отличается тем, что температуры в течение стадии гидротермальной кристаллизации обычно находятся в диапазоне от 150 до 200°С, предпочтительно в диапазоне от 160 до 180°С. Обычно реакцию проводят в течение времени в диапазоне от нескольких часов до нескольких дней, предпочтительно в течение периода времени в диапазоне от 12 часов до 48 часов, более предпочтительно от 20 до 30 часов. Также дополнительно можно добавлять затравочные кристаллы в шихту для синтеза.
В соответствии с вариантом осуществления настоящего изобретения, полученный кристаллический силикалит титана-1 отделяют от реакционной суспензии, т.е. от маточного раствора, необязательно промывают и сушат.
Могут использоваться все способы, известные для отделения кристаллического силиката титана-1 от суспензии. Среди прочего, следует упомянуть фильтрацию, ультрафильтрацию, диафильтрацию и центрифугирование.
В случае, если полученный кристаллический силикалит титана-1 промывают, указанная стадия промывки может выполняться с использованием любого подходящего промывочного вещества, такого как, например, вода, спирты, такие как, например, метанол, этанол или метанол и пропанол, или этанол и пропанол, или метанол и этанол и пропанол, или смеси воды и, по меньшей мере, одного спирта, такого как, например, вода и этанол или вода и метанол, или вода и этанол, или вода и пропанол, или вода и метанол и этанол, или вода и метанол и пропанол, или вода и этанол и пропанол, или вода и этанол и метанол и пропанол. Воду или смесь воды и, по меньшей мере, одного спирта, предпочтительно воду и этанол используют в качестве промывного вещества.
Сушку кристаллического силикалита титана-1 осуществляют при температурах, как правило, в диапазоне от 80 до 160°С, предпочтительно от 90 до 145°С, особенно предпочтительно от 100 до 130°С.
Вместо вышеуказанных способов отделения, таких как, среди прочего, способы фильтрации, ультрафильтрации, диафильтрации и центрифугирования, суспензия может, в соответствии с альтернативным вариантом осуществления изобретения, также подвергаться распылительным способам, как, например, распылительной грануляции и распылительной сушке.
Если отделение кристаллического силикалита титана-1 осуществляется с помощью способа распыления, то стадии отделения и сушки могут быть объединены в одну стадию. В таком случае, могут использоваться либо реакционная суспензия как таковая, либо концентрированная реакционная суспензия. Кроме того, в суспензию можно добавлять подходящую добавку, как например, по меньшей мере, одно подходящее связующее вещество и/или, по меньшей мере, один порообразующий агент, либо в реакционную суспензию как таковую, либо в концентрированную суспензию перед распылительной сушкой или распылительной грануляцией. Подходящие связующие вещества описаны подробно ниже. В качестве порообразующего агента могут использоваться все порообразующие агенты, описанные выше. В случае, если суспензию подвергают распылительной сушке, порообразующий агент, если добавляют, может быть добавлен двумя способами. Во-первых, порообразующий агент может быть добавлен в реакционную смесь перед распылительной сушкой. Тем не менее, также можно добавить часть порообразующего агента в реакционную смесь перед распылительной сушкой, а остальную часть порообразующего агента добавляют в высушенный распылением материал.
В случае, если суспензию сначала концентрируют для повышения содержания силикалита титана-1 в суспензии, концентрация может быть достигнута, например, посредством выпаривания, как, например, с помощью выпаривания при пониженном давлении, или посредством фильтрации в поперечном потоке. Аналогичным образом, суспензия может концентрироваться посредством разделения указанной суспензии на две фракции, где твердые вещества, содержащиеся в одной из обеих фракций, отделяют с помощью способов фильтрации, диафильтрации, ультрафильтрации или центрифугирования, и суспендируют после необязательной стадии промывки и/или стадии сушки в другой фракции суспензии. Таким образом полученная концентрированная суспензия затем может быть подвергнута воздействию распылительных способов, таких как, например, распылительная грануляция и распылительная сушка.
Согласно альтернативному варианту, концентрация достигается с помощью отделения, по меньшей мере, одного титанового цеолита от суспензии, и ресуспендирования титанового цеолита, необязательно вместе с, по меньшей мере, одной подходящей добавкой, как уже было описано выше, где титановый цеолит может быть подвергнут, по меньшей мере, одной стадии промывки и/или, по меньшей мере, одной стадии сушки перед ресуспендированием. Ресуспендированный титановый цеолит затем может использоваться в распылительных способах, предпочтительно распылительной сушке.
Распылительная сушка представляет собой прямой метод сушки шламов, суспензий или растворов посредством загрузки шлама, суспензии или раствора с надлежаще диспергированными жидкой и твердой фазами, часто дополнительно содержащих связующее вещество, в распылитель с последующей флэш-сушкой в потоке горячего воздуха. Распылитель может быть нескольких различных типов. Наиболее распространенным является центробежное распыление, в котором используют высокоскоростное вращение рабочего колеса или диска для разбивания суспензии в капли, которые отрываются от рабочего колеса вследствие вращения, попадая в камеру, и подвергаются мгновенной сушке до того, как они ударяются о стенки камеры. Распыление может также достигаться с помощью однокомпонентных жидкостных сопел, которые основываются на гидростатическом давлении для продавливания суспензии через небольшое сопло. Также используются многокомпонентные жидкостные сопла, в которых давление газа используют для продавливания суспензии через сопло. Распыленный материал, полученный с помощью способов распылительной сушки и распылительной грануляции, например, с помощью сушки в псевдоожиженном слое, может содержать твердые и/или полые сферы и может, по существу, состоять из таких сфер, которые имеют, например, диаметр в диапазоне от 5 до 500 мкм или от 5 до 300 мкм. Могут использоваться однокомпонентные или многокомпонентные сопла. Использование вращающегося распылителя также возможно. Возможные температуры на входе для используемого газа-носителя находятся, например, в диапазоне от 200 до 600°С, предпочтительно в диапазоне от 300 до 500°С. Температура на выходе газа-носителя находится, например, в диапазоне от 50 до 200°С. В качестве газов-носителей могут быть упомянуты воздух, обедненный воздух или смеси кислорода-азота с содержанием кислорода до 10 об. %, предпочтительно до 5 об. %, более предпочтительно менее 5 об. %, например, до 2 об. %. Распылительные способы могут осуществляться в противотоке или в параллельном потоке.
Предпочтительно, силикалит титана-1 отделяют от реакционной суспензии посредством обычной фильтрации или центрифугирования, необязательно сушат и/или обжигают, и ресуспендируют, предпочтительно в смеси, предпочтительно в водной смеси, по меньшей мере, одного связующего вещества и/или одного порообразующего агента. Полученную суспензию затем предпочтительно подвергают распылительной сушке или распылительной грануляции. Полученный распыленный материал может быть подвергнут стадии дополнительной промывки, причем указанную стадию промывки проводят, как описано выше. Необязательно промытый распыленный материал затем высушивают и обжигают, причем сушку и обжиг предпочтительно проводят, как описано выше.
Согласно альтернативному варианту, кристаллизацию силикалита титана-1 осуществляют не раньше, чем описанная выше суспензия будет высушена посредством распылительной сушки. Таким образом, сначала получают суспензию, содержащую источник оксида кремния, предпочтительно диоксид кремния, источник оксида титана, и соединение-шаблон, способное образовывать силикалит титана-1. Затем суспензию подвергают распылительной сушке, после чего, необязательно дополнительный порообразующий агент добавляют к высушенному распылением силикалиту титана-1.
Высушенный распылением силикалит титана 1, полученный в соответствии с упомянутыми выше способами, могут, необязательно, подвергать, по меньшей мере, одному способу промывки. Если осуществляют, по меньшей мере, один способ промывки, за ним следуют, предпочтительно, по меньшей мере, одна стадия сушки и/или, по меньшей мере, одна стадия обжига.
Силикалит титана 1, необязательно полученный посредством способов распыления, может быть дополнительно подвергнут, по меньшей мере, одной стадии обжига, которую проводят в соответствии с предпочтительным вариантом осуществления настоящего изобретения, после стадии сушки, или вместо стадии сушки. По меньшей мере, одна стадия обжига осуществляется при температурах, как правило, в диапазоне от 350-750°С, предпочтительно 400-700°С, особенно предпочтительно от 450-650°С.
Обжиг силикалита титана-1 может осуществляться в любой подходящей газообразной атмосфере, причем воздух и/или обедненный воздух являются предпочтительными. Кроме того, обжиг предпочтительно проводят в муфельной печи, вращающейся конусной и/или конвейерной обжиговой печи, при этом обжиг, как правило, проводят в течение одного часа или более, например, в течение периода времени в диапазоне от 1 до 24 часов или от 4 до 12 часов. Возможно в способе согласно настоящему изобретению, например, обжигать силикалит титана-1 один раз, дважды или более часто, в каждом случае, по меньшей мере, в течение одного часа, например, в каждом случае от 4 часов до 12 часов, предпочтительно от 4 часов до 8 часов, при этом можно поддерживать температуру во время стадии обжига постоянной или изменять температуры непрерывно или прерывисто. Если обжиг осуществляется два раза или чаще, температуры обжига на отдельных стадиях могут отличаться или быть идентичными.
Таким образом, предпочтительный вариант настоящего изобретения относится к способу, как описано выше, который отличается тем, что силикалит титана-1 отделяют от суспензии, например, посредством фильтрования или распылительной сушки, промывают подходящим промывочным веществом, и затем подвергают, по меньшей мере, одной стадии сушки. Сушка осуществляется при температурах, как правило, в диапазоне от 80 до 160°С, предпочтительно от 90 до 145°С, особенно предпочтительно от 100 до 130°С. Наиболее предпочтительно, после сушки осуществляют стадию обжига. Стадия обжига осуществляется при температурах, как правило, в диапазоне от 350-750°С, предпочтительно 400-700°С, особенно предпочтительно от 450-650°С.
Силикалит титана-1, полученный как описано выше, как правило, может непосредственно использоваться в качестве катализатора на стадиях (i) и (iii). Тем не менее, особенно предпочтительно использовать катализатор с неподвижным слоем на обеих стадиях (i) и (iii), т.е. использовать не кристаллический цеолитный материал как таковой в качестве катализатора, а кристаллический материал, подвергнутый обработке для получения формованного изделия, содержащего силикалит титана-1. Таким образом, в соответствии с предпочтительным вариантом осуществления настоящего изобретения, формованное изделие, содержащее силикалит титана-1, как описано выше, используют в качестве катализатора.
Как правило, в случае, если формованное изделие используется в качестве катализатора, указанный катализатор может содержать все возможные другие соединения, в дополнение к силикалиту титана-1 согласно настоящему изобретению, например, среди прочего, по меньшей мере, одно связующее вещество и/или, по меньшей мере, один порообразующий агент. Кроме того, катализатор может содержать, по меньшей мере, один клеящий агент вместо, по меньшей мере, одного связующего вещества и/или, по меньшей мере, одного порообразующего агента, или в дополнение к, по меньшей мере, одному связующему веществу и/или, по меньшей мере, одному порообразующему агенту.
В качестве связующего вещества подходящими являются все соединения, которые обеспечивают адгезию и/или сцепление между силикалитом титана-1, подлежащим формованию, которое выходит за рамки физической адсорбции, которая может присутствовать без связующего вещества. Примерами таких связующих веществ являются оксиды металлов, такие как, например, SiO2, Al2O3, TiO2, ZrO2 или MgO или глины или смеси двух или более из этих соединений. Глинистые минералы и природные или синтетически полученные оксиды алюминия, такие как, например, альфа-, бета-, гамма-, дельта-, эта-, каппа-, хи- или тета-оксиды алюминия и их неорганические или металлоорганические соединения-предшественники, такие как, например, гиббсит, байерит, бемит или псевдобемит, или триалкоксиалюминаты, такие как, например, триизопропилат алюминия, являются особенно предпочтительными в качестве связующих веществ на основе Al2O3. Другие предпочтительные связующие вещества представляют собой амфифильные соединения, имеющие полярные и неполярные фрагменты, и графит. Также связующими веществами являются, например, глины, такие как, например, монтмориллониты, каолины, метакаолин, гекторит, бентониты, галлуазиты, диккиты, накриты или анаукситы.
Эти связующие вещества могут использоваться как таковые. Также в объеме настоящего изобретения можно использовать соединения, из которых образуется связующее вещество, по меньшей мере, на одной дополнительной стадии при получении формованных изделий. Примерами таких предшественников связующих веществ являются тетраалкоксисиланы, тетраалкоксититанаты, тетраалкоксицирконаты или смесь двух или более различных тетраалкоксисиланов, или смесь двух или более различных тетраалкоксититанатов, или смесь двух или более различных тетраалкоксицирконатов, или смесь, по меньшей мере, одного тетраалкоксисилана и, по меньшей мере, одного тетраалкоксититаната или, по меньшей мере, одного тетраалкоксисилана и, по меньшей мере, одного тетраалкоксицирконата или, по меньшей мере, одного тетраалкоксититаната и, по меньшей мере, одного тетраалкоксицирконата или смесь, по меньшей мере, одного тетраалкоксисилана и, по меньшей мере, одного тетраалкоксититаната и, по меньшей мере, одного тетраалкоксицирконата.
В контексте настоящего изобретения связующие вещества, которые полностью или частично содержат SiO2, или которые являются предшественником SiO2, от которого образуется SiO2, по меньшей мере, на одной дополнительной стадии, являются особенно предпочтительными. В этом контексте, могут использоваться как коллоидный диоксид кремния, так и так называемый диоксид кремния по ''мокрому способу'' и так называемый диоксид кремния по ''сухому способу''. Особенно предпочтительно данный диоксид кремния является аморфным диоксидом кремния, при этом размер частиц диоксида кремния находится, например, в диапазоне от 5 до 100 нм, а площадь поверхности частиц диоксида кремния находится в диапазоне от 50 до 500 м2/г.
Коллоидный диоксид кремния, предпочтительно в виде щелочного и/или аммиачного раствора, более предпочтительно в виде аммиачного раствора, является коммерчески доступным, среди прочего, например, как Ludox®, Syton®, Nalco® или Snowtex®. Диоксид кремния по ''мокрому способу'' является коммерчески доступным, среди прочего, например, как Hi-Sil®, Ultrasil®, Vulcasil®, Santocel®, Valron-Estersil®, Tokusil® или Nipsil®. Диоксид кремния по ''сухому способу'' является коммерчески доступным, среди прочего, например, как Aerosil®, Reolosil®, Cab-O-Sil®, Fransil® или ArcSilica®. Среди прочего, аммиачный раствор коллоидного диоксида кремния является предпочтительным в настоящем изобретении. Соответственно, настоящее изобретение также описывает катализатор, содержащий формованное изделие, как описано выше, причем указанное формованное изделие содержит силикалит титана-1, как описано выше, и дополнительно SiO2 в качестве связующего материала, где связующее вещество, используемое согласно стадии (i), представляет собой связующее вещество, содержащее или образовывающее SiO2. Как правило, титановый цеолит может также подвергаться формованию без использования связующего вещества.
Таким образом, настоящее изобретение также относится к способу, который отличается тем, что на стадиях (i) и (iii), катализатор на основе силикалита титана-1 получают посредством формования силикалита титана-1 с получением формованного изделия, содержащего силикалит титана-1, и предпочтительно, по меньшей мере, одно связующее вещество, в частности связующее вещество на основе диоксида кремния.
При необходимости, по меньшей мере, один порообразующий агент может быть добавлен к смеси силикалита титана-1 и, по меньшей мере, одного связующего вещества или, по меньшей мере, предшественника связующего вещества, для дальнейшей обработки и для образования формованного изделия катализатора, для использования в качестве катализатора с неподвижным слоем. Порообразующие агенты, которые могут быть использованы, представляют собой все соединения, которые, по отношению к полученному формованному изделию, обеспечивают определенный размер пор и/или определенное распределение пор по размеру, и/или определенные объемы пор. В частности, порообразующие агенты, которые обеспечивают, по отношению к полученному формованному изделию, микропоры и/или микропоры, в частности мезопоры и микропоры.
Таким образом, настоящее изобретение также относится к способу, который отличается тем, что на стадиях (i) и (iii), катализатор на основе силикалита титана-1 получают посредством формования силикалита титана-1 с получением формованного изделия, содержащего силикалит титана-1, и предпочтительно, по меньшей мере, одно связующее вещество, в частности связующее вещество на основе диоксида кремния, при этом формованное изделие, в частности, имеет микропоры и мезопоры.
Что касается примеров порообразующих агентов, которые могут быть использованы, делается ссылка на порообразующие агенты, уже упомянутые выше. Предпочтительно, порообразующие агенты, используемые в способе формования согласно изобретению, представляют собой полимеры, которые являются диспергируемыми, суспендируемыми или эмульгируемыми в воде или в смесях водных растворителей. Особенно предпочтительными полимерами являются полимерные виниловые соединения, такие как, например, полиалкиленоксиды, такие как полиэтиленоксиды, полистирол, полиакрилаты, полиметакрилаты, полиолефины, полиамиды и полиэфиры, углеводы, такие, как, например, целлюлоза или производные целлюлозы, такие как, например, метилцеллюлоза, или сахара или натуральные волокна. Другими подходящими порообразующими агентами являются, например, пульпа или графит.
При необходимости достижения распределения пор по размерам можно использовать смесь двух или более порообразующих агентов. В особенно предпочтительном варианте осуществления способа согласно изобретению, как описано ниже, порообразующие агенты удаляют посредством обжига с целью получения формованного тела пористого катализатора. Предпочтительно, порообразующие агенты, которые обеспечивают мезопоры и/или микропоры, в частности предпочтительно мезопоры, добавляют к смеси, по меньшей мере, одного связующего вещества и силикалита титана-1 для формования силикалита титана-1. Как правило, силикалит титана-1 также может подвергаться формованию с целью получения формованного тела катализатора без использования порообразующего агента.
Кроме связующего вещества и необязательно порообразующего агента, также можно добавлять дополнительные компоненты, например, по меньшей мере, один клеящий агент, к смеси, которую формуют для получения формованного тела катализатора.
Если, по меньшей мере, один клеящий агент используют в способе согласно изобретению, указанный клеящий агент используют либо вместо, либо в дополнение к, по меньшей мере, одному порообразующему агенту. В частности, соединения, которые также действуют как порообразующие агенты, могут использоваться в качестве клеящего агента. Клеящие агенты, которые могут быть использованы, представляют собой все соединения, известные как подходящие для этой цели. Они представляют собой предпочтительно органические, в частности гидрофильные полимеры, такие как, например, целлюлоза, производные целлюлозы, такие как, например, метилцеллюлоза, а также крахмал, такой как, например, картофельный крахмал, обойный клей, полиакрилаты, полиметакрилаты, поливиниловый спирт, поливинилпирролидон, полиизобутилен или политетрагидрофуран. Следует упомянуть использование воды, спиртов или гликолей или их смесей, таких как смеси воды и спирта или воды и гликоля, таких как, например, воды и метанола, или воды и этанола, или воды, пропанола, или воды и пропиленгликоля в качестве клеящих агентов. Предпочтительно, целлюлоза, производные целлюлозы, вода и смеси двух или более из этих соединений, таких как вода и целлюлоза или вода и производные целлюлозы используются в качестве клеящего агента. В особенно предпочтительном варианте осуществления способа согласно изобретению, по меньшей мере, один клеящий агент удаляют посредством обжига, как далее описано ниже, для получения формованного изделия.
Согласно еще одному варианту настоящего изобретения, по меньшей мере, одну кислотную добавку можно добавить к смеси, которую формуют для получения формованного изделия. Если используют кислотную добавку, органические кислотные соединения, которые могут быть удалены с помощью обжига, являются предпочтительными. В этом контексте могут быть упомянуты карбоновые кислоты, такие как, например, муравьиная кислота, щавелевая кислота и/или лимонная кислота. Можно также использовать два или более из этих кислотных соединений.
Порядок добавления компонентов в смесь, которую подвергают формованию с целью получения формованного изделия, не является критическим. Если например, используют комбинацию связующего вещества, порообразующего агента, клеящего агента и необязательно, по меньшей мере, одно кислотное соединение, можно сначала добавить, по меньшей мере, одно связующее вещество, по меньшей мере, один порообразующий агент, по меньшей мере, одно кислотное соединение и, наконец, по меньшей мере, один клеящий агент, а также поменять последовательность в отношении, по меньшей мере, одного связующего вещества, по меньшей мере, одного порообразующего агента, по меньшей мере, одного кислотного соединения и, по меньшей мере, одного клеящего агента.
После добавления, по меньшей мере, одного связующего вещества и/или, по меньшей мере, одного клеящего агента и/или, по меньшей мере, одного порообразующего агента и/или, по меньшей мере, одной кислотной добавки к смеси, содержащей силикалит титана-1, смесь, как правило, гомогенизируют в течение от 10 до 180 минут. В частности, смесители, бегуны или экструдеры особенно предпочтительно используется для гомогенизации. Смесь предпочтительно перемешивают. В промышленном масштабе, для гомогенизации предпочтительным является помол в бегунах. Гомогенизацию, как правило, проводят при температурах в диапазоне от около 10°С до точки кипения клеящего агента и при атмосферном давлении или давлении слегка выше атмосферного. Необязательно, по меньшей мере, одно из соединений, описанных выше, может быть добавлено. Таким образом, полученную смесь гомогенизируют, предпочтительно перемешивают до тех пор, пока не образуется экструдируемый пластический материал.
Гомогенизированную смесь затем подвергают формованию для получения формованного изделия. Все известные подходящие способы формования, такие как экструзия, распылительная сушка, распылительная грануляция, брикетирование, т.е. механическое сжатие с добавлением или без добавления дополнительного связующего вещества, или гранулирование, т.е. уплотнение с помощью круговых и/или вращательных движений, могут быть использованы.
Предпочтительными способами формования являются те, в которых используют обычные экструдеры для формования смеси, содержащей силикалит титана-1. Таким образом, получают, например, экструдаты, имеющие диаметр от 1 до 10 мм и предпочтительно от 2 до 5 мм. В дополнение к использованию экструдера, для получения формованных изделий также может использоваться экструзионный пресс. Форма формованных изделий, полученных в соответствии с настоящим изобретением, может быть выбрана по желанию. В частности, среди прочего, возможными являются сферы, овальные формы, цилиндры или таблетки. Аналогично, полые структуры, как, например, полые цилиндры или формованные структуры в виде сот, или также структуры с геометрий в форме звезды могут быть упомянуты.
Формование может осуществляться при атмосферном давлении или при давлении выше, чем давление окружающей среды, например, в диапазоне давлений от 1 бара до нескольких сотен бар. Кроме того, уплотнение может осуществляться при температуре окружающей среды или при более высокой температуре, чем температура окружающей среды, например, в диапазоне температур от 20 до 300°С. Если сушка и/или обжиг являются частью стадии формования, температуры до 600°С являются возможными. Наконец, уплотнение может осуществляться в окружающей атмосфере или в регулируемой атмосфере. Регулируемая атмосфера представляет собой, например, атмосферу инертного газа, восстановительную атмосферу и/или окислительную атмосферу.
Стадия формования предпочтительно сопровождается, по меньшей мере, одной стадией сушки. Данную, по меньшей мере, одну стадию сушки проводят при температурах в диапазоне, как правило, от 80 до 160°С, предпочтительно от 90 до 145°С и особенно предпочтительно от 100 до 130°С, обычно в течение 6 часов или более, например, в диапазоне от 6 до 24 часов. Тем не менее, в зависимости от содержания влаги в материале, подлежащем сушке, более короткое время сушки, такое как, например, около 1, 2, 3, 4 или 5 часов, также возможно.
До и/или после стадии сушки, предпочтительно полученный экструдат может, например, быть измельчен. Предпочтительно, таким образом получают гранулы или стружку, имеющие диаметр частиц от 0,1 до 5 мм, в частности от 0,5 до 2 мм.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения, сушка формованных изделий, соответственно, предпочтительно сопровождается, по меньшей мере, одной стадией обжига. Обжиг проводят при температурах, как правило, в диапазоне от 350-750°С, предпочтительно 400-700°С, особенно предпочтительно от 450-650°С. Обжиг может осуществляться в любой подходящей газообразной атмосфере, при этом предпочтительными являются воздух и/или обедненный воздух. Кроме того, обжиг предпочтительно проводят в муфельной печи, вращающейся печи и/или конвейерной обжиговой печи, при этом обжиг, как правило, проводят в течение 1 часа или более, например, в течение периода времени в диапазоне от 1 до 24 часов или от 3 до 12 часов. В способе согласно изобретению, можно, например, обжигать формованное тело катализатора один раз, дважды или более часто, в каждом случае, по меньшей мере, в течение 1 часа, например, в каждом случае от 3 часов до 12 часов, при этом можно поддерживать температуру во время стадии обжига постоянной или изменять температуру непрерывно или прерывисто. Если обжиг осуществляется два раза или чаще, температуры обжига на отдельных стадиях могут отличаться или быть идентичными.
Согласно особенно предпочтительному варианту осуществления настоящего изобретения, формованное тело катализатора подвергают гидротермальной обработке. Гидротермальная обработка может осуществляться с использованием любого подходящего способа, известного специалистам в данной области техники. Таким образом, катализатор или формованное тело катализатора, как правило, контактирует с водой или водяным паром. Как правило, указанную гидротермальную обработку осуществляют посредством загрузки катализатора, или согласно изобретению вместе с водой, в автоклав, и нагревания суспензии до температуры в диапазоне от 100 до 200°С, предпочтительно в диапазоне от 120 до 150°С при давлении в диапазоне от 1,5 до 5 бар, предпочтительно в диапазоне от 2 до 3 бар, в течение периода в диапазоне от 1 до 48 часов, предпочтительно в диапазоне от 24 до 48 часов. Как правило, затем следует, по меньшей мере, одна стадия промывки, предпочтительно водой в качестве промывочного вещества. После обработки водой катализатор предпочтительно сушат и/или обжигают, при этом сушку и обжиг проводят, как уже было описано выше. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, гидротермальную обработку осуществляют посредством перемешивания формованного тела катализатора в автоклаве, при этом скорость перемешивания доводят до такой скорости перемешивания, чтобы избежать истирания, насколько это возможно. Если катализатор используют в виде цилиндрических экструдатов, тем не менее, некоторое истирание является желательным для получения цилиндрических экструдатов, имеющих закругленные края. С помощью таких экструдатов, имеющих закругленные края, может быть достигнута более высокая объемная плотность, например, для использования экструдатов в качестве катализатора с неподвижным слоем в трубчатом реакторе R1 и/или в шахтном реакторе R2. Кроме того, снижается пылеобразование при использовании указанных катализаторов в способе эпоксидирования на стадиях (i) и (iii).
Кроме того, в способе эпоксидирования согласно настоящему изобретению, используют катализатор на основе силикалита титана-1, как описано выше, имеющий микропоры и мезопоры и содержащий от 49,5 до 80 вес. %, предпочтительно от 69,5 до 80 вес. % силикалита титана-1, в расчете на общий вес катализатора, и от 19,5 до 50 вес. %, предпочтительно от 19,5 до 30 вес. %, по меньшей мере, одного связующего вещества, предпочтительно связующего вещества на основе диоксида кремния, в расчете на общий вес формованного тела катализатора.
Если TS-1 используется в качестве каталитически активного материала согласно настоящему изобретению, предпочтительно, чтобы органический растворитель содержал, предпочтительно, по существу, состоял из метанола.
Таким образом, настоящее изобретение относится к способу регенерации катализатора, содержащего TS-1, в качестве каталитически активного материала, причем указанный катализатор использовался в способе получения оксида олефина, который включает в себя:
(i) обеспечение смеси, содержащей метанол, олефин, эпоксидирующий агент и, по меньшей мере, частично растворенную калийсодержащую соль;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей метанол и оксид олефина, и с получением катализатора, содержащего осажденную на нем калиевую соль;
причем указанный способ регенерации включает в себя:
(a) отделение смеси, полученной на стадии (ii), от катализатора;
(b) промывку катализатора, полученного на стадии (а), с помощью жидкой водной системы;
(c) необязательно сушку катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре менее 300°С;
(d) обжиг катализатора, полученного на стадии (с), в газовом потоке, содержащем кислород, при температуре, по меньшей мере, 300°С.
Особенно предпочтительно, настоящее изобретение предпочтительно относится к способу регенерации катализатора, содержащего TS-1, в качестве каталитически активного материала, причем указанный катализатор использовался в непрерывном способе получения пропиленоксида, который включает в себя:
(i) обеспечение смеси, содержащей метанол, пропен, перекись водорода, воду, необязательно пропан и, по меньшей мере, частично растворенную калийсодержащую соль, который отличается тем, что калийсодержащую соль выбирают из группы, состоящей из дигидрофосфата калия, гидроортофосфата калия, формиата калия и смеси двух или более из них;
(ii) воздействие на смесь, обеспеченную на стадии (i), в реакторе посредством условий эпоксидирования в присутствии катализатора с получением смеси, содержащей метанол, пропиленоксид, воду, необязательно пропен, необязательно пропан, и с получением катализатора, содержащего осажденную на нем калиевую соль,
который отличается тем, что смесь согласно стадии (i) содержит калийсодержащую соль с молярным соотношением калия, содержащегося в калийсодержащей соли, по отношению к перекиси водорода в диапазоне от 10×10-6:1 до 1500×10-6:1, предпочтительно от 20×10-6:1 до 1300×10-6:1, более предпочтительно от 30×10-6:1 до 1000×10-6:1,
причем указанный способ регенерации включает в себя:
(a) отделение смеси, полученной на стадии (ii), от катализатора;
(b) промывку катализатора, полученного на стадии (а), с помощью жидкой водной системы, которая содержит, по меньшей мере, 99,9 вес. % воды, более предпочтительно, по меньшей мере, 99,99 вес. % воды, более предпочтительно, по меньшей мере, 99,999 вес. % воды, в расчете на общий вес жидкой водной системы, при давлении в диапазоне от 0,8 до 1,5 бар, предпочтительно от 1,0 до 1,4 бар, и температуре в диапазоне от 40 до 90°С, предпочтительно от 60 до 80°С;
(c) необязательно сушку катализатора, полученного на стадии (b), в газовом потоке, содержащем инертный газ, при температуре в диапазоне от 25 до 100°С, предпочтительно от 30 до 50°С;
(d) обжиг катализатора, полученного на стадии (b) или (с), предпочтительно на стадии (с), в газовом потоке, содержащем кислород, используемом на стадии (d), содержит кислород в диапазоне от 3 до 40 об. %, предпочтительно от 5 до 50 об. % в расчете на общий объем газового потока при температуре в диапазоне от 375 до 525°С, предпочтительно от 400 до 500°С.
Реакция эпоксидирования
Реакцию могут проводить в периодическом режиме или непрерывном режиме, причем непрерывный режим является предпочтительным. Соответствующим образом, реактор содержит гетерогенный катализатор, расположенный в нем, и оснащен средствами для регулирования температуры реакции, такими как охлаждающая рубашка.
Соответствующим образом, степень конверсии исходного продукта может контролироваться посредством регулирования температуры, давления, среднечасовой скорости подачи исходных продуктов, и тому подобное. В качестве примера, температура реакции может быть отрегулирована таким образом, что, по меньшей мере, 90% эпоксидирующего агента подвергается конверсии. Количества исходного вещества, присутствующего в реакционной смеси перед и после реакции эпоксидирования, могут быть проанализированы с помощью любой подходящей методики, например хроматографии.
Как будет объяснено более подробно ниже, постепенное снижение активности катализатора, содержащего титансодержащий цеолит в качестве каталитически активного материала, может быть компенсировано на протяжении определенного периода времени, посредством увеличения температуры реакции. Температура реакции на стадии (ii), как правило, находится в диапазоне от 20 до 50°С, в зависимости от мгновенной активности используемого катализатора.
Как правило, непрерывную реакцию эпоксидирования на стадии (ii) могут проводить любым соответствующим способом. Предпочтительно, реакцию на стадии (ii) проводят, по меньшей мере, в одном реакторе непрерывного действия, таком как трубчатый реактор или кожухотрубный реактор, который предпочтительно содержит, по меньшей мере, одну охлаждающую рубашку, окружающую, по меньшей мере, одну трубку. Если реакцию на стадии (ii) проводят в таком реакторе, содержащем, по меньшей мере, одну охлаждающую рубашку, термин ''температура реакции'', используемый здесь, относится к температуре охлаждающей среды на входе в охлаждающую рубашку.
Катализатор, содержащий титановый цеолит, может быть использован в любой возможной форме, описанной выше, включая порошок, микропорошок, предпочтительно распылительный порошок, в виде формованного изделия, содержащего порошок, или в виде формованного изделия, содержащего микропорошок, предпочтительно распылительный порошок. Предпочтительно, катализатор, содержащий титановый цеолит, используют в виде формованного изделия, содержащего порошок или микропорошок, предпочтительно распылительный порошок, более предпочтительно в виде формованного изделия, содержащего микропорошок, предпочтительно распылительный порошок.
Катализатор, используемый на стадия (ii) настоящего изобретения, может быть размещен в реакторе любым подходящим способом. Предпочтительно, катализатор выполнен в виде псевдоожиженного слоя или в виде неподвижного слоя, более предпочтительно в виде неподвижного слоя.
Как упоминалось выше, жидкий поток поступающего материала, обеспеченный на стадии (i), подают в реактор на стадии (i), содержащий катализатор, предпочтительно находящийся в виде неподвижного слоя. Во время реакции эпоксидирования, нагрузка катализатора предпочтительно находится в диапазоне от 0,05 до 1,25 ч-1, предпочтительно от 0,1 до 1 ч-1, более предпочтительно от 0,2 до 0,7 ч-1, где нагрузка катализатора определена как отношение массового расхода (скорости потока) в кг/ч эпоксидирующего агента, предпочтительно перекиси водорода, содержащейся в жидком потоке поступающего материала, обеспеченного на стадии (i), разделенное на количество в кг катализатора, содержащего титановый цеолит, находящегося в реакторе эпоксидирования на стадии (ii). Термин ''условия эпоксидирования включают в себя'', как используется в данном контексте настоящего изобретения, относится к реакции эпоксидирования на стадии (ii), которая отличается тем, что, по меньшей мере, 90%, предпочтительно, по меньшей мере, 95% слоя катализатора в реакторе и в течение, по меньшей мере, 90%, предпочтительно, по меньшей мере, 95% общего времени реакции, нагрузка катализатора находится в указанных выше пределах.
Во время реакции эпоксидирования на стадии (ii), температуру реакционной смеси в реакторе предпочтительно контролируют, более предпочтительно поддерживают в предпочтительных диапазонах. Для того чтобы контролировать температуру реакционной смеси, могут использоваться внутренние и/или внешние средства регулирования температуры. Термин ''внутренние средства регулирования температуры'', как используется в данном контексте настоящего изобретения, относятся к средствам, которые расположены в реакторе. Термин ''внешние средства регулирования температуры'', как используется в данном контексте настоящего изобретения, относятся к средствам, которые расположены снаружи реактора. Предпочтительно, температуру реакционной смеси регулируют с помощью внешних средств регулирования температуры, более предпочтительно с помощью теплоносителя, который предпочтительно пропускают через подходящую рубашку, причем данная рубашка предпочтительно окружает реактор. В случае использования кожухотрубного реактора в качестве реактора, рубашка предпочтительно окружает все трубки пучка труб.
Предпочтительно, во время реакции эпоксидирования на стадии (ii), температура реакции находится в диапазоне от 20 до 100°С, более предпочтительно от 25 до 90°С, более предпочтительно от 30 до 80°С, более предпочтительно от 35 до 70°С, более предпочтительно от 40 до 60°С. Термин ''температура реакции'', как используется в данном контексте настоящего изобретения, относится к температуре теплоносителя до регулирования температуры реакционной смеси, предпочтительно к температуре теплоносителя на входе в рубашку реактора эпоксидирования, через рубашку которого пропускают теплоноситель. Таким образом, настоящее изобретение относится к способу, как описано выше, который отличается тем, что на стадии (ii), условия эпоксидирования содержат, предпочтительно включают в себя температуру реакции эпоксидирования в диапазоне от 20 до 100°С, предпочтительно от 30 до 80°С, более предпочтительно от 40 до 60°С, где температура реакции эпоксидирования определена как температура теплоносителя до регулирования температуры реакционной смеси, предпочтительно как температура теплоносителя на входе в рубашку реактора эпоксидирования. Термин ''условия эпоксидирования включают в себя'', как используется в данном контексте настоящего изобретения, относится к реакции эпоксидирования на стадии (ii), которая отличается тем, что, по меньшей мере, 98%, предпочтительно, по меньшей мере, 99%, более предпочтительно, по меньшей мере, 99,9% общего времени реакции, температура реакции находится в указанных выше пределах. Термин ''общее время реакции'', как используется в данном контексте настоящего изобретения, относится ко времени реакции, в течение которого данный слой катализатора используется перед тем как, его либо выбракуют, либо подвергнут регенерации. В частности, в начале реакции эпоксидирования на стадии (ii), когда катализатор является ''свежим'', то есть при пуске реакции эпоксидирования на стадии (ii), температура реакции может находится за пределами вышеуказанных диапазонов в течение короткого периода времени. Предпочтительно, скорость потока теплоносителя выбирают таким образом, чтобы перепад температур между его температурой на входе и температурой на выходе составлял не более 3 K, более предпочтительно не более 2 K, более предпочтительно не более 1 K.
Предпочтительно, во время реакции эпоксидирования на стадии (ii), давление реакции эпоксидирования находится в диапазоне от 14 до 100 бар, более предпочтительно от 14,5 до 50 бар, более предпочтительно от 15 до 32 бар, более предпочтительно от 15 до 25 бар. Термин ''давление реакции эпоксидирования'', как используется в данном контексте настоящего изобретения, относится к давлению на выходе из реактора эпоксидирования, где выходящий поток удаляют из реактора в соответствии со стадией (iii). Таким образом, настоящее изобретение относится к способу, как описано выше, который отличается тем, что на стадии (ii), условия эпоксидирования содержат, предпочтительно включают в себя давление реакции эпоксидирования в диапазоне от 14 до 100 бар, предпочтительно от 15 до 32 бар, более предпочтительно от 15 до 25 бар. Термин ''условия эпоксидирования включают в себя'', как используется в данном контексте настоящего изобретения, относится к реакции эпоксидирования на стадии (ii), которая отличается тем, что, по меньшей мере, 98%, предпочтительно, по меньшей мере, 99%, более предпочтительно, по меньшей мере, 99,9% общего времени реакции, температура реакции находится в указанных выше пределах. Термин ''общее время реакции'', как используется в данном контексте настоящего изобретения, относится ко времени реакции, в течение которого данный слой катализатора используется перед тем как, его либо выбракуют, либо подвергнут регенерации.
Предпочтительно, реакция эпоксидирования согласно стадии (ii) настоящего изобретения осуществляется при, по существу, постоянной конверсии эпоксидирующего агента, предпочтительно конверсии перекиси водорода. Предпочтительно, чтобы определить конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода, молярный расход эпоксидирующего агента, предпочтительно перекиси водорода в выходящем потоке, удаляемом на стадии (iii), упоминаемом здесь как mout, сравнивают с молярным расходом эпоксидирующего агента, предпочтительно перекиси водорода, в жидком потоке поступающего материала, обеспеченном на стадии (i), упоминаемом здесь как min, и где конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода, определяют как 100×(1-mout/min). Предпочтительно, температуру теплоносителя на входе, описанную выше, регулируют в вышеупомянутых предпочтительных диапазонах, для поддержания конверсии эпоксидирующего агента, предпочтительно конверсии перекиси водорода, по существу, постоянной в диапазоне от 80 до 100%, более предпочтительно от 90 до 100%, более предпочтительно от 95 до 100%, более предпочтительно от 99 до 100%, более предпочтительно от 99,5 до 100%, более предпочтительно от 99,9 до 100%. Термин ''условия эпоксидирования включают в себя'', как используется в данном контексте настоящего изобретения, относится к реакции эпоксидирования на стадии (ii), которая отличается тем, что, по меньшей мере, 98%, предпочтительно, по меньшей мере, 99%, более предпочтительно, по меньшей мере, 99,9% общего времени реакции, конверсия эпоксидирующего агента, предпочтительно конверсия перекиси водорода, находится в указанных выше пределах. Термин ''общее время реакции'', как используется в данном контексте настоящего изобретения, относится ко времени реакции, в течение которого данный слой катализатора используется перед тем как, его либо выбракуют, либо подвергнут регенерации. В частности, в начале реакции эпоксидирования на стадии (ii), когда катализатор является ''свежим'', то есть при пуске реакции эпоксидирования на стадии (ii), конверсия эпоксидирующего агента, предпочтительно конверсия перекиси водорода, может находиться за пределами вышеуказанных диапазонов в течение короткого периода времени. Предпочтительно, температуру реакции не поддерживают постоянной в ходе реакции, но регулируют непрерывно или ступенчато, чтобы обеспечить постоянную конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода. Как правило, вследствие определенной дезактивации катализатора, температура реакции непрерывно или ступенчато повышается. Предпочтительно, температура реакции непрерывно или ступенчато повышается не более, чем на 1 K/сутки (Кельвин/сутки), более предпочтительно менее, чем на 1 K/сутки.
Предпочтительно, реакционная смесь, которая находится в реакторе на стадии (ii) представляет собой жидкость в условиях эпоксидирования. Предпочтительно, реакционная смесь состоит из одной единственной жидкой фазы, двух жидких фаз или трех, или более жидких фаз. Предпочтительно, реакционная смесь в реакторе на стадии (ii) состоит из одной единственной жидкой фазы или двух жидких фаз, более предпочтительно из одной единственной жидкой фазы.
Как правило, реактор, используемый на стадии (ii) настоящего изобретения, может быть расположен горизонтально или вертикально. Предпочтительно, реактор расположен вертикально. В предпочтительно расположенном вертикально реакторе, жидкий поток поступающего материала, обеспеченный на стадии (i), может подаваться в режиме с восходящим потоком или в режиме с нисходящим потоком, причем режим с восходящим потоком является предпочтительным. Предпочтительно, по сравнению с направлением жидкого потока поступающего материала, теплоноситель пропускают через рубашку в режиме параллельного потока.
Как правило, реакция эпоксидирования на стадии (ii) может осуществляться в одном или нескольких реакторах, причем данные реакторы могут быть расположены параллельно или последовательно. Предпочтительно, реакцию на стадии (ii) проводят в одном реакторе или, по меньшей мере, в двух реакторах, предпочтительно, двух реакторах, которые расположены последовательно, причем между двумя реакторами, расположенными последовательно, может осуществляться подходящая промежуточная обработка. Если реакцию проводят в двух реакторах, расположенных последовательно, то предпочтительно, чтобы первый реактор работал, как описано выше, т.е., как изотермический реактор, а второй реактор, т.е. последующий реактор, работал как адиабатический или, по существу, адиабатический реактор. Термин ''реактор'', как используется здесь, также включает в себя два или более реакторов, расположенных параллельно, где подаваемый поток поступающего материала разделен на два или более субпотока, каждый субпоток подается в реактор, а выходящие потоки, удаляемые из реакторов, объединяются для получения общего отходящего потока. Таким образом, реакция эпоксидирования может быть проведена, по меньшей мере, в одном первом реакторе, например, двух или более первых реакторах, например, 2, 3, 4 первых реакторах, которые расположены параллельно и, которые предпочтительно представляют собой изотермические реакторы, и, по меньшей мере, в одном втором реакторе, например, двух или более вторых реакторах, например, 2, 3, 4 вторых реакторах, которые расположены параллельно и, которые предпочтительно представляют собой адиабатические или, по существу, адиабатические реакторы.
Если реакцию эпоксидирования согласно стадии (ii) проводят в двух реакторах, расположенных последовательно, то предпочтительно, чтобы в первом реакторе, который является предпочтительно изотермическим реактором, конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода, поддерживали, по существу, постоянной в диапазоне от 80 до 99%, предпочтительно от 85 до 98%, более предпочтительно от 90 до 97%, а во втором реакторе, который предпочтительно выполнен в виде адиабатического или, по существу, адиабатического реактора, общую конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода, т.е. конверсию эпоксидирующего агента, предпочтительно конверсию перекиси водорода, с учетом конверсии в первом и во втором реакторе, доводят до значения более 99%, предпочтительно, по меньшей мере, 99,5%, более предпочтительно, по меньшей мере, 99,9%.
Отработанный катализатор
Как правило, после длительного периода использования свежеприготовленного катализатора, содержащего титансодержащий цеолит, в способе получения оксида олефина, наблюдается снижение его каталитической активности по сравнению со свежеприготовленным катализатором. Такое постепенное снижение каталитической активности может быть компенсировано в некоторой степени за счет увеличения температуры реакции. Каталитическая активность может отслеживаться посредством определения степени конверсии, по меньшей мере, одного исходного вещества в ходе реакции при данной температуре. В случае, если наблюдается падение степени конверсии в ходе процесса, температуру реакции увеличивают. Соответственно, катализатор, содержащий титансодержащий цеолит, может быть подвергнут регенерации, когда температура реакции достигла заданного верхнего предела температуры, выше которого процесс становится экологически и экономически неэффективным. Например, катализатор, содержащий титансодержащий цеолит, может подвергаться регенерации, когда температура реакции на стадии (ii), необходимая для поддержания степени конверсии одного из исходных веществ выше, например, 90%, составляет 70°С или выше, предпочтительно 60°С или выше, предпочтительно более 50°С или выше.
В альтернативном варианте катализатор, содержащий титансодержащий цеолит и осажденную на нем калиевую соль, после стадий (i) и (ii), может быть подвергнут регенерации согласно стадиям (а)-(d), если его селективность отклоняется более чем на определенный процент по сравнению с селективностью свежего катализатора, содержащего титансодержащий цеолит. В данном случае, селективность катализатора, содержащего титансодержащий цеолит, определяется общей конверсией одного исходного вещества, разделенной на конверсию указанного исходного вещества в необходимый продукт. Например, катализатор, содержащий титансодержащий цеолит, может подвергаться регенерации после стадий (i) и (ii), когда селективность катализатора по отношению к оксиду олефина, определенная на стадии (ii), отклоняется на 2% или более от соответствующей селективности свежего катализатора, содержащего титансодержащий цеолит, при прочих равных условиях реакции.
В соответствии с настоящим изобретением, было обнаружено, что катализатор, содержащий титансодержащий цеолит, предназначенный для регенерации, который отвечает одному из описанных выше критериев после стадий (i) и (ii) обычно имеет содержание калия выше 0,5 вес. %, предпочтительно в диапазоне от 0,6 до 1,3 вес. %. Также дополнительно, было обнаружено, что после одной последовательности стадий (а)-(d) в соответствии с настоящим изобретением, регенерированный катализатор, содержащий титансодержащий цеолит, полученный на стадии (d), имеет содержание калия не более 0,5 вес. %, предпочтительно не более 0,4 вес. %, более предпочтительно не более 0,3 вес. %, в расчете на общий вес катализатора и определенное с помощью элементного анализа.
Регенерированный катализатор, полученный в соответствии со способом настоящего изобретения, может быть использован для любого варианта применения. Предпочтительно, катализатор, содержащий титансодержащий цеолит, полученный на стадии (d), используют в способе получения оксида олефина, предпочтительно в способе эпоксидирования олефина, который включает в себя:
(i') обеспечение смеси, содержащей органический растворитель, олефин, эпоксидирующий агент и фосфатсодержащее соединение;
(ii') воздействие на смесь, обеспеченную на стадии (i'), в реакторе посредством условий эпоксидирования в присутствии катализатора, полученного на стадии (d), с получением смеси, содержащей органический растворитель и оксид олефина.
Предпочтительные варианты стадий (i') и (ii') осуществляют, как подробно описано для стадий (i) и (ii) выше.
Настоящее изобретение дополнительно относится к катализатору, содержащему титансодержащий цеолит, в качестве каталитически активного материала, получаемый или полученный посредством способа регенерации согласно настоящему изобретению.
Предпочтительно, чтобы катализатор, регенерированный в соответствии с настоящим изобретением, имел при осуществлении способа получения оксида олефина, разность температур конверсии не более 5 K, где разность температур конверсии определяется как абсолютная разница между:
(А1) температурой, при которой заранее заданная конверсия эпоксидирующего агента достигается в указанном способе получения оксида олефина, в котором регенерированный катализатор используют в качестве катализатора, и
(В1) температурой, при которой заранее заданная конверсия эпоксидирующего агента достигается в указанном способе получения оксида олефина, в котором соответствующий свежий катализатор используют в качестве катализатора при прочих равных условиях реакции эпоксидирования.
Как уже было отмечено ранее, после определенного времени работы наблюдается снижение каталитической активности катализатора, содержащего титансодержащий цеолит в качестве каталитически активного материала, в реакции эпоксидирования. Сниженная каталитическая активность напрямую связана со снижением степени конверсии, по меньшей мере, одного из исходных веществ, т.е. олефина и/или эпоксидирующего агента, при этом сниженная степень конверсии может быть компенсирована за счет увеличения общей температуры реакции. Это означает, что при продолжении работы катализатора требуется постепенное увеличение температуры реакции по сравнению с начальной температурой, что делает способ эпоксидирования все менее эффективным.
Тем не менее, посредством воздействия на катализатор, содержащий титансодержащий цеолит, отработанный в реакции эпоксидирования, с помощью способа регенерации согласно настоящего изобретения, его начальная каталитическая активность может быть восстановлена. Начальная каталитическая активность относится здесь к каталитической активности свежеприготовленного катализатора. Поскольку каталитическая активность соответственно напрямую связана с температурой реакции при прочих равных условиях реакции, коэффициент полезного действия регенерации отработанного катализатора может быть выведен из температуры реакции, необходимой для поддержания заданной степени конверсии. В данном случае, катализатор, регенерированный в соответствии с настоящим изобретением, выгодно отличается в способе получения оксида олефина температурой конверсии, которая отклоняется не более, чем на 5 K от температуры конверсии свежего катализатора при прочих равных условиях эпоксидирования.
Кроме того, также предпочтительно, чтобы катализатор, регенерированный в соответствии со способом настоящего изобретения, имел при осуществлении способа получения оксида олефина, разность селективности не более 2, где разность селективности определяется как абсолютная разница в % между:
(А2) селективностью на основе эпоксидирующего агента в указанном способе получения оксида олефина, в котором регенерированный катализатор используют в качестве катализатора, и
(В2) селективностью на основе эпоксидирующего агента в указанном способе получения оксида олефина, в котором соответствующий свежий катализатор используют в качестве катализатора при прочих равных условиях реакции эпоксидирования,
где селективность на основе эпоксидирующего агента определяется как число молей полученного эпоксида, разделенное на число молей использованного эпоксидирующего агента × 100.
Качество катализатора, содержащего титансодержащий цеолит, регенерированного согласно способу настоящего изобретения, также может быть количественно определено посредством сравнения селективности регенерированного катализатора с селективностью свежего катализатора при прочих равных условиях эпоксидирования. После длительного использования также, как правило, наблюдается снижение селективности катализатора. Выгодно, в данном случае, после воздействия с помощью способа регенерации согласно настоящему изобретению, катализатор, содержащий титансодержащий цеолит, обладает селективностью, которая отклоняется не более, чем на 2% от селективности свежего катализатора при прочих равных условиях реакции эпоксидирования.
Описание фигур
Фигура 1 показывает количество калия и фосфора, осажденное на отработанном катализаторе, по отношению к общему содержанию кремния. Фракция 1 представляет собой образец, взятый из первого метра в кубовой части камеры реактора, фракция 2 представляет собой образец, взятый на расстоянии от 1 до 2 м от кубовой части камеры реактора и фракция 3 представляет собой образец, взятый на расстоянии от 2 до 3 м от кубовой части камеры реактора.
Фигура 2 показывает количество углерода и азота, осажденное на отработанном катализаторе, по отношению к общему содержанию кремния. Фракция 1 представляет собой образец, взятый из первого метра в кубовой части камеры реактора, фракция 2 представляет собой образец, взятый на расстоянии от 1 до 2 м от кубовой части камеры реактора и фракция 3 представляет собой образец, взятый на расстоянии от 2 до 3 м от кубовой части камеры реактора.
Фигура 3 показывает каталитические характеристики отработанного катализатора, регенерированного в соответствии со способом известного уровня техники, по сравнению с каталитическими характеристиками свежего катализатора при прочих равных условиях эпоксидирования. Указаны степень конверсии на основе перекиси водорода, нормированные значения селективности на основе перекиси водорода и пропена отработанного катализатора и свежего катализатора, а также температура реакции (°С) отработанного катализатора и свежего катализатора.
Фигура 4 показывает ИК-спектр с Фурье-преобразованием свежего катализатора. Ось х показывает волновые числа в см-1 и ось у показывает поглощающую способность (А).
Фигура 5 показывает ИК-спектр с Фурье-преобразованием отработанного катализатора после циклов регенерации, каждый цикл включает в себя стадии (а)-(b) в соответствии с изобретением. Ось х показывает волновые числа в см-1 и ось у показывает поглощающую способность (А).
Фигура 6 показывает каталитические характеристики отработанного катализатора, подвергнутого воздействию пяти циклов регенерации, включающих в себя стадии (а)-(b) согласно настоящему изобретению, по сравнению с каталитическими характеристиками свежего катализатора при прочих равных условиях эпоксидирования. Указаны степень конверсии на основе перекиси водорода, нормированные значения селективности на основе перекиси водорода и пропена регенерированного катализатора и свежего катализатора, а также температура реакции (°С), применяемая при использовании регенерированного катализатора и свежего катализатора.
Настоящее изобретение дополнительно проиллюстрировано следующими справочными примерами, примерами и сравнительными примерами.
ПРИМЕРЫ
Справочный пример 1: Получение катализатора, содержащего титансодержащий цеолит (ZnTiMWW)
1.1 Получение борсодержащего MWW
470,4 кг деионизированной воды загрузили в сосуд. При перемешивании при 70 оборотах в минуту (об./мин), 162,5 кг борной кислоты суспендировали в воде. Суспензию перемешивали в течение еще 3 часов. Затем добавили 272,5 кг пиперидина и смесь перемешивали в течение еще одного часа. В полученный раствор добавили 392,0 кг Ludox® AS-40 и полученную смесь перемешивали при 70 об./мин в течение еще одного часа. Полученную в результате смесь перенесли в кристаллизационный сосуд и нагревали до 170°С в течение 5 часов при автогенном давлении и при перемешивании (50 об./мин). Температуру 170°С поддерживали по существу постоянной в течение 120 часов; в данный промежуток 120 часов смесь перемешивали при 50 об./мин. Затем смесь охлаждали до температуры 50-60°С в течение 5 часов. Водная суспензия, содержащая B-MWW, имела рН 11,3, как определено с помощью измерения рН-электродом. Из указанной суспензии, B-MWW отделили фильтрованием. Осадок на фильтре промывали деионизированной водой до тех пор, пока промывочная вода не имела электрическую проводимость меньше, чем 700 мкСм/см. Полученный таким образом осадок на фильтре подвергли распылительной сушке в распылительной башне с использованием технического азота в качестве сушильного газа. Высушенный распылением материал затем подвергли обжигу при температуре 650°С в течение 2 часов. Обожженный материал имел содержание бора (В) 1,9 вес. %, содержание кремния (Si) 41 вес. % и общее содержание органического углерода (ТОС) 0,18 вес. %.
1.2 Получение не содержащего бора MWW
На основе высушенного распылением материала, полученного в соответствии с разделом 1.1 выше, подготовили 4 партии не содержащего бора цеолита MWW. В каждой из первых 3 партий, использовали 35 кг материала, высушенного распылением, полученного в соответствии с разделом 1.1, и 525 кг воды. В четвертой партии, использовали 32 кг материала, высушенного распылением, полученного в соответствии с разделом 1.1, и 480 кг воды. В общей сложности были использованы 137 кг высушенного распылением материала, полученного в соответствии с разделом 1.1, и 2025 кг воды. Для каждой партии, соответствующий объем воды подавали в сосуд, оснащенный конденсатором флегмы. При перемешивании при 40 об./мин, данное количество материала, высушенного распылением, суспендировали в воде. Затем сосуд закрыли и запустили в работу конденсатор флегмы. Скорость перемешивания увеличили до 70 об./мин. При перемешивании при 70 об./мин, содержимое сосуда нагревали до 100°С в течение 10 часов и выдерживали при этой температуре в течение 10 часов. Затем содержимое сосуда охладили до температуры ниже 50°С. Полученный не содержащий бора цеолитный материал структурного типа MWW отделили от суспензии посредством фильтрации под давлением азота 2,5 бар и промыли четыре раза деионизованной водой. После фильтрации осадок на фильтре сушили в потоке азота в течение 6 часов. Не содержащий бора цеолитный материал, полученный в 4 партиях (в общей сложности 625,1 кг осадка на фильтре, высушенного азотом), имел остаточную влажность 79%, как было определено с помощью инфракрасной (ИК) шкалы при 160°С. Используя осадок на фильтре, высушенный азотом, имеющий остаточную влажность 79%, полученный выше, получили водную суспензию с деионизированной водой, с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне с использованием технического азота в качестве сушильного газа. Высушенный распылением полученный MWW материал имел содержание бора (В) 0,08 вес. %, содержание кремния (Si) 42 вес. % и общее содержание органического углерода (ТОС) 0,23 вес. %.
1.3 Получение TiMWW
На основе не содержащего бора материала MWW, полученного в соответствии с разделом 1.2 выше, был получен цеолитный материал структурного типа MWW, содержащий титан (Ti), называемый далее как TiMWW. 54,16 кг не содержащего бора цеолитного материала структурного типа MWW загрузили в первый сосуд А. Во второй сосуд В загрузили 200,00 кг деионизированной воды и перемешивали при 80 об./мин. При перемешивании добавили 118,00 кг пиперидина и во время добавления температуру смеси увеличили приблизительно на 15°С. Затем добавили 10,90 кг тетрабутилортотитаната и 20,00 кг деионизированной воды. Перемешивание продолжали в течение 60 минут. Затем смесь сосуда В перенесли в сосуд А, и начали перемешивание в сосуде А (70 об./мин). В сосуд А поместили 24,00 кг деионизированной воды и перенесли ее в сосуд В. Затем смесь в сосуде В перемешивали в течение 60 минут при 70 об./мин. В начале перемешивания, рН смеси в сосуде В составлял 12,6, как определено с помощью рН-электрода. После указанного перемешивания при 70 об./мин, частоту снизили до 50 об./мин. и смесь в сосуде В нагревали до температуры 170°С в течение 5 часов. При постоянной скорости перемешивания 50 об./мин температуру смеси в сосуде В выдерживали, по существу, при постоянной температуре 170°С в течение 120 часов при автогенном давлении. Во время данной кристаллизации TiMWW наблюдалось повышение давления до 10,6 бар. Затем полученную суспензию, содержащую TiMWW, имеющую рН 12,6 охлаждали в течение 5 часов. Охлажденную суспензию подвергли фильтрации и отделенный маточный раствор направили на сброс сточных вод. Осадок на фильтре промыли четыре раза деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 6 часов. Используя 246 кг указанного фильтровального осадка, получили водную суспензию с деионизированной водой с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне с использованием технического азота в качестве сушильного газа. Высушенный распылением материал TiMWW, полученный в результате первого опыта, имел содержание кремния (Si) 37 вес. %, содержание титана (Ti) 2,4 вес. % и общее содержание органического углерода (ТОС) 7,5 вес. %.
1.4 Кислотная обработка TiMWW
Высушенный распылением материал TiMWW, полученный в разделе 1.3 выше, подвергли кислотной обработке с последующей распылительной сушкой и обжигом, как описано ниже. 670,0 кг деионизированной воды загрузили в сосуд. Добавили 900 кг азотной кислоты и 53,0 кг высушенного распылением TiMWW добавили при перемешивании при 50 об./мин. Полученную смесь перемешивали в течение еще 15 минут. Затем скорость перемешивания увеличили до 70 об./мин. В течение 1 часа смесь в сосуде нагревали до 100°С и выдерживали при этой температуре и при автогенном давлении в течение 20 часов при перемешивании. Полученную таким образом смесь затем охлаждали в течение 2 часов до температуры менее 50°С. Охлажденную смесь фильтровали и осадок на фильтре промывали шесть раз деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 10 часов. Промывочная вода после шести стадий промывки имела рН около 2,7. Было получено 225,8 кг высушенного отфильтрованного осадка. Используя полученный отфильтрованный осадок, получили водную суспензию с деионизированной водой с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне с использованием технического азота в качестве сушильного газа. Высушенный распылением обработанный кислотой материал TiMWW имел содержание кремния (Si) 42 вес. %, содержание титана (Ti) 1,6 вес. % и общее содержание органического углерода (ТОС) 1,7 вес. %. Материал, высушенный распылением, затем подвергали обжигу при температуре 650°С во вращающейся печи в течение 2 часов. Обожженный материал имел содержание Si 42,5 вес. %, содержание Ti 1,6 вес. % и содержание ТОС 0,15 вес. %. Поверхность по методу Ленгмюра (Langmuir), определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 612 м2/г, удельная площадь поверхности по многоточечному методу BET (Brunauer - Emmett - Teller), определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 442 м2/г. Общая пористость, определяемая с помощью метода ртутной порометрии в соответствии с DIN 66133, составляла 4,9 мл/г (миллилитр/грамм), соответствующая суммарная площадь пор 104,6 м2/г. Степень кристаллизации, определяемая с помощью рентгенодифракционного (XRD) анализа, составляла 80%, средний размер кристаллитов - 31 нм.
1.5 Пропитка TiMWW цинком (Zn)
Обработанный кислотой, высушенный распылением и обожженный материал, полученный согласно разделу 1.4 затем подвергли стадии пропитки. Пропитку проводили в 3 партиях а)-с) следующим образом:
a) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 840 кг деионизированной воды и 5,13 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 28 кг обожженного материала Ti-MWW, полученного в соответствии с разделом 1.4 суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
b) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 840 кг деионизированной воды и 5,13 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 28 кг обожженного материала Ti-MWW, полученного в соответствии с разделом 1.4 суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
c) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 930 кг деионизированной воды и 5,67 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 31 кг обожженного материала Ti-MWW, полученного в соответствии с разделом 1.4 суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
Во всех партиях а)-с), полученную смесь в сосуде нагревали до 100°С в течение 1 часа и выдерживали с обратным холодильником в течение 4 часов при скорости перемешивания 70 об./мин. Затем смесь охлаждали в течение 2 часов до температуры менее 50°С. В каждой партии а)-с), охлажденную суспензию подвергли фильтрации, а маточный раствор направили на сброс сточных вод. Осадок на фильтре промыли пять раз деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 10 часов. В партии а) в конечном итоге получили 106,5 кг отфильтрованного осадка, высушенного азотом. В партии b) в конечном итоге получили 107,0 кг отфильтрованного осадка, высушенного азотом. В партии с) в конечном итоге получили 133,6 кг отфильтрованного осадка, высушенного азотом. Полученный таким образом высушенный материал TiMWW, пропитанный Zn (ZnTiMWW), для каждой партии, имел содержание Si 42 вес. %, содержание Ti 1,6 вес. %, содержание Zn 1,4 вес. % и ТОС 1,4 вес. %.
1.6 Получение формованного изделия
Используя обожженный высушенный распылением материал ZnTiMWW, полученный выше, получили, высушили и подвергли обжигу формованное изделие. Для этой цели 22 партии, каждая с использованием 3,4 кг обожженного высушенного распылением материала ZnTiMWW, полученного в Примере 1, 0,220 кг Walocel™ (Walocel MW 15000 GB, Wolff Cellulosics GmbH & Co. KG, Germany), 2,125 кг Ludox® AC-40 и 6,6 л деионизированной воды, подготовили следующим образом: 3,4 кг ZnTiMWW и 0,220 кг Walocel подвергли разминанию в бегунковой мельнице в течение 5 минут. Затем во время дальнейшего разминания непрерывно добавляли 2,125 кг Ludox. Спустя еще 10 минут начали добавлять 6 л деионизированной воды. Еще через 30 минут, добавили дополнительно 0,6 л деионизированной воды. После общего времени подготовки 50 минут, замешанная масса стала пригодной для экструзии. Затем замешанную массу подвергли экструзии при 65-80 бар, при этом экструдер охлаждали водой в процессе экструзии. Для каждой партии, время экструзии находилась в диапазоне от 15 до 20 минут. Потребляемая электрическая мощность на каждую партию во время экструзии составляла 2,4 А. Экструзионную головку использовали для получения цилиндрических заготовок диаметром 1,7 мм. На выпускном отверстии экструзионной головки, заготовки не подвергали резке по длине. Заготовки, полученные таким образом, сушили в течение 16 часов при 120°С в сушильной камере на воздухе. В целом (сумма 22 партий) получили 97,1 кг белых заготовок диаметром 1,7 мм. 65,5 кг высушенных заготовок подвергли обжигу во вращающейся печи при 550°С в течение 1 часа на воздухе с выходом 62,2 кг обожженных заготовок. После этого заготовки пропустили через сито (размер ячейки 1,5 мм) и выход после просеивания составил 57,7 кг.
Определение характеристик полученных заготовок
Полученные таким образом формованные изделия имели объемную плотность 322 г/л (грамм на литр) и имели содержание Zn 1,2 вес. %, содержание Ti 1,4 вес. %, содержание Si 43 вес. % и содержание С 0,13 вес. %. Содержание натрия (Na) составляло 0,07 вес. %. Мезопоры микропорошка имели средний диаметр пор (4V/A) 20,1 нм, как определено методом ртутной порометрии в соответствии с DIN 66133. Макропоры микропорошка имели средний диаметр пор (4V/A) 46,8 нм, как определено методом ртутной порометрии в соответствии с DIN 66133. Степень кристаллизации, определяемая с помощью рентгенодифракционного (XRD) анализа, составляла 74 +/- 10%, средний размер кристаллитов - 38,0 нм +/- 10%. Поверхность по методу Ленгмюра (Langmuir), определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 499 м2/г, удельная площадь поверхности по многоточечному методу BET (Brunauer - Emmett - Teller), определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 361 м2/г. Общая пористость, определяемая с помощью метода ртутной порометрии в соответствии с DIN 66133, составляла 1,2 мл/г (миллилитр/грамм), соответствующая суммарная площадь пор 92,2 м2/г.
1.7 Последующая обработка формованного изделия
Используя обожженные заготовки, полученные в соответствии с разделом 1.6 выше, стадию последующей обработки выполнили следующим образом: 590 кг деионизированной воды загрузили в сосуд. Затем добавили 29,5 кг обожженных формованных изделий, полученных в соответствии с Примером 2. Сосуд закрыли (герметично) и полученную смесь нагревали до температуры 145°С в течение 1,5 часов и выдерживали при этой температуре при автогенном давлении (около 3 бар) в течение 8 часов. Затем смесь охлаждали в течение 2 часов. Обработанные водой заготовки подвергли фильтрации и промыли деионизованной водой. Полученные заготовки нагревали в сушильной камере на воздухе в течение 1 часа до температуры 120°С и выдерживали при данной температуре в течение 16 часов. Затем, высушенный материал нагревали на воздухе до температуры 450°С в течение 5,5 часов и выдерживали при данной температуре в течение 2 часов. После этого заготовки просеивали (размер ячейки 1,5 мм) и выход после просеивания составил 27,5 кг.
Определение характеристик полученных заготовок
Полученные таким образом обработанные водой формованные изделия имели объемную плотность 340 г/л (грамм на литр) и имели содержание Zn 1,3 вес. %, содержание Ti 1,4 вес. %, содержание Si 43 вес. % и содержание С 0,10 вес. %. Мезопоры микропорошка имели средний диаметр пор (4V/A) 20,2 нм, как определено методом ртутной порометрии в соответствии с DIN 66133. Таким образом, обработка водой согласно изобретению не имеет практически никакого влияния на характеристики мезопор формованного изделия (ср. формованное изделие, описанное выше, имеет соответствующий средний диаметр пор 20,1 нм). Макропоры микропорошка имели средний диаметр пор (4V/A) 45,9 нм, как определено методом ртутной порометрии в соответствии с DIN 66133. Таким образом, обработка водой согласно изобретению не имеет практически никакого влияния на характеристики макропор формованного изделия (ср. формованное изделие, описанное выше, имеет соответствующий средний диаметр пор 46,8 нм). Степень кристаллизации, определяемая с помощью рентгенодифракционного (XRD) анализа, составляла 64 +/- 10%, средний размер кристаллитов - 39,4 нм +/- 10%. Поверхность по методу Ленгмюра (Langmuir), определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 418,1 м2/г, удельная площадь поверхности по многоточечному методу BET, определяемая с помощью адсорбции азота при 77 K в соответствии с DIN 66131, составляла 299,8 м2/г. Общая пористость, определяемая с помощью метода ртутной порометрии в соответствии с DIN 66133, составляла 1,1322 мл/г (миллилитр/грамм), соответствующая суммарная площадь пор 92,703 м2/г.
Справочный пример 2: Получение пропиленоксида в больших масштабах с использованием катализатора ZnTiMWW
Эпоксидирование пропена в пропиленоксид с использованием катализатора ZnTiMWW, полученного как описано в Справочном примере 1, проводили, как описано в Справочном примере 3. Питающую смесь водной перекиси водорода смешивали с 390 мкмоль K2PO4 добавки на 1 моль Н2О2 на поток (3). Далее реакцию проводили при таких условиях, чтобы степень конверсии перекиси водорода составляла, по меньшей мере, 91% в любое время, что потребовало постепенного увеличения температуры реакции.
В данном случае, чтобы компенсировать потерю активности катализатора ZnTiMWW, начальную температуру водяного охлаждения, т.е. температуру реакции, 30°С постепенно увеличивали до 55°С во время проведения реакции. Эпоксидирование проводили в течение 2100 часов в общей сложности.
Спустя 2100 часов катализатор ZnTiMWW удалили из камеры (трубок) реактора и 12 образцов отобрали для элементного анализа. Отобрали образец катализатора, расположенный на каждом метре реактора.
Образцы 1-12 анализировали с помощью метода индуктивно-связанной плазмы (ИСП). На Фиг. 1 показано количество калия и фосфора, осажденное на отработанном катализаторе, представленное в виде графика как молярные соотношения по отношению к содержанию кремния, которое, по существу, не изменяется в ходе реакции. Это соответствует приблизительно от 0,5 до 2 вес. % калия и приблизительно от 0,5 до 2 вес. % фосфора по отношению к общему количеству катализатора. Фиг. 2 показывает количество углерода и азота, осажденное на отработанном катализаторе, представленное в виде графика как молярные соотношения по отношению к содержанию кремния.
Справочный пример 3: Схема реакции эпоксидирования (в больших масштабах)
Согласно крупномасштабной схеме, реакцию эпоксидирования проводили следующим образом:
а) Эпоксидирование в главном реакторе эпоксидирования (установка эпоксидирования А)
Главный реактор А представляет собой вертикально установленный кожухотрубный реактор с 5 трубками (длина трубок: 12 м, внутренний диаметр трубки: 38 мм), каждая трубка оснащается аксиально размещенной многоточечной термопарой с 10 равномерно расположенными точками измерения, заключенными в подходящем термокармане с диаметром 18 мм. Каждую трубку заполнили 17,5 кг формованными изделиями, содержащими катализатор ZnTiMWW, полученными согласно Справочному примеру 1, раздел 1.7 (формованные изделия, подвергнутые последующей обработке). Свободное пространство, оставшееся в конечном итоге, было заполнено стеатитовыми сферическими гранулами (диаметром 3 мм). Теплоту реакции отводили посредством циркуляции термостатированного теплоносителя (водно-гликолевая смесь) на стороне межтрубного пространства в потоке, параллельном загрузке. Скорость потока теплоносителя регулировали таким образом, чтобы разность температур между входом и выходом не превышала 1°С. Температуру реакции, упоминаемую здесь ниже, определяли как температуру теплоносителя, поступающего в кожух реактора. На выходе реактора, давление регулировали посредством регулятора давления и поддерживали постоянным на уровне 20 бар.
Реактор загружали снизу жидким однофазным потоком (1). Поток 1 получали посредством смешивания трех потоков (2), (3) и (4). Температуру потока (1) активно не регулировали, но, как правило, она находилась в диапазоне от 20 до 40°С:
- Поток (2) имел скорость потока 85 кг/ч. По меньшей мере, 99,5 вес. % потока (2) состоит из ацетонитрила, пропена и воды. Данный поток (2) поступает из кубовой части перегонной установки рециркуляции ацетонитрила (J).
- Поток (3), имеющий скорость потока 15 кг/ч, представляет собой водный раствор перекиси водорода, имеющей концентрацию перекиси водорода 40 вес. % (''сырой/промытый'' сорт от Solvay с общим содержанием органического углерода (ТОС) в диапазоне от 100 до 400 мг/кг. Водный раствор перекиси водорода подают из резервуара для хранения, что позволяет осуществлять непрерывную подачу, и подается с помощью подходящего дозирующего насоса.
- Поток (4) представляет собой подпиточный поток чистого ацетонитрила (химический сорт, от Ineos, чистота 99,9%, содержащий 70-180 вес. частей на миллион пропионитрила, 5-20 вес. ч.н.м. ацетамида и менее 100 вес. ч.н.м. воды в качестве примесей). Достаточно свежий ацетонитрил добавляют, чтобы компенсировать потери в процессе. В обычных условиях, добавляют в среднем от 100 до 150 г/ч подпиточного ацетонитрила.
Из выходного потока, на выходе установки эпоксидирования А, каждые 20 минут отбирали образцы, чтобы определить концентрацию перекиси водорода с помощью метода на основе сульфата титанила, и рассчитать конверсию перекиси водорода. Конверсия перекиси водорода была определена как 100×(1-mout/min), где min представляет собой молярный расход Н2О2 в питающей смеси реактора и mout представляет собой молярный расход Н2О2 на выходе реактора. На основе соответственно полученных значений конверсии перекиси водорода, регулируют температуру теплоносителя на входе, чтобы поддерживать конверсию перекиси водорода, по существу, постоянной в диапазоне от 90 до 92%. Температуру теплоносителя на входе установили на уровне 30°С в начале данного прогона свежей партии катализатора эпоксидирования и увеличивали, при необходимости, для поддержания конверсии перекиси водорода в указанном диапазоне. Необходимое повышение температуры, как правило, составляло мене 1°С/сутки.
b) Промежуточное удаление пропиленоксида (перегонная установка В)
После сброса давления, поток, выходящий из установки эпоксидирования А (поток (5)), направляли в колонну промежуточного удаления пропиленоксида (перегонная установка В), работающую при давлении около 1,1 бара. Колонна имела высоту 6 м, диаметр 200 мм была оборудована 30 барботажными тарелками, испарителем и конденсатором. Питающую смесь подавали в колонну над барботажной тарелкой 25 (считая сверху). Головной поток, выходящий из колонны при 50°С, в основном содержал пропиленоксид, непрореагировавший пропен и небольшое количество кислорода, образовавшегося как побочный продукт. Этот поток частично конденсировали (Т=15-25°С), и конденсированную жидкость использовали в качестве внутреннего потока флегмы, тогда как газообразную часть (поток (6)) направляли в разделительную колонну легких фракций (перегонная установка D).
Температура в кубовой части колонны промежуточного удаления пропиленоксида составляла около 80°С. Кубовый поток (поток (7)) почти не содержал пропиленоксида (<300 вес. частей на миллион) и представлял собой смесь ацетонитрила (около 78-80 вес. %), воды (около 18-20 вес. %), непрореагировавшего эпоксида-водорода и тяжелокипящих соединений, имеющих нормальную точку кипения выше 100°С, причем основным высококипящим соединением является пропиленгликоль. Данный кубовый поток (7) затем охладили до 35°С и перекачивали насосом в реактор финишной обработки (установка эпоксидирования С, см. раздел с) ниже) с использованием подходящего дозирующего насоса.
с) Эпоксидирование в реакторе финишной обработки (установка эпоксидирования С)
Общий питающий поток для реактора финишной обработки С получали посредством смешивания потока (7), полученного в соответствии с разделом b) выше, с потоком (8) жидкого пропена полимерного сорта, содержащего пропан (чистота ≥ 99,5%, скорость подачи: 0,9 кг/ч, при температуре окружающей среды). Оба потока (7) и (8) смешивали с помощью статического смесителя и подавали в кубовую часть реактора финишной обработки С.
Реактор финишной обработки С представляет собой реактор с неподвижным слоем, работающий адиабатически. В данном контексте, термин ''адиабатический'' относится к режиму работы, в соответствии с которым активное охлаждение не осуществляется, и в соответствии с которым реактор финишной обработки соответствующим образом изолирован, чтобы минимизировать тепловые потери. Реактор финишной обработки С имел длину 4 м и диаметр 100 мм. Реактор был заполнен 9 кг такого же катализатора эпоксидирования, который был использован в главном реакторе эпоксидирования А. Свободное пространство было заполнено стеатитовыми сферическими гранулами (диаметром 3 мм). Рабочее давление в реакторе финишной обработки С составляло 10 бар и поддерживалась постоянным с помощью соответствующего регулятора давления на выходе реактора. На выходе реактора финишной обработки С каждые 20 минут отбирали образцы, чтобы определить концентрацию перекиси водорода с помощью метода на основе сульфата титанила.
Поток, выходящий из реактора финишной обработки С, поток (9), подвергали снижению давления в испарительном барабане, и оба потока - жидкость и газ, из данного барабана подавали в разделительную колонну легкокипящих соединений (перегонная установка D).
Поток (6), полученный из верхней части колонны промежуточного удаления пропиленоксида (перегонная установка В), и поток (9), полученный в качестве отходящего потока из реактора финишной обработки С (установка эпоксидирования С), в совокупности образуют отходящий поток реакции эпоксидирования.
Справочный Пример 4: Схема реакции эпоксидирования (микроустановка)
В трубчатый реактор (длина: 1,4 м, внутренний диаметр: 7 мм), оснащенный рубашкой для термостатирования, загружали 15 г необходимого катализатора в виде заготовок диаметром 1,5 мм, как описано в приведенных ниже примерах. Оставшийся объем реактора был заполнен инертным материалом (стеатитовые сферические гранулы диаметром 2 мм, до высоты примерно 5 см на нижнем конце реактора, а оставшуюся часть в верхнем конце реактора). Реактор подвергали термостатированию посредством пропускания теплоносителя (смесь воды и этиленгликоля) через рубашку. Теплоноситель подавали с нижнего конца рубашки так, чтобы он протекал в параллельном потоке к содержимому реактора. Температуру теплоносителя на входе рубашки определяли как температуру реакции. Скорость потока теплоносителя регулировали таким образом, чтобы разность температур между входом и выходом не превышала 1°С. Давление в реакторе регулировали посредством соответствующего клапана регулирования давления и поддерживали постоянным на уровне 20 бар (абс).
Поток поступающего в реактор материала состоит из трех отдельных питающих потоков, которые дозируются с помощью отдельных дозирующих насосов. Первый поток состоит из ацетонитрила (скорость потока: 68 г/час). Второй поток состоит из сжиженного пропилена полимерного сорта (скорость потока: 11 г/час) и третий поток состоит из водного раствора перекиси водорода с концентрацией 40 вес. % (скорость потока: 17 г/час). Добавку калиевой соли, используемой в опытах, растворили в растворе перекиси водорода. Три питающих потока предварительно смешали перед их подачей при температуре окружающей среды в кубовую часть трубчатого реактора. При используемых условиях питающая смесь является жидкой и присутствует только одна жидкая фаза.
Опыты проводили в непрерывном режиме. В начале прогона (t=0 определено в момент запуска Н2О2 дозирующего насоса) температуру реакции установили на 30°С. Со свежим катализатором такие условия обеспечивают первоначально 100% конверсию перекиси водорода. После определенного периода времени (обычно в течение 100 часов на поток) конверсия перекиси водорода начинает падать. Затем температуру регулировали (обычно достаточно от одного раза до двух раз за сутки), чтобы поддерживать конверсию перекиси водорода от 85 до 95%. В течение большей части времени на потоке конверсия остается в диапазоне от 88 до 92%. Выходящий из реактора поток после клапана регулирования давления собирали, взвешивали и анализировали.
Органические компоненты (за исключением гидропероксипропанолов) и О2 анализировали в двух отдельных газовых хроматографах. Перекись водорода определяли колориметрическим методом на основе сульфата титанила. Содержание гидропероксипропанолов (смесь 1-гидропероксипропанола-2 и 2-гидропероксипропанола-1) определяли посредством измерения общего содержания пероксида (йодометрически), а затем вычитания содержания перекиси водорода. Дополнительно, концентрация гидропероксипропанола может также подвергаться перекрестной проверке посредством определения количества пропиленгликоля до и после восстановления с избытком трифенилфосфана. Разница между этими двумя значениями дает величину гидропероксипропанолов, присутствующих в невосстановленном образце.
Селективность пропиленоксида задана по отношению к Н2О2 и была рассчитана как 100-кратное соотношение числа молей пропиленоксида в отходящем потоке реактора, разделенное на сумму молей пропиленоксида плюс пропиленгликоля плюс удвоенное число молей гидропероксипропанолов и удвоенное число молей О2 (множитель два отражает стехиометрию реакций, ведущих к получению этих продуктов: 2 Н2О2 → 2 Н2О + О2 и пропилен + 2 Н2О2 → гидропероксипропанол + Н2О).
Сравнительный пример 1 Традиционная регенерация катализатора ZnTiMWW
Отработанный катализатор ZnTiMWW фракций 1-3 Справочного примера 2 регенерировали посредством воздействия с помощью термической обработки. В частности, 30 г отработанного катализатора перенесли в печь. Катализатор ZnTiMWW контактировал с азотом при температуре 120°С с целью удаления летучих соединений реакции, после чего катализатор ZnTiMWW подвергали обжигу в печи на воздухе при 450°С в течение 5 часов.
Сравнительный пример 2 Каталитические характеристики катализатора ZnTiMWW, регенерированного традиционным способом
После регенерации согласно Сравнительному примеру 1, каталитические характеристики регенерированного катализатора ZnTiMWW сравнили с каталитическими характеристиками свежего катализатора ZnTiMWW.
Две отдельных реакции эпоксидирования провели в соответствии со схемой, как описано в Справочном примере 3, с использованием 15 г свежего катализатора ZnTiMWW и 15 г регенерированного традиционным способом катализатора ZnTiMWW, соответственно, при прочих равных условиях реакции.
Эпоксидирование с использованием свежего катализатора ZnTiMWW было прекращено спустя 405 часов, тогда как эпоксидирование с использованием регенерированного традиционным способом катализатора ZnTiMWW было прекращено спустя 500 часов. Температуры реакции (т.е. температуру охлаждающей воды) регулировали в каждом опыте таким образом, чтобы в любое время степень конверсии перекиси водорода составляла, по меньшей мере, 91%.
На Фиг. 3, сравнивали селективность на основе перекиси водорода, селективность на основе пропена (С3), степень конверсии на основе перекиси водорода и температуру реакции, необходимую для поддержания степени конверсии перекиси водорода, по меньшей мере, 91%, свежего катализатора ZnTiMWW и регенерированного традиционным способом катализатора ZnTiMWW. Количества полученных продуктов и количество конвертированного исходного вещества определяли с помощью газовой хроматографии.
Для свежего катализатора ZnTiMWW, температура реакции может поддерживаться при 35°С в течение большей части времени реакции, чтобы поддерживать степень конверсии на основе перекиси водорода, по меньшей мере, 91%. Кроме того, селективность на основе перекиси водорода и пропена оставалась выше 98% в течение периода времени, пока эпоксидирование проводили со свежим катализатором ZnTiMWW.
Катализатор ZnTiMWW, регенерированный традиционным способом, как описано в Сравнительном примере 1, потребовал значительного увеличения температуры реакции до 64°С для поддержания степени конверсии на основе перекиси водорода, по меньшей мере, 91%. В то время как селективность на основе пропена также оставалась выше 98% подобно свежему катализатору ZnTiMWW, селективность на основе перекиси водорода упала до 94% спустя 400 часов использования катализатора ZnTiMWW, регенерированного только с помощью нагрева.
Пример 1: Однократная регенерация катализатора ZnTiMWW согласно изобретению
Две отдельных регенерации согласно изобретению проводили при двух различных температурах промывки. Одну регенерацию проводили посредством промывки катализатора при 50°С, а другую регенерацию проводили посредством промывки катализатор при 70°С. Для каждого опыта использовали 40 г отработанного катализатора ZnTiMWW из фракций 1-3 Примера 1.
Промывку катализатора ZnTiMWW проводили в обоих опытах с использованием стеклянной трубки с двойной рубашкой водяного охлаждения в качестве реактора длиной 1 м и внутренним диаметром 20 мм. Температуру воды регулировали с помощью термостата для поддержания постоянной температуры в течение соответствующей процедуры промывки. Воду вводили в рубашку реактора с помощью насоса с расходом 4 мл/мин (что соответствует среднечасовой скорости подачи (WHSV) 7 ч-1) в восходящем потоке.
При 50°С, промывку проводили в течение 420 минут. При 70°С, промывку проводили в течение 410 минут. В обоих опытах, промывку проводили до тех пор, пока электрическая проводимость промывочной воды на выходе из реактора в его головной части не составляла около 200 микросименс/см. Электрическую проводимость определяли с помощью кондуктометра (WTW, LF320) со стандартной измерительной ячейкой электрической проводимости (Tetra Con 325).
После промывки катализатор ZnTiMWW сушили в обоих опытах в стеклянном реакторе с двойной рубашкой в потоке газообразного азота 100 л/час при 40°С в течение 16 часов, после чего катализатор ZnTiMWW удаляли из реактора и обжигали в печи при 450°С на воздухе в течение 5 часов.
После регенераций катализатора ZnTiMWW при температурах 50°С и 70°С, отдельные композиции были определены с помощью элементного анализа. Элементный анализ проводили, как указано в Справочном примере 2, и полученные результаты приведены в Таблице 1 ниже.
В строке №1 указаны количества соединений K, Р, Si, Ti и Zn в 40 г общего количества катализатора ZnTiMWW до проведения регенерации. В строках №2-4 указаны общие количества в граммах соединений K, Р, Si, Ti и Zn в собранной промывочной воде в разные периоды времени (строка №2:. с 0 до 180 минуты; строка №3: с 181 до 300 минуты; строка №4: с 301 до 420 минуты). Небольшие потери Si, Ti и Zn, наблюдаемые во время промывки, как полагают, связаны с образованием мелкодисперсных фракций. В строке №5 указаны общие количества в граммах этих соединений в остаточной воде, удаленной из стеклянного трубчатого реактора, после завершения промывки. В строке №. 6 указаны общие количества в граммах указанных соединений в катализаторе ZnTiMWW после завершения регенерации, т.е. промывки, сушки и обжига.
Следовательно, после приблизительно 7 часов промывки с последующей сушкой и обжигом, общие количества K и Р были снижены преимущественно приблизительно на 60% посредством промывки при 50°С и еще более преимущественно приблизительно на 82% и 67%, соответственно, посредством промывки при 70°С.
При обеих температурах удаление отложений является удовлетворительным. Тем не менее, очевидно, что посредством промывки при 70°С калий и фосфор, осажденные на катализаторе ZnTiMWW, могут быть удалены быстрее и более тщательно.
Пример 2: Многократная регенерация катализатора ZnTiMWW, осуществляемая согласно изобретению
34,3 г отработанного катализатора ZnTiMWW фракций 1-3 Справочного примера 2 подвергли 5 последовательным регенерациям, как описано в Примере 1. Промывку проводили каждый раз при 70°С.После каждого цикла определяли точный состав катализатора ZnTiMWW и его другие свойства, в частности, его поверхность, объем пор, прочность на раздавливание. Кроме того, проводили тест на выход пропиленоксида, который является индикатором каталитической активности катализатора ZnTiMWW.
Общие количества K, Р, Ti, Zn и Si в катализаторе ZnTiMWW определяли, как описано в Примере 1, с помощью элементного анализа.
Площадь поверхности по методу Ленгмюра была определена с помощью адсорбции азота при 77 K в соответствии с DIN 66131. Объем пор определяли с помощью метода ртутной порометрии в соответствии с DIN 66133.
Предел прочности на раздавливание, как указано в контексте настоящего изобретения, следует понимать как параметр, определенный с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS1S, поставщик Zwick GmbH & Co., D-89079 Ulm, Germany (Германия). Что касается основных характеристик и принципов работы данной установки, ссылка делается на соответствующие руководство по эксплуатации ''Register 1: Betriebsanleitung / Sicherheitshandbuch  die Material-
die Material-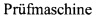 Z2.5/TS1S'', version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. Титульный лист руководства по эксплуатации показан на Фиг. 9. С помощью указанной установки, таблетку катализатора ZnTiMWW подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Установка работала с предварительным усилием 0,5 Н и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено испытуемое формованное изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
Z2.5/TS1S'', version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. Титульный лист руководства по эксплуатации показан на Фиг. 9. С помощью указанной установки, таблетку катализатора ZnTiMWW подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Установка работала с предварительным усилием 0,5 Н и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено испытуемое формованное изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
В тесте на выход ПО (тест на выход пропиленоксида), катализатор ZnTiMWW, регенерированный согласно способу настоящего изобретения, тестировали в миниавтоклаве посредством реакции пропена с водным раствором перекиси водорода (30 вес. %) с получением пропиленоксида. В частности, 0,63 г катализатора ZnTiMWW вместе с 79,2 г ацетонитрила и 12,4 г пропена при комнатной температуре, и 22,1 г перекиси водорода (30 вес. % в воде) поместили в стальной автоклав. После проведения реакции в течение 4 часов при 40°С, смесь охладили и подвергали снижению давления, а жидкую фазу анализировали с помощью газовой хроматографии на предмет содержания в ней пропиленоксида. Содержание пропиленоксида в жидкой фазы (в вес. %) представляет собой результат теста на выход ПО.
Результаты приведены в Таблице 2 ниже.
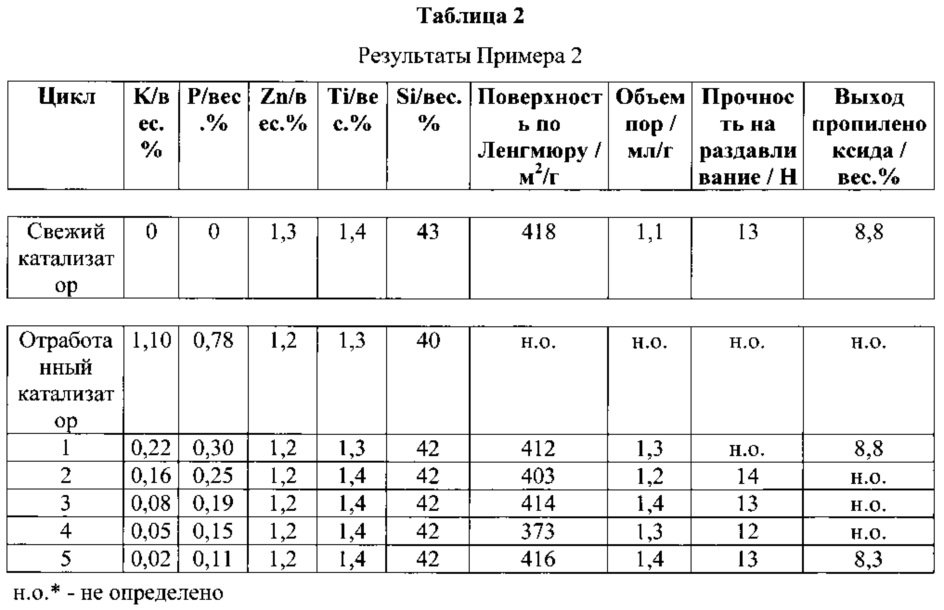
Результаты, представленные в Таблице 2, показывают, что количество отложений калия и фосфора, может быть дополнительно уменьшено при осуществлении способа регенерации согласно настоящему изобретению последовательно несколько раз.
Из Таблицы 2 также становится очевидным, что содержание Zn, Ti и Si в катализаторе ZnTiMWW не изменилось в способе, который включает в себя пять циклов регенерации, по сравнению со свежим катализатором ZnTiMWW. Небольшие определяемые вариации рассматриваются как находящиеся в пределах погрешности измерения.
Кроме того, Таблица 2 также показывает, что поверхность по методу Ленгмюра, объем пор и прочность на раздавливание катализатора ZnTiMWW не изменились при многократном осуществлении способа регенерации по сравнению со свежим катализатором ZnTiMWW. Аналогично, наблюдаемые вариации в отношении определенных значений рассматриваются как находящиеся в пределах погрешности измерения.
Тест на выход ПО также показал, что выход при использовании многократно регенерированного катализатора ZnTiMWW существенно не изменился после пяти циклов регенерации.
Кроме того, ИК-спектр свежего катализатора ZnTiMWW, показанный на Фиг. 4, можно сравнить с ИК-спектром, зарегистрированным для пятикратно регенерированного катализатора ZnTiMWW, представленным на Фиг. 5. Спектры являются во многом идентичными, за исключением полосы, видимой приблизительно при 3500 см-1 в спектре регенерированного катализатора ZnTiMWW. Этот показатель свидетельствует об уменьшении числа внутренних силанольных групп вследствие регенерации. Тем не менее, такое изменение не оказывает влияния на активность цеолитного катализатора, как подтверждено результатами, представленными в Таблице 2.
Подводя итог, следует отметить, что эти результаты показывают, последовательно, что настоящий способ регенерации катализатора, содержащего титановый цеолит в качестве активного материала, является достаточно эффективным, таким образом, что первоначальная каталитическая активность восстанавливается, даже без последующих нескольких циклов регенерации катализатор не изменяется структурно существенным образом.
Пример 3: Каталитические характеристики многократно регенерированного катализатора согласно изобретению
После многократной регенерации в соответствии с Примером 2, каталитические характеристики регенерированного катализатора ZnTiMWW сравнили с каталитическими характеристиками свежего катализатора ZnTiMWW.
Две отдельных реакции эпоксидирования провели в микроустановке с использованием 15 г свежего катализатора ZnTiMWW и 15 г многократно регенерированного катализатора ZnTiMWW, соответственно, при прочих идентичных условиях реакции.
Микроустановка состояла из реакторных труб с водяным охлаждением длиной 1,4 м и внутренним диаметром 7 мм. Загрузка питающего материала, которую осуществляли в восходящем потоке, в каждом случае состояла из 68 г/час ACN, 16 г/час Н2О2 (40 вес. % в воде), 10,8 г/час пропена и имела концентрацию 130 мкмоль KH2PO4 на 1 моль Н2О2. Эпоксидирование с использованием свежего катализатора ZnTiMWW прекратили спустя 500 часов, тогда как эпоксидирование с использованием многократно регенерированного катализатора ZnTiMWW прекратили спустя 310 часов. Температуры реакции (т.е. температуры охлаждающей воды) регулировали в каждом опыте так, чтобы в любое время степень конверсии перекиси водорода составляла, по меньшей мере, 91%.
На Фиг. 6, сравнивают селективность на основе перекиси водорода, селективность на основе пропена, степень конверсии на основе перекиси водорода и температуры реакции (т.е. температуры охлаждающей воды), необходимые для поддержания степени конверсии, по меньшей мере, 91%, полученные для свежего и многократно регенерированного катализаторов, соответственно. Селективность и степень конверсии определяли, как указано в Сравнительном примере 2, с помощью газовой хроматографии.
Совершенно очевидно, что температуры реакции, необходимые для поддержания степени конверсии перекиси водорода выше 91%, являются преимущественно, по существу, идентичными при сравнении свежего катализатора ZnTiMWW и пятикратно регенерированного катализатора ZnTiMWW. В обоих случаях степень конверсии при приблизительно 45°С оставалась значительно выше 91%, за исключением резко отклоняющегося значения непосредственно ниже 90°С спустя приблизительно 255 часов, как наблюдалось в отношении многократно регенерированного катализатора ZnTiMWW.
Кроме того, селективность как на основе перекиси водорода, так и на основе пропена, осталась, по существу, неизменной, отличаясь преимущественно высоким значением приблизительно 99% в ходе реакции эпоксидирования, при сравнении катализатора ZnTiMWW, регенерированного пять раз, согласно изобретению, и свежего катализатора ZnTiMWW.
Пример 4: Регенерация на месте отработанного катализатора ZnTiMWW согласно изобретению
Регенерация в соответствии с настоящим изобретением была выполнена в отношении катализатора ZnTiMWW внутри реактора, используемого для эпоксидирования в Справочном примере 2.
Отработанный катализатор ZnTiMWW промывали в 12 м реакторных трубах водой со скоростью потока 130 л/час при 60°С в течение 17,7 часов, с последующей промывкой водой со скоростью потока 130 л/час при 75°С в течение 4,5 часов. Воду подавали в нисходящем потоке в верхней части труб реактора.
Затем катализатор ZnTiMWW высушили в реакторе в потоке газообразного азота, который подавали в кубовую часть труб реактора. Азот подавали со скоростью потока 12 м3/час при температуре 60°С в течение 96 часов, с последующей подачей азота со скоростью потока 14 м3/час при 65°С в течение 1 часа, и далее с последующей подачей азота со скоростью потока 13 м3/час при 70°С в течение 354,5 часов. В конце стадии сушки, влажность азота на выходе из реактора, как определено с помощью датчика влажности (GE, HygroPro), составляла 243 объемных частей на миллион, что соответствует влажности газообразного азота перед подачей в реактор.
После завершения стадии сушки, катализатор подвергали обжигу в реакторе в течение 6,5 часов при 450°С, при этом температуру обжига постепенно повышали со скоростью 0,5°С/мин.
Свойства катализатора после регенерации при повторном использовании в способе эпоксидирования были аналогичными результатам, полученным в Примерах 1 и 3.
Справочный Пример 5: Определение характеристик катализатора
Справочный пример 5.1: Определение значений Dv10, Dv50 и Dv90
1,0 г микропорошка суспендировали в 100 г деионизированной воды и перемешивали в течение 1 минуты. Образец подвергли измерению в аппарате, используя следующие параметры: Mastersizer S, исполнение с удлиненной станиной 2.15, серийный №33544-325; поставщик: Malvern Instruments GmbH, Herrenberg, Germany (Германия); фокусное расстояние - 300RF мм; длина луча - 10,00 мм; модуль - MS17; затенение - 16,9%; дисперсионная модель - 3$$D; аналитическая модель - полидисперсная, коррекция - отсутствует.
Справочный Пример 5.2: Определение концентрации силанольных групп в формованных изделиях согласно настоящему изобретению
Для определения концентрации силанольных групп, опыты согласно методу ядерного магнитного резонанса при вращении образца под магическим углом 29Si MAS ЯМР проводили при комнатной температуре на спектрометре VARIAN Infinityplus-400 с использованием 5,0 мм ZrO2 роторов. Спектры 29Si MAS ЯМР регистрировали при 79,5 МГц с использованием 1,9 мкс π/4 (микросекунды пи/4) импульса с 10 сек. задержкой повторного цикла и 4000 сканов. Все 29Si-спектры регистрировали на образцах, с вращением при 6 кГц, а химические сдвиги были определены относительно 4,4-диметил-4-силапентан сульфоната натрия (DSS). Для определения концентрации силанольных групп, данный спектр 29Si MAS ЯМР восстанавливали из свертки соответствующих Гауссовых-Лоренцевых форм линий. Концентрацию силанольных групп по отношению к общему числу атомов Si получили посредством интегрирования восстановленных из свертки спектров 29Si MAS ЯМР.
Справочный пример 5.3: Определение прочности формованных изделий на раздавливание
Предел прочности на раздавливание, как указано в контексте настоящего изобретения, следует понимать как параметр, определенный с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS1S, поставщик Zwick GmbH & Co., D-89079 Ulm, Germany (Германия). Что касается основных характеристик и принципов работы данной установки, ссылка делается на соответствующие руководство по эксплуатации ''Register 1: Betriebsanleitung / Sicherheitshandbuch  die Material- Z2.5/TS1S'', version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. С помощью указанной установки, данную заготовку подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Установка работала с предварительным усилием 0,5 Н и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено испытуемое формованное изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
die Material- Z2.5/TS1S'', version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. С помощью указанной установки, данную заготовку подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Установка работала с предварительным усилием 0,5 Н и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено испытуемое формованное изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
Справочный пример 5.4: Спектры 29Si-ЯMP в твердом состоянии в отношении структур Q3 и Q4
Эффект от обработки водой согласно изобретению формованного изделия в отношении структур Q3 и Q4 в материале определяли посредством сравнения изменений в спектрах 29Si-ЯМР в твердом состоянии в сопоставимых условиях. Все опыты 29Si ЯМР твердого тела проводили с использованием спектрометра Bruker Avance при ларморовой частоте 300 МГц 1Н (Bruker BioSpin, Germany). Образцы были упакованы в 7 мм ZrO2 роторы и измерялись при вращении под магическим углом при 5 кГц при комнатной температуре. Прямые спектры поляризации 29Si были получены с использованием (пи/2)-импульсного возбуждения с 5 мкс длительностью импульса, 29Si несущей частотой, соответствующей -65 ч.н.м. в спектре, и задержкой повторного цикла сканирования 120 сек. Сигнал получали за 25 мс при 45 кГц протонной развязки высокой мощности и накапливали в течении 10-17 часов. Спектры обрабатывали с использованием Bruker TopSpin с 30 Гц экспоненциальным уширением линии, ручным фазированием и ручной коррекцией базовой линии по всей ширине спектра. Спектры определяли относительно полимера Q8M8 в качестве внешнего вторичного стандарта, задав резонанс М группы триметилсилила как 12,5 частей на миллион. Спектры затем согласовывали с набором гауссовых форм линий, в соответствии с числом различаемых резонансов. Относительно текущих анализируемых спектров, были использованы 6 линий в общей сложности, что составляет пять различных максимумов пиков (приблизительно при -118, -115, -113, -110 и -104 ч.н.м.) плюс четко видимое плечо при -98 ч.н.м. Корректировку проводили с использованием DMFit (Massiot et al., Magnetic Resonance in Chemistry, 40 (2002) pp 70-76). Пики вручную установили на видимых максимумах пиков или плече. Затем положение пика и ширину линии оставили незафиксированными, т.е. откорректированные пики не были зафиксированы в определенном положении. Результат корректировки был численно устойчивым, то есть, искажения в начальной настройке корректировки, как описано выше, приводили к аналогичным результатам. Площади откорректированных пиков в дальнейшем использовали как нормализованные согласно корректировки посредством DMFit. После обработки водой согласно изобретению, наблюдалось уменьшение интенсивности сигнала ''по левую руку'' спектра, область, которая включает силанольные структуры Q3 (здесь особенно: около и выше -104 ч.н.м., т.е. ''слева'' от -104 ч.н.м.). Кроме того, наблюдалось увеличение интенсивности сигнала ''по правую руку'' спектра (здесь ниже -110 ч.н.м., т.е. ''справа'' от -110 ч.н.м.), область которого включает в себя исключительно структуры Q4. Для количественной оценки изменений спектра, было рассчитано соотношение, которое отражает изменения площадей пиков ''по левую руку'' и ''по правую руку'', следующим образом. Шесть пиков, как описано выше, были маркированы как 1, 2, 3, 4, 5 и 6, а соотношение Q было рассчитан по формуле 100*{[a1+а2]/[а4+а5+а6]}/а3. В данной формуле ai, i=i..6 представляет собой площадь откорректированного пика, которому было присвоен данный номер.
Справочный пример 5.5: Адсорбция / десорбция воды - Водопоглощение
Измерения изотерм адсорбции/десорбции воды проводились с помощью прибора VTI SA от ТА Instruments согласно программы ступенчатых изотерм. Опыт состоял из прогона или серии прогонов, выполненных на образце материала, который поместили в чашу микровесов внутри прибора. Перед началом измерений, из образца удалили остаточную влагу посредством нагревания образца до 100°С (скорость нагрева 5°С/минута) и выдерживали образец в течение 6 часов в потоке N2. После программы сушки, температуру в камере снизили до 25°С и выдерживали изотермический в течение измерений. Микровесы были откалиброваны и вес высушенного образца измерили (максимальное отклонение веса 0,01 вес. %). Поглощение воды образцом измеряли как увеличение веса по сравнению с весом сухого образца. Во-первых, кривую адсорбции измеряли посредством увеличения относительной влажности (RH) (выраженной в вес. % воды в атмосфере внутри камеры), воздействию которой подвергали образцы, и посредством измерения поглощения воды образцом при равновесии. Относительную влажность увеличивали с шагом 10 вес. % от 5 до 85% и на каждой стадии система контролировала значение RH и осуществляла мониторинг веса образца до достижения условий равновесия, регистрируя поглощение веса. Общий объем адсорбированной образцом воды был поглощен после того, как образец подвергли воздействию 85 вес. % RH. Во время измерения десорбции, RH снижали с 85 вес. % до 5 вес. % с шагом 10% и изменение веса образца (водопоглощения) контролировали и регистрировали.
Справочный пример 5.6.: Измерения инфракрасной спектроскопии на основе преобразования Фурье
Измерения инфракрасной спектроскопии на основе преобразования Фурье (Фурье-ИК-спектроскопия) были выполнены с помощью спектрометра Nicolet 6700. Формованное изделие было измельчено в порошок, который затем прессовали в самоподдерживающийся осадок без использования каких-либо добавок. Осадок ввели в ячейку высокого вакуума (HV), размещенную в приборе Фурье-ИК-спектроскопии. Перед измерением образец предварительно обрабатывали в условиях высокого вакуума (10-5 мбар) в течение 3 часов при 300°С. Спектры были получены после охлаждения ячейки до 50°С. Спектры регистрировали в диапазоне от 4000 до 800 см-1 с разрешением 2 см-1. Полученные спектры представлены на графике, имеющем на оси х волновые числа (см-1) и на оси у поглощающую способность (в условных единицах, у.е.). Для количественного определения высот пиков и соотношения между этими пиками проводилась коррекция базовой линии. Изменения в области от 3000 до 3900 см-1 были проанализированы и для сравнения нескольких образцов в качестве эталона была взята полоса при 1880±5 см-1.
Справочный пример 5.7: Определение степени кристалличности с помощью рентгенодифракционного (XRD) анализа
Степень кристалличности цеолитных материалов в соответствии с настоящим изобретением определяли с помощью рентгенодифракционного (XRD) анализа. Данные собирали с помощью стандартного дифрактометра Брэгга-Брентано (Bragg-Brentano) с рентгеновской трубкой с медным анодом в качестве источника рентгеновских лучей и энергодисперсионным точечным детектором. Угловой диапазон от 2° до 70° (2 тета) сканировали с размером шага 0,02°, при этом переменная щель расходимости была задана для постоянной длины выборки 20 мм с подсветкой.
Затем данные анализировали с использованием программного обеспечения TOPAS V4, с помощью которого были смоделированы острые дифракционные пики с использованием метода Паули (Pawley), содержащие элементарную ячейку со следующими исходными параметрами: а = 14,4 ангстрем (1 ангстрем = 10-10 м) и с = 25,2 ангстрем в пространственной группе Р6/mmm. Они были доработаны, чтобы соответствовать данным. Независимые пики были вставлены в следующих положениях. 8.4°, 22,4°, 28,2° и 43°. Они были использованы для описания аморфного содержания. Содержание кристаллической структуры описывает интенсивность кристаллического сигнала по отношению к общей интенсивности рассеянного излучения. Модель также включала в себя линейный фон, коррекции по фактору Лоренца (Lorentz) и поляризации, параметры кристаллической решетки, пространственную группу и размер кристаллитов.
СПИСОК ЛИТЕРАТУРЫ
- WO-A 98/55229
- WO-A 2011/064191
- ЕР-А 0934116
- ЕР-А 079007
- ЕР-А 1371414
- ЕР-А 1221442
- WO-A 2005/000827
- WO-A 2007/013739
- ЕР-А 1122249
- US 2003/0187284 А1
- US 2012/142950 A1
- WO 2011/115234 A1
- US 2004/058798 A1
- US 5916835 A
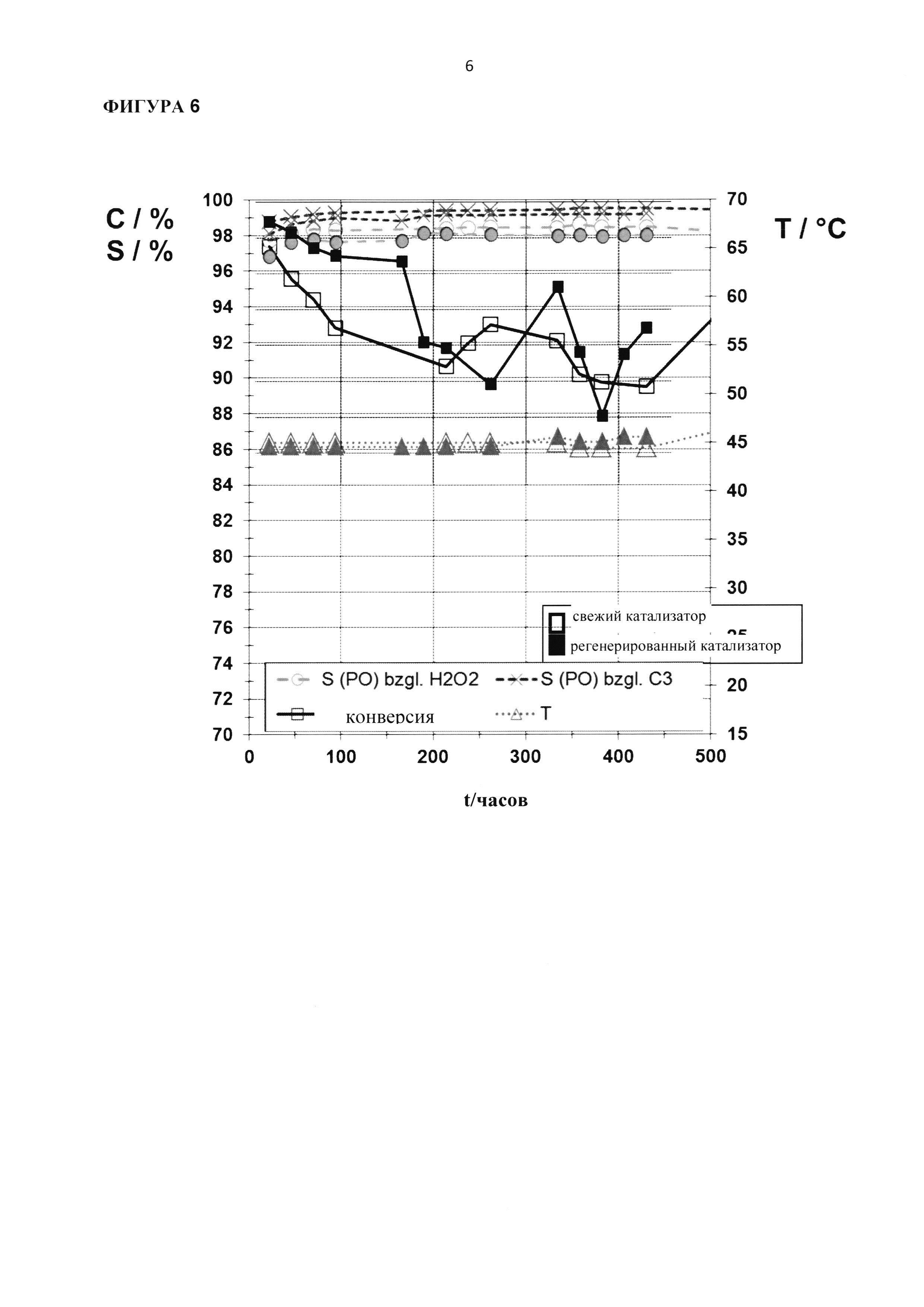
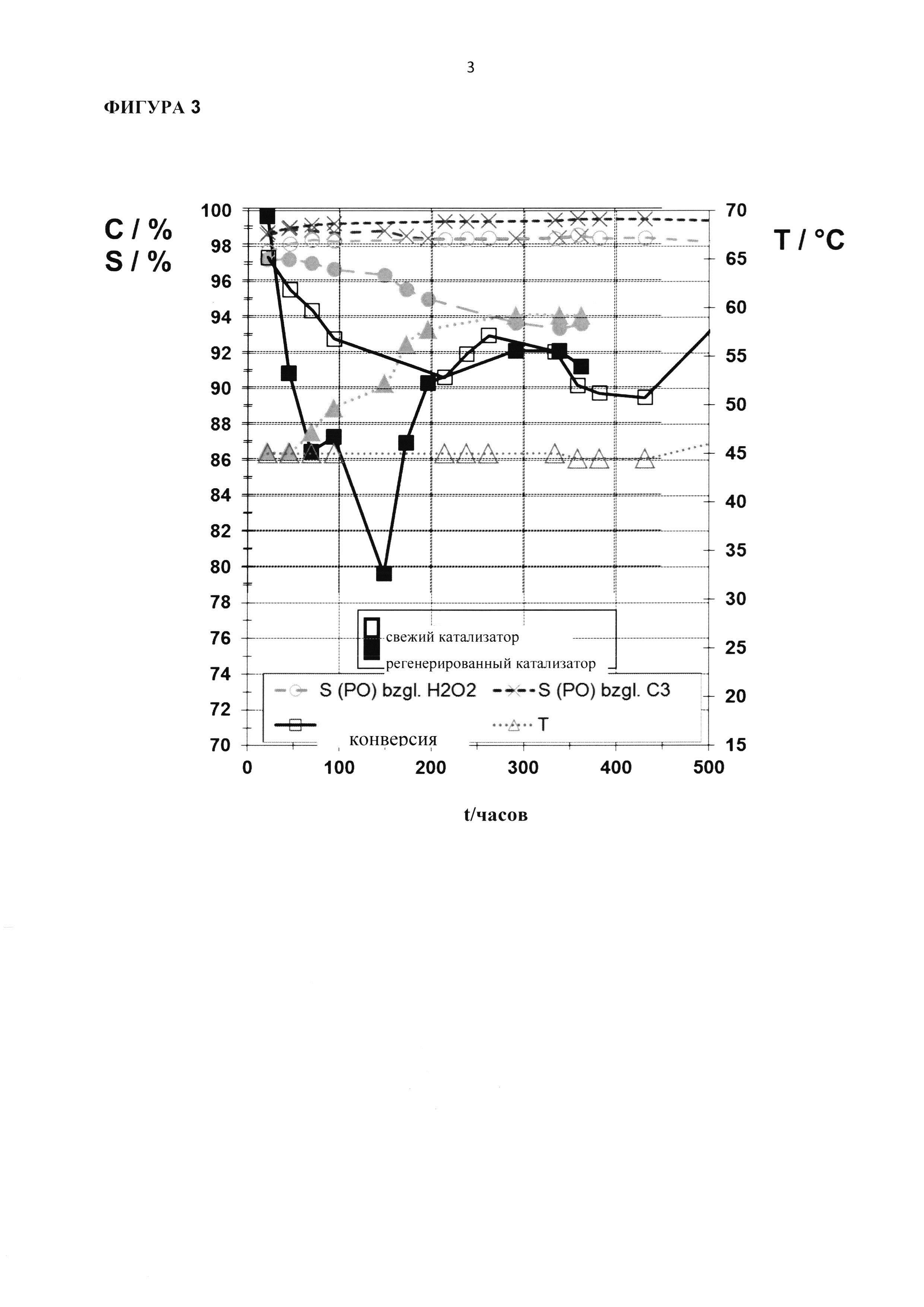
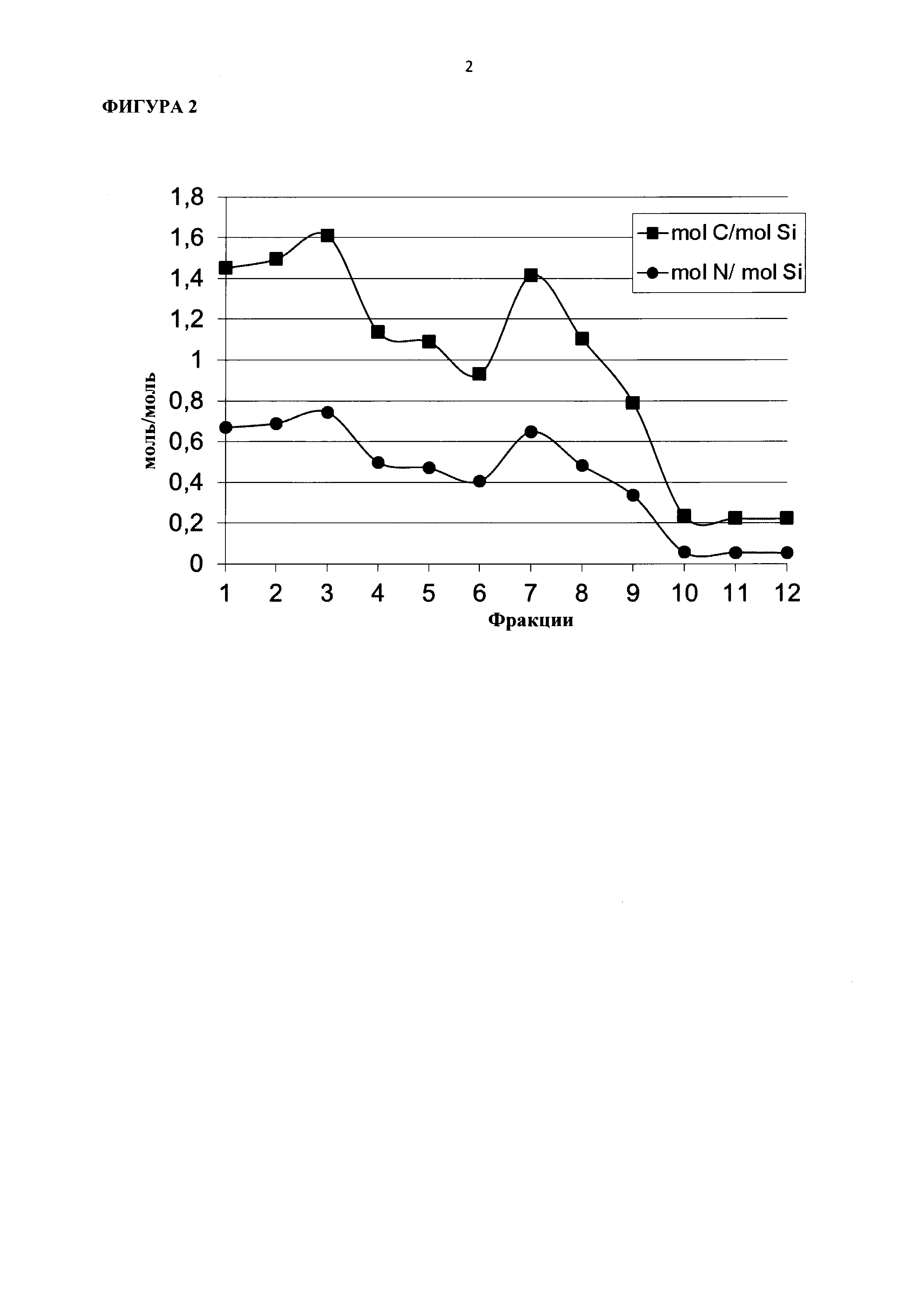
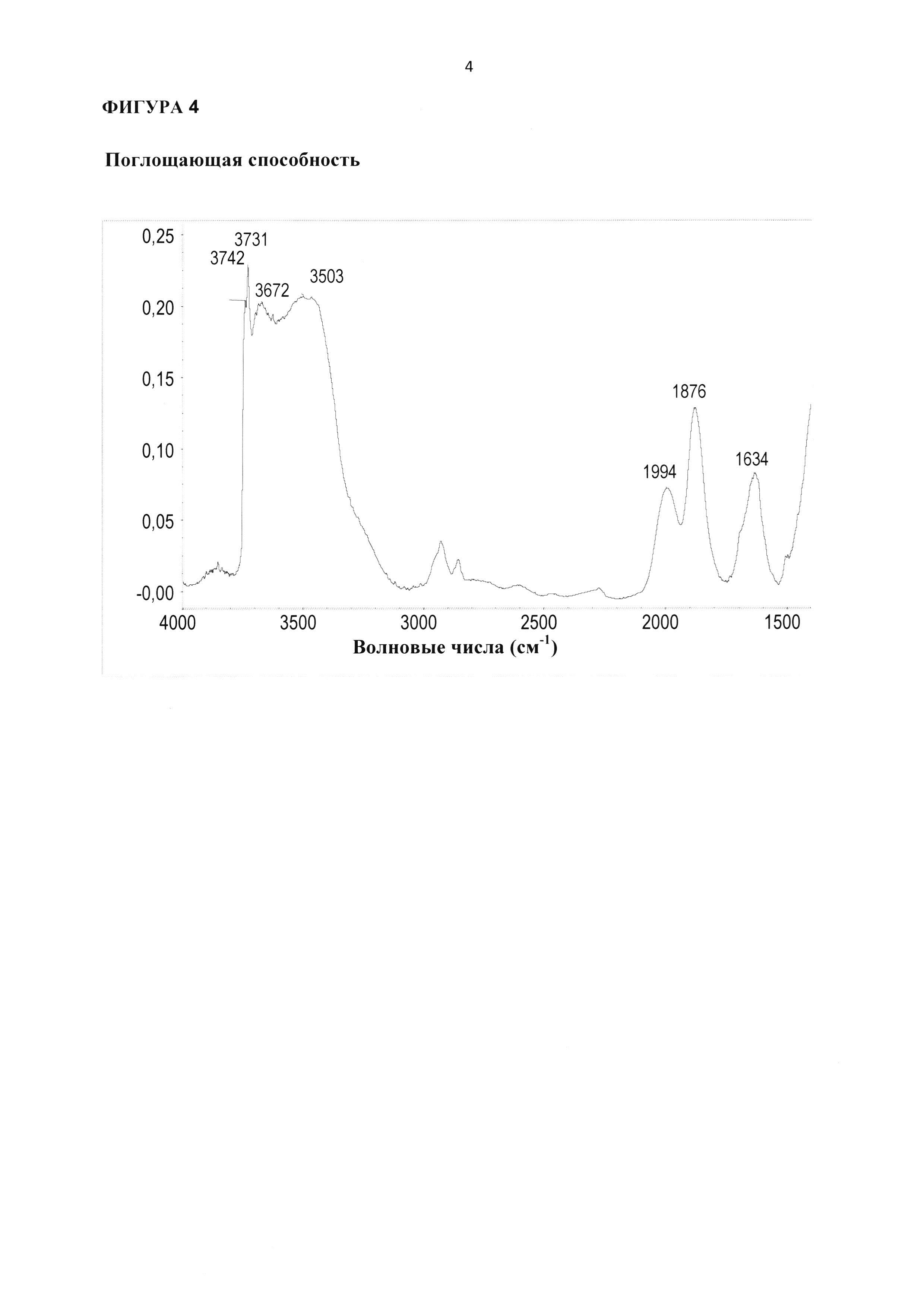
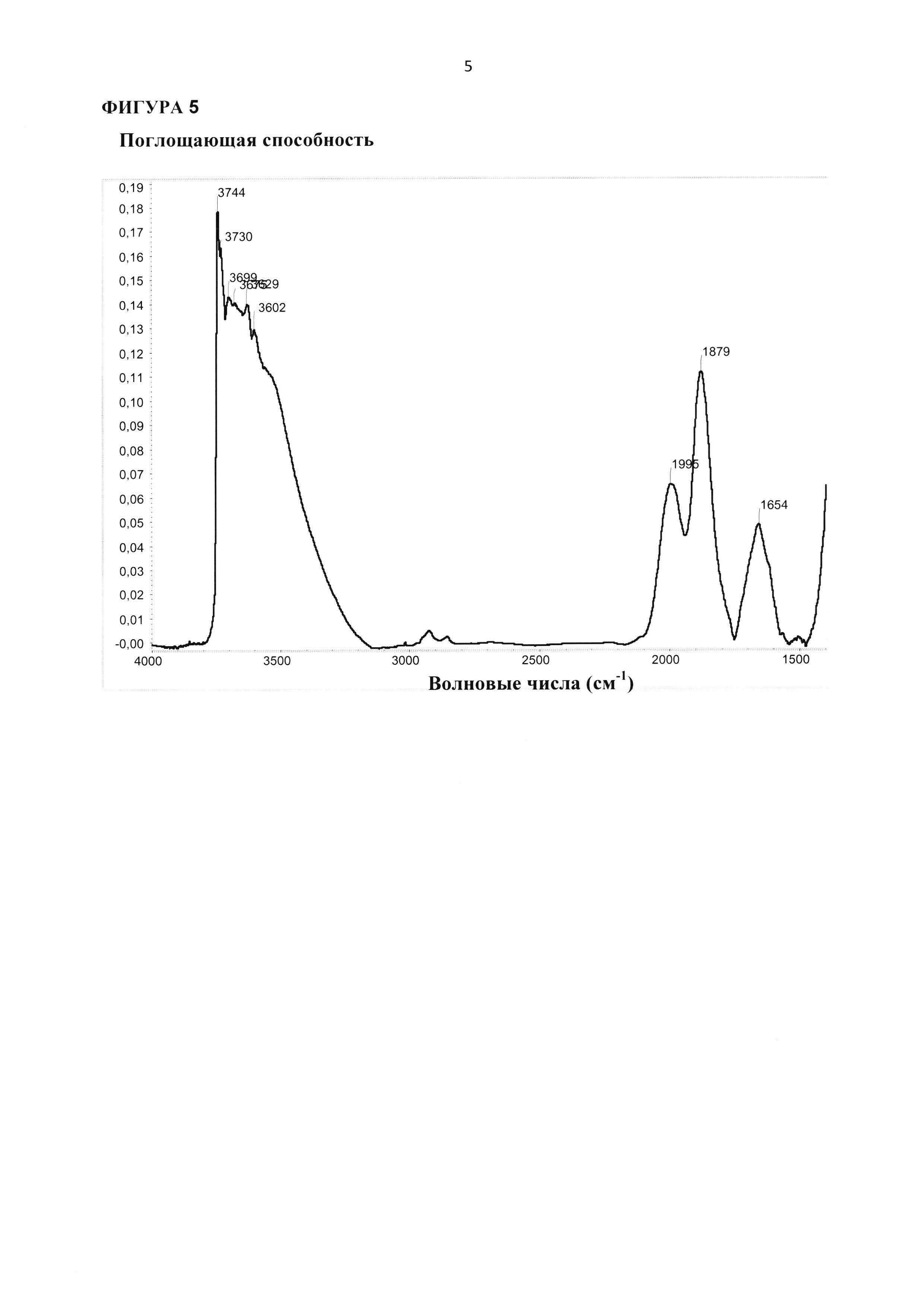
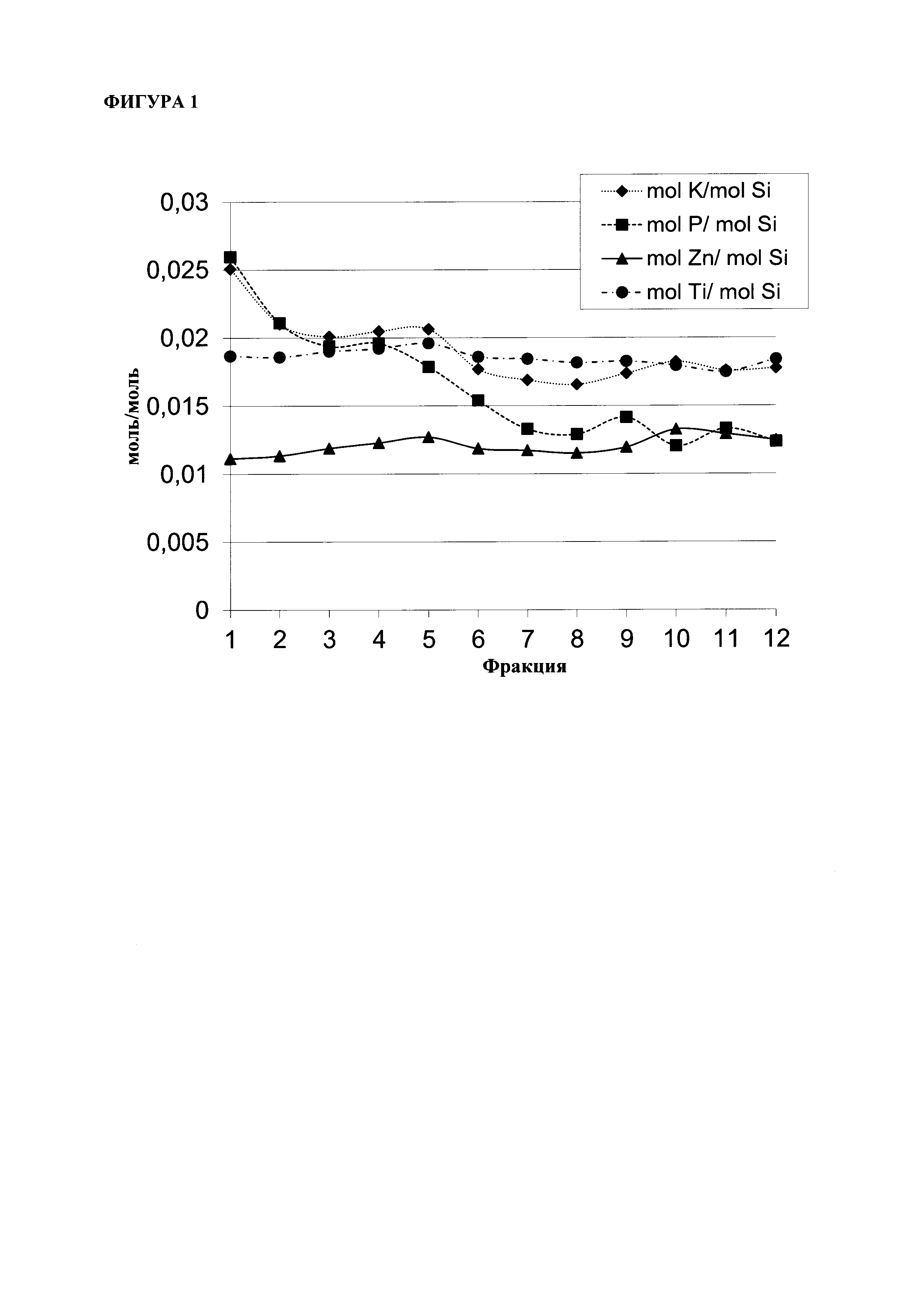
Способ хранения жидкой в условиях хранения мономерной фазы
Способ транспортировки жидкой мономерной фазы, извлеченной из резервуара для хранения, в резервуаре автозаправщика или танкера
Способ получения механически стабильных водопоглощающих полимерных частиц
Способ получения акриловой кислоты
Способ получения триэтилентетрамина (тэта) через этилендиаминдиацетонитрил (эддн)
Способ получения смеси этиленаминов
Средство для нанесения покрытий на вспенивающиеся частицы стирольного полимеризата
Способ для обнаружения и подсчета жизнеспособных микроорганизмов вида legionella pneumophila и набор для его осуществления
Способ получения смесей этиленаминов
Эластичный пеноматериал из частиц на основе смесей полиолефина/полимера стирола
Способ производства изопропанола жидкофазным гидрированием
Способ отделения неразветвленных углеводородов от их разветвленных изомеров
Способ получения олефиноксида
Способ производства пропиленоксида
Способ отделения ацетонитрила от воды
Композиция для улучшения свойств холодной текучести топливных масел
Способ получения катализатора на основе титанового цеолита
Металлорганические скелетные материалы на основе 2,5-фурандикарбоновой или 2,5-тиофендикарбоновой кислоты
Полимер (мет)акрилата для повышения индекса вязкости
Способ получения пропиленоксида

