Результат интеллектуальной деятельности: Способ получения аминофуразанов
Вид РИД
Изобретение
Предлагаемое изобретение относится к области органической химии, а именно к способу получения 4-замещенных 3-аминофуразанов, которые являются предшественниками ряда ценных биологически активных веществ и, в частности, служат необходимыми исходными соединениями в синтезе антибактериальных препаратов [R.М. Christoff, G.L. Murray, X.P. Kostoulias, A.Y. Peleg, В.M. Abbott, Synthesis of novel 1,2,5-oxadiazoles and evaluation of action against Acinetobacter baumannii // Bioorg. Med. Chem., 2017, Vol. 25, Issue 24, P. 6267-6272] и цитотоксических агентов [D.-S. Shin, D. Masciocchi, A. Gelain, S. Villa, D. Barlocco, F. Meneghetti, A. Pedretti, Y.-M. Han, D.C. Han, B.-M. Kwon, L. Legnani, L. Toma, Synthesis, modeling, and crystallographic study of 3,4-disubstituted 1,2,5-oxadiazoles and evaluation of their ability to decrease STAT3 activity // Med. Chem. Commun., 2010, Vol. 1, P. 156-164].
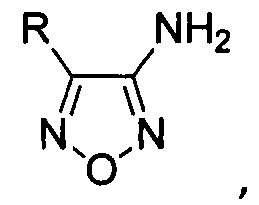
Известен способ получения 4-замещенных 3-аминофуразанов общей формулы:
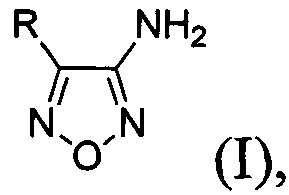
где R - низший алкил либо алкил-, алкокси-, галоген- или трифторметилзамещенный или незамещенный фенил на основе каскадных трансформаций ароматических 1,3-кетоэфиров [А.Б. Шереметев, Однореакторный синтез 3-амино-4-арил- и 3-амино-4-гетарилфуразанов // Изв. АН, Сер. хим., 2005, №4, С. 1007-1012; 1030-1032], принятый за прототип. Выходы целевых 3-амино-4-арилфуразанов на исходные 1,3-кетоэфиры составляют 45-75%. Процесс проводят в одном реакторе и способ включает в себя обработку соответствующих исходных 1,3-кетоэфиров раствором NaOH в воде при 0°С, смесь перемешивают 12 часов, затем добавляют нитрит натрия и 20% водный раствор хлорной кислоты при температуре ниже 10°С. Затем температуру реакционной смеси доводят до комнатной и оставляют получившуюся смесь на 14 часов. После этого осуществляют одновременное параллельное добавление к реакционной смеси водных растворов NH2OH⋅HCl и NaOH, поддерживая температуру реакционной массы ниже 30°С. Затем реакционную смесь нагревают до 95°С, перемешивают 2 часа, добавляют мочевину и кипятят еще 3 часа. После этого, выпавший осадок продукта отфильтровывают, промывают водой, сушат и перекристаллизовывают из смеси растворителей хлороформ : петролейный эфир 1:1.
Однако данный способ имеет ряд существенных недостатков, затрудняющих его применение для практических целей. Из недостатков способа следует, в первую очередь, указать на недостаточно высокий выход продукта реакции, который зависит от заместителя в ароматическом фрагменте (интервал выходов 45-75%); сложность контроля за ходом реакции, поскольку процесс включает в себя однореакторную последовательность трансформаций 1,3-кетоэфиров, состоящую из десяти стадий, полноту протекания каждой из которых невозможно оценить, что неизбежно влечет за собой увеличение количества технологических отходов; сложность обработки реакционной смеси, включающую последовательное добавление большого количества реагентов через различные промежутки времени при строго контролируемой температуре, причем в зависимости от реагента требуется либо охлаждение реакционной смеси до 0°С, либо нагревание до 100°С, что существенно затрудняет масштабирование процесса; высокая стоимость исходных ароматических 1,3-кетоэфиров, что приводит к существенному удорожанию процесса; использование больших количеств сильных минеральных кислот и щелочей, что значительно усложняет технологию производства и снижает экологичность процесса.
Технической задачей настоящего изобретения является повышение выхода целевого продукта, а также упрощение технологии его получения.
Поставленная техническая задача достигается предлагаемым способом получения 4-замещенных 3-аминофуразанов общей формулы:
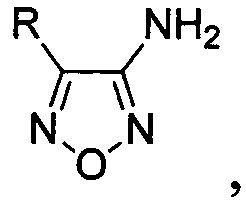
где R - низший алкил либо алкил-, алкокси-, галоген- или трифторметилзамещенный или незамещенный фенил, заключающимся в том, что соответствующие 3-нитрофуроксаны подвергают взаимодействию с двухлористым оловом в присутствии соляной кислоты при мольном соотношении 3-нитрофуроксаны : двухлористое олово 1:8-13.
Процесс протекает по следующей схеме:
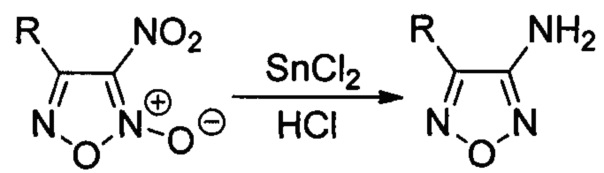
Процесс проводят при комнатной температуре в течение короткого времени (40 минут), затем реакционную массу разбавляют водой, полученные целевые аминофуразаны выпадают в виде мелкокристаллического порошка, которые отделяют фильтрованием с последующей перекристаллизацией из метанола.
В качестве исходных соединений используют соответствующие 4-замещенные 3-нитрофуроксаны общей формулы:
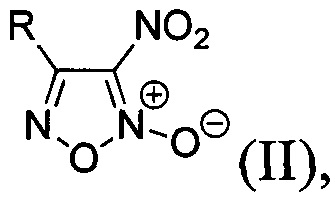
где R - замещенный или незамещенный фенил или низший алкил, простой и масштабируемый способ получения которых был ранее разработан на основе однореакторных трансформаций коммерчески доступных альдоксимов [L.L. Fershtat, М.A. Epishina, I.V. Ovchinnikov, М.I. Struchkova, A.A. Romanova, I.V. Ananyev, N.N. Makhova, Side-chain prototropic tautomerism of 4-hydroxyfuroxans in methylation reactions // Tetrahedron Lett., 2016, Vol. 57, Issue 50, P. 5685-5689]. Процесс проводят в одном реакторе и способ включает в себя хлорирование соответствующих исходных альдоксимов в диметилформамиде под действием N-хлорсукцинимида (NCS). Реакционную смесь перемешивают 1 час, затем охлаждают до 0°С и добавляют натриевую соль динитрометана. Реакционную смесь перемешивают 30 минут и убирают в холодильник на 36 часов. Затем при температуре 0-5°С к реакционной смеси добавляют безводный ацетат натрия, перемешивают 30 минут и приливают порциями уксусную кислоту. После этого добавляют нитрит натрия, перемешивают 30 мин при температуре 0-5°С, доводят температуру реакционной смеси до комнатной и перемешивают еще 3 часа. Затем реакционную смесь выливают в воду, выпавший осадок продукта отфильтровывают, промывают водой и сушат на воздухе. Таким образом, получают 4-замещенные 3-нитрофуроксаны с выходами 78-96% общей формулы II.
Процесс протекает по следующей схеме:
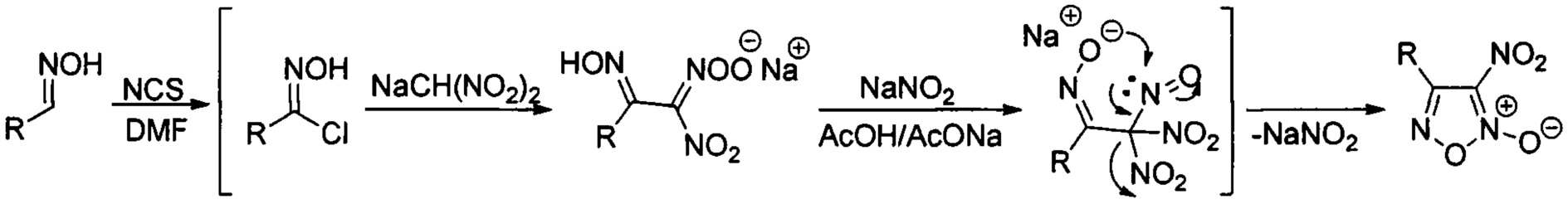
Существенным отличием предложенного способа является одновременное восстановление фуроксанового цикла до фуразанового и нитрогруппы до аминогруппы, что позволяет за одну синтетическую стадию получать целевые аминофуразаны из доступных 3-нитрофуроксанов. Были оптимизированы условия данного процесса, в результате чего наилучшие выходы продуктов достигались при использовании 8-13-кратного избытка двухлористого олова и применении соляной кислоты, которая в данном случае играет роль среды и катализатора. Стоит отметить, что до настоящего времени полностью отсутствовали примеры одновременного восстановления нитрогруппы и N-оксидного фрагмента 1,2,5-оксадиазольного цикла под действием двухлористого олова. По-видимому, именно подбор условий реакции, включающий подбор оптимального соотношения исходного 3-нитрофуроксана к двухлористому олову и использование соляной кислоты в качестве среды и катализатора, привел к восстановлению и нитрогруппы, и N-оксидного фрагмента, благодаря чему реакция пошла в нужном направлении. Изменение условий, например, проведение аналогичной реакции в другой среде, например, этаноле приводит не к целевым аминофуразанам, а к продуктам раскрытия цикла - аминоглиоксимам. Предложенный способ позволяет получать аминофуразаны как с ароматическими, так и с алифатическими заместителями, причем во всех случаях выходы целевых продуктов довольно высокие (77-93% на реакцию восстановления или 76-81% в расчете на коммерчески доступные альдоксимы). Кроме того, методика проведения реакции и выделения продуктов довольно проста. Реакция проводится при комнатной температуре в течение короткого времени (40 минут), затем при разбавлении реакционной массы водой целевые аминофуразаны выпадают в виде мелкокристаллического порошка и отделяются фильтрованием с последующей перекристаллизацией из метанола.
Существенным отличием предлагаемого способа является использование 4-замещенных 3-нитрофуроксанов вместо ранее используемых дорогостоящих ароматических 1,3-кетоэфиров. При этом исходные 3-нитрофуроксаны представляют собой устойчивые соединения, легко синтезируемые в одну стадию из коммерчески доступных альдоксимов. Также существенным отличием от прототипа является легкая реализация процесса в мягких условиях при комнатной температуре и процедура выделения и очистки продукта реакции, которая представляет собой простое фильтрование и перекристаллизацию. Кроме того, в предлагаемом методе увеличены выходы целевых соединений до 77-93% против 45-75% в прототипе. Совокупность вышеуказанных особенностей предлагаемого изобретения дает возможность осуществить способ получения аминофуразанов в более крупном масштабе по сравнению с прототипом.
Техническим результатом предлагаемого изобретения является увеличение выхода целевого продукта до 77-93% против 45-75% в прототипе, а также упрощение процесса за счет проведения его при комнатной температуре в одну стадию и процедуре выделения и очистки продукта реакции, которая представляет собой простое фильтрование и перекристаллизацию, а также использованием доступных 3-нитрофуроксанов вместо дорогостоящих ароматических 1,3-кетоэфиров.
Изобретение иллюстрируется следующими примерами, не ограничивающими его объем.
Пример 1.
А. Получение 3-нитро-4-фенилфуроксана
К раствору бензальдоксима (14.5 г, 0.12 моль) в диметилформамиде (100 мл) добавляют N-хлорсукцинимид (16.0 г, 0.12 моль) при комнатной температуре. Реакционную смесь подогревают до 40°С, перемешивают 10 минут (до инициирования реакции), затем охлаждают до комнатной температуры. Реакционную смесь перемешивают 1 час, затем охлаждают до 0°С и добавляют натриевую соль динитрометана (30.7 г, 0.24 моль). Реакционную смесь перемешивают 30 минут и убирают в холодильник на 36 часов. Затем при температуре 0-5°С к реакционной смеси добавляют безводный ацетат натрия (29.5 г, 0.36 моль), перемешивают 30 минут и приливают порциями уксусную кислоту (150 мл). Затем добавляют нитрит натрия (41.4 г, 0.6 моль), перемешивают 30 мин при температуре 0-5°С, убирают охлаждающую баню, доводят температуру реакционной смеси до комнатной и перемешивают еще 3 часа. Затем реакционную смесь выливают в воду (600 мл), выпавший осадок продукта отфильтровывают, промывают водой и сушат на воздухе. Выход 3-нитро-4-фенилфуроксана составил 22.4 г (90%). Светло-желтый порошок. Т. пл. 109-110°С. 1Н ЯМР (300 МГц, CDCl3): 7.59-7.73 (м, 5Н, Ph). 13С ЯМР (75.5 МГц, CDCl3): 124.0, 129.0, 129.1, 132.3, 151.4. 14N ЯМР (21.7 МГц, CDCl3): -39.7 (уш. c, NO2).
Б. Получение 3-амино-4-фенилфуразана
К суспензии 3-нитро-4-фенилфуроксана (20.7 г, 0.1 моль) в соляной кислоте (400 мл) добавляют хлорид олова(II) (190 г, 1 моль) при комнатной температуре, перемешивают реакционную массу в течение 40 минут, затем выливают в воду (900 мл), выпавший осадок отфильтровывают, промывают водой и перекристаллизовывают из метанола. Выход 3-амино-4-фенилфуразана составил 14.5 г (90%). Белые кристаллы. Т. пл. 100-101°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.17 (уш. с, 2Н, NH2), 7.55 (уш. с, 3H, Ph), 7.76 (уш. с, 2Н, Ph). 13С ЯМР (75.5 МГц, ДМСО-d6): 125.5, 127.6, 129.1, 130.2, 146.8, 155.2.
Общий выход 3-амино-4-фенилфуразана на исходный бензальдоксим составил 81%.
Пример 2.
Аналогично примеру 1, но с использованием 8-кратного избытка двухлористого олова. К суспензии 4-фенил-3-нитрофуроксана (0.1 моль) в соляной кислоте (400 мл) добавляют хлорид олова(II) (152 г, 0.8 моль) при комнатной температуре, перемешивают реакционную массу в течение 90 минут, затем выливают в воду (900 мл), выпавший осадок отфильтровывают, промывают водой и перекристаллизовывают из метанола. Выход 3-амино-4-фенилфуразана составил 14.0 г (87%).
Общий выход 3-амино-4-фенилфуразана на исходный бензальдоксим составил 78%.
Пример 3.
Аналогично примеру 1, но с использованием 13-кратного избытка двухлористого олова. К суспензии 4-фенил-3-нитрофуроксана (0.1 моль) в соляной кислоте (400 мл) добавляют хлорид олова(II) (247 г, 1.3 моль) при комнатной температуре, перемешивают реакционную массу в течение 90 минут, затем выливают в воду (1000 мл), выпавший осадок отфильтровывают, промывают водой и перекристаллизовывают из метанола. Выход 3-амино-4-фенилфуразана 13.5 г (84%).
Общий выход 3-амино-4-фенилфуразана на исходный бензальдоксим составил 76%.
Пример 4.
А. Получение 4-(4-метоксифенил)-3-нитрофуроксана
Аналогично примеру 1, но с использованием оксима 4-метоксибензальдегида (18.1 г, 0.12 моль) в качестве исходного соединения. Выход 4-(4-метоксифенил)-3-нитрофуроксана составил 26.2 г (92%). Ярко-желтый порошок. Т. пл. 75-76°С. 1Н ЯМР (300 МГц, CDCl3): 3.91 (с, 3H, OMe), 7.07 (д, 2Н, 3J=8.9 Гц, Н Ar), 7.66 (д, 2Н, 3J=8.9 Гц, Н Ar). 13С ЯМР (75.5 МГц, CDCl3): 55.6, 114.6, 115.8, 126.8, 130.6, 151.0, 162.7. 14N ЯМР (21.7 МГц, CDCl3): -38.4 (уш. с, NO2).
Б. Получение 3-амино-4-(4-метоксифенил)фуразана
Аналогично примеру 1, но с использованием 4-(4-метоксифенил)-3-нитрофуроксана (23.7 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-метоксифенил)фуразана составил 16.4 г (86%). Белые кристаллы. Т. пл. 105-106°С. 1Н ЯМР (300 МГц, ДМСО-d6): 3.83 (с, 3H, OMe), 6.14 (уш. с, 2Н, NH2), 7.10 (д, 2Н, Н Ar, 3J=8.9 Гц), 7.72 (д, 2Н, Н Ar, 3J=8.9 Гц). 13С ЯМР (75.5 МГц, ДМСО-d6): 55.8, 115.0, 118.2, 129.7, 147.0, 155.7, 161.2.
Общий выход 3-амино-4-(4-метоксифенил)фуразана на исходный оксим 4-метоксибензальдегида составил 79%.
Пример 5.
А. Получение 3-нитро-4-(2-фторфенил)фуроксана
Аналогично примеру 1, но с использованием оксима 2-фторбензальдегида (16.7 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(2-фторфенил)фуроксана составил 23.0 г (85%). Светло-желтый порошок. Т. пл. 112-113°С. 1Н ЯМР (300 МГц, CDCl3): 7.28 (т, 1Н, 3J=8.9 Hz, Н Ar), 7.40 (т, 1H, 3J=8.9 Hz, Н Ar), 7.64-7.72 (м, 2Н, Н Ar). 13С ЯМР (75.5 МГц, CDCl3): 112.8 (d, J=13.9 Hz), 116.4 (d, J=20.4 Hz), 125.2 (d, J=3.5 Hz), 130.3 (d, J=1.3 Hz), 134.5 (d, J=8.6 Hz), 147.3, 158.7, 162.0. 14N ЯМР (21.7 MHz, CDCl3): -38.2 (c, NO2). 19F ЯМР (282.4 MHz, CDCl3): -112.8.
Б. Получение 3-амино-4-(2-фторфенил)фуразана
Аналогично примеру 1, но с использованием 3-нитро-4-(2-фторфенил)фуроксана (22.5 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(2-фторфенил)фуразана составил 15.9 г (89%). Белые кристаллы. Т. пл. 114-115°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.20 (уш. с, 2Н, NH2), 7.36-7.50 (м, 2Н, Н Ar), 7.61-7.67 (м, 2Н, Н Ar). 13С ЯМР (75.5 МГц, ДМСО-d6): 113.3 (д, 3J=14.8 Гц), 116.4 (д, 3J=20.7 Гц), 125.0 (д, 3J=3.6 Гц), 131.1 (д, 3J=2.2 Гц), 132.7 (д, 3J=7.8 Гц), 143.2, 155.8, 159.8 (д, 3J=250.0 Гц). 19F ЯМР (282.4 МГц, ДМСО-d6): -113.4.
Общий выход 3-амино-4-(2-фторфенил)фуразана на исходный оксим 2-фторбензальдегида составил 76%.
Пример 6.
А. Получение 3-нитро-4-(3-фторфенил)фуроксана
Аналогично примеру 1, но с использованием оксима 3-фторбензальдегида (16.7 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(3-фторфенил)фуроксана составил 25.9 г (96%). Белый порошок. Т. пл. 76-77°С. 1Н ЯМР (300 МГц, CDCl3): 7.35-7.40 (м, 1Н, Н Ar), 7.45-7.62 (м, 3H, Н Ar). 13С ЯМР (75.5 МГц, CDCl3): 116.3 (д, 3J=24.4 Гц), 119.5 (д, 3J=21.0 Гц), 124.9 (д, 3J=3.3 Гц), 125.6, 125.7, 130.9 (д, 3J=8.2 Гц), 150.2, 162.6 (д, 3J=249.0 Гц). 14N ЯМР (21.7 МГц, CDCl3): -35.5 (уш. с, NO2). 19F ЯМР (282.4 МГц, CDCl3): -111.1 (с). Найдено, %: С 42.59; Н 1.92; N 18.55. C8H4FN3O4. Вычислено, %: С 42.68; Н 1.79; N 18.66.
Б. Получение 3-амино-4-(3-фторфенил)фуразана
Аналогично примеру 1, но с использованием 3-нитро-4-(3-фторфенил)фуроксана (22.5 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(3-фторфенил)фуразана составил 14.5 г (81%). Белые кристаллы. Т. пл. 106-107°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.28 (уш. с, 2Н, NH2), 7.37-7.46 (м, 1H, Н Ar), 7.57-7.65 (м, 3H, Н Ar). 13С ЯМР (75.5 МГц, ДМСО-d6): 115.1 (д, 3J=23.4 Гц), 117.7 (д, 3J=21.0 Гц), 124.4 (д, 3J=2.9 Гц), 128.0 (д, 3J=8.6 Гц), 131.7 (д, 3J=8.4 Гц), 146.4, 155.7, 162.7 (д, 3J=245.0 Гц). 19F ЯМР (282.4 МГц, ДМСО-d6): -112.5. Найдено, %: С 53.48; Н 3.50; N 23.33. C8H6FN3O. Вычислено, %: С 53.63; Н 3.38; N 23.46.
Общий выход 3-амино-4-(3-фторфенил)фуразана на исходный оксим 3-фторбензальдегида составил 78%.
Пример 7.
А. Получение 3-нитро-4-(4-фторфенил)фуроксана
Аналогично примеру 1, но с использованием оксима 4-фторбензальдегида (16.7 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(4-фторфенил)фуроксана составил 25.7 г (95%). Белый порошок. Т. пл. 88-89°С. 1Н ЯМР (300 МГц, CDCl3): 7.26-7.31 (м, 2Н, Н Ar), 7.73-7.77 (м, 2Н, Н Ar). 13С ЯМР (75.5 МГц, CDCl3): 116.5 (д, 3J=22.4 Гц), 120.0 (2 С), 131.4 (д, 3J=9.1 Гц), 150.5, 165.0 (д, 3J=254.4 Гц). 14N ЯМР (21.7 МГц, CDCl3): -35.1 (уш. с, NO2). 19F ЯМР (282.4 МГц, CDCl3): -107.0 (с). Найдено, %: С 42.79; Н 1.90; N 18.52. C8H4FN3O4. Вычислено, %: С 42.68; Н 1.79; N 18.66.
Б. Получение 3-амино-4-(4-фторфенил)фуразана
Аналогично примеру 1, но с использованием 3-нитро-4-(4-фторфенил)фуроксана (22.5 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-фторфенил)фуразана составил 14.9 г (83%). Белые кристаллы. Т. пл. 135-136°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.22 (уш. с, 2Н, NH2), 7.36-7.44 (м, 2Н, Н Ar), 7.80-7.86 (м, 2Н, Н Ar). 13С ЯМР (75.5 МГц, ДМСО-d6): 116.7 (д, 3J=22.0 Гц), 122.5 (д, 3J=3.3 Гц), 130.7 (д, 3J=8.8 Гц), 146.7, 155.7, 163.7 (д, 3J=248.0 Гц). 19F ЯМР (282.4 МГц, ДМСО-d6): -111.1.
Общий выход 3-амино-4-(4-фторфенил)фуразана на исходный оксим 4-фторбензальдегида составил 79%.
Пример 8.
А. Получение 3-нитро-4-(4-(трифторметил)фенил)фуроксана
Аналогично примеру 1, но с использованием оксима 4-(трифторметил)бензальдегида (22.7 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(4-(трифторметил)фенил)фуроксана составил 32.3 г (98%). Светло-желтый порошок. Т. пл. 72-73°С. 1Н ЯМР (300 МГц, CDCl3): 7.79-7.86 (м, 4Н, Н Ar). 13С ЯМР (75.5 МГц, CDCl3): 120.5, 125.7 (кв, 3J=3.8 Hz), 127.4, 128.1 (д, 3J=35.1 Hz), 129.5, 130.1, 133.7 (д, 3J=35.1 Hz), 150.1. 14N ЯМР (21.7 MHz, CDCl3): -35.5 (с, NO2). 19F NMR (282.4 MHz, CDCl3): -64.1.
Б. Получение 3-амино-4-(4-(трифторметил)фенил)фуразана
Аналогично примеру 1, но с использованием 3-нитро-4-(4-(трифторметил)фенил)фуроксана (27.5 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-(трифторметил)фенил)фуразана составил 17.6 г (77%). Белые кристаллы. Т. пл. 109-110°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.32 (уш. с, 2Н, NH2), 7.91 (д, 2Н, Н Ar, 3J=8.3 Гц), 8.00 (д, 2Н, Н Ar, 3J=8.3 Гц). 13С ЯМР (75.5 МГц, ДМСО-d6): 126.2 (кв, 3J=Гц), 129.1, 130.7 (д, 3J=32.2 Гц), 146.5, 152.1, 155.8, 166.7. Найдено, %: С 47.29; Н 2.53; N 18.48. C9H6F3N3O. Вычислено, %: С 47.17; Н 2.64; N 18.34.
Общий выход 3-амино-4-(4-(трифторметил)фенил)фуразана на исходный оксим 4-(трифторметил)бензальдегида составил 76%.
Пример 9.
А. Получение 3-нитро-4-(4-хлорфенил)фуроксана
Аналогично примеру 1, но с использованием оксима 4-хлорбензальдегида (18.7 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(4-хлорфенил)фуроксана составил 27.2 г (94%). Желтый порошок. Т. пл. 65-66°С. 1Н ЯМР (300 MHz, CDCl3): 7.54 (д, 2Н, 3J=8.5 Hz, Н Ar), 7.65 (д, 2Н, 3J=8.5 Hz, Н Ar). 13С ЯМР (75.5 MHz, CDCl3): 122.1, 129.3, 129.5, 130.2, 138.6, 150.3. 14N ЯМР (21.7 MHz, CDCl3): -39.0 (с, NO2).
Б. Получение 3-амино-4-(4-хлорфенил)фуразана
Аналогично примеру 1, но с использованием 3-нитро-4-(4-хлорфенил)фуроксана (24.2 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-хлорфенил)фуразана составил 16.0 г (82%). Светло-желтые кристаллы. Т. пл. 137-138°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.24 (уш. с, 2Н, NH2), 7.61 (д, 2Н, Н Ar, 3J=7.4 Гц), 7.79 (д, 2Н, Н Ar, 3J=7.4 Гц). 13С ЯМР (75.5 МГц, ДМСО-d6): 124.9, 129.7, 130.0, 135.6, 146.6, 155.7.
Общий выход 3-амино-4-(4-хлорфенил)фуразана на исходный оксим 4-хлорбензальдегида составил 77%.
Пример 10.
А. Получение 3-нитро-4-(4-бромфенил)фуроксана
Аналогично примеру 1, но с использованием оксима 4-бромбензальдегида (24.0 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(4-бромфенил)фуроксана составил 32.9 г (96%). Желтый порошок. Т. пл. 80-81°С. 1H ЯМР (300 MHz, CDCl3): 7.58 (д, 2Н, 3J=8.2 Hz, Н Ar), 7.71 (д, 2Н, 3J=8.2 Hz, Н Ar). 13С ЯМР (75.5 MHz, CDCl3): 122.7, 126.5, 127.2, 130.5, 131.7, 150.5. 14N ЯМР (21.7 MHz, CDCl3): -38.7 (с, NO2).
Б. Получение 3-амино-4-(4-бромфенил)фуразана
Аналогично примеру 1, но с использованием 4-(4-бромфенил)-3-нитрофуроксана (28.6 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-бромфенил)фуразана составил 19.0 г (79%). Светло-желтые кристаллы. Т. пл. 145-146°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.24 (уш. с, 2Н, NH2), 7.73 (д, 2Н, Н Ar, 3J=7.0 Гц), 7.76 (д, 2Н, Н Ar, 3J=7.0 Гц). 13С ЯМР (75.5 МГц, ДМСО-d6): 125.2, 130.3, 132.3, 132.6, 146.7, 155.7.
Общий выход 3-амино-4-(4-бромфенил)фуразана на исходный оксим 4-бромбензальдегида составил 76%.
Пример 11.
А. Получение 4-(4-метилфенил)-3-нитрофуроксана
Аналогично примеру 1, но с использованием оксима 4-метилбензальдегида (16.2 г, 0.12 моль) в качестве исходного соединения. Выход 3-нитро-4-(4-метилфенил)фуроксана составил 23.9 г (90%). Светло-желтый порошок. Т. пл. 95-96°С. 1Н ЯМР (300 MHz, CDCl3): 2.48 (с, 3H, Me), 7.37 (д, 2Н, 3J=8.0 Hz, Н Ar), 7.59 (д, 2Н, 3J=8.0 Hz, Н Ar). 13С ЯМР (75.5 MHz, CDCl3): 21.6, 121.0, 128.8, 129.8, 143.0 (2 С), 151.3. 14N ЯМР (21.7 MHz, CDCl3): -38.6 (с, NO2).
Б. Получение 3-амино-4-(4-метилфенил)фуразана
Аналогично примеру 1, но с использованием 4-(4-метилфенил)-3-нитрофуроксана (22.1 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-(4-метилфенил)фуразана составил 15.6 г (89%). Светло-желтые кристаллы. Т. пл. 143-144°С. 1Н ЯМР (300 МГц, ДМСО-d6): 6.20 (уш. с, 2Н, NH2), 7.25 (д, 2Н, Н Ar, 3J=7.2 Гц), 7.52 (д, 2Н, Н Ar, 3J=7.2 Гц). 13С ЯМР (75.5 МГц, ДМСО-d6): 20.3, 122.1, 129.6, 130.1, 132.6, 146.9, 154.8.
Общий выход 3-амино-4-(4-метилфенил)фуразана на исходный оксим 4-метилбензальдегида составил 80%.
Пример 12.
А. Получение 4-метил-3-нитрофуроксана
Аналогично примеру 1, но с использованием ацетальдоксима (7.1 г, 0.12 моль) в качестве исходного соединения. Выход 4-метил-3-нитрофуроксана составил 14.8 г (85%). Белый порошок. Т. пл. 41-42°С. 1Н ЯМР (300 МГц, CDCl3): 2.73 (с, 3H, Me). 13С ЯМР (75.5 МГц, CDCl3): 13.0, 126.3, 149.7. 14N ЯМР (21.7 МГц, CDCl3): -37.3. Найдено, %: С 24.72; Н 1.99; N 29.12. C3H3N3O4. Вычислено, %: С 24.84; Н 2.08; N 28.96.
Б. Получение 3-амино-4-метилфуразана
Аналогично примеру 1, но с использованием 4-метил-3-нитрофуроксана (14.5 г, 0.1 моль) в качестве исходного соединения. Выход 3-амино-4-метилфуразана составил 9.2 г (93%). Белые кристаллы. Т. пл. 73-74°С. 1Н ЯМР (300 МГц, ДМСО-d6): 2.27 (с, 3H, Me), 6.11 (уш. с, 2Н, NH2). 13С ЯМР (75.5 МГц, ДМСО-d6): 8.0, 146.0, 157.1.
Общий выход 3-амино-4-метилфуразана на исходный ацетальдоксим составил 79%.
Исходные соединения 4-(3-фторфенил)-3-нитрофуроксан, 4-(4-фторфенил)-3-нитрофуроксан и 4-метил-3-нитрофуроксан, а также целевые соединения 4-(3-фторфенил)-3-аминофуразан и 4-(4-(трифторметил)фенил)-3-аминофуразан являются новыми и в литературе не описаны.
3-(тринитрометил-onn-азокси)-4-нитраминофуразаны и способы их получения
Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием
Катализатор для окислительного разложения хлорорганических соединений в газах и способ его получения
Способ приготовления катализатора для получения синтез-газа, катализатор, приготовленный по этому способу, и способ получения синтез-газа с его использованием
Способ приготовления катализатора для получения синтез-газа
Способ получения замещенных 2,3,5,6-тетраоксабицикло[2.2.1]гептанов
Способ приготовления катализатора для получения 3-ацетилгептан-2,6-диона и способ получения 3-ацетилгептан-2,6-диона с использованием полученного катализатора
Катализатор для селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов и способ селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов с его использованием
Способ приготовления катализатора для получения бензола из метана, катализатор, приготовленный по этому способу, и способ получения бензола из метана с использованием полученного катализатора
Катализатор для получения этилбензола из бензола и этана и способ получения этилбензола с его использованием

