Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАЗОЛКАРБОНОВОЙ КИСЛОТЫ
Вид РИД
Изобретение
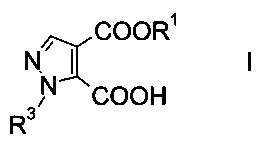
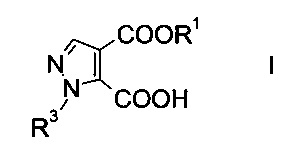
Настоящее изобретение относится к новому способу получения производного пиразолкарбоновой кислоты формулы
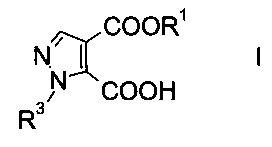
где R1 представляет собой C1-7-алкил, и R3 представляет собой C1-7-алкил, который возможно замещен галогеном или C1-4-алкокси.
Производное пиразолкарбоновой кислоты формулы I можно использовать в качестве структурного элемента при получении фармацевтически активных компонентов, например соединений, выполняющих функцию ингибиторов фосфодиэстеразы (ФДЭ), особенно ингибиторов ФДЭ10. Ингибиторы ФДЭ10 обладают способностью лечить психотические расстройства, подобные шизофрении (межд. публикация патента WO 2011/117264).
Подход к синтезу производного пиразолкарбоновой кислоты формулы I описан на схеме 3 межд. публикации патента WO 2011/117264, использующего способ, раскрытый у Hanzlowsky et al., J. Heterocyclic Chem. 2003, 40(3), 487-489.
Однако при описанных условиях циклоконденсации, катализируемой кислотой, кроме требуемого изомера также образуется значительное количество нежелательного N-1 замещенного изомера. Во многих случаях, особенно в большом масштабе, этот нежелательный изомер является основным продуктом в реакционной смеси при соотношении вплоть до 70:30 в пользу нежелательного изомера, что приводит к фактическим выходам приблизительно 30% нежелательного изомера вместе с приблизительно 25% требуемого изомера.
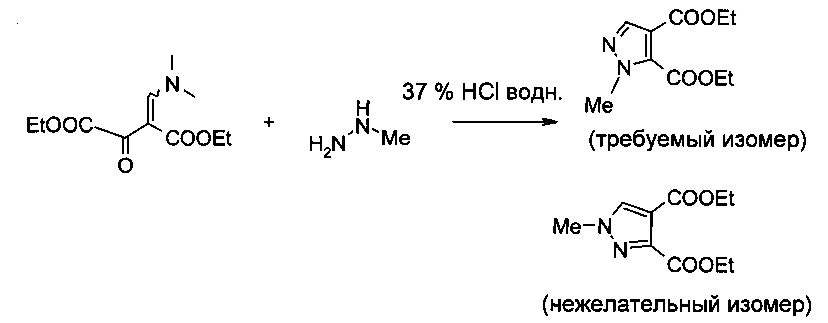
Отделение требуемого изомера от нежелательного изомера, например в описанном выше примере, можно осуществить только с помощью хроматографических методов. Подобные методы нежелательны для синтеза в промышленном масштабе по экономическим и экологическим соображениям.
Следовательно, цель настоящего изобретения заключалась в обнаружении синтетического подхода, который позволит более избирательно и более масштабно получать требуемое производное пиразолкарбоновой кислоты формулы I.
Цель была достигнута с помощью способа по настоящему изобретению, как описано ниже.
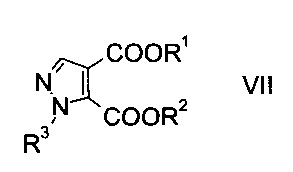
Данный способ получения производного пиразолкарбоновой кислоты формулы
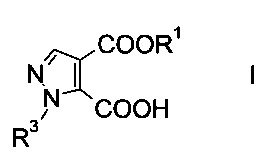
где R1 представляет собой C1-7-алкил и R3 представляет собой C1-7-алкил, который возможно замещен галогеном или C1-4-алкокси, включает стадии, согласно которым
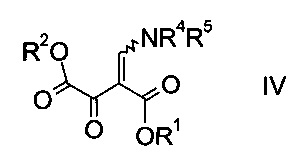
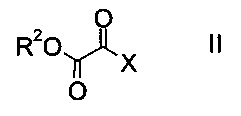
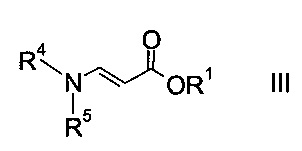
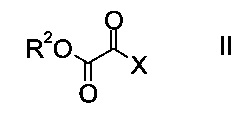
a) осуществляют взаимодействие оксоацетата формулы
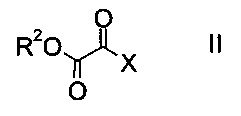
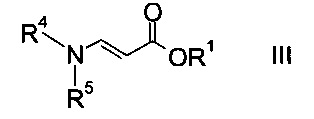
где R2 представляет собой C1-7-алкил, и X представляет собой галоген, с акрилатом формулы
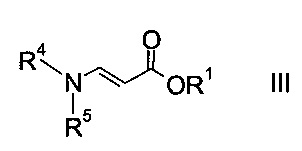
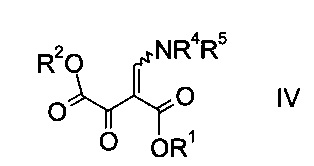
где R1 является таким, как описано выше, и R4 и R5 представляют собой C1-7-алкил, в присутствии основания с образованием эфира аминометилен-янтарной кислоты формулы
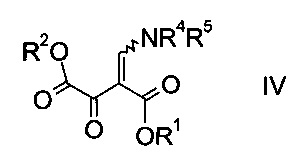
где R1, R2, R4 и R5 являются такими, как описано выше;
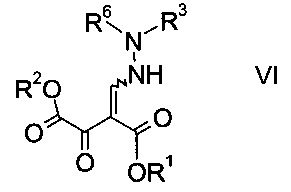
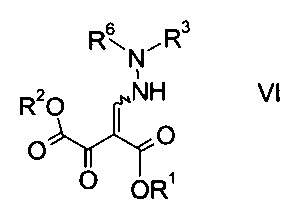
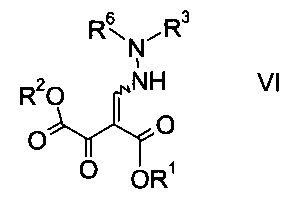
b) связывают эфир аминометилен-янтарной кислоты формулы IV с N-защищенным производным гидразина формулы
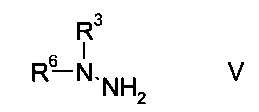
где R3 является таким, как описано выше, и R6 представляет собой амино-защитную группу, с образованием эфира гидразинометилен-янтарной кислоты формулы
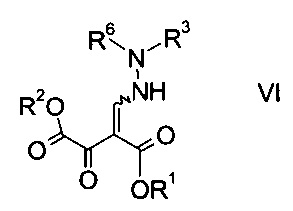
где R1, R2, R3 и R6 являются такими, как описано выше;
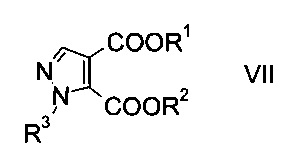
c) замыкают кольцо эфира гидразинометилен-янтарной кислоты формулы VI при кислотных условиях с образованием эфира пиразолдикарбоновой кислоты формулы
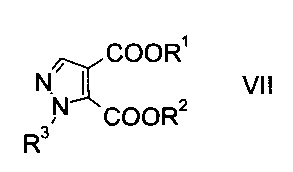
где R1, R2 и R3 являются такими, как описано выше, и
d) подвергают гидролизу эфир пиразолдикарбоновой кислоты формулы VII в 3-положении с основанием с образованием производного пиразолкарбоновой кислоты формулы I.
Если не указано иное, то следующие определения приводятся для иллюстрации и установления значения и объема разных терминов, используемых для описания изобретения в данном документе.
Термин C1-7-алкил отдельно или в сочетании с другими группами относится к разветвленному или неразветвленному одновалентному насыщенному алифатическому углеводородному радикалу от одного до семи атомов углерода. Примерами данного термина могут быть радикалы, подобные метилу, этилу, н-пропилу, изопропилу, н-бутилу, втор-бутилу, трет-бутилу, пентилу, гексилу и гептилу, и их изомеры.
Таким же образом термин C1-4-алкил отдельно или в сочетании с другими группами относится к разветвленному или неразветвленному одновалентному насыщенному алифатическому углеводородному радикалу от одного до четырех атомов углерода. Примерами данного термина могут быть радикалы, подобные метилу, этилу, н-пропилу, изопропилу, н-бутилу, втор-бутилу или трет-бутилу.
Термин C1-4-алкокси означает C1-4-алкильную группу, как определено выше, которая присоединена к радикалу кислорода. Примерами данного термина могут быть радикалы, подобные метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси или трет-бутокси.
Термин "амино-защитная группа" относится к быстрореагирующему заместителю на основе кислоты или кислоты Льюиса, традиционно используемому для замедления реакционной способности аминогруппы. Подходящие быстрореагирующие амино-защитные группы на основе кислоты или кислоты Льюиса описаны у Green Т., "Protective Groups in Organic Synthesis", 4th Ed. by Wiley Interscience, 2007, Chapter 7, 696 ff. Следовательно подходящие амино-защитные группы для R6 можно выбрать из Boc (трет-бутоксикарбонил), Fmoc (флуоренилметоксикарбонил), Cbz (бензилоксикарбонил), Moz (п-метоксибензилкарбонил), Troc (2,2,2-трихлорэтоксикарбонил), Teoc (2-(триметилсилил)этоксикарбонил), Adoc (адамантоксикарбонил), формила, ацетила или циклобутоксикарбонила. В особенности используют Boc.
Термин галоген относится к фтору, хлору, брому или йоду, особенно к фтору, хлору или брому.
На графических изображениях соединений формул IV и VI волнистая линия указывает на существование двух возможных изомеров, E- и Z-, относительно присоединенной двойной связи. В этом случае изображение относится к обоим, E- или Z-изомерам, в виде отдельных изомеров или в виде их смесей.
Стадия a)
На стадии a) проводят реакцию оксоацетата формулы II с акрилатом формулы III с образованием эфира аминометилен-янтарной кислоты формулы IV.
Как оксоацетаты формулы II, так и акрилаты формулы III являются исходными соединениями, которые либо имеются в продаже, либо могут быть синтезированы способами, известными в данной области техники.
Этил-2-хлор-2-оксоацетат (X=Cl и R2=этил) и этил 3-(диметиламино)акрилат (R4, R5=метил и R1=этил) являются особенно полезными в качестве исходных веществ.
Реакцию проводят в присутствии основания, которое можно выбрать из C1-4-триалкиламина, превосходно сочетающегося с каталитическим количеством 4-(диметиламино)-пиридина, или из пиридина. Особенно полезными C1-4-триалкиламинами являются триметиламин, диизопропилэтиламин или триэтиламин.
Как правило, реакцию проводят в апротонном органическом растворителе, таком как 2-метилтетрагидрофуран, дихлорметан, толуол, трет-бутилметиловый эфир или тетрагидрофуран, или в их смесях при температуре реакции между -20°C и 40°C, особенно между -5°C и 30°C.
Эфир аминометилен-янтарной кислоты формулы IV можно выделить из реакционной смеси, используя способы, известные квалифицированному специалисту в данной области техники, однако в конкретном воплощении изобретения эфир янтарной кислоты формулы IV не выделяют, т.е. стадии синтеза a) и b) объединяют.
Стадия b)
На стадии b) связывают эфир аминометилен-янтарной кислоты формулы IV с N-защищенным производным гидразина формулы V с образованием эфира гидразинометилен-янтарной кислоты формулы VI.
N-защищенное производное гидразина формулы V либо имеется в продаже, либо может быть синтезировано способами, известными в данной области техники, например, как описано в межд. публ. патента WO 2011/140425 или у Park et al. в European Journal of Organic Chemistry 2010, pages 3815-3822, или аналогичными способами, известными квалифицированному специалисту в данной области техники.
Как указано выше, когда реакция на стадии a) завершается, стадию b) можно выполнить без выделения продукта реакции со стадии a).
Согласно определению амино-защитной группы R6, как указано выше, подходящим образом защищенные производные гидразина формулы V можно выбрать из, но не ограничиваясь этим, N-Вос-N-метилгидразина, N-Boc-N-этилгидразина, N-Вос-N-н-пропилгидразина, N-Cbz-N-метилгидразина, N-Fmoc-N-метилгидразина, N-Moz-N-метилгидразина, N-Troc-N-метилгидразина, N-Teoc-N-метилгидразина, N-Adoc-N-метилгидразина, N-формил-N-метилгидразина, N-ацетил-N-метилгидразина, N-циклобутоксикарбонил-N-метилгидразина.
Особенно используют N-Вос-N-метилгидразин.
Реакцию можно проводить в полярном апротонном или протонном органическом растворителе, таком как 2-метилтетрагидрофуран, этанол, метанол, этилацетат, изопропилацетат, тетрагидрофуран, трет-бутилметиловый эфир, уксусная кислота, или их смесях при температуре реакции между -10°C и 60°C, особенно между 0°C и 40°C.
Если стадии a) и b) объединяют, то реакцию можно проводить в полярном апротонном органическом растворителе, таком как 2-метилтетрагидрофуран, тетрагидрофуран, трет-бутилметиловый эфир, или их смесях при температуре реакции между -10°C и 60°C, особенно между 0°C и 40°C.
Предпочтительно, можно добавить каталитическое или стехиометрическое количество кислоты, которая не может влиять на амино-защитную группу, такую как фосфорная кислота или уксусная кислота.
Реакционную смесь можно концентрировать в вакууме при температуре между 10°C и 50°C, особенно между 15° и 35°C, чтобы завершить реакцию.
Образующийся в результате эфир гидразинометилен-янтарной кислоты формулы VI можно получить в кристаллической форме после концентрирования реакционной смеси.
Кроме того, можно выполнить очистку, растворяя кристаллический остаток в низшем алифатическом спирте, таком как метанол, и добавляя воду, чтобы вызвать кристаллизацию, или в ходе перекристаллизации из органического растворителя, такого как трет-бутилметиловый эфир.
Эфиры гидразинометилен-янтарной кислоты формулы
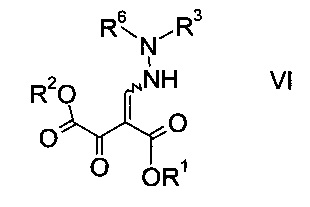
где R1 и R2 представляют собой C1-7-алкил и R3 представляет собой C1-7-алкил, который возможно замещен галогеном или C1-4-алкокси, и R6 означает амино-защитную группу, являются соединениями, не описанными в данной области техники, и таким образом представляют собой дополнительное воплощение настоящего изобретения.
Особыми эфирами гидразинометилен-янтарной кислоты формулы VI являются те, где R1, R2 и R3 представляют собой C1-4-алкил и R6 представляет собой амино-защитную группу, выбранную из Boc, Fmoc, Cbz, Moz, ацетила или формила.
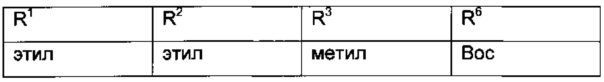
Более конкретные соединения формулы VI имеют следующий образец замещения:

Стадия c)
На стадии c) замыкают кольцо эфира гидразинометилен-янтарной кислоты формулы VI при кислотных условиях с образованием эфира пиразолдикарбоновой кислоты формулы VIII.
Замыкание кольца обычно проводят с неорганической кислотой, органической кислотой или кислотой Льюиса в полярном растворителе, таком как этилацетат, этанол, метанол, вода, тетрагидрофуран, диоксан, уксусная кислота или их смесях при температуре реакции между 0°C и 60°C, в особенности между 10°C и 50°C.
Подходящими неорганическими или органическими кислотами являются, например, хлористоводородная кислота, бромистоводородная кислота, трифторуксусная кислота или п-толуолсульфоновая кислота. Подходящей кислотой Льюиса является, например, триметилсилилиодид. Обычно используют хлористоводородную кислоту, которую можно получить в реакционной смеси, например, добавляя низший алифатический спирт, например этанол, к раствору ацетилхлорида в подходящем полярном растворителе, например этилацетате.
Эфир пиразолдикарбоновой кислоты формулы VII можно выделить из реакционной смеси, используя способы, известные квалифицированному специалисту в данной области техники, например, добавляя воду к реакционной смеси и в ходе последующей экстракции продукта реакции с помощью подходящего растворителя, такого как этилацетат.
Стадия d:
На стадии d) осуществляют гидролиз эфира пиразолдикарбоновой кислоты формулы VII в 3-положении с основанием с образованием производного пиразолкарбоновой кислоты формулы I.
Как правило, основание представляет собой водный раствор гидроксида щелочного металла, выбранного из гидроксида лития, натрия, калия или цезия, или гидрокарбоната щелочного металла, выбранного из гидрокарбоната натрия или калия. Особенно используют гидроксид лития.
Полярный апротонный или протонный растворитель, подобный тетрагидрофурану, N-метилпирролидону, этанолу или метанолу или их смеси можно использовать для растворения эфира пиразолдикарбоновой кислоты формулы VII.
Гидролиз можно проводить при температуре реакции между -20°C и 80°C, особенно между -10°C и 30°C.
По окончании реакции требуемый продукт можно выделить в кристаллической форме способами, известными квалифицированному специалисту в данной области техники, например, подкисляя водную фазу, которую предварительно промывают подходящим растворителем, таким как дихлорметан.
Примеры
Общая часть
Все растворители и реактивы получали от производителей и использовали без обработки. Все реакции контролировали с помощью анализа ТСХ (тонкослойной хроматографии, пластины для ТСХ F254, Merck), ЖХ (жидкостной хроматографии) или ГХ (газовой хроматографии). Спектры протонного ядерного магнитного резонанса (1Н ЯМР) получали на приборах Bruker 300, 400 или 600 МГц с химическими сдвигами (δ в ppm), зарегистрированными относительно тетраметилсилана в качестве внутреннего стандарта, в следующем формате: химический сдвиг в ppm (форма пика, константы взаимодействия, если применимо, интеграл). В случае смеси изомеров оба пика представлены в формате химический сдвиг пика 1 и пика 2 в ppm (формы пиков, константы взаимодействия, если применимо, интеграл, изомеры). Сокращения ЯМР являются следующими: s, синглет; d, дублет; t, триплет; q, квадруплет; quint, квинтет; sext, секстет; hept, гептет; m, мультиплет; br, уширенный. Чистоту анализировали с помощью обращенно-фазовой ВЭЖХ или ГХ. Масс-спектры регистрировали на спектрометре Agilent 6520 QTOF для ЭСИ (ионизации электрораспылением) и ХИАД (химической ионизации при атмосферном давлении), что выполняется одновременно (многорежимный), и на приборе Agilent 5975 для режима ЭИ (электронной ионизации), с детектированием либо положительно (стандартный случай, особо не отмеченный), либо отрицательно (отр.) заряженных ионов. Дополнительно используемыми сокращениями являются: ВТК, внутренний технологический контроль; ДМАП, 4-(диметиламино)пиридин.
Пример 1:
Диэтиловый эфир 2-[1-диметиламино-метилиден]-3-оксо-янтарной кислоты
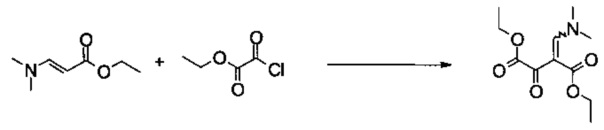
Этил 2-хлор-2-оксоацетат (99 г, 725 ммоль) растворяли в 2-метилтетрагидрофуране (800 мл) и добавляли 4-(диметиламино)-пиридин (1,25 г, 10,0 ммоль). Смесь охлаждали до -5°C и добавляли раствор триэтиламина (76,2 г, 753 ммоль) и (E)-этил 3-(диметиламино)акрилата (106 г, 740 ммоль) в 2-метилтетрагидрофуране (70 мл) с помощью капельной воронки. Смесь перемешивали в течение 3 часов приблизительно при 0°C. После того как добавляли 5% (масс/масс) водный раствор хлорида натрия (250 мл), смесь концентрировали в вакууме, чтобы удалить 2-метилтетрагидрофуран. Добавляли этилацетат (800 мл) и 5% (масс/масс) водный раствор хлорида натрия (250 мл), органическую фазу промывали 5% (масс/масс) водным раствором хлорида натрия (4×250 мл), объединенные водные слои повторно экстрагировали этилацетатом (2×300 мл) и объединенные органические экстракты концентрировали в вакууме. Остаток фильтровали через силикагель (500 г, элюируя этилацетатом / н-гептаном 3:2 (об/об)) и объединенные фильтраты концентрировали в вакууме, получая 146 г неочищенного продукта в виде оранжевого масла. Неочищенный продукт растворяли при комнатной температуре в трет-бутилметиловом эфире (1 л) и охлаждали до 1°C. Кристаллизация начиналась приблизительно при 13°C. Суспензию фильтровали и промывали небольшим количеством холодного трет-бутилметилового эфира, получая 116,6 г указанного в заголовке соединения в виде светло-желтых кристаллов (66%, чистота 99,9% согласно ВЭЖХ). МС (ГХ-разделение): m/z=243 [M]+. 1Н ЯМР (CDCl3, 600 МГц); δ 1,26 (t, J=7,1 Гц, 3H), 1,36 (t, J=7,1 Гц, 3H), 3,03 (s, 3H), 3,36 (s, 3H), 4,17 (q, J=7,1 Гц, 2Н), 4,30 (q, J=7,1 Гц, 2Н), 7,85 (s, 1Н). Продукт выделяли в виде отдельного изомера.
Пример 2:
Диэтиловый эфир 2-(N'-трет-бутоксикарбонил-N'-метилгидразинометилен)-3-оксо-янтарной кислоты
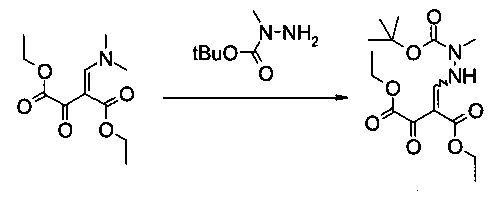
В 1500 мл контролируемой реакционной колбе с рубашкой, оборудованной механической мешалкой, холодильником и внутренним термометром, растворяли диэтиловый эфир 2-[1-диметиламино-мет-(Z)-илиден]-3-оксо-янтарной кислоты (73,2 г, 301 ммоль) в этилацетате (700 мл) и раствор охлаждали до -5°C. Раствор N-трет-бутоксикарбонил-N-метилгидразина (61,5 г, 421 ммоль) в этилацетате (60 мл) добавляли по каплям в течение 45 минут. Реакционную смесь перемешивали в течение 30 минут при -5°C. Затем ее концентрировали в вакууме до объема 100 мл и при постоянном объеме растворитель заменяли трет-бутилметиловым эфиром (1,6 л), что давало в результате густую суспензию. Добавляли еще трет-бутилметиловый эфир (400 мл), суспензию перемешивали в течение 1 часа при 0°C, фильтровали и осадок промывали холодным трет-бутилметиловым эфиром (200 мл). После сушки в вакууме (45°C, 20 мбар) получали указанное в заголовке соединение в виде бесцветного кристаллического твердого вещества (93,2 г, 90%). МС (ЭСИ и ХИАД, отр.): m/z=343,15 [M-H]- 1Н ЯМР (CDCl3, 600 МГц); δ 1,29 (t, J=7,1 Гц, 3H), 1,37 и 1,37 (2t, J=7,1 Гц, 3H, изомеры), 1,48 и 1,48 (2s, 9Н, изомеры), 3,23 и 3,24 (2s, 3H, изомеры), 4,22 и 4,24 (2q, J=7,1 Гц, 2Н, изомеры), 4,31 и 4,35 (2q, J=7,1 Гц, 2Н, изомеры), 8,07 и 8,12 (2d, J=10,3 Гц и 11,6 Гц, 1Н, изомеры), 11,51 и 11,53 (2br, 1H, изомеры). Выделенный продукт представляет собой смесь (E)- и (Z)-изомеров.
Пример 3:
Диэтиловый эфир 2-(N'-трет-бутоксикарбонил-N'-метилгидразинометилен)-3-оксо-янтарной кислоты (однореакторный способ)
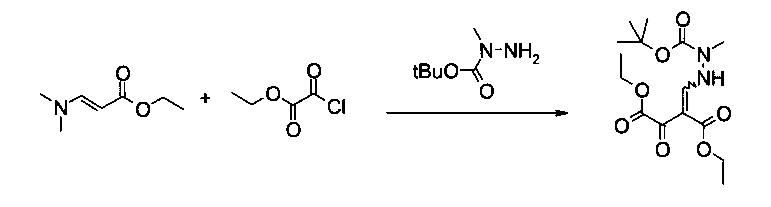
Вариант способа 1: В 12 л контролируемом сосуде с рубашкой, оборудованном механической мешалкой, холодильником, внутренним термометром и подводом инертного газа, растворяли этил 2-хлор-2-оксоацетат (192 г, 158 мл, 1,38 моль) в атмосфере аргона при 20°C в 2-метилтетрагидрофуране (1,34 л). Добавляли ДМАП (2,41 г, 19,3 ммоль) в виде твердого вещества до прозрачного бесцветного раствора. Смесь охлаждали до внутренней температуры 2°C. Готовили раствор (E)-этил 3-(диметиламино)акрилата (179 г, 1,24 моль) в 2-метилтетрагидрофуране (960 мл) и триэтиламина (154 г, 212 мл, 1,51 моль) в отдельной колбе в ходе последующего добавления при комнатной температуре и добавляли к раствору этил 2-хлор-2-оксоацетата и ДМАП со скоростью, чтобы внутренняя температура поддерживалась приблизительно при 2°C (необходимо охлаждение). Смесь становилась мутной, затем густой кристаллической массой (еще перемешиваемой). Через 30 минут перемешивания при 2°C смесь нагревали до комнатной температуры, фильтровали, осадок промывали 2-метилтетрагидрофураном (2 л). N-трет-Бутоксикарбонил-N-метилгидразин (250 г, 254 мл, 1,66 моль) добавляли к объединенному фильтрату при 20°C и полученную в результате смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали в вакууме до оранжевого кристаллического остатка. Остаток растворяли в метаноле (4 л, темно-красный раствор) и добавляли воду (4 л). Продукт кристаллизовался самопроизвольно, взвесь перемешивали в течение ночи при комнатной температуре. Смесь фильтровали, кристаллический осадок последовательно промывали водой (8 л) и гептаном (8 л) и сушили в течение ночи при 50°C и 12 мбар, получая 352 г требуемого продукта в виде белого порошка (83%). Т.пл. 130,2-131,3°C. МС (ЭСИ и ХИАД, отр.): m/z=343,15 [M-H]-. 1Н ЯМР (CDCl3, 600 МГц); δ 1,29 (t, J=7,1 Гц, 3H), 1,37 и 1,37 (2t, J=7,1 Гц, 3H, изомеры), 1,48 и 1,48 (2s, 9Н, изомеры), 3,23 и 3,24 (2s, 3H, изомеры), 4,22 и 4,24 (2q, J=7,1 Гц, 2Н, изомеры), 4,31 и 4,35 (2q, J=7,1 Гц, 2Н, изомеры), 8,07 и 8,12 (2d, J=10,3 Гц и 11,6 Гц, 1Н, изомеры), 11,51 и 11,53 (2brs, 1Н, изомеры). Выделенный продукт представляет собой смесь (E)- и (Z)-изомеров.
Вариант способа 2: 300 л реактор, оборудованный терморегулятором и вакуумной системой, заполняли в атмосфере азота (E)-этил 3-(диметиламино)акрилатом (10,0 кг, 69,8 моль), тетрагидрофураном (80 кг), триэтиламином (8,6 кг, 85,0 моль) и ДМАП (0,14 кг, 1,25 моль) и полученный в результате раствор охлаждали до -5-0°C. Раствор этил 2-хлор-2-оксоацетата (11,0 кг, 80,6 моль) в тетрагидрофуране (9 кг) добавляли по каплям к смеси со скоростью, чтобы внутренняя температура сохранялась при -5-0°C (в течение приблизительно 3 часов). Затем смесь нагревали до 15-25°C и перемешивали в течение 40 минут или до тех пор, пока ВТК показывал полный расход (E)-этил 3-(диметиламино)акрилата. Добавляли N-трет-бутоксикарбонил-N-метилгидразин (13,5 кг, 85,7 моль) к смеси в течение приблизительно 5 минут. Растворитель удаляли выпариванием и смесь нагревали приблизительно до 30-35°C. Выпаривание останавливали, когда прекращалась перегонка тетрагидрофурана (после приблизительно 4 часов). Полученное полутвердое вещество охлаждали до 20-25°C. Добавляли метанол (39,6 кг) и смесь перемешивали в течение 10 минут. Добавляли воду (110 кг) при внутренней температуре 15-20°C в течение 10 минут. Смесь перемешивали в течение 2 часов при 15-25°C, фильтровали и отфильтрованный осадок последовательно промывали водой (2×25 кг) и н-гептаном (2×16,7 кг). Затем его сушили при 50-55°C в течение 10 часов, получая указанное в заголовке соединение в виде белого твердого вещества (21,0 кг, 85,0%, чистота 99,2% согласно ВЭЖХ). Выделенный продукт представляет собой смесь (Е)- и (Z)-изомеров, подлинность продукта подтверждали с помощью 1Н ЯМР и МС.
Пример 4:
Диэтиловый эфир 2-метил-2Н-пиразол-3,4-дикарбоновой кислоты
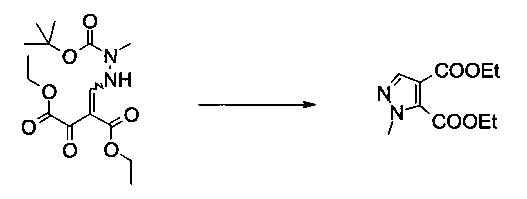
Вариант способа 1: 12 л контролируемый сосуд с рубашкой, оборудованный механической мешалкой, холодильником, внутренним термометром и подводом инертного газа, заполняли этилацетатом (2,21 кг, 2,45 л, 25,0 моль) в атмосфере аргона при 20°C. Добавляли ацетилхлорид (564 г, 511 мл, 7,11 моль) (слабый экзотермический эффект, прозрачный бесцветный раствор). Добавляли этанол (656 г, 826 мл, 14,2 моль) со скоростью, чтобы внутренняя температура поддерживалась при 20-25°C (контролируемый процесс, сильно экзотермический, необходимо эффективное охлаждение). Суспензию (Z)-диэтил 2-((2-(трет-бутоксикарбонил)-2-метилгидразинил)метилен)-3-оксосукцината (350 г, 1,02 моль) в этилацетате (1,05 л) добавляли с помощью насоса при 20°C к безводному раствору хлористоводородной кислоты в этилацетате / этаноле. Полученная в результате белая суспензия становилась зеленоватым раствором без экзотермического эффекта. Смесь перемешивали при 50°C в течение 2 часов. После этого смесь охлаждали до 20°C и добавляли воду (6 л) (слабый экзотермический эффект, внутренняя темп. 34°C, быстрое разделение фаз). После фазового разделения водную фазу экстрагировали этилацетатом (2×1 л). Объединенные органические экстракты сушили (сульфат натрия) и концентрировали в вакууме (температура рубашки 50°C, 10 мбар), получая 236 г неочищенного продукта в виде желтого масла (99%, чистота 96,8% согласно ВЭЖХ). МС (ЭСИ и ХИАД): m/z=227,1 [М+Н]+. 1Н ЯМР (CDCl3, 600 МГц); δ 1,34 (t, J=7,1 Гц, 3H), 1,41 (t, J=7,1 Гц, 3H), 4,02 (s, 3H), 4,30 (q, J=7,1 Гц, 2Н), 4,44 (q, J=7,1 Гц, 2Н), 7,82 (s, 1Н).
Вариант способа 2: 300 л реактор, оборудованный терморегулятором и вакуумной системой, заполняли раствором хлороводорода в этаноле (58,6 кг, содержание: 38,6% масс/масс, 620 моль) и раствор охлаждали приблизительно до 0-5°C. (Z)-Диэтил 2-((2-(трет-бутоксикарбонил)-2-метилгидразинил)метилен)-3-оксосукцинат (58,6 кг, 171 моль) добавляли к раствору частями в течение 50 минут при 0-15°C. Затем смесь нагревали до 15-25°C и перемешивали в течение 3 часов или до тех пор, пока ВТК показывал полный расход исходного вещества, трет-Бутилметиловый эфир (87,9 кг) добавляли к смеси и смесь переносили в 500 л реактор. Воду (175,8 кг) добавляли к раствору со скоростью, чтобы внутренняя температура сохранялась ниже 25°C. После разделения фаз водный слой переносили в 1000 л реактор и экстрагировали трет-бутилметиловым эфиром (2×87,9 кг). Органический слой добавляли в 500 л реактор и промывали последовательно водой (87,9 кг) и раствором гидрокарбоната натрия (4,7 кг) в воде (87,9 кг), и сушили над сульфатом натрия (39,3 кг). Смесь фильтровали и фильтрат выпаривали в вакууме при 30-55°C, получая указанное в заголовке соединение в виде желтой жидкости (36,7 кг, 95,3%, чистота 99,6% согласно ВЭЖХ). Подлинность продукта подтверждали с помощью 1Н ЯМР и МС.
Пример 5:
4-Этиловый эфир 2-метил-2Н-пиразол-3,4-дикарбоновой кислоты
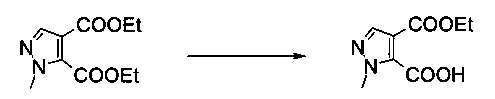
Вариант способа 1: В 63 л стальном/эмалированном сосуде, оборудованном обратным холодильником, объединенным с термометром, механической мешалкой и подводом инертного газа, диэтиловый эфир 2-метил-2Н-пиразол-3,4-дикарбоновой кислоты (2,84 кг, 12,6 моль) растворяли в смеси тетрагидрофурана (20,0 л) и этанола (8,5 л) в атмосфере азота при комнатной температуре. Смесь охлаждали до -5°C и добавляли раствор моногидрата гидроксида лития (0,53 кг, 12,6 моль) в воде (10,0 л) в течение 90 минут при -5°C. Капельную воронку промывали водой (1,4 л). Реакционную смесь перемешивали в течение 95 минут при -4°C до -6°C. После этого смесь разбавляли дихлорметаном (10,0 л) и водой (10,0 л) при -5°C до 0°C и перемешивали в течение 10 минут. Органический слой отделяли. Водную фазу промывали дихлорметаном (2×10,0 л). Водную фазу подкисляли до pH<2, добавляя соляную кислоту (2,75 кг, содержание: 25% масс/масс, 18,8 моль) в воде (2,0 л) в течение 15 минут при 20°C до 25°C. Полученную в результате кристаллическую суспензию перемешивали в течение 17 часов при 22°C. Затем кристаллическую суспензию фильтровали через воронку со стеклянным фильтром. Осадок на фильтре промывали последовательно водой (7,0 л) и н-гептаном (4,0 л). Белые кристаллы сушили в вакууме при 50°C / <5 мбар в течение 70 часов, получая 1,99 кг указанного в заголовке соединения в виде белых кристаллов (80%). МС (ЭСИ и ХИАД): m/z=199,1 [М+Н]+. 1Н ЯМР (D6-ДМСО, 600 МГц); δ 1,25 (t, J=7,1 Гц, 3H), 3,94 (s, 3H), 4,22 (q, J=7,1 Гц, 2Н), 7,85 (s, 1Н), 14,18 (brs, 1Н).
Вариант способа 2: 1000 л реактор, оборудованный терморегулятором и вакуумной системой, заполняли диэтиловым эфиром 2-метил-2Н-пиразол-3,4-дикарбоновой кислоты (36,5 кг, 161 моль), тетрагидрофураном (253 кг) и этанолом (20,0 л) в атмосфере азота при комнатной температуре. Смесь охлаждали до -10-5°C. В другом 300 л реакторе раствор моногидрата гидроксида лития (6,47 кг, 154 моль) в воде (135,8 кг) предварительно охлаждали до 5-10°C и добавляли по каплям в 1000 л реактор со скоростью, чтобы внутренняя температура сохранялась при -10 - -5°C (приблизительно 3 часа). Смесь перемешивали при -10 - -5°C в течение 3 часов или пока ВТК ни удовлетворял требованиям спецификации (т.е. диэтиловый эфир 2-метил-2Н-пиразол-3,4-дикарбоновой кислоты <10% согласно ВЭЖХ и побочный продукт 2-метил-2Н-пиразол-3,4-дикарбоновая кислота <4% согласно ВЭЖХ). Затем добавляли дихлорметан (190,8 кг) и воду (146,8 кг) и смесь перемешивали в течение 20 минут. Органический слой отделяли, водную фазу промывали дихлорметаном (2×190,8 кг), после этого водный слой фильтровали через 8 см вставку из целита и фильтрат переносили в 500 л реактор. Охлаждали до 5-10°C, хлористоводородную кислоту (18% масс/масс) добавляли по каплям в течение 50 минут при 5-15°C до рН=1-2 (приблизительно 30 кг). Продукт кристаллизовался постепенно в виде белого твердого вещества. Суспензию перемешивали при 25-30°C в течение 10 часов. Осадок центрифугировали, промывали водой (69,4 кг) и н-гептаном (2×29 кг) и сушили в вакууме при 40-55°C в течение 48 часов, получая указанное в заголовке соединение в виде белого твердого вещества (22,2 кг, 69,4%, чистота 99,7% согласно ГХ). Подлинность продукта подтверждали с помощью 1Н ЯМР и МС.
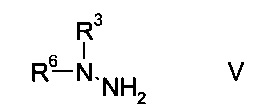
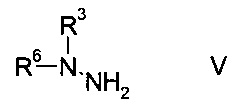
Производные мочевины и их применение в качестве ингибиторов белка, связывающего жирные кислоты
Способ отбора и получения селективных и мультиспецифических терапевтических молекул с заданными свойствами, включающих по меньшей мере две различные нацеливающие группировки, и их применения
Новые бициклические соединения тиофениламида
Производные имидазопиридина, стимулирующие нейрогенез
Способ получения и отбора молекул, включающих по меньшей мере две различные группировки, и их применение
Гетероарил пиридоны и азапиридоны с электрофильной функциональностью
Неаннелированные тиофениламиды в качестве ингибиторов белков, связывающих жирные кислоты fabp 4 и/или 5
Бензимидазолы в качестве активных агентов для центральной нервной системы
Ингибиторы тирозинкиназы брутона
Комбинированная терапия с использованием антитела против cd20 типа ii и селективного ингибитора bcl-2
Производные дипептида лизин-глутаминовая кислота
Производные мочевины и их применение в качестве ингибиторов белка, связывающего жирные кислоты
Способ отбора и получения селективных и мультиспецифических терапевтических молекул с заданными свойствами, включающих по меньшей мере две различные нацеливающие группировки, и их применения
Новые бициклические соединения тиофениламида
Производные имидазопиридина, стимулирующие нейрогенез
Способ получения и отбора молекул, включающих по меньшей мере две различные группировки, и их применение
2-фенилимидазо[1,2-а]пиримидины в качестве визуализирующих средств
Имидазо[1,2-а]пиридин-7-амины в качестве средств визуализации
Новые производные пирролидин-3,4-дикарбоксамида
Новые 2-аминооксазолины в качестве лигандов taar1

