Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ИЛИ ЖИРНОАРОМАТИЧЕСКИХ КЕТОНОВ (ВАРИАНТЫ)
Вид РИД
Изобретение
Область техники
Группа изобретений относится к способу получения ароматических или жирноароматических кетонов по реакции ароматических хлоридов, или бромидов, или йодидов с алифатическими или ароматическими нитрилами, включая внутримолекулярные реакции содержащих нитрильную группу ароматических хлоридов, бромидов или йодидов, причем первоначально образующееся соединение со связью C=N подвергается последующему гидролизу с образованием целевого продукта.
Ароматические и жирноароматические кетоны и их производные широко используются в качестве исходных соединений и полупродуктов в производстве органических соединений, в том числе обладающих биологической активностью.
Уровень техники
Известен способ получения ароматических и жирноароматических кетонов с использованием в качестве исходных соединений алкил-, арил- или гетарилгалогенидов и ароматических или алифатических нитрилов. Для этого соответствующий галогенид предварительно превращают в металлоорганическое соединение, затем проводят реакцию присоединения металлоорганического соединения к нитрилу, а в заключение осуществляют гидролиз образовавшейся соли имина и металла. В качестве металлоорганических соединений для этой цели чаще всего используют реактивы Гриньяра [US 2012088748, кл. A61K 31/553 и др., опубликовано 12.04.2012; US 2013217733, кл. A61K 31/4245, опубликовано 22.08.2013]] и литийорганические соединения [WO 200808059, кл. A61K 31/44, опубликовано 17.01.2008; US 201004171, кл. A61K 31/165, опубликовано 18.02.2010]:
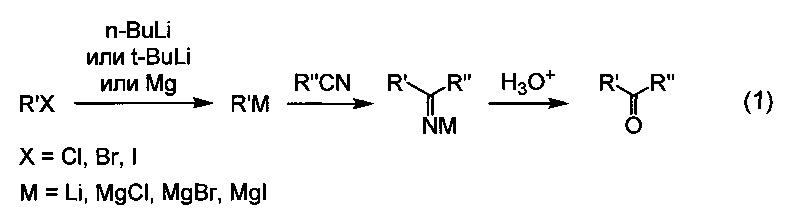
Недостатком данного способа является необходимость предварительного получения металлоорганического соединения, что является дополнительной стадией синтеза, усложняющей процесс. Другим недостатком метода является высокая реакционная способность органических соединений лития и магния, что приводит к возможности протекания побочных реакций. Вследствие этого существуют ограничения для использования в качестве исходных реагентов соединений, содержащих группы, способные к реакциям с органическими соединениями лития и магния, например соединений, содержащих сложноэфирную группу. Еще одним недостатком этого метода является высокая пожароопасность литий- и магнийорганических соединений.
Известен способ получения циклических ароматических и жирноароматических кетонов по катализируемой комплексами палладия внутримолекулярной реакции с участием арилйодидов, содержащих нитрильную группу [TetrahedronLett., 2002, 43, 2133-2136; J. Org. Chem., 2002, 67, 9428-9438]. Недостатком данного способа является использование дорогих и труднодоступных арилйодидов в качестве исходных соединений, а также дорогостоящего палладиевого катализатора. Другим недостатком данного способа является исключительно внутримолекулярный характер реакции, что ограничивает спектр возможных продуктов только лишь циклическими ароматическими и жирноароматическими кетонами. Еще одним недостатком являются ограничения, связанные с возможностью вводить в указанную реакцию только α,α-дизамещенные или ароматические нитрилы, что позволяет получать лишь α,α-дизамещенные жирноароматические или ароматические циклические кетоны, соответственно, в качестве продуктов.
Наиболее близким к предлагаемому изобретению является способ получения ароматических и жирноароматических кетонов, описанный в статье [Org. Lett., 2012, 14, 1283]. По этому способу ароматические кетоны получают реакцией между арилйодидами и алкил- либо арилнитрилами в присутствии каталитической системы, включающей NiCl2(диметоксиэтан), лиганд (1,3-бис-дифенилфосфинопропан), металлический цинк и воду. При этом мольное отношение арилйодид:NiCl2(диметоксиэтан):лиганд:цинк:вода составляет 1:0,1:0,1:2,0:1,2. Реакцию предпочтительно проводят при 100°C в течение 24-36 ч в диметоксиэтане (ДМЭ), который выступает в качестве растворителя. Недостатком данного способа является необходимость использования только труднодоступных арилйодидов в качестве исходных соединений. В указанных условиях более дешевые и легкодоступные арилбромиды и арилхлориды в реакцию не вступают.
Раскрытие изобретения
Задачей данного изобретения является разработка технологичного способа получения ароматических или жирноароматических кетонов по реакции между ароматическими хлоридами, или бромидами, или йодидами и ароматическими либо алифатическими нитрилами с последующим гидролизом первоначально образующихся соединений со связью C=N, что позволяет с высокими выходами получать целевые продукты, содержащие различные заместители.
В частности, в способе происходят внутримолекулярные реакции ароматических хлоридов, или бромидов, или йодидов, содержащих нитрильную группу, которая находится в положении, допускающем замыкание нового цикла посредством образования связи между атомом углерода нитрильной группы и атомом углерода, связанным с атомом галогена. Последующий гидролиз первоначально образующихся соединений со связью C=N позволяет получать циклические ароматические или жирноароматические кетоны, в которых кетогруппа является частью вновь образованного цикла.
Техническим результатом является расширение арсенала средств, т.е. способов получения ароматических или жирноароматических кетонов. Дополнительно преимуществами изобретения по сравнению с прототипом являются: (а) возможность использования в качестве исходных соединений арилхлоридов и арилбромидов, более дешевых и легкодоступных по сравнению с арилйодидами, которые используются в прототипе; (б) использование по крайней мере в два раза более низких загрузок катализатора по сравнению с прототипом, что при применении изобретения в промышленности снижает экологические риски за счет уменьшения количества соединений никеля в отходах производства; (в) возможность проведения реакции при более низкой температуре, чем в прототипе (например, при 20°C, в то время как в прототипе реакция проводится при 100°C), что позволяет снизить энергозатраты на проведение реакции; (г) возможность проведения реакции за меньшее время, чем в прототипе (например, за 3 часа, в то время как в прототипе реакция проводится не менее чем за 24 часа), что также позволяет снизить энергозатраты на проведение реакции.
Технический результат достигается тем, что способ получения ароматических или жирноароматических кетонов общей формулы R1C(O)R2, где R1 - арил или гетарил, а R2 - арил, или гетарил, или алкил, заключается в реакции ароматических галогенидов R1X, где R1 - арил или гетарил, а X=Cl, Br, I с нитрилами R2CN, где R2 - арил, или гетарил, или алкил, которую проводят в присутствии катализатора, включающего атом никеля, координированный с хелатным лигандом, содержащим 1,4-диазабутадиеновый фрагмент (N=C-C=N), и восстановителя в среде эфирного растворителя при мольных соотношениях:
R1X:R2CN, находящемся в пределах от 2:1 до 1:20,
Ni:(хелатный лиганд), находящемся в пределах от 1:1 до 1:2,
Ni:R1X, находящемся в пределах от 1:200 до 1:2,
восстановитель:R1X, находящемся в пределах от 1:2 до 10:1,
а объем растворителя по отношению к количеству галогенида R1X находится в пределах от 0,5 до 20 мл/ммоль,
при температуре реакции 0-120°C, и последующий гидролиз первоначально образующегося соединения со связью C=N.
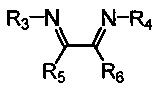
Хелатный лиганд, содержащий 1,4-диазабутадиеновый фрагмент, возможно выбрать из соединений вида
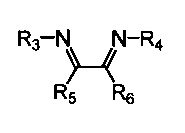
где:
- R3, R4 – алкильные, и/или алкенильные, и/или алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R3, R4 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
- R5, R6 - атом(ы) водорода, и/или алкильные, и/или алкенильные, алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R5, R6 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
при этом любая пара из радикалов R3, R4, R5 и R6 может быть связана между собой с образованием карбоцикла или гетероцикла.
Катализатор возможно получить in situ из исходного соединения никеля и хелатного лиганда, содержащего 1,4-диазабутадиеновый фрагмент.
В качестве соединения никеля возможно использовать NiBr2(ДМЭ) или NiCl2(ДМЭ).
В качестве катализатора возможно использовать предварительно полученный комплекс галогенида никеля с хелатным лигандом, содержащим 1,4-диазабутадиеновый фрагмент при мольном отношении Ni:(хелатный лиганд), равном 1:1.
В качестве хелатного лиганда возможно использовать N,N'-бис(2,6-диизопропилфенил)-1,4-диаза-1,3-бутадиен, или бис(2,6-диизопропилфенил)аценафтенхинондиимин или бис(2,6-бис(1-этилпропил)фенил)аценафтенхинондиимин.
В качестве восстановителя возможно использовать металлический цинк.
В качестве растворителей возможно использовать тетрагидрофуран, 2-метилтетрагидрофуран или 1,4-диоксан.
Реакцию предпочтительно ведут при температуре 20-70°C.
Технический результат также достигается тем, что способ получения ароматических или жирноароматических циклических кетонов общей формулы R1C(O)R2, где R1 - арил или гетарил, а R2 - арил, или гетарил, или алкил, при этом R1 и R2 связаны между собой, образуя карбоцикл или гетероцикл, заключается во внутримолекулярной реакции ароматических соединений вида XR1-R2CN, где R1 - арил или гетарил, X=Cl, Br, I, R2 - алкил, или арил, или гетарил, при этом содержащая в качестве заместителя нитрильная группа находится в положении, допускающем замыкание нового цикла посредством образования связи между атомом углерода нитрильной группы и атомом углерода, связанным с атомом галогена, которую проводят в присутствии катализатора, включающего атом никеля, координированный с хелатным лигандом, содержащим 1,4-диазабутадиеновый фрагмент (N=C-C=N), и восстановителя в среде эфирного растворителя
при мольных соотношениях:
Ni:(хелатный лиганд), находящемся в пределах от 1:1 до 1:2,
Ni:XR1-R2CN, находящемся в пределах от 1:200 до 1:2,
восстановитель:XR1-R2CN, находящемся в пределах от 1:2 до 10:1,
а объем растворителя по отношению к количеству соединения XR1-R2CN в пределах от 0,5 до 20 мл/ммоль,
при температуре реакции 0-120°C,
и последующий гидролиз первоначально образующегося соединения со связью C=N.
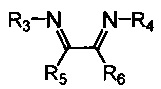
Хелатный лиганд, содержащий 1,4-диазабутадиеновый фрагмент, возможно выбрать из соединений вида
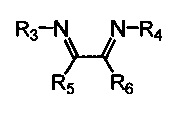
где:
- R3, R4 – алкильные, и/или алкенильные, и/или алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R3, R4 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
- R5, R6 - атом(ы) водорода, и/или алкильные, и/или алкенильные, и/или алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R5, R6 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
при этом любая пара из радикалов R3, R4, R5 и R6 может быть связана между собой с образованием карбоцикла или гетероцикла.
Катализатор возможно получить in situ из исходного соединения никеля и хелатного лиганда, содержащего 1,4-диазабутадиеновый фрагмент.
В качестве соединения никеля возможно использовать NiBr2(ДМЭ) или NiCl2(ДМЭ).
В качестве катализатора возможно использовать предварительно полученный комплекс галогенида никеля с хелатным лигандом, содержащим 1,4-диазабутадиеновый фрагмент при мольном отношении Ni:(хелатный лиганд), равном 1:1.
В качестве хелатного лиганда возможно использовать N,N'-бис(2,6-диизопропилфенил)-1,4-диаза-1,3-бутадиен, или бис(2,6-диизопропилфенил)аценафтенхинондиимин или бис(2,6-бис(1-этилпропил)фенил)аценафтенхинондиимин.
В качестве восстановителя возможно использовать металлический цинк.
В качестве растворителей возможно использовать тетрагидрофуран, 2-метилтетрагидрофуран или 1,4-диоксан.
Реакцию предпочтительно ведут при температуре 20-70°C.
Осуществление изобретения
Представленный способ получения ароматических или жирноароматических кетонов общей формулы R1C(O)R2, где R1 - арил или гетарил, R2 - арил, или гетарил, или алкил, заключается в реакции ароматических галогенидов R1X, где R1 - арил или гетарил, а X=Cl, Br, I с нитрилами R2CN, где R2 - арил, или гетарил, или алкил, которую проводят в присутствии катализатора, а также восстановителя в среде эфирного растворителя при температуре 0-120°C. В ходе реакции образуется цинковая соль ароматического или жирноароматического имина, которая легко гидролизуется с образованием ароматического или жирноароматического кетона, соответственно.
В качестве катализатора используется комплекс, включающий атом никеля, координированный с азотсодержащим хелатным лигандом, либо указанный комплекс получают in situ из исходного соединения никеля и азотсодержащего хелатного лиганда. В качестве азотсодержащих хелатных лигандов используются соединения (2), содержащие 1,4-диазабутадиеновый фрагмент (N=C-C=N):
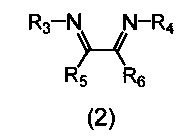
где:
- R3, R4 – алкильные, и/или алкенильные, и/или алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R3, R4 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
- R5, R6 - атом(ы) водорода, и/или алкильные, и/или алкенильные, и/или алкинильные, и/или арильные радикалы, в том числе циклоалифатические, и/или гетероциклические, и/или ароматические, и/или гетероароматические радикалы; при этом R5, R6 могут содержать заместители, такие как алкильная, арильная, гетарильная группа, AlkO, ArO, NR2, SiR3, галоген и другие заместители;
при этом любая пара из радикалов R3, R4, R5 и R6 может быть связана между собой с образованием карбоцикла или гетероцикла.
В качестве исходного соединения никеля могут быть использованы NiCl2, NiBr2, NiI2, NiCl2(ДМЭ), NiBr2(ДМЭ), Ni(циклооктадиен)2, Ni(ацетилацетонат)2 и другие соединения никеля.
В расчете на 1 моль никеля используется не менее 1 моль хелатного лиганда.
В качестве растворителей используются соединения класса простых эфиров, такие как тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран, диоксан и другие.
В качестве восстановителя используют металлический цинк.
Наилучшие результаты были достигнуты при использовании в качестве лигандов N,N'-бис(2,6-диизопропилфенил)-1,4-диаза-1,3-бутадиена (L1), бис(2,6-диизопропилфенил)аценафтенхинондиимина (L2) и бис(2,6-бис(1-этилпропил)фенил)аценафтенхинондиимина (L3), а в качестве соединения никеля - NiBr2(ДМЭ) или NiCl2(ДМЭ), в качестве восстановителя -металлического цинка, а в качестве растворителя - тетрагидрофурана (ТГФ, при проведении реакции при температуре 70°C в течение 17 ч (3):
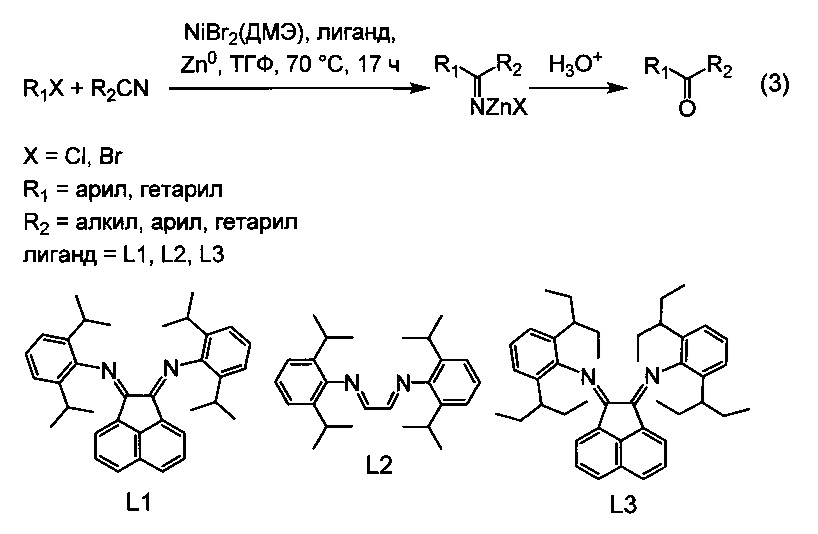
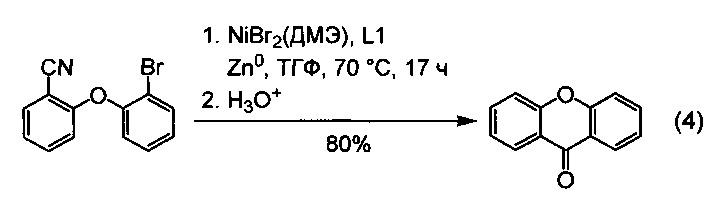
Частным случаем применения настоящего изобретения является получение циклических ароматических и жирноароматических кетонов, то есть кетонов общей формулы R1C(O)R2, в которых радикалы R1 и R2 связаны между собой, образуя карбоцикл или гетероцикл. Это достигается использованием в качестве исходных реагентов соединений вида XR1-R2CN, где R1 - арил или гетарил, X=Cl, Br, I, а R2 - алкил, или арил, или гетарил, то есть, ароматических хлоридов, или бромидов, или йодидов, содержащих в качестве заместителя нитрильную группу в положении, допускающем образование связи между атомом углерода нитрильной группы и атомом углерода, связанным с атомом галогена. При этом реакция протекает внутримолекулярно, что приводит к замыканию нового цикла, содержащего после гидролиза вновь образованную кетогруппу. Частный случай получения циклического кетона по внутримолекулярной реакции проиллюстрирован уравнением (4).
Возможность осуществления изобретения подтверждается следующими примерами синтеза. Приведенные ниже примеры конкретного осуществления изобретения приведены для предоставления специалистам в данной области техники полного описания проведения и применения анализа по изобретению и подразумевают, что приведенные примеры не ограничивают предполагаемый авторами изобретения объем изобретения.
Пример 1.
Синтез проводят в инертной атмосфере. В стеклянный сосуд объемом 8 мл, снабженный магнитной мешалкой, поместили 74,8 мг (1,15 ммоль) цинковой пыли, 15,5 мг (0,05 ммоль) NiBr2(ДМЭ), 25,1 мг (0,05 ммоль) бис(2,6-диизопропил фенил)аценафтенхинондиимина, 157 мг (1,0 ммоль) бромбензола, 103 мг (1,0 ммоль) бензонитрила и 5 мл ТГФ. Реакцию вели при температуре 70°C в течение 17 ч при перемешивании с помощью магнитной мешалки. Затем смесь охладили до комнатной температуры, добавили 2 мл метанола и отфильтровали. К фильтрату добавили 2 мл 6 М раствора соляной кислоты и полученную смесь перемешивали при 40°C в течение 2 ч. Затем смесь нейтрализовали водным раствором гидроксида натрия и экстрагировали 3×10 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия, после чего растворители упарили в вакууме с помощью роторного испарителя. Продукт выделили методом колоночной хроматографии на силикагеле, элюент - гексан-этилацетат. Выход бензофенона составил 157 мг (86%).
Пример 2. Синтез проводили, как в примере 1, но вместо бис(2,6-диизопропилфенил)аценафтенхинондиимина использовали N,N'-бис(2,6-диизопропилфенил)-1,4-диаза-1,3-бутадиен (18,9 мг, 0,05 ммоль). Выход бензофенона составил 122 мг (67%).
Пример 3. Синтез проводили, как в примере 1, но вместо бис(2,6-диизопропилфенил)аценафтенхинондиимина использовали бис(2,6-бис(1-этилпропил)фенил)аценафтенхинондиимин (30,7 мг, 0,05 ммоль). Выход бензофенона составил 124 мг (68%).
Пример 4. Синтез проводили, как в примере 1, но вместо NiBr2(ДМЭ) использовали NiCl2(ДМЭ) (11,0 мг, 0,05 ммоль). Выход бензофенона составил 171 мг (94%).
Пример 5. Синтез проводят как в примере 1, но вместо NiBr2(ДМЭ) используют NiI2 (15,6 мг, 0,05 ммоль). Выход бензофенона составил 146 мг (80%).
Пример 6. Синтез проводили, как в примере 1, но вместо NiBr2(ДМЭ) использовали NiBr2 (11,0 мг, 0,05 ммоль). Выход бензофенона составил 129 мг (71%).
Пример 7. Синтез проводили, как в примере 1, но вместо NiBr2(ДМЭ) использовали Ni(циклооктадиен)2 (13,8 мг, 0,05 ммоль). Выход бензофенона составил 147 мг (81%).
Пример 8. Синтез проводили, как в примере 1, но вместо NiBr2(ДМЭ) использовали Ni(ацетилацетонат)2 (12,9 мг, 0,05 ммоль). Выход бензофенона составил 116 мг (64%).
Пример 9. Синтез проводили, как в примере 1, но вместо NiBr2(ДМЭ) и бис(2,6-диизопропилфенил)аценафтенхинондиимина использовали предварительно полученный комплекс [бис(2,6-диизопропилфенил)аценафтенхинондиимин]NiBr2 (36,0 мг, 0,05 ммоль). Выход бензофенона составил 155 мг (85%).
Пример 10. Синтез проводили, как в примере 1, но вместо 0,05 ммоль NiBr2(ДМЭ) использовали 0,01 ммоль NiBr2(ДМЭ) (3,1 мг), а реакцию вели 48 ч. Выход бензофенона составил 95 мг (52%).
Пример 11. Синтез проводили, как в примере 4, но реакцию вели в течение 1 часа вместо 17 часов. Выход бензофенона составил 113 мг (62%).
Пример 12. Синтез проводили, как в примере 4, но реакцию вели в течение 3 часов вместо 17 часов. Выход бензофенона составил 146 мг (80%).
Пример 13. Синтез проводили, как в примере 1, но вместо 1,15 ммоль цинковой пыли использовали 0,75 ммоль цинковой пыли (49,1 мг). Выход бензофенона составил 119 мг (65%).
Пример 14. Синтез проводили, как в примере 1, но вместо 1,15 ммоль цинковой пыли использовали 4,0 ммоль цинковой пыли (262 мг). Выход бензофенона составил 135 мг (74%).
Пример 15. Синтез проводили, как в примере 1, но при реакции вели при температуре 50°C вместо 70°C. Выход бензофенона составил 157 мг (86%).
Пример 16. Синтез проводили, как в примере 1, но реакцию вели при температуре 20°C вместо 70°C и в течение 48 ч вместо 17 часов. Выход бензофенона составил 149 мг (82%).
Пример 17. Синтез проводили, как в примере 14, но в качестве растворителя вместо ТГФ использовали 2-метилтетрагидрофуран (5 мл). Выход бензофенона составил 144 мг (79%).
Пример 18. Синтез проводили, как в примере 14, но в качестве растворителя вместо ТГФ использовали 1,4-диоксан (5 мл). Выход бензофенона составил 104 мг (57%).
Пример 19. Синтез проводили, как в примере 1, но вместо бромбензола использовали хлорбензол (113 мг, 1,0 ммоль). Выход бензофенона составил 171 мг(94%).
Пример 20. Синтез проводили, как в примере 1, но вместо бромбензола использовали йодбензол (204 мг, 1,0 ммоль). Выход бензофенона составил 153 мг (84%).
Пример 21. Синтез проводили, как в примере 1, но вместо бромбензола использовали орто-бромтолуол (171 мг, 1,0 ммоль). Выход 2-метилбензофенона составил 190 мг (97%).
Пример 22. Синтез проводили, как в примере 1, но вместо бромбензола использовали орто-хлортолуол (126 мг, 1,0 ммоль). Выход 2-метилбензофенона составил 188 мг (96%).
Пример 23. Синтез проводили, как в примере 21, но вместо N,N'-бис(2,6-диизопропилфенил)аценафтенхинондиимина использовали N,N'-бис(циклогексил)аценафтенхинондиимин (18,7 мг, 0,05 ммоль). Выход 2-метилбензофенона составил 94 мг (48%).
Пример 24. Синтез проводили, как в примере 1, но вместо бромбензола использовали 2,6-диметилхлорбензол (141 мг, 1,0 ммоль). Выход 2,6-диметилбензофенона составил 200 мг (95%).
Пример 25. Синтез проводили, как в примере 1, но вместо бромбензола использовали 2,4,6-триметилбромбензол (199 мг, 1,0 ммоль). Выход 2,4,6-триметилбензофенона составил 190 мг (85%).
Пример 26. Синтез проводили, как в примере 1, но вместо бромбензола использовали орто-броманизол (187 мг, 1,0 ммоль). Выход 2-метоксибензофенона составил 150 мг (71%).
Пример 27. Синтез проводили, как в примере 1, но вместо бромбензола использовали 3-бромфторбензол (175 мг, 1,0 ммоль). Выход 3-фторбензофенона составил 134 мг (67%).
Пример 28. Синтез проводили, как в примере 1, но вместо бромбензола использовали 2-бромбифенил (233 мг, 1,0 ммоль). Выход 2-фенилбензофенона составил 217 мг (84%).
Пример 29. Синтез проводили, как в примере 1, но вместо бромбензола использовали 3-бромтрифторметилбензол (225 мг, 1,0 ммоль). Выход 3-трифторметилбензофенона составил 128 мг (51%).
Пример 30. Синтез проводили, как в примере 1, но вместо бромбензола использовали 3-хлортиофен (119 мг, 1,0 ммоль). Выход 3-бензоилтиофена составил 103 мг (55%).
Пример 31. Синтез проводили, как в примере 1, но вместо бензонитрила использовали 2-метилбензонитрил (117 мг, 1,0 ммоль). Выход 2-метилбензофенона составил 176 мг (90%).
Пример 32. Синтез проводили, как в примере 1, но вместо бензонитрила использовали 2-нафтонитрил (153 мг, 1,0 ммоль). Выход 2-нафтилфенилкетона составил 197 мг (85%).
Пример 33. Синтез проводили, как в примере 1, но вместо бензонитрила использовали 2,6-дифторбензонитрил (139 мг, 1,0 ммоль). Выход 2,6-дифторбензофенона составил 153 мг(70%).
Пример 34. Синтез проводили, как в примере 1, но вместо бензонитрила использовали 2,4-дифторбензонитрил (139 мг, 1,0 ммоль). Выход 2,4-дифторбензофенона составил 159 мг (73%).
Пример 35. Синтез проводили, как в примере 1, но вместо бензонитрила и бромбензола использовали 2-(2-бромбензилокси)бензонитрил (288 мг, 1,0 ммоль). При этом протекает внутримолекулярная реакция, приводящая к образованию циклического ароматического кетона. Выход дибензо[b,e]оксепин-11(6H)-она составил 168 мг (80%).
Пример 36, уравнение (4). Синтез проводили, как в примере 34, но вместо 2-(2-бромбензилокси)бензонитрил использовали 2-(2-бромфенокси)бензонитрил (274 мг, 1,0 ммоль). Выход 9H-ксантен-9-она составил 186 мг (95%).
Пример 37. Синтез проводили, как в примере 1, но вместо бензонитрила использовали каприлонитрил (125 мг, 1,0 ммоль). Выход гептилфенилкетона составил 204 мг (84%).
Пример 38. Синтез проводили, как в примере 36, но вместо N,N'-6ис(2,6-диизопропилфенил)аценафтенхинондиимина использовали N-(1-метил-1H-имидазол-2-илметилен)-2,6-диизопропиланилин (13,5 мг, 0,05 ммоль), а реакцию вели в течение 48 ч. Выход гептилфенилкетона составил 98 мг (48%).
Пример 39. Синтез проводили, как в примере 37, но вместо N,N'-6nc(2,6-диизопропилфенил)аценафтенхинондиимина использовали фенантролин (9,0 мг, 0,05 ммоль). Выход гептилфенилкетона составил 63 мг (31%).
Пример 40. Синтез проводили, как в примере 1, но вместо бензонитрила использовали изобутиронитрил (69 мг, 1,0 ммоль). Выход изопропилфенилкетона составил 114 мг (77%).
Новые редокс медиаторы для электролитов фотоэлектрохимических солнечных элементов
Способ визуализации областей деформации, скрытых под поверхностью твердого тела
Способ получения рекомбинантного белка sav-rgd
Магнитный гаситель самостоятельного дугового разряда
Способ получения легированных поли[(r)карбинов] (r=h, алкил, арил)
Способ электрохимического стереоселективного α-гидроксиалкилирования глицина
Полимерные наночастицы состава фермент-поликатион-полианион, содержащие антиоксидантный фермент, для применения в медицине и способ их получения
Способ получения искусственных алмазов
Ферментный биокатализатор для нейтрализации фосфорорганических соединений in vivo
Способ получения наноразмерных порошков лекарственных веществ и устройство для его осуществления
Многоканальный оптоволоконный нейроинтерфейс для мультимодальной микроскопии мозга животных

