Результат интеллектуальной деятельности: Хинуклидиновые эфиры 1-азагетероциклилуксусной кислоты в качестве антимускариновых средств, способ их получения и их лекарственные композиции
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к аналогам хинуклидинового эфира 1-азагетероциклилуксусной кислоты, действующим в качестве антагонистов мускаринового рецептора, к способу их получения, к содержащей их композиции и к их терапевтическому применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Соли четвертичного аммония, действующие в качестве лекарственных средств-антагонистов мускаринового (М) рецептора, применяют в настоящее время в терапии для индукции бронходилатации для лечения респираторных заболеваний. Примеры хорошо известных антагонистов М рецептора представлены ипратропия бромидом и тиотропия бромидом.
Несколько химических классов, действующих в качестве лекарственных средств, являющихся селективными антагонистами мускаринового рецептора М3, разработано для лечения воспалительных или обструктивных заболеваний дыхательных путей, таких как астма и хроническая обструктивная болезнь легких (ХОБЛ).
Производные хинуклидинкарбамата и их применение в качестве антагонистов М3 раскрыты, например, в WO 02/051841, WO 03/053966 и WO 2008/012290.
В WO 2010/015324 раскрыты карбонатные производные и их применение в качестве антагонистов М3.
Спазмолитическая активность 1-этил-3-пиперидинилового эфира возможно замещенной альфа-фенил-1-пиперидин-/1-пирролидин-/4-морфолин-уксусной кислоты по отношению к ацетилхолину раскрыта в US 2952685 и в Bull Soc Chim France 355-359, 1958.
1-Метил-3-пиперидиниловый эфир альфа-фенил-1-пиперидинуксусной кислоты и его аналоги получены и протестированы в качестве потенциальных психотропных лекарственных средств в связи с их психодислептическими свойствами в статье Chim Ther 7, 408-414, 1966.
Соли четвертичного аммония хинуклидиновых эфиров альфа-фенил-альфа-метил-1-пиперидинуксусной кислоты и их аналогов раскрыты в следующих документах: WO 2008/075005, WO 2009/153536 и Bioorg Med Chem Lett (2011), doi:10.1016/j.bmcl.2011.10.002. Данные соединения, имеющие метильную группу вместо гидроксильной группы хорошо известных антагонистов М3 предшествующего уровня техники (тиотропия, гликопирролата, аклидиния бромида), должны обладать более высокой вероятностью связывания с белками плазмы.
Тем не менее, в высокой степени желательно разработать антагонисты рецептора М3 для введения путем ингаляции, способные действовать локально, при этом обладающие высокой эффективностью и большой продолжительностью действия. Данные лекарственные средства сразу после всасывания должны разлагаться до неактивных соединений, лишенных каких-либо системных побочных эффектов, характерных для мускариновых антагонистов.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены аналоги хинуклидинового эфира 1-азагетероциклилуксусной кислоты общей формулы (I) в качестве селективных антагонистов рецептора М3, способных к введению путем ингаляции и действовать локально.
Соединение согласно изобретению способно производить устойчивый бронходилатирующий эффект в легком, но стабильно и быстро преобразовываться в легком в неактивные метаболиты после прохождения в плазму человека. Такие свойства дают значительные преимущества в отношении безопасности.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В частности, изобретение направлено на хинуклидиновый эфир 1-азагетероциклилуксусной кислоты общей формулы (I)
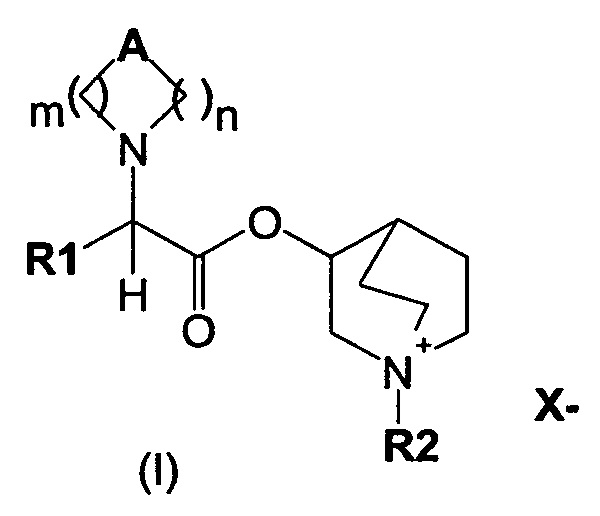
где А может представлять собой простую связь, двойную связь, О, S, SO, SO2, NR3, C(R3)R4, CO, С(O)N(R3), N(R3)С(O)O, SO2N(R3), N(R3)С(O), ОС(O)N(R3), N(R3)SO2, С(R3)=С(R4) и С(R3)-(СН2)-С(R4);
m представляет собой целое число от 1 до 4;
n равно 0 или представляет собой целое число от 1 до 4;
R1 выбран из группы, состоящей из (С1-С10)алкила, арила, (С3-C8)-циклоалкила, гетероарила, арил(С1-С6)алкила и гетероарил(С1-С6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, ОН, оксо (=O), SH, NO2, CN, CON(R3)2, СООН, CO2R3, CF3, (С1-С10)алкоксикарбонила, (C1-С10)алкилсульфанила, (С1-С10)алкилсульфинила, (С1-С10)алкилсульфонила, (С1-С10)алкила, (С1-С10)алкоксила, арилокси и гетероарила;
где X- представляет собой физиологически приемлемый анион;
R2 представляет собой группу формулы (Y)
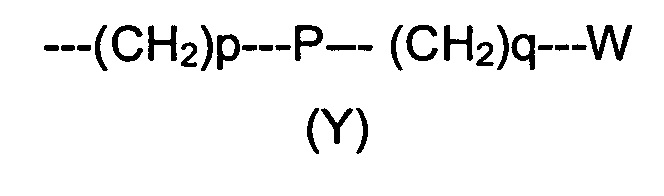
где p равно 0 или представляет собой целое число от 1 до 4;
q равно 0 или представляет собой целое число от 1 до 4;
P отсутствует или выбран из группы, состоящей из О, S, SO, SO2, СО, NR3, СН=СН, N(R3)SO2, N(R3)СОО, N(R3)С(O), SO2N(R3), СО(O)N(R3) и C(O)N(R3);
W выбран из группы, состоящей из Н, (С1-С10)алкила, (С1-С10)алкоксила, (С3-С8)-циклоалкила, арила, гетероарила, (С5-С10)гетероциклоалкила, возможно замещенных одним или более чем одним заместителем выбранным из группы, состоящей из атомов галогена, ОН, оксо (=O), SH, NO2, CN, CON(R3)2, СООН, NH2, NHCOR3, CO2R3, (С1-С10)алкоксикарбонила, (С1-С10)алкилсульфанила, (C1-С10)алкилсульфинила, (С1-С10)алкилсульфонила, (С1-С10)алкила, (C1-С10)алкоксила, (С1-С10)алканоила и арила;
R3 и R4 независимо выбраны из группы, состоящей из Н, атомов галогена, CONH2, (С1-С10)алкила, (С2-С6)алкинила, (С2-С6)алкенила, (С1-С10)алканоила, (С3-С8)циклоалкила, гетероарила и арила, возможно замещенных одним или более чем одним заместителем, выбранный из группы, состоящей из атомов галогена, ОН, оксо (=O), SH, NO2, CN, CONH2, СООН, (C1-С10)алкоксикарбонила, (С1-С10)алкилсульфанила, (С1-С10)алкилсульфинила, (С1-С10)алкилсульфонила, (С1-С10)алкила, (С1-С10)алкоксила и (С3-С7)циклоалкила.
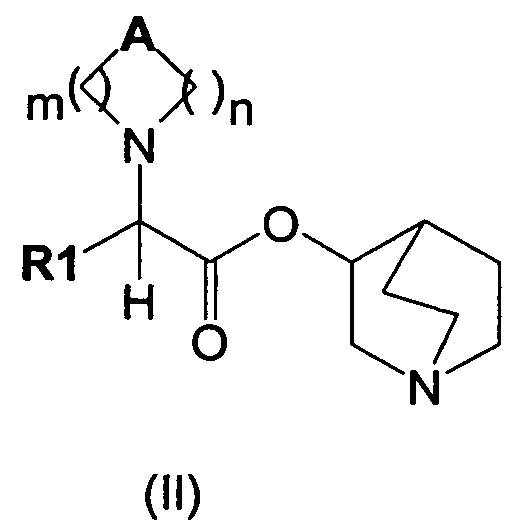
Настоящее изобретение также направлено на соединение общей формулы (II):
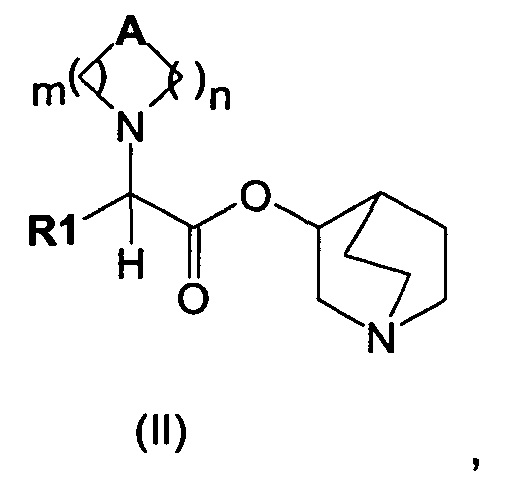
где A, R1, m и n являются такими, как описано выше, и их фармацевтически приемлемую соль.
Изобретение также относится к способу получения соединения формулы (I), как описано на схеме 1, следующим путем:
(а) сочетание соединения формулы (IX), в котором K может представлять собой алкоксигруппу, гидроксигруппу или атом галогена, такой как атом хлора, а А, R1 являются такими, как определено выше, с соединением формулы (X), в котором J представляет собой Н, Na, Li или К, с получением соединений формулы (II),
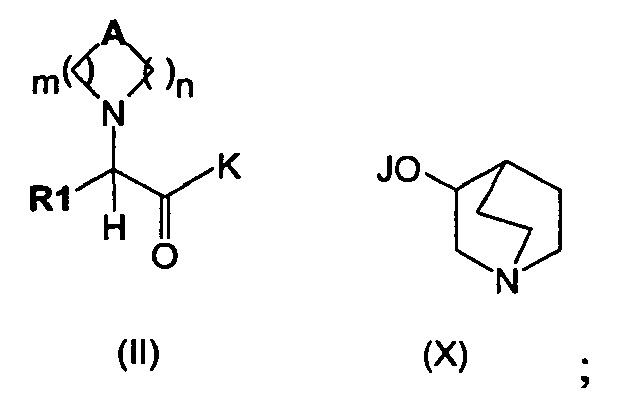
(b) алкилирование соединения формулы (II) алкилирующими агентами общей формулы (XI)
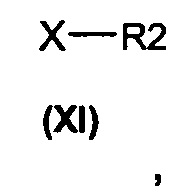
в которых R2 является таким, как определено выше, а Х представляет собой подходящую уходящую группу, выбранную из группы, состоящей из атомов галогена и сложных эфиров сульфокислоты, включающих тозилат, трифлаты или мезилат, с получением соединений общей формулы (I).
В изобретении также предложены фармацевтические композиции соединений общей формулы (I) или общей формулы (II), отдельно, либо в комбинации или в смеси с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом.
В изобретении также предложены соединения общей формулы (I) или (II), применяемые в качестве лекарственного средства.
В следующем аспекте в изобретении предложено применение соединений формулы (I) или общей формулы (II) для получения лекарственного средства для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, либо хронического бронхита, либо хронической обструктивной болезни легких (ХОБЛ).
В изобретении также предложен способ предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, либо хронического бронхита, либо хронической обструктивной болезни легких (ХОБЛ), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы (I) или (II).
В изобретении также предложены фармацевтические препараты соединений общей формулы (I) или общей формулы (II), подходящие для введения путем ингаляции, такие как ингаляционные порошки, управляемые пропеллентом ингаляторы отмеренных доз с распылением сжатым воздухом или ингаляционные препараты без пропеллента.
Изобретение также относится к устройству, которое может представлять собой ингалятор сухого порошка однократной или многократной дозы, ингалятор отмеренной дозы и небулайзер с мягким распылением, содержащее соединения общей формулы (I) или (II).
Изобретение также относится к набору, содержащему фармацевтические композиции соединений общей формулы (I) или (II) отдельно, либо в комбинации или в смеси с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом, и устройство, которое может представлять собой ингалятор сухого порошка однократной или многократной дозы, ингалятор отмеренной дозы и небулайзер с мягким распылением, содержащее соединение общей формулы (I) или (II).
В настоящем описании, если не указано иное, термин "атом галогена" включает атомы фтора, хлора, брома и йода.
Выражение "(С1-С10)алкил" относится к прямоцепочечным или разветвленным алкильным группам, где число атомов углерода составляет от 1 до 10. Примерами этих групп являются следующие группы: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, гексил, гептил, октил, нонил, децил и тому подобное.
Выражение "(С2-С6)алкенил" относится к прямоцепочечным или разветвленным углеродным цепям с одной или более чем одной двойной связью. Примерами этих групп являются следующие группы: этенил, пропенил, бутенил, пентенил, гексенил и тому подобное.
Выражение "(С1-С10)алкоксил" относится к вышеупомянутым алкилокси (например, алкокси) группам. Примерами этих групп являются следующие группы: метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, гексокси и тому подобное.
Выражение "(С1-С10)алканоил" относится к карбонильному остатку, замещенному атомом водорода или прямоцепочечной или разветвленной алкильной группой из атомов углерода в количестве от 1 до 9. Примерами этих групп являются формил, ацетил, пропаноил, бутаноил, изобутаноил и пивалоил.
Выражения "(С1-С10)алкилсульфанил", "(С1-С10)алкилсульфинил" или "(С1-С10)алкилсульфонил" также относятся, соответственно, к следующим группам: алкил-S-, алкил-SO- или алкил-SO2-.
Выражение "(С3-С8)циклоалкил" относится к циклическим неароматическим углеводородным группам, имеющим от 3 до 8 атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и тому подобное.
Выражение "(С5-С10)гетероциклоалкил" относится к циклическим неароматическим системам, имеющим от 5 до 10 кольцевых атомов, в которых по меньшей мере один кольцевой атом представляет собой гетероатом (например, N, NH, S или О).
Выражение "арил" относится к моно-, либо би-, либо трициклическим кольцевым системам, имеющим от 6 до 20 кольцевых атомов, предпочтительно от 6 до 15, и где по меньшей мере одно кольцо является ароматическим.
Выражения "арил(С1-С6)алкил", "гетероарил(С1-С6)алкил" относятся к (C1-С6)алкильным группам, дополнительно замещенным арильными или гетероарильными кольцами.
Выражение "арилокси" относится к вышеописанной арилокси-группе. Пример может представлять собой фенилокси.
Выражение "гетероарил" относится к моно-, би- или трициклическим кольцевым системам, имеющим от 5 до 20 кольцевых атомов, предпочтительно от 5 до 15, в которых по меньшей мере одно кольцо является ароматическим, и в которых по меньшей мере один кольцевой атом представляет собой гетероатом или гетероароматическую группу (например, N, NH, S или О).
Примеры подходящих арильных или гетероарильных моноциклических систем включают, например, тиенильные, фенильные, пирролильные, пиразолильные, имидазолильные, изоксазолильные, оксазолильные, оксадиазолильные, изотиазолильные, тиазолильные, пиридинильные, имидазолидинильные и фуранильные остатки и тому подобное.
Примеры подходящих арильных или гетероарильных бициклических систем включают бензодиоксольные, нафталиновые, дифениленовые, пуриновые, птеридиновые, бензотриазольные, хинолиновые, изохинолиновые, индольные, изоиндольные, бензотиофеновые, дигидробензодиоксиновые, дигидробензодиоксепиновые и бензооксазиновые остатки и тому подобное.
Предпочтительно физиологически приемлемые анионы X- включают анионы, выбранные из хлора, брома, йода, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата, ацетата, цитрата, фумарата, тартрата, оксалата, сукцината, бензоата и пара-толуолсульфоната.
Помимо присутствия аниона X-, в том случае, когда дополнительные основные аминогруппы присутствуют в соединениях формулы (I) или (II), могут присутствовать дополнительные физиологически и/или фармацевтически приемлемые анионы, среди указанных ранее, с образованием соли присоединения кислоты с неорганической или органической кислотой. Также в присутствии кислотных групп, таких как СООН группы, могут также присутствовать соответствующие физиологические катионные соли, например, включающие ион щелочного или щелочноземельного металла или ион аммония.
Подходящая неорганическая кислота для образования соли присоединения кислоты соединения формулы (I) или (II) выбрана из галогеноводородных кислот, таких как фтористоводородная кислота, соляная кислота, бромисто-водородная кислота и йодистоводородная кислота, но также из азотной кислоты, серной кислоты и фосфорной кислоты. Подходящие органические кислоты для образования соли присоединения кислоты соединения формулы (I) или (II) выбраны из следующих кислот: алифатических монокарбоновых кислот, таких как муравьиная кислота, уксусная кислота, трифторуксусная кислота и пропионовая кислота; алифатических гидроксикислот, таких как молочная, лимонная, винная и яблочная кислоты; дикарбоновых кислот, таких как малеиновая или янтарная кислота; ароматических карбоновых кислот, таких как бензойная кислота; ароматических гидроксикислот и сульфоновых кислот.
Первая предпочтительная группа соединений общей формулы (I) или (II) представляет собой группу, где А выбран из О, S, N(R3) или C(R3)R4, R1 выбран из группы, состоящей из арила, арил(С1-С6)алкила и гетероарила, возможно замещенных одним или более чем одним заместителем, выбранным из атомов галогена, (С1-С10)алкила, (С1-С10)алкоксила, арилокси и гетероарила; и R3 является таким, как определено выше.
В пределах этого класса еще более предпочтительны соединения общей формулы (I) или (II), где А представляет собой С(R3)R4, R1 выбран из группы, состоящей из арила и гетероарила, возможно замещенных одним или более чем одним заместителем, выбранным из атомов галогена, (С1-С10)алкила, (C1-С10)алкоксила, арилокси и гетероарила; m и n оба равны 2, и R3 представляет собой группу формулы (Y), где р равно 0, 1 и 3, Р представляет собой СО, q равно 0, W выбран из группы, состоящей из (С1-С10)алкила, арила, гетероарила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, (С1-С10)алкила, (С1-С10)алкоксила, ОН и (С1-С10)алканоила.
В пределах этого класса еще более предпочтительны соединения общей формулы (I), где W выбран из группы, состоящей из фенила, бензотиоксола, тиофенила и тиазолила, возможно замещенных одним или более чем одним атомом галогена, ОН, метилом и ацетилом.
Специалистам в данной области техники очевидно, что соединения общей формулы (I) и (II) содержат асимметрические центры. Следовательно, изобретение также включает оптические стереоизомеры и их смеси.
Активные соединения общей формулы (I) и (II) проявляют по меньшей мере два хиральных центра, которые, соответственно, представлены хинуклидиновым атомом углерода, несущим кислород сложноэфирной группы, и атомом углерода, несущим группу R1. Эти соединения общей формулы (I) и (II) могут быть получены в чистых S-R, R-R, R-S, S-S конфигурациях или в виде смесей диастереоизомеров.
Кроме того, в зависимости от значений R1, R2, R3 и А ясно, что в соединениях формулы (I) и (II) могут присутствовать дополнительные асимметрические центры. Следовательно, изобретение также включает любой из оптических стереоизомеров, диастереоизомеров и их смесей в любом соотношении.
Согласно конкретным воплощениям в настоящем изобретении предложены соединения, приведенные ниже:




Соединения общей формулы (I) и (II) могут быть получены в соответствии с известными способами. Некоторые из способов, которые можно использовать, описаны ниже и представлены на схеме 1.
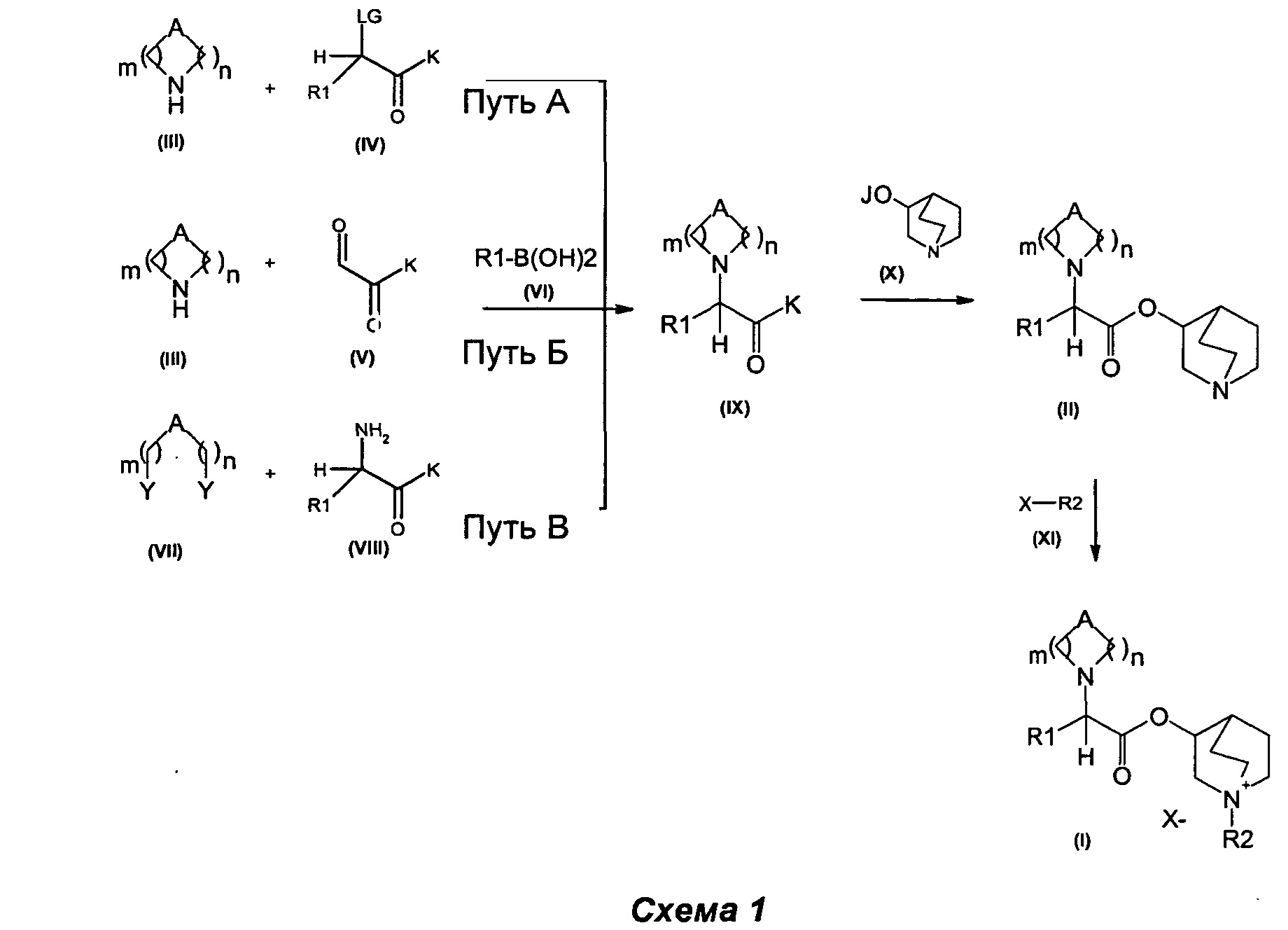
Метод получения соединений Формулы (I) и (II)
Соединения общей формулы (IX) могут быть получены в соответствии со стандартными методами, широко представленными в литературе (например, следуя методу, представленному авторами Kiskinen et al. в статье Tetrahedron, 1983, 39/9, 1627; Haurena et al. в J.O.C., 2010, 75/8, 264; Najer, Bulletin de la Societe Chimique de France, 1958, 1189; Duan et al. Bioorganic and Medicinal Chemistry Letters, 2009, 19/6, 1610). Наиболее предпочтительно соединения общей формулы (IX) могут быть получены в соответствии с тремя различными путями: А, Б и В.
Путь А - Соединения общей формулы (IX) могут быть получены посредством алкилирования реагента общей формулы (III) соединением общей формулы (IV), в котором LG представляет собой подходящую уходящую группу (галогенид, такой как бромид, или сульфоновую сложноэфирную группу, такую как мезилат), и K представляет собой карбоксильную группу, в возможно защищенной форме (в характерном случае включающую карбоксиалкильную сложноэфирную группу (например, K=O(С1-С6)алкил), такую как карбоксиметил (K=ОМе). Данное алкилирование может быть выполнено, следуя одному из стандартных методов, широко представленными в литературе (например, Melloni et al., European Journal of Medicinal Chemistry, 1984, 19/3, 235; Duran et al., Journal of Medicinal Chemistry, 1965, 8, 598; Venkatesan, A.M. et al., Journal of Medicinal Chemistry, 2004, 47/25, 6255; Wlasislaw, В. et al. Synthesis, 1997, 4, 420).
При типовом методе реакцию алкилирования активируют присутствием основания, например амина, выбранного из группы, состоящей из триэтиламина, диизопропилэтиламина, пиридина и 4-диметиламинопиридина, или неорганического основания, такого как карбонат калия или гидрид натрия. Эту реакцию, как правило, проводят в подходящем растворителе (например, в ацетонитриле, тетрагидрофуране (THF), N,N-диметилформамиде (DMF)) в диапазоне температуры от примерно 0°С до примерно 130°С в течение периода времени от примерно 1 часа до примерно 74 часов. Реакцию можно проводить при традиционном нагревании (используя масляную баню) или при микроволновом нагревании. Реакцию можно проводить в открытом сосуде или в запаянной трубке.
Реагенты общей формулы (IV) имеются в продаже или могут быть традиционно получены в соответствии со стандартными методами, широко представленными в литературе. Например, соединения общей формулы (IV), в которых уходящая группа (LG; от англ. leaving group) представляет собой атом галогена, такой как атом брома, могут быть получены путем галогенирования соответствующим образом замещенного сложного эфира фенилуксусной кислоты (например, следуя методу, описанному Epstein, J.W. в статье J. Med. Chem., 1981, 24/5, 481). Альтернативно соединения общей формулы (IV) могут быть получены, начиная с соответствующим образом замещенного производного (IV) миндальной кислоты, используя известные методы (обзор подходящих реакций приведен в книге Larock, L.C., Comprehensive Organic Transformation, Second edition (1999), John Wiley & Son Inc, pg 689-700). Производное миндальной кислоты общей формулы (IV) можно подвергать прямому сочетанию с реагентом общей формулы (III) посредством реакции Мицунобу (Kumara Swamy, K.C. et al. Chem. Rev. 2009, 109, 2551; Powell, N.A. et al. Bioorganic and Medicinal Chemistry 2007, 15/17, 5912). Эту реакцию в характерном случае проводили в присутствии фосфина (например, трифенилфосфина) и азодикарбоксилата (например, диизопропилазодикарбоксилата или диэтилазодикарбоксилата) в подходящем растворителе (таком как дихлорметан (DCM) и THF) и в диапазоне температуры от -10°С до 110°С в течение периода от 1 часа вплоть до 74 часов.
Согласно пути Б соединения общей формулы (IX) могут быть получены посредством реакции Петасиса-Манниха, следуя любому из различных методов, описанных в литературе (например: Petasis N.A., Akritopoulou I., Tetrahedron Lett. 1993, 34, 583; Follmann, M., Synlett, 2005, 6, 1009; Kausik K.N., Tetrahedron Letters, 2005, 46, 2025). При характерном методе эквимолярную смесь амина (III), глиоксиловой кислоты (V) и бороновой кислоты (VI) растворяли в подходящем растворителе (например, в дихлорметане, ацетонитриле) и перемешивали. Обычно данную реакцию проводят в диапазоне температуры от примерно 0°С до примерно 110°С в течение периода времени от примерно 1 часа до примерно 74 часов. Реакцию можно проводить при традиционном нагревании (используя масляную баню) или при микроволновом нагревании. Реакцию можно проводить в открытом сосуде или в запаянной трубке.
Путь В - Соединения общей формулы (IX) могут быть получены, начиная с подходящих производных аминокислот общей формулы (VIII). Эти реагенты могут быть преобразованы в соединения общей формулы (IX), используя известные протоколы (например, Benasutti et al., Tetrahedron Asymmetry, 2006, 17/5, 842; Benson et al., J.O.C., 1988, 53/22, 5335; Cuevas et al., Synlett, 2007, 1, 119; Juarez et al., Tetrahedron Asymmetry, 1997, 8/2, 203). Эти протоколы включают, но не ограничены ими, восстановительное аминирование, образование амида или алкилирование производных аминокислот формулы (VIII) реагентами общей формулы (VII), в которой R1, А, K, m и n являются такими, как определено выше, и Y может представлять собой альдегидную группу (СНО), карбоновую кислоту (СООН), ацильную группу (например, Y=COCl) или подходящую уходящую группу (галогенид, такой как бромид, или сложноэфирную группу сульфоновой кислоты, такую как мезилат).
Затем соединения формулы (IX) могут быть преобразованы в соединения общей формулы (II) путем сочетания с соединениями общей формулы (X). Данное сочетание можно проводить несколькими путями, при которых К может представлять собой алкоксигруппу, гидроксильную группу или атом галогена, такой как атом хлора (обзор подходящих реакций приведен в книге Carey, F.A. and Sundeberg, R.J. Advanced Organic Chemistry, Third Edition (1990), Plenum Press, New York and London, pg 145).
В частности, в случае, когда К представляет собой защищенную гидроксильную группу, такую как алкоксигруппа (например, K=ОМе, OEt или OtBu), сложный эфир (IX) можно либо подвергать взаимодействию с подходящими солями хинуклидин-3-ола, либо подвергать гидролизу с получением соответствующего производного кислоты (IX; K=ОН).
В первом случае соединения формулы (II) получают, подвергая взаимодействию соединения общей формулы (IX), в которых К представляет собой алкоксигруппу (например, K=ОМе), с солями хинуклидин-3-ола (X), в которых J представляет собой Na, Li или К. При характерном методе раствор сложных эфиров (IX) и подходящих солей хинуклидин-3-ола (полученных ранее или образованных in situ) в подходящем растворителе (например, в толуоле, DMF или N-метилпирролидоне (NMP)) нагревают при температуре, находящейся в диапазоне от примерно 80°С до примерно 200°С в течение периода от примерно 1 часа до примерно 74 часов.
Во втором случае гидролиз сложноэфирной группировки в (IX) (например, K=OMe) можно проводить, обрабатывая эти соединения подходящим водным раствором основания, выбранного из группы, состоящей из гидроксида натрия, лития и калия, в подходящих растворителях (например, в тетрагидрофуране, диоксане, воде и т.д.). Реакция протекает при комнатной температуре (KT) в течение периода от 1 часа вплоть до 36 часов. Полученную в результате карбоновую кислоту можно подвергать сочетанию с хинуклидин-3-олом в соответствии с несколькими протоколами.
Первая альтернатива. При типичном методе соединения (II) могут быть получены путем конденсации между спиртом (X) (J=H) и кислотой (IX) (K=ОН) в стандартных условиях амидирования и пептидного сочетания. Например, обработка кислоты (IX) одним или более чем одним эквивалентом имеющегося в продаже агента конденсации, такого как карбодиимид (например, гидрохлорид 1-(3-диметиламино)пропил)-3-этилкарбодиимида (EDC) и тому подобное), например, в присутствии N-гидроксибензотриазола (HOBt), с последующим взаимодействием активированного промежуточного соединения со спиртом (X) приводит в результате к образованию соединений (II). В реакционной смеси может также присутствовать органическое основание, такое как триэтиламин. Активированное промежуточное соединение можно либо выделить, либо образовать предварительно, либо образовать in situ. Подходящие растворители для сочетания включают, но не ограничены ими, галогенуглеродные растворители (например, дихлорметан), тетрагидрофуран, диоксан и ацетонитрил. Реакция протекает в диапазоне температуры от 0°С вплоть до 170°С в течение периода времени в диапазоне от примерно 1 часа вплоть до 72 часов. Эту реакцию можно проводить при традиционном нагревании (используя масляную баню) или при микроволновом нагревании. Реакцию можно проводить в открытом сосуде или в запаянной трубке.
Вторая альтернатива. В случае, где К представляет собой атом галогена, такой как атом хлора, спирт (X) (J=H) подвергают взаимодействию с подходящим ацилгалогенидом (IX), используя известные способы. Эта реакция может быть активирована основанием, таким как триэтиламин, пиридин и 4-диметиламинопиридин, в подходящем растворителе (например, в дихлорметане). Эту реакцию проводят в диапазоне температуры от 0°С до 130°С в течение периода от 1 часа вплоть до 74 часов. Эту реакцию можно проводить при традиционном нагревании (используя масляную баню) или при микроволновом нагревании. Реакцию можно проводить в открытом сосуде или в запаянной трубке.
В некоторых воплощениях настоящего изобретения необходимый ацилгалогенид (IX) может быть легко получен из соответствующей кислоты (IX) (K=ОН). Данную активацию можно осуществлять в соответствии с любым из стандартных методов, представленных в литературе. Например, в результате обработки кислоты (IX) (K=ОН) одним или более чем одним эквивалентом оксалилхлорида в присутствии каталитического количества диметилформамида (DMF) в галогенуглеродном растворителе, таком как дихлорметан, при температуре в диапазоне от 0°С до 35°С получают требуемый ацилхлорид (IX) (K=Cl).
Третья альтернатива. Альтернативно ацилирование спирта (X) (J=H) с получением соединений общей формулы (IX) может быть выполнено, используя методы, преобразующие in situ кислоту (IX) (K=ОН) в соответствующие ацилгалогениды. Например, спирты (X) подвергают взаимодействию с кислотами (IX) (K=ОН) в присутствии трифенилфосфина и галогенуглеродного растворителя, такого как тетрахлорид углерода или дихлорметан, примерно при KT в течение максимального периода времени 16 часов (Lee, J.B. J. Am. Chem. Soc., 1966, 88, 3440).
Четвертая альтернатива. При другом способе для получения соединений по настоящему изобретению кислоту (IX) (K=ОН) можно активировать другими имеющимися в продаже активирующими агентами, такими как гексафторфосфат бромтрипирролидинофосфония (PyBrOP) или карбонилимидазол, в подходящем апротонном растворителе (например, в дихлорметане, тетрагидрофуране), примерно при KT. В результате последующего взаимодействия активированного промежуточного соединения со спиртом (X) получают желаемое соединение формулы (II). Для взаимодействия может также требоваться использование органического основания, такого как диизопропилэтиламин, и обычно протекает примерно при KT.
Пятая альтернатива. При другом способе для получения соединений по настоящему изобретению соединения (II) могут быть эффективно получены путем конденсации между кислотами (IX) (K=ОН) и спиртом (X) (J=H) в характерных условиях Мицунобу (Kumara Swamy, K.C., Chem. Rev. 2009, 109, 2551-2651). Например, кислоты (IX) и спирт (X) подвергают взаимодействию в присутствии фосфина (например, трифенилфосфина) и сложного эфира азодикарбоксилата (например, диэтилазодикарбоксилата или диизопропилазодикарбоксилата) в апротонном растворителе, таком как тетрагидрофуран. В характерном случае эта реакция протекает в диапазоне температуры от 0°С вплоть до 100°С в течение периода времени, находящегося в диапазоне от примерно 30 минут вплоть до 72 часов.
Соединение общей формулы (II), в котором R1, A, m и n являются такими, как определено в данном описании выше, может быть получено либо в виде отдельного диастереоизомера, либо в виде смеси диастереоизомеров. Хинуклидин-3-ол может характеризоваться либо R, либо S конфигурацией. При использовании R-энантиомера соединение (II) может быть получено в S-R конфигурации, в R-R конфигурации или в виде смеси диастереоизомеров (R-R и S-R конфигурации).
При получении смеси диастереоизомеров она может быть преобразована в соединения формулы (I) Схемы 1 или чаще всего могут быть разделены с получением двух отдельных диастереоизомеров, которые, в свою очередь, могут быть преобразованы в соединения формулы (I) Схемы 1. Это разделение может быть выполнено, используя известные методы. Эти методы включают, но не ограничены ими, хроматографическую очистку, очистку препаративной ВЭЖХ (высокоэффективная жидкостная хроматография) и кристаллизацию. Например, два диастереоизомера можно разделить с помощью флэш-хроматографии на силикагеле, элюируя подходящими растворителями или смесью растворителей, например, DCM и метанолом и тому подобным. При другом способе по настоящему изобретению разделение диастереоизомеров может быть получено, используя колонку, наполненную хиральной стационарной фазой, например, колонку Chiralpack AY, либо Chiralcel OD или Chiralcel OZ, и элюируя, например, ацетонитрилом и/или смесями ацетонитрила и спирта. Альтернативно разделение диастереоизомеров может быть легко достигнуто путем кристаллизации из подходящего растворителя (например, этилового эфира) в виде свободного основания или после образования подходящей соли (например, соли (+)-винной кислоты)).
В результате алкилирования соединений общей формулы (II)
алкилирующими агентами общей формулы (XI)
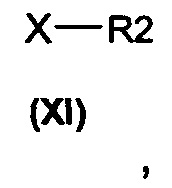
в которых Х представляет собой подходящую уходящую группу, выбранную из группы, состоящей из галогенида (например, атома брома, йода, хлора) и сложного эфира сульфоната (например, тозилата, трифлатов, мезилата) получают соединения общей формулы (I).
Данный вид реакции широко описан в литературе в нескольких различных условиях, например, реакцию можно проводить в чистом виде или в подходящем растворителе, выбранном из группы, состоящей из ацетонитрила, DMF, DMSO и тетрагидрофурана. Эта реакция в характерном случае протекает в диапазоне температур от 0°С вплоть до 170°С в течение периода времени, находящегося в диапазоне от нескольких минут вплоть до 72 часов. Эту реакцию можно проводить при традиционном нагревании (используя масляную баню) или при микроволновом нагревании. Реакцию можно проводить в открытом сосуде или в запаянной трубке.
Соединение общей формулы (I) и (II) на Схеме 1 можно рассматривать либо в качестве конечного продукта, либо в качестве промежуточного соединения для получения других соединений общей формулы (I) и (II). Таким образом, часть группы R1, R2 или А в общей формуле (I) и (II) может претерпевать реакции окисления, восстановления или расщепления (например, для удаления защитной группы) с получением других конечных соединений общей формулы (I) и (II).
В настоящем изобретении также предложена фармацевтическая композиция соединения общей формулы (I) или (II) в смеси с одним или более чем одним фармацевтически приемлемым носителем или эксципиентом, например, описанным в руководстве Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению можно осуществлять в соответствии с потребностями пациента, например, перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, внутригрудинно и путем инфузии), путем ингаляции, ректально, вагинально, местно, локально, трансдермально и путем глазного введения.
Для введения соединений по изобретению можно применять различные твердые пероральные дозируемые формы, включая такие твердые формы, как таблетки, желатиновые капсулы, капсулы, каплетки, гранулы, лепешки и нерасфасованные порошки. Соединения по настоящему изобретению можно вводить отдельно или объединять с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и эксципиентами, известными в данной области техники, включающими, но не ограниченными ими, суспендирующие агенты, солюбилизаторы, буферные агенты, связующие агенты, разрыхлители, консерванты, красители, ароматизаторы, смазывающие агенты и тому подобное. Капсулы, таблетки и гели с замедленным высвобождением также обладают преимуществом при введении соединений по настоящему изобретению.
Для введения соединений по изобретению можно также применять различные жидкие пероральные дозируемые формы, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие дозируемые формы могут также содержать подходящие инертные разбавители, известные в данной области техники, такие как вода, и подходящие эксципиенты, известные в данной области техники, такие как консерванты, увлажняющие агенты, подсластители, ароматизаторы, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по настоящему изобретению можно инъецировать, например, внутривенно, в форме изотонического стерильного раствора. Возможны также другие препараты.
Суппозитории для ректального введения соединений по настоящему изобретению могут быть получены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли.
Препараты для вагинального введения могут находиться в форме препарата в виде крема, геля, пасты, пенки или спрея, содержащего в дополнение к активному ингредиенту такие подходящие носители, которые известны в данной области техники.
Для местного введения фармацевтическая композиция может находиться в форме кремов, мазей, жидких мазей, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для введения на кожу, в глаз, ухо или нос. Местное введение может также включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри.
Для лечения заболеваний дыхательных путей соединения согласно изобретению предпочтительно вводят путем ингаляции.
Ингаляционные препараты включают порошки для ингаляции, аэрозольных ингаляторов отмеренных доз с распылением сжатым воздухом, активируемым пропеллентом, или небулизированных препаратов без пропеллента.
Для введения сухого порошка можно использовать ингаляторы однократной или многократной дозы, известные из предшествующего уровня техники. В этом случае соединение по изобретению в форме порошка можно заполнять в желатиновые, полимерные или другие капсулы, картриджи или блистерные упаковки, либо в резервуар.
К порошкообразному соединению по изобретению можно добавлять разбавитель или носитель, как правило, нетоксичный и химически инертный по отношению к соединению по изобретению, например, лактозу или любую другую добавку, подходящую для улучшения вдыхаемой фракции.
Аэрозольные ингаляторы отмеренных доз с распылением сжатым воздухом, активируемым пропеллентом, содержат соединение по изобретению либо в растворе, либо в диспергируемой форме по меньшей мере в пропеллентном газе, таком как гидрофторалкан. Препараты, активируемые пропеллентом, могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и возможно другие эксципиенты.
Ингаляционные препараты без пропеллента, содержащие соединение по изобретению, могут находиться в форме растворов или суспензий в водной, спиртовой или водно-спиртовой среде, и их можно доставлять с помощью струйных или ультразвуковых небулайзеров или с помощью небулайзеров мягкого распыления.
Соединение по изобретению можно вводить в виде единственного активного агента или в комбинации с одним или более чем одним фармацевтически активным ингредиентом, применяемым в настоящее время при лечении обструктивных воспалительных заболеваний дыхательных путей, выбранным из классов бета-2-агонистов, кортикостероидов и антихолинергических или антимускариновых агентов.
Дозировки соединений по настоящему изобретению зависят от ряда факторов, включающих конкретное заболевание, подлежащее лечению, тяжесть симптомов, путь введения, частоту интервалов дозирования, конкретное применяемое соединение, эффективность, токсикологический профиль и фармацевтический профиль соединения.
Предпочтительно соединения формулы (I) и (II) можно вводить, например, в дозе, составляющей от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки.
При введении соединений формулы (I) и (II) путем ингаляции их предпочтительно дают в дозе, составляющей от 0,001 до 500 мг/сутки, предпочтительно от 0,1 до 200 мг/сутки.
Соединения формулы (I) и (II) можно вводить для предупреждения и/или лечения любого заболевания, где активны антагонисты М3. Это заболевание включает заболевания, в которые вовлечено воспаление, такие как астма и ХОБЛ, острый ринит; заболевания, в которые вовлечен желудочно-кишечный тракт, такие как пептическая язва; заболевания, в которые вовлечена сердечно-сосудистая система, такие как острый инфаркт миокарда; заболевания, в которые вовлечены мочеполовые пути, такие как почечная колика; в качестве антихоли нэстеразного средства и при отравлении грибами; применения при анестезии; применения в офтальмологии.
Эти заболевания также включают неврологические и психиатрические расстройства, такие как паркинсонизм и укачивание.
Предпочтительно соединения формулы (I) и (II) можно применять для предупреждения и/или лечения респираторных заболеваний, такие как состояния астмы и ХОБЛ от легкой до острой тяжелой степени.
Другие респираторные заболевания включают следующие заболевания: бронхит, бронхиолит, бронхоэктаз, острый ринофарингит, острый и хронический синусит, гайморит, фарингит, тонзиллит, ларингит, трахеит, эпиглоттит, круп, хроническое заболевание миндалин и аденоидов, гипертрофия миндалин и аденоидов, перитонзиллярный абсцесс, ринит, абсцесс или язву носа, пневмонию, вирусную и бактериальную пневмонию, бронхопневмонию, грипп, экзогенно-аллергический альвеолит, пневмокониоз шахтеров, асбестоз, пневмокониоз, пневмопатию, респираторные состояния вследствие химических паров, пары и другие экзогенные агенты, эмфизему, плеврит, пневмоторакс, абсцесс легкого и средостения, застой и гипостаз легких, фиброз легких после воспаления, другую альвеолярную и париетоальвеолярную пневмопатию, идиопатический фиброзирующий альвеолит, синдром Хаммена-Рича, ателектаз, респираторный дистресс-синдром взрослых (РДСВ), острую дыхательную недостаточность, медиастинит.
Теперь настоящее изобретение дополнительно описано приведенными ниже примерами.
I - промежуточные соединения
С - соединения
ПРИМЕР 1
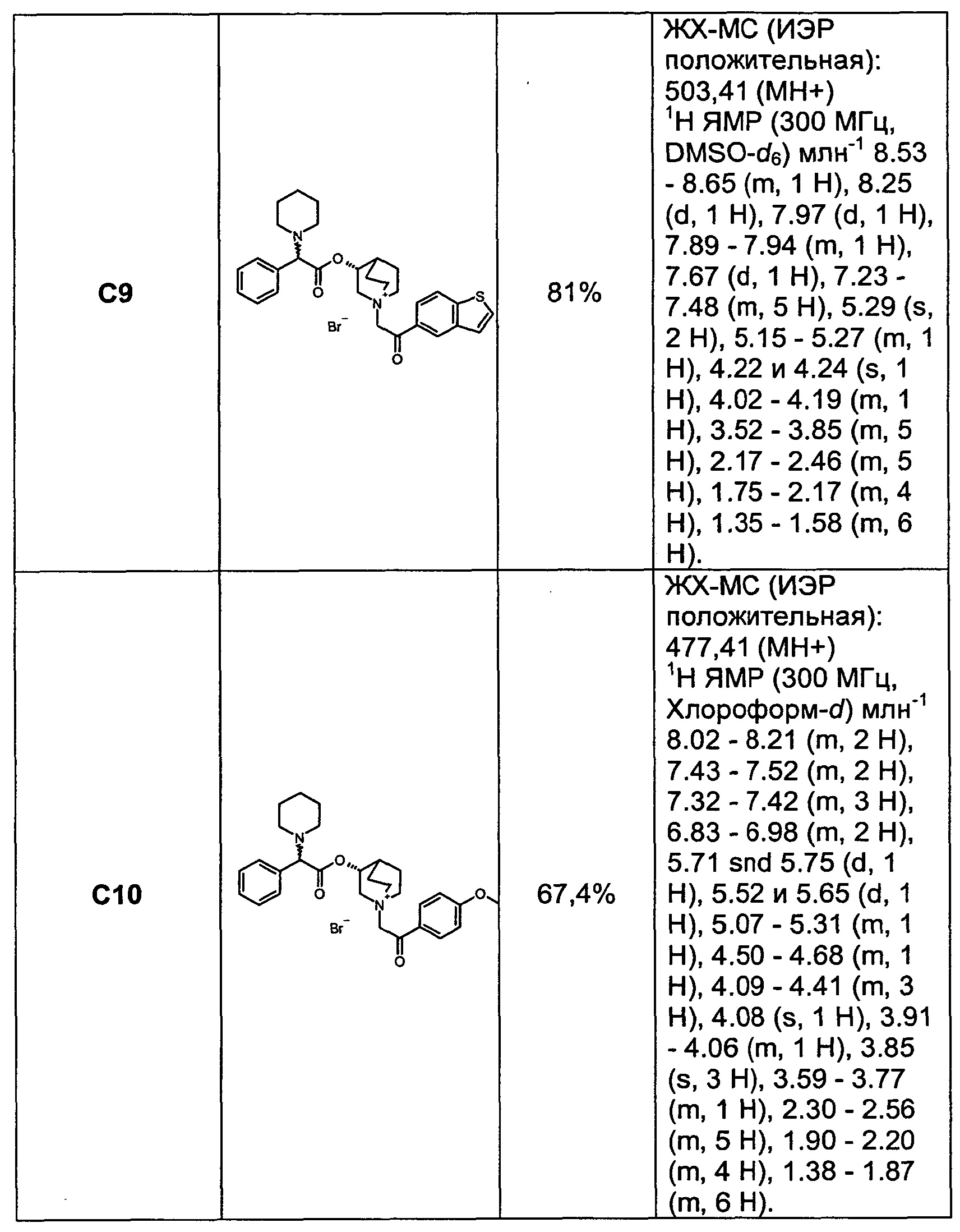
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (С4)
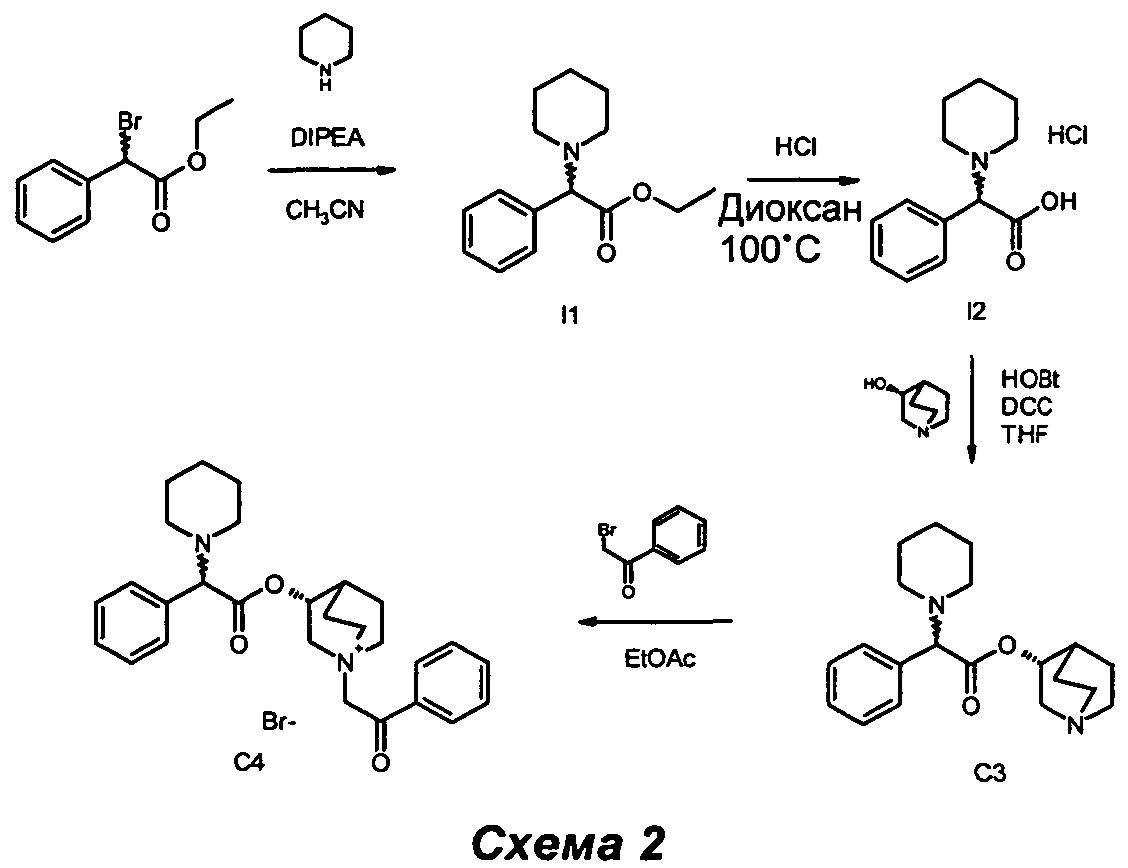
Получение этил-2-фенил-2-(пиперидин-1-ил)ацетата (I1)
Диизопропилэтиламин (DIPEA) (0,86 мл, 4,94 ммоль) и пиперидин (0,49 мл, 4,94 ммоль) последовательно добавляли к раствору этил-2-бром-2-фенилацетата (0,72 мл, 4,11 ммоль) в ацетонитриле (13 мл). Бледно-желтый раствор перемешивали при комнатной температуре, проводя мониторинг с помощью ТСХ (соотношение петролейный эфир/EtOAc равно 9/1). После 1,5 часов обнаружили полное преобразование в желаемый продукт. Растворитель выпаривали, и остаток (бледно-желтое твердое вещество) растирали с Et2O (30 мл). Твердое вещество отфильтровывали, и эфирный раствор выпаривали до сухого состояния. Остаток очищали флэш-хроматографией (петролейный эфир/EtOAc равно 95/5), собирая этил 2-фенил-2-(пиперидин-1-ил)ацетат (990 мг, 97% выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.03-7.54 (m, 5H), 4.05 (s, 1H), 3.93-4.20 (m, 2H), 2.23-2.45 (m, 4H), 1.29-1.59 (m, 6H), 1.13 (t, 3H).
Получение 2-фенил-2-(пиперидин-1-ил)уксусной кислоты гидрохлорида (I2)
Этил-2-фенил-2-(пиперидин-1-ил)ацетат (985 мг, 3,98 ммоль) растворяли в диоксане (33 мл) и медленно добавляли 37%-й HCl (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 48 часов, а затем ее выпаривали при пониженном давлении. Остаток растирали с ацетонитрилом (15 мл), и твердое вещество собирали фильтрованием с получением гидрохлорида 2-фенил-2-(пиперидин-1-ил)уксусной кислоты (876 мг, 86% выход) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 10.57 (br. s., 1H), 7.33-7.67 (m, 5H), 5.27 (s, 1H), 2.84-3.21 (m, 4H), 1.64-1.98 (m, 4H), 1.31-1.64 (m, 2H).
Получение (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (С3)
Гидрохлорид 2-фенил-2-(пиперидин-1-ил)уксусной кислоты (0,87 г, 3,40 ммоль) суспендировали в сухом THF (34,0 мл) и, перемешивая при комнатной температуре, последовательно добавляли дициклогексилкарбодиимид (DCC) (1,40 г, 6,80 ммоль), гидроксибензотриазол (НОВТ) (1,04 г, 6,80 ммоль) и (R)-хинуклидин-3-ол (1,30 г, 10,21 ммоль). Белую суспензию перемешивали при той же температуре в течение ночи (сверхэффективная жидкостная хроматография с масс-спектрометрией (СЭЖХ-МС): полное преобразование). THF выпаривали, и остаток растворяли EtOAc (30 мл) и промывали водой (20 мл), а затем насыщенным раствором NaHCO3 (20 мл). Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно от 98/2/0,2 до 97/3/0,3) с получением (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (40 мг, 40% выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.26-7.44 (m, 5H) 4.64-4.77 (m, 1H) 4.07 (s, 1H) 2.97-3.15 (m, 1H) 2.55-2.71 (m, 3H) 2.40-2.48 (m, 2H) 2.31-2.40 (m, 4H) 1.71-1.95 (m, 1H) 1.33-1.63 (m, 9H) 1.18-1.32 (m, 1H).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (С4)
2-Бром-1-фенилэтанон (91,0 мг, 0,46 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (150 мг, 0,46 ммоль) в EtOAc (10 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Осадок собирали фильтрованием с аспирацией с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (191,7 г, 80% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.90-8.06 (m, 2H), 7.70-7.83 (m, 1H), 7.51-7.69 (m, 2H), 7.23-7.51 (m, 5H), 5.21 и 5.22 (s, 1H), 5.04-5.36 (m, 2H), 4.21 и 4.23 (s, 1H), 4.05-4.19 (m, 1H), 3.47-3.83 (m, 5H), 2.31-2.45 (m, 4H), 2.17-2.31 и 2.30-2.38 (m, 1H), 1.97-2.15 (m, 2H), 1.68-1.97 (m, 2H), 1.46-1.59 (m, 4H),1.33-1.46 (m, 2H).
13С ЯМР (75 МГц, DMSO-d6) млн-1 191.59 (s, 1С), 171.05 (s, 1С), 136.36 (s, 1С), 135.17 (s, 1С), 134.93 (s, 1С), 129.45 (s, 2С), 128.98 (s, 4С), 128.63 и 128.69 (s, 1С), 128.49 (s, 2С), 73.48 и 73.66 (s, 1С), 68.10 и 68.25 (s, 1С), 65.59 (s, 1С), 60.48 и 60.63 (br. s., 1С), 55.22 (s, 1С), 54.96 (s, 1С), 51.76 и 51.86 (s, 2С), 26.06 (s, 2С), 24.44 (s, 1С), 23.70 и 23.82 (s, 1С), 20.79 (s, 1С), 18.11 и 18.27(s, 1С).
СЭЖХ-МС (ионизация электрораспылением (ИЭР) положительная): 447,13(М+).
ПРИМЕР 2
Получение (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (С5)
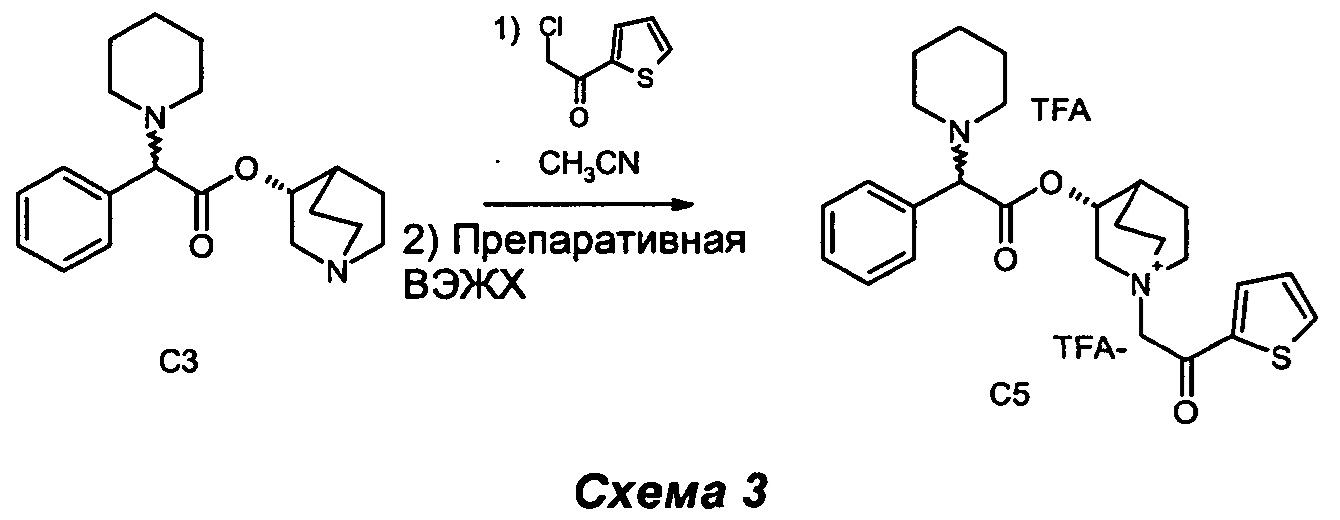
2-Хлор-1-(тиофен-2-ил)этанон (32 мг, 0,22 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (60 мг, 0,18 ммоль) в ацетонитриле (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи (СЭЖХ-МС: полное преобразование). Сырой продукт очищали препаративной высокоэффективной жидкостной хроматографией с масс-спектрометрией (ВЭЖХ-МС) с получением (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (43,3 мг, 35% выход) в виде бесцветного масла.
1H ЯМР (400 МГц, DMSO-d6) млн-1 8.15-8.25 (m, 1H), 7.99-8.09 (m, 1H), 7.46-7.66 (m, 5H), 7.27-7.41 (m, 1H), 5.28-5.41 (m, 1H), 4.92-5.12 (m, 3H), 4.00-4.23 (m, 1H), 3.20-3.92 (m, 5H), 2.70-3.14 (m, 4H), 1.52-2.46 (m, 11H);
СЭЖХ-МС (ИЭР положительная) 453,25 (М+).
ПРИМЕР 3
Получение (3R)-1-(2-(4-хлорфенил)-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (С6)
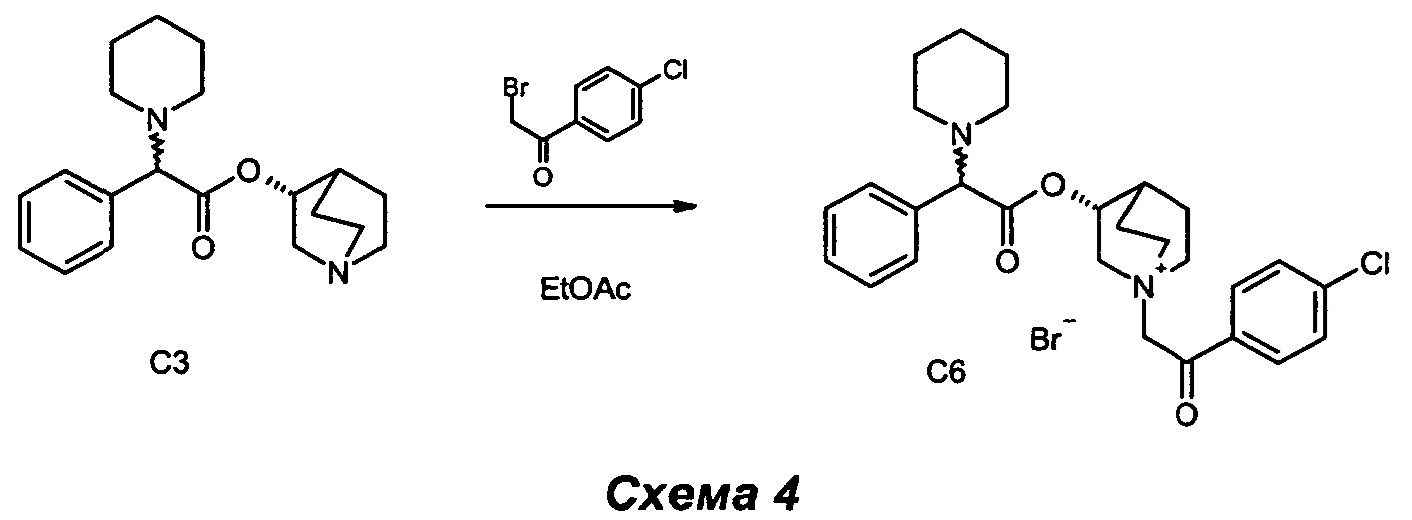
2-Бром-1-(4-хлорфенил)этанон (49,8 мг, 0,21 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (70 мг, 0,21 ммоль) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи (СЭЖХ-МС: полное преобразование). Добавляли Et2O (1 мл), и белый осадок собирали фильтрованием с аспирацией и высушивали в вакууме с получением (3R)-1-(2-(4-хлорфенил)-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (93,5 мг, 78% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.85-8.06 (m, 2H), 7.62-7.77 (m, 2H), 7.29-7.52 (m, 5H), 5.22 (d, 1H), 5.17 и 5.18 (s, 2H), 4.20 и 4.23 (s, 1H), 4.05-4.18 (m, 1H), 3.51-3.81 (m, 5H), 2.19-2.47 (m, 5H), 1.69-2.15 (m, 4H), 1.31-1.63(m, 6H).
СЭЖХ-МС (ИЭР положительная) 481,31 (М+).
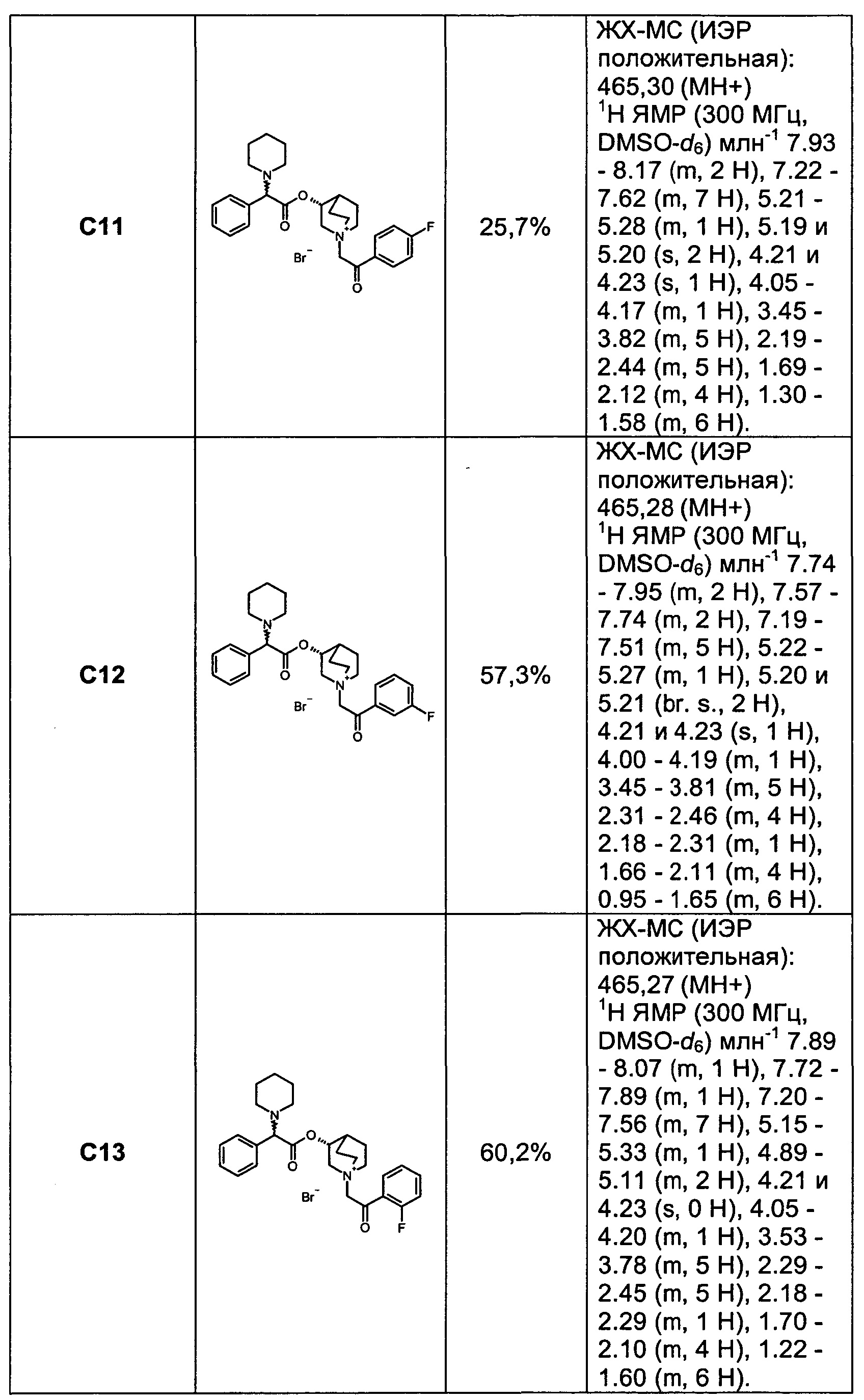
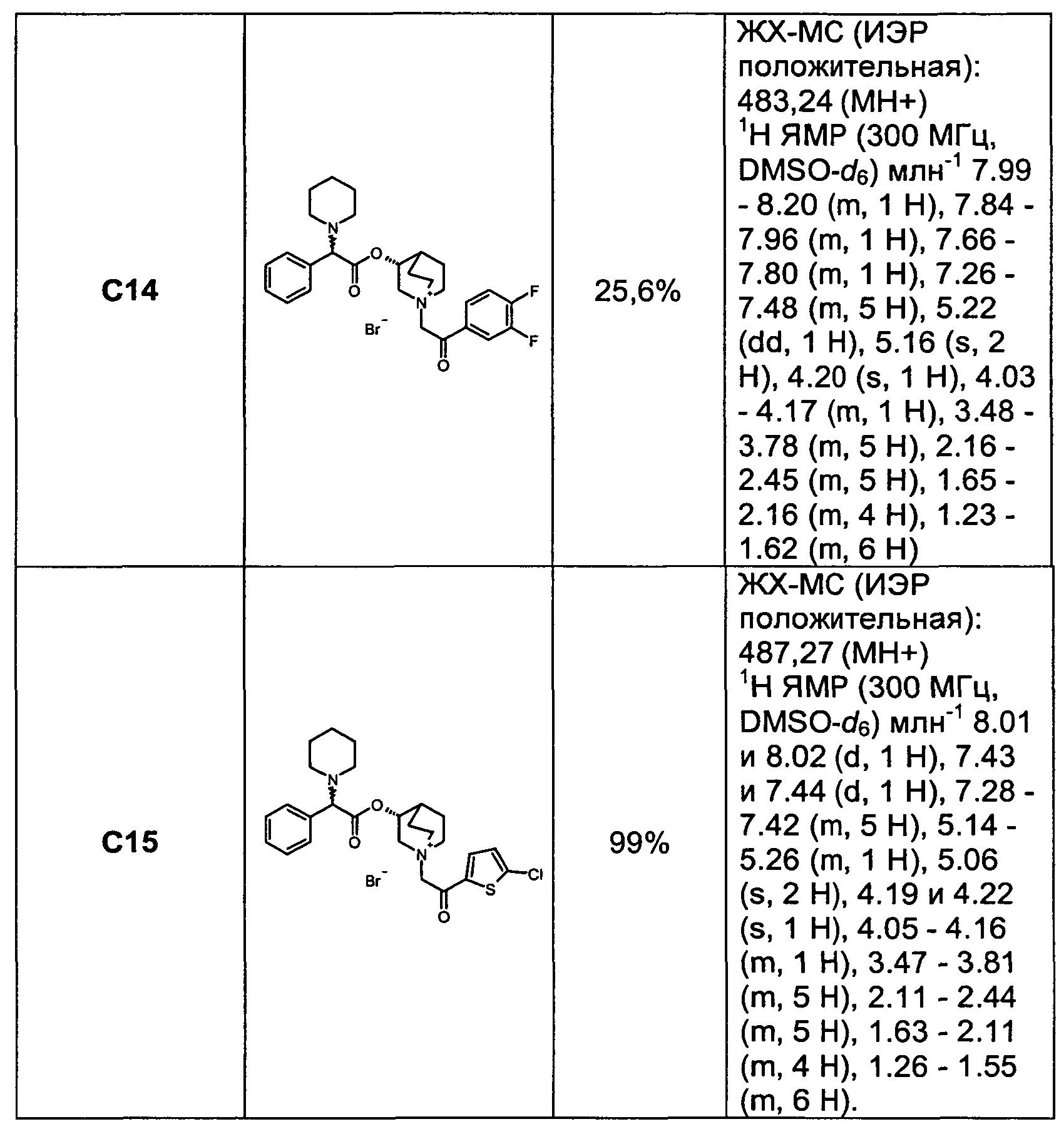
Соединения, перечисленные в таблице 1, были получены, как описано выше для С6, путем алкилирования С3 подходящим имеющимся в продаже алкилбромидом.
ПРИМЕР 4
Получение (3R)-1-(2-трет-бутокси-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикпо[2.2.2]октанбромида (С16)
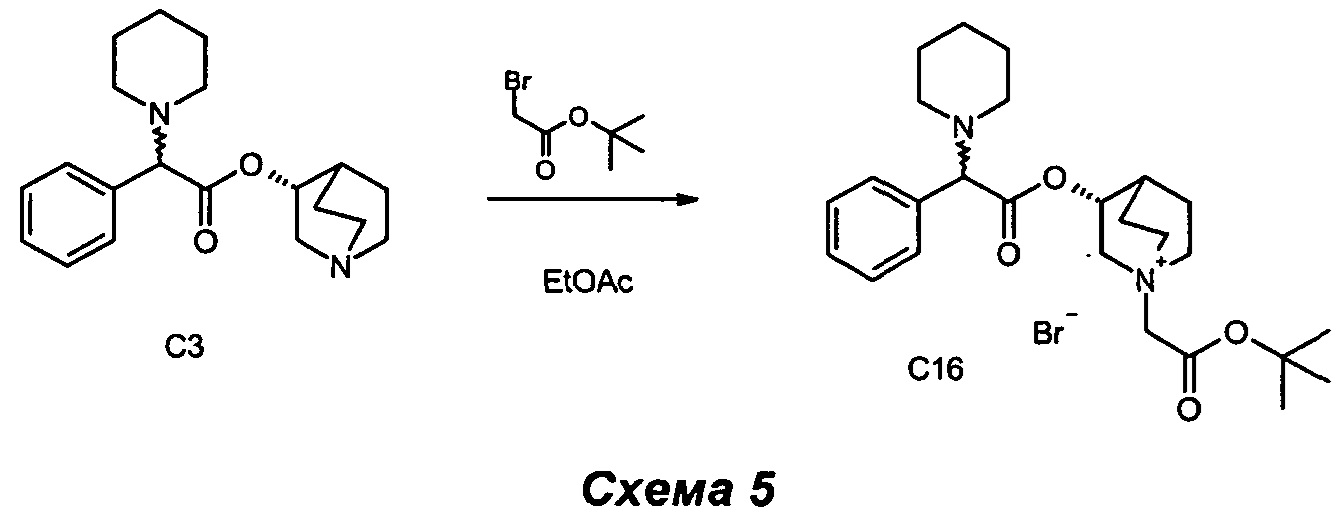
(R)-Хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетат (120 мг, 0,36 ммоль) растворяли в этилацетате (3,65 мл) и добавляли 2-бром-1-(тиазол-2-ил)этанон (83 мг, 0,42 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/MeOH равно от 95/5 до 9/1) с получением (3R)-1-(2-оксо-2-(тиазол-2-ил)этил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (125 мг, 0,234 ммоль, 64,0% выход) в виде светло-коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.19-7.53 (m, 5H), 5.05-5.25 (m, 1H), 4.25 и 4.27 (s, 2H), 4.17 и 4.20 (s, 1H), 3.90-4.12 (m, 1H), 3.39-3.72 (m, 5H), 2.15-2.45 (m, 5H), 1.65-2.08 (m, 4H), 1.47 (s, 9H), 1.35-1.59 (m, 6H);
СЭЖХ-МС (ИЭР положительная) 443,05 (М+).
Соединения, перечисленные в таблице 2, были получены, как описано выше для С16, путем алкилирования С3 подходящим имеющимся в продаже алкилбромидом.
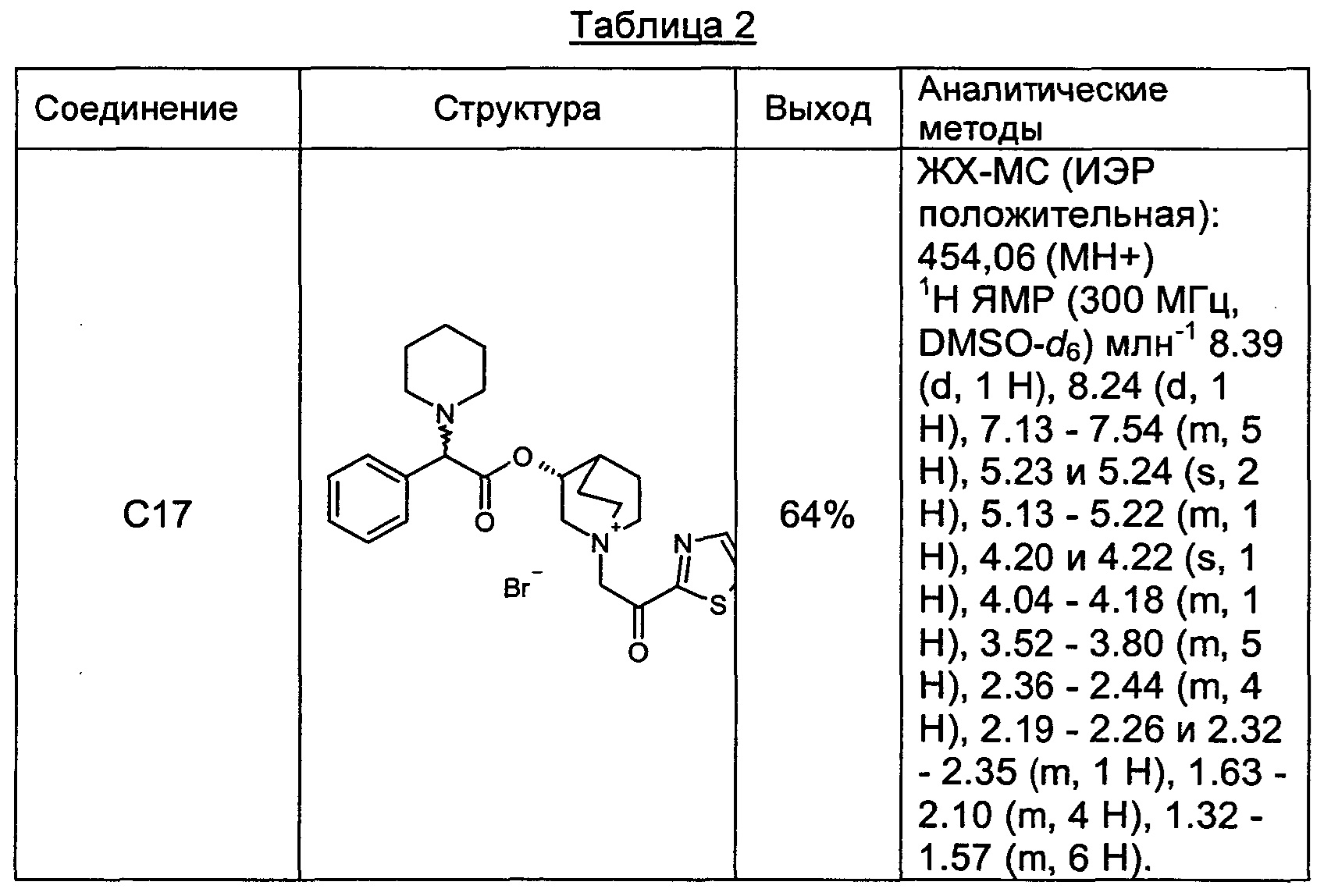
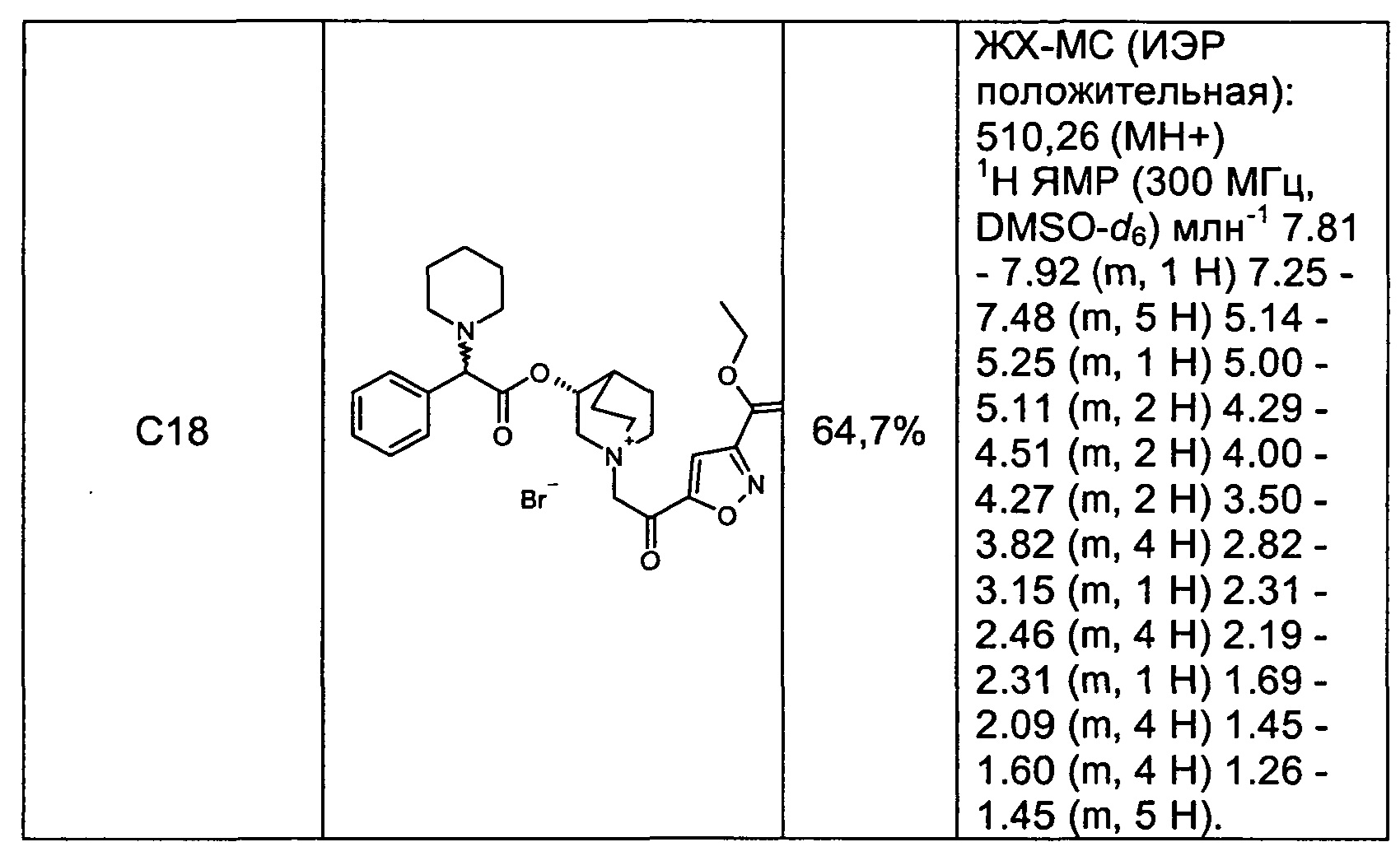
ПРИМЕР 5
Получение (3R)-1-(2-оксо-2-(тиофен-3-ил)этил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (С19).
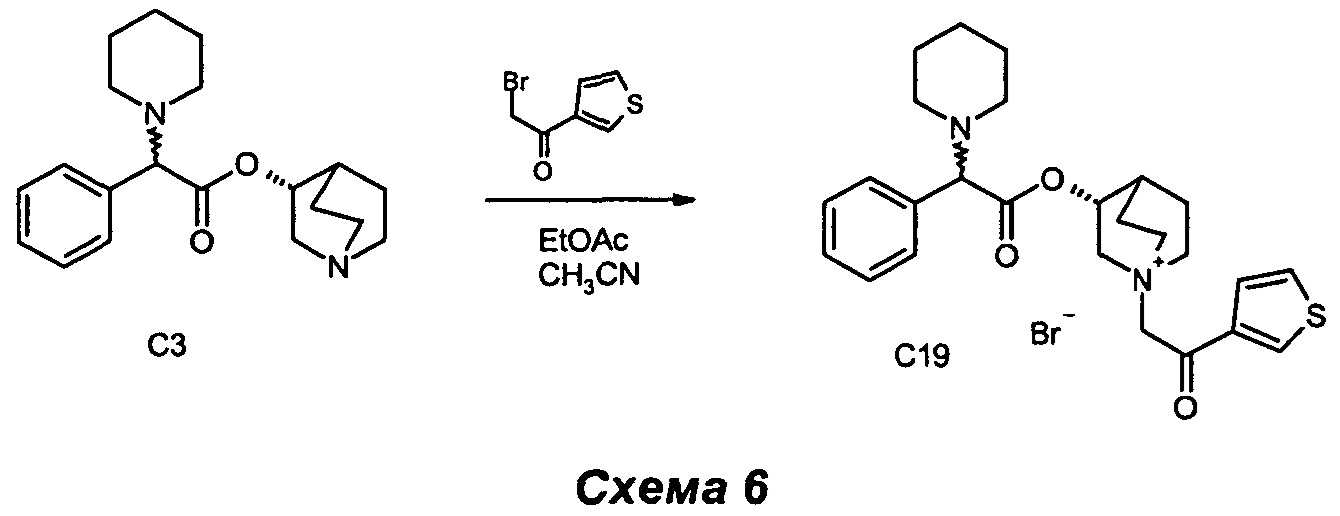
2-Бром-1-(тиофен-3-ил)этанон (54,9 мг, 0,27 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (80 мг, 0,24 ммоль) в этилацетате (1,2 мл) и ацетонитриле (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч (СЭЖХ-МС: полное преобразование). Растворители выпаривали, и остаток очищали сначала флэш-хроматографией (соотношение DCM/MeOH равно 92/8), а затем растиранием с Et2O с получением соединения (3R)-1-(2-оксо-2-(тиофен-3-ил)этил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (96 мг, 73,9% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.64 (dd, 1H) 7.74 (ddd, 1H) 7.56 (dd, 1H) 7.28-7.48 (m, 5H) 5.15-5.26 (m, 1H) 5.09 (s, 2H) 4.05-4.26 (m, 2H) 3.49-3.79 (m, 5H) 2.14-2.46 (m, 5H) 1.69-2.09 (m, 4H) 1.29-1.59 (m, 6H).
СЭЖХ-МС (ИЭР положительная) 453,32 (М+).
ПРИМЕР 6
Получение (3R)-1-((5-фенил-1,2,4-оксадиазол-3-ил)метил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С21)
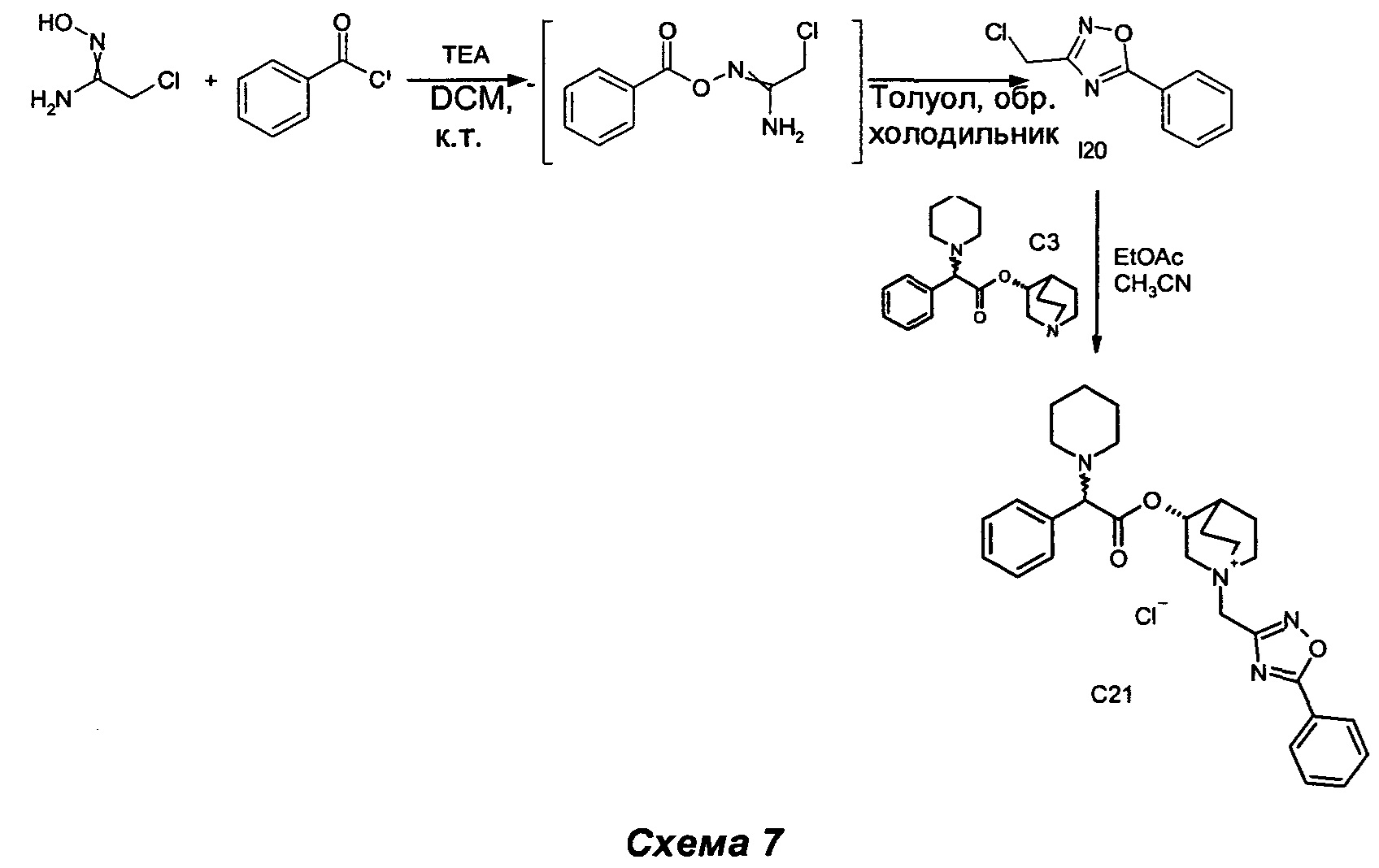
Получение 3-(хлорметил)-5-фенил-1,2,4-оксадиазола (I20)
Бензоилхлорид (1,60 мл, 13,8 ммоль) добавляли к суспензии 2-хлор-N'-гидроксиацетимидамида (1,0 г, 9,21 ммоль) в DCM (25 мл), перемешивая при комнатной температуре. Через 30 мин к белой суспензии добавляли триэтиламин (TEA) (1,41 мл, 10,1 ммоль), и смесь перемешивали дополнительно в течение 30 мин (СЭЖХ-МС: полное преобразование). Раствор разбавляли DCM (20 мл) и добавляли воду (30 мл). Водную фазу экстрагировали три раза DCM (15 мл × 3), а затем объединенные органические фазы высушивали (Na2SO4) и выпаривали. Сырой продукт суспендировали в толуоле (25 мл), и смесь нагревали до образования флегмы в течение 6 ч (СЭЖХ-МС: полное преобразование). Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 95/5) с получением 3-(хлорметил)-5-фенил-1,2,4-оксадиазола (803 мг, 44,8% выход) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.06-8.20 (m, 2H), 7.69-7.79 (m, 1H), 7.57-7.69 (m, 2H), 4.96 (s, 2H).
Получение (3R)-1-((5-фенил-1,2,4-оксадиазол-3-ил)метил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С21)
К раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (100 мг, 0,30 ммоль) в EtOAc (2 мл) и ацетонитрила (1 мл) добавляли 3-(хлорметил)-5-фенил-1,2,4-оксадиазол (71,1 мг, 0,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 ч, а затем растворители выпаривали. Остаток растирали с EtOAc (10 мл), и твердое вещество собирали фильтрованием с аспирацией с получением соединения, указанного в заголовке (113 мг, выход 71%), в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.05-8.28 (m, 2H), 7.74-7.91 (m, 1H), 7.61-7.74 (m, 2H), 7.17-7.51 (m, 5H), 5.03-5.21 (m, 1H), 4.89 (s, 2H), 4.17 и 4.21 (s, 1H), 4.02-4.16 (m, 1H), 3.34-3.87 (m, 5H), 2.13-2.43 (m, 5H), 1.68-2.04 (m, 4H), 1.28-1.60 (m, 6H).
СЭЖХ-МС (ИЭР положительная) 487,11 (М+).
ПРИМЕР 7
Получение (3R)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-((2-фенилоксазол-4-ил)метил)-1-азониабицикло[2.2.2]октанхлорида (С23)
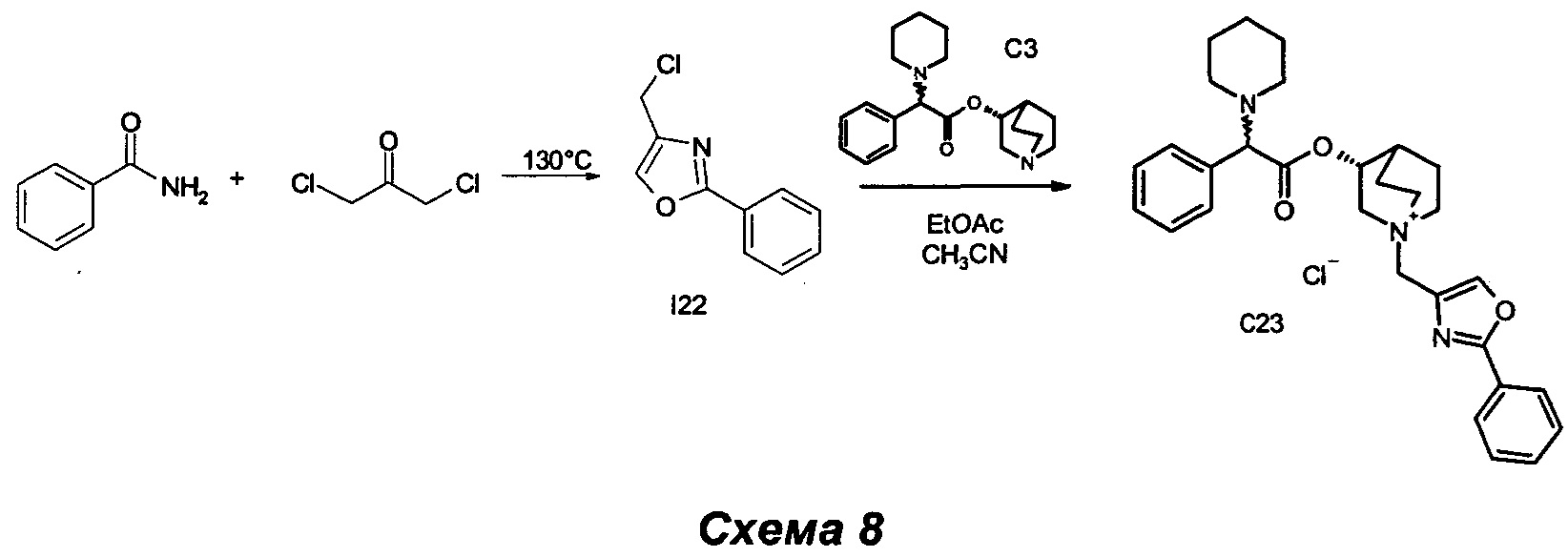
Получение 4-(хлорметил)-2-фенилоксазола (I22)
Смесь бензамида (0,80 г, 6,60 ммоль) и 1,3-дихлорпропан-2-она (1,01 г, 7,92 ммоль) нагревали до 130°С в течение 1 ч в атмосфере азота. Сырой продукт очищали кристаллизацией из ацетонитрила (25 мл): суспензию нагревали до образования флегмы и получали коричневый раствор с белым нерастворимым твердым веществом. Этот раствор фильтровали и охлаждали его до комнатной температуры. Образовался осадок, и суспензию фильтровали на воронке Бюхнера, промывая ацетонитрилом (8 мл). Твердое вещество удаляли с фильтра, растворяли в EtOAc (20 мл) и промывали 1 н. NaHCO3 (15 мл). Органическую фазу высушивали (Na2SO4) и выпаривали с получением 4-(хлорметил)-2-фенилоксазола (315 мг, выход 25%) в виде светло-коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.27 (s, 1Н), 7.89-8.09 (m, 2Н), 7.39-7.68 (m, 3Н), 4.74 (s, 2Н).
Получение (3R)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-((2-фенилоксазол-4-ил)метил)-1-азониабицикло[2.2.2]октанхлорида (С23)
К раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (100 мг, 0,30 ммоль) в EtOAc (2 мл) и ацетонитрила (1 мл) добавляли 4-(хлорметил)-2-фенилоксазол (70,7 мг, 0,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 ч, а затем растворители выпаривали. В результате очистки флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с последующим растиранием с Et2O получили (3R)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-((2-фенилоксазол-4-ил)метил)-1-азониабицикло[2.2.2]октанхлорид (55 мг, 34,6% выход) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.46 и 8.47 (s, 1Н), 7.95-8.09 (m, 2Н), 7.52-7.67 (m, 3Н), 7.21-7.40 (m, 5Н), 4.97-5.26 (m, 1Н), 4.43-4.74 (m, 2Н), 4.15 и 4.17 (s, 1Н), 3.88-4.01 (m, 1Н), 3.13-3.72 (m, 5Н), 2.24-2.42 (m, 4Н), 2.12-2.21 и 2.26-2.36 (m, 1Н), 1.68-2.02 (m, 4Н), 1.28-1.55 (m, 6Н).
СЭЖХ-МС (ИЭР положительная) 486,27 (М+).
ПРИМЕР 8
Получение (3R)-1-(2-(изоксазол-3-иламино)-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорид (С25)
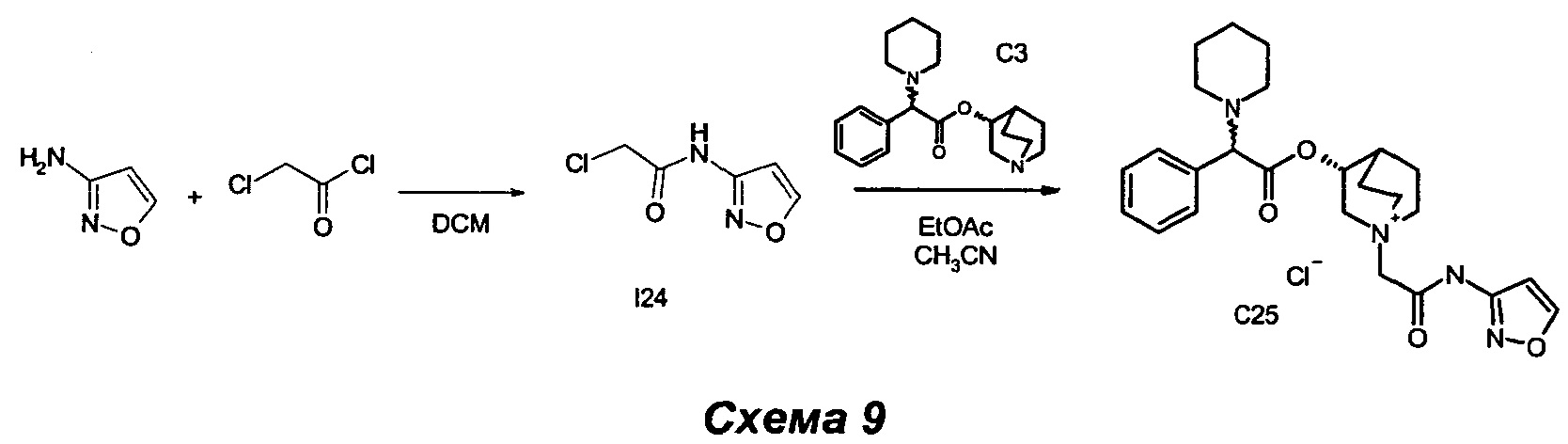
Получение 2-хлор-N-(изоксазол-3-ил)ацетамида (I24)
2-Хлорацетилхлорид (0,21 мл, 2,66 ммоль) растворяли в DCM (10 мл), и раствор охлаждали до 0°C с помощью ледяной бани. Добавляли по каплям изоксазол-3-амин (0,41 мл, 5,58 ммоль), и получали белую суспензию. Смесь перемешивали при комнатной температуре в течение 2 часов, затем суспензию выпаривали, и остаток очищали флэш-хроматографией (соотношение эфир/EtOAc равно от 8/2 до 7/3) с получением 2-хлор-N-(изоксазол-3-ил)ацетамида (367 мг, 86% выход) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 11.38 (br. s., 1H), 8.82 (d, 1H), 6.91 (d, 1H), 4.32 (s, 2H).
(3R)-1-(2-(изоксазол-3-иламино)-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорид (С25)
К раствору (R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетата (100 мг, 0,30 ммоль) в EtOAc (2 мл) и ацетонитрила (1 мл) добавляли 2-хлор-N-(изоксазол-3-ил)ацетамид (58,7 мг, 0,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 ч, а затем растворители выпаривали.
В результате очистки флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с последующим растиранием с Et2O получили соединение, указанное в заголовке (55 мг, 36,9% выход), в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 11.86 (br. s., 1H) 8.88 (d, 1H) 7.18-7.55 (m, 5H) 6.89 (d, 1H) 5.03-5.31 (m, 1H) 4.38 (d, 2H) 4.19 (s, 1H) 4.08-4.24 (m, 1H) 3.44-3.83 (m, 5H) 2.30-2.46 (m, 4H) 2.13-2.28 (m, 1H) 1.69-2.08 (m, 4H) 1.45-1.61 (m, 4H) 1.40 (m, 2H).
СЭЖХ-МС (ИЭР положительная) 453,15 (М+).
ПРИМЕР 9
Получение (3R)-1-(4-фторфенетил)-3-(2-фенил-2-(пиперидиний-1-ил)ацетокси)-1-азониабицикло[2.2.2]октан 2,2,2-трифторацетата (С26)
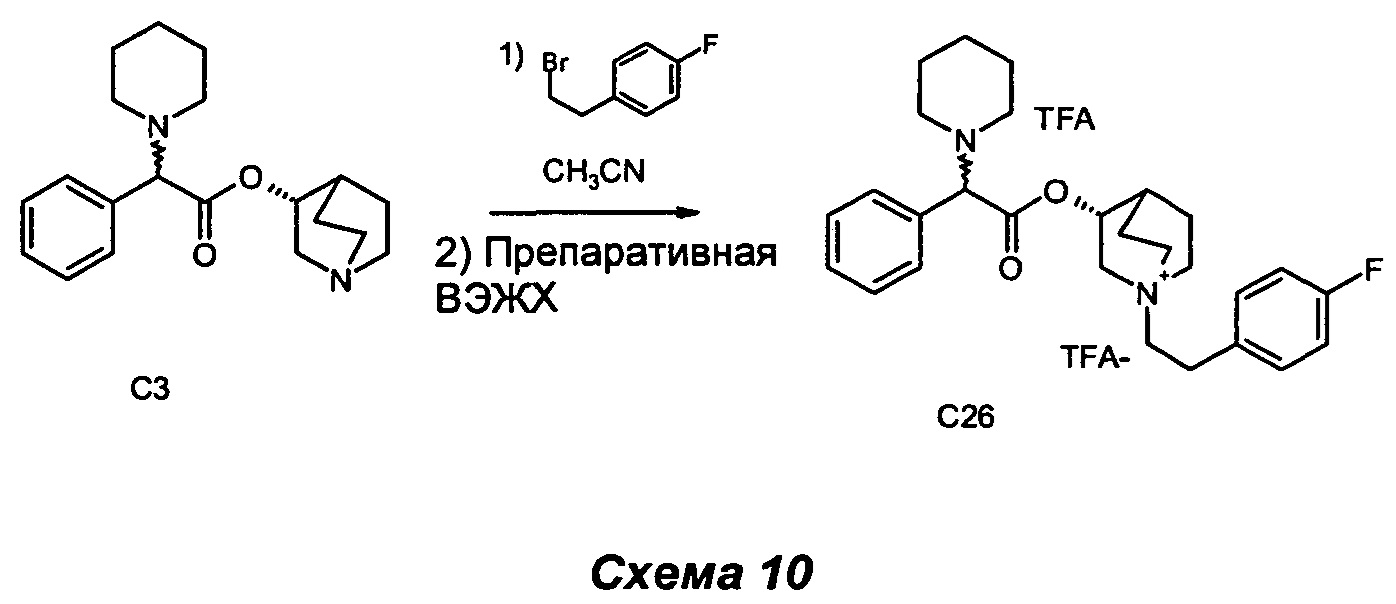
(R)-хинуклидин-3-ил-2-фенил-2-(пиперидин-1-ил)ацетат (200 мг, 0,61 ммоль) растворяли в ацетонитриле (3,1 мл) и добавляли 1-(2-бромэтил)-4-фторбензол (0,17 мл, 1,22 ммоль). Реакционную смесь нагревали при 100°С в течение 1 ч при микроволновом облучении. Раствор выпаривали, и неочищенный продукт очищали сначала флэш-хроматографией (DCM/MeOH равно 9/1), а затем препаративной ВЭЖХ с получением соединения, указанного в заголовке (77 мг, 19% выход), в виде бледно-желтого клейкого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.39-7.58 (m, 5H) 7.28-7.39 (m, 2H) 7.05-7.22 (m, 2H) 5.17-5.32 (m, 1H) 4.71 (br. s., 1H) 3.85-4.06 (m, 1H) 3.14-3.67 (m, 7H) 2.90-3.09 (m, 2H) 2.62-2.84 (m, 4H) 2.20 (d, 1H) 1.72-2.12 (m, 4H) 1.59-1.71 (m, 4H) 1.41-1.56 (m, 2H).
СЭЖХ-МС (ИЭР положительная) 451,25 (М+).
ПРИМЕР 10
Получение (3R)-3-(2-(4-метилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С33)
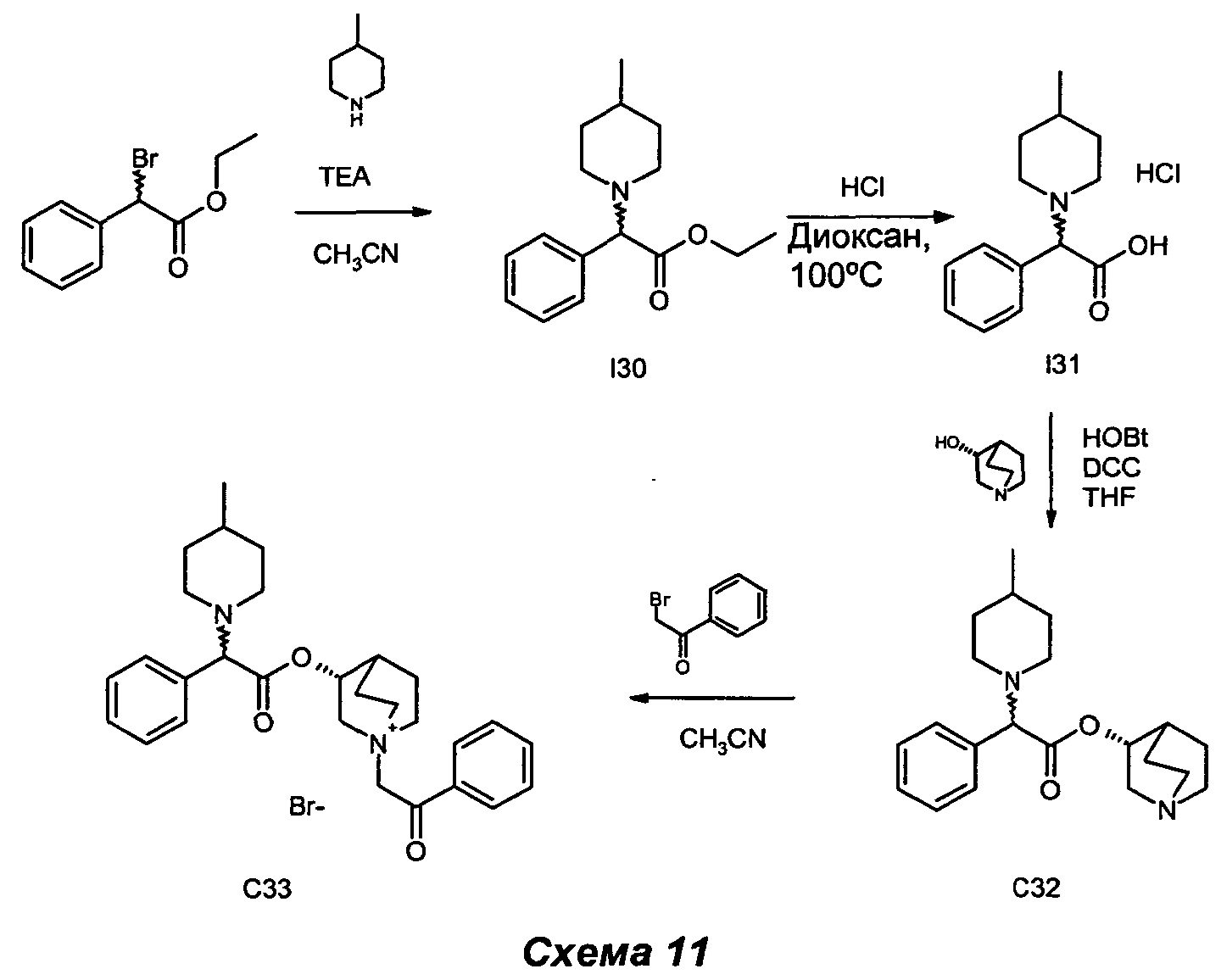
Получение этил-2-(4-метилпиперидин-1-ил)-2-фенилацетата (I30)
4-Метилпиперидин (0,17 мл, 1,48 ммоль), этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) и TEA (0,21 мл, 1,48 ммоль) растворяли в ацетонитриле (6 мл) и перемешивали при KT в течение 48 ч. Летучие вещества выпаривали, и неочищенный остаток очищали флэш-хроматографией (петролейный эфир/AcOEt равно 95/5) с получением этил-2-(4-метилпиперидин-1-ил)-2-фенилацетата (225 мг, 69,8% выход) в виде бесцветного масла.
СЭЖХ-МС (ИЭР положительная) 262,2 (М+).
Получение 2-(4-метилпиперидин-1-ил)-2-фенилуксусной кислоты гидрохлорида (I31)
Этил-2-(4-метилпиперидин-1-ил)-2-фенилацетат (225 мг, 0,86 ммоль) и 37%-й водный HCl (0,52 мл, 17,2 ммоль) растворяли в диоксане (7 мл) и нагревали при микроволновом облучении в герметично закрытом флаконе при 100°С в течение 8 ч. Растворители выпаривали, остаток суспендировали в EtOAc и выпаривали. Остаток суспендировали в EtOAc/Et2O (1/1), обрабатывали ультразвуком и отфильтровывали с аспирацией с получением гидрохлорида 2-(4-метилпиперидин-1-ил)-2-фенилуксусной кислоты (223 мг, 96% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 10.29 (br. s., 1H), 7.27-7.80 (m, 5H), 5.21 (s, 1H), 2.79-3.17 (m, 4H), 1.66-1.91 (m, 2H), 1.39-1.66 (m, 3H), 0.92 (d, 3H).
Получение (R)-хинуклидин-3-ил-2-(4-метилпиперидин-1-ил)-2-фенилацетата (С32)
Гидрохлорид 2-(4-метилпиперидин-1-ил)-2-фенилуксусной кислоты (105 мг, 0,39 ммоль), НОВТ (119 мг, 0,78 ммоль) и DCC (161 мг, 0,78 ммоль) растворяли в сухом THF (5 мл). Добавляли (R)-хинуклидин-3-ол (149 мг, 1,17 ммоль), и реакционную смесь перемешивали при KT в течение 16 ч. THF выпаривали, остаток растворяли в EtOAc и промывали насыщенным раствором NaHCO3, водой и соляным раствором. Органический слой высушивали над Na2SO4, фильтровали и выпаривали. Неочищенный продукт очищали флэш-хроматографией (DCM/MeOH равно 9/1) с получением (R)-хинуклидин-3-ил-2-(4-метилпиперидин-1-ил)-2-фенилацетата (58 мг, 43,5% выход).
Получение (3R)-3-(2-(4-метилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С33)
(R)-Хинуклидин-3-ил-2-(4-метилпиперидин-1-ил)-2-фенилацетат (58 мг, 0,17 ммоль) и 2-бром-1-фенилэтанон (37,1 мг, 0,19 ммоль) растворяли в ацетонитриле (3 мл) и перемешивали при KT в течение ночи. Растворитель выпаривали, и остаток растирали с Et2O и фильтровали в вакууме. Выделенное твердое вещество дополнительно очищали препаративной ВЭЖХ с получением (3R)-3-(2-(4-метилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (5 мг, 5,5% выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.90-8.03 (m, 2H), 7.70-7.82 (m, 1H), 7.58-7.67 (m, 2H), 7.53 (br. s., 5H), 5.27-5.42 (m, 1H), 5.15 (s, 2H), 4.03-4.22 (m, 1H), 3.69-3.88 (m, 5H), 2.77-3.44 (m, 4H), 2.18-2.24 (m, 1H), 1.14-2.16 (m, 10H),0.92 (d, 3H).
СЭЖХ-МС (ИЭР положительная) 461,08 (М+).
ПРИМЕР 11
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пирролидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С37)
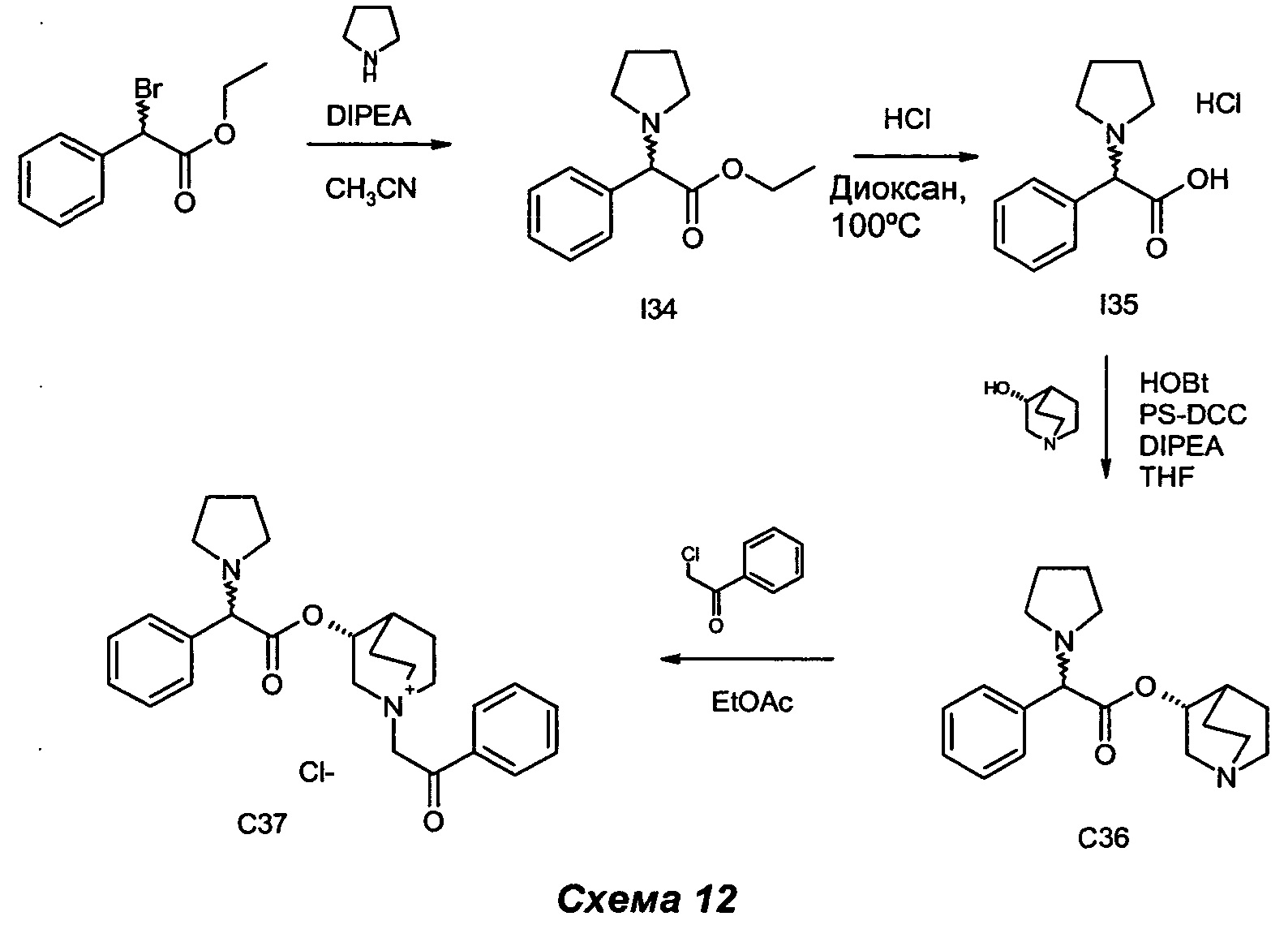
Получение этил-2-фенил-2-(пирролидин-1-ил)ацетата (I34)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) растворяли в ацетонитриле (4,0 мл). Последовательно добавляли DIPEA (0,26 мл, 1,48 ммоль) и пирролидин (0,12 мл, 1,48 ммоль), и раствор перемешивали при комнатной температуре в течение 1,5 ч. Ацетонитрил выпаривали, и остаток очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 9/1) с получением этил-2-фенил-2-(пирролидин-1-ил)ацетата (315 мг, количественный выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.39-7.47 (m, 2H), 7.26-7.39 (m, 3H), 4.09 (dq, 1H), 4.03 (dq, 1H), 3.98 (s, 1H), 2.24-2.48 (m, 4H), 1.58-1.82 (m, 4H), 1.11 (t, 3H).
Получение гидрохлорида 2-фенил-2-(пирролидин-1-ил)уксусной кислоты (I35)
Этил-2-фенил-2-(пирролидин-1-ил)ацетат (0,31 г, 1,33 ммоль) растворяли в диоксане (11 мл) и добавляли по каплям 37%-й HCl (1,09 мл, 13,3 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение ночи. Затем снова добавляли 37%-й HCl (1,09 мл, 13,3 ммоль), и реакционную смесь кипятили с обратным холодильником дополнительно в течение 24 ч. Растворитель выпаривали, остаток растирали с ацетонитрилом (10 мл), и суспензию фильтровали на воронке Бюхнера. Твердое вещество выделяли с получением гидрохлорида 2-фенил-2-(пирролидин-1-ил)уксусной кислоты (0,25 г, 79% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 11.12 (br. s., 1H), 7.56-7.71 (m, 2H), 7.38-7.55 (m, 3H), 5.30 (s, 1H), 3.19-3.65 (m, 2H), 2.88-3.19 (m, 2H), 1.73-2.09 (m, 4H).
Получение (R-хинуклидин-3-ил-2-фенил-2-(пирролидин-1-ил)ацетата (С36)
PS-DCC (загрузка: 1,25 ммоль/г; 0,99 г, 1,24 ммоль) суспендировали в сухом THF (12 мл). Последовательно добавляли НОВТ (0,19 г, 1,24 ммоль), гидрохлорид 2-фенил-2-(пирролидин-1-ил)уксусной кислоты (0,15 г, 0,62 ммоль) и (R)-хинуклидин-3-ол (0,24 г, 1,86 ммоль). Смесь перемешивали в течение ночи, а затем PS-DCC фильтровали, промывая EtOAc и THF. Раствор выпаривали, и остаток растворяли в EtOAc (30 мл) и промывали водой (15 мл) и насыщенным раствором NaHCO3 (20 мл). Органическую фазу высушивали (Na2SO4), фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 92/8 до 85/15) с получением (R)-хинуклидин-3-ил-2-фенил-2-(пирролидин-1-ил)ацетата (53 мг, 27,2% выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.40-7.53 (m, 2H), 7.18-7.40 (m, 3H), 4.53-4.88 (m, 1H), 4.01 (s, 1H), 2.92-3.14 (m, 1H), 2.55-2.70 (m, 5H), 2.12-2.41 (m, 4H), 1.73-1.92 (m, 1H), 1.65-1.72 (m, 4H), 1.08-1.63 (m, 4H).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пирролидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С37)
(R)-Хинуклидин-3-ил-2-фенил-2-(пирролидин-1-ил)ацетат (51 мг, 0,16 ммоль) растворяли в этилацетате (1,6 мл) и добавляли 2-хлор-1-фенилэтанон (25,1 мг, 0,16 ммоль). Раствор перемешивали при комнатной температуре в течение 3,5 суток. Суспензию выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/MeOH равно от 9/1 до 85/15) с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(пирролидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (44 мг, 57,8% выход) в виде желтого стекловидного твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.86-8.05 (m, 2H), 7.70-7.83 (m, 1H), 7.57-7.66 (m, 2H), 7.44-7.53 (m, 2H), 7.29-7.44 (m, 3H), 5.23 и 5.25 (s, 1H), 5.11-5.23 (m, 1H), 4.13 и 4.14 (s, 1H), 4.00-4.23 (m, 1H), 3.45-3.83 (m, 5H), 2.53-2.59 (m, 1H), 2.32-2.46 (m, 4H), 2.15-2.34 (m, 1H), 1.46-2.12 (m, 8H).
СЭЖХ-МС (ИЭР положительная) 433,13 (М+).
ПРИМЕР 12
Получение (3R)-3-(2-морфолино-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С41)
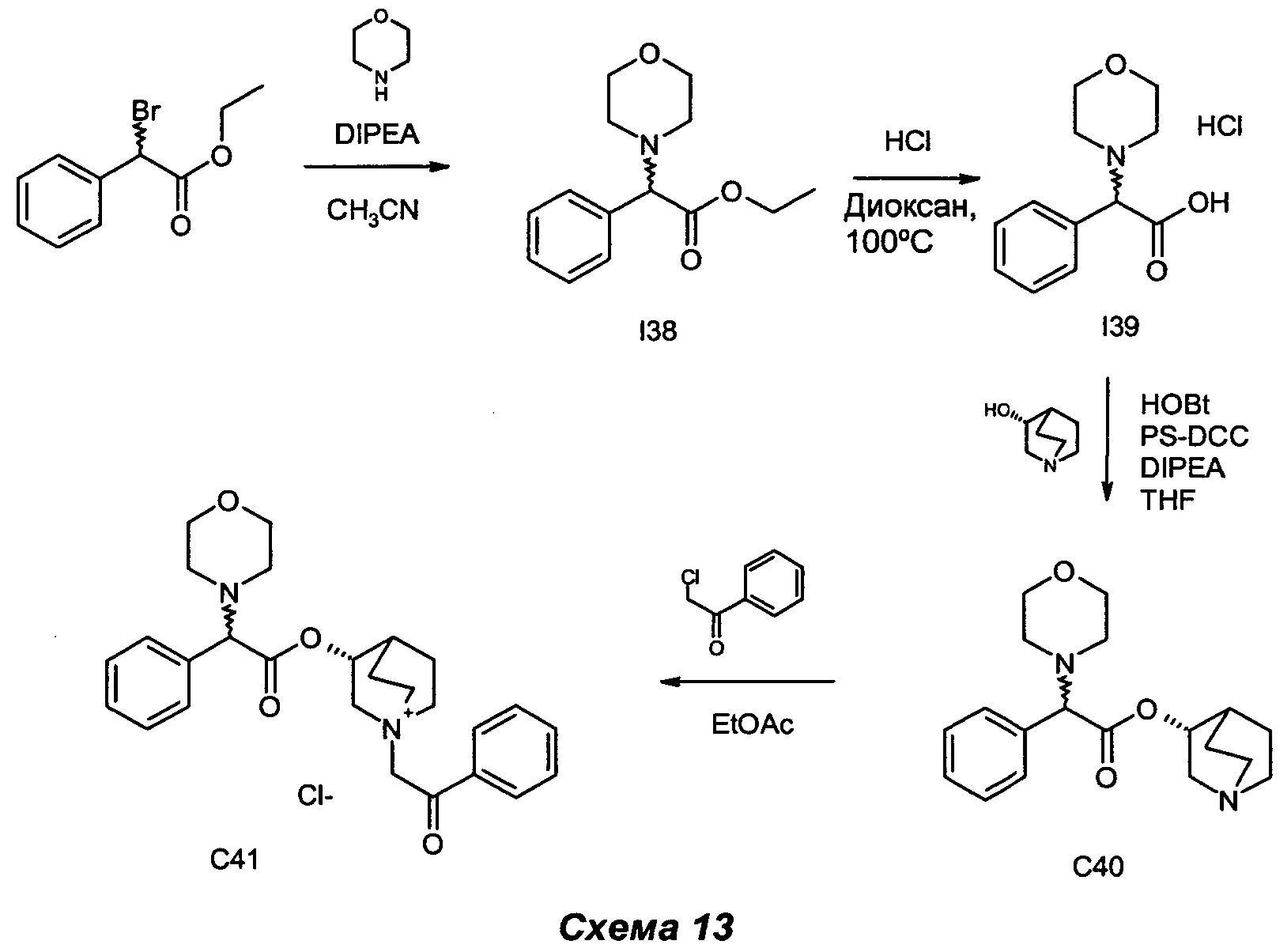
Получение этил-2-морфолино-2-фенилацетата (I38)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) растворяли в ацетонитриле (4,0 мл) и последовательно добавляли DIPEA (0,26 мл, 1,48 ммоль) и морфолин (0,13 мл, 1,481 ммоль). Раствор перемешивали при комнатной температуре в течение 1,5 ч. Ацетонитрил выпаривали, и остаток очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 85/15) с получением этил-2-морфолино-2-фенилацетата (345 мг, количественный выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.20-7.46 (m, 5H), 4.09 (s, 1H), 4.12 (dq, 1H), 4.06 (dq, 1H), 3.47-3.70 (m, 4H), 2.31-2.41 (m, 4H), 1.13 (t, 3H).
Получение гидрохлорида 2-морфолино-2-фенилуксусной кислоты (I39)
Этил-2-морфолино-2-фенилацетат (0,34 г, 1,36 ммоль) растворяли в диоксане (11,4 мл) и добавляли по каплям 37%-й HCl (1,1 мл, 13,6 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение ночи. Добавляли 37%-й HCl (1,1 мл, 13,6 ммоль), и реакционную смесь кипятили с обратным холодильником дополнительно в течение 24 ч. Растворитель выпаривали, и остаток растирали с ацетонитрилом (10 мл) с получением гидрохлорида 2-морфолино-2-фенилуксусной кислоты (0,22 г, выход 63,7%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 11.79 (s, 1H), 7.56-7.72 (m, 2H), 7.37-7.56 (m, 3H), 5.25 (s, 1H), 3.60-4.10 (m, 4H), 3.27 (br. s., 2H), 2.96 (br. s., 2H).
Получение (R)-хинуклидин-3-ил-2-морсфолино-2-фенилацетата (С40)
PS-DCC (загрузка: 1,25 ммоль/г; 0,93 г, 1,16 ммоль) суспендировали в сухом THF (11,6 мл) и последовательно добавляли НОВТ (0,18 г, 1,16 ммоль), гидрохлорид 2-морфолино-2-фенилуксусной кислоты (0,15 г, 0,58 ммоль) и (R)-хинуклидин-3-ол (0,22 г, 1,75 ммоль). Смесь перемешивали в течение ночи, а затем PS-DCC отфильтровывали, промывая EtOAc и THF. Раствор выпаривали, и остаток растворяли в EtOAc (30 мл) и промывали водой, а затем насыщенным раствором NaHCO3. Органическую фазу высушивали (Na2SO4, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 95/5 до 85/15) с получением (R)-хинуклидин-3-ил-2-морфолино-2-фенилацетата (45 мг, выход 23,4%) в виде бесцветного масла.
1H ЯМР (300 МГц. DMSO-d6) млн-1 7.11-7.51 (m, 5H), 4.60-4.82 (m, 1H), 4.10 (s, 1H), 3.48-3.66 (m, 4H), 2.91-3.18 (m, 1H), 2.54-2.78 (m, 4H), 2.18-2.45 (m, 5H), 1.71-1.93 (m, 1H), 1.05-1.69 (m, 4H)
Получение (3R)-3-(2-морфолино-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С41)
(R)-Хинуклидин-3-ил-2-морфолино-2-фенилацетат (43 мг, 0,13 ммоль) растворяли в этилацетате (1,3 мл) и добавляли 2-хлор-1-фенилэтанон (22,1 мг, 0,14 ммоль). Раствор перемешивали при комнатной температуре в течение двух суток. Растворитель выпаривали, и остаток растирали с Et2O с получением (3R)-3-(2-морфолино-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (53 мг, 84% выход) в виде бледно-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.85-8.04 (m, 2H), 7.70-7.84 (m, 1H), 7.55-7.69 (m, 2H), 7.27-7.54 (m, 5H), 5.25 и 5.27 (s, 2H), 5.11-5.25 (m, 1H), 4.23 и 4.26 (s, 1H), 4.04-4.22 (m, 1H), 3.62-3.83 (m, 5H), 3.54-3.62 (m, 4H), 2.39-2.47 (m, 4H), 2.16-2.25 (m, 1H), 1.59-2.15 (m, 4H).
СЭЖХ-МС (ИЭР положительная) 449,24 (М+).
ПРИМЕР 13
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-тиоморфолиноацетокси)-1-азониабицикло[2.2.2]октанхлорида (С45)
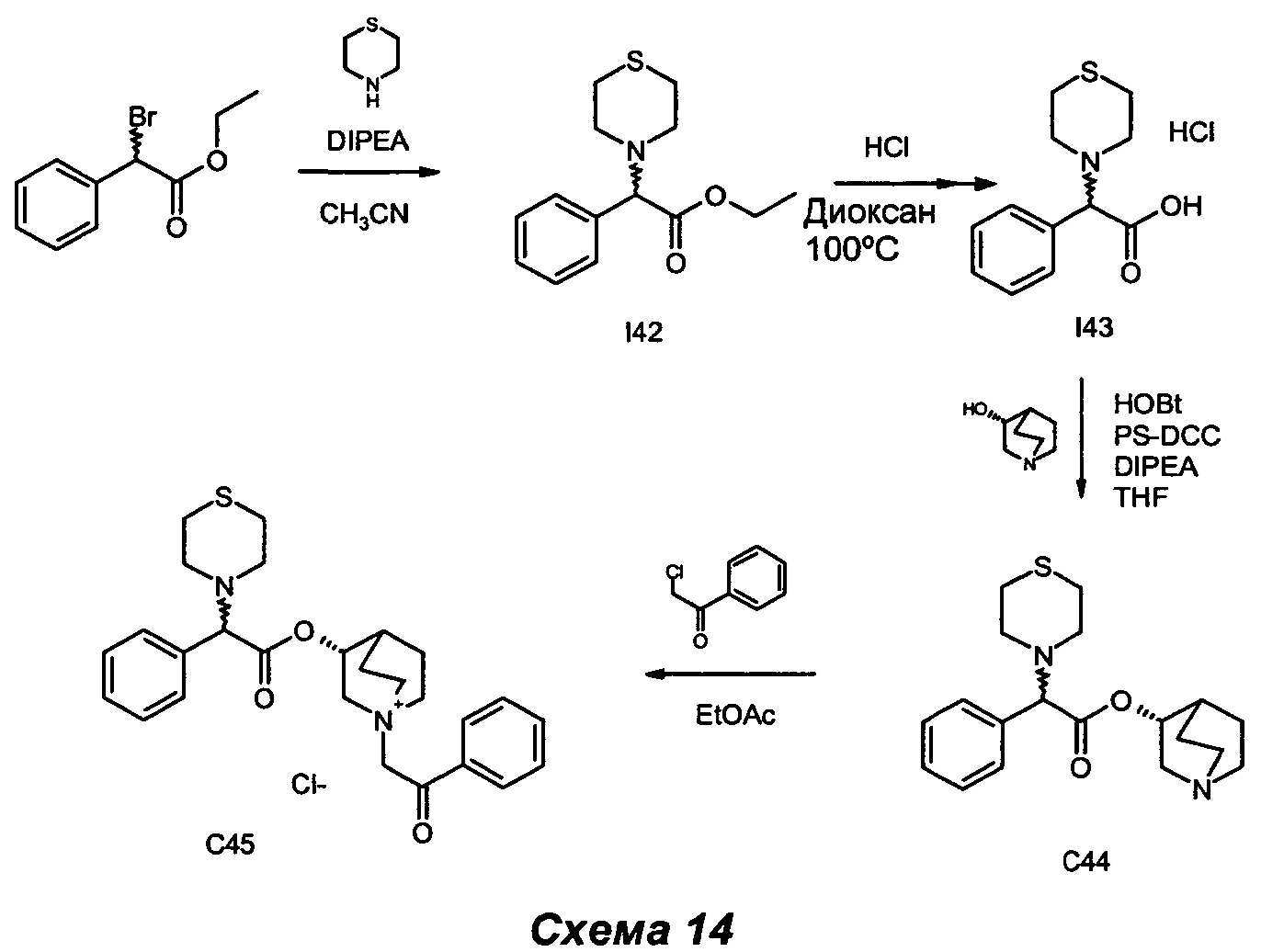
Получение этил-2-фенил-2-тиоморфолиноацетата (I42)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) растворяли в ацетонитриле (4,0 мл). Последовательно добавляли DIPEA (0,26 мл, 1,481 ммоль) и тиоморфолин (0,15 мл, 1,41 ммоль), и раствор перемешивали при комнатной температуре в течение 1,5 ч. Ацетонитрил выпаривали, и остаток очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 95/5) с получением этил-2-фенил-2-тиоморфолиноацетата (362 мг, количественный выход) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.10-7.83 (m, 5H), 4.31 (s, 1H), 4.14 (dq, 1H), 4.09(dq, 1H), 2.55-2.87 (m, 8H), 1.15 (t, 3H).
Получение гидрохлорида 2-фенил-2-тиоморфолиноуксусной кислоты (I43)
37%-ю водный HCl (1,1 мл, 13,5 ммоль) добавляли по каплям к раствору этил-2-фенил-2-тиоморфолиноацетата (0,36 г, 1,35 ммоль) в диоксане (11 мл). Смесь перемешивали при кипячении с обратным холодильником в течение ночи. Затем снова добавляли 37%-ю водный HCl (1,1 мл, 13,5 ммоль), и реакционную смесь кипятили с обратным холодильником дополнительно в течение 24 ч. Растворители выпаривали, остаток растирали с ацетонитрилом и собирали фильтрованием с аспирацией с получением гидрохлорида 2-фенил-2-тиоморфолиноуксусной кислоты (0,13 г, выход 34,4%) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.09-7.78 (m, 5H), 5.26 (s, 1H), 3.09-3.41 (m, 4H), 2.98 (br. s., 4H).
Получение (R)-хинуклидин-3-ил-2-фенил-2-тиоморфолиноацетата (С44)
PS-DCC (загрузка: 1,25 ммоль/г; 0,73 г, 0,91 ммоль) суспендировали в сухом THF (9 мл). Последовательно добавляли НОВТ (0,14 г, 0,91 ммоль), гидрохлорид 2-фенил-2-тиоморфолиноуксусной кислоты (0,12 г, 0,46 ммоль) и (R)-хинукпидин-3-ол (0,17 г, 1,37 ммоль). Смесь перемешивали в течение ночи. PS-DCC отфильтровывали и промывали EtOAc и THF. Раствор выпаривали, и остаток распределяли между EtOAc и водой. Органическую фазу промывали насыщенным раствором NaHCO3, высушивали (Na2SO4), фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 95/5 до 85/15) с получением (R)-хинуклидин-3-ил-2-фенил-2-тиоморфолиноацетата (70 мг, выход 44,2%) в виде бесцветного масла.
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-тиоморфолиноацетокси)-1-азониабицикло[2.2.2]октанхлорида (С45)
(R)-Хинуклидин-3-ил-2-фенил-2-тиоморфолиноацетат (68 мг, 0,20 ммоль) растворяли в этилацетате (2 мл) и добавляли 2-хлор-1-фенилэтанон (33,4 мг, 0,22 ммоль). Раствор перемешивали при комнатной температуре в течение двух суток. Растворитель выпаривали, и остаток растирали с Et2O. Суспензию фильтровали на воронке Бюхнера с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-тиоморфолиноацетокси)-1-азониабицикло[2.2.2]октанхлорида (89 мг, 0,178 ммоль, выход 91%) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.89-8.08 (m, 2H), 7.71-7.83 (m, 1H), 7.56-7.67 (m, 2H), 7.27-7.49 (m, 5H), 5.27 (s, 2H), 5.20-5.25 (m, 1H), 4.46 (s, 1H), 4.01-4.28 (m, 1H), 3.48-3.82 (m, 5H), 2.57-2.88 (m, 8H), 2.23-2.42 (m, 1H), 1.58-2.16 (m, 4H).
СЭЖХ-МС (ИЭР положительная) 465,09 (М+).
ПРИМЕР 14
Получение (3R)-3-(2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С53)
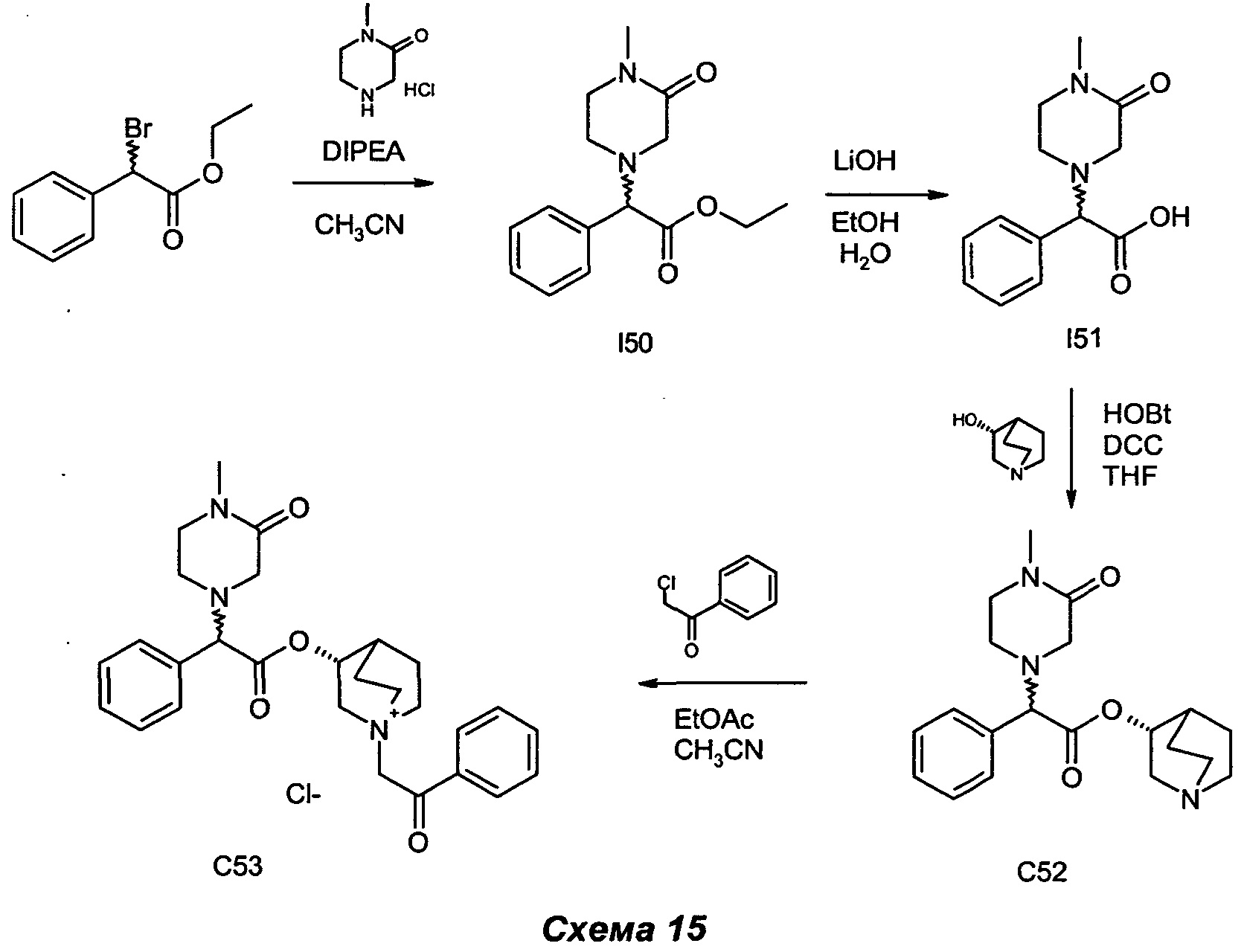
Получение этил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетата (I50)
DIPEA (0,52 мл, 2,96 ммоль) и гидрохлорид 1-метилпиперазин-2-она (0,22 г, 1,48 ммоль) последовательно добавляли к раствору этил-2-бром-2-фенилацетата (0,22 мл, 1,23 ммоль) в ацетонитриле (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Снова добавляли DIPEA (0,13 мл, 0,74 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/ацетон равно 9/1) с получением этил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетата (256 мг, выход 75%) в виде желтого масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 6.91-7.66 (m, 5H), 4.28 (s, 1H), 3.99-4.22 (m, 2H), 3.23 (t, 2H), 3.00 (s, 2H), 2.80 (s, 3H), 2.61-2.71 (m, 2H), 1.14 (t, 3H).
Получение 2-(4-метил-3-оксопиперазин-1-ил)-2-фенилуксусной кислоты (I51)
Этил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетат (250 мг, 0,90 ммоль) растворяли в этаноле (8,6 мл) и воде (4,3 мл). Добавляли гидроксид лития (28,2 мг, 1,18 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение трех суток. Добавляли вторую порцию гидроксида лития (4,33 мг, 0,18 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. EtOH выпаривали, и к водному раствору добавляли 1 н. HCl до рН 7. Воду выпаривали, и остаток суспендировали в ацетонитриле и фильтровали на воронке Бюхнера, промывая ацетонитрилом, с получением 170 мг желаемого соединения в виде белого твердого вещества. Это соединение использовали в следующей стадии без какой-либо дополнительной очистки.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.34-7.51 (m, 2H), 7.08-7.34 (m, 3H), 3.66 (s, 1H), 3.18 (t, 2H), 3.14 (d, 1H), 2.89 (d, 1H), 2.79 (s, 3H), 2.55-2.71 (m, 2H).
Получение (R)-хинуклидин-3-ил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетата (С52)
2-(4-Метил-3-оксопиперазин-1-ил)-2-фенилуксусную кислоту (170 мг, 0,68 ммоль) суспендировали в сухом THF (6 мл) и последовательно добавляли DCC (283 мг, 1,37 ммоль), НОВТ (185 мг, 1,37 ммоль) и (R)-хинуклидин-3-ол (174 мг, 1.37 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение двух суток, а затем нерастворимое вещество отфильтровывали. Раствор выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 96/4/0,4) с получением (R)-хинуклидин-3-ил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетата (175 мг, выход 54,1% за 2 стадии) в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.09-7.60 (m, 5H), 4.64-4.87 (m, 1H), 4.28 (s, 1H), 3.24 (t, 2H), 3.04-3.14 (m, 1H), 3.01 (s, 2H), 2.81 (s, 3H), 2.66-2.72 (m, 2H), 2.54-2.65 (m, 4H), 2.35-2.47 (m, 1H), 1.70-1.81 и 1.85-1.95 (m, 1H),1.06-1.69(m, 4H).
Получение (3R)-3-(2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С53)
(R)-Хинуклидин-3-ил-2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетат (169 мг, 0,47 ммоль) растворяли в смеси этилацетата (3 мл) и ацетонитрила (1,5 мл). Добавляли 2-хлор-1-фенилэтанон (80 мг, 0,52 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Растворитель выпаривали, и остаток растирали с EtOAc (10 мл). Продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 9/1 до 85/15) с получением (3R)-3-(2-(4-метил-3-оксопиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (99 мг, 0,193 ммоль, выход 40,9%) в виде бледно-желтого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.90-8.12 (m, 2H), 7.68-7.85 (m, 1H), 7.55-7.68 (m, 2H), 7.27-7.51 (m, 5H), 5.28-5.38 (m, 2H), 5.17-5.27 (m, 1H), 4.41 и 4.45 (s, 1H), 4.10-4.28 (m, 1H), 3.51-3.86 (m, 5H), 3.21-3.29 (m, 2H), 3.11 (d, 1H), 3.02 (d, 1H), 2.82 (s, 3H), 2.67-2.77 (m, 2H), 2.17-2.25 и 2.34-2.42 (m, 1H), 1.52-2.13 (m, 4H);
СЭЖХ-МС (ИЭР положительная) 476,09 (М+).
ПРИМЕР 15
Получение (3R)-3-(2-(4-ацетилпиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С57)
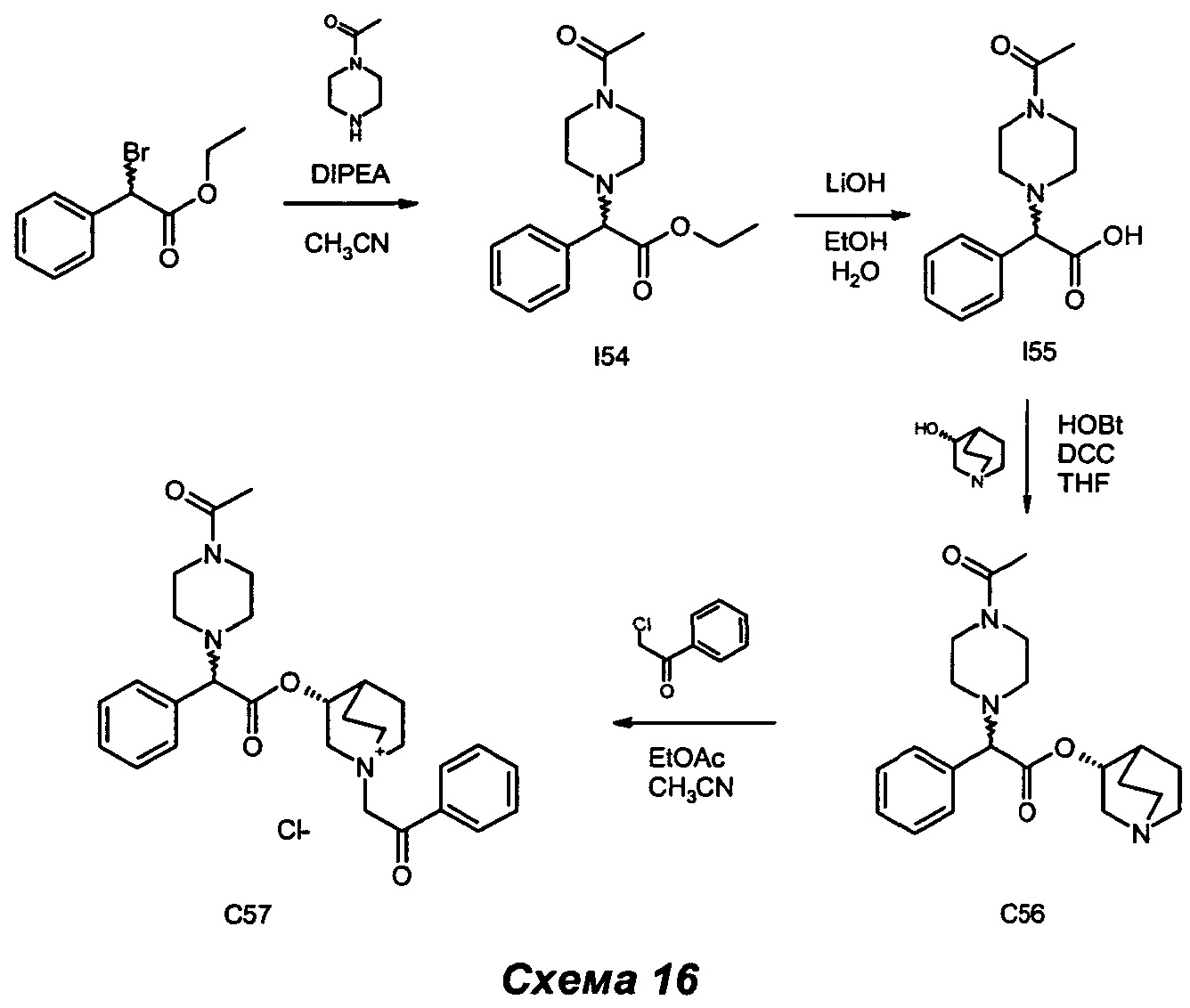
Получение этил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетата (I54)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) растворяли в ацетонитриле (4 мл) и последовательно добавляли DIPEA (0,28 мл, 1,60 ммоль) и 1-(пиперазин-1-ил)этанон (0,21 г, 1,60 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/ацетон равно 9/1) с получением этил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетата (341 мг, выход 95%) в виде бледно-желтого масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 6.80-7.63 (m, 5H), 4.18 (s, 1H), 4.00-4.16 (m, 2H), 3.33-3.49 (m, 4H), 2.26-2.45 (m, 4H), 1.95 (s, 3H), 1.13 (t, 3H).
Получение 2-(4-ацетилпиперазин-1-ил)-2-фенилуксусной кислоты (I55)
Этил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетат (285 мг, 0,98 ммоль) растворяли в смеси EtOH (6,5 мл) и воды (3,3 мл). Добавляли LiOH (23,5 мг, 0,98 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Снова добавляли LiOH (23,5 мг, 0,982 ммоль), и смесь перемешивали дополнительно в течение 24 ч. EtOH выпаривали, и к водному раствору добавляли по каплям 1 н. HCl до рН 6-7, после чего выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 75/25) с получением 2-(4-ацетилпиперазин-1-ил)-2-фенилуксусной кислоты (246 мг, выход 96%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.37-7.49 (m, 2H), 7.20-7.37 (m, 3H), 3.90 (s, 1H), 3.26-3.50 (m, 4H), 2.15-2.47 (m, 4H), 1.95 (s, 3H).
Получение (R)-хинуклидин-3-ил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетата (С56)
2-(4-Ацетилпиперазин-1-ил)-2-фенилуксусную кислоту (238 мг, 0,91 ммоль) суспендировали в сухом THF (9 мл) и, перемешивая при комнатной температуре в атмосфере азота, последовательно добавляли DCC (374 мг, 1,81 ммоль), НОВТ (245 мг, 1,81 ммоль) и (R)-хинуклидин-3-ол (231 мг, 1,81 ммоль). Реакционную смесь подвергали взаимодействию при комнатной температуре в течение трех суток. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/МеОН/NH4OH равно 97/3/0,3). Продукт суспендировали в THF/ацетонитриле (2/1; 10 мл), и белое твердое вещество отфильтровывали. Бесцветный раствор выпаривали с получением (R)-хинуклидин-3-ил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетата (218 мг, выход 64,7%) в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.22-7.55 (m, 5H), 4.61-4.80 (m, 1H), 4.19 (s, 1H), 3.36-3.54 (m, 4H). 3.04-3.19 (m, 1H), 2.54-2.78 (m, 4H), 2.28-2.46 (m, 5H), 1.95 (s, 3H), 1.68-1.80 (m, 1H), 1.02-1.67 (m, 4H)
Получение (3R)-3-(2-(4-ацетилпиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С57)
(R)-Хинуклидин-3-ил-2-(4-ацетилпиперазин-1-ил)-2-фенилацетат (210 мг, 0,56 ммоль) растворяли в смеси EtOAc (3,8 мл) и ацетонитрила (1,8 мл). Добавляли 2-хлор-1-фенилэтанон (96 мг, 0,62 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением (3R)-3-(2-(4-ацетилпиперазин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (178 мг, выход 59,9%) в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.89-8.08 (m, 2H), 7.70-7.87 (m, 1H), 7.55-7.69 (m, 2H), 7.30-7.52 (m, 5H), 5.29 (s, 2H), 5.17-5.28 (m, 1H), 4.34 (s, 1H), 4.08-4.25 (m, 1H), 3.37-3.86 (m, 9H), 2.31-2.47 (m, 4H), 2.17-2.29 (m, 1H), 1.98-2.12 (m, 2H), 1.96 (s, 3H), 1.59-1.94 (m, 2H).
СЭЖХ-МС (ИЭР положительная) 490,26 (М+).
ПРИМЕР 16
Получение (3R)-3-(2-(4-карбамоилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикпо[2.2.2]октанхлорида (С61)
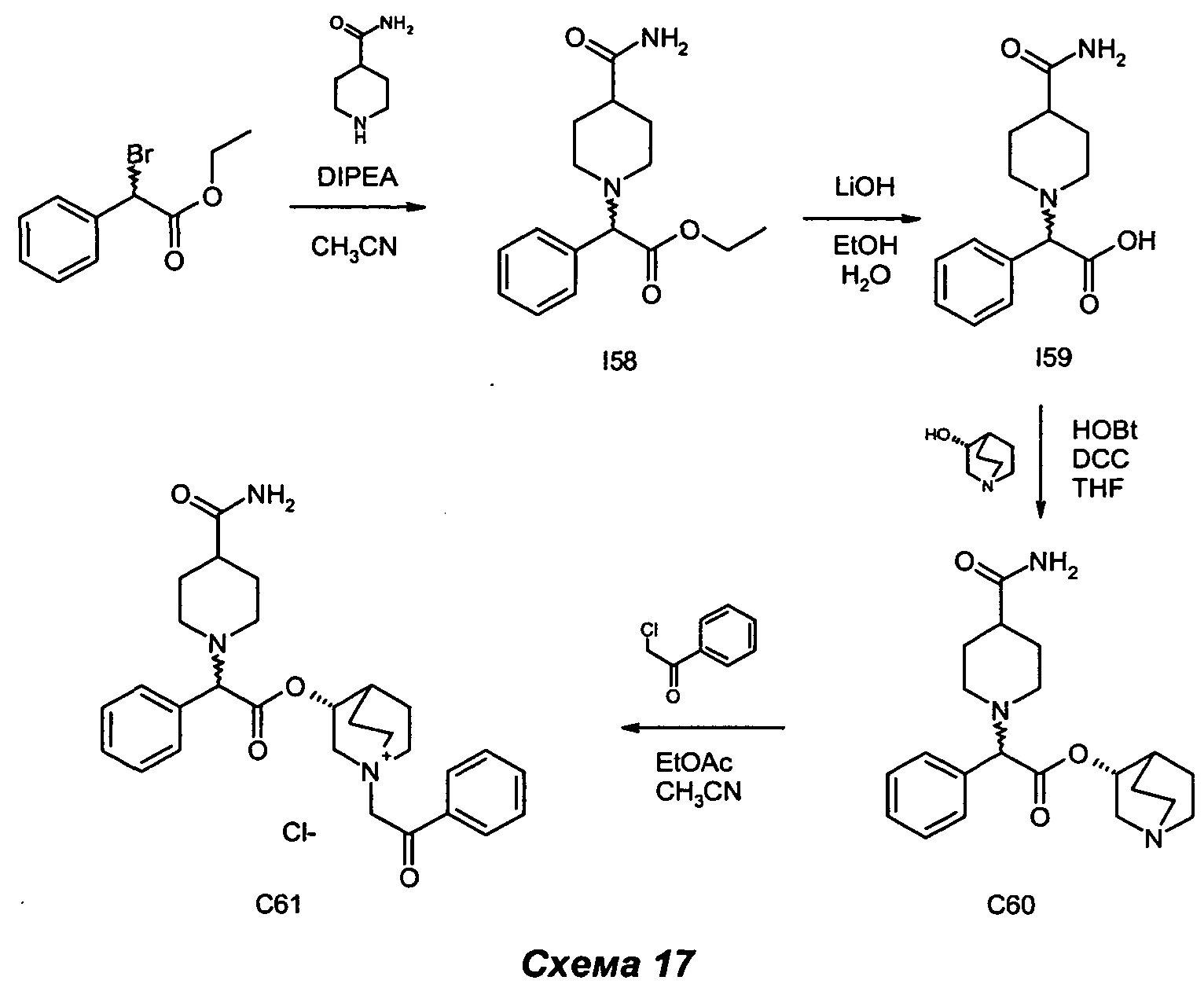
Получение этил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетата (I58)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль) растворяли в ацетонитриле (4 мл). Последовательно добавляли DIPEA (0,28 мл, 1,60 ммоль) и пиперидин-4-карбоксамид (0,21 г, 1,60 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/Ацетон равно 8/2). Твердое вещество растирали с i-Pr2O с получением этил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетата (239 мг, выход 66,7%) в виде бледно-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.26-7.47 (m, 5H), 7.15 (br. s., 1H), 6.66 (br. s., 1H), 3.95-4.27 (m, 3H), 2.75-2.90 (m, 1H), 2.59-2.75 (m, 1H), 2.12 (td, 1H), 1.97-2.06 (m, 1H), 1.92 (td, 1H), 1.44 -1.77 (m, 4H), 1.14 (t, 3H).
Получение 2-(4-карбамоилпиперидин-1-ил)-2-фенилуксусной кислоты (I59)
Этил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетат (202 мг, 0,69 ммоль) растворяли в смеси EtOH (4,6 мл) и воды (2,3 мл) и добавляли гидроксид лития (50,0 мг, 2,09 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. LiOH (33,3 мг, 1,39 ммоль) добавляли в двух порциях в течение двух последующих суток. EtOH выпаривали, и к водному раствору добавляли по каплям 1 н. HCl до рН 6-7. Раствор выпаривали, и остаток очищали флэш-хроматографией (соотношение DCM/MeOH равно от 7/3 до 6/4) с получением 2-(4-карбамоилпиперидин-1-ил)-2-фенилуксусной кислоты (155 мг, выход 85%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.38-7.48 (m, 2Н), 7.25-7.38 (m, 3Н), 7.20 (br. s., 1Н), 6.70 (br. s., 1Н), 3.96 (s, 1Н), 3.05-3.24 (m, 1Н), 2.62-2.79 (m, 1H), 2.26(td, 1H), 1.98-2.18(m, 2H), 1.52-1.89(m, 4H).
Получение (R)-хинуклидин-3-ил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетата (С60)
2-(4-Карбамоилпиперидин-1-ил)-2-фенилуксусную кислоту (145 мг, 0,55 ммоль) суспендировали в сухом THF (5,5 мл) и последовательно добавляли DCC (240 мг, 1,16 ммоль), НОВТ (157 мг, 1,16 ммоль) и (R)-хинуклидин-3-ол (148 мг, 1,16 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 24 ч. Белое твердое вещество отфильтровывали, и раствор выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 95/5/0,5) с получением (R)-хинуклидин-3-ил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетата (78 мг, выход 38%) в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.26-7.46 (m, 5Н) 7.16 (br. s., 1Н) 6.66 (br. s., 1Н) 4.58-4.80 (m, 1Н) 4.11 (s, 1Н) 2.94-3.16 (m, 1Н) 2.78-2.94 (m, 1Н) 2.54-2.76 (m, 4Н) 2.33-2.47 (m, 1Н) 1.72-2.30 (m, 5Н) 1.35-1.72 (m, 7Н) 1.07-1.35(m, 1H).
Получение (3R)-3-(2-(4-карбамоилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С61)
(R)-Хинуклидин-3-ил-2-(4-карбамоилпиперидин-1-ил)-2-фенилацетат (72 мг, 0,19 ммоль) растворяли в смеси EtOAc (1,3 мл) и ацетонитрила (0,6 мл). Добавляли 2-хлор-1-фенилэтанон (33,0 мг, 0,21 ммоль), и смесь подвергали взаимодействию при той же температуре в течение трех суток. Смесь выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 85/15 до 8/2) с получением (3R)-3-(2-(4-карбамоилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (34 мг, выход 33,3%) в виде бледно-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.94-8.04 (m, 2H), 7.69-7.82 (m, 1H), 7.55-7.67 (m, 2H), 7.30-7.49 (m, 5H), 7.20 (br. s., 1H), 6.68 (br. s., 1H), 5.32 (s, 2H), 5.18-5.28 (m, 1H), 4.24 (s, 1H), 4.10-4.33 (m, 1H), 3.51-3.88 (m, 5H), 2.83-2.97 (m, 1H), 2.69-2.82 (m, 1H), 1.38-2.40 (m, 12H).
СЭЖХ-МС (ИЭР положительная) 490,23 (М+).
ПРИМЕР 17
Получение (3R)-3-(2-(3,5-диметилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С64)
Получение этил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетата (I62)
Этил-2-бром-2-фенилацетат (0,22 мл, 1,23 ммоль), 3,5-диметилпиперидин (0,20 мл, 1,48 ммоль) и DIPEA (0,26 мл, 1,48 ммоль) растворяли в ацетонитриле (12 мл) и перемешивали при комнатной температуре в течение 16 часов. Ацетонитрил выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 95/5) с получением этил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетата (203 мг, выход 59,7%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.24-7.42 (m, 5H) 3.99-4.19 (m, 3H) 2.69-2.80 (m, 1H) 2.57-2.68 (m, 1H) 1.49-1.73 (m, 4H) 1.36-1.48 (m, 1H) 1.13 (t, 3H) 0.79 (d, 3H) 0.72 (d, 3H) 0.39-0.56 (m, 1H).
Получение (R)-хинуклидин-3-ил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетата (С63)
(R)-хинуклидин-3-ол (175 мг, 1,38 ммоль) и NaH (60%-я дисперсия в минеральном масле, 52,3 мг, 1,31 ммоль) суспендировали в сухом толуоле (9 мл) в атмосфере азота и перемешивали при комнатной температуре в течение 20 минут. Добавляли этил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетат (200 мг, 0,73 ммоль), и полученную в результате смесь перемешивали при 100°С в течение 24 часов. Снова добавляли NaH (60% дисперсия в минеральном масле, 8,72 мг, 0,36 ммоль), и смесь кипятили с обратным холодильником еще в течение 24 ч. Реакционную смесь охлаждали и распределяли между водой и этиловым эфиром. Органический слой собирали и высушивали над Na2SO4, фильтровали и выпаривали. Сырое соединение очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением (R)-хинуклидин-3-ил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетата (125 мг, выход 48,3%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.22-7.46 (m, 5H) 4.63-4.80 (m, 1H) 4.10 (s, 1H) 2.97-3.15 (m, 1H) 2.75-2.88 (m, 1H) 2.54-2.71 (m, 5H) 2.14-2.46 (m, 1H) 1.73-1.94 (m, 1H) 1.37-1.73 (m, 8H) 1.05-1.37 (m, 1H) 0.79 (d, 3H) 0.72 (d, 3H) 0.39-0.57 (m, 1H).
Получение (3R)-3-(2-(3,5-диметилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С64)
(R)-хинуклидин-3-ил-2-(3,5-диметилпиперидин-1-ил)-2-фенилацетат (120 мг, 0,34 ммоль) и 2-бром-1-фенилэтанон (73,7 мг, 0,37 ммоль) растворяли в ацетонитриле (5 мл) и перемешивали при комнатной температуре в течение 16 часов. Ацетонитрил выпаривали, остаток растворяли небольшим количеством EtOAc (2 мл), а затем добавляли Et2O. Белое твердое вещество, выпавшее в осадок, выделяли фильтрованием с аспирацией и промывали на фильтровальной бумаге Et2O. Твердое вещество высушивали с получением (3R)-3-(2-(3,5-диметилпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (115,8 мг, выход 61,9%).
1Н ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.04 (m, 2H), 7.71-7.83 (m, 1H), 7.57-7.68 (m, 2H). 7.20-7.49 (m, 5H), 5.23-5.26 (m, 1H), 5.22 (br. s., 2H), 4.29 (s, 1H), 4.07-4.20 (m, 1H), 3.49-3.86 (m, 5H), 2.63-2.91 (m, 2H), 2.20-2.42 (m, 1H), 1.39-2.17 (m, 9H), 0.82 (d, 3H), 0.75 (d, 3H), 0.38-0.64 (m, 1H);
СЭЖХ-МС (ИЭР положительная) 475,26 (М+).
ПРИМЕР 18
Получение (3R)-3-(2-(4,4-дифторпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С70)
Получение 2-(4,4-дифторпиперидин-1-ил)-2-фенилуксусной кислоты (I68)
Смесь фенилбороновой кислоты (400 мг, 3,28 ммоль), 4,4-дифторпиперидина (397 мг, 3,28 ммоль) и 2-оксоуксусной кислоты гидрата (302 мг, 3,28 ммоль) в DCM (30 мл) перемешивали при комнатной температуре в течение ночи. DCM выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 8/2) с получением соединения, указанного в заголовке (512 мг, выход 61,1%), в виде белого твердого вещества.
СЭЖХ-МС (ИЭР положительная) 256,3 (МН+).
Получение (R)-хинуклидин-3-ил-2-(4,4-дифторпиперидин-1-ил)-2-фенилацетата (С69)
Смесь 2-(4,4-дифторпиперидин-1-ил)-2-фенилуксусной кислоты (512 мг, 2,01 ммоль), (R)-хинуклидин-3-ола (306 мг, 2,41 ммоль), DCC (497 мг, 2,41 ммоль) и НОВТ (369 мг, 2,41 ммоль) в THF (20 мл) перемешивали при комнатной температуре в течение выходных дней. THF выпаривали, неочищенный продукт растворяли EtOAc и дважды промывали 2 М К2СО3, а затем соляным раствором. Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением соединения, указанного в заголовке (348 мг, выход 47,6%), в виде желтого липкого масла.
СЭЖХ-МС (ИЭР положительная) 365,2 (МН+).
Получение (3R)-3-(2-(4,4-дифторпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С70)
2-Хлор-1-фенилэтанон (31,0 мг, 0,20 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(4,4-дифторпиперидин-1-ил)-2-фенилацетата (24b) (73 мг, 0,20 ммоль) в этилацетате (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали при пониженном давлении, и остаток растирали с Et2O (4 мл). Образовывался белый осадок, и его собирали фильтрованием с аспирацией и высушивали в вакууме при 40°С в течение ночи с получением (3R)-3-(2-(4,4-дифторпиперидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (81 мг, выход 78%).
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.93-8.04 (m, 2H) 7.71-7.83 (m, 1H) 7.57-7.66 (m, 2H) 7.32-7.50 (m, 5H) 5.15-5.31 (m, 2H) 4.48 и 4.52 (s, 1H) 4.08-4.23 (m, 1H) 3.54-3.80 (m, 5H) 2.54-2.70 (m, 4H) 2.21-2.31 и 2.23-2.30 (m, 1H) 1.67-2.13 (m, 8H).
СЭЖХ-МС (ИЭР положительная) 483,24 (М+).
ПРИМЕР 19
Получение (3R)-3-(2-(азепан-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабииикло[2.2.2]октанхлорида (C73)
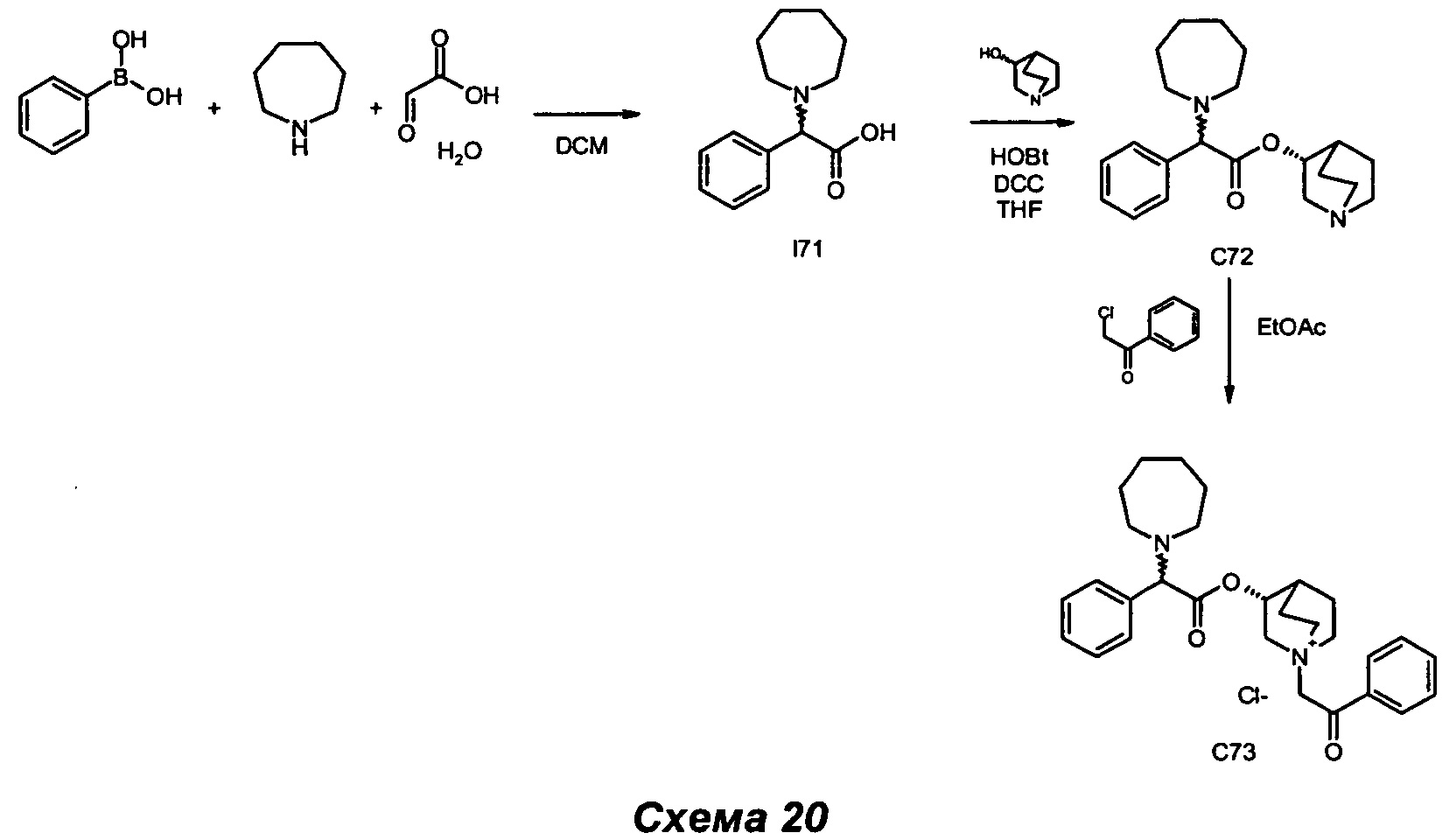
Получение 2-(азепан-1-ил)-2-фенилуксусной кислоты (I71)
Смесь фенилбороновой кислоты (400 мг, 3,28 ммоль), азепана (0,37 мл, 3,28 ммоль) и гидрата 2-оксоуксусной кислоты (302 мг, 3,28 ммоль), растворенную в DCM (30 мл), перемешивали при комнатной температуре в течение, ночи. DCM удаляли в вакууме, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 8/2) с получением соединения, указанного в заголовке (476 мг, выход 62,2%), в виде белого твердого вещества.
Получение (R)-хинуклидин-3-ил-2-(азепан-1-ил)-2-фенилацетата (С72)
Смесь 2-(азепан-1-ил)-2-фенилуксусной кислоты (476 мг, 2,04 ммоль), (R)-хинуклидин-3-ола (311 мг, 2,45 ммоль), НОВТ (375 мг, 2,45 ммоль) и DCC (505 мг, 2,45 ммоль) в THF (20 мл) перемешивали при комнатной температуре в течение выходных дней. THF выпаривали, неочищенный продукт растворяли EtOAc и дважды промывали 2 М K2CO3, а затем соляным раствором. Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение DCM/МеОН равно 95/5) с получением (R)-хинуклидин-3-ил-2-(азепан-1-ил)-2-фенилацетата (110 мг, выход 15,7%) в виде белого твердого вещества.
Получение (3R)-3-(2-(азепан-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (C73)
2-Хлор-1-фенилэтанон (46,9 мг, 0,30 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(азепан-1-ил)-2-фенилацетата (104 мг, 0,30 ммоль) в этилацетате (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли Et2O (2 мл), суспензию обрабатывали ультразвуком, фильтровали и высушивали в вакууме при 40°С в течение ночи с получением соединения, указанного в заголовке (99 мг, выход 65,6%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.08 (m, 2H) 7.70-7.84 (m, 1H) 7.56-7.65 (m, 2H) 7.25-7.49 (m, 5H) 5.13-5.39 (m, 2H) 4.59 и 4.61 (s, 1H) 4.06-4.30 (m, 1H) 3.48-3.83 (m, 4H) 2.58-2.79 (m, 4H) 2.25-2.33 и 2.33-2.42 (m, 1H) 1.76-2.20 (m, 4H) 1.40-1.70 (m, 8H).
СЭЖХ-МС (ИЭР положительная) 461,25 (М+).
ПРИМЕР 20
Получение (3R)-3-(2-((R-2-метилпирролидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С76)
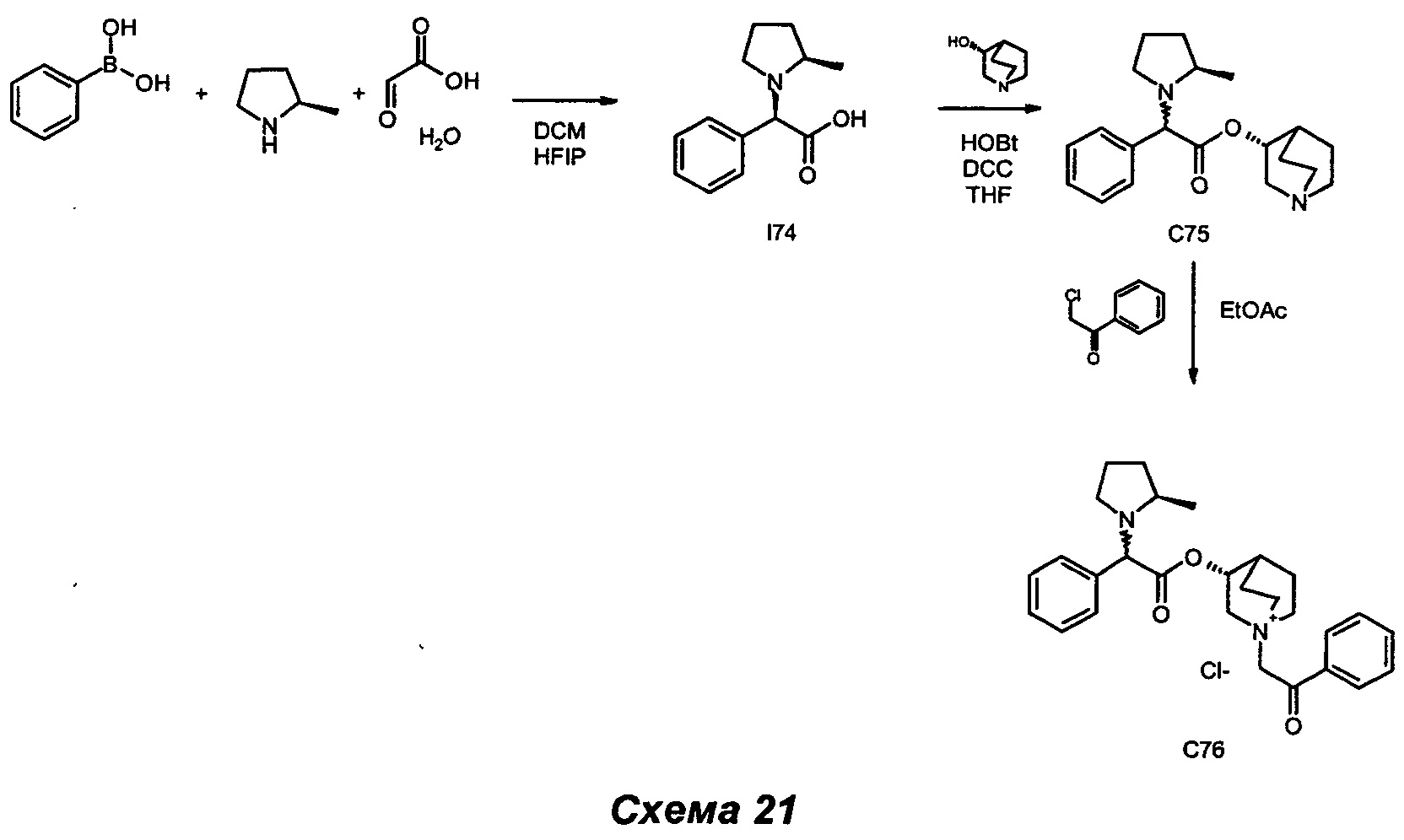
Получение (R)-2-((R)-2-метилпирролидин-1-ил)-2-фенилуксусной кислоты (I74)
(R)-2-Метилпирролидин (0,33 мл, 3,26 ммоль) и фенилбороновую кислоту (397 мг, 3,26 ммоль) последовательно добавляли к раствору гидрата 2-оксоуксусной кислоты (300 мг, 3,26 ммоль) в DCM (14,7 мл) и 1,1,1,3,3,3-гексафторпропан-2-ола (1,6 мл). Смесь перемешивали при комнатной температуре в течение суток, затем растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 8/2), в результате чего собирали соединение, указанное в заголовке (615 мг, выход 86%), в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.14-7.78 (m, 5H), 4.36 (s, 1H), 3.30 (tq, 1H), 3.03 (ddd, 1H), 2.69 (dt, 1H), 1.94-2.21 (m, 1H), 1.64-1.94 (m, 2H), 1.45-1.64 (m, 1H), 1.23(d, 3H).
СЭЖХ-МС (ИЭР положительная) 220,0 (МН+).
Получение (R)-хинуклидин-3-ил-2-((R)-2-метилпирролидин-1-ил)-2-фенилацетата (С75)
К суспензии (R2)-2-((R)-2-метилпирролидин-1-ил)-2-фенилуксусной кислоты 22 с (150 мг, 0,68 ммоль) в сухом THF (6,8 мл) добавляли DCC (282 мг, 1,37 ммоль), НОВТ (185 мг, 1,37 ммоль) и (R)-хинуклидин-3-ол (174 мг, 1,37 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 24 часов. Бледно-желтую суспензию фильтровали, промывая THF (5 мл), и. желтый раствор выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно от 95/5 до 9/1, а затем DCM/MeOH/NH4OH равно 95/5/0,2), в результате чего собирали соединение, указанное в заголовке (48 мг, выход 21,4%), в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.22-7.47 (m, 10H) 4.77 (dt, 1H) 4.62-4.72 (m, 1H) 4.54 (s, 1H) 4.37 (s, 1H) 3.13 (ddd, 1H) 2.90-3.07 (m, 2H) 2.55-2.86 (m, 10H) 2.45 (br. s., 1H) 2.31-2.44 (m, 1H) 2.18-2.29 (m, 1H) 1.76-2.00 (m, 5H) 1.41-1.72 (m, 10H) 1.13-1.41 (m, 5H) 0.95 (d, 3H) 0.91 (d, 3H).
СЭЖХ-МС (ИЭР положительная) 329,0 (МН+).
Получение (3R)-3-(2-((R)-2-метилпирролидин-1-ил)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С76)
(R)-Хинуклидин-3-ил-2-((R)-2-метилпирролидин-1-ил)-2-фенилацетат (24с) (44 мг, 0,13 ммоль) растворяли в смеси этилацетата (0,9 мл) и ацетонитрила (0,4 мл). Добавляли 2-хлор-1-фенилэтанон (22,8 мг, 0,15 ммоль), и смесь перемешивали при комнатной температуре в течение 48 ч. Раствор выпаривали, и остаток очищали сначала флэш-хроматографией (соотношение DCM/MeOH равно от 90/10 до 85/15), а затем растиранием со смесью Et2O/DCM (примерно 5/1) с получением соединения, указанного в заголовке (23 мг, выход 35,5%), в виде бледно-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.10 (m, 2H), 7.70-7.83 (m, 1H), 7.54-7.70 (m, 2H), 7.22-7.52 (m, 5H), 5.23 и 5.26 (s, 2H), 5.24-5.34 (m, 1H), 5.13-5.22 (m, 1H), 4.53 и 4.66 (s, 1H), 4.02-4.29 (m, 1H), 3.48-3.85 (m, 5H), 2.62-3.09 (m, 2H), 2.31-2.42 (m, 1H), 1.49-2.31 (m, 7H), 1.28-1.46 (m, 1H), 0.95 и 0.99 (d, 3H).
СЭЖХ-МС (ИЭР положительная) 447,07 (М+).
ПРИМЕР 21
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(2-оксопирролидин-1-ил)-2-фенилацетокси)-1-азониабицикло[2.2.2]октанхлорида (С80)
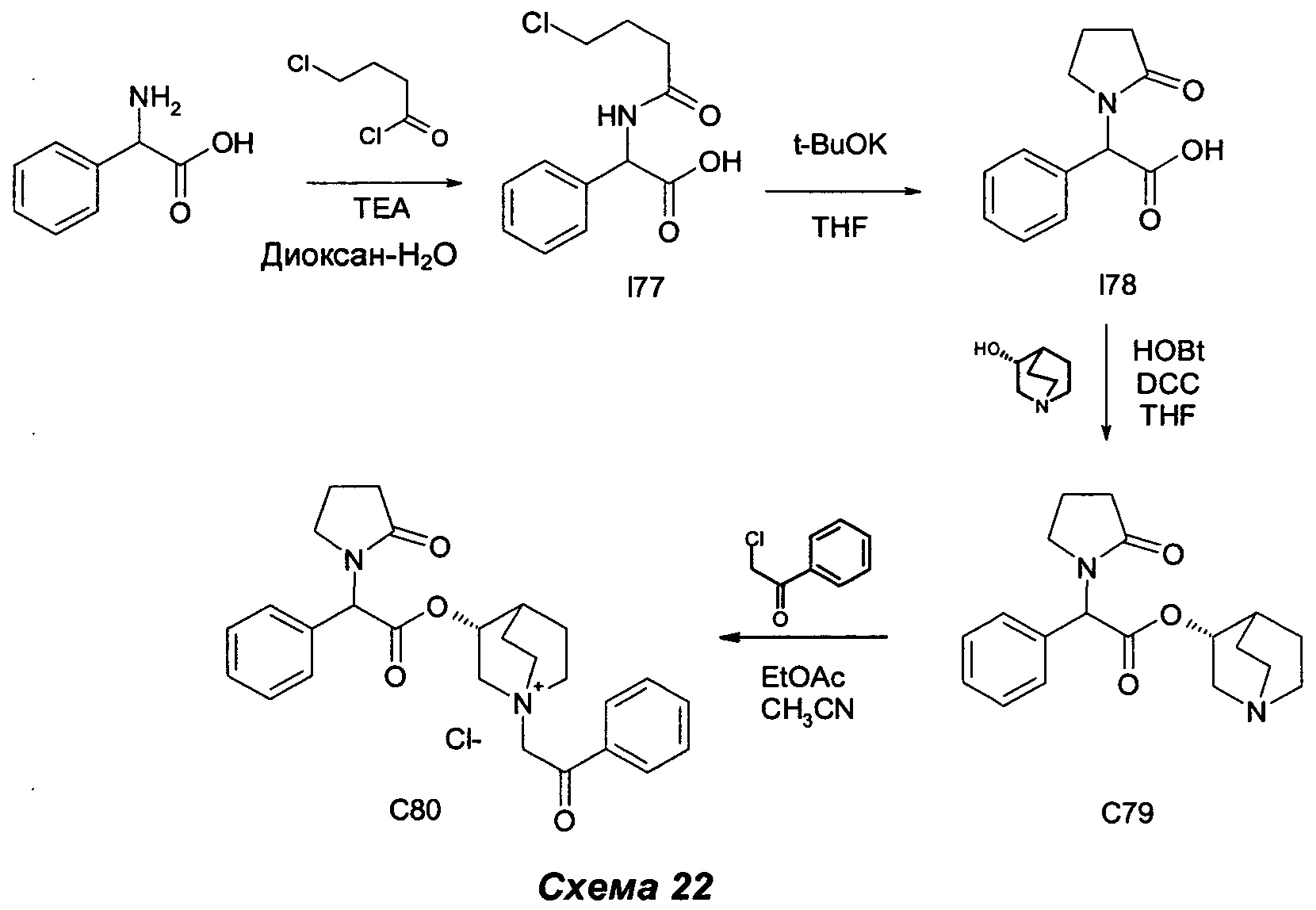
Получение 2-(4-хлорбутанамидо)-2-фенилуксусной кислоты (I77)
2-Амино-2-фенилуксусную кислоту (1,00 г, 6,62 ммоль) и TEA (2,77 мл, 19,8 ммоль) растворяли в смеси диоксана (13,2 мл) и воды (13,2 мл). Смесь охлаждали при 0°C с помощью ледяной бани и добавляли по каплям 4-хлорбутаноилхлорид (814 мкл, 7,28 ммоль). Смесь перемешивали при той же температуре в течение 1 часа, а затем диоксан выпаривали. Водный раствор подкисляли 1 н. HCl, а затем экстрагировали три раза EtOAc (20 мл × 3). Органическую фазу высушивали (Na2SO4) и выпаривали. Сырое бледно-желтое масло очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1), и собранное соединение растирали с Et2O (примерно 15 мл) с получением соединения, указанного в заголовке (658 мг, выход 38,9%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.59 (d, 1Н), 7.09-7.57 (m, 5Н), 5.33 (d, 1Н), 3.63 (t, 2Н), 2.24-2.45 (m, 2Н), 1.94 (quin, 2Н).
СЭЖХ-МС (ИЭР) 255,9 m/z (MH+).
Получение 2-(2-оксопирролидин-1-ил)-2-фенилуксусной кислоты (I78)
2-(4-Хлорбутанамидо)-2-фенилуксусную кислоту (650 мг, 2,54 ммоль) растворяли в сухом THF (8,5 мл), и раствор охлаждали до 0°С с помощью ледяной бани, перемешивая в атмосфере азота. Добавляли mpem-бутилат калия (599 мг, 5,34 ммоль) тремя порциями, и получили белую суспензию. Смесь перемешивали при той же температуре в течение 15 мин. Добавляли по каплям 1 н. HCl до рН 2-3, а затем смесь разбавляли водой (10 мл) и экстрагировали три раза EtOAc (15 мл × 3). Органическую фазу высушивали (Na2SO4) и выпаривали с получением соединения, указанного в заголовке (352 мг, выход 63,2%) в виде бледно-желтого губчатого липкого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 13.05 (br. s., 1Н) 7.32-7.47 (m, 3Н) 7.21-7.32 (m, 2Н) 5.68 (s, 1Н) 3.51 (td, 1Н) 2.86 (td, 1Н) 2.20-2.34 (m, 2Н) 1.70-2.03(m, 2H).
СЭЖХ-МС (ИЭР) 219,9 m/z (MH+).
Получение (R)-хинуклидин-3-ил-2-(2-оксопирролидин-1-ил)-2-фенилацетата (С79)
Смесь 2-(2-оксопирролидин-1-ил)-2-фенилуксусной кислоты (345 мг, 1,57 ммоль), DCC (649 мг, 3,15 ммоль), НОВТ (425 мг, 3,15 ммоль) и (R)-хинуклидин-3-ола (400 мг, 3,15 ммоль) в сухом THF (15,7 мл) перемешивали при комнатной температуре в течение 24 ч. Желтую суспензию фильтровали, промывая белое твердое вещество THF. Желтый раствор выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 96/4 + 0,4% NH4OH), в результате чего собрали соединение, указанное в заголовке (482 мг, выход 93%), в виде бледно-желтого масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.17-7.50 (m, 5H), 5.75 (s, 1H), 4.74-4.91 (m, 1H), 3.43-3.54 (m, 1H), 3.05-3.20 (m, 1H), 2.92 (td, 1H), 2.53-2.76 (m, 5H), 2.30 (t, 2H), 1.77-2.05 (m, 3H), 1.11-1.74 (m, 4H);
СЭЖХ-МС (ИЭР) 329,0 m/z (MH+).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(2-оксопирролидин-1-ил)-2-фенилацетокси)-1-азониабицикло[2.2.2]октанхлорида (С80)
2-Хлор-1-фенилэтанон (62,1 мг, 0,40 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(2-оксопирролидин-1-ил)-2-фенилацетата (120 мг, 0,36 ммоль) в EtOAc (2,4 мл) и ацетонитриле (1,2 мл). Бледно-желтый раствор перемешивали при комнатной температуре в течение трех суток. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1), в результате чего собрали соединение, указанное в заголовке (118 мг, выход 66,9%), в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.92-8.09 (m, 2H), 7.70-7.83 (m, 1H), 7.53-7.70 (m, 2H), 7.23-7.53 (m, 5H), 5.83 (s, 1H), 5.26-5.39 (m, 3H), 4.10-4.43 (m, 1H), 3.40-3.97 (m, 6H), 2.85-3.08 (m, 1H), 2.26-2.46 (m, 3H), 1.63-2.15 (m, 6H).
СЭЖХ-МС (ИЭР) 447,21 m/z (M+).
ПРИМЕР 22
Получение (3R)-3-(2-(3-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С84)
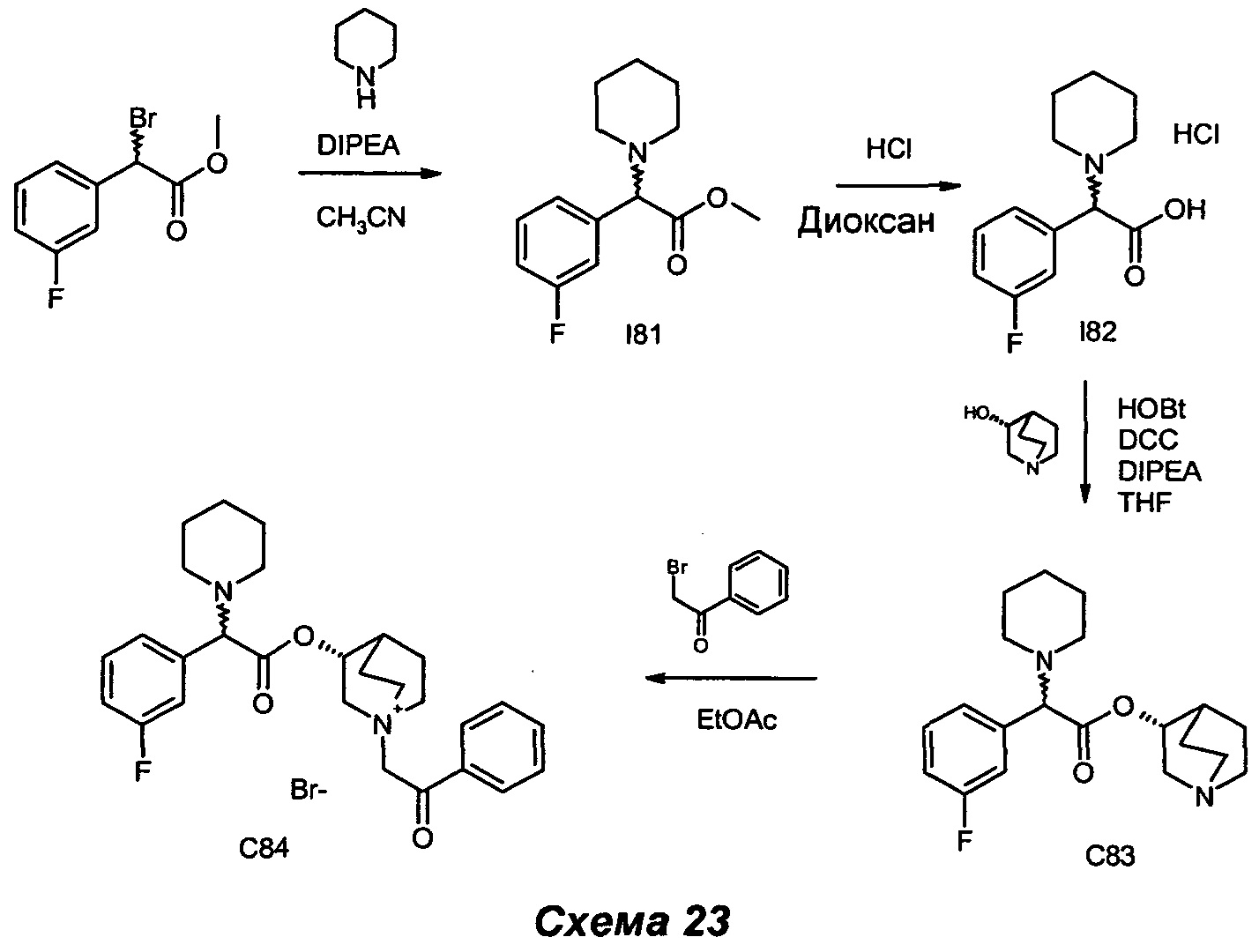
Получение метил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетата (I81)
Метил-2-бром-2-(3-фторфенил)ацетат (310 мг, 1,25 ммоль), пиперидин (0,15 мл, 1,51 ммоль) и DIPEA (0,26 мл, 1,51 ммоль) растворяли в ацетонитриле (15 мл) и перемешивали при комнатной температуре в течение 16 ч. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 9/1) с получением метил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетата (224 мг, выход 71%) в виде бесцветного масла.
СЭЖХ-МС (ИЭР) 252,1 m/z (MH+).
Получение гидрохлорида 2-(3-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (I82)
Метил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетат (224 мг, 0,89 ммоль) и 37%-й HCl (813 мкл, 26,7 ммоль) растворяли в диоксане (3 мл) и перемешивали при микроволновом облучении при 100°С в течение 6 часов. Растворители выпаривали, и остаток растирали с Et2O и обрабатывали ультразвуком. Твердое вещество собирали фильтрованием с аспирацией, промывали Et2O и высушивали в вакууме в течение ночи, в результате чего собрали гидрохлорид 2-(3-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (230 мг, выход 94%).
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.58 (td, 1Н), 7.50 (ddd, 1H), 7.44 (ddd, 1H), 7.38 (dddd, 1H), 5.31 (s, 1H), 2.90-3.07 (m, 4H), 1.39-1.92 (m, 6H). Получение (R)-хинуклидин-3-ил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетата (С83)
Гидрохлорид 2-(3-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (225 мг, 0,82 ммоль), DCC (339 мг, 1,64 ммоль) и НОВТ (252 мг, 1,64 ммоль) растворяли в сухом THF (9 мл). Добавляли (R)-хинуклидин-3-ол (314 мг, 2,47 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. THF выпаривали, остаток растворяли EtOAc и промывали насыщенным раствором NaHCO3, водой и соляным раствором. Органический слой высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением (R)-хинуклидин-3-ил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетата (166 мг, выход 58,3%).
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.31-7.53 (m, 1H), 7.01-7.31 (m, 3H), 4.73 (td, 1H), 4.15 (s, 1H), 2.98-3.20 (m, 1H), 2.55-2.78 (m, 4H), 2.30-2.47 (m, 5H), 1.74-1.85 и 1.85-1.95(m, 1H), 1.01-1.68 (m, 10H).
Получение (3R)-3-(2-(3-Фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С84)
(R)-Хинуклидин-3-ил-2-(3-фторфенил)-2-(пиперидин-1-ил)ацетат (164 мг, 0,47 ммоль) и 2-бром-1-фенилэтанон (104 мг, 0,52 ммоль) растворяли в ацетонитриле (5 мл) и перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, и неочищенный продукт растирали с Et2O, фильтровали с аспирацией и промывали EtOAc и Et2O (1/1) с получением (3R)-3-(2-(3-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (180,5 мг; выход 69,9%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.08 (m, 2H), 7.71-7.88 (m, 1H), 7.56-7.70 (m, 2H), 7.38-7.53 (m, 1H), 7.08-7.37 (m, 3H), 5.24-5.31 (m, 1H), 5.23 и 5.24 (br. s., 2H), 4.30 и 4.31 (s, 1H), 4.04-4.23 (m, 1H), 3.52-3.89 (m, 5H), 2.22-2.47 (m, 5H), 1.76-2.17 (m, 4H), 1.28-1.63 (m, 6H);
СЭЖХ-МС (ИЭР) 465,18 m/z (M+).
ПРИМЕР 23
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-пара-толуилацетокси)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (С88)
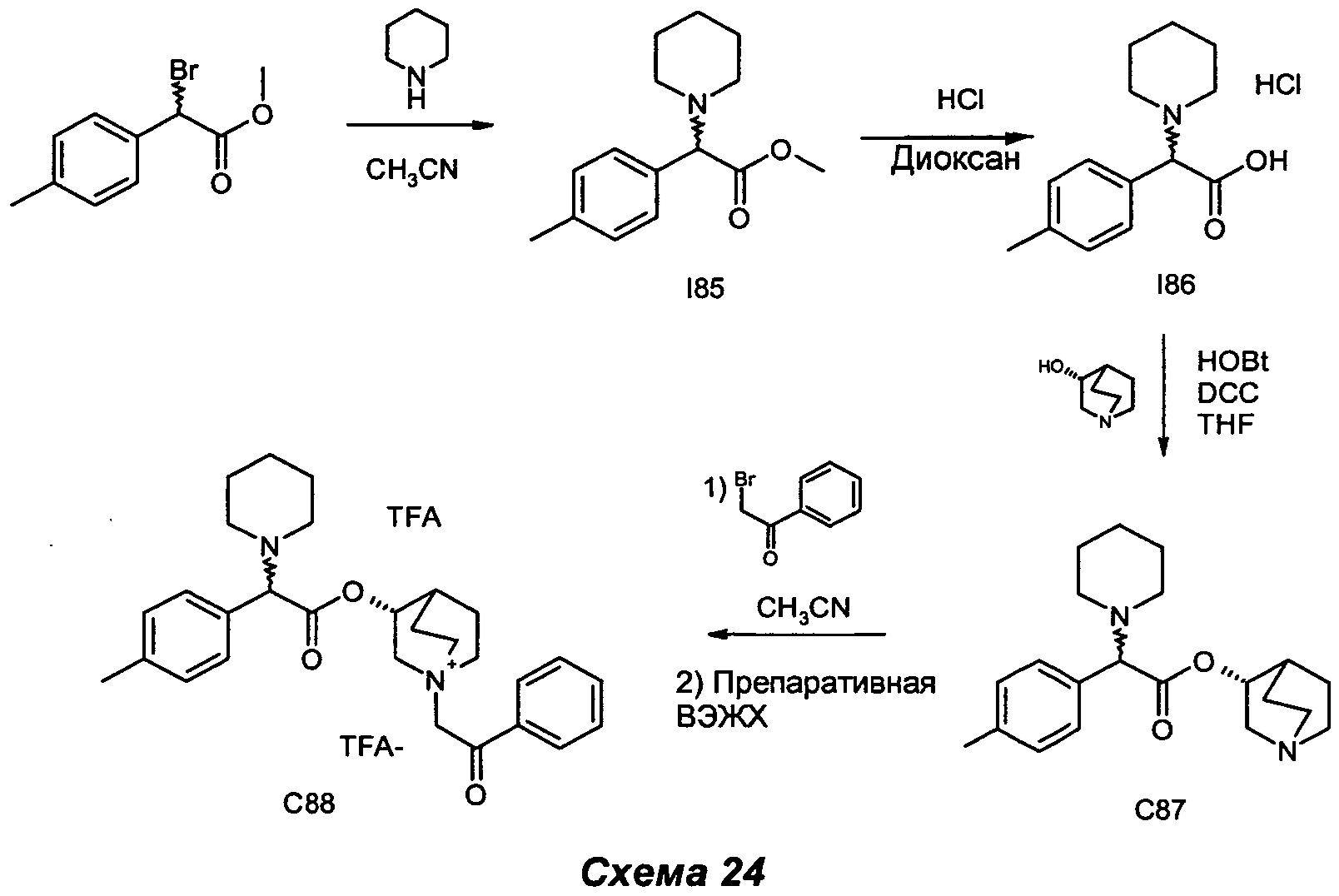
Получение метил-2-(пиперидин-1-ил)-2-пара-толуилацетата (I85)
К раствору метил-2-бром-2-пара-толуилацетата (2,50 г, 10,3 ммоль) в ацетонитриле добавляли пиперидин (2,06 мл, 20,6 ммоль), и смесь подвергали взаимодействию в течение 2 часов при комнатной температуре. Растворитель выпаривали, и полученный в результате неочищенный продукт распределяли между водой и EtOAc. Водную фазу экстрагировали EtOAc, органические фазы объединяли, высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 9/1) с получением метил-2-(пиперидин-1-ил)-2-пара-толуилацетата (273 мг, выход 21%) в виде бесцветного масла.
СЭЖХ-МС (ИЭР) 248,2 m/z (MH+).
Получение 2-(пиперидин-1-ил)-2-пара-толуилуксусной кислоты гидрохлорида (I86)
К раствору метил-2-(пиперидин-1-ил)-2-лара-толуилацетата (273 мг, 1,10 ммоль) в диоксане (2 мл) добавляли 37%-й HCl (3,35 мл, 110 ммоль).Реакционную смесь нагревали при микроволновом облучении при 100°С в течение 4 часов. Растворители выпаривали, и остаток растирали с Et2O, в результате чего собрали 2-(пиперидин-1-ил)-2-пара-толуилуксусной кислоты гидрохлорид (242 мг, выход 81%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 10.27 (br. s., 1H), 7.39-7.52 (m, 2H), 7.21-7.39 (m, 2H), 5.19 (s, 1H), 2.80-3.18 (m, 4H), 2.34 (s, 3H), 1.79 (br. s., 4H), 1.50(br. s., 2H).
Получение (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-лара-толуилацетата (С87)
Гидрохлорид 2-(пиперидин-1-ил)-2-пара-толуилуксусной кислоты (242 мг, 0,90 ммоль), DCC (370 мг, 1,79 ммоль) и НОВТ (275 мг, 1,79 ммоль) растворяли в сухом THF (10 мл). Добавляли (R)-хинуклидин-3-ол (342 мг, 2,69 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. THF выпаривали, а затем остаток растворяли EtOAc и промывали насыщенным раствором NaHCO3, водой и соляным раствором. Органический слой выделяли, высушивали над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-пара-толуилацетата (169 мг, выход 55%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.23-7.38 (m, 2H), 7.06-7.23 (m, 2H), 4.56-4.78 (m, 1H), 4.00 и 4.01 (s, 1H), 3.03 и 3.09 (ddd, 1H), 2.54-2.77 (m, 4H), 2.39-2.47 (m, 1H), 2.30-2.39 (m, 4H), 2.29 (s, 3H), 1.73-1.82 и 1.85-1.93(m, 1H), 1.08-1.74 (m, 10H).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-пара-толуилацетокси)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (С88)
(R)-Хинуклидин-3-ил-2-(пиперидин-1-ил)-2-пара-толуилацетат (160 мг, 0,47 ммоль) и 2-бром-1-фенилэтанон (102 мг, 0,51 ммоль) растворяли в ацетонитриле (5 мл) и перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, и неочищенный остаток растирали с Et2O и собирали фильтрованием с аспирацией. Затем твердое вещество очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) и окончательно очищали препаративной ВЭЖХ с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-пара-толуилацетокси)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (114 мг, выход 35,4%) в виде белого порошка.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.02 (m, 2H), 7.71-7.81 (m, 1H), 7.55-7.67 (m, 2H), 7.42-7.51 (m, 2H), 7.29-7.42 (m, 2H), 5.30-5.44 (m, 2H), 5.16 и 5.19 (s, 2H), 4.01-4.29 (m, 1H), 3.36-3.91 (m, 5H), 2.69-3.11 (m, 4H), 2.36 (s, 3H), 2.22 и 2.45 (br. s., 1H), 1.24-2.16 (m, 10H);
СЭЖХ-МС (ИЭР) 461,13 m/z (M+).
ПРИМЕР 24
Получение (3R)-3-(2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (С92)
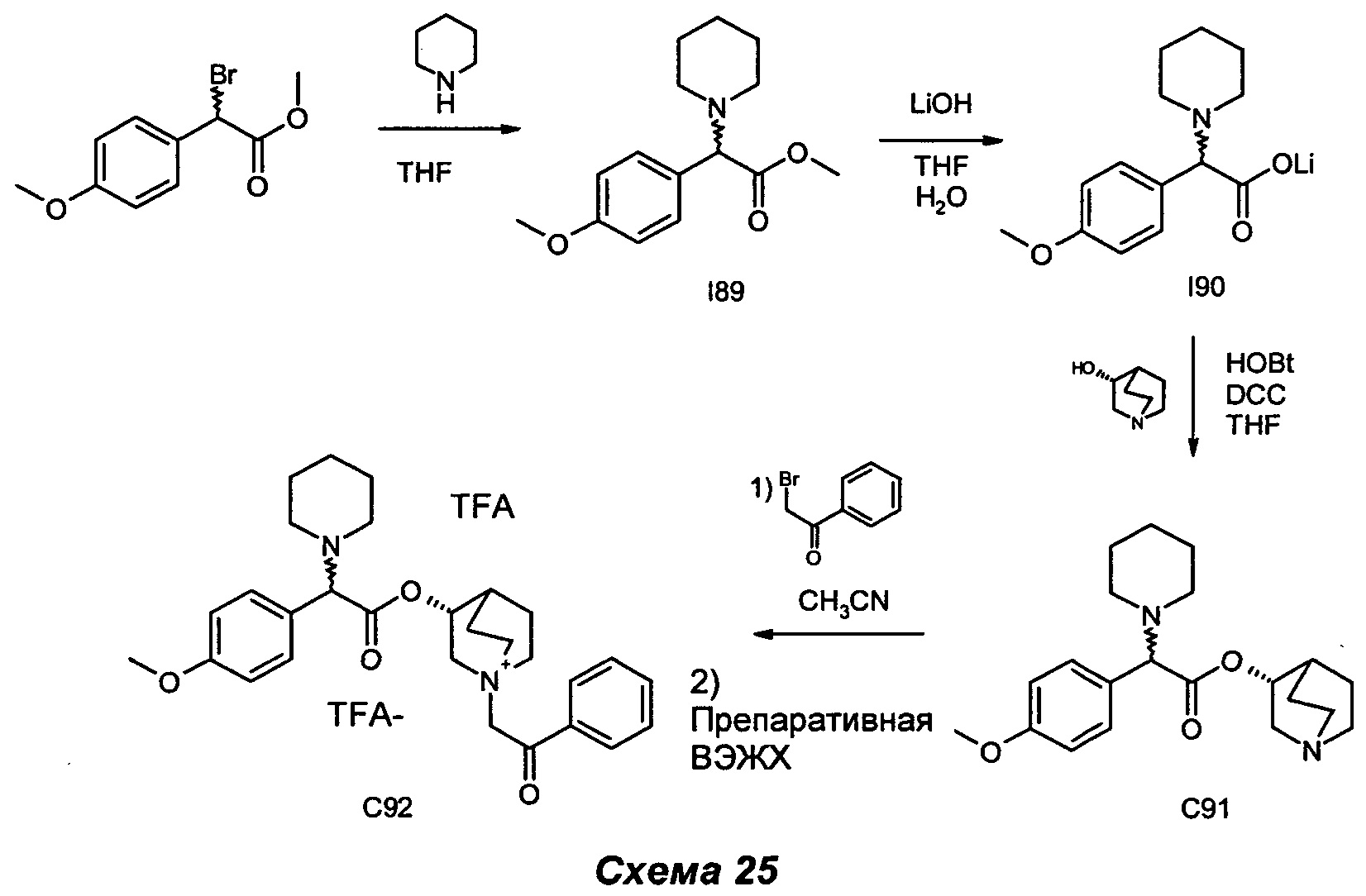
Получение метил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата (I89)
К раствору метил-2-бром-2-(4-метоксифенил)ацетата (2,5 г, 9,65 ммоль) в THF (50 мл) добавляли пиперидин (1,64 г, 19,3 ммоль), и смесь подвергали взаимодействию в течение 2 часов при комнатной температуре. Растворитель выпаривали, и полученный в результате неочищенный продукт распределяли между водой и EtOAc. Водную фазу экстрагировали EtOAc, органические фазы объединяли, высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 85/15) с получением метил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата (1,81 г, выход 71,4%).
СЭЖХ-МС (ИЭР) 264,2 m/z (MH+).
Получение 2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата лития (I90)
Метил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетат (400 мг, 1,52 ммоль) растворяли в смеси THF/вода (12 мл / 4 мл). Добавляли гидрат гидроксида лития (127 мг, 3,04 ммоль), и раствор перемешивали при комнатной температуре в течение 8 часов. Затем добавляли вторую порцию гидрата гидроксида лития (63,7 мг, 1,52 ммоль), и смесь перемешивали при 40°С в течение 16 часов, затем при 50°С в течение 7 часов. Растворители выпаривали, остаток растворяли Et2O, и нерастворимое вещество отфильтровывали. Прозрачный раствор выпаривали с получением 2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата лития (384 мг, выход 99%) в виде белого твердого вещества.
Получение (R)-хинуклидин-3-ил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата (С91)
2-(4-Метоксифенил)-2-(пиперидин-1-ил)ацетат лития (384 мг, 1,50 ммоль), DCC (621 мг, 3,01 ммоль) и НОВТ (461 мг, 3,01 ммоль) растворяли в сухом THF (16 мл). Добавляли (R)-хинуклидин-3-ол (574 мг, 4,51 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. THF выпаривали, и неочищенный продукт растворяли в EtOAc, промывали насыщенным раствором NaHCO3, водой и соляным раствором. Органический слой высушивали над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 9/1/0,02) с получением (R)-хинуклидин-3-ил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетата (207 мг, выход 38,4%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.23-7.37 (m, 2H), 6.80-6.99 (m, 2H), 4.55-4.84 (m, 1H), 3.97 (s, 1H), 3.74 и 3.75 (s, 3H), 3.02 и 3.07 (ddd, 1H), 2.54-2.70 (m, 5H), 2.17-2.47 (m, 4H), 1.72-1.81 и 1.84-1.93 (m, 1H), 1.06-1.70 (m, 10H).
Получение (3R)-3-(2-(4-метоксиФенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октантрифторацетата, трифторацетатный анион (С92)
(R)-Хинуклидин-3-ил-2-(4-метоксифенил)-2-(пиперидин-1-ил)ацетат (207 мг 0,58 ммоль) и 2-бром-1-фенилэтанон (126 мг, 0,63 ммоль) растворяли в ацетонитриле (6 мл) и перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали, и остаток растирали с Et2O, а затем очищали флэш-хроматографией (соотношение DCM/MeOH равно 88/22) и препаративной ВЭЖХ, в результате чего собрали соединение, указанное в заголовке (51 мг, выход 12,5%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 10.48 (br. s., 1H), 7.89-8.11 (m, 2H), 7.69-7.84 (m, 1H), 7.56-7.69 (m, 2H), 7.42-7.56 (m, 2H), 6.91-7.25 (m, 2H), 5.32-5.54 (m, 2H), 5.17 и 5.20 (s, 2H), 3.99-4.32 (m, 1H), 3.81 (s, 3H), 3.37-3.74 (m, 5H), 2.61-3.15 (m, 4H), 2.19-2.30 и 2.40-2.55 (m, 1H), 1.21-2.14(m, 10Н);
СЭЖХ-МС (ИЭР) 477,23 m/z (M+).
ПРИМЕР 25
Получение (3R)-3-(2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С95)
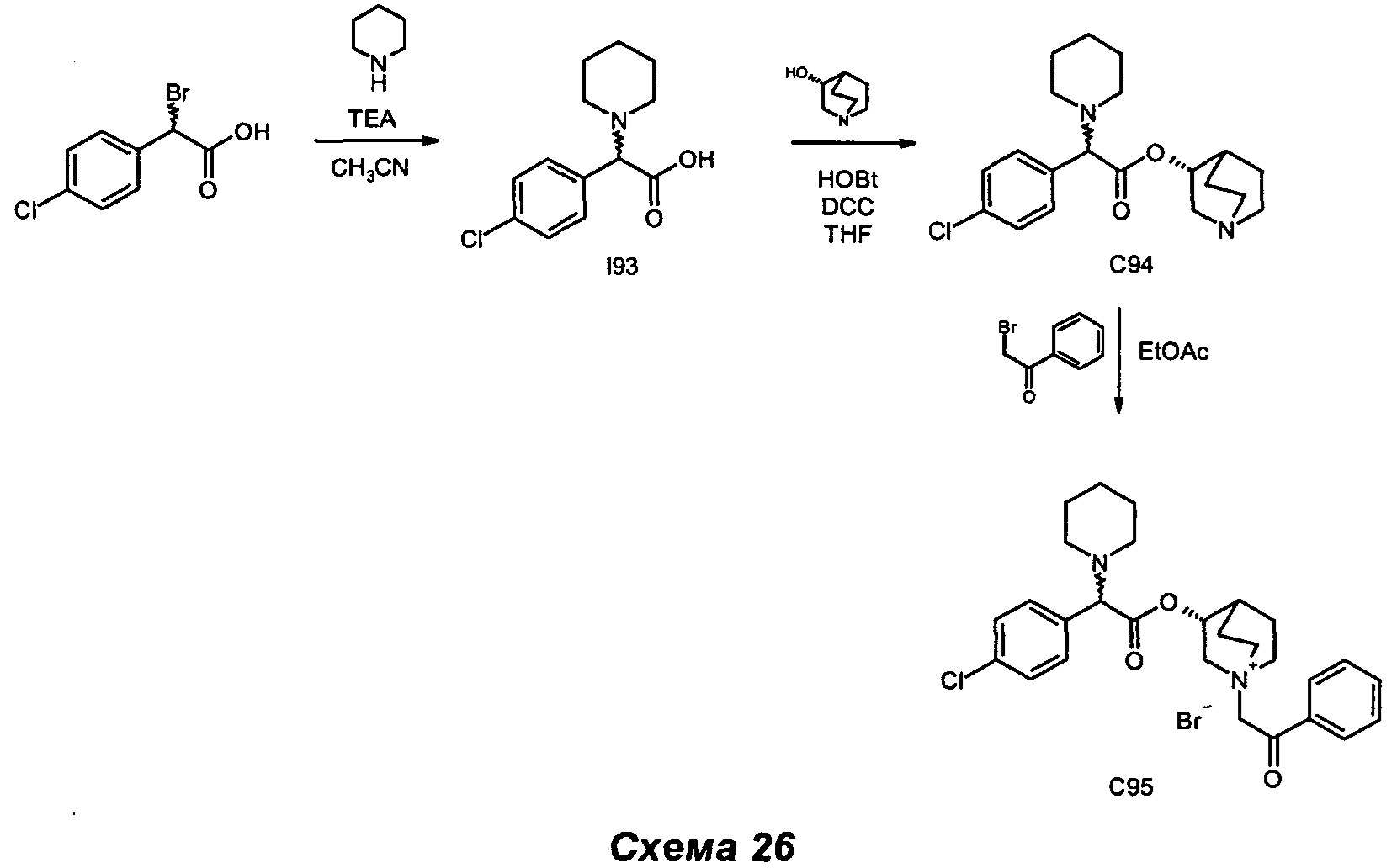
Получение 2-(4-хлорфенил)-2-(пиперидин-1-ил)уксусной кислоты (193) 2-Бром-2-(4-хлорфенил)уксусную кислоту (300 мг, 1,20 ммоль), пиперидин (125 мкл, 1,26 ммоль) и TEA (503 мкл, 3,61 ммоль) растворяли в ацетонитриле (6 мл). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Ацетонитрил выпаривали, остаток растирали с ацетонитрилом/петролейным эфиром (9/1) и фильтровали с аспирацией, в результате чего собирали 2-(4-хлорфенил)-2-(пиперидин-1-ил)уксусную кислоту (190 мг, выход 62,3%) в виде белого твердого вещества.
Получение (R)-хинуклидин-3-ил-2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетата (С94)
2-(4-Хлорфенил)-2-(пиперидин-1-ил)уксусную кислоту (190 мг, 0,75 ммоль), DCC (309 мг, 1,50 ммоль) и НОВТ (229 мг, 1,50 ммоль) растворяли в сухом THF (8 мл). Добавляли (R)-хинуклидин-3-ол (286 мг, 2,25 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. THF выпаривали, и остаток растворяли в EtOAc и промывали 1 М NaHCO3, водой и соляным раствором. Органический слой отделяли, высушивали над Na2SO4, фильтровали и выпаривали. Сырое соединение очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 95/5/0,1) с получением (R)-хинуклидин-3-ил-2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетата (167 мг, выход 61,5%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.33-7.47 (m. 4H), 4.57-4.95 (m, 1H), 4.12 (s, 1H), 3.04 и 3.09 (ddd, 1H), 2.56-2.78 (m, 4H), 2.18-2.47 (m, 5H), 1.72-1.83 и 1.85-1.95 (m, 1H), 1.09-1.70 (m, 10H).
Получение (3R)-3-(2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (С95)
(R)-хинуклидин-3-ил-2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетат (100 мг, 0,28 ммоль) и 2-бром-1-фенилэтанон (60,3 мг, 0,30 ммоль) растворяли в ацетонитриле (4 мл) и перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, и полученный в результате остаток очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1) с получением (3R)-3-(2-(4-хлорфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанбромида (98 мг, выход 63,3%) в виде белого порошка.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.94-8.03 (m, 2H) 7.71-7.81 (m, 1H) 7.57-7.67 (m, 2H) 7.41-7.51 (m, 4H) 5.14-5.29 (m, 3H) 4.24-4.30 (m, 1H) 4.05-4.21 (m, 1H) 3.52-3.82 (m, 5H) 2.34-2.45 (m, 4H) 2.20-2.34 (m, 1H) 1.78-2.16 (m, 4H) 1.29-1.66 (m, 6H);
СЭЖХ-МС (ИЭР) 481,25 m/z (M+).
ПРИМЕР 26
Получение (3R)-3-(2-(4-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С99)
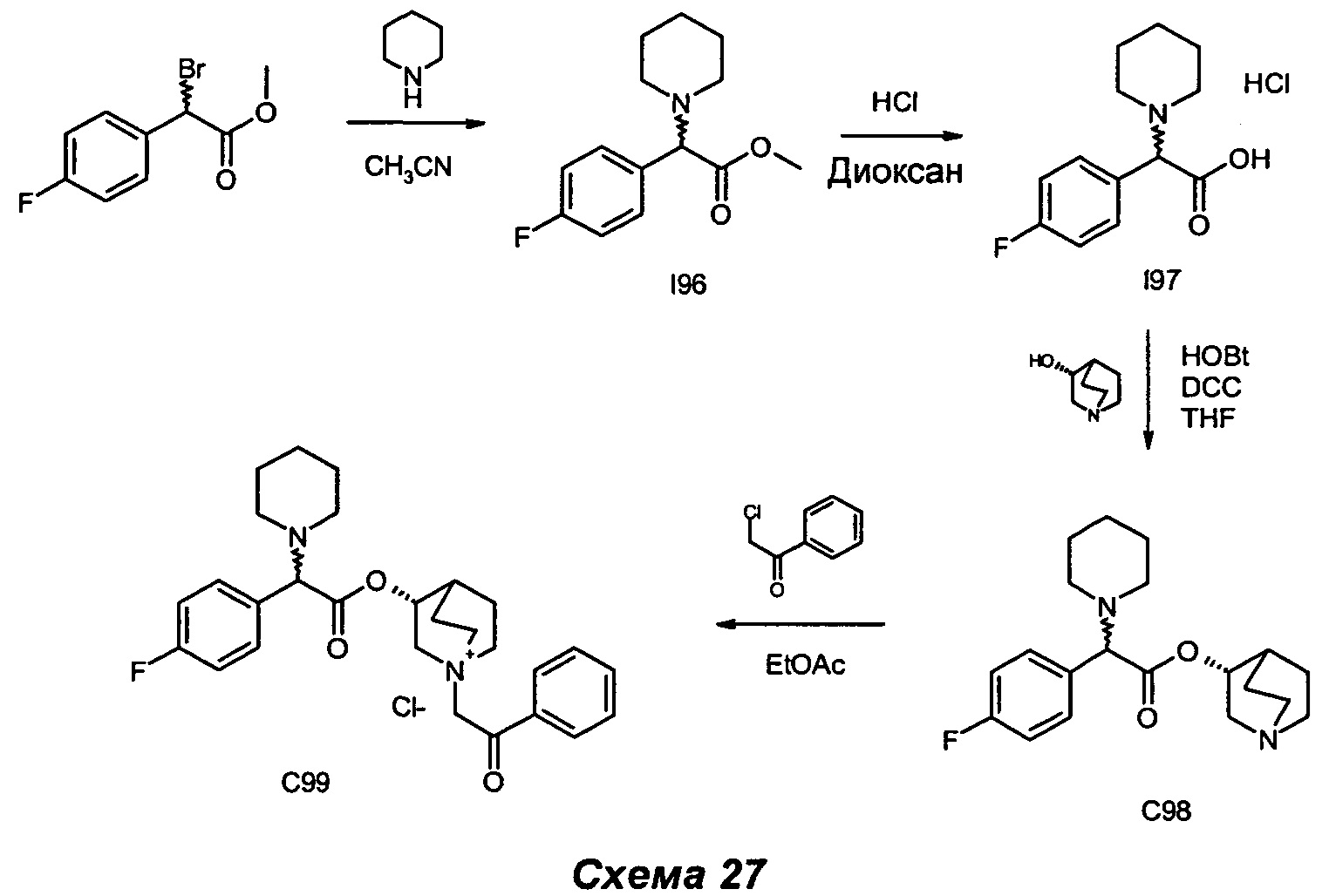
Получение метил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (I96)
Метил-2-бром-2-(4-фторфенил)ацетат (538 мг, 2,18 ммоль) растворяли в CH3CN (6,6 мл) и последовательно добавляли пиперидин (258 мкл, 2,61 ммоль) и N-этил-N-изопропилпропан-2-амин (456 мкл, 2,61 ммоль). Раствор перемешивали при комнатной температуре в течение 3 ч, затем растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение петролейный эфир/EtOAc равно 9/1) в результате чего собирали метил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетат (320 мг, выход 58.5%) в виде бесцветного масла.
Получение гидрохлорида 2-(4-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (I97)
37%-й хлорид водорода (4,18 мл, 50,9 ммоль) добавляли к раствору метил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (320 мг, 1,27 ммоль) в диоксане (5 мл). Реакционную смесь перемешивали при микроволновом облучении при 100°С в течение 5 ч. Растворитель выпаривали, и твердое вещество растирали с ацетонитрилом с получением гидрохлорида 2-(4-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (255 мг, выход 73%) в виде белого твердого вещества.
Получение (R)-хинуклидин-3-ил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (С98)
Гидрохлорид 2-(4-фторфенил)-2-(пиперидин-1-ил)уксусной кислоты (255 мг, 0,93 ммоль) растворяли в сухом THF (10 мл) и добавляли (R)-хинуклидин-3-ол (355 мг, 2,79 ммоль), DCC (384 мг, 1,86 ммоль), HOBt (285 мг, 1,86 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. THF выпаривали, и остаток растворяли EtOAc и промывали насыщенным раствором NaHCO3, водой и соляным раствором. Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 98/2/0,2) с получением (R)-хинуклидин-3-ил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (126 мг, выход 39,0%) в виде белого твердого вещества.
Получение (3R)-3-(2-(4-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С99)
2-Хлор-1-фенилэтанон (29,5 мг, 0,19 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (60 мг, 0,17 ммоль) в EtOAc (3 мл). Добавляли 2-Хлор-1-фенилэтанон (8,85 мг, 0,06 ммоль), и перемешивание поддерживали дополнительно в течение 4 часов. Добавляли Et2O, и осадок собирали фильтрованием с аспирацией с получением (3R)-3-(2-(4-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (49,9 мг, выход 57,5%) в виде смолистого желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.91-8.09 (m, 2H), 7.69-7.80 (m, 1H), 7.56-7.69 (m, 2H), 7.39-7.55 (m, 2H), 7.10-7.30 (m, 2H), 5.24 и 5.26 (s, 2H), 5.10-5.23 (m, 1H), 4.24 и 4.25 (s, 1H), 4.09-4.22 (m, 1H), 3.51-3.82 (m, 5H), 2.20-2.45 (m, 4H), 1.71-2.16 (m, 5H), 1.33-1.61 (m, 6H)
СЭЖХ-МС (ИЭР) 465,24 m/z (MH+).
ПРИМЕР 27
Получение (3R)-1-(2-(4-хлорфенил)-2-оксоэтил)-3-(2-фенил-2-(пиперидин-1-ил)ацетокси)-1-азониабицикло[2.2.2]октанбромида (С100)
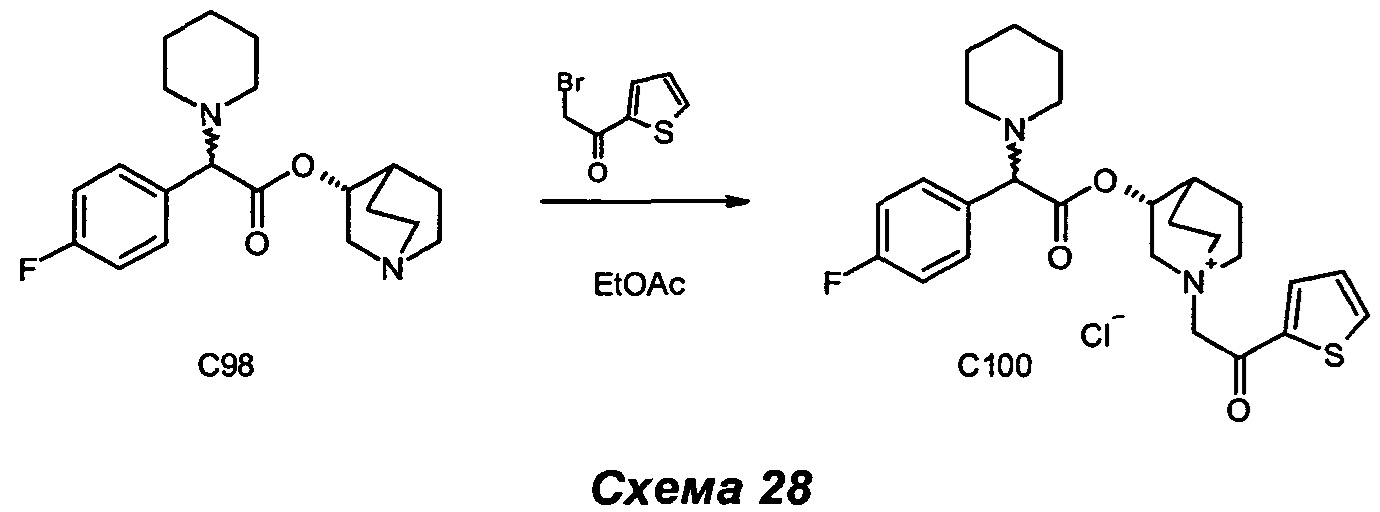
2-Хлор-1-(тиофен-2-ил)этанон (30,6 мг, 0,19 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(4-фторфенил)-2-(пиперидин-1-ил)ацетата (60 мг, 0,17 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли Et2O, осадок собирали фильтрованием и очищали флэш-хроматографией (соотношение DCM/MeOH/NH4OH равно 97/3/0,3) с получением (3R)-3-(2-(4-фторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азониабицикло[2.2.2]октанхлорида (23 мг, выход 26,2%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 8.18-8.31 (m, 2H) 8.03-8.18 (m, 1H) 7.79 (br. s., 1H) 7.40-7.62 (m, 1H) 7.31-7.40 (m, 2H) 7.03-7.31 (m, 1H) 5.06-5.41 (m, 2H) 3.97-4.31 (m, 2H) 3.51-3.88 (m, 5H) 2.33-2.46 (m, 4H) 1.71-2.08 (m, 5H) 1.29-1.67 (m, 6H);
СЭЖХ-МС (ИЭР) 471,15 m/z (M+).
ПРИМЕР 28
Получение (3R)-3-(2-(2,4-дифторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С103)
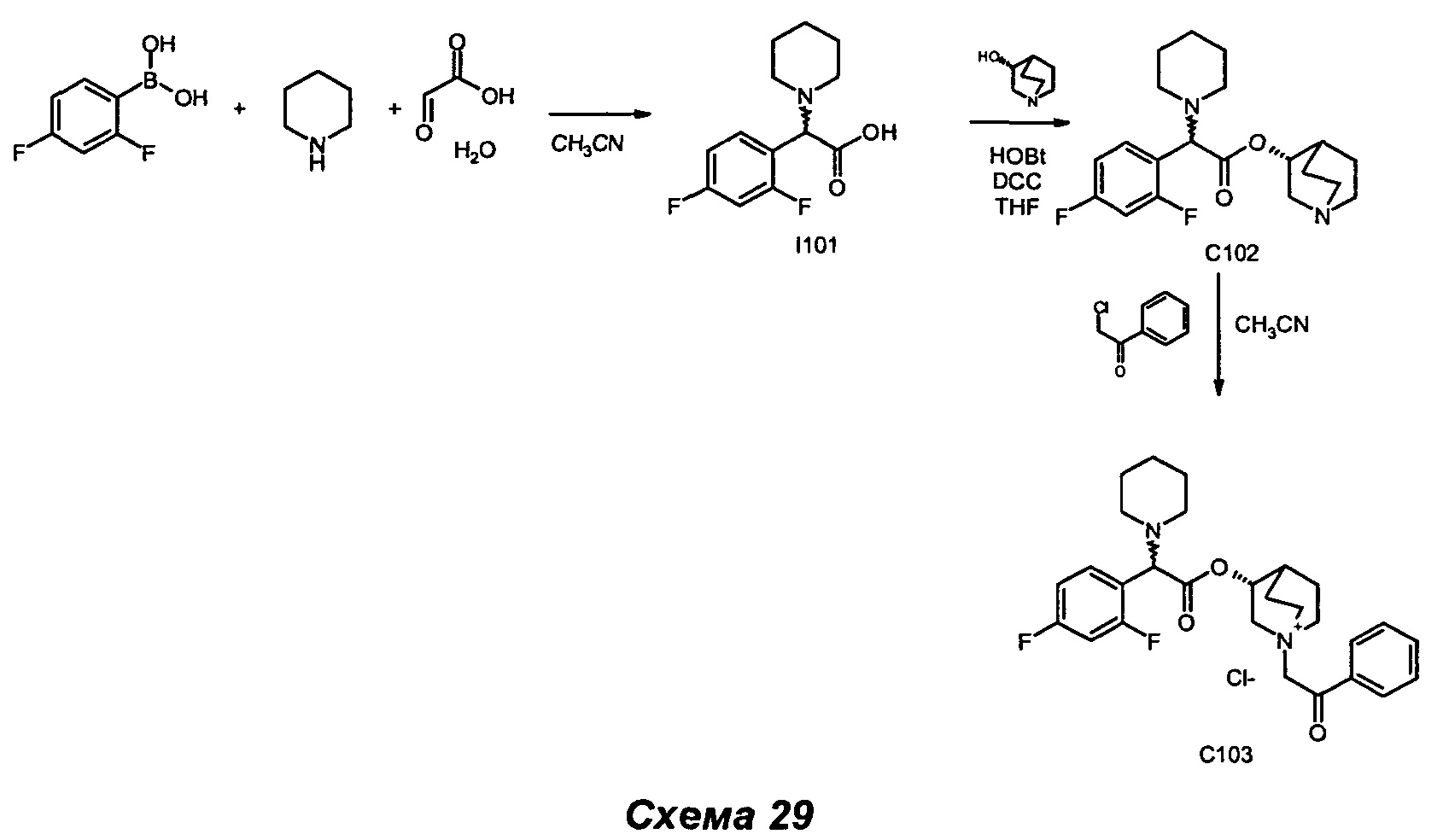
Получение 2-(2,4-дифторфенил)-2-(пиперидин-1-ил)уксусной кислоты (I101)
2-Оксоуксусную кислоту (141 мг, 1,90 ммоль) и пиперидин (0,16 мл, 1,90 ммоль) растворяли в ацетонитриле (10 мл). Добавляли 2,4-дифторфенилбороновую кислоту (300 мг, 1,90 ммоль), и смесь перемешивали при кипячении с обратным холодильником в течение 4 ч. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 85/25) с получением соединения, указанного в заголовке (307 мг, выход 63%), в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.58 (td, 1Н), 7.25 (ddd, 1H), 7.13 (m, 1H), 4.39 (s, 1H), 2.56-2.82 (m, 4H), 1.52-1.70 (m, 4H), 1.35-1.52 (m, 2H).
Получение (R)-хинуклидин-3-ил-2-(2.4-дифторфенил)-2-(пиперидин-1-ил)ацетата (С102)
2-(2,4-Дифторфенил)-2-(пиперидин-1-ил)уксусную кислоту (304 мг, 1,19 ммоль), DCC (491 мг, 2,38 ммоль) и НОВТ (365 мг, 2,38 ммоль) растворяли в сухом THF (12 мл). Добавляли (R)-хинуклидин-3-ол (454 мг, 3,57 ммоль), и смесь перемешивали при комнатной температуре в течение 16 ч. THF выпаривали, и остаток растворяли EtOAc и промывали насыщенным раствором NaHCO3. Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали до сухого состояния. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1 + 0,1% NH4OH) с получением продукта, указанного в заголовке, в виде бесцветного масла (280 мг, выход 64,5%).
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.44-7.66 (m, 1H), 7.19-7.36 (m, 1H), 7.03-7.18 (m, 1H), 4.63-4.83 (m, 1H), 4.48 (s, 1H), 2.97-3.18 (m, 1H), 2.56-2.79 (m, 5H), 2.21-2.47 (m, 4H), 1.74-1.85 и 1.84-1.94 (m, 1H), 1.14-1.69 (m, 10H).
Получение (3R)-3-(2-(2,4-дифторфенил)-2-(пиперидин-1-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азониабицикло[2.2.2]октанхлорида (С103)
(R)-хинуклидин-3-ил-2-(2,4-дифторфенил)-2-(пиперидин-1-ил)ацетат (275 мг, 0,75 ммоль) и 2-хлор-1-фенилэтанон (128 мг, 0,83 ммоль) растворяли в ацетонитриле (8 мл) и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, и остаток растирали с Et2O и обрабатывали ультразвуком. Полученный в результате осадок выделяли фильтрованием с аспирацией и промывали на фильтровальной бумаге EtOAc и Et2O с получением соединения, указанного в заголовке (261 мг, выход 66,6%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.94-8.03 (m, 2H) 7.71-7.80 (m, 1H) 7.49-7.66 (m, 3H) 7.24-7.35 (m, 1H) 7.10-7.21 (m, 1H) 5.21-5.32 (m, 3H) 4.62 (s, 1H) 4.09-4.25 (m, 1H) 3.56-3.81 (m, 5H) 2.32-2.47 (m, 4H) 2.24-2.31 (m, 1H) 2.00-2.14 (m, 2H) 1.76-1.97 (m, 2H) 1.46-1.61 (m, 4H) 1.33-1.46 (m, 2H).
СЭЖХ-МС (ИЭР) 483,24 m/z (M+).
ПРИМЕР 29
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-(тиофен-2-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С106)
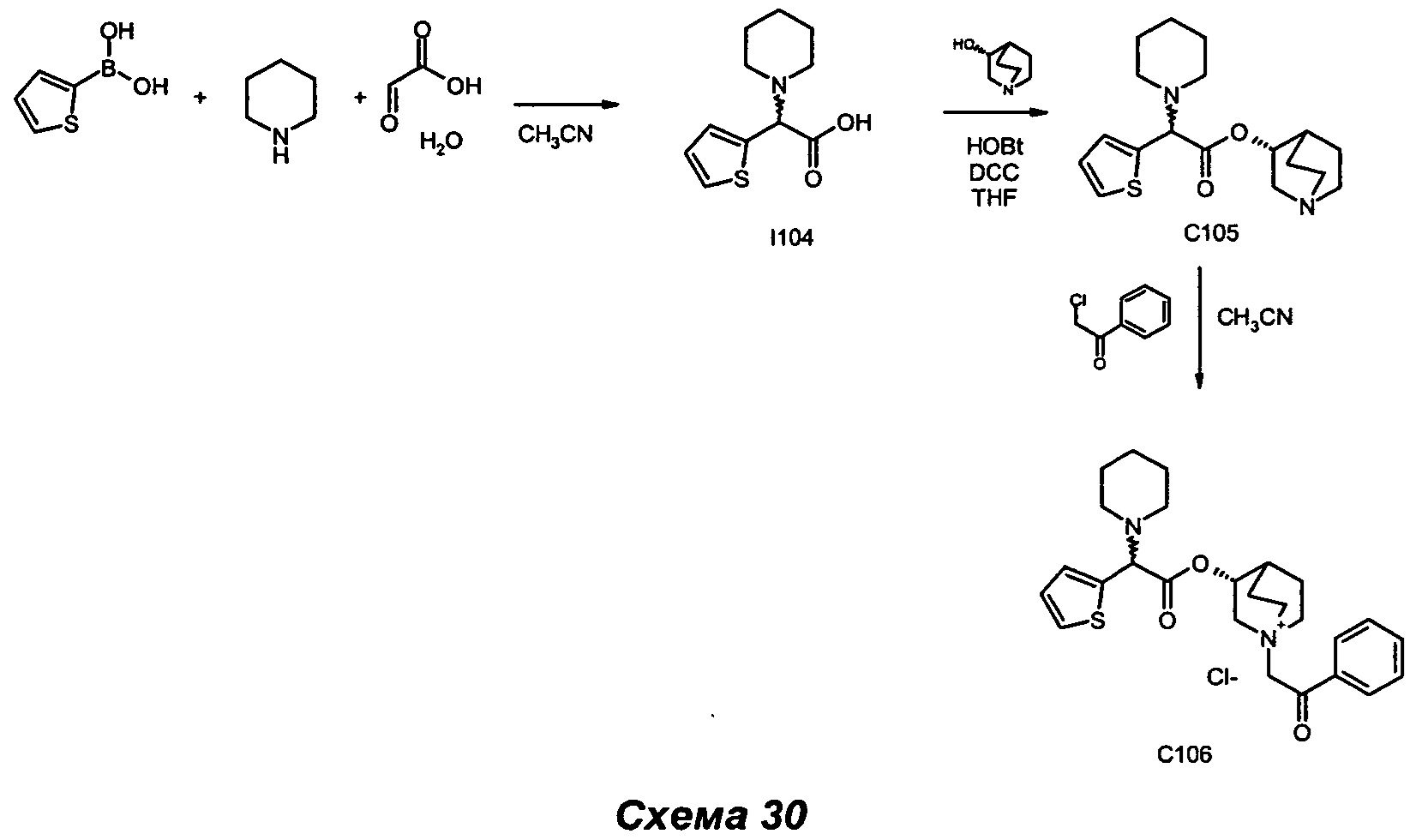
Получение (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-(тиофен-2-ил)ацетата (I104)
Смесь гидрата 2-оксоуксусной кислоты (216 мг, 2,34 ммоль), пиперидина (0,23 мл, 2,34 ммоль) и тиофен-2-илбороновой кислоты (300 мг, 2,34 ммоль) в ацетонитриле (11,7 мл) нагревали до образования флегмы в течение 2,5 часов. Реакционную смесь охлаждали при комнатной температуре, и суспензию фильтровали на воронке Бюхнера, промывая ацетонитрилом (10 мл). Твердое вещество удаляли с фильтра с получением соединения, указанного в заголовке (528 мг, количественный выход), в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.54 (dd, 1Н) 7.14 (dd, 1H) 7.02 (dd, 1H) 4.55 (s, 1H) 2.61-2.90 (m, 4H) 1.52-1.73 (m, 4H) 1.43 (q, 2H);
СЭЖХ-МС (ИЭР) 225,9 m/z (MH+).
Получение (R)-хинукпидин-3-ил-2-(пиперидин-1-ил)-2-(тиофен-2-ил)ацетата (С105)
Смесь 2-(пиперидин-1-ил)-2-(тиофен-2-ил)уксусной кислоты (250 мг, 1,11 ммоль), DCC (458 мг, 2,22 ммоль), НОВТ (300 мг, 2,22 ммоль) и (R)-хинуклидин-3-ола (282 мг, 2,22 ммоль) в сухом THF (11,1 мл) перемешивали при комнатной температуре в атмосфере азота в течение 24 часов. Бледно-розовую суспензию фильтровали, промывая THF (5 мл), и бледно-розовый раствор выпаривали. Сырой продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 95/5 + 0,4% NH4OH) с получением соединения, указанного в заголовке (132 мг, выход 35,6%) в виде бесцветного масла.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.51 (dd, 1Н) 7.02-7.07 (m, 1H) 6.95-7.02 (m, 1H) 4.78 (dd, 1H) 4.43-4.54 (m, 1H) 3.05-3.20 (m, 1H) 2.56-2.83 (m, 4H) 2.37-2.48 (m, 4H) 1.91 (dq, 1H) 1.54-1.76 (m, 2H) 1.43-1.54 (m, 5H) 1.23-1.43(m, 4H).
СЭЖХ-МС (ИЭР) 335,0 m/z (MH+).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-(тиофен-2-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С106)
2-Хлор-1-фенилэтанон (63,0 мг, 0,41 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-(тиофен-2-ил)ацетата (124 мг, 0,37 ммоль) в этилацетате (2,5 мл) и ацетонитриле (1,2 мл). Бледно-желтый раствор перемешивали при комнатной температуре в течение 24 ч. Растворитель выпаривали, и остаток растирали с Et2O (8 мл). Суспензию фильтровали на воронке Бюхнера, промывая Et2O (5 мл), и твердое вещество удаляли с фильтра, в результате чего собирали соединение, указанное в заголовке (88 мг, выход 48,5%) в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.92-8.08 (m, 2H), 7.71-7.83 (m, 1H), 7.47-7.69 (m, 3H), 7.07-7.16 (m, 1H), 6.93-7.07 (m, 1H), 5.33 и 5.34 (s, 2H), 5.22-5.32 (m, 1H), 4.63 и 4.64 (br. s., 1H), 4.11-4.34 (m, 1H), 3.57-3.93 (m, 5H), 2.22-2.48 (m, 5H), 1.85-2.22 (m, 4H), 1.31-1.67 (m, 6H).
СЭЖХ-МС (ИЭР) 453,09 m/z (MH+).
ПРИМЕР 30
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-(тиофен-3-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С109)
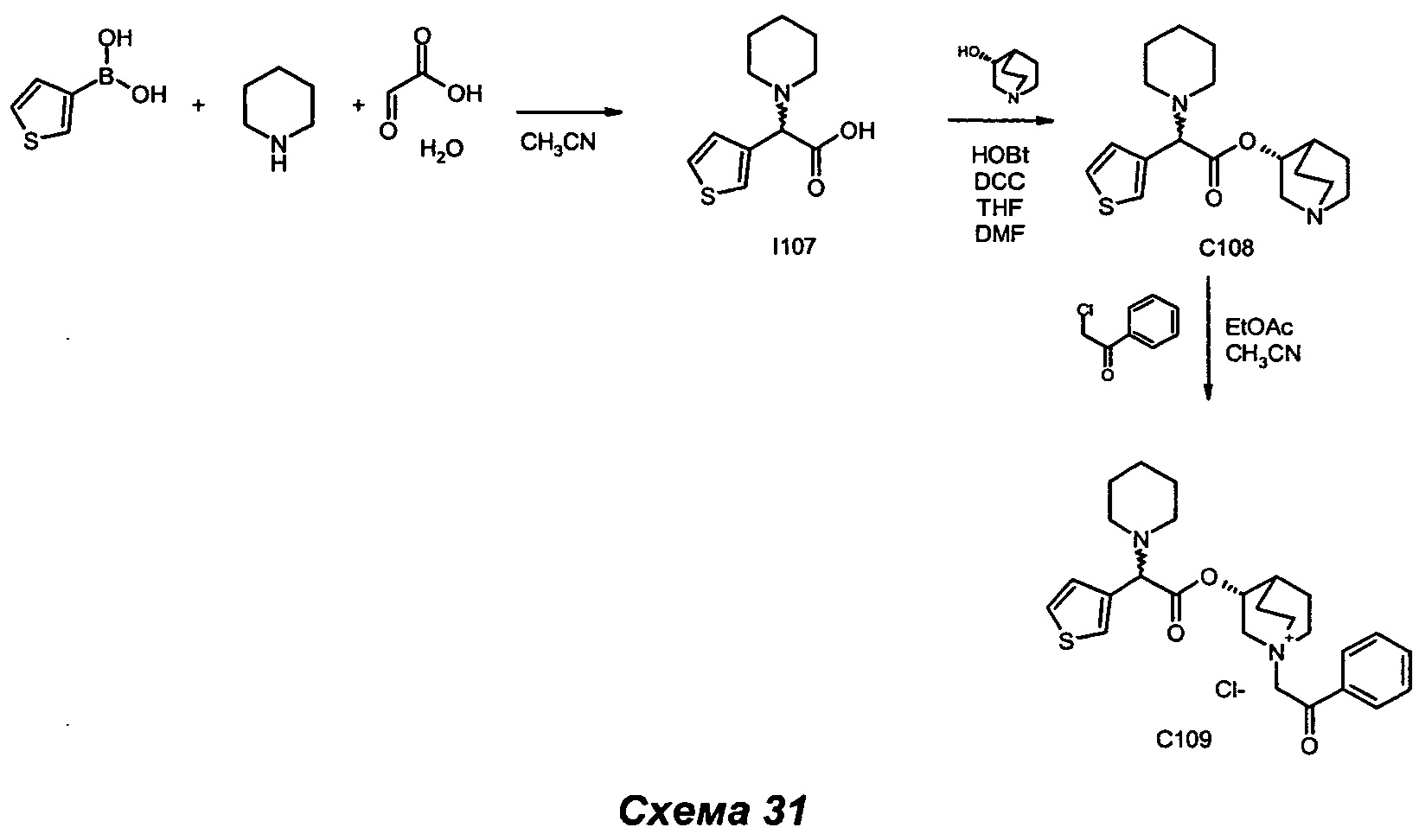
Получение 2-(пиперидин-1-ил)-2-(тиофен-3-ил)уксусной кислоты (I107)
Тиофен-3-илбороновую кислоту (300 мг, 2,34 ммоль) добавляли к раствору гидрата 2-оксоуксусной кислоты (216 мг, 2,34 ммоль) и пиперидина (0,23 мл, 2,34 ммоль) в ацетонитриле (11,7 мл). Смесь нагревали до образования флегмы в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, и суспензию фильтровали на воронке Бюхнера, промывая ацетонитрилом (8 мл). Соединение, указанное в заголовке (528 мг, количественный выход), собирали с фильтра в виде беловатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.42-7.62 (m, 2H), 7.01-7.23 (m, 1H), 4.39 (s, 1H), 2.79-2.97 (m, 2H), 2.60-2.79 (m, 2H), 1.52-1.84 (m, 4H), 1.31-1.52(m, 2H).
СЭЖХ-МС (ИЭР) 225,9 m/z (MH+).
Получение (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-(тиофен-3-ил)ацетата (С108)
Смесь 2-(пиперидин-1-ил)-2-(тиофен-3-ил)уксусной кислоты 21c (250 мг, 1,11 ммоль), DCC (458 мг, 2,22 ммоль), НОВТ (340 мг, 2,22 ммоль) и (R)-хинуклидин-3-ола (282 мг, 2,22 ммоль) в сухом THF (11,1 мл) и сухом DMF (5,5 мл) перемешивали при комнатной температуре в атмосфере азота в течение 48 часов. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 97/3 + 0,3% NH4OH). Продукт растирали с THF (10 мл) с получением соединения, указанного в заголовке (217 мг, выход 58,5%), в виде бесцветного масла.
1Н ЯМР (300 МГц, DMSO-d6) δ млн-1 7.48-7.55 (m, 1H) 7.39-7.46 (m, 1H) 7.10 (dd, 1H) 4.67-4.80 (m, 1H) 4.26 (s, 1H) 2.99-3.18 (m, 1H) 2.55-2.75 (m, 4H) 2.30-2.46 (m, 5H) 1.80-1.96 (m, 1H) 1.24-1.63 (m, 10H).
СЭЖХ-МС (ИЭР) 335,0 m/z (MH+).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-(пиперидин-1-ил)-2-(тиофен-3-ил)ацетокси)-1-азониабицикло[2.2.2]октанхлорида (С109)
2-Хлор-1-фенилэтанон (107 мг, 0,69 ммоль) добавляли к раствору (R)-хинуклидин-3-ил-2-(пиперидин-1-ил)-2-(тиофен-3-ил)ацетата (210 мг, 0,63 ммоль) в EtOAc (4,2 мл) и ацетонитриле (2,1 мл). Смесь перемешивали при комнатной температуре в течение 48 ч. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией (соотношение DCM/MeOH равно 9/1), в результате чего собирали соединение, указанное в заголовке (117 мг, выход 38,1%) в виде беловатого губчатого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) млн-1 7.90-8.13 (m, 2H), 7.71-7.86 (m, 1H), 7.44-7.71 (m, 4H), 7.08-7.22 (m, 1H), 5.30 и 5.31 (s, 2H), 5.16-5.29 (m, 1H), 4.40 и 4.42 (s, 1H), 4.12-4.28 (m, 1H), 3.53-3.89 (m, 5H), 2.33-2.47 (m, 4H), 2.23-2.33 (m, 1H), 1.78-2.15 (m, 4H), 1.28-1.64 (m, 6H);
СЭЖХ-МС (ИЭР) 453,18 m/z (M+).
Условные обозначения
*ЯМР
s - синглет
d - дублет
t - триплет
q - квартет
dd - дублет дублетов
m - мультиплет
br - широкий
БИОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА
ПРИМЕР 31
Связывание радиоактивного лиганда для клонированных мускариновых рецепторов человека
Клональные клетки СНО-K1, экспрессирующие рецепторы человека М1, М2, М3 (Euroscreen, Swissprot P11229, Р08172, Р20309, Genbank: J02960 соответственно) собирали в фосфатно-солевом буферном растворе без Са++/Mg++ и собирали их центрифугированием при 1500 об/мин в течение 10 мин при 4°С. Осадки ресуспендировали в охлажденном во льду буферном растворе А (15 мМ Трис-HCl рН 7,4, 2 мМ MgCl2, 0,3 мМ этилендиаминтетрауксусная кислота (ЭДТА), 1 мМ этиленгликоль-тетрауксусная кислота (ЭГТА)). Клонированные клетки, экспрессирующие рецепторы М1, М2 и М3, гомогенизировали гомогенизатором марки Polytron PBI (установка 5 в течение 15 с). Неочищенную мембранную фракцию собирали с помощью двух последовательных стадий центрифугирования при 40000 g в течение 20 мин при 4°С, разделенных стадией промывания в буфере А.
Осадки, полученные из трех линий клеток, окончательно ресуспендировали в буфере С (75 мМ Трис-HCl рН 7,4, 12,5 мМ MgCl2, 0,3 мМ ЭДТА, 1 мМ ЭГТА, 250 мМ сахароза), и аликвоты хранили при - 80°С.
На сутки эксперимента замороженные мембраны с рецепторами М1, М2 и М3 ресуспендировали в буфере D (50 мМ Трис-HCl рН 7,4, 2,5 мМ MgCl2, 1 мМ ЭДТА). Неселективный мускариновый радиоактивный лиганд [3H]-N-метилскополамин (Mol. Pharmacol. 45: 899-907) использовали для мечения сайтов связывания М1, М2 и М3. Эксперименты по связыванию проводили в двух повторах (кривые концентрации по десяти точкам) в 96-луночных планшетах при концентрации радиоактивного лиганда, составляющей 0,1-0,3 нМ. Неспецифическое связывание определяли в присутствии "холодного" (немеченого) N-метилскополамина 10 мкМ. Образцы (конечный объем 0,75 мл) инкубировали при KT в течение 120 мин для анализа связывания М1, 60 мин для М2 и 90 мин для М3.
Реакционную смесь останавливали быстрым фильтрованием через планшеты GF/B Unifilter и двумя отмывками холодным буфером (0,75 мл), используя коллектор Packard Filtermate Harvester. Радиоактивность на фильтрах измеряли с помощью сцинтилляционного счетчика для микропланшетов TopCount NXT (Canberra Packard).
В настоящих анализах значения Ki для тестируемых соединений определяли на основании наблюдаемых значений IC50 в соответствии с известными способами. Более низкое значение Ki указывает на то, что тестируемое соединение обладает более высоким сродством связывания с рецептором.
Взаимодействие с мускариновыми рецепторами М3 можно оценить на основании результатов исследований in vitro, в которых оценивали эффективность тестируемых соединений и прекращение ингибиторной активности, полученное после отмывания антагонистов в изолированной трахее морской свинки, и на основании продолжительности действия в живом организме против бронхоспазма, индуцированного ацетилхолином, у морской свинки.
ПРИМЕР 32
Взаимодействие in vitro с рецепторами М3 морской свинки
Эффективность антагонистической активности в изолированной трахее морской свинки исследовали, следуя способу, описанному ранее в статье Haddad ЕВ et al. в Br J Pharmacol 127, 413-420, 1999, с небольшими модификациями.
Кумулятивную кривую концентрация-ответ на тестируемые антагонисты строили на препаратах, предварительно подвергнутых сокращению с помощью карбахолина до достижения полного ингибирования тонуса гладкой мышцы. В данном биологическом анализе концентрацию антагониста, вызывающую 50%-ю инверсию индуцированного карбахолином тонического сокращения (IC50), принимали за меру его эффективности.
В экспериментах, нацеленных на оценку прекращения ингибиторных эффектов, производимых тестируемыми соединениями, минимальную концентрацию тестируемых соединений, известных как производящие максимальный ингибиторный эффект, вводили в препараты, предварительно подвергнутые сокращению с помощью карбахолина. Как только тоническое сокращение было полностью инверсировано, раствор в инкубаторе органов обновляли, и препараты тщательно промывали свежим раствором Кребса. Карбахолин (0,3 мкМ) вводили снова (через 30-минутный интервал между промыванием и следующим введением) в течение следующих 4 часов.
После введения карбахолина ингибиторные эффекты соединений по изобретению, введенных при концентрации 10 нМ, выражали в виде процента восстановления сократительного ответа на карбахолин. Процент восстановления через четыре часа после отмывания был ниже 50%.
Значения (IC50) ингибиторной активности М3, протестированной на соединениях С3 - С109, составляли от 0,39 до 130 нМ.
ПРИМЕР 33
Исследования in vivo на индуцированном ацетилхолином бронхоспазме у морских свинок
Тесты in vivo на индуцированном ацетилхолином бронхоспазме у морской свинки проводили согласно публикации Konzett H and Rössler F Arch Exp Path Pharmacol 195, 71-74, 1940. Водные растворы тестируемых соединений закапывали интратрахеально анестезированным, искусственно вентилируемым морским свинкам. Бронхиальный ответ на провокацию внутривенным ацетилхолином определяли до и после введения лекарственного средства, и изменения в легочной сопротивляемости в нескольких временных точках выражали в виде процента ингибирования бронхоспазма.
Бронходилататорная активность тестируемых соединений оставалась неизменной в течение вплоть до 24 часов после введения.
ПРИМЕР 34
Исследования стабильности в плазме человека
Чтобы продемонстрировать, что соединения разлагаются, соединения по изобретению тестировали на стабильность в плазме человека через 1 час и 5 часов. Кратко, 10 мкл исходного раствора 250 мкМ соединения в ацетонитриле добавляли к 1 мл плазмы человека, и образцы инкубировали при 37°С. Плазму (50 мкл) брали после 0, 1 и 5 часов инкубации и добавляли к 140 мкл ацетонитрила с добавлением верапамила в качестве внутреннего стандарта (250 нг/мл). Образцы анализировали с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрией и масс-спектрометрии ВЭЖХ-МС/МС.
Стабильность в плазме вычисляют в виде процента остатка через 1 и 5 часов, путем деления площади пика через 1 час или 5 часов на площадь пика в момент времени 0.
После 1 часа и 5 часов инкубации результат стабильности в плазме, протестированной для нескольких репрезентативных соединений по изобретению, составлял от 10 до 70% остаточного соединения, что указывает на то, что соединения по изобретению нестабильны в плазме человека по сравнению с подобными соединениями предшествующего уровня техники, показывающими через 1 и 5 ч процент соединения, остающегося в плазме человека, очень близкий к 100%.
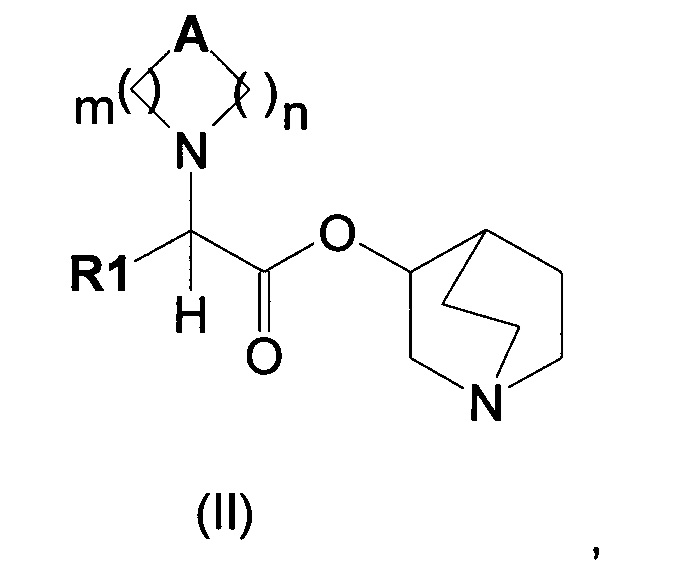
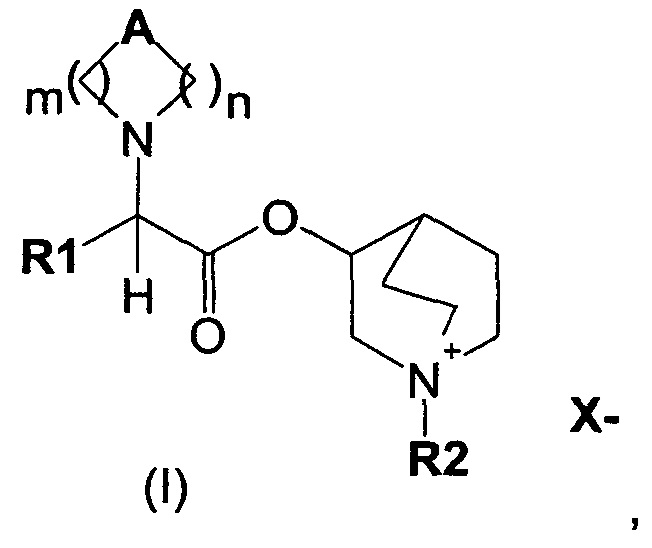
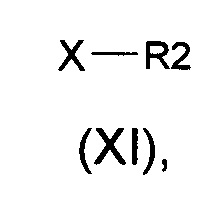
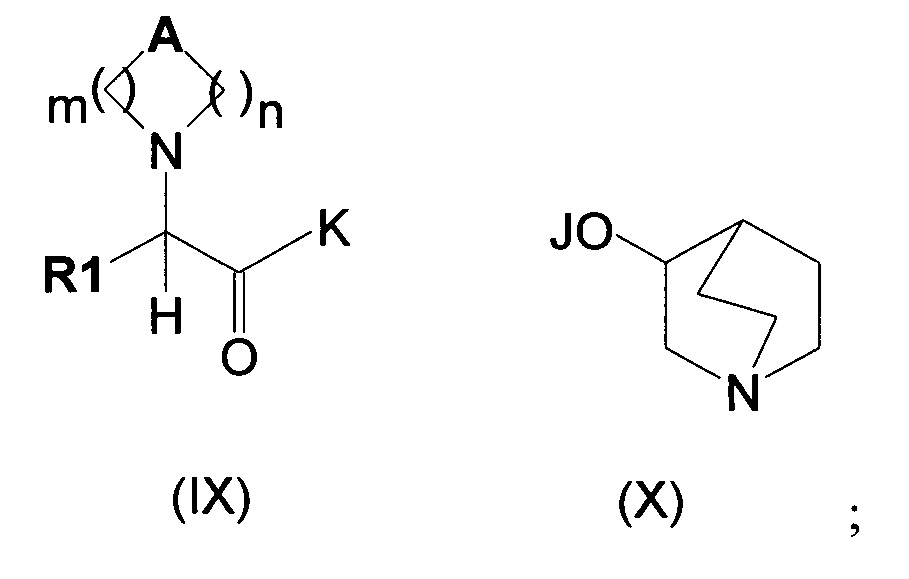
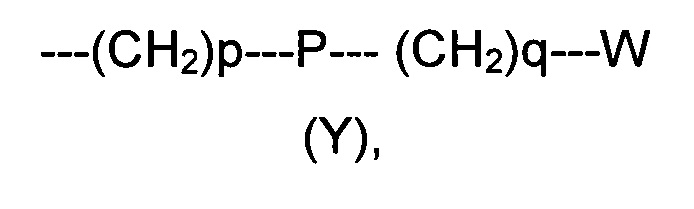
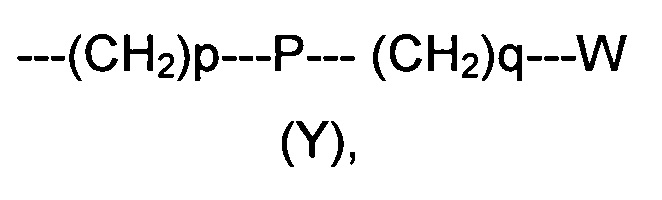
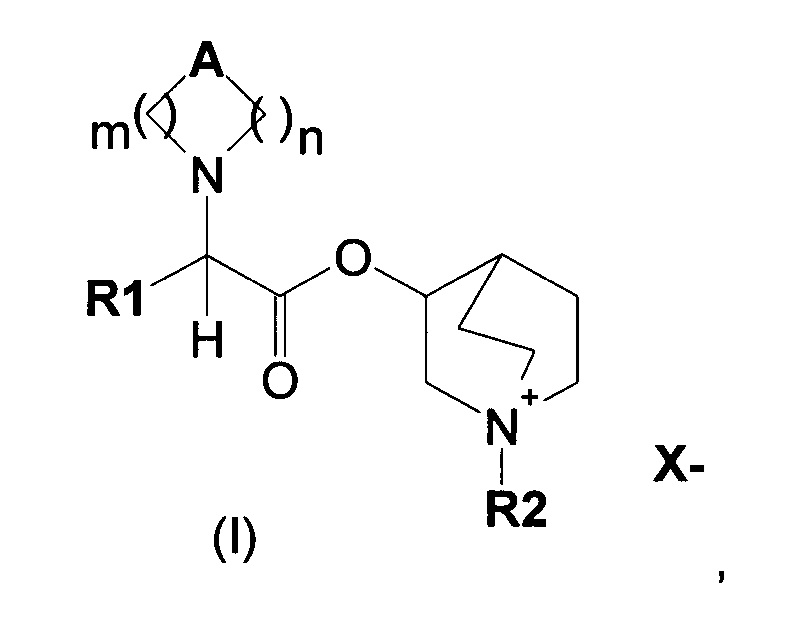
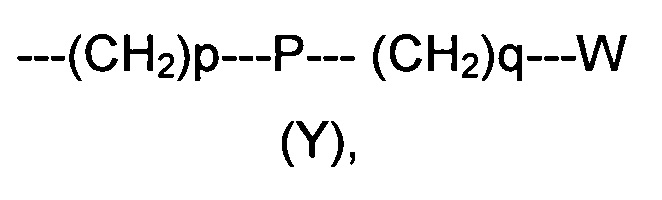
Способ получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты
Терапевтическая комбинация, содержащая легочный сурфактант и стероид
Способ получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты
Производные сложного аминоэфира алкалоида и их лекарственные композиции
Алкалоидные производные сложных аминоэфиров и включающие их лекарственные композиции
Алкалоидные производные на основе сложных аминоэфиров и композиции лекарственных средств, содержащие их
Производные пиримидина и их применение в лечении респираторных заболеваний, таких как copd
Производные 4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил-мочевины и их применение в лечении, среди прочего, заболеваний дыхательного тракта
Производные глицина и их применение в качестве антагонистов мускариновых рецепторов
Производные пиримидина и их применение в лечении респираторных заболеваний, таких как copd
Способ получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты
Терапевтическая комбинация, содержащая легочный сурфактант и стероид
Способ получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты
Производные сложного аминоэфира алкалоида и их лекарственные композиции
Алкалоидные производные сложных аминоэфиров и включающие их лекарственные композиции
Алкалоидные производные на основе сложных аминоэфиров и композиции лекарственных средств, содержащие их
Производные пиримидина и их применение в лечении респираторных заболеваний, таких как copd
Производные 4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил-мочевины и их применение в лечении, среди прочего, заболеваний дыхательного тракта
Производные глицина и их применение в качестве антагонистов мускариновых рецепторов
Производные пиримидина и их применение в лечении респираторных заболеваний, таких как copd

