СОЕДИНЕНИЯ ЗАМЕЩЕННОЙ ТРИАЗОЛБОРОНОВОЙ КИСЛОТЫ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к органическим соединениям, полезным для лечения и/или профилактики воспалительного заболевания или нарушения у млекопитающих, и, в частности, к соединениям замещенной триазолбороновой кислоты для лечения ревматоидного артрита, волчанки и синдрома раздраженного кишечника (СРК), получению этих соединений, содержащим их фармацевтическим композициям и применению их в качестве ингибиторов иммунопротеасомной субъединицы LMP7.
Предшествующий уровень техники
Субъединица LMP7 является важным компонентом иммунопротеасомы, которая экспрессируется главным образом в иммунных клетках, таких как Т/В-лимфоциты и моноциты, а также в не являющихся иммунными клетках, которые, были подвергнуты воздействию цитокинов воспаления, включая интерферон гамма (ИФН-γ) и фактор некроза опухолей (ФНОα). Иммунопротеасома играет существенную роль в генерации спектра антигенных пептидов и формировании CD8+ Т-клеточного ответа, ограниченного классом I молекул главного комплекса гистосовместимости (молекулы МНС-I). Moebius J. et al. European Journal of Immunology. 2010; Basler, M. et al. Journal of Immunology. 2004. 3925-34. Новые данные свидетельствуют о том, что LMP7 также регулирует продукцию цитокинов воспаления и функции иммунных клеток вне пределов регуляции презентации антигена, опосредованной молекулами MHC-I.
Как было показано, PR-957, небольшая молекула ингибитора LMP7, эффективно блокирует Th1/17дифференциацию, эффекторные функции В клеток и продукцию цитокинов воспаления (IL-6, TNF-α, IL-23). Muchamuel T. et al. Natural Medicine. 2009. 15, 781-787; Basler M. et al. Journal of Immunology. 2010, 634-41.
Кроме того, в нескольких моделях преклинической стадии аутоиммунных заболеваний была продемонстрирована блокада LMP7 посредством PR-957 с целью получения терапевтического эффекта. Во-первых, как было показано, PR-957 значительно уменьшает индекс, оценивающий тяжесть заболевания в моделях индуцированного коллагеновыми антителами артрита (CAIA) и индуцированного коллагеном артрита (CAI) на мышах, с признаками значительного уменьшения воспаления и эрозии кости. Muchamuel Т. et al. Natural Medicine. 2009. 15, 781-787. Кроме того, PR-957 снижал число плазматических клеток и уровни анти-дцДНК IgG антител в модели на мышах линии MRL/Ipr, подверженных развитию волчанки и предотвращал прогрессирование болезни у этих мышей. Ichikawa НТ, et al. Arthritis & Rheumatism. 2012. 64, 493-503. Кроме того, PR-957 уменьшал воспаление и деструкцию ткани в модели DSS-индуцированного колита на мышах. Basler М. et al. Journal of Immunology. 2010, 634-41. И наконец, также было показано на моделях синдрома раздраженного кишечника (СРК), что нокаутные по LMP7 мыши были защищены от развития заболевания. Schmidt N. et al. Gut 2010. 896-906.
Взятые в совокупности, эти данные убедительно показывают, что активность субъединицы LMP7 тесно связана с функциями В/Т-лимфоцитов и продукцией цитокинов воспаления, все из которых представляют собой клинически подтвержденные цели/пути патогенеза ревматоидного артрита, волчанки и синдрома раздраженного кишечника (СРК). Таким образом, существующие данные обеспечивают строгое обоснование для направленного воздействия LMP7 при показаниях к лечению аутоиммунных заболеваний. Ввиду потенциальных недостатков ковалентного ингибитора во время длительного его применении при таких хронических заболеваниях, как аутоиммунные заболевания, небольшая молекула ингибитора LMP7 с обратимой ковалентной или с нековалентной связью является особенно рекомендуемой при показаниях к лечению аутоиммунных заболеваний.
Краткое описание изобретения
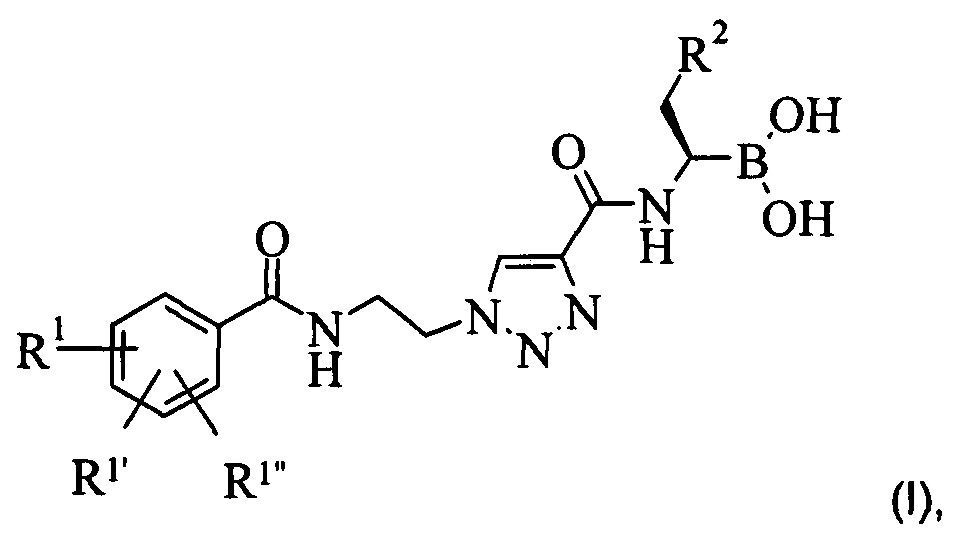
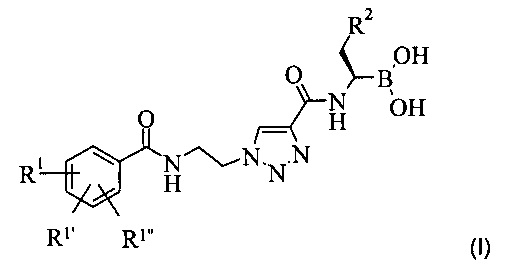
Настоящее изобретение обеспечивает соединение формулы (I):
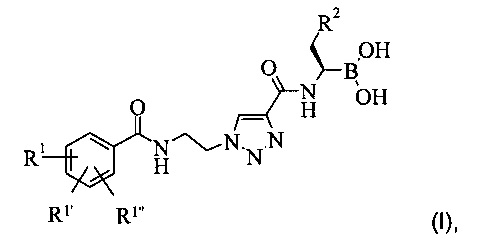
в которой:
остатки R1, R1' и R1'' независимо друг от друга представляют собой водород, алкоксигруппу, галоген или группу -CF3; а остаток R2 представляет собой С1-7 алкил или фенил, или фармацевтически приемлемую соль этого соединения.
Изобретение также относится к фармацевтическим композициям, содержащим эти соединения, способам применения этих соединений и способам получения этих соединений.
Все документы, которые цитируются в настоящем изобретении или на которых основывается настоящее изобретение специально включены в это изобретение путем ссылки.
Подробное описание изобретения
Если не указано иное толкование, следующие ниже специфические термины и фразы, использующиеся в описании и формуле изобретения, определяются следующим образом:
Термин «остаток» относится к атому или группе химически связанных атомов, которая присоединена к другому атому или молекуле одной или более химическими связями и тем самым составляющая часть этой молекулы. Например, остатки R формулы I обозначают остатки, которые связаны с основной структурой формулы I ковалентной связью.
При ссылке на отдельный остаток с одним или более атомов водорода, термин «замещенный» относится к тому факту, что по меньшей мере один из атомов водорода этого остатка заменен на другой заместитель или группу. Например, термин «C1-7 алкил, замещенный галогеном» (например, трифторметил, дифторметил, фторметил, хлорметил, и т.д.) относится к тому факту, что один или несколько атомов водорода C1-7 алкила (как определено ниже) заменен на один или несколько атомов галогена.
Термин «алкил» относится к насыщенному углеводородному остатку с алифатической линейной или разветвленной цепью, имеющему от 1 до 20 атомов углерода. В конкретных формах осуществления настоящего изобретения алкил имеет от 1 до 10 атомов углерода, в частности от 1 до 7 атомов углерода.
Термин «C1-7 алкил» относится к алкильному остатку, имеющему от 1 до 7 атомов углерода. Примеры C1-7 алкилов включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил.
Термин «алкоксигруппа» обозначает группу с формулой -O-R', где R' представляет собой алкильную группу. Примеры алкокси групп включают метоксигруппу, этоксигруппу, изопропоксигруппу и трет-бутоксигруппу.
«Арил» означает одновалентный циклический ароматический углеводородный остаток, имеющий моноциклическую, бициклическую или трициклическую ароматическую структуру. Арильная группа может быть необязательно замещена, как определено в данном изобретении. Примеры арильных групп включают, без ограничений, фенил, нафтил, фенантрил, флуоренил, инденил, пенталенил, азуленил, оксидифенил, бифенил, метилендифенил, аминодифенил, дифенилсульфидил, дифенилсульфонил, дифенилизопропилиденил, бензодиоксанил, бензофуранил, бензодиоксилил, бензопиранил, бензоксазинил, бензоксазинонил, бензопиперадинил, бензопиперазинил, бензопирролидинил, бензоморфолинил, метилендиоксифенил, этилендиоксифенил и т.п., включая их частично гидрированные производные, каждый из которых необязательно замещен.
Термины «гало-», «галоген» и «галоид», которые могут быть использованы взаимозаменяемо, относятся к атомам фтора, хлора, брома или йода в качестве заместителей.
Если не указано иное толкование, термин «водород» или «гидро-» относится к остатку атома водорода (-Н), но не к молекуле Н2.
Если не указано иное толкование, термин «соединение формулы», или «соединение согласно формуле», или «соединения формулы», или «соединения согласно формуле» относится к любому соединению, выбранному из рода соединений, определенных формулой (включая любую фармацевтически приемлемую соль или сложный эфир любого из таких соединений, если не указано иное толкование).
Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, не являющихся биологически или иным образом нежелательными. Соли могут быть образованы неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., предпочтительно хлористоводородная кислота, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, салициловая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, N-ацетилцистеин и т.п. Кроме того, соли могут быть получены добавлением неорганического или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, без ограничений, натриевые, калиевые, литиевые, аммониевые, кальциевые, магниевые соли и т.п. Соли, полученные из органических оснований, включают, без ограничений, соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе природных замещенных аминов, циклических аминов и основных ионообменных смол, такие как изопропиламиновые, триметиламиновые, диэтиламиновые, триэтиламиновые, трипропиламиновые, этаноламиновые, лизиновые, аргининовые, N-этилпиперидиновые, пиперидиновые, полиаминовые смолы и т.п.
Соединения по настоящему изобретению могут присутствовать в виде фармацевтически приемлемых солей. Соединения по настоящему изобретению могут также присутствовать в форме фармацевтически приемлемых сложных эфиров (например, метиловых и этиловых эфиров кислот формулы I для использования их в качестве пролекарств). Соединения по настоящему изобретению также могут быть сольватированными, т.е. гидратированными. Сольватация может быть осуществлена в ходе производственного процесса или может происходить как следствие гигроскопических свойств первоначально безводного соединения формулы I (гидратация).
Соединения, которые имеют одну и ту же молекулярную формулу, но различаются природой или последовательностью связывания их атомов или расположением их атомов в пространстве, называются «изомеры» и входят в объем настоящего изобретения. Изомеры, которые различаются по расположению атомов в пространстве, называются «стереоизомеры». Диастереомеры представляют собой стереоизомеры с противоположной конфигурацией в одном или более хиральных центров, не являющиеся энантиомерами. Стереоизомеры, имеющие один или более асимметрических центров, которые являются несовместимыми зеркальными отражениями друг друга, называют «энантиомерами». Если соединение имеет асимметрический центр, например, когда атом углерода связан с четырьмя различными группами, то возможна пара энантиомеров. Энантиомер может быть охарактеризован абсолютной конфигурацией его асимметрического центра или центров и описывается при помощи правил последовательности R- и S-конфигураций Кана, Ингольда и Прелога, либо способом, при котором молекула вращает плоскость поляризации света и обозначается при этом как правовращающая или левовращающая (т.е. как (+) или (-)-изомер соответственно). Хиральное соединение может существовать в виде индивидуального энантиомера или в виде их смеси. Смесь, содержащая равные пропорции энантиомеров, называется «рацемической смесью».
Термин «терапевтически эффективное количество» соединения означает количество соединения, которое является эффективным для предотвращения, облегчения или улучшения симптомов болезни или для продления жизни пациента, подлежащего лечению. Определение терапевтически эффективного количества находится в пределах квалификации в данной области техники. Терапевтически эффективное количество или дозировка соединения по настоящему изобретению может варьироваться в широких пределах и может быть определена способом, известным в данной области техники. Такие дозировки должны будут соответствовать индивидуальным требованиям в каждом конкретном случае, включая специфику назначенного соединения (соединений), способ введения, состояние, подлежащее лечению, а также тип пациента, которого лечат. В общем случае при пероральном или парентеральном введении взрослому человеку массой приблизительно 70 кг может быть целесообразна суточная доза от примерно 0,1 мг до примерно 5000 мг, от 1 мг до примерно 1000 мг или от 1 мг до 100 мг, хотя нижние и верхние пределы могут быть выше, если так назначено. Суточная доза может быть введена в виде разовой дозы или разделена на отдельные дозы, или при парентеральном введении она может быть введена в виде непрерывного вливания.
Термин «фармацевтически приемлемый носитель» предназначен для того, чтобы включать в себя все и любые материалы, совместимые с введением в виде фармацевтического препарата, включая растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые вещества, изотонические и замедляющие всасывание вещества и другие материалы и соединения, совместимые с введением в виде фармацевтического препарата. В композициях по изобретению предусматривается применение упомянутых выше носителей, за исключением случаев, когда какая-либо обычная среда или вещество несовместимы с активным соединением. В эти композиции также могут быть включены дополнительные активные соединения.
Использующиеся фармацевтические носители для приготовления упомянутых выше композиций могут быть твердыми, жидкими или газообразными; следовательно, композиции могут иметь форму таблеток, пилюль, капсул, суппозиториев, порошков, лекарственных форм, имеющих энтеральное покрытие или защищенных другим способом (например, связанных с ионообменными смолами или упакованных в белково-липидные везикулы), лекарственных форм с замедленным высвобождением, растворов, суспензий, эликсиров, аэрозолей и т.п. Носитель может быть выбран из различных масел, включая масла полученные из нефти, животного, растительного или синтетического сырья, например, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Вода, физиологический раствор, водный раствор декстрозы и гликоли являются предпочтительными жидкими носителями, особенно (когда они изотоничны крови) для инъекционных растворов. Например, лекарственные формы для внутривенного введения включают стерильные водные растворы активного ингредиента (ингредиентов), которые получают путем растворения твердого активного ингредиента (ингредиентов) в воде с получением водного раствора, а также приведением этого раствора в стерильное состояние. Подходящие фармацевтические наполнители включают крахмал, целлюлозу, тальк, глюкозу, лактозу, тальк, желатин, солод, рисовую муку, муку, мел, диоксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и т.п. Композиции могут быть подвергнуты действию с обычных фармацевтических добавок, таких как консерванты, стабилизаторы, смачивающие вещества или эмульгаторы, соли для регулирования осмотического давления, буферы и т.п. Подходящие фармацевтические носители и их состав описаны в руководстве Remington's Pharmaceutical Sciences by Ε.W. Martin. Такие композиции будут, в любом случае, содержать эффективное количество активного соединения вместе с подходящим носителем для того, чтобы получить соответствующую лекарственную форму для надлежащего введения реципиенту.
При практическом осуществлении способа по настоящему изобретению, эффективное количество любого из соединений по данному изобретению, или комбинацию любого из соединений по данному изобретению, или его фармацевтически приемлемую соль или сложный эфир, вводят либо по отдельности, либо в комбинации, с помощью любых обычных и приемлемых методов, известных в данной области техники. Соединения или композиции могут, таким образом, быть введены перорально (например, в ротовую полость), сублингвально, парентерально (например, внутримышечно, внутривенно или подкожно), ректально (например, в виде суппозиториев или промываний), трансдермально (например, при помощи электропорации кожи) или путем ингаляции (например, с помощью аэрозоля) и в виде дозированных твердых, жидких или газообразных лекарственных форм, включая таблетки и суспензии. Введение может быть выполнено в виде непрерывной терапии при помощи одной стандартной лекарственной формы или в виде терапии с помощью однократных доз на усмотрение исполнителя. Терапевтическая композиция может быть также в форме эмульсии или дисперсии в сочетании с липофильной солью, например, памовой кислоты, или в виде биоразлагаемой композиции с замедленным высвобождением для подкожного или внутримышечного введения.
Более подробно, настоящее изобретение обеспечивает соединение формулы (I):
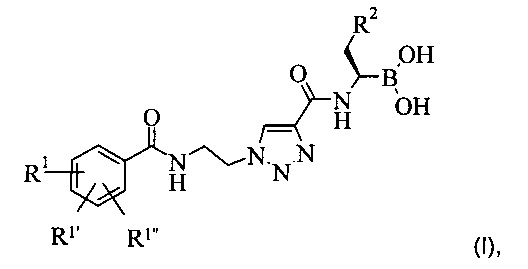
где остатки R1, R1' и R1'' независимо друг от друга представляют собой водород, алкоксигруппу, галоген или группу -CF3; а остаток R2 представляет собой С1-7 алкил или фенил, или фармацевтически приемлемую соль этого соединения.
В другой форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где остатки R1, R1' и R1'' независимо друг от друга представляют собой водород, метоксигруппу, фтор или группу -CF3.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где один из остатков R1, R1' или R1'' независимо друг от друга представляет собой водород, а два других независимо друг от друга являются алкоксигруппой, галогеном или группой -CF3.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где один из остатков R1, R1' или R1'' является водородом, а два других независимо друг от друга представляют собой метоксигруппу, фтор или группу -CF3.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где один из остатков R1, R1' или R1'' представляет собой метоксигруппу в орто-положении, еще один остаток является метоксигруппой в мета-положении и еще один остаток является метоксигруппой в пара-положении.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где один из остатков R1, R1' или R1'' обозначает фтор в орто-положении, еще один остаток является атомом водорода в мета-положении и еще один остаток является группой -CF3 в пара-положении.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где один из остатков R1, R1' или R1'' является метоксигруппой в орто-положении, один остаток представляет собой водород в мета-положении и еще один остаток является группой -CF3 в пара-положении. В другой форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где остаток R2 является метилом.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где остаток R2 является фенилом.
В другой форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I), где это соединение представляет собой:
(R)-3-метил-1-(1-(2-(2,3,4-триметоксибензамидо)этил)-1H-1,2,3-триазол-4-карбоксамидо)бутилбороновую кислоту;
(R)-2-фенил-1-(1-(2-(2,3,4-тримето?сибензамидо)этил)-1H-1,2,3-триазол-4-карбоксамидо)этилбороновую кислоту;
(R)-1-(1-(2-(2-фтор-4-(трифторметил)бензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновую кислоту; или
(R)-1-(1-(2-(2-метокси-4-(трифторметил)бензамидо)этил)-1H-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновую кислоту; или
их фармацевтически приемлемые соли.
В еще одной форме своего осуществления настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения согласно формуле (I) и фармацевтически приемлемый носитель.
В другой форме своего осуществления настоящее изобретение обеспечивает соединение согласно формуле (I) для применения в качестве терапевтически активного вещества.
В еще одной форме своего осуществления настоящее изобретение обеспечивает применение соединения согласно формуле (I) для лечения или профилактики воспалительного заболевания или нарушения, выбранного из ревматоидного артрита, волчанки и синдрома раздраженного кишечника.
В другой форме своего осуществления настоящее изобретение обеспечивает применение соединения согласно формуле (I) с целью получения лекарственного средства для лечения или профилактики воспалительного заболевания или нарушения, выбранного из ревматоидного артрита, волчанки и синдрома раздраженного кишечника.
В еще одной форме своего осуществления настоящее изобретение обеспечивает соединение формулы (I) для лечения или профилактики воспалительного заболевания или нарушения, выбранного из ревматоидного артрита, волчанки и синдрома раздраженного кишечника.
В другой форме своего осуществления настоящее изобретение обеспечивает способ лечения воспалительного заболевания или нарушения, выбранного из ревматоидного артрита, волчанки и синдрома раздраженного кишечника, включающий стадию введения терапевтически эффективного количества соединения согласно формуле (I) нуждающемуся в этом пациенту.
Другая форма осуществления настоящего изобретения обеспечивает изобретение, как описано выше.
Синтез
Исходные материалы и реагенты, используемые при получении этих соединений, как правило, либо доступны для приобретения от коммерческих поставщиков, таких как компания Aldrich Chemical Co., либо могут быть получены способами, известными специалистам в данной области техники при помощи следующих процедур, изложенных в таких источниках, как Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1991, Volumes 1-15; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, Volumes 1-5 and Supplemental; and Organic Reactions, Wiley & Sons: New York, 1991, Volumes 1-40.
Следующие ниже схемы реакций синтеза только иллюстрируют некоторые способы, с помощью которых могут быть синтезированы соединения по настоящему изобретению, и могут быть сделаны различные модификации этих схем реакций синтеза и предложены специалистам в данной области техники, обращающимся к изобретению, которое содержится в данной заявке.
Исходные материалы и промежуточные соединения в схемах реакций синтеза могут быть, при необходимости, выделены и очищены с использованием обычных методов, включая, без ограничений, фильтрование, перегонку, кристаллизацию, хроматографию и т.п.Такие материалы могут быть охарактеризованы с использованием обычных средств, включая физические константы и спектральные данные.
Если не указано иное толкование, реакции, описанные в настоящем изобретении, проводят предпочтительно в инертной атмосфере, под атмосферным давлением при температуре реакции, составляющей от примерно -78°С до примерно 150°С, более предпочтительно от примерно 0°С до примерно 125°С и наиболее предпочтительно и удобно при температуре примерно равной комнатной температуре (или температуре окружающей среды), например, примерно 20°С.
Соединения по настоящему изобретению могут быть получены при помощи любого количества обычных средств. Например, они могут быть получены в соответствии со способами, изложенными в следующих ниже схемах.
Схема 1
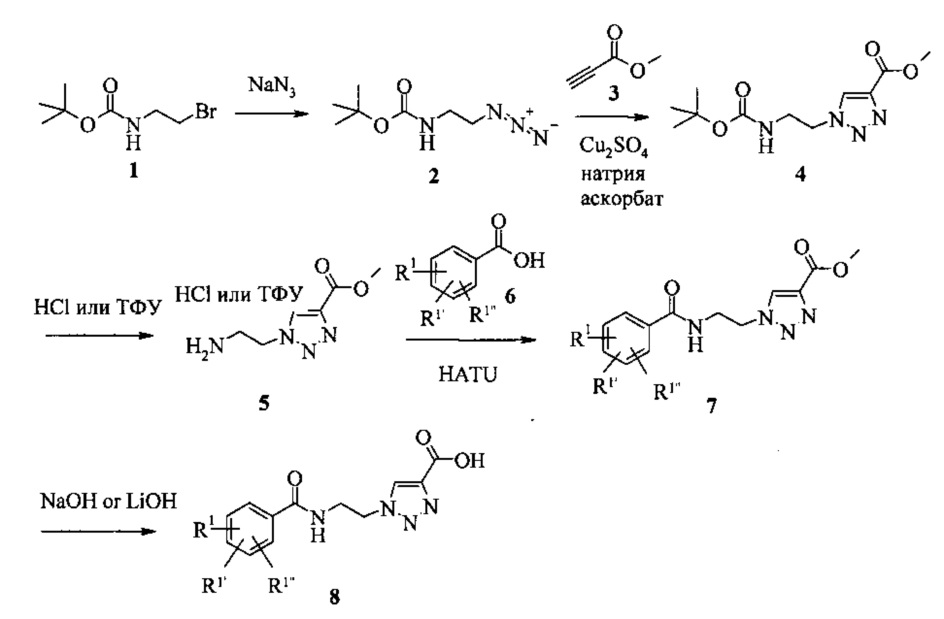
Как видно на Схеме 1, бромид 1 может быть превращен в азид 2 с использованием азида натрия, затем он может вступить в реакцию с метилпропиолатом 3 в присутствии меди (II) сульфата и аскорбата натрия с получением 1,2,3-триазола 4 региоселективным путем. N-трет-бутоксикарбонильную защитную группу (N-Boc) можно удалить с помощью сильной кислоты, такой как HCl или трифторуксусная кислота (ТФУ). Полученная соль амина 5 может быть подвергнута реакции конденсации с различными замещенными кислотами 6 с помощью активирующего реагента, такого как 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид (HATU) для того, чтобы получить сложный эфир 7. Гидролиз в щелочной среде дает кислоту 8. Остатки R1, R1' и R1'', независимо друг от друга, могут представлять собой, например, водород, алкоксигруппу, галоген или группу -CF3. Остаток R2 может представлять собой, например, C1-7 алкил или фенил.
Схема 2
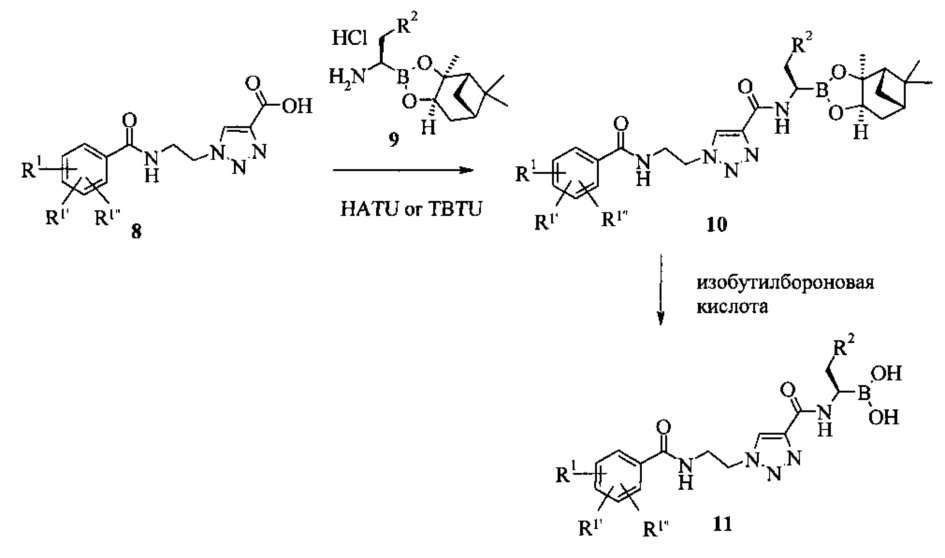
Согласно схеме 2, кислота 8 может вступать в реакцию конденсации с различными замещенными сложных эфиров пинандиола и бороновой кислоты 9 с помощью активирующего реагента, такого как HATU или O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурон тетрафторборат (TBTU) для того, чтобы получить триазол 10. Путем переэтерификации с изобутилбороновой кислотой можно получить целевую бороновую кислоту 11. Остатки R1, R1' и R1'', независимо друг от друга, могут представлять собой, например, водород, алкоксигруппу, галоген или группу -CF3. Остаток R2 может представлять собой, например, С1-7 алкил или фенил.
Примеры
Хотя некоторые формы осуществления настоящего изобретения изображены и раскрыты в данном описании, соединения по настоящему изобретению могут быть получены с использованием соответствующих исходных материалов согласно методам, раскрытым, главным образом, в данном описании и/или с помощью методов, доступных любому специалисту в данной области техники. Все реакции с участием чувствительных к воздушной среде реагентов проводили в инертной атмосфере. Реагенты были применены в том виде, в каком были получены от коммерческих поставщиков, если не указано иное.
Промежуточное соединение 1
1-(2-амино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира гидрохлорид
2-(трет-бутоксикарбониламино)этил бромид (5,0 г, 22,3 ммоль) растворяли в 50 мл диметилформамида (ДМФ) и добавляли азид натрия (1,6 г, 24,5 ммоль). Реакционную смесь перемешивали при 80°С в течение 12 ч. Реакционную смесь разбавляли диэтиловым эфиром (200 мл) и промывали водой (3 раза) и насыщенным солевым раствором (2 раза). Органическую фазу высушивали над сульфатом натрия и концентрировали при пониженном давлении с получением 3,9 г (94%) трет-бутилового эфира (2-азидо-этил)-карбаминовой кислоты в виде бесцветного вязкого масла. По данным газовой хроматографии/масс-спектроскопии, ГХ/МС: (М+Н)+=187,191.
Трет-бутиловый эфир (2-азидо-этил)-карбаминовой кислоты (3,9 г, 20,8 ммоль) и метилпропионат (3,5 г, 3,71 мл, 41,7 ммоль) растворяли в 50 мл трет-бутанола. Добавляли 1,0 M водный раствор меди (II) сульфата пентагидрата (4.17 мл, 4.17 ммоль) с последующим добавлением 1,0 M водного раствора аскорбата натрия (16,7 мл, 16,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 60 часов. Реакцию в реакционной смеси останавливали 150 мл воды и экстрагировали этилацетатом (3 раза по 80 мл). Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на 70 г силикагеля, используя как элюент этилацетат/дихлорметан (градиент: 0-40% этилацетата). Все фракции, содержащие продукт, объединяли и концентрировали с получением 3,2 г (57%) 1-(2-трет-бутоксикарбониламино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира в виде белого с желтоватым оттенком твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М+Н)+=271,1401.
1-(2-трет-бутоксикарбониламино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метиловый эфир (1,75 г, 6,47 ммоль) растворяли в растворе 4N HCl в диоксане (16,2 мл, 64,7 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Растворитель выпаривали с получением 1,32 г (99%) 1-(2-амино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира гидрохлорида в виде белого твердого вещества.
Пример 1
(R)-3-метил-1-(1-(2-(2,3,4-триметоксибензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо) бутилбороновая кислота
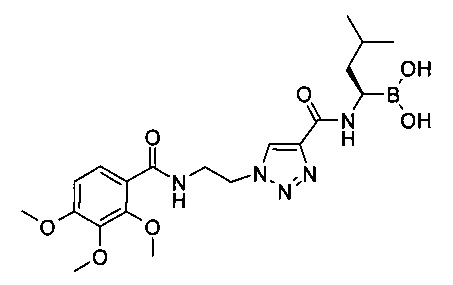
В колбу помещали 2,3,4-триметоксибензойную кислоту (1,29 г, 6,08 ммоль), 57 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (2,9 мл, 16,9 ммоль). Реакционную смесь охлаждали до 0°С. Добавляли HATU (2,83 г, 7,45 ммоль) и реакционную смесь перемешивали при 0°С в течение 1 ч. Добавляли 1-(2-амино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира гидрохлорид (1,4 г, 6,78 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию в реакционной смеси останавливали 1,0 М HCl и экстрагировали этилацетатом. Органический слой промывали водным раствором КНСО3, водой и насыщенным солевым раствором, затем концентрировали и высушивали под высоким вакуумом. Остаток растирали с диэтиловым эфиром с получением 1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира в виде светло-коричневого полутвердого продукта, который использовали без дальнейшей очистки.
В колбу помещали 1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты метиловый эфир (2,22 г, 6,1 ммоль) и 60 мл метанола. Затем добавляли 1,0 М NaOH (24 мл, 24 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь частично концентрировали, затем обрабатывали водой, подкисляли 1,0 М HCl и экстрагировали дважды 200 мл этилацетата. Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток растворяли в 70 мл дихлорметана, 40 мл этилацетата и 10 мл метанола и затем концентрировали до объема ~30 мл. Добавляли диэтиловый эфир и полученную суспензию отфильтровывали, промывали диэтиловым эфиром и высушивали под высоким вакуумом с получением 1,8 г (84%) 1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты в виде белого твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М+Н)+=351,1293.
1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновую кислоту (160 мг, 0,46 ммоль), TBTU (161 мг, 0,50 ммоль) и (R)-боролейцин(+)-пинандиола гидрохлорид (138 мг, 0,46 ммоль) суспендировали в 6 мл дихлорметана при 0°С. Добавляли по каплям растворенный в 1 мл дихлорметана Ν,Ν-диизопропилэтиламин (0,17 мл, 1,00 ммоль) при 0°С в течение 15 мин. Реакционную смесь перемешивали при 0°С и при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли 50 мл дихлорметана и промывали 50 мл 1М HCl, 50 мл 2М КНСО3 и 50 мл воды. Органический слой высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на 20 г силикагеле, используя как элюент этилацетат/дихлорметан (градиент: 0-50% этилацетата). Все фракции, содержащие продукт, объединяли и концентрировали с получением 111 мг (41%) 1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-3-метил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-бутил]-амида в виде белой пены. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М+Н)+=460,2359.
1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-3-метил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-бутил]-амид (109 мг, 0,18 ммоль), изобутилбороновую кислоту (52 мг, 0,51 ммоль) и 2N HCl (0,15 мл, 0,30 ммоль) растворяли в 1,5 мл метанола и 1,5 мл гептана. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Метанольный слой отделяли и промывали дважды 3 мл гептана. Метанольный слой обрабатывали 7 мл этилацетата и концентрировали. Остаток растворяли в 7 мл этилацетата и концентрировали. Остаток растирали с диэтиловым эфиром. Полученное белое твердое вещество экстрагировали дихлорметаном и водой. Органический слой высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток растирали с диэтиловым эфиром с получением 21 мг (25%) (R)-3-метил-1-(1-(2-(2,3,4-триметоксибензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)бутилбороновой кислоты в виде белого твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М-Н)-=462,2161.
Пример 2
(R)-2-фенил-1-(1-(2-(2,3,4-триметоксибензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо) этилбороновая кислота
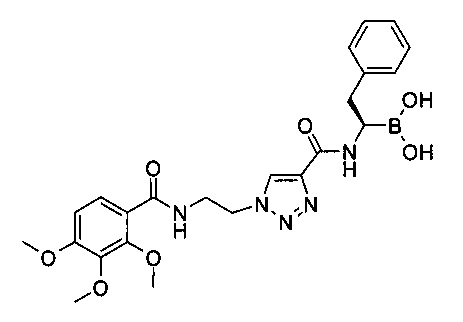
1-(2-(2,3,4-триметоксибензамидо)этил)-1Н-1,2,3-триазол-4-карбоновой кислоты (150 мг, 0,43 ммоль) и (R)-борофенилаланин-(+)-пинандиола гидрохлорид (158 мг, 0,47 ммоль) растворяли в 3 мл ДМФ в круглодонной колбе объемом 10 мл и охлаждали до 0°С. Добавляли по каплям N,N-диизопропилэтиламин (0,19 мл, 1,09 ммоль) при 0°С, затем HATU (179 мг, 0,47 ммоль). После завершения добавления ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали водой и экстрагировали два раза смесью диэтиловый эфир/этилацетат в соотношении 1:1 (40 мл). Органические слои дважды промывали водой и один раз насыщенным солевым раствором, затем объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на 25 г силикагеле, используя как элюент этилацетат/дихлорметан (градиент: 0-50% этилацетата). Все фракции, содержащие продукт, объединяли и концентрировали с получением 153 мг (57%) 1-[2-(2,3,4-триметоксифенил бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амида в виде белой с желтоватым оттенком пены. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (М-Н)-=630.
1-[2-(2,3,4-триметоксифенил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амид (151 мг, 0,24 ммоль) и изобутилбороновую кислоту (70 мг, 0,69 ммоль) растворяли в 1,2 мл метанола и 2,4 мл гексана в круглодонной колбе объемом 10 мл. Добавляли 1,0 M соляную кислоту (0,60 мл, 0,60 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли 10 мл метанола и экстрагировали гексаном. Гексановый слой подвергали реэкстракции 10 мл метанола. Метанольный слой дважды промывали гексаном, затем объединяли и концентрировали. Остаток растворяли в 20 мл дихлорметана и промывали смесью 2 мл воды и 2 мл насыщенного раствора NaHCO3. Водный слой экстрагировали дихлорметаном. Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на 4 г силикагеля, используя как элюент метанол/дихлорметан (градиент: 0-10% метанола, затем 10% метанол/хлороформ). Все фракции, содержащие продукт, объединяли и концентрировали. Остаток растирали с диэтиловым эфиром с получением 24 мг (20%) (R)-2-фенил-1-(1-(2-(2,3,4-триметоксибензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)этилбороновой кислоты в виде белого порошка. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (M+Na)+=520; 1Н ЯМР (400 МГц, CDCl3) δ: 8,23 (t, J=5,7 Hz, 1H), 8,18 (s, 1H), 7,88 (d, J=9,0 Hz, 1H), 7,72 (br. s., 1H), 7,18-7,33 (m, 5H), 6,77 (d, J=9,0 Hz, 1H), 4,71 (t, J=5,6 Hz, 2H), 3,98 (d, J=5,6 Hz, 2H), 3,91 (s, 3H), 3,84 (s, 3H), 3,82 (s, 3H), 3,36 (br. s., 1H), 3,06 (dd, J=14,1, 4,8 Hz, 1H), 2,87 (dd, J=14,1, 9,3 Hz, 1H),
Пример 3
(R)-1-(1-(2-(2-фторо-4-(трифторметил)бензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновая кислота
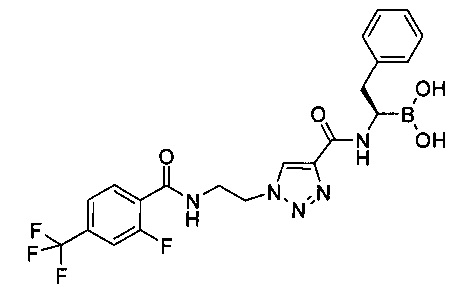
2-фтор-4-(трифторметил)бензойную кислоту (2,31 г, 11,1 ммоль) растворяли в 160 мл М,1Ч-диметилацетамида. Добавляли N,N-диизопропилэтиламин (4,75 мл, 27,8 ммоль), реакционную смесь охлаждали до 0°С и добавляли HATU (4,64 г, 12,2 ммоль). После перемешивания в течение 1 ч при 0°С добавляли 1-(2-амино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира гидрохлорид (2,29 г, 11,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем останавливали реакцию 1М MCl и экстрагировали этилацетатом. Органическую фазу промывали водным раствором KHCO3, водой и насыщенным солевым раствором, затем концентрировали. Неочищенный остаток растирали с диэтиловым эфиром с получением 2,79 г (70%) 1-[2-(2-фтор-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира в виде белого с желтоватым оттенком твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М+Н)+=361,0921.
1-[2-(2-фтор-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты метиловый эфир (2,79 г, 7,74 ммоль) суспендировали в 60 мл метанола. К густой суспензии добавляли 1,0М NaOH (31 мл, 31,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, во время перемешивания реакционная смесь становилась гомогенной. Метанол выпаривали, затем остаток растворяли в воде, подкисляли водным раствором HCl и дважды экстрагировали этилацетатом. Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением небольшого количества твердого вещества. Водные слои объединяли, подщелачивали водным раствором NaOH и концентрировали. Остаток охлаждали до 0°С и подкисляли концентрированной HCl. Полученный осадок собирали фильтрованием и высушивали под высоким вакуумом. Две порции объединяли с получением 2,0 г (75%) 1-[2-(2-фтор-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3] триазол-4-карбоновой кислоты в виде белого твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии высокого разрешения, ЖХ/МС-ВР: (М+Н)+=347,0760.
1-(2-(2-фтор-4-(трифторметил)бензамидо) этил)-1Н-1,2,3-триазол-4-карбоновую кислоту (125 мг, 0,36 ммоль) и (R)-борофенилаланин-(+)-пинандиола гидрохлорид (133 мг, 0,40 ммоль) растворяли в 2,5 мл ДМФ в круглодонной коле объемом 10 мл и охлаждали до 0°С. Добавляли по каплям Ν,Ν-диизопропилэтиламин (0,16 мл, 0,92 ммоль) при 0°С, затем добавляли HATU (151 мг, 0,40 ммоль). После завершения добавления ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали водой и экстрагировали два раза смесью диэтиловый эфир/этилацетат в соотношении 1:1 (40 мл). Органические слои дважды промывали водой и один раз насыщенным солевым раствором, затем объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на 25 г силикагеле, используя элюент этилацетат/дихлорметан (градиент: 0-40% этилацетата). Все фракции, содержащие продукт, объединяли и концентрировали с получением 124 мг (49%) 1-[2-(2-фтор-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амида в виде бесцветного масла. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (М-Н)-=626.
1-[2-(2-фтор-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амид (117 мг, 0,17 ммоль) и изобутилбороновую кислоту (50 мг, 0,49 ммоль) растворяли в 0,8 мл метанола и 1,6 мл гексана в круглодонной колбе объемом 10 мл. Добавляли 1,0 M соляную кислоту (0,42 мл, 0,42 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли 10 мл метанола и экстрагировали гексаном. Гексановый слой подвергали реэкстракции 10 мл метанола. Метанольный слой дважды промывали гексаном, затем объединяли и концентрировали. Остаток растворяли в 20 мл дихлорметана и промывали смесью 2 мл воды и 2 мл насыщенного раствора NaHCO3. Водный слой экстрагировали дихлорметаном. Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на 4 г силикагеля, используя элюент метанол/хлороформ (градиент: 0-10% метанола, затем 10% метанол/этилацетат). Все фракции, содержащие продукт, объединяли и концентрировали. Остаток растирали с диэтиловым эфиром с получением 19 мг (21%) (R)-1-(1-(2-(2-фторо-4-(трифторметил)бензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновой кислоты в виде белого с желтоватым оттенком порошка. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (M+Na)+=516; 1Н ЯМР (400 МГц, CDCl3) δ: 8.21 (br. s., 1Н), 8,02 (t, J=7,6 Hz, 1Н), 7,77 (br. s., 1H), 7,41 (d, J=8,1 Hz, 1H), 7,06-7,30 (m, 7H), 4,55-4,63 (m, 2H), 3,92 (d, J=4,5 Hz, 2H), 3,22 (br. s., 1H), 2,90-2,98 (m, 1H), 2,71-2,82 (m, 1H).
Пример 4
(R)-1-(1-(2-(2-метокси-4-(трифторметил)бензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновая кислота
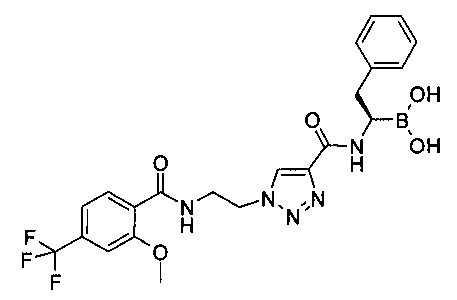
В круглодонной колбе объемом 50 мл суспендировали 1-(2-трет-бутоксикарбониламино-этил)-1Н-[1,2,3]триазол-4-карбоновой кислоты метиловый эфир (500 мг, 1,85 ммоль) в 7 мл дихлорметана. Медленно добавляли трифторуксусную кислоту (4,0 мл, 52 ммоль), в результате чего все твердые вещества растворялись. Реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч, затем концентрировали и высушивали под высоким вакуумом. Остаток растворяли в смеси 5 мл ДМФ и 2-метокси-4-(трифторметил)бензойной кислоты (390 мг, 1,77 ммоль). Добавляли по каплям N,N-диизопропилэтиламин (1,5 мл, 8,6 ммоль), затем добавляли HATU (741 мг, 1,95 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем останавливали реакцию водой и разбавляли петролейным эфиром. Полученную суспензию фильтровали, промывали водой и небольшим количеством петролейного эфира, затем высушивали под высоким вакуумом с получением 557 мг (84%) 1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты метилового эфира в виде белого с желтоватым оттенком твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (M+Na)+=373; 1Н ЯМР (400 МГц, CDCl3) δ: 8,30 (dd, J=8,1, 0,8 Hz, 1H), 8,19 (br. s., 1H), 8,14 (s, 1H), 7,37 (dd, J=8,1, 0,8 Hz, 1H), 7,20 (s, 1H), 4,69-4,76 (m, 2H), 4,02-4,09 (m, 2H), 4,00 (s, 3H), 3,98 (s, 3H).
1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1H-[1,2,3]триазол-4-карбоновой кислоты метиловый эфир (555 мг, 1,49 ммоль) суспендировали в 3 мл метанола и 30 мл тетрагидрофурана (ТГФ) в круглодонной колбе объемом 50 мл. Добавляли гидроксид лития (161 мг, 6,71 ммоль) с последующим добавлением 3 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем органические растворители выпаривали. Водный остаток охлаждали до 0°С и подкисляли 1,0 M HCl до рН~ 2, что приводило к выпадению осадка. Суспензию фильтровали и промывали водой, затем высушивали под высоким вакуумом с получением 452 мг (84%) 1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты в виде белого с желтоватым оттенком твердого вещества. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (М+Н)+=359.
1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновую кислоту (150 мг, 0,42 ммоль) и (R)-борофенилаланин-(+)-пинандиола гидрохлорид (155 мг, 0,46 ммоль) растворяли в 2,5 мл ДМФ в круглодонной колбе объемом 10 мл и охлаждали до 0°С. Добавляли по каплям Ν,Ν-диизопропилэтиламин (0,19 мл, 1,09 ммоль) при 0°С, затем добавляли HATU (175 мг, 0,46 ммоль). После завершения добавления ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали водой и экстрагировали два раза смесью диэтиловый эфир/этилацетат в соотношении 1:1 (40 мл). Органические слои дважды промывали водой и один раз насыщенным солевым раствором, затем объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на 25 г силикагеля, используя элюент этилацетат/дихлорметан (градиент: 0-50% этилацетата). Все фракции, содержащие продукт, объединяли и концентрировали с получением 195 мг (66%) 1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амида в виде бесцветного масла. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (М-Н)-=638.
1-[2-(2-метокси-4-трифторметил-бензоиламино)этил]-1Н-[1,2,3]триазол-4-карбоновой кислоты [(R)-2-фенил-1-((1S,2S,6R,8S)-2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)-этил]-амид (189 мг, 0,27 ммоль) и изобутилбороновую кислоту (78 мг, 0,77 ммоль) растворяли в 1,3 мл метанола и 2,6 мл гексана в круглодонной колбе объемом 10 мл. Добавляли 1,0 M соляную кислоту (0,67 мл, 0,67 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли 10 мл метанола и экстрагировали гексаном. Гексановый слой подвергали реэкстракции 10 мл метанола. Метанольный слой дважды промывали гексаном, затем объединяли и концентрировали. Остаток растворяли в 20 мл дихлорметана и промывали смесью 2 мл воды и 2 мл насыщенного раствора NaHCO3. Водный слой экстрагировали дихлорметаном. Органические слои объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток растирали со смесью диэтиловый эфир/этилацетат с получением 51 мг (38%) (R)-1-(1-(2-(2-метокси-4-(трифторметил)бензамидо)этил)-1Н-1,2,3-триазол-4-карбоксамидо)-2-фенилэтилбороновой кислоты в виде белого порошка. По данным жидкостной хроматографии/масс-спектроскопии, ЖХ/МС: (M+Na)+=528; 1Н ЯМР (400 МГц, CDCl3) δ: 8,26 (d, J=8,1 Hz, 1H), 8,14 (s, 1H), 7,97 (t, J=5,7 Hz, 1H), 7,71 (br. s., 1H), 7,34 (dd, J=8.1, 0,9 Hz, 1H), 7,24-7,28 (m, 4H), 7,16-7,23 (m, 1H), 7,14 (s, 1H), 4,70 (t, J=5,4 Hz, 2H), 3,94-4,05 (m, 2H), 3,89 (s, 3H), 3,30-3,40 (m, 1H), 3,07 (dd, J=14,3, 5,1 Hz, 1H), 2,86 (dd, J=14,3, 10,0 Hz, 1H).
Пример 5
Протоколы анализа и результаты
Клеточный анализ активности/селективности протеасом
Клеточный анализ активности/селективности протеасомных субъединиц представлял собой панель из 5 флуорогенных анализов, при помощи которых независимо друг от друга измеряли протеазную активность субъединиц β5c или β5i (химотрипсинподобная активность), β2c/2i (трипсиноподобная активность) и β1c или β1i (каспазоподобная активность), связанную с протеасомным комплексом в культивируемых клетках. В частности, использовали следующие субстраты для определения активности соответствующих субъединиц: β1i: (PAL)2Rh110, β1c: (LLE)2Rh110, β2c/2i: (KQL)2Rh110, β5c: (WLA)2Rh110, β5i: (ANW)2Rh110. Затем проводили следующую процедуру:
Подготовка клеток: Высевали на планшет с половинным объемом лунок (PerkinElmer Cat 6005569) 25 мкл клеток линии Ramos (2×106 на мл в фосфатно-солевом буфере Дюльбекко (ФСБ-Д)) до конечной концентрации 5×104 клеток на лунку. Прибавляли в каждую лунку по 0,5 мкл на лунку 100×4-кратных серийных разведений соединения или ДМСО. Высшая концентрация испытуемого соединения составляла 20 мкМ, таким образом, серийное разведение соединения начинали с 200 мМ. Инкубировали в течение 30 минут при 37°С. Затем уравновешивали при комнатной температуре в течение 15 минут. Добавляли 25 мкл 2-кратного разведения реакционной смеси, состоящей из 0,025% дигитонина, 20 мкМ каждого из субстратов и 0,5М сахарозы в ФСБ-Д. Встряхивали в течение одной минуты при 700 оборотах в минуту. Инкубировали в течение 120 мин при комнатной температуре. Затем считывали планшеты с помощью планшетного анализатора Envision MultiLabel (PerkinElmer) с использованием длины волны возбуждения 500 нм и длины волны излучения 519 нм.
Модифицированный анализ активности протеасом мононуклеарных клеток периферической крови (МКПК
Клеточный анализ активности протеасом был аналогичен предыдущим клеточным анализам с использованием клеток линии Ramos как субстрата, но в нем применяли человеческие мононуклеарные клетки периферической крови (МКПК) в условиях полной питательной среды RPMI с 10% бычьей эмбриональной сыворотки (ЭБС) в качестве реакционного буфера. Этот анализ был разработан для того, чтобы оценить уровень проникновения испытуемых соединений в клетки на примере первичных человеческих клеток. Затем проводили следующую процедуру: свежие изолированные МКПК от здоровых доноров высевали в концентрации 1×105 клеток на лунку в 100 мкл полной среды RPMI с 10% ЭБС в 96-луночный планшет с V-образным дном лунок. Добавляли по 1 мкл на лунку 100×4-кратных серийных разведений соединения и инкубировали в течение 1 часа. Высшая концентрация испытуемого соединения составляла 20 мкМ (начиная с 2 мМ рабочего раствора, представляющего собой 100-кратное разведение исходного раствора). Центрифугировали клетки при 2000 оборотах в минуту в течение 5 мин. Удаляли все супернатант. Затем клетки ресуспендировали в 25 мкл ФСБ-Д и переносили клетки на новый планшет с половинным объемом лунок (PerkinElmer Cat 6005569). Конечный реакционный объем составлял 50 мкл, включая 25 мкл суспензии клеток, 0,5 мкл 100-кратного разведения ингибитора или ДМСО, 25 мкл смеси субстрата, содержащей 0,025% дигитонина, 20 мкМ субстрата (субстрат: (PAL)2Rh110, (LLE)2Rh110, (KQL)2Rh110 (WLA)2Rh110 или (ANW)2Rh110) в смеси 10% ЭБС и 0,5 M сахарозы. Встряхивали в течение одной минуты (при 700 оборотах в минуту). Инкубировали в течение 2 часов, а затем считывали планшеты с помощью планшетного анализатора Envision с использованием длины волны возбуждения 500 нм и длины волны излучения 519 нм.
Анализ уровня цитокина IP-10 в МКПК
МКПК выделяли из цельной крови следующим образом: кровь собирали в стерильных условиях в гепаринизированные пробирки. Кровь разбавляли равным объемом смеси натрий-фосфатного буфера (НФБ) и 2% фетальной телячьей сыворотки (ФТС) и 30 мл образовавшейся смеси прибавляли в пробирки ACCUSPIN, содержащие 15 мл Histopaque-1077 и предварительно подвергнутые центрифугированию при 800g в течение 30 секунд, и нагревали до комнатной температуры. Затем пробирки центрифугировали при 800g в течение 20 минут при комнатной температуре, без торможения. Полоску мононуклеарных клеток, чуть выше полиэтиленовой пористой перегородки, удаляли пипеткой Пастера. Эти мононуклеарные клетки промывали три раза стерильным НФБ, подсчитывали и ресуспендировали в среде RPMI 1640 с добавлением 10% прогретой инактивированной ФТС, 10 мМ HEPES, 1 мМ пирувата натрия, пенициллина (50 ед/мл), стрептомицин (50 мкг/мл) и глютамина (2 мМ) до концентрации примерно 1,5×106 на 1 мл. Приблизительно 2×105 клеток на лунку высевали в 96-луночные планшеты для культивирования тканей (BD Falcon 353072), и подвергали предварительной инкубации в течении 60 мин при 37°С и титрованию соединений в ДМСО с конечной концентрацией 1%. Затем клетки стимулировали олигонуклеотидом CpG типа A (Invivogen, Cat # tlrl-2216; ODN 2216) до конечной концентрации 2,5 мкМ. Клетки инкубировали в течение ночи и удаляли супернатанты. Жизнеспособность МКПК клеток, оставшихся в лунке, измеряли с помощью АТФ люминесцентного анализа ATPIite (Perkin-Elmer) в соответствии с инструкциями изготовителя. Люминесценцию измеряли на приборе Perkin-Elmer Envision, используя люминесцентный фильтр. Уровень цитокина IP10 измеряли с использованием набора CXCL10/IP10 AlphaLISA kit (Perkin-Elmer) в соответствии с инструкциями изготовителя, за исключением уменьшения наполовину всех объемов. Флуоресценцию измеряли с помощью планшетного анализатора MultiLabel, используя стандартные настройки AlphaScreen.
Результаты:
Результаты приведенных выше анализов для репрезентативных соединений по изобретению представлены ниже в Таблице 1, в которой значения активностей в виде средней ингибирующей концентрации (ИК50) и средней эффективной концентрации (ЭК50) даны в мкМ:
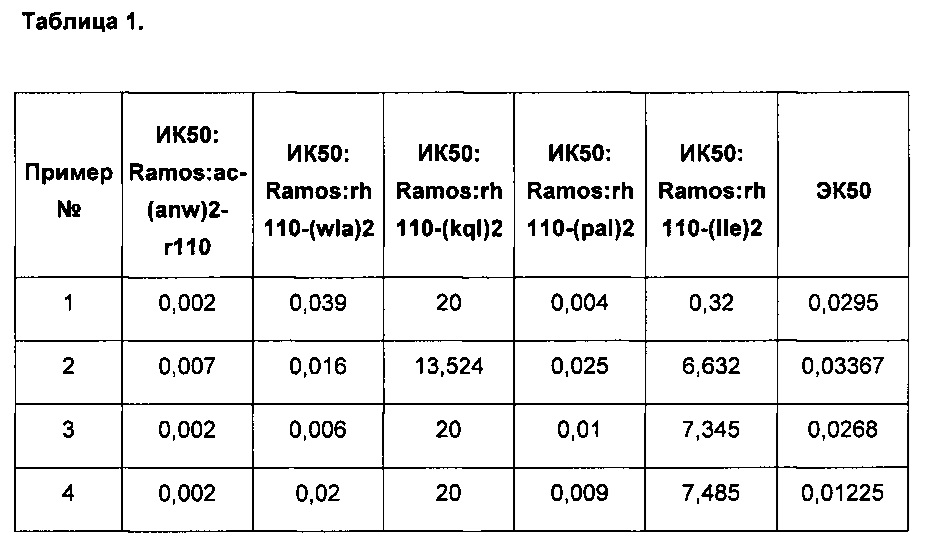
Следует понимать, что настоящее изобретение не ограничивается конкретными формами осуществления изобретения, описанными выше, поскольку могут быть сделаны изменения конкретных форм осуществления изобретения, которые, тем не менее, находятся в пределах прилагаемой формулы изобретения.
Спиро-5,6-дигидро-4н-2,3,5,10в-тетрааза-бензо[е]азулены
2-амино-5,5-дифтор-5,6-дигидро-4н-оксазины в качестве ингибиторов васе 1 и(или) васе 2
Ароиламино- и гетероароиламино-замещенные пиперидины в качестве ингибиторов glyt-1
Способ идентификации модуляторов катехол о-метилтрансферазы
N-пиридин-3-ил или n-пиразин-2-ил карбоксамиды в качестве агентов, повышающих уровень холестерина лпвп
Способ создания магазина аналитических средств
Контейнер для хранения медицинских или фармацевтических жидкостей
Система и способ для анализа биологической жидкости
Производные пирролидина для применения в качестве ингибиторов катепсина
Азациклические спиро-соединения
Способ синтеза производных амино-метилтетралина
Спиро-5,6-дигидро-4н-2,3,5,10в-тетрааза-бензо[е]азулены
2-амино-5,5-дифтор-5,6-дигидро-4н-оксазины в качестве ингибиторов васе 1 и(или) васе 2
Ароиламино- и гетероароиламино-замещенные пиперидины в качестве ингибиторов glyt-1
Способ идентификации модуляторов катехол о-метилтрансферазы
N-пиридин-3-ил или n-пиразин-2-ил карбоксамиды в качестве агентов, повышающих уровень холестерина лпвп
Способ создания магазина аналитических средств
Контейнер для хранения медицинских или фармацевтических жидкостей
Система и способ для анализа биологической жидкости
Производные пирролидина для применения в качестве ингибиторов катепсина

