МАРГАНЕЦСОДЕРЖАЩИЕ ФОСФАТЫ МЕТАЛЛОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к новому содержащему марганец (Mn) фосфату одного металла типа Mn3(PO4)2·3H2О или фосфату нескольких металлов типа (MnxMety)3(PО4)2·3H2О, причем x+y=1, и Met представляет один или несколько металлов, выбранных среди Fe, Co, Ni, Sc, Ti, V, Cr, Cu, Zn, Be, Mg, Ca, Sr, Ba, Al, Zr, Hf, Re, Ru, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu. Кроме того, изобретение относится к способу получения фосфата, а также к его применению.
УРОВЕНЬ ТЕХНИКИ
Перезаряжаемые Li-ионные аккумуляторы представляют собой широко распространенные накопители энергии, в частности, в области мобильных электронных устройств. В качестве катодных материалов хорошо зарекомендовали себя смешанные оксиды лития и еще одного металла, например, такие как LiCoО2, LiNiО2, LiNi1-хCoxО2 и LiMn2О4. Наряду с оксидами, были разработаны также содержащие литий фосфаты с оливиновой структурой, например, такие как LiFePO4 (LFP), которые пригодны в качестве катодных материалов. Эти материалы отличаются хорошей производительностью, высокой удельной емкостью, а также очень высокой стабильностью.
Кроме LFP, имеются и другие литийсодержащие фосфаты, которые обсуждаются как полезные для промышленного производства катодные материалы, например, такие как LiMnPO4, LiCoPO4 или LiNiPO4. Более того, обсуждаются также соединения смешанных металлов типа LiAxByCzPO4 ((x+y+z)=1), например, такие как сплавы LiNiPО4 и LiCoPО4 в форме LiNixCox-1PO4 или LiFexMn1-хPO4.
В качестве соединений, пригодных для замены чистого LiFePO4 (LFP) в катодных материалах, прежде всего, обсуждаются LiFexMnyPO4 и LiFexMnyMzPO4 (LFMP), причем M представляет катион металла, например, такого как Mg. Благодаря более высокому рабочему напряжению соединений, содержащих марганец или никель и, соответственно, кобальт, сравнительно с содержащими железо оливинами, может быть достигнута более высокая плотность аккумулируемой энергии.
Патентный документ DE 102009001204 описывает способ получения кристаллического дигидрата ортофосфата железа(III) (FOP) с кристаллической структурой фосфосидерита, или, соответственно, меташтренгита II, который благодаря способу получения и свойствам материала хорошо пригоден в качестве соединения - прекурсора для получения LFP согласно описанным в литературе способам.
Патентные документы WO 97/40541, US 5910382 и WO 00/60680 описывают получение смешанных фосфатов лития и других металлов, причем, как правило, сначала получаются физические смеси из различных солей металлов, или также металлоорганические соединения, которые в последующей стадии классических методов твердотельного синтеза при высоких температурах и, при необходимости, с регулированием атмосферы, подвергаются кальцинированию. При этом исходные соединения по большей части разлагаются таким образом, что в реакционной системе остаются только желательные ионы для построения целевого соединения.
Для достижения идеального изотропного распределения различных катионов в кристаллической матрице при термических процессах, таких как кальцинирование, в принципе нужно подводить в реакционную систему достаточно высокую энергию, чтобы обеспечить эффективную диффузию ионов. Как правило, предварительно проводится интенсивное перемешивание всех применяемых исходных веществ, чтобы снизить расход энергии и затраты времени. Для перемешивания сырьевых материалов пригодны, в частности, способы сухого или мокрого механического перемешивания, например, размалывание в шаровой мельнице. Однако этим путем получаются только механические смеси частиц или кристаллов различных солей металлов. Поэтому при последующем кальцинировании должно быть гарантировано, что необходимые для формирования желательной кристаллической фазы ионы диффундируют через первичные границы зерен. Обычно для этого требуются температуры от свыше 700 до 800°С, и продолжительности кальцинирования более 15 часов. Также обычной практикой является отжиг физических смесей сначала при более низких температурах (300-400°С), чтобы вызвать начальное разложение. Затем эти промежуточные продукты еще раз подвергаются дополнительным измельчению и интенсивному перемешиванию, чтобы в целом достигнуть хороших результатов в смысле фазовой чистоты, кристалличности и однородности. Поэтому известные термические способы характеризуются большими затратами энергии и времени.
Более того, к применяемым исходным веществам для получения катодных материалов для литий-ионных батарей предъявляются особенно строгие требования в отношении чистоты, так как все компоненты и загрязняющие примеси, которые не разлагаются, остаются в реакционной системе и тем самым в продукте. При разложении катионов и анионов, используемых в качестве исходных соединений металлических соединений (например, NH4+, C2О42-, (CH3)(CH2)nCOO-, CО32-, и т.д.) к тому же образуются газы, которые вследствие потенциально опасных свойств (например, CO, NH3, NOx, и т.д.) должны быть подвергнуты дорогостоящей обработке в потоке отходящих газов.
Патентный документ CA 02443725 описывает получение LiXYPО4 (X, Y = металл, например, Fe, Mn, и т.д.) с использованием сульфата железа, сульфата марганца и фосфата лития, а также дополнительно гидроксида лития в качестве исходных веществ, из которых сначала получается не охарактеризованная более подробно смесь твердых веществ, которая в заключение переводится в желательный продукт путем кальцинирования при температуре от 300 до 1000°С.
Введение определенных металлов в форме их сульфатов в эквимолярном количестве относительно фосфата обычно требует того, чтобы продукт был подвергнут интенсивному промыванию для сокращения содержания сульфата до допустимой степени. Как известно, сульфат вследствие коррозионного воздействия является нежелательной загрязняющей примесью в литий-ионных батареях. Однако в ходе процесса интенсивного промывания также может в значительной мере вымываться литий, поскольку среди ортофосфатов лития только нормальный ортофосфат лития (с тремя катионами лития) имеет очень низкую растворимость. Если продукт согласно патентному документу CA 02443725 подвергается такой обработке в процессе промывания, необходимо учитывать вымывание лития. Правда, в патентном документе CA 02443725 процесс промывания не упоминается, что опять же имело бы результатом высокое загрязнение продукта сульфатом.
С использованием гидротермических или сольвотермических способов в принципе могут быть достигнуты вполне однородные распределения катионов, когда растворимости и константы комплексообразования и, соответственно, коэффициенты роста кристаллов вносимых катионов и анионов на протяжении реакционного процесса в выбранной матрице могут быть контролируемыми и регулируемыми таким образом, что образуются исключительно желательные вещества в выделяемой форме. Здесь зачастую используются поверхностно-активные вещества, или также вспомогательные материалы, которые содействуют формированию определенной кристаллической фазы или росту в предпочтительном направлении, так называемые темплейты, которые известны специалисту, чтобы управлять процессом роста кристаллов. В таких способах работа часто проводится в замкнутых системах за пределами температуры кипения реакционной матрицы, вследствие чего возникают высокие давления. Это предъявляет строгие требования к реакторной технологии. Тем не менее, полученные продукты должны быть многократно, или, соответственно, дополнительно подвергнуты заключительному кальцинированию, чтобы обеспечить необходимую кристалличность. Кроме того, должны быть количественно удалены поверхностно-активные вспомогательные вещества, чтобы не вызывать никаких негативных влияний в последующем применении. Это также достигается нагреванием, причем эти вещества выгорают или, соответственно, обугливаются или превращаются в сажу.
Также описаны способы, действующие без давления, причем всегда сообщаются продолжительности кристаллизации желательных продуктов, занимающие время от многих дней до недель. Это делает сомнительной экономичность при использовании в промышленном масштабе.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задача настоящего изобретения состоит в создании новых фосфатов одного металла или нескольких металлов, которые, например, пригодны для получения катодных материалов для литий-ионных батарей, в частности, таких, из которых могут быть изготовлены катодные материалы с высокими плотностями аккумулирования энергии, а также в разработке способа их получения, который является сравнительно энергосберегающим и простым в исполнении, и с помощью которого могут быть получены фосфаты с высокой чистотой, чтобы они, по сравнению с прототипом, были лучше пригодны в качестве соединений-предшественников (прекурсоров) для получения литиированных катодных материалов для литий-ионных аккумуляторов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задача изобретения решена с помощью содержащего марганец (Mn) фосфата одного металла типа Mn3(PO4)2·3H2О или фосфата нескольких металлов типа (MnxMety)3(PО4)2·3H2О, причем x+y=1, и Met представляет один или несколько металлов, выбранных из Fe, Co, Ni, Sc, Ti, V, Cr, Cu, Zn, Be, Mg, Ca, Sr, Ba, Al, Zr, Hf, Re, Ru, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu, отличающихся тем, что фосфат в дифрактограмме порошкового рентгеноструктурного анализа имеет пики при 10,96±0,05, 12,78±0,17, 14,96±0,13, 17,34±0,15, 18,98±0,18, 21,75±0,21, 22,07±0,11, 22,97±0,10, 25,93±0,25, 26,95±0,30, 27,56±0,10, 29,19±0,12, 29,84±0,21, 30,27±0,12, 34,86±0,21, 35,00±0,20, 35,33±0,30, 35,58±0,10, 35,73±0,12, 42,79±0,45, 43,37±0,45, 44,7±≤±0,15 и 44,93±0,20 градусов 2θ, на основе CuKα-излучения.
Соответствующий изобретению содержащий марганец (Mn) фосфат имеет новый структурный тип, который характеризуется положением его пиков в дифрактограмме порошкового рентгеноструктурного анализа. Новый структурный тип здесь также обозначается как «Mn3(PO4)2·3H2О-структурный тип». В литературе этот структурный тип неизвестен. Он может быть получен как в виде фосфата одного металла типа Mn3(PO4)2·3H2О, так и в форме фосфата нескольких металлов типа (MnxMety)3(PО4)2·3H2О, с вышеуказанными характеристическими положениями пиков в дифрактограмме порошкового рентгеноструктурного анализа. При этом отдельные пики, в зависимости от состава металлических компонентов, могут варьировать с незначительными сдвигами в пределах указанного символом «±» углового диапазона, приведенного в градусах 2θ.
Соответствующий изобретению содержащий марганец (Mn) фосфат нового структурного типа предпочтительно имеет орторомбическую элементарную ячейку с параметрами кристаллической решетки 13,2±0,2, 8,6±0,2 и 8,1±0,2 Ангстрем.
В одном предпочтительном варианте осуществления изобретения содержащий марганец (Mn) фосфат присутствует в виде углеродного композита и содержит от 1 до 10% по весу углерода, предпочтительно от 1,5 до 5% по весу углерода, в особенности предпочтительно от 1,8 до 4% по весу углерода, в расчете на совокупный вес фосфата и углерода.
Такой фосфатно-углеродный композит получают добавлением источника углерода во время получения соответствующего изобретению фосфата, который будет более подробно описан ниже в связи с соответствующим изобретению способом. Введение углерода в соответствующий изобретению продукт позволяет обеспечить электрическую проводимость материала как такового, и/или продукта, который может быть изготовлен из материала, например, катодных материалов для литий-ионных аккумуляторов. Вариацией количества и типа вводимого во время получения источника углерода можно произвольно регулировать достигаемое содержание углерода и тем самым проводимость в известных пределах. Слишком высокое содержание углерода имеет тот недостаток, что сокращается максимально возможное количество активного катодного материала в последующем применении для литий-ионных батарей. При содержании углерода ниже 1% по весу достаточное повышение проводимости уже более не достигается.
В еще одном предпочтительном варианте осуществления изобретения содержащий марганец (Mn) фосфат имеет пластинчатую морфологию, предпочтительно с толщиной пластинок (= наименьшей пространственной протяженностью) в диапазоне от 10 до 100 нм, в особенности предпочтительно в диапазоне от 20 до 70 нм, наиболее предпочтительно в диапазоне от 30 до 50 нм.
Соответствующая изобретению предпочтительная пластинчатая морфология имеет особенное преимущество, например, при применении соответствующего изобретению фосфата для получения литиированного (Li-содержащего) катодного материала для Li-ионных аккумуляторов. При этом пластинчатая форма с наномасштабной толщиной пластинок первичных кристаллитов обеспечивает наименьшие из возможных расстояния (пути) диффузии и продолжительности диффузии при литиировании посредством простого и экономичного способа кальцинирования. Имеющее место при соответствующем изобретению материале идеально изотропное распределение металлических ионов при этом к тому же снижает необходимые температуры кальцинирования и продолжительности кальцинирования, поскольку не требуется никакая диффузия металлических ионов через границы зерен. Определенная кристаллическая структура обеспечивает однозначные и воспроизводимые пути реакции при кальцинировании и при изготовлении катодных материалов.
В одном дополнительном предпочтительном варианте осуществления изобретения содержащий марганец (Mn) фосфат представляет собой фосфат нескольких металлов типа (MnxMety)3(PО4)2·3H2О, в котором соотношение «Mn:(Mn+Met)=х:(х+y)» составляет ≥0,15, предпочтительно ≥0,4, в особенности предпочтительно ≥0,5. Соотношение «Mn:(Mn+Met)» означает атомное отношение марганца к сумме всех содержащихся согласно изобретению металлов, включая марганец. В применяемом для соответствующего изобретению фосфата нескольких металлов способе написания «(MnxMety)3(PО4)2·3H2О» с х+y=1 соотношение «Mn: (Mn+Met)» также может быть выражено отношением «х:(х+y)». В соответствующем изобретению фосфате одного металла соотношение х:(х+y)=1, так как в этом случае y=0. При слишком низком содержании марганца в фосфате нескольких металлов новый «Mn3(PO4)2·3H2О-структурный тип» может быть не получен.
Настоящее изобретение также включает способ получения содержащего марганец (Mn) фосфата одного металла типа Mn3(PO4)2·3H2О или фосфата нескольких металлов типа (MnxMety)3(PО4)2·3H2О, причем x+y=1, а = от 0 до 9, и Met представляет один или многие металлы, выбранные среди Fe, Co, Ni, Sc, Ti, V, Cr, Cu, Zn, Be, Mg, Ca, Sr, Ba, Al, Zr, Hf, Re, Ru, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu, причем способ отличается тем, что
а) готовится водный раствор (I), который содержит по меньшей мере двухвалентные катионы марганца (Mn2+), и, необязательно, один или более из металлов Fe, Со и/или Ni, в качестве двухвалентных катионов, для чего оксидные соединения металла(II), металла(III) и/или металла(IV), или их смеси или соединения со смешанными степенями окисления, выбранные из гидроксидов, оксидов, оксигидроксидов, оксигидратов, карбонатов и гидроксикарбонатов, по меньшей мере одного из металлов Mn, Fe, Со и/или Ni, вносятся вместе с элементарными формами или сплавами по меньшей мере одного из металлов Mn, Fe, Со и/или Ni в водную среду, содержащую фосфорную кислоту, и оксидные соединения металлов с элементарными формами или сплавами металлов (в окислительно-восстановительной реакции) преобразуются в двухвалентные ионы металлов, причем по меньшей мере одно из оксидных соединений металлов и/или по меньшей мере один компонент из элементарных форм или сплавов металла включает марганец,
b) при необходимости, из фосфорнокислотного водного раствора (I) удаляются содержащиеся твердые вещества,
с) когда фосфат представляет собой фосфат нескольких металлов, и, дополнительно к введенным в стадии а) в раствор металлам содержит металл, выбранный согласно обозначению «Met», к водному раствору (I), кроме того, добавляется по меньшей мере одно соединение по меньшей мере одного металла «Met» в форме водного раствора или как твердое вещество в форме соли, причем по меньшей мере одно соединение выбирается из гидроксидов, оксидов, оксигидроксидов, оксигидратов, карбонатов, гидроксикарбонатов, карбоксилатов, сульфатов, хлоридов или нитратов металла,
d) готовится затравочный раствор (II), полученный из водного раствора фосфорной кислоты нейтрализацией водным раствором гидроксида щелочного металла, или образованный из водного раствора одного или нескольких фосфатов щелочных металлов, с величиной рН от 5 до 8,
е) водный раствор (I) добавляется в затравочный раствор (II), и одновременно добавляется основный водный раствор гидроксида щелочного металла таким образом, что значение рН полученной реакционной смеси поддерживается в диапазоне от 5 до 8, предпочтительно от 6 до 7, причем фосфат типа Mn3(PO4)2·3H2О или (MnxMety)3(PО4)2·3H2О выпадает в осадок,
f) выпавший в осадок фосфат отделяется от реакционного раствора.
Введенные в стадии а) в раствор (I) металлы здесь также называются «основными металлами». Основные металлы включают по меньшей мере марганец (Mn) и, необязательно, Fe, Со и/или Ni. Введенные в стадии с) в раствор (I) необязательные металлы, выбранные из Fe, Co, Ni, Sc, Ti, V, Cr, Cu, Zn, Be, Mg, Ca, Sr, Ba, Al, Zr, Hf, Re, Ru, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu, здесь также называются «легирующими металлами». В стадии с) также может быть внесен дополнительный марганец (Mn). Легирующие металлы могут присутствовать в растворе в форме двухвалентных металлических ионов, но они также могут находиться в растворе в форме трехвалентных или четырехвалентных металлических ионов. Некоторые из легирующих металлов предпочтительно присутствуют в трехвалентной форме. Если соответствующий изобретению фосфат, например, в дальнейшем перерабатывается для получения катодного материала, то эти не являющиеся двухвалентными металлические ионы образуют в структуре квазидефекты, что может оказывать благоприятное влияние на производительность катодного материала.
Соответствующий изобретению способ получения фосфата одного металла или нескольких металлов, по сравнению с прототипом, является простым в исполнении и экономичным. Дополнительное преимущество соответствующего изобретению способа состоит в том, что водный фосфорнокислотный раствор (I) содержит только желательные металлические ионы, и исключительно или преимущественно фосфат-анионы или, соответственно, фосфорную кислоту. Тем самым не требуется трудоемкое удаление чужеродных анионов, таких как сульфаты, нитраты, хлориды и другие, в последующем процессе получения соответствующих изобретению продуктов. Если в стадии с) соответствующего изобретению способа легирующие металлы вносятся, например, в форме своих сульфатов, нитратов или хлоридов, то это производится в незначительных количествах, которые еще допустимы в изготовленном продукте, и свойства продукта не ухудшаются вообще, или ущерб причиняется только в допустимой мере. Тем самым соответствующие изобретению фосфаты обладают высокой чистотой, благодаря чему они, например, особенно пригодны для получения литиированных катодных материалов. Литиирование может быть выполнено в реакционной стадии простой термической обработки (кальцинирования), причем здесь, в зависимости от сорта фосфатного материала, должна добавляться подходящая соль лития.
На основе соответствующего изобретению способа появляется в распоряжении исключительно гибкий реакционный принцип, с помощью которого могут быть получены многочисленные фосфатные системы описываемого здесь типа, например, (псевдо)бинарные, (псевдо)тройные и (псевдо)четверные системы.
Соответствующий изобретению способ обеспечивает возможность надлежащим выбором условий осаждения, таких как значение рН, концентрации, температура, и т.д., регулировать определенные параметры материала, такие как кристаллическая фаза и распределение катионов, морфология, размер кристаллитов и вторичных частиц, а также химическую чистоту полученных продуктов. При этом вышеописанные продукты предпочтительно находятся в состоянии с пластинчатой морфологией, имеющей единообразную кристаллическую фазу и изотропное распределение катионов.
В первой реакционной стадии соответствующего изобретению способа оксидные соединения металла(II), металла(III) и/или металла(IV) вводятся во взаимодействие с элементарным металлом или сплавами в фосфорнокислотной водной среде в условиях окислительно-восстановительной реакции с образованием двухвалентных металлических ионов. Течение описываемой окислительно-восстановительной реакции между элементарными металлами и оксидными компонентами зависит от их конкретных удельных площадей поверхности, так как перенос электронов происходит на границе раздела фаз. В качестве побочной реакции, конкурирующей с переносом электронов от металлов в элементарной форме на металлы в окисленной форме, следует принимать во внимание образование газообразного водорода. При этом происходит перенос электронов с металлов в элементарной форме на протоны с образованием радикалов, которые в результате рекомбинации радикалов образуют газообразный водород. Поэтому размеры частиц применяемых металлов в элементарной и оксидной формах должны быть согласованными между собой, чтобы подавлять побочную реакцию и извлекать по возможности максимальную пользу из растворения недорогого металла в оксидной форме. В общем и целом справедливо, что чем мельче металл в элементарной форме, тем больше это благоприятствует побочной реакции, когда оксидная форма не обеспечивает достаточно большой площади активной поверхности.
В зависимости от состава реакционного раствора, в растворе могут оставаться непрореагировавшие компоненты в виде твердого осадка. Если в полученном реакционном растворе еще содержатся твердые вещества, они предпочтительно отделяются от фосфорнокислотного водного раствора. Отделение твердых веществ может быть проведено всеми подходящими известными методами разделения жидкостей и твердых веществ, например, фильтрованием, центрифугированием, седиментацией, и т.д.
Когда полученный согласно изобретению фосфат представляет собой фосфат нескольких металлов, и дополнительно к введенным в стадии а) в раствор металлам содержит другие металлы, выбранные из поименованных как «Met», к водному раствору (I) перед прибавлением к затравочному раствору (II) в стадии е) добавляется по меньшей мере одно соединение по меньшей мере одного из выбранных среди «Met» металлов, по выбору также марганец, в форме водного раствора или твердого вещества в виде соли, причем по меньшей мере одно соединение предпочтительно выбирается из гидроксидов, оксидов, оксигидроксидов, оксигидратов, карбонатов, гидроксикарбонатов, карбоксилатов, сульфатов, хлоридов или нитратов металла. Добавление этих легирующих металлов целесообразнее производится в стадии с) способа, после того, как по обстоятельствам содержащиеся твердые вещества были отделены от фосфорнокислотного водного раствора (I). В альтернативном варианте, описываемое добавление легирующих металлов также может быть проведено непосредственно после получения раствора (I) в стадии а) и перед отделением содержащихся по обстоятельствам твердых веществ. Отделение содержащихся при известных условиях твердых веществ тогда производится по завершении добавления легирующих металлов.
Добавлением подходящих солей металлов (легирующих металлов) в вышеуказанной форме может быть очень точно отрегулировано желательное содержание металла и, соответственно, соотношение металлов между собой, в полученном фосфате. Это справедливо, прежде всего, для металлов, которые вводятся в сравнительно небольшом количестве. Целесообразнее должны вводиться соединения металлов, которые в последующем технологическом процессе не вносят в смесь мешающих анионов, чтобы гарантировать наивысшую чистоту продукта. Они представляют собой, в частности, гидроксиды, оксиды, оксигидроксиды, оксигидраты, карбонаты и гидроксикарбонаты, которые в преобладающих кислотных условиях реагируют с образованием воды или, соответственно, разлагаются. При необходимости могут быть введены привычные специалисту буферные реагенты, чтобы предотвратить нежелательное преждевременное или, соответственно, неконтролируемое осаждение. Также пригодны карбоксилаты, так как остающиеся в смеси доли органических кислот, как правило, разлагаются при последующем кальцинировании продукта. Добавление металлов в форме их сульфатов, хлоридов или нитратов также может быть пригодным для легирующих металлов, когда в результате этого содержание сульфатов, хлоридов или нитратов в продукте не превышает определенных предельных значений, которые могут рассматриваться как приемлемые для конкретного применения.
Затравочный раствор (II) для последующего осаждения соответствующих изобретению фосфатов также представляет собой раствор фосфата со значением рН, забуференным в диапазоне от 5 до 8. Затравочный раствор готовится либо из водного фосфорнокислотного раствора нейтрализацией водным раствором гидроксида щелочного металла, либо непосредственно из водного раствора одного или многих фосфатов щелочных металлов. Для осаждения соответствующих изобретению фосфатов водный раствор (I) прибавляется к затравочному раствору (II). Вследствие более низкого значения рН фосфорнокислотного раствора (I) при этом одновременно прибавляется основный водный раствор гидроксида щелочного металла, чтобы поддерживать величину рН полученной реакционной смеси в диапазоне от 5 до 8. Слишком низкое значение рН затравочного раствора (II) и, соответственно, полученной в результате этого реакционной смеси на уровне менее 5 имеет тот недостаток, что наряду с желательной соответствующей изобретению кристаллической фазой могут образовываться и другие кристаллические фазы, например, гидро- или дигидрофосфаты металлов. Слишком высокое значение рН затравочного раствора (II) свыше величины рН 8 имеет тот недостаток, что могут оставаться следы гидроксидов металлов, которые представляют собой нежелательное загрязнение соответствующих изобретению продуктов. Основный водный раствор гидроксида щелочного металла предпочтительно прибавляется таким образом, чтобы при прибавлении раствора (I) в реакционную смесь значение рН поддерживалось в диапазоне от 6 до 7. Это имеет то преимущество, что образуется исключительно соответствующая изобретению кристаллическая фаза.
После выпадения соответствующего изобретению фосфата в осадок проводится отделение его от реакционного раствора. Это выполняется также общеизвестными способами, например, фильтрованием, центрифугированием, седиментацией, и т.д. Целесообразно, чтобы отделенный от реакционного раствора фосфат затем высушивался, то есть подвергался обезвоживанию. Высушивание может быть по выбору выполнено в условиях окружающей атмосферы, атмосферы защитного газа, и/или при пониженном давлении, и/или при повышенной температуре (свыше комнатной температуры, 25°С). Пригодные для этого способы являются привычными специалисту в этой области и не нуждаются в более подробном описании. В дополнение, можно сослаться на нижеследующие примеры. При высушивании из остатка, отделенного от реакционного раствора, удаляется свободная вода. Но в зависимости от желательного продукта, также удаляется связанная кристаллизационная вода путем высушивания до достижения желательной степени гидратации продукта.
В одном предпочтительном варианте осуществления изобретения осажденный и отделенный от реакционного раствора фосфат высушивается до степени гидратации Mn3(PO4)2·3H2О или (MnxMety)3(PО4)2·3H2О с 0≤а≤9, в особенности предпочтительно с «а», равным 0, 3 или 7, наиболее предпочтительно с а=3. Степень гидратации с а=3 имеет новый соответствующий изобретению структурный тип. Она стабильна в широком температурном диапазоне. Также достаточно стабильными являются степени гидратации с а=0 и а=7.
В одном предпочтительном варианте исполнения полученный соответствующим изобретению способом содержащий марганец (Mn) фосфат представляет собой фосфат нескольких металлов, который, кроме марганца (Mn), содержит по меньшей мере один дополнительный металл (Met), причем фосфат предпочтительно содержит не более 7 различных металлов. Во многих случаях целесообразным является получение фосфата нескольких металлов соответствующего изобретению типа с двумя, тремя или четырьмя различными металлами. Зачастую желательно получение фосфата нескольких металлов, который, кроме марганца (Mn), содержит один или два различных металла, выбранных из Fe, Со и Ni, с высоким уровнем содержания, в качестве так называемых основных металлов, и/или один или многие металлы с низким в каждом случае содержанием в качестве так называемых легирующих металлов. Например, может быть преимущественным соответствующий изобретению фосфат, содержащий марганец в качестве основного металла, с незначительной долей дополнительного металла, например, Mg, Al, Cu, или одного из металлов лантаноидного ряда, как это будет продемонстрировано в нижеследующих примерах.
В особенности предпочтительно содержащий марганец (Mn) фосфат нескольких металлов включает, в расчете на все содержащиеся металлы, по меньшей мере 40 атомных процентов Mn, предпочтительно по меньшей мере 60 атомных процентов Mn, в особенности предпочтительно 80 атомных процентов Mn, наиболее предпочтительно 90 атомных процентов Mn.
В одном альтернативном предпочтительном варианте исполнения полученный соответствующим изобретению способом содержащий марганец (Mn) фосфат представляет собой фосфат одного металла, который, наряду с обусловленными способом загрязняющими примесями, в качестве металла содержит только марганец (Mn).
Соответствующий изобретению способ, в частности, при получении фосфатов нескольких металлов имеет значительное преимущество сравнительно с прототипом в отношении эффективности, технологических затрат, расхода энергии и достижимой чистоты продукта. Более того, содержание различных металлов в фосфате нескольких металлов может очень просто и точно регулироваться. Кроме того, соответствующий изобретению способ позволяет надлежащим выбором условий осаждения, таких как значение рН, концентрации, температура, и т.д., регулировать определенные параметры материала, такие как кристаллическая фаза и распределение катионов, морфология, размер кристаллитов и вторичных частиц, а также химическую чистоту полученных продуктов. Это невозможно или достигается только в ограниченной степени в случае известных способов, в которых фосфаты металлов и другие соли металлов смешиваются и затем подвергаются термическому преобразованию путем кальцинирования, и которые, как правило, проводятся со значительно более высоким расходом энергии.
В особенности предпочтительно полученный согласно изобретению содержащий марганец (Mn) фосфат описываемого здесь нового «Mn3(PO4)2⋅3H2О-структурного типа» имеет в дифрактограмме порошкового рентгеноструктурного анализа пики при 10,96±0,05, 12,78±0,17, 14,96±0,13, 17,34±0,15, 18,98±0,18, 21,75±0,21, 22,07±0,11, 22,97±0,10, 25,93±0,25, 26,95±0,30, 27,56±0,10, 29,19±0,12, 29,84±0,21, 30,27±0,12, 34,86±0,21, 35,00±0,20, 35,33±0,30, 35,58±0,10, 35,73±0,12, 42,79±0,45, 43,37±0,45, 44,70±0,15 и 44,93±0,20 градусов 2θ, на основе CuKα-излучения.
Этот соответствующий изобретению содержащий марганец (Mn) фосфат нового структурного типа предпочтительно имеет орторомбическую элементарную ячейку с параметрами кристаллической решетки 13,2±0,2, 8,6±0,2 и 8,1±0,2 Ангстрем.
В одном предпочтительном варианте исполнения соответствующего изобретению способа осаждение содержащего марганец (Mn) фосфата в стадии е) проводится при температуре в диапазоне от 5 до 105°C, предпочтительно в диапазоне от 10 до 40°C. При этом температура может поддерживаться постоянной с помощью подходящего регулирующего устройства в диапазоне +/-5°C относительно желательного уровня. Более высокие температуры, как правило, ведут к более выраженной кристалличности продукта. Температуры ниже 5°C, хотя и возможны, но требуют ненужного охлаждения. Наиболее благоприятным является проведение осаждения при комнатной температуре, или, соответственно, при отрегулированной сообразно реакционным условиям температуре. При температурах свыше 105°C реакционная смесь закипает, что является нежелательным и может быть вредным. В особенности предпочтительно проводить осаждение фосфата в стадии е) при температуре в диапазоне от 10 до 40°C, так как это является наиболее экономически целесообразным.
В еще одном предпочтительном варианте исполнения соответствующего изобретению способа в водном растворе (I) перед прибавлением к затравочному раствору (II) в стадии d) диспергируется источник углерода, причем источник углерода включает элементарный углерод или состоит исключительно из элементарного углерода, и предпочтительно выбирается из графита, расширенного графита, сажи, такой как технический углерод или сосновая сажа, углеродных нанотрубок (CNT), фуллеренов, графена, стекловидного угля (стеклообразного углерода), углеродных волокон, активированного угля, или их смесей, или вышеуказанный источник углерода, наряду с элементарным углеродом, включает органические соединения, причем органические соединения предпочтительно выбираются из углеводородов, спиртов, альдегидов, карбоновых кислот, поверхностно-активных веществ, олигомеров, полимеров, углеводов, или их смесей.
Добавление источника углерода к водному раствору (I) в соответствующем изобретению способе позволяет получить фосфатно-углеродные композиты, благодаря чему можно обеспечить электрическую проводимость материала как такового, и/или изготовленного из материала продукта, например, для изготовления катодных материалов для литий-ионных аккумуляторов. Вариацией количества и типа источника углерода, вводимого непосредственно в раствор (I), можно произвольно регулировать достигаемое содержание углерода и тем самым проводимость в известных пределах. Целесообразным является добавление источника углерода в водный раствор (I) в количестве от 1 до 10% по весу углерода, предпочтительно от 1,5 до 5% по весу углерода, в особенности предпочтительно от 1,8 до 4% по весу углерода, в расчете на вес осажденного вместе с углеродом фосфата. Слишком высокое содержание углерода имеет тот недостаток, что сокращается максимально возможное количество активного катодного материала в последующем применении для литий-ионных батарей. При содержании углерода ниже 1% по весу достаточное повышение проводимости уже более не достигается.
Для повышения стабильности дисперсии углеродного компонента в растворе, в зависимости от сорта источника углерода, может быть предпочтительным тонкое распределение источника углерода под воздействием механических сил в растворе. Для этого, наряду с известными методами приложения высоких сдвиговых нагрузок, пригодно применение бисерных мельниц. При использовании бисерной мельницы, кроме тонкого распределения источника углерода, также может быть модифицирован его средний размер частиц и, соответственно, величина агломератов. Так, например, средний размер зерен графита сокращается до величины <300 нм. Полученные дисперсии очень устойчивы, и даже после многих дней едва ли проявляют тенденцию к седиментации твердых частиц графита, хотя он, как правило, обусловливает, прежде всего, гидрофобные свойства материала. Путем описываемой обработки и при избытке свободного фосфата или, соответственно, фосфорной кислоты в смеси поверхности графита модифицируются, и твердое вещество в дисперсии стабилизируется. Известны также способы гидрофилизации углерода и, соответственно, графита, которые могут быть предпочтительно применены, например, частичное окисление поверхности. Кроме того, стабильность дисперсии источника углерода в растворе (I) может быть эффективно повышена добавлением поверхностно-активных веществ.
К раствору, дополнительно или альтернативно другим источникам углерода, может быть также добавлен полимер или биополимер в качестве источника углерода. Здесь преимущество обеспечивается растворимостью источников углерода при преобладающих в растворе (I) кислотных условиях. Если компоненты должны быть нерастворимыми, распределение в растворе также может быть улучшено воздействием сдвиговых нагрузок.
В одном дополнительном предпочтительном варианте исполнения соответствующего изобретению способа, содержащая фосфорную кислоту водная среда для получения водного раствора (I) содержит фосфорную кислоту в молярном избытке относительно суммы молярных количеств катионов металлов, вводимых в раствор в составе оксидных соединений металлов, и вводимых в элементарной форме или в виде сплава металлов. Без избытка фосфорной кислоты окислительно-восстановительный процесс не протекает или идет с настолько низкой скоростью, что способ может быть уже неинтересным для промышленного применения.
Концентрация фосфорной кислоты в водном растворе (I) в стадии а) целесообразно составляет от 5% до 85%, предпочтительно от 10% до 40%, в особенности предпочтительно от 15% до 30%, наиболее предпочтительно от 20% до 25%, в расчете на вес водного раствора (I).
В одном дополнительном предпочтительном варианте исполнения соответствующего изобретению способа, затравочный раствор (II) содержит фосфат-ионы, в пересчете на Р2О5, с концентрацией в диапазоне от 0,35 до 1,85 моль/литр. Концентрация фосфат-ионов ниже 0,35 моль/литр Р2О5 имеет тот недостаток, что имеет место ненужное разбавление реакционной смеси, и в случае промышленного применения нужно проводить бесполезную обработку больших объемов фильтрата. Концентрация фосфат-ионов свыше 1,85 моль/литр имеет тот недостаток, что реакционная смесь вследствие большого содержания твердого вещества и обусловленной этим высокой вязкости не может оптимально перемешиваться. Вследствие этого могут возникать локальные концентрационные градиенты, что опять же является неблагоприятным для образования желательной кристаллической фазы.
В одном дополнительном предпочтительном варианте исполнения соответствующего изобретению способа, взаимодействие оксидных соединений металлов с металлами в элементарной форме или в виде сплавов в стадии а) проводится при температуре в диапазоне от 5°C до 105°C, предпочтительно в диапазоне от 10°C до 75°C, в особенности предпочтительно в диапазоне от 20°C до 50°C. При температурах в пределах соответствующего изобретению диапазона взаимодействие с различными металлическими компонентами может быть проведено гладко и с удовлетворительной скоростью, без того, что это приведет к явлениям окисления кислородом воздуха.
Кроме того, взаимодействие оксидных соединений металлов с металлами в элементарной форме или в виде сплавов в стадии а) предпочтительно проводится при интенсивном перемешивании, чтобы обеспечить равномерный ход реакции и предотвратить локальные избыточные концентрации внутри реакционного раствора. Это также справедливо для последующей стадии осаждения.
Взаимодействие оксидных соединений металлов с металлами в элементарной форме или в виде сплавов в стадии а) целесообразно в течение периода времени от 1 минуты до 240 минут, предпочтительно от 5 минут до 120 минут, в особенности предпочтительно от 30 минут до 90 минут. Необходимая продолжительность реакции для достаточно полного преобразования зависит от природы реактантов и условий реакции, и может быть без труда определена специалистом в немногих простых экспериментах. Как правило, при слишком кратковременной реакции превращение является недостаточно полным, и остается слишком много непрореагировавших исходных веществ. Но продолжительность реакции также не должна быть слишком длительной, поскольку тогда способ оказывается менее экономичным. Полнота превращения обусловлена стремлением получить определенный состав металлов. Как было описано выше, концентрация отдельных металлов в растворе по обстоятельствам регулируется добавлением подходящих солей металлов. Однако это означает дополнительные расходы и повышает стоимость процесса, а также опасность загрязнения недопустимыми анионами.
Изобретение включает также содержащий марганец (Mn) фосфат одного металла типа Mn3(PO4)2·3H2О или фосфат нескольких металлов типа (MnxMety)3(PО4)2·3H2О, причем x+y=1, и Met представляет один или многие металлы, выбранные среди Fe, Co, Ni, Sc, Ti, V, Cr, Cu, Zn, Be, Mg, Ca, Sr, Ba, Al, Zr, Hf, Re, Ru, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu, причем фосфат, имеющий в дифрактограмме порошкового рентгеноструктурного анализа пики при 10,96±0,05, 12,78±0,17, 14,96±0,13, 17,34±0,15, 18,98±0,18, 21,75±0,21, 22,07±0,11, 22,97±0,10, 25,93±0,25, 26,95±0,30, 27,56±0,10, 29,19±0,12, 29,84±0,21, 30,27±0,12, 34,86±0,21, 35,00±0,20, 35,33±0,30, 35,58±0,10, 35,73±0,12, 42,79±0,45, 43,37±0,45, 44,70±0,15 и 44,93±0,20 градусов 2θ, на основе CuKα-излучения, может быть получен или получается согласно описанному здесь способу согласно изобретению.
Изобретение также включает применение соответствующего изобретению фосфата для получения литиированного (Li-содержащего) катодного материала для Li-ионных аккумуляторов, например, согласно описанному в литературе способу. Применение соответствующего изобретению фосфата в качестве прекурсора для получения литиированного катодного материала, сравнительно с использованием для этой цели известных способов, имеет то преимущество, что в соответствующем изобретению фосфате разнообразные желательные металлические катионы уже присутствуют в идеально изотропно распределенной форме в высокочистом прекурсоре, что в отношении его кристаллической фазы, состава и морфологии может быть однозначно охарактеризовано простыми и известными методами. Соответствующая изобретению предпочтительная наномасштабная пластинчатая форма первичных кристаллитов при этом обеспечивает наименьшие из возможных дистанции диффузии и продолжительности диффузии при литиировании посредством простого и экономичного способа кальцинирования. Уже имеющееся идеально изотропное распределение металлических ионов при этом к тому же снижает необходимые температуры кальцинирования и продолжительности кальцинирования, поскольку не требуется никакая диффузия металлических ионов через границы зерен. Определенная кристаллическая структура обеспечивает однозначные и воспроизводимые пути реакции при кальцинировании и при изготовлении катодных материалов. Трудоемкость при прецизионном получении прекурсорных смесей сравнительно с известными способами является отчетливо сниженной, так как существенные компоненты уже имеются в определенном соединении. Высокая чистота соответствующего изобретению фосфата, в частности, практически полное отсутствие или, соответственно, очень низкое содержание, анионных загрязнений, таких как сульфаты, нитраты, хлориды, и т.д., имеет следствием при последующем применении в батарее отчетливо повышенные стабильность цикличности и срок службы, что повышает экономические показатели литий-ионных батарей и обеспечивает возможность использования, например, в электромобилях.
Кроме того, изобретение включает литиированный (Li-содержащий) катодный материал для Li-ионных аккумуляторов, изготовленный с применением соответствующего изобретению содержащего марганец (Mn) фосфата.
Кроме того, изобретение включает Li-ионный аккумулятор, который содержит соответствующий изобретению литиированный (Li-содержащий) катодный материал.
ОПИСАНИЕ ФИГУР
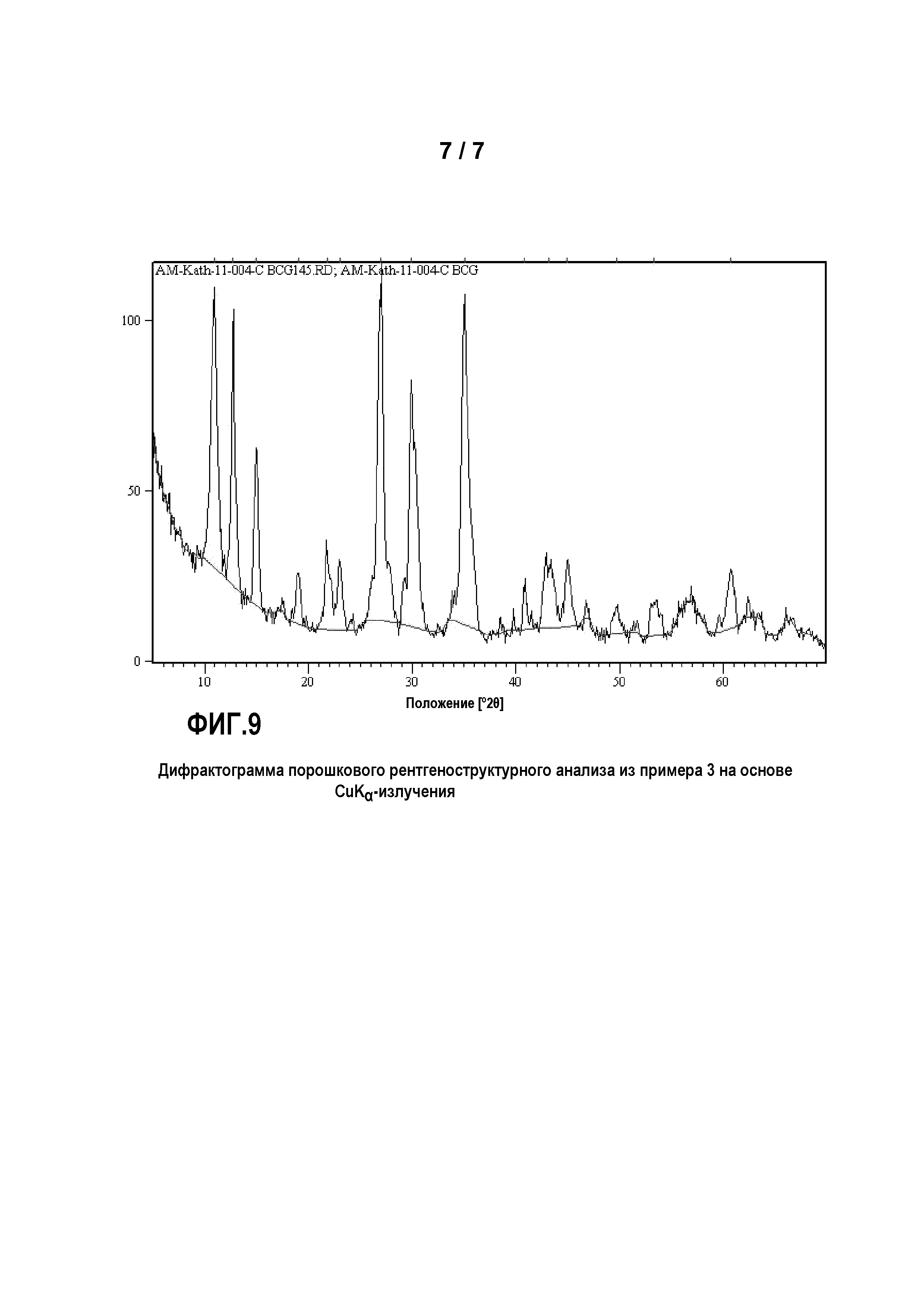
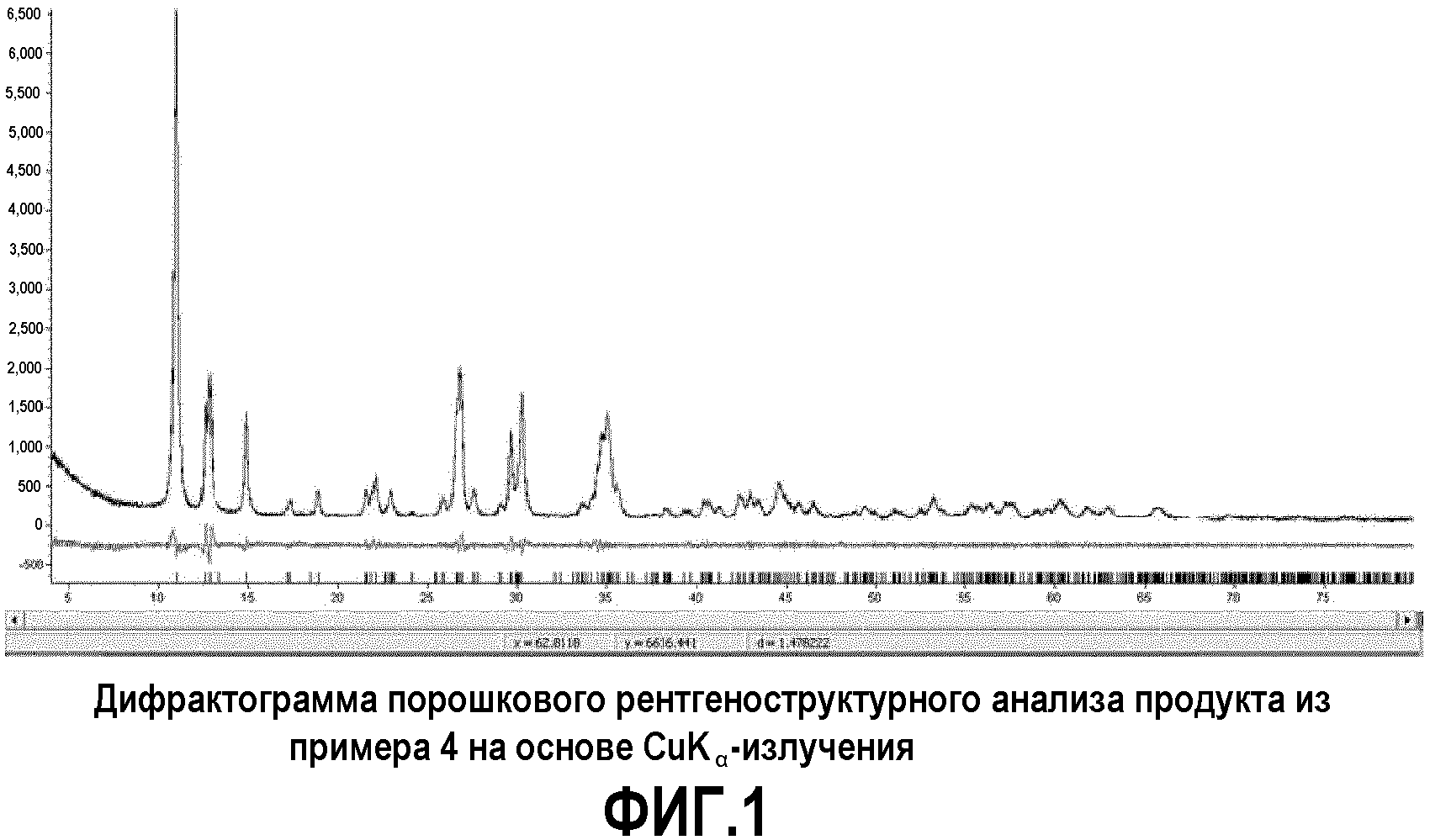
Фиг. 1: представляет дифрактограмму порошкового рентгеноструктурного анализа продукта из примера 4 на основе CuKα-излучения;
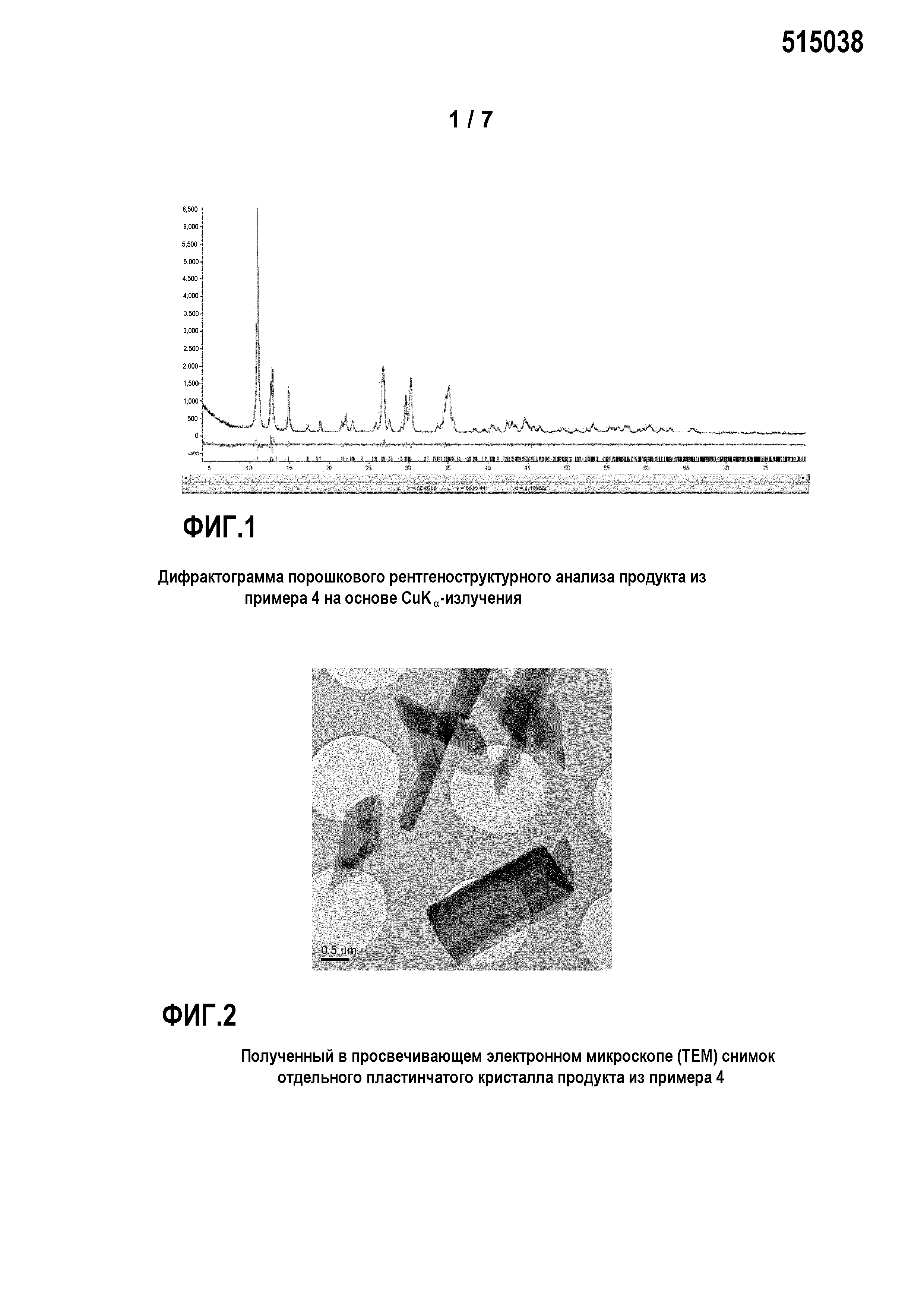
Фиг. 2: представляет полученный в просвечивающем электронном микроскопе (ТЕМ) снимок отдельного пластинчатого кристалла продукта из примера 4;
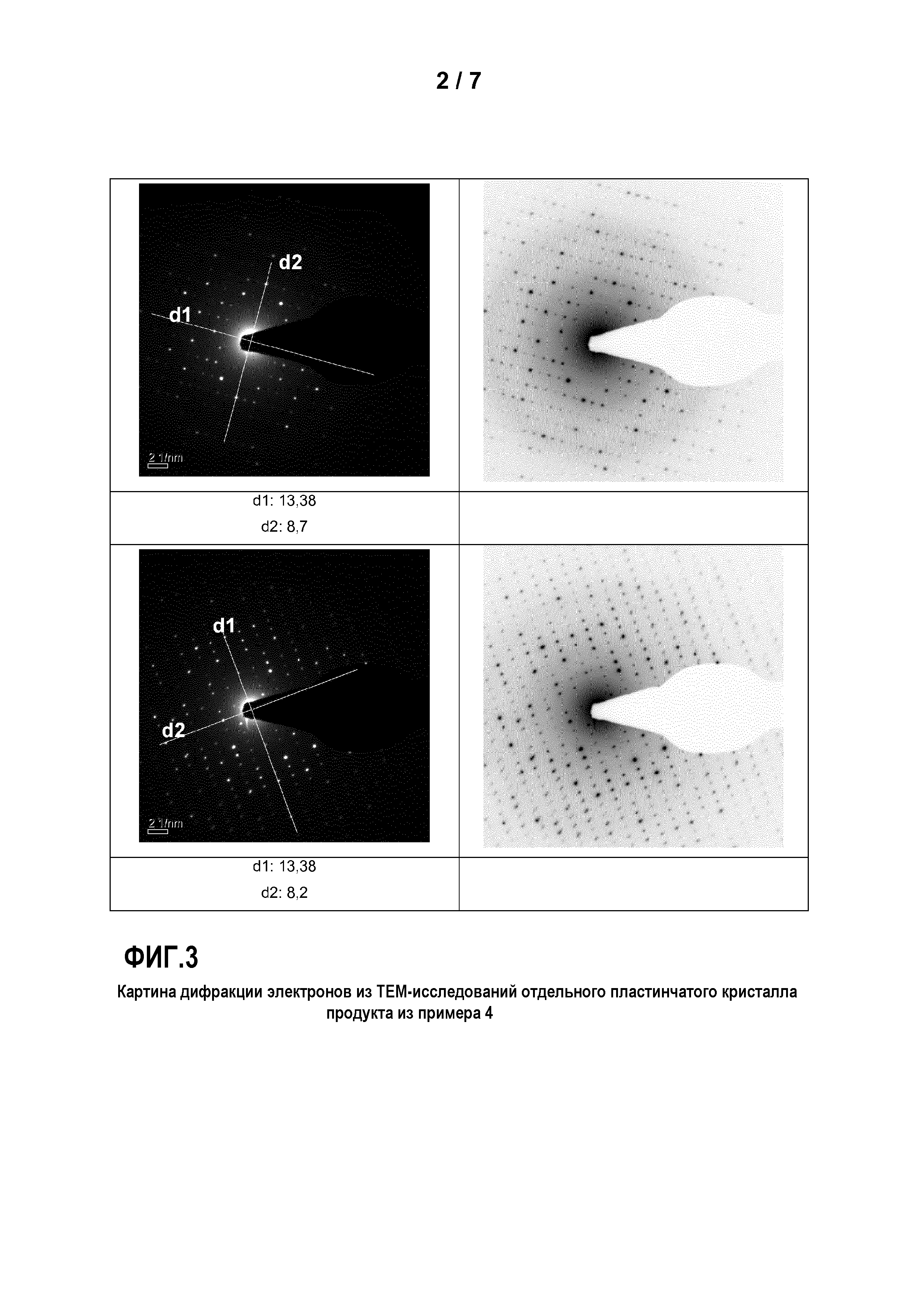
Фиг. 3: представляет картину дифракции электронов из ТЕМ-исследований отдельного пластинчатого кристалла продукта из примера 4;
Фиг. 4: представляет полученный в электронном микроскопе снимок продукта из примера 1;
Фиг. 5: представляет полученный в электронном микроскопе снимок продукта из примера 3;
Фиг. 6: представляет полученный в электронном микроскопе снимок продукта из примера 18;
Фиг. 7: представляет дифрактограмму порошкового рентгеноструктурного анализа продукта из примера 17 на основе CuKα-излучения, полностью идентифицируемого согласно файлам PDF 75-1186 (Fe3(PО4)2 × 8H2О), и, соответственно, 41-0375 (Co3(PО4)2 × 8H2О);
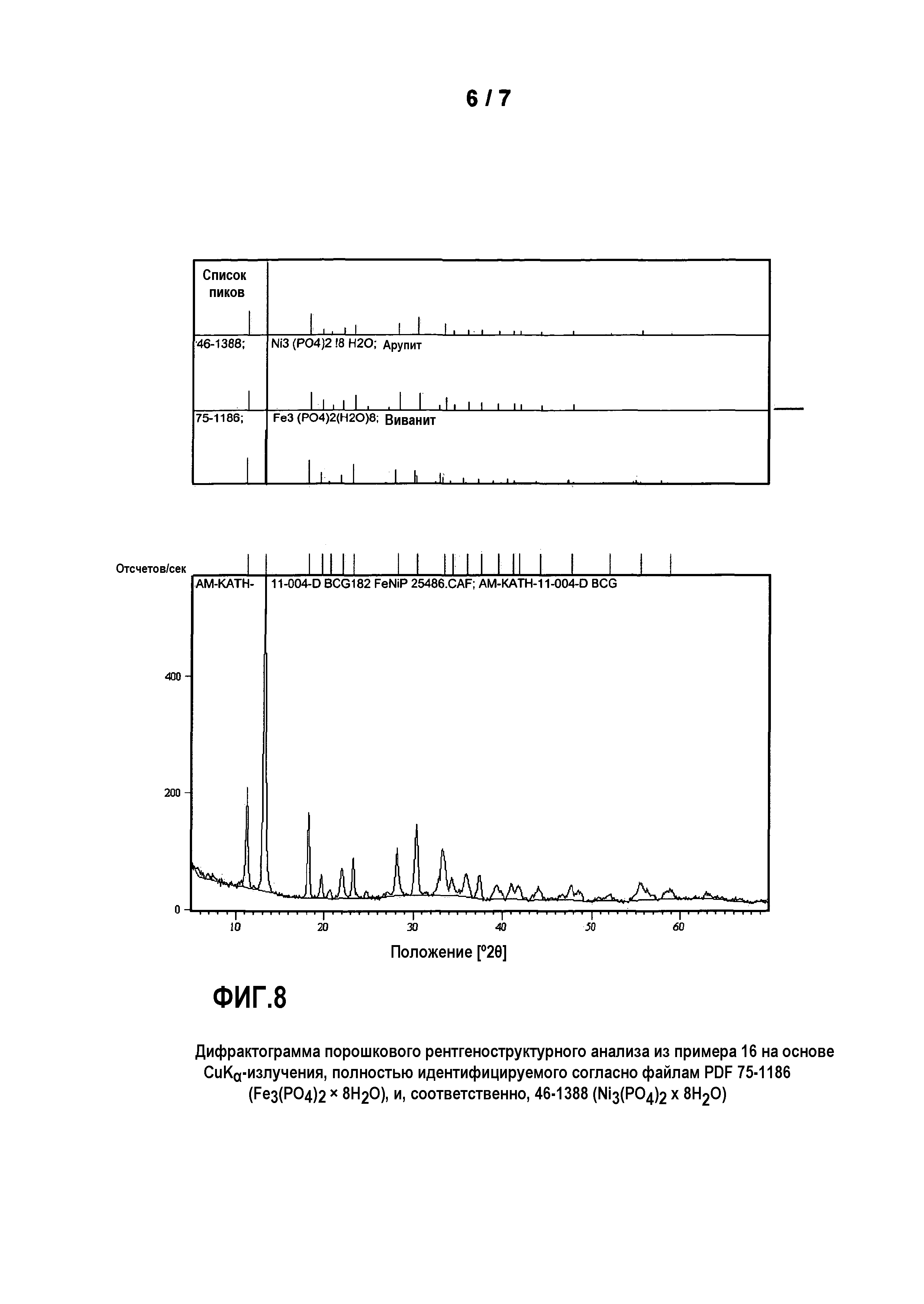
Фиг. 8: представляет дифрактограмму порошкового рентгеноструктурного анализа продукта из примера 16 на основе CuKα-излучения, полностью идентифицируемого согласно файлам PDF 75-1186 (Fe3(PО4)2 × 8H2О), и, соответственно, 46-1388 (Ni3(PО4)2 × 8H2О);
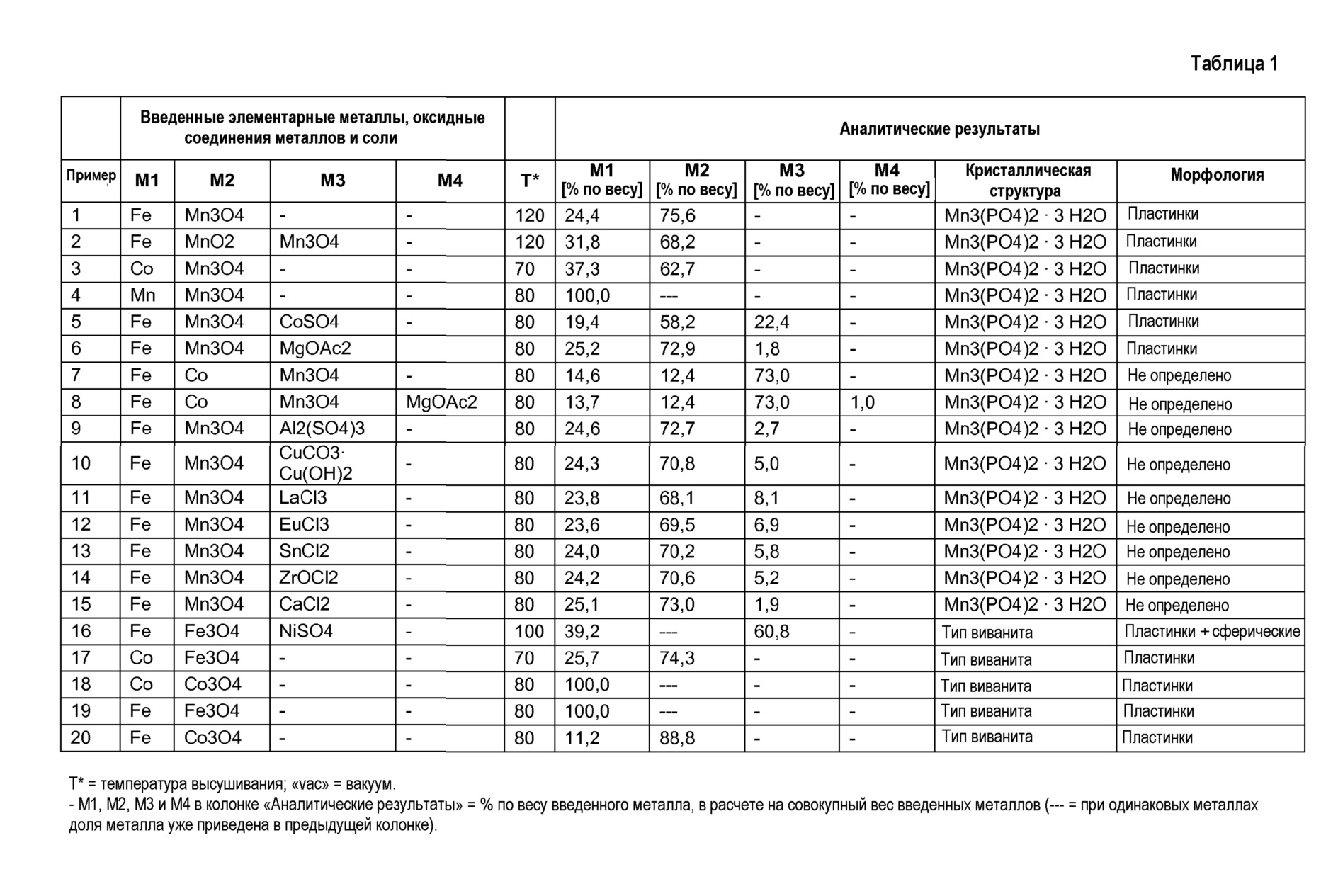
Фиг.9: представляет дифрактограмму порошкового рентгеноструктурного анализа продукта из примера 3 на основе CuKα-излучения.
ПРИМЕРЫ
Пример 1
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 40 г NаОН и 1000 г деминерализованной воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 120°C.
Пример 2
Был приготовлен фосфорнокислотный раствор (I) из 230 г 75%-ной Н3РО4 и 460 г деминерализованной воды. В этот раствор (I) были добавлены 8,9 г MnО2, а также 30,1 г Mn3О4 и 13,1 г Fe. Раствор (I) перемешивался в течение 60 минут при комнатной температуре и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 120 г NаОН и 3000 г деминерализованной воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 90°C.
Пример 3
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 3,8 г Со. Раствор (I) перемешивался в течение 60 минут при температуре 60°C и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 40,4 г NаОН и 229 г воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 70°C.
Пример 4
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,1 г Mn3О4 и 4,5 г Mn. Раствор (I) перемешивался в течение 90 минут при температуре 20°C и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 17,6 г NаОН и 158,7 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись 100 г фосфорнокислотного Ме2+-раствора (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 5
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем были добавлены 17,7 г CoSO4·6H2O, растворенные в 20 г воды. Затем полученный раствор был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 40 г NаОН и 1000 г воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 6
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 90 минут при температуре 60°C, и затем были добавлены 2,6 г Mg(СН3СОО)2⋅6H2O, растворенные в 20 г воды. Затем полученный раствор был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°С.
Пример 7
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 2,2 г Fe, а также 1,5 г Со. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 40 г NаОН и 1000 г деминерализованной воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был разделен на части, и каждая часть была высушена в сушильном шкафу с циркуляцией воздуха при температуре 60°C и, соответственно, 120°C.
Пример 8
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,3 г Mn3О4 и 2,2 г Fe, а также 1,5 г Со. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки. К этому раствору затем были добавлены 2,6 г Mg(СН3СОО)2⋅6H2O, растворенные в 20 г воды.
Кроме того, был приготовлен основный раствор из 40 г NаОН и 1000 г деминерализованной воды. Затем в реакционный сосуд были помещены 25 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был разделен на части, и каждая часть была высушена в сушильном шкафу с циркуляцией воздуха при температуре 60°C и, соответственно, 120°C.
Пример 9
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 1,94 г Al2(SO4)3⋅18H2O, растворенные в 20 г воды, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 10
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 0,65 г CuСО3·Cu(ОН)2⋅0,5H2O, растворенные в 20 мл разбавленной HCl, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 11
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 1,09 г LaCl3⋅7H2O, растворенные в 20 мл воды, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 12
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 1,12 г EuCl3·7H2O, растворенные в 20 мл воды, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 13
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 0,66 г SnCl2⋅2H2O, растворенные в 20 мл разбавленной HCl, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 14
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 0,95 г ZrOCl2, растворенные в 20 мл разбавленной HCl, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 15
Был приготовлен фосфорнокислотный раствор (I) из 1090 г 75%-ной Н3РО4 и 2380 г деминерализованной воды. В этот раствор (I) были добавлены 209 г Mn3О4 и 51 г Fe. Раствор (I) перемешивался в течение 90 минут при комнатной температуре, и затем к 100 г этого раствора были добавлены 0,33 г СаCl2, растворенные в 20 мл разбавленной HCl, для растворения, и был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 450 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 16 (сравнительный)
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,1 г Fe3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 60 минут при температуре 60°C, и затем были добавлены 33,1 г NiSO4⋅6H2O, растворенные в 100 г воды. Полученный раствор был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 500 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 100°C.
Пример 17 (сравнительный)
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,1 г Fe3О4 и 3,8 г Со. Раствор (I) перемешивался в течение 60 минут при температуре 60°C, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 50 г NаОН и 500 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 70°С.
Пример 18 (сравнительный)
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,4 г Со3О4 и 3,8 г Со. Раствор (I) перемешивался в течение 60 минут при комнатной температуре, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 41,9 г NаОН и 376,8 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 19 (сравнительный)
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,1 г Fe3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 60 минут при температуре 60°C, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 17,6 г NаОН и 158,7 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись 100 г фосфорнокислотного Ме2+-раствора (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Пример 20 (сравнительный)
Был приготовлен фосфорнокислотный раствор (I) из 80 г 75%-ной Н3РО4 и 160 г деминерализованной воды. В этот раствор (I) были добавлены 14,4 г Со3О4 и 3,5 г Fe. Раствор (I) перемешивался в течение 60 минут при комнатной температуре, и затем был профильтрован, чтобы удалить из раствора возможные оставшиеся твердые остатки.
Кроме того, был приготовлен основный раствор из 41,9 г NаОН и 376,8 г воды. Затем в реакционный сосуд были помещены 10 г Н3РО4 со 100 г воды, и нейтрализованы основным раствором до величины рН 7, с получением затравочного раствора (II). К нейтрализованному затравочному раствору (II) одновременно прибавлялись фосфорнокислотный Ме2+-раствор (I) и основный раствор при перемешивании таким образом, что значение рН затравочного раствора (II) все время поддерживалось между 6,5 и 7. По завершении прибавления раствор перемешивался в течение последующих 5 минут. Затем выпавший осадок твердого вещества был отделен отсасыванием через нутч-фильтр и промыт деминерализованной водой. Осадок на фильтре был высушен в сушильном шкафу с циркуляцией воздуха при температуре 80°C.
Таблица 1 обобщает примеры 1-20 и результаты аналитических исследований каждого продукта.
Примеры 1-15 показывают, что соответствующим изобретению способом получены содержащие марганец (Mn) фосфаты одного металла и нескольких металлов «Mn3(PO4)2⋅3H2О-структурного типа». Отношение металла к фосфату (РО4) в полученных продуктах составляет примерно 3 к 2. Металлы марганец (Mn) и, если содержатся, металлы Fe, Ni и Со присутствуют в продуктах в своей двухвалентной форме. Возможно, что очень незначительные количества этих металлов находятся в другой степени окисления, например, Fe может в незначительной мере окисляться на поверхностях частиц, например, при высушивании и высоких температурах. Такие маловажные отклонения от двухвалентной формы рассматриваются в смысле настоящего изобретения как неизбежные загрязнения, что не выходит за пределы области правовой защиты изобретения. Легирующие металлы могут присутствовать в форме своих стабильных и, соответственно, известных степеней окисления.
Примеры (сравнительные примеры) 16-20 показывают, что согласно сравнимому способу, однако без добавления элементарного марганца (Mn) или содержащих марганец оксидных соединений, получены фосфаты одного металла или нескольких металлов, которые не имеют «Mn3(PO4)2·3H2О-структурного типа». Все продукты примеров 16-20 по результатам рентгеновского дифракционного анализа могли быть отнесены к кристаллическому структурному типу виванита [Fe3(PO4)2⋅8H2О] или его производным разной степени дегидратации.
Температура высушивания оказывала влияние на содержание связанной кристаллизационной воды. Чем выше были температуры высушивания, и чем длительнее была продолжительность высушивания, тем ниже было содержание кристаллизационной воды. Пониженное парциальное давление воды ускоряло высушивание.
Все соответствующие изобретению продукты примеров 1-15 показывает одинаковую картину рентгеновского дифракционного анализа с пиками при 10,96±0,05, 12,78±0,17, 14,96±0,13, 17,34±0,15, 18,98±0,18, 21,75±0,21, 22,07±0,11, 22,97±0,10, 25,93±0,25, 26,9±50,30, 27,56±0,10, 29,19±0,12, 29,84±0,21, 30,27±0,12, 34,86±0,21, 35,00±0,20, 35,33±0,30, 35,58±0,10, 35,73±0,12, 42,79±0,45, 43,37±0,45, 44,70±0,15 и 44,93±0,20 градусов 2θ, на основе CuKα-излучения. Только положения пиков, в зависимости от сорта и концентрации различных металлов в фосфатах нескольких металлов, проявляют незначительные сдвиги, которые обусловливаются различными ионными радиусами и вариацией степени заселенности катионных центров в кристаллической решетке элементарной ячейки.
Порошковые рентгенографические анализы, а также анализы дифракции электронов в просвечивающем электронном микроскопе подтверждают для продуктов примеров 1-15 орторомбическую элементарную ячейку с параметрами кристаллической решетки 13,2 +/- 0,2, 8,6 +/- 0,2 и 8,1 +/- 0,2 Ангстрем.
Полученные для соответствующих изобретению продуктов дифрактограммы порошкового рентгеноструктурного анализа с вышеуказанными пиками, и определенные для продуктов параметры элементарной ячейки с указанными в зависимости от состава металлических компонентов слегка варьирующими в заданных диапазонах значениями, до сих пор не были известны для соединений состава Mn3(PO4)2·3H2О, а также его (псевдо)бинарных, (псевдо)тройных и (псевдо)четверных вариантов в соответствующих базах данных. Для соответствующих изобретению продуктов был идентифицирован новый «Mn3(PO4)2⋅3H2О-структурный тип». Структура наблюдается, когда соответствующий изобретению продукт в качестве металла содержит исключительно Mn (см. пример 3), но также когда содержатся дополнительные металлы.
Для соединения типа Mn3(PO4)2·3H2О имеется база данных в ICDD (Международном центре дифракционных данных) под номером 003-0426 PDF-файла (файла порошковой дифракции), правда, между депонированными данными и определенными здесь экспериментально значениями для соответствующих изобретению продуктов «Mn3(PO4)2⋅3H2О-структурного типа» нет никаких соответствий в отношении положения, числа и интенсивности описанных рефлексов. Для описанного в ICDD-базе данных соединения к тому же не приведены никакие кристаллографические сведения, которые подробнее описывали бы кристаллическую структуру. Соответствующие изобретению продукты приведенного здесь «Mn3(PO4)2⋅3H2О-структурного типа» тем самым до сих пор не были описаны.
Соответствующие изобретению продукты преимущественно имеют пластинчатую морфологию первичных кристаллитов, причем толщина пластинок может быть определена с помощью растрового электронного микроскопа с порядком величины от около 10 до 50 нм.
Пластинчатая морфология соответствующих изобретению продуктов в принципе обеспечивает возможность плотной упаковки кристаллитов, то есть пластинки могут быть уложены в стопку при меньших объемах полостей, чем в случае круглых сферических частиц. Сформированные слоистыми агрегаты и, соответственно, агломераты этого материала позволяют с помощью общеупотребительных методов под действием сдвиговых нагрузок легко переводить их в дисперсии первичных частиц.
Незначительная толщина пластинчатых кристаллов соответствующих изобретению продуктов обеспечивает высокую скорость реакции при литиировании фосфатов с образованием активных катодных материалов, так как ионы лития при взаимодействии должны проходить лишь короткие дистанции диффузии. Это также ведет к улучшенной работоспособности готовых катодных материалов, поскольку дистанции и продолжительности диффузии Li-ионов могут быть явственно сокращены по сравнению с традиционным материалом.



Ортофосфат железа(iii) для литий-ионных аккумуляторов
Огнестойкий полимерный материал
Получение ортофосфата железа
Снабженные противомикробной защитой материалы
Извлечение фосфата из осадка сточных вод
Вспенивающие агенты для пластмасс
Композит из ортофосфата железа(iii) и углерода
Питательная композиция для биологических систем
Фосфаты металлов и способ их получения
Пригодная для микроволновой обработки пищевая масса
Ортофосфат железа(iii) для литий-ионных аккумуляторов
Безгалогеновый антипирен
Огнестойкий полимерный материал
Получение ортофосфата железа
Снабженные противомикробной защитой материалы
Извлечение фосфата из осадка сточных вод
Вспенивающие агенты для пластмасс
Композит из ортофосфата железа(iii) и углерода
Питательная композиция для биологических систем
Фосфаты металлов и способ их получения

