Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ 1,1-ДИФТОРЭТАНА
Вид РИД
Изобретение
Изобретение относится к химической технологии, а именно к способам получения 1,1-дифторэтана (хладон 152а, R152a, HFC 152а) - озонобезопасного фторуглеводорода, применяемого в качестве хладагента, пропеллента, а также в качестве порообразователя при получении пенопластов.
Известен способ получения 1,1-дифторэтана путем жидкофазного фторирования хлористого винила фтористым водородом в присутствии четыреххлористого олова в качестве катализатора с последующей очисткой 1,1-дифторэтана от хлористого винила, образующего с 1,1-дифторэтаном азеотропную смесь, путем дистилляции в присутствии безводного фтористого водорода (АС СССР №341788, кл. С07С 19/08, опубл. 07.07.1972). Способ позволяет получать 1,1-дифторэтан с содержанием хлористого винила около 0,2%.
Известен способ очистки 1,1-дифторэтана от примеси винилхлорида с помощью экстрактивной дистилляции с применением экстрагента из группы углеводородов, спиртов, хлорпроизводных углеводородов с точкой кипения от 10 до 120°С (Патент РФ №2188815, кл. С07С 17/386, С07С 19/08, опубл. 10.09.2002). Данный способ позволяет снизить содержание примеси винилхлорида в 1,1-дифторэтане до 50 ppm (0,005%).
Известен способ получения 1,1-дифторэтана с низким содержанием непредельных примесей, в том числе винилхлорида, который включает газофазное фторирование 1,1-дихлорэтана фтористым водородом на катализаторе, дистилляцию с выделением последовательно фракции хлористого водорода, фракции 1,1-дифторэтана и фракции, содержащей фтористый водород, 1,1-дихлорэтан, 1-фтор-1-хлорэтан; фракцию 1,1-дифторэтана контактируют с катализатором фторирования, фракцию фтористого водорода возвращают в голову процесса (Патент JP 2005-206584, кл. С07С 19/08, опубл. 04.08.2005, патент US №2007/0135617, кл. 528/502 R, опубл. 14.06.2007). Использование эффективных катализаторов фторирования на стадии очистки фракции 1,1-дифторэтана от непредельных примесей позволяет получить 1,1-дифторэтан с содержанием винилхлорида на уровне 1-2 ppm (0,0002%), однако при фторировании 1,1-дихлорэтана в газовой фазе наряду с винилхлоридом продукт фторирования загрязнен другими непредельными примесями, а именно винилфторидом, винилиденхлоридом, этиленом.
Аналогичный уровень содержания примеси винилхлорида в 1,1-дифторэтане (1-2 ppm) достигнут в способе получения 1,1-дифторэтана, описанном в патенте РФ №2150452, МПК7 С07С 17/08, С07С 17/20, С07С 19/08, опубл. 10.06.2000. Этот способ получения 1,1-дифторэтана по совокупности существенных признаков наиболее близок предлагаемому. Способ включает жидкофазное фторирование винилхлорида фтористым водородом в присутствии четыреххлористого олова в качестве катализатора, дистилляцию в присутствии безводного фтористого водорода, очистку от хлористого водорода путем ректификации под давлением, дополнительную очистку продуктов фторирования от примеси винилхлорида путем контактирования с жидким четыреххлористым оловом, с последующим отделением 1,1-дифторэтана от фтористого водорода ректификацией. Благодаря осуществлению процесса фторирования винилхлорида в жидкой фазе продукт загрязнен минимальным количеством непредельных примесей. Двухстадийная очистка от примеси винилхлорида позволяет получать 1,1-дифторэтан с минимальным содержанием винилхлорида.
К недостаткам способа следует отнести сложность оборудования на стадии дополнительной очистки от винилхлорида. Эта стадия включает емкостный реактор, заполненный четыреххлористым оловом, оборудованный обратным холодильником для предотвращения уноса катализатора из реактора доочистки. Кроме того, после стадии дополнительной очистки от винилхлорида необходима еще одна стадия ректификации для отделения сырца 1,1-дифторэтана от избыточного фтористого водорода, возвращаемого на синтез.
Технический результат предлагаемого изобретения заключается в снижении потерь фтористого водорода и упрощении способа за счет исключения стадии разделения сырца 1,1-дифторэтана и избыточного фтористого водорода. Снижение потерь фтористого водорода и исключение стадии разделения сырца 1,1-дифторэтана и возвратного фтористого водорода осуществлено благодаря оптимальному режиму дистилляции в присутствии фтористого водорода и высокой эффективности стадии дополнительной очистки от винилхлорида.
Указанный технический результат достигается тем, что в способе получения 1,1-дифторэтана, включающем жидкофазное фторирование винилхлорида фтористым водородом в присутствии четыреххлористого олова, дистилляцию продуктов фторирования в присутствии безводного фтористого водорода с отбором в легкую фракцию хлористого водорода, сырца 1,1-дифторэтана и частично фтористого водорода и возвратом высококипящих органических продуктов и основного количества фтористого водорода на стадию фторирования, ректификацию легкой фракции после дистилляции с выделением в легкую фракцию хлористого водорода и в кубовую фракцию сырца 1,1-дифторэтана, дополнительную очистку кубовой фракции от винилхлорида с последующим выделением целевого продукта, согласно изобретению на стадии дистилляции в присутствии фтористого водорода выделяют фракцию с содержанием фтористого водорода от 0,3 до 3% мол., и органических продуктов, выкипающих при температуре выше, чем 1,1-дифторэтан, не более 1% мол., и дополнительную очистку от винилхлорида проводят в газовой фазе путем контактирования с катализатором на основе оксида алюминия.
Существует вариант, в котором катализатор содержит компонент из группы, включающей хром, никель или сурьму.
Существует также вариант, в котором дополнительную очистку от винилхлорида проводят при температуре в интервале от 150 до 250°С.
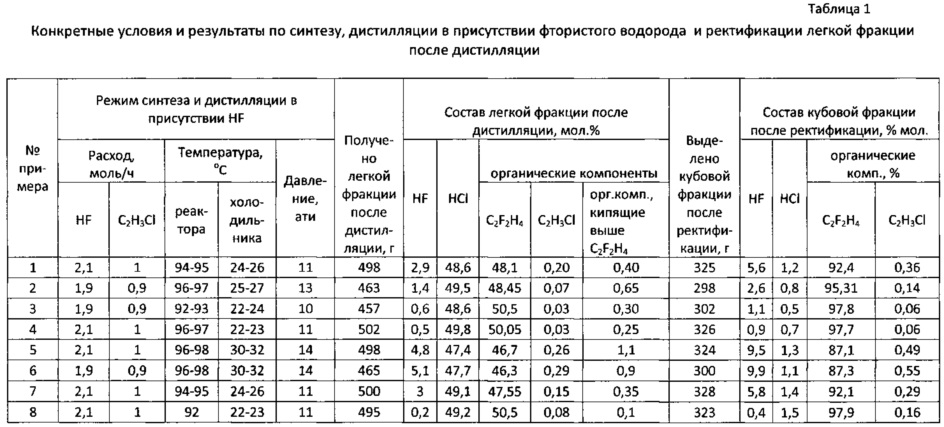
Предлагаемый способ проверен в лабораторных условиях. Продолжительность каждого опыта составляла 5 часов.
Пример 1
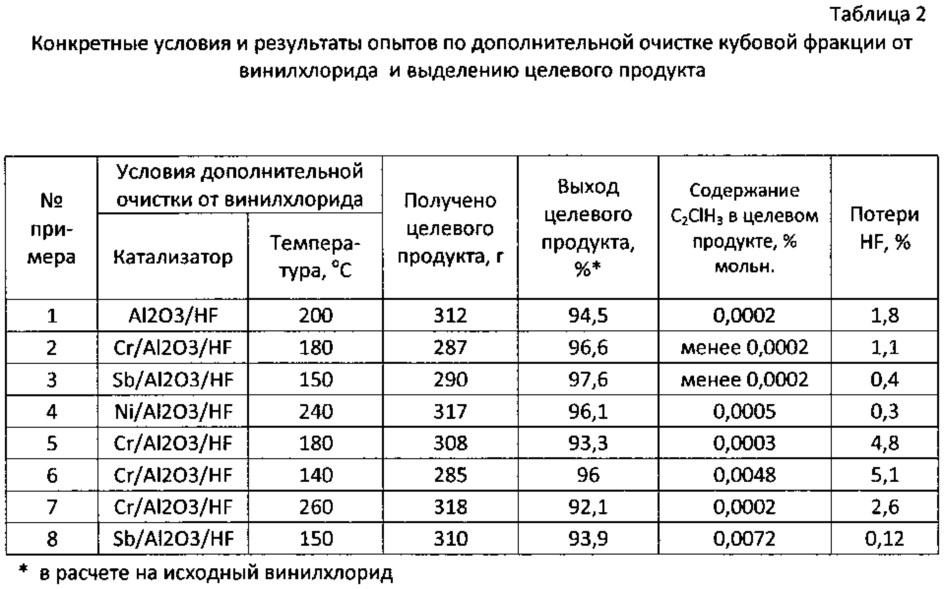
Синтез 1,1-дифторэтана проводили на установке, включающей реактор из стали 12Х18Н10Т емкостью 1 л, снабженный обогревающей рубашкой, гильзой для термопары, дозаторами для подачи винилхлорида и фтористого водорода. Над реактором устанавливали колонку дистилляции эффективностью 10 теоретических тарелок с обратным холодильником; последний был оборудован манометром, предохранительной мембраной, термогильзой и игольчатым вентилем для отбора легкой фракции, включающей хлористый водород, сырец 1,1-дифторэтана и частично фтористый водород. В реактор предварительно загружали четыреххлористое олово, реактор герметизировали, в рубашку обратного холодильника подавали холодную воду, в рубашку реактора - теплоноситель из термостата, нагревали реактор до температуры 95°С. Включали подачу фтористого водорода давлением азота в среднюю часть колонки дистилляции и открывали подачу винилхлорида по сифону в слой четыреххлористого олова. По достижении заданного режима (температура в реакторе и обратном холодильнике, расходы винилхлорида и фтористого водорода, давление) открывали отбор легкой фракции после дистилляции (после обратного холодильника). Полученную фракцию собирали в баллоне из стали 12Х18Н10Т, охлаждаемом жидким азотом. Расход фтористого водорода составлял 2,1 моль/ч, расход винилхлорида 1 моль/ч. Температура в обратном холодильнике составляла 24-26°С, температура в реакторе 94-95°С, давление в реакторе равнялось 11 ати. За время опыта синтезировано и сконденсировано в баллон 498 г легкой фракции после дистилляции в присутствии фтористого водорода состава (мол. %): HF 2,9%, НС1 48,6%, органические компоненты 48,5%, в том числе 1,1-дифторэтан 48,10%, суммарное содержание примесей, выкипающих при температуре выше, чем 1,1-дифторэтан, 0,40%, в том числе винилхлорид 0,20%. Далее проводили ректификацию под давлением на колонке эффективностью около 30 теоретических тарелок для отделения хлористого водорода. Высота насадочной части колонки 60 см, колонка заполнена стальной спирально-призматической насадкой. Колонка оборудована обогреваемым кубом емкостью 1 л и обратным холодильником, охлаждаемым рассолом «минус 40». В куб колонки загружали легкую фракцию после синтеза и дистилляции в присутствии фтористого водорода и проводили ректификацию с выделением в легкую фракцию хлористого водорода. Выделение хлористого водорода проводили при температуре в кубе колонки 18-20°С, верха колонки -14÷-16°С, давлении 13-14 ати. После отделения хлористого водорода выгрузили (переконденсировали в отдельный баллон) кубовую фракцию в количестве 325 г состава (мол. %): HCl 1,2%, HF 5,6%, органические компоненты 93,2%. Данную кубовую фракцию из газовой фазы направляли на дополнительную очистку от примеси винилхлорида. Очистку проводили в реакторе из стали 12Х18Н10Т диаметром 24 мм и длиной 350 мм, снабженном наружным электрообогревом, термогильзой и заполненном катализатором. Катализатор представлял собой гранулы α-оксиды алюминия размером 2-3 мм с удельной поверхностью 8 м2/г, предварительно высушенные в азоте при 400°С и обработанные фтористым водородом при 300°С, объем катализатора составлял 100 см3. Очистку от примеси винилхлорида проводили при температуре 200°С при скорости подачи кубовой фракции в слой катализатора 12 л/ч. После контактирования с катализатором продукт отмывали от кислых примесей водой и водным раствором гидроксида натрия, сушили дегидратированным хлоридом кальция, конденсировали в баллон, охлаждаемый жидким азотом, после чего ректифицировали с выделением 1,1-дифторэтана; всего было выделено 312 г 1,1-дифторэтана с содержанием основного вещества более 99%, содержание винилхлорида в полученном таким образом 1,1-дифторэтане составило 0,0002%. Выход целевого продукта в расчете на поданный винилхлорид составил 94,5%. Потери фтористого водорода составили 1,8%.
Пример 2
Синтез 1,1-дифторэтана и дистилляцию в присутствии фтористого водорода проводили на установке, описанной в примере 1, но при температуре в реакторе синтеза 96-97°С, давлении 13 ати, температуре в обратном холодильнике 25-27°С. Расход винилхлорида составлял 0,9 моль/ч, фтористого водорода 1,9 моль/ч. За время опыта синтезировано 463 г продукта, содержащего (мол. %): HF 1,4%, HCl 49,5%, органические компоненты 49,1%, в том числе 1,1-дифторэтан 48,45%, суммарное содержание примесей, выкипающих при температуре выше, чем 1,1-дифторэтан, 0,65%, в том числе винилхлорид 0,07%. После ректификации полученного продукта на колонке, описанной в примере 1, в легкую фракцию выделили хлористый водород, который направили на поглощение водой с получением соляной кислоты, а из куба колонки выгрузили 298 г кубовой фракции состава (мол. %): HCl 0,8%, HF 2,6%, органические компоненты 96,6%. Эту фракцию направили на дополнительную очистку от примеси винилхлорида на установку, описанную в примере 1, но в качестве катализатора использовали α-оксид алюминия, пропитанный бихроматом аммония, высушенный в токе азота при 400°С и обработанный безводным фтористым водородом при 300°С; содержание хрома составило 3,2% масс. Дополнительную очистку от винилхлорида проводили при температуре 180°С и скорости подачи кубовой фракции в слой катализатора 12 л/ч. После дополнительной очистки от винилхлорида и последующих операций (отмывки от кислых примесей, осушки и ректификации) было выделено 287 г очищенного 1,1-дифторэтана с содержанием основного вещества более 99%, выход целевого продукта составил 96,6% от исходного винилхлорида, содержание винилхлорида составило менее 0,0002%. Потери фтористого водорода составили 1,1%.
Пример 3
Синтез 1,1-дифторэтана, дистилляцию в присутствии фтористого водорода и выделение хлористого водорода проводили на установке, описанной в примере 2, но варьировали температуру в реакторе синтеза и обратном холодильнике над колонкой дистилляции в присутствии фтористого водорода, а также давление в системе синтеза и дистилляции в присутствии фтористого водорода. Конкретные условия синтеза, дистилляции в присутствии фтористого водорода и ректификации с отделением хлористого водорода приведены в табл. 1. За время опыта получено после синтеза и дистилляции в присутствии фтористого водорода 457 г продукта, содержащего (мол. %): HF 0,6%, HCl 48,6%, органические компоненты 50,8%, в том числе 1,1-дифторэтан 50,50%, суммарное содержание примесей, выкипающих при температуре выше, чем 1,1-дифторэтан, 0,26%, в том числе винилхлорид 0,03%. После отделения хлористого водорода получена кубовая фракция в количестве 302 г состава (мол. %): HCl 0,5%, HF 1,1%; органические компоненты 98,4%, в том числе 1,1-дифторэтан 97,8%, винилхлорид 0,06%). Эту фракцию направили на дополнительную очистку от примеси винилхлорида на установку, описанную в примере 1, но в качестве катализатора использовали α-оксид алюминия, пропитанный пентахлоридом сурьмы, высушенный в токе азота при 200°С и обработанный безводным фтористым водородом при 200°С; содержание сурьмы составило 0,7% масс. После дополнительной очистки от винилхлорида при температуре 160°С и скорости подачи кубовой фракции 15 л/ч, а также последующих операций было выделено 290 г очищенного 1,1-дифторэтана с содержанием основного вещества более 99%, выход продукта составил 97,6% от исходного винилхлорида, содержание винилхлорида составило менее 0,0002%.
Примеры 4-8
Варьировали условия синтеза и дистилляции в присутствии фтористого водорода. В примере 4 на стадии дополнительной очистки от винилхлорида использовали катализатор, представляющий собой оксид никеля, нанесенный на α-оксид алюминия, высушенный в азоте при 400°С и обработанный безводным фтористым водородом при 300°С. Содержание никеля составило 5,8% масс.
Опыты 5-8 проведены для обоснования граничных условий.
Конкретные условия и результаты опытов приведены в табл. 1 и 2.
Как видно из приведенных примеров, в оптимальных условиях (примеры 1-4) достигнут высокий выход целевого продукта, 94,5-97%, при этом потери фтористого водорода не превысили 3%, несмотря на отсутствие стадии ректификационного разделения сырца 1,1-дифторэтана и возвратного фтористого водорода. Стадия дополнительной очистки целевого продукта от винилхлорида характеризуется высокой эффективностью благодаря использованию активных катализаторов при повышенной температуре.
При увеличении содержания фтористого водорода в фракции после дистилляции в присутствии фтористого водорода более 3% возрастают потери фтористого водорода (примеры 5-6), при снижении содержания фтористого водорода в фракции после дистилляции в присутствии фтористого водорода менее 0,3% снижается эффективность очистки целевого продукта от примеси винилхлорида (пример 8). Эффективность очистки снижается также при снижении температуры на стадии дополнительной очистки ниже 150°С (пример 6). Повышение температуры на стадии дополнительной очистки выше 250°С нецелесообразно, поскольку снижается выход очищенного 1,1-дифторэтана вследствие образования побочных продуктов при очистке, в частности винилфторида (пример 7).
Таким образом, предлагаемый способ за счет оптимизации режима стадии дистилляции в присутствии фтористого водорода позволяет снизить содержание фтористого водорода и высококипящих органических продуктов (1,1-фторхлорэтан, 1,1-дихлорэтан) в легкой фракции после дистилляции в присутствии фтористого водорода до оптимальных значений, а использование эффективных катализаторов и проведение процесса дополнительной очистки от винилхлорида в газовой фазе при повышенной температуре обеспечивает высокую эффективность очистки. Как видно из примеров, по предлагаемому способу обеспечивается высокая степень очистки от примеси винилхлорида, сравнимая с прототипом. Потери фтористого водорода в оптимальных условиях составили 0,3-1,8%, в опытах по прототипу - 1,9-6,3% (см. табл. 6 прототипа). Кроме того, не требуется проведение дополнительной ректификации для разделения фракции 1,1-дифторэтана и фракции фтористого водорода, что приводит к упрощению способа, снижению материало- и энергозатрат.
Способ получения перфторированных простых эфиров с концевыми функциональными группами
Способ получения 1,1-дифторхлорэтанов
Способ получения пентафторэтана
Способ получения сополимеров тетрафторэтилена
Способ получения перфторированных циклосодержащих третичных аминов
Способ получения перфторированных простых эфиров с концевыми функциональными группами
Способ получения 1,1-дифторхлорэтанов
Способ получения пентафторэтана
Способ получения сополимеров тетрафторэтилена
Способ получения тетрафторэтилена
Способ получения 1,1-1,6-гексаметилен-3,3,3',3'-тетракис(2-оксиэтил)-бисмочевины
Способ получения дийодперфторбутана
Способ получения перфторциклоалканов
Дымовая камера горизонтально вентилируемая
Способ получения перфторированных циклосодержащих третичных аминов

