Результат интеллектуальной деятельности: Способ получения органических нитросоединений
Вид РИД
Изобретение
Изобретение относится к области химии органических нитросоединений, а именно к способу получения нитроэфиров и нитраминов, которые широко применяются в промышленности, в том числе в производстве высокоэнергетических изделий, лекарств, красителей, полимеров, других полезных соединений и материалов.
Известен способ получения органических нитросоединений, в частности нитроэфиров, путем нитрования спиртов под действием смесей азотной и серной кислот различного состава при различных условиях [Urbanski Т. Chemistry and Technology of Explosives. Vol. II. - Oxford: Pergamon Press, 1965, 517 с.]. Недостатками этого способа являются взрывоопасность, образование значительных количеств отработанных кислот и стоков, требующих утилизации, а также кислотные выбросы в атмосферу.
Известен также способ получения органических нитросоединений, в частности нитраминов, взаимодействием соответствующих аминов с оксидом азота (V) в среде четыреххлористого углерода при температуре -20÷-30°C с выходами нитраминов 64-97% [Emmons W.D., Pragano A.S., Stevens Т.Е. Nitration of Amines with Dinitrogen Pentoxide. Journal of Organic Chemistry, 1958, 23, 311-313]. Недостатками данного способа являются необходимость использования криогенной техники и токсичных хлорорганических растворителей, требующих очистки и утилизации.
Известен способ получения вторичных нитраминов нитролизом N,N'-диалкиламидов азотной кислотой в среде трифторуксусного ангидрида [Robson J.H., Reinhart J. The Synthesis of Secondary Nitramines by the Nitrolysis of N,N-Disubstituted Amides. Journal of American Chemical Society, 1955, 77, 2453-2457]. Данный способ требует использование в качестве реакционной среды дорогостоящего трифторуксусного ангидрида и является потенциально взрывоопасным.
Известен также способ получения органических нитросоединений путем нитрования органических соединений азотной кислотой и оксидом азота (V) в среде жидкого и сверхкритического диоксида углерода [Farncomb R.E., Nauflett G.W. Nitration of organics in carbon dioxide. US Patent №6177033, 2001]. Способ используют в основном для получения нитроэфиров, таких как нитрат γ-циклодекстрина, нитроцеллюлоза и 3-нитрометил-3-метилоксетан. Процесс ведут при температуре -10°C и давлении диоксида углерода 68 атм. Однако в указанном патенте отсутствует описание процесса получения заявленных нитраминов. При этом способ требует использования низких температур и высокого давления.
Известен принятый за прототип общий способ получения органических нитросоединений, таких как нитрамины и нитроэфиры, с выходами 35-94% путем нитрования силилнитраминов R1R2N-Si(R3)3 или силиловых эфиров R4O-Si(R3)3 оксидом азота (V) (1.1 эквивалент на каждую группу -Si(R3)3) при температуре от -10 до +10°C в течение 0.5-24 часов в дихлорметане [Millar R.W., Philbin S.P. Clean nitrations: Novel syntheses of nitramines and nitrate esters by nitrodesilylation reactions using dinitrogen pentoxide (N2O5). Tetrahedron 1997, 53 (12), 4371-4386]. Недостатками указанного способа являются необходимость использования для получения исходных соединений токсичных триалкилхлорсиланов, в частности триметилхлорсилана, и образование в качестве побочных продуктов нитрования силилнитратов O2NOSi(R3)3, которые являются токсичными, летучими и легковоспламеняющимися соединениями. Кроме того, применяемый в качестве растворителя дихлорметан также токсичен и может вызвать острое отравление персонала (ПДК в рабочей зоне 50 мг/м3).
Технической задачей настоящего изобретения является разработка пожаро- и взрывобезопасного универсального способа получения органических нитросоединений с использованием доступных и нетоксичных реагентов.
Техническим результатом, достигаемым при реализации заявленного способа получения органических нитросоединений, является уменьшение пожаро- и взрывоопасности, а также улучшение экологических характеристик процесса нитрования благодаря исключению из него кремнийсодержащих исходных соединений и токсичных органических растворителей, а также отсутствие трудноутилизируемых отходов. Предлагаемый способ универсальный и позволяет расширить ассортимент получаемых соединений, т.е. предусматривает получение как известных, так и новых веществ с высоким выходом. Способ характеризуется приемлемыми для промышленности условиями проведения реакций (температура и давление).
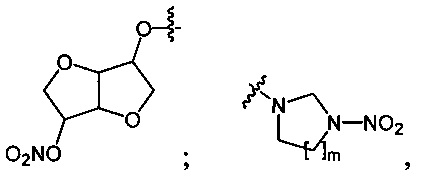
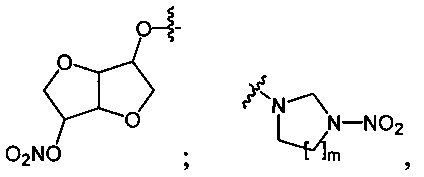
Данный технический результат обеспечивается предлагаемым способом получения органических нитросоединений общей формулы RNO2, где R=n-C4H9O-; O2NO(CH2)2O-; O2NO(CH2)2O(CH2)2O-; O2NOCH2CH(ONO2)CH2O-; (O2NOCH2)3CCH2O-;
 где m=1, 2;
где m=1, 2; 

 где n=2, 3;
где n=2, 3; 
заключающимся в том, что соответствующие спирты либо производные аминов подвергают нитрованию оксидом азота (V) либо его смесью со 100%-ной азотной кислотой в среде низших фторуглеводородов и процесс проводят при давлении 3-60 бар и температуре 5-40°C при мольном соотношении соответствующего исходного соединения и нитрующего агента 1÷(1.1-4.4).
В качестве низших фторуглеводородов используют фреоны (хладоны), например трифторметан или 1,1,1,2-тетрафторэтан.
До настоящего времени фреоны не использовали в процессах нитрования.
Фреоны используют в жидком либо при нагревании свыше 25°C сверхкритическом состоянии.
В качестве нитрующего агента можно использовать оксид азота (V) либо, с целью повышения выхода продуктов реакции при деструктивном нитровании, его смесь со 100%-ной азотной кислотой.
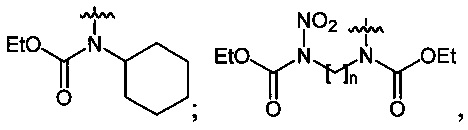
В качестве исходных спиртов используют, например, одноатомные и многоатомные спирты, а в качестве производных аминов используют, например, N-ациламины, N,N'-диалкилоксамиды или эфиры N-алкилкарбаминовой кислоты.
Выходы целевых продуктов достигают 56-99%.
Использование в качестве растворителей доступных и малотоксичных фреонов, которые ранее не применяли в процессах нитрования, может уменьшить экологические и технологические риски. Являясь газами при нормальных условиях, трифторметан R23 и тетрафторэтан R134a негорючи, термически стабильны и инертны к действию большинства нитрующих агентов. Они имеют высокую теплоемкость (трифторметан CF3H Ср 217 Дж⋅моль-1⋅К-1 (20°C, 45 бар), тетрафторэтан CF3CH2F Ср 140 Дж⋅моль-1⋅К-1 (20°C, 45 бар), а дихлорметан CH2Cl2 Ср 102 Дж⋅моль-1⋅К-1 (20°C)) и вследствие этого способны эффективно отводить тепло из зоны реакции, что значительно снижает взрыво- и пожароопасность процесса. Кроме того, они переходят в жидкое и сверхкритическое состояние при значительно меньших значениях давления, чем диоксид углерода (трифторметан CHF3 РКР=48.4 бар, тетрафторэтан CF3CH2F РКР=40.6 бар, диоксид углерода CO2, PКР=73.8 бар), что уменьшает стоимость необходимого оборудования.
Одноатомные и многоатомные спирты 1 превращаются под действием оксида азота (V) в среде фреонов, преимущественно при давлении 3-45 бар и температуре 5-20°C, в нитроэфиры (полинитроэфиры) 2 с выходами 80-99% (Схема 1). Реакции завершаются за 30 минут при минимальном количестве нитрующего агента (1.1 экв. на каждую вводимую нитрогруппу). Процесс протекает по следующей схеме:
Схема 1

Некоторые из полученных соединений используют как компоненты энергоемких составов [Энергетические конденсированные системы: Краткий энциклопедический словарь, под ред. Жукова Б.П., Янус, Москва, 2000, 596 с.], а также в медицине в качестве антиангинальных, сосудорасширяющих, коронародилатирующих препаратов [Koenig А., Roegler С., Lange K., Daiber A., Glusa Е., Lehmann J., NO donors. Part 18: Bioactive metabolites of GTN and PETN - Synthesis and vasorelaxant properties. Bioorganic & Medicinal Chemistry Letters, 2009, 17, 3141-3144].
Предложенный способ нитрования в среде фторуглеводородов эффективен в процессах превращения циклических ацетамидов 3 в соответствующие нитрамины 4 (Схема 2). В данном случае реакции целесообразно проводить под действием смеси оксида азота (V) со 100%-ной азотной кислотой (нитроолеум) различного состава, преимущественно в соотношении 1:1, при давлении 3-60 бар и температуре 5-40°C в течение 120 мин. Выходы продуктов составляют 56-83% в зависимости от условий реакции. Процесс протекает по следующей схеме:
Схема 2

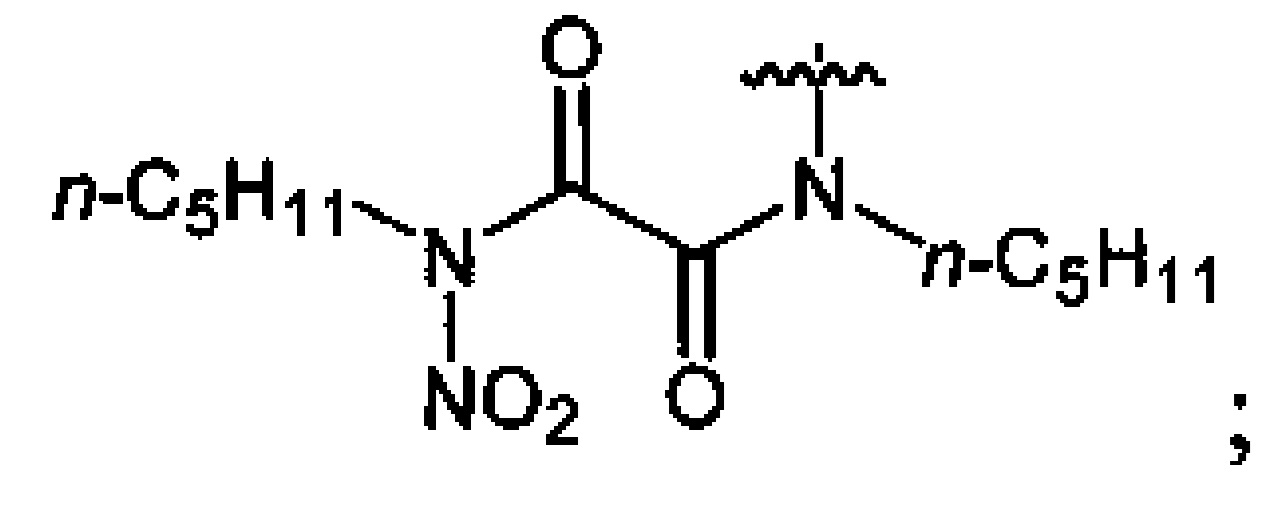
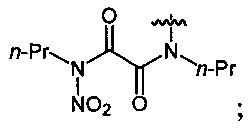
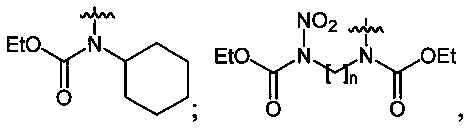
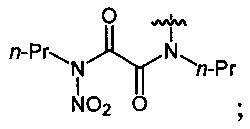
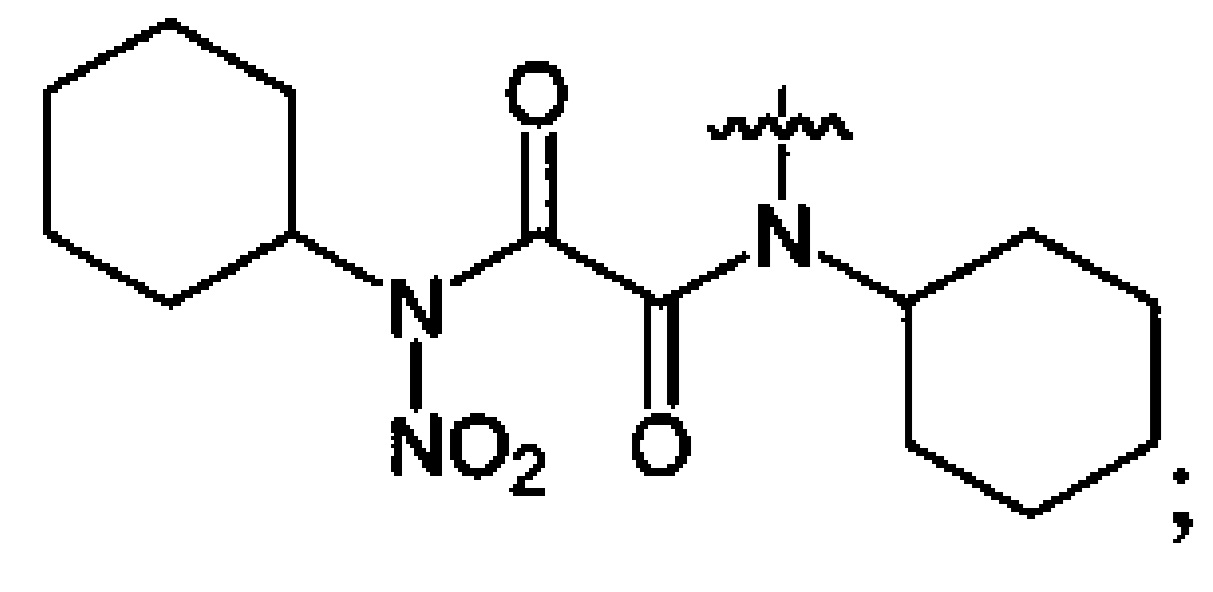
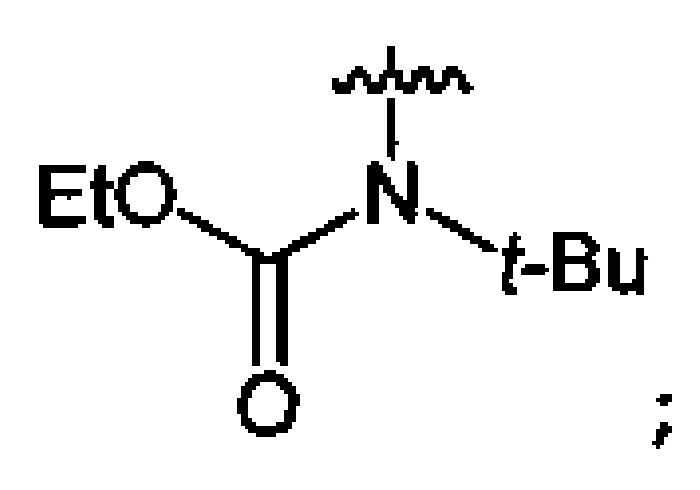
Нитрование карбаматов 5, 7 и оксамидов 9 оксидом азота (V) в среде фреонов дает соответствующие N-нитро- (6) и N,N'-динитрокарбаматы (8), а также N,N'-динитрооксамиды (10) с выходами (65-95%) (Схемы 3 и 4). Нитрамиды 6, 8 и 10 являются непосредственными предшественниками первичных нитраминов, применяемых для получения практически важных компонентов энергоемких составов, содержащих нитраминные группы. Процесс протекает по следующим схемам:
Схема 3

Схема 4

Соединения этил-N-нитро-N-циклогексилкарбамат (6c), диэтил-N,N'-динитро-N,N'-пропандиил-бис-карбамат (8b) и N,N'-дипентил-N,N'-динитрооксамида (10b) являются новыми и в литературе не описаны.
Таким образом, предложенный метод нитрования оксидом азота (V) или его смесями со 100%-ной азотной кислотой в среде низших фреонов является универсальным и может быть пригоден для получения различных классов нитросоединений. Предложенный способ характеризуется меньшей взрыво- и пожароопасностью, чем известные методы нитрования, и позволяет получать различные классы нитросоединений с высокими выходами (56-99%). Следует подчеркнуть, что значения температуры и давления в этих процессах легко достижимы в условиях промышленного производства, что делает возможным создание на их основе новых экологически безвредных технологий получения нитросоединений.
Изобретение иллюстрируется следующими примерами, не ограничивающими его объем.
Пример 1. Получение н-бутилнитрата (2a) в среде жидкого 1,1,1,2-тетрафторэтана
В стальной автоклав объемом 22 см3, снабженный сапфировыми смотровыми окнами, магнитной мешалкой и датчиками давления и температуры, помещают 1-бутанол (1а) (0.67 г, 0.009 моль). Автоклав закрывают, заполняют при комнатной температуре (20°C) жидким 1,1,1,2-тетрафторэтаном на 1/3 объема (P(20°C)=6 бар) и охлаждают до 5°C при перемешивании (600 об/мин) (P(5°C)=3 бар). В емкость-дозатор объемом 13 см3 помещают оксид азота (V) (1.08 г, 0.010 моль), емкость закрывают и заполняют жидким 1,1,1,2-тетрафторэтаном на 1/2 объема (P(20°C)=6 бар). Под действием разности давлений в сосудах раствор нитрующего агента медленно при перемешивании в течение 10 мин добавляют в автоклав при контроле температуры (допустим рост температуры не более чем на 5°C). После добавления всего нитрующего агента емкость-дозатор дважды промывают, набирая 1/3 объема и передавливая флюид в автоклав. Реакционную массу перемешивают при комнатной температуре и давлении 6 бар в течение 0.5 ч, после чего к ней добавляют воду (2 мл) для разрушения избытка нитрующего агента. После этого 1,1,1,2-тетрафторэтан удаляют декомпрессией, автоклав открывают и в смесь продуктов вливают ледяную воду (20 мл). Полученную массу нейтрализуют раствором гидрокарбоната натрия и экстрагируют этилацетатом (4×30 мл). Органический слой сушат над безводным сульфатом магния. Этилацетат отгоняют при пониженном давлении (50 мбар), получают 0.85 г (80%) н-бутилнитрата (2а). Бесцветная жидкость. 1H NMR (CDCl3) δ: 4.44 (t, 2Н, OCH2, J 6.6 Hz), 1.70 (pent, 2Н, OCH2CH2, J 7.2 Hz), 1.42 (sex, 2H, CH3CH2, J 7.4 Hz), 0.95 (t, 3H, CH3, J 7.4 Hz).
Пример 2. Получение динитрата этиленгликоля (2b) в среде жидкого 1,1,1,2-тетрафторэтана
Аналогично примеру 1, этиленгликоль (1b) (0.43 г, 0.007 моль) нитруют оксидом азота (V) (1.66 г, 0.015 моль) в среде жидкого тетрафторэтана. Получают 1.05 г (99%) динитрата этиленгликоля (2b). Желтоватое масло. 1H NMR (CDCl3) δ: 4.74 (s, 4Н, CH2).
Пример 3. Получение динитрата диэтиленгликоля (2c) в среде трифторметана
Аналогично примеру 1, диэтиленгликоль (1c) (0.64 г, 0.006 моль) нитруют оксидом азота (V) (1.42 г, 0.013 моль) в среде жидкого трифторметана при комнатной температуре и давлении 45 бар. Получают 1.17 г (99%) динитрата диэтиленгликоля (2c). 1H NMR (CDCl3) δ: 4.76 (s, 4Н, CH2ONO2).
Пример 4. Получение динитрата изосорбида (2d) в среде жидкого трифторметана
Аналогично примеру 1, изосорбид (1d) (0.73 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде жидкого трифторметана при комнатной температуре и давлении 45 бар. Получают 1.1 г (93%) динитрата изосорбида (2d). Желтоватое масло. 1H NMR (CDCl3) δ: 5.41-5.34 (m, 4Н, CH2), 4.99 (t, 2Н, O2NOCH, J 5.4 Hz), 4.57 (d, 2Н, OCHCH, J 5.2 Hz).
Пример 5. Получение нитроглицерина (2е) в среде жидкого 1,1,1,2-тетрафторэтана
Аналогично примеру 1, глицерин (1e) (0.74 г, 0.008 моль) нитруют оксидом азота (V) (2.81 г, 0.026 моль) в среде жидкого трифторметана. Получают 1.35 г (99%) нитроглицерина (2е). Желтая жидкость. 1H NMR (CDCl3) δ: 5.56-5.48 (m, Н, CH), 4.87-4.61 (m, 4Н, CH2).
Пример 6. Получение тетранитрата пентаэритрита (2f) в среде жидкого 1,1,1,2-тетрафторэтана
Аналогично примеру 1, пентаэритрит (1f) (0.54 г, 0.004 моль) нитруют оксидом азота (V) (1.90 г, 0.018 моль) в среде жидкого тетрафторэтана. После нейтрализации продукт отфильтровывают, сушат и получают 1.16 г (92%) тетранитрата пентаэритрита (2f). Бесцветные кристаллы, Tпл=141-142°C. 1H NMR (DMSO-d6) δ: 4.70 (s, 8Н, CH2).
Пример 7. Получение 1,3-динитроимидазолидина (4а) в среде жидкого 1,1,1,2-тетрафторэтана
В стальной автоклав объемом 22 см3, снабженный сапфировыми смотровыми окнами, магнитной мешалкой и датчиками давления и температуры, помещают 1,3-диацетилимидазолидин (3а) (0.62 г, 0.004 моль). Автоклав закрывают, заполняют при комнатной температуре (20°C) жидким 1,1,1,2-тетрафторэтаном на 1/3 объема (P(20°C)=6 бар) и охлаждают до 5°C при перемешивании (600 об/мин) (P(5°C)=3 бар). В емкость-дозатор объемом 13 см3 помещают оксид азота (V) (0.86 г, 0.008 моль) и 100%-ную азотную кислоту (0.50 г, 0.008 моль), закрывают и заполняют жидким 1,1,1,2-тетрафторэтаном на 1/2 объема (P(20°C)=6 бар). Под действием разности давлений в сосудах раствор нитрующего агента медленно при перемешивании в течение 10 мин добавляют в автоклав при контроле температуры (допустим рост температуры не более чем на 5°C). После добавления всего нитрующего агента емкость-дозатор дважды промывают, набирая 1/3 объема и передавливая флюид в автоклав. Реакционную массу перемешивают при давлении 6 бар и при комнатной температуре 2 ч, после чего добавляют к ней воду (2 мл) для разрушения избытка нитрующего агента. 1,1,1,2-Тетрафторэтан удаляют декомпрессией, автоклав открывают и к смеси продуктов добавляют ледяную воду (20 мл). Полученную массу нейтрализуют раствором гидрокарбоната натрия, отфильтровывают и получают 0.54 г (83%) 1,3-динитроимидазолидина (4а). Бесцветные кристаллы, Tпл=134°C. 1H NMR (DMSO-d6) δ: 5.51 (s, 2Н, NCH2N), 4.16 (s, 4Н, NCH2CH2N).
Пример 8. Получение 1,3-динитроимидазолидина (4а) в среде сверхкритического трифторметана
Аналогично примеру 7, 1,3-диацетилимидазолидин (3а) (0.62 г, 0.004 моль) нитруют смесью оксида азота (V) (0.86 г, 0.008 моль) и 100%-ной азотной кислоты (0.50 г, 0.008 моль) в течение 2 ч в среде сверхкритического трифторметана при температуре 40°C и давлении 60 бар. Получают 0.44 г (69%) 1,3-динитроимидазолидина (4а). Физико-химические свойства полученного продукта соответствуют описанным в примере 7.
Пример 9. Получение 1,3-динитроимидазолидина (4а) в среде жидкого трифторметана
Аналогично примеру 7, 1,3-диацетилимидазолидин (3а) (0.62 г, 0.004 моль) нитруют смесью оксида азота (V) (0.86 г, 0.008 моль) и 100%-ной азотной кислоты (0.50 г, 0.008 моль) в течение 2 ч в среде жидкого трифторметана при температуре 20°C и давлении 45 бар. Получают 0.45 г (70%) 1,3-динитроимидазолидина (4а). Физико-химические свойства полученного продукта соответствуют описанным в примере 7.
Пример 10. Получение 1,3-динитротетрагидропиримидина (4b) в среде жидкого трифторметана
Аналогично примеру 7, 1,3-диацетилтетрагидропиримидин (3b) (0.51 г, 0.003 моль) нитруют смесью оксида азота (V) (0.75 г, 0.007 моль) и 100%-ной азотной кислоты (0.38 г, 0.006 моль) в течение 2 ч в среде жидкого трифторметана при температуре 20°C и давлении 45 бар. Получают 0.30 г (56%) 1,3-динитротетрагидропиримидина (4b). Бесцветные кристаллы, Tпл=89°C. 1H NMR (DMSO-d6) δ: 5.88 (s, 2Н, NCH2N), 3.97 (t, 4Н, NCH2CH2, J 5.7 Hz), 1.89 (pent, 2H, CH2CH2CH2, J 5.7).
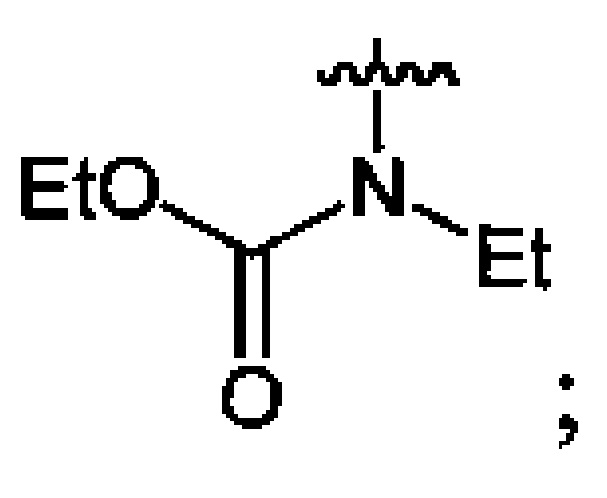
Пример 11. Получение этил-N-этил-N-нитрокарбамата (6а) в среде жидкого трифторметана
В стальной автоклав объемом 22 см3, снабженный сапфировыми смотровыми окнами, магнитной мешалкой и датчиками давления и температуры, помещают этил-N-этилкарбамат (5а) (1.17 г, 0.010 моль). Автоклав закрывают, заполняют при комнатной температуре (20°C) жидким трифторметаном на 1/3 объема (P(20°C)=42 бар) и охлаждают до 5°C при перемешивании (600 об/мин) (P(5°C)=28 бар). В емкость-дозатор объемом 13 см3 помещают оксид азота (V) (1.19 г, 0.011 моль), емкость закрывают и заполняют жидким трифторметаном на 1/2 объема (P(20°C)=42 бар). Под действием разности давлений в сосудах раствор нитрующего агента медленно при перемешивании в течение 10 мин добавляют в автоклав при контроле температуры (допустим рост температуры не более чем на 5°C). После добавления всего нитрующего агента емкость-дозатор дважды промывают, набирая 1/3 объема и передавливая флюид в автоклав. Реакционную массу выдерживают при комнатной температуре, давлении 45 бар и перемешивании 0.5 ч, после чего к ней добавляют воду (2 мл) для разрушения избытка нитрующего агента. Трифторметан удаляют декомпрессией, автоклав открывают и в смесь продуктов вливают холодную воду (20 мл). Полученную массу нейтрализуют раствором гидрокарбоната натрия и экстрагируют этилацетатом (4×30 мл). Органический слой сушат безводным сульфатом магния, этилацетат отгоняют при пониженном давлении (50 мбар), получают 1.54 г (95%) этил-N-этил-N-нитрокарбамата (6а). Бледно-желтая жидкость. 1H NMR (DMSO-d6) δ: 4.32 (q, 2Н, NCH2CH3, J 7.1 Hz), 4.05 (q, 2H, OCH2CH3, J 7.0 Hz), 1.29 (t, 3H, NCH2CH3, 7.1 Hz), 1.21 (t, 3Н, OCH2CH3, J 7.0 Hz)
Пример 12. Получение этил-N-нитро-N-трет-бутилкарбамата (6b) в среде жидкого трифторметана
Аналогично примеру 11, этил-N-трет-бутилкарбамат (5b) (1.16 г, 0.008 моль) нитруют оксидом азота (V) (1.08 г, 0.010 моль) в течение 2 ч в среде жидкого трифторметана. Получают 1.14 г (76%) этил-N-нитро-N-трет-бутилкарбамата (6b). Желтая жидкость, Tкип=49.0-49.5°C (0.8 Torr), nD20=1.4335. 1H NMR (CDCl3) δ: 4.32 (q, 2Н, CH2, J 7.1 Hz), 1.50 (s, 9H, CCH3), 1.33 (t, 3H, CH2CH3, J 7.2 Hz). 13C NMR (CDCl3) δ: 151.59 (CO), 64.66 (OCH2), 62.22 (CCH3), 27.22 (CCH3), 13.91 (CH3CH2). 14N NMR (CDCl3) δ: -37.29 (NO2).
Пример 13. Получение этил-N-нитро-N-циклогексилкарбамата (6c) в среде жидкого трифторметана
Аналогично примеру 11, этил-N-циклогексилкарбамат (5c) (1.03 г, 0.006 моль) нитруют оксидом азота (V) (0.76 г, 0.007 моль) в течение 2 ч в среде жидкого трифторметана. Получают 1.21 г (93%) этил-N-нитро-N-циклогексилкарбамата (6c). Бесцветная жидкость, Tкип=93-95°C (0.6 Torr), nD20=1.4650. 1H NMR (CDCl3) δ: 4.34 (q, 2Н, CH2CH3, J 7.1 Hz), 1.95-1.06 (m, 10H, CH2CH2CH2CH2CH2), 1.35 (t, 3H, CH2CH3, J 7.1 Hz). 13C NMR (CDCl3) δ: 151.31 (CO), 64.60 (OCH2), 61.20 (CH), 29.49 (CHCH2), 25.94 (CHCH2CH2), 25.10 (CHCH2CH2CH2), 14.02 (CH3CH2). 14N NMR (CDCl3) δ: -40.54 (NO2). Вычислено: C, 49.99; H, 7.46; N, 12.96. Найдено: C, 50.40; H, 7.39; N, 12.94.
Пример 14. Получение диэтил-N,N'-динитро-N,N'-этандиил-бис-карбамата (8а) в среде жидкого трифторметана
Аналогично примеру 11, диэтил-N,N'-этандиил-бис-карбамат (7а) (0.95 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч в среде жидкого трифторметана. Получают 1.34 г (96%) диэтил-N,N'-динитро-N,N'-этандиил-бис-карбамата (8а). Бесцветные кристаллы, Tпл=81-82°C. 1H NMR (DMSO-d6) δ: 4.39 (s, 4Н, CH2NNO2), 4.25 (q, 4Н, OCH2CH3, J 7.1 Hz), 1.24 (t, 6Н, OCH2CH3, J 7.1 Hz).
Пример 15. Получение диэтил-N,N'-динитро-N,N'-пропандиил-бис-карбамата (8b) в среде жидкого трифторметана
Аналогично примеру 11, диэтил-N,N'-пропандиил-бис-карбамат (7b) (1.09 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч в среде жидкого трифторметана. Получают 1.41 г (91%) диэтил-N,N'-динитро-N,N'-пропандиил-бис-карбамата (8b). Желтая жидкость, Tкип=131-135°C (0.7 Torr), nD20 1.4789. 1H NMR (CDCl3) δ: 4.36 (q, 4H, CH2Me, J 7.1 Hz), 4.13 (t, 4H, NHCH2, J 7.1 Hz), 2.10 (pent, 2H, CH2CH2CH2, J 7.1 Hz), 1.36 (t, 6H, CH2CH3, J 7.2 Hz). 13C NMR (CDCl3) δ: 150.40 (CO), 65.04 (OCH2), 46.63 (NCH2), 25.59 (CH2CH2CH2), 14.10 (CH3). 14N NMR (CDCl3) δ: -43.94 (NO2). Вычислено: C, 35.70; H, 5.23; N, 18.18. Найдено: C, 35.98; H, 5.45; N, 18.22.
Пример 16. Получение диэтил-N,N'-динитро-N,N'-пропандиил-бис-карбамата (8b) в среде жидкого 1,1,1,2-тетрафторэтана
Аналогично примеру 15, диэтил-N,N'-пропандиил-бис-карбамат (7b) (1.09 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде жидкого 1,1,1,2-тетрафторэтана при комнатной температуре и давлении 6 бар в течение 2 ч. Получают 1.45 г (94%) диэтил-N,N'-динитро-N,N'-пропандиил-бис-карбамата (8b). Физико-химические свойства полученного продукта соответствуют описанным в примере 16.
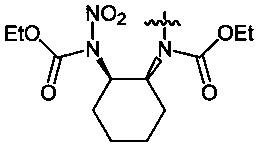
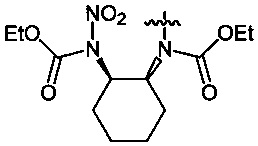
Пример 17. Получение диэтил-циклогексан-1,2-диил-бис-(N-нитрокарбамата) (8c) в среде жидкого 1,1,1,2-тетрафторэтана
Аналогично примеру 11, диэтил-циклогексан-1,2-диил-бис-карбамат (7c) (1.29 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль в среде жидкого 1,1,1,2-тетрафторэтана при комнатной температуре и давлении 6 бар в течение 2 ч. Получают 1.65 г (95%) диэтил-циклогексан-1,2-диил-бис-(N-нитрокарбамата) (8c). Желтая жидкость, Tкип=94-95°C (0.65 Torr), nD20 1.4837. 1H NMR (CDCl3) δ: 5.10-4.98 (m, 2H, CH), 4.33 (q, 4H, OCH2, J 7.1 Hz), 2.16-1.78 (m, 8H, CHCH2CH2), 1-34 (t, 6H, CH3, J 7.2 Hz). 13C NMR (CDCl3) δ: 150.95 (CO), 65.09 (OCH2), 61.24 (CH), 29.31 (CHCH2CH2), 24.94 (CH2CH2CH2), 13.96 (CH3). 14N NMR (CDCl3) δ: -42.79 (NO2). Вычислено: C, 41.38; H, 5.79; N, 16.09. Найдено: C, 41.33; H, 5.81; N, 16.25.
Пример 18. Получение N,N'-дипропил-N,N'-динитрооксамида (10а) в среде жидкого трифторметана
Аналогично примеру 11, N,N'-дипропилоксамид (9а) (0.86 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде жидкого трифторметана в течение 2 ч. Получают 1.22 г (93%) N,N'-дипропил-N,N'-динитрооксамида (10а). Бесцветные кристаллы, Tпл=41-42°C. 1H NMR (CDCl3) δ: 4.23-4.02 (m, 4Н, NHCH2), 1.77 (sex, 4Н, CH2CH2CH3, J 7.4 Hz), 1.00 (t, 6Н, CH3, J 7.4 Hz). 13C NMR (CDCl3) δ: 159.01 (CO), 47.55 (NCH2), 19.78 (CH2CH2CH3), 10.84 (CH3). 14N NMR (CDCl3) δ: -44.77 (NO2).
Пример 19. Получение N,N'-дипентил-N,N'-динитрооксамида (10b) в среде жидкого трифторметана
Аналогично примеру 11, N,N'-дипентилоксамид (9b) (1.14 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде жидкого трифторметана в течение 2 ч. Получают 1.48 г (93%) N,N'-дипентил-N,N'-динитрооксамида (10b). Светло-желтая жидкость, Tкип=124-126°C (0.65 Torr). 1H NMR (CDCl3) δ: 3.15 (dt, 4Н, NHCH2, J 7.7 Hz, J 7.4 Hz), 1.73 (pent, 4H, NHCH2CH2, J 7.1 Hz), 1.44-1.24 (m, 8H, CH2CH2CH3), 0.91 (t, 6H, CH3, J 6.6 Hz). 13C NMR (CDCl3) δ: 158.93 (CO), 46.13 (NHCH2), 28.46 (NHCH2CH2), 25.87 (CH2CH2CH3), 22.20 (CH2CH3), 13.89 (CH3). 14N NMR (CDCl3) δ: -44.75 (NO2). Вычислено: C, 45.28; H, 6.97; N, 17.6. Найдено: C, 45.1; H, 7.28; N, 15.06.
Пример 20. Получение N,N'-дициклогексил-N,N'-динитрооксамида (8c) в среде жидкого трифторметана
Аналогично примеру 11, N,N'-дициклогексилоксамид (9c) (1.26 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде жидкого трифторметана в течение 4 ч. После выделения по описанной методике получают 1.2 г смеси непрореагировавшего N,N'-дициклогексилоксамида и N,N'-дициклогексил-N,N'-динитрооксамида. Целевой продукт экстрагируют диэтиловым эфиром, экстракт упаривают, получают 0.87 г (65%) N,N'-дициклогексил-N,N'-динитрооксамида (8c). Бесцветные кристаллы, Tпл=128.3°C. 1H NMR (CDCl3) δ: 4.66 (t, 2Н, CH, J 12.2 Hz, J 3.6 Hz), 2.25-1.16 (m, 20H, CH2CH2CH2CH2CH2). 13C NMR (CDCl3) δ: 159.31 (CO), 59.71 (CH), 27.89 (CHCH2), 26.09 (CHCH2CH2), 24.93 (CHCH2CH2CH2). 14N NMR (CDCl3) δ: -39.08 (NO2). Вычислено: C, 49.12; H, 8.39; N, 16.37. Найдено: C, 49.05; H, 6.59; N, 16.33.
3-(тринитрометил-onn-азокси)-4-нитраминофуразаны и способы их получения
Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием
Катализатор для окислительного разложения хлорорганических соединений в газах и способ его получения
Способ приготовления катализатора для получения синтез-газа, катализатор, приготовленный по этому способу, и способ получения синтез-газа с его использованием
Способ приготовления катализатора для получения синтез-газа
Способ получения замещенных 2,3,5,6-тетраоксабицикло[2.2.1]гептанов
Устройство разделения элементов конструкций
Способ приготовления катализатора для получения 3-ацетилгептан-2,6-диона и способ получения 3-ацетилгептан-2,6-диона с использованием полученного катализатора
Катализатор для селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов и способ селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов с его использованием
Способ приготовления катализатора для получения бензола из метана, катализатор, приготовленный по этому способу, и способ получения бензола из метана с использованием полученного катализатора
Способ получения гипсового вяжущего
3-(тринитрометил-onn-азокси)-4-нитраминофуразаны и способы их получения
Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием
Катализатор для окислительного разложения хлорорганических соединений в газах и способ его получения
Способ приготовления катализатора для получения синтез-газа, катализатор, приготовленный по этому способу, и способ получения синтез-газа с его использованием
Способ приготовления катализатора для получения синтез-газа
Способ получения замещенных 2,3,5,6-тетраоксабицикло[2.2.1]гептанов
Устройство разделения элементов конструкций
Способ приготовления катализатора для получения 3-ацетилгептан-2,6-диона и способ получения 3-ацетилгептан-2,6-диона с использованием полученного катализатора
Катализатор для селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов и способ селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов с его использованием

