Результат интеллектуальной деятельности: Способ получения первичных алифатических нитраминов
Вид РИД
Изобретение
Изобретение относится к области химии органических нитросоединений, а именно, к способу получения первичных алифатических нитраминов, которые применяются в качестве малочувствительных энергоемких соединений и полупродуктов для их получения, а также для получения пластификаторов энергоемких составов.
Известен способ получения первичных нитраминов из дихлоралкиламинов путем нитрования последних смесью азотной кислоты и уксусного ангидрида с последующей обработкой полученного N-хлорнитрамина водными раствором щелочи [Smart G.N.R., Wright G.F. Catalyzed nitration of amines VII. A new method for preparation of primary nitramines. Canadian Journal of Research, Section B: Chemical Sciences, 1948, 26, 284-293]. Недостатками метода являются использование хлорсодержащих реагентов для получения исходных дихлораминов и применение в качестве нитрующего агента пожаро- и взрывоопасной смеси азотной кислоты и уксусного ангидрида.
Известен способ получения первичных алифатических нитраминов взаимодействием соответствующих аминов с н-бутиллитием при -78°С в среде диэтилового эфира с последующей обработкой полученного металлорганического полупродукта этилнитратом [Winters L.J., Learn D.B., Desai S.C. A preparation of primary aliphatic nitramines. Journal of Organic Chemistry, 1965, 30, 2471-2472]. Недостатками этого метода являются пожароопасность н-бутиллития (воспламеняется на воздухе), необходимость использования абсолютно безводных условий и низких температур, а также низкие выходы продуктов (4-58%).
Известен способ получения первичных нитраминов путем аминолиза N-нитро-N-алкилтолуолсульфонамидов под действием вторичных аминов в среде ацетонитрила при 5-10°С с последующей обработкой продуктов водным раствором щелочи [Emmons W.D., Freeman J.P. The aminolysis of N-nitrotoluenesulfonamide. Journal of the American Chemical Society, 1955, 77; 6062-6063]. Способ позволяет получать первичные нитрамины с выходами 81-96%. Однако суммарные выходы, с учетом стадии получения исходных соединений нитрованием соответствующих толуолсульфонамидов, составляют лишь 46-91%. Кроме того, побочными продуктами реакции аминолиза являются сульфамиды п-толуолсульфокислоты с вторичными аминами, утилизация которых требует дополнительных технических решений.
Наиболее близким к предлагаемому изобретению является способ получения первичных алифатических нитраминов, включающий стадии нитрования уретанов (этиловых эфиров N-алкилкарбаминовой кислоты) в среде концентрированной азотной кислоты, экстракции образующегося нитрамида из нейтрализованного карбонатом натрия водного раствора диэтиловым эфиром и обработки полученного эфирного раствора газообразным аммиаком [Franchimont A.P.N., Klobbie Е.А. Quelques nitramines et leur preparation. Recueil des Travaux Chimiques des Pays-Bas, 1888, 7, 343-357]. Многостадийность и необходимость выделения полупродуктов, образующихся на всех стадиях, делает процесс длительным и ресурсозатратным. Кроме того, процесс проводят в среде азотной кислоты, то есть в условиях значительного избытка нитрующего агента. Выходы не указаны, но, как показывает нижеприведенный сравнительный пример 14 получения алифатических нитраминов в условиях прототипа, он не превышает 45%.
Техническая задача, решаемая заявляемым изобретением, состоит в упрощении способа получения первичных алифатических нитраминов, в повышении выхода целевого продукта, а также в уменьшении его пожаро- и взрывоопасности.
Техническим результатом, достигаемым при реализации заявленного способа получения первичных алифатических нитраминов, является упрощение его технологии, которое заключается в уменьшении количества стадий, сокращении продолжительности процесса за счет проведения его в одном реакторе без выделения промежуточных соединений, в повышении выхода целевого продукта, а также в уменьшении пожаро- и взрывоопасности процесса за счет проведения его в среде негорючих и малотоксичных низших фторуглеводородов. Преимуществом предлагаемого способа также является использование минимального избытка нитрующего агента (не более 10%).
Данный технический результат обеспечивается предлагаемым способом получения первичных алифатических нитраминов общей формулы RNHNO2, где R=CH3, СН3СН2, СН3(СН2)2, СН3(СН2)4, циклогексил, O2NNH(CH2)2, O2NNH(CH2)3, H2NC(O)O(CH2)2, 2-нитрамино-циклогексан-1-ил, включающим нитрование соответствующих N,N'-диалкилоксамидов или эфиров N-алкилкарбаминовой кислоты нитрующим агентом и обработку полученного при этом промежуточного нитросоединения аммиаком, и отличающимся тем, что в качестве нитрующего агента используют оксид азота (V) и процесс проводят в среде низшего фторуглеводорода при давлении 6-45 бар, температуре 5-20°С при мольном соотношении соответствующего исходного соединения и нитрующего агента 1:(1,1-2,2).
Процесс проводят без выделения промежуточного нитросоединения в одном реакторе. Выходы целевого продукта достигают 53-95%.
В качестве низшего углеводорода используют, например, трифторметан или 1,1,1,2-тетрафторэтан.
Процесс протекает по следующим схемам:
Схема 1

Схема 2
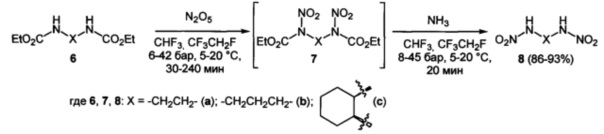
Схема 3

Используемый в предложенном способе получения первичных алифатических нитраминов оксид азота (V) (N2O5) является экологически чистым нитрующим агентом, эффективным в процессах С-, О-, N-нитрования, в том числе деструктивного [Agrawal J.P., Hodgson R.D. Organic chemistry of explosives. - New York: Wiley Interscience, 2007]. Процессы с его участием, как правило, характеризуются использованием меньшего количества нитрующего агента, чем соответствующие реакции под действием серно-азотных смесей, что значительно уменьшает количество отходов. При этом единственным побочным продуктом является азотная кислота, которую можно вновь превращать в N2O5 или использовать как самостоятельный нитрующий агент. Газообразные при нормальных условиях низшие фторуглеводороды (в частности, трифторметан R23 и тетрафторэтан R 134а) ранее не применяли как реакционные среды в процессах нитрования. При этом они не горючи, термически стабильны и инертны к действию как окислителей (нитрующие агенты), так и аммиака. Кроме того, низшие фторуглеводороды обладают более высокой теплоемкостью, чем обычные органические растворители (трифторметан CF3H Ср 217 Дж⋅моль-1⋅К-1 (20°С, 45 бар), тетрафторэтан CF3CH2F Ср 140 Дж⋅моль-1⋅К-1 (20°С, 45 бар), а дихлорметан СН2Сl2 Ср 102 Дж⋅моль-1⋅К-1 (20°С)) и, вследствие этого, способны эффективно отводить тепло из зоны реакции, что значительно снижает взрыво- и пожароопасность реакций, а также уменьшает сопутствующие им экологические и технологические риски.
Оксамиды 1 и карбаматы 3, 6 или 9 образуют под действием оксида азота (V) (1.1 эквивалент на каждую нитрогруппу) в среде трифторметана или тетрафторэтана при давлении 6÷45 бар и температуре 5÷20°С соответствующие динитрооксамиды 2 и нитроуретаны 4, 7 и 10. Последние, без выделения в том же автоклаве, путем добавления к реакционной массе жидкого аммиака, превращаются в соответствующие первичные нитрамины 5 и 11 и динитрамины 8 (Схемы 1-3). Выходы продуктов 5, 11 в расчете на исходные соединения 1, 3 и 9 составляют 53-95%, а выходы продуктов 8 в расчете на исходные бис-карбаматы 6 - 86-93%.
Выходы алифатических нитраминов, полученных по предлагаемому способу, превосходят выходы описанных ранее соединений, синтезированных известными методами [Emmons W.D., Freeman J.P. The aminolysis of N-nitrotoluenesulfonamide. Journal of the American Chemical Society, 1955, 77; 6062-6063; Bachmann W.E., Horton W.J., Jenner E.L., MacNaughton N.W. Maxwell C.E. The nitration of derivatives of ethylenediamine. Journal of the American Chemical Society, 1950, 72; 3132-3134]. Некоторые из полученных соединений используют как полупродукты для получения компонентов энергоемких составов, в частности, пластификаторов [Патент РФ №2169140] и высокоэнергетических полимерных связующих [Herman H.L. Encyclopedia of Explosives and Related Items. Vol. 8, Ed. S. M. Kaye, ARRADCOM, Dover, New Jersey, 138, 1978]. Способ безвреден для окружающей среды. Побочными продуктами двухстадийной реакции являются амид щавелевой и эфир карбаминовой кислоты, а также аммонийная селитра, которые используют как азотные удобрения. Алифатические нитрамины 5, 8 и 11 могут быть легко выделены в индивидуальном виде с помощью декомпрессии. При этом отработанные фторуглеводороды, путем повторной компрессии, могут быть вновь возвращены в процесс нитрования с использованием установки, принцип действия которой аналогичен бытовому холодильнику.
Изобретение иллюстрируется следующими примерами, не ограничивающими его объем.
Пример 1. Получение N-метилнитрамина (5а) в среде трифторметана.
В стальной автоклав объемом 22 см3, снабженный сапфировыми смотровыми окнами, магнитной мешалкой и датчиками давления и температуры, помещают N,N'-диметилоксамид (1а) (0.58 г, 0.005 моль). Автоклав закрывают, заполняют при комнатной температуре (19-20°С) жидким трифторметаном на 1/3 объема (Р=42 бар) и охлаждают до 5°С при перемешивании (600 об/мин), при этом давление в автоклаве уменьшается до 28-29 бар. В емкость-дозатор объемом 13 см3 помещают оксид азота (V) (1.19 г, 0.011 моль), емкость закрывают и заполняют жидким трифторметаном на 1/2 объема (Р=42 бар). Под действием разности давлений в сосудах раствор нитрующего агента медленно при перемешивании в течение 10 мин добавляют в автоклав при контроле температуры (допустим рост температуры не более 5°С). После добавления всего нитрующего агента емкость-дозатор дважды промывают, набирая 1/3 объема жидкого трифторметана и передавливая флюид в автоклав. Реакционную массу выдерживают при давлении 42 бар, комнатной температуре и перемешивании 2 ч. Затем автоклав охлаждают до 5°С и при интенсивном перемешивании с помощью шприцевого насоса высокого давления добавляют жидкий аммиак (3.01 г, 0.177 моль) со скоростью 0.1÷0.2 мл/мин (допустимый рост температуры не более 10°С). Реакционную смесь выдерживают 20 мин при давлении 45 бар и комнатной температуре. Трифторметан и избыток аммиака удаляют декомпрессией, автоклав открывают, добавляют этанол (40 мл) и интенсивно перемешивают при 50°С 10 мин, затем спирт отгоняют на роторном испарителе под вакуумом (70 мбар). К сухому остатку добавляют диэтиловый эфир (40 мл), перемешивают, осадок отфильтровывают. Эфирный раствор упаривают при пониженном давлении (400 мбар), получают 0.61 г (80%) N-метилнитрамина (1а). Бесцветные кристаллы, Тпл=36.3°С. 1Н NMR (CDCl3) δ: 9.01 (br s, Н, NH), 3.18 (s, 3H, CH3). (CDCl3) δ: 32.59 (СН3). 14N NMR (CDCl3) δ: -25.64 (NO2). Анал. расчет: С, 15.79; H, 5.3; N, 36.83. Найдено: С, 15.77; Н, 5.23; N, 36.67.
Пример 2. Получение N-этилнитрамина (5b) в среде трифторметана.
Аналогично примеру 1, этил-N-этилкарбамат (3b) (1.17 г, 0.010 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) при комнатной температуре в течение 1 ч, затем добавляют жидкий аммиак (2.70 г, 0.16 моль). Трифторметан и избыток аммиака удаляют декомпрессией, автоклав открывают, остаток экстрагируют диэтиловым эфиром (4×15 мл). Объединенный эфирный раствор упаривают при пониженном давлении (400 мбар), получают этилкарбамат (0.85 г, 98%). К оставшейся смеси аммониевой соли N-этилнитрамина 5b⋅NH3 и нитрата аммония добавляют этанол (40 мл) и интенсивно перемешивают при 55°С 10 мин. Этанол отгоняют на роторном испарителе (30 мбар), сухой остаток экстрагируют диэтиловым эфиром (40 мл). Эфирный раствор упаривают при пониженном давлении, получают N-этилнитрамин (5b) (0.83 г, 93%). Бесцветная жидкость, Ткип=82.0-83.0°С (7.8 мм рт.ст.), nD20 1.4545. 1H NMR (CDCl3) δ: 9.14 (br. s, NH), 3.59 (dq, 2H, CH2, J 7.0, J 1.9), 1.22 (t, 3H, CH3, J 7.2 Hz).
Пример 3. Получение N-этилнитрамина 5b в среде 1,1,1,2-тетрафторэтана.
Аналогично примеру 2, за исключением того, что реакцию проводят в среде жидкого 1,1,1,2-тетрафторэтана при давлении 6 бар (нитрование) и 8 бар (аммонолиз). Получают N-этилнитрамин (5b) (0.83 г, 92%), физико-химические характеристики которого совпадают с соответствующими показателями продукта, описанными в примере 2.
Пример 4. Получение N-пропилнитрамина (5с) в среде трифторметана.
Аналогично примеру 1, N,N'-дипропилоксамид (1с) (0.86 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в среде трифторметана. Получают 0.96 г (92%) N-пропилнитрамина (5с). Бесцветная жидкость, Ткип=62.0-62.5°С при 0.8 мм рт.ст., nD20 1.4582. 1H NMR (CDCl3) δ: 8.61 (br s, Н, NH), 3.61-3.48 (m, 2H, NHCH2), 1.65 (sex, 2H, CH2CH2CH3, J 7.3 Hz), 0.98 (t, 3H, CH3, J 7.4 Hz).
Пример 5. Получение N-пропилнитрамина (5c) в среде 1,1,1,2-тетрафторэтана.
Аналогично примеру 4, реакцию проводят в среде жидкого 1,1,1,2-тетрафторэтана при давлении 6 бар (нитрование) и 8 бар (аммонолиз). Получают 1.02 г (82%) N-пропилнитрамина (5с), физико-химические характеристики которого совпадают с соответствующими показателями продукта, описанными в примере 4.
Пример 6. Получение N-пентилнитрамина (5d) в среде трифторметана.
Аналогично примеру 1, N,N'-дипентилоксамид (1d) (1.14 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч, затем добавляют жидкий аммиак (3.01 г, 0.177 моль) и получают 1.29 г (89%) N-амилнитрамина (5d). Бесцветная жидкость, Ткип=86.0°С при 0.8 мм рт.ст., nD20=1.4595. lH NMR (CDCl3) δ: 8.80 (br s, Н, NH), 3.62-3.48 (m, 4H, NHCH2), 1.60 (pent, 4H, NHCH2CH2, J 6,8 Hz), 1.39-1.27 (m, 8H, CH2CH2CH3), 0.89 (t, 6H, CH3, J 6.2 Hz). 13C NMR (CDCl3) δ: 46.39 (NHCH2), 28.80 (NHCH2CH2), 26.55 (CH2CH2CH3), 22.21 (CH2CH3), 13.86 (CH3). 14N NMR (CDCl3) δ: -22.56 (NO2). Вычислено: С, 45.44; H, 9.15; N, 21.20. Найдено: С, 45.97; Н, 8.96, N, 21.25.
Пример 7. Получение N-циклогексилнитрамина (5е) в среде трифторметана.
Аналогично примеру 1, N,N'-дициклогексилоксамид (1е) (1.26 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 4 ч, затем добавляют жидкий аммиак (3.01 г, 0.177 моль) и получают 0.94 г (65%) N-циклогексилнитрамина (5е). Желтая маслянистая жидкость с Ткип=98.0°С при 0.7 мм рт.ст, nD20 1.5000. 1Н NMR (CDCl3) δ: 8.75 (br s, Н, NH), 4.02-3.88 (m, H, CH), 2.07-1.15 (m, 10Н, CH2CH2CH2CH2CH2). 13С NMR (CDCl3) δ: 55.48 (С, CH), 30.32 (2C, CHCH2), 25.28 (2C, CHCH2CH2), 24.35 (C, CH2CH2CH2CH). 14N NMR (CDCl3) δ: -28.85 (NO2). Вычислено: С, 49.98; H, 8.39; N, 19.43. Найдено: С, 50.00; Н, 8.42, N, 19.48.
Пример 8. Получение N-циклогексилнитрамина (5е) в среде трифторметана.
Аналогично примеру 2, этил-N-циклогексилкарбамат (3е) (1.03 г, 0.006 моль) нитруют оксидом азота (V) (0.76 г, 0.007 моль) в течение 2 ч, затем добавляют жидкий аммиак (1.83 г, 0.108 моль). Получают 0.82 г (95%) N-циклогексилнитрамина (5е).
Пример 9. Получение этилендинитрамина (8а) в среде трифторметана.
Аналогично примеру 2, диэтил-N,N'-этандиил-бис-карбамат (6а) (1.02 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч и добавляют жидкий аммиак (3.01 г, 0.177 моль). Отделяют диэтиловым эфиром этилкарбамат (0.85 г, 98%). К оставшейся сухой реакционной массе добавляют воду (10 мл) и при перемешивании прикапывают 2 мл 10 N соляной кислоты. Белый творожистый осадок отфильтровывают, сушат. Получают 0.70 г (93%) этилендинитрамина (8а). Бесцветные кристаллы, Тпл=177.0°С. 1H NMR (DMSO-d6) δ: 12.08 (br s, Н, NH), 3.59 (s, 4H, СH2). 14N NMR δ: -26.04 (NO2)
Пример 10. Получение этилендинитрамина (8а) в среде 1,1,1,2-тетрафторэтана.
Аналогично примеру 9, реакцию проводят в среде жидкого 1,1,1,2-тетрафторэтана при давлении 6 бар (нитрование) и 8 бар (аммонолиз). Получают 0.71 г (95%) этилендинитрамина (8а), физико-химические характеристики которого совпадают с соответствующими показателями продукта, описанными в примере 9.
Пример 11. Получение пропилендинитрамина (8b) в среде трифторметана.
Аналогично примеру 2, диэтил-N,N'-пропан-1,3-диил-бис-карбамат (6b) (1.09 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч, затем добавляют жидкий аммиак (3.01 г 0.177 моль). Отделяют этилкарбамат (0.82 г, 94%) диэтиловым эфиром. К сухому остатку добавляют воду (10 мл) и при перемешивании прикапывают раствор 1 N соляной кислоты до рН=1÷2. Водный раствор экстрагируют диэтиловым эфиром (4×15 мл). Органический слой сушат безводным сульфатом магния, эфир отгоняют при пониженном давлении (400 мбар), получают 0.71 г (86%) 1,3-пропилендинитрамина. Бесцветные кристаллы с Тпл=66.0-67.0°С. 1Н NMR (DMSO-d6) δ: 12.00 (br s, Н, NH), 3.45 (t, 4H, NHCH2, J 7.0 Hz), 1.76 (pent, 2H, CH2CH2CH2, J 6.9 Hz). 14N NMR (DMSO-d6) δ: -25.56 (NO2)
Пример 12. Получение 1,2-бис-(нитрамино)циклогексана (8с) в среде трифторметана.
Аналогично примеру 2, диэтил-циклогексан-1,2-диил-бис-карбамат (6с) (1.29 г, 0.005 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч, затем добавляют жидкий аммиак (3.01 г, 0.177 моль). Отделяют диэтиловым эфиром этилкарбамат 0.84 г (96%). К сухому остатку добавляют воду (10 мл) и при перемешивании прикапывают раствор 1 N соляной кислоты до рН=1÷2, фильтруют, сушат и получают 0.89 г (87%) 1,2-бис-нитраминоциклогексана (8с). Бесцветные кристаллы, Тпл=149.8°С. 1Н NMR (DMSO-d6) δ: 12.13 (br s, 2Н, NH), 4.02-3.81 (m, 2Н, CH), 2.14-1.89 (m, 2H, СНСH(В)СН2), 1.79-1.55 (m, 2H, CHCH(A)CH2), 1.4-1.11 (m, 4H, CHCH2CH2). 13C NMR (DMSO-d6) δ: 55.71 (2C, CH), 29.53 (2C, CHCH2CH2), 23.52 (2C, CH2CH2CH2). 14N NMR (DMSO-d6) δ: -27.28 (NO2). Вычислено: С, 35.29; H, 5.92; N, 27.44. Найдено: С, 33.73; Н, 5.97; N, 27.43
Пример 13. Получение (2-нитраминоэтил)карбамата (11) в среде трифторметана.
Аналогично примеру 1, 1,3-оксазолидин-2-он (9) (0.87 г, 0.010 моль) нитруют оксидом азота (V) (1.19 г, 0.011 моль) в течение 2 ч, затем добавляют жидкий аммиак (3.01 г, 0.177 моль). Трифторметан удаляют декомпрессией, автоклав открывают, к реакционной массе добавляют воду (25 мл) и при перемешивании прикапывают 10 N соляную кислоту до рН=1. Полученный водный раствор экстрагируют этилацетатом (4×30 мл). Органический слой сушат безводным сульфатом магния, этилацетат отгоняют при пониженном давлении (50 мбар), получают 1.10 г (94%) (2-нитраминоэтил)-карбамата (11). Бесцветные кристаллы, Тпл=83.9°С. 1Н NMR (DMSO-d6) δ: 12.10 (br s, Н, NH), 6.54 (br s, 2H, NH2), 4.04 (t, 2H, OCH2, J 5.3 Hz), 3.61 (q, 2H, NHCH2, J 4.8 Hz). 13C NMR (DMSO-d6) δ: 156.37 (C, CO), 59.61 (C, OCH2), 44.63 (C, NHCH2). 14N NMR (DMSO-d6) δ: -25.92 (NO2). Вычислено: С, 24.17; Н, 4.73; N, 28.18. Найдено: С, 24.18; Н, 4.77; N, 28.05.
Пример 14. (Сравнительный) Получение N-этилнитрамина (5b) в среде органического растворителя в условиях прототипа.
В трехгорлую колбу объемом 50 см3, снабженную термометром, магнитной мешалкой, обратным холодильником Аллина и капельной воронкой с компенсатором давления объемом 10 см3, помещают 6.30 г (0.100 моль) 100%-ной азотной кислоты. Колбу охлаждают до 5°С и прикапывают при перемешивании (600 об/мин.) этил-N-этилкарбамат (3b) (1.17 г, 10 ммоль). Реакционную массу выдерживают при комнатной температуре и перемешивании в течение 1 ч. Затем реакционную массу выливают в ледяную воду (100 мл), нейтрализуют карбонатом натрия до рН=8-9 и экстрагируют диэтиловым эфиром (4×20 мл). Органические фракции объединяют и барботируют при перемешивании газообразным аммиаком (150 см3/мин) в течение 2 часов. Выпавший кристаллический осадок отфильтровывают, промывают диэтиловым эфиром (50 мл) и кипятят в этаноле (50 мл) в течении 1 часа. Этанол отгоняют на роторном испарителе (30 мбар) сушат в эксикаторе над серной кислотой при пониженном давлении. Получают N-этилнитрамин (5b) (0.41 г, 45%). Физико-химические свойства полученного продукта совпадают с описанными в примере 2.
3-(тринитрометил-onn-азокси)-4-нитраминофуразаны и способы их получения
Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием
Катализатор для окислительного разложения хлорорганических соединений в газах и способ его получения
Способ приготовления катализатора для получения синтез-газа, катализатор, приготовленный по этому способу, и способ получения синтез-газа с его использованием
Способ приготовления катализатора для получения синтез-газа
Способ получения замещенных 2,3,5,6-тетраоксабицикло[2.2.1]гептанов
Устройство разделения элементов конструкций
Способ приготовления катализатора для получения 3-ацетилгептан-2,6-диона и способ получения 3-ацетилгептан-2,6-диона с использованием полученного катализатора
Катализатор для селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов и способ селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов с его использованием
Способ приготовления катализатора для получения бензола из метана, катализатор, приготовленный по этому способу, и способ получения бензола из метана с использованием полученного катализатора
Способ получения гипсового вяжущего
3-(тринитрометил-onn-азокси)-4-нитраминофуразаны и способы их получения
Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием
Катализатор для окислительного разложения хлорорганических соединений в газах и способ его получения
Способ приготовления катализатора для получения синтез-газа, катализатор, приготовленный по этому способу, и способ получения синтез-газа с его использованием
Способ приготовления катализатора для получения синтез-газа
Способ получения замещенных 2,3,5,6-тетраоксабицикло[2.2.1]гептанов
Устройство разделения элементов конструкций
Способ приготовления катализатора для получения 3-ацетилгептан-2,6-диона и способ получения 3-ацетилгептан-2,6-диона с использованием полученного катализатора
Катализатор для селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов и способ селективной очистки этиленовых мономеров от примесей ацетиленовых углеводородов с его использованием

