Результат интеллектуальной деятельности: МОДУЛЯТОРЫ РЕЦЕПТОРОВ МЕЛАНОКОРТИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ И КОСМЕТИЧЕСКИХ ПРЕПАРАТАХ ДЛЯ ЧЕЛОВЕКА
Вид РИД
Изобретение
Настоящее изобретение относится к новым соединениям в качестве продуктов, модулирующих один или несколько рецепторов меланокортина. Изобретение также относится к способу их получения и их терапевтическому применению.
Меланокортины образуют семейство регуляторных пептидов, которые синтезируются через пост-трансляционный процесс гормона пропиомеланокортина (POMC - 131 аминокислота). POMC приводит к получению трех классов гормонов: меланокортинов, гормона адренокортикотропина и различных эндорфинов, например, лигнотропина (Cone, et al., Recent Prog. Norm. Res., 51: 287-317, (1996); Cone et al., Ann. N.Y. Acad. Sc., 31: 342-363, (1993).
Рецепторы меланокортина (MCR) образуют часть суперсемейства GPCR с семью трансмембранными доменами. На сегодняшний день у млекопитающих было идентифицировано пять подтипов рецептора MC1-5R. Эндогенная группа пептидов связывается с MCR с агонистическими или антагонистическими эффектами, например меланоцитостимулирующими гормонами (MSH), адренокортикотропным гормоном (ACTH) и белками Агути и их производными. Однако исключение составляет рецептор MC2R, который связывается только с ACTH (основное фармакологическое отличие рецептора ACTH от других рецепторов меланокортина, Schioth et al., Life Sciences (1996), 59(10), 797-801).
MCR выполняют различные физиологические роли. MC1R регулирует образование меланина в коже и участвует в регуляции иммунной системы. MC2R регулирует продукцию кортикостероидов в надпочечниках. Рецепторы MC3R и MC4R играют роль в контроле приема пищи и сексуального поведения. MC5R участвует в регуляции экзокринных желез (Wikberg, Jarl E.S., Melanocortin receptors: perspectives for novel drugs. European Journal Pharmacology (1999), 375(1-3), 295-310. Wikberg, Jarl E.S., Melanocortin receptors: new opportunities в drug discovery. Expert Opinion on Therapeutic Patents (2001), 11 (1), 61-76).
Потенциальное использование MCR в качестве мишеней для лекарственных средств для лечения основных патологий, таких как ожирение, сахарный диабет, воспалительные процессы и сексуальная дисфункция, обуславливает необходимость в соединениях, которые демонстрируют высокую специфичность в отношении конкретного подтипа. Однако моделирование селективных лекарственных средств для несколько различающихся подтипов рецептора является трудной задачей, которая будет упрощена в свете подробных знаний о детерминантах взаимодействия лиганд-рецептор.
Заявителем в настоящее время обнаружено, удивительно и неожиданно, что новые соединения общей формулы (I) определенные ниже в настоящей заявке, демонстрируют очень хорошую активность на рецепторы меланокортина и, в частности, некоторые соединения являются активными в отношении MC1-R и обладают физико-химическими свойствами, подходящими для местного введения.
Было, в частности, продемонстрировано, что MC1-R является одним из ключевых белков в регулировании синтеза меланина в меланоцитах.
MC1-R экспрессируется в меланоцитах и вовлечен в пигментацию кожи, окраску меха животных и функции меланоцитов. Меланокортины, таким образом, можно использовать для лечения расстройств гипопигментации и гиперпигментации. Данные о полиморфизме для MC1-R гена связывают с фенотипом рыжих волос и с злокачественными и незлокачественными раковыми заболеваниями кожи (Xu X. et al., Nat. Genet. 1996; 14: 384; Van Der Velden P.A. et al., Am. J. Hum. Genet. 2001 ; 69; 774-779; Valverde P. et al., Hum. Mol. Genet. 1996; 5; 1663-1666; Schioth H. B., Biochem. Biophys. Res. Commun. 1999; 260: 488-491 ; Scott M. C. et al., J. Cell Sci. 2002; 1 15; 2349-2355). Таким образом, существует связь между MC1-R и меланомой и, как результат, MC1-R может играть важную роль в профилактики и лечении некоторых форм рака кожи (Stockfleth E. et al., Recent Results in Cancer Res. 2002; 160; 259-268; Stander et al., Exp. Dermatol. 2002; 11 :42-51). MC1-R также экспрессируется в макрофагах и моноцитах (Star et al., Proc. Natl. Acad. Sci. USA 92; 8016-8020; Hartmeyer et al., J. Immunol. 159; 1930-1937), нейтрофилах (Catania et al., Peptides 17; 675-679), эндотелиальных клетках (Hartmeyer et al., J. Immunol. 159; 1930-1937), глиомальных клетках и астроцитах (Wong et al., Neuroimmunomodulation 4, 37-41), фибробластах (Boston and Cone, Endocrinology 137, 2043-2050) и кератиноцитах (Luger et al., J. Invest. Dermatol. Symp. Proc. 2, 87-93). Локализацию MC1-R в этих клетках связывают со способностью пептидов на основе MSH ингибировать воспалительные процессы. В частности, α-MSH показал сильное ингибирование воспаления в моделях хронического воспаления кишечника, артрита, ишемии, контактной аллергии и дерматита, а также способность к индукции толерантности к гаптенам (Ceriana et al., Neuroimmunomodulation, 1, 28-32; Chiao et al., Clin. Invest. 99, 1165-1172; Huh and Lipton, Neurosurgery, 40, 132-139; Luger et al J. Invest. Dermatol. Symp. Proc. 2, 87-93; Rajora et al., Peptides 18, 381-385; J. Neurosci. 17, 2181-2196; Lipton et al., Neuroimmunomodulation, 5, 178-183). Меланокортины, таким образом, можно использовать для лечения воспалительных расстройств и иммунных расстройств. Также было сделано предположение, что MC1-R сигнальный путь играет определенную роль в ощущении боли, и что функциональная изменчивость MC1-R связана с высокой толерантностью к боли (Mogil et al., J. Med. Genet. 2005 JuI; 42(7): 583-7).
Существует сильная взаимосвязь между цветом человеческих волос и изменчивостью MC1-R (Valverde et al., Nat. Genet. 1995 Nov; 11 (3): 328-30). Функциональную изменчивость MC1-R связывают с рыжим цветом волос.
Также известно, что сальная железа экспрессирует как MC1-R (Ganceviciene et al., Exp. Dermatol. 2007 Jul; 16(7): 547-52), так и MC5-R (Zhang et al., Peptides, 2006 Feb; 27(2):413-20). Также было сообщение о том, что MC1-R чрезмерно экспрессируется в сальной железе в случае акне.
Таким образом, соединения в соответствии с настоящим изобретением находят применение в медицине человека, особенно в дерматологии и в области косметики.
Патенты WO 96/35713, WO 96/38471 и WO 99/58501 раскрывают некоторые дипептиды и их применение для стимуляции синтеза гормона роста.
Публикация в Journal Medicinal Chemistry (2003), 46, 1123-1126 описывает "открытие небольших, сильных и селективных молекул агониста MC1-R рецептора на основе тирозина, которые обладают противовоспалительными свойствами".
Патенты WO 02/070511, WO 02/079146 и WO 02/069905 заявляют применение соединений в качестве модуляторов рецепторов меланокортина, более конкретно, MC1-R и MC4-R.
Патент WO 05/047253 описывает соединения и их применение в качестве агонистов рецептора меланокортина.
Автором настоящего изобретения, удивительно и неожиданно, было обнаружено, что некоторые соединения структуры (I), являющейся объектом настоящего изобретения, являются модуляторами одного или нескольких рецепторов меланокортина и, в частности, некоторые соединения являются активными на MC1-R и имеют физико-химические свойства, подходящие для местного применения.
В частности, соединения в соответствии с настоящим изобретением имеют некоторые преимущества, обусловленные их физико-химическими свойствами, что делает возможным местное введение и, в частности, делает возможным достижение мишени быстро и направленным образом для снижения количества активного средства, необходимого для активности на мишени, для снижения системного воздействия, а также для меньших побочных эффектов.
Таким образом, настоящее изобретение относится к соединениям общей формулы (I), представленной ниже:
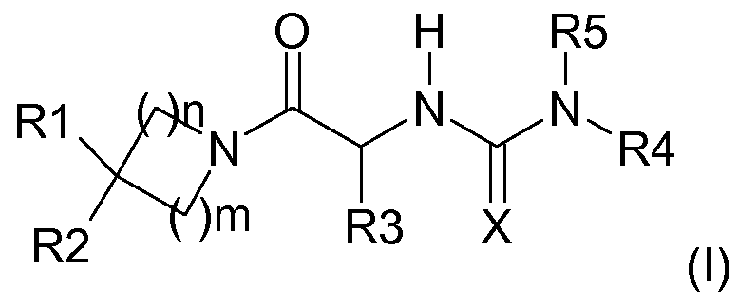
где
R1 представляет собой атом водорода, арил, замещенный арил, алкил, циклоалкил, циклоалкилалкил или циклоалкилалкилалкил,
R2 представляет собой атом водорода, гидроксил, низший алкил, замещенный низший алкил, высший алкил, замещенный высший алкил, циклоалкил, циклоалкилалкил, низший алкокси, замещенный низший алкокси, высший алкокси, замещенный высший алкокси, циклоалкилалкокси, ацилокси, ацил, алкоксикарбонил, карбоксамид, карбоновую кислоту, циано или амино, дизамещенный ацилом и арилом или алкилом,
R3 представляет собой аралкил или замещенный аралкил,
R4 представляет собой гетероаралкил или замещенный гетероаралкил,
R5 представляет собой атом водорода или алкил,
X представляет собой атом кислорода или атом серы,
n, m могут быть равны 1 или 2;
и также соответствующим солям и энантиомерам.
Среди аддитивных солей соединений общей формулы (I) с фармацевтически приемлемыми кислотами, могут быть указаны, предпочтительно, соли с органическими кислотами или с минеральными кислотами.
Подходящими минеральными кислотами являются, например, галогенводородные кислоты, например хлористоводородная кислота или бромистоводородная кислота, серная кислота и азотная кислота.
Подходящими органическими кислотами являются, например, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, щавелевая кислота и винная кислота.
Соединения общей формулы (I) также могут существовать в виде гидратов или сольватов с водой или с растворителем.
Подходящие растворители для образования сольватов или гидратов представляют собой, например, спирты, например, изопропанол, или этанол, или воду.
В соответствии с настоящим изобретением, термин "арил" обозначает незамещенный фенил или нафтил.
В соответствии с настоящим изобретением, термин "замещенный арил" означает фенил или нафтил, замещенный одной или несколькими группами атомов, выбранных из алкила, алкокси, галогена, гидроксила, циано, трифторметила и нитро.
В соответствии с настоящим изобретением, термин "циклоалкил" означает насыщенную циклическую углеводородную цепь, содержащую от 3 до 7 атомов углерода.
В соответствии с настоящим изобретением, термин "гидроксил" означает OH группу.
В соответствии с настоящим изобретением, термин "амино" означает NH2 группу.
В соответствии с настоящим изобретением, термин "циано" означает CN группу.
В соответствии с настоящим изобретением, термин "карбоновая кислота" означает CO2H группу.
В соответствии с настоящим изобретением, термин "ацил" означает формил или карбонил, замещенный алкилом, циклоалкилом или циклоалкилалкилом.
В соответствии с настоящим изобретением, термин "алкил" означает замещенный или незамещенный низший алкил или высший алкил.
В соответствии с настоящим изобретением, термин "низший алкил" означает линейную или разветвленную, насыщенную или ненасыщенную углеводородную цепь, содержащую от 1 до 4 атомов углерода, или ненасыщенную цепь, содержащую от 2 до 4 атомов углерода, и, в частности, например, метил, этил, пропил, изопропил и бутил.
В соответствии с настоящим изобретением, термин "замещенный низший алкил" означает линейную или разветвленную, насыщенную или ненасыщенную углеводородную цепь, содержащую от 1 до 4 атомов углерода и замещенную одним или несколькими атомами галогена или гидроксилом, или ненасыщенную углеводородную цепь, содержащую от 2 до 4 атомов углерода и замещенную одним или несколькими атомами галогена или гидроксилом.
В соответствии с настоящим изобретением, термин "высший алкил" означает линейную или разветвленную, насыщенную или ненасыщенную углеводородную цепь, содержащую от 5 до 10 атомов углерода.
В соответствии с настоящим изобретением, термин "замещенный высший алкил" означает линейную или разветвленную, насыщенную или ненасыщенную углеводородную цепь, содержащую от 5 до 10 атомов углерода и замещенную одним или несколькими атомами галогена или гидроксилом.
В соответствии с настоящим изобретением, термин "атом галогена" означает атомы хлора, фтора, йода и брома.
В соответствии с настоящим изобретением, термин "циклоалкилалкил" означает алкил, замещенный циклоалкилом.
В соответствии с настоящим изобретением, термин "низший алкокси" означает атом кислорода, замещенный низшим алкилом, и, в частности, например, метокси, этокси, пропокси, изопропокси или бутокси.
В соответствии с настоящим изобретением, термин "замещенный низший алкокси" означает атом кислорода, замещенный замещенным низшим алкилом.
В соответствии с настоящим изобретением, термин "высший алкокси" означает атом кислорода, замещенный высшим алкилом.
В соответствии с настоящим изобретением, термин "замещенный высший алкокси" означает атом кислорода, замещенный замещенным высшим алкилом.
В соответствии с настоящим изобретением, термин "циклоалкилалкокси" означает атом кислорода, замещенный циклоалкилалкилом.
В соответствии с настоящим изобретением, термин "ацилокси" означает атом кислорода, замещенный ацилом.
В соответствии с настоящим изобретением, термин "алкоксикарбонил" означает карбонил, замещенный группой алкокси, циклоалкокси или циклоалкилалкокси.
В соответствии с настоящим изобретением, термин "карбоксамид" означает карбонил, замещенный группой моноалкиламино или диалкиламино.
В соответствии с настоящим изобретением, термин "аралкил" означает алкил, замещенный арилом.
В соответствии с настоящим изобретением, термин "замещенный аралкил" означает алкил, замещенный замещенным арилом.
В соответствии с настоящим изобретением, термин "гетероцикл" означает насыщенную или ненасыщенную, циклическую или бициклическую углеводородную цепь, содержащую один или несколько гетероатомов, выбранных из O, S и N.
В соответствии с настоящим изобретением, термин "замещенный гетероцикл" означает насыщенную или ненасыщенную, циклическую или бициклическую углеводородную цепь, содержащую один или несколько гетероатомов, выбранных из O, S и N, замещенную одной или несколькими алкильными группами.
В соответствии с настоящим изобретением, термин "гетероарил" означает ароматический гетероцикл.
В соответствии с настоящим изобретением, термин "замещенный гетероарил" означает ароматический гетероцикл, замещенный одной или несколькими алкильными группами.
В соответствии с настоящим изобретением, термин "гетероаралкил" означает алкил, замещенный гетероарилом.
В соответствии с настоящим изобретением, термин "замещенный гетероаралкил" означает алкил, замещенный замещенным гетероарилом.
Среди соединений общей формулы (I), включенных в контекст настоящего изобретения, можно особенно указать следующие:
1-[(S)-2-(4-Бутирил-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(4-оксо-1-фенил-1,3,8-триазаспиро[4,5]дец-8-ил)-ил)этил]мочевина
1-[2-(4-Циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(4-фенилпиперидин-1-ил)этил]мочевина
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-пиперидин-1-ил-этил]мочевина
Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
N-{1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-ил}-N-фенилпропионамид
1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]-3-фенил-азетидин-3-илбутират
Этил 1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат
1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[4-(2-метоксифенил)-пиперидин-1-ил]-2-оксоэтил}мочевина
1-[2-(3-Бутокси-3-фенилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксиламид
1-[2-(3-Циклогексанкарбонилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-[2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
N-Циклопропил-N-{1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-ил}пропионамид
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3H-имидазол-4-ил)этил]мочевина
1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-2-фенилацетил)-пиперидин-4-карбоксилат
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-этоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-2-фенилацетил)-пиперидин-4-карбоксилат
Этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
4-Циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоновая кислота
1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[3-(2-метилциклогексил)-3-пропоксиазетидин-1-ил]-2-оксоэтил}мочевина
Пропил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(3-пентил-3-фенил-азетидин-1-ил)этил]мочевина
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
Этил 1-((S)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевина
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклопропилметил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
Пропил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-Бензил-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол- 4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3-метил-3H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-хлорбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)пропил]-уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]тиомочевина
1-[(R)-2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]тиомочевина
1-[(R)-1-Бензил-2-(4-циклогексил-4-пропоксипиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]тиомочевина
1-[(R)-1-Бензил-2-(4-циклогексил-4-пропоксипиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]тиоуреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}-3-фенил-пропионил)пиперидин-4-карбоксилат
1-[(R)-2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3-метил-3H-имидазол-4-ил)этил]мочевина
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}-пропионил)-4-циклогексилпиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-фторфенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]-уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-фторфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-фторфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}пропионил)пиперидин-4-карбоксилат
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]тиоуреидо}-пропионил)-4-циклогексилпиперидин-4-карбоксилат
1-[(R)-2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-(4-Хлорбензил)-2-(4-циклогексил-4-пропоксипиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
и также их соответствующие соли и энантиомеры.
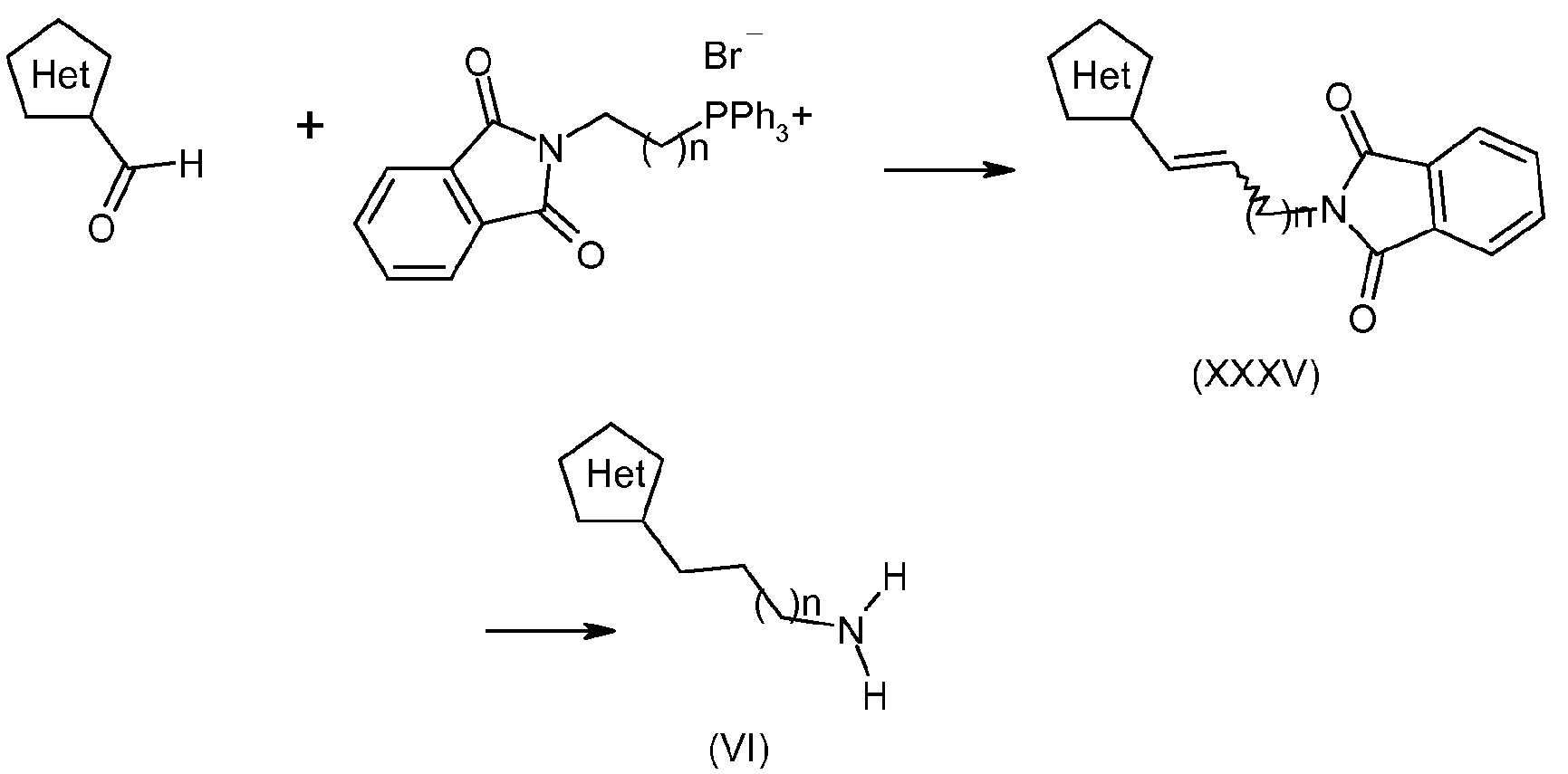
Соединения общей формулы (I) получают в соответствии с общими схемами реакций, представленными на Фиг. 1.
Используя схему реакции 1 (Фиг. 1):
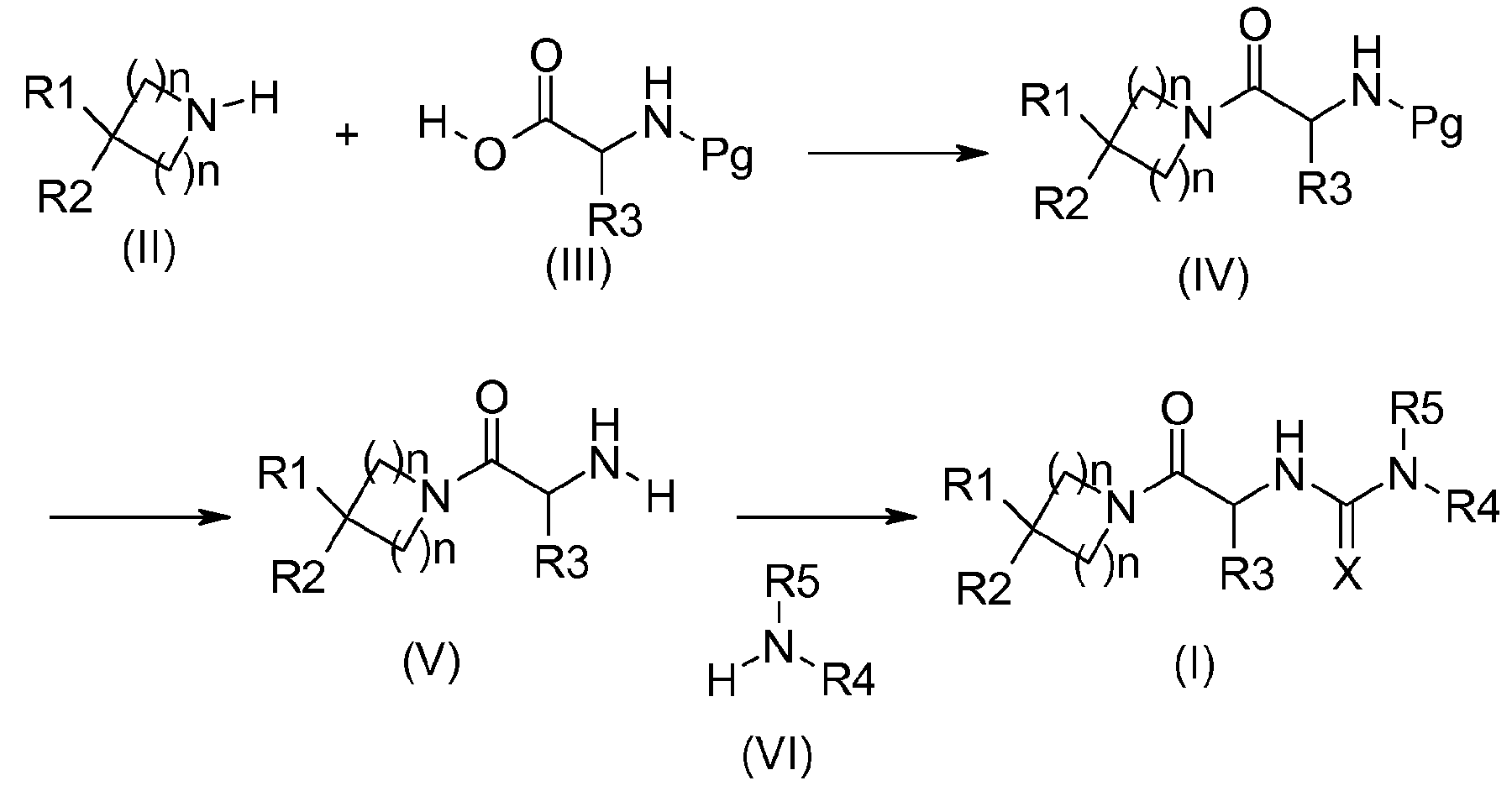
В соответствии со схемой 1, соединения формулы (IV) можно получить путем взаимодействия между промежуточными соединениями формулы (II) и аминокислотой формулы (III), где функциональная группа амина защищена защитной группой Pg (например Boc, CBz или Fmoc группы), в стандартных условиях пептидного связывания (Han, S., Kim, Y. Tetrahedron, 2004, 60, 2447-2467; Albericio, F. Current Opinion в Chemical Biology, 2004, 8, 211-221; Humphrey, J., Chamberlin, R. Chem. Rev., 1997, 97, 2243-2266), используя, например, в качестве связывающего реагента 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид, гидроксибензотриазол или TBTU и в качестве основания триэтиламин или диизопропилэтиламин, в растворителе, таком как дихлорметан или диметилформамид.
Аминокислоты общей формулы (IV) являются коммерчески доступными, или их можно получить способами, описанными в литературе (Williams, R. M., Synthesis optically active α-amino acids, Pergamon Press, Oxford, 1989).
Соединения формулы (V) получают путем удаления защиты у функциональной аминогруппы соединений формулы (IV), используя способы, выбранные из тех, которые известны специалистам в данной области. Они включают, в частности, использование трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетате, например, в случае защиты с использованием Boc-группы, гидрирования с соответствующим металлом в тетрагидрофуране или метаноле, например, в случае защиты с использованием CBz-группы, и пиперидина в ацетонитриле, например, в случае защиты с использованием Fmoc-группы.
На конечной стадии соединения формулы (I) можно получить, например, путем добавления аминов формулы (VI) к изоцианатам или изотиоцианатам, полученным из соединений (V), в дихлорметане или диметилформамиде. Изоцианаты можно получить, например, из аминов (V) в присутствии фосгена, дифосгена или трифосгена. Изотиоцианаты можно получить, например, из аминов (V) в присутствии тиофосгена (Nowick J. S. et al., JOC (1996) 3929-3934) или бис(2-пиридил)тионокарбоната (WO 2008/008954). Соединения формулы (I) также можно синтезировать, например, путем добавления аминов формулы (VI) к активированным карбаматам, полученным из аминов (V), в дихлорметане или диметилформамиде. Термин "активированный карбамат" означает, например, пара-нитрофенилкарбаматную группу (Igarashi, T., Synlett (2007) 1436), которая может быть получена путем добавления пара-нитрофенилхлорформиата к амину (V) в присутствии основания, которое может представлять собой, например, триэтиламин, в дихлорметане или диметилформамид.
Соединения формулы (II) являются коммерчески доступными, или их можно получить в соответствии со способами, описанными в литературе или известными специалистам в данной области, адаптированными в зависимости от природы заместителей R1 и R2. Схемы 1-9, представленные ниже, представляют примеры получения соединений формулы (II).
Например, когда R2 содержит ацилокси или карбоксамидную цепь, получение соединения (II, n, m=2) можно осуществить в соответствии со схемой 1:
Схема 1:
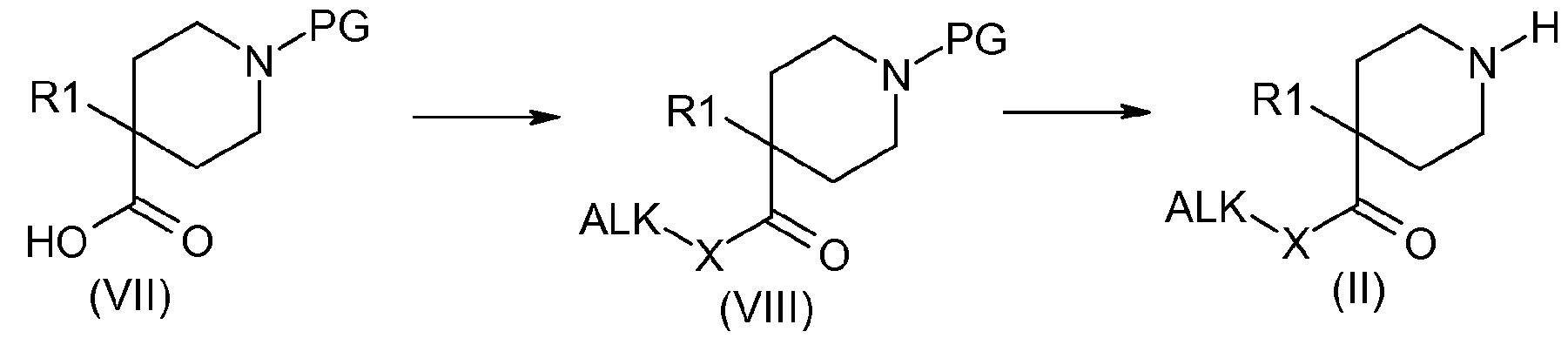
Соединения формулы (VIII) получают:
- когда X представляет собой кислород, например, путем этерификации карбоновокислотной функциональной группы соединений (VII), используя способы, описанные в литературе, или
- когда X представляет собой азот, например, путем добавления к амину или к хлорангидриду кислоты, полученному из карбоновой кислоты (VII), с использованием способов, выбранных из тех, которые известны специалистам в данной области. Лучше всего использовать оксалилхлорид или тионилхлорид в растворителях, таких как дихлорметан или диметилформамид.
Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (VIII), с использованием способов, выбранных из тех, которые известны специалистам в данной области. Они включают, в частности, использование трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетата, например, в случае защиты с использованием Boc-группы, гидрирование с использованием соответствующего металла в тетрагидрофуране или метаноле, например, в случае защиты с использованием CBz-группы, и пиперидина в ацетонитриле, например, в случае защиты с использованием Fmoc-группы.
Например, когда R1 содержит алкильную, циклоалкильную или циклоалкилалкильную группу, и R2 содержит ацилокси-цепь, получение соединения (II, n, m=2) можно осуществить в соответствии со схемой 2:
Схема 2:
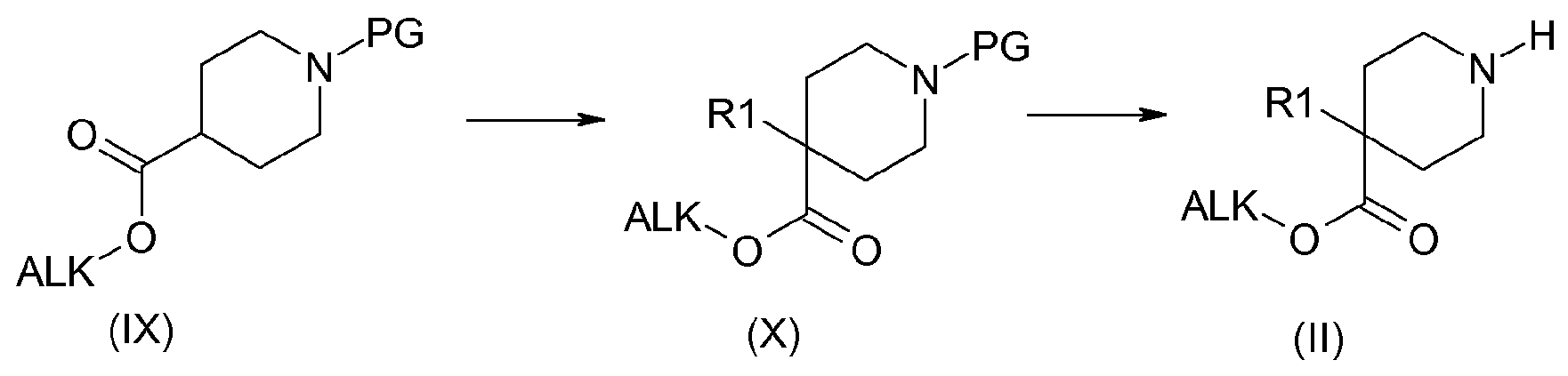
Введение группы R1 можно осуществить, например, путем альфа-депротонирования сложноэфирной функциональной группы соединения (IX) в присутствии основания, такого как диизопропиламид лития или гексаметилдисилазид лития, в растворителях, таких как дихлорметан или тетрагидрофуран. Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (X), с использованием способов, выбранных из тех, которые известны специалистам в данной области. Они включают, в частности, использование трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетата, например, в случае защиты с использованием Boc-группы, гидрирование с использованием соответствующего металла в тетрагидрофуране или метаноле, например, в случае защиты с использованием CBz-группы.
Например, когда R2 содержит алкокси или алкоксикарбонильную цепь, получение соединения (II, n, m=1) можно осуществить в соответствии со схемой 3:
Схема 3:
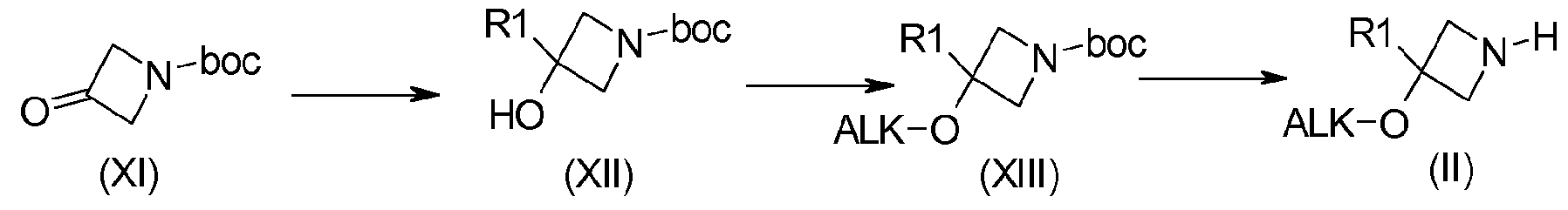
Соединения формулы (XII) получают, например, путем добавления галогенида магния, полученного из R1, к N-Boc-азетидинону (XI) с последующим алкилированием или ацилированием третичного спирта в соответствии со способами, стандартно описанными в литературе, что приводит к получению соединений (XIII). Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (XIII), например, в присутствии трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетате.
Например, когда R2 содержит ацильную группу, получение соединения (II, n=m=1) можно осуществить в соответствии со схемой 4:
Схема 4:
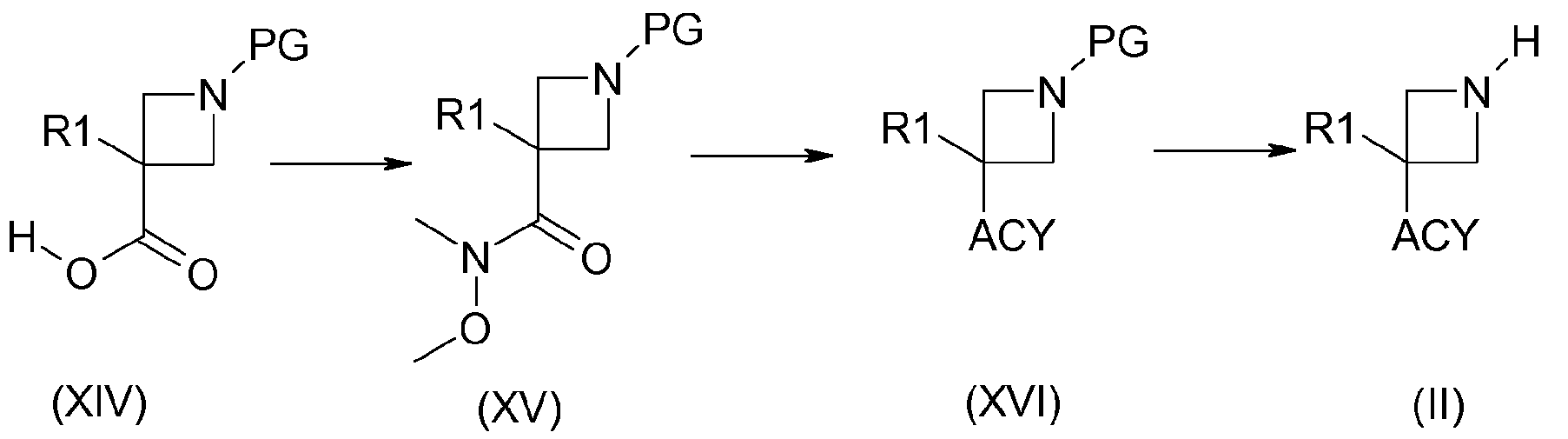
Соединения формулы (XV) можно получить в условиях пептидного связывания между соединениями с карбоновой кислотой (XIV) и амином Вайнреба, используя, например, в качестве агента сочетания 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид, гидроксибензотриазол или TBTU и, в качестве основания, триэтиламин или диизопропилэтиламин, в растворителе, таком как дихлорметан или диметилформамид. Соединения формулы (XVI) получают, например, путем добавления галогенида магния, полученного из R1, к производному амида Вайнреба (XV). Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (X), с использованием способов, выбранных из тех, которые известны специалистам в данной области. Они включают, в частности, использование трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетата, например, в случае удаления защитной Вос-группы, гидрирование с использованием соответствующего металла в тетрагидрофуране или метаноле, например, в случае защиты при помощи CBz-группы.
Например, когда R2 содержит дизамещенный амин, получение соединения (II, n=m=2) можно осуществить в соответствии со схемой 5:
Схема 5:
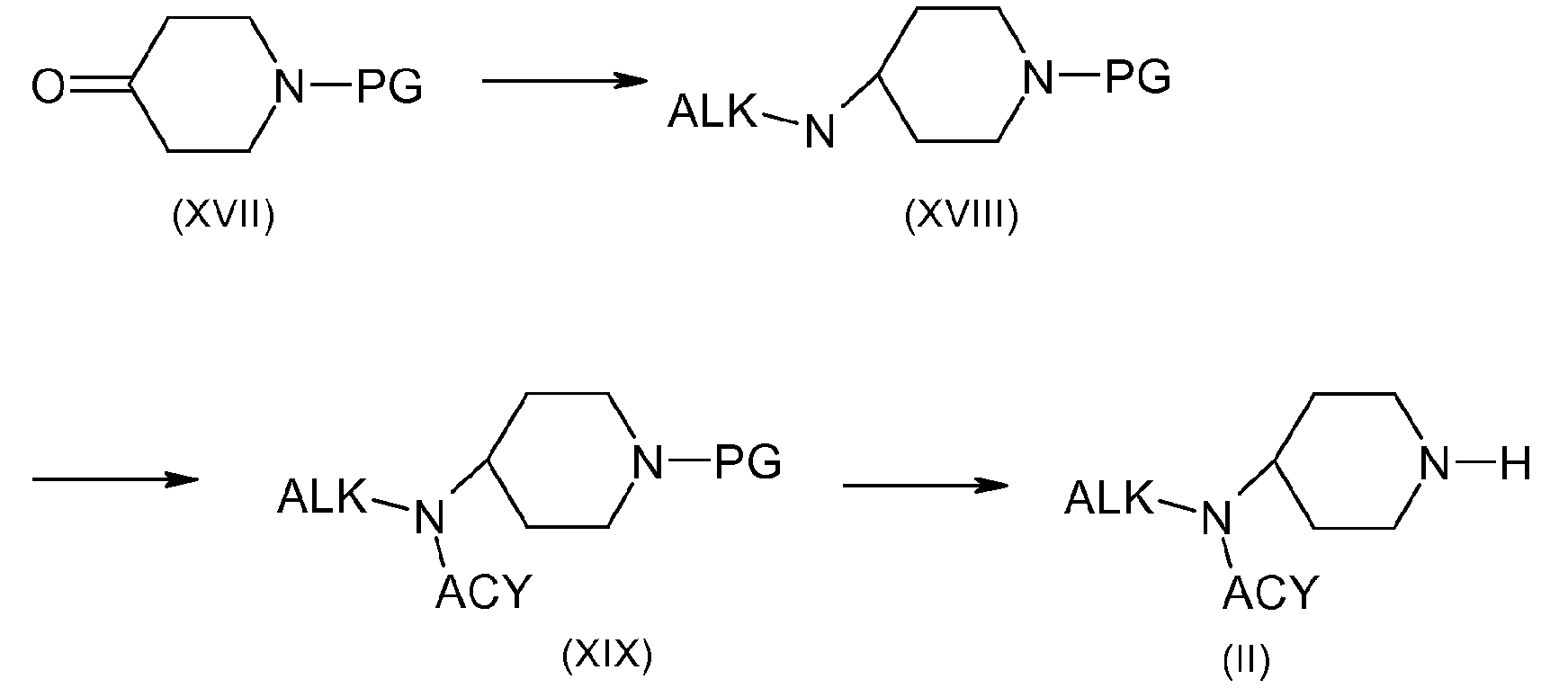
Соединения формулы (XVIII) можно получить, например, в условиях восстановительного аминирования между коммерчески доступным кетоном (XVII) и амином в присутствии боргидрида натрия или цианоборгидрида натрия. Вторичные амины (XVIII) затем могут быть ацилированы в присутствии основания, такого как триэтиламин, и хлорангидрида кислоты, например, с получением соединений (XIX). Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (VIII) с использованием способов, выбранных из тех, которые известны специалистам в данной области. Они включают, в частности, использование трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетате, например, в случае защиты с использованием Boc-группы, гидрирование с использованием соответствующего металла в тетрагидрофуране или метаноле, например, в случае защиты с использованием CBz-группы, и пиперидина в ацетонитриле, например, в случае защиты с использованием Fmoc-группы.
Например, когда R1 содержит циклогексил и R2 содержит ацильную группу, получение соединения (II, n=m=2) можно осуществить в соответствии со схемой 6:
Схема 6:
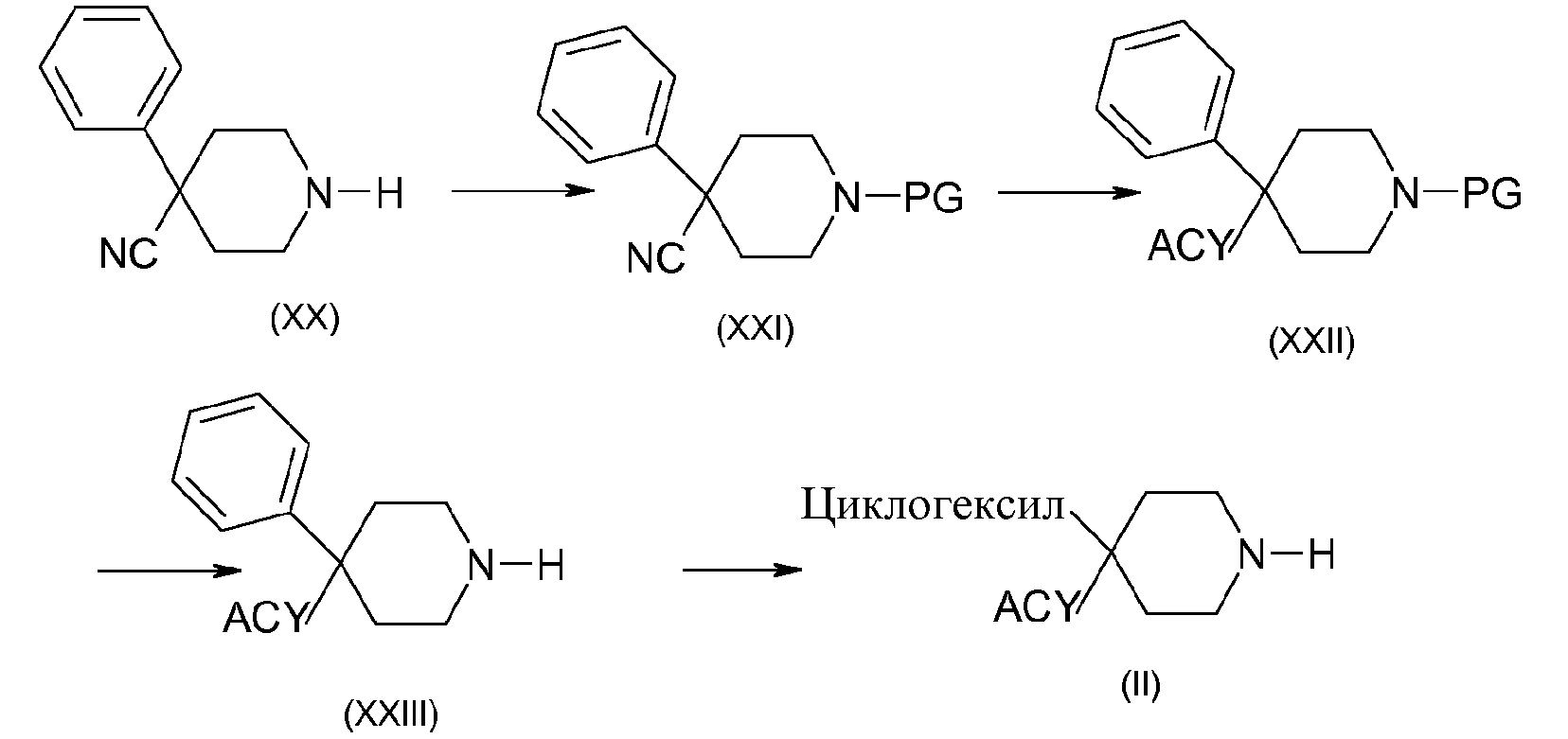
После защиты коммерчески доступного амина (XX) при помощи тозилатной группы, например, путем взаимодействия тозилхлорида в присутствии основания, такого как триэтиламин, в дихлорметане, получают соединения (XXI). Соединения формулы (XXII) получают, например, путем добавления галогенида магния, полученного из алкила в толуоле, к нитрильной функциональной группе производных (XXI), с последующим гидролизом в кислой среде промежуточного имина, например, с использованием хлористоводородной кислоты. Соединения формулы (XXIII) получают путем удаления защиты у функциональной аминогруппы в кислой среде, которая может представлять собой серную кислоту, в случае тозилатной группы. Соединения (II) получены, например, путем гидрирования соединения (X) в присутствии катализатора, который может представлять собой, например, родий на оксиде алюминия или оксид платины в диоксане.
Например, когда R1 содержит циклогексил и R2 содержит алкоксигруппу, получение соединения (II, n=m=2) можно осуществить в соответствии со схемой 7:
Схема 7:
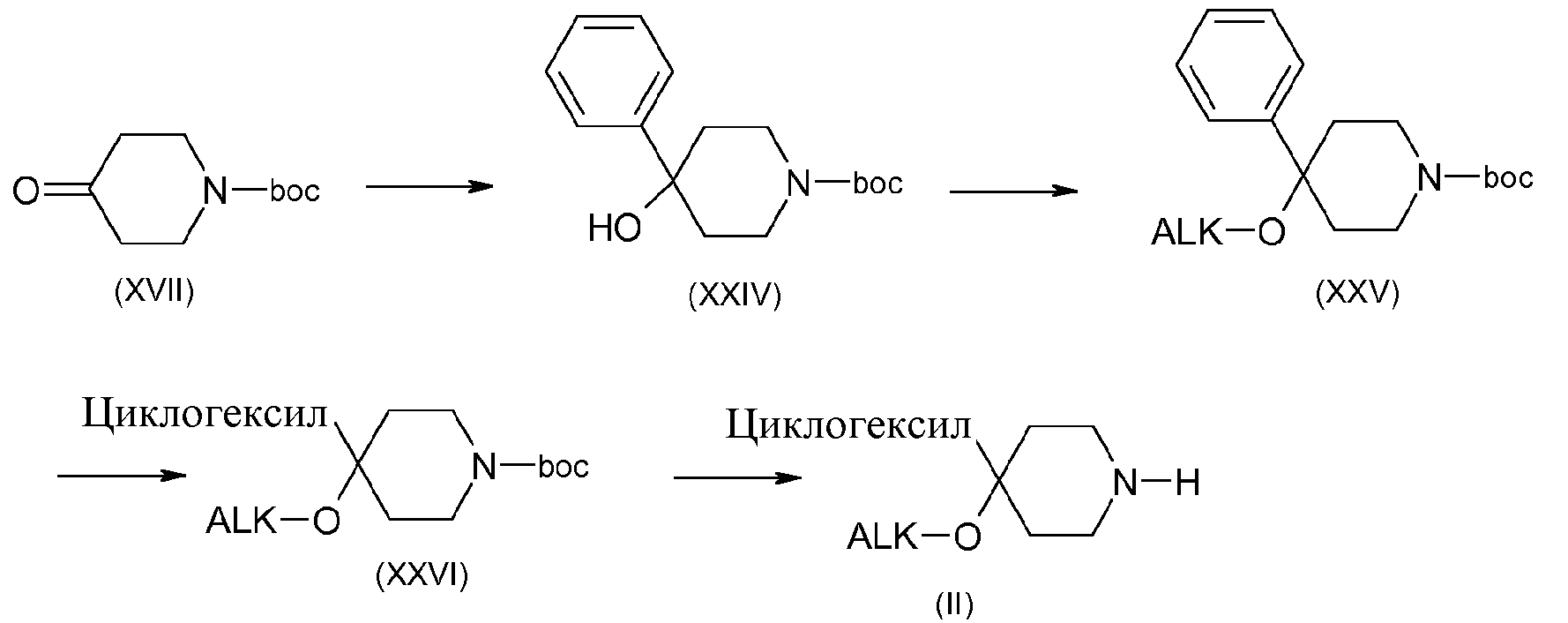
Соединения формулы (XXIV) получены, например, путем добавления галогенида магния, полученного из фенила, к коммерчески доступному кетону (XVII) с последующим алкилированием третичного спирта в соответствии со способами, стандартно описанными в литературе, с получением соединений (XXV). Соединения (XXVI) получают, например, путем гидрирования соединения (XXV) в присутствии катализатора, который может представлять собой, например, родий на оксиде алюминия или оксид платины в диоксане. Соединения формулы (II) получают путем удаления защиты у функциональной аминогруппы соединений формулы (XXVI), например, в присутствии трифторуксусной кислоты или хлористоводородной кислоты в дихлорметане или этилацетате.
Например, когда R1 представляет собой арильную группу и R2 содержит алкильную цепь, получение соединения (II, n=m=1) можно осуществить в соответствии со схемой 8:
Схема 8:
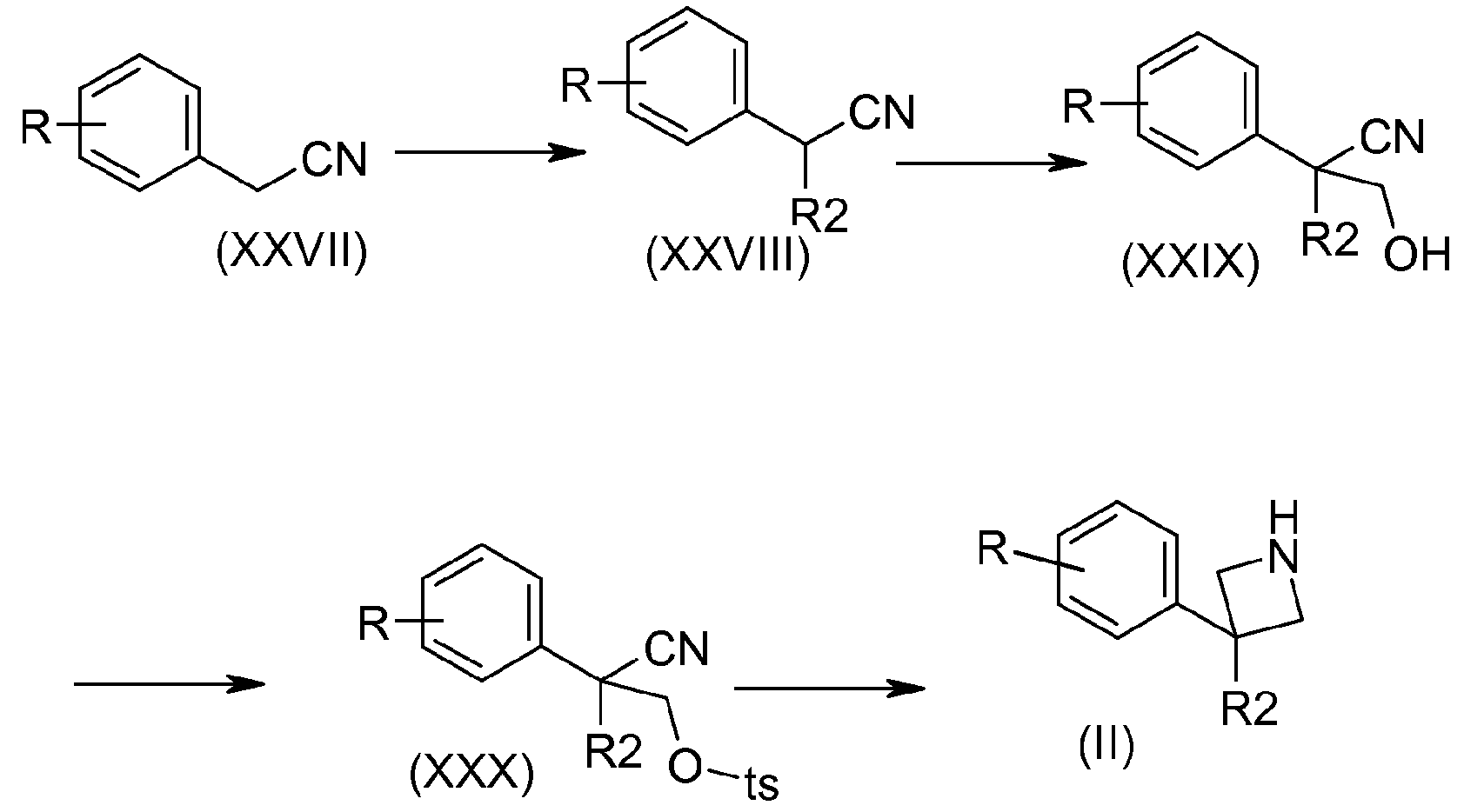
Соединения формулы (XXVIII) можно получить, например, путем добавления основания, такого как гидрид натрия, в присутствии галогенированного производного, полученного из R2, к коммерчески доступным нитрильным производным (XXVII). Первичные спирты (XXIX) можно синтезировать из нитрильных производных (XXVIII) в присутствии основания, например, гидрида натрия и параформальдегида. Функциональная группа первичного спирта соединений (XXIX) может быть преобразована в сульфонат в присутствии основания, которое может представлять собой, например, триэтиламин и тозилхлорид. Азетидиновые соединения (II) можно синтезировать путем внутримолекулярной циклизации между аминовой функциональной группой, полученной после восстановления нитрильной функциональной группы, например, в присутствии литийалюминийгидрида и тозилатной функциональной группы.
Соединения формулы (VI) являются коммерчески доступными, или их можно получить в соответствии со способами, описанными в литературе или известными специалистам в данной области, адаптированными в зависимости от природы заместителей R4 и R5. Схемы 9-11, представленные ниже, представляют примеры получения соединений формулы (VI).
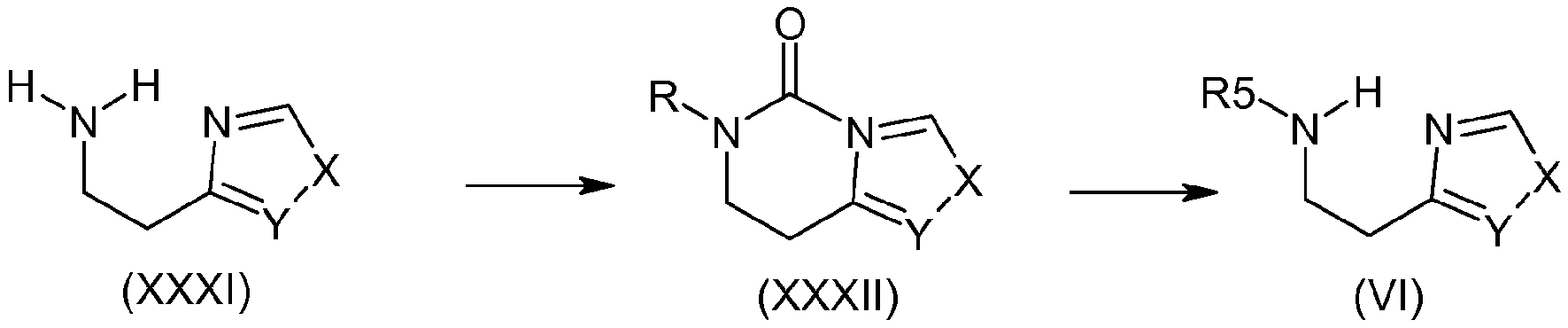
Например, когда R5 представляет собой алкильную группу и R4 содержит гетероаралкильную группу, получение соединения (VI) можно осуществить, например, следуя протоколу, описанному в литературе (Durant G.J., Emmet J. C, Ganellin C. R., Roe A.M., (1973) Br. Pat. 1 341 375), как описано на схеме 9:
Схема 9:
Например, когда R4 содержит 1,2,3-триазольный гетероцикл, получение соединения (VI) можно осуществить в соответствии со схемой 10:
Схема 10:
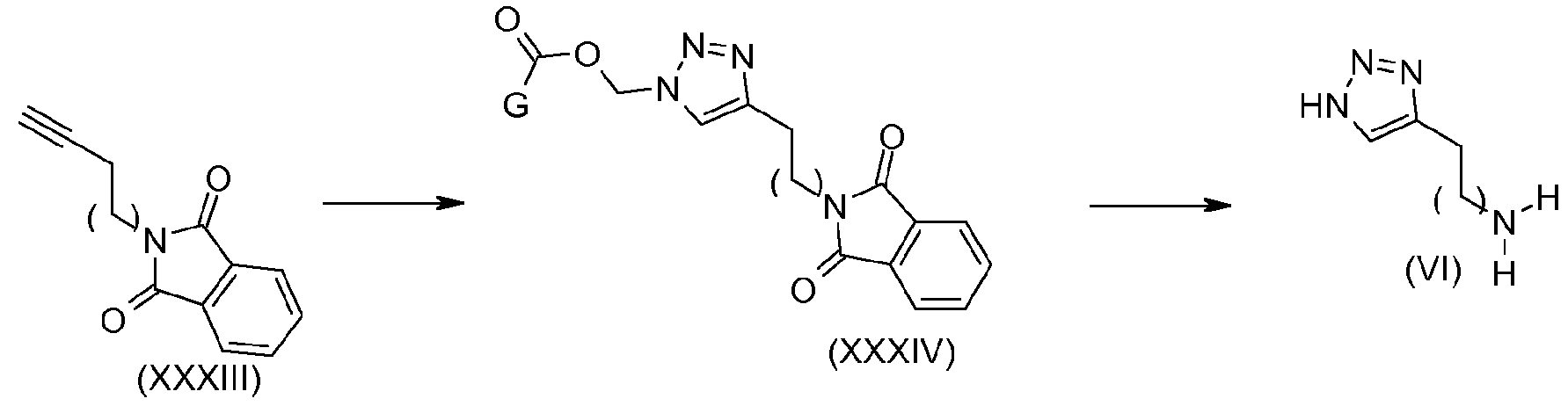
Соединения формулы (XXXIV) можно получить с использованием способов, описанных в литературе (Loren J. C, Synlett, 2005, 2847-2850), с последующим расщеплением в щелочной среде в присутствии, например, гидроксида натрия, с получением соединений (VI).
Например, когда R4 содержит гетероаралкил, получение соединения (VI) можно осуществить в соответствии со схемой 11:
Схема 11:
Соединения формулы (VI) можно получить при помощи способов, описанных в литературе (Wolin R., BOMCL, 1998, 2157) с использованием реакции Виттига между альдегидами, которые замещены гетероарилами, и коммерчески доступными илидами с образованием алкенов (XXXV), с последующим гидрированием двойной связи и гидразинолизом фталимида с получением соединений (VI).
В соответствии с настоящим изобретением, соединения общей формулы (I), которые являются особенно предпочтительными, представляют собой такие, для которых:
R1 представляет собой атом водорода, арил, замещенный арил, алкил, циклоалкил или циклоалкилалкил,
R2 представляет собой атом водорода, низший алкил, замещенный низший алкил, высший алкил, замещенный высший алкил, циклоалкил, циклоалкилалкил, низший алкокси, замещенный низший алкокси, высший алкокси, замещенный высший алкокси, циклоалкилалкокси, ацилокси, ацил, алкоксикарбонил, карбоксамид или циано,
R3 представляет собой аралкил или замещенный аралкил,
R4 представляет собой гетероаралкил или замещенный гетероаралкил,
R5 представляет собой атом водорода,
X представляет собой атом кислорода или атом серы,
n, m могут быть равны 1 или 2; а также соответствующие соли и энантиомеры.
Среди соединений общей формулы (I), входящих в контекст настоящего изобретения, особенно можно выделить следующие:
1-[(S)-2-(4-Бутирил-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(4-фенилпиперидин-1-ил)этил]мочевина
Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]-пиперидин-4-карбоксилат
1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[4-(2-метоксифенил)-пиперидин-1-ил]-2-оксоэтил}мочевина
1-[2-(3-Бутокси-3-фенилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксиламид
1-[2-(3-Циклогексанкарбонилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-[2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3H-имидазол-4-ил)этил]мочевина
1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-2-фенилацетил)-пиперидин-4-карбоксилат
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-этоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-2-фенилацетил)-пиперидин-4-карбоксилат
Этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[3-(2-метилциклогексил)-3-пропоксиазетидин-1-ил]-2-оксоэтил}мочевина
Пропил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(3-пентил-3-фенил-азетидин-1-ил)этил]мочевина
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
Этил 1-((S)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевина
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклопропилметил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
Пропил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-Бензил-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3-метил-3H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-хлорбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)пропил]-уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
и также их соответствующие соли и энантиомеры.
В соответствии с настоящим изобретением, соединения общей формулы (I), которые являются особенно предпочтительными, представляют собой те, для которых:
R1 представляет циклоалкил или циклоалкилалкил,
R2 представляет низший алкокси, циклоалкилалкокси, ацил, алкоксикарбонил или циано,
R3 представляет аралкил или замещенный аралкил,
R4 представляет гетероаралкил или замещенный гетероаралкил,
R5 представляет атом водорода,
X представляет атом кислорода или атом серы,
n, m равны 2; и также соответствующие соли и энантиомеры.
Предпочтительные соединения представляют собой:
Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3H-имидазол-4-ил)этил]мочевина
1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-этоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Пропил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевина
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклопропилметил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
Пропил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-Бензил-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3-метил-3H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-[1,2,3]триазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-хлорбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)пропил]-уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
и также их соответствующие соли и энантиомеры.
В соответствии с настоящим изобретением, соединения общей формулы (I), которые являются особенно предпочтительными, представляют собой те, для которых:
R1 представляет циклоалкил,
R2 представляет низший алкокси, ацил или алкоксикарбонил,
R3 представляет аралкил или замещенный аралкил,
R4 представляет гетероаралкил,
R5 представляет атом водорода,
X представляет атом кислорода или атом серы,
n, m равны 2; и также соответствующие соли и энантиомеры.
Соединения, которые являются особенно предпочтительными, представляют собой следующие:
Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3H-имидазол-4-ил)этил]мочевина
1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-этоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[2-(4-Циклогексил-4-пропоксипиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Пропил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Пропил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
Этил 4-циклопентил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-Бензил-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3-метил-3H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-хлорбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат,
а также их соответствующие соли и энантиомеры.
В соответствии с настоящим изобретением, соединения общей формулы (I), которые являются особенно предпочтительными, представляют собой такие, для которых:
R1 представляет собой циклоалкил,
R2 представляет собой ацил или алкоксикарбонил,
R3 представляет собой аралкил или замещенный аралкил,
R4 представляет собой замещенный или незамещенный имидазол,
R5 представляет собой атом водорода,
X представляет собой атом кислорода или атом серы,
n, m равны 2; а также соответствующие соли и энантиомеры.
Соединения, которые являются особенно предпочтительными, представляют собой следующие:
Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(3H-имидазол-4-ил)этил]мочевина
Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Пропил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат 1-[2-(4-Циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
Пропил 4-циклогексил-1-(2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-фенилпропионил)-пиперидин-4-карбоксилат
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-фторбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-1-Бензил-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-хлорбензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
Этил 4-циклогексил-1-((R)-3-(4-метоксифенил)-2-{3-[2-(3-метил-3H-имидазол-4-ил)этил]уреидо}пропионил)пиперидин-4-карбоксилат
Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат и также их соответствующие соли и энантиомеры.
Соединения в соответствии с настоящим изобретением обладают свойствами модуляции рецепторов меланокортина. Выражение "свойства модуляции рецепторов меланокортина" означает агонистические или антагонистические свойства в отношении рецепторов меланокортина. Такую активность в отношении рецепторов MCR измеряют в испытании трансактивации и количественно определяют по 50% эффективной концентрации (EC 50), как описано в Примере 10.
Предпочтительно, соединения являются, по меньшей мере, модуляторами MCR рецепторов и обладают свойствами, подходящими для местного введения, т.е. они имеют период полужизни меньше чем или равный 10 минутам в человеческих микросомах, они имеют log D при pH 6,5 больше чем или равный 3, и они активны при местном применении в in vivo моделях.
Предпочтительно, соединения по настоящему изобретению имеют 50% эффективную концентрацию (EC50) в отношении рецептора MC1 меньше чем или равную 10 мкМ, и более предпочтительно меньше чем или равную 1 мкМ.
Настоящее изобретение, таким образом, направлено на применение, по меньшей мере, одного соединения общей формулы (I), определенного выше, для получения фармацевтической или косметической композиции, в которой указанное соединение обладает модуляторной активностью на один или несколько рецепторов меланокортина и, в частности, на подтипы 1, 3, 4 и 5.
В одном конкретном варианте воплощения настоящего изобретения, некоторые соединения формулы (I) по настоящему изобретению обладают активностью в отношении MC1R рецептора и являются особенно полезными для лечения нарушений пигментации и воспалительных и иммунных расстройств. Некоторые соединения по настоящему изобретению обладают активностью в отношении MC4R рецептора и являются особенно полезными для лечения расстройств пищевого поведения и метаболизма, а также нейродегенеративных расстройств.
Настоящее изобретение также относится к терапевтическому или косметическому способу лечения, включающему введение фармацевтической или косметической композиции, включающей указанное соединение в качестве модулятора одного или нескольких рецепторов меланокортина и, в частности, подтипов 1, 3, 4 и 5. В одном конкретном варианте воплощения, настоящее изобретение также относится к терапевтическому или косметическому способу, включающему введение фармацевтической или косметической композиции, включающей указанное соединение, для лечения нарушений пигментации и воспалительных и иммунных расстройств. В одном конкретном варианте воплощения настоящего изобретения, соединения являются модуляторами подтипа 1 и обладают свойствами, подходящими для местного введения.
Настоящее изобретение также относится к применению соединения общей формулы (I), определенного выше, а также его соответствующих солей и энантиомеров для получения лекарственного препарата для лечения расстройств, связанных с дисфункцией MC1R рецептора.
Наконец, соединения, используемые в соответствии с настоящим изобретением, являются особенно подходящими для лечения и/или профилактики расстройств и/или заболеваний, таких как воспалительные заболевания:
- пищеварительного аппарата, особенно включая кишечник (и, в частности, толстую кишку в случае синдрома раздраженной толстой кишки, язвенно-геморрагический проктоколита или болезни Крона); панкреатит, гепатит (острый и хронический), воспалительные патологии мочевого пузыря и гастрит;
- опорно-двигательного аппарата, включая ревматоидный артрит, остеоартрит, остеопороз, травматический артрит, постинфекционный артрит, мышечная дегенерация и дерматомиозит;
- мочеполового аппарата и, в частности, гломерулонефрита;
- сердечного аппарата и, в частности, перикардита и миокардита и заболеваний, включающих такие, в основе которых такой фактор, как воспаление. Эти заболевания включают, но не ограничиваются этим, атеросклероз, атеросклероз в результате трансплантации, заболевание периферических сосудов, воспалительные сосудистые заболевания, перемежающаяся хромота или прихрамывание, рестеноз, нарушения мозгового кровообращения, преходящее нарушение мозгового кровообращения, ишемия миокарда и инфаркт миокарда. Эти соединения также можно использовать для лечения гипертонии, гиперлипидемии, сердечно-сосудистых заболеваний, нестабильной стенокардии (или стенокардии), тромбоза, агрегации тромбоцитов, индуцированной тромбином и/или последствиями тромбоза и/или образованием атеросклеротических бляшек;
- респираторного и ORL аппарата, в частности, включая астму, острый респираторный дистресс-синдром, сенную лихорадку, аллергический ринит и хроническое обструктивное заболевание легких. Соединения в соответствии с настоящим изобретением также можно использовать для лечения аллергии;
- центральной нервной системы и, в частности, таких как болезнь Альцгеймера и любые другие формы деменции, болезнь Паркинсона, болезнь Крейтцфельдта-Якоба, рассеянный склероз и менингит;
- кожи и, в частности, таких как крапивница, склеродермия, контактный дерматит, атопический дерматит, псориаз, ихтиоз, акне и другие формы фолликулита, розовые угри и алопеция;
- также иммунных заболеваний и, в частности, таких как красная волчанка, заболевания щитовидной железы, аутоиммунные заболевания надпочечников и аутоиммунный гастрит, витилиго и очаговая алопеция;
- воспалений, сопровождающих бактериальные, вирусные или грибковые инфекции, в частности, таких как туберкулез, сепсис, лихорадка, ВИЧ, независимо от места инфекции, герпес, цитомегаловирус и гепатиты A, B и C;
- трансплантации или отторжения трансплантата, такого как трансплантат почки, печени, сердца, легкого, поджелудочной железы, костного мозга, роговицы, кишечника или кожи (кожный аллотрансплантат, гомотрансплантат или гетеротрансплантат, и т.д.).
Кроме того, эти соединения можно использовать для лечения боли, независимо от ее происхождения: послеоперационной боли, нейромышечной боли, головной боли, раковой боли, зубной боли, костно-суставной боли.
Эти соединения можно использовать для модуляции пигментации и, как результат, для:
- лечения заболеваний с нарушениями пигментации и особенно доброкачественных дерматозов, таких как витилиго, альбинизм, меланодермия, лентиго, веснушки, меланоцитарный невус и все пост-воспалительные пигментациии; а также пигментированных опухолей, таких как меланомы и их локальные (проникающие молекулы), региональные или системные метастазы;
- защиты от солнца с целью предотвращения:
- вредных эффектов солнечных лучей, таких как актиническая эритема, кожное старение, рак кожи (спиноцеллюлярный, базоцеллюлярный и меланома), и, в частности, заболеваний, которые ускоряют их возникновение (пигментная ксеродерма, синдром базоцеллюлярного невуса и семейная меланома);
- фотодерматозов, вызванных экзогенными фотосенсибилизаторами, и, в частности, тех, которые вызваны контактными фотосенсибилизаторами (например, фурокумаринами, галогенированными салициланилидами и производными и местными сульфамидами и производными), или тех, которые вызваны системными фотосенсибилизаторами (например, псораленами, тетрациклинами, сульфамидами, фенотиазинами, налидиксовой кислотой и трициклическими антидепрессантами);
- обострения дерматозов с фоточувствительностью и особенно
- усугубляющихся при свете дерматозов, (например, красная волчанка, повторяющийся герпес, врожденный пойкилодермические или телеангиэктатические состояния с фоточувствительностью (синдром Блума, синдром Кокейна или синдром Ротмунда-Томсона), актинический красный плоский лишай, актиническая гранулема, поверхностный диссеминирующий актинический порокератоз, розовые угри, юношеские угри, буллезный дерматоз, болезнь Дарье, лимфома кожи, псориаз, атопический дерматит, контактная экзема, фолликулярный муциноз, полиморфная эритема, стойкая лекарственная эритема, кожная лимфоцитома, ретикулярный эритематозный муциноз и меланодермия);
- дерматозов с фоточувствительностью из-за дефицита защитной системы с аномалиями образования или распределения меланина (например, глазнично-кожный альбинизм, фенилкетонурия, гипопитуитаризм, витилиго и «пегая кожа»), с дефицитом систем репарации ДНК (например, пигментная ксеродерма и синдром Кокейна),
- дерматозов с фоточувствительностью из-за метаболических аномалий, например, кожная порфирия (например, поздняя кожная порфирия, смешанная порфирия, эритропоэтическая фотопорфирия, врожденная эритропоэтическая порфирия (болезнь Гюнтера) и эритропоэтическая корпопорфирия), пеллагра или пеллагроидная эритема (например, пеллагра, пеллагроидная эритема и нарушения метаболизма триптофана);
- обострений идиопатического фотодерматоза, и, в частности, такого как PMLE (полиморфный фотодерматоз), доброкачественный летний фотодерматоз, актинический пруриго, хроническая чувствительность к свету (актинический ретикулоид, остаточная фоточувствительность и фоточувствительная экзема), солнечная крапивница, световая оспа, юношеские весенние высыпания и солнечный зуд);
- изменения цвета кожи или волос на голове и волос на теле, и, в частности, путем тонирования кожи за счет увеличения синтеза меланина или ее обесцвечивания путем вмешательства в синтез меланина, а также путем предотвращения обесцвечивания или поседения волос на голове или волос на теле (например, поседение и «пегая кожа»);
- изменения цвета волос на голове и волос на теле в косметических целях.
Эти соединения могут быть полезными для модификации функции сальных желез для:
- лечения гиперсеборейных заболеваний и, в частности, акне, себорейного дерматита, жирной кожи и жирных волос, гиперсебореи при болезни Паркинсона и эпилепсии и гиперандрогенизма;
- лечения заболеваний, связанных со снижением секреции сальных желез, и, в частности, патологической сухости кожи и всех форм сухой кожи;
- регулирования доброкачественной или злокачественной пролиферации себоцитов и сальных желез;
- лечения воспалительных заболеваний, таких как сальные фолликулы, и, в частности, акне, фурункулы, карбункулы и фолликулит.
Настоящее изобретение также относится к применению соединения общей формулы (I), определенного выше, для получения лекарственного препарата для лечения расстройства, связанного с дисфункцией MC4R рецептора.
Соединения по настоящему изобретению также можно использовать для лечения нейродегенеративных расстройств, включая депрессию, беспокойство, компульсивные расстройства (такие, как обсессвино-компульсивные расстройства), неврозы, психозы, бессонницу и нарушение сна, синдром апноэ во сне и злоупотребление лекарственными средствами.
Эти соединения можно использовать для лечения мужской или женской сексуальной дисфункции. Мужские сексуальные дисфункции включают, но не ограничиваются этим, импотенцию, потерю либидо и эректильную дисфункцию.
Женские сексуальные дисфункции включают, но не ограничиваются этим, расстройства сексуальной стимуляции или расстройства, связанные с желанием, сексуальной восприимчивостью, оргазмом, и расстройства основных точек сексуальной функции. Женские сексуальные дисфункции также могут включать боли, преждевременные роды, дисменорею, чрезмерно обильные менструации и эндометриоз.
Соединения в соответствии с настоящим изобретением также можно использовать для лечения расстройств, связанных с массой тела, включая, но не ограничиваясь этим, ожирение и анорексию (например, изменение или ухудшение аппетита, метаболизм селезенки, или неконтролируемое потребление жира или углеводов); сахарный диабет (путем толерантности к дозам глюкозы и/или снижения резистентности к инсулину).
Соединения также можно использовать для лечения рака и, в частности, рака легкого, рака предстательной железы, рака кишечника, рака молочной железы, рака яичника, рака кости или ангиогенеза, включая образование или рост солидных опухолей.
Объектом настоящего изобретения также является фармацевтическая композиция, предназначенная, в частности, для лечения указанных выше состояний, которая отличается тем, что содержит, в фармацевтически приемлемом носителе, который является совместимым со способом введения, выбранным для этого, соединение общей формулы (I) в одной из его таутомерных форм или его соль с фармацевтически приемлемой кислотой.
Термин "фармацевтически приемлемый носитель" означает среду, которая является совместимой с кожей, слизистыми оболочками и наружными покровами.
Введение композиции в соответствии с настоящим изобретением можно осуществить перорально, энтерально, парентерально, местным путем или глазным путем. Предпочтительно, фармацевтическая композиция упакована в форму, подходящую для местного применения.
Для введения пероральным путем, композиция может быть в форме таблеток, гелевых капсул, таблеток с покрытием, сиропов, суспензий, растворов, порошков, гранул, эмульсий, суспензий, или микросфер, наносфер или липидных или полимерных везикул, которые делают возможным контролируемое высвобождение. Для введения парентеральным путем, композиция может быть в форме растворов или суспензий для перфузии или для инъекции.
Соединения в соответствии с настоящим изобретением, как правило, вводят при суточной дозе от около 0,01 мг/кг до 100 мг/кг массы тела, для введения одной или несколькими дозами.
Соединения применяют системно при концентрации, как правило, в пределах от 0,001% до 10% масс. и, предпочтительно, от 0,01% до 5% масс. в расчете на массу композиции.
Для введения местным путем, фармацевтическая композиция в соответствии с настоящим изобретением в большей степени предназначена для лечения кожи и слизистых оболочек и может быть в жидкой, пастообразной или твердой форме и, более конкретно, в форме мазей, кремов, молочка, помад, порошков, пропитанных прокладок, повязок, растворов, гелей, спреев, муссов, суспензий, карандашей, шампуней или моющих основ. Они также могут быть в форме суспензий микросфер или наносфер или липидных или полимерных везикул, или полимерных или гелевых пластырей, которые делают возможным контролируемое высвобождение.
Композиции, используемые для местного применения, имеют концентрацию соединения в соответствии с настоящим изобретением, как правило, в пределах от 0,001% до 10% масс. и, предпочтительно, в пределах от 0,01% до 5% масс. в расчете на общую массу композиции.
Соединения общей формулы (I) в соответствии с настоящим изобретением также находят применение в косметической области, в частности, для защиты против вредного воздействия солнечного света, для профилактики и/или борьбы со светоиндуцированным или возрастным старением кожи и наружных покровов.
Объектом настоящего изобретения, таким образом, также является композиция, включающая, в косметически приемлемом носителе, по меньшей мере, одно соединение общей формулы (I). Термин "косметически приемлемая среда" означает среду, которая является совместимой с кожей, слизистыми оболочками и наружными покровами.
Объектом настоящего изобретения также является косметическое применение композиции, включающей, по меньшей мере, одно соединение общей формулы (I), для профилактики и/или лечения признаков старения кожи.
Объектом настоящего изобретения также является косметическое применение композиции, включающей, по меньшей мере, одно соединение общей формулы (I), для гигиены тела или волос.
Косметическая композиция в соответствии с настоящим изобретением, содержащая, в косметически приемлемом носителе, соединение общей формулы (I) или его таутомерную форму или его соль с фармацевтически приемлемой кислотой, может быть, в частности, в форме крема, молочка, геля, суспензий микросфер или наносфер или липидных или полимерных везикул, пропитанных прокладок, растворов, спреев, муссов, карандашей, мыл, моющих основ или шампуней.
Концентрация соединения общей формулы (I) в косметической композиции, предпочтительно находится в пределах от 0,001% до 10% масс. в расчете на общую массу композиции.
Фармацевтические и косметические композиции, описанные выше, также могут содержать инертные добавки или даже фармакодинамически активные добавки, что касается фармацевтических композиций, или комбинации таких добавок и, в частности:
- смачивающие вещества;
- усилители вкуса и аромата;
- консерванты, такие как сложные эфиры пара-оксибензойной кислоты;
- стабилизаторы;
- регуляторы влажности;
- регуляторы pH;
- модификаторы осмотического давления;
- эмульгаторы;
- УФ-A и УФ-B фильтры;
- антиоксиданты, такие как α-токоферол, бутилгидроксианизол или бутилгидрокситолуол, супероксид-дисмутаза или убихинол;
- смягчающие вещества;
- увлажнители, такие как глицерин, ПЭГ-400, тиаморфолинон и его производные, или мочевина;
- средства против себореи или акне, такие как S-карбоксиметилцистеин, S-бензилцистеамин их соли или их производные, или бензоилпероксид.
Безусловно, специалист в данной области сможет выбрать необязательное соединение(соединения) для добавления в эти композиции таким образом, чтобы предусматриваемое добавление не влияло, или существенно не влияло, негативным образом на выгодные свойства, неотъемлемо связанные с настоящим изобретением.
Далее некоторые примеры получения соединений общей формулы (I) в соответствии с настоящим изобретением и результаты испытаний биологической активности этих соединений, представлены в качестве иллюстрации, без какого-либо ограничения их сущности.
Следующие далее примеры описывают получение некоторых соединений в соответствии с настоящим изобретением. Эти примеры не являются ограничивающими и служат исключительно для иллюстрации настоящего изобретения. Номера представленных соединений соответствуют номерам, указанным в таблице, представленной ниже, в которой представлены химические структуры и физические свойства ряда соединений в соответствии с настоящим изобретением.
Использованы следующие сокращения:
-DMAP: Диметиламинопиридин
-EDC: Гидрохлорид 1-этил-(3-Диметиламинопропил)карбодиимида
-HOBt: 1-Гидрокси-1,2,3-бензотриазол
-TBTU: Тетрафторборат N,N,N',N'-тетраМетил-O-(бензотриазол-1-ил)урония
-Fmoc: 6-Флуоренилметоксикарбонил
-DBU: 1,5-Диазабицикло(5,4,0)ундец-5-ен
-NaBH3CN: Цианоборгидрид натрия
-LiAlH4: Литийалюминийгидрид
-Rh/Al2O3: Родий на оксиде алюминия
-NaHCO3: Гидрокарбонат натрия
-NH4Cl: Хлорид аммония
-NaCl: Хлорид натрия
-MgSO4: Сульфат магния
-Na2SO4: Сульфат натрия
-CuSO4: Сульфат меди
-NaOH: Гидроксид натрия
-EtOAc: Этилацетат
-ДХМ: Дихлорметан
-ДМФА : Диметилформамид
-MeOH: Метанол
-ТГФ: Тетрагидрофуран
-TLC: Тонкослойная хроматография
Материалы и методы:
Метод препаративной ВЭЖХ:
Колонка Modulo-cart strategy C18 100×21,2 мм, 5 мкм
УФ детектор: 210-400 нм
Скорость потока: 17 мл/мин
Растворитель A: H2O+0,05 ТФУК
Растворитель B: CH3CN+0,05 ТФУК
Градиент:
|
Методы ВЭЖХ:
Метод A1
Колонка Atlantis C18 150×2,1 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,3 мл/мин
Растворитель A: CH3CN+0,1 ТФУК
Растворитель B: H2O+0,1 ТФУК
Градиент:
|
Метод A
Колонка Gemini 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,05 ТФУК
Растворитель B: H2O+0,05 ТФУК
Градиент:
|
Метод B
Колонка Gemini 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,05 ТФУК
Растворитель B: H2O+0,05 ТФУК
Градиент:
|
Метод C
Колонка Gemini 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Метод D
Колонка Gemini 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Метод E
Колонка
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: MeOH+0,1 ТФУК
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Метод F
Колонка Xbridge phenyl 250*4,6 мм 5 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,6 мл/мин
Растворитель A: MeOH+25 мМ NH4OAc
Растворитель B: H2O+25 мМ NH4OAc
Градиент:
|
Метод G
Колонка Xbridge phenyl 150*2,1 мм 3,5 мкм
УФ детектор: 190-450 нм
Скорость потока: 1,0 мл/мин
Растворитель A: MeOH 95%+ H2O 5% +25 мМ NH4OAc
Растворитель B: H2O+25 мМ NH4OAc
Градиент:
|
Метод I
Колонка Gemini 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,02 ТФУК
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Метод J
Колонка Atlantis dC18 250×4,6 мм, 5 мкм
УФ детектор: 190-450 нм
Скорость потока: 1,0 мл/мин
Растворитель A: CH3CN+0,02 ТФУК
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Метод K
Колонка Gemini C18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,3 мл/мин
Растворитель A: MeOH 94% + H2O 6% + 10 мМ (NH4)2CO3
Растворитель B: H2O + 10 мМ (NH4)2CO3
Градиент:
|
Метод L
Колонка Gemini C18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,3 мл/мин
Растворитель A: MeOH 94% + H2O 6% + 10 мМ (NH4)2CO3
Растворитель B: H2O + 10 мМ (NH4)2CO3
Градиент:
|
Метод M
Колонка Gemini С18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,05 ТФУК
Растворитель B: H2O+0,05 ТФУК
Градиент:
|
Метод N
Колонка Gemini С18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,1% HCOOH
Растворитель B: H2O+0,1% HCOOH
Градиент:
|
Метод O
Колонка Gemini С18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,3 мл/мин
Растворитель A: CH3CN+0,05 ТФУК
Растворитель B: H2O+0,05 ТФУК
Градиент:
|
Метод P
Колонка Phenomenex Gemini C6-Phenyl 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: H2O+0,05 ТФУК
Растворитель B: CH3CN+0,05 ТФУК
Градиент:
|
Метод Q
Колонка Gemini С18 150×3 мм, 3 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: CH3CN+0,1% HCOOH
Растворитель B: H2O+0,1% HCOOH
Градиент:
|
Метод R
Колонка Thermohypersil Hypurity С18 150×4,6 мм, 5 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,5 мл/мин
Растворитель A: H2O+0,05 ТФУК
Растворитель B: CH3CN+0,05 ТФУК
Градиент:
|
Метод S
Колонка Atlantis T3 150×4,6 мм, 5 мкм
УФ детектор: 190-450 нм
Скорость потока: 0,3 мл/мин
Растворитель A: H2O+0,05 ТФУК
Растворитель B: CH3CN+0,05 ТФУК
Градиент:
|
Метод U
Колонка Atlantis T3 С18 150×2,1 мм, 3 мкм
УФ детектор: 190-900 нм
Скорость потока: 0,3 мл/мин
Растворитель A: CH3CN+0,02 ТФУК
Растворитель B: H2O+0,02 ТФУК
Градиент:
|
Пример 1: 1-[(S)-2-(4-Бутирил-4-фенилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 1)
1-1 1-{1-[(S)-2-Амино-3-(4-метоксифенил)пропионил]-4-фенилпиперидин-4-ил}бутан-1-он
К раствору, содержащему 11,9 г (28,5 ммоль) (S)-2-Fmoc-амино-3-(4-метокси-фенил)пропионовой кислоты, растворенной в 120 мл дихлорметана и 10 мл диметилформамида, добавляли 5,51 г (38,9 ммоль) EDC и 5,25 г (38,9 ммоль) HOBt. После перемешивания в течение 30 минут при комнатной температуре добавляли раствор 6,95 г (26,1 ммоль) гидрохлорида 1-(4-фенилпиперидин-4-ил)-бутан-1-она и 18 мл триэтиламина в 150 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов и затем добавляли насыщенный водный раствор гидрокарбоната натрия. Органические соединения экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния и затем фильтровали и растворители выпаривали. Неочищенный продукт хроматографировали на силикагеле (элюент: смесь 9/1 дихлорметан/метанол). Получали 4,7 г 1-{1-[(S)-2-амино-3-(4-метоксифенил)-пропионил]-4-фенилпиперидин-4-ил}бутан-1-она в форме оранжевого масла с выходом 44%.
1-2 1-[(S)-2-(4-Бутирил-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
К раствору, содержащему 100 мг (0,245 ммоль) 1-{1-[(S)-2-амино-3-(4-метокси-фенил)пропионил]-4-фенилпиперидин-4-ил}бутан-1-она в 10 мл дихлорметана, добавляли 64 мкл диизопропилэтиламина и затем 74 мг (0,368 ммоль) 4-нитрофенилхлорформиата. Смесь перемешивали при комнатной температуре в течение 1 часа 30 минут. К этой смеси добавляли раствор, содержащий 90 мг (0,489 ммоль) гистаминдигидрохлорида и 0,15 мл диизопропилэтиламина в 5 мл дихлорметана и 2 мл диметилформамида.
После перемешивания при комнатной температуре в течение 3 часов растворители выпаривали и полученный неочищенный продукт очищали при помощи препаративной ВЭЖХ. Получали 37 мг трифторацетата 1-[(S)-2-(4-бутирил-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины в форме белого порошка с выходом 37%.
ВЭЖХ: (метод A1); время удерживания: 15,87 минут, 98%, M+H: 545
Пример 2: 1-[2-(4-Циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 3)
2-1 Метил (S)-2-амино-3-(4-метоксифенил)пропионат
К 10 г (33,8 ммоль) (S)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)пропионовой кислоты по каплям в течение 30 минут добавляли 75 мл метанола и затем 10 мл серной кислоты. Через 30 часов реакционную смесь подщелачивали до pH 8-9 путем добавления 10 н. водного раствора гидроксида натрия с последующим добавлением насыщенного раствора гидрокарбоната натрия. Органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния и затем фильтровали и растворители выпаривали. Получали 6,36 г метил (S)-2-амино-3-(4-метоксифенил)пропионата в форме коричневого масла с выходом 90%.
2-2 Метил (S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропаноат
К 5,08 г (24,3 ммоль) метил (S)-2-амино-3-(4-метоксифенил)пропионата добавляли 15 мл дихлорметана. Реакционную смесь погружали в ванну с холодной водой. Добавляли 7,34 г (36,4 ммоль) 4-нитрофенилхлорформиата с последующим добавлением 6,33 мл диизопропилэтиламина. После нагревания до комнатной температуры реакционную смесь перемешивали в течение 2 часов. Реакцию останавливали путем добавления воды с последующей экстракцией дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе. Получали 12 г желтого масла. К этим 12 г добавляли 10 мл диметилформамида и смесь затем нагревали до 80°C. Добавляли 8,95 г (48,6 ммоль) гистаминдигидрохлорида с последующим добавлением по каплям 14,8 мл (85,1 ммоль) диизопропилэтиламина. После охлаждения до комнатной температуры растворители выпаривали и неочищенный продукт хроматографировали на силикагеле (элюент: смесь 85/15 дихлорметан/метанол). Получали 5,6 г метил (S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропаноата в форме желтого масла с выходом 67%.
2-3 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропановая кислота
К 500 мг (1,44 ммоль) метил (S)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропаноата добавляли 500 мг гидроксида лития, 7 мл тетрагидрофурана и 2 мл воды. Реакционную смесь помещали в микроволновой реактор при перемешивании при 100°C в течение 10 минут. Осуществляли еще семь идентичных испытаний. Различные образцы для испытаний объединяли и концентрировали досуха. Полученный неочищенный продукт очищали при помощи фильтрации на слое силикагеля (элюент: смесь 1/1 дихлорметан/метанол). Получали 2,73 г 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропановой кислоты в форме бледно-желтого порошка с выходом 70%.
2-4 1-[2-(4-Циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил] мочевина
К 300 мг (0,90 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропановой кислоты, растворенной в 3,5 мл дихлорметана и 1,5 мл диметилформамида, добавляли 0,46 мл (2,7 ммоль) диизопропилэтиламина, 318 мг (0,99 ммоль) TBTU и 220 мг (0,99 ммоль) 4-циано-4-фенилпиперидингидрохлорида. Через 16 часов раствор промывали насыщенным раствором гидрокарбоната натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи фильтрации на слое силикагеля (элюент: смесь 7/3 дихлорметан/метанол). Получали 75 мг 1-[2-(4-циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины в форме белого порошка с выходом 17%.
1H ЯМР/DMSOD6 100°C: δ = 1,92-2,13 (м, 2H); 2,62 (т, J=7,2 Гц, 2H); 2,72-2,99 (м, 8H); 3,26 (ушир.кв, J=5,6-7,2 Гц, 2H); 3,67 (с, 3H); 4,87 (ушир.кв, J=8,4 Гц, 1H); 5,95 (ушир.т, 1H); 6,10 (д, J=8,4 Гц, 1H); 6,74 (с, 1H); 6,83 (д, J=8,8 Гц, 2H); 7,11-7,14 (м, 2H); 7,34-7,53 (м, 7H).
Пример 3: Этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат трифторацетат (соединение 6)
3-1-1 Этил пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-карбоксилат
К 2,00 г (6,42 ммоль) пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-карбоновой кислоты в 10 мл толуола добавляли 1,92 мл (12,8 ммоль) DBU и 1,04 мл (12,8 ммоль) йодоэтана. Смесь перемешивали в микроволновом реакторе в течение 10 минут при 120°C. К реакционной смеси добавляли дихлорметан и органическую фазу промывали насыщенным водным раствором NaHCO3. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 9/1 гептан/этилацетат). Получали 1,92 г этил пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-карбоксилата в форме бесцветного масла с выходом 88%.
3-1-2 Этил пиперидин-4-циклогексил-4'-карбоксилат
К 1,90 г (5,60 ммоль), растворенным в 8 мл дихлорметана, при 0°C добавляли 6 мл трифторуксусной кислоты. Через 4 часа растворители выпаривали и реакционную смесь обрабатывали при помощи EtOAc для ее поглощения и затем промывали 1 н. раствором гидроксида натрия. Органическую фазу сушили над MgSO4, фильтровали и упаривали. Получали 1,21 г этил пиперидин-4-циклогексил-4'-карбоксилата в форме белого порошка с выходом 90%.
3-2 Трифторацетат этил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилата
К 2,0 г (6,02 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропановой кислоты (см. получение 2-3), растворенной в 90 мл диметилформамида, добавляли 1,93 г (6,02 ммоль) TBTU, 2,1 мл (12,0 ммоль) диизопропилэтиламина и 2,05 г (5,80 ммоль) этил пиперидин-4-циклогексил-4'-карбоксилата. Через 6 часов раствор промывали 1 н. раствором гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи препаративной ВЭЖХ. Получали 530 мг трифторацетата 1-[2-(4-циано-4-фенилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]-мочевины в форме белого порошка с выходом 14%.
1H ЯМР/DMSOD6 100°С: δ = 0,87-0,90 (м, 2H); 1,07-1,37 (м, 13Н); 1,57-1,64 (м, 3H); 1,72-1,75 (м, 2H), 1,92-1,96 (м, 2H); 2,66-2,82 (м, 4H); 3,29-3,35 (м, 2H); 3,74 (с, 3H); 4,13 (кв, J=6,8-7,2 Гц, 2H); 4,80 (м, 1H); 6,00-6,10 (м, 2H); 6,82 (д, J=8,4 Гц, 2H); 7,07 (д, J=8,4 Гц, 2H); 7,30 (с, 1H); 8,80 (с, 1H).
Пример 4: 1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]-3-фенилазетидин-3-ил бутират (соединение 8)
4-1-1 3-Гидрокси-3-фенилазетидин-1-трет-бутоксикарбонил
В раствор, погруженный в ванну при -500C, содержащий 500 мг (2,92 ммоль) 3-оксо-азетидин-1-трет-бутоксикарбонила в 10 мл ТГФ, по каплям добавляли 3,9 мл (11,7 ммоль) 3M раствора фенилмагнийбромида в диэтиловом эфире. Смесь перемешивали в течение 1 часа при -50°C и гидролизовали путем добавления насыщенного раствора хлорида аммония. После нагревания до комнатной температуры добавляли 1 н. раствор хлористоводородной кислоты с последующей экстракцией этилацетатом. Органическую фазу сушили и упаривали досуха. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 253 мг в форме белого порошка с выходом 35%.
4-1-2 3-Бутирилокси-3-фенилазетидин-1-трет-бутоксикарбонил
К раствору, содержащему 200 мг (0,80 ммоль) 3-гидрокси-3-фенилазетидин-1-трет-бутоксикарбонила в 4 мл дихлорметана, добавляли 98 мг (0,80 ммоль) DMAP и 0,13 мл пиридина. После перемешивания при комнатной температуре в течение 10 минут добавляли 0,26 мл масляного ангидрида. Через 4 часа добавляли насыщенный раствор хлорида аммония с последующей экстракцией дихлорметаном. Органическую фазу сушили и упаривали досуха. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 186 мг в форме масла с выходом 73%.
4-1-3 3-Фенилазетидин-3-илтрифторацетатбутират
К раствору, содержащему 183 мг (0,57 ммоль) 3-бутирилокси-3-фенилазетидин-1-трет-бутоксикарбонила, растворенного в 8 мл дихлорметана, добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 90/10 дихлорметан/метанол). Получали 88 мг в форме бледно-желтого масла с выходом 46%.
4-2 1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]-3-фенилазетидин-3-илбутират
К 80 мг (0,24 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), растворенной в 2,75 мл дихлорметана и 1,25 мл диметилформамида, добавляли 0,08 мл (0,48 ммоль) диизопропилэтиламина, 84 мг (0,26 ммоль) TBTU и 88 мг (0,26 ммоль) 3-фенилазетидин-3-илбутираттрифторацетата. Через 2 часа раствор промывали 1 н. раствором гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали при помощи препаративной ТСХ (элюент: смесь 9/1 дихлорметан/метанол). Получали 5,4 мг 1-[2-{3-[2-(1H-имидазол-4-ил)этил]-уреидо}-3-(4-метоксифенил)пропионил]-3-фенилазетидин-3-илбутирата с выходом 2%.
ВЭЖХ: (метод B); время удерживания: 12,15 минут, 86%, M+H: 534.
Пример 5 1-[2-(3-Бутокси-3-фенилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 11)
5-1-1 3-Гидрокси-3-фенилазетидин-1-трет-бутоксикарбонил
В раствор, погруженный в ванну при -50°C, содержащий 500 мг (2,92 ммоль) 3-оксо-азетидин-1-трет-бутоксикарбонила в 10 мл ТГФ, по каплям добавляли 3,9 мл (11,7 ммоль) 3M раствора фенилмагнийбромида в диэтиловом эфире. Смесь перемешивали в течение 1 часа при -50°C и гидролизовали путем добавления насыщенного раствора хлорида аммония. После нагревания до комнатной температуры добавляли 1 н. раствор хлористоводородной кислоты с последующей экстракцией этилацетатом. Органическую фазу сушили и упаривали досуха. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 253 мг в форме белого порошка с выходом 35%.
5-1-2 3-Бутокси-3-фенилазетидин-1-трет-бутоксикарбонил
К суспензии 300 мг 60% раствора NaH в 3 мл ДМФА, погруженной в ванну, при 0°C по каплям добавляли раствор 1 г (4,0 ммоль) 3-гидрокси-3-фенилазетидин-1-трет-бутоксикарбонила, растворенного в 5 мл ДМФА. По каплям добавляли 2,5 мл н-йодобутана. Реакционную смесь перемешивали при 0°C в течение 15 минут и в течение 72 часов при комнатной температуре. Смесь гидролизовали путем добавления насыщенного раствора хлорида аммония с последующей экстракцией этилацетатом. Органическую фазу сушили и упаривали досуха. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 500 мг в форме бледно-желтого масла с выходом 41%.
5-1-3 3-Бутокси-3-фенилазетидинтрифторацетат
К раствору, содержащему 500 мг (1,64 ммоль) 3-бутокси-3-фенилазетидин-1-трет-бутоксикарбонила, растворенного в 5 мл дихлорметана, добавляли 1 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 90/10 дихлорметан/метанол). Получали 400 мг в форме бледно-желтого порошка с выходом 76%.
5-2 1-[2-(3-Бутокси-3-фенилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
К 57 мг (0,17 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), растворенной в 4 мл дихлорметана и 1,5 мл диметилформамида, добавляли 0,12 мл (0,68 ммоль) диизопропилэтиламина, 61 мг (0,19 ммоль) TBTU и 55 мг (0,17 ммоль) 3-бутокси-3-фенилазетидинтрифторацетата. Через 3 часа раствор промывали 1 н. раствором гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи препаративной ТСХ (элюент: смесь 9/1 дихлорметан/метанол). Получали 3 мг 1-[2-(3-бутокси-3-фенилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины с выходом 3%.
ВЭЖХ: (метод C); время удерживания: 11,6 минут, 93%, M+H: 520.
Пример 6: Метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксиламид (соединение 12)
6-1 4-Метилкарбамоил-4-циклогексилпиперидин
К 70 мг (0,22 ммоль) пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-метилкарбамоила, растворенного в 2 мл дихлорметана, при 0°C добавляли 1 мл трифторуксусной кислоты. Через 1 час растворители выпаривали и реакционную смесь обрабатывали дихлорметаном для ее поглощения и промывали 1 н. раствором гидроксида натрия. Органическую фазу сушили над MgSO4, фильтровали и упаривали. Получали 44 мг 4-метилкарбамоил-4-циклогексилпиперидина в форме желтого масла с выходом 90%.
6-2 Метил 4-циклогексил-1[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксиламид
К 66 мг (0,20 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), растворенной в 2 мл дихлорметана и 0,5 мл диметилформамида, добавляли 0,06 мл (0,40 ммоль) диизопропилэтиламина, 71 мг (0,22 ммоль) TBTU и 44 мг (0,20 ммоль) 4-метилкарбамоил-4-циклогексилпиперидина. Через 2 часа раствор промывали 1 н. раствором гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи препаративной ТСХ (элюент: смесь 8/2 дихлорметан/метанол). Получали 11 мг метил 4-циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксиламида в форме белого порошка с выходом 10%.
ВЭЖХ: (метод D); время удерживания: 11,82 минуты, 97%, M+H: 539.
Пример 7: 1-[2-(3-Циклогексанкарбонилазетидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 13):
7-1-1 3-(Метоксиметилкарбамоил)азетидин-1-трет-бутоксикарбонил
2 г (9,94 ммоль) 1-трет-бутоксикарбонил-3-азетидинкарбоновой кислоты растворяли в 8 мл дихлорметана и добавляли 3,19 г (9,94 ммоль) TBTU и затем 4 мл диметилформамида. Добавляли 0,97 г (9,94 ммоль) N,O-диметилгидроксиламина в 10 мл дихлорметана и 5,17 мл (29,8 ммоль) диизопропилэтиламина. После перемешивания в течение 2 часов добавляли дихлорметан и смесь промывали насыщенным раствором NaHCO3 и затем 5% раствором лимонной кислоты. Органическую фазу сушили над MgSO4, фильтровали и концентрировали на роторном испарителе. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 1,9 г в форме бесцветного масла с выходом 78%.
7-1-2 3-Циклогексанкарбонилазетидин-1-трет-бутоксикарбонил
К 301 мг (1,23 ммоль) 3-(метоксиметилкарбамоил)азетидин-1-трет-бутоксикарбонила в 5 мл ТГФ при 0°C добавляли 1,48 мл (1,48 ммоль) 1 M раствора циклогексилмагнийбромида в ТГФ. Далее добавляли 3,35 мл (3,35 ммоль) реагента магния, необходимого для исчезновения исходного материала. Добавляли дихлорметан, органические фазы промывали насыщенным раствором NH4Cl и затем насыщенным раствором NaCl и затем сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 205 мг в форме бесцветного масла с выходом 62%.
7-1-3 3-Циклогексанкарбонилазетидин
К раствору, содержащему 192 мг (0,72 ммоль) 3-циклогексанкарбонилазетидин-1-трет-бутоксикарбонила, растворенного в 8 мл дихлорметана, добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали. Реакционную смесь экстрагировали дихлорметаном в присутствии 1 н. раствора гидроксида натрия. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Получали 95 мг в форме белого порошка с выходом 79%.
7-2 1-[2-(3-Циклогексанкарбонилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
К 189 мг (0,57 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропановой кислоты (см. получение 2-3), растворенной в 3,5 мл дихлорметана и 1,0 мл диметилформамида, добавляли 0,2 мл (1,14 ммоль) диизопропилэтиламина, 202 мг (0,63 ммоль) TBTU и 95 мг (0,57 ммоль) 3-циклогексанкарбонилазетидина. Через 5 часов раствор промывали 1 н. раствором гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи препаративной ТСХ (смесь 9/1 дихлорметан/метанол). Получали 8,5 мг 1-[2-(3-циклогексанкарбонилазетидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины с выходом 3%.
ВЭЖХ: (метод E); время удерживания: 12,36 минут, 86%, M+H: 482.
Пример 8: Этил 4-циклогексил-1-[2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]-уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат (соединение 14):
8-1-1 2,6,7,8-Тетрагидроимидазо[1,5-c]пиримидин-5-он
К суспензии, содержащей 500 мг (4,5 ммоль) гистамина в 5 мл ацетонитрила, добавляли 730 мг (4,5 ммоль) N,N'-карбонилдиимидазола и смесь нагревали при 80°C в течение 16 часов. После охлаждения до комнатной температуры растворитель выпаривали и добавляли 2,5 мл этанола. Продукт осаждался, и его оставляли при перемешивании при 0°C в течение 1 часа. Продукт отфильтровывали, промывали ледяным этанолом и сушили в течение ночи в вакуумной печи. Получали 370 мг 2,6,7,8-тетрагидроимидазо[1,5-c]пиримидин-5-она с выходом 60%.
8-1-2 6-Этил-7,8-дигидро-6H-имидазо[1,5-c]пиримидин-5-он
К 500 мг (3,6 ммоль) 5,6,7,8-тетрагидро-5-оксоимидазо[1,5-c]пиримидина, растворенного в 5 мл ДМФА, при комнатной температуре осторожно добавляли 173 мг (4,32 ммоль) гидрида натрия. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, и затем по каплям добавляли 0,35 мл (4,32 ммоль) йодоэтана. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, и растворители затем выпаривали. Остаток обрабатывали 1 н. раствором NaHCO3 и экстрагировали хлороформом. Объединенные органические фазы сушили при помощи MgSO4, фильтровали и упаривали. Полученное масло использовали непосредственно на следующей стадии.
8-1-3 Этил[2-(1H-имидазол-4-ил)этил]амингидрохлорид
К полученному неочищенному продукту с предыдущей стадии добавляли водный раствор гидроксида калия (173 мг (6,16 ммоль) в 5 мл воды) и смесь кипятили с обратным холодильником в течение одного часа. После охлаждения до комнатной температуры добавляли раствор концентрированной хлористоводородной кислоты до уровня pH 1. Воду выпаривали и остаток брали для поглощения в этанол. Этанольный раствор нагревали до 80°C и фильтровали в горячем состоянии. Фильтрат упаривали. Полученное твердое вещество затем осаждали из смеси 1/1 эфир/этанол и затем отфильтровывали в атмосфере азота. Получали 160 мг этил[2-(1H-имидазол-4-ил)этил]амингидрохлорида с выходом 21% за последние две стадии.
8-2 Метил (S)-3-(4-метоксифенил)-2-(4-нитрофеноксикарбониламино)пропионат
1 г (4,8 ммоль) метил (S)-2-амино-3-(4-метоксифенил)пропионата (см. получение 2-1) разбавляли в 30 мл дихлорметана. Раствор охлаждали в ванне с холодной водой и затем добавляли 1,4 г (7,2 ммоль) 4-нитрофенилхлорформиата и 1,2 мл (7,2 ммоль) диизопропилэтиламина. После нагревания до комнатной температуры реакционную смесь перемешивали в течение 2 часов. Раствор выливали в воду и затем экстрагировали дихлорметаном. Органическую фазу сушили и затем упаривали. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки.
8-3 Метил (S)-2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионат
0,14 г (0,38 ммоль) метил (S)-3-(4-метоксифенил)-2-(4-нитрофеноксикарбониламино)пропионата разбавляли в 2 мл ДМФА. Раствор нагревали до 80°C и затем добавляли 0,16 г (0,76 ммоль) этил[2-(1H-имидазол-4-ил)этил]амингидрохлорида и 0,13 мл (1,3 ммоль) диизопропилэтиламина. Через 5 минут реакционную смесь охлаждали до комнатной температуры и перемешивали в течение 15 минут. Добавляли толуол и растворители выпаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 8/2 дихлорметан/метанол). Получали 0,15 г метил (S)-2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионата в форме бесцветного масла с выходом 100%.
8-4 2-{3-Этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионовая кислота
150 мг (0,4 ммоль) метил (S)-2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионата разбавляли в 4 мл ТГФ и 1 мл воды и добавляли 150 мг (6 ммоль) гидроксида лития. Смесь нагревали в течение 10 минут при 100°C в микроволновом реакторе. Растворители выпаривали и остаток затем очищали на слое силикагеля (элюент: смесь 1/1 дихлорметан/метанол). Получали 100 мг 2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионовой кислоты в форме бледно-желтого порошка с выходом 69%.
8-5 Этил 4-циклогексилпиперидин-4-карбоксилаттрифторацетат
0,34 г (1 ммоль) этил пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-карбоксилата (см. получение 3-1-1) растворяли в 4 мл смеси 80/20 дихлорметан/трифторуксусная кислота. Раствор перемешивали в течение 2 часов при комнатной температуре и затем упаривали в атмосфере азота. Получали 0,5 г в форме бесцветного масла и использовали на следующей стадии без дополнительной очистки.
8-6 Этил 4-циклогексил-1-[2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
66 мг (0,19 ммоль) этил 4-циклогексилпиперидин-4-карбоксилаттрифторацетата разбавляли в 3 мл дихлорметана и 3 мл диметилформамида и добавляли 67 мг (0,21 ммоль) TBTU и 75 мг (0,21 ммоль) 2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионовой кислоты и 0,06 мл (0,38 ммоль) диизопропилэтиламина. Через 2 часа перемешивания при комнатной температуре добавляли водный 1 н. раствор NaOH и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили и затем упаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 95/5 дихлорметан/метанол). Получали 34 мг этил 4-циклогексил-1-[2-{3-этил-3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилата в форме желтого масла с выходом 30%.
ВЭЖХ: (метод F); время удерживания: 20,72 минут, 91%, M+H: 582.
Пример 9: N-Циклопропил-N-{1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-ил}пропионамид (соединение 15)
9-1-1 трет-Бутил 4-циклопропиламинопиперидин-1-карбоксилат
1 г (5 ммоль) трет-бутил 4-оксопиперидин-1-карбоксилата растворяли в 40 мл этанола с 10% уксусной кислотой. Добавляли 0,28 мл (4,2 ммоль) циклопропиламина. Через 30 минут добавляли 0,53 г (36 ммоль) NaBH3CN и реакционную смесь перемешивали в течение одного часа при комнатной температуре. Реакцию останавливали путем добавления водного раствора аммиака, и органические продукты затем экстрагировали этилацетатом. Органическую фазу сушили и затем упаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 55/45 гептан/этилацетат). Получали 0,8 г трет-бутил 4-циклопропиламино-пиперидин-1-карбоксилата в форме бесцветного масла с выходом 79%.
9-1-2 трет-Бутил 4-(циклопропилпропиониламино)пиперидин-1-карбоксилат
0,8 г (3,3 ммоль) трет-бутил 4-циклопропиламинопиперидин-1-карбоксилата, 0,48 мл (3,3 ммоль) триэтиламина и 0,56 мл (3,3 ммоль) пропионилхлорида растворяли в 50 мл ТГФ. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Реакцию останавливали путем добавления воды и органические продукты затем экстрагировали дихлорметаном. Органическую фазу сушили и затем упаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 6/4 гептан/этилацетат). Получали 0,87 г трет-бутил 4-(циклопропилпропиониламино)пиперидин-1-карбоксилата в форме бесцветного масла с выходом 89%.
9-1-3 N-Циклопропил-N-пиперидин-4-илпропионамид
0,87 г (2,9 ммоль) трет-бутил 4-(циклопропилпропиониламино)пиперидин-1-карбоксилата растворяли в 8 мл смеси 80/20 ДХМ/MeOH. Раствор перемешивали в течение 2 часов при комнатной температуре и затем вливали в раствор 1 н. NaOH и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом магния и затем упаривали. Получали 290 мг неочищенного продукта и использовали на следующей стадии без дополнительной очистки.
9-2 N-Циклопропил-N-{1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-ил}пропионамид
100 мг (0,51 ммоль) N-циклопропил-N-пиперидин-4-илпропионамида, 170 мг (0,51 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропановой кислоты (см. получение 2-3), 108 мг (0,56 ммоль) EDC и 76 мг (0,56 ммоль) HOBT растворяли в 5 мл ДМФА в атмосфере азота. Смесь перемешивали при комнатной температуре в течение ночи, и раствор затем промывали 5%-ным водным раствором лимонной кислоты и экстрагировали этилацетатом. Органическую фазу промывали водным 1 н. раствором NaOH и затем сушили и упаривали. Полученный неочищенный продукт очищали при помощи препаративной ВЭЖХ. Получали 2 мг N-циклопропил-N-{1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-ил}-пропионамида с выходом 8%.
ВЭЖХ: (метод G); время удерживания: 12,46 минут, 92%, M+H: 511.
Пример 10: 1-[-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевинатрифторацетат (соединение 17):
10-1-1 4-Фенил-1-(толуол-4-сульфонил)пиперидин-4-карбонитрил
17,2 г (89,8 ммоль) 4-метилбензолсульфонилхлорида, растворенного в 150 мл дихлорметана, добавляли к раствору 20 г (89,8 ммоль) 4-фенилпиперидин-4-карбонитрила и 28 мл триэтиламина в 200 мл дихлорметана. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакцию останавливали путем добавления 200 мл воды и затем экстрагировали дихлорметаном. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем брали для поглощения в диэтиловый эфир и отфильтровывали. Получали 30,3 г 4-фенил-1-(толуол-4-сульфонил)пиперидин-4-карбонитрила в форме белого порошка с выходом 99%.
10-1-2 1-[4-Фенил-1-(толуол-4-сульфонил)пиперидин-4-ил]-бутан-1-он
88 мл (176 ммоль) н-пропилмагнийхлорида добавляли к раствору 30 г (88 ммоль) 4-фенил-1-(толуол-4-сульфонил)пиперидин-4-карбонитрила в 500 мл толуола. Реакционную смесь перемешивали в течение 6 часов при 65-70°C и затем в течение ночи при комнатной температуре. Реакцию останавливали путем добавления 100 мл тетрагидрофурана и затем гидролизовали 1 н. раствором хлористоводородной кислоты и экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем брали для поглощения в диэтиловый эфир и отфильтровывали. 1,38 г исходного материала восстанавливали. Фильтрат концентрировали досуха и затем хроматографировали на силикагеле (элюент: смесь 80/20 гептан/этилацетат). Получали 17,8 г 1-[4-фенил-1-(толуол-4-сульфонил)пиперидин-4-ил]-бутан-1-она в форме белого порошка с выходом 50%.
10-1-3 4-Бутирил-4-фенилпиперидингидрохлорид
10 г (26 ммоль) 1-[4-фенил-1-(толуол-4-сульфонил)пиперидин-4-ил]-бутан-1-она суспендировали в 64 мл серной кислоты и 32 мл воды. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 48 часов. Реакцию контролировали при помощи ВЭЖХ. Для гомогенизации реакционной смеси добавляли 50 мл этанола с последующим добавлением 40 мл серной кислоты и нагревание продолжали в течение 24 часов. Реакцию останавливали путем добавления льда и подщелачивали при помощи раствора гидроксида натрия и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем брали для поглощения в диэтиловый эфир и осаждали при помощи 4н раствора хлороводорода в этилацетате. Получали 3,8 г 4-бутирил-4-фенилпиперидингидрохлорида в форме белого порошка с выходом 55%.
10-1-4 1-(4-Циклогексилпиперидин-4-ил)бутан-1-он
В аппарате Парра при давлении водорода 6 бар к раствору 100 мг (0,5 ммоль) 4-бутирил-4-фенилпиперидингидрохлорида в 10 мл диоксана добавляли 100 мг родия на оксиде алюминия и 0,2 мл уксусной кислоты. Реакционную смесь нагревали при 80°C в течение 12 часов. Реакцию останавливали и затем фильтровали через целит и промывали дихлорметаном. Растворители выпаривали и остаток переносили для поглощения в воду, подщелачивали при помощи 1 н. раствора гидроксида натрия и экстрагировали этилацетатом. Органические фазы сушили над сульфатом натрия. Растворители выпаривали и остаток хроматографировали на силикагеле (элюент: смесь 9/1 дихлорметан/метанол). Получали 51,2 мг 1-(4-циклогексилпиперидин-4-ил)-бутан-1-она в виде белого порошка с выходом 61%.
10-2 1-[2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевинатрифторацетат
29 мг (0,22 ммоль) HOBt и 41 мг (0,22 ммоль) EDC добавляли к раствору 62,5 мг (0,19 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропановой кислоты (см. получение 2-3) и 43,7 мг (0,19 ммоль) 1-(4-циклогексилпиперидин-4-ил)бутан-1-она в 2 мл диметилформамида. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакцию останавливали путем добавления 10 мл воды и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем очищали при помощи препаративной ВЭЖХ. Получали 63,7 мг 1-[2-(4-бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевинатрифторацетата в форме белой пены с выходом 59%.
ВЭЖХ: (метод J); время удерживания: 21,49 минут, 97%, M+H: 552.
Пример 11: 1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 18):
11-1-1 4-Гидрокси-4-фенилпиперидин-1-трет-бутоксикарбонил
2,5 г (12,5 ммоль) трет-бутил 4-оксопиперидин-1-карбоксилата растворяли в 100 мл ТГФ и погружали в ванну при -50°C. Добавляли 40 мл (40 ммоль) раствора фенилмагнийбромида. После перемешивания в течение 1 часа 30 минут добавляли насыщенный раствор NH4Cl и реакционную смесь затем экстрагировали этилацетатом. Органическую фазу промывали 5% лимонной кислотой и затем сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/этилацетат). Получали 1,84 г в форме белого порошка с выходом 53%.
11-1-2 4-Бутокси-4-фенилпиперидин-1-трет-бутоксикарбонил
Раствор 500 мг (1,80 ммоль) 4-гидрокси-4-фенилпиперидин-1-трет-бутоксикарбонила, растворенный в 7,5 мл ДМФА, погружали в ванну при 0°C, к раствору добавляли 196 мг (4,90 ммоль) 60% раствора NaH и затем 1 мл (8,7 ммоль) н-йодобутана. После перемешивания в течение 3 часов добавляли дополнительные 206 мг 60% раствора NaH и 1 мл н-йодобутана. Через 5 дней реакционную смесь экстрагировали этилацетатом и промывали насыщенным раствором NH4Cl. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 9/1 Гептан/этилацетат). Получали 233 мг в форме бесцветного масла с выходом 39%.
11-1-3 4-Бутокси-4-циклогексилпиперидин-1-трет-бутоксикарбонил
Раствор, содержащий 178 мг 5% Rh/Al2O3, 0,35 мл уксусной кислоты и 220 мг (0,66 ммоль) 4-бутокси-4-фенилпиперидин-1-трет-бутоксикарбонила в 18 мл диоксана, помещали под давление водорода 6 бар при 80°C на 18 часов. Реакционную смесь фильтровали, промывали дихлорметаном и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 95/5 гептан/EtOAc). Получали 139 мг в форме бесцветного масла с выходом 62%.
11-1-4 4-Бутокси-4-циклогексилпиперидинтрифторацетат
К раствору, содержащему 139 мг (0,41 ммоль) 4-бутокси-4-циклогексилпиперидин-1-трет-бутоксикарбонила, растворенного в 8 мл дихлорметана, добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали. Получали 241 мг соединения в форме бесцветного масла и использовали на следующей стадии без дополнительной очистки.
11-2 1-[2-(4-Бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
К 69 мг (0,21 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), растворенной в 2 мл ДМФА, добавляли раствор 71 мг (0,20 ммоль) 4-бутокси-4-циклогексилпиперидинтрифторацетата в 1,5 мл ДМФА, 44 мг (0,23 ммоль) EDC и 33 мг (0,24 ммоль) HOBt. Через 2 часа 30 минут реакционную смесь гидролизовали 2%-ным водным раствором лимонной кислоты и затем экстрагировали дихлорметаном. Органическую фазу затем промывали водным 1 н. раствором гидроксида натрия. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Это масло хроматографировали на силикагеле (элюент: смесь 8/2 ДХМ/MeOH). Получали 25 мг 1-[2-(4-бутокси-4-циклогексилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины с выходом 22%.
1H ЯМР/DMSOD6 100°C: δ = 0,87-0,94 (м, 4H); 1,06-1,23 (м, 5H); 1,33-1,68 (м, 12H); 1,72-1,80 (м, 3H); 2,63 (т, J=7,6 Гц, 2H); 2,71-2,83 (м, 4H); 2,91-3,09 (м, 2H); 3,19-3,28 (м, 4H); 3,73 (с, 3H); 4,84 (ушир.кв, J=6,4-8,4 Гц, 1H); 5,96-6,02 (м, 2H); 6,79 (д, J=5,6 Гц, 2H); 6,82 (с, 1H); 7,08 (д, J=8,0 Гц, 2H); 7,58 (с, 1H).
Пример 12: Этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат (соединение 24):
12-1-1 трет-Бутил 4-этил-4-этоксикарбонилпиперидин-1-карбоксилат
0,2 г (0,78 ммоль) трет-бутил 4-этоксикарбонилпиперидин-1-карбоксилата растворяли в 4 мл ТГФ при -10°C. По каплям добавляли 0,8 мл (1,56 ммоль) диизопропиламида лития. Через 15 минут добавляли 0,09 мл (1,2 ммоль) йодоэтана и смесь нагревали до комнатной температуры. Эту смесь перемешивали в течение 30 минут и затем вливали в насыщенный раствор хлорида аммония и экстрагировали этилацетатом. Органическую фазу сушили и затем концентрировали досуха. Полученное масло хроматографировали на силикагеле (элюент: смесь 95/5 гептан/этилацетат). Получали 183 мг трет-бутил 4-этил-4-этоксикарбонилпиперидин-1-карбоксилата в форме желтого масла с выходом 82%.
12-1-2 4-Этоксикарбонил-4-этилпиперидинтрифторацетат
183 мг (0,64 ммоль) трет-бутил 4-этил-4-этоксикарбонилпиперидин-1-карбоксилата разбавляли в 4 мл смеси 8/2 дихлорметан/трифторуксусная кислота. Раствор перемешивали при комнатной температуре в течение 1 часа и растворители затем выпаривали. Получали 140 мг 4-этоксикарбонил-4-этилпиперидинтрифторацетата в форме масла с выходом 73%.
12-2 Этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилат
0,194 г (0,59 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), 140 мг (0,47 ммоль) 4-этоксикарбонил-4-этилпиперидинтрифторацетата, 124 мг (0,65 ммоль) EDC и 88 мг (0,65 ммоль) HOBt растворяли в 4 мл ДМФА. Смесь перемешивали при комнатной температуре в течение 72 часов. Реакционную смесь промывали 1 н. водным раствором NaOH и затем экстрагировали дихлорметаном. Органическую фазу сушили и упаривали.
Полученное масло хроматографировали на силикагеле (элюент: смесь 90/10 ДХМ/MeOH). Получали 89 мг этил 4-этил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоксилата в виде бесцветного масла с выходом 30%.
ВЭЖХ: (метод M); время удерживания: 9,32 минут, 93%, M+H: 500.
Пример 13: Трифторацетат циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоновой кислоты (соединение 26)
13-1 Трифторацетат 4-циклогексилпиперидин-4-карбоновой кислоты
К раствору, содержащему 480 мг (1,54 ммоль) пиперидин-1-трет-бутоксикарбонил-4-циклогексил-4'-карбоновой кислоты, растворенной в 3 мл дихлорметана, добавляли при комнатной температуре 1 мл трифторуксусной кислоты. После выпаривания растворителей полученное масло использовали на следующей стадии.
13-2 Трифторацетат циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропионил]пиперидин-4-карбоновой кислоты
К раствору, содержащему 93 мг (0,28 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропановой кислоты и 100 мг (0,31 ммоль) трифторацетата 4-циклогексил-пиперидин-4-карбоновой кислоты в 0,8 мл диметилформамида, добавляли 0,16 мл диизопропилэтиламина и 90 мг (0,28 ммоль) TBTU. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакцию останавливали путем добавления воды и органические соединения экстрагировали дихлорметаном. Органическую фазу сушили и упаривали. Полученное масло очищали при помощи препаративной ВЭЖХ. Получали 35 мг трифторацетата циклогексил-1-[2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоновой кислоты с выходом 19%.
ВЭЖХ: (метод L); время удерживания: 14,75 минут, 96%, M+H: 526.
Пример 14: Трифторацетат 1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[3-(2-метилциклогексил)-3-пропоксиазетидин-1-ил]-2-оксоэтил}мочевины (соединение 27)
14-1-1 трет-Бутил 3-(2-метилциклогексил)-3-пропоксиазетидин-1-карбоксилат
170 мг (0,56 ммоль) трет-бутил 3-(2-метилциклогексил)-3-пропоксиазетидин-1-карбоксилата, полученного как описано в Примере 6-1-2, 48 мг 5% родия на оксиде алюминия, 5 мл метанола и 0,6 мл уксусной кислоты вводили в аппарат Парра. Смесь перемешивали при давлении водорода 4 бар при 90°C в течение 7 дней. Реакционную смесь фильтровали и промывали дихлорметаном и затем концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле (элюент: смесь 95/5 гептан/этилацетат). Получали 96 мг трет-бутил 3-(2-метилциклогексил)-3-пропокси-азетидин-1-карбоксилата в виде бесцветного масла с выходом 55%.
14-1-2 3-(2-Метилциклогексил)-3-пропоксиазетидин трифторацетат
91 мг (0,29 ммоль) трет-бутил 3-(2-метилциклогексил)-3-пропоксиазетидин-1-карбоксилата растворяли в 8 мл дихлорметана. Реакционную смесь погружали в ледяную ванну. Добавляли 2 мл трифторуксусной кислоты. Через 1 час растворители выпаривали. 113 мг в виде коричневого масла использовали на следующей стадии без дополнительной очистки.
14-2 Трифторацетат 1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[3-(2-метилциклогексил)-3-пропоксиазетидин-1-ил]-2-оксоэтил}мочевины
К 97 мг (0,29 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)-пропановой кислоты (см. получение 2-3), растворенной в 2 мл ДМФА, добавляли 94 мг (0,29 ммоль) 3-(2-метилциклогексил)-3-пропоксиазетидинтрифторацетата, 70 мг (0,37 ммоль) EDC и 45 мг (0,33 ммоль) HOBt. Через 3 часа реакционную смесь гидролизовали водным 1 н. раствором гидроксида натрия и затем экстрагировали дихлорметаном. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт очищали при помощи препаративной ВЭЖХ. Получали 33 мг трифторацетата 1-[2-(1H-имидазол-4-ил)этил]-3-{1-(4-метоксибензил)-2-[3-(2-метилциклогексил)-3-пропоксиазетидин-1-ил]-2-оксоэтил}мочевины с выходом 18%.
1H ЯМР/DMSOD6 100°С: δ = 0,79-0,86 (м, 3Н); 0,88-0,95 (м, 3Н); 1,10-1,30 (м, 2H); 1,40-1,50 (м, 8H); 1,63-1,73 (м, 2H); 1,98-2,05 (м, 1H); 2,78 (ушир.кв, J=6,8-8,8 Гц, 5H); 3,22-3,38 (м, 2H); 3,56-3,70 (м, 3H); 3,75 (с, 3H); 3,80-4,20 (м, 1H); 4,34 (ушир.т, J=7,2 Гц, 1H); 5,90-6,20 (м, 2H); 6,85 (ущир.д, 2H); 7,10 (д, J=8,0 Гц, 2H); 7,31 (с, 1H); 8,82 (с, 1H).
Пример 15: 1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(3-пентил-3-фенилазетидин-1-ил)этил]мочевина (соединение 29)
15-1-1 2-Фенилгептаннитрил
К суспензии 2 г 60% NaH в 50 мл ДМФА, охлажденной до 0°C, по каплям добавляли 5 г (42,7 ммоль) фенилацетонитрила. После перемешивания в течение 30 минут при 0°C добавляли по каплям 5,32 мл (42,7 ммоль) бромпентана. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре и затем добавляли лед и экстрагировали этиловым эфиром. Органическую фазу промывали насыщенным водным раствором гидрокарбоната натрия, сушили над сульфатом натрия, фильтровали и упаривали под давлением. Получали оранжево-желтое масло и очищали при помощи фракциональной дистилляции при пониженном давлении (70-75°C при 1×10-1 мбар). Получали 5,14 г в виде масла оранжевого цвета с выходом 64%.
15-1-2 2-Гидроксиметил-2-фенилгептаннитрил
К суспензии 1,34 г 60% NaH в 50 мл ДМФА, охлажденной до 0°C, добавляли 5,14 г (27,4 ммоль) 2-фенилгептаннитрила. После перемешивания в течение 30 минут порциями добавляли 7,6 г (220 ммоль) параформальдегида. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов и затем гидролизовали льдом и экстрагировали диэтиловым эфиром. Органическую фазу промывали насыщенным водным раствором гидрокарбоната натрия, сушили над сульфатом натрия, фильтровали и упаривали под давлением. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 8/2 гептан/этилацетат). Получали 4,24 г в форме бледно-желтого масла с выходом 71%.
15-1-3 2-Циано-2-фенилгептиловый эфир толуол-4-сульфоновой кислоты
К раствору, содержащему 4,24 г (19,5 ммоль) 2-гидроксиметил-2-фенил-гептаннитрила в 25 мл дихлорметана, добавляли 4,1 г (21,5 ммоль) пара-толуолсульфонилхлорида и 6 мл триэтиламина. Реакционную смесь перемешивали в течение 15 часов при комнатной температуре и затем обрабатывали 1 н. раствором хлористоводородной кислоты и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали под давлением. Получали желтое масло и осаждали из смеси 9/1 гептан/диизопропиловый эфир. Образовавшийся осадок отфильтровывали и промывали диизопропиловым эфиром. Получали 5,26 г в форме порошка бежевого цвета с выходом 72%.
15-1-4 3-Пентил-3-фенилазетидин
К раствору, содержащему 5,26 г (14,1 ммоль) 2-циано-2-фенилгептилового эфира толуол-4-сульфоновой кислоты в 25 мл ТГФ, в атмосфере азота осторожно добавляли 600 мг (15,5 ммоль) порошка LiAlH4. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и затем обрабатывали пастой сульфата натрия (горячая вода + Na2SO4). После перемешивания в течение 30 минут при комнатной температуре образовавшиеся соли отфильтровывали и фильтрат упаривали при пониженном давлении. Остаток обрабатывали дихлорметаном для поглощения и промывали водным 1 н. раствором гидроксида натрия. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Получали 2,90 г бесцветного масла и использовали на следующей стадии без дополнительной очистки.
15-2 1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(3-пентил-3-фенил-азетидин-1-ил)этил]мочевина
170 мг (0,837 ммоль) 3-пентил-3-фенилазетидина растворяли в 2 мл ДМФА с 50 мг 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропановой кислоты (см. получение 2-3). К этому раствору добавляли 177 мг (0,920 ммоль) EDC и затем 125 мг (0,920 ммоль) HOBt. Реакционную смесь перемешивали в течение 4 часов и затем обрабатывали 1 н. раствором гидроксида натрия и экстрагировали дихлорметаном. Объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 90/10 ДХМ/метанол). Получали 180 мг 1-[2-(1H-имидазол-4-ил)этил]-3-[1-(4-метоксибензил)-2-оксо-2-(3-пентил-3-фенилазетидин-1-ил)этил]мочевины в форме белого порошка с выходом 41%.
1H ЯМР/DMSOD6 100°C: δ = 7,45 (с, 1H); 7,30 (м, ЗН); 7,22 (м, 1H); 7,19 (м, 4H); 6,82 (м, 2Н); 6,72 (с, 1Н); 5,99 (д, J=9,2 Гц, 1H); 5,90 (м, 1H); 4,38 (кв, J=7,2 Гц, 1H); 3,73 (м, 4H); 3,23 (м, 2H); 2,93 (м, 3H); 2,75 (д, J=6 Гц, 2H); 2,61 (т, J=6,80 Гц, 2H); 1,18 (м, 4H); 0,98 (м, 2H); 0,78 (т, J=6,4 Гц, 3H).
Пример 16: Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат (соединение 30):
16-1 Этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-хлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
3,1 г (10 ммоль) (R)-2-трет-бутоксикарбониламино-3-(4-хлорфенил)пропионовой кислоты, 1,9 г (11 ммоль) EDC, 1,5 г (11 ммоль) HOBt и 1,7 мл (20 ммоль) диизопропилэтиламина растворяли в 80 мл ДМФА. Смесь перемешивали при комнатной температуре в течение 15 минут и затем добавляли 2,6 г (10 ммоль) этил пиперидин-4-циклогексил-4'-карбоксилата (см. получение 9-2), разбавленного в 20 мл ДМФА. После перемешивания в течение 2 часов добавляли еще 1,7 мл (20 ммоль) диизопропилэтиламина. Реакцию останавливали путем добавления 5% водного раствора лимонной кислоты. Органические продукты экстрагировали при помощи этилацетата и органическую фазу промывали водным 1 н. раствором NaOH. Органическую фазу сушили и упаривали. Получали 5,2 г этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-хлорфенил)пропионил]-4-циклогексил-пиперидин-4-карбоксилата в форме бесцветного масла с выходом 75%.
16-2 Этил 1-[(R)-2-амино-3-(4-хлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
0,26 г (0,5 ммоль) этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-хлорфенил)-пропионил]-4-циклогексилпиперидин-4-карбоксилата разбавляли в 4 мл ДХМ и 1 мл трифторуксусной кислоты и раствор перемешивали в течение 2 часов при комнатной температуре и затем гасили путем добавления 1 н. водного раствора NaOH и экстрагировали дихлорметаном. Органическую фазу сушили и затем упаривали. Получали 200 мг этил 1-[(R)-2-амино-3-(4-хлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата в форме бесцветного масла с выходом 95%.
16-3 Этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилат
200 мг (0,48 ммоль) этил 1-[(R)-2-амино-3-(4-хлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата и 105 мг (0,52 ммоль) пара-нитрофенилхлорформиата растворяли в 5 мл дихлорметана. Смесь перемешивали в течение 2 часов при комнатной температуре и раствор затем вливали в 20% водный раствор NH4OH и экстрагировали дихлорметаном. Органическую фазу промывали водой и затем сушили над сульфатом натрия и упаривали. Полученное бесцветное масло разбавляли в 5 мл ДМФА, смесь нагревали до 80°C, затем добавляли 58 мг (0,52 ммоль) гистамина и раствор перемешивали в течение 5 минут при 80°C и в течение 10 минут при комнатной температуре. Реакцию останавливали путем добавления водного 5% раствора лимонной кислоты и органические продукты экстрагировали при помощи ДХМ. Органическую фазу промывали водным 1 н. раствором NaOH и затем водой. После сушки над сульфатом натрия и упаривания полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 80/20 ДХМ/MeOH). Получали 40 мг этил 1-((R)-3-(4-хлорфенил)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}пропионил)-4-циклогексилпиперидин-4-карбоксилата в форме бесцветного масла с выходом 15%.
ВЭЖХ: (метод N); время удерживания: 11,01 минут, 96%, M+H: 558.
Пример 17: Трифторацетат 1-[2-(4-Ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевины (соединение 32):
17-1-1 1-[4-Фенил-1-(толуол-4-сульфонил)пиперидин-4-ил]пропан-1-он
3,9 мл (2,94 ммоль) 3M раствора этилмагнийбромида добавляли к раствору 2 г (5,9 ммоль) 4-фенил-1-(толуол-4-сульфонил)пиперидин-4-карбонитрила в 40 мл толуола. Реакционную смесь перемешивали в течение ночи при 65-70°C. Реакцию останавливали и гидролизовали 1 н. раствором хлористоводородной кислоты и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем обрабатывали этилацетатом для поглощения, в эту смесь добавляли 1 н. раствор хлористоводородной кислоты и смесь перемешивали при комнатной температуре в течение 72 часов. Получали 2,16 г 1-[4-фенил-1-(толуол-4-сульфонил)-пиперидин-4-ил]пропан-1-она в форме желтого масла с выходом 99%.
17-1-2 Гидрохлорид 1-(4-Фенилпиперидин-4-ил)пропан-1-она
2,1 г (5,6 ммоль) 1-[4-фенил-1-(толуол-4-сульфонил)пиперидин-4-ил]пропан-1-она суспендировали в 14 мл серной кислоты и 7 мл воды. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 72 часов. Реакцию останавливали путем добавления в лед и подщелачивали при помощи раствора гидроксида натрия и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем обрабатывали диэтиловым эфиром для поглощения и осаждали при помощи 4н раствора хлороводорода в этилацетате. Получали 1,04 г гидрохлорида 1-(4-фенилпиперидин-4-ил)пропан-1-она в форме порошка бежевого цвета, с выходом 73%.
17-1-3 трет-Бутил 4-фенил-4-пропионилпиперидин-1-карбоксилат
470 мг (2,17 ммоль) ди-трет-бутилдикарбоната, растворенного в 5 мл дихлорметана, добавляли к раствору 500 мг (1,97 ммоль) гидрохлорида 1-(4-фенилпиперидин-4-ил)пропан-1-она и 0,7 мл (4,93 ммоль) триэтиламина в 5 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Реакцию останавливали путем добавления 10 мл воды. Органическую фазу сушили над сульфатом натрия. Растворители выпаривали и остаток затем хроматографировали на силикагеле (элюент: смесь 80/20 гептан/этилацетат). Получали 582 мг трет-бутил 4-фенил-4-пропионилпиперидин-1-карбоксилата в форме белого порошка с выходом 93%.
17-1-4 трет-Бутил 4-циклогексил-4-пропионилпиперидин-1-карбоксилат
Следующие соединения вводили в аппарат Парра: 582 мг (1,83 ммоль) трет-бутил 4-фенил-4-пропионилпиперидин-1-карбоксилата, 300 мг родия на оксиде алюминия и 0,5 мл уксусной кислоты в 15 мл диоксана. Реакционную смесь помещали под давление водорода 6 бар и перемешивали при 80°C в течение 16 часов. Реакцию останавливали, фильтровали через целит и промывали дихлорметаном. Растворители выпаривали и остаток затем хроматографировали на силикагеле (элюент: смесь 95/5 гептан/этилацетат). Получали 549 мг трет-бутил 4-циклогексил-4-пропионилпиперидин-1-карбоксилата в форме бесцветного масла с выходом 93%.
17-1-5 4-Циклогексил-4-пропионилпиперидингидрохлорид
549 мг (1,7 ммоль) трет-бутил 4-циклогексил-4-пропионилпиперидин-1-карбоксилата растворяли в 2 мл (17 ммоль) 4н раствора хлороводорода в этилацетате. Реакционную смесь перемешивали в течение 6 часов при комнатной температуре. Растворители выпаривали. Осуществляли поглощение остатка простым эфиром и отфильтровывали. Получали 395 мг 4-циклогексил-4-пропионилпиперидингидрохлорида в форме белого порошка с выходом 90%.
17-2 Трифторацетат 1-[2-(4-ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевины
58 мг (0,42 ммоль) HOBt и 81 мг (0,42 ммоль) EDC и затем 0,2 мл (1,15 ммоль) диизопропилэтиламина добавляли к раствору 128 мг (0,38 ммоль) 2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионовой кислоты (см. получение 2-3) и 62,9 мг (0,38 ммоль) 4-циклогексил-4-пропионилпиперидингидрохлорида в 5 мл диметилформамида. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакцию останавливали путем добавления 10 мл воды и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем очищали при помощи препаративной ВЭЖХ. Получали 98,5 мг трифторацетата 1-[2-(4-ацетил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)-мочевины в форме белого порошка с выходом 48%.
ВЭЖХ: (метод N); время удерживания: 9,98 минут, 97%, M+H: 538.
Пример 18: Трифторацетат 1-[2-(4-циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевины (соединение 33):
18-1-1 1-Тритил-1H-имидазол-4-карбальдегид:
К раствору, содержащему 1 г (10,4 ммоль) 1H-имидазол-4-карбальдегида и 3,18 г (11,4 ммоль) тритилхлорида, суспендированному в 28 мл ацетонитрила, по каплям добавляли 2,5 мл (17,7 ммоль) триэтиламина. После перемешивания в течение 2 часов при комнатной температуре добавляли 30 мл воды и неочищенный реакционный продукт фильтровали. Получали 3,2 г соединения в форме порошка бежевого цвета и использовали на следующей стадии без дополнительной очистки.
18-1-2 Пиперидин-1-ил-(1-тритил-1H-имидазол-4-ил-метилен)амин:
К раствору, содержащему 342 мг (1,01 ммоль) 1-тритил-1H-имидазол-4-карбальдегида и 100 мг (1,0 ммоль) пиперидин-1-иламина, добавляли 3,5 мл этанола и 1,5 мл дихлорметана. Реакционную смесь нагревали при 80°C в течение 1 часа. Растворители выпаривали и получали 450 мг порошка и использовали на следующей стадии без дополнительной очистки.
18-1-3 C-(1H-имидазол-4-ил)метиламиндигидрохлорид
К раствору, содержащему 450 мг пиперидин-1-ил-(1-тритил-1H-имидазол-4-ил-метилен)амина в 3,5 мл 3н раствора хлороводорода в этилацетате, добавляли 90 мг 10% Pd/C, 0,5 мл ТГФ и 0,9 мл этанола. Реакционную смесь помещали под давление водорода 6 бар и нагревали при 80°C в течение 3 часов. После того, как катализатор отфильтровывали и выпаривали растворители, полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 6/4 ДХМ/MeOH). Получали 50 мг C-(1H-имидазол-4-ил)метиламиндигидрохлорида в форме белого порошка с выходом 29%.
18-2 Метил (S)-2-[3-(1H-имидазол-4-ил-метил)уреидо]-3-(4-метоксифенил)-пропаноат
1,1 г (0,37 ммоль) метил (S)-3-(4-метоксифенил)-2-(4-нитрофеноксикарбонил-амино)пропаноата (см. получение 2-2) и 0,7 мл (0,37 ммоль) диизопропилэтиламина в 5 мл диметилформамида добавляли при 80°C к раствору 500 мг (0,37 ммоль) C-(1H-имидазол-4-ил)метиламиндигидрохлорида в 15 мл диметилформамида. Реакционную смесь перемешивали в течение 2 часов при 80°C. Реакцию останавливали путем добавления 30 мл воды и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем хроматографировали на силикагеле (элюент: смесь 80/20 дихлорметан/метанол). Получали 279 мг метил (S)-2-[3-(1H-имидазол-4-ил-метил)-уреидо]-3-(4-метоксифенил)пропаноата в форме желтого масла с выходом 63%.
18-3 2-[3-(1H-имидазол-4-илметил)уреидо]-3-(4-метоксифенил)пропионовая кислота
351 мг (84 ммоль) моногидрата гидроксида лития добавляли к раствору 278 мг (8,4 ммоль) метил (S)-2-[3-(1H-имидазол-4-илметил)уреидо]-3-(4-метокси-фенил)пропаноата в 5 мл тетрагидрофурана и 1 мл воды. Реакционную смесь нагревали в течение 10 минут при 100°C в микроволновом реакторе. Реакцию останавливали путем добавления 2 мл воды и 0,5 мл уксусной кислоты и затем экстрагировали этилацетатом. Водную фазу концентрировали досуха и остаток затем хроматографировали на силикагеле (элюент: смесь 7/3 дихлорметан/метанол). Получали 261 мг 2-[3-(1H-имидазол-4-илметил)уреидо]-3-(4-метоксифенил)пропионовой кислоты в форме белой пены с выходом 98%.
18-4 Трифторацетат 1-[2-(4-циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевины
70 мг (0,52 ммоль) HOBt и 99,4 мг (0,52 ммоль) EDC и затем 0,25 мл (1,4 ммоль) диизопропилэтиламина добавляли к раствору 150 мг (0,47 ммоль) 2-[3-(1H-имидазол-4-илметил)уреидо]-3-(4-метоксифенил)пропионовой кислоты и 123 мг (0,47 ммоль) 1-(4-циклогексилпиперидин-4-ил)пропан-1-она в 5 мл диметилформамида. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакцию останавливали путем добавления 10 мл воды и затем экстрагировали этилацетатом. Органические фазы объединяли и сушили над сульфатом натрия. Растворители выпаривали и остаток затем очищали при помощи препаративной ВЭЖХ. Получали 8,5 мг трифторацетата 1-[2-(4-циклогексил-4-пропионилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-(1H-имидазол-4-илметил)мочевины в форме бесцветного масла с выходом 4%.
ВЭЖХ: (метод N); время удерживания: 9,93 минут, 98%, M+H: 524.
Пример 19: Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]-уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат (соединение 34):
19-1 Этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
К 3,22 г (10,9 ммоль) (R)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)-пропионовой кислоты, растворенной в 20 мл ДМФА, добавляли 2,29 г (12,0 ммоль) EDC, 1,62 г (12,0 ммоль) HOBt, 2,62 г (10,9 ммоль) этил 4-циклогексилпиперидин-4-карбоксилата (см. получение 3-1-2) и 4,6 мл (32,7 ммоль) триэтиламина. После перемешивания в течение 2 часов 30 минут реакцию останавливали путем добавления 1 н. водного раствора гидроксида натрия и органические продукты экстрагировали дихлорметаном. Органическую фазу сушили над MgSO4, фильтровали и упаривали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 7/3 гептан/EtOAc). Получали 3,70 г этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)пропионил]-4-циклогексил-пиперидин-4-карбоксилата в форме белого порошка с выходом 66%.
19-2 Этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
К 3,7 г (7,16 ммоль) этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-метокси-фенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата, растворенного в 30 мл ДХМ, погруженному в ванну при 0°C, добавляли 10 мл трифторуксусной кислоты. После перемешивания в течение 1 часа 30 минут растворители выпаривали. Реакционную смесь промывали водным 1 н. раствором гидроксида натрия и экстрагировали при помощи ДХМ. Органическую фазу затем промывали водой и затем сушили над MgSO4, фильтровали и концентрировали. Получали 2,74 г этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата в форме белого порошка с выходом 92%.
19-3 Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилат
К 2,35 г (5,64 ммоль) этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата, растворенного в 25 мл ДХМ, погруженного в ванну с холодной водой, добавляли 1,25 г (6,21 ммоль) 4-нитрофенилхлорформиата. После нагревания до комнатной температуры реакционную смесь перемешивали в течение 1 часа. Реакцию останавливали путем добавления водного раствора NH4OH и органические соединения экстрагировали при помощи ДХМ. Органическую фазу затем промывали водой и затем сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт растворяли в 25 мл ДМФА при 80°C и затем добавляли 0,70 г (6,3 ммоль) гистамина. Через 15 минут реакцию останавливали путем добавления 1 н. водного раствора гидроксида натрия и органические соединения экстрагировали при помощи ДХМ. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 86/14 ДХМ/MeOH). Получали 1,48 г этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]уреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилата в форме белого порошка с выходом 47%.
1H ЯМР/DMSOD6 100°С: δ = 0,87-1,28 (м, 10Н); 1,50-1,70 (м, 3H); 1,71-1,74 (м, 2Н); 1,80-2,00 (м, 2H); 2,54-2,63 (м, 2H); 2,71-2,83 (м, 2H); 3,24 (ушир.кв, J=6,4-6,8 Гц, 1H); 3,74 (с, 3H); 3,75-4,05 (м, 4H); 4,12 (ушир.кв, J=6,8-7,2 Гц, 2H); 4,81 (ушир.кв, J=6,8-8,0 Гц, 1H); 5,93 (ушир.т, J=4,8-5,6 Гц, 1H); 6,01 (ушир.д, J=8,0 Гц, 1H); 6,73 (с, 1H); 6,82 (ушир.д, J=8,4 Гц, 2H); 7,08 (ушир.д, J=7,6 Гц, 2H); 7,44 (с, 1H); 11,45 (ушир.с, 1H).
Пример 20: 1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метокси-бензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина (соединение 40):
20-1 1-(4-Циклогексилпиперидин-4-ил)бутан-1-он
2,5 г (7,4 ммоль) трет-бутил 4-бутирил-4-циклогексилпиперидин-1-карбоксилата (см. получение 17-1-4) разбавляли в смеси, содержащей 40 мл дихлорметана и 8 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 2 часов и реакцию затем останавливали путем добавления 1 н. водного раствора NaOH и органические соединения экстрагировали дихлорметаном. Органическую фазу сушили и затем концентрировали. Получали 2,1 г 1-(4-циклогексилпиперидин-4-ил)бутан-1-она в форме бесцветного масла с выходом 100%.
20-2 трет-Бутил [(R)-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]карбамат
2,1 г (8,9 ммоль) 1-(4-циклогексилпиперидин-4-ил)бутан-1-она, 2,6 г (8,9 ммоль) (R)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)пропионовой кислоты, 1,88 г (9,8 ммоль) EDC и 1,2 г (9,8 ммоль) HOBT растворяли в 30 мл ДМФА. Смесь перемешивали при комнатной температуре в течение 2 часов. Раствор промывали водным 2,5% раствором лимонной кислоты и экстрагировали этилацетатом. Органическую фазу промывали 10 н. водным раствором NaOH и затем сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт хроматографировали на силикагеле (элюент: смесь 70/30 гептан/этилацетат). Получали 1,4 г трет-бутил [(R)-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]карбамата в форме бесцветного масла с выходом 31%.
20-3 1-{1-[(R)-2-Амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-ил}-бутан-1-он
1,4 г (2,7 ммоль) трет-бутил [(R)-2-(4-бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]карбамата помещали в смесь, состоящую из 40 мл ДХМ и 8 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре и реакцию затем останавливали путем добавления 1 н. водного раствора гидроксида натрия. После экстракции дихлорметаном органическую фазу сушили над сульфатом натрия и затем фильтровали и концентрировали. Получали 1,24 г 1-{1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-ил}бутан-1-она в форме бесцветного масла с выходом 100%.
20-4 1-[(R)-2-(4-Бутирил-4-циклогексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевина
1,24 г (3 ммоль) 1-{1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексил-пиперидин-4-ил}бутан-1-она и 660 мг (3,3 ммоль) пара-нитрофенилхлорформиата растворяли в 40 мл дихлорметана при 0°C. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и реакцию затем останавливали путем добавления 20% водного раствора аммиака и органические продукты экстрагировали дихлорметаном. Органическую фазу промывали водой и затем сушили над сульфатом натрия, фильтровали и концентрировали. Полученное белое твердое вещество растворяли в 20 мл ДМФА, смесь нагревали до 80°C, затем добавляли 0,367 г (3,3 ммоль) гистамина и раствор перемешивали в течение 5 минут при 80°C и в течение 10 минут при комнатной температуре. Реакцию останавливали путем добавления 1 н. водного раствора NaOH и реакционную смесь экстрагировали при помощи ДХМ. Органическую фазу промывали водой и затем сушили над сульфатом натрия, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 80/20 ДХМ/MeOH). Получали 1,1 г 1-[(R)-2-(4-бутирил-4-цикло-гексилпиперидин-1-ил)-1-(4-метоксибензил)-2-оксоэтил]-3-[2-(1H-имидазол-4-ил)этил]мочевины в форме белого порошка с выходом 67%.
1H ЯМР/DMSOD6 100°C: δ = 0,87-1,22 (м, 10Н); 1,38-1,54 (м, 5H); 1,59-1,62 (м, 2H); 1,70-1,73 (м, 2H); 1,85-1,98 (м, 2H); 2,40 (т, J=7,2 Гц, 2H); 2,61 (т, J=7,2 Hz, 2H); 2,70-2,94 (м, 2H); 3,24 (ушир.кв, J=6,4-6,8 Гц, 2H); 3,73 (с, 3H); 3,75-4,05 (м, 4H); 4,79 (ушир.кв, J=6,8-8,0 Гц, 1H); 5,92 (ушир.т, J=4,8-5,6 Гц, 1H); 5,99 (ушир.д, J=8,0 Гц, 1H); 6,72 (с, 1H); 6,81 (ушир.д, J=8,4 Гц, 2H); 7,07 (ушир.д, J=7,6 Гц, 2H); 7,44 (с, 1H); 11,45 (ушир.с, 1H).
Пример 21: Этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)пропил]уреидо}пропионил)пиперидин-4-карбоксилат (соединение 45):
21-1 Этил 1-[(R)-2-трет-бутоксикарбониламино-3-(3,4-дихлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
0,5 г (2,1 ммоль) этил пиперидин-4-циклогексил-4'-карбоксилат (см. получение 3-1-2), 0,7 г (2,1 ммоль) Boc-3,4-дихлор-D-фенилаланина, 0,31 г (2,3 ммоль) HOBT, 0,44 г (2,3 ммоль) EDC и 0,73 мл (4,2 ммоль) диизопропилэтиламина помещали в 5 мл диметилформамида. Смесь перемешивали в течение 2 часов при комнатной температуре и затем промывали водным 5% раствором лимонной кислоты и экстрагировали этилацетатом. Органическую фазу промывали водным 1 н. раствором гидроксида натрия и затем водой и сушили над сульфатом натрия, фильтровали и окончательно концентрировали досуха. Полученное масло хроматографировали (элюент: смесь 5/5 гептан/этилацетат). Получали 300 мг этил 1-[(R)-2-трет-бутоксикарбониламино-3-(3,4-дихлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата с выходом 26%.
21-2 Этил 1-[(R)-2-амино-3-(3,4-дихлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилат
300 мг (0,54 ммоль) этил 1-[(R)-2-трет-бутоксикарбониламино-3-(3,4-дихлор-фенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата разбавляли в 5 мл раствора 2/1 дихлорметан/трифторуксусная кислота. Через 2 часа смесь вливали в водный 1 н. раствор гидроксида натрия и затем экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия и затем фильтровали и концентрировали досуха. Получали 210 мг этил 1-[(R)-2-амино-3-(3,4-дихлорфенил)пропионил]-4-цикло-гексилпиперидин-4-карбоксилата с выходом 86%.
21-3 Бис(трифторацетат)4-(3-аминопропил)-1H-имидазол-1-иума
0,5 г (1,4 ммоль) 3-(1-тритил-1H-имидазол-4-ил)пропиламина растворяли в 8 мл дихлорметана с 2 мл трифторуксусной кислоты. Через 2 часа смесь концентрировали. Полученное твердое вещество бежевого цвета брали для поглощения в EtOAc и воду и водную фазу затем концентрировали досуха. Получали 0,3 г бис(трифторацетата) 4-(3-аминопропил)-1H-имидазол-1-иума с выходом 61%.
21-4 Этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)-пропил]уреидо}пропионил)пиперидин-4-карбоксилат
0,07 г (0,15 ммоль) этил 1-[(R)-2-амино-3-(3,4-дихлорфенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата и 34 мг (0,17 ммоль) пара-нитрофенилхлорформиата растворяли в 5 мл дихлорметана. Смесь перемешивали в течение 2 часов при комнатной температуре и раствор затем вливали в воду и экстрагировали дихлорметаном. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали досуха. Полученное бесцветное масло разбавляли в 5 мл диметилформамида, смесь нагревали до 80°C и добавляли 57 мг (0,16 ммоль) бис(трифторацетат) 3-(1H-имидазол-4-ил)пропиламина с 0,05 мл (0,3 ммоль) диизопропиламина. Раствор перемешивали в течение 5 минут при 80°C и в течение 16 часов при комнатной температуре. Реакцию останавливали путем добавления 1 н. водного раствора гидроксида натрия и затем экстрагировали дихлорметаном. Органическую фазу промывали водой и затем сушили над сульфатом натрия, фильтровали и упаривали досуха. Полученное масло хроматографировали (элюент: смесь 90/10 ДХМ/MeOH). Получали 40 мг этил 4-циклогексил-1-((R)-3-(3,4-дихлорфенил)-2-{3-[3-(1H-имидазол-4-ил)пропил]уреидо}-пропионил)пиперидин-4-карбоксилата в форме бесцветного масла с выходом 44%.
1H ЯМР/DMSOD6100°C: δ = 0,92-1,20 (м, 6H); 1,22 (т, ЗН, 7,2 Hz); 1,27 (м, 2H); 1,61 (м, 2H); 1,72-1,84 (м, 3H); 1,98 (м, 3H); 2,82-3,22 (м, 6H); 3,64 (м, 4H); 3,93 (т, 2H, 6,8 Гц); 4,14 (кварт, 2H, 7,2 Гц); 4,87 (кварт, 1H, 6,4 Гц); 6,01 (м, 2H); 6,87 (с, 1H); 7,07 (с, 1H); 7,18 (д, 1H, 6,8 Гц); 7,40 (д, 1H, 2,4 Гц); 7,47 (д, 1H, 8,4 Гц); 7,52 (с, 1H).
Пример 22: Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]-тиоуреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат (соединение 47):
22-1 Трифторацетат этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата
К 10 г (19,4 ммоль) этил 1-[(R)-2-трет-бутоксикарбониламино-3-(4-метоксифенил)-пропионил]-4-циклогексилпиперидин-4-карбоксилата (см. получение 19-1) в 80 мл дихлорметана при комнатной температуре добавляли 20 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 2 часов и растворители затем выпаривали. К полученному маслу добавляли диэтиловый эфир и несколько капель дихлорметана и полученное белое твердое вещество отфильтровывали и затем сушили. Получали 8 г трифторацетата этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата с выходом 78%.
22-2 Этил 4-циклогексил-1-[(R)-2-изотиоцианато-3-(4-метоксифенил)пропионил]-пиперидин-4-карбоксилат
К 500 мг (0,94 ммоль) трифторацетата этил 1-[(R)-2-амино-3-(4-метоксифенил)пропионил]-4-циклогексилпиперидин-4-карбоксилата, растворенного в 5 мл CH2Cl2 и 0,5 мл (2,8 ммоль) диизопропилэтиламина, добавляли 219 мг (0,94 ммоль) O,O-бис(2-пиридил)ового эфира тиокарбоновой кислоты. После перемешивания реакционной смеси в течение 2 часов при комнатной температуре растворители выпаривали и полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 6/4 гептан/EtOAc). Получали 421 мг этил 4-циклогексил-1-[(R)-2-изотиоцианато-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилата в форме бесцветного масла с выходом 97%.
22-3 Этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилат
К 400 мг (0,87 ммоль) этил 4-циклогексил-1-[(R)-2-изотиоцианато-3-(4-метокси-фенил)пропионил]пиперидин-4-карбоксилата в 5 мл ДМФА добавляли 97 мг (0,87 ммоль) гистамина и затем 0,130 мл (0,87 ммоль) 1,8-диазабицикло[5,4,0]ундец-7-ена. Реакционную смесь перемешивали в течение 2 часов и затем гидролизовали путем добавления водного 5% раствора лимонной кислоты. Органические соединения экстрагировали дихлорметаном. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный неочищенный продукт хроматографировали на силикагеле (элюент: смесь 80/20 CH2Cl2/MeOH). Получали 265 мг этил 4-циклогексил-1-[(R)-2-{3-[2-(1H-имидазол-4-ил)этил]тиоуреидо}-3-(4-метоксифенил)пропионил]пиперидин-4-карбоксилата в форме бледно-желтого порошка с выходом 53%.
ВЭЖХ: (метод U); время удерживания: 16,05 минут, 95%, M+H: 569.
Таблица I иллюстрирует примеры соединений в соответствии с настоящим изобретением.
В данной таблице:
- в колонке "соль", ТФУК представляет соединение в форме трифторацетата
- чистота (%) представляет чистоту продукта, полученную по данным спектра ВЭЖХ
- масса (M+H) представляет массу + H, полученную из данных масс-спектра, ассоциируемую с ВЭЖХ пиком ожидаемого продукта.
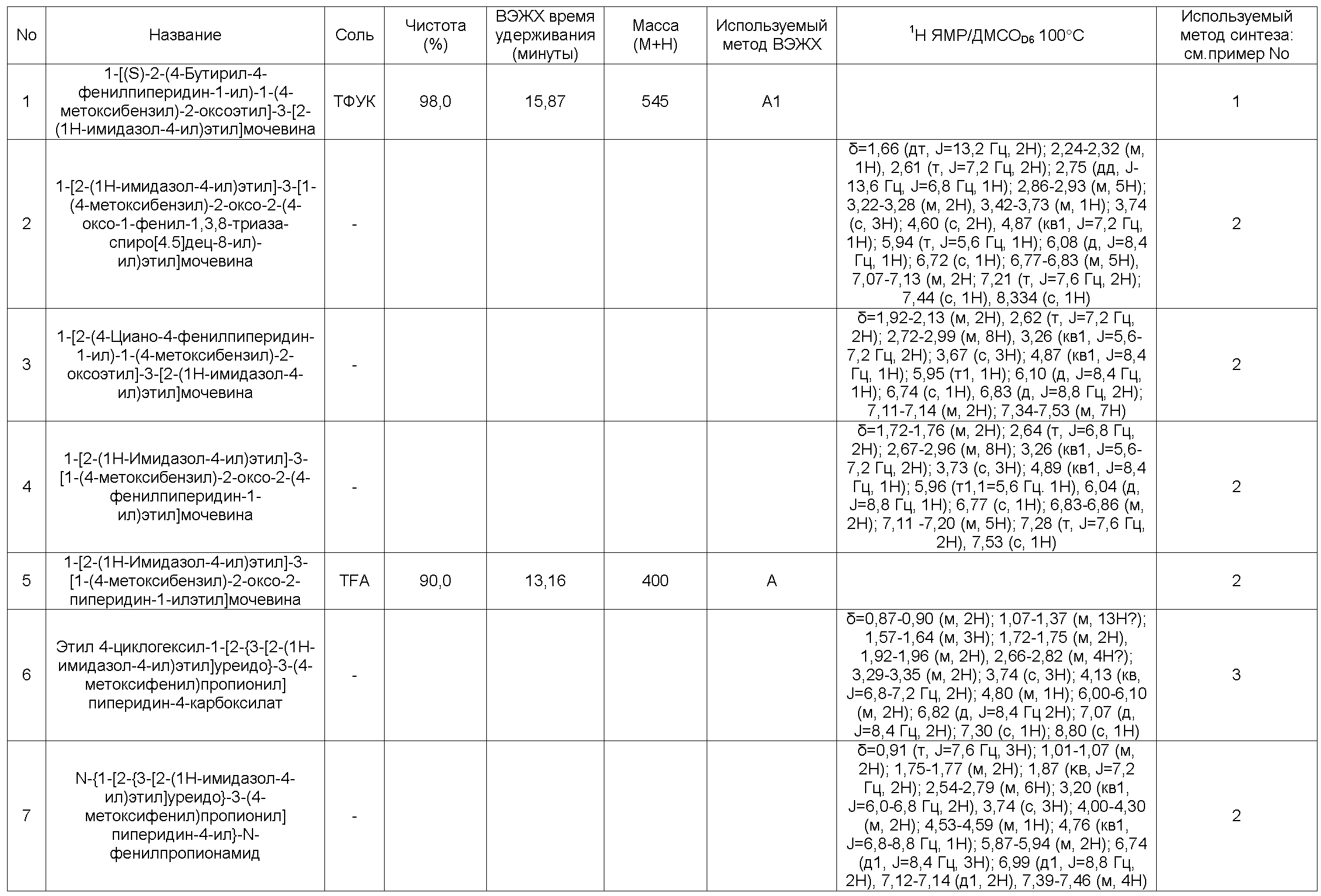


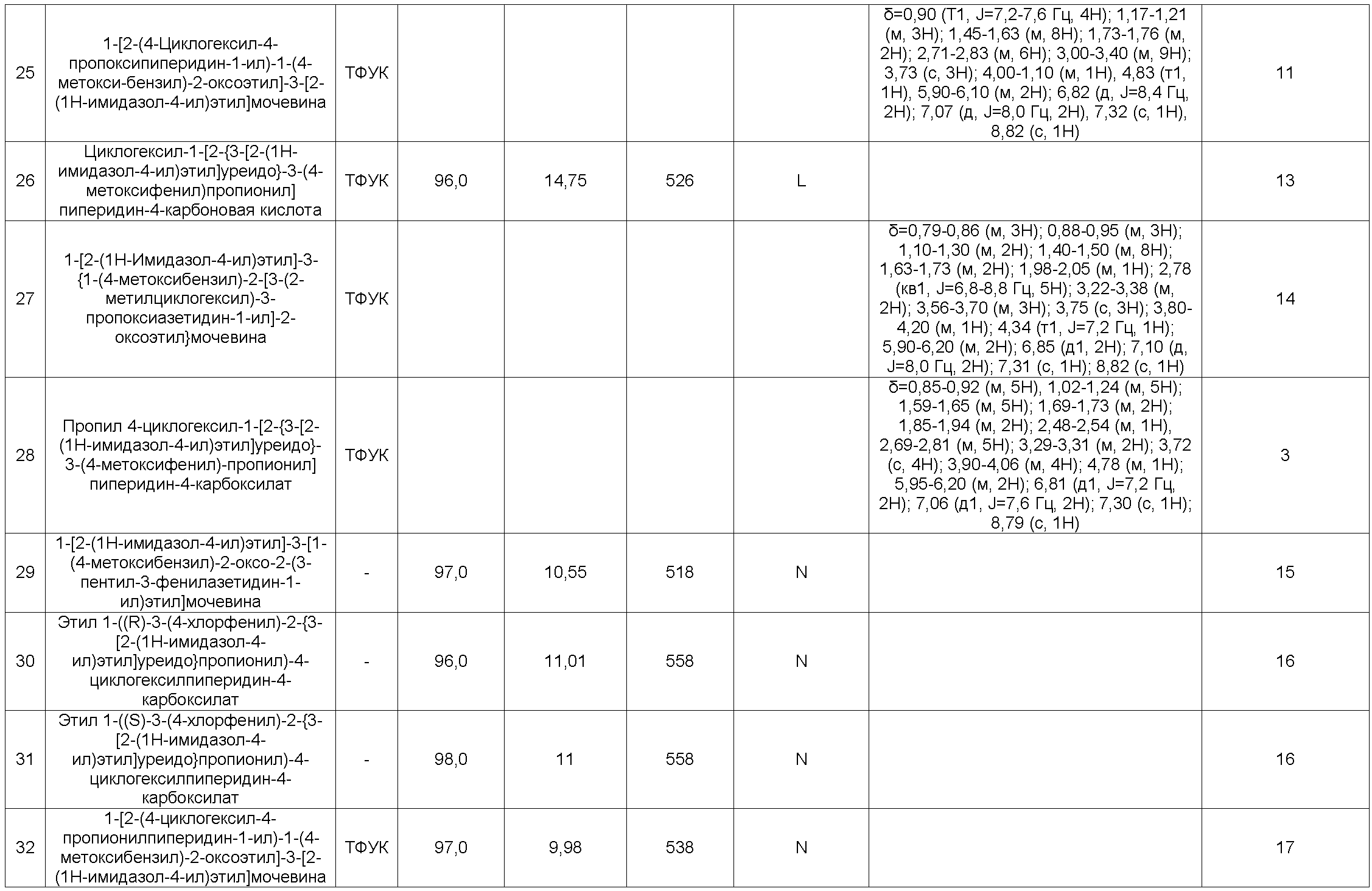
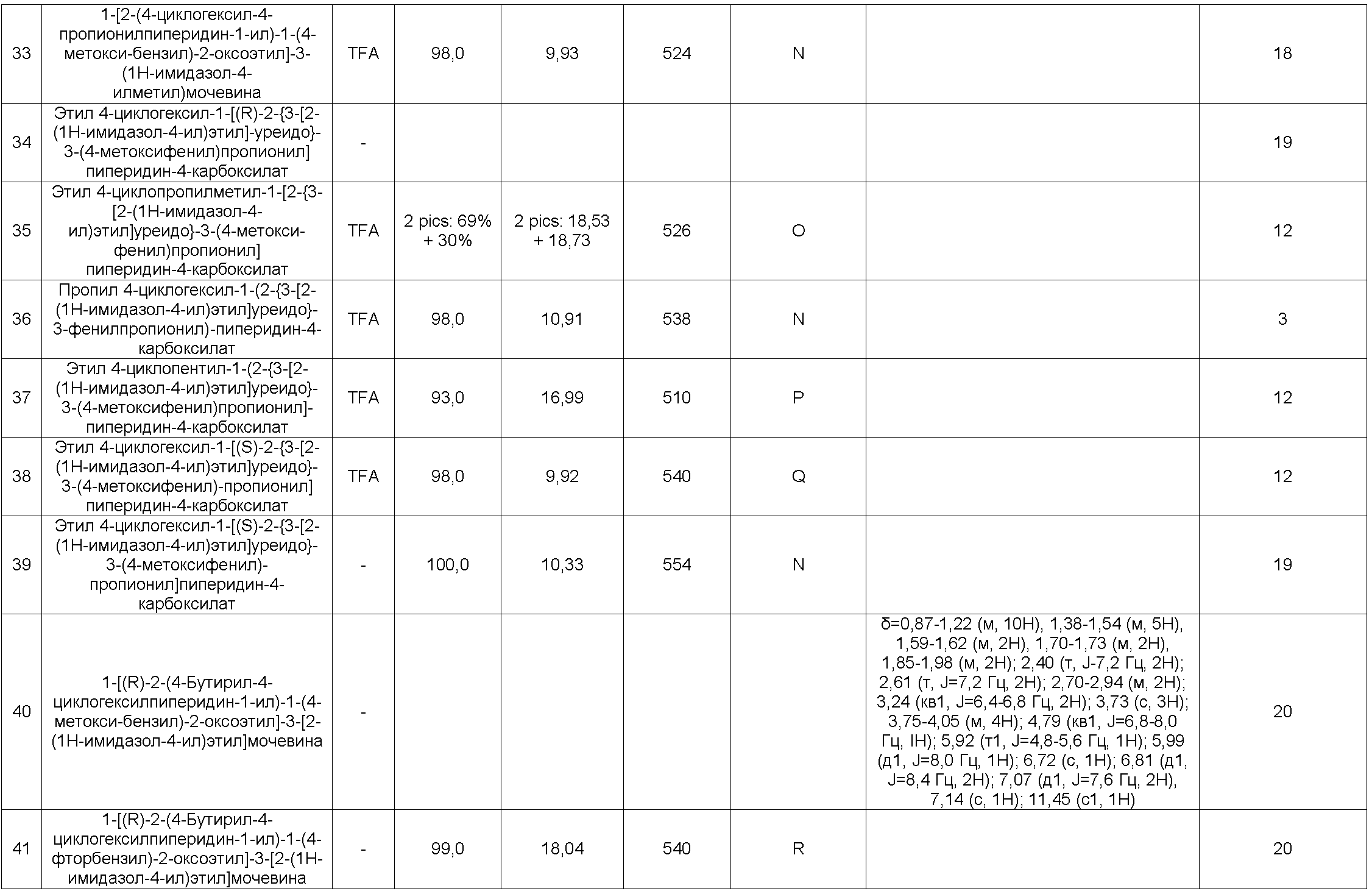
Пример 23: Испытание трансактивации
Клетки: Линии HEK293 трансфицируют векторами pCRE-Luc и hMC1R. Клетки культивируют при 37°C и 5% CO2 в среде DMEM, дополненной 10% фетальной телячьей сывороткой.
Принцип испытания: В присутствии активатора (агонист), рецептор меланокортина будет активировать cAMP путь, что, через вектор CRE-Luc, приведет к синтезу люциферазы. Путем добавления лизисного буфера, содержащего люминисцентный люциферазный субстрат, можно измерить люминисценцию, пропорциональную степени активации или ингибированию рецептора.
Испытание продуктов: Продукты растворяют при 10 мМ в DMSO. Их испытывают как дозу, вызывающую ответ, при конечной концентрации DMSO 0,1%. Диапазон, включающий 10 точек и ноль, начинается с 10 мкМ с четырехкратными разведениями. Для испытания агонистов продукты испытывают отдельно. Для определения антагонистического поведения, представляющие интерес продукты испытывают в присутствии 1 нМ NDP-MSH (ссылочный агонист). Клетки инокулируют при плотности 5000 клеток на лунку (384-луночный планшет) в бессывороточной DMEM среде и инкубируют в течение ночи при 37°C и 5% CO2.
Продукты и ссылочный лиганд (NDP-MSH) добавляют на следующий день и планшеты снова инкубируют в течение 6 часов при 37°C и 5% CO2. После добавления лизисного буфера, содержащего люциферин, планшеты считывают в Top-Count считывающем устройстве. Результаты нормализуют как процент активности, используя 100% (клетки + NDP-MSH при 10 нМ) и 0% (только клетки) контроли. Значение EC50 рассчитывают для каждого продукта с использованием программы XLFit. Результаты представлены в нМ.
|
Пример 24: Испытание метаболической стабильности в присутствии микросом печени:
Микросомы печени: Микросомы представляют собой везикулы клеточной эндоплазматической сети, полученные после очистки из ткани печени. Они содержат мембранные ферменты (Цитохромы P450), участвующие в окислительном метаболизме (фаза I).
Принцип испытания: В забуференной реакционной среде при 37°C испытываемый продукт инкубируют в присутствии микросом от различных видов. Образцы инкубата отбирают в разные точки времени (кинетика). Количественный ЖХ/МС/МС анализ дает возможность измерить исчезновение исходного продукта, связанное с фазой I печеночного метаболизма.
Испытание продуктов: Продукты растворяют при концентрации 10 мМ в DMSO. Их инкубируют при 10 мкМ в присутствии микросом (0,5 мг/мл). Реакционную смесь получают из 100 мМ фосфатного буфера при pH 7,4, G6PDH (0,4 Ед./мл), 0,01% плюроновой кислоты и кофакторов MgCl2 5 мМ, NADP и G6P (1,3 мМ и 3,3 мМ, соответственно). Образцы реакционной смеси отбирают в точках времени 0, 5, 10, 15, 30 и 60 минут. В каждой временной точке ферментативную реакцию останавливают путем добавления метанола. После центрифугирования (3000 об/мин, 30 минут) образцы анализируют методом ЖХ/МС/МС. Концентрацию оставшегося исходного продукта количественно определяют с течение времени, и это позволяет рассчитать период полужизни для каждого продукта.
|
Пример 25: Log P и Log D по данным ВЭЖХ-MS
Определение:
LogP, также известный как Log Kow, (коэффициент распределения октанол/вода) позволяет охарактеризовать липофильную природу молекулы.
LogP = Log(Cоктанол/Cвода)
В отличие от LogP, Log D является определением заданного pH.
Получение образцов
Молекулу при 10-2M в DMSO помещают в смесь октанол-вода (50/50 об/об).
После перемешивания в течение десяти часов, образец центрифугируют и две фазы разделяют для анализа. Октанольную фазу разбавляют в метаноле перед анализом.
Концентрации испытываемой молекулы, присутствующей в каждой фазе, определяют при помощи ЖХ/МС/МС анализа.
LogP = Log (площадьокт *Коэффициент разведения/площадьвод)
|
Пример 26: Модуляция пигментации волос в процессе вырастания после депиляции у мышей: (результаты в таблице Фиг. 2)
Самок 8-недельных B6.Cg-Ay мышей брили и затем подвергали депиляции холодным воском под газовой анестезией для синхронизации всех волосяных фолликул к началу анагенной фазы.
После депиляции MC1R агонисты, разбавленные до 5% в этаноле, применяли местно каждый день на участке депиляции в течение 11 дней. Через девятнадцать дней после депиляции пигментацию волос измеряли при помощи колориметрии на площади вырастания волос: значение L представляет собой измерение цвета по шкале от черного до белого (чем меньше значение L, тет темнее волосы).
|
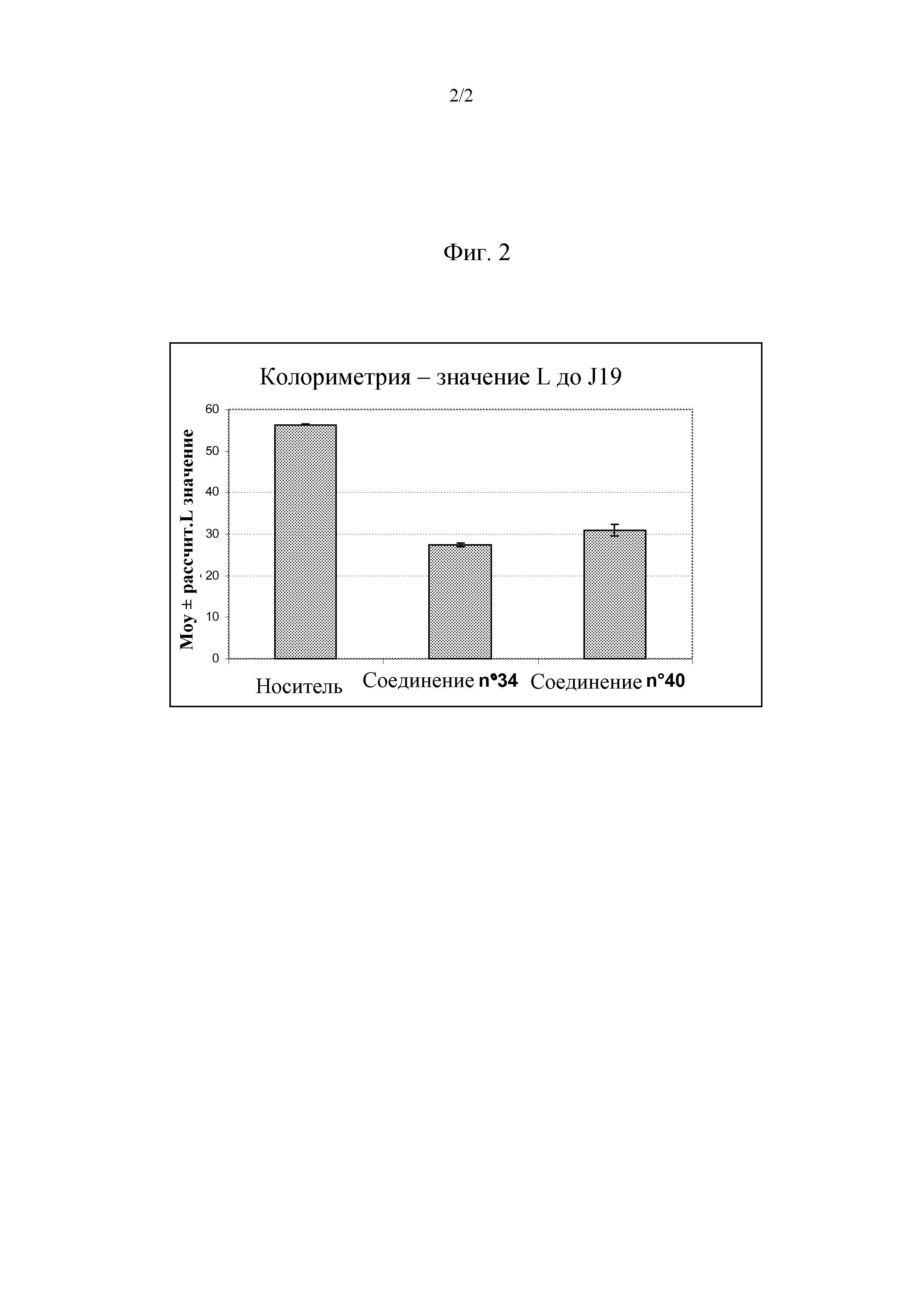
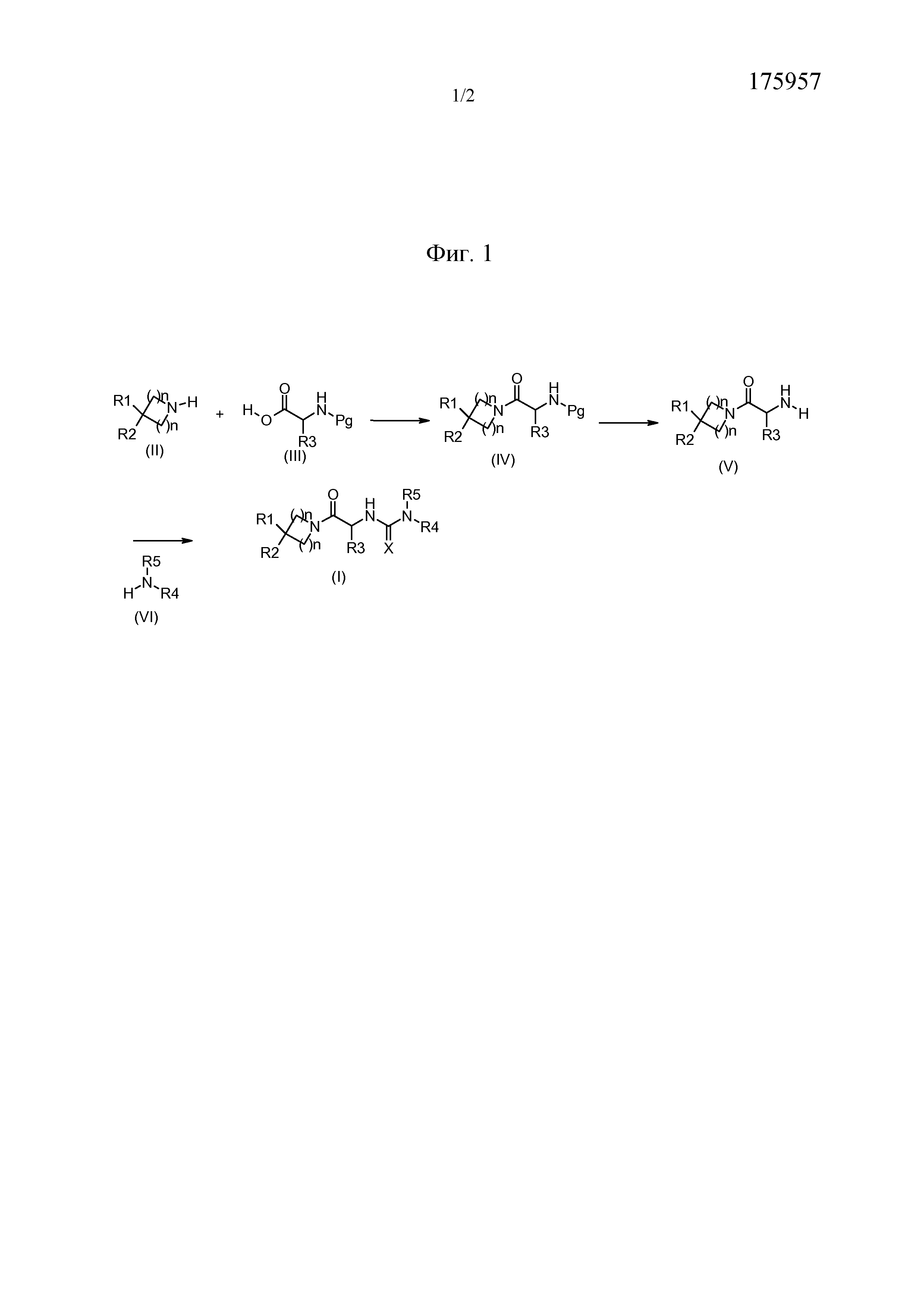
Способ получения адапаленовых гелей
Комбинация, включающая пирролидон-5-карбоновую кислоту и, по меньшей мере, одно соединение из цитруллина, аргинина и аспарагина, и их применение при лечении атопического дерматита
Применение адапалена и пероксида бензоила для продолжительного лечения угрей обыкновенных
Новые 4-(гетероциклоалкил)бензол-1,3-диольные соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека, а также в косметических средствах
Схема лечения заболеваний, связанных с акне
Новые 4-(азациклоалкил)бензол-1,3-диоловые соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека и в косметических средствах
Усовершенствованные способы и композиции для безопасного и эффективного лечения эритемы
Увлажняющий агент для кожи и его применение
Композиции, включающие по меньшей мере одно производное нафтойной кислоты, бензоилпероксид и по меньшей мере один пленкообразующий компонент, способы их получения и их применения
Новые бензолсульфонамидные соединения, способ их получения и применение в терапии и косметике
Способ получения адапаленовых гелей
Комбинация, включающая пирролидон-5-карбоновую кислоту и, по меньшей мере, одно соединение из цитруллина, аргинина и аспарагина, и их применение при лечении атопического дерматита
Способ синтеза(z)-3-[2-бутокси-3'-(3-гептил-1-метилуреидо)бифенил-4-ил]-2-метоксиакриловой кислоты
Применение адапалена и пероксида бензоила для продолжительного лечения угрей обыкновенных
Новые 4-(гетероциклоалкил)бензол-1,3-диольные соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека, а также в косметических средствах
Схема лечения заболеваний, связанных с акне
Новые 4-(азациклоалкил)бензол-1,3-диоловые соединения в качестве ингибиторов тирозиназ, способ их получения и их применение в лечении человека и в косметических средствах
Усовершенствованные способы и композиции для безопасного и эффективного лечения эритемы
Увлажняющий агент для кожи и его применение
Композиции, включающие по меньшей мере одно производное нафтойной кислоты, бензоилпероксид и по меньшей мере один пленкообразующий компонент, способы их получения и их применения

