Результат интеллектуальной деятельности: СПОСОБ РАЗДЕЛЕНИЯ ЭТИЛЕНГЛИКОЛЯ И 1,2-БУТАНДИОЛА
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к способу разделения смеси, содержащей этиленгликоль и 1,2-бутандиол, в особенности к способу выделения и очистки этиленгликоля из жидкофазного продукта, полученного путем гидрирования оксалата.
Уровень техники изобретения
Этиленгликоль является важным основным органическим химическим веществом и главным образом используется для сополимеризации с терефталевой кислотой для получения полиэтиленгликольтерефталата (ПЭТ). Кроме того, этиленгликоль также может использоваться для производства антифриза, смазочного вещества, пластификатора, неионогенного поверхностно-активного вещества и взрывчатого вещества и т.д. и имеет разнообразные применения. Китай является крупным потребителем этиленгликоля. В последние годы, в связи с конструированием и эксплуатацией ряда крупных ПЭТ-устройств, потребность в этиленгликоле нарастает быстрыми темпами. В настоящее время отечественное производство этиленгликоля далеко от возможностей удовлетворить эту потребность. В 2010 объем импорта Китаем этиленгликоля составлял 6644 миллиона тонн, и предполагалось, что к 2011 объем импорта этиленгликоля Китаем превысит 7 миллионов тонн. Поэтому этиленгликолевая промышленность Китая имеет хорошие перспективы развития.
Существуют различные технологические методы, использующие уголь в качестве сырья для производства этиленгликоля, и один метод, имеющий наилучшую промышленную перспективу, заключается в получении оксалата посредством дегидроконденсации синтез-газа и затем в производстве этиленгликоля путем гидрирования оксалата. Продукт реакции, полученный при гидрировании оксалата для получения этиленгликоля, содержит, в дополнение к веществам, имеющим более низкие точки кипения, таким как метанол, сложный эфир гликолевой кислоты и т.д., небольшое количество веществ, таких как 1,2-пропиленгликоль и 1,2-бутандиол, которые имеют точки кипения, близкие к таковым для этиленгликоля и которые могут легко подвергаться азеотропии с этиленгликолем и трудно разделимы путем обычной ректификации, где 1,2-бутандиол имеет точку кипения, наиболее близкую к этиленгликолю, и таким образом наиболее трудно отделить 1,2-бутандиол от этиленгликоля. Поэтому суть проблемы заключается в том, как отделить и удалить 1,2-бутандиол из этиленгликоля.
Кроме того, метод, использующий зерно в качестве сырья для производства этиленгликоля, 1,2-пропиленгликоля и 1,2-бутандиола посредством биотрансформации, также привлекает внимание различных стран. Для получения различных химических спиртовых продуктов, имеющих высокую чистоту, включая этиленгликоль и 1,2-бутандиол, по-прежнему необходимо решать техническую проблему, заключающуюся в том, что обычная ректификация требует многих теоретических тарелок и крупных капиталовложений, поскольку разница точек кипения этиленгликоля и 1,2-бутандиола очень мала.
Существует немного патентов по разделению этиленгликоля и 1,2-бутандиола, как внутри страны, так и за рубежом. CN101928201 раскрывает очистку синтез-газа посредством технического решения, использующего реакцию омыления, удаление метанола, реакцию гидрирования, трехколонную ректификацию и абсорбционную очистку для получения сырого этиленгликолевого продукта. Техническое решение, относящееся к этому патенту, не приводит к полному отделению 1,2-бутандиола от этиленгликоля, поскольку 1,2-бутандиол подвержен азеотропии с этиленгликолем в процессе разделения и очистки при трехколонной ректификации. Более того, оно также вызывает потерю этиленгликолевого продукта и снижает выход продукта. Патент США 4966658 раскрывает использование этилбензола, 3-гептанона и диизобутилкетона и т.д., в качестве азеотропных агентов для разделения этиленгликоля и 1,2-бутандиола или 1,3-бутандиола путем азеотропной ректификации, и число теоретических тарелок ректификационной колонны составляет 30. Однако азеотропный агент, описанный в этом патенте, требует при своем использовании очень высокой степени вакуума (например, 8 кПа) или очень долгого времени пребывания (например, 5-12 часов) в случае меньшей степени вакуума, чтобы получить этиленгликоль повышенной чистоты. И содержание этиленгликоля в азеотропной смеси верхней части колонны является относительно низким, не более 15%, и получающийся конечный продукт этиленгликоля все еще содержит приблизительно 100 частей на миллион (ч/млн) 1,2-бутандиола и незначительное количество азеотропных агентов, таких как этилбензол, 3-гептанон и диизобутилкетон и т.д. Поскольку данные азеотропные агенты отличаются повышенной абсорбцией в оптической ультрафиолетовой области, УФ-пропускание этиленгликолевого продукта недостаточно высокое, чтобы соответствовать стандартам продукта высшего сорта, и таким образом упомянутый продукт не пригоден для промышленного производства.
Содержание изобретения
Техническая проблема, требующая решения в настоящем изобретении, заключается в обеспечении нового способа выделения этиленгликоля и 1,2-бутандиола из смеси, содержащей этиленгликоль и 1,2-бутандиол, в ответ на проблему в известном уровне техники, заключающуюся в разделении этиленгликоля и 1,2-бутандиола: проблему крупных капиталовложений и высокого энергопотребления, вызванных обычной ректификацией, требующей очень высокого флегмового числа и большого количества теоретических тарелок, а также трудных условий разделения или неудовлетворительных результатов разделения, вызванных обычными азеотропными агентами в случае применения азеотропной ректификации. Данный новый способ имеет преимущества небольших капиталовложений и низкого энергопотребления, так же как и высокой чистоты и высокого значения УФ-пропускания получающегося этиленгликолевого продукта.
Для того чтобы решить вышеупомянутую техническую проблему, техническое решение, которое может быть использовано по настоящему изобретению, заключается в следующем: способ разделения смеси, содержащей этиленгликоль и 1,2-бутандиол, включающий перегонку смеси с азеотропным агентом, имеющим следующую структурную формулу,

где R1 является H атомом или алкилом, содержащим 1-4 атома углерода, предпочтительно H атомом или алкилом, содержащим 1-2 атома углерода; R2 является H атомом или алкилом, содержащим 1-4 атома углерода, предпочтительно H атомом или алкилом, содержащим 1-2 атома углерода; R3 является H атомом или алкилом, содержащим 1-8 атомов углерода, предпочтительно H атомом или алкилом, содержащим 1-5 атомов углерода; R4 является H атомом или алкилом, содержащим 1-8 атомов углерода, предпочтительно H атомом или алкилом, содержащим 1-5 атомов углерода, для получения материального потока, содержащего этиленгликоль и азеотропный агент, делающий упомянутый материальный поток неподвижным для расслоения, чтобы выделить смесь, обогащенную этиленгликолем, и далее перегонку упомянутой смеси для получения этиленгликоля. Предпочтительно вышеупомянутую перегонку, использующую азеотропный агент, осуществляют при помощи разделительной колонны, и предпочтительное число теоретических тарелок упомянутой разделительной колонны составляет 8-30, рабочее давление составляет 30-101,3 кПа на основе абсолютного давления, флегмовое число R=0,8-5, и молярное отношение азеотропного агента к этиленгликолю в сырье составляет 0,1-10:1. Предпочтительно содержание в массовых процентах легких компонентов, таких как метанол, этанол и сложный эфир гликолевой кислоты в смеси сырья, содержащей этиленгликоль и 1,2-бутандиол, составляет менее 1%.
В одном варианте осуществления настоящего изобретения настоящее изобретение относится к способу разделения этиленгликоля и 1,2-бутандиола из смеси, содержащей этиленгликоль и 1,2-бутандиол, включающему следующие стадии:
a) факультативно, материальный поток 1 смеси, содержащей этиленгликоль и 1,2-бутандиол, поступает в нижнюю часть средней части первой разделительной колонны C1, материальный поток 2, содержащий главным образом легкие компоненты, отгоняют из верхней части колонны, и материальный поток 3, содержащий главным образом этиленгликоль и 1,2-бутандиол, отводят из кубовой части колонны;
b) факультативно, материальный поток 3 поступает в нижнюю часть средней части второй разделительной колонны C2, материальный поток 4, содержащий главным образом легкие компоненты, отгоняют из верхней части колонны, и материальный поток 5, содержащий главным образом этиленгликоль и 1,2-бутандиол, отводят из кубовой части колонны;
c) материальный поток 5 поступает в нижнюю часть средней части третьей разделительной колонны C3, материальный поток 6, содержащий упомянутый азеотропный агент, добавляют в верхнюю часть разделительной колонны С3, материальный поток 7 азеотропной смеси, образованной главным образом азеотропным агентом и этиленгликолем, отгоняют из верхней части колонны, и материальный поток 8, содержащий главным образом 1,2-бутандиол, получают в кубовой части колонны;
d) после конденсации материальный поток 7 поступает в фазовый сепаратор D1 и разделяется на верхний материальный поток 9, обогащенный азеотропным агентом, и нижний материальный поток 10, обогащенный этиленгликолем, и материальный поток 9 факультативно возвращается в верхнюю часть третьей разделительной колонны C3, чтобы продолжать образовывать азеотроп;
e) материальный поток 10 поступает в верхнюю часть средней части четвертой разделительной колонны C4, материальный поток 11, содержащий азеотропный агент, отгоняют из верхней части колонны, и этиленгликолевый продукт с чистотой более 99,9% получают в кубовой части колонны.
В одном предпочтительном варианте осуществления вышеупомянутого технического решения первая разделительная колонна C1 является колонной для удаления первого легкого компонента, которая главным образом удаляет метанол в сырье и имеет 10-30 теоретических тарелок; рабочее давление является атмосферным давлением; и флегмовое число R=0,1-5. Вторая разделительная колонна С2 является колонной для удаления второго легкого компонента, которая главным образом удаляет эфирные соединения в сырье и имеет 20-50 теоретических тарелок; рабочее давление составляет 40-101 кПа на основе абсолютного давления; и флегмовое число R=0,3-6. Третья разделительная колонна C3 является колонной азеотропной ректификации и имеет 8-30 теоретических тарелок; рабочее давление составляет 30-101,3 кПа на основе абсолютного давления, флегмовое число R=0,8-5; молярное соотношение азеотропного агента к этиленгликолю в сырье составляет 0,1-10:1, и азеотропный агент и этиленгликоль являются несмешиваемыми. Четвертая разделительная колонна С4 является колонной очистки этиленгликоля и имеет 60-120 теоретических тарелок; рабочее давление составляет 10-101 кПа на основе абсолютного давления; и флегмовое число R=3-60. После очистки чистота этиленгликоля в массовых процентах составляет не менее 99,9%, и степень извлечения этиленгликоля составляет не менее 90%.
Впоследствии спирты и альдегиды, кетоны могут быть подвергнуты реакции конденсации под действием катализатора протонной кислоты для формирования ацеталевых и кеталевых соединений. Эти соединения обычно используются в качестве парфюмерных материалов, а также могут быть использованы в качестве растворителей специальных реакций. Азеотропный агент, предлагаемый в настоящем изобретении, принадлежит к ацеталевым и кеталевым соединениям и может быть синтезирован при помощи известных методов альдольной и гидроксикетонной конденсации. Например, см. следующие стадии для сведения: реагент I взаимодействует с реагентом II в течение 0,1-10 часов под действием катализатора протонной кислоты III в температурных условиях 80°C-250°C и при давлении реакции 30-100 кПа на основе абсолютного давления для получения упомянутого азеотропного агента, где молярное отношение реагента I к реагенту II составляет 1-20:1, концентрация катализатора III составляет 0,01-10% на основе молярного состава реакционной смеси, и структурная формула реагента I следующая:
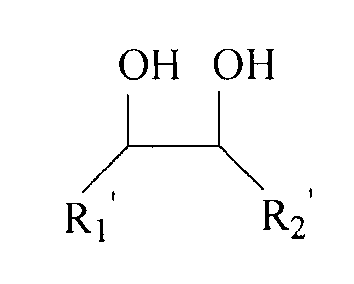
где R1' является H атомом или алкилом, содержащим 1-4 атома углерода, предпочтительно H атомом или алкилом, содержащим 1-2 атома углерода; R2' является H атомом или алкилом, содержащим 1-4 атома углерода, предпочтительно H атомом или алкилом, содержащим 1-2 атома углерода, и структурная формула реагента II является следующей:

где R3' является H атомом или алкилом, содержащим 1-8 атомов углерода, предпочтительно H атомом или алкилом, содержащим 1-5 атомов углерода; и R4' является H атомом или алкилом, содержащим 1-8 атомов углерода, предпочтительно H атомом или алкилом, содержащим 1-5 атомов углерода. Молекула реагента I предпочтительно содержит 2-6 атомов углерода, а молекула реагента II предпочтительно содержит 2-11 атомов углерода. Катализатор III предпочтительно отбирают из по меньшей мере одного из следующих веществ: концентрированной серной кислоты, концентрированной азотной кислоты, п-толуолсульфокислоты, фосфорновольфрамовой кислоты и каталитической смоляной кислоты. Предпочтительный диапазон концентраций катализатора III составляет 0,05-5% на основе молярной концентрации реакционной смеси.
Азеотропный агент, используемый в настоящем изобретении, обладает способностью формировать гетерогенный азеотроп, имеющий наименьшую азеотропную температуру с этиленгликолем, и он может существенно повысить относительную летучесть этиленгликоля и 1,2-бутандиола, чтобы в значительной степени сократить количество теоретических тарелок и флегмовое число ректификационной колонны, так чтобы достичь снижения капиталовложений и энергопотребления. Поскольку азеотропный агент и этиленгликоль не являются полностью смешиваемыми, после формирования азеотропной смеси с этиленгликолем упомянутый азеотропный агент может быть легко отделен от этиленгликоля при помощи простой сепарации и повторно использован. Более того, поскольку этиленгликоль имеет очень низкую растворимость в азеотропном агенте, настоящее изобретение снижает потери этиленгликоля и имеет более высокую эффективность. Способ настоящего изобретения может быть использован для разделения материального потока, содержащего этиленгликоль и 1,2-бутандиол, включая выделение и очистку этиленгликоля из жидкофазного продукта, полученного путем гидрирования оксалата, а также разделения и очистки этиленгликоля и 1,2-бутандиола в процессе производства этиленгликоля, использующего зерно в качестве сырья. При использовании способа по настоящему изобретению этиленгликоль после очистки имеет чистоту в массовых процентах не менее 99,9% и степень извлечения не менее 90%. Таким образом, способ по настоящему изобретению достигает лучших технических результатов.
Описание чертежей
Фигура 1 является схемой технологического процесса по настоящему изобретению.
На фигуре 1 C1 является первой разделительной колонной (колонной удаления первого легкого компонента); C2 является второй разделительной колонной (колонной удаления второго легкого компонента); C3 является третьей разделительной колонной (колонной азеотропной ректификации); C4 является четвертой разделительной колонной (колонной очистки); D1 является фазовым сепаратором. Материальный поток 1 является материальным потоком, содержащим этиленгликоль и 1,2-бутандиол; материальный поток 2 представляет легкие компоненты с более низкими точками кипения в материальном потоке 1; материальный поток 3 является материальным потоком, содержащим этиленгликоль и 1,2-бутандиол после того, как легкие компоненты с более низкими точками кипения удалены из материального потока 1; материальный поток 4 представляет легкие компоненты с более высокими точками кипения в материальном потоке 1; материальный поток 5 является материальным потоком, главным образом содержащим этиленгликоль и 1,2-бутандиол, после того, как легкие компоненты с более высокими точками кипения удалены из материального потока 3; материальный поток 6 является свежим азеотропным агентом; материальный поток 7 является азеотропной смесью, образованной этиленгликолем и азеотропным агентом; материальный поток 8 является материальным потоком, который не участвует в азеотропии и который содержит 1,2-бутандиол и другие компоненты материального потока 1; материальный поток 9 является верхним материальным потоком фазового сепаратора, обогащенным азеотропным агентом; материальный поток 10 является нижним материальным потоком фазового сепаратора, обогащенным этиленгликолем; материальный поток 11 представляет небольшое количество азеотропного агента и других соединений материального потока 10; материальный поток 12 является конечным этиленгликолевым продуктом после очистки материального потока 10.
Согласно технологической схеме процесса, как показано на фигуре 1, из материального потока 1, содержащего этиленгликоль и 1,2-бутандиол, удаляют материальный поток 2, т.е. легкие компоненты с более низкими точками кипения, при помощи первой разделительной колонны C1, и удаляют материальный поток 4, т.е. легкие компоненты с более высокими точками кипения, при помощи второй разделительной колонны C2, и далее получают материальный поток 5, содержащий главным образом этиленгликоль и 1,2-бутандиол, в третьей разделительной колонне C3; этиленгликоль в материальном потоке 5 формирует азеотропную смесь 7 с азеотропным агентом материального потока 6, добавляемого с верхней части колонны, и упомянутая азеотропная смесь 7 отгоняется из верхней части третьей разделительной колонны C3 и поступает в фазовый сепаратор D1 после конденсации; материальный поток 8, который содержит главным образом 1,2-бутандиол, получают в кубовой части колонны, и он может образовывать 1,2-бутандиольный продукт после дальнейшей очистки; в фазовом сепараторе D1 верхний материальный поток 9, обогащенный азеотропным агентом, возвращается в верхнюю часть третьей разделительной колонны C3 для продолжения образования азеотропа, и нижний материальный поток 10, обогащенный этиленгликолем, поступает в четвертую разделительную колонну C4 для дальнейшей очистки, где, после того как материальный поток 11, содержащий азеотропный агент, отгоняют из верхней части колонны, этиленгликолевый продукт с чистотой не менее 99,9% по массе получают в кубовой части колонны.
Настоящее изобретение будет далее конкретизировано при помощи следующих примеров.
ПРИМЕРЫ
Пример 1
Использована технологическая схема, показанная на фигуре 1. Материальный поток 1 является жидкофазным продуктом, полученным путем гидрирования оксалата, и содержит следующие компоненты в массовых процентах: метанол - 85,65%, этанол - 0,20%, метилгликолят - 0,15%, диметилоксалат - 0,45%, 1,2-пропиленгликоль - 0,21%, 1,2-бутандиол - 0,40%, этиленгликоль - 12,20%, диэтиленгликоль и другие легкие и тяжелые компоненты - 0,84%.
Первая разделительная колонна C1 являлась колонной для удаления первого легкого компонента и имела 20 теоретических тарелок, материальный поток 1 подавался на 15-ю теоретическую тарелку, рабочее давление являлось атмосферным давлением, флегмовое число составляло 0,5, температура в верхней части колонны составляла 64,2°C, температура в кубовой части колонны составляла 92,7°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 3 в кубовой части колонны, после того как 97% метанола было отведено колонной С1 для удаления первого легкого компонента, составляли: 73,2%, 0,61%.
Вторая разделительная колонна C2 являлась колонной удаления второго легкого компонента и имела 50 теоретических тарелок, материальный поток 3 подавался на 35-ю теоретическую тарелку, рабочее давление являлось атмосферным давлением, флегмовое число составляло 1,5, температура в верхней части колонны составляла 69,3°C, температура в кубовой части колонны составляла 196,7°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 5 в кубовой части колонны, после того как эфирные соединения, имеющие низкие точки кипения, такие как метилгликолят, были отведены колонной удаления второго легкого компонента, составляли: 94,54%, 0,79%.
Третья разделительная колонна C3 являлась колонной азеотропной ректификации и имела 20 теоретических тарелок, материальный поток 5 подавали на 15-ю теоретическую тарелку, материальный поток 6, содержащий свежий азеотропный агент AZ1 (где замещающими группами R1, R2, R3 и R4 были соответственно -H, -H, -CH3, -(CH2)4CH3), подавали из верхней части колонны, молярное отношение азеотропного агента AZ1 к этиленгликолю в материальном потоке 5 было 1,5:1, рабочее давление было атмосферным давлением, флегмовое число составляло 2, температура в верхней части колонны была 166,7°C, температура в кубовой части колонны была 200,5°C, азеотропная смесь 7, образованная этиленгликолем и азеотропным агентом, отгонялась из верхней части колонны и поступала в фазовый сепаратор D1 после конденсации. После отделения фазы верхний продукт D1 являлся материальным потоком 9, обогащенным азеотропным агентом, который возвращался в верхнюю часть колонны азеотропной ректификации, чтобы продолжать участвовать в азеотропии, а нижний продукт D1 был материальным потоком 10, обогащенным этиленгликолем, а именно неочищенным этиленгликолевым продуктом, который не содержал 1,2-бутандиол, и массовая доля этиленгликоля составляла 89,95%.
Четвертая разделительная колонна C4 была колонной очистки этиленгликоля и имела 100 теоретических тарелок, материальный поток 10 подавался на 30-ю теоретическую тарелку, рабочее давление составляло 30 кПа на основе абсолютного давления, флегмовое число было 50, температура в верхней части колонны была 137,5°C, температура в кубовой части колонны была 171,8°C, и после очистки этиленгликоль имел чистоту в массовых процентах 99,91% и общую степень извлечения 99,10%.
Пример 2
Использована технологическая схема, показанная на фигуре 1. Материальный поток 1 являлся раствором, содержащим этиленгликоль и 1,2-бутандиол, и содержал следующие компоненты в массовых процентах: метанол - 29,46%, диметилоксалат - 3,15%, 1,2-пропиленгликоль - 1,18%, 1,2-бутандиол - 0,33%, этиленгликоль - 65,09% и другие легкие и тяжелые компоненты - 0,79%.
Первая разделительная колонна C1 имела 10 теоретических тарелок, материальный поток 1 подавался на 8-ю теоретическую тарелку, рабочее давление было атмосферным давлением, флегмовое число составляло 0,3, температура в верхней части колонны была 64,2°C, температура в кубовой части колонны была 192,5°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 3, после удаления 95% метанола колонной удаления первого легкого компонента С1, были: 92,33%, 0,47%.
Вторая разделительная колонна C2 имела 30 теоретических тарелок, материальный поток 3 подавали на 20-ю теоретическую тарелку, рабочее давление было атмосферным давлением, флегмовое число составляло 5, температура в верхней части колонны была 97,8°C, температура в кубовой части колонны составляла 196,6°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 5, после того как эфирные соединения, имеющие низкие точки кипения, такие как метилгликолят, были отведены колонной удаления второго легкого компонента, составляли: 96,59%, 0,51%.
Третья разделительная колонна C3 являлась колонной азеотропной ректификации и имела 20 теоретических тарелок, материальный поток 5 подавали на 15-ю теоретическую тарелку, материальный поток 6, содержащий свежий азеотропный агент AZ2 (где замещающими группами R1, R2, R3 и R4 были соответственно -CH3, -CH3, -CH2CH(CH3)2, -CH2CH(CH3)2), подавали из верхней части колонны, молярное отношение азеотропного агента AZ2 к этиленгликолю в материальном потоке 5 было 0,5:1, рабочее давление составляло 50 кПа на основе абсолютного давления, флегмовое число составляло 3, температура в верхней части колонны была 156°C, температура в кубовой части колонны была 185°C, азеотропная смесь 7, образованная этиленгликолем и азеотропным агентом, отгонялась из верхней части колонны и поступала в фазовый сепаратор D1 после конденсации, нижний материальный поток 10 (а именно неочищенный этиленгликолевый продукт) после отделения фазы не содержал 1,2-бутандиола, и массовое содержание этиленгликоля составляло 89,65%.
Четвертая разделительная колонна C4 была колонной очистки этиленгликоля и имела 120 теоретических тарелок, материальный поток 10 подавался на 50-ю теоретическую тарелку, рабочее давление составляло 30 кПа на основе абсолютного давления, флегмовое число было 40, температура в верхней части колонны была 136,8°C, температура в кубовой части колонны была 171,8°C, и после очистки этиленгликоль имел чистоту в массовых процентах 99,95% и общую степень извлечения 93,15%.
Пример 3
Использована технологическая схема, показанная на фигуре 1. Материальный поток 1 являлся раствором, содержащим этиленгликоль и 1,2-бутандиол, и содержал следующие компоненты в массовых процентах: метанол - 67,75%, этанол - 0,10%, диметилоксалат - 0,5%, 1,2-пропиленгликоль - 0,16%, 1,2-бутандиол - 0,45%, этиленгликоль - 30,10% и другие легкие и тяжелые компоненты - 0,94%.
Первая разделительная колонна C1 имела 15 теоретических тарелок, материальный поток 1 подавался на 10-ю теоретическую тарелку, рабочее давление было атмосферным давлением, флегмовое число составляло 0,5, температура в верхней части колонны была 64,2°C, температура в кубовой части колонны была 95,8°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 3, после удаления 93% метанола колонной удаления первого легкого компонента С1, составляли: 81,76%, 1,23%.
Вторая разделительная колонна C2 имела 40 теоретических тарелок, материальный поток 3 подавали на 30-ю теоретическую тарелку, рабочее давление составляло 60 кПа на основе абсолютного давления, флегмовое число составляло 3, температура в верхней части колонны была 54,2°C, температура в кубовой части колонны составляла 185,1°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 5, после того как эфирные соединения, имеющие низкие точки кипения, такие как метилгликолят, были отведены колонной удаления второго легкого компонента, составляли: 97,74%, 1,47%.
Третья разделительная колонна C3 являлась колонной азеотропной ректификации и имела 15 теоретических тарелок, материальный поток 5 подавали на 10-ю теоретическую тарелку, материальный поток 6, содержащий свежий азеотропный агент AZ3 (где замещающими группами R1, R2, R3 и R4 были соответственно -H, -H, -CH2CH3, -(CH2)2CH3), подавали из верхней части колонны, молярное отношение азеотропного агента AZ3 к этиленгликолю в материальном потоке 5 было 3:1, рабочее давление составляло 30 кПа на основе абсолютного давления, флегмовое число составляло 5, температура в верхней части колонны была 136°C, температура в кубовой части колонны была 162°C, азеотропная смесь 7, образованная этиленгликолем и азеотропным агентом, отгонялась из верхней части колонны и поступала в фазовый сепаратор D1 после конденсации, нижний материальный поток 10 (а именно неочищенный этиленгликолевый продукт) после отделения фазы не содержал 1,2-бутандиола, и массовое содержание этиленгликоля составляло 88,33%.
Четвертая разделительная колонна C4 была колонной очистки этиленгликоля и имела 100 теоретических тарелок, материальный поток 10 подавался на 50-ю теоретическую тарелку, рабочее давление составляло 20 кПа на основе абсолютного давления, флегмовое число было 50, температура в верхней части колонны была 122,0°C, температура в кубовой части колонны была 164,5°C, и после очистки этиленгликоль имел чистоту в массовых процентах 99,92% и общую степень извлечения 95,36%.
Пример 4
Использована технологическая схема, показанная на фигуре 1. Материальный поток 1 являлся раствором, содержащим этиленгликоль и 1,2-бутандиол, и содержал следующие компоненты в массовых процентах: метанол - 52,75%, этанол - 0,05%, диметилоксалат - 0,65%, 1,2-пропиленгликоль - 0,53%, 1,2-бутандиол - 0,45%, этиленгликоль - 45,03% и другие легкие и тяжелые компоненты - 0,59%.
Первая разделительная колонна C1 имела 15 теоретических тарелок, материальный поток 1 подавался на 10-ю теоретическую тарелку, рабочее давление было атмосферным давлением, флегмовое число составляло 0,3, температура в верхней части колонны была 64,2°C, температура в кубовой части колонны была 167,8°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 3, после удаления 96% метанола первой колонной для удаления легких компонентов С1, составляли: 93,75%, 0,94%.
Вторая разделительная колонна C2 имела 40 теоретических тарелок, материальный поток 3 подавали на 30-ю теоретическую тарелку, рабочее давление составляло 80 кПа на основе абсолютного давления, флегмовое число составляло 5, температура в верхней части колонны была 85,3°C, температура в кубовой части колонны составляла 191,3°C, и массовые доли компонентов этиленгликоля и 1,2-бутандиола в материальном потоке 5, после того как эфирные соединения, имеющие низкие точки кипения, такие как метилгликолят, были отведены колонной удаления второго легкого компонента, составляли: 97,90%, 0,96%.
Третья разделительная колонна C3 являлась колонной азеотропной ректификации и имела 30 теоретических тарелок, материальный поток 5 подавали на 20-ю теоретическую тарелку, материальный поток 6, содержащий свежий азеотропный агент AZ4 (где замещающими группами R1, R2, R3 и R4 были соответственно -CH2CH3, -CH2CH3, -H, -CH3) подавали из верхней части колонны, молярное отношение азеотропного агента AZ4 к этиленгликолю в материальном потоке 5 было 2:1, рабочее давление было атмосферным давлением, флегмовое число составляло 2, температура в верхней части колонны была 161°C, температура в кубовой части колонны была 198°C, азеотропная смесь 7, образованная этиленгликолем и азеотропным агентом, отгонялась из верхней части колонны и поступала в фазовый сепаратор D1 после конденсации, нижний материальный поток 10 (а именно неочищенный этиленгликолевый продукт) после отделения фазы не содержал 1,2-бутандиола, и массовое содержание этиленгликоля составляло 85,16%.
Четвертая разделительная колонна C4 была колонной очистки этиленгликоля и имела 80 теоретических тарелок, материальный поток 10 подавали на 50-ю теоретическую тарелку, рабочее давление составляло 15 кПа на основе абсолютного давления, флегмовое число было 60, температура в верхней части колонны была 112,8°C, температура в кубовой части колонны была 160,0°C, и после очистки этиленгликоль имел чистоту в массовых процентах 99,92% и общую степень извлечения 92,81%.
Пример 5
Смесь, содержащая следующие компоненты в массовых процентах: метанол - 0,23%, диметилоксалат - 0,09%, 1,2-пропиленгликоль - 0,79%, 1,2-бутандиол - 3,43%, этиленгликоль - 95,32% и другие легкие и тяжелые компоненты - 0,14%, была отобрана в качестве сырья и разделена третьей разделительной колонной C3 и четвертой разделительной колонной C4, как показано на фигуре 1. Третья разделительная колонна C3 являлась колонной азеотропной ректификации и имела 15 теоретических тарелок, сырье подавалось на 10-ю теоретическую тарелку, материальный поток 6, содержащий свежий азеотропный агент AZ1 (где замещающими группами R1, R2, R3 и R4 были соответственно -H, -H, -CH3, -(CH2)4CH3), подавали из верхней части колонны, молярное отношение азеотропного агента AZ1 к этиленгликолю в сырье было 1,5:1, рабочее давление было атмосферным давлением, флегмовое число составляло 2, температура в верхней части колонны была 166,7°C, температура в кубовой части колонны была 200,5°C, азеотропная смесь 7, образованная этиленгликолем и азеотропным агентом, отгонялась из верхней части колонны и поступала в фазовый сепаратор D1 после конденсации. После отделения фазы верхний продукт D1 являлся материальным потоком 9, обогащенным азеотропным агентом, который возвращался в верхнюю часть колонны азеотропной ректификации, чтобы продолжать участвовать в азеотропии, а нижний продукт D1 был материальным потоком 10, обогащенным этиленгликолем, а именно неочищенным этиленгликолевым продуктом, который не содержал 1,2-бутандиола, и массовое содержание этиленгликоля составляло 88,56%.
Четвертая разделительная колонна C4 была колонной очистки этиленгликоля и имела 100 теоретических тарелок, материальный поток 10 подавался на 30-ю теоретическую тарелку, рабочее давление составляло 25 кПа на основе абсолютного давления, флегмовое число было 60, температура в верхней части колонны была 130,3°C, температура в кубовой части колонны была 168,4°C, и после очистки этиленгликоль имел чистоту в массовых процентах 99,96% и общую степень извлечения 98,30%.
Сравнительный пример 1
Жидкофазный продукт, полученный при гидрировании оксалата в примере 1, был отобран в качестве сырьевого материального потока 1 и был разделен по технологической схеме, как показано на фигуре 1, где рабочие условия первой разделительной колонны C1 и второй разделительной колонны C2 были теми же самыми, что и в примере 1, в третью разделительную колонну С3 не добавляли азеотропного агента и для разделения использовали метод обычной ректификации, другие рабочие условия были такими же, что и в примере 1, температура в верхней части колонны была 197,1°C и температура в кубовой части колонны была 225,8°C. Результаты эксперимента показали, что массовые доли компонентов этиленгликоля и 1,2-бутандиола в дистилляте верхней части третьей разделительной колонны C3 составляли 89,91% и 3,67%, что показало, что этиленгликоль и 1,2-бутандиол не были эффективно разделены; дистиллят дальше поступал в четвертую разделительную колонну C4 для разделения, где число теоретических тарелок разделительной колонны C4 было 120, рабочее давление составляло 25 кПа на основе абсолютного давления, флегмовое число было 130, температура в верхней части колонны была 140,0°C, температура в кубовой части колонны была 168,0°C, и этиленгликоль в кубовой части колонны, полученный после очитки, имел чистоту в массовых процентах 99,04% и общую степень извлечения 49,56%.
Сравнительный пример 2
Жидкофазный продукт, полученный при гидрировании оксалата в примере 1, был взят в качестве сырьевого материального потока 1 и разделен по технологической схеме, как показано на фигуре 1, где рабочие условия первой разделительной колонны C1 и второй разделительной колонны C2 были такими же, что и в примере 1, этилбензол добавляли в третью разделительную колонну С3 в качестве азеотропного агента, и молярное отношение этилбензола к этиленгликолю в материальном потоке 5 составляло 3:1, другие рабочие условия были теми же самыми, что и в примере 1, температура в верхней части колонны была 132,0°C и температура в кубовой части колонны была 198,0°C. Экспериментальные результаты показали: азеотропная смесь из верхней части третьей разделительной колонны C3 была направлена в фазовый сепаратор D1 после конденсации, массовые доли компонентов этиленгликоля и 1,2-бутандиола в нижнем материальном потоке после отделения фазы составляли 94,35% и 2,64%, что показало, что этиленгликоль и 1,2-бутандиол не были эффективно разделены; дистиллят далее поступал в четвертую разделительную колонну C4 для разделения, рабочие условия были такими же, что и в сравнительном примере 1, этиленгликоль в кубовой части колонны, полученный после очистки, имел чистоту в массовых процентах 99,72% и УФ-пропускания были 78, 92 и 99, соответственно, при соответственных длинах волн 220 мм, 275 мм и 350 мм, и после того, как этиленгликолевые продукты, полученные в примерах 1 и 2, были смешаны, УФ-пропускания составляли 86, 95 и 100 соответственно.

Способ отделения побочных продуктов в водной фазе синтеза фишера-тропша
Способ селективного гидрирования фенилацетилена в присутствии стирола с использованием композитного слоя
Обессеривающий адсорбент, способ его приготовления и использования
Способ селективного гидрирования фенилацетилена в присутствии стирола
Нагруженный металлом катализатор и способ его приготовления
Добавка и способ для обрыва полимеризации и/или снижения вязкости раствора полимера
Каталитический компонент для полимеризации олефинов и катализатор, включающий таковой
Способ получения оксалата монооксидоуглеродным газофазным способом
Компонент катализатора, применяемый для полимеризации олефинов, способ его получения и катализатор, содержащий такой компонент
Способ обработки серосодержащего газа и используемый для данных целей катализатор гидрирования
Способ отделения побочных продуктов в водной фазе синтеза фишера-тропша
Способ селективного гидрирования фенилацетилена в присутствии стирола с использованием композитного слоя
Обессеривающий адсорбент, способ его приготовления и использования
Способ селективного гидрирования фенилацетилена в присутствии стирола
Нагруженный металлом катализатор и способ его приготовления
Добавка и способ для обрыва полимеризации и/или снижения вязкости раствора полимера
Каталитический компонент для полимеризации олефинов и катализатор, включающий таковой
Способ получения оксалата монооксидоуглеродным газофазным способом
Компонент катализатора, применяемый для полимеризации олефинов, способ его получения и катализатор, содержащий такой компонент
Способ обработки серосодержащего газа и используемый для данных целей катализатор гидрирования

