КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ СЛАБООСНОВНЫЕ ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА, И ЛЕКАРСТВЕННЫЕ ФОРМЫ С КОНТРОЛИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка заявляет приоритет предварительной заявки на патент США №61/045170, поданной 15 апреля 2008 г., которая ссылкой включается в настоящее описание полностью во всех отношениях.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Многие терапевтические средства наиболее эффективны тогда, когда они становятся доступны с постоянными скоростями в центрах абсорбции или поблизости от них. Абсорбция терапевтических средств, сделанная, таким образом, доступной, как правило, приводит к требуемым концентрациям в плазме, ведущим к максимальной эффективности и минимуму токсических побочных эффектов. Множество усилий было направлено на разработку сложных систем доставки лекарственных веществ, таких как осмотические устройства для перорального применения. Однако существуют случаи, в которых простые системы доставки лекарственных веществ, такие как матричные таблетки, включающие полимеры, контролирующие скорость растворения, или монолитные или многочастичные системы, покрытые функциональными полимерами, неспособны обеспечить целевые фармакокинетические (ФК) профили, пригодные для одно- или двухразовых режимов дневного дозирования.
Для соответствующей абсорбции лекарственного вещества из желудочно-кишечного тракта, лекарственное вещество должно высвободиться из лекарственной формы и стать доступным в форме раствора в центре абсорбции или поблизости от него. Скорость, с которой лекарственное вещество переходит в раствор и высвобождается из лекарственной формы, важна для кинетики абсорбции лекарственного вещества. Лекарственная форма и, следовательно, активный ингредиент подвержены в ходе транзита различным значениям рН, изменяющимся от рН 1,2 (на голодный желудок) до около 7,0 (желчь и кишечник). Кроме того, время транзита лекарственной формы по отдельным частям пищеварительного тракта может значительно изменяться в зависимости от размера лекарственной формы и локальных условий внутри пищеварительного тракта. Другие факторы, влияющие на абсорбцию лекарственного вещества, включают физико-химические свойства самого лекарственного вещества, такие как рКа, растворимость, энергия кристаллической решетки и удельная площадь поверхности. Превалирующие локальные условия внутри пищеварительного тракта, играющие важную роль, включают свойства содержимого полостей (рН, поверхностное натяжение, объем, перемешивание и буферная емкость), а также изменения, происходящие вследствие приема пищи. Соответственно, часто трудно достигнуть высвобождения лекарственного вещества с постоянными скоростями, особенно в случае чрезвычайно растворимых или свободно растворимых слабоосновных лекарственных веществ, которые быстро высвобождаются при кислотных рН среды, что, таким образом, приводит к выбросу дозы. Функциональные полимерные мембраны, включающие подходящие сочетания синтетических полимеров, таких как водорастворимые полимеры (например, повидон), нерастворимые в воде полимеры (например, этилцеллюлозу) и энтеросолюбильные полимеры (например, устойчивый в желудке гипромеллозы фталат), наносятся на ядра таблеток или пеллет, включающие лекарственное вещество, для достижения замедленных профилей высвобождения с ограниченным успехом.
Перорально распадающиеся лекарственные формы неуклонно набирают популярность как более удобная и потенциально более безопасная альтернатива традиционным таблеткам и капсулам. Эти быстро диспергирующиеся лекарственные формы распадаются в ротовой полости и легко проглатываются с водой. Они являются благоприятными для 50% населения, испытывающих трудности с проглатыванием традиционных таблеток и капсул (обычные для гериатрических и педиатрических пациентов), людей, не имеющих свободного доступа к воде (например, прикованных к постели или неподвижных пациентов, или активных людей, часто находящихся вдали от дома), а также сиделок, чьи пациенты неохотно принимают медикаменты. Перорально распадающиеся лекарственные формы помогают улучшить соблюдение пациентом режимов пероральной дозировки, поскольку они легко вводятся, удобны для осторожного приема где угодно и сложны для отказа от приема после введения. Однако от этих лекарственных форм требуется не только быстрый распад при контакте со слюной в ротовой полости, но и приемлемые органолептические (т.е. приятный вкус) и фармакокинетические (т.е. скорость и продолжительность высвобождения лекарственного вещества) свойства, соответствующие конкретному лекарственному веществу и заболеванию, лечение которого производится. Эти свойства часто взаимно противоположны. Таким образом, разработка перорально распадающихся таблеток (ПРТ), содержащих слабоосновные лекарственные вещества, которые свободно растворимы в интервале физиологических значений рН 1,2-6,8 для одно- и двухразовых режимов дневной дозировки, является чрезвычайно многообещающей.
Слабоосновные лекарственные вещества быстро высвобождаются в кислой среде и поэтому часто не способны обеспечить профили ФК, пригодные для одно- или двухразового режима дневной дозировки. Кроме того, основные лекарственные вещества трудны в работе с ними, если для получения терапевтического эффекта требуются высокие дозы, поскольку их растворимость уменьшается на 1-2 порядка при транзите через желудок в толстую кишку. Если нанести чрезвычайно плотные полимерные покрытия с намерением замедлить высвобождение лекарственного вещества в области более низких значений рН, т.е. при рН 1,2-6,8, лекарственное вещество будет высвобождаться путем диффузии через мембрану покрытия со скоростью, настолько медленной, что не будет обладать практической применимостью.
Совместно рассматриваемые опубликованная заявка на патент США №11/668167 (опубликована как заявка №2007/0190145) и заявка на патент США №11/668408 (опубликована как заявка №2007/0196491, поданные 29 января 2007 г., раскрывают фармацевтические композиции, включающие раздельные слои слабоосновных лекарственных веществ и органической кислоты.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном варианте осуществления данное изобретение относится к фармацевтической композиции, включающей множество частиц с контролированным высвобождением, где каждая частица включает ядро, включающее слабоосновное вещество, слой щелочного буфера, расположенный над ядром лекарственного вещества, и покрытие контролированного высвобождения, включающее нерастворимый в воде полимер.
В одном варианте осуществления данное изобретение относится к фармацевтической композиции, включающей множество частиц с контролированным высвобождением, где каждая частица включает ядро, включающее слабоосновное вещество, содержащее, по меньшей мере, один азотсодержащий фрагмент с рКа от около 5 до около 14, с растворимостью, по меньшей мере, 200 мг/мл при комнатной температуре в водном растворе при рН около 1,2-6,8 и растворимостью не более чем около 10 мг/мл при рН 8 и выше, слой щелочного буфера, расположенный над ядром лекарственного вещества, и покрытие контролированного высвобождения, расположенное над слоем щелочного буфера, где покрытие контролированного высвобождения включает нерастворимый в воде полимер.
В другом варианте осуществления данное изобретение относится к способу изготовления фармацевтической композиции, где способ включает (а) изготовление ядра, включающего слабоосновное лекарственное вещество, (b) покрытие ядра этапа (а) слоем, включающим щелочной буфер, (с) покрытие ядра, покрытого слоем щелочного буфера, этапа (b) слоем контролированного высвобождения.
В другом варианте осуществления данное изобретение относится к фармацевтической лекарственной форме, включающей множество частиц. Каждая частица включает ядро, включающее слабоосновное лекарственное вещество, слой щелочного буфера, расположенный над ядром, и покрытие контролированного высвобождения, расположенное над слоем щелочного буфера, где покрытие контролированного высвобождения включает нерастворимый в воде полимер, необязательно, в сочетании с энтеросолюбильным или водорастворимым полимером.
Еще в одном варианте осуществления данное изобретение относится к фармацевтической лекарственной форме, включающей, по меньшей мере, две совокупности частиц лекарственного вещества. Первая совокупность частиц лекарственного вещества включает ядра, включающие слабоосновное лекарственное вещество, а вторая совокупность частиц лекарственного вещества включает ядра, включающие слабоосновное лекарственное вещество, слой щелочного буфера, расположенный над ядром, и покрытие контролированного высвобождения, расположенное над слоем щелочного буфера, где покрытие контролированного высвобождения включает нерастворимый в воде полимер, необязательно, в сочетании с энтеросолюбильным или водорастворимым полимером.
Еще в одном варианте осуществления данное изобретение относится к фармацевтической лекарственной форме, включающей, по меньшей мере, две совокупности частиц лекарственного вещества. Одна совокупность частиц лекарственного вещества включает ядра, включающие слабоосновное лекарственное вещество, а вторая совокупность частиц лекарственного вещества включает ядра, включающие слабоосновное лекарственное вещество, слой щелочного буфера, расположенный над ядром лекарственного вещества, и покрытие контролированного высвобождения, расположенное над слоем щелочного буфера, где покрытие контролированного высвобождения включает нерастворимый в воде полимер сам по себе или в сочетании с энтеросолюбильным или водорастворимым полимером.
В другом варианте осуществления данное изобретение относится к способу изготовления фармацевтической лекарственной формы. В одном из вариантов фармацевтическая лекарственная форма изготавливается путем смешивания микрочастиц, описываемых в настоящем описании, с быстро диспергирующимися гранулами, включающими сахарид и/или сахарный спирт в сочетании с дезинтегрантом, с образованием смеси для прессования, а также прессование смеси в таблетку. В другом варианте фармацевтическая лекарственная форма изготавливается путем заполнения капсулы описанными в настоящем описании микрочастицами.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
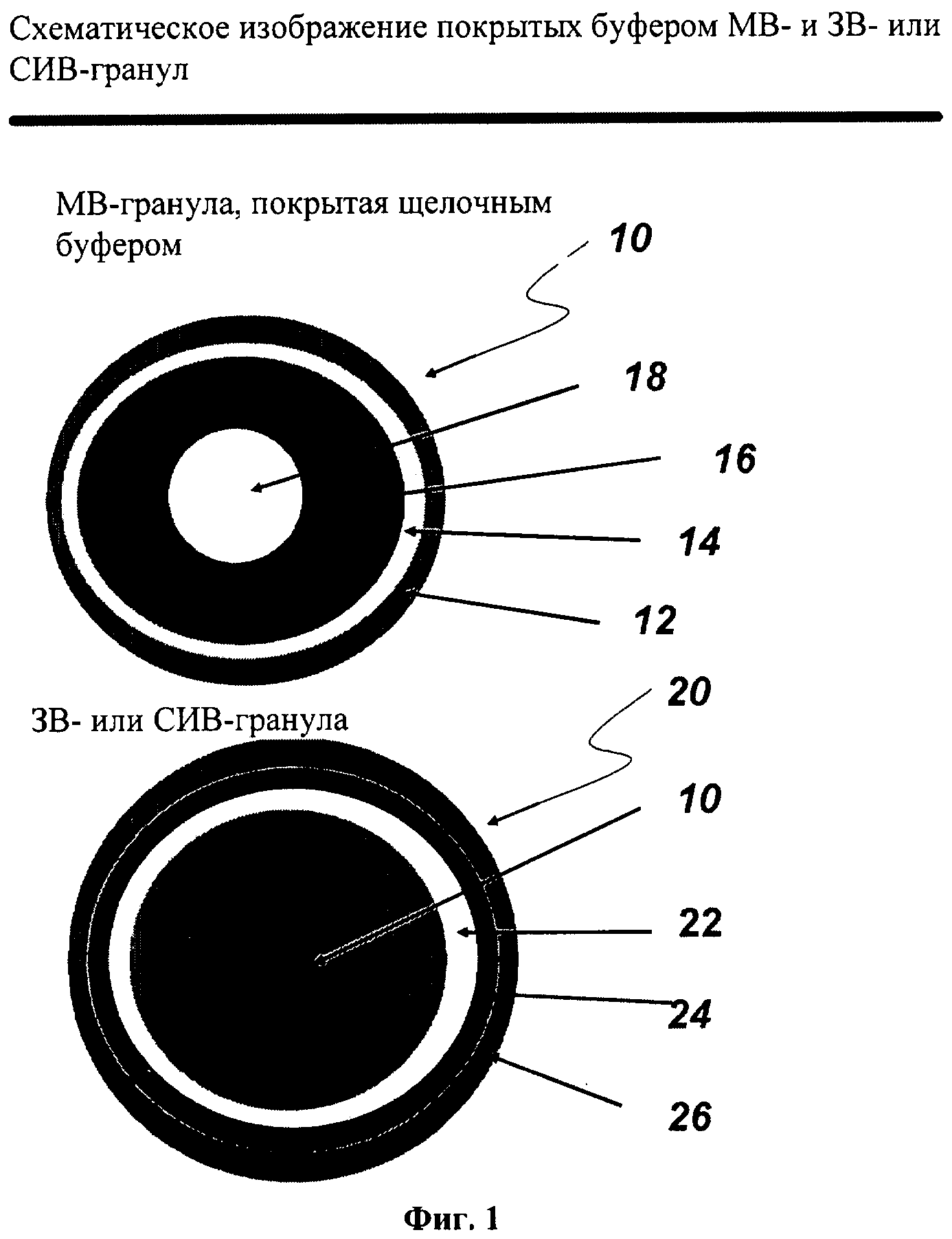
ФИГ. 1 иллюстрирует поперечные сечения МВ-гранул, покрытых щелочным буфером (верхняя иллюстрация), и ЗВ- и СИВ-гранул, включающих покрытую щелочным буфером МВ-гранулу, включающую слабоосновное лекарственное вещество, в соответствии со специфическими вариантами осуществления данного изобретения (нижняя иллюстрация). На Фиг.1 (верхнее схематическое изображение) покрытая щелочным буфером МВ-гранула 10 включает слой щелочного буфера 72, расположенный на защитном герметизирующем слое 14, который располагается на слое слабоосновного лекарственного вещества 16, расположенного на инертном ядре 18, включающем сферическую частицу сахара или лактозы, микрокристаллическую целлюлозу, маннит-микрокристаллическую целлюлозу или диоксид кремния. На той же фигуре (нижнее схематическое изображение), ЗВ- или СИВ-гранула 20 включает сжимаемый покрывающий слой 26, расположенный на покрытии контролированного высвобождения (ЗВ- или СИВ-слое) 24, который расположен на герметизирующем слое 22, расположенном на покрытой слоем щелочного буфера МВ-грануле 10.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Все цитируемые документы ссылкой включаются в настоящее описание полностью во всех отношениях; цитирование любого документа не следует истолковывать как признание его прототипом по отношению к настоящему изобретению.
Выражения «лекарственное вещество», «активный» или «активный фармацевтический ингредиент», используемые в настоящем описании, включают фармацевтически приемлемое и терапевтически эффективное соединение, его фармацевтически приемлемые соли, стереоизомеры и смеси стереоизомеров, сольваты (включая гидраты), полиморфы и/или сложные эфиры. Ссылаясь на лекарственное вещество в описаниях различных вариантов осуществления изобретения ссылка охватывает основное лекарственное вещество, его фармацевтически приемлемые соли, стереоизомеры и смеси стереоизомеров, сольваты (включая гидраты), полиморфы и/или сложные эфиры.
Выражения «перорально распадающаяся таблетка» или «ПРТ» относятся к таблетке, которая быстро распадается в ротовой полости пациента после ее введения без, например, необходимости пережевывания. Скорость распада может варьироваться, однако она больше, чем скорость распада традиционных твердых лекарственных форм (например, таблеток или капсул), которые, как предусматривается, должны проглатываться сразу после введения, или жевательных твердых лекарственных форм.
Выражение «около» в том смысле, как он используется в настоящем описании для отнесения численной величины, включает «точно». Например, «около 60 секунд» включает 60 секунд точно, а также значения, близкие к 60 секундам (например, 50 секунд, 55 секунд, 59 секунд, 61 секунда, 65 секунд, 70 секунд и т.д.).
Выражение «слабоосновное лекарственное вещество» охватывает лекарственные вещества, содержащие один или несколько азотных фрагментов с рКа в интервале от около 5 до около 14, которые очень хорошо растворимы или легко растворимы в условиях кислотных и нейтральных значений рН (т.е. при рН от около 1,2 до около 6,8), но плохо растворимы выше рН 6,8. Выражения, относящиеся к растворимости (например, «очень хорошо растворимый», «легкорастворимый», «малорастворимый» и т.д.) имеют то же значение, что и в Фармакопее США (Т.26, NF 21, 2003) с учетом понимания того, что приводимые пределы растворимости представляют приблизительные границы. Например, «очень хорошо растворимый» означает - имеющий растворимость не менее 1 г растворенного вещества в 1 мл воды или водного раствора при комнатной температуре и указанном рН; «легкорастворимый» означает имеющий растворимость от около 100 мг до около 1000 мг растворенного вещества на 1 мл воды или водного раствора при комнатной температуре и указанном рН; «малорастворимый» означает имеющий растворимость от менее чем около 100 мг растворенного вещества на 1 мл воды при комнатной температуре.
В том смысле, как он используется в настоящем описании, выражение покрытие «контролированного высвобождения» охватывает покрытия, которые задерживают высвобождение, замедляют высвобождение, предотвращают высвобождение и/или как-либо иначе продлевают высвобождение лекарственного вещества из частицы, покрытой покрытием контролированного высвобождения. Выражение «контролированное высвобождение» охватывает выражения «замедленное высвобождение» и «синхронное импульсное высвобождение». Покрытие контролированного высвобождения также может называться в настоящем описании покрытием, обеспечивающим «время запаздывания».
В том смысле, как он используется в настоящем описании, выражение «ядро с мгновенным высвобождением» относится к ядру, содержащему лекарственное вещество, необязательно, покрытому слоем герметизирующего вещества, но не покрытому покрытием контролированного высвобождения. Выражение «ядро с мгновенным высвобождением» может включать кристаллы лекарственного вещества (или аморфные частицы), гранулы лекарственного вещества с одним или несколькими вспомогательными веществами или инертное ядро (например, сферическую частицу сахара), покрытое слоем лекарственного вещества (и, необязательно, связывающего вещества), защитное герметизирующее покрытие и, необязательно, слой щелочного буфера. «Ядра с мгновенным высвобождением» обладают свойствами мгновенного высвобождения, как описано в настоящем описании. Частицы с продленным высвобождением (например, ЗВ-частицы, СИВ-частицы и т.д.) могут изготавливаться путем покрытия ядер с мгновенным высвобождением покрытием продленного высвобождения.
В том смысле, как он используется в настоящем описании, выражение «мгновенное высвобождение», или MB, относится к высвобождению более чем или равному около 50% (особенно в случае маскировки вкуса для введения в лекарственные формы перорально распадающихся таблеток), предпочтительно - более чем около 75%, более предпочтительно - более чем около 90%, и, в соответствии с некоторыми вариантами осуществления изобретения, более чем около 95%, активного вещества в течение около 2 ч, конкретнее - в течение около 1 ч, после введения лекарственной формы.
Выражение «СИВ-частица» или «СИВ-гранула» относится к частице, содержащей лекарственное вещество, например, к грануле, покрытой слоем лекарственного вещества, грануле, содержащей лекарственное вещество, или частице лекарственного вещества, покрытой СИВ-покрытием (покрытием «синхронного импульсного высвобождения»), СИВ-покрытие предусматривает импульс мгновенного высвобождения лекарственного вещества или профиль замедленного высвобождения лекарственного вещества после истечения заранее определенного времени запаздывания. Выражение «время запаздывания» относится к временному периоду после перорального введения частицы, содержащей лекарственное вещество, или после выдерживания в двухступенчатой растворяющей среде или имитированной (-ых) биологической(-их) жидкости(-ях), во время которого из частицы, содержащей лекарственное вещество, высвобождается менее чем около 10% лекарственного вещества. В одном варианте осуществления изобретения выражение «время запаздывания» относится к временному периоду, во время которого лекарственное вещество практически не высвобождается из частицы, или после выдерживания в двухступенчатой растворяющей среде или имитированных биологических жидкостях. В некоторых вариантах осуществления изобретения время запаздывания, составляющее около 1-10 ч, достигается путем покрытия частицы, например, комбинацией, по меньшей мере, одного нерастворимого в воде полимера и, по меньшей мере, одного энтеросолюбильного полимера (например, комбинацией этилцеллюлозы и гипромеллозы фталата). В одном варианте осуществления изобретения время запаздывания находится в диапазоне от около 2 дней до около 10 дней. СИВ-слой, необязательно, может содержать пластификатор.
Выражение покрытие «замедленного высвобождения» или «ЗВ-покрытие», относится к покрытию, обеспечивающему свойства замедленного высвобождения, например, к покрытию, которое замедляет высвобождение лекарственного вещества из частицы, содержащей лекарственное вещество, однако не обеспечивает существенного «времени запаздывания». В одном варианте осуществления изобретения ЗВ-покрытие включает нерастворимый в воде полимер и, необязательно, водорастворимый полимер.
Выражение «в значительной степени распадается» означает уровень распада, составляющий, по меньшей мере, около 50%, по меньшей мере, около 60%, по меньшей мере, около 70%, по меньшей мере, около 80%, по меньшей мере, около 90% или около 100% растворения ПРТ-композиции.
Выражение «в значительной степени маскирует вкус» в отношении маскирующего вкус слоя МВ-частиц (если он присутствует) относится к способности маскирующего вкус слоя в значительной степени предотвращать высвобождение лекарственного вещества, имеющего горький вкус, в ротовой полости пациента. Маскирующий вкус слой, который «в значительной степени маскирует» вкус лекарственного вещества, как правило, высвобождает менее чем около 10% лекарственного вещества в ротовой полости пациента, в других вариантах осуществления изобретения - менее чем около 5%, менее чем около 1%, менее чем около 0,5%, менее чем около 0,1%, менее чем около 0,05%, менее чем около 0,03%, менее чем около 0,01% лекарственного вещества. Маскирующие вкус свойства маскирующего вкус слоя композиций согласно настоящему изобретению могут измеряться т vivo (например, с использованием традиционных органолептических методов тестирования, известных в данной области) или in vitro (например, с использованием описываемых в настоящем описании испытаний растворения). Специалистам в данной области известно, что количество высвобождаемого лекарственного вещества, связанное с маскирующим вкус слоем, который «в значительной степени маскирует» вкус лекарственного; вещества, не ограничено интервалами, явно раскрытыми в настоящем описании, и может варьироваться в зависимости от других факторов, таких как восприимчивость к горькому вкусу лекарственного вещества и, например, присутствие ароматизирующих веществ в композиции.
Выражения «временная зависимость концентрация в плазме», «Cmax», «AUC», «Тщах» и «период полувыведения» имеют свои обычные значения, определяемые FDA Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products (Март 2003 г.).
Если не обусловлено иное, количества различных покрытий или слоев, описываемые в настоящем описании («вес покрытия»), представляется как процентный привес частиц или гранул, оснащенных высушенным покрытием, по отношению к исходному весу частиц или гранул перед наслоением покрытия. Таким образом, вес покрытия 10% относится к высушенному покрытию, которое увеличивает вес частицы на 10%. Если не обусловлено противное, отношения приводятся в вес.%.
В одном варианте осуществления данное изобретение относится к фармацевтической композиции, включающей множество частиц с контролированным высвобождением, где каждая частица включает ядро, включающее слабоосновное вещество, слой щелочного буфера, расположенный над ядром лекарственного вещества, и покрытие контролированного высвобождения, расположенное над слоем щелочного буфера. В конкретных вариантах осуществления изобретения покрытие контролированного высвобождения включает нерастворимый в воде полимер. В соответствии с некоторыми вариантами осуществления настоящего изобретения, фармацевтическая композиция охватывает любые слабоосновные лекарственные вещества, имеющие, по меньшей мере, один азотсодержащий фрагмент, рКа от около 5 до около 14, растворимость, по меньшей мере, 200 мг/мл при комнатной температуре в водном растворе при рН около 1,2-6,8 и растворимость менее чем около 10 мг/мл при рН 8 и выше. Без привязки к теории, описывающей механизм регулирования высвобождения лекарственного вещества, слой щелочного буфера, расположенный на ядре, содержащем слабоосновное лекарственное вещество, создает микроокружение с щелочным рН на границе с лекарственным веществом, где лекарственное вещество, в лучшем случае, малорастворимо даже тогда, когда наружное окружение гранулы с покрытием, регулирующим высвобождение, является кислым, и лекарственное вещество в нем легкорастворймо, что, таким образом, позволяет избежать выброса дозы при пероральном введении.
В некоторых вариантах осуществления слабоосновные лекарственные вещества согласно настоящему изобретению могут выбираться из следующих неограничивающих примеров классов лекарственных веществ: анальгетики, противосудорожные средства, противодиабетические средства, противомикробные средства, противоопухолевые средства, противопаркинсонические средства, противоревматические средства, сердечно-сосудистые средства, стимуляторы центральной нервной системы (ЦНС), агонисты допаминовых рецепторов, противорвотные средства, желудочно-кишечные средства, психотерапевтические средства (например, нейролептики), опиоидные агонисты, опиоидные антагонисты, противоэпилептические средства, гистаминовые H2 антагонисты, противоастматические средства, релаксанты скелетных мышц.
Примеры слабоосновных лекарственных веществ включают в качестве неограничивающих примеров производные бутирофенона, содержащие азотный фрагмент, фениламиноимидазолин (например, клонидин, антигипертензивное средство), дигидроксифенилизопропиламиноэтан (например, фенотерол, бронхолитическое средство), феноксибутиламинопропанол (например, β-адренолитический динитродол), феноксиаминопропан (например, антиаритмический мексилетин), аминоэтилоксазолоазепин (антигипертензивное и антиангинальное средство), или их фармацевтически приемлемые соли, сольваты, сложные эфиры и полиморфы, а также их смеси. В некоторых вариантах осуществления изобретения слабоосновное лекарственное вещество имеет период полувыведения от около 2 ч до около 7 ч.
Выражение «расположенный над» означает, что второй материал наносится на первый материал, где второй материал может находиться или не находиться в физическом контакте с первым материалом. Таким образом, возможно, но не является необходимым, чтобы между первым и вторым материалами лежал промежуточный материал.
Слой щелочного буфера, как ожидается, создает щелочное микроокружение на поверхности лекарственного вещества внутри частицы с контролированным высвобождением. Поскольку слабоосновное лекарственное вещество имеет более низкую растворимость в таком микроокружении, слой щелочного буфера эффективно задерживает высвобождение лекарственного вещества в условиях кислых и нейтральных рН желудочно-кишечного тракта, при которых лекарственное вещество иначе должно было бы растворяться быстро. Введением слоя щелочного буфера в композиции согласно настоящему изобретению можно достичь фармакокинетических профилей, пригодных для одно- и двух разовых режимов дневной дозировки. Неограничивающие примеры щелочных буферов, пригодных для композиций согласно настоящему изобретению, включают натрия гидроксид, мононатрия дигидрофосфат, динатрия гидрофосфат, тринатрия фосфат, натрия ацетат, натрия карбонат или бикарбонат, монокалия дигидрофосфат, дикалия гидрофосфат, трикалия фосфат, калия ацетат, калия карбонат или бикарбонат, магния фосфат, магния ацетат, магния карбонат, магния оксид, магния гидроксид, натрия силикат, кальция силикат, сложный метасиликат магния-алюминия, а также их смеси. Слой щелочного буфера, необязательно, содержит полимерное связывающее вещество. Полимерное связывающее вещество может выбираться из группы, состоящей из гидрокиспропилцеллюлозы, повидона, метилцеллюлозы, гидроксипропилметилцеллюлозы, карбоксиалкилцеллюлозы, полиэтиленоксида и полисахарида.
Внутри частиц с контролированным высвобождением слой щелочного буфера располагается на слое герметизирующего вещества, который, в свою очередь, расположен на ядре, включающем слабоосновное лекарственное вещество. В одном варианте осуществления изобретения слой щелочного буфера может включать полимерное связывающее вещество, если это необходимо. Неограничивающие примеры пригодных полимерных связывающих веществ включают гидроксипропилцеллюлозу, повидон, метилцеллюлозу, гидроксипропилметилцеллюлозу, карбоксиалкилцеллюлозу, полиэтиленоксид, крахмал и полисахарид. В некоторых вариантах осуществления изобретения соотношение щелочного буфера и слабоосновного лекарственного вещества находится в диапазоне от около 5:1 до коло 1:5, включая от около 3:1 до около 1:3.
Композиции согласно настоящему изобретению могут, в некоторых вариантах осуществления изобретения, включать герметизирующий слой, расположенный на ядре, содержащем лекарственное вещество, под слоем щелочного буфера. Данный защитный герметизирующий слой отделяет ядро, содержащее лекарственное вещество, от слоя щелочного буфера и может обеспечивать одно или несколько преимуществ: предотвращение (или минимизацию) контакта между лекарственным веществом и щелочным буфером в ходе обработки или хранения, предотвращение (или минимизацию) статического свойства, предотвращение (или минимизацию) истирания частиц, избежание потенциальной нестабильности, которая может являться результатом нахождения лекарственного вещества поблизости от щелочного буфера в ходе укладки лекарственного вещества и хранения (например, образования дополнительного соединения между кислотой и буфером), гарантию, что щелочной буфер и слабоосновное лекарственное вещество не образуют непосредственный контакт, пока лекарственная форма не образует контакт с растворяющей средой или биологической жидкостью в ходе перорального приема внутрь. В одном варианте осуществления изобретения герметизирующий слой включает гидрофильный полимер. Неограничивающие примеры пригодных гидрофильных полимеров включают гидрофильную гидроксипропилцеллюлозу (например, Klucel® LF), гидроксипропилцеллюлозу, или гипромеллозу, (например, Opadry® Clear или Pharmacoat™ 603), сополимер винилпирролдидона и винилацетата (например, Kollidon® VA 64, BASF) и этилцеллюлозу малой вязкости (например, с вязкостью 10 сП или менее в 5% растворе в смеси 80/20 толуол/спирт при 25°С при измерении с использованием вискозиметра Ubbelohde). Герметизирующий слой может состоять из от около 1% до около 20% по весу содержащего лекарственное вещество и покрытого защитным слоем ядра, например, около 1%, около 2%, около 3%, около 4%, около 5%, около 7%, около 10%, около 12%, около 15%, около 17%, около 20%, включая все интервалы и подинтервалы между ними.
В некоторых вариантах осуществления микрочастицы согласно настоящему изобретению включают покрытие контролированного высвобождения, включающее нерастворимый в воде полимер, наслоенное на слой щелочного буфера. В некоторых вариантах осуществления изобретения покрытие контролированного высвобождения включает нерастворимый в воде полимер в отсутствие водорастворимого или энтеросолюбильного полимера. В этом последнем варианте осуществления изобретения покрытие контролированного высвобождения замедляет высвобождение лекарственного вещества более чем на от около 8 ч до около 20 ч при испытании по методу двухступенчатого растворения (700 мл 0,1 н. НСl (соляной кислоты) в течение первых 2 ч, а затем - 900 мл при рН 6,8, полученном путем добавления 200 мл модификатора рН), и является пригодным для одно- и двухразового режима дневной дозировки.
Неограничивающие примеры пригодных нерастворимых в воде полимеров включают этилцеллюлозу, целлюлозы ацетат, целлюлозы ацетат-бутират, поливинилацетат, нейтральные сополимеры метакриловой кислоты и метилметакрилата, а также их смеси. В одном варианте осуществления изобретения нерастворимый в воде полимер включает этилцеллюлозу. В другом варианте осуществления изобретения нерастворимый в воде полимер включает этилцеллюлозу со средней вязкостью 10 сП в 5% растворе в смеси 80/20 толуол/спирт, измеренной при 25°С вискозиметром Ubbelohde. Нерастворимый в воде полимер покрытия контролированного высвобождения обеспечивает привес от около 3% до около 30%, включая около 3%, около 5%, около 7%, около 10%, около 12%, около 15%, около 17%, около 20%, около 22%, около 25%, около 27%, около 30%, около 35% и около 40%, включая все интервалы и подинтервалы между ними. В одном варианте осуществления изобретения микрочастица с замедленным высвобождением может содержать покрытие замедленного высвобождения из пластифицированного нерастворимого в воде полимера, такого как этилцеллюлоза (ЕС-10), в количестве около 5-50% по весу для замедления высвобождения лекарственного вещества на более чем около 4-20 ч.
В одном варианте осуществления нерастворимый в воде полимер покрытия контролированного высвобождения дополнительно включает пластификатор. Неограничивающие примеры пригодных пластификаторов включают триацетин, трибутилцитрат, триэтилцитрат, ацетил-три-н-бутилцитрат, диэилфталат, касторовое масло, дибутилсебацинат, моноацетилированные и диацетилированные диглицериды (например, Myvacet® 9-45), а также их смеси. При использовании в одном варианте осуществления настоящего изобретения пластификатор может составлять от около 3% до около 30% по весу в расчете на нерастворимый в воде полимер. В другом варианте осуществления изобретения пластификатор составляет от около 10% до около 25% по весу в расчете на нерастворимый в воде полимер. В других вариантах осуществления изобретения количество пластификатора по отношению к весу нерастворимого в воде полимера составляет около 3%, около 5%, около 7%, около 10%, около 12%, около 15%, около 17%, около 20%, около 22%, около 25%, около 27% и около 30%, включая все интервалы и подинтервалы между ними. Специалисту в данной области понятно, что тип(-ы) и количество(-а) пластификатора(-ов) выбираются на основе полимера, или полимеров, и природы системы покрытия (например, покрытия на основе воды или растворителя, раствора или дисперсии, а также/общего содержания твердых веществ). В одном варианте осуществления изобретения, если пластификатор используется в покрытии контролированного высвобождения, пластификатор не содержит фталатов.
В одном варианте осуществления настоящего изобретения в каждом из слоев покрытий, где присутствует пластификатор, пластификатор(-ы) не содержит фталатов.
В некоторых вариантах осуществления покрытие контролированного высвобождения, расположенное на слое щелочного буфера, включает водорастворимый полимер в комбинации с водорастворимым полимером и обеспечивает замедленное высвобождение лекарственного вещества. В одном варианте осуществления соотношение нерастворимого в воде полимера и водорастворимого полимера находится в диапазоне от около 95/5 до около 50/50, включая интервал от около 90/10 до около 60/40. В другом варианте осуществления нерастворимые в воде полимеры в сочетании с водорастворимыми полимерами составляют от около 3% до около 50% по весу в расчете на покрытое ядро, включая интервалы от около 10% до около 50%, от около 3% до около 30%, а также от около 5% до около 30%. В других вариантах осуществления изобретения количество нерастворимого в воде полимера в сочетании с водорастворимым полимером составляет около 3%, около 5%, около 7%, около 10%, около 12%, около 15%, около 17%, около 20%, около 22%, около 25%, около 27%, около 30%, около 35%, около 40%, около 45%, около 50% по весу в расчете на ядро с мгновенным высвобождением, включая все интервалы и подинтервалы между ними.
Водорастворимые полимеры, используемые в соответствии с некоторыми вариантами осуществления настоящего изобретения, охватывают водорастворимые полимеры. Неограничивающие примеры пригодных водорастворимых полимеров включают поливинилпирролидон (например, Povidone K-25), полиэтиленгликоль (например, ПЭГ 400), гидроксипропилметилцеллюлозу и гидроксипропилцеллюлозу. В одном варианте осуществления покрытие замедленного высвобождения обеспечивает высвобождение лекарственного вещества, замедленное на более чем около 12 ч до около 16 ч при испытании по методу двухступенчатого растворения (700 мл 0,1 н. НСl (соляной кислоты) в течение первых 2 ч, а затем - 900 мл при рН 6,8 путем добавления 200 мл модификатора рН), и пригодно для одно- или двухразового режима дневной дозировки.
В другом варианте осуществления изобретения покрытие контролированного высвобождения включает нерастворимый в воде полимер в сочетании с растворимым в желудке порообразователем и обеспечивает замедленное высвобождение лекарственного вещества. Примером растворимого в желудке порообразователя является кальция карбонат. Другие пригодные растворимые в желудке порообразователи включают натрия хлорид, кальция карбонат, кальция фосфат, кальция сахарид, кальция сукцинат, кальция тартрат, железа ацетат, железа гидроксид, железа фосфат, магния карбонат, магния цитрат, магния гидроксид, магния фосфат и т.д.
В некоторых вариантах осуществления покрытие контролированного высвобождения включает нерастворимый в воде полимер в сочетании с энтеросолюбильным полимером и обеспечивает отложенное, или синхронизированное импульсное высвобождение (СИВ) лекарственного вещества. Данный тип покрытия контролированного высвобождения (т.е., сочетание нерастворимого в воде полимера и энтеросолюбильного полимера) здесь также называется покрытием, обеспечивающим «время запаздывания», а микрочастицы, покрытые покрытием, обеспечивающим время запаздывания, могут также называться СИВ-частицами. Выражение «время запаздывания» относится к временному периоду после перорального введения частицы, содержащей лекарственное вещество, или после выдерживания в двухступенчатой растворяющей среде или имитированной(-ых) биологической(-их) жидкости(-ях), во время которого из частицы, содержащей лекарственное вещество, высвобождается менее чем около 10% лекарственного вещества. В одном варианте осуществления выражение «время запаздывания» относится к временному периоду, во время которого лекарственное вещество практически не высвобождается из частицы, или после выдерживания в двухступенчатой растворяющей среде или имитированной(-ых) биологической(-их) жидкости(-ях). В одном варианте осуществления покрытие, обеспечивающее время запаздывания, наносится непосредственно на слой щелочного буфера. В другом варианте осуществления покрытие, обеспечивающее время запаздывания, наносится непосредственно на один или несколько слоев (например, герметизирующий слой), которые наслоены на слой щелочного буфера. В некоторых вариантах осуществления изобретения соотношение нерастворимого в воде полимера и энтеросолюбильного полимера находится в диапазоне от около 10:1 до около 1:4, включая интервалы от около 9:1 до около 1:3 и от около 3:1 до около 1:1. В других вариантах осуществления нерастворимый в воде и энтеросолюбильный полимеры в сочетании составляют от около 5% до коло 60% по весу в расчете на ядро с мгновенным высвобождением, включая интервалы от около 10% до около 60%, а также от около 10% до около 50%. Неограничивающие примеры пригодных энтеросолюбильных полимеров включают целлюлозы ацетат-фталат, гидроксипропилметилцеллюлозы фталат, гидроксипропилметилцеллюлозы фталат, гидроксипропилметилцеллюлозы ацетат-сукцинат, поливинилацетат-фталат, чувствительные к рН сополимеры метакриловой кислоты и метилметакрилата, шеллак, а также их смеси. (Выражение «чувствительный к рН» относится к полимерам с зависящей от рН растворимостью). Данные энтеросолюбильные полимеры могут применяться в виде сухого порошка или водной дисперсии. Некоторыми имеющимися в продаже материалами, которые могут использоваться, являются сополимеры метакриловой кислоты, имеющиеся в продаже под торговой маркой Eudragit (L100, S100, L30D), производимые Rohm Pharma, Cellacefate (целлюлозы ацетат-фталат) от Eastman Chemical Co., Aquateric (водная дисперсия целлюлозы ацетата-фталата) от FMC Corp.и Aqoat (водная дисперсия гидроксипропилметилцеллюлозы ацетата-сукцината) от Shin Etsu K.K. В одном варианте осуществления СИВ-покрытие включает этилцеллюлозу (например, ЕС-10) в качестве нерастворимого в воде полимера и гипромеллозы фталата (например, HP-55) в качестве энтеросолюбильного полимера.
В одном варианте осуществления СИВ-микрочастицы могут обеспечивать время запаздывания от около 1 ч до около 10 ч, включая от около 2 ч до около 7 ч, от около 2 ч до около 4 ч («малое время запаздывания»), а также от около 7 ч до около 8 ч («длительное время запаздывания»). В другом варианте осуществления СИВ-микрочастицы высвобождают лекарственное вещество в течение периода времени от около 4 ч до около 16 ч в желудочно-кишечном тракте после истечения времени запаздывания от около 1 ч до около 10 ч после перорального введения.
В другом варианте осуществления микрочастицы содержат наружное покрытие, обеспечивающее время запаздывания, которое располагается на покрытии контролированного высвобождения. В варианте осуществления изобретения этого типа высвобождение лекарственного вещества начинается при более высоких рН в кишечнике с последующим замедленным высвобождением лекарственного вещества.
Профили высвобождения лекарственного вещества 3 В- и СИВ-микрочастицами могут определяться испытанием на растворение согласно Фармакопее США в Приборе 1 или 2 с использованием двухступенчатой растворяющей среды (первые 2 часа - в 700 мл 0,1 н. НСl при 37°С, с последующим испытанием на растворение при рН 6,8 путем добавления 200 мл модификатора рН). Высвобождение лекарственного вещества с течением времени можно определять различными методами, например методом ВЭЖХ на образцах, отобранных в выбранные моменты времени.
ЗВ или СИВ-покрытие вносит вклад в регулирование растворения лекарственного вещества на поверхности лекарственного вещества и, следовательно, высвобождение лекарственного вещества из микрочастиц. Достигаемое время запаздывания, или время замедленного высвобождения, зависит от композиции и толщины покрытия замедленного высвобождения и/или от композиции и толщины покрытия, обеспечивающего время запаздывания. Специфические факторы, которые влияют на достижение оптимальных двух- или одноразовых дневных лекарственных форм, включают в качестве неограничивающих примеров рКа терапевтического средства (и его растворимость, т.е. лекарственное вещество, легкорастворимое в условиях кислотных и нейтральных рН, малорастворимо при рН 8,0 и выше), период полувыведения, а также снижение растворимости в микроокружении с щелочным рН, создаваемым щелочным буфером.
В другом варианте осуществления микрочастицы содержат сжимаемое покрытие, расположенное на покрытии контролированного высвобождения (или размещено на самом наружном покрытии, если покрытие контролированного высвобождения дополнительно покрыто СИВ-покрытием). Сжимаемое покрытие включает гидрофильный полимер. В одном варианте осуществления изобретения гидрофильный полимер выбирается из группы, которая состоит из гидроксипропилцеллюлозы, поли(винилацетата и винилпирролидона), поливинилацетата и пластифицированной латексной дисперсии этилцеллюлозы с низкой вязкостью. Это покрытие может наноситься, например, путем покрытия в псевдоожиженном слое пластифицированной водной дисперсией этилцеллюлозы. Его назначением является поддержание целостности мембраны в ходе прессования с быстро диспергирующимися микрогранулами.
Ядро микрочастицы включает слабоосновное лекарственное вещество. В некоторых вариантах осуществления изобретения ядро может принимать форму инертной гранулы, микрогранулы или кристаллов лекарственного вещества. В одном варианте осуществления ядро включает инертную гранулу, покрытую слоем лекарственного вещества, включающим слабоосновное лекарственное вещество. Инертная гранула может включать сахар, микрокристаллическую целлюлозу, маннит-микрокристаллическую целлюлозу, диоксид кремния и т.д. Ядро имеет средний размер частиц не более 400 мкм или, в другом варианте осуществления изобретения, не более 350 мкм. В одном варианте осуществления слой лекарственного вещества включает полимерное связывающее вещество. Полимерное связывающее вещество может выбираться из группы, которая содержит гидроксипропилцеллюлозу, повидон, метилцеллюлозу, гидроксипропилметилцеллюлозу, карбоксиалкилцеллюлозу, полиэтиленоксид, крахмал (например, кукурузный крахмал и желатинизированный кукурузный крахмал) и полисахарид. Соотношение лекарственного вещества и полимерного связывающего вещества может находиться в интервале от около 85:15 до около 100:0 (связывающее вещество отсутствует).
Описываемые в настоящем описании фармацевтические композиции также могут включать быстро диспергирующиеся гранулы, включающие сахарид и/или сахарный спирт в сочетании с дезинтегрантом. Дезинтегрант может быть выбран из группы, которая включает кросповидон, натрия крахмалгликолят, сшитую натрия карбоксиметилцеллюлозу и гидроксипропилцеллюлозу с низкой степенью замещения. Сахарид и/или сахарный спирт может выбираться из группы, которая включает лактозу, сукралозу, сахарозу, мальтозу, маннит, сорбит, ксилит и мальтит.Соотношение дезинтегранта и сахарида и/или сахарного спирта в быстро диспергирующихся микрогранулах находится в диапазоне от около 1/99 до около 10/90, а в некоторых вариантах осуществления изобретения составляет около 5/95 (по весу). В некоторых вариантах осуществления дезинтегрант или сахарид и/или сахарный спирт, или оба, может присутствовать в форме микрочастиц, имеющих средний размер около 30 мкм и менее. Соотношение микрочастиц, содержащих лекарственное вещество, и быстро диспергирующихся гранул может находиться в интервале от около 1:6 до около 1:2.
В другом варианте осуществления данное изобретение относится к фармацевтическим лекарственным формам, включающим описываемые здесь микрочастицы. Фармацевтические лекарственные формы включают перорально распадающиеся таблетки (ПРТ), традиционные таблетки и капсулы (например, капсулы из твердого желатина, гидроксипропилметилцеллюлозы (НРМС) или полисахаридов). Если фармацевтическая лекарственная форма принимает форму ПРТ, ПРТ в значительной степени распадается в течение около 60 секунд после контакта со слюной в оральной полости или с имитированной слюнной жидкостью. В другом варианте осуществления ПРТ в значительной степени распадается в течение около 30 секунд. Распад испытывается в соответствии с испытанием распада №701 согласно Фармакопее США. В одном варианте осуществления ПРТ включает терапевтически эффективное количество слабоосновного лекарственного вещества, где после введения ПРТ в значительной степени распадается в ротовой полости пациента с образованием однородной, легко проглатываемой суспензии, не обладающей песчаной консистенцией или привкусом, и обеспечивает требуемый профиль ФК (т.е. график зависимости концентрации в плазме от времени) указанного слабоосновного лекарственного вещества, пригодный для одно- или двухразового режима дневной дозировки.
Если фармацевтической лекарственной формой является таблетка, она предпочтительно обладает истираемостью менее чем около 1%. Если фармацевтической лекарственной формой является ПРТ, таблетка может также включать фармацевтически приемлемые вспомогательные вещества, пригодные для использования в составах распадающихся таблеток, такие как сжимаемые разбавители, наполнители, красящие средства и, необязательно, смазочное вещество.
В некоторых вариантах осуществления вес ПРТ составляет не более чем около 2000 мг, например, 2000 мг или менее, 1500 мг или менее, 1000 мг или менее, 500 мг или менее. В некоторых вариантах осуществления вес ПРТ составляет не более чем около 1600 мг.В другом варианте осуществления вес ПРТ составляет не более чем около 800 мг. В другом варианте осуществления вес ПРТ составляет не более чем около 500 мг.
ПРТ включают одну или несколько совокупностей 3 В- и/или одну или несколько совокупностей СИВ-микрочастиц, описанных в настоящем описании, или их смесей, в сочетании с быстро диспергирующимися микрочастицами. ПРТ могут дополнительно включать МВ-частицы. Например, фармацевтическая лекарственная форма может включать: ЗВ-микрочастицы в сочетании с быстро диспергирующимися гранулами; СИВ-микрочастицы в сочетании с быстро диспергирующимися гранулами, МВ-микрочастицы, ЗВ-микрочастицы и быстро, диспергирующиеся гранулы; МВ-микрочастицы, СИВ-микрочастицы и быстро диспергирующиеся гранулы; МВ-микрочастицы, ЗВ-микрочастицы и одну или несколько совокупностей СИВ-микрочастиц, которые могут иметь одинаковые или различные времена запаздывания (например, СИВ-микрочастицы с малым временем запаздывания и СИВ-частицы с длительным временем запаздывания), в сочетании с быстро диспергирующимися гранулами. Такие различные комбинации микрочастиц могут достигать различных требуемых профилей высвобождения лекарственного вещества. Например, лекарственная форма для одноразовой дневной дозировки активного вещества с периодом полувыведения около 7 ч может содержать смесь совокупности МВ-гранул, обеспечивающей импульс мгновенного высвобождения, вторую совокупность 3 В- или СИВ-гранул с малым временем запаздывания (около 2-4 ч), обеспечивающую быстрый профиль замедленного высвобождения, и третью совокупность СИВ-гранул с длительным временем запаздывания (около 7-8 ч), которая, позволяет получить профиль отложенного, замедленного высвобождения через около 8-12 ч, для поддержания допустимых концентраций в плазме в течение 12-24 ч.
Если в фармацевтической лекарственной форме присутствуют МВ-частицы, соотношение МВ-частиц и 3 В- и/или СИВ-частиц находится в диапазоне от около 0:100 (МВ-частицы отсутствуют) до около 50:50. Вкус МВ-частиц может маскироваться путем наслоения маскирующего вкус слоя, который в значительной степени маскирует вкус лекарственного вещества, содержащегося в частице. Данные МВ-частицы с маскировкой вкуса высвобождают не более 10% в течение 3 мин (наиболее длительное время удержания, ожидаемое для ПРТ в ротовой полости) при испытании растворения в имитированной слюнной жидкости (рН ~ 6,8), в то время как около 75% дозы высвобождается в течение около 60 минут при испытании растворения в 0,1 н. НСl.
МВ-частицы включают ядро, содержащее лекарственное вещество, необязательно, покрытое нерастворимым в воде полимером (например, этилцеллюлозой), обеспечивающим маскирующий вкус слой. Покрытие из нерастворимого в воде полимера может включать пластификатор. Кроме того, оно может включать растворимый в желудке порообразователь (например, кальция карбонат), например, в соответствии с раскрытием в совместно рассматриваемой заявке на патент США №11/213266, поданной 26 августа 2005 г.(опубликованная заявка №2006/0105038, опубликованная 18 мая 2006 г.), или путем наслоения в псевдоожиженном слое нерастворимого в воде полимера (например, этилцеллюлозы со средней вязкостью 10 сП) самого по себе или в сочетании с растворимым в желудке полимером (например, Eudragit El 00 или ЕРО), например, в соответствии с раскрытием в совместно рассматриваемой заявке на патент США №11/248596, поданной 12 октября 2005 г.(опубликованная заявка №2006/0078614, опубликованная 13 апреля 2006 г.), или растворимым в желудке порообразователем (например, кальция карбонатом), например, в соответствии с раскрытием в совместно рассматриваемой заявке на патент США №11/256653, поданной 12 октября 2005 г.(опубликованная заявка №2006/0105039, опубликованная 18 мая 2006 г.). Каждая из изложенных заявок ссылкой включается в настоящее описание полностью во всех отношениях.
Описываемые в настоящем описании ПРТ могут обладать одним или несколькими следующими преимуществами: (i) распадаются при контакте со слюной в ротовой полости в течение около 60 секунд, образуя однородную, легко проглатываемую суспензию, включающую частицы с маскировкой вкуса и/или содержащие лекарственное вещество; (ii) распадаются в течение 30 секунд при испытании растворения в соответствии с испытанием распада №701 согласно Фармакопее США; (iii) МВ-частицы с маскировкой вкуса, если они присутствуют, обеспечивают быстрое, в значительной степени полное высвобождение дозы при попадании в желудок (например, как правило, более чем около 75% в течение 60 мин); (iv) ЗВ- или СИВ-частицы обеспечивают замедленное и/или отложенное высвобождение лекарственного вещества в желудочно-кишечном тракте.
В другом варианте осуществления данное изобретение относится к способам изготовления фармацевтической композиции из микрочастиц, описанных в настоящем описании. В одном варианте осуществления способ включает: (а) изготовление ядра, содержащего слабоосновное лекарственное вещество, (b) покрытие ядра, содержащего лекарственное вещество, этапа (а) герметизирующим слоем, (с) покрытие ядра, покрытого изолирующим слоем, этапа (b) слоем, включающим щелочной буфер, и (d) покрытие ядра, покрытого слоем щелочного буфера, этапа (с) слоем контролированного высвобождения с образованием микрочастиц. Этап изготовления ядра может осуществляться любым из известных на данном уровне развития техники способов; например, путем наслоения на инертную гранулу (например, сахара, микрокристаллической целлюлозы, маннит-микрокристаллической целлюлозы, диоксида кремния и т.д.) раствора, включающего лекарственное вещество и, необязательно, полимерное связывающее вещество (например, в псевдоожиженном слое или в дражировочном котле); гранулирования лекарственного вещества с соответствующим разбавителем (например, микрокристаллической целлюлозой и/или лактозой); экструзии и сферонизации смеси, содержащей лекарственное вещество; прессования лекарственного вещества в минитаблетки диаметром около 1-2 мм; или получения кристаллов лекарственного вещества с требуемым средним размером частиц (например, около 50-500 мкм, включая 100-400 мкм).
В одном варианте осуществления используется способ изготовления микрочастицы с покрытием замедленного высвобождения. В этом варианте покрытие контролированного высвобождения этапа (d) включает покрытие нерастворимым в воде полимером и, необязательно, водорастворимым полимером до привеса от около 3% до около 30% с образованием ЗВ-микрочастицы. В другом варианте осуществления используется способ изготовления микрочастиц с покрытием синхронизированного импульсного высвобождения (СИВ-покрытием). В этом варианте осуществления покрытие контролированного высвобождения этапа (d) включает нерастворимый в воде полимер и энтеросолюбильный полимер до привеса от около 10% до около 60% с образованием СИВ-микрочастиц. В другом варианте осуществления используется способ изготовления микрочастиц с покрытием замедленного высвобождения, лежащим под наружным покрытием синхронизированного импульсного высвобождения. В данном варианте осуществления покрытие контролированного высвобождения этапа (d) включает наслоение покрытия нерастворимого в воде полимера и, необязательно, водорастворимого полимера до привеса от около 3 до около 30% с образованием частицы с замедленным высвобождением. Микрочастица с замедленным высвобождением затем покрывается слоем, включающим нерастворимый в воде полимер и энтеросолюбильный полимер с образованием ЗВ/СИВ-микрочастицы.
В другом варианте осуществления данное изобретение относится к способу изготовления фармацевтической лекарственной формы, где способ включает: смешивание микрочастиц, описываемых в настоящем описании, с быстро диспергирующимися гранулами, включающими сахарид и/или сахарный спирт в сочетании с дезинтегрантом; и прессование результирующей смеси в таблетку с образованием ПРТ. Еще в одном варианте осуществления фармацевтическая лекарственная форма может быть изготовлена путем заполнения твердой желатиновой капсулы описанными в настоящем описании микрочастицами.
В одном варианте осуществления изобретения способ включает следующие этапы:
a) изготовление частиц слабоосновного лекарственного вещества (кристаллов, микрогранул, гранул или пеллет со средним размером частиц 50-500 мкм, в особенности, 100-400 мкм, в частности, - 100-350 мкм) и наслоение защитного герметизирующего слоя на гранулы, покрытые слоем лекарственного вещества, с образованием МВ-гранул;;
b) наслоение слоя щелочного буфера на МВ-гранулы из раствора полимерного связывающего вещества, если это необходимо, и наслоение защитного герметизирующего покрытия на буферный слой;
c) наслоение покрытия замедленного высвобождения, включающего нерастворимый в воде полимер или нерастворимый в воде полимер в сочетании с водорастворимым полимером до привеса около 3-30% с образованием ЗВ-частиц;
и/или
d) наслоение покрытия, обеспечивающего время запаздывания, на ЗВ-частицы или частицы, покрытые слоем щелочного буфера, комбинации нерастворимого в воде полимера и энтеросолюбильного полимера в весовом соотношении около 10:1-1:4 до привеса около 10-60% по весу в расчете на покрытую' гранулу с образованием СИВ-частиц;
e) необязательно, наслоение слоя гидрофильного полимера на ЗВ-слой или СИВ-слой; и
f) заполнение соответствующими количествами 3 В- и СИВ-частиц в присутствии или в отсутствие МВ-частиц твердых желатиновых капсул; или их прессование в традиционные или перорально распадающиеся таблетки (ПРТ) после смешивания с фармацевтически приемлемыми вспомогательными веществами и одной или несколькими совокупностями гранул (например, комбинации МВ-гранул, 3 В-гранул и/или СИВ-гранул в требуемом соотношении).
Возможно изготовления пеллет, покрытых слоем лекарственного вещества, с использованием Granurex путем управляемой сферонизации и создания слоя щелочного буфера, расположенного над покрытыми изолирующим покрытием МВ-пеллетами в том же Granurex, а затем - наслоение покрытия контролированного высвобождения в оборудовании с псевдоожиженным слоем с образованием ЗВ-, ПВ- или СИВ-гранул.
ПРИМЕРЫ
Пример 1:
Развернутые профили высвобождения лекарственного вещества in vitro для слабоосновного лекарственного вещества
Фармакокинетический анализ был предпринят для идентификации набора теоретических профилей высвобождения лекарственного вещества in vitro, который учитывал бы одно- или двухразовую дневную дозировку лекарственной формы слабоосновных лекарственных веществ. Использовали данные о временной зависимости концентрации в плазме человека при пероральном одноразовом введении, или при гомеостазе, и/или при внутривенном (ВВ) профиле, одно- или двухкомпартментная фармакокинетическая (ФК) модель (например, двухкомпонентная модель показана на схематическом изображении ниже). К ФК модели могут одновременно быть аппроксимированы как пероральные (ПО), так и ВВ-данные. С использованием программного обеспечения WinNonlin оценки параметра ФК и предсказания данных ПО- и/.или ВВ-данных осуществлены с целью генерирования уравнений для модельных профилей. Разработаны составы с профилями высвобождения лекарственного Вещества in vitro, которые имитируют модельные, т.е. развернутые, профили in vitro или охватывают окно целевого профиля. Эти составы затем испытываются в ФК исследованиях на взрослых здоровых объектах.
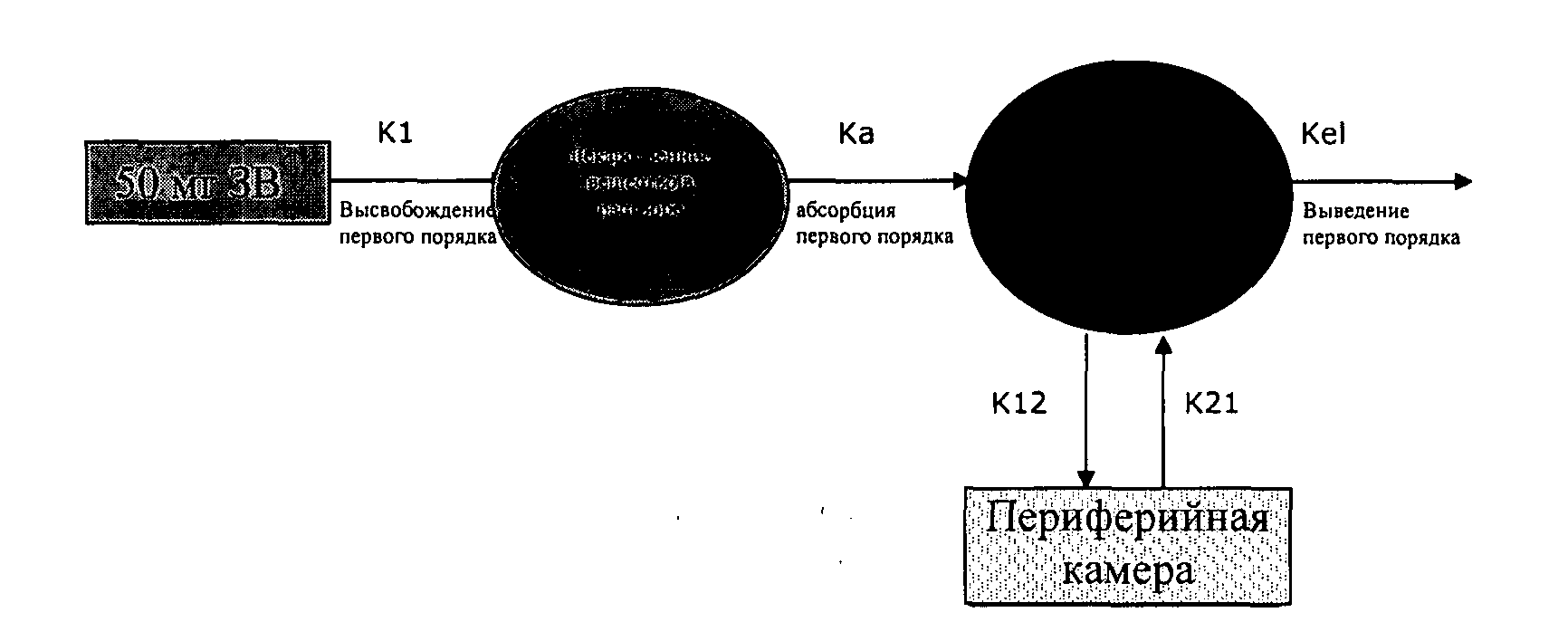
Пример 2
2.А МВ-гранулы, содержащие слабоосновное лекарственное вещество
Связывающий полимер медленно добавляли к системе растворителей (например, вода, ацетон, этанол или их смеси) для изготовления раствора связывающего вещества. Слабоосновное лекарственное вещество медленно добавляли к системе растворителей до растворения. Раствор связывающего вещества затем добавляли к раствору лекарственного вещества и затем перемешивали. В альтернативном варианте связывающее вещество и лекарственное вещество последовательно добавляли до растворения. В установку для наслоения покрытий в псевдоожиженном слое, например, Glatt GPCG 3 (например, оснащенную 7 13/16"колонной Вюрстера с 7" нижним соплом, 'С" пластиной распределения воздуха снизу, покрытой ситом для удерживания продукта размером 200 меш), загружали сферическими частицами сахара (например, размером 60-80 меш), которые затем опрыскивали раствором связывающего вещества и лекарственного вещества. Покрытые сферические частицы сахара затем высушивались для удаления остаточных растворителей (включая влагу) и просеивались (например, через сита 35 и 80 меш) для избавления от частиц большего и меньшего размера.
2.В Наслоение буферного слоя безводного динатрия фосфата (DP А)
Безводный динатрия фосфат добавлялся к очищенной воде при перемешивании до растворения. В установку для наслоения покрытий в псевдоожиженном слое, например, Glatt GPCG 3 (например, оснащенную 13 6" колонной Вюрстера с нижним соплом 8", "С" пластиной распределения воздуха снизу, покрытой ситом для удерживания продукта размером 200 меш и форсункой с диаметром отверстия 1,0 мм) загружали МВ-гранулы (например, из Примера 2.А). На МВ-гранулы распылялся буферный раствор. После, необязательно, ополаскивания растворителем покрытых буфером гранул, наносилось около 2% по весу изолирующего покрытия. Высушенные МВ-гранулы можно просеять (например, с использованием сит 35 и 80 меш) для избавления от гранул большего и меньшего размера.
2.С ЗВ-гранулы. содержащие слабоосновное лекарственное вещество
Покрытые буфером гранулы из Примера 2.В покрывались в установке для наслоения покрытий в псевдоожиженном слое ЗВ-покрытием из, необязательно, платифицированного (например, 10% вес/вес триэтилцитрата от этилцеллюлозы) нерастворимого в воде полимера (например, этилцеллюлозы). Раствор сжимаемого покрытия (например, гидроксипропилцеллюлозы, такой как Klucel® LF), растворенного в растворителе, распылялся на покрытые буфером гранулы до привеса около 2% по весу. Полученные в результате ЗВ-гранулы можно высушить для удаления остаточных растворителей.
2.D Быстро диспергирующиеся микрогранулы
Быстро диспергирующиеся микрогранулы изготавливаются в соответствии с процедурой, раскрытой в совместно рассматриваемой заявке на патент США №10/827106 (опубликована как опубликованная заявка на патент США №2005/0232988, 20 октября 2005 г., содержание которой ссылкой включается в настоящее описание во всех отношениях). D-маннит со средним размером частиц около 20 мкм или менее (например, Pearlitol 25, Roquette, Франция) смешивался с 8 кг поперечно-сшитого повидона (например, Crospovidone XL-10, ISP) в грануляторе с большим усилием сдвига (GMX 600, Vector) и гранулировался с очищенной водой и подвергался мокрому помолу с использованием Comil (Quadro) и высушивался в лотках до потери на высушивание (LOD) менее чем около 1%. Высушенные гранулы просеивались, и материал большего размера размалывался с образованием быстро диспергирующихся микрогранул со средним размером частиц в интервале приблизительно 175-300 мкм.
2.Е ПРТ с контролированным высвобождением, содержащие ЗВ-гранулы
Быстро диспергирующиеся микрогранулы (-1200 г) смешивались с 3 В-гранулами слабоосновного лекарственного вещества (-850 г) и другими фармацевтически приемлемыми ингредиентами, такими как корригент (-25 г), подсластитель (например, сукралоза, -10 г), дополнительный кросповидон (-125 г) и микрокристаллическая целлюлоза (например, Avicel PHI 01, -250 г), с соотношением быстро диспергирующихся микрогранул и ЗВ-гранул около 3:2 в V-образном блендере с двойным корпусом в течение времени, достаточного для образования однородно распределенной смеси для прессования. ПРТ, включающие 50 мг слабоосновного лекарственного вещества в виде ЗВ-гранул прессовались с использованием промышленного таблеточного пресса, оснащенного внешней системой смазки, в следующих условиях: инструмент - круг 15 мм, плоская поверхность, краевой радиус; сжимающая сила - 16 кН; средний вес - 1000 мг; средняя твердость: 46 Н; и истираемость: 0,28%. Полученные таким образом ПРТ (дозировка 50 мг) быстро распадаются в ротовой полости, образуя однородную, легко проглатываемую суспензию, которая включает покрытые гранулы и обеспечивает профиль высвобождения лекарственного вещества пригодный для одноразового режима дневной дозировки.
Пример 3
3.А МВ-гранулы, содержащие слабоосновное лекарственное вещество
Связывающий полимер медленно добавляли к системе растворителей (например, вода, ацетон, этанол или их смеси) для изготовления раствора связывающего вещества. Слабоосновное лекарственное вещество, клонидин, производное фениламиноимидазолина, медленно добавляли к раствору связывающего вещества при перемешивании до растворения. В установку для наслоения покрытий в псевдоожиженном слое, например, Glatt GPCG 3 (например, оснащенную 7 13/16"колонной Вюрстера с 7" нижним соплом, 'С" пластиной распределения воздуха снизу, покрытой ситом для удерживания продукта размером 200 меш), загружали сферическими частицами микрокристаллической целлюлозы (например, Cellets 100, Glatt), которые затем опрыскивали раствором связывающего вещества и лекарственного вещества. МВ-гранулы затем высушивали для удаления остаточных растворителей (включая влагу) и просеивали (например, через сита 40 и 100 меш) для избавления от гранул большего и меньшего размера.
3.В Наслоение буферного слоя безводного динатрия фосфата (DPA)
Безводный динатрий фосфат добавлялся к очищенной воде при перемешивании до растворения. В установку для наслоения покрытий в псевдоожиженном слое, например, Glatt GPCG 3 (например, оснащенную 13 6" колонной Вюрстера с нижним соплом 8", "С" пластиной распределения воздуха снизу, покрытой ситом для удерживания продукта размером 200 меш и форсункой с диаметром отверстия 1,0 мм) загружали МВ-гранулы (например, из Примера З.А). На МВ-гранулы распылялся буферный раствор. После, необязательно, ополаскивания растворителем на покрытые буфером гранулы наносилось около 2% по весу изолирующего покрытия. Высушенные МВ-гранулы можно просеять (например, с использованием сит 35 и 80 меш) для избавления от гранул большего и меньшего размера.
3.С Покрытие СИВ-слоем:
Покрытые буфером гранулы из Примера З.В покрывались в установке для наслоения покрытий в псевдоожиженном слое СИВ-покрытием, включающим этилцеллюлозу (Ethocel Premium, 10 сП), гипромеллозы фталат (HP-55) и ТЕС (триэтилцитрат) в соотношении 55/30/15, растворенными в смеси 90/10 ацетон/вода, до привеса 30% по весу в расчете на покрытую гранулу. Раствор сжимаемого покрытия (например, гидроксипропилцеллюлозы, такой как Klucel® LF), растворенного в растворителе, распылялся на покрытые буфером гранулы до привеса около 2% по весу. Полученные в результате СИВ-гранулы можно высушить для удаления остаточных растворителей.
3.D ПРТ с контролированным высвобождением, содержащие СИВ-гранулы:
Быстро диспергирующиеся микрогранулы из Примера 2.D смешивались с СИВ-гранулами из Примера З.С и другими фармацевтически приемлемыми ингредиентами, такими как корригент, подсластитель (например, сукралоза), дополнительный кросповидон и микрокристаллическая целлюлоза (например, Avicel PHI 01), с соотношением быстро диспергирующихся микрогранул и СИВ-гранул около 3:2 в V-образном блендере с двойным корпусом в течение времени, достаточного для образования однородно распределенной смеси для прессования. ПРТ, включающие 50 мг слабоосновного лекарственного вещества в виде СИВ-гранул, прессовались с использованием промышленного таблеточного пресса, оснащенного внешней системой смазки. Полученные таким образом ПРТ (дозировка 50 мг) быстро распадаются в ротовой полости, образуя однородную, легко проглатываемую суспензию, которая включает покрытые гранулы и обеспечивает профиль высвобождения лекарственного вещества, пригодный для одноразового режима дневной дозировки. Пример 4
4.А МВ-гранулы. содержащие слабоосновное лекарственное вещество:
Раствор для наслоения покрытия лекарственного вещества в соответствующей системе растворителей приготавливался путем добавления, в первую очередь, полимерного связывающего вещества до растворения, а затем слабоосновного лекарственного вещества. Затем раствор наносили на Cellets 100 (сферические частицы микрокристаллической целлюлозы со средним размером частиц 100-200 мкм). Полученные МВ-гранулы затем высушивались для удаления остаточных растворителей (включая влагу) и просеивались (например, через сита 40 и 100 меш) для избавления от частиц большего и меньшего размера.
4.В Наслоение буферного слоя оксида магния:
Тонкоизмельченный оксид магния добавлялся к раствору полимерного связывающего вещества в системе растворителей на Основе этанола при перемешивании до образования однородной дисперсии. Устройство для наслоения покрытий в псевдоожиженном слое, например, Glatt GPCG 3, загружали МВ-гранулами (например, из Примера 4.А), и распыляли раствор оксида магния и полимерного связывающего вещества на МВ-гранулы. После, необязательно, ополаскивания растворителем покрытых буфером гранул, наносилось около 2% по весу изолирующего покрытия. Высушенные МВ-гранулы можно просеять для избавления от гранул большего и меньшего размера.
4.С Покрытие СИВ-слоем:
Покрытые буфером гранулы из Примера 4.В покрывались в установке для наслоения покрытий в псевдоожиженном слое ЗВ-покрытием, включающим этилцеллюлозу (ЕС-10) и ТЕС в соотношении 90/10, растворенными в смеси 95/5 ацетон/вода, до привеса 10% по весу в расчете на покрытую гранулу.
Гранулы, покрытые ЗВ-покрытием, затем дополнительно покрывались раствором СИВ-покрытия, включающим ЕС-10, HP-55 и ТЕС в соотношении 60/30/10, а затем - сжимаемым покрытием Klucel® LF до привеса около 2% по весу. Полученные СИВ-гранулы можно высушить для удаления остаточных растворителей.
4.D МВ-гранулы с маскировкой вкуса
МВ-гранулы из примера 4.А подвергались маскировке вкуса путем покрытия ЕС-10, Eudragit® El 00, ТЕС и магния стеаратом в установке Glatt GPCG 3 до привеса около 15% по весу.
i 4.Е ПРТ с контролированным высвобождением, содержащие MB- и СИВ-гранулы:
Быстро диспергирующиеся микрогранулы из Примера 2.D, СИВ-гранулы и МВ-гранулы слабоосновного лекарственного вещества с маскировкой вкуса из Примера 4.D в соотношении 2:1 смешивались с другими фармацевтически приемлемыми ингредиентами, такими как корригент, подсластитель (например, сукралоза), дополнительный кросповидон и микрокристаллическая целлюлоза (например, Avicel PHI 01). СИВ-гранулы и МВ-гранулы с маскировкой вкуса объединяли с быстро диспергирующимися гранулами в соотношении быстро диспергирующихся микрогранул и покрытых гранул около 3:2 в V-образном блендере с двойным корпусом в течение времени, достаточного для образования однородно распределенной смеси для прессования. ПРТ, включающие 50 мг слабоосновного лекарственного вещества в форме 3 В- и СИВ-гранул, прессовались с использованием промышленного таблеточного пресса, оснащенного внешней системой смазки. Полученные таким образом ПРТ (дозировка 50 мг) быстро распадаются в ротовой полости, образуя однородную, легко проглатываемую суспензию, которая включает покрытые гранулы и обеспечивает профиль высвобождения лекарственного вещества, пригодный для одноразового режима дневной дозировки.
Пример 5
5.А МВ-гранулы, содержащие пропиверина НСl
Пропиверина НС1 (308 г) медленно добавляли к очищенной воде (2054,7 г) при перемешивании до растворения. В предварительно прогретую установку Glatt 3 загружали Cellets 100 (900 г) и опрыскивали раствором лекарственного вещества со скоростью 4 мл/мин со ступенчатым возрастанием скорости до 12 мл/мин и с объемом поступающего воздуха 8 куб. футов в мин, температура продукта 50±2°С.Распыляющую систему затем ополаскивали 40 г воды, наносили 2% покрытия Opadry Clear (6% твердых веществ в воде), полученные МВ-гранулы высушивали для удаления остаточных растворителей (включая влагу) и просеивали (например, через сита 40 и 100 меш) для избавления от частиц большего и меньшего размера.
5.В Наслоение буферного слоя динатрия фосфата
Динатрия фосфат (113,9 г) медленно добавляли к полимерному связывающему веществу, водному раствору (2278 г воды) повидона (2,3 г), при перемешивании до растворения. В предварительно прогретую установку Glatt GPCG 3 загружали МВ-гранулами (например, из Примера 5.А, 1000 г) и распыляли на МВ-грануды буферный раствор аналогично раскрытию из Примера 3.Б. После, необязательно, ополаскивания системы распыления 40 г воды, наносилось 2 вес.% изолирующего покрытия Opadry Clear. Высушенные МВ-гранулы можно просеять для избавления от гранул большего и меньшего размера. 5. С ЗВ-гранулы пропиверина (30% покрытия)
Покрытые буфером гранулы (900 г) из Примера 5.В покрывались в предварительно прогретой установке для наслоения покрытий в пресвдоожиженном слое ЗВ-слоем, включающим этилцеллюлозу (357,4 г) и ТЕС (39,7 г) в соотношении 90/10, растворенными в смеси ацетон (3375 г)/вода (596 г), до привеса 30% по весу в расчете на покрытую гранулу. Гранулы с ЗВ-покрытием дополнительно покрывались сжимаемым покрытием Klucel® LF (26,5 г) до привеса около 2% по весу. Полученные ЗВ-гранулы высушивались для удаления остаточных растворителей. ЗВ-гранулы с 20%, 25% и 30% по весу покрытия подвергались испытанию на растворение по методу двухступенчатого растворения (Фармакопея США, Прибор 2, лопасти при 50 об./мин, растворяющая среда: 700 мл 0,1 н. HCl первые 2 часа, затем - при pH 6,8, полученном путем добавления 200 мл модификатора буфера при 37°C). Данные растворения представлены ниже в Таблице 2. Из таблицы видно, что уровень покрытия необходимо существенно снизить.
|
Пример 6
6.А МВ-гранулы, содержащие пропиверин·HCl
Пропиверина HCl(256,5 г) медленно добавляли к смеси 50/50 ацетон/вода (по 855 г каждого) при перемешивании до растворения, а затем добавляли натрия стеарилфумарат (PRUV, 28,5 г) при интенсивном перемешивании до равномерного диспергирования. Предварительно прогретый Glatt 3 загрузили сферическими частицами сахара 45-60 меш (972 г) и раствор лекарственного вещества (непрерывно перемешиваемый в ходе распыления) распыляли со скоростью 4 мл/мин со ступенчатым возрастанием до 8 мл/мин и с объемом поступающего воздуха 10 куб. футов в мин, температура продукта 45±2°C. После ополаскивания распыляющей системы 40 г ацетона наносили изолирующее покрытие при 2% Opadry Clear (6% твердых веществ в воде), полученные МВ-гранулы высушивали для удаления остаточных растворителей (включая влагу) и просеивали (например, через сита 40 и 80 меш) для избавления от частиц большего и меньшего размера.
6.В Наслоение буферного слоя двухосновного натрия фосфата
Слой двухосновного натрия фосфата (113,9 г) наслаивали на МВ-гранулы (например, из Примера 5.А, 1000 г) в предварительно прогретой Glatt GPCG 3 в соответствии с процедурами, раскрытыми в Примере 5.В. После, факультативно, ополаскивания системы распыления 40 г ацетона наносили 2 вес.% изолирующее покрытия с Opadry Clear. Высушенные МВ-гранулы можно просеять для избавления от гранул большего и меньшего размера. 6.С ЗВ-гранулы пропиверина (10% покрытия)
Покрытые буфером гранулы (850 г) из Примера 6.В покрывались в предварительно прогретой установке для покрытия в псевдоожиженном слое 3 В-слоем, включающим этилцеллюлозу (86,9 г) и ТЕС (9,7 г) в соотношении 90/10, растворенными в смеси ацетон (821 г)/вода (145 г), для привеса на 10 вес.% в расчете на покрытую гранулу. Покрытые 3 В гранулы затем покрывались сжимаемым покрытием с Klucel® LF (19,3 г) для привеса около 2 вес.%. Полученные ЗВ-гранулы высушивались для удаления остаточных растворителей.
6.D ПРТ пропиверина»НС1 CR
Быстро диспергирующиеся микрогранулы (43,68 частей) из примера 2.D, ЗВ-гранулы пропиверина«НС1 (34,97 частей) из примера 6.С и предварительно подготовленная смесь (микрокристаллическая целлюлоза (Ceolus KG 802+Avicel PHI 01, 7,5 частей каждого), кросповидон (5 частей), сукралоза (0,35 частей), корригент со вкусом мяты перечной (1,0 часть) смешивались и пропускались через сито 40 меш для получения гомогенной смеси) смешивались в V-образном блендере как раскрыто в Примере 4.Е. Таблетки ПРТ, включающие 50 мг пропиверина*НС1 в форме ЗВ-гранул прессовались с помощью промышленного таблеточного пресса, оснащенного внешней системой смазки: полученные таким образом ПРТ (дозировка 50 мг) быстро распадаются в ротовой полости, образуя однородную, легко проглатываемую суспензию, включающую покрытые гранулы, а время распада, при испытании способом №701 согласно Фармакопее США, составляло менее 30 секунд.
Пример 7
7.А Пеллеты пропиверина»НС1 путем регулируемой сферонизации
Повидон (PVP К-30, 111,1 г) и пропиверин*НС1 (распределение размеров частиц - D(0,l): 2,6 мкм; D(0,5): 10,38 мкм; D(0,9): 42,52 мкм; 1000 г) смешивали совместно и загружали в чашу Granurex GX-35 от Vector Corporation (Айова, США). Во вращающуюся подложку для материала с регулируемой скоростью распылялась очищенная вода. Оптимизированные параметры во время формовки пеллет: температура воздуха процесса - -19-20°С; температура продукта 16±2°С; скорость ротора - 425 об./мин; внешний подвод воздуха- 150 л/мин; скорость распыления - 15 об./мин (~8 мл/мин), падение давления от края до края щели - 1,3-11 мм в воде; и во время высушивания пеллет: объем воздуха процесса- 30 куб. футов/мин., температура воздуха процесса - ~60°С, температура продукта - 35°С (к моменту прекращения высушивания), скорость ротора - 180 об./мин, объем воздуха, подаваемого через щель - 10 куб. футов/мин, время обработки - 40 мин. Изготовленные таким образом пеллеты содержат около 65% частиц в размерном диапазоне 40-80 мещ.
7.В МВ-пеллеты пропиверина»НС1 с маскировкой вкуса
Пеллеты (970 г) из Примера 7.А покрывались изолирующим покрытием Klucel LF (30 г), растворенным в смеси ацетон/вода (7,5% твердых веществ), для привеса 3%. Этилцеллюлозу (ЕС-10, Ethocel Premium 10, от Dow Chemicals, 159,1 г) медленно добавляли к смеси 85/15 ацетон/вода (10% твердых веществ) при постоянном перемешивании до растворения. Триэтилцитрат (ТЕС, 15,9 г) медленно добавляли до растворения. Маскировку вкуса МВ-пеллет осуществляли в Glatt GPCG 3 путем опрыскивания вышеописанным раствором для привеса 20%.
7.С Наслоение слоя оксида магния на пеллеты пропиверина путем порошкового наслаивания
Повидон (11,9 г) медленно добавляли к очищенной воде (5% твердых веществ) при перемешивании до растворения. Пеллеты пропиверина из Примера 7.А (2000 г) или пеллеты пропиверина, покрытые изолирующим покрытием, из примера 7.В (2000 г) загружались в чашу Granurex GX-35 от Vector Corporation (Айова, США). Во вращающуюся подложку для материала с регулируемой скоростью распылялся раствор повидона, в то время как одновременно с ним порошок (229,3 г оксида магния) распылялся в аппарат при помощи устройства порошкового наслоения (К-Тгоп) с регулируемой скоростью. Оптимизированные параметры в ходе порошкового наслоения: температура продукта: 22-25°С; скорость ротора: 300 об./мин; внешняя подача воздуха: 150-320 л/мин и температура - 100°С; падение давления от края до края щели - 1-2 мм воды; скорость распыления раствора - 3-5 мл/мин (воздух через форсунку: 20 фунтов/кв. фут); и скорость распыления порошка- 5 г/мин (давление воздуха: 12,5 фунтов/кв. фут). Пеллеты покрывались изолирующим покрытием путем распыления того же раствора связывающего вещества или раствора Opandry Clear (5% твердых веществ) при рабочем объеме воздуха 70 куб. футов/мин, а покрытые изолирующим покрытием с наслоенным буферным слоем пеллеты высушивались около 5 мин для снижения содержания влаги.
Молотая или тонкоизмельченная лекарственная субстанция может быть смешана с сыпучими вспомогательными веществами, такими как коллоидный кремнезем или магния стеарат.Связывающее вещество в количестве до 10% может быть частично смешано с порошком лекарственного вещества и частично растворено в распыляемой жидкости. Растворители (например, ацетон, этанол или смесь) могут применяться в пригодной для работы с растворителями аппарате Granurex. Также могут применяться альтернативные связывающие вещества, такие как Klucel LF, гипромеллоза. На сферонизированные пеллеты в самом Granurex или в установке для наслоения покрытий в псевдоожиженном слое, как раскрыто в Примера 6.В выше, может наноситься защитное изолирующее покрытие с Opadry Clear или Klucel LF.
7.D CR-пеллеты пропиверина«НС1 (25% СИВ/10% ЗВ-покрытие)
Как раскрыто в Примере 2.С, покрытые буфером пеллеты пропиверина из Примера 7. В (900 г) покрывались в предварительно прогретом устройстве для наслоения покрытий в псевдоожиженном слое, GPCG 3, ЗВ-покрытием при 10 вес.%, включая этилцеллюлозу (Ethocel Premium 10 сП; 128,6 г), пластифицированную триэтилцитратом (14,3 г), а затем покрывали СИВ-покрытием, включающим этилцеллюлозу (214,3 г), гипромеллозы фталат (HP-55, 107,2 г) и ТЕС (триэтилцитрат, 35,7 г) в соотношении 60/30/10, растворенными в 85/15 ацетон/вода, для привеса 25 вес.% в расчете на покрытый пеллет.Раствор сжимаемого покрытия Klucel® LF (7,5% твердых веществ) распыляли на покрытые СИВ пеллеты для привеса около 2 вес.%. Полученные в результате CR-пеллеты в течение 5 минут высушивались в аппарате для удаления остаточных растворителей.
7.Е CR-ПРТ пропиверина'НС1. 200 мг
Быстро диспергирующиеся микрогранулы (57,2 части) из примера 2.D, CR-пеллеты (15,6 части) из Примера 6.С и МВ-пеллеты с маскировкой вкуса (12,8 части) из Примера 6.В смешивались с предварительно подготовленной смесью, включающей другие фармацевтически приемлемые ингредиенты, такие как корригент (1 часть), подсластитель (например, сукралоза, 0,35 части), дополнительный кросповидон (5 частей) и микрокристаллическая целлюлоза (например, Avicel PHI 01, 10 частей) и прессовались в 200 мг CR-ПРТ-таблетки весом около 1250 мг с использованием промышленного таблеточного пресса, оснащенного внешней системой смазки: в результате полученные таким образом ПРТ (дозировка 100 мг) могут быстро распадаться в ротовой полости, образуя однородную, легко проглатываемую суспензию, включающую покрытые гранулы и обеспечивают ожидаемый профиль высвобождения лекарственного вещества, пригодный для режима дозировки один раз в день.
Специалисты в данной области должны понимать, что приведенные выше процедуры и композиции могут быть должным образом модифицированы для обеспечения соответствующей дозировки слабоосновного лекарственного вещества.
Системы доставки лекарственного средства, содержащие слабощелочной селективный 5-ht, серотониновый блокатор и органические кислоты
Системы доставки лекарственных веществ, включающие в себя слабоосновные лекарственные вещества и органические кислоты
Орально распадающиеся таблеточные композиции темазепама
Получение лекарственных форм миорелаксантов скелетных мышц с контролируемым высвобождением
Системы доставки лекарственного средства, содержащие слабощелочной селективный 5-ht, серотониновый блокатор и органические кислоты
Системы доставки лекарственных веществ, включающие в себя слабоосновные лекарственные вещества и органические кислоты
Орально распадающиеся таблеточные композиции темазепама
Получение лекарственных форм миорелаксантов скелетных мышц с контролируемым высвобождением

