Результат интеллектуальной деятельности: PDE10 ИНГИБИТОРЫ И СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка заявляет приоритет в соответствии с 35 U.S.C. §119(e) предварительной патентной заявкой США № 61/313544, поданной 12 марта 2010, и предварительной патентной заявкой США № 61/430841, поданной 7 января 2010, данные заявки включены в настоящее описание полностью с помощью ссылки.
УРОВЕНЬ ТЕХНИКИ
Область техники, к которой относится настоящее изобретение
В общем, настоящее изобретение относится к соединениям, обладающим активностью в качестве PDE10 ингибиторов, и к содержащим их композициям, а также способам лечения различных заболеваний путем введения данных соединений нуждающемуся в этом теплокровному животному.
Описание родственного уровня техники
Фосфодиэстеразы циклических нуклеотидов (PDE) представлены большим суперсемейством ферментов. Известно, что PDE имеют модульное строение, с консервативным каталитическим доменом, близким к карбоксильному концу, и регуляторными доменами или мотивами, часто вблизи аминоконца. В настоящее время PDE суперсемейство включает более двадцати различных генов, разделенных на одиннадцать PDE семейств (Lugnier, C., "Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents". Pharmacol Ther. 2006 Mar; 109(3):366-98).
О недавно описанной PDE, PDE10, одновременно сообщалось тремя независимыми группами (Fujishige et al., "Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A)", J Biol Chem 1999, 274:18438-18445; Loughney et al., "Isolation and characterization of PDE10A, a novel human 3',5'-cyclic nucleotide phosphodiesterase," Gene 1999, 234:109-117; Soderling et al., "Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A", Proc Natl Acad Set USA 1999, 96:7071-7076). PDE10 обладает способностью гидролизовать и цАМФ, и цГМФ; однако, K m для цАМФ составляет приблизительно 0,05 мкМ, тогда как K M для цАМФ составляет 3 мкМ. Кроме того, V max для цАМФ гидролиза в пять раз меньше, чем для цГМФ. Из-за данной кинетики цГМФ гидролиз PDE10 эффективно ингибируется цАМФ in vitro, предполагая, что PDE10 может функционировать как цАМФ-ингибированная цГМФ фосфодиэстераза in vivo. В отличие от PDE8 или PDE9, PDE10 ингибируется IBMX с IC50 (концентрация, ингибирующая 50%) 2,6 мкМ (см. Soderling and Beavo, "Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions", Current Opinion in Cell Biology, 2000, 12:174-179).
PDE10 содержит два аминоконцевых домена, которые являются аналогичными цГМФ-связывающим доменам PDE2, PDE5 и PDE6, которые представляют собой домены, консервативные для широкого спектра белков. Из-за большой консервативности данного домена в настоящее время его называют GAF доменом (для GAF белков: цГМФ связывающие фосфодиэстеразы; аденилатциклаза цианобактерии Anabaena; и транскрипционный регулятор fh1A Escherichia coli). Хотя в PDE2, PDE5 и PDE6 GAF домены связывают цГМФ, это, вероятно, не основная функция данного домена во всех случаях (например, считается, что E. coli не синтезирует цГМФ). Интересно, что in vitro исследования связывания PDE10 показывают константу диссоциации (Kd) для цГМФ связывания, значительно большую 9 мкМ. Поскольку считается, что in vivo концентрации цГМФ не достигают таких больших значений в большинстве клеток, по-видимому вероятно, что либо сродство PDE10 к цГМФ увеличивается регуляцией, либо первичная функция GAF домена в PDE10 может быть несколько отличной от цГМФ связывания.
Повсеместно ведется поиск ингибиторов PDE семейства ферментов для широкого диапазона показаний терапевтических применений. Описанные в литературе терапевтические применения PDE ингибиторов включают аллергию, обструктивное заболевание легких, гипертонию, рак почки, стенокардию, сердечную недостаточность, депрессию и эректильную дисфункцию (WO 01/41807 A2). Другие ингибиторы PDE описывают для лечения ишемических заболеваний сердца (патент США № 5693652). Более конкретно, ингибиторы PDE10 описывают как пригодные для лечения определенных неврологических и психических расстройств, включая, болезнь Паркинсона, болезнь Хантингтона, шизофрению, бредовые расстройства, психоз, вызванный употреблением наркотиков, и панические и обсессивно-компульсивные расстройства (патентная заявка США № 2003/0032579). Было показано, что PDE10 присутствует при высокой концентрации в нейронах в областях мозга, которые тесно связаны со многими неврологическими и психиатрическими расстройствами. Посредством ингибирования PDE10 активности концентрации цАМФ и цГМФ увеличиваются в нейронах, и способность данных нейронов правильно функционировать посредством этого улучшается. Таким образом, считается, что ингибирование PDE10 является пригодным для лечения широкого диапазона заболеваний или расстройств, для которых было бы полезно увеличивать концентрации цАМФ и цГМФ в нейронах, включая те неврологические, психотические, тревожные расстройства и/или двигательные расстройства, которые упомянуты выше.
Тогда как успех был достигнут относительно ингибирования PDE10, в данной области еще остается необходимость в ингибиторах PDE10, а также необходимость лечения различных заболеваний и/или состояний, для которых могло бы быть полезно данное ингибирование.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Вкратце, настоящее изобретение, в общем, относится к соединениям, которые обладают активностью в качестве PDE10 ингибиторов, а также способам их получения и применения, и к содержащим их фармацевтическим композициям.
В одном варианте осуществления соединения имеют следующую общую структуру (I):
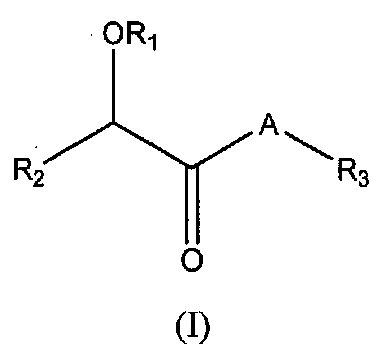
включая их фармацевтически приемлемые соли, стереоизомеры, сольваты и пролекарства, в которых A, R1, R2 и R3 такие, как определено ниже.
Соединения настоящего изобретения можно применять в широком диапазоне терапевтических применений и их можно применять для лечения широкого диапазона заболеваний или расстройств, для которых было бы полезно увеличение концентрации цАМФ и цГМФ, особенно в нейронах, включая (но не ограничиваясь) неврологические расстройства, такие как психотические расстройства, тревожные расстройства, двигательные расстройства и/или неврологические расстройства, такие как болезнь Паркинсона, болезнь Хантингтона, болезнь Альцгеймера, энцефалит, фобия, эпилепсия, афазия, паралич Белла, церебральный паралич, нарушения сна, боль, синдром Туретта, шизофрения, бредовые расстройства, биполярные расстройства, посттравматические стрессовые расстройства, психоз, вызванный употреблением наркотиков, панические расстройства, обсессивно-компульсивные расстройства, синдром дефицита внимания, расстройство социального поведения, аутизм, депрессия, деменция, когнитивные расстройства, эпилепсия, бессонница и рассеянный склероз.
Способы настоящего изобретения включают введение эффективного количества соединения вышеуказанной структуры, обычно в виде фармацевтической композиции, нуждающемуся в этом млекопитающему, включая человека. Таким образом, в следующем варианте осуществления описывают фармацевтические композиции, содержащие одно или более соединений вышеуказанной структуры в комбинации с фармацевтически приемлемым носителем или разбавителем.
Данные и другие аспекты настоящего изобретения будут очевидны со ссылкой на следующее подробное описание. С этой целью в настоящем описании указаны различные ссылки, которые описывают более подробно определенную вспомогательную информацию, методики, соединения и/или композиции, и каждая из ссылок вводится в настоящее описание полностью с помощью ссылки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ.1 показывает, что соединение 1-1 настоящего изобретения (Пример 1), введенное внутрибрюшинной инъекцией, значительно снижает гиперактивность у мышей в модели психоза, вызванного психостимулятором (PCP), при сравнении с контролем с носителем.
ФИГ.2 показывает, что соединение 1-1 настоящего изобретения (Пример 1), введенное пероральным способом введения, значительно снижает гиперактивность у мышей в модели психоза, вызванного психостимулятором (PCP), при сравнении с контролем с носителем.
ФИГ.3 показывает, что соединение 2-1 настоящего изобретения (Пример 2), введенное внутрибрюшинной инъекцией, значительно снижает гиперактивность у мышей в модели психоза, вызванного психостимулятором (PCP), при сравнении с контролем с носителем.
ФИГ.4 показывает, что соединение 2-1 настоящего изобретения (Пример 2), введенное пероральным способом введения, значительно снижает гиперактивность у мышей в модели психоза, вызванного психостимулятором (PCP), при сравнении с контролем с носителем.
ФИГ.5 показывает, что соединение 2-1 настоящего изобретения (Пример 2) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.6 показывает, что соединение 11-1 настоящего изобретения (Пример 11), введенное внутрибрюшинной инъекцией, значительно снижает гиперактивность у мышей в модели психоза, вызванного психостимулятором (PCP), при сравнении с контролем с носителем.
ФИГ.7 показывает, что соединение 34-1 настоящего изобретения (Пример 34) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.8 показывает, что соединение 36-1 настоящего изобретения (Пример 36) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.9 показывает, что соединение 47-1 настоящего изобретения (Пример 47) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.10 показывает, что соединение 61-1 настоящего изобретения (Пример 61) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.11 показывает, что соединение 63-1 настоящего изобретения (Пример 63) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.12 показывает, что соединение 49-1 настоящего изобретения (Пример 49) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ФИГ.13 показывает, что соединение 65-10 настоящего изобретения (Пример 65, таблица 1) значительно снижает условную реакцию избегания (CAR) у мышей, подготовленных в CAR модели психоза при сравнении с контролем с носителем.
ПОДРОБНОЕ ОПИСАНИЕ
Как упоминается выше, в общем, настоящее изобретение относится к соединениям, пригодным в качестве PDE10 ингибиторов, а также способам их получения и применения и к содержащим их фармацевтическим композициям.
В одном варианте осуществления PDE10 ингибиторы имеют следующую структуру (I):
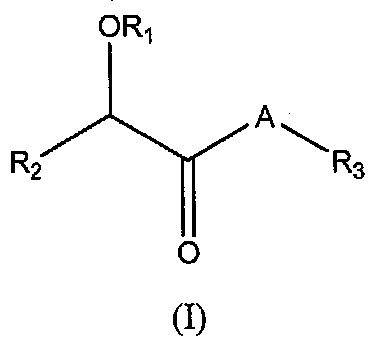
или их фармацевтически приемлемая соль, стереоизомер, сольват или пролекарство,
в которой:
A представляет собой:
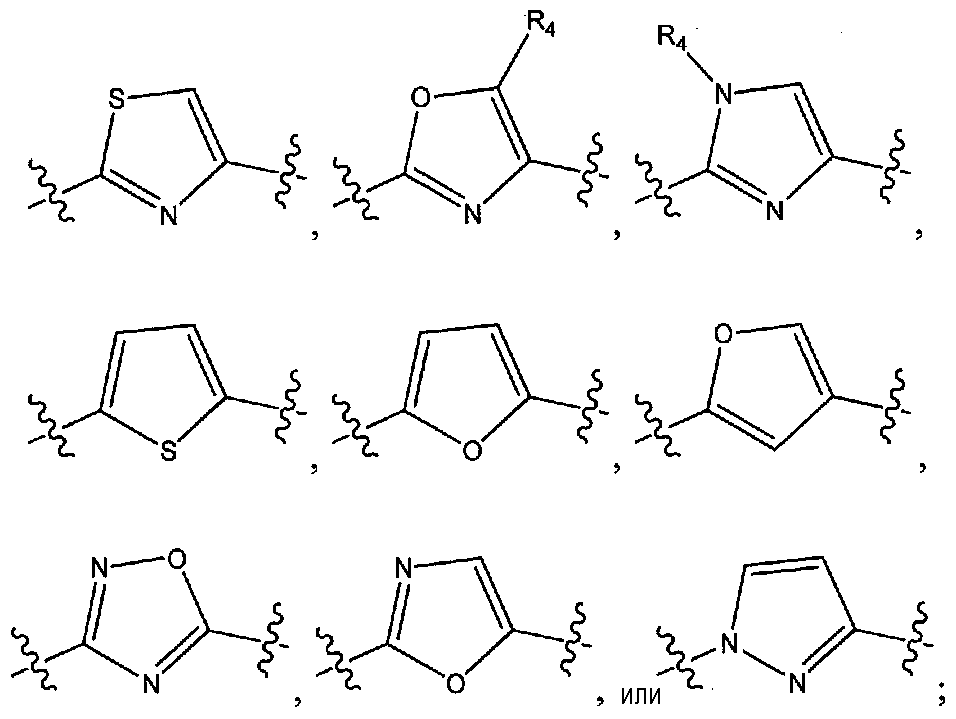
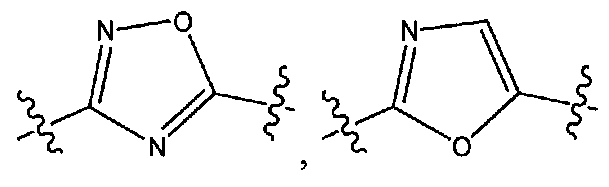 или
или 
R1 представляет собой C1-6алкил, C1-6галогеналкил, C1-6аралкил, арил, -(CH2)nO(CH2)mCH3 или -(CH2)nN(CH3)2;
R2 представляет собой (i) замещенный или незамещенный арил или (ii) замещенный или незамещенный гетероциклил;
R3 представляет собой замещенный или незамещенный арил;
R4 представляет собой водород, C1-6алкил или C1-6галогеналкил;
n равно 1, 2, 3, 4, 5 или 6; и
m равно 0, 1, 2, 3, 4, 5 или 6.
Как используется в настоящем описании, вышеуказанные термины имеют следующие значения:
"Амино" относится к -NH2 радикалу.
"Циано" относится к -CN радикалу.
"Гидрокси" или "гидроксил" относится к -OH радикалу.
"Имино" относится к =NH заместителю.
"Нитро" относится к -NO2 радикалу.
"Оксо" относится к =O заместителю.
"Тиоксо" относится к =S заместителю.
"C1-6алкил" относится к линейному или разветвленному, нециклическому или циклическому, ненасыщенному или насыщенному алифатическому углеводородному радикалу, содержащему от 1 до 6 атомов углерода. Примеры насыщенных алкилов с прямой цепью включают метил, этил, н-пропил, н-бутил, н-пентил, н-гексил и подобные; тогда как насыщенные разветвленные алкилы включают изопропил, втор-бутил, изобутил, трет-бутил, изопентил и подобные. Примеры насыщенных циклических алкилов включают циклопропил, циклобутил, циклопентил, циклогексил и подобные; тогда как ненасыщенные циклические алкилы включают циклопентенил и циклогексенил и подобные. Ненасыщенные алкилы содержат, по меньшей мере, одну двойную или тройную связь между соседними атомами углерода (называемые "алкенил" или "алкинил" соответственно). Примеры алкенилов с прямой или разветвленной цепью включают этиленил, пропиленил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил и подобные; тогда как примеры алкинилов с прямой или разветвленной цепью включают ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-бутинил и подобные.
"C1-6алкилен" или "C1-6алкиленовая цепь" относится к прямой или разветвленной двухвалентной углеводородной цепи, присоединяющей остаток молекулы к радикалу, состоящей только из атомов углерода и водорода, которая является насыщенной или ненасыщенной (т.е. содержит одну или более двойных и/или тройных связей), и содержащей от одного до шести атомов углерода, например метилен, этилен, пропилен, н-бутилен, этенилен, пропенилен, н-бутенилен, пропинилен, н-бутинилен и подобные. Алкиленовую цепь присоединяют к остатку молекулы через одинарную или двойную связь и к радикалу через одинарную или двойную связь. Положения присоединения алкиленовой цепи к остатку молекулы и к радикалу можно осуществлять через один атом углерода или любые два атома углерода в цепи.
"C1-6алкокси" относится к радикалу формулы -ORa, где Ra представляет собой алкильный радикал, как определено выше, например метокси, этокси и подобные.
"Арил" относится к углеводородной кольцевой системе, включающей водород, 6-18 атомов углерода и, по меньшей мере, одно ароматическое кольцо. Арильный радикал может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую кольцевую систему, которая может содержать конденсированные или мостиковые кольцевые системы. Арильные радикалы включают, но не ограничиваются ими, арильные радикалы, полученные из ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, фторантена, флуорена, as-индацена, s-индацена, индана, индена, нафталина, феналена, фенантрена, плеиадена, пирена и трифенилена.
"C1-6аралкил" обозначает радикал формулы -Rb-Rc, где Rb представляет собой алкиленовую цепь, как определено выше, и Rc представляет собой один или более арильных радикалов, как определено выше, например бензил, дифенилметил и подобные.
"Циклоалкил" или "карбоциклическое кольцо" относится к стабильному неароматическоу моноциклическому или полициклическому углеводородному радикалу, состоящему только из атомов углерода и водорода, который может содержать конденсированные или мостиковые кольцевые системы, содержащие от трех до пятнадцати атомов углерода, предпочтительно содержащие от трех до десяти атомов углерода, и который может быть насыщенным или ненасыщенным и присоединенным к остатку молекулы одинарной связью. Моноциклические радикалы включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Полициклические радикалы включают, например, адамантил, норборнил, декалинил, 7,7-диметил-бицикло[2,2,1]гептанил и подобные.
"Гало" или "галоген" относится к брому, хлору, фтору или йоду.
"C1-6галогеналкил" относится к C1-6алкильному радикалу, как определено выше, который замещен одним или более галогеновыми радикалами, как определено выше, например трифторметил, дифторметил, трихлорметил, 2,2,2-трифторэтил, 1,2-дифторэтил, 3-бром-2-фторпропил, 1,2-дибромэтил и подобные.
"Гетероцикл" или "гетероциклил" относится к 4-7-членному моноциклическому или 7-10-членному бициклическому, гетероциклическому кольцу, который является либо насыщенным, ненасыщенным, либо ароматическим, и который содержит от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, и в котором гетероатомы азота или серы могут быть необязательно окислены, и гетероатом азота может быть необязательно кватернизирован, включая бициклические кольца, в которых любой из вышеуказанных гетероциклов конденсирован с бензольным кольцом. Гетероцикл может быть присоединен через любой гетероатом или атом углерода. Ароматический гетероцикл называют в настоящем описании "гетероарил", и он включает (но не ограничивается) фурил, бензофуранил, тиофенил, бензотиофенил, пирролил, индолил, изоиндолил, азаиндолил, пиридил, хинолинил, изохинолинил, оксазолил, изоксазолил, бензоксазолил, пиразолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, оксадиазолил, тиадиазолил, бензизоксазолил, триазолил, тетразолил, индазолил и хиназолинил. В добавление к перечисленным выше гетероарилам, гетероциклы также включают морфолинил, пирролидинонил, пирролидинил, пиперидинил, пиперазинил и подобные. Кроме того, гетероциклы также включают бензотиофен-2-ил, 2,3-дигидробензо-1,4-диоксин-6-ил, бензо-1,3-диоксол-5-ил и подобные.
Термин "замещенный", как применяют в настоящем описании (например, в контексте замещенного гетероциклила или замещенного арила), обозначает то, что, по меньшей мере, один атом водорода замещен заместителем. "Заместители" в контексте настоящего изобретения включают галоген, гидрокси, оксо, циано, нитро, имино, тиоксо, амино, алкиламино, диалкиламино, алкил, алкокси, алкилтио, галогеналкил, арил, аралкил, гетероарил, гетероарилалкил, гетероцикл и гетероциклалкил, а также -NRaRb, -NRaC(=O)Rb, -NRaC(=O)NRaNRb, -NRaC(=O)ORb, - NRaSO2Rb, -C(=O)Ra, -C(=O)ORa, -C(=O)NRaRb, -OC(=O)NRaRb, -ORa, -SRa, -SORa, -S(=O)2Ra, -OS(=O)2Ra, -S(=O)2ORa, =NSO2Ra и -SO2NRaRb. В вышеуказанных заместителях Ra и Rb в данном контексте могут быть одинаковыми или различными и независимо представлять собой водород, алкил, галогеналкил, циклоалкил, арил, аралкил, гетероциклил. Кроме того, вышеуказанные заместители могут быть дополнительно замещены одним или более из указанных выше заместителей.
В следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-A):
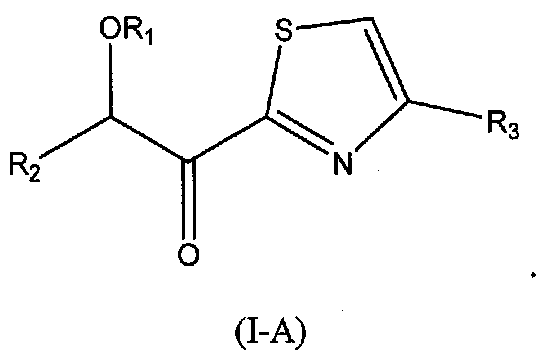
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-B):
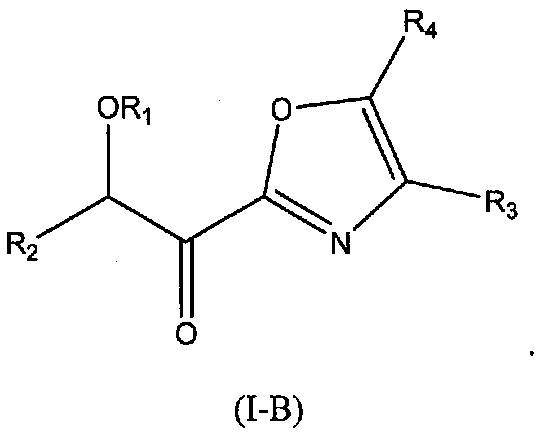
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-С):
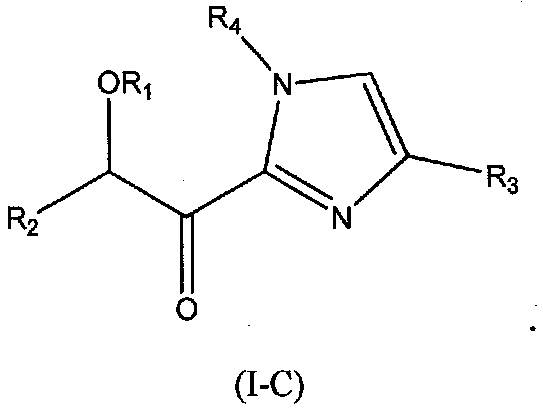
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-D):
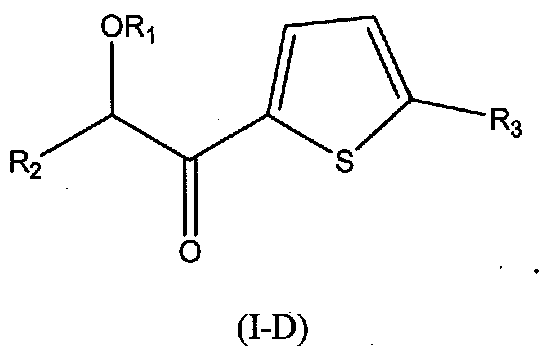
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-E):
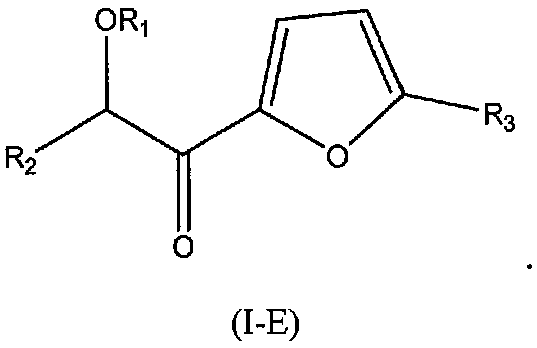
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-F):
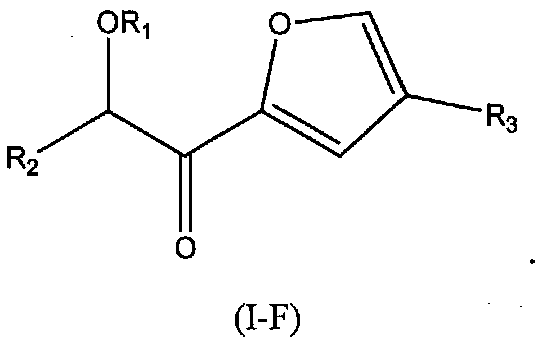
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-G):
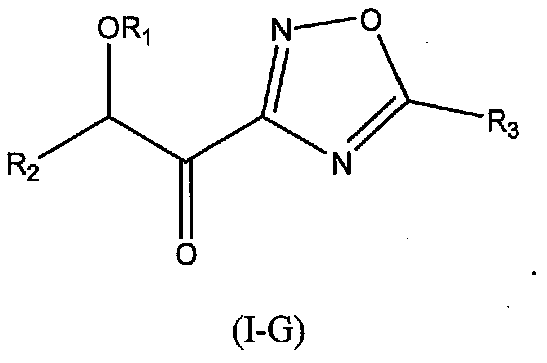
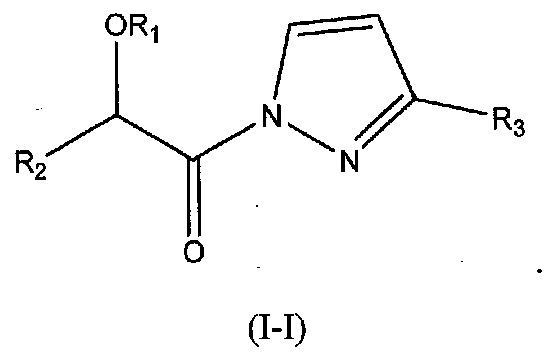
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-H):
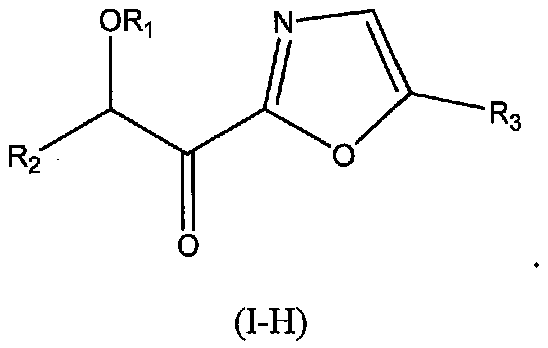
В других следующих вариантах осуществления структуры (I) соединение имеет следующую структуру (I-I):
В других следующих вариантах осуществления структуры (I), в частности структурах (I-B) и (I-C), R4 представляет собой водород или R4 представляет собой C1-6алкил (такой как, например, метил).
В других следующих вариантах осуществления структуры (I) R1 представляет собой C1-6алкил (такой как, например, R1 представляет собой метил или этил).
В других следующих вариантах осуществления структуры (I) R3 представляет собой замещенный или незамещенный фенил (такой как, например, 4-бром-3,5-диметоксифенил, 4-хлор-3,5-диметоксифенил или 3,4,5-триметоксифенил).
В других следующих вариантах осуществления структуры (I) R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил. В более конкретных вариантах осуществления, в которых R2 представляет собой замещенный фенил, R2 представляет собой фенил, замещенный C1-6алкокси, или R2 представляет собой фенил, замещенный замещенным или незамещенным гетероциклилом (такой как, например, 4-(5-метил-1,3,4-оксадиазол-2-ил)фенил, 4-(5-метил-1,3,4-тиадиазол-2-ил)фенил или 4-морфолинофенил).
В других следующих вариантах осуществления структуры (I) R2 представляет собой замещенный или незамещенный гетероциклил.
Соединения настоящего изобретения можно обычно применять в виде свободной кислоты или свободного основания. Альтернативно, соединения настоящего изобретения можно применять в виде солей присоединения кислоты или основания. Соли присоединения кислоты свободных аминосоединений настоящего изобретения можно получить способами, хорошо известными в данной области техники, и их можно получить из органических и неорганических кислот. Подходящие органические кислоты включают малеиновую, фумаровую, бензойную, аскорбиновую, янтарную, метансульфоновую, уксусную, трифторуксусную, щавелевую, пропионовую, винную, салициловую, лимонную, глюконовую, молочную, миндальную, коричную, аспарагиновую, стеариновую, пальмитиновую, гликолевую, глютаминовую и бензолсульфоновую кислоты. Подходящими неорганическими кислотами являются хлороводородная, бромоводородная, серная, фосфорная и азотная кислота. Соли присоединения основания включают те соли, которые образуются с карбоксилатным анионом, и включают соли, полученные с органическими и неорганическими катионами, такие как соли, выбранные из солей щелочных и щелочноземельных металлов (например, лития, натрия, калия, магния, бария и кальция), а также аммониевым ионом и его замещенными производными (например, дибензиламмоний, бензиламмоний, 2-гидроксиэтиламмоний и подобные). Таким образом, предполагается, что термин "фармацевтически приемлемая соль" структуры (I) включает любую и все приемлемые солевые формы.
Кроме того, пролекарства также включены в контекст настоящего изобретения. Пролекарства представляют собой любые ковалентно-связанные носители, которые высвобождают соединение структуры (I) in vivo при введении данного пролекарства пациенту. Пролекарства обычно получают модификацией функциональных групп таким способом, чтобы данная модификация расщеплялась, или обычными способами или in vivo, давая исходное соединение. Пролекарства включают, например, соединения настоящего изобретения, в которых гидрокси, амино или сульфгидрильная группа соединены с любой группой, которая при введение пациенту расщепляется, образуя гидрокси, амино или сульфгидрильную группы. Таким образом, типичные примеры пролекарств включают (но не ограничиваются ими) ацетатные, формиатные и бензоатные производные спиртовой и аминовой функциональной группы соединения структуры (I). Далее, в случае карбоновой кислоты (-COOH) можно применять эфиры, такие как метиловый эфир, этиловый эфир и подобные.
Предусматривается, что настоящее изобретение, описанное в настоящем описании, также включает все фармацевтически приемлемые соединения структуры (I), изотопно меченные одним или более атомами, замещенными атомами, имеющими различный атомный вес и массовое число. Примеры изотопов, которые можно вводить в описанные соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl, 123I и 125I соответственно. Данные радиомеченные соединения могут быть пригодны для того, чтобы помочь определить или измерить эффективность соединений, характеризуя, например, место или способ действия, или связывающую способность фармакологически важного места действия. Определенные изотопно-меченные соединения структуры (I), например соединения с введенным радиоактивным изотопом, являются пригодными для исследований распределения в тканях лекарственного средства и/или субстрата. Радиоактивные изотопы тритий, т.е. 3H, и углерод-14, т.е. 14C, являются особенно пригодными для данной цели из-за легкости введения и готовых способов детекции. Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может давать определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например увеличения in vivo периода полураспада или снижения требуемых доз, и, следовательно, оно может быть предпочтительным в некоторых обстоятельствах. Замещение позитронно-активными изотопами, такими как 11C, 18F, 15O и 13N, может быть пригодным в исследованиях с помощью позитронно-эмиссионной томографии (PET) для изучения степени занятости рецептора субстратом. Изотопно-меченные соединения структуры (I) обычно можно получить общепринятыми способами, известными специалисту в данной области техники, или способами, аналогичными способам, описанным в примерах, как указано ниже, применяя подходящий изотопно-меченный реагент вместо немеченого реагента, применяемого ранее.
Что касается стереоизомеров, соединения структуры (I) могут содержать хиральные центры и могут существовать в виде рацематов, рацемических смесей и в виде индивидуальных энантиомеров или диастереомеров. Все данные изомерные формы включены в объем настоящего изобретения, включая их смеси. Кроме того, некоторые кристаллические формы соединений структуры (I) могут существовать в виде полиморфов, которые включены в объем настоящего изобретения. Кроме того, некоторые соединения структуры (I) могут также образовывать сольваты с водой или другими органическими растворителями. Аналогично, данные сольваты включены в объем настоящего изобретения.
В другом варианте осуществления настоящего изобретения описывают фармацевтические композиции, содержащие одно или более из соединений структуры (I). Для целей введения соединения настоящего изобретения можно формулировать в виде фармацевтических композиций. Фармацевтические композиции настоящего изобретения содержат одно или более из соединений настоящего изобретения и фармацевтически приемлемый носитель и/или разбавитель. PDE10 ингибитор присутствует в композиции в количестве, которое является эффективным для лечения конкретного заболевания, т.е. в количестве, достаточном для достижения требуемого PDE10 ингибирования, и предпочтительно с приемлемой токсичностью для теплокровного животного. Обычно фармацевтические композиции настоящего изобретения могут содержать PDE10 ингибитор в количестве от 0,1 мг до 250 мг на одну дозу, в зависимости от пути введения, и более обычно от 1 мг до 60 мг. Подходящие концентрации и дозы может легко определить специалист в данной области техники.
В общих чертах, стандартная дневная доза может находиться в диапазоне от приблизительно 1 мкг/кг до 100 мг/кг, предпочтительно 0,01-100 мг/кг, более предпочтительно 0,1-70 мг/кг, в зависимости от типа и тяжести заболевания, например, одним или более отдельными введениями. Для повторяющегося введения в течение нескольких дней или более в зависимости от заболевания лечение осуществляют до требуемого подавления возникающих симптомов заболевания. Однако могут быть пригодны другие режимы дозирования. Прогресс данной терапии может быть отслежен стандартными способами и анализами. Характеристики единичных лекарственных форм настоящего изобретения определяются и непосредственно зависят от уникальных характеристик активного соединения и конкретного терапевтического эффекта, которого требуется достичь, и ограничений, присущих области составления композиций активного соединения для лечения индивидов.
Фармацевтически приемлемый носитель и/или разбавители являются известными специалисту в данной области техники. Что касается композиций, полученных в виде жидких растворов, приемлемые носители и/или разбавители включают соляной раствор и стерильную воду и могут необязательно содержать антиоксиданты, буферы, бактериостаты и другие стандартные добавки. Композиции можно также формулировать в виде пилюль, капсул, гранул или таблеток, которые содержат в добавление к PDE10 ингибитору разбавители, диспергирующие агенты и поверхностно-активные вещества, связующие и смазывающие вещества. Кроме того, специалист в данной области техники может формулировать PDE10 ингибитор подходящим способом и согласно принятой практике, такой как та, что описана в Remington's Pharmaceutical Sciences, Gennaro, Ed., Mack Publishing Co., Easton, PA 1990.
В другом варианте осуществления настоящее изобретение относится к способу лечения заболевания, такого как (но не ограничиваясь ими) психотические расстройства, тревожные расстройства, двигательные расстройства и/или неврологические расстройства, такие как болезнь Паркинсона, болезнь Хантингтона, болезнь Альцгеймера, энцефалит, фобия, эпилепсия, афазия, паралич Белла, церебральный паралич, нарушения сна, боль, синдром Туретта, шизофрения, бредовые расстройства, биполярные расстройства, посттравматические стрессовые расстройства, психоз, вызванный употреблением наркотиков, панические расстройства, обсессивно-компульсивные расстройства, синдром дефицита внимания, расстройства социального поведения, аутизм, депрессия, деменция, когнитивные расстройства, эпилепсия, бессонница и рассеянный склероз, как описано выше. Данные способы включают введение соединения настоящего изобретения теплокровному животному в количестве, достаточном для лечения заболевания. В данном контексте "лечить" включает профилактическое введение. Данный способ включает системное введение PDE10 ингибитора настоящего изобретения, предпочтительно в виде фармацевтической композиции, как обсуждается выше. Как применяют в настоящем изобретении, системное введение включает пероральные и парентеральные способы введения, включая подкожное, внутримышечное, внутричерепное, внутриглазничное, офтальмологическое, внутрижелудочковое, интракапсулярное, внутрисуставное, интраспинальное, интрацистернальное, внутрибрюшинное, интраназальное, аэрозолем, внутривенное, внутрикожное, ингаляционное, трансдермальное, трансмукозальное и ректальное введение.
Что касается перорального введения, подходящие фармацевтические композиции PDE10 ингибиторов включают порошки, гранулы, пилюли, таблетки и капсулы, а также жидкости, сиропы, суспензии и эмульсии. Данные композиции могут также содержать ароматизаторы, консерванты, суспендирующие агенты, загустители и эмульгаторы и другие фармацевтически приемлемые добавки и наполнители. Что касается парентерального введения, соединения настоящего изобретения могут быть получены в виде водных инъецируемых растворов, которые могут содержать, кроме PDE10 ингибитора, буферы, антиоксиданты, бактериостаты и другие добавки и вспомогательные вещества, обычно применяемые в данных растворах. Композиции настоящего изобретения можно доставлять в системе доставки для обеспечения замедленного высвобождения или повышенного поглощения или активности терапевтического соединения, такой как липосомальная или гидрогельная система для инъекции, микрочастицы, наносистема или мицелловая система для пероральной или парентеральной доставки, или капсула с поэтапным высвобождением для пероральной доставки.
В качестве дополнительного преимущества настоящего изобретения ожидается, что у соединений структуры (I) будут отсутствовать или снижены метаболические побочные эффекты, связанные с общепринятыми противопсихотическими препаратами, в частности инцидента терапевтически вызванного ожирения. Например, продолжительное применение оланзапина (Zyprexa®), наиболее часто прописываемого лекарственного препарата для лечения шизофрении, и родственных нестандартных протиопсихотических препаратов, связано со значительными метаболическими побочными эффектами, включая ожирение и связанные с ним заболевания, такие как диабет.
У животных субхроническое лечение оланзапином стимулирует потребление пищи и увеличивает вес тела, также как у человека. Кроме того, оланзапин резко снижает концентрацию лептина в крови. Лептин представляет собой гормон сытости, получаемый в адипозных тканях, и снижение концентрации лептина стимулирует аппетит. Существует теория, что оланзапин может стимулировать потребление пищи, по меньшей мере, частично снижением концентрации лептина. Однократное введение оланзапина также изменяет ответную реакцию животного на концентрацию глюкозы и инсулина в тестах толерантности к глюкозе, что может также быть напрямую связано с эффектом оланзапина на потребление пищи и увеличение массы тела. Исследование острого эффекта PDE10 ингибиторов настоящего изобретения на метаболизм, такой как изменения, связанные с лептином, инсулином и глюкозой в процессе метаболизма в стандартных животных моделях, а также хронического эффекта PDE10 ингибиторов настоящего изобретения на потребление пищи, вес тела и энергетический гомеостаз, в сравнении с оланзапином должно обосновать фармацевтическое преимущество PDE10 ингибиторов в качестве антипсихотических препаратов с точки зрения побочных эффектов.
Композиции настоящего изобретения можно вводить в комбинации с одним или более дополнительными терапевтическими агентами, в комбинации с или путем одновременного или последовательного введения. Подходящие дополнительные агенты (т.е. вспомогательные вещества) могут включать стандартные противопсихотические препараты, которые блокируют допаминовые D2-рецепторы и серотониновые 5HT2 рецепторы, например галоперидол, флуфеназин, хлорпромазин и нестандартные противопсихотические препараты, например клозапин, оланзапин, рисперидон, кветиамин, зипразидог.
Соединения настоящего изобретения могут быть проанализированы для определения их IC50 величин посредством модификации двухстадийного способа Thompson и Appleman (Biochemistry 10; 311-316; 1971). Вкратце, цАМФ дополняют (3H)цАМФ и выдерживают с PDE10 и различными концентрациями соединений структуры (I). После подходящего периода выдерживания реакцию прекращают нагреванием. Затем смесь подвергают обработке фосфатазой змеиного яда. Фосфатаза гидролизует любой АМФ в смеси, но оставляет непрореагировавший цАМФ незатронутым. Таким образом, выделением цАМФ из смеси и определением ее концентрации (радиографией) можно определить процент ингибирования. IC50 величины можно рассчитать проведением эксперимента при различных концентрациях, применяя стандартные графические способы. Подробное описание данного способа, применяемого для анализов IC50, приведено в следующих примерах. В связи с этим PDE10 ингибиторы настоящего изобретения обладают IC50 100 мкМ или меньшей, обычно меньшей чем 10 мкМ и обычно меньшей чем 1 мкМ.
Соединения настоящего изобретения можно получить известными способами органического синтеза, включая способы, описанные подробно в следующих примерах. Следующие примеры приведены с целью иллюстрации, а не ограничения.
ПРИМЕРЫ
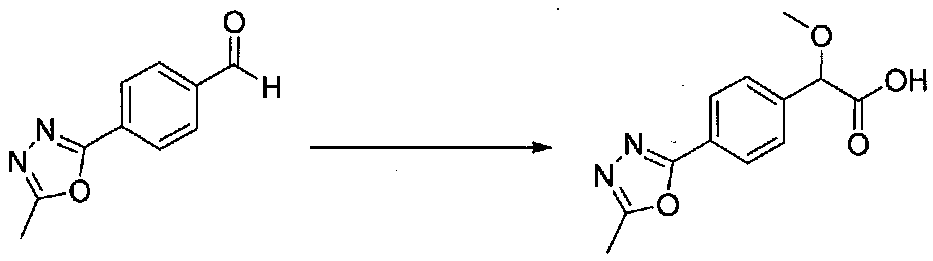
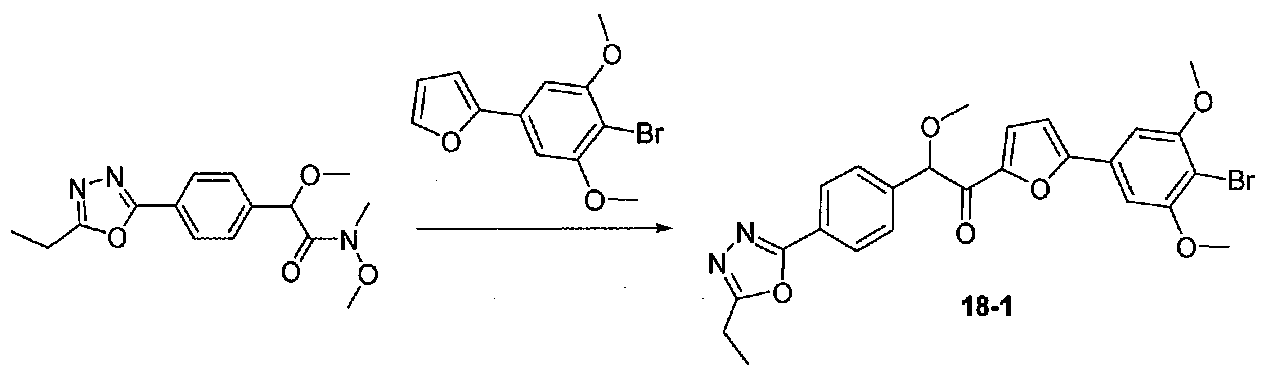
ПРИМЕР 1
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ТИАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
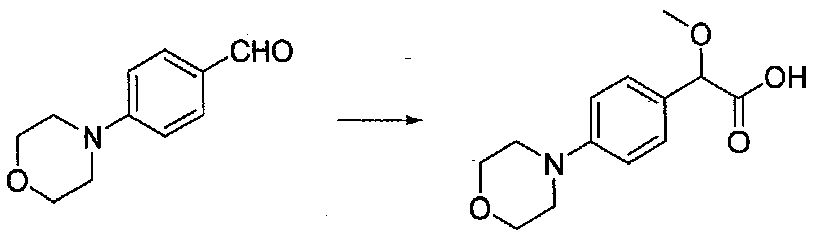
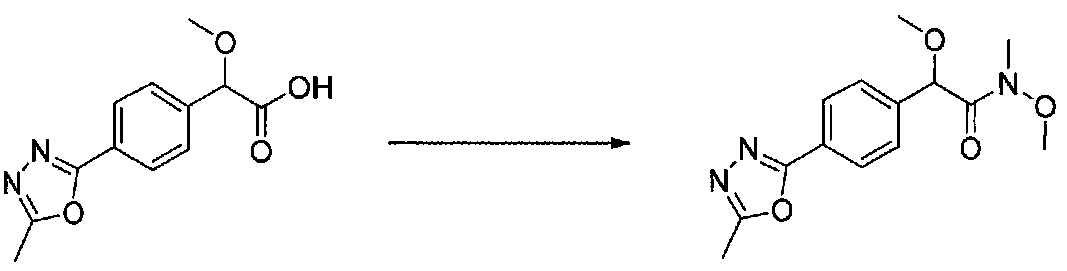
В колбу, высушенную в сушильном шкафу, загружали 4-(4-морфолинил)бензальдегид (10,1 г, 53 ммоль), безводный метанол (60 мл) и безводный диоксан (60 мл), затем оснащали ее капельной воронкой. В капельную воронку загружали раствор KOH (14,8 г, 264 ммоль) в безводном метаноле (60 мл), и аликвоту (~2 мл) добавляли к реакционной смеси. Добавляли к реакционной смеси бромоформ (5,8 мл, 67,1 ммоль), затем оставшийся KOH/MeOH раствор добавляли по каплям в течение 10 минут. После перемешивания в течение 18 часов смесь фильтровали через целит и промывали метанолом. Фильтрат собирали и концентрировали в вакууме. Затем остаток разбавляли насыщенным водным NH4Cl и экстрагировали EtOAc. Затем дополнительное количество EtOAc применяли для экстракции водной фазы, при этом медленно доводя pH с ~8 до ~2, используя концентрированную HCl. В сумме приблизительно 1,5 л EtOAc применяли для экстракции. Объединенные EtOAc экстракты сушили над Na2SO4 и фильтровали. Концентрирование фильтрата в вакууме давало 2-метокси-2-(4-морфолинофенил)уксусную кислоту в виде желтовато-коричневого твердого вещества (7,25 г, 58%).
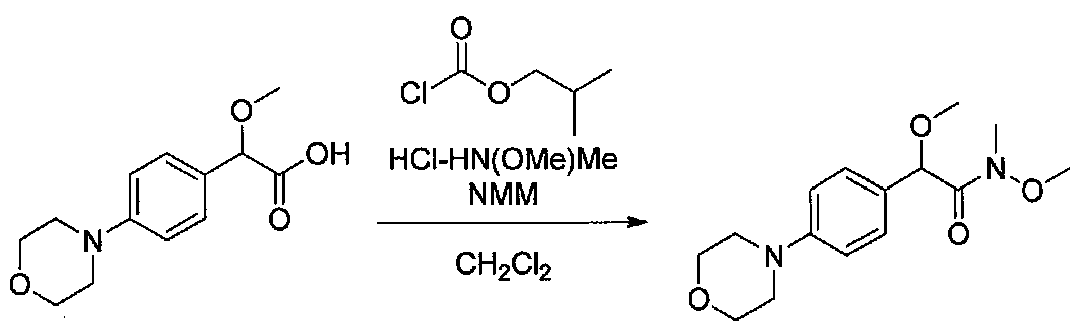
К суспензии 2-метокси-2-(4-морфолинофенил)уксусной кислоты (2,97 г, 11,8 ммоль) в безводном CH2Cl2 (66 мл) в колбе, высушенной в сушильном шкафу, в атмосфере аргона добавляли N-метилморфолин (3 мл, 27,3 ммоль) и полученный в результате раствор охлаждали на льду. Добавляли по каплям изобутилхлорформиат (1,8 мл, 13,76 ммоль). После перемешивания в течение 50 минут добавляли N,O-диметилгидроксиламингидрохлорид (1,5 г, 15,3 ммоль) и смесь медленно нагревали до комнатной температуры. После перемешивания в течение 16 часов добавляли насыщенный водный NaHCO3 и смесь перемешивали в течение >15 минут. Смесь разбавляли CH2Cl2 и слои разделяли. Органический слой промывали соляным раствором, сушили над MgSO4/Na2SO4 и концентрировали. Очистка хроматографией (60-85% EtOAc-гексаны) давала N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамид в виде не совсем белого твердого вещества (3,15 г, выход 90%).
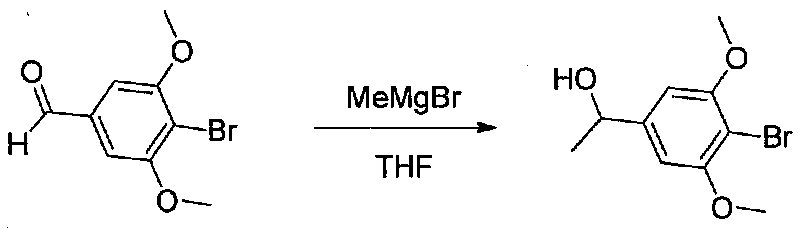
В колбу, высушенную в сушильном шкафу, в атмосфере аргона загружали 4-бром-3,5-диметоксибензальдегид (10,08 г, 41,1 ммоль) и безводный THF (70 мл). Смесь охлаждали до -78°C на бане, затем добавляли по каплям из капельной воронки в течение 45 минут раствор MeMgBr (3,0 M в диэтиловом эфире, 17,8 мл, 53,4 ммоль). После перемешивания в течение 20 минут смесь нагревали до комнатной температуры и перемешивали в течение 19 часов. После прекращения реакции раствором водного NH4Cl ее разбавляли H2O и EtOAc, затем охлаждали на бане со льдом. После охлаждения смеси слои разделяли. Органический слой промывали H2O и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Остаток растворяли в дихлорметане и концентрировали в вакууме снова с получением 1-(4-бром-3,5-диметоксифенил)этанола в виде белого твердого вещества (10,8 г, количественный выход). Продукт использовали без дополнительной очистки.
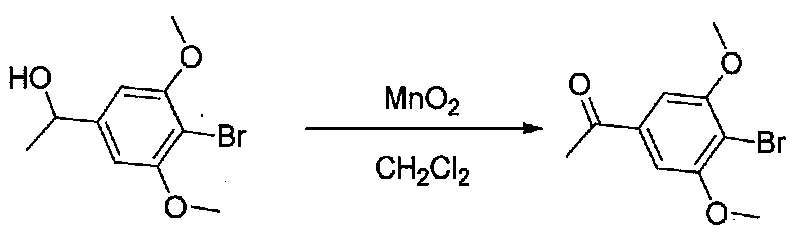
К раствору 1-(4-бром-3,5-диметоксифенил)этанола (10,8 г, 41,1 ммоль) в безводном CH2Cl2 (150 мл) добавляли MnO2 (48 г, 552 ммоль). Реакционный сосуд со смесью закрывали хлоркальциевой трубкой и смесь перемешивали при комнатной температуре в течение 22 часов, фильтровали через слой целита и силикагеля и промывали EtOAc. Концентрирование фильтрата в вакууме давало 1-(4-бром-3,5-диметоксифенил)этанон в виде белого твердого вещества (10,3 г, выход 97%). Продукт использовали без дополнительной очистки.
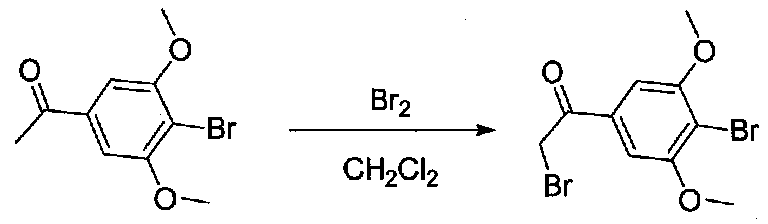
К раствору 1-(4-бром-3,5-диметоксифенил)этанона (0,895 г, 3,45 ммоль) в безводном CH2Cl2 (5 мл) под хлоркальциевой трубкой добавляли по каплям свежеприготовленный раствор Br2 в CH2Cl2 (1,95 M, 1,9 мл, 3,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем нейтрализовали раствором насыщенного водного NaHCO3. Смесь разбавляли CH2Cl2 и слои разделяли. Органический слой промывали насыщенным водным NaHCO3 и соляным раствором, затем сушили над MgSO4/Na2SO4 и концентрировали в вакууме. Неочищенный продукт наносили на силикагель (2,9 г) в виде CH2Cl2 раствора. Очистка хроматографией (0-20% EtOAc-гексаны) давала 2-бром-1-(4-бром-3,5-диметоксифенил)этанон в виде белого твердого вещества (0,737 г, выход 63%). Крупномасштабный синтез 2-бром-1-(4-бром-3,5-диметоксифенил)этанона осуществляли без хроматографической очистки бромида.
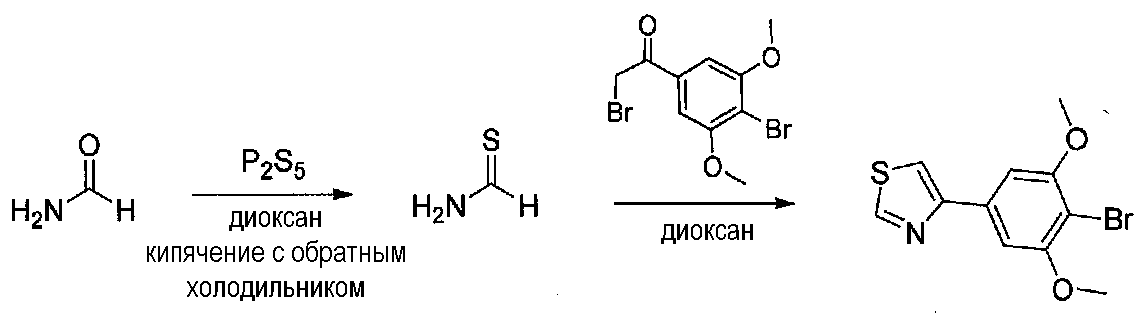
В колбу, высушенную в сушильном шкафу, в атмосфере аргона загружали P2S5 (0,53 г, 1,2 ммоль), безводный диоксан (5 мл) и формамид (0,53 мл, 13,3 ммоль). Реакционную колбу оснащали противоточным холодильником и хлоркальциевой трубкой и кипятили с обратным холодильником в течение 2,25 часов. В отдельную высушенную в сушильном шкафу колбу в атмосфере аргона загружали 2-бром-1-(4-бром-3,5-диметоксифенил)этанон (0,313 г, 0,93 ммоль) и безводный диоксан (6 мл). Тиоформамидную смесь (выше) декантировали в реакционную колбу, оставляя твердые вещества внутри. Реакционную колбу оснащали противоточным холодильником, помещали под хлоркальциевую трубку и кипятили с обратным холодильником в течение 3 часов, затем охлаждали до комнатной температуры. После перемешивания в течение ночи смесь подщелачивали добавлением водного раствора 2 M Na2CO3, разбавляли H2O, затем экстрагировали EtOAc три раза. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали. Неочищенный твердый остаток растворяли в CH2Cl2 и наносили на силикагель. Очистка хроматографией (0-35% EtOAc-гексаны) давала 4-(4-бром-3,5-диметоксифенил)тиазол в виде белого твердого вещества (0,20 г, выход 73%).
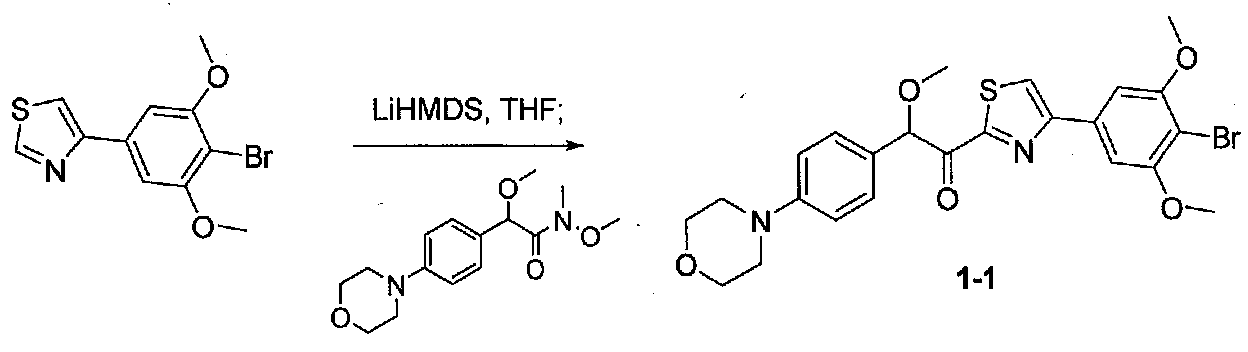
К охлажденному до -78°C раствору 4-(4-бром-3,5-диметоксифенил)тиазола (0,096 г, 0,32 ммоль) в безводном THF (2 мл) в атмосфере аргона добавляли по каплям раствор LiHMDS (1,0 M в THF, 0,35 ммоль). После перемешивания в течение 30 минут добавляли по каплям раствор N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида (0,121 г, 0,41 ммоль) в безводном THF (1,5 мл, 1,0 мл). После перемешивания в течение 35 минут ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры. Смесь гасили соляным раствором и разбавляли EtOAc. Слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали. Очистка хроматографией (25-45% EtOAc-гексаны) давала 1-(4-(4-бром-3,5-диметоксифенил)тиазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон в виде желтого твердого вещества (0,050 г, выход 29%). МС: m/z 533,1 [M+H]+.
ПРИМЕР 2
1-(4-(4-БРОМО-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
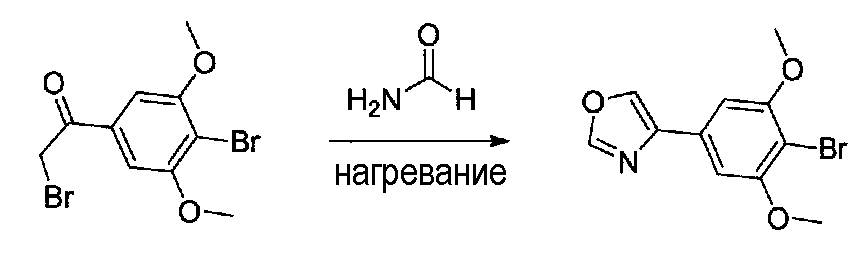
Раствор 2-бром-1-(4-бром-3,5-диметоксифенил)этанона (0,8 г, 0,96 ммоль) в формамиде (7 мл) в колбе, высушенной в сушильном шкафу, в атмосфере аргона нагревали при 100°C в течение 10 часов, затем при 110°C в течение 5 часов. После охлаждения до комнатной температуры осторожно добавляли EtOAc и насыщенный водный NaHCO3 и смесь перемешивали в течение 15 минут. Затем ее экстрагировали EtOAc дважды и объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали. Очистка хроматографией (20-40% EtOAc-гексаны) давала 4-(4-бром-3,5-диметоксифенил)оксазол в виде желтого твердого вещества (0,387 г, выход 58%).
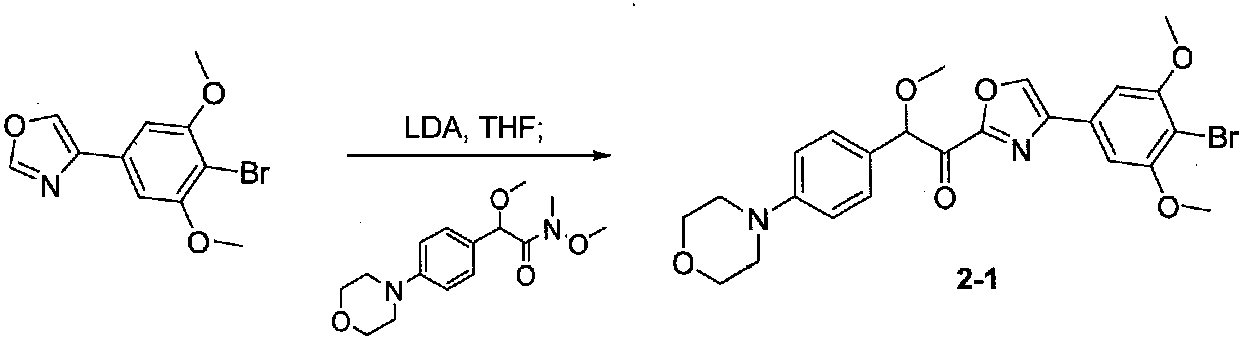
К охлажденному до -20°C раствору 4-(4-бром-3,5-диметоксифенил)оксазола (0,158 г, 0,56 ммоль) в безводном THF (2 мл) в колбе, высушенной в сушильном шкафу, в атмосфере аргона добавляли по каплям раствор LDA (2,0 M в THF/гептан/этилбензол; 0,37 мл, 0,74 ммоль). Смесь перемешивали при от -20 до -10°C в течение 50 минут, затем охлаждали до -20°C. Добавляли раствор N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида (0,245 г, 0,83 ммоль) в безводном THF (3 мл), затем смесь медленно нагревали до комнатной температуры и перемешивали в течение 21 часа. Реакционную смесь гасили H2O и экстрагировали EtOAc. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали. Очистка хроматографией (50-60% EtOAc-гексаны) давала 1-(4-(4-бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон в виде желтого твердого вещества (0,097 г, выход 34%). МС: m/z 517,1 [M+H]+.
ПРИМЕР 3
2-(4-(1H-ПИРАЗОЛ-1-ИЛ)ФЕНИЛ)-1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ТИАЗОЛ-2-ИЛ)-2-МЕТОКСИЭТАНОН
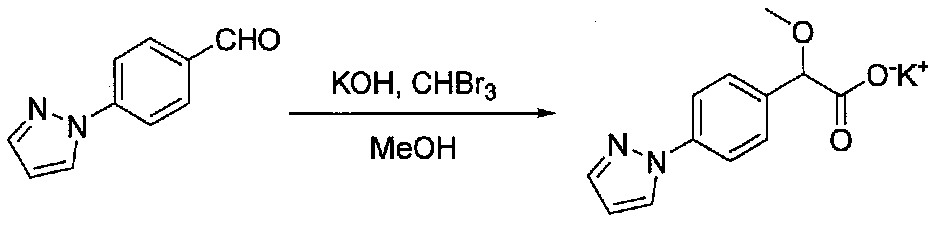
К перемешиваемому раствору 4-(1H-пиразол-1-ил)бензальдегида (1,3 г, 7,55 ммоль) и бромоформа (0,85 мл, 9,75 ммоль) в MeOH (10 мл) и диоксане (10 мл) добавляли по каплям раствор гидроксида калия (2,2 г, 39 ммоль) в MeOH (10 мл) в течение 15 минут. Затем перемешивание продолжали в течение 23 часов. Смесь фильтровали через целит, промывали EtOAc и концентрировали при пониженном давлении с получением 2-(4-(1-пиразол-1-ил)фенил)-2-метоксиацетата калия в виде светло-желтого твердого вещества (3,2 г), который использовали без дополнительной очистки. См. патент США № 7129238.
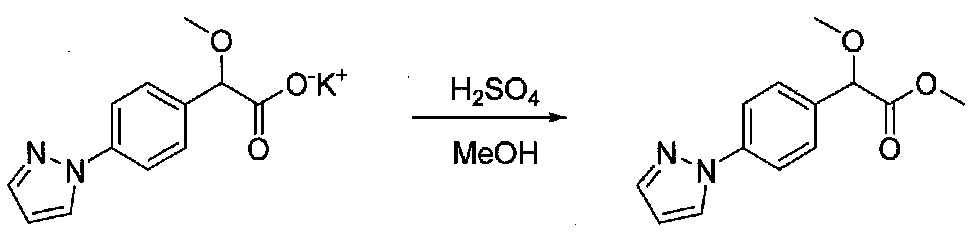
К перемешиваемому раствору 2-(4-(1H-пиразол-1-ил)фенил)-2-метоксиацетата калия (~7,55 ммоль) в сухом MeOH в атмосфере аргона добавляли по каплям серную кислоту (2,0 мл). Смесь нагревали при 80°C в течение 17 часов. После охлаждения до комнатной температуры добавляли воду, затем смесь подщелачивали добавлением насыщенного водного NaHCO3. Водную фазу экстрагировали EtOAc и объединенные органические слои промывали водой и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (20-35% EtOAc-гексаны) давала метил 2-(4-(1H-пиразол-1-ил)фенил)-2-метоксиацетат в виде бесцветного масла (0,88 г, выход 47% для двух стадий).
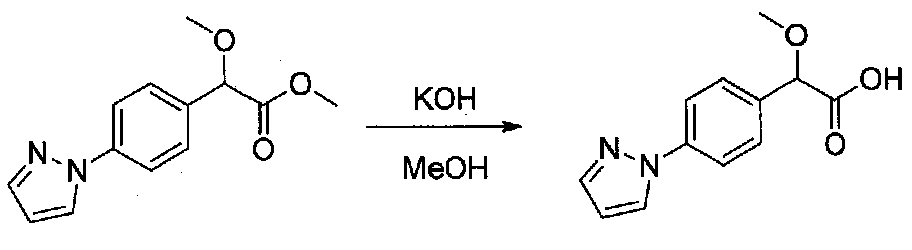
К перемешиваемому раствору метил 2-(4-(1H-пиразол-1-ил)фенил)-2-метоксиацетата (0,204 г, 0,83 ммоль) в сухом MeOH (5 мл) в атмосфере аргона добавляли раствор KOH в MeOH (1,6 мл 0,5 M раствора, 8,3 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и летучие компоненты удаляли при пониженном давлении. Остаток разбавляли насыщенным водным NH4Cl и экстрагировали EtOAc. Затем применяли дополнительное количество EtOAc для экстракции водной фазы, по мере того как pH доводили от ~8 до ~2, используя концентрированную HCl. Объединенные органические слои сушили над Na2SO4, фильтровали и растворители удаляли при пониженном давлении с получением 2-(4-(1H-пиразол-1-ил)фенил)-2-метоксиуксусной кислоты (0,18 г, 95%), которую использовали без дополнительной очистки.
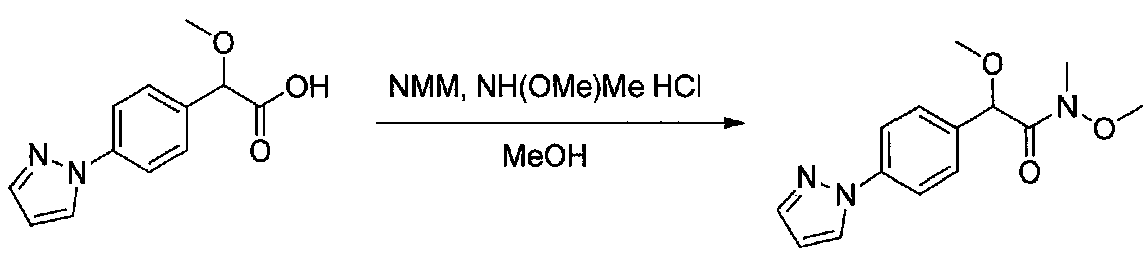
К раствору метил 2-(4-(1H-пиразол-1-ил)фенил)-2-метоксиуксусной кислоты (0,18 г, 0,77 ммоль) в сухом дихлорметане (5 мл) в атмосфере аргона добавляли N-метилморфолин (0,18 мл, 1,7 ммоль). Реакционную смесь охлаждали до 0°C, добавляли изобутилхлорформиат (0,11 мл, 0,85 ммоль) и смесь перемешивали в течение 40 минут. Затем добавляли одной порцией N,O-диметилгидроксиламингидрохлорид (0,098 г, 1 ммоль) и медленно нагревали до комнатной температуры и перемешивали в течение 18 часов. Смесь разбавляли насыщенным водным NaHCO3 и экстрагировали EtOAc. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-80% EtOAc-гексаны) давала 2-(4-(1H-пиразол-1-ил)фенил)-N,2-диметокси-N-метилацетамид (0,16 г, выход 76%).
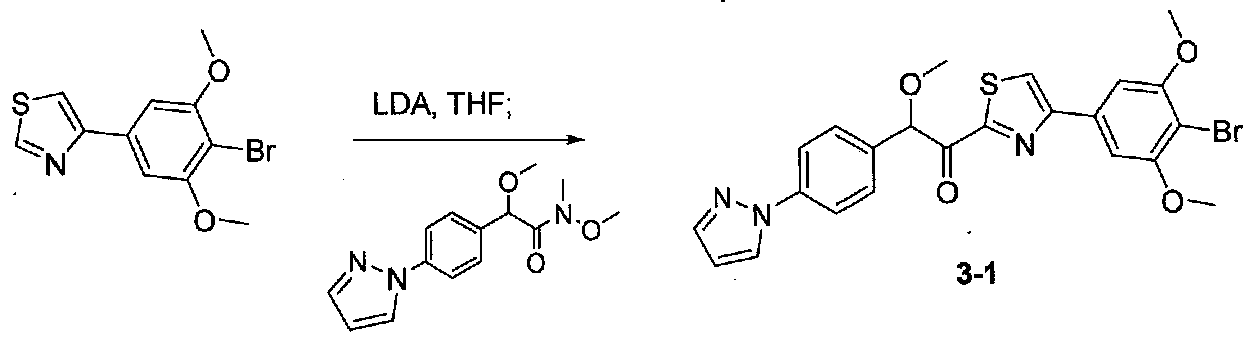
К охлажденному до -20°C раствору 4-(4-бром-3,5-диметоксифенил)тиазола (0,092 г, 0,305 ммоль) в безводном THF (2 мл) в атмосфере аргона добавляли по каплям раствор LDA (0,18 мл 2,0 M раствора в THF/гептан/этилбензол, 0,37 ммоль). После перемешивания в течение 30 минут реакционную смесь охлаждали до -78°C и добавляли по каплям раствор 2-(4-(1H-пиразол-1-ил)фенил)-N,2-диметокси-N-метилацетамида (0,15 г, 0,55 ммоль) в безводном THF (1,5 мл, 1,0 мл). После перемешивания в течение 90 минут смесь гасили соляным раствором и разбавляли EtOAc. Слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали. Очистка хроматографией (0-35% EtOAc-гексаны) давала 2-(4-(1H-пиразол-1-ил)фенил)-1-(4-(4-бром-3,5-диметоксифенил)тиазол-2-ил)-2-метоксиэтанон в виде желтого твердого вещества (0,030 г, выход 20%). МС: m/z 514,0 [M+H]+.
ПРИМЕР 4
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)-1-МЕТИЛ-1H-ИМИДАЗОЛ-2-ил)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
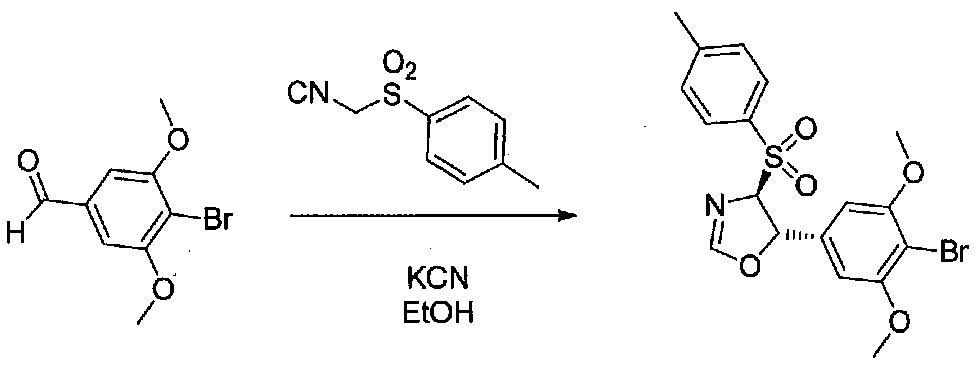
В колбу, высушенную в сушильном шкафу, в атмосфере аргона добавляли 4-бром-3,5-диметоксибензальдегид (1,0 г, 4,08 ммоль), абсолютный EtOH (34 мл), п-толуолсульфонилметилизоцианид (0,78 г, 4,0 ммоль) и KCN (0,035 г, 0,54 ммоль). Смесь перемешивали в течение 19 часов при комнатной температуре, затем концентрировали в вакууме с получением 5-(4-бром-3,5-диметоксифенил)-4-тозил-4,5-дигидрооксазола, который использовали на следующей стадии синтеза без очистки.

В высушенную в сушильном шкафу колбу, выдерживающую высокое давление, добавляли 5-(4-бром-3,5-диметоксифенил)-4-тозил-4,5-дигидрооксазол (0,4 г, 0,91 ммоль), THF раствор метиламина (1,8 мл 2,0 M раствора, 3,6 ммоль) и ксилол (5 мл). Колбу герметично закрывали в атмосфере аргона, затем нагревали при 135°C в течение 15 часов. После охлаждения до комнатной температуры реакционную смесь переносили и концентрировали в вакууме, затем распределяли между EtOAc и H2O. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические слои промывали соляным раствором дважды, сушили над Na2SO4 и концентрировали. Очистка хроматографией (50-100% EtOAc-гексаны, затем 2-5% MeOH-EtOAc) давала 4-(4-бром-3,5-диметоксифенил)-1-метил-1H-имидазол в виде светло-желтого твердого вещества (0,056 г, выход 21%).
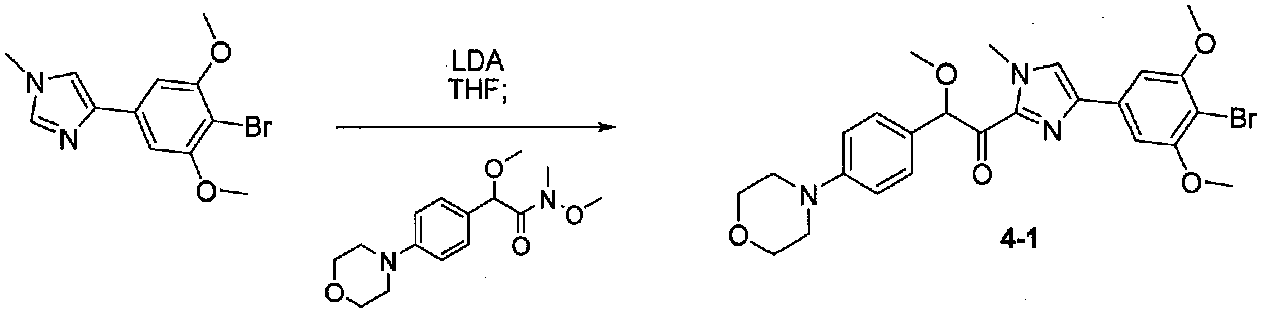
Способ, применяемый для конечной стадии конденсации примера 3, применяли с модификацией. Реакционную смесь нагревали при комнатной температуре в течение ночи. Очистка хроматографией (40-55% EtOAc-гексаны) давала 1-(4-(4-бром-3,5-диметоксифенил)-1-метил-1H-имидазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон (0,0039 г, выход 4%). МС: m/z 530,1 [M+H]+.
ПРИМЕР 5
2-(4-(1H-ПИРАЗОЛ-1-ИЛ)ФЕНИЛ)-1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИЭТАНОН
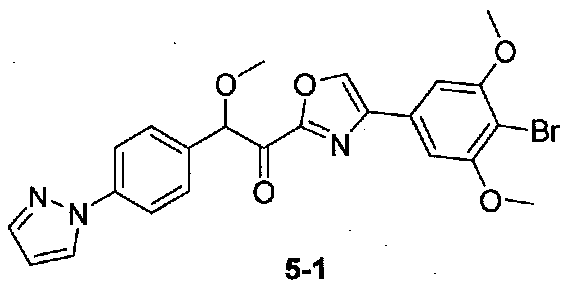
2-(4-(1H-Пиразол-1-ил)фенил)-1-(4-(4-бром-3,5-диметоксифенил)оксазол-2-ил)-2-метоксиэтанон получали из 2-(4-(1H-пиразол-1-ил)фенил)-N,2-диметокси-N-метилацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола способом, аналогичным способу, применяемому в примере 2, за исключением того, что амид добавляли к реакционной смеси при -50°C. МС: m/z 498,1 [M+H]+.
ПРИМЕР 6
1-(4-(4-БРОМО-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-(ПИПЕРИДИН-1-ИЛ)ФЕНИЛ)ЭТАНОН
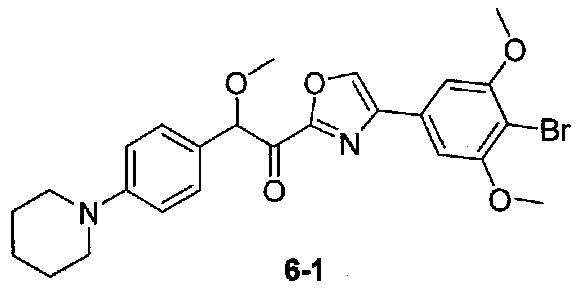
N,2-Диметокси-N-метил-2-(4-(пиперидин-1-ил)фенил)ацетамид получали в две стадии из 4-(пиперидин-1-ил)бензальдегида, следуя способу, применяемому в примере 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(4-(пиперидин-1-ил)фенил)этанон получали из N,2-диметокси-N-метил-2-(4-(пиперидин-1-ил)фенил)ацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому в примере 5. МС: m/z 515,1 [M+H]+.
ПРИМЕР 7
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-(ПИРРОЛИДИН-1-ИЛ)ФЕНИЛ)ЭТАНОН
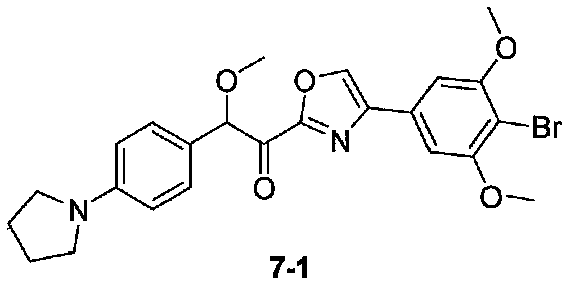
N,2-диметокси-N-метил-2-(4-(пирролидин-1-ил)фенил)ацетамид получали из 4-(пирролидин-1-ил)бензальдегида, следуя способу, применяемому в получении примера 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(4-(пирролидин-1-ил)фенил)этанон получали из 2-диметокси-N-метил-2-(4-(пирролидин-1-ил)фенил)ацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому в примере 2, за исключением того, что амид добавляли к реакционной смеси при -45°C. МС: m/z 501,1 [M+H]+.
ПРИМЕР 8
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-(4-ИЗОПРОПОКСИФЕНИЛ)-2-МЕТОКСИЭТАНОН
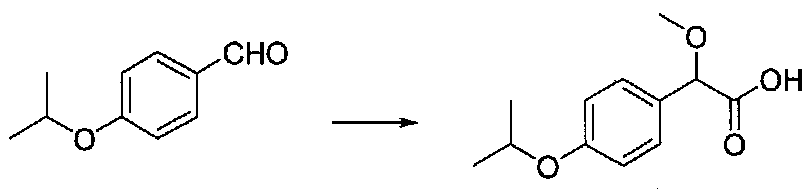
В колбу, высушенную в сушильном шкафу, загружали 4-изопропоксибензальдегид (4,9 г, 29,9 ммоль), безводный метанол (30 мл) и безводный диоксан (30 мл), затем оснащали капельной воронкой. В капельную воронку загружали раствор KOH (8,4 г, 149,5 ммоль) в безводном метаноле (30 мл) и аликвоту (~2 мл) добавляли к реакционной смеси. Добавляли к реакционной смеси бромоформ (3,4 мл, 38,8 ммоль), затем добавляли по каплям в течение 10 минут оставшийся KOH/MeOH раствор. После перемешивания в течение 18 часов смесь концентрировали в вакууме. Остаток разбавляли водой, и pH доводили до ~2, используя концентрированную HCl, затем экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и фильтровали. Концентрирование фильтрата в вакууме давало 2-(4-изопропоксифенил)-2-метоксиуксусную кислоту в виде светло-желтого твердого вещества (6,8 г), которое использовали без дополнительной очистки.
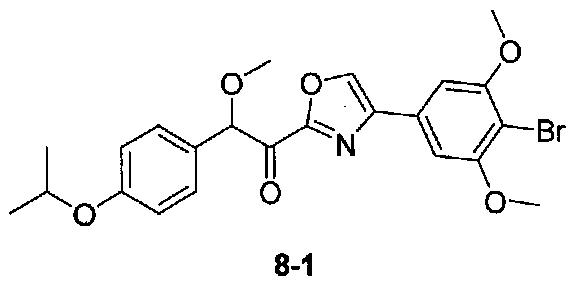
2-(4-Изопропоксифенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-изопропоксифенил)-2-метоксиуксусной кислоты, следуя способу, применяемому для получения примера 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-(4-изопропоксифенил)-2-метоксиэтанон получали из 2-(4-изопропоксифенил)-N,2-диметокси-N-метилацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому для получения примера 7. МС: m/z 490,1 [M+H]+.
ПРИМЕР 9
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(ХИНОЛИН-5-ИЛ)ЭТАНОН
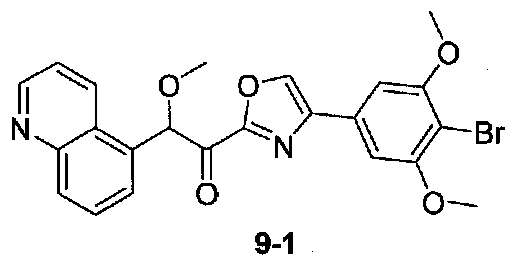
N,2-Диметокси-N-метил-2-(хинолин-5-ил)ацетамид получали из хинолин-5-карбальдегида в две стадии, следуя способу, применяемому для получения примера 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(хинолин-5-ил)этанон получали из N,2-диметокси-N-метил-2-(хинолин-5-ил)ацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому в примере 2. МС: m/z 483,0 [M+H]+.
ПРИМЕР 10
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(ХИНОЛИН-3-ИЛ)ЭТАНОН
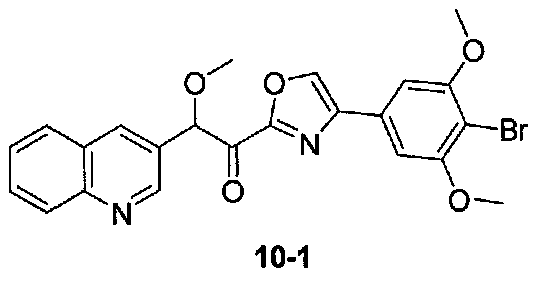
N,2-Диметокси-N-метил-2-(хинолин-3-ил)ацетамид получали из хинолин-3-карбальдегида в две стадии, следуя способу, применяемому для получения примера 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(хинолин-3-ил)этанон получали из N,2-диметокси-N-метил-2-(хинолин-3-ил)ацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому в примере 2. МС: m/z 483,1 [M+H]+.
ПРИМЕР 11
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН

N,2-Диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамид получали из 4-(5-метил-1,3,4-оксадиазол-2-ил)бензальдегида в две стадии, следуя способу, применяемому для получения примера 1. 1-(4-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида и 4-(4-бром-3,5-диметоксифенил)оксазола, следуя способу, применяемому в примере 2, за исключением того, что амид добавляли к реакционной смеси при -30°C. МС: m/z 514,0 [M+H]+.
ПРИМЕР 12
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
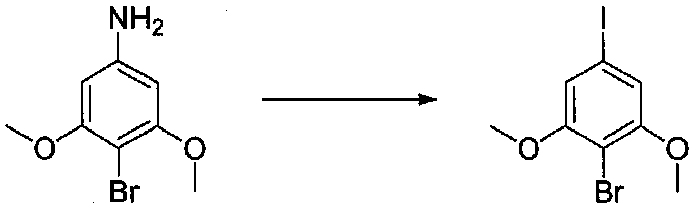
В колбу, содержащую суспензию 4-бром-3,5-диметоксианилина (1,99 г, 8,56 ммоль; полученного согласно US2006/128695) в H2O (60 мл), медленно добавляли концентрированную H2SO4 (10 мл). Экзотермический эффект контролировали охлаждением на льду, затем реакционную смесь охлаждали до от -10 до -8°C (температура бани). Затем добавляли по каплям в течение 6 минут раствор NaNO2 (0,70 г, 10 ммоль) в H2O (3,5 мл), и смесь перемешивали в течение 70 минут. Добавляли по каплям раствор KI (2,8 г, 16,9 ммоль) в H2O (3,5 мл) и смесь перемешивали при от -10 до -5°C в течение 30 минут. После удаления бани со льдом смесь перемешивали в течение 80 минут, затем добавляли EtOAc и смесь перемешивали в течение дополнительных 40 минут. Слои разделяли и водный слой экстрагировали EtOAc несколько раз. Объединенные органические слои промывали 1 M NaOH дважды, 10% Na2S2O3 дважды и соляным раствором, затем сушили над Na2SO4. Очистка хроматографией (0-20% EtOAc-гексаны) давала 2-бром-5-йод-1,3-диметоксибензол (1,86 г, выход 63%).
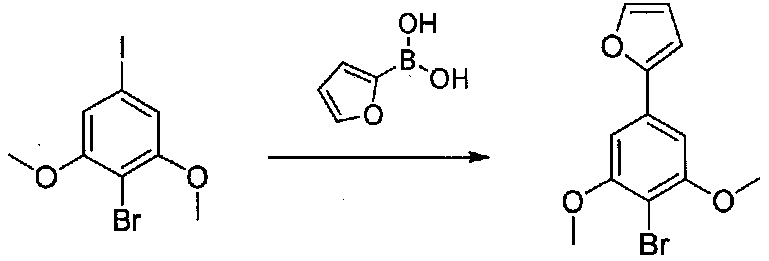
2-(4-Бром-3,5-диметоксифенил)фуран получали согласно методике, приведенной в WO 2008/040669, следующим образом. В круглодонную колбу, содержащую 3,5-диметокси-4-бром-йодбензол (7,9 г, 85% чистота, 19,6 ммоль), 2-фурилбороновую кислоту (3,4 г, 30,4 ммоль), трифенилфосфин (0,358 г, 1,37 ммоль), тетрабутиламмонийбромид (7,94 г, 14,6 ммоль) и Na2CO3 (4,9 г, 46,2 ммоль), добавляли THF (87 мл) и H2O (87 мл). Смесь дегазировали чередующимся помещением под вакуум и аргон три раза в течение нескольких минут каждый. Добавляли 10% Pd/C (1,36 г), и смесь нагревали при 60°C в течение 17 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь фильтровали через целит и промывали THF и EtOAc. Фильтратные слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали. Очистка колоночной хроматографией (0-25% Et2O-гексаны) давала 2-(4-бром-3,5-диметоксифенил)фуран в виде белого твердого вещества (5,06 г, выход 91%). Продукт ТСХ Rf 0,35 (15% EtOAc-гексаны, элюент ТСХ).
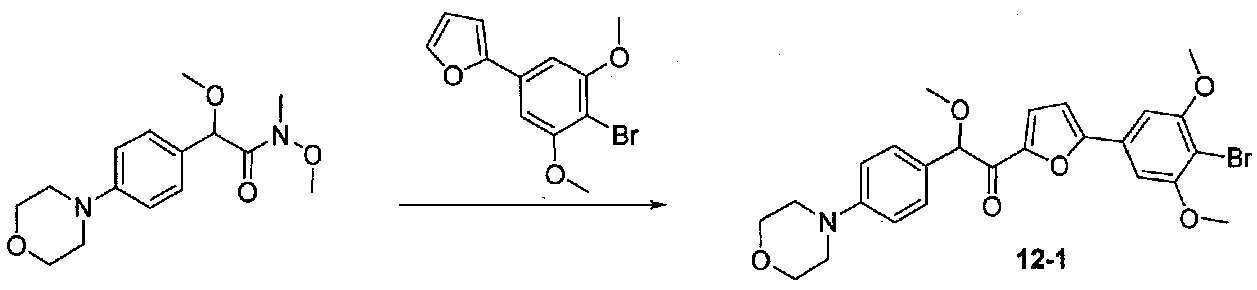
К раствору 2-(4-бром-3,5-диметоксифенил)фурана (0,203 г, 0,72 ммоль) в безводном THF (2 мл) в атмосфере аргона в колбе, высушенной в сушильном шкафу, охлажденном до -78°C, добавляли по каплям раствор диизопропиламида лития (2,0 M в THF/гептан/этилбензол; 0,4 мл, 0,8 ммоль). После перемешивания в течение 35 минут при -78°C добавляли по каплям раствор N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамид (0,315 г, 1,1 ммоль) в THF (2 мл). После перемешивания в течение 25 минут смесь нагревали до комнатной температуры при перемешивании в течение 2 часов. Реакционную смесь гасили добавлением насыщенного водного NH4Cl, затем добавляли соляной раствор и EtOAc. Слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка колоночной хроматографией (35-65% EtOAc-гексаны) давала масло, которое растирали с ΜeΟΗ-H2O (1:1) при обработке ультразвуком. Твердое вещество собирали на воронку Бюхнера, промывали MeOH-Et2O (1:1) и сушили в вакууме с получением 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)этанона в виде желтого твердого вещества. Вторую порцию собирали из MeOH-Et2O (~10%) для получения дополнительного количества продукта (0,220 г в сумме, выход 59%). МС: m/z 516,1 [M+H]+.
ПРИМЕР 13
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
К раствору 4-(5-метил-1,3,4-оксадиазол-2-ил)бензальдегида (5,12 г, 27,2 ммоль) в безводном MeOH (27 мл) и безводном диоксане (27 мл) при от -15 до -10°C (температура бани) добавляли несколько капель раствора KOH (7,6 г, 135,4 ммоль) в MeOH (27 мл). Добавляли бромоформ (3 мл, 34,4 ммоль), затем добавляли оставшийся раствор KOH/MeOH в течение 20 минут. Смесь перемешивали в течение 1 часа и ледяную баню удаляли. После перемешивания в течение 30 минут реакционную смесь помещали на ледяную баню и нагревали медленно до комнатной температуры в течение ночи, затем концентрировали досуха. После растворения в минимальном количестве H2O остаток подкисляли до pH 1 с помощью 6 M HCl. Водную смесь экстрагировали EtOAc несколько раз при добавлении соляного раствора к водному слою в процессе экстракции. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусной кислоты в виде полутвердого вещества (6,8 г, количественный выход). Продукт использовали без дополнительной очистки.
К охлажденному на льду раствору 2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусной кислоты (6,8 г, 27,4 ммоль) в безводном CH2Cl2 (270 мл) и диизоопропилэтиламина (17 мл, 97 ммоль) в атмосфере аргона добавляли по каплям трифторид бис(2-метоксиэтил)аминосеры (5,6 мл, 30,3 ммоль). После перемешивания над ледяной баней в течение 45 минут добавляли N,O-диметилгидроксиламингидрохлорид (3,40 г, 34,8 ммоль) тремя порциями в течение 15 минут. Смесь перемешивали в течение 15 минут, затем баню со льдом удаляли. Через 3 часа добавляли насыщенный водный NaHCO3 и перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные органические слои промывали насыщенным водным NaHCO3, H2O и соляным раствором, сушили над MgSO4, фильтровали через целит и концентрировали в вакууме. Очистка хроматографией (75-100% EtOAc-гексаны, затем 0-5% EtOH-EtOAc) давала N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамид в виде масла, которое становилось твердым при стоянии (2,06 г, выход 26%).
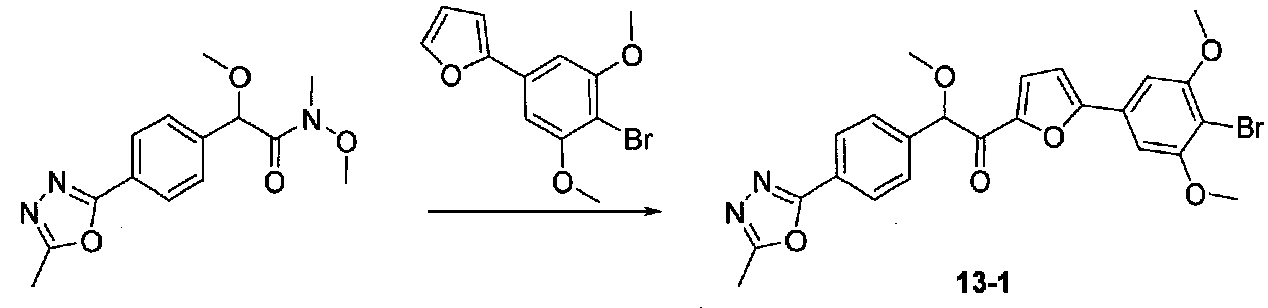
2-(4-Бром-3,5-диметоксифенил)фуран конденсировали с N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамидом согласно способу, применяемому для получения примера 12. Очистка хроматографией (50-80% EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон в виде желтой пены (0,356 г, выход 40%). МС: m/z 513,2 [M+H]+.
ПРИМЕР 14
2-(4-(1H-ПИРАЗОЛ-1-ИЛ)ФЕНИЛ)-1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИЭТАНОН
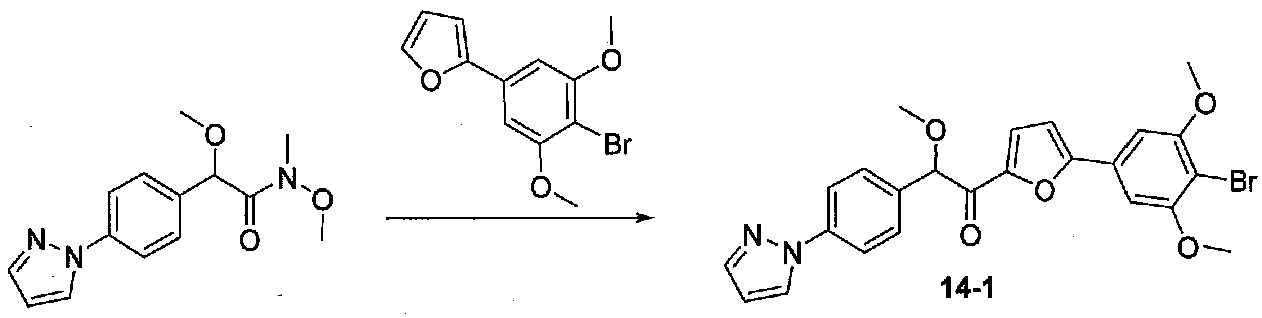
Применяли способ, применяемый для конечной стадии конденсации примера 12. Очистка хроматографией (20-55% EtOAc-гексаны) давала 2-(4-(1H-пиразол-1-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон в виде светло-желтого твердого вещества (0,0625 г, выход 37%). МС: m/z 497,2 [M+H]+.
ПРИМЕР 15
2-(4-(1H-ПИРАЗОЛ-4-ИЛ)ФЕНИЛ)-1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИЭТАНОН
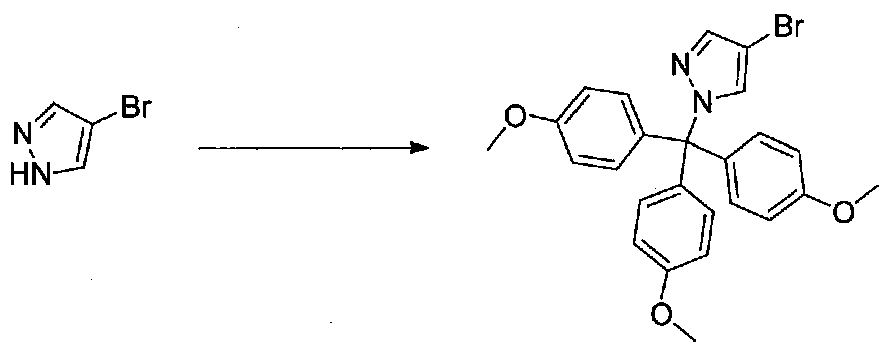
К суспензии 4-бромпиразола (1,5 г, 10,2 ммоль) и 4,4',4"-триметокситритилхлорида (4,5 г, 12,2 ммоль) в безводном DMF (20 мл) в атмосфере аргона добавляли триэтиламин (3 мл, 21,5 ммоль), и смесь охлаждали на бане со льдом. После перемешивания в течение 10 минут баню со льдом удаляли, и реакционную смесь перемешивали в течение 2,5 часов. Смесь разбавляли H2O и экстрагировали EtOAc. Объединенные органические фракции промывали H2O три раза, затем насыщенным водным NaHCO3 и соляным раствором. Раствор сушили над Na2SO4 и концентрировали в вакууме. Неочищенное масло перекристаллизовывали из изопропанола с получением 4-бром-1-(трис(4-метоксифенил)метил)-1H-пиразола в виде грязно-белых кристаллов (две порции; 2,55 г, выход 52%).
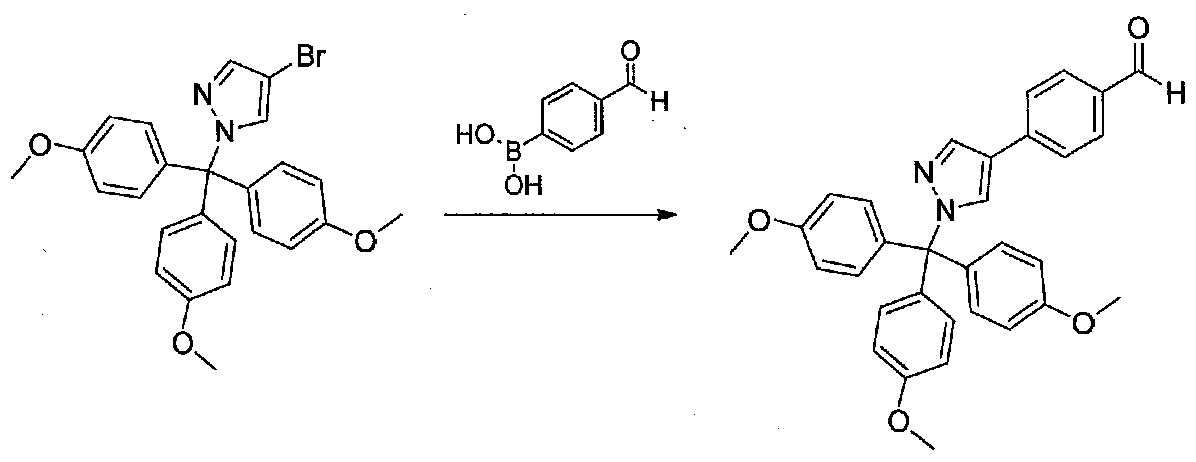
Смесь 4-бром-1-(трис(4-метоксифенил)метил)-1H-пиразола (1,47 г, 3,1 ммоль), (4-формилфенил)бороновой кислоты (0,94 г, 6,3 ммоль) и K2CO3 (0,65 г, 4,7 ммоль) в DME-H2O (25 мл, 4:1) дегазировали чередующимся помещением ее в вакуум и в атмосферу аргона три раза в течение нескольких минут каждый. Затем добавляли тетракис(трифенилфосфин)палладий (0,35 г, 0,3 ммоль), смесь снова дегазировали. После нагревания в течение 16,5 часов при 80°C и охлаждения до комнатной температуры добавляли H2O. Смесь экстрагировали EtOAc и объединенные органические слои промывали H2O, насыщенным водным NaHCO3 и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (20-30% EtOAc-гексаны; EtOAc, содержащий 1% Et3N) давала 4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)бензальдегид (1,31 г, полученный из двух реакций, 33%).
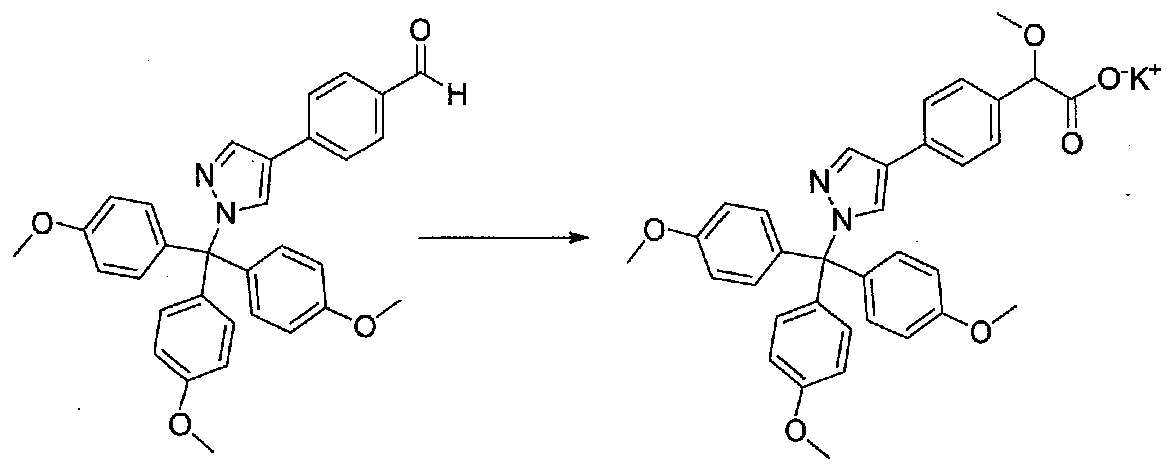
Применяли способ, применяемый для получения примера 1, за исключением того, что выделяли соль калия. Проводя реакцию при комнатной температуре, добавляли EtOAc и смесь фильтровали через целит и концентрировали в вакууме. Остаток растворяли в EtOAc, фильтровали через целит и концентрировали в вакууме. Продукт растворяли в смеси EtOAc-толуол и концентрировали с получением 2-метокси-2-(4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)фенил)ацетата калия (1,75 г, количественный выход), который использовали без дополнительной очистки.
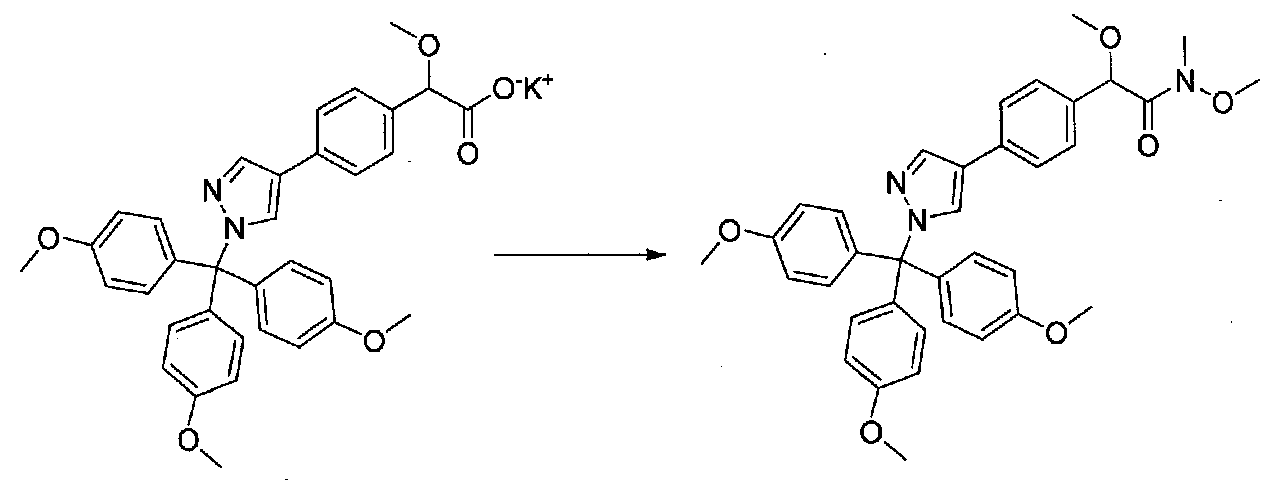
К охлажденному на льду раствору 2-метокси-2-(4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)фенил)ацетата калия (1,25 г, 2,1 ммоль) в безводном DMF (10 мл) в атмосфере аргона добавляли по каплям диизопропилэтиламин (0,54 мл, 3,1 ммоль) и трифторид бис(2-метоксиэтил)аминосеры (0,46 мл, 2,5 ммоль). Реакционную смесь перемешивали в течение 30 минут, затем добавляли N,Ο-диметилгидроксиламингидрохлорид (0,303 г, 3,1 ммоль). После перемешивания в течение дополнительных 15 минут на бане со льдом смесь нагревали до комнатной температуры и перемешивали в течение 3,5 часов. Добавляли H2O и смесь экстрагировали EtOAc. Объединенные органические слои промывали H2O, насыщенным водным NH4Cl, насыщенным водным NaHCO3 и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (45-90% EtOAc-гексаны; EtOAc, содержащий 1% Et3N) давала N,2-диметокси-N-метил-2-(4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)фенил)ацетамид (0,478 г из двух реакций, суммарный выход 32%).
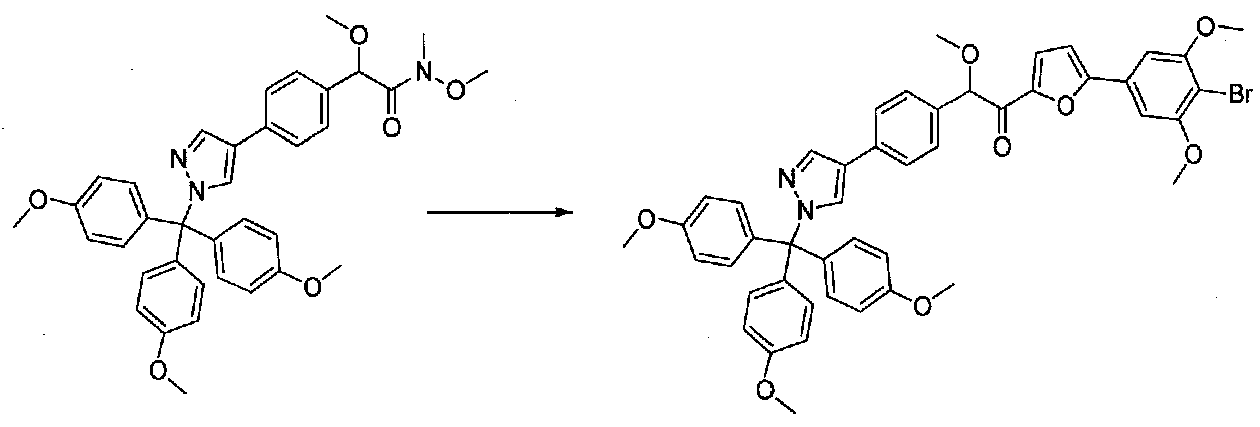
Применяли способ, применяемый для конечной стадии конденсации примера 12, за исключением более короткого времени реакции при комнатной температуре 70 минут. Очистка хроматографией (35-90% EtOAc-гексаны; EtOAc, содержащий 1% Et3N) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)фенил)этанон в виде твердого вещества (0,10 г, выход 18%).
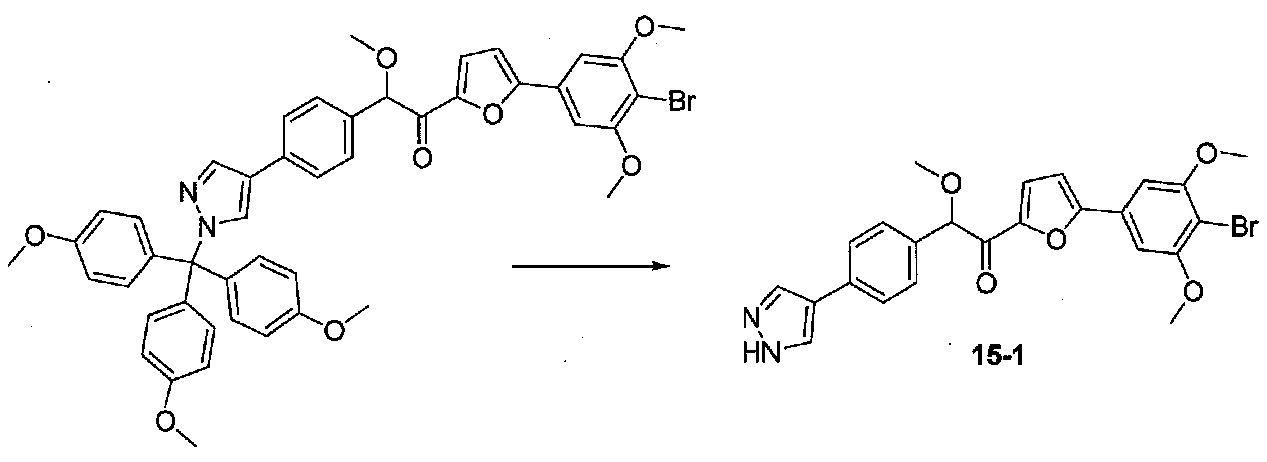
К суспензии 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(1-(трис(4-метоксифенил)метил)-1H-пиразол-4-ил)фенил)этанона (0,10 г, 0,12 ммоль) в MeOH-H2O (22 мл, 10:1) добавляли пара-толуолсульфонат пиридиния (0,046 г, 0,16 ммоль). После перемешивания в течение 18 часов при комнатной температуре добавляли насыщенный водный NaHCO3 и летучие компоненты удаляли в вакууме. Остаток разбавляли небольшим количеством H2O, затем экстрагировали EtOAc. Объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (50-100% EtOAc-гексаны) давала 2-(4-(1H-пиразол-4-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон в виде желтой пены (0,010 г, выход 17%). МС: m/z 497,0 [M+H]+.
ПРИМЕР 16
1-(5-(4-БРОМО-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛФУРАН-2-ИЛ)ФЕНИЛ)ЭТАНОН
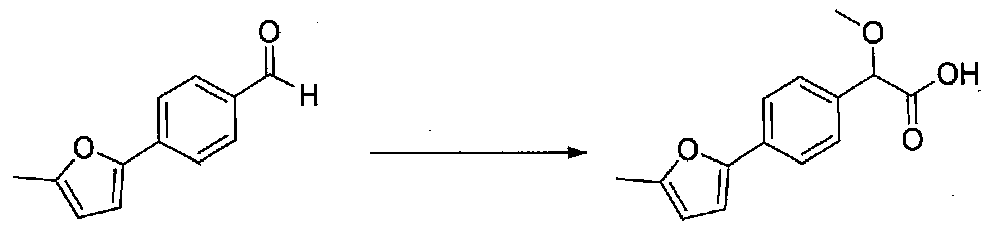
2-Метокси-2-(4-(5-метилфуран-2-ил)фенил)уксусную кислоту получали из 4-(5-метилфуран-2-ил)бензальдегида, применяя способ, описанный в примере 1 (3,3 г, количественный выход). Продукт использовали без дополнительной очистки.
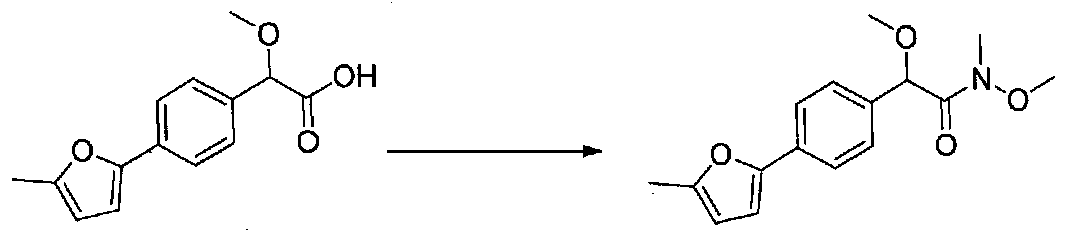
N,2-Диметокси-N-метил-2-(4-(5-метилфуран-2-ил)фенил)ацетамид получали из 2-метокси-2-(4-(5-метилфуран-2-ил)фенил)уксусной кислоты, следуя способу, применяемому в примере 13. Продукт выделяли в виде оранжевого масла (0,693 г, выход 18%).
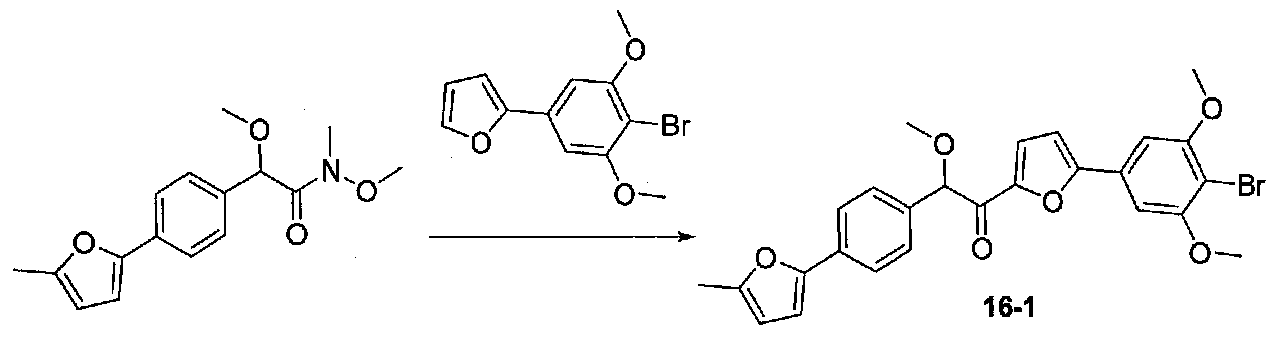
2-(4-Бром-3,5-диметоксифенил)фуран конденсировали с N,2-диметокси-N-метил-2-(4-(5-метилфуран-2-ил)фенил)ацетамидом, следуя способу, применяемому для конечной стадии конденсации примера 12. Очистка хроматографией (30-60% EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метилфуран-2-ил)фенил)этанон в виде бледно-оранжевого твердого вещества (0,172 г, выход 47%). МС: m/z 511,0 [M+H]+.
ПРИМЕР 17
2,3-ДИМЕТОКСИ-5-(5-(2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)АЦЕТИЛ)ФУРАН-2-ИЛ)БЕНЗОНИТРИЛ
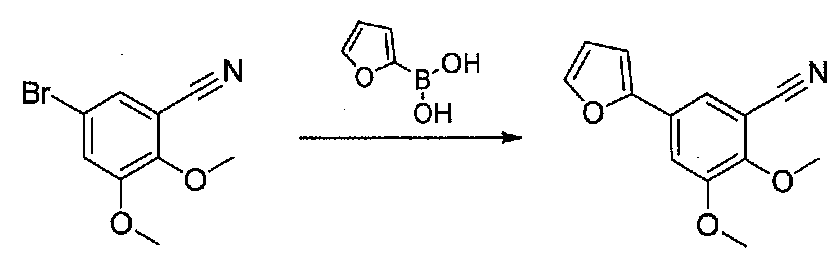
К смеси 5-бром-2,3-диметоксибензонитрила (0,958 г, 4,0 ммоль), 2-фурилбороновой кислоты (0,53 г, 4,7 ммоль), диоксана (24 мл), H2O (8 мл) и Na2CO3 (1,1 г, 10,4 ммоль) добавляли тетракис(трифенилфосфин)палладий (0,23 г, 0,2 ммоль). Смесь дегазировали чередующимся помещением в вакуум и в атмосферу аргона три раза в течение нескольких минут каждый, затем нагревали при 85°C в атмосфере аргона в течение 16,5 часов. После охлаждения до комнатной температуры смесь разбавляли H2O и экстрагировали EtOAc. Объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-20% EtOAc-гексаны) давала 5-(фуран-2-ил)-2,3-диметоксибензонитрил в виде белого твердого вещества (0,85 г, выход 94%).
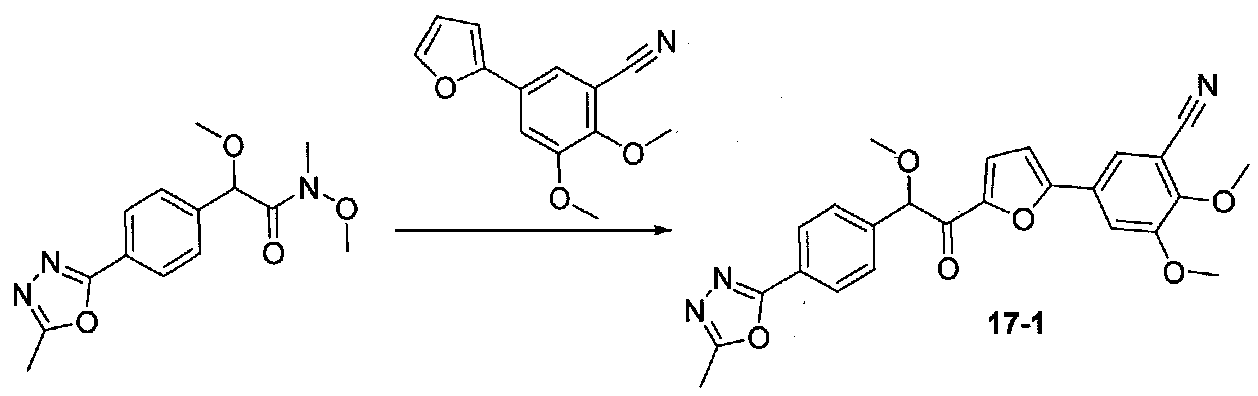
Следовали способу, применяемому для конечной стадии конденсации примера 12, для реакции 5-(фуран-2-ил)-2,3-диметоксибензонитрила и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида, за исключением того, что реакцию осуществляли при от -40 до -25°C, затем нагревали до комнатной температуры. Очистка хроматографией (20-100% EtOAc-гексаны) давала 2,3-диметокси-5-(5-(2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетил)фуран-2-ил)бензонитрил (0,040 г, выход 13%). МС: m/z 460,2 [M+H]+.
ПРИМЕР 18
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-(4-(5-ЭТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)-2-МЕТОКСИЭТАНОН
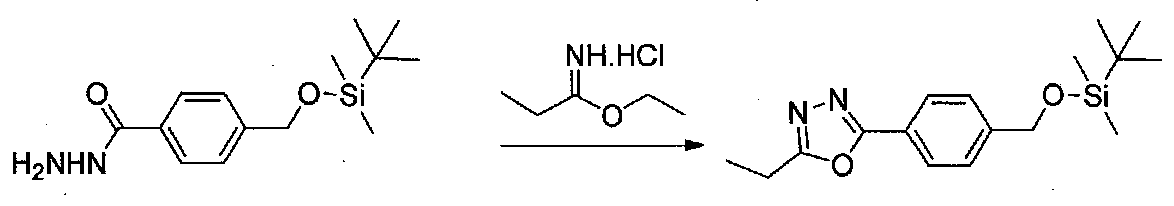
К раствору 4-(((трет-бутилдиметилсилил)окси)метил)бензогидразида (6,0 г, 21,4 ммоль; Tanaka, A. et al. J. Med. Chem. 1998, 41, 2390) и этилпропионимидатгидрохлорида (3,5 г, 25,7 ммоль; получение приводится в WO2007/73299 A1) в EtOH (120 мл) добавляли Et3N (2,59 г, 25,7 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали в вакууме. Остаток распределяли между EtOAc и H2O и органический слой промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (EtOAc-гексаны) давала 2-(4-(((трет-бутилдиметилсилил)окси)метил)фенил)-5-этил-1,3,4-оксадиазол в виде коричневого масла (3,0 г, выход 44%).
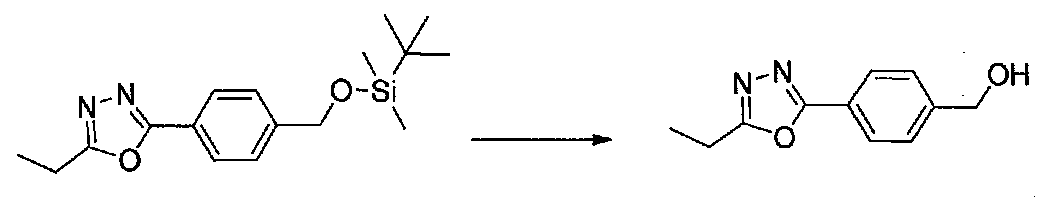
К охлажденному на льду раствору 2-(4-(((трет-бутилдиметилсилил)окси)метил)фенил)-5-этил-1,3,4-оксадиазола (3,0 г, 9,4 ммоль) в MeOH (30 мл) добавляли по каплям 1 M HCl (20 мл, 20 ммоль). Реакционную смесь перемешивали на льду в течение 1 часа, затем концентрировали. Остаток гасили насыщенным водным NaHCO3 и экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением (4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)метанола в виде не совсем белого твердого вещества (1,92 г, выход 93%).
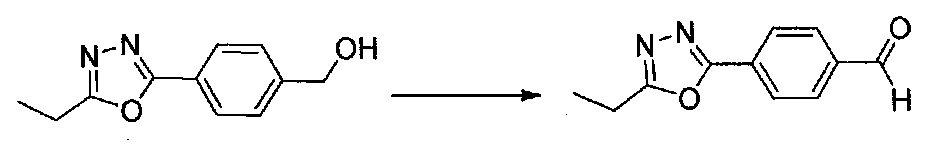
К раствору (4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)метанола (1,8 г, 8,8 ммоль) в CH2Cl2 (50 мл) добавляли молекулярные сита (1,5 г, 4 Å) и смесь охлаждали до 0°C. Добавляли порциями к реакционной смеси хлорхромат пиридиния (2,27 г, 10,5 ммоль), затем ее нагревали до комнатной температуры. После перемешивания в течение 2 часов смесь фильтровали через целит и промывали дополнительным количеством CH2Cl2. Фильтрат концентрировали в вакууме. Очистка хроматографией (EtOAc-гексаны) давала 4-(5-этил-1,3,4-оксадиазол-2-ил)бензальдегид в виде не совсем белого твердого вещества (1,3 г, 70%).
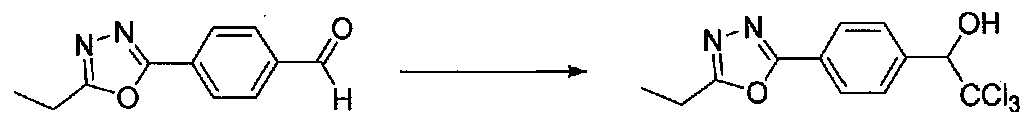
К раствору 4-(5-этил-1,3,4-оксадиазол-2-ил)бензальдегида (1,6 г, 7,9 ммоль) в безводном DMF (7 мл) добавляли CHCl3 (2,13 г, 17,8 ммоль). После охлаждения до -10°C добавляли по каплям в течение 20 минут раствор KOH (0,31 г, 5,5 ммоль) в безводном MeOH (1,5 мл). После перемешивания при -10°C в течение 1 часа реакционную смесь гасили 1 M HCl. Образовавшееся твердое вещество собирали на воронку Бюхнера, промывали H2O и сушили в вакууме с получением 2,2,2-трихлор-1-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)этанола в виде белого твердого вещества (2,0 г, выход 80%).
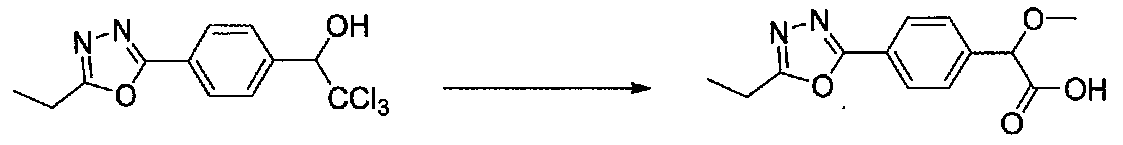
К раствору 2,2,2-трихлор-1-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)этанола (2,0 г, 6,25 ммоль) в безводном 1,4-диоксане (12,5 мл) и безводном MeOH (15 мл) добавляли раствор NaOH (1,25 г, 31,3 ммоль) в безводном MeOH (15 мл). После перемешивания в течение 4 часов при 55°C смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток нейтрализовали насыщенным водным NH4Cl, осторожно подкисляли 1 M HCl и экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток растирали с Et2O с получением 2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиуксусной кислоты в виде не совсем белого твердого вещества (1,2 г, выход 73%).
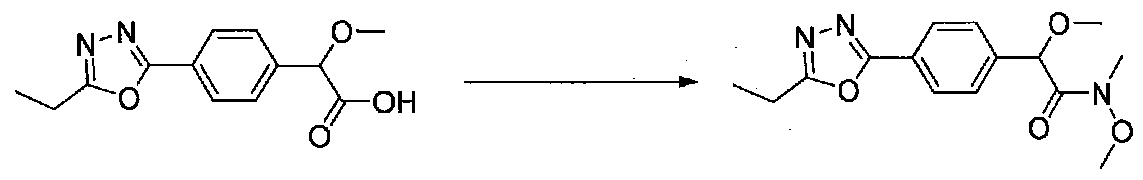
Следовали способу, применяемому для получения примера 1, с получением 2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамида из 2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиуксусной кислоты. Очистка хроматографией (20% EtOAc-гексаны) давала 2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамид в виде аморфного твердого вещества (0,55 г, выход 40%).
Следовали способу, применяемому для конечной стадии конденсации примера 12, для реакции 2-(4-бром-3,5-диметоксифенил)фурана с 2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамидом. Очистка хроматографией (20-100% EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-(4-(5-этил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиэтанон в виде желтой пены (0,037 г, выход 12%). МС: m/z 527,1 [M+H]+.
ПРИМЕР 19
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-(4-(1,1-ДИОКСИДОИЗОТИАЗОЛИДИН-2-ИЛ)ФЕНИЛ)-2-МЕТОКСИЭТАНОН
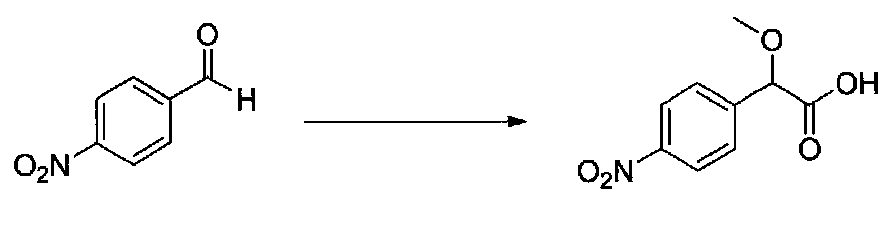
Следовали способу, применяемому для получения примера 1, с получением 2-метокси-2-(4-нитрофенил)уксусной кислоты из 4-нитробензальдегида. Кислоту, выделенную после экстракции, использовали без дополнительной очистки (5,3 г, выход 76%).
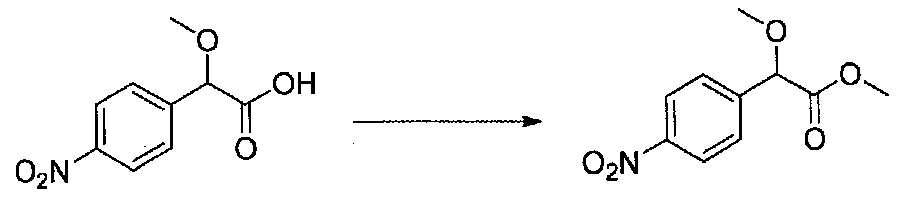
2-Метокси-2-(4-нитрофенил)уксусную кислоту этерифицировали согласно методике получения примера 3. Очистка хроматографией (15-22% EtOAc-гексаны) давала метил 2-метокси-2-(4-нитрофенил)ацетат в виде желтого масла (2,15 г, выход 38%).
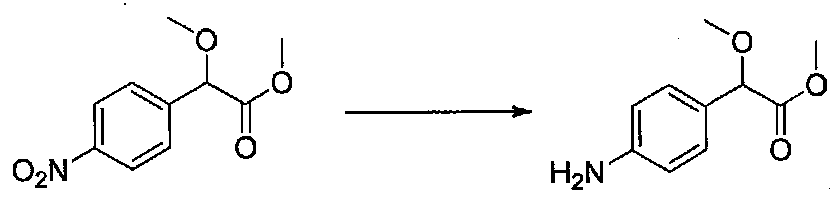
Раствор метил 2-метокси-2-(4-нитрофенил)ацетата (0,42 г, 1,87 ммоль) в абсолютном EtOH (15 мл) дегазировали чередующимся помещением в вакуум и в атмосферу аргона три раза в течение нескольких минут каждый, затем добавляли 10% Pd/C (0,19 г). Смесь перемешивали в атмосфере H2 (1 атм) в течение 3 часов, затем разбавляли EtOAc и фильтровали через слой целита и силикагеля. Фильтрат концентрировали в вакууме. Очистка хроматографией (25-40% EtOAc-гексаны) давала метил 2-(4-аминофенил)-2-метоксиацетат в виде желтого масла, содержащего примеси (0,6 г для двух порций, выход 77%). Соединение использовали без дополнительной очистки.
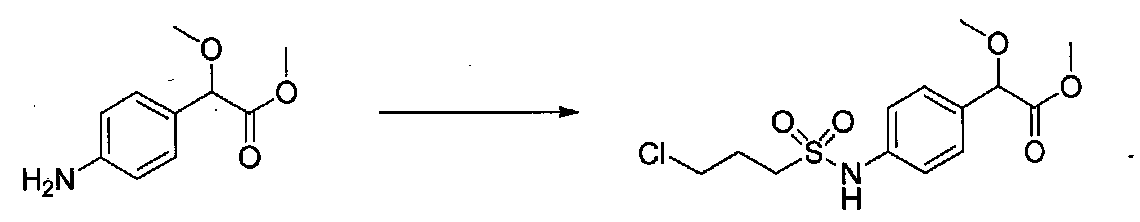
К раствору метил 2-(4-аминофенил)-2-метоксиацетата (0,6 г, 3,07 ммоль) в безводном пиридине (6 мл) в атмосфере аргона добавляли по каплям 3-хлорпропан-1-сульфонилхлорид (0,5 мл, 4,11 ммоль). Экзотермическую реакцию быстро охлаждали на бане с ледяной H2O. После перемешивания в течение 1 часа реакционную смесь разбавляли H2O, 1 M HCl и соляным раствором и экстрагировали EtOAc. Объединенные органические слои промывали 1 M HCl, H2O и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме с получением метил 2-(4-(3-хлорпропилсульфонамидо)фенил)-2-метоксиацетата в виде оранжевого масла (1,03 г, количественный выход). Продукт использовали без дополнительной очистки.
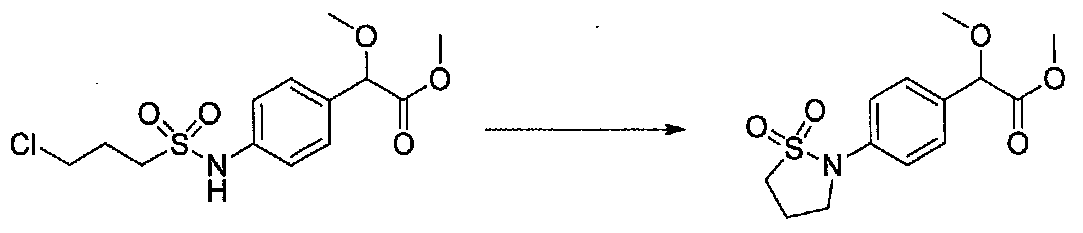
К раствору метил 2-(4-(3-хлорпропилсульфонамидо)фенил)-2-метоксиацетата (1,03 г, 3,07 ммоль) в безводном DMF (10 мл) добавляли N,N-диизопропилэтиламин (2,0 мл, 11,5 ммоль). Смесь нагревали при 50°C в атмосфере аргона в течение 17 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли H2O, 1 HCl и соляным раствором, затем экстрагировали EtOAc. Объединенные органические слои промывали 1 M HCl, H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток растирали с Et2O-EtOAc. После перемешивания в течение ночи раствор декантировали от твердых веществ, затем концентрировали в вакууме. Очистка хроматографией (50-70% EtOAc-гексаны) давала метил 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-2-метоксиацетат в виде неочищенного масла (0,69 г, выход 75%). Продукт использовали на следующей стадии синтеза без дополнительной очистки.
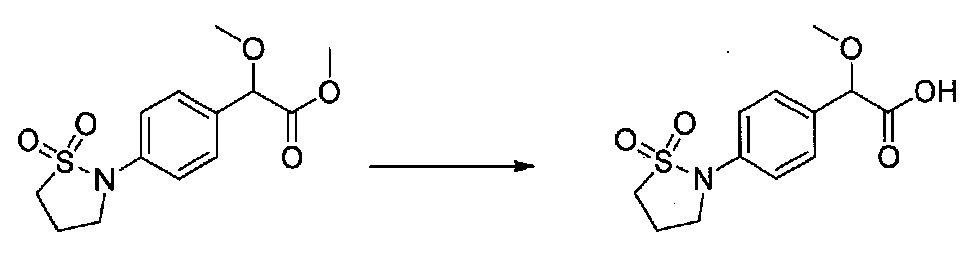
К раствору метил 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-2-метоксиацетата (0,48 г, 1,6 ммоль) в MeOH (21 мл) медленно добавляли 1 M NaOH (7 мл, 7 ммоль). После перемешивания при комнатной температуре в течение 23 часов летучие компоненты удаляли в вакууме и остаток растворяли в H2O. К водному раствору добавляли EtOAc и насыщенный водный NH4Cl и медленно добавляли 1 M HCl до pH ~3. Смесь экстрагировали EtOAc, затем водный слой подкисляли до pH 1 и снова экстрагировали EtOAc. Объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-2-метоксиуксусной кислоты в виде желтой пены (0,378 г, выход 82%). Продукт использовали на следующей стадии синтеза без дополнительной очистки.
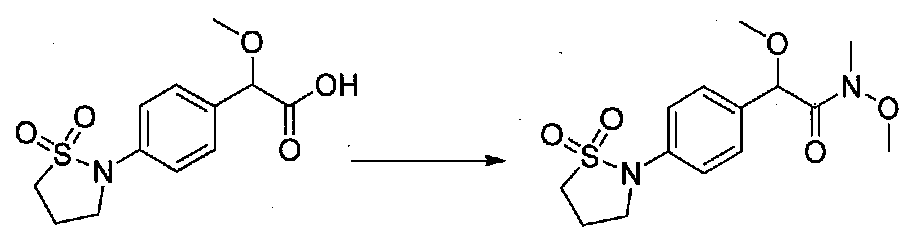
2-(4-(1,1-Диоксидоизотиазолидин-2-ил)фенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-2-метоксиуксусной кислоты, следуя методике для получения примера 13. Очистка хроматографией (0-4% EtOH-EtOAc) давала масло, которое растирали с Et2O. Твердое вещество сушили в вакууме с получением чистого 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-N,2-диметокси-N-метилацетамида в виде светло-желтого порошка (0,182 г, выход 64%).
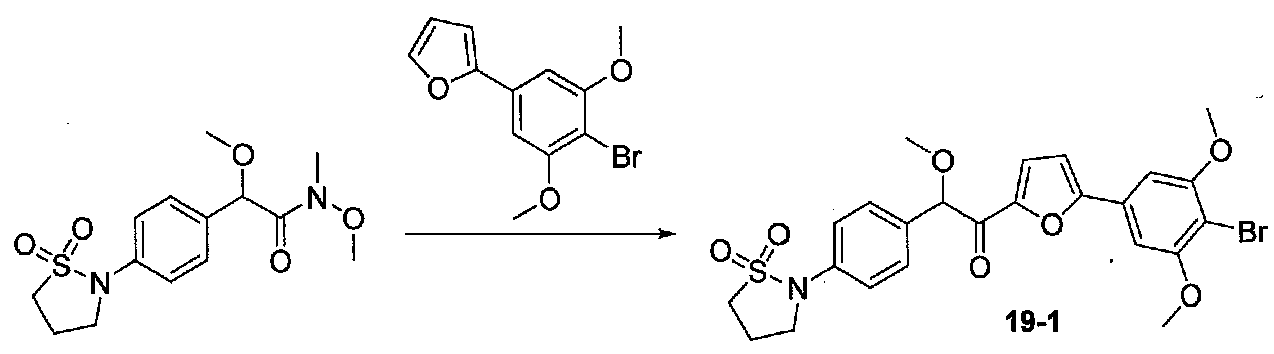
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-2-метоксиэтанон получали из 2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)-N,2-диметокси-N-метилацетамида и 2-(4-бром-3,5-диметоксифенил)фурана, следуя методике, применяемой для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала соединение примера 19 в виде желтого твердого вещества (0,111 г, выход 37%). МС: m/z 550,1 [M+H]+.
ПРИМЕР 20
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(ХИНОЛИН-5-ИЛ)ЭТАНОН
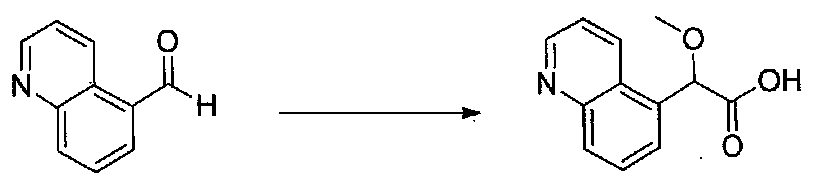
К охлажденному на льду раствору хинолин-5-карбальдегида (3,5 г, 22,3 ммоль) в безводном MeOH (30 мл) и безводном диоксане (30 мл) добавляли несколько капель раствора KOH (6,2 г, 113,4 ммоль) в MeOH (30 мл). Добавляли бромоформ (2,5 мл, 30 ммоль), затем добавляли в течение 10 минут оставшийся раствор KOH/MeOH. После перемешивания в течение 30 минут реакционную смесь медленно нагревали до комнатной температуры в течение ночи, затем концентрировали досуха. После растворения в минимальном количестве H2O остаток подкисляли до pH 1 концентрированной HCl. Водную смесь экстрагировали EtOAc несколько раз, добавляя соляной раствор к водному слою в процессе экстракции. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2-метокси-2-(хинолин-5-ил)уксусной кислоты в виде полутвердого вещества (2,8 г, выход 58%). Продукт использовали без дополнительной очистки.

К охлажденному на льду раствору 2-метокси-2-(хинолин-5-ил)уксусной кислоты (2,8 г, 12,9 ммоль) в безводном CH2Cl2 (50 мл) и NMM (3,1 мл, 29 ммоль) в атмосфере аргона добавляли по каплям изобутилхлорформиат (1,9 мл, 14 ммоль). После перемешивания на бане со льдом в течение 40 минут добавляли тремя порциями N,O-диметилгидроксиламингидрохлорид (1,63 г, 16,8 ммоль) в течение 15 минут. Смесь перемешивали в течение 15 минут, затем баню со льдом удаляли. Через 24 часа добавляли насыщенный водный NaHCO3 и перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные органические слои промывали насыщенным водным NaHCO3, H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-100% EtOAc-гексаны) давала N,2-диметокси-N-метил-2-(хинолин-5-ил)ацетамид в виде масла, которое кристаллизовалось при стоянии (1,8 г, выход 60%).

1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(хинолин-5-ил)этанон получали из N,2-диметокси-N-метил-2-(хинолин-5-ил)ацетамида и 2-(4-бром-3,5-диметоксифенил)фурана, следуя методике, применяемой для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(хинолин-5-ил)этанон в виде светло-желтой пены (0,102 г, выход 46%). МС: m/z 482,1 [M+H]+.
ПРИМЕР 21
1-(5-(3,5-ДИМЕТОКСИ-4-МЕТИЛФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
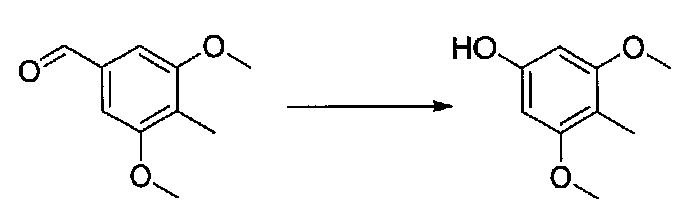
К раствору 3,5-диметокси-4-метилбензальдегида (3,0 г, 16,8 ммоль) в CH2Cl2 (35 мл) добавляли мета-хлорпероксибензойную кислоту (чистота 77%; 5,8 г, 25,9 ммоль). После перемешивания при комнатной температуре в течение 19 часов добавляли дополнительное количество мета-хлорпероксибензойной кислоты (чистота 77%; 3,5 г, 15,6 ммоль). После нагревания в течение 4 часов при 40°C и охлаждения до комнатной температуры добавляли 10% водный Na2S2O3 и смесь перемешивали в течение 30 минут. Смесь разбавляли CH2Cl2 и слои разделяли. Органический слой промывали 10% водным Na2S2O3, ~5% водным NaHCO3 и соляным раствором, затем сушили над MgSO4 и концентрировали с получением 3,5-диметокси-4-метилфенилформиата в виде желтого твердого вещества (2,9 г, выход 88%).
К раствору 3,5-диметокси-4-метилфенилформиата (2,9 г, 14,8 ммоль) во влажном MeOH (81 мл) добавляли K2CO3 (8,1 г, 58,6 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре, затем добавляли H2O (2-3 мл). После перемешивания в течение 21 часов смесь разбавляли H2O, подкисляли 6M HCl до pH 3-4 и экстрагировали EtOAc. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-35% EtOAc-гексаны) давала 3,5-диметокси-4-метилфенол в виде неочищенного желтого порошка (0,71 г, выход 29%).
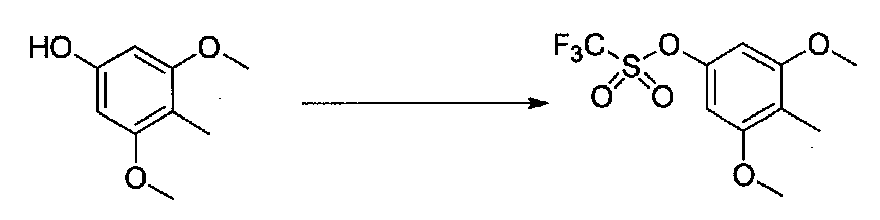
К суспензии 3,5-диметокси-4-метилфенола в безводном CH2Cl2 (10 мл) в атмосфере аргона добавляли сухой пиридин (0,4 мл, 4,9 ммоль), затем смесь охлаждали на бане со льдом. Добавляли по каплям ангидрид трифторметансульфокислоты (0,52 г, 3,1 ммоль) и реакционную смесь перемешивали в течение 1 часа. Добавляли насыщенный водный NaHCO3 и перемешивали, затем ее нагревали до комнатной температуры. Смесь разбавляли CH2Cl2 и слои разделяли. Органический слой промывали H2O и насыщенным водным NaHCO3, затем сушили над MgSO4 и концентрировали в вакууме с получением 3,5-диметокси-4-метилфенилтрифторметансульфоната в виде желтого масла (0,508 г, выход 60%). Продукт использовали без дополнительной очистки.

К раствору 3,5-диметокси-4-метилфенилтрифторметансульфоната (0,50 г, 1,67 ммоль) в DME (15 мл) добавляли 2-фурилбороновую кислоту (0,245 г, 2,2 ммоль), LiCl (0,149 г, 3,5 ммоль), 2 M водный Na2CO3 (1,8 мл, 3,6 ммоль) и тетракис(трифенилфосфин)палладий (0,098 г, 0,085 ммоль) и смесь дегазировали, как описано ранее. Реакционную смесь нагревали при 80°C в атмосфере аргона в течение 22 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли H2O и экстрагировали EtOAc. Объединенные органические слои промывали H2O, насыщенным водным NH4Cl, H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-25% Et2O-гексаны) давала 2-(3,5-диметокси-4-метилфенил)фуран в виде белого твердого вещества (0,28 г, выход 77%).
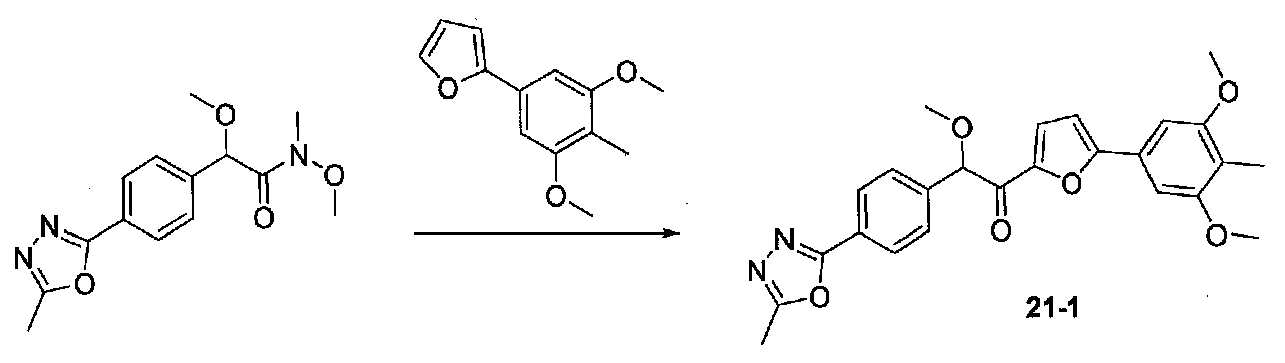
N,2-Диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамид взаимодействовал с 2-(3,5-диметокси-4-метилфенил)фураном согласно методике, применяемой для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала 1-(5-(3,5-диметокси-4-метилфенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон в виде желтой пены (0,148 г, выход 29%). МС: m/z 449,2 [M+H]+.
ПРИМЕР 22
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-(4-(5-ЦИКЛОПРОПИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)-2-МЕТОКСИЭТАНОН
4-(((трет-Бутилдиметилсилил)окси)метил)бензогидразид взаимодействовал с гидрохлоридом этилциклопропанкарбимидата согласно методике, применяемой для получения примера 18. Очистка хроматографией (EtOAc-гексаны) давала 2-(4-(((трет-бутилдиметилсилил)окси)метил)фенил)-5-циклоропил-1,3,4-оксадиазол в виде коричневого масла (3,7 г, выход 40%).
(4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)метанол получали из 2-(4-(((трет-бутилдиметилсилил)окси)метил)фенил)-5-циклопропил-1,3,4-оксадиазола согласно методике примера 18. (4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)метанол выделяли в виде не совсем белого твердого вещества и использовали на следующей стадии синтеза без дополнительной очистки (2,2 г, выход 84%).
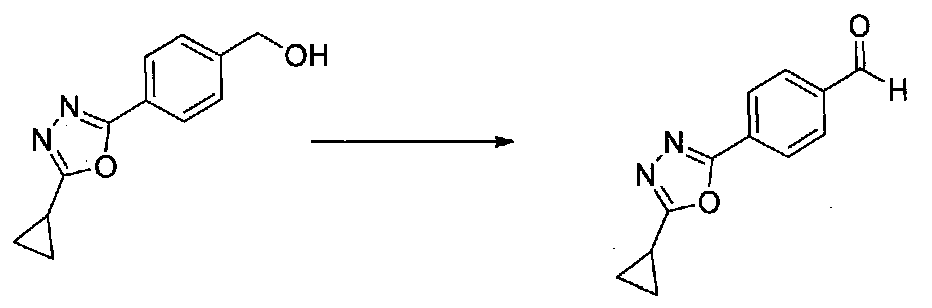
4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)бензальдегид получали из (4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)метанола согласно методике примера 18. Очистка хроматографией (EtOAc-гексаны) давала 4-(5-циклопропил-1,3,4-оксадиазол-2-ил)бензальдегид в виде белого твердого вещества (1,8 г, выход 82%).

2,2,2-Трихлор-1-(4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)этанол получали из 4-(5-циклопропил-1,3,4-оксадиазол-2-ил)бензальдегида согласно методике для получения примера 18. Продукт выделяли в виде белого твердого вещества и использовали без дополнительной очистки (2,5 г, выход 89%).
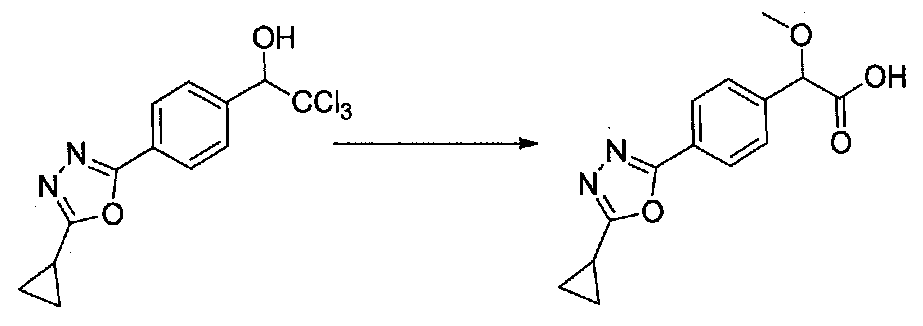
2-(4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиуксусную кислоту получали из 2,2,2-трихлор-1-(4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)этанола, следуя методике, применяемой для получения примера 18. Продукт выделяли в виде желтого полутвердого вещества и использовали на следующей стадии синтеза без дополнительной очистки (1,8 г, выход 90%).
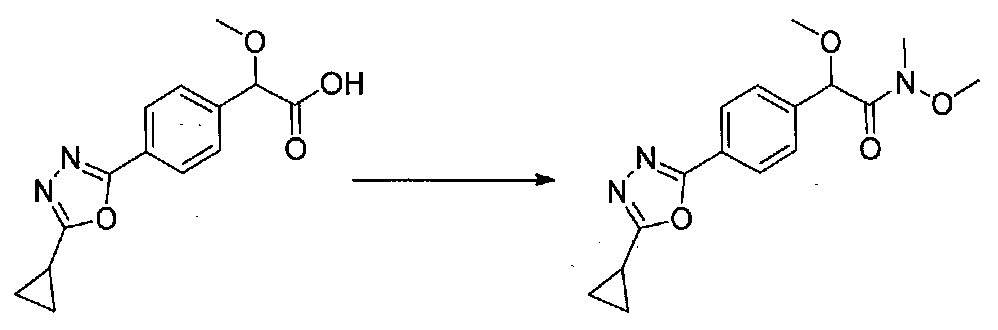
2-(4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиуксусной кислоты согласно методике для получения примера 18. Продукт выделяли в виде белого полутвердого вещества (0,56 г, выход 27%).

2-(4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамид взаимодействовал с 2-(4-бром-3,5-диметоксифенил)фураном согласно методике для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-(4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)-2-метоксиэтанон в виде желтой пены (0,126 г, выход 31%). МС: m/z 539,2 [M+H]+.
ПРИМЕР 23
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(6-(ПИПЕРИДИН-1-ИЛ)ПИРИДИН-3-ИЛ)ЭТАНОН
2-Метокси-2-(6-(пиперидин-1-ил)пиридин-3-ил)уксусную кислоту получали из 6-(пиперидин-1-ил)никотинальдегида согласно методике, применяемой для получения примера 1, за исключением того, что реакционную смесь охлаждали на льду перед добавлением раствора KOH-MeOH. Продукт выделяли в виде светло-бежевой пены и использовали без дополнительной очистки (0,569 г, выход 52%).
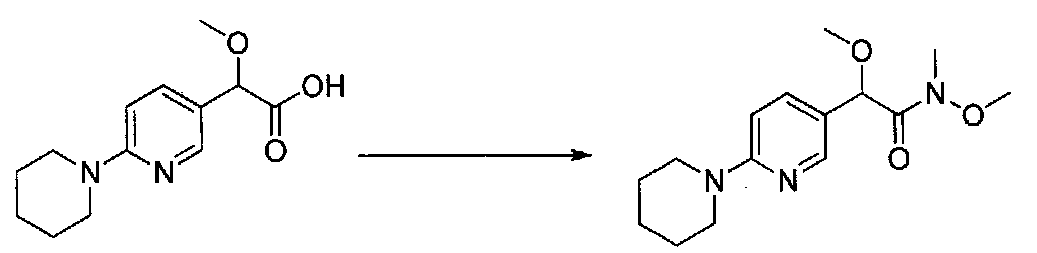
N,2-Диметокси-N-метил-2-(6-(пиперидин-1-ил)пиридин-3-ил)ацетамид получали из 2-метокси-2-(6-(пиперидин-1-ил)пиридин-3-ил)уксусной кислоты согласно методике, применяемой для получения примера 13. Очистка хроматографией (50-100% EtOAc-гексаны) давала продукт в виде оранжевого масла (0,295 г, выход 46%).
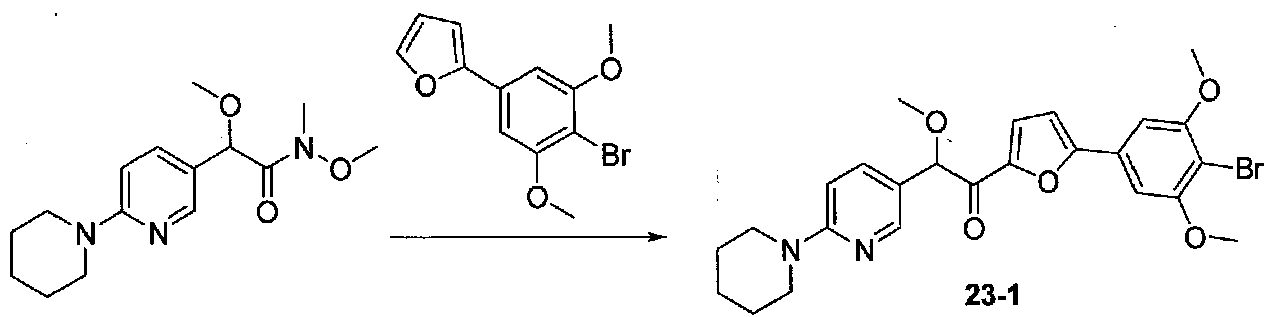
N,2-Диметокси-N-метил-2-(6-(пиперидин-1-ил)пиридин-3-ил)ацетамид взаимодействовал с 2-(4-бром-3,5-диметоксифенил)фураном согласно методике, применяемой в примере 12. Очистка хроматографией (20-75% EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(6-(пиперидин-1-ил)пиридин-3-ил)этанон в виде светло-желтого твердого вещества (0,328 г, выход 65%). МС: m/z 515,4 [M+H]+.
ПРИМЕР 24
2,6-ДИМЕТОКСИ-4-(5-(2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)АЦЕТИЛ)ФУРАН-2-ИЛ)ФЕНИЛБЕНЗОАТ
4-Бром-2,6-диметоксифенол (Lee, H.; et al. Tetrahedron Letters, 2004, 45, 1019) конденсировали с 2-фурилбороновой кислотой согласно методике, применяемой в получении примера 21. Очистка хроматографией (20-40% EtOAc-гексаны) давала 4-(фуран-2-ил)-2,6-диметоксифенол в виде светло-желтого твердого вещества (1,95 г, выход 79%).
К раствору 4-(фуран-2-ил)-2,6-диметоксифенола (1,3 г, 5,9 ммоль) в безводном CH2Cl2 (20 мл) в атмосфере аргона добавляли Et3N (1,6 мл, 11,5 ммоль), затем смесь охлаждали на бане со льдом. Добавляли по каплям бензоилхлорид (0,75 мл, 6,4 ммоль) и реакционную смесь перемешивали в течение 3 часов. Добавляли 10% водный NaHCO3 и CH2Cl2, перемешивали и слои разделяли. Органический слой промывали H2O и насыщенным водным NaHCO3, сушили над MgSO4 и концентрировали в вакууме. Очистка хроматографией (0-25% EtOAc-гексаны) давала 4-(фуран-2-ил)-2,6-диметоксифенилбензоат в виде светло-желтого твердого вещества (0,54 г, выделенные после хроматографии приблизительно половины неочищенного продукта; выход 57%).
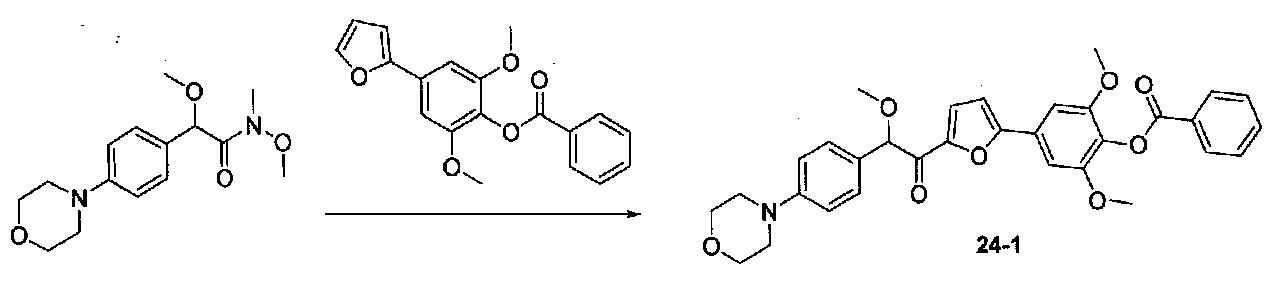
4-(Фуран-2-ил)-2,6-диметоксифенилбензоат взаимодействовал с N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамидом согласно методике, описанной для получения примера 12, за исключением того, что реакционную смесь нагревали только до 0°C. Очистка хроматографией (EtOAc-гексаны) давала 2,6-диметокси-4-(5-(2-метокси-2-(4-морфолинофенил)ацетил)фуран-2-ил)фенилбензоат в виде желтого твердого вещества (0,257 г, выход 50%). МС: m/z 558,2 [M+H]+.
ПРИМЕР 25
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(1-МЕТИЛ-1H-БЕНЗО[d][1,2,3]ТРИАЗОЛ-5-ИЛ)ЭТАНОН
2-Метокси-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)уксусную кислоту получали из 1-метил-1H-бензо[d][1,2,3]триазол-5-карбальдегида согласно методике для получения примера 23. Продукт выделяли в виде желтого твердого вещества (1,5 г, выход 96%).
N,2-Диметокси-N-метил-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)ацетамид получали из 2-метокси-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)уксусной кислоты согласно способу, применяемому для получения примера 13. Очистка хроматографией (60-100% EtOAc-гексаны) давала N,2-диметокси-N-метил-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)ацетамид в виде желтого твердого вещества (1,02 г, выход 57%).
N,2-Диметокси-N-метил-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)ацетамид конденсировали с 2-(4-бром-3,5-диметоксифенил)фураном согласно методике для получения примера 13. Очистка хроматографией давала 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(1-метил-1H-бензо[d][1,2,3]триазол-5-ил)этанон в виде светло-желтой пены (0,153 г, выход 34%). МС: m/z 486,1 [M+H]+.
ПРИМЕР 26
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(МОРФОЛИНОМЕТИЛ)ФЕНИЛ)ЭТАНОН
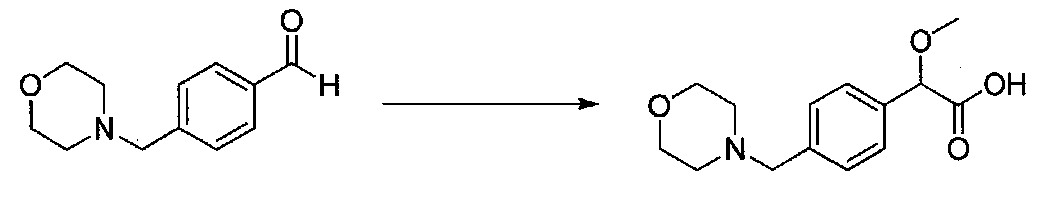
2-Метокси-2-(4-(морфолинометил)фенил)уксусную кислоту получали из 4-(морфолинометил)бензальдегида, следуя методике для получения примера 23. После реакции летучие компоненты удаляли в вакууме. Остаток повторно растворяли в минимальном количестве H2O и подкисляли 1 M HCl до pH 3. Водный слой концентрировали в вакууме и остаток суспендировали в MeOH, обрабатывали ультразвуком и медленно нагревали. Смесь фильтровали через целит и концентрировали в вакууме с получением белого твердого вещества (3 г, количественный выход). Продукт использовали на следующей стадии синтеза без очистки.

N,2-Диметокси-N-метил-2-(4-(морфолинометил)фенил)ацетамид получали из 2-метокси-2-(4-(морфолинометил)фенил)уксусной кислоты, следуя методике, применяемой в примере 13. Очистка хроматографией (0-7% раствор NH3-MeOH в CH2Cl2; 0,7 M NH3-MeOH) давала N,2-диметокси-N-метил-2-(4-(морфолинометил)фенил)ацетамид в виде оранжевого масла (0,743 г, выход 41%).
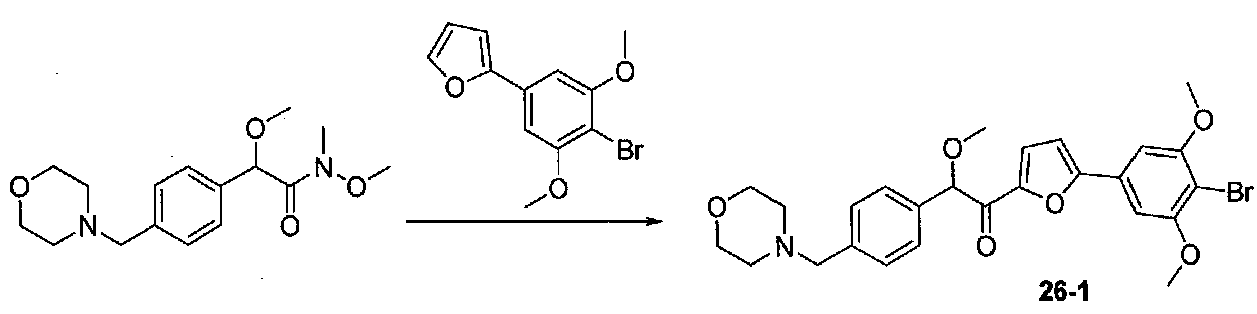
N,2-Диметокси-N-метил-2-(4-(морфолинометил)фенил)ацетамид конденсировали с 2-(4-бром-3,5-диметоксифенил)фураном, следуя методике примера 12. Очистка хроматографией (использовали 0-9% NH3-MeOH в CH2Cl2; 0,7 M NH3-MeOH) давала смесь, которую обрабатывали EtOAc при 30-40°C. После охлаждения до комнатной температуры кристаллы собирали на воронке Бюхнера, промывали EtOAc и сушили в вакууме с получением 1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(морфолинометил)фенил)этанона в виде белого твердого вещества. Также выделяли вторую порцию (0,125 г для двух порций, выход 13%). МС: m/z 530,1 [M+H]+.
ПРИМЕР 27
1-(5-(3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
1-Бром-3,5-диметоксибензол конденсировали с 2-фурилбороновой кислотой, следуя методике примера 21. 2-(3,5-Диметоксифенил)фуран выделяли в виде бесцветной жидкости (0,746 г, выход 79%).
N,2-Диметокси-N-метил-2-(4-морфолинофенил)ацетамид конденсировали с 2-(3,5-диметоксифенил)фураном, следуя методике примера 12. 1-(5-(3,5-Диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)этанон выделяли в виде желтой маслянистой пены (0,343 г, выход 62%). МС: m/z 438,2 [M+H]+.
ПРИМЕР 28
1-(5-(4-(ДИФТОРМЕТОКСИ)-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
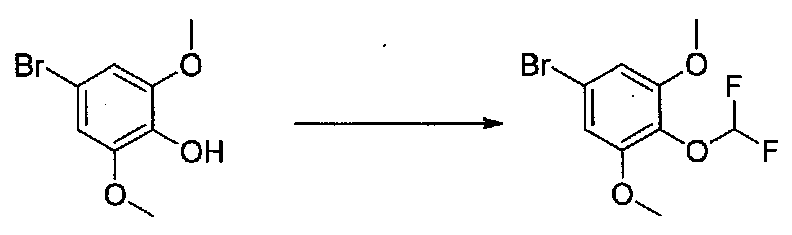
5-Бром-2-(дифторметокси)-1,3-диметоксибензол получали из 4-бром-2,6-диметоксифенола согласно методике Zafrani, Y. Tetrahedron 2009, 65, 5278 следующим образом: к раствору 4-бром-2,6-диметоксифенола (1,08 г, 4,6 ммоль) в MeCN (27 мл) добавляли раствор KOH (5,0 г, 89 ммоль) в H2O (27 мл). Смесь сразу охлаждали до -78°C на бане и добавляли диэтил(бромдифторметил)фосфонат (1,6 мл, 8,9 ммоль). Колбу герметично закрывали септой и удаляли баню со льдом. Смесь перемешивали в течение в сумме 3,5 часов, после которых септу удаляли с колбы. Реакционную смесь разбавляли EtOAc и слои разделяли. Водный слой экстрагировали EtOAc и объединенные органические слои промывали 1 M NaOH, H2O и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-30% EtOAc-гексаны) давала 5-бром-2-(дифторметокси)-1,3-диметоксибензол в виде белого твердого вещества (0,808 г, выход 62%).
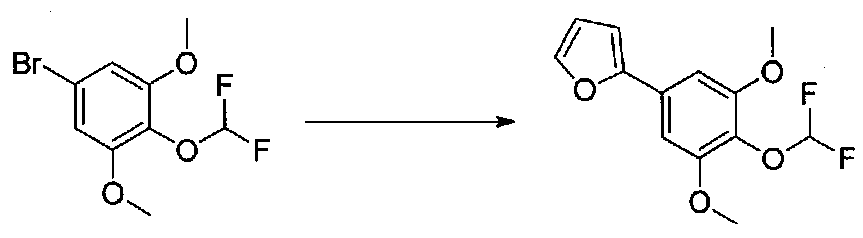
5-Бром-2-(дифторметокси)-1,3-диметоксибензол конденсировали с 2-фурилбороновой кислотой, следуя методике для получения примера 21. Очистка хроматографией (0-30% EtOAc-гексаны) давала 2-(4-(дифторметокси)-3,5-диметоксифенил)фуран в виде белого кристаллического вещества (0,555 г, выход 67%).
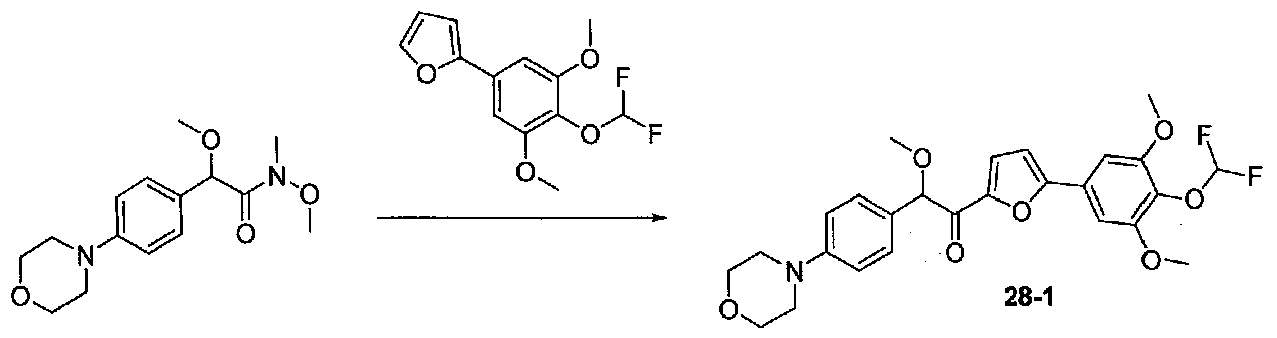
2-(4-(Дифторметокси)-3,5-диметоксифенил)фуран конденсировали с N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамидом, следуя методике, применяемой для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала 1-(5-(4-(дифторметокси)-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)-этанон в виде коричневого масла (0,021 г, выход 6%). МС: m/z 504,2 [M+H]+.
ПРИМЕР 29
1-(5-(4-ЭТОКСИ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
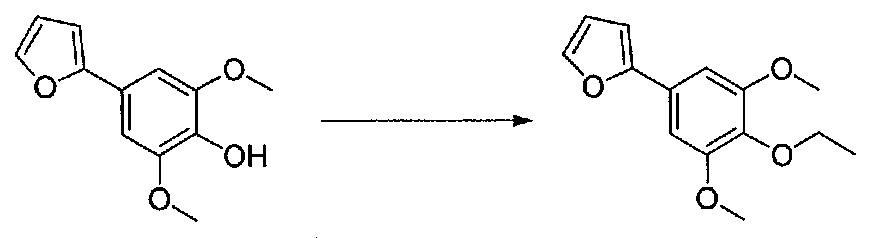
К суспензии 4-(фуран-2-ил)-2,6-диметоксифенола (0,493 г, 2,24 ммоль) в безводном DMF (10 мл) в атмосфере аргона добавляли Cs2CO3 (1,2 г, 3,7 ммоль) и йодэтан (0,22 мл, 2,7 ммоль). Смесь перемешивали в течение 20 минут, затем нагревали при 80°C в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли H2O и EtOAc, затем ее подкисляли добавлением 6 M HCl. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические слои разбавляли гексанами и промывали H2O и соляным раствором, сушили над Na2SO4 и фильтровали через слой силикагеля на целите. Фильтрат концентрировали в вакууме с получением 2-(4-этокси-3,5-диметоксифенил)фурана в виде бежевого твердого вещества (0,513 г, выход 92%). Продукт использовали без дополнительной очистки.
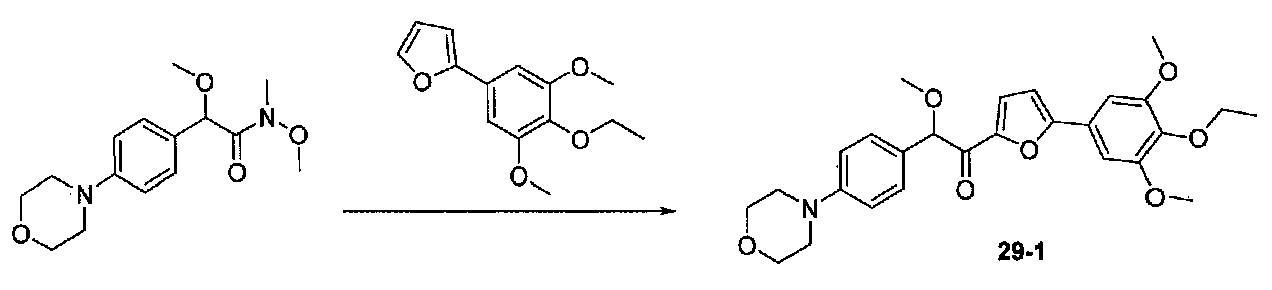
2-(4-этокси-3,5-диметоксифенил)фуран конденсировали с N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамидом согласно методике для получения примера 12. Очистка хроматографией (EtOAc-гексаны) давала 1-(5-(4-этокси-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)этанон в виде желтой пены (0,263 г, выход 58%). МС: m/z 482,2 [M+H]+.
ПРИМЕР 30
2-(4-(2H-1,2,3-ТРИАЗОЛ-2-ИЛ)ФЕНИЛ)-1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИЭТАНОН
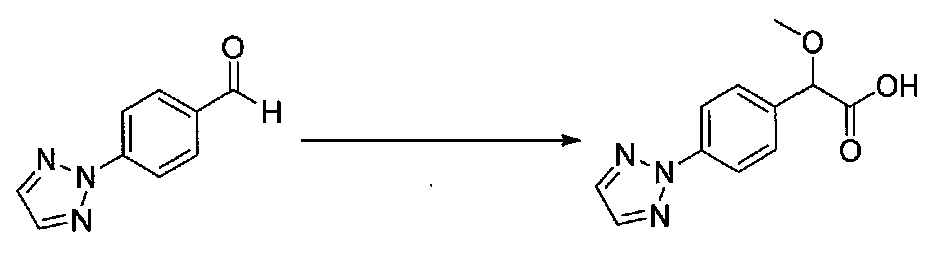
К раствору 4-(2H-1,2,3-триазол-2-ил)бензальдегида (1,0 г, 5,77 ммоль) в безводном MeOH (15 мл) при 0°C (температура бани) добавляли бромоформ (1,82 г, 7,21 ммоль) при перемешивании. Добавляли порциями в течение 10 минут твердый KOH (1,62 г, 28,9 ммоль). Смесь перемешивали в течение 1 часа и баню со льдом удаляли. После перемешивания в течение 30 минут реакционную смесь медленно нагревали до комнатной температуры в течение ночи, затем концентрировали досуха. После растворения в минимальном количестве H2O остаток подкисляли до pH 1 с помощью 6 M HCl. Водную смесь экстрагировали EtOAc несколько раз, добавляя соляной раствор к водному слою в процессе экстракции. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2-(4-(2H-1,2,3-триазол-2-ил)фенил)-2-метоксиуксусной кислоты в виде масла (1,19 г, выход 88%). Продукт использовали без дополнительной очистки.
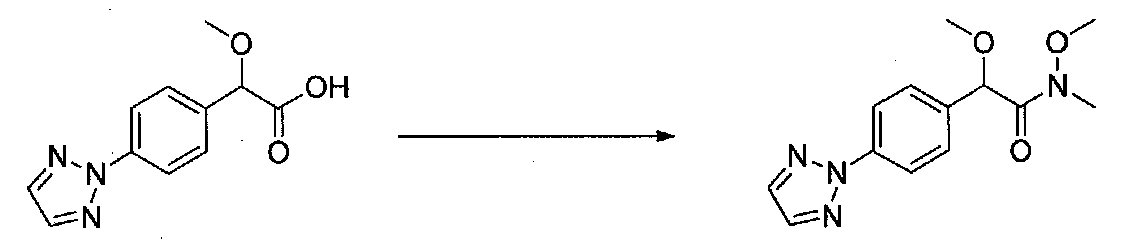
К раствору 2-(4-(2H-1,2,3-триазол-2-ил)фенил)-2-метоксиуксусной кислоты (1,19 г, 5,1 ммоль) в безводном CH2Cl2 (15 мл) добавляли 4-метилморфолин (1,55 г, 15,3 ммоль) и ее охлаждали на бане со льдом. Добавляли изобутилхлорформиат (0,84 г, 6,12 ммоль) и перемешивали в течение 45 минут. Добавляли N,O-диметилгидроксиламин HCl (0,746 г, 7,65 ммоль) и смесь перемешивали в течение ночи при нагревании до комнатной температуры. Добавляли насыщенный водный NaHCO3 (15 мл) и перемешивали в течение 5 минут. Органический слой сушили над Na2SO4 и упаривали досуха. Очистка хроматографией (100% EtOAc) давала 2-(4-(2H-1,2,3-триазол-2-ил)фенил)-N,2-диметокси-N-метилацетамид в виде белого твердого вещества (0,63 г, выход 44%).

К раствору 2-(4-бром-3,5-диметоксифенил)фурана (0,20 г, 0,71 ммоль) в безводном THF (10 мл) в атмосфере аргона в колбе, высушенной в сушильном шкафу, охлажденном до -78°C, добавляли по каплям диизопропиламид лития (2,0 M в THF/гептан/этилбензол; 0,43 мл, 0,85 ммоль). После перемешивания в течение 1 часа при -78°C добавляли по каплям раствор 2-(4-(2H-1,2,3-триазол-2-ил)фенил)-N,2-диметокси-N-метилацетамида (0,195 г, 0,71 ммоль) в THF (2 мл). После перемешивания в течение 25 минут смесь нагревали до комнатной температуры при перемешивании в течение 2 часов. Реакционную смесь гасили добавлением насыщенного водного NH4Cl и добавляли EtOAc. Слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (30% EtOAc-гексаны) давала 2-(4-(2H-1,2,3-триазол-2-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон в виде желтого твердого вещества (0,07 г, выход 10%). МС: m/z 497,9 [M+H]+.
ПРИМЕР 31
2-(4-(1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ)-1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИЭТАНОН
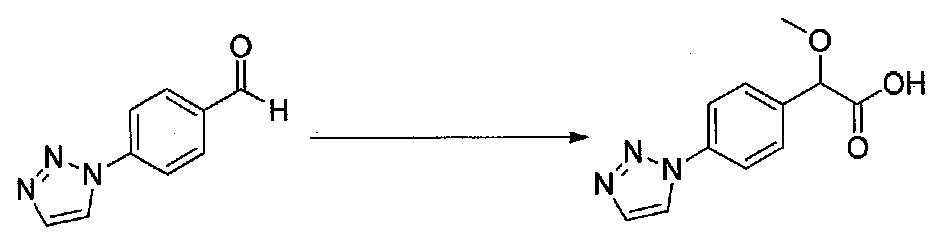
2-(4-(1H-1,2,3-Триазол-1-ил)фенил)-2-метоксиуксусную кислоту получали из 4-(1H-1,2,3-триазол-1-ил)бензальдегида согласно методике, применяемой в примере 30. Продукт получали в виде масла и использовали без дополнительной очистки (1,02 г, выход 76%).
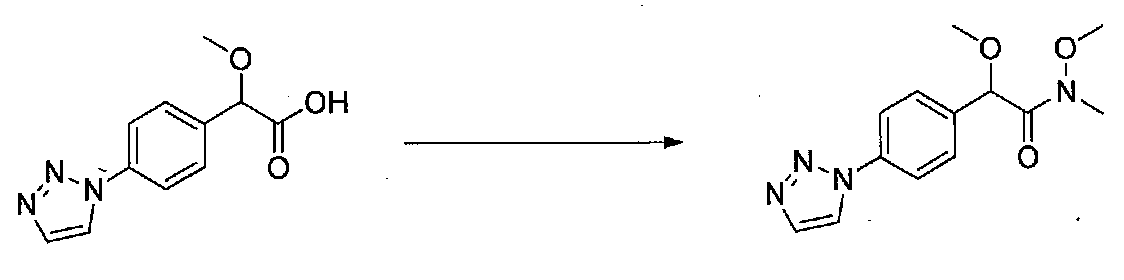
2-(4-(1H-1,2,3-Триазол-1-ил)фенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-(1H-1,2,3-триазол-1-ил)фенил)-2-метоксиуксусной кислоты согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде масла (0,73 г, выход 58%).
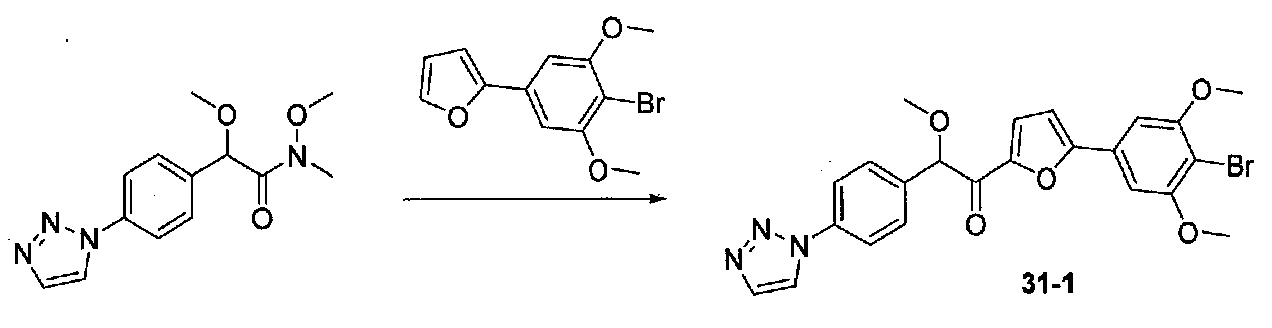
2-(4-(1H-1,2,3-Триазол-1-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-(4-(1H-1,2,3-триазол-1-ил)фенил)-N,2-диметокси-N-метилацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (50% EtOAc гексаны) давала 2-(4-(1H-1,2,3-триазол-1-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон в виде желтого твердого вещества (0,025 г, выход 7%). МС: m/z 498,2 [M+H]+.
ПРИМЕР 32
2-(4-(1H-1,2,4-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ)-1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИЭТАНОН
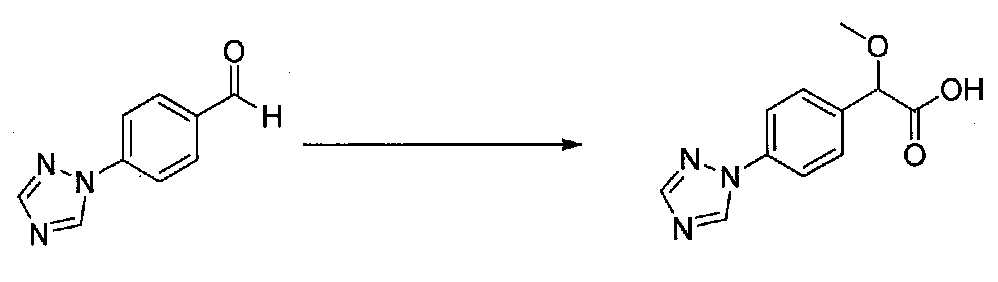
2-(4-(1H-1,2,4-Триазол-1-ил)фенил)-2-метоксиуксусную кислоту получали из 4-(1H-1,2,4-триазол-1-ил)бензальдегида согласно методике, применяемой в примере 30. Неочищенный продукт получали в виде белого твердого вещества и использовали без дополнительной очистки (1,9 г, выход 47%).
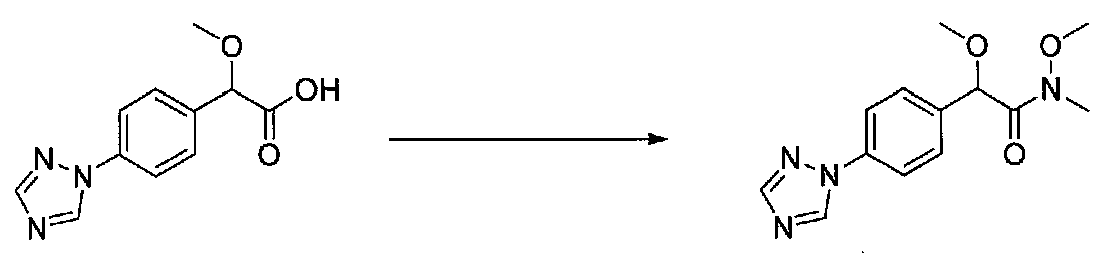
2-(4-(1H-1,2,4-Триазол-1-ил)фенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-(1H-1,2,4-триазол-1-ил)фенил)-2-метоксиуксусной кислоты согласно методике, применяемой в примере 30. Очистка хроматографией (100% EtOAc) давала продукт в виде не совсем белого твердого вещества (0,30 г, выход 26%).
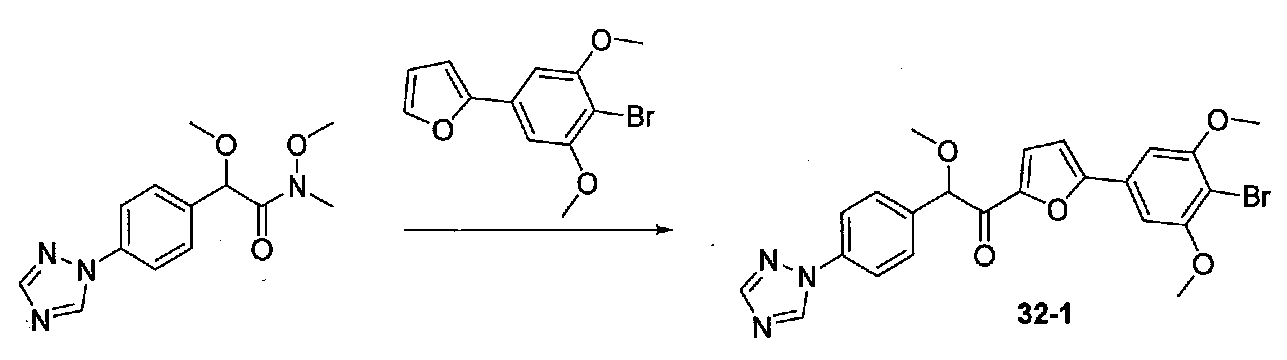
2-(4-(1H-1,2,4-Триазол-1-ил)фенил-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-(4-(1H-1,2,4-триазол-1-ил)фенил)-N,2-диметокси-N-метилацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) с последующей другой очисткой (40% ацетон-гексаны) давала 2-(4-(1H-1,2,4-триазол-1-ил)фенил)-1-(5-(4-бром-3,5-диметоксифенил)фуран-2-ил)-2-метоксиэтанон в виде бледно-желтого твердого вещества (0,078 г, выход 11%). МС: m/z 498,2 [M+H]+.
ПРИМЕР 33
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(2-МЕТИЛ-2H-ТЕТРАЗОЛ-5-ИЛ)ФЕНИЛ)ЭТАНОН
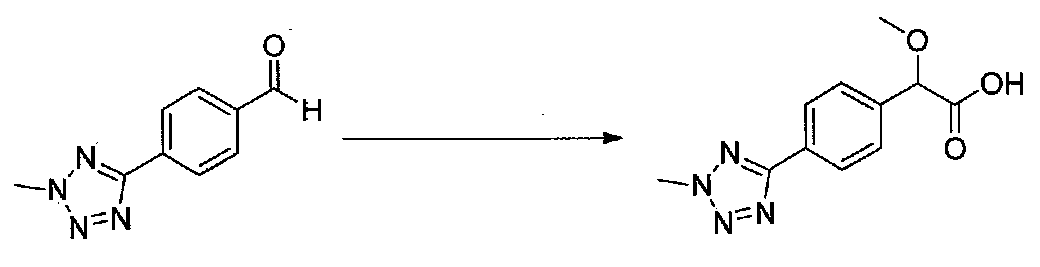
2-Метокси-2-(4-(2-метил-2H-тетразол-5-ил)фенил)уксусную кислоту получали из 4-(2-метил-2H-тетразол-5-ил)бензальдегида согласно методике, применяемой в примере 30. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (0,88 г, выход 86%).
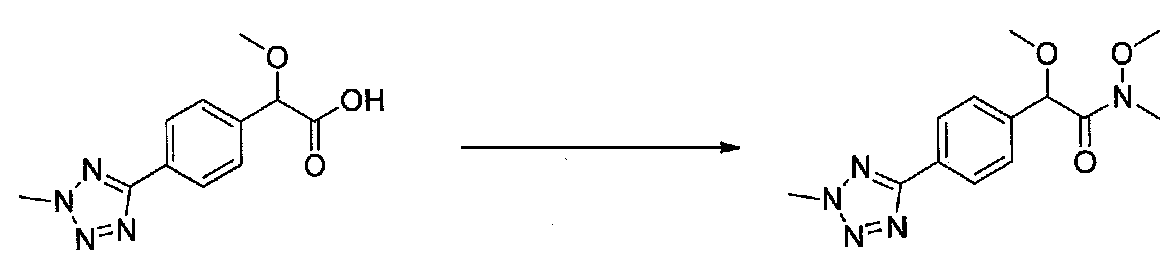
К раствору 2-метокси-2-(4-(2-метил-2H-тетразол-5-ил)фенил)уксусной кислоты (0,87 г, 3,5 ммоль) в безводном CH2Cl2 (10 мл) добавляли N,N-диизопропилэтиламин (1,36 г, 10,5 ммоль) при охлаждении на бане со льдом. Добавляли трифторид бис(2-метоксиэтил)аминосеры (0,93 г, 4,2 ммоль) и перемешивали в течение 15 минут. Добавляли N,Ο-диметилгидроксиламин HCl (0,512 г, 5,25 ммоль) и смесь перемешивали в течение ночи при нагревании до комнатной температуры. Добавляли насыщенный водный NaHCO3 (15 мл) и перемешивали в течение 5 минут. Органический слой сушили над Na2SO4 и упаривали досуха. Очистка хроматографией (60% EtOAc-гексаны) давала N,2-диметокси-N-метил-2-(4-(2-метил-2H-тетразол-5-ил)фенил)ацетамид в виде прозрачного масла (0,425 г, выход 42%).
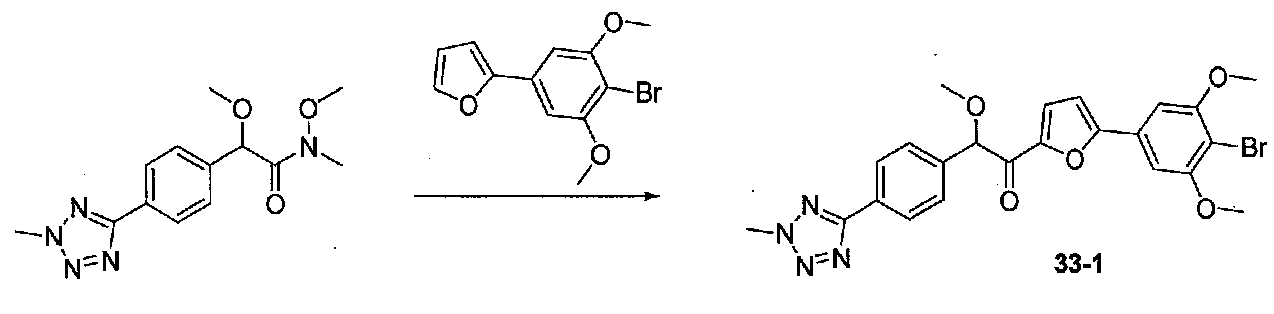
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(2-метил-2H-тетразол-5-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(2-метил-2H-тетразол-5-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (40% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,106 г, выход 29%). МС: m/z 513,3 [M+H]+.
ПРИМЕР 34
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ТИАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
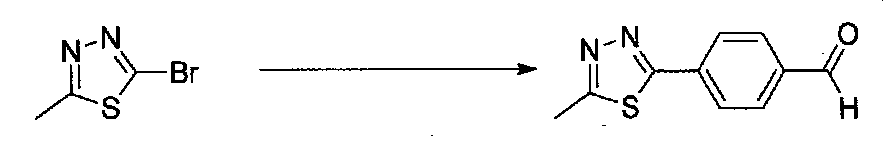
К раствору 2-бром-5-метил-1,3,4-тиадиазола (2,0 г, 11,17 ммоль) в диоксане (40 мл) добавляли 4-формилбензолбороновую кислоту (3,35 г, 22,34 ммоль) и 2M Na2CO3 (23 мл). Данную смесь дегазировали потоком аргона в течение 2 минут. Добавляли тетракис(трифенилфосфин)палладий (0,636 г, 0,55 ммоль) и данную смесь кипятили в течение ночи в атмосфере аргона. После охлаждения до комнатной температуры добавляли H2O (30 мл) и EtOAc (50 мл) и перемешивали в течение 5 минут. Органический слой отделяли и промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (10-20% EtOAc/гексаны) давала 4-(5-метил-1,3,4-тиадиазол-2-ил)бензальдегид в виде бледно-желтой жидкости (1,16 г, выход 51%).

2-Метокси-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)уксусную кислоту получали из 4-(5-метил-1,3,4-тиадиазол-2-ил)бензальдегида согласно методике, применяемой в примере 30. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (1,12 г, выход 75%).
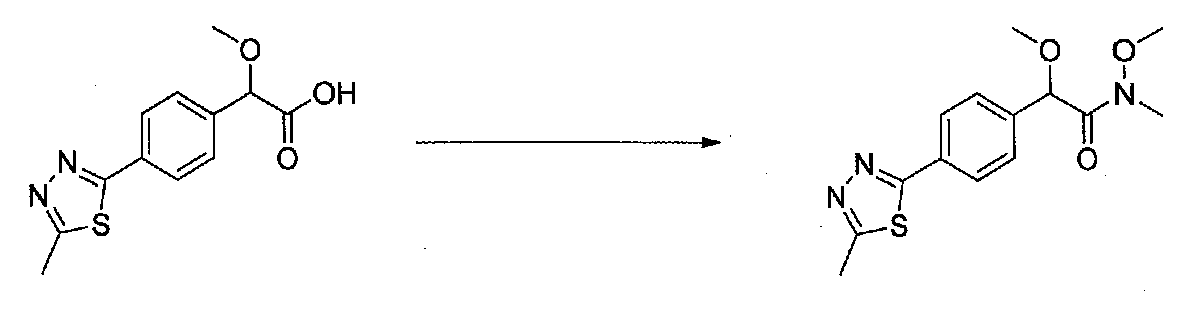
N,2-Диметокси-N-метил-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)ацетамид получали из 2-метокси-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (100% EtOAc) давала продукт в виде бледно-желтого масла (0,177 г, выход 26%).
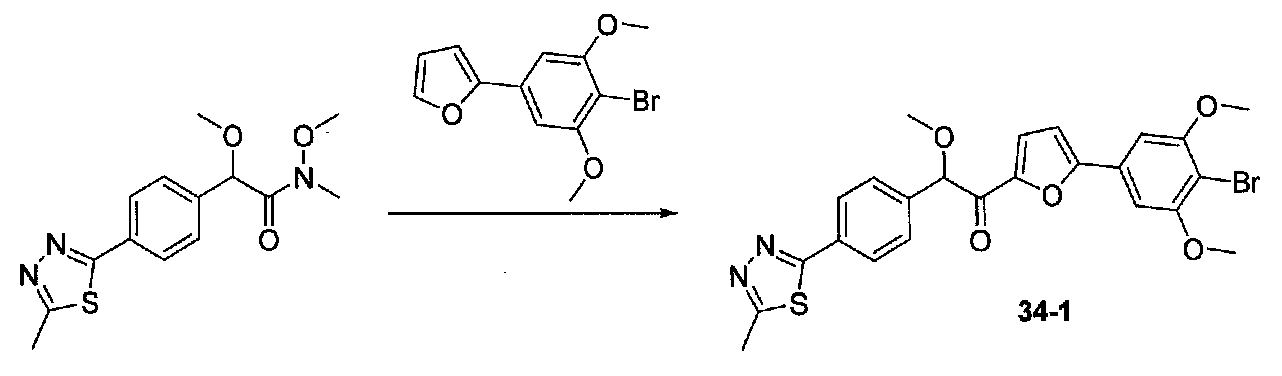
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (70% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,044 г, выход 15%). МС: m/z 529,3 [M+H]+.
ПРИМЕР 35
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(2-МЕТИЛТИАЗОЛ-4-ИЛ)ФЕНИЛ)ЭТАНОН
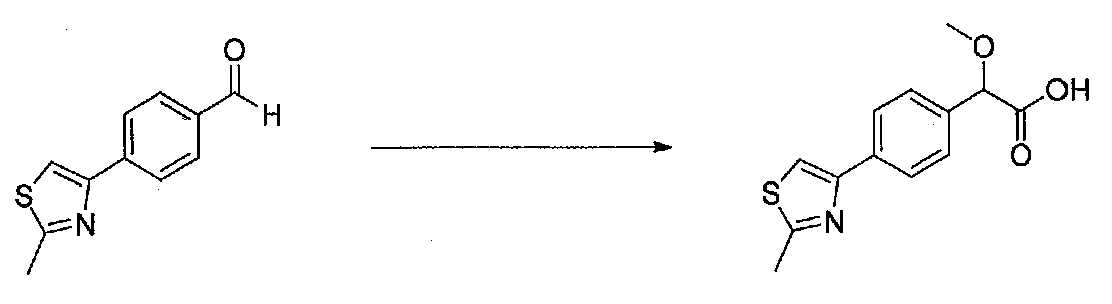
2-Метокси-2-(4-(2-метилтиазол-4-ил)фенил)уксусную кислоту получали из 4-(2-метилтиазол-4-ил)бензальдегида согласно методике, применяемой в примере 30. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (0,972 г, выход 78%).
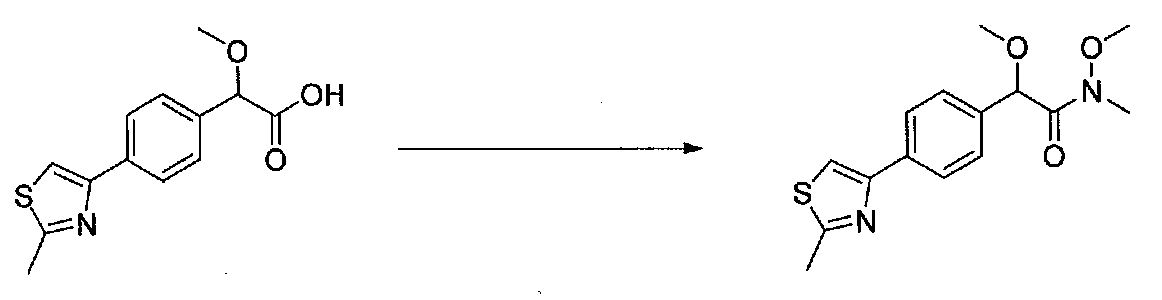
N,2-Диметокси-N-метил-2-(4-(2-метилтиазол-4-ил)фенил)ацетамид получали из 2-метокси-2-(4-(2-метилтиазол-4-ил)фенил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (70% EtOAc/гексаны) давала продукт в виде масла (0,516 г, выход 46%).
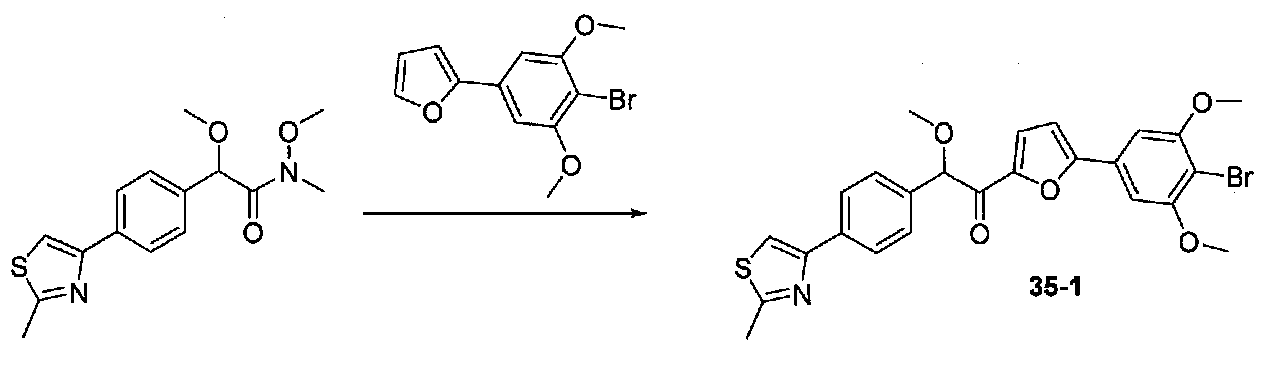
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(2-метилтиазол-4-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(2-метилтиазол-4-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде не совсем белого твердого вещества (0,039 г, выход 10%). МС: m/z 528,2 [M+H]+.
ПРИМЕР 36
2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)-1-(5-(3,4,5-ТРИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)ЭТАНОН
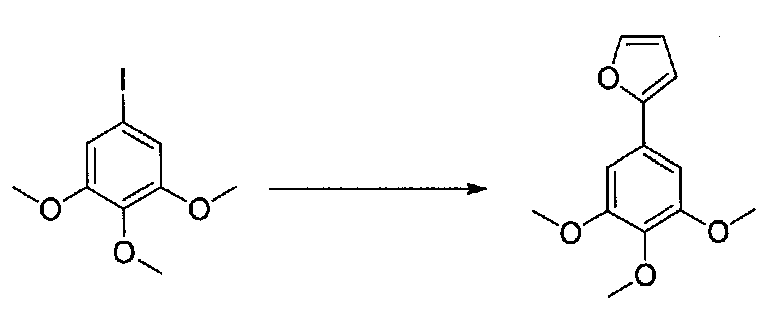
2-(3,4,5-Триметоксифенил)фуран получали из 5-йод-1,2,3-триметоксибензола согласно методике, применяемой в примере 12. Очистка хроматографией (10% EtOAc/гексаны) давала продукт в виде не совсем белого твердого вещества (1,2 г, выход 60%).
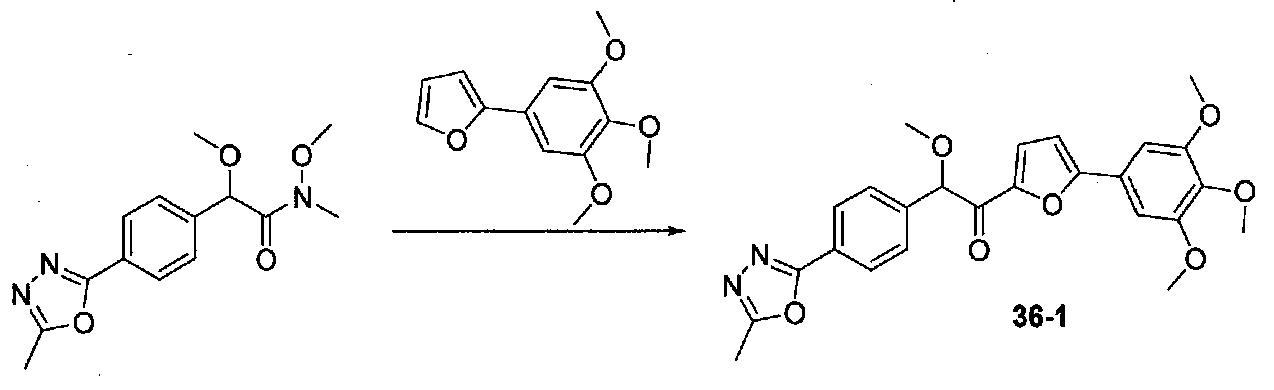
К раствору 2-(3,4,5-триметоксифенил)фурана (1,4 г, 5,98 ммоль) в безводном THF (70 мл) в атмосфере аргона в колбе, высушенной в сушильном шкафу, охлажденном до -78°C, добавляли по каплям н-бутиллитий (2,5 M в гексанах; 2,63 мл, 6,28 ммоль). После перемешивания в течение 1 часа при -78°C добавляли по каплям раствор N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида (1,75 г, 6,0 ммоль) в THF (5 мл). После перемешивания в течение 25 минут смесь нагревали до комнатной температуры при перемешивании в течение 2 часов. Реакционную смесь гасили добавлением насыщенного водного NH4Cl, затем соляным раствором и добавляли EtOAc. Слои разделяли и органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка колоночной хроматографией (60% EtOAc/гексаны) давала 2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)-1-(5-(3,4,5-триметоксифенил)фуран-2-ил)этанон в виде рыхлого желтого твердого вещества (0,81 г, выход 29%). МС: m/z 465,3 [M+H]+.
ПРИМЕР 37
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(ПИРИДИН-3-ИЛ)ФЕНИЛ)ЭТАНОН
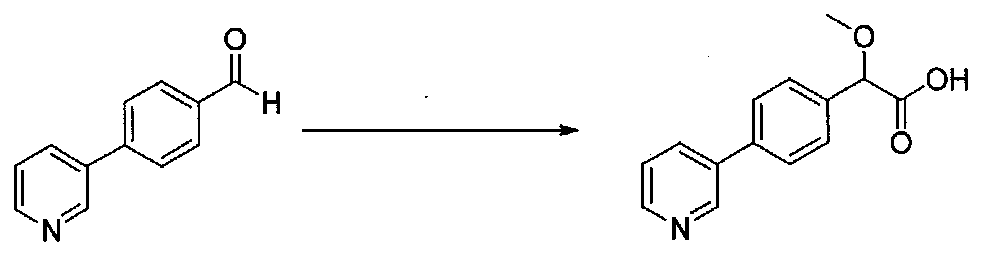
2-Метокси-2-(4-(пиридин-3-ил)фенил)уксусную кислоту получали из 4-(пиридин-3-ил)бензальдегида согласно методике, применяемой в примере 30, за исключением того, что pH 4 применяли в процессе экстракции. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (0,415 г, выход 33%).
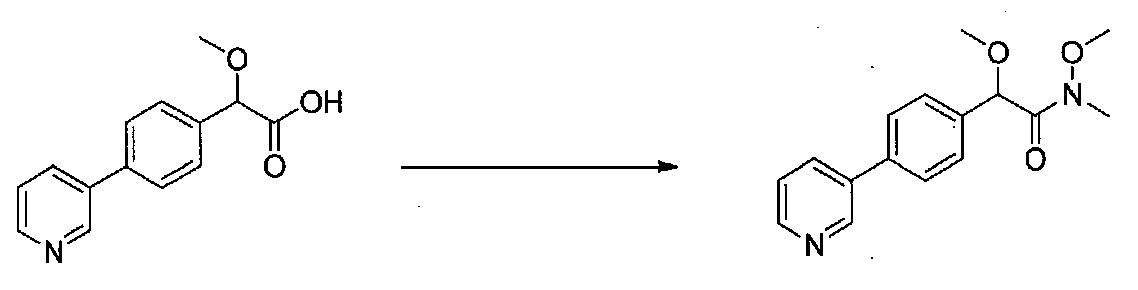
N,2-Диметокси-N-метил-2-(4-(пиридин-3-ил)фенил)ацетамид получали из 2-метокси-2-(4-(пиридин-3-ил)фенил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (100% EtOAc) давала продукт в виде масла (0,232 г, выход 48%).
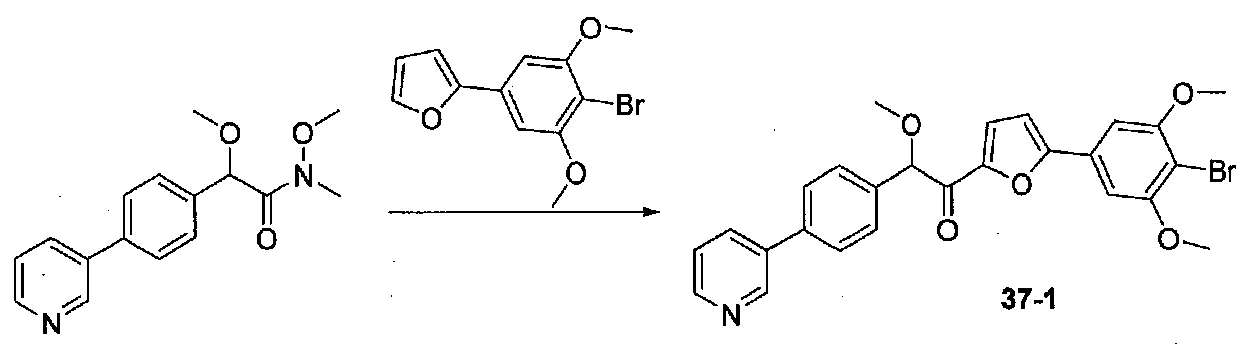
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(пиридин-3-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(пиридин-3-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (50% EtOAc/гексаны) давала продукт в виде не совсем белого твердого вещества (0,126 г, выход 35%). МС: m/z 508,2 [M+H]+.
ПРИМЕР 38
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-ЭТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
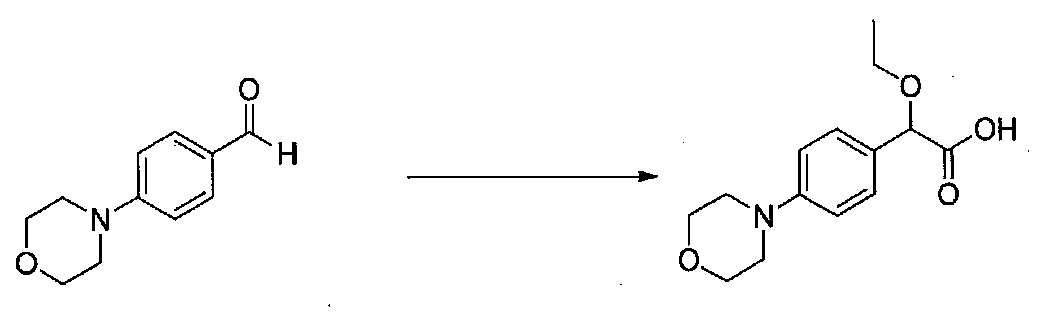
2-Этокси-2-(4-морфолинофенил)уксусную кислоту получали из 4-морфолинобензальдегида согласно методике, применяемой для получения примера 1, за исключением того, что EtOH применяли в качестве растворителя. Продукт выделяли в виде бледно-желтого масла и использовали без дополнительной очистки.
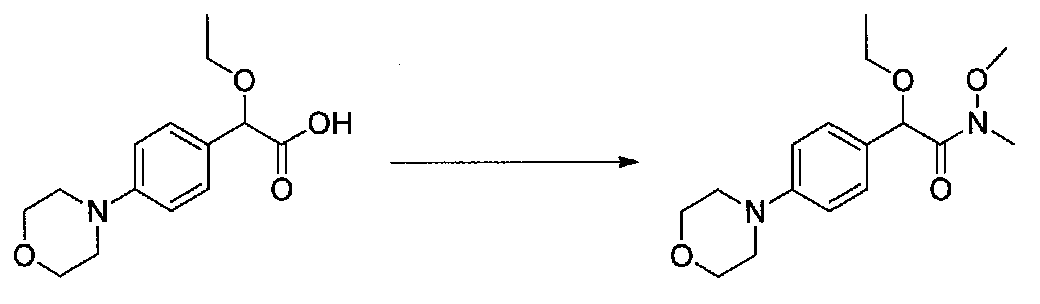
2-Этокси-N-метокси-N-метил-2-(4-морфолинофенил)ацетамид получали из 2-этокси-2-(4-морфолинофенил)уксусной кислоты, следуя методике, применяемой в примере 13. Очистка хроматографией (EtOAc-гексаны) давала продукт в виде желтого твердого вещества (1,0 г, выход 31% для двух стадий).
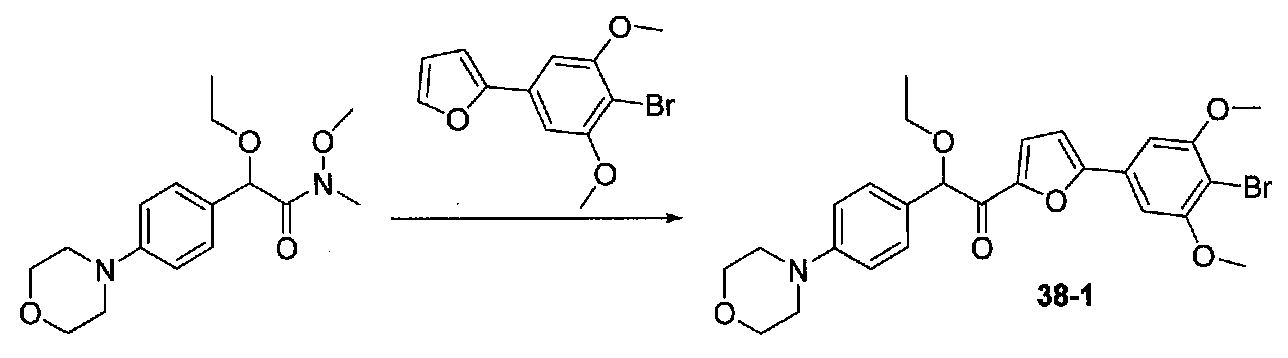
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-этокси-2-(4-морфолинофенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-этокси-N-метокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (50% EtOAc/гексаны) давала продукт в виде желтого твердого вещества (0,129 г, выход 34%). МС: m/z 530,2 [M+H]+.
ПРИМЕР 39
1-(5-(3-БРОМ-4,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН

3-Бром-4,5-диметоксифенол получали из 3-бром-4,5-диметоксибензальдегида согласно методике, применяемой в примере 21. Кристаллизация из смеси эфир/гексаны давала продукт в виде белого твердого вещества (2,02 г, выход 39%).
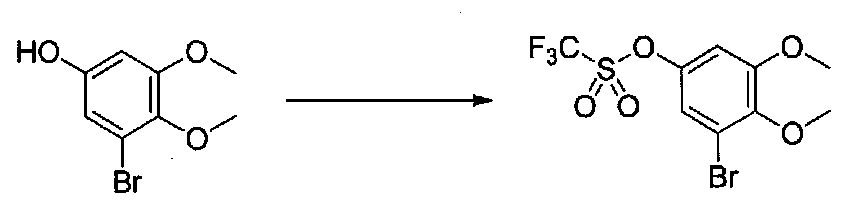
3-Бром-4,5-диметоксифенилтрифторметансульфонат получали из 3-бром-4,5-диметоксифенола согласно методике, применяемой в примере 21. Упаривание досуха давало продукт в виде бледно-желтого масла (3,3 г, выход 100%).
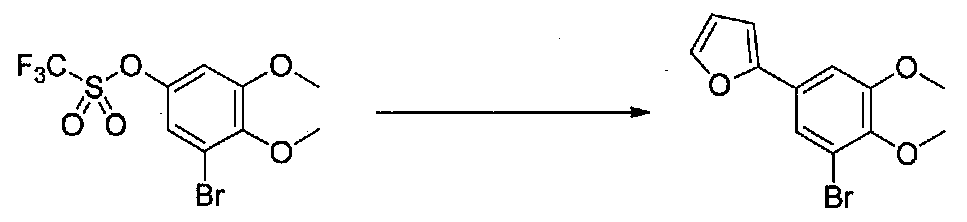
2-(3-Бром-4,5-диметоксифенил)фуран получали из 3-бром-4,5-диметоксифенилтрифторметансульфоната согласно методике, применяемой в примере 21. Очистка хроматографией (10% эфир/гексаны) давала продукт в виде бледно-желтого масла (1,7 г, выход 73%).
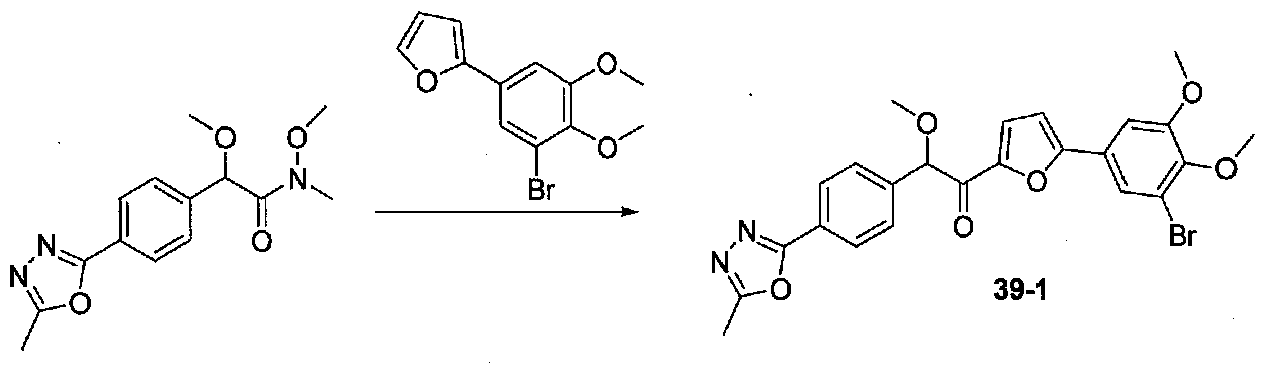
1-(5-(3-Бром-4,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(3-бром-4,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (70% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,037 г, выход 10%). МС: m/z 513,1 [M+H]+.
ПРИМЕР 40
1-(5-(3-ХЛОР-4,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
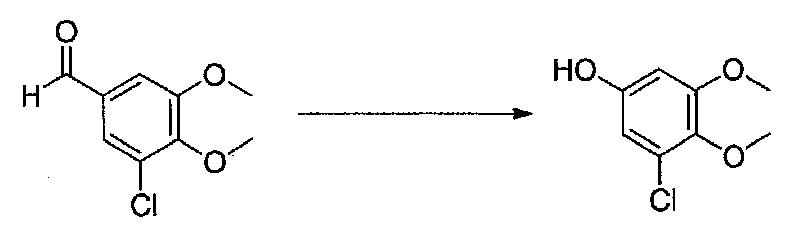
3-Хлор-4,5-диметоксифенол получали из 3-хлор-4,5-диметоксибензальдегида согласно методике, применяемой для получения примера 21. Полученное неочищенное твердое вещество обрабатывали ~10% EtOAc-гексаны при 40°C в течение 1 часа. После охлаждения до комнатной температуры твердое вещество собирали на воронке Бюхнера и сушили в вакууме с получением бежевых кристаллов (1,44 г, выход 38% на две стадии).
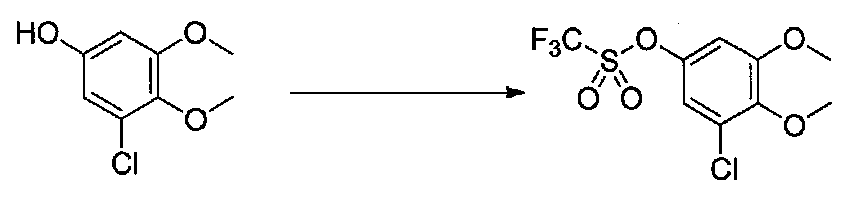
3-Хлор-4,5-диметоксифенилтрифторметансульфонат получали из 3-хлор-4,5-диметоксифенола согласно методике для получения примера 21. Продукт выделяли в виде бледно-желтой жидкости и использовали на следующей стадии синтеза без дополнительной очистки (2,33 г, выход 96%).
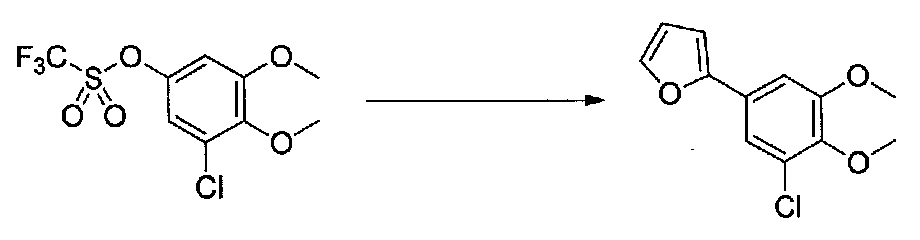
2-(3-Хлор-4,5-диметоксифенил)фуран получали из 3-хлор-4,5-диметоксифенилтрифторметансульфоната согласно методике, применяемой в примере получения 21. Очистка хроматографией (10% Et2O-гексаны) давала продукт в виде бледно-желтого масла (1,6 г, выход 94%).

l-(5-(3-Хлор-4,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(3-хлор-4,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (70% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,051 г, выход 12%). МС: m/z 469,2 [M+H]+.
ПРИМЕР 41
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-(ЦИКЛОПРОПИЛМЕТОКСИ)-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
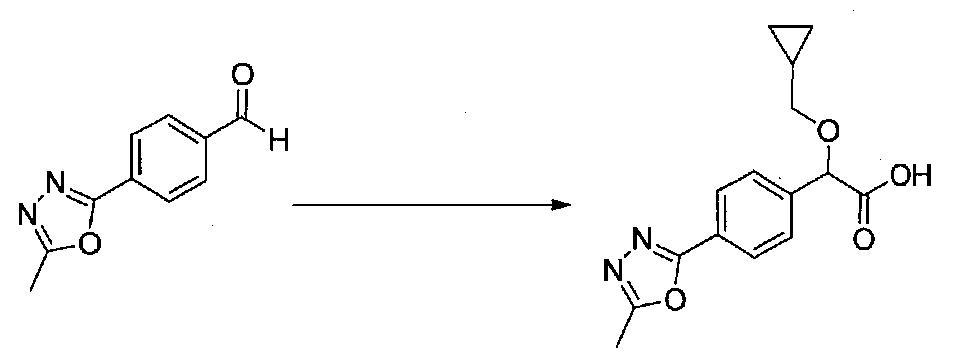
К раствору 4-(5-метил-1,3,4-оксадиазол-2-ил)бензальдегида (5,3 г, 27,7 ммоль) в циклопропилметаноле (25 мл) и безводном диоксане (25 мл) при 0°C (температура бани) добавляли несколько капель раствора KOH (7,77 г, 138,5 ммоль) в циклопропилметаноле (35 мл). Добавляли бромоформ (3,1 мл, 35,2 ммоль), затем добавляли в течение 20 минут оставшийся раствор KOH/циклопропилметанол. После перемешивания в течение 30 минут реакционную смесь нагревали медленно до комнатной температуры в течение ночи, затем концентрировали досуха. После растворения в минимальном количестве H2O остаток подкисляли до pH 1 концентрированной HCl. Водную смесь экстрагировали EtOAc несколько раз, добавляя соляной раствор к водному слою в процессе экстракции. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2-(циклопропилметокси)-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусной кислоты в виде полутвердого вещества, которое затем перекристаллизовали из Et2O (0,5 г, выход 7%).
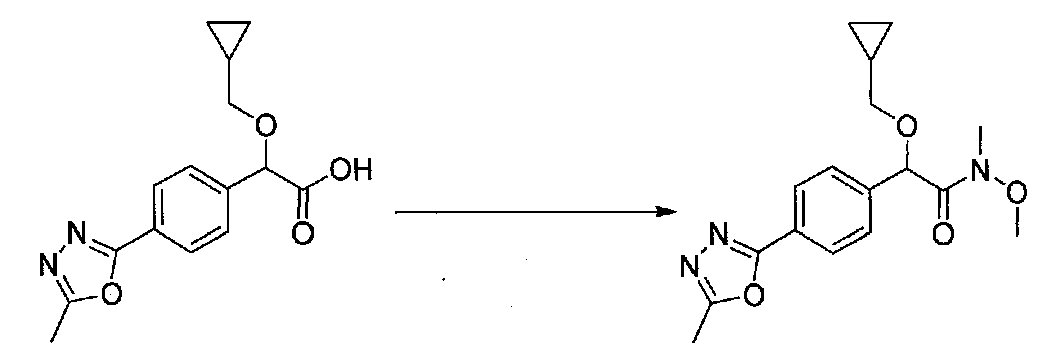
2-(Циклопропилметокси)-N-метокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамид получали из 2-(циклопропилметокси)-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусной кислоты согласно методике, применяемой в примере получения 13. Очистка хроматографией (0-70% EtOAc-гексаны) давала продукт в виде масла (0,48 г, выход 77%).
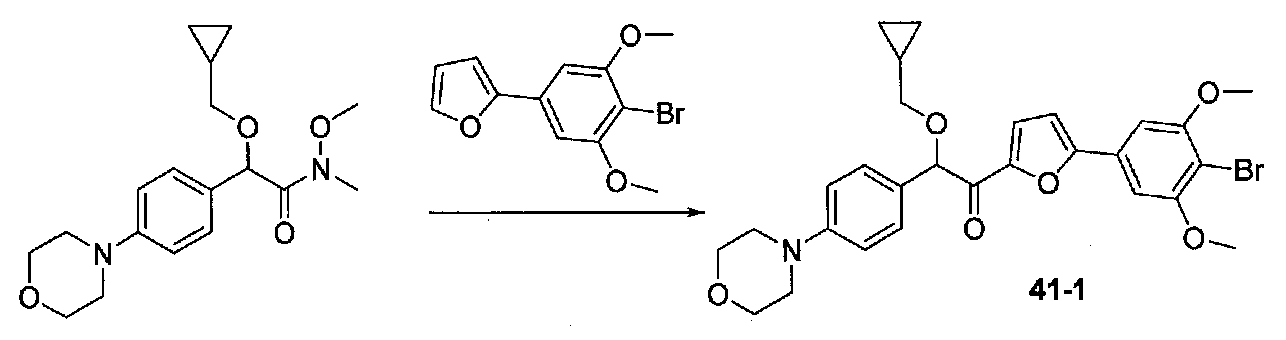
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-(циклопропилметокси)-2-(4-морфолинофенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-(циклопропилметокси)-N-метокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (40% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,092 г, выход 23%). МС: m/z 556,3 [M+H]+.
ПРИМЕР 42
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(6-МЕТОКСИПИРИДИН-3-ИЛ)ЭТАНОН
2-Метокси-2-(6-метоксипиридин-3-ил)уксусную кислоту получали из 6-метоксиникотинальдегида согласно методике, применяемой в примере 37. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (5,67 г, выход 79%).
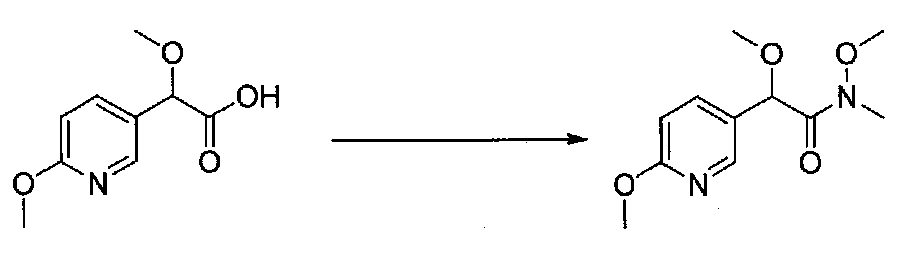
N,2-Диметокси-2-(6-метоксипиридин-3-ил)-N-метилацетамид получали из 2-метокси-2-(6-метоксипиридин-3-ил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде масла (0,97 г, выход 40%).

1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(6-метоксипиридин-3-ил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-2-(6-метоксипиридин-3-ил)-N-метилацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (50% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,092 г, выход 28%). МС: m/z 462,2 [M+H]+.
ПРИМЕР 43
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-(4-((ДИМЕТИЛАМИНО)МЕТИЛ)ФЕНИЛ)-2-МЕТОКСИЭТАНОН
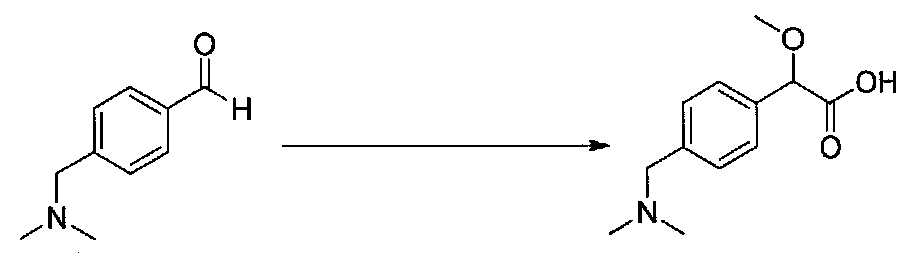
2-(4-((Диметиламино)метил)фенил)-2-метоксиуксусную кислоту получали из 4-((диметиламино)метил)бензальдегида согласно методике, применяемой в примере 37. После того как pH доводили до 4, водный раствор упаривали досуха. Добавляли MeOH (20 мл) при перемешивании и вещество фильтровали. Упаривание досуха давало неочищенный продукт, который использовали без дополнительной очистки (1,3 г, выход 95%).

2-(4-((Диметиламино)метил)фенил)-N,2-диметокси-N-метилацетамид получали из 2-(4-((диметиламино)метил)фенил)-2-метоксиуксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (100% EtOAc) давала продукт в виде масла (0,212 г, выход 14%).
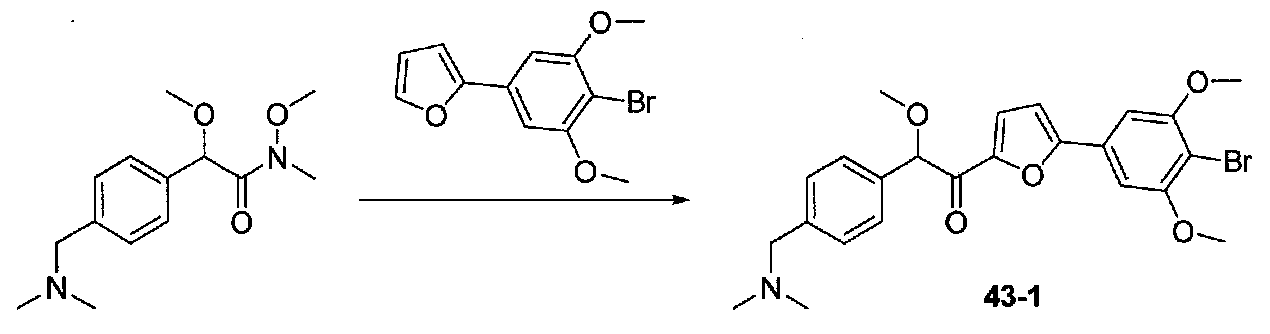
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-(4-((диметиламино)метил)фенил)-2-метоксиэтанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-(4-((диметиламино)метил)фенил)-N,2-диметокси-N-метилацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (4% MeOH/CH2Cl2) давала продукт в виде бледно-желтого масла (0,004 г, выход 1%). МС: m/z 488,3 [M+H]+.
ПРИМЕР 44
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(6-МОРФОЛИНОПИРИДИН-3-ИЛ)ЭТАНОН
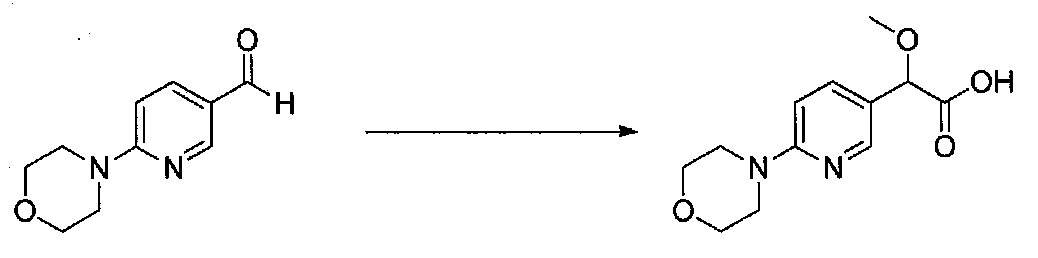
2-Метокси-2-(6-морфолинопиридин-3-ил)уксусную кислоту получали из 6-морфолиноникотинальдегида согласно методике, применяемой в примере 30, за исключением того, что водную смесь доводили до pH 4 в процессе экстракции. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (1,17 г, выход 89%).
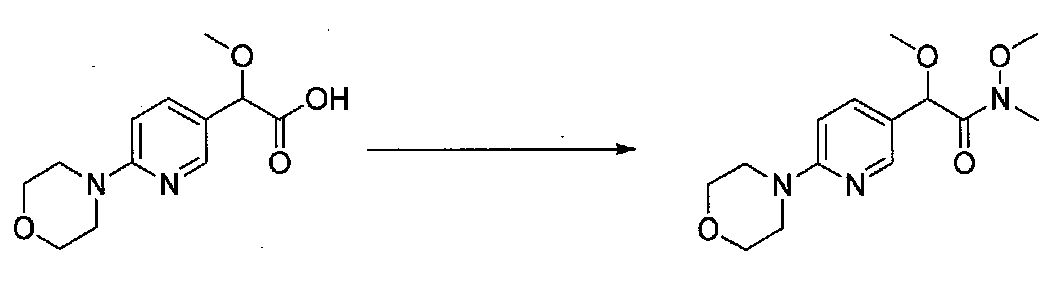
N,2-Диметокси-N-метил-2-(6-морфолинопиридин-3-ил)ацетамид получали из 2-метокси-2-(6-морфолинопиридин-3-ил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (80% EtOAc/гексаны) давала продукт в виде масла (0,692 г, выход 51%).
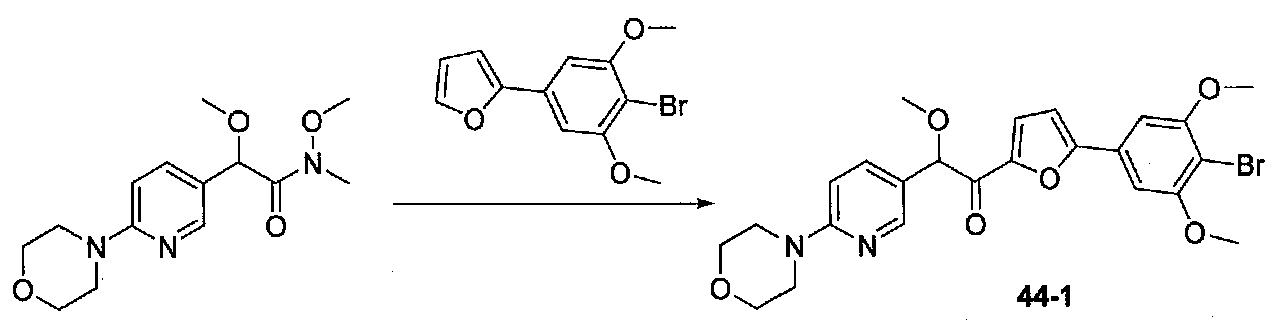
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(6-морфолинопиридин-3-ил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(6-морфолинопиридин-3-ил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,04 г, выход 11%). МС: m/z 517,3 [M+H]+.
ПРИМЕР 45
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(ПИРАЗИН-2-ИЛ)ФЕНИЛ)ЭТАНОН

2-Метокси-2-(4-(пиразин-2-ил)фенил)уксусную кислоту получали из 4-(пиразин-2-ил)бензальдегида согласно методике, применяемой в примере 30, за исключением того, что водную смесь доводили до pH 4 в процессе экстракции. Неочищенный продукт получали в виде масла и использовали без дополнительной очистки (1,21 г, выход 91%).
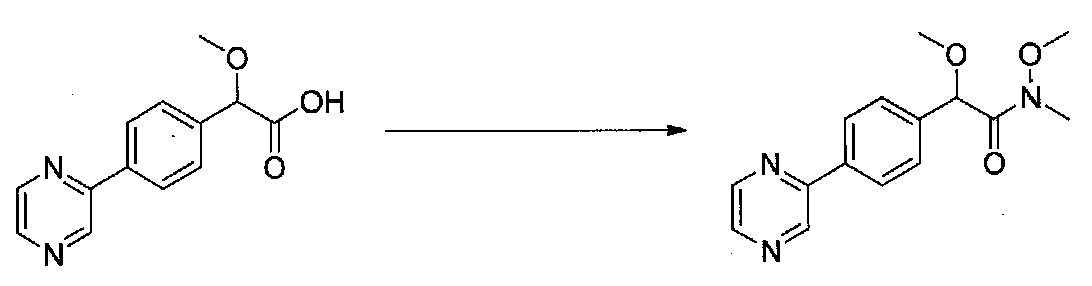
N,2-Диметокси-N-метил-2-(4-(пиразин-2-ил)фенил)ацетамид получали из 2-метокси-2-(4-(пиразин-2-ил)фенил)уксусной кислоты согласно методике, применяемой в примере 33. Очистка хроматографией (80% EtOAc/гексаны) давала продукт в виде масла (0,493 г, выход 35%).
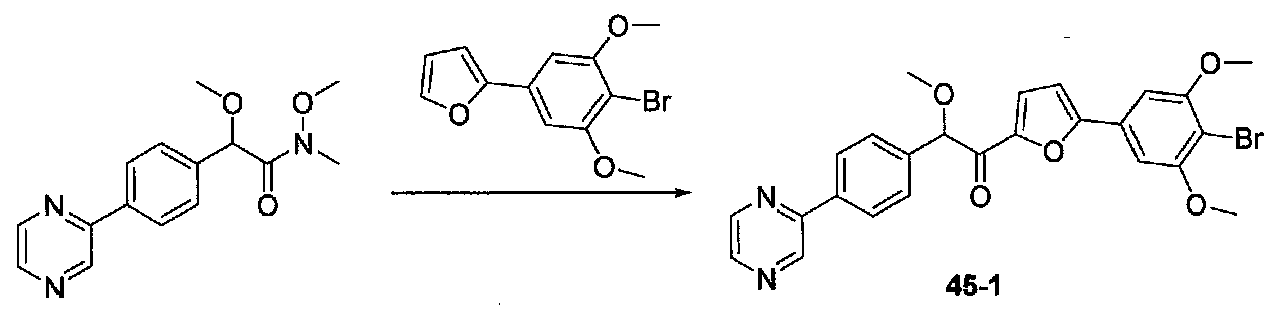
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(пиразин-2-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(пиразин-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,060 г, выход 17%). МС: m/z 509,3 [M+H]+.
ПРИМЕР 46
2-ЭТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)-1-(5-(3,4,5-ТРИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)ЭТАНОН
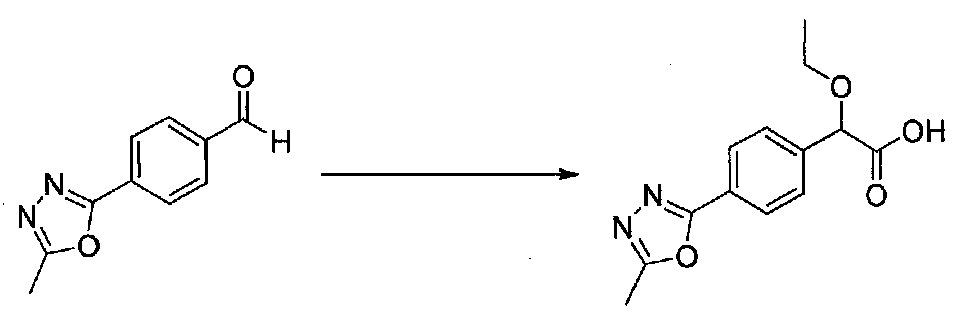
2-Этокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусную кислоту получали из 4-(5-метил-1,3,4-оксадиазол-2-ил)бензальдегида, следуя методике примера 47. Продукт выделяли в виде полутвердого вещества и использовали без дополнительной очистки (2,6 г, выход 93%).
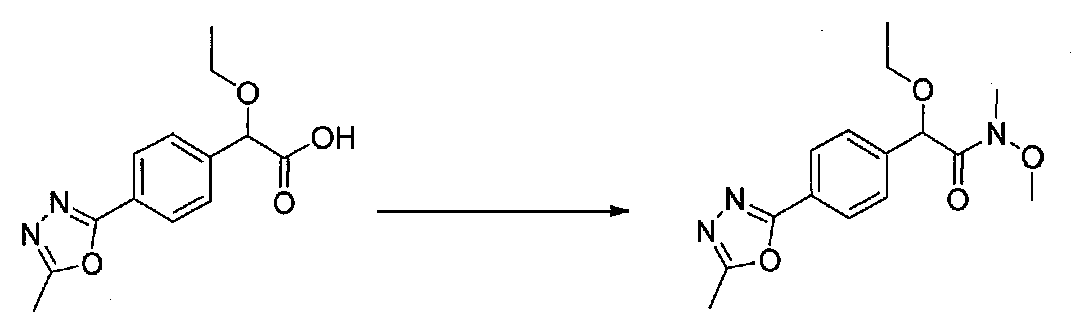
2-Этокси-N-метокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамид получали из 2-этокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)уксусной кислоты согласно методике для получения примера 41. Очистка хроматографией (0-60% EtOAc-гексаны) давала продукт в виде масла (1,0 г, выход 30%).
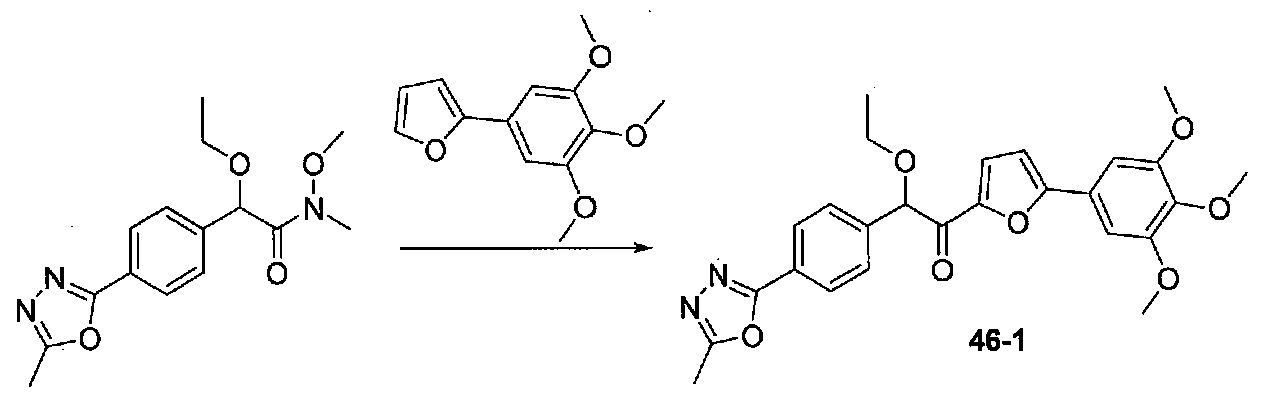
2-Этокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)-1-(5-(3,4,5-триметоксифенил)фуран-2-ил)этанон получали из 2-(3,4,5-триметоксифенил)фурана и 2-этокси-N-метокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,060 г, выход 15%). МС: m/z 479,4 [M+H]+.
ПРИМЕР 47
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-ЭТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
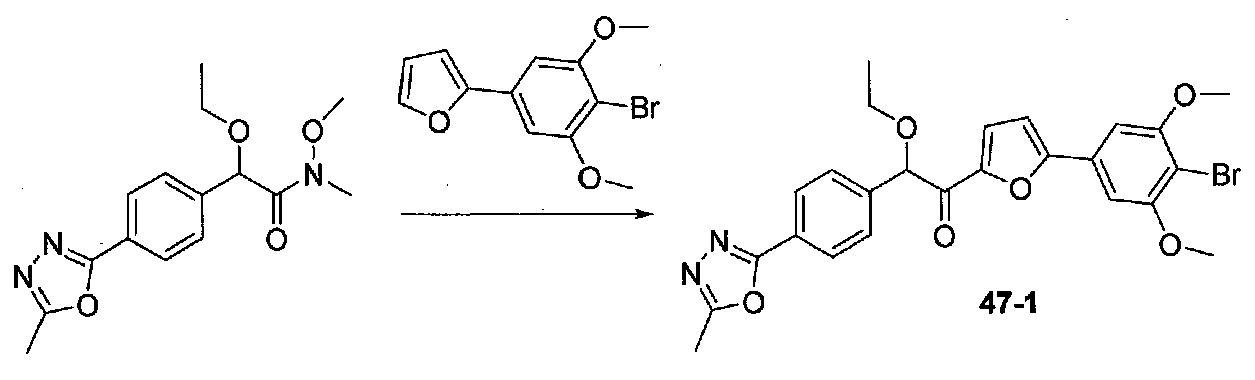
1-(5-(4-Бром-3,5-диметоксифенил)фуран-2-ил)-2-этокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)фурана и 2-этокси-N-метокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,070 г, выход 19%). МС: m/z 527,3 [M+H]+.
ПРИМЕР 48
1-(5-(4-ФТОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
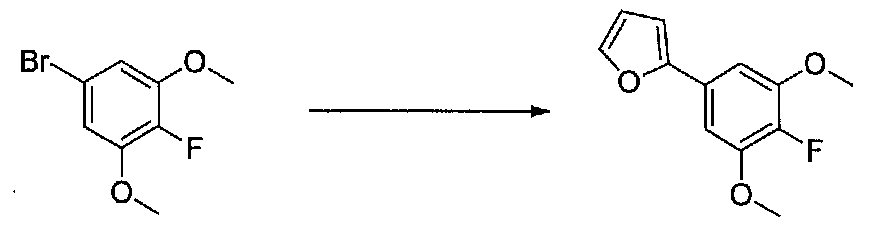
2-(4-Фтор-3,5-диметоксифенил)фуран получали из 5-бром-2-фтор-1,3-диметоксибензола (US6177154 B1) согласно методике, применяемой в примере 15. Очистка хроматографией (10% EtOAc/гексаны) давала продукт в виде белого твердого вещества (2,1 г, выход 58%).
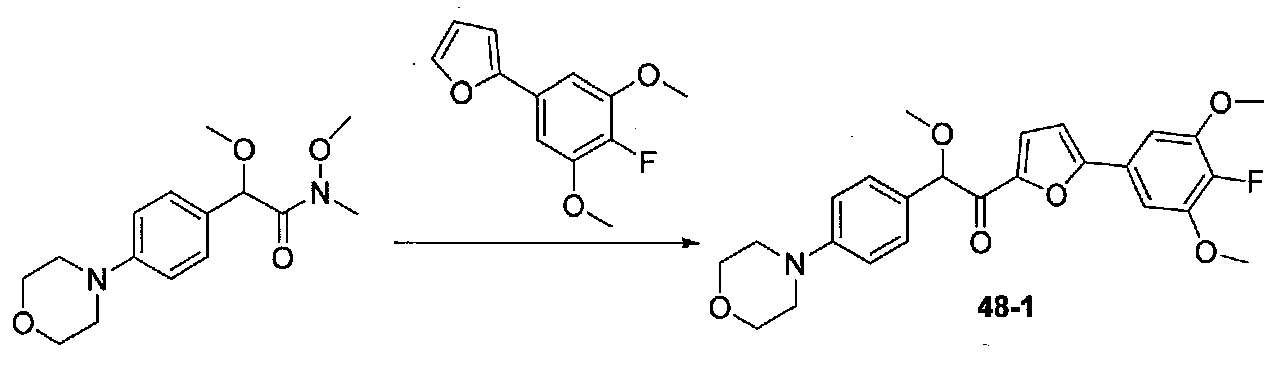
1-(5-(4-Фтор-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 2-(4-фтор-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,072 г, выход 16%). МС: m/z 456,4 [M+H]+.
ПРИМЕР 49
1-(5-(4-ХЛОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ТИАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
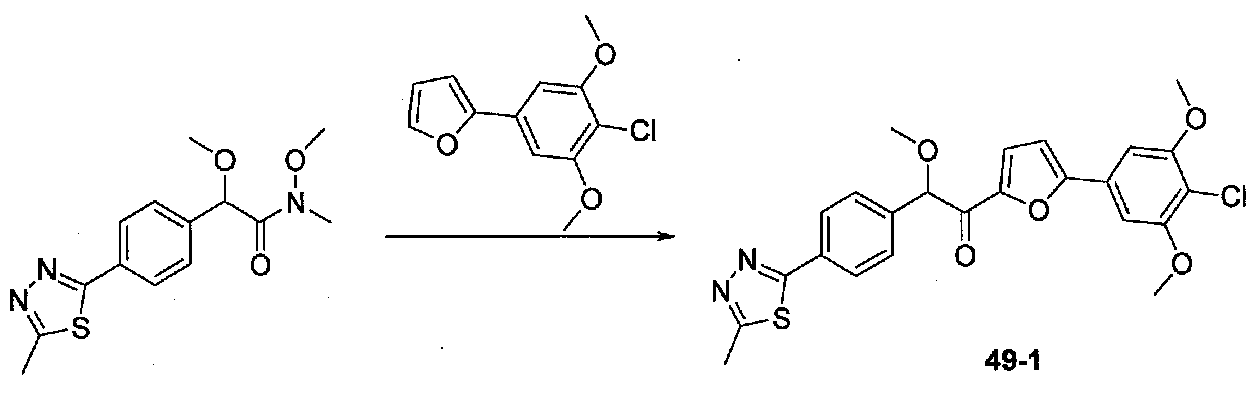
1-(5-(4-Хлор-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)этанон получали из 2-(4-хлор-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-тиадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (80% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,106 г, выход 26%). МС: m/z 485,4 [M+H]+.
ПРИМЕР 50
1-(5-(4-ХЛОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
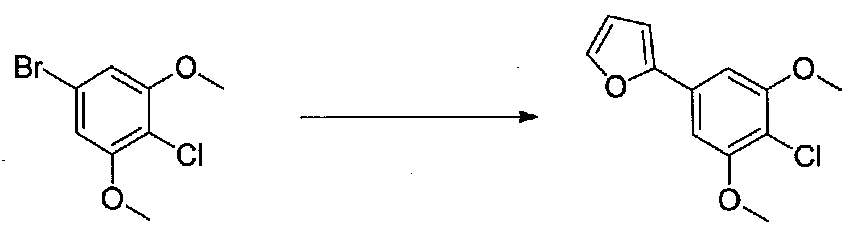
2-(4-Хлор-3,5-диметоксифенил)фуран получали из 5-бром-2-хлор-1,3-диметоксибензола (EP1568691 A1) согласно методике, применяемой в примере 15. Очистка хроматографией (10% эфир/гексаны) давала продукт в виде белого твердого вещества (1,24 г, выход 75%).
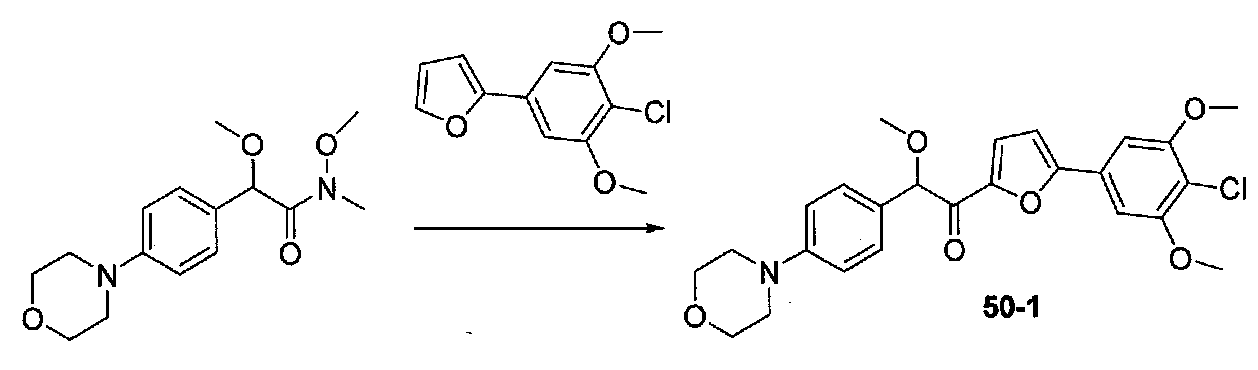
1-(5-(4-хлор-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 2-(4-хлор-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (70% EtOAc/гексаны) давала продукт в виде бледно-желтого твердого вещества (0,201 г, выход 43%). МС: m/z 472,2 [M+H]+.
ПРИМЕР 51
1-(3-(3,4-ДИМЕТОКСИФЕНИЛ)-1H-ПИРАЗОЛ-1-ИЛ)-2-(4-ФТОРФЕНИЛ)-2-МЕТОКСИЭТАНОН
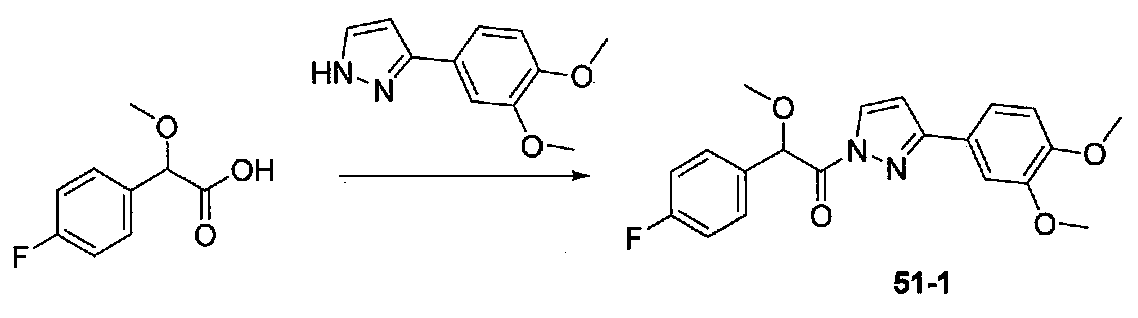
К раствору 2-(4-фторфенил)-2-метоксиуксусной кислоты (0,11 г, 0,61 ммоль) в безводном THF (2 мл) при комнатной температуре добавляли одной порцией DCC (0,14 г, 0,67 ммоль). После перемешивания в течение 10 минут добавляли одной порцией 3-(3,4-диметоксифенил)-1H-пиразол (0,14 г, 0,67 ммоль). Через 48 часов реакционную смесь разбавляли EtOAc и твердые вещества удаляли фильтрацией. Фильтрат концентрировали в вакууме. Очистка хроматографией (0-30% EtOAc-гексаны) давала продукт в виде масла (0,12 г, выход 46%). МС: m/z 371,1 [M+H]+.
ПРИМЕР 52
1-(3-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)-1H-ПИРАЗОЛ-1-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
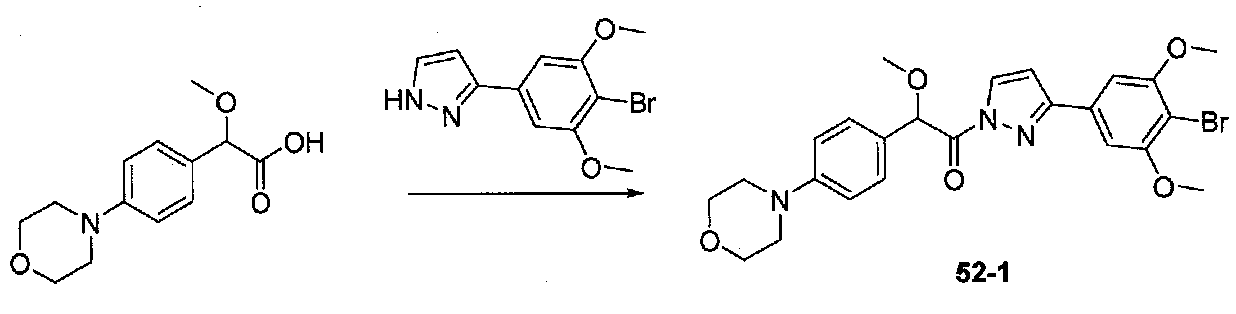
1-(3-(4-Бром-3,5-диметоксифенил)-1H-пиразол-1-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 3-(4-бром-3,5-диметоксифенил)-1H-пиразола (полученного согласно Journal of Org. Chem., 2003, 68, 5381) и 2-метокси-2-(4-морфолинофенил)уксусной кислоты, применяя аналогичную методику, как в примере 51 с получением 1-(3-(4-бром-3,5-диметоксифенил)-1H-пиразол-1-ил)-2-метокси-2-(4-морфолинофенил)этанона в виде твердого вещества (0,065 г, выход 22%). МС: m/z 516,1 [M+H]+.
ПРИМЕР 53
1-(3-(3,4-ДИМЕТОКСИФЕНИЛ)-1H-ПИРАЗОЛ-1-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
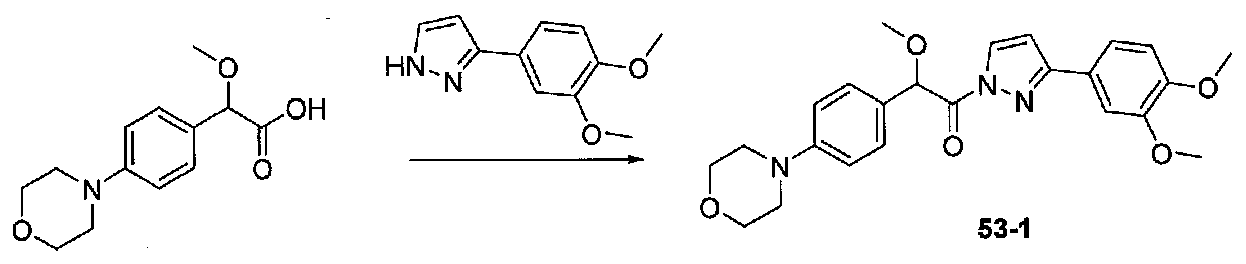
К раствору 2-метокси-2-(4-морфолинофенил)уксусной кислоты (0,1 г, 0,42 ммоль) и 3-(3,4-диметоксифенил)-1H-пиразола (0,86 г, 0,42 ммоль) в безводном DMF (4 мл) при комнатной температуре добавляли диизопропилэтиламин (0,22 мл, 1,26 ммоль), затем гексафторфосфат бромтрипирролидинофосфония (0,23 г, 0,50 ммоль). Через 24 часа добавляли насыщенный водный NaHCO3. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (0-60% EtOAc-гексаны) давала 1-(3-(3,4-диметоксифенил)-1H-пиразол-1-ил)-2-метокси-2-(4-морфолинофенил)этанон в виде белого твердого вещества (0,90 г, выход 50%). МС: m/z 438,2 [M+H]+.
ПРИМЕР 54
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)-1,2,4-ОКСАДИАЗОЛ-3-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
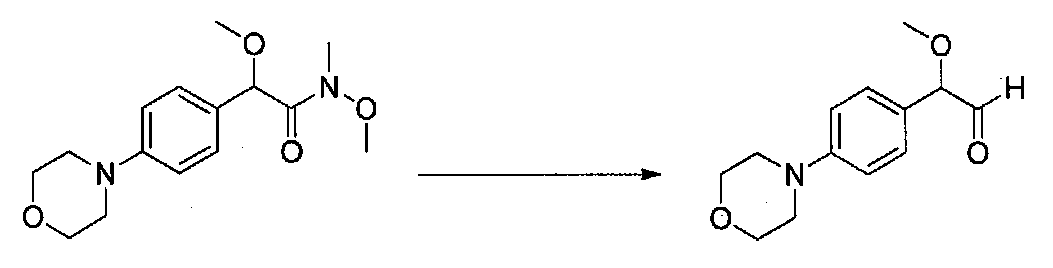
К раствору N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида (1,5 г, 5,1 ммоль) в безводном CH2Cl2 (12 мл) и безводном толуоле (6 мл) при -78°C добавляли раствор DIBALH (1 M в гексанах, 7,8 мл, 7,8 ммоль) по каплям в течение 5 минут. После перемешивания при -78°C в течение 1 часа реакционную смесь гасили добавлением по каплям EtOAc. Смесь перемешивали 2 минуты, затем добавляли Εt2Ο и насыщенный водный NH4Cl и смесь нагревали до комнатной температуры. После перемешивания в течение 30 минут смесь разбавляли EtOAc и H2O и слои разделяли. Добавляли к водному слою 10% водный тартрат калия натрия и продукт экстрагировали EtOAc/Et2O. Объединенные органические слои промывали насыщенным водным NH4Cl и сушили над Na2SO4 с получением 2-метокси-2-(4-морфолинофенил)ацетальдегида в виде желтого масла (1,27 г). Продукт использовали на следующей стадии синтеза без дополнительной очистки.
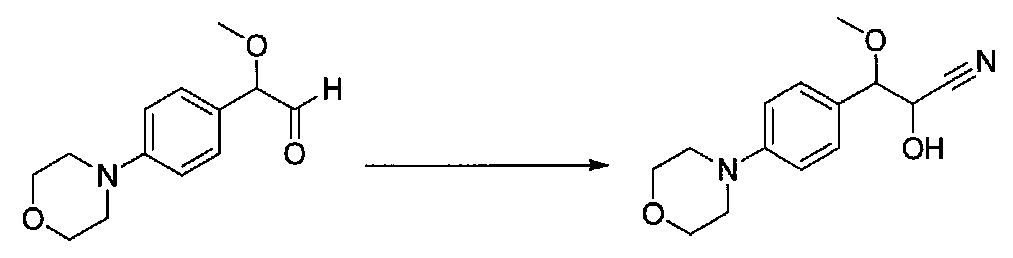
К раствору 2-метокси-2-(4-морфолинофенил)ацетальдегида (1,27 г, ~5,1 ммоль) в Et2O (35 мл) под хлоркальциевой трубкой добавляли триметилсилилцианид (1 мл, 8 ммоль) и ZnI2 (50 мг, 0,16 ммоль). После перемешивания при комнатной температуре в течение 15,5 часов добавляли насыщенный водный NaHCO3 и смесь перемешивали несколько часов. Смесь разбавляли EtOAc и H2O и слои разделяли. Органические слои промывали насыщенным водным NaHCO3, сушили над Na2SO4 и концентрировали в вакууме с получением 2-гидрокси-3-метокси-3-(4-морфолинофенил)пропаннитрила в виде оранжевой пены (1,26 г). Продукт использовали без дополнительной очистки.
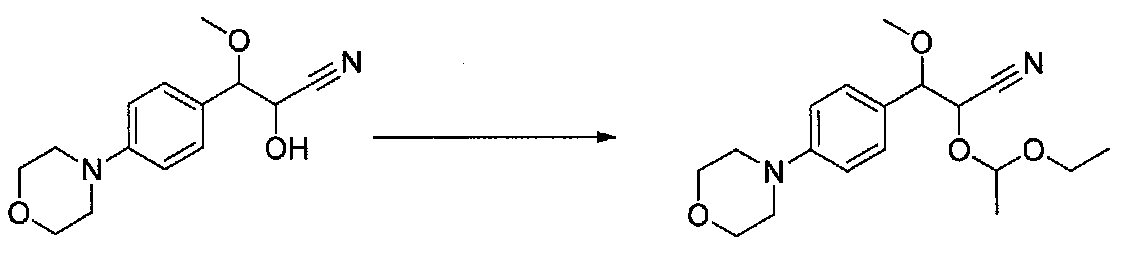
К раствору 2-гидрокси-3-метокси-3-(4-морфолинофенил)пропаннитрила (~5,1 ммоль) в безводном CH2Cl2 (17 мл) в атмосфере аргона добавляли пара-толуолсульфонат пиридиния (0,094 г, 0,37 ммоль) и этилвиниловый эфир (17 мл, 178 ммоль). Смесь помещали под хлоркальциевую трубку и перемешивали при комнатной температуре в течение 16 часов. Добавляли дополнительные количества этилвинилового эфира (4 мл, 42 ммоль) и пара-толуолсульфоната пиридиния (0,11 г, 0,44 ммоль) и смесь перемешивали в течение 24 часов. Добавляли к смеси насыщенный водный NaHCO3, затем ее разбавляли H2O и CH2Cl2. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные органические слои промывали соляным раствором, сушили над MgSO4 и концентрировали в вакууме. Очистка хроматографией (20-35% EtOAc-гексаны, содержащий 1% Et3N) давала 2-(1-этоксиэтокси)-3-метокси-3-(4-морфолинофенил)пропаннитрил (0,606 г, 35% для трех стадий).
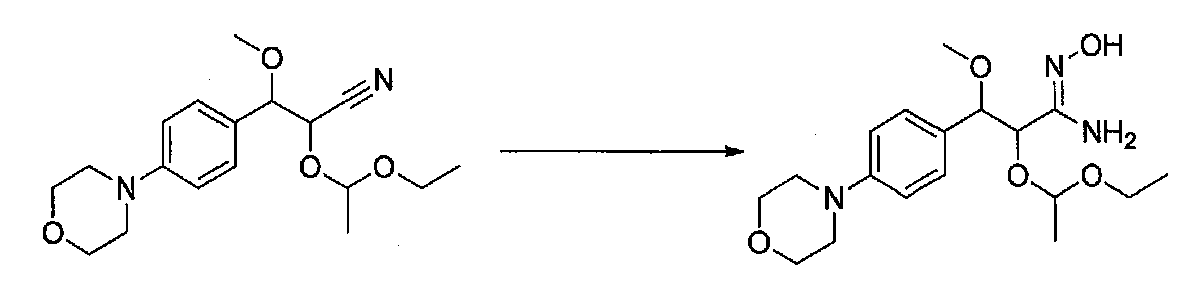
К раствору 2-(1-этоксиэтокси)-3-метокси-3-(4-морфолинофенил)пропаннитрила (0,60 г, 1,8 ммоль) в безводном MeOH (10 мл) в атмосфере аргона добавляли NH2OH·HCl (0,175 г, 2,5 ммоль) и NaHCO3 (0,234 г, 2,8 ммоль). Реакционную смесь быстро нагревали до 75°C, затем нагревали при 60°C в течение 16 часов. После охлаждения до комнатной температуры смесь концентрировали в вакууме с получением (Z)-2-(1-этоксиэтокси)-N'-гидрокси-3-метокси-3-(4-морфолинофенил)пропанимидамида, который использовали на следующей стадии синтеза без дополнительной очистки (0,777 г).
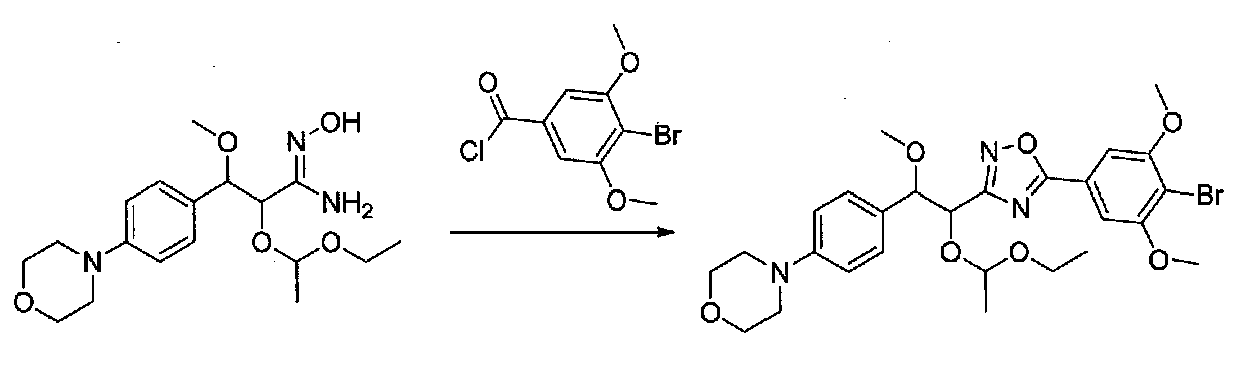
К охлажденной на льду суспензии (Z)-2-(1-этоксиэтокси)-N'-гидрокси-3-метокси-3-(4-морфолинофенил)пропанимидамида (~1,41 ммоль) в безводном CH2Cl2 в атмосфере аргона добавляли Et3N (0,8 мл, 5,7 ммоль), 4-бром-3,5-диметоксибензоилхлорид (0,434 г, 1,6 ммоль) и 4-диметиламинопиридин (0,015 г, 0,12 ммоль). Реакционную смесь перемешивали в течение 1 часа на бане со льдом, затем нагревали до комнатной температуры. После перемешивания в течение 3 часов при комнатной температуре добавляли дополнительное количество 4-бром-3,5-диметоксибензоилхлорида (0,047 г, 0,17 ммоль) и смесь перемешивали в течение дополнительного часа. Добавляли 10% водный раствор NaHCO3 и перемешивали в течение 20 минут. Слои разделяли и органический слой промывали H2O, соляным раствором, сушили над MgSO4 и концентрировали в вакууме с получением не совсем белой пены.
Данное полученное промежуточное соединение растворяли в безводном DMF (15 мл) в атмосфере аргона, затем нагревали при 120°C в течение 7 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли H2O и экстрагировали EtOAc. Объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (25-35% EtOAc-гексаны, содержащий 1% Et3N) давала 4-(4-(2-(5-(4-бром-3,5-диметоксифенил)-1,2,4-оксадиазол-3-ил)-2-(1-этоксиэтокси)-1-метоксиэтил)фенил)морфолин в виде светло-желтого масла (0,335 г, выход 40%).
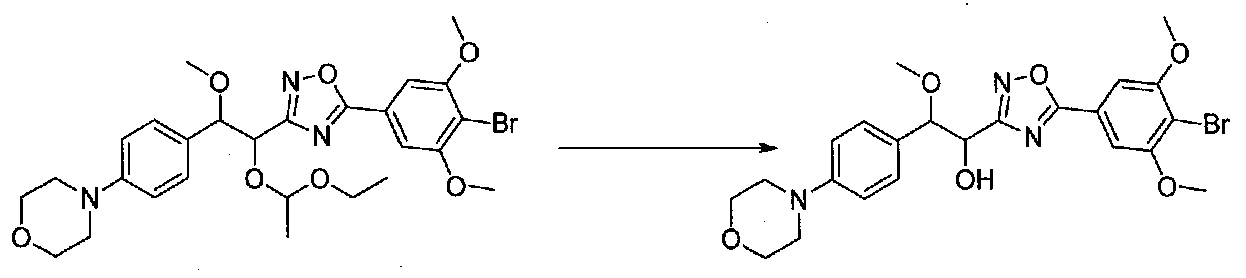
К раствору 4-(4-(2-(5-(4-бром-3,5-диметоксифенил)-1,2,4-оксадиазол-3-ил)-2-(1-этоксиэтокси)-1-метоксиэтил)фенил)морфолина (0,335 г, 0,57 ммоль) в MeOH (15 мл) добавляли пара-толуолсульфонат пиридиния (0,15 г, 0,6 ммоль) и смесь перемешивали при комнатной температуре в течение 16,5 часов. Затем реакционную смесь нагревали при 40°C в течение 6 часов, охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растворяли в EtOAc и промывали H2O, насыщенным NaHCO3 и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (50-60% EtOAc-гексаны) давала 1-(5-(4-бром-3,5-диметоксифенил)-1,2,4-оксадиазол-3-ил)-2-метокси-2-(4-морфолинофенил)этанол в виде бесцветного остатка (0,234, выход 79%).
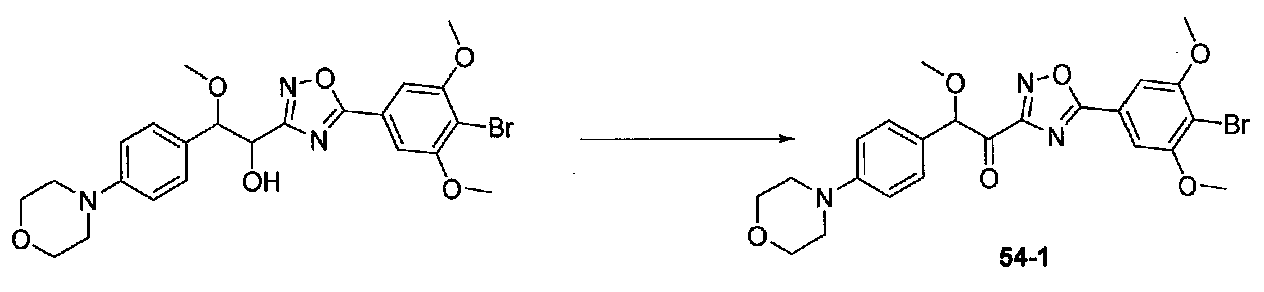
К раствору 1-(5-(4-бром-3,5-диметоксифенил)-1,2,4-оксадиазол-3-ил)-2-метокси-2-(4-морфолинофенил)этанола (0,18 г, 0,35 ммоль) в безводном CH2Cl2 (4 мл) в атмосфере аргона добавляли 1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензодиоксол-3-(1H)-он (0,198 г, 0,47 ммоль). После перемешивания при комнатной температуре в течение 50 минут смесь разбавляли Et2O, насыщенным водным NaHCO3 и 20% Na2S2O3. Слои разделяли и органический слой промывали насыщенным NaHCO3, сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (40-50% EtOAc-гексаны) давала продукт в виде ярко-желтого твердого вещества (0,1249 г, выход 70%). МС: m/z 518,1 [M+H]+.
ПРИМЕР 55
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)-5-МЕТИЛОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
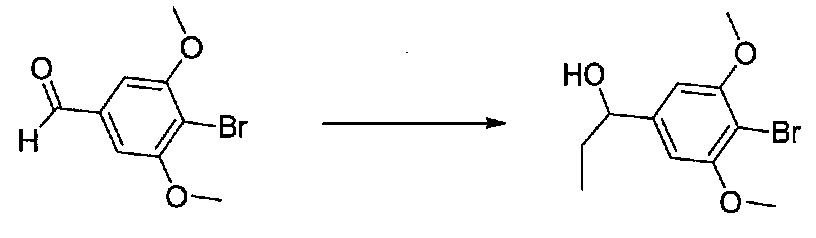
В колбу, высушенную в сушильном шкафу, в атмосфере аргона загружали 4-бром-3,5-диметоксибензальдегид (5,0 г, 20,4 ммоль) и безводный THF (30 мл). Смесь охлаждали на льду, затем добавляли по каплям из капельной воронки в течение 45 минут раствор этилмагнийбромида (3,0 M в диэтиловом эфире, 8,2 мл, 24,5 ммоль). После перемешивания в течение 20 минут смесь нагревали до комнатной температуры и перемешивали в течение 19 часов. После прекращения реакции раствором водного NH4Cl реакционную смесь разбавляли H2O и EtOAc, затем охлаждали на бане со льдом. После охлаждения смеси слои разделяли. Органический слой промывали H2O и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Остаток растворяли в CH2Cl2 и снова концентрировали в вакууме с получением 1-(4-бром-3,5-диметоксифенил)пропан-1-ола в виде прозрачного масла (5,43 г, выход 97%). Продукт использовали без дополнительной очистки.
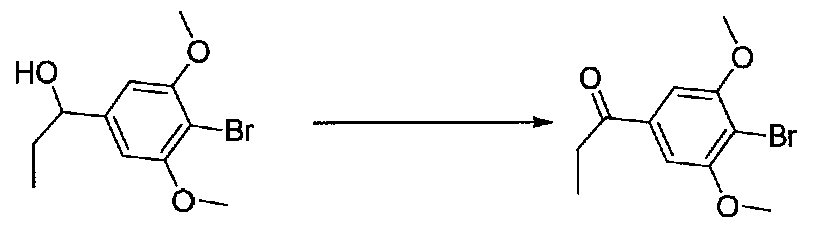
К раствору 1-(4-бром-3,5-диметоксифенил)пропан-1-ола (5,4 г, 19,6 ммоль) в безводном CH2Cl2 (75 мл) добавляли MnO2 (17 г, 196 ммоль). После того как смесь помещали под хлоркальциевую трубку и перемешивали при комнатной температуре в течение 22 часов, ее фильтровали через слой целита и силикагеля и промывали EtOAc. Концентрирование фильтрата в вакууме давало 1-(4-бром-3,5-диметоксифенил)пропан-1-он в виде белого твердого вещества (5,4 г, выход 100%). Продукт использовали без дополнительной очистки.
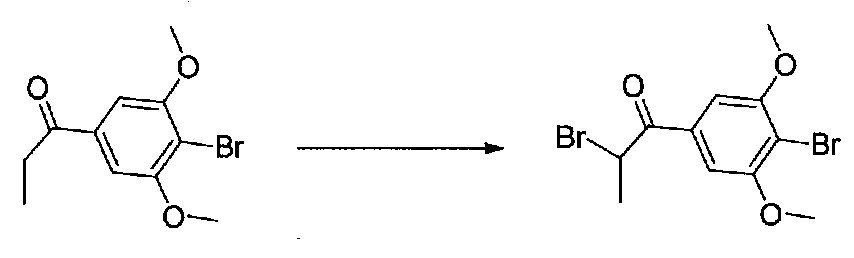
К раствору 1-(4-бром-3,5-диметоксифенил)пропан-1-она (1,50 г, 5,49 ммоль) в безводном THF (20 мл) добавляли трибромид пиридиния (1,93 г, 6,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем нейтрализовали раствором насыщенного водного NaHCO3. Смесь экстрагировали EtOAc, объединенные органические слои промывали насыщенным водным NaHCO3 и соляным раствором, затем сушили над Na2SO4 и концентрировали в вакууме. Очистка хроматографией (10-20% EtOAc-гексаны) давала 2-бром-1-(4-бром-3,5-диметоксифенил)пропан-1-он в виде оранжевого масла (1,09 г, выход 56%).
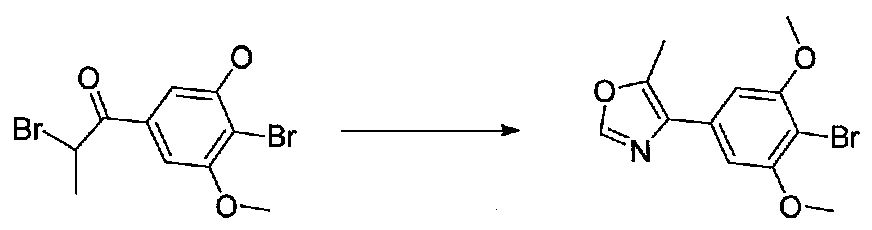
Раствор 2-бром-1-(4-бром-3,5-диметоксифенил)пропан-1-она (1,07 г, 3,04 ммоль) в формамиде (10 мл) в колбе, высушенной в сушильном шкафу, в атмосфере аргона нагревали при 110°C в течение 16 часов. После охлаждения до комнатной температуры осторожно добавляли EtOAc и насыщенный водный NaHCO3 и смесь перемешивали в течение 15 минут. Затем ее экстрагировали дважды EtOAc и объединенные органические слои промывали H2O и соляным раствором, сушили над Na2SO4 и концентрировали. Очистка хроматографией (30% EtOAc-гексаны) давала 4-(4-бром-3,5-диметоксифенил)-5-метилоксазол в виде желтого твердого вещества (0,496 г, выход 55%).
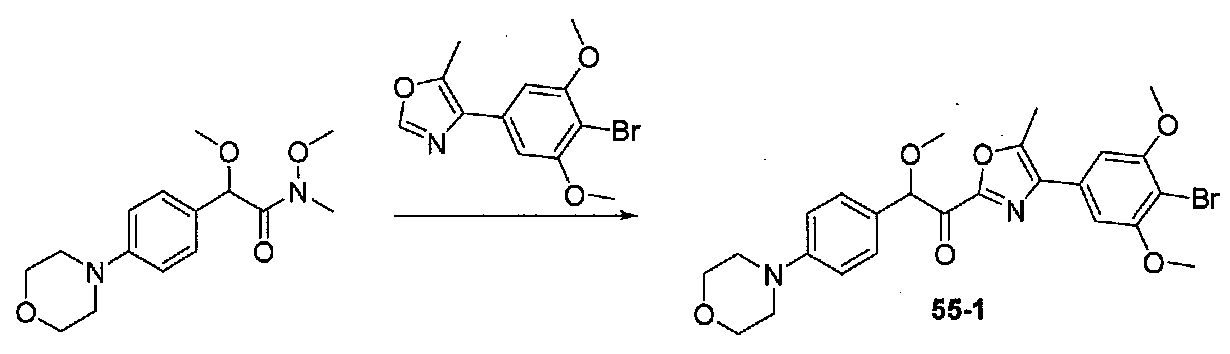
1-(4-(4-Бром-3,5-диметоксифенил)-5-метилоксазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 4-(4-бром-3,5-диметоксифенил)-5-метилоксазола и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (40% EtOAc-гексаны) давала продукт в виде желтого твердого вещества (0,048 г, выход 13%). МС: m/z 531,1 [M+H]+.
ПРИМЕР 56
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
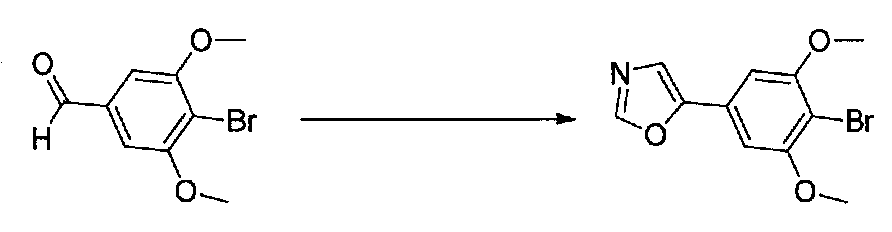
Раствор 4-бром-3,5-диметоксибензальдегида (5,04 г, 20,57 ммоль) и толуолсульфонилметилизоцианида (4,22 г, 21,6 ммоль) в MeOH (50 мл) кипятили с обратным холодильником в течение 3 часов. После упаривания практически досуха добавляли при перемешивании H2O (50 мл) и EtOAc (200 мл). Органический слой отделяли и промывали соляным раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Добавляли при перемешивании Et2O (50 мл) и продукт собирали фильтрацией, промывали Et2O (2×25 мл) и сушили, получая 5-(4-бром-3,5-диметоксифенил)оксазол в виде бледно-желтого твердого вещества (2,25 г, выход 39%).
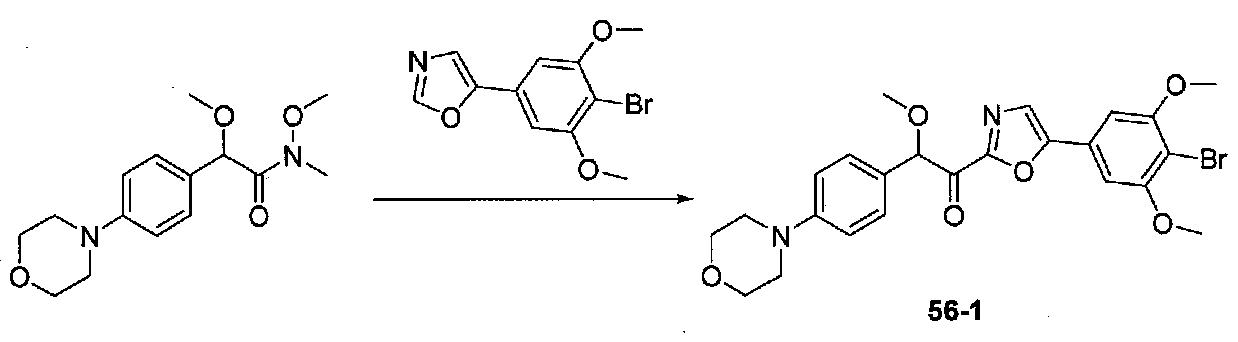
1-(5-(4-Бром-3,5-диметоксифенил)оксазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 5-(4-бром-3,5-диметоксифенил)оксазола и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Перекристаллизация из EtOAc давала продукт в виде желтого твердого вещества (0,308 г, выход 56%). МС: m/z 517,3 [M+H]+.
ПРИМЕР 57
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ТИОФЕН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН

2-(4-Бром-3,5-диметоксифенил)тиофен получали из 2-бром-5-йод-1,3-диметоксибензола и тиофен-2-илбороновой кислоты согласно методике, применяемой в примере 12. Очистка хроматографией (0-10% EtOAc-гексаны) давала желтое твердое вещество (0,624 г, выход 55%).
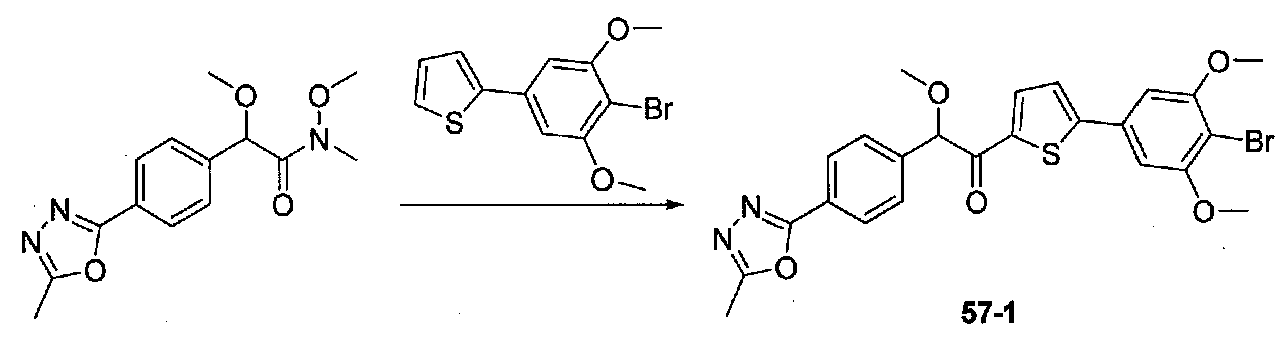
1-(5-(4-Бром-3,5-диметоксифенил)тиофен-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)тиофена и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (70% EtOAc-гексаны) давала продукт в виде желтой пены (0,031 г, выход 11%). МС: m/z 529,2 [M+H]+.
ПРИМЕР 58
1-(4-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)-5-(ТРИФТОРМЕТИЛ)ОКСАЗОЛ-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
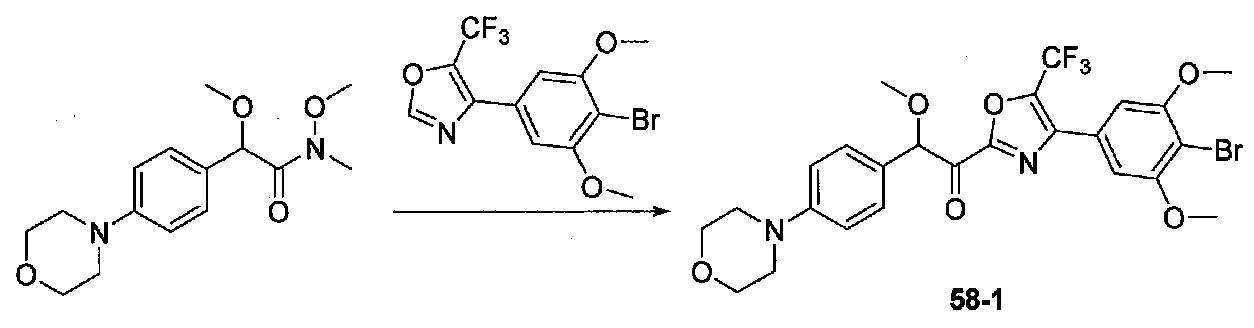
1-(4-(4-Бром-3,5-диметоксифенил)-5-(трифторметил)оксазол-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 4-(4-бром-3,5-диметоксифенил)-5-(трифторметил)оксазола (полученного из 4-бром-3,5-диметоксибензальдегида согласно Heterocycles, 1992, 34, 1047) и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (20-40% EtOAc-гексаны) давала продукт в виде желтого твердого вещества (0,032 г, выход 14%). МС: m/z 585,3 [M+H]+.
ПРИМЕР 59
1-(5-(4-БРОМ-3,5-ДИМЕТОКСИФЕНИЛ)ТИОФЕН-2-ИЛ)-2-МЕТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
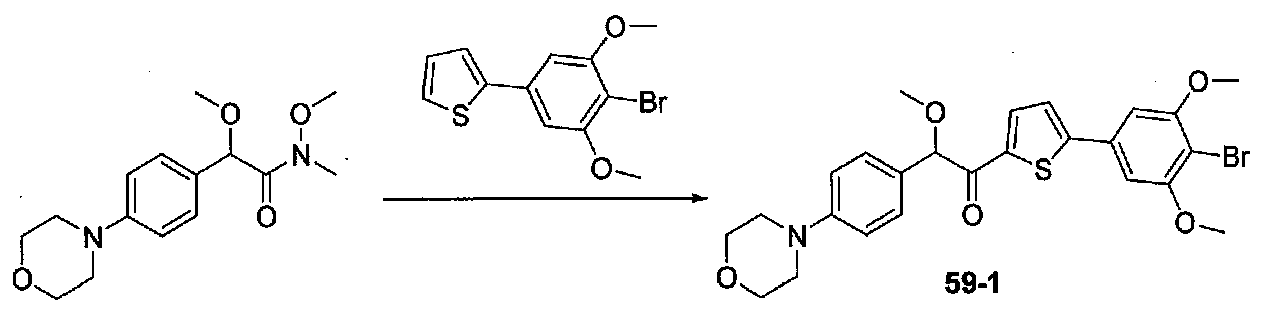
1-(5-(4-Бром-3,5-диметоксифенил)тиофен-2-ил)-2-метокси-2-(4-морфолинофенил)этанон получали из 2-(4-бром-3,5-диметоксифенил)тиофена и N,2-диметокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (40% EtOAc-гексаны) давала продукт в виде желтого твердого вещества (0,122 г, выход 46%). МС: m/z 532,4 [M+H]+.
ПРИМЕР 60
2-(4-(5-ЦИКЛОПРОПИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)-2-МЕТОКСИ-1-(5-(3,4,5-ТРИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)ЭТАНОН
2-(4-(5-Циклопропил-1,3,4-оксадиазол-2-ил)фенил)-2-метокси-1-(5-(3,5-триметоксифенил)фуран-2-ил)этанон получали из 2-(3,4,5-триметоксифенил)фурана и 2-(4-(5-циклопропил-1,3,4-оксадиазол-2-ил)фенил)-N,2-диметокси-N-метилацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,129 г, выход 42%). МС: m/z 491,1 [M+H]+.
ПРИМЕР 61
1-(5-(4-ХЛОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
1-(5-(4-Хлор-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(4-хлор-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,116 г, выход 25%). МС: m/z 469,1 [M+H]+.
ПРИМЕР 62
1-(5-(4-ФТОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-МЕТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
1-(5-(4-Фтор-3,5-диметоксифенил)фуран-2-ил)-2-метокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(4-фтор-3,5-диметоксифенил)фурана и N,2-диметокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,036 г, выход 8%). МС: m/z 453,2 [M+H]+.
ПРИМЕР 63
1-(5-(4-ХЛОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-ЭТОКСИ-2-(4-(5-МЕТИЛ-1,3,4-ОКСАДИАЗОЛ-2-ИЛ)ФЕНИЛ)ЭТАНОН
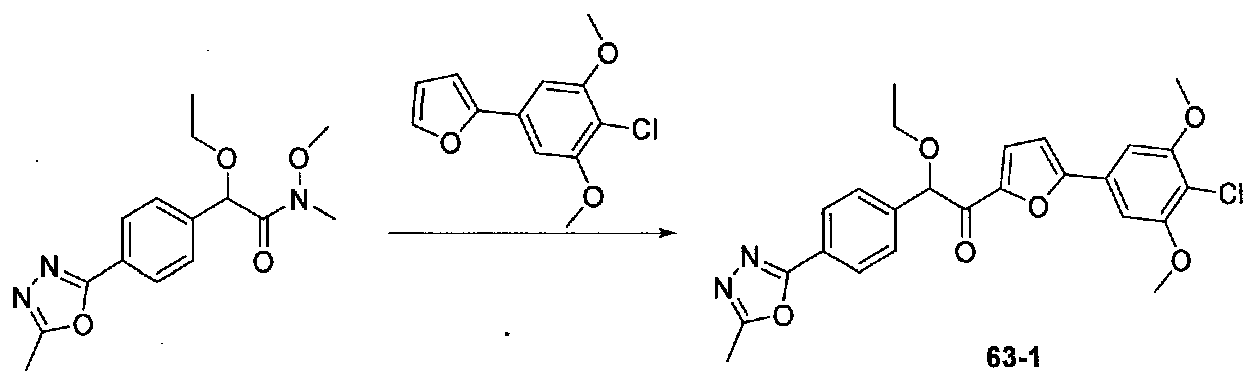
1-(5-(4-Хлор-3,5-диметоксифенил)фуран-2-ил)-2-этокси-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)этанон получали из 2-(4-хлор-3,5-диметоксифенил)фурана и 2-этокси-N-метокси-N-метил-2-(4-(5-метил-1,3,4-оксадиазол-2-ил)фенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,122 г, выход 25%). МС: m/z 483,1 [M+H]+.
ПРИМЕР 64
1-(5-(4-ХЛОР-3,5-ДИМЕТОКСИФЕНИЛ)ФУРАН-2-ИЛ)-2-ЭТОКСИ-2-(4-МОРФОЛИНОФЕНИЛ)ЭТАНОН
1-(5-(4-Хлор-3,5-диметоксифенил)фуран-2-ил)-2-этокси-2-(4-морфолинофенил)этанон получали из 2-(4-хлор-3,5-диметоксифенил)фурана и 2-этокси-N-метокси-N-метил-2-(4-морфолинофенил)ацетамида согласно методике, применяемой в примере 30. Очистка хроматографией (60% EtOAc-гексаны) давала продукт в виде бледно-желтого твердого вещества (0,127 г, выход 26%). МС: m/z 486,5 [M+H]+.
ПРИМЕР 65
ПОЛУЧЕНИЕ ДОПОЛНИТЕЛЬНЫХ ПРИМЕРНЫХ СОЕДИНЕНИЙ
Следующие примерные соединения в таблице 1 получали согласно (i) вышеуказанным способам, выбирая подходящие исходные вещества, и (ii) известными способами органического синтеза.
|
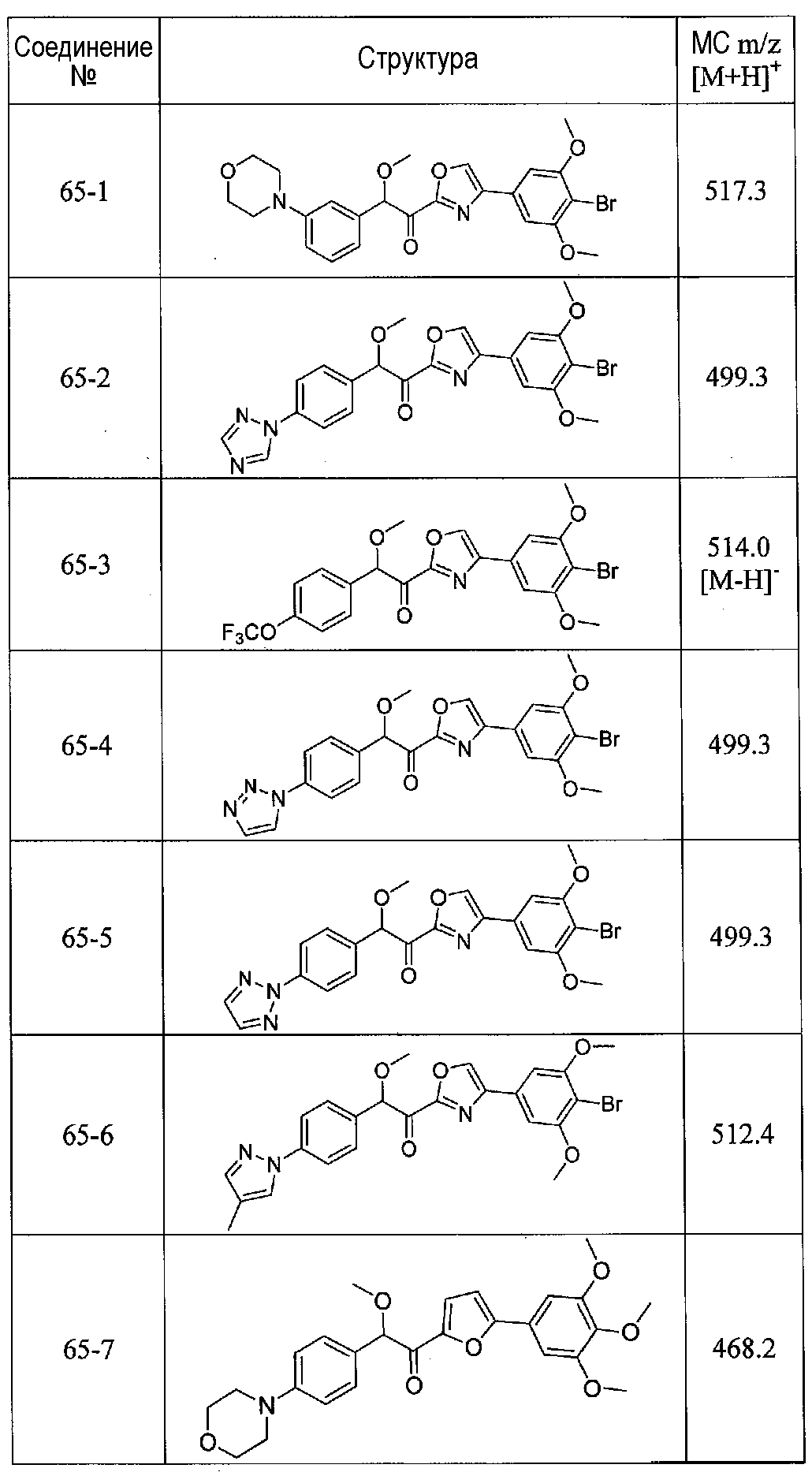
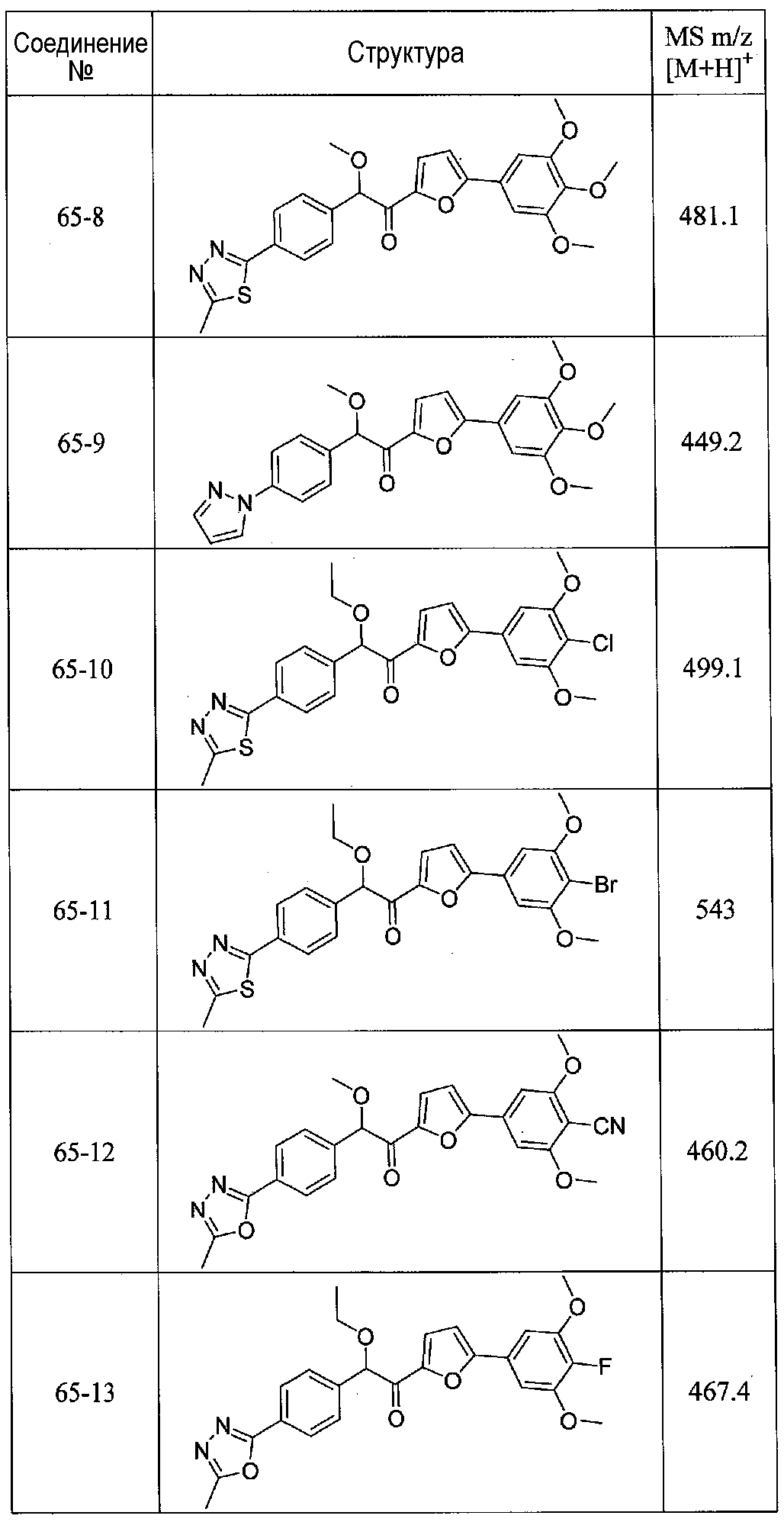
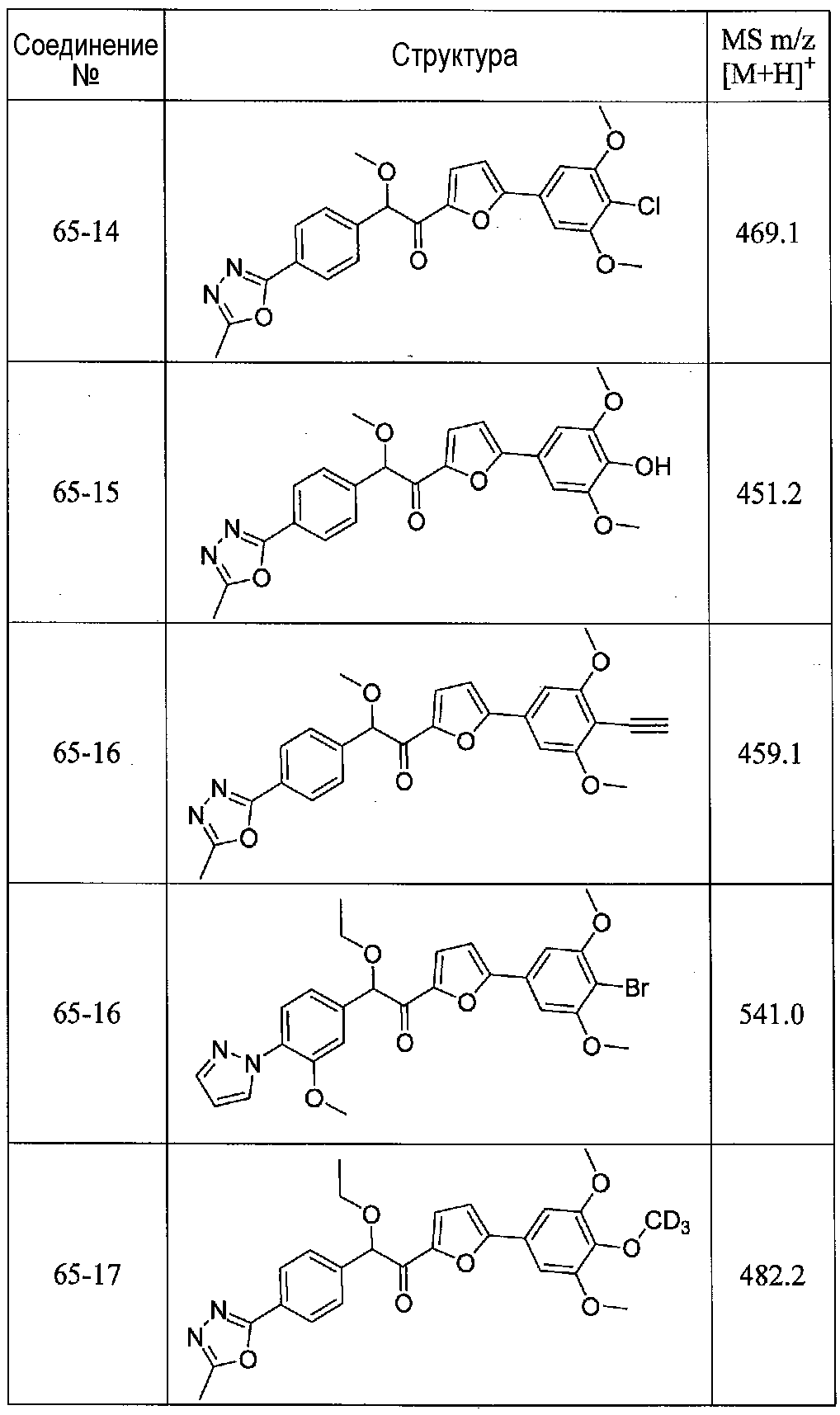
Следующие примерные соединения в таблице 2 получали согласно (i) вышеуказанным способам, выбирая подходящие исходные вещества, и (ii) известными способами органического синтеза.
|
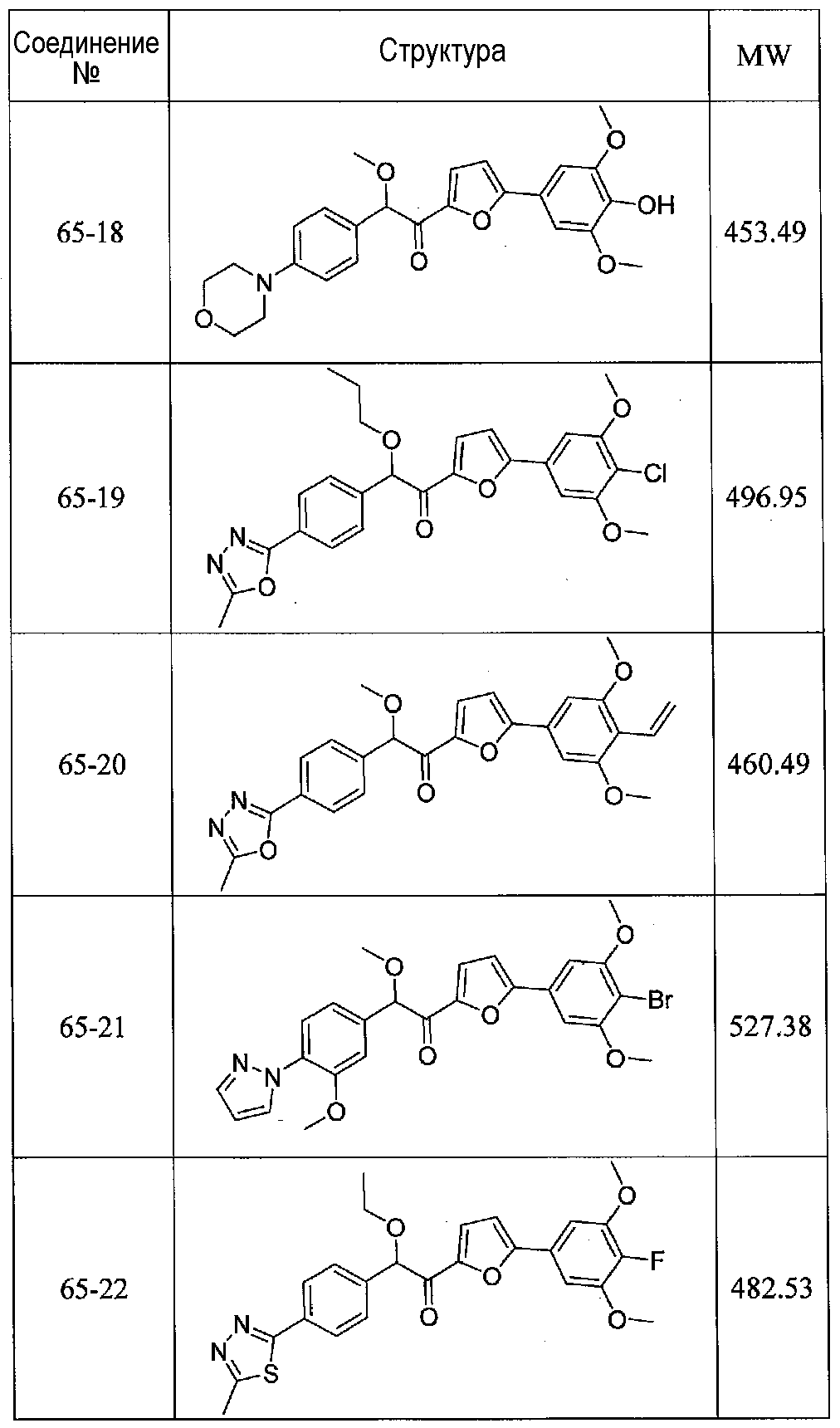
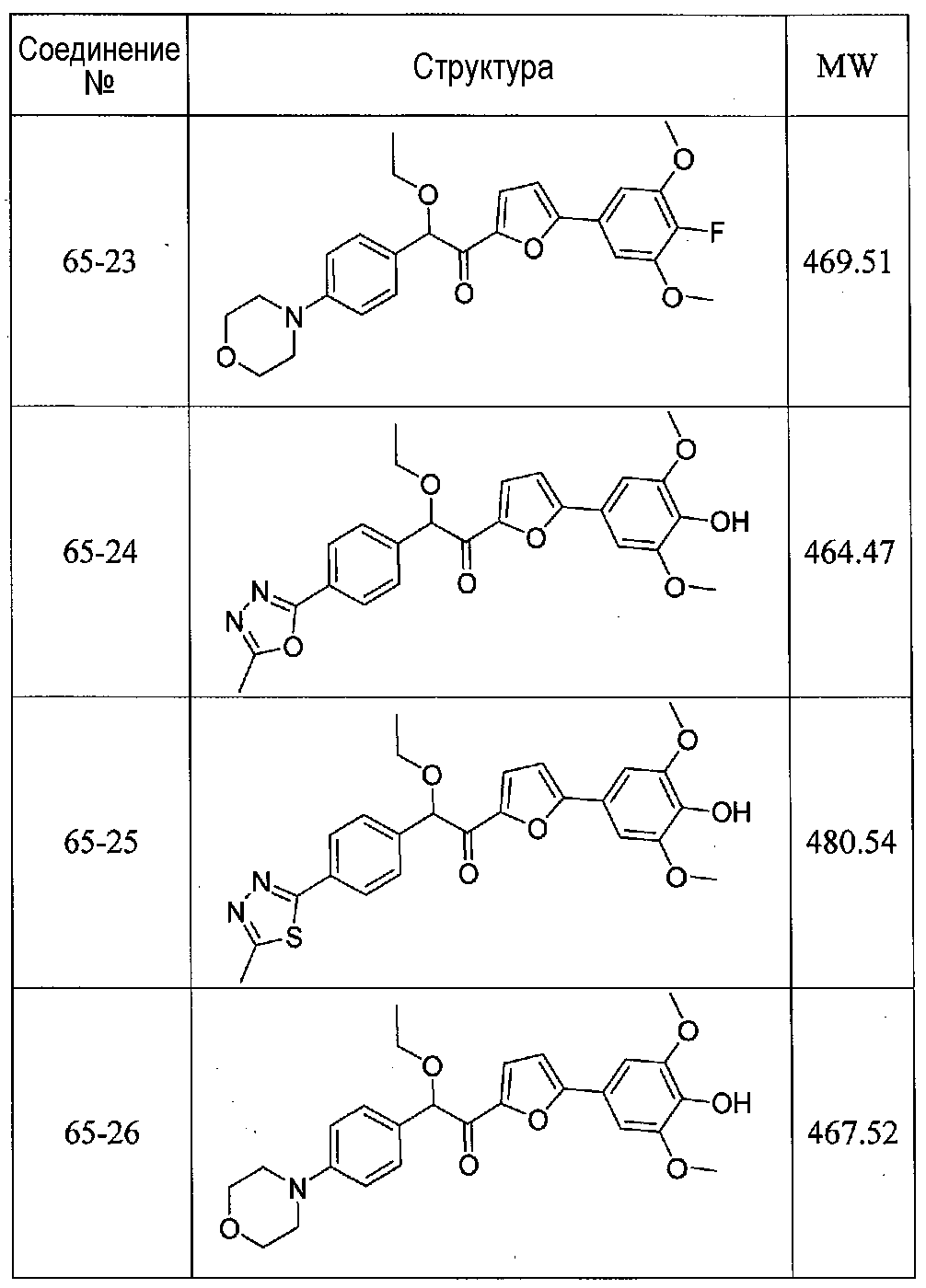
ПРИМЕР 66
АНАЛИЗ СОЕДИНЕНИЙ
PDE 10 биохимический анализ
Фосфодиэстеразный (PDE) анализ осуществляли, применяя рекомбинантные человеческие PDE 1A3, 2A3, 3 каталитический участок, 4 каталитический участок, 5 каталитический участок, 7A, 8A, 9A2, 10A1 и 11A1 ферменты, экспрессируемые в бакуловирусной системе, применяя Sf9 клетки. PDE активность измеряли, применяя модификацию двухстадийного способа Thompson и Appleman, описанного выше, который приспосабливали для формата 96-луночного планшета. Эффект PDE ингибиторов определяли исследованием фиксированного количества фермента в присутствии концентраций испытуемого соединения и концентраций субстрата, меньших чем Km, так чтобы Ki равнялась IC50. Конечный объем для анализа составлял 110 мкл с буфером для анализа (10 мМ MgCl2; 40 мМ Трис·HCl; pH 7,4). Реакции начинали с ферментом и выдерживали с (3H)-субстратом и веществом в течение 20 минут при 30°C. Реакцию прекращали денатурацией фермента (нагревание реакционной смеси при 70°C в течение 2 минут). Затем реакционную смесь охлаждали до 4°C в течение 10 минут перед добавлением змеиного яда (Crotalus atrox, 0,2 мг/мл) в течение 10 минут при 30°C, таким образом обеспечивая неспецифический гидролиз насыщенного тритием субстрата. Отделение оставшегося негидролизованного циклического нуклеотида осуществляли комплексным связыванием смеси с активированной анионообменной смолой Dowex (200 мкл). Анионообменная смола связывала заряженные нуклеотиды, оставляя только гидролизованный (3H) субстрат в растворимой фракции. Затем растворимую фракцию (50 мкл) добавляли к microscint-20 (200 мкл) и считали на планшетном ридере Top Count. Единицы радиоактивности наносили на график относительно концентрации ингибитора и IC50 величины получали, применяя программное обеспечение Graph Pad Prism.
Альтернативно, активность фосфодиэстеразы измеряли сцинтилляционным анализом сближения (SPA) с [3H]-цГМФ в качестве субстрата. Очищенный PDE10 разбавляли и хранили в 25 мМ Трис-Cl (pH 8,0)/100 мМ NaCl/0,05% Tween 20/50% глицерин/3 мМ DTT. Анализ включал (конечные концентрации): 50 мМ Трис-Cl (pH 7,5)/8,3 мМ MgCl2/1,7 мМ EGTA/0,5 мг/мл BSA/5% ДМСО и 2 нг PDE10 в конечном объеме 0,1 мл. Ингибирование оценивали при 8 концентрациях в двух экземплярах. Реакции начинали добавлением фермента и прекращали через 20 минут при 30°C добавлением 50 мкл SPA гранул, содержащих Zn++. Смесь встряхивали, позволяли осесть в течение 3 часов и считали на планшетном счетчике Wallac. Результаты (общий ед./мин) приспосабливали к логистической модели с четырьмя параметрами, применяя Excel Solver®.
Далее, ингибирование других PDE ферментов PDE10 ингибиторами оценивали при тех же условиях, описанных выше для PDE10, за исключением того, что количество добавляемого фермента оптимизировали для каждого PDE. Парциальное ингибирование оценивали при четырех концентрациях (0,1, 1, 10 и 100 мкМ). В случаях, когда ингибирование при наибольшей концентрации было меньше чем 50%, величину низшего предела в логистической модели устанавливали на 0% активности.
В вышеуказанном анализе соединения настоящего изобретения представляли собой PDE10 ингибиторы с IC50 100 мкМ или менее, обычно меньше чем 10 мкМ и обычно меньше чем 1 мкМ. В связи с этим было обнаружено, что соединения 1-1, 2-1, 3-1, 4-1, 5-1, 6-1, 7-1, 8-1, 9-1, 10-1, 11-1, 12-1, 13-1, 14-1, 15-1, 16-1, 17-1, 18-1, 19-1, 20-1, 21-1, 22-1, 23-1, 25-1, 26-1, 27-1, 28-1, 29-1, 30-1, 31-1, 32-1, 33-1, 34-1, 35-1, 36-1, 37-1, 38-1, 39-1, 40-1, 41-1, 42-1, 43-1, 44-1, 45-1, 46-1, 47-1, 48-1, 49-1, 50-1, 51-1, 52-1, 53-1, 54-1, 55-1, 56-1, 57-1, 58-1, 59-1, 60-1, 61-1, 62-1, 63-1, 64-1, 65-1, 65-2, 65-3, 65-4, 65-5, 65-6, 65-7, 65-8, 65-9, 65-10, 65-11, 65-12, 65-13, 65-14 и 65-15, например, имеют IC50 величины, менее чем или равные 1 мкМ.
ПРИМЕРЫ 67-77
ОЦЕНКА ПРЕДСТАВИТЕЛЕЙ СОЕДИНЕНИЙ В МОДЕЛЯХ ПОВЕДЕНИЯ
Шизофрения связана с нарушением допаминэргической, глутаматэргической и серотонинэргической нейропередачи. Психостимуляторы в данных трех классах, допаминэргические агонисты (такие, как амфетамин и апоморфин), глутаматэргические антагонисты (такие, как фенциклидин (PCP) и кетамин) и серотонинэргические агонисты (такие, как LSD и MDMA) все вызывают психотомиметические состояния (например, гиперактивность и нарушение преимпульсного ингибирования) у животных, которые сильно напоминают симптомы шизофрении у людей. Известные противопсихотические лекарственные средства, включая и стандартные противопсихотические лекарственные средства (например, галоперидол), и нестандартные противопсихотические лекарственные средства (например, оланзапин), обращают данные психотомиметические состояния у животных. Примеры 67-77, описанные ниже, оценивают представителей соединений настоящего изобретения в моделях поведения животных, что позволяет сравнить полученный в результате эффект с эффектом известных противопсихотических лекарственных средств. Способы, применяемые в примерах 67-77, являются следующими.
Введение дозы соединений осуществляли внутрибрюшинной (i.p.) инъекцией или пероральным способом введения (p.o.). Внутрибрюшинная инъекция сопровождается удерживанием животного, оголением брюха и вставлением иглы прямо над коленями на правой стороне мыши. Пероральный способ введения осуществляют удерживанием животного таким способом, чтобы его голова была отклонена назад, и пищевод находился в относительно прямом положении. Иглу для принудительного введения (20G ×1,5", Cadence Science) вставляют в пасть параллельно телу и аккуратно опускают вдоль пищевода и в желудок. Если возникает сопротивление, иглу удаляют и вставляют повторно.
Вызванную психостимулятором гиперактивность измеряли введением животным PCP и отслеживанием степени активности животных в камерах VersaMax (Accuscan Instruments, Columbus, OH) с размерами 40×40 см. Двигательную активность определяли фотоинфракрасными датчиками, как только животное пересекает каждый луч. Животное помещали в центр поля и оставляли нетронутым в течение 20 минут для измерения его непроизвольной активности в новом окружении. Измерения, применяемые для оценки двигательной активности, включали: горизонтальную активность, суммарное пройденное расстояние, вертикальную активность (подъем на задние лапы - животное поднимается на задние конечности), вращение, сохранение определенного положения тела и расстояние, пройденное в центре, по сравнению с суммарным пройденным расстоянием (отношение центр: суммарное расстояние). NMDA антагонист PCP вызывает проявление психозоподобного симптома в виде гиперактивности и усиливает кататонию. Известные противопсихотические лекарственные средства способны обращать вызванную психостимулятором гиперактивность и кататонию.
Условная реакция избегания (CAR) представляет собой поведенческий тест для оценки противопсихотического эффекта испытуемого соединения. В нем применяют челночную коробку (Med Associates, St. Albans, VT) с двумя одинаковыми камерами, разделенными убирающейся дверью. Каждая камера оснащена полом из металлической сетки, который способен осуществлять независимо электрический удар. Компьютерную программу применяют для выполнения тестового подхода, а также записи движений животного между двумя камерами через сенсоры с инфракрасными лучами. Тестовый подход был следующим. Мышь помещают в одну камеру. Включают свет (условный стимул, CS). Через пять секунд подают умеренный электрический удар (0,4 мА; (безусловный стимул, US) в камеру, в которой находится мышь (как определено инфракрасными лучами) до того момента, как мышь перейдет в соседнюю камеру или как пройдет 10 секунд. US и CS всегда прекращают одновременно. С рандомизированными интервалами между испытаниями в среднем 15 секунд проводили 30 данных CS-US спаренных испытаний с каждой мышью каждый день. Для каждого испытания реакцию бегства регистрируют, если мышь переходит в другую камеру после электрического удара (т.е. в течение 10-секундного US периода), и реакцию избегания регистрируют, если мышь переходит в другую камеру в течение первых 5 секунд только CS периода. Животных обучали в данном подходе в течение 30-40 дней, в течение которых средний процент реакций избегания будет увеличиваться до 80-90%. Это показывает, что животные научились избегать воздействия на лапы электрического тока перемещением в противоположную камеру при активации CS (свет). Затем данных обученных мышей применяют для испытания соединений в этом же подходе. Было найдено, что известные противопсихотические лекарственные средства подавляют условную реакцию избегания, и считается, что способность новых соединений подавлять данную реакцию предсказывает противопсихотический эффект у людей.
ПРИМЕР 67
СНИЖЕНИЕ PCP-ВЫЗВАННОЙ ГИПЕРАКТИВНОСТИ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 1-1
Было обнаружено, что соединение 1-1 (пример 1) снижает PCP-вызванную гиперактивность, как показано на фиг.1 и 2. C57BL/6 мужским особям мышей давали либо соединение 1-1, либо носитель внутрибрюшинной инъекцией (ФИГ.1) или пероральным способом введения (ФИГ.2). Через двадцать минут (для внутрибрюшинного способа введения) или сорок минут (для перорального способа введения) животным вводили PCP (5 мг/кг, внутрибрюшинно). Через десять минут мышей помещали в камеры для оценки активности и их двигательную активность в горизонтальном направлении отслеживали инфракрасными датчиками в течение 20 минут (5 последовательных 4-минутных интервала (INT), как указано). ФИГ.1 показывает, что соединение 1-1 (10 мг/кг) значительно снижает гиперактивность, вызванную PCP, как видно при сравнении с контролем с введением носитель+PCP (p=0,0088, n=8 на группу, t-критерий Стьюдента для независимой выборки). ФИГ.2 показывает, что соединение 1-1 является также эффективным при пероральном способе введения (p=0,0045, n=8 на группу, t-критерий Стьюдента для независимой выборки).
ПРИМЕР 68
СНИЖЕНИЕ PCP-ВЫЗВАННОЙ ГИПЕРАКТИВНОСТИ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 2-1
Было обнаружено, что соединение 2-1 (Пример 2) снижает PCP-вызванную гиперактивность, как показано на фиг.3 и 4. C57BL/6 мужским особям мышей давали либо соединение 2-1, либо носитель внутрибрюшинной инъекцией (ФИГ.3) или пероральным способом введения (ФИГ.4). Через двадцать минут (для внутрибрюшинного способа введения) или сорок минут (для перорального способа введения) животным вводили PCP (5 мг/кг, внутрибрюшинно). Через десять минут мышей помещали в камеры для оценки активности и их двигательную активность в горизонтальном направлении отслеживали инфракрасными датчиками в течение 20 минут (5 последовательных 4-минутных интервала (INT), как указано). ФИГ.3 показывает, что соединение 2-1 (10 мг/кг) значительно снижает гиперактивность, вызванную PCP, как видно при сравнении с контролем с введением носитель+PCP (p=0,0021, n=8 на группу, t-критерий Стьюдента для независимой выборки). ФИГ.4 показывает, что соединение 2-1 также является эффективным при пероральном способе введения (p=0,000005 для дозы 10 мг/кг, n=8 на группу, t-критерий Стьюдента для независимой выборки).
ПРИМЕР 69
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 2-1
Было обнаружено, что соединение 2-1 (Пример 2) снижает условную реакцию избегания (CAR), как показано на фиг.5. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25 реакций избегания на 30 испытаний каждый день. Затем мышам давали либо носитель, либо соединение 2-1 пероральным способом введения за 30 минут до испытаний в течение 30 испытаний в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем не изменяла реакции избегания данных тренированных животных. ФИГ.5 показывает, что соединение 2-1 (12 мг/кг) значительно уменьшает число реакций избегания (p=0,0048, n=7 на группу, двусторонний t-критерий Стьюдента).
ПРИМЕР 70
СНИЖЕНИЕ PCP-ВЫЗВАННОЙ ГИПЕРАКТИВНОСТИ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 11-1
Было обнаружено, что соединение 11-1 (Пример 11) снижает PCP-вызванную гиперактивность, как показано на фиг.6. C57BL/6 мужским особям мышей давали либо соединение 11-1, либо носитель внутрибрюшинной инъекцией. Через пять минут животным вводили PCP (5 мг/кг, внутрибрюшинно). Через десять минут мышей помещали в камеры для оценки активности и их двигательную активность в горизонтальном направлении отслеживали инфракрасными датчиками в течение 20 минут (5 последовательных 4-минутных интервала (INT), как указано). ФИГ.6 показывает, что соединение 11-1 (10 мг/кг) значительно снижает гиперактивность, вызванную PCP, как видно при сравнении с контролем с введением носитель+PCP (p=0,00040, n=8 на группу, t-критерий Стьюдента для независимой выборки).
ПРИМЕР 71
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 34-1
Было обнаружено, что соединение 34-1 (Пример 34) снижает условную реакцию избегания (CAR), как показано на фиг.7. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 25 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.7 показывает, что соединение 34-1 значительно уменьшает число реакций избегания при 10 мг/кг (p=0,0003, n=7 на группу).
ПРИМЕР 72
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 36-1
Было обнаружено, что соединение 36-1 (Пример 36) снижает условную реакцию избегания (CAR), как показано на фиг.8. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 25 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.8 показывает, что соединение 36-1 значительно уменьшает число реакций избегания при 15 мг/кг (p=0,000014, n=5 на группу) и показывает тенденцию, которая не достигает уровня значимости при 5 и 10 мг/кг.
ПРИМЕР 73
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 47-1
Было обнаружено, что соединение 47-1 (Пример 47) снижает условную реакцию избегания (CAR), как показано на фиг.9. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 55 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.9 показывает, что соединение 47-1 значительно уменьшает число реакций избегания при 5 и 10 мг/кг (p=0,0002 и p=3,3 E-10, соответственно, n=8 на группу) и показывает тенденцию, которая не достигает уровня значимости при 2 мг/кг.
ПРИМЕР 74
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 61-1
Было обнаружено, что соединение 61-1 (Пример 61) снижает условную реакцию избегания (CAR), как показано на фиг.10. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 55 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.10 показывает, что соединение 61-1 значительно уменьшает число реакций избегания при 2, 5 и 10 мг/кг (p=0,015, p=0,00008 и p=2,1 E-7, соответственно, n=5 на группу).
ПРИМЕР 75
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 63-1
Было обнаружено, что соединение 63-1 (Пример 63) снижает условную реакцию избегания (CAR), как показано на фиг.11. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 55 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.11 показывает, что соединение 63-1 значительно уменьшает число реакций избегания при 2, 5 и 10 мг/кг (p=0,0099, p=0,00011 и p=6,6 E-16, соответственно, n=6 на группу).
ПРИМЕР 76
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 49-1
Было обнаружено, что соединение 49-1 (Пример 49) снижает условную реакцию избегания (CAR), как показано на фиг.12. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 55 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.12 показывает, что соединение 49-1 значительно уменьшает число реакций избегания при 10 мг/кг (p=5,7 E-9, n=8 на группу).
ПРИМЕР 77
СНИЖЕНИЕ УСЛОВНОЙ РЕАКЦИИ ИЗБЕГАНИЯ ПОСРЕДСТВОМ СОЕДИНЕНИЯ 65-10
Было обнаружено, что соединение 65-10 (Пример 65, таблица 1) снижает условную реакцию избегания (CAR), как показано на фиг.13. C57BL/6 мужских особей тренировали в CAR подходе для предсказания и избегания болевого стимула (электрический удар по лапам), достигая стабильного значения приблизительно 25-28 реакций избегания на 30 испытаний ("тренировочное плато") каждый день. Затем мышам давали либо носитель, либо соединение (за 55 минут до испытания) пероральным способом введения и затем испытывали в 30 испытаниях в CAR подходе. Обработку носителем и обработку соединением осуществляли на тех же животных через день и эффект соединения на снижение реакции избегания анализировали с помощью внутригруппового сравнения (двусторонний t-критерий Стьюдента). Обработка носителем ("носитель") не изменяла реакции избегания данных тренированных животных. ФИГ.13 показывает, что соединение 65-10 значительно уменьшает число реакций избегания при 5 и 10 мг/кг (p=0,0145 и p=0,00011; n=8 на группу).
Должно быть ясно, что, хотя конкретные варианты осуществления описываются в настоящем изобретении с целью иллюстрации, можно осуществлять различные модификации, не выходя за пределы объема и сущности настоящего изобретения. Соответственно, настоящее изобретение не ограничивается, за исключением прилагаемой формулы изобретения.
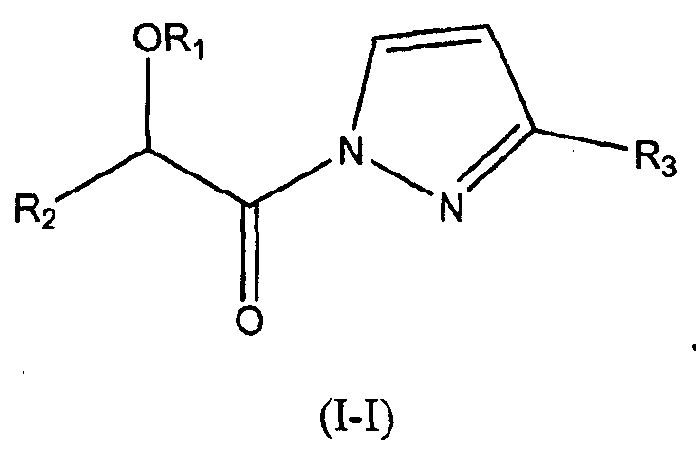
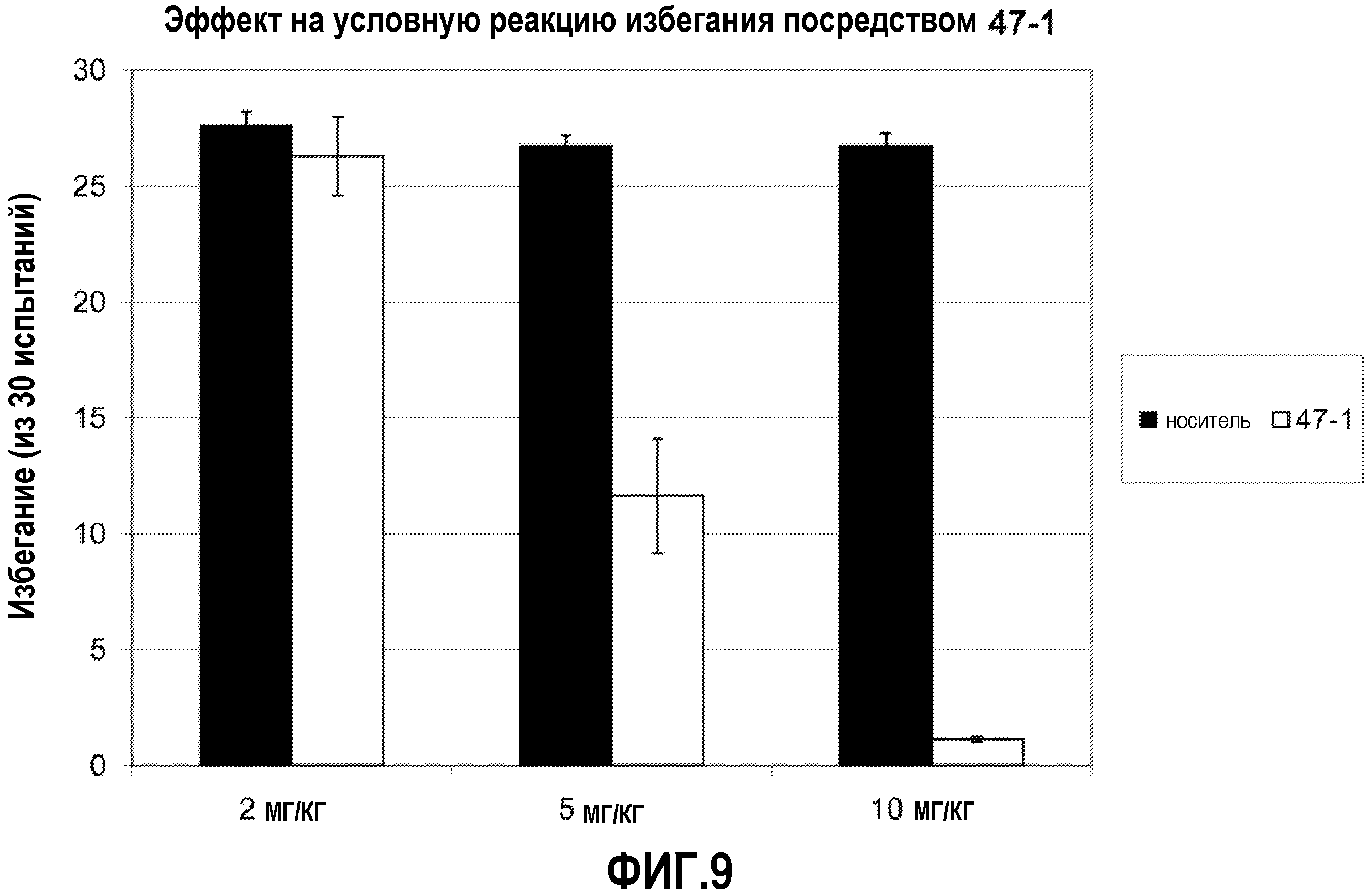
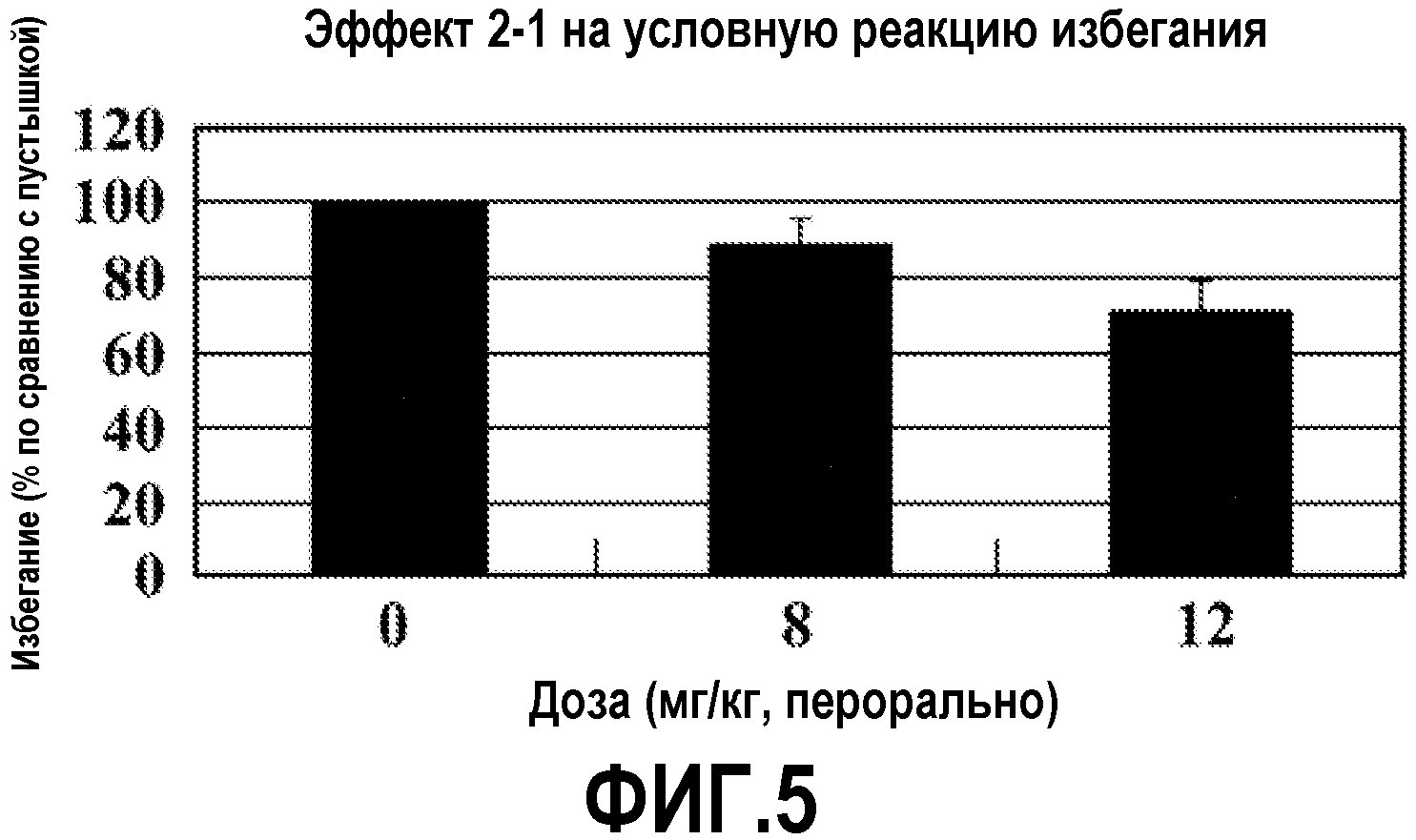
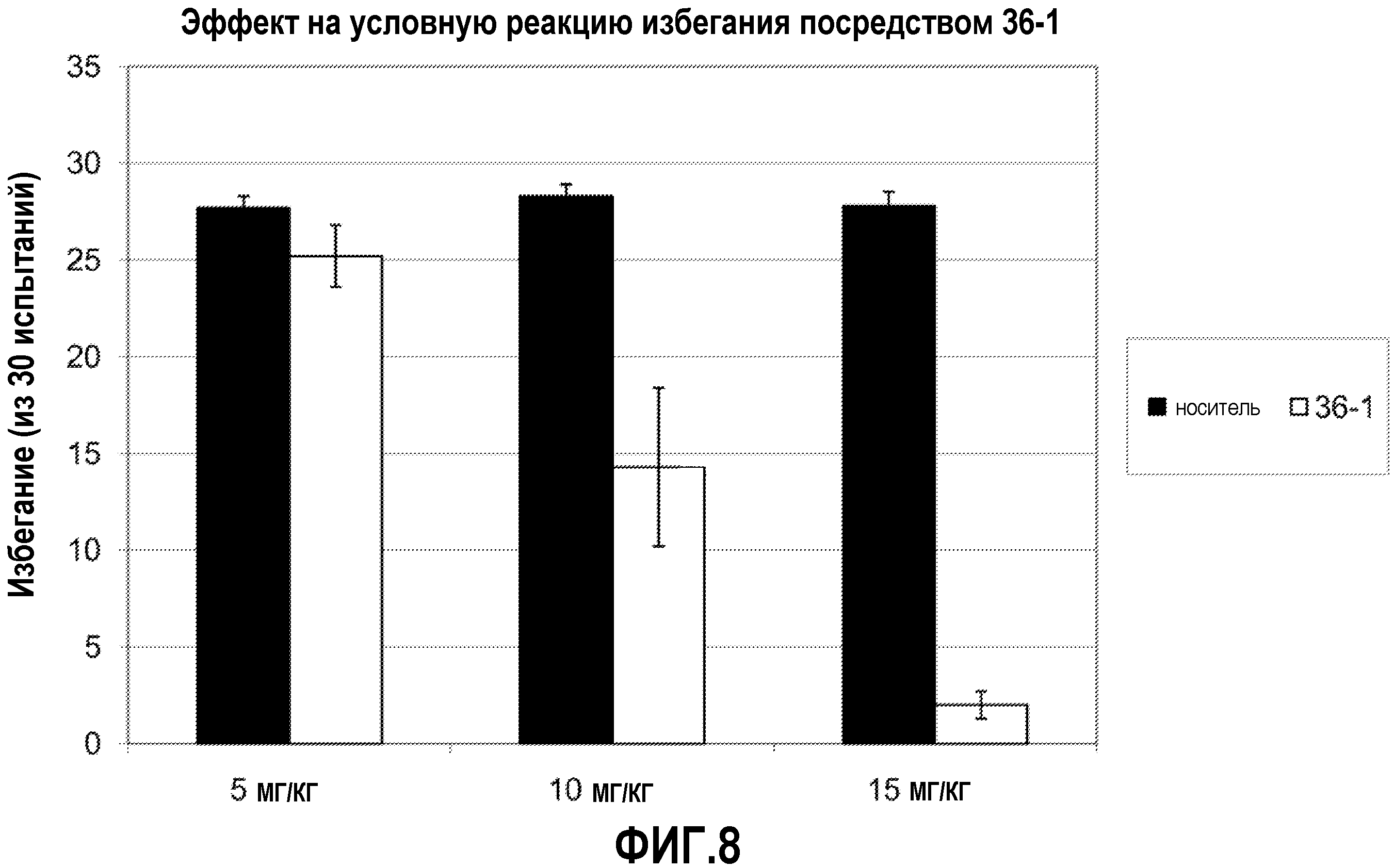
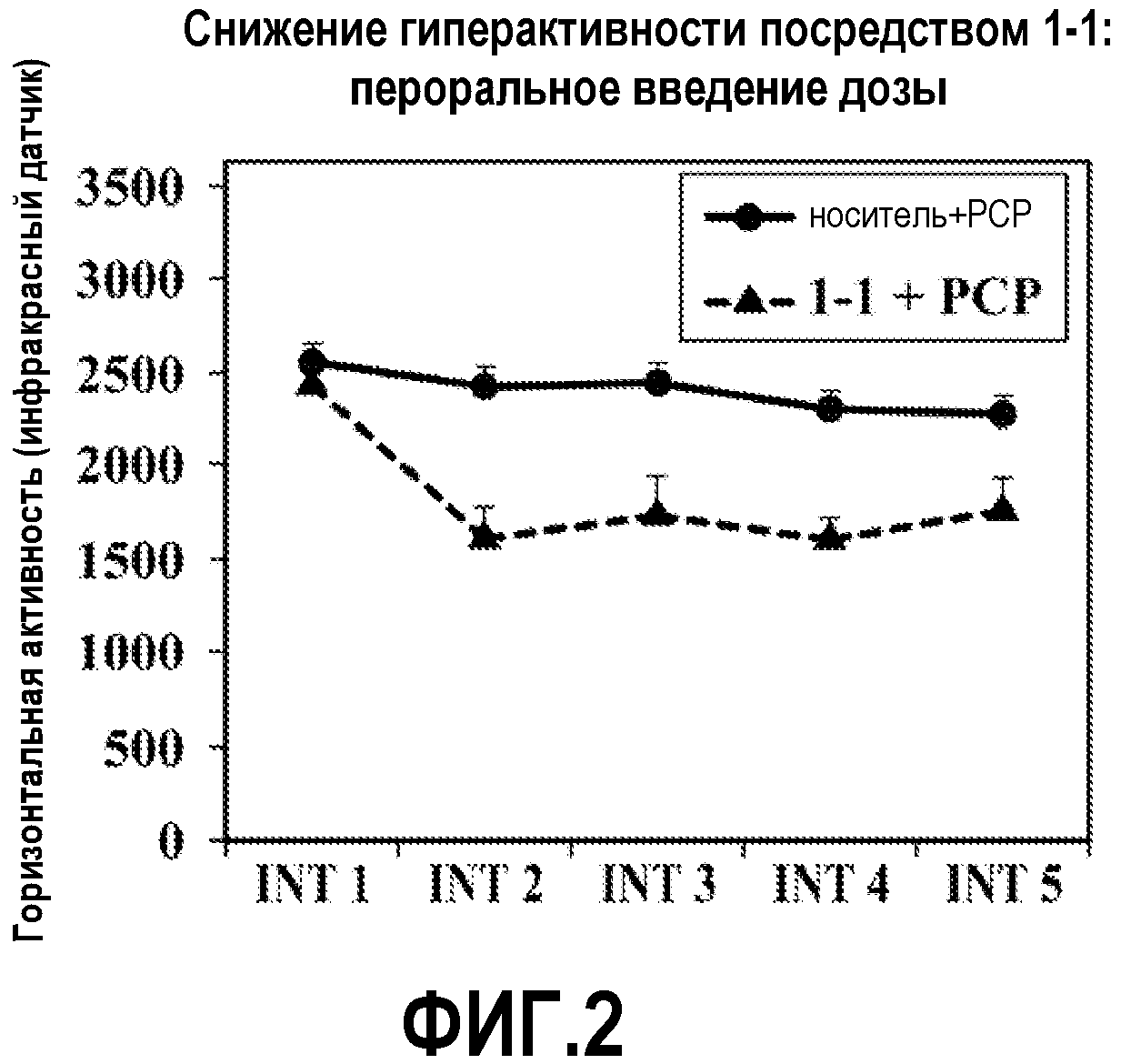
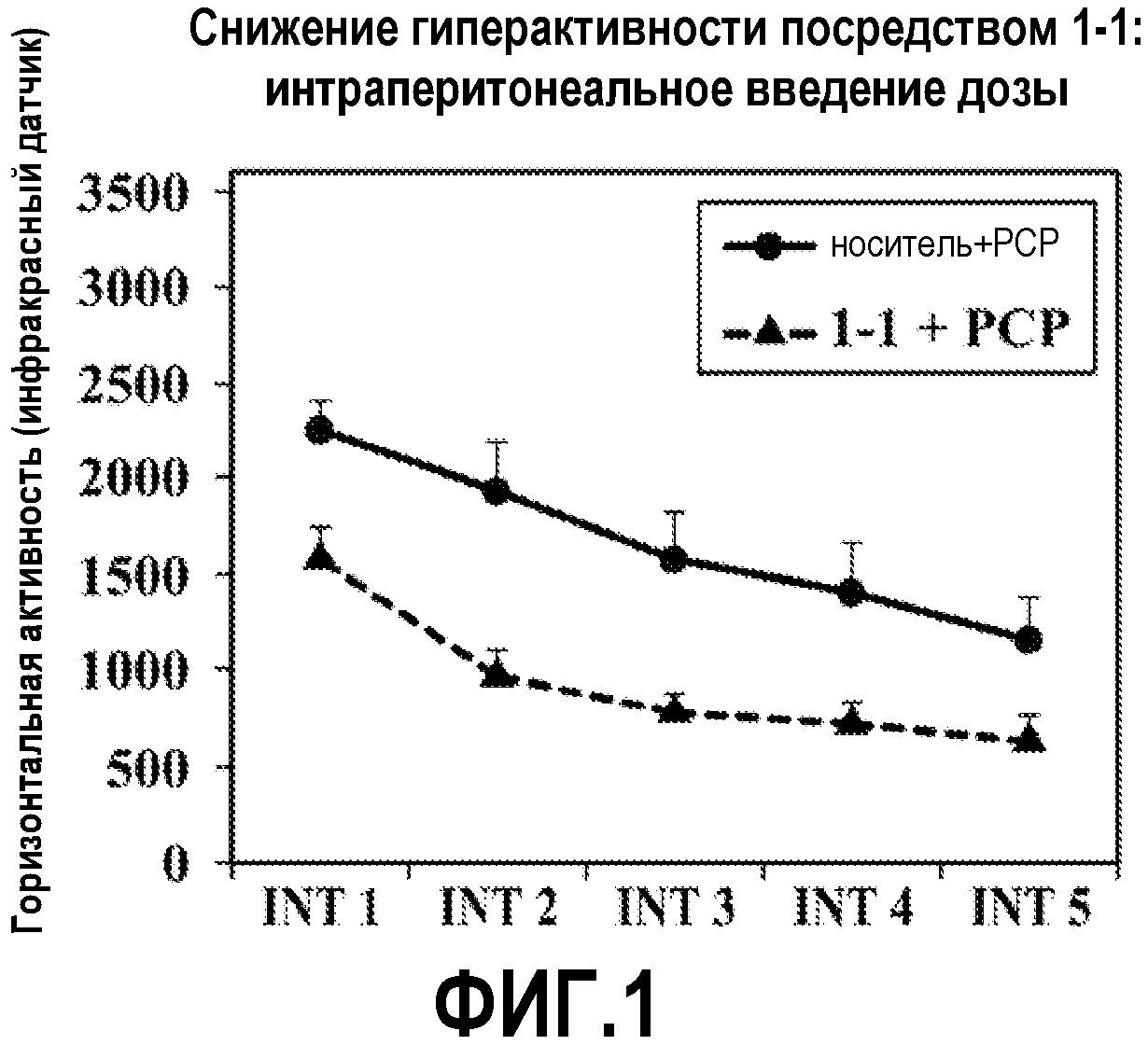
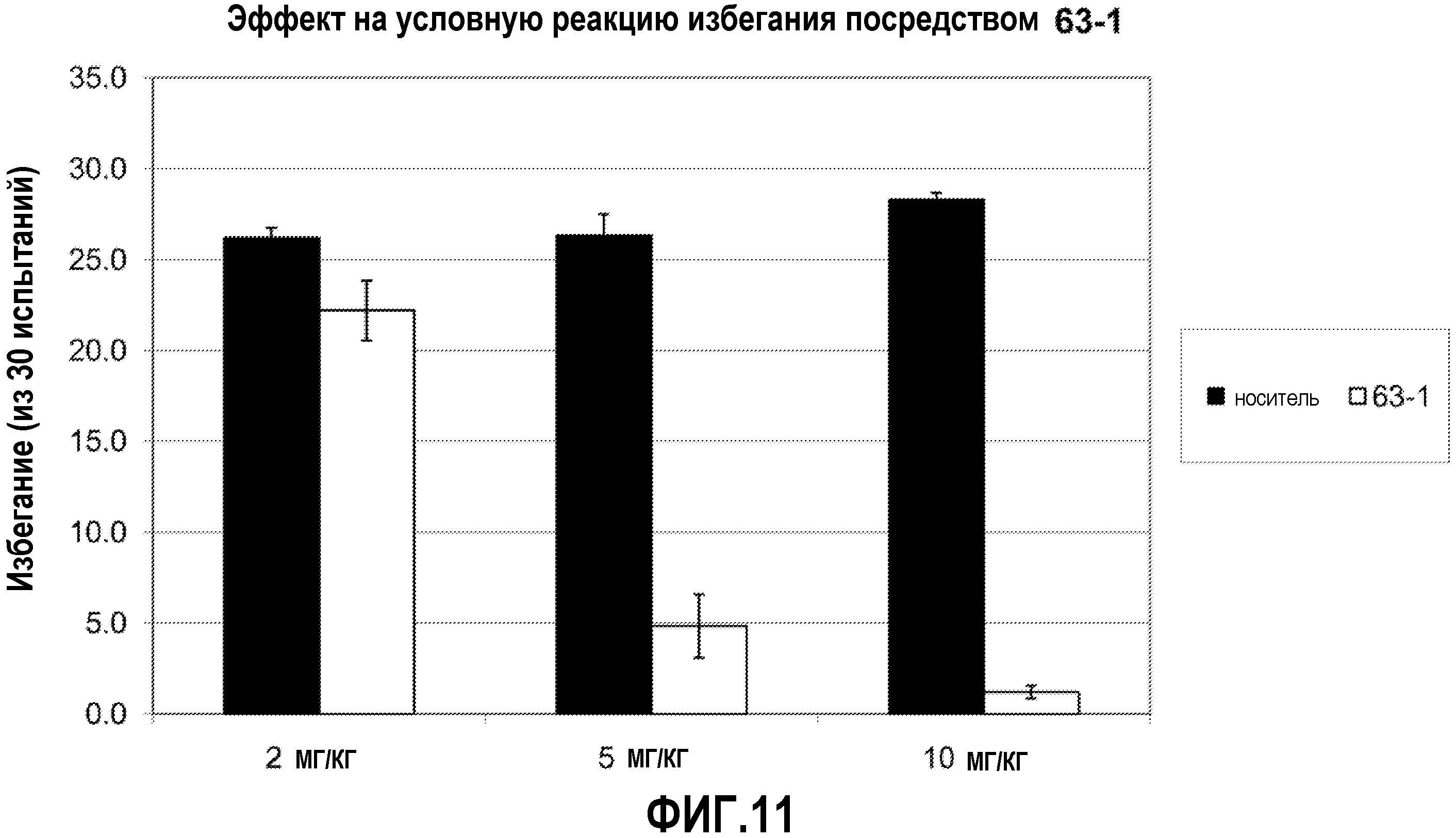
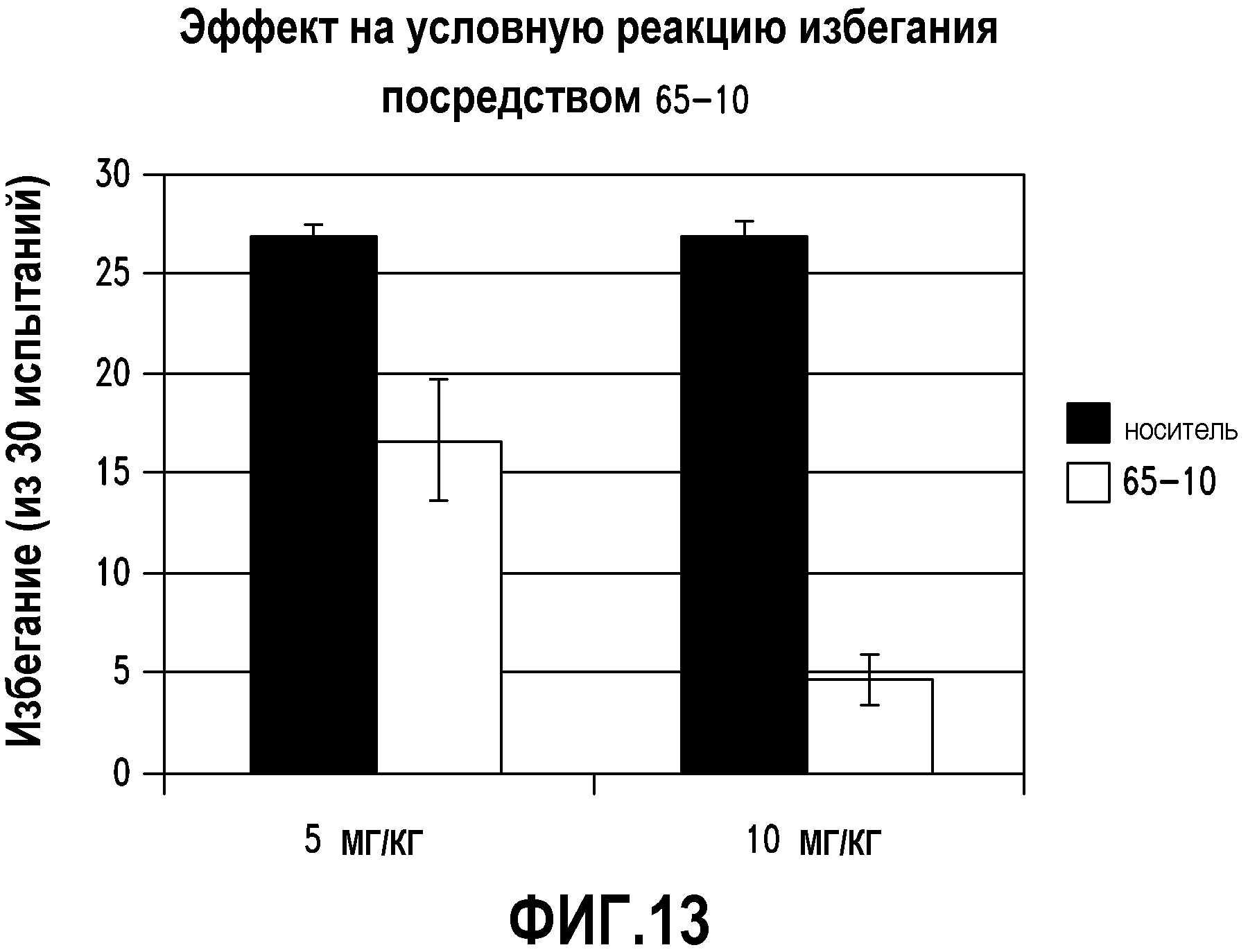
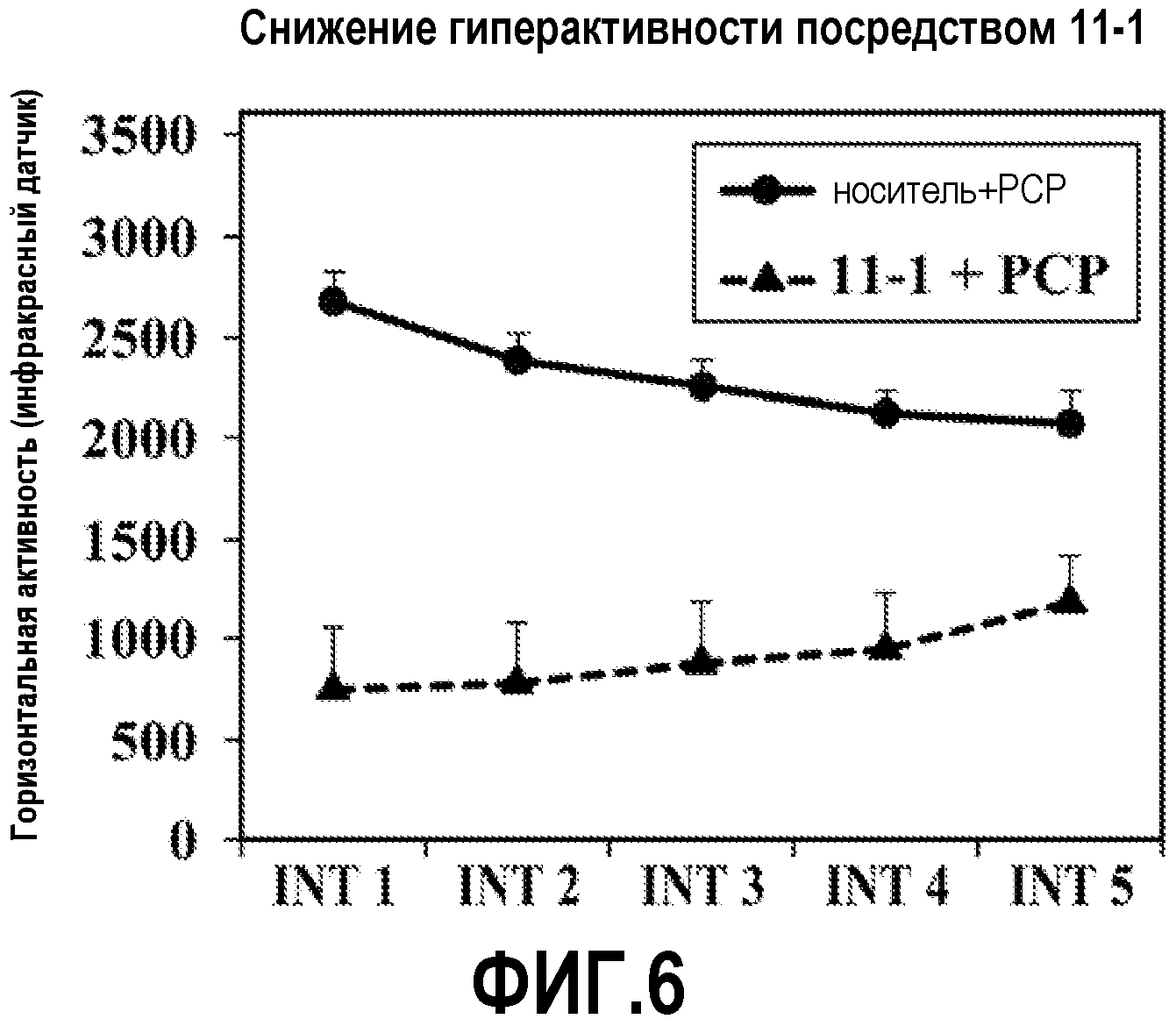
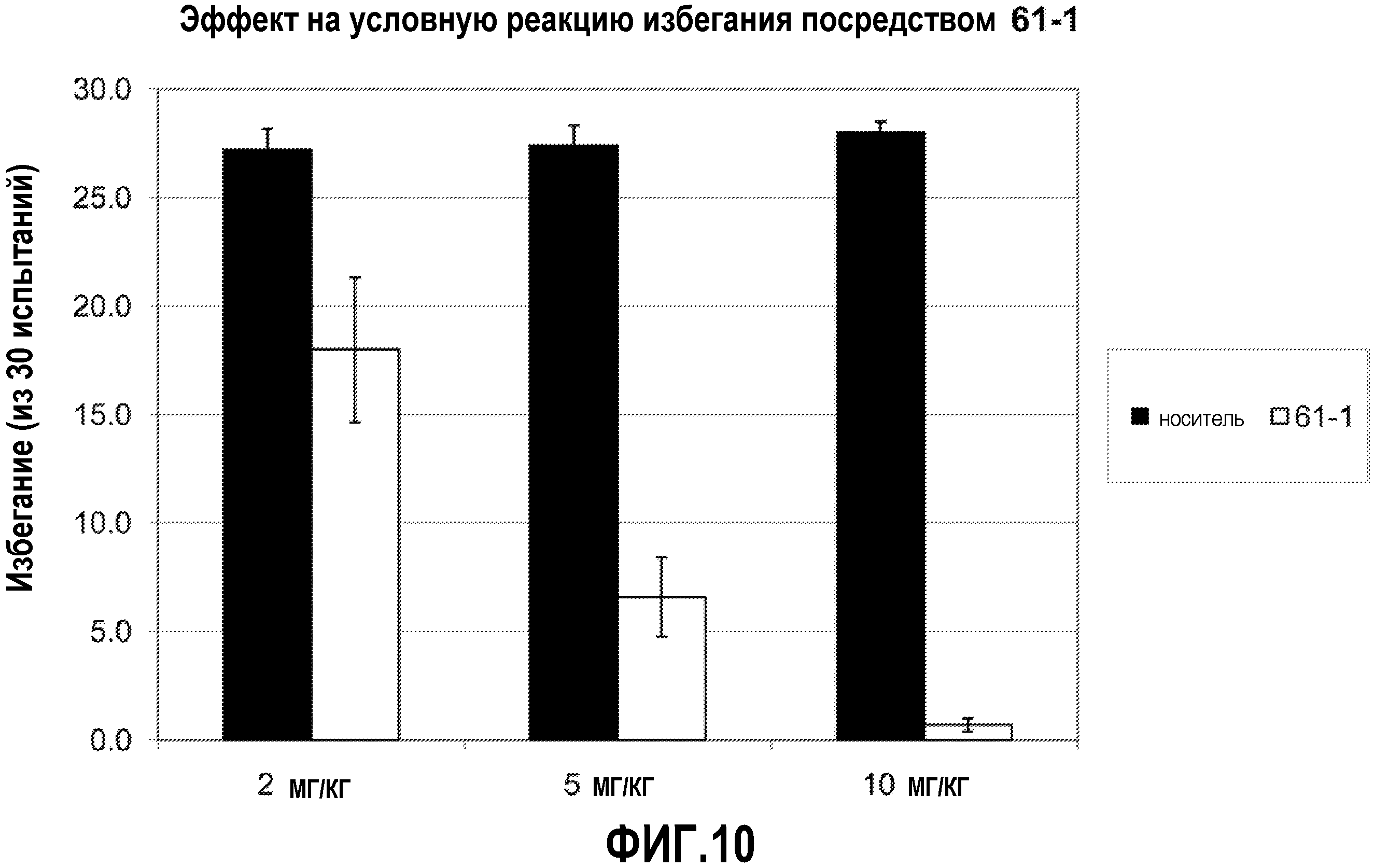
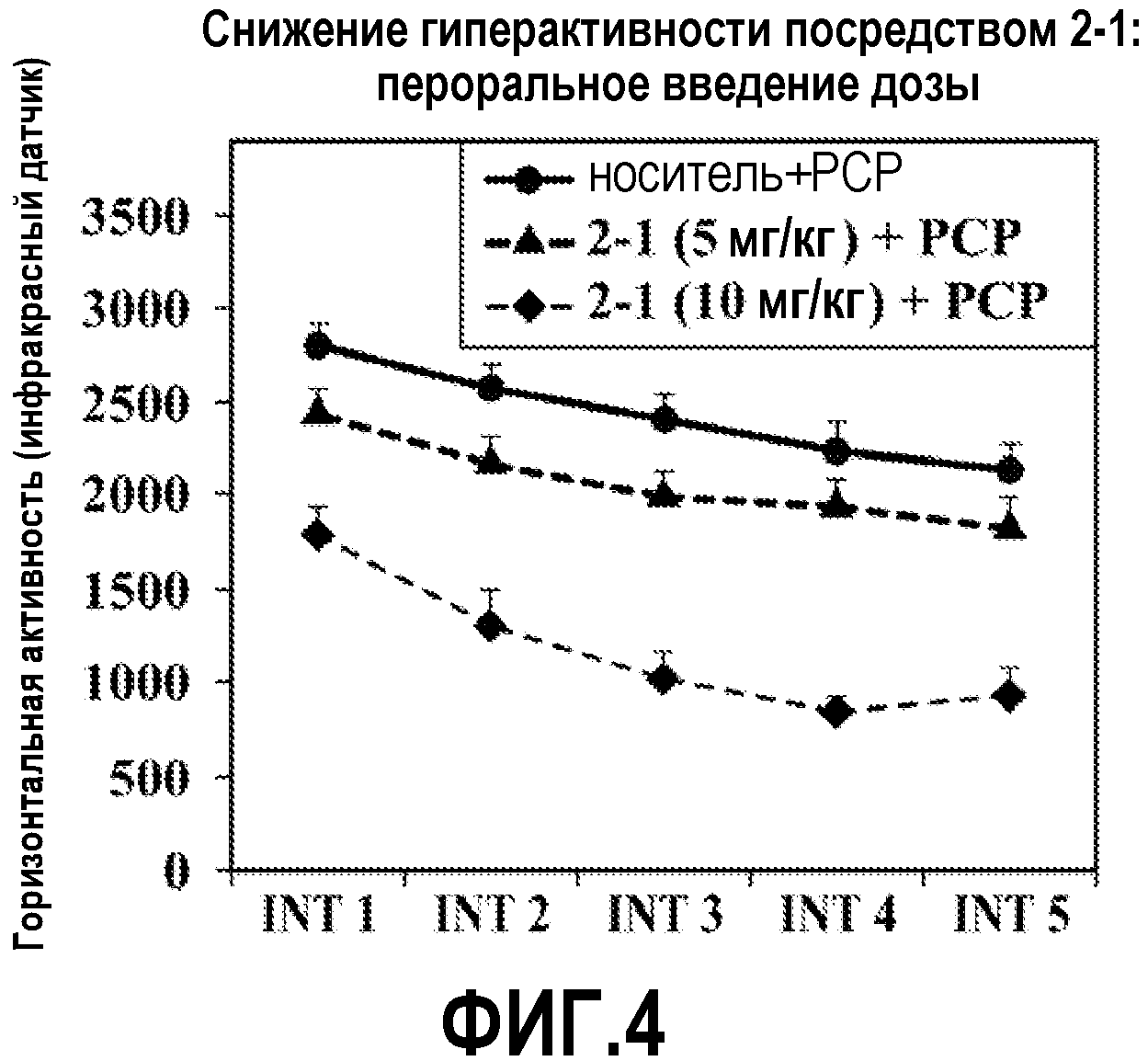
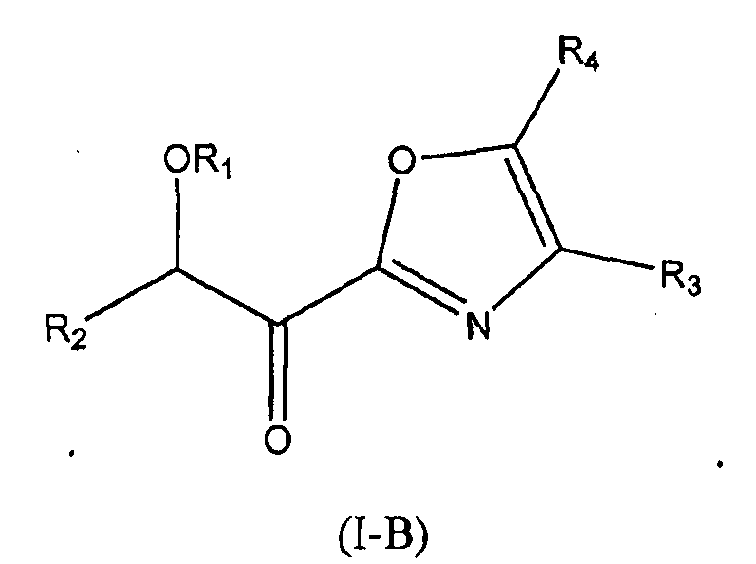
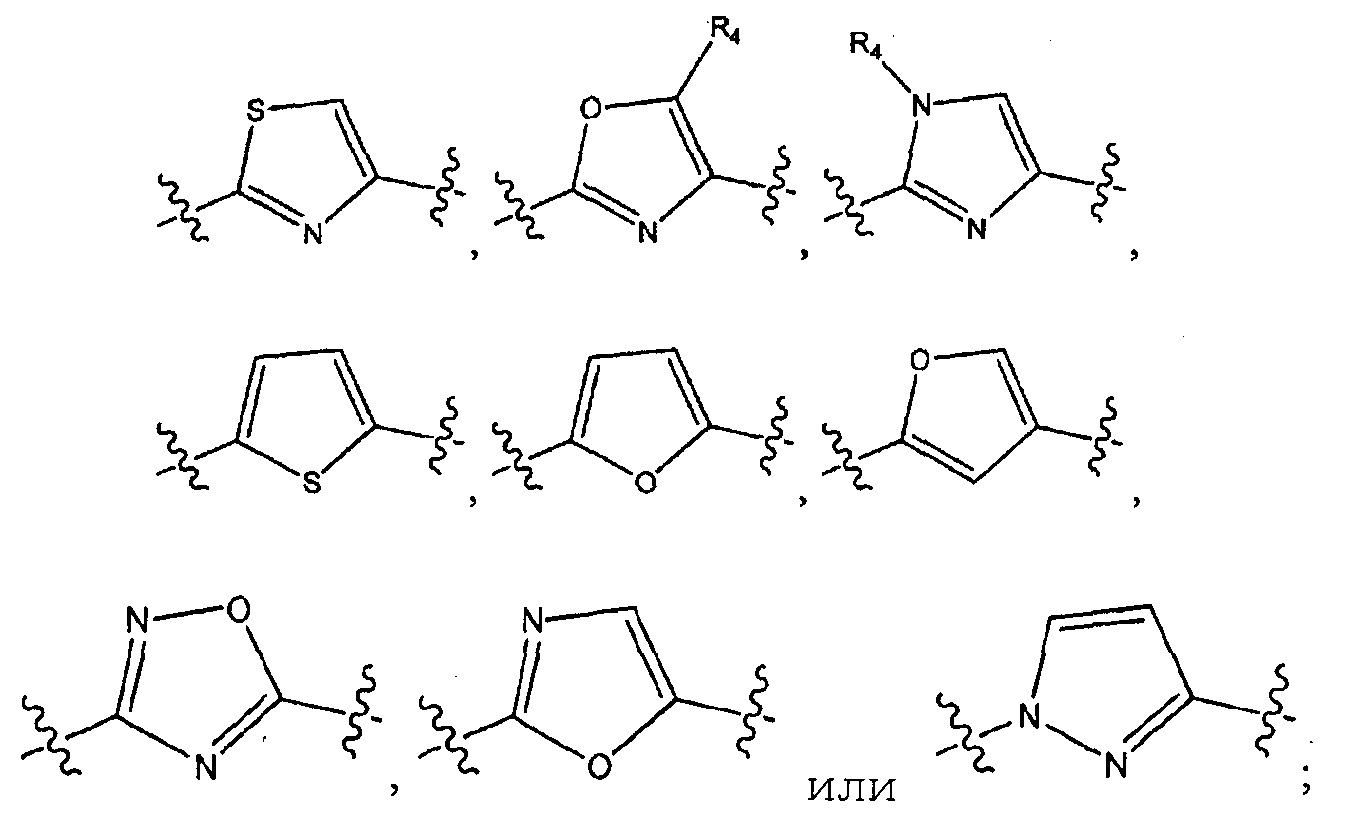
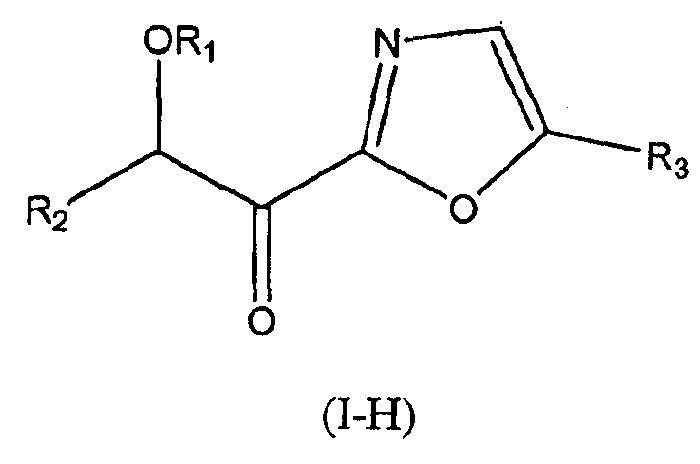
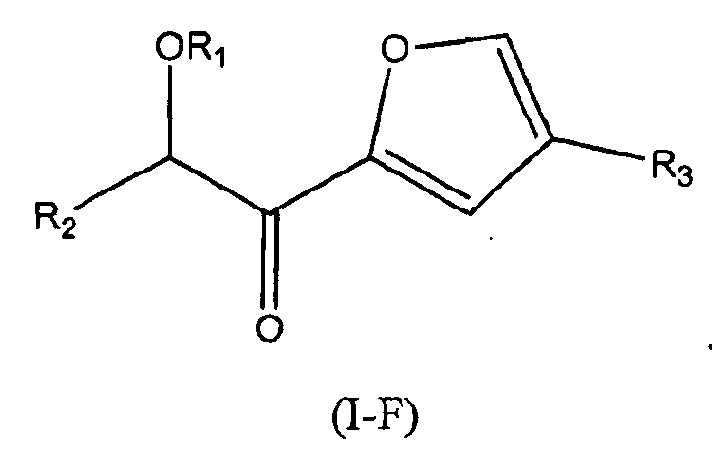
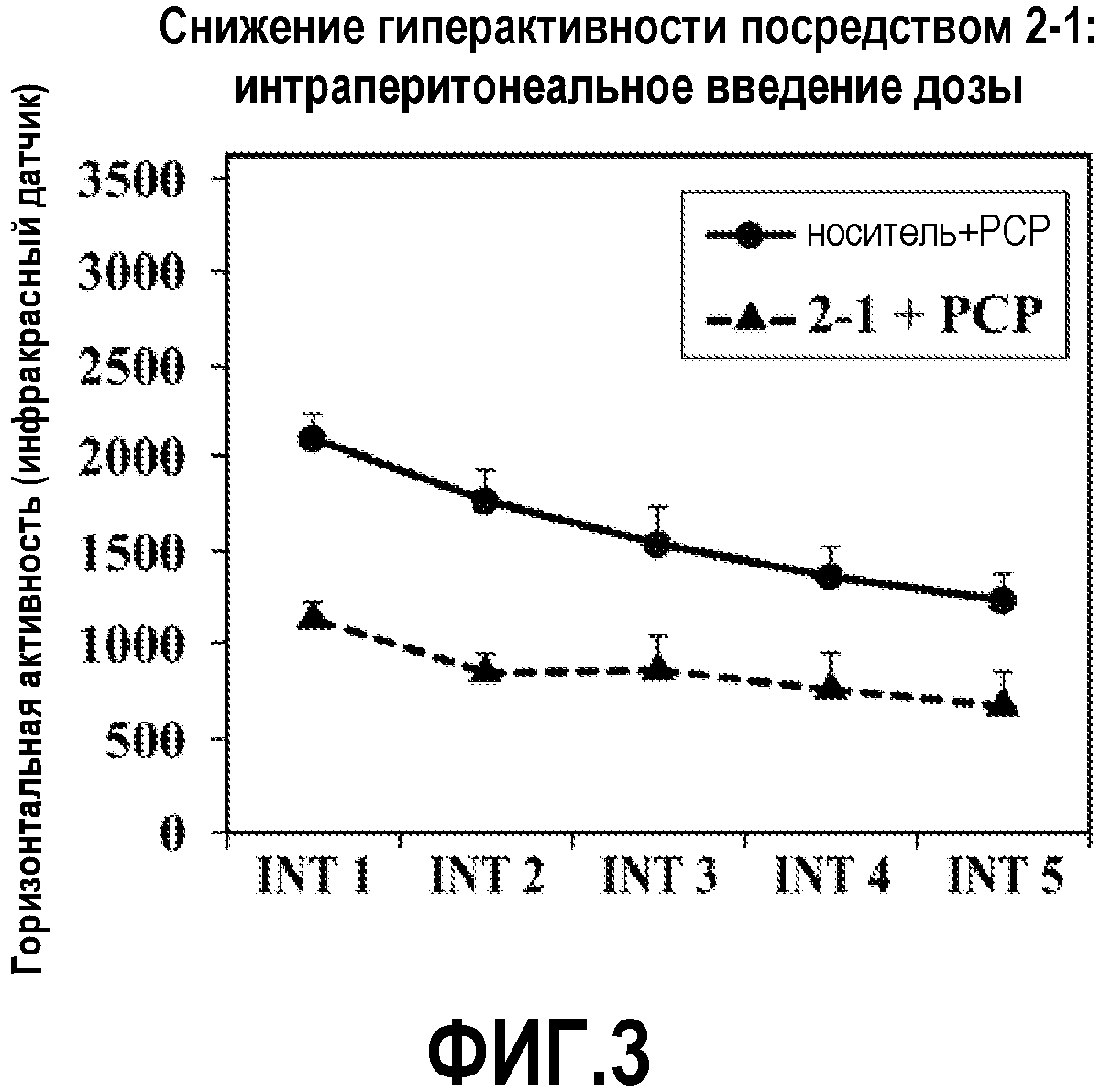
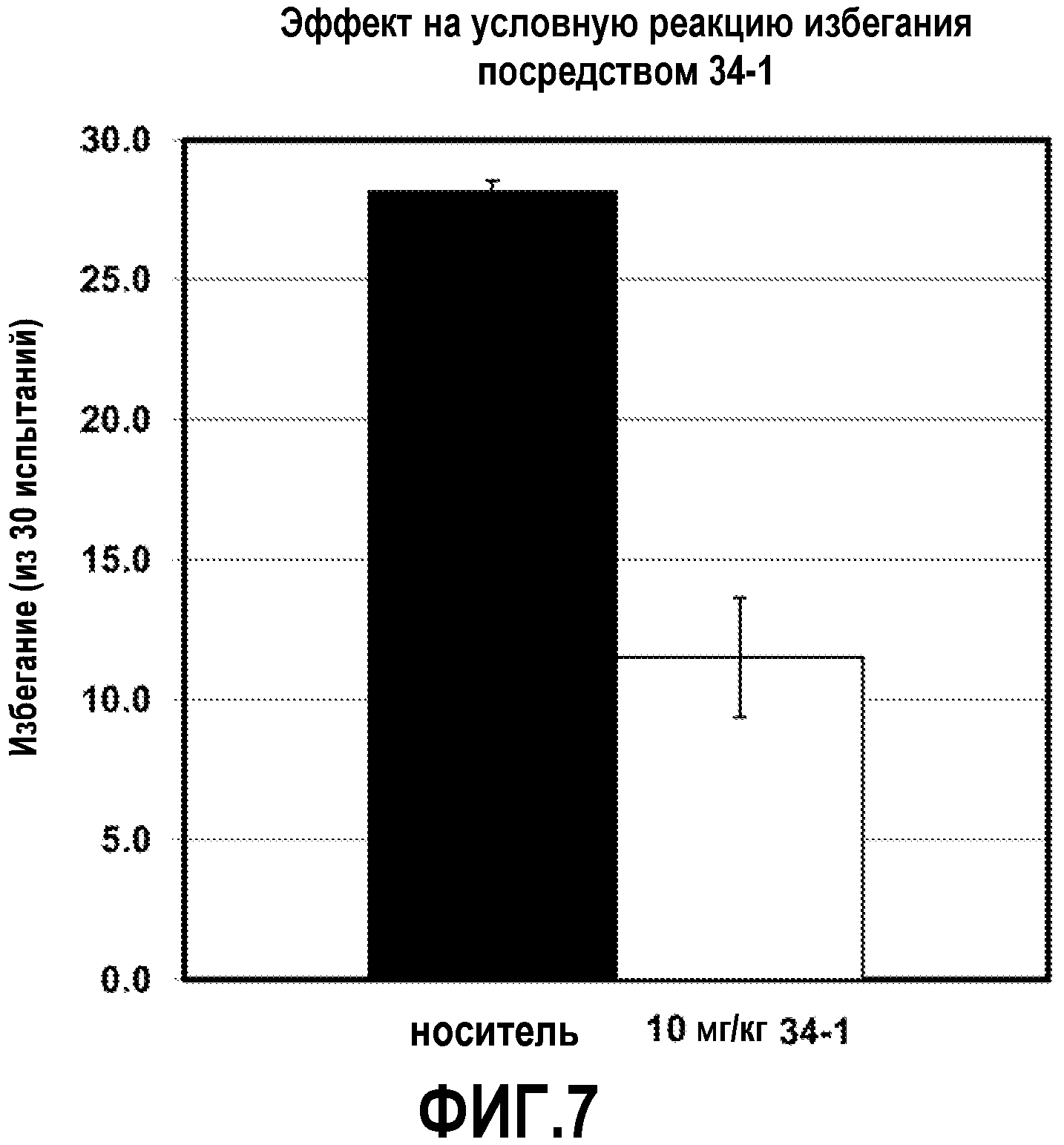
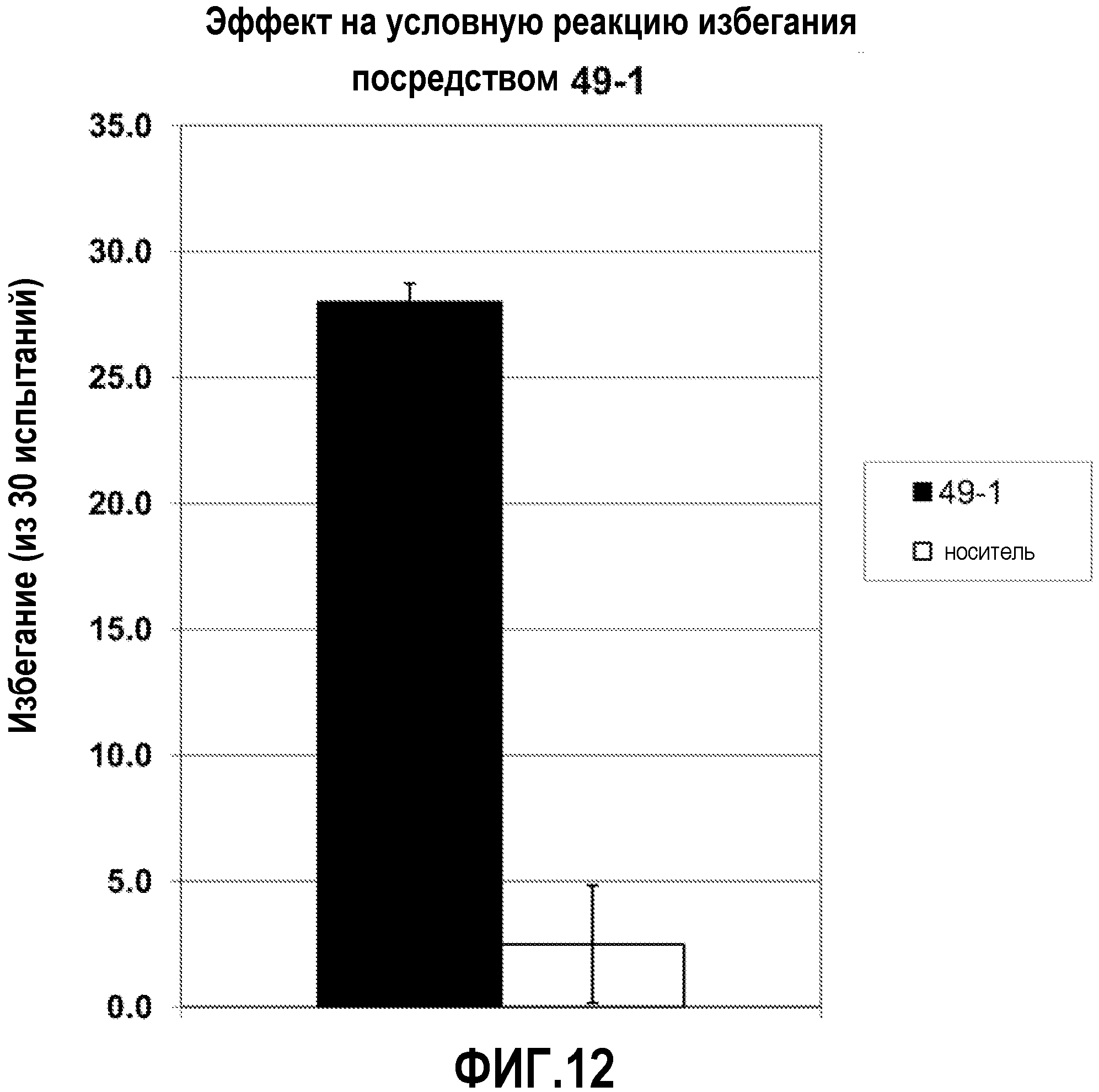
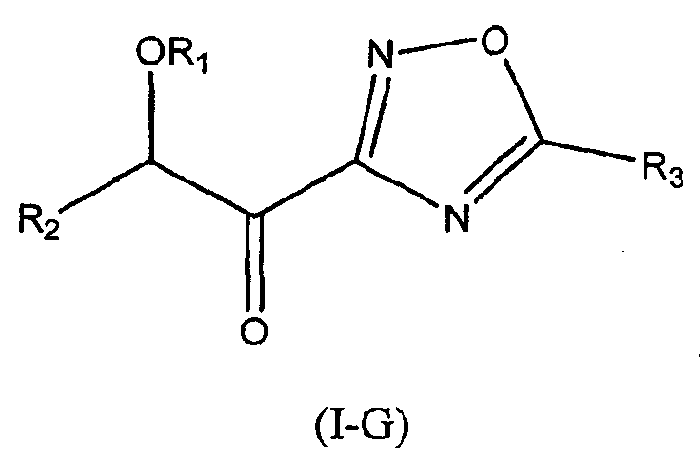
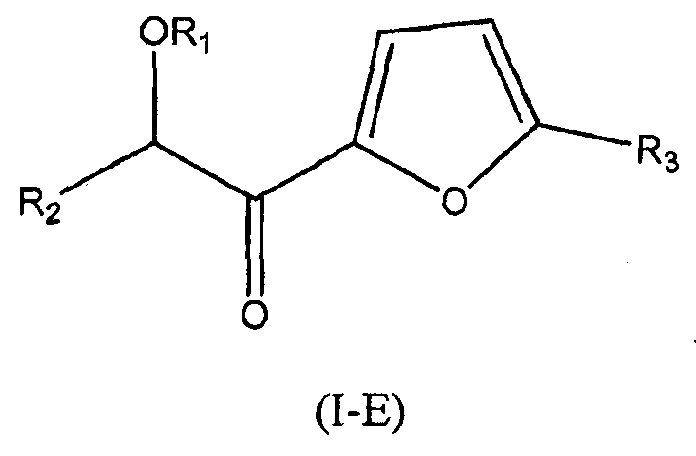
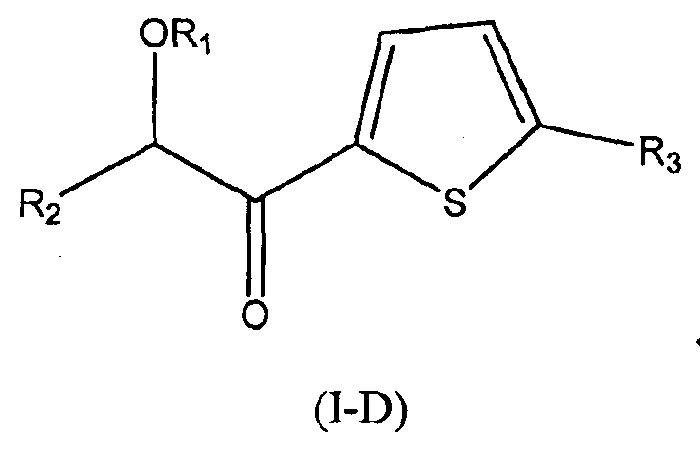
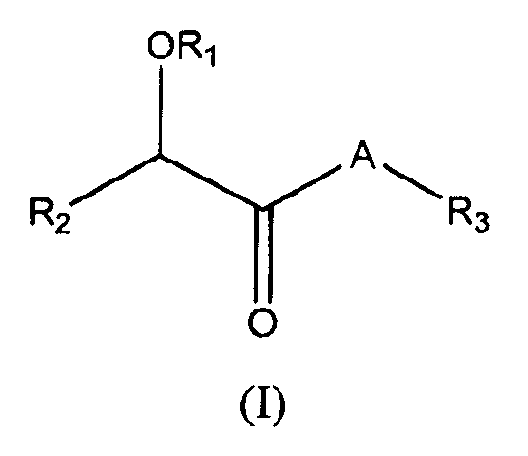
Композиции и способы профилактики и лечения зависимостей
Стабильные противовоспалительные растворы для инъекции
Композиции для ингибирования masp-2-зависимой активации комплемента
Лечение зависимости и расстройства побуждений с применением ингибиторов фдэ7
Способы лечения состояний, связанных с masp-2 зависимой активацией комплемента
Лечение зависимости и расстройств побуждений с применением ингибиторов фдэ7
Композиции и способы ингибирования masp-1, и/или masp-2, и/или masp-3 для лечения различных заболеваний и нарушений
Способы и промежуточные соединения для получения ингибитора pde10
Способы лечения состояний, ассоциированных с masp-2-зависимой активацией комплемента
Композиции для ингибирования masp-2-зависимой активации комплемента
Композиции и способы профилактики и лечения зависимостей
Стабильные противовоспалительные растворы для инъекции
Композиции для ингибирования masp-2-зависимой активации комплемента
Способы и промежуточные соединения для получения ингибитора pde10

