Результат интеллектуальной деятельности: СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ
Вид РИД
Изобретение
Настоящее изобретение относится к лечению аутоиммунных заболеваний. Изобретение относится к средству, такому как гуманизированное моноклональное антитело, которое можно вводить в более высоких дозах, чем ранее описанные дозы. В частности, такое ведение эффективно для пациентов, страдающих заболеванием или имеющих симптомы, для эффективного лечения которых требуются более высокие дозы. Изобретение относится к фармацевтической композиции, содержащей средство, такое как антитело, в эффективной концентрации, а также к применению и способам лечения, при которых используются композиции и лекарственные препараты, содержащие указанное средство.
Аутоиммунная реакция представляет собой неспособность организма распознавать собственные составные части (вплоть до субмолекулярных уровней) как “свое”, что приводит к иммунному ответу против собственных клеток и тканей. Любое заболевание, которое является следствием такого патологического иммунного ответа, называют аутоиммунным заболеванием. Аутоиммунные заболевания включают рассеянный склероз (MS), ревматоидный артрит (RA), псориаз, псориатический артрит, язвенный колит, болезнь Крона, миастению гравис (MG), аутоиммунный полигландулярный синдром типа II (APS-II), тиреоидит Хашимото (НТ), диабет 1 типа (T1D), системную красную волчанку (SLE) и аутоиммунный лимфопролиферативный синдром (ALS).
Аутоиммунное заболевание возникает, когда Т-клетки распознают “собственные” молекулы, т.е. молекулы, продуцируемые клетками хозяина, и взаимодействуют с ними. Активация "аутореактивных" T-клеток путем презентации аутоантигенов, процессированных антиген-презентирующими клетками (APC), приводит к увеличению их числа и миграции в определенные ткани, где они вызывают воспаление и разрушение ткани.
В норме T-клетки толерантны к аутологичной ткани и реагируют только на презентацию гетерологичных структур. Центральная толерантность и периферическая толерантность включает два механизма, посредством которых иммунная система препятствует индукции разрушительных функций аутореактивными T-клетками. Центральная толерантность опосредуется негативной селекцией. Этот процесс включает элиминацию через клональную делецию аутореактивных T-клеток в процессе онтогенного развития тимуса.
Периферическая толерантность представляет собой резерв, доступный в том случае, если центральная толерантность является недостаточной и аутореактивные клетки выходят из тимуса. Этот механизм толерантности действует постоянно на протяжении всей жизни, сохраняя иммунное безразличие (анергии), периферическую делецию и/или активную супрессию в отношении аутореактивных клеток.
Регуляторные T-клетки (Treg, ранее называемые "супрессорными клетками") в качестве части активной супрессии поддерживают периферическую толерантность и регулируют аутоиммунные реакции (Suri-Payer et al., J. Immunol. 157: 1799-1805 (1996); Asano et al., J. Exp. Med. 184: 387-396 (1996); Bonomo et al., J. Immunol. 154: 6602-6611 (1995); Willerford et al., Immunity 3: 521-530 (1995); Takahashi et al., Int. Immunol. 10: 1969-1980 (1998); Salomon et al., Immunity 12: 431-440 (2000); Read et al., J. Exp. Med. 192: 295-302 (2000). Как правило, регуляторные T-клетки ингибируют активацию и/или функцию T-хелперов 1 типа (TH1) и эффекторных клеток TH2. Нарушение регуляции частоты или функционирования клеток Treg может привести к истощающим аутоиммунным заболеваниям (Baecher-Allan et al., Immunol. Review 212: 203-216 (2006); Shevach, Annu. Rev. Immunol. 18: 423-449 (2000); Salomon et al., Immunity 12: 431-440 (2000); Sakaguchi et al., Immunol. Rev. 182: 18-32 (2001)).
Охарактеризовано несколько подгрупп регуляторных T-клеток. Семейство Treg состоит из двух основных подгрупп: появляющиеся естественным образом, например CD4+CD25+ Treg, и периферически индуцированные, Tr1- и Th3-Treg. Более того, у человека и грызунов были описаны NKTreg и CD8+ Treg (Fehervari et al., J. Clin. Investigation 114: 1209-1217 (2004)).
Происходящие из тимуса клетки Treg (встречающиеся в природе CD4+CD25+Treg) являются главными регуляторными клетками, вовлеченными в регуляцию аутоиммунных реакций или патогенных иммунных ответов.
i) они представляют собой CD4+ T-клетки и составляют 5-10% от периферических CD4+ T-клеток
ii) они созревают в тимусе
iii) они, главным образом, характеризуются объединенной экспрессией рецептора IL-2 (CD25), низкомолекулярной изоформы молекулы CD45, CD152 (CTLA-4) и фактора транскрипции FoxP3.
Роль Treg наилучшим образом можно продемонстрировать на экспериментах, касающихся реконструкции иммунодефицитных “голых” мышей с клетками CD4+ и истощением клеток CD25+. У “голых” мышей CD4+CD25- с восстановлением развиваются различные орган-специфические аутоиммунные заболевания, такие как гастрит, оофорит, орхит и тиреоидит (Suri-Payeret et al.; J. Immunol. 160: 1212-1218 (1998)).
Введение подгруппы CD4+CD25+ в эксперименты по реконструкции с “голыми” мышами препятствует возникновению этих заболеваний (Sakaguchi et al., J Immunol. 155: 1151-1164 (1995)). Защитное значение клеток CD4+CD25+ против органоспецифических аутоиммунных реакций также было показано на нескольких других моделях аутоиммунных реакций (например, модели аутоиммунного гастрита, простатита, оофорита, гломерулонефрита, эпидимита и тириоидита), вызванного неонатальной тимэктомией, проведенной через 3 суток после рождения (d3Tx), или воспалительного заболевания кишечника, вызванного реконструкцией у мышей SCID T-клетками CD45RBhigh, CD4+CD25-. Введение антитела против CD25 мышам in vivo также индуцирует органоспецифическое аутоиммунное заболевание.
Открытие важности регулятора транскрипции FoxP3 для реализации функции регуляторных T-клеток CD4+CD25+ у мышей и предшествующие наблюдения за пациентами с синдромом IPEX (нарушение иммунной регуляции, полиэндокринопатия, энтеропатия и X-сцепленное наследование), у которых тяжелое воспалительное заболевание, сходное с заболеванием, наблюдаемым у мышей с дефицитом регуляторных клеток CD4+CD25+ ("scurfy"-синдром), связано с мутацией FoxP3, выявило прямую связь между аутоиммунной моделью животных, регуляторными T-клетками мыши и аутоиммунным заболеванием человека (Sakaguchi et al., J. Immunol. 155: 1151-1164 (1995)).
Фармацевтический механизм регуляторных T-клеток не полностью ясен. Treg CD4+CD25+ ингибируют поликлональную и антиген-специфическую активацию T-клеток. Супрессия опосредуется зависимым от клеточных контактов механизмом, который требует активации Treg CD4+CD25+ через TCR, но Treg не демонстрируют пролиферативного ответа при активации или стимуляции TCR митогенными антителами (анергическими) (Shevach, Nature Rev. Immunol 2: 389 (2002). После стимуляции они являются компетентными для супрессии независимым от антигена образом ответа CD4+ T-клеток и CD8+ T-клеток, а также ингибирования активации B-клеток и их клональной экспансии.
Существуют дополнительные данные, указывающие на то, что супрессорная активность CD4+CD25+ Treg также частично основана на противовоспалительных цитокинах, таких как TGF-β (Kingsley et al., J. Immunol. 168: 1080 (2002); Nakamura et al., J. Exp. Med. 194: 629-644 (2001)). Функциональное значение секреции TGF-β, более того, подтверждается данными о том, что у мышей с дефицитом TGF-β развивается аутоиммунное заболевание и что введение нейтрализующих антител против TGF-β снимает in vivo профилактику аутоиммунных реакций или индуцирующую толерантность активность CD4+ T-клеток в некоторых моделях.
В подгруппе CD4+ T-клеток могут существовать по меньшей мере еще 2 различных типа клеток с супрессивной функцией, индукция которых происходит после воздействия специфического экзогенного антигена (называемые "адаптивными или индуцибельными регуляторными T-клетками"): регуляторные T-клетки 1 типа (Tr1) и клетки Th3. Эти типы клеток, по-видимому, отличаются от CD4+CD25+ Treg характером продукции цитокинов. Однако взаимосвязь между этими различными типами неясна и их способы действия не перекрываются.
Tr1-клетки индуцируются повторной стимуляцией TCR в присутствии IL-10, и было показано, что они, главным образом, подавляют иммунные ответы за счет получения высоких уровней IL-10 совместно с умеренными количествами TGF-β (Chen et al., J. Immunol. 171: 733-744 (2003)).
Th3-клетки (идентифицированные на модели EAE после пероральной доставки антигена) продуцируют высокие количества TGF-β и различные количества IL-4 и IL-10. Было показано, что IL-4 сам по себе является основным фактором для дифференцировки Th3-клеток, в противоположность Tr1-клеткам, которые дифференцируются при помощи IL-10 (Chen et al., Science 265:1237-1240 (1994)).
Супрессия функции T-клеток с использованием иммуносупрессивных лекарственных средств является основной терапевтической стратегией, которую успешно использовали для лечения аутоиммунных заболеваний. Однако эти лекарственные средства индуцируют системную иммуносупрессию из-за плохой селективности, что приводит к ингибированию не только вредных функций иммунной системы, но также и полезных. Вследствие этого могут возникнуть различные типы риска, такие как риск инфекции, злокачественной опухоли и токсичности лекарственного средства.
Средства, препятствующие функции T-клеток, представляют собой основу терапии различных аутоиммунных заболеваний.
До настоящего времени было показано, что подход с использованием средств, нацеленных на активацию регуляторных T-клеток, для терапии аутоиммунных заболеваний, является крайне затруднительным. Активация Treg через TCR с использованием антитела-агониста против CD3 OKT-3 (Abramowicz et al., N Engl. J Med. 1992 Sep 3; 327(10):736) или через костимуляторную молекулу CD28 с использованием антитела-суперагониста против CD28 TGN1412 приводит к полному истощению популяции регуляторных T-клеток, а также других традиционных T-клеток и к системной индукции и высвобождению избыточных количеств провоспалительных цитокинов, включая IFN-γ, TNF-α, IL-1 и IL-2, что приводит к клинически выраженному синдрому высвобождения цитокинов (CRS) у человека (Suntharalingam et al., N Engl. J Med. 2006 Sep 7; 355(10):1018-28).
После первых двух или трех инъекций по 5 мг моноклонального антитела OKT3 у большинства пациентов развивается синдром высвобождения цитокинов с высокими уровнями фактора некроза опухоли-альфа, интерлейкина-2 и гамма-интерферона, появляющимися в кровотоке в течение 1-2 ч у реципиентов трансплантата почки (Abramowicz et al., Transplantation. 1989 Apr; 47(4):606-8). Это приводит к узкому терапевтическому окну, которое ограничивает использование этого антитела для лечения аутоиммунного заболевания.
Лечение общей дозой 5-10 мг TGN1412 (0,1 мг антитела против CD28 на килограмм массы тела) приводит к системному воспалительному ответу с полиорганной недостаточностью в течение 90 минут после введения однократной внутривенной дозы TGN1412 (Suntharalingam et al., N Engl. J Med. 2006 Sep 7; 355(10):1018-28).
Общеизвестно, что CD4 T-клетки играют основную роль в инициации и поддержании аутоиммунных реакций. Таким образом, было предложено использовать mAb против поверхностных молекул CD4 T-клеток, и, в частности, mAb против CD4, в качестве иммуносупрессивных средств. Несмотря на то, что множество клинических испытаний подтвердило потенциальный интерес этого подхода, они также выявили несколько трудностей, которые необходимо устранить, чтобы сделать mAb против CD4 более подходящими для применения в рутинной клинической практике.
Было предложено несколько различных механизмов действия mAb против CD4, включая: (1) антагонизм взаимодействий CD4-MHC II, приводящий к ингибированию активации T-клеток, (2) модулирование рецептора CD4, определяемое по снижению экспрессии на клеточной поверхности CD4, (3) частичную передачу сигнала через рецептор CD4 в отсутствие связывания T-клеточного рецептора, которая может подавлять последующую активацию T-клеток и инициировать апоптотическую гибель CD4 T-клеток, (4) Fc-опосредуемую комплемент-зависимую цитотоксичность (CDC) или антитело-зависимую клеточную цитотоксичность (ADCC), приводящие к истощению CD4 T-клеток, и (5) стимуляцию регуляторных T-клеток.
Fc-опосредуемая комплемент-зависимая цитотоксичность (CDC) или антитело-зависимая клеточная цитотоксичность (ADCC), приводящие к истощению CD4 T-клеток, представляют собой главные наблюдаемые механизмы и они, главным образом, показаны для антител подкласса IgG1. Только нескольким антителам против CD4 свойственны другие механизмы, таким как TRX-1, TNX-355, IDEC-151, OKTcdr4A, причем только TRX-1 представляет собой IgG1 (Schulze-Koops et al., J Rheumatol. 25(11): 2065-76 (1998); Mason et al., J Rheumatol. 29(2): 220-9 (2002); Choy et al., Rheumatology 39(10): 1139-46 (2000); Herzyk et al., Infect Immun. 69(2): 1032-43 (2001); Kon et al., Eur Respir J. 18(1): 45-52 (2001); Mourad et al., Transplantation 65(5): 632-41 (1998); Skov et al., Arch Dermatol. 139(11): 1433-9 (2003); Jabado et al., J Immunol. 158(1): 94-103 (1997)).
В случае нескольких антител против CD4 (Mason et al., J. Rheumatol. 29 (2): 220-229 (2002); Kon et al., Eur. Respir J. 18(1):45-52 (2001)) и HuMax-CD4 (Skov et al., Arch Dermatol. 139(11): 1433-1439 (2003), Choy et al., Rheumatology 41 (10): 1142-8 (2002)) было отмечено дозозависимое истощение CD4+ T-клеток при "высоких" дозах (несколько циклов с дозировками >100 мг) и временная секвестрация (кратковременное истощение) при "более низких" дозах (несколько циклов при дозировках >10 мг). Несмотря на их активность в отношении истощения, mAb против CD4 не обеспечивают клиническую эффективность и постоянную активность при исследуемых аутоиммунных заболеваниях, например ревматоидном артрите (Strand et al., Nature Reviews 6: 75-92 (2007)). Более того истощение CD4+ T-клеток, главным образом, рассматривают как сценарий, который может привести к тяжелой иммуносупрессии.
Антитело B-F5 (IgG1 мыши против CD4 человека) тестировали в отношении различных аутоиммунных заболеваний.
Антитело мыши B-F5 использовали для лечения небольшого количества пациентов с тяжелым псориазом и были описаны некоторые положительные эффекты (Robinet et al., Eur J. Dermatol 1996: 6: 141-6, и Robinet et al., J. Am. Acad. Dermatol. 1997; 36: 582-8).
У пациентов с ревматоидным артритом результаты, наблюдаемые в плацебо-контролируемом испытании с суточной дозой B-F5, не показали значимого улучшения (Wendling et al. J Rheumatol; 25(8):1457-61, 1998).
У пациентов с рассеянным склерозом (MS) наблюдали некоторые положительные эффекты после лечения в течение 10 суток пациентов с ремитирующими формами, некоторые из которых не имели обострений в течение 6 месяцев после терапии (Racadot et al., J Autoimmun, 6(6):771-86, 1993). Сходные эффекты наблюдали Rumbach et al. (Mutt Scler; 1(4):207-12, 1996).
При тяжелой болезни Крона не наблюдали значимого улучшения у пациентов, получавших B-F5 в течение 7 последовательных суток (Canva-Delcambre et al., Aliment Pharmacol Ther 10(5):721-7, 1996).
При профилактике отторжения аллотрансплантата было описано, что биодоступность B-F5 не была достаточной, чтобы позволить его применение для профилактики отторжения аллотрансплантата (Dantal et al. Transplantation, 27; 62(10): 1502-6, 1996).
Из вышесказанного очевидно, что первой проблемой, которую необходимо решить, является необходимость применения высоких доз mAb для достижения клинического эффекта. Это, помимо прочего, может быть результатом плохой доступности лимфоцитов для mAb в тканях-мишенях. Применение более высоких доз может привести к избыточному действию на лимфоциты крови, вызывая нежелательные побочные эффекты.
Другим недостатком терапии моноклональными антителами у человека является то, что эти антитела, главным образом, получают из клеток мыши, и они вызывают противомышиные ответы у реципиентов-людей. Это приводит не только к более низкой эффективности лечения и особенно при любом последующем лечении моноклональными антителами мыши, но также к повышенному риску анафилаксии.
Этого недостатка, в принципе, можно избежать путем использования гуманизированных антител, полученных пересадкой определяющих комплементарность областей (CDR) моноклонального антитела мыши, которые определяют антигенсвязывающую специфичность, в каркасные области (FR) молекулы иммуноглобулина человека. Целью гуманизации является получение рекомбинантного антитела, имеющего те же антигенсвязывающие свойства, что и моноклональное антитело мыши, из которого были получены последовательности CDR, и являющегося значительно менее иммуногенным у человека.
В некоторых случаях замена CDR человека на CDR антитела мыши в каркасных областях человека является достаточной для переноса антигенсвязывающих свойств (включая не только специфичность, но также аффинность к антигену). Однако во многих антителах некоторые остатки FR важны для связывания антигена, поскольку они напрямую контактируют с антигеном в комплексе антитело-антиген или поскольку они влияют на конформацию CDR и, таким образом, на их антиген-связывающие характеристики.
Таким образом, в большинстве случаев также необходимо заменять один или несколько остатков FR человека соответствующими каркасными остатками из антитела мыши. Поскольку для предотвращения противомышиных реакций количество замещенных остатков должно быть настолько малым, насколько это возможно, задачей является определить, какой аминокислотный остаток(ки) важен для сохранения антиген-связывающих свойств. Для прогноза более подходящих для замены участков были предложены различные способы. Несмотря на то, что эти способы обеспечивают общие принципы, которые могут помочь на первых стадиях гуманизации, конечный результат изменяется от одного антитела против другого. Таким образом, для заданного антитела, очень трудно прогнозировать, какие замены обеспечат желаемый результат.
Ранее предпринимались попытки гуманизировать B-F5 мыши, и был достигнут успех в получении гуманизированного B-F5 (в дальнейшем в настоящем документе называемого hB-F5), имеющего сходные свойства связывания CD4 по сравнению с родительским B-F5 мыши.
Таким образом, в WO 2004/083247 описано, что гуманизированное антитело BT061 (гуманизированное B-F5 или просто hB-F5) является эффективным для лечения аутоиммунных заболеваний, таких как псориаз и ревматоидный артрит. В указанной патентной заявке описаны композиции для парентерального введения, полученные для введения дозы 0,1-10 мг, предпочтительно 1-5 мг. Предусмотренные схемы дозирования представляют собой внутривенную дозу 1 мг в сутки и дозу 5 мг раз в двое суток для пациентов с ревматоидным артритом в течение 10 суток. Таким образом, наиболее высокая описанная доза составляет 5 мг за один раз и 25 мг в течение 10 суток.
Это исследование также было описано Wijdenes et al., в реферате и постере, представленном на конференции EULAR, в июне 2005 года. Было описано лечение 11 пациентов, страдающих ревматоидным артритом. Пациентов лечили 5 внутривенными инфузиями по 5 мг BT061 один раз в двое суток с параллельным лечением 150 мг диклофенака.
Антитело, описанное в этом исследовании, не раскрыто в качестве подходящего для применения в более высоких дозах, и по-прежнему сохраняется необходимость в способах лечения при более высоких дозах, с тем, чтобы лечить большее количество пациентов.
Учитывая указанный выше уровень техники, целью настоящего изобретения является лечение пациентов, страдающих аутоиммунным заболеванием, которые не отвечают в должной степени на существующие способы лечения. В частности, целью настоящего изобретения является поиск лекарственных средств для лечения аутоиммунных заболеваний, которые можно использовать у пациентов в высоких дозах, для повышения ответа на лечение у не отвечающих на текущей момент пациентов.
Удивительно, что эксперименты, проведенные авторами изобретения, показали, что IgG1-антитело BT061 в противоположность другим mAb против CD4, после связывания с CD4 клеток-мишеней не индуцирует ни ADCC, ни CDC, ни апоптоз.
Таким образом, настоящее изобретение относится к фармацевтической композиции для лечения аутоиммунного заболевания, содержащей фармацевтически приемлемый носитель и средство, способное активировать CD4+CD25+ регуляторные T-клетки, причем композицию вводят индивиду в дозе от 10 мг до 200 мг средства.
Кроме того, изобретение относится к фармацевтической композиции для лечения аутоиммунного заболевания, содержащей фармацевтически приемлемый носитель и средство, способное активировать CD4+CD25+ регуляторные T-клетки, причем композицию вводят индивиду с дозой средства от 5 до 60 мг/м2.
Кроме того, изобретение относится к фармацевтической композиции для лечения аутоиммунного заболевания, содержащей фармацевтически приемлемый носитель и средство, способное активировать CD4+CD25+ регуляторные T-клетки, где композицию вводят с дозой средства от 1 до 500 мкг/кг.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и средство, способное активировать CD4+CD25+ регуляторные T-клетки, где средство находится в концентрации от 10 до 150 мг/мл.
В предпочтительном аспекте изобретения средство представляет собой гуманизированное антитело против CD4, или его фрагмент, или производное.
Изобретение также относится к применению средства по настоящему изобретению для получения лекарственного средства для лечения аутоиммунного заболевания, где средство вводят индивиду в дозе, как определено в настоящем документе. Кроме того, изобретение относится к средству по настоящему изобретению, которое можно использовать для лечения аутоиммунного заболевания, где средство вводят индивиду в дозе, как определено в настоящем документе.
Исходя из указанных выше доз понятно, что авторы изобретения неожиданно обнаружили, что гуманизированное антитело BT061 (гуманизированное B-F5 или просто hB-F5) по существу не модулирует и не индуцирует высвобождение провоспалительных цитокинов по сравнению с другими взаимодействующими с T-клетками антителами, например антителами против CD3. Кроме того, антитело не вызывает существенного длительного истощения CD4+ лимфоцитов.
Концентрация средства по изобретению конкретно не ограничена, при условии, что оно находится в концентрации выше известных концентраций. Однако, предпочтительно, концентрация средства составляет от 10 (или более чем 10) до 150 мг/мл, от 15 до 150 мг/мл, от 15 до 100 мг/мл, от 15 (или более чем 15) до 75 мг/мл или от 20 до 60 мг/мл. Наиболее предпочтительно, концентрация средства является (приблизительно) любой из 10 мг/мл, 12,5 мг/мл, 20 мг/мл, 25 мг/мл, 50 мг/мл, 60 мг/мл, 70 мг/мл, 80 мг/мл, 90 мг/мл или 100 мг/мл.
Объем дозы, вводимой индивиду с использованием композиции, конкретно не ограничен, при условии, что он доставляет высокую общую дозу по сравнению с уже известными дозами, и, таким образом, подходит для лечения индивидов, для которых такие дозы могут быть эффективны, например, но не ограничиваясь ими, индивидов с тяжелыми случаями и длительным анамнезом заболевания и недостаточным ответом на используемые лекарственные средства. В частности, концентрация средства в величине доз может изменяться для обеспечения требуемых доз, которые описаны в этой заявке.
Величина доз может изменяться в зависимости от способа введения. Предпочтительным является парентеральное введение. Примерами парентерального введения являются внутримышечное введение, внутривенное введение или подкожное введение. Если композицию вводят внутривенной инфузией, то величина дозы может составлять от 0,1 или 0,5 мл до 500 мл, предпочтительно от 15 до 25 мл, и, как правило, приблизительно 20 мл. Если композицию вводят подкожной или внутримышечной инъекцией, то величина дозы дозировки может составлять от 0,1 до 3 мл, предпочтительно от 0,5 до 1,5 мл и, как правило, приблизительно 1 мл.
Однако в некоторых вариантах осуществления композиция может находиться в концентрированной форме и разбавлена до концентрации, требуемой для конкретных индивидов. Предпочтительно, в этих случаях композицию предоставляют в относительно небольших объемах приблизительно 1, 2, 3, 4 или 5 мл. В альтернативных вариантах осуществления, композицию предоставляют в требуемой концентрации с величиной дозы, описанной выше (т.е. в готовом для введения виде). В одном из конкретных вариантов осуществления фармацевтические композиции для подкожного введения предоставляют в готовой для введения форме, так что их может легко вводить лицо, не являющееся медицинским персоналом.
Как указано выше, до настоящего времени не было известно, что средства с эффектом в отношении аутоиммунных заболеваний можно вводить в высоких дозах, которые предусмотрены настоящим изобретением. Хотя известные дозы средств с эффектом в отношении аутоиммунных заболеваний являются эффективными у некоторых индивидов или при некоторых типах заболеваний, понимание того, что их можно успешно использовать в более высоких дозах, открыло путь для более эффективного лечения некоторых аутоиммунных заболеваний и классов пациентов.
Изобретение проиллюстрировано в качестве примера, с помощью следующих чертежей, где:
На фигуре 1 показан эффект BT061 на синтез цитокинов в культурах цельной крови 3 здоровых доноров. Культуры стимулировали в отдельных экспериментах 4 различными типами активаторов: CD3=антитела против CD3; LPS=липополисахарид; PHA=фитогемагглютинин+антитела против CD28; SEB=стафилококковый энтеротоксин B+антитела против CD28. Для выявления эффектов на различные субпопуляции лейкоцитов определяли различные цитокины: Treg-клетки (CD3: TGF-β, IL-10); моноциты/макрофаги (LPS: IL-10, TNFα, IL-1β); Th2-клетки (PHA: IL-4, IL-5, IL-13); Th1-клетки (SEB: IL-2, IFNγ);
На фигуре 2 показан эффект BT061 в культурах цельной крови человека (доноры с ревматоидным артритом) на синтез цитокинов, инициируемый различными стимуляторами;
На фигуре 3 показана нуклеотидная последовательность, кодирующая VH-область B-F5 мыши (SEQ ID NO: 5);
На фигуре 4 показана нуклеотидная последовательность, кодирующая Vk-область B-F5 мыши (SEQ ID NO: 6);
На фигуре 5 показана нуклеотидная последовательность (SEQ ID NO: 3) фрагмента плазмиды, кодирующая VH-область гуманизированного BF-5. Последовательность, кодирующая V-область, подчеркнута, и соответствующая полипептидная последовательность (SEQ ID NO: 17) указана ниже нуклеотидной последовательности;
На фигуре 6 приведена нуклеотидная последовательность (SEQ ID NO: 4) фрагмента плазмиды, кодирующая VK-область гуманизированного BF-5. Последовательность, кодирующая V-область, подчеркнута, и соответствующая полипептидная последовательность (SEQ ID NO: 2) указана ниже нуклеотидной последовательности;
На фигурах 7A и 7B соответственно приведено высвобождение TFNα и IL-6, наблюдаемое в клиническом испытании с BT061 (однократная внутривенная инфузия или подкожная инъекция) у здоровых добровольцев по сравнению с уровнями, описанными для моноклональных антител против CD3. В чертежи включены уровни доз и время до выздоровления. Результаты для TRX4, указанные на чертежах в качестве "2)", описаны в Keymeulen et al., 2005 N. Engl. J. Med. Type 1 Diabetes patients. Результаты для теплизумаба, указанные на чертежах в качестве "3)", описаны в Herold et al., 2002 N. Engl. J. Med. Type 1 Diabetes patients. Нормальные значения, указанные на чертежах в качестве "4)", описаны в Straub et al., 2007, Athr. & Rheumat. "*)" соответствует однократной дозе, "**)" соответствует совокупной дозе до достижения пиковой концентрации.
На фигуре 8 показаны уровни IL-2 и IFN-γ в плазме после введения однократной внутривенной или подкожной дозы BT061 у здоровых добровольцев. ULN=верхняя граница нормы; LLN=нижняя граница нормы.
На фигуре 9 показана кинетика количества CD4-клеток (количество клеток на мл плазмы) у добровольцев, которым ввели однократную подкожную дозу BT061. Указаны средние значения для 3 пациентов на группу. Пунктирная линия указывает на верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
На фигуре 10 показана кинетика количества CD4-клеток (количество клеток на мл плазмы) у добровольцев, которым ввели однократную внутривенную дозу BT061. Указаны средние значения для 3 пациентов на группу. Пунктирная линия указывает на верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
На фигуре 11, части A-H, приведены графики, на которых показаны данные клинических испытаний у пациентов с псориазом дозовой группы I, как описано в примере 7, в которой пациентам проводили внутривенную инъекцию 0,5 мг BT061 или плацебо. В частях A-H фигуры 8 показаны графики показателей PASI для пациентов 1-8 в дозовой группе I, соответственно.
На фигуре 12, части A-H, приведены графики, на которых показаны данные клинических испытаний у пациентов с псориазом дозовой группы II, как описано в примере 7, в которой пациентам проводили внутривенную инъекцию 2,5 мг BT061 или плацебо. В частях A-H фигуры 9 приведены графики показателей PASI для пациентов 1-8 в дозовой группе II, соответственно.
На фигуре 13, части A и B, приведены фотографии клинического испытания у пациентов с псориазом, как описано в примере 7. Фотографии являются фотографиями пациента, являющегося пациентом, получающим дозу группы II. Фотография, показанная в части A, получена перед введением. Фотография, показанная в части B, получена через 28 суток после введения.
На фигуре 14 приведены результаты клинического испытания у пациентов с ревматоидным артритом, как описано в примере 8. На чертеже приведена гистограмма процента пациентов из дозовых групп, которым подкожно вводили 1,25 мг, 6,25 мг, 12,5 мг и 25 мг BT061, достигших по меньшей мере ответа ACR20. Шести пациентам в каждой группе вводили дозу антитела, в то время как двум пациентам вводили плацебо.
На фигурах 15A и 15B приведены результаты клинического испытания у пациентов с ревматоидным артритом, как описано в примере 8. На фигуре 15A приведена гистограмма числа болезненных суставов у пациентов из дозовой группы, в которой подкожно вводили 25 мг BT061. На фигуре 15B приведена гистограмма для числа увеличенных суставов у пациентов из той же дозовой группы. Шести пациентам в каждой группе вводили дозу антитела, в то время как двум пациентам вводили плацебо.
На фигурах 16A и 16B приведены результаты клинического испытания у пациентов с ревматоидным артритом, как описано в примере 8. На чертежах приведены изменения индивидуальных параметров (в %) для одного отвечающего пациента (фигура 16A) и одного неотвечающего пациента (фигура 16B) из группы с подкожным введением дозы 25 мг. На чертежах "Pat GA" и "Phy GA" относятся к общей оценке пациентом и общей оценке врачом, соответственно. Термин "PA боли" относится к оценке пациентом боли.
На фигурах 17A и 17B приведены дополнительные результаты клинического испытания у пациентов с ревматоидным артритом, как описано в примере 8. На чертежах приведено количество болезненных суставов у пациентов из группы с подкожным введением дозы 1,25 мг (фигура 17A) и из группы с подкожным введением дозы 6,25 мг (фигура 17B).
На фигурах 18A и 18B приведены дополнительные результаты клинического испытания у пациентов с ревматоидным артритом, как описано в примере 8. На чертежах приведено количество болезненных суставов у пациентов из группы с подкожным введением дозы 50 мг (фигура 18A) и из группы внутривенной дозы 6,25 мг (фигура 18B).
На фигуре 19 приведено выравнивание полипептидных последовательностей VK B-F5 мыши (SEQ ID NO:8), FK-001 (SEQ ID NO:9, 10, 11 и 12), L4L (SEQ ID NO: 18) и L4M (SEQ ID NO:2) при конструировании гуманизированной формы B-F5 (т.е. BT061).
На фигуре 20 приведено выравнивание полипептидных последовательностей VH B-F5 мыши (SEQ ID NO:7), M26 (SEQ ID NO:13, 14, 15 и 16), H37L (SEQ ID NO:1) и H37V (SEQ ID NO:17) при конструировании гуманизированной формы B-F5.
Далее изобретение описано более подробно.
Средства, которые могут использоваться в настоящем изобретении, представляют собой средства, способные активировать CD4+CD25+ регуляторные T-клетки. Средство может представлять собой полипептид, белок или антитело. Если средство представляет собой антитело, то оно может представлять собой моноклональное антитело. Предпочтительно, антитело представляет собой моноклональное антитело против CD4. Также антитело может, предпочтительно, представлять собой IgG1-антитело и оно может представлять собой немодифицированное IgG1-антитело.
В предпочтительном аспекте изобретения средство не вызывает существенного повышения уровня провоспалительных цитокинов в плазме крови индивида после введения по сравнению с антителами против CD3. В частности, уровни IFN-γ, TNF-α, IL-6 и/или IL-2 после введения средства по существу не повышаются по сравнению с уровнями плазмы, измеренными у здоровых индивидов (см. таблицу A1). Конкретно, если ULN для конкретного цитокина, приведенную в таблице A1, принять за X, тогда в пределах 96 часов после введения средства по изобретению X увеличивается менее чем в 20 раз. Предпочтительно X увеличивается менее чем в 10 раз. Более предпочтительно эти уровни представляют собой уровни в период, составляющий от 10 минут после начала введения до 96 часов после завершения введения.
Возможно, что у пациентов с аутоиммунным заболеванием уровни цитокинов перед введением средства уже выше, чем уровни, наблюдаемые у здоровых индивидов (ULN, приведенная в таблице A1), например, вследствие модифицированного статуса активации иммунных клеток по сравнению со статусом активации клеток у здоровых индивидов. В этих случаях, за X принимают концентрацию конкретного цитокина непосредственно перед введением, и в пределах 96 часов после введения средства по изобретению X увеличивается менее чем в 20 раз. Предпочтительно X увеличивается менее чем в 10 раз. Более предпочтительно эти уровни представляют собой уровни в течение периода, составляющего 10 минут после начала введения до 96 часов после завершения введения.
|
В следующем предпочтительном аспекте изобретения средство не вызывает существенного длительного снижения числа CD4+ лимфоцитов в плазме крови индивида. Конкретно, в период от 72 до 96 часов после введения число CD4+ лимфоцитов в плазме крови индивида может составлять более 250 клеток/мкл (или по меньшей мере 250 клеток/мкл).
Предпочтительно эффекты на цитокины и CD4+ лимфоциты, описанные выше, наблюдают по меньшей мере у 80% пациентов, получающих лечение.
Для предотвращения отрицательного влияния на иммунную систему, например снижения числа лимфоцитов или индукции высвобождения цитокинов, в данной области известно применение антител (особенно взаимодействующих с T-клетками антител) подкласса IgG2, IgG3 или IgG4, поскольку антитела подкласса IgG1 проявляют более высокие взаимодействия с Fc-рецептором. Также в данной области известна модификация антител (особенно взаимодействующих с T-клетками антител) путем мутации, дегликозилирования, модификации углеводной части или инженерии углеводной части Fc для уменьшения взаимодействий с Fc-рецептором.
В экспериментах, описанных в настоящем документе, авторы настоящего изобретения выявили, что для средства по настоящему изобретению не обязательно избегать антител подкласса IgG1 и модификации. В частности, данные, приведенные в этой патентной заявке, указывают на то, что средство по настоящему изобретению не проявляет существенного или длительного истощения CD4+ клеток или не индуцируют существенного высвобождения цитокинов по сравнению с антителами против CD3.
Таким образом, в предпочтительном аспекте изобретения средство представляет собой немодифицированное антитело IgG1, т.е. антитело, которое не включает мутацию Fc и не подвергнуто дегликозилированию, модификации углеводной части или инженерии углеводной части для уменьшения взаимодействий с Fc-рецептором, или его фрагмент, или производное.
Антитела, которые наиболее эффективны для применения в настоящем изобретении, представляют собой гуманизированные антитела против CD4, или их фрагменты, или производные, которые способны активировать CD4+CD25+ регуляторные T-клетки. Примеры антител, которые способны активировать CD4+CD25+ регуляторные T-клетки, рассмотрены в Becker et al. (European Journal of Immunology (2007), Vol. 37: pages 1217-1223).
Как правило, антитело по настоящему изобретению, кроме того, содержит константную область (Fc) человека. Эту константную область можно выбирать из константных доменов любого класса иммуноглобулинов, включая IgM, IgG, IgD, IgA и IgE, и любого изотипа, включая IgG1, IgG2, IgG3 и IgG4. Предпочтительные константные области выбирают из константных доменов IgG, в частности IgG1.
Также в объем настоящего изобретения включены любые фрагменты антитела, содержащий его V-области. В частности, в объем изобретения включены Fab-, Fab'-, F(ab)'2-, Fv- и scFv-фрагменты.
В особенно предпочтительном аспекте настоящего изобретения антитело представляет собой гуманизированное антитело против CD4 или его фрагмент или производное, полученное из моноклонального антитела против CD4 мыши B-F5. Примером такого антитела является антитело BT061.
Антитело BT061, его фрагменты и производные.
Гуманизированное антитело BT061 (hB-F5) получают из mAb B-F5 мыши, и оно имеет V-домены, определенные следующими полипептидными последовательностями:
- V-домен H-цепи: EEQLVESGGGLVKPGGSLRLSCAASGFSFSDCRMYWLRQAPGKGLEWIGVISVKSENYGANYAESVRGRFTISRDDSKNTVYLQMNSLKTEDTAVYYCSASYYRYDVGAWFAYWGQGTLVTVSS (SEQ ID NO:1)
- V-домен L-цепи: DIVMTQSPDSLAVSLGERATINCRASKSVSTSGYSYIYWYQQKPGQPPKLLIYLASILESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQHSRELPWTFGQGTKVEIK (SEQ ID NO:2).
Производные этого антитела также могут использоваться по настоящему изобретению. Производные включают производные с V-доменами, определенными полипептидными последовательностями, которые по меньшей мере на 80%, предпочтительно по меньшей мере на 90%, наиболее предпочтительно по меньшей мере на 95% идентичны последовательности SEQ ID NO:1 или SEQ ID NO:2.
Особенно предпочтительные антитела представляют собой антитела, которые содержат определяющие комплементарность области (CDR) mAb B-F5 мыши, и сохраняют способность hB-F5 активировать CD4+CD25+ регуляторные T-клетки. Расположение CDR в VH- и VK-доменах приведено на фигурах 19 и 20. Такие антитела необязательно могут иметь варианты в последовательности CDR, которые по существу не влияют на специфичность и/или аффинность связывания.
Как правило, антитело hB-F5 по изобретению, кроме того, содержит константную область (Fc) человека. Как указано выше, эта константная область может быть выбрана из константных доменов иммуноглобулинов любого класса, включая IgM, IgG, IgD, IgA и IgE, и любого изотипа, включая IgG1, IgG2, IgG3 и IgG4. Предпочтительные константные области выбраны из константных доменов IgG, в частности IgG1.
Также настоящее изобретение относится к любому фрагменту антитела BT061, содержащему его V-области. Изобретение относится, в частности, к Fab-, Fab'-, F(ab)'2-, Fv- и scFv-фрагменты.
Полинуклеотид, кодирующий V-домен H-цепи или L-цепи антитела BT061, можно слить с полинуклеотидом, кодирующим константную область H- или L-цепи антитела, для экспрессии полноразмерных H- и L-цепей, полученных таким образом; также можно добавлять последовательность, кодирующую сигнальный пептид, позволяющий секрецию белка.
Также для изобретения используются экспрессирующие кассеты, где полинуклеотид, как описано выше, связан с соответствующими контрольными последовательностями, регулирующими его транскрипцию и трансляцию в выбранной клетке-хозяине, и рекомбинантные векторы, содержащие полинуклеотид или экспрессирующую кассету по изобретению.
Эти рекомбинантные конструкции ДНК можно получать и вводить в клетки-хозяева хорошо известными способами рекомбинантных ДНК и генной инженерии.
Также для изобретения используют клетку-хозяина, трансформированную полинуклеотидом по изобретению. Подходящие клети-хозяева в рамках настоящего изобретения могут представлять собой прокариотические или эукариотические клетки. Среди подходящих эукариотических клеток в качестве примера могут быть упомянуты клетки растений, клетки дрожжей, таких как Saccharomyces, клетки насекомых, таких как Drosophila или Spodoptera, и клетки млекопитающих, такие как HeLa, CHO, 3T3, C127, BHK, COS и т.д.
Конструирование экспрессирующих векторов по настоящему изобретению и трансформацию клеток-хозяев можно проводить стандартными способами молекулярной биологии.
Антитело BT061 (hB-F5) по изобретению можно получать культивированием клеток-хозяев, содержащих экспрессирующий вектор, содержащий последовательность нуклеиновой кислоты, кодирующую указанное антитело, в условиях, подходящих для его экспрессии, и выделение указанного антитела из культуры клеток-хозяев.
Конструирование гуманизированного B-F5
Конструирование гуманизированных V H - и V K -областей B-F5
Последовательности ДНК, кодирующие VH- и VK-области B-F5 мыши, соответственно, приведены на фигуре 3 и фигуре 4 и под идентификаторами последовательностей SEQ ID NO:5 и SEQ ID NO:6. VH и VK человека, в которые пересажены CDR мыши, отбирали поиском в базах данных VH человека, наиболее сходных с оригинальными VH и VK B-F5 мыши. VH-область антител человека (M26; регистрационный номер A36006) имела наиболее высокую гомологию с VH B-F5. VK-область другого антитела человека (FK-001; NAKATANI et al., Biotechnology, 7 (1989), 805-810)) имела наиболее высокую гомологию с VK B-F5.
Было сконструировано два типа VK, отличающихся между собой тем, что их 4-й остаток представлял собой лейцин или метионин, и их обозначили как L4L и L4M. Было сконструировано два типа VH, отличающихся между собой тем, что их 37-й аминокислотный остаток представлял собой лейцин или валин, и обозначили их как H37L и H37V. Выравнивание полипептидных последовательностей B-F5, FK-001, L4L и L4M приведено на фигуре 19. Выравнивание полипептидных последовательностей B-F5, M26, H37L и H37V приведено на фигуре 20. Остатки FR, ранее описанные в качестве важных для сворачивания CDR (Chothia et al., Nature 342(1989), 877; Foote et al., J. Mol. Biol., 224(1992), 487) заключены в рамки.
Комбинированием этих VH и VK было сконструировано 4 варианта V-областей.
Экспрессия гуманизированного B-F5
Последующие стадии получения гуманизированного B-F5 были такими же, как и стадии, раскрытые в патенте США 5886152 для гуманизированного B-B10.
В кратком изложении, экспрессирующие плазмиды для H-цепи (гуманизированная VH-область, слитая с константной областью y-1-цепи человека (TAKAHASHI et al., Cell, 29 (1982), 671-679)) и L-цепи (гуманизированная VK-область, слитая с константной областью K-цепи FK-001) гуманизированного B-F5 конструировали по отдельности. В этих плазмидах экспрессия гуманизированного B-F5 инициируется промотором/энхансером гена моноклонального IgM человека, FK-001. На фигурах 5 и 6, соответственно, приведены фрагменты плазмиды, кодирующей VH- и VK-области гуманизированного BF-5. Последовательности, кодирующие V-область, подчеркнуты, и соответствующие полипептидные последовательности указаны под нуклеотидной последовательностью. Обе плазмиды и pSV2neo одновременно вводили в клетки миеломы мыши Sp2/0 (ATCC CRL-1581) с использованием Lipofectineni. Трансфектомы, продуцирующие IgG человека, отбирали посредством ELISA с использованием антитела против IgG человека (y-цепь) и антитела против K-цепи Ig человека.
Охарактеризация различных вариантов гуманизированного B-F5
Оценка связывающей активности CD4
Культуральные супернатанты трансфектов четырех вариантов hB-F5 собирали и концентрировали. Различные антитела очищали из культуральных супернатантов аффинной хроматографией с использованием сефарозы с белком A и оценивали в отношении их активности связывания CD4 путем измерения, способами конкурентного ELISA, их ингибиторной активности в отношении связывания биотинилированного mB-F5 с растворимым CD4, нанесенным на микропланшеты для титрования. Время инкубации составляло 2 часа при 37°C и ночь при 4°C.
Относительная связывающая активность hB-F5 (связывающую активность mB-F5 приняли за 100%) представлена в таблице A ниже:
|
Из результатов, представленных в таблице A, следует, что 37 остаток лейцина, является критическим для поддержания активности связывания hB-F5 с CD4, поскольку активности связывания CD4 в несколько раз снижается при преобразовании 37Leu в 37Val. Напротив, выявлено, что 4-й остаток VK не является настолько важным для активности связывания CD4. Поскольку структурное различие между 37Leu и 37Val в VH не продемонстрировано явно путем молекулярного моделирования, превосходство H37L над H37V в активности связывания CD4 было неожиданным.
Для оценки были выбраны H37L/L4L и H37L/L4M.
Исследование in vitro видов биологической активности гуманизированного B-F5
Оценивали виды биологической активности in vitro B-F5 мыши и гуманизированных B-F5 (H37L/L4M IgG1 и H37L/L4L IgG1). Также тестировали гуманизированные B-F5 типа IgG2 (H37L/L4M IgG2 и H37L/L4L IgG2).
Виды биологической активности mB-F5 и четырех типов hB-F5 оценивали in vitro с использованием мононуклеарных клеток периферической крови (PBMC) от здоровых доноров. PBMC активировали посредством ConA (2,5 пг/мл, 3 суток) или PPD (10 пг/мл, 4 суток) в присутствии B-F5 мыши или hB-F5 и проводили мониторинг их пролиферативных ответов по включению 3H-тимидина.
B-F5 мыши и hB-F5 могли умеренно ингибировать индуцированную ConA пролиферацию, но их виды активности варьировали от антитела против антитела и/или от донора против донора. Также B-F5 мыши и hB-F5 были способны ингибировать Ag-специфическую пролиферацию PBMC, индуцированную PPD.
hB-F5 типа IgG1 ингибировало индуцируемую PPD пролиферацию более эффективно (ингибирование до 70%), чем mB-F5. Тип IgG1 оказался более эффективным, чем тип IgG2, ингибиторная активность которого была практически такой же, как и у mB-F5. В случае типа IgG1, H37L/L4M было не более эффективным, чем H37L/L4L. Тип IgG2 H37L/L4M и H37L/L4L имел практически одинаковую ингибиторную активность. Вкратце, ингибиторная активность B-F5 в отношении индуцированной PPD пролиферации PBMC была следующей: H37L/L4M IgG1>H37L/L4L IgG1>H37L/L4M IgG2=H37L/L4L IgG2=mB-F5.
Учитывая эффективность биологической активности in vitro и меньшее количество аминокислот мыши, для дальнейшей оценки было выбрано H37L/L4M IgG1, и оно представляет собой антитело, которое названо BT061 и используется для демонстрации настоящего изобретения в разделе "Примеры", предоставленном в данной заявке.
Композиция и применения
Как указано, фармацевтическая композиция и лекарственные средства по настоящему изобретению, предпочтительно, эффективны для лечения аутоиммунного заболевания у пациентов, для которых эффективными являются более высокие дозы. Такие пациенты включают, но ими не ограничиваются, тяжелые случаи с длительным анамнезом заболевания.
В одном из аспектов настоящее изобретение также относится к применению гуманизированного антитела против CD4 или его фрагмента или производного для получения лекарственного средства, эффективного при аутоиммунном заболевании, где гуманизированное антитело способно активировать CD4+CD25+ регуляторные T-клетки, и где лекарственное средство содержит антитело в концентрации от 10 до 150 мг/мл, предпочтительно от 15 до 75 мг/мл, наиболее предпочтительно от 20 до 60 мг/мл.
Кроме того, изобретение относится к применению гуманизированного антитела против CD4 или его фрагмента или производного для получения лекарственного средства, эффективного для лечения аутоиммунного заболевания, где гуманизированное антитело способно активировать CD4+CD25+ регуляторные T-клетки и где лекарственное средство вводят индивиду в однократной дозе или во множестве доз с антителом в количестве от 10 до 200 мг на дозу.
Также настоящее изобретение относится к фармацевтической композиции для лечения аутоиммунного заболевания, содержащей фармацевтически приемлемый носитель и средство, способное активировать CD4+CD25+ регуляторные T-клетки, где композицию также вводят индивиду с дозой средства от 10 мг до 100 мг, от 10 мг до 80 мг, от 15 мг до 80 мг, от 20 мг до 75 мг, предпочтительно от более чем 20 мг до 60 мг и наиболее предпочтительно от 25 мг до 60 мг.
В одном из аспектов изобретения индивиду вводят множество доз. В этих случаях является подходящим, чтобы доза в течение периода 10 суток превышала 25 мг, но была меньше или равна 200 мг, более предпочтительно дозировка составляет между 28 мг и 100 мг и наиболее предпочтительно между 30 мг и 100 мг. Кроме того, доза в течение периода 5 суток, предпочтительно, превышает 15 мг, но меньше или равна 100 мг, более предпочтительно, дозировка составляет между 18 мг и 100 мг и наиболее предпочтительно между 20 мг и 100 мг. В этом аспекте изобретения особенно предпочтительно, чтобы дозу вводили подкожно.
Дозу также можно вычислять, исходя из площади поверхности тела (BSA) индивида. Площадь поверхности тела (BSA) можно вычислять любым известным способом. Примеры способов вычисления BSA являются следующими:
формула Mosteller: (BSA(м2)=([рост(см)×масса тела (кг)]/36001/2
(Mosteller R.D: Simplified Calculation of Body Surface Area. N Engl J Med 1987 Oct 22; 317(17):1098)
Формула DuBois и DuBois: BSA (м2)=0,20247×рост(м)0,725×масса тела (кг)0,425
(DuBois D.; DuBois E.F.: A formula to estimate the approximate surface area if height and weight be known. Arch Int Med 1916 17:863-71).
Формула Haycock: BSA(м2)=0,024265×рост(см)0,3964×масса тела(кг)0,5378
(Haycock G.B., Schwartz G.J., Wisotsky D.H. Geometric method for measuring body surface area: A height weight formula validated in infants, children and adults. The Journal of Pediatrics 1978 93:1:62-66)
Формула Gehan и George: BSA(м2)=0,0235×рост (см)0,42246×масса тела (кг)0,51456
(Gehan E.A., George S.L., Estimation of human body surface area from height and weight. Cancer Chemother Rep 1970 54:225-35)
Формула Boyd: BSA (м2)=0,0003207×рост(см)0,3×масса тела(грамм)(0,7285-(0,0188 ×LOG(граммы))
Согласно изобретению доза средства для индивида составляет от 5 до 60 мг/м2 площади поверхности тела пациента, предпочтительно от 6 до 50 мг/м2 и наиболее предпочтительно от 8 до 40 мг/м2.
Кроме того, дозу можно вычислять исходя из массы тела индивида. Согласно изобретению доза средства для индивида составляет от 0,1 до 2 мг/кг, предпочтительно от 0,15 до 1,5 мг/кг и наиболее предпочтительно от 0,2 до 1 мг/кг.
В этих аспектах изобретения, когда доза основана на площади поверхности тела или на массе тела индивида, предпочтительно, чтобы доза в течение периода 10 суток составляла между 10 мг/м2 и 120 мг/м2, более предпочтительно между 16 мг/м2 и 120 мг/м2, или между 0,2 мг/кг и 4 мг/кг, более предпочтительно между 0,4 мг/кг и 4 мг/кг. Особенно предпочтительным является введение доз подкожно.
Частота введения конкретно не ограничена, при условии, что она не препятствует эффективности лечения. Для изобретения предпочтительно, чтобы множество доз вводили по меньшей мере одним из следующих способов: раз в сутки, раз в двое суток, раз в неделю, раз в 4 недели, раз в 6 недель, раз в 12 недель, раз в 24 недели, раз в календарный месяц, раз в 3 календарных месяца, раз в 6 календарных месяцев или раз в год. Таким образом, дозы могут быть разделены по меньшей мере на одни сутки, или альтернативно по меньшей мере на одну неделю, по меньшей мере на один месяц, по меньшей мере на 3 месяца, по меньшей мере на 6 месяцев или по меньшей мере на один год (что означает, что дозы вводят по меньшей мере каждый день, каждую неделю, каждый месяц, каждые 6 месяцев или каждый год). В следующей альтернативе, множество доз вводят каждые 1-31 суток или каждые 1-12 месяцев.
Длительность лечения конкретно не ограничена, и, как правило, при лечении аутоиммунных заболеваний лечение протекает неопределенно долго или до тех пор пока симптомы не снизятся до допустимого уровня для пациента. Как правило, дозу вводят индивиду в течение по меньшей мере 1 месяца.
Также изобретение относится к набору для применения, как определено выше, где набор содержит множество дозировок лекарственных средств, как определено выше, для одновременного, последовательного или разделенного введения индивиду.
Также изобретение относится к способу лечения аутоиммунного заболевания, который включает введение фармацевтической композиции, как определено выше, индивиду.
Также предусматривается способ лечения аутоиммунного заболевания, который включает введение лекарственного средства индивиду, где лекарственное средство включает средство, способное активировать CD4+CD25+ регуляторные T-клетки, и где лекарственное средство вводят индивиду в количестве, как описано выше.
Предпочтительно, средство представляет собой гуманизированное антитело против CD4 или его фрагмент или производное, полученное из моноклонального антитела против CD4 мыши B-F5.
Как указано, фармацевтическая композиция и лекарственные средства по настоящему изобретению, предпочтительно, способны лечить аутоиммунное заболевание у пациентов, для которых являются полезными более высокие дозы. Такие пациенты включают, но ими не ограничиваются, тяжелые случаи с длительным анамнезом заболевания.
Предпочтительно, аутоиммунное заболевание выбрано из группы, состоящей из псориаза, ревматоидного артрита, рассеянного склероза, диабета 1 типа, воспалительных заболеваний кишечника, болезни Крона, тиреоидита Хашимото, аутоиммунного тиреоидита, аутоиммунной миастении, системной красной волчанки, язвенного колита, атопического дерматита, миокардита и связанных с трансплантацией заболеваний, таких как реакции "трансплантат против хозяина" или "хозяин против трансплантата", или общие проблемы толерантности органов.
В особенно предпочтительном аспекте изобретения фармацевтические композиции предназначены для лечения аутоиммунного заболевания псориаза. В частности, такие фармацевтические композиции следует вводить внутривенно или подкожно в дозе, указанной в настоящем документе.
Псориаз представляет собой нарушение, которое приводит к псориатическим очагам повреждения или бляшкам на коже больного.
Показатель псориатического индекса площади и тяжести (PASI) широко используется для оценки и регистрации уровня псориаза, проявляемого больными. Оценка PASI вовлекает оценку эритемы (E), инфильтрации (I) и десквамации (D) и вовлечение площади поверхности тела (A) в 4 областях тела (голова (h), туловище (t), верхние (u) и нижние (l) конечности). В таблице B, ниже, показано, как работает оценочная система.
|
Поскольку голова, верхние конечности, туловище и нижние конечности соответствуют приблизительно 10, 20, 30 и 40% площади поверхности тела, соответственно, показатель PASI вычисляют по формуле:
PASI=0,1(Eh+Ih+Dh)Ah+0,2(Eu+Iu+Du)Au+0,3(Et+It+Dt)At+0,4(El+Il+Dl)Al
Показатель PASI находится в диапазоне 0-72. Показатель 0 означает отсутствие псориаза, а показатель 72 соответствует наиболее тяжелому псориазу.
В предпочтительном варианте осуществления этого аспекта фармацевтическая композиция по настоящему изобретению эффективна для лечения псориаза путем обеспечения по меньшей мере 40%, и предпочтительно по меньшей мере 50%, улучшения показателя PASI у пациента. Предпочтительно индивид перед лечением имеет показатель PASI по меньшей мере 10. Эти эффекты могут наблюдаться по меньшей мере через 56 суток после введения, более предпочтительно по меньшей мере через 75 суток после введения. В частности, эти эффекты можно наблюдать по меньшей мере у 80% пациентов, подвергнутых лечению.
В следующем аспекте настоящего изобретения фармацевтические композиции предназначены для лечения ревматоидного артрита.
Ревматоидный артрит представляет собой аутоиммунное заболевание, которое вызывает хроническое воспаление суставов и окружающих тканей, и также может поражать другие ткани и органы.
Улучшение при ревматоидном артрите, проявляемое подвергнутым лечению пациентом, обычно оценивают с использованием основного набора параметров American College of Rheumatology (ACR) (Felson et al., Arthritis & Rheumatism, 1995, 38(6), 727-735). Эта система определяет величину ACR20 как 20% улучшение показателя болезненных и увеличенных суставах и 20% улучшение 3 из 5 остальных основных показателей ACR: общая оценка пациента и врача, боль, нетрудоспособность и реактант острой фазы, такой как C-реактивный белок (CRP).
В частности, фармацевтические композиции для лечения ревматоидного артрита, предпочтительно, вводят внутримышечно или подкожно в дозировках, указанных в настоящем документе.
Современное лечение артрита включает лекарственные средства первой линии для устранения боли и воспаления, классифицируемые как противовоспалительные лекарственные средства (NSAID), например аспирин, ибупрофен, напроксен и т.д. Вторичное лечение артрита включает кортикостероиды (например, преднизон и дексаметазон), антиревматические лекарственные средства замедленного действия (SAARD) или модифицирующие заболевание антиревматические лекарственные средства (DMARD), например метотрексат, пенициллинамин, циклофосфамид, соли золота, азотиоприн, лефлуномид и т.д.
Кортикостероиды, синтетические варианты гормона - кортизона в организме, используют для ингибирования прогрессирования RA (например, преднизон и дексаметазон).
Также для лечения RA была разработана другая группа лекарственных средств, называемая модификаторами биологического ответа (BRM), включающая антагонисты TNF-альфа (адалимумаб, инфликсимаб, этанерцепт), которые действуют путем связывания с его рецептором или прямого связывания с белком TNF-альфа.
В одном из вариантов осуществления этого аспекта изобретения композиции вводят в сочетании с лекарственными средствами, в настоящее время используемыми для лечения ревматоидного артрита. В частности, композиции вводят с одним из лекарственных средств, упомянутых выше, предпочтительно метотрексатом.
Известные лекарственные средства, такие как метотрексат, и фармацевтическую композицию по настоящему изобретению можно вводить одновременно, последовательно или по отдельности.
Далее изобретение описано в виде конкретных вариантов осуществления.
ПРИМЕРЫ
ПРИМЕР 1 - Исследование иммуномодулирующих способностей BT061, связанных с пролиферацией T-клеток
Способ
Культуры цельной крови получали из взятой свежей периферической крови. В кратком изложении, с использованием игл 19G проводили взятие крови от трех здоровых добровольцев в гепаринизированные шприцы. Кровь высевали в 96-луночные культуральные планшеты не позднее чем через 60 мин после донирования.
Антитело по настоящему изобретению (BT061, партия 40588 или партия 70A0013B) добавляли в культуры до стимуляции лейкоцитов в 5 различных концентрациях (см. "Тестируемые вещества", ниже). Клеткам позволяли взаимодействовать с антителом в течение 90 мин при 37°С, 5% CO2 в увлажненной атмосфере, затем в отдельные культуры добавляли четыре различных стимулятора:
(a) антитела против CD3 (R&D Systems; 50 нг/мл)
(b) фитогемагглютинин (PHA, Biochrom KG; 3 пг/мл) вместе с антителами против CD28 (Becton-Dickinson; 1 мкг/мл);
(c) липополисахарид (LPS, подтип 055:B15 от Sigma Aldrich; 1 мкг/мл);
(d) SE-B (Bernhard-Nocht-Institut; 25 нг/мл) вместе с антителами против CD28 (Becton-Dickinson; 1 мкг/мл).
Все культуры цельной крови инкубировали в течение 24 ч при 37°C, 5% CO2 (увлажненная атмосфера). Затем культуральные супернатанты собирали для определения конечных уровней цитокинов, за исключением культур, стимулированных PHA/антиCD28, которые инкубировали в течение 48 ч для обеспечения достаточной стимуляции Th2-клеток.
Результаты приведены на фигуре 1.
Результаты
BT061 не проявлял выраженного эффекта на основные виды активности моноцитов/макрофагов и на виды активности Th1, а также Th2, в культурах цельной крови от здоровых добровольцев. Существовал зависимый от концентрации эффект на Treg-клетки (продемонстрированный в качестве увеличения высвобождения TGF-бета).
В частности, результаты подтверждают, что:
- Отсутствует модулирование воспалительного цитокина IL-2 в концентрациях вплоть до 50 мкг/мл, соответствующих применению высокой дозы у пациентов вплоть до 150 мг
- Отсутствует индукция воспалительного цитокина IFN-гамма в концентрациях вплоть до 50 мкг/мл, соответствующих применению у пациентов высокой дозы вплоть до 150 мг
- Отсутствует модулирование Th1/Th2-цитокинов в концентрациях вплоть до 50 мкг/мл, соответствующих применению у пациентов высокой дозы
- Происходит только случайная активация IL-6 с крайне низким возрастанием, вовлечение которой является противоречивым
- Происходит повышение высвобождения TGF-бета (Treg-клетки)
- Отсутствует существенный эффект на большинство регуляторных видов активности моноцитов/макрофагов и на виды активности Th1 и Th2.
ПРИМЕР 2 - Тестирование влияния BT061 на культивированные PBMC человека, стимулированные для ответа на противостолбнячный токсоид, и анализ цитокинов
Способ
Анализ пролиферации
Свежие выделенные PBMC культивировали в 96-луночных микропланшетах для титрования с плоским дном в объеме 200 мкл/лунка (4×105 клеток/лунка). Объект тестирования (антитело против CD4 AK BT061) использовали в концентрациях 20 мкг/мл, 4 мкг/мл и 0,8 мкг/мл (кроме того, 40 мкг/мл в предварительном тесте); столбнячный токсоид использовали в концентрациях 25 мкг/мл, 5 мкг/мл и 1 мкг/мл. Для отрицательного контроля брали среду для культивирования клеток. Все культивирование проводили в трех параллелях.
Для стимуляции ConA использовали концентрацию 2,5 мкг/мл и объем 200 мкл/лунка. PBMC доводили до плотности 1×106/мл и распределяли в объеме 100 мкл на лунку.
В конце периода культивирования проводили детекцию пролиферации клеток добавлением 0,4 мкКи 3H-тимидина на лунку в течение шестнадцати часов. В конце периода культивирования клетки открепляли от поверхности с использованием раствора EDTA и собирали на фильтрах из стеклянных волокон с использованием устройства для сбора клеток “Scatron”. Величину радиоактивности, включенной в ДНК в каждой лунке, измеряли в сцинтилляционном счетчике, и она пропорциональна количеству пролиферирующих клеток, которая, в свою очередь, является функцией количества лейкоцитов, которые стимулировали для вхождения в S-фазу клеточного цикла. Определяемыми параметрами были число импульсов в минуту (cpm) и индекс стимуляции (SI) для каждой концентрации, определенный как cpmсоединения/cpmпустого образца.
Анализы цитокинов
Все цитокины количественно определяли в культуральных супернатантах с использованием коммерческих наборов для ELISA, согласно соответствующим инструкциям изготовителей. Использованные реагенты указаны в таблице 1, ниже:
|
Уровни интерлейкина (IL)-1 в культуральных супернатантах определяли с использованием тестового набора для IL-1 человека ELISA Set A (Bender), согласно инструкциям изготовителя. Указанный диапазон теста, определенный стандартом, доставляемым с набором, составлял от 1,3 до 130 пг/мл для неразбавленных образцов.
Уровни IL-4, 5, 6 и 10 в культуральных супернатантах, а также трансформирующего фактора роста (TGF) β1 и фактора некроза опухоли (TNF) α определяли с использованием тестового набора OptEIA (BD biosciences), согласно инструкциям изготовителей. Указанный диапазон тестов, определенный стандартами, предоставленными с наборами, составлял от 3,8 до 330 пг/мл для IFN-γ, от 6,3 до 616 пг/мл (для IL-4, 5, 10 и TNF) при измерении неразбавленных образцов и от 7,6 до 660 пг/мл для IL-6 с использованием двухкратных разведений образцов.
В вычисление среднего значения и стандартного отклонения были включены два (для культур моноцитов) или восемь (для культур PBMC) определений цитокинов с помощью ELISA, каждое из независимых микрокультур. Для вычисления, титрам выше верхнего диапазона теста (например, 616 пг/мл в случае TNF) присваивали это значение. Перед вычислением из каждого среднего значения вычитали нижний предел диапазона теста.
Результаты
BT061 (также называемое гуманизированным B-F5 или просто hB-F5) способно подавлять дозозависимым образом специфичную к столбнячному токсоиду пролиферацию T-клеток; отсутствовал эффект на общее число T-клеток. Была показана общая супрессия высвобождения цитокинов.
В таблице 2, ниже, представлено влияние mAb против CD4 BT061 на специфичную к столбнячному токсоиду пролиферацию T-клеток в анализе, измеренную в трех параллелях.
Представлены средние значения и SD для включения 3H-Tdr, измеренного в трех параллелях, а также индекс стимуляции (SI, определенный как cpmсоединения/cpmнуля) для каждой концентрации и уровень значимости для непарного двухстороннего t-критерия относительно контроля, представляющего собой среду (n.s.: не значимо; *: p<0,05; **: p<0,01; ***: p<0,001).
|
|
|
Данные в таблицах 2-4 демонстрируют следующее:
- Дозозависимая супрессия индуцированной столбнячным токсоидом пролиферации T-клеток (вторичный ответ) была продемонстрирована даже при высоких дозах BT061, указывая на то, что BT061 не устраняет весь иммунный ответ. Использованные дозы относятся к соответствующему применению у пациентов высокой дозы вплоть до 60 мг.
- Общая супрессия высвобождения цитокинов (дозозависимое снижение уровней IFN-гамма, IL-5 и TNF-альфа без изменения уровней IL-1, IL-4, IL-6, IL-10) и без влияния на баланс Th1/Th2.
- Увеличение высвобождения TGF-бета
ПРИМЕР 3 - Тест проточной цитометрией, индуцируемой BT061 (mAb против CD4) ADCC (антитело-зависимой клеточно-опосредуемой цитотоксичности)
Клетки-мишени HuT 78 метили BT061 (hB-F5) и инкубировали с клетками PBMC в качестве эффекторов. Погибшие клетки можно было выявить вследствие захвата красителя ДНК - йодида пропидия - после инкубации в течение 30 минут. Результаты представлены в таблице 5.
|
|
Данные в таблице демонстрируют, что не происходит индукции ADCC посредством BT061 (hB-F5) даже при высоких концентрациях.
ПРИМЕР 4 - Апоптоз
В тесте проточной цитометрией индуцируемого BT061 (mAb против CD4) апоптоза PBMC из цельной крови инкубировали с BT061 или с положительным контролем.
После инкубации в течение 7 суток проводили детекцию апоптотических клеток окрашиванием апоптотических клеток аннексин-V-флуоресцеином. Результаты представлены в таблице 6.
|
Эти данные демонстрируют, что не происходило индукции апоптоза даже при высоких концентрациях BT061.
ПРИМЕР 5 - Связывание комплемента
В тесте проточной цитометрией связывания фактора комплемента C1q, PBMC выделяли и инкубировали с BT061 (mAb против CD4), с последующей инкубацией с очищенным рекомбинантным C1q.
ATG (Tecelac) служил в качестве положительного контроля.
Детекцию проводили с помощью меченного FITC антитела для детекции против C1q. Результаты представлены в таблице 7, ниже.
|
Данные указывают на то, что связывание комплемента не может наблюдаться даже при высоких концентрациях.
ПРИМЕР 6 - Безопасность и переносимость возрастающих доз BT061
Проводили исследование для мониторинга безопасности и переносимости BT061 с использованием возрастающих доз антитела у здоровых мужчин- и женщин-добровольцев в возрасте от ≥18 до ≤75 лет.
Тридцати добровольцам вводили BT061 путем внутривенного введения в 10 дозовых группах, по 3 добровольца на группу. Кроме того, в 5 дозовых группах, также по 3 добровольца на группу, 15 добровольцам вводили BT061 путем подкожного введения. Внутривенное введение BT061 проиллюстрировано в таблице 8, ниже:
|
Каждую дозу разбавляли 0,9% хлоридом натрия для инъекций вплоть до общего объема 20 мл. Дозу вводили в виде однократной непрерывной внутривенной инфузии в течение 2 часов.
Введение BT061 подкожно проиллюстрировано в таблице 9, ниже:
|
Каждую дозу инъецировали в виде однократной болюсной инъекции.
Добровольцев оценивали в течение 3 месяцев после инъекции.
Для подкожного применения проводили взятие образцов плазмы до введения и через 3, 6, 12, 24, 36, 48, 56, 72, 88, 96, 120, 144 и 168 часов после введения и на 75 сутки.
Для внутривенного применения взятие образцов плазмы проводили до введения и через 30 минут, 1, 2, 3, 6, 12, 24, 36, 48, 72, 96, 120, 144 и 168 часов после введения.
Образцы плазмы анализировали с использованием стандартных способов ELISA для определения уровней цитокинов. Соответствующие проанализированные цитокины включали: IFN-γ, TNF-α, IL-6 и IL-2.
Также образцы плазмы анализировали с использованием стандартных способов проточной цитометрии для определения количества CD4+ лимфоцитов.
Результаты
Было выявлено, что внутривенные и подкожные дозы вплоть до 60 мг, главным образом, хорошо переносились.
Уровни цитокинов
Индукция высвобождения цитокинов является частым немедленным осложнением, происходящим при применении взаимодействующих с T-клетками терапевтических антител, таких как ATG, OKT3, CAMPATH-1H и гуманизированные mAb против CD3 (TRX4, висилизумаб и теплизумаб). Симптомы, главным образом, включают умеренную лихорадку, головные боли и самоограничивающиеся желудочно-кишечные проявления. Побочные эффекты, коррелирующие с индукцией цитокинов после введения антитела, требуют применения дополнительных лекарственных средств, таких как антигистамин дифенгидрамина гидрохлорид и/или противовоспалительное средство ибупрофен.
При использовании OKT3 (муромонаб-CD3), специфичного против CD3 терапевтического моноклонального антитела мыши, была описана гибель и тяжелые побочные эффекты, ограничивающие клиническое применение этого антитела, главным образом, пациентами с иммуносупрессией.
Несмотря на то, что гуманизированные не связывающие FcR специфичные против CD3 моноклональные антитела, которые в настоящее время используют в клинике для лечения аутоиммунного заболевания (теплизумаб и TRX4), проявляют уменьшение побочных эффектов, индуцированных активацией T-клеток и/или активацией экспрессирующих Fc-рецептор, клеток после первой дозы, по сравнению со связывающими FcR специфичными против CD3 антителами, такими как OKT3, тем не менее наблюдается некоторая степень активации T-клеток и активации экспрессирующих Fc-рецептор клеток, которая ведет к высвобождению цитокинов, как правило, связанному с зависимыми от цитокинов побочными эффектами.
В настоящем исследовании было неожиданно выявлено, что индукция цитокинов, наблюдаемая у здоровых добровольцев после внутривенного или подкожного введения BT061, была низкой и временной по сравнению с антителами против CD3. Индукция цитокинов, как правило, возрастала при увеличении дозировки. Однако даже при наиболее высоких дозировках от 40 до 60 мг индукция цитокинов являлась значительно более низкой, чем индукция, наблюдаемая в случае других взаимодействующих с T-клетками моноклональных антител (фигура 7A и B).
Срединные пиковые концентрации для цитокинов, наблюдаемые в любой момент времени в пределах 96 часов после введения, при использовании наиболее высоких доз (от 40 мг до 60 мг BT061) приведены на фигурах 7 и 8.
Срединную пиковую концентрацию для каждого цитокина вычисляют следующим образом: медиана наиболее высоких концентраций цитокинов, наблюдаемых после введения антитела.
На фигуре 7A и B приведено высвобождение TNFα и IL-6, наблюдаемое у здоровых добровольцев после внутривенного или подкожного введения BT061 по сравнению с высвобождением после введения моноклональных антител против CD3, теплизумаба и TRX4. Нормальные значения для этих цитокинов были взяты из Straub et al., (2007, Arthr. & Rheumat). На фигуре 8 приведены уровни IL-2 и IFN-γ после внутривенного или подкожного введения BT061. Срединные пиковые уровни вычисляли в дозовых группах 40 и 60 мг, измеряя их в течение 4 суток после инъекции антитела. Верхнюю границу нормы (ULN) вычисляли, исходя из уровней цитокинов, определенных у 39 здоровых индивидов, где ULN=среднее значение+2×стандартное отклонение.
По сравнению с теплизумабом и TRX4 (результаты взяты из Herold et al., 2002, New Engl. J. Med, и Keymeulen et al., 2005 New Engl. J. Med, соответственно) BT061 индуцировал только крайне малое и временное высвобождение цитокинов. Уровни TNF-α и IL-6 немного повышались. На фигуре 7 показано, что медианные пиковые значения уровней цитокинов IL-6 и TNFα, выявленные в плазме после применения BT061 (40 и 60 мг), были ниже уровней, наблюдаемых после введения специфичных против CD3 терапевтических антител теплизумаба и TRX4.
Кроме того, в противоположность mAb против CD3, BT061 не приводит к существенному повышению уровней IFN-γ и IL-2 (фигура 8), как было описано для применения TRX4 (Keymeulen et al. 2005).
CD4+ лимфоциты
Кроме того, испытание также включало исследование количеств CD4-положительных лимфоцитов в собранных образцах плазмы.
Результаты внутривенного введения приведены ниже в таблицах 9, 10 и 11. В таблице 12 приведены результаты испытания с подкожным введением. Результаты графически приведены на фигурах 9 и 10.
|
|
В частности, на фигуре 9 приведено количество CD4 клеток (количество клеток на мл плазмы) у добровольцев, которым вводили однократную внутривенную дозу BT061. Данные представляют собой средние значения для 3 пациентов в каждой дозовой группе. Пунктирной линией указана верхняя граница нормы (ULN) и нижняя граница нормы (LLN). ULN и LLN (среднее значение+ (или -) стандартное отклонение) были вычислены исходя из количества клеток здоровых добровольцев, как 443 CD4 клеток на мкл (нижняя граница нормы; LLN) и 1324 CD4 клеток на мкл (верхняя граница нормы; ULN).
На фигуре 10 приведено количество CD4 клеток (количество клеток на мл плазмы) у добровольцев, которым вводили однократную подкожную дозу BT061. Как и на фигуре 9, данные представляют собой средние значения для 3 пациентов в каждой дозовой группе. Пунктирная линия указывает на верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
|
|
Многие специфичные против CD4 моноклональные антитела, известные в данной области (такие как антитела, рассмотренные в Strand et al., 2007) вызывают иммуносупрессию путем истощения CD4-положительных лимфоцитов. Недостатком этих антител является то, что индивиды, получающие лечение, становятся иммунодефицитными и чувствительными к другим инфекциям.
В противоположность этому данное исследование показало, что BT061 не индуцировало массового длительного истощения CD4-положительных клеток. Однако наблюдали временное снижение уровня CD4-положительных лимфоцитов с восстановлением до нормальных значений в периферической крови в пределах 72 ч после введения антитела.
В момент времени 72 ч после введения BT061, для количеств CD4 клеток у четырех здоровых добровольцев в группах внутривенных доз были показаны уровни CD4, которые были ниже этих нормальных значений следующим образом: 1 доброволец с внутривенной дозой 100 мкг:400 CD4 клеток на мкл; 1 доброволец группы 5 мг:419 CD4 клеток на мкл; 1 доброволец группы 10 мг:440 CD4 клеток на мкл и 1 доброволец группы 20 мг:392 CD4 клетки на мкл.
Однако эти значения были только немного ниже нормальных значений. Количества CD4 клеток у остальных 26 добровольцев в группах внутривенных доз находились в пределе нормальных значений через 72 часа после введения BT061.
В группах подкожных доз через 72 ч только у одного из 15 добровольцев было выявлено количество CD4 клеток ниже нормальных значений.
В заключение, в противоположность истощающим специфичным против CD4 mAb, даже при высоких дозах BT061 индуцировало только временное снижение уровней CD4-положительных клеток с последующим общим восстановлением. Из временного снижения и быстрого общего восстановления до нормальных значений можно заключить, что происходило временное перераспределение CD4-положительных клеток, а не истощение этих клеток.
ПРИМЕР 7 - Клиническое испытание BT061 у пациентов с хроническим псориазом от умеренного до тяжелого
Способность hB-F5 BT061 лечить аутоиммунное заболевание тестируют на 56 пациентах, страдающих хроническим псориазом от умеренного до тяжелого. Испытание включает исследование с возрастающей однократной дозой для оценки безопасности и эффективности hB-F5.
Условия испытания являются следующими.
56 пациентов разделяют на семь дозовых групп по восемь индивидуумов в каждой. В пяти дозовых группах (дозовые группы I-V) вводят антитело или плацебо путем внутривенного введения и в двух дозовых группах (дозовые группы VI и VII) вводят антитело или плацебо путем подкожного введения. Двум пациентам в каждой дозовой группе вводят плацебо, а остальным шести пациентам в каждой дозовой группе вводят дозу BT061. В дозовой группе I шести пациентам внутривенно вводят 0,5 мг BT061. В дозовых группах II-V шести пациентам вводят 2,5 мг, 5 мг, 10 мг или 20 мг BT061, соответственно. В дозовых группах VI и VII, где введение является подкожным, шести пациентам вводят 12,5 мг или 25 мг BT061, соответственно.
Для внутривенного введения антитело/плацебо инфузируют в вену предплечья согласно принятым в медицине процедурам. В данном случае общий объем вводят в виде однократной непрерывной внутривенной инфузии в течение 2 часов через перфузатор (Fresenius Pilot C, Fresenius AG, Germany). Каждую дозу антитела разбавляют инъекционным 0,9% хлоридом натрия (B. Braun Melsungen AG, Germany) до общего объема 20 мл.
Для подкожного введения антитело вводят в виде однократной подкожной инъекции. Ту же процедуру проводят для плацебо.
Уровень псориаза, который проявляет каждый пациент, регистрируют с использованием шкалы псориатического индекса площади и тяжести (PASI). Как описано выше, более высокие показатели PASI соответствуют более высокому уровню псориаза. Пациенты, включенные в испытание, имеют хронический псориаз от умеренного до тяжелого, т.е. показатель PASI, равный 10 или выше.
Показатель PASI пациента оценивают до испытания для получения "исходного" значения на 0 сутки и повторно в ходе испытания на 5, 7, 14, 21, 28, 42, 56 и 75 сутки.
Дозовая группа I
Шести пациентам из дозовой группы I проводили однократное внутривенное введение 0,5 мг BT061, в то время как двум пациентам из дозовой группы I вводили плацебо. Доза на массу тела и доза на площадь поверхности тела (BSA) для каждого пациента представлены в таблице 13. Площадь поверхности тела вычисляли по формуле Mosteller, описанной в настоящем документе.
Показатели PASI для пациентов в дозовой группе I представлены в таблице 13 вместе с процентным улучшением показателя PASI относительно исходного уровня.
Дозовая группа II
Шести пациентам из дозовой группы II проводили однократную внутривенную инъекцию 2,5 мг BT061, в то время как двум пациентам из дозовой группы II вводили плацебо. Доза на массу тела и доза на площадь поверхности тела (BSA) для каждого пациента представлены в таблице 14.
Показатели PASI для пациентов в дозовой группе II представлены в таблице 14 вместе с процентным улучшением показателя PASI относительно исходного уровня.
Дозовая группа III
Шести пациентам из дозовой группы III проводили однократную внутривенную инъекцию 5,0 мг BT061, в то время как двум пациентам из дозовой группы III вводили плацебо. Доза на массу тела и доза на площадь поверхности тела (BSA) для каждого пациента представлены в таблице 14B.
Показатели PASI для пациентов в дозовой группе III представлены в таблице 14B вместе с процентным улучшением показателя PASI относительно исходного уровня.
Дозовая группа IV
Шести пациентам из дозовой группы IV проводили однократную внутривенную инъекцию 10,0 мг BT061, в то время как двум пациентам из дозовой группы IV вводили плацебо. Доза на массу тела и доза на площадь поверхности тела (BSA) для пациентов представлены в таблице 14C.
Показатели PASI для пациентов в дозовой группе IV представлены в таблице 14C вместе с процентным улучшением показателя PASI относительно исходного уровня.
|
|
|
|
Кроме того, показатели PASI с течением времени для отдельных пациентов приведены в графической форме на фигурах 11A-11H и на фигурах 12A-12H. На графиках, показанных на фигурах 11A-11H, приведены показатели PASI для пациентов из дозовой группы I, а на графиках, показанных на фигурах 12A-12H, приведены показатели PASI для пациентов из дозовой группы II.
Как можно видеть из результатов, приведенных в таблицах 13 и 14, 75% всех пациентов из дозовой группы I и дозовой группы II демонстрируют явное улучшение их показателей PASI, т.е. по меньшей мере 40% улучшение относительно исходного значения, после однократной дозы. Следует отметить, что 25% пациентов в дозовой группе I и в дозовой группе II вводили плацебо.
Действительно, в обеих дозовых группах у 50% пациентов было выявлено по меньшей мере 50% улучшение показателей PASI, а у одного пациента в дозовой группе II показано 88% улучшение показателя PASI на 56 сутки (т.е. у пациента 3 в таблице 14). Более того, терапевтический эффект является длительным даже при этих низких дозах, причем в конце испытания, через 75 суток после введения, улучшения наблюдались у многих пациентов.
У пациентов в дозовой группе III также выявлено улучшение их показателей PASI, причем у шести из восьми пациентов было показано более чем 20% улучшение и у двух из них было показано более чем 30% улучшение после лечения. Однако улучшение не было настолько существенным, как наблюдалось у пациентов из дозовой группы I и дозовой группы II, которым вводили более низкую дозу антитела. Некоторую эффективность также наблюдали у пациентов дозовой группы IV. В частности у пациентов 1, 4, 5 и 8 в этой дозовой группе (как показано в таблице 14C) выявлено явное улучшение их показателей PASI, хотя оно являлось ограниченным по сравнению с пациентами дозовых групп I-III.
Количество пациентов, у которых было показано по меньшей мере 40%, 50%, 60% и 75% улучшение показателя PASI, представлено в таблице 15.
|
На фигурах 13A и 13B предоставлены фотографии, указывающие на повышение уровня псориаза до и после введения. На фигуре 13A показана область псориаза на коже пациента в дозовой группе II перед введением. На фигуре 13B показана та же область псориаза через 28 суток после введения. Области улучшения показаны на фигуре 13B черными рамками.
Из этих результатом можно явно видеть, что BT061 обеспечивает эффективное лечение умеренного и тяжелого хронического псориаза.
Результаты этого исследования, в сочетании с результатами, описанными выше в примере 6, в котором показано, что высокие дозы антитела по изобретению, как правило, являются хорошо переносимыми у человека, демонстрируют способность фармацевтических композиций по изобретению обеспечивать эффективное лечение аутоиммунных заболеваний в дозах, описанных в настоящем документе.
ПРИМЕР 8 - Клиническое испытание BT061 у пациентов с ревматоидным артритом
Способность BT061 лечить ревматоидный артрит тестируют у пациентов, страдающих этим заболеванием. Испытание включает испытание с многократными дозами, вовлекающее 96 пациентов, подразделенных на 12 групп. В каждой группе двум пациентам вводят плацебо, а 6 пациентам вводят BT061. Пациентам вводят дозу раз в неделю в течение 6 недель.
Пациентов подразделяют на пациентов, которым вводят антитело подкожно, и на пациентов, которым вводят антитело внутривенно. Группы с подкожным введением доз представляют собой: 1,25 мг, 6,25 мг, 12,5 мг, 25 мг, 50 мг, 75 мг и 100 мг. Группы внутривенных доз представляют собой: 0,5 мг, 2 мг, 6,25 мг, 12,5 мг и 25 мг.
В группе подкожной дозы 1,25 мг пациентов нумеруют как 101, 102, 103, 104, 105, 106, 107 и 108. В группе подкожной дозы 6,25 мг пациентов нумеруют как 201-208. В группе подкожной дозы 12,5 мг пациентов нумеруют как 301-308. В группе подкожной дозы 25 мг пациентов нумеруют как 401-408. В группе подкожной дозы 50 мг пациентов нумеруют как 501-508. В группе внутривенной дозы 6,25 мг пациентов нумеруют как 601-608.
Способ внутривенного и подкожного введения был таким же, как описано в примере 7 для испытания псориаза.
Уровень ревматоидного артрита регистрируют раз в неделю путем оценки параметров ACR и, в частности, исследования количества болезненных и увеличенных суставов и установления уровней C-реактивного белка (CRP) и скорости оседания эритроцитов (ESR). Эти параметры оценивают до испытания для получения "исходного" значения на 0 сутки и повторно в течение периода испытания и после этого через 8, 22 и 43 суток после окончания периода введения (т.е. в ходе наблюдения (FU) на 8 сутки, на 22 сутки FU и на 43 сутки FU).
В таблицах ниже представлены данные, полученные после испытания. Конкретно, в таблицах 16-21 представлено количество болезненных и увеличенных суставов в ходе испытания.
|
|
|
|
|
|
На фигуре 14 приведен процент пациентов из дозовых групп, в которых подкожно вводили 1,25 мг, 6,25 мг, 12,5 мг и 25 мг BT061, достигших по меньшей мере 20% улучшения соответствующих параметров ACR в ходе испытания, и процент пациентов, достигших по меньшей мере ответа ACR20 на 7 неделе.
В частности, можно видеть, что 50% пациентов в группе подкожной дозы 25 мг (т.е. 4 из 8 пациентов, где 2 пациентам вводили плацебо) достигли по меньшей мере 20% улучшения соответствующих параметров ACR на 6 неделе. Эти показатели возросли до 5 из 8 пациентов на 7 неделе, т.е. 5 из 8 пациентов достигли по меньшей мере ACR20. Один пациент в этой дозовой группе достиг более чем 50% улучшения соответствующих параметров ACR на 5 и 6 неделях (полный набор данных не представлен).
Положительные результаты также были получены пациентами в других дозовых группах. Один пациент в группе подкожной дозы 6,25 мг достиг по меньшей мере 50% улучшения соответствующих параметров ACR на 4 неделе, в то время как другой достиг по меньшей мере 70% улучшения соответствующих параметров ACR на 3 неделе (полный набор данных не представлен).
На фигуре 15A и 15B приведены результаты для количества болезненных и увеличенных суставов у пациентов из группы подкожной дозы 25 мг BT061 в течение шести недель. У нескольких пациентов было выявлено снижение количества болезненных и увеличенных суставов в ходе лечения. Результаты для одного отвечающего пациента и одного неотвечающего пациента и этой дозовой группы приведены на фигурах 16A и 16B, соответственно. У “отвечающего” пациента выявлено значимое улучшение в количестве болезненных и увеличенных суставов и уровнях боли.
Снижение количества болезненных и увеличенных суставов также наблюдается у пациентов из других дозовых групп. На фигурах 17A, 17B, 18A и 18B приведено количество болезненных суставов в группах подкожных доз 1,25 мг, 6,25 мг, 50 мг и внутривенной дозы 6,25 мг, соответственно, в течение испытания и в последующие недели.
Эти результаты демонстрируют эффективность средства по настоящему изобретению в отношении лечения ревматоидного артрита в диапазонах доз, описанных в настоящем документе.
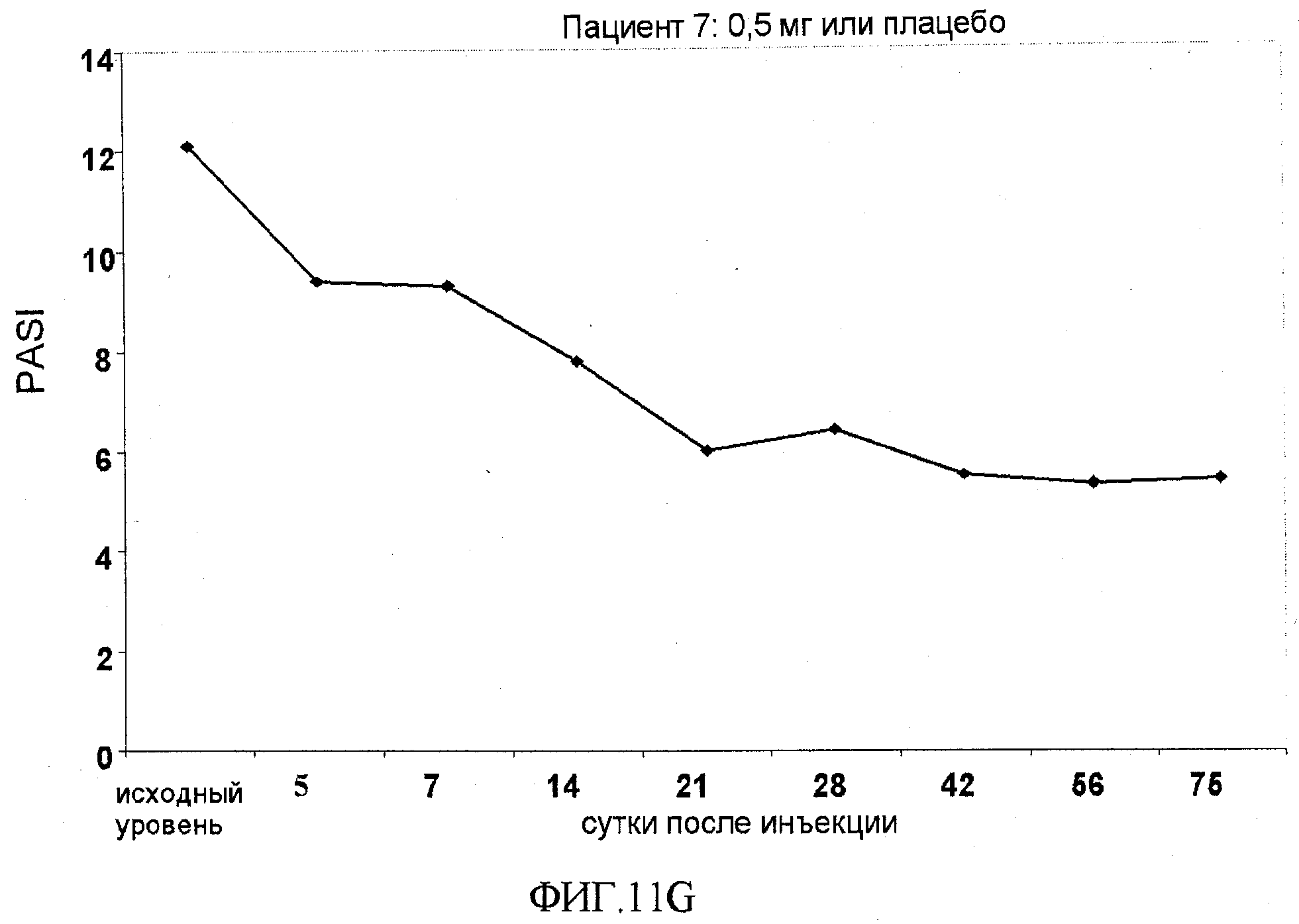
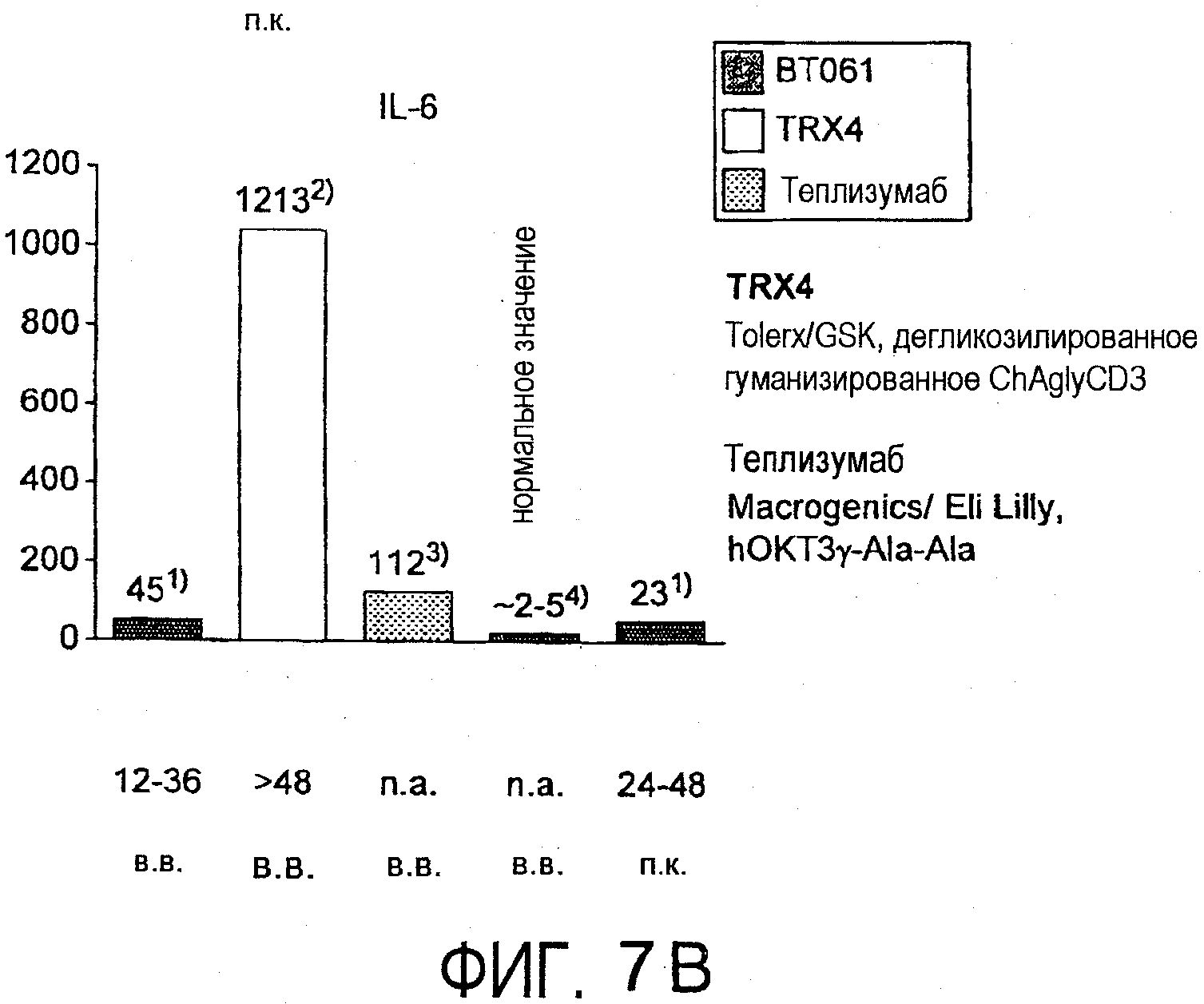
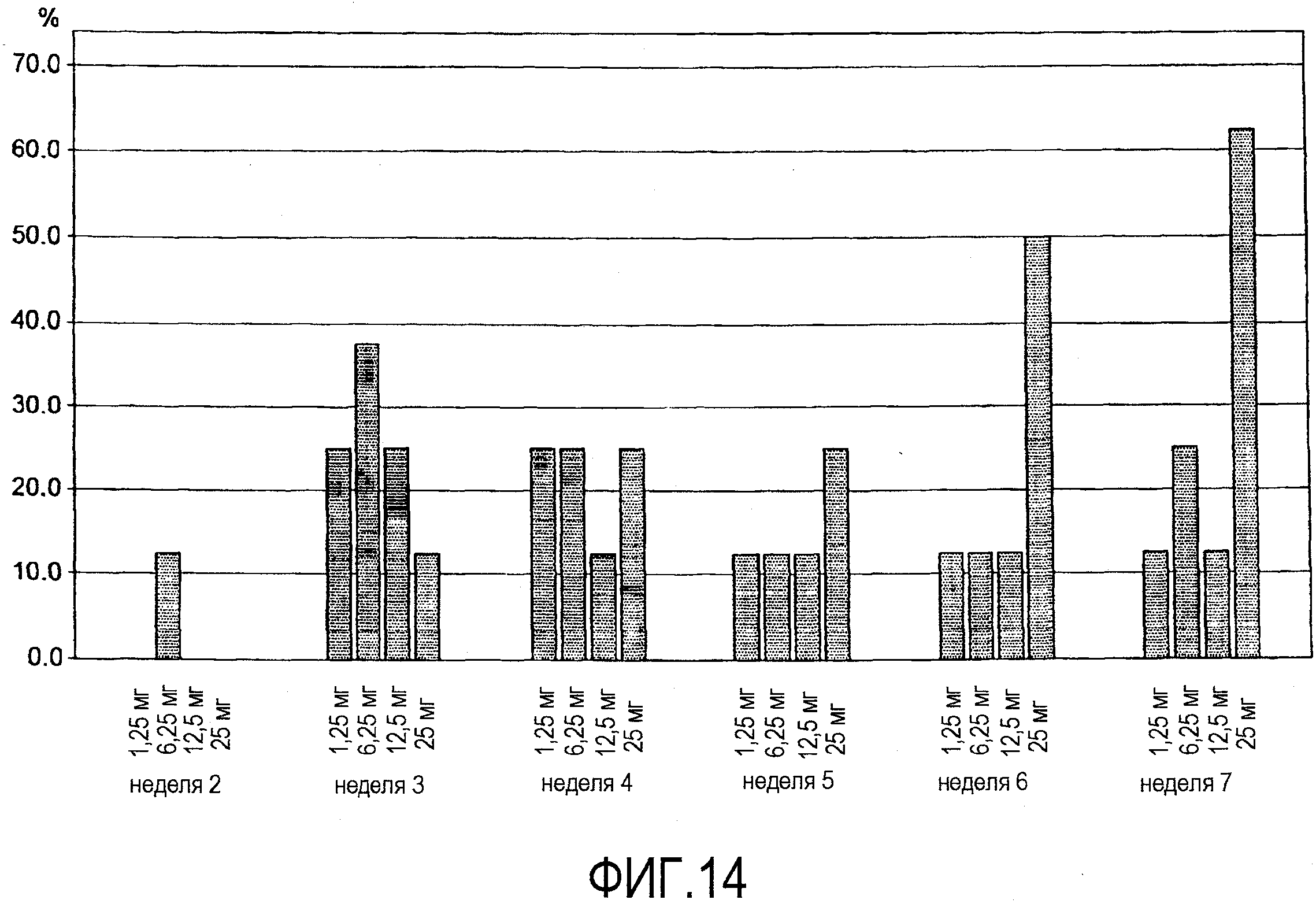
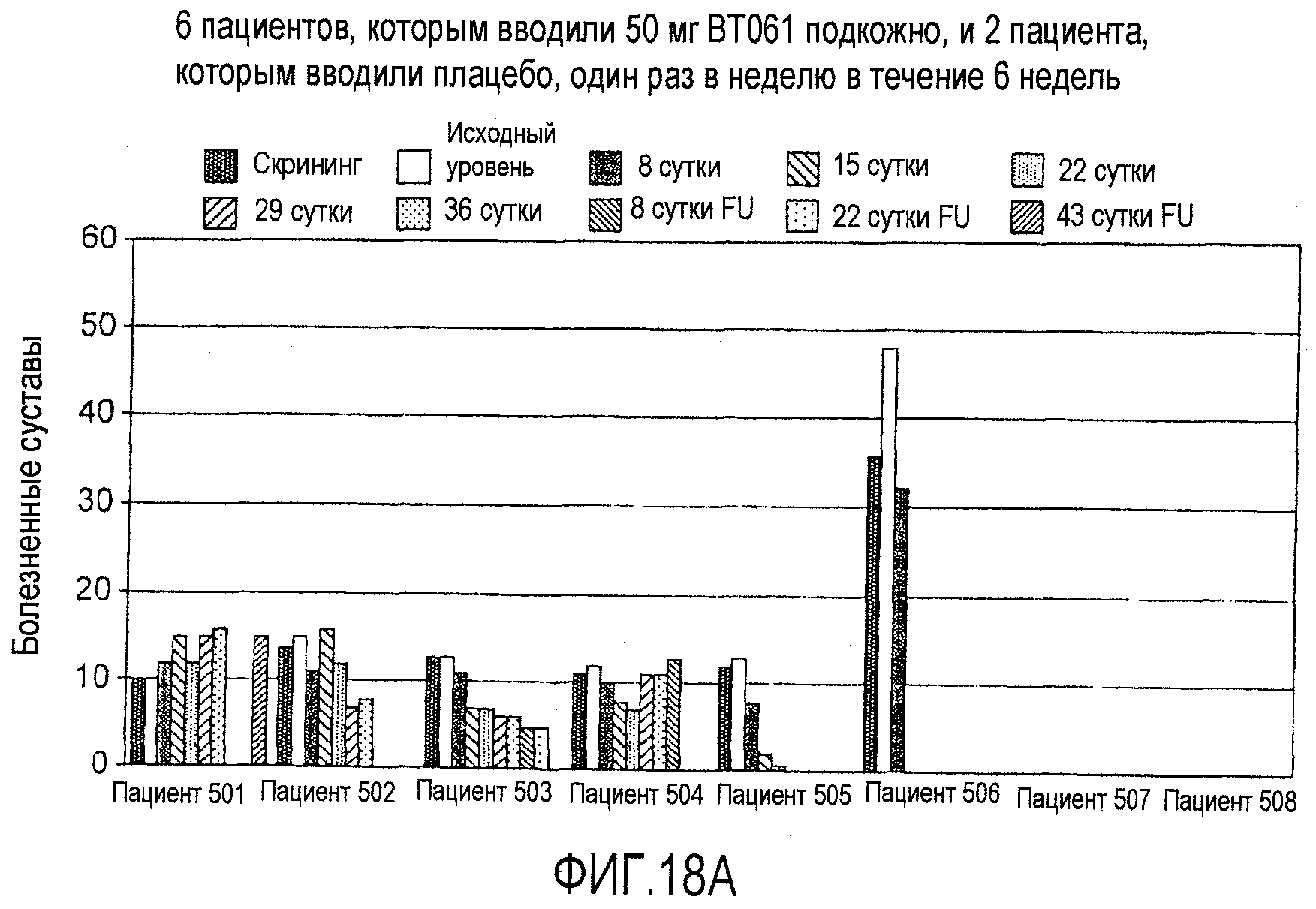
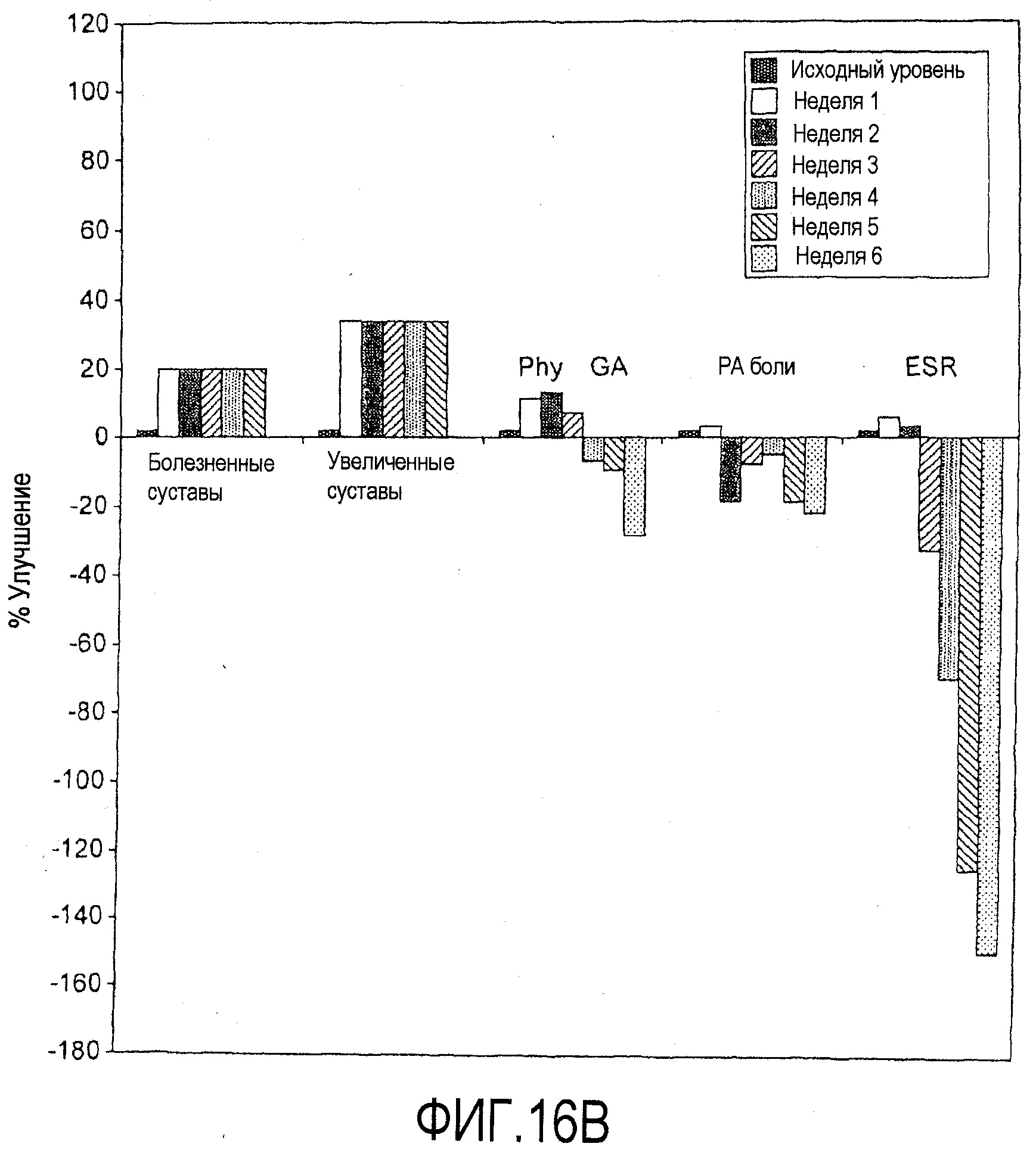

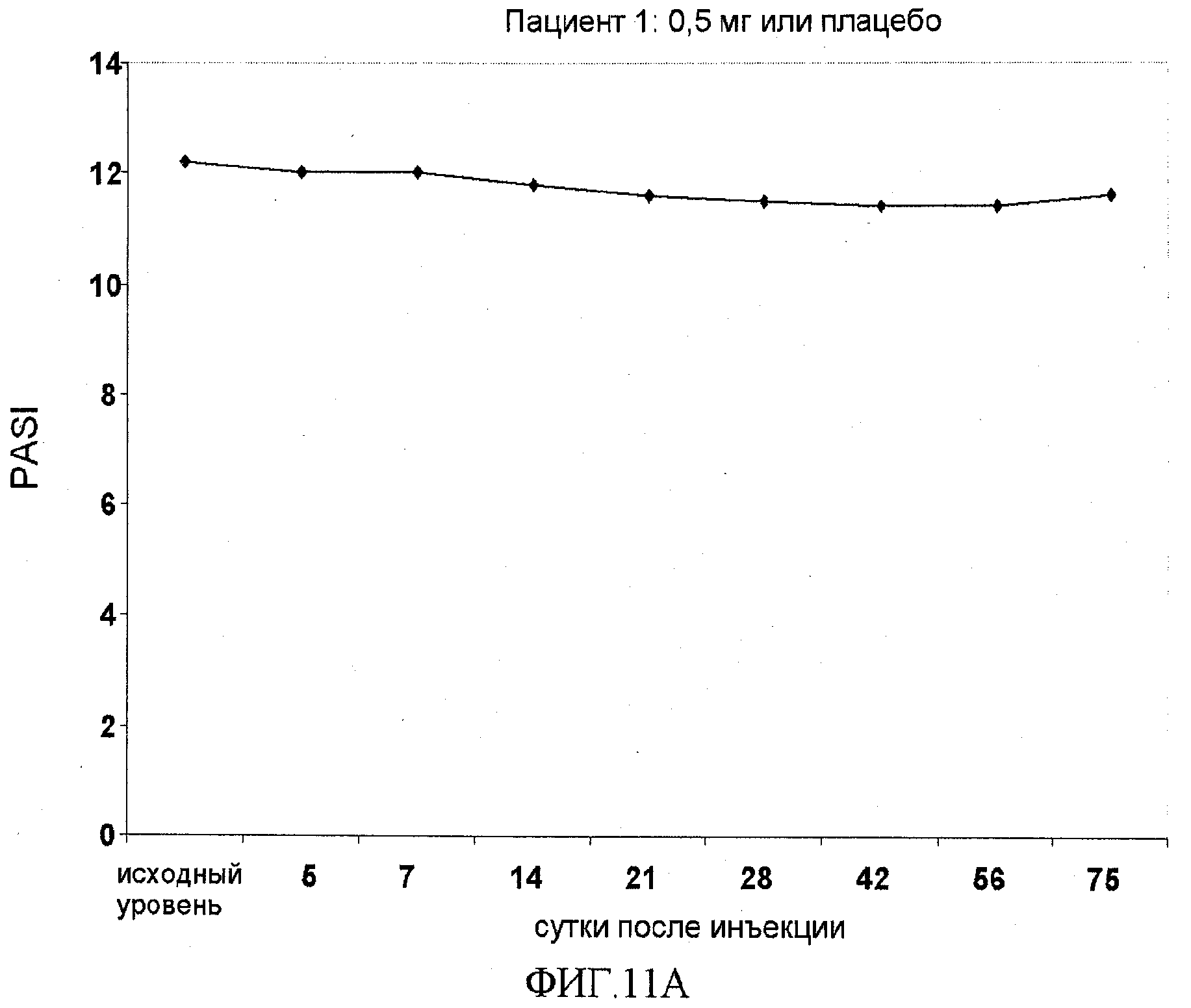
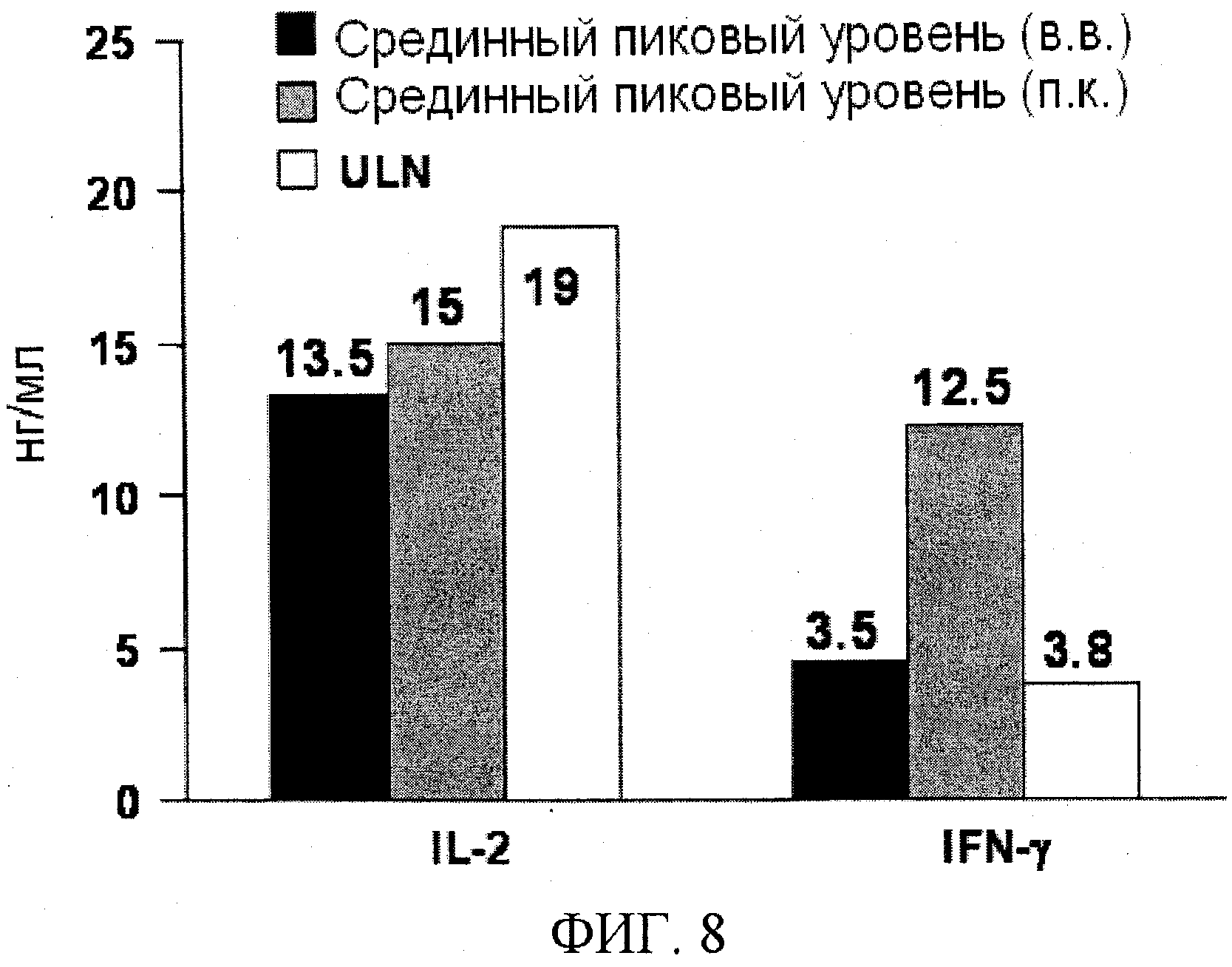
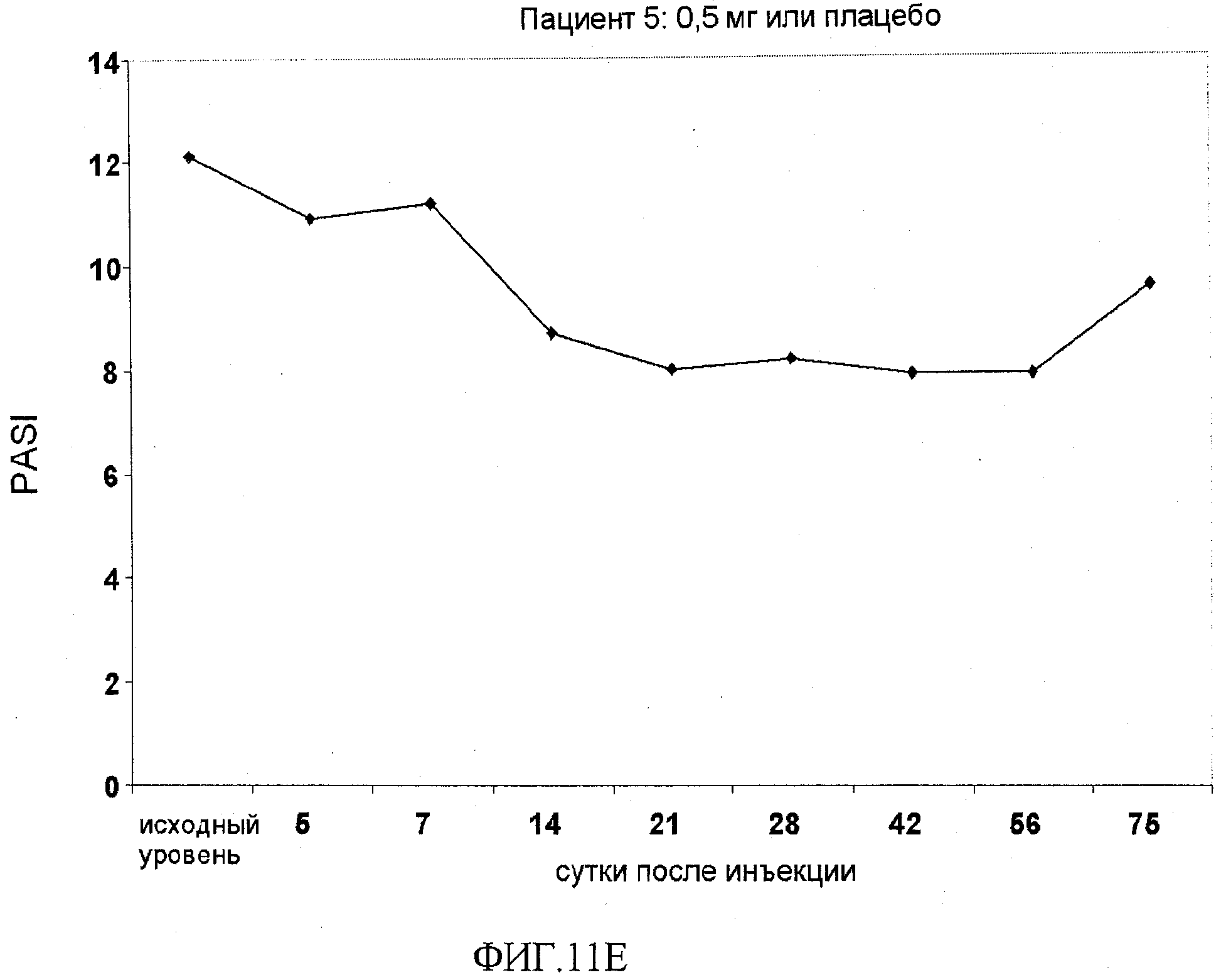

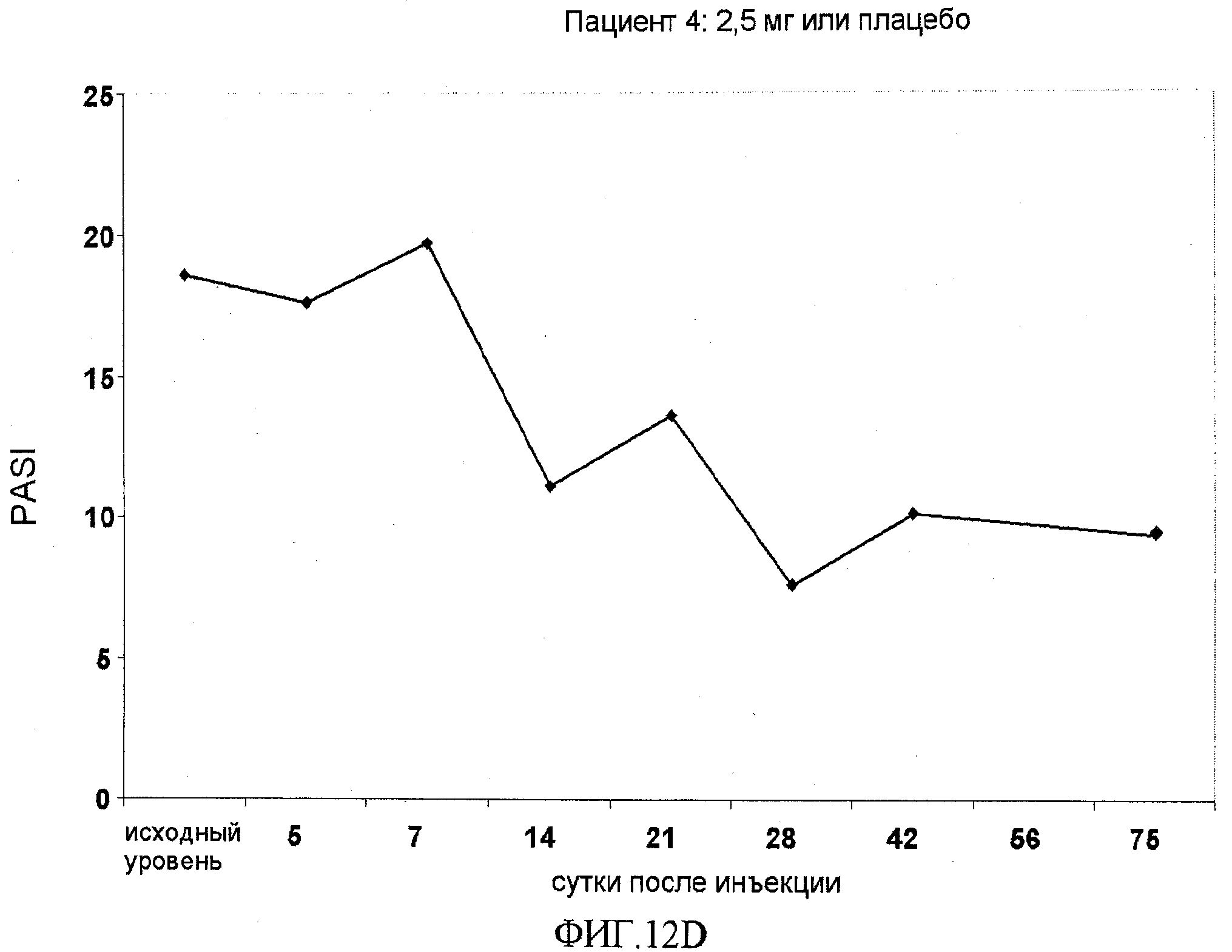
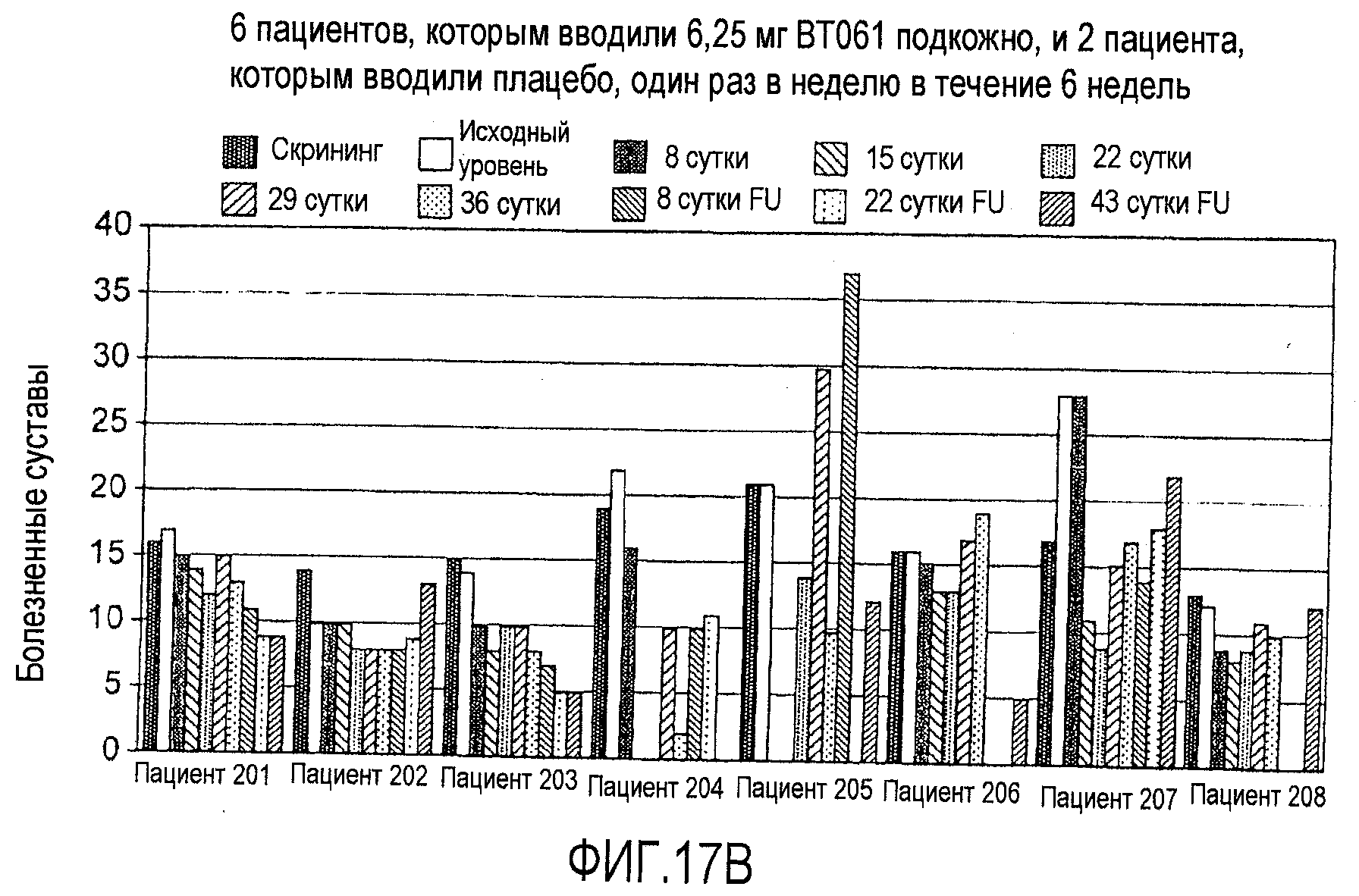

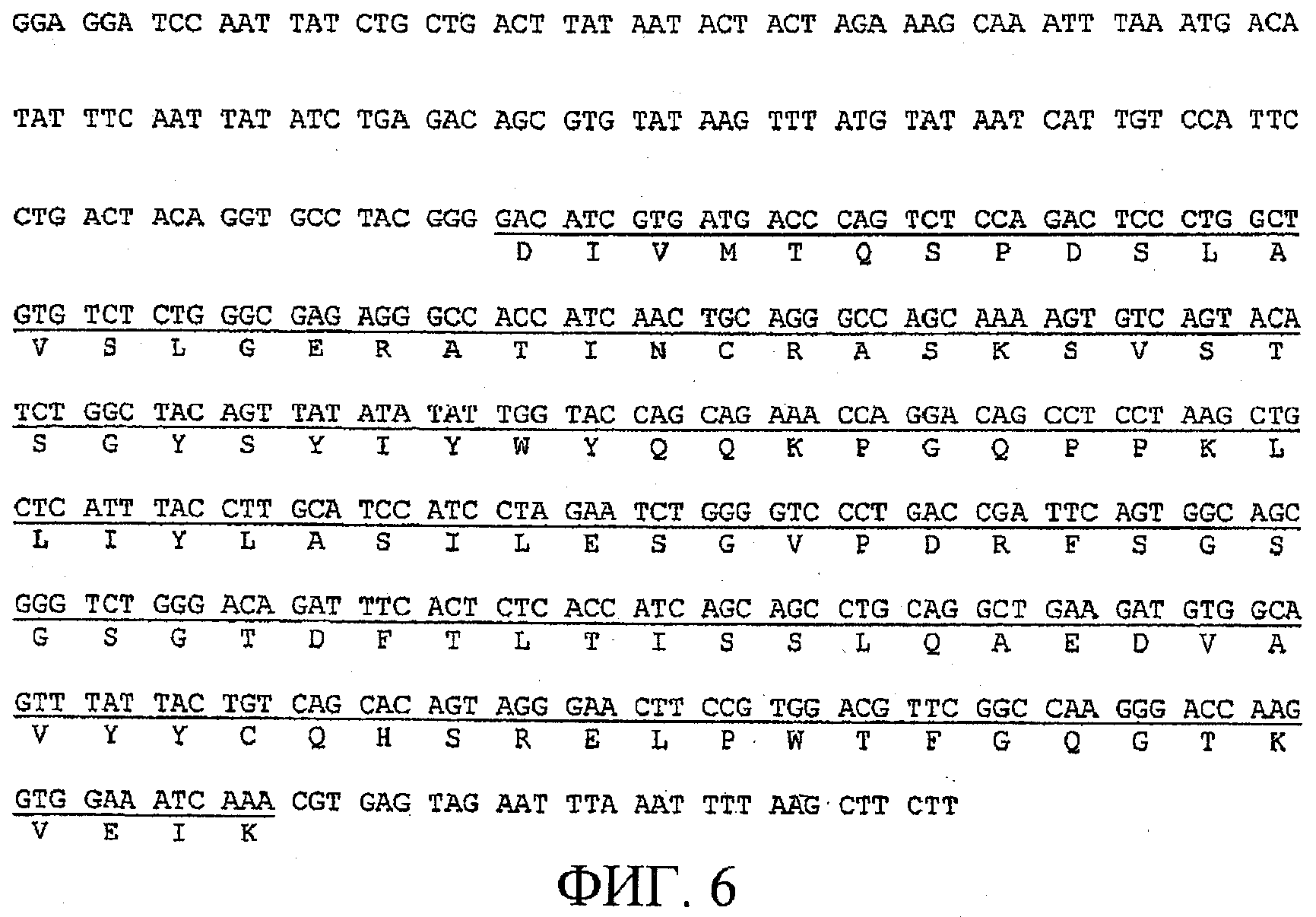
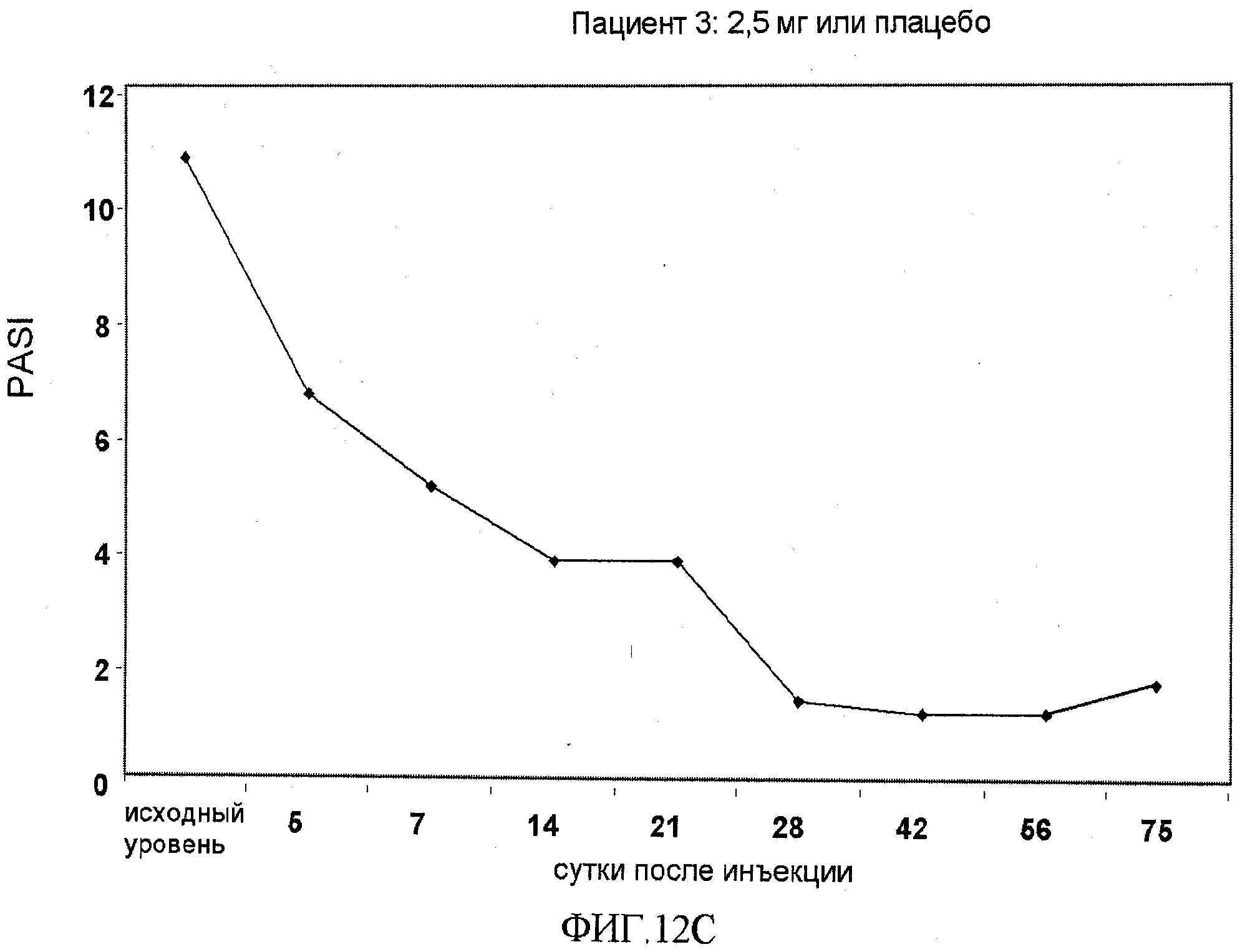
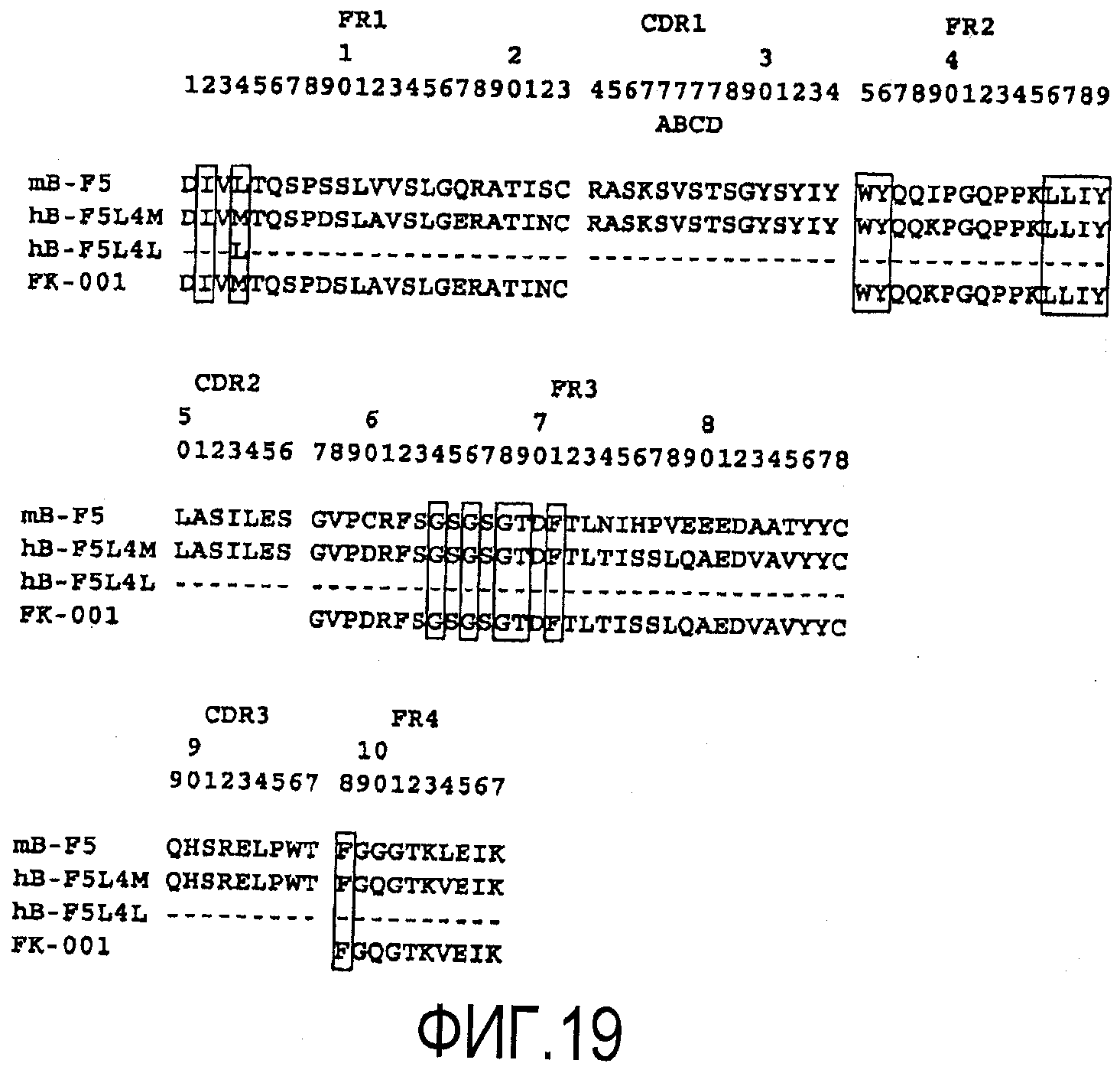
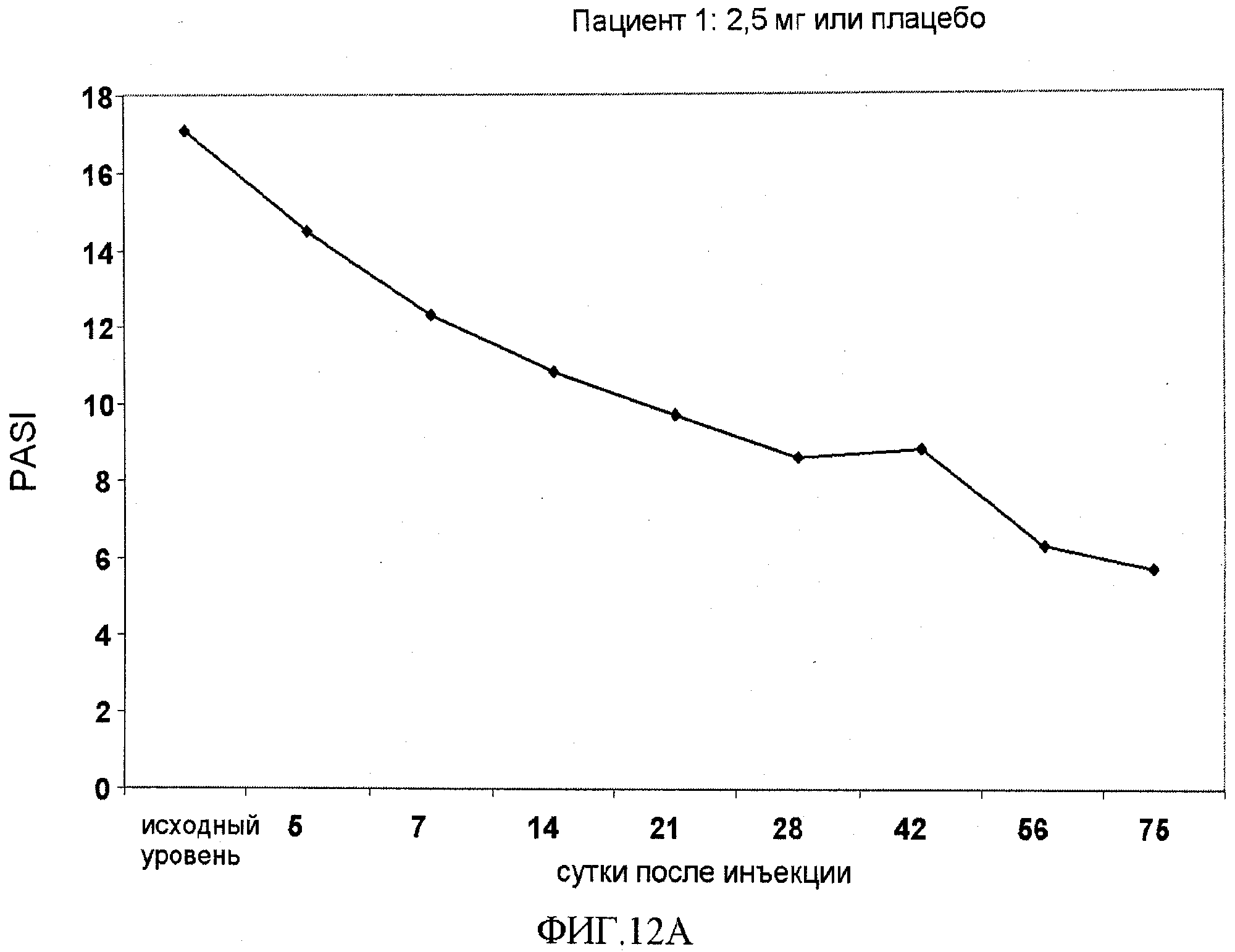
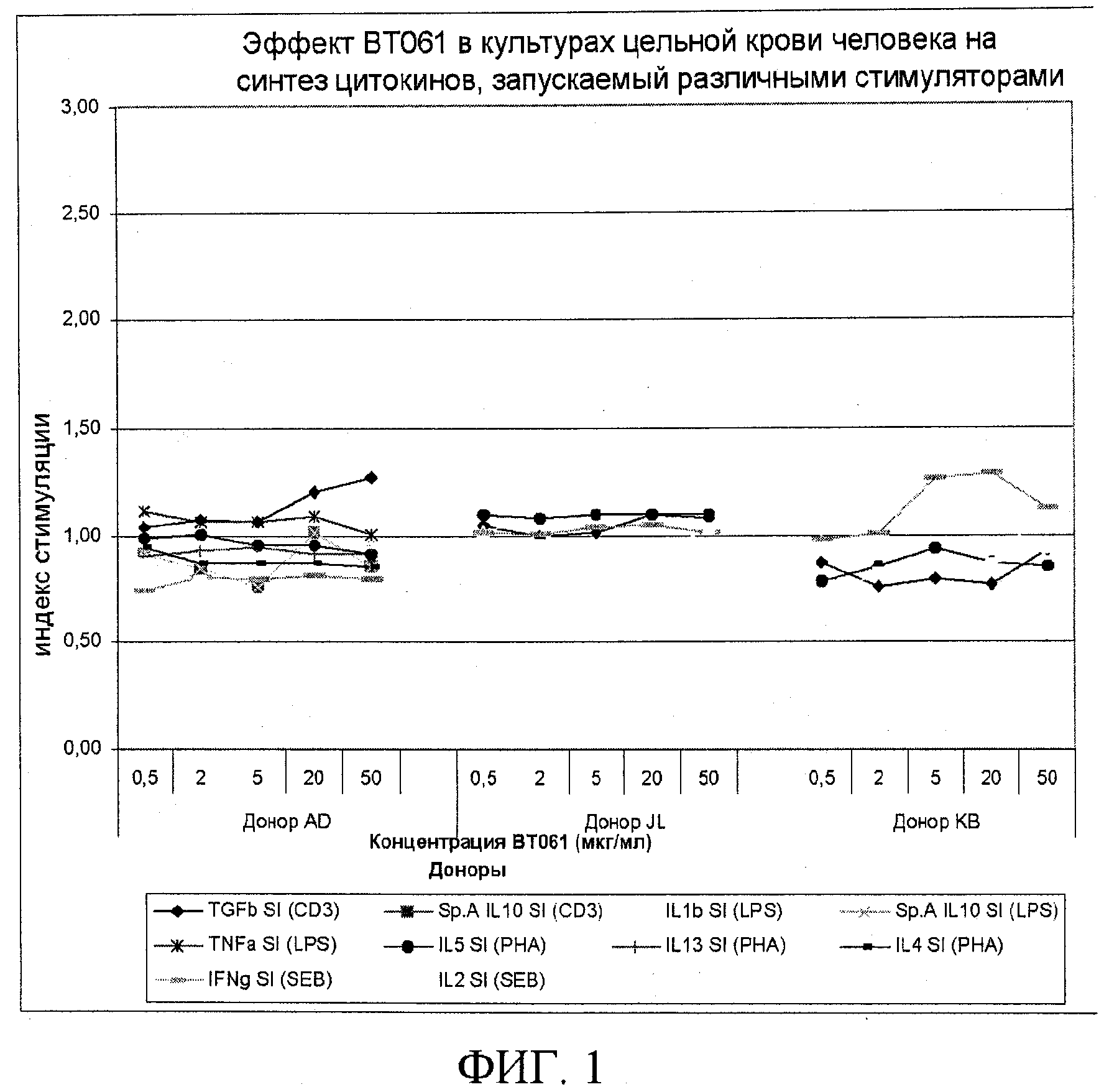


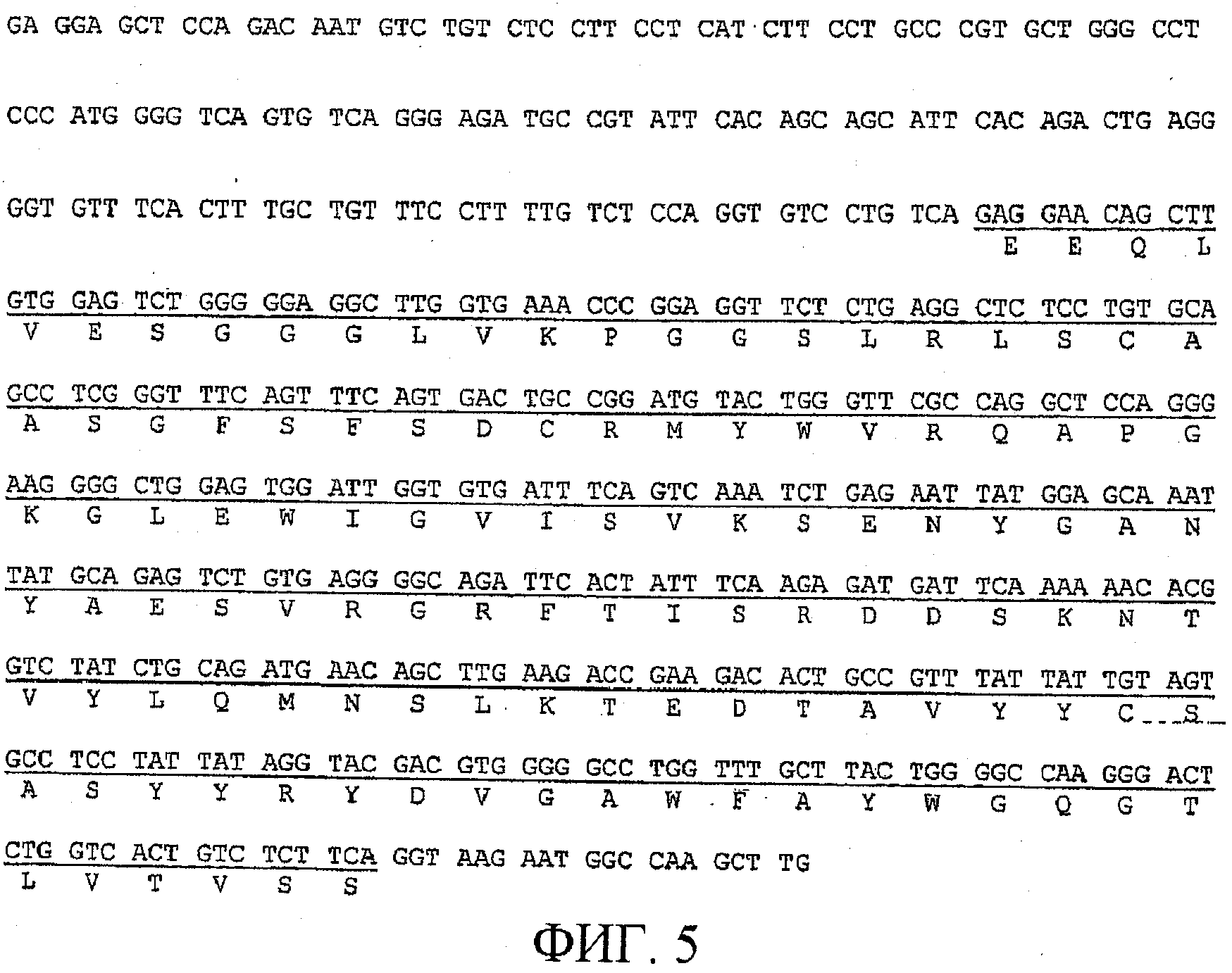
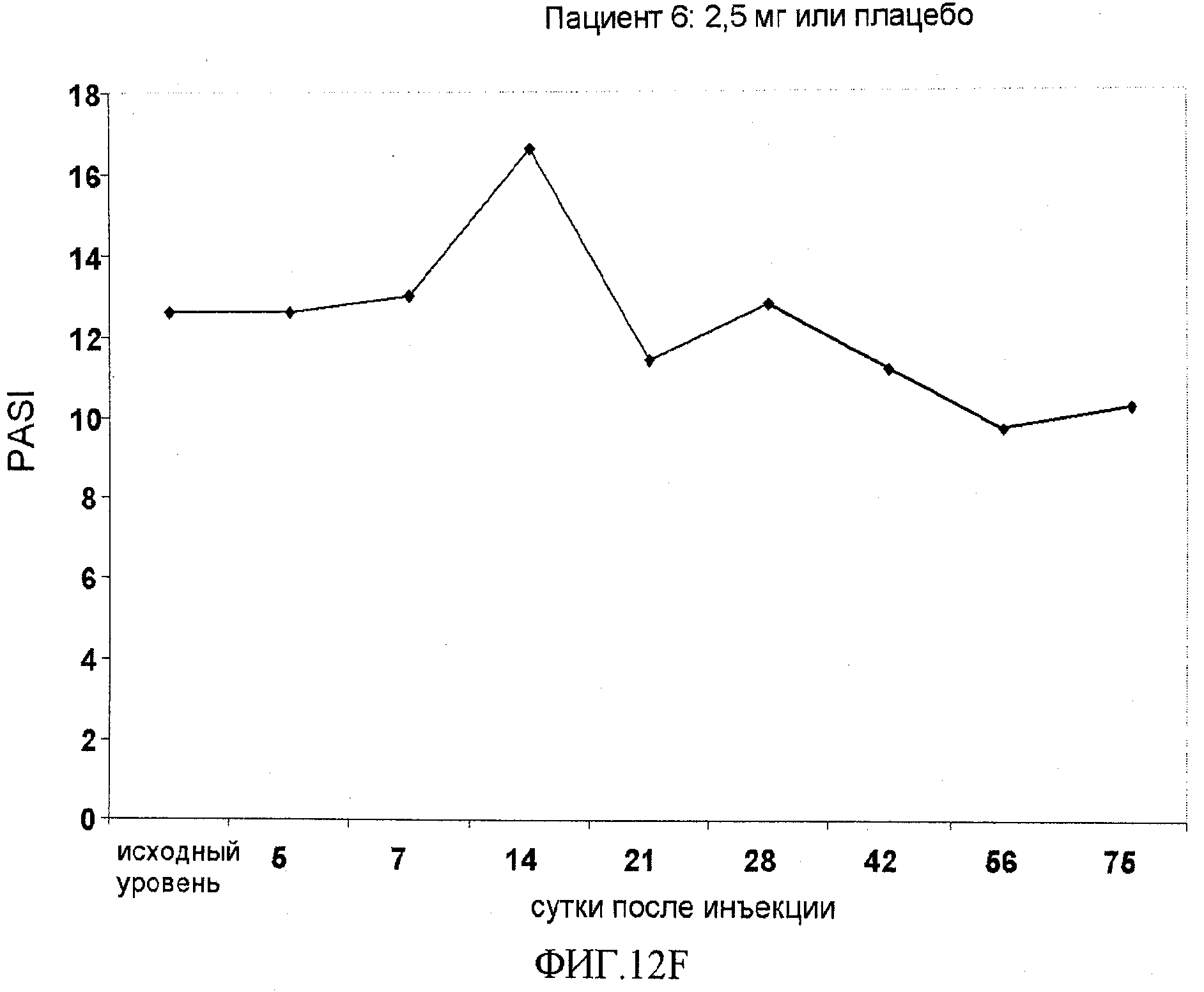

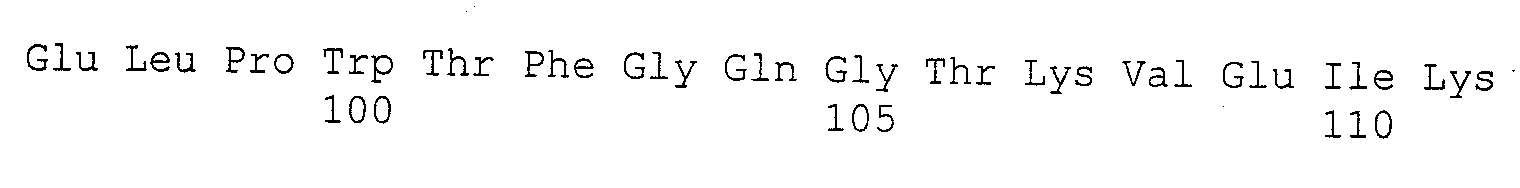
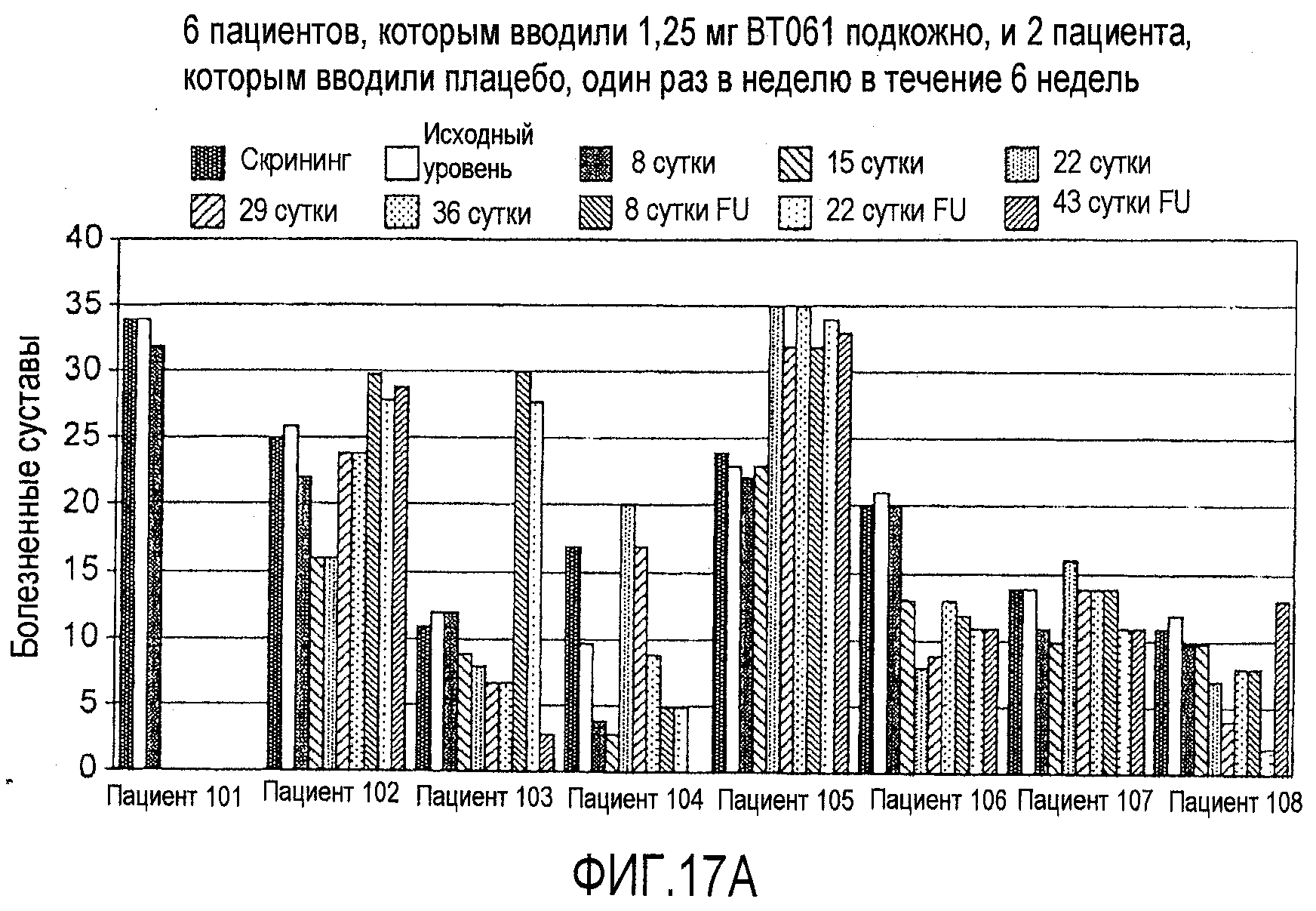

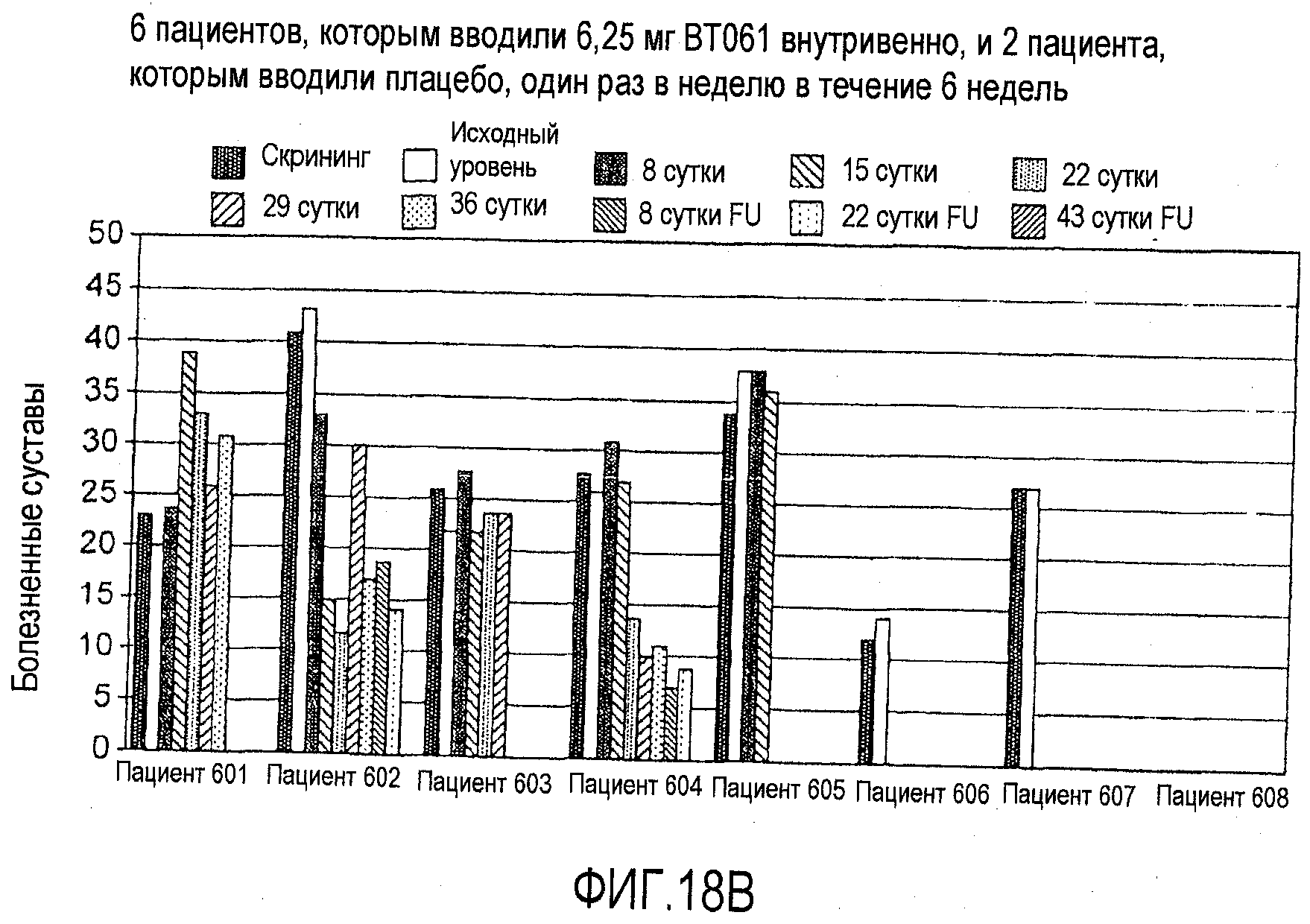
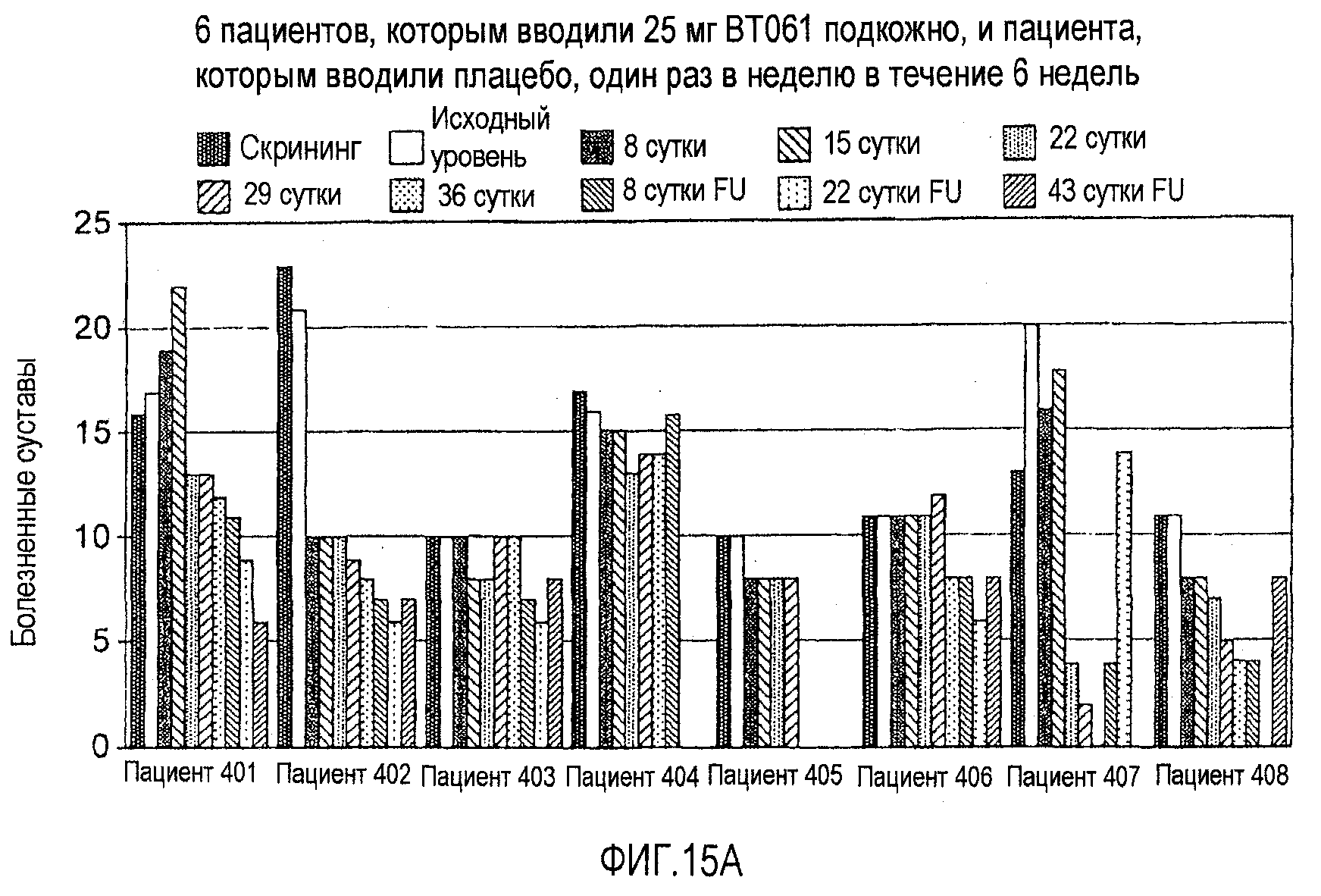
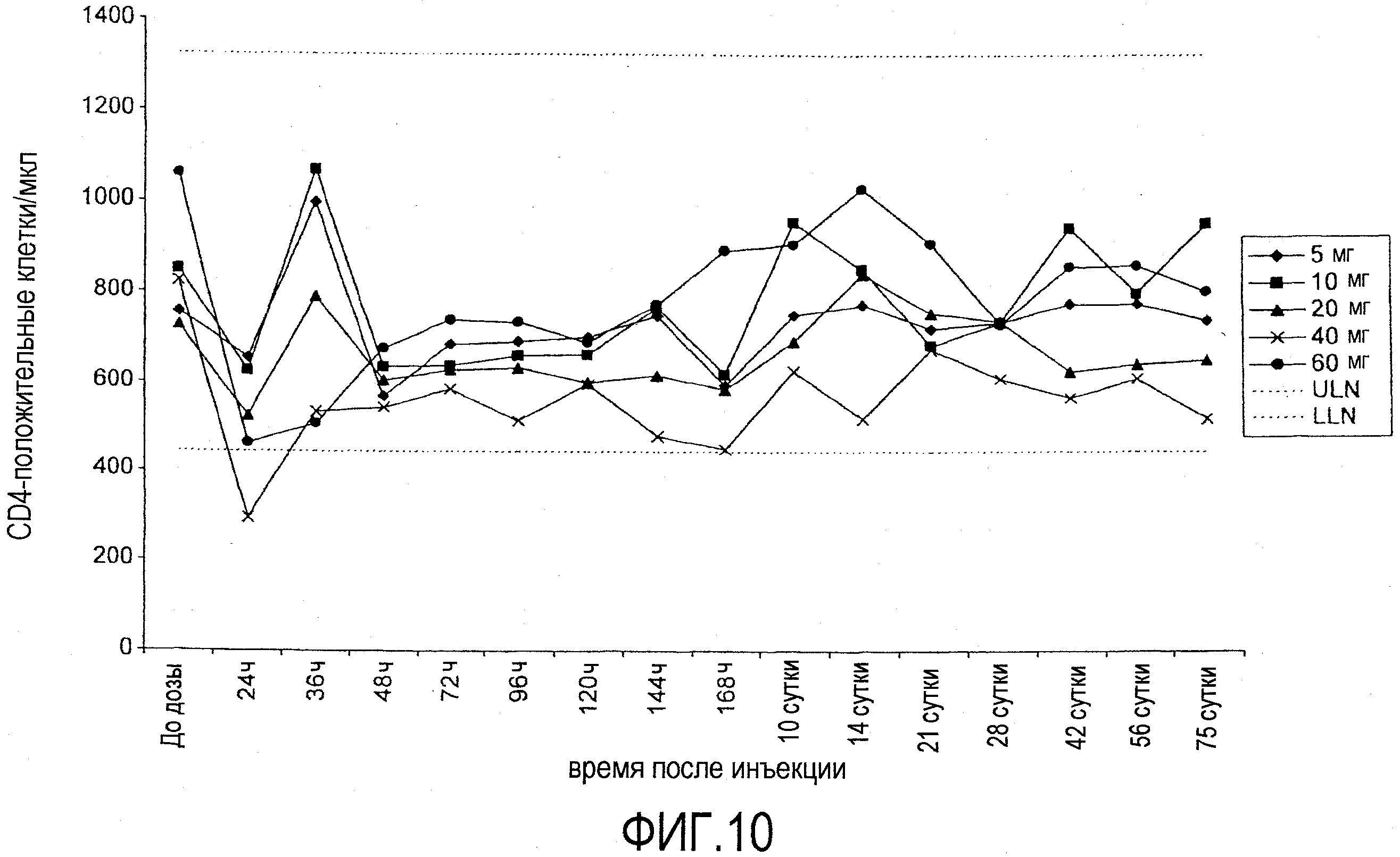
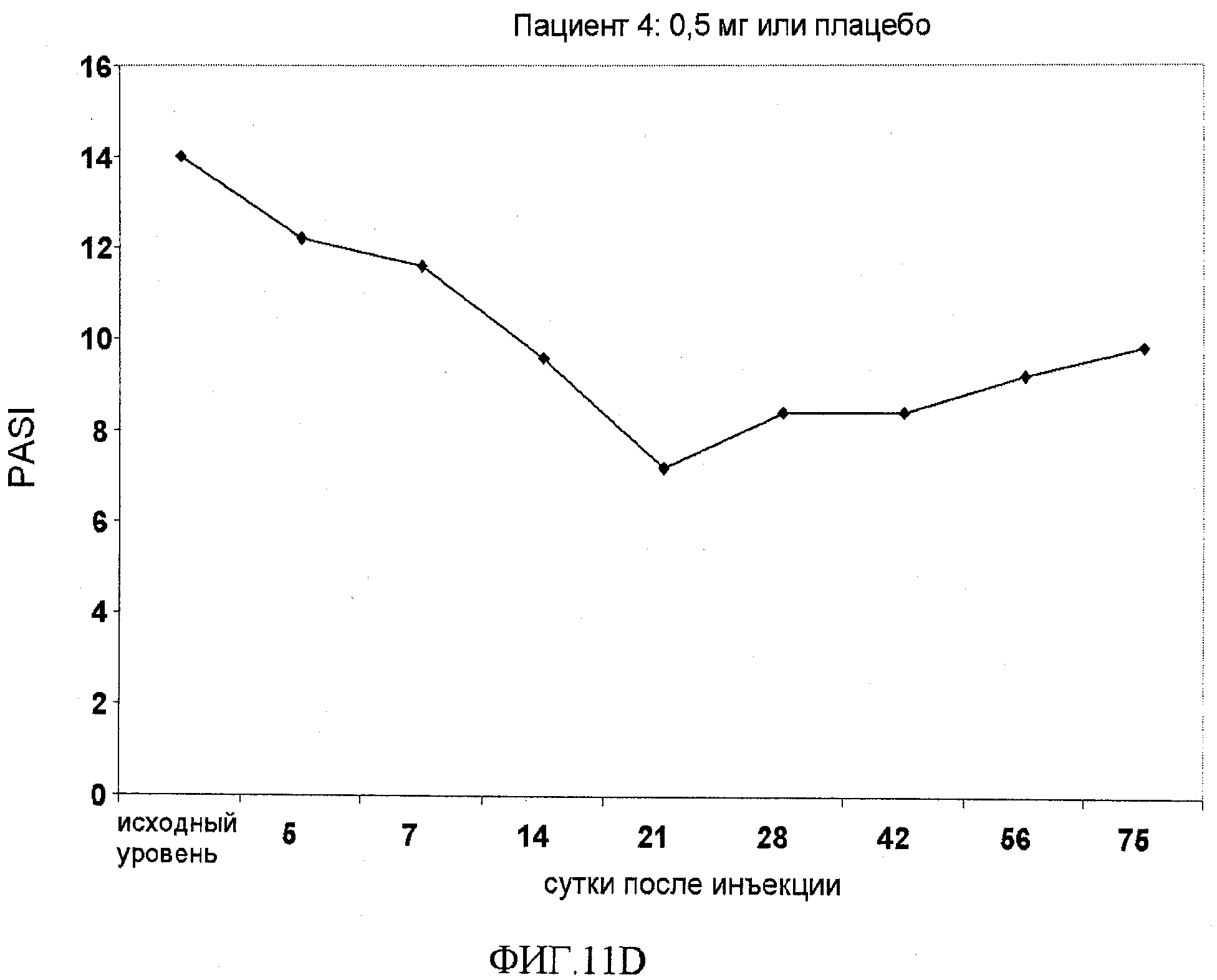
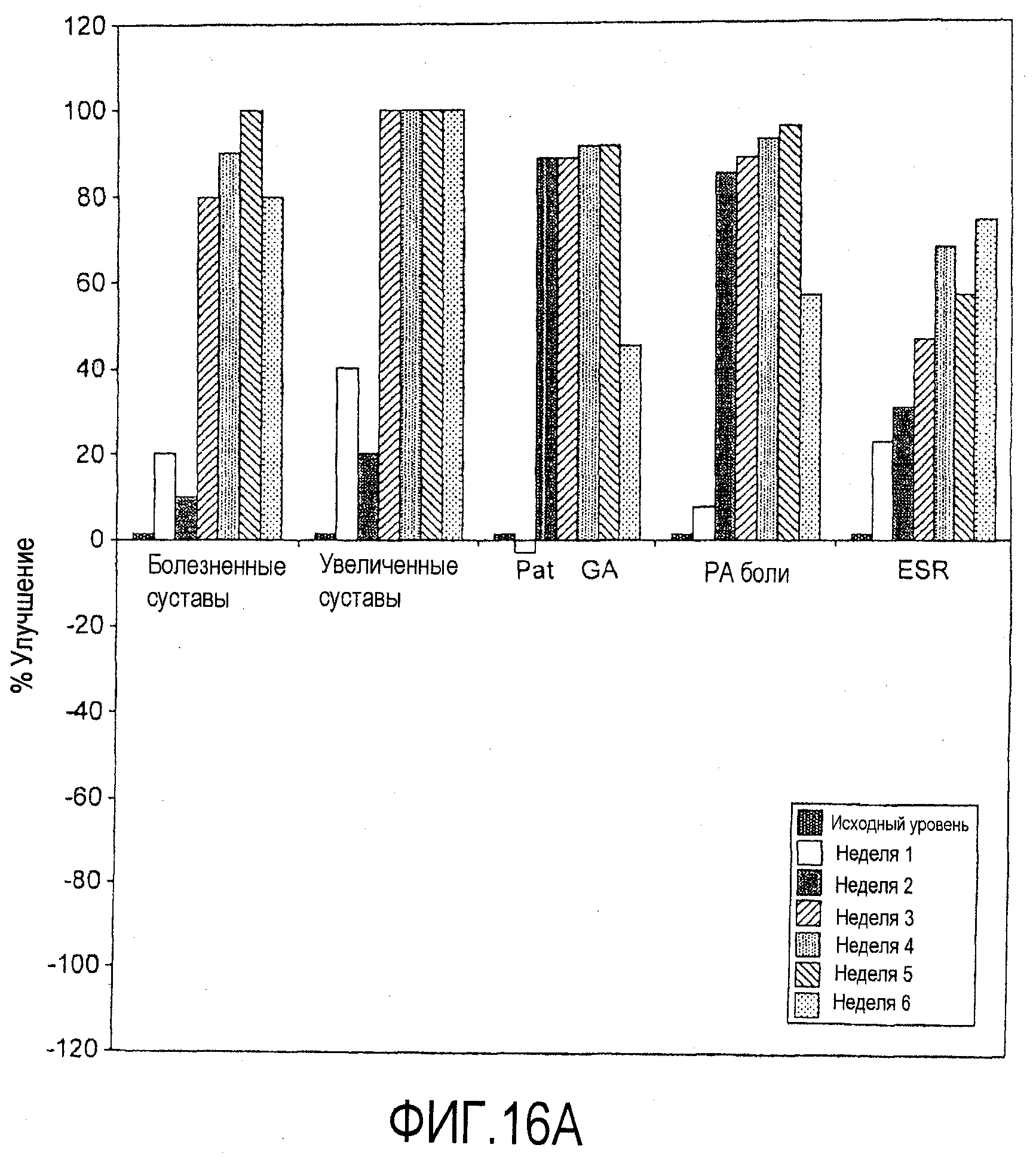
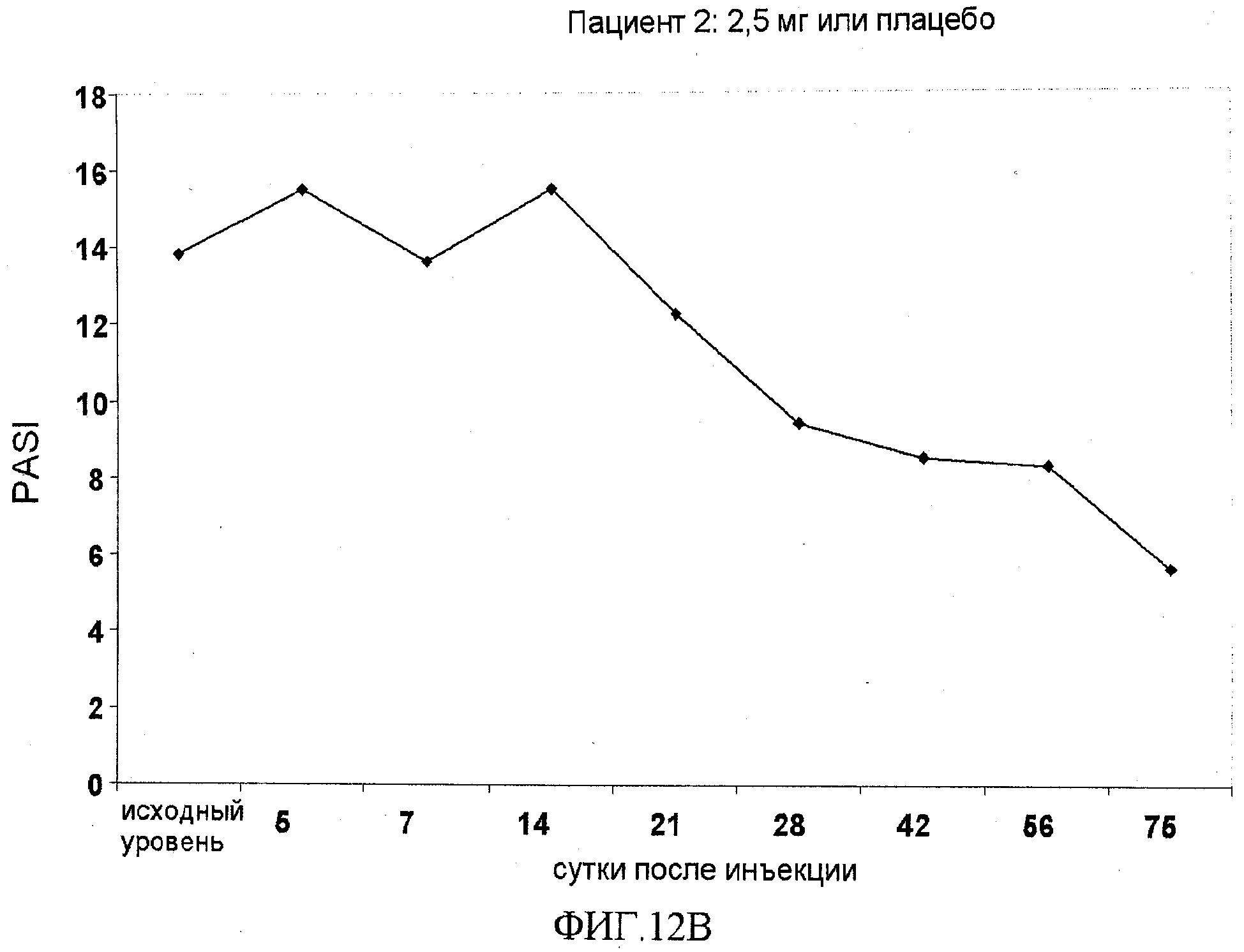
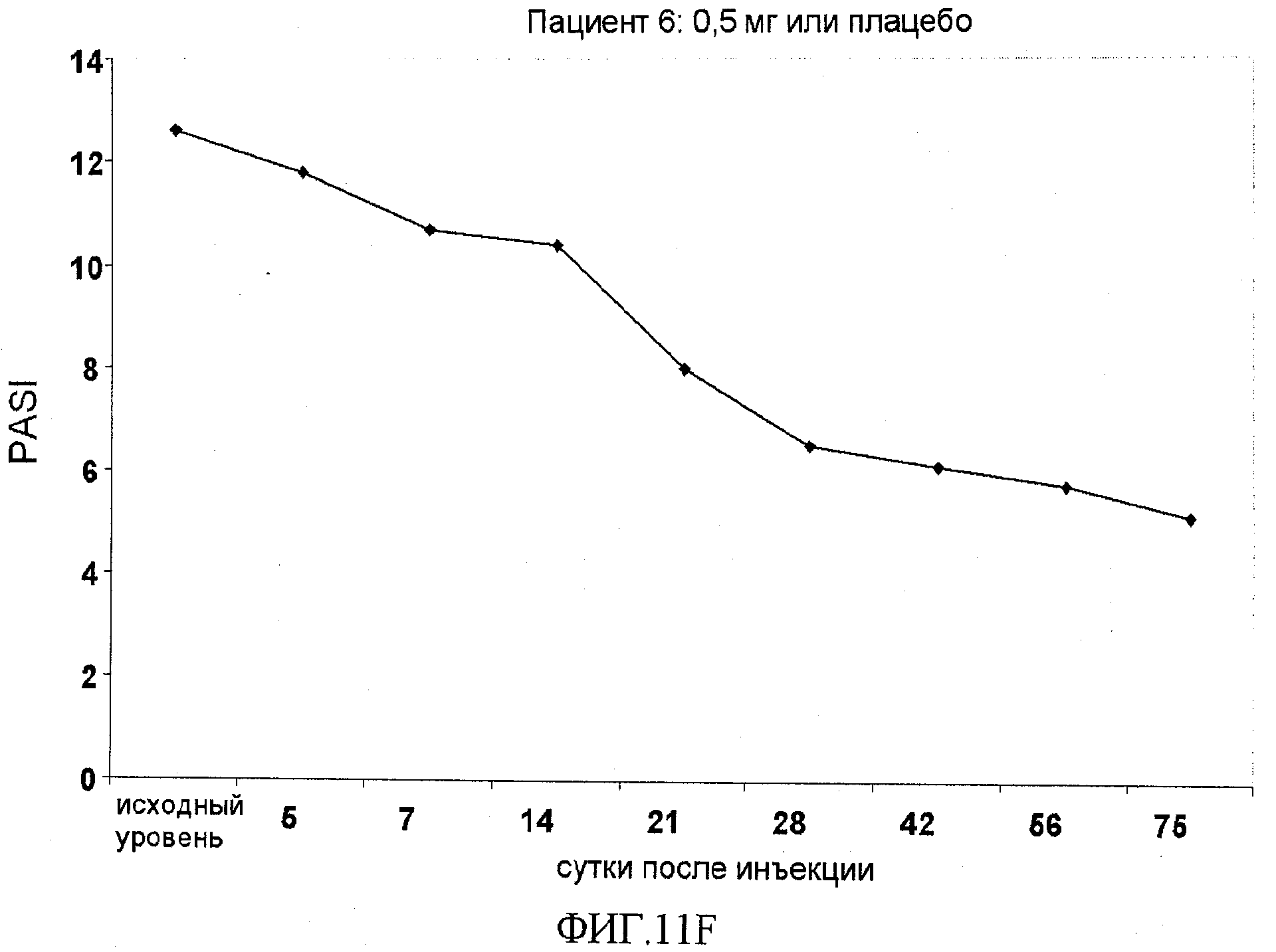
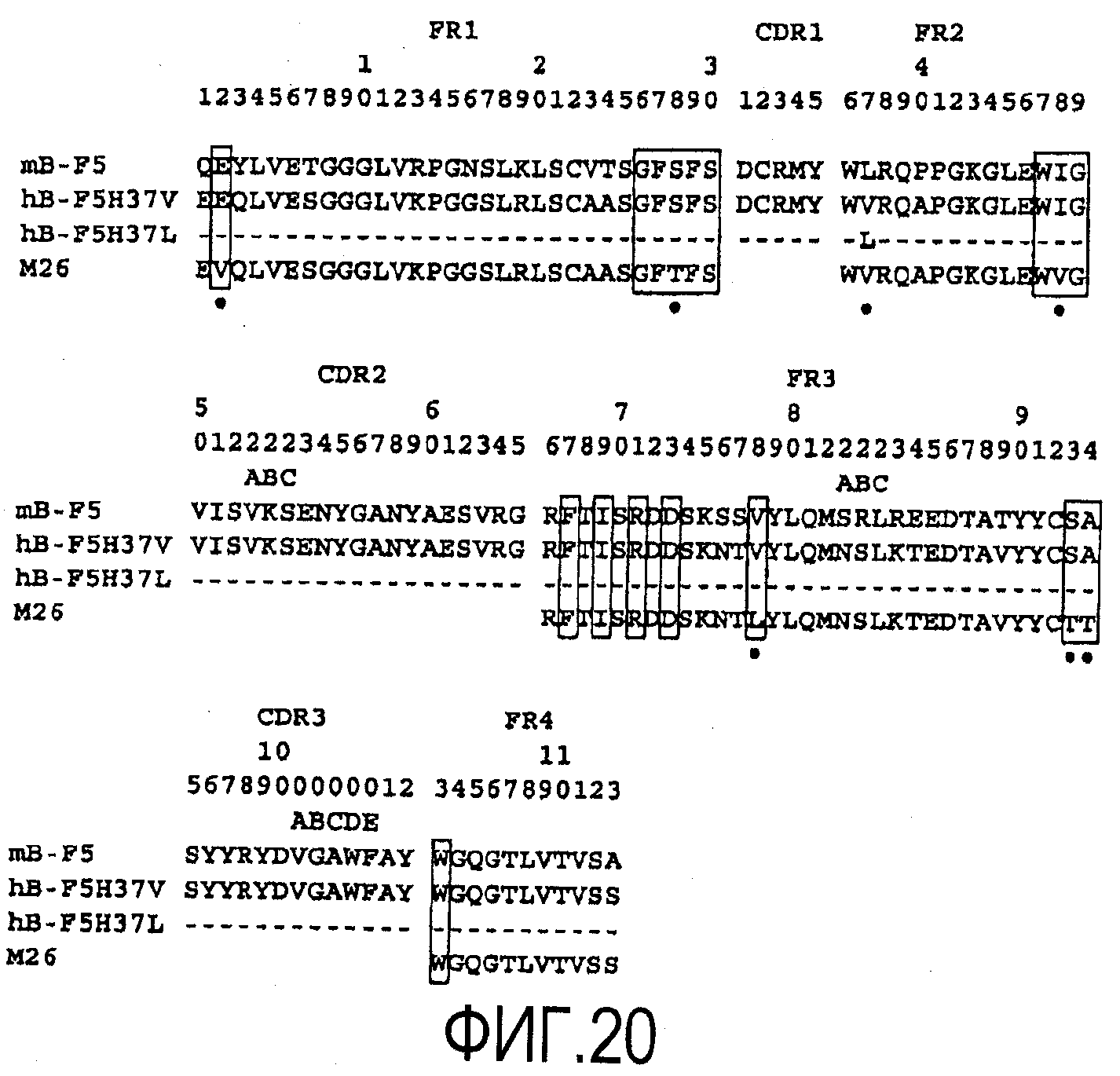
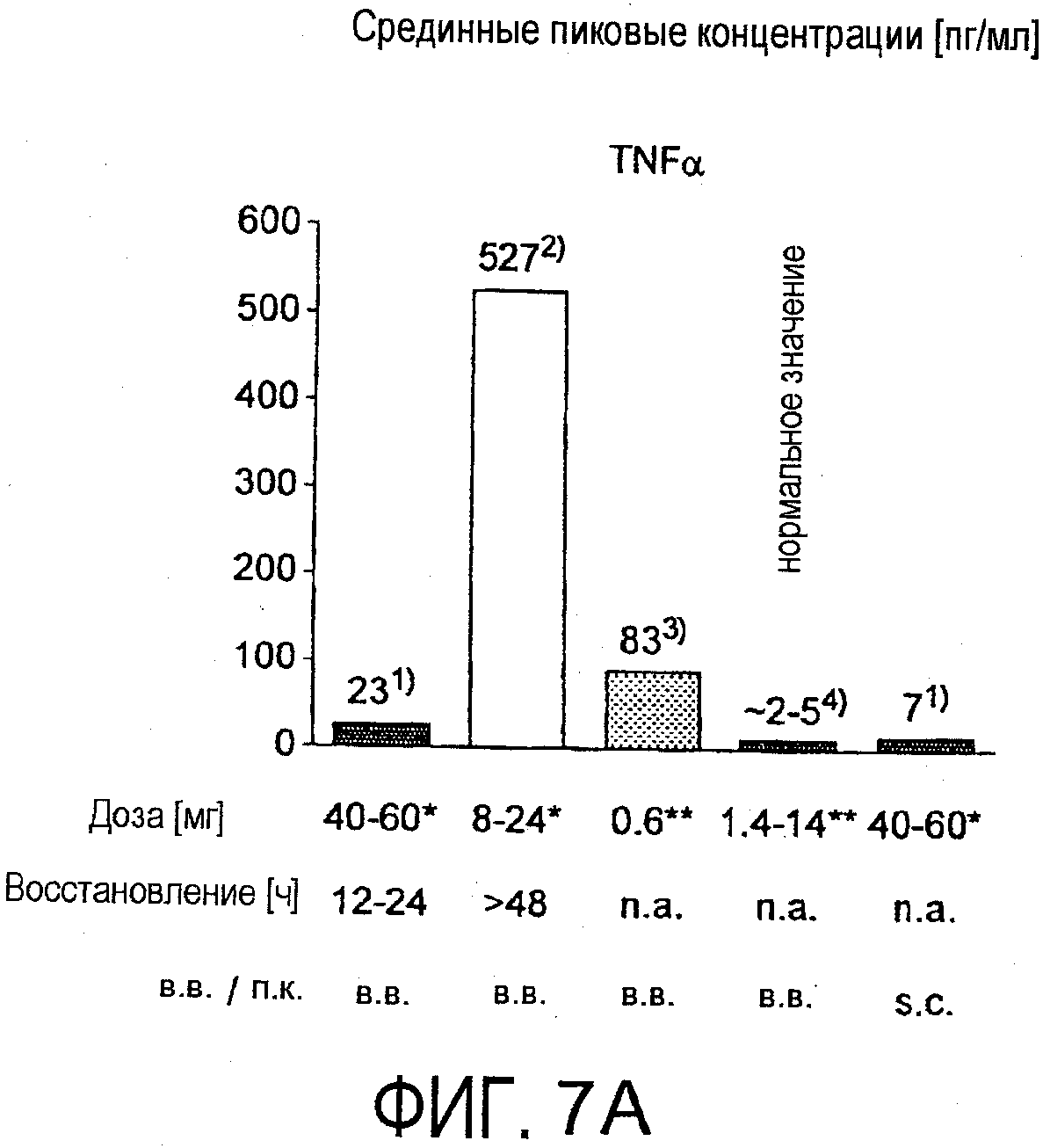

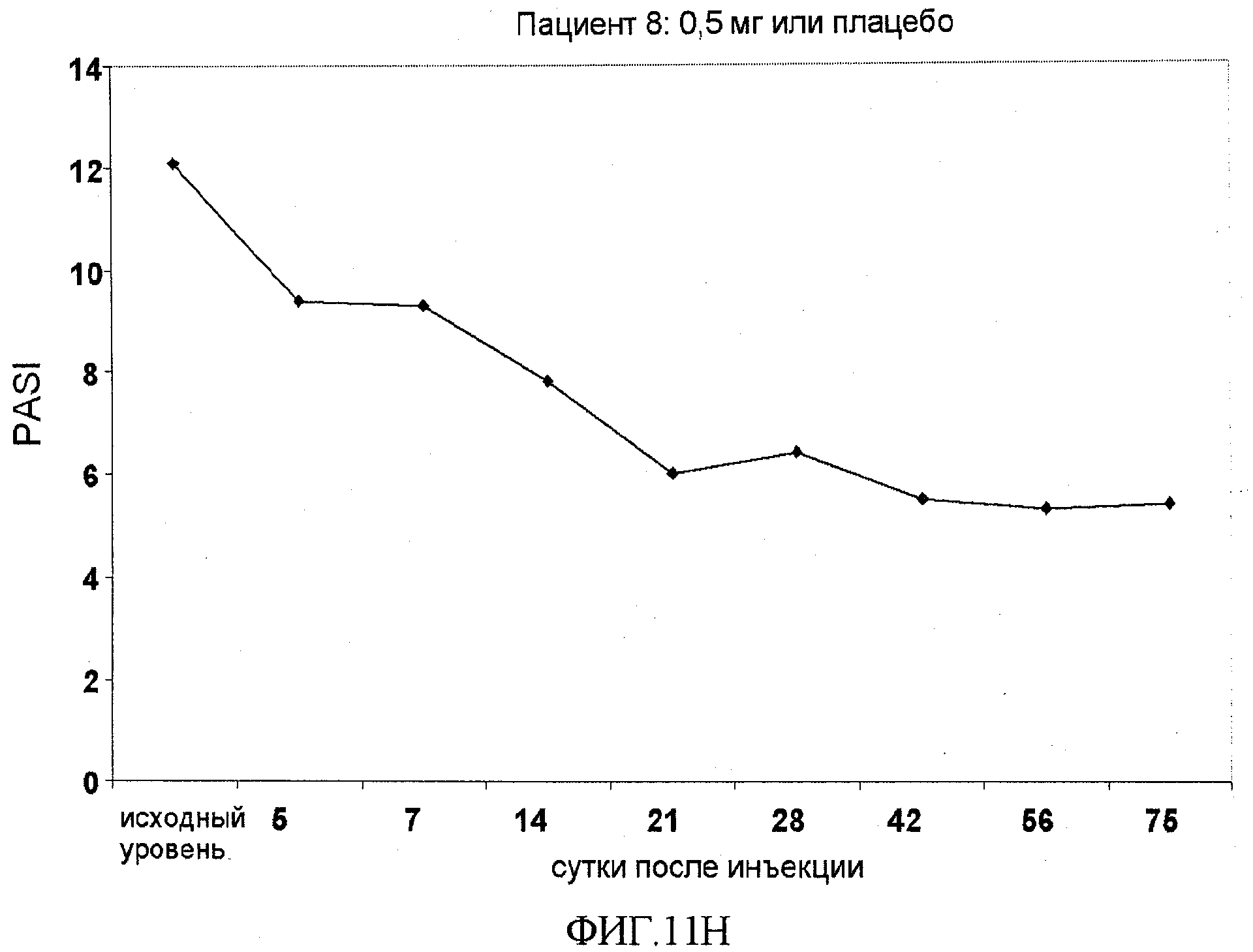
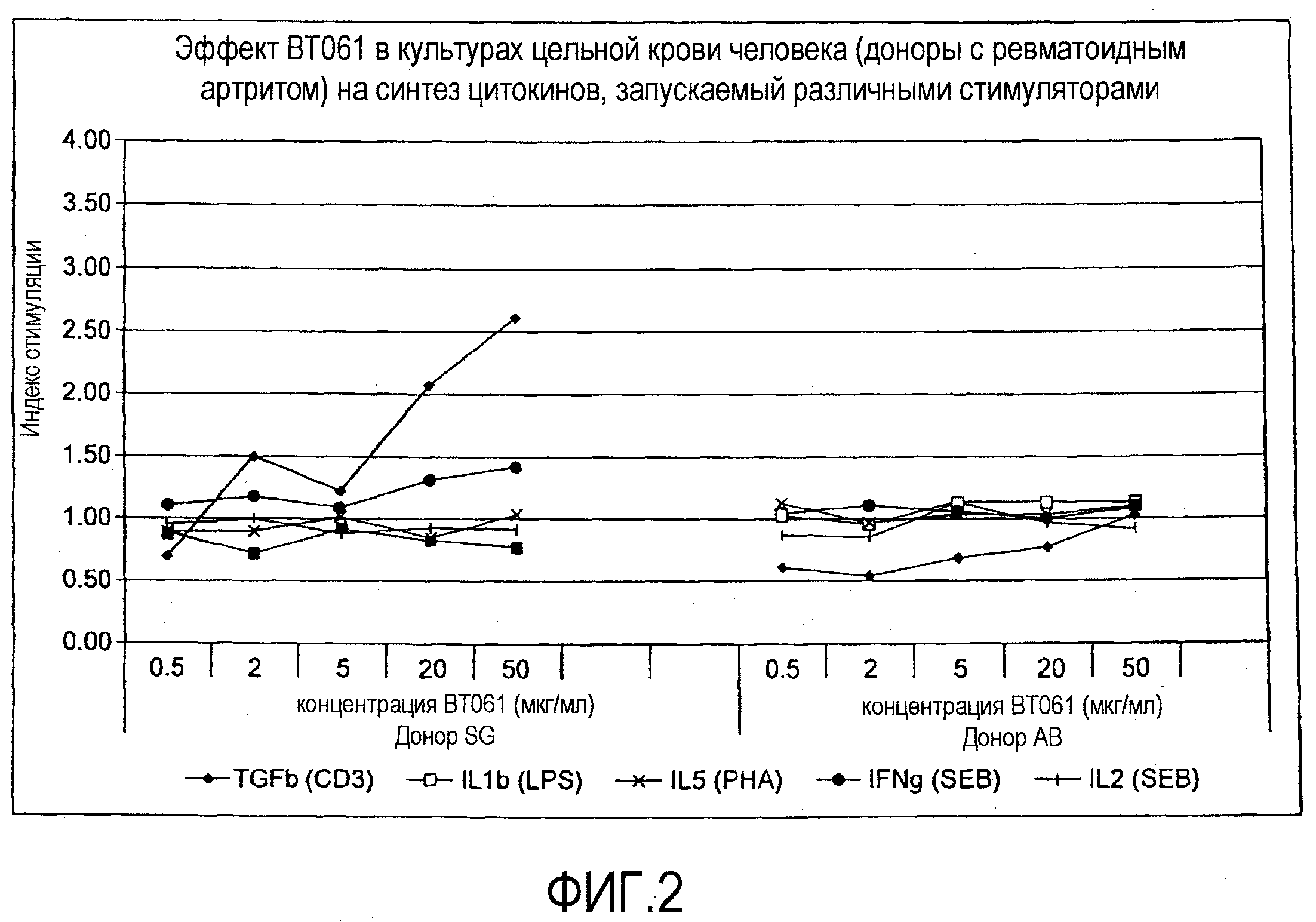
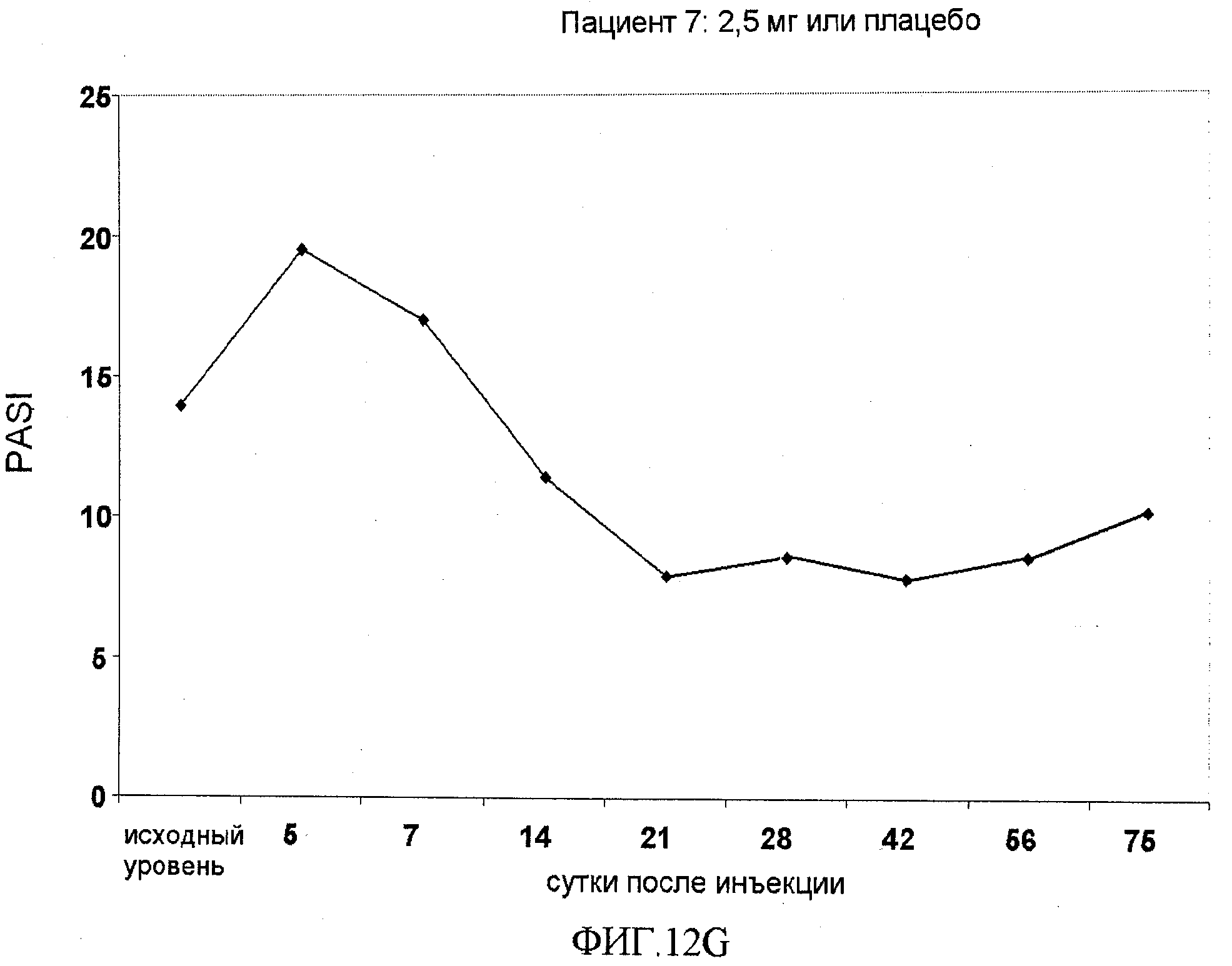

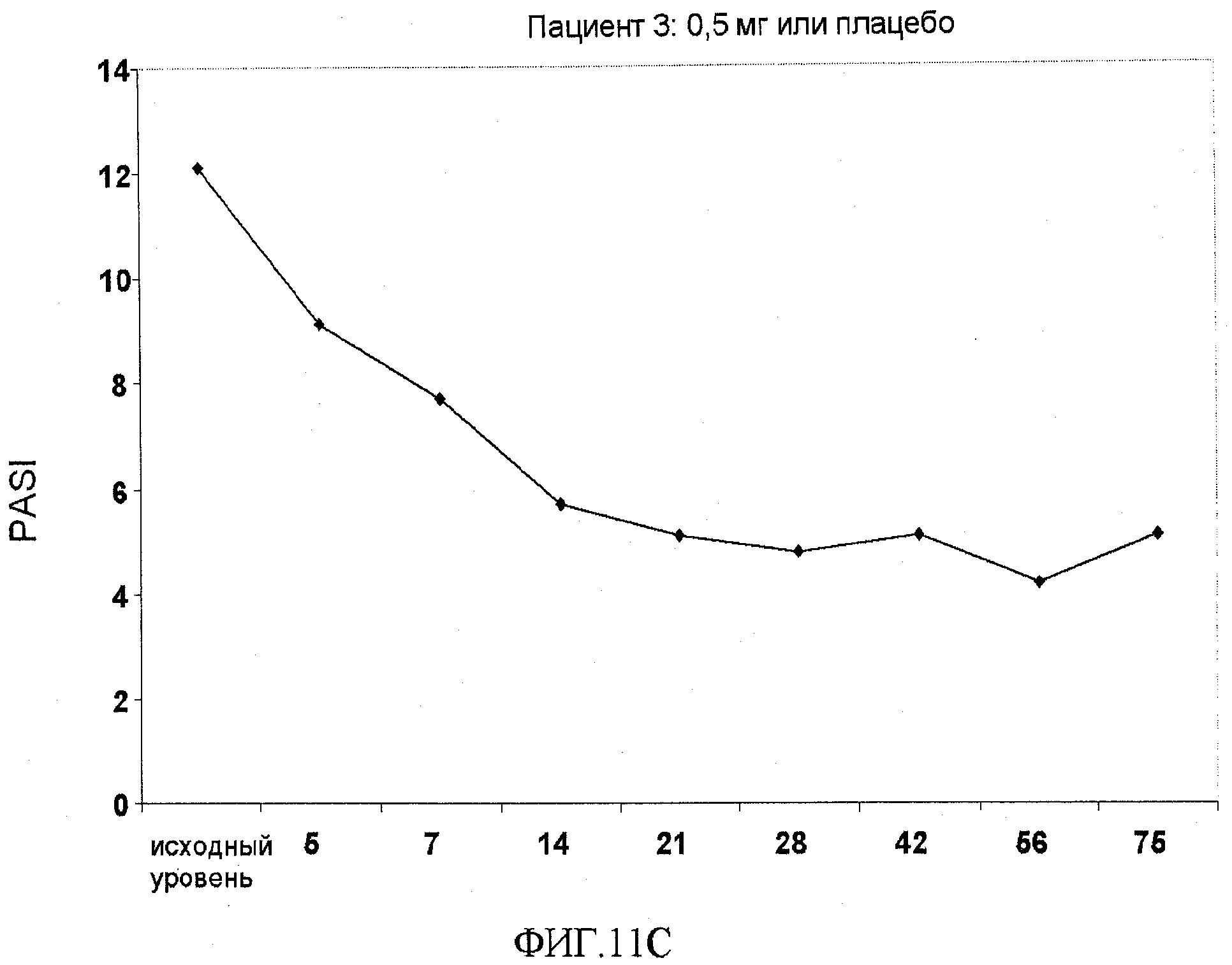
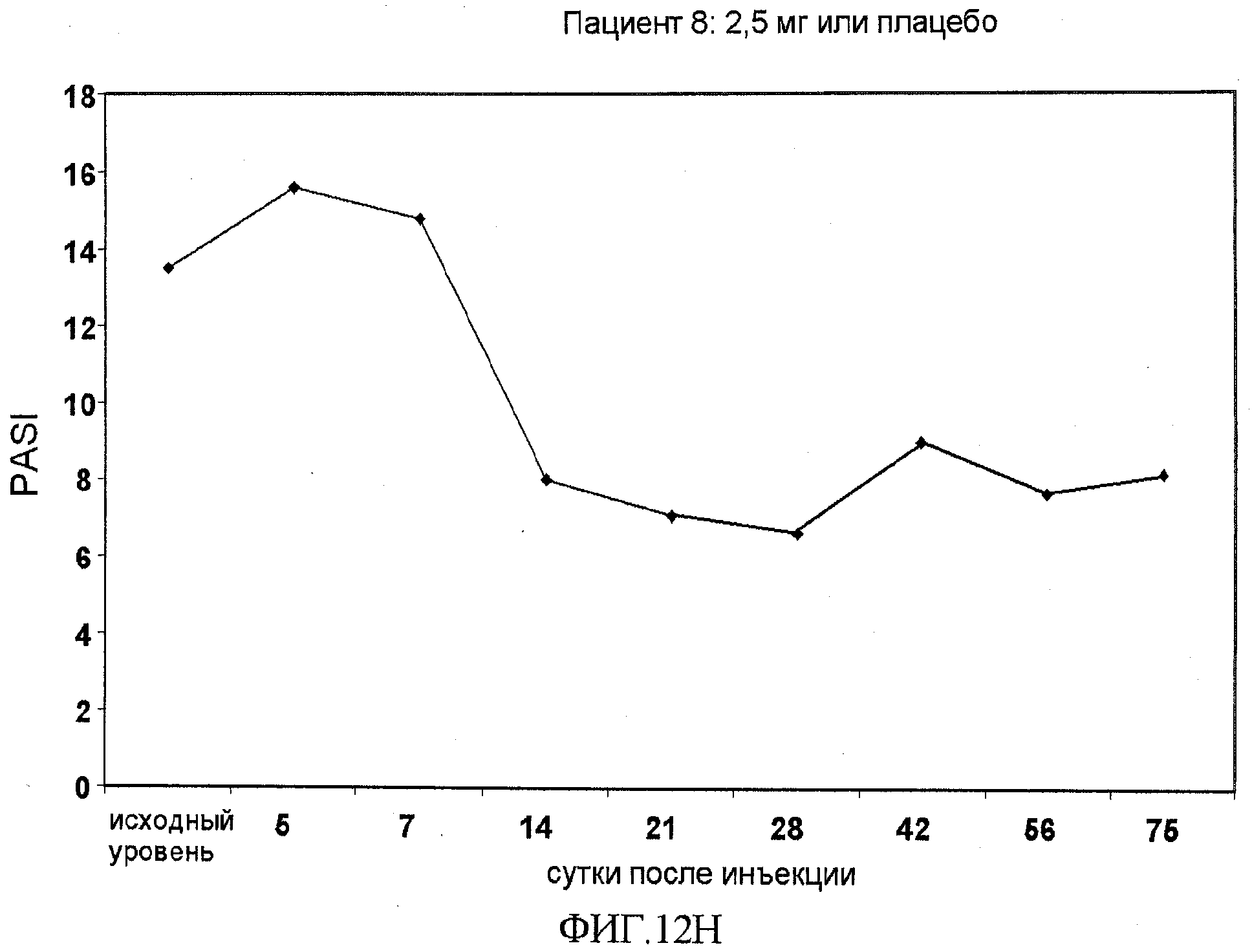

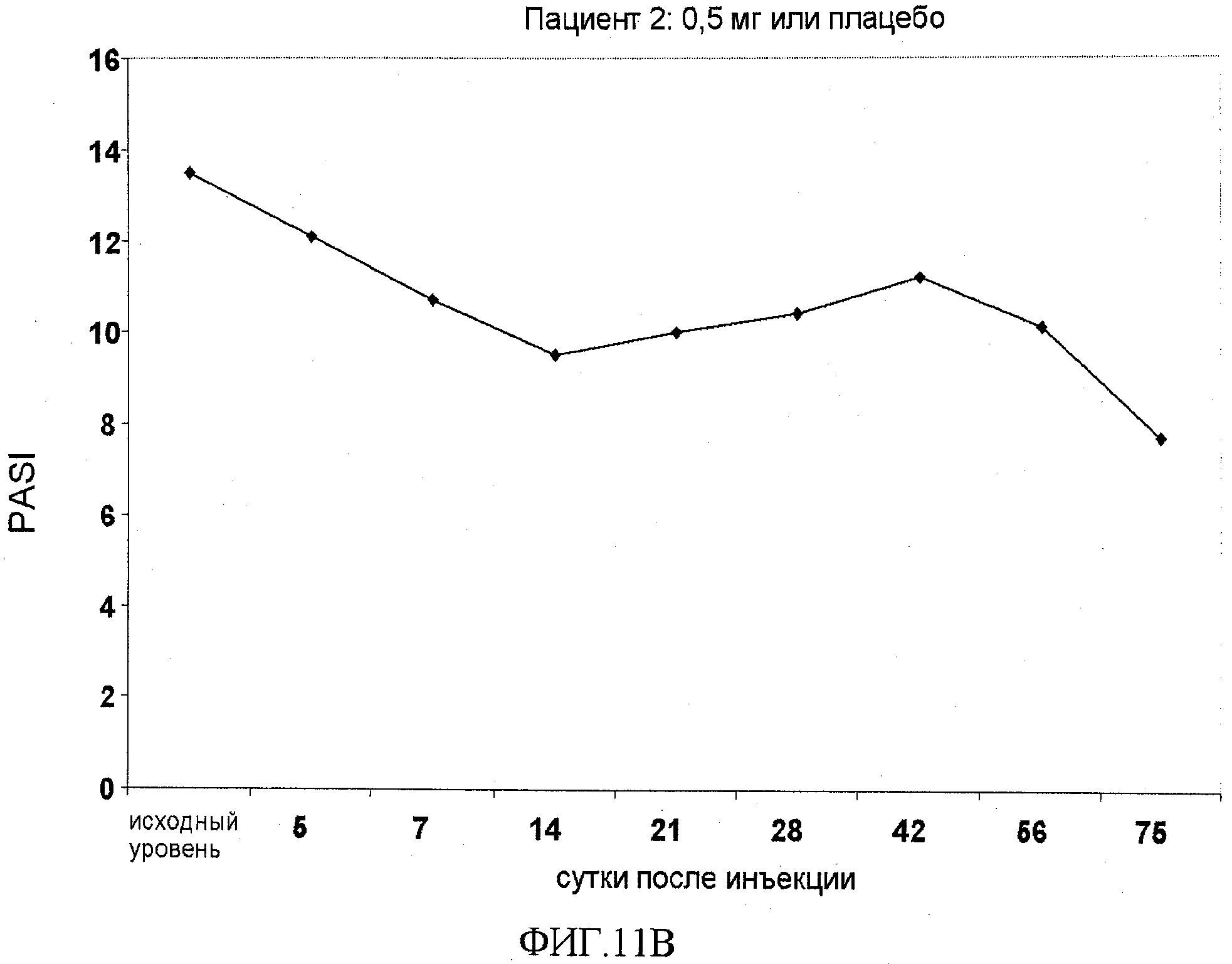
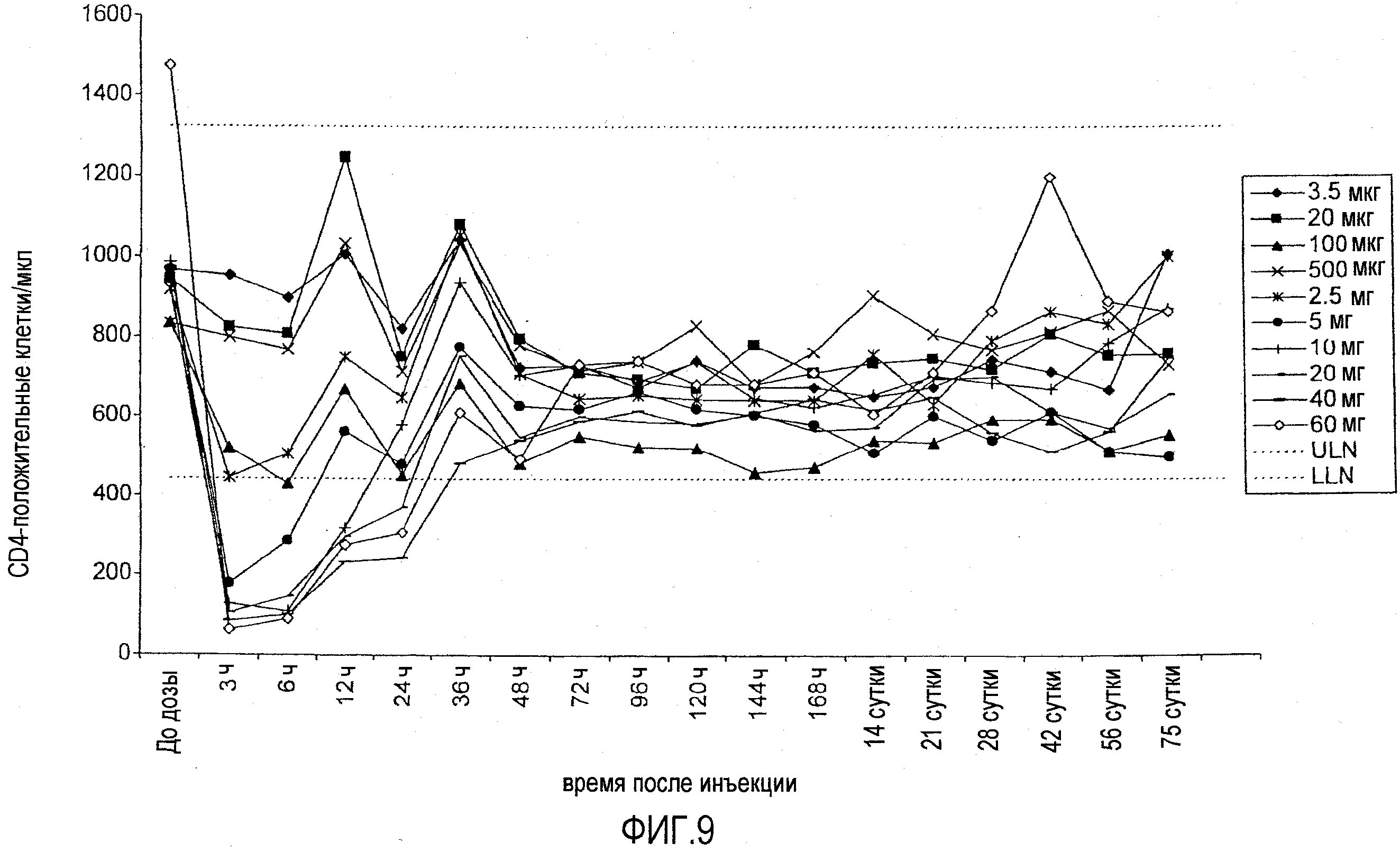


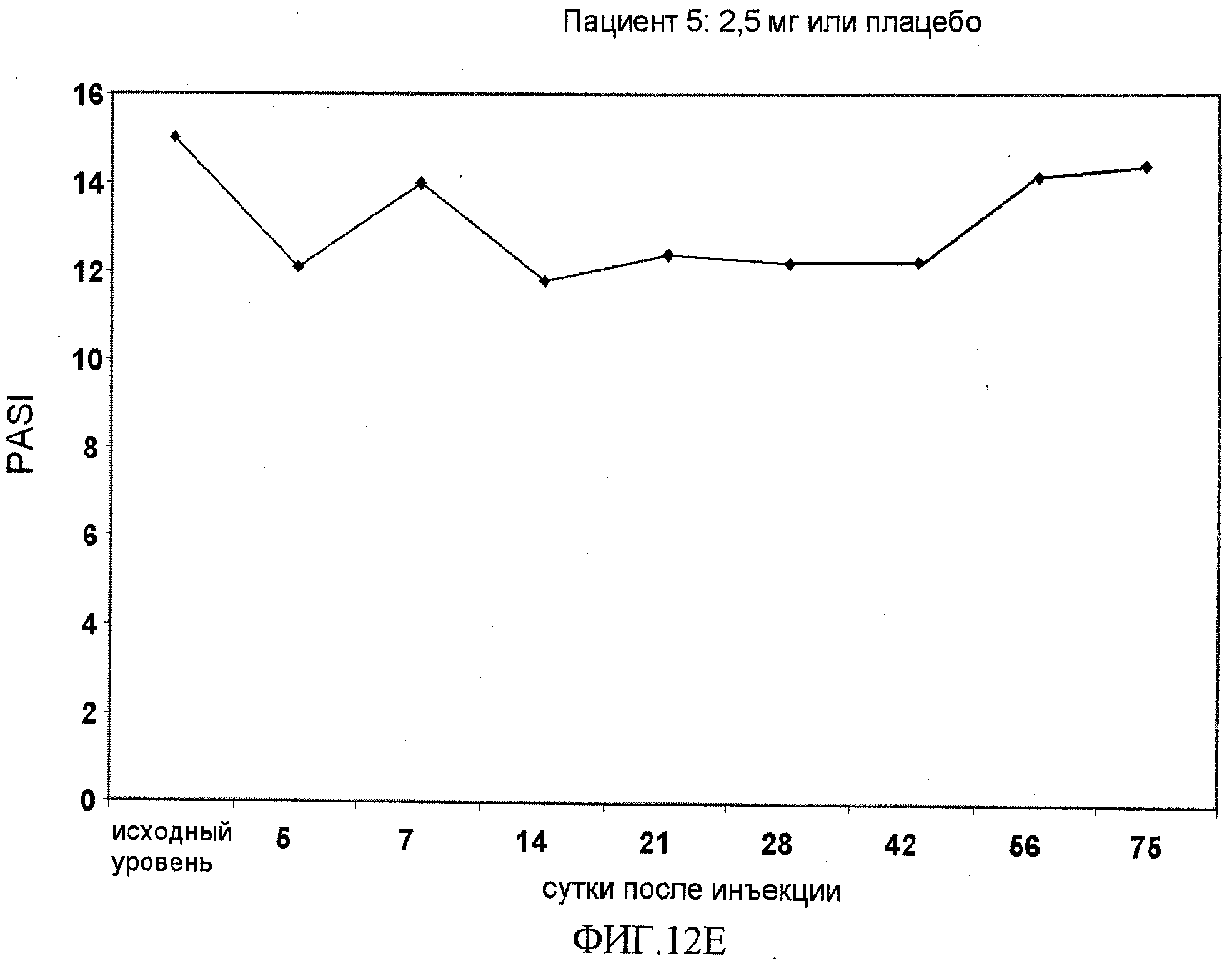
Способы улучшения направленного воздействия на cd138-экспрессирующие опухолевые клетки и агенты для их осуществления
Композиция для лечения заболевания
Агенты против клетки-мишени, нацеленные на cd138, и их применение
Способ лечения аутоиммунного заболевания (варианты)
Средство для лечения заболевания
Иммуноконъюгаты, направленные на cd138, и их применение
Применения иммуноконъюгатов, мишенью которых является cd138
Гуманизированные антитела против il-10 для лечения системной красной волчанки (sle)
Гуманизированные антитела против il-10 для лечения системной красной волчанки (sle)
Средства для лечения заболевания
Способы улучшения направленного воздействия на cd138-экспрессирующие опухолевые клетки и агенты для их осуществления
Композиция для лечения заболевания
Агенты против клетки-мишени, нацеленные на cd138, и их применение
Способ лечения аутоиммунного заболевания (варианты)
Средство для лечения заболевания
Иммуноконъюгаты, направленные на cd138, и их применение
Применения иммуноконъюгатов, мишенью которых является cd138
Гуманизированные антитела против il-10 для лечения системной красной волчанки (sle)
Гуманизированные антитела против il-10 для лечения системной красной волчанки (sle)
Средства для лечения заболевания

