СПОСОБ ПОЛУЧЕНИЯ 5-БИФЕНИЛ-4-АМИНО-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ
Вид РИД
Изобретение
Настоящее изобретение относится к пирролидин-2-онам, соответствующим формуле (1), или к их солям
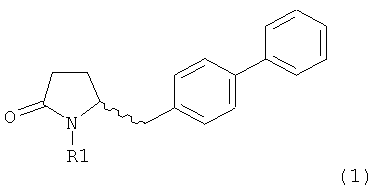 ,
,
где R1 представляет собой водород или азотзащитную группу, определенную в настоящем описании, к способам их получения или к их применению для получения NEP-ингибиторов, в частности, для получения этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соли.
Эндогенные предсердные натрийуретические пептиды (ANP), также называемые предсердными натрийуретическими факторами (ANF), у млекопитающих обладают диуретическим, натрийуретическим и сосудорасширяющим действием. Природные ANF-пептиды являются метаболически инактивированными, в частности, при помощи разрушающих ферментов, которые, как установлено, соответствуют фермент-нейтральной эндопептидазе (NEP, ЕС 3.4.24.11), которая также отвечает, например, за метаболическую инактивацию энкефалинов.
Из уровня техники известны биарилзамещенные производные фосфоновой кислоты, пригодные в качестве ингибиторов нейтральной эндопептидазы (NEP), например, в качестве ингибиторов ANF-разрушающего фермента у млекопитающих, таким образом, способствуя пролонгированию и эффективности диуретических, натрийуретических и сосудорасширяющих свойств ANF у млекопитающих, посредством ингибирования их разложения в менее активные метаболиты. Таким образом, NEP-ингибиторы особенно пригодны для лечения состояний и расстройств, чувствительных к ингибированию нейтральной эндопептидазы (ЕС 3.4.24.11), в частности, сердечно-сосудистых расстройств, таких как гипертензия, почечной недостаточности, включая водянку и отложение солей, отека легких и ишемической болезни сердца.
Способы получения NEP-ингибиторов являются известными.
В US 5217996 описаны биарилзамещенные производные амида 4-аминомасляной кислоты, пригодные в качестве ингибиторов нейтральной эндопептидазы (NEP), например, в качестве ингибиторов ANF-разрушающего фермента у млекопитающих. В качестве предпочтительного воплощения US 5217996 раскрыт этиловый эфир N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты и способ его получения.
Некоторые ингибиторы нейтральной эндопептидазы дипептиддикарбоновой кислоты (NEP) описаны G.M. Ksander и др. в J. Med. Chem. 1995, 38, 1689-1700, "Dicarboxylic Acid Dipeptide Neutral Endopeptidase Inhibitors". Среди прочих раскрыт этиловый эфир N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты и способ его получения.
Таким образом, задачей настоящего изобретения является создание альтернативного реакционного пути для способа получения NEP-ингибиторов или их пролекарств, в частности, задачей является создание альтернативного реакционного пути для способа получения этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соли.
В US 5217996 раскрыто получение этилового эфира N-(3-карбоксил-1-оксопропил)-(4R)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты. При получении данного соединения этиловый эфир N-трет-бутоксикарбонил-(4S)-(n-фенилфенилметил)-4-амино-2-метил-2-бутеновой кислоты подвергают гидрированию в присутствии палладия на активированном угле. Основным недостатком данного процесса является то, что такая стадия гидрирования является не очень селективной и приводит к получению этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты в виде смеси диастереомеров (80:20). Кроме того, в данном способе требуется использование D-тирозина, в качестве исходного вещества, который является неприродной аминокислотой и не является легко доступным.
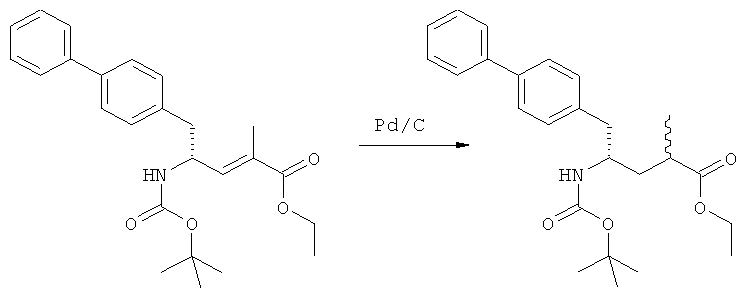
Таким образом, задачей настоящего изобретения создание альтернативного реакционного пути для способа получения этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соли, предпочтительно, реакционного пути, лишенного упомянутых выше недостатков известного способа. В частности, задачей является создание альтернативного реакционного пути для способа получения этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соли, в котором упомянутая выше стадия гидрирования отсутствует.
Другой задачей настоящего изобретения является создание способа получения этилового эфира N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соли, имеющего высокое диастереомерное соотношение, в котором (2R, 4S)-конфигурация, соответствующая формуле (3-а), является предпочтительной. В частности, подходящее диастереомерное соотношение составляет более, чем 60:40, предпочтительно - более, чем 70:30, особенно предпочтительно - более, чем 80:20. Наиболее предпочтительным является диастереомерное соотношение, составляющее более, чем 90:10. Диастереомерное соотношение может составлять до 99:1, предпочтительно - 100:1. Предпочтительные диастереомерные соотношения относятся к соотношению диастереомеров (3-а) и (3-b), или их солей,
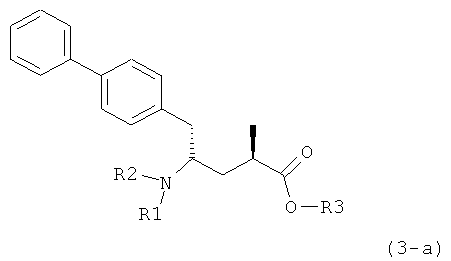 ,
,
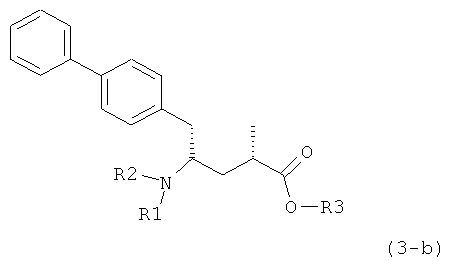 ,
,
где R1 представляет собой Н, R2 представляет собой трет-бутоксикарбонил и R3 представляет собой этил. Превращение соединения формулы (3)
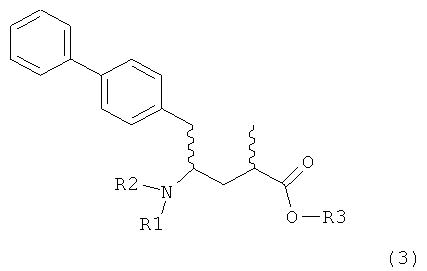 ,
,
в которой R1 представляет собой Н, R2 представляет собой трет-бутоксикарбонил и R3 представляет собой этил, в NEP-ингибитор или его пролекарство, в частности, в этиловый эфир N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соль, описан, например, в Journal of Medicinal Chemistry, 1995, 38, 1689.
Также задачей изобретения является альтернативный способ, в котором этиловый эфир N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты или его соль может быть получен в чистой или даже в кристаллической форме.
Кроме того, задачей настоящего изобретения является создание способа, в котором в качестве исходных веществ могут быть использованы легкодоступные соединения, например, природные аминокислоты или их производные, а неприродные аминокислоты не используются. Предпочтительно задача настоящего изобретения заключается в создании способа, в котором исходные материалы доступны из массива коммерчески доступных хиральных соединений.
Цели настоящего изобретения могут быть достигнуты путем получения определенного лактама, в качестве ключевого промежуточного соединения. Исходя из данного лактама возможно осуществление выгодного реакционного пути получения желаемых NEP-ингибиторов и их пролекарств.
Таким образом, объектом настоящего изобретения является пирролидин-2-он, соответствующий формуле (1) или его соль,
,
где R1 представляет собой водород или азотзащитную группу, как определено далее. Соединение, соответствующее формуле (1), или его соль, далее называют, как «Ключевой Лактам(1)».
Изобретение включает следующие разделы:
|
Особым образом изобретение относится к способам, описанным в каждом разделе. Более того, изобретение относится, независимо, к каждой отдельной стадии, описанной в последовательностях способа в соответствующем разделе. Поэтому каждая и всякая отдельная стадия любого способа, содержащая последовательность стадий и описанная в настоящем описании, сама по себе является предпочтительным вариантом осуществления настоящего изобретения. Таким образом, изобретение также относится к таким вариантам осуществления способа, в соответствии с которыми соединение, полученное в качестве промежуточного на любой стадии способа, используется в качестве исходного вещества.
Более того, изобретение относится к новым исходным веществам, которые специальным образом разработаны для получения соединений настоящего изобретения, к их применению и к способам их получения.
Необходимо отметить, что в настоящей заявке обычно толкования, приведенные в одном из разделов, также применимы и для других разделов, пока не указано иное. Например, толкование остатка R1 в формуле (1), данное в разделе А, также применимо, в случае если формула (1) встречается в разделах Б, В, Г и Д, пока не указано иное. Что касается соединений, описанных в настоящем изобретении, следует понимать, что тоже относится и к их солям. В зависимости от выбора исходного материала и процедур соединения могут находиться в форме одного из возможных изомеров или в виде их смеси, например, в виде чистых оптических изомеров, или в виде смеси изомеров, таких как рацематы и смеси диастереомеров, в зависимости от числа асимметричных атомов углерода.
Раздел А: Ключевой Лактам (1)
Объектом настоящего изобретения является лактам, соответствующий формуле (1), или его соль,
,
где R1 представляет собой водород или азотзащитную группу, как определено далее.
В соответствии с формулой (1), возможны два энантиомера, отвечающие формулам (1-а) и (1-b), или их соли.
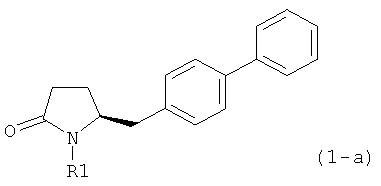
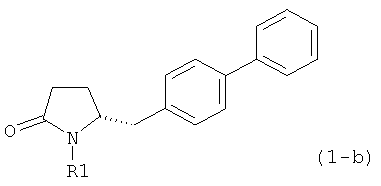 .
.
Согласно настоящему изобретению, соединения, отвечающие формуле (1-а) (=S-энантиомеры), являются предпочтительными. В формулах (1), (1-а) и (1-b), или их солях, остаток R1 представляет собой водород или азотзащитную группу, как показано в дальнейшем; предпочтительно азотзащитной группой является пивалоил, пирролидинилметил, трет-бутоксикарбонил, бензил, силил (например, ТЭС), ацетил, бензилоксикарбонил (Cbz) и триметилсилилэтоксиметил (СЭМ), более предпочтительны пивалоил, пирролидинилметил, трет-бутоксикарбонил, бензил и силил (например, ТЭС).
В целом, в рамках настоящей заявки, все соединения пирролидин-2-она, или их соли, обычно представлены в кето-форме. Однако, принимая во внимание возможность кето-енольной таутомерии, настоящее изобретение касается также описанных соединений, или их солей, в соответствующей енольной форме, как показано ниже, где * обозначает точку присоединения к остатку молекулы.
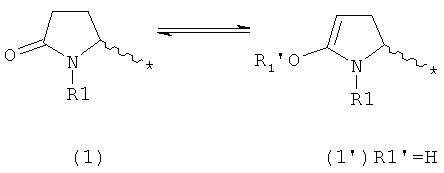
В случае формулы (1) соответствующее енольное производное представлено формулой (1'):
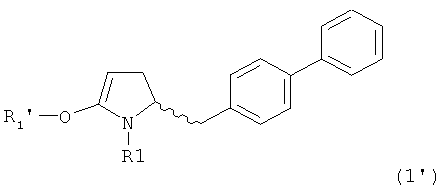 ,
,
где R1 представляет собой водород или азотзащитную группу, как будет показано ниже, a R1' представляет собой водород или кислородзащитную группу, как будет показано ниже.
Вышеизложенное применимо ко всем соответствующим соединениям настоящего изобретения, имеющим структуру пирролидин-2-она, в частности, к соединениям, отвечающим формулам (1), (2), (4), (5), (12) и (13), или их солям, а также к соединениям, имеющим предпочтительную конфигурацию, приведенную в формулах (1-а), (2-а), (4-а), (5-а), (12-а) и (13-а), или их солям.
В настоящей заявке термин «азотзащитная группа» включает в себя, в общем случае, любую группу, которая способна обратимо защищать функциональную группу азота, предпочтительно амино- и/или амидогруппу. Термин «кислородзащитная группа» включает в себя, в общем случае, любую группу, способную обратимо защищать функциональную группу кислорода.
Предпочтительно, азотзащитная группа представляет собой амино- и/или амидозащитную группу. Подходящие группы традиционно применяются в химии белков и описаны, например, в соответствующих главах справочников, например в J.F.W.McOmie, «Protective Groups in Organic Chemistry», Plenum Press, London and New York, 1973; T.W.Green and P.G.M.Wuts, "Protective Groups in Organic Synthesis", 3-е издание, Wiley, New York 1999, in "The Peptides"; том 3 (под редакцией E. Gross и J.Meienhofer), Academic Press, London and New York 1981, а также в "Methoden der organischen Chemie" (Methods of Organic Chemistry), Houben Weyl, 4-е издание, том 15/1, Georg Thieme Verlag, Stuttgart 1974.
Предпочтительные азотзащитные группы, в основном, включают:
C1-С6-алкил, предпочтительно С1-С4-алкил, более предпочтительно С1-С2-алкил, наиболее предпочтительно - C1-алкил, который является моно-, ди- или тризамещенным триалкилсилил-С1-С7-алкоксигруппой (например, триметилсилилэтоксигруппой), арилом, предпочтительно фенилом, или гетероциклической группой, предпочтительно пирролидинильной группой, где арильное или гетероциклическое кольцо является незамещенным или замещенным одним или более, например, двумя или тремя, остатками, например, выбранными из группы, состоящей из С1-С7-алкила, гидроксигруппы, С1-С7-алкоксигруппы, С2-С8-алканоилоксигруппы, галогена, нитрогруппы, цианогруппы и CF3;
арил-С1-С2-алкоксикарбонил (предпочтительно фенил-С1-С2-алкоксикарбонил, например, бензилоксикарбонил); С1-10алкенилоксикарбонил; C1-6алкилкарбонил (например, ацетил или пивалоил); С6-10арилкарбонил; С1-6алкоксикарбонил (например, трет-бутоксикарбонил); С6-10арилС1-6алкоксикарбонил; аллил или циннамил; сульфонил или сульфенил; сукцинимидильную группу, силил, например, триарилсилил или триалкилсилил (например, триэтилсилил).
Примерами предпочтительных азотзащитных групп являются ацетил, бензил, кумил, бензгидрил, тритил, бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), бензилоксиметил (БОМ), пивалоилоксиметил (ПОМ), трихлорэтоксикарбонил (Troc), 1-адамантилоксикарбонил (Adoc), аллил, аллилоксикарбонил, триметилсилил, трет-бутилдиметилсилил, триэтилсилил (ТЭС), триизопропилсилил, триметилсилилэтоксиметил (СЭМ), трет-бутоксикарбонил (БОК), трет-бутнп, 1-метил-1,1-диметилбензил, (фенил)метилбензол, пиридинил и пивалоил. Наиболее предпочтительными азотзащитными группами являются ацетил, бензил, бензилоксикарбонил (Cbz), триэтилсилил (ТЭС), триметилсилилэтоксиметил (СЭМ), трет-бутоксикарбонил (БОК), пирролидинилметил и пивалоил.
Примерами более предпочтительных азотзащитных групп являются пивалоил, пирролидинилметил, трет-бутоксикарбонил, бензил и силильные группы, в частности, силильные группы, соответствующие формуле SiR7R8R9, в которой R7, R8 и R9, независимо друг от друга, представляют собой алкил или арил. Предпочтительными примерами R7, R8 и R9 являются метил, этил, изопропил, трет-бутип и фенил.
Особенно предпочтительными азотзащитными группами являются пивалоил и трет-бутоксикарбонил (БОК).
Предпочтительными кислородзащитными группами являются силильные группы, соответствующие формуле SiR7R8R9, в которой R7, R8 и R9, независимо друг от друга, представляют собой алкил или арил. Предпочтительными примерами R7, R8 и R9 являются метил, этил, изопропил, трет-бутил и фенил.
В частности, R7, R8 и R9 представляют собой этил или метил. Особенно предпочтительными кислородзащитными группами являются SiMe3 и SiEt3.
Алкил, являющийся радикалом или частью радикала, представляет собой прямолинейную или разветвленную (один или, если необходимо и возможно, более раз) углеродную цепь, и, в частности, обозначает С1-С7-алкил, предпочтительно - С1-С4алкил.
Термин "С1-С7-" означает фрагмент, содержащий до и включающий максимум 7, особенно - содержащий до и включающий максимум 4, атомов углерода, при этом данный фрагмент является разветвленным (один или более раз) или прямолинейным, и присоединяется через терминальный или нетерминальный атом углерод.
Циклоалкил, например, представляет собой С3-С7-циклоалкил и обозначает, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклопентил и циклогексил являются предпочтительными.
Алкоксигруппа, например, представляет собой С1-С7-алкоксигруппу и обозначает, например, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, а также включает соответствующие пентилокси-, гексилокси- и гептилоксирадикалы. С1-4алкоксигруппа является предпочтительной.
Алканоил, например, представляет собой С2-С8-алканоил и обозначает, например, ацетил [-С(=O)Ме], пропионил, бутирил, изобутирил или пивалоил. С2-С5-Алканоил является предпочтительным, особенно - ацетил.
Гало или галоген предпочтительно представляет собой фтор, хлор, бром или йод, наиболее предпочтительно - фтор, хлор или бром.
Гало-алкил представляет собой, например, гало-С1-С7алкил и, в частности, обозначает гало-С1-С4алкил, такой как трифторметил, 1,1,2-трифтор-2-хлорэтил или хлорметил. Предпочтительным гало-С1-С7алкилом является трифторметил.
Алкенил может быть линейным или разветвленным алкилом, содержащим двойную связь и предпочтительно включающим от 2 до 12 атомов углерода, особенно предпочтительно - от 2 до 10 атомов углерода. Особенно предпочтительным является линейный С2-С4алкенил. Некоторые примеры алкильных групп представляют собой этил и изомеры пропила, бутила, пентила, гексила, гептила, октила, нонила, децила, ундецила, додецила, тетрадецила, гексадецила, октацила и эйкозила, каждый из которых содержит двойную связь. Особенно предпочтительным является аллил.
Алкилен представляет собой бивалентный радикал, полученный из С1-С7алкила и, в частности, обозначающий С2-С7алкилен или С2-С7алкилен, включающий один или несколько О, NR14 или S, где R14 представляет собой алкил, каждый из которых может быть незамещенным или замещенным одним или более заместителями, независимо выбранными, например, из С1-С7-алкила, С1-С7-алкокси-С1-С7-алкила или С1-С7алкоксигруппы.
Алкинилен представляет собой бивалентный радикал, полученный из С2-С7алкенила, и может включать один или несколько О, NR14 или S, где R14 представляет собой алкил, и является незамещенным или замещенным одним или более, например, тремя, заместителями, предпочтительно независимо выбранными из заместителей, перечисленных выше для алкилена.
Арил, являющийся радикалом или частью радикала, представляет собой, например, С6-С10арил, и предпочтительно обозначает моно- или полициклический, в частности, моноциклический, бициклический или трициклический арильный фрагмент, содержащий от 6 до 10 атомов углерода, предпочтительно - фенил, и который может быть незамещенным или замещенным одним или более заместителями, независимо выбранными, например, из C1-С7-алкила, С1-С7-алкокси-С1-С7-алкила или С1-С7-алкоксигруппы.
Арилоксигруппа относится к арил-О, где арил имеет значения, определенные выше.
Незамещенный или замещенный гетероциклил представляет собой моно- или полициклическую, предпочтительно - моно-, би- или трициклическую, наиболее предпочтительно - моноциклическую, ненасыщенную, частично насыщенную, насыщенную или ароматическую кольцевую систему, содержащую предпочтительно от 3 до 14 (более предпочтительно - от 5 до 14) кольцевых атомов, и один или более, предпочтительно от одного до четырех, гетероатомов, независимом выбранных из азота, кислорода, серы, S(=O)- или S-(=O)2, и являющуюся незамещенной или замещенной одним или более, например, до трех, заместителями, предпочтительно независимо выбранными из предпочтительных заместителей, выбранных из группы, состоящей из С1-С7-алкила, гало-С1-С7-алкила, С1-С7-алкоксигруппы, гало- С1-С7-алкоксигруппы, такой как трифторметоксигруппа, и С1-С7-алкокси- С1-С7-алкоксигруппы. В случае, когда гетероциклил представляет собой ароматическую кольцевую систему, он также относится к гетероарилу.
Ацетил представляет собой -С(=O)С1-С7алкил, предпочтительно -С(=O)Ме.
Силил представляет собой SiRR'R'', где R, R' и R'', независимо друг от друга, обозначают С1-С7алкил, арил или фенил-С1-С4алкил.
Сульфонил представляет собой (незамещенный или замещенный) С1-С7-алкилсульфонил, такой как метилсульфонил, (незамещенный или замещенный) фенил- или нафтил-С1-С7-алкилсульфонил, такой как фенилметансульфонил, или (незамещенный или замещенный) фенил- или нафтилсульфонил; при этом в случае наличия более одного заместителя, например, от одного до трех заместителей, заместители независимо выбраны из цианогруппы, гало, гало-C1-С7-алкила, гало-С1-С7-алкилоксигруппы и С1-С7-алкилоксигруппы. Особенно предпочтительными являются С1-С7-алкилсульфонил, такой как метилсульфонил, и (фенил- или нафтил)- С1-С7-алкилсульфонил, такой как фенилметансульфонил.
Сульфенил представляет собой (незамещенный или замещенный) С6-С10арил-С1-С7-алкилсульфенил или (незамещенный или замещенный) С6-С10арилсульфенил, при этом в случае наличия более одного заместителя, например, от одного до трех заместителей, заместители независимо выбраны из нитрогруппы, гало, гало- С1-С7алкила и гало-С1-С7-алкилоксигруппы.
Под термином "омыляющий реагент" следует понимать основание, которое способно гидролизовать сложный эфир с образованием спирта и соли карбоновой кислоты, например, гидроксид щелочного металла, такой как КОН или NaOH.
Под термином "группа, которая может быть омылена" следует понимать сложноэфирную группу -CO2R, в которой R представляет собой алкил, арил или арилалкил, которая может быть гидролизована, например, в основных условиях (например, в присутствии основания на основе щелочного металла, такого как NaOH, LiOH или КОН) или в кислых условиях (например, при использовании минеральных кислот, таких как HCl, H2SO4, HBr, Н3РО4), с получение карбоновой кислоты. Дополнительно термин "группа, которая может быть омылена" может также включать сложноэфирную группу -CO2R, в которой R представляет собой алкил, арил или арилалкил, которая может вступать в реакцию при использовании гетерогенного катализатора (например, Pd/C, Pt/C, Rh/C, Pd/Al2O3, PtO2), в присутствии кислоты (например, уксусной кислоты) или основания (например, триэтиламина), или в нейтральных условиях, с получением карбоновой кислоты.
Под термином "группа, которая может быть декарбоксилирована" следует понимать группу -CO2R, в которой R представляет собой водород, алкил, арил или арилалкил, которая может быть замещена водородом в реакционных условиях, таких как нагревание, необязательно в присутствии растворителя, предпочтительно инициируемое кипением. Дополнительно данное определение может включать сложноэфирную группу -СО2М, в которой М представляет собой щелочной металл, например, Na или К, в присутствии краун-эфира, например, 18-краун-6. Подходящими растворителями, например, являются толуол, о-/м-/n-ксилол, бензол, ТГФ, 1,4-диоксан, ДМФА, вода, трет-бутилметиловый эфир. Предпочтительно используют высококипящие растворители, оптимально - растворители, с температурой кипения, составляющей при атмосферном давлении, более, чем 50°С. Более предпочтительными являются растворители, с температурой кипения, составляющей более 100°С.
Термин "таутомер" относится, в частности, к енольному таутомеру фрагмента пирролидин-2-она соединений настоящего изобретения.
Термин "бифенил" или "бифенильный", представленный в настоящем описании, как "бифенилмагний галогенид" или "бифенильное соединение", следует понимать как обозначающий 4-бифенил или 4-бифенильный, также называемый пара-бифенип или пара-бифенильный, например, 4-бифенилмагний бромид или 4-бромбифенил.
Термины "PG", "PG1" и "PG2" независимо относятся к азотзащитной группе, как определено в настоящем описании.
В формулах настоящей заявки термин " " на C-sp3 представляет ковалентную связь, при этом стереохимия данной связи не определена. Это означает, что термин "" на C-sp3 включает (5)-конфигурацию, также как и (S)-конфигурацию, соответствующего хирального центра. Кроме того, данный термин также включает смеси конфигураций.
" на C-sp3 представляет ковалентную связь, при этом стереохимия данной связи не определена. Это означает, что термин "" на C-sp3 включает (5)-конфигурацию, также как и (S)-конфигурацию, соответствующего хирального центра. Кроме того, данный термин также включает смеси конфигураций.
В формулах настоящей заявки термин " " на C-sp3 указывает на абсолютную стереохимию - или (R), или (S).
" на C-sp3 указывает на абсолютную стереохимию - или (R), или (S).
В формулах настоящей заявки термин " " на C-sp3 указывает на абсолютную стереохимию - или (R), или (S).
" на C-sp3 указывает на абсолютную стереохимию - или (R), или (S).
Соли представляют собой, главным образом, фармацевтически приемлемые соли или, в общем случае, соли любых упомянутых промежуточных продуктов, причем соли не исключаются по причинам химического характера, которые будут ясны специалисту в данной области. Они могут образовываться при наличии солеобразующих групп, например - кислотных или основных групп, которые могут существовать в диссоциированной форме по меньшей мере частично, например, в диапазоне pH водных растворов от 4 до 10, или могут быть выделены, главным образом, в твердой, особенно - в кристаллической форме.
Такие соли образуются, например, как кислые соли присоединения, предпочтительно - с органическими или неорганическими кислотами, из соединений, или любых указанных здесь промежуточных соединений, с основным атомом азота (например, имино- или аминогруппы), в особенности фармацевтически приемлемые соли. Подходящими неорганическими кислотами являются, например, галогенводородные кислоты, например - соляная кислота, серная кислота или фосфорная кислота. Подходящие органические кислоты, например - карбоновые, фосфорные, сульфоновые или сульфаминовые кислоты, например - уксусная кислота, пропионовая кислота, молочная кислота, фумаровая кислота, янтарная кислота, лимонная кислота, такие аминокислоты, как глютамовая или аспарагиновая кислоты, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, бензойная кислота, метан- или этансульфоновая кислота, этан-1,2-дисульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 1,5-нафталиндисульфоновая кислота, N-циклогексилсульфаминовая кислота, N-метил-, N-этил- или N-пропилсульфаминовая кислота, или прочие органические протонные кислоты, например, аскорбиновая кислота.
В присутствии отрицательно-заряженных радикалов, например карбоксильных или сульфорадикалов, соли могут также быть образованы основаниями, например соли металлов или аммония, такие как соли щелочных или щелочно-земельных металлов, например соли натрия, калия, магния или кальция, или соли аммония с аммиаком или подходящими органическими аминами, например третичными моноаминами, такими как триэтиламин или три(2-гидроксиэтил)амин, или гетероциклическими основаниями, например, N-этилпиперидном или N,N'-диметилпиперазином.
Когда в одной молекуле имеются основная и кислотная группы, любые из упомянутых здесь промежуточных соединений могут также образовывать внутренние соли.
Для целей выделения или очистки упомянутых здесь промежуточных соединений, представляется возможным использовать фармацевтически неприемлемые соли, например, пикраты или перхлораты.
Принимая во внимание тесную связь между соединениями и промежуточными соединениями в свободном виде и в виде их солей, включая такие соли, которые могут быть использованы как интермедиаты, например, при очистке или определении соединений или их солей, любое указание на «соединения», «исходные материалы» и «интермедиаты» в тексте настоящей заявки следует понимать, как относящееся к одной или более соли или смеси соответствующих свободного соединения, интермедиата или исходного материала, и одной или более их солей, каждое из которых также включает в себя, подходящим образом, любой сольват или любую соль одного или более из них, если явным образом не указано иное. Могут быть получены различные кристаллические формы, которые также включены в настоящее описание.
При использовании множественного числа при описании соединений, исходных материалов, интермедиатов, солей, фармацевтических препаратов, заболеваний, расстройств и т.п., следует понимать, что одно (предпочтительное) или более отдельных соединений, солей, фармацевтических препаратов, заболеваний, расстройств и т.п., которые употребляются как в единственном, так и во множественном числе, не подразумевает исключения множественного их числа, а просто единственное число является предпочтительным.
Соединения настоящего изобретения могут иметь один или более асимметричных центров. Предпочтительные абсолютные конфигурации имеют вид, специальным образом показанный в настоящей заявке. Однако, любые возможные чистые энантиомеры, чистые диастереомеры, или их смеси, например, такие смеси энантиомеров, как рацематы, включаются в объем настоящего изобретения.
Ключевой Лактам, соответствующий формуле (1), или его соль, где R1 представляет собой водород, может быть превращен в Ключевой Лактам, соответствующий формуле (1),или его соль, где R1 является защитной азотсодержащей группой, как определено выше, в соответствии со стандартными методиками, известными из области органической химии, в частности, это касается способов с использованием защитной азотсодержащей группы, описанных в J.F.W.McOmie, "Protective groups in organic chemistry", Plenum Press, London & New York, 1973, в T.W.Greene и P.G.M.Wuts, "Protective groups in organic synthesis", 3-е издание, Wiley, New York, 1999, а также в Richard С.Larock "Comprehensive organic transformations:a guide to functional group preparations", 2-е издание, Wiley-VCH Verlag GmbH, 2000.
Все вышесказанное относится и к Ключевому Лактаму, соответствующему формуле (1'), или его соли, где R1' представляет собой водород. Превращение R1, представляющего собой водород, в кислородзащитную группу, как определено выше, может быть осуществлено известными методами; стандартные условия для осуществления данных методов описаны, например, в справочниках, упомянутых выше.
В первом предпочтительном воплощении изобретения R1' представляет собой водород и R1 представляет собой силильную защитную группу, как описано далее. Во втором воплощении изобретения оба R1' и R1 представляют собой силильную защитную группу, как описано далее. Получение соединений, в соответствии с указанными воплощениями, может быть осуществлено, например, так как описано в патенте US 4604383. В соответствии со вторым воплощением изобретения получают соединения, соответствующие формуле (1''), или их соли
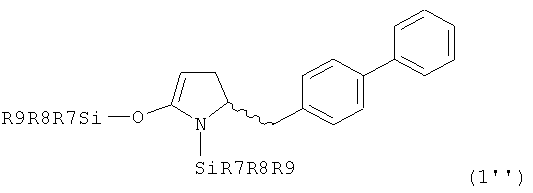 ,
,
где R7, R8 и R9, независимо друг от друга, представляют собой арил или алкил, предпочтительно - метил или этил. Предпочтительными примерами R7, R8 и R9 являются метил, этил, изопропил, н-бутил, фенил. В частности, R7, R8 и R9 представляют собой этил или метил. Особенно предпочтительными защитными группами являются SiMe3 и SiEt3.
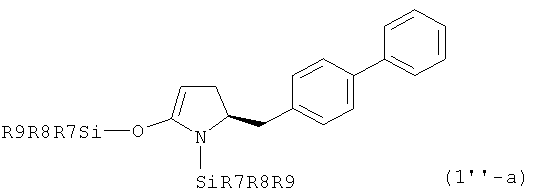
Как было упомянуто выше предпочтительная стереохимическая конфигурация не зависит от того, находятся ли соединения в кетоформе или в виде енольных производных. Так в предпочтительном воплощении изобретения соединения, соответствующие формуле (1''), или их соли, находятся в виде S-энантиомеров, соответствующих формуле (1'-а)
 ,
,
где R7, R8 и R9 имеют значения, определенные выше.
В предпочтительном воплощении изобретения соединения, соответствующие формуле (1''-а), или их соли, могут быть получены путем взаимодействия соединения, соответствующего формуле (1), или его соли, где R1 представляет собой водород, с соединением R7R8R9SiX, где R7, R8 и R9 имеют значения, определенные выше, и X представляет собой уходящую группу, предпочтительно, хлор, бром, трифлат или тозилат. Предпочтительно соединение формулы R7R8R9SiX представляет собой триметилсилилхлорид или триэтилсилил.
Предпочтительно взаимодействие осуществляют в присутствии основания. Предпочтительным основанием является триэтиламин, диэтиламин, лютидин и их смеси. Примерами других подходящих оснований являются ЛДА и KHMDS.
Получение силиленольного производного, соответствующего формуле (1''), или его соли, может быть осуществлено при термодинамическом контроле. Кроме того, реакция может быть проведена до завершения.
Раздел Б: Применение Ключевого Лактама при получении NEP-ингибиторов
Объектом настоящего изобретения является применение Ключевого Лактама, соответствующего формуле (1), или его соли,
где R1 представляет собой водород и азотзащитную группу, как определено выше, при получении NEP-ингибитора или его пролекарства. Предпочтительно используемый Ключевой Лактам имеет S-конфигурацию, соответствующую формуле (1-а).
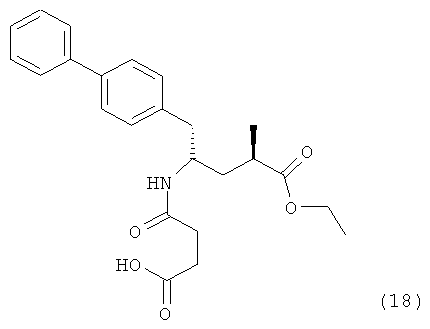
В предпочтительном воплощении изобретения пролекарством NEP-ингибитора является этиловый эфир N-(3-карбоксил-1-оксопропил)-(4S)-(n-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты, как показано в формуле (18), или его соль:
 .
.
Раздел Б настоящего изобретения включает три подраздела:
|
Подраздел Б-1: Взаимодействие соединения (1) с получением метилированного лактама (2)
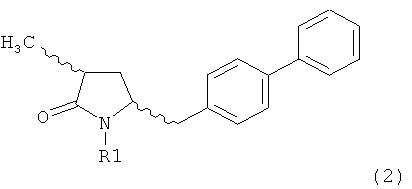
Другим объектом настоящего изобретения является способ получения соединения, соответствующего формуле (2), или его соли
 ,
,
где R1 представляет собой водород или азотзащитную группу, как определено ранее, включающий метилирование соединения, соответствующего формуле (1), или его соли, предпочтительно метилирование соединения формулы (1-а) или его соли. В основном все пояснения, приведенные ранее в отношении предпочтительных воплощений ключевого лактама (1) также применимы в данном разделе.
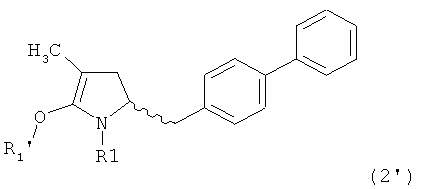
Как установлено выше соединение, соответствующее формуле (2), или его соль, представлено в кето-форме. Однако настоящее изобретение также охватывает соответствующие енольные формы, соответствующие формуле (2')
 ,
,
где R1 представляет собой водород или азотзащитную группу, как определено ранее, a R1' представляет собой водород или кислородзащитную группу, как определено ранее. В предпочтительном воплощении изобретения соединение формулы (2') или его соль соответствуют формуле (2'-а)
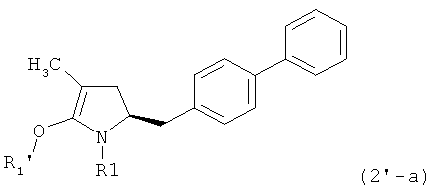 .
.
В основном вышеописанную реакцию метилирования проводят в присутствии метилирующего агента. Обычно подходящим является любой метилирующий агент, известный из уровня техники. Примерами подходящих метилирующих агентов являются метилйодид, метилбромид, метилхлорид, метилфторид, диметилсульфат, метилтрифлат (MeOTf), 4-метилсульфонилтолуол и их смеси. Предпочтительно используют метилйодид или диметилсульфат, или их смеси.
Реакцию метилирования предпочтительно осуществляют в широком температурном интервале, например, между -100°С и+50°С.Предпочтительно реакцию проводят между -80°С и +20°С, более предпочтительно реакцию осуществляют между -10°С и +10°С, еще более предпочтительно реакцию проводят при 0°С. Реакцию можно проводить в различных растворителях, например, тетрагидрофуране (ТГФ), трет-бутилметиловом эфире (ТБМЭ), 1,2-диметоксиэтане, диэтиловом эфире, толуоле и их смесях. Предпочтительно используют ТГФ или толуол.
В предпочтительном воплощении изобретения реакцию осуществляют в присутствии основания. Основанием является, например, RcRdNM, где Rc и Rd независимо выбраны из алкила, циклоалкила, гетероциклила или силила, а М представляет собой щелочной метал, такой как Na, Li или К. Примерами подходящих оснований являются литий бис(триметилсилил)амид (LHMDS), натрий бис(триметилсилил)амид (NaHMDS), калий бис(триметилсилил)амид (KHMDS), литий диизопропиламид (ЛДА), н-/втор-/трет-бутиптпий, изопропилмагний хлорид, фениллитий и их смеси. Как вариант, походящими основаниями являются литий дициклогексиламин и литий тетраметилпиперидин. Предпочтительно используют ЛДА, KHMDS или их смеси. Более предпочтительно основанием является литий тетраметилпиперидин и калий бис(триметилсилил)амид.
Также предпочтительным является добавление "усилителя реакции". В основном в качестве соединений, усиливающих реакцию, пригодны соединения улучшающие растворимость образующегося продукта или способствующие деагрегации основания, делая его более реакционноспособным. Подходящие усилители реакции описаны в соответствующих главах в FA Carey, RJ Sundberg, Organische Chemie, VCH, Weinheim, 1995 (немецкий перевод английского оригинального текста). Примерами предпочтительных усилителей реакции являются гексаметилфосфорамид (ГМФА), N,N'-диметилпропиленмочевина (ДИПМ), тетраметилэтилендиамин (ТМЭД), диметилсульфоксид (ДМСО) или их смеси. Также для этой цели пригодны краун-эфиры или хиральные краун-эфиры.
В предпочтительном воплощении изобретения реакция может быть осуществлена в две стадии. На первой стадии соединение, соответствующее формуле (1'), или его соль, где R1 представляет собой водород, вступает в реакцию, с образованием соединения, соответствующего формуле (1), или его соли, где R1 представляет собой азотзащитную группу, как определено выше. На второй стадии соединение, соответствующее формуле (1), или его соль, где R1 представляет собой азотзащитную группу, как определено выше, вступает в реакцию с образованием соединения, соответствующего формуле (2), или его соли, где R1 представляет собой азотзащитную группу, как определено выше.
Как вариант, соединение, соответствующее формуле (1), или его соль, где R1 представляет собой водород, может вступать в реакцию непосредственно, например, в присутствии втор-BuLi, в качестве основания, и метилйодида, в качестве метилирующего агента, с получением соединения, соответствующего формуле (2), или его соли, где R1 представляет собой водород.
Соединение, соответствующее формуле (2), или его соль, где R1 представляет собой азотзащитную группу, как определено выше, может быть депротектировано (т.е. защитную группу удаляют таким образом, чтобы R1 представляло собой водород) или непосредственно превращено в соединение, соответствующее формуле (3), или его соль, где R1 и R2 независимо друг от друга представляют собой водород или азотзащитную группу, как определено выше, и R3 представляет собой водород или алкил (данная реакция детально описана далее в подразделе Б-2). Депротектированное соединение, соответствующее формуле (2) (т.е., в которой R1 представляет собой водород), или его соль, может также вступать в реакцию, с получением соединения, соответствующего формуле (3), или его соли, как детально описано далее, где R1 и R2 независимо друг от друга представляют собой водород или азотзащитную группу, как определено выше, и R3 представляет собой водород или алкил.
Описанные выше реакционные пути показаны на реакционной Схеме 1, где "PG" обозначает азотзащитную группу, как определено выше, предпочтительно бензил, бензилоксикарбонил (Cbz), триэтилсилил (ТЭС), триметилсилилэтоксиметил (СЭМ), трет-бутоксикарбонил (БОК), пирролидинилметил и пивалоил, более предпочтительно - бензил, триметилсилилэтоксиметил, пирролидинилметил и пивалоил, наиболее предпочтительно - пивалоил или трет-бутоксикарбонил (БОК):
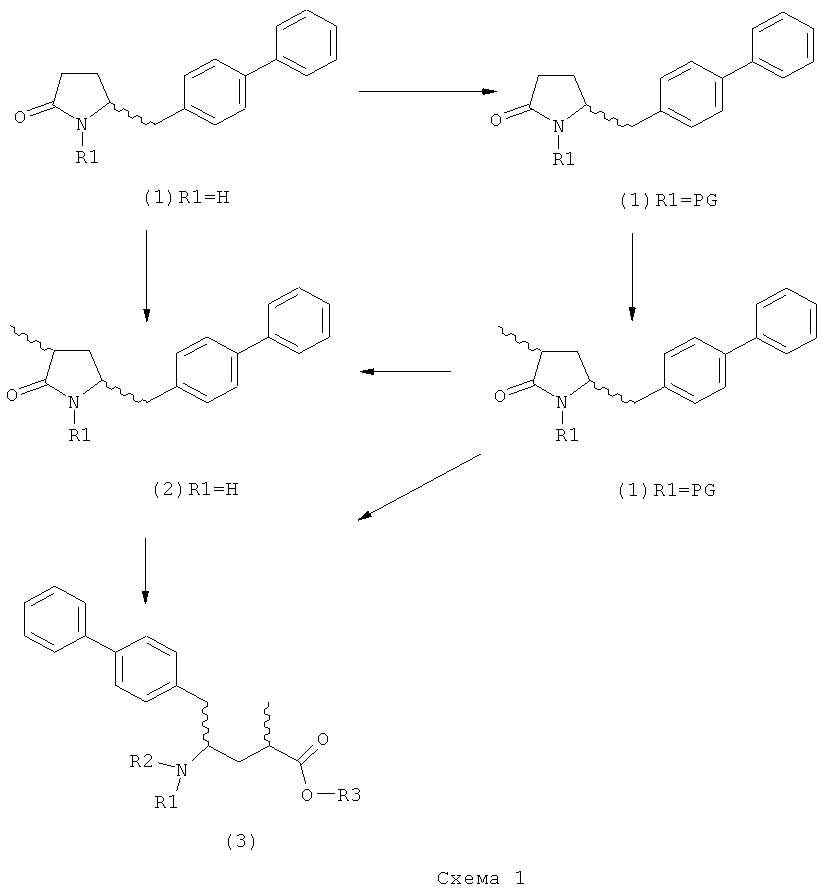
В другом воплощении настоящее изобретение относится к полной последовательности реакций, описанной на Схеме 1, а также изобретение относится к каждой реакционной стадии. В другом воплощении настоящее изобретение относится к продукту, полученному в соответствии с полной последовательностью реакций, описанной на Схеме 1, а также изобретение относится к продукту, полученному на каждой реакционной стадии, показанной на Схеме 1.
Если в воплощении изобретения требуется удалить азотзащитную группу, как описано ранее, удаление обычно осуществляют, используя известные методы. Предпочтительно азотзащитную группу, как описано ранее, удаляют, используя кислотные или основные условия. Примерами кислотных условий являются соляная кислота, трифторуксусная кислота, серная кислота. Примерами основных условий являются гидроксид лития, этоксид натрия. Также могут быть использованы нуклеофилы, такие как боргидрид натрия.
В случае N-бензила, как азотзащитной группы, то он может быть удален посредством гидрирования или при использовании некоторых подходящих окислителей, например, церий аммонийнитрата (ЦАН) или 2,3-дихлор-5,6-дициано-n-бензохинона (ДДХ).
В другом предпочтительном воплощении изобретения соединение, соответствующее формуле (1'), или его соль, где R7, R8 и R9 имеют значения, определенные выше, метилируют с получением соединения, соответствующего формуле (2), или его соли, где R1 представляет собой водород, как показано на Схеме 2. Соединения формулы (1''), или их соли, могут быть получены из соединений формулы (1), или их солей, согласно способам, хорошо известным из уровня техники, как описано, например, в релевантных частях обычных ссылок, таких как J.F.W.McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, и Т.W.Greene, P.G.M.Wuts, "Protective Groups in Organic Synthesis", 3-е издание, Wiley, New York 1999.
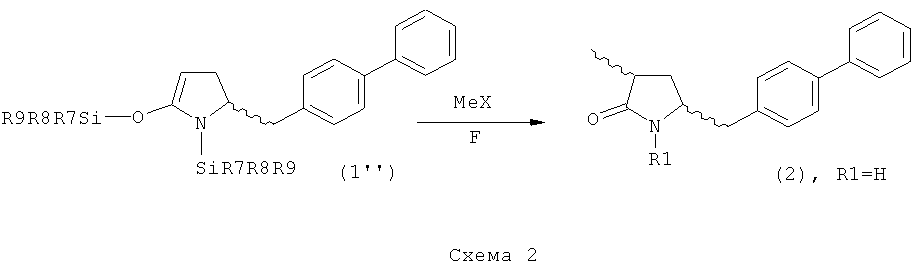
Реакцию метилирования проводят в присутствии метилирующего агента (например, МеХ). Предпочтительными метилирующими агентами являются агенты, описанные выше. Кроме того, реакцию предпочтительно проводят в присутствии источника фтора. Предпочтительными источниками фтора являются фторсодержащие соли щелочных или щелочно-земельных металлов (например, LiF, CaF2, CsF, KF) или прочие фторидные соли, например, фторид тетрабутиламмония (ТБАФ). Источник фтора может использоваться в каталитических или стехиометрических количествах. Предпочтительно, используют фторид калия или ТБАФ, в частности, в каталитических количествах.
В предпочтительном варианте осуществления изобретения, превращение соединения формулы (1'') в соединение формулы (2) может осуществляться двухстадийным способом, а именно - реакцией (1'') с метилирующим агентом и последующим взаимодействием полученного метилированного продукта с источником фтора (например, солью щелочного или щелочно-земельного металла, такой как LiF, CaF2, CsF и KF, или иной фторидной солью, например, ТБАФ.
Использование соединения формулы (1''), или его соли, в качестве исходного материала, может иметь то преимущество, что удается избежать раздельной защиты N-группы, поскольку N-силильная группа может быть удалена in situ в условиях вышеописанной реакции метилирования.
Может представлять интерес стереохимия вышеописанных реакций, приведенных на схемах 1 и 2. В предпочтительно варианте осуществления, соединение формулы (1) на Схеме 1, или его соль, характеризуется тем, что имеет конфигурацию, соответствующую формуле (1-а) (S-энантиомер). Аналогично, в предпочтительном воплощении, соединение формулы (1''), или его соль, на схеме 2, характеризуется тем, что имеет конфигурацию, соответствующую формуле (1'-а) (S-энантиомер).
Если соединение формулы (1-а), или его соль, используется в качестве исходного материала, то могут быть получены два соединения формулы (2), или их соли, а именно - два диастереомера формул (2-а) и (2-b), или их соли,
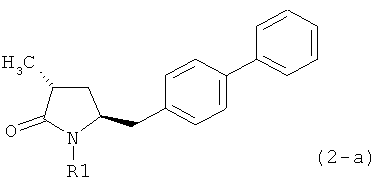
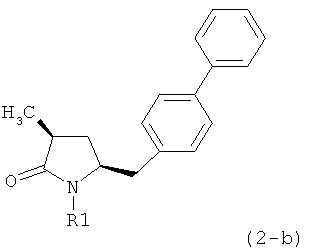 ,
,
где R1 представляет собой водород, или вышеуказанную азотзащитную группу. Более того, если соединение формулы (1''-а), или его соль, используется в качестве исходного материала, то могут быть получены два соединения формулы (2), или их соли, а именно - два диастереомера, соответствующих формулам (2-а) и (2-b), или их соли, где R1 представляет собой водород. В предпочтительном осуществлении изобретения, соединение формулы (2) или (3) на Схеме 1, или его соли, имеет формулу (2-а) или (3-а), соответственно. Как вариант, в предпочтительном варианте осуществления, соединение формулы (1'') или (2) на Схеме 2, или его соли, имеет формулу (1''-а) или (2-а), соответственно.
Получаемое соотношение диастереомеров зависит от выбранных условий реакции, в частности, от азотзащитной группы и используемого основания. В предпочтительном варианте осуществления изобретения, образуется соединение формулы (2-а). В частности, соединение формулы (1-а) используют в качестве исходного материала, и получают соединение формулы (2-а) при соотношении диастереомеров более, чем 60:40, предпочтительно более, чем 70:30, особенно предпочтительно - более, чем 80:20. Более предпочтительным является соотношение диастереомеров свыше 90:10. Соотношение диастереомеров может достигать 99:1, предпочтительно - 100:0.
Было обнаружено, что при использовании способа настоящего изобретения алкилирование соединения формулы (1), или его соли, может быть проведено с высокой диастереоселективностью. Способ настоящего изобретения обеспечивает получение соединения формулы (2), или его соли, с высокой диастереоселективностью, путем взаимодействия соединения формулы (1) с основанием и метилирующим агентом, как описано ранее. В частности, метилирование соединения формулы (1-а), согласно этому варианту осуществления изобретения, приводит к соединению формулы (2), причем соотношение диастереомеров (2-а) и (2-b) составляет, по меньшей мере, 80:20, более предпочтительно, по меньшей мере, 85:15, еще более предпочтительно, по меньшей мере, 91:9. При таком предпочтительном осуществлении реакции метилирования основание представляет собой, например, RcRdNM, где Rc и Rd, независимо, выбраны из алкила, циклоалкила, гетероциклила или силила, а М представляет собой такой щелочной металл, как Na, Li или К. Предпочтительными основаниями являются диизопропиламид лития, дициклогексиламин лития, тетраметилпиперидин лития, бис(триметилсилил)амид лития и бис(триметилсилил)амид калия; более предпочтительными являются тетраметилпиперидин лития и бис(триметилсилил)амид калия. Метилирующий агент предпочтительно представляет собой диметилсульфат, метилйодид или метилбромид, предпочтительно - метилйодид или диметилсульфат, более предпочтительно - диметилсульфат. Предпочтительно, метилирование проводят при температуре между -78 и 20°С, предпочтительно- между -10 и 20°С, более предпочтительно между -10 и 0°С. Неожиданно было обнаружено, что реакция метилирования протекает при 0°С с высокой диастереоселективностью и высоким выходом. Обычно, метилирование проводят в растворителе, как описано выше, предпочтительно в тетрагидрофуране, толуоле или их смесях.
Еще в одном варианте осуществления изобретения, метилирование соединения формулы (1) или (1''), предпочтительно формулы (1-а) или (1'-а), или его солей, метилирующим агентом и основанием, как было показано выше, может приводить к соединению формулы (2''), предпочтительно формулы (2''-а), или его солям,
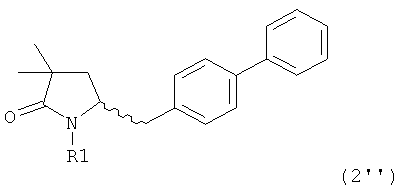
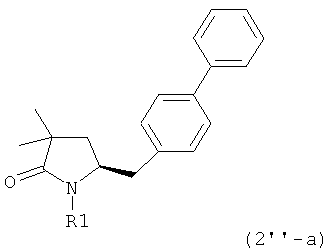 ,
,
где R1 представляет собой водород или азотзащитную группу, как определено ранее.
В еще одном варианте осуществления, соединение формулы (2''), предпочтительно формулы (2''-а), или его соли, где R1 представляет собой водород или азотзащитную группу, как определено ранее, может быть также получено обработкой соединения формулы (2), предпочтительно формулы (2-а), или его солей, где R1 - водород или азотзащитная группа, метилирующим агентом и основанием, как описано выше.
Соединения формулы (2-а), или их соли, могут быть получены в виде кристаллического твердого вещества. Предпочтительно, R1 представляет собой пивалоил или трет-бутоксикарбонил, более предпочтительно R1 является пивалоилом. При необходимости, соединения формулы (2-а), или их соли, могут быть очищены путем кристаллизации.
В предпочтительном варианте осуществления изобретения, выход желаемого изомера может быть увеличен. В этом случае проводят стадии реакции в соответствии со Схемой 3. В одном осуществлении, согласно схеме 3, когда R1 представляет собой PG, предпочтительным является пивалоил или трет-бутоксикарбонил.
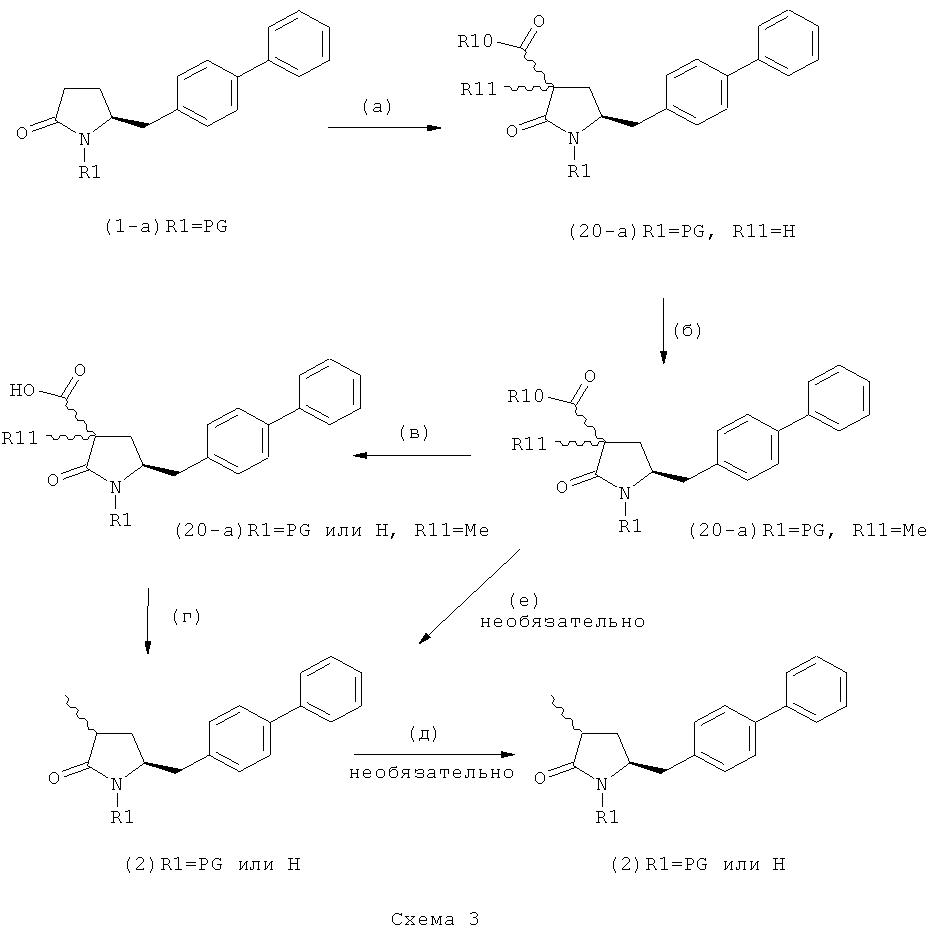
В другом варианте осуществления настоящее изобретение относится к полной последовательности стадий взаимодействия, показанных на Схеме 3, а также к каждой из стадий взаимодействия. Еще в одном варианте осуществления, настоящее изобретение касается продукта, получаемого согласно полной последовательности реакций, показанных на Схеме 3, а также касается продукта, получаемого в соответствии с каждой из стадий взаимодействия, приведенных на Схеме 3.
PG на Схеме 3 обозначает азотзащитную группу, как указывалось выше. R10 представляет собой любую группу, которая может подходящим образом омыляться и/или декарбоксилироваться. Предпочтительно R10 представляет собой -О-алкил, или -О-арил, в частности -O-Et, -О-фенил или -О-алкиларил, такой как -О-бензил; предпочтительно R10 представляет собой -O-Et или -О-фенил. Для каждой из реакции Схемы 3, предпочтительно используют следующие условия взаимодействия:
(а): Обработка основанием (например, NaH, NaHMDS или KHMDS, предпочтительно NaH или NaHMDS), а затем дополнительным реагентом таким образом, что после удаления уходящей группы, получают группу -С(=O), например, группу -C(=O)OR, в которой R представляет собой алкил, арил или алкиларил, предпочтительно алкил или арил. Такими дополнительными реагентами являются предпочтительно карбонаты формулы (RO)(R'O)CO или соединения формулы XCOOR, где R и R' независимо друг от друга представляют собой алкил, арил или арилалкил, предпочтительно алкил или арил, а X представляет собой галоген, в частности, хлор; предпочтительно дополнительными реагентами формулы (RO)(R'O)CO или формулы XCOOR являются (МеО)2СО, (EtO)2CO, (BnO)2СО, ClCO2Me, ClCO2Et, ClCO2 Bn; наиболее предпочтительно дополнительными реагентами являются (МеО)2СО, (EtO)2СО, ClCO2Me или ClCO2Et;
(б): Обработка основанием, предпочтительно таким, как описано на стадии (а), и метилирующим агентом, как описано выше;
(в): Обработка омыляющим реагентом (таким как основание), например, гидроксидом натрия, или обработка в условиях реакции гидрирования, например, Pd/C и водород; предпочтительно - обработка омыляющим реагентом. Если в данных условиях реакции омыления также одновременно происходит снятие защиты с атома азота, то образуются соединения формулы (20-а), в которой R1=Н. При необходимости перед стадией (г) может быть осуществлена повторная защита атома азота, таким образом, что R1=PG. Повторная защита может быть осуществлена при помощи обработки подходящим азотзащитным агентом, как описано ранее, при этом PG может быть такой же, или отличной от первоначально используемой PG;
(г): Обработка в условиях реакции декарбоксилирования, например, нагревание, предпочтительно в присутствии растворителя, более предпочтительно - инициированное кипением;
(д): Обработка подходящим агентом для снятия защиты с атома азота, предпочтительно, агентом для снятия защиты с амина, например, обработка кислотой или основанием, предпочтительно обработка n-толуолсульфоновой кислотой; или обработка подходящим азотзащитным агентом, предпочтительно аминозащитным агентом, как описано ранее, для защиты N-защитной группой PG, которая может такой же, или отличной от первоначально используемой PG. Стадия (д) является необязательной.
(е): Обработка в условиях реакции декарбоксилирования, например, нагревание, предпочтительно в присутствии растворителя, более предпочтительно - инициированное кипением, необязательно в присутствии основания, например, так, как описано для соединения 3с на Схеме 3 в Org. Lett., 2004, 6(25), 4727
Стадия декарбоксилирования (г) является средством для получения соединения формулы (2) или его соли диастереоселективным способом. В одном из вариантов осуществления изобретения при декарбоксилировании соединения формулы (20-а), в которой R1 представляет собой водород или азотзащитную группу, предпочтительно пивалоил, и R11 представляет собой метил, получают соединение формулы (2), в которой R1 представляет собой водород или азотзащитную группу, предпочтительно пивалоил, при соотношении диастереомеров (2-а) и (2-b), равном, по меньшей мере, 55:45. В другом варианте осуществления изобретения при декарбоксилировании соединения формулы (20-а), в которой R1 представляет собой водород или азотзащитную группу, предпочтительно водород, и R11 представляет собой метил, получают соединение формулы (2), в которой R1 представляет собой водород или азотзащитную группу, предпочтительно водород, при соотношении диастереомеров (2-а) и (2-b), равном, по меньшей мере, 29:79.
Стадия декарбоксилирования (е) также является средством для диастереоселективного получения соединения формулы (2), в которой R1 представляет собой водород или азотзащитную группу, или его соли.
В предпочтительном варианте осуществления изобретения соединения формулы (2), получаемые через стадии (г), (д) или (е) на Схеме 3, соответствуют формуле (2-а). В другом предпочтительном варианте осуществления стадии (г) R представляет собой пивалоил или водород. В еще одном варианте осуществления изобретения R1 представляет собой азотзащитную группу для соединений формулы (2-а) и/или (2).
Стадии (в) или (г) могут быть осуществлены так как, например, описано в Org. Biomol. Chem 2007, 5, 143 и в Org. Lett. 2003, 5, 353.
Подраздел Б-2: Взаимодействие метилированного лактама, соответствующего формуле (2), с получением промежуточного соединения, соответствующего формуле (3)
Другим объектом настоящего изобретения является способ получения соединения, соответствующего формуле (3), или его соли
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано выше, и R3 представляет собой водород или алкил,
включающий взаимодействие соединения, соответствующего формуле (2), или его соли
,
где R1 представляет собой водород или азотзащитную группу, с агентом раскрытия цикла, определенным далее. В предпочтительном варианте осуществления изобретения R1 и R2 представляют собой водород, a R3 представляет собой этильную группу.
Как вариант, в формуле (3) или ее соли, R1 и R2, вместе с атомом N, к которому они присоединены, могут образовывать циклическую кольцевую систему, предпочтительно пятичленное кольцо (и, таким образом, образуют бифункциональную циклическую азотзащитную группу, которая вместе с упомянутым атомом N, приводит, например, к пятичленной кольцевой сукцинимидной или малеимидной структуре). Соединение, соответствующее формуле (2), или его соль, предпочтительно получают посредством реакций, описанных выше в подразделе Б-1.
Предпочтительно в формуле (3), или ее соли, R1 представляет собой водород или азотзащитную группу, как описано выше, a R2 представляет собой водород. Кроме того, R3 предпочтительно обозначает водород или этил.
В основном, после описанной выше реакции метилирования, соединение формулы (2) или его соль, предпочтительно соединение формулы (2-а) или его соль, может вступать в реакцию с агентом раскрытия кольца, с получением соединения формулы (3) или его соли. Реакция раскрытия лактамового кольца может проходить в основных, нейтральных или кислотных условиях.
Примерами агентов раскрытия цикла являются нуклеофильные основания, такие как гидроксиды щелочных металлов (например, гидроксид натрия или гидроксид лития) или нейтральные соединения, такие как гидропероксиды (такие как гидропероксид лития). Другими примерами являются кислоты Льюиса или Бренстеда, предпочтительно в присутствии воды. Предпочтительными кислотами являются минеральные кислоты, такие как серная, перхлорная и соляная кислота. Сульфоновые кислоты, такие как пара-толуолсульфоновая кислота, также пригодны в качестве кислот, связывающихся с полимерами, такими как Amberlyst®. В основном в качестве агента раскрытия цикла используют соляную кислоту.
Агент раскрытия цикла может быть использован в каталитических количествах или стехиометрически. Предпочтительно агент раскрытия цикла используют в количестве от 1 до 10 эквивалентов.
Соединения, соответствующие формуле (3), могут существовать в виде солей, например, в виде карбоксилатных солей или кислотно-аддитивных солей. Кислотно-аддитивные соли являются предпочтительными. В основном различные кислоты пригодны для получения кислотно-аддитивной соли. Предпочтительными являются минеральные кислоты, в частности, серная кислота, соляная кислота, бромистоводородная кислота или перхлорная кислота. Также пригодны сульфоновые кислоты, такие как пара-толуолсульфоновая кислота. В основном используют соляную кислоту. Как вариант, соединения, соответствующие формуле (3), могут существовать в виде свободного основания (цвиттерионы).
Реакцию раскрытия цикла осуществляют в широком температурном интервале, например, между -10°С и +150°С. Предпочтительно реакцию проводят при температуре между +20°С и +125°С. Реакция может быть проведена в различных растворителях, например, в воде или этаноле, или в их смесях. Также могут быть использованы дополнительные растворители, такие как толуол, изопропилацетат, тетрагидрофуран или трет-бутилметиловый эфир. Предпочтительно используют этанол и/или воду.
В предпочтительном воплощении изобретения реакцию раскрытия цикла проводят так, что образуется соединение, имеющее конфигурацию, соответствующую формуле (3-а), или его соль,
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как определено выше, и R3 представляет собой водород или алкил. В предпочтительном варианте осуществления изобретения R1 и R2 представляет собой водород и R3 представляет собой этил. Соединение, соответствующее формуле (3-а), или его соль, может быть получено, если в качестве исходного вещества используют соединение, соответствующее формуле (2-а), или его соль.
Соединение, соответствующее формуле (3-а), или его соль, представляет собой, так называемый, 2R,4S-диастереоизомер. Как вариант, также могут быть получены 2R,4R-диастереоизомер, 2S,4S-диастереоизомер и 2S,4R-диастереоизомер.
Реакция получения соединения (3) или его соли из соединения (2) или его соли может быть осуществлена различными способами. Например, в качестве исходного вещества может быть использовано соединение, соответствующее формуле (2), или его соль, где R1 представляет собой водород. Также может быть использовано соединение, соответствующее формуле (2), или его соль, где R1 представляет собой азотзащитную группу, как определено выше, предпочтительно - пивалоильную группу или БОК-группу. Если соединение, соответствующее формуле (2), или его соль, где R1 представляет собой азотзащитную группу, как определено выше, используется в качестве исходного вещества, то, предпочтительно, азотзащитная группа удаляется при реакции раскрытия цикла. Это означает, что предпочтительно получают соединения, соответствующие формуле (3), или его соль, где R1 и R2 представляют собой водород.
При необходимости, соединение формулы (3) или его соль, где R1 и R2 представляют собой водород, снова может быть превращено в соединение формулы (3), или его соль, где R1 и/или R2 независимо друг от друга обозначают азотзащитную группу, как определено выше. Это возможно, если R3 в формуле (3), представляющий собой водород, необходимо заменить на алкильный остаток. На Схеме 4, приведенной ниже, показаны примеры предпочтительных вариантов осуществления изобретения.
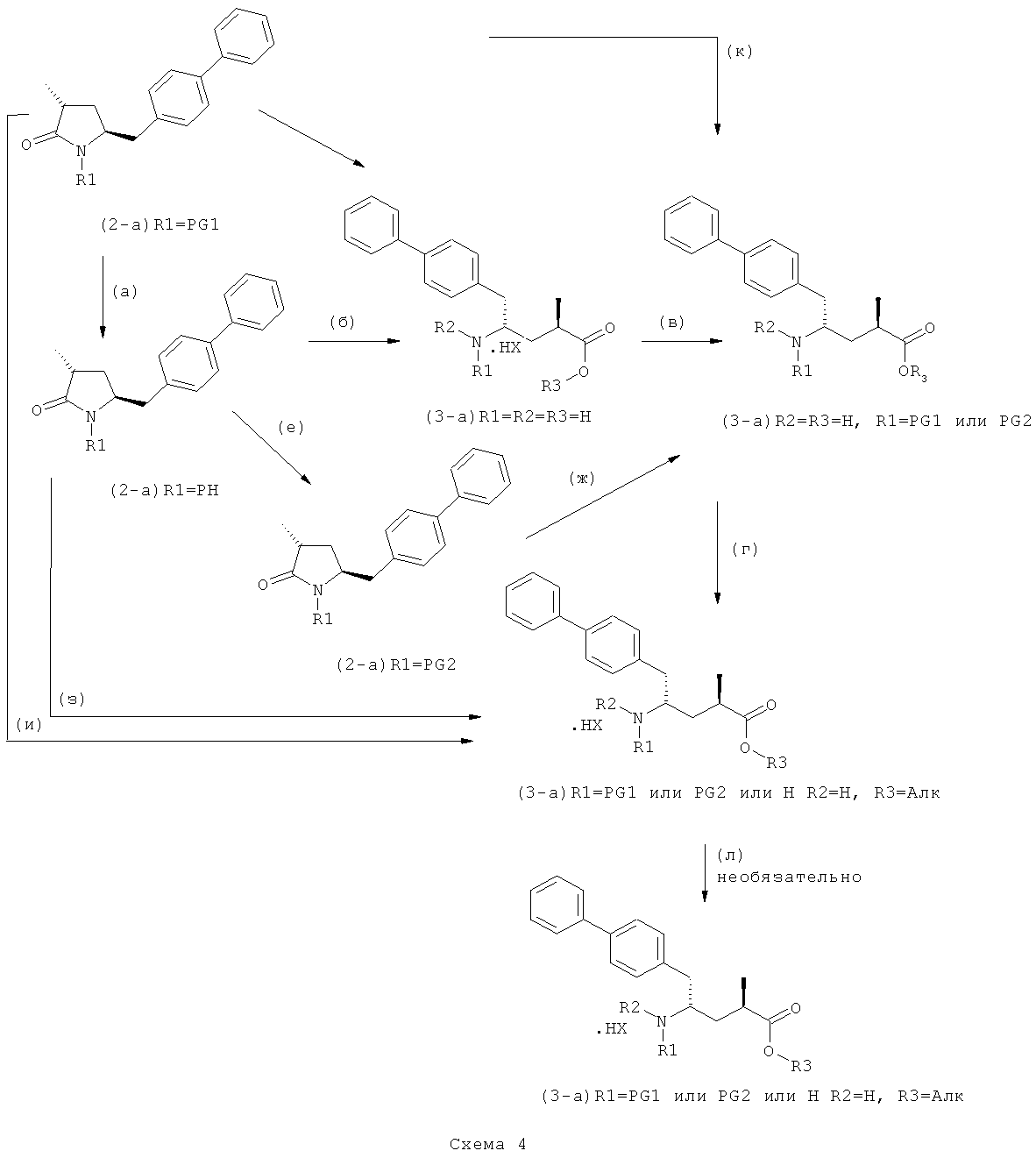
В другом варианте осуществления настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 4, превращения соединения формулы (2-а), как определено в настоящем описании, в соединение формулы (3-а), как определено в настоящем описании, и также относится к каждой реакционной стадии. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, представленной на Схеме 4, а также - к продукту, полученному на каждой реакционной стадии, показанной на Схеме 4.
На Схеме 4 "PG1" обозначает азотзащитную группу, как определено выше, предпочтительно - пивалоил. "PG2" обозначает азотзащитную группу, как определено выше, предпочтительно отличную от PG1, в частности, БОК. "Алк" обозначает алкильную группу, предпочтительно, этил. Обозначение ".НХ" указывает на то, что соединение предпочтительно находится в виде кислотно-аддитивной соли, в частности, в форме HCl.
Реакции, представленные на Схеме 4, не ограничены конкретной стехиометрией. Напротив, они могут также быть осуществлены с соединениями, имеющими любую другую возможную конфигурацию.
В общем реакции (а)-(к) могут быть осуществлены с использованием различных условий. Предпочтительные условия для каждой из реакций (а)-(к), показанных на Схеме 4, даны ниже:
(а): Обработка подходящим агентом, снимающим защиту с атома азота, предпочтительно, снимающим защиту с аминогруппы, например - обработка кислотой или основанием, предпочтительно- обработка n-толуолсульфокислотой;
(б): Обработка описанными ранее агентами раскрытия цикла, предпочтительно соляной кислотой или смесью соляной и уксусной кислот;
(в): Обработка подходящим азотзащитным агентом, предпочтительно аминозащитным агентом, более предпочтительно - дн-трет-бутилдикарбонатом;
(г): Обработка алифатическим спиртом, предпочтительно этанолом. Необязательно в присутствии тионилхлорида или такой кислоты, как минеральная кислота, например HCl, H2SO4, H3PO4 или HBr; предпочтительно, необязательно в присутствии тионилхлорида. Необязательно, когда PG1 или PG2 являются кислотно-лабильными, данная стадия может включать в себя обработку подходящим алкилирующим агентом, например алкилгалогенидом (таким как этилхлорид, этилбромид или этилйодид, предпочтительно - этилйодид) в присутствии основания (например, NaH, Cs2CO3).
(д): Обработка кислотой или основанием в присутствии воды, предпочтительно соляной кислотой в воде. Эта стадия может включать в себя дополнительную обработку подходящим агентом, снимающим защиту с азота, например Pd/C и водородом, когда PG1 не является ни кислотой, ни основно-лабильной N-защитной группой. Обработку проводят кислотой или щелочью в присутствии воды;
(е): Обработка подходящей азотзащитной группой, предпочтительно аминозащитной группой, более предпочтительно - БОК;
(ж): Обработка агентом раскрытия цикла, как описано выше, предпочтительно - гидроксидом лития;
(з): Обработка кислотой или основанием в спиртовом растворе, предпочтительно - соляной кислотой в этаноле;
(и): Обработка кислотой или основанием в спиртовом растворе, предпочтительно - соляной кислотой в этаноле;
(к): Обработка агентом раскрытия цикла, как описано выше, предпочтительно гидроксидом лития; необязательно, когда PG1 представляет собой основно-лабильную, N-защитную группу, данная стадия может дополнительно включать в себя дальнейшую обработку подходящим азотзащитным агентом, который может быть таким же, или отличаться от используемого исходного PG1;
(л): Обработка подходящим азотзащитным агентом, как определено ранее, предпочтительно агентом, снимающим защиту с аминогруппы, предпочтительно в присутствии основания, такого как триэтиламин; или обработка подходящим агентом, снимающим защиту с азота, предпочтительно агентом, снимающим защиту с аминогруппы, например - обработка Pd/C и водородом, или обработка основанием. Стадия (л) является необязательной.
Защитная группа предпочтительно замещается по схеме реакции (а)/(е) и (д)/(в), например, из PG1 в PG2. Защитная группа предпочтительно не претерпевает изменений при осуществлении реакции по схеме (к).
В предпочтительно варианте осуществления изобретения, R1 и R2 в соединении формулы (3-а), показанном на Схеме 4, представляют собой водород.
Является предпочтительным, чтобы реакции (г), (з) и (и) на Схеме 4 приводили к соединению, в котором «Алк» представляет собой этил. Это соединение предпочтительно используют для получения NEP- ингибитора, как более подробно описано ниже, в подразделе Б-3.
Еще в одном варианте осуществления, настоящее изобретение относится к стадии (г), на которой предпочтительно соединение формулы (3-а), в котором R1 и R3 представляют собой водород, a R2 является азотзащитной группой, предпочтительно - кислотно-лабильной азотзащитной группой, такой как БОК, превращается в соединение формулы (3-а) или его соль, где R1 и R2 представляют собой водород, a R3 представляет собой алкил, предпочтительно этил, посредством обработки тионилхлоридом и алифатическим спиртом, предпочтительно - этанолом. В предпочтительном варианте осуществления изобретения, соединение формулы (3-а), полученное согласно данному способу, в котором R1 и R2 - водород, a R3 - алкил, представляет собой этиловый эфир (2R, 45)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты, или его соль.
Схема 4 включает в себя предпочтительные абсолютные конфигурации соединений формул (2) и (3). Однако настоящее изобретение также касается полной последовательности реакций и каждой стадии реакции, где любое из соединений (продукт или исходный материал), или их смесь, является чистым диастереомером, или энантиомером, или их смесями, например, смесями энантиомеров, такими как рацематы. В предпочтительном варианте осуществления стадия (г) обеспечивает получение соединения формулы (3), предпочтительно, где R1 и R2 представляют собой водород, a R3 - этил, или его соли, при диастереомерном соотношении (3-а) к (3-b), по меньшей мере, 60:40, предпочтительно, по меньшей мере, 70:30, более предпочтительно, по меньшей мере, 80:20, еще более предпочтительно, по меньшей мере, 90:10, наиболее предпочтительно, по меньшей мере, 99:1.
Подраздел Б-3: Взаимодействие промежуточного соединения (3) для получения NEP-ингибитора или его пролекарства, предпочтительно пролекарства NEP-ингибитора согласно формуле (18)
Соединение, соответствующее формуле (3), или его соль, особенно соединение формулы (3-а), или его соль, может быть использовано для получения NEP-ингибитора или его пролекарства.
Термин «NEP-ингибитор» описывает соединение, которое ингибирует активность фермент-нейтральной эндопептидазы (NEP, ЕС 3.4.24.11) и, как следует понимать, включает в себя его соли.
Термин «пролекарство» описывает фармакологическую субстанцию, которую вводят в неактивной (или менее активной) форме. Будучи однажды введенным, пролекарство подвергается метаболизму в организме in vivo до активного соединения.
Таким образом, один вариант осуществления способа согласно настоящему изобретению включает в себя одну или более дополнительных стадий, на которых соединение формулы (1) дополнительно реагирует с образованием NEP-ингибитора или его пролекарства.
В настоящем изобретении термины «NEP-ингибитор» или «пролекарство NEP-ингибитора» относятся к веществам как таковым, или к их солям, предпочтительно, к их фармацевтически приемлемым солям. Примерами являются соли натрия, калия, магния, кальция или аммония. Предпочтительными являются соли кальция.
Предпочтительно, соединения формулы (1-а), или их соли, реагируют далее с образованием NEP-ингибитора или его пролекарства. Особенно предпочтительным является соединение формулы (3-а), или его соль, в котором R1 и R2 представляют собой водород, a R3 - этил.
В предпочтительном варианте осуществления, соединение формулы (3-а) подвергают дальнейшему взаимодействию с получением пролекарства NEP-ингибитора этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соли, в соответствии с формулой (18) (известный из литературы как AHU 377), или его солью.
Таким образом, другим объектом настоящего изобретения является способ получения соединения, соответствующего формуле (18), или его соли.
,
включающий стадии:
а) предоставление соединения, соответствующего формуле (1-а), или его соли,
,
б) метилирование указанного соединения формул (1-а), или его соли, с получением соединения формулы (2-а), или его соли,
,
в) взаимодействие соединения формулы (2-а), или его соли, с раскрывающим цикл реагентом, с получением соединения формулы (3-а), или его соли,
,
г) взаимодействие соединения формулы (3-а), или его соли, с получением соединения, соответствующего формуле (18), или его соли,
где в вышеуказанных формулах R1 и R2 являются, независимо друг от друга, водородом или азотзащитной группой, как описано выше, a R3 представляет собой водород или алкил.
В другом варианте осуществления, настоящее изобретение относится к указанному способу, включающему в себя стадии (а)-(г), а также к каждой из реакционных стадий (а)-(г). В еще одном варианте осуществления, настоящее изобретение также относится продукту, полученному в соответствии с каждой из стадий (а)-(г), и к продукту, полученному в соответствии с полной последовательностью реакций (а)-(г).
Превращения соединения (1-а), или его соли, в (2-а) или его соль, и соединения (2-а), или его соли, в (3-а) или его соль, описаны ранее в предшествующих подразделах Б-1 и Б-2, соответственно.
В общем случае, настоящее изобретение включает в себя любые фармацевтически приемлемые соли этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, причем предпочтительной является кальциевая соль.
Пролекарство NEP-ингибитора этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соль, необязательно дополнительно взаимодействует с получением активного NEP-ингибитора N-(3-карбокси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соли.
В предпочтительном варианте осуществления настоящего изобретения синтез этилового эфира N-(3-карбоуси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соли, проводят согласно Схеме 5:
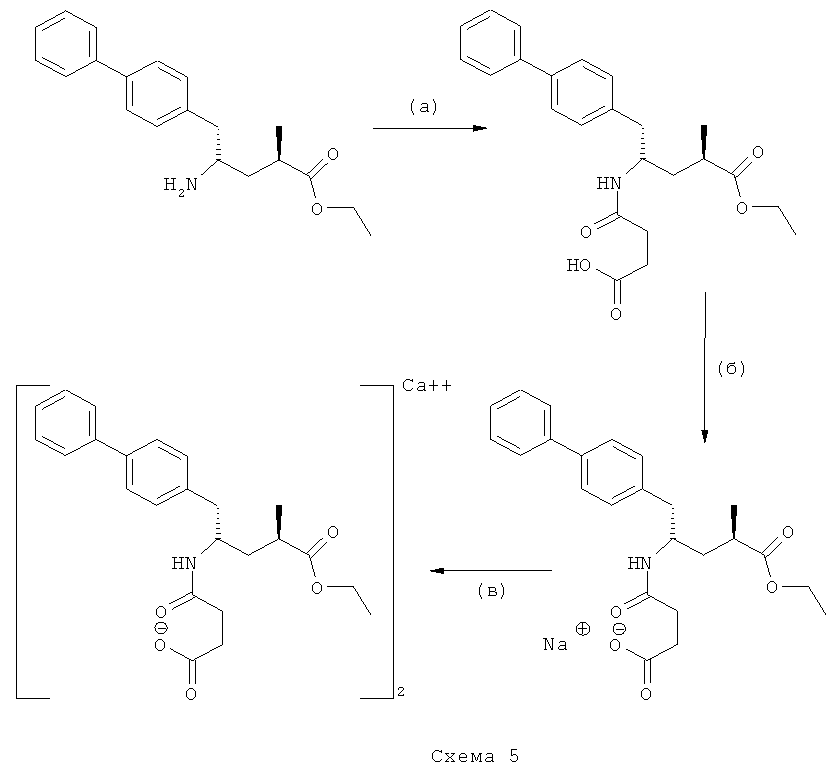
Как правило, реакции (а)-(в) могут быть осуществлены в различных условиях. Стадии (б) и (в), обеспечивающие получение соответственно натриевой и кальциевой солей, являются необязательными стадиями. Предпочтительные условия для каждой из реакций (а)-(в) Схемы 5 даны ниже.
(а): Обработка сукциновым ангидридом, предпочтительно в присутствии основания. Предпочтительными основаниями являются триэтиламин, пиридин, карбонат натрия, гидрокарбонат натрия и карбонат калия; более предпочтительным основанием является триэтиламин;
(б): Обработка натриевым основанием, предпочтительно NaOH.
(в): Обработка солью кальция, предпочтительно CaCl2.
В предпочтительном варианте осуществления, исходный материал для синтеза этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-n-фенилфенилметил)-4-амино-(2R)-метилмасляной кислоты, или его соли, в соответствии со Схемой 5 находится в форме кислотно-аддитивной соли НХ, предпочтительно HCl. В таком предпочтительном осуществлении изобретения, стадия (а) соответственно Схеме 5 требует наличия основания, такого как триэтиламин, пиридин, карбонат натрия, гидрокарбонат натрия или карбонат калия, предпочтительно, основанием является триэтиламин.
Раздел В: Способы получения Ключевого Лактама (1)
Настоящее изобретение включает в себя семь способов получения Ключевого Лактама (1), или его соли, которые описываются далее, в подразделах В-1-В-7.
Подраздел В-1: Способ 1
В одном варианте осуществления, объектом настоящего изобретения является способ получения соединения формулы (1), или его соли,
где R1 имеет значения, приведенные выше,
содержащий следующие стадии:
а) предоставление соединения, соответствующего формуле (4), или его соли,
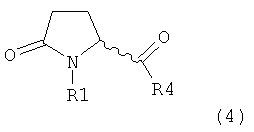 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, a R4 является СО-активирующей группой, как определено ниже,
б) взаимодействие соединения формулы (4), или его соли, с бифенильным соединением, с получением соединения формулы (5)
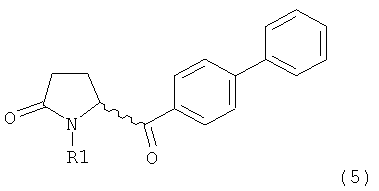 ,
,
где R1 представляет собой водород или азотзащитную группу, как определено выше, и
в) восстановление, например, обработкой водородом или с использованием известного из литературы восстановителя (например, гидрида, такого как боргидрид натрия), предпочтительно - обработкой водородом, соединения формулы (5), или его соли, с получением соединения, соответствующего формуле (1), или его соли.
В другом варианте осуществления, настоящее изобретение относится к вышеуказанному способу, включающему стадии (а)-(в), а также к любой из реакционных стадий (а)-(в). В еще одном варианте осуществления, настоящее изобретение также относится к продукту, полученному в соответствии с каждой из стадий взаимодействия (а)-(в), и к продукту, полученному в соответствии со всей последовательностью реакций (а)-(в).
Разъяснения, касающиеся стадий (4)->(5):
Соединения формулы (4), или их соли, легко получаются из глютамовой кислоты и/или пироглютамовой кислоты, или их производных, то есть из массива хиральных соединений.
В формуле (4) R4 представляет собой СО-активирующую группу. Подходящая СО-активирующая группа обычно является любой группой, которая может действовать, как уходящая. Примерами групп, которые могут действовать как СО-активирующие, являются -NR2, -OR, -SR или галоген, где R представляет собой водород или необязательно замещенный алкил или необязательно замещенный арил.
Предпочтительно, следующие группы являются пригодными в качестве СО-активирующей группы в формуле (4):
a) R4 может быть аминогруппой, в частности, -NR12R13, где R12 и R13
- независимо выбраны из группы, состоящей из алкила, алкоксигруппы, арила, арилоксигруппы, арилалкила и арилалкоксигруппы; предпочтительно, R12 является алкилом (например, метилом), a R13 выбрана из группы, состоящей из алкоксигруппы (например, метокси- или этоксигруппы), арилоксигруппы (например, фенилоксигруппы) и арилалкоксигруппы (например, бензилоксигруппы); или
- совместно являются замещенным или незамещенным алкиленом, или незамещенным или замещенным алкениленом; например, пиперидинилом, морфолинилом, 1-алкилпиперазинилом (например, 1-метилпиперазинилом), 2-, 3-, 4-алкилпиперидинилом, 1,2,3,6-тетрагидропиридинилом, пирролидинилом или имидазолилом; или
- R12 представляет собой алкил (например, метил), a R13 является -X-R14, где X представляет собой S, a R14 обозначает алкил (например, метил или этил), арил (например, фенил) или арилалкил (например, бензил); или
- R12 представляет собой алкил (например, метил), a R13 обозначает -NRaRb, где Ra и Rb, независимо, выбраны из алкила (например, метила или этила), арила (например, фенила) или арилалкила (например, бензила).
Предпочтительная группа R4 является диалкилированной аминогруппой, которая может быть циклической (например, морфолинил или имидазолил) или ациклической (например, диметиламиновая группа). Циклические аминогруппы предпочтительно содержат 5- или 6-членный цикл, имеющий, или не имеющий, дополнительные заместители, в частности, замещения относятся к одному или более заместителей, выбранных из группы, состоящей из галогена, алкила, алкоксигруппы, арила, арилоксигруппы, алкиларила и арилалкоксигруппы. Также, подходящими являются алкилариламиногруппы (например, фенилметиламиногруппа) или диариламиногруппы (например, дифениламиногруппа). Другими подходящими группами являются так называемые производные Вайнреба (то есть производные метилметоксиамина), в частности- NR12R13, где R12 представляет собой метил или метоксигруппу, a R13 независимо выбрана из алкила, алкоксигруппы; арила, арилоксигруппы, арилалкила или арилалкоксигруппы. Кроме того, пригодными являются аминогруппы, обладающие алкильной/арильной группой и координирующей группой, например, алкоксигруппой,алкилтиогруппой.
б) R4 может быть группой, имеющей формулу -X-R, где X представляет собой О или S, a R обозначает алкил или арил. Кроме того, R4 может быть группой, имеющей формулу -O-CO-R, где R обозначает алкил или арил.
в) R4 может быть галогеном, предпочтительно хлором.
г) R4 может быть -O-R15, где R15 представляет собой -NR12R13, как определено выше, или R15 представляет собой замещенный или незамещенный гетероциклил.
Предпочтительно, СО-активирующую группу выбирают из диметиламиногруппы, морфолинила, имидазолила,
метилметоксиаминогруппы, -О-метила, -О-этила, хлора, брома,пивалоила и ацетила. В частности, СО-активирующей группой является морфолин.
Если СО-активирующая группа выбрана из вышеуказанных групп а) или б) в формуле (4), остаток R1, предпочтительно, представляет собой азотзащитную группу, как описано выше, или, как вариант, водород. Если СО-активирующую группу выбирают из вышеуказанной группы в) в формуле (4), остаток R1 представляет собой, предпочтительно, водород.
Соединение формулы (4), или его соль, реагирует с бифенильным соединением.
В предпочтительном варианте осуществления изобретения бифенильное соединение может быть активированным. Подходящим способом активации является получение органометаллического комплекса, содержащего бифенильный лиганд.
Предпочтительными активированными бифенильными соединениями являются:
дифенилмагний галогенид или ди(бифенил)магний (реактивы Гриньяра). Подходящие галогениды обычно представляют собой хлорид, бромид и йодид, причем бромид является особенно предпочтительным.
Дополнительными примерами активированных бифенильных соединений являются бифениллитий, бифенилкупрат (купраты низшего и высшего порядков) и бифенилцинк. Эти соединения могут использоваться по отдельности, или в присутствии другого металла, например меди, цинка, палладия, платины, железа, иридия или рутения.
Обычно используют от 2,0 до 2,5 эквивалентов бифенилмагний галогенида или ди(бифенил)магния. В некоторых вариантах осуществления, начальное депротонирование N-H-группы при помощи, например, другого реактива Гриньяра (например, хлорида изопропилмагния) или основания (например, гидрида натрия), может быть осуществлено до добавления активированного бифенильного соединения для восстановления требуемого количества галогенида бифенилмагния или ди(бифенил)магния. В данном случае, используют от 0,7 до 1,5 эквивалентов, предпочтительно от 1,0 до 1,25 эквивалентов.
Обычно для проведения вышеуказанных реакций существует два предпочтительных варианта осуществления изобретения:
1) Взаимодействие соединения формулы (4), или его соли, где R4 (СО-активирующая группа) выбирают из вышеуказанных групп а) или б). В данном случае, предпочтительно используют активированное (например, металлированное) дбфенильное соединение, в частности - галогенид бифенилмагния.
2) Взаимодействие соединения формулы (4), или его соли, где R4 (СО-активирующая группа) выбирают из вышеуказанной группы в). В данном случае, в качестве бифенильного соединения предпочтительно используют бифенил. Реакцию предпочтительно проводят в присутствии подходящей кислоты Льюиса, например, трихлорида алюминия. Альтернативно, бифенильное соединение может быть активировано подходящей функциональной группой (например, пара-силилом), для обеспечения более мягких условий при осуществлении ацилирования по Фриделю-Крафцу. Также, дается ссылка на способ ацилирования по Фриделю-Крафцу, описанный в J.Am.Chem.Soc, том 103, №20, 1981, 6157.
Таким образом, является предпочтительным, чтобы R4 формулы (4) представлял собой морфолинил, а бифенильное соединение, используемое на стадии б), представляло собой галогенид бифенилмагния,
или
R4 формулы (4) представляет собой хлорид и бифенильное соединение, используемое на стадии б), является бифенилом.
Варианты осуществления 1) и 2) поясняются ниже на схеме 6.
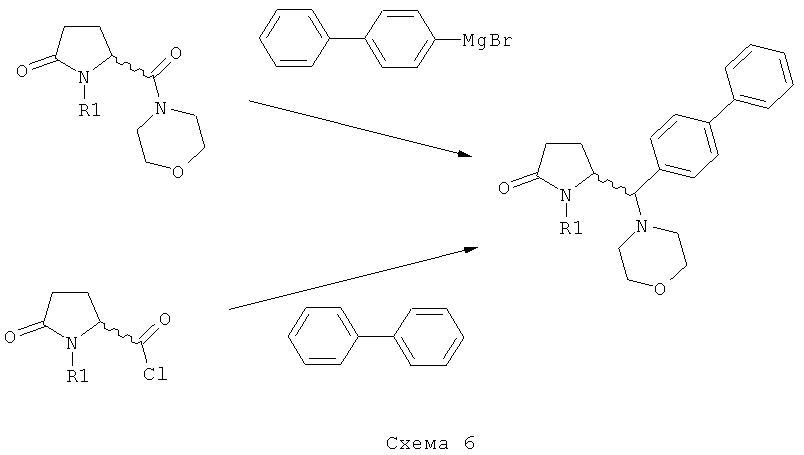
Пояснения к стадии (5)->(1):
Восстановление карбонильной группы соединения формулы (5), или его соли, например, обработкой водородом или с использованием известного из уровня техники восстановителя (например, гидридного реагента, такого как боргидрид натрия), приводит к образованию соединения, соответствующего формуле (1), или его соли. Предпочтительно, восстановление соединения формулы (5), или его соли, завершается гидрированием. В зависимости от условий, реакция может проводиться непосредственно, или через соответствующий спирт формулы (13), или его соль, в качестве промежуточного соединения
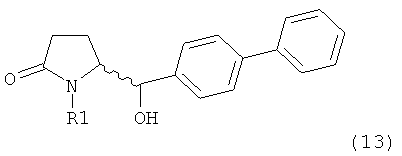 ,
,
где R1 представляет собой водород или азотзащитную группу, как определено выше.
Полное восстановление карбонильной группы (т.е. реакция (5)->(1)) может быть достигнуто при использовании катализатора гидрирования, такого как палладий на угле (здесь и далее обозначаемый, как Pd/C). Восстановление может проводиться в присутствии, или в отсутствии кислоты. Предпочтительным является Pd/C, выбранный из группы, состоящей из 10% Pd/C типа К-0218 (коммерчески доступный от Heraeus GmbH), 10% Pd/C типа PD 4505 D/R (коммерчески доступный от BASF), 5% Pd/C типа 39, 10% Pd/C типа 39, 10% Pd/C типа 39 (7200), 20% Pd/C типа 91, 10% Pd/C типа 338, 10% Pd/C типа 394, 10% Pd/C типа 394 (6065), 10% Pd/C типа 394 (6249), 10% Pd/C типа 395, 10% Pd/C типа 395 (6002), 10% Pd/C типа Mod (72595), 15% Pd/C типа A101023 и 15% Pd/C типа А502085 (коммерчески доступные от Johnson Matthey); более предпочтительно - 10% Pd/C типа 338, 10% Pd/C Mod (72595), 10% Pd/C типа 39, 10% Pd/C типа 394 (6065) и 10% Pd/C типа 395; наиболее предпочтительно - 10% Pd/C типа 39 и 10% Pd/C типа 394 (6065).
В одном из воплощений изобретения гидрирование обычно проводят при температуре между 0°С и 60°С, предпочтительно - между 20°С и 50°С.Используемое давление водорода обычно лежит в интервале между 1 бар и 30 бар, предпочтительно - между 2 бар и 25 бар. Время реакции обычно лежит в интервале между 1 ч и 30 ч, предпочтительно - между 5 ч и 20 ч.
В другом воплощении изобретения гидрирование обычно проводят при температуре от 0°С до 100°С, предпочтительно - от 20°С до 80°С, более предпочтительно - от 40°С до 80°С, наиболее предпочтительно - от 50°С до 80°С.
В основном используют растворители известные из уровня техники. Предпочтительными растворителями, например, являются изопропилацетат, метилтетрагидрофуран, толуол или моновалентный спирт, такой как метанол или этанол. Более предпочтительно используют толуол. Количество используемого растворителя может быть таким, чтобы концентрация субстрата находилась в интервале от 0,1 до 1,5 М, предпочтительно - от 0,2 до 0,8 М.
Количество катализатора гидрирования по отношению к количеству субстрата, обычно используемое в способе, может находиться в интервале от 1 до 30 влажн. мас.%, предпочтительно - от 2 до 25 влажн. мас.%, более предпочтительно - от 5 до 20 влажн. мас.%.
В зависимости от условий реакции гидрирование может быть завершено на образовании соответствующего вторичного спирта (13) (в виде смеси диастереоизомеров), который затем может быть выделен. Восстановление карбонила в спирт, в основном, может быть осуществлено с использованием восстановителя, известного из уровня техники (например, боргидрида натрия). Превращение ОН в уходящую группу, такую как галоген, методами, хорошо известными специалисту в данной области, и последующая обработка гидридным реагентом (например, боргидридом натрия или диизобутилалюминий гидридом) приведут к образованию соединения, соответствующего формуле (1). При необходимости, спиртовое промежуточное соединения, соответствующее формуле (13), или его соль, также могут быть получены в виде единственного диастереоизомера (или R, или S), например, при использовании энантиоселективного/диатереоселективного катализатора гидрирования, например, так как описано в Angew. Chem. Int. Ed 2001, 40, 40-73, в частности, так как представлено на Схеме 36.
В реакциях (4)→(5) и (5)→(1) может быть важной стереохимия. В предпочтительном воплощении изобретения соединения формул (4), (5) и (1), или их соли, соответствуют формулам (4-а), (5-а) и (1-а)
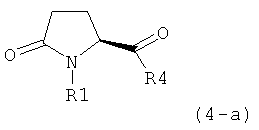 ,
,
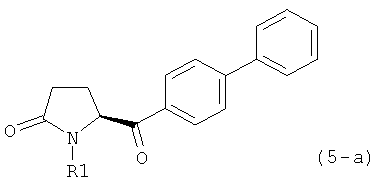 ,
,
,
***
в которых R1 представляет собой водород или азотзащитную группу, как описано ранее, и R4 представляет собой СО-активирующую группу, как описано выше.
В том случае, если в качестве промежуточного соединения образуется вторичный спирт, соответствующий формуле (13), или его соль, то данный спирт, предпочтительно, имеет конфигурацию, как показана для формулы (13-а)
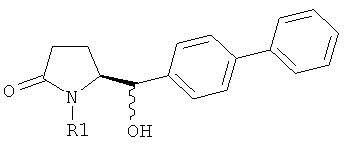 ,
,
в которой R1 представляет собой водород или азотзащитную группу, как описано ранее.
Подраздел В-2: Способ 2
В другом воплощении объектом настоящего изобретения является способ получения соединения, соответствующего формуле (1), или его соли
,
где R1 представляет собой водород или азотзащитную группу, как описано ранее,
включающий следующие стадии:
а) предоставление соединения, соответствующего формуле (7), или его соли
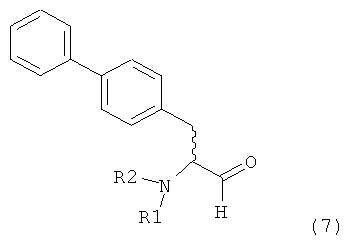 ,
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано ранее,
б) взаимодействие соединения, соответствующего формуле (7), или его соли, в условиях реакции Виттига, с получением соединения, соответствующего формуле (8), или его соли
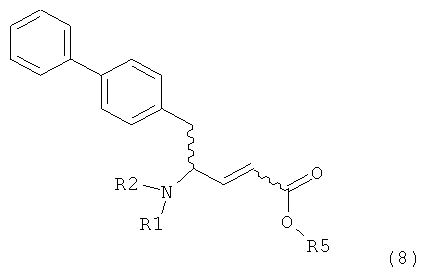 ,
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано ранее, и R5 представляет собой водород или алкил,
в) восстановление соединения, соответствующего формуле (8), или его соли, с получением соединения, соответствующего формуле (9), или его соли
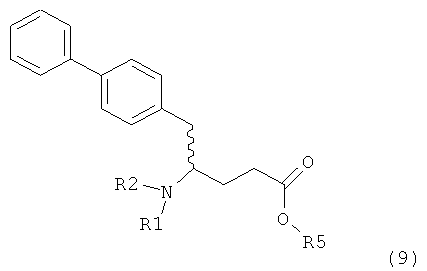 ,
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано ранее, и R5 представляет собой водород или алкил,
г) необязательно удаление азотзащитных групп, с получением соединения, соответствующего формуле (10), или его соли
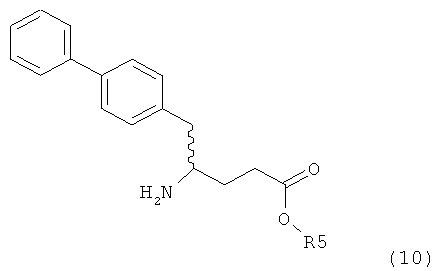 ,
,
где R5 представляет собой водород или алкил, и
д) взаимодействие соединения, соответствующего формуле (9) или (10), или его соли, предпочтительно, взаимодействие соединения формулы (10), в условия реакции замыкания цикла, с получение соединения, соответствующего формуле (1), или его соли, где R1 представляет собой водород или азотзащитную группу, предпочтительно R1 представляет собой водород.
Настоящее изобретение относится к описанному выше способу, включающему стадии (а)-(д), а также относится к каждой из реакционных стадий (а)-(д). Кроме того, настоящее изобретение относится к продукту, полученному в соответствии с каждой из реакционных стадий (а)-(д), и к продукту, полученному в соответствии с полной последовательностью реакций (а)-(д).
Необязательно, соединение, соответствующее формуле (1), или его соль, где R1 представляет собой водород, полученное при взаимодействии соединения, соответствующего формуле (10), или его соли, в условиях реакции замыкания цикла, обрабатывают подходящим азотзащитным агентом, как определено выше, получая соединение формулы (1), или его соль, где R1 представляет собой азотзащитную группу.
В предпочтительном варианте осуществления изобретения в формуле (7) R1 представляет собой азотзащитную группу (как определено выше в разделе А) и R2 представляет собой водород. Данные определения также относятся и к формулам (8) и (9).
Соединение, соответствующее формуле (7), или его соль, взаимодействует в условиях реакции Виттига, с получением соединения, соответствующего формуле (8), или его соли. Обычно в условиях реакции Виттига альдегид, соответствующий формуле (7), или его соль, обрабатывают илидом фосфора (также называемым фосфораном), с получением олефина формулы (8) или его соли. Илиды фосфора обычно получают посредством обработки солей фосфония основанием, а соли фосфония обычно получают из фосфора и алкилгалогенида, методами, хорошо известными специалисту в данной области техники.
Предпочтительно в настоящей заявке соединение, соответствующее формуле (Ar)3Р=СН-СО2-К, в которой Ar представляет собой арил и R представляет собой алкил, используют в реакции Виттига. В частности, в реакции Виттига используют Ph3P=СН-СО2-С2Н5.
Обозначение " " на олефине настоящего изобретения представляет ковалентную связь, включающую как (E)-стереоизомер, так и (Z)-стереоизомер соответствующего олефина. Кроме того, данное обозначение охватывает смеси (Е)- и (Z)-стереоизомеров.
" на олефине настоящего изобретения представляет ковалентную связь, включающую как (E)-стереоизомер, так и (Z)-стереоизомер соответствующего олефина. Кроме того, данное обозначение охватывает смеси (Е)- и (Z)-стереоизомеров.
Кроме того, в формуле (8) остаток R5 означает водород или алкил. Предпочтительно R5 представляет собой С1-С6алкил, более предпочтительно - этил. Данные определения также относятся и к формулам (9) и (10).
Двойную связь соединения, соответствующего формуле (8), или его соли, гидрируют с получением соединения, соответствующего формуле (9), или его соли. В основном гидрирование может быть осуществлено с использованием известных способов. Предпочтительно гидрирование проводят в присутствии Pd/C в качестве катализатора.
Обычно гидрирование проводят при температуре между 0°С и 60°С, предпочтительно между 20°С и 50°С. Применяемое давление водорода обычно находится в интервале между 1 бар и 30 бар, предпочтительно - между 2 бар и 25 бар. Время реакции обычно находится в интервале между 1 ч и 30 ч, предпочтительно - между 5 ч и 20 ч.
Если, по меньшей мере, один из остатков R1 и R2 формулы (9) представляет собой азотзащитную группу, как определено ранее, он может быть удален на необязательной реакционной стадии, методами, хорошо известными из уровня техники, в частности, так как описано в соответствующих главах справочников, упомянутых выше, с получение соединения, соответствующего формуле (10). Если при воплощении изобретения требуется удаление азотзащитной группы, то удаление обычно осуществляют, используя известные способы. Предпочтительно, азотзащитную группу удаляют при помощи SOCl2, соляной кислоты, серной кислоты или трифторуксусной кислоты. В случае, когда азотзащитной группой является N-бензильная группа, она может быть удалена посредством гидрирования или обработки некоторыми подходящими окислителями, например, нитратом аммония-церия (CAN) или 2,3-дихлор-5,6-дициано-n-бензохиноном (ДДХ).
В условиях, обеспечивающих замыкание цикла, из соединения формулы (10), или его соли, получают соединение формулы (1), или его соль. Подходящие условия, обеспечивающие замыкание цикла, представляют собой основную среду. Предпочтительными используемыми основаниями являются алкиламины или алкоксиды щелочных металлов. В частности, предпочтительными являются триэтиламин или метоксид натрия.
В вышеуказанных реакциях стереохимия может играть важную роль. В предпочтительном осуществлении изобретения, используемые соединения, или их соли, имеют конфигурацию, как показано в формулах (7-а), (8-а), (9-а) и (10-а).
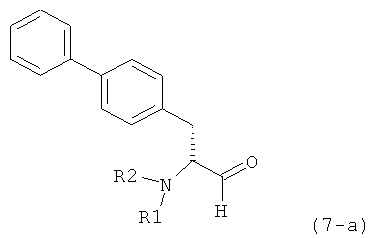
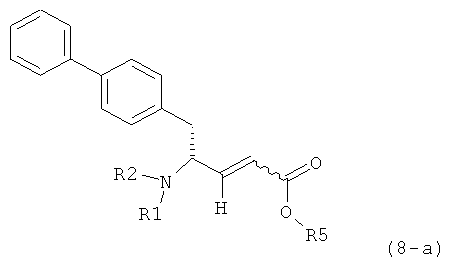
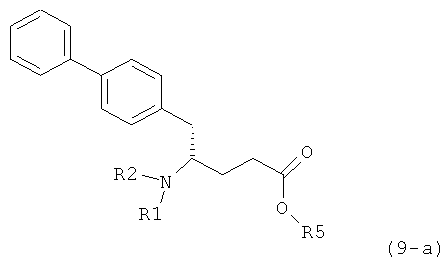
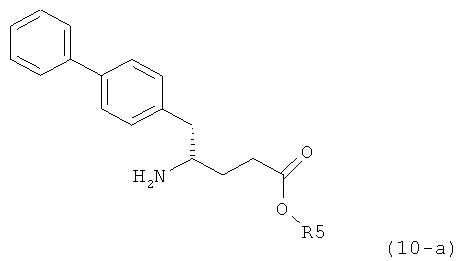 ,
,
где в вышеуказанных формулах R1, R2 и R5 такие, как определено выше для формул (7)-(10). Кроме того, предпочтительно получают соединение формулы (1-а), или его соль, как описано выше.
Предпочтительный вариант осуществления изобретения получения Ключевого Лактама (1), или его соли, исходя из соединения формулы (7), или его соли, показан ниже на Схеме 7.
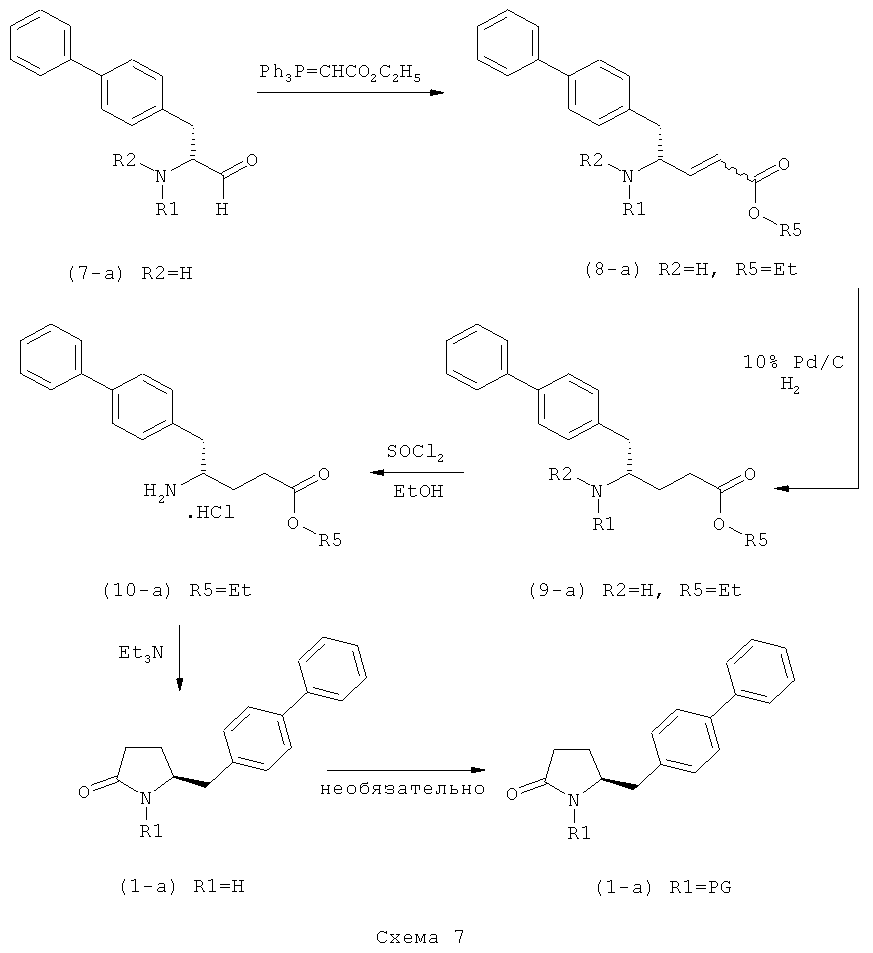
На Схеме 7 "R1" представляет собой азотзащитную группу, предпочтительно БОК.
В другом варианте осуществления настоящее изобретение относится к полной последовательности реакций, показанной на Схеме 7, превращения соединения формулы (7-а), как определено здесь, в описанное здесь соединение формулы (1-а), а также касается каждой из реакционных стадий. Еще в одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной последовательностью реакций, приведенной на Схеме 7, а также к продукту, полученному согласно каждой из реакционных стадий, показанных на Схеме 7.
Подраздел В-3: Способ 3
В качестве альтернативы восстановлению арилкетона формулы (5), который образуется в способе 1, может быть проведено присоединение бифенильного аниона к яр3-гибридизованной уходящей группе.
Таким образом, еще одним объектом настоящего изобретения является способ получения соединения формулы (1), или его соли,
,
где R1 представляет собой водород или азотзащитную группу, как показано выше,
включающий взаимодействие соединения формулы (11), или его соли
 ,
,
где R1 обозначает водород или азотзащитую группу, как определено выше, a R6 представляет собой уходящую группу, как показано ниже, с активированным (например, металлированным) бифенильным соединением, предпочтительно - с галогенидом бифенилмагния.
R1 может быть азотзащитной группой, как описано выше в разделе А: Ключевой Пактам (1), или его соль. Предпочтительно, R1 является водородом.
В общем случае R6 является подходящей уходящей группой. Примерами подходящих уходящих групп являются аминогруппа, алкоксигруппа (например, метоксигруппа, этокигруппа), карбоксилат, галоген (например, фтор, хлор, бром, йод), азид, тиоцианат, нитрогруппа, цианид, тозилат, трифлат и мезилат.Предпочтительно R6 представляет собой йод, тозилат или мезилат.
Относительно термина "активированное бифенильное соединение", то данный термин относится к определениям, данным выше для способа 1 в подразделе В-1. Предпочтительно активированным бифенильным соединением является бифенилмагний галогенид, в частности, бифенилмагний бромид.
Реакция может быть осуществлена в соответствии со следующими предпочтительными вариантами осуществления изобретения, где R1 представляет собой водород.
Исходя из известного пироглютамина (CAS №17342-08-4), замещение спиртовой группы уходящей группой (например, йодом [CAS №29266-73-7], тозилатом [CAS №51693-17-5] или мезилатом), в соответствии с хорошо известными методами, например, так, как описано Richard С.Larock в "Comprehensive Organic Transformations: A Guide to Functional Group Preparations", Second Edition, Wiley-VCH Verlag GmbH, 2000, в частности, так, как описано в соответствующих главах данного источника информации, приводит к образованию соединения, соответствующего формуле (11). Последующее добавление бифенилмагний галогенида, в частности, бифенилмагний бромида (или, как вариант, металлированного бифенила, например, Li, Zn), необязательно в присутствии другого металла (например, меди, цинка, палладия), в каталитическом или стехиометрическом количестве, приводит к образованию соединения, соответствующего формуле (1).
В другом варианте осуществления изобретения сочетание соединения, соответствующего формуле (11), или его соли, где R6 представляет собой уходящую группу, с бифенилмагний галогенидом, осуществляют при катализе Fe или Mn, в условиях реакции кросс-сочетания, например, при использовании FeCl3, Fe(acac)3 или MnCl2, так, как описано, например, в Angew. Chem. Int. Ed., 2004, 43, 3955-3957, в Org. Lett, 2004, 6, 1297-1299, в Chem. Commun., 2004, 2822-2823, в J. Am. Chem. Soc, 2004, 126, 3686-3687, в Synlett, 2001, 1901-1903 или в Synthesis, 1998, 1199-1205. Реакция кросс-сочетания, катализируемая Fe, предпочтительно протекает при использовании FeCl3.
Еще в одном варианте осуществления изобретения сочетание соединения формулы (11) или его соли, где R6 представляет собой уходящую группу, с бифенилмагний галогенидом, протекает в присутствии добавки - соли металла, которую используют в каталитическом или стехиометрическом количестве. Предпочтительной солью металла является, например, соль меди(I), меди(II), цинка(II), серебра(I), кадмия(II), ртути(II), алюминия(III), галлия(III), индия(III), олова(IV), титана(IV) или циркония(IV). Примерами таких солей являются соответствующие хлорид, бромид, йодид, карбонат, гидроксид, оксид, С1-С7-алканоаты, такие как ацетат и пропионат, С1-С7-алкоксиды, такие как метоксид и этоксид, трифторацетат, ацетилаценоат, нитрат, цианид, сульфат, трифторметансульфонат, метансульфонат, бензолсульфонат или пара-толуолсульфонат. Предпочтительными добавками - солями металлов, являются соли меди, такие как цианид меди, который, предпочтительно используют в стехиометрическом количестве.
Как правило, использованию более, чем одного эквивалента реактива Гриньяра (или другого металлсодержащего вещества), может препятствовать добавление дополнительного основания (например, хлорида изопропилмагния или NaH) для удаления протона NH-группы, если R1 представляет собой водород.
Стереохимия реакции может представлять интерес. Предпочтительно, используют соединение, или его соль, имеющее конфигурацию, соответствующую формуле (11-а)
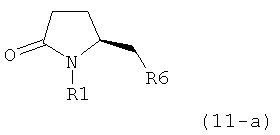 ,
,
где R1 и R6 определены выше для соединений формулы (11), или их солей. Кроме того, предпочтительно получают соединение формулы (1-а), или его соль.
В дополнение к соединению формулы (1), предпочтительно - формулы (1-а), соответствующий азиридин формулы (19), предпочтительно - формулы (19-а), или его соль, может образовываться, когда R1 в соединении формулы (11), предпочтительно - (11-а), представляет собой водород.
 .
.
Соединение формулы (19), описанное в J.Org.Chem., 1988, 53, 4006, или его соль, может быть выделено, обеспечивая постадийное превращение соединения формулы (11) в соединение формулы (1). Как вариант, соединение формулы (19) может генерироваться in situ. Подраздел В-4: Способ 4
Альтернативный способ получения Ключевого Лактама (1) или его соли заключается в окислении первичного спирта пироглютамина в альдегид, с последующим добавлением активированного (например, металлированного) бифенила, и удалением вторичного спирта (например, путем восстановления или гидрирования). Добавление активированного (например, металлированного) бифенила к альдегиду формулы (12), или его соли, может осуществляться в соответствии с методиками, известными из уровня техники, например, так, как это описано в J.Org.Chem., 1987, 52, 4352. Кроме того, способы получения соединений формулы (12), или их солей, описаны в литературе, например в Biochemistry, 1985, 24, 3907.
Таким образом, еще одним объектом настоящего изобретения является способ получения соединения формулы (1), или его соли
,
где R1 представляет собой водород или азотзащитную группу, как определено выше,
включающий следующие стадии:
а) предоставление соединения, соответствующего формуле (12), или его соли,
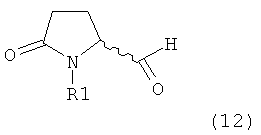 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше,
б) взаимодействие соединения формулы (12), или его соли, с активированным (например, металлированным) бифенильным соединением с получением соединения формулы (13), или его соли
,
где R1 представляет собой водород или азотзащитную группу, как определено выше, и
в) восстановление соединения формулы (13), или его соли, с получением соединения, соответствующего формуле (1), или его соли.
В другом варианте осуществления настоящее изобретение относится к вышеуказанному способу, включающему стадии (а)-(в), а также относится к каждой из реакционных стадий (а)-(в). Еще в одном варианте осуществления настоящее изобретение также относится к продукту, полученному в соответствии с каждой из стадий реакции (а)-(в), и к продукту, полученному в соответствии с полной последовательностью реакций (а)-(в).
R1 может быть азотзащитной группой, как описано выше, в разделе А: Ключевой Пактам (1), или его соль. Предпочтительно, R1 представляет собой водород.
Относительно термина «активированное бифенильное соединение», следует обратить внимание на приведенные выше пояснения, данные для способа 1 в разделе В-1. Предпочтительно, активированное бифенильное соединение представляет собой металлированный бифенил, например галогенид бифенилмагния, особенно - бромид бифенилмагния.
Вторичный спирт формулы (13), или его соль, обычно может быть восстановлен известными способами. В предпочтительном варианте осуществления изобретения ОН-группа превращается в уходящую группу. Предпочтительные уходящие группы описаны выше, в подразделе В-4. Последующая обработка восстановителем приводит к соединению формулы (1), или его соли. Подходящие восстановители описаны, например, ранее в подразделе В-1.
В предпочтительном варианте осуществления соединения (12) и (13), или их соли, имеют конфигурацию согласно формулам (12-а) и (13-а)
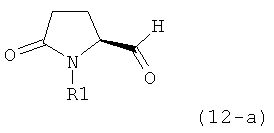
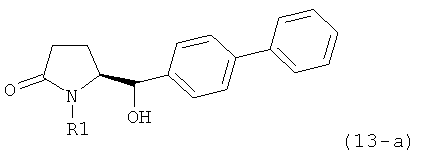 ,
,
где R1 тако, как определено выше для соединений (12) и (13). Более того, предпочтительно получается соединение формулы (1-а), или его соль.
Получение соединений формулы (12) описано в литературе, например, в Biochemistry, 1985, 24, 3907.
В предпочтительном варианте осуществления, взаимодействие соединения формулы (12), с получением соединения формулы (13), является аналогичным присоединению по Гриньяру, описанному в JOC, 1987, 52, 4352.
Подраздел В-5: Способ 5
Альтернативный способ получения Ключевого Лактама (1) представляет собой взаимодействие, исходя из 1,5-дигидропиррол-2-она, или его соли, с использованием хемо- и энантиоселективного гидрирования, в качестве ключевой стадии.
Таким образом, еще одним объектом настоящего изобретения является способ получения соединения формулы (1), или его соли
где R1 представляет собой водород или азотзащитную группу, как определено выше,
включающий следующие стадии:
а) предоставление соединения, соответствующего формуле (14), или его соли,
б) взаимодействие соединения формулы (14), или его соли, с 4-формилбифенилом, с получением соединения формулы (15), или его соли
в) гидрирование соединения формулы (15), или его соли, с получением соединения, соответствующего формуле (16), или его соли,
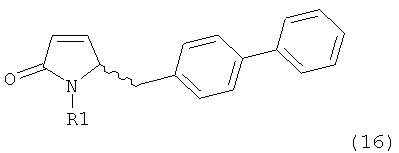
и
г) восстановление соединения формулы (16), или его соли, с получением соединения формулы (1), или его соли,
где в формулах (14)-(16) R1 представляет собой водород или азотзащитную группу, как определено выше.
В другом варианте осуществления настоящее изобретение относится к описанному выше способу, включающему стадии (а)-(г), а также к каждой из реакционных стадий (а)-(г). В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с каждой из реакционных стадий (а)-(г), а также к продукту, полученному в соответствии с полной реакционной последовательностью.
R1 может быть азотзащитной группой, так как описано выше в разделе А: Ключевой Пактам (1) или его соль. Предпочтительно R1 представляет собой водород.
Реакцию соединения, соответствующего формуле (14), или его соли, с 4-формилбифенилом, предпочтительно, осуществляют в присутствии основания, в частности, гидроксида натрия.
Гидрирование соединения (15) или его соли, с получение соединения (16) или его соли, предпочтительно, проводят посредством энантиоселективного гидрирования, предпочтительно, так как описано далее.
Гидрирование соединения (16) или его соли, с получением соединения (1) или его соли, предпочтительно осуществляют с палладием на угле, в качестве катализатора. Как вариант, соединение формулы (16) или его соль может быть превращено в соединение формулы (1) или его соль в условиях восстановления, например, при использовании гидридного восстановителя, такого как NaBH4-NiCl2, NaBH4-BiCl3 или NaBH4-InCl3.
В предпочтительном воплощении изобретения конфигурация соединения (16) или его соли соответствует формуле (16-а)
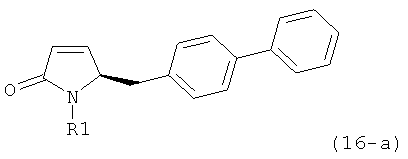 ,
,
в которой R1 имеет значения, определенные выше для формулы (16).
Кроме того, предпочтительно получают соединение формулы (1) или его соль.
Предпочтительное воплощение способа 5 представлено на нижеприведенной Схеме 8.
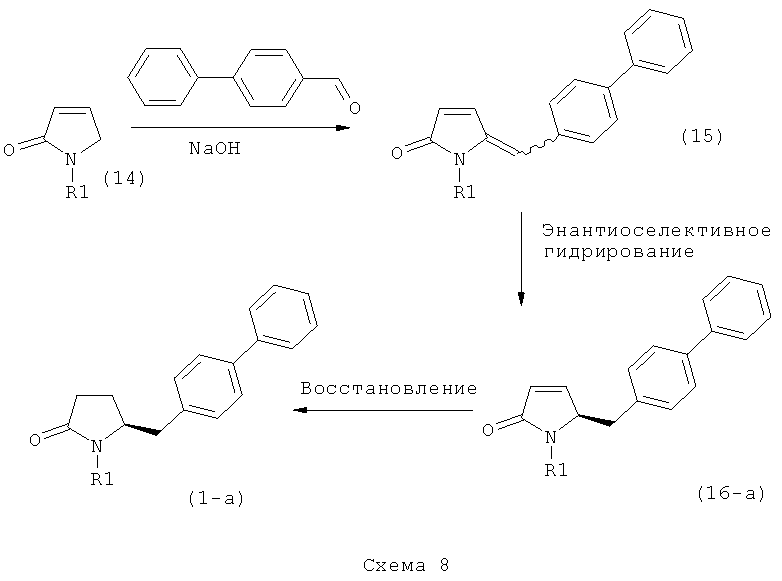
В другом варианте осуществления настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 8, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 8. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 8, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 8.
Соединение формулы (14) или его соль, где R1 представляет собой водород, является коммерчески доступным, например, от производителей, таких как J & W PharmaLab LLC. Обработка соединения формулы (14), в которой R1 представляет собой Н, подходящим азотзащитным агентом, как описано в настоящей заявке, для защиты N защитной группой PG, может быть осуществлена в соответствии с вышеописанными методами.
Превращение соединения формулы (14) или его соли, где R1 представляет собой водород или азотзащитную группу, в соединение формулы (15) или его соль, где R1 представляет собой водород или азотзащитную группу, может быть достигнуто посредством реакции с бифенил-4-карбальдегидом в присутствии основания, такого как основание на основе щелочного металла (например, NaOH или КОН), как описано в J. Org. Chem., 2002, 67 (14), 4702, в Synthesis, 2004, (10), 1560 и в Synth. Commun., 2002, 32 (7), 1031.
Хемоселективное и энантиоселективное гидрирование соединения формулы (15) или его соли, где R1 представляет собой водород или азотзащитную группу, в соединение формулы (16-а) или его соль, где R1 представляет собой водород или азотзащитную группу, может быть осуществлено при использовании, например, условий ассиметричного гидрирования, с использованием енамидных субстратов, таких, как условия, описанные в Tetrahedron Asymm., 1991, 2(1), 51, в частности, для соединений 8а и 8b, или как описано в J Org Chem 1994, 59(2), 297.
Восстановление соединения формулы (16-а) или его соли, где R1 представляет собой водород или азотзащитную группу, в соединение формулы (1-а) или его соль, где R1 представляет собой водород или азотзащитную группу, может быть проведено, например, в условиях гидрирования, например, при использовании Pd/C, как описано, например, в Synthesis, 1993, 216, или при использовании восстановителя, например, при использовании гидридного реагента, такого как NaBH4, как описано, например, в J. Org. Chem, 2006, 71(5), 2173, в частности, как описано для превращения соединения 8 в соединение 15.
Предпочтительное воплощение способа, представленного на Схеме 8, относится к соединениям, в которых R1 представляет собой водород. Другое предпочтительное воплощение способа, представленного на Схеме 8, относится к соединениям, для которых R1 представляет собой водород и где восстановление соединения формулы (16-а) в (1-а) протекает при использовании Pd/C и водорода.
Как вариант, соединение формулы (14) или его соль, может быть превращено в соединение формулы (1), предпочтительно - формулы (1-а), или его соль, как детально представлено на нижеприведенных Схемах 9-12. В предпочтительном воплощении способов, представленных на Схемах 9-12, R1 для соединений представляет собой водород.
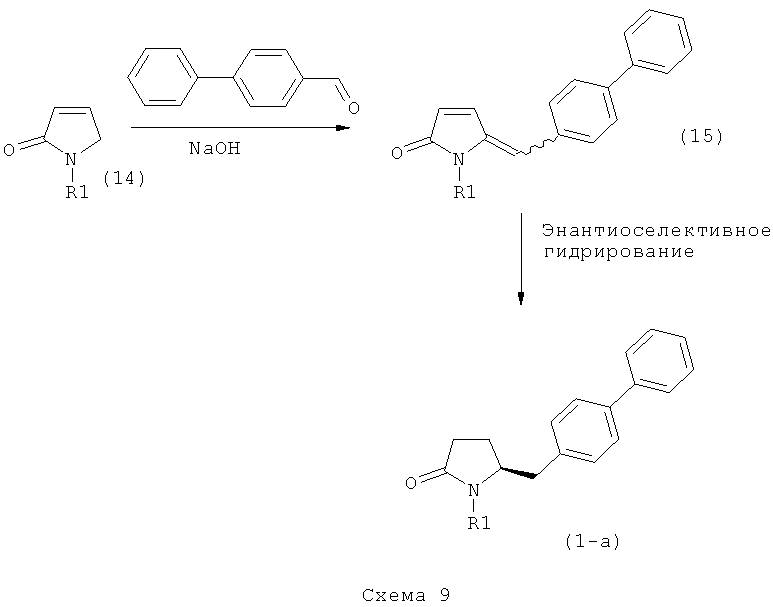
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 9, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 9. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 9, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 9.
Схема 9 охватывает частное воплощение Схемы 8, где энантиоселективное гидрирование соединения формулы (15), как детально описано выше, непосредственно приводит к соединению формулы (1-а). Т.е. данное воплощение изобретения относится к энантиоселективному гидрированию соединения формулы (15) или его соли, которое не является стереоселективным и, таким образом, обе эндо- и экзо-С=С-связи пирролидинового остатка восстанавливаются в условиях энантиоселективного гидрирования.
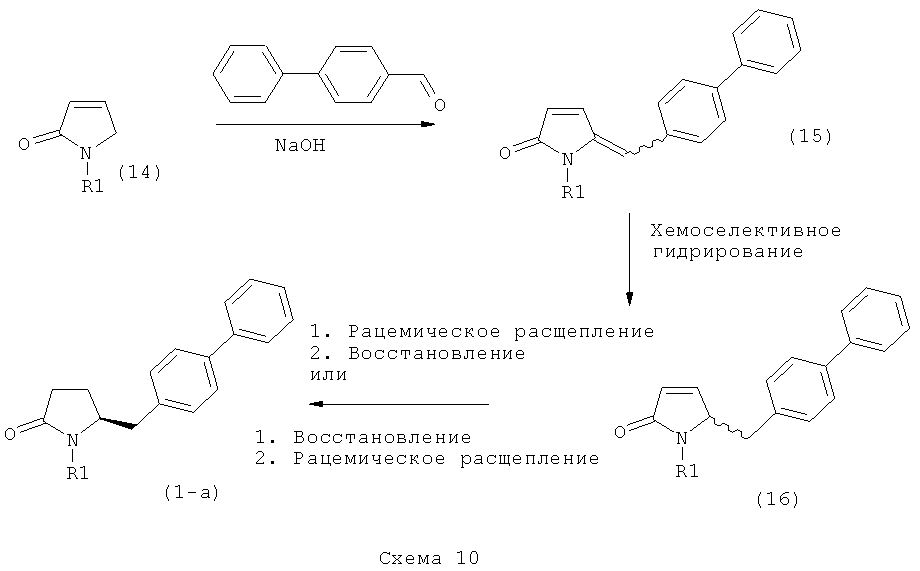
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 10, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 10. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 10, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 10.
На Схеме 10 описан альтернативный путь, в котором восстановление соединения формулы (15) или его соли, где R1 представляет собой водород или азотзащитную группу, является хемоселективным и приводит к соединению формулы (16) или его соли, где R1 представляет собой водород или азотзащитную группу. Хемоселективное гидрирование может быть осуществлено, например, в условиях, при которых используются Na2(S2O4) и NaHCO3 в ДМФА, как описано в Monatshefte fuer Chemie 1995, 126(3), 355, в Monatshefte fuer Chemie 1986, 117(2), 185 и в Liebigs Annalen der Chemie, 1986, 1241. Как вариант, гидрирование может быть осуществлено при использовании Pd/C, например, так как описано в Helv. Chim. Acta, 1987, 70(8), 2098. Рацемическое расщепление соединения формулы (16) или его соли, например, как описано для соединения формулы 5 в Helv Chim Acta 1987, 70, 2098, и последующее восстановление полученного соединения формулы (16-а), например, в тех же реакционных условиях, что описаны на Схеме 8, может привести к соединению формулы (1-а) или его соли. Две последние стадии - рацемическое расщепление и восстановление, могут быть проведены в обратном порядке, т.е. восстановление соединения формулы (16) или его соли, например, в тех же реакционных условиях, что описаны выше для соединения формулы (16-а) на Схеме 8, а затем рацемическое расщепление соединения формулы (1) или его соли, например, в тех же реакционных условиях, что описаны выше для соединения формулы (16).
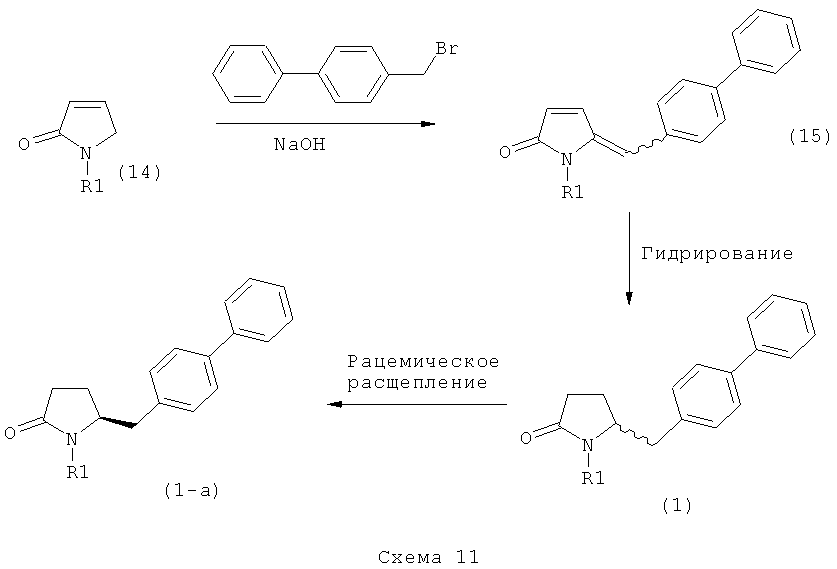
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 11, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 11. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 11, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 11.
Другой альтернативный путь превращения формулы (14) или его соли в соединение формулы (1-а) или его соль детально описан на Схеме 11. В соответствии с данным путем восстановление соединения формулы (15), описанного выше, или его соли, непосредственно приводит к соединению формулы (1), описанному в настоящей заявке, или к его соли. Данное восстановление может быть осуществлено, например, в стандартных условиях гидрирования, таких как Pd/C и водород. Последующее рацемическое расщепление, как описано выше, приводит к соединению формулы (1-а), описанному в настоящей заявке, или к его соли.
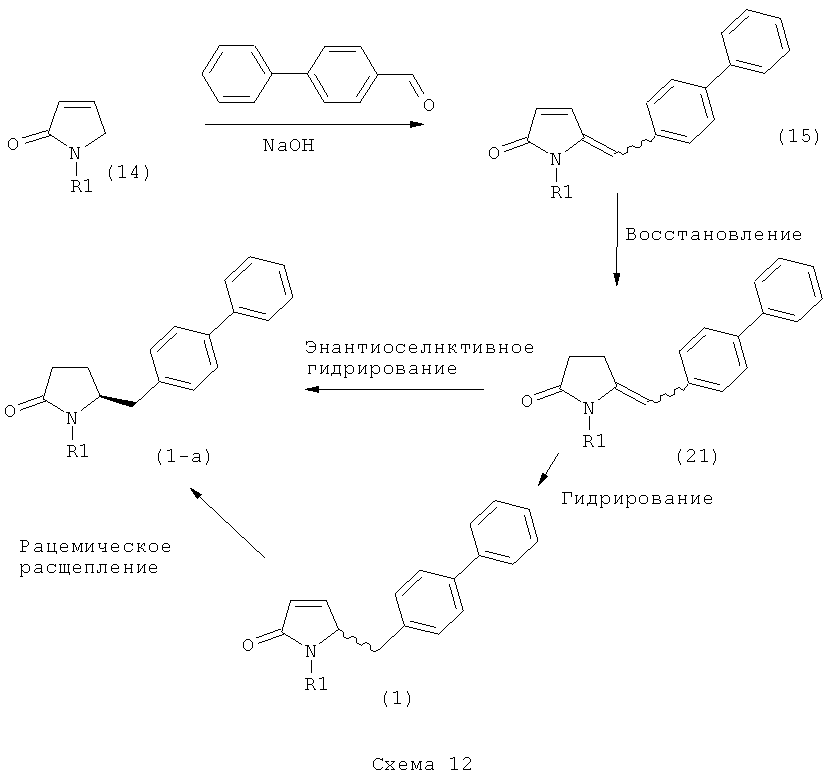
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 12, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 12. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 12, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 12.
В соответствии со Схемой 12 соединение формулы (15), как описано в настоящей заявке, или его соль, как вариант, может быть хемоселективно восстановлено в соединение формулы (21) или его соль, где R1 представляет собой водород или азотзащитную группу. Данное восстановление может быть осуществлено, например, так как описано для соединения формулы (16-а) на Схеме 8. Затем соединение формулы (21), как описано в настоящей заявке, или его соль может быть превращено в соединение формулы (1-а) или его соль, в условиях реакции энантиоселективного гидрирования, например, так как описано в J. Chem. Soc, Perkin Trans 1, 1998, 1403, в частности, так как описано для соединений 7 a-d. Как вариант, стандартное гидрирование, например, при использовании Pd/C и водорода, соединения формулы (21) или его соли, приводит к соединению формулы (1) или его соли. Рацемическое расщепление, как описано выше, приводит к соединению формулы (1-а), как описано в настоящей заявке, или его соли.
Подраздел В-6: Способ 6
Способ 6 для получения Ключевого Лактама (1) или его соли также включает реакционную стадию восстановления соединения, соответствующего формуле (16), или его соли. Однако способ получения соединения формулы (16) или его соли отличается от способа 5.
Способ 6 включает синтез, исходя из 1,5-дигидропиррол-2-она или его соли, необязательно с использованием хирального катализатора фазового переноса, с получением требуемой конфигурации хирального центра соединения формулы (16). Последующее восстановление, например, посредством гидрирования, например, так как описано выше для соединения формулы (16-а) на Схеме 8, дает Ключевой Лактам (1) или его соль.
Таким образом, еще одним объектом настоящего изобретения является способ получения соединения, соответствующего формуле (16), или его соли
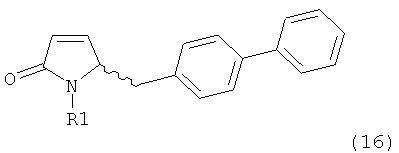 ,
,
включающему взаимодействие соединения, соответствующего формуле (14), или его соли
,
с активированным 4-метилбифенилом, с получением соединения, соответствующего формулы (16) или его соли, в присутствии основания, предпочтительно, в присутствии основания и хирального катализатора фазового переноса, где в формулах (14) и (16) R1 представляет собой водород или азотзащитную группу, как описано выше.
R1 может быть азотзащитной группой, как описано выше в разделе А: Ключевой Пактам (1) или его соль. Предпочтительно R1 представляет собой водород.
Предпочтительно для получения соединения формулы (16) или его соли, имеющего конфигурацию, как показана для формулы (16-а), используют хиральный катализатор фазового переноса. Предпочтительно получают Ключевой Пактам или его соль, имеющий конфигурацию, соответствующую формуле (1-а).
В основном подходящими являются известные катализаторы фазового переноса. Примеры хиральных катализаторов фазового переноса включают хиральные краун-эфиры или, более предпочтительно, алкалоиды цинхона. Конкретные примеры подходящих хиральных катализаторов фазового переноса даны в номере 17, на стр.1-15 "Industrial Phase-Transfer Catalysis ", опубликованном РТС Communications, Inc., 900 Briggs Road, Suite 145, Mt. Laurel, NJ 08054 USA.
Активированный 4-метилбифенил представляет собой соединение формулы Ph-Ph-СН2-Х, в которой X представляет собой уходящую группу. В основном используются примеры уходящих групп, приведенные выше в подразделе В-3. Предпочтительно X является галогеном, в частности, бромом.
В предпочтительном варианте осуществления изобретения основание, используемое на данной стадии, представляет собой алкиллитиевое основание, такое как BuLi, или гидроксид щелочного металла, такой как КОН, или представляет собой ПДА; например, как описано в Synlett, 2003, (2), 271, в Synlett, 2004, (2), 247 и в Perkin Transactions 1, 1990, (8), 2350.
В частном воплощении данного пути соединения формулы (16) или (1), или их соли, могут быть расщеплены, как детально описано в подразделе В-5, с получением соединений формулы (16-а) или (1-а), или их солей, соответственно.
Предпочтительное воплощение способа 6 представлено ниже на Схеме 13.
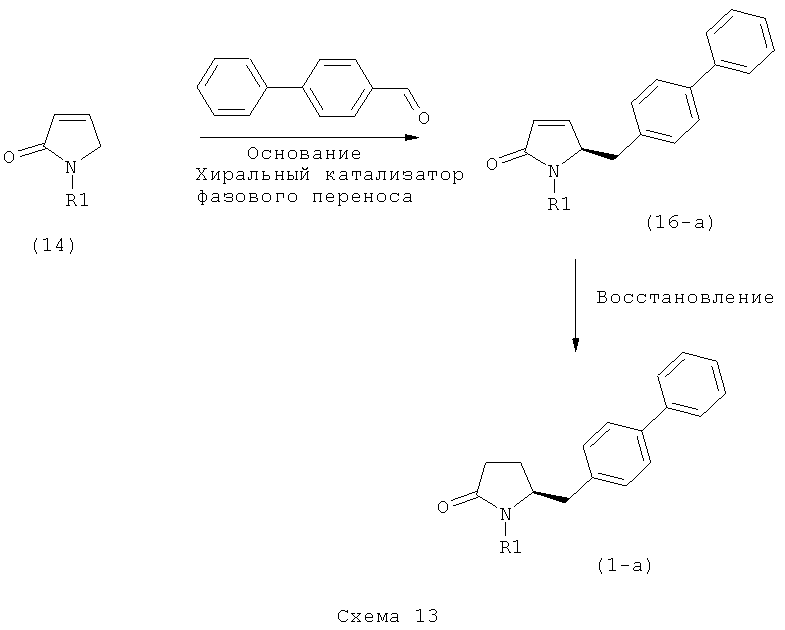
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 13, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 13. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 13, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 13.
Предпочтительное воплощение способа, представленное на Схеме 13, относится к соединениям, в которых R1 представляет собой водород. Другое предпочтительное воплощение способа, представленное на Схеме 13, относится к соединениям, в которых R1 представляет собой водород, и где восстановление соединения формулы (16-а) в (1-а) протекает в присутствии Pd/C и водорода.
В частности, предпочтительные основания и катализаторы фазового переноса первой стадии (превращение соединения формулы (14) или его соли с соединение формулы (16-а) или его соли), данного варианта осуществления изобретения выбирают, например, из группы оснований и хиральных катализаторов, упомянутых выше. Последующее восстановление соединения формулы (16-а) или его соли, с получением соединения формулы (1-а) или его соли, может быть осуществлено, например, в условиях гидрирования при использовании Pd/C или так, как описано в предыдущем подразделе В-5.
Подраздел В-7: Способ 7
Способ 7 для получения Ключевого Лактама (1) или его соли также включает реакционные стадии гидрирования соединения, соответствующего формуле (15) или его соли, с получением соединения (16) или его соли, и восстановления соединения, соответствующего формуле (16) или его соли, с получением Ключевого Лактама (1) или его соли. Однако способ получения соединения (15) или его соли отличается от способа 5.
Способ 7 начинается с N-защищенного сукцинимида, с последующим хемо- и энантиоселективным гидрированием, в качестве ключевой стадии.
Таким образом, еще одним объектом настоящего изобретения является способ получения соединения, соответствующего формуле (15) или его соли
,
где R1 представляет собой водород или азотзащитную группу, как описано выше,
включающий взаимодействие соединения, соответствующего формулы (17) или его соли
 ,
,
с металлорганическим реагентом, полученным из 4-метилбифенила, с получением соединения, соответствующего формуле (15) или его соли. В предпочтительном воплощении, соответствующем данному способу, соединение формулы (15) обладает следующей стереохимией
 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше.
Предпочтительно R1 представляет собой водород или азотзащитную группу, как описано выше в разделе А: Ключевой Пактам (1) или его соль. В частности, R1 представляет собой пивалоил или БОК.
Металлорганический реагент, полученный из 4-метилбифенила, предпочтительно представляет собой соединение формулы Ph-Ph-CH2-Y, в которой Y обозначает нуклеофильную группу. Предпочтительными соединениями формулы Ph-Ph-CH2-Y, предпочтительно - 4-бифенил-СНг-У, являются, например, 4-бифенилметилмагний галогениды (реактивы Гриньяра). Нуклеофильная группа Y, предпочтительно, представляет собой группу MX, в которой X представляет собой галоген и М представляет собой Mg или Zn. В основном предпочтительными галогенидами являются хлорид, бромид и йодид, при этом бромид является особенно предпочтительным.
Другими примерами металлорганических реагентов, полученных из 4-метилбифенила, являются 4-бифенилметиллитий, 4-бифенилметилкупрат (купраты низшего или высшего порядка), 4-бифенилметилцинк. Данные соединения могут быть использованы сами по себе или в присутствии другого металла, например, меди, цинка, палладия, железа, иридия или рутения.
Предпочтительно в выше приведенной формуле Y представляет собой MgBr (4-бифенилметилмагний бромид).
Предпочтительное воплощение способа 7 представлено ниже на Схеме 14.
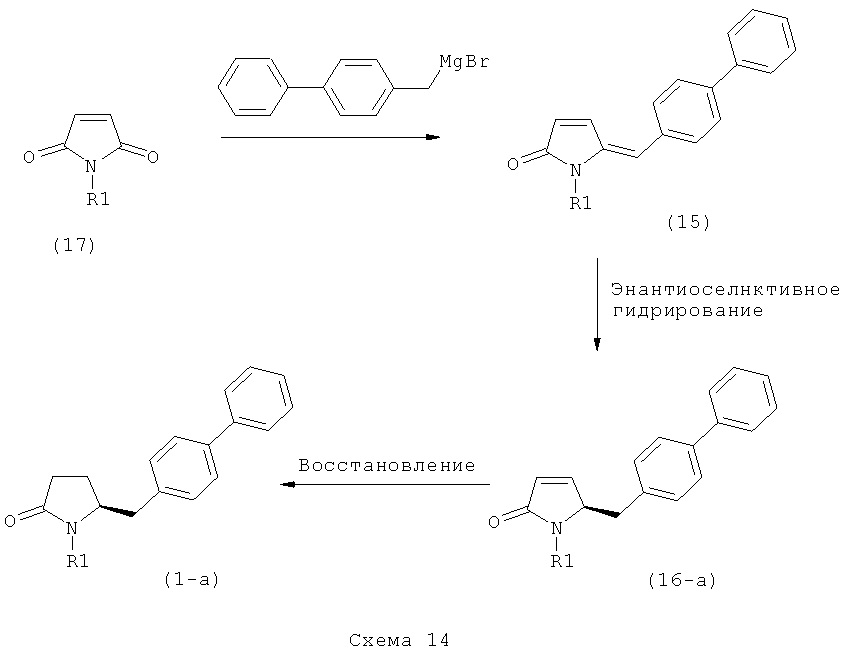
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 14, превращения соединения формулы (14), описанного в настоящей заявке, в соединение формулы (1-а), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 14. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 14, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 14.
В данном воплощении настоящего изобретения соединение формулы (15) или его соль получают из соединения формулы (17) или его соли, в условиях реакции Гриньяря, например, аналогично описанному в Perkin Transactions 1, 1993, (21), 2567. Далее соединение формулы (15) или его соль может быть превращено в соединение формулы (16-а) или его соль, через энантиоселективное гидрирования, например, так как описано выше. Последующее восстановление соединения формулы (16-а) или его соли, приводящее к образованию соединения формулы (1-а) или его соли, может быть проведено, например, в условия реакции гидрирования. В предпочтительном варианте осуществления изобретения восстановление соединения формулы (16-а) или его соли осуществляют, используя Pd/C.
Как вариант, соединение формулы (15), которое получают из соединения формулы (17), как описано выше, может быть превращено в соединение формулы (1), предпочтительно - формулы (1-а), как представлено на Схемах 9-12.
Подраздел В-8: Способ 8
В еще одном варианте осуществления настоящее изобретения относится к соединению формулы (21) или его соли, где R1 представляет собой водород или азотзащитную группу, как описано выше, которое может быть получено из соединения формулы (6) или его соли, где R1 представляет собой водород или азотзащитную группу, например, так как показано на Схеме 15.
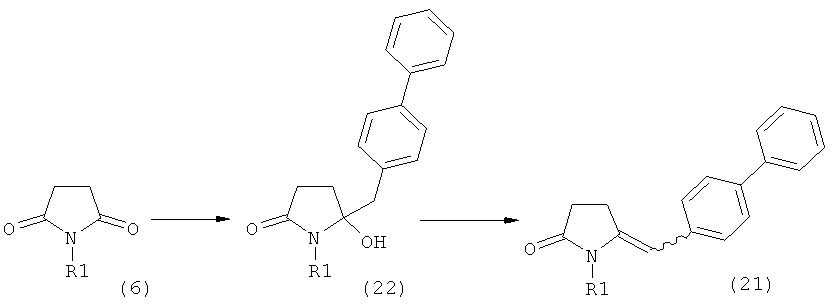
В другом воплощении настоящее изобретение относится к полной реакционной последовательности, представленной на Схеме 15, превращения соединения формулы (6), описанного в настоящей заявке, в соединение формулы (21), описанное в настоящей заявке, а также относится к каждой из реакционных стадий, показанных на Схеме 15. В еще одном варианте осуществления настоящее изобретение относится к продукту, полученному в соответствии с полной реакционной последовательностью, приведенной на Схеме 15, а также к продукту, полученному в соответствии с каждой из реакционных стадий, показанных на Схеме 15.
В данном воплощении соединение формулы (22) или его соль, где R1 представляет собой водород или азотзащитную группу, получают из соединения формулы (6) или его соли, где R1 представляет собой водород или азотзащитную группу, например, при использовании металлорганического реагента, полученного из 4-метилбифенила, как описано выше. Далее соединение формулы (22) или его соль превращают в соединение формулы (21), в которой R1 имеет значения, приведенные выше, или его соль, в условиях реакции дегидратации, например, при использовании реактива Бургесса. Как вариант, третичный спирт может быть активирован, например, посредством мезилирования, а последующая обработка основанием, например, NaOH, приводит к соединению формулы (21) или его соли.
В предпочтительном воплощении, соответствующем способу, показанному на Схеме 15, соединение формулы (21) имеет следующую стереохимию
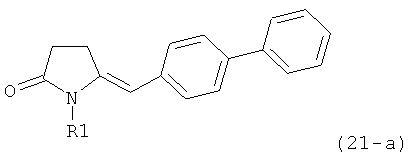 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше.
Соединение формулы (21) или его соль, предпочтительно - формулы (21-а), может быть превращено в соединение формулы (1) или его соль, предпочтительно - формулы (1-а) или его соль, например, так как описано выше.
Раздел Г: Новые и соответствующие условию изобретательского уровня соединения, встречающиеся в одном из предшествующих разделов
Способы, приведенные выше, включают несколько новых и соответствующих изобретательскому уровню соединений. Поэтому другими объектами настоящего изобретения являются соединения, показанные ниже.
Соединение, соответствующее формуле (2), или его соль
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, предпочтительно, имеющее конфигурацию, соответствующую формуле (2-а)
.
В предпочтительном воплощении формулы (2-а) R1 представляет собой водород или азотзащитную группу, выбранную из пивалоила или трет-бутоксикарбонила (БОК).
Соединение, соответствующее формуле (4), или его соль
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, и R4 представляет собой морфолин, предпочтительно имеющее конфигурацию, соответствующую формуле (4-а)
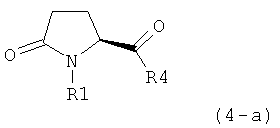 ,
,
в которой R1 и R4 имеют значения, как описано выше для формулы (4).
Соединение, соответствующее формуле (8), или его соль
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано выше, a R5 представляет собой водород или алкил, предпочтительно, имеющее конфигурацию, соответствующую формуле (8-а)
.
Соединение, соответствующее формуле (9), или его соль
,
где R1 и R2, независимо друг от друга, представляют собой водород или азотзащитную группу, как описано выше, a R5 представляет собой водород или алкил, предпочтительно, имеющее конфигурацию, соответствующую формуле (9-а)
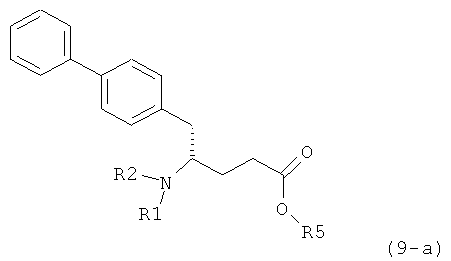 .
.
Соединение, соответствующее формуле (10), или его соль
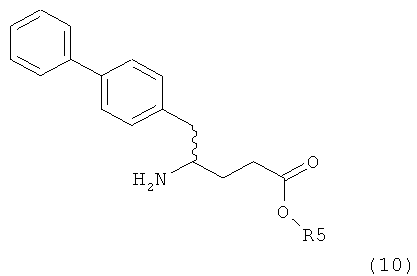 ,
,
где R5 представляет собой водород или алкил, предпочтительно, имеющее конфигурацию, соответствующую формуле (10-а)
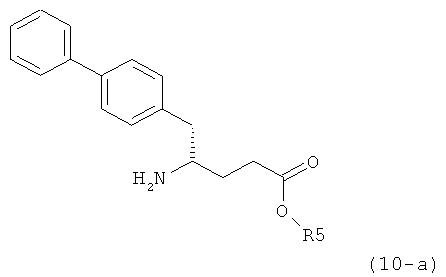 .
.
Соединение, соответствующее формуле (13), или его соль
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, предпочтительно, имеющее конфигурацию, соответствующую формуле (13-а)
.
Соединение, соответствующее формуле (15), или его соль
 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше.
Соединение, соответствующее формуле (16), или его соль
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, предпочтительно, имеющее конфигурацию, соответствующую формуле (16-а)
.
Соединение, соответствующее формуле (20), или его соль
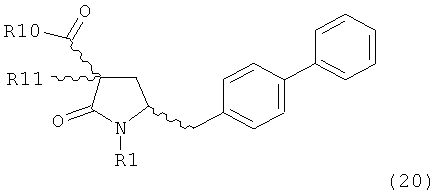 ,
,
предпочтительно имеющее конфигурацию, соответствующую формуле (20-а)
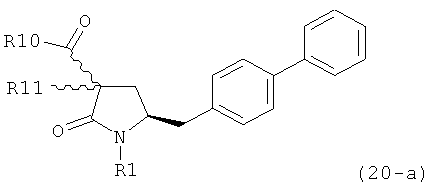 ,
,
где в приведенных выше формулах R1 представляет собой водород или азотзащитную группу, как описано выше, R10 представляет собой группу, которая может быть подвергнута омылению и/или декарбоксилированию, как определено в настоящей заявке, a R11 представляет собой водород или метил. Предпочтительно R10 представляет собой -О-алкил, в частности, -O-Et или -О-Ме, предпочтительно - -O-Et, или -О-арил, в частности, арил представляет собой фенил. В другом предпочтительном варианте осуществления изобретения -О-алкиларил, например, -О-бензил.
Соединение, соответствующее формуле (21), или его соль
 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше.
Соединение, соответствующее формуле (22), или его соль
 ,
,
где R1 представляет собой водород или азотзащитную группу, как описано выше.
Соединение, соответствующее формуле (5), или его соль, в кристаллической форме
,
где R1 представляет собой водород или азотзащитную группу, как описано выше, предпочтительно, имеющее конфигурацию, соответствующую формуле (5-а)
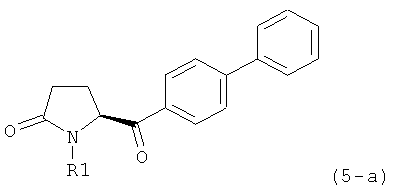 .
.
В предпочтительном воплощении изобретения кристаллические соединения, соответствующие формуле (5-а), или их соли, включают моноклинную кристаллическую систему. Другое предпочтение кристаллических продуктов изобретения заключается в том, что они включают пространственную группу Р21.
В другом воплощении изобретение относится к соединению формулы (1), (1-а), (1-b), (1'), (1''), (1'-а), (1''-а), (2), (2-а), (2-b), (2'), (2'-а), (2''), (2''-а), (3), (3-а), (3-b), (4), (4-а), (5), (5-а), (7), (7-а), (8), (8-а), (9), (9-а), (10), (10-а), (11), (11-а), (12), (12-а), (13), (13-а), (14), (15), (15-а), (16), (16-а), (17), (18), (19), (20), (20-а), (21), (21-а) или (22), или к их солям, как определено в настоящем описании выше или ниже, более предпочтительно - к соединения формулы (1), (1-а), (1-b), (1'), (1''), (1'-а), (2), (2-а), (2-b), (2'), (2'а), (3), (3-а), (4), (4-а), (5), (5-а), (7), (7-а), (8), (8-а), (9), (9-а), (10), (10-а), (13), (13-а), (15), (16), (16-а), (20), (20-а), (21) или (22), или к их солям, как определено в настоящем описании выше или ниже; в частности, в которых R1 выбран из предпочтительных определений, определенных выше.
В еще одном воплощении изобретение относится к соединению формулы (1), (2), (3) или (10), предпочтительно к соединениям (1-а), (2-а) или (10-а), или к их солям, в кристаллической форме, как определено выше или ниже.
Частные воплощения изобретения представлены в примерах - таким образом, изобретение, в очень предпочтительных вариантах осуществления изобретения, относится к соединениям формул, упомянутых выше, или к их солям, выбранным из соединений, приведенных в примерах, также как и к их применению в соответствии с изобретением.
Раздел Д: Примеры
Следующие примеры служат иллюстрацией изобретения, без ограничения его объема, а с другой стороны представляют предпочтительные варианты осуществления реакционных стадий, промежуточных соединений и/или способов настоящего изобретения.
Сокращения:
|
|
При цитировании данных ЯМР используются следующие сокращения: с - синглет; д - дублет; т - триплет; кв. - квартет; квинт. - квинтет; м - мультиплет.
Пример 1-1: (S)-5-(Морфолин-4-карбонил)пирролидин-2-он
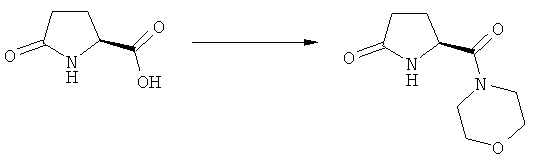
Способ 1
К смеси 48 г дициклогексилкарбодиимида, 30 г коммерчески доступной L-пироглютаминовой кислоты и 3,1 г гидроксибензотриазола добавляют 20,2 г морфолина в 170 мл дихлорметана при температуре около -15°С. Затем смеси позволяют нагреться до комнатной температуры. Суспензию фильтруют и разбавляют 290 мл ТГФ. После этого смесь концентрируют с получением суспензий, фильтрование которой приводит к 41 г (E)-5-(морфолин-4-карбонил)пирролидин-2-она.
Температура плавления (в настоящем описании рассматривается как "т.пл"): 130-132°С. 1Н-ЯМР (ДМСО): 1,87 (1Н); 2,19 (1Н); 2,28 (1Н); 3,35-3,65 (8Н); 4,51 (1Н); 7,70 (1Н). МС (ESI, m/е) 199 [М+Н]+; 397 [2М+Н]+.
Энантиомерный избыток (ее): 98,0% (4-а, R1=Н; R4 = морфолин), как определено ГХ.
Способ 2
Суспензию 5 г.S-глютаминовой кислоты (38,737 ммоля) в 50 мл ТГФ и 250 мкл ДМФА охлаждают до 0°С, а затем добавляют 3,09 мл (5,07 г, 42,611 ммоля) тионилхлорида в течение 5 мин. После этого реакционной смеси в течение 2 ч позволяют нагреться до комнатной температуры. Полученный прозрачный раствор снова охлаждают до 0°С, а затем обрабатывают 7,25 мл (7,25 г, 170,1 ммоля) морфолина. После нагревания реакционной смеси до температуры окружающей среды в течение 2 ч суспензию отфильтровывают. Фильтрат концентрируют при пониженном давлении с получением сырого остатка. Энантиомерный избыток (ее): 98,0% (4-а, R1=Н; R4 = морфолин), как определено ГХ.
Способ 3
Реакционный сосуд заполняют 30 г L-пироглютаминовой кислоты и 4,71 г гидрата 1-гидроксибензотриазола. После этого твердое вещество суспендируют в 190 мл безводного ацетонитрила, а затем смесь нагревают до 45°С. После этого, через капельную воронку, в течение 20 мин добавляют 20,24 мл морфолина, и капельную воронку, затем, промывают 5 мл безводного ацетонитрила. Полученную реакционную смесь нагревают до 65°С, а затем в течение 1 ч через капельную воронку добавляют 29,32 г N,N'-диизопропилкарбодиимида. После завершения добавления капельную воронку промывают 5 мл безводного ацетонитрила, а реакционную смесь перемешивают при 65°С еще 30 мин. Затем полученную суспензию охлаждают до температуры окружающей среды, фильтруют, и остаток на фильтре промывают 3×30 мл ацетонитрила. Фильтрат концентрируют при нагревании и пониженном давлении до приблизительно ½ от первоначального объема и в остаток, затем, добавляют 16 мг затравки (S)-5-(морфолин-4-карбонил)пирролидин-2-она. Концентрирование при пониженном давлении продолжают до тех пор, пока будет достигнут приблизительно 1/3 от первоначального объема. В течение дальнейшего концентрирования при пониженном давлении и удаления приблизительно 95 мл дистиллята одновременно добавляют 210 мл 2-метилтетрагидрофурана. После этого суспензию охлаждают до 0°С и выделяют фильтрованием твердое вещество. Остаток на фильтре промывают 3×20 мл охлажденного 2-метилтетрагидрофурана и сушат при 55°С под вакуумом, получая 42,54 г (S)-5-(морфолин-4-карбонил)пирролидин-2-она, в виде чистых, белых кристаллов (чистота 98,0% по данным ВЭЖХ, ее (ВЭЖХ)>99,6% [4-а, R1=Н; R4 = морфолин]).
Пример 1-2: (R)-5-(Морфолин-4-карбонил)пирролидин-2-он
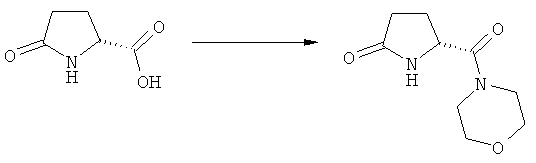
К смеси 30 г R-пироглютаминовой кислоты и 4,71 г гидроксибензотриазола в 200 мл ацетонитрила добавляют 20,24 г морфолина 30 при 50°С. Полученную смесь нагревают до 60°С и добавляют 29,32 г N,N'-диизопропилкарбодиимида. По истечении 1 ч суспензию охлаждают до комнатной температуры и фильтрованием удаляют осадок. Маточный раствор подвергают дистилляции при пониженном давлении. В течение дистилляции добавляют метилтетрагидрофуран. Полученную суспензию охлаждают до 0°С, а затем фильтруют. Остаток на фильтре промывают метилтетрагидрофураном и сушат при пониженном давлении, с получением (7?)-5-(морфолин-4-карбонил)пирролидин-2-он. 1Н-ЯМР (ДМСО): 1,88 (1Н); 2,11 (1Н); 2,32 (1Н); 3,46 (4Н); 3,59 (4Н); 4,54 (1Н, дд, J=3,3, 8,8); 7,72 (1Н). МС (ESI, т/е) 199 [М+Н]+; 397 [2М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3341; 2860; 1709; 1694; 1639; 1462; 1270; 1255; 1116; 655. Энантиомерный избыток (ее): 99,6% (4-b, R1=Н; R4 = морфолин), как определено ГХ.
Пример 1-3: (R/S)-5-(Морфолин-4-карбонил)пирролидин-2-он
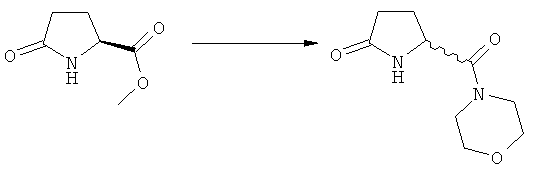
Смесь 143 г метилового эфира пироглютаминовой кислоты (1 моль) и 139 г морфолина (1,60 моля) в 900 мл ксилола нагревают до 140°С в течение 20 ч. Затем ксилол выпаривают до объема 500 мл. Из полученной суспензии отфильтровывают осадок, промывают его ТГФ и после сушки под вакуумом получают требуемое соединение. Из фильтрата полностью выпаривают растворитель. Полученный сырой продукт [(R/S)-5-(морфолин-4-карбонил)пирролидин-2-он] фильтруют через 800 г силикагеля, с использованием ТГФ в качестве растворителя, и выпаривают до суха, получая вторую порцию требуемого соединения. Данное соединение непосредственно используют на следующей стадии. По данным ВЭЖХ-анализа полученный продукт имеет энантиомерное соотношение: 55,8: 44,2 [(4-а, R1=Н; R4 = морфолин): (4-b, R1=Н; R4 = морфолин), соответственно].
Пример 1-4: (S)-5-(Морфолнн-4-карбонил)пирролидин-2-он
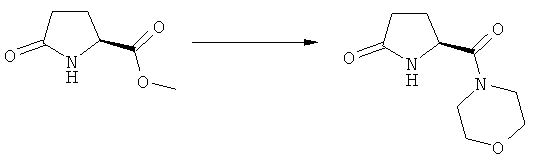
Метиловый эфир L-пироглютаминовой кислоты (23,91 г, 177 ммоля), морфолин (21,8 г, 250 ммоля) и 4-диметиламинопиридин (0,2 г, 1,64 ммоля) кипятят с обратным холодильником в толуоле (130 мл) в течение 12 ч. Полученный продукт осаждается в виде масла. Масло отделяют от толуола, 5 разбавляют дихлорметаном (100 мл), а затем последовательно промывают 0,1 М HCl, 0,1 М NaOH и водой (по 50 мл каждого). Дихлорметановую фазу концентрируют с получением суспензии, которую отфильтровывают, получая сырой продукт. Сырой продукт растворяют в ТГФ (200 мл) при 45°С, после охлаждения на ледяной бане, (S)-5-(морфолин-4-карбонил)пирролидин-2-он 10 выпадает в осадок в виде белых кристаллов. Энантиомерная чистота (ЭЧ): 88,7% (S)-энантиомера.
Пример 1-5: (R/S)-5-(Морфолин-4-карбонил)пирролидин-2-он
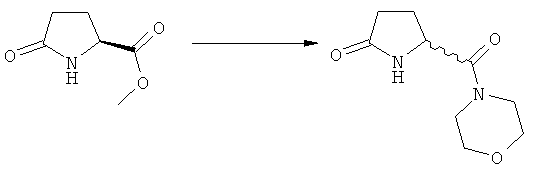
Смесь 14,5 г метилового эфира пироглютаминовой кислоты (100 мл) и 15 13,1 г морфолина (150 мл) в 80 мл толуола кипятят с обратным холодильником (110°С) в течение 48°С. В течение данного времени реакции образующийся метанол отгоняют из реакционной смеси. В итоге образуется две фазы. Верхнюю, толуоловую фазу, отделяют, а нижнюю фазу продукта фильтруют через силикагель (180 г), с использованием смеси этилацетата и метанола (70:20 30). Затем растворитель выпаривают, получая продукт в виде масла. Энантиомерное соотношение - 93:7 [(4-а, R1=Н; R4=морфолин): (4-b, R1=Н; R4 = морфолин) и чистота, по данным ГХ, составляет 96,4%.
Метод ВЭЖХ (пример 1):
Колонка: Chirobiotic-T; 100×4,6 мм; 5 мкм. Подвижная фаза А (0,01 М 25 NH4OAc в МеОН + 0,1% ТФУК). Изократический режим: 0 мин (100%); 10 мин (100% А). Скорость потока: 1,0 мл мл-1. Длина волны: 210 нм. Температура в колонке: 10°С.
Время удерживания:
4-а (R1=Н; R4 = морфолин): 2,3 мин
4-b (R1=Н; R4 = морфолин): 2,8 мин
Метод ГХ (пример 1):
Колонка: Fused-Silica-Capillary, CHIRALDEX G-BP; 20 м × 0,25 мм. Предколонка: Деактивированный плавленый кварц, 1 м × 0,53 мм. Температура блока ввода пробы: 250°С. Температура детектора: 30°С. Газ-носитель: гелий, 3,0 мл мин-1, непрерывный поток. Объем вводимой пробы: 2,0 мкл. Коэффициент распределения: 20:1. Температура термостата: 200°С (начальная), изократический режим в течение 50 мин.
Время удерживания:
4-а (R1=Н; R4 = морфолин): 27,5 мин
4-b (R1=Н; R4 = морфолин): 30,1 мин
Пример 2-1: (S)-5-(Бифенил-4-карбонил)пирролидин-2-он
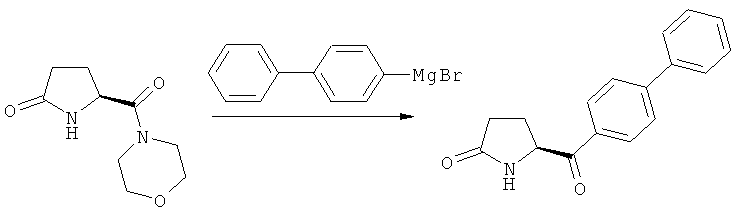
Способ 1
39 г (S)-5-(морфолин-4-карбонил)пирролидин-2-она, полученного в примере 1, суспендируют в 300 мл сухого, дегазированного ТГФ. Полученный раствор охлаждают до температуры около -15°С. После этого в течение приблизительно 20 мин добавляют 2,2 М раствор 4-бифенилмагний бромида в ТГФ. Полученной смеси позволяют нагреться до комнатной температуры около 19 ч. Затем смесь резко охлаждают, добавляя ледяную 2 М соляную кислоту, и отделяют органический слой. Частичное концентрирование приводит к образованию осадка, который собирают при помощи фильтрации, промывают водой и толуолом, получая 44 г (S)-5-(бифенил-4-карбонил)пирролидин-2-она, Тпл 163-203°С (разл.). 1Н ЯМР (ДМСО): 1,88 (1H); 2,15 (2Н); 2,53 (1H); 5,30 (1Н); 7,34-7,60 (3Н); 7,77 (2Н); 7,90 (3Н); 8,11 (2Н). Энантиомерный избыток (ее): 98,08% (5-а, R1=Н), как определено ВЭЖХ.
Рентгеновская структура полученных кристаллов показана на фиг.4. Соответствующие кристаллографические данные:
Эмпирическая формула C17H15NO2, формульная масса 265,30, температура 100(2) К, длина волны 1,54178 Å; моноклинная кристаллическая система, пространственная группа Р21,
параметры ячейки: а=10,362(3) Å, а=90°, b=8,236(2) Å, b=90,872(15)°, с=15,583(4) Å, g=90°; объем 1329,7(6) Å3; Z 4; плотность (расчетная) 1,325 мг/м; коэффициент поглощения 0,698 мм-1; F(000) 560.
Рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 26 в градусах дано с погрешностью ±0,2): градусы 2θ: 3,8, 5,6, 8,3, 10,1, 13,4, 14,2, 14,6, 17,6, 18,9, 19,8, 20,4, 20,7, 21,1, 22,7. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 2
50 г (5)-5-(морфолин-4-карбонил)пирролидин-2-она суспендируют в 450 мл ТГФ. Затем раствор охлаждают до приблизительно -5°С. Далее, в течение 30 мин добавляют 127 мл раствора хлорида изопропилмагния в ТГФ (1,90 М). Смесь перемешивают еще 30 минут, а затем добавляют 300 мл раствора бромида 4-бифенилмагния в ТГФ (1,01 М). Смесь нагревают до комнатной температуры. Перемешивание продолжают в течение 10 ч. Затем смесь добавляют к 585 мл соляной кислоты (2М) при 0°С. Затем смесь нагревают до комнатной температуры. Затем добавляют 162 мл гидроксида натрия (2М). Смесь нагревают до 60°С и удаляют летучие растворители под вакуумом. К остатку добавляют 250 мл толуола и смесь перемешивают в течение 30 минут. Смесь охлаждают до комнатной температуры. Твердое вещество отделяют фильтрованием и промывают 150 мл толуола и 150 мл воды с получением, после высушивания, (S)-5-(бифенил-4-карбонил)пирролидин-2-она. Энантиомерный избыток (ее): 99,8% (5-а, R1=Н), как определено ВЭЖХ.
Пример 2-2: (R)-5-(Бнфенил-4-карбонил)пирролидин-2-он
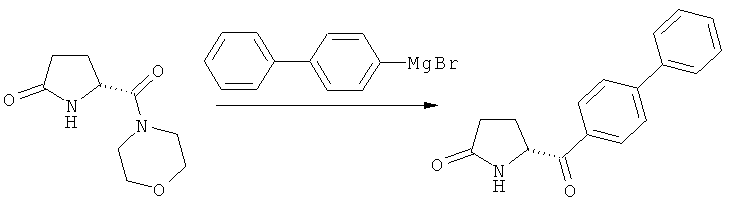
35,48 г (R)-5-(морфолин-4-карбонил)пирролидин-2-она суспендируют в 302 мл сухого, обезгаженого ТГФ. Раствор охлаждают до -5°С. В течение 30 мин добавляют 92,3 мл раствора хлорида изопропилмагния в тетрагидрофуране (1,9 М). Затем в течение 20 мин добавляют 210 мл раствора бромида 4-дифенилмагния в ТГФ (1,0 М). В течение 19 часов смесь нагревается до комнатной температуры. Смесь резко охлаждают путем добавления ледяной 2М соляной кислоты и удаляют органический растворитель при пониженном давлении. К образующейся суспензии добавляют 133 мл толуола, и промывают осадок, который отделяют фильтрацией, водой и толуолом. Осадок на фильтре сушат при пониженном давления, получая (R)-5-(бифенил-4-карбонил)пирролидин-2-он. 1Н ЯМР (ДМСО): 1,93 (1Н); 2,17 (2Н); 2,55 (1Н); 5,33 (1Н, дд, J=4,5, 9,6); 7,45 (1Н); 7,53 (2Н, т, J=7,6); 7,77 (2Н, д, J=7,l); 7,89 (3Н, t, J=8,5); 8,11 (2Н, д, J=8,5). МС (ESI, m/е) 266 [М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3203; 3103; 2880; 1699; 1684; 1604; 1407; 1238; 980; 769; 733; 698. Энантиомерный избыток (ее): 99,0% (5-b, R1=Н), по данным ВЭЖХ.
Пример 2-3: (R)-5-(Бифенил-4-карбонил)пирролидин-2-он и (S)-5-(Бифенил-4-карбонил)пирролидин-2-он
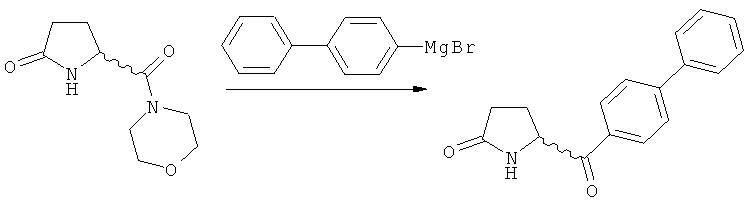
К 32 г магниевой стружки добавляют раствор 289 г 4-бромбифенила в 1100 мл ТГФ. Добавление исходного 4-бромбифенила осуществляют со скоростью, при которой реакционная смесь находится в состоянии кипения. После завершения добавления смесь кипятят еще в течение 1,5 ч. Затем к раствору 100 г (R/S)-5-(морфолин-4-карбонил)пирролидин-2-она в 1000 мл ТГФ при температуре <- 50°С добавляют раствор Гриньяра. Охлаждающую баню удаляют и реакционную смесь перемешивают в течение ночи. После этого реакционную смесь быстро охлаждают, добавляя смесь H2O/NH4Cl и 2М HCl. pH реакционной смеси поддерживают на уровне pH=8. Затем реакционную смесь концентрируют до объема 360 мл, а образующийся осадок удаляют фильтрованием. После этого осадок перемешивают при 70°С в 400 мл толуола, а затем фильтруют, получая, после высушивания, (S)-5-(бифенил-4-карбонил)пирролидин-2-он и (R)-5-(бифенил-4-карбонил)пирролидин-2-он, в соотношении 64: 38, соответственно, как определено ВЭЖХ. 1Н-ЯМР: (400 МГц; CDCl3): 2,04-2,16 (м, 1H), 2,26-2,45 (мм, 2H), 2,54-2,67 (м, 1Н), 5,08-5,13 (м, 1Н), 7,32-7,94 (м, 9Н).
Метод ВЭЖХ (пример 2):
Колонка: Chiralpak AS-RH (DAICEL); 150×4,6 мм; 5 мкм. Подвижная фаза А (вода); Подвижная фаза Б (ацетонитрил). Изократический режим: 0 мин (50% Б); 15 мин (50% Б). Скорость потока: 0,8 мл мин-1. Длина волны: 285 нм. Температура: 25°С.
Время удерживания:
|
Пример 3-1: (S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]
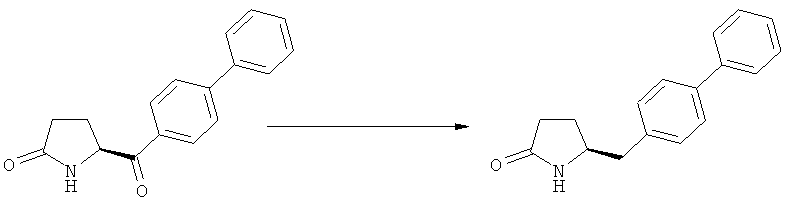
Способ 1
200 мг (S)-5-(бифенил-4-карбонил)пирролидин-2-она, полученного в примере 2, суспендируют в 3 мл ТГФ. Затем добавляют 20 мг серной кислоты и 5 мг палладия на угле (10 м/м) (W.С.Heraeus GmbH, тип К-0218), и смесь перемешивают под давлением водорода 3 бар в течение 20 ч. После этого смесь нейтрализуют 50 мг карбоната натрия, а полученный осадок удаляют фильтрованием. Фильтрат концентрируют до суха, получая (S)-5-бифенил-4-илметилпирролидин-2-он. 1Н-ЯМР (ДМСО): 1,66 (1Н, м, 4-CHH); 1,94 (1Н, м, 4-CHH); 2,01 (2Н, м, 3-СН2); 2,65 (1Н, дд, 1-CHH); 2,84 (1Н, дд, 1-CHH); 3,78 (1Н, м, 5-СН); 7,30 (2Н, д, ароматический); 7,33 (1Н, м, ароматический); 7,43 (2Н, т, ароматический); 7,57 (2Н, д, ароматический); 7,63 (2Н, м, ароматический); 7,81 (1Н, с, NH). m/z: 252 (МН+, 100%).
Рентгеновская структура полученных кристаллов показана на фиг.1.
Кристаллографические данные [полученные при 100(2) К]
|
|
Способ 2
100 г (S)-5-(бифенил-4-карбонил)пирролидин-2-она суспендируют в 860 мл ТГФ. Затем добавляют 2,4 г серной кислоты и 10 г палладия на угле (10 м/м) (W.С.Heraeus GmbH, тип К-0218). Полученную смесь нагревают до 40°С и подают водородный газ при давлении 3 бар. По истечении 10 ч смесь охлаждают до комнатной температуры, фильтруют, и остаток на фильтре промывают 300 мл ТГФ. После этого к объединенным фильтратам добавляют 100 мл воды, а затем -21,9 г раствора гидроксида натрия (2 М). Полученную смесь нагревают до 60°С и летучие растворители удаляют под вакуумом. Полученную суспензию фильтруют, а остаток на фильтре промывают 200 мл воды. Затем твердое вещество помещают в 820 мл изопропилацетата при 65°С и частично концентрируют. После этого смесь охлаждают до 3°С, и фильтрованием собирают твердое вещество, получая, после высушивания, (S)-5-бифенил-4-илметилпирролидин-2-он.
Способ 3
(S)-5-Бифенил-4-илметилпирролидин-2-он (106 мг) помещают в толуол (2 мл) и добавляют 10% Pd/C (10,6 мг, загрузка 10 влажн. мас.%, тип катализатора 394 (6249), Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 94%.
Способ 4
(S)-5-Бифенил-4-илметилпирролидин-2-он (106 мг) помещают в толуол (2 мл) и добавляют 10% Pd/C (10,6 мг, загрузка 10 влажн. мас.%, тип катализатора 388, Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 98%.
Способ 5
(S)-5-Бифенил-4-илметилпирролидин-2-он (106 мг) помещают в толуол (2 мл) и добавляют 10% Pd/C (10,6 мг, загрузка 10 влажн. мас.%, тип катализатора Mod (72595), Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 96%.
Способ 6
(S)-5-Бифенил-4-илметилпирролидин-2-он (848 мг) помещают в толуол (4 мл) и добавляют 10% Pd/C (85 мг, загрузка 10 влажн. мас.%, тип катализатора 39, Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 97%.
Способ 7
(S)-5-Бифенил-4-илметилпирролидин-2-он (5,07 мг) помещают в толуол (24 мл) и добавляют 10% Pd/C (0,25 г, загрузка 5 влажн. мас.%, тип катализатора 39, Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. Катализатор отфильтровывают. Продукт получают посредством осаждения из гептана. По данным ВЭЖХ-анализа чистота составляет 97%.
Способ 8
(S)-5-Бифенил-4-илметилпирролидин-2-он (848 мг) помещают в толуол (4 мл) и добавляют 10% Pd/C (85 мг, загрузка 5 влажн. мас.%, тип катализатора 394 (6065), Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 97%.
Способ 9
(S)-5-Бифенил-4-илметилпирролидин-2-он (848 мг) помещают в толуол (4 мл) и добавляют 10% Pd/C (42 мг, загрузка 10 влажн. мас.%, тип катализатора 394 (6065), Johnson Matthey). Подают водород под давлением 30 psi, и смесь нагревают до 70°С. По истечении 16 ч смесь охлаждают до комнатной температуры. По данным ВЭЖХ-анализа чистота составляет 97%.
Пример 3-2: (R)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-b, R1=Н)]
14 г (R)-5-(бифенил-4-карбонил)пирролидин-2-она суспендируют в 80 мл тетрагидрофурана. Затем добавляют 331 мг серной кислоты, и реакционный сосуд продувают аргоном. После этого добавляют 1,4 г палладия на угле (W.С.Heraeus GmbH, тип К-0218), и смесь перемешивают в атмосфере водорода (32 бар) при 30°С в течение 20 ч. Черную суспензию отфильтровывают, а остаток на фильтре промывают тетрагидрофураном. После добавления 50 мл воды pH полученной эмульсии поддерживают на уровне pH=5,5, используя раствор гидроксида натрия (2М). Затем при пониженном давлении удаляют органический растворитель и добавляют 100 мл изопропилацетата. Водный слой отделяют, а оставшийся органический слой концентрируют до приблизительно половины от первоначального объема. Затем суспензию охлаждают до -15°С и отфильтровывают. Остаток на фильтре промывают холодным изопропилацетатом, а затем сушат при пониженном давлении, получая (R)-5-бифенил-4-илметилпирролидин-2-он в виде бесцветных кристаллов. 1Н-ЯМР (CDCl3): 1,93 (1Н); 2,37 (3Н); 2,80 (1Н, дд, J=8,5, 13,5); 2,92 (1H, дд, J=5,4, 13,5); 3,95 (1Н); 5,70 (1H); 7,29 (1Н, д, J=7,5); 7,38 (1Н); 7,47 (2Н); 7,59 (4Н). МС (ESI, m/е) 252 [М+Н]+; 503 [2М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3191; 3055; 2930; 1692; 1489; 1272; 762; 692.
Пример 3-3: (R/S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-b, R1=Н)]
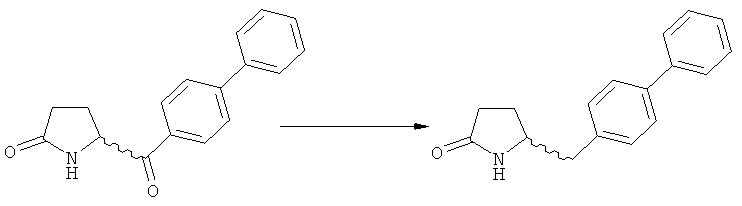
Смесь 26,5 г (100 мл) (R/S)-5-(бифенил-4-карбонил)пирролидин-2-она, 3,0 г Pd/C (10% катализатор PD CP 2505 D/R, BASF), 5 г конц. H2SO4 в 300 мл ТГФ и 300 мл МеОН гидрируют при давлении 1 атм и нагревании при 50°С. По истечении 10 ч подачу водорода прекращают. Катализатор отфильтровывают и промывают метанолом, а полученный фильтрат концентрируют под вакуумом. Остаток выпаривания растворяют в толуоле и промывают водным раствором Na2CO3. Часть продукта, выпавшего в осадок, отфильтровывают и сушат под вакуумом (96,5% ВЭЖХ). Толуоловую фазу дважды промывают H2O и полностью выпаривают, получая дополнительное количество сырого продукта. Сырой продукт очищают хроматографией на силикагеле, с использованием смеси толуол/метанол = 4:1, получая требуемый продукт. Данный продукт перемешивают в 70 мл толуола при 60°С в течение 2 ч. Затем добавляют 70 мл гептановой фракции и полученный осадок отфильтровывают. Осадок сушат под вакуумом, получая (R/S)-5-бифенил-4-илметилпирролидин-2-он (96,5% ВЭЖХ). 1Н ЯМР (ДМСО): 1,69 (1Н), 1,96-2,05 (2Н), 2,67 (1Н), 2,84 (1Н), 3,80 (1Н), 7,31 (2Н), 7,44 (2Н), 7,57 (2Н), 7,63 (2Н), 7,74 (1Н).
Метод 1 ВЭЖХ (пример 3):
Колонка: YMC-Pack ODS-AQ HP; 150×3,0 мм; 3 мкм. Подвижная фаза А (10 мМ КН2РО4 в воде); Подвижная фаза Б (ацетонитрил). Градиент: 0 мин (25% Б); 7 мин (40% Б); 10 мин (40% Б); 12 мин (80% Б); 20 мин (80% Б); 20,1 мин (25% Б). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 45°С.
Время удерживания:
|
****
Метод 2 ВЭЖХ (пример 3):
Колонка: Chiralpak AS-RH (DAICEL); 150×4,6 мм; 5 мкм. Подвижная фаза А (вода); Подвижная фаза Б (ацетонитрил). Изократический режим: 0 мин (60% Б); 15 мин (60% Б). Скорость потока: 0,8 мл мин-1. Длина волны: 254 нм. Температура: 25°С.
|
Пример 4: (s)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил)
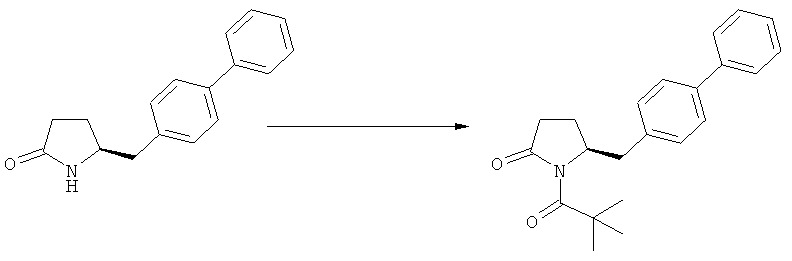
Способ 1
20 г (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н), полученного в примере 3, растворяют в 200 мл тетрагидрофурана. Полученную смесь охлаждают до приблизительно -78°С и добавляют 55 мл н-бутиллития (1,6 М). По истечении приблизительно 0,5 ч добавляют 11,8 мл пивалоилхлорида. По истечении еще 1 ч реакцию гасят добавлением 210 мл раствора хлорида аммония и реакционную смесь экстрагируют 70 мл этилацетата. Затем смесь концентрируют до суха, получая 26,7 г (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1, R1 = пивалоил). 1Н-ЯМР (CDCl3): 1,40 (9Н, с, С(СН3)3); 1,91 (1Н, м, 4-CHH); 2,03 (1Н, м, 4-CHH); 2,47 (1Н, м, 3-CHH); 2,55 (1Н, м, 3-CHH); 2,74 (1Н, дд, 1-CHH); 3,18 (1Н, дд, 1-CHH); 4,67 (1Н, м, 5-СН); 7,34 (2Н, м, ароматический); 7,36 (1Н, м, ароматический); 7,46 (2Н, м, ароматический); 7,58 (4Н, м, ароматический), m/z: 336 (МН+, 100%).
Рентгеновская структура полученных кристаллов показана на фиг.9. Монокристалл, для данного исследования, получен при использовании в качестве растворителя диизопропилового эфира.
Кристаллографические данные [полученные при 100(2) К]
|
|
Способ 2
(S)-5-Бифенил-4-илметилпирролидин-2-он (1-а, R1=Н) (100 г, 398 ммоля) и триэтиламин (166 мл, 1,2 моля) добавляют к толуолу (1 л) при комнатной температуре. Полученную смесь нагревают до 60°С. Затем в течение 2 ч добавляют пивалоилхлорид (73,5 мл, 597 ммоля). По истечении еще 1 ч добавляют лимонную кислоту (237 мг в 1 л) и фазы разделяют. Водную фазу промывают толуолом (0,5 л). Органические фазы объединяют, промывают водой (0,5 л) и сушат (MgSO4). Смесь концентрируют под вакуумом. Остаток суспендируют в гептане (550 мл) и кипятят с обратным холодильником. Затем смесь охлаждают до комнатной температуры. По истечении 1 ч смесь охлаждают до 0°С, а затем фильтруют, получая (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1, R1 = пивалоил).
Пример 5-1: (3R, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил)
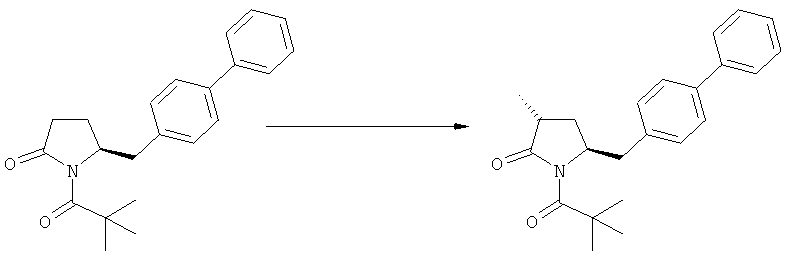
Способ 1
0,37 мл н-бутиллития (1,6 М) добавляют к раствору 88 мкл дииопропиламина в 1 мл тетрагидрофурана при температуре около 0°С. По истечении приблизительно 15 мин добавляют 200 мг (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1-а, R1 = пивалоил), полученного в примере 4, в 2 мл тетрагидрофурана. По истечении приблизительно 15 мин добавляют 59 мкл диметилсульфата. По истечении приблизительно 2 ч при температуре около 0°С смесь разбавляют раствором хлорида аммония, экстрагируют этилацетатом и концентрируют до суха (196 мг). В соответствии с НЯМР - анализом соотношение диастереоизомеров составляет 83:17 [(3R, 5S): (3S, 5S)]. Полученное вещество очищают посредством хроматографии, получая 43 мг (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она. 1Н-ЯМР (ДМСО): (2-а, R1 = пивалоил) 1,07 (3H, д, 1-СН3); 1,29 (9Н, с, С(СН3)3); 1,63 (1Н, м, 4-CHH); 2,03 (1Н, м, 4-CHH); 2,81 (2Н, м, 3-СН, 1-CHH); 2,94 (1Н, дд, 1-CHH); 4,45 (1Н, м, 5-СН); 7,34 (3H, м, ароматический); 7,44 (2Н, м, ароматический); 7,61 (4Н, м, ароматический), m/z: 350 (МН+, 100%). Данные спектрального анализа для (2-b, R1 = пивалоил) такие же, как и в примере 5-2.
Рентгеновская структура предпочтительного диастереоизомера (соединения, соответствующего формуле (2-а)) показана на фиг.2.
Кристаллографические данные [полученные при 100(2) К]
|
|
Способ 2
10 г (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1-а, R1 = пивалоил) растворяют в 150 мл толуола. Полученную смесь охлаждают до приблизительно 0°С и добавляют 71,5 мл раствора бис(триметилсилил)амида калия (0,5 М в толуоле). По истечении 15 мин добавляют 11 мл диметилсульфата, и смесь перемешивают еще в течение 1 ч. Затем реакцию гасят раствором хлорида аммония и реакционную смесь экстрагируют этилацетатом. Объединенные органические фазы концентрируют до суха, получая 15,3 г сырого (3R, 55 г)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она. По данным НЯМР - анализа сырого продукта соотношение диастереоизомеров составляет 83: 17 [(3R, 5S): (3S, 5S)]. Данные спектрального анализа для (2-а, R1 = пивалоил) представлены в примере 5-1, способ 1. Данные спектрального анализа для (2-b, R1 = пивалоил) представлены в примере 5-2.
Способ 3
65 мкл (60 мг, 0,323 ммоля) дициклогексиламина растворяют в 1 мл сухого ТГФ и полученный раствор охлаждают до 0°С. После добавления 197 мкл бутиллития в гексане (1,59 М) раствор перемешивают при 0°С в течение 15 мин. Затем по каплям добавляют раствор 100 мг (2,84 ммоля) (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1-а, R1 = пивалоил) в 1 мл ТГФ и перемешивают при 0°С еще 15 мин. После этого в течение 20 мин добавляют 20 мкл (47 мг, 0,328 ммоля) метилйодида. Полученную смесь перемешивают при 0°С в течение 2 ч. Затем реакцию гасят посредством добавления 2 мл насыщенного раствора NH4Cl, 2 мл воды и 20 мл изопропилацетата. Органический слой отделяют, сушат над MgSO4, фильтруют и выпаривают. Методом ВЭЖХ остатка обнаружено два метилированных диастереомерных соединения 2-а (R1=Piv) и 2-b (R1=Piv). Соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 85:15.
Способ 4
65 мкл (60 мг, 0,323 ммоля) 2,2,6,6-тетраметипиперидина растворяют в 1 мл сухого ТГФ и полученный раствор охлаждают до 0°С. После добавления 197 мкл бутиллития в гексане (1,59 М) раствор перемешивают при 0°С в течение 15 мин. Затем по каплям добавляют раствор 100 мг (2,84 ммоля) (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1-а, R1 = пивалоил) в 1 мл ТГФ и смесь перемешивают при 0°С еще 15 мин. После этого в течение 20 мин добавляют 20 мкл (47 мг, 0,328 ммоля) метилйодида. Полученную смесь перемешивают при 0°С в течение 2,5 ч. Затем реакцию гасят посредством добавления 2 мл насыщенного раствора NH4Cl, 2 мл воды и 20 мл изопропилацетата. Органический слой отделяют, сушат над MgSO4, фильтруют и выпаривают. Методом ВЭЖХ остатка обнаружено два метилированных диастереомерных соединения 2-а (R1=Piv) и 2-b (R1=Piv); соотношение (3R,5S): (3S,5S) составляет 88: 12.
Способ 5
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (10 г, 29,8 ммоля) растворяют в толуоле (50 мл) и охлаждают до 0°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (77,5 мл, 38,7 ммоля). После этого в течение 0,5 ч добавляют диметилсульфат (4,2 мл, 44,7 ммоля). По истечении 15 мин добавляют 1 М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют и органическую фазу промывают 1М NaOH (50 мл), а затем водой (50 мл). После этого из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт. Остаток содержит (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил). Соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 86:14.
Остаток кипятят с обратным холодильником в метаноле (50 мл). Затем добавляют воду (5 мл) и смесь охлаждают до комнатной температуры. По истечении 1 ч смесь охлаждают до 0°С и перемешивают еще 1 ч. Твердое вещество собирают фильтрованием, промывают МеОН/Н2О (5 мл, 9:1), а затем сушат под вакуумом, получая дополнительное количество очищенного продукта. Соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 86:14.
Способ 6
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (5 г, 14,9 ммоля) растворяют в толуоле (25 мл) и охлаждают до -10°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (38,8 мл, 19,4 ммоля). После этого в течение 40 мин добавляют диметилсульфат (2,1 мл, 22,4 ммоля). По истечении 30 мин добавляют раствор NH4Cl (25 мл) и воду (25 мл). Фазы разделяют, и водную фазу промывают толуолом (25 мл). Объединенные органические фазы сушат (MgSO4). Затем под вакуумом из органической фазы удаляют растворитель, получая остаток, содержащий продукт: Соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 92:8.
Способ 7
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (10 г, 29,8 ммоля) растворяют в толуоле (50 мл) и охлаждают до 0°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (77,5 мл, 38,7 ммоля). После этого в течение 0,5 ч добавляют диметилсульфат (4,2 мл, 44,7 ммоля). По истечении 15 мин добавляют 1 М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют и органическую фазу промывают 1М NaOH (50 мл), а затем водой (50 мл). После этого из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт. Остаток содержит (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил): соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 86:14.
Остаток кипятят с обратным холодильником в метаноле (50 мл). Затем добавляют воду (5 мл) и смесь охлаждают до комнатной температуры. По истечении 1 ч смесь охлаждают до 0°С и перемешивают еще 1 ч. Твердое вещество собирают фильтрованием, промывают МеОН/Н2О (5 мл, 9:1), а затем сушат под вакуумом, получая дополнительное количество очищенного продукта. Соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 86:14.
Способ 8
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (5 г, 14,9 ммоля) растворяют в толуоле (25 мл) и охлаждают до -10°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (19,4 ммоля, 38,8 мл). После этого в течение 20 мин добавляют диметилсульфат (2,1 мл, 22,4 ммоля), растворенный в ТГФ (6 мл). По истечении 30 мин добавляют морфолин (2,0 мл, 22,4 ммоля). По истечении 1 ч добавляют 1М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют, и органическую фазу промывают водой (3×50 мл). Фазы разделяют. После этого из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт: соотношение (3R,5S): (3S,5S), по данным ВЭЖХ, составляет 85:15.
Способ 9
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (5 г, 14,9 ммоля) растворяют в толуоле (25 мл) и охлаждают до -10°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (19,4 ммоля, 38,8 мл). После этого в течение 30 мин полученную смесь помещают в раствор диметилсульфата (2,1 мл, 22,4 ммоля) в толуоле (2 мл) при 0°С. По истечении 30 мин добавляют морфолин (2,0 мл, 22,4 ммоля). По истечении 1 ч добавляют 1М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют и органическую фазу промывают водой (3×50 мл). Фазы разделяют и, после этого, из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт: соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 91:9.
Способ 10
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (5 г, 14,9 ммоля) растворяют в толуоле (25 мл) и охлаждают до -10°С. Полученный раствор переносят в сосуд, содержащий 0,5 М раствор бис(триметилсилил)амида калия в толуоле (19,4 мл, 38,8 ммоля), поддерживая температуру 0°С. Затем смесь в течение 30 мин помещают в раствор диметилсульфата (2,1 мл, 22,4 ммоля) в толуоле (2 мл) при 0°С. По истечении 30 мин добавляют морфолин (2,0 мл, 22,4 ммоля). По истечении 1 ч добавляют 1М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют и органическую фазу промывают водой (3×50 мл). Фазы разделяют и, после этого, из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт: соотношение (3R,5S):(3S,5S), по данным ВЭЖХ, составляет 91:9.
Способ 11
(S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-он (1-а, R1 = пивалоил) (10 г, 29,8 ммоля) растворяют в толуоле (50 мл) и охлаждают до 0°С. Затем в течение 30 мин добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (58,6 мл, 38,7 ммоля). После этого в течение 0,5 ч добавляют диметилсульфат (4,2 мл, 44,7 ммоля). По истечении приблизительно 15 мин добавляют морфолин (3,9 мл, 44,7 ммоля). По истечении 0,5 ч добавляют 1 М HCl (50 мл) и смеси позволяют нагреться до комнатной температуры. Фазы разделяют и органическую фазу промывают 1М NaOH (50 мл), а затем водой (50 мл). После этого из органической фазы под вакуумом удаляют растворитель, получая остаток, содержащий продукт: соотношение (3R,5S): (3S,5S), по данным ВЭЖХ, составляет 87:13.
Пример 5-2: (3S, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-b, R1 = пивалоил)
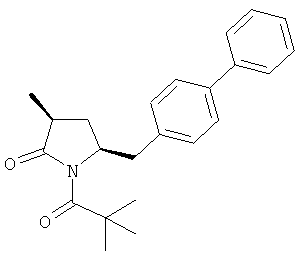
Получают в результате реакции (1)→(2), как показано в примере 5-1, способ 1. Разделение осуществляют посредством хроматографии, элюлируемой смесью гептан/эти л ацетат (7:1) [Rf (2-а=0,33); (2-b=0,26)]. 1Н-ЯМР (ДМСО): 1,10 (3H, д, СН3); 1,29 (9Н, с, С(СН3)3); 1,46 (1Н, м, 4-CHH); 2,15 (1Н, м, 4-CHH); 2,57 (2Н, м, 3-СН, 1-CHH); 3,19 (1Н, м, 1-CHH); 4,31 (1Н, м, 5-СН); 7,29-7,63 (9Н, 4×м, ароматический), m/z: 350 (МН+, 100%); 320 (11); 266 (10).
Рентгеновская структура другого диастереоизомера (соединения, соответствующего формуле (2-b)) показана на фиг.3. Рентгеноструктурный анализ также показывает наличие некоторого количества соединения (2-а), которое совместно кристаллизуется, и, таким образом, образец представляет собой смесь диастероизомеров. Однако чистое соединение может быть получено при использовании колоночной хроматографии.
Кристаллографические данные [полученные при 100(2) К]
|
|
Пример 5-3: (R)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3,3-диметилпирролидин-2-он
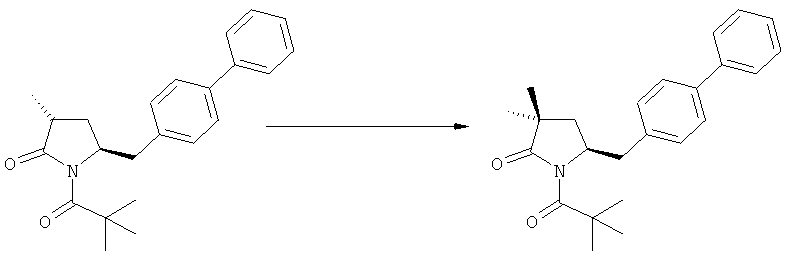
10 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она (2-а, R1 = пивалоил) добавляют к ТГФ (100 мл). Полученную смесь охлаждают до -30°С. Затем добавляют литий диизопропиламид (17,16 мл, 2 М) и смесь перемешивают в течение 1 ч. После этого добавляют метилйодид (5,4 мл) и смесь перемешивают в течение 3 ч. Затем добавляют аминоэтилэтаноламин (6,1 мл) и смесь нагревают до 40°С и перемешивают в течение 15 мин. После этого добавляют серную кислоту (12 г). Смесь концентрируют под вакуумом, а остаток помещают в толуол. Фазы разделяют. Органическую фазу концентрируют под вакуумом и помещают в метанол (300 мл) при кипении. После охлаждения фильтрованием собирают осадок, получая (R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3,3-диметилпирролидин-2-он. 1Н ЯМР (ДМСО): 1,04 (3H), 1,15 (3H), 1,28 (9Н), 1,72 (1H), 1,38 (1Н), 2,59 (1H), 3,14 (1Н), 4,37 (1H), 7,31 (3H), 7,45 (2Н), 7,62 (4Н).
Пример 5-4: (R)-5-Бифенил-4-илметил-3,3-диметилпирролидин-2-он
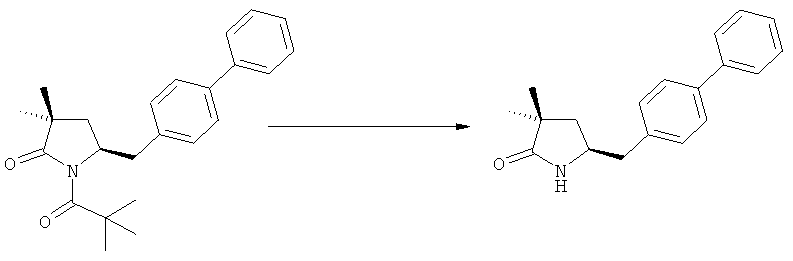
6 г (R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она добавляют в ТГФ (6 мл). После этого добавляют гидроксид лития (15,6 мл, 3 моля л-1), а затем - тетра-бутиламмоний бромид (0,15 г). Полученную смесь добавляют к смеси пероксида водорода (4,54 г) и ТГФ (12 мл) при 0°С. По истечении 2,5 ч добавляют раствор бисульфита натрия (12 г, 38-40%). ТГФ удаляют под вакуумом. После этого добавляют толуол (70 мл) и фазы разделяют. Органическую фазу промывают водой (15 мл). Фазы разделяют. Органическую фазу концентрируют под вакуумом, затем добавляют гептан (60 мл) и смесь охлаждают до 0°С. Осадок собирают фильтрованием и сушат под вакуумом, получая (R)-5-бифенил-4-илметил-3,3-диметилпирролидин-2-он. 1Н ЯМР (ДМСО): 0,96 (3H), 0,97 (3H), 1,52 (1Н), 1,78 (1Н), 2,61 (1Н), 2,93 (1Н), 3,75 (1Н), 7,32 (3H), 7,45 (2Н), 7,58 (2Н), 7,63 (2Н), 7,70 (1Н).
Метод ВЭЖХ (примеры 5-1-5-4):
Колонка: Gemini С6 Phenyl (Phenomenex); 150×3,0 мм; 3 мкм. Подвижная фаза А (0,01 М (NH4)H2PO4 pH 6,6); Подвижная фаза Б (ацетонитрил). Градиент: 0 мин (40% А; 60% Б); 15 мин (40% А; 60% Б); 20 мин (20% А; 80% Б); 23 мин (20% А; 80% Б); 23,1 мин (40% А; 60% Б); 26 мин (40% А; 60% Б). Скорость потока: 0,8 мл мин"1. Длина волны: 254 нм.
Время удерживания:
|
Соотношение диастероизомеров (3R,5S):(3S,5S) [-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она (2-а)] определено на основании площади пиков для 2-а (R1=Piv) [8,6 мин] и 2-b (R1=Piv) [8,2 мин].
Пример 6: (3R, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Н)
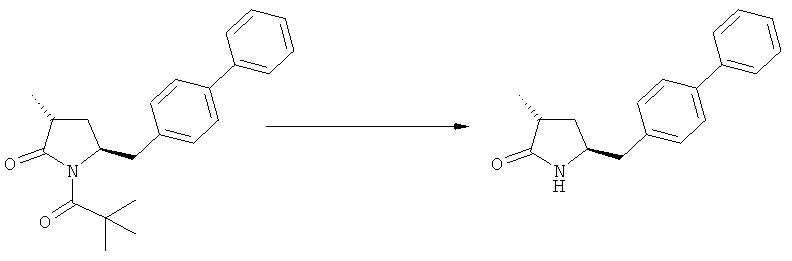
2 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она, полученного в примере 5, м 2 г пара-толуолсульфокислоты в 40 мл кипятят с обратным холодильников около 1 ч. Затем раствор охлаждают до комнатной температуры и нейтрализуют 10 мл разбавленного раствора карбоната натрия. Органическую фазу отделяют, промывают водой и концентрируют до суха. Остаток кристаллизуют из смеси изо-пропилацетат/гептан, получая 1,2 г (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-она (2-а, R1=Н). 1Н-ЯМР (CDCl3): 1,90 (3H, с, СН3); 1,83 (1H, м, 4-CHH); 2,12 (1Н, м, 4-CHH); 2,42 (1Н, м, 3-Н); 2,82 (2Н, м, 1-СН2); 3,87 (1Н, м, 5-СН); 7,10 (1Н, с, NH); 7,26 (2Н, д, ароматический); 7,34 (1Н, т, ароматический); 7,43 (2Н, м, ароматический); 7,54 (2Н, м, ароматический); 7,58 (2Н, м, ароматический), m/z: 266 (МН+, 100%).
Рентгеновская структура полученных кристаллов показана на фиг.7. Монокристалл для данного определения получают при использовании в качестве растворителя изопропилацетата.
Кристаллографические данные [полученные при 100(2) К]
|
|
Пример 7: Гидрохлорид (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н)
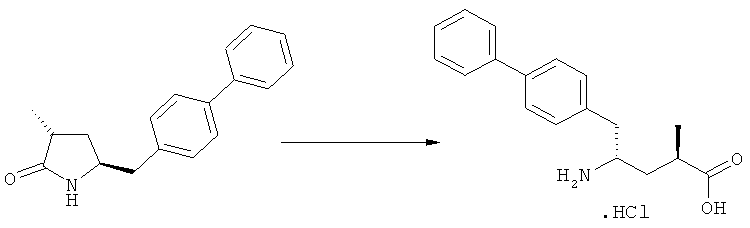
5 г (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-она, полученного в примере 6, смешивают со смесью уксусной кислоты и концентрированной соляной кислоты (50 мл, соотношение 1: 1), и перемешивают при кипении с обратным холодильником около 20 ч. Затем раствор концентрируют под вакуумом, а остаток кристаллизуют из смеси уксусная кислота/этилацетат, получая 4,7 г гидрохлорида (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н). 1Н-ЯМР (ДМСО): 1,10 (3H, с, СН3); 1,58 (1Н, м, 3-CHH); 1,88 (1Н, м, CHH); 2,64 (1Н, м, 4-СН); 2,88 (1Н, дд, 5-CHH); 3,01 (1Н, дд, 5-CHH); 3,45 (1H, м, 2-СН); 7,38 (3H, м, ароматический); 7,47 (2Н, м, ароматический); 7,66 (4Н, м, ароматический); 8,07 (2Н, с, NH); 12,25 (1Н, с, CO2H). m/z: 284 (МН+, 100%); 267 (25); 249 (47); 221 (13); 193 (24).
Рентгеновская структура полученных кристаллов показана на фиг.5. Монокристалл для данного определения получают при использовании в качестве растворителя смеси ацетонитрил/метанол.
Кристаллографические данные [полученные при 100(2) К]
|
|
Пример 8: (2R,4S)-5-Бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановая кислота (3-а, R1=БОК, R2=R3=Н)
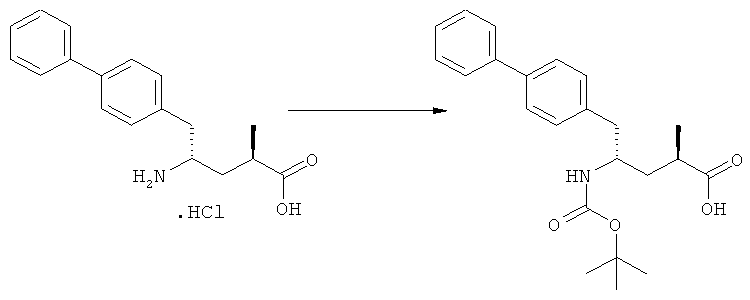
3,2 г гидрохлорида (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н) смешивают с 3,2 г ди-трет-бутилдикарбоната, 5 г карбоната калия и 50 мл смеси вода/изо-пропанол (1:1), и перемешивают при комнатной температуры в течение 1 ч. Затем смесь подкисляют разбавленной фосфорной кислотой, экстрагируют изо-пропилацетатом, промывают водой, концентрируют и кристаллизуют из смеси изо-пропилацетат/гептан, получая (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановую кислоту (3-а, R1=БОК, R2=R3=Н). Т. пл.=146-147°С; δН (500 МГц; ДМСО) 1,07 (3H, д, J 7,0, 1-СН3), 1,34 (9Н, с, (СН3)3), 1,38 (1Н, м, 3-НА), 1,77 (1Н, м, 3-НВ), 2,43 (1H, м, 2-Н), 2,70 (2Н, д, J 7,0, 5-Н), 3,69 (1Н, м, 4-Н), 6,74 (1Н, д, J 9,0, NH), 7,27 (2Н, д, J 8,0, Ar-орто-H(Ph)), 7,36 (1Н, т, J 7,0, Ar-(Ph)-пара-H), 7,46 (2Н, т, J 7,5, Ar-(Ph)-мета-H), 7,57 (2Н, д, J 8,0, Ar-мета-H(Ph), 7,64 (2Н, д, J 7,5, Ar-(Ph)-орто-H), 12,01 (1H, с, CO2H); 5С (500 МГц, ДМСО) 18,1 (1-СН3), 28,3 [(СН3)3], 35,9 (2-С), 37,9 (3-С), 40,7 (5-С), 50,0 (4-С), 77,4 [(С(СН3)3], 126,3, 126,5, 127,2, 128,9, 129,8 (Ar-СН), 137,7 (Ar-ипсо-C(Ph)), 138,3 (Ar-пара-C(Ph)), 140,1 (Ar-(Ph)-ипсо-С), 155,2 (NCO), 177,2 (CO2H); m/z (+ESI) 406 ([MNa]+, 6%), 384 ([MH]+, 31), 328 (100), 284 (19); Найдено: [МН]+, 384,21691. C23H30NO4 требуемое МН 384,21693.
Пример 9-1: Гидрохлорид этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et)
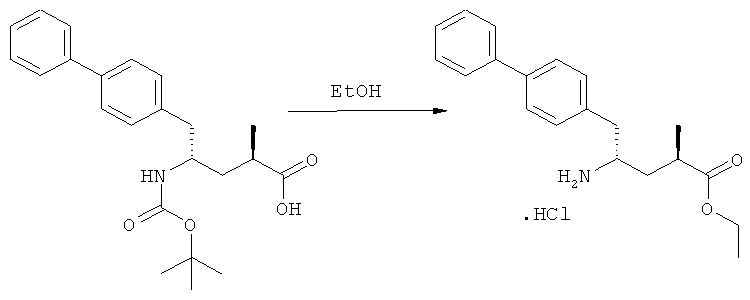
Способ 1
150 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) растворяют в 1500 мл этанола при 70°С. Затем около 1 ч добавляют 43 мл тионилхлорида. Полученную смесь перемешивают еще 2 ч. После этого смесь концентрируют до суха, а затем суспендируют в 3400 мл гептана. Осадок собирают фильтрованием, получая 133 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et). 1Н-ЯМР (ДМСО): 1,05 (3H, т, 1-СН3); 1,08 (3H, т, CH2CH3); 1,61 (1Н, м, 3-CHH); 1,85 (1H, м, 3-CHH); 2,74 (1Н, м, 2-СН); 2,81 (1Н, дд, 5-CHH); 3,08 (1Н, дд, 5-CHH); 3,36 (1Н, м, 4-СН); 3,95 (2Н, кв., CH2CH3); 7,31 (1Н, м, ароматический); 7,35 (2Н, м, ароматический); 7,43 (2Н, м, ароматический); 7,62 (4Н, м, ароматический); 8,30 (3H, с, NH3 +). m/z 312 (МН+, 100%)
Способ 2
150 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) добавляют 1500 мл этанола при комнатной температуре. Смесь затем нагревают до температуры 60-70°С. После этого к реакционной смеси в течение 1 ч добавляют тионилхлорид. Затем смесь перемешивают еще 2 ч. 810 мл растворителя удаляют дистилляцией при пониженном давлении. После этого добавляют 1460 мл гептановой фракции. Затем дистилляцией при пониженном давлении удаляют 1310 мл растворителя. После этого добавляют 1460 мл гептановой фракции. Затем дистилляцией при пониженном давлении удаляют 520 мл растворителя. После этого добавляют 1460 мл гептановой фракции. Смесь охлаждают до комнатной температуры в течение 1 ч. Затем смесь перемешивают при комнатной температуре в течение 2 ч. После этого твердое вещество собирают посредством фильтрования, промывают гептановой фракцией (600 мл) и сушат, получая гидрохлорид этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et). Спектральные данные соответствуют данным, приведенным в примере 9-1, способ 1.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 20: 5,7, 6,2, 7,7, 11,3, 12,5, 17,1, 22,4, 22,9. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Рентгеновская структура полученных кристаллов показана на фиг.6а и фиг.66. Монокристалл для данного определения получают при использовании в качестве растворителя ацетонитрила.
Кристаллографические данные [полученные при 293(2) К]
|
Пример 9-2: Гидрохлорид этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et)
(3-а, R1=R2=Н, R3=Et) получают в соответствии с примером 9-1 и кристаллизуют в соответствии со следующими способами:
Способ 1
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 3 мл толуола при комнатной температуре. Затем смесь нагревают до 75°С и перемешивают при данной температуре до полного растворения вещества. После этого смесь охлаждают до комнатной температуры и перемешивают в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 29: 16,9, 18,2, 22,2, 22,7, 24,0. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 2
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 3 мл ксилола при комнатной температуре. Затем смесь нагревают до 80°С и перемешивают при данной температуре до полного растворения вещества. После этого смесь охлаждают до комнатной температуры и перемешивают в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 20: 16,9, 18,2, 22,2, 22,7, 23,9. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 3
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 5 мл этилацетацетата при комнатной температуре. Затем смесь нагревают до 80°С и перемешивают при данной температуре до полного растворения вещества. После этого смесь охлаждают до комнатной температуры и перемешивают в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 29: 7,7, 17,2, 18,5, 22,4, 22,9, 24,0. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 4
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 5 мл этилацетата при комнатной температуре. Затем смесь кипятят с обратным холодильником. После этого смесь охлаждают до комнатной температуры и перемешивают в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 20: 7,5, 17,0, 18,3, 22,2, 22,8, 24,0. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 5
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 5 мл ксилола при комнатной температуре. Затем смесь нагревают на масляной бане при температуре 120°С и перемешивают при данной температуре до полного растворения вещества. После этого смесь охлаждают до комнатной температуры и перемешивают в течение 4 ч. Затем смесь охлаждают до 0°С и перемешивают в течение 1 ч. После этого смесь еще перемешивают при комнатной температуре в течение 72 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 20: 7,6, 17,1, 18,3, 19,7, 22,4, 22,8, 24,0. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 6
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 5 мл ксилола при комнатной температуре. Затем смесь нагревают на масляной бане при температуре 120°С и перемешивают при данной температуре до полного растворения вещества. После этого смесь в течение нескольких часов медленно охлаждают до комнатной температуры. Затем смесь перемешивают при комнатной температуре в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 29: 7,5, 17,0, 18,3, 19,7, 22,3, 22,7, 24,0. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Способ 7
500 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) добавляют к 5 мл этилацетацетата при комнатной температуре. Затем смесь нагревают на масляной бане при температуре 120°С и перемешивают при данной температуре до полного растворения вещества. Затем смесь охлаждают до комнатной температуры и перемешивают в течение 16 ч. Полученный осадок собирают фильтрованием.
Значимые рефлексы на рентгенограмме соответствуют следующим межплоскостным расстояниям (среднее значение 2θ в градусах дано с погрешностью ±0,2): градусы 29: 7,5, 16,1, 16,9, 18,2, 20,2, 22,2, 22,7, 23,9. Данные получены на дифрактометре Bruker D8 Advance, с использованием Cu-Кα излучения.
Пример 10: (S)-5-((S,R)-Бифенил-4-илгидроксиметил)пирролидин-2-он (13, R1=Н)
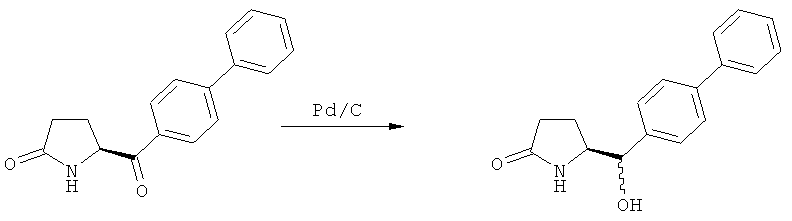
2 г (S)-5-(бифенил-4-карбонил)пирролидин-2-она растворяют в 40 мл ТГФ. Затем добавляют 200 мг палладия на угле, и смесь перемешивают в атмосфере водорода в течение 14 ч. Удаляют фильтрованием катализатор и концентрируют фильтрат до суха, получая требуемый продукт (13) в виде смеси спиртовых диастереоизомеров. 1Н ЯМР (CDCl3): 1,71-2,35 (4Н); 3,66-3,87 (1Н); 4,45 (приблизительно 0,7Н) и 4.60 (приблизительно 0,3H); 5,70 (приблизительно 0,3H) и 6,24 (приблизительно 0,7Н); 6,85-7,66 (9Н). Полученный спирт затем может быть превращен в (S)-5-бифенил-4-илметилпирролидин-2-она при использовании, например, условий, подобных условиям примера 3. Вычисленное соотношение диастереоизомеров составляет 70: 30 (1Н ЯМР), на основании суммирования сигналов при 4,45 м.д. (0,7 Н) и 4,60 (0,3H).
Пример 11: (2-а, R1 = пивалоил) в (3-а, R1=R2=R3=Н)
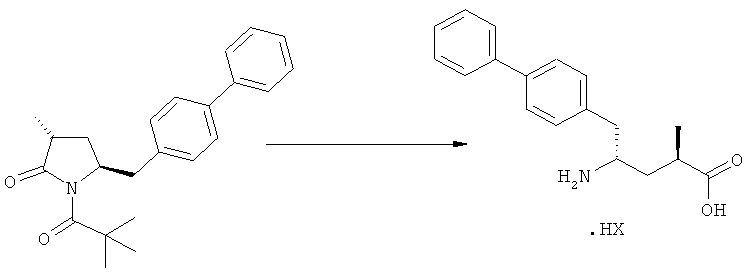
Способ 1
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она в 2,5 мл воды, 2,5 мл концентрированной соляной кислоты и 2 мл этилацетата нагревают при температуре около 80°С приблизительно 15 ч. Затем смесь выпаривают до суха, получая гидрохлорид (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н, гидрохлоридная соль). Спектральные данные соответствуют данным, приведенным в примере 7.
Способ 2
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она в 5 мл бромистоводородной кислоты (48%) и 4 мл этилацетата нагревают при температуре около 80°С приблизительно 15 ч. Затем смесь выпаривают до суха, получая гидрохлорид (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н, гидрохлоридная соль). МС (ES+): 284 ([МН]+, 100%), 267 (17), 249 (18), 221 (3), 194 (3), 193 (25), 167 (4).
Пример 12: (2-а, R1 = пивалоил) в (3-а, R1=R2=Н, R3=Et)
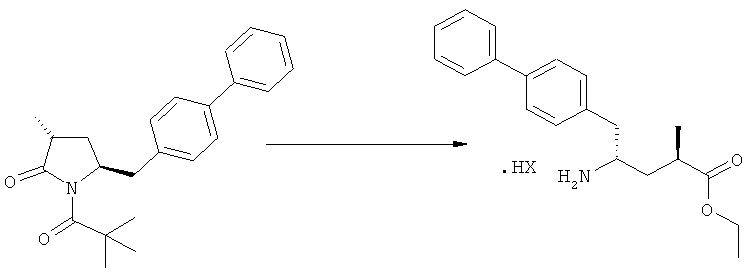
Способ 1
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она, 5 мл этанола и 0,5 мл концентрированной соляной кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (гидрохлоридная соль). Спектральные данные соответствуют данным, приведенным в примере 9.
Способ 2
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она, 5 мл этанола и 0,3 мл концентрированной серной кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (гидросульфатная соль). Спектральные данные соответствуют данным, приведенным в примере 44.
Способ 3
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она, 5 мл этанола и 0,3 мл перхлорной кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (перхлорат). 1Н ЯМР (CDCl3): 1,17 (3H), 1,20 (3H), 1,98 (2Н), 2,76 (1Н), 2,96 (1Н), 3,29 (1Н), 3,82 (1Н), 3,96 (2Н), 7,32-7,59 (13Н), 8,21 (3H).
Способ 4
0,5 г (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-она, 5 мл этанола и 1,1 г пара-толуолсульфоновой кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (n-толуолсульфонатная соль). 1Н ЯМР (CDCl3): 1,08 (3H), 1,16 (3H), 1,87 (1Н), 1,95 (1H), 2,38 (3H), 2,77 (1Н), 2,92 (1Н), 3,15 (1Н), 3,69 (1H), 4,07 (2Н), 7,16-7,77 (13Н), 9,89 (3H).
Пример 13: (2-а, R1=Н) в (3-а, R1=R2=Н, R3=Et)
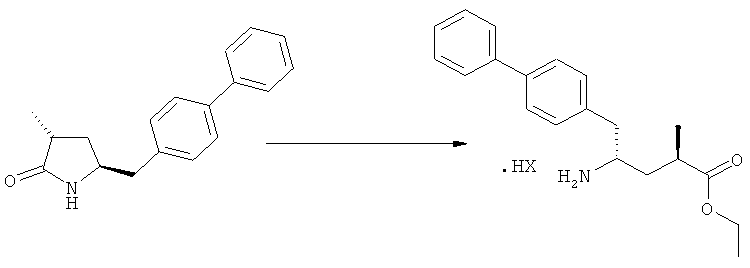
1 г (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-она (2-а, R1=Н), 10 мл этанола и 1,4 мл концентрированной соляной кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (гидрохлоридная соль). Спектральные данные соответствуют данным, приведенным в примере 9.
Способ 2
1 г (3R, 55 г)-5-бифенил-4-илметил-3-метилпирролидин-2-она, 10 мл этанола и 0,4 мл концентрированной серной кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (гидросульфатная соль). Спектральные данные соответствуют данным, приведенным в примере 44.
Способ 3
1 г (3R, 55 г)-5-бифенил-4-илметил-3-метилпирролидин-2-она, 10 мл этанола и 1,4 г пара-толуолсульфоновой кислоты нагревают при температуре около 80-120°С приблизительно 24 ч. Затем смесь выпаривают до суха, получая этиловый эфир (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (n-толуолсульфонатная соль). Спектральные данные соответствуют данным, приведенным в примере 12.
Пример 14: (2-а, R1=Н) в трет-бутиловый эфир (3R, 5S)-6-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=БОК)
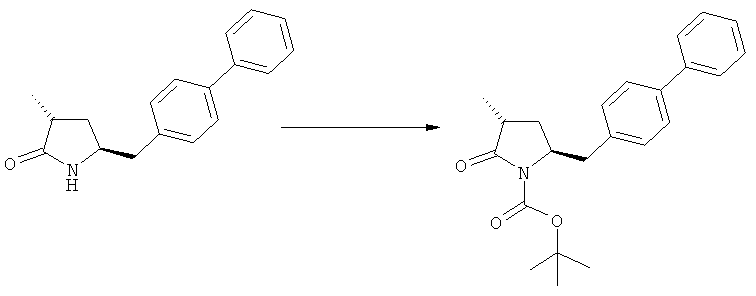
5 г (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-она (2-а, R1=Н) в растворе 50 мл этилацетата смешивают 6,6 г ди-трет-бутилдикарбоната, 3,5 г триэтиламина и 1 г диметиламинопиридина. По истечении 1 ч при 50°С раствор смешивают с водой. После разделения слоев органическую фазу концентрируют под вакуумом и разбавляют гептаном. Полученное кристаллическое вещество собирают и сушат под вакуумом, получая около 5,5 г трет-бутилового эфира (3R, 5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=БОК).
Данное вещество может быть очищено колоночной хроматографией, элюлируемой смесью этилацетат/гептан (6: 1).
1Н-ЯМР (ДМСО): 0,95 (3H, д, 1-СН3); 1,42 (9Н, с, С(СН3)3); 1,56 (1Н, м, 4-CHH); 1,90 (1Н, м, 4-CHH); 2,50-2,57 (2Н, м, 3-СН2); 2,78 (1Н, дд, 1-CHH); 2,99 (1H, дд, 1-CHH); 4,14 (1Н, м, 5-СН); 7,25-7,31 (3H, м, ароматический); 7,39 (2Н, м, ароматический); 7,58 (4Н, м, ароматический).
Пример 15: (2-а, R1=БОК) в [(2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановую кислоту] (3-а, R1=БОК, R2=R3=Н)
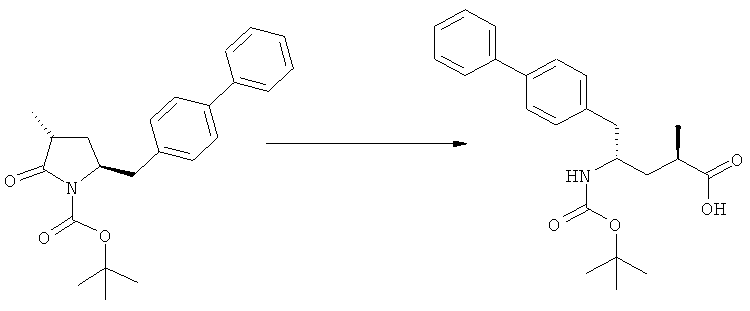
5 г трет-бутилового эфира (3R, 5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=БОК) растворяют в 50 мл смеси ТГФ и 2 М раствора гидроксида лития (соотношение 1:1). По истечении 1 ч при командной температуре добавляют фосфорную кислоту для нейтрализации избытка гидроксида лития. Суспензию концентрируют под вакуумом для удаления наибольшего количества растворителя, и экстрагируют изо-пропилацетатом. Органическую фазу затем промывают водой, частично концентрируют под вакуумом и подвергают кристаллизации после добавления гептана. Полученное кристаллическое твердое вещество собирают и сушат под вакуумом, получая около 3,3 г [(2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты] (3-а, R1=БОК, R2=R3=Н). Т. пл.=146-147°С. δН (500 МГц; ДМСО) 1,07 (3H, д, J 7,0, 1-СН3), 1,34 (9Н, с, (СН3)3), 1,38 (1Н, м, 3-HA), 1,77 (1Н, м, 3-НВ), 2,43 (1Н, м, 2-Н), 2,70 (2Н, д, J 7,0, 5-Н), 3,69 (1Н, м, 4-Н), 6,74 (1Н, д, J 9,0, NH), 7,27 (2Н, д, J 8,0, Ar-орто-H(Ph)), 7,36 (1Н, т, J 7,0, Ar-(Ph)-пара-Н), 7,46 (2Н, т, J 7,5, Ar-(Ph)-мета-H), 7,57 (2Н, д, J 8,0, Ar-мета-H(Ph), 7,64 (2Н, д, J 7,5, Ar-(Ph)-орто-H), 12,01 (1H, с, CO2H); 5 с (500 МГц, ДМСО) 18,1 (1-СН3), 28,3 [(СН3)3], 35,9 (2-С), 37,9 (3-С), 40,7 (5-С), 50,0 (4-С), 77,4 [(С(СН3)3], 126,3, 126,5, 127,2, 128,9, 129,8 (Ar-СН), 137,7 (Ar-мисо-С(Ph)), 138,3 (Ar-пара-C(Ph)), 140,1 (Ar-(Ph)-ипсо-С), 155,2 (NCO), 177,2 (CO2H); m/z (+ESI) 406 ([MNa]+, 6%), 384 ([МН]+, 31), 328 (100), 284 (19); Найдено: [МН]+, 384,21691. C23H30NO4 требуется МН 384,21693.
Пример 16: трет-Бутиловый эфир (S)-5-бифенил-4-илметил-2-оксопирролидин -1-карбоновой кислоты (1-а, R1=БОК)
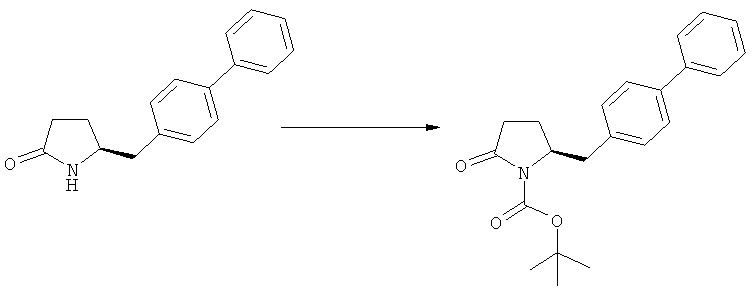
Способ 1
520 мг ди-трет-бутилдикарбоната и 12 мг 4-(диметиламино)пиридина добавляют к суспензии 500 мг (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) в 3 мл ацетонитрила. По истечении приблизительно 20 ч смесь концентрируют до суха. Затем смесь распределяют между этилацетатом и водным гидросульфатом калия, органический слой отделяют и выпаривают до суха, получая 630 мг трет-бутиповото эфира (5')-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК). 1Н-ЯМР (CDCl3): 1,62 (9Н, с, С(СН3)3); 1,88 (1Н, м, 4-CHH); 2,03 (1Н, м, 4-CHH); 2,34-2,43 (2Н, м, 3-СН2); 2,80 (1Н, дд, 1-CHH); 3,21 (1Н, дд, 1-CHH); 4,42 (1Н, м, 5-СН); 7,28 (2Н, м, ароматический); 7,76 (1Н, м, ароматический); 7,46 (2Н, м, ароматический); 7,59 (4Н, м, ароматический), m/z: 352 (МН+, 14%); 337 (16); 296 (100); 293 (13); 252 (25).
Рентгеновская структура полученных кристаллов показана на фиг.10. Монокристалл для данного исследования получают при использовании в качестве растворителя диэтилового эфира.
Кристаллографические данные [полученные при 100(2) К]
|
Способ 2
8,6 г (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) (34,22 ммоля) растворяют в 135 мл метиленхлорида. Затем добавляют 4,2 г ДМАП (34,22 ммоля). Полученную смесь еще раз разбавляют 10 мл метиленхлорида. После этого добавляют 14,9 г БОК2О (68,44 ммоля) и 10 мл метиленхлорида Полученную реакционную смесь перемешивают при кипении с обратным холодильником в течение 7,5 ч. Затем добавляют еще 3,7 г БОК2О. По истечении общего реакционного времени 24 ч при кипении с обратным холодильником полностью выпаривают растворитель. После этого остаток выпаривания фильтруют через 460 г силикагеля, с использованием в качестве элюента смеси толуол: этилацетат (4:1). Фракции продукта концентрируют под вакуумом, получая сырой продукт, перекристаллизация которого из смеси метиленхлорид/гептановая фракция (1:6), приводит к трет-бутиловому эфиру (S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК).
Способ 3
(S)-5-Бифенил-4-илметилпирролидин-2-он (1-а, R1=Н) (100 г, 398 ммоля) добавляют к толуолу (1 л) при комнатной температуре. Затем добавляют N,N-диметиламинопиридин (4,9 г, 29,8 ммоля), а после этого - триэтиламин (72 мл, 517 ммоля). Полученную смесь нагревают до 65°С. Затем в течение 0,5 ч добавляют ди-трет-бутилдикарбонат (104 г, 478 ммоля). По истечении 0,5 ч смесь концентрируют под вакуумом. Образовавшийся остаток растворяют в метаноле (1 л) и нагревают до 60°С. После этого удаляют 400 мл растворителя. Затем добавляют воду (100 мл) и смесь охлаждают до комнатной температуры. По истечении 2 ч смесь охлаждают до 0°С. По истечении 1 ч смесь фильтруют и твердое вещество промывают смесью метанол-вода (5:1, 30 мл × 3). Затем твердое вещество сушат под вакуумом, получая трет-бутиловый эфир (S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК).
Пример 17: (S)-5-Бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (1-а, R1 = метилпирролидин)
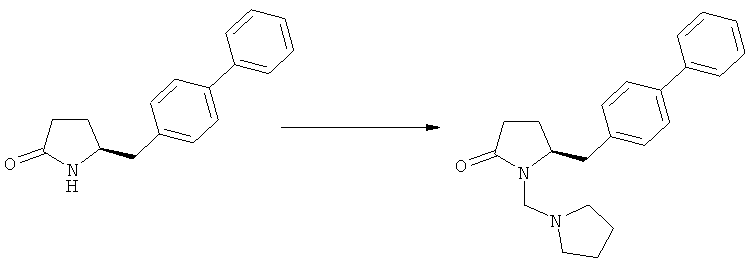
Смесь 500 мг (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н), 329 мкл пирролидина и 415 мкл формальдегида в 3,5 мл этанола кипятя с обратным холодильником в течение 3 ч. Затем добавляют еще 164 мкл пирролидина и 148 мкл формальдегида и смесь кипятя с обратным холодильником около 24 ч. После этого смесь концентрируют до суха и очищают хроматографией, получая 533 мг (.5)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-она (1-а, R1 = метилпирролидин). 1H ЯМР (ДМСО): 1,69 (4Н, м, 2×NCH2CH2); 1,68-2,15 (4Н, м, 3-СН2, 4-СН2); 2,50 (4Н, 2×NCH2); 2,66 (1Н, дд, 1-CHH); 3,12 (1Н, дд, 1-CHH); 3,94 (1Н, д, NCHHN); 4,21 (1Н, д, NCHHN); 7,29 (2Н, д, ароматический); 7,33 (1Н, т, ароматический); 7,44 (2Н, т, ароматический); 7,59 (2Н, д, ароматический); 7,64 (2Н, д, ароматический), m/z: 335 (МН+, 100%).
Пример 18: трет-Бутиловый эфир (3R, 5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=БОК)
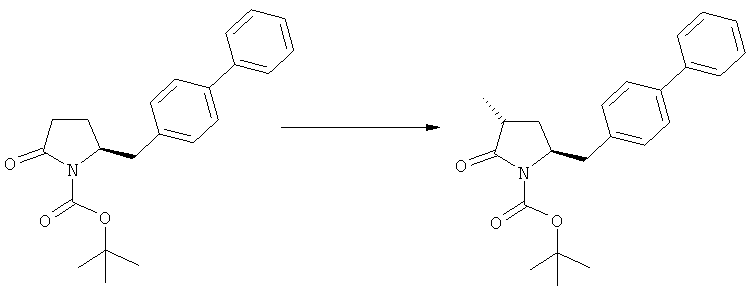
Способ 1
46 мкл диизопропиламина растворяют в 2 мл тетрагидрофурана при 0°С. Затем добавляют 0,2 мл н-бутиллития (1,6 М в гексанах) и полученную смесь перемешивают около 15 мин. После этого смесь охлаждают -78°С. Затем добавляют 100 мг исходного вещества (1-а, R1=БОК), растворенного в 1 мл тетрагидрофурана. По истечении 15 мин добавляют 71 мкл метилйодида и смесь перемешивают при -78°С еще 5 ч. Затем реакцию гасят, добавляя аммоний хлорид, и смесь экстрагируют этилацетатом. Органическую фазу отделяют и концентрируют до суха, получая смесь (2R,5S):(3S,5S)-диастереоизомеров (57: 43), соответственно. Соотношение определено посредством ВЭЖХ-анализа.
Спектральные данные для основного диастереомера (2-а, R1=БОК) соответствуют данным, приведенным в примере 14 (2-а, R1=БОК).
Способ 2
459 мкл (331 мг, 3,27 ммоля) диизопропиламина растворяют в 5 мл сухого ТГФ и охлаждают до 0°С. После добавления 1,97 мл бутиллития в гексане (1,59 М), раствор перемешивают при 0°С в течение 15 мин. Затем реакционную смесь охлаждают до -78°С и в течение 15 мин добавляют раствор 1 г (2,84 ммоля) трет-бутиловый эфира (S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК) и 1,03 мл (1,09 г, 8,52 ммоля) 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинона (ДМП) в 5 мл ТГФ. После перемешивания в течение 15 мин в течение 10 мин добавляют 708 мкл (1,61 г, 11,38 ммоля) метилйодида. Полученную смесь перешивают при -78°С в течение 3 ч. Затем реакцию гасят путем добавления 1 мл морфолина, а затем - 1 мл насыщенного раствора NH4Cl и 15 мл изопропилацетата. Органическую фазу отделяют и промывают водой (3×10 мл). Органический слой отделяют, сушат над MgSO4, фильтруют и выпаривают. ВЭЖХ остатка указывает на наличие двух метилированных диастереомерных соединений - трет-бутилового эфира [(3R, 5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=БОК) и трет-бутилового эфира (3S, 5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-b, R1=БОК)]. По данным Н-ЯМР соотношение двух диастереоизомеров составляет 50: 50. (Спектральные данные для диастереоизомерной смеси). ЯМР (300 МГц, ДМСО-d6, δ/ppm): 7,50 (м, 4H); 7,37 (м, 2H); 7,27 (м, 1H); 7,18 (м, 2H); 4,29-4,12 (м, 1H); 3,45 (м, 1H); 3,10 (м, 1H); 2,73-2,34 (м, 2H); 2,17-1,97 (м, 1H); 1,54, 1,51 (2×с, 9Н); 1,13, 1,09 (2×g при соотношении приблизительно 1:1, J=7,2, 7,0, 3H). MS (ESI, m/e) 366 [М+Н]+.
Способ 3
100 мг (0,284 ммоля) трет-бутиловый эфира (5,)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК) растворяют в 2 мл ТГФ и раствор охлаждают до -78°С. Затем в течение 5 мин добавляют 312 мкл литий бис(триметилсилил)амида в ТГФ (1 М). После перемешивания в течение 15 мин добавляют 71 мкл (161 мг, 1,136 ммоля) метилйодида. Полученную смесь перемешивают 5 ч, а затем реакцию гасят морфолином и водой. В соответствии с ВЭЖХ-анализом соотношение диастереоизомеров составляет 67: 32.
Метод (1) ВЭЖХ:
Колонка: СС 125/3 Nucleosil 10-3. Подвижная фаза А (вода); Подвижная фаза Б (ацетонитрил). Градиент: 0 мин (90% А; 10% Б); 20 мин (10% А; 90% Б); 25 мин (0% А; 100% Б); 25,1 мин (90% А; 10% Б). Скорость потока: 1,0 мл мин-1. Длина волны: 254 нм.
Время удерживания:
|
В данных условиях ВЭЖХ не наблюдается разделение 2-а, R1=БОК и 2-Ь, R1=БОК. Таким образом, остатки, полученные в результате реакций, перед ВЭЖХ-анализом обрабатывают трифторуксусной кислотой для того, чтобы удалить БОК-защитную группу.
Время удерживания:
|
Метод (2) ВЭЖХ:
Колонка: Chiralpak AD-RH, 150×2,6 мм, 5,0 мкм. Подвижная фаза А (вода); Подвижная фаза Б (ацетонитрил). Изократический режим: 0 мин (80% Б; 15 мин (80% Б). Скорость потока: 0,5 мл мин-1. Длина волны: 210 нм.
Время удерживания:
|
Пример 19: (3R, 5S)-5-Бифенил-4-илметил-3-метил-1-пирролидин-1-илметилпирролидин-2-он (2-а, R1=метилпирролидин)
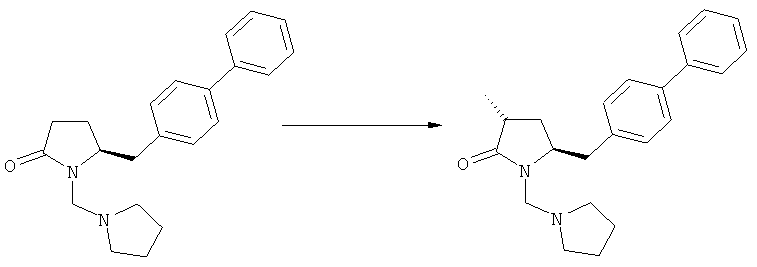
Способ 1
93 мкл диизопропиламина растворяют в 2 мл тетрагидрофурана при 0°С. Затем добавляют 0,4 мл н-бутиллития (1,6 М в гексанах) и смесь перемешивают около 30 мин. После этого добавляют 200 мг исходного вещества (1-а, R1 = метилпирролидин), растворенного в 1 мл тетрагидрофурана. По истечении 30 мин добавляют 41 мкл метилйодида и смесь перемешивают при 0°С еще 2 ч. После этого реакцию гасят, добавляя раствор хлорида аммония, и реакционную смесь экстрагируют этилацетатом. Органическую фазу отделяют и концентрируют до суха, получая 188 мг сырого продукта.
В соответствии с ЯМР-анализом диастереомерное соотношение 2-а R1 = метилпирролидин: 2-b R1 = метилпирролидин составляет 85: 15 (суммирование сигналов при 3,06 и 3,40 м.д.).
1Н-ЯМР (ДМСО, Основной стереоизомер): 0,99 (3H, д, 1-СН3); 1,55 (1Н, м, 4-CHH); 1,69 (4Н, м, 2×NCH2CH2); 2,00 (1Н, м, 4-CHH); 2,20 (1Н, м, 3-СН); 2,50 (4Н, 2×NCH2); 2,69 (1Н, дд, 1-CHH); 3,06 (1Н, дд, 1-CHH); 3,90 (1Н, м, 5-СН); 3,93 (1Н, м, NCHHN); 4,22 (1Н, м, NCHHN); 7,30 (2Н, д, ароматический); 7,34 (1Н, т, ароматический); 7,44 (2Н, т, ароматический); 7,60 (2Н, д, ароматический); 7,65 (2Н, д, ароматический), m/z: 349 (МН+, 100%).
Способ 2
(1-а, R1 = метилпирролидин) (200 мг, 0,6 ммоля) растворяют в ТГФ (3,4 мл). Полученную смесь охлаждают до 0°С. Затем добавляют литий бис(триметилсислил)амид (0,66 мл, 1 М в ТГФ) и смесь перемешивают в течение 1 ч. После этого добавляют метилйодид (40,9 мкл, 0,65 ммоля) и полученную смесь перемешивают в течение 4 ч при 0°С. Реакцию гасят, добавляя насыщенный раствор хлорида аммония (2 мл), воду (1 мл) и этилацетат (1 мл). Фазы разделяют. Органический фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом (222 мг сырого продукта). В соответствии с ЯМР-анализом диастереомерное соотношение 2-а R1 = метилпирролидин: 2-b R1 = метилпирролидин составляет 66: 34.
Пример 20: (3R, 3S)-5-Бифенил-4-илметил-3-метил-1-пирролидин-1-илметилпирролидин-2-он (2-а, R1 = метилпирролидин)
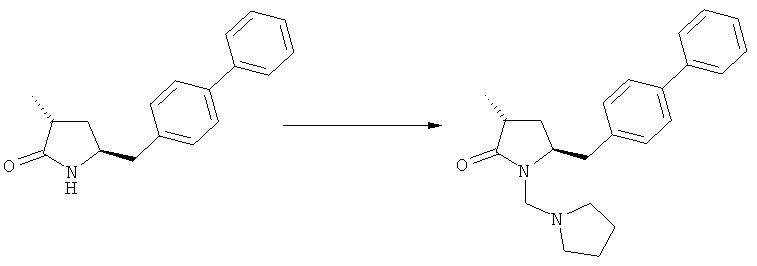
(3R, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Н) (1 г, 3,8 ммоля) добавляют к этанолу (7 мл). Затем добавляют пирролидин (312 мкл, 3,8 ммоля) и водный формальдегид (393 мкл, 5,3 ммоля). Полученную смесь кипятят с обратным холодильником в течение 3 ч. Затем смесь охлаждают до комнатной температуры и концентрируют под вакуумом, получая (3R, 5S)-5-бифенил-4-илметил-3-метил-1-пирролидин-1-илметилпирролидин-2-он (2-а, R1 = метилпирролидин). Спектральные данные соответствуют данным, представленным в примере 19.
Пример 21: (3R, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1=Piv)
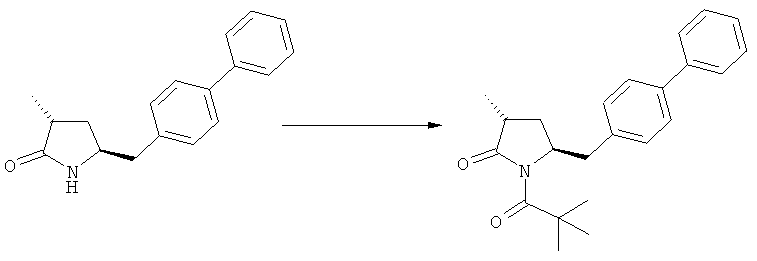
(3R, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Н) (500 мг, 1,9 ммоля) растворяют в ТГФ (5 мл) при комнатной температуре. Затем смесь охлаждают до -78°С и добавляют бутиллитий (1,3 мл, 1,6 М). По истечении 0,5 ч добавляют пивалоилхлорид (278 мкл, 2,3 ммоля) и смесь нагревают до комнатной температуры. По истечении 0,5 ч смесь разбавляют этилацетатом и реакцию гасят, добавляя раствор насыщенного хлорида аммония, а затем - воду. Фазы разделяют. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Сырое вещество очищают колоночной хроматографией, элюлируемой смесью этилацетат/гептан (1: 6), получая (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1=Piv). Спектральные данные соответствуют данным, представленным в примере 5-1, способ 1.
Пример 22: ((S)-2-Бифенил-4-илметил-5-оксопирролидин-1-ил)ацетонитрил (1-а, R1 = цианометил)
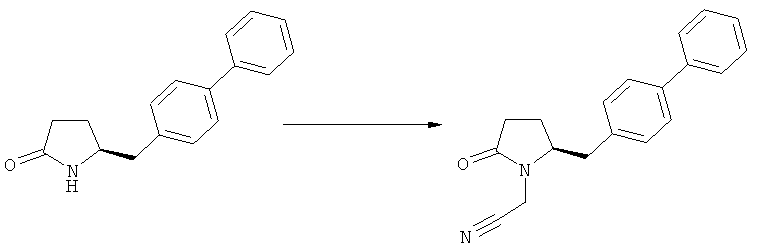
1,5 г (5,97 ммоля) (S)-5-бифенил-4-илметилпирролидин-2-она (1, R1=Н) растворяют в 15 мл сухого ТГФ и охлаждают до -78°С. После добавления 4,13 мл бутиллития в гексане (1, 59 М) полученный желтый раствор перемешивают при -78°С в течение 30 мин. Затем добавляют 475 мкл (855 мг, 7,16 ммоля) бромацетонитрила и смесь нагревают до комнатной температуры в течение ночи. Затем реакцию гасят, добавляя 10 мл насыщенного раствора NH4Cl, с последующим добавлением 6 мл воды, и смесь, затем, экстрагируют 2×40 мл этилацетатом. Объединенные органические слои отделяют, сушат над MgSO4, фильтруют и выпаривают. Полученный остаток очищают колоночной хроматографией (дихлорметан: метанол = 99:1), получая ((S)-2-бифенил-4-илметил-5-оксопирролидин-1-ил)ацетонитрил (1-а, R1 = цианометил) в виде грязно-белого твердого вещества. ЯМР (400 МГц, ДМСО-d6, δ/ч.м.): 7,65 (м, 4H); 7,47 (т, J=7,4, 2H); 7,40 (м, 3H); 4,57 (д, J=17,7, 1H); 4,44 (д, J=17,7, 1H); 3,91 (м, 1H); 3,19 (дд, J=3,9, 13,8, 1H); 2,70 (дд, J=9,2, 13,8, 1Н); 2,18 (м, 2H); 1,95 (м, 1H); 1,77 (м, 1H). МС (ESI, m/е) 291 [М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3025; 2985; 2257; 1697; 1688; 1486; 1421; 1323; 1271; 1182; 760; 699.
Пример 23: (S)-1-Ацетил-5-бифенил-4-илметилпирролидин-2-он (1-а, R1=Ас)
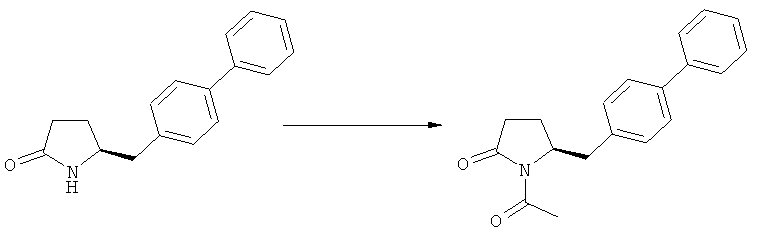
(S)-5-Бифенил-4-илметилпирролидин-2-он (1, R1=Н) (5 г, 19,9 ммоля) растворяют в ТГФ (50 мл). Смесь охлаждают до -78°С и добавляют н-бутиллитий (14 мл, 1,6 М). По истечении 0,5 ч добавляют ацетилхлорид (1,7 мл, 24 ммоля) и смеси позволяют нагреться до комнатной температуры. По истечении 1 ч реакцию гасят, добавляя насыщенный раствор хлорида аммония (40 мл, воду (10 мл) и этилацетат (20 мл). Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, получая (S)-1-ацетил-5-бифенил-4-илметилпирролидин-2-он (1-а, R1=Ас). 1Н ЯМР (CDCl3): 1,84 (1Н), 1,93 (1Н), 2,30 (1Н), 2,32 (1Н), 2,50 (3H), 2,72 (1Н), 3,09 (1H), 4,56 (1Н), 7,20 (2Н), 7,28 (1H), 7,37 (2Н), 7,49 (4Н).
Пример 24: (S)-5-Бифенил-4-илметил-1-триэтилсиланилпирролидин-2-он (1-а, R1=ТЭС)
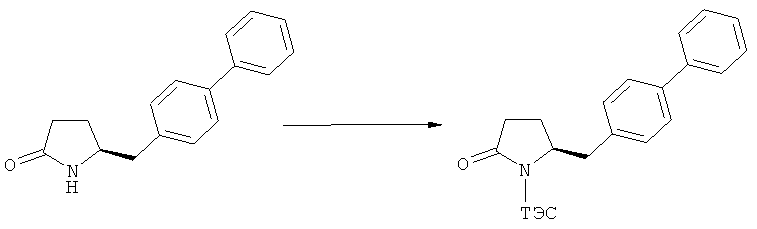
В сухую колбу помещают 1,256 г (5 ммоля) (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) и 15 мл сухого ТГФ. К полученному прозрачному раствору добавляют 2,02 г (20 ммоля) триэтиламина, а затем -904 мг (6 ммоля) триэтилхлорсилана (ТЭС-С1). Полученную реакционную смесь перемешивают при комнатной температуре в течение 4 ч, а затем реакцию гасят, добавляя 5 мл насыщенного раствора NaHCO3, 10 мл воды и 10 мл изопропилацетата. Слои разделяют, и водный слой экстрагируют 10 мл изопропилацетата. Объединенные органические слои сушат над MgSO4, фильтруют и выпаривают. Полученный остаток очищают колоночной хроматографией (дихлорметан + 1% об./об. триэтиламина), получая (S)-5-бифенил-4-илметил-1-триэтилсиланилпирролидин-2-она (1-а, R1=ТЭС), в виде желтого масла. ЯМР (300 МГц, ДМСО-d6, δ/м.д.): 7,63 (м, 4H); 7,55 (м, 2H); 7,32 (м, 3H); 3,80 (м, 1H); 2,85 (м, 1H), 2,70 (м, 1H), 2,10-1,70 (м, 4H), 1,02-0,80 (м, 15Н). МС (ESI, m/е) 366 [М+Н]+; 731 [2М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3426; 3047; 2957; 1698; 1672; 1487; 1378; 1242; 1114; 1008.
Пример 25: (3R,5S)-5-Бифенил-4-илметил-3-метил-1-триэтилсиланилпирролидин-2-он (2-а, R1=ТЭС) и (3S,5S)-5-Бифенил-4-илметил-3-метил-1-триэтилсиланилпирролидин-2-он (2-b, R1=ТЭС)
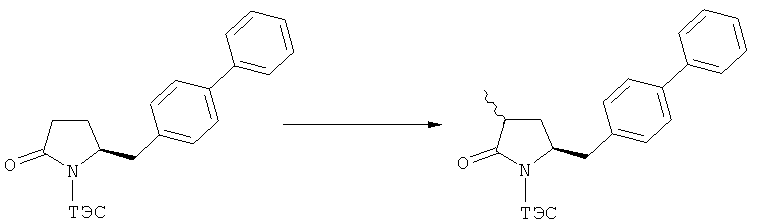
109 г (0,2981 ммоля) (S)-5-бифенил-4-илметил-1-триэтилсиланилпирролидин-2-она (1-а, R1=ТЭС) растворяют в 1,5 мл толуола и раствор охлаждают до 0°С. После медленного добавления 628 мл калия бис(триэтйлсилил)амида в толуоле (0,57 М) раствор перемешивают при 0°С в течение 15 мин. Затем к суспензии в течение 5 мин добавляют 113 мкл (150 мг, 1,19 ммоля) диметилсульфата. После этого смесь перемешивают при 0°С в течение 1 ч. Реакцию гасят, добавляя 2 мл насыщенного раствора NH4Cl, 2 мл воды, а затем 20 мл изопропилацетата. Органическую фазу промывают водой (3×10 мл), отделяют, сушат над MgSO4, фильтруют и выпаривают. ВЭЖХ остатка указывает на превращение в два метилированных диастереомерных соединения [(3R,5S)-5-бифенил-4-илметил-3-метил-1-триэтилсиланилпирролидин-2-он (2-а, R1=ТЭС) и (3R,5S)-5-бифенил-4-илметил-3-метил-1-триэтилсиланилпирролидин-2-он (2-b, R1=ТЭС)].
Пример 26: Бензиловый эфир (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (1-а, R1=Cbz)
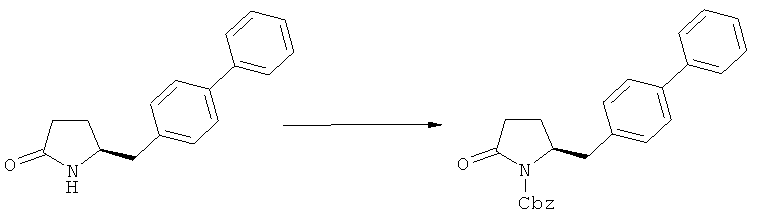
1 г (4 ммоля) (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) растворяют в 10 мл сухого ТГФ и охлаждают до -78°С. После добавления 2,76 мл бутиллития в гексане (1, 59 М) полученный желтый раствор перемешивают при -78°С в течение 30 мин. Затем добавляют 676 мкл (820 мг, 4,8 ммоля) бензилформиата (Cbz-Cl) и продолжают перемешивание при -78°С еще 2 ч. Затем реакцию гасят, добавляя 12 мл насыщенного раствора NH4Cl, с последующим добавлением 10 мл воды, и смесь, затем, экстрагируют 2×40 мл изопропилацетатом. Слои разделяют, и органический слой сушат над MgSO4, фильтруют и выпаривают. Полученный остаток (1,73 г) очищают колоночной хроматографией (дихлорметан: метанол=99,5: 0,5), получая бензиловый эфир (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (1-а, R1=Cbz) в виде грязно-белого твердого вещества. ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,66 (м, 2H); 7,60 (м, 2H); 7,51-7,34 (м, 8 Н); 7,26 (д, J=8,2, 2H); 5,28 (с, 2H); 4,37 (м, 1H); 3,05 (дд, J=3,3, 13,1, 1H); 2,88 (дд, J=9,0, 13,1, 1H); 2,45 (м, 1H); 2,28 (м, 1H); 2,01 (м, 1H); 1,77 (м, 1H). МС (ESI, m/е) 386 [М+Н]+, 788 [2M+NH4 +]. ИК (раствор в CH2Cl2, v/см-1): 3092; 2957; 1756; 1705; 1488; 1396; 1304; 1287; 1231; 1139; 1043; 952;750.
Пример 27: Бензиловый эфир (3R,5S)-6-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=Cbz) и Бензиловый эфир (3S,5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-b, R1=Cbz)
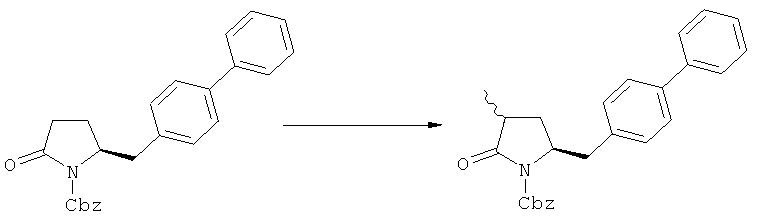
Способ 1
119 мг (0,309 ммоля) бензилового эфира (5}-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (1-а, R1=Cbz) растворяют в 1,5 мл толуола и раствор охлаждают до -78°С. После добавления 665 мкл калия бис(триметилсилил)амида в толуоле (0,57 М) раствор перемешивают при -78°С в течение 20 мин. Затем в течение 5 мин к полученному оранжевому раствору добавляют 117 мкл (155 мг, 1,23 ммоля) диметилсульфата. Полученную смесь перемешивают при -78°С в течение 3 ч, а затем при 0°С в течение 1 ч. После этого реакцию гасят, добавляя 2 мл насыщенного раствора NH4Cl, 2 мл воды, с последующим добавлением 20 мл изопропилацетата. Органическую фазу промывают водой (3×10 мл), сушат над MgSO4, фильтруют и выпаривают. ВЭЖХ и ЖХ-МС остатка указывает на образование двух метилированных диастереомерных соединений [бензилового эфира (3R,5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=Cbz) и бензилового эфира (3R,5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-b, R1=Cbz)], которые неотделимы друг от друга. МС (ESI): 399 [МН]+, 816 [2M+NH4]+.
Способ 2
53 мкл (38 мг, 0,379 ммоля) диизопропиламина растворяют в 1 мл сухого ТГФ и охлаждают до 0°С. После добавления 226 мкл бутиллитя в гексане (1,59 М) раствор перемешивают при 0°с в течение 10 мин. Затем в течение 5 мин добавляют раствор бензилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (1-а, R1=Cbz) в 1 мл ТГФ. Полученную смесь перемешивают при 0°С в течение 2 ч. После этого реакцию гасят, добавляя 2 мл насыщенного раствора NH4Cl, 1 мл воды и 5 мл изопропилацетата. Органическую фазу отделяют и промывают водой (3×5 мл). Затем органический слой сушат над MgSO4, фильтруют и выпаривают. ВЭЖХ и ЖХ-МС остатка указывает на образование двух метилированных диастереомерных соединения [бензилового эфира (3R,5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-а, R1=Cbz) и бензилового эфира (3S,5S)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1-карбоновой кислоты (2-b, R1=Cbz)], которые неотделимы друг от друга.
Пример 28: (S)-5-Бифенил-4-илметил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-он (1-а, R1=СЭМ)
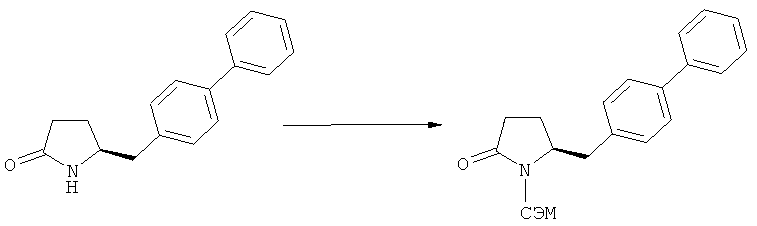
804 мг (3,2 ммоля) (S)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) растворяют в 16 мл сухого ТГФ и охлаждают до -78°С. После добавления 2,21 мл бутиллития в гексане (1,59 М) полученный желтый раствор перемешивают при -78°С в течение 30 мин. Затем добавляют 680 мкл (640 мг, 3,84 ммоля) (2-хлорметоксиэтил)триметилсилана (СЭМ-С1) и продолжают перемешивание при -20°С еще 5 ч. После этого смеси позволяют нагреться до комнатной температуры в течение ночи. Затем реакцию гасят, добавляя 10 мл насыщенного раствора NH4Cl, с последующим добавлением 10 мл воды, и смесь, затем, экстрагируют 2×40 мл изопропилацетатом. Слои разделяют, и органический слой сушат над MgSO4, фильтруют и выпаривают. Полученный остаток (1,33 г) очищают колоночной хроматографией (дихлорметан: триэтиламин=99,5: 0,5 → дихлорметан: метанол: триэтиламин = 98,5: 1: 0,5), получая (5}-5-бифенил-4-илметил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-он (1-а, R1=СЭМ) в виде желтого масла. ЯМР (400 МГц, CDCl3, 5/м.д.): 7,58 (м, 4H); 7,47 (м, 2H); 7,37 (м, 1H); 7,30 (м, 2H); 5,04 (д, J=10,6, 1H); 4,70 (д, J=10.6, 1H); 4,03 (м, 1Н); 3,59 (м, 2H); 3,24 (дд, J=4,2, 13,4, 1H); 2,67 (дд, J=9,1, 13,4, 1H); 2,33 (т, J=8,2, 2H); 2,10 (м, 1H); 1,81 (м, 1H); 1,00 (м, 2H); 0,06 (с, 9 Н). МС (ESI, m/е) 382 [М+Н]+; 763 [2М+Н]+. ИК (раствор в CH2Cl2, v/см-1): 3057; 2951; 1702; 1487; 1414; 1249; 1073; 859; 836; 759; 697.
Пример 29: (3R,5S)-5-Бифенил-4-илметил-3-метил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-он (2-а, R1=СЭМ) и (3S,5S)-5-Бифенил-4-илметил-3-метил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-он (2-b, R1=СЭМ)
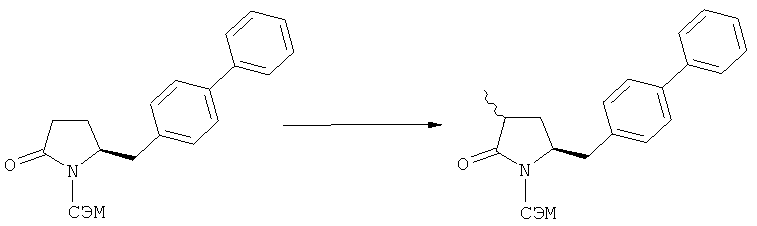
118 мг (0,309 ммоля) (5 г)-5-бифенил-4-илметил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-она (1-а, R1=СЭМ) растворяют в 1,5 мл толуола и раствор охлаждают до 0°С на ледяной бане. После медленного добавления 664 мкл калия бис(триметилсилил)амида в толуоле (0,57 М) раствор перемешивают при 0°С в течение 15 мин. Затем в течение 5 мин к полученному оранжевому раствору добавляют 114 мкл (151 мг, 1,2 ммоля) диметилсульфата. Полученную смесь перемешивают при 0°С в течение 2 ч. После этого реакцию гасят, добавляя 2 мл насыщенного раствора NH4Cl, 2 мл воды и 20 мл изопропилацетата. Органическую фазу отделяют и промывают водой (3×10 мл), сушат над MgSO4, фильтруют и выпаривают. ВЭЖХ остатка указывает на образование двух метилированных диастереомерных соединений [(3R,5S)-5-бифенил-4-илметил-3-метил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-она (2-а, R1=СЭМ) и (3S,5S)-5-бифенил-4-илметил-3-метил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-она (2-b, R1=СЭМ)] и 25% неизрасходованного исходного вещества - (S)-5-бифенил-4-илметил-1-(2-триметилсиланилэтоксиметил)пирролидин-2-она (1-а, R1=СЭМ). По данным ЯМР-анализа соотношение диастереоизомеров составляет 80: 20. (Спектральные данные для смеси) ЯМР (300 МГц, CDCl3, δ/м.д.): 7,58 (м, 4H); 7,47 (м, 2H); 7,35 (м, 1H); 7,25 (м, 2H); 5,00 (м, 1H); 4,63 (м, 1H); 3,96 (м, 1H); 3,55 (м, 2H); 3,15 (м, 1H); 2,60 (м, 1H); 2,30 (м, 1H); 2,05 (м, 1H); 1,65 (м, 1H); 1,16, 1,13 (2×g, с соотношением 2: 8, J=7,2, 7,0, 3H); 0,90 (м, 2H); 0,06 (с, 9 Н). МС (ESI, ш/е) 395 [М+Н]+, 791 [2М+Н]+.
Пример 30: (S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]
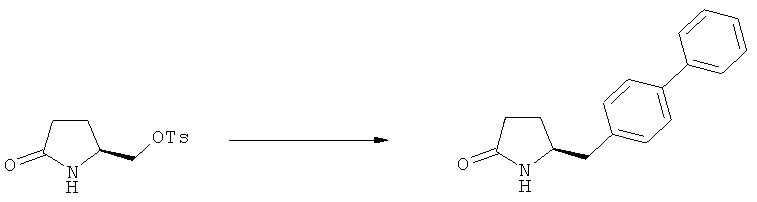
11 г магниевой стружки (452 ммоля) помещают в реактор. Затем добавляют 93,2 г 4-бромбифенила (400 ммоля), растворенного в 380 мл ТГФ. Образование соответствующего реактива Гриньяра начинается после добавления 5% от общего объема вышеупомянутого раствора и после добавления 10 мг 12. Раствор 4-бромбифенила в ТГФ добавляют с такой скоростью, чтобы температура держалась на уровне 50-55°С (в течение добавления: 1 ч). После завершения добавления реакционную смесь кипятя с обратным холодильником в течение 1,5 ч. Затем смесь охлаждают до кт и добавляют 800 мл ТГФ, а после этого - 22 г 1,4-диоксана. Полученную смесь охлаждают на ледяной бане и добавляют 18,0 г CuCN (200 ммоля). После этого реакционную смесь охлаждают до -40°С, а затем в течение 40 мин добавляют раствор 27 г (100 ммоля) (S)-5-оксопирролидин-2илметилового эфира толуол-4-сульфокислоты в 270 мл ТГФ. После завершения добавления раствора тозилата смесь нагревают в течение 0,5 ч до температуры 35°С. Реакционную смесь перемешивают при данной температуре в течение ночи. После этого реакционную смесь охлаждают до 20°С и добавляют 200 мл 25% NH3 (водн.), а затем - 900 мл NH4Cl 29% (водн.). Фазы разделяют, и водную фазу реэкстрагируют 250 мл ТГФ. Объединенные органические фазы дважды промывают 200 мл 15% раствора NaCl и концентрируют под вакуумом, получая 74,5 г сырого продукта. Сырой продукт дважды очищают на силикагеле (элюент: смесь толуол: метанол, 93: 7), получая (S)-5-бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]. Спектральные данные соответствуют данным, приведенным в примере 3.
Пример 31: (S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]
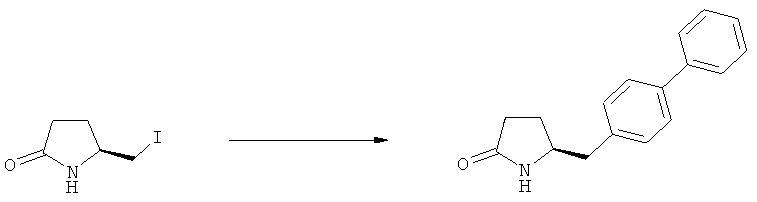
(S)-5-Йодметилпирродин-2-он (225 мг) добавляют к раствору безводного хлорида железа (III) (9,5 мл, 0,1 М в ТГФ). Полученную смесь охлаждают до 0°С. Затем в течение 0,5 ч добавляют бифенилмагний бромид (5 мл, 0,5 М в ТГФ) и ТМЭДА (180 мкл). Полученную смесь перемешивают еще 10 мин. После этого добавляют воду (2 мл). Полученную смесь экстрагируют дихлорметаном (3×5 мл). Объединенные органические экстракты промывают 2 М HCl (2×5 мл), сушат (Na2SO4), а затем концентрируют под вакуумом. При очистке колоночной хроматографией получают (S)-5-бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]. Спектральные данные соответствуют данным, приведенным в примере 3.
Пример 32: (S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]
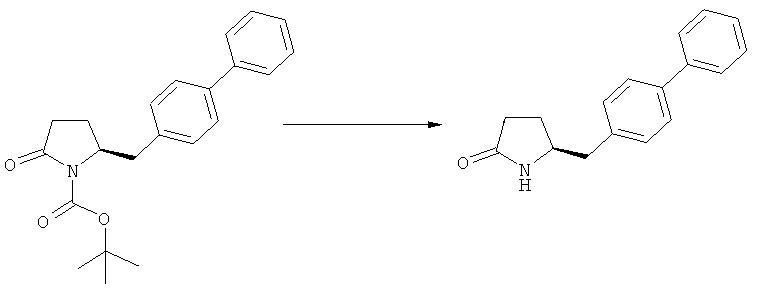
8,9 г трет-Бутилового эфира (S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (1-а, R1=БОК) растворяют в 90 мл метиленхлорида. Затем добавляют 5 мл CF3COOH. Полученную смесь перемешивают при кт в течение 1,5 ч. После этого реакционную смесь концентрируют. К полученному остатку добавляют гептановую фракцию. Продукт кристаллизуют. Полученный продукт растворяю в толуоле, а затем промывают раствором NaHCO3 (водн.). Органический слой выпаривают, получая (S)-5-бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]. Спектральные данные соответствуют данным, приведенным в примере 3.
Пример 33: (S)-1-Бензил-5-бифенил-4-илметилпирролидин-2-он (1-а, R1=Bn)
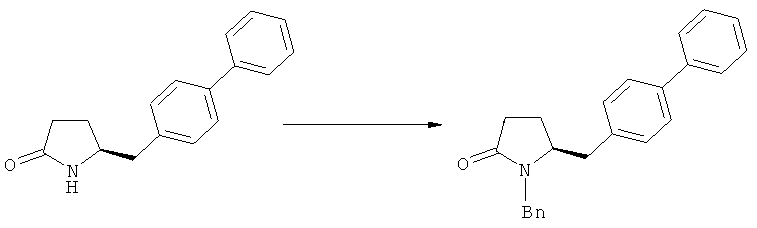
6,1 г (S)-5-бифенил-4-илметилпирролидин-2-она [Ключевой Лактам (1-а, R1=Н)] (24 ммоля) растворяют в 100 мл ТГФ. Затем добавляют 4,5 г бензилбромида. Полученную смесь охлаждают на ледяной бане и добавляют 1,2 г NaH. Ледяную баню удаляют и реакционную смесь перемешивают в течение ночи при кт. После этого добавляют 50 мл Н2О, а затем - толуол, и фазы разделяют. Объединенные органические фазы промывают Н2О и растворитель выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией на 150 г силикагеля, получая (S)-1-бензил-5-бифенил-4-илметилпирролидин-2-он (1-а, R1=Bn). 1Н ЯМР (400 МГц, ДМСО): 1,79 (1Н), 1,93 (1Н), 2,31 (2Н), 2,61 (1Н), 3,03 (1Н), 3,69 (1Н), 4,02 (1Н), 5,10 (1Н), 7,12 (2Н), 7,26 (2Н), 7,33 (4Н), 7,42 (2Н), 7,49 (2Н), 7,56 (2Н).
Пример 34: (3R,5S)-1-Бензил-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Bn)
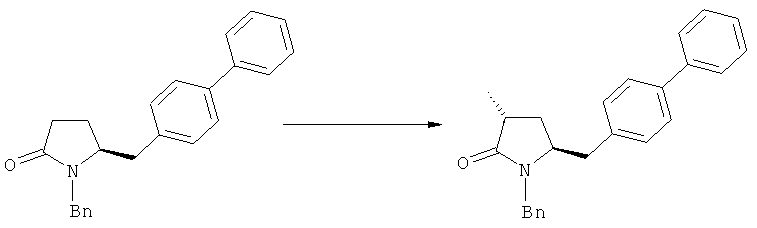
Способ 1
Под атмосферой Ar 15 мл 1М LiN(TMS)2 добавляют к 20 мл ТГФ. Полученный раствор охлаждают до - 78°С. Затем в течение 10 минут при -78°С добавляют раствор 3,6 г (S)-1-бензил-5-бифенил-4-илметилпирролид-2-она (1-а, R1=Bn) в 20 мл ТГФ. Воронку промывают 3 мл ТГФ. Полученную смесь нагревают до 0°С и перемешивают в течение 10 мин при данной температуре. Затем реакционную смесь охлаждают до - 78°С в течение 5 мин добавляют раствор 1,87 г метилйодида в 0,5 мл ТГФ. Воронку промывают 0,5 мл ТГФ. Полученную реакционную смесь перемешивают 18 ч при -78°С. После этого реакцию гасят, добавляя 18 мл насыщенного NH4Cl (водн.). Затем добавляют 36 мл толуола и 9 мл Н2О. Фазы разделяют. Водную фазу реэкстрагируют 10 мл толуола, а объединенные органические фазы дважды промывают 25 мл Н2О. Затем при пониженном давлении выпаривают растворитель, получая сырой продукт. ЯМР-анализ указывает на образование смеси диастереомеров, 77: 33 (2-а, R1=Bn/2-b, R1=Bn). При очистке колоночной хроматографией на силикагеле, с использованием смеси этилацетат: гептановая фракция (4:6), получают (3R,5S)-1-бензил-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Bn). 1Н ЯМР (400 МГц, CDCl3): 1,20 (3H), 1,59 (1Н), 2,07 (1Н), 2,40 (1Н), 2,66 (1Н), 2,99 (1Н), 3,63 (1Н), 4,02 (1Н), 5,11 (1Н), 7,14 (2Н), 7,24 (2Н), 7,33 (4Н), 7,43 (2Н), 7,50 (2Н), 7,56 (2Н).
Способ 2
Взаимодействие осуществляют в соответствии с процедурой, приведенной в способе 1, со следующими изменениями: после добавления метилйодида и промывания воронки ТГФ смесь перемешивают в течение 4 ч при -78°С, вместо 18 ч, указанных в способе 1. Реакцию гасят, и смесь обрабатывают с использованием условий, соответствующих условиям, используемым в способе 1. В данных условиях, по данным ЯМР-анализа, соотношение диастереоизомеров составляет 88:12 (2-а, R1=Bn/2-b, R1=Bn).
Пример 35: Этиловый эфир (Е/Z)-(R)-5-бифеннл-4-ил-4-трет-бутоксикарбониламинопент-2-еновая кислота (8-а, R1=БОК, R2=Н, R5=Et)
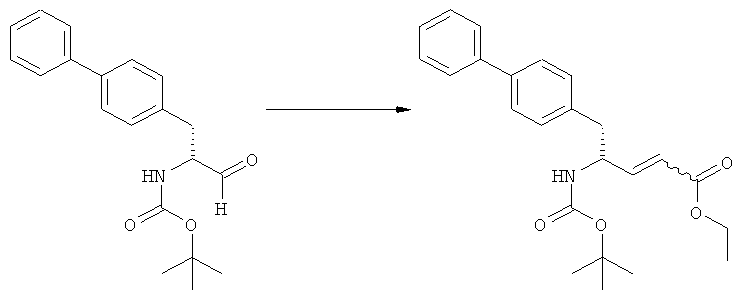
К раствору 159 г трет-бутилового эфира (2-бифенил-4-ил-1-формилэтил)карбаминовой кислоты (7-а, R1=БОК, R2=Н) в изопропилацетате (3,2 л) добавляют этил(трифенилфосфоранилиден)ацетат (199 г). Смесь перемешивают в течение 2 ч. Затем к смеси добавляют раствор лимонной кислоты (79 г) в воде (400 мл). По истечении 1 ч фазы разделяют и органическую фазу концентрируют до суха. Сырую смесь очищают колоночной хроматографией (смесь гептан/этилацетат), получая этиловый эфир (E/Z)-(R)-5-бифенил-4-ил-4-трет-бутоксикарбониламинопент-2-еновой кислоты (8-а, R1=БОК, R2=Н, R5=Et), в виде смеси цис- и транс-изомеров. 1Н ЯМР (ДМСО) основной (транс-изомер по двойной связи): 1,19 (3H, т, СН3); 1,30 (9Н, с, С(СН3)3); 2,74 (1Н, м, 5-CHH); 2,90 (1Н, дд, 5-CHH); 4,11 (2Н, м, CH2CH3); 4,41 (1Н, м, 4-СН); 5,85 (1Н, д, 2-СН); 6,89 (1Н, дд, 3-СН); 7,32 (3H, м, ароматический); 7,43 (2Н, м, ароматический); 7,57 (2Н, м, ароматический); 7,62 (2Н, м, ароматический), m/z: 413 (MNH4 +, 100%), 396 (МН+, 17); 340 (60); 296 (96); 250 (11).
Пример 36: Этиловый эфир (S)-5-бифенил-4-ил-4-трет-бутоксикарбониламинопентановой кислоты [9-а, R1=БОК, R2=Н, R5=Et]
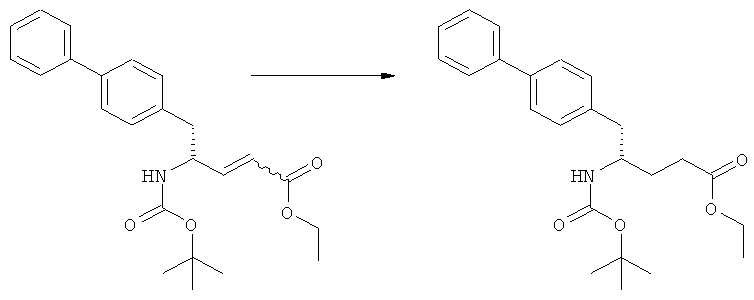
115 г этилового эфира (E/Z)-(R)-5-бифенил-4-ил-4-трет-бутоксикарбониламинопент-2-еновой кислоты (8-а, R1=БОК, R2=Н, R5=Et) растворяют в изопропилацетате (1,2 л). Затем добавляют палладий на угле (10%, загрузка = 11,5 г) и в сосуд подают водород под давлением. По истечении 1 ч сосуд наполняют аргоном, а катализатор удаляют фильтрованием. Оставшийся раствор концентрируют до суха, получая этиловый эфир (S)-5-бифенил-4-ил-4-трет-бутоксикарбониламинопентановой кислоты [9-а, R1=БОК, R2=Н, R5=Et]. 1Н-ЯМР (ДМСО): 1,14 (3H, т, СН3); 1,31 (9Н, с, С(СН3)3); 1,56 (1Н, м, 3-CHH); 1,71 (1Н, м, 3-CHH); 2,28 (2Н, м, 2-СН2); 2,69 (2Н, м, 5-СН2); 3,62 (1H, м, 4-СН); 4,01 (2Н, кв., CH2CH3); 6,75 (1H, д, NH); 7,25 (2Н, д, ароматический); 7,33 (1Н, т, ароматический); 7,43 (2Н, т, ароматический); 7,55 (2Н, т, ароматический); 7,62 (2Н, т, ароматический), m/z: 398 (МН+, 100%); 342 (52); 298 (59).
Пример 37: Гидрохлорид этилового эфира (S)-4-амино-5-бифенил-4-илпентановой кислоты [10-а, R5=Et]
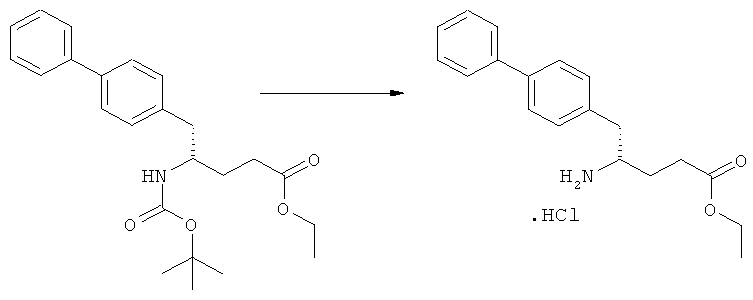
К раствору этилового эфира (5')-5-бифенил-4-ил-4-т/?ет-бутоксикарбониламинопентановой кислоты [9-а, R1=БОК, R2=Н, R5=Et] (115 г) в этаноле (1,1 л) при 70°С добавляют тионилхлорид (32 мл) в течение 45 мин. По истечении 1,5 ч смесь концентрируют до суха. Сырое вещество суспендируют в этилацетате и фильтруют, получая гидрохлорид этилового эфира (S)-4-амино-5-бифенил-4-илпентановой кислоты [10-а, R5=Et]. 1Н ЯМР (ДМСО): 1,08 (3H, д, СН3); 1,73 (2Н, м, 3-СН2); 2,35-2,52 (2Н, м, 2-СН2); 2,79 (1Н, дд, 5-CHH); 2,97 (1Н, дд, CHH); 3,38 (1Н, м, 4-СН); 3,97 (2Н, кв, СН2СН3); 7,30 (3H, м, ароматический); 7,40 (2Н, м, ароматический); 7,58 (4Н, м, ароматический); 8,15 (3H, с, NH3 +). m/z: 298 (МН+, 100%); 281 (4); 235 (3).
Рентгеновская структура полученных кристаллов показана на фиг.11. Монокристалл для данного исследования получают при использовании в качестве растворителя метилового спирта.
Кристаллографические данные [полученные при 100(2) К]
|
Пример 38: (S)-5-Бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]
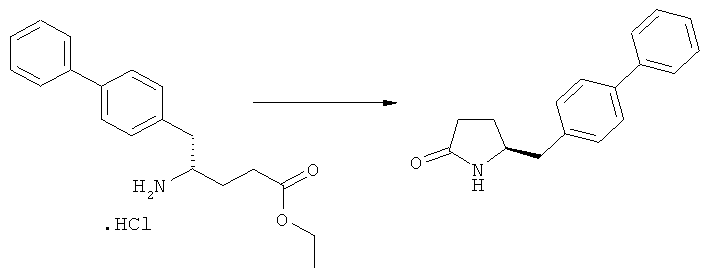
К смеси гидрохлорида этилового эфира (S)-4-амино-5-бифенил-4-илпентановой кислоты [10-а, R5=Et] (86 г) в изопропилацетате (1 л) добавляют триэтиламин (43 г). Полученную смесь перемешивают при температуре около 55°С в течение 1 ч, а затем фильтруют. Фильтрат кипятят с обратным холодильником в течение 24 ч. После этого к смеси добавляют насыщенный раствор хлорида аммония и фазы разделяют. Органический слой концентрируют до суха и кристаллизуют из изопропилацетата, получая (S)-5-бифенил-4-илметилпирролидин-2-он [Ключевой Лактам (1-а, R1=Н)]. Спектральные данные соответствуют данным, приведенным в примере 3.
Пример 39: (2R,4S)-5-Бифенил-4-ил-4-[3-(2-бромэтоксикарбонил)пропиониламино]-2-метилпентановая кислота (3-а, R1=2-бромметиловый эфир 4-оксопентановой кислоты, R2=Н, R3=Et)
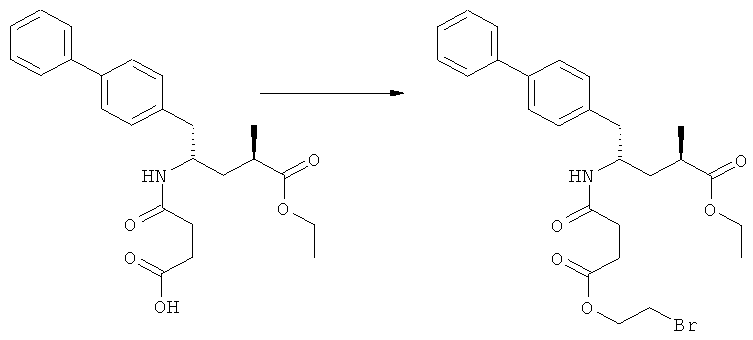
Этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (3-а, R1=4-оксопентановая кислота, R2=Н, R3=Et) растворяют в толуоле (33 мл) при комнатной температуре. Затем добавляют диметилформамид (0,1 мл), а потом - тионилхлорид (2,4 мл). Полученный раствор охлаждают до 0°С и добавляют 2-бромэтанол (0,94 ммоля). Полученную смесь перемешивают при 0°С в течение 2 ч, а затем при комнатной температуре еще 1 ч. После этого добавляют дополнительное количество 2-бромэтанола (0,94 мл) и смесь перемешивают в течение 0,5 ч при комнатной температуре. После этого еще добавляют дополнительное количество 2-бромэтанола (0,94 мл) и смесь перемешивают при комнатной температуре в течение 16 ч. Затем смесь концентрируют под вакуумом, получая сырой продукт. При очистке колоночной хроматографией, с использованием смеси гептан/EtOAc (2: 1), получают (2R,4S)-5-бифенил-4-ил-4-[3-(2-бромэтоксикарбонил)пропиониламино]-2-метилпентановую кислоту (3-а, R1=2-бромметиловый эфир 4-оксопентановой кислоты, R2=Н, R3=Et). 1Н ЯМР (ДМСО): 1,07 (3H), 1,14 (3H), 1,41 (1Н), 1,79 (1Н), 2,37 (2Н), 2,48 (1Н), 2,50 (2Н), 2,71 (2Н), 3,66 (2Н), 3,94 (1Н), 3,99 (2Н), 4,33 (2Н), 7,25 (2Н), 7,35 (1H), 7,46 (2Н), 7,57 (2Н), 7,64 (2Н), 7,78 (1Н).
Пример 40: Этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(2,5-диоксопирролидин-1-ил)-2-метилпентановой кислоты
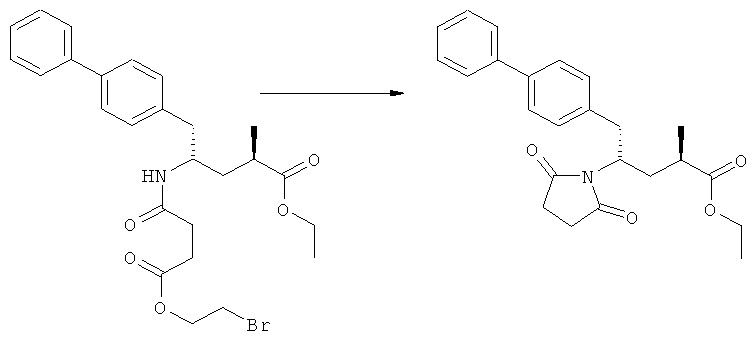
(2R,4S)-5-Бифенил-4-ил-4-[3-(2-бромэтоксикарбонил)пропиониламино]-2-метилпентановую кислоту (3-а, R1=2-бромметиловый эфир 4-оксопентановой кислоты, R2=Н, R3=Et) (6 г, 11,6 ммоля) растворяют в ДМФА (30 мл). Затем добавляют карбонат цезия (8,2 г, 23,2 ммоля) и смесь перемешивают при 50°С в течение 2 ч. После этого добавляют воду (150 мл), а затем - этилацетат (150 мл). Затем фазы разделяют. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Смесь очищают посредством колоночной хроматографией: смесь гептан/EtOAc (2: 1), получая этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(2,5-диоксопирролидин-1-ил)-2-метилпентановой кислоты. 1Н ЯМР (ДМСО): 1,05 (3H), 1,14 (3H), 1,94 (1Н), 2,20 (1Н), 2,39 (1Н). 2,51 (4Н), 2,98 (1Н), 3,12 (1Н), 4,01 (2H), 4,29 (1H), 7,18 (2H), 7,32 (1H), 7,43 (2H), 7,56 (2H), 7,62 (2Н).
Пример 41: (2R,4S)-5-Бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановая кислота (3-а, R1=4-оксопентановая кислота, R2=Н, R3=Н)
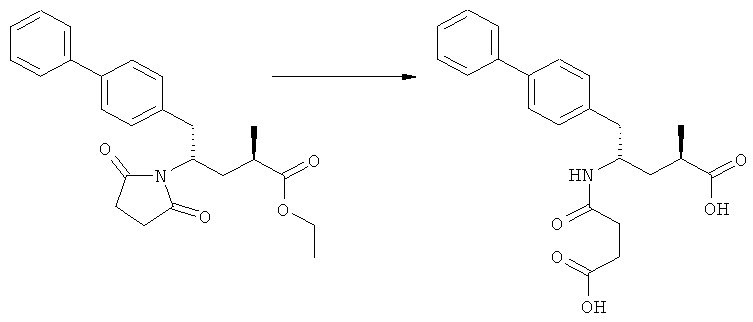
Этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(2,5-диоксопирролидин-1-ил)-2-метилпентановой кислоты (590 мг) добавляют к смеси 1 М NaOH (2,75 мл), ТГФ (9 мл) и этанол (9 мл). Полученную смесь перемешивают в течение 16 ч. Затем смесь разбавляют водой (20 мл) и экстрагируют изопропилацетатом. Водную фазы подкисляют 1 М HCl (5 мл) и экстрагируют изопропилацетатом. Объединенные органические фазы сушат (MgSO4) и концентрируют под вакуумом, получая (2R,4S)-5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановую кислоту (3-а, R1=4-оксопентановая кислота, R2=Н, R3=Н). 1Н ЯМР (ДМСО): 1,05 (2Н), 1,35 (1Н), 1,79 (1Н), 2,29 (2Н), 2,39 (3H), 2,70 (2Н), 3,97 (1Н), 7,25 (2Н), 7,34 (1Н), 7,44 (2Н), 7,56 (2Н), 7,63 (2Н), 7,74 (1Н), 12,01 (2Н).
Пример 42: Этиловый эфир (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=Н, R3=Et)
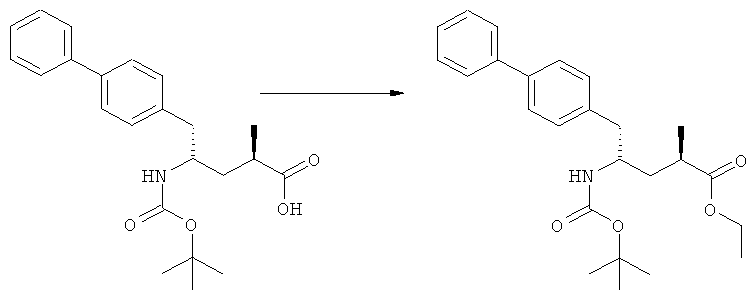
50 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) добавляют к диметилформамиду (80 мл). Затем добавляют карбонат цезия (69 г), а потом - этилйодид (13,6 г), и полученную смесь перемешивают в течение ночи при комнатной температуре. После этого к смеси добавляют воду (200 мл) и смесь экстрагируют изопропилацетатом (2×200 мл). Объединенные органические фазы промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, получая этиловый эфир (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=Н, R3=Et) (56 г). 1Н ЯМР (ДМСО): 1,12 (3H), 1,19 (3H), 1,36 (9Н), 1,53 (1Н), 1,84 (1H), 2,55 (1Н), 2,75 (2Н), 3,74 (1Н), 4,05 (2Н), 6,04 (1Н), 7,23 (2Н), 7,30 (1Н), 7,41 (2Н), 7,50 (2Н), 7,57 (2Н). m/z (ES+): 412 ([МН]+, 100%), 356 (63), 312 (73).
Пример 43: Гидробромид этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et)
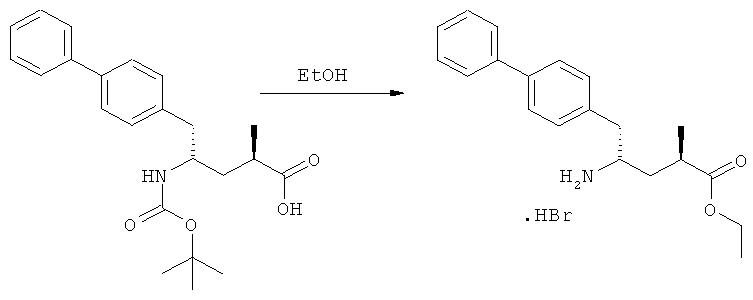
10 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) добавляют к этанолу (100 мл). Полученную смесь нагревают до 65°С, а затем в течение 0,5 ч добавляют 3 мл тионилбромид. Затем смесь перемешивают еще 1 ч. После этого этанол удаляют и добавляют гептан. С использованием гептана проводят азеотропную дистилляцию для удаления остатков этанола. После этого растворитель удаляют под вакуумом, получая гидробромид этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et). 1Н ЯМР (ДМСО): 1,11 (6 Н), 1,61 (1Н), 1,87 (1Н), 2,73 (1Н), 2,84 (1Н), 3,04 (1Н), 3,42 (1Н), 4,01 (2Н), 7,36 (3H), 7,47 (2Н), 7,65 (4Н), 8,03 (3H). m/z (ES+) 312 ([МН]+, 100%).
Пример 44: Гидросульфат этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et)
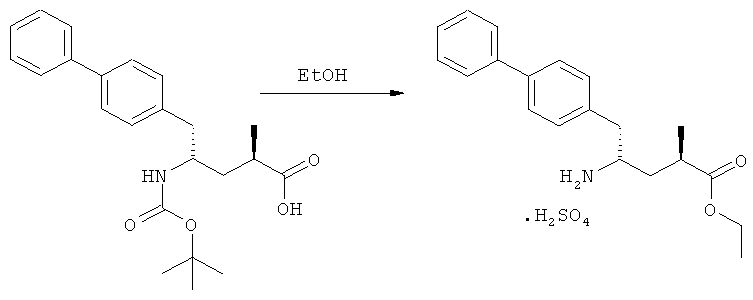
10 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) добавляют к этанолу (100 мл). Полученную смесь нагревают до 65°С, а затем в течение 0,5 ч добавляют 2 мл концентрированной серной кислоты. Затем смесь перемешивают в течение ночи. После этого этанол удаляют и добавляют гептан. С использованием гептана проводят азеотропную дистилляцию для удаления остатков этанола. После этого растворитель удаляют под вакуумом, получая гидросульфат этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et). 1Н ЯМР (ДМСО): 1,12 (6Н), 1,56 (1Н), 1,87 (1Н), 2,67 (1Н), 2,78 (1Н), 2,98 (1Н), 3,76 (2Н), 7,34 (3H), 7,47 (2Н), 7,64 (4Н), 8,57 (3H). m/z (ES+) 312 ([МН]+, 100.
Пример 45: Гидрохлорид (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н)
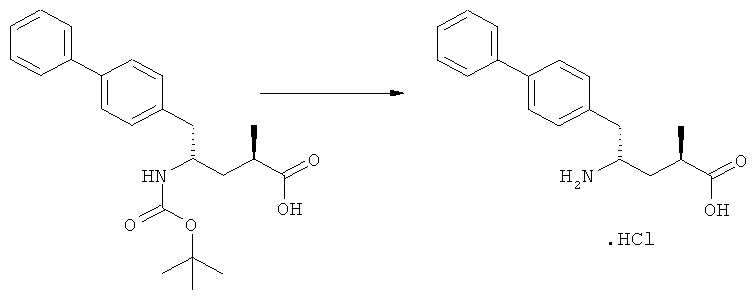
Смесь 37% соляной кислоты (85 мл), этилацетата (100 мл) и воды (100 мл) нагревают на масляной бане при 130°С. Затем добавляют 50 г (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилпентановой кислоты (3-а, R1=БОК, R2=R3=Н) в этилацетате (100 мл) и смесь перемешивают в течение 45°С, а затем еще 1 ч. После этого смеь охлаждают до 0°С и твердое вещество собирают фильтрованием, получая гидрохлорид (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=R3=Н). Спектральные данные соответствуют данным, приведенным в примере 7.
Пример 46: Этиловый эфир (2R,4S)-5-Бифенил-4-ил-4-(2,2-диметилпропиониламино)-2-метилпентановой кислоты (3-а, R1=Piv, R2=Н, R3=Et)
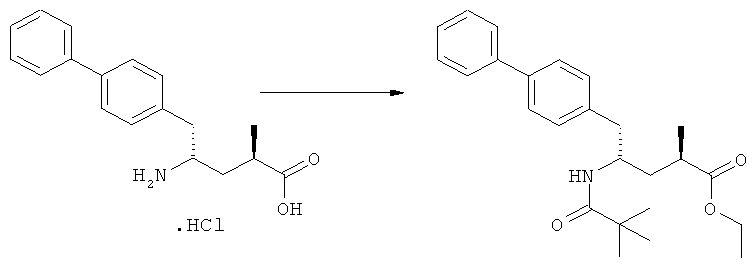
10 г гидрохлорида этилового эфира (2R, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-а, R1=R2=Н, R3=Et) в изопропилацетате (50 мл) добавляют к смеси пивалоилхлорида (4,1 мл) в изопропилацетате (50 мл). Полученную смесь перемешимивают в течение 40 мин при комнатной температуре. После этого в течение 1 ч добавляют триэтиламин (10,3 мл) в изопропилацетате (30 мл). Полученную смесь перемешивают в течение 16 ч. Затем добавляют лимонную кислоту (7,5 г), растворенную в воде (30 мл), и фазы разделяют. Органическую фазу промывают дважды водой (30 мл) и концентрируют под вакуумом, получая этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(2,2-диметилпропиониламино)-2-метилпентановой кислоты (3-а, R1=Piv, R2=Н, R3=Et). ЯМР (ДМСО): 1,05 (9Н), 1,09 (3H), 1,15 (3H), 1,54 (1Н), 1,78 (1Н), 2,48 (1Н), 2,72 (2Н), 3,97 (1Н), 4,00 (2Н), 7,15 (1Н), 7,25 (2Н), 7,34 (1Н), 7,45 (2Н), 7,55 (2Н), 7,62 (2Н).
Пример 47: (3S, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-b, R1=Н)
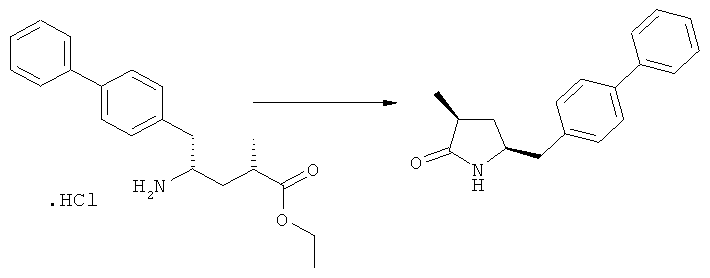
К смеси гидрохлорида этилового эфира (25, 4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (3-b, R1=R2=Н, R3=Et) [диастереомерное соотношение = 9:1 [(2S, 4S): (2R, 4S)]] в изопропилацетате (10 мл) добавляют триэтиламина (418 мг). Смесь перемешивают при 55°С в течение 1 ч, а затем фильтруют. Фильтрат кипятят с обратным холодильником в течение 24 ч. Затем к смеси добавляют насыщенный раствор хлорида аммония и фазы разделяют. Органический слой концентрируют до суха. Остаток очищают колоночной хроматографией, элюлируемой вначале смесью ИПА/гептан (2: 1), а затем - смесью (1:1) ИПА/гептан, получая (3S, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-b, R1=Н) и (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-b, R1=Н). Соотношение диастереоизомеров, по данным 1Н ЯМР-анализа, составляет 9:1 [(3S, 5S): (3R, 5S)]. 1Н ЯМР (CDCl3) для (2-b, R1=H): 1,15 (3H), 1,38 (1Н), 2,42 (2Н), 2,63 (1Н), 2,82 (1Н), 3,75 (1H), 5,51 (1Н), 7,17 (2Н), 7,28 (1Н), 7,37 (2Н), 7,49 (4Н). Спектральные данные для (2-а, R1=H) соответствуют данным, приведенным в примере 6.
***
Пример 48: (3S, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-b, R1 = пивалоил)
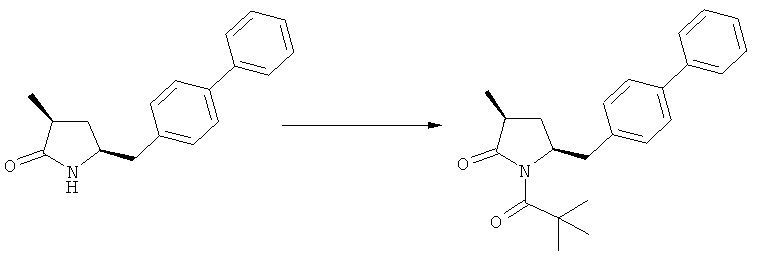
146 мг (3S, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-она (2-b, R1=Н) [смесь диастереоизомеров (9:1) [(2S, 4S): (2R, 4S)]] добавляют к 10 мл ТГФ. Полученную смесь охлаждают до -78°С и добавляют 381 мкл бутиллития (1,59 М в гексане) После этого добавляют 81 млк пивалоилхлорида. По истечении 4 ч смесь нагревают до комнатной температуры. После этого реакцию гасят, добавляя насыщенный раствор хлорида аммония и изопропилацетат. Фазы разделяют, и органическую фазу сушат (MgSO4), а затем концентрируют под вакуумом. Остаток очищают колоночной хроматографией, элюлируемой смесью изопропилацетат/гексан (от 3:1 до 1:0), получая (3S, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-b, R1 = пивалоил) и (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил). Соотношение диастереоизомеров, по данным 1Н ЯМР-анализа, составляет 4: 1 [(3S, 5S): (3R, 5S)]. 1Н ЯМР (CDCl3) для (2-b, R1=Piv): 1,09, 1,14 (3H), 1,30 (9Н), 1,34 (1H), 2,01-2,66 (3H), 3,03, 3,27 (1Н), 4,31, 4,50 (1Н), 7,21-7,53 (9Н). Диастереомерное соотношение определяют, суммируя пары сигналов при [3,03 м.д. (2-а, R1=Piv) и 3,27 м.д. (2-b, R1=Piv)] или при [4,31 м.д. (2-b, R1=Piv) и 4,50 м.д. (2-а, R1=Piv)], исходя из 1Н ЯМР-спектра. Спектральные данные для (2-а, R1=Piv) соответствуют данным, приведенным в примере 5.
Пример 49: 1-Бензил-5-бифенил-4-илметил-5-гидроксипирролидин-2-он
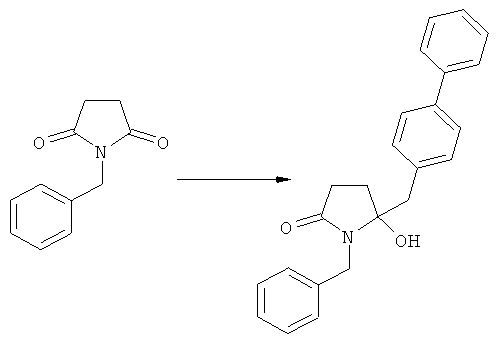
9,5 г N-бензилсукцинимида добавляют к 120 мл ТГФ и смесь охлаждают до -78°С. Затем добавляют раствор 4-метилбифенилмагний хлорида в ТГФ (1,3 экв.). После этого смесь перемешивают при -78°С в течение 2 ч. Затем смесь нагревают до 10°С и добавляют 100 мл насыщенного раствора хлорида аммония. Фазы разделяют, и водную фазу экстрагируют толуолом. Объединенные органические фазы промывают водой, а затем рассолом, и коцентрируют под вакуумом. Сырое вещество кристаллизуют из толуола, получая 1-бензил-5-бифенил-4-илметил-5-гидроксипирролидин-2-он. 1Н ЯМР (ДМСО): 1,72 (1Н), 1,91 (1Н), 2,27 (2Н), 2,72 (1Н), 2,97 (2Н), 4,40 (1Н), 4,55 (1Н), 7,21-7,66 (14Н).
Пример 50: 1-Бензил-5-[1-бифенил-4-илмет-(E/Z)-илиден]пирролидин-2-он
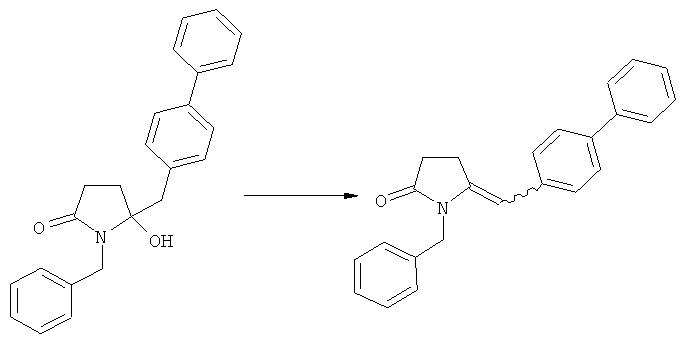
5,5 г 1-бензил-5-бифенил-4-илметил-5-гидроксипирролидин-2-она добавляют к дихлорметану (55 мл) при комнатной температуре. Затем добавляют 22 мл трифторуксусной кислоты и полученной смеси позволяют нагреваться при перемешивании в течение ночи. После этого смесь фильтруют и концентрируют под вакуумом. Затем к остатку добавляют толуол (100 мл) и насыщенный раствор гидрокарбоната натрия (50 мл). Фазы разделяют, и органическую фазу концентрируют под вакуумом. Остаток добавляют к 30 мл метанола и кипятят с обратным холодильником. После этого смесь охлаждают до комнатной температуры, фильтруют и концентрируют под вакуумом, получая 1-бензил-5-[1-бифенил-4-илмет-(E/Z)-илиден]пирролидин-2-он. 1Н ЯМР (ДМСО): (E-изомер): 2,67 (2Н), 3,07 (2Н), 4,80 (2Н), 5,83 (1Н), 7,26-7,36 (8Н), 7,44 (2Н), 7,59 (2Н), 7,63 (2Н). m/е (ES+): 340 ([МН]+, 100%), 262 (28), 249 (63).
Пример 51: (R)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-1,5-дигидропиррол-2-он
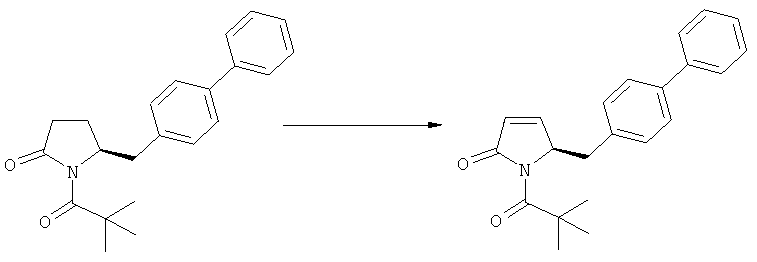
1,68 г (5)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1, R1 = пивалоил) добавляют к 10 мл толуола. Полученную смесь охлаждают до -15°С, а затем добавляют 5,5 мл литий
бис(триметилсилил)амида (1 M в ТГФ). По истечении 1 ч добавляют смесь 1,3 г фенилселенилбромида в 10 мл толуола. По истечении еще 30 мин добавляют 100 мл воды. Фазы разделяют, и органическую фазу концентрируют под вакуумом. Остаток помещают в 25 мл этилацетата, и затем к нему добавляют 5,1 мл пероксида водорода (37%) при комнатной температуре. По истечении 1 ч фазы разделяют и органическую фазу промывают насыщенным раствором гидрокарбоната натрия, а затем сушат (MgSO4). Смесь концентрируют под вакуумом и очищают колоночной хроматографией, элюлируемой смесью гептан/этилацетат (5:1), получая (R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-1,5-дигидропиррол-2-он. 1Н ЯМР (CDCl3): 1,28 (9Н), 2,70 (1Н), 3,30 (1Н), 4,97 (1Н), 5,89 (1Н), 7,06 (2Н), 7,19 (2Н), 7,31 (2Н), 7,41 (4Н).
Пример 52: Этиловый эфира (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me) и Этиловый эфира (3S, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me)
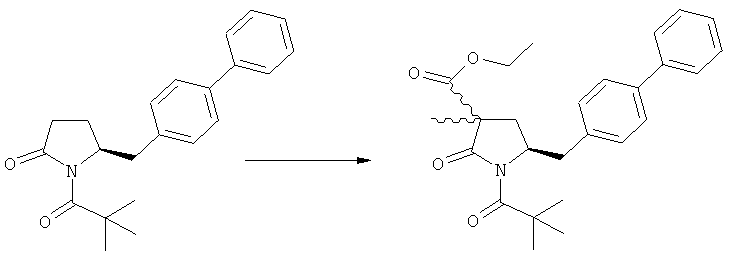
2,0 г (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1, R1 = пивалоил) в 7,5 мл толуола добавляют к 26,2 мл калия бис(триметилсилил)амида (0,5 М в толуоле) при -10°С. По истечении 1 ч добавляют 569 мкл этилхлорформиата и смесь перемешивают в течение 1,5 ч при температурах от -5°С до 0°С. После этого добавляют 733 мкл диметилсульфата и смесь перемешивают при комнатной температуре в течение 1,5 ч. Затем добавляют 8 мл насыщенного раствора хлорида аммония, вместе с 10 мл воды и 20 мл этилацетата. Водную фазу экстрагируют этилацетатом, а объединенные органические фазы промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. По данным НЯМР-анализа соотношение диастереоизомеров составляет 62:38. Остаток очищают колоночной хроматографией, элюлируемой смесью этилацетат/гептан (1:12), получая этиловый эфир (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me) и этиловый эфир (3S, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R1 1=Ме). Фракции, содержащие оба диастереоизомера, объединяют. Под данным НЯМР-анализа соотношение диастереоизомеров составляет 62:38. 1Н ЯМР (CDCl3) Основной диастереомер: 1,11 (3H), 1,17 (9Н), 1,23 (3H), 1,62 (1Н), 2,26 (2Н), 2,95 (1Н), 4,07 (2Н), 4,30 (1Н), 7,03-7,37 (9Н). 1Н ЯМР (CDCl3) Неосновной диастереомер: 0,99 (3H), 1,16 (9Н), 1,25 (3H), 1,51 (1Н), 2,20 (2Н), 3,13 (1Н), 3,91 (2Н), 4,34 (1Н), 7,03-7,37 (9Н). Данные для смеси диастереоизомеров.
Пример 53: 1-трет-Бутиловый эфир 3-этиловый эфир (3R, 5R)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1,3-дикарбоновой кислоты
(R1=БОК; R10=OEt; R11=Me) и 1-трет-Бутиловый эфир 3-этиловый эфир (3S, 5R)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-1,3-дикарбоновой кислоты (R1=BOK; R10=OEt; R11=Me)
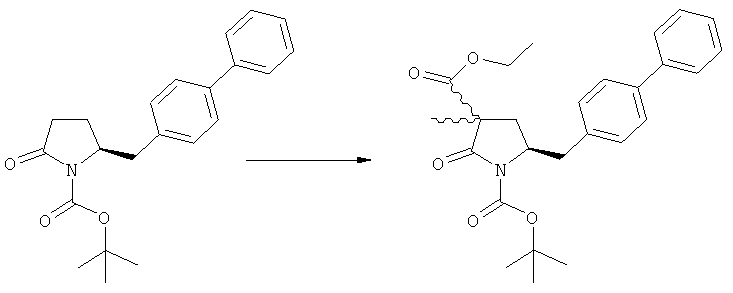
2,0 г (S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)пирролидин-2-она (1, R1 = пивалоил) в 7,5 мл толуола добавляют к 25 мл калия бис(триметилсилил)амида (0,5 М в толуоле) при -10°С. По истечении 1 ч добавляют 542 мкл этилхлорформиата и смесь перемешивают в течение 1,5 ч при температурах от -5°С до 0°С.После этого добавляют 733 мкл диметилсульфата и смесь перемешивают при комнатной температуре в течение 1,5 ч. Затем добавляют 8 мл насыщенного раствора хлорида аммония, вместе с 10 мл воды и 20 мл этилацетата. Водную фазу экстрагируют этилацетатом, а объединенные органические фазы промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. По данным НЯМР-анализа соотношение диастереоизомеров составляет 55:45. Остаток очищают колоночной хроматографией, элюлируемой смесью этилацетат/гептан (1:6). Очищенные образцы каждого полученного диастереоизомера анализируют. 1Н ЯМР (CDCl3) Основной диастереоизомер (Rf 0,13): 1,28 (3H), 1,41 (2Н), 1,54 (9Н), 1,72 (1Н), 2,43 (1Н), 2,59 (1Н), 3,28 (1Н), 4,16 (1H), 4,19 (2Н), 7,22 (3H), 7,37 (2Н), 7,47 (4Н). 1Н ЯМР (CDCl3) Неосновной диастереоизомер (Rf 0,17): 1,15 (3H), 1,36 (3H), 1,54 (9Н), 1,58 (1Н), 2,36 (1Н), 2,59 (1Н), 3,41 (1Н), 4,04 (2Н), 4,31 (1Н), 7,16 (2Н), 7,25 (1Н), 7,34 (2Н), 7,46 (4Н).
Пример 54: (3R, 5R)-5-Бифенил-4-илметил-3-метил-2-оксопирролидин-3-карбоновая кислота (R1=H; R10=OH; R11=Me) и (3S, 5R)-5-Бифенил-4-илметил-3-метил-2-оксопирролидин-3-карбоновая кислота (R1=H; R10=OH; R11=Me)
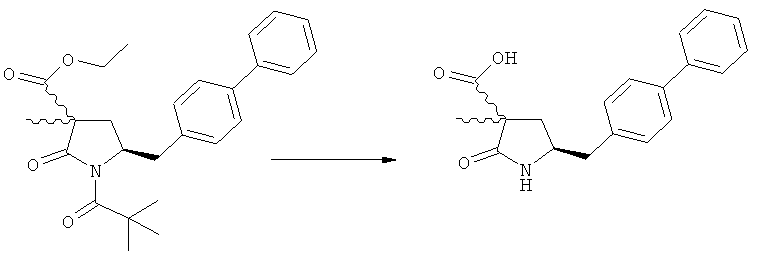
252 мг этилового эфира (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me) [смесь С3-изомеров, 62:38] добавляют к 1 мл ацетонитрила при 0°С. Затем добавляют 0,3 мл 3М раствора гидроксида натрия и смесь перемешивают при комнатной температуре в течение 20 ч. После этого добавляют две дополнительные порции (по 50 мл) ЗМ гидроксида натрия. Перемешивание продолжают еще 2 ч. Затем смесь концентрируют под вакуумом. К остатку добавляют 2,5 мл воды и дважды экстрагируют 1 мл толуола. После этого к водной фазе добавляют 5 мл этилацетата, которую затем охлаждают до 0°С. Затем добавляют 500 мкл 2М соляной кислоты и фазы разделяют. Водную фазу экстрагируют этилацетатом. Объединенные органические фазы собирают, сушат (MgSO4) и концентрируют под вакуумом, получая (3R, 5R)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-3-карбоновую кислоту (R1=H; R10=OH; R11=Me) и (35, 5Д)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-3-карбоновую кислоту (R1=H; R10=OH; R11=Ме). По данным Н-ЯМР-анализа получают смесь С3-стереоизомеров (66: 34). Идентификация основного изомера не осуществлялась. 1Н-ЯМР (ДМСО) [смесь стереоизомеров]: 1,21 (3H), 1,59, 1,84, 2,20, 2,29 (общий 2Н), 2,66 (1Н), 2,93 (1Н), 3,81 (1Н), 7,31 (3H), 7,44 (2Н), 7,60 (4Н), 8,10, 8,15 (общий 1Н), 12,58 (1Н).
Пример 55: (3R, 5R)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновая кислота (R1=Piv; R10=OH; R11=Me) и (3S, 5R)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновая кислота (R1=Piv; R10=OH; R11=Me)
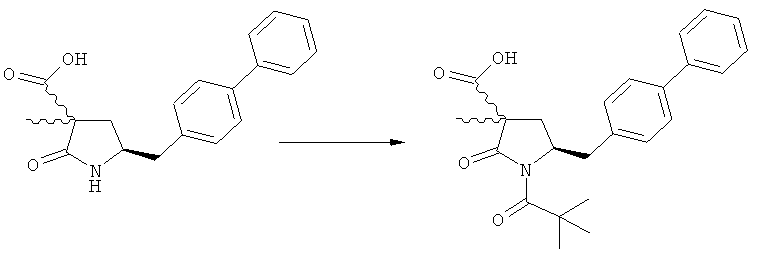
44 мг (3R/S, 5R)-5-бифенил-4-илметил-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=H; R10=OH; R11=Me) [66: 34, смесь С3-стереоизомеров] добавляют к 10 мл толуола. Затем добавляют 119 мкл триэтиламина и полученную смесь нагревают до 60°С. Затем добавляют 52 мкл пивалоилхлорида и смесь перемешивают 4 ч. После этого смесь охлаждают до комнатной температуры. Добавляют 250 мг лимонной кислоты в воде (5 мл) и фазы разделяют.Органическую фазу промывают водой, сушат (MgSO4) и концентрируют под вакуумом, получая (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновую кислоту (R1=Piv; R10=OH; R11=Me) и (3S, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновую кислоту (R1=Piv; R10=OH; R1 1=Ме). По данным Н-ЯМР-анализа получают смесь С3-стереоизомеров (65: 35). Идентификация основного изомера не осуществлялась. 1Н-ЯМР (ДМСО) [смесь стереоизомеров]: 1,19 (9Н), 1,21 (3H), 1,73-1,91 (1Н), 2,23 (1Н), 2,50 (2Н), 3,12-3,33 (1H), 4,45-4,60 (1Н), 7,19 (1H), 7,27 (2Н), 7,37 (2Н), 7,46 (4Н).
Пример 56: (31?, 55)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил) и (3S, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-b, R1 = пивалоил)
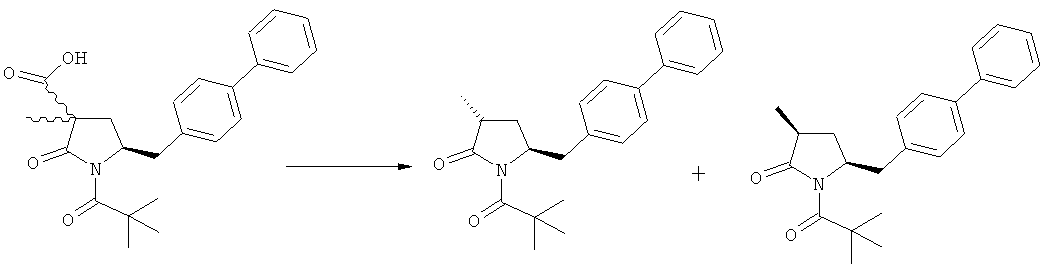
63 мг (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me) [смесь С3-изомеров, 65:35] добавляют к 25 мл толуола. Полученную смесь кипятят с обратным холодильником и перемешивают в течение 16 ч. Затем смесь охлаждают до комнатной температуры и последовательно промывают 10 мл водного гидрокарбоната натрия, рассолом и водой. Органическую фазу сушат (MgSO/t) и концентрируют под вакуумом, получая (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил) и (3S, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-b, R1 = пивалоил), в виде смеси диастереомеров (55:45), соотвественно, в соотвествии с 1Н ЯМР-спектром.
Пример 57: (3R, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Н) и (35, 5S)-5-Бифенил-4-илметил-3-метилпирролидин-2-он (2-b, R1=Н)
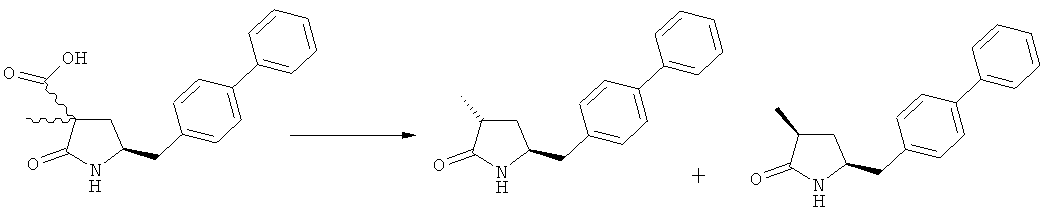
15 мг (3R, 5R)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метил-2-оксопирролидин-3-карбоновой кислоты (R1=Piv; R10=OEt; R11=Me) [смесь С3-изомеров, 66:34] добавляют к 25 мл толуола. Полученную смесь кипятят с обратным холодильником и перемешивают в течение 16 ч. Затем смесь охлаждают до комнатной температуры и последовательно промывают 10 мл водного гидрокарбоната натрия, рассолом и водой. Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, получая (3R, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-а, R1=Н) и (35, 5S)-5-бифенил-4-илметил-3-метилпирролидин-2-он (2-b, R1=Н), в виде смеси диастереомеров (29: 79), соотвественно, в соотвествии с 1Н ЯМР-спектром.
Пример 58: 1-Бензил-5-[1-бифенил-4-илмет-(E/Z)-илиден]-1,5-дигидропиррол-2-он
3,55 г N-Бензилмалеимида добавляют к 35 мл ТГФи смесь охлаждают до 0°С. Затем в течение 30 мин добавляют раствор 4-метилбифенилмагний хлорида в ТГФ (5,6 г, 0,69 М). После этого смесь перемешивают при комнатной температуре в течение 1,5 ч. Затем добавляют насыщенный раствор хлорида аммония (50 мл) и смесь перемешивают в течение 20 мин. Фазы разделяют, и водную фазу экстрагируют толуолом. Объединенные органические фазы промывают водой и рассолом, а затем концентрируют под вакуумом. Осаток помещают в дихлорметан (35 мл). После этого в течение 5 мин добавляют трифторуксусную кислоту, и смесь перемешивают в течение 3 ч при комнатной температуре. Затем смесь концентрируют под вакуумом. Добавляют толуол (50 мл) и насыщенный раствор гидрокарбоната натрия (50 мл), и фазы разделяют. Органическую фазу промывают водой, а затем концентрируют под вакуумом. К остатку добавляют метанол (2 мл) и кипятят с обратным холодильником и осуществляют горячее фильтрование. Фильтрат концентрируют под вакуумом, получая 1-бензил-5-[1-бифенил-4-илмет-(E/Z)-илиден]-1,5-дигидропиррол-2-он. 1Н ЯМР (ДМСО): 4,96 (2Н), 6,50 (1Н), 6,70 (1H), 7,26-7,76 (15 Н).
Пример 59: 1-Бензил-5-бифенил-4-илметилпирролидин-2-он
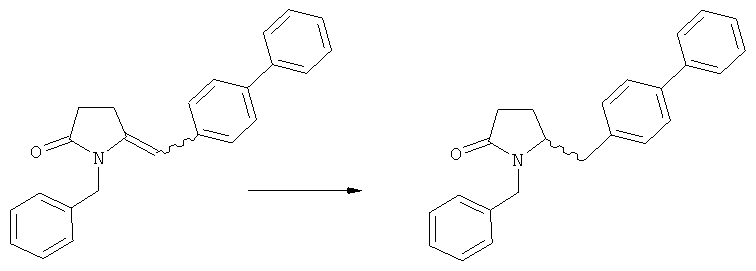
300 мг 1-бензил-5-[1-бифенил-4-илмет-(Е/Z)-илиден]-1,5-дигидропиррол-2-она добавляют к этанолу (3 мл) при комнатной температуре. Затем добавляют 10% Pd/C (50% влажн.) (30 мг) и в сосуд подают водородный газ. Полученную смесь перемешивают в течение 72 ч при комнатной температуре. Затем фильтрованием удаляют катализатор, а фильтрат концентрируют под вакуумом, получая 1-бензил-5-бифенил-4-илметилпирролидин-2-он. 1Н ЯМР (ДМСО): 1,74 (1Н), 1,86 (1Н), 2,16 (2Н), 2,63 (1Н), 3,02 (1Н), 3,63 (1Н), 4,21 (1Н), 4,82 (1Н), 7,23 (2Н), 7,30 (3H), 7,35 (3H), 7,45 (2Н), 7,57 (2Н), 7,64 (2Н).
Пример 60: 1-Бензил-5-бифенил-4-илметилпирролидин-2-он
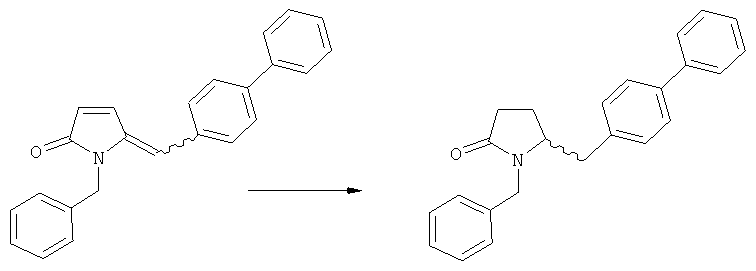
50 мг 1-бензил-5-[1-бифенил-4-илмет-(E/Z)-илиден]-1,5-дигидропиррол-2-она добавляют к метанолу (1,5 мл) при комнатной температуре. Затем добавляют 10% Pd/C (50% влажн.) (15 мг) и в сосуд подают водородный газ. Полученную смесь перемешивают в течение 1 ч при комнатной температуре. Затем фильтрованием удаляют катализатор, а фильтрат концентрируют под вакуумом, получая 1-бензил-5-бифенил-4-илметилпирролидин-2-он. Спектральные данные соответствуют данным, приведенным в примере 59.
Пример 61: (3R, 5S)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил) и (35, 55)-5-Бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил)
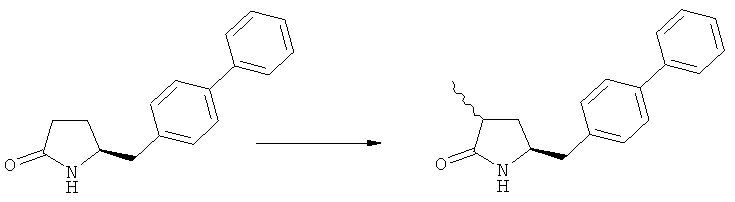
250 мг (5)-5-бифенил-4-илметилпирролидин-2-она (1-а, R1=Н) добавляют к ТГФ (4 мл). Полученную смесь охлаждают до -78°С. Затем добавляют 1,68 мл втор-бутиллития (1,3 М в циклогексане) и полученную смесь перемешивают в течение 0,5 ч. После этого добавляют 68 мкл метилйодида и смесь перемешивают в течение 2 ч при -78°С.Затем добавляют насыщенный раствор аммония хлорида (5 мл), воды (3 мл) и этилацетата (5 мл), и смесь нагревают до комнатной температуры. Фазы разделяют, и органическую фазу промывают рассолом, отделяют, сушат (MgSCu) и концентрируют под вакуумом, получая (3R, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил) и (3S, 5S)-5-бифенил-4-илметил-1-(2,2-диметилпропионил)-3-метилпирролидин-2-он (2-а, R1 = пивалоил), в виде смеси (2-а) и (2-b), при соотношении 20: 80, по данным 1Н ЯМР. Спектральные данные для (2-а, R1=H) соответствуют данным, приведенным в примере 6. Спектральные данные для (2-b, R1=H) соответствуют данным, приведенным в примере 47.
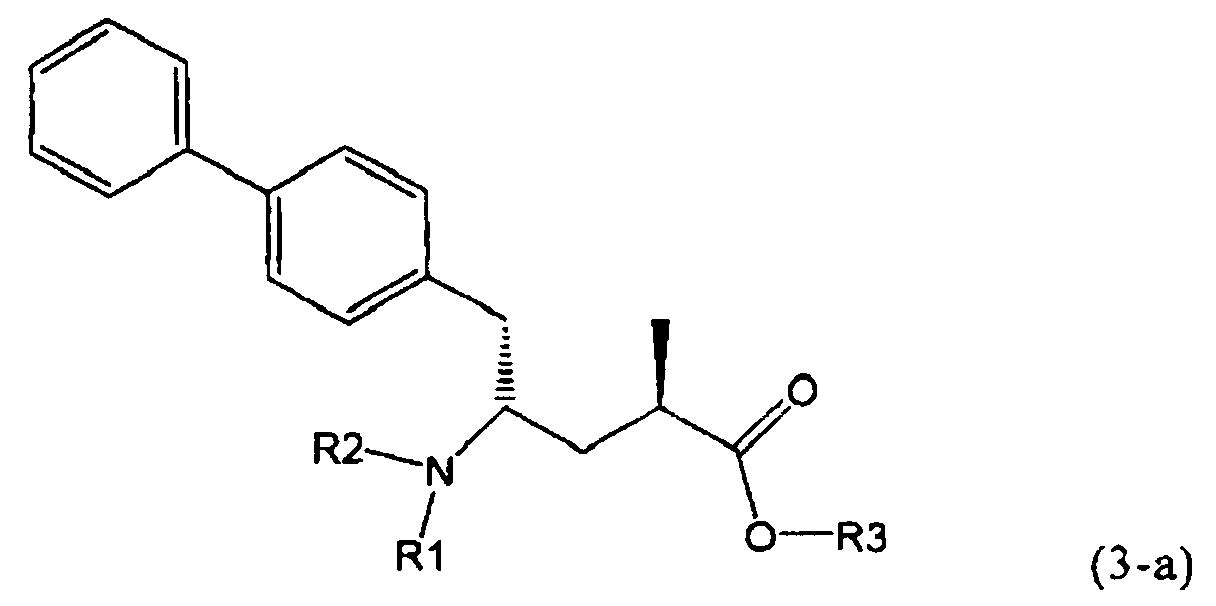
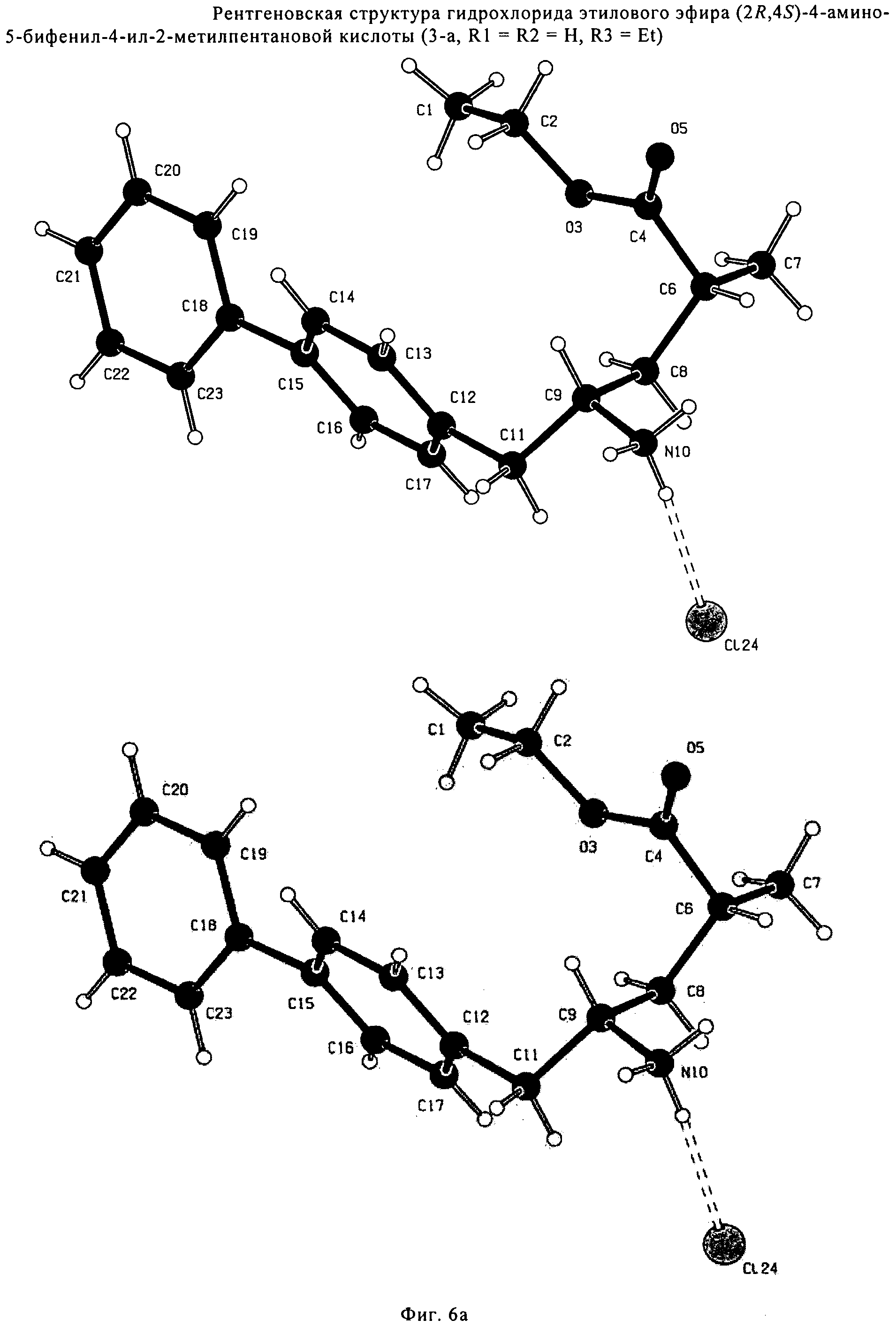
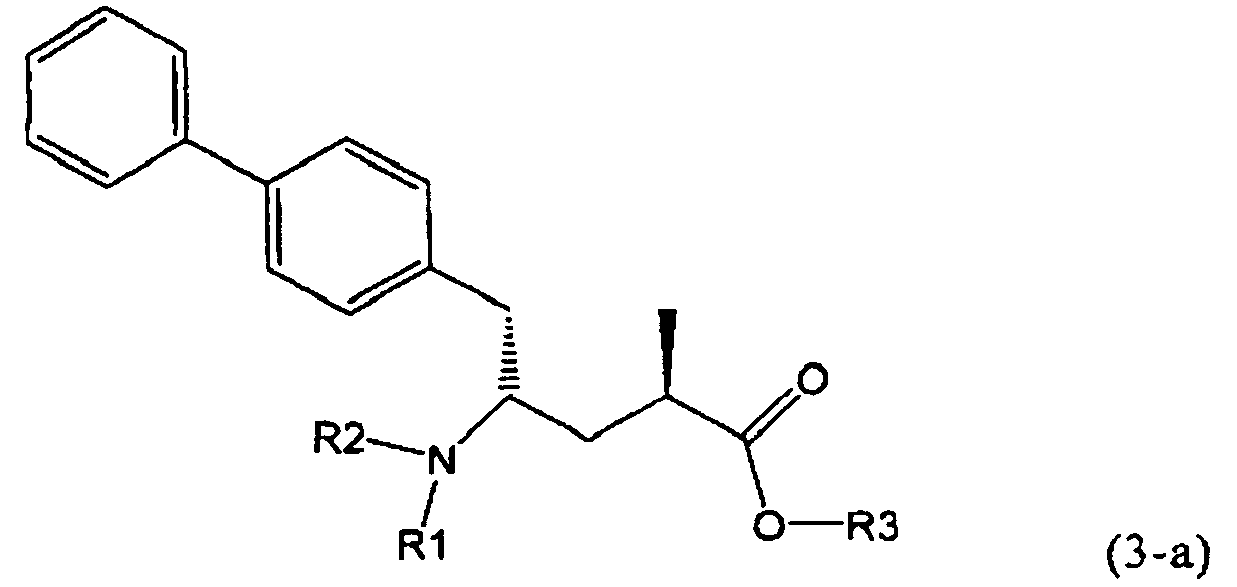
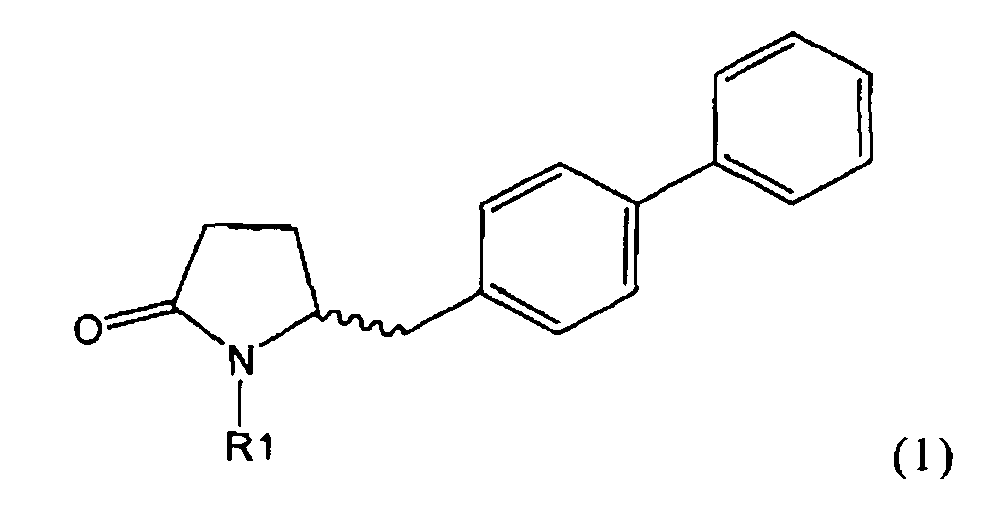
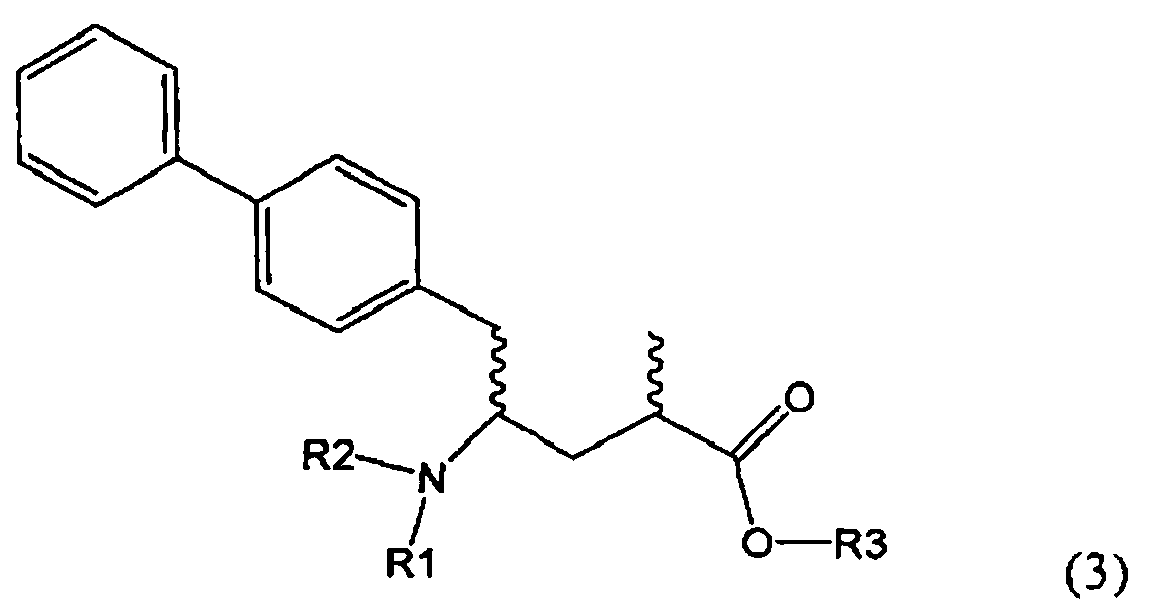
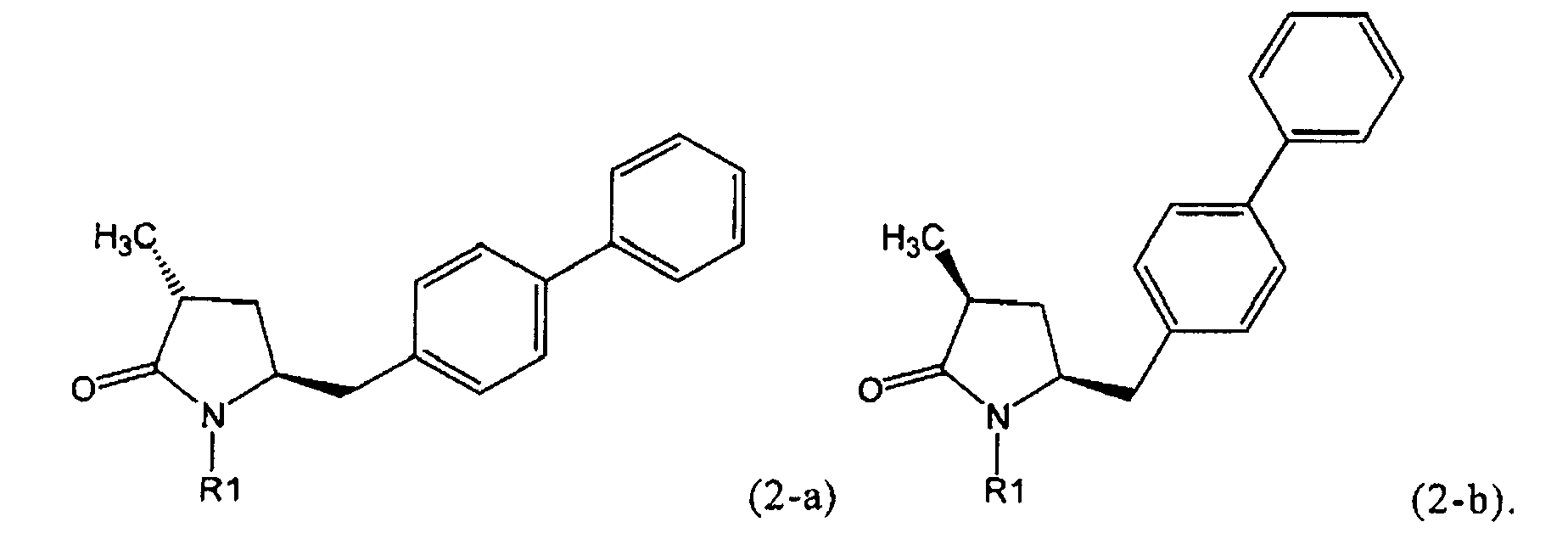
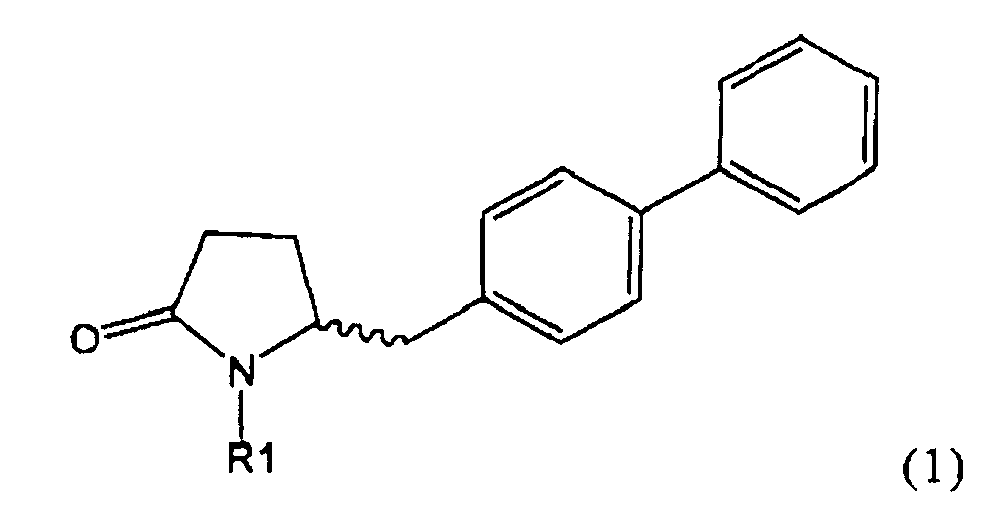
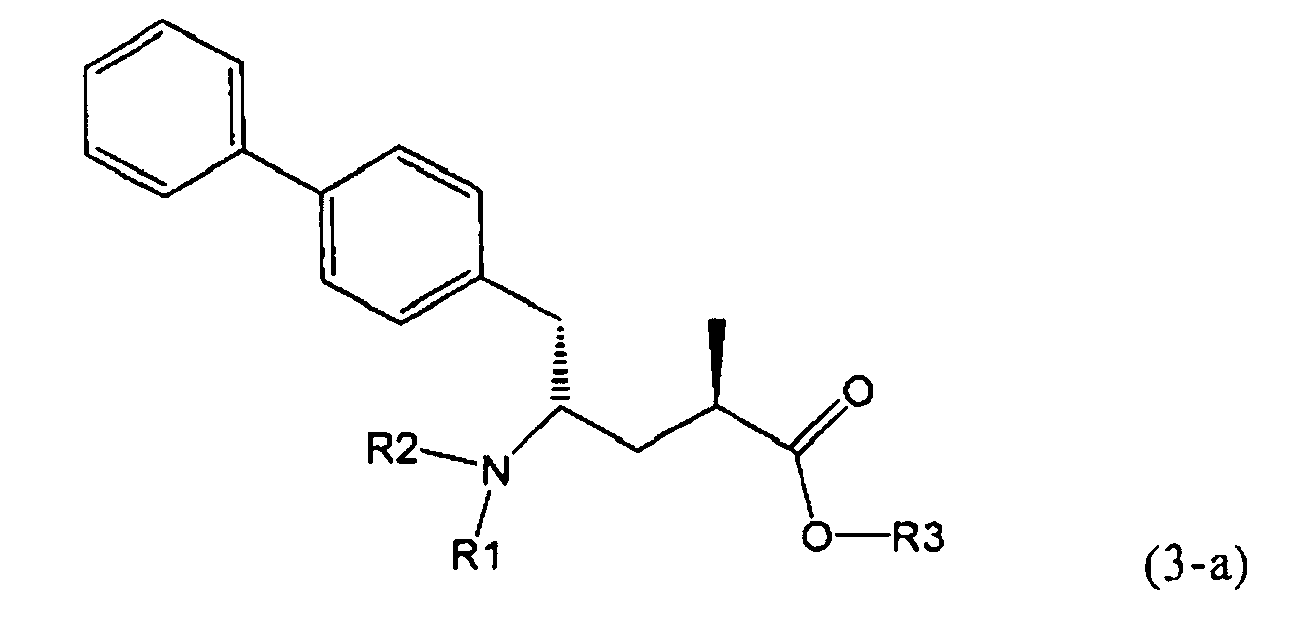
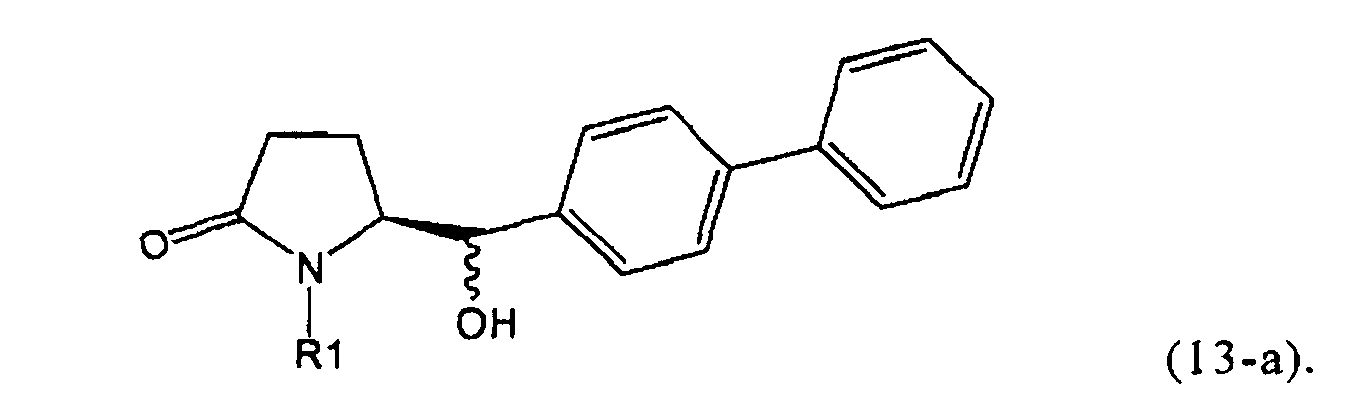
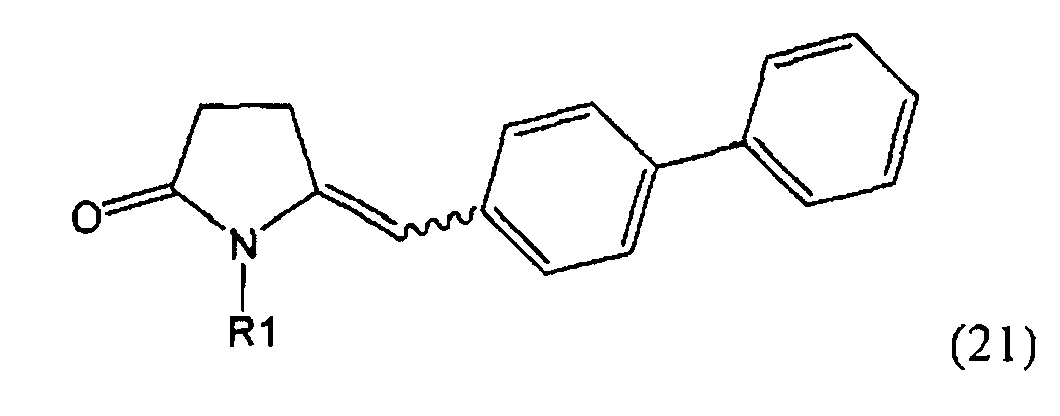
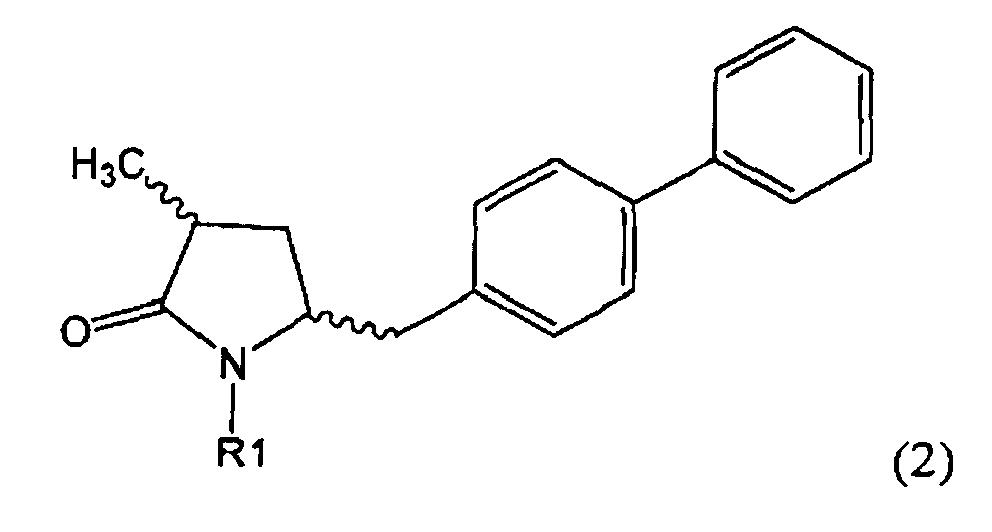
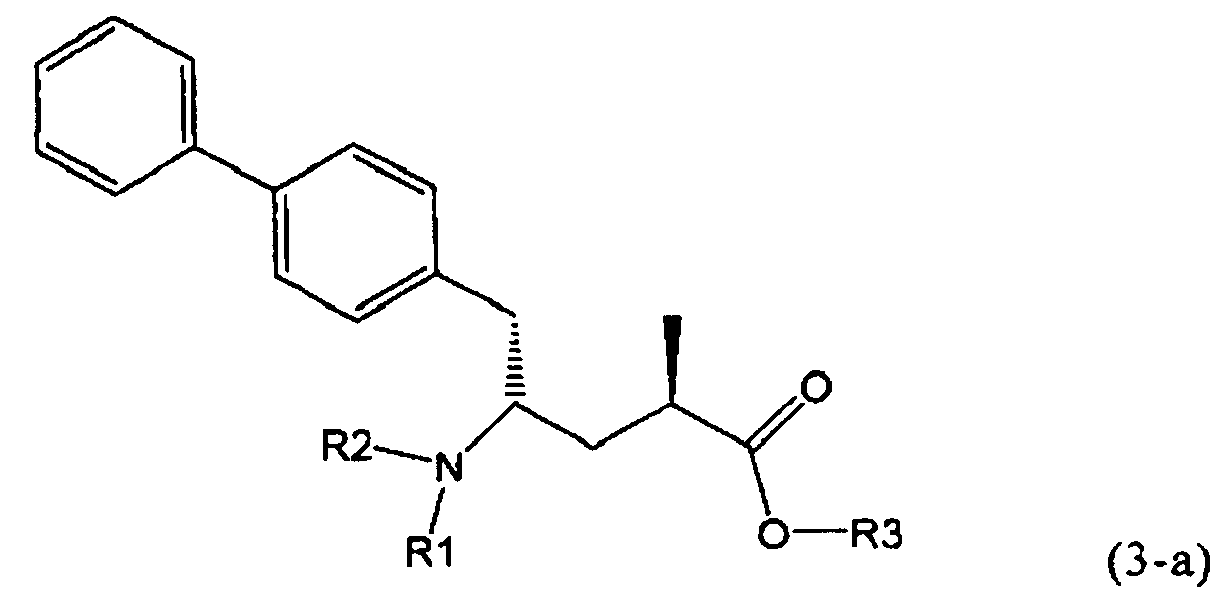
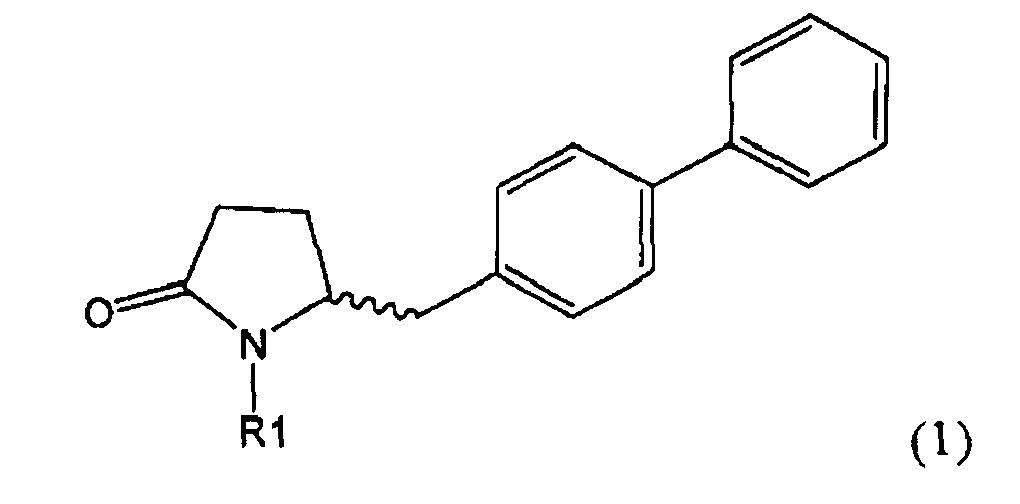
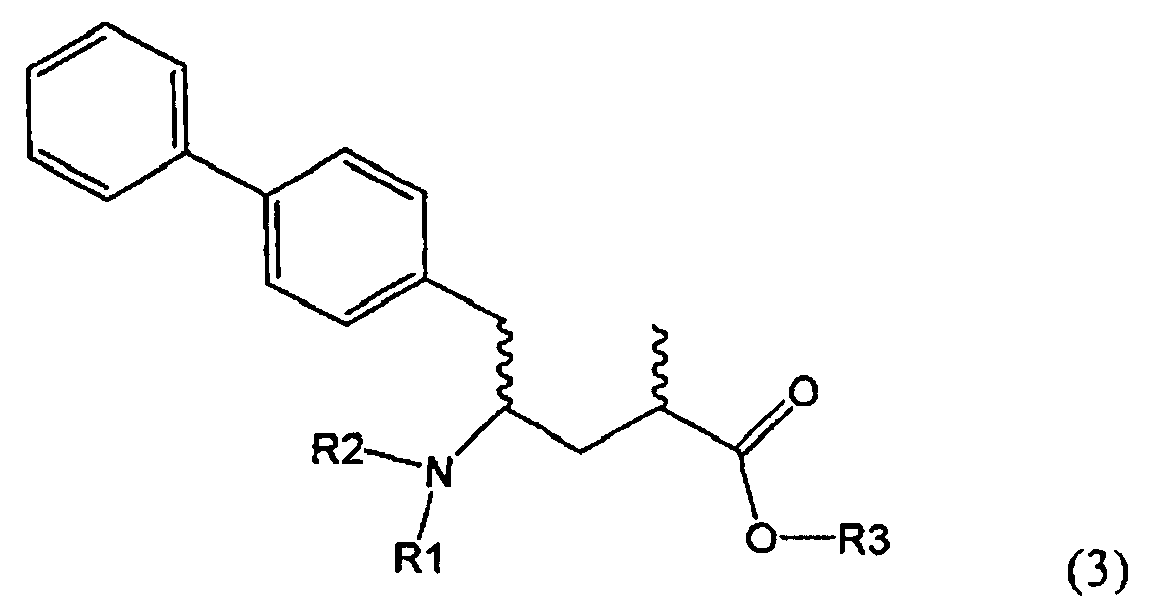
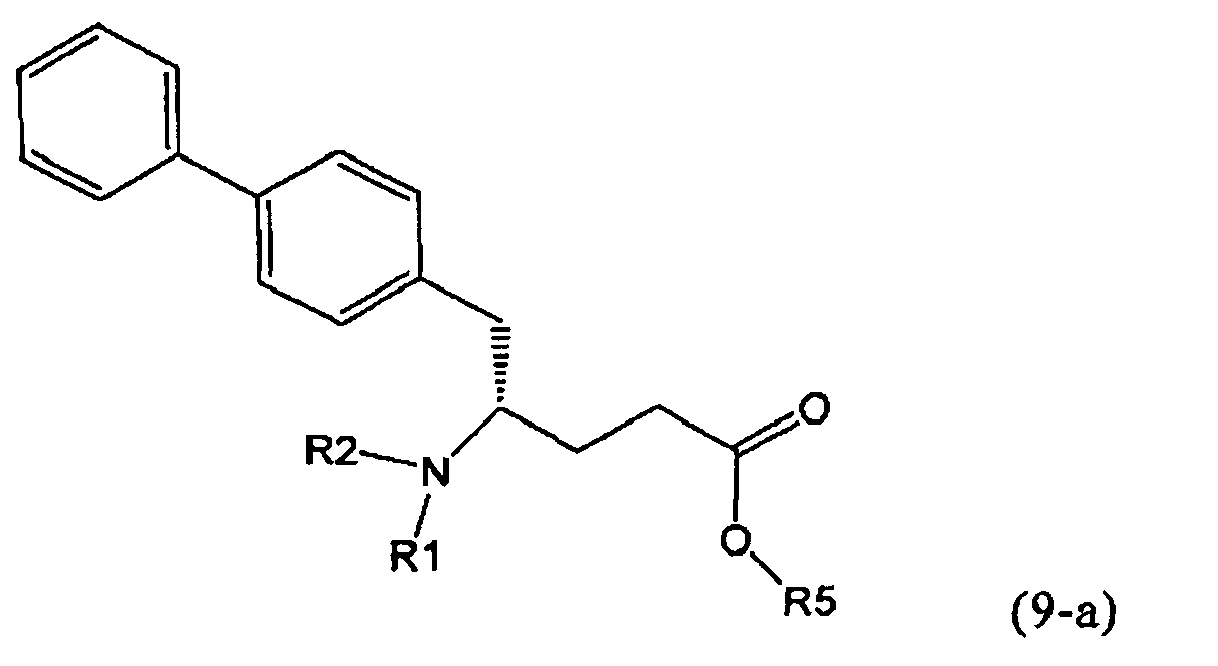
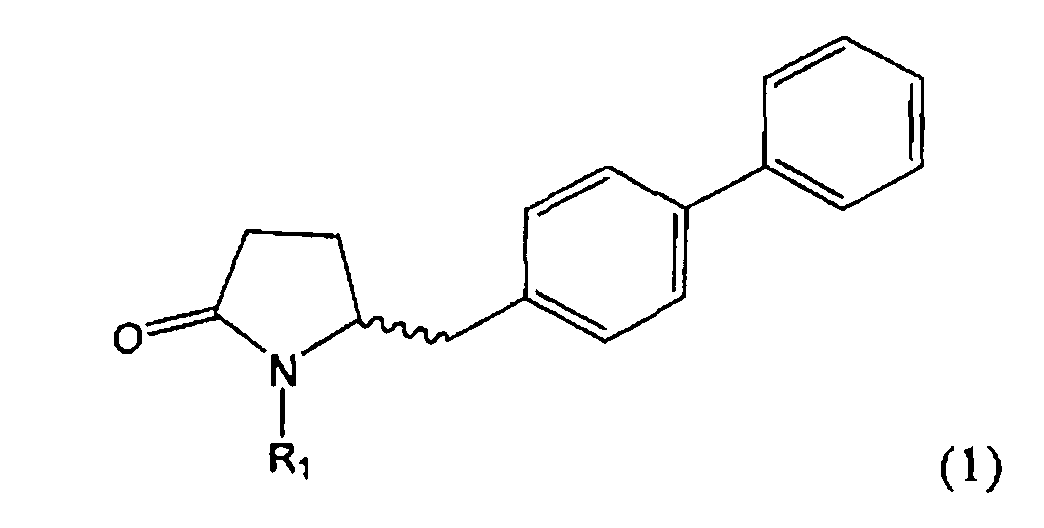
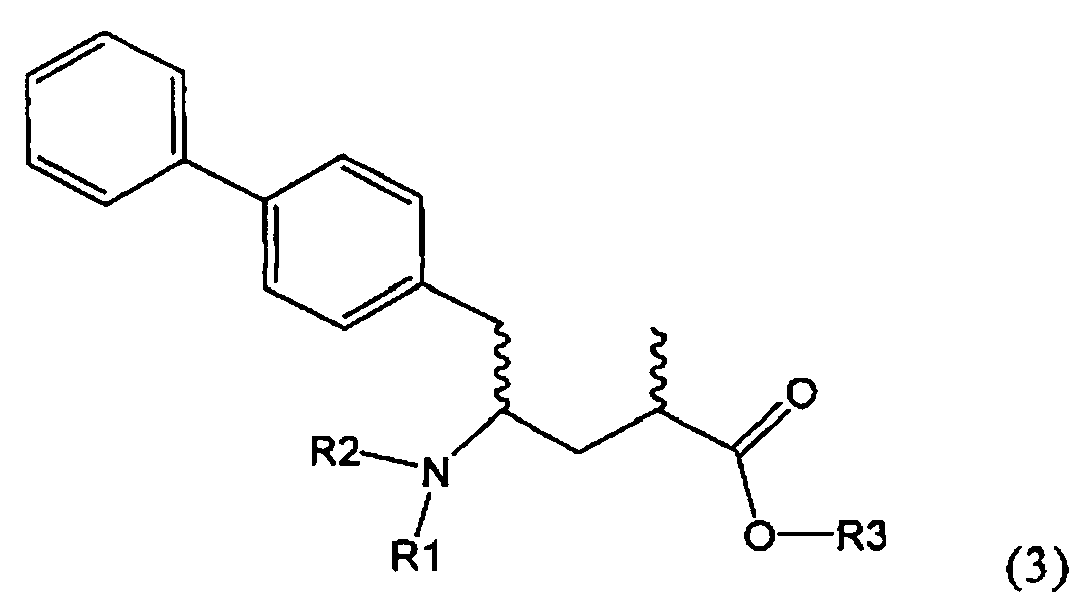
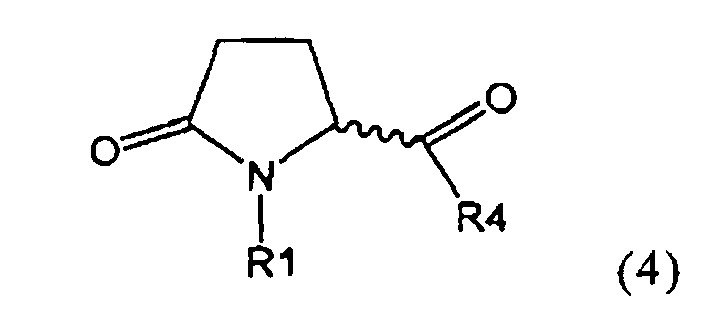
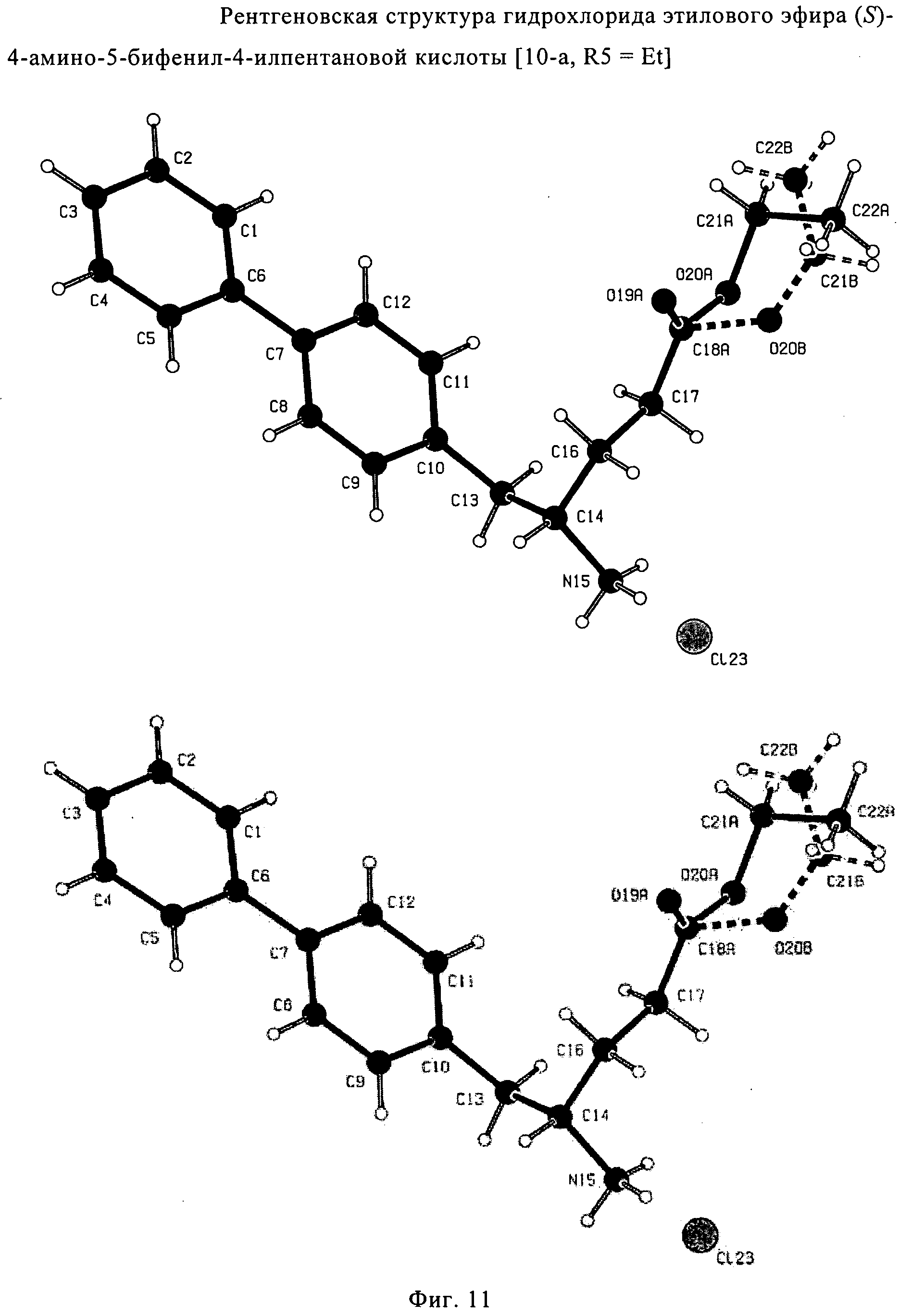
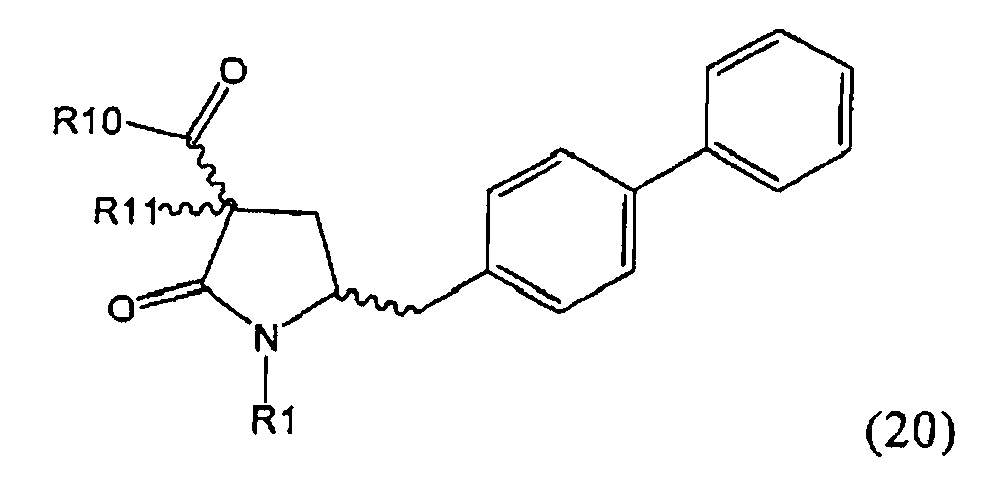
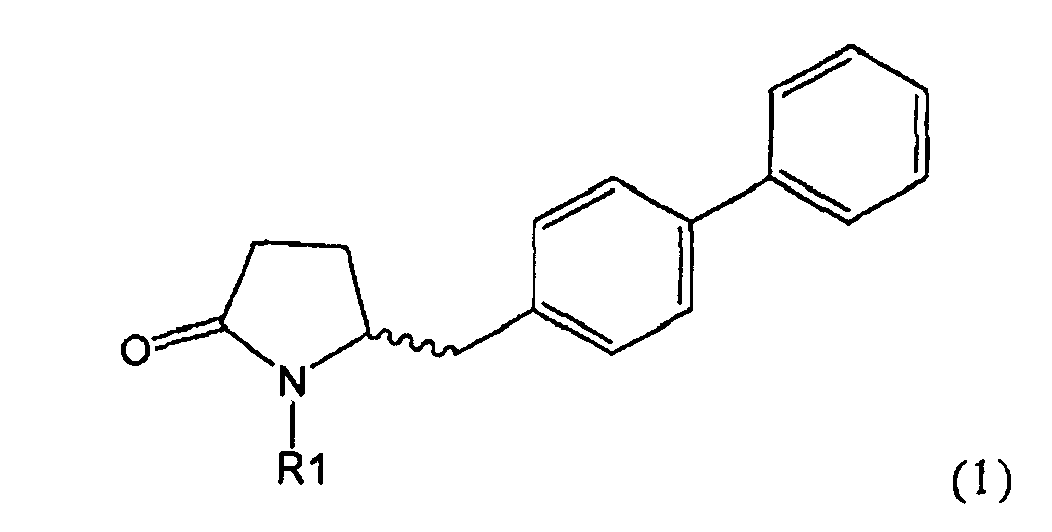
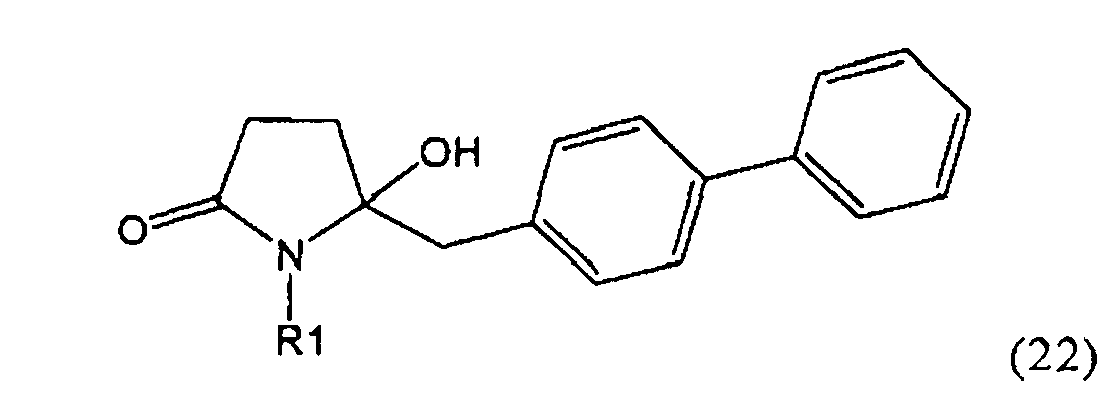
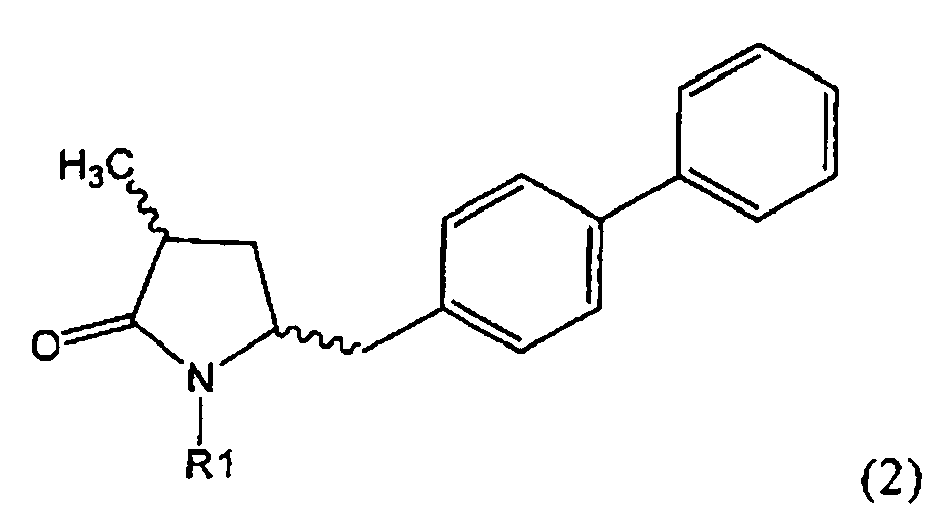
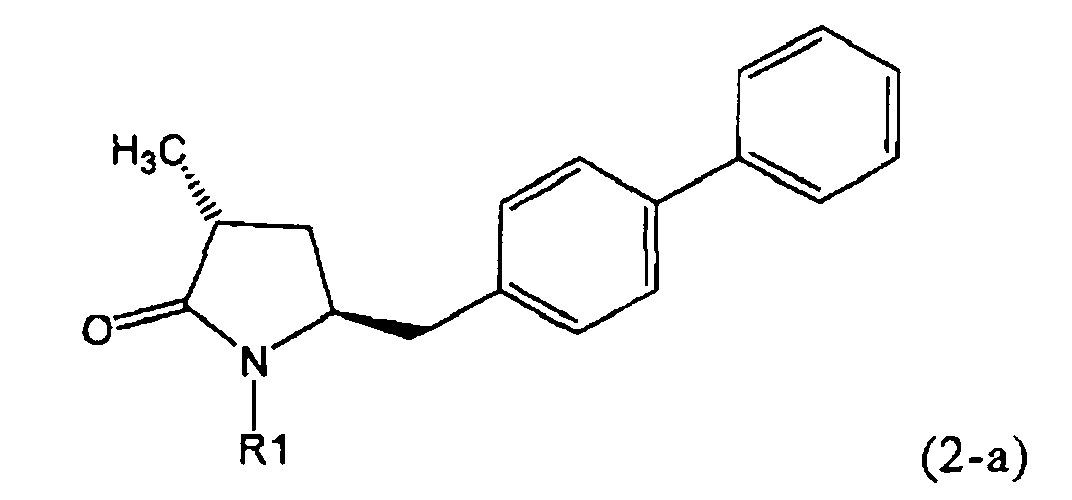
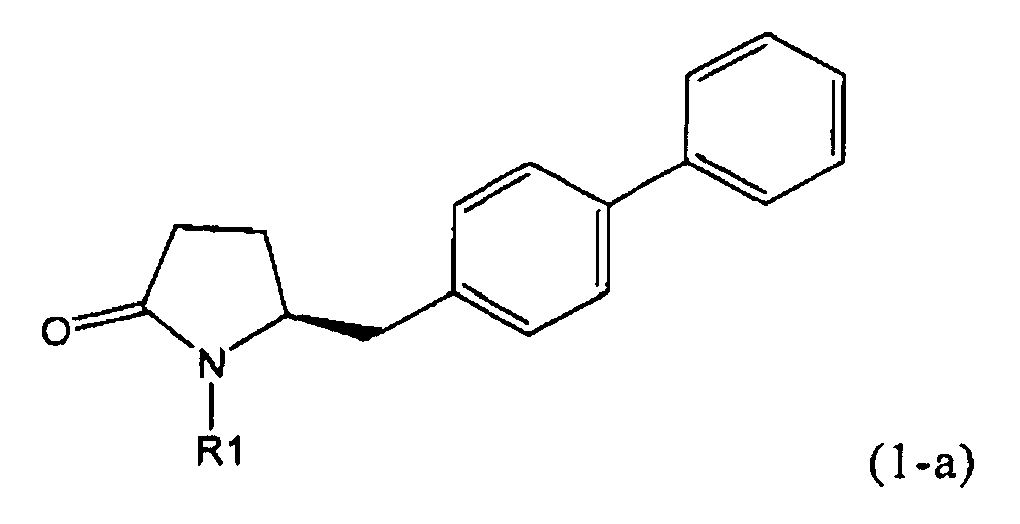
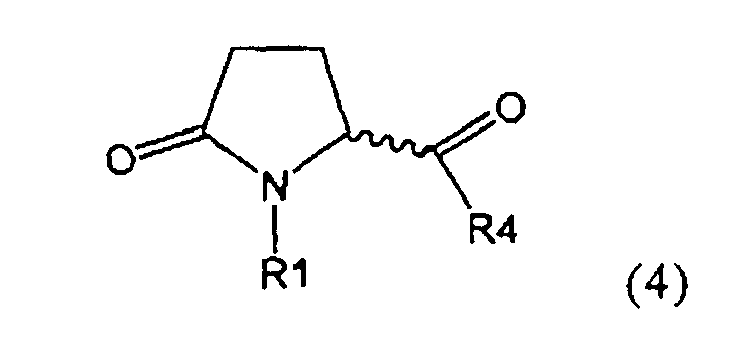
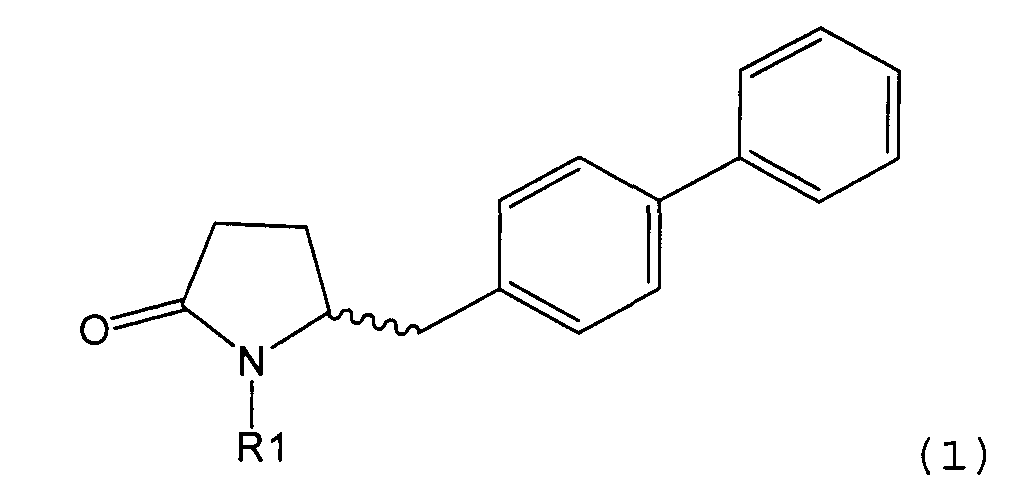
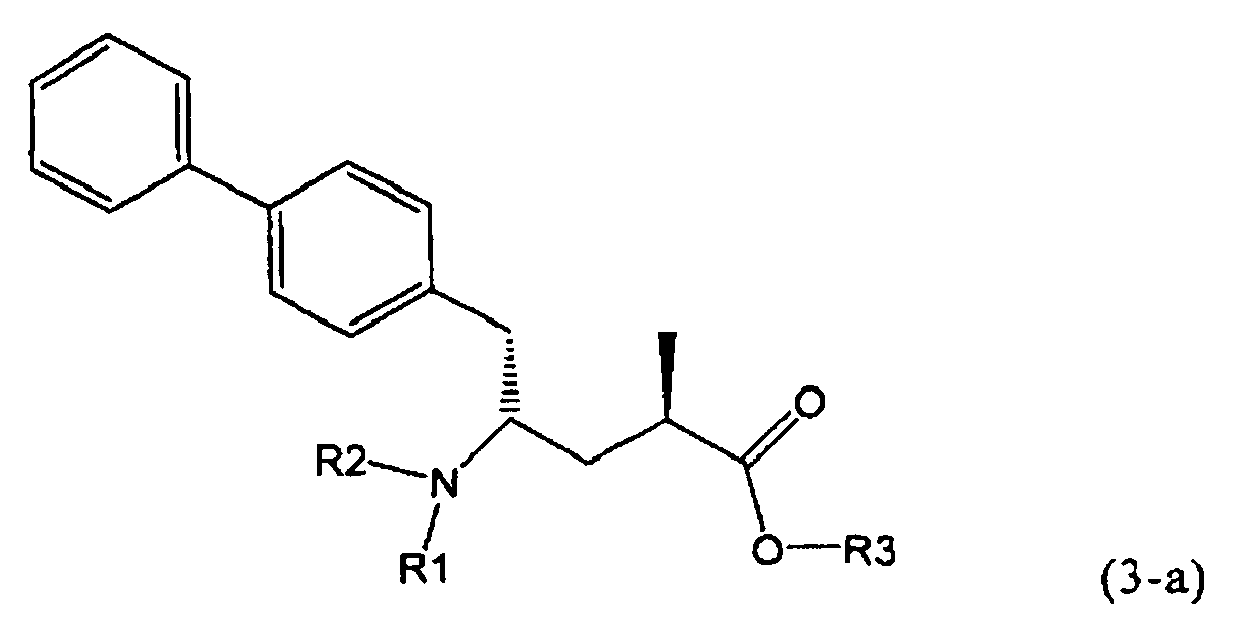
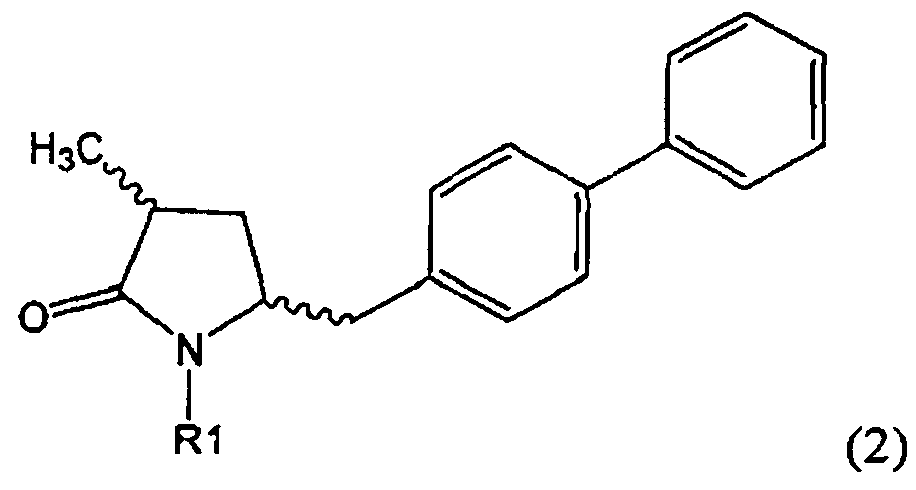
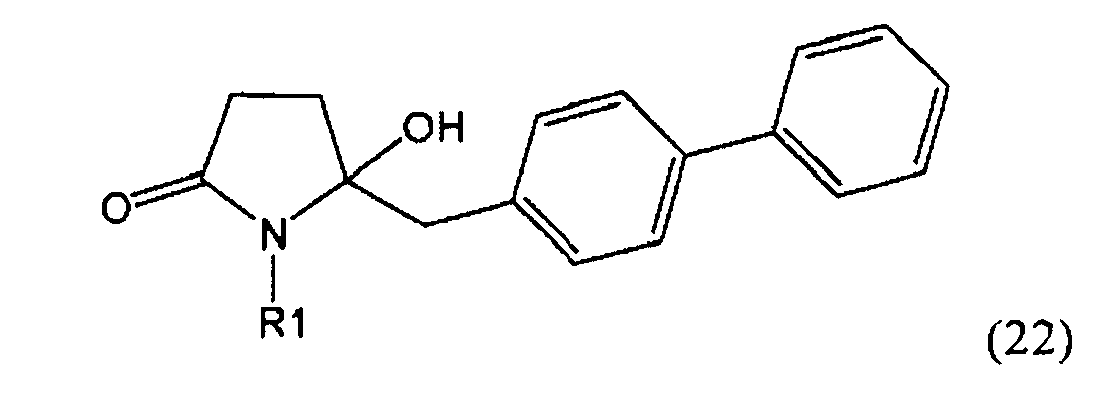
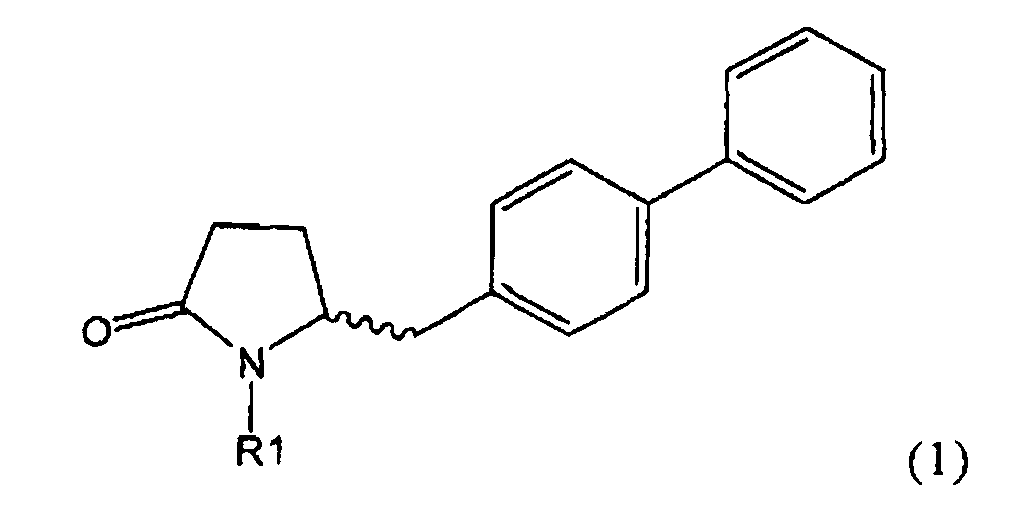
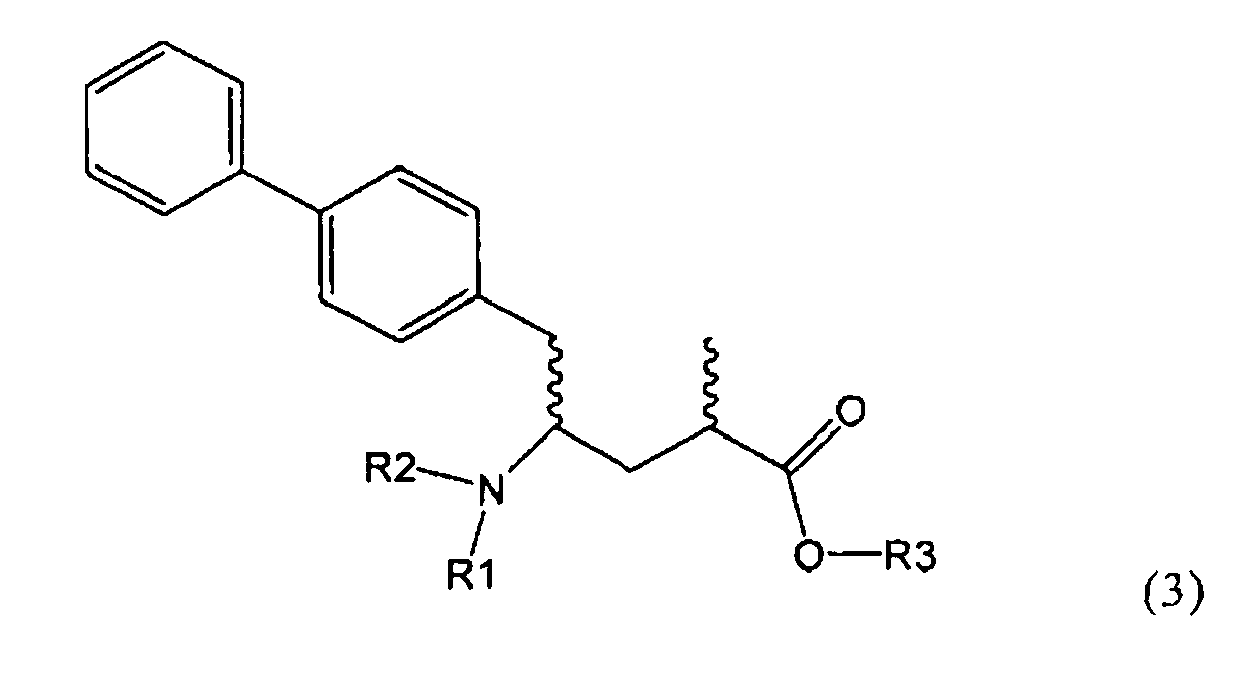
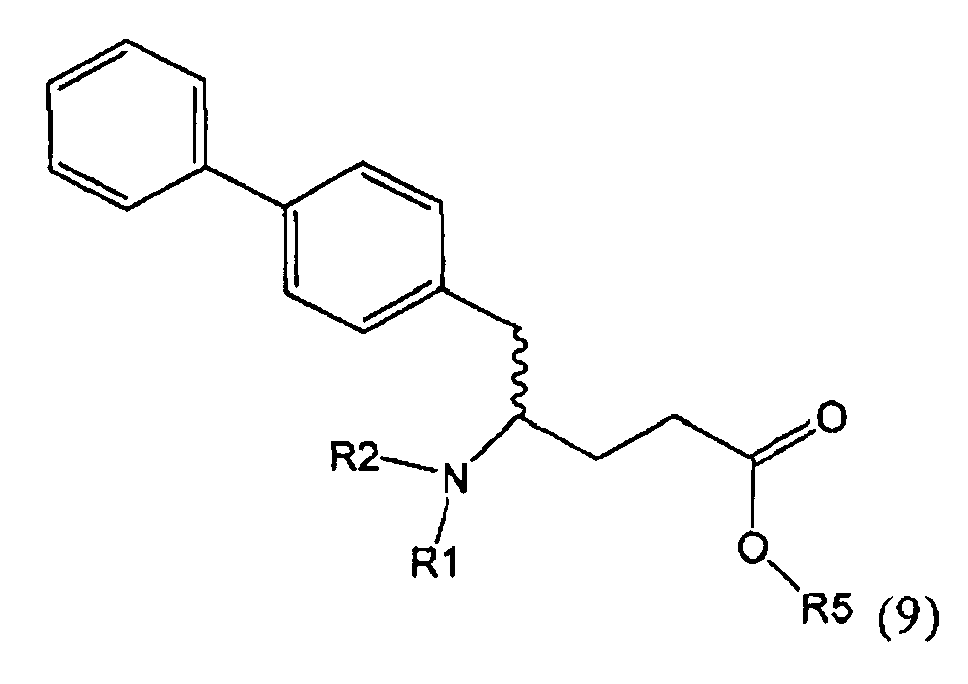
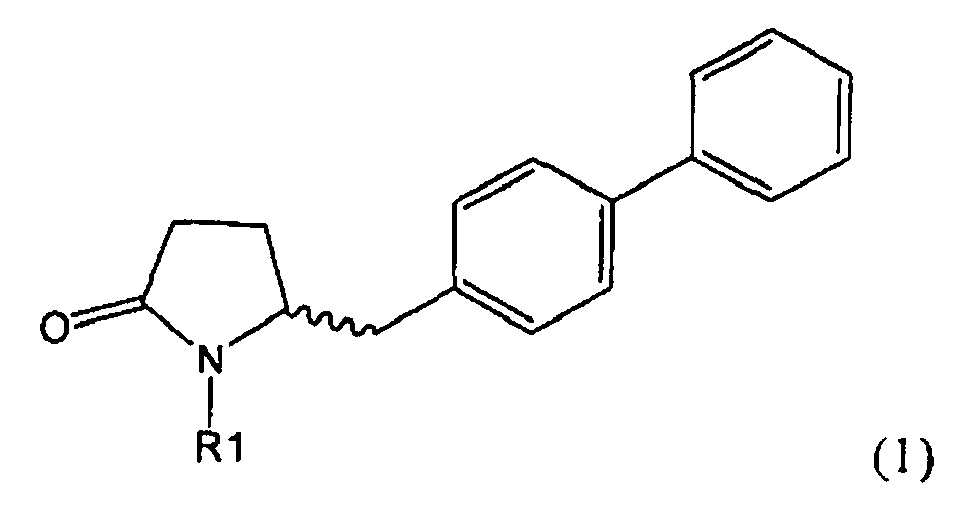
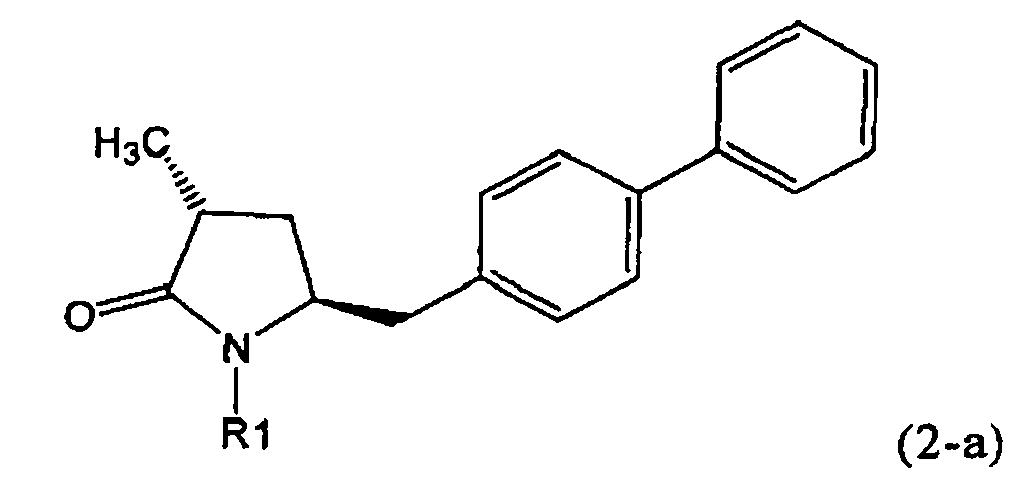
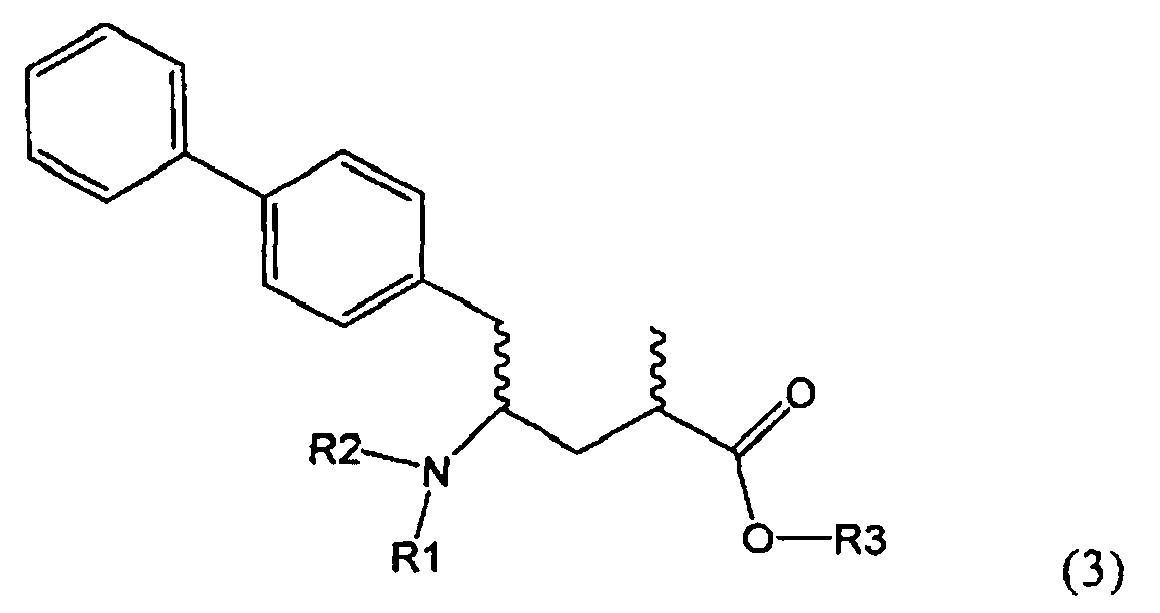
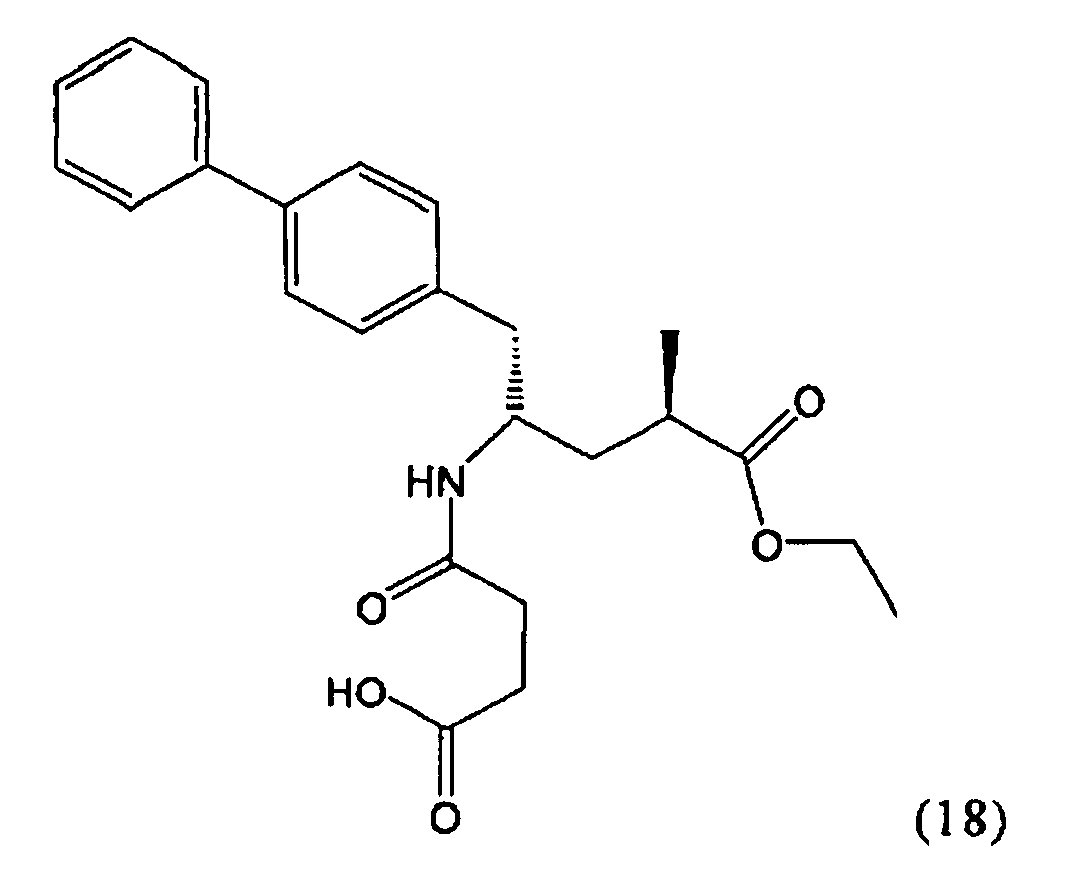
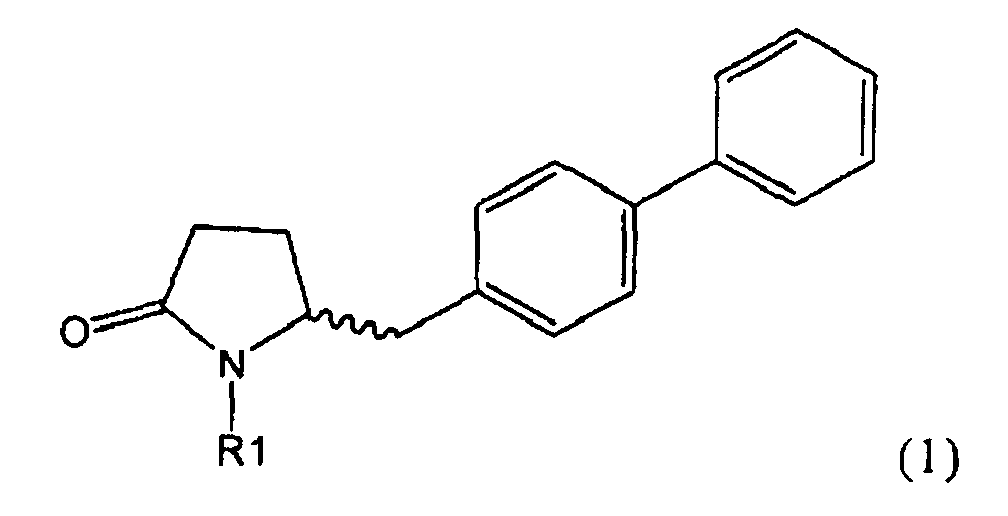
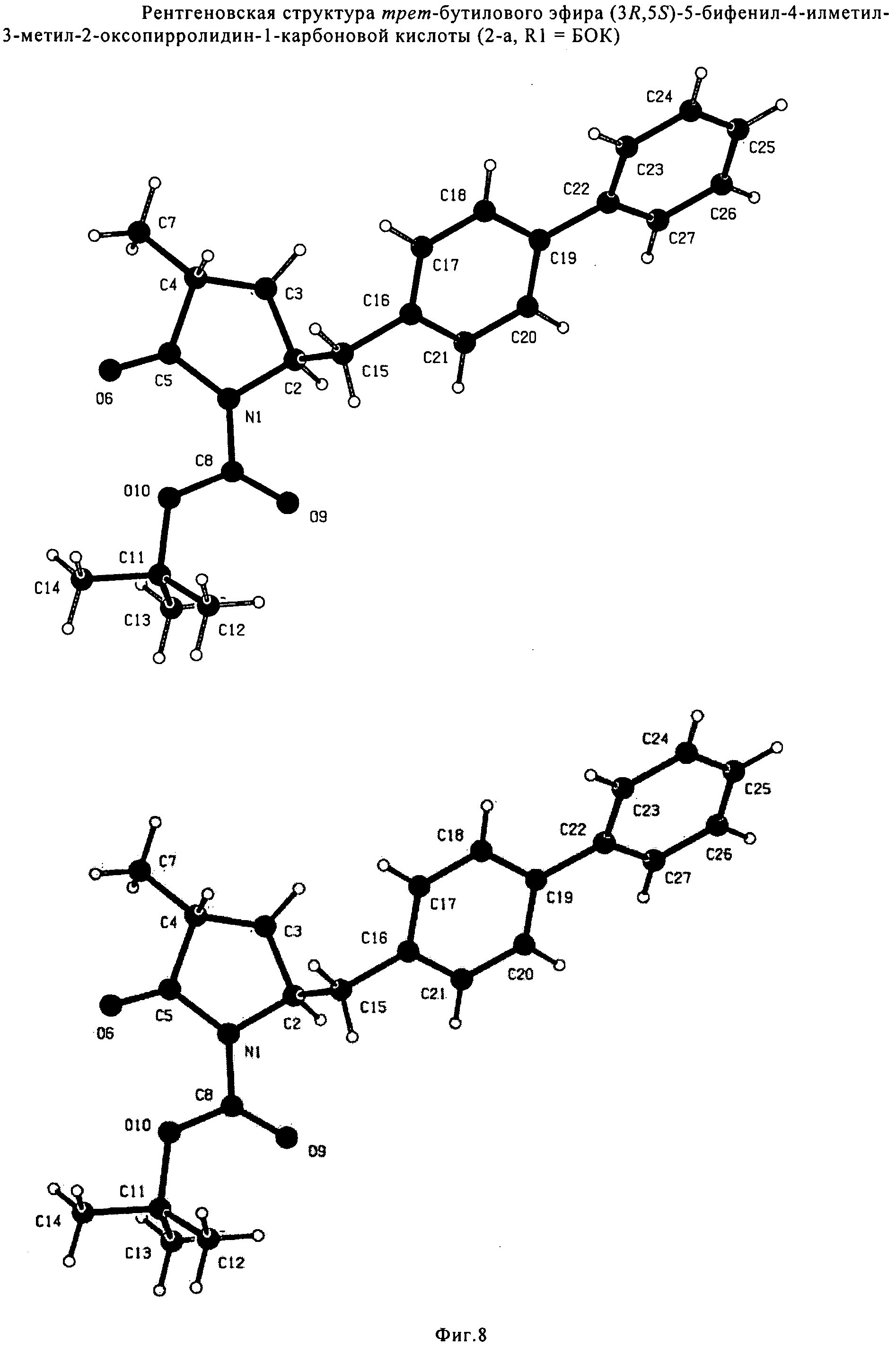
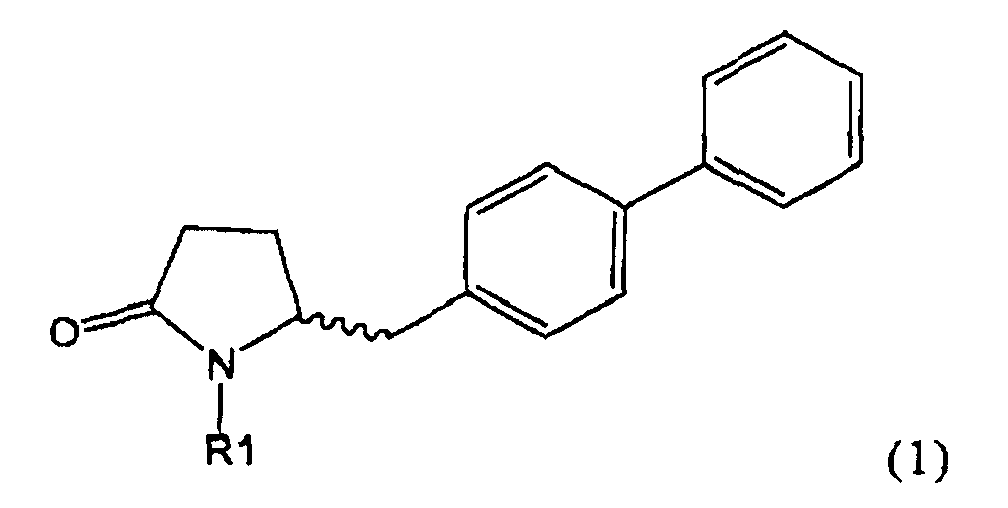
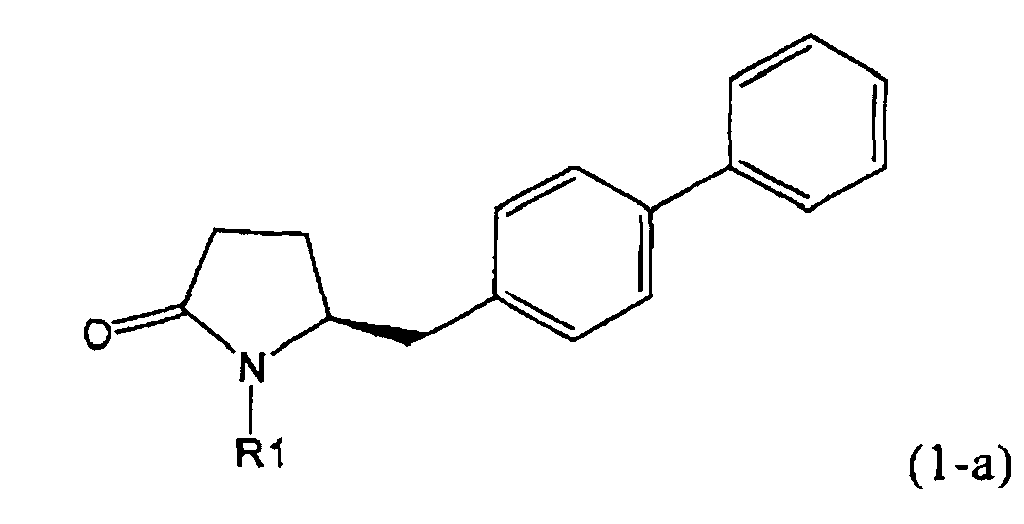
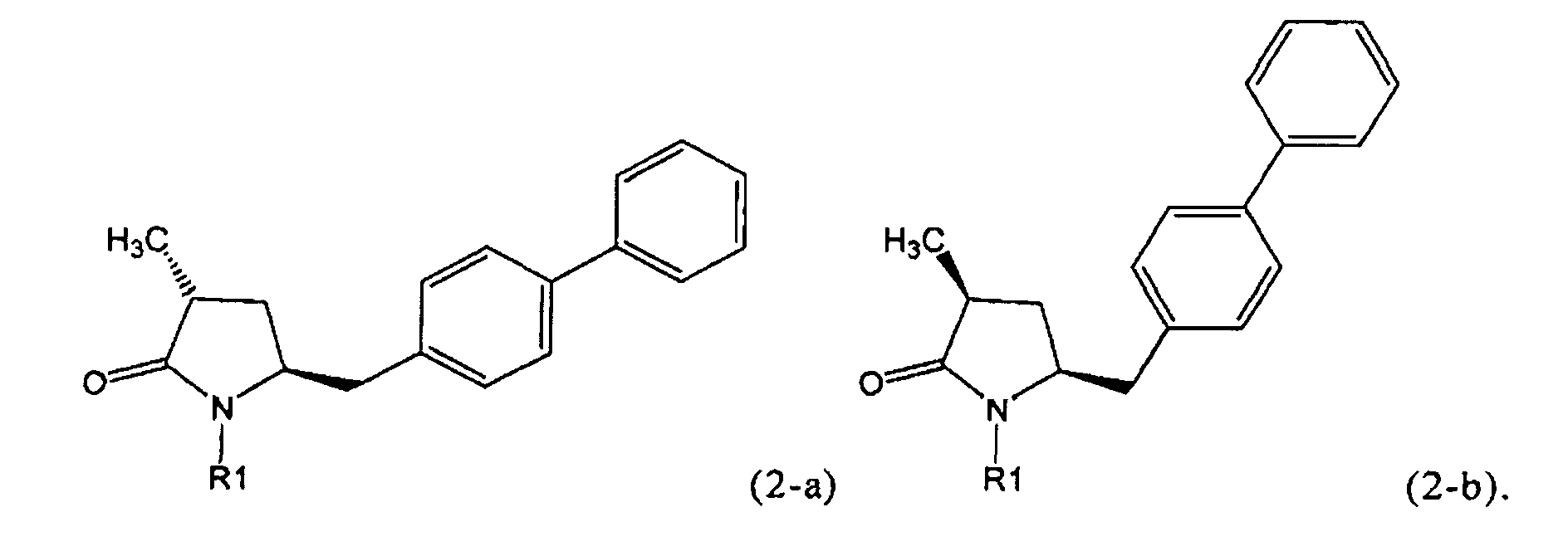
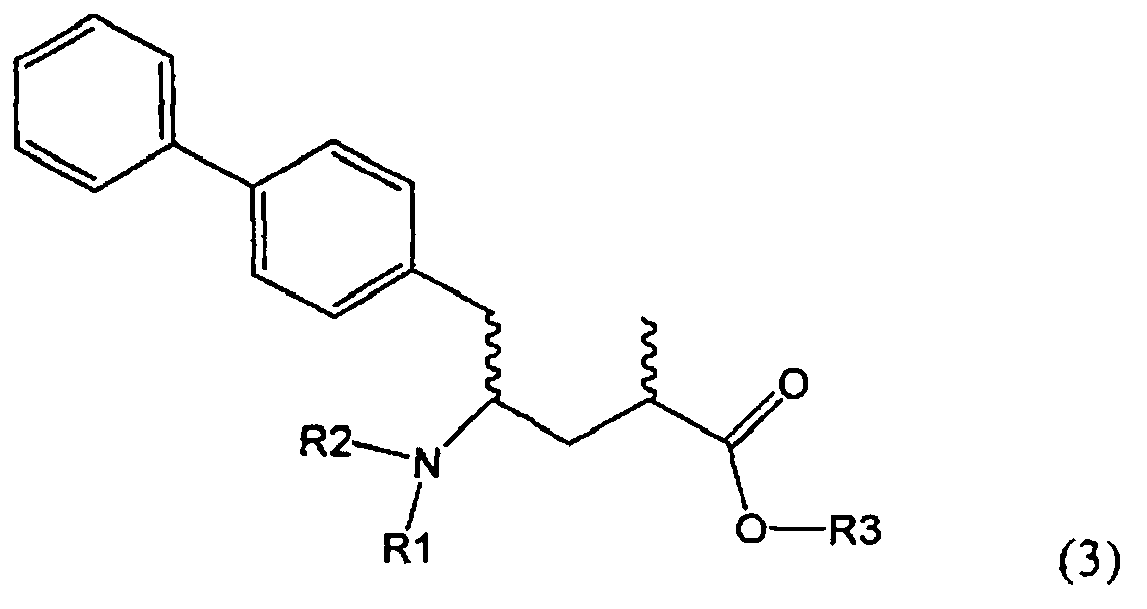
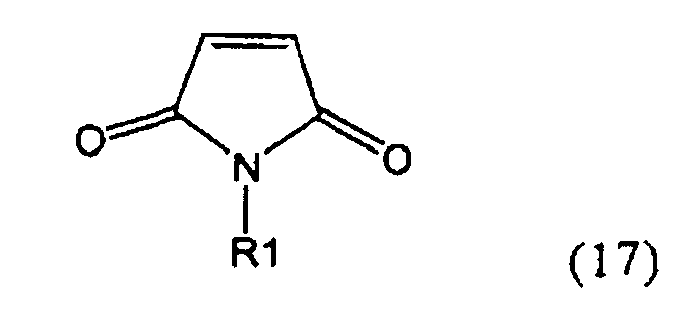
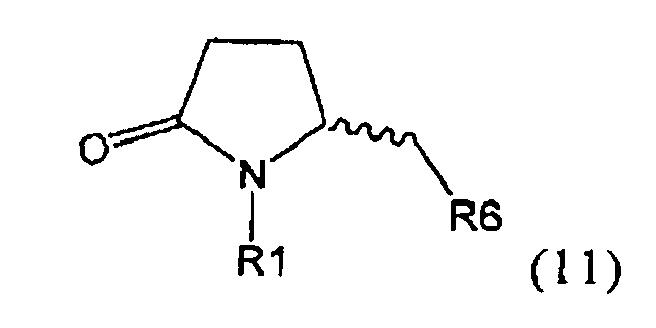
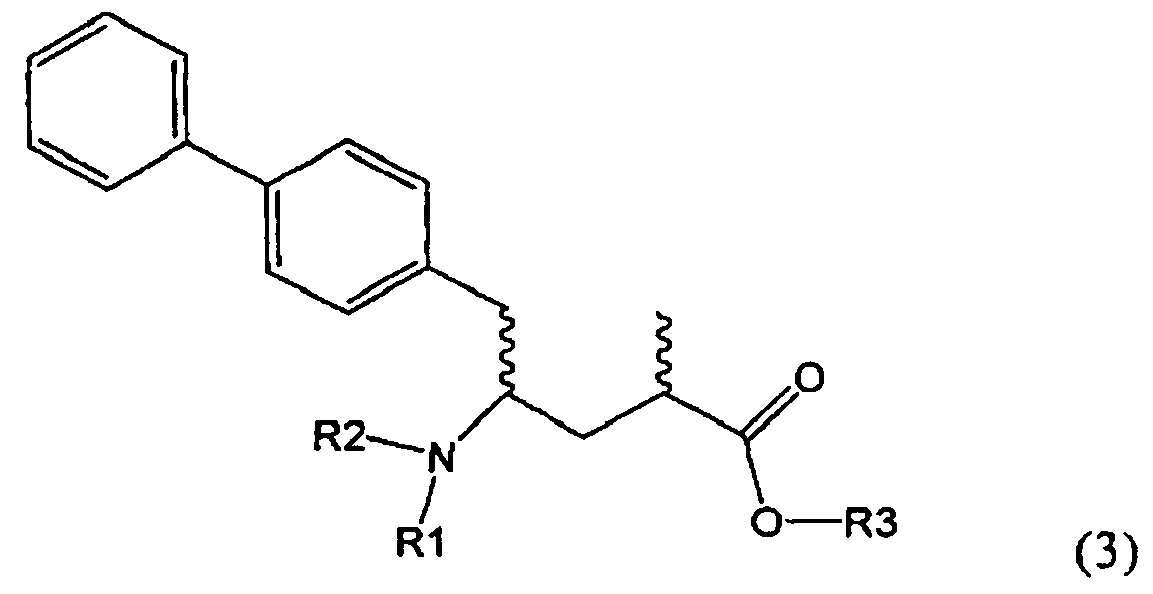
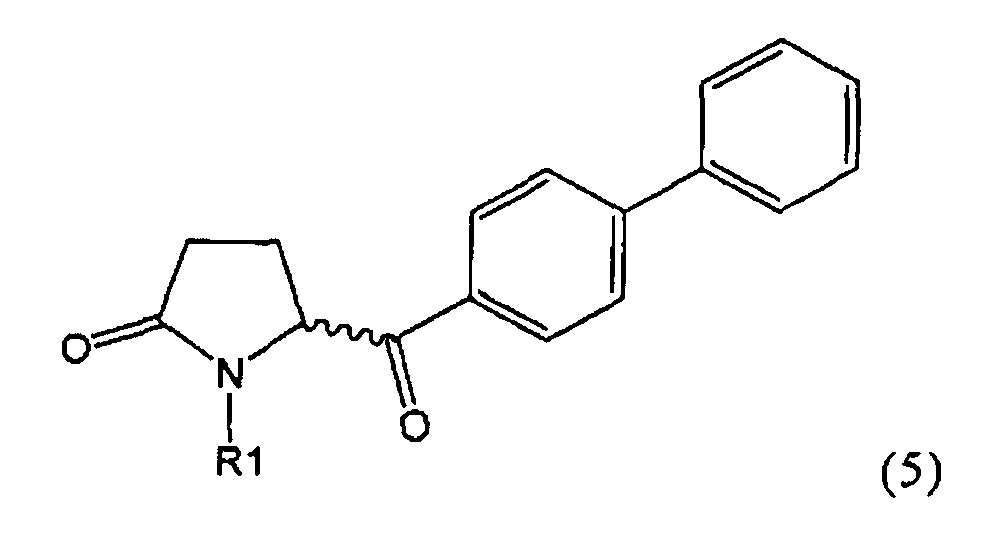
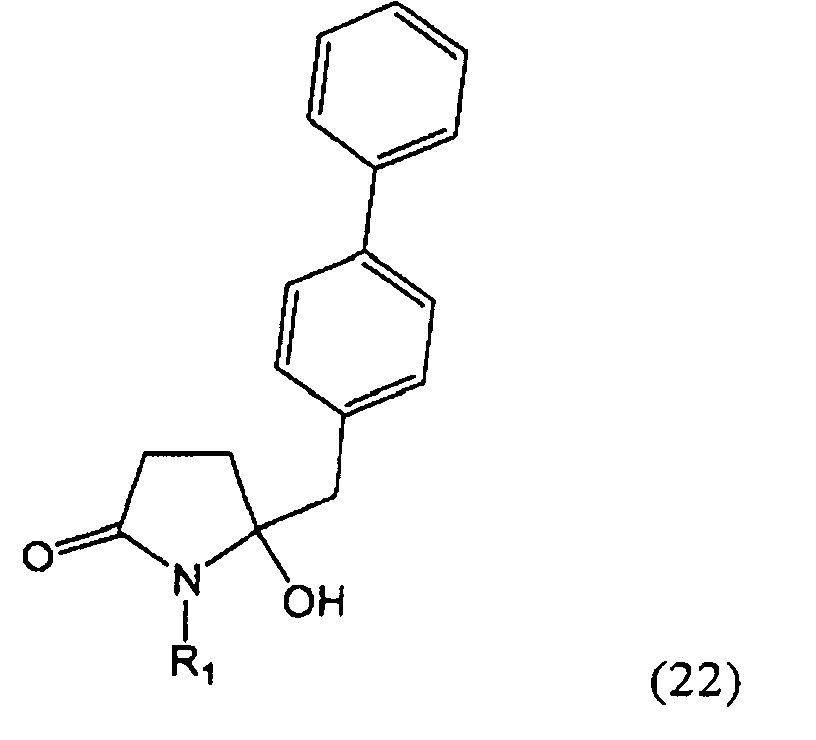
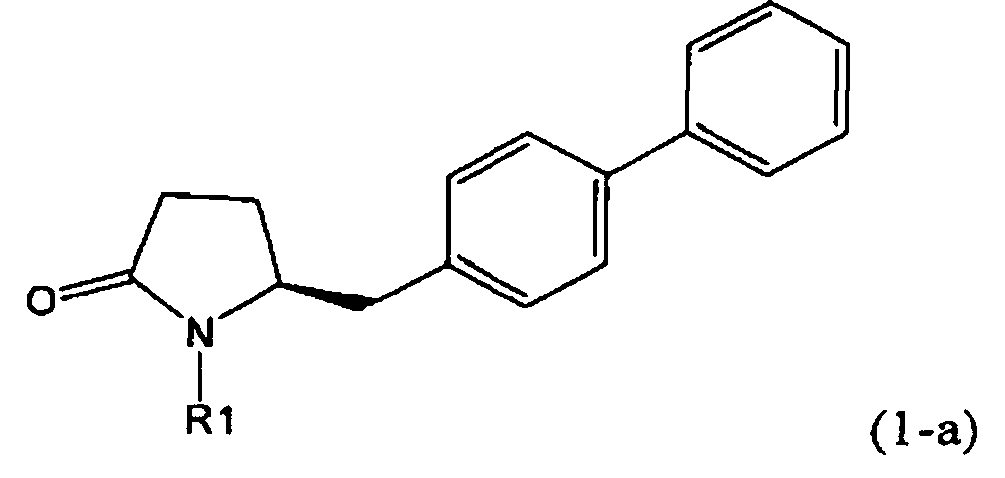
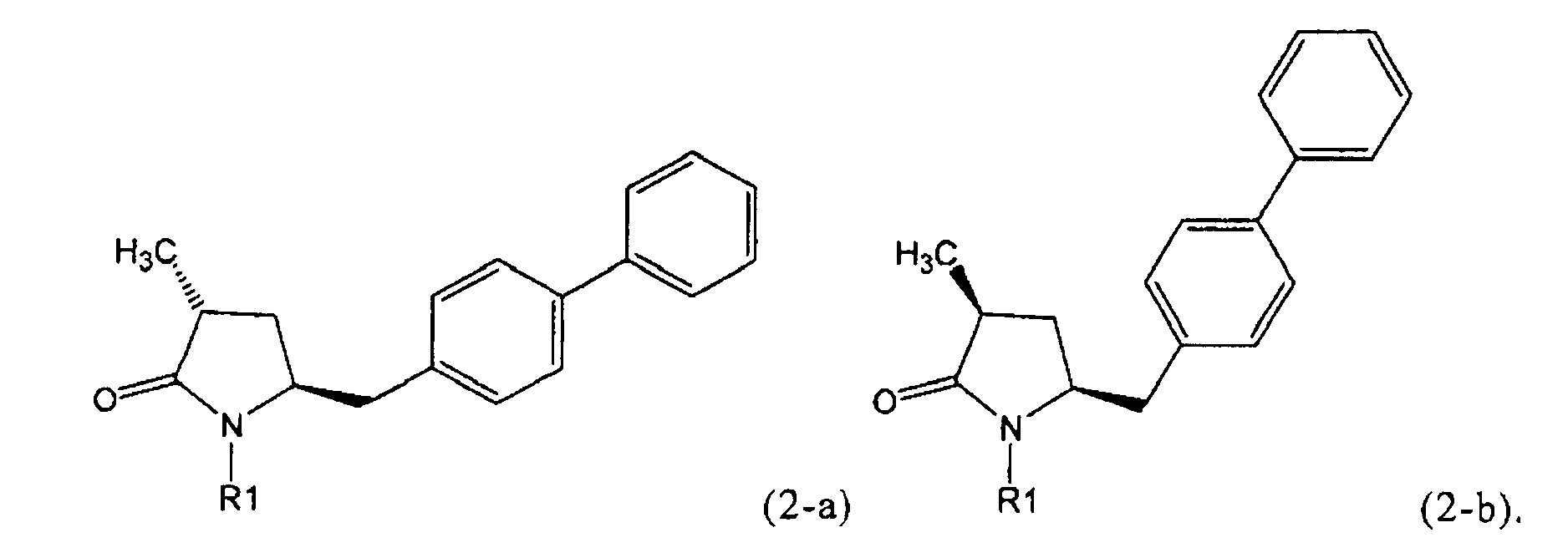
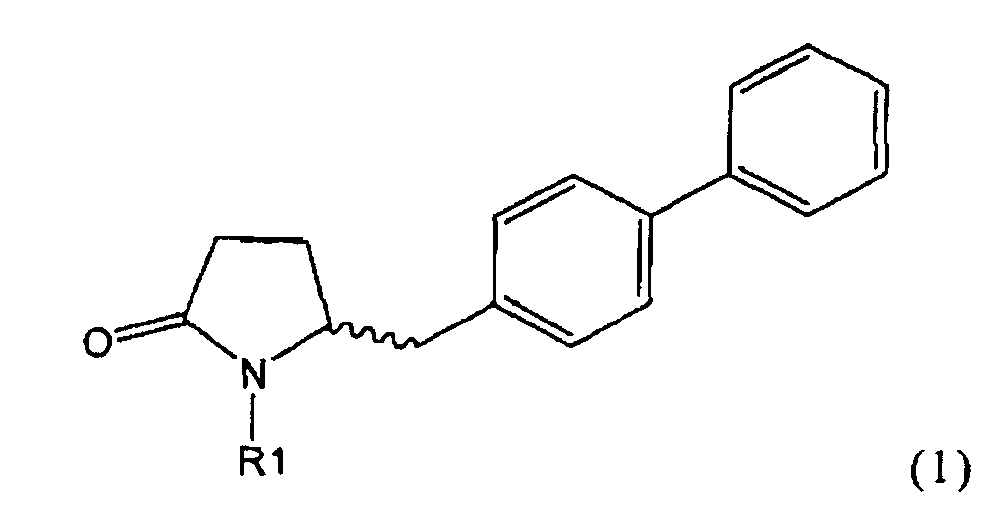
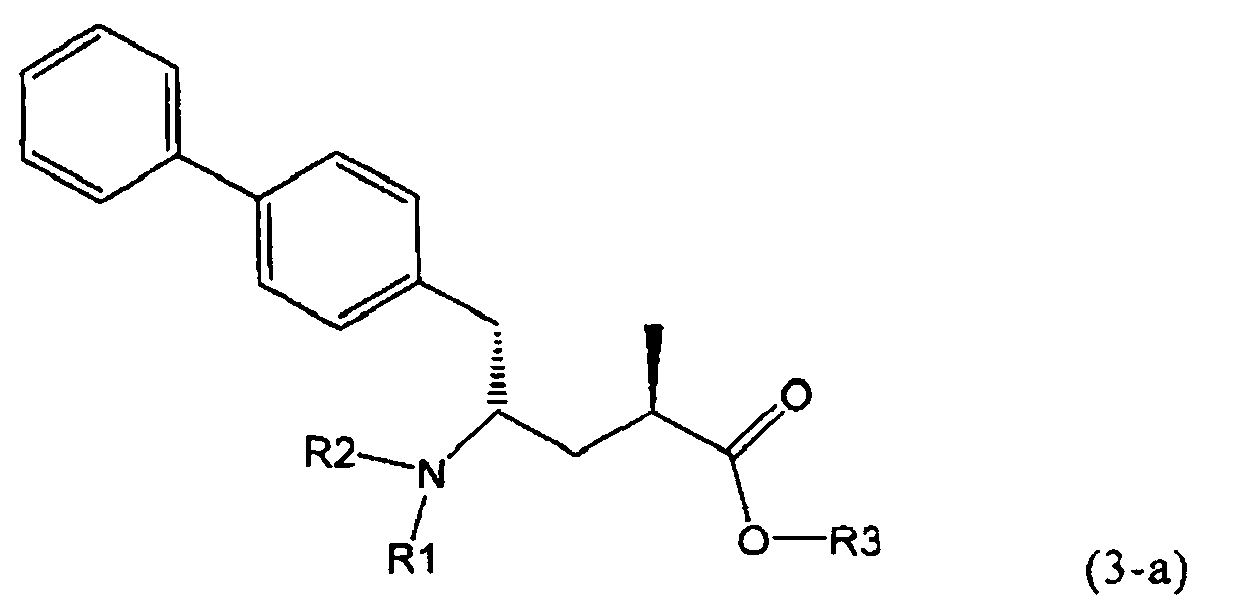
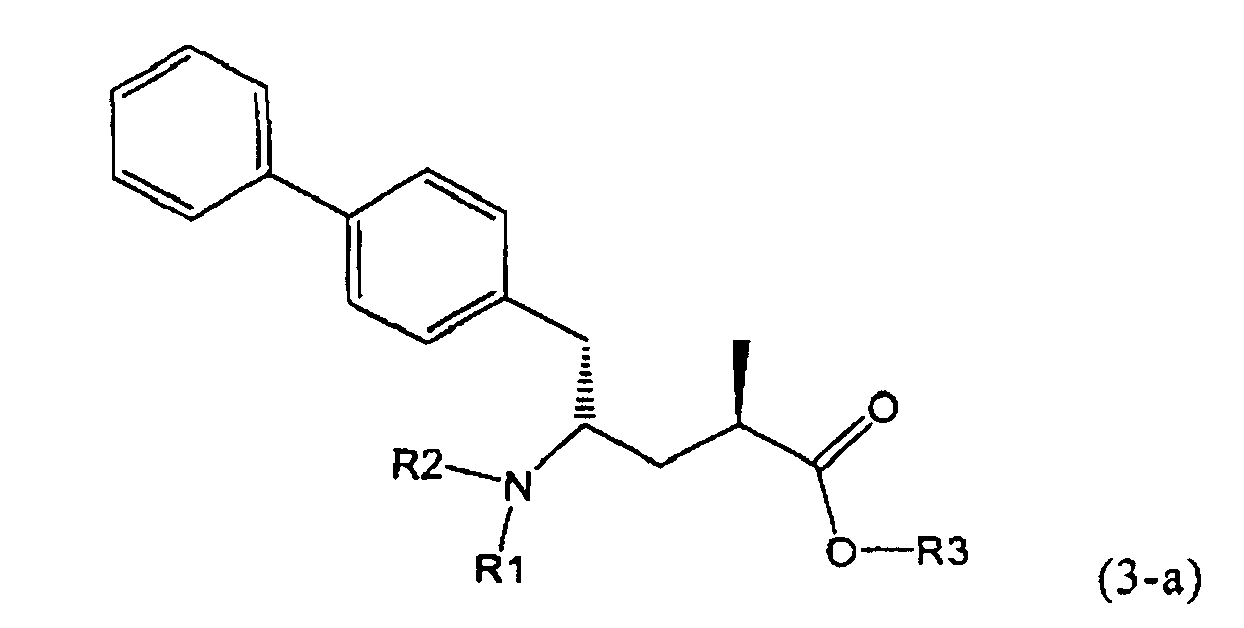
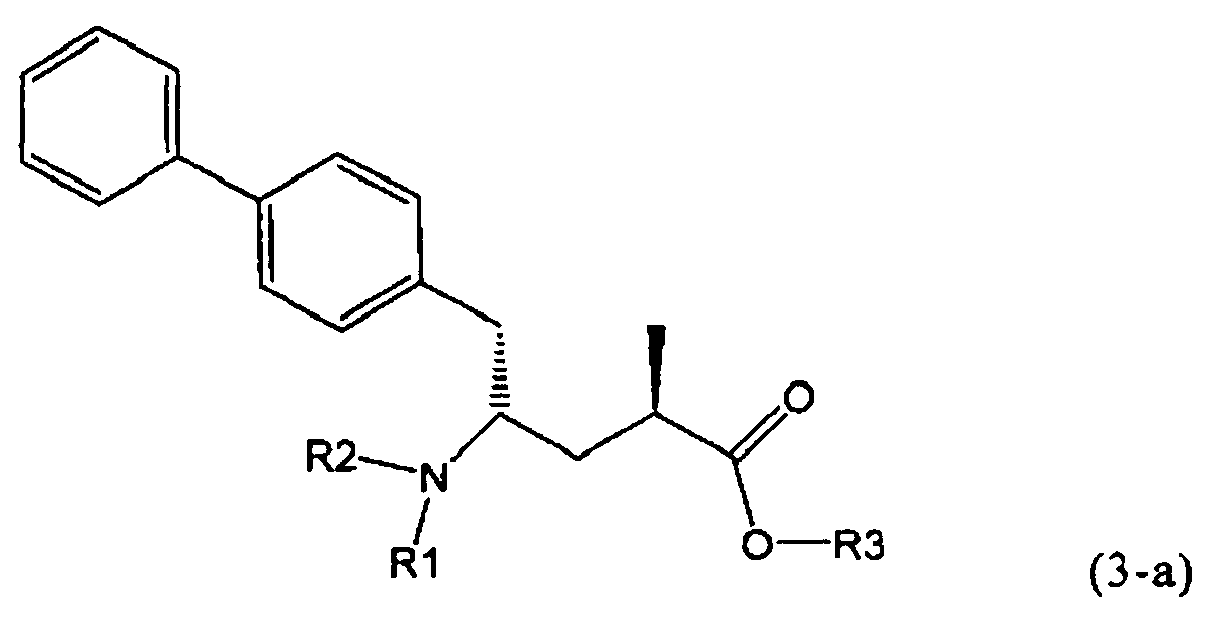
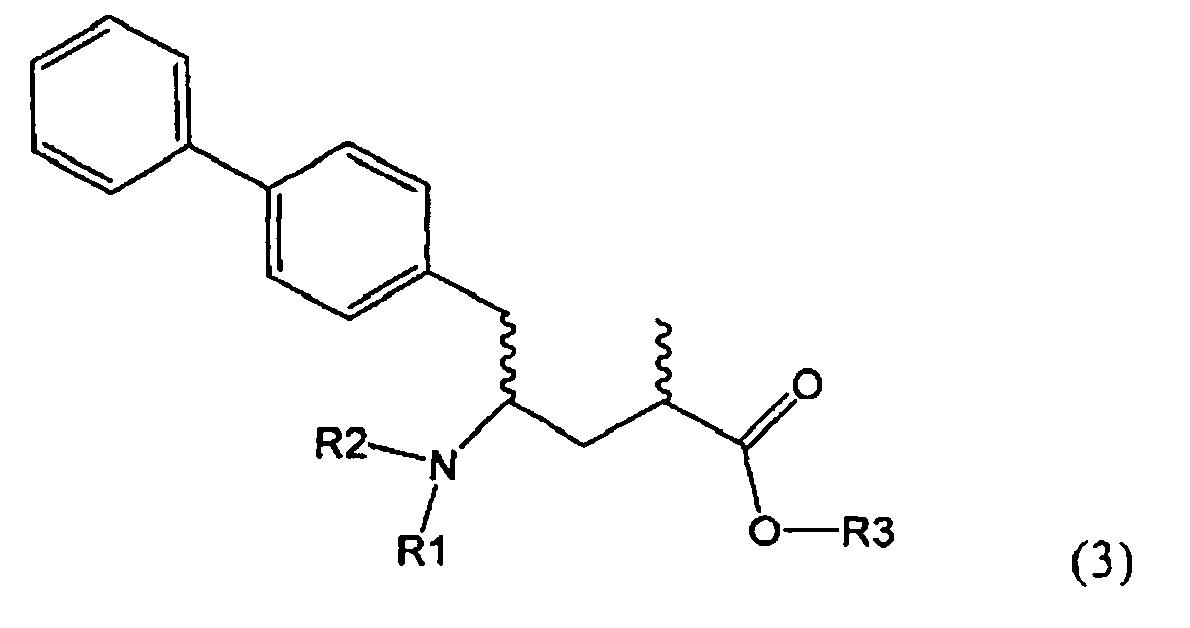
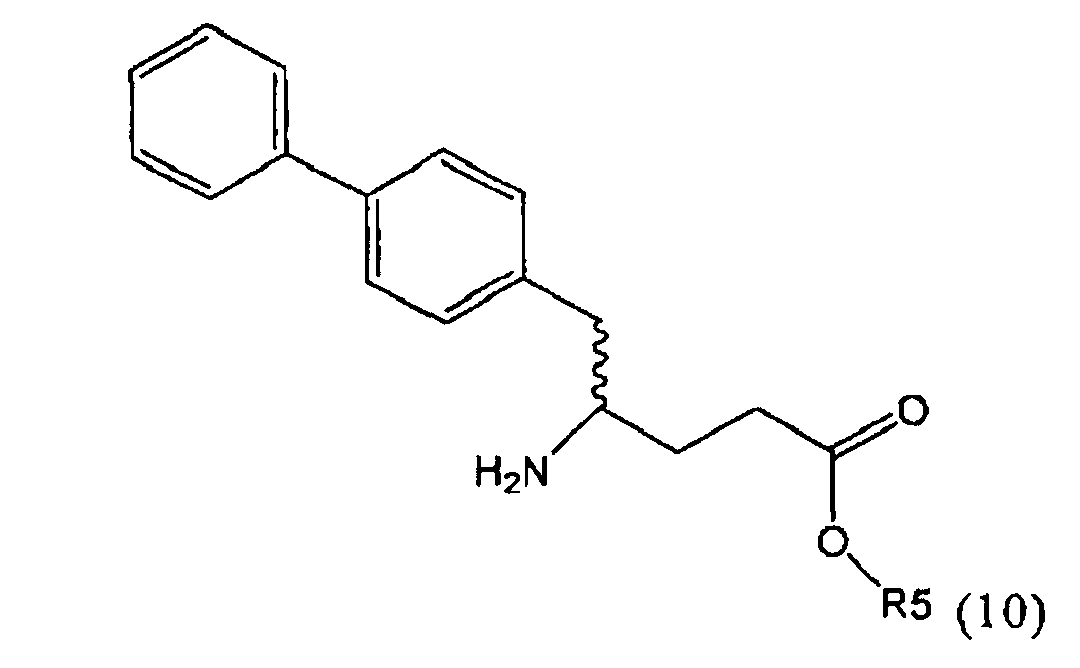
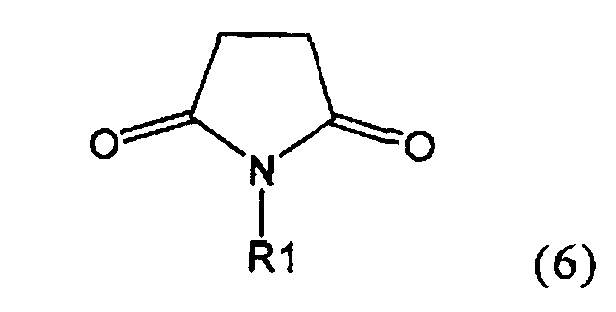
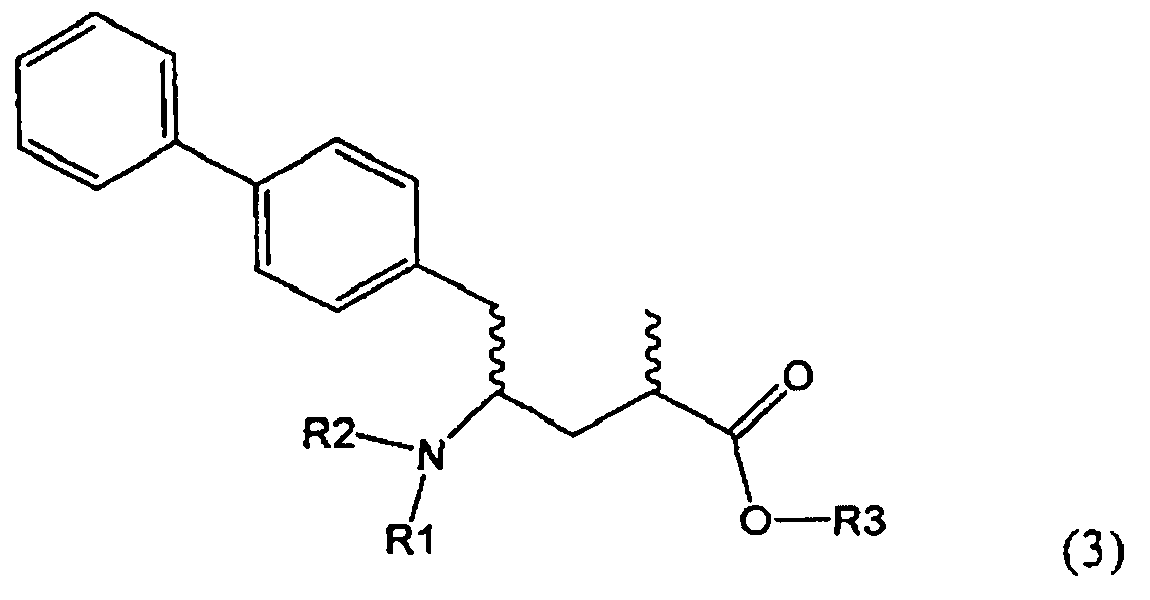
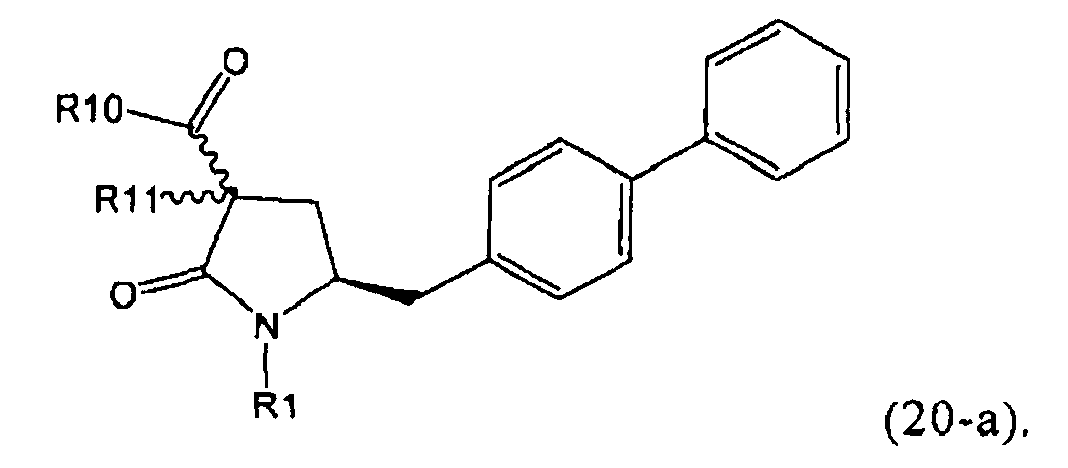
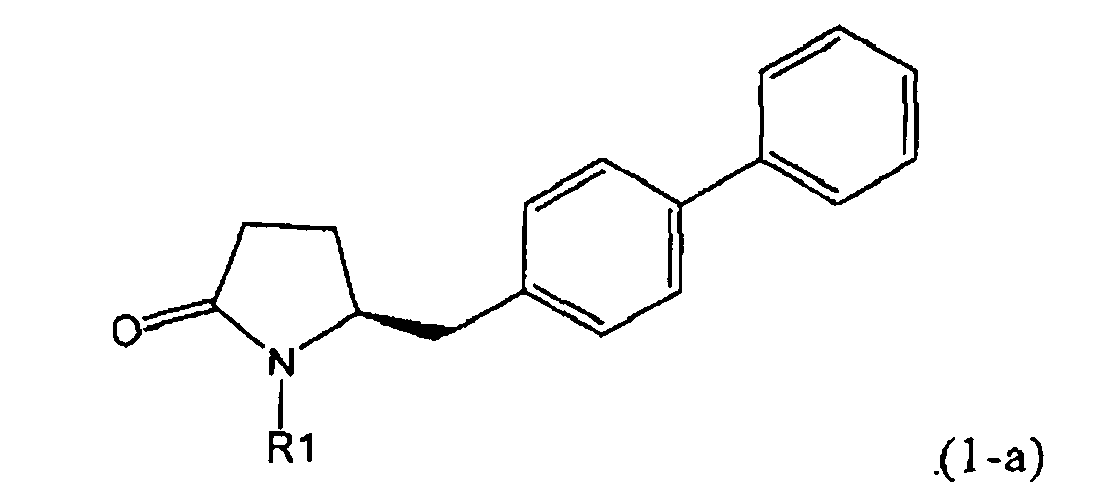
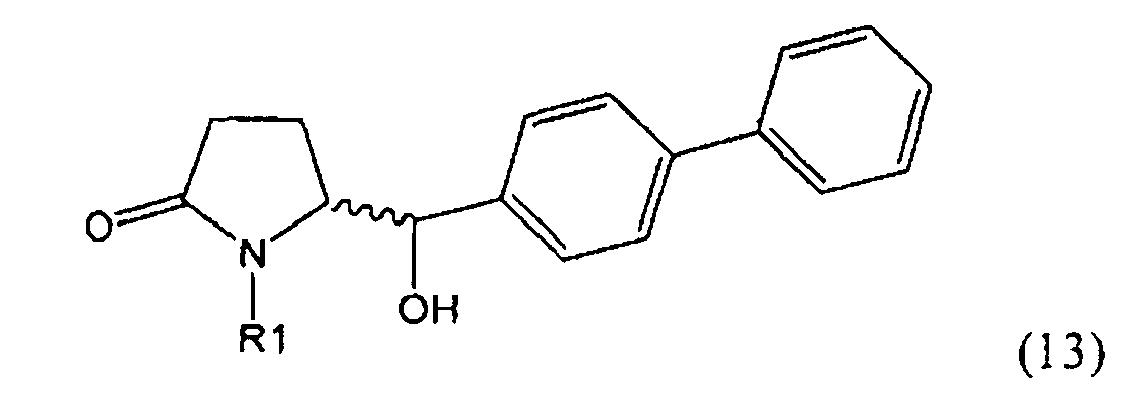
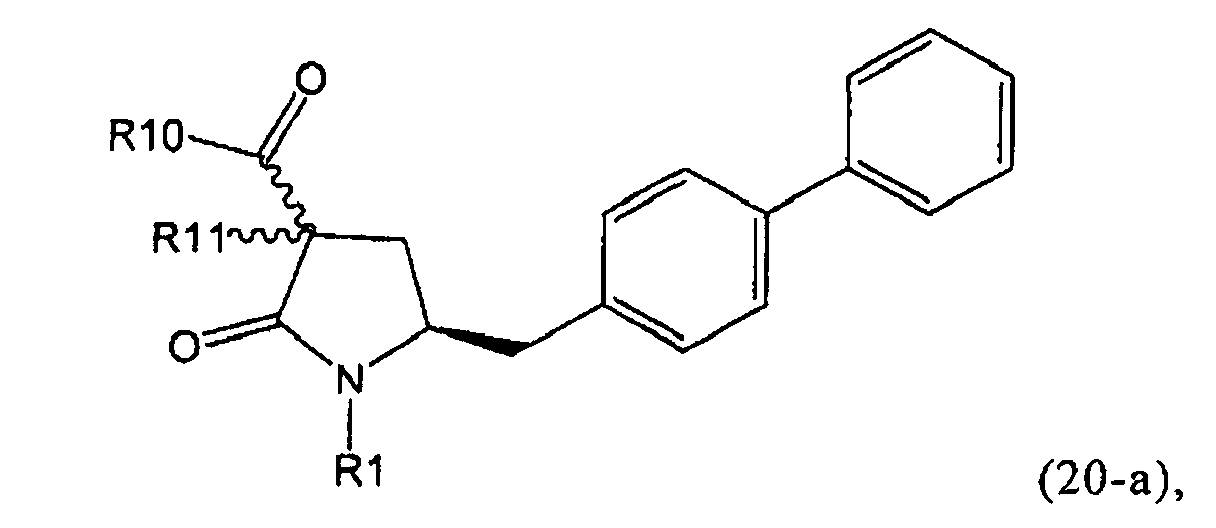
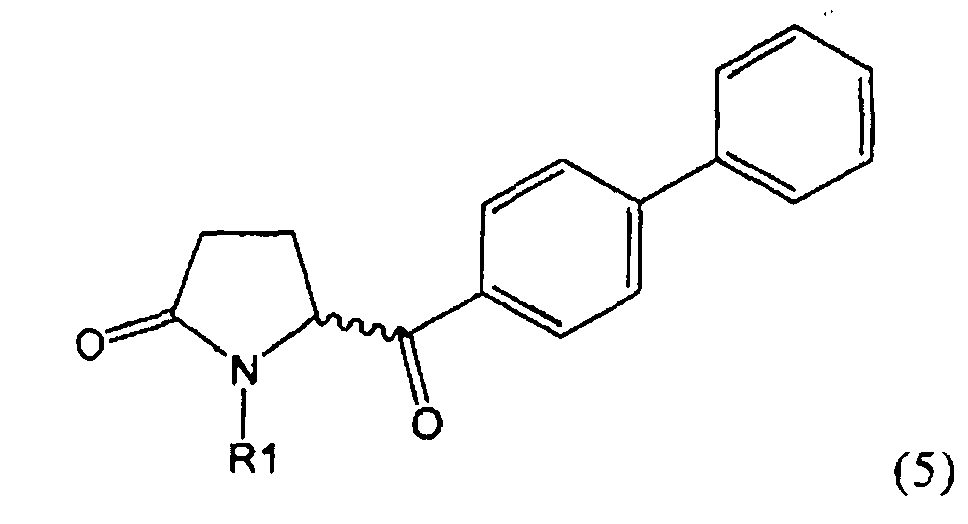
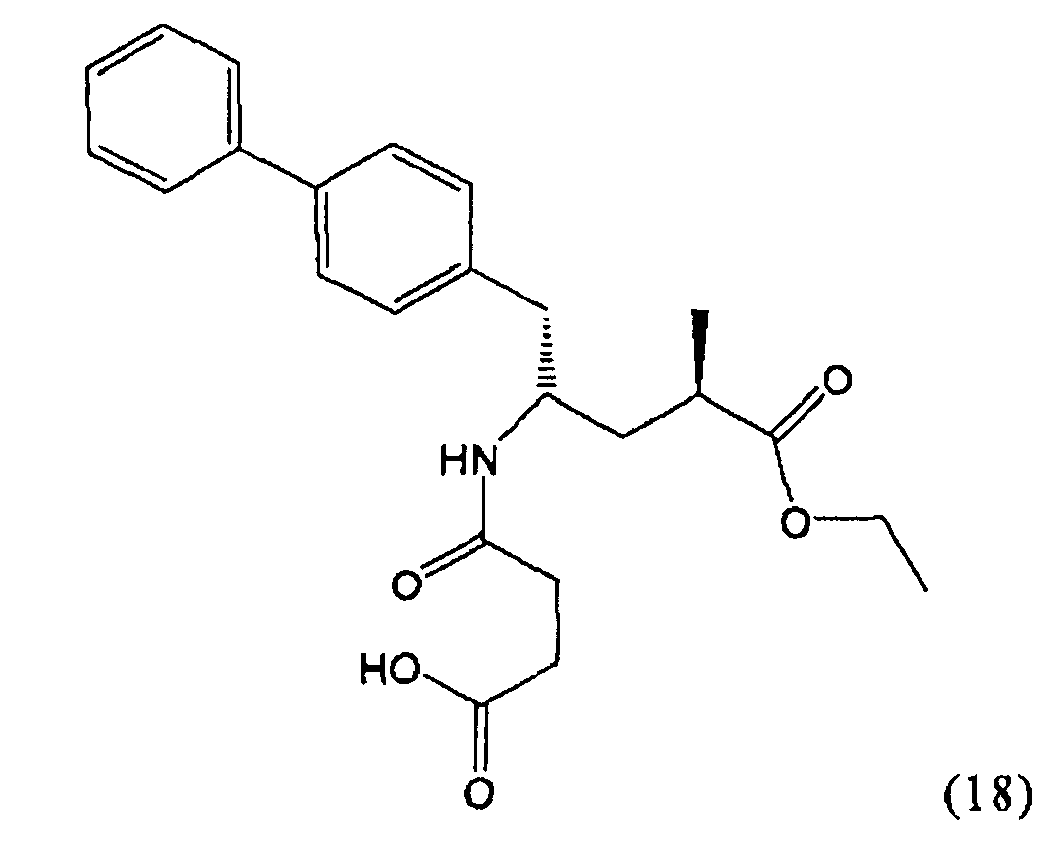
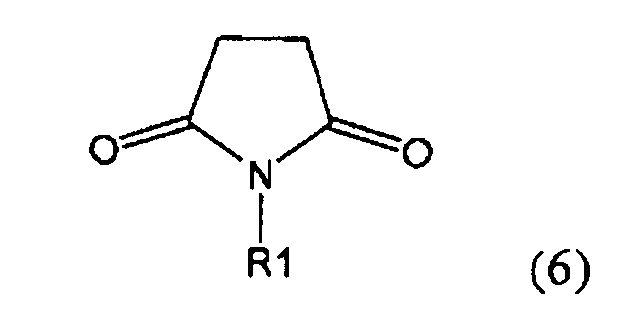
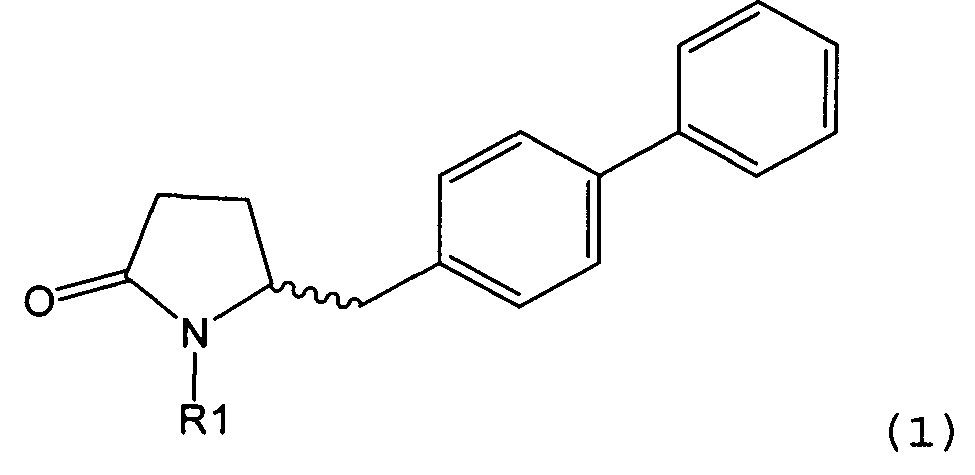
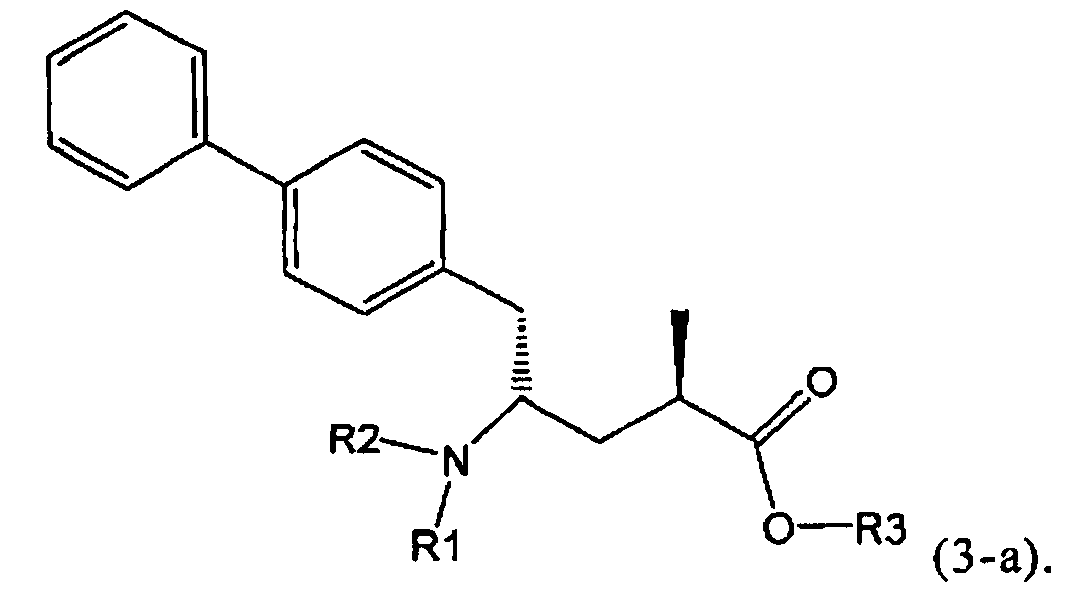
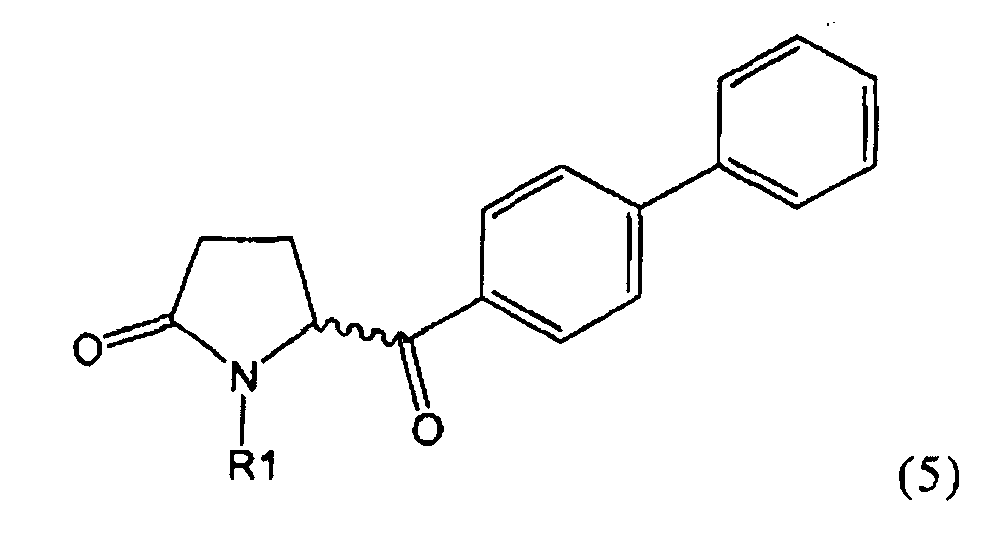
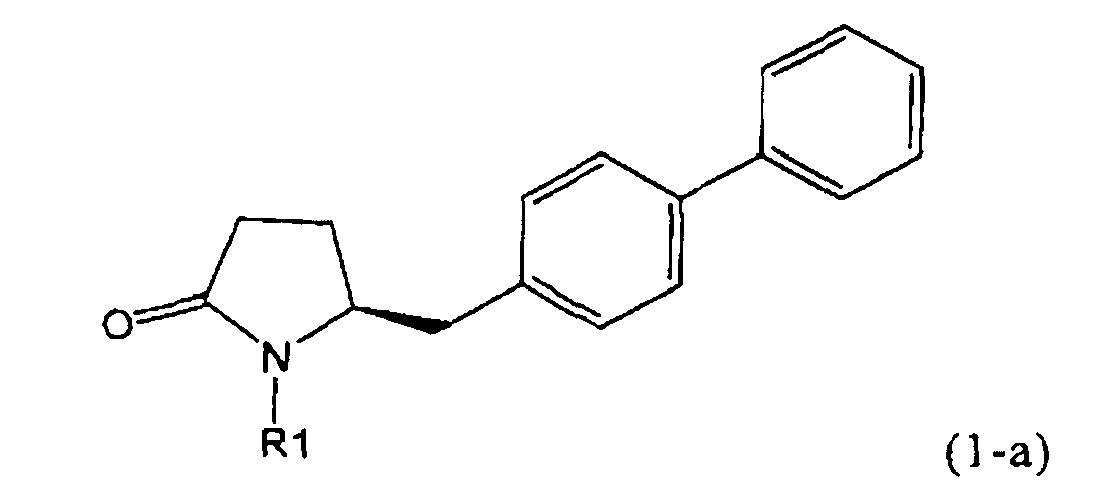
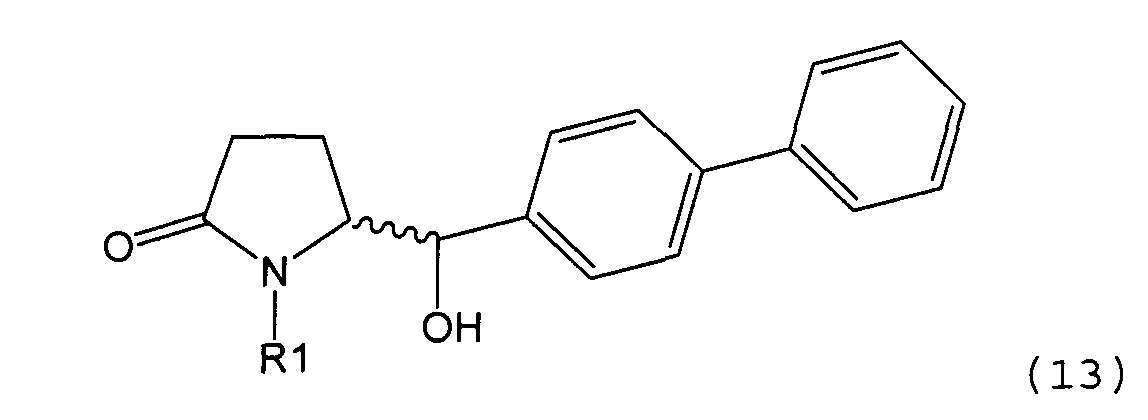
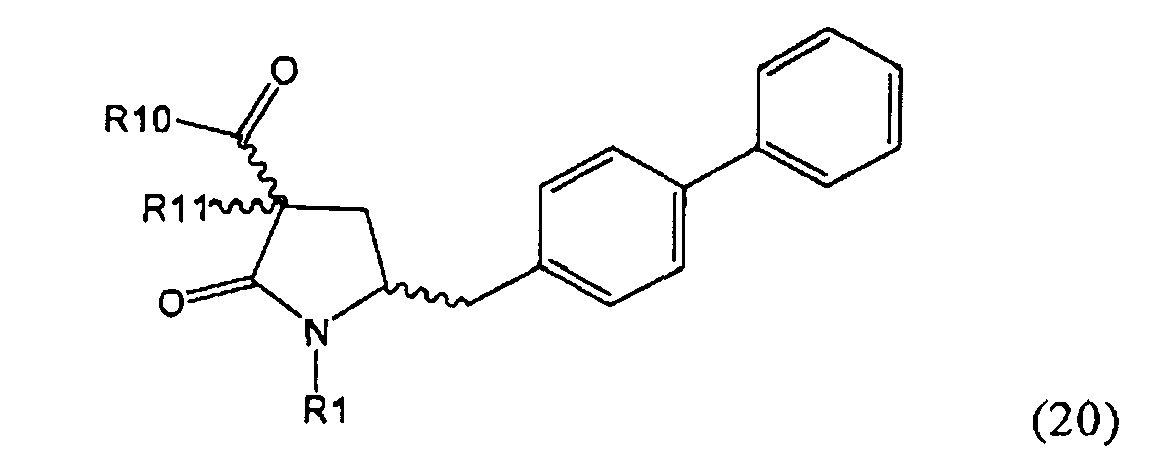
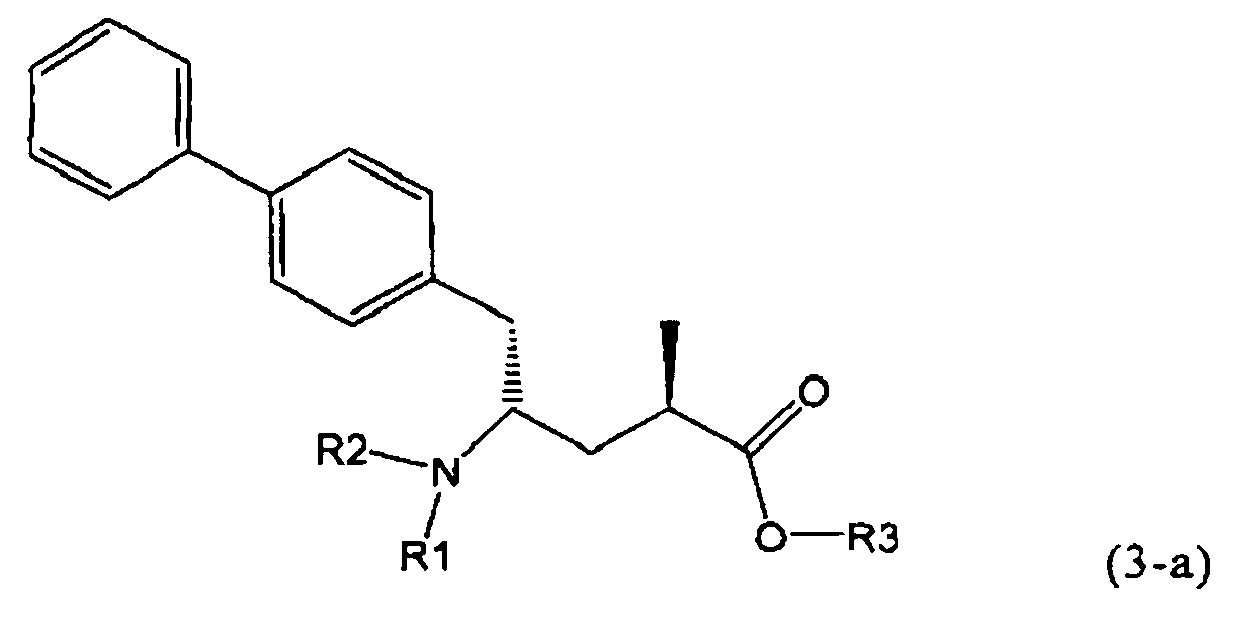
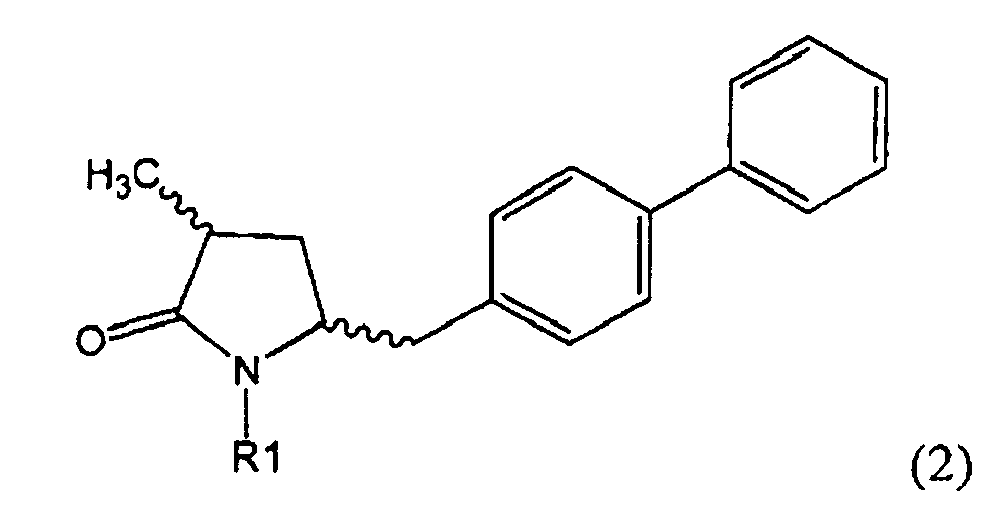
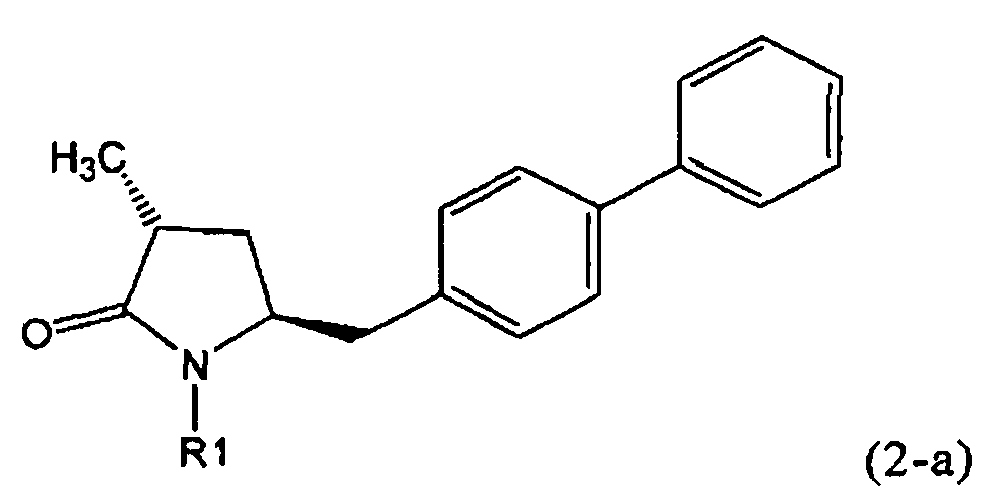
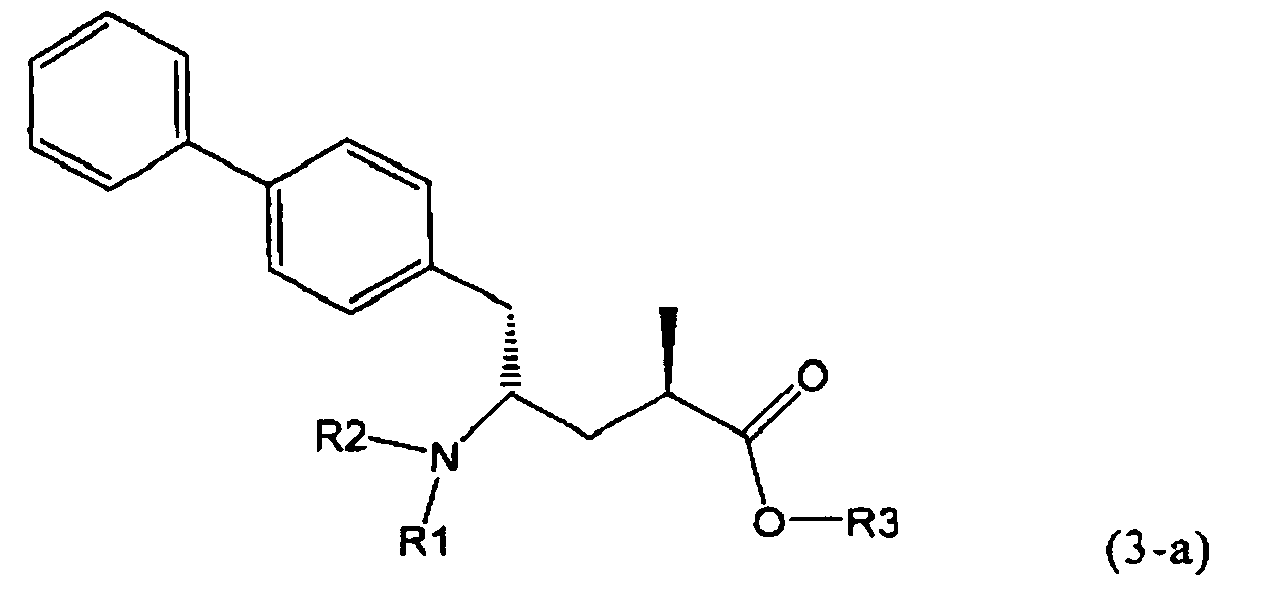
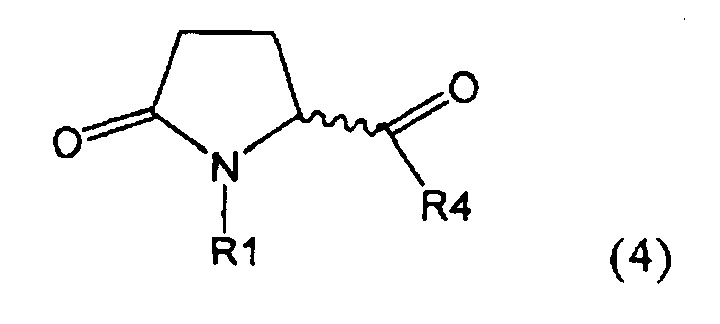
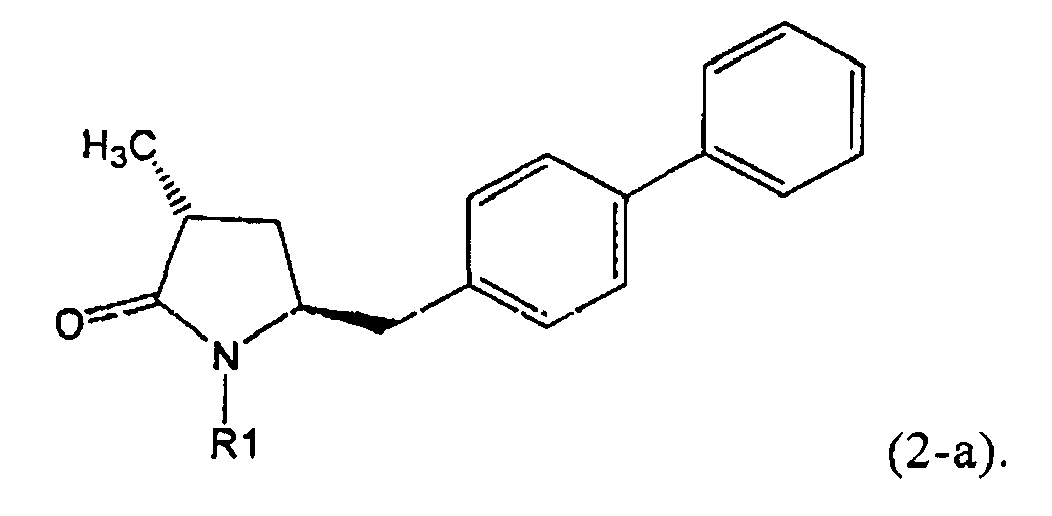
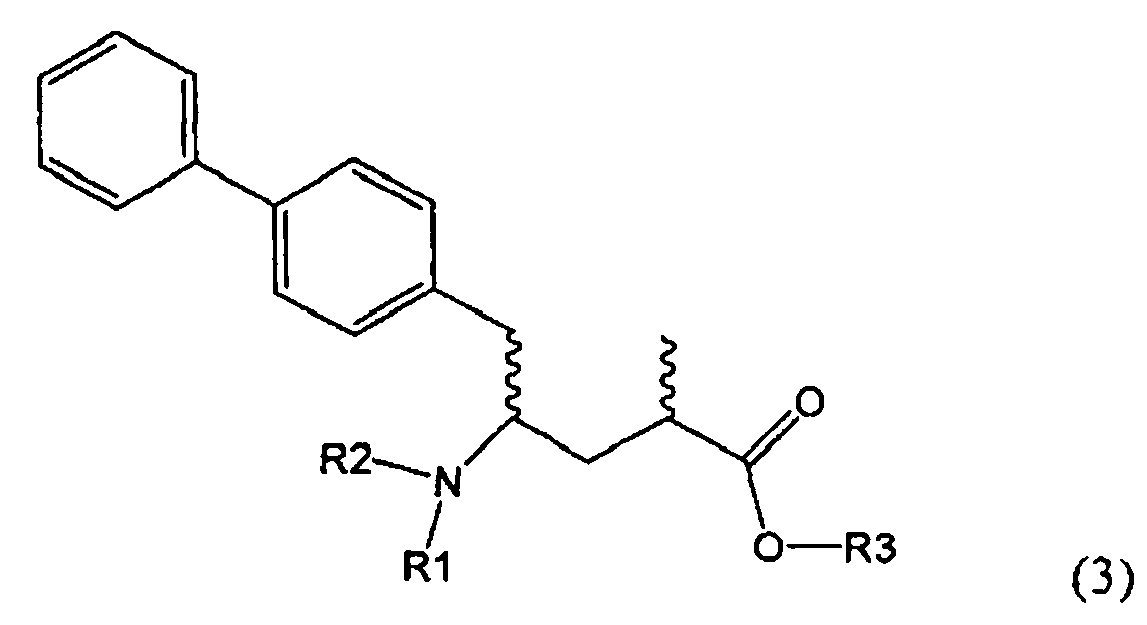
1н-хиназолин-2,4-дионы
Соли 4-метил-n-[3-(4-метилимидазол-1-ил)-5-трифторметилфенил]-3-(4-пиридин-3-илпиримидин-2-иламино)бензамида
Способ и промежуточные соединения для получения производных 5-бифенил-4-ил-2-метилпентановой кислоты
Улучшенные молекулы, связывающиеся с nogo-а и их фармацевтическое применение
Способы получения метилового эфира 4-оксооктагидроиндол-1-карбоновой кислоты и ее производные
Способ обработки упаковки с однократной дозой лекартвенного препарата
Гемифумаратная соль 1-[4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил] азетидин-3-карбоновой кислоты
Органические соединения
Биомаркеры и биомаркерные признаки почечной сохранности, обладающие предсказательной силой, для использования в мониторинге состояния почек
Применение антагонистов smoothened для лечения связанных с путем hedgehog нарушений
Композиция для местного введения
Внутрилегочное введение флуорохинолона
Кристаллические формы и две сольватные формы солей молочной кислоты 4-амино-5-фтор-3-[5-(4-метилпиперазин-1-ил)-1 - н-бензимидазол-2-ил]хинолин-2(1н)она
Средство борьбы с миопией
Использование модулятора рецептора s1p
Набор для получения иммуногенной композиции против neisseria meningitidis серологической группы в
Мутантные формы стрептолизина о
Способ изготовления силиконовых гидрогелевых контактных линз
Таблетируемые жевательные резинки
Органические соединения

